WO2013094759A1 - 重合体粉体、硬化性樹脂組成物及びその硬化物 - Google Patents
重合体粉体、硬化性樹脂組成物及びその硬化物 Download PDFInfo
- Publication number
- WO2013094759A1 WO2013094759A1 PCT/JP2012/083361 JP2012083361W WO2013094759A1 WO 2013094759 A1 WO2013094759 A1 WO 2013094759A1 JP 2012083361 W JP2012083361 W JP 2012083361W WO 2013094759 A1 WO2013094759 A1 WO 2013094759A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polymer powder
- mass
- curable resin
- polymer
- resin composition
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
- C08L33/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, which oxygen atoms are present only as part of the carboxyl radical
- C08L33/10—Homopolymers or copolymers of methacrylic acid esters
- C08L33/12—Homopolymers or copolymers of methyl methacrylate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F20/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride, ester, amide, imide or nitrile thereof
- C08F20/02—Monocarboxylic acids having less than ten carbon atoms, Derivatives thereof
- C08F20/10—Esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/10—Esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/10—Esters
- C08F220/12—Esters of monohydric alcohols or phenols
- C08F220/16—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms
- C08F220/18—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms with acrylic or methacrylic acids
- C08F220/1808—C8-(meth)acrylate, e.g. isooctyl (meth)acrylate or 2-ethylhexyl (meth)acrylate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F265/00—Macromolecular compounds obtained by polymerising monomers on to polymers of unsaturated monocarboxylic acids or derivatives thereof as defined in group C08F20/00
- C08F265/04—Macromolecular compounds obtained by polymerising monomers on to polymers of unsaturated monocarboxylic acids or derivatives thereof as defined in group C08F20/00 on to polymers of esters
- C08F265/06—Polymerisation of acrylate or methacrylate esters on to polymers thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
- C08L33/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, which oxygen atoms are present only as part of the carboxyl radical
- C08L33/08—Homopolymers or copolymers of acrylic acid esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L51/00—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L51/00—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers
- C08L51/003—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers grafted on to macromolecular compounds obtained by reactions only involving unsaturated carbon-to-carbon bonds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L51/00—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers
- C08L51/06—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers grafted on to homopolymers or copolymers of aliphatic hydrocarbons containing only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J163/00—Adhesives based on epoxy resins; Adhesives based on derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K3/00—Materials not provided for elsewhere
- C09K3/10—Materials in mouldable or extrudable form for sealing or packing joints or covers
- C09K3/1006—Materials in mouldable or extrudable form for sealing or packing joints or covers characterised by the chemical nature of one of its constituents
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01B—CABLES; CONDUCTORS; INSULATORS; SELECTION OF MATERIALS FOR THEIR CONDUCTIVE, INSULATING OR DIELECTRIC PROPERTIES
- H01B3/00—Insulators or insulating bodies characterised by the insulating materials; Selection of materials for their insulating or dielectric properties
- H01B3/18—Insulators or insulating bodies characterised by the insulating materials; Selection of materials for their insulating or dielectric properties mainly consisting of organic substances
- H01B3/30—Insulators or insulating bodies characterised by the insulating materials; Selection of materials for their insulating or dielectric properties mainly consisting of organic substances plastics; resins; waxes
- H01B3/40—Insulators or insulating bodies characterised by the insulating materials; Selection of materials for their insulating or dielectric properties mainly consisting of organic substances plastics; resins; waxes epoxy resins
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01B—CABLES; CONDUCTORS; INSULATORS; SELECTION OF MATERIALS FOR THEIR CONDUCTIVE, INSULATING OR DIELECTRIC PROPERTIES
- H01B3/00—Insulators or insulating bodies characterised by the insulating materials; Selection of materials for their insulating or dielectric properties
- H01B3/18—Insulators or insulating bodies characterised by the insulating materials; Selection of materials for their insulating or dielectric properties mainly consisting of organic substances
- H01B3/30—Insulators or insulating bodies characterised by the insulating materials; Selection of materials for their insulating or dielectric properties mainly consisting of organic substances plastics; resins; waxes
- H01B3/44—Insulators or insulating bodies characterised by the insulating materials; Selection of materials for their insulating or dielectric properties mainly consisting of organic substances plastics; resins; waxes vinyl resins; acrylic resins
- H01B3/447—Insulators or insulating bodies characterised by the insulating materials; Selection of materials for their insulating or dielectric properties mainly consisting of organic substances plastics; resins; waxes vinyl resins; acrylic resins from acrylic compounds
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/04—Manufacture or treatment of semiconductor devices or of parts thereof the devices having at least one potential-jump barrier or surface barrier, e.g. PN junction, depletion layer or carrier concentration layer
- H01L21/50—Assembly of semiconductor devices using processes or apparatus not provided for in a single one of the subgroups H01L21/06 - H01L21/326, e.g. sealing of a cap to a base of a container
- H01L21/52—Mounting semiconductor bodies in containers
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L24/00—Arrangements for connecting or disconnecting semiconductor or solid-state bodies; Methods or apparatus related thereto
- H01L24/01—Means for bonding being attached to, or being formed on, the surface to be connected, e.g. chip-to-package, die-attach, "first-level" interconnects; Manufacturing methods related thereto
- H01L24/26—Layer connectors, e.g. plate connectors, solder or adhesive layers; Manufacturing methods related thereto
- H01L24/28—Structure, shape, material or disposition of the layer connectors prior to the connecting process
- H01L24/29—Structure, shape, material or disposition of the layer connectors prior to the connecting process of an individual layer connector
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/10—Esters
- C08F220/12—Esters of monohydric alcohols or phenols
- C08F220/16—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms
- C08F220/18—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms with acrylic or methacrylic acids
- C08F220/1804—C4-(meth)acrylate, e.g. butyl (meth)acrylate, isobutyl (meth)acrylate or tert-butyl (meth)acrylate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2201/00—Properties
- C08L2201/02—Flame or fire retardant/resistant
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2201/00—Properties
- C08L2201/10—Transparent films; Clear coatings; Transparent materials
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/02—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group
- C08L2205/025—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group containing two or more polymers of the same hierarchy C08L, and differing only in parameters such as density, comonomer content, molecular weight, structure
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L2224/00—Indexing scheme for arrangements for connecting or disconnecting semiconductor or solid-state bodies and methods related thereto as covered by H01L24/00
- H01L2224/01—Means for bonding being attached to, or being formed on, the surface to be connected, e.g. chip-to-package, die-attach, "first-level" interconnects; Manufacturing methods related thereto
- H01L2224/26—Layer connectors, e.g. plate connectors, solder or adhesive layers; Manufacturing methods related thereto
- H01L2224/28—Structure, shape, material or disposition of the layer connectors prior to the connecting process
- H01L2224/29—Structure, shape, material or disposition of the layer connectors prior to the connecting process of an individual layer connector
- H01L2224/29001—Core members of the layer connector
- H01L2224/29099—Material
- H01L2224/2919—Material with a principal constituent of the material being a polymer, e.g. polyester, phenolic based polymer, epoxy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L2224/00—Indexing scheme for arrangements for connecting or disconnecting semiconductor or solid-state bodies and methods related thereto as covered by H01L24/00
- H01L2224/01—Means for bonding being attached to, or being formed on, the surface to be connected, e.g. chip-to-package, die-attach, "first-level" interconnects; Manufacturing methods related thereto
- H01L2224/26—Layer connectors, e.g. plate connectors, solder or adhesive layers; Manufacturing methods related thereto
- H01L2224/28—Structure, shape, material or disposition of the layer connectors prior to the connecting process
- H01L2224/29—Structure, shape, material or disposition of the layer connectors prior to the connecting process of an individual layer connector
- H01L2224/29001—Core members of the layer connector
- H01L2224/29099—Material
- H01L2224/29198—Material with a principal constituent of the material being a combination of two or more materials in the form of a matrix with a filler, i.e. being a hybrid material, e.g. segmented structures, foams
- H01L2224/29199—Material of the matrix
- H01L2224/2929—Material of the matrix with a principal constituent of the material being a polymer, e.g. polyester, phenolic based polymer, epoxy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L2224/00—Indexing scheme for arrangements for connecting or disconnecting semiconductor or solid-state bodies and methods related thereto as covered by H01L24/00
- H01L2224/80—Methods for connecting semiconductor or other solid state bodies using means for bonding being attached to, or being formed on, the surface to be connected
- H01L2224/83—Methods for connecting semiconductor or other solid state bodies using means for bonding being attached to, or being formed on, the surface to be connected using a layer connector
- H01L2224/8319—Arrangement of the layer connectors prior to mounting
- H01L2224/83191—Arrangement of the layer connectors prior to mounting wherein the layer connectors are disposed only on the semiconductor or solid-state body
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L2224/00—Indexing scheme for arrangements for connecting or disconnecting semiconductor or solid-state bodies and methods related thereto as covered by H01L24/00
- H01L2224/80—Methods for connecting semiconductor or other solid state bodies using means for bonding being attached to, or being formed on, the surface to be connected
- H01L2224/83—Methods for connecting semiconductor or other solid state bodies using means for bonding being attached to, or being formed on, the surface to be connected using a layer connector
- H01L2224/838—Bonding techniques
- H01L2224/8385—Bonding techniques using a polymer adhesive, e.g. an adhesive based on silicone, epoxy, polyimide, polyester
- H01L2224/83855—Hardening the adhesive by curing, i.e. thermosetting
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L23/00—Details of semiconductor or other solid state devices
- H01L23/28—Encapsulations, e.g. encapsulating layers, coatings, e.g. for protection
- H01L23/29—Encapsulations, e.g. encapsulating layers, coatings, e.g. for protection characterised by the material, e.g. carbon
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L24/00—Arrangements for connecting or disconnecting semiconductor or solid-state bodies; Methods or apparatus related thereto
- H01L24/80—Methods for connecting semiconductor or other solid state bodies using means for bonding being attached to, or being formed on, the surface to be connected
- H01L24/83—Methods for connecting semiconductor or other solid state bodies using means for bonding being attached to, or being formed on, the surface to be connected using a layer connector
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L2924/00—Indexing scheme for arrangements or methods for connecting or disconnecting semiconductor or solid-state bodies as covered by H01L24/00
- H01L2924/10—Details of semiconductor or other solid state devices to be connected
- H01L2924/11—Device type
- H01L2924/12—Passive devices, e.g. 2 terminal devices
- H01L2924/1204—Optical Diode
- H01L2924/12041—LED
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L2924/00—Indexing scheme for arrangements or methods for connecting or disconnecting semiconductor or solid-state bodies as covered by H01L24/00
- H01L2924/10—Details of semiconductor or other solid state devices to be connected
- H01L2924/11—Device type
- H01L2924/12—Passive devices, e.g. 2 terminal devices
- H01L2924/1204—Optical Diode
- H01L2924/12042—LASER
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L33/00—Semiconductor devices with at least one potential-jump barrier or surface barrier specially adapted for light emission; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof
- H01L33/48—Semiconductor devices with at least one potential-jump barrier or surface barrier specially adapted for light emission; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof characterised by the semiconductor body packages
- H01L33/52—Encapsulations
- H01L33/56—Materials, e.g. epoxy or silicone resin
Definitions
- the present invention relates to polymer powder, a curable resin composition containing the same, and a cured product thereof.
- thermosetting resins or active energy ray curable resins such as epoxy resins, polyimide resins, acrylic curable resins, oxetane curable resins, and the like, which are excellent in heat resistance and insulating properties, is rapidly increasing.
- Most of light emitting devices such as optical semiconductors that are put to practical use in various display boards, image reading light sources, traffic display units, etc. are manufactured by resin sealing.
- a sealing resin an epoxy resin is widely used because it is excellent in mechanical properties, electrical insulation, and adhesiveness.
- epoxy resins are frequently used as sealing resins for optical semiconductors because of their excellent transparency and heat resistance.
- epoxy resins that are liquid at room temperature can be cast and applied at room temperature, and thus are used as various paste-like or film-like materials.
- the viscosity of the epoxy resin composition is highly temperature dependent, the viscosity is significantly reduced by increasing the temperature until it is cured. Therefore, precise injection and application by a dispenser, precise pattern formation by screen printing, high film Precision processing such as coating on the film with thickness accuracy is difficult.
- the demand for high-precision processing is increasing year by year, and the epoxy resin composition to be used does not decrease in viscosity even when the temperature rises, and there is a demand for the shape to be stabilized early. Is extremely strong.
- a specific vinyl polymer is blended in the epoxy resin composition to impart gelation properties.
- There is a method used as an agent hereinafter referred to as “pregelling agent”.
- the method of using a specific vinyl polymer as a pregelling agent is proposed.
- Patent Documents 2 to 4 propose a method of blending acrylic rubber particles as a method of reducing the elastic modulus of the cured epoxy resin.
- Patent Document 5 proposes a method for improving impact resistance, adhesiveness, and gelation properties by blending a specific (meth) acrylate polymer into an epoxy resin composition.
- Patent Document 1 gelation property can be imparted to the epoxy resin composition, but since the glass transition temperature of the cured epoxy resin compounded with the pregelling agent tends to decrease, it has high heat resistance. This is not suitable for applications that require high performance. Further, Patent Document 1 does not mention anything about transparency. On the other hand, in the methods proposed in Patent Documents 2 to 4, the crack resistance of the cured epoxy resin is improved, but the viscosity of the epoxy resin composition is remarkably lowered by the temperature rise until curing. It was difficult to apply and pattern with high accuracy.
- Patent Document 5 Although the method proposed in Patent Document 5 has an effect on the gelation property of the epoxy resin composition and the low elastic modulus of the cured product, the gelation and high gel in a very short time required for high-precision processing. The chemical conversion was not achieved, and it was not a satisfactory state. Furthermore, since acrylic rubber particles have a low glass transition temperature, when blended in a large amount in an epoxy resin composition, the glass transition temperature of the cured epoxy resin may decrease, and for applications that require high heat resistance. It was unsuitable. Moreover, since the content of ionic impurities is not particularly taken into consideration, there is a concern that the electrical properties of the cured epoxy resin are deteriorated when blended in the epoxy resin composition.
- An object of the present invention is to provide a polymer powder and a powder that rapidly bring the curable resin composition into a gel state by heating at a predetermined temperature for a short time, and further reduce the elastic modulus of the resulting cured product. It is providing the curable resin composition to contain and its hardened
- the above-mentioned problems include (i) a (meth) acrylic acid ester polymer (A1) having a glass transition temperature of 0 ° C. or lower, the acetone content of the polymer powder is 5% by mass or more, and acetone is acceptable.
- the polymer powder (P1) having a mass average molecular weight of not less than 100,000 and (ii) an acetone soluble content of not less than 2 mass% and not more than 35 mass%, and a mass average molecular weight of acetone soluble content of 10
- the polymer powder (P) includes (i) a (meth) acrylic acid ester polymer (A1) having a glass transition temperature of 0 ° C. or less, and the acetone soluble content of the polymer powder is The polymer powder (P) according to [1], which is 5% by mass or more and is a polymer powder (P1) having an acetone-soluble mass average molecular weight of 100,000 or more. [3] The polymer powder (P) according to [2], further including a polymer (B1) having a glass transition temperature exceeding 0 ° C. [4] The polymer powder (P) according to [3], wherein the polymer (B1) is a (meth) acrylate polymer.
- the content of the (meth) acrylate polymer (A1) having a glass transition temperature of 0 ° C. or lower is 30 to 90% by mass, and the content of the polymer (B1) is 70 to 10% by mass (total) 100% by mass) of the polymer powder (P) according to [3] or [4].
- Dv volume average primary particle diameter
- the polymer powder (P) described in 1. [8] The polymer powder (P) according to any one of [2] to [7], wherein the content of alkali metal ions is 10 ppm or less. [9] The polymer powder (P) according to any one of [2] to [8], wherein the proportion of particles having a particle diameter of 10 ⁇ m or less is less than 30 vol% and has the following friability. (Crushability) (1) The polymer powder (P) is diluted with ion exchange water. (2) Ultrasonic irradiation is performed at a frequency of 42 kHz and an output of 40 W for 5 minutes.
- the glass transition temperature of the (meth) acrylic acid ester polymer (A1) is 0 ° C. or lower, and the acetone-soluble content of the (meth) acrylic acid ester polymer powder is 5% by mass or higher.
- a stress relaxation agent / pregelling agent for a curable resin comprising the polymer powder (P) according to any one of [2] to [10].
- the stress relaxation agent / pregelling agent for curable resin according to [11], wherein the ratio (G′B / G′A) of the storage elastic modulus G′B at a gelation temperature + 20 ° C. is 10 or more.
- a curable resin composition comprising the polymer powder (P) according to any one of [2] to [10] and a curable resin.
- a semiconductor sealing material using the curable resin composition according to any one of [13] to [15].
- a sheet-like article using the curable resin composition according to any one of [13] to [15].
- the polymer powder (P) has (ii) an acetone-soluble content of 2% by mass or more and 35% by mass or less, an acetone-soluble content of mass average molecular weight of 100,000 or more, and a volume average primary.
- the polymer powder (P) according to [20] wherein the acetone soluble content of the polymer powder (P2) is less than 30% by mass.
- a polymer powder (P2) is obtained by polymerizing a monomer raw material (a2) containing 70 to 99% by mass of methyl methacrylate and 30 to 1% by mass of a monomer raw material other than methyl methacrylate.
- the polymer powder (P) according to any one of [20] to [24], which is a methyl methacrylate polymer powder obtained by pulverizing a vinyl polymer.
- the monomer raw material other than methyl methacrylate is at least one functional group-containing monomer selected from a carboxyl group-containing vinyl monomer, a hydroxyl group-containing vinyl monomer, and a glycidyl group-containing vinyl monomer.
- the polymer powder (P) according to [25] which is contained.
- the monomer raw material other than methyl methacrylate is at least one functional group-containing monomer selected from a carboxyl group-containing vinyl monomer, a hydroxyl group-containing vinyl monomer, and a glycidyl group-containing vinyl monomer.
- the polymer powder (P) is diluted with ion exchange water.
- Ultrasonic irradiation is performed for 3 minutes at a frequency of 42 kHz and an output of 40 W.
- the particle size distribution is measured by laser diffraction / scattering particle size distribution measurement.
- the proportion of particles having a particle diameter of 10 ⁇ m or less is 30 vol% or more.
- a pregelling agent for a curable resin comprising the polymer powder (P) according to any one of [20] to [29].
- a curable resin composition comprising the vinyl polymer powder (P) according to any one of [20] to [29] and a curable resin.
- the curable resin composition according to [32] wherein the curable resin is an epoxy resin.
- the polymer powder of the present invention can quickly bring the curable resin composition into a gel state by heating at a predetermined temperature for a short time.
- a cured product obtained by curing the curable resin composition of the present invention has a low elastic modulus.
- the curable resin composition of the present invention and the cured product thereof are suitable for semiconductor sealing materials.
- the polymer powder of this invention can make a curable resin composition into a gel state rapidly by heating for a short time at predetermined temperature.
- the cured product obtained by curing the curable resin composition of the present invention does not impair transparency.
- the cured product of the present invention is excellent in heat resistance. For this reason, the curable resin composition of the present invention and the cured product thereof are suitable for applications requiring transparency and heat resistance, such as an optical semiconductor material or an optical semiconductor sealing material.
- the polymer powder (P) of the present invention is (i) including a (meth) acrylic acid ester polymer (A1) having a glass transition temperature of 0 ° C. or less, the acetone soluble content of the polymer powder being 5% by mass or more, and the mass of the acetone soluble content Polymer powder (P1) having an average molecular weight of 100,000 or more, and (ii) Polymer powder having an acetone-soluble content of 2% by mass or more and 35% by mass or less, an acetone-soluble component having a mass average molecular weight of 100,000 or more, and a volume average primary particle size (Dv) of 200 nm or more.
- Body (P2) It is a polymer powder (P) selected from the group consisting of:
- the polymer powder (P1) of the present invention is a polymer powder containing a (meth) acrylic acid ester polymer (A1) having a glass transition temperature of 0 ° C. or less, and the polymer powder can be acetone.
- the dissolved content is 5% by mass or more, and the mass average molecular weight of the acetone-soluble component is 100,000 or more.
- (meth) acryl means acryl or methacryl.
- the (meth) acrylic acid ester polymer (A1) is obtained by polymerizing a monomer mixture (a1) containing a (meth) acrylic acid ester monomer (a11), It is a polymer having a glass transition temperature (Tg) determined by the Fox formula of 0 ° C. or less.
- Tg glass transition temperature
- the Tg of the (meth) acrylic acid ester polymer (A1) can be obtained by the Fox equation using the Tg value of the above homopolymer.
- the Fox formula is shown below.
- Tg is the copolymer Tg (° C.)
- wi is the mass fraction of the monomer i
- Tg i is Tg of the homopolymer obtained by polymerizing a monomer i (° C.).
- the numerical value of the Tg of the homopolymer the numerical value described in POLYMER HANDBOOK THIRD EDITION (WILEY INTERSCIENCE) was used.
- the monomer mixture (a1) contains the crosslinkable monomer (a13) and / or the graft crossing agent (a14), the monomer excluding the crosslinkable monomer and the graft crossing agent is used. , Tg is obtained.
- formula of the (meth) acrylic-ester type polymer (A1) is 0 degrees C or less, the elasticity modulus of the hardened
- the Tg obtained from the Fox formula of the (meth) acrylic acid ester polymer (A1) is preferably ⁇ 20 ° C. or lower.
- the polymer (A1) having a Tg determined by the Fox formula of 0 ° C. or less contains, for example, 50% by mass or more of a (meth) acrylic acid ester monomer (a11) having a Tg of a homopolymer of 0 ° C. or less. It can be obtained by polymerizing the monomer mixture (a1).
- Examples of the (meth) acrylic acid ester monomer (a11) having a homopolymer Tg of 0 ° C. or less include ethyl acrylate (Tg of homopolymer: ⁇ 24 ° C.), n-propyl acrylate (same as above). : -37 ° C), n-butyl acrylate (same as -54 ° C), i-butyl acrylate (same as -49 ° C), 2-ethylhexyl acrylate (same as -50 ° C).
- 2-ethylhexyl acrylate (same as ⁇ 50 ° C.) is preferable because it is excellent in lowering the elastic modulus of the cured product.
- 2-ethylhexyl acrylate is preferably contained in an amount of 50% by mass or more in the monomer mixture (a1).
- These monomers (a11) may be used individually by 1 type, and may use 2 or more types together.
- the monomer mixture (a1) is a (meth) acrylic acid ester single monomer, as necessary, within a range where the Tg determined by the Fox formula of the (meth) acrylic acid ester polymer (A1) is 0 ° C. or lower. You may contain other monomers (a12) other than a body (a11).
- Examples of the other monomer (a12) include aromatic vinyl monomers such as styrene, ⁇ -methylstyrene, and alkyl-substituted styrene; methyl (meth) acrylate, ethyl methacrylate, n-propyl methacrylate, methacryl (Meth) acrylic acid ester monomers other than (a11) such as n-butyl acid; vinyl cyanide monomers such as acrylonitrile and methacrylonitrile; vinyl units having a glycidyl group such as glycidyl (meth) acrylate And vinyl monomers having a hydroxyl group such as 2-hydroxyethyl (meth) acrylate, and (meth) acrylic acid-modified silicones. These monomers (a12) may be used individually by 1 type, and may use 2 or more types together.
- the content of the other monomer (a12) in the monomer mixture (a1) (100% by mass) is preferably in the range of 0 to 20% by mass. If the content of the other monomer (a12) in the monomer mixture (a1) (100% by mass) is 20% by mass or less, the elasticity of the cured product obtained by curing the curable resin composition The rate drops.
- the monomer mixture (a1) may contain a crosslinkable monomer (a13) and / or a graft crossing agent (a14) as necessary.
- crosslinkable monomer (a13) examples include ethylene glycol di (meth) acrylate, propylene glycol di (meth) acrylate, 1,3-butylene recall di (meth) acrylate, and di (meth) acrylic acid.
- examples thereof include 1,4-butylene glycol, divinylbenzene, and polyfunctional (meth) acrylic group-modified silicone.
- graft crossing agent (a14) examples include allyl (meth) acrylate, triallyl cyanurate, and triallyl isocyanurate.
- allyl (meth) acrylate can also be used as a crosslinkable monomer (a13).
- crosslinkable monomers (a13) and / or graft crossing agents (a14) may be used alone or in combination of two or more.
- a (meth) acrylic acid ester polymer (A1) having a crosslinked structure can be obtained.
- the content of the crosslinkable monomer (a13) and / or the graft crossing agent (a14) is preferably 0.001 to 15% by mass in total in the monomer mixture (a1) (100% by mass). More preferably, the content is 0.01 to 5% by mass. If the content of the crosslinkable monomer (a13) and / or the graft crossing agent (a14) in the monomer mixture (a1) (100% by mass) is 15% by mass or less, the resulting cured product is low. Elastic modulus.
- the (meth) acrylic acid ester polymer (A1) may be used in combination of two or more different compositions.
- the polymerization method of the monomer mixture (a1) is not particularly limited as long as it is a polymerization method capable of obtaining a particulate polymer, but a polymerization method that is easy to obtain true spherical particles is preferable, for example, an emulsion polymerization method.
- Examples include soap-free emulsion polymerization, dispersion polymerization, swelling polymerization, miniemulsion polymerization, and fine suspension polymerization.
- a soap-free emulsion polymerization method and a fine suspension polymerization method are preferred because a polymer having a particle size excellent in dispersibility in a curable resin and corresponding to fine pitch can be obtained.
- Polymerization aid When polymerizing the monomer mixture (a1), a polymerization initiator, an emulsifier, a dispersion stabilizer, and a chain transfer agent can be used.
- Polymerization initiator As a polymerization initiator used for the polymerization of the monomer mixture (a1), a known polymerization initiator can be used. Since there is no residual metal ion when the (meth) acrylic acid ester polymer is powdered by spray drying, it is preferable to use a polymerization initiator that does not contain metal ions.
- polymerization initiator not containing metal ions examples include 2,2′-azobisisobutyronitrile, 4,4′-azobis- (4-cyanovaleric acid), 2,2′-azobis- Azo compounds such as [N- (2-carboxyethyl) -2-methylpropionamidine]; persulfates such as ammonium persulfate; diisopropylbenzene hydroperoxide, p-menthane hydroperoxide, cumene hydroperoxide, t- Examples thereof include organic peroxides such as butyl hydroperoxide; redox initiators containing the persulfuric acid compound or the organic peroxide as one component. These polymerization initiators may be used alone or in combination of two or more.
- ammonium persulfate 2,2′-azobisisobutyronitrile, 4,4′-azobis- (4-cyanovaleric acid), 2,2′-azobis- [N- ( 2-carboxyethyl) -2-methylpropionamidine] is preferred.
- the polymerization temperature depends on the kind of the polymerization initiator, it can be suitably carried out, for example, in the range of about 40 to 90 ° C.
- emulsifier As the emulsifier used for the polymerization of the monomer mixture (a1), known emulsifiers can be used, for example, alkali metal salts or ammonium salts of higher fatty acids such as disproportionated rosin acid, oleic acid, stearic acid, and dodecyl. Examples include sulfonic acid alkali metal salts such as benzenesulfonic acid, ammonium salts, and nonionic emulsifiers. Among these emulsifiers, it is preferable to use ammonium salt type anionic emulsifiers and nonionic emulsifiers that do not contain metal ions because no metal ions remain in the polymer powder (P1).
- ammonium salt type anionic emulsifier ammonium lauryl sulfate and ammonium di- (2-ethylhexyl) sulfosuccinate are preferable because of excellent stability of emulsion polymerization.
- nonionic emulsifier polyoxyethylene (85) monotetradecyl ether and polyoxyethylene distyrenated phenyl ether are preferable because of excellent stability of emulsion polymerization.
- dispersion stabilizer examples include, for example, poorly water-soluble inorganic salts such as calcium phosphate, calcium carbonate, aluminum hydroxide, and starch silica; nonionic compounds such as polyvinyl alcohol, polyethylene oxide, and cellulose derivatives Polymer compounds; and anionic polymer compounds such as polyacrylic acid or salts thereof, polymethacrylic acid or salts thereof, and copolymers of methacrylic acid esters and methacrylic acid or salts thereof. Of these, nonionic polymer compounds are preferred because of their excellent electrical characteristics. Also, two or more types of dispersion stabilizers can be used in combination depending on the purpose from the viewpoint of compatibility with polymerization stability.
- poorly water-soluble inorganic salts such as calcium phosphate, calcium carbonate, aluminum hydroxide, and starch silica
- nonionic compounds such as polyvinyl alcohol, polyethylene oxide, and cellulose derivatives Polymer compounds
- anionic polymer compounds such as polyacrylic acid or salts thereof, polymethacrylic
- (Chain transfer agent) In the polymerization of the monomer mixture (a1), a chain transfer agent such as t-dodecyl mercaptan, n-octyl mercaptan, or ⁇ -methylstyrene may be used as necessary.
- the (meth) acrylic acid ester polymer (A1) of the present invention may have a single layer or a multilayer structure of two or more layers. Two or more (meth) acrylic acid ester polymers (A1) may be combined.
- the polymer powder (P1) can contain a polymer (B1) other than the (meth) acrylate polymer (A1) having a glass transition temperature of 0 ° C. or lower.
- the polymer (B1) is obtained by polymerizing a monomer mixture (b1) containing a vinyl monomer (b11) capable of radical polymerization.
- the polymer (B) preferably has a Tg determined by the Fox formula exceeding 0 ° C.
- Tg is determined for the monomer excluding the crosslinkable monomer.
- the polymer (B1) having a Tg of 0 ° C or higher determined by the Fox formula polymerizes a monomer mixture (b1) containing 50% by mass or more of a monomer having a Tg of a homopolymer exceeding 0 ° C. Can be obtained.
- Examples of the radically polymerizable vinyl monomer (b11) include methyl (meth) acrylate, ethyl (meth) acrylate, n-propyl (meth) acrylate, i-propyl (meth) acrylate, (meth ) N-butyl acrylate, t-butyl (meth) acrylate, i-butyl (meth) acrylate, n-hexyl (meth) acrylate, n-octyl (meth) acrylate, t- (meth) acrylic acid Butylcyclohexyl, isobornyl (meth) acrylate, dicyclopentadienyl (meth) acrylate, N, N-dimethylaminoethyl (meth) acrylate, N-methyl-2,2,6,6 (meth) acrylate -(Meth) acrylic acid ester monomers such as tetramethylpiperidyl; vinyl cyan
- the monomer mixture (b1) may contain a crosslinkable monomer (b12) as necessary.
- the crosslinkable monomer (b12) include ethylene glycol di (meth) acrylate, propylene glycol di (meth) acrylate, 1,3-butylene glycol di (meth) acrylate, and di (meth) acrylic acid. Examples include 1,4-butylene glycol, divinylbenzene, polyfunctional (meth) acryl group-modified silicone, and allyl (meth) acrylate.
- crosslinkable monomers (b12) may be used individually by 1 type, and may use 2 or more types together.
- the content of the crosslinkable monomer (b12) is preferably 0.001 to 15% by mass, and 0.01 to 5% by mass in the monomer mixture (b1) (100% by mass). Is more preferable.
- the content of the crosslinkable monomer (b12) in the monomer mixture (b1) (100% by mass) is 15% by mass or less, the gelling property-imparting effect is excellent.
- the polymer (B1) may have a single layer or a multilayer structure of two or more layers.
- the method for polymerizing the monomer mixture (b1) is not particularly limited as long as it is a polymerization method capable of obtaining a particulate polymer, but a polymerization method that facilitates obtaining spherical particles is preferable, for example, an emulsion polymerization method.
- Soap-free emulsion polymerization, dispersion polymerization, swelling polymerization, miniemulsion polymerization, and fine suspension polymerization are preferable because a polymer having excellent dispersibility and a particle size corresponding to fine pitch can be obtained.
- the polymer is a spherical particle
- the viscosity of the curable resin composition can be suppressed from increasing, and the curable resin composition having excellent fluidity. Things are obtained.
- Polymerization aid In polymerizing the monomer mixture (b1), a polymerization initiator, an emulsifier, a dispersion stabilizer, and a chain transfer agent can be used.
- Polymerization initiator As a polymerization initiator used for the polymerization of the monomer mixture (b1), the same polymerization initiator as used for the polymerization of the monomer mixture (a1) can be used.
- emulsifier As the emulsifier used for the polymerization of the monomer mixture (b1), the same emulsifier as used for the polymerization of the monomer mixture (a1) can be used.
- Dispersion stabilizer As the emulsifier used for the polymerization of the monomer mixture (b1), the same dispersion stabilizer as that used for the polymerization of the monomer mixture (a1) can be used.
- Chain transfer agent In the polymerization of the monomer mixture (b1), a chain transfer agent such as t-dodecyl mercaptan, n-octyl mercaptan, or ⁇ -methylstyrene may be used as necessary.
- ⁇ Polymer powder (P1)> Composition ratio
- the acrylic ester polymer (A1) is preferably 30 to 90% by mass and the polymer (B1) 70 to 10% by mass. If the content of the (meth) acrylic acid ester polymer (A1) in the whole polymer (100% by mass) is 30% by mass or more, a cured product obtained by curing the curable resin composition is obtained.
- blending a graft copolymer with resin can be suppressed.
- the content of the polymer (B1) in the whole polymer (100% by mass) is 10% by mass or more, the increase over time when the polymer powder (P1) is blended with the curable resin. Viscosity can be suppressed and the gelling property-imparting effect is excellent.
- the content rate of a polymer (B1) is 70 mass% or less, it is excellent in the low elastic modulus reduction of the hardened
- a known pulverization method can be used, and examples thereof include a spray drying method, a freeze drying method, and a coagulation method.
- the spray drying method is preferable because the polymer powder (P1) is excellent in dispersibility in the curable resin.
- the emulsion is sprayed in the form of fine droplets and dried by applying hot air to the emulsion.
- the method for generating micro droplets include a rotating disk type, a pressurized nozzle type, a two-fluid nozzle type, and a pressurized two-fluid nozzle type.
- the capacity of the dryer is not particularly limited, and any capacity from a small scale used in a laboratory to a large scale used industrially can be used.
- the temperature of the hot air introduced into the apparatus that is, the maximum temperature of the hot air that can come into contact with the polymer is excellent in the dispersibility of the polymer powder (P1) in the resulting curable resin composition.
- the temperature is preferably 100 to 200 ° C, more preferably 120 to 180 ° C.
- the polymer emulsion during spray drying may be one kind or a mixture of a plurality of emulsions.
- inorganic fillers such as silica, talc and calcium carbonate, polyacrylate, polyvinyl alcohol, polyacrylamide and the like may be added.
- the acetone soluble content of the polymer powder (P1) is 5% by mass or more. If the acetone soluble content of the polymer powder is 5% by mass or more, sufficient gelling property can be imparted to the curable resin composition, and the flow of the curable resin is suppressed even at high temperatures.
- the acetone soluble content of the polymer powder (P1) is preferably 10% by mass or more, and more preferably 15% by mass or more because it can impart high gelling properties even when the viscosity of the curable resin is extremely low. In particular, in applications that are used at a low viscosity, it is required that a high gelation property can be imparted with a small addition amount.
- the acetone-soluble content of the polymer powder (P1) is preferably 50% by mass or less, more preferably 40% by mass or less, and more preferably 30% by mass or less because the storage stability of the resulting curable resin composition is excellent. Further preferred.
- the acetone-soluble component refers to that obtained by a method for measuring an acetone-soluble component described later.
- the acetone-soluble content is determined based on the content of the crosslinkable monomer (a13) and the graft crossing agent (a14) in the monomer mixture (a1), the (meth) acrylate polymer (A1) and the polymer ( It can be appropriately set by adjusting the composition ratio of B1) and the content of the crosslinkable monomer (b12) in the monomer mixture (b1).
- the content of the crosslinkable monomer (a13) and the graft crossing agent (a14) in the monomer mixture (a1) is decreased, or the polymer ( It is preferable to increase the content of B1) or decrease the content of the crosslinkable monomer (b12) in the monomer mixture (b1).
- the mass average molecular weight of acetone-soluble component is 100,000 or more
- the mass average molecular weight of the acetone-soluble component of the polymer powder (P1) is 100,000 or more. If the mass average molecular weight of the acetone soluble part of the polymer powder (P1) is 100,000 or more, high gelling property can be imparted with a small addition amount, and the flow of the curable resin is suppressed even at high temperatures.
- the mass average molecular weight is preferably 20 million or less because the solubility in the curable resin does not decrease and a sufficient gel state can be obtained in a short time.
- the mass average molecular weight of the acetone-soluble component of the polymer powder (P1) is preferably 150,000 or more, more preferably 200,000 or more, since high gelation property can be imparted even when the viscosity of the curable resin is extremely low. More preferably 400,000 or more. Moreover, since it can be efficiently made into a gel state at a fixed temperature, 10 million or less is more preferable, 5 million or less is more preferable, and 2 million or less is especially preferable. In the present invention, the gel state can be evaluated by gelation temperature and gelation performance obtained by the measurement method described later. Moreover, in this invention, a mass average molecular weight means what was obtained by the measuring method of the mass average molecular weight mentioned later.
- the mass average molecular weight can be appropriately set by adjusting the type of the polymerization initiator, the polymerization initiator amount, the polymerization temperature, and the chain transfer agent amount. In order to increase the mass average molecular weight, it is preferable to reduce the polymerization initiator amount, lower the polymerization temperature, or lower the chain transfer agent amount.
- the volume average primary particle diameter (Dv) of the polymer powder (P1) is preferably 200 nm or more, and more preferably 500 nm or more.
- the powder obtained by spray drying, wet coagulation or the like is an agglomerated powder in which a large number of primary particles are aggregated.
- the agglomerated powder is It is easy to disperse in the primary particles, and the dispersibility of the present powder becomes better when blended with a curable resin such as a liquid epoxy resin.
- the volume average primary particle diameter (Dv) is 200 nm or more, the total surface area of the particles can be sufficiently reduced, so that the viscosity of the curable resin composition is hardly increased.
- the volume average primary particle diameter (Dv) of the polymer powder (P1) is preferably 8 ⁇ m or less, more preferably 5 ⁇ m or less, and 1 ⁇ m or less. More preferably, 800 nm or less is particularly preferable.
- the volume average primary particle diameter (Dv) can be appropriately adjusted according to the polymerization method. For example, particles having a particle diameter of ⁇ 250 nm are obtained by the emulsion polymerization method, ⁇ 1 mm by the soap-free emulsion polymerization method, and ⁇ 10 mm by the fine suspension polymerization method.
- the polymer powder (P1) of the present invention may have any properties or structure as a powder.
- a large number of primary particles obtained by polymerization may be aggregated to form an agglomerated powder (secondary particles), or a higher order structure may be formed.
- secondary particles agglomerated powder
- the polymer powder (P1) preferably has a small volume average primary particle diameter (Dv) and has a good monodispersity because the dispersibility in the curable resin is good.
- the monodispersity of the polymer powder (P1) is the ratio (Dv / Dn) between the volume average primary particle diameter (Dv) and the number average primary particle diameter (Dn) of the polymer powder (P1).
- the Dv / Dn of the polymer powder (P1) is preferably 3.0 or less, more preferably 2.0 or less, and even more preferably 1.5 or less.
- the content of alkali metal ions in the polymer powder (P1) is preferably 10 ppm or less. If the content of alkali metal ions is 10 ppm or less, the insulating properties of the cured product obtained by curing the curable resin composition of the present invention will be excellent.
- the content of alkali metal ions is more preferably 5 ppm or less, and further preferably 1 ppm or less.
- the content of alkali metal ions in the polymer powder (P1) is the total amount of Na ions and K ions, which is obtained by a method for measuring the content of alkali metal ions described later.
- the proportion of particles having a particle size of 10 ⁇ m or less is preferably less than 30 vol%, and from the viewpoint of handling properties, it is preferably 20 vol% or less.
- the particle diameter of the polymer powder (P1) means the particle diameter of an aggregate obtained by a spray drying method or a wet coagulation method. At this time, a large number of primary particles of the polymer powder (P1) are aggregated to form an aggregate.
- the polymer powder (P1) of the present invention preferably has a state in which the primary particles are not firmly bonded to each other and are loosely aggregated, and after irradiation with ultrasonic waves having a frequency of 42 kHz and an output of 40 w for 5 minutes.
- the proportion of particles having a particle diameter of 10 ⁇ m or less is preferably 30 vol% or more. Further, the proportion of particles having a particle diameter of 10 ⁇ m or less is preferably increased by 10 vol% or more after ultrasonic irradiation than before ultrasonic irradiation.
- the above-described ultrasonic irradiation is performed by diluting the obtained polymer powder (P1) with ion-exchanged water.
- a laser diffraction / scattering particle size distribution analyzer manufactured by Shimadzu Corporation, “SALD”). -7100 "
- SALD Shimadzu Corporation
- -7100 " the proportion of particles having a particle diameter of 10 ⁇ m or less is measured on a volume basis.
- the sample concentration of the polymer powder (P1) was appropriately adjusted so as to be within an appropriate range in the scattered light intensity monitor attached to the apparatus.
- the volume average secondary particle diameter (Dv) of the polymer powder (P1) is preferably 6 to 300 ⁇ m, more preferably 10 to 220 ⁇ m, and further preferably 20 to 200 ⁇ m.
- the volume average secondary particle diameter of the polymer powder (P1) is 6 ⁇ m or more, the polymer powder (P1) hardly aggregates in the curable resin, and when it is 300 ⁇ m or less, the polymer powder (P1) is heavy in the curable resin.
- the combined powder (P1) has excellent dispersibility.
- As the volume average secondary particle diameter of the polymer powder (P1) in the curable resin a measurement value obtained by the same measurement method as that of the volume average primary particle diameter can be adopted.
- the ratio of moisture contained in the polymer powder (P1) is preferably 1.5% or less, and more preferably 1.0% or less. If the ratio of the moisture contained in the polymer powder (P1) is 1.5% or less, there is little risk of cracks occurring when the curable resin composition is molded.
- the content of sulfate ions (SO 4 2 ⁇ ) in the polymer powder (P1) is preferably 500 ppm or less, more preferably 300 ppm or less, and even more preferably 100 ppm or less.
- the curable resin composition used in electronic materials is used in an environment where it comes into contact with metal wires such as copper and aluminum, circuit wiring, etc., so if sulfate ions remain, it causes metal corrosion, causing conduction failure and malfunction. It may become. If the content of sulfate ion in the polymer powder (P1) is 100 ppm or less, it can be used for a wide range of applications.
- a sulfate ester or a sulfonic acid compound may be used in addition to the sulfate.
- the sulfonate ion, sulfinate ion and sulfate ester ion contained in these compounds may also cause metal corrosion. Therefore, at the time of polymerization of the monomer mixture, it is preferable to reduce the use amount of sulfate ester, sulfonic acid compound and the like.
- the polymer powder (P1) of the present invention can be used by adding to a curable resin such as an epoxy resin.
- the polymer powder (P1) of the present invention can be used as a stress relaxation agent / pregelling agent for curable resins.
- the stress relaxation agent is an additive that lowers the elastic modulus of the cured product of the curable resin composition
- the pregelling agent is an additive that imparts gelling properties to the curable resin composition.
- the amount of additive added can be reduced, and the initial viscosity, gelling property, and curing property can be reduced.
- a curable resin composition excellent in reducing the elastic modulus of the product can be obtained.
- examples of the curable resin include a thermosetting resin and an active energy ray curable resin.
- examples of the thermosetting resin include epoxy resin, phenol resin, melamine resin, urea resin, oxetane resin, unsaturated polyester resin, alkyd resin, polyurethane resin, acrylic resin, and polyimide resin. These can be used alone or in combination of two or more.
- examples of the active energy ray-curable resin include resins that are cured by irradiation with ultraviolet rays or electron beams.
- active energy ray-curable acrylic resins, active energy ray-curable epoxy resins, and active energy ray-curable oxetane resins are used. Can be mentioned.
- the curable resin a hybrid curing (dual cure) type of thermal curing and active energy ray curing can be used according to the purpose.
- a hybrid curing (dual cure) type of thermal curing and active energy ray curing can be used according to the purpose.
- an epoxy resin, a phenol resin, a polyimide resin, and an oxetane resin are preferable because they have high insulating properties and excellent electrical characteristics and are suitable for the electronic material field.
- epoxy resin examples include bisphenol A type epoxy resins such as JER827, JER828, JER834 (manufactured by Mitsubishi Chemical Corporation), RE-310S (manufactured by Nippon Kayaku Co., Ltd.); JER806L (manufactured by Mitsubishi Chemical Corporation).
- Bisphenol F type epoxy resins such as RE303S-L (manufactured by Nippon Kayaku Co., Ltd.); naphthalene type epoxy resins such as HP-4032 and HP-4032D (manufactured by Dainippon Ink and Chemicals); NC-3000 (Japan) Biphenyl type epoxy resins such as YX4000 (manufactured by Mitsubishi Chemical Corporation); crystalline epoxy resins such as YDC-1312, YSLV-80XY, YSLV-120TE (manufactured by Tohto Kasei Co., Ltd.); Cycloaliphatic epoxy resins such as YX8000 (manufactured by Mitsubishi Chemical Corporation), CEL2021P (manufactured by Daicel Chemical Industries, Ltd.); E N-501H, EPN-501HY, include EPN-502H (manufactured by Nippon Kayaku Co., Ltd.) heat-resistant epoxy resin or the like.
- hydrogenated bisphenol A type epoxy resin bisphenol AD type epoxy resin, bisphenol E type epoxy resin, dicyclopentadiene type epoxy resin, phenol novolac type epoxy resin, cresol novolac type epoxy resin, brominated epoxy resin and glycidylamine Type epoxy resin.
- the epoxy resin a prepolymer of the epoxy resin, a polyether-modified epoxy resin, a copolymer of the epoxy resin such as a silicone-modified epoxy resin and another polymer, and a part of the epoxy resin are epoxy groups. And those substituted with a reactive diluent having
- Examples of the reactive diluent include resorcing ricidyl ether, t-butylphenyl glycidyl ether, 2-ethylhexyl glycidyl ether, allyl glycidyl ether, phenyl glycidyl ether, 3-glycidoxypropyltrimethoxysilane, and 3-glycidide.
- Monoglycidyl compounds such as amines; diglycidyl compounds such as neopentyl glycol diglycidyl ether, 1,6-hexanediol diglycidyl ether, propylene glycol diglycidyl ether; and 2- (3,4) -epoxycyclohexyl) Mono alicyclic epoxy compounds such as Le trimethoxysilane.
- the epoxy resin can be used alone or in combination of two or more.
- the epoxy resin is an epoxy resin that is liquid at room temperature or a liquid that is solid at room temperature but does not sufficiently cure when heated, in terms of imparting gelling properties to the epoxy resin composition. What contains 50 mass% or more of epoxy resin to convert is preferable.
- examples of the epoxy resin include bisphenol A type epoxy resin, hydrogenated bisphenol A type epoxy resin, bisphenol F type epoxy resin, and bisphenol S type.
- the curable resin composition of the present invention contains the aforementioned polymer powder (P1) and a curable resin.
- the amount of the polymer powder (P1) in the curable resin composition is preferably 5 parts by mass or more, more preferably 10 parts by mass, and even more preferably 15 parts by mass or more.
- the blending ratio of the polymer powder (P1) is 5 parts by mass or more, the effect as an additive of the polymer powder (P1) is sufficiently exhibited.
- a compounding rate of polymer powder (P1) 50 mass parts or less are preferable, and 40 mass parts or less are more preferable. If the blending ratio of the polymer powder (P1) is 50 parts by mass or less, an increase in the paste viscosity of the curable resin composition can be suppressed, and the workability and workability may be reduced depending on the application. Can be suppressed.
- additives can be blended with the curable resin composition of the present invention within a range not impairing the effects of the present invention.
- additives include conductive fillers such as silver powder, gold powder, nickel powder, and copper powder; insulating fillers such as aluminum nitride, calcium carbonate, silica, and alumina; thixotropic agents, fluidity improvers, flame retardants, and heat stabilizers , Antioxidants, ultraviolet absorbers, ion adsorbents, coupling agents, mold release agents and stress relaxation agents.
- a known one such as phosphorus, halogen, or inorganic flame retardant may be used.
- the heat stabilizer include phenolic antioxidants, sulfur antioxidants, and phosphorus antioxidants. The antioxidants can be used alone, but it is preferable to use two or more kinds in combination, such as phenol / sulfur or phenol / phosphorus.
- a well-known kneading apparatus When preparing the curable resin composition of this invention, a well-known kneading apparatus can be used.
- the kneading apparatus for obtaining the curable resin composition include a raking machine, an attritor, a planetary mixer, a dissolver, a three roll, a ball mill, and a bead mill. Two or more of these can be used in combination.
- the order of blending is not particularly limited, but in order to fully demonstrate the effects of the present invention, the polymer powder (P1) is kneaded at the end as much as possible. It is preferable to do.
- the curable resin composition of the present invention seals a liquid sealing material such as a primary mounting underfill material, a secondary mounting underfill material, a grab top material in wire bonding; Sealing sheet; Pre-dispensing type underfill material; Sealing material for semiconductors such as a sealing sheet encapsulated at the wafer level, optical adhesive; Liquid crystal display such as sealing materials for various flat panel displays such as liquid crystal and organic EL Device sealant, adhesive layer for three-layer copper-clad laminate; adhesive layer for die bond film, die attach film, interlayer insulating film, coverlay film, etc .; die bond paste, interlayer insulating paste, conductive paste, anisotropic conductive paste, etc.
- a liquid sealing material such as a primary mounting underfill material, a secondary mounting underfill material, a grab top material in wire bonding
- Sealing sheet Pre-dispensing type underfill material
- Sealing material for semiconductors such as a sealing sheet encapsulated at the wafer level, optical adhesive
- a sealing sheet that collectively seals various chips on the substrate a sealing sheet that collectively seals at the wafer level
- a sealing sheet that collectively seals at the wafer level a sealing sheet that collectively seals at the wafer level
- a die bond film a sealing sheet that collectively seals at the wafer level
- a die attach film a sealing sheet that collectively seals at the wafer level
- a die attach film a sealing sheet that collectively seals at the wafer level
- a die bond film a sealing sheet that collectively seals at the wafer level
- a die bond film a sealing sheet that collectively seals at the wafer level
- a die bond film a sealing sheet that collectively seals at the wafer level
- a die bond film a sealing sheet that collectively seals at the wafer level
- a die attach film a sealing sheet that collectively seals at the wafer level
- an interlayer insulating film a coverlay film
- the cured product of the present invention is obtained by curing a curable resin composition.
- the curable resin composition can be cured using a curing agent such as an acid anhydride, an amine compound, or a phenol compound.
- a curing agent such as an acid anhydride, an amine compound, or a phenol compound.
- Examples of the acid anhydride include phthalic anhydride, methyltetrahydrophthalic anhydride, methylhexahydrophthalic anhydride, hexahydrophthalic anhydride, tetrahydrophthalic anhydride, trialkyltetrahydrophthalic anhydride, methyl hymic anhydride, methylcyclohexene Tetracarboxylic anhydride, trimellitic anhydride, pyromellitic anhydride, benzophenone tetracarboxylic anhydride, ethylene glycol bistrimellitate, glycerol tris trimellitate, dodecenyl succinic anhydride, poly azelaic anhydride and poly (ethyl octadecane) And diacid) anhydride.
- methylhexahydrophthalic anhydride and hexahydrophthalic anhydride are preferred for uses that require weather resistance, light resistance, heat resistance, and the like.
- amine compound examples include aliphatic polyamines such as ethylenediamine, diethylenetriamine, triethylenetetramine, tetraethylenepentamine, hexamethylenediamine, trimethylhexamethylenediamine, m-xylenediamine, 2-methylpentamethylenediamine, and diethylaminopropylamine; Isophorodiamine, 1,3-bisaminomethylcyclohexane, methylenebiscyclohexanamine, norbornenediamine, 1,2-diaminocyclohexane, bis (4-amino-3-methyldicyclohexyl) methane, diaminodicyclohexylmethane, 2,5 ( 2,6) -bis (aminomethyl) bicyclo [2,2,1] heptane and other alicyclic polyamines; diaminodiethyldiphenylmethane, diaminophenylmethane, di Mino diphenyl
- 2,5 (2,6) -bis (aminomethyl) bicyclo [2,2,1] heptane and isophoronediamine are preferred for applications requiring weather resistance, light resistance, heat resistance and the like. These can be used alone or in combination of two or more.
- the phenol compound include phenol novolac resins, cresol novolac resins, bisphenol A, bisphenol F, bisphenol AD, and derivatives of diallysates of these bisphenols. Of these, bisphenol A is preferred because of its excellent mechanical strength and curability. These can be used alone or in combination of two or more.
- the amount of the curing agent used is preferably 20 to 120 parts by mass and more preferably 60 to 110 parts by mass with respect to 100 parts by mass of the epoxy resin because the cured product is excellent in heat resistance and curability.
- the amount of the curing agent used is preferably an equivalent ratio of 0.7 to 1.3 equivalents, more preferably 0.001 equivalents of acid anhydride groups per equivalent of epoxy groups.
- the active hydrogen is preferably 0.3 to 1.4 equivalent, more preferably about 0.4 to 1.2 equivalent.
- the active hydrogen is preferably about 0.3 to 0.7 equivalent, more preferably about 0.4 to 0.6 equivalent.
- a hardening accelerator when hardening an epoxy resin, a hardening accelerator, a latent hardening agent, etc. can be used as needed.
- the curing accelerator a known one used as a thermosetting catalyst for epoxy resins can be used.
- imidazole compounds such as 2-methylimidazole and 2-ethyl-4-methylimidazole;
- examples include adducts of epoxy resins; organophosphorus compounds such as triphenylphosphine; borates such as tetraphenylphosphine tetraphenylborate; and diazabicycloundecene (DBU).
- DBU diazabicycloundecene
- the curing accelerator is usually added in an amount of 0.1 to 8 parts by mass, preferably 0.5 to 6 parts by mass with respect to 100 parts by mass of the epoxy resin.
- the latent curing agent is solid at room temperature, and liquefies when the epoxy resin is heated and cured to act as a curing agent.
- latent curing agents include dicyandiamide, carbohydrazide, oxalic acid dihydrazide, malonic acid dihydrazide, succinic acid dihydrazide, iminodiacetic acid dihydrazide, adipic acid dihydrazide, pimelic acid dihydrazide, suberic acid dihydrazide, azelaic acid dihydrazide dihydrazide, Dodecanedihydrazide, hexadecanedihydrazide, maleic acid dihydrazide, fumaric acid dihydrazide, diglycolic acid dihydrazide, tartaric acid dihydrazide, malic acid dihydrazide, isophthalic acid dihydrazide, terephthalic acid dihydrazide, 2,6-naphtho
- an oxetane resin for example, a curing agent such as an acid anhydride, or a curing catalyst capable of initiating ring opening and polymerization of the oxetane ring by heat is blended and cured.
- a curing agent such as an acid anhydride
- a curing catalyst capable of initiating ring opening and polymerization of the oxetane ring by heat is blended and cured.
- the oxetane resin include EHO, OXBP, OXMA, and OXTP (manufactured by Ube Industries, Ltd.).
- the amount of the curing agent or curing catalyst used is the same as that for the epoxy resin.
- the curing condition is, for example, 80 to 180 ° C. for about 10 minutes to 5 hours.
- active energy ray curable resin as an active energy ray to be used, an electron beam, an ultraviolet-ray, a gamma ray, and infrared rays are mentioned, for example.
- a known ultraviolet irradiation device including a high-pressure mercury lamp, an excimer lamp, a metal halide lamp, or the like can be used when curing with ultraviolet rays.
- the amount of ultraviolet irradiation is about 50 to 1,000 mJ / cm 2 .
- a known electron beam irradiation apparatus can be used, and the amount of electron beam irradiation is about 10 to 100 kGy.
- the cured product obtained by curing the curable resin composition of the present invention can be used for various applications including electronic materials, and applications that require a low elastic modulus such as semiconductor sealing materials and adhesives. Is particularly suitable.
- Polymer powder (P2) The polymer powder (P2) of the present invention has an acetone-soluble content of 2% by mass to 35% by mass, an acetone-soluble content mass average molecular weight of 100,000 or more, and a volume average primary particle diameter (Dv ) Is a polymer powder of 200 nm or more.
- the acetone-soluble component means a dissolved mass (%) after dissolving a predetermined amount of the polymer powder (P2) in 50 times mass of acetone and refluxing at 70 ° C. for 6 hours. To do. More specifically, it is obtained by a method for measuring acetone-soluble matter described later. If the acetone soluble content of the polymer powder (P2) is 35% by mass or less, the decrease in the glass transition temperature of the cured product when the polymer powder (P2) is blended with the curable resin composition is suppressed. can do.
- the acetone-soluble content of the polymer powder (P2) is 2% by mass or more, preferably 5% by mass or more, preferably 8% by mass, because it can impart high gelling properties even when the viscosity of the curable resin is extremely low.
- the above is more preferable.
- cured material can be suppressed, it is 35 mass% or less, less than 30 mass% is preferable, 25 mass% or less is more preferable, and 20 mass% or less is still more preferable.
- the acetone-soluble component can be appropriately set by adjusting the content of the crosslinkable monomer in the monomer raw material. In order to increase the acetone-soluble content, the content of the crosslinkable monomer is decreased. To decrease the acetone-soluble content, the content of the crosslinkable monomer may be increased.
- Mass average molecular weight of acetone-soluble matter If the mass average molecular weight of the acetone soluble part of the polymer powder (P2) is 100,000 or more, high gelling property can be imparted with a small addition amount, and the flow of the curable resin is suppressed even at a high temperature. Moreover, since the solubility to curable resin does not fall and it can be made into a sufficient gel state for a short time, the mass average molecular weight of acetone soluble part is 20 million or less.
- the mass average molecular weight of the acetone-soluble component of the polymer powder (P2) is preferably 200,000 or more, more preferably 300,000 or more, since high gelling property can be imparted even when the viscosity of the curable resin is extremely low. More preferably 400,000 or more. Moreover, since it can be efficiently made into a gel state at a fixed temperature, 10 million or less is more preferable, 5 million or less is still more preferable, and 2 million or less is especially preferable. In the present invention, the gel state can be evaluated by gelation temperature and gelation performance obtained by the measurement method described later. Moreover, in this invention, a mass average molecular weight means what was obtained by the measuring method of the mass average molecular weight mentioned later.
- the mass average molecular weight can be appropriately set by adjusting the type of the polymerization initiator, the polymerization initiator amount, the polymerization temperature, and the chain transfer agent amount. In order to increase the mass average molecular weight, it is preferable to reduce the polymerization initiator amount, lower the polymerization temperature, or lower the chain transfer agent amount.
- the volume average primary particle diameter (Dv) of the polymer powder (P2) is 200 nm or more, preferably 300 nm or more, more preferably 500 nm or more, and further preferably 600 nm or more. If the volume average primary particle diameter (Dv) is 200 nm or more, the total surface area of the particles can be sufficiently reduced, so that the viscosity of the curable resin composition is hardly increased. Further, since it is possible to cope with fine pitch and thin film, the volume average primary particle diameter (Dv) of the polymer powder (P2) is preferably 8 ⁇ m or less, more preferably 5 ⁇ m or less, and 1 ⁇ m or less. Further preferred.
- the volume average primary particle diameter (Dv) can be appropriately adjusted according to the polymerization method.
- particles having a particle diameter of ⁇ 250 nm are obtained by the emulsion polymerization method, ⁇ 1 mm by the soap-free emulsion polymerization method, and ⁇ 10 mm by the fine suspension polymerization method.
- the amount of emulsifiers when superposing
- the polymer powder (P2) preferably has good monodispersity.
- the monodispersity of the polymer powder (P2) is the ratio (Dv / Dn) of the volume average primary particle diameter (Dv) and the number average primary particle diameter (Dn) of the polymer powder (A).
- the Dv / Dn of the polymer powder (P2) is preferably 3.0 or less, more preferably 2.0 or less, and even more preferably 1.5 or less.
- the polymer powder (P2) may have any properties or structure as a powder.
- a large number of primary particles obtained by polymerization may be aggregated to form an agglomerated powder (secondary particles), or a higher order structure may be formed.
- secondary particles agglomerated powder
- the primary particles are not firmly bonded to each other and are loosely aggregated. Thereby, primary particles are finely and uniformly dispersed in the curable resin.
- the powder obtained by spray drying or wet coagulation is usually an agglomerated powder in which a large number of primary particles are aggregated.
- the diameter (Dv) is 200 nm or more, the aggregated powder is easily dispersed in the primary particles, and the dispersibility in the epoxy resin is improved.
- the refractive index of the polymer powder (P2) is not particularly limited, but the refractive index at 20 ° C. is preferably 1.48 or more and 1.51 or less.
- the refractive index of each layer is preferably 1.48 or more and 1.51 or less.
- the refractive index is within the above range, the cured product obtained by blending the polymer powder (P2) has excellent transparency.
- the refractive index of the polymer powder (P2) is preferably 1.49 or more and more preferably 1.50 or more from the viewpoint of transparency of the cured product. In the present invention, the refractive index at 20 ° C.
- the refractive index of the copolymer can be calculated from the volume ratio.
- the glass transition temperature of the polymer powder (P2) is not particularly limited, but the glass transition temperature is preferably 0 ° C. or higher.
- the glass transition temperature of each layer is preferably 0 ° C. or higher. If the glass transition temperature is 0 ° C. or higher, even when blended in a large amount in the curable resin composition, the glass transition temperature of the cured product can be suppressed from being lowered, and can be used in applications where high heat resistance is required. it can. Further, the glass transition temperature is preferably 250 ° C. or lower because the gel state can be efficiently achieved at a constant temperature.
- the glass transition temperature of the vinyl polymer can be determined by the Fox equation.
- the vinyl polymer is a homopolymer
- standard analytical values described in the “Polymer Data Handbook” edited by the Polymer Science Society can be adopted. It can be calculated by the formula (1) from the Tg of the unit homopolymer.
- Tg is Tg (° C.) of the copolymer
- wi is a mass fraction of the monomer i
- Tgi is Tg (° C.) of a homopolymer obtained by polymerizing the monomer i.
- the content of alkali metal ions in the polymer powder (P2) is preferably 10 ppm or less, more preferably 5 ppm or less, and even more preferably 1 ppm or less.
- the curable resin composition is used for various applications, the presence of a small amount of ionic impurities may cause insulation failure. Therefore, if the content of alkali metal ions is within the above range, it can be used for a wide range of applications. It can also be used in applications that require a large amount of pregelling agent.
- the content of alkali metal ions in the polymer powder (P2) is the total amount of Na ions and K ions, which is obtained by a method for measuring the content of alkali metal ions described later.
- the content of sulfate ion (SO 4 2 ⁇ ) in the polymer powder (P2) is preferably 20 ppm or less.
- the curable resin composition used in electronic materials is used in an environment where it comes into contact with metal wires such as copper and aluminum, circuit wiring, etc., so if sulfate ions remain, it causes metal corrosion, causing conduction failure and malfunction. It may become. If the content of sulfate ions in the polymer powder (P2) is 20 ppm or less, it can be used for a wide range of applications.
- a sulfate ester or a sulfonic acid compound may be used in addition to the sulfate.
- the sulfonate ion, sulfinate ion and sulfate ester ion contained in these compounds may also cause metal corrosion. Therefore, at the time of polymerization of the vinyl monomer raw material (a2), it is preferable to reduce the amount of sulfuric acid ester, sulfonic acid compound and the like used.
- the vinyl polymer (A2) is obtained by polymerizing a radically polymerizable vinyl monomer raw material (a2).
- the radically polymerizable vinyl monomer raw material (a2) include methyl (meth) acrylate, ethyl (meth) acrylate, n-propyl (meth) acrylate, i-propyl (meth) acrylate, ( N-butyl (meth) acrylate, t-butyl (meth) acrylate, i-butyl (meth) acrylate, n-hexyl (meth) acrylate, n-octyl (meth) acrylate, (meth) acrylic acid 2 -Ethylhexyl, cyclohexyl (meth) acrylate, benzyl (meth) acrylate, phenyl (meth) acrylate, nonyl (meth) acrylate, de
- (meth) acrylic acid ester is preferable because radical polymerization is easy and emulsion polymerization is easy. Furthermore, among these, since the polymerization stability is excellent, it is possible to use a monomer raw material (a2) containing 70 to 99% by mass of methyl methacrylate and 30 to 1% by mass of a vinyl monomer raw material other than methyl methacrylate. preferable.
- (meth) acryl means “acryl” or “methacryl”.
- the monomer raw material (a2) includes butyl methacrylate, a carboxyl group-containing vinyl monomer, a hydroxyl group-containing vinyl monomer, and a glycidyl group-containing vinyl monomer as vinyl monomer raw materials other than methyl methacrylate. It is preferable to contain at least one functional group-containing monomer selected from a monomer. Thereby, transparency of the hardened
- the content of at least one functional group-containing monomer selected from a carboxyl group-containing vinyl monomer, a hydroxyl group-containing vinyl monomer, and a glycidyl group-containing vinyl monomer in the monomer raw material (a2) is as follows. In view of transparency of the cured product, it is preferably 2% by mass or more, more preferably 4% by mass or more, and further preferably 6% by mass or more. Further, from the viewpoint of polymerization stability, the content of at least one functional group-containing monomer is preferably 30% by mass or less, and more preferably 20% by mass or less.
- carboxyl group-containing vinyl monomer methacrylic acid is preferable because radical polymerization is easy and emulsion polymerization is easy.
- hydroxyl group-containing vinyl monomer 2-hydroxyethyl (meth) acrylate is preferable because radical polymerization is easy and emulsion polymerization is easy.
- glycidyl group-containing vinyl monomer glycidyl (meth) acrylate is preferable because radical polymerization is easy and emulsion polymerization is easy.
- a carboxyl group-containing vinyl monomer, a hydroxyl group-containing vinyl monomer and glycidyl are used as monomer raw materials in each stage. It is preferable to use a monomer raw material (a2) containing 2% by mass or more of at least one functional group-containing monomer selected from group-containing vinyl monomers.
- the composition of the monomer raw material in each stage in the multistage polymerization may be the same or different.
- the polymerization method of the vinyl monomer raw material (a2) is easy to obtain spherical particles and easy to control the particle morphology. Therefore, emulsion polymerization, soap-free emulsion polymerization, swelling polymerization, miniemulsion A polymerization method, a dispersion polymerization method and a fine suspension polymerization method are preferred. Among these, a soap-free emulsion polymerization method is more preferable because a polymer having excellent dispersibility and a particle size corresponding to fine pitch can be obtained.
- the vinyl polymer (A2) is preferably spherical particles because the viscosity of the curable resin composition does not increase and the fluidity is excellent.
- the internal morphology of the vinyl polymer (primary particles) (A2) is not particularly limited, and even if various factors such as polymer composition, molecular weight, glass transition temperature, solubility parameter are uniform, the core-shell structure and gradient It may have various generally recognized particle morphologies such as structure.
- Examples of a method for controlling the internal morphology of the vinyl polymer (A2) include a method of forming multilayer structure particles having different solubility parameters and molecular weights on the inside and outside of the particle. This method is preferable because the storage stability (pot life) of the curable resin composition and the gelation rate are easily compatible.
- An industrially highly practical method for controlling the internal morphology of the vinyl polymer (A2) includes, for example, a method in which vinyl monomer mixtures having different compositions are successively dropped in multiple stages.
- a method for determining whether or not the vinyl polymer (A2) has a core-shell structure for example, the particle diameter of the polymer particles sampled in the polymerization process is surely growing, and the sample is sampled in the polymerization process. Confirming that the minimum film-forming temperature (MFT) and solubility in various solvents of the polymer particles are satisfied at the same time.
- MFT minimum film-forming temperature
- a method of confirming the presence or absence of a concentric structure by observing a section of the vinyl polymer with a transmission electron microscope (TEM), or a section of the frozen and broken polymer section with a scanning electron microscope (cryo SEM) And confirming the presence or absence of a concentric structure.
- TEM transmission electron microscope
- crystal SEM scanning electron microscope
- ⁇ Polymerization aid> When polymerizing the vinyl monomer raw material (a2), a polymerization initiator, an emulsifier, a dispersion stabilizer, and a chain transfer agent can be used.
- a known polymerization initiator can be used as the polymerization initiator.
- the vinyl polymer is pulverized by spray drying, there is no residue of metal ions, so it is preferable to use a polymerization initiator that does not contain metal ions.
- polymerization initiator not containing metal ions examples include 2,2′-azobisisobutyronitrile, 4,4′-azobis- (4-cyanovaleric acid), 2,2′-azobis- Azo compounds such as [N- (2-carboxyethyl) -2-methylpropionamidine]; persulfates such as ammonium persulfate; diisopropylbenzene hydroperoxide, p-menthane hydroperoxide, cumene hydroperoxide, t- Examples thereof include organic peroxides such as butyl hydroperoxide; redox initiators containing the persulfuric acid compound or the organic peroxide as one component. These polymerization initiators can be used alone or in combination of two or more.
- ammonium persulfate 2,2′-azobisisobutyronitrile, 4,4′-azobis- (4-cyanovaleric acid), 2,2′-azobis- [N- ( 2-carboxyethyl) -2-methylpropionamidine] is preferred.
- the polymerization temperature depends on the kind of the polymerization initiator, it can be suitably carried out, for example, in the range of about 40 to 90 ° C.
- emulsifier As the emulsifier, known emulsifiers can be used, for example, alkali metal salts or ammonium salts of higher fatty acids such as disproportionated rosin acid, oleic acid, stearic acid, alkali metal salts of sulfonic acid such as dodecylbenzenesulfonic acid, Examples thereof include ammonium salts and nonionic emulsifiers.
- the emulsifier can be used alone or in combination of two or more.
- ammonium salt type anionic emulsifiers and nonionic emulsifiers that do not contain metal ions because no metal ions remain in the polymer powder (P2).
- ammonium salt type anionic emulsifier ammonium lauryl sulfate and ammonium di- (2-ethylhexyl) sulfosuccinate are preferable because of excellent stability of emulsion polymerization.
- nonionic emulsifier polyoxyethylene (85) monotetradecyl ether and polyoxyethylene distyrenated phenyl ether are preferable because of excellent stability of emulsion polymerization.
- Dispersion stabilizer examples include poorly water-soluble inorganic salts such as calcium phosphate, calcium carbonate, aluminum hydroxide, and starch silica; nonionic polymer compounds such as polyvinyl alcohol, polyethylene oxide, and cellulose derivatives; and polyacrylic acid or a salt thereof. And anionic polymer compounds such as polymethacrylic acid or a salt thereof, and a copolymer of a methacrylic acid ester and methacrylic acid or a salt thereof. Of these, nonionic polymer compounds are preferred because of their excellent electrical characteristics. Also, two or more types of dispersion stabilizers can be used in combination depending on the purpose from the viewpoint of compatibility with polymerization stability.
- poorly water-soluble inorganic salts such as calcium phosphate, calcium carbonate, aluminum hydroxide, and starch silica
- nonionic polymer compounds such as polyvinyl alcohol, polyethylene oxide, and cellulose derivatives
- anionic polymer compounds such as polymethacrylic acid or a salt thereof,
- Chain transfer agent examples include mercaptans such as n-dodecyl mercaptan, t-dodecyl mercaptan, n-octyl mercaptan, t-octyl mercaptan, n-tetradecyl mercaptan, n-hexyl mercaptan, and n-butyl mercaptan; carbon tetrachloride And halogen compounds such as ethylene bromide; and ⁇ -methylstyrene dimer.
- chain transfer agents can be used alone or in combination of two or more.
- a known pulverization method can be used, and examples thereof include a spray drying method, a freeze drying method, and a coagulation method.
- the spray drying method is preferable because the polymer powder (P2) is excellent in dispersibility in the curable resin.
- the emulsion is sprayed into fine droplets and dried by applying hot air to the emulsion.
- the method for generating fine droplets include a rotating disk type, a pressurized nozzle type, a two-fluid nozzle type, and a pressurized two-fluid nozzle type.
- the capacity of the dryer is not particularly limited, and any capacity from a small scale used in a laboratory to a large scale used industrially can be used.
- the positions of the inlet part which is the supply part of the heating gas for drying and the outlet part which is the outlet for the heating gas for drying and the dry powder in the dryer are not particularly limited, and may be the same as the spray drying apparatus used normally.
- the temperature of hot air introduced into the apparatus that is, the maximum temperature of hot air that can come into contact with the vinyl polymer is excellent in dispersibility of the polymer powder (P2) in the resulting curable resin composition.
- inlet temperature that is, the maximum temperature of hot air that can come into contact with the vinyl polymer is excellent in dispersibility of the polymer powder (P2) in the resulting curable resin composition.
- 100 to 200 ° C is preferable, and 120 to 180 ° C is more preferable.
- a vinyl polymer emulsion may be used alone or a plurality of emulsions may be mixed.
- inorganic fillers such as silica, talc and calcium carbonate, polyacrylate, polyvinyl alcohol, polyacrylamide and the like may be added.
- the proportion of particles having a particle size of 10 ⁇ m or less is preferably less than 30 vol%, and from the viewpoint of handling properties, it is preferably 20 vol% or less.
- the particle diameter of the polymer powder (P2) means the particle diameter of an aggregate obtained by a spray drying method or a wet coagulation method. At this time, a large number of primary particles of the polymer powder (P2) are aggregated to form an aggregate.
- the polymer powder (P2) of the present invention has a state in which the primary particles are not firmly bonded to each other and are loosely aggregated, after being irradiated with ultrasonic waves having a frequency of 42 kHz and an output of 40 w for 3 minutes, 10 ⁇ m
- the proportion of particles having the following particle diameter is 30 vol% or more.
- the proportion of particles having a particle diameter of 10 ⁇ m or less is preferably increased by 10 vol% or more after ultrasonic irradiation than before ultrasonic irradiation.
- the proportion of particles having a particle diameter of 10 ⁇ m or less is preferably 40 vol% or more, and more preferably 50 vol% or more.
- the above-described ultrasonic irradiation is performed by diluting the obtained polymer powder (P2) with ion-exchanged water.
- a laser diffraction / scattering particle size distribution measuring apparatus manufactured by Shimadzu Corporation, “SALD” -7100 "
- SALD laser diffraction / scattering particle size distribution measuring apparatus
- the ratio of particles having a particle diameter of 10 ⁇ m or less is measured on a volume basis.
- the sample concentration of the polymer powder (P2) was appropriately adjusted to be within an appropriate range in the scattered light intensity monitor attached to the apparatus.
- the polymer powder (P2) of the present invention can be used by adding to a curable resin.
- the curable resin used in the present invention examples include bisphenol A type epoxy resin, bisphenol F type epoxy resin, bisphenol S type epoxy resin, orthocresol novolac type epoxy resin, alicyclic epoxy resin, triglycidyl isocyanurate, aliphatic And epoxy resins such as epoxy resins. These may be used alone or in combination of two or more.
- the epoxy resin is an epoxy resin that is liquid at room temperature or a liquid that is solid at room temperature but does not sufficiently cure when heated, in terms of imparting gelling properties to the epoxy resin composition. What contains 50 mass% or more of epoxy resin to convert is preferable. Further, among these epoxy resins, alicyclic epoxy resins are preferable because they are excellent in transparency and heat resistance and are suitable for optical semiconductor materials.
- alicyclic epoxy resins examples include 3,4-epoxycyclohexylmethyl-3 ′, 4′-epoxycyclohexanecarboxylate (Celoxide 2021), 3,4-epoxycyclohexylmethyl-3 ′, 4′-epoxycyclohexanecarboxyl.
- Dimer adduct of selenium and ⁇ -caprolactone (Celoxide 2081), 1,2,8,9-diepoxy limonene (Celoxide 3000) (both manufactured by Daicel), bisphenol A type hydrogenated cycloaliphatic epoxy Resin (YX-8000, YX-8034: manufactured by Mitsubishi Chemical Corporation, EPICLON 750: manufactured by Dainippon Ink & Chemicals, Inc.).
- the curable resin composition of the present invention contains polymer powder (P2) and a curable resin.
- the amount of the polymer powder (P2) in the curable resin composition is preferably 5 parts by mass or more, more preferably 10 parts by mass or more, and still more preferably 15 parts by mass or more.
- the blending amount of the polymer powder (P2) is 5 parts by mass or more, a sufficient gel state can be realized, and bleeding of the curable resin composition, pattern disturbance, and the like can be suppressed.
- a compounding quantity of polymer powder (P2) 50 mass parts or less are preferable, and 30 mass parts or less are more preferable.
- a plurality of polymer powders (P2) having different gelation temperatures may be used in combination.
- the polymer powder (P2) of the present invention can be used as a pregelling agent for a curable resin that is added to the curable resin to realize a gel state.
- additives can be blended with the curable resin composition of the present invention within a range not impairing the effects of the present invention.
- additives include silicone-based and fluorine-based antifoaming agents, leveling agents, silane coupling agents such as ⁇ -glycidoxypropyltrimethoxysilane, surfactants, fillers, flame retardants, colorants, and antioxidants.
- Conventional additives such as an agent, an ultraviolet absorber, an ion adsorbent, a plasticizer, a pigment, a release agent, glass beads, inorganic particles, and a stress relaxation agent can be used.
- antioxidants examples include a phenol-based antioxidant, a sulfur-based antioxidant, and a phosphorus-based antioxidant.
- the antioxidants can be used alone, but it is preferable to use two or more kinds in combination, such as phenol / sulfur or phenol / phosphorus.
- plasticizer known ones can be used, for example, phthalate ester, aliphatic dibasic ester, polyester plasticizer, epoxy plasticizer, phosphate ester plasticizer, trimellitic acid plasticizer, and the like. Can be mentioned.
- the flame retardant does not deviate from the object of the present invention, a known one such as phosphorus, halogen, or inorganic flame retardant may be used.
- the inorganic particles include conductive fillers such as silver powder, gold powder, nickel powder, and copper powder; and insulating fillers such as aluminum nitride, calcium carbonate, silica, and alumina.
- kneading apparatus When preparing the curable resin composition of this invention, a well-known kneading apparatus can be used.
- the kneading apparatus for obtaining the curable resin composition include a raking machine, an attritor, a planetary mixer, a dissolver, a three roll, a ball mill, and a bead mill. Moreover, these can use 2 or more types together.
- the order of blending is not particularly limited, but the polymer powder (P2) is kneaded at the end as much as possible in order to fully exhibit the effects of the present invention. It is preferable to do. Further, when the temperature in the system is increased due to shearing heat generation or the like due to kneading, it is preferable to devise measures not to increase the temperature during kneading.
- the total light transmittance at 23 ° C. of the 3 mm thick curable resin obtained by curing is preferably 70.0% or more, and 80.0% or more. More preferably.
- the total light transmittance at 120 ° C. of the cured curable resin having a thickness of 3 mm obtained by curing is preferably 70.0% or more, and 80.0% More preferably.
- the elastic modulus of the cured product obtained by curing the curable resin composition of the present invention by blending the polymer powder (P2) decreases.
- the curable resin composition of the present invention includes a transparent underfill material, a transparent liquid sealing material, a transparent sealing sheet, a transparent die attach film, a transparent die attach paste, a light emitting diode sealing material, a transparent adhesive, and an optical adhesive. It can be used for various applications such as various sealing materials such as various flat panel displays such as an agent, liquid crystal, and organic EL. Especially, it can use suitably for sealing materials for optical semiconductors, such as a transparent underfill material, a transparent liquid sealing material, a transparent sealing sheet, and a light emitting diode sealing material.
- the cured product of the present invention is obtained by curing the curable resin composition of the present invention.
- the curing conditions can be appropriately determined depending on the type and content of each component constituting the curable resin composition.
- the curing temperature is generally 80 to 180 ° C.
- the curable resin composition can be cured using a curing agent such as an acid anhydride, an amine compound, or a phenol compound.
- the curable resin composition of the present invention as an encapsulating material for optical semiconductors, those with relatively little coloration are preferable.
- an acid anhydride curing agent and alicyclic acid anhydride curing.
- An agent is more preferable.
- Specific examples include hexahydrophthalic anhydride, methylhexahydrophthalic anhydride, tetrahydrophthalic anhydride, and hydrogenated methylnadic acid anhydride. These may be used alone or in combination of two or more.
- the amount of the curing agent to be used is preferably 50 to 150 parts by mass, more preferably 60 to 140 parts by mass with respect to 100 parts by mass of the curable resin because the cured resin is excellent in heat resistance and curability.
- the acid anhydride group equivalent per equivalent of epoxy group is preferably 0.7 to 1.3 equivalent, more preferably 0.8 to 1.1 equivalent.
- the active hydrogen equivalent per equivalent of epoxy group is preferably 0.3 to 1.4 equivalent, and more preferably 0.4 to 1.2 equivalent.
- the active hydrogen equivalent per equivalent of epoxy group is preferably 0.3 to 0.7 equivalent, more preferably 0.4 to 0.6 equivalent.
- the curing accelerator has an action of promoting the reaction between the curable resin and the curing agent.
- the cured product is colored.
- a curing accelerator with less is preferred.
- the curing accelerator include organic phosphine curing accelerators such as triphenylphosphine and diphenylphosphine; imidazole curing accelerators such as 2-methylimidazole, 2-phenyl-4-methylimidazole and 2-phenylimidazole; 1 , 8-diazabicyclo (5,4,0) undecene-7, tertiary amine curing accelerators such as triethanolamine and benzylmethylamine; tetraphenylborate curing accelerators such as tetraphenylphosphonium and tetraphenylborate It is done. These may be used alone or in combination of two or more.
- the mixing ratio of the curing accelerator is preferably 0.05 to 5% by mass with respect to 100% by mass of the curable resin.
- the cured product of the present invention can be suitably used as a sealing material for electric / electronic parts, for example, an insulating material. Furthermore, since it is excellent in transparency and heat resistance, it is suitable as a sealing material for optical semiconductors, an optical adhesive, and various sealing agents.
- volume average secondary particle size (Dv) was determined in the same manner as the volume average primary particle size except that the polymer powder (P1) was diluted with ion-exchanged water.
- the dispersion is filtered through a 0.2 ⁇ m cellulose mixed ester membrane filter (manufactured by Advantech Toyo Co., Ltd., model number: A020A025A), and 100 ml of the filtrate is used as a polymer powder.
- the alkali metal ion in (P1) was measured under the following conditions.
- content of alkali metal ion measured the total amount of Na ion and K ion.
- ICP emission analyzer manufactured by Thermo, IRIS “Intrepid II XSP" Quantitative method: Absolute calibration curve method using samples with known concentrations (4 points of 0 ppm, 0.1 ppm, 1 ppm and 10 ppm) Measurement wavelength: Na; 589.5 nm and K; 766.4 nm
- the polymer powder (P1) is diluted with ion-exchanged water, and is then measured using a laser diffraction / scattering particle size distribution measuring device (“SALD-7100” manufactured by Shimadzu Corporation). The ratio of particles of 10 ⁇ m or less before and after sonication (frequency 42 kHz, output 40 W, irradiation for 5 minutes) was measured on a volume basis.
- SALD-7100 laser diffraction / scattering particle size distribution measuring device
- G ′ B / G ′ A is 100 or more ⁇ : G ′ B / G ′ A is 10 or more and less than 100 ⁇ : G ′ B / G ′ A is 5 or more and less than 10 ⁇ : G ′ B / G ′ A is Less than 5 If G ′ B / G ′ A is 10 or more, the flow of the curable resin is suppressed even at a high temperature.
- (9) Dispersibility The dispersion state of the polymer powder (P1) in the epoxy resin composition was measured according to JIS K-5600 using a particle gauge, and the dispersibility was evaluated according to the following criteria. ⁇ : 2 ⁇ m or less ⁇ : Over 2 ⁇ m and 10 ⁇ m or less If the dispersion state of the polymer powder (P1) in the epoxy resin composition is 2 ⁇ m or less, it is possible to cope with fine pitch and thin film.
- Polymer powders (P1-1) to (P1-10) were produced according to the following Examples 1 to 6 and Comparative Examples 1 to 4. In Examples 1 to 6 and Comparative Examples 1 to 4, the following raw materials were used.
- Methyl methacrylate Mitsubishi Rayon Co., Ltd., trade name “Acryester M” N-Butyl methacrylate: Mitsubishi Rayon Co., Ltd., trade name “Acryester B” N-butyl acrylate: manufactured by Mitsubishi Chemical Corporation Allyl methacrylate: manufactured by Mitsubishi Rayon Co., Ltd., trade name “Acryester A” 2-ethylhexyl acrylate: manufactured by Mitsubishi Chemical Co., Ltd. Methacrylic acid: manufactured by Mitsubishi Rayon Co., Ltd., trade name “Acryester MAA” n-octyl mercaptan: Katayama Chemical Co., Ltd.
- Ammonium persulfate 2,2′-azobis- [N- (2-carboxyethyl) -2-methylpropionamidine] manufactured by Wako Pure Chemical Industries, Ltd., trade name “VA-057” manufactured by Wako Pure Chemical Industries, Ltd.
- Di- (2-ethylhexyl) ammonium sulfosuccinate trade name “Rikasurf M-300” manufactured by Shin Nippon Rika Co., Ltd.
- Example 1 Production of polymer powder (P1-1) In a separable flask equipped with a Max blend stirrer, a reflux condenser, a temperature controller, a dropping pump and a nitrogen introduction tube, 78.00 parts of ion-exchanged water, methacryl 2.83 parts of methyl acid and 2.17 parts of n-butyl methacrylate were added, and nitrogen gas was bubbled for 30 minutes while stirring at 120 rpm. Thereafter, the temperature was raised to 80 ° C. in a nitrogen atmosphere, and an aqueous solution of 0.04 part of ammonium persulfate and 2.00 parts of ion-exchanged water prepared in advance was put together and held for 60 minutes to form seed particles.
- Table 1 shows the glass transition temperature (Tg) determined by the Fox formula of the (meth) acrylic acid ester polymer (A1). Next, 45.15 parts methyl methacrylate, 3.00 parts n-butyl methacrylate, 1.00 parts n-butyl acrylate, 0.85 parts methacrylic acid, 0.50 ammonium di- (2-ethylhexyl) sulfosuccinate
- the monomer mixture used for the second stage polymerization obtained by emulsifying 25.0 parts of ion-exchanged water with a disper mixer was added dropwise over 150 minutes, and then maintained for 60 minutes. An emulsion was obtained. Table 1 shows the evaluation results of the volume average primary particle diameter of the obtained polymer.
- the obtained polymer emulsion was spray-dried under the following conditions using an L-8 type spray dryer manufactured by Okawara Chemical Industries Co., Ltd. to obtain a polymer powder (P1-1).
- the obtained polymer powder (P1-1) has an acetone-soluble component, an acetone-soluble component molecular weight (Mw, Mn), an alkali metal ion content, and evaluation results of the powder crushability in Table 1-1. Show.
- Spray system Rotating disk type Disk rotation speed: 25,000 rpm Hot air temperature Inlet temperature: 150 ° C Outlet temperature: 65 ° C
- MMA methyl methacrylate n-BMA: n-butyl methacrylate n-BA: n-butyl acrylate AMA: allyl methacrylate 2-EHA: 2-ethylhexyl acrylate MAA: methacrylic acid n-OM: n-octyl mercaptan emulsifier : Ammonium di- (2-ethylhexyl) sulfosuccinate VA-057: 2,2′-azobis- [N- (2-carboxyethyl) -2-methylpropionamidine]
- the obtained kneaded material was used in a three roll mill (manufactured by EXAKT, “M-80E”), with a roll speed of 200 rpm, a roll interval of 20 ⁇ m ⁇ 10 ⁇ m, 1 pass, 10 ⁇ m / 5 ⁇ m, 1 pass, 5 ⁇ m, 5 ⁇ m, 1 pass. Pass processing.
- a planetary motion vacuum mixer (trade name “Awatake Netaro ARV-310LED” manufactured by Shinky Corporation)
- the mixture was kneaded and degassed for 2 minutes under a reduced pressure of 3 KPa and a rotational speed of 1200 rpm.
- An epoxy resin composition was obtained.
- the resulting epoxy resin composition was evaluated for gelation temperature and gelation performance. The evaluation results are shown in Tables 2-1 and 2-2.
- the obtained kneaded material was used in a three roll mill (manufactured by EXAKT, “M-80E”), with a roll speed of 200 rpm, a roll interval of 20 ⁇ m ⁇ 10 ⁇ m, 1 pass, 10 ⁇ m / 5 ⁇ m, 1 pass, 5 ⁇ m, 5 ⁇ m, 1 pass. Pass processing.
- a planetary motion vacuum mixer (trade name “Awatake Netaro ARV-310LED” manufactured by Shinky Corporation)
- the mixture was kneaded and degassed for 2 minutes under a reduced pressure of 3 KPa and a rotational speed of 1200 rpm.
- An epoxy resin composition was obtained.
- the obtained epoxy resin composition was evaluated for dispersibility, initial viscosity, gelation temperature, and gelation performance. The evaluation results are shown in Tables 3-1 and 3-2.
- the epoxy resin composition thus obtained was cured with an acid anhydride curing agent (trade name “Ricacid MH-700”, manufactured by Shin Nippon Rika Co., Ltd.) and 2-ethyl-4-methylimidazole (Shikoku) as a curing accelerator. (Made by Kasei Kogyo Co., Ltd.) at the ratios shown in Tables 3-3 and 3-4, and using a planetary motion vacuum mixer (made by Shinky Corporation, trade name “Awatori Netaro ARV-310LED”) The mixture was kneaded and degassed for 2 minutes under reduced pressure at a rotation speed of 1200 rpm to obtain an epoxy resin composition.
- an acid anhydride curing agent trade name “Ricacid MH-700”, manufactured by Shin Nippon Rika Co., Ltd.
- 2-ethyl-4-methylimidazole Shikoku
- a PET film (manufactured by Toyobo Co., Ltd., trade name: TN200) is attached to one side of each of the two tempered glass plates of 300 mm long ⁇ 300 mm wide ⁇ 5 mm thick, and the PET film surfaces face each other.
- a mold was prepared by sandwiching a Teflon (registered trademark) spacer having a thickness of 3 mm between tempered glass plates.
- the epoxy resin composition is poured into the mold, fixed with a clamp, precured at 100 ° C. for 3 hours, then cured at 120 ° C. for 4 hours, and taken out of the mold to obtain a cured product having a thickness of 3 mm.
- the obtained cured product was annealed at 180 ° C. for 6 hours and used for evaluation of flexural modulus. The measurement results are shown in Tables 3-3 and 3-4.
- the epoxy resin compositions of Examples 7 to 12 to which the polymer powders (P1-1) to (P1-6) of the present invention were added were gels. Excellent chemical properties.
- the epoxy resin compositions of Comparative Examples 6 to 8 to which the polymer powder (P1-10) having a mass average molecular weight lower than the range of the present invention was added had a low gelling property. It was.
- the polymer powder (P1-1) of the present invention and the polymer powder (P1-9) excellent in stress relaxation ability (the mass average molecular weight of the acetone-soluble component and the acetone-soluble component is outside the range of the present invention)
- the epoxy resin compositions of Examples 14 and 15 used in combination were excellent in the balance between the initial viscosity, gelation characteristics, and the effect of lowering the elastic modulus of the cured product.
- the epoxy resin composition of Example 15 since the epoxy resin composition of Example 15 has a low initial viscosity, it can be expected to improve workability, apply with high accuracy, and form a pattern.
- Polymer powder (P1-1) and polymer powder (P1-2) have different molecular weights of acetone-soluble components.
- Examples 13 and 16, Examples 14 and 17, and Examples 15 and 18 are compared, Examples 13 to 15 to which the polymer powder (P1-1) is added are superior in gelling performance. It can be seen that the gelation performance is greatly influenced by the molecular weight of the acetone-soluble component.
- the polymer powder (P1-3) is a (meth) acrylic acid ester polymer (A1) in which n-butyl acrylate units are 50% by mass or more.
- the epoxy resin composition of Example 14 has a higher effect of lowering the elastic modulus of the cured product. It can be seen that the (meth) acrylic acid ester polymer (A1) having a 2-ethylhexyl acrylate unit of 50% by mass or more is more excellent in the effect of lowering the elastic modulus of the cured product.
- Example 20 polymer powders (P1-4), (P1-5), (P1-5), (M1) having a different composition ratio of the (meth) acrylate polymer (A1) to the polymer (B1) were used.
- Example 21 in which the polymer powder (P1-7) (outside the scope of the present invention) is used in combination with the polymer powder (P1-4), which is excellent in gelation characteristics, further has an initial viscosity, gelation characteristics, elasticity It was found that the balance of the rate reduction effect was excellent.
- the polymer powder (P1-6) with a low content of the (meth) acrylic acid ester polymer (A1) has a low elastic modulus effect of the cured product when used alone, but has a low stress relaxation ability. It was found that when the excellent polymer powder (P1-9) (outside the scope of the present invention) was used in combination (Example 23), the balance between the initial viscosity, the gelation characteristics, and the elastic modulus lowering effect was excellent.
- the epoxy resin composition of Comparative Example 9 to which the polymer powder of the present invention was not added showed no gelling property, and the cured product obtained had a high elastic modulus.
- the epoxy resin composition of Comparative Example 10 to which the polymer powder (P1-7) in which the glass transition temperature of the (meth) acrylic acid ester polymer (A1) exceeds 0 ° C. is added has high gelling performance. The effect of lowering the elastic modulus of the cured product was not observed.
- the epoxy resin composition of Comparative Example 11 to which the polymer powder (P1-8) having a low acetone-soluble content was added showed no gelling property, and the effect of lowering the elastic modulus of the resulting cured product was low. It was.
- the epoxy resin composition of Comparative Example 12 to which the polymer powder (P1-9) having a low acetone-soluble content and a low mass-average molecular weight of the acetone-soluble component was added did not show gelation properties.
- the epoxy resin composition of Comparative Example 13 to which the polymer powder (P1-7) and the polymer powder (P1-9) were added has a high gelling performance and an elastic modulus lowering effect, but has a large initial viscosity. Therefore, it was difficult to apply and pattern with high accuracy.
- the epoxy resin composition of Comparative Example 14 to which the polymer powder (P1-10) having a low molecular weight of acetone soluble component was added had a high initial viscosity and a low gelling property.
- monodispersity (Dv / Dn) was determined from the values of Dv and Dn.
- the monodispersity was evaluated according to the following criteria.
- the sample concentration of the vinyl polymer emulsion was appropriately adjusted so as to be within an appropriate range in the scattered light intensity monitor attached to the apparatus.
- HLC8220 manufactured by Tosoh Corporation Column: TSKgel SuperHZM-M manufactured by Tosoh Corporation (inner diameter 4.6 mm ⁇ length 15 cm) Number: 4; Exclusion limit: 4 ⁇ 10 6 Temperature: 40 ° C Carrier liquid: Tetrahydrofuran Flow rate: 0.35 ml / min Sample concentration: 0.1% by mass Sample injection volume: 10 ⁇ l Standard: Polystyrene
- the alkali metal ions therein were measured under the following conditions. In addition, content of alkali metal ion measured the total amount of Na ion and K ion.
- ICP emission analyzer manufactured by Thermo, IRIS "Intrepid II XSP" Quantitative method: Absolute calibration curve method using samples with known concentrations (4 points of 0 ppm, 0.1 ppm, 1 ppm and 10 ppm) Measurement wavelength: Na; 589.5 nm and K; 766.4 nm
- the polymer powder (P2) is diluted with ion-exchanged water, and ultra-fine using a laser diffraction / scattering particle size distribution measuring device (manufactured by Shimadzu Corporation, “SALD-7100”). The ratio of particles of 10 ⁇ m or less before and after sonication (frequency 42 kHz, output 40 W, 3 minutes irradiation) was measured on a volume basis.
- SALD-7100 laser diffraction / scattering particle size distribution measuring device
- the dispersion state of the polymer powder (P2) in the epoxy resin composition was measured according to JIS K-5600 using a particle gauge, and the dispersibility was evaluated according to the following criteria. ⁇ : 5 ⁇ m or less ⁇ : More than 5 ⁇ m If the dispersion state of the polymer powder (P2) in the epoxy resin composition is 5 ⁇ m or less, it is possible to cope with fine pitch and thin film.
- Methacrylic acid manufactured by Mitsubishi Rayon Co., Ltd., trade name “Acryester MAA” 2-Hydroxyethyl methacrylate: Mitsubishi Rayon Co., Ltd., trade name “Acryester HO” Allyl methacrylate: Mitsubishi Rayon Co., Ltd., trade name “Acryester A” Ammonium di-2-ethylhexylsulfosuccinate, manufactured by Toho Chemical Industry Co., Ltd., trade name “Rikasurf M-300” Ammonium persulfate: manufactured by Wako Pure Chemical Industries, Ltd. Potassium persulfate: manufactured by Wako Pure Chemical Industries, Ltd. 2,2'-azobis (2,4-dimethylvaleronitrile): manufactured by Wako Pure Chemical Industries, Ltd. V-65 "(10-hour half-life temperature 51 ° C)
- Example 31 Production of polymer powder (P2-1) 78.00 parts by mass of ion-exchanged water in a separable flask equipped with a Max blend stirrer, a reflux condenser, a temperature controller, a dropping pump, and a nitrogen introduction tube, 2.83 parts by mass of methyl methacrylate and 2.17 parts by mass of n-butyl methacrylate were added, and nitrogen gas was bubbled for 30 minutes while stirring at 120 rpm. Thereafter, the temperature was raised to 80 ° C. in a nitrogen atmosphere, and 0.02 parts by mass of ammonium persulfate and 2.00 parts by mass of ion-exchanged water prepared in advance were added together and held for 60 minutes to form seed particles. .
- the obtained vinyl polymer emulsion was spray-dried under the following conditions using an L-8i type spray dryer manufactured by Okawara Chemical Industries, Ltd. to obtain a polymer powder (P2-1).
- Table 4-1 shows the evaluation results of acetone-soluble content, molecular weight (Mw, Mn) of acetone-soluble content, alkali metal ion content and powder crushability of the obtained polymer powder (P2-1). Shown in
- Spray system Rotating disk type Disk rotation speed: 25,000 rpm Hot air temperature Inlet temperature: 150 ° C Outlet temperature: 65 ° C
- Examples 32 to 40 and Comparative Example 31 Production of Polymer Powders (P2-2) to (P2-10) and (P2′-1) Examples 32 to 40 and Comparative Example 31 are shown in Table 4-1.
- Polymer powders (P2-2) to (P2-10) and (P2′-1) were obtained in the same manner as in Example 31, except that the raw material composition and polymerization conditions shown in 4-2 were used.
- the evaluation results of the primary particle size of the obtained vinyl polymer emulsion are shown in Tables 4-1 and 4-2.
- the obtained polymer powders (P2-2) to (P2-10) and (P2′-1) have an acetone-soluble content, an acetone-soluble molecular weight (Mw, Mn), an alkali metal ion content and
- the evaluation results of the powder crushability are shown in Tables 4-1 and 4-2.
- the evaluation results of the primary particle diameter of the obtained vinyl polymer emulsion are shown in Table 4-2.
- the obtained vinyl polymer emulsion was spray-dried in the same manner as in Example 31 to obtain a polymer powder (P2′-2).
- the obtained polymer powder (P2′-2) has an acetone-soluble content, molecular weight (Mw, Mn) of acetone-soluble content, alkali metal ion content, and evaluation results of powder crushability. It is shown in 2.
- MMA methyl methacrylate
- n-BMA n-butyl methacrylate
- n-BA n-butyl acrylate
- St styrene
- AMA divinylbenzene
- MAA methacrylic acid
- HEMA 2-hydroxyethyl methacrylate emulsifier: di -2-Ethylhexyl ammonium sulfosuccinate
- V-65 2,2'-azobis (2,4-dimethylvaleronitrile)
- Examples 41 to 50, Comparative Examples 33 to 35 An alicyclic epoxy resin (manufactured by Daicel, “Celoxide 2021”) and polymer powder were blended in the proportions shown in Table 5, and a planetary motion type vacuum mixer (trade name “Awatake Kentaro ARV-310LED, manufactured by Sinky Corporation”). )), And kneading was performed for 3 minutes under atmospheric pressure at a rotation speed of 1200 rpm to obtain a kneaded product.
- a planetary motion type vacuum mixer trade name “Awatake Kentaro ARV-310LED, manufactured by Sinky Corporation”.
- the obtained kneaded material was used in a three roll mill (manufactured by EXAKT, “M-80E”), with a roll speed of 200 rpm, a roll interval of 20 ⁇ m ⁇ 10 ⁇ m, 1 pass, 10 ⁇ m / 5 ⁇ m, 1 pass, 5 ⁇ m, 5 ⁇ m, 1 pass. Pass processing. Then, again using a planetary motion vacuum mixer (trade name “Awatake Kentaro ARV-310LED” manufactured by Shinky Corporation), the mixture was kneaded and degassed for 2 minutes under a reduced pressure of 3 KPa and a rotational speed of 1200 rpm. An epoxy resin composition was obtained. The obtained epoxy resin composition was evaluated for dispersibility, gelation temperature, and gelation performance. The evaluation results are shown in Table 5.
- the mixture was kneaded and degassed for 2 minutes under the condition of a rotational speed of 1200 rpm to obtain an epoxy resin composition.
- a PET film manufactured by Toyobo Co., Ltd., trade name: TN200
- a mold was prepared by sandwiching a Teflon (registered trademark) spacer having a thickness of 3 mm between tempered glass plates.
- the epoxy resin composition is poured into the mold, fixed with a clamp, precured at 100 ° C. for 3 hours, then cured at 120 ° C. for 4 hours, and taken out of the mold to obtain a cured product having a thickness of 3 mm. Obtained.
- Table 6 shows the evaluation results of the appearance, total light transmittance, glass transition temperature, and dielectric constant of the cured epoxy resin.
- Example 61 and 62 Comparative Examples 39 to 41
- Bisphenol A type hydrogenated cycloaliphatic epoxy resin Mitsubishi Chemical Co., Ltd., “YX-8000”
- polymer powder were blended in the proportions shown in Table 7, and an epoxy resin composition was obtained in the same manner as in Example 41. It was. Evaluation of dispersibility, gelation temperature, and gelation performance of the obtained epoxy resin composition was performed. Table 7 shows the evaluation results.
- Example 63 and 64 Comparative Examples 42 to 44
- an acid anhydride curing agent (trade name “Ricacid MH-700”, manufactured by Shin Nippon Rika Co., Ltd.) as a curing agent
- tetrabutylphosphonium diethylphosphodithionate (Japan) as a curing accelerator.
- “Hishicolin PX-4ET” manufactured by Chemical Industry Co., Ltd.” was blended in the proportions shown in Table 8, and a cured epoxy resin was obtained in the same manner as in Example 51.
- Table 8 shows the evaluation results of the appearance, total light transmittance, glass transition temperature, and dielectric constant of the obtained cured epoxy resin.
- the epoxy resin composition to which the polymer powders (P2-1) to (P2-10) of the present invention are added has excellent dispersibility and high gelling performance.
- the cured epoxy resin obtained by curing the epoxy resin composition to which the polymer powders (P2-1) to (P2-10) are added has excellent appearance and high transparency.
- polymer powders having carboxyl groups (P2-2, P2-3, P2-4, P2-9), polymer powders having hydroxyl groups (P2-5, P2-6, P2-7, P2- The cured epoxy resin obtained by adding 10) has high transparency (Examples 52 to 57, 59, 60, 64).
- the cured epoxy resin obtained by adding polymer powders (P2-8) to (P2-10) having a refractive index of 1.50 has a small difference in refractive index between the cured epoxy resin and the vinyl polymer.
- high transparency (Examples 58 to 60).
- by introducing a carboxyl group or a hydroxyl group it is possible to maintain high transparency even under high temperature conditions (Examples 59 and 60).
- the cured epoxy resin obtained by adding the polymer powders (P2-1) to (P2-10) of the present invention has a small decrease in glass transition temperature, so that it is used in applications requiring high heat resistance. Can also be used. Furthermore, since the dielectric constant is low, it is also suitable for the electronic material field.
- the cured epoxy resin added with the polymer powder (P2′-1) whose acetone-soluble content is outside the scope of the present invention has a large amount of acetone-soluble content, so the polymer powder (P2) of the present invention Compared with the cured product to which is added, the glass transition temperature is greatly reduced.
- the polymer powder of the present invention is excellent in dispersibility in a curable resin, particularly an epoxy resin, and quickly turns the curable resin composition into a gel state by heating at a predetermined temperature for a short time. Since its elastic modulus is low, it can be used as a stress relaxation agent and pregel agent for electronic materials.
- the curable resin composition obtained by adding the polymer powder of the present invention is excellent in the balance of initial viscosity, gelation characteristics, and reduced modulus of elasticity of the cured product.
- liquid sealing material such as grab top material in wire bond, sealing sheet for sealing various chips on the substrate at once, pre-dispensing type underfill material, sealing at wafer level Sealing sheet to be stopped, adhesive layer for three-layer copper-clad laminate, die-bonding film, die-attach film, interlayer insulating film, adhesive layer such as coverlay film, die-bonding paste, interlayer insulating paste, conductive paste, anisotropic conductive paste Adhesive paste, etc., sealing materials for light emitting diodes, optical adhesives, liquid crystal, sealing of various flat panel displays such as organic EL It can be used for various applications like.
Abstract
Description
種々の表示板、画像読み取り用光源、交通ディスプレイ用ユニット等に実用化されている光半導体等の発光装置は、その大部分が樹脂封止によって製造されている。封止用樹脂としては、エポキシ樹脂が、機械的性質、電気的絶縁性、接着性に優れるため、広く使用されている。中でも、脂環式エポキシ樹脂は透明性、耐熱性に優れるため、光半導体用の封止用樹脂として多用されている。
特に、常温で液状のエポキシ樹脂は、常温で注型、塗布できることから、各種のペースト状又はフィルム状の材料として使用されている。しかしながら、エポキシ樹脂組成物は粘度の温度依存性が高いため、硬化するまでの温度上昇により粘度が顕著に低下することから、ディスペンサーによる精密な注入や塗布、スクリーン印刷による精密なパターン形成、高い膜厚精度でのフィルム上へのコーティング等、精密加工が困難な状況にある。特に電子材料分野においては、年々高精度加工への要求が高まっており、使用するエポキシ樹脂組成物には温度上昇しても粘度が低下しないことや、早期に形状が安定化することへの要望が極めて強い。
エポキシ樹脂組成物に上記のような特性を付与する方法として、加熱により速やかにエポキシ樹脂組成物をゲル状態とするために、エポキシ樹脂組成物に特定のビニル重合体を配合し、ゲル化性付与剤(以下、「プレゲル化剤」という。)として用いる方法がある。
特許文献1では、特定のビニル重合体をプレゲル化剤として用いる方法が提案されている。
一方、脂環式エポキシ樹脂は高い透明性と耐熱性を有するが、硬化して得られるエポキシ樹脂硬化物は耐クラック性が悪く、例えば冷熱サイクルによって亀裂破壊を生じやすいといった特徴を有しており、長期間の信頼性が要求される用途には不向きであった。クラックの発生は、エポキシ樹脂硬化物と無機材料との線膨張係数の差及びエポキシ樹脂硬化物の弾性率が大きな要因であり、エポキシ樹脂硬化物のガラス転移温度の上昇、線膨張係数及び弾性率の低下により抑制できると考えられている。特許文献2~4では、エポキシ樹脂硬化物の弾性率を低下する方法として、アクリル系ゴム粒子を配合する方法が提案されている。
さらに、特許文献5では、エポキシ樹脂組成物に特定の(メタ)アクリレート系重合体を配合し、耐衝撃性、接着性、及びゲル化性を改良する方法が提案されている。
一方、特許文献2~4で提案されている方法では、エポキシ樹脂硬化物の耐クラック性は改善されるが、硬化するまでの温度上昇によりエポキシ樹脂組成物の粘度が顕著に低下することから、高精度な塗布やパターン形成が困難であった。
特許文献5で提案されている方法では、エポキシ樹脂組成物のゲル化性及び硬化物の低弾性率化に効果は認められるものの、高精度加工で求められる極めて短時間でのゲル化や高いゲル化性は達成できておらず、満足できる状態とはいえなかった。
さらに、アクリル系ゴム粒子はガラス転移温度が低いため、エポキシ樹脂組成物に多量に配合した場合、エポキシ樹脂硬化物のガラス転移温度が低下する場合があり、高い耐熱性が要求される用途には不向きであった。また、イオン性不純物の含有量についても特に考慮されていないため、エポキシ樹脂組成物に配合した場合の、エポキシ樹脂硬化物の電気特性の悪化が懸念される。
したがって、エポキシ樹脂組成物のゲル化性付与及びエポキシ樹脂硬化物の透明性及び耐熱性を満足する材料は、従来まで提案されていないのが実状である。
本発明の目的とするところは、所定の温度で短時間の加熱によって速やかに硬化性樹脂組成物をゲル状態とし、さらに得られる硬化物の弾性率を低下させる重合体粉体、同粉体を含有する硬化性樹脂組成物及びその硬化物を提供することである。
また本発明の他の目的は、硬化性樹脂組成物への分散性に優れ、所定の温度で短時間の加熱によって速やかに硬化性樹脂組成物をゲル状態とし、得られる硬化性樹脂硬化物の透明性及び耐熱性を損なわない光半導体材料に適したプレゲル化剤として有用な重合体粉体、同粉体を含有する硬化性樹脂組成物及びその硬化物を提供することである。
すなわち、本発明は以下を提供する。
[1](i)ガラス転移温度が0℃以下である(メタ)アクリル酸エステル系重合体(A1)を含み、重合体粉体のアセトン可溶分が5質量%以上であり、アセトン可溶分の質量平均分子量が10万以上である重合体粉体(P1)、及び(ii)アセトン可溶分が2質量%以上35質量%以下であり、アセトン可溶分の質量平均分子量が10万以上であり、体積平均一次粒子径(Dv)が200nm以上である重合体粉体(P2)からなる群より選択される重合体粉体(P)。
[2]前記重合体粉体(P)が、(i)ガラス転移温度が0℃以下である(メタ)アクリル酸エステル系重合体(A1)を含み、重合体粉体のアセトン可溶分が5質量%以上であり、アセトン可溶分の質量平均分子量が10万以上である重合体粉体(P1)である、[1]に記載の重合体粉体(P)。
[3]ガラス転移温度が0℃を超える重合体(B1)をさらに含む[2]に記載の重合体粉体(P)。
[4]重合体(B1)が(メタ)アクリル酸エステル系重合体である[3]に記載の重合体粉体(P)。
[5]ガラス転移温度が0℃以下である(メタ)アクリル酸エステル系重合体(A1)の含有量が30~90質量%、重合体(B1)の含有量が70~10質量%(合計100質量%)である[3]又は[4]に記載の重合体粉体(P)。
[6]重合体粉体(P1)の体積平均一次粒子径(Dv)が200nm以上である[2]~[5]のいずれか一つに記載の重合体粉体(P)。
[7]ガラス転移温度が0℃以下である(メタ)アクリル酸エステル系重合体(A1)がアクリル酸2-エチルヘキシル単位を50質量%以上含有する[2]~[6]のいずれか一つに記載の重合体粉体(P)。
[8]アルカリ金属イオンの含有量が10ppm以下であ[2]~[7]のいずれか一つに記載の重合体粉体(P)。
[9]粒子径10μm以下の粒子の占める割合が30vol%未満であり、以下の解砕性を有する[2]~[8]のいずれか一つに記載の重合体粉体(P)。
(解砕性)
(1)重合体粉体(P)をイオン交換水で希釈する。
(2)超音波照射を周波数42kHz、出力40Wで5分間行う。
(3)レーザー回折/散乱式粒子径分布測定により粒子径分布を測定する。
(4)10μm以下の粒子径の粒子の占める割合が30vol%以上である。
[10](メタ)アクリル酸エステル系重合体(A1)の存在下で単量体混合物(b1)を重合し、粉体化して得られる(メタ)アクリル酸エステル系重合体粉体であって、(メタ)アクリル酸エステル系重合体(A1)のガラス転移温度が0℃以下であり、(メタ)アクリル酸エステル系重合体粉体のアセトン可溶分が5質量%以上であり、アセトン可溶分の質量平均分子量が10万以上である[2]~[9]のいずれか一つに記載の重合体粉体(P)。
[11][2]~[10]のいずれか一つに記載の重合体粉体(P)からなる硬化性樹脂用応力緩和剤兼プレゲル化剤。
[12]硬化性樹脂100質量部に対して重合体粉体(P)を20質量部添加して得られる硬化性樹脂組成物の、ゲル化温度-20℃での貯蔵弾性率G‘Aと、ゲル化温度+20℃での貯蔵弾性率G’Bの比率(G‘B/G’A)が10以上となる[11]に記載の硬化性樹脂用応力緩和剤兼プレゲル化剤。
[13][2]~[10]のいずれか一つに記載の重合体粉体(P)と硬化性樹脂とを含有する硬化性樹脂組成物。
[14]硬化性樹脂がエポキシ樹脂である[13]に記載の硬化性樹脂組成物。
[15]硬化性樹脂100質量部に対して重合体粉体(P)を5質量部以上含有する[13]に記載の硬化性樹脂組成物。
[16][13]~[15]のいずれか一つに記載の硬化性樹脂組成物を硬化して得られる硬化物。
[17][13]~[15]のいずれか一つに記載の硬化性樹脂組成物を用いた半導体用封止材料。
[18][13]~[15]のいずれか一つに記載の硬化性樹脂組成物を用いたシート状物品。
[19][13]~[15]のいずれか一つに記載の硬化性樹脂組成物を用いた液晶表示素子用シール剤。
[20]前記重合体粉体(P)が、(ii)アセトン可溶分が2質量%以上35質量%以下であり、アセトン可溶分の質量平均分子量が10万以上であり、体積平均一次粒子径(Dv)が200nm以上である重合体粉体(P2)である、[1]に記載の重合体粉体(P)。
[21]重合体粉体(P2)のアセトン可溶分が30質量%未満である[20]に記載の重合体粉体(P)。
[22]ビニル重合体粉体(A)のアセトン可溶分が25質量%以下である[20]に記載のビニル重合体粉体(A)。
[23]重合体粉体(P2)の20℃での屈折率が1.48以上1.51以下である[20]~[22]のいずれか一つに記載の重合体粉体(P)。
[24]重合体粉体(P2)のガラス転移温度が0℃以上である[20]~[23]のいずれか一つに記載の重合体粉体(P)。
[25]重合体粉体(P2)が、メタクリル酸メチル70~99質量%、メタクリル酸メチル以外の単量体原料30~1質量%を含む単量体原料(a2)を重合して得られたビニル重合体を粉体化して得られたメタクリル酸メチル系重合体粉体である[20]~[24]のいずれか一つに記載の重合体粉体(P)。
[26]メタクリル酸メチル以外の単量体原料が、カルボキシル基含有ビニル単量体、水酸基含有ビニル単量体及びグリシジル基含有ビニル単量体から選ばれる少なくとも1種の官能基含有単量体を含有する、[25]に記載の重合体粉体(P)。
[27]メタクリル酸メチル以外の単量体原料が、カルボキシル基含有ビニル単量体、水酸基含有ビニル単量体及びグリシジル基含有ビニル単量体から選ばれる少なくとも1種の官能基含有単量体を2質量%以上含有する[26]に記載の重合体粉体(P)。
[28]アルカリ金属イオンの含有量が10ppm以下である[20]~[27]のいずれか一つに記載の重合体粉体(P)。
[29]粒子径10μm以下の粒子の占める割合が30vol%未満であり、以下の解砕性を有する[20]~[28]のいずれか一つに記載の重合体粉体(P)。
(解砕性)
(1)重合体粉体(P)をイオン交換水で希釈する。
(2)超音波照射を周波数42kHz、出力40Wで3分間行う。
(3)レーザー回折/散乱式粒子径分布測定により粒子径分布を測定する。
(4)10μm以下の粒子径の粒子の占める割合が30vol%以上である。
[30][20]~[29]のいずれか一つに記載の重合体粉体(P)からなる硬化性樹脂用プレゲル化剤。
[31]硬化性樹脂100質量部に対して重合体粉体(P)を20質量部添加して得られる硬化性樹脂組成物の、ゲル化温度-20℃での貯蔵弾性率G‘Aと、ゲル化温度+20℃での貯蔵弾性率G’Bの比率(G‘B/G’A)が100以上となる[30]に記載の硬化性樹脂用プレゲル化剤。
[32][20]~[29]のいずれか一つに記載のビニル重合体粉体(P)と硬化性樹脂とを含有する硬化性樹脂組成物。
[33]硬化性樹脂がエポキシ樹脂である[32]に記載の硬化性樹脂組成物。
[34]エポキシ樹脂が脂環式エポキシ樹脂である[33]に記載の硬化性樹脂組成物。
[35]重合体粉体(P)と前記硬化性樹脂とを含有する硬化性樹脂組成物を硬化させて得られる厚さ3mmの硬化物の23℃における全光線透過率が70%以上である[32]~[34]のいずれか一つに記載の硬化性樹脂組成物。
[36][32]~[35]のいずれか一つに記載の硬化性樹脂組成物を硬化して得られる硬化物。
[37][32]~[35]のいずれか一つに記載の硬化性樹脂組成物を用いた光半導体用封止材料。
また、本発明の重合体粉体は、所定の温度で短時間の加熱によって速やかに硬化性樹脂組成物をゲル状態とすることができる。本発明の硬化性樹脂組成物を硬化して得られる硬化物は透明性を損なわない。さらに、本発明の硬化物は、耐熱性に優れる。このため、本発明の硬化性樹脂組成物及びその硬化物は、光半導体材料あるいは光半導体用封止材料等の透明性及び耐熱性が要求される用途に好適である。
本発明の重合体粉体(P)は、
(i)ガラス転移温度が0℃以下である(メタ)アクリル酸エステル系重合体(A1)を含み、重合体粉体のアセトン可溶分が5質量%以上であり、アセトン可溶分の質量平均分子量が10万以上である重合体粉体(P1)、及び
(ii)アセトン可溶分が2質量%以上35質量%以下であり、アセトン可溶分の質量平均分子量が10万以上であり、体積平均一次粒子径(Dv)が200nm以上である重合体粉体(P2)
からなる群より選択される重合体粉体(P)である。
本発明の重合体粉体(P1)は、ガラス転移温度が0℃以下である(メタ)アクリル酸エステル系重合体(A1)を含む重合体粉体であって、重合体粉体のアセトン可溶分が5質量%以上であり、アセトン可溶分の質量平均分子量が10万以上である。尚、本発明書において、(メタ)アクリルは、アクリル又はメタクリルを意味する。
本発明において、(メタ)アクリル酸エステル系重合体(A1)は、(メタ)アクリル酸エステル単量体(a11)を含有する単量体混合物(a1)を重合して得られるものであり、Fox式で求めたガラス転移温度(Tg)が0℃以下の重合体である。
(メタ)アクリル酸エステル系重合体(A1)のTgは、上記の単独重合体のTgの数値を用い、Fox式により求めることができる。Fox式を以下に示す。
1/(273+Tg)=Σ(wi/(273+Tgi))
式中、Tgは共重合体のTg(℃)、wiは単量体iの質量分率、Tgiは単量体iを重合して得られる単独重合体のTg(℃)である。
ここで、単独重合体のTgの数値としては、POLYMER HANDBOOK THIRD EDITION(WILEY INTERSCIENCE)に記載の数値を用いた。
尚、単量体混合物(a1)が架橋性単量体(a13)及び/又はグラフト交叉剤(a14)を含有する場合には、架橋性単量体及びグラフト交叉剤を除いた単量体について、Tgを求めることとする。
Fox式で求めたTgが0℃以下の重合体(A1)は、例えば、単独重合体のTgが0℃以下となる(メタ)アクリル酸エステル単量体(a11)を50質量%以上含有する単量体混合物(a1)を重合することにより得ることができる。
その他の単量体(a12)としては、例えば、スチレン、α-メチルスチレン、アルキル置換スチレン等の芳香族ビニル単量体;(メタ)アクリル酸メチル、メタクリル酸エチル、メタクリル酸n-プロピル、メタクリル酸n-ブチル等の(a11)以外の(メタ)アクリル酸エステル単量体;アクリロニトリル、メタクリロニトリル等のシアン化ビニル単量体;(メタ)アクリル酸グリシジル等のグリシジル基を有するビニル単量体、(メタ)アクリル酸2-ヒドロキシエチル等のヒドロキシル基を有するビニル単量体、(メタ)アクリル酸変性シリコーンが挙げられる。これらの単量体(a12)は1種を単独で用いてもよく、2種以上を併用してもよい。
単量体混合物(a1)は、必要に応じて、架橋性単量体(a13)及び/又はグラフト交叉剤(a14)を含有してもよい。
これらの架橋性単量体(a13)及び/又はグラフト交叉剤(a14)は、1種を単独で用いてもよく、2種以上を併用してもよい。これらの架橋性単量体(a13)及び/又はグラフト交叉剤(a14)を用いることで、架橋構造を有する(メタ)アクリル酸エステル系重合体(A1)を得ることができる。
架橋性単量体(a13)及び/又はグラフト交叉剤(a14)の含有率は、単量体混合物(a1)(100質量%)中、合計で0.001~15質量%であることが好ましく、0.01~5質量%であることがより好ましい。単量体混合物(a1)(100質量%)中の、架橋性単量体(a13)及び/又はグラフト交叉剤(a14)の含有量が15質量%以下であれば、得られる硬化物が低弾性率となる。
(メタ)アクリル酸エステル系重合体(A1)は組成の異なる2種以上を併用しても良い。
単量体混合物(a1)の重合方法としては、粒子状の重合体を得ることができる重合方法であれば特に制限されないが、真球状粒子を得やすい重合方法が好ましく、例えば、乳化重合法、ソープフリー乳化重合法、分散重合法、膨潤重合法、ミニエマルション重合法、微細懸濁重合法が挙げられる。この中でも、硬化性樹脂中での分散性に優れ、ファインピッチ化にも対応した粒子径を有する重合体が得られることから、ソープフリー乳化重合法、微細懸濁重合法が好ましい。
単量体混合物(a1)を重合する際には、重合開始剤、乳化剤、分散安定剤、連鎖移動剤を用いることができる。
単量体混合物(a1)の重合に用いる重合開始剤としては、公知の重合開始剤を用いることができる。(メタ)アクリル酸エステル系重合体を噴霧乾燥で粉体化する際に金属イオンの残留がないため、金属イオンが含まれていない重合開始剤を用いることが好ましい。
金属イオンが含まれていない重合開始剤としては、例えば、2,2’-アゾビスイソブチロニトリル、4,4’-アゾビス-(4-シアノバレリックアシッド)、2,2’-アゾビス-[N-(2-カルボキシエチル)-2-メチルプロピオンアミジン]等のアゾ系化合物;過硫酸アンモニウム等の過硫酸化合物;ジイソプロピルベンゼンヒドロパーオキシド、p-メンタンヒドロパーオキシド、クメンヒドロパーオキシド、t-ブチルヒドロパーオキシド等の有機過酸化物;前記過硫酸化合物又は前記有機過酸化物を一成分としたレドックス系開始剤が挙げられる。これらの重合開始剤は、1種を単独で用いてもよく、2種以上を併用してもよい。これらの重合開始剤の中でも、過硫酸アンモニウム、2,2’-アゾビスイソブチロニトリル、4,4’-アゾビス-(4-シアノバレリックアシッド)、2,2’-アゾビス-[N-(2-カルボキシエチル)-2-メチルプロピオンアミジン]が好ましい。
重合温度は重合開始剤の種類にもよるが、例えば、40~90℃程度の範囲で適宜行なうことができる。
単量体混合物(a1)の重合に用いる乳化剤としては、公知の乳化剤を用いることができ、例えば、不均化ロジン酸、オレイン酸、ステアリン酸等の高級脂肪酸のアルカリ金属塩やアンモニウム塩、ドデシルベンゼンスルホン酸等のスルホン酸アルカリ金属塩やアンモニウム塩、ノニオン系乳化剤が挙げられる。
これらの乳化剤の中でも、重合体粉体(P1)への金属イオンの残留がないことから、金属イオンが含まれていないアンモニウム塩型アニオン系乳化剤、ノニオン系乳化剤を用いることが好ましい。
アンモニウム塩型アニオン系乳化剤としては、乳化重合の安定性に優れることから、ラウリル硫酸アンモニウム、ジ-(2-エチルヘキシル)スルホコハク酸アンモニウムが好ましい。ノニオン系乳化剤としては、乳化重合の安定性に優れることから、ポリオキシエチレン(85)モノテトラデシルエーテル、ポリオキシエチレンジスチレン化フェニルエーテルが好ましい。
単量体混合物(a1)の重合に用いる分散安定剤としては、例えば、リン酸カルシウム、炭酸カルシウム、水酸化アルミニウム、澱粉末シリカ等の水難溶性無機塩;ポリビニルアルコール、ポリエチレンオキサイド、セルロース誘導体等のノニオン系高分子化合物;及びポリアクリル酸又はその塩、ポリメタクリル酸又はその塩、メタクリル酸エステルとメタクリル酸又はその塩との共重合体等のアニオン系高分子化合物が挙げられる。これらの中では、電気特性に優れることからノニオン系高分子化合物が好ましい。また、重合安定性との両立の観点から目的に応じて2種以上の分散安定剤を併用することができる。
単量体混合物(a1)の重合の際に、必要に応じて、t-ドデシルメルカプタン、n-オクチルメルカプタン、α-メチルスチレン等の連鎖移動剤を用いることもできる。
本発明の(メタ)アクリル酸エステル系重合体(A1)は、単層、又は2層以上の多層構造を有するものであっても良い。また、2種以上の(メタ)アクリル酸エステル系重合体(A1)を複合させても良い。
本発明において、重合体粉体(P1)は、ガラス転移温度が0℃以下である(メタ)アクリル酸エステル系重合体(A1)以外の重合体(B1)を含むことができる。
本発明において、重合体(B1)は、ラジカル重合可能なビニル単量体(b11)を含有する単量体混合物(b1)を重合して得られる。本発明において、重合体(B)は、Fox式で求めたTgが0℃を超えることが好ましい。尚、単量体混合物(b1)が架橋性単量体(b12)を含有する場合には、架橋性単量体を除いた単量体について、Tgを求めることとする。Fox式で求めたTgが0℃を超える重合体(B1)は、例えば、単独重合体のTgが0℃を超える単量体を50質量%以上とする単量体混合物(b1)を重合することにより得ることができる。
ラジカル重合可能なビニル単量体(b11)としては、例えば、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸n-プロピル、(メタ)アクリル酸i-プロピル、(メタ)アクリル酸n-ブチル、(メタ)アクリル酸t-ブチル、(メタ)アクリル酸i-ブチル、(メタ)アクリル酸n-ヘキシル、(メタ)アクリル酸n-オクチル、(メタ)アクリル酸t-ブチルシクロヘキシル、(メタ)アクリル酸イソボルニル、(メタ)アクリル酸ジシクロペンタジエニル、(メタ)アクリル酸N,N-ジメチルアミノエチル、(メタ)アクリル酸N-メチル-2,2,6,6-テトラメチルピペリジル等の(メタ)アクリル酸エステル単量体;(メタ)アクリロニトリル等のシアン化ビニル単量体;スチレン、α-メチルスチレン、ビニルトルエン等の芳香族ビニル単量体;アクリル酸、メタクリル酸、クロトン酸、マレイン酸、イタコン酸、フマル酸、イソクロトン酸、サリチル酸ビニロキシ酢酸、アリロキシ酢酸、2-(メタ)アクリロイルプロパン酸、3-(メタ)アクリロイルブタン酸、4-ビニル安息香酸等のカルボキシル基含有ビニル単量体;(メタ)アクリル酸ヒドロキシメチル、(メタ)アクリル酸2-ヒドロキシエチル、(メタ)アクリル酸2-ヒドロキシプロピル、(メタ)アクリル酸2-ヒドロキシブチル、(メタ)アクリル酸4-ヒドロキシブチル等の水酸基含有ビニル単量体;(メタ)アクリル酸グリシジル等のグリシジル基含有ビニル単量体;(メタ)アクリルアミド;ビニルピリジン、ビニルアルコール、ビニルイミダゾール、ビニルピロリドン、酢酸ビニル、1-ビニルイミダゾール等のビニル単量体;モノメチルイタコネート、モノエチルイタコネート、モノプロピルイタコネート、モノブチルイタコネート、ジメチルイタコネート、ジエチルイタコネート、ジプロピルイタコネート、ジブチルイタコネート等のイタコン酸エステル;モノメチルフマレート、モノエチルフマレート、モノプロピルフマレート、モノブチルフマレート、ジメチルフマレート、ジエチルフマレート、ジプロピルフマレート、ジブチルフマレート等のフマル酸エステル;及びモノメチルマレート、モノエチルマレート、モノプロピルマレート、モノブチルマレート、ジメチルマレート、ジエチルマレート、ジプロピルマレート、ジブチルマレート等のマレイン酸エステルが挙げられる。これらの単量体(b11)は1種を単独で用いてもよく、2種以上を併用してもよい。これらの中で、ラジカル重合が容易であり、且つ乳化重合が容易であることから、(メタ)アクリル酸エステルが好ましい。
単量体混合物(b1)は、必要に応じて、架橋性単量体(b12)を含有してもよい。
架橋性単量体(b12)としては、例えば、ジ(メタ)アクリル酸エチレングリコール、ジ(メタ)アクリル酸プロピレングリコール、ジ(メタ)アクリル酸1,3-ブチレングリコール、ジ(メタ)アクリル酸1,4-ブチレングリコール、ジビニルベンゼン、多官能(メタ)アクリル基変性シリコーン、(メタ)アクリル酸アリルが挙げられる。これらの架橋性単量体(b12)は、1種を単独で用いてもよく、2種以上を併用してもよい。
架橋性単量体(b12)の含有率は、単量体混合物(b1)(100質量%)中、0.001~15質量%であることが好ましく、0.01~5質量%であることがより好ましい。単量体混合物(b1)(100質量%)中の、架橋性単量体(b12)の含有率量が15質量%以下であれば、ゲル化性の付与効果に優れる。
本発明において重合体(B1)は、単層、又は2層以上の多層構造を有するものであっても良い。
単量体混合物(b1)を重合する方法としては、粒子状の重合体を得ることができる重合方法であれば特に制限されないが、真球状粒子を得やすい重合方法が好ましく、例えば、乳化重合法、ソープフリー乳化重合法、分散重合法、膨潤重合法、ミニエマルション重合法、微細懸濁重合法が挙げられる。この中でも、分散性に優れ、ファインピッチ化にも対応した粒子径を有する重合体が得られることから、ソープフリー乳化重合法、微細懸濁重合法が好ましい。重合体が球状粒子であると、重合体粉体(P1)を硬化性樹脂に配合する際に、硬化性樹脂組成物の粘度が上昇するのを抑制でき、流動性に優れた硬化性樹脂組成物が得られる。
単量体混合物(b1)を重合する際には、重合開始剤、乳化剤、分散安定剤、連鎖移動剤を用いることができる。
単量体混合物(b1)の重合に用いる重合開始剤としては、単量体混合物(a1)の重合に用いる重合開始剤と同様のものを用いることができる。
単量体混合物(b1)の重合に用いる乳化剤としては、単量体混合物(a1)の重合に用いる乳化剤と同様のものを用いることができる。
単量体混合物(b1)の重合に用いる乳化剤としては、単量体混合物(a1)の重合に用いる分散安定剤と同様のものを用いることができる。
単量体混合物(b1)の重合の際に、必要に応じて、t-ドデシルメルカプタン、n-オクチルメルカプタン、α-メチルスチレン等の連鎖移動剤を用いることもできる。
(組成比)
本発明の重合体粉体(P1)は、重合体の全体((メタ)アクリル酸エステル系重合体(A1)と重合体(B1)の合計)を100質量%とした場合に、(メタ)アクリル酸エステル系重合体(A1)30~90質量%、重合体(B1)70~10質量%であることが好ましい。
重合体の全体(100質量%)中の、(メタ)アクリル酸エステル系重合体(A1)の含有率が30質量%以上であれば、硬化性樹脂組成物を硬化して得られる硬化物が低弾性率となり、90質量%以下であれば、グラフト共重合体を樹脂に配合した際の、経時的な増粘を抑制することができる。
重合体の全体(100質量%)中の、重合体(B1)の含有率が10質量%以上であれば、重合体粉体(P1)を硬化性樹脂に配合した際の、経時的な増粘を抑制することができ、さらにゲル化性付与効果に優れる。また、重合体(B1)の含有率が70質量%以下であれば、硬化性樹脂組成物を硬化して得られる硬化物の低弾性率化に優れる。
本発明において、重合体のエマルションを粉体化する方法としては、公知の粉体化方法を用いることができ、例えば、噴霧乾燥法、凍結乾燥法、凝固法が挙げられる。これらの粉体化方法の中でも、硬化性樹脂中での重合体粉体(P1)の分散性に優れることから、噴霧乾燥法が好ましい。
噴霧乾燥法は、エマルションを微小液滴状に噴霧し、これに熱風を当てて乾燥するものである。微小液滴を発生させる方法としては、例えば、回転ディスク式、加圧ノズル式、二流体ノズル式、加圧二流体ノズル式が挙げられる。乾燥機容量は、特に制限はなく、実験室で用いるような小規模なスケールから、工業的に用いるような大規模なスケールまで、いずれも用いることができる。乾燥機における乾燥用加熱ガスの供給部である入口部、乾燥用加熱ガス及び乾燥粉末の排出口である出口部の位置も、特に制限はなく、通常用いる噴霧乾燥装置と同様でよい。装置内に導入する熱風の温度(入口温度)、即ち重合体に接触し得る熱風の最高温度は、得られる硬化性樹脂組成物中の重合体粉体(P1)の分散性に優れることから、100~200℃であることが好ましく、120~180℃であることがより好ましい。
噴霧乾燥の際の重合体のエマルションは、1種でもよく、複数のエマルションの混合物でもよい。また、噴霧乾燥時のブロッキング、嵩比重等の粉体特性を向上させるために、シリカ、タルク、炭酸カルシウム等の無機質充填剤や、ポリアクリレート、ポリビニルアルコール、ポリアクリルアミド等を添加してもよい。また、必要に応じて、酸化防止剤や添加剤等を加えて噴霧乾燥してもよい。
本発明において、重合体粉体(P1)のアセトン可溶分は5質量%以上である。重合体粉体のアセトン可溶分が5質量%以上であれば、硬化性樹脂組成物に充分なゲル化性を付与することができ、高温においても硬化性樹脂の流動が抑制される。
重合体粉体(P1)のアセトン可溶分は、硬化性樹脂の粘度が極めて低い場合でも高いゲル化性を付与できることから、10質量%以上が好ましく、15質量%以上がより好ましい。特に、低粘度で使用される用途では、少ない添加量で高いゲル化性を付与できることが要求されるため、アセトン可溶分が多いほど幅広い用途に使用できる。
重合体粉体(P1)のアセトン可溶分は、得られる硬化性樹脂組成物の貯蔵安定性に優れることから、50質量%以下が好ましく、40質量%以下がより好ましく、30質量%以下がさらに好ましい。
尚、本発明において、アセトン可溶分は、後述するアセトン可溶分の測定法により得られたものをいう。
アセトン可溶分は、単量体混合物(a1)中の架橋性単量体(a13)及びグラフト交叉剤(a14)の含有率、(メタ)アクリル酸エステル系重合体(A1)と重合体(B1)の組成比、並びに単量体混合物(b1)中の架橋性単量体(b12)の含有率を調整することで、適宜設定することができる。
アセトン可溶分を増やすには、単量体混合物(a1)中の架橋性単量体(a13)及びグラフト交叉剤(a14)の含有率を少なくする、又は重合体の全体中の重合体(B1)の含有率を多くする、又は単量体混合物(b1)中の架橋性単量体(b12)の含有率を少なくすることがよい。
本発明において、重合体粉体(P1)のアセトン可溶分の質量平均分子量は10万以上である。重合体粉体(P1)のアセトン可溶分の質量平均分子量が10万以上であれば、少ない添加量で高いゲル化性を付与でき、高温においても硬化性樹脂の流動が抑制される。硬化性樹脂への溶解性が低下することがなく、短時間で充分なゲル状態にできることから、質量平均分子量は2000万以下が好ましい。
重合体粉体(P1)のアセトン可溶分の質量平均分子量は、硬化性樹脂の粘度が極めて低い場合でも高いゲル化性を付与できることから、15万以上が好ましく、20万以上がより好ましく、40万以上がさらに好ましい。また、一定温度で効率的にゲル状態にできることから、1000万以下がより好ましく、500万以下がさらに好ましく、200万以下が特に好ましい。
本発明においてゲル状態は、後述する測定法により得られたゲル化温度及びゲル化性能で評価することができる。
また、本発明において、質量平均分子量は後述する質量平均分子量の測定法により得られたものをいう。
質量平均分子量は、重合開始剤の種類、重合開始剤量、重合温度、連鎖移動剤量を調整することで、適宜設定することができる。質量平均分子量を高くするには、重合開始剤量を減らす、又は重合温度を低くする、又は連鎖移動剤量を低くすることがよい。
重合体粉体(P1)の体積平均一次粒子径(Dv)は200nm以上であることが好ましく、500nm以上であることがより好ましい。通常、噴霧乾燥法や湿式凝固法等によって得られる粉体は、一次粒子が多数集合した凝集粉体であるが、体積平均一次粒子径(Dv)が200nm以上の場合は、この凝集粉体が一次粒子に分散し易く、液状エポキシ樹脂等の硬化性樹脂に配合した際の本粉体の分散性が良好となる。また、体積平均一次粒子径(Dv)が200nm以上であれば、粒子が持つ総表面積を充分に小さくできるため、硬化性樹脂組成物の粘度が上昇しにくいという利点を持つ。
体積平均一次粒子径(Dv)は、重合方法によって適宜調整することができる。例えば乳化重合法によれば~250nm、ソープフリー乳化重合法によれば~1mm、微細懸濁重合法によれば~10mmの粒子径のものが得られる。また、乳化重合法で重合する場合には、乳化剤量を調整することで、適宜設定することができる。
本発明の重合体粉体(P1)は、粉体としての性状や構造は問わない。例えば、重合で得られた一次粒子が多数集合して凝集粉体(二次粒子)を形成していてもよく、それ以上の高次構造を形成していても良い。但し、このような凝集粉体の場合、一次粒子同士が強固に結合せず、緩く凝集している状態が好ましい。これにより、硬化性樹脂中で一次粒子が微細、且つ均一に分散される。
また、重合体粉体(P1)は、硬化性樹脂中での分散性が良好となることから、体積平均一次粒子径(Dv)の小さな粒子が少ないものが好ましく、単分散性の良好なものが好ましい。
本発明において、重合体粉体(P1)の単分散性は、重合体粉体(P1)の体積平均一次粒子径(Dv)と個数平均一次粒子径(Dn)との比(Dv/Dn)で示される。重合体粉体(P1)のDv/Dnとしては3.0以下が好ましく、2.0以下がより好ましく、1.5以下がさらに好ましい。重合体粉体(P1)の単分散性が高い(Dv/Dnが1に近い)ほど、硬化性樹脂組成物のゲル化が短時間で急速に進行し、硬化性樹脂組成物の貯蔵安定性との両立がし易くなる傾向にある。
重合体粉体(P1)中のアルカリ金属イオンの含有量は10ppm以下であることが好ましい。アルカリ金属イオンの含有量は10ppm以下であれば、本発明の硬化性樹脂組成物を硬化して得られる硬化物の絶縁特性が優れたものとなる。アルカリ金属イオンの含有量は5ppm以下であることがより好ましく、1ppm以下であることがさらに好ましい。硬化性樹脂組成物は様々な用途に用いられるが、半導体ウェハーに直接触れる用途では、特に高い電気特性が要求される。また電子機器の薄型化に伴い、僅かなイオン性不純物の存在が絶縁不良を生じる場合もある。従って、アルカリ金属イオンの含有量が上記の範囲内であれば、幅広い用途に使用できる。
本発明において、重合体粉体(P1)のアルカリ金属イオンの含有量はNaイオン及びKイオンの合計量であり、後述するアルカリ金属イオンの含有量の測定法により得られたものをいう。
本発明の重合体粉体(P1)は、10μm以下の粒子径の粒子の占める割合が30vol%未満であることが好ましく、ハンドリング性の観点から、20vol%以下であることが好ましい。ここで、重合体粉体(P1)の粒子径とは、噴霧乾燥法や湿式凝固法等によって得られた凝集体の粒子径を意味する。このとき、重合体粉体(P1)の一次粒子同士は多数集合して凝集体を形成している。
本発明の重合体粉体(P1)は一次粒子同士が強固に結合せず、緩く凝集している状態を有していることが好ましく、周波数42kHz、出力40wの超音波を5分間照射した後に、10μm以下の粒子径の粒子の占める割合が30vol%以上となることが好ましい。さらに、10μm以下の粒子径の粒子の占める割合は、超音波照射前より超音波照射後が10vol%以上増加することが好ましい。
上記の超音波の照射は、得られた重合体粉体(P1)をイオン交換水で希釈して行うものであり、例えばレーザー回折/散乱式粒子径分布測定装置(島津製作所社製、「SALD-7100」)を用いて、5分間超音波を照射後、粒子径10μm以下の粒子の割合を体積基準で測定する。
重合体粉体(P1)の試料濃度は、装置に付属の散乱光強度モニターにおいて適正範囲となるよう適宜調整した。
重合体粉体(P1)が上記の粉体解砕性を有するためには、例えば、体積平均一次粒子径(Dv)が200nm以上、好ましくは400~900nm、より好ましくは500~800nmの重合体エマルションを噴霧回収法等で粉体化することが好ましい。
重合体粉体(P1)の体積平均二次粒子径(Dv)は、6~300μmであることが好ましく、10~220μmであることがより好ましく、20~200μmであることがさらに好ましい。重合体粉体(P1)の体積平均二次粒子径が6μm以上であると、硬化性樹脂中で重合体粉体(P1)が凝集しにくく、300μm以下であると、硬化性樹脂中で重合体粉体(P1)が分散性に優れたものとなる。硬化性樹脂中で重合体粉体(P1)の体積平均二次粒子径は、体積平均一次粒子径と同様の測定方法による測定値を採用することができる。
重合体粉体(P1)が含有する水分の比率は、1.5%以下が好ましく、1.0%以下がより好ましい。重合体粉体(P1)が含有する水分の比率が1.5%以下であれば、硬化性樹脂組成物を成形した際に、クラックが発生するおそれが少ない。
重合体粉体(P1)中の硫酸イオン(SO4 2-)の含有量は500ppm以下が好ましく、300ppm以下がより好ましく、100ppm以下がさらに好ましい。電子材料に用いる硬化性樹脂組成物は、銅やアルミニウム等の金属製のワイヤーや回路配線等と接触する環境で用いられることから、硫酸イオンが残存すると金属腐食を引き起こし、導通不良や誤動作の原因となる場合がある。重合体粉体(P1)中の硫酸イオンの含有量が100ppm以下であれば、幅広い用途に使用できる。
重合体粉体(P1)を得るため、乳化重合法や懸濁重合法で単量体混合物を重合する場合、硫酸塩以外に、硫酸エステルやスルホン酸化合物等を用いることがある。これらの化合物に含まれる、スルホン酸イオン、スルフィン酸イオン、硫酸エステルイオンも、金属腐食を引き起こす場合がある。
従って、単量体混合物の重合時には、硫酸エステルやスルホン酸化合物等の使用量を減らすことが好ましい。
本発明の重合体粉体(P1)はエポキシ樹脂等の硬化性樹脂に添加して使用することができる。本発明の重合体粉体(P1)は硬化性樹脂用応力緩和剤兼プレゲル化剤として使用することができる。尚、応力緩和剤とは硬化性樹脂組成物の硬化物の弾性率を低下させる添加剤であり、プレゲル化剤とは硬化性樹脂組成物にゲル化性を付与する添加剤である。
硬化性樹脂組成物に、応力緩和剤、プレゲル化剤をそれぞれ添加して、硬化性樹脂組成物にゲル化性を付与し、硬化物の弾性率を低下する方法があるが、硬化性樹脂組成物に各種添加剤を配合すると、硬化性樹脂組成物の粘度は上昇する。そのため、特に硬化性樹脂組成物の低粘度化が要求される用途においては、添加剤の配合量を削減したいという要求が強い。
本発明の重合体粉体(P1)からなる応力緩和剤兼プレゲル剤を使用することで、硬化性樹脂組成物に配合する添加剤量を削減することができ、初期粘度、ゲル化性、硬化物の低弾性率化に優れた硬化性樹脂組成物を得ることができる。
熱硬化性樹脂としては、例えば、エポキシ樹脂、フェノール樹脂、メラミン樹脂、尿素樹脂、オキセタン樹脂、不飽和ポリエステル樹脂、アルキド樹脂、ポリウレタン樹脂、アクリル樹脂及びポリイミド樹脂が挙げられる。これらは、1種を単独で又は2種以上を併用することができる。
活性エネルギー線硬化性樹脂としては、紫外線や電子線等の照射により硬化する樹脂が挙げられ、例えば、活性エネルギー線硬化性アクリル樹脂、活性エネルギー線硬化性エポキシ樹脂及び活性エネルギー線硬化性オキセタン樹脂が挙げられる。
これらの中で硬化性樹脂としては、絶縁性が高く電気特性に優れ電子材料分野に好適であることから、エポキシ樹脂、フェノール樹脂、ポリイミド樹脂及びオキセタン樹脂が好ましい。
また、エポキシ樹脂としては、上記エポキシ樹脂のプレポリマーや、ポリエーテル変性エポキシ樹脂、シリコーン変性エポキシ樹脂のような前記エポキシ樹脂と他の重合体との共重合体及びエポキシ樹脂の一部がエポキシ基を有する反応性希釈剤で置換されたものも挙げられる。
本発明においては、エポキシ樹脂としては、エポキシ樹脂組成物にゲル化性を付与する点で、常温で液体のエポキシ樹脂か、又は常温で固体であるが加熱時に硬化が充分に進行する前に液体化するエポキシ樹脂を50質量%以上含有するものが好ましい。
本発明の硬化性樹脂組成物は、前述の重合体粉体(P1)及び硬化性樹脂を含有するものである。
硬化性樹脂組成物中の重合体粉体(P1)の配合量としては5質量部以上が好ましく、10質量部がより好ましく、15質量部以上がさらに好ましい。重合体粉体(P1)の配合率が5質量部以上であれば、重合体粉体(P1)の添加剤としての効果が充分に発現する。また、重合体粉体(P1)の配合率としては50質量部以下が好ましく、40質量部以下がより好ましい。重合体粉体(P1)の配合率が50質量部以下であれば、硬化性樹脂組成物のペースト粘度が上昇するのを抑制することができ、用途によって加工性・作業性が低下する可能性を抑制することができる。
本発明の硬化性樹脂組成物には、本発明の効果を損なわない範囲内で、各種添加剤を配合することができる。
添加剤として、例えば、銀粉、金粉、ニッケル粉、銅粉等の導電性フィラー;窒化アルミニウム、炭酸カルシウム、シリカ、アルミナ等の絶縁フィラー;チキソ付与剤、流動性向上剤、難燃剤、耐熱安定剤、酸化防止剤、紫外線吸収剤、イオン吸着体、カップリング剤、離型剤及び応力緩和剤が挙げられる。
難燃剤は、本発明の目的を逸脱しない範囲であれば、リン系、ハロゲン系、無機系難燃剤等、公知のものを用いればよい。
耐熱安定剤としては、例えば、フェノール系酸化防止剤、イオウ系酸化防止剤、リン系酸化防止剤が挙げられる。酸化防止剤は、それぞれ単独で使用できるが、フェノール系/イオウ系、又はフェノール系/リン系のように2種以上を併用することが好ましい。
本発明の硬化性樹脂組成物を調製する際には、公知の混練装置を用いることができる。
硬化性樹脂組成物を得るための混練装置としては、例えば、らいかい機、アトライタ、プラネタリミキサ、ディゾルバー、三本ロール、ボールミル及びビーズミルが挙げられる。これらは2種以上を併用することができる。
本発明の硬化性樹脂組成物に添加剤等を配合する場合、配合する順番は特に問わないが、本発明の効果を充分に発揮するために、重合体粉体(P1)はできるだけ最後に混練することが好ましい。また、混練による剪断発熱等で系内の温度が上がるような場合には、混練中に温度を上げない工夫をすることが好ましい。
本発明の硬化性樹脂組成物は、一次実装用アンダーフィル材、二次実装用アンダーフィル材、ワイヤーボンドにおけるグラブトップ材等の液状封止材;基板上の各種チップ類を一括で封止する封止用シート;プレディスペンス型のアンダーフィル材;ウエハーレベルで一括封止する封止シートといった半導体用封止材料,光学接着剤;液晶、有機EL等の各種フラットパネルディスプレイのシーリング材といった液晶表示素子用シール剤,3層銅張積層板用の接着層;ダイボンドフィルム、ダイアタッチフィルム、層間絶縁フィルム、カバーレイフィルム等の接着層;ダイボンドペースト、層間絶縁ペースト、導電ペースト、異方導電ペースト等の接着性ペースト;発光ダイオードの封止材等の各種用途に使用することができる。中でも、基板上の各種チップ類を一括で封止する封止用シート、ウエハーレベルで一括封止する封止シート、ダイボンドフィルム、ダイアタッチフィルム、層間絶縁フィルム、カバーレイフィルムといったシート状物品に好適である。
本発明の硬化物は硬化性樹脂組成物を硬化して得られるものである。
本発明においては、硬化性樹脂としてエポキシ樹脂を使用する場合、例えば、酸無水物、アミン化合物、フェノール化合物等の硬化剤を使用して硬化性樹脂組成物を硬化させることができる。硬化剤を使用することにより、エポキシ樹脂の硬化性及び硬化物特性を調整することができる。特に、硬化剤として酸無水物を使用する場合、本硬化物の耐熱性や耐薬品性を向上させることができ、好ましい。
フェノール化合物としては、例えば、フェノールノボラック樹脂、クレゾールノボラック樹脂、ビスフェノールA、ビスフェノールF、ビスフェノールAD及びこれらビスフェノール類のジアリル化物の誘導体が挙げられる。これらの中で、機械強度及び硬化性に優れることからビスフェノールAが好ましい。これらは、1種を単独で又は2種以上を併用することができる。
硬化促進剤としては、エポキシ樹脂の熱硬化触媒として用いられている公知のものを使用することができ、例えば、2-メチルイミダゾール、2-エチル-4-メチルイミダゾール等のイミダゾール化合物;イミダゾール化合物とエポキシ樹脂のアダクト;トリフェニルホスフィン等の有機リン化合物;テトラフェニルホスフィンテトラフェニルボレート等のボレート類;及びジアザビシクロウンデセン(DBU)が挙げられる。これらは、1種を単独で又は2種以上を併用することができる。
潜在性硬化剤としては、例えば、ジシアンジアミド、カルボヒドラジド、シュウ酸ジヒドラジド、マロン酸ジヒドラジド、コハク酸ジヒドラジド、イミノジ酢酸ジヒドラジド、アジピン酸ジヒドラジド、ピメリン酸ジヒドラジド、スベリン酸ジヒドラジド、アゼライン酸ジヒドラジド、セバシン酸ジヒドラジド、ドデカンジヒドラジド、ヘキサデカンジヒドラジド、マレイン酸ジヒドラジド、フマル酸ジヒドラジド、ジグリコール酸ジヒドラジド、酒石酸ジヒドラジド、リンゴ酸ジヒドラジド、イソフタル酸ジヒドラジド、テレフタル酸ジヒドラジド、2,6-ナフトエ酸ジヒドラジド、4,4’-ビスベンゼンジヒドラジド、1,4-ナフトエ酸ジヒドラジド、アミキュアVDH及びアミキュアUDH(いずれも商品名、味の素(株)製)、クエン酸トリヒドラジド等の有機酸ヒドラジド及び各種のアミンアダクト系化合物が挙げられる。これらは、1種を単独で又は2種以上を併用することができる。
硬化剤又は硬化触媒の使用量は、エポキシ樹脂の場合と同様である。
また、硬化性樹脂として活性エネルギー線硬化性樹脂を使用する場合、使用する活性エネルギー線としては、例えば、電子線、紫外線、ガンマ線及び赤外線が挙げられる。また、活性エネルギー線の硬化条件としては、紫外線で硬化させる場合、高圧水銀灯、エキシマランプ、メタルハライドランプ等を備えた公知の紫外線照射装置を使用することができる。
紫外線照射量としては50~1,000mJ/cm2程度である。電子線で硬化させる場合、公知の電子線照射装置を使用することができ、電子線照射量としては10~100kGy程度である。
本発明の硬化性樹脂組成物を硬化して得られる硬化物は、電子材料を始めとする各種用途に用いることができ、半導体封止材料や接着剤等の低弾性率化が要求される用途に特に好適である。
本発明の重合体粉体(P2)は、アセトン可溶分が2質量%以上35質量%以下であり、アセトン可溶分の質量平均分子量が10万以上であり、体積平均一次粒子径(Dv)が200nm以上である重合体粉体である。
本発明において、アセトン可溶分とは、所定量の重合体粉体(P2)を、50倍の質量のアセトンに溶解させ、70℃で6時間還流した後の溶解した質量(%)を意味する。より詳細には、後述するアセトン可溶分の測定法により得られるものをいう。
重合体粉体(P2)のアセトン可溶分が、35質量%以下であれば、重合体粉体(P2)を硬化性樹脂組成物に配合した際の硬化物のガラス転移温度の低下を抑制することができる。
重合体粉体(P2)のアセトン可溶分は、硬化性樹脂の粘度が極めて低い場合でも高いゲル化性を付与できることから、2質量%以上であり、5質量%以上が好ましく、8質量%以上がより好ましい。また、硬化物のガラス転移温度の低下を抑制できることから、35質量%以下であり、30質量%未満が好ましく、25質量%以下がより好ましく、20質量%以下が更に好ましい。
アセトン可溶分は、単量体原料中の架橋性単量体の含有率を調整することで、適宜設定することができる。アセトン可溶分を増やすには架橋性単量体の含有率を減らし、アセトン可溶分を減らすには架橋性単量体の含有率を増やせば良い。
重合体粉体(P2)のアセトン可溶分の質量平均分子量が10万以上であれば、少ない添加量で高いゲル化性を付与でき、高温においても硬化性樹脂の流動が抑制される。また、硬化性樹脂への溶解性が低下することがなく、短時間で充分なゲル状態にできることから、アセトン可溶分の質量平均分子量は2000万以下が好ましい。
重合体粉体(P2)のアセトン可溶分の質量平均分子量は、硬化性樹脂の粘度が極めて低い場合でも高いゲル化性を付与できることから、20万以上が好ましく、30万以上がより好ましく、40万以上が更に好ましい。また、一定温度で効率的にゲル状態にできることから、1000万以下がより好ましく、500万以下が更に好ましく、200万以下が特に好ましい。
本発明においてゲル状態は、後述する測定法により得られたゲル化温度及びゲル化性能で評価することができる。
また、本発明において、質量平均分子量は後述する質量平均分子量の測定法により得られたものをいう。
質量平均分子量は、重合開始剤の種類、重合開始剤量、重合温度、連鎖移動剤量を調整することで、適宜設定することができる。質量平均分子量を高くするには、重合開始剤量を減らす、又は重合温度を低くする、又は連鎖移動剤量を低くすることがよい。
重合体粉体(P2)の体積平均一次粒子径(Dv)は200nm以上であり、300nm以上が好ましく、500nm以上がより好ましく、600nm以上が更に好ましい。体積平均一次粒子径(Dv)が200nm以上であれば、粒子が持つ総表面積を充分に小さくできるため、硬化性樹脂組成物の粘度が上昇しにくいという利点を持つ。また、ファインピッチ化や薄膜化への対応が可能であることから、重合体粉体(P2)の体積平均一次粒子径(Dv)は、8μm以下が好ましく、5μm以下がより好ましく、1μm以下が更に好ましい。
体積平均一次粒子径(Dv)は、重合方法によって適宜調整することができる。例えば乳化重合法によれば~250nm、ソープフリー乳化重合法によれば~1mm、微細懸濁重合法によれば~10mmの粒子径のものが得られる。また、乳化重合法で重合する場合には、乳化剤量を調整することで、適宜設定することができる。
また、重合体粉体(P2)は単分散性の良好なものが好ましい。本発明において、重合体粉体(P2)の単分散性は、重合体粉体(A)の体積平均一次粒子径(Dv)と個数平均一次粒子径(Dn)との比(Dv/Dn)で示される。重合体粉体(P2)のDv/Dnとしては3.0以下が好ましく、2.0以下がより好ましく、1.5以下が更に好ましい。重合体粉体(A)の単分散性が高い(Dv/Dnが1に近い)ほど、硬化性樹脂組成物のゲル化が短時間で急速に進行し、硬化性樹脂組成物の貯蔵安定性との両立がし易くなる傾向にある。
重合体粉体(P2)は、粉体としての性状や構造は問わない。例えば、重合で得られた一次粒子が多数集合して凝集粉体(二次粒子)を形成していてもよく、それ以上の高次構造を形成していてもよい。ただし、このような凝集粉体の場合、一次粒子同士が強固に結合せず、緩く凝集している状態が好ましい。これにより、硬化性樹脂中で一次粒子が微細且つ均一に分散される。
さらに、重合体粉体(P2)を粉体として回収する際、通常、噴霧乾燥法や湿式凝固法等によって得られる粉体は一次粒子が多数集合した凝集粉体であるが、体積平均一次粒子径(Dv)が200nm以上の場合には、この凝集粉体が一次粒子に分散し易く、エポキシ樹脂への分散性が良好となる。
本発明において、重合体粉体(P2)の屈折率は特に限定されないが、20℃での屈折率が1.48以上1.51以下であることが好ましい。ビニル重合体が多層構造粒子である場合は、各層の屈折率が1.48以上1.51以下であることが好ましい。屈折率が上記の範囲内であれば、重合体粉体(P2)を配合して得られる硬化物の透明性が優れたものとなる。重合体粉体(P2)の屈折率は、硬化物の透明性の観点から、1.49以上が好ましく、1.50以上がより好ましい。
尚、本発明において、重合体粉体(P2)の20℃での屈折率は、「POLYMER HAND BOOK」(Wiley Interscience社)に記載されている、20℃におけるホモポリマーの屈折率の値(ポリメチルメタクリレート:1.490、ポリn-ブチルメタクリレート:1.483、ポリスチレン:1.591等)を用いる。また、共重合体の屈折率については、その体積比率により算出できる。
本発明において、重合体粉体(P2)のガラス転移温度は特に限定されないが、ガラス転移温度は0℃以上であることが好ましい。ビニル重合体が多層構造粒子である場合は、各層のガラス転移温度が0℃以上であることが好ましい。ガラス転移温度が0℃以上であれば、硬化性樹脂組成物に多量に配合した場合でも、硬化物のガラス転移温度の低下が抑制でき、高い耐熱性が要求される用途においても使用することができる。また、一定温度で効率的にゲル状態にできることから、ガラス転移温度は250℃以下であることが好ましい。
尚、本発明において、ビニル重合体のガラス転移温度は、Fox式により求めることができる。ビニル重合体が単独重合体の場合は、高分子学会編「高分子データハンドブック」等に記載されている標準的な分析値を採用することができ、共重合体の場合は、各単量体単位の単独重合体のTgから式(1)により算出することができる。
Fox式を以下に示す。
1/(273+Tg)=Σ(wi/(273+Tgi)) 式(1)
式中、Tgは共重合体のTg(℃)、wiは単量体iの質量分率、Tgiは単量体iを重合して得られる単独重合体のTg(℃)である。
重合体粉体(P2)中のアルカリ金属イオンの含有量は10ppm以下が好ましく、5ppm以下がより好ましく、1ppm以下がさらに好ましい。硬化性樹脂組成物は様々な用途に用いられるが、僅かなイオン性不純物の存在が絶縁不良を生じる場合もある。
従って、アルカリ金属イオンの含有量が上記の範囲内であれば、幅広い用途に使用できる。また、プレゲル化剤を多量に必要とする用途でも使用できる。
本発明において、重合体粉体(P2)のアルカリ金属イオンの含有量はNaイオン及びKイオンの合計量であり、後述するアルカリ金属イオンの含有量の測定法により得られたものをいう。
重合体粉体(P2)中の硫酸イオン(SO4 2-)の含有量は20ppm以下が好ましい。電子材料に用いる硬化性樹脂組成物は、銅やアルミニウム等の金属製のワイヤーや回路配線等と接触する環境で用いられることから、硫酸イオンが残存すると金属腐食を引き起こし、導通不良や誤動作の原因となる場合がある。重合体粉体(P2)中の硫酸イオンの含有量が20ppm以下であれば、幅広い用途に使用できる。
重合体粉体(P2)を得るため、乳化重合法や懸濁重合法でビニル単量体原料を重合する場合、硫酸塩以外に、硫酸エステルやスルホン酸化合物等を用いることがある。これらの化合物に含まれる、スルホン酸イオン、スルフィン酸イオン、硫酸エステルイオンも、金属腐食を引き起こす場合がある。
従って、ビニル単量体原料(a2)の重合時には、硫酸エステルやスルホン酸化合物等の使用量を減らすことが好ましい。
本発明においてビニル重合体(A2)は、ラジカル重合可能なビニル単量体原料(a2)を重合して得られる。
ラジカル重合可能なビニル単量体原料(a2)としては、例えば、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸n-プロピル、(メタ)アクリル酸i-プロピル、(メタ)アクリル酸n-ブチル、(メタ)アクリル酸t-ブチル、(メタ)アクリル酸i-ブチル、(メタ)アクリル酸n-ヘキシル、(メタ)アクリル酸n-オクチル、(メタ)アクリル酸2-エチルヘキシル、(メタ)アクリル酸シクロヘキシル、(メタ)アクリル酸ベンジル、(メタ)アクリル酸フェニル、(メタ)アクリル酸ノニル、(メタ)アクリル酸デシル、(メタ)アクリル酸ドデシル、(メタ)アクリル酸ステアリル、(メタ)アクリル酸t-ブチルシクロヘキシル、(メタ)アクリル酸イソボルニル、(メタ)アクリル酸トリシクロ[5.2.1.02.6]デカン-8-イル-メタクリレート、ジシクロペンタジエニル、(メタ)アクリル酸N,N-ジメチルアミノエチル、(メタ)アクリル酸N-メチル-2,2,6,6-テトラメチルピペリジル等の(メタ)アクリル酸エステル;(メタ)アクリロニトリル等のシアン化ビニル単量体;スチレン、α-メチルスチレン、ビニルトルエン等の芳香族ビニル単量体;アクリル酸、メタクリル酸、クロトン酸、マレイン酸、イタコン酸、フマル酸、イソクロトン酸、サリチル酸ビニロキシ酢酸、アリロキシ酢酸、2-(メタ)アクリロイルプロパン酸、3-(メタ)アクリロイルブタン酸、4-ビニル安息香酸等のカルボキシル基含有ビニル単量体;(メタ)アクリル酸ヒドロキシメチル、(メタ)アクリル酸2-ヒドロキシエチル、(メタ)アクリル酸2-ヒドロキシプロピル、(メタ)アクリル酸2-ヒドロキシブチル、(メタ)アクリル酸4-ヒドロキシブチル、(メタ)アクリル酸グリセロールモノ等の水酸基含有ビニル単量体;(メタ)アクリル酸グリシジル等のグリシジル基含有ビニル単量体;(メタ)アクリルアミド;ビニルピリジン、ビニルアルコール、ビニルイミダゾール、ビニルピロリドン、酢酸ビニル、1-ビニルイミダゾール等のビニル単量体;モノメチルイタコネート、モノエチルイタコネート、モノプロピルイタコネート、モノブチルイタコネート、ジメチルイタコネート、ジエチルイタコネート、ジプロピルイタコネート、ジブチルイタコネート等のイタコン酸エステル;モノメチルフマレート、モノエチルフマレート、モノプロピルフマレート、モノブチルフマレート、ジメチルフマレート、ジエチルフマレート、ジプロピルフマレート、ジブチルフマレート等のフマル酸エステル;及びモノメチルマレート、モノエチルマレート、モノプロピルマレート、モノブチルマレート、ジメチルマレート、ジエチルマレート、ジプロピルマレート、ジブチルマレート等のマレイン酸エステルが挙げられる。
尚、本発明において、「(メタ)アクリル」は、「アクリル」又は「メタクリル」を示す。
単量体原料(a2)中のカルボキシル基含有ビニル単量体、水酸基含有ビニル単量体及びグリシジル基含有ビニル単量体から選ばれる少なくとも1種の官能基含有単量体の含有量は、本硬化物の透明性の点で、好ましくは2質量%以上、より好ましくは4質量%以上、さらに好ましくは6質量%以上である。また、重合安定性の観点から少なくとも1種の官能基含有単量体の含有量は、30質量%以下であることが好ましく20質量%以下であることがより好ましい。
水酸基含有ビニル単量体としては、ラジカル重合が容易であり、且つ乳化重合が容易であることから、(メタ)アクリル酸2-ヒドロキシエチルが好ましい。
グリシジル基含有ビニル単量体としては、ラジカル重合が容易であり、且つ乳化重合が容易であることから、(メタ)アクリル酸グリシジルが好ましい。
これらのカルボキシル基含有ビニル単量体、水酸基含有ビニル単量体及びグリシジル基含有ビニル単量体は1種を単独で又は2種以上を併用することができる。
本発明においてビニル単量体原料(a2)の重合方法としては、真球状粒子を得やすいこと及び粒子モルフォロジーを制御しやすいことから、乳化重合法、ソープフリー乳化重合法、膨潤重合法、ミニエマルション重合法、分散重合法及び微細懸濁重合法が好ましい。この中でも、分散性に優れ、ファインピッチ化にも対応した粒子径を持つ重合体が得られることから、ソープフリー乳化重合法がより好ましい。
ビニル重合体(A2)は、硬化性樹脂組成物の粘度が上昇せず流動性に優れることから、真球状の粒子が好ましい。
ビニル重合体(一次粒子)(A2)の内部モルフォロジーについては特に限定されるものではなく、重合体組成、分子量、ガラス転移温度、溶解度パラメーター等の各種因子が均一であっても、コアシェル構造やグラディエント構造等、一般的に認識されている様々な粒子モルフォロジーを有していてもよい。
ビニル重合体(A2)の内部モルフォロジーを制御する方法としては、例えば、粒子の内側と外側で溶解度パラメーターや分子量の異なる多層構造粒子にする方法が挙げられる。この方法は、硬化性樹脂組成物の貯蔵安定性(ポットライフ)とゲル化速度とを両立し易くなることから好ましい。
ビニル重合体(A2)の内部モルフォロジーを制御するための工業的に実用性の高い手法としては、例えば、異なる組成のビニル単量体混合物を多段階で逐次的に滴下重合する方法が挙げられる。
ビニル重合体(A2)がコアシェル構造を有しているかどうかの判定方法としては、例えば、重合過程でサンプリングされる重合体粒子の粒子径が確実に成長していること、及び重合過程でサンプリングされる重合体粒子の最低造膜温度(MFT)や各種溶剤への溶解度が変化していることを同時に満足することを確認することが挙げられる。
また、透過型電子顕微鏡(TEM)によりビニル重合体の切片を観察して、同心円状の構造の有無を確認する方法、又は凍結破断された本重合体の切片を走査型電子顕微鏡(クライオSEM)で観察して、同心円状の構造の有無を確認する方法が挙げられる。
ビニル単量体原料(a2)を重合する際には、重合開始剤、乳化剤、分散安定剤、連鎖移動剤を用いることができる。
重合開始剤としては、公知の重合開始剤を用いることができる。ビニル重合体を噴霧乾燥で粉体化する際に金属イオンの残留がないため、金属イオンが含まれていない重合開始剤を用いることが好ましい。
金属イオンが含まれていない重合開始剤としては、例えば、2,2’-アゾビスイソブチロニトリル、4,4’-アゾビス-(4-シアノバレリックアシッド)、2,2’-アゾビス-[N-(2-カルボキシエチル)-2-メチルプロピオンアミジン]等のアゾ系化合物;過硫酸アンモニウム等の過硫酸化合物;ジイソプロピルベンゼンヒドロパーオキシド、p-メンタンヒドロパーオキシド、クメンヒドロパーオキシド、t-ブチルヒドロパーオキシド等の有機過酸化物;前記過硫酸化合物又は前記有機過酸化物を一成分としたレドックス系開始剤が挙げられる。
これらの重合開始剤は、1種を単独で又は2種以上を併用することができる。
重合温度は重合開始剤の種類にもよるが、例えば、40~90℃程度の範囲で適宜行なうことができる。
乳化剤としては、公知の乳化剤を用いることができ、例えば、不均化ロジン酸、オレイン酸、ステアリン酸等の高級脂肪酸のアルカリ金属塩やアンモニウム塩、ドデシルベンゼンスルホン酸等のスルホン酸アルカリ金属塩やアンモニウム塩、ノニオン系乳化剤が挙げられる。
乳化剤は、1種を単独で又は2種以上を併用することができる。
分散安定剤としては、例えば、リン酸カルシウム、炭酸カルシウム、水酸化アルミニウム、澱粉末シリカ等の水難溶性無機塩;ポリビニルアルコール、ポリエチレンオキサイド、セルロース誘導体等のノニオン系高分子化合物;及びポリアクリル酸又はその塩、ポリメタクリル酸又はその塩、メタクリル酸エステルとメタクリル酸又はその塩との共重合体等のアニオン系高分子化合物が挙げられる。これらの中では、電気特性に優れることからノニオン系高分子化合物が好ましい。また、重合安定性との両立の観点から目的に応じて2種以上の分散安定剤を併用することができる。
連鎖移動剤としては、例えば、n-ドデシルメルカプタン、t-ドデシルメルカプタン、n-オクチルメルカプタン、t-オクチルメルカプタン、n-テトラデシルメルカプタン、n-ヘキシルメルカプタン、n-ブチルメルカプタン等のメルカプタン;四塩化炭素、臭化エチレン等のハロゲン化合物;及びα-メチルスチレンダイマーが挙げられる。
これらの連鎖移動剤は、1種を単独で又は2種以上を併用することができる。
本発明においてビニル重合体(A2)のエマルションを粉体化する方法としては、公知の粉体化方法を用いることができ、例えば、噴霧乾燥法、凍結乾燥法、凝固法が挙げられる。これらの粉体化方法の中でも、硬化性樹脂中での重合体粉体(P2)の分散性に優れることから、噴霧乾燥法が好ましい。
また、必要に応じて、酸化防止剤や添加剤等を加えて噴霧乾燥してもよい。
重合体粉体(P2)は、10μm以下の粒子径の粒子の占める割合が30vol%未満であることが好ましく、ハンドリング性の観点から、20vol%以下であることが好ましい。ここで、重合体粉体(P2)の粒子径とは、噴霧乾燥法や湿式凝固法等によって得られた凝集体の粒子径を意味する。このとき、重合体粉体(P2)の一次粒子同士は多数集合して凝集体を形成している。
重合体粉体(P2)の試料濃度は、装置に付属の散乱光強度モニターにおいて適正範囲となるよう適宜調整した
本発明の重合体粉体(P2)は硬化性樹脂に添加して使用することができる。
本発明で用いる硬化性樹脂としては、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ビスフェノールS型エポキシ樹脂、オルトクレゾールノボラック型エポキシ樹脂、脂環式エポキシ樹脂、トリグリシジルイソシアヌレート、脂肪族系エポキシ樹脂等のエポキシ樹脂が挙げられる。
これらは、1種を単独で用いてもよく2種以上を併用してもよい。
本発明においては、エポキシ樹脂としては、エポキシ樹脂組成物にゲル化性を付与する点で、常温で液体のエポキシ樹脂か、又は常温で固体であるが加熱時に硬化が充分に進行する前に液体化するエポキシ樹脂を50質量%以上含有するものが好ましい。さらにこれらのエポキシ樹脂の中でも、透明性及び耐熱性に優れ、光半導体材料に好適であることから、脂環式エポキシ樹脂が好ましい。
本発明の硬化性樹脂組成物は、重合体粉体(P2)及び硬化性樹脂を含有するものである。
本発明の重合体粉体(P2)は、硬化性樹脂に添加してゲル状態を実現する硬化性樹脂用プレゲル化剤として使用することができる。
本発明の硬化性樹脂組成物には、本発明の効果を損なわない範囲内で、各種添加剤を配合することができる。
添加剤としては、例えば、シリコーン系やフッ素系消泡剤、レベリング剤、γ-グリシドキシプロピルトリメトキシシランなどのシランカップリング剤、界面活性剤、充填剤、難燃剤、着色剤、酸化防止剤、紫外線吸収剤、イオン吸着体、可塑剤、顔料、離型剤、ガラスビーズ、無機粒子、応力緩和剤等の慣用の添加剤を使用することができる。
無機粒子としては、例えば、銀粉、金粉、ニッケル粉、銅粉等の導電性フィラー;窒化アルミニウム、炭酸カルシウム、シリカ、アルミナ等の絶縁フィラー等が挙げられる。
本発明の硬化性樹脂組成物を調製する際には、公知の混練装置を用いることができる。
硬化性樹脂組成物を得るための混練装置としては、例えば、らいかい機、アトライタ、プラネタリミキサ、ディゾルバー、三本ロール、ボールミル及びビーズミルが挙げられる。また、これらは2種以上を併用することができる。
透過率が上記範囲を下回る場合には、光半導体用封止材料として使用した際に、発光効率が低下し、性能が低下する場合がある。
本発明の硬化性樹脂組成物は、透明アンダーフィル材、透明液状封止材、透明封止用シート、透明ダイアタッチフィルム、透明ダイアタッチペースト、発光ダイオードの封止材、透明接着剤、光学接着剤、液晶、有機EL等の各種フラットパネルディスプレイ等の各種シーリング材等の各種用途に使用することができる。中でも、透明アンダーフィル材、透明液状封止材、透明封止用シート、発光ダイオードの封止材といった光半導体用封止材料に好適に使用することができる。
本発明の硬化物は、本発明の硬化性樹脂組成物を硬化して得られる。
硬化条件としては、硬化性樹脂組成物を構成する各成分の種類や含有量等によって適宜決めることができる。硬化温度としては、80~180℃が一般的である。
本発明においては、硬化性樹脂組成物を、例えば、酸無水物、アミン化合物、フェノール化合物等の硬化剤を使用して硬化させることができる。
これらは、1種を単独で用いてもよく2種以上を併用してもよい。
本発明においては、本発明の硬化物の透明性を損なわない範囲で硬化促進剤を使用してもよい。
硬化促進剤としては、例えば、トリフェニルホスフィン、ジフェニルホスフィン等の有機ホスフィン系硬化促進剤;2-メチルイミダゾール、2-フェニル-4-メチルイミダゾール、2-フェニルイミダゾール等のイミダゾール系硬化促進剤;1,8-ジアザビシクロ(5,4,0)ウンデセン-7、トリエタノールアミン、ベンジルメチルアミン等の三級アミン系硬化促進剤;テトラフェニルホスホニウム・テトラフェニルボレート等のテトラフェニルボレート系硬化促進剤が挙げられる。
これらは、1種を単独で用いてもよく2種以上を併用してもよい。
本発明の硬化物は電気・電子部品用の封止材料、例えば絶縁材料として好適に使用できる。さらに、透明性、耐熱性に優れるため、光半導体用封止材料、光学用接着剤、各種シーリング剤として好適である。
(1)一次粒子径及び単分散性
重合体エマルションをイオン交換水で希釈し、レーザー回折/散乱式粒子径分布測定装置(島津製作所社製、「SALD-7100」)を用い、体積平均一次粒子径(Dv)及び個数平均一次粒子径(Dn)を測定した。
屈折率は仕込みモノマー組成から算出される屈折率を用いた。
いずれも平均径としてはメジアン径を用いた。また、Dv及びDnの値より単分散性(Dv/Dn)を求めた。
重合体エマルションの試料濃度は、装置に付属の散乱光強度モニターにおいて適正範囲となるよう適宜調整した。
重合体粉体(P1)をイオン交換水で希釈した他は、体積平均一次粒子径と同様にして体積平均二次粒子径(Dv)を求めた。
重合体粉体(P1)1gをアセトン50gに溶解させ、70℃で6時間還流及び抽出した後、遠心分離装置((株)日立製作所製、「CRG SERIES」)を用いて、4℃にて14,000rpmで30分間遠心分離した。分離したアセトン可溶分をデカンテーションで取り除き、アセトン不溶分を真空乾燥機にて50℃で24時間乾燥させて質量を測定した。アセトン可溶分(%)は以下の式にて算出した。
(アセトン可溶分)=(1-アセトン不溶分の質量)×100
ゲルパーミエーションクロマトグラフィを用いて、下記の条件で重合体粉体(P1)のアセトン可溶分の質量平均分子量(Mw)を測定した。また、併せて数平均分子量(Mn)も測定した。
装置 :東ソー(株)製HLC8220
カラム:東ソー(株)製TSKgel SuperHZM-M(内径4.6mm×長さ15cm)、本数;4本、排除限界;4×106
温度 :40℃
キャリアー液:テトラヒドロフラン
流量 :0.35ml/分
サンプル濃度 :0.1%
サンプル注入量:10μl
標準 :ポリスチレン
重合体粉体(P1)20gをガラス製耐圧容器に量り取り、これにメスシリンダーを用いてイオン交換水200mlを加え、蓋をして強く振り混ぜて均一に分散させ、重合体粉体(P1)の分散液を得た。この後、得られた分散液を95℃のギヤーオーブン内に20時間静置して重合体粉体(P1)中のイオン分の抽出を行なった。
次いで、ガラス容器をオーブンから取り出して冷却した後、分散液を0.2μmセルロース混合エステル製メンブレンフィルター(アドバンテック東洋(株)製、型番:A020A025A)で濾過し、濾液100mlを用いて重合体粉体(P1)中のアルカリ金属イオンを下記の条件で測定した。尚、アルカリ金属イオンの含有量はNaイオン及びKイオンの合計量を測定した。
ICP発光分析装置:Thermo社製、IRIS「Intrepid II XSP」
定量法:濃度既知試料(0ppm、0.1ppm、1ppm及び10ppmの4点)による絶対検量線法
測定波長:Na;589.5nm及びK;766.4nm
重合体粉体(P1)をイオン交換水で希釈し、レーザー回折/散乱式粒子径分布測定装置(島津製作所社製、「SALD-7100」)を用いて、超音波照射(周波数42kHz、出力40W、5分間照射)前後の10μm以下の粒子の割合を体積基準で測定した。
エポキシ樹脂組成物を動的粘弾性測定装置(ユービーエム(株)製、「Rheosol G-3000」、パラレルプレート直径40mm、ギャップ0.4mm、周波数1Hz、捻り角度1度)を用い、開始温度40℃、終了温度200℃及び昇温速度4℃/分の条件で粘弾性の温度依存性を測定した。
また、測定開始時に20以上である、貯蔵弾性率G’と損失弾性率G”との比(G”/G’=tanδ)が昇温開始後ある温度にて、20を下回るようになった場合にゲル化が進行したと判断し、tanδ=20となる温度をもってゲル化温度とした。
上記のエポキシ樹脂組成物のゲル化温度の測定において、ゲル化温度-20℃での貯蔵弾性率G’をG’A、ゲル化温度+20℃での貯蔵弾性率G’をG’B(到達弾性率)とし、その比率(G’B/G’A)を求めて、下記の基準でゲル化性能を評価した。
◎:G’B/G’Aが100以上
○:G’B/G’Aが10以上100未満
△:G’B/G’Aが5以上10未満
×:G’B/G’Aが5未満
G’B/G’Aが10以上であれば、高温においても硬化性樹脂の流動が抑制される。
エポキシ樹脂組成物中の重合体粉体(P1)の分散状態を、粒ゲージを用いてJIS K-5600に準拠して測定し、下記の基準で分散性を評価した。
○:2μm以下
△:2μmを超え、10μm以下
エポキシ樹脂組成物中での重合体粉体(P1)の分散状態が2μm以下であれば、ファインピッチ化や薄膜化への対応が可能である。
エポキシ樹脂組成物を調製後、直ちに25℃に調温し、BM型粘度計(東京計器(株)製B型粘度計、ローターNo.4、回転数60rpm)を用いて粘度を測定し、エポキシ樹脂組成物の初期粘度とした。下記の基準で初期粘度を評価した。
◎:30000以下。
○:30000を超え、35000以下。
△:35000を超え、40000以下。
×:40000を超える。
エポキシ樹脂組成物の初期粘度が35000以下であれば作業性が低下することなく、高精度な塗布、パターン形成ができる。
得られた成形体を、3mm×10mm×60mmに切断して試験片とし、引張圧縮試験機(ストログラフ T:(株)東洋精機製作所製)を用い、JIS K 7171に準拠し、温度23℃で負荷を測定し、以下の基準で評価した。
○:2400MPa以下
△:2400MPaを超え、2500MPa以下
×:2500MPaを超える。
下記の実施例1~6、比較例1~4に従い、重合体粉体(P1-1)~(P1-10)を製造した。実施例1~6、比較例1~4では下記の原料を使用した。
メタクリル酸メチル :三菱レイヨン(株)製、商品名「アクリエステルM」
メタクリル酸n-ブチル:三菱レイヨン(株)製、商品名「アクリエステルB」
アクリル酸n-ブチル :三菱化学(株)製
メタクリル酸アリル :三菱レイヨン(株)製、商品名「アクリエステルA」
アクリル酸2-エチルヘキシル:三菱化学(株)製
メタクリル酸 :三菱レイヨン(株)製、商品名「アクリエステルMAA」
n-オクチルメルカプタン:片山化学(株)製(試薬特級品)
過硫酸アンモニウム:和光純薬工業(株)製
2,2’-アゾビス-[N-(2-カルボキシエチル)-2-メチルプロピオンアミジン]:和光純薬(株)製、商品名「VA-057」
ジ-(2-エチルヘキシル)スルホコハク酸アンモニウム:新日本理化(株)製、商品名「リカサーフM-300」
マックスブレンド攪拌機、還流冷却管、温度制御装置、滴下ポンプ及び窒素導入管を備えたセパラブルフラスコにイオン交換水78.00部、メタクリル酸メチル2.83部、及びメタクリル酸n-ブチル2.17部を投入し、120rpmで攪拌しながら窒素ガスのバブリングを30分間行った。その後、窒素雰囲気下で80℃に昇温し、予め調製した過硫酸アンモニウム0.04部及びイオン交換水2.00部の水溶液を一括投入して60分間保持し、シード粒子を形成させた。
上記のシード粒子が形成されたフラスコ内に、アクリル酸2-エチルヘキシル49.66部、及びメタクリル酸アリル0.34部、2,2’-アゾビス-[N-(2-カルボキシエチル)-2-メチルプロピオンアミジン]0.10部、ジ-2-エチルヘキシルスルホコハク酸アンモニウム0.50部及びイオン交換水25.00部をホモジェナイザー(IKA社製、「ウルトラタラックスT-25」、25000rpm)で乳化処理して得られた混合物を150分かけて滴下して、その後60分間保持し、(メタ)アクリル酸エステル系重合体(A1)(第1段目)の重合を終了した。(メタ)アクリル酸エステル系重合体(A1)のFox式で求めたガラス転移温度(Tg)を表1に示す。
次いで、メタクリル酸メチル45.15部、メタクリル酸n-ブチル3.00部、アクリル酸n-ブチル1.00部、メタクリル酸0.85部、ジ-(2-エチルヘキシル)スルホコハク酸アンモニウム0.50部、イオン交換水25.0部をディスパーミキサーで乳化処理して得られた第2段目の重合に用いる単量体混合物を150分かけて滴下して、その後60分間保持し、重合体のエマルションを得た。得られた重合体の体積平均一次粒子径の評価結果を表1に示す。
得られた重合体のエマルションを、大川原化工機(株)製、L-8型スプレードライヤーを用い、下記条件で噴霧乾燥処理して重合体粉体(P1-1)を得た。得られた重合体粉体(P1-1)のアセトン可溶分、アセトン可溶分分子量(Mw、Mn)、アルカリ金属イオンの含有量及び粉体解砕性の評価結果を表1-1に示す。
噴霧方式:回転ディスク式
ディスク回転数:25,000rpm
熱風温度 入口温度:150℃
出口温度:65℃


MMA:メタクリル酸メチル
n-BMA:メタクリル酸n-ブチル
n-BA:アクリル酸n-ブチル
AMA:メタクリル酸アリル
2-EHA:アクリル酸2-エチルヘキシル
MAA:メタクリル酸
n-OM:n-オクチルメルカプタン
乳化剤:ジ-(2-エチルヘキシル)スルホコハク酸アンモニウム
VA-057:2,2’-アゾビス-[N-(2-カルボキシエチル)-2-メチルプロピオンアミジン]
重合体粉体(P1-2)~(P1-10)の製造は、表1-1及び表1-2に示す原料組成とする以外は、実施例1と同様にして重合体粉体(P1-2)~(P1-10)を得た。得られた重合体の体積平均一次粒子径の評価結果を表1-1及び表1-2に示す。得られた重合体粉体(P1-2)~(P1-10)のアセトン可溶分、アセトン可溶分分子量(Mw、Mn)、アルカリ金属イオンの含有量及び粉体解砕性の評価結果を表1-1及び表1-2に示す。
エポキシ樹脂(商品名「JER828」:三菱化学(株)製)及び重合体粉体を表2-1及び表2-2に示す割合で配合し、遊星運動式真空ミキサー(シンキー社製、商品名「泡取り練太郎ARV-310LED」)を使用して、大気圧下で回転数1200rpmの条件で3分間混練を行い、混練物を得た。得られた混練物を3本ロールミル(EXAKT社製、「M-80E」)を使用し、ロール回転数200rpm、ロール間隔20μm・10μmで1パス、10μm・5μmで1パス、5μm・5μmで1パス処理した。
その後、再び遊星運動式真空ミキサー(シンキー社製、商品名「泡取り練太郎ARV-310LED」)を使用して、3KPaの減圧下で回転数1200rpmの条件で2分間混練・脱泡を行い、エポキシ樹脂組成物を得た。得られたエポキシ樹脂組成物についてゲル化温度、ゲル化性能の評価を実施した。評価結果を表2-1,2-2に示す。


エポキシ樹脂(商品名「JER828」:三菱化学(株)製)及び重合体粉体を表3-1及び表3-2に示す割合で配合し、遊星運動式真空ミキサー(シンキー社製、商品名「泡取り練太郎ARV-310LED」)を使用して、大気圧下で回転数1200rpmの条件で3分間混練を行い、混練物を得た。得られた混練物を3本ロールミル(EXAKT社製、「M-80E」)を使用し、ロール回転数200rpm、ロール間隔20μm・10μmで1パス、10μm・5μmで1パス、5μm・5μmで1パス処理した。
その後、再び遊星運動式真空ミキサー(シンキー社製、商品名「泡取り練太郎ARV-310LED」)を使用して、3KPaの減圧下で回転数1200rpmの条件で2分間混練・脱泡を行い、エポキシ樹脂組成物を得た。得られたエポキシ樹脂組成物について分散性、初期粘度、ゲル化温度、ゲル化性能の評価を実施した。評価結果を表3-1及び3-2に示す。



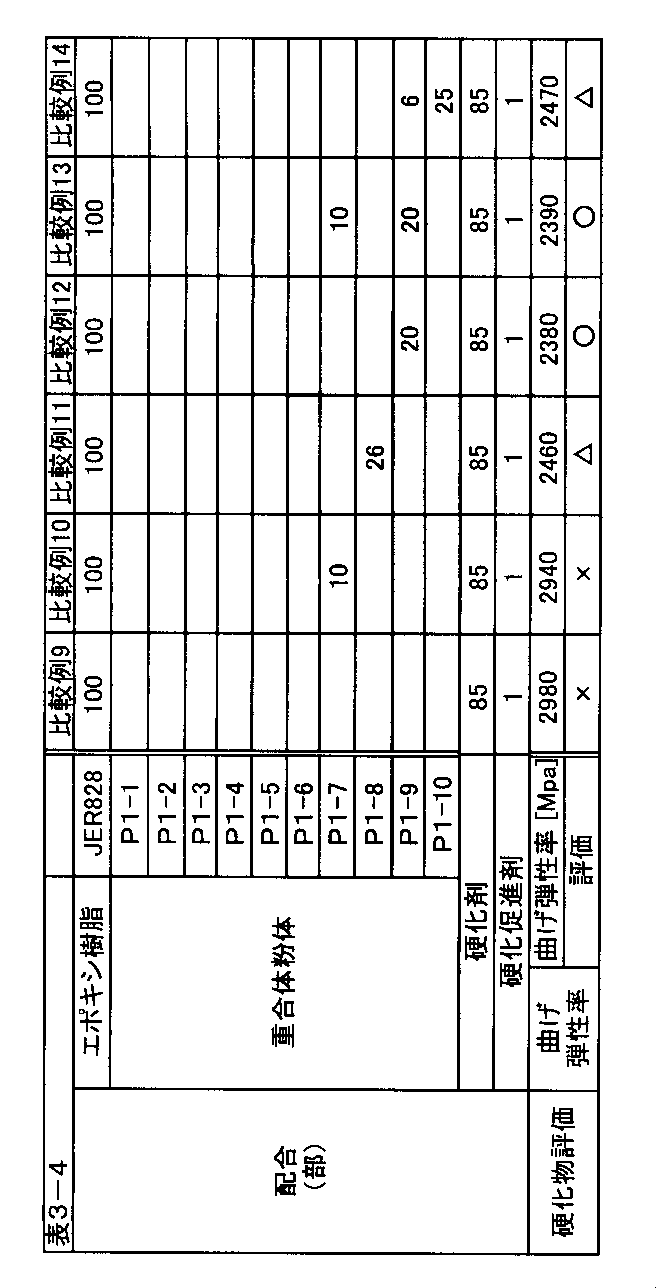
目的とするゲル化特性、弾性率に応じて、本発明の重合体粉体(P1)の他にプレゲル化剤、応力緩和剤を併用することで、さらなる初期粘度、ゲル化特性、硬化物の弾性率低下効果のバランスの向上が期待できる。
本発明の重合体粉体を添加していない比較例9のエポキシ樹脂組成物は、ゲル化性が見られず、さらに得られる硬化物の弾性率も高かった。
(メタ)アクリル酸エステル系重合体(A1)のガラス転移温度が0℃を超える重合体粉体(P1-7)を添加した比較例10のエポキシ樹脂組成物は、高いゲル化性能を有するものの、硬化物の弾性率低下効果は見られなかった。
アセトン可溶分が少なく、アセトン可溶分の質量平均分子量が低い重合体粉体(P1-9)を添加した比較例12のエポキシ樹脂組成物は、ゲル化性が見られなかった。
重合体粉体(P1-7)と重合体粉体(P1-9)とを添加した比較例13のエポキシ樹脂組成物は、高いゲル化性能及び弾性率低下効果を有するが、初期粘度が大幅に高くなっており、高精度な塗布・パターン形成が困難であった。
アセトン可溶分分子量が低い重合体粉体(P1-10)を添加した比較例14のエポキシ樹脂組成物は、初期粘度が高く、ゲル化特性も低位であった。
(1)一次粒子径及び単分散性
重合体のエマルションをイオン交換水で希釈し、レーザー回折/散乱式粒子径分布測定装置(島津製作所社製、「SALD-7100」)を用い、体積平均一次粒子径(Dv)及び個数平均一次粒子径(Dn)を測定した。
屈折率は仕込みモノマー組成から算出される屈折率を用いた。粒子がコアシェル構造等の多層構造を持つ場合には、各層毎の屈折率を算出し、層毎の質量比で全体平均を算出して用いた。
いずれも平均径としてはメジアン径を用いた。また、Dv及びDnの値より単分散性(Dv/Dn)を求めた。下記の基準で単分散性を評価した。
ビニル重合体エマルションの試料濃度は、装置に付属の散乱光強度モニターにおいて適正範囲となるよう適宜調整した。
重合体粉体(P2)1gをアセトン50gに溶解させ、70℃で6時間還流及び抽出した後、遠心分離装置((株)日立製作所製、「CRG SERIES」)を用いて、4℃にて14,000rpmで30分間遠心分離した。分離したアセトン可溶分をデカンテーションで取り除き、アセトン不溶分を真空乾燥機にて50℃で24時間乾燥させて質量を測定した。アセトン可溶分(質量%)は以下の式にて算出した。
(アセトン可溶分)=(1-アセトン不溶分の質量)×100
上記のアセトン可溶分の測定で得られたアセトン可溶分からアセトンを留去してアセトン可溶分の固形物を得た。この固形物についてゲルパーミエーションクロマトグラフィを用いて下記の条件で質量平均分子量(Mw)を測定した。また、併せて数平均分子量(Mn)も測定した。
装置 :東ソー(株)製HLC8220
カラム:東ソー(株)製TSKgel SuperHZM-M(内径4.6mm×長さ15cm)
本数;4本、排除限界;4×106
温度 :40℃
キャリアー液:テトラヒドロフラン
流量 :0.35ml/分
サンプル濃度 :0.1質量%
サンプル注入量:10μl
標準 :ポリスチレン
重合体粉体(P2)20gをガラス製耐圧容器に量り取り、これにメスシリンダーを用いてイオン交換水200mlを加え、蓋をして強く振り混ぜて均一に分散させ、ビニル重合体の分散液を得た。この後、得られた分散液を95℃のギヤーオーブン内に20時間静置して重合体粉体中のイオン分の抽出を行なった。
次いで、ガラス容器をオーブンから取り出して冷却した後、分散液を0.2μmセルロース混合エステル製メンブレンフィルター(アドバンテック東洋(株)製、型番:A020A025A)で濾過し、濾液100mlを用いて重合体粉体中のアルカリ金属イオンを下記の条件で測定した。尚、アルカリ金属イオンの含有量はNaイオン及びKイオンの合計量を測定した。
ICP発光分析装置:Thermo社製、IRIS「Intrepid II XSP」
定量法:濃度既知試料(0ppm、0.1ppm、1ppm及び10ppmの4点)による絶対検量線法
測定波長:Na;589.5nm及びK;766.4nm
重合体粉体(P2)をイオン交換水で希釈し、レーザー回折/散乱式粒子径分布測定装置(島津製作所社製、「SALD-7100」)を用いて、超音波照射(周波数42kHz、出力40W、3分間照射)前後の10μm以下の粒子の割合を体積基準で測定した。
エポキシ樹脂組成物中の重合体粉体(P2)の分散状態を、粒ゲージを用いてJIS K-5600に準拠して測定し、下記の基準で分散性を評価した。
○:5μm以下
△:5μmを超える
エポキシ樹脂組成物中での重合体粉体(P2)の分散状態が5μm以下であれば、ファインピッチ化や薄膜化への対応が可能である。
得られたエポキシ樹脂組成物を動的粘弾性測定装置(ユービーエム(株)製、「Rheosol G-3000」、パラレルプレート直径40mm、ギャップ0.4mm、周波数1Hz、捻り角度1度)を用い、開始温度40℃、終了温度200℃及び昇温速度4℃/分の条件で粘弾性の温度依存性を測定した。
また、測定開始時に10以上である、貯蔵弾性率G’と損失弾性率G”との比(G”/G’=tanδ)が昇温開始後ある温度にて、10を下回るようになった場合にゲル化が進行したと判断し、tanδ=10となる温度をもってゲル化温度とした。
上記のエポキシ樹脂組成物のゲル化温度の測定において、ゲル化温度-20℃での貯蔵弾性率G’をG’A、ゲル化温度+20℃での貯蔵弾性率G’をG’B(到達弾性率)とし、その比率(G’B/G’A)を求めて、下記の基準でゲル化性能を評価した。
○:G’B/G’Aが100以上
×:G’B/G’Aが100未満
G’B/G’Aが100以上であれば、高温においてもエポキシ樹脂組成物の流動が抑制される。
エポキシ樹脂組成物を硬化して得られた厚さ3mmのエポキシ樹脂硬化物について、外観を下記の基準で評価した。
○:目視で凝集物(ブツ)なし
△:目視で凝集物(ブツ)あり
エポキシ樹脂組成物を硬化して得られたエポキシ樹脂硬化物を、3mm×30mm×30mmに切断して試験片とし、ヘイズメーター(村上色彩研究所製、「HR-100」)を用い、23℃及び120℃の条件で硬化物の全光線透過率を測定し、下記の基準で評価した。
尚、120℃の全光線透過率評価は、120℃のオーブン中で30分間加熱した試験片を用いた。
○:全光線透過率が80%以上
△:全光線透過率が50%以上80%未満
×:全光線透過率が50%未満
エポキシ樹脂組成物を硬化して得られたエポキシ樹脂硬化物を、3mm×10mm×60mmに切断して試験片とし、動的機械的特性解析装置(機種名「EXSTAR DMS6100」、セイコーインスツル(株)製)により、両持ち曲げモード、昇温速度2℃/分、周波数10Hzの条件でtanδ曲線を測定し、転移点に対応した温度をガラス転移温度とし計測した。
エポキシ樹脂組成物を硬化して得られたエポキシ樹脂硬化物を、3mm×30mm×30mmに切断して試験片とし、インピーダンスアナライザー(機種名「E4991A」、アジレントテクノロジーズ社製)を用い、周波数100メガヘルツ時の比誘電率を測定した。比誘電率の値が低いほど絶縁性が良好となる。
下記の実施例31~40、比較例31に従い、重合体粉体(P2-1)~(P2-10)、(P2‘-1)を製造した。実施例31~40、比較例31では下記の原料を使用した。
メタクリル酸メチル :三菱レイヨン(株)製、商品名「アクリエステルM」
メタクリル酸n-ブチル:三菱レイヨン(株)製、商品名「アクリエステルB」
アクリル酸n-ブチル :三菱化学(株)製
スチレン :新日鐵化学(株)製
ジビニルベンゼン :新日鐵化学(株)製
メタクリル酸 :三菱レイヨン(株)製、商品名「アクリエステルMAA」
メタクリル酸2-ヒドロキシエチル:三菱レイヨン(株)製、商品名「アクリエステルHO」
メタクリル酸アリル :三菱レイヨン(株)製、商品名「アクリエステルA」
ジ-2-エチルヘキシルスルホコハク酸アンモニウム:東邦化学工業(株)製、商品名「リカサーフM-300」
過硫酸アンモニウム:和光純薬工業(株)製
過硫酸カリウム :和光純薬工業(株)製
2,2'-アゾビス(2,4-ジメチルバレロニトリル):和光純薬(株)製、商品名「V-65」(10時間半減期温度51℃)
マックスブレンド攪拌機、還流冷却管、温度制御装置、滴下ポンプ及び窒素導入管を備えたセパラブルフラスコにイオン交換水78.00質量部、メタクリル酸メチル2.83質量部、及びメタクリル酸n-ブチル2.17質量部を投入し、120rpmで攪拌しながら窒素ガスのバブリングを30分間行なった。
その後、窒素雰囲気下で80℃に昇温し、予め調製した過硫酸アンモニウム0.02質量部及びイオン交換水2.00質量部の水溶液を一括投入して60分間保持し、シード粒子を形成させた。
上記のシード粒子が形成されたフラスコ内に、メタクリル酸メチル92.94質量部、メタクリル酸n-ブチル2.00質量部、メタクリル酸アリル0.06質量部、ジ-2-エチルヘキシルスルホコハク酸アンモニウム1.00質量部、2,2'-アゾビス(2,4-ジメチルバレロニトリル)0.02質量部及びイオン交換水50.00質量部をホモジェナイザー(IKA社製、「ウルトラタラックスT-25」、25000rpm)で乳化処理して得られた混合物を300分かけて滴下して、その後1時間保持し、重合を終了した。得られたビニル重合体エマルションの一次粒子径の評価結果を表4-1に示す。
噴霧方式:回転ディスク式
ディスク回転数:25,000rpm
熱風温度
入口温度:150℃
出口温度:65℃
実施例32~40,比較例31は、表4-1及び4-2に示す原料組成及び重合条件とする以外は、実施例31と同様にして重合体粉体(P2-2)~(P2-10)、(P2’-1)を得た。得られたビニル重合体エマルションの一次粒子径の評価結果を表4-1及び4-2に示す。得られた重合体粉体(P2-2)~(P2-10)、(P2’-1)のアセトン可溶分、アセトン可溶分の分子量(Mw、Mn)、アルカリ金属イオンの含有量及び粉体解砕性の評価結果を表4-1及び4-2に示す。
マックスブレンド攪拌機、還流冷却管、温度制御装置、滴下ポンプ及び窒素導入管を備えたセパラブルフラスコにイオン交換水157.57質量部、アクリル酸n-ブチル3.05質量部、スチレン0.83質量部、ジビニルベンゼン0.13質量部、及びジ-2-エチルヘキシルスルホコハク酸アンモニウム0.20質量部を投入し、120rpmで攪拌しながら窒素ガスのバブリングを30分間行なった。
その後、窒素雰囲気下で80℃に昇温し、予め調製した過硫酸カリウム0.01質量部及びイオン交換水3.22質量部の水溶液を一括投入して60分間保持し、シード粒子を形成させた。
得られたビニル重合体エマルションは、実施例31と同様に噴霧乾燥処理して、重合体粉体(P2’-2)を得た。得られた重合体粉体(P2’-2)のアセトン可溶分、アセトン可溶分の分子量(Mw、Mn)、アルカリ金属イオンの含有量及び粉体解砕性の評価結果を表4-2に示す。

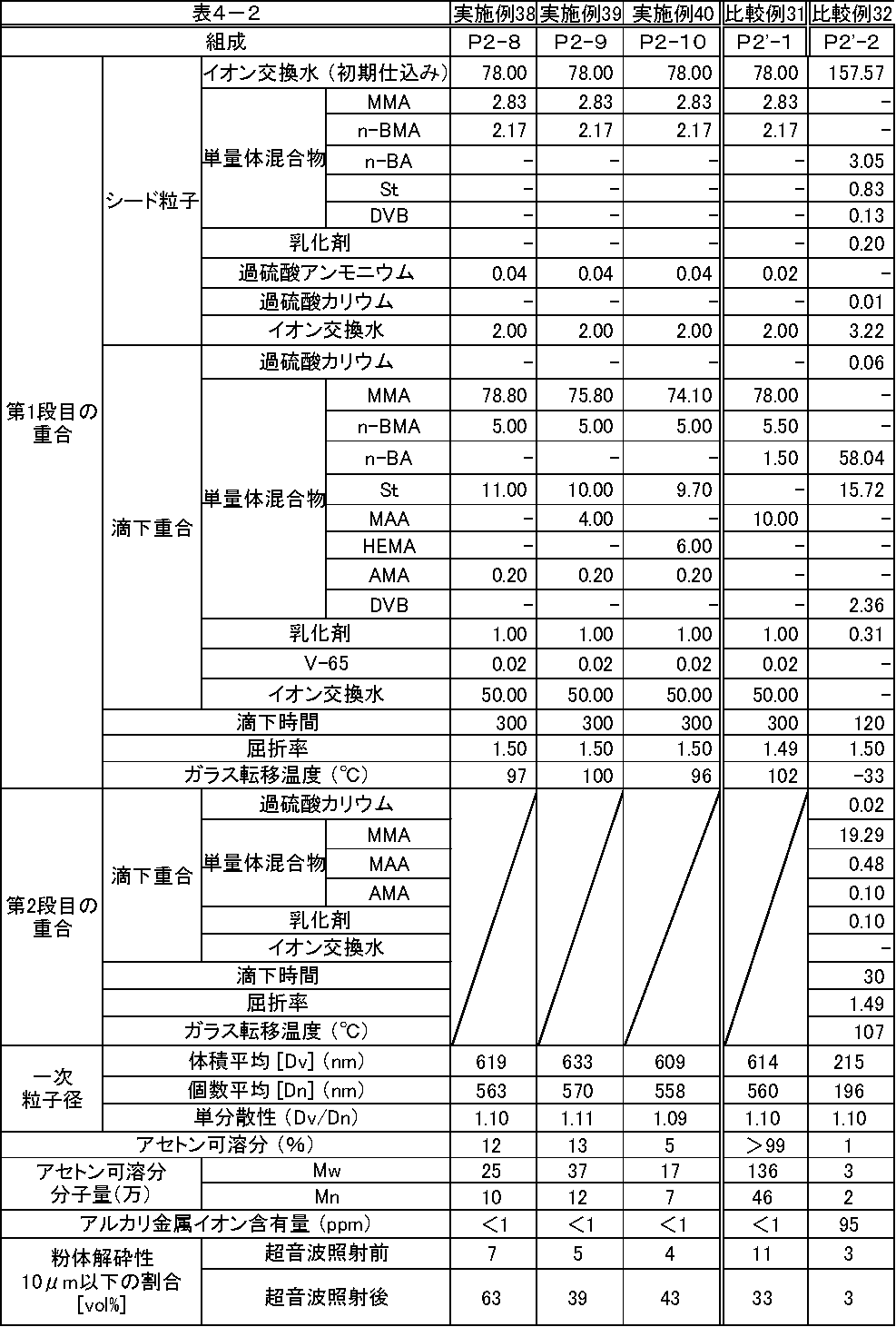
MMA :メタクリル酸メチル
n-BMA:メタクリル酸n-ブチル
n-BA :アクリル酸n-ブチル
St :スチレン
DVB :ジビニルベンゼン
AMA :メタクリル酸アリル
MAA :メタクリル酸
HEMA :メタクリル酸2-ヒドロキシエチル
乳化剤 :ジ-2-エチルヘキシルスルホコハク酸アンモニウム
V-65 :2,2’-アゾビス(2,4-ジメチルバレロニトリル)
脂環式エポキシ樹脂(ダイセル社製、「セロキサイド2021」)及び重合体粉体を表5で示す割合で配合し、遊星運動式真空ミキサー(シンキー社製、商品名「泡取り練太郎ARV-310LED」)を使用して、大気圧下で回転数1200rpmの条件で3分間混練を行ない、混練物を得た。得られた混練物を3本ロールミル(EXAKT社製、「M-80E」)を使用し、ロール回転数200rpm、ロール間隔20μm・10μmで1パス、10μm・5μmで1パス、5μm・5μmで1パス処理した。
その後、再び遊星運動式真空ミキサー(シンキー社製、商品名「泡取り練太郎ARV-310LED」)を使用して、3KPaの減圧下で回転数1200rpmの条件で2分間混練・脱泡を行ない、エポキシ樹脂組成物を得た。
得られたエポキシ樹脂組成物について分散性、ゲル化温度、ゲル化性能の評価を実施した。評価結果を表5に示す。
得られたエポキシ樹脂組成物に、硬化剤として酸無水物系硬化剤(商品名「リカシッドMH-700」、新日本理化(株)製)、硬化促進剤としてテトラブチルホスホニウムジエチルホスホジチオネート(日本化学工業社製、「ヒシコーリンPX-4ET」)を表6に示す割合で配合し、遊星運動式真空ミキサー(シンキー社製、商品名「泡取り練太郎ARV-310LED」)を用い、3KPaの減圧下で回転数1200rpmの条件で2分間混練・脱泡を行い、エポキシ樹脂組成物を得た。
次いで、長さ300mm×幅300mm×厚さ5mmの強化ガラス板2枚の、それぞれの強化ガラス板の片面にPETフィルム(東洋紡(株)製、商品名:TN200)を貼り、PETフィルム面が向き合うように並べ、強化ガラス板の間に厚み3mmのテフロン(登録商標)製のスペーサーを挟んで型を作製した。この型の中に上記のエポキシ樹脂組成物を流し込み、クランプで固定し100℃で3時間予備硬化を行なった後、120℃で4時間硬化を行い、型から取り出して厚さ3mmの硬化物を得た。エポキシ樹脂硬化物の外観、全光線透過率、ガラス転移温度、比誘電率の評価結果を表6に示す。


ビスフェノールA型水素化脂環式エポキシ樹脂(三菱化学社製、「YX-8000」)及び重合体粉体を表7で示す割合で配合し、実施例41と同様にしてエポキシ樹脂組成物を得た。得られたエポキシ樹脂組成物の分散性、ゲル化温度、ゲル化性能の評価を実施した。評価結果を表7に示す。
得られたエポキシ樹脂組成物に、硬化剤として酸無水物系硬化剤(商品名「リカシッドMH-700」、新日本理化(株)製)、硬化促進剤としてテトラブチルホスホニウムジエチルホスホジチオネート(日本化学工業社製、「ヒシコーリンPX-4ET」)を表8に示す割合で配合し、実施例51と同様にしてエポキシ樹脂硬化物を得た。得られたエポキシ樹脂硬化物の外観、全光線透過率、ガラス転移温度、比誘電率の評価結果を表8に示す。


特に、カルボキシル基を有する重合体粉体(P2-2、P2-3、P2-4,P2-9)、水酸基を有する重合体粉体(P2-5,P2-6,P2-7,P2-10)を添加して得られるエポキシ樹脂硬化物は、高い透明性を有している(実施例52~57,59,60,64)。さらに、高温条件下でも透明性の低下が少ない(実施例52~57,59,60)。そのため、高い透明性及び高温条件下での透明性が要求される光半導体用封止材料等の用途においても使用可能である。
屈折率が1.50である重合体粉体(P2-8)~(P2-10)を添加して得られるエポキシ樹脂硬化物は、エポキシ樹脂硬化物とビニル重合体の屈折率差が小さいため、高い透明性を有している(実施例58~60)。さらに、カルボキシル基、水酸基を導入することにより、高温条件下でも高い透明性を維持することが可能である(実施例59,60)。
また、本発明の重合体粉体(P2-1)~(P2-10)を添加して得られるエポキシ樹脂硬化物は、ガラス転移温度の低下が少ないため、高い耐熱性が要求される用途においても使用が可能である。更に、比誘電率が低いことから電子材料分野にも好適である。
アセトン可溶分が本発明の範囲外である、重合体粉体(P2’-1)を添加したエポキシ樹脂硬化物は、アセトン可溶分が多いため、本発明の重合体粉体(P2)を添加した硬化物と比較するとガラス転移温度が大幅に低下している。(比較例36、比較例42)
アセトン可溶分、アセトン可溶分分子量が本発明の範囲外である、重合体粉体(P2’-2)を添加したエポキシ樹脂組成物は、ゲル化性が見られなかった。(比較例34、40)
また、重合体粉体(P2’-2)はアルカリ金属イオン量が多いため、添加して得られるエポキシ樹脂硬化物の電気特性が低位であった(比較例37)。さらに、重合体粉体(P2’-2)はガラス転移温度が低いため、添加して得られるエポキシ樹脂硬化物のガラス転移温度の低下が見られた(比較例37、比較例43)。
ビニル重合体を添加していない比較例35、41のエポキシ樹脂組成物は、ゲル化性が見られなかった。
特に、本発明の重合体粉体を添加して得られる硬化性樹脂組成物は、初期粘度、ゲル化特性、硬化物の弾性率低下効果のバランスに優れるため、一次実装用アンダーフィル材、二次実装用アンダーフィル材、ワイヤーボンドにおけるグラブトップ材等の液状封止材、基板上の各種チップ類を一括で封止する封止用シート、プレディスペンス型のアンダーフィル材、ウエハーレベルで一括封止する封止シート、3層銅張積層板用の接着層、ダイボンドフィルム、ダイアタッチフィルム、層間絶縁フィルム、カバーレイフィルム等の接着層、ダイボンドペースト、層間絶縁ペースト、導電ペースト、異方導電ペースト等の接着性ペースト、発光ダイオードの封止材、光学接着剤、液晶、有機EL等の各種フラットパネルディスプレイのシーリング材等の各種用途に使用することができる。
Claims (27)
- (i)ガラス転移温度が0℃以下である(メタ)アクリル酸エステル系重合体(A1)を含み、重合体粉体のアセトン可溶分が5質量%以上であり、アセトン可溶分の質量平均分子量が10万以上である重合体粉体(P1)、及び(ii)アセトン可溶分が2質量%以上35質量%以下であり、アセトン可溶分の質量平均分子量が10万以上であり、体積平均一次粒子径(Dv)が200nm以上である重合体粉体(P2)からなる群より選択される重合体粉体(P)。
- 前記重合体粉体(P)が、(i)ガラス転移温度が0℃以下である(メタ)アクリル酸エステル系重合体(A1)を含み、重合体粉体のアセトン可溶分が5質量%以上であり、アセトン可溶分の質量平均分子量が10万以上である重合体粉体(P1)である、請求項1に記載の重合体粉体(P)。
- ガラス転移温度が0℃を超える重合体(B1)をさらに含む請求項2に記載の重合体粉体(P)。
- 重合体(B1)が(メタ)アクリル酸エステル系重合体である請求項3に記載の重合体粉体(P)。
- ガラス転移温度が0℃以下である(メタ)アクリル酸エステル系重合体(A1)の含有量が30~90質量%、重合体(B1)の含有量が70~10質量%(合計100質量%)である請求項3に記載の重合体粉体(P)。
- 粒子径10μm以下の粒子の占める割合が30vol%未満であり、以下の解砕性を有する請求項2に記載の重合体粉体(P)。
(解砕性)
(1)重合体粉体(P)をイオン交換水で希釈する。
(2)超音波照射を周波数42kHz、出力40Wで5分間行う。
(3)レーザー回折/散乱式粒子径分布測定により粒子径分布を測定する。
(4)10μm以下の粒子径の粒子の占める割合が30vol%以上である。 - (メタ)アクリル酸エステル系重合体(A1)の存在下で単量体混合物(b1)を重合し、粉体化して得られる(メタ)アクリル酸エステル系重合体粉体であって、(メタ)アクリル酸エステル系重合体(A1)のガラス転移温度が0℃以下であり、(メタ)アクリル酸エステル系重合体粉体のアセトン可溶分が5質量%以上であり、アセトン可溶分の質量平均分子量が10万以上である請求項2に記載の重合体粉体(P)。
- 請求項2に記載の重合体粉体(P)からなる硬化性樹脂用応力緩和剤兼プレゲル化剤。
- 硬化性樹脂100質量部に対して重合体粉体(P)を20質量部添加して得られる硬化性樹脂組成物の、ゲル化温度-20℃での貯蔵弾性率G‘Aと、ゲル化温度+20℃での貯蔵弾性率G’Bの比率(G‘B/G’A)が10以上となる請求項8に記載の硬化性樹脂用応力緩和剤兼プレゲル化剤。
- 請求項2に記載の重合体粉体(P)と硬化性樹脂とを含有する硬化性樹脂組成物。
- 硬化性樹脂がエポキシ樹脂である請求項10に記載の硬化性樹脂組成物。
- 請求項10に記載の硬化性樹脂組成物を硬化して得られる硬化物。
- 請求項10に記載の硬化性樹脂組成物を用いた半導体用封止材料。
- 請求項10に記載の硬化性樹脂組成物を用いた液晶表示素子用シール剤。
- 前記重合体粉体(P)が、(ii)アセトン可溶分が2質量%以上35質量%以下であり、アセトン可溶分の質量平均分子量が10万以上であり、体積平均一次粒子径(Dv)が200nm以上である重合体粉体(P2)である、請求項1に記載の重合体粉体(P)。
- 重合体粉体(P2)のアセトン可溶分が30質量%未満である請求項15に記載の重合体粉体(P)。
- 重合体粉体(P2)の20℃での屈折率が1.48以上1.51以下である請求項15に記載の重合体粉体(P)。
- 重合体粉体(P2)が、メタクリル酸メチル70~99質量%、メタクリル酸メチル以外の単量体原料30~1質量%を含む単量体原料(a2)を重合して得られたビニル重合体を粉体化して得られたメタクリル酸メチル系重合体粉体である請求項15に記載の重合体粉体(P)。
- 粒子径10μm以下の粒子の占める割合が30vol%未満であり、以下の解砕性を有する請求項15に記載の重合体粉体(P)。
(解砕性)
(1)重合体粉体(P)をイオン交換水で希釈する。
(2)超音波照射を周波数42kHz、出力40Wで3分間行う。
(3)レーザー回折/散乱式粒子径分布測定により粒子径分布を測定する。
(4)10μm以下の粒子径の粒子の占める割合が30vol%以上である。 - 請求項15に記載の重合体粉体(P)からなる硬化性樹脂用プレゲル化剤。
- 硬化性樹脂100質量部に対して重合体粉体(P)を20質量部添加して得られる硬化性樹脂組成物の、ゲル化温度-20℃での貯蔵弾性率G‘Aと、ゲル化温度+20℃での貯蔵弾性率G’Bの比率(G‘B/G’A)が100以上となる請求項20に記載の硬化性樹脂用プレゲル化剤。
- 請求項15に記載のビニル重合体粉体(P)と硬化性樹脂とを含有する硬化性樹脂組成物。
- 硬化性樹脂がエポキシ樹脂である請求項22に記載の硬化性樹脂組成物。
- エポキシ樹脂が脂環式エポキシ樹脂である請求項23に記載の硬化性樹脂組成物。
- 重合体粉体(P)と前記硬化性樹脂とを含有する硬化性樹脂組成物を硬化させて得られる厚さ3mmの硬化物の23℃における全光線透過率が70%以上である請求項22に記載の硬化性樹脂組成物。
- 請求項22に記載の硬化性樹脂組成物を硬化して得られる硬化物。
- 請求項22に記載の硬化性樹脂組成物を用いた光半導体用封止材料。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12860338.8A EP2796482B1 (en) | 2011-12-21 | 2012-12-21 | Polymer powder, curable resin composition, and cured product thereof |
| EP17185269.2A EP3263612B1 (en) | 2011-12-21 | 2012-12-21 | Polymer powder, curable resin composition and cured material thereof |
| CN201280063503.2A CN104011098B (zh) | 2011-12-21 | 2012-12-21 | 聚合物粉体、固化性树脂组合物及其固化物 |
| JP2013502735A JP6086062B2 (ja) | 2011-12-21 | 2012-12-21 | 重合体粉体、硬化性樹脂組成物及びその硬化物 |
| KR1020147014385A KR101685775B1 (ko) | 2011-12-21 | 2012-12-21 | 중합체 분체, 경화성 수지 조성물 및 그의 경화물 |
| US14/364,417 US9193812B2 (en) | 2011-12-21 | 2012-12-21 | Polymer powder, curable resin composition and cured material thereof |
| US14/931,431 US9522997B2 (en) | 2011-12-21 | 2015-11-03 | Polymer powder, curable resin composition and cured material thereof |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-279344 | 2011-12-21 | ||
| JP2011279344 | 2011-12-21 | ||
| JP2012035161 | 2012-02-21 | ||
| JP2012-035161 | 2012-02-21 | ||
| JP2012217760A JP2013028813A (ja) | 2012-02-21 | 2012-09-28 | ビニル重合体粉体、エポキシ樹脂組成物及びその硬化物 |
| JP2012-217760 | 2012-09-28 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/364,417 A-371-Of-International US9193812B2 (en) | 2011-12-21 | 2012-12-21 | Polymer powder, curable resin composition and cured material thereof |
| US14/931,431 Continuation US9522997B2 (en) | 2011-12-21 | 2015-11-03 | Polymer powder, curable resin composition and cured material thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013094759A1 true WO2013094759A1 (ja) | 2013-06-27 |
Family
ID=48668636
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/083361 WO2013094759A1 (ja) | 2011-12-21 | 2012-12-21 | 重合体粉体、硬化性樹脂組成物及びその硬化物 |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US9193812B2 (ja) |
| EP (2) | EP2796482B1 (ja) |
| JP (1) | JP6086062B2 (ja) |
| KR (1) | KR101685775B1 (ja) |
| CN (1) | CN104011098B (ja) |
| TW (2) | TWI532752B (ja) |
| WO (1) | WO2013094759A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013253183A (ja) * | 2012-06-08 | 2013-12-19 | Namics Corp | 先供給型液状半導体封止樹脂組成物 |
| CN103980592A (zh) * | 2014-04-30 | 2014-08-13 | 中国科学院化学研究所 | 一种用于3d打印的高填充量微纳粉体/聚合物复合材料及其制备方法和制品 |
| WO2015122115A1 (ja) * | 2014-02-12 | 2015-08-20 | 昭和電工株式会社 | 半導体パッケージの製造方法 |
| CN104952565A (zh) * | 2015-06-18 | 2015-09-30 | 西安高强绝缘电气有限责任公司 | 一种大直径多芯组合芯棒的制作方法 |
| WO2016060166A1 (ja) * | 2014-10-16 | 2016-04-21 | 三菱レイヨン株式会社 | 樹脂組成物およびそのプレス成形体 |
| JP2018110106A (ja) * | 2016-12-28 | 2018-07-12 | 株式会社タムラ製作所 | 異方性導電ペーストおよび電子基板の製造方法 |
| US10195621B2 (en) | 2013-07-19 | 2019-02-05 | Graco Minnesota Inc. | Pump changeover algorithm for spray system |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013112777A1 (en) * | 2012-01-25 | 2013-08-01 | Kemet Electronics Corporation | Polymerization method for preparing conductive polymer |
| FR3031106B1 (fr) | 2014-12-24 | 2017-02-03 | Arkema France | Composition de polymeres a phases multiples, son procede de preparation, son utilisation et composition la comprenant |
| US9574021B1 (en) * | 2015-08-14 | 2017-02-21 | King Abdulaziz City for Science and Technology (KACST) | Polymer nanoparticle preparation by miniemulsion polymerization |
| WO2018012234A1 (ja) * | 2016-07-15 | 2018-01-18 | 三菱ケミカル株式会社 | ホットメルト接着剤用アクリル系樹脂粉体ならびに樹脂組成物、及びその製造方法 |
| US10623846B2 (en) * | 2016-12-06 | 2020-04-14 | Bose Corporation | Earpieces employing viscoelastic materials |
| PL3369787T3 (pl) | 2017-03-03 | 2019-10-31 | Evonik Roehm Gmbh | Utwardzalne kompozycje żywicy (met)akrylowej o zwiększonej lepkości |
| ES2733471T3 (es) | 2017-03-03 | 2019-11-29 | Roehm Gmbh | Composiciones de resina termoendurecible curable con propiedades mecánicas mejoradas |
| WO2019021618A1 (ja) | 2017-07-26 | 2019-01-31 | 三菱ケミカル株式会社 | アクリル系重合体、プラスチゾル、およびテキスタイルインク |
| DE102017125177A1 (de) * | 2017-10-26 | 2019-05-02 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e. V. | Elektrisches Bauteil mit Isolationsschicht und Verfahren zu dessen Herstellung |
| CN111902461A (zh) * | 2018-03-26 | 2020-11-06 | 三菱化学株式会社 | 丙烯酸树脂粉体、树脂组合物、包含丙烯酸树脂粉体的热熔胶粘剂组合物及其制造方法 |
| WO2019221044A1 (ja) * | 2018-05-14 | 2019-11-21 | 積水化学工業株式会社 | 液晶表示素子用シール剤、上下導通材料、及び、液晶表示素子 |
| CN110669463A (zh) * | 2018-07-03 | 2020-01-10 | 北京天山新材料技术有限公司 | 环氧树脂胶粘剂及其应用 |
| JP6589172B1 (ja) * | 2019-01-11 | 2019-10-16 | 吉川工業株式会社 | 積層鉄心 |
| CN112909198B (zh) * | 2019-11-19 | 2024-02-02 | 利诺士尖端材料有限公司 | 能常温下贴合的有机电子装置用封装材料及有机电子装置 |
| KR102325733B1 (ko) * | 2019-11-19 | 2021-11-12 | (주)이녹스첨단소재 | 상온에서 합착이 가능한 유기전자장치용 봉지재 및 이를 포함하는 유기전자장치 |
| TWI780827B (zh) * | 2021-07-21 | 2022-10-11 | 緯創資通股份有限公司 | 膠化時間感測裝置、膠化時間感測方法、扭力閾值決定方法及面積收縮率閾值決定方法 |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS565391A (en) | 1979-06-25 | 1981-01-20 | Nat Jutaku Kenzai | Manufacture of heat insulating material |
| JPH0565391A (ja) * | 1991-09-04 | 1993-03-19 | Nissan Motor Co Ltd | エポキシ樹脂系接着性組成物 |
| JPH0995599A (ja) * | 1995-09-29 | 1997-04-08 | Nippon Zeon Co Ltd | エポキシ樹脂組成物及びそれを用いてなるプリプレグ及び成形品 |
| JPH11505871A (ja) * | 1995-05-24 | 1999-05-25 | インペリアル ケミカル インダストリーズ パブリック リミティド カンパニー | 耐衝撃性が改良された(メタ)アクリルポリマー |
| WO2000001748A1 (en) * | 1998-07-01 | 2000-01-13 | Mitsubishi Rayon Co., Ltd. | Fine acrylic polymer particles and plastisol containing the same |
| JP2006233145A (ja) | 2005-02-28 | 2006-09-07 | Nippon Zeon Co Ltd | 液状エポキシ樹脂組成物 |
| JP2010053199A (ja) | 2008-08-27 | 2010-03-11 | Daicel Chem Ind Ltd | 光半導体封止用樹脂組成物 |
| WO2010090246A1 (ja) | 2009-02-05 | 2010-08-12 | 三菱レイヨン株式会社 | ビニル重合体粉体、硬化性樹脂組成物及び硬化物 |
| WO2010104055A1 (ja) | 2009-03-10 | 2010-09-16 | 三菱レイヨン株式会社 | (メタ)アクリレート系重合体、樹脂組成物及び成形体 |
| JP2011208123A (ja) * | 2010-01-29 | 2011-10-20 | Kaneka Corp | 導電性樹脂組成物、及びその成形体 |
| JP2013028813A (ja) * | 2012-02-21 | 2013-02-07 | Mitsubishi Rayon Co Ltd | ビニル重合体粉体、エポキシ樹脂組成物及びその硬化物 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4125857A1 (de) | 1991-08-03 | 1993-02-04 | Roehm Gmbh | Mattierte polymethacrylat-folie |
| US5290857A (en) | 1991-09-04 | 1994-03-01 | Nippon Zeon Co., Ltd. | Epoxy resin adhesive composition |
| IT1271767B (it) * | 1993-05-14 | 1997-06-09 | Toyo Seikan Kaisha Ltd | Composizione di plastisol |
| US20110214717A1 (en) | 2008-11-12 | 2011-09-08 | Pythagoras Solar Inc. | Light curable photovoltaic cell encapsulant |
| WO2012165413A1 (ja) * | 2011-05-30 | 2012-12-06 | 三菱レイヨン株式会社 | エポキシ樹脂組成物、硬化物及び光半導体封止材料 |
-
2012
- 2012-12-21 EP EP12860338.8A patent/EP2796482B1/en active Active
- 2012-12-21 TW TW101149157A patent/TWI532752B/zh active
- 2012-12-21 WO PCT/JP2012/083361 patent/WO2013094759A1/ja active Application Filing
- 2012-12-21 TW TW105101938A patent/TWI580698B/zh active
- 2012-12-21 EP EP17185269.2A patent/EP3263612B1/en active Active
- 2012-12-21 CN CN201280063503.2A patent/CN104011098B/zh active Active
- 2012-12-21 US US14/364,417 patent/US9193812B2/en active Active
- 2012-12-21 KR KR1020147014385A patent/KR101685775B1/ko active IP Right Grant
- 2012-12-21 JP JP2013502735A patent/JP6086062B2/ja active Active
-
2015
- 2015-11-03 US US14/931,431 patent/US9522997B2/en active Active
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS565391A (en) | 1979-06-25 | 1981-01-20 | Nat Jutaku Kenzai | Manufacture of heat insulating material |
| JPH0565391A (ja) * | 1991-09-04 | 1993-03-19 | Nissan Motor Co Ltd | エポキシ樹脂系接着性組成物 |
| JPH11505871A (ja) * | 1995-05-24 | 1999-05-25 | インペリアル ケミカル インダストリーズ パブリック リミティド カンパニー | 耐衝撃性が改良された(メタ)アクリルポリマー |
| JPH0995599A (ja) * | 1995-09-29 | 1997-04-08 | Nippon Zeon Co Ltd | エポキシ樹脂組成物及びそれを用いてなるプリプレグ及び成形品 |
| WO2000001748A1 (en) * | 1998-07-01 | 2000-01-13 | Mitsubishi Rayon Co., Ltd. | Fine acrylic polymer particles and plastisol containing the same |
| JP2006233145A (ja) | 2005-02-28 | 2006-09-07 | Nippon Zeon Co Ltd | 液状エポキシ樹脂組成物 |
| JP2010053199A (ja) | 2008-08-27 | 2010-03-11 | Daicel Chem Ind Ltd | 光半導体封止用樹脂組成物 |
| WO2010090246A1 (ja) | 2009-02-05 | 2010-08-12 | 三菱レイヨン株式会社 | ビニル重合体粉体、硬化性樹脂組成物及び硬化物 |
| WO2010104055A1 (ja) | 2009-03-10 | 2010-09-16 | 三菱レイヨン株式会社 | (メタ)アクリレート系重合体、樹脂組成物及び成形体 |
| JP2011208123A (ja) * | 2010-01-29 | 2011-10-20 | Kaneka Corp | 導電性樹脂組成物、及びその成形体 |
| JP2013028813A (ja) * | 2012-02-21 | 2013-02-07 | Mitsubishi Rayon Co Ltd | ビニル重合体粉体、エポキシ樹脂組成物及びその硬化物 |
Non-Patent Citations (3)
| Title |
|---|
| "POLYMER HANDBOOK", WILEY INTERSCIENCE |
| "POLYMER HANDBOOK", WILEY INTERSCIENCE CORP. |
| See also references of EP2796482A4 |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013253183A (ja) * | 2012-06-08 | 2013-12-19 | Namics Corp | 先供給型液状半導体封止樹脂組成物 |
| US10195621B2 (en) | 2013-07-19 | 2019-02-05 | Graco Minnesota Inc. | Pump changeover algorithm for spray system |
| US11148152B2 (en) | 2013-07-19 | 2021-10-19 | Graco Minnesota Inc. | Pump changeover algorithm for spray system |
| WO2015122115A1 (ja) * | 2014-02-12 | 2015-08-20 | 昭和電工株式会社 | 半導体パッケージの製造方法 |
| CN103980592A (zh) * | 2014-04-30 | 2014-08-13 | 中国科学院化学研究所 | 一种用于3d打印的高填充量微纳粉体/聚合物复合材料及其制备方法和制品 |
| CN103980592B (zh) * | 2014-04-30 | 2016-02-24 | 中国科学院化学研究所 | 一种用于3d打印的高填充量微纳粉体/聚合物复合材料及其制备方法和制品 |
| WO2016060166A1 (ja) * | 2014-10-16 | 2016-04-21 | 三菱レイヨン株式会社 | 樹脂組成物およびそのプレス成形体 |
| JP6094686B2 (ja) * | 2014-10-16 | 2017-03-15 | 三菱レイヨン株式会社 | 樹脂組成物およびそのプレス成形体 |
| JPWO2016060166A1 (ja) * | 2014-10-16 | 2017-04-27 | 三菱レイヨン株式会社 | 樹脂組成物およびそのプレス成形体 |
| US10363724B2 (en) | 2014-10-16 | 2019-07-30 | Mitsubishi Chemical Corporation | Resin composition and compression-molded article of same |
| CN104952565A (zh) * | 2015-06-18 | 2015-09-30 | 西安高强绝缘电气有限责任公司 | 一种大直径多芯组合芯棒的制作方法 |
| JP2018110106A (ja) * | 2016-12-28 | 2018-07-12 | 株式会社タムラ製作所 | 異方性導電ペーストおよび電子基板の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2796482A1 (en) | 2014-10-29 |
| EP2796482B1 (en) | 2017-09-13 |
| CN104011098B (zh) | 2016-08-24 |
| EP2796482A4 (en) | 2015-05-20 |
| EP3263612B1 (en) | 2020-08-05 |
| CN104011098A (zh) | 2014-08-27 |
| KR20140095520A (ko) | 2014-08-01 |
| TW201335196A (zh) | 2013-09-01 |
| JP6086062B2 (ja) | 2017-03-01 |
| US9193812B2 (en) | 2015-11-24 |
| TWI580698B (zh) | 2017-05-01 |
| US20140350186A1 (en) | 2014-11-27 |
| EP3263612A1 (en) | 2018-01-03 |
| US9522997B2 (en) | 2016-12-20 |
| TWI532752B (zh) | 2016-05-11 |
| KR101685775B1 (ko) | 2016-12-12 |
| TW201615670A (zh) | 2016-05-01 |
| US20160096955A1 (en) | 2016-04-07 |
| JPWO2013094759A1 (ja) | 2015-04-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6086062B2 (ja) | 重合体粉体、硬化性樹脂組成物及びその硬化物 | |
| JP5736776B2 (ja) | ビニル重合体粉体、硬化性樹脂組成物及び硬化物 | |
| JP5979006B2 (ja) | ビニル重合体粉体、硬化性樹脂組成物及び硬化物 | |
| JP2013028813A (ja) | ビニル重合体粉体、エポキシ樹脂組成物及びその硬化物 | |
| JP2012077129A (ja) | 樹脂組成物、および、それを用いた封止材 | |
| TW200908166A (en) | Adhesive for electronic component, semiconductor chip stacking method, and semiconductor device | |
| JP4541186B2 (ja) | 液状エポキシ樹脂組成物 | |
| TWI535738B (zh) | (甲基)丙烯酸酯系聚合物、樹脂組成物及成形體 | |
| WO2012165413A1 (ja) | エポキシ樹脂組成物、硬化物及び光半導体封止材料 | |
| JP5971609B2 (ja) | 硬化性樹脂組成物及びこれを硬化した硬化物 | |
| JP2013076092A (ja) | 光半導体用封止シート用エポキシ樹脂組成物、光半導体用封止シート及び光半導体装置 | |
| JP2015218317A (ja) | 硬化性樹脂用応力緩和剤、硬化性樹脂組成物及び成形体 | |
| JP2013095860A (ja) | 硬化性樹脂用応力緩和剤、硬化性樹脂組成物及び成形体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2013502735 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12860338 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20147014385 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14364417 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012860338 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012860338 Country of ref document: EP |