WO2012002455A1 - ポリアクリル酸系吸水性樹脂及びその製造方法 - Google Patents
ポリアクリル酸系吸水性樹脂及びその製造方法 Download PDFInfo
- Publication number
- WO2012002455A1 WO2012002455A1 PCT/JP2011/064951 JP2011064951W WO2012002455A1 WO 2012002455 A1 WO2012002455 A1 WO 2012002455A1 JP 2011064951 W JP2011064951 W JP 2011064951W WO 2012002455 A1 WO2012002455 A1 WO 2012002455A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- water
- absorbent resin
- polymerization
- monomer
- aqueous solution
- Prior art date
Links
Images








Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
- C08J9/12—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a physical blowing agent
- C08J9/122—Hydrogen, oxygen, CO2, nitrogen or noble gases
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/22—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons containing macromolecular materials
- A61L15/24—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/42—Use of materials characterised by their function or physical properties
- A61L15/60—Liquid-swellable gel-forming materials, e.g. super-absorbents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/01—Processes of polymerisation characterised by special features of the polymerisation apparatus used
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F20/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride, ester, amide, imide or nitrile thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/04—Acids; Metal salts or ammonium salts thereof
- C08F220/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
- C08J3/245—Differential crosslinking of one polymer with one crosslinking type, e.g. surface crosslinking
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/16—Making expandable particles
- C08J9/20—Making expandable particles by suspension polymerisation in the presence of the blowing agent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/36—After-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2203/00—Foams characterized by the expanding agent
- C08J2203/06—CO2, N2 or noble gases
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2205/00—Foams characterised by their properties
- C08J2205/04—Foams characterised by their properties characterised by the foam pores
- C08J2205/05—Open cells, i.e. more than 50% of the pores are open
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/02—Homopolymers or copolymers of acids; Metal or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2335/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical, and containing at least one other carboxyl radical in the molecule, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Derivatives of such polymers
- C08J2335/02—Characterised by the use of homopolymers or copolymers of esters
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/29—Coated or structually defined flake, particle, cell, strand, strand portion, rod, filament, macroscopic fiber or mass thereof
- Y10T428/2982—Particulate matter [e.g., sphere, flake, etc.]
Definitions
- the present invention relates to a polyacrylic acid-based water absorbent resin and a method for producing the same. More specifically, the present invention relates to use of the water-absorbent resin for sanitary goods such as paper diapers and sanitary napkins, and has a foam-like polyacrylic acid-based water-absorbent resin with improved water absorption performance (particularly water absorption speed) and its It relates to a manufacturing method.
- Water-absorbing resins are widely used in various applications such as disposable diapers, sanitary napkins, sanitary products such as incontinence products for adults, and water retention agents for soil, and are produced and consumed in large quantities.
- the water-absorbing resin include a crosslinked polyacrylic acid partially neutralized product, a hydrolyzate of starch-acrylic acid graft polymer, a saponified product of vinyl acetate-acrylic acid ester copolymer, an acrylonitrile copolymer, or an acrylamide.
- a hydrolyzate of a copolymer or a crosslinked product thereof, and a crosslinked product of a cationic monomer are known.
- These water-absorbing resins are, for example, a method of polymerizing an aqueous solution containing a hydrophilic monomer while crushing a polymer gel by stirring (Patent Document 1), or a method of standing polymerization of an aqueous solution containing a monomer ( Patent Documents 2 to 6), manufactured by reverse phase suspension polymerization, dropping polymerization and the like.
- Patent Documents 7 to 33 a method in which a large amount of bubbles are dispersed in an aqueous monomer solution, and a surfactant or, in some cases, a large amount of a bubble stabilizer is used so that the bubbles do not disappear by the start of polymerization (Patent Documents 8, 20, and 21), or an aqueous monomer solution.
- Patent Documents 7 and 9 a method in which a large amount of bubbles are dispersed in an aqueous monomer solution, and a surfactant or, in some cases, a large amount of a bubble stabilizer is used so that the bubbles do not disappear by the start of polymerization
- Patent Documents 8, 20, and 21 have been proposed (Patent Documents 7 and 9) and methods for producing foamed water-absorbing resins (Patent Documents 8, 20, 21, 30 to 33).
- foam polymerization is carried out for the purpose of improving the water absorption rate by increasing the surface area.
- a technique using carbonate as a foaming agent used for the monomer Patent Documents 10 to 17.
- Patent Literatures 18 and 19 Technology using an organic solvent
- Patent Literature 22 technology using an inert gas
- Patent Literature 23 and 24 technology using an azo compound
- Patent Document 25 technology using an insoluble inorganic powder
- Patent Document 26 a technique of foaming and crosslinking after polymerization
- Patent Document 27 a technique of using water-insoluble particles for polymerization
- Patent Document 28 a technique of polymerizing in a state where a precipitate of sodium acrylate is dispersed
- Patent Documents 8, 20, 21, 30 to 32 it is disclosed that “avoidance of boiling during polymerization” is recommended (Patent Documents 8, 20, 21, 30 to 32), or “polymerization at 65 ° C. or lower”. (Patent Document 33).
- a technique for controlling the maximum temperature to be low for the purpose of reducing soluble components in polymerization other than foam polymerization is disclosed.
- a technique for setting the maximum temperature to 95 ° C. or less Patent Document 34
- a technique for polymerizing at a polymerization temperature of 20 to 70 ° C. Patent Document 35
- a technique for polymerizing at a polymerization temperature of 20 to 95 ° C. Patent Document 36
- the method of dispersing a large amount of bubbles using a large amount of surfactant is polymerized to form at least open cells, but requires a relatively long time to disperse the bubbles.
- the amount of surfactant (or surface) tension is lowered by a large amount of surfactant, which obstructs the liquid absorption characteristics of sanitary products and increases the amount of return (Re-Wet). is there.
- the method of keeping the aqueous monomer solution at a low temperature requires a long time for polymerization, so that the productivity is inferior, and there is almost no communication of bubbles (open cells) in the resulting porous polymer, and the bulk density.
- the water-absorbing resin is generally in the form of powder (particulate), when actually used in paper diapers, sanitary napkins, tampons, etc., it is mixed with fiber materials and adhesives, if necessary, and then in powder form (particulate) It is necessary to process the water-absorbent resin into a sheet shape, a cylindrical shape, and the like. Such processing not only increases the cost, but also decreases the water absorption rate and breaks the powder (generates fine powder).
- Patent Document 29 discloses a coloring prevention method using ammonium acrylate as a monomer and a coloring prevention method in which a compound containing a phosphorus atom or a sulfur-based reducing agent is added a plurality of times.
- the hydrogel crosslinked polymer obtained by polymerizing an aqueous monomer solution requires large equipment and thermal energy for drying, and further requires a longer drying time, while the water-absorbing resin is deteriorated due to prolonged drying.
- the conventional water-absorbent resin is mainly in powder form, but when used as absorbent articles (final consumer goods) such as paper diapers, a process of fixing or incorporating the water-absorbent resin powder into the absorbent article is required. There are also problems such as the use of an expensive adhesive for immobilization and the falling off and movement of powder.
- Patent Documents 37 and 38 sheet-like water-absorbing resin moldings and composites have also been proposed (Patent Documents 37 and 38), but complicated processes and expensive auxiliary materials are required for molding.
- Patent Documents 37 and 38 since the specific surface area of a molded product such as a sheet is significantly lower than that of powder, the water absorption speed and air permeability are insufficient, which causes leakage of paper diapers and stuffiness.
- the problem to be solved by the present invention is to improve the water absorption rate of the water-absorbent resin, particularly to improve the water absorption rate by foaming or making it porous. More preferably, the formation of communication holes (open cells) in the foamed polymer (foamed water absorbent resin) is promoted by a simpler method, and the drying step of the water absorbent resin is simplified. is there.
- the object is to provide a hygroscopic article in the form of a sheet (molded product) or a powder, and a water-absorbing resin having a high water absorption speed and a manufacturing method thereof with high efficiency.
- the method for producing a water-absorbent resin of the present invention comprises a step (A) of obtaining an aqueous acrylic acid monomer solution in which bubbles are dispersed, and polymerizing the aqueous monomer solution.
- a method for producing a polyacrylic acid-based water-absorbing resin comprising: a step (B) for obtaining a foamed polymer; and a step (C) for drying the foamed polymer by heating.
- the bubbles are contained so that the volume expansion ratio defined by; exceeds 1.1 times, and the following formula (2):
- a method for producing a polyacrylic acid-based water-absorbing resin comprising boiling-polymerizing an aqueous monomer solution having a monomer concentration of 40% by weight or more defined by the above at a temperature of 100 ° C. or more.
- the present invention provides a polyacrylic acid water-absorbing resin having an open cell ratio of 5% or more.
- the water absorption speed of the water absorbent resin is improved by foaming.
- an open-cell water-absorbing resin can also be obtained.
- the water absorbent resin of the present invention since the open cell bubble ratio is high, the water absorption speed and air permeability of the water absorbent resin are improved.
- FIG. 1 is a flow diagram showing an example of a foam regulating step of circulating an aqueous monomer solution containing an inert gas in a circulation tank.
- FIG. 2 is a flow diagram showing another example of a foam regulating step of circulating an aqueous monomer solution containing an inert gas in a circulation tank.
- FIG. 3 is a schematic view showing a mixing zone having irregularities and fillers.
- 4 is an SEM (scanning electron microscope) photograph image of the dried water absorbent resin (1 ′) in Example 1.
- FIG. FIG. 5 is a SEM (scanning electron microscope) photographic image of the dried water absorbent resin (5 ′) in Example 5.
- FIG. 6 shows a typical shape (sheet shape) of the water-absorbent resin molding.
- FIG. 7 is a conceptual diagram of open bubbles and closed cells.
- FIG. 8 is a schematic view showing an example of a method for determining the true specific gravity of the water absorbent resin.
- the present invention is not limited to the following embodiments, and various modifications are possible within the scope shown in the claims, and various technical means disclosed in different embodiments are appropriately combined. The obtained embodiment is also included in the technical scope of the present invention.
- Water absorbent resin The “water-absorbing resin” in the present invention means a water-swellable, water-insoluble polymer gelling agent, and the “water-absorbing resin powder” means a water-absorbing resin that has been pulverized into a powder form.
- Water swellability means that the CRC (absorption capacity under no pressure) specified by ERT441.2-02 is 5 [g / g] or more, preferably 10 to 100 [g / g]. More preferably, it is 20 to 80 [g / g], and “water-insoluble” means that Ext (water-soluble content) defined by ERT470.2-02 is 0 to 50% by weight. It is preferably 0 to 30% by weight, more preferably 0 to 20% by weight, particularly preferably 0 to 10% by weight.
- the water-absorbent resin can be appropriately designed according to its use and is not particularly limited, but may be a hydrophilic cross-linked polymer obtained by cross-linking an unsaturated monomer having a carboxyl group. preferable. Moreover, the whole quantity (100 weight%) is not limited to the form which is a polymer, In the range which maintains the said performance, an additive etc. may be included as other components other than a water absorbing resin. That is, even a water absorbent resin composition containing a water absorbent resin powder and an additive is generically referred to as a water absorbent resin in the present invention.
- the content of the polyacrylic acid (salt) water-absorbing resin is preferably 70 to 99.9% by weight, more preferably 80 to 99.7% by weight, and still more preferably 90 to 99%. .5% by weight.
- water is preferable from the viewpoint of the water absorption speed and the impact resistance of the powder (particles), and an additive is included if necessary.
- polyacrylic acid water-absorbing resin “foam-like water-absorbing resin”, “water-absorbing resin molding”
- the “polyacrylic acid-based water-absorbent resin” in the present invention optionally contains a graft component, and has a water-absorbing component mainly composed of acrylic acid and / or a salt thereof (hereinafter referred to as acrylic acid (salt)) as a repeating unit. It means a functional resin.
- the salt as a polymer essentially contains a water-soluble salt, preferably a monovalent salt, more preferably an acrylic metal salt or an ammonium salt, particularly an alkali metal salt, and further a sodium salt.
- the form of the water-absorbing resin is foam (foamed) or porous and is referred to as “foam-shaped water-absorbing resin”, and the shape (sheet, block, powder, etc.) does not matter.
- a water-absorbent resin dried product is obtained by heating and drying a hydrogel.
- the water-absorbing resin having a certain shape such as a sheet shape, a block shape, or a cylindrical shape with respect to the powdered (particulate) water-absorbing resin may be hereinafter referred to as a water-absorbing resin molded product. Includes a sheet-like water-absorbing resin.
- open bubbles and “closed bubbles”
- “open bubbles” refers to bubbles that are not completely surrounded by the wall of the water absorbent resin (bubbles that are mutually connected to the outside of the water absorbent resin
- bubbles refers to bubbles that are surrounded by the walls of the water-absorbent resin, and is a conceptual diagram of open bubbles and closed cells.
- EDANA European Disposables and Nonwovens Associations
- ERT is an abbreviation for a method for measuring water-absorbent resin (EDANA Recommended Test Methods), which is a European standard (almost world standard). is there.
- the physical properties of the water-absorbent resin powder are measured according to the ERT original (known document: revised in 2002).
- CRC is an abbreviation for Centrifugation Retention Capacity (centrifuge retention capacity) and means water absorption capacity without pressure (hereinafter also referred to as “water absorption capacity”). Specifically, with respect to 0.2 g of the water-absorbent resin powder in the non-woven bag, the water absorption ratio (unit; [G / g]).
- AAP is an abbreviation for Absorption Against Pressure, which means water absorption capacity under pressure. Specifically, 0.9 g of the water-absorbent resin was swollen under a load of 2.06 kPa (0.3 psi, 21 [g / cm 2 ]) for 1 hour against a large excess of 0.9 wt% sodium chloride aqueous solution. It is a subsequent water absorption ratio (unit: [g / g]). In ERT442.2-02, “Absorption Under Pressure” is described, but the contents are substantially the same. In some cases, only the load condition is changed to 4.83 kPa (0.7 psi, 50 [g / cm 2 ]).
- Ext is an abbreviation for Extractables and means a water-soluble component (water-soluble component amount). Specifically, after 1 g of water-absorbent resin powder was stirred at 500 rpm for 16 hours with respect to 200 g of 0.9 wt% sodium chloride aqueous solution, the amount of dissolved polymer was measured by pH titration (unit: wt%). is there.
- PSD is an abbreviation for Particle Size Distribution, and means a particle size distribution measured by sieve classification.
- the weight average particle size (D50) and the particle size distribution width are measured by the same method as “(1) Average Particle Diameter and Distribution of Particle Diameter” described in International Publication No. 2004/69915 pamphlet.
- liquid permeability The flow of liquid flowing between particles of the swollen water-absorbent resin powder under load or no load is referred to as “liquid permeability”.
- Typical measurement methods for this “liquid permeability” include SFC (Saline Flow Conductivity / Saline Flow Inductivity) and GBP (Gel Bed Permeability / Gel Bed Permeability).
- SFC Seline Flow Inducibility
- GBP gel bed permeability
- X to Y indicating a range means “X or more and Y or less”.
- t (ton) as a unit of weight means “Metric ton”, and “ppm” means “weight ppm” unless otherwise noted.
- mass and weight weight, “mass%” and “wt%”, “mass part” and “part by weight” are synonymous, and there is no particular notice regarding measurement of physical properties and the like. In this case, measurement is performed at room temperature (20 to 25 ° C.) / Relative humidity 40 to 50%. Further, “ ⁇ acid (salt)” means “ ⁇ acid and / or salt thereof”, and “(meth) acryl” means “acryl and / or methacryl”.
- polyacrylic acid-based water absorbent resin according to the present invention can be produced, for example, by the following method. Such a production method can be preferably applied to continuous production.
- the gas for generating bubbles used in the present invention is a solid foaming agent (eg, carbonate, azo) which is a gas compound at room temperature before use and generates gas during polymerization.
- a solid foaming agent eg, carbonate, azo
- the acrylic monomer aqueous solution in which bubbles are dispersed is a gas, preferably an inert gas, and the monomer aqueous solution or its raw material (individual monomer or solvent) directly. Mix.
- Anionic unsaturated monomers such as sulfonic acid, 2- (meth) acryloylethanesulfonic acid, 2- (meth) acryloylpropanesulfonic acid, 2-hydroxyethyl (meth) acryloyl phosphate and salts thereof; Saturated monomers; phenolic hydroxyl group-containing unsaturated monomers; amide group-containing unsaturated monomers such as (meth) acrylamide, N-ethyl (meth) acrylamide, N, N-dimethyl (meth) acrylamide; N-dimethylaminoethyl (
- Monomers used in combination include water-soluble or hydrophobic unsaturated monomers such as 2-hydroxyethyl (meth) acrylate, methoxypolyethylene glycol (meth) acrylate, polyethylene glycol (meth) acrylate, isobutylene and lauryl (meth) acrylate. What uses a monomer etc. as a copolymerization component is also contained.
- acrylic acid and / or a salt thereof for example, sodium salt, lithium salt
- Potassium salts, ammonium salts, amines and the like are preferred, and sodium salts are more preferred from the viewpoint of cost.
- polyacrylic acid-based water-absorbing resins using acrylic monomers are preferred.
- the amount of acrylic acid and / or salt thereof used as the polyacrylic acid-based water-absorbing resin is preferably 50 mol% or more, more preferably 70 mol% or more, based on the total monomer components (excluding the internal cross-linking agent described later). Preferably it is 80 mol% or more, More preferably, it is 90 mol% or more, Most preferably, it is 95 mol% or more (an upper limit is 100 mol%), Most preferably, it is substantially 100 mol%.
- polyacrylic acid is the concept containing polyacrylate (especially monovalent salt).
- a monomer containing 200 ppm or less of methoxyphenol in terms of acrylic acid is preferably used.
- the main component of this monomer may be acrylic acid, and may be acrylic acid and acrylate.
- Specific examples of the methoxyphenols include o, m, p-methoxyphenol, and methoxyphenols having one or more substituents such as a methyl group, a t-butyl group, and a hydroxyl group.
- p-methoxyphenol is used in the present invention.
- the content of methoxyphenols is preferably 200 ppm by weight or less, more preferably 10 to 200 ppm, further preferably 10 to 120 ppm, particularly preferably 10 to 90 ppm, and most preferably 20 to 90 ppm in terms of acrylic acid. is there. If content of methoxyphenol is 200 ppm or less, coloring (yellowing / yellowing) of the obtained water absorbing resin can be suppressed. In addition, when the content of methoxyphenols is less than 10 ppm, that is, when methoxyphenols that are polymerization inhibitors are removed by purification such as distillation, there is only a risk that polymerization will occur before intentionally starting the polymerization. In other words, the weather resistance of the water-absorbent resin obtained using acrylic acid (salt) as the main raw material may deteriorate.
- the main component of the monomer is acrylic acid and / or acrylate, but the molecular weight is different between acrylic acid and acrylate.
- the value in terms of acrylic acid is defined in the present invention.
- the value in terms of acrylic acid is the content ratio (weight ratio) of the above-mentioned trace component weight to the weight of acrylic acid when all acrylates are converted to be equimolar unneutralized acrylic acid. That is, for example, neutralized sodium acrylate (molecular weight 94) is converted to acrylic acid (molecular weight 72) by weight, and the content of methoxyphenols by weight after conversion to acrylic acid (94 converted to 72) (weight) Ratio) and the like.
- the conversion value for acrylic acid is all of the partially neutralized or completely neutralized polyacrylate. It can be calculated in terms of equimolar unneutralized polyacrylic acid.
- the partial neutralization means that the neutralization rate is more than 0 mol% and less than 100 mol%.
- the said complete neutralization means that the neutralization rate is 100 mol%.
- the said non-neutralized means that the neutralization rate is 0 mol%.
- the method for producing a water-absorbent resin of the present invention is preferably a method for producing a water-absorbent resin through polymerization of an aqueous monomer solution containing 200 ppm or less of methoxyphenols (particularly p-methoxyphenol). . Further, through the polymerization step (concentration, initiator, temperature) and drying step (temperature, time, solid content, air volume, etc.), a predetermined amount of methoxyphenol is consumed, and methoxyphenols (especially p-methoxy) are consumed.
- a water-absorbing resin containing 60 ppm or less of phenol), more preferably 5 to 60 ppm, particularly a water-absorbing resin containing methoxyphenols uniformly within the polymer can be obtained.
- the production method of the present invention by using a monomer having a methoxyphenol (particularly p-methoxyphenol) content of 200 ppm or less, the methoxyphenols of the water-absorbent resin obtained by the polymerization step and the drying step (in particular, the production method may be such that the content of p-methoxyphenol is 60 ppm or less.
- the methoxyphenol at the time of polymerization is based on acrylic acid (molecular weight 72)
- the acrylic acid salt obtained by neutralization if necessary has an increased molecular weight (for example, a molecular weight of 88.5 for 75 mol% neutralized sodium salt).
- the content of methoxyphenols is reduced.
- the content of methoxyphenols in the acrylate before polymerization is 200 ppm or less, and the methoxyphenols in the obtained polyacrylate are 60 ppm or less. Is preferred.
- control method of methoxyphenols in the water-absorbent resin is not limited to the above example, and other methods can be exemplified as follows, and these may be used in combination.
- Production Method 1 A method of polymerizing with the absence of methoxyphenol or a monomer containing less than 10 ppm, and further adding a predetermined amount of methoxyphenol after drying.
- Method 2 A method of removing a predetermined amount of methoxyphenol by washing before drying after polymerization into a water-absorbent resin with a monomer containing excess methoxyphenol.
- water or a hydroalcoholic liquid mixture can be used for washing.
- the monomer used in the present invention may use a polymerization inhibitor other than methoxyphenols in the production process, or the polymerization inhibitor may be used in combination with methoxyphenols.
- polymerization inhibitors other than methoxyphenols for example, phenothiazine, hydroquinone, copper salt, manganese acetate, methylene blue and the like are effective.
- these polymerization inhibitors unlike methoxyphenols, inhibit polymerization, so the smaller the final, the better.
- the concentration in the monomer is preferably 0.01 to 10 ppm. .
- the above components can be quantified by liquid chromatography or gas chromatography.
- the value measured by the method of the following example is adopted as the content of methoxyphenols.
- the monomer concentration at the time of polymerization is 40% by weight or more, more preferably 45% by weight or more, and further preferably 50% by weight or more and 53% by weight or more.
- the upper limit is not particularly limited, but is preferably 80% by weight or less, and more preferably 75% by weight or less.
- the monomer concentration is defined by the following formula (2).
- (monomer weight) represents the total weight of monomers present in the monomer aqueous solution.
- (monomer weight) represents the total weight of monomers present in the monomer aqueous solution.
- total weight of acrylic acid and sodium acrylate present in the monomer aqueous solution is (weight of monomer) in the above formula (2).
- acrylic acid and sodium acrylate are used in preparing the monomer aqueous solution, the total amount of acrylic acid and sodium acrylate is (weight of monomer) in the above formula (2).
- the (solvent weight) in the above formula (2) represents the amount of the solvent when supplied in a solution state such as an aqueous solution among the raw materials supplied to the polymerization system. Therefore, the below-mentioned surfactant, hydrophilic polymer, etc. used if necessary are not taken into account in the above-mentioned definition (calculation) of the monomer concentration.
- the monomer concentration during polymerization is 40% by weight or higher, preferably 42% by weight or higher, more preferably 43% by weight or higher, still more preferably 45% by weight or higher, particularly preferably 50% by weight or higher.
- the present invention is particularly advantageous.
- the monomer solvent is water, and in addition to water, a small amount (for example, in the range of more than 0% by weight to 30% by weight, or more in the range of more than 0% by weight to 10% by weight).
- An organic solvent may be used in combination.
- the organic solvent include alcohols such as methyl alcohol, ethyl alcohol, n-propyl alcohol, isopropyl alcohol, n-butyl alcohol, isobutyl alcohol, and t-butyl alcohol.
- polymerization with a monomer slurry may reduce physical properties (water absorption capacity, soluble content, residual monomer, etc.). is there. Therefore, in the present invention, when the monomer is an acid group-containing monomer, the neutralization rate is such that the neutralized salt does not precipitate in the aqueous monomer solution. That is, in the present invention, an acrylic acid monomer aqueous solution is polymerized, not an aqueous dispersion of an acrylic acid monomer.
- Precipitation of the neutralized salt depends on the solubility of the neutralized salt in water, monomer concentration, neutralization rate, temperature, pressure, neutralizing base, and dispersant (surfactant, other monomer used as necessary). Therefore, the solubility of the same monomer increases as the temperature of the aqueous monomer solution increases, depending on the conditions.
- the polymerization is performed in a relatively short time while the bubbles are dispersed in the monomer aqueous solution to increase the surface area of the liquid and the water is evaporated. It is not preferred that sodium acid is precipitated because many of its salts remain until after polymerization without dissolving. Also, if the salt that precipitates during polymerization is dissolved, the evaporation time of water must be suppressed, and the polymerization time must be relatively long because it is necessary to take the dissolution time of the salt. The connectivity (open cell property) of the bubble which is a preferred embodiment is significantly reduced. In the neutralization, the polymer gel may be neutralized after the polymerization, if necessary.
- the neutralization rate of the monomer or polymer gel is preferably 40 to 90 mol%, more preferably 50 to 80 mol%.
- an internal crosslinking agent In the polymerization, an internal crosslinking agent is used as necessary.
- an internal crosslinking agent a conventionally known internal crosslinking agent can be used. Specifically, for example, N, N′-methylenebis (meth) acrylamide, (poly) ethylene glycol di (meth) acrylate, (poly) propylene glycol di (meth) acrylate, trimethylolpropane tri (meth) acrylate, glycerin Tri (meth) acrylate, glycerin acrylate methacrylate, ethylene oxide modified trimethylolpropane tri (meth) acrylate, pentaerythritol hexa (meth) acrylate, triallyl cyanurate, triallyl isocyanurate, triallyl phosphate, triallylamine, poly (meta ) Allyloxyalkane, (poly) ethylene glycol diglycidyl ether, glycerol dig
- the amount of the internal cross-linking agent used can be appropriately determined depending on the desired physical properties of the water-absorbent resin, but is usually preferably 0.0001 to 3 mol%, preferably 0.0005 to 2 based on the monomer component amount. The mol% is more preferable, and 0.001 to 1 mol% is more preferable. If the amount of the internal cross-linking agent used is 0.0001 mol% or more, the water-soluble content of the resulting water-absorbent resin powder is appropriate, so that a sufficient amount of water absorption under pressure can be secured.
- a crosslinking density will also be moderate and the water absorption amount of the water-absorbent resin powder obtained will become sufficient.
- the internal crosslinking agent may be added all at once to the reaction system or may be added in divided portions.
- Bubbles may be dispersed in the acrylic monomer aqueous solution by introducing bubbles into the prepared acrylic acid monomer aqueous solution, or by individually introducing bubbles into the raw material of the acrylic monomer aqueous solution.
- an acrylic acid monomer aqueous solution may be prepared from the raw material containing bubbles.
- bubbles are introduced into the raw material, bubbles are dispersed in unneutralized acrylic acid, solvent (water, etc.), acrylate aqueous solution, acrylic acid monomer aqueous solution, cross-linked An agent etc. are mentioned, Preferably, it is a mixture thereof, and bubbles are dispersed in water, an aqueous solution of acrylate, or an aqueous solution of an acrylic acid monomer to be polymerized prepared therewith.
- gas examples of the gas used for the dispersion of the bubbles include air, exhaust gas, oxygen, nitrogen, carbon dioxide, argon, helium, ozone and a mixture thereof, preferably from nitrogen, carbon dioxide,
- inert gases such as argon are used, and among these, inexpensive nitrogen is particularly preferable.
- the ratio of the inert gas is 80% by volume or more, more preferably 99% by volume or more, more preferably 99.9% by volume or more, and particularly preferably 99.99% by volume or more. As appropriate.
- the temperature of the inert gas is also appropriately determined and is not particularly limited. From the viewpoint of the effect, the boiling point of the gas (for example, ⁇ 210 ° C. for nitrogen) to 1000 ° C., preferably 0 to 100 ° C., 10 to 50 ° C. It is a range. By cooling or heating the temperature of the gas, the temperature of the monomer aqueous solution may be controlled, or the solubility and dispersibility of the gas may be adjusted.
- carbon dioxide gas is dispersed in the monomer, solid CO 2 (dry ice, melting point ⁇ 79 ° C.) or carbonate (for example, sodium carbonate, sodium hydrogen carbonate, magnesium carbonate, etc.) is neutralized with acrylic acid. CO 2 gas is generated), solid urea (N 2 gas is generated) or the like may be used as a gas generation source.
- a gaseous gas particularly a gas having the above temperature is preferably used. used.
- the method for dispersing bubbles (particularly gaseous gas) in the present invention is preferably such that the volume expansion ratio after dispersion with respect to the aqueous monomer solution before dispersion of bubbles is 1 by at least one method selected from the following and / or a combination thereof. It is carried out to exceed 1 time. Furthermore, a surfactant is preferably used for the stable dispersion of the bubbles.
- the method of dispersing the bubbles may be a batch type or a continuous type, and may be performed in a single stage or two or more stages, but from the viewpoint of the stability of the bubbles, it is preferably performed in a single continuous stage. That is, it is preferable to continuously supply the gas to the continuous fluid of the monomer aqueous solution and other raw materials, and in particular, the gas is continuously fed into the pipe through which the monomer aqueous solution and other raw materials are fed. Preferably it is supplied. By doing so, bubbles are dispersed in the aqueous monomer solution. When an inert gas is used as the gas, it is also preferable to degas the monomer aqueous solution in advance (dissolved oxygen is 1 mg / l or less) in order to accelerate the polymerization.
- a method for dispersing bubbles by introducing an aqueous monomer solution and a gas into a mixing region having irregularities and / or fillers and mixing them is used. .
- the bubbles can be uniformly and stably dispersed in the aqueous monomer solution.
- Examples of the mixing area having irregularities and fillings include a mixing area as shown in FIG. In FIG. 3, by passing the monomer aqueous solution containing bubbles mixed between the gaps having the protrusions, the bubbles are stably and uniformly dispersed, and the volume expansion ratio is 1 as compared with the state where the bubbles are not dispersed. A monomer aqueous solution exceeding 1 time is obtained. More specifically, using FIG. 3, the monomer aqueous solution 4 prepared in the monomer preparation tank 8 passes through the mixing zone 2 having the unevenness 3 using the aspirator 6 and the pump 9 and contains bubbles. The monomer aqueous solution 7 is sent to the storage tank 10. Before the mixing zone 2, the gas 5 and the monomer aqueous solution 4 are mixed. Examples of the apparatus having such a mixing zone include “Whipping Auto (trade name)” manufactured by Hanskrat, Germany, “Zanomat (trade name)” manufactured by F. Feichinger, Germany.
- a rotating blade may be provided as the mixing zone to stir the aqueous monomer solution in which bubbles are dispersed.
- fine bubbles can be more uniformly and stably dispersed in the aqueous monomer solution.
- S1 mixer etc. are mentioned.
- the depressurized and supersaturated gas is mainly microbubbles from the liquid. And released.
- the degree of supersaturation is preferably 1.01 to 10 times, more preferably 1.05 to 5 times, and further preferably 1.06 to 3 times the saturation solubility of the gas at a predetermined temperature.
- the method for dispersing bubbles in the present invention preferably includes formation of monomer aqueous solution and gas swirl flow.
- This method is a method in which gas-liquid two-phase fluid is swirled and bubbles are dispersed at the outlet (discharge port of the mixer).
- the ratio of the gas flow rate to the liquid flow rate is preferably 1/7 to 1/15, and the swirling speed Is preferably 10 to 10,000 revolutions per second, more preferably 100 to 1,000 revolutions per second.
- swirling microbubble generator examples include, for example, International Publication No. 00/69550, Japanese Patent Publication “JP 2003-205228”, “JP 2000-447”, and “JP 2006-”. No. 116365 ”etc., but is not particularly limited.
- the bubbles may be dispersed by mixing the monomer aqueous solution and the inert gas with a microbubble generator.
- One or more methods (a) to (c) and (1) to (8) described later can be applied to the generation of microbubbles.
- (a) or (b) is applied, and if necessary, more easily.
- a shearing force is applied to the gas-liquid mixture composed of the aqueous solution of the monomer and the bubbles.
- adopted by this invention is not specifically limited, What is marketed can be used. An example of a commercial product is illustrated below.
- OHR line mixer OHR Fluid Engineering Laboratory Co., Ltd.
- M-type microbubble generator Na planet Research Laboratories
- Microbubble generator SMB-450 for business use Ishimaru Shoyuki Co., Ltd.
- Microbubble generator Mbelife Kansai Autome Equipment Co., Ltd.
- Sphere built-in type bubble generator MBG type Nishida Tekko Co., Ltd.
- Pompator Teikoku Electric Co., Ltd.
- the microbubble generator has a water inlet and a water outlet. When a liquid (water or monomer) flows into the water inlet at a certain pressure or higher, the gas mixed inside the water Are collected in the center due to the density difference, and a gas axis is formed.
- the number average diameter of bubbles containing microbubbles generated by a microbubble generator or other methods is preferably 5 to 1000 ⁇ m, more preferably 10 to 500 ⁇ m.
- the average diameter of the bubbles is less than 5 ⁇ m, the communication of the bubbles after polymerization is lowered, and the water absorption rate tends to be inferior.
- the average diameter exceeds 1 mm, the strength becomes brittle, and it becomes difficult to pulverize the polymer gel after drying into a powder mostly having a size of 150 ⁇ m or more.
- the throughput of the microbubble generator can be appropriately set depending on the desired properties of the water-absorbent resin powder, but it is desirable to increase the flow rate of the monomer aqueous solution.
- the flow rate of the aqueous monomer solution is preferably 500 [kg / hr] or more, more preferably 1000 [kg / hr] or more, and still more preferably 2000 [kg / hr] or more.
- the production amount per hour is not limited to the use of the microbubble generator, and the production method of the present invention can generally be suitably applied to industrial huge scale production.
- Static mixer system A static mixer that has no moving parts and is mixed when fluid passes through an element fixed inside the pipe, a spiral flow guiding part inside the circular pipe, and a mushroom-like protrusion attached inside the pipe As a result, a gas-liquid two-phase flow flowing in a swirling manner is blasted to generate bubbles.
- an OHR mixer can be mentioned.
- Cavitation method This is a method of generating bubbles by deforming the flow path so that cavitation is intentionally generated in the gas distributor.
- Venturi method There is a method in which, when gas and liquid are simultaneously flowed through the straw section (throttle), large bubbles are blasted by a shock wave generated by a rapid change in the liquid flow velocity, and bubbles are generated.
- Rotating type A method in which a stirring blade is rotated at a high speed, gas is self-supplied, and bubbles are generated.
- Ultrasonic method A method of generating bubbles by appropriately setting an ultrasonic frequency, a pressure amplitude and the like can be mentioned.
- Electrolytic decomposition method There is a method of generating micro-order bubbles by electrolysis of water.
- the gas-liquid mixture comprising the monomer aqueous solution and the bubbles is subjected to a shearing treatment.
- a shearing method (3) A combination of a centrifugal pump and a swirling microbubble generator, or a static mixer having both shearing and swirling flows represented by an OHR mixer is used.
- volume expansion ratio of monomer aqueous solution (Volume expansion ratio of monomer aqueous solution)
- degree of bubble dispersion in the aqueous acrylic acid monomer solution is defined as the volume expansion ratio after dispersion in the aqueous monomer solution before dispersion of the bubbles. That is, the volume expansion ratio is defined by the following formula (1).
- the volume expansion ratio is more than 1.1 times, preferably more than 1.1 times and 10 times or less, more preferably more than 1.1 times and 8 times or less, still more preferably 1.2 to 5 times.
- the production method of the present invention is characterized in that the bubbles in the polymer gel are easily communicated with a relatively low bubble dispersion degree in which the volume expansion ratio exceeds 1.1 times, preferably 1.2 times or more.
- the higher the volume expansion ratio the easier it can be to open cells, but the higher the ratio, the larger the pore size of the bubbles and the worse the stability of the bubbles in the aqueous monomer solution. It becomes difficult.
- the volume expansion ratio is 10 times or less in order to avoid excessive reduction in handling property and bulk specific gravity and impact resistance as powder particles. It should be 8 times or less, more preferably 5 times or less.
- the conditions of the bubble generating apparatus are set as appropriate; an additive for improving the bubble stability such as a surfactant or a hydrophilic polymer is used. It is added to a monomer aqueous solution;
- Step of foaming monomer aqueous solution (D) (Foam regulation step)
- a foam regulating step and a defoaming step may be further included.
- the foaming time used is 5 seconds or more, 10 seconds to 60 minutes, preferably 30 seconds to 30 minutes, particularly 60 seconds to 20 minutes.
- a method using a circulation tank shown in FIG. there is a method of holding for a predetermined time at a normal pressure after the introduction of bubbles, or a method of repeating it, and the holding temperature is 0 to 100 ° C., more preferably 20 to 50 ° C.
- the foam regulating step used in the present invention may be a known technique, such as the method described in US Pat. No. 6,667,372, or the method described in Techno Systems Publishing Co., Ltd. “Foam Engineering First Edition”, pages 759-774. Etc.
- a preferable foam regulating step is to circulate a monomer containing a circulating air flow (bubbles) to the circulation tank, and the foam regulating step preferably contains 1% by volume or more of oxygen in the upper space of the circulation tank. It is preferable to perform the process (B) which superpose
- FIGS. 1 and 2 Such a defoaming step (foaming step) is illustrated in FIGS. 1 and 2, respectively.
- the circulation pump, the heat exchanger, and the like are omitted.
- the monomer entering the tank 1 flows out from the lower part of the tank 1 to the circulation line, flows through the circulation line while introducing the inert gas, and flows into the tank 1 again from the upper part of the tank. It is circulating by that.
- the monomer since the monomer circulates while the inert gas is introduced, the gas is dissolved and / or dispersed in the monomer. Then, the monomer in which the gas is dissolved and / or dispersed stays in the tank 1 until it flows into the tank 1 and again flows out into the circulation line, so that foam regulation proceeds.
- the neutralized monomer that has entered the tank 1 has an inert gas from the bottom of the tank 1. It flows out while being introduced, and it can circulate and perform foam regulation by flowing back into the tank 1 from the upper part.
- the foamed monomer may be extracted as it is and used in the next step, or as shown in FIG. 2, with a neutralizing agent or a crosslinking agent added. It may be extracted.
- the circulation tank is also exemplified in International Publication Nos. 2007/28746, 2007/28747, and 2009/123197, in the present invention, as shown in FIG. 1 and FIG.
- the inside of the circulation tank may be filled with an inert gas, but is preferably filled with oxygen, particularly air, from the stability of the monomer.
- polymerization starts in a monomer aqueous solution that stably contains fine bubbles after a predetermined time.
- Polymerization may be carried out by adding an agent or irradiation with energy rays such as ultraviolet rays, infrared rays or microwaves.
- the size of the bubble is determined by (a) laser diffraction scattering method (also known as static light scattering method), (b) dynamic light scattering method, (c) electrical detection band method (common name: Coulter counter method), (d ) Particle counter method (light scattering method, light blocking method), (e) Visualization method by camera photographing, (f) Measurement by laser beam and interference image method using CCD camera, and the like.
- laser diffraction scattering method also known as static light scattering method
- dynamic light scattering method also known as static light scattering method
- electrical detection band method common name: Coulter counter method
- Particle counter method light scattering method, light blocking method
- e Visualization method by camera photographing
- CCD camera Measurement by laser beam and interference image method using CCD camera, and the like.
- the monomer aqueous solution and the inert gas are preferably mixed in the presence of a surfactant. Bubbles can be stably dispersed by using a surfactant. Further, by appropriately adjusting the type and amount of the surfactant, a water-absorbing resin having desired physical properties can be obtained.
- the use of the surfactant is optional, even in the absence. Applicable.
- Such a surfactant is not particularly limited. For example, anionic surfactants, nonionic surfactants, cationic surfactants, amphoteric surfactants, fluorosurfactants, organometallic surfactants, etc. There is. These surfactants may be used alone or in combination.
- polyglycerin fatty acid esters preferably trimeric or more, more preferably 6-10 mer polyglycerin fatty acid esters, the fatty acids having 6 to 28 carbon atoms, More preferred is a fatty acid having a linear or branched chain of 12 to 24, particularly preferably 16 to 20.
- polyglycerol fatty acid esters include tetraglyceryl monostearate, tetraglyceryl monooleate, tetraglyceryl tristearate, tetraglyceryl pentastearate, tetraglyceryl pentaoleate, tetraglyceryl monolaurate, and monomyristic acid.
- Tetraglyceryl Tetraglyceryl, hexaglyceryl monostearate, hexaglyceryl monooleate, hexaglyceryl tristearate, hexaglyceryl pentastearate, hexaglyceryl pentaoleate, hexaglyceryl polyricinoleate, decaglyceryl monolaurate, decaglyceryl monostearate, Decaglyceryl monomyristate, decaglyceryl monoisostearate, decaglyceryl monooleate, monolinoleate Glyceryl, decaglyceryl distearate, decaglyceryl diisostearate, decaglyceryl tristearate, decaglyceryl trioleate, decaglyceryl trioleate, decaglyceryl pentastearate, decaglyceryl pentaisostearate, decaglyceryl pentaoleate, h
- nonionic surfactants include nonylphenol polyethylene oxide adducts; block polymers of ethylene oxide and propylene oxide; sorbitan monolaurate, sorbitan monomyristate, sorbitan monopalmitate, sorbitan monostearate, sorbitan Sorbitan fatty acid esters such as tristearate, sorbitan monooleate, sorbitan trioleate, sorbitan sesquioleate, sorbitan distearate; glycerol monostearate, glycerol monooleate, diglycerol monooleate, self-emulsifying glycerol monostearate Glycerin fatty acid ester such as rate; polyoxyethylene lauryl ether, polyoxyethylene cetyl ether, polyoxyethylene Polyoxyethylene alkyl ethers such as stearyl ether, polyoxyethylene oleyl ether and polyoxyethylene higher alcohol ether; polyoxyethylene alkyl aryl ethers such
- alkyl sulfate salts such as sodium dodecyl sulfate, potassium dodecyl sulfate, ammonium alkyl sulfate, etc .
- sodium dodecyl polyglycol ether sulfate sodium dodecyl polyglycol ether sulfate
- Sodium sulforicinoates alkyl sulfonates such as sulfonated paraffin salts; sodium decyl benzene sulfonates
- alkyl sulfonates such as alkali phenol hydroxyethylene alkali metal sulfates; high alkyl naphthalene sulfonates; naphthalene sulfonic acid formalin condensates, sodium Fatty acid salts such as laurate and triethanolamine oleate; polyoxyalkyl ether sulfate ester Reactive anionic emulsifier
- the amount of these surfactants used is preferably as small as possible relative to the monomers used. Preferably it is less than 10% by weight, more preferably less than 5% by weight, even more preferably less than 1% by weight, particularly preferably less than 0.1% by weight.
- the amount of the surfactant is large, the amount of the surfactant that is eluted when the resulting water-absorbent resin is in contact with the aqueous liquid increases. Therefore, in actual use with sanitary materials such as diapers, body fluid Therefore, it is desirable to use a surfactant amount as small as possible.
- surfactants with reactive unsaturated groups that can be polymerized with acrylic acid (salt) in the structure, or silicone surfactants with epoxy groups or amino groups that are highly reactive with carboxyl groups of acrylic acid, etc.
- a so-called reactive surfactant or using a high molecular weight surfactant elution of the surfactant upon water absorption can be suppressed.
- the surface tension of the water absorbent resin measured in (5-8) described later is preferably 55 [mN / m] or more, more preferably 60 [mN / m] or more, particularly preferably. Is controlled to be 65 [mN / m] or more, more preferably 70 [mN / m] or more. The upper limit is usually about 75 [mN / m]. Since the decrease in surface tension is suppressed, the liquid absorption characteristics of sanitary goods are improved, and the return amount (Re-Wet) is reduced.
- a step of polymerizing an aqueous monomer solution to obtain a foamed polymer (B) (polymerization step)
- This step is a step of obtaining a foamed polymer by polymerizing the monomer aqueous solution that has undergone the step (A) and, if necessary, the step (D). It is desired that the aqueous monomer solution in which bubbles are dispersed at a predetermined volume expansion ratio is supplied to the polymerization apparatus as quickly as possible so that a certain amount of bubbles is maintained, preferably within 5 minutes, It is preferable to supply the polymerization apparatus within 3 minutes, particularly preferably within 1 minute.
- Polymerization initiator There is no restriction
- a thermal decomposition type initiator for example, persulfate such as sodium persulfate, potassium persulfate, ammonium persulfate; peroxide such as hydrogen peroxide, t-butyl peroxide, methyl ethyl ketone peroxide; 2-carbamoylazoiso Azonitrile compounds such as butyronitrile, azoamidine compounds such as 2,2′-azobis (2-methylpropionamidine) dihydrochloride, 2,2′-azobis-2- (2-imidazolin-2-yl) propane hydrochloride Cyclic azoamidine compounds, azoamide compounds, alkylazo compounds, 2,2′-azobis (2-amidinopropane) dihydrochloride, 2,2′-azobis [2- (2-imidazolin-2-yl) propane] dihydrochloride, etc.
- persulfate such as sodium persulfate, potassium persulfate, ammonium persulfate
- Azo compounds; etc. and photodegradable initiators eg benzoin derivatives, benzyl derivatives
- Conductor acetophenone derivatives such as 2-hydroxy-2-methyl-1-phenyl-propan-1-one, benzophenone derivatives, azo compounds, etc.
- an azo polymerization initiator preferably a water-soluble azo polymerization initiator such as 2,2′-azobis (2-methylpropionamidine) dihydrochloride
- bubbles may be further included.
- the decomposition of these polymerization initiators can be promoted by using a reducing agent in combination. Therefore, it can also be used as a redox initiator combining both.
- the reducing agent is not particularly limited.
- reducing metal (salt) such as (heavy) sulfite (salt) such as sodium sulfite and sodium bisulfite, L-ascorbic acid (salt), and ferrous salt.
- amines reducing metal (salt) such as (heavy) sulfite (salt) such as sodium sulfite and sodium bisulfite, L-ascorbic acid (salt), and ferrous salt.
- amines amines.
- an oxidative polymerization initiator and a reducing agent are used like a redox initiator, each may be combined with the monomer aqueous solution, or the reducing agent may be mixed in advance with the monomer aqueous solution.
- the amount of the polymerization initiator used is preferably 0.0001 to 1 mol%, more preferably 0.0005 to 0.5 mol%, based on the total amount of monomers. If it is 0.0001 mol% or more, the amount of residual monomers is reduced, and it is preferable.
- a water-soluble polymer or water-absorbing resin or water-insoluble fine particles generally known as a thickener is used in the aqueous monomer solution before polymerization.
- starch starch derivatives (eg, etherified starch, esterified starch, etc.), cellulose, cellulose derivatives (eg, carboxymethylcellulose, hydroxyethylcellulose, etc.), high molecular polysaccharides such as guar gum, or polyvinyl alcohol
- water-soluble polymer such as hydrophilic polymer such as polyacrylic acid (salt), crosslinked polyacrylic acid (salt), or water-insoluble fine particles such as silicon dioxide (silica), zeolite, talc, titanium dioxide
- the fine powder described later of the water-absorbent resin (preferably, fine powder containing 70% by weight or more of powder having a particle diameter of 150 ⁇ m or less) is recycled to the polymerization step to obtain a monomer aqueous solution. May be thickened.
- a water-soluble polymer or a water-absorbing resin particularly a nonionic water-soluble polymer (especially starch, PVA, hydroxyethyl cellulose) or a polyacrylic acid (salt) water-absorbing resin is used as a thickener. Is done.
- an inorganic reducing agent such as hypophosphorous acid (salt) and a polymerization improving agent such as a chelating agent may be added.
- a polymerization improving agent such as a chelating agent
- a known plasticizer other than water for example, a polyol such as glycerin, polyethylene glycol, or polypropylene glycol may be used.
- the use amount of the above-mentioned additives is 0 to 50 parts by weight, further 0.01 to 20 parts by weight with respect to 100 parts by weight of the monomer.
- the polymerization method employed in the present invention is not particularly limited, and those employed in ordinary water-absorbing resin production methods are employed, and examples thereof include an aqueous solution polymerization method.
- examples of the aqueous solution polymerization method include a static polymerization method in which an aqueous monomer solution is polymerized in a static state (without mechanical stirring), a stirring polymerization method in which polymerization is performed in a stirrer, and the like.
- a stationary polymerization method is employed.
- These polymerization methods include a batch method and a continuous method, respectively, but a continuous method is preferred. In these polymerization methods, a belt polymerization apparatus, a tank (silo) polymerization apparatus, and a stirring polymerization apparatus are usually employed.
- the apparatus for producing a water-absorbing resin according to the present invention is not particularly limited as long as it is an apparatus capable of continuously polymerizing a monomer aqueous solution continuously supplied by the above-described method.
- a polymerization apparatus or a continuous stirring polymerization apparatus is preferred.
- the monomer aqueous solution in which bubbles are dispersed at a predetermined volume expansion ratio is desired to start polymerization as soon as possible after being supplied to the polymerization apparatus so that a certain amount of bubbles is maintained.
- Polymerization is initiated by irradiation with energy rays or heating within 5 minutes after feeding, more preferably within 3 minutes, and particularly preferably within 1 minute.
- a preferable form of the polymerization apparatus is an endless belt type continuous polymerization apparatus, in which the belt is made of fluororesin or the surface is coated with fluororesin. Furthermore, a device having a heating device or a heat retaining device and having a system for recovering and reusing water and / or monomer vapor generated during polymerization is preferable. Further, the belt preferably has a lower horizontal or monomer mixture supply part to prevent the backflow of the monomer mixture, and between the time when the polymer gel is discharged from the belt to the monomer mixture supply port, A belt cleaning process is desirable.
- the thickness of the belt polymerization is appropriately determined according to the purpose and the heat removal during the polymerization.
- the polymerization is performed with a gel thickness of 0.1 to 30 cm, further 0.5 to 20 cm, and 1 to 10 cm.
- the size of the belt is determined by the production amount, and industrially, for example, the belt width is selected from about 0.1 to 10 m, about 1 to 5 m, and the length is selected from about 5 to 200 m.
- a tank of 0.1 to 300 m 3 , 1 to 100 m 3 is used.
- the polymer gel obtained by the stationary polymerization may be formed into a molded product by cutting, surface polishing, compression, etc., which will be described later, if necessary, or may be powdered (non-molded product).
- a uniaxial stirring apparatus or a stirring apparatus having a plurality of stirring shafts such as a continuous kneader can be adopted, but a multi-shaft stirring apparatus is preferably used from the viewpoint of productivity.
- boiling polymerization (above the boiling point of water as a solvent) is used to increase bubbles in the water-absorbent resin, and at least one stage of the polymerization step is set to 100 ° C. or more. That is, the upper limit of the temperature during the polymerization is 100 ° C. or more, preferably 100 to 140 ° C., more preferably 102 to 130 ° C., still more preferably 104 to 120 ° C., and particularly preferably 105 to 118 ° C. Is called boiling polymerization.
- boiling point of water 100 ° C.
- pressure polymerization rise in molar boiling point of a monomer dissolved in water, or the like. This is called boiling polymerization.
- the maximum temperature reached during the polymerization is within the above range, and may be appropriately heated or cooled during the polymerization, or may be within the above range only by the polymerization heat.
- Polymerization temperature can be measured with a contact thermometer or a non-contact thermometer (for example, an infrared thermometer), and with belt polymerization, the surface temperature of the polymer gel can be measured.
- a contact thermometer or a non-contact thermometer for example, an infrared thermometer
- the surface temperature of the polymer gel can be measured.
- an open-cell polymer gel is obtained, and water as a solvent is boiled at the time of polymerization and water is evaporated, so that the polymer gel boils to a substantially uniform temperature on the surface and inside. Therefore, the maximum temperature of the polymer gel may be measured internally or on the surface, but is preferably defined in the interior (center). What is necessary is just to measure with the contact thermometer, such as a thermocouple, for the maximum temperature measurement of the center part of superposition
- a PC card type data collection system NR-1000 manufactured by Keyence Co., Ltd. is used to measure the temperature of a system with a rapid temperature change.
- the pair can be placed in the center of the polymerization system and measured with a sampling period of 0.1 seconds. From the obtained temperature-time chart, the polymerization start temperature and peak temperature (maximum temperature reached) can be read.
- the pressure during polymerization may be reduced pressure polymerization (especially reduced pressure exceeding 10% of atmospheric pressure) or pressurized polymerization (pressurized exceeding 10%), but the simplicity and cost of the apparatus, and the foaming efficiency, etc.
- reduced pressure polymerization especially reduced pressure exceeding 10% of atmospheric pressure
- pressurized polymerization pressurized exceeding 10%
- the polymerization may be carried out at a substantially normal pressure within the above range.
- the time for the polymerization to be 100 ° C. or higher is appropriately determined depending on the polymerization method, and is preferably 1 second or longer, 5 seconds or longer, and further 30 seconds or longer. It is preferable to adjust to.
- Long-time boiling polymerization not only does not contribute to foaming, but may also reduce physical properties (water absorption capacity and soluble content). Within 30 minutes, within 10 minutes, within 5 minutes, especially within 1 minute.
- the maximum attainment temperature (peak temperature) of polymerization exceeds 140 ° C.
- the physical properties of the water absorbent resin powder may be deteriorated.
- it is 130 degrees C or less, More preferably, it is 120 degrees C or less.
- the amount of water evaporation during the polymerization varies depending on the difference in the starting temperature, etc., but the increase in the solid content concentration of the polymer gel relative to the solid content concentration in the aqueous monomer solution is 2% by weight or more (the upper limit is 20% by weight, the polymerization Later, a gel-like product, in particular a range in which the solid content concentration is 80% by weight or less, is preferable.) Further, it is preferable that the increase range is 3 to 20% by weight, particularly 5 to 20% by weight.
- the solid content concentration in the monomer aqueous solution is defined by the following formula.
- the weight of the nonvolatile additive for example, hydrophilic polymer, surfactant, etc.
- the nonvolatile additive is used at the following solid content concentration. Stipulated including the weight of Moreover, it is estimated that generation
- the polymerization is carried out in the presence of an air flow such as an inert gas in a polymerization machine. Therefore, even if the temperature during the polymerization is less than 100 ° C. and the polymerization time is long, water evaporation occurs.
- the communication of bubbles disclosed in the invention has no effect and is not intended.
- bubbles are dispersed in an aqueous monomer solution and the surface area is increased by more than 1.1 times to increase the surface area.
- the ratio of open pores (open cells) in the foamed polymer (foam polymer) can be improved.
- the polymerization start temperature is preferably high temperature start higher than room temperature, specifically, the polymerization start temperature in the polymerization step is 40 ° C or higher, more preferably 50 ° C or higher, the upper limit is 100 ° C or lower, Furthermore, it is 90 degrees C or less, Most preferably, it is 85 degrees C or less. If the starting temperature is less than 40 ° C., it is difficult to reach 100 ° C., and if it is too high, it is difficult to stably disperse bubbles, which is not preferable.
- the polymerization time may be appropriately determined according to the type of monomer and polymerization initiator, the polymerization temperature, etc., but it is preferable to shorten the time from the start of polymerization until the polymerization temperature reaches the maximum temperature. Specifically, it is preferably within 20 minutes, more preferably within 10 minutes, further preferably within 5 minutes, particularly preferably within 2 minutes, and most preferably within 1 minute. When the time is longer, for example, 1 hour or more, it is preferable because bubbles dispersed during polymerization disappear, and the generated water vapor hardly breaks through the gel partition walls, and the dispersed bubbles are less likely to become continuous cells. Absent. Under the conditions of the present invention, it is considered that the balance between the bubble stability during polymerization, the strength of the polymer gel forming the cell walls during polymerization, and the evaporation of moisture is advantageous for the formation of open cells.
- the concentration of the acrylic acid monomer aqueous solution is preferably as high as possible in the polymerization step, and is 40 wt% or more, 45 wt% or more, 50 wt% or more (the upper limit is usually 80 wt% or less, and further 70 wt%). Less than).
- concentration is 40% by weight or more, dispersed bubbles can exist stably even at 40 ° C. or more, whereas it is less than 40% by weight, for example, under the conditions disclosed in the examples of Patent Document 23. Although the bubbles can be stably dispersed at room temperature, the bubbles become unstable and disappear in a short time when the temperature rises.
- a polymer gel in the form of a continuous foam is obtained. Further, in the tank type (silo type) stationary polymerization, a foamed polymer gel having a tank (silo) shape is obtained.
- the foamed polymer gel can be used as it is, or can be finely crushed for use and / or further use (the following fragmentation step).
- the foamed polymer gel has an advantage that the load on the apparatus during pulverization tends to be smaller than that of a high solid content plate-like gel containing no bubbles.
- the cutting pulverizer advantageous for pulverizing the high solid content gel disclosed in JP-A No.
- this is an extrusion pulverizer represented by a chopper, which was impossible with a plate-like gel. Can be applied.
- the foamed polymer gel may be further dried and pulverized to obtain a water absorbent resin powder, or the foam polymer gel may be cut into a predetermined sheet shape and dried to obtain a water absorbent resin molded product.
- the molded water-absorbing resin or water-absorbing resin powder is further subjected to surface cross-linking treatment, granulated, water preparation and various modifiers added, and used as a water-absorbing resin product.
- surface cross-linking treatment granulated, water preparation and various modifiers added, and used as a water-absorbing resin product.
- Known techniques may be employed for the pulverization and drying of the polymer gel and the surface crosslinking treatment.
- Patent Document 20 discloses that in the production of a foam-like water-absorbent resin, “the polymerization temperature is preferably the boiling of the polymerizable aqueous mixture. "Adjust to avoid.” Patent Documents 8, 21, and 30 to 32 also disclose that “boiling during polymerization” is avoided in the production of a foamed water-absorbent resin as in Patent Document 20. Furthermore, Patent Document 33 discloses that “open cell foam increases when foam is produced at 65 ° C. or lower”.
- a technique for controlling the maximum temperature to be low for the purpose of reducing soluble components in polymerization other than foam polymerization is disclosed.
- a technique for setting the maximum temperature to 95 ° C. or lower Patent Document 34
- a technique for polymerizing at a polymerization temperature of 20 to 70 ° C. Patent Document 35
- a technique for polymerizing at a polymerization temperature of 20 to 95 ° C. Patent Document 35
- boiling polymerization has been avoided in the production of a (continuous) foam-like water-absorbent resin in Patent Documents 8, 20, 21, 29-33, etc., but in the present invention, it has been avoided in the conventional production of a foam-like water-absorbent resin. It has been found that the boiling polymerization that has been provided efficiently provides a water-absorbing resin having open cells at a concentration of 40% by weight or more and a volume expansion ratio of 1.1 times or more.
- the bubbles are dispersed in a monomer aqueous solution having a monomer concentration of 40% by weight or more until the volume expansion ratio becomes 1.1 times or more, and then subjected to boiling polymerization, whereby the independent dispersion dispersed in the monomer aqueous solution is obtained.
- Bubbles for example, spherical bubbles such as nitrogen gas expand in volume, and volume expansion of closed cells dispersed in water vapor and monomer aqueous solution generated by boiling water when the monomer aqueous solution is gelated by boiling polymerization
- the continuous bubbles are generated by breaking through the partition of gel bubbles (spherical closed cells) in the initial stage of polymerization generated by polymerization of the monomer aqueous solution.
- boiling polymerization is necessary for the breakage of the gel partition due to the generation of water vapor, and the monomer aqueous solution having a monomer concentration of 40% by weight or more is polymerized from the gel hardness and polymerization rate derived from the solid content.
- the initial gel bubbles (spherical closed cells) break through the partition walls and easily generate open cells.
- a subdividing step of pulverizing and subdividing at the same time during polymerization, or crushing and finely pulverizing the foamed polymer after polymerization may be performed.
- gel pulverization particularly by gel pulverization, particularly kneading, it is possible to achieve both water absorption speed and liquid permeability, and further improve impact resistance.
- the gel is crushed by gel during polymerization, and when the polymerization process is continuous belt polymerization, the gel is crushed by gel after polymerization.
- gel kneading of the gel can be performed during the polymerization by the kneader, so that the subdivision step is included in the kneader polymerization step.
- the gel crusher that can be used is not particularly limited.
- a gel crusher equipped with a plurality of rotary stirring blades such as a batch type or continuous double arm kneader, a single screw extruder, a twin screw extruder, and a meat chopper.
- a screw type extruder having a perforated plate at the tip is preferable, and for example, a screw type extruder disclosed in Japanese Patent Application Laid-Open No. 2000-63527 can be given.
- the temperature of the water-containing gel at the time of gel pulverization (pulverization) is preferably 40 to 120 ° C., more preferably 50 to 110 ° C. from the viewpoint of physical properties.
- the gel temperature is 40 to 120 ° C., the hardness of the hydrogel is appropriate, and the particle shape and particle size distribution can be easily controlled during gel grinding.
- the gel temperature can be controlled by the temperature during polymerization, heating or cooling after polymerization, and the like.
- the weight average particle diameter (D50) of the particulate hydrogel after gel pulverization (pulverization) is preferably 0.5 to 4 mm, more preferably 0.5 to 3 mm, and still more preferably 0.6 to 2 mm. If the weight average particle diameter (D50) of the particulate hydrogel is 0.5 mm or more, an increase in residual monomer (monomer) and an effect of improving the water absorption rate (FSR) can be expected, and the weight average particle diameter ( If D50) is 4 mm or less, the drying time is short, and an increase in the water-soluble component (Ext) can be suppressed.
- the proportion of the particulate hydrogel having a particle size of 5 mm or more is preferably 0 to 10% by weight, more preferably 0 to 5% by weight, based on the entire particulate hydrogel.
- the particle size of the particulate water-containing gel is determined by classification with a sieve having a specific opening, similarly to the particle size of the water absorbent resin powder after the pulverization step. Further, the weight average particle diameter (D50) can be determined in the same manner.
- the wet classification method described in paragraph [0091] of JP-A-2000-63527 is used. To measure.
- gel pulverization described in Japanese Patent Application No. 2010-088993 International Application PCT / JP2011 / 058829
- GGE gel pulverization energy
- the gel grinding energy has an upper limit of preferably 60 [J / g] or less, more preferably 50 [J / g] or less, and still more preferably 40 [J / g] or less.
- 18 [J / g] or more is preferable, 20 [J / g] or more is more preferable, and 25 [J / g] or more is still more preferable.
- Step of heating and drying the foamed polymer (C) (heating and drying step)
- the foamed polymer gel obtained as described above is dried to obtain a dry polymer.
- the resin solid content determined from the loss on drying (1 g of powder or particles heated at 180 ° C. for 3 hours) is preferably 80% by weight or more, more preferably 85 to 99% by weight, and still more preferably 90 to 98% by weight.
- the flexibility may be adjusted by adding water or a plasticizer as necessary.
- the drying temperature is not particularly limited, but is preferably in the range of 100 to 300 ° C, more preferably in the range of 150 to 250 ° C.
- the foamed polymer gel of the present invention can be easily dried as it is or pulverized, and the drying method disclosed in JP-A-2000-212215 is preferably applied.
- the gel tends to expand and deform when the coarsely pulverized gel is dried at high temperature, but the gel of the present invention has many open cells, so there is almost no gel expansion during high temperature drying. Less deformation is also an advantageous feature.
- the gel of the present invention since the gel of the present invention has many open cells, it is also an advantageous feature that the drying time until a dry polymer is obtained is shortened.
- drying of the hydrogel after polymerization requires large equipment, thermal energy, and a long drying time.
- the deterioration and coloring of the water-absorbing resin due to this long-time drying, and further the production of the water-absorbing resin The problem of increased cost has occurred, but the present invention solves this problem and achieves shortening of drying time, prevention of coloring, improvement of water absorption performance, and the like.
- the water-absorbent resin that has undergone the above-described heat drying step may be used in the form of a sheet or block, but it is preferable to obtain a powder having a predetermined particle size by pulverization and / or classification.
- the weight average particle diameter (D50) in the case of the water absorbent resin powder is 200 to 600 ⁇ m, preferably 300 ⁇ m or more and less than 600 ⁇ m, more preferably 200 to 550 ⁇ m, still more preferably 250 to 500 ⁇ m, particularly from the viewpoint of improving physical properties. Preferably, it is adjusted to 350 to 450 ⁇ m. Further, the smaller the particle size is less than 150 ⁇ m, the better, and it is usually adjusted to 0 to 5% by weight, preferably 0 to 3% by weight, particularly preferably 0 to 1% by weight.
- the surface cross-linking is preferably performed at a ratio of 850 to 150 ⁇ m, more preferably 710 to 150 ⁇ m at 95% by weight or more, further 98% by weight or more (upper limit 100% by weight).
- the particle size before surface crosslinking is preferably applied to the final product (also known as particulate water-absorbing agent) after surface crosslinking.
- the foamed polymer gel obtained as described above and its dried polymer may be powdered, but the foamed polymer gel obtained after stationary polymerization such as belt polymerization or tank polymerization. Or its dry polymer may be used as it is, or may be molded. That is, the present invention includes a molding step simultaneously with the polymerization step or after the polymerization step. After the polymerization step, a dried polymer that has been heat-dried may be formed into a molded product, or may be heat-dried simultaneously with the polymerization step or after the polymerization step and then through a molding step.
- the foam-like polymer gel obtained by the belt polymerization is in the form of a band, and the tank-like polymer gel (for example, a cylindrical gel) is obtained in the tank polymerization, so they may be used as a molded product as they are, If necessary, cutting, surface polishing, compression or the like may be performed.
- the aqueous monomer solution may be put into a three-dimensional mold in accordance with a predetermined shape such as a napkin to obtain a water-absorbent resin molded product in the final product form simultaneously with polymerization.
- a sheet-shaped polymer gel having a thickness of 0.1 to 30 cm, further 0.5 to 20 cm, and 1 to 10 cm is continuously obtained.
- the obtained sheet-shaped polymer gel may be cut, surface-polished, hollowed out, compressed, etc. in the thickness direction, width direction, and length direction as appropriate to obtain a molded product.
- a polymer gel for example, a cylindrical gel, a cubic gel, etc.
- a volume of 0.1 to 300 m 3 or 1 to 100 m 3 can be obtained. Further, it may be molded by performing surface polishing, compression or the like.
- the shape of the molded product is appropriately determined according to the purpose, but for the purpose of a napkin or a diaper, the thickness is 0.1 to 2 cm, and further 0.2 to 1 cm, and the sheet is a sheet having an area of 5 cm 2 or more. It is preferable.
- the sheet-like material may be molded up to the final napkin or the absorbent layer of the diaper, or may be in the form of a roll or a carpet so that it can be further molded (cut or cut). Moreover, you may give a pattern, a hole, and embossing on the surface of the said sheet-like material.
- a surface cross-linking step may be further included after drying.
- a covalent bond surface cross-linking agent and / or an ionic cross-linkable surface cross-linking agent are used, and these cross-linking agents are preferably used in combination.
- Surface cross-linking is an operation to make the surface highly cross-linked compared to the inside.
- Surface cross-linking is performed by radical cross-linking with a radical polymerization initiator (for example, persulfate or photoinitiator) or by adding a monomer to the particle surface.
- a crosslinking agent capable of reacting with the carboxyl group of the polyacrylic acid (salt) water-absorbing resin is preferably used, and will be described below, but the surface crosslinking of the present invention is not limited to the following.
- the surface cross-linking agent that can be used in the present invention include various organic or inorganic cross-linking agents, but organic surface cross-linking agents can be preferably used.
- the surface crosslinking agent is preferably a polyhydric alcohol compound, an epoxy compound, an oxetane compound, a polyvalent amine compound or a condensate thereof with a haloepoxy compound, an oxazoline compound, a (mono, di, or poly) oxazolidinone compound, an alkylene.
- a dehydration-reactive cross-linking agent composed of a polyhydric alcohol compound, an alkylene carbonate compound, or an oxazolidinone compound, which is a carbonate compound and requires a reaction at a high temperature, can be used.
- a dehydration-reactive crosslinking agent is not used, more specifically, compounds exemplified in U.S. Pat. Nos. 6,228,930, 6071976, 6254990 and the like can be mentioned.
- mono-, di-, tri-, tetra- or higher multimeric propylene glycol 1,3-propanediol, glycerin, 1,4-butanediol, 1,3-butanediol, 1,5-pentanediol, Polyhydric alcohol compounds such as 1,6-hexanediol and sorbitol; Epoxy compounds such as ethylene glycol diglycidyl ether and glycidol; Alkylene carbonate compounds such as ethylene carbonate; Oxetane compounds; Cyclic urea compounds such as 2-imidazolidinone It is done.
- the liquid permeability may be improved by using a polyamine polymer or a polyvalent metal salt as an ion-binding surface cross-linking agent.
- a polyamine polymer or a polyvalent metal salt as an ion-binding surface cross-linking agent.
- the polyvalent metal salt (inorganic surface crosslinking agent) used are divalent or higher, preferably trivalent to tetravalent polyvalent metal salts (organic salts or inorganic salts) or hydroxides.
- the polyvalent metal that can be used include aluminum and zirconium, and examples thereof include aluminum lactate and aluminum sulfate.
- the amount of the surface cross-linking agent used is appropriately determined from 0.001 to 10 parts by weight and 0.01 to 5 parts by weight with respect to 100 parts by weight of the water absorbent resin powder.
- Water can preferably be used in accordance with the surface cross-linking agent.
- the amount of water used is in the range of 0.5 to 20 parts by weight, more preferably 0.5 to 10 parts by weight with respect to 100 parts by weight of the water absorbent resin powder.
- 0.001 to 10 parts by weight and 0.01 to 5 parts by weight are used together with respect to 100 parts by weight of the water-absorbent resin powder.
- a hydrophilic organic solvent may be used, and the amount thereof is in the range of 0 to 10 parts by weight, preferably 0 to 5 parts by weight with respect to 100 parts by weight of the water absorbent resin powder. Further, when mixing the crosslinking agent solution into the water-absorbent resin powder, the range does not hinder the effect of the present invention, for example, 0 to 10 parts by weight or less, preferably 0 to 5 parts by weight, more preferably 0 to 1 part by weight.
- An acid catalyst, a basic catalyst, water-insoluble fine particles, and a surfactant may coexist. The surfactant used and the amount of use thereof are exemplified in US Pat. No. 7,473,739.
- the water-absorbent resin after mixing the surface cross-linking agent is subjected to heat treatment, and then subjected to cooling treatment if necessary.
- the heating temperature is 70 to 300 ° C, preferably 120 to 250 ° C, more preferably 150 to 250 ° C, and the heating time is preferably 1 minute to 2 hours.
- the water absorption capacity under pressure (AAP) described later is preferably increased to 20 [g / g] or more, and further to 23 to 30 [g / g]. .
- Fine powder recycling step The water-absorbent resin after the polymerization step, preferably after the heat drying step, is adjusted to the above particle size through a pulverization and classification step, if necessary.
- coarse particles for example, 1 mm or more
- fine particles for example, less than 150 ⁇ m, or even less than 106 ⁇ m
- the production method of the present invention may preferably include a fine powder recycling step.
- the fine powder recycling process is the process of separating the fine powder generated in the drying process and, if necessary, the pulverizing and classification processes (particularly the fine powder containing 70% by weight or more of the powder having a particle diameter of 150 ⁇ m or less), or in the state of hydration or It is granulated and recycled before the pulverization step, preferably the step of recycling to the polymerization step, the step of fragmenting the foamed polymer, or the heat drying step.
- the fine powder may be fine powder before surface cross-linking or fine powder after surface cross-linking, and the amount of fine powder recycled is appropriately set at 1 to 40% by weight, further 5 to 30% by weight of the dried product.
- the fine powder recycling method preferably used in the present invention is a method in which a water-absorbent resin fine powder, a hydrate or granulated product thereof, and, if necessary, inorganic fine particles are mixed with an aqueous monomer solution during polymerization or a hydrogel during polymerization. . Further, the fine powder to be recycled may be used to increase the viscosity of the monomer during polymerization to promote foaming.
- a second classification step an evaporation monomer recycling step, a granulation step, a fine powder removal step, and the like may be provided as necessary.
- the following chelating agents and / or reducing agents may be used in the monomer or polymer thereof for the purpose of color stability over time, prevention of gel degradation, and the like. That is, in order to prevent coloring and deterioration, the present invention preferably includes a step of adding a chelate and / or a reducing agent.
- the water-absorbent resin having open cells of the present invention preferably contains a chelating agent from the viewpoint of urine resistance and prevention of coloring.
- a chelating agent that is not disclosed in Patent Document 20 and the like, a water-absorbing resin having open cells and excellent in urine resistance and coloring prevention is provided.
- the chelating agent of the present invention a polymer compound or a non-polymer compound, particularly a non-polymer compound is preferable from the viewpoint of effects, and specifically, an amino polyvalent carboxylic acid, an organic polyvalent phosphoric acid, an inorganic polyvalent phosphoric acid. A compound selected from aminopolyphosphoric acid and salts thereof is preferred.
- the chelating agent preferably has a molecular weight of 100 to 5,000, more preferably 200 to 1,000.
- polyvalent has a plurality of the functional groups in one molecule, preferably 2 to 30, more preferably 3 to 20, and 4 to 10 functional groups.
- These chelating agents are preferably water-soluble chelating agents, specifically, water-soluble chelating agents that dissolve in 1 g or more, more preferably 10 g or more, in 100 g (25 ° C.) of water.
- the contents described in paragraphs [0104] to [0111] of WO 2011/040530 pamphlet are applied mutatis mutandis.
- the water-absorbent resin having open cells according to the present invention preferably contains an organic or inorganic reducing agent, more preferably an inorganic reducing agent, and more preferably an inorganic reducing agent as a reducing inorganic, from the viewpoint of urine resistance and coloring prevention.
- a water-soluble inorganic compound having an element or a water-soluble organic compound having a reducing inorganic element is included.
- the “water-soluble” means that 1 g or more, further 5 g or more, particularly 10 g or more is dissolved in 100 g of water at 25 ° C.
- the water-absorbent resin powder may include oxidizing agents, antioxidants, water, polyvalent metal compounds, water-insoluble inorganic or organic powders such as silica and metal soap, deodorants, antibacterial agents, pulp and heat.
- Plastic fibers or the like may be added to the water absorbent resin powder in an amount of 0 to 3% by weight, preferably 0 to 1% by weight.
- a preferable amount of the surfactant in the water-absorbent resin powder is within the above range.
- the present invention also provides an open-celled polyacrylic acid-based water-absorbing resin, particularly a polyacrylic acid-based water-absorbing resin having an open cell ratio of 5% or more, preferably 5 to 98%, more preferably 5 to 90%.
- the open cell rate of the “water absorbent resin” is defined by the open cell rate of the “foamed polymer (hydrous gel)” or “water absorbent resin (powder)”.
- the open cell ratio particularly preferable for solving the problem is preferably 5 to 30%, more preferably 5 to 15% in the case of a water absorbent resin (powder).
- a water-absorbent resin molding particularly a sheet-like molding
- it is preferably 5 to 98%, more preferably 10 to 90%.
- the open cell ratio of the foamed polymer (hydrous gel) before drying is preferably 5% or more, and more preferably 5 to 90%. Those containing predominantly open cells show little change in shape (ie, volume) after drying, while those containing many closed cells expand and increase in volume, and the shape also approaches a sphere.
- the content of closed cells in the water absorbent resin (powder) is not particularly limited, but is preferably 5% or more, more preferably 5 to 25%. More preferably, it is ⁇ 25%. Further, the closed cell ratio of the foamed polymer (hydrogel) before drying is preferably 0% or more, and more preferably 0 to 20%.
- the total of open cell rate and closed cell rate is preferably 10 to 90% in the case of water-absorbent resin powder. More preferably, it is 10 to 50%, particularly preferably 10 to 40%. In the case of a water-absorbent resin molded product (particularly a sheet-shaped molded product), it is preferably 10 to 98%, more preferably 20 to 90%, and particularly preferably 50 to 90%.
- sanitary materials especially paper diapers
- the production method of the present invention can be suitably applied to the following water-absorbent resin powder production method, but is preferably applicable to the control and improvement of the water absorption rate (FSR).
- FSR water absorption rate
- the physical properties of the following and examples are defined by the EDANA method unless otherwise specified.
- a polyacrylic acid-based water-absorbent resin powder having a water absorption rate index defined by the following formula of 90 or more and a bulk specific gravity of 0.3 to 0.8 [g / cm 3 ] is preferable.
- the polyacrylic acid water-absorbing resin powder having a water absorption rate index of 90 or more and a bulk specific gravity of 0.3 to 0.8 [g / cm 3 ] defined by the following formula may be surface-crosslinked. preferable.
- FSR indicates the water absorption rate at 20 times swelling in physiological saline.
- the water absorption rate index is preferably as high as 90, 95, 100, 105, 110, 115, 120 in order, and 150 or 140 is sufficient as the upper limit.
- a water-absorbent resin powder is excellent in liquid permeability and impact resistance and can be suitably used for absorbent articles such as paper diapers. When the water absorption rate index is low or too high, it tends to be unsuitable for actual use.
- Such a water-absorbent resin powder has a foam structure (also known as a porous structure), and the porous structure can be discriminated by confirming the particle surface with an electron micrograph.
- the average pore diameter on the particle surface is preferably 200 ⁇ m or less, more preferably 0.1 to 150 ⁇ m and 1 to 100 ⁇ m.
- the main component of each powder is porous particles.
- a complicated shape is included in addition to the above.
- AAP Water absorption capacity under pressure
- the water absorption capacity (AAP) with respect to a 0.9% by weight sodium chloride aqueous solution under a pressure of 2.06 kPa and further under a pressure of 4.83 kPa Is preferably 20 [g / g] or more, more preferably 22 [g / g] or more, and still more preferably 24 [g / g] or more.
- the upper limit value of AAP is not particularly limited, but is preferably 40 [g / g] or less in view of balance with other physical properties.
- the water absorption capacity (CRC) under no pressure is preferably 10 [g / g] or more, more preferably 20 [g / g] or more, still more preferably 25 [g / g] or more, particularly preferably 30 [g. / G] or more.
- the upper limit of CRC is preferably as high as possible, but the upper limit is not particularly limited. However, from the balance of other physical properties, it is preferably 50 [g / g] or less, more preferably 45 [g / g] or less, and still more preferably 40 [g / g]. It is as follows.
- the water-absorbing resin When the CRC is less than 10 [g / g], the water-absorbing resin has a low water absorption amount and may not be suitable for use in absorbent articles such as paper diapers. When the CRC exceeds 50 [g / g], if such a water-absorbent resin is used for the absorbent body, there is a possibility that a sanitary product having an excellent liquid uptake rate cannot be obtained.
- 0.69 wt% saline flow conductivity is 1 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g -1] or more, preferably 20 [ ⁇ 10 -7 ⁇ cm 3 ⁇ s ⁇ g -1] or more, more preferably 50 [ ⁇ 10 -7 ⁇ cm 3 ⁇ s ⁇ g - 1 ] or more, more preferably 70 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] or more, and particularly preferably 100 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] or more.
- the upper limit of SFC is not particularly limited, but is preferably 3000 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] or less, more preferably 2000 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] or less. preferable.
- SFC exceeds 3000 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] if such a water absorbent resin is used for the water absorbent, there is a possibility that liquid leakage occurs in the water absorbent.
- SFC is a well-known measurement method and can be defined, for example, in US Pat. No. 5,562,646.
- the present invention can be suitably applied to the improvement of liquid permeability, in particular, to a method for producing a highly liquid-permeable water-absorbent resin powder having SFC20 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] or more.
- the water-soluble content is preferably 0 to 35% by weight or less, more preferably 25% by weight or less, further preferably 15% by weight or less, and particularly preferably 10% by weight or less.
- the Ext exceeds 35% by weight, the gel strength of the obtained water-absorbent resin is weak and the liquid permeability may be inferior.
- a water absorbing resin with little liquid return (rewetting) when pressure is applied to the water absorbing body cannot be obtained.
- FSR Water absorption speed
- the water absorption rate (FSR) of 1 g of water-absorbent resin powder with respect to 20 g of physiological saline is usually 0.1 [g / g / s] or more, 0.15 [g / g / s] or more, and 0.20 [g. / G / s] or more, further 0.25 [g / g / s] or more, 0.35 [g / g / s] or more, or 0.45 [g / g / s] or more.
- the upper limit is 20 [g / g / s].
- the measurement method of FSR is defined in the following examples.
- the L value of the water absorbent resin is preferably 87 or more, and more preferably 90 or more. Since the water-absorbent resin which is one embodiment of the present invention has a high open cell ratio of 5% or more, the L value becomes high. Further, when the content of p-methoxyphenol is 60 ppm or less, the L value becomes higher.
- the bulk specific gravity of the water-absorbent resin powder is 0.3 to 0.8 [g / cm 3 ], preferably 0.4 to 0.7 [g / cm 3 ], more preferably Is 0.5 to 0.7 [g / cm 3 ].
- it has a foamed structure (other name; porous structure), and has a lower bulk specific gravity than ordinary non-foamed particles.
- the surface tension (specified by the measurement method in the examples) is preferably 50 [mN / m] or more, more preferably 55 [mN / m] or more, 60 [mN / m] or more, 65 [MN / m] or more, more preferably 70 [mN / m] or more, particularly 72 [mN / m] or more. If it is 72 [mN / m] or more, there is no substantial decrease in surface tension. The upper limit is usually 75 [mN / m].
- the water-absorbent resin may be a molded product or a non-molded product (powder), but in the case of powder, it is preferable to have a particle size distribution in the range of (2-5) above. .
- the physical properties are particularly preferably defined when the shape is powder.
- the use of the water-absorbent resin of the present invention is not particularly limited.
- the water-absorbent resin for agricultural and horticultural use, waste liquid solidification, industrial use, sanitary material use, etc.
- the water-absorbent resin is excellent in air permeability and water absorption speed and is white, it is preferably absorbent articles such as paper diapers, sanitary napkins, incontinence pads, more preferably absorbent articles for sanitary materials, particularly preferably paper diapers. Can be used.
- the water-absorbent resin of the present invention is a sheet, it can be an absorbent body (absorbing core) having shape retention as it is. Furthermore, even if it is powdery, there is no excessive decrease in bulk specific gravity, so it can be used for thin paper diapers.
- the content (core concentration) of the water-absorbent resin powder in the absorbent body optionally containing other absorbent materials (pulp fibers and the like) in this absorbent article is 30 to 100% by weight, preferably 40 to 100%.
- the effects of the present invention are exhibited by weight%, more preferably 50 to 100 weight%, further preferably 60 to 100 weight%, particularly preferably 70 to 100 weight%, and most preferably 75 to 95 weight%.
- each step in each example is carried out at a substantially normal pressure ( ⁇ 5% of atmospheric pressure, more preferably within 1%). It was carried out without any changes. Physical properties were measured at room temperature (20 to 25 ° C.) and relative humidity 40 to 50% RH unless otherwise specified.
- the sample volume (Va) [cm 3 ] of the cubic sample was measured using a dry density meter (manufactured by Shimadzu Corporation; Accupic II-1340). Note that the sample volume (Va) is obtained by subtracting the volume of open bubbles (including those in which closed cells became open bubbles at the time of preparing the cubic sample) from the geometric (circumferential shape) volume (va). Become.
- the cubic sample is further cut into fine pieces, and the geometric volume (v′a) [cm 3 ] and the surface area (s′a) [cm 2 ] are measured with a caliper. It was determined using the measured dimensions.
- the sample volume (V′a) [cm 3 ] was determined using a dry density meter.
- (va ⁇ Voa) and (v′a ⁇ Voa) are the open cell volume [cm 3 ], and (sa ⁇ Vca) and (s′a ⁇ Vca) are opened during the sample preparation process.
- the open (continuous) cell ratio (Voa ⁇ 100 [%]) of the foamed polymer can be obtained.
- the particulate water-absorbing resin is applied using JIS standard sieves (JIS Z8801-1 (2000)) having openings of 850 ⁇ m, 710 ⁇ m, 600 ⁇ m, 500 ⁇ m, 425 ⁇ m, 300 ⁇ m, 212 ⁇ m, 150 ⁇ m, and 45 ⁇ m. Sieving was performed, and fractions having a particle size of 500 ⁇ m or more and less than 600 ⁇ m and 300 ⁇ m or more and less than 425 ⁇ m were taken out.
- JIS standard sieves JIS Z8801-1 (2000)
- the geometrical (circumscribed shape) volume (vb, v′b) [cm 3 ] and surface area (sb, s′b) [cm 2 ] in the above two types of fractions are assumed to be spheres. And calculated. That is, a fraction having a particle diameter of 500 ⁇ m or more and less than 600 ⁇ m was assumed to be a sphere having a diameter of 550 ⁇ m, and a fraction having a particle diameter of 300 ⁇ m or more to less than 425 ⁇ m was assumed to be a sphere having a diameter of 362.5 ⁇ m.
- sample volume (Vb, V′b) [cm 3 ] per one particle of each fraction was measured using a dry densitometer as in the case of the foamed polymer. That is, by measuring the weight of each whole fraction and the weight of a known number (for example, 100 particles), the weight per particle can be obtained, and the number of particles in each fraction can be grasped. Then, the sample volume (Vb, V′b) [cm 3 ] per grain is determined from the value obtained by the dry densitometer.
- (vb ⁇ Vob) and (v′b ⁇ Vob) are the open cell volume [cm 3 ], and (sb ⁇ Vcb) and (s′b ⁇ Vcb) are opened in the sample preparation process. (Assumed) (independent) bubble volume [cm 3 ].
- the moisture content ⁇ a [wt%] of the cubic sample was determined by the method disclosed in (5-5). Subsequently, the dried cubic sample after obtaining the water content is finely pulverized to 45 ⁇ m or less by the method disclosed in (5-13) to obtain the true density [g / cm 3 of the water-absorbent resin part. ] was obtained (see FIG. 8). Furthermore, from the specific gravity of water (1.00 [g / cm 3 ]), the true density Da [g / cm 3 ] of the cubic sample was calculated according to the following formula.
- Vga geometrical (circumferential shape) volume [cm 3 ], weight Wa [g], true density Da [g / cm 3 ] of the above cubic sample and foaming determined in the above (5-1).
- the closed cell rate (Vc [%]) of the foamed polymer was determined according to the following formula.
- the particulate water-absorbing resin is applied using JIS standard sieves (JIS Z8801-1 (2000)) having openings of 850 ⁇ m, 710 ⁇ m, 600 ⁇ m, 500 ⁇ m, 425 ⁇ m, 300 ⁇ m, 212 ⁇ m, 150 ⁇ m, and 45 ⁇ m. Sieving was performed, and fractions having a particle size of 500 ⁇ m or more and less than 600 ⁇ m and less than 45 ⁇ m were taken out.
- JIS standard sieves JIS Z8801-1 (2000)
- the density [g / cm 3 ] is obtained from the total weight [g] and volume [cm 3 ], and this is defined as the true density of the water absorbent resin portion of the water absorbent resin (powder).
- the true density Db [g / cm 3 ] of the water absorbent resin (powder) was calculated from the water content ⁇ b [wt%] by the method described in (5-5) according to the following formula.
- Vgb is the volume of a sphere having a diameter of 550 ⁇ m
- Wb [g] the true density Db [g / cm 3 ]
- release of the water absorbent resin (powder) obtained in (5-1) above
- the closed cell rate (Vd [%]) of the water-absorbent resin (powder) was determined from the continuous cell rate (Vob [%]) according to the following formula.
- Weight average particle diameter (D50) and logarithmic standard deviation of particle size distribution ( ⁇ ) were measured according to the following procedure.
- solid content can be calculated
- the water-absorbing resin (powder) is put into a cylindrical glass container (diameter 32 to 34 mm, height 50 mm) with an open top so that the upper surface of the water-absorbing resin (powder) is horizontal. did. At this time, if necessary, the upper surface of the water-absorbent resin (powder) may be leveled by carefully hitting the bottom of the glass container.
- a glass beaker containing the physiological saline whose surface tension was measured was charged with a thoroughly washed 25 mm long fluororesin rotor and 0.5 g of a water absorbent resin (powder), and at 500 rpm for 4 minutes. Stir. After 4 minutes, stirring was stopped and the water-absorbing water-absorbing resin (powder) was allowed to settle. Thereafter, the surface tension of the supernatant was measured by performing the same operation as described above.
- the platinum plate method was adopted, and the platinum plate was thoroughly washed with deionized water before each measurement and heated and washed with a gas burner.
- Liquid permeability (SFC) is a well-known measurement method, and was measured according to the method disclosed in US Pat. No. 5,562,646.
- a water-absorbing resin Powder
- a powder / paste sample container having an inner diameter of 30 mm and a height of 12 mm
- L-value Lightness: Brightness index
- This value is evaluated as “lightness index (initial)”, and the larger this value is, the whiter the color becomes.
- the a value and b value were also measured. The smaller the a and b values, the lower the color and the closer the color becomes substantially white.
- reflection measurement was selected as the measurement condition, and a standard round white plate for powder / paste was used as a standard. 2 and a 30 ⁇ light projecting pipe was used.
- the color tone of the water-absorbent resin (powder) immediately after production or the water-absorbent resin (powder) within a storage period of 1 year was evaluated as an initial color tone under conditions of 30 ° C. or less and a relative humidity of 50% RH or less.
- the sample container for powder / paste is filled with about 5 g of water-absorbing resin (powder), it is adjusted to an atmosphere of 70 ⁇ 1 ° C. and relative humidity 65 ⁇ 1% RH (manufactured by ESPEC Corporation). ; Left in a small environmental tester (model SH-641) for 7 days (coloring acceleration test). Thereafter, the L value, a value, and b value of the surface of the water-absorbent resin (powder) were measured as the color tone after time-dependent coloring (time-dependent color tone) using the spectral color difference meter.
- Methoxyphenols in water-absorbent resin (powder) were measured by the following method.
- the filtrate obtained by the above operation is analyzed by high performance liquid chromatography under the following conditions to quantify methoxyphenols (unit: ppm (vs. water absorbent resin (powder))) in the water absorbent resin (powder). be able to.
- the chelating agent (unit: ppm (vs. water-absorbing resin (powder))) in the water-absorbing resin (powder) can be quantified by analyzing the filtrate obtained by the above operation by high performance liquid chromatography.
- water absorbent resin (powder) 15 0.0 g and 400 g of cylindrical magnetic balls (diameter 13 mm, length 13 mm) were placed and pulverized at 60 Hz for 2 hours using a ball mill, and more than 70% by weight passed through a JIS standard sieve with an opening of 45 ⁇ m.
- a powder was obtained.
- the fine powder pulverized to 45 ⁇ m or less was measured using a dry density meter (manufactured by Shimadzu Corporation; Accupic II-1340), and the obtained value was defined as the true density of the water absorbent resin (powder).
- Example 1 224.0 g of acrylic acid having a p-methoxyphenol content adjusted to 70 ppm, 0.48 g of polyethylene glycol diacrylate (number average molecular weight 522) and 2-hydroxy-2-methyl-1-phenyl-propan-1-one Solution (A) in which 13 g was mixed, 153.8 g of 48.5 wt% sodium hydroxide aqueous solution was diluted with 113.2 g of ion-exchanged water, and 0.03 g of diethylenetriaminepentaacetic acid / pentasodium was added as a chelating agent.
- the solution (A) was stirred with a magnetic stirrer, and the solution (B) was mixed while removing heat in an open system, and a monomer aqueous solution (1 ′) at 45 ° C. was added. Obtained.
- the foamed polymer (1) was taken out of the stainless steel bat-shaped container, and the open cell rate and the closed cell rate were measured and calculated.
- the open cell rate was 14% and the closed cell rate was Was 16%.
- the foamed polymer (1) had innumerable fine bubbles, and white foamed bread-like foam gel was formed by the bubbles.
- the foamed polymer (1) was cut into a cube of 5 mm on a side with a knife, hot air temperature 180 ° C., wind speed 2.0 [m / S] with a hot air dryer for 30 minutes to obtain a dried water-absorbent resin (1 ′).
- the foamed polymer (1) obtained by the above operation was cut into a cube of 2 mm on a side with a knife, and heated with a hot air dryer at a hot air temperature of 180 ° C. and a wind speed of 2.0 [m / s]. Drying for 3 minutes gave a cubic water-absorbent resin dried product (1).
- the dried water absorbent resin (1) is further pulverized with a roll mill and then classified with a JIS standard sieve having openings of 600 ⁇ m and 300 ⁇ m, whereby a water absorbent resin powder (1) having a weight average particle diameter (D50) of 460 ⁇ m.
- Table 1 shows properties of the water absorbent resin powder (1) obtained.
- the open cell ratio and closed cell ratio of the water absorbent resin powder (1) are separately applied to the dried water absorbent resin (1) by the method described in (5-1) and (5-2) above. (Measured in the same manner in the following Examples and Comparative Examples). As a result, the open cell rate was 6% and the closed cell rate was 16%.
- Example 2 (Foam control step added) The same operation as in Example 1 was performed to obtain a monomer aqueous solution (2) in which nitrogen gas bubbles were dispersed (monomer concentration: 53.2% by weight, volume expansion ratio: 1.4 times). .
- the monomer aqueous solution (2) was circulated while continuously mixing nitrogen gas, and the same operation as in Example 1 was performed except that foam regulation was performed (see FIG. 1). Combined (2) was obtained.
- the maximum temperature reached 107 ° C. was recorded about 40 seconds after the start of the polymerization.
- the foamed polymer (2) had an open cell ratio of 12% and a closed cell ratio of 14%. Further, the foamed polymer (2) had innumerable fine bubbles, and white foamed bread-like foam gel was formed by the bubbles.
- the foamed polymer (2) was cut into a 5 mm side cube with a knife and dried with hot air in the same manner as in Example 1 to absorb water.
- Product dried resin (2 ′) was obtained.
- the foamed polymer (2) (5 mm piece) easily evaporates from water from the open cells, so that gel expansion due to drying is not observed, and substantial deformation does not occur except for some drying shrinkage. There wasn't.
- SEM scanning electron microscope
- the foamed polymer (2) obtained by the above operation is cut into a cube having a side of 2 mm with a knife and dried with hot air in the same manner as in Example 1 to obtain a dried cubic water-absorbent resin. (2) was obtained.
- Example 2 Thereafter, the dried water absorbent resin (2) was further pulverized and classified in the same manner as in Example 1 to obtain a water absorbent resin powder (2) having a weight average particle diameter (D50) of 460 ⁇ m.
- Table 1 shows properties of the water absorbent resin powder (2) obtained.
- the open cell ratio of the water absorbent resin powder (2) was 6%, and the closed cell ratio was 13%.
- Example 3 (Volume expansion ratio 1.3 times)
- the amount of 30% by weight polyoxyethylene sorbitan monostearate aqueous solution (manufactured by Kao Corporation) was used from 4.4 g (Example 1) to 0.80 g (content per monomer was 0.00). 09% by weight), and at the same time 2.65 g of hydroxyethyl cellulose (Wako First Grade / manufactured by Wako Pure Chemical Industries, Ltd.) was added, and the monomer aqueous solution (3) (monomer concentration: 53.5% by weight, volume)
- a foamed polymer (3) was obtained in the same manner as in Example 1 except that the expansion ratio was 1.3 times.
- the maximum temperature reached 107 ° C.
- the foamed polymer (3) had an open cell ratio of 10% and a closed cell ratio of 11%. Further, the foamed polymer (3) had innumerable fine bubbles, and white foamed bread-like foam gel was formed by the bubbles.
- the foamed polymer (3) was cut into a cube with a side of 5 mm with a knife, followed by hot air drying similar to Example 1, A water absorbent resin dried product (3 ′) was obtained.
- the foamed polymer (3) obtained by the above operation was cut into a 2 mm side cube with a knife and dried with hot air in the same manner as in Example 1 to obtain a dried cubic water absorbent resin. (3) was obtained.
- Example 2 Thereafter, the dried water absorbent resin (3) was further pulverized and classified in the same manner as in Example 1 to obtain a water absorbent resin powder (3) having a weight average particle diameter (D50) of 430 ⁇ m.
- Table 1 shows properties of the water absorbent resin powder (3) obtained.
- the open cell ratio of the water absorbent resin powder (3) was 5%, and the closed cell ratio was 10%.
- surface crosslinking comprising 0.48 parts by weight of 1,4-butanediol, 0.75 parts by weight of propylene glycol and 4.0 parts by weight of deionized water with respect to 100 parts by weight of the water absorbent resin powder (3).
- the agent solution was uniformly sprayed and mixed. Thereafter, heat treatment was performed at a temperature of 180 ° C. for 45 minutes, and further passed through a JIS standard sieve having an opening of 600 ⁇ m, whereby surface-crosslinked water-absorbing resin particles (3) were obtained.
- the water-absorbent resin particles mean a water-absorbent resin powder that has been surface-crosslinked.
- the obtained water-absorbent resin (3a) has a CRC of 26.5 [g / g], an FSR of 0.45 [g / g / s], and an SFC of 117 [ ⁇ 10 ⁇ 7 ⁇ s ⁇ cm 3 ⁇ g ⁇ 1 ]. Further, the open cell rate of the water absorbent resin (3a) was 5%, and the closed cell rate was 10%, and no change in the bubble rate due to the surface treatment was observed.
- Example 4 (Volume expansion ratio 1.2 times)
- Example 1 instead of using a 30% by weight polyoxyethylene sorbitan monostearate aqueous solution (manufactured by Kao Corporation), instead of 5.30 g of hydroxyethyl cellulose (manufactured by Wako First Grade / manufactured by Wako Pure Chemical Industries, Ltd.)
- the same procedure as in Example 1 was performed except that the monomer aqueous solution (4) (monomer concentration: 53.5 wt%, volume expansion ratio: 1.2 times) was used. 4) was obtained.
- the maximum temperature reached 107 ° C. was recorded about 40 seconds after the start of the polymerization.
- the foamed polymer (4) had an open cell ratio of 7% and a closed cell ratio of 9%. Further, the foamed polymer (4) had innumerable fine bubbles, and white foamed bread-like foam gel was formed by the bubbles.
- the foamed polymer (4) was cut into a cube having a side of 5 mm with a knife, followed by hot air drying as in Example 1, A water absorbent resin dried product (4 ′) was obtained.
- the foamed polymer (4) obtained by the above operation was cut into a 2 mm cube with a knife and dried with hot air in the same manner as in Example 1 to obtain a dried cubic water absorbent resin. (4) was obtained.
- Example 2 Thereafter, the dried water absorbent resin (4) was further pulverized and classified in the same manner as in Example 1 to obtain a water absorbent resin powder (4) having a weight average particle diameter (D50) of 440 ⁇ m.
- Table 1 shows properties of the water absorbent resin powder (4) obtained.
- the open cell ratio of the water absorbent resin powder (4) was 5%, and the closed cell ratio was 9%.
- Example 5 (monomer concentration: 42.6% by weight)
- a solution (C) obtained by mixing 177.50 g of acrylic acid, 0.77 g of polyethylene glycol diacrylate (number average molecular weight 522) and 0.11 g of 2-hydroxy-2-methyl-1-phenyl-propan-1-one, 48. After diluting 121.89 g of a 5 wt% aqueous sodium hydroxide solution with 180.30 g of ion-exchanged water and further adding 0.02 g of diethylenetriaminepentaacetic acid and 0.02 g of sodium, each solution (C) was prepared. The solution (D) was mixed while removing heat in an open system while stirring with a magnetic stirrer to obtain a monomer aqueous solution (5 ′) at 45 ° C.
- aqueous polyoxyethylene sorbitan monostearate manufactured by Kao Corporation
- hydroxyethyl cellulose manufactured by Wako First Grade / manufactured by Wako Pure Chemical Industries, Ltd.
- the volume of 400 g of the obtained monomer aqueous solution (5) was 345 ml. Thereafter, the mixture was further deaerated with nitrogen gas for 20 minutes.
- Example 1 400 g of the monomer aqueous solution (5) that passed through the whip auto was put into the stainless steel bat-type container used in Example 1 that had been heated to 90 ° C. with a hot plate in an open air system. Immediately thereafter, UV irradiation with a black light mercury lamp was performed in the same manner as in Example 1 to initiate the polymerization reaction.
- the foamed polymer (5) was taken out from the stainless steel bat-shaped container, and the open cell rate and the closed cell rate were measured and calculated.
- the open cell rate was 87% and the closed cell rate was Was 0%.
- the foamed polymer (5) was cut into a cube of 5 mm on a side with a knife, and hot air drying similar to that in Example 1 was performed. A water absorbent resin dried product (5 ′) was obtained.
- Solution (E) prepared by mixing 150.5 g of acrylic acid, 1.86 g of trimethylolpropane triacrylate and 0.09 g of 2-hydroxy-2-methyl-1-phenyl-propan-1-one, 48.5% by weight of water After diluting 129.2 g of an aqueous sodium oxide solution with 205.1 g of ion-exchanged water and adding 0.02 g of diethylenetriaminepentaacetic acid / 0.05 g of diethylenetriamine, each solution (E) was prepared with a magnetic stirrer. Under stirring, the solution (F) was mixed while removing heat in an open system to obtain a comparative monomer aqueous solution (1 ′) at 30 ° C.
- Example 1 100 g of the comparative monomer aqueous solution (1) that passed through the whip auto was introduced into the stainless steel bat-type container that had been heated to 90 ° C. with a hot plate in Example 1 in an open air system. did. Immediately thereafter, UV irradiation with a black light mercury lamp was performed in the same manner as in Example 1 to initiate the polymerization reaction.
- the comparative polymer (1) was taken out from the stainless steel bat-shaped container.
- the foam did not become a white foam gel by air bubbles as in Examples 1 to 5, but Since it did not contain, it was an almost transparent gel. From the record of the temperature change of the polymerization system, the maximum temperature reached during polymerization was 97 ° C. (about 100 seconds after the start of polymerization).
- the comparative polymer (1) was cut into a cube with a side of 5 mm with a knife, followed by hot air drying similar to Example 1, A comparative water absorbent resin dried product (1 ′) was obtained.
- Example 2 Volume expansion ratio 1.0 times
- a 30% by weight polyoxyethylene sorbitan monostearate aqueous solution manufactured by Kao Corporation
- a comparative monomer aqueous solution (2) (monomer concentration; 53.5% by weight, volume)
- a comparative polymer (2) was obtained in the same manner as in Example 1 except that the expansion ratio was 1.0.
- the highest temperature reached during the polymerization was 107 ° C. (about 40 seconds after the start of the polymerization) from the record of the temperature change of the polymerization system.
- the comparative polymer (2) was polymerized in the same manner as in Example 1. However, the comparative polymer (2) was not a white foam-like gel due to air bubbles, and was an almost transparent gel because it did not contain air bubbles.
- the comparative polymer (2) was cut into a cube with a side of 5 mm with a knife and dried with hot air as in Example 1.
- a comparative water absorbent resin dried product (2 ′) was obtained.
- the comparative polymer (2) obtained by the above operation was cut into a 2 mm side cube with a knife and dried with hot air in the same manner as in Example 1 to dry the cubic comparative water absorbent resin. A product (2) was obtained.
- the comparative water absorbent resin dried product (2) was further pulverized and classified in the same manner as in Example 1 to obtain a comparative water absorbent resin powder (2) having a weight average particle diameter (D50) of 470 ⁇ m.
- Table 1 shows properties of the comparative water absorbent resin powder (2) obtained.
- the comparative water absorbent resin powder (2) had an open cell ratio of 1.5% and a closed cell ratio of 4%.
- Example 3 the same surface treatment as in Example 3 was performed on the comparative water absorbent resin powder (2) to obtain a comparative water absorbent resin (2).
- the comparative water absorbent resin (2) obtained has a CRC of 26.3 [g / g], an FSR of 0.18 [g / g / s], and an SFC of 135 [ ⁇ 10 ⁇ 7 ⁇ s ⁇ cm 3 ⁇ g. ⁇ 1 ]. Further, the open cell ratio of the comparative water absorbent resin (2) was 1.3%, and the closed cell ratio was 4.2%.
- Example 3 (Volume expansion ratio 1.0 times)
- the comparative monomer aqueous solution (3) (monomer concentration: 53.2% by weight, volume expansion ratio) ; Comparative Example (3) was obtained in the same manner as in Example 1, except that the ratio was 1.0).
- the highest temperature reached during the polymerization was 107 ° C. (about 40 seconds after the start of the polymerization) from the record of the temperature change of the polymerization system.
- the comparative polymer (3) was polymerized in the same manner as in Example 1. However, the comparative polymer (3) was not a white foam gel due to air bubbles, and was almost transparent because it did not contain air bubbles.
- the comparative polymer (3) was cut into a cube having a side of 5 mm with a knife, followed by hot air drying similar to Example 1, A comparative water absorbent resin dried product (3 ′) was obtained.
- the comparative polymer (3) (5 mm piece) does not contain open cells, it is difficult to evaporate water vapor from the inside, and gel expansion is confirmed by drying, and about 1 to several cm after drying. Deformation occurred in a balloon shape.
- SEM scanning electron microscope
- Example 4 In Example 1, a comparative monomer aqueous solution (4) was added using Whip Auto (trade name) without adding 3.0% by weight of 2,2′-azobis (2-methylpropionamidine) dihydrochloride aqueous solution. And nitrogen gas were fluidly mixed, and bubbles of nitrogen gas were dispersed in a comparative monomer aqueous solution (4) (monomer concentration: 53.6% by weight, volume expansion ratio: 1.4 times).
- Example 1 400 g of the comparative monomer aqueous solution (4) that passed through the whip auto was charged into the stainless steel bat-type container used in Example 1 in an open air system. Immediately thereafter, UV irradiation was performed with a black light mercury lamp in the same manner as in Example 1 to initiate the polymerization reaction.
- the polymerization is completed by repeating the operation of stopping UV irradiation when the monomer temperature rises to 80 ° C. and restarting UV irradiation when the monomer temperature drops to 60 ° C. 4) was obtained.
- the comparative polymer (4) was cut into a cube with a side of 5 mm with a knife, followed by hot air drying similar to that in Example 1, A comparative water absorbent resin dried product (4 ′) was obtained.
- the comparative polymer (4) obtained by the above operation was cut into a 2 mm cube with a knife and dried with hot air in the same manner as in Example 1 to dry the cubic comparative water absorbent resin. A product (4) was obtained.
- the comparative water absorbent resin dried product (4) was further pulverized and classified in the same manner as in Example 1 to obtain a comparative water absorbent resin powder (4) having a weight average particle diameter (D50) of 470 ⁇ m.
- Table 1 shows properties of the comparative water absorbent resin powder (4) obtained.
- the comparative water absorbent resin powder (4) had an open cell ratio of 2% and a closed cell ratio of 5%.
- the desktop whipped cream machine “Whipped Auto (product) Name) ” is used to fluidly mix the comparative monomer aqueous solution (5) and nitrogen gas, and nitrogen gas bubbles are generated in the comparative monomer aqueous solution (5) (monomer concentration: 35.2 wt%). Dispersed.
- the volume of 400 g of the comparative monomer aqueous solution (5) in which the nitrogen gas is dispersed is 500 ml
- Example 1 400 g of the comparative monomer aqueous solution (5) that passed through the whip auto was put into a stainless steel bat-type container that had been heated to 90 ° C. with a hot plate in Example 1 in an open air system. did. Immediately thereafter, UV irradiation with a black light mercury lamp was performed in the same manner as in Example 1 to initiate the polymerization reaction. The temperature of the comparative monomer aqueous solution (5) at the start of the polymerization was 30 ° C.
- the comparative polymer (5) was taken out from the stainless steel bat-shaped container.
- the foam did not become a white foam-like gel with bubbles as in Examples 1-5. Since it did not contain, it was an almost transparent gel. From the record of the temperature change of the polymerization system, the maximum temperature reached during polymerization was 92 ° C. (about 120 seconds after the start of polymerization).
- the comparative polymer (5) was cut into a cube having a side of 5 mm with a knife, followed by hot air drying as in Example 1, A comparative water absorbent resin dried product (5 ′) was obtained.
- Example 2 using the desktop whipped cream machine “Whipped Auto (trade name)” in the same manner as in Example 1, the comparative monomer aqueous solution (6) and nitrogen gas were fluidly mixed to prepare the comparative monomer aqueous solution (6). Nitrogen gas bubbles were dispersed in the sample. The volume expansion ratio of the comparative monomer aqueous solution (6) after passing through Whip Auto (trade name) was 1.2 times.
- the polymerization form is a static polymerization for 1 hour at a temperature of 25 to 95 ° C. (maximum temperature 95 ° C. (15 minutes after the start of polymerization), polymerization starts 7 minutes after supplying the polymerization apparatus), A sponge-like comparative polymer (6) containing was obtained.
- the comparative polymer (6) was cut into a cube having a side of 5 mm with a knife, followed by hot air drying as in Example 1, A comparative water absorbent resin dried product (6 ′) was obtained.
- the comparative polymer (6) (5 mm piece) does not contain open cells, it is difficult to evaporate water vapor from the inside, and gel expansion is confirmed by drying, and about 1 to several cm after drying. The balloon was deformed.
- the obtained comparative water absorbent resin dried product (6 ′) was cut and an SEM (scanning electron microscope) photograph image of the fractured surface was observed, no open cells were observed and there were many closed cells. confirmed.
- Patent Document 20 Polymerization was carried out according to Example 2 of Patent Document 20 (US Pat. No. 6,136,873).
- Patent Document 20 has a description that “the polymerization temperature is preferably adjusted so as to avoid boiling of the polymerizable aqueous mixture”.
- the comparative monomer aqueous solution (7) was put into a polypropylene container (size: 15 cm ⁇ 19 cm ⁇ 18 cm), but the defoaming speed was high and the comparative monomer aqueous solution (7 ) Volume was almost halved. Although microwave irradiation was continued, it was confirmed that defoaming further progressed. In Comparative Example 7, the polymerization temperature was adjusted so as to avoid boiling of the comparative monomer aqueous solution (7).
- the comparative polymer (7) was taken out from the container, but it was not a foam polymer as defined in the present invention.
- the comparative monomer dispersion (8) was a slurry stock solution having a neutralization rate of 75 mol% and a monomer concentration of 54.9 wt%.
- the comparative monomer dispersion (8) was cast into a layer having a thickness of about 2 cm in a polymerization vessel purged with nitrogen, and heated from the bottom. Polymerization started immediately after heating, and the polymerization was accompanied by uniform foaming, and a foamed rubber plate polymer having a thickness of about 5 cm was obtained in 20 minutes.
- the polymer obtained by the above operation was not sticky, and was cut into a sheet by cutting to a thickness of 5 mm using a slicer. Furthermore, after cutting into 5 mm square and shape
- Comparative Example 1 (monomer concentration: 37.3% by weight), Comparative Example 5 (monomer concentration: 35.2% by weight), Comparative Example 2 and Comparative Example 3 (volume expansion ratio: 1.0 times) ), Comparative Example 4 (temperature during polymerization; 60 to 80 ° C.), Comparative Example 8 (slurry / non-aqueous solution), etc., all three constituent requirements of the present invention are not satisfied, and open cells are not formed. Alternatively, it can be seen that the open cell rate is less than 5%, and a high water absorption rate cannot be achieved.
- boiling polymerization which has been conventionally avoided in the method for producing a (continuous) foam-like water-absorbing resin, has a monomer concentration of 40% by weight or more and a volume expansion ratio of 1.1 times or more. It has been found to provide open cells efficiently.
- Example 6 (monomer concentration 45.4% by weight, MQ 70 ppm) 50.
- Solution (I) obtained by mixing 190.18 g of acrylic acid, 0.83 g of polyethylene glycol diacrylate (number average molecular weight 522) and 0.11 g of 2-hydroxy-2-methyl-1-phenyl-propan-1-one
- a solution (J) was prepared by diluting 130.60 g of a 5% by weight aqueous sodium hydroxide solution with 163.46 g of ion-exchanged water, and further adding 0.02 g of diethylenetriaminepentaacetic acid and 5 sodium salt.
- the solution (J) was mixed while removing heat in an open system while stirring with a magnetic stirrer to obtain a monomer aqueous solution (6 ′) at 45 ° C.
- Acrylic acid having a p-methoxyphenol content (hereinafter sometimes simply referred to as “MQ”) adjusted to 70 ppm was used.
- the foamed polymer (6) which is a white foam gel obtained in the boiling polymerization, slightly expanded in volume as in Example 1, but there was almost no volume change.
- the temperature of the aqueous monomer solution (6) at the start of polymerization was 42 ° C. due to heating from a hot plate, etc., but then the temperature rapidly increased with the progress of polymerization, and about 50 seconds after the start of polymerization. Recorded a maximum temperature of 102 ° C.
- the foamed polymer (6) was taken out from the stainless steel bad-type container, and the open cell rate and the closed cell rate were measured and calculated.
- the open cell rate was 53% and the closed cell rate was Was 8%.
- the foamed polymer (6) had innumerable fine bubbles, and white foamed bread-like foam gel was formed by the bubbles.
- Example 2 the foamed polymer (6) was cut into a cube of 5 mm on a side with a knife, and hot air drying similar to that in Example 1 was performed. A water absorbent resin dried product (6 ′) was obtained.
- the foamed polymer (6) obtained by the above operation was cut into a 2 mm side cube with a knife and dried with hot air in the same manner as in Example 1 to obtain a dried cubic water absorbent resin. (6) was obtained.
- Example 2 shows properties of the water absorbent resin powder (6).
- the open cell ratio of the water absorbent resin powder (6) was 15%, and the closed cell ratio was 24%. Further, the p-methoxyphenol content in the water absorbent resin powder (6) was 20 ppm.
- Example 7 (MQ 120 ppm) A foamed polymer (7) was obtained in the same manner as in Example 6, except that acrylic acid having a p-methoxyphenol (MQ) content adjusted to 120 ppm was used. Further, the foamed polymer (7) was subjected to the same operation as in Example 6 to obtain a water absorbent resin powder (7) having a weight average particle diameter (D50) of 460 ⁇ m. Table 2 shows properties of the water absorbent resin powder (7). The open cell ratio of the water absorbent resin powder (7) was 13%, and the closed cell ratio was 23%. Furthermore, the content of p-methoxyphenol in the water absorbent resin powder (7) was 35 ppm.
- Example 8 (MQ 200 ppm) A foamed polymer (8) was obtained in the same manner as in Example 6, except that acrylic acid having a p-methoxyphenol (MQ) content adjusted to 200 ppm was used. Further, the foamed polymer (8) was subjected to the same operation as in Example 6 to obtain a water absorbent resin powder (8) having a weight average particle diameter (D50) of 450 ⁇ m. Table 2 shows properties of the water absorbent resin powder (8) obtained. The open cell ratio of the water absorbent resin powder (8) was 13%, and the closed cell ratio was 21%. Furthermore, the content of p-methoxyphenol in the water absorbent resin powder (8) was 56 ppm.
- Example 9 (MQ 250 ppm) A foamed polymer (9) was obtained in the same manner as in Example 6, except that acrylic acid having a p-methoxyphenol (MQ) content adjusted to 250 ppm was used. Further, the foamed polymer (9) was subjected to the same operation as in Example 6 to obtain a water absorbent resin powder (9) having a weight average particle diameter (D50) of 460 ⁇ m. Table 2 shows properties of the water absorbent resin powder (9) obtained. The open cell ratio of the water absorbent resin powder (9) was 14%, and the closed cell ratio was 22%. Further, the content of p-methoxyphenol in the comparative water absorbent resin powder (9) was 67 ppm.
- Example 9 (Volume expansion ratio 1.0 times, MQ 70 ppm)
- a comparative monomer aqueous solution ( 9) A comparative polymer (9), which is a transparent gel, was obtained in the same manner as in Example 6 except that the volume expansion ratio was 1.0. Further, the comparative polymer (9) was subjected to the same operation as in Example 6 to obtain a comparative water absorbent resin powder (9) having a weight average particle diameter (D50) of 470 ⁇ m.
- Table 2 shows properties of the comparative water absorbent resin powder (9).
- the comparative water absorbent resin powder (9) had an open cell ratio of 1.3% and a closed cell ratio of 3%. Further, the content of p-methoxyphenol in the comparative water absorbent resin powder (10) was 20 ppm.
- Comparative Example 10 (Volume expansion ratio 1.0 times, MQ 120 ppm) A comparative water-absorbent resin having a weight average particle diameter (D50) of 470 ⁇ m was obtained in the same manner as in Comparative Example 9, except that acrylic acid having a p-methoxyphenol content adjusted to 120 ppm was used. A powder (10) was obtained. Table 2 shows properties of the comparative water absorbent resin powder (10). In addition, the comparative water absorbent resin powder (10) had an open cell ratio of 1.5% and a closed cell ratio of 3.5%. Further, the content of p-methoxyphenol in the comparative water absorbent resin powder (10) was 36 ppm.
- Comparative Example 11 (Volume expansion ratio 1.0 times, MQ 200 ppm) A comparative water absorbent resin having a weight average particle diameter (D50) of 480 ⁇ m was obtained by performing the same operation as in Comparative Example 9, except that acrylic acid having a p-methoxyphenol content adjusted to 200 ppm was used in Comparative Example 9. A powder (11) was obtained. Table 2 shows properties of the comparative water absorbent resin powder (11) obtained. The comparative water absorbent resin powder (11) had an open cell ratio of 2.1% and a closed cell ratio of 3.1%. Further, the content of p-methoxyphenol in the comparative water absorbent resin powder (11) was 55 ppm.
- Comparative Example 12 (Volume expansion ratio 1.0 times, MQ 250 ppm) A comparative water absorbent resin having a weight average particle diameter (D50) of 460 ⁇ m was obtained by performing the same operation as in Comparative Example 9 except that acrylic acid having a p-methoxyphenol content adjusted to 250 ppm was used in Comparative Example 9. A powder (12) was obtained. Table 2 shows properties of the comparative water absorbent resin powder (12). The comparative water absorbent resin powder (12) had an open cell ratio of 1.8% and a closed cell ratio of 2.8%. Further, the content of p-methoxyphenol in the comparative water absorbent resin powder (12) was 68 ppm.
- FSR water absorption rate
- CRC water absorption rate
- the initial color tone is also improved (L value is large and a value / b value is small).
- drying rate of the hydrogel is improved (drying time is shortened) because the solid content (% by weight) is high in the method of the present invention.
- the conventional water absorption speed improving method such as Patent Document 20 does not disclose the prevention of coloring, and the water absorption speed (FSR) of the present invention is improved in each stage as compared with the conventional coloring prevention method described in Patent Document 29 or the like. To do.
- Example 13 (Monomer concentration: 37.3% by weight)
- the same operation as in Example 6 was performed except that the ion exchange water used for the preparation of the monomer aqueous solution was changed to 271.60 g, so that the monomer concentration was 37.3% by weight.
- a comparative polymer (13) was obtained.
- the maximum temperature reached during the polymerization was 97 ° C. (about 100 seconds after the start of the polymerization) from the record of the temperature change of the polymerization system.
- Example 6 the comparative polymer (13) obtained by the above operation was dried, ground and classified in the same manner as in Example 6 to obtain a comparative water absorbent resin powder (13).
- Table 3 shows the physical properties of the comparative water absorbent resin powder (13).
- Example 10 (monomer concentration 50.5% by weight)
- Example 6 the same operation as in Example 6 was performed except that the monomer concentration was changed to 50.5% by weight by changing the ion exchange water used for the preparation of the aqueous monomer solution to 113.49 g.
- a foamed polymer (10) was obtained.
- the maximum temperature reached during the polymerization was 105 ° C. (about 45 seconds after the start of the polymerization) from the record of the temperature change of the polymerization system.
- Example 6 the foamed polymer (10) obtained by the above operation was dried, ground and classified in the same manner as in Example 6 to obtain a water absorbent resin powder (10).
- Table 3 shows the physical properties of the water absorbent resin powder (10) obtained.
- Example 14 (Maximum temperature 98 ° C)
- the maximum temperature reached during polymerization was controlled at 98 ° C. (about 50 seconds after the start of polymerization) by enhancing the cooling after the start of polymerization, to obtain a comparative polymer (14).
- Example 6 the comparative polymer (14) obtained by the above operation was dried, ground and classified in the same manner as in Example 6 to obtain a comparative water absorbent resin powder (14).
- Table 3 shows the physical properties of the comparative water absorbent resin powder (14).
- Example 15 (Volume expansion ratio 1.04)
- the monomer aqueous solution (6) having a volume expansion ratio of 2.5 times was left as it was for 10 minutes to obtain a volume expansion ratio of 1.04, and then the same polymerization as in Example 6 was performed.
- a comparative polymer (15) was obtained.
- the maximum temperature reached during the polymerization was 102 ° C. (about 50 seconds after the start of the polymerization) from the record of the temperature change of the polymerization system.
- Example 6 the comparative polymer (15) obtained by the above operation was dried, ground and classified in the same manner as in Example 6 to obtain a comparative water absorbent resin powder (15).
- Table 3 shows the physical properties of the comparative water absorbent resin powder (15).
- Example 11 (Drying experiment) The foamed polymer (6), which is the white foam gel obtained in Example 6, was cut to 2 to 3 mm and placed on a punching plate, and hot air having a temperature of 180 ° C. and a dew point of 5 ° C. was applied at a wind speed of 1.6 [m / S], air was passed upward from the bottom of the punching plate. At this time, when the drying rate was measured, the drying time up to 95% by weight or more of the solid content was 20 minutes, and it was found that the drying rate was fast in the method of the present invention.
- Comparative Example 16 (Drying Experiment) The comparative polymer (9), which was a transparent gel obtained in Comparative Example 9, was cut into 2 to 3 mm, and then the drying rate was measured in the same manner as in Example 11. As a result, it was found that the solid content was 95% by weight or more. The time was 25 minutes.
- Example 12 Water absorbing sheet
- the foamed polymer (6) which is the white foam gel obtained in Example 6, was cut into a diameter of 90 mm to obtain a water absorbent sheet (12) (molded product).
- the return amount (Re-Wet) was measured by the following evaluation method according to paper diaper evaluation.
- the mini-absorber is a paper diaper model and is an example of the absorbent article of the present invention.
- mini-absorber 12 Place the mini-absorber 12 (90 mm in diameter) on the bottom of an SUS petri dish with an inner diameter of 90 mm, place a non-woven fabric with a diameter of 90 mm on it, and apply a load of 4.8 kPa evenly on the mini-absorber 12 An adjusted piston and weight were placed.
- the piston and weight used were those having a 5 mm diameter liquid inlet at the center.
- the water absorbent sheet (12) was excellent in air permeability and almost free of stuffiness.
- Comparative Example 17 Water Absorbing Sheet
- the comparative polymer (9), which is a transparent gel obtained in Comparative Example 9 was cut in the same manner as in Example 12 to obtain a comparative water absorbent sheet (17) (molded product).
- the return amount (Re-Wet) was measured by the same method as in Example 12. As a result, it was 14.6 g.
- the comparative water absorbent sheet (18) had a large amount of return, was inferior in air permeability, and had a feeling of stuffiness.
- the water-absorbent resin of the present invention gives an excellent absorbent article (especially sanitary material such as paper diapers) with little urine return, excellent air permeability, and no stuffiness. I understand.
- a water-absorbing resin having a high water absorption rate can be easily obtained, and such a water-absorbing resin can be widely used including sanitary materials such as disposable diapers.
Abstract
【課題】より簡便な方法で発泡重合体(フォーム状吸水性樹脂)中の連通孔(連続気泡)の形成を促進することであり、衛生用品等にシート形態や、粉末状でも高吸水速度の吸水性樹脂を高効率で製造する方法を提供すること。 【解決手段】気泡を分散させたアクリル酸系単量体水溶液を得る工程(A)と、該単量体水溶液を重合して発泡重合体を得る工程(B)と、該発泡重合体を加熱乾燥する工程(C)と、を含む、ポリアクリル酸系吸水性樹脂の製造方法であって、 下記式(1);;で定義される体積膨張倍率が1.1倍を超えるように気泡を含有させ、下記式(2);;で定義される単量体濃度が40重量%以上の単量体水溶液を100℃以上の温度で重合することを特徴とする、ポリアクリル酸系吸水性樹脂の製造方法。
Description
本発明は、ポリアクリル酸系吸水性樹脂及びその製造方法に関する。さらに詳しくは、該吸水性樹脂の紙オムツや生理用ナプキン等の衛生用品等への使用に関するものであり、吸水性能(特に吸水速度)の改善されたフォーム状ポリアクリル酸系吸水性樹脂及びその製造方法に関するものである。
吸水性樹脂は、紙オムツや生理用ナプキン、成人向け失禁用製品等の衛生用品、土壌用保水剤等、各種用途に幅広く利用され、大量に生産、消費されている。かかる吸水性樹脂としては、例えば、ポリアクリル酸部分中和物架橋体、澱粉-アクリル酸グラフト重合体の加水分解物、酢酸ビニル-アクリル酸エステル共重合体の鹸化物、アクリロニトリル共重合体若しくはアクリルアミド共重合体の加水分解物又はこれらの架橋体、及びカチオン性モノマーの架橋体等が知られている。これらの吸水性樹脂は、例えば、親水性単量体を含む水溶液を攪拌により重合ゲルを砕断しながら重合する方法(特許文献1)や、単量体を含む水溶液を静置重合する方法(特許文献2~6)、逆相懸濁重合、滴下重合等により製造されている。
近年において、綿状パルプ含有量が少ない薄型の衛生用品が市販されているが、その分、吸水性樹脂の使用量が増加し、更には吸水性樹脂の高物性化が要求されている。このような状況の下、吸水性樹脂の高吸水速度化が望まれており、各吸水性樹脂メーカーから種々の技術が提案されている(特許文献7~33)。例えば、モノマー水溶液に多量に気泡を分散させ、さらに重合開始時までに気泡が消滅しないように界面活性剤や場合により気泡安定剤を多量に用いる方法(特許文献8、20、21)やモノマー水溶液を低温に保つ方法(特許文献7、9)、フォーム状吸水性樹脂を製造する方法(特許文献8、20、21、30~33)が提案されている。
また、表面積増大による吸水速度向上を目的として発泡重合が行われるが、当該発泡重合において、単量体に使用する発泡剤として、具体的には、炭酸塩を使用する技術(特許文献10~17)、有機溶媒を使用する技術(特許文献18、19)、不活性ガスを使用する技術(特許文献22)、アゾ化合物を使用する技術(特許文献23、24)、不溶性無機粉末を使用する技術(特許文献25)等が知られている。また、重合後に発泡及び架橋する技術(特許文献26)や水不溶性粒子を重合に使用する技術(特許文献27)、アクリル酸ナトリウム塩の沈殿が分散した状態で重合する技術(特許文献28)等が提案されている。
更に、フォーム状吸水性樹脂を製造する方法では、「重合時の沸騰の回避」を推奨する旨(特許文献8、20、21、30~32)を開示したり、「65℃以下での重合」(特許文献33)を開示したりする。
また、発泡重合以外の重合において、可溶分低減等の目的のため、最高温度を低く制御する技術が開示されている。具体的は、最高温度を95℃以下とする技術(特許文献34)、重合温度を20~70℃で重合する技術(特許文献35)、重合温度を20~95℃で重合する技術(特許文献36)が提案されている。
しかし、界面活性剤を多量に用いて多量の気泡を分散させる方法は、重合して少なくとも連続気泡は形成されるものの、気泡の分散に比較的長時間を要し、その方法により製造した吸水性樹脂を衛生用品に使用した際、多量の界面活性剤によって体液の界面(表面)張力を低下させるため、衛生用品の吸液特性を阻害し、戻り量(Re-Wet)が増加するという問題がある。又、モノマー水溶液を低温に保つ方法は、重合に長時間を要することとなるため、生産性が劣るばかりか、得られる多孔質重合体中の気泡の連通(連続気泡)がほとんどなく、嵩密度の低下のわりに吸水速度向上が低いという問題がある。更に、従来から上記特許文献等で提案されてきた単量体中に気泡を分散させて単量体の重合を行う発泡重合では、過度の嵩密度の低下を引き起こし、輸送コストや保管コストを増加させるのみならず、吸水性樹脂粉末の耐衝撃性の低下による物性低下や粉塵発生の問題も有する。
以上、特許文献7~33等、多くの吸水速度の向上方法が提案されているが、高価な製造設備が必要であったり、生産性が低かったり、多量の界面活性剤の使用によるコストアップや吸水性樹脂の表面張力の低下(に伴うオムツの戻り量の増加)を伴うだけでなく、これら方法は、いまだ発泡の形成が不十分であって、さらに、発泡させることで吸水特性(例;加圧下吸水倍率、通液性、水可溶成分、残存モノマー)が損なわれたり、発泡による体積膨張倍率が高いために、嵩比重が過度に低下にして微粉が増加したりするものであった。又、特許文献8、20、21、30~33等で推奨されている「重合時における沸騰の回避」は、生産性の低下や高価な冷却設備等を必要とするものであった。
また、吸水性樹脂は一般に粉末状(粒子状)であるため、紙オムツや生理用ナプキン、タンポン等で実使用する場合、必要により繊維材料や接着剤と混合した後、粉末状(粒子状)吸水性樹脂をシート状や筒状等に加工する必要があり、かかる加工はコストアップのみにならず、吸水速度の低下や粉末破壊(微紛発生)を伴うものであった。
また、吸水速度の問題に加えて、吸水性樹脂は衛生材料に主に使用されるが、衛生材料ひとつあたりの使用量(gまたは重量%)の増加に伴い、吸水性樹脂自体の白色度がより重視され、その着色問題も大きくなっている。そこで、特許文献29は、アクリル酸アンモニウム塩を単量体に用いる着色防止方法やリン原子を含む化合物又は硫黄系還元剤を複数回添加する着色防止方法を開示する。
更に、単量体水溶液を重合して得られる含水ゲル状架橋重合体は、その乾燥に大きな設備や熱エネルギー、更に長い乾燥時間が必要となる一方、長時間の乾燥による吸水性樹脂の劣化や着色、更には吸水性樹脂の製造コストの増大という問題も生じている。今日、吸水性樹脂において、乾燥時間の短縮はコスト面、物性面、着色面から、重要な課題である。
また、従来の吸水性樹脂は粉末状が主流であるが、紙オムツ等の吸収物品(最終消費財)して使用する場合、吸水性樹脂粉末を吸収物品中に固定化したり、組み込む工程が必要であったり、固定化のための高価な接着剤の使用や、また、粉末の脱落や移動という問題もあった。
かかる粉末の問題を解決するため、シート状等の吸水性樹脂成型物や複合体も提案(特許文献37、38)されているが、成型のために複雑な工程や高価な副原料が必要であるうえに、粉末に比べてシート状等の成型物では比表面積が大きく低下するため、吸水速度や通気性が不十分であり、紙オムツ等の漏れや蒸れの原因となっていた。
本発明が解決しようとする課題は、吸水性樹脂の吸水速度を向上すること、特に発泡や多孔質化によって吸水速度を向上することである。さらに好ましくは、より簡便な方法で発泡重合体(フォーム状吸水性樹脂)中の連通孔(連続気泡)の形成を促進することであり、又、吸水性樹脂の乾燥工程を簡便化することである。
即ち、衛生用品等にシート形態(成型物)や、粉末状でも高吸水速度の吸水性樹脂及びその高効率での製造方法を提供することにある。
上記課題を解決するために、重合時の単量体への気泡の分散と重合方法に着目し、特定の方法を使用することで上記課題を解決されることを見いだし、本発明を完成させた。
すなわち、上記課題を解決するために、本発明の吸水性樹脂の製造方法は、気泡を分散させたアクリル酸系単量体水溶液を得る工程(A)と、該単量体水溶液を重合して発泡重合体を得る工程(B)と、該発泡重合体を加熱乾燥する工程(C)と、を含む、ポリアクリル酸系吸水性樹脂の製造方法であって、
下記式(1);
下記式(1);
;で定義される体積膨張倍率が1.1倍を超えるように気泡を含有させ、下記式(2);
で定義される単量体濃度が40重量%以上の単量体水溶液を100℃以上の温度で沸騰重合することを特徴とする、ポリアクリル酸系吸水性樹脂の製造方法を提供する。
さらに本発明は、開放気泡率が5%以上であるポリアクリル酸系吸水性樹脂を提供する。
本発明にかかる吸水性樹脂の製造方法によると、発泡によって吸水性樹脂の吸水速度が向上する。好ましい実施形態として、連続気泡の吸水性樹脂も得ることができる。また、本発明の吸水性樹脂によると、開泡気泡率が高いため、吸水性樹脂の吸水速度や通気性が向上する。
以下、本発明に係るポリアクリル酸系吸水性樹脂の製造方法について詳しく説明するが、本発明の範囲はこれらの説明に拘束されることなく、以下の例示以外についても、本発明の趣旨を損なわない範囲で適宜変更、実施し得る。
具体的には、本発明は下記の実施形態に限定されるものではなく、請求項に示した範囲で種々の変更が可能であり、異なる実施形態にそれぞれ開示された技術的手段を適宜組み合わせて得られる実施形態についても、本発明の技術的範囲に含まれる。
〔1〕用語の定義
(1-1)「吸水性樹脂」
本発明における「吸水性樹脂」とは、水膨潤性水不溶性の高分子ゲル化剤を意味し、「吸水性樹脂粉末」とは、粉砕されて粉末状になった吸水性樹脂のことをいう。なお、「水膨潤性」とは、ERT441.2-02で規定するCRC(無加圧下吸水倍率)が5[g/g]以上であることをいい、好ましくは10~100[g/g]、更に好ましくは20~80[g/g]であり、また、「水不溶性」とは、ERT470.2-02で規定するExt(水可溶分)が0~50重量%であることをいい、好ましくは0~30重量%、更に好ましくは0~20重量%、特に好ましくは0~10重量%である。
(1-1)「吸水性樹脂」
本発明における「吸水性樹脂」とは、水膨潤性水不溶性の高分子ゲル化剤を意味し、「吸水性樹脂粉末」とは、粉砕されて粉末状になった吸水性樹脂のことをいう。なお、「水膨潤性」とは、ERT441.2-02で規定するCRC(無加圧下吸水倍率)が5[g/g]以上であることをいい、好ましくは10~100[g/g]、更に好ましくは20~80[g/g]であり、また、「水不溶性」とは、ERT470.2-02で規定するExt(水可溶分)が0~50重量%であることをいい、好ましくは0~30重量%、更に好ましくは0~20重量%、特に好ましくは0~10重量%である。
上記吸水性樹脂は、その用途に応じて適宜設計可能であり、特に限定されるものではないが、カルボキシル基を有する不飽和単量体を架橋重合させた、親水性架橋重合体であることが好ましい。また、全量(100重量%)が重合体である形態に限定されず、上記性能を維持する範囲内において、吸水性樹脂以外のその他の成分として添加剤等を含んでもよい。すなわち、吸水性樹脂粉末および添加剤を含む吸水性樹脂組成物であっても、本発明では吸水性樹脂と総称する。ポリアクリル酸(塩)系吸水性樹脂の含有量は、好ましくは全体に対して70~99.9重量%であり、より好ましくは80~99.7重量%であり、さらに好ましくは90~99.5重量%である。吸水性樹脂以外のその他の成分としては、吸水速度や粉末(粒子)の耐衝撃性の観点から水が好ましく、必要により添加剤が含まれる。
(1-2)「ポリアクリル酸系吸水性樹脂」、「フォーム状吸水性樹脂」、「吸水性樹脂成型物」
本発明における「ポリアクリル酸系吸水性樹脂」とは、任意にグラフト成分を含み、繰り返し単位として、アクリル酸および/またはその塩(以下、アクリル酸(塩)と称する)を主成分とする吸水性樹脂を意味する。
本発明における「ポリアクリル酸系吸水性樹脂」とは、任意にグラフト成分を含み、繰り返し単位として、アクリル酸および/またはその塩(以下、アクリル酸(塩)と称する)を主成分とする吸水性樹脂を意味する。
具体的には、重合に用いられる総単量体(架橋剤を除く)のうち、アクリル酸(塩)を50~100モル%含む重合体をいい、好ましくは70~100モル%、より好ましくは90~100モル%、特に好ましくは実質100モル%を含む吸水性樹脂をいう。また、重合体としての塩は、必須に水溶性塩を含み、好ましくは一価塩、さらに好ましくはアクリル金属塩またはアンモニウム塩、特にアルカリ金属塩、さらにはナトリウム塩を含む。
また、該吸水性樹脂の形態がフォーム(発泡)状あるいは多孔質状のものを「フォーム状吸水性樹脂」といい、形状(シート状、ブロック状、粉末状等)は問わない。なお、本発明では、含水ゲルを加熱乾燥したものを吸水性樹脂乾燥物とする。
なお、粉末状(粒子状)吸水性樹脂に対して、シート状、ブロック状、筒状など一定形状を有する吸水性樹脂については、以下、特に吸水性樹脂成型物と呼ぶことがあり、代表的にはシート状吸水性樹脂が挙げられる。
(1-3)「開放気泡」及び「独立気泡」
本発明において、「開放気泡」(連続気泡ともいう)は、吸水性樹脂の壁によって完全には取り囲まれていない気泡(吸水性樹脂の外部と互いに連結している気泡をいい、「独立気泡」とは、吸水性樹脂の壁によって吸水性樹脂内部に全て囲まれている気泡を指す。図7に、開放気泡と独立気泡の概念図を示す。
本発明において、「開放気泡」(連続気泡ともいう)は、吸水性樹脂の壁によって完全には取り囲まれていない気泡(吸水性樹脂の外部と互いに連結している気泡をいい、「独立気泡」とは、吸水性樹脂の壁によって吸水性樹脂内部に全て囲まれている気泡を指す。図7に、開放気泡と独立気泡の概念図を示す。
(1-4)「EDANA」及び「ERT」
「EDANA」は、欧州不織布工業会(European Disposables and Nonwovens Assoiations)の略称であり、「ERT」は、欧州標準(ほぼ世界標準)である吸水性樹脂の測定方法(EDANA Recommended Test Metods)の略称である。
「EDANA」は、欧州不織布工業会(European Disposables and Nonwovens Assoiations)の略称であり、「ERT」は、欧州標準(ほぼ世界標準)である吸水性樹脂の測定方法(EDANA Recommended Test Metods)の略称である。
なお、本発明においては、特に断りのない限り、ERT原本(公知文献:2002年改定)に準拠して、吸水性樹脂粉末の物性を測定する。
(a)「CRC」(ERT441.2-02)
「CRC」は、Centrifuge Retention Capacity(遠心分離機保持容量)の略称であり、無加圧下吸水倍率(以下、「吸水倍率」と称することもある)を意味する。具体的には、不織布袋中の吸水性樹脂粉末0.2gについて、大過剰の0.9重量%塩化ナトリウム水溶液に対する30分間の自由膨潤後さらに遠心分離機で水切りした後の吸水倍率(単位;[g/g])である。
「CRC」は、Centrifuge Retention Capacity(遠心分離機保持容量)の略称であり、無加圧下吸水倍率(以下、「吸水倍率」と称することもある)を意味する。具体的には、不織布袋中の吸水性樹脂粉末0.2gについて、大過剰の0.9重量%塩化ナトリウム水溶液に対する30分間の自由膨潤後さらに遠心分離機で水切りした後の吸水倍率(単位;[g/g])である。
(b)「AAP」(ERT442.2-02)
「AAP」は、Absorption Against Pressureの略称であり、加圧下吸水倍率を意味する。具体的には、0.9gの吸水性樹脂を、大過剰の0.9重量%塩化ナトリウム水溶液に対する1時間、2.06kPa(0.3psi、21[g/cm2])での荷重下膨潤後の吸水倍率(単位;[g/g])である。なお、ERT442.2-02では、Absorption Under Pressureと表記されているが、実質的に同一内容である。また、荷重条件のみを4.83kPa(0.7psi、50[g/cm2])に変更して測定することもある。
「AAP」は、Absorption Against Pressureの略称であり、加圧下吸水倍率を意味する。具体的には、0.9gの吸水性樹脂を、大過剰の0.9重量%塩化ナトリウム水溶液に対する1時間、2.06kPa(0.3psi、21[g/cm2])での荷重下膨潤後の吸水倍率(単位;[g/g])である。なお、ERT442.2-02では、Absorption Under Pressureと表記されているが、実質的に同一内容である。また、荷重条件のみを4.83kPa(0.7psi、50[g/cm2])に変更して測定することもある。
(c)「Ext」(ERT470.2-02)
「Ext」は、Extractablesの略称であり、水可溶分(水可溶成分量)を意味する。具体的には、0.9重量%塩化ナトリウム水溶液200gに対して、吸水性樹脂粉末1gを500rpmで16時間攪拌した後、溶解したポリマー量をpH滴定で測定した値(単位;重量%)である。
「Ext」は、Extractablesの略称であり、水可溶分(水可溶成分量)を意味する。具体的には、0.9重量%塩化ナトリウム水溶液200gに対して、吸水性樹脂粉末1gを500rpmで16時間攪拌した後、溶解したポリマー量をpH滴定で測定した値(単位;重量%)である。
(d)「PSD」(ERT420.2-02)
「PSD」とは、Particle Size Disributionの略称であり、ふるい分級により測定される粒度分布を意味する。なお、重量平均粒子径(D50)および粒子径分布幅は国際公開第2004/69915号パンフレットに記載された「(1) Average Particle Diameter and Distribution of Particle Diameter」と同様の方法で測定する。
「PSD」とは、Particle Size Disributionの略称であり、ふるい分級により測定される粒度分布を意味する。なお、重量平均粒子径(D50)および粒子径分布幅は国際公開第2004/69915号パンフレットに記載された「(1) Average Particle Diameter and Distribution of Particle Diameter」と同様の方法で測定する。
(1-5)「通液性」
荷重下又は無荷重下での膨潤した吸水性樹脂粉末の粒子間を流れる液の流れを「通液性」という。この「通液性」の代表的な測定方法として、SFC(Saline Flow Conductivity/生理食塩水流れ誘導性)や、GBP(Gel Bed Permeability/ゲル床透過性)がある。
荷重下又は無荷重下での膨潤した吸水性樹脂粉末の粒子間を流れる液の流れを「通液性」という。この「通液性」の代表的な測定方法として、SFC(Saline Flow Conductivity/生理食塩水流れ誘導性)や、GBP(Gel Bed Permeability/ゲル床透過性)がある。
「SFC(生理食塩水流れ誘導性)」は、荷重2.07kPa(0.3psi)における吸水性樹脂粉末0.9gに対する0.69重量%生理食塩水の通液性をいう。米国特許第5669894号明細書に記載されたSFC試験方法に準じて測定される。
「GBP(ゲル床透過性)」は、荷重下または自由膨張における吸水性樹脂粉末に対する0.69重量%生理食塩水の通液性をいう。国際公開第2005/016393号パンフレットに記載されたGBP試験方法に準じて測定される。
(1-6)その他
本明細書において、範囲を示す「X~Y」は「X以上Y以下」を意味する。また、重量の単位である「t(トン)」は、「Metric ton(メトリック トン)」を意味し、更に、特に注釈のない限り、「ppm」は「重量ppm」を意味する。また、本願明細書において、「質量」と「重量」、「質量%」と「重量%」及び「質量部」と「重量部」は同義語であり、物性等の測定に関しては特に断りがない場合は室温(20~25℃)/相対湿度40~50%で測定する。更に、「~酸(塩)」は「~酸及び/又はその塩」を意味し、「(メタ)アクリル」は「アクリル及び/又はメタクリル」を意味する。
本明細書において、範囲を示す「X~Y」は「X以上Y以下」を意味する。また、重量の単位である「t(トン)」は、「Metric ton(メトリック トン)」を意味し、更に、特に注釈のない限り、「ppm」は「重量ppm」を意味する。また、本願明細書において、「質量」と「重量」、「質量%」と「重量%」及び「質量部」と「重量部」は同義語であり、物性等の測定に関しては特に断りがない場合は室温(20~25℃)/相対湿度40~50%で測定する。更に、「~酸(塩)」は「~酸及び/又はその塩」を意味し、「(メタ)アクリル」は「アクリル及び/又はメタクリル」を意味する。
〔2〕ポリアクリル酸系吸水性樹脂の製造方法
本発明に係るポリアクリル酸系吸水性樹脂は、例えば、以下の方法によって製造することが出来る。かかる製造方法は好適に連続製造に適用できる。
本発明に係るポリアクリル酸系吸水性樹脂は、例えば、以下の方法によって製造することが出来る。かかる製造方法は好適に連続製造に適用できる。
ここで、本発明で使用される気泡生成のための気体は、使用前にそれ自身が常温で気体の化合物をさし、重合時などにガスを発生する固体発泡剤(例;炭酸塩、アゾ化合物)とは異なる概念である。よって、本発明で、気泡を分散させたアクリル酸系単量体水溶液とは、気体、好ましくは不活性気体と、単量体水溶液ないしその原料(個々の単量体や溶媒)と、を直接混合させてなる。
(2-1)気泡を分散させたアクリル酸系単量体水溶液を得る工程(A)(分散工程)
(単量体の組成)
本発明で用いられるモノマー(単量体)は、重合により吸水性樹脂となりうるものであれば特に限定されないが、物性面からアクリル酸(塩)を用い、さらに以下に示すようなものが挙げられる。例えば、(メタ)アクリル酸、(無水)マレイン酸、イタコン酸、ケイ皮酸、ビニルスルホン酸、アリルトルエンスルホン酸、ビニルトルエンスルホン酸、スチレンスルホン酸、2-(メタ)アクリルアミド-2-メチルプロパンスルホン酸、2-(メタ)アクリロイルエタンスルホン酸、2-(メタ)アクリロイルプロパンスルホン酸、2-ヒドロキシエチル(メタ)アクリロイルフォスフェート等のアニオン性不飽和単量体及びその塩;メルカプト基含有不飽和単量体;フェノール性水酸基含有不飽和単量体;(メタ)アクリルアミド、N-エチル(メタ)アクリルアミド、N,N-ジメチル(メタ)アクリルアミド等のアミド基含有不飽和単量体;N,N-ジメチルアミノエチル(メタ)アクリレート、N,N-ジメチルアミノプロピル(メタ)アクリレート、N,N-ジメチルアミノプロピル(メタ)アクリルアミド等のアミノ基含有不飽和単量体等が挙げられる。
(単量体の組成)
本発明で用いられるモノマー(単量体)は、重合により吸水性樹脂となりうるものであれば特に限定されないが、物性面からアクリル酸(塩)を用い、さらに以下に示すようなものが挙げられる。例えば、(メタ)アクリル酸、(無水)マレイン酸、イタコン酸、ケイ皮酸、ビニルスルホン酸、アリルトルエンスルホン酸、ビニルトルエンスルホン酸、スチレンスルホン酸、2-(メタ)アクリルアミド-2-メチルプロパンスルホン酸、2-(メタ)アクリロイルエタンスルホン酸、2-(メタ)アクリロイルプロパンスルホン酸、2-ヒドロキシエチル(メタ)アクリロイルフォスフェート等のアニオン性不飽和単量体及びその塩;メルカプト基含有不飽和単量体;フェノール性水酸基含有不飽和単量体;(メタ)アクリルアミド、N-エチル(メタ)アクリルアミド、N,N-ジメチル(メタ)アクリルアミド等のアミド基含有不飽和単量体;N,N-ジメチルアミノエチル(メタ)アクリレート、N,N-ジメチルアミノプロピル(メタ)アクリレート、N,N-ジメチルアミノプロピル(メタ)アクリルアミド等のアミノ基含有不飽和単量体等が挙げられる。
上記に加え、得られるフォーム状重合体(発泡重合体)の柔軟性等の性状を改良するため、その他の単量体を併用してもよい。併用される単量体としては、2-ヒドロキシエチル(メタ)アクリレート、メトキシポリエチレングリコール(メタ)アクリレート、ポリエチレングリコール(メタ)アクリレート、イソブチレン、ラウリル(メタ)アクリレート等の水溶性または疎水性不飽和単量体等を共重合成分とするものも含まれる。
これらのモノマーは単独で用いても、2種類以上を併用してもよいが、得られる吸水性樹脂粉末の性能やコストの点から、アクリル酸及び/又はその塩(例えば、ナトリウム塩、リチウム塩、カリウム塩、アンモニウム塩、アミン類等の塩が好ましく、中でもコスト面からナトリウム塩がより好ましい。)を含むアクリル酸系単量体を用いたポリアクリル酸系吸水性樹脂が好ましくは挙げられる。
ポリアクリル酸系吸水性樹脂としてアクリル酸及び/又はその塩の使用量は、全モノマー成分(後述する内部架橋剤は除く)に対して50モル%以上、さらには70モル%以上が好ましく、より好ましくは80モル%以上、さらに好ましくは90モル%以上、特に好ましくは95モル%以上であり(上限は100モル%である)、最も好ましくは実質100モル%である。なお、本発明でポリアクリル酸とはポリアクリル酸塩(特に一価塩)を含む概念である。
(メトキシフェノール類)
本発明に係る吸水性樹脂の製造方法においては、対アクリル酸換算値で、メトキシフェノール類を200ppm以下含有する単量体が好ましく用いられる。この単量体の主成分は、アクリル酸であってもよく、アクリル酸及びアクリル酸塩であってもよい。上記メトキシフェノール類としては、具体的には、o,m,p-メトキシフェノールや、それらに更にメチル基、t-ブチル基、水酸基等の1個又は2個以上の置換基を有するメトキシフェノール類が例示され、特に好ましくは、p-メトキシフェノールが本発明では使用される。
本発明に係る吸水性樹脂の製造方法においては、対アクリル酸換算値で、メトキシフェノール類を200ppm以下含有する単量体が好ましく用いられる。この単量体の主成分は、アクリル酸であってもよく、アクリル酸及びアクリル酸塩であってもよい。上記メトキシフェノール類としては、具体的には、o,m,p-メトキシフェノールや、それらに更にメチル基、t-ブチル基、水酸基等の1個又は2個以上の置換基を有するメトキシフェノール類が例示され、特に好ましくは、p-メトキシフェノールが本発明では使用される。
メトキシフェノール類の含有量は、対アクリル酸換算値で、好ましくは200重量ppm以下、より好ましくは10~200ppm、さらに好ましくは10~120ppm、特に好ましくは10~90ppm、最も好ましくは20~90ppmである。メトキシフェノール類の含有量が200ppm以下であれば、得られた吸水性樹脂の着色(黄ばみ/黄変)を抑制することができる。また、メトキシフェノール類の含有量が10ppm未満の場合、すなわち、蒸留等の精製によって重合禁止剤であるメトキシフェノール類を除去した場合、意図的に重合を開始させる前に重合が起きる危険があるのみならず、アクリル酸(塩)を主原料として得られた吸水性樹脂の耐候性が悪くなる場合がある。
単量体の主成分は、アクリル酸及び/又はアクリル酸塩であるが、これらアクリル酸とアクリル酸塩とでは、分子量が異なる。この分子量の相違を考慮して、本発明において、対アクリル酸換算値が定義される。対アクリル酸換算値とは、アクリル酸塩が全て等モルの未中和アクリル酸であるとして換算した場合における、アクリル酸の重量に対する上記微量成分の重量の含有割合(重量比)である。すなわち、例えば、中和後のアクリル酸ナトリウム(分子量94)はアクリル酸(分子量72)に重量換算されて、アクリル酸換算後(94を72に換算)の重量でメトキシフェノール類の含有割合(重量比)等が規定される。すなわち、重合後の吸水性樹脂において、部分中和又は完全中和のアクリル酸塩がポリマーとなっている場合、対アクリル酸換算値は、部分中和又は完全中和のポリアクリル酸塩が全て等モルの未中和のポリアクリル酸であると換算して計算され得る。上記部分中和とは、中和率が0モル%を超えて100モル%未満であることを意味する。上記完全中和とは、中和率が100モル%であることを意味する。上記未中和とは、中和率が0モル%であることを意味する。
本発明の吸水性樹脂の製造方法は、上述したように、メトキシフェノール類(特にp-メトキシフェノール)を200ppm以下含有する単量体水溶液の重合を経る吸水性樹脂の製造方法であることが好ましい。更に、当該重合工程(濃度、開始剤、温度)及び乾燥工程(温度、時間、固形分、風量等)を経ることで、メトキシフェノール類の所定量が消費され、メトキシフェノール類(特にp-メトキシフェノール)を好ましくは60ppm以下、より好ましくは5~60ppmで含有する吸水性樹脂、特に重合体内部にメトキシフェノール類を均一に含有する吸水性樹脂を得ることができる。
即ち、本発明の製造方法では、メトキシフェノール類(特にp-メトキシフェノール)含有量が200ppm以下の単量体を用いることによって、上記重合工程及び乾燥工程によって得られる吸水性樹脂のメトキシフェノール類(特にp-メトキシフェノール)含有量を60ppm以下とする製造方法であってもよい。
重合時のメトキシフェノール類はアクリル酸(分子量72)を基準とする場合、必要により中和して得られたアクリル酸塩は分子量が増大(例えば、75モル%中和ナトリウム塩では分子量88.5)し、メトキシフェノール類の含有量は低減する。また、重合時の消費も鑑み、本発明では、重合前のアクリル酸塩におけるメトキシフェノール類の含有量200ppm以下に対して、得られたポリアクリル酸塩中のメトキシフェノール類は60ppm以下とすることが好ましい。
尚、吸水性樹脂中のメトキシフェノール類の制御方法は、上記一例に限定はされず、その他の手法として下記方法が例示でき、これらを併用してもよい。
製法その1;メトキシフェノール類の不存在又は10ppm未満含有する単量体で重合し、更には乾燥後に所定量のメトキシフェノール類を添加する方法。
製法その2;メトキシフェノール類を過剰に含む単量体で吸水性樹脂へと重合後、乾燥前に所定量のメトキシフェノール類を洗浄によって除去する方法。尚、洗浄には水又は水アルコール混合液が使用できる。
また、本発明で用いられる単量体は、製造工程でメトキシフェノール類以外の重合禁止剤を使用してもよいし、当該重合禁止剤をメトキシフェノール類と併用してもよい。
メトキシフェノール類以外の重合禁止剤としては、例えば、フェノチアジン、ハイドロキノン、銅塩、酢酸マンガン、メチレンブルー等が有効である。但し、これらの重合禁止剤はメトキシフェノール類と異なり、重合を阻害するため、最終的には少ないほどよく、メトキシフェノール類と併用する場合、単量体中の濃度は0.01~10ppmが好ましい。
尚、上記成分の定量は液体クロマトグラフィー又はガスクロマトグラフィーで行うことができる。特に本発明において、メトキシフェノール類の含有量は、下記実施例の方法により測定した値を採用する。
(単量体濃度)
重合時における単量体濃度は40重量%以上であり、45重量%以上がより好ましく、50重量%以上、53重量%以上がさらに好ましい。上限は特に限定されるものではないが、80重量%以下、さらに75重量%以下であることが好ましい。なお、本発明で単量体濃度は下記式(2)で定義される。
重合時における単量体濃度は40重量%以上であり、45重量%以上がより好ましく、50重量%以上、53重量%以上がさらに好ましい。上限は特に限定されるものではないが、80重量%以下、さらに75重量%以下であることが好ましい。なお、本発明で単量体濃度は下記式(2)で定義される。
上記式(2)における(単量体の重量)は、単量体水溶液中に存在する単量体の総重量を表す。例えば、下記実施例のように、単量体水溶液を作製する際、アクリル酸及びアクリル酸を中和するための水酸化ナトリウムを用いる場合、アクリル酸及び水酸化ナトリウムの合計量から中和反応で生成した水の量を引いたもの(単量体水溶液中に存在するアクリル酸及びアクリル酸ナトリウムの総重量)が上記式(2)における(単量体の重量)となる。また、単量体水溶液を作製する際に、アクリル酸及びアクリル酸ナトリウムを用いる場合、アクリル酸及びアクリル酸ナトリウムの合計量が上記式(2)における(単量体の重量)となる。
又、上記式(2)における(溶媒の重量)は、重合系に供給される原料のうち、水溶液等の溶液状態で供給される場合の溶媒量を表す。したがって、必要により使用する後述の界面活性剤、親水性高分子等は上記単量体濃度の規定(計算)において考慮されない。
重合時における単量体濃度が40重量%未満の場合、生産性が低くなるばかりか、単量体水溶液中に分散した気泡の安定性が悪く、さらに重合中の気泡も消泡しやすいため好ましくない。なお、重合時における単量体濃度が40重量%以上、好ましくは42重量%以上、より好ましくは43重量%以上、更に好ましくは45重量%以上、特に好ましくは50重量%以上という高濃度の領域で、本発明で規定するその他条件において、気泡の連通性(連続気泡性)が高まるため、特に、本発明の優位性がある。単量体の溶媒は水であり、水に加えて少量(例えば、全溶媒中で0重量%を超え30重量%以下の範囲、さらには、0重量%を超え10重量%以下の範囲)の有機溶媒を併用してもよい。有機溶媒としては、メチルアルコール、エチルアルコール、n-プロピルアルコール、イソプロピルアルコール、n-ブチルアルコール、イソブチルアルコール、t-ブチルアルコール等のアルコール類が例示される。
特許文献28(特開平1-318021号)のように単量体のスラリー(アクリル酸塩の水分散液)での重合は物性(吸水倍率、可溶分、残存モノマーなど)が低下することがある。よって、本発明においては上記単量体が酸基含有単量体の場合、その中和率は、単量体水溶液中に中和塩が析出しない程度とする。すなわち、アクリル酸系単量体の水分散液でなく、本発明では、アクリル酸系単量体水溶液が重合される。中和塩の析出は、中和塩の水に対する溶解度、単量体濃度、中和率、温度、圧力、中和塩基、および必要により使用される分散剤(界面活性剤、他の単量体、水溶性高分子)などにより異なるため、同じ単量体でも単量体水溶液の温度上昇でその溶解度が上がるなど、適宜決定され、それらの条件に依存する。
本発明においては、単量体水溶液に気泡を分散させ液の表面積を大きくした状態で、かつ水を蒸発させながら比較的短時間で重合を行うため、重合前に単量体の塩、例えばアクリル酸ナトリウムが析出していると、その塩の多くが溶解することなく重合後まで残存するため好ましくない。また、重合中に析出する塩を溶解させるなら、水の蒸発を抑制し、塩の溶解時間を取る必要から重合時間を比較的長くしなければならず、この場合には発泡の度合い、中でも、好ましい実施形態である気泡の連通性(連続気泡性)が著しく低下してしまう。上記中和は、必要に応じて、重合後に重合ゲルが中和されてもよい。しかし、衛生用品等人体に触れる可能性のある用途では、重合後の中和はなるべく避けられる。そして、衛生用品等での使用では、単量体または重合ゲルの中和率は、40~90モル%が好ましく、50~80モル%がより好ましい。
(内部架橋剤)
上記重合に際して、必要に応じて内部架橋剤が用いられる。かような内部架橋剤としては、従来公知の内部架橋剤を用いることができる。具体的には、例えば、N,N’-メチレンビス(メタ)アクリルアミド、(ポリ)エチレングリコールジ(メタ)アクリレート、(ポリ)プロピレングリコールジ(メタ)アクリレート、トリメチルロールプロパントリ(メタ)アクリレート、グリセリントリ(メタ)アクリレート、グリセリンアクリレートメタクリレート、エチレンオキサイド変性トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリトールヘキサ(メタ)アクリレート、トリアリルシアヌレート、トリアリルイソシアヌレート、トリアリルホスフェート、トリアリルアミン、ポリ(メタ)アリロキシアルカン、(ポリ)エチレングリコールジグリシジルエーテル、グリセロールジグリシジルエーテル、エチレングリコール、ポリエチレングリコール、プロピレングリコール、グリセリン、1,4-ブタンジオール、ペンタエリスリトール、エチレンジアミン、エチレンカーボネート、プロピレンカーボネート、ポリエチレンイミン、グリシジル(メタ)アクリレート等を挙げることができる。これらの中から、反応性を考慮して、1種または2種以上を用いることができる。なかでも、2個以上の重合性不飽和基を有する化合物を用いることが好ましい。
上記重合に際して、必要に応じて内部架橋剤が用いられる。かような内部架橋剤としては、従来公知の内部架橋剤を用いることができる。具体的には、例えば、N,N’-メチレンビス(メタ)アクリルアミド、(ポリ)エチレングリコールジ(メタ)アクリレート、(ポリ)プロピレングリコールジ(メタ)アクリレート、トリメチルロールプロパントリ(メタ)アクリレート、グリセリントリ(メタ)アクリレート、グリセリンアクリレートメタクリレート、エチレンオキサイド変性トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリトールヘキサ(メタ)アクリレート、トリアリルシアヌレート、トリアリルイソシアヌレート、トリアリルホスフェート、トリアリルアミン、ポリ(メタ)アリロキシアルカン、(ポリ)エチレングリコールジグリシジルエーテル、グリセロールジグリシジルエーテル、エチレングリコール、ポリエチレングリコール、プロピレングリコール、グリセリン、1,4-ブタンジオール、ペンタエリスリトール、エチレンジアミン、エチレンカーボネート、プロピレンカーボネート、ポリエチレンイミン、グリシジル(メタ)アクリレート等を挙げることができる。これらの中から、反応性を考慮して、1種または2種以上を用いることができる。なかでも、2個以上の重合性不飽和基を有する化合物を用いることが好ましい。
上記内部架橋剤の使用量は、所望する吸水性樹脂の物性により適宜決定することができるが、通常、上記モノマー成分量に対して、0.0001~3モル%が好ましく、0.0005~2モル%がより好ましく、0.001~1モル%がさらに好ましい。内部架橋剤の使用量が0.0001モル%以上であれば、得られる吸水性樹脂粉末の水可溶分の割合が適切であるため、加圧下における吸水量を充分に確保でき、また、内部架橋剤の使用量が3モル%以下であれば架橋密度も適度であり、得られる吸水性樹脂粉末の吸水量が充分なものとなる。なお、内部架橋剤は、反応系に一括添加されても、分割添加されてもよい。
(気泡の分散場所)
アクリル酸系単量体水溶液への気泡の分散方法は、調製後のアクリル酸系単量体水溶液に気泡を入れてもよく、アクリル酸系単量体水溶液の原料に個別に気泡を投入して、気泡を含有した原料からアクリル酸系単量体水溶液を調製してもよい。原料に気泡を投入する場合、気泡の分散場所としては、未中和のアクリル酸、溶媒(水等)、アクリル酸の中和物であるアクリル酸塩水溶液、アクリル酸系単量体水溶液、架橋剤等が挙げられ、好ましくは、これらの混合物であり、水、アクリル酸塩水溶液やそれらで調製した重合されるアクリル酸系単量体水溶液へ気泡が分散される。
アクリル酸系単量体水溶液への気泡の分散方法は、調製後のアクリル酸系単量体水溶液に気泡を入れてもよく、アクリル酸系単量体水溶液の原料に個別に気泡を投入して、気泡を含有した原料からアクリル酸系単量体水溶液を調製してもよい。原料に気泡を投入する場合、気泡の分散場所としては、未中和のアクリル酸、溶媒(水等)、アクリル酸の中和物であるアクリル酸塩水溶液、アクリル酸系単量体水溶液、架橋剤等が挙げられ、好ましくは、これらの混合物であり、水、アクリル酸塩水溶液やそれらで調製した重合されるアクリル酸系単量体水溶液へ気泡が分散される。
(気体)
かかる気泡の分散のために使用される気体は、空気、排ガス、酸素、窒素、炭酸ガス、アルゴン、ヘリウム、オゾンやそれらの混合物等が挙げられるが、重合性から好ましくは、窒素、炭酸ガス、アルゴン等の不活性ガスの一種以上が使用され、この中でも、安価である窒素が特に好ましい。不活性ガスの割合は80容積%以上、さらに好ましくは99容積%以上、より好ましくは99.9容積%以上、特に好ましくは99.99容積%以上であり、圧力は常圧、加圧、減圧で適宜決定される。
かかる気泡の分散のために使用される気体は、空気、排ガス、酸素、窒素、炭酸ガス、アルゴン、ヘリウム、オゾンやそれらの混合物等が挙げられるが、重合性から好ましくは、窒素、炭酸ガス、アルゴン等の不活性ガスの一種以上が使用され、この中でも、安価である窒素が特に好ましい。不活性ガスの割合は80容積%以上、さらに好ましくは99容積%以上、より好ましくは99.9容積%以上、特に好ましくは99.99容積%以上であり、圧力は常圧、加圧、減圧で適宜決定される。
不活性ガスの温度も適宜決定され特に問わないが、効果の面からも好ましくは、気体の沸点(例えば、窒素で-210℃)~1000℃、さらには0~100℃、10~50℃の範囲である。気体の温度を冷却または加熱することで、単量体水溶液の温度を制御したり、気体の溶解度や分散性を調整したりしてもよい。なお、単量体に炭酸ガスを分散させる場合、固体のCO2(ドライアイス、融点-79℃)や炭酸塩(例えば、炭酸ナトリウム、炭酸水素ナトリウム、炭酸マグネシウム等。アクリル酸との中和でCO2ガスを発生)、固体の尿素(N2ガスを発生)等をガスの発生源として使用してもよいが、効果とコストの面から、好ましくは気体のガス、特に上記温度のガスが使用される。
以下、主に気体のガスをそのまま使用する方法について説明する。
(気泡の分散方法)
本発明における気泡(特に気体のガス)の分散方法は、好ましくは以下から選ばれる少なくとも一つの方法及び/又はそれらの併用により、気泡分散前の単量体水溶液に対する分散後の体積膨張倍率が1.1倍を超えるように実施される。さらに、気泡の安定的な分散のために、好ましくは界面活性剤が使用される。
本発明における気泡(特に気体のガス)の分散方法は、好ましくは以下から選ばれる少なくとも一つの方法及び/又はそれらの併用により、気泡分散前の単量体水溶液に対する分散後の体積膨張倍率が1.1倍を超えるように実施される。さらに、気泡の安定的な分散のために、好ましくは界面活性剤が使用される。
本発明において、気泡の分散方法はバッチ式でも連続式でもよく、又、一段でも二段以上で行ってもよいが、気泡の安定性の観点から、連続式の一段で行うことが好ましい。即ち、単量体水溶液やその他の原料の連続的な流体に、気体を連続的に供給することが好ましく、特に単量体水溶液やその他の原料が送液される配管中に気体が連続的に供給されることが好ましい。こうすることで、単量体水溶液に気泡が分散される。当該気体として不活性ガスを使用する場合、単量体水溶液を予め脱気(溶存酸素を1[mg/l]以下)しておくことも、重合を促進する上で好ましい。
(凹凸及び/又は充填物を有する混合域での気液混合・気泡の分散)
本発明における気泡の分散方法として、好ましくは、単量体水溶液と気体とを、凹凸及び/又は充填物を有する混合域に導入することにより、両者を混合させることによる気泡の分散方法が用いられる。流体の流れを阻害する凹凸、突起、羽、邪魔板、充填物等を有する混合域で混合することにより、気泡を単量体水溶液中に均一かつ安定に分散させることができる。
本発明における気泡の分散方法として、好ましくは、単量体水溶液と気体とを、凹凸及び/又は充填物を有する混合域に導入することにより、両者を混合させることによる気泡の分散方法が用いられる。流体の流れを阻害する凹凸、突起、羽、邪魔板、充填物等を有する混合域で混合することにより、気泡を単量体水溶液中に均一かつ安定に分散させることができる。
凹凸や充填物を有する混合域としては、例えば、図3に示すような混合域が挙げられる。図3では突起を有する隙間の間に流体混合した気泡を含む単量体水溶液を通すことにより、気泡が安定かつ均一に分散し、気泡の分散していない状態と比較して体積膨張倍率が1.1倍を超えた単量体水溶液が得られる。さらに具体的に図3を用いて説明すると、単量体調製槽8で調製された単量体水溶液4は、アスピレーター6およびポンプ9を用いて凹凸3を有する混合域2を通過し、気泡含有単量体水溶液7が貯蔵槽10へと送られる。混合域2の手前で、気体5と単量体水溶液4とが混合する。こうした混合域を有する装置としては、例えば、ドイツ・ハンスクラット社製「ホイップオート(商品名)」やドイツ・F・ファイヒンガー社製「ザノマット(商品名)」等が挙げられる。
本発明では、前記混合域として回転翼を設け、気泡の分散した単量体水溶液を攪拌させることもできる。攪拌により、単量体水溶液中に微細な気泡をより均一かつ安定に分散させることができる。具体的には、S1ミキサー等が挙げられる
(a)単量体水溶液および気体の加圧による気体の溶解とその後の放圧
本発明における気泡の分散方法として、好ましくは、単量体水溶液および気体の加圧による気体の溶解とその後の放圧による分散方法が用いられる。液中に気体を100~1000kPa、さらには200~400kPa程度に加圧して溶解させ、減圧弁を通して液中にフラッシュ操作すなわち放圧すると、減圧され過飽和となった気体が液中から主にマイクロバブルとなり放出される。気体の液への溶解度はヘンリーの法則(p=HC)に従い、温度と圧力で決定される。かかる加圧によって一旦溶解させた気体を経て、分散させた気泡が得られる。
(a)単量体水溶液および気体の加圧による気体の溶解とその後の放圧
本発明における気泡の分散方法として、好ましくは、単量体水溶液および気体の加圧による気体の溶解とその後の放圧による分散方法が用いられる。液中に気体を100~1000kPa、さらには200~400kPa程度に加圧して溶解させ、減圧弁を通して液中にフラッシュ操作すなわち放圧すると、減圧され過飽和となった気体が液中から主にマイクロバブルとなり放出される。気体の液への溶解度はヘンリーの法則(p=HC)に従い、温度と圧力で決定される。かかる加圧によって一旦溶解させた気体を経て、分散させた気泡が得られる。
過飽和の程度としては、該気体の所定温度における飽和溶解度に対して1.01~10倍が好ましく、1.05~5倍がより好ましく、1.06~3倍がさらに好ましい。
(b)単量体水溶液及び気体の旋回流の形成
上記の方法に加えて、本発明における気泡の分散方法として、好ましくは、単量体水溶液および気体の旋回流の形成が挙げられる。該方法は、気液二相流体を旋回させて出口(混合機の吐出口)で気泡を分散させる方法であり、ガス流量と液流量の比は1/7~1/15が好ましく、旋回速度は毎秒10~10000回転が好ましく、さらには毎秒100~1000回転であることがより好ましい。
上記の方法に加えて、本発明における気泡の分散方法として、好ましくは、単量体水溶液および気体の旋回流の形成が挙げられる。該方法は、気液二相流体を旋回させて出口(混合機の吐出口)で気泡を分散させる方法であり、ガス流量と液流量の比は1/7~1/15が好ましく、旋回速度は毎秒10~10000回転が好ましく、さらには毎秒100~1000回転であることがより好ましい。
旋回式微細気泡発生装置は、例えば、国際公開第00/69550号、日本国公開特許公報「特開2003-205228号公報」、同「特開2000-447号公報」、同「特開2006-116365号公報」等に例示されるが、特に限定されない。
(c)細孔を通した気体の単量体水溶液への混合
各種多孔質物質、膜、フィルター等の細孔から気泡を生成させる方法であり、多孔質ガラス(Na2O-CaO-Al2O3-B2O3-SiO2系ガラス)等が使用され、上記方法は、例えば、木下理化工業株式会社製木下式ガラスボールフィルターを用いて行うことが出来る。
各種多孔質物質、膜、フィルター等の細孔から気泡を生成させる方法であり、多孔質ガラス(Na2O-CaO-Al2O3-B2O3-SiO2系ガラス)等が使用され、上記方法は、例えば、木下理化工業株式会社製木下式ガラスボールフィルターを用いて行うことが出来る。
(マイクロバブル発生装置による気泡の分散)
本発明における気泡の分散は、単量体水溶液と不活性ガスとをマイクロバブル発生装置で混合させて行ってもよい。マイクロバブル発生には上記(a)~(c)や後述の(1)~(8)の1種以上の手法が適用でき、好ましくは(a)または(b)が適用され、必要によりさらに単量体水溶液及び気泡からなる気液混合物に対してせん断力が適用される。なお、本発明で採用されるマイクロバブル発生装置は、特に限定はされず、市販されているものを使用できる。市販品の一例を以下に例示する。
本発明における気泡の分散は、単量体水溶液と不活性ガスとをマイクロバブル発生装置で混合させて行ってもよい。マイクロバブル発生には上記(a)~(c)や後述の(1)~(8)の1種以上の手法が適用でき、好ましくは(a)または(b)が適用され、必要によりさらに単量体水溶液及び気泡からなる気液混合物に対してせん断力が適用される。なお、本発明で採用されるマイクロバブル発生装置は、特に限定はされず、市販されているものを使用できる。市販品の一例を以下に例示する。
OHRラインミキサー(株式会社OHR流体工学研究所)
M型マイクロバブル発生装置(株式会社ナノプラネット研究所)
業務用マイクロバブル発生装置SMB―450型(石丸商行有限会社)
マイクロバブル発生装置Mbelife(関西オートメ機器株式会社)
球体内蔵型気泡発生装置MBG型(西田鉄工株式会社)
ポンパレーター(株式会社帝国電気製作所)
マイクロバブルの発生器には入水口と出水口があり、この入水口に、ある一定以上の圧力で液体(水や単量体)を流入させた場合、内部では水の中に混ざっている気体が密度差により中心部に集められ、気体軸が形成される。これによってマイクロバブル発生器の内部には外周と中心部の間で圧力勾配が生じる。この時、気体軸の中心部はほぼ真空状態となり、一方では加圧され噴出しようとする水と、真空状態(超負圧の状態)の気体軸へと流入しようとする水が衝突し、また旋回しながら気体軸がこの間を通り抜ける時に気体はせん断され微細化してマイクロバブルとなるのである。
M型マイクロバブル発生装置(株式会社ナノプラネット研究所)
業務用マイクロバブル発生装置SMB―450型(石丸商行有限会社)
マイクロバブル発生装置Mbelife(関西オートメ機器株式会社)
球体内蔵型気泡発生装置MBG型(西田鉄工株式会社)
ポンパレーター(株式会社帝国電気製作所)
マイクロバブルの発生器には入水口と出水口があり、この入水口に、ある一定以上の圧力で液体(水や単量体)を流入させた場合、内部では水の中に混ざっている気体が密度差により中心部に集められ、気体軸が形成される。これによってマイクロバブル発生器の内部には外周と中心部の間で圧力勾配が生じる。この時、気体軸の中心部はほぼ真空状態となり、一方では加圧され噴出しようとする水と、真空状態(超負圧の状態)の気体軸へと流入しようとする水が衝突し、また旋回しながら気体軸がこの間を通り抜ける時に気体はせん断され微細化してマイクロバブルとなるのである。
本発明において、マイクロバブル発生装置やその他の手法により発生したマイクロバブルを含む気泡の数平均直径は、5~1000μmが好ましく、10~500μmがより好ましい。気泡の平均直径が5μm未満の場合、重合後の気泡の連通が低下し、吸水速度の劣ったものになり易い。平均直径が1mmを超える場合には強度が脆くなり、乾燥後の重合ゲルを粉砕して大部分が150μm以上の粉末にするのが困難となる。
また、マイクロバブル発生装置の処理量は、所望する吸水性樹脂粉末の物性等によって、適宜設定することができるが、モノマー水溶液の流速を大きくすることが望ましい。該モノマー水溶液の流速としては、好ましくは500[kg/hr]以上であり、1000[kg/hr]以上がより好ましく、2000[kg/hr]以上が更に好ましい。なお、かかる時間あたり生産量はマイクロバブル発生装置の使用に限らず、工業的な巨大スケールの生産として、本発明の製造方法が一般に好適に適用できる。
(その他必要により併用される方法)
気泡の分散方法として、上述した凹凸及び/又は充填物を有する混合域での気液混合・気泡の分散方法、方法(a)~(c)、マイクロバブル発生装置による気泡の分散方法に加えて、下記(1)~(8)の方法を使用ないし併用することができる。
気泡の分散方法として、上述した凹凸及び/又は充填物を有する混合域での気液混合・気泡の分散方法、方法(a)~(c)、マイクロバブル発生装置による気泡の分散方法に加えて、下記(1)~(8)の方法を使用ないし併用することができる。
(1)スタティックミキサー方式
可動部分がなく、流体が管内部に固定されたエレメントを通過する際に混合されるスタティックミキサー、円管内部に螺旋流誘導部と管内部に取り付けられたキノコ状の突起により旋回状に流れる気液2相流を発破して気泡が発生される。具体的には、OHRミキサーが挙げられる。
可動部分がなく、流体が管内部に固定されたエレメントを通過する際に混合されるスタティックミキサー、円管内部に螺旋流誘導部と管内部に取り付けられたキノコ状の突起により旋回状に流れる気液2相流を発破して気泡が発生される。具体的には、OHRミキサーが挙げられる。
(2)キャビテーション方式
ガス分散器内に意図的にキャビテーションが発生するように流路を変形させて気泡を発生させる方法である。
ガス分散器内に意図的にキャビテーションが発生するように流路を変形させて気泡を発生させる方法である。
(3)遠心ポンプと旋回流式マイクロバブル発生器の組み合わせ
ポンプによる渦流攪拌作用とポンプでの昇圧により、液中に気体を加圧溶解させ、溶解しきれない気体を旋回流式マイクロバブル発生器でマイクロ化させる方法である。
ポンプによる渦流攪拌作用とポンプでの昇圧により、液中に気体を加圧溶解させ、溶解しきれない気体を旋回流式マイクロバブル発生器でマイクロ化させる方法である。
(4)ベンチュリー方式
ストロー部(絞り)に気液を同時に流すと液流速の急激な変化により生成した衝撃波により大気泡を発破させ、気泡を発生させる方法が挙げられる。
ストロー部(絞り)に気液を同時に流すと液流速の急激な変化により生成した衝撃波により大気泡を発破させ、気泡を発生させる方法が挙げられる。
(5)回転式
攪拌翼を高速回転され、ガスを自給させ気泡を発生させる方法が挙げられる。
攪拌翼を高速回転され、ガスを自給させ気泡を発生させる方法が挙げられる。
(6)超音波式
超音波周波数、圧力振幅などを適宜設定して気泡を発生させる方法が挙げられる。
超音波周波数、圧力振幅などを適宜設定して気泡を発生させる方法が挙げられる。
(7)相変化式
気体(窒素ガス)と水蒸気の混合ガスを液中に細いノズルから吹き込むと、水蒸気が凝集し、凝集しない気体(窒素ガス)の気泡が残る。
気体(窒素ガス)と水蒸気の混合ガスを液中に細いノズルから吹き込むと、水蒸気が凝集し、凝集しない気体(窒素ガス)の気泡が残る。
(8)電解分解法
水の電気分解でマイクロオーダーの気泡を発生させる方法が挙げられる。
水の電気分解でマイクロオーダーの気泡を発生させる方法が挙げられる。
これらの中でも、効果の面から好ましくは、さらに、アクリル酸系単量体水溶液を得る工程で単量体水溶液および気泡からなる気液混合物がせん断処理されてなり、せん断方法としては、(3)遠心ポンプと旋回流式マイクロバブル発生器の組み合わせ、または、OHRミキサーに代表されるせん断と旋回流を併せ持ったスタティックミキサーが使用される。
(単量体水溶液の体積膨張倍率)
本発明において、アクリル酸系単量体水溶液への気泡の分散の程度は、気泡分散前の単量体水溶液に対する分散後の体積膨張倍率として規定される。すなわち、体積膨張倍率とは下記式(1)で定義される。
本発明において、アクリル酸系単量体水溶液への気泡の分散の程度は、気泡分散前の単量体水溶液に対する分散後の体積膨張倍率として規定される。すなわち、体積膨張倍率とは下記式(1)で定義される。
体積膨張倍率は1.1倍を超え、好ましくは1.1倍を超え10倍以下、より好ましくは1.1倍を超え8倍以下、さらに好ましくは1.2~5倍である。本発明の製造方法では、体積膨張倍率が1.1倍を超える、好ましくは1.2倍以上という比較的低い気泡分散程度で重合ゲル中の気泡が連通し易いという特徴を持つ。体積膨張倍率が高いほど容易に連続気泡となりえるが、上記倍率が高いほど気泡の孔径が大きくなる傾向にあり、単量体水溶液中での気泡安定性も悪くなるため、均一な気泡分散の操作が困難となる。多孔質ゲルを乾燥、粉砕して粉末として使用する場合には、粉末状の粒子としての取扱い性とかさ比重や耐衝撃性の過度の低下を避けるためにも、体積膨張倍率は10倍以下、8倍以下、さらには5倍以下とするのがよい。
体積膨張倍率が1.1倍を超えるようにする制御方法としては、上記気泡の発生装置の条件を適宜設定する;界面活性剤や親水性高分子等、気泡安定性を向上させる添加剤を単量体水溶液に添加する;脱泡して膨張倍率を調整するなどが挙げられる。
(2-2)単量体水溶液を整泡する工程(D)(整泡工程)
本発明では必要により整泡工程や脱泡工程を更に含んでもよい。整泡工程を含むことで、大きな気泡から順次単量体から除去され、過度の発泡を抑制し、均一な気泡とすることで、気泡の安定性を向上させる。用いられる整泡時間は5秒以上、10秒~60分、好ましくは30秒~30分、特に60秒~20分である。
本発明では必要により整泡工程や脱泡工程を更に含んでもよい。整泡工程を含むことで、大きな気泡から順次単量体から除去され、過度の発泡を抑制し、均一な気泡とすることで、気泡の安定性を向上させる。用いられる整泡時間は5秒以上、10秒~60分、好ましくは30秒~30分、特に60秒~20分である。
好ましい整泡方法として、後述する図1や図2の循環タンクを用いる方法が挙げられる。また、好ましい整泡方法として、気泡の導入後に常圧で所定時間保持、あるいは、それを繰り返す方法があり、保持温度は0~100℃、さらに好ましくは20~50℃である。
本発明で用いられる整泡工程は公知の技術を採用すればよく、米国特許第6667372号明細書に記載の方法や、株式会社テクノシステム出版「泡のエンジニアリング初版」759~774頁に記載の方法等がある。
好ましい整泡工程は循環式タンクへ循環気流(気泡)を含む単量体を循環させることであり、整泡工程は好ましくは循環式タンクの上部空間に酸素を1容積%以上含む。整泡工程の後に単量体水溶液を重合する工程(B)を行うことが好ましい。この際、循環ラインから少なくとも一部の単量体水溶液を、必要により中和した後に重合することが好ましい。その他、重合開始までの時間を配管中や重合装置中で、一定時間もたせることで、気泡を集積させて整泡をしてよい。
具体的に、このような脱泡工程(製泡工程)について、図1及び図2にそれぞれ例示する。尚、図1及び図2においては、循環ポンプや熱交換器等については省略している。
図1に示すように、タンク1に入った単量体は、タンク1の下部から循環ラインへと流出し、不活性気体が導入されながら循環ラインを流れ、タンク上部からタンク1へ再度流入することによって循環している。ここで、単量体は、不活性気体が導入されながら循環するため、当該気体が単量体中に溶解および/または分散することになる。そして、気体が溶解および/または分散した当該単量体は、タンク1へ流入し再度循環ラインへ流出するまでの間、タンク1内に滞留することになり、整泡が進むことになる。
また、単量体を中和しながら整泡する場合も同様に、図2に示すように、タンク1に入った、中和された単量体は、タンク1の底から、不活性気体が導入されながら流出し、上部からタンク1へ再度流入することによって循環して整泡を行うことができる。
また、整泡された単量体は、図1に示すように、そのまま抜き出されて次工程に用いてもよいし、図2に示すように、中和剤や架橋剤を添加した状態で抜き出されてもよい。
なお、循環タンクは国際公開第2007/28746号、同第2007/28747号、同第2009/123197号にも例示されているが、本発明では、図1および図2に示すように、循環前の単量体水溶液に不活性ガスを溶解または分散させ、さらにタンクに循環することで、気泡を均一化および安定化することができる。ここで、循環タンク内は不活性ガスで満たされていてもよいが、単量体の安定性から酸素、特に空気で満たされていることが好ましい。
また、上部が開放された重合装置に気泡を含有した単量体水溶液を投入後、大きな泡を中心に脱泡後、細かな気泡を安定的に含有する単量体水溶液に所定時間後に重合開始剤の添加や紫外線、赤外線、あるいはマイクロ波などのエネルギー線の照射を行って重合をさせてもよい。
気泡の大きさは、(a)レーザー回折散乱法(別称;静的光散乱法)、(b)動的光散乱法、(c)電気的検知帯法(通称;コールターカウンター法)、(d)パーティクルカウンター法(光り散乱方式、光遮断方式)、(e)カメラ撮影による可視化法、(f)レーザー光とCCDカメラによる干渉画像法等による測定が挙げられる。重合前の単量体水溶液での測定が困難な場合には、好ましくは、重合後のフォーム状重合体を、必要により半固体状のスライスし易い水分に調整してから切断し薄片を採取した後、顕微鏡を用いて目視か画像解析ソフトにより平均孔径を測定することができる。
個数の測定には、可能ならば(c)電気的検知帯法や(d)パーティクルカウンター法が用いられ、ナノオーダーの気泡を測定するには(b)動的光散乱法、(a)レーザー回折散乱法(別称;静的光散乱法)が適宜使用される。
(界面活性剤)
本発明において、単量体水溶液と不活性ガスとの混合を、好ましくは界面活性剤の存在下で行う。界面活性剤を用いることで気泡を安定的に分散させることができる。また、界面活性剤の種類や量を適宜調整することにより、所望の物性を有する吸水性樹脂を得ることができるが、本発明においては、界面活性剤の使用は任意であり、不存在下でも適用できる。かような界面活性剤は、特に限定されないが、例えば、アニオン性界面活性剤、ノニオン性界面活性剤、カチオン性界面活性剤、両性界面活性剤、フッ素系界面活性剤、有機金属界面活性剤等がある。これら界面活性剤は、単独で使用してもよいし、併用してもよい。
本発明において、単量体水溶液と不活性ガスとの混合を、好ましくは界面活性剤の存在下で行う。界面活性剤を用いることで気泡を安定的に分散させることができる。また、界面活性剤の種類や量を適宜調整することにより、所望の物性を有する吸水性樹脂を得ることができるが、本発明においては、界面活性剤の使用は任意であり、不存在下でも適用できる。かような界面活性剤は、特に限定されないが、例えば、アニオン性界面活性剤、ノニオン性界面活性剤、カチオン性界面活性剤、両性界面活性剤、フッ素系界面活性剤、有機金属界面活性剤等がある。これら界面活性剤は、単独で使用してもよいし、併用してもよい。
具体的には、ポリグリセリン脂肪酸エステルが挙げられ、好ましくは3量体以上、より好ましくは6~10量体のポリグリセリンの脂肪酸エステルであり、その脂肪酸としては、炭素原子数が6~28、より好ましくは12~24、特に好ましくは16~20の直鎖、または分岐を有している脂肪酸である。ポリグリセリン脂肪酸エステルとしては、具体的には、モノステアリン酸テトラグリセリル、モノオレイン酸テトラグリセリル、トリステアリン酸テトラグリセリル、ペンタステアリン酸テトラグリセリル、ペンタオレイン酸テトラグリセリル、モノラウリン酸テトラグリセリル、モノミリスチン酸テトラグリセリル、モノステアリン酸ヘキサグリセリル、モノオレイン酸ヘキサグリセリル、トリステアリン酸ヘキサグリセリル、ペンタステアリン酸ヘキサグリセリル、ペンタオレイン酸ヘキサグリセリル、ポリリシノール酸ヘキサグリセリル、モノラウリン酸デカグリセリル、モノステアリン酸デカグリセリル、モノミリスチン酸デカグリセリル、モノイソステアリン酸デカグリセリル、モノオレイン酸デカグリセリル、モノリノール酸デカグリセリル、ジステアリン酸デカグリセリル、ジイソステアリン酸デカグリセリル、トリステアリン酸デカグリセリル、トリオレイン酸デカグリセリル、トリオレイン酸デカグリセリル、ペンタステアリン酸デカグリセリル、ペンタイソステアリン酸デカグリセリル、ペンタオレイン酸デカグリセリル、ヘプタステアリン酸デカグリセリル、ヘプタオレイン酸デカグリセリル、デカステアリン酸デカグリセリル、デカイソステアリン酸デカグリセリル、デカオレイン酸デカグリセリル等がある。
ノニオン性界面活性剤としては、具体的には、ノニルフェノールポリエチレンオキサイド付加物;エチレンオキサイドとプロピレンオキサイドのブロックポリマー;ソルビタンモノラウレート、ソルビタンモノミリスチレート、ソルビタンモノパルミテート、ソルビタンモノステアレート、ソルビタントリステアレート、ソルビタンモノオレエート、ソルビタントリオレエート、ソルビタンセスキオレエート、ソルビタンジステアレート等のソルビタン脂肪酸エステル;グリセロールモノステアレート、グリセロールモノオレエート、ジグリセロールモノオレエート、自己乳化型グリセロールモノステアレート等のグリセリン脂肪酸エステル;ポリオキシエチレンラウリルエーテル、ポリオキシエチレンセチルエーテル、ポリオキシエチレンステアリルエーテル、ポリオキシエチレンオレイルエーテル、ポリオキシエチレン高級アルコールエーテル等のポリオキシエチレンアルキルエーテル;ポリオキシエチレンノニルフェニルエーテル等のポリオキシエチレンアルキルアリールエーテル;ポリオキシエチレンソルビタンモノラウレート、ポリオキシエチレンソルビタンモノラウレート、ポリオキシエチレンソルビタンモノパルミテート、ポリオキシエチレンソルビタンモノステアレート、ポリオキシエチレンソルビタントリステアレート、ポリオキシエチレンソルビタンモノオレエート、ポリオキシエチレンソルビタントリオレエート等のポリオキシエチレンソルビタン脂肪酸エステル;テトラオレイン酸ポリオキシエチレンソルビット等のポリオキシエチレンソルビトール脂肪酸エステル;ポリエチレングリコールモノラウレート、ポリエチレングリコールモノステアレート、ポリエチレングリコールジステアレート、ポリエチレングリコールモノオレエート等のポリオキシエチレン脂肪酸エステル;ポリオキシエチレンアルキルアミン;ポリオキシエチレン硬化ヒマシ油;アルキルアルカノールアミド等がある。
アニオン性界面活性剤としては、アニオン部と油溶性部とを有するものが好ましく使用でき、例えば、ナトリウムドデシルサルフェート、カリウムドデシルサルフェート、アンモニウムアルキルサルフェート等の如きアルキルサルフェート塩;ナトリウムドデシルポリグリコールエーテルサルフェート;ナトリウムスルホリシノエート;スルホン化パラフィン塩等の如きアルキルスルホネート;ナトリウムドデシルベンゼンスルホネート、アルカリフェノールヒドロキシエチレンのアルカリ金属サルフェート等の如きアルキルスルホネート;高アルキルナフタレンスルホン酸塩;ナフタレンスルホン酸ホルマリン縮合物、ナトリウムラウレート、トリエタノールアミンオレエート等の如き脂肪酸塩;ポリオキシアルキルエーテル硫酸エステル塩;ポリオキシエチレンカルボン酸エステル硫酸エステル塩、ポリオキシエチレンフェニルエーテル硫酸エステル塩;コハク酸ジアルキルエステルスルホン酸塩;ポリオキシエチレンアルキルアリールサルフェート塩等の如き二重結合を持った反応性アニオン乳化剤等がある。上記以外にも、特開平10-251310号公報に列記された界面活性剤が挙げられる。
これら界面活性剤の使用量は、使用されるモノマーに対して、なるべく少ない量が望まれる。好ましくは10重量%未満、より好ましくは5重量%未満、さらに好ましくは1重量%未満、特に好ましくは0.1重量%未満である。界面活性剤量が多いと、得られる吸水性樹脂が吸収する水性液と接した場合に、溶出する界面活性剤の量も多くなるため、オムツ等の衛生材料での実使用の場合には体液の界面張力を低下させうるため、できる限り少ない界面活性剤量の使用が望まれる。そのため、構造中にアクリル酸(塩)と重合できる反応性不飽和基を持つ界面活性剤、あるいはアクリル酸のカルボキシル基との反応性が高いエポキシ基やアミノ基を持つシリコーン系界面活性剤などのいわゆる反応性界面活性剤を使用したり、あるいは、高分子量の界面活性剤を用いたりするなどして、吸水時の界面活性剤の溶出を抑制することもできる。特に、気泡の安定分散のために、後述の親水性高分子等を併用し界面活性剤の使用量を減らして、上記の界面張力の低下を抑制することが好ましい。
本発明において、具体的には、後述の(5-8)で測定された吸水性樹脂の表面張力が好ましくは55[mN/m]以上、さらに好ましくは60[mN/m]以上、特に好ましくは65[mN/m]以上、更に好ましくは70[mN/m]以上となるように制御される。上限は通常75[mN/m]程度で十分である。表面張力の低下が抑制されるため、衛生用品の吸液特性が向上し、戻り量(Re-Wet)が低下する。
(2-3)単量体水溶液を重合して発泡重合体を得る工程(B)(重合工程)
本工程は、工程(A)、必要により工程(D)を経た単量体水溶液を重合して、発泡重合体を得る工程である。所定の体積膨張倍率に気泡を分散させた単量体水溶液は、一定量の気泡が維持されるように重合装置にできるだけ速やかに重合装置に供給することが望まれ、好ましくは5分以内、さらに好ましくは3分以内、特に好ましくは1分以内に重合装置に供給することが好ましい。
本工程は、工程(A)、必要により工程(D)を経た単量体水溶液を重合して、発泡重合体を得る工程である。所定の体積膨張倍率に気泡を分散させた単量体水溶液は、一定量の気泡が維持されるように重合装置にできるだけ速やかに重合装置に供給することが望まれ、好ましくは5分以内、さらに好ましくは3分以内、特に好ましくは1分以内に重合装置に供給することが好ましい。
(重合開始剤)
本発明で用いられる重合開始剤としては、特に制限はなく、重合させるモノマーの種類、重合条件等に合わせて、通常の吸水性樹脂の製造において利用されているものの中から1種又は2種以上を選択して使用することができる。例えば、熱分解型開始剤(例えば、過硫酸ナトリウム、過硫酸カリウム、過硫酸アンモニウム等の過硫酸塩;過酸化水素、t-ブチルパーオキシド、メチルエチルケトンパーオキシド等の過酸化物;2-カルバモイルアゾイソブチロニトリル等のアゾニトリル化合物、2,2’-アゾビス(2-メチルプロピオンアミジン)二塩酸塩等のアゾアミジン化合物、2,2’-アゾビス-2-(2-イミダゾリン-2-イル)プロパン塩酸塩等の環状アゾアミジン化合物、アゾアミド化合物、アルキルアゾ化合物、2,2’-アゾビス(2-アミジノプロパン)ジヒドロクロリド、2,2’-アゾビス[2-(2-イミダゾリン-2-イル)プロパン]ジヒドロクロリド等のアゾ化合物;等)や、光分解型開始剤(例えば、ベンゾイン誘導体、ベンジル誘導体、2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン等のアセトフェノン誘導体、ベンゾフェノン誘導体、アゾ化合物等)等を挙げることができる。これら重合開始剤の中でも、重合時に窒素ガスを発生するアゾ重合開始剤(好ましくは、水溶性アゾ重合開始剤、例えば、2,2’-アゾビス(2-メチルプロピオンアミジン)二塩酸塩)を使用して、さらに気泡を含有させてもよい。
本発明で用いられる重合開始剤としては、特に制限はなく、重合させるモノマーの種類、重合条件等に合わせて、通常の吸水性樹脂の製造において利用されているものの中から1種又は2種以上を選択して使用することができる。例えば、熱分解型開始剤(例えば、過硫酸ナトリウム、過硫酸カリウム、過硫酸アンモニウム等の過硫酸塩;過酸化水素、t-ブチルパーオキシド、メチルエチルケトンパーオキシド等の過酸化物;2-カルバモイルアゾイソブチロニトリル等のアゾニトリル化合物、2,2’-アゾビス(2-メチルプロピオンアミジン)二塩酸塩等のアゾアミジン化合物、2,2’-アゾビス-2-(2-イミダゾリン-2-イル)プロパン塩酸塩等の環状アゾアミジン化合物、アゾアミド化合物、アルキルアゾ化合物、2,2’-アゾビス(2-アミジノプロパン)ジヒドロクロリド、2,2’-アゾビス[2-(2-イミダゾリン-2-イル)プロパン]ジヒドロクロリド等のアゾ化合物;等)や、光分解型開始剤(例えば、ベンゾイン誘導体、ベンジル誘導体、2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン等のアセトフェノン誘導体、ベンゾフェノン誘導体、アゾ化合物等)等を挙げることができる。これら重合開始剤の中でも、重合時に窒素ガスを発生するアゾ重合開始剤(好ましくは、水溶性アゾ重合開始剤、例えば、2,2’-アゾビス(2-メチルプロピオンアミジン)二塩酸塩)を使用して、さらに気泡を含有させてもよい。
また、還元剤を併用することで、これら重合開始剤の分解を促進しうる。そこで、両者を組み合わせたレドックス系開始剤として使用することもできる。前記の還元剤としては、特に限定されないが、例えば、亜硫酸ナトリウム、亜硫酸水素ナトリウム等の(重)亜硫酸(塩)、L-アスコルビン酸(塩)、第一鉄塩等の還元性金属(塩)、アミン類等が挙げられる。レドックス系開始剤のように酸化性重合開始剤と還元剤を用いる場合、それぞれを単量体水溶液に合流させてもよいし、還元剤は予め単量体水溶液に混合しておいてもよい。
重合開始剤の使用量は、単量体全量に対して、0.0001~1モル%が好ましく、0.0005~0.5モル%がより好ましい。0.0001モル%以上であれば、残存モノマー量が少なくなるため好ましく、また1モル%以下であれば、吸水性樹脂の色調に影響を与えることが少ない。
(親水性高分子等)
上記重合に際しては、更に必要に応じて、重合前の単量体水溶液中に、一般に増粘剤として知られる、水溶性高分子または吸水性樹脂、水不溶性微粒子が使用される。
上記重合に際しては、更に必要に応じて、重合前の単量体水溶液中に、一般に増粘剤として知られる、水溶性高分子または吸水性樹脂、水不溶性微粒子が使用される。
具体的には、澱粉、澱粉誘導体(例えば、エーテル化澱粉、エステル化澱粉など)、セルロース、セルロース誘導体(例えば、カルボキシメチルセルロース、ヒドロキシエチルセルロースなど)、グアーガムなどの高分子多糖類、または、ポリビニルアルコール、ポリアクリル酸(塩)、ポリアクリル酸(塩)架橋体等の親水性高分子などの水溶性高分子、あるいは、二酸化ケイ素(シリカ)、ゼオライト、タルク、二酸化チタンなどの水不溶性微粒子を添加することにより、単量体水溶液中に分散した気泡の安定性を改良し、発泡を促進させることができる。さらに、微粉低減や発泡の促進のために、吸水性樹脂の後述の微粉(好ましくは、粒子径150μm以下の粉体を70重量%以上含む微粉)を重合工程にリサイクルして、単量体水溶液を増粘させてもよい。好ましくは、増粘剤として、水溶性高分子または吸水性樹脂、特にノニオン性水溶性高分子(特に澱粉、PVA、ヒドロキシエチルセロースなど)、またはポリアクリル酸(塩)系吸水性樹脂が使用される。
また、吸水性能の改良のために、次亜燐酸(塩)等の無機還元剤、キレート剤等の重合改良剤を添加してもよい。なお、無機還元剤およびキレート剤については後に詳述する。さらに、得られるフォーム状重合体の柔軟性を向上させるために、水以外の公知の可塑剤、例えば、グリセリン、ポリエチレングリコール、ポリプロピレングリコールのようなポリオールを使用してもよい。
上記する添加剤の使用量は単量体100重量部に対して0~50重量部、さらには0.01~20重量部である。
(重合方法)
本発明で採用される重合方法としては、特に限定されるものではなく、通常の吸水性樹脂の製造方法で用いられるものが採用され、例えば、水溶液重合法を挙げることができる。該水溶液重合法には、モノマー水溶液を静置状態(機械的な無攪拌状態)で重合させる静置重合法、攪拌装置内で重合する攪拌重合法等があるが、安定してフォーム状重合体を得たい場合には静置重合法が採用される。これらの重合法には、各々回分法と連続法があるが、連続法が好ましい。また、これらの重合法では、通常、ベルト重合装置やタンク式(サイロ式)重合装置や攪拌重合装置が採用されている。
本発明で採用される重合方法としては、特に限定されるものではなく、通常の吸水性樹脂の製造方法で用いられるものが採用され、例えば、水溶液重合法を挙げることができる。該水溶液重合法には、モノマー水溶液を静置状態(機械的な無攪拌状態)で重合させる静置重合法、攪拌装置内で重合する攪拌重合法等があるが、安定してフォーム状重合体を得たい場合には静置重合法が採用される。これらの重合法には、各々回分法と連続法があるが、連続法が好ましい。また、これらの重合法では、通常、ベルト重合装置やタンク式(サイロ式)重合装置や攪拌重合装置が採用されている。
本発明に係る吸水性樹脂の製造装置としては、上記方法によって連続供給される単量体水溶液を、連続的に重合することができる装置であれば、特に限定されるものではないが、連続ベルト重合装置又は連続攪拌重合装置であることが好ましい。所定の体積膨張倍率に気泡を分散させた単量体水溶液は、一定量の気泡が維持されるように重合装置に供給後はできるだけ速やかに重合を開始させることが望まれ、好ましくは重合装置に供給後5分以内、さらに好ましくは3分以内、特に好ましくは1分以内にエネルギー線照射や加熱により重合を開始させる。
なお、連続ベルト重合装置に関しては、特開2000-034305号公報や特開平11-228604号公報、特開昭62-156102号公報等に開示された技術を適用することができる。例えば、特開2002-212204号公報に開示された高温高濃度重合技術が適用されうる。この場合、好ましい重合装置の形態としては、エンドレスベルト式の連続重合装置であって、ベルトがフッ素樹脂製であるか、または表面をフッ素樹脂でコーティングされたベルトであることがよい。更に、加熱装置あるいは保温装置が具備され、重合時に発生する水及び/又はモノマー液の蒸気を回収・再利用するシステムを有する装置がよい。また、ベルトは、モノマー混合液の逆流防止のため、水平またはモノマー混合液供給部が低くなっているのが好ましく、重合ゲルがベルト上から排出されてからモノマー混合液供給口までの間に、ベルト洗浄工程が設置されたものが望ましい。
ベルト重合の厚みは目的や重合時の除熱に応じて適宜決定され、例えば、0.1~30cm、更には0.5~20cm、1~10cmのゲル厚みで重合される。また、ベルトの大きさは生産量によって決定され、工業的には例えば、ベルト幅も0.1~10m、1~5m程度、長さも5~200m程度で選択される。更に、タンク式で重合する場合、生産量によって決定され、工業的には0.1~300m3、1~100m3のタンク等が使用される。これら静置重合で得られた重合ゲルは必要により後述の切断、表面研磨、圧縮等を行って成型物としてもよく、また、粉末(非成型物)してもよい。
更に、連続攪拌重合装置に関しては、一軸攪拌装置でも、連続ニーダー等の複数攪拌軸を持つ攪拌装置でも採用することができるが、生産性の観点から、複数軸攪拌装置が好ましく用いられる。
(重合時の最高到達温度及び測定方法)
上記重合に際して、吸水性樹脂中の気泡を増加させるため沸騰重合(溶媒である水の沸点以上)が用いられ、重合工程の少なくとも一時期が100℃以上とされる。すなわち、重合中の温度の上限を100℃以上、好ましくは100~140℃、より好ましくは102~130℃、更に好ましくは104~120℃、特に好ましくは105~118℃の範囲とし重合中に水分を蒸発させる、いわゆる沸騰重合とする。なお、減圧重合や加圧重合、水に溶解させた単量体などでのモル沸点上昇などで、水の沸点(100℃)が変化する場合も含め、本発明では100℃以上の重合工程を経る場合を沸騰重合と呼ぶ。重合時の最高到達温度は、上記範囲内で、適宜、重合中に加熱や冷却を行ってよく、重合熱だけで上記範囲内としてもよい。
上記重合に際して、吸水性樹脂中の気泡を増加させるため沸騰重合(溶媒である水の沸点以上)が用いられ、重合工程の少なくとも一時期が100℃以上とされる。すなわち、重合中の温度の上限を100℃以上、好ましくは100~140℃、より好ましくは102~130℃、更に好ましくは104~120℃、特に好ましくは105~118℃の範囲とし重合中に水分を蒸発させる、いわゆる沸騰重合とする。なお、減圧重合や加圧重合、水に溶解させた単量体などでのモル沸点上昇などで、水の沸点(100℃)が変化する場合も含め、本発明では100℃以上の重合工程を経る場合を沸騰重合と呼ぶ。重合時の最高到達温度は、上記範囲内で、適宜、重合中に加熱や冷却を行ってよく、重合熱だけで上記範囲内としてもよい。
重合の温度は接触温度計や非接触温度計(例えば赤外線温度計)で測定でき、ベルト重合では重合ゲルの表面温度などで測定できる。本発明の方法では、連続気泡の重合ゲルが得られ、かつ重合時に溶媒である水が沸騰して、水が蒸発しているため、重合ゲルは表面と内部で実質均一な温度に沸騰しているため、重合ゲルの最高温度は内部で測定してもよく、表面で測定してもよいが、好ましくは、内部(中心部)で規定される。重合ゲルの中心部の最高温度測定には、熱伝対等の接触温度計で測定すればよい。
重合時の最高到達温度の好ましい測定方法の一例としては、温度変化の急速な系の測温のために、(株)キーエンス(Keyence)製PCカード型データ収集システムNR-1000を用い、熱伝対を重合系の中心部に置き、サンプリング周期0.1秒で測定できる。得られた温度-時間チャートから重合開始温度、ピーク温度(最高到達温度)を読み取れる。
(その他の重合条件等)
重合時の圧力としては、減圧重合(特に大気圧の10%を超えて減圧)や加圧重合(同10%を超えて加圧)でもよいが、装置の簡便さやコスト、さらには発泡効率などから大気圧の±10%以内、±7%以内、±5%以内、±3%以内、±1%以内、特に±0.1%以内での実質常圧での重合が行われる。ここで、意図的に減圧や加圧が行われなくても、重合時の温度上昇や低下、さらに、重合容器への不活性ガスや空気の導入や排気によって、重合時の圧力が多少変化してもよく、上記範囲で実質常圧での重合が行われる。
重合時の圧力としては、減圧重合(特に大気圧の10%を超えて減圧)や加圧重合(同10%を超えて加圧)でもよいが、装置の簡便さやコスト、さらには発泡効率などから大気圧の±10%以内、±7%以内、±5%以内、±3%以内、±1%以内、特に±0.1%以内での実質常圧での重合が行われる。ここで、意図的に減圧や加圧が行われなくても、重合時の温度上昇や低下、さらに、重合容器への不活性ガスや空気の導入や排気によって、重合時の圧力が多少変化してもよく、上記範囲で実質常圧での重合が行われる。
本発明で重合時に100℃以上となる時間は重合方法により適宜決定され、好ましくは1秒以上、5秒以上、さらには30秒以上であり、下記のゲルの固形分濃度の上昇幅を示すように調整することが好ましい。長時間の沸騰重合は発泡に寄与しにくいだけでなく、物性(吸水倍率や可溶分)を低下させることもあり、よって、重合で100℃以上の時間は、上限は1時間以内、さらには30分以内、10分以内、5分以内、特に1分以内とされる。
なお、重合の最高到達温度(ピーク温度)が140℃を超えると、吸水性樹脂粉末の物性が低下するおそれがある。好ましくは130℃以下、より好ましくは120℃以下とする。重合中の水分蒸発量は、開始温度などの違いにより異なるものの、単量体水溶液中の固形分濃度に対する重合ゲルの固形分濃度の上昇幅が2重量%以上(上限は20重量%で、重合後にゲル状物、特に固形分濃度が80重量%以下である範囲が好ましい。)が好ましく、更に上昇幅3~20重量%、特に5~20重量%となるようにするのが好ましい。ただし、単量体水溶液中の固形分濃度は下記式で定義される。なお、前述の単量体濃度(式2)においては、不揮発性の添加剤の重量(例えば、親水性高分子、界面活性剤等)は考慮されないが、以下の固形分濃度では不揮発性添加剤の重量を含んで規定される。また、上述した水蒸気の発生は、連続気泡の生成に寄与すると推定される。
なお、通常、重合は重合機中で不活性ガス等の気流の存在下で行われるため、重合中の温度が100℃未満でも重合時間が長い場合には水分の蒸発が起こるが、これは本発明で開示する気泡の連通には効果がなく意図するところでない。
本発明においては、単量体水溶液に気泡を分散させ1.1倍を超えて体積膨張をさせて表面積を増やしているため比較的除熱がよく温度が上がりにくいが、重合中の上限温度を上記の範囲となるように調整し、重合中の積極的な水分蒸発が起こるようにすることで、発泡重合体(フォーム状重合体)中の連通孔(連続気泡)の比率を向上させることができ、吸収速度の大きなフォーム状吸水性樹脂またはそれを用いた吸水速度の大きな吸水性樹脂粉末を製造することができるようになった。また、上記のためには、重合の開始温度は室温より高くした高温開始が好ましく、具体的には重合工程で重合開始温度40℃以上、さらには50℃以上が好ましく、上限は100℃以下、さらには90℃以下、特に好ましくは85℃以下である。開始温度が40℃未満では100℃に到達が困難となり、高すぎると気泡の安定分散が困難となり好ましくない。
重合時間については、モノマー及び重合開始剤の種類、重合温度等に応じて、適宜決定されればよいが、重合開始から重合温度が最高温度に到達するまでの時間は短くするのが好ましく、具体的には好ましくは20分以内、より好ましくは10分以内、更に好ましくは5分以内、特に好ましくは2分以内、最も好ましくは1分以内とする。前記時間が例えば、1時間以上のように長くなると、重合中に分散した気泡が消泡したりして、発生した水蒸気がゲルの隔壁を突き破り難くなり、分散した気泡が連続気泡となりにくいため好ましくない。本発明の条件において、重合中の気泡安定性、重合中の気泡壁を形成する重合ゲルの強度及び水分の蒸発のバランスが連続気泡の形成に有利となっていると考えられる。
この場合、重合工程でアクリル酸系単量体水溶液の濃度は高いほど好ましく、40重量%以上、45重量%以上、50重量%以上である(上限は通常80重量%以下、さらには70重量%以下)。かかる濃度が40重量%以上の場合には、分散した気泡が40℃以上でも安定して存在できるのに対して、40重量%未満、例えば、特許文献23の実施例に開示するような条件では、室温では安定した気泡の分散が可能なものの、温度が高くなると気泡が不安定となり短時間のうちに消泡してしまう。これを避けるためには、開始温度を低くし、かつ、重合温度が上がるまでの重合時間を比較的長くして重合率を上げ気泡壁のゲル強度を高める必要があると考えられる。この場合には、分散した気泡は独立した状態でゲル中存在することになろう。
前記した連続ベルト重合では、帯状に連続したフォーム状の重合ゲルが得られる。また、タンク式(サイロ式)の静置重合では、タンク(サイロ)形状のフォーム状の重合ゲルが得られる。フォーム状重合ゲルはそのままで使用することもできるし、細かく解砕して使用ないし/さらに使用することもできる(下記細分化工程)。フォーム状重合ゲルは気泡を含まない高固形分の板状のゲルに比べて粉砕時の装置負荷も小さくなる傾向となる利点がある。このことは、特開2002-212204号に開示する高固形分ゲルの粉砕に有利な切断式粉砕機に加えて、板状のゲルでは不可能であったチョッパーに代表される押出し式粉砕機への適用も可能とする。この場合、フォーム状ゲルを混練しながら粉砕することで、混練しない粉砕の場合に比べて、吸水速度を不利に遅くすることなく吸水性樹脂粉末としたときの嵩比重と耐衝撃性の向上を図ることができよう。フォーム状重合ゲルはさらに乾燥・粉砕して吸水性樹脂粉末としてもよいし、フォーム状重合ゲルを、例えば、所定の形のシート状に切断して乾燥し吸水性樹脂成形体としてもよい。成形された吸水性樹脂または吸水性樹脂粉末はさらに表面架橋処理されたり、造粒されたり、水分調製や各種改質剤を添加されたりして吸水性樹脂製品として使用される。重合ゲルの粉砕や乾燥及び表面架橋処理は公知の技術を採用すればよい。
(従来のフォーム状吸水性樹脂の製造技術と重合時の最高到達温度)
吸水性樹脂の重合時における最高温度を低く制御することは周知の技術であり、特許文献20には、フォーム状吸水性樹脂の製造において、「重合温度は、好ましくは重合性水性混合物の沸騰を避けるように調整する。」旨、開示される。また、特許文献8、21、30~32にも、特許文献20と同様にフォーム状吸水性樹脂の製造において、「重合時の沸騰を回避」する旨が開示される。さらに、特許文献33には、「オープンセルフォームは65℃以下でフォームを製造すると増加する」旨、開示される。
吸水性樹脂の重合時における最高温度を低く制御することは周知の技術であり、特許文献20には、フォーム状吸水性樹脂の製造において、「重合温度は、好ましくは重合性水性混合物の沸騰を避けるように調整する。」旨、開示される。また、特許文献8、21、30~32にも、特許文献20と同様にフォーム状吸水性樹脂の製造において、「重合時の沸騰を回避」する旨が開示される。さらに、特許文献33には、「オープンセルフォームは65℃以下でフォームを製造すると増加する」旨、開示される。
また、発泡重合以外の重合において、可溶分低減等の目的のため、最高温度を低く制御する技術が開示されている。具体的には、最高温度を95℃以下とする技術(特許文献34)、重合温度を20~70℃で重合する技術(特許文献35)、重合温度を20~95℃で重合する技術(特許文献36)が提案されている。
上記したように特許文献8、20、21、29~33等、沸騰重合は(連続)フォーム状吸水性樹脂の製造において避けられてきたが、本発明では従来フォーム状吸水性樹脂の製造で避けられてきた沸騰重合が、40重量%以上の濃度及び1.1倍以上の体積膨張倍率において、効率的に連続気泡を有する吸水性樹脂を提供することを見いだした。
(連続気泡の生成機構)
上記特許文献8、20、21、29~33等に開示のない、本発明の連続気泡の生成機構は、下記のように推測されるが、かかる推定機構の正否は本発明の範囲を制限するものではない。
上記特許文献8、20、21、29~33等に開示のない、本発明の連続気泡の生成機構は、下記のように推測されるが、かかる推定機構の正否は本発明の範囲を制限するものではない。
即ち、単量体濃度40重量%以上の単量体水溶液に、体積膨張倍率1.1倍以上となるまで気泡を分散させたのち、沸騰重合することで、単量体水溶液中に分散した独立気泡(例えば、窒素ガス等の球形気泡)が体積膨張するとともに、単量体水溶液の沸騰重合によるゲル化時に、水の沸騰で発生する水蒸気及び単量体水溶液中に分散した独立気泡の体積膨張によって、単量体水溶液の重合で発生する重合初期のゲルの気泡(球形の独立気泡)の隔壁を突き破って、連続気泡を生成するものとも推定される。ここで、沸騰重合は水蒸気の発生によるゲルの隔壁破壊に必要であり、また、単量体濃度40重量%以上の単量体水溶液は、その固形分由来のゲルの硬さと重合速度から、重合初期のゲルの気泡(球形の独立気泡)の隔壁を突き破って、連続気泡を生成しやすいとも推定される。
具体的に、例えば、単量体濃度45重量%の単量体水溶液(100g、うち固形分45g)が沸騰重合によって固形分50重量%の重合ゲル(うち固形分45g)を与える場合、沸騰重合によって水10gの蒸発、即ち、100℃の水蒸気としても約17Lの水蒸気の蒸発を伴うため、固形分45gに対して、かかる多量の水蒸気がゲル中の気泡(球形の独立気泡)の膨張を伴って、独立気泡の隔壁を突き破って、連続気泡を生成するものとも推定される。
かかる事実は、後述の比較例13~15で、沸騰重合、単量体濃度40重量%以上、体積膨張倍率1.1以上、のひとつが欠如しても、十分な連続気泡を提供しないことからも支持される。ただし、これらの推定機構は、発明の範囲を制限するものではない。
(細分化工程)
重合時に粉砕、細分化を同時に行う、または重合後の発泡重合体を粉砕し細粒化する、細分化工程を必要により行ってもよい。ゲル粉砕、特に混練によってゲル粉砕(細分化)されることで、吸水速度と通液性との両立が図れ、更に耐衝撃性も向上する。具体的には、重合工程が連続ニーダー重合の場合、重合時にゲルがゲル粉砕され、また、重合工程が連続ベルト重合の場合、重合後にゲルがゲル粉砕される。ニーダー重合を行う場合、ニーダーにより重合中にゲルのゲル粉砕を行うことができるため、ニーダー重合工程に細分化工程が含まれる。
重合時に粉砕、細分化を同時に行う、または重合後の発泡重合体を粉砕し細粒化する、細分化工程を必要により行ってもよい。ゲル粉砕、特に混練によってゲル粉砕(細分化)されることで、吸水速度と通液性との両立が図れ、更に耐衝撃性も向上する。具体的には、重合工程が連続ニーダー重合の場合、重合時にゲルがゲル粉砕され、また、重合工程が連続ベルト重合の場合、重合後にゲルがゲル粉砕される。ニーダー重合を行う場合、ニーダーにより重合中にゲルのゲル粉砕を行うことができるため、ニーダー重合工程に細分化工程が含まれる。
使用できるゲル粉砕機は、特に限定されず、例えば、バッチ式又は連続式の双腕型ニーダー等、複数の回転攪拌翼を備えたゲル粉砕機、1軸押出機、2軸押出機、ミートチョッパー等が挙げられる。中でも、先端に多孔板を有するスクリュー型押出機が好ましく、例えば、特開2000-63527号公報に開示されたスクリュー型押出機が挙げられる。
ゲル粉砕(解砕)時の含水ゲルの温度は、物性の面から40~120℃が好ましく、50~110℃がより好ましい。該ゲル温度が40~120℃であると、含水ゲルの硬度が適当でゲル粉砕時に粒子形状や粒度分布の制御を容易に行うことができる。なお、ゲル温度は、重合時の温度や重合後の加熱又は冷却等で制御することができる。
ゲル粉砕(解砕)後の粒子状含水ゲルの重量平均粒子径(D50)は、0.5~4mmが好ましく、0.5~3mmがより好ましく、0.6~2mmが更に好ましい。上記粒子状含水ゲルの重量平均粒子径(D50)が、0.5mm以上であれば、残存モノマー(単量体)の増加や吸水速度(FSR)の向上効果が期待でき、重量平均粒子径(D50)が4mm以下であれば、乾燥時間が短く、水可溶分(Ext)の増加を抑制できる。5mm以上の粒径を有する粒子状含水ゲルの割合は、粒子状含水ゲル全体の0~10重量%が好ましく、0~5重量%がより好ましい。上記粒子状含水ゲルの粒子径は、粉砕工程後の吸水性樹脂粉末の粒子径と同様に、特定の目開きの篩で分級することによって求められる。また、重量平均粒子径(D50)についても、同様に求めることができる。但し、上記粒子状含水ゲルの分級操作が、乾式の分級方法では凝集等により測定が困難である場合は、特開2000-63527号公報の段落〔0091〕に記載の、湿式の分級方法を用いて測定する。
好適には、特願2010-088993号(国際出願PCT/JP2011/058829)に記載されたゲル粉砕、特にゲル粉砕エネルギー(GGE)を18~60[J/g]とするゲル粉砕が、本発明に適用される。ゲル粉砕エネルギーは、上限値として、60[J/g]以下が好ましく、50[J/g]以下がより好ましく、40[J/g]以下が更に好ましい。下限値としては、18[J/g]以上が好ましく、20[J/g]以上がより好ましく、25[J/g]以上が更に好ましい。
(2-4)発泡重合体を加熱乾燥する工程(C)(加熱乾燥工程)
上記して得られたフォーム状重合ゲルは乾燥され、乾燥重合体とされる。その乾燥減量(粉末ないし粒子1gを180℃で3時間加熱)から求められる樹脂固形分は、好ましくは80重量%以上、より好ましくは85~99重量%、さらに好ましくは90~98重量%の範囲に調整され乾燥重合体を得る。粉末以外の成形された吸水性樹脂としての使用の場合には必要により水や可塑剤を添加して柔軟性を調整してもよい。
上記して得られたフォーム状重合ゲルは乾燥され、乾燥重合体とされる。その乾燥減量(粉末ないし粒子1gを180℃で3時間加熱)から求められる樹脂固形分は、好ましくは80重量%以上、より好ましくは85~99重量%、さらに好ましくは90~98重量%の範囲に調整され乾燥重合体を得る。粉末以外の成形された吸水性樹脂としての使用の場合には必要により水や可塑剤を添加して柔軟性を調整してもよい。
乾燥温度は、特に限定されるものではないが、好ましくは100~300℃の範囲内、より好ましくは150~250℃の範囲内とすればよい。本発明のフォーム状重合ゲルは、そのままでも粉砕されたものでも乾燥が容易であり、特開2000-212215に開示の乾燥方法を好ましく適用される。重合ゲル中の独立気泡が多い場合には、粗粉砕ゲルを高温乾燥する時にゲルが膨張し変形しやすいが、本発明のゲルは連続気泡が多いため高温乾燥時のゲル膨張がほとんど見られず変形が少ないのも有利な特徴である。また、本発明のゲルは連続気泡が多いため、乾燥重合体を得るまでの乾燥時間が短くなることも有利な特徴である。
即ち、従来、重合後の含水ゲルの乾燥には大きな設備や熱エネルギー、更には長い乾燥時間が必要であり、この長時間の乾燥による吸水性樹脂の劣化や着色、更には吸水性樹脂の製造コストの増大という問題が生じていたが、本発明ではかかる問題を解消し、乾燥時間の短縮、着色防止、吸水性能の向上等を達成する。
(2-5)粉砕工程/分級工程、粒度分布
(粒度)
上記の加熱乾燥工程を経た吸水性樹脂はシート状やブロック状で使用してもよいが、粉砕および/または分級されることで、所定の粒度の粉末とすることが好ましい。
(粒度)
上記の加熱乾燥工程を経た吸水性樹脂はシート状やブロック状で使用してもよいが、粉砕および/または分級されることで、所定の粒度の粉末とすることが好ましい。
(粉末)
吸水性樹脂粉末とした場合の重量平均粒子径(D50)としては、物性向上の観点から、200~600μm、好ましくは300μm以上600μm未満、より好ましくは200~550μm、さらに好ましくは250~500μm、特に好ましくは350~450μmに調整される。また、150μm未満の粒子が少ないほどよく、通常0~5重量%、好ましくは0~3重量%、特に好ましくは0~1重量%に調整される。さらに、850μm以上(さらには710μm以上)の粒子が少ないほどよく、通常0~5重量%、好ましくは0~3重量%、特に好ましくは0~1重量%に調整される。また、本発明では好ましくは850~150μmの割合、さらには710~150μmの割合が95重量%以上さらには98重量%以上(上限100重量%)で表面架橋される。
吸水性樹脂粉末とした場合の重量平均粒子径(D50)としては、物性向上の観点から、200~600μm、好ましくは300μm以上600μm未満、より好ましくは200~550μm、さらに好ましくは250~500μm、特に好ましくは350~450μmに調整される。また、150μm未満の粒子が少ないほどよく、通常0~5重量%、好ましくは0~3重量%、特に好ましくは0~1重量%に調整される。さらに、850μm以上(さらには710μm以上)の粒子が少ないほどよく、通常0~5重量%、好ましくは0~3重量%、特に好ましくは0~1重量%に調整される。また、本発明では好ましくは850~150μmの割合、さらには710~150μmの割合が95重量%以上さらには98重量%以上(上限100重量%)で表面架橋される。
これらの測定方法については、標準篩を用いて、例えば、国際公開第2004/69915号パンフレットやEDANA-ERT420.2-02に記載されている。上記表面架橋前の粒度は好ましくは表面架橋後さらには最終製品(別称;粒子状吸水剤)にも適用される。
(2-6)成型物および成型工程
上記して得られたフォーム状重合ゲルやその乾燥重合体は粉末としてもよいが、ベルト重合やタンク重合などの静置重合後に得られたフォーム状重合ゲルやその乾燥重合体は、そのまま使用してもよく、成型してもよい。すなわち、本発明では重合工程と同時に、または重合工程後、成型工程を含む。重合工程の後、加熱乾燥を経た乾燥重合体を成型物としてもよいし、重合工程と同時にまたは重合工程後、成型工程を経て加熱乾燥を行ってもよい。ベルト重合で得られたフォーム状重合ゲルは帯状であり、タンク重合ではタンク形状のフォーム状重合ゲル(例えば円筒状ゲル)が得られるため、それらをまま成型物として使用してもよく、また、必要により切断、表面研磨、圧縮などを行ってよい。また、また、ナプキンなどの所定形状に合わせて立体的な型に単量体水溶液を入れて重合と同時に最終製品形態の吸水性樹脂成型物を得てもよい。
上記して得られたフォーム状重合ゲルやその乾燥重合体は粉末としてもよいが、ベルト重合やタンク重合などの静置重合後に得られたフォーム状重合ゲルやその乾燥重合体は、そのまま使用してもよく、成型してもよい。すなわち、本発明では重合工程と同時に、または重合工程後、成型工程を含む。重合工程の後、加熱乾燥を経た乾燥重合体を成型物としてもよいし、重合工程と同時にまたは重合工程後、成型工程を経て加熱乾燥を行ってもよい。ベルト重合で得られたフォーム状重合ゲルは帯状であり、タンク重合ではタンク形状のフォーム状重合ゲル(例えば円筒状ゲル)が得られるため、それらをまま成型物として使用してもよく、また、必要により切断、表面研磨、圧縮などを行ってよい。また、また、ナプキンなどの所定形状に合わせて立体的な型に単量体水溶液を入れて重合と同時に最終製品形態の吸水性樹脂成型物を得てもよい。
上記したベルト重合では、例えば、厚み0.1~30cm、さらには0.5~20cm、1~10cmのシート状重合ゲルが連続的に得られ、その幅もベルチ幅により0.1~10m、1~5m程度となるが、得られたシート状重合ゲルを厚み方向、幅方向、長さ方向に適宜、切断、表面研磨、くりぬき、圧縮などを行って、成型物とすればよい。また、タンク式で重合する場合、体積で0.1~300m3、1~100m3の重合ゲル(例えば、円筒状ゲル、立法体状ゲルなど)が得られるが、かかる重合ゲルは必要により切断、表面研磨、圧縮などを行って成型物としてもよい。
成型物の形状は目的に応じて適宜決定されるが、ナプキンやおむつを目的とする場合、厚み0.1~2cm、さらには0.2~1cmで面積5cm2以上のシート状物とされることが好ましい。シート状物は最終のナプキンやおむつの吸収層にまで成型されてもよく、さらに成型(切断、裁断)できるようにロール状や絨毯状としてもよい。また、上記シート状物の表面に模様や孔、エンボスを持たせてもよい。
(2-7)表面架橋工程
(架橋剤)
本発明では乾燥後に表面架橋工程をさらに含んでもよい。表面架橋には、共有結合性表面架橋剤および/またはイオン架橋性の表面架橋剤が使用され、好ましくはそれら架橋剤が併用される。
(架橋剤)
本発明では乾燥後に表面架橋工程をさらに含んでもよい。表面架橋には、共有結合性表面架橋剤および/またはイオン架橋性の表面架橋剤が使用され、好ましくはそれら架橋剤が併用される。
なお、表面架橋とは表面を内部に比べて高架橋にする操作であり、ラジカル重合開始剤(例えば過硫酸塩や光開始剤)によりラジカル架橋したり、粒子表面に単量体を加えて表面重合したりしてもよいが、好ましくはポリアクリル酸(塩)系吸水性樹脂のカルボキシル基と反応しうる架橋剤が用いられ、以下に説明するが、本発明の表面架橋は下記に限定されない。
(共有結合性表面架橋剤)
本発明で用いることの出来る表面架橋剤としては、種々の有機または無機架橋剤を例示できるが、有機表面架橋剤が好ましく使用できる。物性面で好ましくは、表面架橋剤として、多価アルコール化合物、エポキシ化合物、オキセタン化合物、多価アミン化合物またはそのハロエポキシ化合物との縮合物、オキサゾリン化合物、(モノ、ジ、またはポリ)オキサゾリジノン化合物、アルキレンカーボネート化合物であり、特に高温での反応が必要な、多価アルコール化合物、アルキレンカーボネート化合物、オキサゾリジノン化合物からなる脱水反応性架橋剤が使用できる。脱水反応性架橋剤を使用しない場合、より具体的には、米国特許第6228930号、同第6071976号、同第6254990号等に例示されている化合物を挙げることが出来る。例えば、モノ,ジ,トリ,テトラまたはそれ以上の多量体のプロピレングリコール、1,3-プロパンジオール、グリセリン、1,4-ブタンジオール、1,3-ブタンジオール、1,5-ペンタンジオール、1,6-ヘキサンジオール、ソルビトール等の多価アルコール化合物;エチレングリコールジグリシジルエーテルやグリシドール等のエポキシ化合物;エチレンカーボネート等のアルキレンカーボネート化合物;オキセタン化合物;2-イミダゾリジノン等の環状尿素化合物等が挙げられる。
本発明で用いることの出来る表面架橋剤としては、種々の有機または無機架橋剤を例示できるが、有機表面架橋剤が好ましく使用できる。物性面で好ましくは、表面架橋剤として、多価アルコール化合物、エポキシ化合物、オキセタン化合物、多価アミン化合物またはそのハロエポキシ化合物との縮合物、オキサゾリン化合物、(モノ、ジ、またはポリ)オキサゾリジノン化合物、アルキレンカーボネート化合物であり、特に高温での反応が必要な、多価アルコール化合物、アルキレンカーボネート化合物、オキサゾリジノン化合物からなる脱水反応性架橋剤が使用できる。脱水反応性架橋剤を使用しない場合、より具体的には、米国特許第6228930号、同第6071976号、同第6254990号等に例示されている化合物を挙げることが出来る。例えば、モノ,ジ,トリ,テトラまたはそれ以上の多量体のプロピレングリコール、1,3-プロパンジオール、グリセリン、1,4-ブタンジオール、1,3-ブタンジオール、1,5-ペンタンジオール、1,6-ヘキサンジオール、ソルビトール等の多価アルコール化合物;エチレングリコールジグリシジルエーテルやグリシドール等のエポキシ化合物;エチレンカーボネート等のアルキレンカーボネート化合物;オキセタン化合物;2-イミダゾリジノン等の環状尿素化合物等が挙げられる。
(イオン結合性表面架橋剤)
また、上記有機表面架橋剤以外にイオン結合性表面架橋剤としてポリアミンポリマーや多価金属塩を使用して通液性などを向上させてもよい。使用される多価金属塩(無機表面架橋剤)は2価以上、好ましくは3価ないし4価の多価金属の塩(有機塩ないし無機塩)ないし水酸化物が例示できる。使用できる多価金属としてはアルミニウム、ジルコニウム等が挙げられ、乳酸アルミニムや硫酸アルミニムが挙げられる。
また、上記有機表面架橋剤以外にイオン結合性表面架橋剤としてポリアミンポリマーや多価金属塩を使用して通液性などを向上させてもよい。使用される多価金属塩(無機表面架橋剤)は2価以上、好ましくは3価ないし4価の多価金属の塩(有機塩ないし無機塩)ないし水酸化物が例示できる。使用できる多価金属としてはアルミニウム、ジルコニウム等が挙げられ、乳酸アルミニムや硫酸アルミニムが挙げられる。
(溶媒)
表面架橋剤の使用量は吸水性樹脂粉末100重量部に対して0.001~10重量部、0.01~5重量部で適宜決定される。表面架橋剤に合わせて好ましくは水が使用され得る。使用される水の量は吸水性樹脂粉末100重量部に対して0.5~20重量部、より好ましくは0.5~10重量部の範囲である。無機表面架橋剤と有機表面架橋剤を併用する場合も吸水性樹脂粉末100重量部に対して各々0.001~10重量部、0.01~5重量部で併用される。また、この際、親水性有機溶媒を使用してもよく、またその量は、吸水性樹脂粉末100重量部に対して、0~10重量部、好ましくは0~5重量部の範囲である。また吸水性樹脂粉末への架橋剤溶液の混合に際し、本発明の効果を妨げない範囲、例えば、0~10重量部以下、好ましくは0~5重量部、より好ましくは0~1重量部で、酸触媒、塩基性触媒、水不溶性微粒子や界面活性剤を共存させてもよい。用いられる界面活性剤やその使用量は米国特許第7473739号等に例示されている。
表面架橋剤の使用量は吸水性樹脂粉末100重量部に対して0.001~10重量部、0.01~5重量部で適宜決定される。表面架橋剤に合わせて好ましくは水が使用され得る。使用される水の量は吸水性樹脂粉末100重量部に対して0.5~20重量部、より好ましくは0.5~10重量部の範囲である。無機表面架橋剤と有機表面架橋剤を併用する場合も吸水性樹脂粉末100重量部に対して各々0.001~10重量部、0.01~5重量部で併用される。また、この際、親水性有機溶媒を使用してもよく、またその量は、吸水性樹脂粉末100重量部に対して、0~10重量部、好ましくは0~5重量部の範囲である。また吸水性樹脂粉末への架橋剤溶液の混合に際し、本発明の効果を妨げない範囲、例えば、0~10重量部以下、好ましくは0~5重量部、より好ましくは0~1重量部で、酸触媒、塩基性触媒、水不溶性微粒子や界面活性剤を共存させてもよい。用いられる界面活性剤やその使用量は米国特許第7473739号等に例示されている。
表面架橋剤を混合後の吸水性樹脂は加熱処理され、必要によりその後冷却処理される。加熱温度は70~300℃、好ましくは120~250℃、より好ましくは150~250℃であり、加熱時間は好ましくは1分~2時間である。
かかる表面架橋によって、吸水性樹脂粉末の場合には後述の加圧下吸水倍率(AAP)を、好ましくは20[g/g]以上、さらには23~30[g/g]にまで向上すればよい。
(2-8)微粉リサイクル工程
重合工程後、好ましくは加熱乾燥工程後の吸水性樹脂は必要により粉砕、分級工程を経て、上記粒度に調整される。また、分級で除去される粗大粒子(例えば1mm以上)は必要により粉砕してもよく、また、分級で除去される微粒子(例えば150μm未満、さらには106μm未満)は廃棄してもよく、他の用途に使用してもよく、微粉リサイクルしてもよい。
重合工程後、好ましくは加熱乾燥工程後の吸水性樹脂は必要により粉砕、分級工程を経て、上記粒度に調整される。また、分級で除去される粗大粒子(例えば1mm以上)は必要により粉砕してもよく、また、分級で除去される微粒子(例えば150μm未満、さらには106μm未満)は廃棄してもよく、他の用途に使用してもよく、微粉リサイクルしてもよい。
すなわち、本発明の製造方法においては、好ましくは微粉リサイクル工程を含んでもよい。微粉リサイクル工程とは、乾燥工程および必要により粉砕、分級工程で発生する微粉(特に粒子径150μm以下の粉体を70重量%以上含む微粉)を分離した後、そのままの状態で、あるいは水和ないし造粒して、粉砕工程以前にリサイクル、好ましくは、重合工程、発泡重合体の細分化工程、または加熱乾燥工程にリサイクルする工程をいう。微粉をリサイクルすることで、ベースポリマーの粒度を制御することができるとともに、微粉の添加によって、より吸水速度を向上することができる。微粉としては表面架橋前の微粉でもよく、表面架橋後の微粉でもよく、微粉リサイクル量は乾燥物の1~40重量%、さらには5~30重量%で適宜設定される。
本発明で好ましく用いられる微粉リサイル方法は、重合時の単量体水溶液や重合途中の含水ゲルに吸水性樹脂微粉やその水和物や造粒物、また必要により無機微粒子を混合する方法である。また、リサイクルされる微粉をもって、重合時の単量体を増粘させて発泡を促進してもよい。
(2-9)その他の工程
上記以外に、必要により、第2の分級工程、蒸発モノマーのリサイクル工程、造粒工程、微粉除去工程等を設けてもよい。さらには、経時での色安定性効果やゲル劣化防止等のために、以下のキレート剤および/または還元剤を単量体ないしその重合物に使用してもよい。すなわち、着色防止および劣化防止のために、本発明では好ましくはキレート及び/又は還元剤の添加工程を含む。
上記以外に、必要により、第2の分級工程、蒸発モノマーのリサイクル工程、造粒工程、微粉除去工程等を設けてもよい。さらには、経時での色安定性効果やゲル劣化防止等のために、以下のキレート剤および/または還元剤を単量体ないしその重合物に使用してもよい。すなわち、着色防止および劣化防止のために、本発明では好ましくはキレート及び/又は還元剤の添加工程を含む。
以下、キレート剤及び還元剤について説明する。
(キレート剤)
本発明の連続気泡を有する吸水性樹脂は、耐尿性、着色防止の観点から、好ましくはキレート剤を含む。上記特許文献20等に開示のないキレート剤を使用することによって、連続気泡を有した耐尿性、着色防止に優れた吸水性樹脂を提供する。
本発明の連続気泡を有する吸水性樹脂は、耐尿性、着色防止の観点から、好ましくはキレート剤を含む。上記特許文献20等に開示のないキレート剤を使用することによって、連続気泡を有した耐尿性、着色防止に優れた吸水性樹脂を提供する。
本発明のキレート剤としては、効果の面から、高分子化合物又は非高分子化合物、中でも非高分子化合物が好ましく、具体的には、アミノ多価カルボン酸、有機多価燐酸、無機多価燐酸、アミノ多価燐酸およびこれらの塩から選ばれる化合物が好ましい。効果の面から、キレート剤の分子量は100~5000であることが好ましく、より好ましくは200~1000である。吸水性樹脂がキレート剤を含むことによって、樹脂の経時着色および劣化が抑制される。
ここで、多価とは1分子中に複数の該官能基を有し、好ましくは2~30個、更には3~20個、4~10個の該官能基を有する。また、これらキレート剤は水溶性キレート剤、具体的には、100g(25℃)の水に1g以上、更には10g以上溶解する水溶性キレート剤であることが好ましい。尚、具体的なキレート剤及びその含有量等については、国際公開第2011/040530号パンフレットの段落[0104]~[0111]の記載内容を準用する。
(有機又は無機還元剤)
本発明に係る連続気泡を有する吸水性樹脂は、耐尿性、着色防止の観点から好ましくは有機又は無機還元剤、より好ましくは無機還元剤を含み、更に好ましくは無機還元剤として、還元性無機元素を有する水溶性無機化合物又は還元性無機元素を有する水溶性有機化合物を含む。尚、上記「水溶性」とは、25℃の水100gに対して1g以上、更には5g以上、特に10g以上溶解することをいう。上記特許文献20等に開示のない有機又は無機還元剤を使用することによって、連続気泡を有した、残存モノマー、着色や劣化の面で優れた吸水性樹脂となる。
本発明に係る連続気泡を有する吸水性樹脂は、耐尿性、着色防止の観点から好ましくは有機又は無機還元剤、より好ましくは無機還元剤を含み、更に好ましくは無機還元剤として、還元性無機元素を有する水溶性無機化合物又は還元性無機元素を有する水溶性有機化合物を含む。尚、上記「水溶性」とは、25℃の水100gに対して1g以上、更には5g以上、特に10g以上溶解することをいう。上記特許文献20等に開示のない有機又は無機還元剤を使用することによって、連続気泡を有した、残存モノマー、着色や劣化の面で優れた吸水性樹脂となる。
なお、本発明における無機還元剤の具体例については、国際公開第2011/040530号パンフレットの段落[0114]~[0121]の記載内容を準用する。
さらに、目的に応じて、吸水性樹脂粉末には酸化剤、酸化防止剤、水、多価金属化合物、シリカや金属石鹸等の水不溶性無機ないし有機粉末、消臭剤、抗菌剤、パルプや熱可塑性繊維等を吸水性樹脂粉末中に0~3重量%、好ましくは0~1重量%添加してもよい。吸水性樹脂粉末中の好ましい界面活性剤量は上記範囲である。
本発明はまた、連続気泡のポリアクリル酸系フォーム状吸水性樹脂、特に開泡気泡率5%以上、好ましくは5~98%、より好ましくは5~90%のポリアクリル酸系吸水性樹脂を提供する。ここで、「吸水性樹脂」の開放気泡率は、「発泡重合体(含水ゲル)」または「吸水性樹脂(粉末)」の開放気泡率を規定したものである。更に、課題を解決する上で特に好ましい開放気泡率は、吸水性樹脂(粉末)の場合には5~30%が好ましく、5~15%がより好ましい。また、吸水性樹脂成型物(特にシート状成型物)の場合には5~98%が好ましく、10~90%がより好ましい。
なお、乾燥前の発泡重合体(含水ゲル)の開放気泡率は、5%以上であることが好ましく、5~90%であることがより好ましい。優位に連続気泡を含むものは、乾燥後に形状(すなわち体積)の変化がほとんど見られず、独立気泡が多いものは膨張して体積が増加し形状も球形に近づく。
なお、本発明では吸水性樹脂(粉末)の独立気泡率の含有量は、特に限定されるものではないが、5%以上であることが好ましく、5~25%であることがより好ましく、10~25%であることがさらに好ましい。また、乾燥前の発泡重合体(含水ゲル)の独立気泡率は、0%以上であることが好ましく、0~20%であることがより好ましい。
なお、独立気泡と開放気泡以外は吸水性樹脂であり、その合計は100%であるため、独立気泡率との開放気泡率の合計は好ましくは吸水性樹脂粉末の場合、好ましくは10~90%、より好ましくは10~50%、特に好ましくは10~40%である。また、吸水性樹脂成型物(特にシート状成型物)の場合、好ましくは10~98%、より好ましくは、20~90%、特に好ましくは50~90%である。
〔3〕ポリアクリル酸系吸水性樹脂(粉末)の物性
衛生材料、特に紙オムツを目的とする場合、上記重合や表面架橋をもって、下記(3-1)~(3-9)の少なくとも1つ、さらにはAAPを含め2つ以上、特に3つ以上に制御されることが好ましい。下記を満たさない場合、後述の高濃度オムツでは十分な性能を発揮しないことがある。
衛生材料、特に紙オムツを目的とする場合、上記重合や表面架橋をもって、下記(3-1)~(3-9)の少なくとも1つ、さらにはAAPを含め2つ以上、特に3つ以上に制御されることが好ましい。下記を満たさない場合、後述の高濃度オムツでは十分な性能を発揮しないことがある。
本発明の製造方法は下記の吸水性樹脂粉末の製造方法に好適に適用できるが、好ましくは、吸水速度(FSR)の制御および向上に適用できる。なお、下記および実施例の物性は断りのない限りEDANA法で規定される。
本発明では、下記式で規定される吸水速度指数が90以上で、かつ、嵩比重が0.3~0.8[g/cm3]のポリアクリル酸系吸水性樹脂粉末であることが好ましい。また、下記式で規定される吸水速度指数が90以上で、かつ、嵩比重が0.3~0.8[g/cm3]のポリアクリル酸系吸水性樹脂粉末を表面架橋することがより好ましい。
ただし、FSRは生理食塩水への20倍膨潤での吸水速度を示す。
吸水速度指数は90、95、100、105、110、115、120の順に高いほど好ましく、上限は150さらには140で十分である。かかる吸水性樹脂粉末は通液性や耐衝性に優れ、紙オムツ等の吸収性物品に好適に使用できる。吸水速度指数が低い場合や逆に高すぎる場合も実使用に適さない傾向にある。
かかる吸水性樹脂粉末は発泡構造(別称;多孔質構造)であり、多孔質構造は電子顕微鏡写真で粒子表面を確認することで判別できる。粒子表面の平均孔径は好ましくは200μm以下、さらに好ましくは0.1~150μm、1~100μmである。個々の粉末の主成分は多孔質粒子である。また、ゲルを混練粉砕機で粉砕した場合には、上記の他にも複雑な形状を含む。
(3-1)AAP(加圧下吸水倍率)
紙オムツでのモレを防止するため、上記重合を達成手段の一例として、2.06kPaの加圧下さらには4.83kPaの加圧下での0.9重量%の塩化ナトリウム水溶液に対する吸水倍率(AAP)が好ましくは20[g/g]以上、よりに好ましくは22[g/g]以上、さらに好ましくは24[g/g]以上に制御される。AAPの上限値は、特に限定されないが、他の物性とのバランスから40[g/g]以下が好ましい。上記AAPが20[g/g]未満の場合、かような吸水性樹脂を吸収体に使用すると、吸収体に圧力が加わった際の液の戻り(通常、「リウェット(Re-Wet)」とも称される)が少ない衛生用品を得ることができないおそれがある。
紙オムツでのモレを防止するため、上記重合を達成手段の一例として、2.06kPaの加圧下さらには4.83kPaの加圧下での0.9重量%の塩化ナトリウム水溶液に対する吸水倍率(AAP)が好ましくは20[g/g]以上、よりに好ましくは22[g/g]以上、さらに好ましくは24[g/g]以上に制御される。AAPの上限値は、特に限定されないが、他の物性とのバランスから40[g/g]以下が好ましい。上記AAPが20[g/g]未満の場合、かような吸水性樹脂を吸収体に使用すると、吸収体に圧力が加わった際の液の戻り(通常、「リウェット(Re-Wet)」とも称される)が少ない衛生用品を得ることができないおそれがある。
(3-2)CRC(無加圧下吸水倍率)
無加圧下吸水倍率(CRC)は、好ましくは10[g/g]以上であり、より好ましくは20[g/g]以上、さらに好ましくは25[g/g]以上、特に好ましくは30[g/g]以上に制御される。CRCは高いほど好ましく上限値は特に限定されないが、他の物性のバランスから、好ましくは50[g/g]以下、より好ましくは45[g/g]以下、さらに好ましくは40[g/g]以下である。上記CRCが10[g/g]未満の場合、吸水性樹脂の吸水量が低く、紙オムツ等、衛生用品中の吸収体への使用に適さないおそれがある。また、上記CRCが50[g/g]を超える場合、かような吸水性樹脂を吸収体に使用すると、液の取込み速度に優れる衛生用品を得ることができないおそれがある。
無加圧下吸水倍率(CRC)は、好ましくは10[g/g]以上であり、より好ましくは20[g/g]以上、さらに好ましくは25[g/g]以上、特に好ましくは30[g/g]以上に制御される。CRCは高いほど好ましく上限値は特に限定されないが、他の物性のバランスから、好ましくは50[g/g]以下、より好ましくは45[g/g]以下、さらに好ましくは40[g/g]以下である。上記CRCが10[g/g]未満の場合、吸水性樹脂の吸水量が低く、紙オムツ等、衛生用品中の吸収体への使用に適さないおそれがある。また、上記CRCが50[g/g]を超える場合、かような吸水性樹脂を吸収体に使用すると、液の取込み速度に優れる衛生用品を得ることができないおそれがある。
(3-3)SFC(生理食塩水流れ誘導性)
紙オムツでのモレを防止するため、上記重合を達成手段の一例として、加圧下での液の通液特性である0.69重量%生理食塩水流れ誘導性(SFC)は1[×10-7・cm3・s・g-1]以上、好ましくは20[×10-7・cm3・s・g-1]以上、より好ましくは50[×10-7・cm3・s・g-1]以上、さらに好ましくは70[×10-7・cm3・s・g-1]以上、特に好ましくは100[×10-7・cm3・s・g-1]以上に制御される。SFCの上限値は、特に限定されないが、3000[×10-7・cm3・s・g-1]以下が好ましく、2000[×10-7・cm3・s・g-1]以下がより好ましい。上記SFCが3000[×10-7・cm3・s・g-1]を超える場合、かような吸水性樹脂を吸水体に使用すると、吸水体で液漏れが発生するおそれがある。SFCは周知の測定法であり、例えば、米国特許第5562646号で規定できる。
紙オムツでのモレを防止するため、上記重合を達成手段の一例として、加圧下での液の通液特性である0.69重量%生理食塩水流れ誘導性(SFC)は1[×10-7・cm3・s・g-1]以上、好ましくは20[×10-7・cm3・s・g-1]以上、より好ましくは50[×10-7・cm3・s・g-1]以上、さらに好ましくは70[×10-7・cm3・s・g-1]以上、特に好ましくは100[×10-7・cm3・s・g-1]以上に制御される。SFCの上限値は、特に限定されないが、3000[×10-7・cm3・s・g-1]以下が好ましく、2000[×10-7・cm3・s・g-1]以下がより好ましい。上記SFCが3000[×10-7・cm3・s・g-1]を超える場合、かような吸水性樹脂を吸水体に使用すると、吸水体で液漏れが発生するおそれがある。SFCは周知の測定法であり、例えば、米国特許第5562646号で規定できる。
本発明では通液性の向上、特に、SFC20[×10-7・cm3・s・g-1]以上の、高通液性の吸水性樹脂粉末の製法に好適に適用できる。
(3-4)Ext(水可溶分)
水可溶分は、好ましくは0~35重量%以下、より好ましくは25重量%以下であり、さらに好ましくは15重量%以下、特に好ましくは10重量%以下である。上記Extが35重量%を超える場合、得られる吸水性樹脂のゲル強度が弱く、液透過性に劣ったものとなるおそれがある。また、かような吸水性樹脂を吸水体に使用すると、吸水体に圧力が加わった際の液の戻り(リウェット)が少ない吸水性樹脂を得ることができないおそれがある。
水可溶分は、好ましくは0~35重量%以下、より好ましくは25重量%以下であり、さらに好ましくは15重量%以下、特に好ましくは10重量%以下である。上記Extが35重量%を超える場合、得られる吸水性樹脂のゲル強度が弱く、液透過性に劣ったものとなるおそれがある。また、かような吸水性樹脂を吸水体に使用すると、吸水体に圧力が加わった際の液の戻り(リウェット)が少ない吸水性樹脂を得ることができないおそれがある。
(3-5)FSR(吸水速度)
20gの生理食塩水に対する吸水性樹脂粉末1gでの吸水速度(FSR)は、通常0.1[g/g/s]以上、0.15[g/g/s]以上、0.20[g/g/s]以上、さらには0.25[g/g/s]以上、0.35[g/g/s]以上、0.45[g/g/s]以上である。上限は20[g/g/s]である。FSRの測定法は下記実施例で規定される。
20gの生理食塩水に対する吸水性樹脂粉末1gでの吸水速度(FSR)は、通常0.1[g/g/s]以上、0.15[g/g/s]以上、0.20[g/g/s]以上、さらには0.25[g/g/s]以上、0.35[g/g/s]以上、0.45[g/g/s]以上である。上限は20[g/g/s]である。FSRの測定法は下記実施例で規定される。
(3-6)初期色調
吸水性樹脂のL値は、87以上であることが好ましく、90以上であることがより好ましい。本発明の一実施形態である吸水性樹脂は開放気泡率が5%以上と高いため、L値が高くなる。さらにp-メトキシフェノールの含有量が60ppm以下であるとよりL値が高くなる。
吸水性樹脂のL値は、87以上であることが好ましく、90以上であることがより好ましい。本発明の一実施形態である吸水性樹脂は開放気泡率が5%以上と高いため、L値が高くなる。さらにp-メトキシフェノールの含有量が60ppm以下であるとよりL値が高くなる。
(3-7)嵩比重
吸水性樹脂粉末の嵩比重は、0.3~0.8[g/cm3]であり、好ましくは0.4~0.7[g/cm3]、さらに好ましくは0.5~0.7[g/cm3]である。本発明では発泡構造(別称;多孔質構造)であり、通常の発泡していない粒子と比べ嵩比重が低くなる。
吸水性樹脂粉末の嵩比重は、0.3~0.8[g/cm3]であり、好ましくは0.4~0.7[g/cm3]、さらに好ましくは0.5~0.7[g/cm3]である。本発明では発泡構造(別称;多孔質構造)であり、通常の発泡していない粒子と比べ嵩比重が低くなる。
(3-8)表面張力
表面張力(実施例の測定法で規定)は、好ましくは50[mN/m]以上、より好ましくは55[mN/m]以上、60[mN/m]以上、65[mN/m]以上、さらに好ましくは70[mN/m]以上、特に72[mN/m]以上である。72[mN/m]以上であれば実質的な表面張力の低下はない。上限は通常75[mN/m]で十分である。
表面張力(実施例の測定法で規定)は、好ましくは50[mN/m]以上、より好ましくは55[mN/m]以上、60[mN/m]以上、65[mN/m]以上、さらに好ましくは70[mN/m]以上、特に72[mN/m]以上である。72[mN/m]以上であれば実質的な表面張力の低下はない。上限は通常75[mN/m]で十分である。
(3-9)粉末の粒度
吸水性樹脂は成型物でもよく、非成型物(粉末状)でもよいが、粉末とする場合、上記(2-5)の範囲の粒度分布とされることが好ましい。上記物性は形状が粉末である場合、特に好適に規定される。
吸水性樹脂は成型物でもよく、非成型物(粉末状)でもよいが、粉末とする場合、上記(2-5)の範囲の粒度分布とされることが好ましい。上記物性は形状が粉末である場合、特に好適に規定される。
(3-10)成型物の形状
吸水性樹脂を成型物とする場合、好ましくは、上記(2-6)に記載したシート状物とされる。当該シートの形状は上記(2-6)に記載の通りであるが、これらに限定されない。図6に、吸水性樹脂成型物の代表的な形状(シート状)を示す。
吸水性樹脂を成型物とする場合、好ましくは、上記(2-6)に記載したシート状物とされる。当該シートの形状は上記(2-6)に記載の通りであるが、これらに限定されない。図6に、吸水性樹脂成型物の代表的な形状(シート状)を示す。
〔4〕ポリアクリル酸系吸水性樹脂の用途
本発明の吸水性樹脂の用途は特に限定されないが、例えば、農園芸向け保水用、廃液固化用、産業用、衛生材料用等であり、本発明の吸水性樹脂は通気性や吸水速度に優れ、白色であるため、好ましくは、紙オムツ、生理ナプキン、失禁パット等の吸収性物品、より好ましくは衛生材料用吸収性物品、特に好ましくは紙オムツに使用され得る。また、本発明の吸水性樹脂がシート状であれば、そのまま保形性を有する吸収体(吸収コア)となり得る。更に、粉末状であっても、過度の嵩比重の低下もないため、薄型の紙オムツに使用され得る。
本発明の吸水性樹脂の用途は特に限定されないが、例えば、農園芸向け保水用、廃液固化用、産業用、衛生材料用等であり、本発明の吸水性樹脂は通気性や吸水速度に優れ、白色であるため、好ましくは、紙オムツ、生理ナプキン、失禁パット等の吸収性物品、より好ましくは衛生材料用吸収性物品、特に好ましくは紙オムツに使用され得る。また、本発明の吸水性樹脂がシート状であれば、そのまま保形性を有する吸収体(吸収コア)となり得る。更に、粉末状であっても、過度の嵩比重の低下もないため、薄型の紙オムツに使用され得る。
本発明の吸水性樹脂は上記の特性を有するため、パルプの使用量を低減することが可能となる。従って、この吸収性物品中の、任意に他の吸収性材料(パルプ繊維等)を含む吸収体における吸水性樹脂粉末の含有量(コア濃度)は、30~100重量%、好ましくは40~100重量%、より好ましくは50~100重量%、さらに好ましくは60~100重量%、特に好ましくは70~100重量%、最も好ましくは75~95重量%で本発明の効果が発揮される。
〔5〕実施例
以下、実施例に従って発明を説明するが、本発明は実施例に限定され解釈させるものではない。尚、特に断りのない限り、各実施例での各工程は実質常圧(大気圧の±5%、更に好ましくは1%以内)で行われ、同一工程では意図的な加圧又は減圧による圧力変化は加えずに実施した。物性等の測定に関しては、特に断りのない限り、室温(20~25℃)、相対湿度40~50%RH下で行った。
以下、実施例に従って発明を説明するが、本発明は実施例に限定され解釈させるものではない。尚、特に断りのない限り、各実施例での各工程は実質常圧(大気圧の±5%、更に好ましくは1%以内)で行われ、同一工程では意図的な加圧又は減圧による圧力変化は加えずに実施した。物性等の測定に関しては、特に断りのない限り、室温(20~25℃)、相対湿度40~50%RH下で行った。
(5-1)開放(連続)気泡率
本発明に係る開放(連続)気泡率は、島津製作所HP記載の方法に準じて行った。
(http://www.shimadzu.co.jp/powder/user/appli/csc221.pdf)
(a)発泡重合体
重合工程を経て得られた発泡重合体を鋭利なナイフできれいに切断し、一辺が5mmの立方体試料を得た。次に、ノギスを用いて、当該立方体試料の幾何学的な(外接形状の)体積(va)[cm3]及び表面積(sa)[cm2]とを、その寸法を正確に測定して求めた。一方、当該立方体試料の試料体積(Va)[cm3]を乾式密度計(株式会社島津製作所製;アキュピックII-1340)を用いて測定した。尚、幾何学的な(外接形状の)体積(va)から開放気泡(上記立方体試料の作成時に独立気泡が開放気泡となったものを含む)の体積を減じたものが試料体積(Va)となる。
本発明に係る開放(連続)気泡率は、島津製作所HP記載の方法に準じて行った。
(http://www.shimadzu.co.jp/powder/user/appli/csc221.pdf)
(a)発泡重合体
重合工程を経て得られた発泡重合体を鋭利なナイフできれいに切断し、一辺が5mmの立方体試料を得た。次に、ノギスを用いて、当該立方体試料の幾何学的な(外接形状の)体積(va)[cm3]及び表面積(sa)[cm2]とを、その寸法を正確に測定して求めた。一方、当該立方体試料の試料体積(Va)[cm3]を乾式密度計(株式会社島津製作所製;アキュピックII-1340)を用いて測定した。尚、幾何学的な(外接形状の)体積(va)から開放気泡(上記立方体試料の作成時に独立気泡が開放気泡となったものを含む)の体積を減じたものが試料体積(Va)となる。
続いて、上記立方体試料を更に切断して細かくし、その幾何学的な(外接形状の)体積(v’a)[cm3]と表面積(s’a)[cm2]とをノギスで測った寸法を用いて求めた。又、試料体積(V’a)[cm3]について乾式密度計を用いて求めた。
上記操作で得られた数値を用いて以下の連立方程式を立て、単位体積あたりの開放気泡の体積(Voa)[cm3/cm3]及び発泡重合体を切断することで開放される単位面積あたりの(独立)気泡の体積(Vca)[cm3/cm2]を求めた。

尚、上式中、(va×Voa)、(v’a×Voa)は開放気泡の体積[cm3]を、(sa×Vca)、(s’a×Vca)は試料準備の過程で開放された(独立)気泡の体積[cm3]である。
以上により、発泡重合体の開放(連続)気泡率(Voa×100[%])が求まる。
(b)吸水性樹脂(粉末)
重合工程を経て得られた発泡重合体を鋭利なナイフできれいに切断し、一辺が2mmの立方体試料を得た。その後、熱風乾燥機(温度180℃、風速2.0[m/s]、30分間)で乾燥し、ロールミルで粉砕して粒子状の吸水性樹脂(粉末)を得た。
重合工程を経て得られた発泡重合体を鋭利なナイフできれいに切断し、一辺が2mmの立方体試料を得た。その後、熱風乾燥機(温度180℃、風速2.0[m/s]、30分間)で乾燥し、ロールミルで粉砕して粒子状の吸水性樹脂(粉末)を得た。
次に、当該粒子状の吸水性樹脂(粉末)を、目開き850μm、710μm、600μm、500μm、425μm、300μm、212μm、150μm、45μmのJIS標準篩(JIS Z8801-1(2000))を用いて篩い分けし、粒子径が500μm以上600μm未満、及び300μm以上425μm未満のフラクションを取り出した。
上記2種類のフラクションにおける幾何学的な(外接形状の)体積(vb,v’b)[cm3]及び表面積(sb,s’b)[cm2]は、各フラクションの粒子を球と仮定して算出した。即ち、粒子径が500μm以上600μm未満のフラクションは直径550μm、粒子径が300μm以上425μm未満のフラクションは直径362.5μmの球と仮定した。
又、各フラクションの1粒あたりの試料体積(Vb,V’b)[cm3]は、上記発泡重合体と同様、乾式密度計を用いて測定した。即ち、各フラクション全体の重量及び既知数(例えば、100個)の粒子の重量を測定することで1粒あたりの重量が求められ、各フラクション全体の粒子数を把握することができる。そして、乾式密度計で得られた値とから、1粒あたりの試料体積(Vb,V’b)[cm3]が求められる。
上記操作で得られた数値を用いて以下の連立方程式を立てて、単位体積あたりの開放気泡の体積(Vob)[cm3/cm3]及び試料準備の過程で開放される(と仮定する)単位面積あたりの(独立)気泡の体積(Vcb)[cm3/cm2]を求めた。
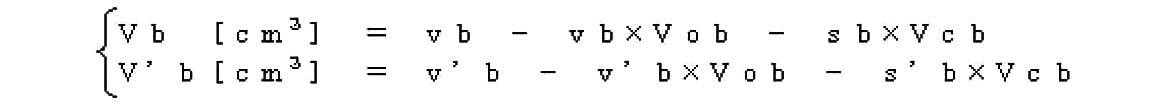
尚、上式中、(vb×Vob)、(v’b×Vob)は開放気泡の体積[cm3]を、(sb×Vcb)、(s’b×Vcb)は試料準備の過程で開放された(と仮定する)(独立)気泡の体積[cm3]である。
以上により、吸水性樹脂(粉末)の開放(連続)気泡率(Vob×100[%])が求まる。
(5-2)独立気泡率
(a)発泡重合体
発泡重合体を鋭利なナイフできれいに切断し、一辺が5mmの立方体試料を得た。次に、ノギスを用いて、当該立方体試料の幾何学的な(外接形状の)体積(Vga)[cm3]をその寸法を正確に測定して求めた。また、当該立方体試料の重量Wa[g]を測定した。
(a)発泡重合体
発泡重合体を鋭利なナイフできれいに切断し、一辺が5mmの立方体試料を得た。次に、ノギスを用いて、当該立方体試料の幾何学的な(外接形状の)体積(Vga)[cm3]をその寸法を正確に測定して求めた。また、当該立方体試料の重量Wa[g]を測定した。
次に、当該立方体試料の含水率αa[重量%]を(5-5)に開示した方法により求めた。続いて、含水率を求めた後の乾燥された当該立方体試料について、(5-13)に開示した方法により45μm以下になるまで微粉砕することで吸水性樹脂部の真密度[g/cm3]を求めた(図8参照)。更に水の比重(1.00[g/cm3])から、次式に従って、立方体試料の真密度Da[g/cm3]を算出した。
更に、上記立方体試料の幾何学的な(外接形状の)体積(Vga)[cm3]、重量Wa[g]、真密度Da[g/cm3]及び上記(5-1)で求めた発泡重合体の開放(連続)気泡率(Voa[%])から、次式に従って、発泡重合体の独立気泡率(Vc[%])を求めた。
(b)吸水性樹脂(粉末)の場合
発泡重合体を鋭利なナイフできれいに切断し、一辺が2mmの立方体試料を得た。その後、熱風乾燥機(温度180℃、風速2.0[m/s]、30分間)で乾燥し、ロールミルで粉砕して粒子状の吸水性樹脂(粉末)を得た。
発泡重合体を鋭利なナイフできれいに切断し、一辺が2mmの立方体試料を得た。その後、熱風乾燥機(温度180℃、風速2.0[m/s]、30分間)で乾燥し、ロールミルで粉砕して粒子状の吸水性樹脂(粉末)を得た。
次に、当該粒子状の吸水性樹脂(粉末)を、目開き850μm、710μm、600μm、500μm、425μm、300μm、212μm、150μm、45μmのJIS標準篩(JIS Z8801-1(2000))を用いて篩い分けし、粒子径が500μm以上600μm未満及び45μm未満のフラクションを取り出した。
粒子径45μm未満のフラクションについて、その全体の重量[g]と体積[cm3]から密度[g/cm3]を求め、これを吸水性樹脂(粉末)の吸水性樹脂部の真密度とした。更に(5-5)に記載した方法で含水率αb[重量%]とから、次式に従って、吸水性樹脂(粉末)の真密度Db[g/cm3]を算出した。
更に、上記粒子径が500μm以上600μm未満のフラクションにおける幾何学的な(外接形状の)体積(Vgb)[cm3]((5-1)(b)と同様、フラクションの粒子を球と仮定した。従って、Vgbは直径550μmの球の体積となる。)、重量Wb[g]、真密度Db[g/cm3]及び上記(5-1)で求めた吸水性樹脂(粉末)の開放(連続)気泡率(Vob[%])から、次式に従って、吸水性樹脂(粉末)の独立気泡率(Vd[%])を求めた。
(5-3)重量平均粒子径(D50)及び粒度分布の対数標準偏差(σζ)
本発明に係る吸水性樹脂(粉末)の重量平均粒子径(D50)及び粒度分布の対数標準偏差(σζ)は以下の手順に従って測定した。
本発明に係る吸水性樹脂(粉末)の重量平均粒子径(D50)及び粒度分布の対数標準偏差(σζ)は以下の手順に従って測定した。
即ち、吸水性樹脂(粉末)10.0gを、室温(23±2℃)、湿度50RH%の条件下で、目開き850μm、710μm、600μm、500μm、425μm、300μm、212μm、150μm、45μmのJIS標準篩(THE IIDA TESTING SIEVE:径8cm/JIS Z8801-1(2000))に仕込み、振動分級器(IIDA SIEVE SHAKER;TYPE ES-65型/SER.No.0501)を回転数60Hz、230rpm/衝撃数60Hz、130rpmで稼働し、5分間分級した。次いで、残留百分率Rを対数確率紙にプロットし、R=50重量%に相当する粒子径を重量平均粒子径(D50)として読み取った。又、粒度分布の対数標準偏差(σζ)は下記式により求められ、σζの値が小さいほど粒度分布が狭いことを意味する。
尚、X1はR=84.1重量%、X2はR=15.9重量%に相当する粒子径をそれぞれ意味する。
(5-4)無加圧下吸水倍率(CRC)
本発明に係る吸水性樹脂(粉末)の無加圧下吸水倍率(CRC)は、ERT441.2-02に準じて測定した。
本発明に係る吸水性樹脂(粉末)の無加圧下吸水倍率(CRC)は、ERT441.2-02に準じて測定した。
即ち、吸水性樹脂(粉末)0.200g(重量W0[g])を秤量し、不織布製の袋(60×85mm)に均一に入れヒートシールした後、25±3℃に調温した0.9重量%塩化ナトリウム水溶液500ml中に浸漬した。30分経過後、袋を引き上げ、遠心分離機(株式会社コクサン社製遠心機:形式H-122)を用いて、250G、3分間の条件で水切りを行った。その後、袋の重量(W1[g])を測定した。
同様の操作を、吸水性樹脂(粉末)を入れずに行い、そのときの袋の重量(W2[g])を測定した。得られたW0[g]、W1[g]、W2[g]から次式にしたがって、無加圧下吸水倍率(CRC)を算出した。
(5-5)含水率及び固形分
本発明に係る吸水性樹脂(粉末)の含水率は、ERT430.2-02に準じて測定した。
本発明に係る吸水性樹脂(粉末)の含水率は、ERT430.2-02に準じて測定した。
即ち、底面の大きさが直径約50mmのアルミカップに、吸水性樹脂(粉末)1.00gを量り取り、試料(吸水性樹脂(粉末)及びアルミカップ)の総重量W3[g]を測定した。次に、雰囲気温度180℃の無風オーブン中に上記試料を静置して、吸水性樹脂(粉末)を乾燥させた。3時間経過後、オーブンから該試料を取り出し、デシケーター中で室温まで冷却した。その後、乾燥後の試料(乾燥後の吸水性樹脂(粉末)及びアルミカップ)の総重量W4[g]を測定し、次式に従って含水率[重量%]を算出した。
尚、固形分は、次式に従って求めることができる。
(5-6)吸水速度(FSR)
本発明に係る吸水性樹脂(粉末)の吸水速度(FSR)は、以下の手順に従って測定した。
本発明に係る吸水性樹脂(粉末)の吸水速度(FSR)は、以下の手順に従って測定した。
即ち、吸水性樹脂(粉末)1.00gを上部が開放された円筒形のガラス製容器(直径32~34mm、高さ50mm)に入れ、吸水性樹脂(粉末)の上面が水平となるようにした。この際、必要により、慎重にガラス製容器の底をたたく等して、吸水性樹脂(粉末)の上面を水平にしてもよい。
次に、23±0.2℃に調温した0.90重量%の塩化ナトリウム水溶液20gを50mlのガラス製ビーカーに量り取り、塩化ナトリウム水溶液及びガラス製ビーカーの合計重量(重量W5[g])を測定した。その後、当該塩化ナトリウム水溶液を吸水性樹脂(粉末)の入ったガラス製容器に素早く丁寧に注いだ。
上記ガラス製容器に注いだ塩化ナトリウム水溶液が吸水性樹脂(粉末)に接触した時点を起点として、塩化ナトリウム水溶液の上面が塩化ナトリウム水溶液を吸収した吸水性樹脂(粉末)の膨潤ゲルに置き換わるまでの時間(時間ts[秒])を測定した。尚、上面状態の確認は約20°の角度から目視によって行った。
次に、塩化ナトリウム水溶液投入後の空となった50mlのガラス製ビーカーの重量(重量W6[g])を測定し、次式に従って吸水速度(FSR)[g/g/s]を算出した。
(5-7)嵩比重
本発明に係る吸水性樹脂(粉末)の嵩比重は、JIS K 3362に準じて嵩比重測定器(蔵持科学機器製作所製)を用いて測定した。
本発明に係る吸水性樹脂(粉末)の嵩比重は、JIS K 3362に準じて嵩比重測定器(蔵持科学機器製作所製)を用いて測定した。
即ち、粒度による偏りを無くすために十分に混合された吸水性樹脂(粉末)100.0gを、ダンパーを閉めた漏斗に入れた後、速やかにダンパーを開け吸水性樹脂(粉末)を内容量100mlの受器(重量W7[g])に落下させた。次いで、当該受器から盛り上がった吸水性樹脂(粉末)をガラス棒ですり落とし、吸水性樹脂(粉末)で充満した受器の重量(重量W8[g])を小数点以下1位の位まで正確に測定し、次式に従って嵩比重[g/ml]を算出した。
(5-8)表面張力
本発明に係る吸水性樹脂(粉末)の表面張力は、以下の手順に従って測定した。
本発明に係る吸水性樹脂(粉末)の表面張力は、以下の手順に従って測定した。
即ち、十分に洗浄した容量100mlのガラス製ビーカーに、20℃に調温した0.9重量%の塩化ナトリウム水溶液(生理食塩水)50mlを入れ、表面張力計(KRUSS社製;K11自動表面張力計)を用いて測定した。尚、本測定では、表面張力が71~75[mN/m]となる必要がある。
次に、上記表面張力を測定した生理食塩水が入ったガラス製ビーカーに、十分に洗浄した25mm長のフッ素樹脂製回転子及び吸水性樹脂(粉末)0.5gを投入し、500rpmで4分間攪拌した。4分後攪拌を止め、吸水した吸水性樹脂(粉末)を沈降させた。その後、上澄み液の表面張力について、上記と同様の操作を行って測定した。尚、本発明では白金プレート法を採用し、白金プレートは各測定前に脱イオン水で十分に洗浄し、かつ、ガスバーナーで加熱洗浄して使用した。
(5-9)通液性(SFC)
通液性(SFC)は周知の測定方法であり、米国特許第5562646号に開示されている方法に準じて測定した。
通液性(SFC)は周知の測定方法であり、米国特許第5562646号に開示されている方法に準じて測定した。
(5-10)色調評価(ハンターLab表色系)
本発明に係る吸水性樹脂(粉末)中の色調評価(ハンターLab表色系)は、分光式色差計(HunterLab社製;LabScan(登録商標)XE)を用いて、下記の手法により測定した。
本発明に係る吸水性樹脂(粉末)中の色調評価(ハンターLab表色系)は、分光式色差計(HunterLab社製;LabScan(登録商標)XE)を用いて、下記の手法により測定した。
即ち、内径30mm、高さ12mmの粉末・ペースト用の試料容器に吸水性樹脂(粉末)約5gを充填し、上記分光式色差計にて、吸水性樹脂(粉末)表面のL値(Lightness:明度指数)を測定した。この値を「明度指数(初期)」として評価し、この値が大きいほど白色となる。又、同時にa値、b値も測定した。これらのa値、b値は小さいほど低着色で実質白色に近づく。又、上記測定条件は反射測定が選択され、標準として粉末・ペースト用標準丸白板No.2を用い、30Φの投光パイプを用いた。尚、製造直後の吸水性樹脂(粉末)、或いは、30℃以下で相対湿度50%RH以下の条件下、保存期間1年以内の吸水性樹脂(粉末)の色調を初期色調として評価した。
一方、上記粉末・ペースト用の試料容器に吸水性樹脂(粉末)約5gを充填した後、70±1℃、相対湿度65±1%RHの雰囲気に調整した恒温恒湿機(エスペック株式会社製;小型環境試験器 形式SH-641)中に7日間静置した(着色促進試験)。その後、経時着色後の色調(経時色調)として、上記分光式色差計を用いて吸水性樹脂(粉末)表面のL値、a値、b値を測定した。
(5-11)吸水性樹脂(粉末)中のメトキシフェノール類
本発明に係る吸水性樹脂(粉末)中のメトキシフェノール類は、下記の手法により測定した。
本発明に係る吸水性樹脂(粉末)中のメトキシフェノール類は、下記の手法により測定した。
即ち、長さ35mmの回転子を入れた容量250mLの蓋付きプラスチック容器に、吸水性樹脂(粉末)1.0gと0.90重量%の塩化ナトリウム水溶液200.0gとを入れ、20~25℃(室温)、相対湿度50±5RH%の雰囲気下で、1時間攪拌を行った。次に、当該液を濾紙(ADVANTEC東洋株式会社、品名:JIS P 3801、No.2、厚さ0.26mm、保留粒子径5μm)1枚を用いて濾過した。
上記操作で得られた濾液について、以下の条件で高速液体クロマトグラフィーで分析することによって吸水性樹脂(粉末)中のメトキシフェノール類(単位;ppm(対吸水性樹脂(粉末)))を定量することができる。

(5-12)吸水性樹脂(粉末)中のキレート剤
本発明に係る吸水性樹脂(粉末)中のキレート剤は、下記の手法により測定した。
本発明に係る吸水性樹脂(粉末)中のキレート剤は、下記の手法により測定した。
即ち、長さ35mmの回転子を入れた容量250mLの蓋付きプラスチック容器に、吸水性樹脂(粉末)1.0gと0.90重量%の塩化ナトリウム水溶液200.0gとを入れ、20~25℃(室温)、相対湿度50±5RH%の雰囲気下で、1時間攪拌を行った。次に、当該液を濾紙(ADVANTEC東洋株式会社、品名:JIS P 3801、No.2、厚さ0.26mm、保留粒子径5μm)1枚を用いて濾過した。
上記操作で得られた濾液について、高速液体クロマトグラフィーで分析することによって吸水性樹脂(粉末)中のキレート剤(単位;ppm(対吸水性樹脂(粉末)))を定量することができる。
(5-13)真密度
本発明に係る吸水性樹脂(粉末)の真密度は、下記の手法により測定した。
本発明に係る吸水性樹脂(粉末)の真密度は、下記の手法により測定した。
即ち、ボールミルポット(株式会社テラオカ製;磁製ボールミルポット、型番No.90/内寸:直径80mm、高さ75mm、外寸:直径90mm、高さ110mm)中に、吸水性樹脂(粉末)15.0g及び400gの円柱状磁製ボール(径13mm、長さ13mm)を入れ、ボールミルを用いて60Hzで2時間粉砕したところ、その70重量%以上が目開き45μmのJIS標準篩を通過する微粉末が得られた。当該45μm以下に粉砕された微粉末について、乾式密度計(株式会社島津製作所製;アキュピックII-1340)を用いて測定し、得られた値を、吸水性樹脂(粉末)の真密度とした。
[実施例1]
p-メトキシフェノール含有量を70ppmに調整したアクリル酸224.0g、ポリエチレングリコールジアクリレート(数平均分子量522)0.48g及び2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン0.13gを混合した溶液(A)、48.5重量%の水酸化ナトリウム水溶液153.8gをイオン交換水113.2gで希釈し、更に、キレート剤としてジエチレントリアミン5酢酸・5ナトリウム0.03gを加えた溶液(B)をそれぞれ作成した後、上記溶液(A)をマグネチックスターラーで攪拌下、上記溶液(B)を開放系で除熱しながら混合し、45℃の単量体水溶液(1’)を得た。
p-メトキシフェノール含有量を70ppmに調整したアクリル酸224.0g、ポリエチレングリコールジアクリレート(数平均分子量522)0.48g及び2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン0.13gを混合した溶液(A)、48.5重量%の水酸化ナトリウム水溶液153.8gをイオン交換水113.2gで希釈し、更に、キレート剤としてジエチレントリアミン5酢酸・5ナトリウム0.03gを加えた溶液(B)をそれぞれ作成した後、上記溶液(A)をマグネチックスターラーで攪拌下、上記溶液(B)を開放系で除熱しながら混合し、45℃の単量体水溶液(1’)を得た。
次いで、上記単量体水溶液(1’)に30重量%のポリオキシエチレンソルビタンモノステアレート水溶液(花王株式会社製)4.4gを加えて攪拌し、単量体水溶液(1)を得た。得られた単量体水溶液(1)400gの体積は340mlであった。その後、更に室温の窒素ガスで20分間脱気した。
続いて、3.0重量%の2,2’-アゾビス(2-メチルプロピオンアミジン)二塩酸塩水溶液4.1gを加えた後、卓上型ホイップクリームマシーン「ホイップオート(商品名)」(ドイツ・ハンスクラット社製/株式会社愛工舎販売)を用いて、単量体水溶液(1)及び窒素ガスを流体混合し、単量体水溶液(1)(単量体濃度;53.2重量%)中に窒素ガスの気泡を分散させた。尚、当該窒素ガスを分散させた単量体水溶液(1)400gの体積は480mlであり、実施例1における単量体水溶液(1)の体積膨張倍率は1.4倍(=480ml/340ml)であった。
次に、上記ホイップオートを通過した単量体水溶液(1)400gを、予め90℃に加熱しておいたステンレス製バット型容器(底面250mm×250mm、高さ30mm、内面;テフロン(登録商標)シート張り)に大気開放系で投入した。尚、上記ステンレス製バット型容器の加熱にはホットプレートを使用した。その後直ちに、UV照射を行って重合反応を開始させた。尚、UV照射はブラックライト水銀ランプ(ピーク波長;352nm、(株)東芝ライテック製 形式H400BL)を使用した。
上記重合反応は、重合開始から約35秒で水蒸気が発生し、100℃以上に沸騰して進行した。当該沸騰重合において、得られる発泡重合体(1)は単量体水溶液(1)から若干体積膨張したが、連続気泡から容易に水蒸気が蒸発するため、単量体水溶液(1)と比べてほとんど体積変化はなかった。尚、重合開始時の単量体水溶液(1)の温度はホットプレートからの加熱等によって40℃であったが、その後、重合の進行に伴い急激に温度上昇し、重合開始から約40秒後には最高到達温度107℃を記録した。
上記UV照射を3分間行った時点で、発泡重合体(1)をステンレス製バット型容器から取り出し、開放気泡率及び独立気泡率を測定・計算したところ、開放気泡率は14%、独立気泡率は16%であった。尚、発泡重合体(1)は無数の微細な気泡を有し、気泡によって白色の蒸しパン状フォームゲルとなっていた。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記発泡重合体(1)を1辺5mmの立方体状にナイフで切断し、熱風温度180℃、風速2.0[m/s]の熱風乾燥機で30分間乾燥して、吸水性樹脂乾燥物(1’)を得た。
当該乾燥時において、発泡重合体(1)(5mm片)は連続気泡から容易に水が蒸発するため、乾燥によるゲル膨張は見られず、若干の乾燥収縮を除いては実質的な変形は起こらなかった。得られた立方体状の吸水性樹脂乾燥物(1’)を切断し、その破断面についてSEM(走査電子顕微鏡)写真画像(図4参照)を観察したところ、連続気泡が形成されていることが分かった。
次に、別途、上記操作で得られた発泡重合体(1)を1辺2mmの立方体状にナイフで切断し、熱風温度180℃、風速2.0[m/s]の熱風乾燥機で30分間乾燥して、立方体状の吸水性樹脂乾燥物(1)を得た。
その後更に、吸水性樹脂乾燥物(1)をロールミルで粉砕し、次いで、目開き600μm及び300μmのJIS標準篩で分級することにより、重量平均粒子径(D50)が460μmの吸水性樹脂粉末(1)を得た。得られた吸水性樹脂粉末(1)の諸物性を表1に示す。又、吸水性樹脂粉末(1)の開放気泡率及び独立気泡率は、別途、吸水性樹脂乾燥物(1)について、上記(5-1)及び(5-2)に記載した方法を適用して測定した(以下の実施例、比較例で、同様にして測定した)。結果、開放気泡率は6%、独立気泡率は16%であった。
[実施例2](整泡工程追加)
実施例1と同様の操作を行って、窒素ガスの気泡を分散させた単量体水溶液(2)(単量体濃度;53.2重量%、体積膨張倍率;1.4倍)を得た。
実施例1と同様の操作を行って、窒素ガスの気泡を分散させた単量体水溶液(2)(単量体濃度;53.2重量%、体積膨張倍率;1.4倍)を得た。
その後、更に窒素ガスを連続的に混合しながら単量体水溶液(2)を循環し、整泡を行ったこと(図1参照)以外は、実施例1と同様の操作を行って、発泡重合体(2)を得た。重合時には、重合開始から約40秒後には最高到達温度107℃を記録した。尚、発泡重合体(2)の開放気泡率は12%、独立気泡率は14%であった。又、発泡重合体(2)は、無数の微細な気泡を有し、気泡によって白色の蒸しパン状フォームゲルとなっていた。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記発泡重合体(2)を1辺5mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行い吸水性樹脂乾燥物(2’)を得た。
当該乾燥時において、発泡重合体(2)(5mm片)は連続気泡から容易に水から蒸発するため、乾燥によるゲル膨張は見られず、若干の乾燥収縮を除いては実質的な変形は起こらなかった。得られた吸水性樹脂乾燥物(2’)を切断し、その破断面のSEM(走査電子顕微鏡)写真画像を観察したところ、連続気泡が形成されていることが分かった。
次に、別途、上記操作で得られた発泡重合体(2)を1辺2mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行って、立方体状の吸水性樹脂乾燥物(2)を得た。
その後更に、吸水性樹脂乾燥物(2)について、実施例1と同様の粉砕及び分級を行うことにより、重量平均粒子径(D50)が460μmの吸水性樹脂粉末(2)を得た。得られた吸水性樹脂粉末(2)の諸物性を表1に示す。又、吸水性樹脂粉末(2)の開放気泡率は6%、独立気泡率は13%であった。
[実施例3](体積膨張倍率1.3倍)
実施例1において、30重量%のポリオキシエチレンソルビタンモノステアレート水溶液(花王株式会社製)の使用量を4.4g(実施例1)から0.80g(単量体あたりの含有量が0.09重量%)に変更し、更に同時にヒドロキシエチルセルロース(和光一級/和光純薬工業株式会社製)2.65gを加え、単量体水溶液(3)(単量体濃度;53.5重量%、体積膨張倍率;1.3倍)とした以外は、実施例1と同様の操作を行って、発泡重合体(3)を得た。重合時には、重合開始から約40秒後には最高到達温度107℃を記録した。尚、発泡重合体(3)の開放気泡率は10%、独立気泡率は11%であった。又、発泡重合体(3)は、無数の微細な気泡を有し、気泡によって白色の蒸しパン状フォームゲルとなっていた。
実施例1において、30重量%のポリオキシエチレンソルビタンモノステアレート水溶液(花王株式会社製)の使用量を4.4g(実施例1)から0.80g(単量体あたりの含有量が0.09重量%)に変更し、更に同時にヒドロキシエチルセルロース(和光一級/和光純薬工業株式会社製)2.65gを加え、単量体水溶液(3)(単量体濃度;53.5重量%、体積膨張倍率;1.3倍)とした以外は、実施例1と同様の操作を行って、発泡重合体(3)を得た。重合時には、重合開始から約40秒後には最高到達温度107℃を記録した。尚、発泡重合体(3)の開放気泡率は10%、独立気泡率は11%であった。又、発泡重合体(3)は、無数の微細な気泡を有し、気泡によって白色の蒸しパン状フォームゲルとなっていた。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記発泡重合体(3)を1辺5mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行い、吸水性樹脂乾燥物(3’)を得た。
当該乾燥時において、実施例1における発泡重合体(1)の乾燥と同様にゲル膨張が見られず、変形は起こらなかった。得られた吸水性樹脂乾燥物(3’)を切断し、その破断面のSEM(走査電子顕微鏡)写真画像を観察したところ、連続気泡が形成されていることが分かった。
次に、別途、上記操作で得られた発泡重合体(3)を1辺2mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行って、立方体状の吸水性樹脂乾燥物(3)を得た。
その後更に、吸水性樹脂乾燥物(3)について、実施例1と同様の粉砕及び分級を行うことにより、重量平均粒子径(D50)が430μmの吸水性樹脂粉末(3)を得た。得られた吸水性樹脂粉末(3)の諸物性を表1に示す。又、吸水性樹脂粉末(3)の開放気泡率は5%、独立気泡率は10%であった。
次に、上記吸水性樹脂粉末(3)100重量部に対して、1,4-ブタンジオール0.48重量部、プロピレングリコール0.75重量部及び脱イオン水4.0重量部からなる表面架橋剤溶液を均一にスプレーして混合した。その後、温度180℃で45分間加熱処理をし、更に目開き600μmのJIS標準篩を通過させることで、表面架橋された吸水性樹脂粒子(3)を得た。尚、吸水性樹脂粒子とは表面架橋された吸水性樹脂の粉末を意味する。
その後更に、表面架橋された吸水性樹脂粒子(3)100重量部に対して、27重量%の硫酸アルミニウム水溶液(酸化アルミニウム換算で8重量%)0.80重量部、60重量%の乳酸ナトリウム水溶液0.134重量部及びプロピレングリコール0.016重量部からなる第2の表面架橋剤混合液を添加した。その後、無風下、温度60℃で1時間乾燥し、更に目開き600μmのJIS標準篩を通過させて、吸水性樹脂(3a)を得た。
得られた吸水性樹脂(3a)のCRCは26.5[g/g]、FSRは0.45[g/g/s]、SFCは117[×10-7・s・cm3・g-1]であった。又、吸水性樹脂(3a)の開放気泡率は5%、独立気泡率は10%であり、表面処理に伴う気泡率の変化は認められなかった。
続いて、上記操作で得られた吸水性樹脂(3a)100重量部に対して、30重量%の亜硫酸ナトリウム水溶液1.66重量部を添加、混合した後、60℃の熱風乾燥機中に30分間放置した。その後、目開き600μmのJIS標準篩を通過させて、吸水性樹脂(3b)を得た。得られた吸水性樹脂(3b)は、吸水性樹脂(3a)に対して、経時着色性及び耐尿性が改善されていた。
[実施例4](体積膨張倍率1.2倍)
実施例1において、30重量%のポリオキシエチレンソルビタンモノステアレート水溶液(花王株式会社製)を使用せずに、代わりに、ヒドロキシエチルセルロース(和光一級/和光純薬工業株式会社製)5.30gを加え、単量体水溶液(4)(単量体濃度;53.5重量%、体積膨張倍率;1.2倍)とした以外は、実施例1と同様の操作を行って、発泡重合体(4)を得た。重合時には、重合開始から約40秒後には最高到達温度107℃を記録した。尚、発泡重合体(4)の開放気泡率は7%、独立気泡率は9%であった。又、発泡重合体(4)は、無数の微細な気泡を有し、気泡によって白色の蒸しパン状フォームゲルとなっていた。
実施例1において、30重量%のポリオキシエチレンソルビタンモノステアレート水溶液(花王株式会社製)を使用せずに、代わりに、ヒドロキシエチルセルロース(和光一級/和光純薬工業株式会社製)5.30gを加え、単量体水溶液(4)(単量体濃度;53.5重量%、体積膨張倍率;1.2倍)とした以外は、実施例1と同様の操作を行って、発泡重合体(4)を得た。重合時には、重合開始から約40秒後には最高到達温度107℃を記録した。尚、発泡重合体(4)の開放気泡率は7%、独立気泡率は9%であった。又、発泡重合体(4)は、無数の微細な気泡を有し、気泡によって白色の蒸しパン状フォームゲルとなっていた。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記発泡重合体(4)を1辺5mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行い、吸水性樹脂乾燥物(4’)を得た。
当該乾燥時において、実施例1における発泡重合体(1)の乾燥と同様にゲル膨張が見られず、変形は起こらなかった。得られた吸水性樹脂乾燥物(4’)を切断し、その破断面のSEM(走査電子顕微鏡)写真画像を観察したところ、連続気泡が形成されていることが分かった。
次に、別途、上記操作で得られた発泡重合体(4)を1辺2mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行って、立方体状の吸水性樹脂乾燥物(4)を得た。
その後更に、吸水性樹脂乾燥物(4)について、実施例1と同様の粉砕及び分級を行うことにより、重量平均粒子径(D50)が440μmの吸水性樹脂粉末(4)を得た。得られた吸水性樹脂粉末(4)の諸物性を表1に示す。又、吸水性樹脂粉末(4)の開放気泡率は5%、独立気泡率は9%であった。
[実施例5](単量体濃度;42.6重量%)
アクリル酸177.50g、ポリエチレングリコールジアクリレート(数平均分子量522)0.77g及び2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン0.11gを混合した溶液(C)、48.5重量%の水酸化ナトリウム水溶液121.89gをイオン交換水180.30gで希釈し、更にジエチレントリアミン5酢酸・5ナトリウム0.02gを加えた溶液(D)をそれぞれ作成した後、上記溶液(C)をマグネチックスターラーで攪拌下、上記溶液(D)を開放系で除熱しながら混合し、45℃の単量体水溶液(5’)を得た。
アクリル酸177.50g、ポリエチレングリコールジアクリレート(数平均分子量522)0.77g及び2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン0.11gを混合した溶液(C)、48.5重量%の水酸化ナトリウム水溶液121.89gをイオン交換水180.30gで希釈し、更にジエチレントリアミン5酢酸・5ナトリウム0.02gを加えた溶液(D)をそれぞれ作成した後、上記溶液(C)をマグネチックスターラーで攪拌下、上記溶液(D)を開放系で除熱しながら混合し、45℃の単量体水溶液(5’)を得た。
次いで、上記単量体水溶液(5’)に30重量%のポリオキシエチレンソルビタンモノステアレート水溶液(花王株式会社製)7.00g及びヒドロキシエチルセルロース(和光一級/和光純薬工業株式会社製)4.20gを加えて攪拌し、単量体水溶液(5)を得た。得られた単量体水溶液(5)400gの体積は345mlであった。その後、更に窒素ガスで20分間脱気した。
続いて、3.0重量%の2,2’-アゾビス(2-メチルプロピオンアミジン)二塩酸塩水溶液8.21gを加えた後、実施例1と同様に卓上型ホイップクリームマシーン「ホイップオート(商品名)」を用いて、単量体水溶液(5)及び窒素ガスとを流体混合し、単量体水溶液(5)(単量体濃度;42.6重量%)中に窒素ガスの気泡を分散させた。尚、当該窒素ガスを分散させた単量体水溶液(5)400gの体積は3300mlであり、実施例5における単量体水溶液(5)の体積膨張倍率は9.6倍(=3300ml/345ml)であった。
次に、ホイップオートを通過した単量体水溶液(5)400gを、実施例1で用いた、予めホットプレートで90℃に加熱しておいたステンレス製バット型容器に大気開放系で投入した。その後直ちに、実施例1と同様にブラックライト水銀ランプによるUV照射を行って、重合反応を開始させた。
上記重合反応は、重合開始から約40秒で水蒸気が発生し、100℃以上に沸騰して進行した。当該沸騰重合において得られる白色の発泡重合体(5)は、単量体水溶液(5)から若干体積膨張したが、連続気泡から容易に水蒸気が蒸発するため、ほとんど体積変化はなかった。尚、重合開始時の単量体水溶液(5)の温度はホットプレートからの加熱等によって40℃であったが、その後、重合の進行に伴い急激に温度上昇し、重合開始時から約50秒後には最高到達温度101℃を記録した。
上記UV照射を3分間行った時点で、発泡重合体(5)をステンレス製バット型容器から取り出し、開放気泡率及び独立気泡率を測定、計算したところ、開放気泡率は87%、独立気泡率は0%であった。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記発泡重合体(5)を1辺5mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行い、吸水性樹脂乾燥物(5’)を得た。
当該乾燥時において、実施例1での発泡重合体(1)の乾燥と同様にゲル膨張が見られず、若干の乾燥収縮を除くと実質的な変形は起こらなかった。得られた吸水性樹脂乾燥物(5’)を切断し、その破断面のSEM(走査電子顕微鏡)写真画像(図5)を観察したところ、連続気泡が形成していることが分かった。
[比較例1](単量体濃度37.3重量%)
アクリル酸150.5g、トリメチロールプロパントリアクリレート1.86g及び2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン0.09gを混合した溶液(E)、48.5重量%の水酸化ナトリウム水溶液129.2gをイオン交換水205.1gで希釈し、更にジエチレントリアミン5酢酸・5ナトリウム0.02gを加えた溶液(F)をそれぞれ作成した後、上記溶液(E)をマグネチックスターラーで攪拌下、上記溶液(F)を開放系で除熱しながら混合し、30℃の比較単量体水溶液(1’)を得た。
アクリル酸150.5g、トリメチロールプロパントリアクリレート1.86g及び2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン0.09gを混合した溶液(E)、48.5重量%の水酸化ナトリウム水溶液129.2gをイオン交換水205.1gで希釈し、更にジエチレントリアミン5酢酸・5ナトリウム0.02gを加えた溶液(F)をそれぞれ作成した後、上記溶液(E)をマグネチックスターラーで攪拌下、上記溶液(F)を開放系で除熱しながら混合し、30℃の比較単量体水溶液(1’)を得た。
次いで、上記比較単量体水溶液(1’)に30重量%のポリオキシエチレンソルビタンモノステアレート水溶液(花王株式会社製)6.2gを加えて攪拌し、比較単量体水溶液(1)を得た。得られた比較単量体水溶液(1)100gの体積は90mlであった。その後、更に窒素ガスで20分間脱気した。
続いて、3.0重量%の2,2’-アゾビス(2-メチルプロピオンアミジン)二塩酸塩水溶液7.0gを加えた後、実施例1と同様に卓上型ホイップクリームマシーン「ホイップオート(商品名)」を用いて、比較単量体水溶液(1)及び窒素ガスを流体混合し、比較単量体水溶液(1)(単量体濃度;37.3重量%)中に窒素ガスの気泡を分散させた。尚、当該窒素ガスを分散させた比較単量体水溶液(1)100gの体積は920mlであり、比較例1における比較単量体水溶液(1)の体積膨張倍率は10.2倍(=920ml/90ml)であった。
次に、上記ホイップオートを通過した比較単量体水溶液(1)100gを、実施例1で用いた、予めホットプレートで90℃に加熱しておいたステンレス製バット型容器に大気開放系で投入した。その後直ちに、実施例1と同様にブラックライト水銀ランプによるUV照射を行って、重合反応を開始させた。
ところが、上記比較単量体水溶液(1)をステンレス製バット型容器に投入した直後から気泡の合一化が進行し、消泡していく様子が確認された。更に、温度上昇に伴って一段と消泡が進行していくことが確認された。
上記UV照射を3分間行った時点で、比較重合体(1)をステンレス製バット型容器から取り出したが、実施例1~5のような気泡による白色のフォーム状ゲルとはならず、気泡を含有しないため、ほぼ透明なゲルであった。重合系の温度変化の記録から、重合時の最高到達温度は97℃(重合開始から約100秒後)であった。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記比較重合体(1)を1辺5mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行い、比較吸水性樹脂乾燥物(1’)を得た。
当該乾燥時において、比較重合体(1)(5mm片)は連続気泡を含まないため、その内部からの水蒸気の蒸発が困難であり、乾燥によってゲル膨張が確認され、乾燥後に約1~数cmの風船状に変形が生じていた。得られた比較吸水性樹脂乾燥物(1’)を切断し、その破断面についてSEM(走査電子顕微鏡)写真画像を観察したところ、連続気泡は認められなかった。
[比較例2](体積膨張倍率1.0倍)
実施例1において、30重量%のポリオキシエチレンソルビタンモノステアレート水溶液(花王株式会社製)を使用せずに、比較単量体水溶液(2)(単量体濃度;53.5重量%、体積膨張倍率;1.0倍)とした以外は、実施例1と同様の操作を行って、比較重合体(2)を得た。重合時において、重合系の温度変化の記録から、重合時の最高到達温度は107℃(重合開始から約40秒後)であった。尚、比較重合体(2)は、実施例1と同様の重合を行ったが気泡による白色のフォーム状ゲルとはならず、気泡を含有しないため、ほぼ透明なゲルであった。
実施例1において、30重量%のポリオキシエチレンソルビタンモノステアレート水溶液(花王株式会社製)を使用せずに、比較単量体水溶液(2)(単量体濃度;53.5重量%、体積膨張倍率;1.0倍)とした以外は、実施例1と同様の操作を行って、比較重合体(2)を得た。重合時において、重合系の温度変化の記録から、重合時の最高到達温度は107℃(重合開始から約40秒後)であった。尚、比較重合体(2)は、実施例1と同様の重合を行ったが気泡による白色のフォーム状ゲルとはならず、気泡を含有しないため、ほぼ透明なゲルであった。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記比較重合体(2)を1辺5mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行い、比較吸水性樹脂乾燥物(2’)を得た。
当該乾燥時において、比較重合体(2)(5mm片)は連続気泡を含まないため、その内部からの水蒸気の蒸発が困難であり、乾燥によってゲル膨張が確認され、乾燥後に約1~数cmの風船状に変形が生じていた。得られた比較吸水性樹脂乾燥物(2’)を切断し、その破断面についてSEM(走査電子顕微鏡)写真画像を観察したところ、連続気泡は認められなかった。
次に、別途、上記操作で得られた比較重合体(2)を1辺2mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行って、立方体状の比較吸水性樹脂乾燥物(2)を得た。
その後更に、比較吸水性樹脂乾燥物(2)について、実施例1と同様の粉砕及び分級を行うことにより、重量平均粒子径(D50)が470μmの比較吸水性樹脂粉末(2)を得た。得られた比較吸水性樹脂粉末(2)の諸物性を表1に示す。又、比較吸水性樹脂粉末(2)の開放気泡率は1.5%、独立気泡率は4%であった。
次に、上記比較吸水性樹脂粉末(2)に対して、実施例3と同様の表面処理を行って、比較吸水性樹脂(2)を得た。
得られた比較吸水性樹脂(2)のCRCは26.3[g/g]、FSRは0.18[g/g/s]、SFCは135[×10-7・s・cm3・g-1]であった。又、比較吸水性樹脂(2)の開放気泡率は1.3%、独立気泡率は4.2%であった。
同様の表面架橋を行った実施例3(FSR;0.45[g/g/s])及び比較例2(FSR;0.18[g/g/s])との対比から、本発明では表面架橋後の吸水速度(FSR)が2.5倍に向上することが分かる。
[比較例3](体積膨張倍率1.0倍)
実施例1において、ホイップオートを使用せずに単量体水溶液中に窒素ガスの気泡を分散させないで、比較単量体水溶液(3)(単量体濃度;53.2重量%、体積膨張倍率;1.0倍)とした以外は、実施例1と同様の操作を行って、比較重合体(3)を得た。重合時において、重合系の温度変化の記録から、重合時の最高到達温度は107℃(重合開始から約40秒後)であった。尚、比較重合体(3)は、実施例1と同様の重合を行ったが気泡による白色のフォーム状ゲルとはならず、気泡を含有しないため、ほぼ透明なゲルであった。
実施例1において、ホイップオートを使用せずに単量体水溶液中に窒素ガスの気泡を分散させないで、比較単量体水溶液(3)(単量体濃度;53.2重量%、体積膨張倍率;1.0倍)とした以外は、実施例1と同様の操作を行って、比較重合体(3)を得た。重合時において、重合系の温度変化の記録から、重合時の最高到達温度は107℃(重合開始から約40秒後)であった。尚、比較重合体(3)は、実施例1と同様の重合を行ったが気泡による白色のフォーム状ゲルとはならず、気泡を含有しないため、ほぼ透明なゲルであった。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記比較重合体(3)を1辺5mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行い、比較吸水性樹脂乾燥物(3’)を得た。
当該乾燥において、比較重合体(3)(5mm片)は連続気泡を含まないため、その内部からの水蒸気の蒸発が困難であり、乾燥によってゲル膨張が確認され、乾燥後に約1~数cmの風船状に変形が生じていた。得られた比較吸水性樹脂乾燥物(3’)を切断し、その破断面についてSEM(走査電子顕微鏡)写真画像を観察したところ、連続気泡は認められなかった。
[比較例4]
実施例1において、3.0重量%の2,2’-アゾビス(2-メチルプロピオンアミジン)二塩酸塩水溶液を添加せずに、ホイップオート(商品名)を用いて比較単量体水溶液(4)及び窒素ガスを流体混合し、比較単量体水溶液(4)(単量体濃度;53.6重量%、体積膨張倍率;1.4倍)中に窒素ガスの気泡を分散させた。
実施例1において、3.0重量%の2,2’-アゾビス(2-メチルプロピオンアミジン)二塩酸塩水溶液を添加せずに、ホイップオート(商品名)を用いて比較単量体水溶液(4)及び窒素ガスを流体混合し、比較単量体水溶液(4)(単量体濃度;53.6重量%、体積膨張倍率;1.4倍)中に窒素ガスの気泡を分散させた。
次に、上記ホイップオートを通過した比較単量体水溶液(4)400gを、実施例1で用いたステンレス製バット型容器に大気開放系で投入した。その後直ちに、実施例1と同様にブラックライト水銀ランプでUV照射を行って、重合反応を開始させた。
当該重合は、単量体温度が80℃まで上昇した時点でUV照射を止め、単量体温度が60℃まで低下した時点でUV照射を再開する操作を繰り返すことで完結させ、比較重合体(4)を得た。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記比較重合体(4)を1辺5mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行い、比較吸水性樹脂乾燥物(4’)を得た。
当該乾燥時において、比較重合体(4)(5mm片)は連続気泡を含まないため、その内部からの水蒸気の蒸発が困難であり、乾燥によってゲル膨張が確認され、乾燥後に約1~数cmの風船状に変形が生じていた。得られた比較吸水性樹脂乾燥物(4’)を切断し、その破断面についてSEM(走査電子顕微鏡)写真画像を観察したところ、連続気泡は認められなかった。
次に、別途、上記操作で得られた比較重合体(4)を1辺2mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行って、立方体状の比較吸水性樹脂乾燥物(4)を得た。
その後更に、比較吸水性樹脂乾燥物(4)について、実施例1と同様の粉砕及び分級を行うことにより、重量平均粒子径(D50)が470μmの比較吸水性樹脂粉末(4)を得た。得られた比較吸水性樹脂粉末(4)の諸物性を表1に示す。又、比較吸水性樹脂粉末(4)の開放気泡率は2%、独立気泡率は5%であった。
[比較例5](単量体濃度35.2重量%)
アクリル酸142.4g、トリメチロールプロパントリアクリレート1.75g及び2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン0.09gを混合した溶液(G)、48.5重量%の水酸化ナトリウム水溶液122.2gをイオン交換水226.3gで希釈し、更にジエチレントリアミン5酢酸・5ナトリウム0.02gを加えた溶液(H)をそれぞれ作成した後、上記溶液(G)をマグネチックスターラーで攪拌下、上記溶液(H)を開放系で除熱しながら混合し、30℃の比較単量体水溶液(5’)を得た。
アクリル酸142.4g、トリメチロールプロパントリアクリレート1.75g及び2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン0.09gを混合した溶液(G)、48.5重量%の水酸化ナトリウム水溶液122.2gをイオン交換水226.3gで希釈し、更にジエチレントリアミン5酢酸・5ナトリウム0.02gを加えた溶液(H)をそれぞれ作成した後、上記溶液(G)をマグネチックスターラーで攪拌下、上記溶液(H)を開放系で除熱しながら混合し、30℃の比較単量体水溶液(5’)を得た。
次いで、上記比較単量体水溶液(5’)に30重量%のポリオキシエチレンソルビタンモノステアレート水溶液(花王株式会社製)0.6gを加えて攪拌し、比較単量体水溶液(5)を得た。得られた比較単量体水溶液(5)400gの体積は360mlであった。その後、更に窒素ガスで20分間脱気した。
続いて、3.0重量%の2,2’-アゾビス(2-メチルプロピオンアミジン)二塩酸塩水溶液6.6gを加えた後、実施例1と同様に卓上型ホイップクリームマシーン「ホイップオート(商品名)」を用いて、比較単量体水溶液(5)及び窒素ガスを流体混合し、比較単量体水溶液(5)(単量体濃度;35.2重量%)中に窒素ガスの気泡を分散させた。尚、当該窒素ガスを分散させた比較単量体水溶液(5)400gの体積は500mlであり、比較例5における比較単量体水溶液(5)の体積膨張倍率は1.4倍(=500ml/360ml)であった。
次に、上記ホイップオートを通過した比較単量体水溶液(5)400gを、実施例1で用いた、予めホットプレートで90℃に加熱しておいたステンレス製バット型容器に大気開放系で投入した。その後直ちに、実施例1と同様にブラックライト水銀ランプによるUV照射を行って、重合反応を開始させた。この重合開始時の比較単量体水溶液(5)の温度は30℃であった。
ところが、上記比較単量体水溶液(5)をステンレス製バット型容器に投入した直後から気泡の合一化が進行し、消泡していく様子が確認された。更に、温度上昇に伴って一段と消泡が進行していくことが確認された。
上記UV照射を3分間行った時点で、比較重合体(5)をステンレス製バット型容器から取り出したが、実施例1~5のような気泡による白色のフォーム状ゲルとはならず、気泡を含有しないためほぼ透明なゲルであった。重合系の温度変化の記録から、重合時の最高到達温度は92℃(重合開始から約120秒後)であった。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記比較重合体(5)を1辺5mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行い、比較吸水性樹脂乾燥物(5’)を得た。
当該乾燥時において、比較重合体(5)(5mm片)は連続気泡を含まないため、その内部からの水蒸気の蒸発が困難であり、乾燥によってゲル膨張が確認され、乾燥後に約1~数cmの風船状に変形が生じていた。得られた比較吸水性樹脂乾燥物(5’)を切断し、その破断面についてSEM(走査電子顕微鏡)写真画像を観察したところ、連続気泡は認められなかった。
[比較例6](特許文献22の追試)
上記特許文献22(米国特許第6107358号)の実施例18に準じて重合を行った。
上記特許文献22(米国特許第6107358号)の実施例18に準じて重合を行った。
即ち、アクリル酸306g、37重量%のアクリル酸ナトリウム3240g、ポリエチレングリコール(n=8)ジアクリレート8.2g、ポリオキシエチレンソルビタンモノステアレート(花王株式会社製)0.3g、純水1420g及び10重量%の過硫酸ナトリウム水溶液10gを混合し、比較単量体水溶液(6)を作成した。
次いで、実施例1と同様に卓上型ホイップクリームマシーン「ホイップオート(商品名)」を用いて、比較単量体水溶液(6)及び窒素ガスを流体混合し、比較単量体水溶液(6)中に窒素ガスの気泡を分散させた。尚、ホイップオート(商品名)通過後の比較単量体水溶液(6)の体積膨張倍率1.2倍であった。
続いて、10重量%の亜硫酸水溶液10gを加えて、単量体濃度を30.2重量%として重合を開始した。当該重合形態は、温度25~95℃で1時間の静置重合(最高到達温度95℃(重合開始から15分後)、重合装置供給後、7分で重合開始)であり、気泡を多量に含むスポンジ状の比較重合体(6)が得られた。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記比較重合体(6)を1辺5mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行い、比較吸水性樹脂乾燥物(6’)を得た。
当該乾燥時において、比較重合体(6)(5mm片)は連続気泡を含まないため、その内部からの水蒸気の蒸発が困難であり、乾燥によってゲル膨張が確認され、乾燥後に約1~数cmの風船状に変形が生じていた。得られた比較吸水性樹脂乾燥物(6’)を切断し、その破断面についてSEM(走査電子顕微鏡)写真画像を観察したところ、連続気泡は認められず、独立気泡が多いものであることが確認された。
[比較例7](特許文献20の追試)
上記特許文献20(米国特許第6136873号明細書)の実施例2に準じて重合を行った。尚、特許文献20には、「重合温度は好ましくは重合性水性混合物の沸騰が避けられるように調整する」との記載がある。
上記特許文献20(米国特許第6136873号明細書)の実施例2に準じて重合を行った。尚、特許文献20には、「重合温度は好ましくは重合性水性混合物の沸騰が避けられるように調整する」との記載がある。
即ち、アクリル酸38.1gにトリメチロールプロパントリアクリレート2.00gを溶解させた後、攪拌しながら37.3重量%のアクリル酸ナトリウム400.0g及びC13/C15-オキソアルコール硫酸半エステルのナトリウム塩10.00gを加え、5時間攪拌して、比較単量体水溶液(7)を得た。その後、市販の調理機(BRAUN Multiquick Professional)を用いて窒素バブリングしながら攪拌した。
続いて、3.0重量%の2,2-アゾビス-(N,N-ジメチレンイソブチルアミン)ジヒドロクロリド水溶液21.22gを加え、5分間攪拌した。尚、気泡含有後の比較単量体水溶液(7)の体積膨張倍率は8倍であった。
その後、当該比較単量体水溶液(7)をポリプロピレン製容器(大きさ:15cm×19cm×18cm)に投入したが、消泡速度が速く、マイクロ波を照射する時点で比較単量体水溶液(7)の体積はほぼ半減していた。引き続きマイクロ波の照射を継続したが、消泡はさらに進行していくことが確認された。なお、比較例7においては、比較単量体水溶液(7)の沸騰を避けるように重合温度を調節した。
上記マイクロ波照射を10分間行った時点で、比較重合体(7)を容器から取り出したが、本願発明で規定するフォーム状の重合体とはなっていなかった。
[比較例8](特許文献28の追試)
上記特許文献28(特開平1-318021号公報)の実施例1に準じて重合した。
上記特許文献28(特開平1-318021号公報)の実施例1に準じて重合した。
即ち、80重量%のアクリル酸水溶液90重量部に、メチレンビスアクリルアミド0.08重量部及びポリオキノエチレン(20)ステアリルエーテル0.1重量部を添加し、窒素気流中で激しく攪拌させながら、42重量%の苛性ソーダ水溶液71重量部を徐々に加えた。冷却後、過硫酸アンモニウム0.15重量部を溶解し、アクリル酸ナトリウム塩の微細析出物が均一に分散した比較単量体分散液(8)を得た。当該比較単量体分散液(8)は中和率75モル%、単量体濃度54.9重量%のスラリー原液であった。
次に、比較単量体分散液(8)を窒素置換した重合容器中に厚さ約2cmの層状に流延し、底部より加熱した。加熱し始めて直ぐに重合が開始し、重合は均一な発泡を伴うものであり、20分間で厚さ約5cmの発泡ゴム板状の重合物が得られた。
上記操作で得られた重合物は粘着性がなく、スライサーを用いて厚さ5mmに切断し、シート状とした。更に、5mm角に切断してペレット状に成型した後、乾燥して比較吸水性樹脂乾燥物(8)とした。当該比較吸水性樹脂乾燥物(8)を切断し、その破断面のSEM(走査電子顕微鏡)写真画像を観察したところ、連続気泡は認められなかった。

(まとめ)
実施例1等及び比較例との対比から、「体積膨張倍率が1.1倍を超えて」、「単量体濃度が40重量%以上」及び「重合時の最高到達温度が100℃以上」の3つの要件を満たすことが、高吸水速度(FSR)の吸水性樹脂を得るために重要であることが分かる。
実施例1等及び比較例との対比から、「体積膨張倍率が1.1倍を超えて」、「単量体濃度が40重量%以上」及び「重合時の最高到達温度が100℃以上」の3つの要件を満たすことが、高吸水速度(FSR)の吸水性樹脂を得るために重要であることが分かる。
即ち、比較例1(単量体濃度;37.3重量%)、比較例5(単量体濃度;35.2重量%)、比較例2及び比較例3(体積膨張倍率;1.0倍)、比較例4(重合時の温度;60~80℃)、比較例8(スラリー/非水溶液)等に示したように、本願発明の構成要件3つすべてを満たさないと、連続気泡とならないか、或いは連続気泡率が5%未満となり、高吸水速度が達成できないことが分かる。
又、従来技術としての比較例6や比較例7においても、連続気泡とならないことが分かる。更に、本発明の方法では固形分(重量%)が高い方が、含水ゲルの乾燥速度が向上(乾燥時間が短縮)していることが分かる。
上記(2-3)に記載したように、特許文献8、20、21、30~33等で推奨する「重合時の沸騰の回避」は、生産性の低下や高価な冷却設備等を伴うものであったが、本発明では、従来、(連続)フォーム状吸水性樹脂の製造方法で避けられてきた沸騰重合が、単量体濃度40重量%以上及び体積膨張倍率1.1倍以上において、効率的に連続気泡を提供することを見いだした。
[実施例6](単量体濃度45.4重量%、MQ70ppm)
アクリル酸190.18g、ポリエチレングリコールジアクリレート(数平均分子量522)0.83g及び2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン0.11gを混合した溶液(I)、48.5重量%の水酸化ナトリウム水溶液130.60gをイオン交換水163.46gで希釈し、さらにジエチレントリアミン5酢酸・5ナトリウム0.02gを加えた溶液(J)をそれぞれ作成した後、上記溶液(I)をマグネチックスターラーで攪拌下、上記溶液(J)を開放系で除熱しながら混合し、45℃の単量体水溶液(6’)を得た。尚、p-メトキシフェノール(以下、単に「MQ」と称することもある)の含有量を70ppmに調整したアクリル酸を用いた。
アクリル酸190.18g、ポリエチレングリコールジアクリレート(数平均分子量522)0.83g及び2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン0.11gを混合した溶液(I)、48.5重量%の水酸化ナトリウム水溶液130.60gをイオン交換水163.46gで希釈し、さらにジエチレントリアミン5酢酸・5ナトリウム0.02gを加えた溶液(J)をそれぞれ作成した後、上記溶液(I)をマグネチックスターラーで攪拌下、上記溶液(J)を開放系で除熱しながら混合し、45℃の単量体水溶液(6’)を得た。尚、p-メトキシフェノール(以下、単に「MQ」と称することもある)の含有量を70ppmに調整したアクリル酸を用いた。
次いで、上記単量体水溶液(6’)に30重量%のポリオキシエチレンソルビタンモノステアレート水溶液(花王株式会社製)3.75gおよびヒドロキシエチルセルロース(和光一級/和光純薬工業株式会社製)2.25gを加えて攪拌し、単量体水溶液(6)(p-メトキシフェノールの含有量70ppm(対アクリル酸))を得た。得られた単量体水溶液(6)400gの体積は340mlであった。その後、更に窒素ガスで20分間脱気した。
続いて、3.0重量%の2,2’-アゾビス(2-メチルプロピオンアミジン)二塩酸塩水溶液8.8gを加えた後、実施例1と同様に卓上型ホイップクリームマシーン「ホイップオート(商品名)」を用いて、単量体水溶液(6)及び窒素ガスを流体混合し、単量体水溶液(6)(単量体濃度;45.4重量%)中に窒素ガスの気泡を分散させた。尚、当該窒素ガスを分散させた単量体水溶液(6)400gの体積は850mlであり、実施例6における単量体水溶液(6)の体積膨張倍率は2.5倍(=850ml/340ml)であった。
次に、ホイップオートを通過した単量体水溶液(6)400gを、実施例1で用いた、予めホットプレートで90℃に加熱しておいたステンレス製バッド型容器に大気開放系で投入した。その後直ちに、実施例1と同様にブラックライト水銀ランプによるUV照射を行って、重合反応を開始させた。
上記重合反応は、重合開始から約40秒で水蒸気が発生し、100℃以上に沸騰して進行した。当該沸騰重合において得られる白色フォーム状ゲルである発泡重合体(6)は、実施例1と同様に若干体積膨張したが、ほとんど体積変化はなかった。尚、重合開始時の単量体水溶液(6)の温度はホットプレートからの加熱等によって42℃であったが、その後、重合の進行に伴い急激に温度上昇し、重合開始から約50秒後には最高到達温度102℃を記録した。
上記UV照射を3分間行った時点で、発泡重合体(6)をステンレス製バッド型容器から取り出し、開放気泡率及び独立気泡率を測定・計算したところ、開放気泡率は53%、独立気泡率は8%であった。尚、発泡重合体(6)は無数の微細な気泡を有し、気泡によって白色の蒸しパン状フォームゲルとなっていた。
次に、SEM(走査電子顕微鏡)写真画像で状態を確認するために、上記発泡重合体(6)を1辺5mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行い、吸水性樹脂乾燥物(6’)を得た。
当該乾燥時において、実施例1における発泡重合体(1)の乾燥と同様にゲル膨張が見られず、変形は起こらなかった。得られた吸水性樹脂乾燥物(6’)を切断し、その破断面のSEM(走査電子顕微鏡)写真画像を観察したところ、連続気泡が形成されていることが分かった。
次に、別途、上記操作で得られた発泡重合体(6)を1辺2mmの立方体状にナイフで切断し、実施例1と同様の熱風乾燥を行って、立方体状の吸水性樹脂乾燥物(6)を得た。
その後更に、吸水性樹脂乾燥物(6)について、実施例1と同様の粉砕及び分級を行うことにより、重量平均粒子径(D50)が460μmの吸水性樹脂粉末(6)を得た。得られた吸水性樹脂粉末(6)の諸物性を表2に示す。又、吸水性樹脂粉末(6)の開放気泡率は15%、独立気泡率は24%であった。更に、吸水性樹脂粉末(6)中のp-メトキシフェノール含有量は20ppmであった。
[実施例7](MQ120ppm)
実施例6において、p-メトキシフェノール(MQ)の含有量を120ppmに調整したアクリル酸を使用した以外は、実施例6と同様の操作を行い、発泡重合体(7)を得た。更に発泡重合体(7)について、実施例6と同様の操作を行うことにより、重量平均粒子径(D50)が460μmの吸水性樹脂粉末(7)を得た。得られた吸水性樹脂粉末(7)の諸物性を表2に示す。又、吸水性樹脂粉末(7)の開放気泡率は13%、独立気泡率は23%であった。更に、吸水性樹脂粉末(7)中のp-メトキシフェノールの含有量は35ppmであった。
実施例6において、p-メトキシフェノール(MQ)の含有量を120ppmに調整したアクリル酸を使用した以外は、実施例6と同様の操作を行い、発泡重合体(7)を得た。更に発泡重合体(7)について、実施例6と同様の操作を行うことにより、重量平均粒子径(D50)が460μmの吸水性樹脂粉末(7)を得た。得られた吸水性樹脂粉末(7)の諸物性を表2に示す。又、吸水性樹脂粉末(7)の開放気泡率は13%、独立気泡率は23%であった。更に、吸水性樹脂粉末(7)中のp-メトキシフェノールの含有量は35ppmであった。
[実施例8](MQ200ppm)
実施例6において、p-メトキシフェノール(MQ)の含有量を200ppmに調整したアクリル酸を使用した以外は、実施例6と同様の操作を行い、発泡重合体(8)を得た。更に発泡重合体(8)について、実施例6と同様の操作を行うことにより、重量平均粒子径(D50)が450μmの吸水性樹脂粉末(8)を得た。得られた吸水性樹脂粉末(8)の諸物性を表2に示す。又、吸水性樹脂粉末(8)の開放気泡率は13%、独立気泡率は21%であった。更に、吸水性樹脂粉末(8)中のp-メトキシフェノールの含有量は56ppmであった。
実施例6において、p-メトキシフェノール(MQ)の含有量を200ppmに調整したアクリル酸を使用した以外は、実施例6と同様の操作を行い、発泡重合体(8)を得た。更に発泡重合体(8)について、実施例6と同様の操作を行うことにより、重量平均粒子径(D50)が450μmの吸水性樹脂粉末(8)を得た。得られた吸水性樹脂粉末(8)の諸物性を表2に示す。又、吸水性樹脂粉末(8)の開放気泡率は13%、独立気泡率は21%であった。更に、吸水性樹脂粉末(8)中のp-メトキシフェノールの含有量は56ppmであった。
[実施例9](MQ250ppm)
実施例6において、p-メトキシフェノール(MQ)の含有量を250ppmに調整したアクリル酸を使用した以外は、実施例6と同様の操作を行い、発泡重合体(9)を得た。更に発泡重合体(9)について、実施例6と同様の操作を行うことにより、重量平均粒子径(D50)が460μmの吸水性樹脂粉末(9)を得た。得られた吸水性樹脂粉末(9)の諸物性を表2に示す。又、吸水性樹脂粉末(9)の開放気泡率は14%、独立気泡率は22%であった。更に、比較吸水性樹脂粉末(9)中のp-メトキシフェノールの含有量は67ppmであった。
実施例6において、p-メトキシフェノール(MQ)の含有量を250ppmに調整したアクリル酸を使用した以外は、実施例6と同様の操作を行い、発泡重合体(9)を得た。更に発泡重合体(9)について、実施例6と同様の操作を行うことにより、重量平均粒子径(D50)が460μmの吸水性樹脂粉末(9)を得た。得られた吸水性樹脂粉末(9)の諸物性を表2に示す。又、吸水性樹脂粉末(9)の開放気泡率は14%、独立気泡率は22%であった。更に、比較吸水性樹脂粉末(9)中のp-メトキシフェノールの含有量は67ppmであった。
[比較例9](体積膨張倍率1.0倍、MQ70ppm)
実施例6において、ポリオキシエチレンソルビタンモノステアレート及びヒドロキシエチルセルロースを使用せずに、更にホイップオートを使用せずに単量体水溶液中に窒素ガスの気泡を分散させないで、比較単量体水溶液(9)(体積膨張倍率;1.0倍)とした以外は、実施例6と同様の操作を行って、透明ゲルである比較重合体(9)を得た。更に比較重合体(9)について、実施例6と同様の操作を行うことにより、重量平均粒子径(D50)が470μmの比較吸水性樹脂粉末(9)を得た。得られた比較吸水性樹脂粉末(9)の諸物性を表2に示す。又、比較吸水性樹脂粉末(9)の開放気泡率は1.3%、独立気泡率は3%であった。更に、比較吸水性樹脂粉末(10)中のp-メトキシフェノールの含有量は20ppmであった。
実施例6において、ポリオキシエチレンソルビタンモノステアレート及びヒドロキシエチルセルロースを使用せずに、更にホイップオートを使用せずに単量体水溶液中に窒素ガスの気泡を分散させないで、比較単量体水溶液(9)(体積膨張倍率;1.0倍)とした以外は、実施例6と同様の操作を行って、透明ゲルである比較重合体(9)を得た。更に比較重合体(9)について、実施例6と同様の操作を行うことにより、重量平均粒子径(D50)が470μmの比較吸水性樹脂粉末(9)を得た。得られた比較吸水性樹脂粉末(9)の諸物性を表2に示す。又、比較吸水性樹脂粉末(9)の開放気泡率は1.3%、独立気泡率は3%であった。更に、比較吸水性樹脂粉末(10)中のp-メトキシフェノールの含有量は20ppmであった。
[比較例10](体積膨張倍率1.0倍、MQ120ppm)
比較例9において、p-メトキシフェノールの含有量を120ppmに調整したアクリル酸を使用した以外は、比較例9と同様の操作を行って、重量平均粒子径(D50)が470μmの比較吸水性樹脂粉末(10)を得た。得られた比較吸水性樹脂粉末(10)の諸物性を表2に示す。又、比較吸水性樹脂粉末(10)の開放気泡率は1.5%、独立気泡率は3.5%であった。更に、比較吸水性樹脂粉末(10)中のp-メトキシフェノールの含有量は36ppmであった。
比較例9において、p-メトキシフェノールの含有量を120ppmに調整したアクリル酸を使用した以外は、比較例9と同様の操作を行って、重量平均粒子径(D50)が470μmの比較吸水性樹脂粉末(10)を得た。得られた比較吸水性樹脂粉末(10)の諸物性を表2に示す。又、比較吸水性樹脂粉末(10)の開放気泡率は1.5%、独立気泡率は3.5%であった。更に、比較吸水性樹脂粉末(10)中のp-メトキシフェノールの含有量は36ppmであった。
[比較例11](体積膨張倍率1.0倍、MQ200ppm)
比較例9において、p-メトキシフェノールの含有量を200ppmに調整したアクリル酸を使用した以外は、比較例9と同様の操作を行って、重量平均粒子径(D50)が480μmの比較吸水性樹脂粉末(11)を得た。得られた比較吸水性樹脂粉末(11)の諸物性を表2に示す。又、比較吸水性樹脂粉末(11)の開放気泡率は2.1%、独立気泡率は3.1%であった。更に、比較吸水性樹脂粉末(11)中のp-メトキシフェノールの含有量は55ppmであった。
比較例9において、p-メトキシフェノールの含有量を200ppmに調整したアクリル酸を使用した以外は、比較例9と同様の操作を行って、重量平均粒子径(D50)が480μmの比較吸水性樹脂粉末(11)を得た。得られた比較吸水性樹脂粉末(11)の諸物性を表2に示す。又、比較吸水性樹脂粉末(11)の開放気泡率は2.1%、独立気泡率は3.1%であった。更に、比較吸水性樹脂粉末(11)中のp-メトキシフェノールの含有量は55ppmであった。
[比較例12](体積膨張倍率1.0倍、MQ250ppm)
比較例9において、p-メトキシフェノールの含有量を250ppmに調整したアクリル酸を使用した以外は、比較例9と同様の操作を行って、重量平均粒子径(D50)が460μmの比較吸水性樹脂粉末(12)を得た。得られた比較吸水性樹脂粉末(12)の諸物性を表2に示す。又、比較吸水性樹脂粉末(12)の開放気泡率は1.8%、独立気泡率は2.8%であった。更に、比較吸水性樹脂粉末(12)中のp-メトキシフェノールの含有量は68ppmであった。
比較例9において、p-メトキシフェノールの含有量を250ppmに調整したアクリル酸を使用した以外は、比較例9と同様の操作を行って、重量平均粒子径(D50)が460μmの比較吸水性樹脂粉末(12)を得た。得られた比較吸水性樹脂粉末(12)の諸物性を表2に示す。又、比較吸水性樹脂粉末(12)の開放気泡率は1.8%、独立気泡率は2.8%であった。更に、比較吸水性樹脂粉末(12)中のp-メトキシフェノールの含有量は68ppmであった。

(まとめ)
実施例6~8及び比較例9~13との対比から、吸水性樹脂粉末中のp-メトキシフェノール(MQ)の含有量が初期着色の観点から重要であることが分かる。
実施例6~8及び比較例9~13との対比から、吸水性樹脂粉末中のp-メトキシフェノール(MQ)の含有量が初期着色の観点から重要であることが分かる。
表2に記載の重合時に同じMQ量を使用する実施例6と比較例9(MQ=70ppm)、実施例7と比較例10(同120ppm)、実施例8と比較例11(同200ppm)、実施例9と比較例12(同250ppm)、のそれぞれの対比において、本発明の方法では吸水速度(FSR)が4倍と飛躍的に大きくなり、さらに、吸水倍率(CRC)も若干向上し、初期色調も改善(L値が大きく、a値/b値が小さく)される。また、それぞれの対比において、本発明の方法では固形分(重量%)も高いことより、含水ゲルの乾燥速度が向上(乾燥時間が短縮)していることが分かる。
特許文献20等の従来の吸水速度向上方法は着色防止を開示しないし、また、特許文献29等に記載の従来の着色防止法に比べて、本発明は吸水速度(FSR)が各段に向上する。
[比較例13](単量体濃度37.3重量%)
実施例6において、単量体水溶液の調整に使用するイオン交換水を271.60gに変更することで、単量体濃度37.3重量%とした以外は、実施例6と同様の操作を行って比較重合体(13)を得た。重合時において、重合系の温度変化の記録から、重合時の最高到達温度は97℃(重合開始から約100秒後)であった。
実施例6において、単量体水溶液の調整に使用するイオン交換水を271.60gに変更することで、単量体濃度37.3重量%とした以外は、実施例6と同様の操作を行って比較重合体(13)を得た。重合時において、重合系の温度変化の記録から、重合時の最高到達温度は97℃(重合開始から約100秒後)であった。
次いで、上記操作で得られた比較重合体(13)について、実施例6と同様の乾燥、粉砕及び分級をすることで、比較吸水性樹脂粉末(13)を得た。得られた比較吸水性樹脂粉末(13)の物性を表3に示す。
[実施例10](単量体濃度50.5重量%)
実施例6において、単量体水溶液の調整に使用するイオン交換水を113.49gに変更することで単量体濃度を50.5重量%とした以外は、実施例6と同様の操作を行って発泡重合体(10)を得た。重合時において、重合系の温度変化の記録から、重合時の最高到達温度は105℃(重合開始から約45秒後)であった。
実施例6において、単量体水溶液の調整に使用するイオン交換水を113.49gに変更することで単量体濃度を50.5重量%とした以外は、実施例6と同様の操作を行って発泡重合体(10)を得た。重合時において、重合系の温度変化の記録から、重合時の最高到達温度は105℃(重合開始から約45秒後)であった。
次いで、上記操作で得られた発泡重合体(10)について、実施例6と同様の乾燥、粉砕及び分級をすることで、吸水性樹脂粉末(10)を得た。得られた吸水性樹脂粉末(10)の物性を表3に示す。
[比較例14](最高温度98℃)
実施例6において、重合開始後の冷却を強化することで、重合時の最高到達温度を98℃(重合開始から約50秒後)に制御して、比較重合体(14)を得た。
実施例6において、重合開始後の冷却を強化することで、重合時の最高到達温度を98℃(重合開始から約50秒後)に制御して、比較重合体(14)を得た。
次いで、上記操作で得られた比較重合体(14)について、実施例6と同様の乾燥、粉砕及び分級をすることで、比較吸水性樹脂粉末(14)を得た。得られた比較吸水性樹脂粉末(14)の物性を表3に示す。
[比較例15](体積膨張倍率1.04)
実施例6において、体積膨張倍率2.5倍の単量体水溶液(6)をそのままの状態で10分間放置して体積膨張倍率1.04とした後、実際例6と同様の重合を行って、比較重合体(15)を得た。重合時において、重合系の温度変化の記録から、重合時の最高到達温度は102℃(重合開始から約50秒後)であった。
実施例6において、体積膨張倍率2.5倍の単量体水溶液(6)をそのままの状態で10分間放置して体積膨張倍率1.04とした後、実際例6と同様の重合を行って、比較重合体(15)を得た。重合時において、重合系の温度変化の記録から、重合時の最高到達温度は102℃(重合開始から約50秒後)であった。
次いで、上記操作で得られた比較重合体(15)について、実施例6と同様の乾燥、粉砕及び分級をすることで、比較吸水性樹脂粉末(15)を得た。得られた比較吸水性樹脂粉末(15)の物性を表3に示す。

(まとめ)
表3に示すように、本願規定の重合最高温度(100℃以上)、単量体濃度(40重量%以上)、体積膨張倍率(1.1倍以上)の3つの要件をすべて満たす本願実施例10(開放気泡率=8.0%,FSR=0.61)に対して、要件のひとつが欠ける、比較例13(単量体濃度37.3重量%)、比較例14(最高温度98℃)、比較例15(1.04倍)では、開放気泡率が2.0~3.5%と低く、かつ吸水速度(FSR)も1/3以下に低下する。また、本発明の方法では固形分(重量%)も高いことより、含水ゲルの乾燥速度が向上(乾燥時間が短縮)していることが分かる。表3から、上記要件3つが必須であることが分かる。
表3に示すように、本願規定の重合最高温度(100℃以上)、単量体濃度(40重量%以上)、体積膨張倍率(1.1倍以上)の3つの要件をすべて満たす本願実施例10(開放気泡率=8.0%,FSR=0.61)に対して、要件のひとつが欠ける、比較例13(単量体濃度37.3重量%)、比較例14(最高温度98℃)、比較例15(1.04倍)では、開放気泡率が2.0~3.5%と低く、かつ吸水速度(FSR)も1/3以下に低下する。また、本発明の方法では固形分(重量%)も高いことより、含水ゲルの乾燥速度が向上(乾燥時間が短縮)していることが分かる。表3から、上記要件3つが必須であることが分かる。
[実施例11](乾燥実験)
実施例6で得られた白色フォーム状ゲルである発泡重合体(6)を2~3mmに裁断した後、パンチングプレート上に載せ、温度180℃、露点5℃の熱風を風速1.6[m/s]で、パンチングプレートの下部から上方に向かって通風させた。このとき、乾燥速度を測定したところ、固形分95重量%以上までの乾燥時間は20分であり、本発明の方法では乾燥速度が速いことが分かる。
実施例6で得られた白色フォーム状ゲルである発泡重合体(6)を2~3mmに裁断した後、パンチングプレート上に載せ、温度180℃、露点5℃の熱風を風速1.6[m/s]で、パンチングプレートの下部から上方に向かって通風させた。このとき、乾燥速度を測定したところ、固形分95重量%以上までの乾燥時間は20分であり、本発明の方法では乾燥速度が速いことが分かる。
[比較例16](乾燥実験)
比較例9で得られた透明ゲルである比較重合体(9)を2~3mmに裁断した後、実施例11と同様の方法で乾燥速度を測定したところ、固形分95重量%以上までの乾燥時間は25分であった。
比較例9で得られた透明ゲルである比較重合体(9)を2~3mmに裁断した後、実施例11と同様の方法で乾燥速度を測定したところ、固形分95重量%以上までの乾燥時間は25分であった。
[実施例12](吸水シート)
実施例6で得られた白色フォーム状ゲルである発泡重合体(6)を直径90mmに切断して、吸水シート(12)(成型物)を得た。得られた吸水シート(ミニ吸収体12)について、紙オムツ評価に準じる下記の評価方法により、戻り量(Re-Wet)を測定した。ここで、ミニ吸収体とは紙オムツのモデルであり、本発明の吸収性物品の一例である。
実施例6で得られた白色フォーム状ゲルである発泡重合体(6)を直径90mmに切断して、吸水シート(12)(成型物)を得た。得られた吸水シート(ミニ吸収体12)について、紙オムツ評価に準じる下記の評価方法により、戻り量(Re-Wet)を測定した。ここで、ミニ吸収体とは紙オムツのモデルであり、本発明の吸収性物品の一例である。
上記ミニ吸収体12(直径90mm)を、内径90mmのSUS製シャーレの底に置き、その上に直径90mmの不織布を載せ、更に4.8kPaの荷重が当該ミニ吸収体12に均等にかかるように調整されたピストンと錘を置いた。尚、該ピストン及び錘は、中心部に直径5mmの液投入口があるものを使用した。
次いで、生理食塩水(0.9重量%塩化ナトリウム水溶液)25mlを該液投入口から注ぎ入れ、ミニ吸収体12に吸液させた。30分経過後、更に生理食塩水25mlを液投入口から注ぎ入れ、吸液させた。30分経過後、上記ピストン及び錘を取り外し、予め総重量(W9[g])を測定した外径90mmの濾紙(トーヨー濾紙株式会社製;No.2)30枚を載せ、更に荷重が均等にかかるピストンと錘(総重量20kg)を素早く置いた。5分経過後、上記濾紙30枚の重量(W10[g])を測定し、次式にしたがってミニ吸収体12の戻り量[g]を算出したところ、3.8gであった。
また、吸水シート(12)は通気性にも優れ、ムレ感も殆どなかった。
[比較例17](吸水シート)
比較例9で得られた透明ゲルである比較重合体(9)を実施例12と同様に切断して、比較吸水シート(17)(成型物)を得た。得られた比較吸水シート(17)について、実施例12と同様の方法により、戻り量(Re-Wet)を測定したところ、14.6gであった。また、比較吸水シート(18)は戻り量が多く、通気性にも劣り、ムレ感も大きかった。
比較例9で得られた透明ゲルである比較重合体(9)を実施例12と同様に切断して、比較吸水シート(17)(成型物)を得た。得られた比較吸水シート(17)について、実施例12と同様の方法により、戻り量(Re-Wet)を測定したところ、14.6gであった。また、比較吸水シート(18)は戻り量が多く、通気性にも劣り、ムレ感も大きかった。
(まとめ)
比較例17と実施例12との対比より、本発明の吸水性樹脂では、尿の戻りも少なく、通気性に優れ、蒸れのない優れた吸収物品(特に紙オムツ等の衛生材料)を与えることが判る。
比較例17と実施例12との対比より、本発明の吸水性樹脂では、尿の戻りも少なく、通気性に優れ、蒸れのない優れた吸収物品(特に紙オムツ等の衛生材料)を与えることが判る。
高吸水速度の吸水性樹脂を簡便に得ることができ、かかる吸水性樹脂は紙おむつなどの衛生材料を含め、広く利用できる。
本出願は、2010年6月30日に出願された日本特許出願番号2010-149907号に基づいており、その開示内容は、参照され、全体として、組み入れられている。
1 タンク
2 混合域
3 凹凸
4 単量体水溶液
5 気体
6 アスピレーター
7 気泡含有単量体水溶液
8 単量体調製槽
9 ポンプ。
2 混合域
3 凹凸
4 単量体水溶液
5 気体
6 アスピレーター
7 気泡含有単量体水溶液
8 単量体調製槽
9 ポンプ。
Claims (28)
- 気泡を分散させたアクリル酸系単量体水溶液を得る工程(A)と、該単量体水溶液を重合して発泡重合体を得る工程(B)と、該発泡重合体を加熱乾燥する工程(C)と、を含む、ポリアクリル酸系吸水性樹脂の製造方法であって、
下記式(1);


- 前記単量体水溶液を整泡する工程(D)を含む、請求項1に記載の製造方法。
- 前記工程(D)の後に前記工程(B)を行う、請求項1または2に記載の製造方法。
- 前記工程(A)において、分散させる気泡が不活性ガスである、請求項1~3のいずれか1項に記載の製造方法。
- 前記工程(A)において、単量体水溶液および気泡からなる気液混合物がせん断処理されて気泡を分散させたアクリル酸系単量体水溶液が得られる、請求項1~4のいずれか1項に記載の製造方法。
- 前記工程(D)において、気泡を含む単量体水溶液が循環式タンクで循環される、請求項2~5のいずれか1項に記載の製造方法。
- 前記工程(D)において、前記循環式タンク上部の空間の酸素濃度が1容積%以上である、請求項6に記載の製造方法。
- 循環ラインから少なくとも一部の単量体水溶液を重合する、または循環ラインから少なくとも一部の単量体水溶液を中和した後に単量体水溶液を重合する、請求項6または7に記載の製造方法。
- 前記アクリル酸系単量体水溶液が界面活性剤を含んでなる、請求項1~8のいずれか1項に記載の製造方法。
- 前記工程(B)において、重合開始温度が40℃以上である、請求項1~9のいずれか1項に記載の製造方法。
- 前記単量体濃度が45重量%以上である、請求項1~10のいずれか1項に記載の製造方法。
- 前記工程(C)の後に、さらに前記発泡重合体を表面架橋する工程を含む、請求項1~11のいずれか1項に記載の製造方法。
- 前記工程(B)において、前記アクリル酸系単量体水溶液を重合装置に供給後、重合が開始されるまでの時間が5分以内である、請求項1~12のいずれか1項に記載の製造方法。
- 前記工程(B)において、重合の開始から最高温度に到達するまでの時間が2分以内である、請求項1~13のいずれか1項に記載の製造方法。
- 前記工程(C)の後に、粉砕工程および/または分級工程、ならびに微粉リサイクル工程を含み、微粉の1~40重量%が分級工程以前にリサイクルされる、請求項1~14のいずれか1項に記載の製造方法。
- 前記工程(C)の後に、粉砕工程および/または分級工程を含み、ポリアクリル酸系吸水性樹脂を粉末とする請求項1~15のいずれか1項に記載の製造方法。
- 前記工程(B)と同時に、または前記工程(B)の後に、成型工程を含む、請求項1~16のいずれか1項に記載の製造方法。
- 前記成型物がシート状である、請求項17に記載の製造方法。
- 前記アクリル酸系単量体水溶液が対アクリル酸系換算値でp-メトキシフェノールを200ppm以下で含む、請求項1~18のいずれか1項に記載の製造方法。
- 開放気泡率が5%以上である、ポリアクリル酸系吸水性樹脂。
- p-メトキシフェノールの含有量が60ppm以下である、請求項20に記載の吸水性樹脂。
- 独立気泡率5%以上である、請求項20または21に記載の吸水性樹脂。
- 金属キレート剤または還元剤の少なくとも何れかを含む、請求項20~22のいずれか1項に記載の吸水性樹脂。
- 吸水速度(FSR)が0.25[g/g/s]以上である、請求項20~23のいずれか1項に記載の吸水性樹脂。
- 共有結合性架橋剤および/またはイオン結合性架橋剤で、表面架橋されてなる、請求項20~24のいずれか1項に記載の吸水性樹脂。
- シート状である、請求項20~25のいずれか1項に記載の吸水性樹脂。
- 重量平均粒子径(D50)が300μm以上600μm未満の粉末状である、請求項20~26のいずれか1項に記載の吸水性樹脂。
- 請求項20~27のいずれか1項に記載の吸水性樹脂を含む、吸収性物品。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012522673A JP6157853B2 (ja) | 2010-06-30 | 2011-06-29 | ポリアクリル酸系吸水性樹脂及びその製造方法 |
| EP20110800917 EP2589613B1 (en) | 2010-06-30 | 2011-06-29 | Polyacrylic acid-based water-absorbing resin and process for producing same |
| US13/807,530 US9074030B2 (en) | 2010-06-30 | 2011-06-29 | Polyacrylic acid-type water absorbent resin and method for producing same |
| US14/723,026 US9315640B2 (en) | 2010-06-30 | 2015-05-27 | Polyacrylic acid-type water absorbent resin and method for producing same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-149907 | 2010-06-30 | ||
| JP2010149907 | 2010-06-30 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/807,530 A-371-Of-International US9074030B2 (en) | 2010-06-30 | 2011-06-29 | Polyacrylic acid-type water absorbent resin and method for producing same |
| US14/723,026 Division US9315640B2 (en) | 2010-06-30 | 2015-05-27 | Polyacrylic acid-type water absorbent resin and method for producing same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012002455A1 true WO2012002455A1 (ja) | 2012-01-05 |
Family
ID=45402159
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/064951 WO2012002455A1 (ja) | 2010-06-30 | 2011-06-29 | ポリアクリル酸系吸水性樹脂及びその製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (2) | US9074030B2 (ja) |
| EP (1) | EP2589613B1 (ja) |
| JP (1) | JP6157853B2 (ja) |
| WO (1) | WO2012002455A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140042364A1 (en) * | 2011-01-28 | 2014-02-13 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water absorbent resin powder |
| JP2015016450A (ja) * | 2013-07-12 | 2015-01-29 | 株式会社日本触媒 | 吸水剤及びその製造方法 |
| WO2015046604A1 (ja) | 2013-09-30 | 2015-04-02 | 株式会社日本触媒 | 粒子状吸水剤の充填方法および粒子状吸水剤充填物のサンプリング方法 |
| KR20160127742A (ko) | 2014-02-28 | 2016-11-04 | 가부시키가이샤 닛폰 쇼쿠바이 | 폴리(메트)아크릴산(염)계 입자상 흡수제 및 제조 방법 |
| KR20180074586A (ko) * | 2016-12-23 | 2018-07-03 | 주식회사 엘지화학 | 다공성 고흡수성 수지의 제조 방법 및 다공성 고흡수성 수지 |
| JPWO2018159801A1 (ja) * | 2017-03-02 | 2019-12-19 | 住友精化株式会社 | 吸水性樹脂、土壌保水材、及び農園芸材料 |
| JPWO2018159803A1 (ja) * | 2017-03-02 | 2019-12-26 | 住友精化株式会社 | 吸水性樹脂及び土嚢 |
| CN114805880A (zh) * | 2022-06-24 | 2022-07-29 | 山东诺尔生物科技有限公司 | 一种多糖接枝共聚高分子吸水膜及其制备方法 |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9074030B2 (en) * | 2010-06-30 | 2015-07-07 | Nippon Shokubai Co., Ltd. | Polyacrylic acid-type water absorbent resin and method for producing same |
| DE102011086522A1 (de) | 2011-11-17 | 2013-05-23 | Evonik Degussa Gmbh | Superabsorbierende Polymere für hochgefüllte oder faserfreie Hygieneartikel |
| DE102011086516A1 (de) | 2011-11-17 | 2013-05-23 | Evonik Degussa Gmbh | Superabsorbierende Polymere mit schnellen Absorptionseigenschaften sowieVerfahren zu dessen Herstellung |
| US9738769B2 (en) | 2012-02-15 | 2017-08-22 | Basf Se | Water-absorbing polymer particles with high free swell rate and high permeability |
| CN104703691B (zh) | 2012-10-03 | 2018-03-02 | 株式会社日本触媒 | 吸水剂及其制造方法 |
| WO2014084068A1 (ja) | 2012-11-29 | 2014-06-05 | 株式会社堀場アドバンスドテクノ | 測定装置 |
| US9302248B2 (en) | 2013-04-10 | 2016-04-05 | Evonik Corporation | Particulate superabsorbent polymer composition having improved stability |
| DE102013208942A1 (de) * | 2013-05-15 | 2014-11-20 | Evonik Industries Ag | Superabsorbierende Polymere mit schnellen Absorptionseigenschaften sowie Verfahren zu dessen Herstellung |
| DE102013209023A1 (de) * | 2013-05-15 | 2014-11-20 | Evonik Industries Ag | Superabsorbierende Polymere mit schnellen Absorptionseigenschaften sowie Verfahren zu dessen Herstellung |
| CN107936189B (zh) | 2013-08-28 | 2021-06-01 | 株式会社日本触媒 | 聚丙烯酸(盐)系吸水性树脂粉末及其制品 |
| KR102297640B1 (ko) | 2013-08-28 | 2021-09-06 | 가부시키가이샤 닛폰 쇼쿠바이 | 겔 분쇄 장치, 폴리아크릴산(염)계 흡수성 수지 분말의 제조 방법, 및 흡수성 수지 분말 |
| WO2015093594A1 (ja) | 2013-12-20 | 2015-06-25 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水剤及びその製造方法 |
| US9896529B2 (en) | 2014-03-03 | 2018-02-20 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water-absorbable resin |
| CN107107027B (zh) | 2015-01-07 | 2020-12-18 | 株式会社日本触媒 | 吸水剂及其制造方法、以及评价方法及测定方法 |
| KR20170132800A (ko) | 2015-03-30 | 2017-12-04 | 가부시키가이샤 닛폰 쇼쿠바이 | 입자상 흡수제 |
| EP3279238B1 (en) | 2015-03-31 | 2021-07-14 | Nippon Shokubai Co., Ltd. | Super absorbent polyacrylic acid (salt)-based resin powder, method for manufacturing same, and method for evaluating same |
| US11535689B2 (en) | 2015-06-19 | 2022-12-27 | Nippon Shokubai Co., Ltd. | Poly (meth) acrylic acid (salt)-based particulate water-absorbing agent and production method therefor |
| WO2017170501A1 (ja) | 2016-03-28 | 2017-10-05 | 株式会社日本触媒 | 吸水剤およびその製造方法、並びに吸水剤を用いた吸収性物品 |
| KR102102459B1 (ko) | 2016-12-20 | 2020-04-20 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR102417078B1 (ko) | 2017-09-05 | 2022-07-04 | 주식회사 엘지화학 | 고흡수성 수지 |
| US11078343B2 (en) | 2017-10-06 | 2021-08-03 | Evonik Operations Gmbh | Absorbent polymeric foam for shoe insoles |
| EP4113099A3 (en) | 2017-10-12 | 2023-03-08 | Nippon Shokubai Co., Ltd. | Particulate absorbent agent |
| KR102566942B1 (ko) * | 2017-10-27 | 2023-08-14 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| CN107722329A (zh) * | 2017-11-03 | 2018-02-23 | 宜兴丹森科技有限公司 | 聚丙烯酸系吸水性树脂、组合物及其制造方法 |
| WO2019124536A1 (ja) | 2017-12-21 | 2019-06-27 | 株式会社日本触媒 | 発熱体組成物用吸水性樹脂粉末、及び発熱体組成物 |
| KR102452567B1 (ko) | 2019-01-07 | 2022-10-06 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| EP4186945A4 (en) * | 2020-12-18 | 2024-02-14 | Lg Chemical Ltd | SUPERABSORBENT POLYMER AND PRODUCTION PROCESS THEREOF |
| JP2022104704A (ja) * | 2020-12-29 | 2022-07-11 | ユニ・チャーム株式会社 | 複合吸収体及び衛生用品 |
| JP2023535949A (ja) * | 2021-02-03 | 2023-08-22 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| WO2023168269A1 (en) * | 2022-03-01 | 2023-09-07 | University Of Maryland, College Park | Systems and methods for making and using superabsorbent porous gels |
| CN115386347A (zh) * | 2022-08-25 | 2022-11-25 | 昆山徽虎冰袋有限公司 | 一种适用于0℃以下的速配凝胶型高焓值相变蓄冷剂 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10168129A (ja) * | 1996-12-16 | 1998-06-23 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造方法 |
| JP2005111474A (ja) * | 2003-09-19 | 2005-04-28 | Nippon Shokubai Co Ltd | 吸水剤およびその製法 |
| WO2009048145A1 (ja) * | 2007-10-10 | 2009-04-16 | Nippon Shokubai Co., Ltd. | 吸水性樹脂組成物およびその製造方法 |
| JP2009247969A (ja) * | 2008-04-04 | 2009-10-29 | Shi Mechanical & Equipment Inc | 撹拌装置 |
Family Cites Families (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5734101A (en) | 1980-08-11 | 1982-02-24 | Nippon Shokubai Kagaku Kogyo Co Ltd | Novel polymerization process |
| DE3239476C2 (de) | 1981-10-26 | 1984-06-20 | Arakawa Kagaku Kogyo K.K., Osaka | Verfahren zur Herstellung eines festen, trockenen und wasserabsorbierenden harzes |
| DE3544770A1 (de) | 1985-12-18 | 1987-06-19 | Stockhausen Chem Fab Gmbh | Verfahren und vorrichtung zum kontinuierlichen herstellen von polymerisaten und copolymerisaten der acrylsaeure und/oder methacrylsaeure |
| KR0130652B1 (ko) | 1987-08-14 | 1998-04-07 | 존 휴즈 | 수분 흡수성 수지의 제조 방법 |
| JPH01318021A (ja) | 1988-06-17 | 1989-12-22 | Kazuo Saotome | 吸水性樹脂成形物の製造方法 |
| JPH03115313A (ja) | 1989-09-28 | 1991-05-16 | Kazuo Saotome | 吸水性樹脂の製造方法 |
| US5145906A (en) | 1989-09-28 | 1992-09-08 | Hoechst Celanese Corporation | Super-absorbent polymer having improved absorbency properties |
| DE69108804T2 (de) | 1990-07-17 | 1995-08-24 | Sanyo Chemical Ind Ltd | Verfahren zur Herstellung von Wasser absorbierenden Harzen. |
| JPH04236203A (ja) | 1991-01-19 | 1992-08-25 | Sanyo Chem Ind Ltd | 吸水性ヒドロゲル成形物の製造法 |
| US5154713A (en) | 1991-10-22 | 1992-10-13 | Nalco Chemical Company | Enhancing absorption rates of superabsorbents by incorporating a blowing agent |
| US5118719A (en) | 1991-10-22 | 1992-06-02 | Nalco Chemical Company | Enhancing absorption rates of superabsorbents by incorporating a blowing agent |
| US5338766A (en) | 1993-03-26 | 1994-08-16 | The Procter & Gamble Company | Superabsorbent polymer foam |
| IL110134A (en) | 1993-07-09 | 1998-07-15 | Stockhausen Chem Fab Gmbh | Polymers capable of adsorbing aqueous liquids and body fluids, their preparation and use |
| US5451613A (en) | 1993-09-17 | 1995-09-19 | Nalco Chemical Company | Superabsorbent polymer having improved absorption rate and absorption under pressure |
| US5314420A (en) | 1993-09-17 | 1994-05-24 | Nalco Chemical Company | Superabsorbent polymer having improved absorption rate and absorption under pressure |
| DE4344224A1 (de) | 1993-12-23 | 1995-06-29 | Stockhausen Chem Fab Gmbh | Vernetzte synthetische Polymerisate mit poröser Struktur, hoher Aufnahmegeschwindigkeit für Wasser, wäßrige Lösungen und Körperflüssigkeiten, ein Verfahren zu ihrer Herstellung und ihre Verwendung zur Absorption und/oder Retention von Wasser und/oder wäßrigen Flüssigkeiten |
| WO1996017884A1 (fr) | 1994-12-08 | 1996-06-13 | Nippon Shokubai Co., Ltd. | Resine absorbant l'eau, son procede de production et composition de resine absorbant l'eau |
| US5750585A (en) | 1995-04-04 | 1998-05-12 | Purdue Research Foundation | Super absorbent hydrogel foams |
| DE19540951A1 (de) | 1995-11-03 | 1997-05-07 | Basf Ag | Wasserabsorbierende, schaumförmige, vernetzte Polymerisate, Verfahren zu ihrer Herstellung und ihre Verwendung |
| DE19607551A1 (de) | 1996-02-28 | 1997-09-04 | Basf Ag | Wasserabsorbierende, schaumförmige, vernetzte Polymerisate, Verfahren zu ihrer Herstellung und ihre Verwendung |
| DE19607529A1 (de) | 1996-02-28 | 1997-09-04 | Basf Ag | Absorberelement aus superabsorbierenden Schäumen mit anisotropem Quellverhalten |
| JPH1057805A (ja) * | 1996-08-23 | 1998-03-03 | Nippon Shokubai Co Ltd | 高吸水速度吸水性樹脂組成物 |
| JPH10251530A (ja) * | 1997-03-14 | 1998-09-22 | Nippon Shokubai Co Ltd | 高吸水速度吸水性樹脂組成物 |
| US6107358A (en) * | 1996-08-23 | 2000-08-22 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and method for production thereof |
| TW473485B (en) | 1997-12-10 | 2002-01-21 | Nippon Catalytic Chem Ind | The production process of a water-absorbent resin |
| DE19809540A1 (de) * | 1998-03-05 | 1999-09-09 | Basf Ag | Wasserabsorbierende, schaumförmige, vernetzte Polymerisate, Verfahren zu ihrer Herstellung und ihre Verwendung |
| DE19909214A1 (de) | 1999-03-03 | 2000-09-07 | Basf Ag | Wasserabsorbierende, schaumförmige, vernetzte Polymerisate mit verbesserter Verteilungswirkung, Verfahren zu ihrer Herstellung und ihre Verwendung |
| US6417425B1 (en) | 2000-02-01 | 2002-07-09 | Basf Corporation | Absorbent article and process for preparing an absorbent article |
| US6906159B2 (en) * | 2000-08-03 | 2005-06-14 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, hydropolymer, process for producing them, and uses of them |
| DE10231356B4 (de) | 2002-07-11 | 2007-02-15 | Stockhausen Gmbh | Wasserabsorbierende, schaumförmige Polymergebilde, Verfahren zu deren Herstellung, deren Verwendung sowie daraus hergestellte Verbunde |
| US7163966B2 (en) | 2003-12-19 | 2007-01-16 | Stockhausen, Inc. | Superabsorbent polymer having increased rate of water absorption |
| WO2006109882A1 (en) | 2005-04-12 | 2006-10-19 | Nippon Shokubai Co., Ltd. | Particulate water absorbing agent including polyacrylic acid (polyacrylate) based water absorbing resin as a principal component, method for production thereof, water-absorbent core and absorbing article in which the particulate water absorbing agent is used |
| CN101175632B (zh) | 2005-05-13 | 2010-04-14 | 旭化成化学株式会社 | 吸收性复合体及其制造方法 |
| EP1837348B9 (en) | 2006-03-24 | 2020-01-08 | Nippon Shokubai Co.,Ltd. | Water-absorbing resin and method for manufacturing the same |
| EP2212081B1 (de) | 2007-11-15 | 2012-04-04 | Basf Se | Superabsorbierender schaum mit grafischen zeichen an der oberfläche |
| US9074030B2 (en) * | 2010-06-30 | 2015-07-07 | Nippon Shokubai Co., Ltd. | Polyacrylic acid-type water absorbent resin and method for producing same |
-
2011
- 2011-06-29 US US13/807,530 patent/US9074030B2/en active Active
- 2011-06-29 EP EP20110800917 patent/EP2589613B1/en active Active
- 2011-06-29 WO PCT/JP2011/064951 patent/WO2012002455A1/ja active Application Filing
- 2011-06-29 JP JP2012522673A patent/JP6157853B2/ja active Active
-
2015
- 2015-05-27 US US14/723,026 patent/US9315640B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10168129A (ja) * | 1996-12-16 | 1998-06-23 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造方法 |
| JP2005111474A (ja) * | 2003-09-19 | 2005-04-28 | Nippon Shokubai Co Ltd | 吸水剤およびその製法 |
| WO2009048145A1 (ja) * | 2007-10-10 | 2009-04-16 | Nippon Shokubai Co., Ltd. | 吸水性樹脂組成物およびその製造方法 |
| JP2009247969A (ja) * | 2008-04-04 | 2009-10-29 | Shi Mechanical & Equipment Inc | 撹拌装置 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2589613A4 * |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140042364A1 (en) * | 2011-01-28 | 2014-02-13 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water absorbent resin powder |
| JP2015016450A (ja) * | 2013-07-12 | 2015-01-29 | 株式会社日本触媒 | 吸水剤及びその製造方法 |
| US10577135B2 (en) | 2013-09-30 | 2020-03-03 | Nippon Shokubai Co., Ltd. | Method for filling particulate water absorbing agent and method for sampling filled particulate water absorbing agent |
| WO2015046604A1 (ja) | 2013-09-30 | 2015-04-02 | 株式会社日本触媒 | 粒子状吸水剤の充填方法および粒子状吸水剤充填物のサンプリング方法 |
| KR20160064113A (ko) | 2013-09-30 | 2016-06-07 | 가부시키가이샤 닛폰 쇼쿠바이 | 입자상 흡수제의 충전 방법 및 입자상 흡수제 충전물의 샘플링 방법 |
| EP4159307A1 (en) | 2013-09-30 | 2023-04-05 | Nippon Shokubai Co., Ltd. | Method for filling particulate water absorbing agent and method for sampling filled particulate water absorbing agent |
| US10934031B2 (en) | 2013-09-30 | 2021-03-02 | Nippon Shokubai Co., Ltd. | Method for filling particulate water absorbing agent and method for sampling filled particulate water absorbing agent |
| KR20160127742A (ko) | 2014-02-28 | 2016-11-04 | 가부시키가이샤 닛폰 쇼쿠바이 | 폴리(메트)아크릴산(염)계 입자상 흡수제 및 제조 방법 |
| US10207250B2 (en) | 2014-02-28 | 2019-02-19 | Nippon Shokubai Co., Ltd. | Poly(meth)acrylic acid (salt)-based particulate absorbent |
| JPWO2015129917A1 (ja) * | 2014-02-28 | 2017-03-30 | 株式会社日本触媒 | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 |
| KR102112834B1 (ko) * | 2016-12-23 | 2020-05-19 | 주식회사 엘지화학 | 다공성 고흡수성 수지의 제조 방법 및 다공성 고흡수성 수지 |
| KR20180074586A (ko) * | 2016-12-23 | 2018-07-03 | 주식회사 엘지화학 | 다공성 고흡수성 수지의 제조 방법 및 다공성 고흡수성 수지 |
| US10934400B2 (en) | 2016-12-23 | 2021-03-02 | Lg Chem, Ltd. | Porous super absorbent polymer and preparation method thereof |
| JPWO2018159801A1 (ja) * | 2017-03-02 | 2019-12-19 | 住友精化株式会社 | 吸水性樹脂、土壌保水材、及び農園芸材料 |
| JPWO2018159803A1 (ja) * | 2017-03-02 | 2019-12-26 | 住友精化株式会社 | 吸水性樹脂及び土嚢 |
| US11332558B2 (en) | 2017-03-02 | 2022-05-17 | Sumitomo Seika Chemicals Co., Ltd. | Water-absorbent resin, and soil |
| JP7288399B2 (ja) | 2017-03-02 | 2023-06-07 | 住友精化株式会社 | 吸水性樹脂、土壌保水材、及び農園芸材料 |
| CN114805880A (zh) * | 2022-06-24 | 2022-07-29 | 山东诺尔生物科技有限公司 | 一种多糖接枝共聚高分子吸水膜及其制备方法 |
| CN114805880B (zh) * | 2022-06-24 | 2022-09-09 | 山东诺尔生物科技有限公司 | 一种多糖接枝共聚高分子吸水膜及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6157853B2 (ja) | 2017-07-05 |
| US20150259494A1 (en) | 2015-09-17 |
| US20130101851A1 (en) | 2013-04-25 |
| US9074030B2 (en) | 2015-07-07 |
| EP2589613A4 (en) | 2014-06-25 |
| EP2589613B1 (en) | 2015-05-13 |
| EP2589613A1 (en) | 2013-05-08 |
| JPWO2012002455A1 (ja) | 2013-08-29 |
| US9315640B2 (en) | 2016-04-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6157853B2 (ja) | ポリアクリル酸系吸水性樹脂及びその製造方法 | |
| JP6359600B2 (ja) | ポリアクリル酸系吸水性樹脂粉末の製造方法 | |
| JP6457067B2 (ja) | ポリアクリル酸(塩)系吸水性樹脂粉末及びその製造方法、並びにその評価方法 | |
| JP5889349B2 (ja) | ポリアクリル酸系吸水性樹脂粉末及びその製造方法 | |
| JP5616437B2 (ja) | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法 | |
| JP5337703B2 (ja) | 吸水性樹脂の製造方法および吸水性樹脂並びにその利用 | |
| WO2015030130A1 (ja) | ゲル粉砕装置、及びポリアクリル酸(塩)系吸水性樹脂粉末の製造方法、並びに吸水性樹脂粉末 | |
| WO2015030129A1 (ja) | ゲル粉砕装置、及びポリアクリル酸(塩)系吸水性樹脂粉末の製造方法、並びに吸水性樹脂粉末 | |
| WO2013002387A1 (ja) | ポリアクリル酸(塩)系吸水性樹脂粉末及びその製造方法 | |
| WO2011126079A1 (ja) | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法及びポリアクリル酸(塩)系吸水性樹脂粉末 | |
| JP6029800B2 (ja) | 吸水性樹脂粒子 | |
| JP7064614B2 (ja) | キレート剤を含む吸水性樹脂の製造方法 | |
| JP7181948B2 (ja) | 吸水剤、及び吸水剤の製造方法 | |
| JPH10251309A (ja) | 高吸水速度吸水性樹脂の製造方法 | |
| WO2022196763A1 (ja) | 吸水性樹脂の製造方法 | |
| WO2022239723A1 (ja) | ポリ(メタ)アクリル酸(塩)系吸水性樹脂、及び吸収体 | |
| JP2023088497A (ja) | 表面架橋された(メタ)アクリル酸(塩)系吸水性樹脂を含む吸水剤および(メタ)アクリル酸(塩)系吸水性樹脂の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11800917 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012522673 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011800917 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13807530 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |