WO2010053052A1 - 2-デオキシ-シロ-イノソース(doi)生産細菌及びこれを用いた2-デオキシ-シロ-イノソース(doi)生産方法 - Google Patents
2-デオキシ-シロ-イノソース(doi)生産細菌及びこれを用いた2-デオキシ-シロ-イノソース(doi)生産方法 Download PDFInfo
- Publication number
- WO2010053052A1 WO2010053052A1 PCT/JP2009/068666 JP2009068666W WO2010053052A1 WO 2010053052 A1 WO2010053052 A1 WO 2010053052A1 JP 2009068666 W JP2009068666 W JP 2009068666W WO 2010053052 A1 WO2010053052 A1 WO 2010053052A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- doi
- escherichia coli
- gene
- producing
- sucrose
- Prior art date
Links
- UJZOBRIPCQJAEY-UHFFFAOYSA-N CC(C1C2C1CC1C2)C1=[N](C)(C)=C Chemical compound CC(C1C2C1CC1C2)C1=[N](C)(C)=C UJZOBRIPCQJAEY-UHFFFAOYSA-N 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N1/00—Microorganisms, e.g. protozoa; Compositions thereof; Processes of propagating, maintaining or preserving microorganisms or compositions thereof; Processes of preparing or isolating a composition containing a microorganism; Culture media therefor
- C12N1/20—Bacteria; Culture media therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P1/00—Preparation of compounds or compositions, not provided for in groups C12P3/00 - C12P39/00, by using microorganisms or enzymes
- C12P1/04—Preparation of compounds or compositions, not provided for in groups C12P3/00 - C12P39/00, by using microorganisms or enzymes by using bacteria
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/02—Monosaccharides
Definitions
- the present invention relates to a bacterium that produces 2-deoxy-siro-inosose (DOI) from sucrose and a DOI production method using the bacterium.
- DOI 2-deoxy-siro-inosose
- 2-Deoxy-siro-inosose (hereinafter also referred to as DOI) is a useful substance used as a raw material for pharmaceuticals or as a chemical industry resource.
- DOI 2-Deoxy-siro-inosose
- G-6- glucose-6-phosphate
- P recombinant DOI synthase obtained using E. coli.
- WO 2006/109479 pamphlet a method for producing DOI from glucose in one step using E. coli expressing DOI synthase has also been developed. Thereby, it became possible to produce DOI from glucose obtained from plant-derived resources.
- coli phosphoglucose isomerase (pgi), glucose-6-phosphate-1-dehydrogenase (zwf), and phosphoglucomutase (pgm). It is shown. Since all metabolic pathways into which glucose enters the glycolysis system are blocked in this way, it has been concluded that a sugar (for example, mannitol) that can be used separately for glycolysis is necessary for the growth / growth of bacterial cells. . Moreover, in order to obtain the same high DOI productivity, it is only necessary to destroy both the pgi gene and the zwf gene at the same time. In this case, too, if glucose is the only carbon source, the cells cannot grow, It has been shown that mannitol is required for cell growth / growth.
- mannitol is required for cell growth / growth.
- sucrose is known as a sugar raw material that is cheaper than glucose.
- Sucrose is the main component of molasses, and Escherichia coli that assimilate sucrose to produce DOI has been expected to be useful in producing inexpensive industrially available DOI.
- Escherichia coli that assimilate sucrose to produce DOI has been expected to be useful in producing inexpensive industrially available DOI.
- sucrose PTS Phosphoenolpyruvate: “Carbohydrate” Phosphotransferase ”System
- sucrose non-PTS sucrose non-PTS
- sucrose PTS Phosphoenolpyruvate: “Carbohydrate” Phosphotransferase ”System
- sucrose non-PTS sucrose non-PTS
- the microorganism takes sucrose as it is, and then breaks it down into glucose and fructose.
- sucrose PTS when the microorganism takes up sucrose, it phosphorylates sucrose and converts it to sucrose-6-phosphate. And it decomposes into glucose-6-phosphate and fructose inside the microorganism.
- fructose derived from sucrose first appears inside the microorganism in a form that is not phosphorylated.
- this non-phosphorylated fructose hereinafter referred to as non-phosphorylated fructose
- the fructose must be isomerized to glucose or phosphorylated.
- an object of the present invention is to provide a DOI-producing Escherichia coli that can efficiently produce DOI from sucrose, which is an inexpensive sugar, and a DOI production method using the same.
- the present invention has been made in view of the above situation, and the DOI-producing Escherichia coli and the DOI producing method of the present invention are as follows.
- a DOI-producing Escherichia coli having a gene encoding at least a sucrose hydrolase (CscA) and having a 2-deoxy-siro-inosose (DOI) production system added or enhanced, among sucrose non-PTS genes.
- CscA sucrose hydrolase
- DOI 2-deoxy-siro-inosose
- the DOI-producing E. coli according to [1] which has only a gene encoding sucrose hydrolase (CscA) in the sucrose non-PTS gene group.
- the DOI-producing Escherichia coli originally contains phosphoglucose isomerase (Pgi), glucose-6-phosphate-1-dehydrogenase (Zwf), phosphoglucomutase (Pgm), and protein synthesis in stationary phase.
- Rmf ribosome modifying factor
- the DOI-producing E. coli according to any one of [1] to [4], wherein the DOI production system is derived from DOI synthase (BtrC) activity.
- a method for producing DOI comprising producing DOI from a plant-derived material containing sucrose using the DOI-producing E. coli according to any one of [1] to [12].
- the DOI-producing Escherichia coli of the present invention has at least a gene encoding sucrose hydrolase (CscA) in the sucrose non-PTS gene group, and a 2-deoxy-siro-inosose (DOI) production system is imparted or enhanced.
- CscA sucrose hydrolase
- DOI 2-deoxy-siro-inosose
- the present invention provides a DOI-producing Escherichia coli having a gene encoding at least a sucrose hydrolase (CscA) and having a 2-deoxy-siro-inosose (DOI) production system added or enhanced, among sucrose non-PTS genes.
- CscA sucrose hydrolase
- DOI 2-deoxy-siro-inosose
- the DOI-producing Escherichia coli of the present invention can phosphorylate sucrose-derived fructose and take it into the cells, and converts this fructose into energy for cell growth / growth using a glycolysis system. be able to.
- a glycolysis system e.g., a glycolysis system for cell growth.
- sucrose hydrolase (CscA) is recognized as an enzyme that is hardly present on the cell membrane of E. coli (Can. J. Microbiol., Vol. 45, pp. 18- 422 (1999)), on the other hand, DOI-producing Escherichia coli is constructed so that non-phosphorylated fructose cannot be incorporated into a glycolytic system, and only phosphorylated fructose can be used for growth (see WO 2006/109479 pamphlet). ).
- fructose When sucrose is decomposed, equal amounts of glucose and fructose are produced. In general, however, it is known that in Escherichia coli, glucose uptake is generally given priority over fructose, and in the presence of glucose, fructose is not fully metabolized. For this reason, for example, it was surprising that fructose could be efficiently used for the growth of E. coli without being affected by metabolic inhibition (catabolite repression) by glucose by adjusting the pH during cell culture. It is.
- a numerical range indicated by using “to” indicates a range including the numerical values described before and after “to” as the minimum value and the maximum value, respectively.
- “assimilation of sucrose” refers to the ability to make sucrose as it is, low molecular weight or high molecular weight, preferably low molecular weight, and take it into a living body, or metabolically convert it into another substance.
- assimilation includes decomposition that lowers the molecular weight of sucrose. Specifically, it includes breaking down sucrose into D-glucose and D-fructose.
- the “sucrose non-PTS gene group” in the present invention refers to a gene group involved in a non-PTS system in the sucrose utilization pathway of microorganisms. Specifically, a gene (cscR) encoding a repressor protein (CscR), a gene (cscA) encoding sucrose hydrolase (CscA), a gene (cscK) encoding fructokinase (CscK), a sucrose permease ( It is a gene group composed of genes (cscB) encoding CscB).
- At least cscA may be included, for example, only cscA, a combination of cscA and cscK, a combination of cscA and cscB, a combination of cscA and cscR, a combination of cscA, cscB and cscR, cscA and cscK, A combination of cscR, a combination of cscA, cscK, and cscB, and a combination of cscA, cscK, cscB, and cscR.
- the “sucrose hydrolase (CscA)” in the present invention is classified into enzyme number 3.2.1.26 based on the report of the International Biochemical Union (I.U.B.) Enzyme Committee. -A generic term for enzymes that catalyze the reaction of producing glucose and D-fructose. This enzyme is not originally held in Escherichia coli such as K-12 and B strains.
- CscA activity is imparted by introducing at least cscA into E. coli among sucrose non-PTS genes, and in particular, only cscA is introduced into E. coli among sucrose non-PTS genes.
- sucrose existing outside the cell is decomposed into glucose and fructose on the cell membrane and released to the outside of the cell, and these are phosphorylated via glucose PTS and fructose PTS originally possessed by Escherichia coli. While taking it into the cytoplasm.
- fructose present outside the cell appears in the microbial cells as fructose-1-phosphate, and then converted to fructose-1,6-phosphate by fructose-1-phosphate kinase (FruK) present in the microbial cells. It is converted and enters the glycolysis system.
- FruK fructose-1-phosphate kinase
- the sucrose hydrolase (CscA) gene (cscA) introduced into the host bacterium of the present invention is a DNA having a base sequence of a gene encoding sucrose hydrolase (CscA) obtained from an organism having this enzyme.
- a synthetic DNA sequence synthesized based on the known base sequence can be used. Preferred are: Erwinia, Porteus, Proteus, Vibrio, Agrobacterium, Rhizobium, Staphylococcus (Staphylococcus) ), Bifidobacterium, and Escherichia. Examples include DNA having the base sequence of a gene derived from Escherichia coli O-157.
- DNA having the base sequence of a gene derived from Escherichia coli O-157 is particularly preferred.
- the signal sequence for transferring CscA to the periplasm of a microbial cell is added to cscA.
- the gene for the repressor protein (CscR) that can be introduced into the host bacterium of the present invention includes DNA having the base sequence of the gene encoding the repressor protein (CscR) obtained from an organism having this enzyme, or its known A synthetic DNA sequence synthesized based on the base sequence can be used. Preferred are: Erwinia, Porteus, Proteus, Vibrio, Agrobacterium, Rhizobium, Staphylococcus (Staphylococcus) ), Bifidobacterium, and Escherichia. Examples include DNA having the base sequence of a gene derived from Escherichia coli O-157. Particularly preferred is DNA having the base sequence of a gene derived from Escherichia coli O-157.
- a synthetic DNA sequence synthesized based on the base sequence can be used.
- Preferred are: Erwinia, Porteus, Proteus, Vibrio, Agrobacterium, Rhizobium, Staphylococcus (Staphylococcus) ), Bifidobacterium, and Escherichia.
- Examples include DNA having the base sequence of a gene derived from Escherichia coli O-157. Particularly preferred is DNA having the base sequence of a gene derived from Escherichia coli O-157.
- the “host bacterium” means the E. coli that becomes the DOI-producing E. coli of the present invention as a result of introduction of one or more genes from outside the cell.
- the “DOI production system” in the present invention refers to a structure for imparting DOI production ability introduced or modified by genetic recombination.
- a DOI production system may be any system that increases the amount of DOI production in the target Escherichia coli.
- the enzyme activity involved in DOI production is imparted or enhanced, or a combination thereof.
- CscA activity DOI can be effectively produced from sucrose even in Escherichia coli that originally has no sucrose utilization ability.
- “addition” or “enhancement” refers to the promoter activity of the enzyme gene possessed by the host bacterium on the genome in addition to introducing the gene encoding the enzyme from outside the host bacterium. Including those in which an enzyme gene is strongly expressed by strengthening or replacing other promoters.
- the term “by gene recombination” means that a change in the base sequence is caused by the insertion of another DNA into the base sequence of the native gene, or the substitution, deletion or combination of a part of the gene. It may be included, for example, as long as it is obtained as a result of mutation.
- the DOI production system in the present invention is preferably derived from DOI synthetase (BtrC) activity from the viewpoint of DOI production efficiency, and such DOI synthetase introduces a DOI synthetase gene (btrC). By doing so, it can be given to E. coli.
- BtrC DOI synthetase
- DOI synthase is an enzyme that catalyzes the reaction of producing 2-deoxy-siro-isonose (DOI) from glucose-6-phosphate, and is a 42 kDa and 23 kDa dimeric peptide comprising cobalt ions It is known that the enzyme activity is promoted in the presence of zinc, the enzyme activity is suppressed in the presence of zinc and copper ions, and the reaction is carried out using NAD + as a coenzyme (for example, JP 2000-236881, WO 2006). / 109479). Any btrC gene may be used as long as it is derived from an organism having this gene, and examples thereof include those derived from Bacillus bacteria. Among them, a gene encoding a 42 kDa subunit derived from Bacillus circulans can be preferably used from the viewpoint of DOI production efficiency (GenBank accession number AB066276).
- the DOI-producing Escherichia coli in the present invention preferably has a sugar uptake capacity enhancing system.
- the sugar uptake ability in the present invention means a sugar transport ability through a biological membrane, and this ability includes any action on the sugar transport from the outside to the inside of the biological membrane or the sugar transport from the inside to the outside of the biological membrane. be able to.
- the sugar in sugar transport includes 5 monosaccharides or 6 monosaccharides. Specifically, glucose, mannose, arabinose, galactose, fructose, etc. are mentioned, Preferably glucose can be mentioned.
- “having a system for enhancing sugar uptake ability” refers to a state in which uptake of sugar from outside the bacterial body into the bacterial body is increasing.
- the “sugar uptake capacity enhancing system” preferably means a structure for improving glucose uptake ability. More preferably, the system for enhancing sugar uptake ability is different from the PTS system or non-PTS system, for example, a system that takes in glucose outside the cell as it is into the cell. The incorporated glucose is then phosphorylated by a phosphorylating enzyme such as glucokinase (Glk) possessed by the microbial cell, and can be used as a substrate for substance production.
- Glk glucokinase
- the sugar uptake capacity enhancing system in the present invention is preferably a glucose transport enhancing protein (Glf) activity enhancing system from the viewpoint of DOI productivity.
- the glucose transport promoting protein (Glf) in the present invention is a generic term for proteins having an action of transporting D-glucose, D-fructose and the like from the outside to the inside of a biological membrane.
- a technique for improving the productivity of mannitol in E. coli into which the glf gene has been introduced see, for example, Japanese Patent Publication No. 2006-503559
- the productivity of L-phenylalanine and shikimic acid are improved (Japanese Patent Publication No. 2002-512802, Applied Microbiology).
- the glucose transport promoting protein gene (glf) introduced into the host bacterium of the present invention includes a DNA having a base sequence of a gene encoding a glucose transport promoting protein (Glf) derived from an organism having this protein, or a known one thereof
- a synthetic DNA sequence synthesized based on the base sequence can be used.
- Preferable examples include those derived from yeasts and Zymomonas bacteria, and more preferably derived from Zymomonas bacteria.
- Examples include DNA having a base sequence of a gene derived from Zymomonas mobilis.
- the Particularly preferred is DNA having a base sequence of a gene derived from Zymomonas mobilis.
- the DOI-producing Escherichia coli has a sucrose hydrolase (CscA) activity derived from a bacterium belonging to the genus Escherichia from the viewpoint of the ability to degrade sucrose.
- CscA sucrose hydrolase
- a glucose transport promoting protein The (Glf) activity can be obtained by introducing a gene encoding each protein derived from the genus Zymomonas.
- sucrose hydrolase is derived from Escherichia coli O-157 bacterium
- glucose transport promoting protein is obtained by introduction of a gene encoding each protein derived from Zymomonas mobilis bacterium.
- the functions of these genes can be reliably expressed.
- a promoter for expressing various genes may be any promoter as long as it can control the expression of any of the above genes, but it is a strong promoter that constantly functions in a microorganism and in the presence of glucose. Promoters that are less susceptible to suppression of expression are preferred, and specific examples include the promoter of glyceraldehyde 3-phosphate dehydrogenase (hereinafter sometimes referred to as GAPDH) and the promoter of serine hydroxymethyltransferase.
- GAPDH glyceraldehyde 3-phosphate dehydrogenase
- E. coli to which each activity is given refers to E. coli to which activity based on an enzyme or protein is given from outside the cell body to the inside of the cell body by some method.
- Escherichia coli can be produced, for example, using a method such as introducing a gene encoding the enzyme and protein from outside the cell body into the cell body using a gene recombination technique.
- Preparation of genomic DNA necessary for introducing a gene from outside the cell into the cell DNA cleavage and ligation, transformation, PCR (Polymerase Chain Reaction), design of oligonucleotides used as primers, synthesis, etc. This can be done by conventional methods well known to those skilled in the art. These methods are described in Sambrook, J. et al. , Et. al. , “Molecular Cloning A Laboratory Manual, Second Edition”, Cold Spring Harbor ab Laboratory Press, (1989).
- the following enzymes involved in the metabolism of glucose-6-phosphate possessed by the host bacterium phosphoglucose isomerase (Pgi), glucose At least one activity selected from the group consisting of -6-phosphate-1-dehydrogenase (Zwf) and phosphoglucomutase (Pgm) and a ribosome modifying factor (Rmf) involved in the control of protein synthesis in the stationary phase, Inactivated or reduced.
- Pgi phosphoglucose isomerase
- Zwf -6-phosphate-1-dehydrogenase
- Pgm phosphoglucomutase
- Rmf ribosome modifying factor
- the genes encoding each of the above are individually gene-disrupted, or two genes are simultaneously gene-disrupted (pgi gene and zwf gene).
- Pgi gene and pgm gene or three of them are simultaneously disrupted, or the gene encoding the RMF protein involved in the control of protein synthesis in the stationary phase (rmf gene) has been disrupted alone, or the above
- the rmf gene has been disrupted against various gene-disrupted strains in which the gene encoding an enzyme involved in the metabolism of glucose-6-phosphate is disrupted.
- DOI-producing bacteria are described in, for example, International Publication No. 2006/109479.
- the DOI-producing E. coli according to the present invention is more preferably E. coli in which the activities of phosphoglucose isomerase (Pgi) and glucose-6-phosphate-1-dehydrogenase (Zwf) are simultaneously inactivated or reduced. Most preferred is Escherichia coli in which the activities of phosphoglucose isomerase (Pgi) and glucose-6-phosphate-1-dehydrogenase (Zwf) and phosphoglucomutase (Pgm) are simultaneously inactivated or reduced.
- any means that is usually used for this purpose can be used without particular limitation, and examples thereof include gene disruption by homologous recombination of a gene encoding the enzyme. Can do.
- gene disruption may be a gene on a chromosome or a plasmid gene.
- Escherichia coli in the present invention may be a strain in which various chromosome / plasmid genes are disrupted in consideration of DOI synthesis.
- “Inactivation” in the present invention refers to a state where the activity of the enzyme measured by an existing measurement system is below the detection limit.
- the “reduction” in the present invention refers to a state in which the activity of the enzyme is significantly reduced by genetic recombination of the gene encoding the enzyme as compared to the state before the treatment.
- the activity of the enzyme in the present invention may be the activity measured by any existing measurement system.
- gene disruption refers to a mutation in the base sequence of a gene, insertion of another DNA, or deletion of a certain part of a gene so that the function of the gene cannot be exhibited. It is shown that. As a result of gene disruption, the gene cannot be transcribed into mRNA and the structural gene is not translated, or the transcribed mRNA is incomplete, resulting in a mutation or deletion in the amino acid sequence of the translated structural protein. The function cannot be performed.
- the gene-disrupted strain can be produced by any method as long as a disrupted strain that does not express the enzyme or protein is obtained.
- Various methods of gene disruption have been reported (natural breeding, addition of mutagen, UV irradiation, irradiation, random mutation, transposon, site-specific gene disruption), but only certain genes can be disrupted Thus, gene disruption by homologous recombination is preferred. Homologous recombination techniques are described in J. Bacteriol. 161, 1219-1221 (1985) and J. MoI. Bacteriol. , 177, 1511-1519 (1995) and Proc. Natl. Acad. Sci. U. S. A, 97, 6640-6645 (2000). These methods and their applications can be easily implemented by engineers in the same industry.
- the Escherichia coli used in the present invention may be any Escherichia coli as long as the above genes can be introduced and changed. More preferably, it is a type of Escherichia coli that does not inherently have sucrose utilization ability. Examples of such E. coli include Escherichia bacteria, which are particularly convenient and have a track record of industrial use. Abundant Escherichia coli (Escherichia coli) is preferably used. For example, K-12 strain, B strain, C strain and its derived strains can be mentioned. Thereby, the use of the kind of E. coli which does not originally have sucrose utilization ability can be expanded.
- the B strain and its derived strain are preferably used.
- high DOI productivity can be obtained using corn steep liquor which is a general-purpose industrial medium.
- Escherichia coli is generally classified into K-12, B and C strains.
- K-12-derived strains and B-derived strains are widely used as industrial material production hosts, both of which can express heterologous proteins by genetic recombination, but B strain is a preferred host for DOI production. There is no known knowledge of this. Therefore, it is an unexpected and unique phenomenon that the B-derived strain exhibits significantly higher productivity than the K-12-derived strain in the production of DOI.
- the method for producing DOI of the present invention comprises producing 2-deoxy-siro-inosose from a plant-derived material containing sucrose using the DOI-producing Escherichia coli, ie, the DOI-producing Escherichia coli and sucrose.
- the method includes a step of bringing a plant-derived raw material into contact with a plant and a recovery step of recovering the DOI obtained by the contact.
- process is not limited to an independent process, and even if it cannot be clearly distinguished from other processes, it is sufficient that the intended action of this process is achieved. include.
- the plant-derived raw material used in the DOI production method is a carbon source obtained from a plant, and is not particularly limited as long as E. coli can be metabolized and converted into DOI.
- it refers to organs such as roots, stems, trunks, branches, leaves, flowers, seeds, plants containing them, degradation products of these plant organs, and further from plant bodies, plant organs, or degradation products thereof.
- the obtained carbon sources those that can be used as a carbon source in culture by microorganisms are also included in plant-derived materials.
- carbon sources included in such plant-derived raw materials in addition to sucrose, saccharides such as starch, glucose, fructose, xylose, and arabinose as a general substance, or herbaceous degradation products and cellulose containing a large amount of these components A hydrolyzate etc. can be illustrated. Furthermore, vegetable oil-derived glycerin and fatty acids also correspond to the carbon source in the present invention.
- crops such as cereals, corn, rice, wheat, soybeans, sugar cane, beet, cotton, etc., or combinations thereof can be preferably mentioned, and the usage form as the materials is unprocessed
- the product, juice, pulverized product, etc. are not particularly limited. Moreover, the form of only the above-mentioned carbon source may be sufficient.
- the contact between the DOI-producing Escherichia coli and the plant-derived raw material in the contacting step is generally performed by culturing the DOI-producing Escherichia coli in a medium containing the plant-derived raw material.
- the contact density between the plant-derived material and the DOI-producing E. coli varies depending on the activity of the DOI-producing Escherichia coli.
- the initial sugar concentration in terms of glucose is 20% of the total mass of the mixture as the concentration of the plant-derived material in the medium. From the viewpoint of E. coli sugar resistance, the initial sugar concentration can be preferably 15% by mass or less.
- Each of these other components may be added in an amount usually added to the microorganism medium, and is not particularly limited.
- the content of DOI-producing Escherichia coli in the medium varies depending on the type and activity of Escherichia coli, but in general, the initial bacterial concentration is preferably 0.1% to 30% by mass with respect to the culture solution, preferably from the viewpoint of controlling culture conditions. May be 1 mass% to 10 mass%.
- the method for producing DOI in the present invention comprises assimilating plant-derived raw materials by culturing DOI-producing E. coli in a mixture containing DOI-producing E. coli and plant-derived raw materials, and distilling DOI secreted into the culture solution after a certain period of time. , Purification methods using known techniques such as membrane separation and extraction.
- the mixture in the production method of DOI should be mainly composed of a basic medium generally used for culturing bacteria, and any medium that is usually used according to the type of DOI-producing Escherichia coli should be used. Can do.
- a basic medium is not particularly limited as long as it is a medium containing a carbon source, a nitrogen source, inorganic ions, and other trace components as required.
- sucrose alone is sufficient, but from the viewpoint of DOI production efficiency, sugars such as glucose, fructose and molasses, organic acids such as fumaric acid, citric acid and succinic acid, methanol, ethanol and glycerol Alcohols such as may be additionally used as appropriate.
- organic nitrogen sources such as organic ammonium salts, inorganic ammonium salts, ammonia gas, ammonia water, and the like, organic nitrogen sources such as protein hydrolysates, and the like are appropriately used.
- inorganic ions magnesium ions, phosphate ions, potassium ions, iron ions, manganese ions, and others are appropriately used as necessary.
- a culture medium used for this invention if the point used for industrial production is considered, a liquid culture medium is preferable.
- the culture conditions vary depending on the prepared bacterial cells and the culture apparatus.
- the culture temperature is 20 ° C. to 40 ° C., and from the viewpoint of DOI production efficiency, it is more preferable to culture at 25 ° C. to 35 ° C. .
- the pH can be adjusted to pH 4 to pH 9, preferably pH 5.0 to pH 7.5, more preferably pH 6.0 to pH 7.0, and may be adjusted with NaOH, NH 3 or the like.
- the culture time is not particularly limited, but is a time required for the bacterial cells to grow sufficiently and to produce DOI.
- a culture tank capable of controlling temperature, pH, aeration conditions, and stirring speed.
- the culturing of the present invention is not limited to using a culture tank.
- seed culture may be performed in advance as a preculture, and this may be inoculated into a medium in a culture tank prepared in advance.
- the microorganisms obtained in the present invention are cultured to produce DOI, it is not necessary to perform aeration at all, but it is better to perform aeration in order to obtain a more preferable result.
- the aeration condition referred to here does not necessarily require air to pass through the culture medium, and depending on the shape of the culture tank, the top surface ventilation may be used to ventilate the air layer above the culture medium while stirring the culture medium appropriately. It means that a gas containing oxygen is allowed to flow into the culture tank.
- the dissolved oxygen concentration changes depending on the combination of internal pressure, stirring blade position, stirring blade shape, and stirring speed. Therefore, the optimum is as follows using the productivity of DOI and the amount of organic acids other than DOI as indicators. Conditions can be determined. For example, when culturing in a relatively small culture vessel such as the culture device BMJ-01 manufactured by ABLE, when using 500 g of culture solution, the air is at a normal pressure of 0.005 L / min to 2 L / min, and the stirring speed is 50 rpm.
- aeration conditions that can be achieved at ⁇ 2000 rpm, more preferably 0.05 L / min to 1 L / min at normal pressure and a stirring speed of 100 rpm to 1000 rpm.
- the aeration conditions described above do not need to be consistently performed from the beginning of the culture to the end, and preferable results can be obtained by performing it in part of the culture process.
- the DOI obtained by this contact is recovered, and is generally performed by recovering the DOI from the culture obtained by the above culture.
- the culture in the present invention refers to the cells produced by the above-described method, the culture solution, and the processed product thereof.
- a conventionally known method can be used from a culture solution. For example, after removing the cells by centrifugation or the like, the culture supernatant is added to an ion exchange resin and eluted with distilled water. While measuring the refractive index, pH, and conductivity, fractions containing no impurities can be collected and the aqueous solution removed to recover DOI.
- the bacterial cells produced by the method of the present invention produce an enzyme group suitable for DOI production, it is also possible to produce and collect DOI using this, and to collect DOI from the culture. Considered part of the method of recovery.
- DOI can be produced at an extremely high level using inexpensive plant-derived sugar as a raw material.
- the present invention is extremely useful for the spread of carbon neutral DOI.
- Example 1 ⁇ Cloning of the vicinity of Escherichia coli pgi gene>
- the entire nucleotide sequence of Escherichia coli genomic DNA is known (GenBank accession number U00096), and the nucleotide sequence of a gene encoding Escherichia coli phosphoglucose isomerase (hereinafter sometimes referred to as pgi) has also been reported.
- CAGGAATTCG CTATATCTGGG CTCTGCACG SEQ ID NO: 1
- CAGTCTAGAG CAATACTCTG CGATTTTGAG SEQ ID NO: 2
- CAGTCTGATCCATGTGTGATG Four types of oligonucleotide primers having a base sequence of GCTGTACGC (SEQ ID NO: 4) were synthesized.
- the primer of SEQ ID NO: 1 has an EcoRI recognition site on the 5 ′ end side
- the primers of SEQ ID NOS: 2 and 3 have an XbaI recognition site on the 5 ′ end side
- the primer of SEQ ID NO: 4 has a PstI recognition site on the 5 ′ end side.
- the genomic DNA of Escherichia coli MG1655 strain was prepared by the method described in Current Protocols in Molecular Biology (JohnWiley & Sons), and 1 ⁇ g of the obtained genomic DNA, a primer having the base sequence of SEQ ID NO: 1, and the base of SEQ ID NO: 2
- a DNA fragment of 0.0 kb (hereinafter sometimes referred to as a pgi-L fragment and a pgi-R fragment) was amplified.
- DNA fragments were separated and collected by agarose electrophoresis, and the pgi-L fragment was digested with EcoRI and XbaI, and the pgi-R fragment was digested with XbaI and PstI, respectively.
- the two digested fragments were mixed with EcoRI and PstI digests of the temperature sensitive plasmid pTH18cs1 (GenBank accession number AB019610), reacted with T4 DNA ligase, and transformed into Escherichia coli DH5 ⁇ competent cell (manufactured by Takara Bio Inc.). After conversion, a plasmid containing two fragments, a 5 ′ upstream neighboring fragment and a 3 ′ downstream neighboring fragment of the gene encoding pgi, was obtained and named pTH ⁇ pgi.
- Example 2 ⁇ Preparation of Escherichia coli MG1655 ⁇ pgi strain>
- the plasmid pTH ⁇ pgi obtained in Example 1 was transformed into Escherichia coli MG1655 strain and cultured overnight on an LB agar plate containing 10 ⁇ g / ml of chloramphenicol at 30 ° C. where the cells can hold a temperature-sensitive plasmid. A conversion was obtained.
- the obtained transformant was cultured at 30 ° C. from LB medium for 3 hours to overnight, diluted with LB liquid medium or physiological saline, and spread on an LB agar plate containing 10 ⁇ g / ml of chloramphenicol.
- This LB agar plate was cultured at 42 ° C. where no temperature-sensitive plasmid could be maintained, and the grown transformant was obtained as a strain in which the entire length of the plasmid was integrated into the Escherichia coli genome by extragenomic-genomic homologous recombination.
- Genomic DNA was obtained from this strain, PCR was performed using this as a template, the presence of the chloramphenicol resistance gene of pTH18cs1 on the genome, and the region near the 5 ′ side of the gene encoding pgi, In addition, it was confirmed that the entire plasmid length was integrated into the Escherichia coli genome by the presence of a region homologous to each of the 3 ′ adjacent regions on the genome.
- a strain in which the entire length of the plasmid was integrated into the Escherichia coli genome was planted in a 100 ml baffled flask containing 20 ml of LB liquid medium not containing chloramphenicol, and this was cultured with shaking at 30 ° C. for 4 hours.
- This culture solution was diluted with an LB liquid medium not containing chloramphenicol and spread on an LB agar medium not containing chloramphenicol.
- Ninety-six colonies grown at 42 ° C. were randomly selected and grown on LB agar medium without chloramphenicol and LB agar medium with chloramphenicol, Chloramphenicol sensitive strains were selected. Further, genomic DNA was obtained from the selected strain, PCR was performed using this as a template, a strain lacking the gene encoding pgi was selected, and this was named MG1655 ⁇ pgi strain.
- Example 3 ⁇ Cloning of the vicinity region of the Escherichia coli zwf gene> The base sequence of a gene encoding Escherichia coli glucose-6-phosphate dehydrogenase (hereinafter sometimes referred to as zwf) has also been reported.
- CAGGAATTCA TGCGGTGCAG CACGATATC SEQ ID NO: 5
- CAGTCTAGAT AACCCCGGTAC TTAAGCCAG SEQ ID NO: 6
- CAGTCTAGAC TGCGCTTTGC CTGT Four types of oligonucleotide primers having the base sequence of (SEQ ID NO: 8) were synthesized.
- the primer of SEQ ID NO: 5 has an EcoRI recognition site on the 5 ′ end side
- the primers of SEQ ID NOS: 6 and 7 have an XbaI recognition site on the 5 ′ end side
- the primer of SEQ ID NO: 8 has a PstI recognition site on the 5 ′ end side.
- a primer having the base sequence of SEQ ID NO: 5 and a primer having the base sequence of SEQ ID NO: 6, a primer having the base sequence of SEQ ID NO: 7 and a primer having the base sequence of SEQ ID NO: 8 By performing PCR under normal conditions using 100 pmol of each of the above primer DNAs, a DNA fragment of about 0.85 kb (hereinafter sometimes referred to as zwf-L fragment) and about 1.0 kb (hereinafter referred to as zwf-L fragment) are used.
- the DNA fragment (sometimes called the zwf-R fragment) was amplified.
- DNA fragments were separated and collected by agarose electrophoresis, and the zwf-L fragment was digested with EcoRI and XbaI, and the zwf-R fragment was digested with XbaI and PstI, respectively.
- the two digested fragments were mixed with EcoRI and PstI digests of the temperature sensitive plasmid pTH18cs1, reacted with T4 DNA ligase, transformed into Escherichia coli DH5 ⁇ competent cell (manufactured by Takara Bio Inc.), zwf
- a plasmid containing two fragments, a 5 ′ upstream neighboring fragment and a 3 ′ downstream neighboring fragment, of the gene to be encoded was obtained and named pTH ⁇ zwf.
- Example 4 ⁇ Preparation of Escherichia coli MG1655 ⁇ pgi ⁇ zwf strain>
- the plasmid pTH ⁇ zwf obtained in Example 3 was transformed into the Escherichia coli MG1655 ⁇ pgi strain obtained in Example 2, and the LB agar plate containing 10 ⁇ g / ml of chloramphenicol at 30 ° C. where the cells can hold the temperature-sensitive plasmid.
- the above was cultured overnight to obtain a transformant.
- the obtained transformant was cultured at 30 ° C.
- Genomic DNA was obtained from this strain, PCR was performed using this as a template, the presence of the chloramphenicol resistance gene of pTH18cs1 on the genome, and the region near the 5 ′ side of the gene encoding zwf, In addition, it was confirmed that the entire plasmid length was integrated into the Escherichia coli genome by the presence of a region homologous to each of the 3 ′ adjacent regions on the genome.
- a strain in which the entire length of the plasmid was integrated into the Escherichia coli genome was planted in a 100 ml baffled flask containing 20 ml of LB liquid medium not containing chloramphenicol, and this was cultured with shaking at 30 ° C. for 4 hours.
- This culture solution was diluted with an LB liquid medium not containing chloramphenicol, and applied on an LB agar medium not containing chloramphenicol.
- Ninety-six colonies grown at 42 ° C. were randomly selected and grown on LB agar medium without chloramphenicol and LB agar medium with chloramphenicol, Chloramphenicol sensitive strains were selected. Further, genomic DNA was obtained from the selected strain, PCR was performed using this as a template, a strain lacking the gene encoding zwf was selected, and this strain was designated as MG1655 ⁇ pgi ⁇ zwf strain.
- the primer of SEQ ID NO: 9 has an NdeI recognition site on the 5 ′ end side
- the primer of SEQ ID NO: 10 has a HindIII, PstI, SalI, BamHI, and SacI recognition site in this order from the 5 ′ end side.
- the PCR method was carried out under normal conditions using the genomic DNA of Escherichia coli MG1655 strain together with the above two types of primers as a template to amplify the DNA fragment.
- the obtained DNA fragment was digested with restriction enzymes NdeI and HindIII to obtain a fragment encoding the GAPDH promoter of about 100 bp.
- the above DNA fragment was mixed with the Escherichia coli DH5 ⁇ competent cell (manufactured by Takara Bio Inc.) after mixing with the above-mentioned DNA fragment and the cloning vector pBR322 (GenBank accession number J01749) digested with NdeI and HindIII and ligase.
- the base sequence of the DOI synthase gene (btrC) possessed by Bacillus circulans has already been reported (GenBank accession number AB066276).
- oligonucleotide primers having the base sequences of CACTGGAGCT, CCGTGGGTGGA, ATATAGACG, ACTAAACAAA, TTTG (SEQ ID NO: 11), and CAGGATCCTT ACAGCCCTTC CCGGATC (SEQ ID NO: 12) were synthesized.
- the primer of SEQ ID NO: 11 has a SacI recognition site and a 13-base GAPDH gene ribosome binding sequence in this order from the 5 'end.
- the primer of SEQ ID NO: 12 has a BamHI recognition site on the 5 'end side.
- a PCR method is performed under normal conditions using the Bacillus circulans genomic DNA as a template together with the above two primers, and the obtained DNA fragment is digested with restriction enzymes SacI and BamHI to obtain an about 1.1 kbp DOI synthase gene (btrC). ) Fragment was obtained.
- This DNA fragment was mixed with a fragment obtained by digesting plasmid pGAP with restriction enzymes SacI and BamHI, ligated with ligase, and then transformed into Escherichia coli DH5 ⁇ competent cell (manufactured by Takara Bio Inc.). A transformant that grew on an LB agar plate containing 50 ⁇ g / mL of ampicillin was obtained.
- the obtained colony was cultured overnight at 30 ° C. in an LB liquid medium containing 50 ⁇ g / mL of ampicillin, and plasmid pGAP-btrC was recovered from the obtained bacterial cells to construct a DOI synthase gene (btrC) expression vector. .
- Example 6 ⁇ Construction of DOI synthase gene (btrC) and sucrose hydrolase gene (cscA) expression vectors under the control of the GAPDH promoter>
- the base sequence of the sucrose hydrolase gene (cscA) possessed by Escherichia coli O-157 has already been reported. That is, it is described in the Escherichia coli O-157 strain genomic sequence 3274383-3275816 described in GenBank accession number AE005174.
- oligonucleotide primers having the base sequences of GCGGATCGCC TGGTGGAATA TATGACGCAA TCTCGGATTGC (SEQ ID NO: 13) and GACGCGTCGA CTTAACCCAG (SEQ ID NO: 14) were respectively synthesized.
- the primer of SEQ ID NO: 13 has a BamHI recognition site and a 13-base GAPDH gene ribosome binding sequence in this order from the 5 ′ end.
- the primer of SEQ ID NO: 14 has a SalI recognition site on the 5 ′ end side.
- the PCR method is performed under normal conditions using the above two primers together with the genomic DNA of Escherichia coli O-157 strain (SIGMA-ALDRICH: IRMM449) as a template, and the resulting DNA fragment is digested with restriction enzymes BamHI and SalI.
- the sucrose hydrolase gene (cscA) fragment of about 1.4 kbp was obtained.
- This DNA fragment was mixed with a fragment obtained by digesting plasmid pGAP-btrC with restriction enzymes BamHI and SacI, ligated with ligase, and transformed into Escherichia coli DH5 ⁇ competent cell (manufactured by Takara Bio Inc.).
- a transformant that grew on an LB agar plate containing 50 ⁇ g / mL of ampicillin was obtained.
- the obtained colonies were cultured overnight in an LB liquid medium containing 50 ⁇ g / mL of ampicillin at 30 ° C., and the plasmid pGAP-btrC-cscA was recovered from the obtained bacterial cells to obtain DOI synthase gene (btrC) and sucrose hydrolysate.
- a degrading enzyme gene (cscA) expression vector was constructed.
- Example 7 ⁇ Construction of DOI synthase gene (btrC), sucrose hydrolase gene (cscA) and glucose transport promoting protein gene (glf) expression vector under control of GAPDH promoter>
- the nucleotide sequence of the glucose transport promoting protein gene (glf) possessed by Zymomonas mobilis (ATCC 29191) has already been reported (GenBank accession number M60615).
- oligonucleotide primers having the base sequences of CCTGTCGACG CTGGTGGAAT ATATGAGTTC TGAAAGTAGT CAGG (SEQ ID NO: 15) and CTACTGCAGC TACTCTGGG (AGCGGCCACA (SEQ ID NO: 16)) were synthesized.
- the primer of SEQ ID NO: 15 has a SalI recognition site and a 13-base GAPDH gene ribosome binding sequence in this order from the 5 ′ end.
- the primer of SEQ ID NO: 16 has a PstI recognition site on the 5 ′ end side.
- a PCR method was performed under normal conditions using the Zymomonas mobilis genomic DNA as a template together with the above two primers, and the obtained DNA fragment was digested with restriction enzymes SalI and PstI to give a glucose transport-promoting protein gene of about 1.4 kbp ( glf) fragment was obtained.
- This DNA fragment was mixed with a fragment obtained by digesting the plasmid pGAP-btrC-cscA with restriction enzymes SalI and PstI, ligated with ligase, and then transferred to Escherichia coli DH5 ⁇ competent cell (manufactured by Takara Bio Inc.).
- the transformant was obtained by transforming and growing on an LB agar plate containing 50 ⁇ g / mL of ampicillin. The obtained colony was cultured overnight at 30 ° C. in an LB liquid medium containing 50 ⁇ g / mL of ampicillin, and the plasmid pGAP-btrC-cscA-glf was recovered from the obtained bacterial cells to obtain a DOI synthase gene (btrC), A sucrose hydrolase gene (cscA) and glucose transport promoting protein gene (glf) expression vector was constructed.
- btrC DOI synthase gene
- cscA sucrose hydrolase gene
- glf glucose transport promoting protein gene
- Example 8 ⁇ Introduction of pGAP-btrC-cscA and pGAP-btrC-cscA-glf into MG1655 ⁇ pgi ⁇ zwf strain>
- the above plasmids pGAP-btrC-cscA and pGAP-btrC-cscA-glf were transformed into MG1655 ⁇ pgi ⁇ zwf strain, respectively, and cultured at 37 ° C. overnight on LB agar plates containing 50 ⁇ g / mL of ampicillin.
- the MG1655 ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strain was obtained.
- Example 9 ⁇ Difference in DOI productivity using MG1655 ⁇ pgi ⁇ zwf / pGAP-btrC-cscA strain and DOI productivity depending on culture pH> Escherichia coli MG1655 ⁇ pgi ⁇ zwf / pGAP-btrC-cscA strain was inoculated into 25 ml of LB Broth, Miller culture solution (Difco244620) placed in an Erlenmeyer flask as a preculture, and after stirring overnight at 120 rpm, a medium having the following composition was cultured. The entire amount was inoculated into a 1 L culture tank (culture device BMJ-01 manufactured by ABLE) containing 475 g.
- Culturing was performed under atmospheric pressure at an aeration rate of 1.0 vvm, a stirring speed of 800 rpm, and a culture temperature of 30 ° C.
- a total of four culture vessels were used, and the pH was adjusted to pH 7.0, pH 6.5, and pH 6.0 using 12.5% aqueous ammonia and 2N hydrochloric acid.
- concentration was measured by measuring the light absorbency of 660 nm.
- the bacterial cell concentration at the start of the culture was all 0.29.
- the amounts of DOI, sucrose, glucose, and fructose in the obtained culture solution were measured by HPLC according to a conventional method.
- concentration was measured by measuring the light absorbency of 660 nm.
- the composition of the medium used for the culture is shown in Table 1 below. The results are shown in Table 2.
- This example confirmed that DOI can be produced from sucrose using the Escherichia coli MG1655 ⁇ pgi ⁇ zwf / pGAP-btrC-cscA strain.
- the bacteria used in this example should be unable to catabolize glucose because the pgi gene and the zwf gene are disrupted at the same time. In the test area, the number of bacteria increased more than 100 times.
- the Escherichia coli of the present invention does not require expensive mannitol or the like for cell proliferation / growth and uses fructose obtained by decomposing sucrose for cell growth / growth. confirmed.
- the DOI accumulation amounts were 31 g / L, 36 g / L, and 33 g / L, and were the highest at pH 6.5.
- the sucrose input was completely degraded under all pH conditions, and the fructose in the medium was the lowest when the pH was 6.5 or lower. From this, it was found that fructose obtained by sucrose decomposition was most efficiently taken into the cells when the pH was 6.5 or lower.
- the uptake of fructose is faster than when pH 7.0, and DOI-producing Escherichia coli takes up glucose and fructose efficiently. I understand that.
- Example 10 ⁇ Production of DOI by MG1655 ⁇ pgi ⁇ zwf / pGAP-btrC-cscA strain and MG1655 ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strain> Escherichia coli MG1655 ⁇ pgi ⁇ zwf / pGAP-btrC-cscA and MG1655 ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strains were separately seeded in 25 ml of LB Broth, Miller culture solution (Difco244620) in an Erlenmeyer flask as a preculture.
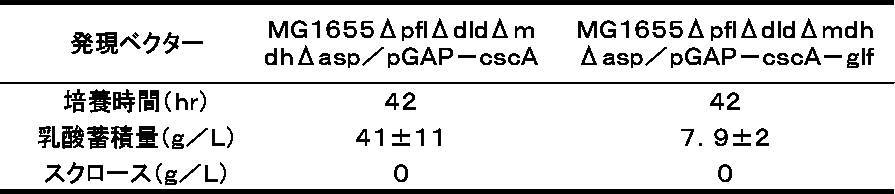
- the whole amount was inoculated in a 1 L culture tank (culture device BMJ-01 manufactured by ABLE) containing 475 g of a medium having the following composition. Cultivation was performed under atmospheric pressure with an aeration rate of 1.0 vvm, a stirring speed of 800 rpm, a culture temperature of 30 ° C., and a pH of 6.5 (adjusted with 12.5% aqueous ammonia). After 48 hours of culturing, the amounts of DOI, sucrose, and glucose in the obtained culture broth were measured by HPLC according to a conventional method. The composition of the medium used for the culture is shown in Table 3 below. The results are shown in Table 4.
- Example 11 ⁇ Production of DOI from sucrose by using B ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strain and MG1655 ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strain using an industrial medium>
- corn steep liquor is used as an alternative to expensive yeast extract or the like as a culture medium component in industrial production.
- the productivity of DOI when using corn steep liquor medium was compared between the MG1655 strain and the B strain, which are subspecies of Escherichia coli.
- the pgi gene and the zwf gene were also disrupted by the method described in Examples 1 to 4 for the Escherichia coli B strain (ATCC 11303), and the resulting strain was named B ⁇ pgi ⁇ zwf strain.
- the expression vector pGAP-btrC-cscA-glf was introduced by the method described in Example 8, and the resulting bacterium was named B ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strain.
- B ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strain and MG1655 ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strain are inoculated in 120 ml overnight in 25 ml of LB Broth and Miller broth (Difco244620) placed in an Erlenmeyer flask as preculture. After stirring culture, the entire amount was inoculated into a 1 L culture tank (culture device BMJ-01 manufactured by ABLE) containing 475 g of a medium having the following composition.
- Cultivation was performed under atmospheric pressure with an aeration rate of 1.5 vvm, a stirring speed of 800 rpm, a culture temperature of 30 ° C., and a pH of 6.2 (adjusted with 12.5% aqueous ammonia). Cultivation was performed while adding a 50% aqueous sucrose solution as a fermentation raw material at a rate of 4 g / hr. The amount of DOI in the obtained culture broth was measured by HPLC according to a conventional method. The composition of the medium used for the culture is shown in Table 5 below. The results are shown in Table 6 and FIG. In Table 6, “-” represents no measured value. In FIG.
- the black circle represents the B ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strain
- the white square represents the MG1655 ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strain.
- Example 12 ⁇ Production of DOI from molasses by B ⁇ pgi ⁇ zwf / pGAP-btrC-cscA strain, B ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strain and B ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-pck strain> Phosphoenolpyruvate carboxykinase (Pck) was selected as a glucose transport promoting protein gene other than glf.
- Pck Phosphoenolpyruvate carboxykinase
- Phosphoenolpyruvate carboxykinase is a protein that produces phosphoenolpyruvate using oxaloacetate as a substrate. Since Escherichia coli originally has a glucose uptake system (PTS) that requires phosphoenolpyruvate, high expression of this protein increases the supply of phosphoenolpyruvate, resulting in an increase in PTS. It is expected that it can be strengthened.
- PTS glucose uptake system
- the base sequence of phosphoenolpyruvate carboxykinase (Pck) possessed by Escherichia coli has already been reported (GenBank accession number M59823).
- primers having the base sequences of TACTGCAGAG CTGGTGGAAT, ATATCGCGGT, TAACAATGGT TTG (SEQ ID NO: 17), and TACTGCAGTT ACAGTTTCGG ACCAGCCG (SEQ ID NO: 18) were prepared.
- the primer of SEQ ID NO: 17 has a PstI recognition site and a 13-base GAPDH gene ribosome binding sequence in this order from the 5 'end.
- the primer of SEQ ID NO: 18 has a PstI recognition site on the 5 'end side.
- PCR is carried out under normal conditions using the genomic DNA of Escherichia coli B strain as a template together with the above two primers, and the resulting DNA fragment is digested with the restriction enzyme PstI to obtain about 1.6 kbp phosphoenolpyruvate carboxy.
- a kinase gene (pck) fragment was obtained.
- This DNA fragment was mixed with a fragment obtained by digesting plasmid pGAP-btrC-cscA with restriction enzyme PstI, ligated with ligase, and transformed into Escherichia coli DH5 ⁇ competent cell (manufactured by Takara Bio Inc.).
- a transformant that grew on an LB agar plate containing 50 ⁇ g / mL of ampicillin was obtained.
- the obtained colonies were cultured overnight in an LB liquid medium containing 50 ⁇ g / mL of ampicillin at 30 ° C., and the plasmid pGAP-btrC-cscA-pck was recovered from the obtained cells, and the DOI synthase gene (btrC), Sucrose hydrolase gene (cscA) and phosphoenolpyruvate carboxykinase gene (pck) expression vectors were constructed.
- the expression vector pGAP-btrC-cscA-pck was introduced into the B ⁇ pgi ⁇ zwf strain by the method described in Example 8 and named B ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-pck.
- Cultivation was performed under atmospheric pressure at an aeration rate of 2.0 vvm, a stirring speed of 1000 rpm, a culture temperature of 32 ° C., and a pH of 6.2 (adjusted with 12.5% aqueous ammonia). Cultivation was performed while adding a 90% molasses aqueous solution as a fermentation raw material at a rate of 6 g / hr. The amount of DOI in the obtained culture broth was measured by HPLC according to a conventional method. The composition of the medium used for the culture is shown in Table 7 below. The results are shown in Table 8 and FIG. In FIG.
- the white rhombus represents the B ⁇ pgi ⁇ zwf / pGAP-btrC-cscA strain
- the black circle represents the B ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-glf strain
- the white triangle represents the B ⁇ pgi ⁇ zwf / pGAP-btrC-cscA-pck strain.
- MG1655 ⁇ pfl ⁇ dld ⁇ mdh ⁇ asp / GAPldhA genome insertion strain refers to pyruvate formate lyase (Pfl), FAD-dependent D-lactate dehydrogenase (Dld), malate dehydrogenase (Mdh), aspartate ammonia lyase (AspA) (EspA) that E. coli MG1655 originally possesses.
- Pfl pyruvate formate lyase
- Dld FAD-dependent D-lactate dehydrogenase
- Mdh malate dehydrogenase
- LidhA NADH-dependent D-lactate dehydrogenase
- cscA Escherichia coli O-157 derived sucrose hydrolase gene
- cscA Escherichia coli O-157 derived sucrose hydrolase gene
- glf zymomonas mobilis derived glucose transport promoting protein gene
- the primer of SEQ ID NO: 9 has an NdeI recognition site on the 5 ′ end side
- the primer of SEQ ID NO: 10 has a HindIII, PstI, SalI, BamHI, and SacI recognition site in this order from the 5 ′ end side.
- the above DNA fragment was mixed with the Escherichia coli DH5 ⁇ competent cell (manufactured by Takara Bio Inc.) after mixing with the above-mentioned DNA fragment and the cloning vector pBR322 (GenBank accession number J01749) digested with NdeI and HindIII, and ligated with ligase.
- pBR322 GenBank accession number J01749
- the obtained colony was cultured overnight at 30 ° C. in an LB liquid medium containing 50 ⁇ g / mL of ampicillin, and the plasmid was recovered from the obtained cells.
- This plasmid was named pGAP.
- the base sequence of the sucrose hydrolase gene (cscA) possessed by Escherichia coli O-157 has already been reported. That is, it is described in the Escherichia coli O-157 strain genomic sequence 3274383-3275816 described in GenBank accession number AE005174.
- primers having the nucleotide sequences of SEQ ID NOS: 13 and 14 were synthesized.
- the primer of SEQ ID NO: 13 has a BamHI recognition site and a 13-base GAPDH gene ribosome binding sequence in this order from the 5 ′ end.
- the primer of SEQ ID NO: 14 has a SalI recognition site on the 5 ′ end side.
- the PCR method is performed under normal conditions using the above two primers together with the genomic DNA of Escherichia coli O-157 strain (SIGMA-ALDRICH: IRMM449) as a template, and the resulting DNA fragment is digested with restriction enzymes BamHI and SalI.
- the sucrose hydrolase gene (cscA) fragment of about 1.4 kbp was obtained.
- This DNA fragment and the fragment obtained by digesting plasmid pGAP with restriction enzymes BamHI and SalI were mixed, ligated using ligase, and then transformed into Escherichia coli DH5 ⁇ competent cell (manufactured by Takara Bio Inc.).
- a transformant that grew on an LB agar plate containing 50 ⁇ g / mL of ampicillin was obtained.
- the obtained colonies were cultured overnight in an LB liquid medium containing 50 ⁇ g / mL of ampicillin at 30 ° C., and the plasmid pGAP-cscA was recovered from the resulting cells to construct a sucrose hydrolase gene (cscA) expression vector.
- cscA sucrose hydrolase gene
- the nucleotide sequence of the glucose transport promoting protein gene (glf) possessed by Zymomonas mobilis has already been reported (GenBank accession number M60615).
- primers having the nucleotide sequences of SEQ ID NOS: 15 and 16 were prepared, respectively.
- the primer of SEQ ID NO: 15 has a SalI recognition site and a 13-base GAPDH gene ribosome binding sequence in this order from the 5 ′ end.
- the primer of SEQ ID NO: 16 has a PstI recognition site on the 5 ′ end side.
- a PCR method was performed under normal conditions using Zymomonas mobilis genomic DNA as a template together with the above two types of primers, and the obtained DNA fragment was digested with restriction enzymes SalI and PstI to give a glucose transport-promoting protein gene of about 1.4 kbp ( glf) fragment was obtained.
- This DNA fragment and the fragment obtained by digesting plasmid pGAP-cscA with restriction enzymes SalI and PstI were mixed, ligated with ligase, and transformed into Escherichia coli DH5 ⁇ competent cell (manufactured by Takara Bio Inc.). Thus, a transformant that grew on an LB agar plate containing 50 ⁇ g / mL of ampicillin was obtained.
- the above plasmids pGAP-cscA and pGAP-cscA-glf are transformed into MG1655 ⁇ pfl ⁇ dld ⁇ mdh ⁇ asp / GAPldhA genome insert competent cells described in patent document WO2005 / 033324, and LB Broth, Miller agar plate containing ampicillin 50 ⁇ g / mL. By culturing at 37 ° C.
- MG1655 ⁇ pfl ⁇ dld ⁇ mdh ⁇ asp / GAPldhA genome inserted strain / pGAP-cscA strain and MG1655 ⁇ pfl ⁇ dld ⁇ mdh ⁇ asp / GAPldhA genomic insert strain / pGAP-cscA-glf strain were obtained.
- the productivity of D-lactic acid by the MG1655 ⁇ pfl ⁇ dld ⁇ mdh ⁇ asp / pGAP-cscA-glf strain was lower than that of MG1655 ⁇ pfl ⁇ dld ⁇ mdh ⁇ asp / pGAP-cscA.
- the productivity improvement effect by introducing the glf gene was not seen at all, but rather a negative effect was shown. This indicates that the introduction of the glf gene does not necessarily increase the production amount in the production process of the substance by fermentation.
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- General Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Tropical Medicine & Parasitology (AREA)
- Medicinal Chemistry (AREA)
- Virology (AREA)
- Mycology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description
従って本発明は、安価な糖であるスクロースから効率よくDOIを生産することができるDOI生産大腸菌及びこれを用いたDOI生産方法を提供することを目的とする。
[2] スクロース非PTS遺伝子群の中で、スクロース加水分解酵素(CscA)をコードする遺伝子のみを有する[1]に記載のDOI生産大腸菌。
[3] 前記DOI生産大腸菌が、糖取り込み能力強化系を有する[1]又は[2]に記載のDOI生産大腸菌。
[4] 前記DOI生産大腸菌が、該大腸菌が本来有しているホスホグルコースイソメラーゼ(Pgi)、グルコース-6-リン酸-1-デヒドロゲナーゼ(Zwf)、ホスホグルコムターゼ(Pgm)及び定常期における蛋白質合成の修飾を担うリボソーム修飾因子(Rmf)からなる群より選択された少なくとも1つの活性が、不活化あるいは低減化されている[3]に記載のDOI生産大腸菌。
[5] 前記DOI生産系が、DOI合成酵素(BtrC)活性に由来する[1]~[4]のいずれか一つに記載のDOI生産大腸菌。
[6] 前記糖取り込み能力強化系が、グルコース輸送促進蛋白質(Glf)活性の強化である[3]~[5]のいずれか一つに記載のDOI生産大腸菌。
[7] 前記スクロース加水分解酵素(CscA)をコードする遺伝子が、エシェリヒア・コリ細菌に由来する[1]~[6]のいずれか一つに記載のDOI生産大腸菌。
[8] 前記エシェリヒア・コリ細菌が、エシェリヒア・コリO-157細菌である[7]に記載のDOI生産大腸菌。
[9] 前記グルコース輸送促進蛋白質(Glf)がザイモモナス属細菌に由来する[6]~[8]のいずれか一つに記載のDOI生産大腸菌。
[10] 前記ザイモモナス属細菌がザイモモナス・モビリス細菌である[9]に記載のDOI生産大腸菌。
[11] 前記DOI生産大腸菌が、本来スクロース資化能を持たない種類の大腸菌である[1]~[10]のいずれか一つに記載のDOI生産大腸菌。
[12] 前記DOI生産大腸菌が、B株又はその由来株である[11]に記載のDOI生産大腸菌。
[13] [1]~[12]のいずれか一つに記載のDOI生産大腸菌を用いて、スクロースを含む植物由来原料からDOIを生産することを含むDOIの生産方法。
即ち、本発明のDOI生産大腸菌は、スクロース由来のフルクトースをリン酸化して菌体内に取り込むことができると共に、このフルクトースを、解糖系を用いて菌体の増殖/生育用のエネルギーへ変換することができる。このように、スクロース由来のフルクトースを菌体増殖の栄養源として利用しながら大腸菌でDOIを生産させた報告例はない。
本明細書において「~」を用いて示された数値範囲は、「~」の前後に記載される数値をそれぞれ最小値及び最大値として含む範囲を示す。
なかでも、DOIを更に効率よく生産するという観点から、cscAのみを有し、その他の遺伝子を含まないことが好ましい。
好ましくは、DOI生産に関与する酵素活性の付与若しくは強化又はこれらの組み合わせを挙げることができる。これにより、上記のCscA活性と組み合わせて、本来はスクロース資化能を有しない大腸菌であっても、スクロースからDOIを効果的に生産することができる。
本発明における糖取り込み能力とは、生体膜を介する糖輸送能力を意味し、生体膜の外側から内側への糖輸送又は生体膜の内側から外側への糖輸送のどちらに対する作用でもこの能力に含むことができる。糖輸送における糖としては5単糖又は6単糖を含む。具体的には、グルコース、マンノース、アラビノース、ガラクトース、フルクトース等が挙げられ、好ましくはグルコースを挙げることができる。
本発明におけるグルコース輸送促進蛋白質(Glf)とは、D-グルコースやD-フルクトース等を生体膜の外側から内側へ輸送する作用をもつ蛋白質の総称を指す。glf遺伝子を導入した大腸菌においてマンニトールの生産性が向上した技術(例えば、特表2006-503559号参照)や、L-フェニルアラニンやシキミ酸の生産性が向上(特表2002-512802号公報、Applied Microbiology and Biotechnology(2004),64,333-339)することは知られているが、グルコース輸送促進蛋白質遺伝子(glf)の導入が、DOI生産大腸菌においても生産性向上の効果を示すかどうかは全く分かっていなかった。なお、後述するようにglf遺伝子の導入は、乳酸の生産性を向上させるためには効果はない。このため、glf遺伝子の導入は糖を原料とした物質生産に有効な共通する技術ではなく、本発明においてglf遺伝子を導入することによりDOIが効率よく生産できたことは意外なことである。
これによって、DOI生産のための直接の基質となるグルコース-6-リン酸の菌による分解代謝が抑制され、また定常期における蛋白質合成によりDOI生産能を高めることができる。このようなDOI生産細菌は、例えば、国際公開2006/109479号パンフレットに記載されている。
また本発明における大腸菌は、DOIの合成を考慮して、種々の染色体/プラスミド遺伝子を破壊した株を用いてもよい。
本発明における「低減化」とは、当該酵素をコードする遺伝子の遺伝子組換えにより、それらの処理を行う前の状態よりも有意に当該酵素の活性が低下している状態を指す。
本発明における酵素の活性は、既存の測定系のいずれによって測定された活性であってもよい。
なお、本明細書において「工程」との語は、独立した工程だけでなく、他の工程と明確に区別できない場合であっても本工程の所期の作用が達成されればよく、本用語に含まれる。
なお、本発明に使用される培地としては、工業的生産に供する点を考慮すれば液体培地が好ましい。
上述した通気条件は培養初期から終了まで一貫して行う必要はなく、培養工程の一部で行うことでも好ましい結果を得ることができる。
本発明における培養物とは、上述した方法により生産された菌体、培養液、及びそれらの処理物を指す。
<エシェリヒア・コリpgi遺伝子の近傍領域のクローニング>
エシェリヒア・コリのゲノムDNAの全塩基配列は公知であり(GenBank accession number U00096)、エシェリヒア・コリのホスホグルコースイソメラーゼ(以下pgiと呼ぶことがある)をコードする遺伝子の塩基配列も報告されている。pgiをコードする遺伝子(1,650bp)の塩基配列近傍領域をクローニングするため、CAGGAATTCG CTATATCTGG CTCTGCACG(配列番号1)、CAGTCTAGAG CAATACTCTT CTGATTTTGA G(配列番号2)、CAGTCTAGAT CATCGTCGAT ATGTAGGCC(配列番号3)及びGACCTGCAGA TCATCCGTCA GCTGTACGC(配列番号4)の塩基配列を有するオリゴヌクレオチドプライマーを4種合成した。配列番号1のプライマーは5’末端側にEcoRI認識部位を、配列番号2及び3のプライマーは5’末端側にXbaI認識部位を、配列番号4のプライマーは5’末端側にPstI認識部位をそれぞれ有している。
<エシェリヒア・コリMG1655Δpgi株の作製>
実施例1で得たプラスミドpTHΔpgiをエシェリヒア・コリMG1655株に形質転換し、細胞が温度感受性プラスミドを保持できる30℃でクロラムフェニコール10μg/mlを含むLB寒天プレート上で一晩培養し、形質転換体を得た。得られた形質転換体をLB培地30℃で3時間から一晩培養後、LB液体培地または生理食塩水で希釈して、クロラムフェニコール10μg/mlを含むLB寒天プレート上に塗布した。このLB寒天プレートを、温度感受性プラスミドを保持できない42℃で培養し、生育した形質転換体をゲノム外-ゲノム間相同組換えによりプラスミド全長がエシェリヒア・コリゲノムに組み込まれた株として得た。
さらに選抜された株からゲノムDNAを取得し、これを鋳型としたPCRを実施してpgiをコードする遺伝子が欠損した株を選抜し、これをMG1655Δpgi株と命名した。
<エシェリヒア・コリzwf遺伝子の近傍領域のクローニング>
エシェリヒア・コリのグルコース-6-リン酸デヒドロゲナーゼ(以下zwfと呼ぶことがある)をコードする遺伝子の塩基配列も報告されている。zwfをコードする遺伝子(1,476bp)の塩基配列近傍領域をクローニングするため、CAGGAATTCA TGCGTTGCAG CACGATATC(配列番号5)、CAGTCTAGAT AACCCGGTAC TTAAGCCAG(配列番号6)、CAGTCTAGAC TGCGCTTATC CTTTATGGT(配列番号7)及びGACCTGCAGT TACCGGTCAT GCGTGTAAC(配列番号8)の塩基配列を有するオリゴヌクレオチドプライマーを4種合成した。配列番号5のプライマーは5’末端側にEcoRI認識部位を、配列番号6及び7のプライマーは5’末端側にXbaI認識部位を、配列番号8のプライマーは5’末端側にPstI認識部位をそれぞれ有している。
<エシェリヒア・コリMG1655ΔpgiΔzwf株の作製>
実施例3で得たプラスミドpTHΔzwfを、実施例2で得たエシェリヒア・コリMG1655Δpgi株に形質転換し、細胞が温度感受性プラスミドを保持できる30℃で、クロラムフェニコール10μg/mlを含むLB寒天プレート上で一晩培養し、形質転換体を得た。得られた形質転換体をLB培地30℃で3時間から一晩培養後、LB液体培地または生理食塩水で希釈して、クロラムフェニコール10μg/mlを含むLB寒天プレート上に塗布した。このLB寒天プレートを、温度感受性プラスミドを保持できない42℃で培養し、生育した形質転換体をゲノム外-ゲノム間相同組換えによりプラスミド全長がエシェリヒア・コリゲノムに組み込まれた株として得た。
さらに選抜された株からゲノムDNAを取得し、これを鋳型としたPCRを実施してzwfをコードする遺伝子が欠損した株を選抜し、これをMG1655ΔpgiΔzwf株と命名した。
<GAPDHプロモーター制御下、DOI合成酵素遺伝子(btrC)発現ベクターの構築>
エシェリヒア・コリのGAPDH遺伝子の塩基配列はすでに報告されている。グリセルアルデヒド3-リン酸デヒドロゲナーゼ(GAPA)プロモーターを取得するため、CGAGCTACAT ATGCAATGAT TGACACGATT CCG(配列番号9)、及びCCAAGCTTCT GCAGGTCGAC GGATCCGAGC TCAGCTATTT GTTAGTGAAT AAAAGG(配列番号10)の塩基配列を有するオリゴヌクレオチドプライマーをそれぞれ合成した。配列番号9のプライマーはその5’末端側にNdeI認識部位を、配列番号10のプライマーはその5’末端側から順にHindIII、PstI、SalI、BamHI、SacI認識部位をそれぞれ有している。
<GAPDHプロモーター制御下、DOI合成酵素遺伝子(btrC)及びスクロース加水分解酵素遺伝子(cscA)発現ベクターの構築>
エシェリヒア・コリO-157株が有するスクロース加水分解酵素遺伝子(cscA)の塩基配列はすでに報告されている。すなわち、GenBank accession number AE005174に記載のエシェリヒア・コリO-157株ゲノム配列の3274383-3275816に記載されている。cscA遺伝子を取得するために、GCGGATCCGC TGGTGGAATA TATGACGCAA TCTCGATTGC(配列番号13)、及びGACGCGTCGA CTTAACCCAG TTGCCAGAGT GC(配列番号14)の塩基配列を有するオリゴヌクレオチドプライマーをそれぞれ合成した。配列番号13のプライマーはその5’末端側から順にBamHI認識部位と13塩基のGAPDH遺伝子のリボソーム結合配列を有している。配列番号14のプライマーはその5’末端側にSalI認識部位を有している。
<GAPDHプロモーター制御下、DOI合成酵素遺伝子(btrC)、スクロース加水分解酵素遺伝子(cscA)及びグルコース輸送促進蛋白質遺伝子(glf)発現ベクターの構築>
Zymomonas mobilis(ATCC 29191)が有するグルコース輸送促進蛋白質遺伝子(glf)の塩基配列はすでに報告されている(GenBank accession number M60615)。glf遺伝子を取得するために、CCTGTCGACG CTGGTGGAAT ATATGAGTTC TGAAAGTAGT CAGG(配列番号15)、及びCTACTGCAGC TACTTCTGGG AGCGCCACA(配列番号16)の塩基配列を有するオリゴヌクレオチドプライマーをそれぞれ合成した。配列番号15のプライマーはその5’末端側から順にSalI認識部位と13塩基のGAPDH遺伝子のリボソーム結合配列を有している。配列番号16のプライマーはその5’末端側にPstI認識部位を有している。
<pGAP-btrC-cscA及びpGAP-btrC-cscA-glfのMG1655ΔpgiΔzwf株への導入>
上記のプラスミドpGAP-btrC-cscAおよびpGAP-btrC-cscA-glfをそれぞれMG1655ΔpgiΔzwf株に形質転換し、アンピシリン50μg/mLを含むLB寒天プレートで37℃一晩培養することによりMG1655ΔpgiΔzwf/pGAP-btrC-cscA株およびMG1655ΔpgiΔzwf/pGAP-btrC-cscA-glf株を得た。
<MG1655ΔpgiΔzwf/pGAP-btrC-cscA株を用いたDOIの生産性と培養pHによるDOIの生産性の違い>
前培養として三角フラスコに入れたLB Broth, Miller培養液(Difco244620)25mlにエシェリヒア・コリMG1655ΔpgiΔzwf/pGAP-btrC-cscA株を植菌し、一晩120rpmで撹拌培養を行った後、下記組成の培地475gを入れた1L容培養槽(ABLE社製培養装置BMJ-01)に全量植菌した。培養は大気圧下、通気量1.0vvm、撹拌速度800rpm、培養温度30℃で行った。培養槽は全部で4台用い、pHを12.5%アンモニア水と2N塩酸を用いてpH7.0、pH6.5、pH6.0に調整した。また、660nmの吸光度を測定することにより、菌体濃度を測定した。培養開始時の菌体濃度は全て0.29であった。培養62時間後、得られた培養液中のDOI、スクロース、グルコース、フルクトースの量をHPLCで定法に従って測定した。また、660nmの吸光度を測定することにより、菌体濃度を測定した。培養に使用する培地の組成を下記表1に記載する。結果を表2に示す。


また、本実施例で用いた菌はpgi遺伝子とzwf遺伝子が同時に破壊されていることからグルコースを異化代謝できないはずであるが、スクロースを唯一の炭素源とした培地で62時間培養した結果、全ての試験区において菌は100倍以上に増えた。このことにより、本発明の大腸菌が、菌体の増殖/生育用に高価なマンニトール等を必要とせず、スクロースを分解して得られるフルクトースを菌体の増殖/生育用に利用していることが確認された。
<MG1655ΔpgiΔzwf/pGAP-btrC-cscA株及びMG1655ΔpgiΔzwf/pGAP-btrC-cscA-glf株によるDOIの生産>
前培養として三角フラスコに入れたLB Broth, Miller培養液(Difco244620)25mlにエシェリヒア・コリMG1655ΔpgiΔzwf/pGAP-btrC-cscA株とMG1655ΔpgiΔzwf/pGAP-btrC-cscA-glf株をそれぞれ植菌し、一晩120rpmで撹拌培養を行った後、下記組成の培地475gを入れた1L容培養槽(ABLE社製培養装置BMJ-01)にそれぞれ全量を植菌した。培養は大気圧下、通気量1.0vvm、撹拌速度800rpm、培養温度30℃、pH6.5(12.5%アンモニア水で調整)で行った。培養48時間後、得られた培養液中のDOIとスクロース、グルコースの量をHPLCで定法に従って測定した。培養に使用する培地の組成を下記表3に記載する。結果を表4に示す。


<BΔpgiΔzwf/pGAP-btrC-cscA-glf株及びMG1655ΔpgiΔzwf/pGAP-btrC-cscA-glf株による、工業用培地を用いたときのスクロースからのDOIの生産>
一般に工業生産では、培養培地成分として高価な酵母エキス等の代替としてコーンスティープリカーを使用する。コーンスティープリカー培地を用いた場合のDOIの生産性をエシェリヒア・コリの亜種であるMG1655株とB株で比較した。
エシェリヒア・コリB株(ATCC11303)に対しても実施例1~4に記載の方法でpgi遺伝子およびzwf遺伝子の破壊を行い、得られた株をBΔpgiΔzwf株と命名した。この株に対して、実施例8に記載の方法で発現ベクターpGAP-btrC-cscA-glfを導入し、得られた菌をBΔpgiΔzwf/pGAP-btrC-cscA-glf株と命名した。


<BΔpgiΔzwf/pGAP-btrC-cscA株、BΔpgiΔzwf/pGAP-btrC-cscA-glf株及びBΔpgiΔzwf/pGAP-btrC-cscA-pck株による廃糖蜜からのDOIの生産>
glf以外のグルコース輸送促進蛋白質遺伝子として、ホスホエノールピルビン酸カルボキシキナーゼ(Pck)を選定した。ホスホエノールピルビン酸カルボキシキナーゼ(Pck)は、オキサロ酢酸を基質としてホスホエノールピルビン酸を産生する蛋白質である。エシェリヒア・コリが本来もっているグルコースの取り込み系(PTS)はホスホエノールピルビン酸を必要とすることから、この蛋白質を高発現することによりホスホエノールピルビン酸の供給量が増加し、結果的にPTSを強化することができると予想される。


glf遺伝子の導入がDOI以外の物質生産にどのような影響を与えるか確認するために、D-乳酸生産細菌であるMG1655ΔpflΔdldΔmdhΔasp/GAPldhAゲノム挿入株にcscA遺伝子とglf遺伝子を導入し、乳酸の生成量を調べた。
なお、MG1655ΔpflΔdldΔmdhΔasp/GAPldhAゲノム挿入株とは、大腸菌MG1655が本来保有するピルベートホルメートリアーゼ(Pfl)、FAD依存性D-乳酸デヒドロゲナーゼ(Dld)、リンゴ酸デヒドロゲナーゼ(Mdh)、アスパラギン酸アンモニアリアーゼ(Asp)の各遺伝子が破壊されていて、NADH依存性D-乳酸デヒドロゲナーゼ(LdhA)の遺伝子が導入されていている株のことであり、WO2005/033324号において、120gのグルコースから98g/LのD-乳酸を生産することが確認されている。
エシェリヒア・コリのGAPDH遺伝子の塩基配列はすでに報告されている。グリセルアルデヒド3-リン酸デヒドロゲナーゼ(GAPDH)プロモーターを取得するため、配列番号9及び配列番号10の塩基配列を有するプライマーを合成した。配列番号9のプライマーはその5’末端側にNdeI認識部位を、配列番号10のプライマーはその5’末端側から順にHindIII、PstI、SalI、BamHI、SacI認識部位を有している。
上記2種のプライマーとともに、エシェリヒア・コリMG1655株のゲノムDNAをテンプレートに用い、通常条件でPCR法を行い、DNA断片を増幅した。得られたDNAフラグメントを制限酵素NdeIとHindIIIで消化することで約100bpのGAPDHプロモーターをコードするフラグメントを得た。次に上記のDNAフラグメントと、NdeI、HindIIIで消化した大腸菌用クローニングベクターpBR322(GenBank accession number J01749)を混合し、リガーゼを用いて結合した後、エシェリヒア・コリDH5αコンピテントセル(タカラバイオ社製)に形質転換し、アンピシリン50μg/mLを含むLB寒天プレートに生育する形質転換体を得た。得られたコロニーをアンピシリン50μg/mLを含むLB液体培地で30℃で一晩培養し、得られた菌体からプラスミドを回収した。このプラスミドをpGAPと命名した。
上記2種のプライマーとともに、エシェリヒア・コリO-157株のゲノムDNA(SIGMA-ALDRICH:IRMM449)をテンプレートとして通常条件でPCR法を行い、得られたDNAフラグメントを制限酵素BamHI及びSalIで消化することで約1.4kbpのスクロース加水分解酵素遺伝子(cscA)フラグメントを得た。このDNAフラグメントとプラスミドpGAPを制限酵素BamHI及びSalIで消化することで得られるフラグメントを混合し、リガーゼを用いて結合した後、エシェリヒア・コリDH5αコンピテントセル(タカラバイオ社製)に形質転換し、アンピシリン50μg/mLを含むLB寒天プレートに生育する形質転換体を得た。得られたコロニーをアンピシリン50μg/mLを含むLB液体培地で30℃で一晩培養し、得られた菌体からプラスミドpGAP-cscAを回収して、スクロース加水分解酵素遺伝子(cscA)発現ベクターを構築した。
上記2種のプライマーとともに、Zymomonas mobilisのゲノムDNAをテンプレートとして通常条件でPCR法を行い、得られたDNAフラグメントを制限酵素SalI及びPstIで消化することで約1.4kbpのグルコース輸送促進蛋白質遺伝子(glf)フラグメントを得た。このDNAフラグメントとプラスミドpGAP-cscAを制限酵素SalI及びPstIで消化することで得られるフラグメントを混合し、リガーゼを用いて結合した後、エシェリヒア・コリDH5αコンピテントセル(タカラバイオ社製)に形質転換し、アンピシリン50μg/mLを含むLB寒天プレートに生育する形質転換体を得た。得られたコロニーをアンピシリン50μg/mLを含むLB液体培地で30℃で一晩培養し、得られた菌体からプラスミドpGAP-cscA-glfを回収して、スクロース加水分解酵素遺伝子(cscA)及びグルコース輸送促進蛋白質遺伝子(glf)発現ベクターを構築した。
前培養として、三角フラスコに50μg/mLのアンピシリンを含むLB Broth, Miller培養液(Difco244620)20mlを入れ、エシェリヒア・コリMG1655ΔpflΔdldΔmdhΔasp/pGAP-cscA株及びMG1655ΔpflΔdldΔmdhΔasp/pGAP-cscA-glf株をそれぞれ植菌し、9時間、30℃、200rpmで撹拌培養を行った。前培養液0.2mLを下記組成の培地20mLを入れた100mL容量のフラスコにn=4で植菌し、撹拌速度90rpm、培養温度35℃で培養を行った。培養に使用する培地の組成を下記表9に記載する。なお、スクロースはフィルター滅菌して用いた。得られた培養液中のD-乳酸とスクロースの定量はHPLCで定法に従って測定した。結果を表10に記載する。

本明細書に記載された全ての文献、特許出願、及び技術規格は、個々の文献、特許出願、及び技術規格が参照により取り込まれることが具体的かつ個々に記された場合と同程度に、本明細書中に参照により取り込まれる。
Claims (13)
- スクロース非PTS遺伝子群の中で、少なくともスクロース加水分解酵素(CscA)をコードする遺伝子を有すると共に、2-デオキシ-シロ-イノソース(DOI)生産系が付与又は強化されたDOI生産大腸菌。
- スクロース非PTS遺伝子群の中で、スクロース加水分解酵素(CscA)をコードする遺伝子のみを有する請求項1に記載のDOI生産大腸菌。
- 前記DOI生産大腸菌が、糖取り込み能力強化系を有する請求項1に記載のDOI生産大腸菌。
- 前記DOI生産大腸菌が、該大腸菌が本来有しているホスホグルコースイソメラーゼ(Pgi)、グルコース-6-リン酸-1-デヒドロゲナーゼ(Zwf)、ホスホグルコムターゼ(Pgm)及び定常期における蛋白質合成の修飾を担うリボソーム修飾因子(Rmf)からなる群より選択された少なくとも1つの活性が、不活化あるいは低減化されている請求項3に記載のDOI生産大腸菌。
- 前記DOI生産系が、DOI合成酵素(BtrC)活性に由来する請求項1に記載のDOI生産大腸菌。
- 前記糖取り込み能力強化系が、グルコース輸送促進蛋白質(Glf)活性の強化である請求項3に記載のDOI生産大腸菌。
- 前記スクロース加水分解酵素(CscA)をコードする遺伝子が、エシェリヒア・コリ細菌に由来する請求項1に記載のDOI生産大腸菌。
- 前記エシェリヒア・コリ細菌が、エシェリヒア・コリO-157細菌である請求項7に記載のDOI生産大腸菌。
- 前記グルコース輸送促進蛋白質(Glf)がザイモモナス属細菌に由来する請求項6に記載のDOI生産大腸菌。
- 前記ザイモモナス属細菌がザイモモナス・モビリス細菌である請求項9に記載のDOI生産大腸菌。
- 前記DOI生産大腸菌が、本来スクロース資化能を持たない種類の大腸菌である請求項1に記載のDOI生産大腸菌。
- 前記DOI生産大腸菌が、B株又はその由来株である請求項11に記載のDOI生産大腸菌。
- 請求項1に記載のDOI生産大腸菌を用いて、スクロースを含む植物由来原料からDOIを生産することを含むDOIの生産方法。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09824752.1A EP2371952B1 (en) | 2008-11-05 | 2009-10-30 | Bacterium capable of producing 2-deoxy-scyllo-inosose (doi), and process for producing 2-deoxy-scyllo-inosose (doi) by using same |
| KR1020117010937A KR101324369B1 (ko) | 2008-11-05 | 2009-10-30 | 2-디옥시-실로-이노소스(doi) 생산 세균 및 이를 이용한 2-디옥시-실로-이노소스(doi) 생산 방법 |
| US13/126,154 US9150868B2 (en) | 2008-11-05 | 2009-10-30 | Bacterium producing 2-deoxy-scyllo-inosose (DOI) and method of producing 2-deoxy-scyllo-inosose (DOI) by using same |
| JP2010536758A JP5254353B2 (ja) | 2008-11-05 | 2009-10-30 | 2−デオキシ−シロ−イノソース(doi)生産細菌及びこれを用いた2−デオキシ−シロ−イノソース(doi)生産方法 |
| CN200980143112.XA CN102197135B (zh) | 2008-11-05 | 2009-10-30 | 生产2-脱氧蟹肌醇(doi)的细菌及使用其生产2-脱氧蟹肌醇(doi)的方法 |
| BRPI0919755A BRPI0919755B8 (pt) | 2008-11-05 | 2009-10-30 | escherichia coli produtora de 2-desóxi-scilo-inosose (doi) e método de produção de doi |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008-284639 | 2008-11-05 | ||
| JP2008284639 | 2008-11-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010053052A1 true WO2010053052A1 (ja) | 2010-05-14 |
Family
ID=42152862
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/068666 WO2010053052A1 (ja) | 2008-11-05 | 2009-10-30 | 2-デオキシ-シロ-イノソース(doi)生産細菌及びこれを用いた2-デオキシ-シロ-イノソース(doi)生産方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9150868B2 (ja) |
| EP (1) | EP2371952B1 (ja) |
| JP (1) | JP5254353B2 (ja) |
| KR (1) | KR101324369B1 (ja) |
| CN (1) | CN102197135B (ja) |
| BR (1) | BRPI0919755B8 (ja) |
| WO (1) | WO2010053052A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012007481A3 (en) * | 2010-07-12 | 2012-03-15 | Universiteit Gent | Metabolically engineered organisms for the production of added value bio-products |
| WO2012082720A2 (en) | 2010-12-13 | 2012-06-21 | Myriant Corporation | Method of producing succinic acid and other chemicals using sucrose-containing feedstock |
| WO2015005451A1 (ja) * | 2013-07-12 | 2015-01-15 | 三井化学株式会社 | 2-デオキシ-シロ-イノソースの生産方法 |
| WO2015093320A1 (ja) | 2013-12-16 | 2015-06-25 | 旭化成ケミカルズ株式会社 | 2-デオキシ-シロ-イノソース還元酵素 |
| JP2016526919A (ja) * | 2013-08-05 | 2016-09-08 | グリーンライト バイオサイエンシーズ インコーポレーテッドGreenlight Biosciences,Inc. | プロテアーゼ切断部位を有する操作されたタンパク質 |
| WO2018180568A1 (ja) | 2017-03-27 | 2018-10-04 | 学校法人 新潟科学技術学園 | 変異型2-デオキシ-シロ-イノソース合成酵素 |
| US10316342B2 (en) | 2017-01-06 | 2019-06-11 | Greenlight Biosciences, Inc. | Cell-free production of sugars |
| WO2020085435A1 (ja) | 2018-10-25 | 2020-04-30 | 株式会社Ihi | トリヒドロキシベンゼンの製造方法 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8647642B2 (en) | 2008-09-18 | 2014-02-11 | Aviex Technologies, Llc | Live bacterial vaccines resistant to carbon dioxide (CO2), acidic PH and/or osmolarity for viral infection prophylaxis or treatment |
| US11926858B2 (en) | 2014-06-27 | 2024-03-12 | Glycom A/S | Oligosaccharide production |
| US10731193B2 (en) | 2014-06-27 | 2020-08-04 | Glycom A/S | Oligosaccharide production |
| US11180535B1 (en) | 2016-12-07 | 2021-11-23 | David Gordon Bermudes | Saccharide binding, tumor penetration, and cytotoxic antitumor chimeric peptides from therapeutic bacteria |
| US11129906B1 (en) | 2016-12-07 | 2021-09-28 | David Gordon Bermudes | Chimeric protein toxins for expression by therapeutic bacteria |
| PL3620510T3 (pl) | 2018-09-06 | 2024-02-19 | Chr. Hansen HMO GmbH | Fermentacyjne wytwarzanie oligosacharydów przez całkowitą fermentację z wykorzystaniem mieszanego surowca |
| EP3702468A1 (en) | 2019-03-01 | 2020-09-02 | Jennewein Biotechnologie GmbH | Fermentative production of carbohydrates by microbial cells utilizing a mixed feedstock |
| CN115803443A (zh) | 2020-07-13 | 2023-03-14 | 格礼卡姆股份公司 | 寡糖的制造 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001346578A (ja) * | 2000-04-26 | 2001-12-18 | Ajinomoto Co Inc | アミノ酸生産菌及びアミノ酸の製造法 |
| JP2002512802A (ja) * | 1998-04-24 | 2002-05-08 | ディーエスエム ビオテック ゲーエムベーハー | 芳香族代謝/iii物質の微生物による製造 |
| JP2006503559A (ja) * | 2002-10-09 | 2006-02-02 | フォルシュングスツェントルム ユーリッヒ ゲゼルシャフト ミット ベシュレンクテル ハフツング | D−マンニトールを製造するための方法並びに微生物 |
| WO2006109479A1 (ja) * | 2005-03-30 | 2006-10-19 | Niigata Bio-Research Park, Inc. | 遺伝子発現カセット及び形質転換体、並びにこの形質転換体を用いた2-デオキシ-シロ-イノソースの製造方法及び2-デオキシ-シロ-イノソースの精製方法 |
| WO2007041269A2 (en) * | 2005-09-29 | 2007-04-12 | E. I. Du Pont De Nemours And Company | Fermentive production of four carbon alcohols |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3122762B2 (ja) | 1999-02-22 | 2001-01-09 | 東京工業大学長 | 2−デオキシ−シロ−イノソース合成酵素、アミノ酸配列、遺伝子塩基配列 |
| CN101970648A (zh) | 2007-12-18 | 2011-02-09 | 韩国科学技术院 | 具有使用蔗糖作为碳源能力的重组微生物 |
-
2009
- 2009-10-30 JP JP2010536758A patent/JP5254353B2/ja active Active
- 2009-10-30 BR BRPI0919755A patent/BRPI0919755B8/pt active IP Right Grant
- 2009-10-30 WO PCT/JP2009/068666 patent/WO2010053052A1/ja active Application Filing
- 2009-10-30 US US13/126,154 patent/US9150868B2/en active Active
- 2009-10-30 EP EP09824752.1A patent/EP2371952B1/en active Active
- 2009-10-30 CN CN200980143112.XA patent/CN102197135B/zh active Active
- 2009-10-30 KR KR1020117010937A patent/KR101324369B1/ko active IP Right Grant
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002512802A (ja) * | 1998-04-24 | 2002-05-08 | ディーエスエム ビオテック ゲーエムベーハー | 芳香族代謝/iii物質の微生物による製造 |
| JP2001346578A (ja) * | 2000-04-26 | 2001-12-18 | Ajinomoto Co Inc | アミノ酸生産菌及びアミノ酸の製造法 |
| JP2006503559A (ja) * | 2002-10-09 | 2006-02-02 | フォルシュングスツェントルム ユーリッヒ ゲゼルシャフト ミット ベシュレンクテル ハフツング | D−マンニトールを製造するための方法並びに微生物 |
| WO2006109479A1 (ja) * | 2005-03-30 | 2006-10-19 | Niigata Bio-Research Park, Inc. | 遺伝子発現カセット及び形質転換体、並びにこの形質転換体を用いた2-デオキシ-シロ-イノソースの製造方法及び2-デオキシ-シロ-イノソースの精製方法 |
| WO2006112000A1 (ja) * | 2005-03-30 | 2006-10-26 | The Niigata Institute Of Science And Technology | 大腸菌変異株による2-デオキシ-シロ-イノソースの合成および精製する方法並びに得られた2-デオキシ-シロ-イノソース |
| WO2007041269A2 (en) * | 2005-09-29 | 2007-04-12 | E. I. Du Pont De Nemours And Company | Fermentive production of four carbon alcohols |
Non-Patent Citations (7)
| Title |
|---|
| JAHREIS, K. ET AL.: "Adaptation of Sucrose Metabolism in the Escherichia coli Wild-type Strain EC3132.", J. BACTERIOL., vol. 184, no. 19, 2002, pages 5307 - 5316, XP002984526 * |
| KAUP, B. ET AL.: "Metabolic Engineering of Escherichia coli: Construction of an Efficient Biocatalyst for D-Mannitol Formation in a Whole -cell Biotransformation.", APPL. MICROBIOL. BIOTECHNOL., vol. 64, no. 3, 2004, pages 333 - 339, XP002268517 * |
| KOGURE, T. ET AL.: "Efficient Production of 2- Deoxy-scyllo-inosose from D-Glucose by Metabolically Engineered Recombinant Escherichia coli.", J. BIOTECHNOL., vol. 129, no. 3, 2007, pages 502 - 509, XP022027139 * |
| OLSON, M.M. ET AL.: "Production of Tyrosine from Sucrose or Glucose Achieved by Rapid Genetic Changes to Phenylalanine-producing Escherichia coli Strains.", APPL. MICROBIOL. BIOTECHNOL., vol. 74, no. 5, 2007, pages 1031 - 1040, XP019489541 * |
| SAHIN-TOTH, M. ET AL.: "Cloning, Sequencing, and Expression of cscA Invertase from Escherichia coli B-62.", CAN. J. MICROBIOL., vol. 45, no. 5, 1999, pages 418 - 422, XP008140063 * |
| See also references of EP2371952A4 * |
| SHUKLA, V.B. ET AL.: "Production of D(-)-Lactate from Sucrose and Molasses.", BIOTECHNOL. LETT., vol. 26, no. 9, 2004, pages 689 - 693, XP002367980 * |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12098403B2 (en) * | 2010-07-12 | 2024-09-24 | Inbiose N.V. | Metabolically engineered organisms for the production of added value bio-products |
| JP2018121641A (ja) * | 2010-07-12 | 2018-08-09 | インビオース エヌ.ヴェ.Inbiose N.V. | 付加価値バイオ生成物の生成のための代謝操作された生物 |
| CN103025874A (zh) * | 2010-07-12 | 2013-04-03 | 根特大学 | 用于生产附加值生物产品的代谢改造的生物 |
| JP2013535962A (ja) * | 2010-07-12 | 2013-09-19 | ユニヴェルシテイト ヘント | 付加価値バイオ生成物の生成のための代謝操作された生物 |
| US11384374B2 (en) | 2010-07-12 | 2022-07-12 | Inbiose N.V. | Metabolically engineered organisms for the production of added value bio-products |
| AU2011278315B2 (en) * | 2010-07-12 | 2015-05-28 | Inbiose N.V. | Metabolically engineered organisms for the production of added value bio-products |
| US20210017558A1 (en) * | 2010-07-12 | 2021-01-21 | Inbiose N.V. | Metabolically engineered organisms for the production of added value bio-products |
| WO2012007481A3 (en) * | 2010-07-12 | 2012-03-15 | Universiteit Gent | Metabolically engineered organisms for the production of added value bio-products |
| US9701992B2 (en) | 2010-07-12 | 2017-07-11 | Universiteit Gent | Metabolically engineered organisms for the production of added value bio-products |
| EP4242320A3 (en) * | 2010-07-12 | 2023-11-29 | Inbiose N.V. | Metabolically engineered organisms for the production of added value bio-products |
| JP2017018093A (ja) * | 2010-07-12 | 2017-01-26 | ユニヴェルシテイト ヘントUniversiteit Gent | 付加価値バイオ生成物の生成のための代謝操作された生物 |
| EP3235907A1 (en) * | 2010-07-12 | 2017-10-25 | Universiteit Gent | Metabolically engineered organisms for the production of added value bio-products |
| US10570430B2 (en) | 2010-07-12 | 2020-02-25 | Inbiose N.V. | Metabolically engineered organisms for the production of added value bio-products |
| CN107603935A (zh) * | 2010-07-12 | 2018-01-19 | 因比奥斯公司 | 用于生产附加值生物产品的代谢改造的生物 |
| US9845513B2 (en) | 2010-12-13 | 2017-12-19 | Myriant Corporation | Method of producing succinic acid and other chemicals using sucrose-containing feedstock |
| WO2012082720A2 (en) | 2010-12-13 | 2012-06-21 | Myriant Corporation | Method of producing succinic acid and other chemicals using sucrose-containing feedstock |
| JPWO2015005451A1 (ja) * | 2013-07-12 | 2017-03-02 | 三井化学株式会社 | 2−デオキシ−シロ−イノソースの生産方法 |
| WO2015005451A1 (ja) * | 2013-07-12 | 2015-01-15 | 三井化学株式会社 | 2-デオキシ-シロ-イノソースの生産方法 |
| JP2016526919A (ja) * | 2013-08-05 | 2016-09-08 | グリーンライト バイオサイエンシーズ インコーポレーテッドGreenlight Biosciences,Inc. | プロテアーゼ切断部位を有する操作されたタンパク質 |
| US10421953B2 (en) | 2013-08-05 | 2019-09-24 | Greenlight Biosciences, Inc. | Engineered proteins with a protease cleavage site |
| WO2015093320A1 (ja) | 2013-12-16 | 2015-06-25 | 旭化成ケミカルズ株式会社 | 2-デオキシ-シロ-イノソース還元酵素 |
| US10577635B2 (en) | 2017-01-06 | 2020-03-03 | Greenlight Biosciences, Inc. | Cell-free production of sugars |
| US10704067B2 (en) | 2017-01-06 | 2020-07-07 | Greenlight Biosciences, Inc. | Cell-free production of sugars |
| US10316342B2 (en) | 2017-01-06 | 2019-06-11 | Greenlight Biosciences, Inc. | Cell-free production of sugars |
| US12110526B2 (en) | 2017-01-06 | 2024-10-08 | Greenlight Biosciences, Inc. | Cell-free production of sugars |
| US11499175B2 (en) | 2017-03-27 | 2022-11-15 | Mitsui Chemicals, Inc. | Mutant type 2-deoxy-scyllo-inosose synthase |
| WO2018180568A1 (ja) | 2017-03-27 | 2018-10-04 | 学校法人 新潟科学技術学園 | 変異型2-デオキシ-シロ-イノソース合成酵素 |
| WO2020085435A1 (ja) | 2018-10-25 | 2020-04-30 | 株式会社Ihi | トリヒドロキシベンゼンの製造方法 |
| US11685704B2 (en) | 2018-10-25 | 2023-06-27 | Mitsui Chemicals, Inc. | Trihydroxybenzene production method |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2371952B1 (en) | 2016-07-27 |
| KR20110084251A (ko) | 2011-07-21 |
| EP2371952A4 (en) | 2012-08-22 |
| JPWO2010053052A1 (ja) | 2012-04-05 |
| BRPI0919755B1 (pt) | 2019-01-22 |
| JP5254353B2 (ja) | 2013-08-07 |
| US9150868B2 (en) | 2015-10-06 |
| BRPI0919755A2 (pt) | 2016-08-23 |
| BRPI0919755B8 (pt) | 2019-06-04 |
| EP2371952A1 (en) | 2011-10-05 |
| US20110207187A1 (en) | 2011-08-25 |
| KR101324369B1 (ko) | 2013-11-01 |
| CN102197135A (zh) | 2011-09-21 |
| CN102197135B (zh) | 2014-08-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5254353B2 (ja) | 2−デオキシ−シロ−イノソース(doi)生産細菌及びこれを用いた2−デオキシ−シロ−イノソース(doi)生産方法 | |
| CA2737428C (en) | Bacterium capable of producing lactic acid, and method for producing lactic acid | |
| JP3692538B2 (ja) | 新規リジンデカルボキシラーゼ遺伝子及びl−リジンの製造法 | |
| TW200306352A (en) | Method for producing L-amino acid | |
| JP5243546B2 (ja) | 植物由来原料から乳酸を生産する方法及び乳酸生産細菌 | |
| CA2947663A1 (en) | Genetically modified microorganism for improved production of fine chemicals on sucrose | |
| JPWO2020208842A5 (ja) | ||
| JPWO2010032698A6 (ja) | 植物由来原料から乳酸を生産する方法及び乳酸生産細菌 | |
| EP3102675A1 (en) | Improved microorganisms for succinic acid production | |
| JP6321645B2 (ja) | 2−デオキシ−シロ−イノソースの生産方法 | |
| WO2016030373A1 (en) | Modified microorganism for improved production of fine chemicals on sucrose | |
| JP5568562B2 (ja) | イソプロピルアルコール生産細菌及びイソプロピルアルコール生産方法 | |
| WO2011052482A1 (ja) | イソプロピルアルコール生産細菌及びイソプロピルアルコール生産方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980143112.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09824752 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010536758 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13126154 Country of ref document: US |
|
| REEP | Request for entry into the european phase |
Ref document number: 2009824752 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009824752 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20117010937 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: PI0919755 Country of ref document: BR Kind code of ref document: A2 Effective date: 20110426 |