WO2011023883A1 - Conjugues de dimeres de pyrrolo [1, 4 ] benzodiazepine en tant qu' anticancereux - Google Patents
Conjugues de dimeres de pyrrolo [1, 4 ] benzodiazepine en tant qu' anticancereux Download PDFInfo
- Publication number
- WO2011023883A1 WO2011023883A1 PCT/FR2010/051709 FR2010051709W WO2011023883A1 WO 2011023883 A1 WO2011023883 A1 WO 2011023883A1 FR 2010051709 W FR2010051709 W FR 2010051709W WO 2011023883 A1 WO2011023883 A1 WO 2011023883A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alk
- ethoxy
- group
- compound
- conjugate
- Prior art date
Links
- IFOHPTVCEBWEEQ-UHFFFAOYSA-N pyrrolo[2,3-i][1,4]benzodiazepine Chemical class N1=CC=NC2=C3C=CN=C3C=CC2=C1 IFOHPTVCEBWEEQ-UHFFFAOYSA-N 0.000 title claims abstract description 14
- 239000002246 antineoplastic agent Substances 0.000 title claims abstract description 8
- 150000001875 compounds Chemical class 0.000 claims description 72
- -1 cyclic amine Chemical class 0.000 claims description 53
- 239000000243 solution Substances 0.000 claims description 52
- 239000003795 chemical substances by application Substances 0.000 claims description 42
- 230000008685 targeting Effects 0.000 claims description 41
- 239000000539 dimer Substances 0.000 claims description 30
- 206010028980 Neoplasm Diseases 0.000 claims description 27
- 238000006243 chemical reaction Methods 0.000 claims description 26
- 238000000034 method Methods 0.000 claims description 26
- 125000000217 alkyl group Chemical group 0.000 claims description 23
- 238000002360 preparation method Methods 0.000 claims description 22
- 125000003118 aryl group Chemical group 0.000 claims description 18
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 10
- 239000007864 aqueous solution Substances 0.000 claims description 10
- 125000001072 heteroaryl group Chemical group 0.000 claims description 9
- 238000001228 spectrum Methods 0.000 claims description 9
- 230000015572 biosynthetic process Effects 0.000 claims description 8
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 8
- 230000021615 conjugation Effects 0.000 claims description 7
- 125000003277 amino group Chemical group 0.000 claims description 6
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 6
- 230000001093 anti-cancer Effects 0.000 claims description 5
- 239000000427 antigen Substances 0.000 claims description 5
- 102000036639 antigens Human genes 0.000 claims description 5
- 108091007433 antigens Proteins 0.000 claims description 5
- 125000004122 cyclic group Chemical group 0.000 claims description 5
- 238000004519 manufacturing process Methods 0.000 claims description 5
- 108090000623 proteins and genes Proteins 0.000 claims description 5
- 102000004169 proteins and genes Human genes 0.000 claims description 5
- 238000002835 absorbance Methods 0.000 claims description 4
- 238000010521 absorption reaction Methods 0.000 claims description 4
- 230000001939 inductive effect Effects 0.000 claims description 4
- 125000001424 substituent group Chemical group 0.000 claims description 4
- 125000003545 alkoxy group Chemical group 0.000 claims description 3
- 238000011026 diafiltration Methods 0.000 claims description 3
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 3
- 230000008569 process Effects 0.000 claims description 3
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 claims description 2
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 2
- SPXOTSHWBDUUMT-UHFFFAOYSA-M 4-nitrobenzenesulfonate Chemical compound [O-][N+](=O)C1=CC=C(S([O-])(=O)=O)C=C1 SPXOTSHWBDUUMT-UHFFFAOYSA-M 0.000 claims description 2
- 206010007134 Candida infections Diseases 0.000 claims description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 claims description 2
- 108091034117 Oligonucleotide Proteins 0.000 claims description 2
- GNVMUORYQLCPJZ-UHFFFAOYSA-M Thiocarbamate Chemical compound NC([S-])=O GNVMUORYQLCPJZ-UHFFFAOYSA-M 0.000 claims description 2
- 125000005843 halogen group Chemical group 0.000 claims description 2
- 239000003446 ligand Substances 0.000 claims description 2
- 229920001542 oligosaccharide Polymers 0.000 claims description 2
- 150000002482 oligosaccharides Chemical class 0.000 claims description 2
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 2
- 210000002536 stromal cell Anatomy 0.000 claims description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 2
- 210000004881 tumor cell Anatomy 0.000 claims description 2
- 238000000108 ultra-filtration Methods 0.000 claims description 2
- 125000002228 disulfide group Chemical group 0.000 claims 1
- 239000012634 fragment Substances 0.000 claims 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 claims 1
- 239000008194 pharmaceutical composition Substances 0.000 claims 1
- 239000000546 pharmaceutical excipient Substances 0.000 claims 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 72
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 55
- 238000004949 mass spectrometry Methods 0.000 description 45
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 40
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 38
- 239000000203 mixture Substances 0.000 description 36
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 29
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 28
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 25
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 24
- 238000005481 NMR spectroscopy Methods 0.000 description 23
- 210000004027 cell Anatomy 0.000 description 20
- 239000000377 silicon dioxide Substances 0.000 description 20
- UQVNRKBFAXNOGA-LWTNMJDUSA-N (E)-tomaymycin Chemical compound CO[C@H]1NC2=CC(O)=C(OC)C=C2C(=O)N2C\C(=C\C)C[C@@H]12 UQVNRKBFAXNOGA-LWTNMJDUSA-N 0.000 description 19
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 17
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 15
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 14
- 238000003818 flash chromatography Methods 0.000 description 14
- 239000012074 organic phase Substances 0.000 description 14
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 12
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- 235000019253 formic acid Nutrition 0.000 description 12
- UQVNRKBFAXNOGA-IUODEOHRSA-N Tomaymycin Natural products CO[C@H]1Nc2cc(O)c(OC)cc2C(=O)N3CC(=CC)C[C@H]13 UQVNRKBFAXNOGA-IUODEOHRSA-N 0.000 description 11
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 11
- 239000002798 polar solvent Substances 0.000 description 11
- 239000002253 acid Substances 0.000 description 10
- 150000002148 esters Chemical class 0.000 description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 10
- 239000011734 sodium Substances 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 230000029936 alkylation Effects 0.000 description 9
- 238000005804 alkylation reaction Methods 0.000 description 9
- 239000008346 aqueous phase Substances 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 8
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 8
- 239000003643 water by type Substances 0.000 description 8
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 7
- 0 CCc(cc(c(*)c1)OC)c1C(*(CC(*)(C1)I)[C@@]1C(N[U])N=C)=O Chemical compound CCc(cc(c(*)c1)OC)c1C(*(CC(*)(C1)I)[C@@]1C(N[U])N=C)=O 0.000 description 7
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 7
- 230000000259 anti-tumor effect Effects 0.000 description 7
- 230000008033 biological extinction Effects 0.000 description 7
- 238000010511 deprotection reaction Methods 0.000 description 7
- 150000002009 diols Chemical class 0.000 description 7
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- 241000699670 Mus sp. Species 0.000 description 6
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 6
- 150000001412 amines Chemical class 0.000 description 6
- 230000022811 deglycosylation Effects 0.000 description 6
- 238000011156 evaluation Methods 0.000 description 6
- 239000001963 growth medium Substances 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- 238000001819 mass spectrum Methods 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 6
- 150000003573 thiols Chemical class 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 6
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 5
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 5
- 101000841411 Homo sapiens Protein ecdysoneless homolog Proteins 0.000 description 5
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 5
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 5
- 102100029090 Protein ecdysoneless homolog Human genes 0.000 description 5
- 229910004298 SiO 2 Inorganic materials 0.000 description 5
- 125000002947 alkylene group Chemical group 0.000 description 5
- 238000003776 cleavage reaction Methods 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 238000006722 reduction reaction Methods 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 230000007017 scission Effects 0.000 description 5
- 230000004580 weight loss Effects 0.000 description 5
- IQFYYKKMVGJFEH-OYDXRQHMSA-N 1-[(2r,4s,5s)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-methylpyrimidine-2,4-dione Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H]([14CH2]O)[C@@H](O)C1 IQFYYKKMVGJFEH-OYDXRQHMSA-N 0.000 description 4
- LCPGZHHBMNINRM-UHFFFAOYSA-N 2,2,3,3-tetraethoxypropanoic acid Chemical compound CCOC(OCC)C(OCC)(OCC)C(O)=O LCPGZHHBMNINRM-UHFFFAOYSA-N 0.000 description 4
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- KCUOMSIICVALRH-UHFFFAOYSA-N [6-(hydroxymethyl)-4-(2-piperazin-1-ylethoxy)pyridin-2-yl]methanol Chemical compound OCC1=NC(CO)=CC(OCCN2CCNCC2)=C1 KCUOMSIICVALRH-UHFFFAOYSA-N 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 125000000753 cycloalkyl group Chemical group 0.000 description 4
- 231100000433 cytotoxic Toxicity 0.000 description 4
- 230000001472 cytotoxic effect Effects 0.000 description 4
- 238000010586 diagram Methods 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 4
- 125000004076 pyridyl group Chemical group 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 4
- SZYFMMNWNDIBGM-UHFFFAOYSA-N 3-[2-[2-[2-[2-[methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]propanoic acid Chemical compound CC(C)(C)OC(=O)N(C)CCOCCOCCOCCOCCC(O)=O SZYFMMNWNDIBGM-UHFFFAOYSA-N 0.000 description 3
- PXCVVFCMJCKILT-UHFFFAOYSA-N 3-acetamido-2,2,3,3-tetraethoxypropanoic acid Chemical compound CCOC(C(=O)O)(C(NC(=O)C)(OCC)OCC)OCC PXCVVFCMJCKILT-UHFFFAOYSA-N 0.000 description 3
- AYFNWSLGBNRXKU-UHFFFAOYSA-N CN(CCCS[S+](C)C)CCOC1=CC(CO)=NC(CO)=C1 Chemical compound CN(CCCS[S+](C)C)CCOC1=CC(CO)=NC(CO)=C1 AYFNWSLGBNRXKU-UHFFFAOYSA-N 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- 101150039048 GCR1 gene Proteins 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 3
- 101100338491 Oryza sativa subsp. japonica HCT1 gene Proteins 0.000 description 3
- 238000011579 SCID mouse model Methods 0.000 description 3
- 101100495309 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) CDH1 gene Proteins 0.000 description 3
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 150000001335 aliphatic alkanes Chemical class 0.000 description 3
- 150000001350 alkyl halides Chemical class 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 239000012062 aqueous buffer Substances 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 125000003636 chemical group Chemical group 0.000 description 3
- 239000007822 coupling agent Substances 0.000 description 3
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 229940127121 immunoconjugate Drugs 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- IADXKDSSQHERAZ-UHFFFAOYSA-N methyl 3-[2-[2-[2-[2-[methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]propanoate Chemical compound COC(=O)CCOCCOCCOCCOCCN(C)C(=O)OC(C)(C)C IADXKDSSQHERAZ-UHFFFAOYSA-N 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- SHNUBALDGXWUJI-UHFFFAOYSA-N pyridin-2-ylmethanol Chemical compound OCC1=CC=CC=N1 SHNUBALDGXWUJI-UHFFFAOYSA-N 0.000 description 3
- 238000006268 reductive amination reaction Methods 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 239000012279 sodium borohydride Substances 0.000 description 3
- 229910000033 sodium borohydride Inorganic materials 0.000 description 3
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- CWMGPICYSNPTAA-UHFFFAOYSA-N tert-butyl 4-[2-[2,6-bis(hydroxymethyl)pyridin-4-yl]oxyethyl]piperazine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCN1CCOC1=CC(CO)=NC(CO)=C1 CWMGPICYSNPTAA-UHFFFAOYSA-N 0.000 description 3
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 150000003512 tertiary amines Chemical class 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- VLBWEJJOETYCSE-UHFFFAOYSA-N 2-Methyl-2-(methyldithio)propanal Chemical compound CSSC(C)(C)C=O VLBWEJJOETYCSE-UHFFFAOYSA-N 0.000 description 2
- DLPZHAZMDMSVQH-UHFFFAOYSA-N 2-[3,5-bis(2-tert-butylsilyloxypropan-2-yl)phenyl]propan-2-ol Chemical compound OC(C)(C)C1=CC(=CC(=C1)C(O[SiH2]C(C)(C)C)(C)C)C(O[SiH2]C(C)(C)C)(C)C DLPZHAZMDMSVQH-UHFFFAOYSA-N 0.000 description 2
- PBVAJRFEEOIAGW-UHFFFAOYSA-N 3-[bis(2-carboxyethyl)phosphanyl]propanoic acid;hydrochloride Chemical compound Cl.OC(=O)CCP(CCC(O)=O)CCC(O)=O PBVAJRFEEOIAGW-UHFFFAOYSA-N 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- AOZSYWPAYARVEC-UHFFFAOYSA-N CCCN(CCOC1=CC(CO)=NC(CO)=C1)S[S+](C)C Chemical compound CCCN(CCOC1=CC(CO)=NC(CO)=C1)S[S+](C)C AOZSYWPAYARVEC-UHFFFAOYSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 2
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- LJLKDIRBXMMZJC-UHFFFAOYSA-N O-[2-[3-(2-tert-butylsilyloxypropan-2-yl)-5-(hydroxymethyl)phenyl]propan-2-yl] ethanethioate Chemical compound C(C)(=S)OC(C)(C)C1=CC(=CC(=C1)CO)C(O[SiH2]C(C)(C)C)(C)C LJLKDIRBXMMZJC-UHFFFAOYSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- DPAQTYREWOHMLW-UHFFFAOYSA-N [3-[[tert-butyl(dimethyl)silyl]oxymethyl]-5-(2-sulfanylpropan-2-yl)phenyl]methanol Chemical compound CC(C)(C)[Si](C)(C)OCC1=CC(CO)=CC(C(C)(C)S)=C1 DPAQTYREWOHMLW-UHFFFAOYSA-N 0.000 description 2
- VAIGWIMILWRXKD-UHFFFAOYSA-N [6-(hydroxymethyl)-4-[2-[methyl-(2-methyl-2-sulfanylpropyl)amino]ethoxy]pyridin-2-yl]methanol Chemical compound CC(S)(C)CN(C)CCOC1=CC(CO)=NC(CO)=C1 VAIGWIMILWRXKD-UHFFFAOYSA-N 0.000 description 2
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 125000003158 alcohol group Chemical group 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 150000001408 amides Chemical group 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 230000003698 anagen phase Effects 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- PFYXSUNOLOJMDX-UHFFFAOYSA-N bis(2,5-dioxopyrrolidin-1-yl) carbonate Chemical compound O=C1CCC(=O)N1OC(=O)ON1C(=O)CCC1=O PFYXSUNOLOJMDX-UHFFFAOYSA-N 0.000 description 2
- 201000008274 breast adenocarcinoma Diseases 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- 150000001945 cysteines Chemical class 0.000 description 2
- GLBVAZGXVWJUGP-UHFFFAOYSA-N diethyl 4-[2-[4-[(2-methylpropan-2-yl)oxycarbonyl]piperazin-1-yl]ethoxy]pyridine-2,6-dicarboxylate Chemical compound CCOC(=O)C1=NC(C(=O)OCC)=CC(OCCN2CCN(CC2)C(=O)OC(C)(C)C)=C1 GLBVAZGXVWJUGP-UHFFFAOYSA-N 0.000 description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- CCNWZGXOWHXIJE-UHFFFAOYSA-N ethoxyethane;oxane Chemical compound CCOCC.C1CCOCC1 CCNWZGXOWHXIJE-UHFFFAOYSA-N 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 238000004191 hydrophobic interaction chromatography Methods 0.000 description 2
- 150000002466 imines Chemical group 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 2
- 125000003588 lysine group Chemical class [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- RSPUCDHRULIVAV-UHFFFAOYSA-N methyl 3-[2-[2-[2-[2-(methylamino)ethoxy]ethoxy]ethoxy]ethoxy]propanoate Chemical compound CNCCOCCOCCOCCOCCC(=O)OC RSPUCDHRULIVAV-UHFFFAOYSA-N 0.000 description 2
- 150000004702 methyl esters Chemical class 0.000 description 2
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000008363 phosphate buffer Substances 0.000 description 2
- 239000003880 polar aprotic solvent Substances 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 238000006257 total synthesis reaction Methods 0.000 description 2
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 238000000870 ultraviolet spectroscopy Methods 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- UAYWVJHJZHQCIE-UHFFFAOYSA-L zinc iodide Chemical compound I[Zn]I UAYWVJHJZHQCIE-UHFFFAOYSA-L 0.000 description 2
- VRDGQQTWSGDXCU-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-iodoacetate Chemical compound ICC(=O)ON1C(=O)CCC1=O VRDGQQTWSGDXCU-UHFFFAOYSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- GUJAGMICFDYKNR-UHFFFAOYSA-N 1,4-benzodiazepine Chemical compound N1C=CN=CC2=CC=CC=C12 GUJAGMICFDYKNR-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 1
- NRKYWOKHZRQRJR-UHFFFAOYSA-N 2,2,2-trifluoroacetamide Chemical compound NC(=O)C(F)(F)F NRKYWOKHZRQRJR-UHFFFAOYSA-N 0.000 description 1
- LOTMLXVUNZRTCR-UHFFFAOYSA-N 2,2,3-triethoxypropanoic acid Chemical compound C(C)OCC(C(=O)O)(OCC)OCC LOTMLXVUNZRTCR-UHFFFAOYSA-N 0.000 description 1
- ZUSJSHCXTGUVAF-UHFFFAOYSA-N 2-[3-bromo-5-(2-tert-butylsilyloxypropan-2-yl)phenyl]propan-2-yloxy-tert-butylsilane Chemical compound BrC1=CC(=CC(=C1)C(O[SiH2]C(C)(C)C)(C)C)C(O[SiH2]C(C)(C)C)(C)C ZUSJSHCXTGUVAF-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- WBDRVMIJNFZLMN-UHFFFAOYSA-N 2-phenylpropane-2-thiol Chemical compound CC(C)(S)C1=CC=CC=C1 WBDRVMIJNFZLMN-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- DKUZHSDZSMQOGQ-UHFFFAOYSA-N 3-[2-[2-[2-(2-aminoethoxy)ethoxy]ethoxy]ethoxy]propanoic acid Chemical compound NCCOCCOCCOCCOCCC(O)=O DKUZHSDZSMQOGQ-UHFFFAOYSA-N 0.000 description 1
- YEIYIPDFZMLJQH-UHFFFAOYSA-N 3-[2-[2-[2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethoxy]ethoxy]ethoxy]ethoxy]propanoic acid Chemical compound CC(C)(C)OC(=O)NCCOCCOCCOCCOCCC(O)=O YEIYIPDFZMLJQH-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- GYFCEVXCVFNSCL-UHFFFAOYSA-N 4-methyl-4-(methyldisulfanyl)pentanoic acid Chemical compound CSSC(C)(C)CCC(O)=O GYFCEVXCVFNSCL-UHFFFAOYSA-N 0.000 description 1
- XTLJJHGQACAZMS-UHFFFAOYSA-N 4-oxo-1h-pyridine-2,6-dicarboxylic acid Chemical compound OC(=O)C1=CC(=O)C=C(C(O)=O)N1 XTLJJHGQACAZMS-UHFFFAOYSA-N 0.000 description 1
- XVMSFILGAMDHEY-UHFFFAOYSA-N 6-(4-aminophenyl)sulfonylpyridin-3-amine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=N1 XVMSFILGAMDHEY-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- MSFVELCIRCUUPJ-UHFFFAOYSA-N CC(C)(CN(C)CCOc1cc(CO)nc(CO)c1)SSC Chemical compound CC(C)(CN(C)CCOc1cc(CO)nc(CO)c1)SSC MSFVELCIRCUUPJ-UHFFFAOYSA-N 0.000 description 1
- KXNZQNVDLFOPLR-DWWXAFPLSA-N CC(C)(CN(C)CCOc1cc(COc(c(OC)c2)cc(N=CC(C3)N4C/C3=C/C)c2C4=O)nc(COc(c(OC)c2)cc(N=C[C@H](C3)N4C/C3=C/C)c2C4=O)c1)SSC Chemical compound CC(C)(CN(C)CCOc1cc(COc(c(OC)c2)cc(N=CC(C3)N4C/C3=C/C)c2C4=O)nc(COc(c(OC)c2)cc(N=C[C@H](C3)N4C/C3=C/C)c2C4=O)c1)SSC KXNZQNVDLFOPLR-DWWXAFPLSA-N 0.000 description 1
- FHHKGBUYCVLUNZ-UHFFFAOYSA-N CC(C)(c1cc(CO)cc(CO)c1)SCC(NCCOCCOCCOCCOCCC(OC)=O)=O Chemical compound CC(C)(c1cc(CO)cc(CO)c1)SCC(NCCOCCOCCOCCOCCC(OC)=O)=O FHHKGBUYCVLUNZ-UHFFFAOYSA-N 0.000 description 1
- NYEXPFUVIHYLQS-UHFFFAOYSA-N CC(CC1=CC(=CC(=C1)CO)CO)SN(CCOCCOCCOCCOCCC(=O)O)C(=O)C Chemical compound CC(CC1=CC(=CC(=C1)CO)CO)SN(CCOCCOCCOCCOCCC(=O)O)C(=O)C NYEXPFUVIHYLQS-UHFFFAOYSA-N 0.000 description 1
- BAUCSWOEMWWJIL-HZGXIKAXSA-N CC/C=C/[C@H](C[C@@H](CO)N)OCCNCCN(CC)[C@@H](CCC(C)(C)SSC)O Chemical compound CC/C=C/[C@H](C[C@@H](CO)N)OCCNCCN(CC)[C@@H](CCC(C)(C)SSC)O BAUCSWOEMWWJIL-HZGXIKAXSA-N 0.000 description 1
- UZWKFZYQVHCWKJ-UHFFFAOYSA-N CC1=C(C=C(C=C1O[Si](C)(C)C(C)(C)C)CO)C(C)(C)S Chemical compound CC1=C(C=C(C=C1O[Si](C)(C)C(C)(C)C)CO)C(C)(C)S UZWKFZYQVHCWKJ-UHFFFAOYSA-N 0.000 description 1
- ITNUMDSKZZATIA-UHFFFAOYSA-N CCCN(CCOC1=CC=C(CO)N=C1CO)S[S+](C)C Chemical compound CCCN(CCOC1=CC=C(CO)N=C1CO)S[S+](C)C ITNUMDSKZZATIA-UHFFFAOYSA-N 0.000 description 1
- LMWYRLSCPUTCRR-UHFFFAOYSA-N CCOC(C(=O)O)(C(NC(=O)C)(OCC)OCC)OC(C)C(C)(NC)SC(C)(C)C Chemical compound CCOC(C(=O)O)(C(NC(=O)C)(OCC)OCC)OC(C)C(C)(NC)SC(C)(C)C LMWYRLSCPUTCRR-UHFFFAOYSA-N 0.000 description 1
- KKLCJSSDKHVYMQ-UHFFFAOYSA-N CCOC(C(=O)O)(C(NC)(OCC)OCC)OCCC(=O)C Chemical compound CCOC(C(=O)O)(C(NC)(OCC)OCC)OCCC(=O)C KKLCJSSDKHVYMQ-UHFFFAOYSA-N 0.000 description 1
- HGPNOKHYDOHLAU-UHFFFAOYSA-N CCOC(C)OC(C)(C(=O)O)OCC Chemical compound CCOC(C)OC(C)(C(=O)O)OCC HGPNOKHYDOHLAU-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 102000005744 Glycoside Hydrolases Human genes 0.000 description 1
- 108010031186 Glycoside Hydrolases Proteins 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 101001024703 Homo sapiens Nck-associated protein 5 Proteins 0.000 description 1
- 101001121386 Homo sapiens Ovochymase-1 Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 1
- 229930190887 Leptomycin Natural products 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- 238000005684 Liebig rearrangement reaction Methods 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 102100036946 Nck-associated protein 5 Human genes 0.000 description 1
- 102000002423 Octamer Transcription Factor-6 Human genes 0.000 description 1
- 108010068113 Octamer Transcription Factor-6 Proteins 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 102100026305 Ovochymase-1 Human genes 0.000 description 1
- 239000002033 PVDF binder Substances 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 229920005654 Sephadex Polymers 0.000 description 1
- 239000012507 Sephadex™ Substances 0.000 description 1
- 241000187747 Streptomyces Species 0.000 description 1
- 239000012505 Superdex™ Substances 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- 102000001742 Tumor Suppressor Proteins Human genes 0.000 description 1
- 108010040002 Tumor Suppressor Proteins Proteins 0.000 description 1
- 238000006959 Williamson synthesis reaction Methods 0.000 description 1
- YSVZGWAJIHWNQK-UHFFFAOYSA-N [3-(hydroxymethyl)-2-bicyclo[2.2.1]heptanyl]methanol Chemical compound C1CC2C(CO)C(CO)C1C2 YSVZGWAJIHWNQK-UHFFFAOYSA-N 0.000 description 1
- MBPDGESBYWXQHH-UHFFFAOYSA-N [3-(hydroxymethyl)-5-(2-sulfanylpropan-2-yl)phenyl]methanol Chemical compound CC(C)(S)C1=CC(CO)=CC(CO)=C1 MBPDGESBYWXQHH-UHFFFAOYSA-N 0.000 description 1
- DYSHJLWOUURIBP-UHFFFAOYSA-N [4-(2-aminoethoxy)-6-(hydroxymethyl)pyridin-2-yl]methanol Chemical compound NCCOC1=CC(CO)=NC(CO)=C1 DYSHJLWOUURIBP-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 238000005377 adsorption chromatography Methods 0.000 description 1
- 238000012382 advanced drug delivery Methods 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 1
- 239000012965 benzophenone Substances 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000010364 biochemical engineering Methods 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- FNXLCIKXHOPCKH-UHFFFAOYSA-N bromamine Chemical compound BrN FNXLCIKXHOPCKH-UHFFFAOYSA-N 0.000 description 1
- 125000001246 bromo group Chemical class Br* 0.000 description 1
- FNXLCIKXHOPCKH-VQEHIDDOSA-N bromoamine Chemical group Br[15NH2] FNXLCIKXHOPCKH-VQEHIDDOSA-N 0.000 description 1
- 239000008366 buffered solution Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000001720 carbohydrates Chemical group 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 238000000451 chemical ionisation Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 231100000599 cytotoxic agent Toxicity 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 150000005690 diesters Chemical class 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 229960001484 edetic acid Drugs 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 230000006862 enzymatic digestion Effects 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 238000000105 evaporative light scattering detection Methods 0.000 description 1
- 238000002270 exclusion chromatography Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000004222 ferrous gluconate Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 125000004836 hexamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000013415 human tumor xenograft model Methods 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229910052588 hydroxylapatite Inorganic materials 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000003978 infusion fluid Substances 0.000 description 1
- JDNTWHVOXJZDSN-UHFFFAOYSA-N iodoacetic acid Chemical compound OC(=O)CI JDNTWHVOXJZDSN-UHFFFAOYSA-N 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 235000018977 lysine Nutrition 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 1
- 231100000682 maximum tolerated dose Toxicity 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- YVWPDYFVVMNWDT-UHFFFAOYSA-N methyl 2-ethoxypropanoate Chemical compound CCOC(C)C(=O)OC YVWPDYFVVMNWDT-UHFFFAOYSA-N 0.000 description 1
- RZZMPRGBOKCCSU-UHFFFAOYSA-N methyl 3-[2-[2-[2-[2-[(2-iodoacetyl)-methylamino]ethoxy]ethoxy]ethoxy]ethoxy]propanoate Chemical compound COC(=O)CCOCCOCCOCCOCCN(C)C(=O)CI RZZMPRGBOKCCSU-UHFFFAOYSA-N 0.000 description 1
- LPNPILLNPLYGMH-UHFFFAOYSA-N methyl 3-[2-[2-[2-[2-[(2-iodoacetyl)amino]ethoxy]ethoxy]ethoxy]ethoxy]propanoate Chemical compound COC(=O)CCOCCOCCOCCOCCNC(=O)CI LPNPILLNPLYGMH-UHFFFAOYSA-N 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- YRCHYHRCBXNYNU-UHFFFAOYSA-N n-[[3-fluoro-4-[2-[5-[(2-methoxyethylamino)methyl]pyridin-2-yl]thieno[3,2-b]pyridin-7-yl]oxyphenyl]carbamothioyl]-2-(4-fluorophenyl)acetamide Chemical compound N1=CC(CNCCOC)=CC=C1C1=CC2=NC=CC(OC=3C(=CC(NC(=S)NC(=O)CC=4C=CC(F)=CC=4)=CC=3)F)=C2S1 YRCHYHRCBXNYNU-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000005935 nucleophilic addition reaction Methods 0.000 description 1
- 238000007344 nucleophilic reaction Methods 0.000 description 1
- 125000001979 organolithium group Chemical group 0.000 description 1
- 125000002734 organomagnesium group Chemical group 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- XYJRXVWERLGGKC-UHFFFAOYSA-D pentacalcium;hydroxide;triphosphate Chemical compound [OH-].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O XYJRXVWERLGGKC-UHFFFAOYSA-D 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- QVLTXCYWHPZMCA-UHFFFAOYSA-N po4-po4 Chemical compound OP(O)(O)=O.OP(O)(O)=O QVLTXCYWHPZMCA-UHFFFAOYSA-N 0.000 description 1
- 239000003495 polar organic solvent Substances 0.000 description 1
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- ZBJSHDVMDCJOEZ-UHFFFAOYSA-N potassium;1h-naphthalen-1-ide Chemical compound [K+].[C-]1=CC=CC2=CC=CC=C21 ZBJSHDVMDCJOEZ-UHFFFAOYSA-N 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 230000004845 protein aggregation Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- KOUKXHPPRFNWPP-UHFFFAOYSA-N pyrazine-2,5-dicarboxylic acid;hydrate Chemical compound O.OC(=O)C1=CN=C(C(O)=O)C=N1 KOUKXHPPRFNWPP-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- UOWVMDUEMSNCAV-WYENRQIDSA-N rachelmycin Chemical compound C1([C@]23C[C@@H]2CN1C(=O)C=1NC=2C(OC)=C(O)C4=C(C=2C=1)CCN4C(=O)C1=CC=2C=4CCN(C=4C(O)=C(C=2N1)OC)C(N)=O)=CC(=O)C1=C3C(C)=CN1 UOWVMDUEMSNCAV-WYENRQIDSA-N 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 238000001542 size-exclusion chromatography Methods 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- VRXIOAYUQIITBU-UHFFFAOYSA-N tert-butyl 4-(2-hydroxyethyl)piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCN(CCO)CC1 VRXIOAYUQIITBU-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- PJEXKHZITJQSGE-UHFFFAOYSA-N tert-butyl(2-phenylpropan-2-yloxy)silane Chemical compound C(C)(C)(C)[SiH2]OC(C)(C)C1=CC=CC=C1 PJEXKHZITJQSGE-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 108700026220 vif Genes Proteins 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
- C07D519/04—Dimeric indole alkaloids, e.g. vincaleucoblastine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
- C07D519/06—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00 containing at least one condensed beta-lactam ring system, provided for by groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00, e.g. a penem or a cepham system
Definitions
- the present invention relates to conjugates of dimers of pyrrolo [1,4] benzodiazepine (PBD), the compositions containing them and their therapeutic application, especially as anticancer agents.
- PBD pyrrolo [1,4] benzodiazepine
- the invention also relates to the process for preparing the conjugates for their application as anti-cancer agents as well as for the dimers themselves. [Technical area]
- Pyrrolo [1,4] benzodiazepine dimers are anticancer agents that act by covalently binding to cell DNA. These derivatives have been described in applications WO 00/12508 and WO 2005/085260 as well as in the following publications: Eur.J.Med.Chem. 2005, 40, 641-654; Teirahedron Leisters 1988, 29 (40), 5105-5108.
- Conjugate chemistry has been known for many years and has been applied to several cytotoxic families such as maytansinoids (WO 04103272), taxanes (WO 06061258), leptomycins (WO 07144709), CC-1065 and the like. (WO 2007102069); see also Conjugates, Monneret C, et al., Cancer Bulletin 2000, 87 (1 1), 829-38; Ricart A.D., et al., Nature Clinical Practice Oncology 2007, 4, 245-255; Singh R. and Rickson H. K., Therapeutic Antibodies: Methods and Protocols, 2009, 525, 445-467.
- G represents a single or double bond or -O-, -S- or -NR-.
- D represents a single bond or one of the following groups: -E-, -E-NR-, -E-NR-F-, -EO-, -EOF-, -E-NR-CO-, -E -NR-CO-F-, -E-CO-, -CO-E-, -E-CO-F, -ES-, -ESF-, -E-NR-CS-, -E-NR- CS- F where E and F are selected from - (OCH 2 CH 2) l alkyl (OCH 2 CH 2) j -, -alkyl (OCH 2 CH 2) r alkyl, - (OCH 2 CH 2) -, - (OCH 2 CH 2 ), cycloalkyl (OCH 2 CH 2 ) r , - (OCH 2 CH 2 ), heterocyclyl (OCH 2 CH 2 ) r , - (OCH 2 CH 2 ), aryl (OCH 2 CH 2 ) r
- G can only be a connection (simple, double, triple) or -O-, -S- or -NR-.
- G can only be a connection (simple, double, triple) or -O-, -S- or -NR-.
- the other compounds of the present invention which are characterized by the -O-ALK-NR 3 -ALK-S- (CH 2 CH 2 O) -ALK- linker, none of the D motifs of WO 07085930 or WO 2009 / 016516 does not provide the combination of an amino group NR 3 and a bond -S-.
- the following dimers are described in WO 2009/016516:
- dimers include a linker similar to those described in the present invention (in particular, no -ALK-S- motif).
- the technical problem to be solved by the present invention is to propose novel conjugates of dimers of pyrrolo [1,4] benzodiazepine.
- conjugate a cell targeting agent to which is covalently attached at least one molecule of a cytotoxic compound
- Cell targeting agent (or "cell binding agent” in English): a molecule having an affinity for a biological target: it may be for example a ligand, a protein, an antibody, more particularly monoclonal, a protein or antibody fragment, a peptide, an oligonucleotide, an oligosaccharide.
- the targeting agent has the function of directing the biologically active compound as a cytotoxic agent to the biological target;
- biological target an antigen (or group of antigens) preferentially localized on the surface of the cancerous cells or stromal cells associated with this tumor; these antigens may be, for example, a growth factor receptor, an oncogene product or mutated "tumor suppressor” gene, a molecule related to angiogenesis. an adhesion molecule;
- Alkyl group a saturated aliphatic hydrocarbon group obtained by removing a hydrogen atom from an alkane.
- the alkyl group can be linear or branched. By way of examples, mention may be made of methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl, 2,2-dimethylpropyl and hexyl groups;
- Cycloalkyl group a cyclic alkyl group comprising between 3 and 8 carbon atoms engaged in the cyclic structure.
- cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl groups By way of examples, mention may be made of cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl groups;
- Aryl group an aromatic group containing no heteroatom, mono- or bicyclic. It is more particularly phenyl and naphthyl groups;
- Heteroaryl group an aromatic group comprising at least one heteroatom (O, S, N) engaged in the ring and connected to the carbon atoms forming the ring, mono- or bicylic. It is more particularly pyridinyl, pyrrolyl, thienyl, furanyl, pyrimidinyl, triazolyl;
- Heterocycloalkyl group a cycloalkyl group comprising at least one heteroatom (O, S, N) engaged in the ring and connected to the carbon atoms forming the ring;
- Alkoxy group an -O-alkyl group, wherein the alkyl group is as defined above;
- Alkanoyloxy group a group -O-CO-alkyl, where the alkyl group is as defined above;
- Alkylene group a divalent saturated group of empirical formula -C m H 2m -, obtained by removing two hydrogen atoms from an alkane.
- the alkane can be linear or branched.
- a linear alkylene group may be more particularly of the formula - (CH 2 ) m -, m being an integer;
- Fig.1 high resolution mass spectrum of the conjugate of ex.1 after deglycosylation
- Fig.2 high resolution mass spectrum of the ex.2 conjugate after deglycosylation
- Fig.3 high resolution mass spectrum of the conjugate of ex.3 after deglycosylation
- Fig.4 high resolution mass spectrum of the conjugate of ex.4 after deglycosylation
- Fig.5 high resolution mass spectrum of the non-deglycosylated conjugate of ex.6. These figures show for each conjugate after deconvolution, the distribution of species carrying 0 to 8 dimers of tomaymycin (D 0 : no dimer, D x : x dimers).
- the invention relates to compounds of formula:
- OH, -OR, -OCOR, -COOR, -OCOOR, -OCONRR ', a cyclic carbamate such as N10 and C11 are included in a ring, -NRCONRR',
- a cyclic thiocarbamate such as N10 and C11 are included in a ring, -SH, -SR, -SOR, -SOOR, -SO 3 " , -NRSOOR ', -NRR', a cyclic amine such as N10 and C11 are included in a ring, -NROR ', -NRCOR', -N 3 , -CN, HaI, a trialkylphosphonium or triarylphosphonium group;
- R 1, R 2 , R 1 ', R 2 ' which are identical or different, represent, independently of each other: H,
- Y and Y ' identical or different, represent independently of each other H or OR;
- M represents CH or N
- ALK and ALK ' which are identical or different, represent, independently of one another, a (C 1 -C 6) alkylene group;
- R and R ' represent, independently of one another, H or a group (CrC ⁇ ) alkyl or aryl optionally substituted with one or more substituents chosen from: HaI, CN, NRR', CF 3 , OR an aryl or heteroaryl group;
- R 3 represents H or a (C 1 -C 6) alkyl group
- i represents an integer from 1 to 40, preferably from 1 to 20, preferably from 1 to 10;
- Z b represents a single bond, -O- or -NH- and R b represents H or a group (C 1 -C 8 alkyl, (C 3 -C 7 ) cycloalkyl, aryl, heteroaryl or (C 3 -C 7 ) heterocycloalkyl or well
- Z b represents a single bond and R b represents HaI.
- the compounds of formula (I), including those exemplified, may exist in the form of bases or addition salts with pharmaceutically acceptable acids or alternatively of hydrates or solvates of these bases or of these salts.
- the two ALK and ALK 'groups attached to the phenyl or pyridinyl ring both denote a methylene group:
- Y and Y ' more particularly represent a (dC 4 ) alkoxy group, especially the methoxy group.
- L may be more particularly chosen from one of the following:
- ALK is more particularly a (dC 4 ) alkylene group.
- ALK can be one of the following: -CH 2 CH 2 -, -CH 2 CMe 2 -, -CH 2 CH 2 CMe 2 -.
- L can also be one of those described in Table I below or in Table II.
- i represents an integer ranging from 1 to 40, preferably from 1 to 20, preferably from 1 to 10.
- i may take each of values ranging from 1 to 40, in particular i may be 3, 4, 5, 6, 7 , 8, 9 or 10.
- the reaction between GCR1 and GCR2 assures the attachment of the compound to the targeting agent by formation of a covalent bond.
- the compound is capable of being conjugated to a targeting agent.
- activated esters which have a good reactivity with respect to the GCR2 groups, especially with respect to the amino groups present on the antibodies, are preferred.
- activated esters are the ')): in which
- Gl represents at least one inductive group such as -NO 2 or -HaI, especially -F. It can be by
- GCR2 By way of example of GCR2, mention may be made of the ⁇ -amino groups of the lysines carried by the side chains of the lysine residues which are present on the surface of an antibody, the saccharide groups of the hinge region or thiols of cysteines by reduction of intra-chain disulfide bonds (Garnett M. C, et al., Advanced Drug Delivery Reviews 2001, 53, 171-216).
- the compounds according to the invention can therefore be used for the preparation of a targeting agent to which is covalently attached in the para position of M the dimer of formula:
- the targeting agent is an antibody. More particularly, the dimer has the formula:
- it is necessary to transform L 3 into a group -LC ( O) Z b R b with the aid of one or several reactions.
- Tomaymycin the latter can be prepared using the strain Streptomyces croceus by following the teaching of FR 1516743 or by total synthesis (see J. Antibiotics 1983, XXXVI (3), 276-282 Z.Tozuka "Studies on tomaymycin, total syntheses of the antitumor antibiotics E- and Z-tomaymycins ").
- Pi / P'i compounds There are also commercial Pi / P'i compounds.
- E and E ' represent independently of each other an -OH group or a leaving group.
- L can more particularly represent one of those described in Schemes 2, 2 ', 3, 3', 3 ", 4, 5, 5 ', 6, 6', 6", 7.
- L * is selected from: -ALK-SH; -O-ALK-NR 3 -ALK-SH; X - f ;
- the term "leaving group” denotes an atom or a group of atoms which, in the heterolytic reaction between P 2 and P 1 or P ' 1 , leaves carrying the electron pair of the covalent bond connecting ALK. and LG or LG '.
- the leaving group is chosen more particularly from a halogen atom, in particular chlorine or bromine, a mesylate, tosylate, nosylate or -OPPh 3 + group .
- an aprotic polar solvent such as DCM: reaction between the amine function and the N-hydroxysuccinimidyl haloacetate and then in situ addition of a coupling agent such as DIC.
- an aprotic polar solvent such as DCM
- Step (iii): alkylation of the amine and saponification of the ester; the reaction is carried out in two successive stages in an anhydrous aprotic polar solvent such as THF: alkylation of the amine by treatment with a base such as NaH in the presence of a reagent bearing a nucleofuge group such as an alkyl halide R 3 HaI then addition of lithium and water; Step (i): Following step (iii), the reactions of step (i) are repeated for the case R 3 H. case where ALK ⁇ CHgCH ⁇
- anhydrous aprotic polar solvent such as THF or DMF
- ALK ALK 1 "ALK M ALK”
- lv cya noborohyclmre "X ALK M ALK”
- Figure 3 i. nucleophilic reaction between one of the -OH functions (the two others being protected by GP and GP 'denoting protecting groups) and a boc protected bromoamine having the formula Br-ALK-NHboc in the presence of a base such as eg. K 2 CO 3 in a polar solvent such as DMF, THF or MeCN (see for example the conditions on page 63 of WO 07085930).
- a base such as eg. K 2 CO 3 in a polar solvent such as DMF, THF or MeCN
- an intermediate complex is formed which is reduced in situ with a reducing agent such as, for example, sodium cyanoborohydride;
- a variant shown in Scheme 3 "consists in introducing the group -ALK-SSMe on the NHboc group, in particular taking inspiration from the method described by Kitagawa T. et al., JACS, 2006, 128 (45), 14448-14449. used to introduce acetylthioalkyl chains on a secondary amine:
- NHS-PEG-maleimide It may be more particularly the compound of N 0 CAS 756525-99-2.
- Figure 5 i. alkylation of the aromatic ring hydroxyl by a mono-protected piperazine at the 1-position and 4-bearing of an alkyl chain terminally functionalized by a nucleofug LG group.
- the nucleofuge group is a mesylate group and the Williamson reaction takes place in the presence of a hydride in an anhydrous aprotic polar solvent such as THF or DMF;
- nucleophilic substitution of bromine can be carried out
- LG and LG starter groups iv. introduction of LG and LG starter groups.
- mesylate group methanesulfonyl chloride is used in the presence of a base such as a tertiary amine (eg TEA).
- a base such as a tertiary amine (eg TEA).
- Compound P 6 can be for example the compound of CAS No. 564476-32-0, which was prepared according to WO 03068144 (see compound 10a in Figure 7) or the compound N 0 CAS 309916-91-4. Analogous compounds of different chain lengths can be prepared according to the same principle of Figure 7 of WO03068144 from the corresponding PEG compound. about Pi
- RbZb-CO-ALK- (OCH 2 CH 2 ) I can be prepared separately
- anhydrous aprotic polar solvent such as DCM
- mesyl chloride in the presence of a base such as TEA.
- an aprotic polar solvent such as acetone
- a sodium halide such as sodium iodide.
- hydrochloric acid eg, dioxane solution
- trifluoroacetic acid e.g, trifluoroacetic acid
- THF tetrahydropyran ether
- the preparation of this type of monoprotected PEG diol is well described in the literature, see e.g. Richard A. et al. Chem. Eur. J. 2005, 11, 7315-7321 or Sakellariou EG, et al. Tetrahedron 2003, 59, 9083-9090.
- the conjugate is obtained by the process of:
- step (ii) then optionally separating the conjugate formed in step (i) from the compound of formula (I) and / or the unreacted targeting agent and / or aggregates that would have formed.
- step (ii) the conjugate formed in step (i) of the unreacted targeting agent and any aggregates present in the solution are separated off.
- step (ii) the conjugate of step (i) is separated from only the unreacted compound of formula (I) and the aggregates that would have formed and left in the solution. the targeting agent who might not have reacted.
- a low reactive or insufficiently reactive chemical group can easily be converted into a more reactive group by one or more chemical reactions known to those skilled in the art.
- the aqueous solution of the targeting agent can be buffered using, for example, buffers such as, for example, potassium phosphate or N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES buffer).
- buffers such as, for example, potassium phosphate or N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES buffer).
- the buffer depends on the nature of the targeting agent.
- the compound of formula (I) is dissolved in a polar organic solvent, for example DMSO or DMA.
- the reaction takes place at a temperature generally between 20 and 40 ° C.
- the reaction time can vary between 1 to 24 hours.
- the reaction between the targeting agent and compound of formula (I) may be followed by SEC with a refractometric and / or ultraviolet detector to determine the progress thereof. If the degree of grafting is insufficient, one can let react longer and / or add compound of formula (I).
- the conjugate can be purified for example by steric exclusion chromatography (SEC), by adsorption chromatography (such as the exchange agent). ions, IEC), by hydrophobic interaction chromatography (HIC), by affinity chromatography, by chromatography on mixed media such as ceramic hydroxyapatite or by HPLC. Purification by dialysis or diafiltration can also be used.
- SEC steric exclusion chromatography
- IEC hydrophobic interaction chromatography
- HPLC hydrophobic interaction chromatography
- purification by dialysis or diafiltration can also be used.
- Aggregates means the associations that can be formed between two or more targeting agents, the targeting agents having been modified or not by conjugation. Aggregates are likely to form under the influence of a large number of parameters such as a high concentration of targeting agent in the solution, the pH of the solution, high shear forces, the number of grafted dimers and their hydrophobic nature, the temperature
- step (i) or (ii) the solution of the conjugate may undergo a step (iii) of ultrafiltration and / or diafiltration. At the end of these steps, the conjugate in aqueous solution is thus obtained.
- the antibody may be selected from those described especially in WO 04043344, WO 08010101, WO 08047242, WO 05009369 (anti-CA6).
- the antibody can be in particular monoclonal, polyclonal or multispecific. It can also be an antibody fragment. It can also be a murine, human, humanized or chimeric antibody. joint
- a conjugate generally comprises on the order of 1 to 10 dimer (s) of pyrrolo [1,4] benzodiazepine attached to the targeting agent (this is the level of grafting or "drug-to-antibody” ratio "or” DAR "in English). This number varies according to the nature of the targeting agent and the dimer as well as the operating conditions used for the conjugation (for example the number of dimer equivalents with respect to the targeting agent, the reaction time, the nature of the solvent and any cosolvent).
- Contacting the targeting agent with the dimer results in a mixture comprising: a plurality of conjugates individually distinguishable from each other by different DARs; optionally the unreacted targeting agent (in the case of an incomplete reaction); possibly aggregates.
- the DAR which is determined on the final solution for example by UV spectroscopy corresponds to an average DAR.
- UV spectroscopy may be a method used to determine the DAR. This method is inspired by that presented in Antony S. Dimitrov (ed), LLC, 2009, “Therapeutic Antibodies and Protocols," Vol. 525, 445, Springer Science. It consists of measuring the absorbance of a conjugate solution after the separation step (ii) at two wavelengths denoted LO1 and LO2. The following molar extinction coefficients of the naked antibody and the pyrrolo [1,4] benzodiazepine dimer measured prior to conjugation are used.
- ALOI (CD X e D LO 1) + (c A xe A L ⁇ 1)
- c D and c A respectively denote the concentrations in the solution of the part of the conjugate relative to the dimer of pyrrolo [1,4] benzodiazepine and the part of the conjugate relating to the antibody;
- • e D LO 1 and e DL o 2 respectively denote the molar absorption coefficients of the pyrrolo [1, 4] benzodiazepine dimer before conjugation at the two wavelengths LO1 and LO2; • e AL o 1 and e A ⁇ _o 2 respectively denote the molar absorption coefficients of the naked antibody at the two wavelengths LO1 and LO2.
- naked antibody is intended to mean the antibody to which no dimer of pyrrolo [1,4] benzodiazepine is attached, that is to say the antibody before the conjugation step.
- CD [( ⁇ A LOI x A LO 2) - ( ⁇ A LO 2 x A LO 1)] / [( ⁇ D LO 2 X e A LO 1) - ( ⁇ A LO 2 X e DL o 1)]
- CA [ALOI - (CD x D LO 1)] / ⁇ A LO 1
- the average DAR then corresponds to c D / c A.
- the average DAR measured on SEC spectrum is preferably between 1 and 10, preferably between 1, 5 and 7.
- the conjugate can be used as an anti-cancer agent. Because of the presence of the targeting agent, the conjugate is made very selective for tumor cells rather than healthy cells.
- the conjugate is formulated in the form of an aqueous buffered solution at a concentration generally of between 1 and 10 mg / ml. This solution can be injected as an infusion such that it can be rediluted to form an infusion solution.
- the spectra were made by chemical ionization (reactant gas: ammonia) on the WATERS GCTof (direct introduction without LC) apparatus.
- Deglycosylation is a technique of enzymatic digestion using glycosidase. It is made from 500 ⁇ l of conjugate + 100 ⁇ l of 50 mM Tris HCl buffer + 10 ⁇ l of glycanase-F enzyme (100 units of freeze-dried enzyme / 100 ⁇ l of water). The mixture is vortexed and kept overnight at 37 ° C. The deglycosylated sample is then ready to be analyzed in HRMS. Depending on the case, the HRMS analysis of the sample can also be performed without deglycosylation prior. In both cases, the mass spectra were obtained on a Waters Q-Tof-2 device in electrospray positive (ES +) mode.
- ES + electrospray positive
- a conjugate is prepared by reacting hu2H1 1 (also referred to as hu53 2H1 1 on page 15 of WO 2008010101, which is an antibody comprising a Vh having the amino acid sequence SED ID No. 24) and the 3 - (2- ⁇ 2- [2- (2- ⁇ 3- [3- (2 - ⁇ [2- (2,6-bis - [(S) -2-eth- (E) -ylidene-7- 1,2,3,11- ⁇ -tetrahydro-pyrrolo [2,1-c] [1,4] benzodiazepin-5-one-8-yloxymethyl] -pyridin-4-yloxy) -ethyl] -methyl-amino ⁇ N-Hydroxysuccinimidyl -1- (1-dimethyl-ethylsulfanyl) -2,5-dioxo-pyrrolidin-1-yl] -propanoylamino ⁇ ethoxy)
- the fractions of interest are pooled and concentrated on Amicon Ultra-15 (Ultracel 50k Millipore) and then filtered on Sephadex G-25 (NAP-5 and NAP-10 GE columns) previously equilibrated in an aqueous buffer of 10 mM concentration containing histidine. 10% sucrose and 5% NMP.
- the mixture obtained is stirred for 24 h at RT and then filtered through silica (Interchrom Puriflash Silica 15 / 35U 2G) using a gradient of 0 to 10% methanol in DCM.
- the fractions containing the desired product are combined, concentrated under RP and then purified by flash chromatography on silica (Interchrom Puriflash Silica 15 / 35U 2G) using a gradient of 0 to 10% MeOH in DCM.
- the fractions containing the desired product are combined and concentrated under RP.
- the mixture is then added 6 ml of ethanol, stirred for 20 min at RT and then added 124 mg of sodium cyanoborohydride. After stirring for 45 minutes, 124 mg of additional sodium cyanoborohydride are added and after 1 hour of stirring, the mixture is concentrated under RP, diluted in AcOEt and water. The resulting precipitate is filtered, dissolved in a 1M aqueous HCl solution. The aqueous phase obtained is brought to basic pH with a 5M aqueous sodium hydroxide solution, extracted three times with DCM and the combined organic phases are concentrated under RP.
- a conjugate is prepared by reacting hu2H1 1 and 3- ⁇ 2- [2- (2- ⁇ 2- [2- (2- ⁇ [2- (2,6-bis) ⁇ [(S) -2-Eth- (E) -ylidene-7-dimethoxy-1,2,3,1 1-tetrahydropyrrolo [2,1 c] [1,4] benzodiazepin-5-one-8-yloxymethyl N-hydroxysuccinimidyl] -pyridin-4-yloxy) -ethyl] -methylamino ⁇ -1,1-dimethylethylsulfanyl) acetylamino] -ethoxy-ethoxy-ethoxy-ethoxy-propanoate.
- a conjugate is prepared as for example 1 by reacting hu2H11 and 3- (2- ⁇ 2- [2- (2- ⁇ 2- [1- (3,5-bis - [(S)] 2-Eth- (E) -ylidene-7-dimethoxy-1,2,3,1a-tetrahydro-pyrrolo [2,1c] [1,4] benzodiazepin-5-one-8-yloxymethyl] -1- N-hydroxysuccinimidyl methyl-ethylsulfanyl] acetylamino-ethoxy-ethoxy-ethoxy-ethoxy-propanoate.
- a conjugate is prepared by reacting hu2H1 1 and 3- ⁇ 2- [2- (2- ⁇ 2- [2- (4- ⁇ 4- [2- (2,6- bis - [(S) -2-eth- (E) -ylidene-7-dimethoxy-1,2,3,1-a-tetrahydropyrrolo [2,1 c] [1,4] benzodiazepin-5-one 8-yloxymethyl] -pyridin-4-yloxy) -ethyl] -piperazin-1-yl ⁇ -1,1-dimethyl-4-oxo-butylsulfanyl) -acetylamino] -ethoxy ⁇ -ethoxy) -ethoxy] -ethoxy ⁇ N-hydroxysuccinimidyl propanoate.
- a conjugate is prepared by reacting hu2H1 1 and 3- ⁇ 2- [2- (2- ⁇ 2- [2- (2- ⁇ [2- (2,6-bis) ⁇ [(S) -2-Eth- (E) -ylidene-7-dimethoxy-1,2,3,1 1-tetrahydropyrrolo [2,1 c] [1,4] benzodiazepin-5-one-8 N -yloxymethyl] -pyridin-4-yloxy) -ethyl] -methyl-amino-1,1-dimethyl-ethylsulfanyl) -acetyl-methylamino-ethoxy-ethoxy-ethoxy-propanoate hydroxy succinimidyl.
- the MDA-MB-231, MDA-A1 or HCT1 16 cells in their exponential growth phase are trypsinized and resuspended in their respective culture medium (DMEM / F12 Gibco # 21331, 10% SVF Gibco # 10500-056, 2 nM Gibco glutamine # 25030 for MDA cells, DMEM Gibco # 1 1960, 10% SVF Gibco # 10500-056, 2 mM Glutamine Gibco # 25030 for HCT1 cells 16).
- the cell suspension is seeded into Cytostar 96-well culture plates (GE Healthcare Europe, # RPNQ0163) in the complete culture medium containing serum at a density of 5000 cells / well (MDA-MB-231, MDA-A1, HCT1 16).
- tomaymycin dimers are added to the triplicate wells for each concentration.
- the cells are cultured for 3 days at 37 ° C. in a 5% CO 2 atmosphere in the presence of cytotoxics.
- the 4 th day 10 .mu.l of a solution of thymidine 14 C (0.1 .mu.Ci / well, Perkin Elmer # NEC56825000) are added to each well.
- the incorporation of 14 C thymidine is measured 96 h after the start of the experiment with a radioactive microbeta counter (Perkin Elmer).
- the data are expressed as a% survival by making the ratio between the count obtained with the cells treated with the cytotoxic and that obtained with the cells of the controlled wells (treated with the culture medium alone).
- the MDA-MB-231 cells in their exponential growth phase are trypsinized and resuspended in their culture medium (DMEM / F12 Gibco # 21331, 10% Gibco SVF # 10500-056, 2 nM Gibco Glutamine # 25030).
- the cell suspension is seeded in Cytostar 96-well culture plates (GE Healthcare Europe, # RPNQ0163) in the complete culture medium containing serum at a density of 5000 cells / well. After 4 hours of incubation, successive dilutions of antibody-cytotoxic immunoconjugates are added to the wells at decreasing concentrations of 10 -7 to 10 -12 M (triplicate for each concentration).
- the cells are cultured at 37 ° C.
- noncleavable hu2H11 conjugates prepared with tomaymycin dimers described in Examples 3 and 5 of the international application WO 09016516 tend to have their monomeric purity decrease over time after several months of storage at 3-5 ° C.
- these conjugates formulated in a pH 6.5 aqueous buffer of 10 mM concentration in histidine containing 10% sucrose and 5% NMP, having an average of 3 to 3.5 dimer tomaymycin per molecule of antibody, of initial monomeric purity of the order of 97.5%, can lose 6 to 15% of monomeric purity in 6 to 8 months.
- C1 prepared with the tomaymycin dimer described in example 5 of the international application WO 09016516
- C2 conjugate of example 2
- the control groups were not treated.
- the doses of the two conjugates are given in ⁇ g of tomaymycin dimer / kg. They were administered at 80, 40, 20 and 10 ⁇ g / kg by intravenous (IV) bolus injection at day 13 for C1 and at day 24 for C2.
- the animals were weighed daily and the tumors measured twice a week using calipers.
- Animal body weight includes the weight of tumors.
- the efficacy parameters are ⁇ T / ⁇ C, mean percent regression, partial and complete regressions (RP and CR), and the number of tumor-free mice at the end of the study (SST).
- the evolution of tumor volume for each treated (T) and control (C) mouse is calculated for each tumor by subtracting the tumor volume at the start of the study to the tumor volume at the specified observation day. .
- the mean ⁇ T is calculated for the treated group and the average ⁇ C is calculated for the control group.
- a dose is considered to be therapeutically active for ⁇ T / ⁇ C less than 40% and very active for ⁇ T / ⁇ C less than 10%.
- the percentage of tumor regression is defined as the percentage decrease in tumor volume in the treated group at the specified day of observation compared to its volume at the first day of treatment. At a specific time and for each animal, the percentage of tumor regression is calculated. The average regression percentage is then calculated for the group.
- RP Partial regression
- mice Tumorless mice are defined as mice with no detectable tumor at the end of the study (beyond 100 days of treatment).
- DMT Maximum Tolerated Dose
- SST Tumor-free mouse at the end of the study
- C1 is very active and induces a weight loss of 9.6% at nadir, a ⁇ T / ⁇ C of 7% and 3 RP for 5 SCID mice. Antitumor activity was also observed at lower doses of 20 and 10 ⁇ g / kg without inducing RP.
- C2 is highly active and induces a weight loss of 5% in nadir, a tumor regression of 96.2% with 6 RP for 6 mice including 1 SST. It is also very active at the lower dose of 20 ⁇ g / kg with a tumor regression of 66.4% and 4 RP for 6 mice. Antitumor activity was also observed at the lowest dose of 10 ⁇ g / kg without inducing regression or PR.
- C2 has a high anticancer activity and has shown better activity than C1 with tumor regression, RP and SST at the DMT, not observed with C1 at the same doses.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description

































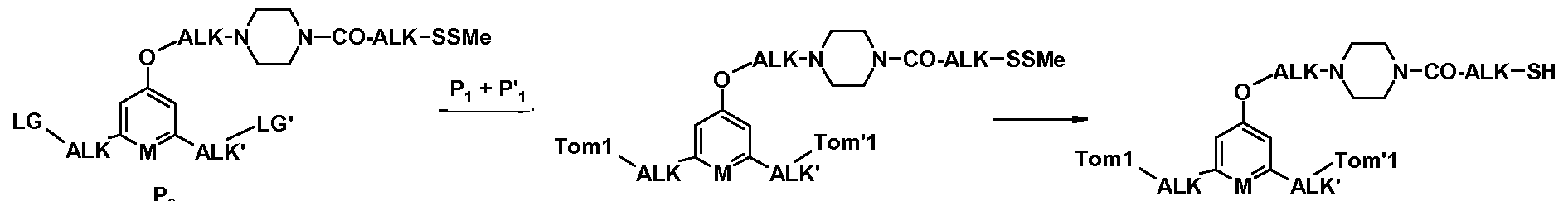



















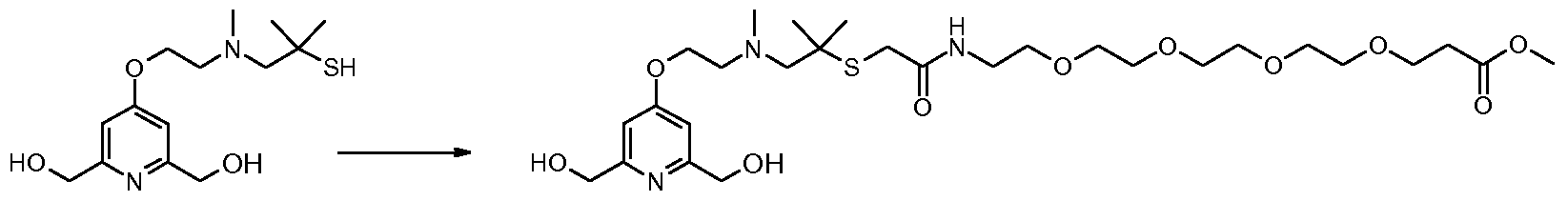


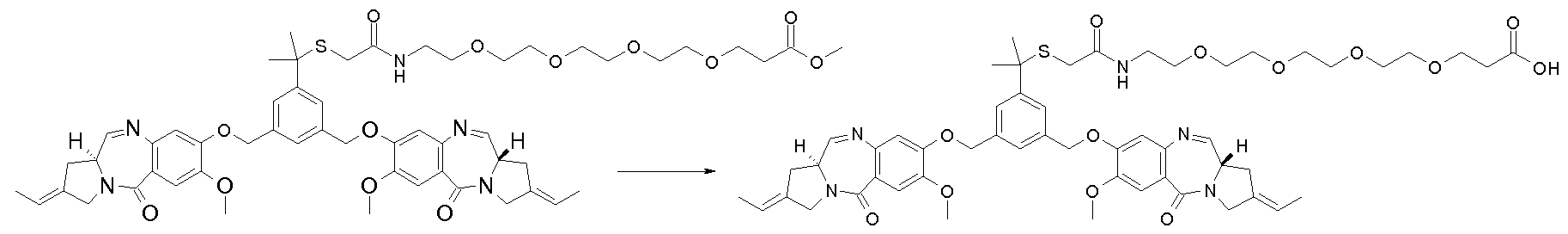



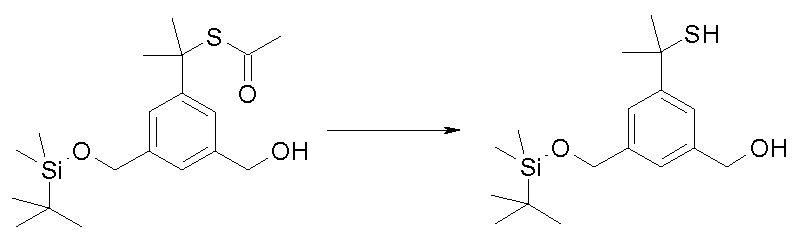


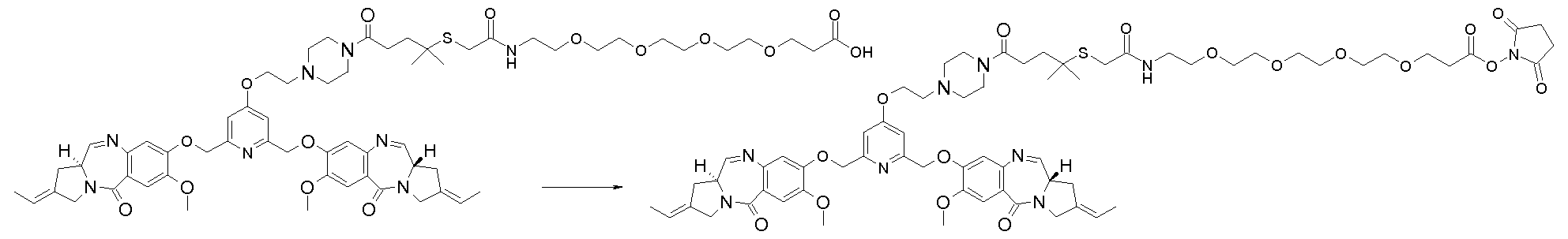






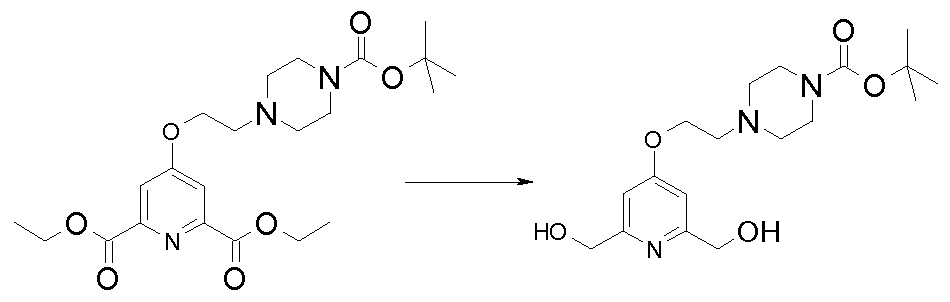













Claims

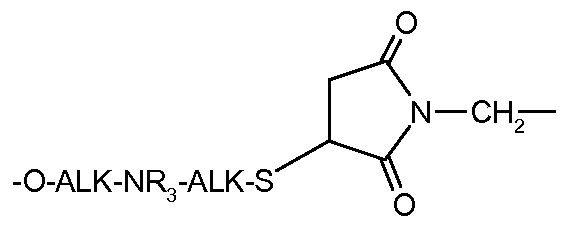
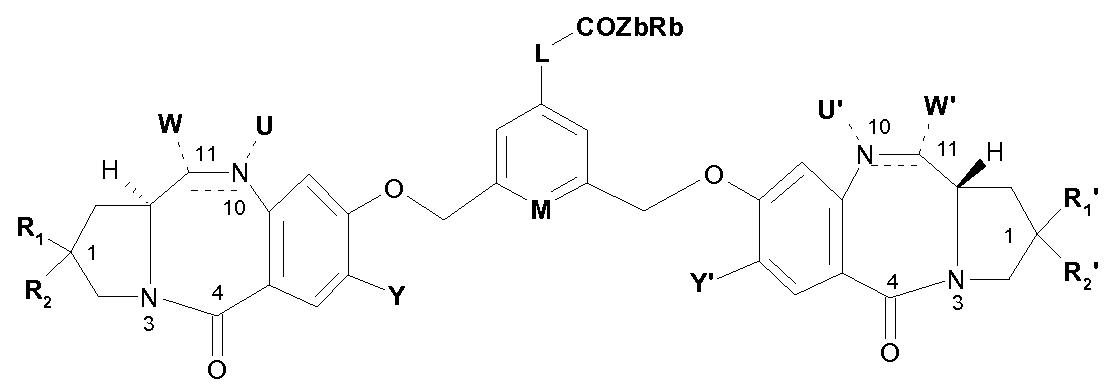
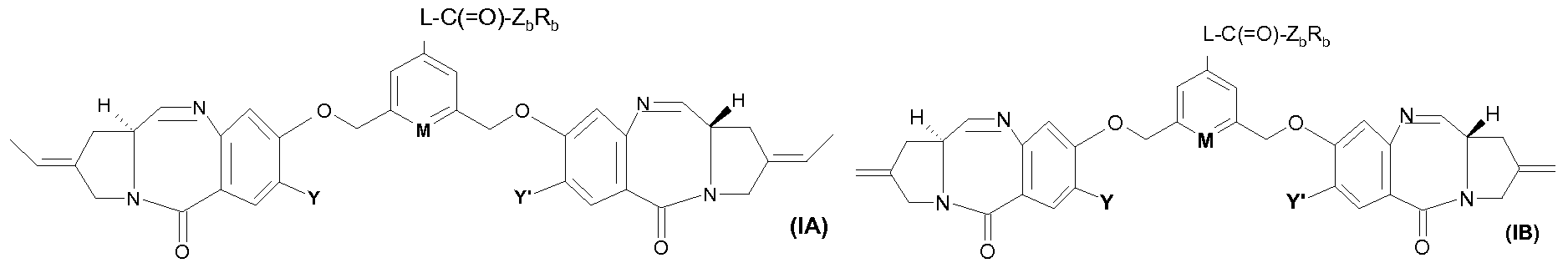
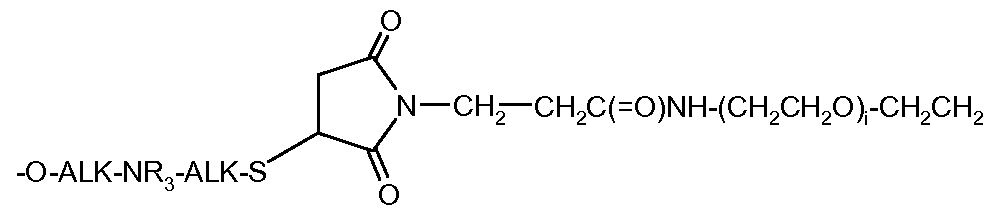











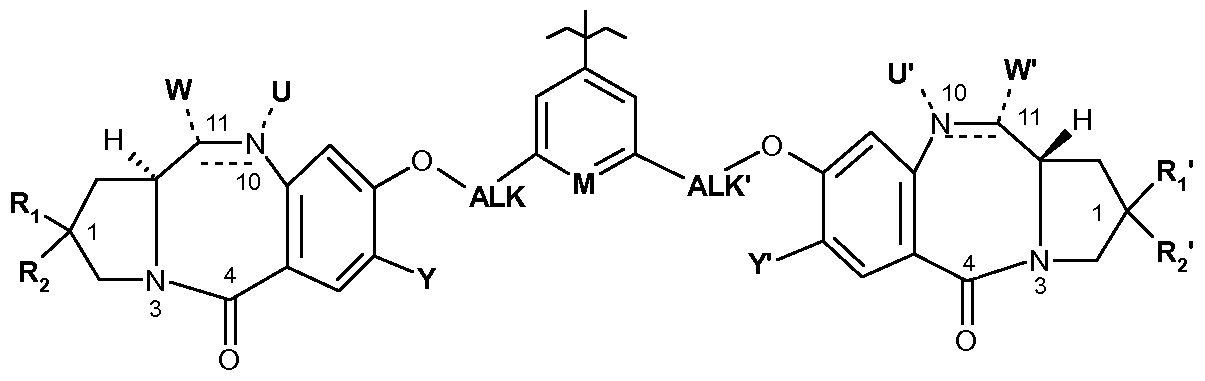


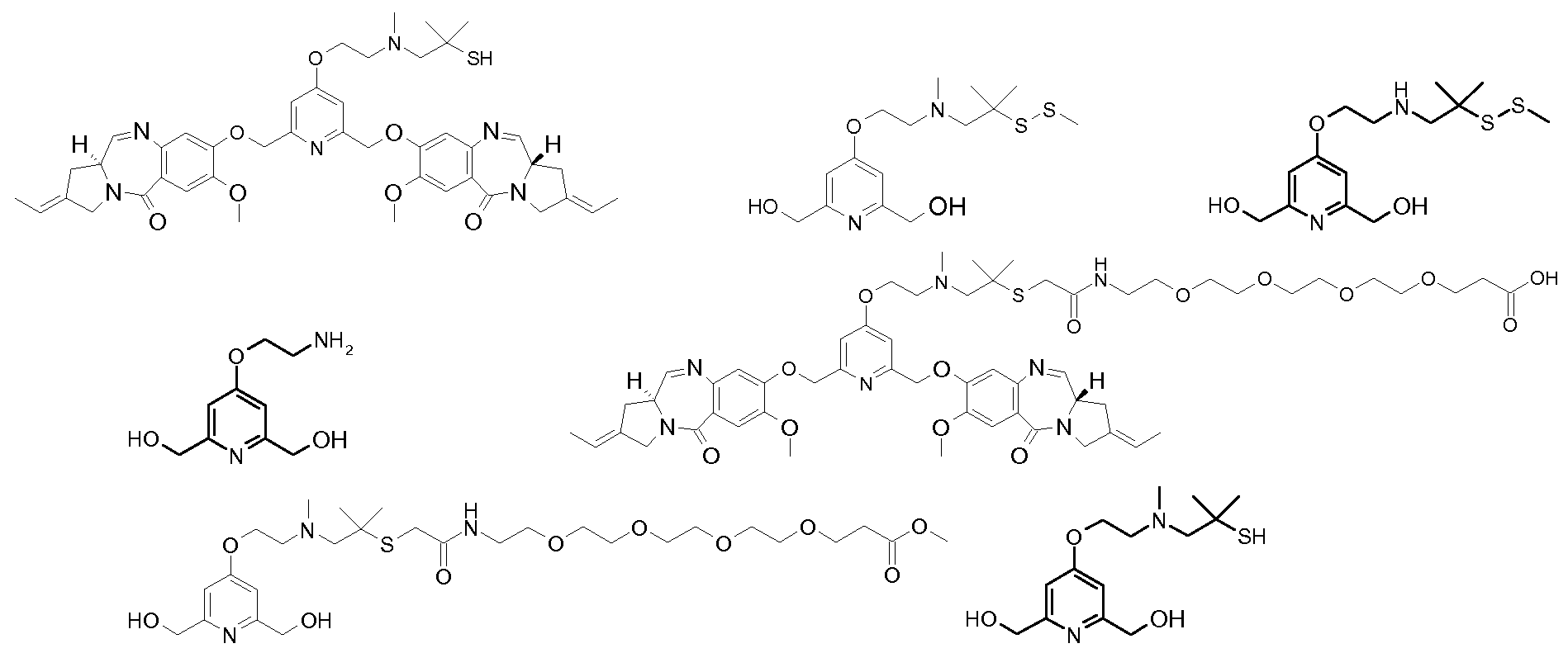

Priority Applications (19)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EA201270308A EA026488B1 (ru) | 2009-08-25 | 2010-08-12 | Конъюгаты димеров пирроло[1,4]бензодиазепина в качестве противоракового средства |
| KR1020127007518A KR20130026405A (ko) | 2009-08-25 | 2010-08-12 | 항암제로서의 피롤로〔1,4〕벤조디아제핀 이량체의 콘쥬게이트 |
| CA2772102A CA2772102C (fr) | 2009-08-25 | 2010-08-12 | Conjugues de dimeres de pyrrolo[1,4] benzodiazepine en tant qu'anticancereux |
| BR112012004146A BR112012004146A2 (pt) | 2009-08-25 | 2010-08-12 | "conjungados de dímeros de pirrol[1,4]benzodiazepina como agentes anticâncer" |
| AU2010288382A AU2010288382B2 (en) | 2009-08-25 | 2010-08-12 | Conjugates of pyrrolo[1,4]benzodiazepine dimers as anticancer agents |
| JP2012526097A JP5889189B2 (ja) | 2009-08-25 | 2010-08-12 | 抗癌剤としてのピロロ[1,4]ベンゾジアゼピン二量体のコンジュゲート |
| NZ598401A NZ598401A (en) | 2009-08-25 | 2010-08-12 | Conjugates of pyrrolo[1,4]benzodiazepine dimers as anticancer agents |
| EP10761055.2A EP2470547B1 (fr) | 2009-08-25 | 2010-08-12 | Conjugues de dimeres de pyrrolo[1,4]benzodiazepine en tant qu'anticancereux |
| SG2012012431A SG178537A1 (en) | 2009-08-25 | 2010-08-12 | Conjugates of pyrrolo[1,4]benzodiazepine dimers as anticancer agents |
| UAA201203443A UA106507C2 (uk) | 2009-08-25 | 2010-08-12 | Кон'югати димерів піроло[1,4]бензодіазепіну як протираковий засіб |
| CN201080048398.6A CN102753559B (zh) | 2009-08-25 | 2010-08-12 | 作为抗癌剂的吡咯并[1,4]苯并二氮杂*二聚物的共轭物 |
| MX2012002434A MX2012002434A (es) | 2009-08-25 | 2010-08-12 | Conjugados de dimeros de pirrolo[1,4] benzodiazepina como agentes anticacerosos. |
| ES10761055.2T ES2484690T3 (es) | 2009-08-25 | 2010-08-12 | Conjugados de dímeros de pirrolo[1,4]benzodiazepina como agentes anticancerígenos |
| TNP2012000053A TN2012000053A1 (fr) | 2009-08-25 | 2012-02-03 | Conjugues de dimeres de pyrrolo [1,4] benzodiazepine en tant qu'anticancereux |
| IL218262A IL218262A0 (en) | 2009-08-25 | 2012-02-22 | Conjugates of pyrrolo [1,4]benzodiazepine dimers as anticancer agents |
| ZA2012/01397A ZA201201397B (en) | 2009-08-25 | 2012-02-24 | Conjugates of pyrrolo[1,4]benzodiazepine dimers as anticancer agents |
| US13/404,872 US8481042B2 (en) | 2009-08-25 | 2012-02-24 | Conjugates of pyrrolo[1,4]benzodiazepine dimers as anticancer agents |
| MA34711A MA33600B1 (fr) | 2009-08-25 | 2012-03-21 | Conjugues de dimeres de pyrrolo [1, 4 ] benzodiazepine en tant qu' anticancereux |
| US13/923,544 US20140155590A1 (en) | 2009-08-25 | 2013-06-21 | Conjugates of Pyrrolo[1,4]Benzodiazepine Dimers As Anticancer Agents |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0904043 | 2009-08-25 | ||
| FR0904043A FR2949469A1 (fr) | 2009-08-25 | 2009-08-25 | Derives anticancereux, leur preparation et leur application en therapeutique |
| FR0904368A FR2949470B1 (fr) | 2009-08-25 | 2009-09-11 | Derives anticancereux, leur preparation et leur application therapeutique |
| FR0904368 | 2009-09-11 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/404,872 Continuation US8481042B2 (en) | 2009-08-25 | 2012-02-24 | Conjugates of pyrrolo[1,4]benzodiazepine dimers as anticancer agents |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011023883A1 true WO2011023883A1 (fr) | 2011-03-03 |
Family
ID=41720559
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR2010/051709 WO2011023883A1 (fr) | 2009-08-25 | 2010-08-12 | Conjugues de dimeres de pyrrolo [1, 4 ] benzodiazepine en tant qu' anticancereux |
Country Status (33)
| Country | Link |
|---|---|
| US (2) | US8481042B2 (fr) |
| EP (1) | EP2470547B1 (fr) |
| JP (1) | JP5889189B2 (fr) |
| KR (1) | KR20130026405A (fr) |
| CN (1) | CN102753559B (fr) |
| AR (1) | AR078143A1 (fr) |
| AU (1) | AU2010288382B2 (fr) |
| BR (1) | BR112012004146A2 (fr) |
| CA (1) | CA2772102C (fr) |
| CL (2) | CL2012000489A1 (fr) |
| CO (1) | CO6612210A2 (fr) |
| CR (1) | CR20120085A (fr) |
| DO (1) | DOP2012000038A (fr) |
| EA (1) | EA026488B1 (fr) |
| EC (1) | ECSP12011688A (fr) |
| ES (1) | ES2484690T3 (fr) |
| FR (2) | FR2949469A1 (fr) |
| GT (1) | GT201200046A (fr) |
| HN (1) | HN2012000437A (fr) |
| IL (1) | IL218262A0 (fr) |
| MA (1) | MA33600B1 (fr) |
| MX (1) | MX2012002434A (fr) |
| MY (1) | MY157469A (fr) |
| NI (1) | NI201200031A (fr) |
| NZ (1) | NZ598401A (fr) |
| PE (1) | PE20120992A1 (fr) |
| SG (1) | SG178537A1 (fr) |
| TN (1) | TN2012000053A1 (fr) |
| TW (1) | TWI477275B (fr) |
| UA (1) | UA106507C2 (fr) |
| UY (1) | UY32854A (fr) |
| WO (1) | WO2011023883A1 (fr) |
| ZA (1) | ZA201201397B (fr) |
Cited By (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013173337A2 (fr) | 2012-05-15 | 2013-11-21 | Seattle Genetics, Inc. | Conjugués de lieurs à auto-stabilisation |
| US8697688B2 (en) | 2010-04-15 | 2014-04-15 | Seattle Genetics Inc. | Pyrrolobenzodiazepines used to treat proliferative diseases |
| WO2014159981A3 (fr) * | 2013-03-13 | 2015-04-09 | Spirogen Sarl | Pyrrolobenzodiazépines et leurs conjugués |
| WO2015057699A2 (fr) | 2013-10-15 | 2015-04-23 | Seattle Genetics, Inc. | Lieurs de médicaments pégylés pour pharmacocinétique de conjugués ligand-médicament améliorée |
| WO2015123265A1 (fr) | 2014-02-11 | 2015-08-20 | Seattle Genetics, Inc. | Réduction sélective de protéines |
| WO2016008392A1 (fr) | 2014-07-16 | 2016-01-21 | Medshine Discovery Inc. | Lieurs et leur application à des conjugués anticorps-médicaments (adc) |
| US9242013B2 (en) | 2010-04-15 | 2016-01-26 | Seattle Genetics Inc. | Targeted pyrrolobenzodiazapine conjugates |
| WO2016044396A1 (fr) * | 2014-09-17 | 2016-03-24 | Genentech, Inc. | Immunoconjugués comprenant des anticorps anti-her2 et des pyrrolobenzodiazépines |
| US9388187B2 (en) | 2011-10-14 | 2016-07-12 | Medimmune Limited | Pyrrolobenzodiazepines |
| US9387259B2 (en) | 2011-10-14 | 2016-07-12 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| US9399641B2 (en) | 2011-09-20 | 2016-07-26 | Medimmune Limited | Pyrrolobenzodiazepines as unsymmetrical dimeric PBD compounds for inclusion in targeted conjugates |
| US9399073B2 (en) | 2011-10-14 | 2016-07-26 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines |
| US9415117B2 (en) | 2012-10-12 | 2016-08-16 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| US9427478B2 (en) | 2013-06-21 | 2016-08-30 | Innate Pharma | Enzymatic conjugation of polypeptides |
| US9504756B2 (en) | 2012-05-15 | 2016-11-29 | Seattle Genetics, Inc. | Self-stabilizing linker conjugates |
| US9526798B2 (en) | 2011-10-14 | 2016-12-27 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| US9649390B2 (en) | 2013-03-13 | 2017-05-16 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| US9717803B2 (en) | 2011-12-23 | 2017-08-01 | Innate Pharma | Enzymatic conjugation of polypeptides |
| US9745303B2 (en) | 2012-10-12 | 2017-08-29 | Medimmune Limited | Synthesis and intermediates of pyrrolobenzodiazepine derivatives for conjugation |
| US9919056B2 (en) | 2012-10-12 | 2018-03-20 | Adc Therapeutics S.A. | Pyrrolobenzodiazepine-anti-CD22 antibody conjugates |
| US9931414B2 (en) | 2012-10-12 | 2018-04-03 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US9931415B2 (en) | 2012-10-12 | 2018-04-03 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US9950078B2 (en) | 2013-10-11 | 2018-04-24 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US9956299B2 (en) | 2013-10-11 | 2018-05-01 | Medimmune Limited | Pyrrolobenzodiazepine—antibody conjugates |
| US9956298B2 (en) | 2013-10-11 | 2018-05-01 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| US10010624B2 (en) | 2013-10-11 | 2018-07-03 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US10036010B2 (en) | 2012-11-09 | 2018-07-31 | Innate Pharma | Recognition tags for TGase-mediated conjugation |
| WO2018138032A2 (fr) | 2017-01-24 | 2018-08-02 | Innate Pharma | Agents de liaison nkp46 |
| WO2018141959A1 (fr) | 2017-02-06 | 2018-08-09 | Innate Pharma | Conjugués médicament anticorps immunomodulateurs se liant à un polypeptide mica humain |
| US10071169B2 (en) | 2013-06-20 | 2018-09-11 | Innate Pharma | Enzymatic conjugation of polypeptides |
| US10132799B2 (en) | 2012-07-13 | 2018-11-20 | Innate Pharma | Screening of conjugated antibodies |
| EP3441072A1 (fr) | 2013-03-13 | 2019-02-13 | Seattle Genetics, Inc. | Filtration par charbon actif pour la purification d'adc de benzodiazépine |
| WO2019092148A1 (fr) | 2017-11-10 | 2019-05-16 | Innate Pharma | Anticorps avec des résidus de glutamine fonctionnalisés |
| US10335497B2 (en) | 2012-10-12 | 2019-07-02 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| US10392393B2 (en) | 2016-01-26 | 2019-08-27 | Medimmune Limited | Pyrrolobenzodiazepines |
| US10420777B2 (en) | 2014-09-12 | 2019-09-24 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| US10543279B2 (en) | 2016-04-29 | 2020-01-28 | Medimmune Limited | Pyrrolobenzodiazepine conjugates and their use for the treatment of cancer |
| US10544223B2 (en) | 2017-04-20 | 2020-01-28 | Adc Therapeutics Sa | Combination therapy with an anti-axl antibody-drug conjugate |
| US10611824B2 (en) | 2013-03-15 | 2020-04-07 | Innate Pharma | Solid phase TGase-mediated conjugation of antibodies |
| US10695439B2 (en) | 2016-02-10 | 2020-06-30 | Medimmune Limited | Pyrrolobenzodiazepine conjugates |
| US10695433B2 (en) | 2012-10-12 | 2020-06-30 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US10736903B2 (en) | 2012-10-12 | 2020-08-11 | Medimmune Limited | Pyrrolobenzodiazepine-anti-PSMA antibody conjugates |
| US10751346B2 (en) | 2012-10-12 | 2020-08-25 | Medimmune Limited | Pyrrolobenzodiazepine—anti-PSMA antibody conjugates |
| US10780096B2 (en) | 2014-11-25 | 2020-09-22 | Adc Therapeutics Sa | Pyrrolobenzodiazepine-antibody conjugates |
| US10799595B2 (en) | 2016-10-14 | 2020-10-13 | Medimmune Limited | Pyrrolobenzodiazepine conjugates |
| CN112119082A (zh) * | 2018-04-12 | 2020-12-22 | 免疫医疗有限公司 | 吡咯并苯并二氮杂*及其缀合物 |
| WO2021060439A1 (fr) | 2019-09-26 | 2021-04-01 | 日油株式会社 | Polyéthylène glycol monodispersé hétérobifonctionnel ayant un lieur peptidique |
| US11059893B2 (en) | 2015-04-15 | 2021-07-13 | Bergenbio Asa | Humanized anti-AXL antibodies |
| WO2021207701A1 (fr) | 2020-04-10 | 2021-10-14 | Seagen Inc. | Lieurs de variants de charge |
| US11160872B2 (en) | 2017-02-08 | 2021-11-02 | Adc Therapeutics Sa | Pyrrolobenzodiazepine-antibody conjugates |
| US11318211B2 (en) | 2017-06-14 | 2022-05-03 | Adc Therapeutics Sa | Dosage regimes for the administration of an anti-CD19 ADC |
| WO2022112356A1 (fr) | 2020-11-25 | 2022-06-02 | Innate Pharma | Traitement du cancer |
| US11352324B2 (en) | 2018-03-01 | 2022-06-07 | Medimmune Limited | Methods |
| US11370801B2 (en) | 2017-04-18 | 2022-06-28 | Medimmune Limited | Pyrrolobenzodiazepine conjugates |
| WO2022198231A1 (fr) | 2021-03-18 | 2022-09-22 | Seagen Inc. | Libération sélective de médicament à partir de conjugués internalisés de composés biologiquement actifs |
| WO2022198232A1 (fr) | 2021-03-18 | 2022-09-22 | Seagen Inc. | Libération sélective de médicament à partir de conjugués internalisés de composés biologiquement actifs |
| US11517626B2 (en) | 2016-02-10 | 2022-12-06 | Medimmune Limited | Pyrrolobenzodiazepine antibody conjugates |
| US11541128B2 (en) | 2016-12-14 | 2023-01-03 | Seagen Inc. | Multi-drug antibody drug conjugates |
| US11583590B2 (en) | 2017-09-29 | 2023-02-21 | Daiichi Sankyo Company, Limited | Antibody-pyrrolobenzodiazepine derivative conjugate and method of use thereof for treating a tumor |
| US11612665B2 (en) | 2017-02-08 | 2023-03-28 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US11649250B2 (en) | 2017-08-18 | 2023-05-16 | Medimmune Limited | Pyrrolobenzodiazepine conjugates |
| US11702473B2 (en) | 2015-04-15 | 2023-07-18 | Medimmune Limited | Site-specific antibody-drug conjugates |
| EP4212181A2 (fr) | 2013-12-19 | 2023-07-19 | Seagen Inc. | Liants à base de carbamate de méthylène à utiliser avec des conjugués de médicaments ciblés |
| US11730822B2 (en) | 2017-03-24 | 2023-08-22 | Seagen Inc. | Process for the preparation of glucuronide drug-linkers and intermediates thereof |
| WO2023227660A1 (fr) | 2022-05-25 | 2023-11-30 | Innate Pharma | Agents de liaison à la nectine-4 |
| US11844839B2 (en) | 2016-03-25 | 2023-12-19 | Seagen Inc. | Process for the preparation of pegylated drug-linkers and intermediates thereof |
| US11944689B2 (en) | 2016-08-09 | 2024-04-02 | Seagen Inc. | Drug conjugates with self-stabilizing linkers having improved physiochemical properties |
| WO2024129756A1 (fr) | 2022-12-13 | 2024-06-20 | Seagen Inc. | Conjugués anticorps-médicament modifiés par la cystéine spécifiques d'un site |
| US12121590B2 (en) | 2023-06-06 | 2024-10-22 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT3191135T (pt) | 2014-09-12 | 2020-11-12 | Genentech Inc | Anticorpos anti-her2 e imunoconjugados |
| CN107231804B (zh) | 2015-01-14 | 2019-11-26 | 百时美施贵宝公司 | 亚杂芳基桥连苯并二氮杂*二聚体、其缀合物及制备和使用方法 |
| CA2973354A1 (fr) | 2015-01-14 | 2016-07-21 | Bristol-Myers Squibb Company | Dimeres de benzodiazepine, conjugues de ceux-ci, et procedes de fabrication et d'utilisation |
| EP3313854A1 (fr) | 2015-06-23 | 2018-05-02 | Bristol-Myers Squibb Company | Formes macrocycliques dimères des benzodiazépines, leurs conjugués, préparation et utilisations |
| MA43345A (fr) | 2015-10-02 | 2018-08-08 | Hoffmann La Roche | Conjugués anticorps-médicaments de pyrrolobenzodiazépine et méthodes d'utilisation |
| CN117875311B (zh) * | 2024-01-11 | 2024-06-21 | 北京领初医药科技有限公司 | 一种缩略语句匹配方法、装置及存储介质 |
Citations (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1516743A (fr) | 1966-04-01 | 1968-02-05 | Rhone Poulenc Sa | Nouvel antibiotique et son procédé de préparation par culture de streptomyces croceus |
| WO2000012507A2 (fr) | 1998-08-27 | 2000-03-09 | Spirogen Limited | Composes |
| WO2000012508A2 (fr) | 1998-08-27 | 2000-03-09 | Spirogen Limited | Composes |
| WO2003068144A2 (fr) | 2001-12-21 | 2003-08-21 | Immunogen, Inc. | Agents cytotoxiques supportant une fraction reactive de polyethyleneglycol, conjugues cytotoxiques comprenant des groupes de liaison de polyethyleneglycol et leurs procedes de fabrication et d'utilisation |
| WO2004043344A2 (fr) | 2002-11-07 | 2004-05-27 | Immunogen, Inc. | Anticorps anti-cd33 et methode de traitement de la leucemie myeloide aigue a l'aide dudit anticorps |
| WO2004103272A2 (fr) | 2003-05-20 | 2004-12-02 | Immunogen, Inc. | Agents cytotoxiques ameliores contenant de nouveaux maytansinoides |
| WO2005009369A2 (fr) | 2003-07-21 | 2005-02-03 | Immunogen, Inc. | Conjugue cytotoxique specifique de l'antigene ca6 et procedes d'utilisation |
| WO2005040170A2 (fr) | 2003-10-22 | 2005-05-06 | Government Of The United States Of America, Represented By The Secretary, Department Of Health And Human Services | Derives de pyrrolobenzodiazepine, compositions comprenant ces derives et methodes s'y rapportant |
| WO2005085260A1 (fr) | 2004-03-09 | 2005-09-15 | Spirogen Limited | Pyrrolobenzodiazepines |
| WO2006061258A2 (fr) | 2004-12-07 | 2006-06-15 | Aventis Pharma S.A. | Agents cytotoxiques comprenant de nouveaux taxanes |
| WO2006069246A2 (fr) | 2004-12-22 | 2006-06-29 | Ambrx, Inc. | Compositions contenant des acides amines non naturels et des polypeptides, procedes impliquant ces acides amines non naturels et polypeptides, et utilisations desdits acides amines non naturels et polypeptides |
| US7230101B1 (en) | 2002-08-28 | 2007-06-12 | Gpc Biotech, Inc. | Synthesis of methotrexate-containing heterodimeric molecules |
| WO2007085930A1 (fr) | 2006-01-25 | 2007-08-02 | Sanofi-Aventis | Agents cytotoxiques comprenant de nouveaux dérivés de tomaymycine et leurs applications thérapeutiques |
| WO2007102069A1 (fr) | 2006-03-07 | 2007-09-13 | Sanofi-Aventis | Promedicaments ameliores d'analogues de cc-1065 |
| WO2007127440A2 (fr) | 2006-04-27 | 2007-11-08 | Intezyne Technologies, Inc. | Poly(éthylène glycol) contenant des groupes amino-protecteurs acides-labiles et leurs utilisations |
| WO2007144709A2 (fr) | 2006-06-09 | 2007-12-21 | Sanofi-Aventis | DÉRIVÉS de LEPTOMYCINE |
| WO2008010101A2 (fr) | 2006-07-18 | 2008-01-24 | Sanofi-Aventis | Anticorps antagoniste destiné au traitement du cancer |
| WO2008047242A2 (fr) | 2006-10-19 | 2008-04-24 | Sanofi-Aventis | Nouveaux anticorps anti-cd38 pour le traitement du cancer |
| WO2009016516A2 (fr) | 2007-07-19 | 2009-02-05 | Sanofi-Aventis | Agents cytotoxiques comprenant de nouveaux dérivés de la tomaymycine et leur utilisation thérapeutique |
| WO2009026274A1 (fr) | 2007-08-22 | 2009-02-26 | Medarex, Inc. | Attachement spécifique d'un site de médicaments ou autres agents à des anticorps synthétisés par génie génétique avec des extensions c-terminales |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8307865D0 (en) * | 1983-03-22 | 1983-04-27 | Fujisawa Pharmaceutical Co | Benzimidazole derivatives |
| GB9205051D0 (en) * | 1992-03-09 | 1992-04-22 | Cancer Res Campaign Tech | Pyrrolobenzodiazepine derivatives,their preparation,and compositions containing them |
| KR19990029749A (ko) * | 1997-09-17 | 1999-04-26 | 미우라 아끼라 | 2가 반응성 수용성 고분자 유도체 및 이들을 함유하는 복합체 |
| ATE424414T1 (de) * | 2001-06-08 | 2009-03-15 | Sod Conseils Rech Applic | Chimere somatostatin-dopamin-analoga |
| PL1945647T3 (pl) * | 2005-11-08 | 2012-04-30 | Immunogen Inc | Procesy wytwarzania maytansinolu |
| EP2276506A4 (fr) * | 2008-04-30 | 2014-05-07 | Immunogen Inc | Conjugués puissants et coupleurs hydrophiles |
| RU2545080C2 (ru) * | 2009-02-05 | 2015-03-27 | Иммьюноджен, Инк. | Новые производные бензодиазепина |
| UY32913A (es) * | 2009-10-02 | 2011-04-29 | Sanofi Aventis | Nuevos maitansinoides y el uso de dichos maitansinoides para preparar conjugados con un anticuero |
| FR2963007B1 (fr) * | 2010-07-26 | 2013-04-05 | Sanofi Aventis | Derives anticancereux, leur preparation et leur application therapeutique |
-
2009
- 2009-08-25 FR FR0904043A patent/FR2949469A1/fr active Pending
- 2009-09-11 FR FR0904368A patent/FR2949470B1/fr not_active Expired - Fee Related
-
2010
- 2010-08-12 CA CA2772102A patent/CA2772102C/fr not_active Expired - Fee Related
- 2010-08-12 AU AU2010288382A patent/AU2010288382B2/en not_active Ceased
- 2010-08-12 JP JP2012526097A patent/JP5889189B2/ja active Active
- 2010-08-12 CN CN201080048398.6A patent/CN102753559B/zh active Active
- 2010-08-12 WO PCT/FR2010/051709 patent/WO2011023883A1/fr active Application Filing
- 2010-08-12 MX MX2012002434A patent/MX2012002434A/es active IP Right Grant
- 2010-08-12 NZ NZ598401A patent/NZ598401A/en not_active IP Right Cessation
- 2010-08-12 KR KR1020127007518A patent/KR20130026405A/ko not_active Application Discontinuation
- 2010-08-12 EP EP10761055.2A patent/EP2470547B1/fr active Active
- 2010-08-12 EA EA201270308A patent/EA026488B1/ru not_active IP Right Cessation
- 2010-08-12 UA UAA201203443A patent/UA106507C2/uk unknown
- 2010-08-12 PE PE2012000267A patent/PE20120992A1/es not_active Application Discontinuation
- 2010-08-12 MY MYPI2012000814A patent/MY157469A/en unknown
- 2010-08-12 SG SG2012012431A patent/SG178537A1/en unknown
- 2010-08-12 ES ES10761055.2T patent/ES2484690T3/es active Active
- 2010-08-12 BR BR112012004146A patent/BR112012004146A2/pt not_active Application Discontinuation
- 2010-08-23 AR ARP100103066A patent/AR078143A1/es unknown
- 2010-08-24 TW TW099128330A patent/TWI477275B/zh not_active IP Right Cessation
- 2010-08-24 UY UY0001032854A patent/UY32854A/es not_active Application Discontinuation
-
2012
- 2012-02-03 TN TNP2012000053A patent/TN2012000053A1/fr unknown
- 2012-02-08 DO DO2012000038A patent/DOP2012000038A/es unknown
- 2012-02-16 GT GT201200046A patent/GT201200046A/es unknown
- 2012-02-17 CR CR20120085A patent/CR20120085A/es unknown
- 2012-02-21 CO CO12029986A patent/CO6612210A2/es unknown
- 2012-02-22 IL IL218262A patent/IL218262A0/en unknown
- 2012-02-22 EC ECSP12011688 patent/ECSP12011688A/es unknown
- 2012-02-24 ZA ZA2012/01397A patent/ZA201201397B/en unknown
- 2012-02-24 NI NI201200031A patent/NI201200031A/es unknown
- 2012-02-24 CL CL2012000489A patent/CL2012000489A1/es unknown
- 2012-02-24 US US13/404,872 patent/US8481042B2/en active Active
- 2012-02-24 HN HN2012000437A patent/HN2012000437A/es unknown
- 2012-03-21 MA MA34711A patent/MA33600B1/fr unknown
-
2013
- 2013-06-05 CL CL2013001609A patent/CL2013001609A1/es unknown
- 2013-06-21 US US13/923,544 patent/US20140155590A1/en not_active Abandoned
Patent Citations (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1516743A (fr) | 1966-04-01 | 1968-02-05 | Rhone Poulenc Sa | Nouvel antibiotique et son procédé de préparation par culture de streptomyces croceus |
| WO2000012507A2 (fr) | 1998-08-27 | 2000-03-09 | Spirogen Limited | Composes |
| WO2000012508A2 (fr) | 1998-08-27 | 2000-03-09 | Spirogen Limited | Composes |
| WO2003068144A2 (fr) | 2001-12-21 | 2003-08-21 | Immunogen, Inc. | Agents cytotoxiques supportant une fraction reactive de polyethyleneglycol, conjugues cytotoxiques comprenant des groupes de liaison de polyethyleneglycol et leurs procedes de fabrication et d'utilisation |
| US7230101B1 (en) | 2002-08-28 | 2007-06-12 | Gpc Biotech, Inc. | Synthesis of methotrexate-containing heterodimeric molecules |
| WO2004043344A2 (fr) | 2002-11-07 | 2004-05-27 | Immunogen, Inc. | Anticorps anti-cd33 et methode de traitement de la leucemie myeloide aigue a l'aide dudit anticorps |
| WO2004103272A2 (fr) | 2003-05-20 | 2004-12-02 | Immunogen, Inc. | Agents cytotoxiques ameliores contenant de nouveaux maytansinoides |
| WO2005009369A2 (fr) | 2003-07-21 | 2005-02-03 | Immunogen, Inc. | Conjugue cytotoxique specifique de l'antigene ca6 et procedes d'utilisation |
| WO2005040170A2 (fr) | 2003-10-22 | 2005-05-06 | Government Of The United States Of America, Represented By The Secretary, Department Of Health And Human Services | Derives de pyrrolobenzodiazepine, compositions comprenant ces derives et methodes s'y rapportant |
| WO2005085260A1 (fr) | 2004-03-09 | 2005-09-15 | Spirogen Limited | Pyrrolobenzodiazepines |
| WO2006061258A2 (fr) | 2004-12-07 | 2006-06-15 | Aventis Pharma S.A. | Agents cytotoxiques comprenant de nouveaux taxanes |
| WO2006069246A2 (fr) | 2004-12-22 | 2006-06-29 | Ambrx, Inc. | Compositions contenant des acides amines non naturels et des polypeptides, procedes impliquant ces acides amines non naturels et polypeptides, et utilisations desdits acides amines non naturels et polypeptides |
| WO2007085930A1 (fr) | 2006-01-25 | 2007-08-02 | Sanofi-Aventis | Agents cytotoxiques comprenant de nouveaux dérivés de tomaymycine et leurs applications thérapeutiques |
| WO2007102069A1 (fr) | 2006-03-07 | 2007-09-13 | Sanofi-Aventis | Promedicaments ameliores d'analogues de cc-1065 |
| WO2007127440A2 (fr) | 2006-04-27 | 2007-11-08 | Intezyne Technologies, Inc. | Poly(éthylène glycol) contenant des groupes amino-protecteurs acides-labiles et leurs utilisations |
| WO2007144709A2 (fr) | 2006-06-09 | 2007-12-21 | Sanofi-Aventis | DÉRIVÉS de LEPTOMYCINE |
| WO2008010101A2 (fr) | 2006-07-18 | 2008-01-24 | Sanofi-Aventis | Anticorps antagoniste destiné au traitement du cancer |
| WO2008047242A2 (fr) | 2006-10-19 | 2008-04-24 | Sanofi-Aventis | Nouveaux anticorps anti-cd38 pour le traitement du cancer |
| WO2009016516A2 (fr) | 2007-07-19 | 2009-02-05 | Sanofi-Aventis | Agents cytotoxiques comprenant de nouveaux dérivés de la tomaymycine et leur utilisation thérapeutique |
| WO2009026274A1 (fr) | 2007-08-22 | 2009-02-26 | Medarex, Inc. | Attachement spécifique d'un site de médicaments ou autres agents à des anticorps synthétisés par génie génétique avec des extensions c-terminales |
Non-Patent Citations (30)
| Title |
|---|
| "Protein Aggregation and Bioprocessing", AAPS JOURNAL, vol. 8, no. 3, 2006, pages E572 - E579 |
| "Therapeutic Antibodies and Protocols", vol. 525, 2009, SPRINGER SCIENCE, pages: 445 |
| ANALYTICAL BIOCHEMISTRY, vol. 212, no. 2, 1993, pages 469 - 480 |
| CARTER P.J. ET AL.: "Antibody-drug conjugates for cancer therapy", CANCER J., vol. 14, 2008, pages 154 - 169 |
| CHARI R.: "Targeted cancer therapy: conferring specificity to cytotoxic drugs", ACC. CHEM. RES., vol. 41, 2008, pages 98 - 107 |
| CHIN J.W. ET AL., JACS, vol. 124, 2002, pages 9026 - 9027 |
| DE GRAAF A.J. ET AL., BIOCONJUGATE CHEM, 3 February 2009 (2009-02-03) |
| DYKES DJ; HOLLINGSHEAD M; SIMPSON-HERREN L; ALLEY MC: "Human tumor xenograft models in NCI drug development : Feibig HH BA", 1999, pages: 101 - 125 |
| EUR.J.MED.CHEM., vol. 40, 2005, pages 641 - 654 |
| G.T.CRISP; P.D.TURNER, TETRAHEDRON, vol. 56, no. 42, 2000, pages 8335 |
| GARNETT M.C. ET AL., ADVANCED DRUG DELIVERY REVIEWS, vol. 53, 2001, pages 171 - 216 |
| GREG T. HERMANSON: "Bioconjugate Techniques", ELSEVIER INC., pages: 721 |
| J. ANTIBIOTICS, vol. XXXVI, no. 3, 1983, pages 276 - 282 |
| J.MEMBRANE SCI., vol. 318, 2008, pages 311 - 316 |
| JACS, vol. 80, 1958, pages 4423 |
| JACS, vol. 82, 1960, pages 4596 |
| JANEWAY ET AL.: "Immunobiology", 2001, GARLAND PUBLISHING |
| JUNUTULA J.R. ET AL., NATURE BIOTECHNOLOGY, vol. 26, 2008, pages 925 - 932 |
| KITAGAWA T. ET AL., JACS, vol. 128, no. 45, 2006, pages 14448 - 14449 |
| LIEBIGS ANNALEN DER CHEMIE, vol. 10, 1991, pages 987 - 988 |
| MONNERET C. ET AL., BULLETIN DU CANCER, vol. 87, no. 11, 2000, pages 829 - 38 |
| MORI M. ET AL., TETRAHEDRON, vol. 42, 1986, pages 3793 - 3806 |
| RICART A.D. ET AL., NATURE CLINICAT PRACTICE ONCOLOGY, vol. 4, 2007, pages 245 - 255 |
| RICHARD A. ET AL., CHEM. EUR. J., vol. 11, 2005, pages 7315 - 7321 |
| SAKELLARIOU E.G. ET AL., TETRAHEDRON, vol. 59, 2003, pages 9083 - 9090 |
| SCRIMIN, P.; TECILLA, P.; TONELLATO, U.; VENDRAME, T., J. ORG. CHEM., vol. 54, 1989, pages 5988 |
| SINGH R.; RICKSON H.K., THERAPEUTIC ANTIBODIES : METHODS AND PROTOCOLS, vol. 525, 2009, pages 445 - 467 |
| TETRAHEDRON LETTERS, vol. 29, no. 40, 1988, pages 5105 - 5108 |
| TETRAHEDRON, vol. 61, no. 7, 2005, pages 1755 - 1763 |
| WESSJOHANN, L. ET AL., SYNTHESIS, vol. 5, 1989, pages 359 - 63 |
Cited By (108)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9242013B2 (en) | 2010-04-15 | 2016-01-26 | Seattle Genetics Inc. | Targeted pyrrolobenzodiazapine conjugates |
| US8697688B2 (en) | 2010-04-15 | 2014-04-15 | Seattle Genetics Inc. | Pyrrolobenzodiazepines used to treat proliferative diseases |
| US10561739B2 (en) | 2010-04-15 | 2020-02-18 | Seattle Genetics Inc. | Targeted pyrrolobenzodiazapine conjugates |
| US9732084B2 (en) | 2010-04-15 | 2017-08-15 | Medimmune Limited | Pyrrolobenzodiazepines used to treat proliferative diseases |
| US9592240B2 (en) | 2010-04-15 | 2017-03-14 | Seattle Genetics Inc. | Targeted pyrrolobenzodiazapine conjugates |
| US9399641B2 (en) | 2011-09-20 | 2016-07-26 | Medimmune Limited | Pyrrolobenzodiazepines as unsymmetrical dimeric PBD compounds for inclusion in targeted conjugates |
| US9707301B2 (en) | 2011-10-14 | 2017-07-18 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| US9526798B2 (en) | 2011-10-14 | 2016-12-27 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| US9388187B2 (en) | 2011-10-14 | 2016-07-12 | Medimmune Limited | Pyrrolobenzodiazepines |
| US9387259B2 (en) | 2011-10-14 | 2016-07-12 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| US10328084B2 (en) | 2011-10-14 | 2019-06-25 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| US9399073B2 (en) | 2011-10-14 | 2016-07-26 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines |
| US10329352B2 (en) | 2011-10-14 | 2019-06-25 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| US9713647B2 (en) | 2011-10-14 | 2017-07-25 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| US9764038B2 (en) | 2011-12-23 | 2017-09-19 | Innate Pharma | Enzymatic conjugation of antibodies |
| US9717803B2 (en) | 2011-12-23 | 2017-08-01 | Innate Pharma | Enzymatic conjugation of polypeptides |
| US10675359B2 (en) | 2011-12-23 | 2020-06-09 | Innate Pharma | Enzymatic conjugation of antibodies |
| WO2013173337A2 (fr) | 2012-05-15 | 2013-11-21 | Seattle Genetics, Inc. | Conjugués de lieurs à auto-stabilisation |
| US9504756B2 (en) | 2012-05-15 | 2016-11-29 | Seattle Genetics, Inc. | Self-stabilizing linker conjugates |
| US10132799B2 (en) | 2012-07-13 | 2018-11-20 | Innate Pharma | Screening of conjugated antibodies |
| US10646584B2 (en) | 2012-10-12 | 2020-05-12 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| US9931414B2 (en) | 2012-10-12 | 2018-04-03 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US9745303B2 (en) | 2012-10-12 | 2017-08-29 | Medimmune Limited | Synthesis and intermediates of pyrrolobenzodiazepine derivatives for conjugation |
| US10780181B2 (en) | 2012-10-12 | 2020-09-22 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US10722594B2 (en) | 2012-10-12 | 2020-07-28 | Adc Therapeutics S.A. | Pyrrolobenzodiazepine-anti-CD22 antibody conjugates |
| US10695433B2 (en) | 2012-10-12 | 2020-06-30 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US10751346B2 (en) | 2012-10-12 | 2020-08-25 | Medimmune Limited | Pyrrolobenzodiazepine—anti-PSMA antibody conjugates |
| US10736903B2 (en) | 2012-10-12 | 2020-08-11 | Medimmune Limited | Pyrrolobenzodiazepine-anti-PSMA antibody conjugates |
| US9919056B2 (en) | 2012-10-12 | 2018-03-20 | Adc Therapeutics S.A. | Pyrrolobenzodiazepine-anti-CD22 antibody conjugates |
| US11690918B2 (en) | 2012-10-12 | 2023-07-04 | Medimmune Limited | Pyrrolobenzodiazepine-anti-CD22 antibody conjugates |
| US9931415B2 (en) | 2012-10-12 | 2018-04-03 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US10994023B2 (en) | 2012-10-12 | 2021-05-04 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| US11701430B2 (en) | 2012-10-12 | 2023-07-18 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| US10335497B2 (en) | 2012-10-12 | 2019-07-02 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| US9415117B2 (en) | 2012-10-12 | 2016-08-16 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| US10799596B2 (en) | 2012-10-12 | 2020-10-13 | Adc Therapeutics S.A. | Pyrrolobenzodiazepine-antibody conjugates |
| US11779650B2 (en) | 2012-10-12 | 2023-10-10 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US11771775B2 (en) | 2012-10-12 | 2023-10-03 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US10036010B2 (en) | 2012-11-09 | 2018-07-31 | Innate Pharma | Recognition tags for TGase-mediated conjugation |
| US10576164B2 (en) | 2013-03-13 | 2020-03-03 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| AU2014229529B2 (en) * | 2013-03-13 | 2018-02-15 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| EP3441072A1 (fr) | 2013-03-13 | 2019-02-13 | Seattle Genetics, Inc. | Filtration par charbon actif pour la purification d'adc de benzodiazépine |
| US9649390B2 (en) | 2013-03-13 | 2017-05-16 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| EA027910B1 (ru) * | 2013-03-13 | 2017-09-29 | Медимьюн Лимитед | Пирролобензодиазепины и их конъюгаты |
| US9821074B2 (en) | 2013-03-13 | 2017-11-21 | Genentech, Inc. | Pyrrolobenzodiazepines and conjugates thereof |
| AU2014244245B2 (en) * | 2013-03-13 | 2017-12-07 | Genentech, Inc. | Pyrrolobenzodiazepines and conjugates thereof |
| WO2014159981A3 (fr) * | 2013-03-13 | 2015-04-09 | Spirogen Sarl | Pyrrolobenzodiazépines et leurs conjugués |
| AU2014244245C1 (en) * | 2013-03-13 | 2018-04-19 | Genentech, Inc. | Pyrrolobenzodiazepines and conjugates thereof |
| KR102057755B1 (ko) | 2013-03-13 | 2019-12-19 | 메디뮨 리미티드 | 피롤로벤조디아제핀 및 그의 컨쥬게이트 |
| US10611824B2 (en) | 2013-03-15 | 2020-04-07 | Innate Pharma | Solid phase TGase-mediated conjugation of antibodies |
| US10071169B2 (en) | 2013-06-20 | 2018-09-11 | Innate Pharma | Enzymatic conjugation of polypeptides |
| US10434180B2 (en) | 2013-06-21 | 2019-10-08 | Innate Pharma | Enzymatic conjugation of polypeptides |
| US9427478B2 (en) | 2013-06-21 | 2016-08-30 | Innate Pharma | Enzymatic conjugation of polypeptides |
| US9950078B2 (en) | 2013-10-11 | 2018-04-24 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US10010624B2 (en) | 2013-10-11 | 2018-07-03 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US9956299B2 (en) | 2013-10-11 | 2018-05-01 | Medimmune Limited | Pyrrolobenzodiazepine—antibody conjugates |
| US9956298B2 (en) | 2013-10-11 | 2018-05-01 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| WO2015057699A2 (fr) | 2013-10-15 | 2015-04-23 | Seattle Genetics, Inc. | Lieurs de médicaments pégylés pour pharmacocinétique de conjugués ligand-médicament améliorée |
| US11103593B2 (en) | 2013-10-15 | 2021-08-31 | Seagen Inc. | Pegylated drug-linkers for improved ligand-drug conjugate pharmacokinetics |
| EP3756663A1 (fr) | 2013-10-15 | 2020-12-30 | Seagen Inc. | Lieurs de médicaments pégylés pour pharmacocinétique de conjugués ligand-médicament améliorée |
| EP4212181A2 (fr) | 2013-12-19 | 2023-07-19 | Seagen Inc. | Liants à base de carbamate de méthylène à utiliser avec des conjugués de médicaments ciblés |
| EP4190809A1 (fr) | 2014-02-11 | 2023-06-07 | Seagen Inc. | Réduction sélective de protéines |
| US11667696B2 (en) | 2014-02-11 | 2023-06-06 | Seagen Inc. | Selective reduction of proteins |
| US10464997B2 (en) | 2014-02-11 | 2019-11-05 | Seattle Genetics, Inc. | Selective reduction of proteins |
| WO2015123265A1 (fr) | 2014-02-11 | 2015-08-20 | Seattle Genetics, Inc. | Réduction sélective de protéines |
| WO2016008392A1 (fr) | 2014-07-16 | 2016-01-21 | Medshine Discovery Inc. | Lieurs et leur application à des conjugués anticorps-médicaments (adc) |
| US10280229B2 (en) | 2014-07-16 | 2019-05-07 | Medshine Discovery Inc. | Linkers and their application towards ADC |
| US10420777B2 (en) | 2014-09-12 | 2019-09-24 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| WO2016044396A1 (fr) * | 2014-09-17 | 2016-03-24 | Genentech, Inc. | Immunoconjugués comprenant des anticorps anti-her2 et des pyrrolobenzodiazépines |
| RU2727663C2 (ru) * | 2014-09-17 | 2020-07-22 | Дженентек, Инк. | Иммуноконъюгаты, содержащие антитела против her2 и пирролбензодиазепины |
| US10314846B2 (en) | 2014-09-17 | 2019-06-11 | Genentech, Inc. | Immunoconjugates comprising anti-HER2 antibodies and pyrrolobenzodiazepines |
| US10780096B2 (en) | 2014-11-25 | 2020-09-22 | Adc Therapeutics Sa | Pyrrolobenzodiazepine-antibody conjugates |
| US11702473B2 (en) | 2015-04-15 | 2023-07-18 | Medimmune Limited | Site-specific antibody-drug conjugates |
| US11059893B2 (en) | 2015-04-15 | 2021-07-13 | Bergenbio Asa | Humanized anti-AXL antibodies |
| US10392393B2 (en) | 2016-01-26 | 2019-08-27 | Medimmune Limited | Pyrrolobenzodiazepines |
| US10695439B2 (en) | 2016-02-10 | 2020-06-30 | Medimmune Limited | Pyrrolobenzodiazepine conjugates |
| US11517626B2 (en) | 2016-02-10 | 2022-12-06 | Medimmune Limited | Pyrrolobenzodiazepine antibody conjugates |
| US11844839B2 (en) | 2016-03-25 | 2023-12-19 | Seagen Inc. | Process for the preparation of pegylated drug-linkers and intermediates thereof |
| US10543279B2 (en) | 2016-04-29 | 2020-01-28 | Medimmune Limited | Pyrrolobenzodiazepine conjugates and their use for the treatment of cancer |
| US11944689B2 (en) | 2016-08-09 | 2024-04-02 | Seagen Inc. | Drug conjugates with self-stabilizing linkers having improved physiochemical properties |
| US10799595B2 (en) | 2016-10-14 | 2020-10-13 | Medimmune Limited | Pyrrolobenzodiazepine conjugates |
| US11541128B2 (en) | 2016-12-14 | 2023-01-03 | Seagen Inc. | Multi-drug antibody drug conjugates |
| WO2018138032A2 (fr) | 2017-01-24 | 2018-08-02 | Innate Pharma | Agents de liaison nkp46 |
| WO2018141959A1 (fr) | 2017-02-06 | 2018-08-09 | Innate Pharma | Conjugués médicament anticorps immunomodulateurs se liant à un polypeptide mica humain |
| US11813335B2 (en) | 2017-02-08 | 2023-11-14 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US11612665B2 (en) | 2017-02-08 | 2023-03-28 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US11160872B2 (en) | 2017-02-08 | 2021-11-02 | Adc Therapeutics Sa | Pyrrolobenzodiazepine-antibody conjugates |
| US11730822B2 (en) | 2017-03-24 | 2023-08-22 | Seagen Inc. | Process for the preparation of glucuronide drug-linkers and intermediates thereof |
| US11370801B2 (en) | 2017-04-18 | 2022-06-28 | Medimmune Limited | Pyrrolobenzodiazepine conjugates |
| US10544223B2 (en) | 2017-04-20 | 2020-01-28 | Adc Therapeutics Sa | Combination therapy with an anti-axl antibody-drug conjugate |
| US11318211B2 (en) | 2017-06-14 | 2022-05-03 | Adc Therapeutics Sa | Dosage regimes for the administration of an anti-CD19 ADC |
| US11938192B2 (en) | 2017-06-14 | 2024-03-26 | Medimmune Limited | Dosage regimes for the administration of an anti-CD19 ADC |
| US11649250B2 (en) | 2017-08-18 | 2023-05-16 | Medimmune Limited | Pyrrolobenzodiazepine conjugates |
| US11628223B2 (en) | 2017-09-29 | 2023-04-18 | Daiichi Sankyo Company, Limited | Antibody-drug conjugates comprising substituted benzo[e]pyrrolo[1,2-α][1,4]diazepines |
| US11583590B2 (en) | 2017-09-29 | 2023-02-21 | Daiichi Sankyo Company, Limited | Antibody-pyrrolobenzodiazepine derivative conjugate and method of use thereof for treating a tumor |
| WO2019092148A1 (fr) | 2017-11-10 | 2019-05-16 | Innate Pharma | Anticorps avec des résidus de glutamine fonctionnalisés |
| US11352324B2 (en) | 2018-03-01 | 2022-06-07 | Medimmune Limited | Methods |
| CN112119082A (zh) * | 2018-04-12 | 2020-12-22 | 免疫医疗有限公司 | 吡咯并苯并二氮杂*及其缀合物 |
| US11524969B2 (en) | 2018-04-12 | 2022-12-13 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof as antitumour agents |
| WO2021060439A1 (fr) | 2019-09-26 | 2021-04-01 | 日油株式会社 | Polyéthylène glycol monodispersé hétérobifonctionnel ayant un lieur peptidique |
| WO2021207701A1 (fr) | 2020-04-10 | 2021-10-14 | Seagen Inc. | Lieurs de variants de charge |
| WO2022112356A1 (fr) | 2020-11-25 | 2022-06-02 | Innate Pharma | Traitement du cancer |
| WO2022198231A1 (fr) | 2021-03-18 | 2022-09-22 | Seagen Inc. | Libération sélective de médicament à partir de conjugués internalisés de composés biologiquement actifs |
| WO2022198232A1 (fr) | 2021-03-18 | 2022-09-22 | Seagen Inc. | Libération sélective de médicament à partir de conjugués internalisés de composés biologiquement actifs |
| US12036286B2 (en) | 2021-03-18 | 2024-07-16 | Seagen Inc. | Selective drug release from internalized conjugates of biologically active compounds |
| WO2023227660A1 (fr) | 2022-05-25 | 2023-11-30 | Innate Pharma | Agents de liaison à la nectine-4 |
| WO2024129756A1 (fr) | 2022-12-13 | 2024-06-20 | Seagen Inc. | Conjugués anticorps-médicament modifiés par la cystéine spécifiques d'un site |
| US12121590B2 (en) | 2023-06-06 | 2024-10-22 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2470547B1 (fr) | Conjugues de dimeres de pyrrolo[1,4]benzodiazepine en tant qu'anticancereux | |
| KR101428112B1 (ko) | 신규 토메이마이신 유도체를 포함하는 세포독성제 및 그의 치료적 용도 | |
| FR2963007A1 (fr) | Derives anticancereux, leur preparation et leur application therapeutique | |
| JP5551070B2 (ja) | 新規トマイマイシン誘導体を含む細胞毒性剤およびこれらの治療的使用 | |
| CA2712629C (fr) | Derives de carboxamides n-azabicycliques, leur preparation et leur application en therapeutique | |
| WO2011001052A1 (fr) | Nouveaux conjugues, leur preparation et leur application en therapeutique | |
| US20230310640A1 (en) | Pyrrolobenzodiazepine derivative and ligand-linker conjugate thereof | |
| PT1680425E (pt) | 6 - [fenil (substituído)] triazolopirimidinas como agentes contra o cancro. | |
| EP3649214B1 (fr) | Complexes de terbium et de dysprosium avec antenne optimisée, utiles comme marqueurs luminescents | |
| WO2022262671A1 (fr) | Composé macro hétérocyclique et son utilisation médicale | |
| CN115028648A (zh) | 三并环化合物及其药物组合物和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080048398.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10761055 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 415/MUMNP/2012 Country of ref document: IN Ref document number: CR2012-000085 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2012000131 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12029986 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2772102 Country of ref document: CA Ref document number: 218262 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 0151112 Country of ref document: KE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12012500391 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012526097 Country of ref document: JP Ref document number: MX/A/2012/002434 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1201000764 Country of ref document: TH |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010288382 Country of ref document: AU Ref document number: 000267-2012 Country of ref document: PE |
|
| ENP | Entry into the national phase |
Ref document number: 2010288382 Country of ref document: AU Date of ref document: 20100812 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010761055 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201203443 Country of ref document: UA |
|
| ENP | Entry into the national phase |
Ref document number: 20127007518 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201270308 Country of ref document: EA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012004146 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013001609 Country of ref document: CL |
|
| ENP | Entry into the national phase |
Ref document number: 112012004146 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120224 |