WO2013044845A1 - 联芳基杂环取代的噁唑烷酮抗菌药 - Google Patents
联芳基杂环取代的噁唑烷酮抗菌药 Download PDFInfo
- Publication number
- WO2013044845A1 WO2013044845A1 PCT/CN2012/082318 CN2012082318W WO2013044845A1 WO 2013044845 A1 WO2013044845 A1 WO 2013044845A1 CN 2012082318 W CN2012082318 W CN 2012082318W WO 2013044845 A1 WO2013044845 A1 WO 2013044845A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- group
- pharmaceutically acceptable
- membered
- methyl
- Prior art date
Links
- 0 CC[C@](C)(C=C*C)*1=CC2(CC2)OC1=O Chemical compound CC[C@](C)(C=C*C)*1=CC2(CC2)OC1=O 0.000 description 2
- YUCSORQLDJFJPA-CQSZACIVSA-N CN(C1=NN(C)N(C)N1)c(cc1)ncc1-c(ccc(N(C[C@H](CO)O1)C1=O)c1)c1F Chemical compound CN(C1=NN(C)N(C)N1)c(cc1)ncc1-c(ccc(N(C[C@H](CO)O1)C1=O)c1)c1F YUCSORQLDJFJPA-CQSZACIVSA-N 0.000 description 1
- NOHCHHFUUKYMTQ-CYBMUJFWSA-N CN(c1n[n](C)nn1)c(cc1)ncc1-c(ccc(N(C[C@H](COP(O)(O)=O)O1)C1=O)c1)c1F Chemical compound CN(c1n[n](C)nn1)c(cc1)ncc1-c(ccc(N(C[C@H](COP(O)(O)=O)O1)C1=O)c1)c1F NOHCHHFUUKYMTQ-CYBMUJFWSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/04—Nitro compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
Definitions
- the present invention relates to the field of medical technology, and particularly relates to a biarylheterocyclic substituted 'oxazolidinone antibacterial agent, a pharmaceutically acceptable salt thereof, an isomer thereof and a prodrug thereof, and a method for preparing the same, which comprises the compound Pharmaceutical compositions and pharmaceutical preparations, and the use of these compounds in the preparation of a medicament for the treatment and/or prevention of infectious diseases and for the treatment and/or prevention of infectious diseases.
- Oxazolidinones antibacterials are a new class of chemically synthetic antibacterial drugs developed after sulfonamides and fluoroquinolones, which have the effect of inhibiting multi-drug resistant Gram-positive bacteria.
- Linezolid is the first B oxazolidinone antibiotic to be marketed.
- Linezolid is mainly used for the treatment of infectious diseases caused by resistant Gram-positive bacteria, and also for the treatment of surgical infectious diseases.
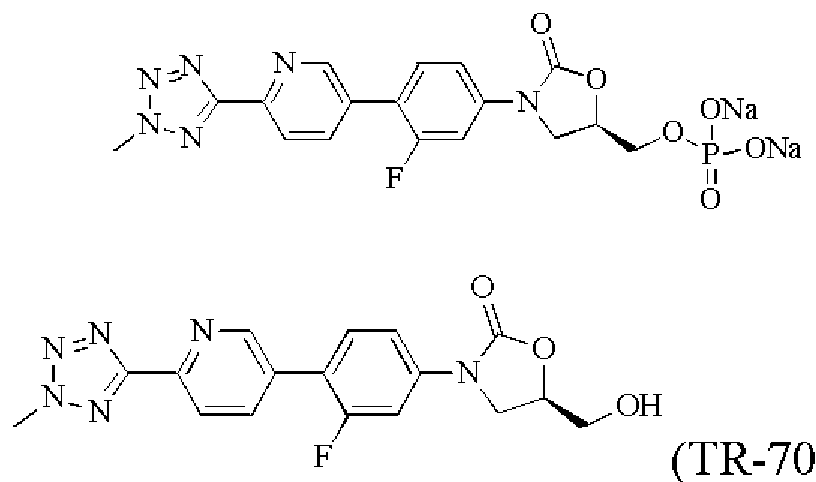
- CN201010508824.1 discloses the clinical phase III drug T-701, TR-700 of Trius Therapeutics Biopharmaceutical Company for infectious diseases caused by Gram-positive bacteria,
- the present invention provides a class of anti-infective compounds having better antibacterial activity, and the specific technical solutions are as follows:
- 1 is selected from the group consisting of (1) -OR 6 , (2) -N 6 6: , (3) -COR 6 , (4) -COO 6 , (5) -OCO 6 , (6) -CON 6 6 ', ( 7)-N 6 CO 6 ', (8) -OCON V, (9) -N 6 COO 6 ' , (10) -N 6 CON 6 6 , (11) CSR 6 , (12) -CSO 6 , ( 13) OCSR 6 , (14) — CSNR 6 R 6 ', (15) — NR 6 CSR 6 ', (16) -OCSN 6 6: , (17)-N 6 CSO 6 ', (18) -N 6 CSN 6 6 , (19) -N 6 C(N 6 )N 6 6 , (20) — S(0) p R 6 , (21)-SO ⁇ 6 R 6 ', or (22)R 6 ,
- p 0, 1 or 2
- R 6 and R 6 ' are selected from the group consisting of: (1) hydrogen, (2) d. 6 alkyl, (3) C 2-6 alkenyl, (4) C 2 . 6 alkynyl, (5) 3-14 Cycloalkyl, (6) 6-14 membered aryl, (7) 3-14 membered heterocyclic group containing one or more heteroatoms selected from N, S, O and/or S0 2 , (8 ) - CO -6 alkyl, (9) - COC 2-6 alkenyl, or (10) - COC 2 - 6 alkynyl;
- RR 3 is independently selected from the group consisting of hydrogen, halogen or alkyl
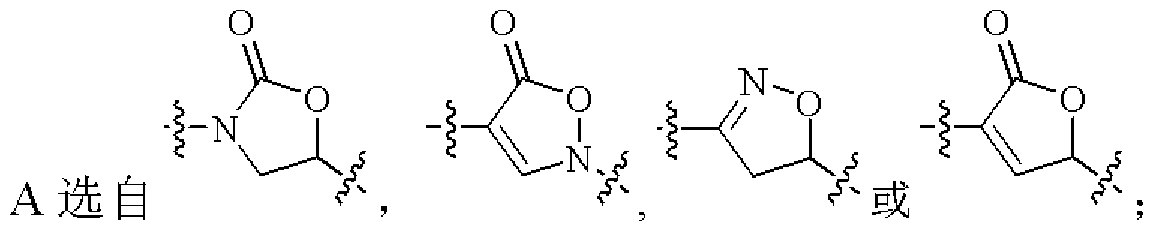
- B is selected from Wherein X, Y, W, and Z are each independently a C atom or an N atom, and Q is independently a CH 2 , NH, O or S atom;
- R 4 is selected from the group consisting of: (1) hydrogen, (2) d alkyl, (3) C 2 - 6 alkenyl, (4) C 2 - 6 alkynyl, (5) 3-14 membered cycloalkyl, (6) a 6-14 membered aryl group, (7) a 3-14 membered heterocyclic group, containing one or more heteroatoms selected from N, S, O and/or S0 2 , (8)-COd alkyl, (9) -COC 2-6 alkenyl, or (10)-COC 2-6 alkynyl;
- R 5 is selected from the following groups which are unsubstituted or substituted by 1-3 R 7 -
- R 7 is selected from the group consisting of halogen, carboxyl, hydroxy, amino, cyano, nitro, alkyl, carboxy d- 6 alkyl, hydroxy CL 6 alkyl, amino d- 6 alkyl, halogenated d- 6 alkyl, d. 6 alkane Oxy, halo d 6 alkoxy, d 6 alkoxy fluorenyl, Ci -6 fluorenylamino, bis(Ci -6 fluorenyl)amino, diindenyl)amino fluorenyl, Ci -6 fluorenyl Base, alkylcarbonyloxy, alkoxycarbonyl, carbamoyl, carbamoyl d.
- alkyl alkylcarbamoyl, bis( -6 alkyl)carbamoyl, aminosulfonyl, aminosulfonyl CL 6 alkyl, d alkylaminosulfonyl, bis(C ⁇ alkyl)aminosulfonyl, d. 6 alkylsulfonylamino, d- 6 alkylsulfonyl, alkylcarbonylamino or fluorenyl.
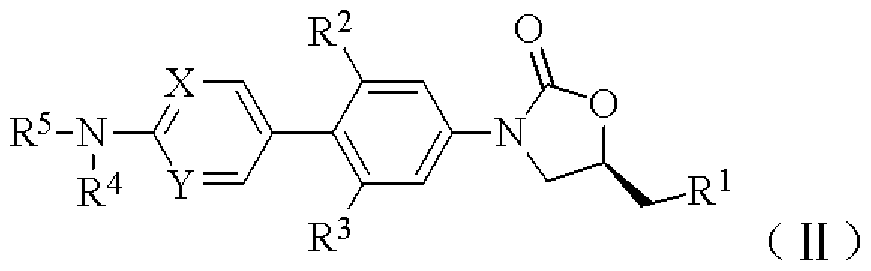
- the formula (I) has a structure represented by the following formula (II) -
- R ⁇ R ⁇ R R R X and Y are as defined in the general formula (I).
- R 1 is selected from the group consisting of -NHCOCH 3 , -OH, -NH 2 , -NHd alkyl,
- RR 3 is independently selected from hydrogen or halogen
- X and Y are each independently a C atom or a N atom
- R 4 is selected from the group consisting of: (1) hydrogen, (2) d- 4 alkyl, (3) C 2-4 alkenyl, (4) C 2-4 alkynyl, - CC ⁇ alkyl, (6) -C (0) C 2 .4 alkenyl, or (7) -C(0)C 2 .4 alkynyl;
- R 5 is selected from a 5-14 membered heteroaryl group which is unsubstituted or substituted with 1 to 3 R 7 groups, and the 5-14 membered heteroaryl group contains one or more selected from N, S, O and/or S0. 2 heteroatoms,
- R 7 is selected from the group consisting of halogen, carboxyl, hydroxy, amino, cyano, nitro, CM alkyl, carboxy d- 4 alkyl, hydroxy CM alkyl, amino CM alkyl, halo CM alkyl, CM alkoxy, halo CM alkoxy, CM alkoxy
- a compound represented by the formula ( ⁇ ), a pharmaceutically acceptable salt thereof, an isomer thereof or a prodrug thereof is preferably:
- R 1 is selected from -NHCOCH 3 or -OH;
- R 3 is independently selected from hydrogen or halogen
- R 4 is selected from hydrogen or CM alkyl
- X and Y are each independently a C atom or a N atom
- R 5 is selected from a 5-8 membered monoheteroaryl group which is unsubstituted or substituted with 1-3 R 7 groups, the 5-8 membered monoheteroaryl group comprising one or more selected N, S, O and/or a hetero atom of S0 2 ,
- R 7 is selected from a C 14 alkyl group or a halogenated C 14 alkyl group.
- R 1 is selected from -NHCOC 3 ⁇ 4 or -OH;
- R 2 is hydrogen
- R 3 is fluorine
- R 4 is hydrogen
- X is a C atom
- Y is a N atom
- R 5 is selected from a 5-6 membered monoheteroaryl group which is unsubstituted or substituted with 1-2 R 7 groups, the 5-6 membered monoheteroaryl group containing one or more selected from N, S or 0 atom,
- R 7 is selected from a .4 alkyl group or a halogenated d. 4 alkyl group.
- R 1 is selected from -NHCOC 3 ⁇ 4 or -OH;
- R 2 is hydrogen
- R 3 is fluorine
- R 4 is hydrogen
- X is a C atom
- Y is a N atom
- R 5 is selected from pyrrolyl, furyl, imidazolyl, 1,2,4-triazolyl, 1,2,3-triazolyl, pyrazolyl which is unsubstituted or substituted by 1-2 R 7 , oxazolyl, iso-oxazolyl, 1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,3,4-thiazide Diazolyl, 1,2,3-oxadiazolyl, 1,2,4-oxadiazole, 1,2,5-oxadiazolyl, pyridyl, pyrimidinyl, thiazolyl, isothiazolyl, 1, 2,3,4-tetrazolyl, pyranyl or pyrazinyl,
- R 7 is a CM alkyl group.
- a compound represented by the formula (III), a pharmaceutically acceptable salt thereof, an isomer thereof or a prodrug thereof is preferably:
- R 1 is selected from the group consisting of -NHCOCH 3 , -OH, -NH 2 , -NHC 1-6 alkyl,
- R 3 is independently selected from hydrogen or halogen
- W and Z are each independently a C atom or an N atom, and Q is independently a CH 2 , NH, O or S atom;
- R 4 is selected from the group consisting of: (1) hydrogen, (2) d- 4 alkyl, (3) C 2-4 alkenyl, (4) C 2-4 alkynyl, - CC ⁇ alkyl, (6) -C (0) C 2 .4 alkenyl, or (7) -C(0)C 2 .4 alkynyl;
- R 5 is selected from a 5-14 membered heteroaryl group which is unsubstituted or substituted with 1 to 3 R 7 groups, and the 5-14 membered heteroaryl group contains one or more selected from N, S, O and/or S0. 2 heteroatoms,
- R 7 is selected from the group consisting of halogen, carboxyl, hydroxy, amino, cyano, nitro, CM alkyl, carboxy CM alkyl, hydroxy CL 4 alkyl, amino CM alkyl, halo CM alkyl, d. 4 alkoxy , halogenated CM alkoxy, d_ 4 alkoxy
- a compound represented by the formula (III), a pharmaceutically acceptable salt thereof, an isomer thereof or a prodrug thereof is preferably:
- R 1 is selected from -NHCOCH 3 or -OH;
- R 3 is independently selected from hydrogen or halogen
- R 4 is selected from hydrogen or C 14 alkyl; W and Z are each independently a C atom or an N atom, and Q is independently an NH, O or S atom; R 5 is selected from a 5-8 membered monoheteroaryl group which is unsubstituted or substituted by 1-3 R 7 The 5-8 membered monoheteroaryl group contains one or more heteroatoms selected from the group consisting of g, S, O, and/or so 2 ,
- R 7 is selected from a .4 alkyl group or a halogenated d. 4 alkyl group. More preferably:
- R 1 is selected from -NHCOCH 3 or -OH;
- R 2 is hydrogen
- R 3 is fluorine
- R 4 is hydrogen
- W and Z are each independently a C atom or an N atom, and Q is independently an O or S atom;
- R 5 is selected from a 5-6 membered monoheteroaryl group which is unsubstituted or substituted by 1 - 2 R 7 groups, the 5-6 membered monoheteroaryl group containing one or more heteroatoms selected from N, S or O ,
- R 7 is selected from a C 14 alkyl group or a halogenated CM alkyl group. Further preferably - wherein
- R 1 is selected from -NHCOCH 3 or -OH;
- R 2 is hydrogen
- R 3 is fluorine
- R 4 is hydrogen
- W is a N atom, Z is a C atom, and Q is an S atom;
- R 5 is selected from pyrrolyl, furyl, imidazolyl, 1,2,4-triazolyl, 1,2,3-triazolyl, pyrazolyl, which are unsubstituted or substituted by 1 - 2 R 7 , oxazolyl, isoxazolyl, 1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,3,4-thiadiazolyl, 1,2,3-oxadiazole 1,2,4-oxadiazolyl, 1,2,5-oxadiazolyl, pyridyl, pyrimidinyl, thiazolyl, isothiazolyl, 1,2,3,4-tetrazolyl, pyranyl Or pyrazinyl,
- R 7 is a CM alkyl group.
- halogen as used in the present invention means a fluorine atom, a chlorine atom, a bromine atom, an iodine atom or the like. Preferred fluorine atom and chlorine Atom.
- Cw alkyl group as used in the present invention means a straight or branched alkyl group having 1 to 6 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, 2-methyl.
- a C 14 alkyl group is preferred.
- the "C 14 alkyl group" of the present invention refers to the above examples containing from 1 to 4 carbon atoms.
- C 2 -6 alkenyl group as used in the present invention means a linear or branched or cyclic alkenyl group having a double bond and having 2 to 6 carbon atoms, such as a vinyl group, a 1-propenyl group, and a 2- Propylene, 1-methylvinyl, 1-butenyl, 2-butenyl, 3-butenyl, 1-methyl-1-propenyl, 2-methyl-1-propenyl, 1- Methyl-2-propenyl, 2-methyl-2-propenyl, 1-pentenyl, 2-pentenyl, 3-pentenyl, 4-pentenyl, 1-methyl-1-butene Alkenyl, 2-methyl-1-butenyl, 3-methyl-1-butenyl, 1-methyl-2-butenyl, 2-methyl-2-butenyl, 3-methyl 2-butenyl, 1-methyl-3-butenyl, 2-methyl-3-butenyl, 3-methyl-3-butenyl, 1,1-dimethyl-2 -propen
- C 2 -6 alkynyl group as used in the present invention means a linear or branched alkynyl group having a triple bond and having 2 to 6 carbon atoms, such as an ethynyl group, a 1-propynyl group or a 2-propyne group.
- CM alkoxy means "CM alkyl-0-" group, respectively, "d .
- alkyl-NH-" group alkyl: 2 N-" group, alkyl-C(0)-0-” group, alkyl-OC(O)-" group, "C 14 alkyl-C(O)-” group, "C 14 alkyl-S0 2 -” group, “C 14 alkyl-S0 2 -NH-” group, "C 14 alkyl-C (0 )-NH-” group, ".4 alkyl-NH-C(O)-” group, alkyl) 2 NC(0)-” group, "C 14 alkyl-NH-S0 2 -” a group, "(d. 4 alkyl) 2 N-S0 2 -” group, wherein “d. 4 alkyl” is as defined above.
- Alkyl "Hydroxyalkyl”, “carboxy d. 6 alkyl”, “amino d- 6 alkyl”, “aminosulfonyl CL 6 alkyl”, “di(C ⁇ alkyl)amino d. 6 according to the invention.
- Alkyl "d alkoxy CL 6 alkyl”, “carbamoyl d. 6 alkyl” means more than one hydroxy, carboxy, amino, aminosulfonyl, bis(C ⁇ alkyl)amino, d, respectively. a group formed by 6 alkoxy, carbamoyl substituted -6 alkyl, wherein "C l alkyl” is as defined above.
- hydroxy C 14 alkyl group means more than one hydroxy group, carboxy group, amino group, aminosulfonyl group, di(c 14 alkyl)amino group, c 14 alkoxy group, amino group, respectively.
- haloalkyl group as used in the present invention means a group formed by substituting one or more "halogen” atoms for "d- 6 alkyl group", and the "halogenated C14 alkyl group” means that one or more "halogen” atoms are substituted for "d_”.
- a group formed by a 4- alkyl group, "Halogen” and “CL 6 alkyl”, “C 1 alkyl” are as defined above.
- haloalkoxy as used in the present invention means a group formed by substituting one or more "halogen” atoms for "d. 6 alkoxy", and the "halo CM alkoxy” means one or more "halogen” atoms.
- the "3-14 membered cycloalkyl group” as used in the present invention means a group derived from a hydrogen atom of a cyclic alkane moiety of 3 to 14 carbon atoms, including a 3-8 membered monocycloalkyl group, 6-14 members. And a cycloalkyl group, preferably a 3-8 membered monocycloalkyl group, a 3-6 membered monocyclic alkyl group, and a 5-6 membered monocycloalkyl group.
- "3-8 membered monocycloalkyl group”, "3-6 membered monocycloalkyl group”, "5-6 membered cyclocycloalkyl group” are respectively 3-8, 3-6, 5 in the following examples. Specific examples of -6 carbon atoms.
- a 3-8 membered monocycloalkyl group examples of which include, but are not limited to: cyclopropyl, cyclobutane, cyclopentyl, cyclohexane, cycloheptyl, cyclooctyl, methylcyclopropane, two Methylcyclopropane, methylcyclobutane, dimethylcyclobutane, methylcyclopentyl, dimethylcyclopentanyl, methylcyclohexane, dimethylcyclohexane Wait.
- 6- 14 membered cycloalkyl which refers to a 6-14 membered cyclic group formed by two or more cyclic structures sharing two adjacent carbon atoms, preferably 6-12 members and naphthenes.
- Base 6-10 yuan and cycloalkyl.
- Examples thereof include, but are not limited to, bicyclo[3.1.0]hexane, bicyclo[4.1.0]heptyl, bicyclo[2.2.0]hexyl, bicyclo[3.2.0]heptyl Bicyclo[4.2.0]octyl, octahydrocyclopentadienyl, octahydro-1H-indenyl, decalinyl, tetrahydrophenanthyl, and the like.
- 7- 12-membered bridged ring group refers to a structure containing 7-12 carbon atoms or/and hetero atoms formed by any two adjacent atoms sharing two non-adjacent atoms, and the hetero atom is selected from N, S, and 0. , CO, SO and / or S0 2 and so on.
- hetero atom is selected from N, S, and 0. , CO, SO and / or S0 2 and so on.
- the 7-12 membered spirocyclic group refers to a structure containing at least two rings sharing one atom and having 7 to 12 carbon atoms or/and hetero atoms selected from N, S, 0, CO, SO and / or S0 2 and so on. Which includes "7-10
- the "6-14 membered aryl group" as used in the present invention means a cyclic aromatic group having a ring atom of 6 to 14 carbon atoms, and includes a 6-8 membered monocyclic aryl group and an 8-14 membered fused ring aryl group.
- the 6-8 membered monocyclic aryl group includes a phenyl group, a cyclooctyltetraenyl group, and the like.
- the 8-14 membered fused ring aryl group means a cyclic group formed by two or more ring structures sharing two adjacent carbon atoms, including a naphthyl group, a fluorenyl group and a phenanthryl group, and the like.
- 8-14 membered partially saturated fused ring aryl group such as benzo 3-8 membered monocyclic cycloalkyl group, and specific examples are 2,3-dihydro-1H-indenyl, 1H-indenyl, 1,2,3 , 4-tetrahydronaphthyl, 1,4-dihydronaphthyl and the like.
- a 6-10 membered aryl group is preferred, and a benzene or a benzo 3-8 membered monocyclic cycloalkyl group is further preferred.
- the "5-14 membered heteroaryl” having a ring atom including one or more hetero atoms in addition to a carbon atom, the "hetero atom” being selected from the group consisting of N, S, 0, CO, SO, and/or S0 2 and so on.
- the heteroaryl group may be bonded through a carbon or a hetero atom. It includes a 5-8 membered monoheteroaryl group and a 8-14 membered fused heteroaryl group.
- 5-8 membered monoheteroaryl preferably 5-6 membered monoheteroaryl, including but not limited to pyrrolyl, imidazolyl, pyrazolyl, 1,2,3-triazolyl, 1,2,4-tri Azyl, pyridyl, furyl, thienyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, 1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,3,4-thiadiazolyl, 1,2,3-, oxadiazolyl, 1,2,4-oxadiazolyl, 1,2,5-.
- 8-14 membered heteroaryl includes, but is not limited to, benzofuranyl, isobenzofuranyl, benzothienyl, indolyl, isodecyl, quinolinyl, isoquinolinyl, pyridazinyl, Carbazolyl, pyridazinyl, quinoxalinyl, quinazolinyl, benzodiazinyl, benzisoxazolyl, benzoxazinyl, benzimidazolyl, pyridopyridyl, pyrazolo Pyridyl, fluorenyl, acridinyl and xanthene.
- the "3-14 membered heterocyclic group” as used in the present invention means a 3-14 membered cyclic group having one to more hetero atoms, the "hetero The atom “selected from N, S, 0, CO, SO and/or S0 2 and the like. It includes a 3-8 membered monoheterocyclic group and a 6-14 membered fused heterocyclic group.
- a 3-8 membered monoheterocyclic group means a monocyclic heterocyclic group having 3 to 8 ring atoms (having at least one hetero atom), preferably a 5-7 membered monoheterocyclic group, and specific examples include, but are not limited to, 2,5-dihydrothienyl, 4,5 -dihydropyrazolyl, 3,4-dihydro-2H-pyranyl, 5,6-dihydro-4H-l, 3-, xyl, aziridine, azetidinyl , thietane, tetrahydrofuranyl, tetrahydropyrrolyl, imidazolidinyl, pyrazolidinyl, tetrahydrofuranyl, 1,4-dioxanyl, 1,3-dioxane Base, 1,3-dithiacyclohexane, morpholinyl, piperazinyl and the like.
- a 6-14-membered fused heterocyclic group refers to a cyclo-ring structure in which 6 to 14 ring atoms (having at least one hetero atom) are bonded by two or more ring-shaped structures sharing two adjacent atoms with each other.
- a structure of a 6-10 membered fused heterocyclic group such as a benzo-3-8 membered monoheterocyclic group, a 3-8 membered monoheterocyclic group and a 3-8 membered monoheterocyclic group. Specific examples include, but are not limited to, 1,3-dihydrobenzofuranyl, benzo[[1.3]dioxolyl, hetero
- the "1-3" as used in the present invention means 1, 2 or 3.
- the "3-8 yuan" as used in the present invention means 3 yuan, 4 yuan, 5 yuan, 6 yuan, 7 yuan or 8 yuan, preferably 5-8 yuan, further preferably 5-7 yuan, still more preferably 5-6. yuan.
- Table 1 Compounds of the invention
- the invention also provides a preparation method of the above compound, but is not limited to the following methods :
- Raw material 1 raw material 2, inorganic base (such as potassium t-butoxide, cesium carbonate, potassium carbonate, etc.) and palladium catalyst (such as Pd(dppf)Cl 2 , Pd 2 (dba) 3 , Pd(PPh 3 ) 4 . Pd (PPh 3 ) 2 Cl 2 , etc.), BINAP was dissolved in toluene and heated to reflux overnight. Water was added, and the mixture was extracted with EtOAc. EtOAc was evaporated.
- inorganic base such as potassium t-butoxide, cesium carbonate, potassium carbonate, etc.
- palladium catalyst such as Pd(dppf)Cl 2 , Pd 2 (dba) 3 , Pd(PPh 3 ) 4 .
- BINAP was dissolved in toluene and heated to reflux overnight. Water was added, and the mixture was extracted with EtOAc. EtOAc was
- the compound of the formula (III) of the present invention can be produced by the method of the compound of the formula (II).
- a “pharmaceutically acceptable salt” of a compound of the invention refers to a base or acid addition salt of a compound of the invention with a pharmaceutically acceptable, non-toxic base or acid, including organic acid salts, inorganic acid salts, Organic alkali salt, inorganic alkali salt.
- Organic acid salts include formates, acetates, propionates, besylate, benzoates, p-toluenesulfonates, 2,3-dihydroxysuccinates, camphorsulfonates, citric acid Salt, methanesulfonate, ethanesulfonate, propanesulfonate, fumarate, gluconate, glutamate, isethionate, lactate, maleate, malate , mandelic acid salt, mucic acid salt, pamoate, pantothenate, succinate, tartrate, etc., particularly preferably benzoate, Toluenesulfonate, p-toluenesulfonate, methanesulfonate, citrate, maleate, fumarate, tartrate.
- the inorganic acid salt includes a hydrochloride, a hydrobromide, a hydroiodide, a sulfate, a phosphate, a nitrate, etc., and particularly preferably a hydrochloride, a hydrobromide, a sulfate, or a phosphate.
- the organic base salt includes an amine salt, including a salt formed with primary, secondary and tertiary amines, a cyclic amine and an alkali ion exchange resin, and may be selected from salts formed with the following organic bases: for example, arginine, betaine, caffeine, gall Base, hydrazine, hydrazine '-dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, hydrazine-ethyl-morpholine, hydrazine-ethyl Piperidine, meglumine, glucosamine, histidine, seabamin, isopropylamine, lysine, methyl glucosamine, morpholine, piperazine, piperidine, procaine, guanidine, cocoa Base, triethylamine, trimethylamine, tripropylamine and tromethamine.
- Inorganic alkali salts include salts with ammonia, alkali metals, alkaline earth metals, such as ammonium salts, and lithium, sodium, potassium, calcium, magnesium, zinc, barium, aluminum, iron, copper,
- the ferrous salt, the manganese salt, and the divalent manganese salt are particularly preferably an ammonium salt, and a sodium salt, a potassium salt, a calcium salt, and a magnesium salt.
- the compound of the formula (I) of the present invention forms a phosphate with phosphoric acid and further forms a metal phosphate of a compound of the formula (I), such as a phosphate disodium salt, with a metal salt.
- a "prodrug" of a compound of the invention refers to a compound (referred to as the original drug) which can be converted to any of the compounds of formula (I) or to a pharmaceutically acceptable salt of a compound of formula (I) under physiological conditions or by solvolysis.
- the prodrug When administered to a patient, the prodrug may be inactive, but it is converted to the active compound in vivo.
- the compound of the formula (I) of the present invention has a hydroxyl group, it can form an ester type prodrug with an amino acid, a phosphoric acid or the like, and the prodrug is stable in water or an acid solution, and dissociates to form a free form by action of an esterase or a phosphatase in the blood.
- Compound Compound.
- the prodrug of the compound of the formula (I) of the present invention has more excellent solubility than the original drug, is more easily absorbed by a passive object or a human body, is better converted into a prodrug compound in the blood, and exerts an antibacterial activity.
- the “isomer” of the compound of the present invention means a compound having the same chemical formula and a different structure, including a conformational isomer (structural isomer) and a stereoisomer (configuration isomer).
- “Stereoisomer” means when the compound of the invention contains one or more asymmetric centers and thus acts as a racemate and a racemic mixture, a single enantiomer, a mixture of diastereomers and a single Diastereomers.
- the compounds of the invention have asymmetric centers, each of which will independently produce two optical isomers, the scope of the invention including all possible optical isomers and mixtures of diastereomers and pure or partially Pure compound. If the compound of the present invention contains an olefinic double bond, the present invention includes a cis isomer and a trans isomer unless otherwise specified.
- the compounds of the present invention may also exist in tautomeric forms which have different hydrogen attachment points by one or more double bond shifts. Each tautomer and mixtures thereof are included in the compounds of the invention.
- the compound of the formula (I) of the present invention and an intermediate thereof in the preparation process represented by R 5
- R 5 When it is interconverted, when one of them is prepared, it is equivalent to the preparation of its tautomer.
- All of the compounds of the present invention and intermediates thereof, which are related to the above, are considered equivalent and are included in the scope of the present invention.
- the present invention also provides a pharmaceutical composition comprising the above-described compound of the formula (I) of the present invention, or a pharmaceutically acceptable salt thereof, an isomer thereof or a prodrug thereof, and one or more pharmaceutically acceptable carriers and / or thinner.
- the composition may be formulated into any of clinically or pharmaceutically acceptable dosage forms, preferably oral preparations and injections.
- the compound of the present invention or a pharmaceutically acceptable salt thereof or an isomer thereof can be administered to a mammal, such as a human, orally, parenterally (intravenously, intramuscularly, subcutaneously or rectally), topically, or the like.
- the compound of the present invention is used in an amount ranging from about 0.1 to 100 mg/kg body weight per day, for example, from 3 to 50 mg/kg body weight per day.
- the compound of the present invention or a pharmaceutically acceptable salt thereof or an isomer thereof may be formulated into an injection, including for intramuscular injection, intravenous injection, intravenous drip, subcutaneous injection, and the like.
- the injection can be produced by a conventional method in the field of the prior art, and an optional aqueous solvent or a non-aqueous solvent can be used.
- aqueous solvent is water for injection, and 0.9% sodium chloride solution or other suitable aqueous solution may also be used;
- the commonly used non-aqueous solvent is vegetable oil, for example, soybean oil for injection, and others, such as ethanol, propylene glycol, polyethylene glycol, etc. Aqueous solution, etc.
- additives may not be added, and appropriate additives may be added according to the nature of the drug, such as osmotic pressure regulators, pH adjusters, solubilizers, fillers, antioxidants, bacteriostatic agents, emulsifiers, suspending agents. Agents, etc.
- Commonly used osmotic pressure adjusting agents include sodium chloride, glucose, potassium chloride, magnesium chloride, calcium chloride, sorbitol, etc., preferably sodium chloride or glucose; commonly used pH adjusting agents include acetic acid - sodium acetate, lactic acid, hydrazine Acid-sodium citrate, sodium bicarbonate-sodium carbonate, etc.; commonly used solubilizing agents include polysorbate 80, propylene glycol, lecithin, polyoxyethylene castor oil, etc.; commonly used fillers include lactose, mannitol, sorbitol, Dextran anhydride; commonly used antioxidants are sodium sulfite, sodium bisulfite, sodium metabisulfite, etc.; commonly used bacteriostatic agents are phenol, cresol, chlorobutanol and the like.
- the pharmaceutical composition may also be formulated in a conventional form for rectal or topical administration, including suppositories, ointments, creams, patches, powders, sprays, inhalants and the like.
- the compound of the present invention or a pharmaceutically acceptable salt thereof or an isomer thereof can be formulated into a conventional solid preparation such as a tablet, a capsule, a pill, a granule, etc. by a conventional method; It can be formulated into oral liquid preparations such as oral solutions, oral suspensions, sugar collectors and the like.
- Tablets are mainly oral tablets, and other tablets, sublingual tablets, oral patches, chewable tablets, dispersible tablets, soluble tablets, effervescent tablets, sustained release tablets, controlled release tablets and enteric dissolution Film and so on.
- Capsules can be classified into hard capsules, soft capsules, sustained release capsules, controlled release capsules and enteric capsules according to their dissolution and release characteristics.
- Pills include dropping pills, sugar pills, pellets, and the like.
- the granules can be classified into soluble granules, suspended granules, effervescent granules, enteric granules, sustained release granules and controlled release granules.
- a suitable filler, binder, disintegrator, lubricant or the like may be added.
- Commonly used fillers include starch, powdered sugar, calcium phosphate, calcium sulfate dihydrate, dextrin, microcrystalline cellulose, lactose, pregelatinized starch, mannitol, etc.
- commonly used binders include sodium carboxymethyl cellulose, PVP -K30, hydroxypropyl cellulose, starch, methyl cellulose, ethyl cellulose, hypromellose, gelatinized starch, etc.
- commonly used disintegrants include kilo starch, crospovidone, cross-linked carboxy Methylcellulose sodium, sodium carboxymethyl starch, low-substituted hydroxypropylcellulose, etc.
- commonly used lubricants include magnesium stearate, talc, sodium lauryl sulfate, micronized silica gel, and the like.
- the present invention provides the use of a compound of the formula (I), or a pharmaceutically acceptable salt thereof, or an isomer thereof, for the manufacture of a medicament for the treatment and/or prevention of an infectious disease.
- the present invention provides a method of treating and/or preventing an infectious disease comprising administering a compound of the formula (I) of the present invention, or a pharmaceutically acceptable salt thereof or an isomer thereof, to such treatment or prevention.
- a compound of the formula (I) of the present invention or a pharmaceutically acceptable salt thereof or an isomer thereof, to such treatment or prevention.
- Mammals such as humans.
- the compounds of the present invention have good antibacterial activity and are useful for the treatment and/or prevention of various infectious diseases.
- the oxazolidinone antibiotic of the invention has good antibacterial activity against Gram-positive bacteria, and also has good antibacterial activity against Gram-positive resistant bacteria, and can be used for treating and/or preventing caused by Gram-positive bacteria.
- the beneficial effects of the compounds of the present invention are further illustrated by the antibacterial activity test, but it should not be construed that the compounds of the present invention have only the following beneficial effects.
- Test species methicillin-resistant Staphylococcus aureus (MRSA), methicillin-sensitive Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus epidermidis (MRSE), methicillin-sensitive golden yellow grapes Coccus (MSSE), Enterococcus faecium, Enterococcus faecalis, Streptococcus pneumoniae.
- MRSA methicillin-resistant Staphylococcus aureus
- MSSA methicillin-sensitive Staphylococcus aureus
- MRSE methicillin-resistant Staphylococcus epidermidis
- MSSE methicillin-sensitive golden yellow grapes Coccus
- Enterococcus faecium Enterococcus faecalis
- Streptococcus pneumoniae Streptococcus pneumoniae.
- Source Qianfoshan Hospital, Yunnan First People's Hospital, Shanghai Renji Hospital, Jilin Provincial People'
- Test substance The compounds of the present invention 1, 3, 4, 6, 8, and 11, were obtained by the methods of the respective examples.
- Test compound Compound 2 3 5 7 9 Prepared according to the example, dissolved in physiological saline.
- I.V intravenous bolus administration
- P.O stands for intragastric administration
- Blood collection IV Blood collection was performed according to the time points at the following table. About 100 ⁇ of whole blood was added to the liver at each time point. The sodium sodium anticoagulant tube was centrifuged at 8000 rpm for 6 minutes at 4 ° C in a low-temperature high-speed centrifuge to separate the blood 3 ⁇ 4, and the blood was stored in a refrigerator at -80 °C.
- Compound 2 Take 20 ⁇ of blood, add 100 ⁇ l of 50 ng/ml Radexolid in methanol, vortex for 3 minutes, centrifuge at 12000 rpm for 5 minutes, and take supernatant 100. ⁇ was added to ⁇ , vortexed for 2 minutes, and analyzed by LC-MS/MS.
- Compound 3 Take 20 ⁇ of blood 3 ⁇ 4, add 800 ⁇ of 10 ng/ml KBP-3957 methyl tert-butyl ether solution, vortex for 10 minutes, centrifuge at 12000 rpm for 5 minutes, take supernatant 400 The ⁇ was added to a 96-well plate, nitrogen was blown, and a 200 ⁇ l methanol:water (7:3) solution was added, vortexed for 10 minutes, and analyzed by LC-MS/MS.
- Trace test compound 2 is a prodrug of compound 1
- compound 5 is a prodrug of compound 4
- compound 7 is a prodrug of compound 6
- compound 9 is a prodrug of compound 8, so that the original drug compound 1 and 4 are separately detected after administration. 6, 6 and 8 PK data.
- AUC inf represents the area under the curve of the drug, 0.
- Vss represents the apparent volume of distribution
- T 1/2 represents half life
- T max represents the peak time of blood medicine ⁇ O
- the compounds of the present invention have good pharmacokinetic properties and are excellent in drug-forming properties. 4, the specific implementation
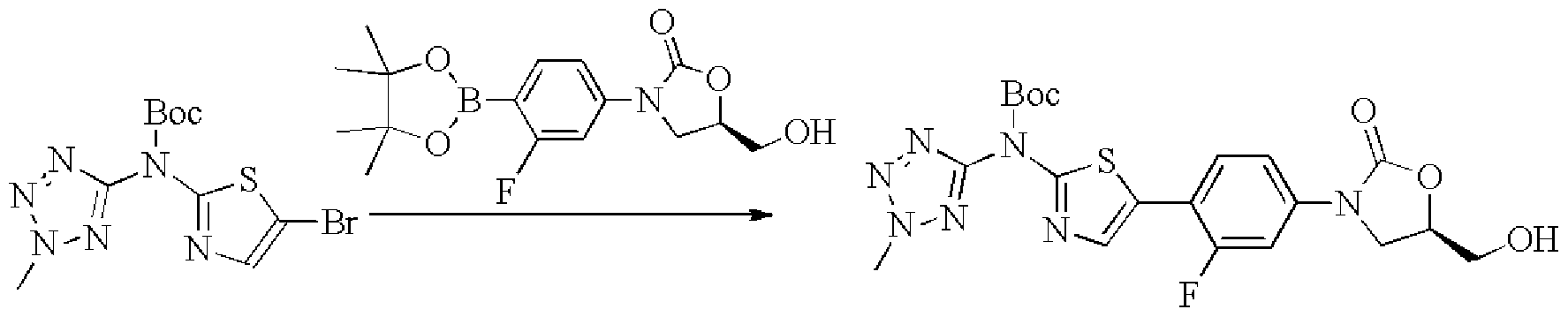
- Example 1 The operation was the same as in Example 1 (1) except that 2-methyltetrazolyl-5-amine was substituted for 2-methyl-2H-tetrazole-5-amine in a yield of 33%.
- Example 1 (2) The same procedure as in Example 1 (2) was followed, except that 5-bromo(1-methyltetrazol-5-yl)pyridin-2-amine was substituted for 5-bromo-W-(2-methyl-2H-tetrazole -5-yl)pyridin-2-amine, yield 11%.
- Methyl dihydrogen phosphate 140 mg 0.3 mmol was dissolved in 15 mL of methanol, sodium methoxide (48 mg 0.9 mmol) was added, and the reaction was carried out for 12 h at room temperature.
- EP fl- -lH- gg please-5-m, oxygen.
- -3- phenyl, -5- (hydroxypyrene yoxane-2 eyebrows (servant) country
- Example 5 (1) The same procedure as in Example 5 (1) was carried out except that 5-bromo(2-methyl-2H-tetrazol-5-yl)pyridin-2-amine was substituted for 5-bromo-W-(l-methyl-1H- Tetrazolium-5-yl)pyridin-2-amine, yield 31%.
- Example 1 The operation was the same as in Example 1 (1) except that 2-methylpyrazol-3-amine was substituted for 2-methyl-2H-tetrazole-5-amine, and the yield was 9%.
- Example 1 The same procedure as in Example 1 (2) was carried out, but 5-bromo-W-(2-methyl-2H-1,2,3-triazol-4-yl) B was substituted for 5-bromo-2-amine. -W-(2-methyl-2H-tetrazol-5-yl)pyridin-2-amine, yield 11%.
- the compound 13 can be produced.
- Example 2 The same procedure as in Example 1 (2) was carried out, but 5-bromo-W-(2-A was replaced by 5-bromothiazol-2-yl(2-methyl-2H-tetrazol-5-yl)carbonic acid tert-butyl ester. Base-2H-tetrazol-5-yl)pyridin-2-amine, 16% yield.
Abstract
Description
































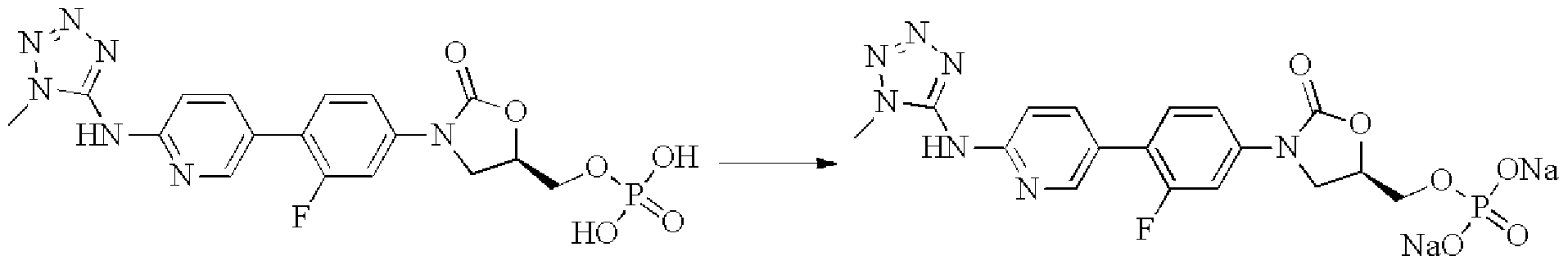



























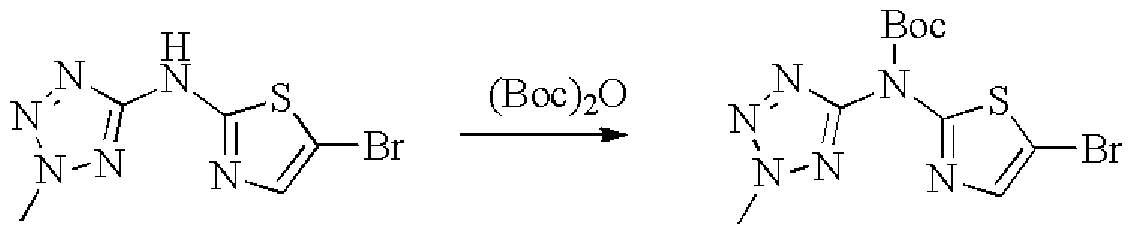




Claims








Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/347,583 US9359344B2 (en) | 2011-09-29 | 2012-09-28 | Biaryl heterocycle substituted oxazolidinone antibacterial agents |
| JP2014532234A JP5796134B2 (ja) | 2011-09-29 | 2012-09-28 | ビアリールヘテロ環で置換されたオキサゾリドン抗菌薬 |
| KR1020147008359A KR101629115B1 (ko) | 2011-09-29 | 2012-09-28 | 바이아릴 헤테로고리로 치환된 옥사졸리디논 항균제 |
| EP12834694.7A EP2762479A4 (en) | 2011-09-29 | 2012-09-28 | OXAZOLIDINONE ANTIBACTERIAL MEDICINE BASED ON BIARYLIC HETEROCYCLE SUBSTITUTED |
| UAA201402653A UA112876C2 (uk) | 2011-09-29 | 2012-09-28 | Біарилгетероциклзаміщені оксазолідинонові антибактеріальні засоби |
| HK14112754.5A HK1199250A1 (zh) | 2011-09-29 | 2014-12-19 | 聯芳基雜環取代的噁唑烷酮抗菌藥 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201110290839 | 2011-09-29 | ||
| CN201110290839.X | 2011-09-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013044845A1 true WO2013044845A1 (zh) | 2013-04-04 |
Family
ID=47994272
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2012/082318 WO2013044845A1 (zh) | 2011-09-29 | 2012-09-28 | 联芳基杂环取代的噁唑烷酮抗菌药 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US9359344B2 (zh) |
| EP (1) | EP2762479A4 (zh) |
| JP (1) | JP5796134B2 (zh) |
| KR (1) | KR101629115B1 (zh) |
| CN (1) | CN103030635B (zh) |
| HK (1) | HK1199250A1 (zh) |
| UA (1) | UA112876C2 (zh) |
| WO (1) | WO2013044845A1 (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103975931A (zh) * | 2014-06-04 | 2014-08-13 | 吴文君 | 一类二氢噻唑类化合物作为农用杀菌剂的用途 |
| WO2016041508A1 (zh) * | 2014-09-17 | 2016-03-24 | 博瑞生物医药技术(苏州)有限公司 | 一种噁唑烷酮类化合物及其中间体的制备方法 |
| CN105524008A (zh) * | 2014-10-21 | 2016-04-27 | 山东轩竹医药科技有限公司 | 噁唑烷酮抗菌药中间体的制备方法 |
| US11555033B2 (en) | 2020-06-18 | 2023-01-17 | Akagera Medicines, Inc. | Oxazolidinone compounds, liposome compositions comprising oxazolidinone compounds and method of use thereof |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111560014A (zh) * | 2020-05-13 | 2020-08-21 | 河南科技大学第一附属医院 | 一种杀菌消毒所用噁唑链接三唑类药物分子及其制备方法和应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005019211A2 (en) * | 2003-06-03 | 2005-03-03 | Rib-X Pharmaceuticals, Inc. | Biaryl heterocyclic compounds and methods of making and using the same |
| CN101560209A (zh) * | 2008-04-15 | 2009-10-21 | 沈阳亿利奥医药科技有限公司 | 含有嘧啶的噁唑烷酮类化合物及其制备方法 |
| CN102153547A (zh) * | 2010-02-11 | 2011-08-17 | 山东轩竹医药科技有限公司 | 含有并环的噁唑烷酮抗菌素 |
| CN102190656A (zh) * | 2010-03-16 | 2011-09-21 | 山东轩竹医药科技有限公司 | 含有五元杂环的噁唑烷酮抗菌素 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0873455A (ja) | 1994-03-15 | 1996-03-19 | Upjohn Co:The | オキサゾリジノン誘導体及びこれを有効成分とする医薬組成物 |
| CN1211384C (zh) * | 1997-07-11 | 2005-07-20 | 法玛西雅厄普约翰美国公司 | 噻二唑基和噁二唑基苯基噁唑烷酮抗菌剂 |
| BR0111280A (pt) * | 2000-06-05 | 2003-06-10 | Dong A Pharm Co Ltd | Novos derivados de oxazolidinona e um processo para a preparação dos mesmos |
| PL363960A1 (en) | 2000-07-17 | 2004-11-29 | Ranbaxy Laboratories Limited | Oxazolidinone derivatives as antimicrobials |
| WO2004014392A1 (en) | 2002-07-29 | 2004-02-19 | Ranbaxy Laboratories Limited | Oxazolidinone derivatives as antimicrobials |
| CN100457742C (zh) * | 2003-06-03 | 2009-02-04 | 瑞伯-X医药品有限公司 | 联芳基杂环化合物的制备和用途 |
| US7687627B2 (en) * | 2003-09-08 | 2010-03-30 | Wockhardt Limited | Substituted piperidino phenyloxazolidinones having antimicrobial activity with improved in vivo efficacy |
| KR100854211B1 (ko) | 2003-12-18 | 2008-08-26 | 동아제약주식회사 | 신규한 옥사졸리디논 유도체, 그의 제조방법 및 이를유효성분으로 하는 항생제용 약학 조성물 |
| WO2005082899A1 (en) * | 2004-01-28 | 2005-09-09 | Ranbaxy Laboratories Limited | Oxazolidinone derivatives as antimicrobials |
| BRPI0909028A2 (pt) | 2008-03-26 | 2015-09-22 | Global Alliance For Tb Drug Dev | nitroimidazóis bicíclicos covalentemente ligados a fenil oxazolidinonas substituídas, composições farmacêuticas e uso dos mesmos |
| WO2009157423A1 (ja) * | 2008-06-24 | 2009-12-30 | 財団法人乙卯研究所 | 縮合環を有するオキサゾリジノン誘導体 |
| CN102276595A (zh) * | 2010-04-16 | 2011-12-14 | 山东轩竹医药科技有限公司 | 含有五元杂环的噁唑烷酮抗菌素 |
-
2012
- 2012-09-28 UA UAA201402653A patent/UA112876C2/uk unknown
- 2012-09-28 KR KR1020147008359A patent/KR101629115B1/ko not_active IP Right Cessation
- 2012-09-28 US US14/347,583 patent/US9359344B2/en not_active Expired - Fee Related
- 2012-09-28 JP JP2014532234A patent/JP5796134B2/ja not_active Expired - Fee Related
- 2012-09-28 EP EP12834694.7A patent/EP2762479A4/en not_active Withdrawn
- 2012-09-28 WO PCT/CN2012/082318 patent/WO2013044845A1/zh active Application Filing
- 2012-09-29 CN CN201210394995.5A patent/CN103030635B/zh active Active
-
2014
- 2014-12-19 HK HK14112754.5A patent/HK1199250A1/zh unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005019211A2 (en) * | 2003-06-03 | 2005-03-03 | Rib-X Pharmaceuticals, Inc. | Biaryl heterocyclic compounds and methods of making and using the same |
| CN101560209A (zh) * | 2008-04-15 | 2009-10-21 | 沈阳亿利奥医药科技有限公司 | 含有嘧啶的噁唑烷酮类化合物及其制备方法 |
| CN102153547A (zh) * | 2010-02-11 | 2011-08-17 | 山东轩竹医药科技有限公司 | 含有并环的噁唑烷酮抗菌素 |
| CN102190656A (zh) * | 2010-03-16 | 2011-09-21 | 山东轩竹医药科技有限公司 | 含有五元杂环的噁唑烷酮抗菌素 |
Non-Patent Citations (2)
| Title |
|---|
| "Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically", 2006, NATIONAL COMMITTEE FOR CLINICAL LABORATORY STANDARDS, pages: M7 - A7 |
| See also references of EP2762479A4 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103975931A (zh) * | 2014-06-04 | 2014-08-13 | 吴文君 | 一类二氢噻唑类化合物作为农用杀菌剂的用途 |
| CN103975931B (zh) * | 2014-06-04 | 2015-08-19 | 吴文君 | 一类二氢噻唑类化合物作为农用杀菌剂的用途 |
| WO2016041508A1 (zh) * | 2014-09-17 | 2016-03-24 | 博瑞生物医药技术(苏州)有限公司 | 一种噁唑烷酮类化合物及其中间体的制备方法 |
| CN105524008A (zh) * | 2014-10-21 | 2016-04-27 | 山东轩竹医药科技有限公司 | 噁唑烷酮抗菌药中间体的制备方法 |
| CN105524008B (zh) * | 2014-10-21 | 2018-09-07 | 山东轩竹医药科技有限公司 | 噁唑烷酮抗菌药中间体的制备方法 |
| US11555033B2 (en) | 2020-06-18 | 2023-01-17 | Akagera Medicines, Inc. | Oxazolidinone compounds, liposome compositions comprising oxazolidinone compounds and method of use thereof |
| US11566023B2 (en) | 2020-06-18 | 2023-01-31 | Akagera Medicines, Inc. | Oxazolidinone compounds, liposome compositions comprising oxazolidinone compounds and method of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| KR101629115B1 (ko) | 2016-06-09 |
| EP2762479A1 (en) | 2014-08-06 |
| US9359344B2 (en) | 2016-06-07 |
| JP5796134B2 (ja) | 2015-10-21 |
| UA112876C2 (uk) | 2016-11-10 |
| KR20140054396A (ko) | 2014-05-08 |
| JP2014528946A (ja) | 2014-10-30 |
| US20140235584A1 (en) | 2014-08-21 |
| CN103030635B (zh) | 2015-11-25 |
| EP2762479A4 (en) | 2015-04-22 |
| HK1199250A1 (zh) | 2015-06-26 |
| CN103030635A (zh) | 2013-04-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5592388B2 (ja) | 疼痛治療用のp2x3受容体アンタゴニスト技術分野 | |
| JP5833764B2 (ja) | 縮合環を有するオキサゾリドン(oxazolidone)類の抗菌物質 | |
| AU2018257203B2 (en) | Novel tetrahydronaphthyl urea derivatives | |
| WO2019101086A1 (zh) | 卤代烯丙基胺类ssao/vap-1抑制剂及其应用 | |
| ES2651484T3 (es) | Compuesto heterocíclico que contiene nitrógeno | |
| WO2017023941A1 (en) | Benzazole compounds and methods for making and using the compounds | |
| TWI785474B (zh) | 用作選擇性Aurora A抑制劑的新型雜環化合物 | |
| TW200906825A (en) | Inhibitors of protein kinases | |
| KR20210150491A (ko) | 포스파티딜이노시톨 3-키나제 저해제 | |
| JP2022532719A (ja) | Acss2阻害剤およびその使用方法 | |
| WO2013044845A1 (zh) | 联芳基杂环取代的噁唑烷酮抗菌药 | |
| TW202003472A (zh) | 鈣蛋白酶(calpain)調節劑及其醫療用途 | |
| CA2933683A1 (en) | Wnt pathway modulators | |
| WO2022089389A1 (zh) | 杂环化合物及其制备方法、药物组合物和应用 | |
| US20240018102A1 (en) | Compounds and compositions for treating conditions associated with lpa receptor activity | |
| WO2020132636A1 (en) | Analogues of pentamidine and uses therefor | |
| TWI640524B (zh) | Aminopyran ring derivatives and compositions and uses thereof | |
| TW201439088A (zh) | 多形體形式 | |
| KR102636651B1 (ko) | 티아졸로피리딘 또는 이의 약학적으로 허용 가능한 염 및 이의 용도 | |
| US11884627B2 (en) | Compounds and compositions for treating conditions associated with LPA receptor activity | |
| CN117616022A (zh) | 晶形 | |
| TW202408509A (zh) | 雜芳基衍生物的藥物組合物及其在醫藥上的應用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12834694 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14347583 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012834694 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20147008359 Country of ref document: KR Kind code of ref document: A Ref document number: 2014532234 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201402653 Country of ref document: UA |