WO2011115076A1 - 末端を修飾したポリアミック酸エステルを含有する液晶配向剤、及び液晶配向膜 - Google Patents
末端を修飾したポリアミック酸エステルを含有する液晶配向剤、及び液晶配向膜 Download PDFInfo
- Publication number
- WO2011115076A1 WO2011115076A1 PCT/JP2011/055971 JP2011055971W WO2011115076A1 WO 2011115076 A1 WO2011115076 A1 WO 2011115076A1 JP 2011055971 W JP2011055971 W JP 2011055971W WO 2011115076 A1 WO2011115076 A1 WO 2011115076A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polyamic acid
- acid ester
- group
- liquid crystal
- added
- Prior art date
Links
- 0 NC(*CI=I)=O Chemical compound NC(*CI=I)=O 0.000 description 12
- CCRQBNJTBJRTJD-UHFFFAOYSA-N CC(N(CCN1C(Cl)=O)C1=O)=O Chemical compound CC(N(CCN1C(Cl)=O)C1=O)=O CCRQBNJTBJRTJD-UHFFFAOYSA-N 0.000 description 1
- XZVYDRLPXWFRIS-UHFFFAOYSA-N CCN(C)C(Cl)=O Chemical compound CCN(C)C(Cl)=O XZVYDRLPXWFRIS-UHFFFAOYSA-N 0.000 description 1
- FJCKSHKHGVIICO-UHFFFAOYSA-N CCOc1ccc(C(CC2)CCC2OCc2cc(C)cc(C)c2)cc1 Chemical compound CCOc1ccc(C(CC2)CCC2OCc2cc(C)cc(C)c2)cc1 FJCKSHKHGVIICO-UHFFFAOYSA-N 0.000 description 1
- NGGGKMHRWNMKIR-UHFFFAOYSA-N Cc1cc(C)cc(COC(CC2)CCC2C(CC2)CCC2OC)c1 Chemical compound Cc1cc(C)cc(COC(CC2)CCC2C(CC2)CCC2OC)c1 NGGGKMHRWNMKIR-UHFFFAOYSA-N 0.000 description 1
- HFJCOFKHYUXNGD-UHFFFAOYSA-N Cc1cc(C)cc(COc2ccc(C(CC3)CCC3OC)cc2)c1 Chemical compound Cc1cc(C)cc(COc2ccc(C(CC3)CCC3OC)cc2)c1 HFJCOFKHYUXNGD-UHFFFAOYSA-N 0.000 description 1
- LHBRJNYLLZOROE-UHFFFAOYSA-N O=C(N(C(CC1)=O)C1=O)Cl Chemical compound O=C(N(C(CC1)=O)C1=O)Cl LHBRJNYLLZOROE-UHFFFAOYSA-N 0.000 description 1
- BIFDXOOJPDHKJH-UHFFFAOYSA-N O=C(N1CCCCC1)Cl Chemical compound O=C(N1CCCCC1)Cl BIFDXOOJPDHKJH-UHFFFAOYSA-N 0.000 description 1
- DPTCOFHATGDORM-UHFFFAOYSA-N O=C(N1CCN(Cc2ccccc2)CC1)Cl Chemical compound O=C(N1CCN(Cc2ccccc2)CC1)Cl DPTCOFHATGDORM-UHFFFAOYSA-N 0.000 description 1
- XOVUITMHPFXQPN-UHFFFAOYSA-N O=C([n]1cncc1)Cl Chemical compound O=C([n]1cncc1)Cl XOVUITMHPFXQPN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/16—Polyester-imides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K19/00—Liquid crystal materials
- C09K19/52—Liquid crystal materials characterised by components which are not liquid crystals, e.g. additives with special physical aspect: solvents, solid particles
- C09K19/54—Additives having no specific mesophase characterised by their chemical composition
- C09K19/56—Aligning agents
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F1/00—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics
- G02F1/01—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour
- G02F1/13—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on liquid crystals, e.g. single liquid crystal display cells
- G02F1/1303—Apparatus specially adapted to the manufacture of LCDs
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F1/00—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics
- G02F1/01—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour
- G02F1/13—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on liquid crystals, e.g. single liquid crystal display cells
- G02F1/133—Constructional arrangements; Operation of liquid crystal cells; Circuit arrangements
- G02F1/1333—Constructional arrangements; Manufacturing methods
- G02F1/1337—Surface-induced orientation of the liquid crystal molecules, e.g. by alignment layers
Definitions
- the present invention relates to a liquid crystal aligning agent containing a terminal polyamic acid ester, a liquid crystal aligning film obtained from the liquid crystal aligning agent, and a liquid crystal display element.
- a liquid crystal alignment film for controlling the alignment state of liquid crystals is usually provided in the element.
- a polyimide liquid crystal alignment film obtained by applying a liquid crystal alignment agent mainly composed of a polyimide precursor such as polyamic acid (polyamic acid) or a solution of soluble polyimide to a glass substrate or the like and baking it is mainly used. It is used.
- liquid crystal alignment films are required to have excellent liquid crystal orientation and stable pretilt angle in addition to the demands of suppressing contrast reduction and afterimage phenomenon.
- the characteristics such as the voltage holding ratio, the suppression of afterimages generated by AC driving, the small amount of residual charges when a DC voltage is applied, and / or the rapid relaxation of the residual charges accumulated by the DC voltage are becoming increasingly important.
- polyamic acid and its imidized polymer contains one carboxylic acid group in the molecule as a liquid crystal alignment film with a high voltage holding ratio and a short time until the afterimage generated by direct current voltage disappears.
- a liquid crystal aligning agent containing a very small amount of a compound selected from a compound containing one carboxylic anhydride group in the molecule and a compound containing one tertiary amino group in the molecule ( For example, see Patent Document 3).
- a liquid crystal alignment agent containing a polyamic acid obtained from a dianhydride and a specific diamine compound or an imidized polymer thereof for example, see Patent Document 4
- a method of suppressing an afterimage caused by alternating current driving in a liquid crystal display element of a lateral electric field driving method a method of using a specific liquid crystal alignment film that has good liquid crystal alignment and large interaction with liquid crystal molecules (patent) Document 5) is known.
- liquid crystal televisions with large screens and high-definition are mainly used, and the demand for afterimages has become more severe, and characteristics that can withstand long-term use in harsh usage environments are required.
- liquid crystal alignment films to be used are required to have higher reliability than conventional liquid crystal alignment films. Not only the initial characteristics of the liquid crystal alignment films are good, but also, for example, they are longer at high temperatures. There is a need to maintain good properties even after time exposure.
- a polyimide-based liquid crystal aligning agent for achieving this has been reported in which the terminal of polyamic acid or polyimide is modified. That is, an imidized polymer having a terminal modified by reacting with a monoacid anhydride, a monoamine compound, and a monoisocyanate compound with the aim of improving liquid crystal alignment, high pretilt angle, small afterimage erasing time, and high reliability. has been proposed (see Patent Document 6). Although the modification methods by these reactions have the advantage that purification after the reaction is unnecessary because there are no by-products, the reaction of the monoacid anhydride and the amine is a reversible reaction, so the end of the polymer is efficiently modified.
- the isocyanate compound is highly reactive, but there is a possibility that the solubility of the polymer in the organic solvent constituting the liquid crystal aligning agent may be lowered due to the influence of the urea bond formed.
- polyamic acid ester is highly reliable, and heat treatment when imidizing it does not cause a decrease in molecular weight. It has been reported that it is excellent in reliability (see Patent Document 7). However, no polyamic acid ester having such a terminal-modified structure has been reported yet.
- An object of the present invention is to provide a liquid crystal aligning agent containing a polyamic acid ester having a specific repeating unit and having a terminal amino group modified to have a specific structure. Moreover, this invention provides the method of modifying the terminal of polyamic acid ester by making the polyamic acid ester which has a specific repeating unit and has an amino group at the terminal react with a chlorocarbonyl compound.
- the inventor conducted research on modification of a polyamic acid ester having an amino group at the terminal, and found that the terminal can be modified with high efficiency by reacting with a chlorocarbonyl compound in the presence of a base. Modification of the terminal by such a method is possible because the polyamic acid ester does not have a carboxyl group and does not cause a side reaction with the existing base. Modification of the terminal amino group in the case of polyamic acid or soluble imide Is not effective.
- polyamic acid ester having a terminal amino group modified has improved solubility in an organic solvent even when it has a high molecular weight.
- a low-viscosity liquid crystal aligning agent contained in a solvent at a high concentration can be obtained, which makes it easy to produce a liquid crystal aligning film by, for example, an inkjet method, and also to produce a thick liquid crystal aligning film. was found to be.
- the present invention is based on the above novel findings and has the following gist. 1.
- a polyamic acid ester having a repeating unit of the following formula (1) and having a terminal amino group modified so that the terminal amino group has the structure of the following formula (2), and an organic solvent Liquid crystal aligning agent.
- X 1 is a tetravalent organic group
- Y 1 is a divalent organic group
- R 1 is an alkyl group having 1 to 5 carbon atoms
- a 1 and A 2 are each independently selected.
- A is a single bond, —O—, —S—, or —NR 3 —
- R 2 and R 3 are each independently an organic group having 1 to 30 carbon atoms, R 2 and R And may be bonded to each other to form a ring structure 3.
- a 3 is a hydrogen atom or an optionally substituted alkyl group, alkenyl group or alkynyl group having 1 to 10 carbon atoms.
- the liquid crystal aligning agent according to 1 or 2 above which contains 0.5 to 15% by mass of the polyamic acid ester whose end is modified with respect to the organic solvent. 4). 4. The polyamic acid ester having a modified end, wherein the terminal amino group of the polyamic acid ester is reacted with a chlorocarbonyl compound having a structure represented by the following formula (3): Liquid crystal aligning agent.
- A is a single bond, -O -, - S-, or -NR 3 - and is, R 2, R 3 are each independently an organic group having 1 to 30 carbon atoms, R 2 And R 3 may be bonded to each other to form a ring structure.
- R 2 and R 3 may be bonded to each other to form a ring structure.
- liquid crystal aligning agent according to any one of 1 to 5, wherein Y 1 in the formula (1) is at least one selected from structures represented by the following formula.
- a method for producing a polyamic acid ester having a terminal modified characterized in that a polyamic acid ester having an amino group at the terminal is reacted with a chlorocarbonyl compound having a structure represented by the following formula (3).
- A is a single bond, -O -, - S-, or -NR 3 - and is, R 2, R 3 are each independently an organic group having 1 to 30 carbon atoms, R 2 and R 3 may combine with each other to form a ring structure.
- 8 The method for producing a polyamic acid ester having a terminal modified as described in 7 above, wherein 0.5 to 60 mol% of a chlorocarbonyl compound is reacted in the presence of a base with respect to one repeating unit of the polyamic acid ester.
- 9. The method for producing a polyamic acid ester having a terminal modified as described in 7 or 8 above, wherein the reaction is carried out at ⁇ 20 ° C. to 150 ° C.
- the novel method of manufacturing the polyamic acid ester which modified the terminal amino group of the polyamic acid ester which has a specific repeating unit with high efficiency, and modified the terminal is provided.
- the terminal polyamic acid ester having a modified terminal has the same structure as that of the polyimide-based liquid crystal aligning agent.
- a liquid crystal aligning agent having a low viscosity can be obtained even at a high concentration. Therefore, for example, a liquid crystal alignment film can be easily manufactured by an ink jet method, and a liquid crystal alignment film having a large thickness can be easily manufactured.
- the polyamic acid ester modified with a terminal having a high solubility in an organic solvent is, for example, a liquid crystal aligning agent mixed with other various liquid crystal aligning compounds having excellent alignment characteristics and electrical characteristics but low solubility in an organic solvent.
- a liquid crystal aligning agent mixed with other various liquid crystal aligning compounds having excellent alignment characteristics and electrical characteristics but low solubility in an organic solvent.
- the polyamic acid ester used for this invention is a polyimide precursor for obtaining a polyimide, and is a polymer which has the site
- the polyamic acid ester contained in the liquid crystal aligning agent of the present invention has a repeating unit of the following formula (1).
- R 1 is an alkyl group having 1 to 5 carbon atoms, preferably 1 to 2 carbon atoms.
- R 1 is particularly preferably a methyl group from the viewpoint of ease of imidization by heat.
- a 1 and A 2 are each independently a hydrogen atom or an alkyl group, alkenyl group, or alkynyl group having 1 to 10 carbon atoms that may have a substituent.
- alkyl group examples include a methyl group, an ethyl group, a propyl group, a butyl group, a t-butyl group, a hexyl group, an octyl group, a decyl group, a cyclopentyl group, and a cyclohexyl group.
- alkenyl group examples include those in which one or more CH 2 —CH 2 structures present in the above alkyl group are replaced with a CH ⁇ CH structure, and more specifically, vinyl groups, allyl groups, 1- Examples include propenyl group, isopropenyl group, 2-butenyl group, 1,3-butadienyl group, 2-pentenyl group, 2-hexenyl group, cyclopropenyl group, cyclopentenyl group, cyclohexenyl group and the like.
- Alkynyl groups include those in which one or more CH 2 —CH 2 structures present in the alkyl group are replaced with C ⁇ C structures, and more specifically, ethynyl groups, 1-propynyl groups, 2 -Propynyl group and the like.
- the above alkyl group, alkenyl group, and alkynyl group may have a substituent as long as it has 1 to 10 carbon atoms as a whole, and may further form a ring structure by the substituent.
- forming a ring structure with a substituent means that the substituents or a substituent and a part of the mother skeleton are bonded to form a ring structure.
- substituents are halogen groups, hydroxyl groups, thiol groups, nitro groups, aryl groups, organooxy groups, organothio groups, organosilyl groups, acyl groups, ester groups, thioester groups, phosphate ester groups, amide groups, alkyls.
- the halogen group as a substituent include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom.
- the aryl group as a substituent include a phenyl group. This aryl group may be further substituted with the other substituent described above.
- the organooxy group which is a substituent can have a structure represented by OR.
- the R may be the same or different, and examples thereof include the alkyl group, alkenyl group, alkynyl group, and aryl group described above. These Rs may be further substituted with the substituent described above.
- alkyloxy group examples include methoxy group, ethoxy group, propyloxy group, butoxy group, pentyloxy group, hexyloxy group, heptyloxy group, octyloxy group and the like.
- the organothio group as a substituent can have a structure represented by —S—R. Examples of R include the aforementioned alkyl group, alkenyl group, alkynyl group, aryl group, and the like. These Rs may be further substituted with the substituent described above.
- alkylthio group examples include a methylthio group, an ethylthio group, a propylthio group, a butylthio group, a pentylthio group, a hexylthio group, a heptylthio group, and an octylthio group.
- the organosilyl group as a substituent can have a structure represented by —Si— (R) 3 .
- the R may be the same or different, and examples thereof include the alkyl group, alkenyl group, alkynyl group, and aryl group described above. These Rs may be further substituted with the substituent described above.
- alkylsilyl group examples include a trimethylsilyl group, a triethylsilyl group, a tripropylsilyl group, a tributylsilyl group, a tripentylsilyl group, a trihexylsilyl group, a pentyldimethylsilyl group, and a hexyldimethylsilyl group.
- the acyl group as a substituent can have a structure represented by —C (O) —R. Examples of R include the above-described alkyl group, alkenyl group, and aryl group. These Rs may be further substituted with the substituent described above.
- acyl group examples include formyl group, acetyl group, propionyl group, butyryl group, isobutyryl group, valeryl group, isovaleryl group, benzoyl group and the like.
- ester group which is a substituent a structure represented by —C (O) O—R or —OC (O) —R can be shown.
- R examples include the aforementioned alkyl group, alkenyl group, alkynyl group, aryl group, and the like. These Rs may be further substituted with the substituent described above.
- the thioester group which is a substituent can have a structure represented by —C (S) O—R or —OC (S) —R.
- R examples include the aforementioned alkyl group, alkenyl group, alkynyl group, aryl group, and the like. These Rs may be further substituted with the substituent described above.
- the phosphate group which is a substituent can have a structure represented by —OP (O) — (OR) 2 .
- the R may be the same or different, and examples thereof include the alkyl group, alkenyl group, alkynyl group, and aryl group described above. These Rs may be further substituted with the substituent described above.
- Examples of the substituent amide group include —C (O) NH 2 , —C (O) NHR, —NHC (O) R, —C (O) N (R) 2 , —NRC (O) R.
- the structure represented by can be shown.
- the R may be the same or different, and examples thereof include the alkyl group, alkenyl group, alkynyl group, and aryl group described above. These Rs may be further substituted with the substituent described above.
- Examples of the aryl group as a substituent include the same aryl groups as described above. This aryl group may be further substituted with the other substituent described above.
- Examples of the alkyl group as a substituent include the same alkyl groups as described above.
- This alkyl group may be further substituted with the other substituent described above.
- Examples of the alkenyl group as a substituent include the same alkenyl groups as described above. This alkenyl group may be further substituted with the other substituent described above.
- Examples of the alkynyl group that is a substituent include the same alkynyl groups as described above. This alkynyl group may be further substituted with the other substituent described above.
- a 1 and A 2 a hydrogen atom or a carbon atom that may have a substituent is 1
- An alkyl group of 1 to 5 is more preferable, and a hydrogen atom, a methyl group or an ethyl group is particularly preferable.
- R 1 is an alkyl group having 1 to 5 carbon atoms, preferably 1 to 2 carbon atoms.
- X 1 is a tetravalent organic group and is not particularly limited. Two or more kinds of X 1 may be mixed in the polyimide precursor. Specific examples of X 1 include X-1 to X-46 shown below independently of each other.
- X 1 is independently X-1, X-2, X-3, X-4, X-5, X-6, X-8, X-16, X-19, X-21, X-25, X-26, X-27, X-28, or X-32 are preferred.
- the amount of these tetracarboxylic dianhydrides having preferable X 1 is preferably 1 to 100 mol%, more preferably 50 to 100 mol% of the total tetracarboxylic dianhydrides.
- Y 1 is a divalent organic group and is not particularly limited. In the polyimide precursor, two or more kinds of Y 1 may be mixed independently. Specific examples of Y 1 include the following Y-1 to Y-103.
- Y 1 is Y-7, Y-8, Y-10 to Y-113.
- a diamine having a long chain alkyl group, an aromatic ring, an aliphatic ring, a steroid skeleton, or a combination thereof in the side chain into the polyamic acid ester.
- the diamine of any of Y-76 to Y-97 is more preferable.
- Y 1 is preferably Y-19, Y-23, Y-25 to Y-27, Y-30 to Y-36, Y-40 to Y-42, Y-44, Y-45, Y- 49, Y-50, Y-51, or Y-61 is more preferable, and Y-31 or Y-40 is particularly preferable.
- the amount of these preferred diamines having Y 1 is preferably 1 to 100 mol%, more preferably 50 to 100 mol% of the total diamine.
- Y 1 is Y-7, Y-8, Y-20 to Y22, Y-28, Y-30, Y-31, Y-40, Y-41, Y-48, Y-72, or Particularly preferred is at least one selected from the structure represented by the following formula: 79.
- the polyamic acid ester represented by the above formula (1) is obtained by reaction of any of the tetracarboxylic acid derivatives represented by the following formulas (6) to (8) with the diamine compound represented by the formula (9). be able to.
- the polyamic acid ester represented by the above formula (1) can be synthesized by the following methods (1) to (3) using the above monomer.
- Polyamic acid ester can be manufactured by esterifying the polyamic acid obtained from tetracarboxylic dianhydride and diamine.
- the polyamic acid and the esterifying agent are reacted in the presence of an organic solvent at ⁇ 20 ° C. to 150 ° C., preferably 0 ° C. to 50 ° C. for 30 minutes to 24 hours, preferably 1 to 4 hours.
- an organic solvent at ⁇ 20 ° C. to 150 ° C., preferably 0 ° C. to 50 ° C. for 30 minutes to 24 hours, preferably 1 to 4 hours.
- an organic solvent ⁇ 20 ° C. to 150 ° C., preferably 0 ° C. to 50 ° C. for 30 minutes to 24 hours, preferably 1 to 4 hours.
- the esterifying agent those that can be easily removed by purification are preferable.
- the addition amount of the esterifying agent is preferably 2 to 6 molar equivalents per 1 mol of the polyamic acid repeating unit.
- the solvent used in the above reaction is preferably N, N-dimethylformamide, N-methyl-2-pyrrolidone or ⁇ -butyrolactone from the solubility of the polymer, and these may be used alone or in combination of two or more. .
- the concentration at the time of production is preferably 1 to 30% by mass and more preferably 5 to 20% by mass from the viewpoint that polymer precipitation is unlikely to occur and a high molecular weight product is easily obtained.
- tetracarboxylic acid diester dichloride and diamine are reacted in the presence of a base and an organic solvent at ⁇ 20 to 150 ° C., preferably 0 to 50 ° C., for 30 minutes to 24 hours, preferably 1 to 4 hours.
- a base pyridine, triethylamine, 4-dimethylaminopyridine and the like can be used, but pyridine is preferable because the reaction proceeds gently.
- the addition amount of the base is preferably 2 to 4 moles relative to the tetracarboxylic acid diester dichloride from the viewpoint that it can be easily removed and a high molecular weight product can be easily obtained.
- the solvent used in the above reaction is preferably N-methyl-2-pyrrolidone or ⁇ -butyrolactone because of the solubility of the monomer and polymer, and these may be used alone or in combination.
- the concentration of tetracarboxylic acid diester dichloride and diamine during production is preferably 1 to 30% by mass and more preferably 5 to 20% by mass from the viewpoint that polymer precipitation is difficult to occur and a high molecular weight product is easily obtained.
- the solvent used for the production of the polyamic acid ester is preferably dehydrated as much as possible, and it is preferable to prevent mixing of outside air in a nitrogen atmosphere.
- Polyamic acid ester can be produced by polycondensation of tetracarboxylic acid diester and diamine. Specifically, tetracarboxylic acid diester and diamine are reacted in the presence of a condensing agent, a base, and an organic solvent at 0 to 150 ° C., preferably 0 to 100 ° C., for 30 minutes to 24 hours, preferably 3 to 15 hours. Can be manufactured.
- condensing agent examples include triphenyl phosphite, dicyclohexylcarbodiimide, 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, N, N′-carbonyldiimidazole, dimethoxy-1,3,5-triazide.
- Nylmethylmorpholinium O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium tetrafluoroborate, O- (benzotriazol-1-yl) -N, N , N ′, N′-tetramethyluronium hexafluorophosphate, (2,3-dihydro-2-thioxo-3-benzoxazolyl) phosphonate diphenyl, and the like.
- the addition amount of the condensing agent is preferably 2 to 3 times the molar amount of the tetracarboxylic acid diester.
- tertiary amines such as pyridine and triethylamine can be used.
- the amount of the base added is preferably 2 to 4 times the molar amount of the diamine component from the viewpoint of easy removal and high molecular weight.
- the reaction proceeds efficiently by adding Lewis acid as an additive.
- the Lewis acid lithium halides such as lithium chloride and lithium bromide are preferable.
- the addition amount of the Lewis acid is preferably 0 to 1.0 times mol with respect to the diamine component.
- the production method of (1) or (2) is particularly preferable.
- the polyamic acid ester solution obtained as described above can be polymerized by pouring into a poor solvent while stirring well. Precipitation is performed several times, and after washing with a poor solvent, a purified polyamic acid ester powder can be obtained at room temperature or by heating and drying.
- a poor solvent is not specifically limited, Water, methanol, ethanol, hexane, butyl cellosolve, acetone, toluene etc. are mentioned.
- the polyamic acid ester of the present invention is a polyamic acid ester in which the terminal obtained by reacting the chlorocarbonyl compound represented by the following formula (3) with the main chain terminal amino group of the polyamic acid ester is modified.
- A is a single bond, —O—, —S—, or —NR 3 —
- R 2 and R 3 are each independently an organic group having 1 to 30 carbon atoms, This organic group is selected from an alkyl group, an alkenyl group, an alkynyl group, an aryl group, and a group obtained by combining these, and these may have a substituent.
- R 2 and R 3 may be bonded to each other to form a monocyclic or polycyclic ring.
- the above alkyl group, alkenyl group, alkynyl group, and aryl group may have a substituent as long as it has 1 to 20 carbon atoms as a whole, and may further form a ring structure by the substituent.
- forming a ring structure with a substituent means that the substituents or a substituent and a part of the mother skeleton are bonded to form a ring structure.
- the alkyl group is preferably one having 1 to 20 carbon atoms, specifically, methyl group, ethyl group, propyl group, butyl group, t-butyl group, hexyl group, octyl group, decyl group, cyclopentyl group, cyclohexyl group. Group, bicyclohexyl group and the like.
- alkenyl group examples include those in which one or more CH 2 —CH 2 structures present in the above alkyl group are replaced with a CH ⁇ CH structure, and more specifically, vinyl groups, allyl groups, 1- Examples include propenyl group, isopropenyl group, 2-butenyl group, 1,3-butadienyl group, 2-pentenyl group, 2-hexenyl group, cyclopropenyl group, cyclopentenyl group, cyclohexenyl group and the like.
- Alkynyl groups include those in which one or more CH 2 —CH 2 structures present in the alkyl group are replaced with C ⁇ C structures, and more specifically, ethynyl groups, 1-propynyl groups, 2 -Propynyl group and the like.
- aryl group examples include a phenyl group, ⁇ -naphthyl group, ⁇ -naphthyl group, o-biphenylyl group, m-biphenylyl group, p-biphenylyl group, 1-anthryl group, 2-anthryl group, 9-anthryl group, 1 -Phenanthryl group, 2-phenanthryl group, 3-phenanthryl group, 4-phenanthryl group, 9-phenanthryl group and the like.
- the above alkyl group, alkenyl group, alkynyl group, and aryl group may have a substituent as long as it has 1 to 20 carbon atoms as a whole, and may further form a ring structure by the substituent.
- forming a ring structure with a substituent means that the substituents or a substituent and a part of the mother skeleton are bonded to form a ring structure.
- this substituent include halogen groups, hydroxyl groups, thiol groups, nitro groups, organooxy groups, organothio groups, organosilyl groups, acyl groups, ester groups, thioester groups, phosphate ester groups, amide groups, aryl groups, alkyls.
- halogen group as a substituent examples include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom.
- the organooxy group as a substituent can have a structure represented by —O—R such as an alkoxy group, an alkenyloxy group, and an aryloxy group. Examples of R include the above-described alkyl group, alkenyl group, and aryl group. These Rs may be further substituted with the substituent described above.
- alkyloxy group examples include a methoxy group, an ethoxy group, a propoxy group, a butoxy group, a pentyloxy group, a hexyloxy group, a heptyloxy group, an octyloxy group, a nonyloxy group, a decyloxy group, and a lauryloxy group.
- the organothio group as a substituent can have a structure represented by —SR, such as an alkylthio group, an alkenylthio group, and an arylthio group.
- R include the above-described alkyl group, alkenyl group, and aryl group. These Rs may be further substituted with the substituent described above.
- alkylthio group examples include a methylthio group, an ethylthio group, a propylthio group, a butylthio group, a pentylthio group, a hexylthio group, a heptylthio group, an octylthio group, a nonylthio group, a decylthio group, and a laurylthio group.
- the organosilyl group as a substituent can have a structure represented by —Si— (R) 3 .
- the R may be the same or different, and examples thereof include the alkyl groups and aryl groups described above. These Rs may be further substituted with the substituent described above.
- alkylsilyl group examples include trimethylsilyl group, triethylsilyl group, tripropylsilyl group, tributylsilyl group, tripentylsilyl group, trihexylsilyl group, pentyldimethylsilyl group, hexyldimethylsilyl group, octyldimethylsilyl group, Examples include decyldimethylsilyl group.
- the acyl group as a substituent can have a structure represented by —C (O) —R.
- R include the above-described alkyl group, alkenyl group, and aryl group. These Rs may be further substituted with the substituent described above.
- Specific examples of the acyl group include formyl group, acetyl group, propionyl group, butyryl group, isobutyryl group, valeryl group, isovaleryl group, benzoyl group and the like.
- As the ester group which is a substituent a structure represented by —C (O) O—R or —OC (O) —R can be shown. Examples of R include the above-described alkyl group, alkenyl group, and aryl group.
- the thioester group which is a substituent can have a structure represented by —C (S) O—R or —OC (S) —R.
- R include the above-described alkyl group, alkenyl group, and aryl group.
- the phosphate group which is a substituent can have a structure represented by —OP (O) — (OR) 2 .
- the R may be the same or different, and examples thereof include the alkyl groups and aryl groups described above. These Rs may be further substituted with the substituent described above.
- Examples of the substituent amide group include —C (O) NH 2 , —C (O) NHR, —NHC (O) R, —C (O) N (R) 2 , —NRC (O) R.
- the structure represented by can be shown.
- the R may be the same or different, and examples thereof include the alkyl groups and aryl groups described above. These Rs may be further substituted with the substituent described above.
- Examples of the aryl group as a substituent include the same aryl groups as described above. This aryl group may be further substituted with the other substituent described above.
- Examples of the alkyl group as a substituent include the same alkyl groups as described above. This alkyl group may be further substituted with the other substituent described above.
- Examples of the alkenyl group as a substituent include the same alkenyl groups as described above. This alkenyl group may be further substituted with the other substituent described above.
- Examples of the alkynyl group that is a substituent include the same alkynyl groups as described above. This alkynyl group may be further substituted with the other substituent described above.
- chlorocarbonyl compound represented by the above formula (1) examples include the following (C-1) to (C-111), but the present invention is not limited thereto.
- the chlorocarbonyl compound used in the present invention is preferably a chlorocarbonyl compound in which A is a single bond in the formula (3), and specifically, C-1, C-2, C-3.
- the terminal of the polyamic acid ester having a repeating unit of the formula (1) having an amino group at the terminal is modified so that the amino group has the structure of the formula (2).
- This terminal-modified polyamic acid ester can be obtained by several methods, but the polyamic acid ester powder having an amino group at the terminal obtained by the above-described method for producing a polyamic acid ester is dissolved in an organic solvent.
- a polyamic acid ester having an amino group is obtained, the polyamic acid ester is added to the reaction system without isolating the polyamic acid ester, and the polyamic acid ester having an amino group at the terminal present in the reaction system
- the method of making it react is mentioned.
- the method of adding a chlorocarbonyl compound to the latter reaction system is more preferable because the polyamic acid ester can be purified by reprecipitation only once and the production process can be shortened.
- the ratio of the diamine component to the tetracarboxylic acid dialkyl ester derivative is preferably 1: 0.7 to 1: 1, and more preferably 1: 0.8 to 1: 1.
- a method of adding a chlorocarbonyl compound to the above reaction system a method of adding a tetracarboxylic acid dialkyl ester derivative and reacting with a diamine, a reaction of a tetracarboxylic acid dialkyl ester derivative and a diamine sufficiently, There is a method of adding a chlorocarbonyl compound after producing a polyamic acid ester in which is an amino group. The latter method is more preferable because the molecular weight of the polymer can be easily controlled.
- the reaction between the polyamic acid ester having an amino group at the terminal and the chlorocarbonyl compound is carried out at ⁇ 20 to 150 ° C., preferably 0 to 50 ° C. in the presence of a base and an organic solvent. 30 minutes to 24 hours, preferably 30 minutes to 4 hours.
- the addition amount of the chlorocarbonyl compound is preferably 0.5 to 60 mol%, more preferably 1 to 40 mol%, based on the repeating unit of the polyamic acid ester having an amino group at the end. When the addition amount is large, unreacted chlorocarbonyl compound remains and is difficult to remove, so that it is more preferably 1 to 20 mol%.
- pyridine triethylamine, and 4-dimethylaminopyridine can be preferably used, but pyridine is preferable because the reaction proceeds gently. If the amount of the base is too large, removal is difficult, and if it is too small, the molecular weight is small. Therefore, the amount is preferably 2 to 4 times the mol of the chlorocarbonyl compound.
- the organic solvent N, N-dimethylformamide, N-methyl-2-pyrrolidone, and ⁇ -butyrolactone are preferable. You may use these 1 type or in mixture of 2 or more types.
- the terminal polyamic acid ester modified is the total amount of the polyamic acid ester contained in the liquid crystal aligning agent. Is not necessarily required, but is preferably 15% by mass or more, more preferably 40% by mass or more, and particularly preferably 60% by mass or more based on the total amount of the polyamic acid ester contained.
- the content of the terminal amino group-modified polyamic acid ester is small, it is not preferable because a sufficient effect intended in the present invention cannot be obtained.
- the liquid crystal aligning agent of the present invention is in the form of a solution in which the polyamic acid ester in which the terminal amino group is modified is dissolved in an organic solvent.
- the molecular weight of the polyamic acid ester modified with the terminal amino group is preferably 2,000 to 500,000, more preferably 5,000 to 300,000 in terms of the weight average molecular weight even when the terminal amino group is not modified. More preferably, it is 10,000 to 100,000.
- the number average molecular weight is preferably 1,000 to 250,000, more preferably 2,500 to 150,000, and still more preferably 5,000 to 50,000.
- the polyamic acid ester in which the terminal amino group is modified has a feature that the solubility in an organic solvent is high even when the average molecular weight is high.
- the viscosity of a solution of a polyamic acid ester having a terminal amino group modified in an organic solvent is preferably higher than that of a polyamic acid ester solution having no terminal modification.
- Such a decrease in viscosity can provide a liquid crystal aligning agent having a higher polymer concentration, can easily obtain a liquid crystal aligning film having a large thickness, and is also clogged when a liquid crystal aligning film is obtained by an inkjet method or the like. This is advantageous because the occurrence of trouble can be suppressed.
- the liquid crystal aligning agent of the present invention has the form of a solution in which the terminal-modified polyamic acid ester is dissolved in an organic solvent, for example, when producing the terminal polyamic acid ester in an organic solvent,
- the resulting reaction solution itself may be used, or the reaction solution may be diluted with an appropriate solvent.
- the polyamic acid ester which modified the terminal is obtained as a powder, it may be dissolved in an organic solvent to form a solution.
- the concentration of the polymer component during production is preferably 10 to 30% by mass, particularly preferably 10 to 15% by mass.
- the heating temperature is preferably 20 ° C to 150 ° C, particularly preferably 20 ° C to 80 ° C.
- the organic solvent contained in the liquid crystal aligning agent of the present invention is not particularly limited as long as the polymer component is uniformly dissolved.
- Specific examples thereof include N, N-dimethylformamide, N, N-diethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidone, N-ethyl-2-pyrrolidone, N-methylcaprolactam, Examples include 2-pyrrolidone, N-vinyl-2-pyrrolidone, dimethyl sulfoxide, dimethyl sulfone, ⁇ -butyrolactone, 1,3-dimethyl-imidazolidinone, 3-methoxy-N, N-dimethylpropanamide and the like. You may use these 1 type or in mixture of 2 or more types. Moreover, even if it is a solvent which cannot melt
- the liquid crystal aligning agent of the present invention may contain a solvent for improving the uniformity of the coating film when the liquid crystal aligning agent is applied to the substrate, in addition to the organic solvent for dissolving the polymer component.
- a solvent for improving the uniformity of the coating film when the liquid crystal aligning agent is applied to the substrate, in addition to the organic solvent for dissolving the polymer component.
- a solvent having a surface tension lower than that of the organic solvent is generally used.
- ethyl cellosolve examples thereof include ethyl cellosolve, butyl cellosolve, ethyl carbitol, butyl carbitol, ethyl carbitol acetate, ethylene glycol, 1-methoxy-2-propanol, 1-ethoxy-2-propanol, 1-butoxy-2 -Propanol, 1-phenoxy-2-propanol, propylene glycol monoacetate, propylene glycol diacetate, propylene glycol-1-monomethyl ether-2-acetate, propylene glycol-1-monoethyl ether-2-acetate, butyl cellosolve acetate, di Propylene glycol, 2- (2-ethoxypropoxy) propanol, lactate methyl ester, lactate ethyl ester, lactate n-propyl ester, lactate n-butyl ester, lactic acid Isoamyl ester, and the like. Two types of
- the content (concentration) of the end-modified polyamic acid ester in the liquid crystal aligning agent of the present invention can be appropriately changed by setting the thickness of the liquid crystal alignment film to be formed, but it is uniform and has no defect. From the viewpoint of forming a film, it is preferably 0.5% by mass or more with respect to the organic solvent, and from the viewpoint of storage stability of the solution, it is preferably 15% by mass or less, particularly preferably 1 to 10% by mass.
- the other liquid crystal aligning agent which is a compound which has liquid crystal aligning property other than the polyamic acid ester which modified the terminal may contain.
- liquid crystal aligning agents examples include various types such as a polyamic acid ester whose terminal amino group is not modified, a soluble polyimide, and / or a liquid crystal aligning agent containing polyamic acid.
- the polyamic acid ester whose end is modified has a high solubility in an organic solvent, so it has excellent alignment characteristics and electrical characteristics, but has a low solubility in an organic solvent, for example, a liquid crystal aligning agent containing polyamic acid or soluble polyimide. It is particularly useful when it is contained.
- the liquid crystal aligning agent of the present invention may contain various additives such as a silane coupling agent and a crosslinking agent.
- the silane coupling agent is added for the purpose of improving the adhesion between the substrate on which the liquid crystal alignment agent is applied and the liquid crystal alignment film formed thereon.
- the specific example of a silane coupling agent is given to the following, the silane coupling agent which can be used for the liquid crystal aligning agent of this invention is not limited to this.
- the polyamic acid ester modified with the terminal of the present invention to which the silane coupling agent is added is heated to promote the reaction between the organic functional group of the silane coupling agent and the polyamic acid ester. By making it, adhesiveness with a base material can be improved more.
- the silane coupling agent is added to a solution obtained by dissolving the polyamic acid ester in the above-mentioned good solvent, and this is added at 20 ° C. to 80 ° C., more preferably 40 ° C.
- An example is a method of stirring at a temperature of from 60 to 60 ° C. for 1 to 24 hours.
- an imidization accelerator may be added to efficiently advance imidization of the polyamic acid ester when the coating film is baked. Specific examples of the imidization accelerator for polyamic acid ester are given below, but the imidization accelerator that can be used in the liquid crystal aligning agent of the present invention is not limited thereto.
- D in the above formulas (B-1) to (B-17) is each independently a tert-butoxycarbonyl group or a 9-fluorenylmethoxycarbonyl group.
- (B-14) to (B-17) there are a plurality of D's in one formula, but these may be the same or different.
- the content of the imidization accelerator is not particularly limited as long as the effect of promoting thermal imidation of the polyamic acid ester is obtained. If the lower limit is deliberately shown, it is preferably 0.01 mol or more, more preferably 0.05 mol or more, still more preferably with respect to 1 mol of the amic acid ester moiety of the following formula (12) contained in the polyamic acid ester. 0.1 mol or more is mentioned.
- the upper limit of the polyamic acid ester of the present invention can be given if the upper limit is shown.
- the imidization accelerator is 2 mol or less, more preferably 1 mol or less, and still more preferably 0.5 mol or less with respect to 1 mol of the amic acid ester moiety of the following formula (12) contained.
- the liquid crystal alignment film of the present invention is a film obtained by applying the liquid crystal aligning agent to a substrate, drying and baking.
- the substrate to which the liquid crystal aligning agent of the present invention is applied is not particularly limited as long as it is a highly transparent substrate, and a glass substrate, a silicon nitride substrate, an acrylic substrate, a plastic substrate such as a polycarbonate substrate, or the like can be used. It is preferable to use a substrate on which an ITO electrode or the like for driving is formed in terms of simplification of the process. Further, in the reflection type liquid crystal display element, an opaque material such as a silicon wafer can be used as long as the substrate is only on one side, and in this case, a material that reflects light such as aluminum can be used.
- Examples of the method for applying the liquid crystal aligning agent of the present invention include a spin coating method, a printing method, and an ink jet method.
- Arbitrary temperature and time can be selected for the drying and baking steps after applying the liquid crystal aligning agent of the present invention.
- it is dried at 50 to 120 ° C. for 1 to 10 minutes and then baked at 150 to 300 ° C. for 5 to 120 minutes.
- the thickness of the coating film after firing is not particularly limited, but if it is too thin, the reliability of the liquid crystal display element may be lowered, and therefore it is 5 to 300 nm, preferably 10 to 200 nm.
- Examples of a method for aligning the obtained liquid crystal alignment film include a rubbing method and a photo-alignment processing method.
- the liquid crystal aligning agent of the present invention is particularly useful when used in the photo-alignment processing method.
- the photo-alignment treatment method there is a method in which the surface of the coating film is irradiated with radiation polarized in a certain direction, and in some cases, a heat treatment is further performed at a temperature of 150 to 250 ° C. to impart liquid crystal alignment ability.
- the radiation ultraviolet rays and visible rays having a wavelength of 100 to 800 nm can be used.
- ultraviolet rays having a wavelength of 100 to 400 nm are preferable, and those having a wavelength of 200 to 400 nm are particularly preferable.
- radiation may be irradiated while heating the coated substrate at 50 to 250 ° C. Dose of the radiation is preferably in the range of 1 ⁇ 10,000mJ / cm 2, and particularly preferably in the range of 100 ⁇ 5,000mJ / cm 2.
- the liquid crystal alignment film produced as described above can stably align liquid crystal molecules in a certain direction.
- the molecular weight of the polyamic acid ester is measured by a GPC (normal temperature gel permeation chromatography) apparatus, and is a number average molecular weight (hereinafter also referred to as Mn) and a weight average molecular weight (hereinafter also referred to as Mw) as polyethylene glycol and polyethylene oxide equivalent values. ) was calculated.
- Mn number average molecular weight
- Mw weight average molecular weight
- GPC device manufactured by Shodex (GPC-101) Column: manufactured by Shodex (series of KD803 and KD805) Column temperature: 50 ° C Eluent: N, N-dimethylformamide (as additives, lithium bromide-hydrate (LiBr ⁇ H 2 O) 30 mmol / L, phosphoric acid / anhydrous crystals (o-phosphoric acid) 30 mmol / L, tetrahydrofuran) (THF) is 10 ml / L) Flow rate: 1.0 ml / min Standard sample for preparing calibration curve: TSK standard polyethylene oxide (weight average molecular weight (Mw) of about 900,000, 150,000, 100,000, 30,000) manufactured by Tosoh Corporation, and polymer laboratory Polyethylene glycol manufactured by the company (peak top molecular weight (Mp) of about 12,000, 4,000, 1,000). In order to avoid the overlapping of peaks, the measurement was performed by mixing four types of 900,000, 100,000, 12,000
- this crystal was found to be compound (3-1), that is, dimethyl-1,3-bis (chlorocarbonyl) -1,3-dimethylcyclobutane-2,4-dicarboxylate (1,3-DM -CBDE-C1) (HPLC relative area 99.5%) (yield 77.2%).
- 1 H NMR (CDCl 3 , ⁇ ppm): 3.78 (s, 6H), 3.72 (s, 2H), 1.69 (s, 6H).
- the obtained polyamic acid ester solution was poured into 1757 g of water with stirring, and the precipitated white precipitate was collected by filtration, followed by 1757 g of water once, 1757 g of ethanol once, and 439 g of ethanol.
- the white polyamic acid ester resin powder 16.63g was obtained by wash
- the obtained polyamic acid ester resin powder 14.8252 was placed in a 200 ml Erlenmeyer flask, NMP99.3048 g was added, and the mixture was stirred and dissolved at room temperature for 24 hours to obtain a polyamic acid ester solution (B-1).
- the obtained polyamic acid ester solution was poured into 1747 g of water with stirring, and the precipitated white precipitate was collected by filtration, and then once with 1747 g of water, once with 1747 g of ethanol, and 437 g of ethanol.
- the white polyamic acid ester resin powder 16.65g was obtained by wash
- Example 1 Take 4.2033 g of the polyamic acid ester solution (A-1) obtained in Production Example 1 in an Erlenmeyer flask, add 0.5991 g of NMP and 1.2099 g of BCS, and stir for 30 minutes with a magnetic stirrer.
- the liquid crystal aligning agent (I) was obtained.
- the viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in the table below.
- Example 2 Take 4.2123 g of the polyamic acid ester solution (A-2) obtained in Production Example 2 in an Erlenmeyer flask, add 0.6077 g of NMP and 1.2077 g of BCS, and stir for 30 minutes with a magnetic stirrer.
- a liquid crystal aligning agent (II) was obtained.
- the viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured.
- the results are shown in the table below.
- (Example 3) Take 4.1955 g of the polyamic acid ester solution (A-3) obtained in Production Example 3 in an Erlenmeyer flask, add 0.6053 g of NMP and 1.1953 g of BCS, and stir for 30 minutes with a magnetic stirrer with a solid content of 7% by mass.
- a liquid crystal aligning agent (III) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in the table below.
- Example 4 Take 4.2010 g of the polyamic acid ester solution (A-4) obtained in Production Example 4 in an Erlenmeyer flask, add 0.5993 g of NMP and 1.2519 g of BCS, and stir for 30 minutes with a magnetic stirrer with a solid content of 7% by mass. A liquid crystal aligning agent (IV) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in the table below.
- Example 5 4.1984 g of the polyamic acid ester solution (A-5) obtained in Production Example 5 is placed in an Erlenmeyer flask, 0.59888 g of NMP and 1.2029 g of BCS are added, and the mixture is stirred for 30 minutes with a magnetic stirrer with a solid content of 7% by mass. A liquid crystal aligning agent (V) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in the table below.
- Example 6 4.1999 g of the polyamic acid ester solution (A-6) obtained in Production Example 6 is placed in an Erlenmeyer flask, 0.5906 g of NMP and 1.0244 g of BCS are added, and the mixture is stirred for 30 minutes with a magnetic stirrer with a solid content of 7% by mass. A liquid crystal aligning agent (VI) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in the table below.
- Example 7 Take 3.0789 g of the polyamic acid ester solution (A-7) obtained in Production Example 7 in an Erlenmeyer flask, add 0.4393 g of NMP and 0.8791 g of BCS, and stir for 30 minutes with a magnetic stirrer. A liquid crystal aligning agent (VII) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in the table below.
- Example 8 Take 3.0791 g of the polyamic acid ester solution (A-8) obtained in Production Example 8 in an Erlenmeyer flask, add 0.4361 g of NMP and 0.8833 g of BCS, and stir for 30 minutes with a magnetic stirrer. A liquid crystal aligning agent (VIII) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in the table below.
- Example 9 Take 3.0231 g of the polyamic acid ester solution (A-9) obtained in Production Example 9 in an Erlenmeyer flask, add 0.4355 g of NMP and 0.8699 g of BCS, and stir with a magnetic stirrer for 30 minutes to obtain a solid concentration of 7% by mass. A liquid crystal aligning agent (IX) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in the table below.
- Comparative Example 1 Take 4.2066 g of the polyamic acid ester solution (B-1) obtained in Comparative Production Example 1 in an Erlenmeyer flask, add 0.5957 g of NMP and 1.1908 g of BCS, and stir with a magnetic stirrer for 30 minutes to obtain a solid content concentration of 7% by mass.
- a liquid crystal aligning agent (BI) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in the table below.
- Comparative Example 2 Take 4.2010 g of the polyamic acid ester solution (B-2) obtained in Comparative Production Example 2 in an Erlenmeyer flask, add 0.5993 g of NMP and 1.2519 g of BCS, and stir for 30 minutes with a magnetic stirrer. The solid content concentration is 7% by mass. A liquid crystal aligning agent (B-II) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in the table below.
- Example 10 The liquid crystal aligning agent (I) obtained in Example 1 was filtered through a 1.0 ⁇ m filter, spin-coated on a glass substrate with a transparent electrode, dried on a hot plate at a temperature of 80 ° C. for 5 minutes, and then at a temperature of 250. An imidized film having a film thickness of 100 nm was obtained after baking at 1 ° C. for 1 hour. The coating surface was irradiated with 100 mJ / cm 2 of 254 nm ultraviolet light through a polarizing plate to obtain a substrate with a liquid crystal alignment film.
- the obtained polyamic acid ester solution was poured into 498 g of ethanol while stirring, and the precipitated white precipitate was collected by filtration, followed by 226 g of ethanol once, 452 g of water twice, and 453 g of ethanol 1 This was washed 3 times with 113 g of ethanol and dried to obtain 4.4587 g of white polyamic acid ester resin powder.
- the yield was 98.53%.
- the obtained polyamic acid ester solution was poured into 500 g of ethanol while stirring, and the precipitated white precipitate was collected by filtration, followed by 227 g of ethanol once, 455 g of water twice, and 455 g of ethanol 1 This was washed 3 times with 114 g of ethanol and dried to obtain 4.2721 g of white polyamic acid ester resin powder.
- the yield was 93.91%.
- the obtained polyamic acid ester solution was poured into 498 g of ethanol with stirring, and the precipitated white precipitate was collected by filtration, followed by 226 g of ethanol once, 453 g of water twice, and 453 g of ethanol 1 This was washed 3 times with 113 g of ethanol and dried to obtain 3.98 g of white polyamic acid ester resin powder.
- the yield was 87.92%.
- the obtained polyamic acid ester solution was poured into 491 g of ethanol while stirring, and the precipitated white precipitate was collected by filtration, followed by 223 g of ethanol once, 446 g of water twice, and 446 g of ethanol 1 This was washed 3 times with 111 g of ethanol and dried to obtain 3.74 g of white polyamic acid ester resin powder.
- the yield was 83.86%.
- the obtained polyamic acid ester solution was poured into 499 g of ethanol while stirring, and the precipitated white precipitate was collected by filtration, followed by 227 g of ethanol once, 454 g of water twice, and 454 g of ethanol once. This was washed 3 times with 114 g of ethanol and dried to obtain 4.5616 g of white polyamic acid ester resin powder. The yield was 99.0%.
- 2.2436 g of the obtained polyamic acid ester resin powder was placed in a 50 ml Erlenmeyer flask, 20.1778 g of NMP was added, and the mixture was stirred and dissolved at room temperature for 24 hours to obtain a polyamic acid ester solution (A-22).
- a 100 mL four-necked flask equipped with a stirrer is placed in a nitrogen atmosphere. I let you. Next, while stirring this diamine solution, 3.0831 g (9.4825 mmol) of 1,3DM-CBDE-Cl was added and reacted for 4 hours under water cooling.
- the obtained polyamic acid ester solution was added to 552 g of ethanol while stirring, and the precipitated white precipitate was collected by filtration, followed by 227 g of ethanol once, 460 g of water twice, and 228 g of ethanol 1 This was washed three times with 115 g of ethanol and dried to obtain 4.24 g of white polyamic acid ester resin powder.
- the yield was 92.2%.
- the obtained polyamic acid ester solution was added to 545 g of ethanol while stirring, and the precipitated white precipitate was collected by filtration, followed by 227 g of ethanol once, 454 g of water twice, and 227 g of ethanol once. This was washed 3 times with 114 g of ethanol and dried to obtain 3.89 g of white polyamic acid ester resin powder. The yield was 85.7%.
- 2.2187 g of the obtained polyamic acid ester resin powder was placed in a 50 ml Erlenmeyer flask, 19.9635 g of NMP was added, and the mixture was stirred and dissolved at room temperature for 24 hours to obtain a polyamic acid ester solution (A-26).
- Example 11 Take 2.8093 g of the polyamic acid ester solution (A-10) obtained in Production Example 10 in a 50 ml Erlenmeyer flask, add 0.4070 g of NMP and 0.7930 g of BCS, and stir for 30 minutes with a magnetic stirrer. % Liquid crystal aligning agent (A1-1) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 12 Take 2.8168 g of the polyamic acid ester solution (A-11) obtained in Production Example 11 in a 50 ml Erlenmeyer flask, add 0.3964 g of NMP and 0.7930 g of BCS, stir for 30 minutes with a magnetic stirrer, solid content concentration 7 mass % Liquid crystal aligning agent (A1-2) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 13 Take 2.8030 g of the polyamic acid ester solution (A-12) obtained in Production Example 12 in a 50 ml Erlenmeyer flask, add 0.3929 g of NMP and 0.8060 g of BCS, stir for 30 minutes with a magnetic stirrer, solid content concentration 7 mass % Liquid crystal aligning agent (A1-3) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 14 Take 2.8092 g of the polyamic acid ester solution (A-13) obtained in Production Example 13 in a 50 ml Erlenmeyer flask, add 0.4024 g of NMP and 0.8047 g of BCS, and stir with a magnetic stirrer for 30 minutes. A mass% liquid crystal aligning agent (A1-4) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 15 Take 2.8121 g of the polyamic acid ester solution (A-14) obtained in Production Example 14 in a 50 ml Erlenmeyer flask, add 0.4002 g of NMP and 0.8154 g of BCS, and stir for 30 minutes with a magnetic stirrer, solid content concentration 7 mass % Liquid crystal aligning agent (A1-5) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 16 Take 2.7915 g of the polyamic acid ester solution (A-15) obtained in Production Example 15 in a 50 ml Erlenmeyer flask, add 0.4162 g of NMP and 0.7968 g of BCS, stir for 30 minutes with a magnetic stirrer, solid content concentration 7 mass % Liquid crystal aligning agent (A1-6) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 17 Take 2.8010 g of polyamic acid ester solution (A-16) obtained in Production Example 16 in a 50 ml Erlenmeyer flask, add 0.3998 g of NMP and 0.7965 g of BCS, and stir for 30 minutes with a magnetic stirrer, solid content concentration 7 mass % Liquid crystal aligning agent (A1-7) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 18 Take 2.8110 g of the polyamic acid ester solution (A-17) obtained in Production Example 17 in a 50 ml Erlenmeyer flask, add 0.4008 g of NMP and 0.7928 g of BCS, stir with a magnetic stirrer for 30 minutes, solid content concentration 7 mass % Liquid crystal aligning agent (A1-8) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 19 Take 2.8184 g of the polyamic acid ester solution (A-18) obtained in Production Example 18 in a 50 ml Erlenmeyer flask, add 0.4152 g of NMP and 0.8100 g of BCS, stir for 30 minutes with a magnetic stirrer, solid content concentration 7 mass % Liquid crystal aligning agent (A1-9) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 20 Take 2.8071 g of the polyamic acid ester solution (A-20) obtained in Production Example 20 in a 50 ml Erlenmeyer flask, add 0.4303 g of NMP and 0.8028 g of BCS, stir with a magnetic stirrer for 30 minutes, solid content concentration 7 mass % Liquid crystal aligning agent (A1-10) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 21 Take 2.8070 g of the polyamic acid ester solution (A-21) obtained in Production Example 21 in a 50 ml Erlenmeyer flask, add 0.3778 g of NMP and 0.8186 g of BCS, and stir for 30 minutes with a magnetic stirrer, solid content concentration 7 mass % Liquid crystal aligning agent (A1-11) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 22 Take 2.8094 g of the polyamic acid ester solution (A-22) obtained in Production Example 22 in a 50 ml Erlenmeyer flask, add 0.3975 g of NMP and 0.8108 g of BCS, stir for 30 minutes with a magnetic stirrer, solid content concentration 7 mass % Liquid crystal aligning agent (A1-12) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 23 Take 2.7974 g of the polyamic acid ester solution (A-23) obtained in Production Example 23 in a 50 ml Erlenmeyer flask, add 0.3942 g of NMP and 0.8015 g of BCS, and stir for 30 minutes with a magnetic stirrer, solid content concentration 7 mass % Liquid crystal aligning agent (A1-13) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 24 Take 2.8076 g of the polyamic acid ester solution (A-24) obtained in Production Example 24 in a 50 ml Erlenmeyer flask, add 0.4283 g of NMP and 0.8107 g of BCS, stir for 30 minutes with a magnetic stirrer, solid content concentration 7 mass % Liquid crystal aligning agent (A1-14) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- Example 25 Take 2.8012 g of the polyamic acid ester solution (A-25) obtained in Production Example 25 in a 50 ml Erlenmeyer flask, add 0.3944 g of NMP and 0.7942 g of BCS, stir for 30 minutes with a magnetic stirrer, solid content concentration 7 mass % Liquid crystal aligning agent (A1-15) was obtained. The viscosity of this liquid crystal aligning agent at a temperature of 25 ° C. was measured. The results are shown in Table 2 below.
- the polyamic acid ester having a modified terminal according to the present invention has high solubility in an organic solvent, for example, liquid crystal mixed with other various liquid crystal alignment compounds having excellent alignment characteristics and electrical characteristics but low solubility in organic solvents.
- the alignment agent By using the alignment agent, it is possible to obtain a further excellent liquid crystal alignment agent.
- the present invention is widely useful for a TN element, an STN element, a TFT liquid crystal element, a vertical alignment type liquid crystal display element, an electric field driving type display element, an FFS driving type liquid crystal display element, and the like.
Landscapes
- Chemical & Material Sciences (AREA)
- Physics & Mathematics (AREA)
- Nonlinear Science (AREA)
- Organic Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Optics & Photonics (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Polymers & Plastics (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Physics & Mathematics (AREA)
- Mathematical Physics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Liquid Crystal (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
液晶表示素子の高精細化に伴い、液晶表示素子のコントラスト低下の抑制や残像現象の低減といった要求から、液晶配向膜においては、優れた液晶配向性や安定したプレチルト角の発現に加えて、高い電圧保持率、交流駆動により発生する残像の抑制、直流電圧を印加した際の残留電荷が少ない、及び/又は直流電圧による蓄積した残留電荷の緩和が早いといった特性が次第に重要となっている。
しかし、近年では大画面で高精細の液晶テレビが主体となり、残像に対する要求はより厳しくなり、且つ過酷な使用環境での長期使用に耐えうる特性が要求されている。それとともに、使用される液晶配向膜は従来よりも信頼性の高いものが必要となってきており、液晶配向膜の諸特性に関しても、初期特性が良好なだけでなく、例えば、高温下に長時間曝された後であっても、良好な特性を維持することが求められている。
これらの反応による修飾法は、副生物がないため反応後の精製が不要であるという利点はあるが、モノ酸無水物とアミンの反応は、可逆反応であるため、ポリマーの末端を効率よく修飾することができないという難点がある上に、イソシアネート化合物は反応性が高いが、生成するウレア結合の影響で液晶配向剤を構成する有機溶媒におけるポリマーの溶解性を下げる可能性がある。
一方、ポリイミド系の液晶配向剤を構成するポリマー成分として、ポリアミック酸エステルは、信頼性が高く、これをイミド化するときの加熱処理により、分子量低下を起こさないために、液晶の配向安定性・信頼性に優れることが報告されている(特許文献7参照)。しかし、かかる末端を修飾した構造を有するポリアミック酸エステルはいまだ報告された例はない。
また、本発明は、特定の繰り返し単位を有し、末端にアミノ基を有するポリアミック酸エステルをクロロカルボニル化合物と反応させることによるポリアミック酸エステルの末端を修飾する方法を提供する。
1.下記式(1)の繰り返し単位を有し、その末端のアミノ基を下記式(2)の構造を有するように末端を修飾したポリアミック酸エステルと、有機溶媒と、を含有することを特徴とする液晶配向剤。


2.前記末端を修飾したポリアミック酸エステルが、含有されるポリアミック酸エステルの15質量%以上含有する上記1に記載の液晶配向剤。
3.前記末端を修飾したポリアミック酸エステルを有機溶媒に対して、0.5~15質量%含有する上記1又は2に記載の液晶配向剤。
4.前記末端を修飾したポリアミック酸エステルが、前記ポリアミック酸エステルの末端のアミノ基を下記式(3)で表される構造のクロロカルボニル化合物と反応させて得られる上記1~3のいずれかに記載の液晶配向剤。

5.前記式(1)におけるX1が下記式で表される構造から選ばれる少なくとも1種類である上記1~4のいずれかに記載の液晶配向剤。


7.末端にアミノ基を有するポリアミック酸エステルを、下記式(3)で表される構造のクロロカルボニル化合物と反応させることを特徴とする末端を修飾したポリアミック酸エステルの製造方法。

8.ポリアミック酸エステルの一つの繰り返し単位に対して、0.5~60mol%のクロロカルボニル化合物を塩基の存在下で反応させる上記7に記載の末端を修飾したポリアミック酸エステルの製造方法。
9.有機溶媒の存在下で-20℃~150℃で反応させる上記7又は8に記載の末端を修飾したポリアミック酸エステルの製造方法。
10.上記1~6のいずれかに記載の液晶配向剤を塗布、焼成して得られる液晶配向膜。
11.上記1~6のいずれかに記載の液晶配向剤を塗布、焼成して得られる被膜に、偏光された放射線を照射して得られる液晶配向膜。
末端を修飾したポリアミック酸エステルは、繰り返し単位はポリイミド系の液晶配向剤と同じ構造を有するが、高い分子量でも有機溶媒に対する溶解性が大きいために、高い濃度でも低い粘度の液晶配向剤が得られるので、例えばインクジェット法による液晶配向膜の製造も容易になり、また、厚みの大きい液晶配向膜の製造も容易になる。
また、有機溶媒に対する溶解性が大きい末端を修飾したポリアミック酸エステルは、例えば、配向特性や電気特性は優れるが、有機溶媒に対する溶解性が小さい他の種々の液晶配向性化合物と混合した液晶配向剤とすることにより、さらに優れた液晶配向剤を得ることも可能になる。
本発明に用いられるポリアミック酸エステルは、ポリイミドを得るためのポリイミド前駆体であり、加熱することによって下記に示すイミド化反応が可能な部位を有するポリマーである。


この置換基の例としてはハロゲン基、水酸基、チオール基、ニトロ基、アリール基、オルガノオキシ基、オルガノチオ基、オルガノシリル基、アシル基、エステル基、チオエステル基、リン酸エステル基、アミド基、アルキル基、アルケニル基、アルキニル基を挙げることができる。
置換基であるハロゲン基としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子が挙げられる。
置換基であるアリール基としては、フェニル基が挙げられる。このアリール基には前述した他の置換基がさらに置換していてもよい。
置換基であるオルガノオキシ基としては、O-Rで表される構造を示すことができる。このRは同一でも異なってもよく、前述したアルキル基、アルケニル基、アルキニル基、アリール基などを例示することができる。これらのRには前述した置換基がさらに置換していてもよい。アルキルオキシ基の具体例としては、メトキシ基、エトキシ基、プロピルオキシ基、ブトキシ基、ペンチルオキシ基、ヘキシルオキシ基、ヘプチルオキシ基、オクチルオキシ基などが挙げられる。
置換基であるオルガノチオ基としては、-S-Rで表される構造を示すことができる。このRとしては、前述したアルキル基、アルケニル基、アルキニル基、アリール基などを例示することができる。これらのRには前述した置換基がさらに置換していてもよい。アルキルチオ基の具体例としては、メチルチオ基、エチルチオ基、プロピルチオ基、ブチルチオ基、ペンチルチオ基、ヘキシルチオ基、ヘプチルチオ基、オクチルチオ基などが挙げられる。
置換基であるアシル基としては、-C(O)-Rで表される構造を示すことができる。このRとしては、前述したアルキル基、アルケニル基、アリール基などを例示することができる。これらのRには前述した置換基がさらに置換していてもよい。アシル基の具体例としては、ホルミル基、アセチル基、プロピオニル基、ブチリル基、イソブチリル基、バレリル基、イソバレリル基、ベンゾイル基などが挙げられる。
置換基であるエステル基としては、-C(O)O-R、又は-OC(O)-Rで表される構造を示すことができる。このRとしては、前述したアルキル基、アルケニル基、アルキニル基、アリール基などを例示することができる。これらのRには前述した置換基がさらに置換していてもよい。
置換基であるチオエステル基としては、-C(S)O-R、又は-OC(S)-Rで表される構造を示すことができる。このRとしては、前述したアルキル基、アルケニル基、アルキニル基、アリール基などを例示することができる。これらのRには前述した置換基がさらに置換していてもよい。
置換基であるリン酸エステル基としては、-OP(O)-(OR)2で表される構造を示すことができる。このRは同一でも異なってもよく、前述したアルキル基、アルケニル基、アルキニル基、アリール基などを例示することができる。これらのRには前述した置換基がさらに置換していてもよい。
置換基であるアリール基としては、前述したアリール基と同じものを挙げることができる。このアリール基には前述した他の置換基がさらに置換していてもよい。
置換基であるアルキル基としては、前述したアルキル基と同じものを挙げることができる。このアルキル基には前述した他の置換基がさらに置換していてもよい。
置換基であるアルケニル基としては、前述したアルケニル基と同じものを挙げることができる。このアルケニル基には前述した他の置換基がさらに置換していてもよい。
置換基であるアルキニル基としては、前述したアルキニル基と同じものを挙げることができる。このアルキニル基には前述した他の置換基がさらに置換していてもよい。
一般に、嵩高い構造を導入すると、アミノ基の反応性や液晶配向性を低下させる可能性があるため、A1及びA2としては、水素原子、又は置換基を有してもよい炭素数1~5のアルキル基がより好ましく、水素原子、メチル基又はエチル基が特に好ましい。




これらの好ましいX1を有するテトラカルボン酸二無水物の使用量は、全テトラカルボン酸二無水物の好ましくは1~100モル%、より好ましくは50~100モル%である。
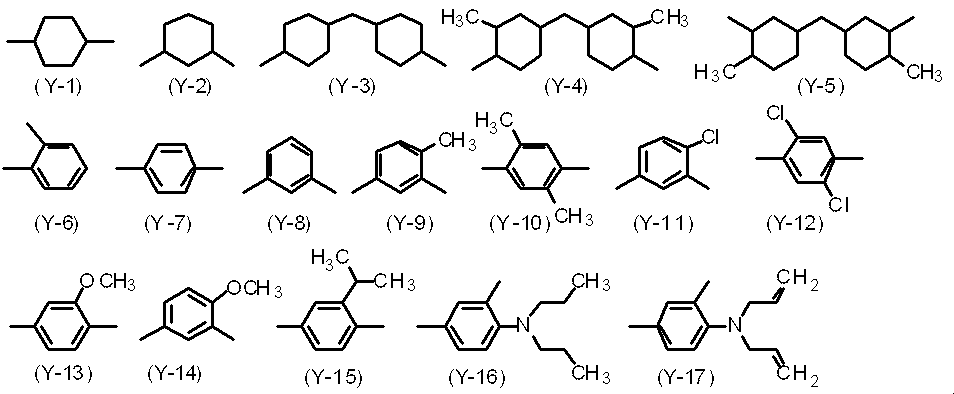











ポリアミック酸エステルの体積抵抗率を低くすることで、直流電圧の蓄積による残像を低減できるため、ヘテロ原子を有する構造、多環芳香族構造、又はビフェニル骨格を有するジアミンをポリアミック酸エステルに導入することが好ましく、Y1としては、Y-19、Y-23、Y-25~Y-27、Y-30~Y-36、Y-40~Y-42、Y-44、Y-45、Y-49、Y-50、Y-51、又はY-61がより好ましく、Y-31、又はY-40が特に好ましい。
これら好ましいY1を有するジアミンの使用量は、全ジアミンの好ましくは1~100モル%、より好ましくは50~100モル%である。

上記式(1)で表されるポリアミック酸エステルは、下記式(6)~(8)で表されるテトラカルボン酸誘導体のいずれかと、式(9)で表されるジアミン化合物との反応によって得ることができる。


上記式(1)で表されるポリアミック酸エステルは、上記モノマーを用いて、以下に示す(1)~(3)の方法で合成することができる。
ポリアミック酸エステルは、テトラカルボン酸二無水物とジアミンから得られるポリアミック酸をエステル化することによって製造することができる。
エステル化剤としては、精製によって容易に除去できるものが好ましく、N,N-ジメチルホルムアミドジメチルアセタール、N,N-ジメチルホルムアミドジエチルアセタール、N,N-ジメチルホルムアミドジプロピルアセタール、N,N-ジメチルホルムアミドジネオペンチルブチルアセタール、N,N-ジメチルホルムアミドジ-t-ブチルアセタール、1-メチル-3-p-トリルトリアゼン、1-エチル-3-p-トリルトリアゼン、1-プロピル-3-p-トリルトリアゼン、4-(4,6-ジメトキシー1,3,5-トリアジンー2-イル)-4-メチルモルホリニウムクロリドなどが挙げられる。エステル化剤の添加量は、ポリアミック酸の繰り返し単位1モルに対して、2~6モル当量が好ましい。
ポリアミック酸エステルは、テトラカルボン酸ジエステルジクロリドとジアミンから製造することができる。
前記塩基には、ピリジン、トリエチルアミン、4-ジメチルアミノピリジンなどが使用できるが、反応が穏和に進行するためにピリジンが好ましい。塩基の添加量は、除去が容易な量で、かつ高分子量体が得やすいという点から、テトラカルボン酸ジエステルジクロリドに対して、2~4倍モルであることが好ましい。
ポリアミック酸エステルは、テトラカルボン酸ジエステルとジアミンを重縮合することにより製造することができる。
具体的には、テトラカルボン酸ジエステルとジアミンを縮合剤、塩基、有機溶剤の存在下で0~150℃、好ましくは0~100℃において、30分~24時間、好ましくは3~15時間反応させることによって製造することができる。
前記塩基には、ピリジン、トリエチルアミンなどの3級アミンが使用できる。塩基の添加量は、除去が容易な量で、かつ高分子量体が得やすいという点から、ジアミン成分に対して2~4倍モルが好ましい。
上記3つのポリアミック酸エステルの製造方法の中でも、高分子量のポリアミック酸エステルが得られるため、上記(1)又は上記(2)の製造法が特に好ましい。
上記のようにして得られるポリアミック酸エステルの溶液は、よく撹拌させながら貧溶媒に注入することで、ポリマーを析出させることができる。析出を数回行い、貧溶媒で洗浄後、常温あるいは加熱乾燥して精製されたポリアミック酸エステルの粉末を得ることができる。貧溶媒は、特に限定されないが、水、メタノール、エタノール、ヘキサン、ブチルセロソルブ、アセトン、トルエン等が挙げられる。
本発明のポリアミック酸エステルは、該ポリアミック酸エステルの主鎖末端アミノ基に下記式(3)で表されるクロロカルボニル化合物を反応させて得られる末端を修飾したポリアミック酸エステルである。

上記のアルキル基、アルケニル基、アルキニル基、アリール基は、全体として炭素数が1~20であれば置換基を有していてもよく、更には置換基によって環構造を形成してもよい。なお、置換基によって環構造を形成するとは、置換基同士又は置換基と母骨格の一部とが結合して環構造となることを意味する。
この置換基の例としてはハロゲン基、水酸基、チオール基、ニトロ基、オルガノオキシ基、オルガノチオ基、オルガノシリル基、アシル基、エステル基、チオエステル基、リン酸エステル基、アミド基、アリール基、アルキル基、アルケニル基、アルキニル基を挙げることができる。
置換基であるオルガノオキシ基としては、アルコキシ基、アルケニルオキシ基、アリールオキシ基など-O-Rで表される構造を示すことができる。このRとしては、前述したアルキル基、アルケニル基、アリール基などを例示することができる。これらのRには前述した置換基がさらに置換していてもよい。アルキルオキシ基の具体例としては、メトキシ基、エトキシ基、プロピオキシ基、ブトキシ基、ペンチルオキシ基、ヘキシルオキシ基、ヘプチルオキシ基、オクチルオキシ基、ノニルオキシ基、デシルオキシ基、ラウリルオキシ基などが挙げられる。
置換基であるオルガノシリル基としては、-Si-(R)3で表される構造を示すことができる。このRは同一でも異なってもよく、前述したアルキル基、アリール基などを例示することができる。これらのRには前述した置換基がさらに置換していてもよい。アルキルシリル基の具体例としては、トリメチルシリル基、トリエチルシリル基、トリプロピルシリル基、トリブチルシリル基、トリペンチルシリル基、トリヘキシルシリル基、ペンチルジメチルシリル基、ヘキシルジメチルシリル基、オクチルジメチルシリル基、デシルジメチルシリル基などが挙げられる。
置換基であるエステル基としては、-C(O)O-R、又は-OC(O)-Rで表される構造を示すことができる。このRとしては、前述したアルキル基、アルケニル基、アリール基などを例示することができる。これらのRには前述した置換基がさらに置換していてもよい。
置換基であるチオエステル基としては、-C(S)O-R、又は-OC(S)-Rで表される構造を示すことができる。このRとしては、前述したアルキル基、アルケニル基、アリール基などを例示することができる。これらのRには前述した置換基がさらに置換していてもよい。
置換基であるアミド基としては、-C(O)NH2、又は、-C(O)NHR、-NHC(O)R、-C(O)N(R)2、-NRC(O)Rで表される構造を示すことができる。このRは同一でも異なってもよく、前述したアルキル基、アリール基などを例示することができる。これらのRには前述した置換基がさらに置換していてもよい。
置換基であるアルキル基としては、前述したアルキル基と同じものを挙げることができる。このアルキル基には前述した他の置換基がさらに置換していてもよい。
置換基であるアルケニル基としては、前述したアルケニル基と同じものを挙げることができる。このアルケニル基には前述した他の置換基がさらに置換していてもよい。
置換基であるアルキニル基としては、前述したアルキニル基と同じものを挙げることができる。このアルキニル基には前述した他の置換基がさらに置換していてもよい。














上記末端にアミノ基を有する式(1)の繰り返し単位をもつポリアミック酸エステルは、そのアミノ基を上記の式(2)の構造を有するようにその末端が修飾される。
この末端を修飾したポリアミック酸エステルは、幾つかの方法で得られるが、上記したポリアミック酸エステルの製造方法で得られる、末端にアミノ基を有するポリアミック酸エステルの粉末を有機溶媒に溶解した後、塩基の存在下にクロロカルボニル化合物を添加して反応させる方法、また、ジアミン成分とテトラカルボン酸ジアルキルエステル誘導体(ビス(クロロカルボニル)化合物、ジアルキルエステルジカルボン酸など)を有機溶媒中で反応させて末端にアミノ基を有するポリアミック酸エステルを得る場合に、該ポリアミック酸エステルを単離することなく、その反応系にクロロカルボニル化合物を加えて、反応系に存在する末端にアミノ基を有するポリアミック酸エステルと反応させる方法、などが挙げられる。なかでも、後者の反応系にクロロカルボニル化合物を添加する方法は、再沈殿によるポリアミック酸エステルの精製が1回でよく、製造工程を短縮できるため、より好ましい。
上記の反応系に対してクロロカルボニル化合物を添加する方法としては、テトラカルボン酸ジアルキルエステル誘導体と同時に添加し、ジアミンと反応させる方法、テトラカルボン酸ジアルキルエステル誘導体とジアミンを十分に反応させて、末端がアミノ基であるポリアミック酸エステルを製造した後に、クロロカルボニル化合物を添加する方法がある。ポリマーの分子量を制御しやすい点から、後者の方法がより好ましい。
クロロカルボニル化合物の添加量は、末端がアミノ基のポリアミック酸エステルの繰り返し単位に対して、0.5~60mol%が好ましく、1~40mol%がより好ましい。添加量が多いと、未反応のクロロカルボニル化合物が残存し、取り除くのが困難であるため、1~20mol%であることがさらに好ましい。
前記塩基には、好ましくはピリジン、トリエチルアミン、4-ジメチルアミノピリジンが使用できるが、反応が穏和に進行するためにピリジンが好ましい。塩基の添加量は、多すぎると除去が難しく、少なすぎると分子量が小さくなるため、クロロカルボニル化合物に対して、2~4倍モルであることが好ましい。
上記有機溶媒としては、N,N-ジメチルホルムアミド、N-メチル-2-ピロリドン、γ-ブチロラクトンが好ましい。これらは1種類又は2種類以上を混合して用いてもよい。
本発明の液晶配向剤は、上記の末端のアミノ基を修飾したポリアミック酸エステルが有機溶媒中に溶解された溶液の形態である。末端のアミノ基を修飾したポリアミック酸エステルの分子量は、末端のアミノ基が修飾されない場合にも、その重量平均分子量で2,000~500,000が好ましく、より好ましくは5,000~300,000であり、さらに好ましくは、10,000~100,000である。また、数平均分子量は、好ましくは、1,000~250,000であり、より好ましくは、2,500~150,000であり、さらに好ましくは、5,000~50,000である。
末端のアミノ基を修飾したポリアミック酸エステルは、上記したように、その平均分子量が高い場合においても有機溶媒に対する溶解性が大きいという特徴を有する。例えば、後記する実施例において示されるように、末端のアミノ基を修飾したポリアミック酸エステルの有機溶媒に溶解した溶液は、末端を修飾しないポリアミック酸エステルの溶液に比べて、その粘度は、好ましくは5~40%小さい粘度を有する。かかる粘度の低下は、より高いポリマー濃度の液晶配向剤を得ることができ、厚みの大きい液晶配向膜を容易に得ることでき、また、インクジェット法などにより液晶配向膜を得る場合にも目詰まりのトラブルの発生も抑制できるので有利である。
本発明の液晶配向剤には、末端を修飾したポリアミック酸エステルのほかに、液晶配向性を有する化合物である他の液晶配向剤が含有されていてもよい。これらの他の液晶配向剤としては、末端のアミノ基が修飾されていないポリアミック酸エステル、可溶性ポリイミド、及び/又はポリアミック酸を含有する液晶配向剤などの種々のものが挙げられる。
なかでも、末端を修飾したポリアミック酸エステルは有機溶媒に対する溶解性が大きいので、配向特性や電気特性に優れるが、有機溶媒に対する溶解性が小さい、例えば、ポリアミック酸や可溶性ポリイミドを含む液晶配向剤を含有させる場合には、特に有用である。
塗膜を焼成する際にポリアミック酸エステルのイミド化を効率よく進行させるために、イミド化促進剤を添加してもよい。以下にポリアミック酸エステルのイミド化促進剤の具体例を挙げるが、本発明の液晶配向剤に使用可能なイミド化促進剤はこれに限定されるものではない。


ポリアミック酸エステルの熱イミド化を促進する効果が得られる範囲であれば、イミド化促進剤の含有量は特に制限されるものではない。あえてその下限を示すならば、ポリアミック酸エステルに含まれる下記式(12)のアミック酸エステル部位1モルに対して、好ましくは0.01モル以上、より好ましくは0.05モル以上、更に好ましくは0.1モル以上が挙げられる。また、焼成後の膜中に残留するイミド化促進剤自体が、液晶配向膜の諸特性に及ぼす悪影響を最小限に留めるという点から、あえてその上限を示すならば、本発明のポリアミック酸エステルに含まれる下記式(12)のアミック酸エステル部位1モルに対して、好ましくはイミド化促進剤が2モル以下、より好ましくは1モル以下、更に好ましくは0.5モル以下が挙げられる。

本発明の液晶配向膜は、上記液晶配向剤を基板に塗布し、乾燥、焼成して得られる膜である。
本発明の液晶配向剤を塗布する基板としては、透明性の高い基板であれば特に限定されず、ガラス基板、窒化珪素基板、アクリル基板、ポリカーボネート基板等のプラスチック基板等を用いることができ、液晶駆動のためのITO電極等が形成された基板を用いることがプロセスの簡素化の点から好ましい。また、反射型の液晶表示素子では片側の基板のみにならばシリコンウエハー等の不透明な物でも使用でき、この場合の電極はアルミ等の光を反射する材料も使用できる。
光配向処理法の具体例としては、前記塗膜表面に、一定方向に偏光した放射線を照射し、場合によってはさらに150~250℃の温度で加熱処理を行い、液晶配向能を付与する方法が挙げられる。放射線としては、100~800nmの波長を有する紫外線および可視光線を用いることができる。このうち、100~400nmの波長を有する紫外線が好ましく、200~400nmの波長を有するものが特に好ましい。また、液晶配向性を改善するために、塗膜基板を50~250℃で加熱しつつ、放射線を照射してもよい。前記放射線の照射量は、1~10,000mJ/cm2の範囲にあることが好ましく、100~5,000mJ/cm2の範囲にあることが特に好ましい。上記のようにして作製した液晶配向膜は、液晶分子を一定の方向に安定して配向させることができる。
以下に、本実施例及び比較例で使用した化合物の略号、及び各特性の測定方法は、以下のとおりである。
1,3DMCBDE-Cl:ジメチル 1,3-ビス(クロロカルボニル)-1,3-ジメチルシクロブタン-2,4-ジカルボキシレート
BDA:1,2,3,4-ブタンテトラカルボン酸二無水物
CBDA:1,2,3,4-シクロブタンテトラカルボン酸二無水物
DDM:4,4-ジアミノージフェニルメタン
NMP:N-メチル-2-ピロリドン
γ-BL:γ-ブチロラクトン
BCS:ブチルセロソルブ
合成例において、ポリアミック酸エステル及びポリアミック酸溶液の粘度は、E型粘度計TVE-22H(東機産業社製)を用い、サンプル量1.1mL、コーンロータTE-1(1°34’、R24)、温度25℃で測定した。
[分子量]
また、ポリアミック酸エステルの分子量はGPC(常温ゲル浸透クロマトグラフィー)装置によって測定し、ポリエチレングリコール、ポリエチレンオキシド換算値として数平均分子量(以下、Mnとも言う。)と重量平均分子量(以下、Mwとも言う。)を算出した。
GPC装置:Shodex社製(GPC-101)
カラム:Shodex社製(KD803、KD805の直列)
カラム温度:50℃
溶離液:N,N-ジメチルホルムアミド(添加剤として、臭化リチウム-水和物(LiBr・H2O)が30mmol/L、リン酸・無水結晶(o-リン酸)が30mmol/L、テトラヒドロフラン(THF)が10ml/L)
流速:1.0ml/分
検量線作成用標準サンプル:東ソー社製 TSK 標準ポリエチレンオキサイド(重量平均分子量(Mw) 約900,000、150,000、100,000、30,000)、及び、ポリマーラボラトリー社製 ポリエチレングリコール(ピークトップ分子量(Mp)約12,000、4,000、1,000)。測定は、ピークが重なるのを避けるため、900,000、100,000、12,000、1,000の4種類を混合したサンプル、及び150,000、30,000、4,000の3種類を混合したサンプルの2サンプルを別々に測定した。
本発明の液晶配向剤を塗膜、焼成して得られた被膜に偏光された254nmの紫外線を照射し、本発明の液晶配向膜を作製した。得られた液晶セルについて、液晶配向性を偏光顕微鏡にて観察し、クロスニコル下での光り抜けがなく、液晶注入口から扇状の流動配向が観察されないものを液晶配向性良好とした。
・ジメチル 1,3-ビス(クロロカルボニル)-1,3-ジメチルシクロブタン-2,4-ジカルボキシレート(1,3DMCBDE-Cl)の合成
a-1:テトラカルボン酸ジアルキルエステルの合成

エバポレーターにて、この反応液から溶媒を留去した後、酢酸エチル1301gを加えて80℃まで加熱し、30分還流させた。その後、10分間に2~3℃の速度で内温が25℃になるまで冷却し、そのまま25℃で30分撹拌した。析出した白色結晶をろ過によって取り出し、この結晶を酢酸エチル141gにて2回洗浄した後、減圧乾燥することで、白色結晶を103.97g得た。
この結晶は、1H NMR分析、及びX線結晶構造解析の結果により、化合物(1-1)であることを確認した(HPLC相対面積97.5%)(収率36.8%)。
1H NMR (DMSO-d6, δppm);12.82 (s, 2H), 3.60 (s, 6H), 3.39 (s, 2H), 1.40 (s, 6H).
a-2.1,3-DM-CBDE-C1の合成

続いて窒素気流下中、3Lの四つ口フラスコに、上記で得られた白色結晶226.09g、及びn-ヘプタン452.18gを仕込んだ後、60℃に加熱撹拌して結晶を溶解させた。その後、25℃まで10分間に1℃の速度で冷却撹拌し、結晶を析出させた。そのまま25℃で1時間撹拌させた後、析出した白色結晶をろ過により取り出し、この結晶をn-ヘキサン113.04gにて洗浄した後、減圧乾燥することで白色結晶を203.91g得た。この結晶は、1H NMR分析結果により、化合物(3-1)すなわち、ジメチル-1,3-ビス(クロロカルボニル)-1,3-ジメチルシクロブタン-2,4-ジカルボキシレート(1,3-DM-CBDE-C1)であるであることを確認した(HPLC相対面積99.5%)(収率77.2%)。
1H NMR (CDCl3, δppm) : 3.78 (s, 6H), 3.72 (s, 2H), 1.69 (s, 6H).
撹拌装置付きの300mL四つ口フラスコを窒素雰囲気とし、4,4´-ジアミノジフェニルメタンを6.40g (32.3mmol)入れ、NMP131g、及び塩基としてピリジン6.16g(77.86mmol) を加え撹拌して溶解させた。次にこのジアミン溶液を撹拌しながら1,3DM-CBDE-Clを9.8641g (27.16mmol)添加し、水冷下4時間反応させた。4時間後、アクリロイルクロリドを0.380g(4.20mmol)加えて、水冷下で30分反応させた。30分後、反応溶液にNMP144.33gを加え、室温(20℃)で15分撹拌した。得られたポリアミック酸エステルの溶液を、1443g の水に撹拌しながら投入し、析出した白色沈殿をろ取し、続いて、1443g の水で1回、1443g のエタノールで1回、361g のエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末14.37gを得た。収率は、99.6%であった。また、このポリアミック酸エステルの分子量はMn=13,335、Mw=23,824であった。
得られたポリアミック酸エステル樹脂粉末3.3076gを50ml三角フラスコにとり、NMP30.4854g を加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(A-1)を得た。
撹拌装置付きの300mL四つ口フラスコを窒素雰囲気とし、4,4´-ジアミノジフェニルメタンを6.40g (32.3mmol)入れ、NMP136.3g、及び塩基としてピリジン6.16g(77.86mmol) を加え撹拌して溶解させた。次にこのジアミン溶液を撹拌しながら1,3DM-CBDE-Clを9.8641g (27.16mmol)添加し、水冷下4時間反応させた。4時間後、クロロぎ酸9-フルオレニルメチルを1.0856g(4.20mmol)加えて、水冷下で30分反応させた。30分後、反応溶液にNMP151.4gを加え、室温(20℃)で15分撹拌した。得られたポリアミック酸エステルの溶液を、1514g の水に撹拌しながら投入し、析出した白色沈殿をろ取し、続いて、1514 の水で1回、1514g のエタノールで1回、378g のエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末15.10gを得た。収率は、99.7%であった。また、このポリアミック酸エステルの分子量はMn=13,379、Mw=24,358であった。
得られたポリアミック酸エステル樹脂粉末3.1938gを50ml三角フラスコにとり、NMP28.7202g を加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(A-2)を得た。
撹拌装置付きの300mL四つ口フラスコを窒素雰囲気とし、4,4´-ジアミノジフェニルメタンを6.40g (32.3mmol)入れ、NMP131g、及び塩基としてピリジン6.16g(77.86mmol) を加え撹拌して溶解させた。次にこのジアミン溶液を撹拌しながら1,3DM-CBDE-Clを9.8644g (27.16mmol)添加し、水冷下4時間反応させた。4時間後、tert-ブチルアセチルクロリドを0.506g(4.20mmol)加えて、水冷下で30分反応させた。30分後、反応溶液にNMP145.6gを加え、室温(20℃)で15分撹拌した。得られたポリアミック酸エステルの溶液を、1456g の水に撹拌しながら投入し、析出した白色沈殿をろ取し、続いて、1456g の水で1回、1456g のエタノールで1回、364g のエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末13.74gを得た。収率は、94.4%であった。また、このポリアミック酸エステルの分子量はMn=15,369、Mw=25,452であった。
得られたポリアミック酸エステル樹脂粉末2.9915gを50ml三角フラスコにとり、NMP26.946gを加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(A-3)を得た。
撹拌装置付きの300mL四つ口フラスコを窒素雰囲気とし、4,4´-ジアミノジフェニルエーテルを6.50g (32.46mmol)入れ、NMP132.3g、及び塩基としてピリジン6.19g(78.3mmol) を加え撹拌して溶解させた。次にこのジアミン溶液を撹拌しながら1,3DM-CBDE-Clを9.9166g (30.51mmol)添加し、水冷下4時間反応させた。4時間後、クロロぎ酸アリルを0.509g(4.21mmol)加えて、水冷下で30分反応させた。30分後、反応溶液にNMP147gを加え、室温(20℃)で15分撹拌した。得られたポリアミック酸エステルの溶液を、1470g の水に撹拌しながら投入し、析出した白色沈殿をろ取し、続いて、1470gの水で1回、1470g のエタノールで1回、368g のエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末13.27gを得た。収率は、90.2%であった。また、このポリアミック酸エステルの分子量はMn=18,947、Mw=32,153であった。
得られたポリアミック酸エステル樹脂粉末3.0630gを50ml三角フラスコにとり、NMP27.5789g を加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(A-4)を得た。
撹拌装置付きの300mL四つ口フラスコを窒素雰囲気とし、4,4´-ジアミノジフェニルエーテルを6.50g (32.46mmol)入れ、NMP132.4g、及び塩基としてピリジン6.19g(78.3mmol) を加え撹拌して溶解させた。次にこのジアミン溶液を撹拌しながら1,3DM-CBDE-Clを9.9205g (30.51mmol)添加し、水冷下4時間反応させた。4時間後、クロロぎ酸をプロピル0.517g(4.22mmol)を加えて、水冷下で30分反応させた。30分後、反応溶液にNMP147.13gを加え、室温(20℃)で15分撹拌した。得られたポリアミック酸エステルの溶液を、1471g の水に撹拌しながら投入し、析出した白色沈殿をろ取し、続いて、1471gの水で1回、1471g のエタノールで1回、368g のエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末14.60gを得た。収率は、99.3%であった。また、このポリアミック酸エステルの分子量はMn=18,535、Mw=32,403であった。
得られたポリアミック酸エステル樹脂粉末3.4512gを50ml三角フラスコにとり、NMP31.0895gを加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(A-5)を得た。
撹拌装置付きの300mL四つ口フラスコを窒素雰囲気とし、4,4´-ジアミノジフェニルエーテルを6.50g (32.46mmol)入れ、NMP135.3g、及び塩基としてピリジン6.19g(78.3mmol) を加え撹拌して溶解させた。次にこのジアミン溶液を撹拌しながら1,3DM-CBDE-Clを9.9208g (30.51mmol)添加し、水冷下4時間反応させた。4時間後、1-アダマンタンカルボニルクロリドを0.8385g(4.22mmol)を加えて、水冷下で30分反応させた。30分後、反応溶液にNMP150.3gを加え、室温(20℃)で15分撹拌した。得られたポリアミック酸エステルの溶液を、1503g の水に撹拌しながら投入し、析出した白色沈殿をろ取し、続いて、1503gの水で1回、1503g のエタノールで1回、376g のエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末13.4gを得た。収率は、89.1%であった。また、このポリアミック酸エステルの分子量はMn=14,731、Mw=28,526であった。
得られたポリアミック酸エステル樹脂粉末3.3428gを50ml三角フラスコにとり、NMP30.0009g を加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(A-6)を得た。
撹拌装置付きの300mL四つ口フラスコを窒素雰囲気とし、4,4´-ジアミノジフェニルメタンを5.00g (25.22mmol)入れ、NMP102g、及び塩基としてピリジン4.81g(60.83mmol) を加え撹拌して溶解させた。次にこのジアミン溶液を撹拌しながら1,3DM-CBDE-Cl7.707g (23.71mmol)添加し、水冷下4時間反応させた。4時間後、2-フラニルクロリド0.4302g(3.30mmol)を加えて、水冷下で30分反応させた。30分後、反応溶液にNMP114gを加え、室温(20℃)で15分撹拌した。得られたポリアミック酸エステルの溶液を、1141g の水に撹拌しながら投入し、析出した白色沈殿をろ取し、続いて、1141gの水で1回、1141g のエタノールで1回、285g のエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末11.12gを得た。収率は、97.5%であった。また、このポリアミック酸エステルの分子量はMn=12,864、Mw=22,513であった。
得られたポリアミック酸エステル樹脂粉末3.1266gを50ml三角フラスコにとり、NMP28.1581g を加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(A-7)を得た。
撹拌装置付きの300mL四つ口フラスコを窒素雰囲気とし、4,4´-ジアミノジフェニルメタンを5.00g (25.22mmol)入れ、NMP103g、及び塩基としてピリジン4.81g(60.83mmol) を加え撹拌して溶解させた。次にこのジアミン溶液を撹拌しながら1,3DM-CBDE-Clを7.7075g (23.71mmol)添加し、水冷下4時間反応させた。4時間後、ベンゾイルクロリド0.4702g(3.35mmol)を加えて、水冷下で30分反応させた。30分後、反応溶液にNMP114gを加え、室温(20℃)で15分撹拌した。得られたポリアミック酸エステルの溶液を、1144gの水に撹拌しながら投入し、析出した白色沈殿をろ取し、続いて、1144gの水で1回、1144g のエタノールで1回、286g のエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末11.10gを得た。収率は、97.0%であった。また、このポリアミック酸エステルの分子量はMn=11,260、Mw=19,060であった。
得られたポリアミック酸エステル樹脂粉末3.6625gを50ml三角フラスコにとり、NMP32.9616g を加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(A-8)を得た。
撹拌装置付きの300mL四つ口フラスコを窒素雰囲気とし、4,4´-ジアミノジフェニルメタンを5.00g (25.22mmol)入れ、NMP103g、及び塩基としてピリジン4.81g(60.83mmol) を加え撹拌して溶解させた。次にこのジアミン溶液を撹拌しながら1,3DM-CBDE-Clを7.7014g (23.70mmol)添加し、水冷下4時間反応させた。4時間後、クロロギ酸フェニル0.5140g(3.28mmol)を加えて、水冷下で30分反応させた。30分後、反応溶液にNMP115gを加え、室温(20℃)で15分撹拌した。得られたポリアミック酸エステルの溶液を、1149g の水に撹拌しながら投入し、析出した白色沈殿をろ取し、続いて、1503gの水で1回、1149g のエタノールで1回、287g のエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末11.01gを得た。収率は、95.8%であった。また、このポリアミック酸エステルの分子量はMn=11,772、Mw=20,564であった。
得られたポリアミック酸エステル樹脂粉末3.6176gを50ml三角フラスコにとり、NMP32.5597g を加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(A-9)を得た。
撹拌装置付きの300mL四つ口フラスコを窒素雰囲気とし、DDMを8.0102g (40.35mmol)入れ、NMP158.1g、及び塩基としてピリジン7.20g (91.03mmol) を加え撹拌して溶解させた。次にこのジアミン溶液を撹拌しながら1,3DM-CBDE-Clを12.3419g (37.93mmol)添加し、水冷下4時間反応させた。得られたポリアミック酸エステルの溶液を、1757g の水に撹拌しながら投入し、析出した白色沈殿をろ取し、続いて、1757g の水で1回、1757g のエタノールで1回、439g のエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末16.63gを得た。収率は、94.6%であった。また、このポリアミック酸エステルの分子量はMn=10,180、Mw=21,476であった。
得られたポリアミック酸エステル樹脂粉末14.8252を200ml三角フラスコにとり、NMP99.3048g を加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(B-1)を得た。
撹拌装置付きの300mL四つ口フラスコを窒素雰囲気とし、4,4´-ジイミノジフェニルエーテルを8.0129 g (35.03mmol)入れ、NMP157.25g、及び塩基としてピリジン7.13g (90.13mmol) を加え撹拌して溶解させた。次にこのジアミン溶液を撹拌しながら1,3DM-CBDE-Clを12.2295g (37.61mmol)添加し、水冷下4時間反応させた。得られたポリアミック酸エステルの溶液を、1747g の水に撹拌しながら投入し、析出した白色沈殿をろ取し、続いて、1747g の水で1回、1747g のエタノールで1回、437g のエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末16.65gを得た。収率は、95.3%であった。また、このポリアミック酸エステルの分子量はMn=13,104、Mw=29,112であった。
得られたポリアミック酸エステル樹脂粉末1.8731gを50ml三角フラスコにとり、NMP16.89gを加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(B-2)を得た。
製造例1で得られたポリアミック酸エステル溶液(A-1)4.2033gを三角フラスコにとり、NMP0.5991g、及びBCS1.2099gを加えてマグネチックスターラーで30分間撹拌し、固形分濃度7質量%の液晶配向剤(I)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表に示す。
(実施例2)
製造例2で得られたポリアミック酸エステル溶液(A-2)4.2123gを三角フラスコにとり、NMP0.6077g、及びBCS1.2077gを加えてマグネチックスターラーで30分間撹拌し固形分濃度7質量%の液晶配向剤(II)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表に示す。
(実施例3)
製造例3で得られたポリアミック酸エステル溶液(A-3)4.1955gを三角フラスコにとり、NMP0.6053g、及びBCS1.1953gを加えてマグネチックスターラーで30分間撹拌し固形分濃度7質量%の液晶配向剤(III)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表に示す。
(実施例4)
製造例4で得られたポリアミック酸エステル溶液(A-4)4.2010gを三角フラスコにとり、NMP0.5993g、及びBCS1.2519gを加えてマグネチックスターラーで30分間撹拌し固形分濃度7質量%の液晶配向剤(IV)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表に示す。
(実施例5)
製造例5で得られたポリアミック酸エステル溶液(A-5)4.1984gを三角フラスコにとり、NMP0.5988g、及びBCS1.2029gを加えてマグネチックスターラーで30分間撹拌し固形分濃度7質量%の液晶配向剤(V)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表に示す。
製造例6で得られたポリアミック酸エステル溶液(A-6)4.1999gを三角フラスコにとり、NMP0.5906g、及びBCS1.2024gを加えてマグネチックスターラーで30分間撹拌し固形分濃度7質量%の液晶配向剤(VI)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表に示す。
(実施例7)
製造例7で得られたポリアミック酸エステル溶液(A-7)3.0789gを三角フラスコにとり、NMP0.4393g、及びBCS0.8791gを加えてマグネチックスターラーで30分間撹拌し固形分濃度7質量%の液晶配向剤(VII)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表に示す。
(実施例8)
製造例8で得られたポリアミック酸エステル溶液(A-8)3.0791gを三角フラスコにとり、NMP0.4361g、及びBCS0.8833gを加えてマグネチックスターラーで30分間撹拌し固形分濃度7質量%の液晶配向剤(VIII)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表に示す。
(実施例9)
製造例9で得られたポリアミック酸エステル溶液(A-9)3.0231gを三角フラスコにとり、NMP0.4355g、及びBCS0.8699gを加えてマグネチックスターラーで30分間撹拌し固形分濃度7質量%の液晶配向剤(IX)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表に示す。
比較製造例1で得られたポリアミック酸エステル溶液(B-1)4.2066gを三角フラスコにとり、NMP0.5957g、及びBCS1.1908gを加えてマグネチックスターラーで30分間撹拌し固形分濃度7質量%の液晶配向剤(B-I)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表に示す。
(比較例2)
比較製造例2で得られたポリアミック酸エステル溶液(B-2)4.2010gを三角フラスコにとり、NMP0.5993g、及びBCS1.2519gを加えてマグネチックスターラーで30分間撹拌し固形分濃度7質量%の液晶配向剤(B-II)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表に示す。

実施例1~9と比較例1~2の結果より、ポリアミック酸エステルの主鎖の末端アミノ基をクロロカルボニル化合物で修飾することにより、ポリマーの溶解性が向上し、高固形分でのより粘度の低い液晶配向剤が得られることが確認された。
実施例1で得られた液晶配向剤(I)を1.0μmのフィルターで濾過した後、透明電極付きガラス基板上にスピンコートし、温度80℃のホットプレート上で5分間乾燥後、温度250℃で1時間の焼成を経て、膜厚100nmのイミド化した膜を得た。この塗膜面に偏光板を介して254nmの紫外線を100mJ/cm2照射し、液晶配向膜付き基板を得た。このような液晶配向膜付き基板を2枚用意し、一方の基板の液晶配向膜面に6μmのスペーサーを散布した後、2枚の基板の配向が逆平行になるように組み合わせ、液晶注入口を残して周囲をシールし、セルギャップが6μmの空セルを作製した。この空セルに液晶(MLC-2041、メルク社製)を常温で真空注入し、注入口を封止して液晶セルとした。この液晶セルについて液晶配向性を偏光顕微鏡で確認した結果、液晶配向性は良好であった。
実施例10の結果より、本発明の末端を修飾したポリアミック酸エステルを含有する液晶配向剤は、液晶配向膜とした場合において、良好な特性を有することが確認された。
攪拌装置付きの四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを40.73 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、2-テノイルクロリドを0.192g (1.3114mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.2499g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、498gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて226gのエタノールで1回、452gの水で2回、453gのエタノールで1回、113gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末4.4587gを得た。収率は、98.53%であった。またこのポリアミック酸エステルの分子量はMn=12256、Mw=21405であった。
得られたポリアミック酸エステル樹脂粉末2.1520gを50ml三角フラスコにとり、NMPを19.3658g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-10)を得た。
(製造例11)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMP を40.42 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、3,3-ジメチルアクリロイルクロライドを0.1555g (1.3114mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを44.9236g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、494gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて225gのエタノールで1回、449gの水で2回、449gのエタノールで1回、112gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末3.9916gを得た。収率は、88.98%であった。またこのポリアミック酸エステルの分子量はMn=13673、Mw=22739であった。
得られたポリアミック酸エステル樹脂粉末2.3883gを50ml三角フラスコにとり、NMPを21.5218g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-11)を得た。
(製造例12)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを 40.94 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、シンナモイルクロリドを0.2185g (1.3114mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.54g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、500gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて227gのエタノールで1回、455gの水で2回、455gのエタノールで1回、114gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末4.2721gを得た。収率は、93.91%であった。またこのポリアミック酸エステルの分子量はMn=13033、Mw=23520であった。
得られたポリアミック酸エステル樹脂粉末2.4517gを50ml三角フラスコにとり、NMPを22.0656g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-12)を得た。
(製造例13)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを40.94 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、イソオキサゾール-5-カルボン酸クロライドを0.1725g (1.3114mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.06g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、495gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて226gのエタノールで1回、451gの水で2回、451gのエタノールで1回、113gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末4.3714gを得た。収率は、96.99%であった。またこのポリアミック酸エステルの分子量はMn=13418、Mw=22819であった。
得られたポリアミック酸エステル樹脂粉末2.2172gを50ml三角フラスコにとり、NMPを19.9964g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-13)を得た。
(製造例14)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMP を40.75 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、2-オキソ-1-イミダゾリジンカルボニルクロリドを0.1948g (1.3114mmol)加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.23g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、498gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて226gのエタノールで1回、453gの水で2回、453gのエタノールで1回、113gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末3.98gを得た。収率は、87.92%であった。またこのポリアミック酸エステルの分子量はMn=12119、Mw=23633であった。
得られたポリアミック酸エステル樹脂粉末2.1446gを50ml三角フラスコにとり、NMPを19.2937g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-14)を得た。
(製造例15)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを40.14 g、塩基としてピリジンを1.9246g (24.3317 mmol)を加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、プロピオニルクロリドを0.1213g (1.3114mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを44.60g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、491gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて223gのエタノールで1回、446gの水で2回、446gのエタノールで1回、111gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末3.74gを得た。収率は、83.86%であった。またこのポリアミック酸エステルの分子量はMn=13082、Mw=23048であった。
得られたポリアミック酸エステル樹脂粉末2.1867gを50ml三角フラスコにとり、NMPを19.6897g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-15)を得た。
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを 40.14 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、4-フルオロベンゾイルクロリドを0.2079g (1.3114mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.39g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、499gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて227gのエタノールで1回、454gの水で2回、454gのエタノールで1回、114gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末3.87gを得た。収率は、85.26%であった。またこのポリアミック酸エステルの分子量はMn=12207、Mw=22609であった。
得られたポリアミック酸エステル樹脂粉末1.9882gを50ml三角フラスコにとり、NMPを17.908g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-16)を得た。
(製造例17)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを41.49 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、4-フェニルベンゾイルクロリドを0.2841g (1.3114mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを46.10g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、507gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて230gのエタノールで1回、461gの水で2回、461gのエタノールで1回、115gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末4.02gを得た。収率は、87.20%であった。またこのポリアミック酸エステルの分子量はMn=11563、Mw=22120であった。
得られたポリアミック酸エステル樹脂粉末2.1231gを50ml三角フラスコにとり、NMPを19.1000g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-17)を得た。
(製造例18)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMP を40.86 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、シクロプロパンカルボニルクロリドを0.2079g (1.3114mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.39g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、499gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて227gのエタノールで1回、454gの水で2回、454gのエタノールで1回、114gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末3.8463gを得た。収率は、84.7%であった。またこのポリアミック酸エステルの分子量はMn=12995、Mw=23470であった。
得られたポリアミック酸エステル樹脂粉末2.3403gを50ml三角フラスコにとり、NMPを21.0717g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-18)を得た。
(製造例19)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを45.39 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、ジフェニルカルバモイルクロリドを0.2079g(0.897mmol)加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.39g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、499gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて227gのエタノールで1回、454gの水で2回、454gのエタノールで1回、114gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末3.7689gを得た。収率は、83.0%であった。またこのポリアミック酸エステルの分子量はMn=9543、Mw=21337であった。
得られたポリアミック酸エステル樹脂粉末2.0849gを50ml三角フラスコにとり、NMPを18.7717g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-19)を得た。
(製造例20)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを40.86 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、アセチルクロリド0.2079g (2.6484mmol)加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.39g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、499gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて227gのエタノールで1回、454gの水で2回、454gのエタノールで1回、114gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末4.2288gを得た。収率は、93.2%であった。またこのポリアミック酸エステルの分子量はMn=13739、Mw=24113であった。
得られたポリアミック酸エステル樹脂粉末2.2812gを50ml三角フラスコにとり、NMPを20.5236g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-20)を得た。
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを40.86 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、メタクリロイルクロリド0.2079g (1.9889mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.39g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、499gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて227gのエタノールで1回、454gの水で2回、454gのエタノールで1回、114gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末4.5616gを得た。収率は、99.0%であった。またこのポリアミック酸エステルの分子量はMn=14046、Mw=23471であった。
得られたポリアミック酸エステル樹脂粉末2.2641gを50ml三角フラスコにとり、NMPを20.3711g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-21)を得た。
(製造例22)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを40.86 g、塩基としてピリジンを1.9246g (24.3317 mmol)を加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、メチルクロロチオフォルメートを0.2079g (1.8804mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.39g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、499gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて227gのエタノールで1回、454gの水で2回、454gのエタノールで1回、114gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末4.2667gを得た。収率は、94.0%であった。またこのポリアミック酸エステルの分子量はMn=13857、Mw=24200であった。
得られたポリアミック酸エステル樹脂粉末2.2436gを50ml三角フラスコにとり、NMPを20.1778g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-22)を得た。
(製造例23)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを40.86 g、塩基としてピリジンを1.9246g (24.3317 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、4-メトキシベンゾイルクロリドを0.2079g (1.2187mmol)加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.39g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、499gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて227gのエタノールで1回、454gの水で2回、454gのエタノールで1回、114gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末4.2667gを得た。収率は、95.7%であった。またこのポリアミック酸エステルの分子量はMn=12439、Mw=23256であった。
得られたポリアミック酸エステル樹脂粉末2.4178gを50ml三角フラスコにとり、NMPを21.7607g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-23)を得た。
(製造例24)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを40.86 g、塩基としてピリジンを2.0759g (26.2443 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、2-ナフチルクロロフォルメートを0.6003g (2.90528mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.98g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、552gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて227gのエタノールで1回、460gの水で2回、228gのエタノールで1回、115gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末4.24gを得た。収率は、92.2%であった。またこのポリアミック酸エステルの分子量はMn=12498、Mw=22829であった。
得られたポリアミック酸エステル樹脂粉末1.9683gを50ml三角フラスコにとり、NMPを17.7163g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-24)を得た。
(製造例25)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを40.86 g、塩基としてピリジンを2.0759g (26.2443 mmol)を加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、2-n-プロピル-n-バレリルクロリドを0.4726g(2.90528mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.44g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、545gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて227gのエタノールで1回、454gの水で2回、227gのエタノールで1回、114gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末3.89gを得た。収率は、85.7%であった。またこのポリアミック酸エステルの分子量はMn=15211、Mw=25954であった。
得られたポリアミック酸エステル樹脂粉末2.6046gを50ml三角フラスコにとり、NMPを18.5329g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-25)を得た。
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを2.0000 g (10.0878mmol)入れ、NMPを40.86 g、塩基としてピリジンを2.0759g (26.2443 mmol)加え攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを3.0831g (9.4825mmol)添加し、水冷下4時間反応させた。4時間後、ジアリルカルバミルクロリドを0.4637g (2.90528mmol) 加えて、水冷下で30分反応させた。30分後、反応溶液にNMPを45.41g加え、室温(20℃)で15分攪拌した。得られたポリアミック酸エステルの溶液を、545gのエタノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて227gのエタノールで1回、454gの水で2回、227gのエタノールで1回、114gのエタノールで3回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末3.83gを得た。収率は、84.3%であった。またこのポリアミック酸エステルの分子量はMn=9243、Mw=20232であった。
得られたポリアミック酸エステル樹脂粉末2.2187gを50ml三角フラスコにとり、NMPを19.9635g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-26)を得た。
(製造例27)
攪拌装置付きの100ml四つ口フラスコに2,4-ビス(メトキシカルボニル)シクロブタン‐1,3-ジカルボン酸を4.9034g(18.84mmol)取り、NMPを68.12g加え、撹拌して溶解させた。続いて、トリエチルアミンを4.45 g(43.98mmol)、p-フェニレンジアミンを1.7315 g(16.01mmol)、4,4'-ジアミノジフェニルメタンを0.7922g(4.00mmol)加え、撹拌して溶解させた。この溶液を撹拌しながら(2,3-ジヒドロキシ-2-チオキソ-3-ベンゾオキサゾイル)ホスホン酸ジフェニルを16.90g(44.08mmol)添加し、更にNMPを9.67g加え、水冷下で4時間反応させた。4時間後、アクリロイルクロリドを0.2607g(2.88mmol)加えて、水冷下で30分間反応させた。得られたポリアミド酸エステル溶液を650gの2-プロパノールに撹拌しながら投入し、析出した沈殿物をろ取し、続いて、210gの2-プロパノールで5回洗浄し、乾燥することでポリアミック酸エステル樹脂粉末を得た。
このポリアミック酸エステルの分子量はMn=3189、Mw=4783であった。
得られたポリアミック酸エステル樹脂粉末2.3389gを50ml三角フラスコに取り、NMPを22.6242g加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(A-27)を得た。
(製造例28)
攪拌装置付きの100mL四つ口フラスコを窒素雰囲気とし、4,4'-ジアミノジフェニルメタンを3.0000g (15.13mmol)、3-{(メチルアミノ)メチル}アニリンを1.38g(10.13mmol)入れ、NMPを94.65g、塩基としてトリエチルアミンを5.75g(56.89mmol)加え、攪拌して溶解させた。次にこのジアミン溶液を攪拌しながら1,3DM-CBDE-Clを7.7149g(23.73mmol)添加し、水冷下4時間反応させた。4時間後、アクリロイルクロリドを0.0.6574g (7.2632mmol)加えて、水冷下で30分反応させた。得られたポリアミック酸エステルの溶液を、450gの2-プロパノールに攪拌しながら投入し、析出した白色沈殿をろ取し、続いて220gの2-プロパノールで5回洗浄し、乾燥することで白色のポリアミック酸エステル樹脂粉末を得た。このポリアミック酸エステルの分子量はMn=8861、Mw=20627であった。
得られたポリアミック酸エステル樹脂粉末1.5913gを50ml三角フラスコにとり、NMPを14.5979g加え、室温で24時間攪拌し溶解させて、ポリアミック酸エステル溶液(A-28)を得た。
(製造例29)
攪拌装置付きの100ml四つ口フラスコに2,5-ビス(メトキシカルボニル)テレフタル酸を1.2654g(4.48mmol)、2,4-ビス(メトキシカルボニル)シクロブタン‐1,3-ジカルボン酸を2.6157g(10.05mmol)取り、NMPを73.16g加え、撹拌して溶解させた。続いて、トリエチルアミンを3.34g(33.01mmol)、4,4'-{プロパン-1,3-ビス(オキシ)}ジアニリンを3.8784g(15.01mmol)加え、撹拌して溶解させた。この溶液を撹拌しながら(2,3-ジヒドロキシ-2-チオキソ-3-ベンゾオキサゾイル)ホスホン酸ジフェニルを12.68g(33.08mmol)添加し、更にNMPを10.05g加え、水冷下で4時間反応させた。4時間後、アクリロイルクロリドを0.1508g(1.07mmol)加えて、水冷下で30分間反応させた。得られたポリアミド酸エステル溶液を650gの2-プロパノールに撹拌しながら投入し、析出した沈殿物をろ取し、続いて、210gの2-プロパノールで5回洗浄し、乾燥することでポリアミック酸エステル樹脂粉末を得た。
このポリアミック酸エステルの分子量はMn=15633、Mw=32874であった。
得られたポリアミック酸エステル樹脂粉末1.2264gを50ml三角フラスコに取り、NMPを11.4164g加え、室温で24時間撹拌し溶解させて、ポリアミック酸エステル溶液(A-28)を得た。
50ml三角フラスコに製造例10で得られたポリアミック酸エステル溶液(A-10)を2.8093 g取り、NMPを0.4070g、BCSを0.7930g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-1)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例12)
50ml三角フラスコに製造例11で得られたポリアミック酸エステル溶液(A-11)を2.8168 g取り、NMPを0.3964g、BCSを0.7930g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-2)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例13)
50ml三角フラスコに製造例12で得られたポリアミック酸エステル溶液(A-12)を2.8030 g取り、NMPを0.3929g、BCSを0.8060g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-3)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例14)
50ml三角フラスコに製造例13で得られたポリアミック酸エステル溶液(A-13)を2.8092 gを取り、NMPを0.4024g、BCSを0.8047g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-4)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例15)
50ml三角フラスコに製造例14で得られたポリアミック酸エステル溶液(A-14)を2.8121 g取り、NMPを0.4002g、BCSを0.8154g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-5)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
50ml三角フラスコに製造例15で得られたポリアミック酸エステル溶液(A-15)を2.7915 g取り、NMPを0.4162g、BCSを0.7968g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-6)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例17)
50ml三角フラスコに製造例16で得られたポリアミック酸エステル溶液(A-16)を2.8010 g取り、NMPを0.3998g、BCSを0.7965g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-7)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例18)
50ml三角フラスコに製造例17で得られたポリアミック酸エステル溶液(A-17)を2.8110 g取り、NMPを0.4008g、BCSを0.7928g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-8)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例19)
50ml三角フラスコに製造例18で得られたポリアミック酸エステル溶液(A-18)を2.8184 g取り、NMPを0.4152g、BCSを0.8100g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-9)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例20)
50ml三角フラスコに製造例20で得られたポリアミック酸エステル溶液(A-20)を2.8071 g取り、NMPを0.4303g、BCSを0.8028g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-10)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
50ml三角フラスコに製造例21で得られたポリアミック酸エステル溶液(A-21)を2.8070 g取り、NMPを0.3778g、BCSを0.8186g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-11)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例22)
50ml三角フラスコに製造例22で得られたポリアミック酸エステル溶液(A-22)を2.8094 g取り、NMPを0.3975g、BCSを0.8108g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-12)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例23)
50ml三角フラスコに製造例23で得られたポリアミック酸エステル溶液(A-23)を2.7974 g取り、NMPを0.3942g、BCSを0.8015g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-13)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例24)
50ml三角フラスコに製造例24で得られたポリアミック酸エステル溶液(A-24)を2.8076 g取り、NMPを0.4283g、BCSを0.8107g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-14)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。
(実施例25)
50ml三角フラスコに製造例25で得られたポリアミック酸エステル溶液(A-25)を2.8012 g取り、NMPを0.3944g、BCSを0.7942g加えてマグネチックスターラーで30分攪拌し、固形分濃度7質量%の液晶配向剤(A1-15)を得た。この液晶配向剤の温度25℃における粘度を測定した。結果は、後述の表2に示す。

なお、2010年3月15日に出願された日本特許出願2010-058555号の明細書、特許請求の範囲、及び要約書の全内容をここに引用し、本発明の明細書の開示として、取り入れるものである。
Claims (11)
- 下記式(1)の繰り返し単位を有し、その末端のアミノ基を下記式(2)の構造を有するように末端を修飾したポリアミック酸エステルと、有機溶媒と、を含有することを特徴とする液晶配向剤。


- 前記末端を修飾したポリアミック酸エステルが、含有されるポリアミック酸エステルの15質量%以上含有する請求項1に記載の液晶配向剤。
- 前記末端を修飾したポリアミック酸エステルを有機溶媒に対して、0.5~15質量%含有する請求項1又は2に記載の液晶配向剤。
- 前記末端を修飾したポリアミック酸エステルが、前記ポリアミック酸エステルの末端のアミノ基を下記式(3)で表される構造のクロロカルボニル化合物と反応させて得られる請求項1~3のいずれかに記載の液晶配向剤。

- 前記式(1)におけるX1が下記式で表される構造から選ばれる少なくとも1種類である請求項1~4のいずれかに記載の液晶配向剤。

- 前記式(1)におけるY1が下記式で表される構造から選ばれる少なくとも1種類である請求項1~5のいずれかに記載の液晶配向剤。

- 末端にアミノ基を有するポリアミック酸エステルを、下記式(3)で表される構造のクロロカルボニル化合物と反応させることを特徴とする末端を修飾したポリアミック酸エステルの製造方法。

- ポリアミック酸エステルの一つの繰り返し単位に対して、0.5~60mol%のクロロカルボニル化合物を塩基の存在下で反応させる請求項7に記載の末端を修飾したポリアミック酸エステルの製造方法。
- 有機溶媒の存在下で-20℃~150℃で反応させる請求項7又は8に記載の末端を修飾したポリアミック酸エステルの製造方法。
- 請求項1~6のいずれかに記載の液晶配向剤を塗布、焼成して得られる液晶配向膜。
- 請求項1~6のいずれかに記載の液晶配向剤を塗布、焼成して得られる被膜に、偏光された放射線を照射して得られる液晶配向膜。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012505683A JP5904119B2 (ja) | 2010-03-15 | 2011-03-14 | 末端を修飾したポリアミック酸エステルを含有する液晶配向剤、及び液晶配向膜 |
| CN201180023615.0A CN102934011B (zh) | 2010-03-15 | 2011-03-14 | 包含对末端进行了修饰的聚酰胺酸酯的液晶取向剂及液晶取向膜 |
| KR1020127026262A KR101818786B1 (ko) | 2010-03-15 | 2011-03-14 | 말단을 수식한 폴리아믹산에스테르를 함유하는 액정 배향제, 및 액정 배향막 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010058555 | 2010-03-15 | ||
| JP2010-058555 | 2010-03-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011115076A1 true WO2011115076A1 (ja) | 2011-09-22 |
Family
ID=44649161
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/055971 WO2011115076A1 (ja) | 2010-03-15 | 2011-03-14 | 末端を修飾したポリアミック酸エステルを含有する液晶配向剤、及び液晶配向膜 |
Country Status (5)
| Country | Link |
|---|---|
| JP (1) | JP5904119B2 (ja) |
| KR (1) | KR101818786B1 (ja) |
| CN (1) | CN102934011B (ja) |
| TW (1) | TWI500658B (ja) |
| WO (1) | WO2011115076A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013147083A1 (ja) * | 2012-03-30 | 2013-10-03 | 日産化学工業株式会社 | ポリイミド系の液晶配向処理剤、液晶配向膜、及び液晶表示素子 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103995382A (zh) * | 2014-04-23 | 2014-08-20 | 河北冀雅电子有限公司 | 一种电焊护目镜的制造方法 |
| CN106415379B (zh) * | 2014-06-02 | 2019-04-30 | Dic株式会社 | 液晶取向膜 |
| JP2017090781A (ja) * | 2015-11-13 | 2017-05-25 | 株式会社ジャパンディスプレイ | 光配向膜用ワニス及び液晶表示装置 |
| KR102202054B1 (ko) | 2018-01-22 | 2021-01-11 | 주식회사 엘지화학 | 액정 배향제 조성물, 이를 이용한 액정 배향막의 제조 방법, 및 이를 이용한 액정 배향막 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001033963A (ja) * | 1999-05-17 | 2001-02-09 | Asahi Chem Ind Co Ltd | 感光性樹脂組成物 |
| JP2001296525A (ja) * | 2000-04-12 | 2001-10-26 | Jsr Corp | 液晶配向剤および液晶表示素子 |
| JP2003026918A (ja) * | 2001-07-13 | 2003-01-29 | Hitachi Ltd | 液晶配向膜用材料、液晶表示素子、その製造方法及び液晶表示装置 |
| JP2006299079A (ja) * | 2005-04-20 | 2006-11-02 | Teijin Ltd | ポリイミド前駆体成形物の製造方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001089426A (ja) | 1999-09-14 | 2001-04-03 | Fuji Photo Film Co Ltd | 新規アセチレン化合物 |
| CN1266192C (zh) * | 2002-07-29 | 2006-07-26 | Jsr株式会社 | 二胺化合物、聚酰胺酸、酰亚胺化聚合物、液晶取向剂及液晶显示元件 |
| JP5013934B2 (ja) * | 2007-04-12 | 2012-08-29 | 旭化成イーマテリアルズ株式会社 | ポリアミドイミド及びネガ型感光性樹脂組成物 |
| WO2008153101A1 (ja) * | 2007-06-15 | 2008-12-18 | Nissan Chemical Industries, Ltd. | 熱硬化膜形成用樹脂組成物 |
| CN101373296B (zh) | 2007-08-24 | 2012-07-04 | 株式会社日立显示器 | 液晶显示装置及其制造方法 |
| CN102067024B (zh) | 2008-06-17 | 2013-08-07 | 日产化学工业株式会社 | 液晶取向处理剂及使用了该处理剂的液晶显示元件、以及新的二胺 |
| KR101699564B1 (ko) * | 2009-04-02 | 2017-01-24 | 닛산 가가쿠 고교 가부시키 가이샤 | 폴리아믹산알킬에스테르를 함유하는 폴리이미드 전구체 조성물 |
-
2011
- 2011-03-14 CN CN201180023615.0A patent/CN102934011B/zh active Active
- 2011-03-14 WO PCT/JP2011/055971 patent/WO2011115076A1/ja active Application Filing
- 2011-03-14 KR KR1020127026262A patent/KR101818786B1/ko active IP Right Grant
- 2011-03-14 JP JP2012505683A patent/JP5904119B2/ja active Active
- 2011-03-15 TW TW100108725A patent/TWI500658B/zh active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001033963A (ja) * | 1999-05-17 | 2001-02-09 | Asahi Chem Ind Co Ltd | 感光性樹脂組成物 |
| JP2001296525A (ja) * | 2000-04-12 | 2001-10-26 | Jsr Corp | 液晶配向剤および液晶表示素子 |
| JP2003026918A (ja) * | 2001-07-13 | 2003-01-29 | Hitachi Ltd | 液晶配向膜用材料、液晶表示素子、その製造方法及び液晶表示装置 |
| JP2006299079A (ja) * | 2005-04-20 | 2006-11-02 | Teijin Ltd | ポリイミド前駆体成形物の製造方法 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013147083A1 (ja) * | 2012-03-30 | 2013-10-03 | 日産化学工業株式会社 | ポリイミド系の液晶配向処理剤、液晶配向膜、及び液晶表示素子 |
| CN104204925A (zh) * | 2012-03-30 | 2014-12-10 | 日产化学工业株式会社 | 聚酰亚胺类的液晶取向处理剂、液晶取向膜及液晶显示元件 |
| JPWO2013147083A1 (ja) * | 2012-03-30 | 2015-12-14 | 日産化学工業株式会社 | ポリイミド系の液晶配向処理剤、液晶配向膜、及び液晶表示素子 |
| KR102058764B1 (ko) * | 2012-03-30 | 2019-12-23 | 닛산 가가쿠 가부시키가이샤 | 폴리이미드계의 액정 배향 처리제, 액정 배향막, 및 액정 표시 소자 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW201206992A (en) | 2012-02-16 |
| JPWO2011115076A1 (ja) | 2013-06-27 |
| CN102934011A (zh) | 2013-02-13 |
| TWI500658B (zh) | 2015-09-21 |
| KR20130038229A (ko) | 2013-04-17 |
| CN102934011B (zh) | 2016-01-20 |
| JP5904119B2 (ja) | 2016-04-13 |
| KR101818786B1 (ko) | 2018-02-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101818788B1 (ko) | 폴리아믹산에스테르와 폴리아믹산을 함유하는 액정 배향제 및 액정 배향막 | |
| KR101820981B1 (ko) | 폴리아믹산에스테르 함유 액정 배향제 및 액정 배향막 | |
| JP5708636B2 (ja) | 末端を修飾したポリアミック酸エステル含有液晶配向剤、及び液晶配向膜 | |
| KR101818787B1 (ko) | 폴리아믹산에스테르 액정 배향제 및 그것을 사용한 액정 배향막 | |
| JP5761183B2 (ja) | 熱脱離性基含有化合物を含有する液晶配向剤、及び液晶配向膜 | |
| JP6064900B2 (ja) | 液晶配向剤、及びそれを用いた液晶配向膜 | |
| WO2011115079A1 (ja) | 光配向処理法用の液晶配向剤、及びそれを用いた液晶配向膜 | |
| JP5998939B2 (ja) | ポリアミック酸エステルとポリアミック酸とを含有する液晶配向剤 | |
| WO2012133829A1 (ja) | 液晶配向剤及びそれを用いた液晶配向膜 | |
| WO2013147083A1 (ja) | ポリイミド系の液晶配向処理剤、液晶配向膜、及び液晶表示素子 | |
| JP5904119B2 (ja) | 末端を修飾したポリアミック酸エステルを含有する液晶配向剤、及び液晶配向膜 | |
| JP6597645B2 (ja) | 液晶配向剤 | |
| WO2014024885A1 (ja) | 液晶配向剤、及びそれを用いた液晶配向膜 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180023615.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11756261 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012505683 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20127026262 Country of ref document: KR Kind code of ref document: A |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11756261 Country of ref document: EP Kind code of ref document: A1 |