WO2009121228A2 - 鲁比前列酮晶体、其制备方法及用途 - Google Patents
鲁比前列酮晶体、其制备方法及用途 Download PDFInfo
- Publication number
- WO2009121228A2 WO2009121228A2 PCT/CN2008/070971 CN2008070971W WO2009121228A2 WO 2009121228 A2 WO2009121228 A2 WO 2009121228A2 CN 2008070971 W CN2008070971 W CN 2008070971W WO 2009121228 A2 WO2009121228 A2 WO 2009121228A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lubiprostone
- crystal
- crystals
- solvent
- ray powder
- Prior art date
Links
- WGFOBBZOWHGYQH-MXHNKVEKSA-N lubiprostone Chemical compound O1[C@](C(F)(F)CCCC)(O)CC[C@@H]2[C@@H](CCCCCCC(O)=O)C(=O)C[C@H]21 WGFOBBZOWHGYQH-MXHNKVEKSA-N 0.000 title claims abstract description 105
- 229960000345 lubiprostone Drugs 0.000 title claims abstract description 104
- 239000013078 crystal Substances 0.000 title claims abstract description 89
- 238000002360 preparation method Methods 0.000 title claims abstract description 16
- 238000000634 powder X-ray diffraction Methods 0.000 claims abstract description 17
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 11
- 208000010643 digestive system disease Diseases 0.000 claims abstract description 6
- 208000018685 gastrointestinal system disease Diseases 0.000 claims abstract description 6
- 239000000203 mixture Substances 0.000 claims description 32
- 238000000034 method Methods 0.000 claims description 25
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 21
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 21
- 239000003814 drug Substances 0.000 claims description 20
- 239000002904 solvent Substances 0.000 claims description 19
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 18
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 16
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 15
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 15
- 208000018522 Gastrointestinal disease Diseases 0.000 claims description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 11
- 239000013543 active substance Substances 0.000 claims description 10
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 9
- 239000003937 drug carrier Substances 0.000 claims description 9
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 8
- 238000011282 treatment Methods 0.000 claims description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 8
- 239000002798 polar solvent Substances 0.000 claims description 5
- 238000001816 cooling Methods 0.000 claims description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 4
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 claims description 3
- 238000001228 spectrum Methods 0.000 abstract description 9
- 206010010774 Constipation Diseases 0.000 abstract description 7
- 239000000243 solution Substances 0.000 description 14
- 229940079593 drug Drugs 0.000 description 12
- 239000007787 solid Substances 0.000 description 9
- 230000037396 body weight Effects 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 230000000968 intestinal effect Effects 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- SUBDBMMJDZJVOS-UHFFFAOYSA-N 5-methoxy-2-{[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl}-1H-benzimidazole Chemical compound N=1C2=CC(OC)=CC=C2NC=1S(=O)CC1=NC=C(C)C(OC)=C1C SUBDBMMJDZJVOS-UHFFFAOYSA-N 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 238000001035 drying Methods 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 229960003174 lansoprazole Drugs 0.000 description 5
- MJIHNNLFOKEZEW-UHFFFAOYSA-N lansoprazole Chemical compound CC1=C(OCC(F)(F)F)C=CN=C1CS(=O)C1=NC2=CC=CC=C2N1 MJIHNNLFOKEZEW-UHFFFAOYSA-N 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 229960000381 omeprazole Drugs 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 229940126409 proton pump inhibitor Drugs 0.000 description 5
- 239000000612 proton pump inhibitor Substances 0.000 description 5
- 238000010521 absorption reaction Methods 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 230000001737 promoting effect Effects 0.000 description 4
- -1 prostaglandin compound Chemical class 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- 238000002329 infrared spectrum Methods 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- 238000002441 X-ray diffraction Methods 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 238000000113 differential scanning calorimetry Methods 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 210000003608 fece Anatomy 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 230000008855 peristalsis Effects 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 150000003180 prostaglandins Chemical class 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- AWQSAIIDOMEEOD-UHFFFAOYSA-N 5,5-Dimethyl-4-(3-oxobutyl)dihydro-2(3H)-furanone Chemical compound CC(=O)CCC1CC(=O)OC1(C)C AWQSAIIDOMEEOD-UHFFFAOYSA-N 0.000 description 1
- 108010062745 Chloride Channels Proteins 0.000 description 1
- 102000011045 Chloride Channels Human genes 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 229940040386 amitiza Drugs 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000003467 chloride channel stimulating agent Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000000295 emission spectrum Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 239000012925 reference material Substances 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/94—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems condensed with rings other than six-membered or with ring systems containing such rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C405/00—Compounds containing a five-membered ring having two side-chains in ortho position to each other, and having oxygen atoms directly attached to the ring in ortho position to one of the side-chains, one side-chain containing, not directly attached to the ring, a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, and the other side-chain having oxygen atoms attached in gamma-position to the ring, e.g. prostaglandins ; Analogues or derivatives thereof
- C07C405/0008—Analogues having the carboxyl group in the side-chains replaced by other functional groups
- C07C405/0025—Analogues having the carboxyl group in the side-chains replaced by other functional groups containing keto groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
Definitions
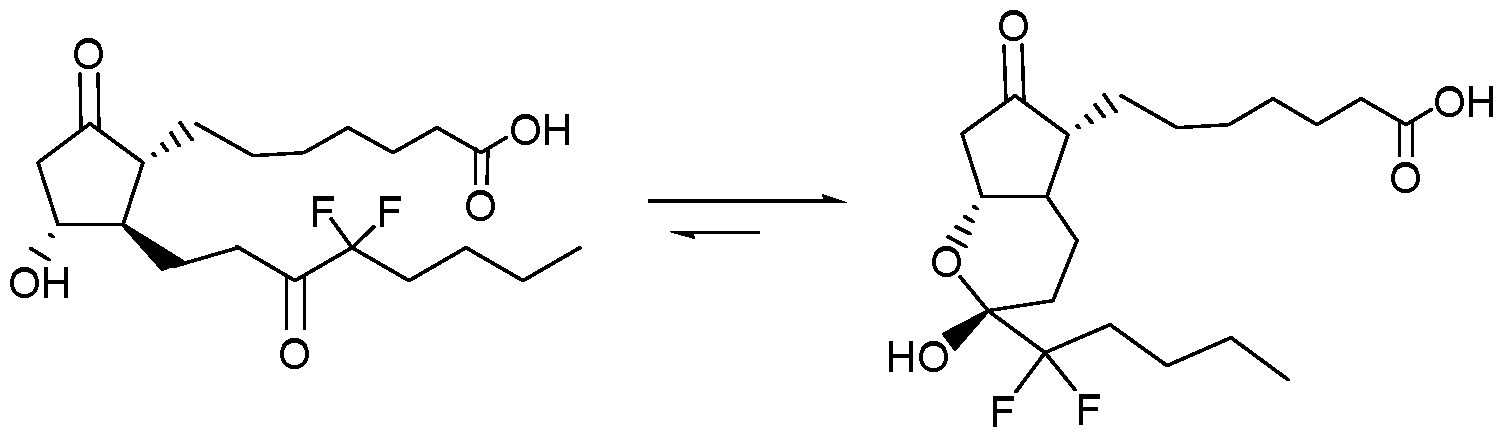
- a pharmaceutical composition comprising: the lubiprostone crystal of the invention or the lubiprostone crystal prepared by the method described above; and pharmaceutically acceptable Carrier.
- the lubiprostone crystal of the present invention has a specific crystal morphology which has a specific characteristic peak in an X-ray diffraction pattern.
- the lubiprostone crystal of the present invention has the following main features 2 ⁇ absorption characteristic peaks: 14.6 ⁇ 0.2 °, 17.0 ⁇ 0.2 °, and 19.6 ⁇ 0.2 °.
- the map also has the following characteristics: 2 ⁇ absorption characteristic peaks: 7.6 ⁇ 0.2 °, 8.5 ⁇ 0.2 °, 10.6 ⁇ 0.2 °, 17.7 ⁇ 0.2 °, 20.1 ⁇ 0.2 °, and 23.4 ⁇ 0.2 °.
- the lubiprostone crystal has an X-ray diffraction pattern substantially identical to that of Figure 1.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Pyrane Compounds (AREA)
Description

Claims
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08748577.7A EP2275419B1 (en) | 2008-04-01 | 2008-05-15 | A lubiprostone crystal, its preparation process and its use |
| US12/935,859 US8748482B2 (en) | 2008-04-01 | 2008-05-15 | Lubiprostone crystal, the use and the method for the preparation thereof |
| JP2011502211A JP2011516430A (ja) | 2008-04-01 | 2008-05-15 | ルビプロストン結晶、その製造方法および用途 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2008100354481A CN101318948B (zh) | 2008-04-01 | 2008-04-01 | 鲁比前列酮晶体、其制备方法及用途 |
| CN200810035448.1 | 2008-04-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009121228A2 true WO2009121228A2 (zh) | 2009-10-08 |
Family
ID=40179250
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2008/070971 WO2009121228A2 (zh) | 2008-04-01 | 2008-05-15 | 鲁比前列酮晶体、其制备方法及用途 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8748482B2 (zh) |
| EP (1) | EP2275419B1 (zh) |
| JP (1) | JP2011516430A (zh) |
| CN (1) | CN101318948B (zh) |
| WO (1) | WO2009121228A2 (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011091513A1 (en) | 2010-01-28 | 2011-08-04 | Apotex Pharmachem Inc. | Polymorphic forms of lubiprostone |
| US8513441B2 (en) | 2008-08-29 | 2013-08-20 | Alphora Research Inc. | Prostaglandin synthesis and intermediates for use therein |
| CN104710398A (zh) * | 2015-02-17 | 2015-06-17 | 齐鲁制药有限公司 | 鲁比前列酮的新晶型及其制备方法 |
| US10253011B1 (en) | 2018-07-13 | 2019-04-09 | Chirogate International Inc. | Lubiprostone crystals and methods for preparing the same |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8569279B2 (en) | 2009-05-27 | 2013-10-29 | Sucampo Ag | Method for modulating claudin mediated functions |
| JP2012528077A (ja) * | 2009-05-27 | 2012-11-12 | スキャンポ・アーゲー | クローディン媒介機能の調節および皮膚疾患の処置に使用するための、プロスタグランジン誘導体を含む医薬組成物 |
| CN102020625B (zh) * | 2009-09-22 | 2013-04-17 | 上海天伟生物制药有限公司 | 一种高纯度的鲁比前列酮及其制备方法和用途 |
| WO2013161973A1 (en) * | 2012-04-23 | 2013-10-31 | Sucampo Ag | Method for treating irritable bowel syndrome with diarrhea |
| CN104667259A (zh) * | 2015-03-26 | 2015-06-03 | 深圳市健元医药科技有限公司 | 治疗慢性便秘的药物组合物胶囊及其制备方法 |
| CN107474033A (zh) * | 2016-06-07 | 2017-12-15 | 北京深蓝海生物医药科技有限公司 | 一种精制鲁比前列酮的方法 |
| US11534404B2 (en) | 2016-10-06 | 2022-12-27 | Sucampo Ag | Multilayer beads for pharmaceutical use |
| CN109280074A (zh) * | 2017-07-20 | 2019-01-29 | 歌礼药业(浙江)有限公司 | 丹诺瑞韦钠晶体及其制备方法 |
| CN109988089B (zh) * | 2018-05-29 | 2022-04-26 | 上海凡秦医药科技有限公司 | 一种前列腺素化合物、制备方法及其用途 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5254588A (en) * | 1989-11-22 | 1993-10-19 | Kabushikikaisha Ueno Seiyaku Oyo Kenkyujo | Treatment of pulmonary dysfunction with 15-ketoprostaglandin compounds |
| JP4332316B2 (ja) | 1999-10-15 | 2009-09-16 | スキャンポ・アーゲー | 二環式化合物組成物およびその安定化方法 |
| US6414016B1 (en) * | 2000-09-05 | 2002-07-02 | Sucampo, A.G. | Anti-constipation composition |
| BR0212233A (pt) * | 2001-08-31 | 2004-10-05 | Sucampo Ag | Abridor de canal de cloreto |
| WO2007091697A2 (en) * | 2006-02-07 | 2007-08-16 | R-Tech Ueno, Ltd. | Method for preparing prostaglandin derivative |
-
2008
- 2008-04-01 CN CN2008100354481A patent/CN101318948B/zh active Active
- 2008-05-15 US US12/935,859 patent/US8748482B2/en active Active
- 2008-05-15 WO PCT/CN2008/070971 patent/WO2009121228A2/zh active Application Filing
- 2008-05-15 EP EP08748577.7A patent/EP2275419B1/en not_active Not-in-force
- 2008-05-15 JP JP2011502211A patent/JP2011516430A/ja active Pending
Non-Patent Citations (1)
| Title |
|---|
| None |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8513441B2 (en) | 2008-08-29 | 2013-08-20 | Alphora Research Inc. | Prostaglandin synthesis and intermediates for use therein |
| WO2011091513A1 (en) | 2010-01-28 | 2011-08-04 | Apotex Pharmachem Inc. | Polymorphic forms of lubiprostone |
| US20130096325A1 (en) * | 2010-01-28 | 2013-04-18 | Apotex Pharmachem Inc. | Polymorphic forms of lubiprostone |
| US8785663B2 (en) * | 2010-01-28 | 2014-07-22 | Alfredo Paul Ceccarelli | Polymorphic forms of Lubiprostone |
| CN104710398A (zh) * | 2015-02-17 | 2015-06-17 | 齐鲁制药有限公司 | 鲁比前列酮的新晶型及其制备方法 |
| US10253011B1 (en) | 2018-07-13 | 2019-04-09 | Chirogate International Inc. | Lubiprostone crystals and methods for preparing the same |
| EP3594210A1 (en) | 2018-07-13 | 2020-01-15 | Chirogate International Inc. | Lubiprostone crystals and methods for preparing the same |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2275419B1 (en) | 2017-09-06 |
| CN101318948A (zh) | 2008-12-10 |
| EP2275419A2 (en) | 2011-01-19 |
| US20110028541A1 (en) | 2011-02-03 |
| EP2275419A4 (en) | 2016-03-30 |
| CN101318948B (zh) | 2011-04-27 |
| US8748482B2 (en) | 2014-06-10 |
| JP2011516430A (ja) | 2011-05-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009121228A2 (zh) | 鲁比前列酮晶体、其制备方法及用途 | |
| TWI619495B (zh) | C-met調節劑醫藥組合物 | |
| WO2013104317A1 (zh) | 一种前列腺素类似物的晶型及其制备方法和用途 | |
| WO2013104318A1 (zh) | 一种前列腺素类似物的晶型及其制备方法和用途 | |
| JP5899214B2 (ja) | 4−[−2−[[5−メチル−1−(2−ナフタレニル)−1h−ピラゾール−3−イル]オキシ]エチル]モルホリン塩酸塩の固体形態 | |
| US20220298117A1 (en) | Crystalline forms of [3-(4- {2-butyl-1-[4-(4-chloro-phenoxy)-phenyl]-1h-imidazol-4-yl} -phenoxy)-propyl]-diethyl-amine | |
| TW201945369A (zh) | 盛格列汀的鹽、其製備方法與用途以及藥物組合物 | |
| KR20180111797A (ko) | 퀴놀론 유사체의 결정형 및 그것의 염 | |
| CN104230912B (zh) | 喹啉衍生物、其制备方法及其用途 | |
| WO2023193563A1 (zh) | 一种噻吩并吡啶化合物的晶型a、制备方法及其药物组合物 | |
| JP6965274B2 (ja) | ナトリウム−グルコース結合輸送体阻害剤のアミン溶媒和物、その調製方法およびその適用 | |
| US12030886B2 (en) | Form of ponatinib | |
| JP7466642B2 (ja) | レンバチニブメシル酸塩結晶形xi及びその調製方法 | |
| US20220251091A1 (en) | Amorphous umbralisib monotosylate | |
| TWI808217B (zh) | 稠合三環γ-胺基酸衍生物之組合物及其製備方法 | |
| US20180170919A1 (en) | Crystaline forms of n-[6-(cis-2,6-dimethylmorpholine -4-yl)pyridine-3-yl]-2-methyl-4'-(trifluoromethoxy) [1,1'-biphenyl]-3-methanamide monophosphate, and process of preparation thereof | |
| JP2022517396A (ja) | Egfr阻害剤の塩、結晶形及びその製造方法 | |
| CN107235960B (zh) | 酰胺类衍生物、其制备方法及其在医药上的用途 | |
| TWI814468B (zh) | 藥用組合物、其製備方法及用途 | |
| WO2023197934A1 (zh) | 阿片受体拮抗剂缀合物的固体盐型、晶型及其制备方法、组合物和用途 | |
| WO2022199707A1 (zh) | 哌马色林药用盐、制备方法、含其的药物组合物及应用 | |
| WO2022161507A1 (zh) | Brepocitinib甲苯磺酸盐的晶型及其制备方法和用途 | |
| WO2018130226A1 (zh) | 利奥西呱的新晶型及其制备方法和用途 | |
| TWI765848B (zh) | 喹諾酮類似物及其鹽的結晶形式 | |
| CN101805337B (zh) | 一类含脯氨酸和异恶唑骨架的化合物、其制备方法和用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08748577 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2029/MUMNP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12935859 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011502211 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2008748577 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008748577 Country of ref document: EP |