WO2013191221A1 - リン酸カルシウム多孔体の製造方法、および当該製造方法で得られたリン酸カルシウム多孔体 - Google Patents
リン酸カルシウム多孔体の製造方法、および当該製造方法で得られたリン酸カルシウム多孔体 Download PDFInfo
- Publication number
- WO2013191221A1 WO2013191221A1 PCT/JP2013/066883 JP2013066883W WO2013191221A1 WO 2013191221 A1 WO2013191221 A1 WO 2013191221A1 JP 2013066883 W JP2013066883 W JP 2013066883W WO 2013191221 A1 WO2013191221 A1 WO 2013191221A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydroxyapatite
- calcium phosphate
- porous body
- fiber
- porous
- Prior art date
Links
Images










Classifications
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F9/00—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments
- D01F9/08—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments of inorganic material
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/32—Phosphates of magnesium, calcium, strontium, or barium
- C01B25/327—After-treatment
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/32—Phosphates of magnesium, calcium, strontium, or barium
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/32—Phosphates of magnesium, calcium, strontium, or barium
- C01B25/321—Methods for converting an alkaline earth metal ortho-phosphate into another ortho-phosphate
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01D—MECHANICAL METHODS OR APPARATUS IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS
- D01D5/00—Formation of filaments, threads, or the like
- D01D5/0007—Electro-spinning
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01D—MECHANICAL METHODS OR APPARATUS IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS
- D01D5/00—Formation of filaments, threads, or the like
- D01D5/0007—Electro-spinning
- D01D5/0015—Electro-spinning characterised by the initial state of the material
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01D—MECHANICAL METHODS OR APPARATUS IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS
- D01D5/00—Formation of filaments, threads, or the like
- D01D5/0007—Electro-spinning
- D01D5/0015—Electro-spinning characterised by the initial state of the material
- D01D5/0023—Electro-spinning characterised by the initial state of the material the material being a polymer melt
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01D—MECHANICAL METHODS OR APPARATUS IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS
- D01D5/00—Formation of filaments, threads, or the like
- D01D5/0007—Electro-spinning
- D01D5/0015—Electro-spinning characterised by the initial state of the material
- D01D5/003—Electro-spinning characterised by the initial state of the material the material being a polymer solution or dispersion
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01D—MECHANICAL METHODS OR APPARATUS IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS
- D01D5/00—Formation of filaments, threads, or the like
- D01D5/0007—Electro-spinning
- D01D5/0015—Electro-spinning characterised by the initial state of the material
- D01D5/003—Electro-spinning characterised by the initial state of the material the material being a polymer solution or dispersion
- D01D5/0038—Electro-spinning characterised by the initial state of the material the material being a polymer solution or dispersion the fibre formed by solvent evaporation, i.e. dry electro-spinning
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01D—MECHANICAL METHODS OR APPARATUS IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS
- D01D5/00—Formation of filaments, threads, or the like
- D01D5/0007—Electro-spinning
- D01D5/0015—Electro-spinning characterised by the initial state of the material
- D01D5/003—Electro-spinning characterised by the initial state of the material the material being a polymer solution or dispersion
- D01D5/0046—Electro-spinning characterised by the initial state of the material the material being a polymer solution or dispersion the fibre formed by coagulation, i.e. wet electro-spinning
-
- D—TEXTILES; PAPER
- D10—INDEXING SCHEME ASSOCIATED WITH SUBLASSES OF SECTION D, RELATING TO TEXTILES
- D10B—INDEXING SCHEME ASSOCIATED WITH SUBLASSES OF SECTION D, RELATING TO TEXTILES
- D10B2101/00—Inorganic fibres
- D10B2101/10—Inorganic fibres based on non-oxides other than metals
Definitions
- the present invention relates to a method for producing a calcium phosphate porous body, and a calcium phosphate porous body obtained by the production method.
- Hydroxyapatite a kind of calcium phosphate, is a ceramic that is the main component of teeth and bones, and has excellent biocompatibility, protein adsorption characteristics, catalytic activity, etc., so biomaterials such as artificial bones and bone prosthetic materials, It is used as a packing material for chromatography and a catalyst for synthesis of polymer alcohol and the like.
- a porous hydroxyapatite material is more suitable from the viewpoint of biocompatibility, adsorption characteristics, reaction characteristics, and the like.
- hydroxyapatite when hydroxyapatite is used as a biomaterial, it is possible to allow the living tissue to enter the pores and to be easily bonded to the living tissue by making it porous.
- a specific surface area becomes large and it becomes possible to improve a separation characteristic and a reaction characteristic.
- the obtained porous body when used as an artificial bone, blood flow can be circulated, and when a new bone is formed inside or used as a chromatographic filler, the sample can be easily passed.
- the bubbles formed in the slurry are not sufficiently connected to each other, and the proportion formed as independent holes in the porous body is high, and the communication holes are not formed. There is a problem that the rate of formation is low.
- it is difficult to control the bubbles there is a problem that it is difficult to control the uniform pore diameter and porosity.
- the inventors of the present invention have intensively studied to solve the above problems.
- calcium phosphate is formed into a fibrous form by electrospinning to form a fibrous structure precursor (hereinafter sometimes referred to as a “fiber assembly”), so that it communicates, in particular, communicates and is uniform.
- the inventors have found that a calcium phosphate porous material having fine pores can be suitably produced, and based on this finding, the present invention has been completed.
- the present invention has the following configuration.
- the scanning electron micrograph which shows the uniformity of the hole of a hydroxyapatite porous body, and the state of communication.
- a scanning electron micrograph showing the state of aggregation and integration of hydroxyapatite fine particles in a hydroxyapatite porous body.
- the scanning electron micrograph which shows the uniformity of the hole of a hydroxyapatite porous body, and the state of communication.
- a scanning electron micrograph showing the state of aggregation and integration of hydroxyapatite fine particles in a hydroxyapatite porous body.
- the method for producing a porous calcium phosphate of the present invention is a method based on a method of electrostatic spinning calcium phosphate.
- the resulting calcium phosphate porous body is referred to as a hydroxyapatite porous body.
- the resulting calcium phosphate porous body is referred to as a tricalcium phosphate porous body.
- calcium phosphate used in the present invention examples include calcium hydrogen phosphate, tricalcium phosphate, and hydroxyapatite. Of these, tricalcium phosphate and hydroxyapatite are preferable from the viewpoint of biocompatibility. In addition, tricalcium phosphate here is used in the meaning containing a tricalcium phosphate precursor.
- hydroxyapatite used for the production of the porous hydroxyapatite (hereinafter sometimes referred to as “hydroxyapatite used for the production of the present invention”) is obtained by reacting, for example, an aqueous calcium ion solution and an aqueous phosphate ion solution. Can be manufactured. Moreover, what is marketed as SHAp (baked hydroxyapatite nanoparticle, the product made from Sofcera) etc. can also be used.
- the hydroxyapatite used in the production of the present invention may be low crystalline or highly crystalline. Low crystallinity is preferable in that the hydroxyapatite aggregates and integrates in the fiber, and the strength of the hydroxyapatite porous body can be improved.
- the degree of crystallinity of this hydroxyapatite can be measured by an X-ray diffraction method, and the crystallinity becomes lower as the full width at half maximum of the peak indicating each crystal plane increases.
- Such a low crystalline hydroxyapatite can be easily produced, for example, by a wet method.
- a wet method for example, an aqueous calcium nitrate solution and an aqueous ammonium phosphate solution are mixed under alkaline conditions, and the temperature of the reaction solution is set to 0 to 100 ° C., the pH is set to 7 to 14, and the reaction time is set to a range of 3 to 48 hours.
- the crystallinity within a desired range.
- the hydroxyapatite used in the present invention has high crystallinity, the strength of the hydroxyapatite porous body is reduced, but a fine irregular shape is formed on the fiber surface, so that the specific surface area can be improved.
- Such highly crystalline hydroxyapatite can be obtained, for example, by firing low crystalline hydroxyapatite synthesized by a wet method. In firing, for example, the crystallinity can be controlled within a desired range by setting the firing temperature within a range of 100 to 1800 ° C. and appropriately setting the firing time based on the hardness of the desired ceramic particles. Is possible.
- an anti-fusing agent may be added and fired for the purpose of preventing fusion between the hydroxyapatite primary particles.
- anti-fusing agents include calcium chloride, calcium oxide, calcium sulfate, calcium nitrate, calcium carbonate, calcium hydroxide, calcium acetate, calcium citrate, and other inorganic compounds, potassium chloride, potassium oxide, sulfuric acid Inorganic compounds containing potassium such as potassium, potassium nitrate, potassium carbonate, potassium hydroxide, potassium phosphate, inorganic compounds containing sodium such as sodium chloride, sodium oxide, sodium sulfate, sodium nitrate, sodium carbonate, sodium hydroxide, sodium phosphate Compound etc. are mentioned.
- the amount used is preferably 1 g or more and 1000 g or less, more preferably 5 g or more and 800 g or less, and most preferably 10 g or more and 500 g or less with respect to 100 g of hydroxyapatite primary particles.
- a polymer compound having any of a carboxyl group, a sulfonic acid group, a phosphoric acid group, or an amino group in the side chain is used as an anti-fusing aid.
- the anti-fusing aid for example, polyacrylic acid, polymethacrylic acid, polyglutamic acid, polyethylene sulfonic acid, polymethacrylic acid alkylsulfonic acid ester, polyacryloylaminomethylphosphonic acid, polypeptide and the like can be used.
- Acrylic acid for example, polyacrylic acid, polymethacrylic acid, polyglutamic acid, polyethylene sulfonic acid, polymethacrylic acid alkylsulfonic acid ester, polyacryloylaminomethylphosphonic acid, polypeptide and the like can be used.
- Acrylic acid for example, polyacrylic acid, polymethacrylic acid, polyglutamic acid, polyethylene sulfonic acid, polymethacrylic acid alkylsulfonic acid ester, polyacryl
- the amount used is preferably 1 g or more and 1000 g or less, more preferably 5 g or more and 800 g or less, and most preferably 10 g or more and 500 g or less with respect to 100 g of hydroxyapatite primary particles.
- the shape of hydroxyapatite used in the production of the present invention is not particularly limited.
- it may be in the form of particles, rods, fibers, or an amorphous polycrystal. From the standpoint of obtaining a homogeneous fiber by closely packing hydroxyapatite in the fiber, particulate hydroxyapatite is preferable.
- the particle size is preferably less than 200 nm, and more preferably less than 50 nm. By setting the size within the above range, the packing density of the particles is further increased, and the hydroxyapatite is easily aggregated and integrated in the obtained fiber, whereby the strength is improved.
- Dispersion medium There are various modes of the spinning solution to be spun, but it is preferable that hydroxyapatite is dispersed in a dispersion medium to form a spinning solution.
- the dispersion medium to be used is not particularly limited as long as hydroxyapatite can be uniformly dispersed.
- a polar solvent from the viewpoint of dispersibility of hydroxyapatite, it is preferable to use a polar solvent, and it is more preferable to use, for example, water, methanol, ethanol, propanol, acetone or N, N-dimethylformamide as a dispersion. .
- the concentration of hydroxyapatite with respect to the dispersion medium is preferably 30% by weight or less, and more preferably 15% by weight or less in order to disperse the hydroxyapatite.
- concentration is 30% by weight or less, hydroxyapatite hardly aggregates in the dispersion medium, and fiberization becomes easy.
- a fiber-forming polymer may be further contained in the dispersion containing hydroxyapatite and the dispersion medium for the purpose of improving the spinnability.
- the fiber-forming polymer is only required to exhibit the action of promoting the fiberization of hydroxyapatite, and is selected from those that can be dissolved in the dispersion medium and decomposed by firing.
- fiber-forming polymers examples include polyvinyl alcohol, polyethylene glycol, polyethylene oxide, polyvinyl pyrrolidone, polyacrylic acid, sodium polyacrylate, polymethacrylic acid, polysodium methacrylate, polyacrylamide, polymethacrylamide, polyvinyl acetate, Polyethylene, polypropylene, polyethylene terephthalate, polylactic acid, polyamide, polyurethane, polystyrene, polyvinylidene fluoride, polyacrylonitrile, polymethyl methacrylate, polyglycolic acid, polycaprolactone, cellulose, cellulose derivatives, chitin, chitosan, collagen, gelatin and these A copolymer etc. can be mentioned. These fiber-forming polymers may be used alone or in combination of two or more.
- the mixing ratio in the case of mixing and using is not particularly limited, and can be appropriately set in view of the required spinnability and dispersibility and the physical properties of the obtained fiber.
- a fiber-forming polymer having a functional group having a high coordination property of hydroxyapatite in addition to improving the spinnability and more highly dispersing hydroxyapatite.
- hydroxyapatite coordination functional groups include amino groups, alkoxysilyl groups, ether groups, hydroxyl groups, carbonyl groups, carboxyl groups, phosphate groups, sulfonyl groups, and the like.
- polyvinyl pyrrolidone polyethylene glycol, polyethylene oxide, polyvinyl alcohol, polyacrylic acid, polyacrylamide, polyvinyl acetate, polyethylene terephthalate, polylactic acid, polyglycolic acid, polycaprolactone, polymethyl methacrylate, polyurethane, polyamide , Cellulose, cellulose derivatives, collagen, gelatin and the like are preferably used.
- the hydroxyapatite / fiber-forming polymer (weight ratio) is 0.3 or more, the shrinkage of the fiber after firing is small, and it is easy for hydroxyapatite to maintain the fiber shape and to form uniform pores. Therefore, it is preferable.
- it is 1.0 or more, the shrinkage of the fiber during firing is particularly suppressed, and processing into a desired shape is facilitated, which is more preferable.
- a surfactant may be further contained in the dispersion.
- Surfactants can be used within a range that does not inhibit the effect of hydroxyapatite.
- general ionic surfactants such as sodium dodecyl sulfate, tetrabutylammonium bromide, tetrabutylammonium chloride, polyoxyethylene sorbitan, etc.
- Common nonionic surfactants such as monolaurate can be mentioned.
- the surfactant does not contain metal ions from the viewpoint of obtaining a high-purity hydroxyapatite porous body. From this viewpoint, tetrabutylammonium bromide, tetrabutylammonium chloride, polyoxyethylene sorbitan mono Examples include laurate.
- the surfactant is preferably dissolved uniformly in the dispersion medium.
- the concentration of the surfactant is appropriately set depending on the dispersion medium to be used, the type of the fiber-forming polymer, and the like, and is not particularly limited, but is preferably in the range of 30% by weight or less with respect to hydroxyapatite. More preferably, it is in the range of% or less.
- concentration of the surfactant is 30% by weight or less, it is preferable because an effect suitable for use is obtained or the influence on the composition of the hydroxyapatite porous body is reduced.
- components other than those described above may also be included as components of the dispersion.
- the method for preparing the hydroxyapatite dispersion is not particularly limited. Examples of the preparation method include methods such as stirring and ultrasonic treatment. Furthermore, it is possible to obtain a highly uniform dispersion by using a dispersing machine such as a bead mill. Also, the order of mixing is not particularly limited, and may be mixed simultaneously or sequentially. In order to obtain a fibrous hydroxyapatite, the viscosity of the dispersion is preferably adjusted in the range of 10 to 10,000 cP, more preferably in the range of 100 to 8,000 cP.
- the viscosity is 10 cP or more, spinnability for forming fibers is obtained, and when the viscosity is 10,000 cP or less, it is easy to discharge the dispersion.
- a viscosity in the range of 100 to 8,000 cP is more preferable because good spinnability can be obtained over a wide range of spinning conditions.
- the viscosity of the dispersion can be adjusted by appropriately changing the hydroxyapatite concentration or the molecular weight and concentration of the fiber-forming polymer.
- the electrostatic spinning method is a method in which a spinning solution is discharged and an electric field is applied to fiberize the discharged spinning solution to obtain fibers on a collector.
- a method of spinning by spinning the spinning solution from a nozzle and applying an electric field a method of spinning by spinning the spinning solution and applying an electric field, and spinning by directing the spinning solution to the surface of a cylindrical electrode and applying an electric field. And the like. According to this method, uniform fibers having a diameter of 10 nm to 10 ⁇ m can be obtained.
- the spinning solution is not particularly limited as long as it has spinnability, but the fiber forming material is dispersed in a dispersion medium, the fiber forming material is dissolved in a solvent, or the fiber forming material is heated or heated. What was melt
- the hydroxyapatite used in the production of the present invention is preferably dispersed in a dispersion medium and electrospun.
- the spinning solution can be spun at normal temperature, or heated and cooled for spinning. Examples of the method of discharging the spinning solution include a method of discharging the spinning solution filled in the syringe using a pump from a nozzle.
- the inner diameter of the nozzle is not particularly limited, but is preferably in the range of 0.1 to 1.5 mm.
- the discharge amount is not particularly limited, but is preferably 0.1 to 10 ml / hr.
- the method for applying an electric field is not particularly limited as long as an electric field can be formed in the nozzle and the collector.
- a high voltage may be applied to the nozzle and the collector may be grounded.
- the voltage to be applied is not particularly limited as long as fibers are formed, but is preferably in the range of 5 to 50 kV.
- the distance between the nozzle and the collector is not particularly limited as long as fibers are formed, but is preferably in the range of 5 to 30 cm.
- the collector is not particularly limited as long as it can collect the spun fibers, and the material and shape thereof are not particularly limited.
- As the collector material a conductive material such as metal is preferably used.
- the shape of the collector is not particularly limited, and examples thereof include a flat plate shape, a shaft shape, and a conveyor shape. When the collector is flat, the fiber aggregate can be collected in a sheet form, and when the collector is a shaft, the fiber aggregate can be collected in a tube form. If it is a conveyor form, the fiber assembly collected by the sheet
- the fiber assembly may be collected in a collector installed between the nozzle and the collector.
- the collector preferably has a volume resistivity of 10 10 ⁇ ⁇ cm or less, and more preferably 10 8 ⁇ ⁇ cm or less.
- a material having a volume resistivity exceeding 10 10 ⁇ ⁇ cm can be suitably used by using it together with a device for eliminating charges, such as an ionizer.
- a fiber assembly can be collected according to the shape of the collector.
- a liquid such as water or alcohol as the collector.
- ⁇ Baking method> The electrospun fiber assembly is fired.
- the bond between the hydroxyapatites in the fiber assembly can be strengthened and the crystallinity can be improved.
- a fiber-forming polymer, a surfactant, or the like these can be thermally decomposed to obtain a high-purity hydroxyapatite porous body.
- the firing method is not particularly limited, but can be performed in an air atmosphere. By firing in air, it is possible to reduce the residue of components other than hydroxyapatite, such as fiber-forming polymers and surfactants.
- the firing temperature is preferably 500 ° C. or higher, more preferably in the range of 600 to 1500 ° C., further preferably in the range of 800 to 1300 ° C., and particularly preferably in the range of 1000 ° C. to 1300 ° C.
- firing temperature is 500 ° C. or higher, firing is sufficient, bonding between hydroxyapatites becomes strong, and components other than hydroxyapatite hardly remain. Hydroxyapatite is decomposed into tricalcium phosphate or the like when the temperature is 1300 ° C. or higher.
- a porous body having such a composition can also be used as an implant material having high bioabsorbability.
- Examples of the tricalcium phosphate that may be contained in the produced hydroxyapatite porous body include ⁇ -tricalcium phosphate and ⁇ -tricalcium phosphate.
- ⁇ -tricalcium phosphate When calcined at 1500 ° C. for 5 hours, it is possible to obtain a hydroxyapatite porous material containing about 30% by weight of ⁇ -tricalcium phosphate in the porous material.
- ⁇ -The content of tricalcium phosphate can be arbitrarily changed.
- the firing temperature is in the range of 800 to 1300 ° C., hydroxyapatite having high crystallinity and high purity can be produced.
- the resulting hydroxyapatite porous body has low crystallinity, but such fibers are also suitable for applications such as bone fillers and early-dissolving cell carriers. You may be able to use it.
- the firing time is not particularly limited, but may be fired for 1 to 24 hours, for example.
- the temperature raising rate is not particularly limited, but can be appropriately changed within the range of 5 to 50 ° C./min.
- the “firing time” herein refers to a time for holding at a preset firing temperature, and does not include a temperature raising time until the firing temperature is reached. In the present invention, it is not excluded to adopt a firing method in which the preset firing temperature is changed in multiple stages, but in this case, the firing time including the time required to change the firing temperature is referred to.
- the pre-baking temperature may be a temperature at which components other than hydroxyapatite do not remain. For example, it is preferably in the range of 300 to 700 ° C., more preferably in the range of 400 to 650 ° C.
- the pre-baking time is not particularly limited as long as components other than hydroxyapatite disappear sufficiently, and examples thereof include 0 to 24 hours.
- the pre-baking time of 0 hour means a direct transition from the pre-baking temperature rising process to the baking temperature rising process.
- the pre-baking temperature is preferably 50 ° C. or higher and lower than 100 ° C. lower than the baking temperature performed thereafter. More preferably.
- the pre-firing step and the calcining step performed before firing may be performed continuously or discontinuously, that is, a time may be provided between the two steps. It is a method to do in. Note that the definition of “pre-baking time” here is substantially the same as the definition of “baking time” described above.
- Hydroxyapatite porous bodies having various shapes can be obtained by forming a fiber assembly obtained by electrostatic spinning into an arbitrary shape and firing it. For example, by forming and firing a fiber assembly into a two-dimensional sheet, a sheet-like hydroxyapatite porous body can be obtained, and by collecting the fiber assembly by winding it around a shaft, A hydroxyapatite porous body can be obtained. It is also possible to obtain a cotton-like hydroxyapatite porous body by collecting fiber aggregates in a liquid, freeze-drying, forming into a cotton-like shape, and firing.
- the strength of the hydroxyapatite porous body is not particularly limited, and as described above, the crystallinity, shape, and size of the hydroxyapatite used in the production of the present invention, the weight ratio of hydroxyapatite / fiber-forming polymer, firing conditions, and It can be adjusted as appropriate by the molding method described above.
- the content of components other than hydroxyapatite in the hydroxyapatite porous body obtained by firing is not particularly limited, but it is generally preferable that the content of hydroxyapatite is sufficiently derived, and it is preferably 50% by weight or less. It is preferable that On the other hand, it can be combined with another functional material from the viewpoint of adding characteristics other than the function of hydroxyapatite.
- the functional material include ⁇ -TCP ( ⁇ -tricalcium phosphate), and the content is preferably in the range of 10 to 90% by weight.
- a sample with a known ratio of ⁇ -tricalcium phosphate to hydroxyapatite is prepared, and a calibration curve function is obtained in advance by X-ray diffraction measurement or the like for each sample. Based on this calibration curve function, the content of ⁇ -tricalcium phosphate in hydroxyapatite can be determined from X-ray diffraction measurement of the hydroxyapatite porous material.
- a calcium phosphate porous body can be manufactured easily. It is particularly suitable for producing a hydroxyapatite porous body or a tricalcium phosphate porous body. Below, although the example of a hydroxyapatite porous body is demonstrated, a tricalcium phosphate porous body can be demonstrated similarly. Further, the present invention is not limited to this.
- the fiber diameter of the hydroxyapatite fiber constituting the hydroxyapatite porous body obtained by firing is not particularly limited, and can be appropriately selected according to the required characteristics and applications of the hydroxyapatite porous body. For example, by controlling the fiber diameter, the hole diameter and mechanical characteristics can be controlled.
- the method for controlling the fiber diameter is not particularly limited, and examples thereof include the type of dispersion medium, the concentration of hydroxyapatite in the dispersion medium, the viscosity of the dispersion, and the electrospinning conditions. By appropriately changing these, It is possible to control the fiber diameter.
- the fiber length of the hydroxyapatite fiber constituting the hydroxyapatite porous body obtained by firing is not particularly limited, but it is preferably a continuous fiber from the viewpoint of obtaining excellent mechanical strength.
- the calcium phosphate porous body of the present invention has continuous and uniform fine pores because the calcium phosphate porous body obtained by forming calcium phosphate into fibers by electrostatic spinning to form a fiber aggregate and firing is used for the fibers.
- the holes to be formed communicate with each other between any surfaces of the porous body, for example, the upper and lower surfaces and the left and right surfaces, and the size of the formed holes is 500 ⁇ m or less, further 200 ⁇ m or less, particularly 50 ⁇ m.
- the CV value of the size is 50% or less. In the case of the present invention, it can be said that the pores communicate with each other if the porosity of the porous body is 30% or more.
- the porosity of the hydroxyapatite porous body obtained by firing is not particularly limited, and can be appropriately selected according to required mechanical properties and applications. When the porosity increases, the mechanical strength tends to decrease. For example, when used as an artificial bone, a bone grafting material, a chromatography packing, or the like, a relatively high porosity is preferable.
- the porosity is preferably in the range of 30 to 95%, more preferably in the range of 50 to 95%, and still more preferably in the range of 80 to 95%.
- the hydroxyapatite fibers constituting the hydroxyapatite porous body obtained by firing may be arranged randomly or may be arranged one-dimensionally or two-dimensionally. When arranged randomly, characteristics such as mechanical strength are isotropically expressed, and when arranged one-dimensionally or two-dimensionally, various characteristics are anisotropically expressed.
- the surface property of the hydroxyapatite fiber constituting the hydroxyapatite porous body obtained by firing is not particularly limited, and may be a smooth structure or an uneven structure.
- hydroxyapatite is preferable in that it aggregates and integrates at a high density and the mechanical strength is high, and in the case of a concavo-convex structure, the mechanical strength is low but is preferable in terms of a larger specific surface area. .
- the calcium phosphate porous material of the present invention has communication, in particular, communication and uniform fine pores, it can be suitably used for various applications.
- cell culture substrates used in cell culture petri dishes biomaterials such as artificial bones for bones and bone and tooth filling materials, chromatographic packing materials with protein adsorption properties, catalysts for polymer alcohol synthesis , Catalyst carriers, filter media, electronic materials, physiologically active substance-immobilized carriers and heavy metal adsorbents.
- the calcium phosphate porous body when used as a cell culture substrate, because of the configuration / features of the calcium phosphate porous body of the present invention, or by taking advantage of the configuration / features, the calcium phosphate porous body may be used alone,
- the calcium phosphate porous body may be combined with other materials and used as a composite material.
- the composite material include a laminate of a metal plate and a calcium phosphate porous body obtained by electrostatic spinning of a fiber assembly on a metal plate and firing the laminate. By using the laminate, an improvement in handling properties can be expected.
- the laminated body enables three-dimensional culture of cells because of the structural feature of the porous body.
- the type of the metal plate is not particularly limited, but for example, a titanium plate can be suitably used in consideration of heat resistance and compatibility with a living body.
- the calcium phosphate porous body of the present invention is obtained by firing an assembly of fibers obtained by electrostatic spinning and collecting in a cotton shape, Since it has flexibility, it can be compensated without cutting according to the shape of the bone defect portion, and an effect of reducing friction between the bone defect portions can be expected.
- a fiber aggregate obtained by electrospinning is baked in a state where it is packed in a cylindrical container, so that a cylindrical calcium phosphate porous material is obtained.
- the body can be manufactured. By using this, the effect of facilitating packing operation of the column or the like can be expected.
- a sheet-like calcium phosphate porous body obtained by firing a sheet-like fiber assembly obtained by electrostatic spinning can be suitably used as a membrane chromatography material.
- the thickness of the membrane can be appropriately adjusted by adjusting the thickness of the fiber assembly sheet before firing or by stacking a plurality of sheet-like calcium phosphate porous bodies obtained by firing.
- a porous granule can be manufactured by grind
- the method for pulverizing the calcium phosphate porous body is not particularly limited, and a general method such as grinding can be employed.
- the size and shape of the granules obtained by pulverization are not particularly limited, and can be appropriately adjusted in consideration of uniform packing properties, packing density, column flow rate, and the like.
- the size (diameter) of the granule is preferably in the range of 5 to 500 ⁇ m, more preferably in the range of 10 to 200 ⁇ m.
- the thus obtained chromatographic material composed of cylindrical, sheet-like, and granular calcium phosphate porous bodies is preferable because it is not compressed or broken even at a high flow rate, and the productivity of separation and purification can be improved.
- the calcium phosphate porous body When using calcium phosphate as a catalyst, the calcium phosphate porous body may be used alone, or the calcium phosphate porous body may be used in combination with other materials.
- the calcium phosphate porous material when used alone, it can be used, for example, as a polymer alcohol synthesis catalyst for synthesizing 1-butanol, hexanol, octanol, decanol and the like from ethanol.
- examples of other materials include transition metals such as ruthenium, palladium, vanadium, silver, and titanium, and these can be exchanged on the surface of the calcium phosphate porous material.
- it by carrying it by adsorption or the like, it can be used as an oxidation reaction catalyst or photocatalyst for alcohol, amine, silane and the like.
- the calcium phosphate porous material When used as a fixed carrier for physiologically active substances, biological substances, microorganisms or microorganism-derived substances, the calcium phosphate porous material is used alone because of the constitution / characteristics of the calcium phosphate porous material of the present invention or by utilizing the constitution / characteristics. It may be used, or the calcium phosphate porous material may be combined with other materials and used as a composite material.
- the physiologically active substance to be immobilized on the carrier include an enzyme. When applied to a recoverable enzyme-immobilized catalyst, the catalytic activity is reduced due to a structural change of the enzyme as seen in an ordinary enzyme-immobilized catalyst. Can be expected to be reduced.
- a biological substance-immobilized carrier when used as a biological substance-immobilized carrier, it can be used as a diagnostic carrier (solid phase) for use in immunoassay of clinical diagnostic agents by utilizing the nonspecific adsorption property of the calcium phosphate porous material.
- a target substance for example, virus
- the anti-virus specific antibody is spotted on the solid phase side to capture the virus, but a calcium phosphate porous body is used as the solid phase.
- solid phase for capturing a plurality of target substances non-specifically.
- the target substance is a nucleic acid (DNA, RNA), it can be similarly captured.
- Example 1 ⁇ Synthesis of hydroxyapatite> The low crystalline hydroxyapatite particles were prepared by the wet method shown below. As Ca (NO 3 ) 2 4H 2 O and (NH 4 ) 2 HPO 4 , calcium nitrate tetrahydrate and diammonium hydrogen phosphate manufactured by Nacalai Tesque Co., Ltd. were used. 25% by weight ammonia water manufactured by Yakuhin Kogyo Co., Ltd. was used, and Milli-Q water was used as pure water.
- Ca (NO 3 ) 2 4H 2 O and (NH 4 ) 2 HPO 4 calcium nitrate tetrahydrate and diammonium hydrogen phosphate manufactured by Nacalai Tesque Co., Ltd. were used. 25% by weight ammonia water manufactured by Yakuhin Kogyo Co., Ltd. was used, and Milli-Q water was used as pure water.
- Hydroxyapatite ethanol dispersion 13 wt%) prepared by the above method, 64.2 parts by weight, polyvinylpyrrolidone (Mw: 1,300,000 manufactured by ALDRICH), 8.3 parts by weight, ethanol (primary; manufactured by Nacalai Tesque) 27 A dispersion comprising 5 parts by weight was prepared.
- the obtained dispersion had a hydroxyapatite concentration of 9.1% by weight with respect to ethanol and a hydroxyapatite / polyvinylpyrrolidone (weight ratio) of 1.0.
- the dispersion liquid is supplied at 1.0 ml / hr to a nozzle having an inner diameter of 0.22 mm by a syringe pump, a voltage of 20 kV is applied to the nozzle, and a fibrous structure precursor (fiber assembly) is applied to a grounded collector.
- a hydroxyapatite porous body having an average fiber diameter of 1 ⁇ m is obtained by heating an electrospun fiber assembly to 900 ° C. at a temperature increase rate of 10 ° C./min in the air, holding it for 1 hour, and then cooling to room temperature. Was made. Scanning electron micrographs of the obtained hydroxyapatite porous material are shown in FIGS.
- the hydroxyapatite porous body had continuous and uniform fine pores. Further, from FIG. 2, it was confirmed that the obtained hydroxyapatite porous body was a fiber having high surface smoothness in which hydroxyapatite fine particles were aggregated and integrated. A scanning electron micrograph before firing is shown in FIG.
- Example 2 ⁇ Production of hydroxyapatite> High crystalline hydroxyapatite nanoparticles were produced by firing the low crystalline hydroxyapatite particles obtained above by the following method.
- 0.5 g of 0.5 g of polyacrylic acid (ALDRICH, weight average molecular weight 15,000 g / mol) and pH 7.0 aqueous solution (hereinafter referred to as “Aqueous Solution A”) is 0.5 g.
- Aqueous Solution A aqueous solution
- calcium acrylate was precipitated on the surface of the particles by adding 500 mL of a saturated aqueous solution of calcium hydroxide [Ca (OH) 2 ] as an anti-fusing agent to the dispersion prepared above.
- the resulting precipitate was collected and dried at 80 ° C. under reduced pressure to collect mixed particles.
- the mixed particles were put in a crucible and fired at a firing temperature of 800 ° C. for 1 hour. At this time, calcium polyacrylate was thermally decomposed to calcium oxide [CaO].
- the fired body obtained in 500 mL of the aqueous solution A prepared above is suspended, separated and washed by centrifugation, further suspended in distilled water, and separated and washed by centrifugation in the same manner, whereby the anti-fusing agent and The anti-fusing aid was removed, and highly crystalline hydroxyapatite nanoparticles were recovered.
- a hydroxyapatite porous body having an average fiber diameter of 1.2 ⁇ m was produced in the same manner as in Example 1 using the hydroxyapatite produced by the above method. Scanning electron micrographs of the obtained hydroxyapatite porous body are shown in FIGS. As is clear from FIG. 3, the hydroxyapatite porous body had continuous and uniform fine pores. Further, from FIG. 4, the hydroxyapatite constituting the fiber is higher in crystallinity than the fiber of Example 1, and the particles are large, so that the aggregation and integration of the hydroxyapatite particles are not sufficient, and the fiber surface In Fig. 4, fine irregularities derived from individual fine particles were observed. From such a shape, it was inferred that the fiber specific surface area was higher than that of Example 1, but the strength was low and fragile.
- Example 3 6.9 parts by weight of an ethanol dispersion (13% by weight) of hydroxyapatite of Example 1, 9.0 parts by weight of polyvinylpyrrolidone (Mw: 1,300,000 ALDRICH), ethanol (primary; manufactured by Nacalai Tesque) 84. A dispersion consisting of 1 part by weight was prepared. The obtained dispersion had a hydroxyapatite concentration of 1.0% by weight with respect to ethanol and a hydroxyapatite / polyvinylpyrrolidone (weight ratio) of 0.1. Next, a hydroxyapatite porous body was produced in the same manner as in Example 1. A scanning electron micrograph of the obtained hydroxyapatite porous material is shown in FIG. It was confirmed that the holes communicated as shown in FIG. However, the fiber contracted, and the cut part, that is, the part where the size of the hole exceeded 50 ⁇ m was observed, and the uniformity of the hole was lacking.
- Example 4 45.6 parts by weight of an ethanol dispersion (13% by weight) of hydroxyapatite of Example 1, 8.5 parts by weight of polyvinylpyrrolidone (Mw: 1,300,000 ALDRICH), ethanol (primary; manufactured by Nacalai Tesque) A dispersion composed of 9 parts by weight was prepared. The obtained dispersion had a hydroxyapatite concentration of 6.5% by weight with respect to ethanol and a hydroxyapatite / polyvinylpyrrolidone (weight ratio) of 0.7. Next, a hydroxyapatite porous body was produced in the same manner as in Example 1. Scanning electron micrographs of the obtained hydroxyapatite are shown in FIGS. As apparent from FIG.
- the hydroxyapatite porous body had continuous and uniform fine pores. Further, from FIG. 8, it was confirmed that the obtained hydroxyapatite porous body was a fiber with high surface smoothness, in which hydroxyapatite fine particles were aggregated and integrated. However, although it was at a level that could be used sufficiently, the shrinkage of the porous body before and after firing was larger than that in Example 1.
- Example 5 A hydroxyapatite porous body was produced in the same manner as in Example 4 except that the firing temperature was 600 ° C.
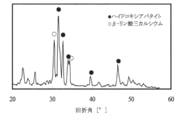
- FIG. 9 shows diffraction patterns obtained by X-ray diffraction measurement of the hydroxyapatite porous material obtained in Example 4 and the hydroxyapatite porous material obtained in Example 5. From FIG. 9, it was confirmed that the hydroxyapatite porous material obtained in Example 5 was slightly lower in crystallinity than that obtained in Example 4.
- Example 6 A hydroxyapatite porous material was produced in the same manner as in Example 4 except that the firing temperature was 1150 ° C. A scanning electron micrograph of the obtained hydroxyapatite is shown in FIG. From FIG. 10, it was confirmed that the hydroxyapatite fine particles were strongly aggregated and integrated as compared with Example 4.
- Example 7 A hydroxyapatite porous body was produced in the same manner as in Example 4 except that the firing was held at 1500 ° C. for 5 hours.
- FIG. 11 shows a diffraction diagram obtained by X-ray diffraction measurement of the obtained hydroxyapatite porous body. From FIG. 11, it was confirmed that the obtained hydroxyapatite porous material contains ⁇ -tricalcium phosphate. Based on a calibration curve described in paragraph Nos. 0090 to 0094 of Japanese Patent Publication No.
- Example 8 In the following virtual example (Example 8), one of the preferred examples of the present invention will be described by describing the production of fibers when tricalcium phosphate is used.
- the eighth embodiment can be easily defined from a method correlated with the first to seventh embodiments.
- Tricalcium phosphate is prepared by the slurry method shown below. Thoroughly pulverize and mix 5.04 g of bis (dihydrogen phosphate) calcium, 2.0 g of calcium carbonate, and 7.36 g of tetracalcium phosphate monooxide as phosphates and calcium salts using a mortar. To make mixture 1 (mixing step). In the mixture 1 adjusted at the above ratio, Ca / P, which is the molar ratio of calcium atoms to phosphorus atoms, is 1.5.
- the dispersion liquid is supplied at 1.0 ml / hr to a nozzle having an inner diameter of 0.22 mm by a syringe pump, a voltage of 20 kV is applied to the nozzle, and a fibrous structure precursor (fiber assembly) is applied to a grounded collector. To collect. The distance between the needle and the collector is 20 cm.
- the electrospun fiber assembly is heated to 900 ° C. at a temperature increase rate of 10 ° C./min in the air, held for 1 hour, and then cooled to room temperature. Can be obtained.
- the calcium phosphate porous material of the present invention has communication, in particular, communication and uniform fine pores, it can be suitably used for various applications.
- cell culture substrates used in cell culture petri dishes, biomaterials such as artificial bones and bone grafting materials for implants, chromatographic fillers with protein adsorption properties, catalysts for polymer alcohol synthesis, catalyst carriers Applications include filter media, electronic materials, physiologically active substance-fixing carriers, and heavy metal adsorbents.
Landscapes
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Textile Engineering (AREA)
- Mechanical Engineering (AREA)
- Inorganic Chemistry (AREA)
- Dispersion Chemistry (AREA)
- Geology (AREA)
- Environmental & Geological Engineering (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Materials For Medical Uses (AREA)
- Inorganic Fibers (AREA)
- Spinning Methods And Devices For Manufacturing Artificial Fibers (AREA)
- Nonwoven Fabrics (AREA)
Abstract
Description
(1)リン酸カルシウムを静電紡糸することを特徴とする、リン酸カルシウム多孔体の製造方法。
(2)前記静電紡糸するリン酸カルシウムがハイドロキシアパタイト、またはリン酸三カルシウムである、前記(1)に記載のリン酸カルシウム多孔体の製造方法。
(3)前記リン酸カルシウム多孔体がハイドロキシアパタイト多孔体、またはリン酸三カルシウム多孔体である、前記(1)または(2)に記載のリン酸カルシウム多孔体の製造方法。
(4)X線回折測定による回折角2θ=46.7°におけるピークの半値幅が0.5°以上であるハイドロキシアパタイトを用いる、前記(1)~(3)のいずれかに記載のリン酸カルシウム多孔体の製造方法。
(5)リン酸カルシウムを分散媒体に分散させて分散液とし、該分散液を静電紡糸した後に、紡糸された繊維を焼成することを特徴とする、前記(1)~(4)のいずれかに記載のリン酸カルシウム多孔体の製造方法。
(6)分散液に、更に、繊維形成性高分子を分散させる、前記(5)に記載のリン酸カルシウム多孔体の製造方法。
(7)焼成を、500℃以上の温度範囲で行う、前記(5)または(6)に記載のリン酸カルシウム多孔体の製造方法。
(8)リン酸カルシウム/繊維形成性高分子(重量比)が、0.3以上である、前記(6)または(7)に記載のリン酸カルシウム多孔体の製造方法。
(9)前記(1)~(8)のいずれかに記載の製造方法で得られた、リン酸カルシウム多孔体。
<リン酸カルシウム多孔体の製造方法>
本発明のリン酸カルシウム多孔体の製造方法は、リン酸カルシウムを静電紡糸する方法を基本とする方法である。前記リン酸カルシウムがハイドロキシアパタイトである場合、得られるリン酸カルシウム多孔体をハイドロキシアパタイト多孔体という。また、前記リン酸カルシウムがリン酸三カルシウムである場合、得られるリン酸カルシウム多孔体をリン酸三カルシウム多孔体という。
本発明に用いるリン酸カルシウムとしては、リン酸水素カルシウム、リン酸三カルシウム、ハイドロキシアパタイトなどを挙げることができる。なかでも生体適合性の点で、リン酸三カルシウム、ハイドロキシアパタイトが好ましい。なお、ここでいうリン酸三カルシウムは、リン酸三カルシウム前駆体を含む意味で用いる。
紡糸する紡糸液の態様は種々の態様があるが、ハイドロキシアパタイトは、分散媒体に分散させて紡糸液とすることが好ましい。使用する分散媒体は、ハイドロキシアパタイトを均一に分散することが可能であれば、特に限定されないが、例えば、水、メタノール、エタノール、プロパノール、アセトン、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、ジメチルスルホキシド、N-メチル-2-ピロリドン、トルエン、キシレン、ピリジン、蟻酸、酢酸、テトラヒドロフラン、ジクロロメタン、クロロホルム、1,1,1,3,3,3-ヘキサフルオロイソプロパノール、及びこれらの混合物などを挙げることができる。これらのうち、ハイドロキシアパタイトの分散性の観点からは、極性溶媒を用いることが好ましく、例えば、水、メタノール、エタノール、プロパノール、アセトンやN,N-ジメチルホルムアミドを用い分散液とすることがさらに好ましい。
ハイドロキシアパタイトと分散媒体を含む分散液中に、曳糸性を向上させる目的で、さらに繊維形成性高分子を含有させてもよい。繊維形成性高分子は、ハイドロキシアパタイトの繊維化を促す作用を奏すればよく、上記分散媒体に溶解可能で、焼成により分解されるものから選ばれる。繊維形成性高分子として、例えば、ポリビニルアルコール、ポリエチレングリコール、ポリエチレンオキシド、ポリビニルピロリドン、ポリアクリル酸、ポリアクリル酸ナトリウム、ポリメタクリル酸、ポリメタクリル酸ナトリウム、ポリアクリルアミド、ポリメタクリルアミド、ポリ酢酸ビニル、ポリエチレン、ポリプロピレン、ポリエチレンテレフタレート、ポリ乳酸、ポリアミド、ポリウレタン、ポリスチレン、ポリフッ化ビニリデン、ポリアクリロニトリル、ポリメタクリル酸メチル、ポリグリコール酸、ポリカプロラクトン、セルロース、セルロース誘導体、キチン、キトサン、コラーゲン、ゼラチンおよびこれらの共重合体などを挙げることができる。これら繊維形成性高分子は1種類で使用してもよく、2種類以上を混合して使用してもよい。混合して使用する場合の混合率は、特に限定されるものではなく、求める曳糸性や分散性、得られる繊維の物性を鑑みて、適宜設定することができる。
分散液中のハイドロキシアパタイトの分散性を促進させる目的で、さらに界面活性剤を分散液に含有させてもよい。界面活性剤は、ハイドロキシアパタイトの効果を阻害しない範囲で用いることができ、例えば、ドデシル硫酸ナトリウムや臭化テトラブチルアンモニウム、塩化テトラブチルアンモニウムなどの一般的なイオン性界面活性剤、ポリオキシエチレンソルビタンモノラウレートなどの一般的な非イオン性界面活性剤などを挙げることができる。界面活性剤には、金属イオンを含まないことが、高純度のハイドロキシアパタイト多孔体を得ることができる点で好ましく、かかる観点からは臭化テトラブチルアンモニウムや塩化テトラブチルアンモニウム、ポリオキシエチレンソルビタンモノラウレートなどを例示できる。また、界面活性剤は、分散媒体中で均一に溶解することが好ましい。
ハイドロキシアパタイトの分散液を調製する方法は特に限定されない。調製方法として、撹拌や超音波処理などの方法を挙げることができる。さらに、ビーズミル等の分散機を用いることで、均一性の高い分散液を得ることが可能である。また、混合の順序も、特に限定されるものではなく、同時に混合しても、逐次に混合してもよい。繊維形状のハイドロキシアパタイトを得るためには、分散液の粘度を、10~10,000cPの範囲に調製することが好ましく、100~8,000cPの範囲であることがより好ましい。粘度が10cP以上であると、繊維を形成するための曳糸性が得られ、10,000cP以下であると、分散液を吐出させるのが容易となる。粘度が100~8,000cPの範囲であれば、広い紡糸条件範囲で良好な曳糸性が得られるのでより好ましい。分散液の粘度は、ハイドロキシアパタイトの濃度あるいは繊維形成性高分子の分子量、濃度を適宜変更することで、調整することができる。
静電紡糸法とは、紡糸液を吐出させるとともに、電界を作用させて、吐出された紡糸液を繊維化し、コレクター上に繊維を得る方法である。例えば、紡糸液をノズルから押し出すとともに電界を作用させて紡糸する方法、紡糸液を泡立たせるとともに電界を作用させて紡糸する方法、円筒状電極の表面に紡糸液を導くとともに電界を作用させて紡糸する方法などを挙げることができる。この方法によれば、直径10nm~10μmの均一な繊維を得ることができる。
静電紡糸された繊維集合体は焼成される。繊維集合体を焼成することによって、繊維集合体中のハイドロキシアパタイト同士の結合を強固にし、結晶性を向上させることが可能となる。また、繊維形成性高分子や界面活性剤等を含む場合、これらを加熱分解することができ、高純度のハイドロキシアパタイト多孔体とすることができる。焼成方法は、特に限定されないが、空気雰囲気中で行うことができる。空気中で焼成することにより、繊維形成性高分子や界面活性剤等のハイドロキシアパタイト以外の成分の残存物を少なくすることが可能である。
本方法を用いれば、リン酸カルシウム多孔体を容易に製造することができる。特にハイドロキシアパタイト多孔体、またはリン酸三カルシウム多孔体の製造に好適である。以下では、ハイドロキシアパタイト多孔体の例について説明するが、リン酸三カルシウム多孔体も同様に説明できる。また、本発明はこれに限定されるものではない。
・X線回折測定による回折角2θ=46.7°におけるピークの半値幅
BRUKER製のX線回折装置(D8 DISCOVER)を使用し、X線回折測定により得られた回折図において、回折角2θ=46.7°におけるピークの半値幅を求めた。
・ハイドロキシアパタイト多孔体における孔の観察
走査型電子顕微鏡(日本電子株式会社製 JSM-5410LV)により、倍率500で写真を撮影し、孔の均一性、連通性を確認した。
・ハイドロキシアパタイト多孔体における繊維構造の観察
走査型電子顕微鏡(日本電子株式会社製 JSM-5410LV)により、倍率5,000で写真を撮影し、繊維の表面外観、及び、繊維内部におけるハイドロキシアパタイトの凝集性を確認した。
・一次粒子の平均粒径の測定方法
走査型電子顕微鏡(日本電子株式会社製 JSM-6301F)により4万倍で撮影した画像中、50点以上の一次粒子の直径を計測し、平均値を算出することで測定した。
<ハイドロキシアパタイトの合成>
低結晶性ハイドロキシアパタイト粒子を以下に示す湿式法で調整した。なお、Ca(NO3)24H2Oおよび(NH4)2HPO4は、ナカライテスク株式会社製の硝酸カルシウム四水和物およびリン酸水素二アンモニウムを用い、25重量%アンモニア水は和光純薬工業株式会社製の25重量%アンモニア水を用い、純水はMilli-Q waterを使用した。先ず、25重量%アンモニア水でpHを12に調整したCa(NO3)2水溶液(42mN、80mL)を、冷却管及び半月状撹拌翼を接続した1Lフラスコに注ぎ入れ、室温(30℃)に保った。このフラスコに、アンモニア水でpHを12に調製した(NH4)2HPO4水溶液(10mN、200mL)を室温にて添加し、10時間反応させた。次に、得られた反応物を遠心分離により分離洗浄することにより、低結晶性ハイドロキシアパタイト粒子を得た。得られたハイドロキシアパタイトは、球形の形状で、一次粒子の平均粒径は20nm、X線回折測定による回折角2θ=46.7°におけるピークの半値幅は、0.51°であった。
上記方法により作製したハイドロキシアパタイトのエタノール分散液(13重量%)64.2重量部、ポリビニルピロリドン(Mw:1,300,000 ALDRICH製)8.3重量部、エタノール(一級;ナカライテスク製)27.5重量部からなる分散液を作製した。得られた分散液は、エタノールに対するハイドロキシアパタイトの濃度が9.1重量%、ハイドロキシアパタイト/ポリビニルピロリドン(重量比)が1.0であった。次いで、シリンジポンプにより内径0.22mmのノズルに分散液を1.0ml/hrで供給すると共に、ノズルに20kVの電圧を印加し、接地されたコレクターに繊維状構造物前駆体(繊維集合体)を捕集した。ニードルとコレクターの距離は20cmとした。静電紡糸された繊維集合体を空気中、10℃/minの昇温速度で900℃まで昇温し、1時間保持した後、室温まで冷却することで、平均繊維径1μmのハイドロキシアパタイト多孔体を作製した。得られたハイドロキシアパタイト多孔体の走査型電子顕微鏡写真を図1および2に示す。図1から明らかなように、ハイドロキシアパタイト多孔体は、連続かつ均一な微細孔を有するものであった。また、図2から、得られたハイドロキシアパタイト多孔体は、ハイドロキシアパタイト微粒子が凝集一体化し、表面平滑性の高い繊維であることが確認できた。焼成前の走査型電子顕微鏡写真を図6として示す。
<ハイドロキシアパタイトの作製>
上記で得られた低結晶性ハイドロキシアパタイト粒子を、次に示す方法によって焼成することで高結晶性ハイドロキシアパタイトナノ粒子を作製した。まず、融着防止助剤として、0.5gのポリアクリル酸(ALDRICH社製、重量平均分子量15,000g/mol)を含む、pH7.0の水溶液(以下、水溶液A)100mLに、0.5gの上記低結晶性ハイドロキシアパタイトナノ粒子を分散させることで、同粒子表面にポリアクリル酸を吸着させた。次に、上記で調製した分散液に、融着防止剤として、水酸カルシウム〔Ca(OH)2〕飽和水溶液500mLを添加することで、同粒子表面にポリアクリル酸カルシウムを析出させた。結果として生じる沈殿物を回収し、減圧下80℃にて乾燥させることで、混合粒子を回収した。上記混合粒子をルツボに入れ、焼成温度800℃にて一時間焼成を行った。この際、ポリアクリル酸カルシウムは熱分解し、酸化カルシウム〔CaO〕となった。次に、上記で調製した水溶液A500mLに得られた焼成体を懸濁し、遠心分離により分離洗浄し、更に蒸留水に懸濁し、同様に遠心分離により分離洗浄を行うことによって、融着防止剤及び融着防止助剤を除去し、高結晶性ハイドロキシアパタイトナノ粒子を回収した。得られたハイドロキシアパタイトは、球形の形状で、一次粒子の平均粒径は40nm、X線回折測定による回折角2θ=46.7°におけるピークの半値幅は、0.29°であった。
上記方法により作製したハイドロキシアパタイトを用い、実施例1と同様にして、平均繊維径1.2μmのハイドロキシアパタイト多孔体を作製した。得られたハイドロキシアパタイト多孔体の走査型電子顕微鏡写真を図3および4に示す。図3から明らかなように、ハイドロキシアパタイト多孔体は、連通しかつ均一な微細孔を有するものであった。また、図4から、実施例1の繊維に比べて、繊維を構成しているハイドロキシアパタイトの結晶性が高く、粒子が大きいために、ハイドロキシアパタイト粒子同士の凝集一体化が十分ではなく、繊維表面には個々の微粒子に由来した微細な凹凸が観察された。このような形状からして、実施例1に比べて、繊維比表面積は高くなるものの、強度が低く脆いことが推察された。
実施例1のハイドロキシアパタイトのエタノール分散液(13重量%)6.9重量部、ポリビニルピロリドン(Mw:1,300,000 ALDRICH製)9.0重量部、エタノール(一級;ナカライテスク製)84.1重量部からなる分散液を作製した。得られた分散液は、エタノールに対するハイドロキシアパタイトの濃度が1.0重量%、ハイドロキシアパタイト/ポリビニルピロリドン(重量比)が0.1であった。次いで、実施例1と同様にして、ハイドロキシアパタイト多孔体を作製した。得られたハイドロキシアパタイト多孔体の走査型電子顕微鏡写真を図5に示す。図5に示すとおり孔は連通していることが確認された。ただ、繊維は収縮し、切断している部分、即ち孔の大きさが50μmを超える箇所も観察され、孔の均一性には欠けていた。
実施例1のハイドロキシアパタイトのエタノール分散液(13重量%)45.6重量部、ポリビニルピロリドン(Mw:1,300,000 ALDRICH製)8.5重量部、エタノール(一級;ナカライテスク製)45.9重量部からなる分散液を作製した。得られた分散液は、エタノールに対するハイドロキシアパタイトの濃度が6.5重量%、ハイドロキシアパタイト/ポリビニルピロリドン(重量比)が0.7であった。次いで、実施例1と同様にして、ハイドロキシアパタイト多孔体を作製した。得られたハイドロキシアパタイトの走査型電子顕微鏡写真を図7および8に示す。図7から明らかなように、ハイドロキシアパタイト多孔体は、連続かつ均一な微細孔を有するものであった。また、図8から、得られたハイドロキシアパタイト多孔体は、ハイドロキシアパタイト微粒子が凝集一体化し、表面平滑性の高い繊維であることが確認できた。ただし、十分に使用できるレベルではあるものの、実施例1に比べて、焼成前後での多孔体の収縮が大きかった。
焼成温度を600℃とした以外は、実施例4と同様にして、ハイドロキシアパタイト多孔体を作製した。実施例4で得られたハイドロキシアパタイト多孔体および実施例5で得られたハイドロキシアパタイト多孔体のX線回折測定により得られた回折図を図9に示す。図9から、実施例5で得られたハイドロキシアパタイト多孔体は、実施例4で得られたものに比べて、やや結晶性が低いことが確認された。
焼成温度を1150℃とした以外は、実施例4と同様にして、ハイドロキシアパタイト多孔体を作製した。得られたハイドロキシアパタイトの走査型電子顕微鏡写真を図10に示す。図10から、実施例4に比べて、ハイドロキシアパタイト微粒子同士が強く凝集一体化している様子が確認された。
焼成を1500℃で5時間保持した以外は、実施例4と同様にして、ハイドロキシアパタイト多孔体を作製した。得られたハイドロキシアパタイト多孔体のX線回折測定により得られた回折図を図11に示す。図11から、得られたハイドロキシアパタイト多孔体にはβ-リン酸三カルシウムが含有していることが確認された。特許公報第4265946号の段落番号0090~0094に記載されている検量線に基づいて、β-リン酸三カルシウム(β-TCP)とハイドロキシアパタイト(HAp)の混合物中におけるβ-リン酸三カルシウムの含有率(β-TCP/(β-TCP+HAp)×100(%))を求めた結果、含有率は28.6%であった。
市販のβ-リン酸三カルシウム(フルカ(株)製 No.21218)およびハイドロキシアパタイト(ペンタックス(株)製、アパセラム:登録商標)を用いて、X線解析法(XRD)により、板状リン酸カルシウム中に含まれるβ-リン酸三カルシウムの含有率を導出する為の検量線関数を以下のようにして求めた。上記市販のβ-リン酸三カルシウム(β-TCP)とハイドロキシアパタイト(HAp)とを、下記式(1)で示される試薬混合物中のβ-リン酸三カルシウムの存在比xが所定の値になるように、試薬混合物を調製した。
x(%)=β-TCP/(β-TCP+HAp)×100 ・・・(1)
当該試薬混合物についてのX線解析の結果から、試薬混合物中のβ-リン酸三カルシウムの強度比yを算出した結果、下記表1のようになった。なお、上記強度比yは、X線解析の結果得られた、ハイドロキシアパタイトの(2 1 1) (2θ=31.92°)の強度を、β-リン酸三カルシウムの(0 2 10) (2θ=31.16°)の強度で除した値を示している。

log10y=-0.02093x+0.7736 ・・・(2)
(0≦x≦100)
<リン酸三カルシウムの合成>
リン酸三カルシウムを以下に示すスラリー法により調整する。
リン酸塩およびカルシウム塩としての、ビス(リン酸二水素)カルシウム5.04g、炭酸カルシウム2.0g、およびテトラカルシウムホスフェイトモノオキサイト7.36gを、乳鉢を用いて十分に細砕・混合し、混合物1とする(混合工程)。上記の割合で調整された混合物1は、リン原子に対するカルシウム原子のモル比であるCa/Pが1.5となる。上記混合物1に、反応促進剤であるリン酸二水素ナトリウム水溶液(0.15M)15mlを添加し、迅速に攪拌して、混合物2とした(混合工程)後、成形容器へ充填する。上記混合物2を1時間乾燥処理し、乳鉢により粉砕し、β-リン酸三カルシウム前駆体を得る。得られたβ-リン酸三カルシウム前駆体を焼成温度800℃にて1時間焼成することによりリン酸三カルシウム粒子を得る。
上記方法により作製したリン酸三カルシウムのエタノール分散液(13重量%)64.2重量部、ポリビニルピロリドン(Mw:1,300,000 ALDRICH製)8.3重量部、エタノール(一級;ナカライテスク製)27.5重量部からなる分散液を作製する。得られる分散液は、エタノールに対するリン酸三カルシウムの濃度が9.1重量%、リン酸三カルシウム/ポリビニルピロリドン(重量比)が1.0である。次いで、シリンジポンプにより内径0.22mmのノズルに分散液を1.0ml/hrで供給すると共に、ノズルに20kVの電圧を印加し、接地されたコレクターに繊維状構造物前駆体(繊維集合体)を捕集する。ニードルとコレクターの距離は20cmとする。静電紡糸された繊維集合体を空気中、10℃/minの昇温速度で900℃まで昇温し、1時間保持した後、室温まで冷却することで、極細繊維によるリン酸三カルシウム多孔体を得ることができる。
Claims (9)
- リン酸カルシウムを静電紡糸することを特徴とする、リン酸カルシウム多孔体の製造方法。
- 前記静電紡糸するリン酸カルシウムがハイドロキシアパタイト、またはリン酸三カルシウムである、請求項1に記載のリン酸カルシウム多孔体の製造方法。
- 前記リン酸カルシウム多孔体がハイドロキシアパタイト多孔体、またはリン酸三カルシウム多孔体である、請求項1または2に記載のリン酸カルシウム多孔体の製造方法。
- X線回折測定による回折角2θ=46.7°におけるピークの半値幅が0.5°以上であるハイドロキシアパタイトを用いる、請求項1~3のいずれか1項に記載のリン酸カルシウム多孔体の製造方法。
- リン酸カルシウムを分散媒体に分散させて分散液とし、該分散液を静電紡糸した後に、紡糸された繊維を焼成することを特徴とする、請求項1~4のいずれか1項に記載のリン酸カルシウム多孔体の製造方法。
- 分散液に、更に、繊維形成性高分子を分散させる、請求項5に記載のリン酸カルシウム多孔体の製造方法。
- 焼成を、500℃以上の温度範囲で行う、請求項5または6に記載のリン酸カルシウム多孔体の製造方法。
- リン酸カルシウム/繊維形成性高分子(重量比)が、0.3以上である、請求項6または7に記載のリン酸カルシウム多孔体の製造方法。
- 請求項1~8のいずれか1項に記載の製造方法で得られた、リン酸カルシウム多孔体。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/409,475 US9896782B2 (en) | 2012-06-20 | 2013-06-19 | Method for producing porous calcium phosphate body |
| EP13807515.5A EP2865791A4 (en) | 2012-06-20 | 2013-06-19 | PROCESS FOR PRODUCING POROUS CALCIUM PHOSPHATE BODY AND POROUS CALCIUM PHOSPHATE BODY PRODUCED THEREBY |
| KR1020157001081A KR102079096B1 (ko) | 2012-06-20 | 2013-06-19 | 인산칼슘 다공체의 제조 방법 및 해당 제조 방법으로 얻어진 인산칼슘 다공체 |
| AU2013278347A AU2013278347C1 (en) | 2012-06-20 | 2013-06-19 | Method for producing porous calcium phosphate body, and porous calcium phosphate body produced thereby |
| CN201380032693.6A CN104411869A (zh) | 2012-06-20 | 2013-06-19 | 磷酸钙多孔体的制造方法、及利用所述制造方法获得的磷酸钙多孔体 |
| JP2013544602A JP5422784B1 (ja) | 2012-06-20 | 2013-06-19 | リン酸カルシウム多孔体の製造方法、および当該製造方法で得られたリン酸カルシウム多孔体 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-139147 | 2012-06-20 | ||
| JP2012139147 | 2012-06-20 | ||
| JP2012226291 | 2012-10-11 | ||
| JP2012-226291 | 2012-10-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013191221A1 true WO2013191221A1 (ja) | 2013-12-27 |
Family
ID=49768815
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/066883 WO2013191221A1 (ja) | 2012-06-20 | 2013-06-19 | リン酸カルシウム多孔体の製造方法、および当該製造方法で得られたリン酸カルシウム多孔体 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9896782B2 (ja) |
| EP (1) | EP2865791A4 (ja) |
| JP (1) | JP5422784B1 (ja) |
| KR (1) | KR102079096B1 (ja) |
| CN (2) | CN104411869A (ja) |
| AU (1) | AU2013278347C1 (ja) |
| WO (1) | WO2013191221A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016069745A (ja) * | 2014-09-29 | 2016-05-09 | 富田製薬株式会社 | リン酸カルシウムの繊維状成型体の製造方法 |
| WO2017043585A1 (ja) * | 2015-09-08 | 2017-03-16 | 日本製紙株式会社 | リン酸カルシウム微粒子と繊維との複合体、および、その製造方法 |
| JP2018095999A (ja) * | 2016-12-13 | 2018-06-21 | 日本バイリーン株式会社 | 無機系繊維シート及びその製造方法 |
| JP2020007190A (ja) * | 2018-07-10 | 2020-01-16 | 白石工業株式会社 | ハイドロキシアパタイトの製造方法 |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110885071B (zh) * | 2019-12-17 | 2021-07-09 | 衢州学院 | 一种微米级超长钙基蠕虫状胶束模板及羟基钙磷灰石晶须 |
| KR102711590B1 (ko) * | 2021-05-06 | 2024-09-30 | 충북대학교 산학협력단 | 약물 담지용 다공성 수산화아파타이트 구조체의 제조 방법, 및 이로부터 제조된 약물 담지체 |
| CN113559317B (zh) * | 2021-06-18 | 2022-04-15 | 东华大学 | 磁响应ha纳米棒及其制备和在齿科修复用树脂上的应用 |
| TWI791260B (zh) * | 2021-08-12 | 2023-02-01 | 行政院農業委員會 | 一種從含鈣生物廢棄物製備雙相磷酸鈣多孔陶瓷的兩階段燒結方法 |
| CN115161885A (zh) * | 2022-08-25 | 2022-10-11 | 中国矿业大学 | 一种含均分散生物驻极体的高过滤效率聚乳酸纳米纤维膜及其制备方法 |
| CN115636399A (zh) * | 2022-09-08 | 2023-01-24 | 昆明川金诺化工股份有限公司 | 一种高活性质量稳定的活性磷酸钙生产工艺 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02167868A (ja) | 1988-09-20 | 1990-06-28 | Asahi Optical Co Ltd | 多孔質セラミックス及びその製造用乾燥体並びにそれらの製造方法 |
| JPH0585666B2 (ja) * | 1984-09-14 | 1993-12-08 | Tonen Corp | |
| JP2004049355A (ja) | 2002-07-17 | 2004-02-19 | Olympus Corp | 骨補填材およびその製造方法 |
| JP2007246299A (ja) * | 2006-03-13 | 2007-09-27 | Japan Science & Technology Agency | リン酸カルシウム透明体およびその製造方法 |
| JP4265946B2 (ja) | 2003-08-29 | 2009-05-20 | 独立行政法人科学技術振興機構 | 板状リン酸カルシウムおよびその製造方法、ならびにそれを用いた医療用材料およびリン酸カルシウム複合体 |
| JP2010189798A (ja) * | 2009-02-18 | 2010-09-02 | Teijin Ltd | ショットを含まない無機繊維及びその製造方法 |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4659617A (en) | 1984-09-11 | 1987-04-21 | Toa Nenryo Kogyo Kabushiki Kaisha | Fibrous apatite and method for producing the same |
| JPH0585666A (ja) | 1991-09-27 | 1993-04-06 | Ricoh Co Ltd | 記録紙検出方法および記録紙検出装置 |
| JP3269589B2 (ja) | 1993-07-19 | 2002-03-25 | 日本特殊陶業株式会社 | リン酸カルシウム系長繊維の製造方法。 |
| CN1203902C (zh) * | 2003-05-28 | 2005-06-01 | 东南大学 | 可吸收超细纤维组织修复材料及其制备方法 |
| CN1751745A (zh) * | 2005-08-02 | 2006-03-29 | 天津大学 | 磷灰石超细纤维材料及其制备方法 |
| US8512741B2 (en) * | 2006-09-01 | 2013-08-20 | Cornell Research Foundations, Inc. | Electrospun calcium phosphate nanofibers |
| WO2008157594A2 (en) * | 2007-06-18 | 2008-12-24 | New Jersey Institute Of Technology | Electrospun ceramic-polymer composite as a scaffold for tissue repair |
| US9931438B2 (en) * | 2008-01-28 | 2018-04-03 | Ngk Spark Plug Co., Ltd. | Article with foamed surface, implant and method of producing the same |
| CN101376567B (zh) * | 2008-10-07 | 2011-09-14 | 中国科学院长春应用化学研究所 | 具有纳米孔的复合生物活性玻璃超细纤维及其制备方法 |
| CN101736441A (zh) * | 2008-11-07 | 2010-06-16 | 北京化工大学 | 生物活性碳纳米纤维及其制备方法 |
| CN101822852B (zh) * | 2009-03-03 | 2013-04-10 | 北京化工大学 | 仿生磷酸钙纤维复合支架材料及制备方法 |
| CN102102242B (zh) | 2010-12-31 | 2012-06-27 | 中国科学院上海硅酸盐研究所 | 一种聚乳酸-非晶磷酸钙复合纳米纤维材料的制备方法 |
| EP2696806B1 (en) * | 2011-04-13 | 2017-12-27 | New Jersey Institute of Technology | System and method for electrospun biodegradable scaffold for bone repair |
| WO2014066884A1 (en) * | 2012-10-26 | 2014-05-01 | Tufts University | Silk-based fabrication techniques to prepare high strength calcium phosphate ceramic scaffolds |
-
2013
- 2013-06-19 EP EP13807515.5A patent/EP2865791A4/en not_active Withdrawn
- 2013-06-19 WO PCT/JP2013/066883 patent/WO2013191221A1/ja active Application Filing
- 2013-06-19 KR KR1020157001081A patent/KR102079096B1/ko active IP Right Grant
- 2013-06-19 CN CN201380032693.6A patent/CN104411869A/zh active Pending
- 2013-06-19 US US14/409,475 patent/US9896782B2/en active Active
- 2013-06-19 AU AU2013278347A patent/AU2013278347C1/en not_active Ceased
- 2013-06-19 CN CN201910316671.1A patent/CN110042502A/zh active Pending
- 2013-06-19 JP JP2013544602A patent/JP5422784B1/ja active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0585666B2 (ja) * | 1984-09-14 | 1993-12-08 | Tonen Corp | |
| JPH02167868A (ja) | 1988-09-20 | 1990-06-28 | Asahi Optical Co Ltd | 多孔質セラミックス及びその製造用乾燥体並びにそれらの製造方法 |
| JP2004049355A (ja) | 2002-07-17 | 2004-02-19 | Olympus Corp | 骨補填材およびその製造方法 |
| JP4265946B2 (ja) | 2003-08-29 | 2009-05-20 | 独立行政法人科学技術振興機構 | 板状リン酸カルシウムおよびその製造方法、ならびにそれを用いた医療用材料およびリン酸カルシウム複合体 |
| JP2007246299A (ja) * | 2006-03-13 | 2007-09-27 | Japan Science & Technology Agency | リン酸カルシウム透明体およびその製造方法 |
| JP2010189798A (ja) * | 2009-02-18 | 2010-09-02 | Teijin Ltd | ショットを含まない無機繊維及びその製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2865791A4 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016069745A (ja) * | 2014-09-29 | 2016-05-09 | 富田製薬株式会社 | リン酸カルシウムの繊維状成型体の製造方法 |
| WO2017043585A1 (ja) * | 2015-09-08 | 2017-03-16 | 日本製紙株式会社 | リン酸カルシウム微粒子と繊維との複合体、および、その製造方法 |
| JPWO2017043585A1 (ja) * | 2015-09-08 | 2018-06-28 | 日本製紙株式会社 | リン酸カルシウム微粒子と繊維との複合体、および、その製造方法 |
| US10737940B2 (en) | 2015-09-08 | 2020-08-11 | Nippon Paper Industries Co., Ltd. | Complexes of calcium phosphate microparticles and fibers as well as processes for preparing them |
| JP2018095999A (ja) * | 2016-12-13 | 2018-06-21 | 日本バイリーン株式会社 | 無機系繊維シート及びその製造方法 |
| JP2020007190A (ja) * | 2018-07-10 | 2020-01-16 | 白石工業株式会社 | ハイドロキシアパタイトの製造方法 |
| JP7109776B2 (ja) | 2018-07-10 | 2022-08-01 | 白石工業株式会社 | ハイドロキシアパタイトの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN110042502A (zh) | 2019-07-23 |
| JP5422784B1 (ja) | 2014-02-19 |
| US9896782B2 (en) | 2018-02-20 |
| AU2013278347C1 (en) | 2017-09-28 |
| CN104411869A (zh) | 2015-03-11 |
| KR20150031291A (ko) | 2015-03-23 |
| EP2865791A4 (en) | 2016-02-24 |
| US20150152573A1 (en) | 2015-06-04 |
| EP2865791A1 (en) | 2015-04-29 |
| KR102079096B1 (ko) | 2020-02-19 |
| AU2013278347A1 (en) | 2015-02-05 |
| AU2013278347B2 (en) | 2017-04-20 |
| JPWO2013191221A1 (ja) | 2016-05-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5422784B1 (ja) | リン酸カルシウム多孔体の製造方法、および当該製造方法で得られたリン酸カルシウム多孔体 | |
| KR100814730B1 (ko) | 나노-매크로 사이즈의 계층적 기공구조를 가지는 다공성 생체활성유리 및 이의 합성방법 | |
| Maheshwari et al. | Fabrication and evaluation of (PVA/HAp/PCL) bilayer composites as potential scaffolds for bone tissue regeneration application | |
| US7799839B2 (en) | Porous material having hierarchical pore structure and preparation method thereof | |
| JP5424354B2 (ja) | 無機系繊維構造体及びその製造方法 | |
| JP4463719B2 (ja) | 有機−無機複合多孔体、繊維状有機物の製造方法、及び有機−無機複合多孔体の製造方法 | |
| JP4959780B2 (ja) | 繊維状有機物の製造方法 | |
| Sánchez-Salcedo et al. | Design and preparation of biocompatible zwitterionic hydroxyapatite | |
| KR100751504B1 (ko) | 나노-마크로 사이즈의 계층적 기공구조를 가지는 생체재료및 이의 합성 방법 | |
| WO2008075824A1 (en) | Bioactive glass nanofibers and method of manufacturing the same | |
| JP5463504B2 (ja) | 非球形微粒子および非球形微粒子の製造方法 | |
| KR20110036072A (ko) | 의료용 조성물 및 의료용 키트 | |
| KR101047897B1 (ko) | 단백질 결합형 생체활성유리 나노섬유 및 이의 제조방법 | |
| KR101003025B1 (ko) | 다공성 티타늄-수산화인회석 복합지지체 및 이의 제조방법 | |
| Santos et al. | Calcium phosphate granules for use as a 5-Fluorouracil delivery system | |
| Mahanta et al. | Fabrication and characterization of hybrid nanofibers from poly (vinyl alcohol), milk protein and metal carbonates | |
| JP4683590B2 (ja) | 新規なリン酸カルシウム多孔体およびその製造方法 | |
| Alsenany et al. | Controlled compositions of tellurium/vanadium co-doped into hydroxyapatite/ε-polycaprolactone for wound healing applications | |
| JP2004284933A (ja) | 繊維状リン酸カルシウム | |
| EP4215271A1 (en) | Porous permanently polarized hydroxyapatite, a process for its production and uses thereof | |
| JP2004284898A (ja) | リン酸カルシウム多孔体およびその製法 | |
| JPS61174460A (ja) | アパタイト不織布 | |
| Tian et al. | Hydrothermally doping valve metal Nb into Titanate nanofibers structure for potentially engineering bone tissue | |
| Bielec et al. | Microemulsion Synthesis of Mesoporous β-tricalcium Phosphate Powder with a Novel System | |
| KR890003068B1 (ko) | 인회석 부직포/면형상체 및 그 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2013544602 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13807515 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14409475 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013807515 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20157001081 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2013278347 Country of ref document: AU Date of ref document: 20130619 Kind code of ref document: A |