WO2007042325A1 - 5-chinolinderivate mit antibakterieller aktivität - Google Patents
5-chinolinderivate mit antibakterieller aktivität Download PDFInfo
- Publication number
- WO2007042325A1 WO2007042325A1 PCT/EP2006/009932 EP2006009932W WO2007042325A1 WO 2007042325 A1 WO2007042325 A1 WO 2007042325A1 EP 2006009932 W EP2006009932 W EP 2006009932W WO 2007042325 A1 WO2007042325 A1 WO 2007042325A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- solution
- alkenyl
- hydroxy
- Prior art date
Links
- 230000000844 anti-bacterial effect Effects 0.000 title abstract description 6
- 150000001875 compounds Chemical class 0.000 claims abstract description 136
- -1 heteroalkyloxy Chemical group 0.000 claims description 121
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 28
- 239000000203 mixture Substances 0.000 claims description 27
- 125000000217 alkyl group Chemical group 0.000 claims description 24
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 23
- 125000004404 heteroalkyl group Chemical group 0.000 claims description 21
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 19
- 125000005843 halogen group Chemical group 0.000 claims description 19
- 125000003342 alkenyl group Chemical group 0.000 claims description 18
- 125000001072 heteroaryl group Chemical group 0.000 claims description 18
- 125000000304 alkynyl group Chemical group 0.000 claims description 17
- 125000003118 aryl group Chemical group 0.000 claims description 17
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 16
- 229910052739 hydrogen Inorganic materials 0.000 claims description 16
- 239000001257 hydrogen Substances 0.000 claims description 16
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 14
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 14
- 150000003839 salts Chemical class 0.000 claims description 11
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 10
- 125000005119 alkyl cycloalkyl group Chemical group 0.000 claims description 10
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 9
- 125000004475 heteroaralkyl group Chemical group 0.000 claims description 9
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 8
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 7
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 6
- 125000003277 amino group Chemical group 0.000 claims description 6
- 238000009472 formulation Methods 0.000 claims description 5
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 5
- 239000004480 active ingredient Substances 0.000 claims description 4
- 125000004474 heteroalkylene group Chemical group 0.000 claims description 4
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- 125000004193 piperazinyl group Chemical group 0.000 claims description 4
- 125000006296 sulfonyl amino group Chemical group [H]N(*)S(*)(=O)=O 0.000 claims description 4
- 150000003573 thiols Chemical class 0.000 claims description 4
- 125000004450 alkenylene group Chemical group 0.000 claims description 3
- 125000003545 alkoxy group Chemical group 0.000 claims description 3
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 3
- 125000002947 alkylene group Chemical group 0.000 claims description 3
- 125000004419 alkynylene group Chemical group 0.000 claims description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 3
- 239000012453 solvate Substances 0.000 claims description 3
- 239000002671 adjuvant Substances 0.000 claims description 2
- 150000001540 azides Chemical class 0.000 claims description 2
- 125000000000 cycloalkoxy group Chemical group 0.000 claims description 2
- 229910052736 halogen Inorganic materials 0.000 claims description 2
- 150000002367 halogens Chemical class 0.000 claims description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 5
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims 1
- 208000035143 Bacterial infection Diseases 0.000 claims 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims 1
- 208000022362 bacterial infectious disease Diseases 0.000 claims 1
- GBXQPDCOMJJCMJ-UHFFFAOYSA-M trimethyl-[6-(trimethylazaniumyl)hexyl]azanium;bromide Chemical compound [Br-].C[N+](C)(C)CCCCCC[N+](C)(C)C GBXQPDCOMJJCMJ-UHFFFAOYSA-M 0.000 claims 1
- 108010054814 DNA Gyrase Proteins 0.000 abstract description 2
- 108010041052 DNA Topoisomerase IV Proteins 0.000 abstract description 2
- 108010046308 Type II DNA Topoisomerases Proteins 0.000 abstract description 2
- 102000007537 Type II DNA Topoisomerases Human genes 0.000 abstract description 2
- 239000003534 dna topoisomerase inhibitor Substances 0.000 abstract 1
- 239000002271 gyrase inhibitor Substances 0.000 abstract 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 240
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 191
- 239000000243 solution Substances 0.000 description 186
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 140
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 114
- 239000000047 product Substances 0.000 description 97
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 75
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 70
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 63
- 239000000741 silica gel Substances 0.000 description 62
- 229910002027 silica gel Inorganic materials 0.000 description 62
- 238000003818 flash chromatography Methods 0.000 description 58
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 57
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 57
- 239000012074 organic phase Substances 0.000 description 55
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 47
- 150000001299 aldehydes Chemical class 0.000 description 47
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 39
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 38
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N DMSO Substances CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 35
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 34
- 239000011541 reaction mixture Substances 0.000 description 32
- 239000011780 sodium chloride Substances 0.000 description 32
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 31
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 30
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 28
- 229910052938 sodium sulfate Inorganic materials 0.000 description 28
- 235000011152 sodium sulphate Nutrition 0.000 description 28
- 150000002118 epoxides Chemical class 0.000 description 27
- 125000004432 carbon atom Chemical group C* 0.000 description 26
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 24
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 23
- 239000000706 filtrate Substances 0.000 description 22
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 21
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 21
- 150000001412 amines Chemical class 0.000 description 21
- 239000008346 aqueous phase Substances 0.000 description 20
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 19
- 238000001816 cooling Methods 0.000 description 19
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 18
- 235000019341 magnesium sulphate Nutrition 0.000 description 18
- 239000007787 solid Substances 0.000 description 18
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 16
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 16
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 15
- 235000017557 sodium bicarbonate Nutrition 0.000 description 15
- 239000012043 crude product Substances 0.000 description 14
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 14
- 229910052717 sulfur Inorganic materials 0.000 description 14
- KBRAGXDAPQXUCZ-UHFFFAOYSA-N 3-methoxy-5-(2-piperazin-1-ylethoxy)quinoline Chemical compound C12=CC(OC)=CN=C2C=CC=C1OCCN1CCNCC1 KBRAGXDAPQXUCZ-UHFFFAOYSA-N 0.000 description 13
- 239000012071 phase Substances 0.000 description 13
- 238000003756 stirring Methods 0.000 description 13
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 229910052731 fluorine Inorganic materials 0.000 description 12
- 229910052757 nitrogen Inorganic materials 0.000 description 12
- 229910052760 oxygen Inorganic materials 0.000 description 12
- 229910000027 potassium carbonate Inorganic materials 0.000 description 12
- 239000000725 suspension Substances 0.000 description 12
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 11
- 239000001301 oxygen Substances 0.000 description 11
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 11
- 239000012298 atmosphere Substances 0.000 description 10
- 229910052801 chlorine Inorganic materials 0.000 description 10
- GAGAICHLGQDUTL-UHFFFAOYSA-N 4h-thiazin-3-one Chemical compound O=C1CC=CSN1 GAGAICHLGQDUTL-UHFFFAOYSA-N 0.000 description 9
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 239000011737 fluorine Substances 0.000 description 9
- 238000010992 reflux Methods 0.000 description 9
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 8
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 8
- 229910021529 ammonia Inorganic materials 0.000 description 8
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 8
- 239000000460 chlorine Substances 0.000 description 8
- 150000002148 esters Chemical class 0.000 description 8
- 125000006413 ring segment Chemical group 0.000 description 8
- WDYVUKGVKRZQNM-UHFFFAOYSA-N 6-phosphonohexylphosphonic acid Chemical compound OP(O)(=O)CCCCCCP(O)(O)=O WDYVUKGVKRZQNM-UHFFFAOYSA-N 0.000 description 7
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 7
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 7
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 7
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 7
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 7
- 229910052794 bromium Inorganic materials 0.000 description 7
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 7
- FLQPQOIGLVLFDY-UHFFFAOYSA-N 3-methoxy-5-(oxiran-2-yl)quinoline Chemical compound C12=CC(OC)=CN=C2C=CC=C1C1CO1 FLQPQOIGLVLFDY-UHFFFAOYSA-N 0.000 description 6
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 6
- 125000005605 benzo group Chemical group 0.000 description 6
- 150000002009 diols Chemical class 0.000 description 6
- MHCFAGZWMAWTNR-UHFFFAOYSA-M lithium perchlorate Chemical compound [Li+].[O-]Cl(=O)(=O)=O MHCFAGZWMAWTNR-UHFFFAOYSA-M 0.000 description 6
- 229910001486 lithium perchlorate Inorganic materials 0.000 description 6
- 150000003254 radicals Chemical class 0.000 description 6
- 239000012279 sodium borohydride Substances 0.000 description 6
- 229910000033 sodium borohydride Inorganic materials 0.000 description 6
- 239000011593 sulfur Substances 0.000 description 6
- LFKDJXLFVYVEFG-UHFFFAOYSA-N tert-butyl carbamate Chemical compound CC(C)(C)OC(N)=O LFKDJXLFVYVEFG-UHFFFAOYSA-N 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 5
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 5
- 229910052796 boron Inorganic materials 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 239000002808 molecular sieve Substances 0.000 description 5
- 229910052698 phosphorus Inorganic materials 0.000 description 5
- 239000011574 phosphorus Substances 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 5
- 125000004434 sulfur atom Chemical group 0.000 description 5
- KLXZPUSMSLXPAY-UHFFFAOYSA-N 1,4-dioxine-2-carbaldehyde Chemical compound O=CC1=COC=CO1 KLXZPUSMSLXPAY-UHFFFAOYSA-N 0.000 description 4
- CWKXDPPQCVWXAG-UHFFFAOYSA-N 2,3-dihydro-1,4-benzodioxine-6-carbaldehyde Chemical compound O1CCOC2=CC(C=O)=CC=C21 CWKXDPPQCVWXAG-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- XEKAWZARUWARND-UHFFFAOYSA-N 6h-oxazin-3-one Chemical compound O=C1NOCC=C1 XEKAWZARUWARND-UHFFFAOYSA-N 0.000 description 4
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 4
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- 125000001153 fluoro group Chemical group F* 0.000 description 4
- 150000002576 ketones Chemical class 0.000 description 4
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 4
- SATCULPHIDQDRE-UHFFFAOYSA-N piperonal Chemical compound O=CC1=CC=C2OCOC2=C1 SATCULPHIDQDRE-UHFFFAOYSA-N 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 229910052711 selenium Inorganic materials 0.000 description 4
- 239000011669 selenium Substances 0.000 description 4
- 229910052710 silicon Inorganic materials 0.000 description 4
- 239000010703 silicon Substances 0.000 description 4
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 4
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 3
- PUQJEKAPFRAZOG-UHFFFAOYSA-N 2-(4-aminopiperidin-1-yl)-1-(3-methoxyquinolin-5-yl)ethanol Chemical compound C12=CC(OC)=CN=C2C=CC=C1C(O)CN1CCC(N)CC1 PUQJEKAPFRAZOG-UHFFFAOYSA-N 0.000 description 3
- ZTQNUTNKGQGWCM-UHFFFAOYSA-N 2-methoxyquinoline Chemical compound C1=CC=CC2=NC(OC)=CC=C21 ZTQNUTNKGQGWCM-UHFFFAOYSA-N 0.000 description 3
- RLCHCSCEUDTQNC-UHFFFAOYSA-N 2h-thiazine-6-carboxylic acid Chemical compound OC(=O)C1=CC=CNS1 RLCHCSCEUDTQNC-UHFFFAOYSA-N 0.000 description 3
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 125000001309 chloro group Chemical group Cl* 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 150000002431 hydrogen Chemical class 0.000 description 3
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 3
- MKIJJIMOAABWGF-UHFFFAOYSA-N methyl 2-sulfanylacetate Chemical compound COC(=O)CS MKIJJIMOAABWGF-UHFFFAOYSA-N 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 229920005862 polyol Polymers 0.000 description 3
- 150000003077 polyols Chemical class 0.000 description 3
- JUJWROOIHBZHMG-UHFFFAOYSA-N pyridine Substances C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 3
- 239000012312 sodium hydride Substances 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 3
- FYFGDDQFBUPCNC-UHFFFAOYSA-N tert-butyl n-[1-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperidin-4-yl]carbamate Chemical compound C12=CC(OC)=CN=C2C=CC=C1C(O)CN1CCC(NC(=O)OC(C)(C)C)CC1 FYFGDDQFBUPCNC-UHFFFAOYSA-N 0.000 description 3
- 235000015112 vegetable and seed oil Nutrition 0.000 description 3
- 239000008158 vegetable oil Substances 0.000 description 3
- KJPRLNWUNMBNBZ-QPJJXVBHSA-N (E)-cinnamaldehyde Chemical compound O=C\C=C\C1=CC=CC=C1 KJPRLNWUNMBNBZ-QPJJXVBHSA-N 0.000 description 2
- PNYDVKRZZLUMCJ-OWOJBTEDSA-N (e)-3-(2,5-difluorophenyl)prop-2-enal Chemical compound FC1=CC=C(F)C(\C=C\C=O)=C1 PNYDVKRZZLUMCJ-OWOJBTEDSA-N 0.000 description 2
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 2
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 2
- DTYLXDLAOLOTKT-UHFFFAOYSA-N 1,4-dihydroquinoline-3-carboxylic acid Chemical compound C1=CC=C2CC(C(=O)O)=CNC2=C1 DTYLXDLAOLOTKT-UHFFFAOYSA-N 0.000 description 2
- HYYXHBZBLHCDDP-UHFFFAOYSA-N 1-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-piperazin-1-ylethanol Chemical compound C=1C=C2OCCOC2=CC=1C(O)CN1CCNCC1 HYYXHBZBLHCDDP-UHFFFAOYSA-N 0.000 description 2
- DHFHACZDNMQQCC-UHFFFAOYSA-N 1-(3-methoxyquinolin-5-yl)-2-[4-(quinoxalin-2-ylmethylamino)piperidin-1-yl]ethanol Chemical compound C1=CC=CC2=NC(CNC3CCN(CC3)CC(O)C3=CC=CC4=NC=C(C=C43)OC)=CN=C21 DHFHACZDNMQQCC-UHFFFAOYSA-N 0.000 description 2
- NZVZVGPYTICZBZ-UHFFFAOYSA-N 1-benzylpiperidine Chemical compound C=1C=CC=CC=1CN1CCCCC1 NZVZVGPYTICZBZ-UHFFFAOYSA-N 0.000 description 2
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Chemical compound C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 2
- ODLUBLURAWVYJD-UHFFFAOYSA-N 2-(2-bromoethylsulfanyl)thiophene Chemical compound BrCCSC1=CC=CS1 ODLUBLURAWVYJD-UHFFFAOYSA-N 0.000 description 2
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 2
- CDAWCLOXVUBKRW-UHFFFAOYSA-N 2-aminophenol Chemical compound NC1=CC=CC=C1O CDAWCLOXVUBKRW-UHFFFAOYSA-N 0.000 description 2
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 2
- IMRWILPUOVGIMU-UHFFFAOYSA-N 2-bromopyridine Chemical compound BrC1=CC=CC=N1 IMRWILPUOVGIMU-UHFFFAOYSA-N 0.000 description 2
- XUGNJOCQALIQFG-UHFFFAOYSA-N 2-ethenylquinoline Chemical compound C1=CC=CC2=NC(C=C)=CC=C21 XUGNJOCQALIQFG-UHFFFAOYSA-N 0.000 description 2
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 2
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 2
- ALHUXMDEZNLFTA-UHFFFAOYSA-N 2-methylquinoxaline Chemical compound C1=CC=CC2=NC(C)=CN=C21 ALHUXMDEZNLFTA-UHFFFAOYSA-N 0.000 description 2
- PJKVFARRVXDXAD-UHFFFAOYSA-N 2-naphthaldehyde Chemical compound C1=CC=CC2=CC(C=O)=CC=C21 PJKVFARRVXDXAD-UHFFFAOYSA-N 0.000 description 2
- CMWKITSNTDAEDT-UHFFFAOYSA-N 2-nitrobenzaldehyde Chemical compound [O-][N+](=O)C1=CC=CC=C1C=O CMWKITSNTDAEDT-UHFFFAOYSA-N 0.000 description 2
- SLAMLWHELXOEJZ-UHFFFAOYSA-N 2-nitrobenzoic acid Chemical compound OC(=O)C1=CC=CC=C1[N+]([O-])=O SLAMLWHELXOEJZ-UHFFFAOYSA-N 0.000 description 2
- IQUPABOKLQSFBK-UHFFFAOYSA-N 2-nitrophenol Chemical compound OC1=CC=CC=C1[N+]([O-])=O IQUPABOKLQSFBK-UHFFFAOYSA-N 0.000 description 2
- ZPSJGADGUYYRKE-UHFFFAOYSA-N 2H-pyran-2-one Chemical compound O=C1C=CC=CO1 ZPSJGADGUYYRKE-UHFFFAOYSA-N 0.000 description 2
- AGIJRRREJXSQJR-UHFFFAOYSA-N 2h-thiazine Chemical compound N1SC=CC=C1 AGIJRRREJXSQJR-UHFFFAOYSA-N 0.000 description 2
- HRHFUHGNTSSIFS-UHFFFAOYSA-N 2h-thiazine 1-oxide Chemical compound O=S1NC=CC=C1 HRHFUHGNTSSIFS-UHFFFAOYSA-N 0.000 description 2
- IBGBGRVKPALMCQ-UHFFFAOYSA-N 3,4-dihydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1O IBGBGRVKPALMCQ-UHFFFAOYSA-N 0.000 description 2
- IBADFXOMCWHDMS-UHFFFAOYSA-N 3,5-dibromoquinoline Chemical compound C1=CC=C(Br)C2=CC(Br)=CN=C21 IBADFXOMCWHDMS-UHFFFAOYSA-N 0.000 description 2
- CIYUDMUBYRVMLP-UHFFFAOYSA-N 3-oxo-4h-1,4-benzoxazine-6-carboxylic acid Chemical compound O1CC(=O)NC2=CC(C(=O)O)=CC=C21 CIYUDMUBYRVMLP-UHFFFAOYSA-N 0.000 description 2
- CGTCULUUVYBAPX-UHFFFAOYSA-N 3-oxo-4h-1,4-benzoxazine-6-sulfonyl chloride Chemical compound O1CC(=O)NC2=CC(S(=O)(=O)Cl)=CC=C21 CGTCULUUVYBAPX-UHFFFAOYSA-N 0.000 description 2
- SZHYPQDQYNLZJP-UHFFFAOYSA-N 3-oxo-4h-pyrido[3,2-b][1,4]thiazine-6-carboxylic acid Chemical compound S1CC(=O)NC2=NC(C(=O)O)=CC=C21 SZHYPQDQYNLZJP-UHFFFAOYSA-N 0.000 description 2
- YGCZTXZTJXYWCO-UHFFFAOYSA-N 3-phenylpropanal Chemical compound O=CCCC1=CC=CC=C1 YGCZTXZTJXYWCO-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- RGHHSNMVTDWUBI-UHFFFAOYSA-N 4-hydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1 RGHHSNMVTDWUBI-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- UIRSLBDCOYTZPH-UHFFFAOYSA-N 4h-1,4-thiazin-3-one Chemical compound O=C1CSC=CN1 UIRSLBDCOYTZPH-UHFFFAOYSA-N 0.000 description 2
- JOFKQPXLVBANEF-UHFFFAOYSA-N 5-[2-[4-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)piperazin-1-yl]ethoxy]-3-methoxyquinoline Chemical compound O1CCOC2=CC(CN3CCN(CC3)CCOC3=CC=CC4=NC=C(C=C43)OC)=CC=C21 JOFKQPXLVBANEF-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 229920002556 Polyethylene Glycol 300 Polymers 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 241000191967 Staphylococcus aureus Species 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- AXMVYSVVTMKQSL-UHFFFAOYSA-N UNPD142122 Natural products OC1=CC=C(C=CC=O)C=C1O AXMVYSVVTMKQSL-UHFFFAOYSA-N 0.000 description 2
- 108010059993 Vancomycin Proteins 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 125000004423 acyloxy group Chemical group 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 239000000908 ammonium hydroxide Substances 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 239000004599 antimicrobial Substances 0.000 description 2
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 2
- 229940073608 benzyl chloride Drugs 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 229940117916 cinnamic aldehyde Drugs 0.000 description 2
- KJPRLNWUNMBNBZ-UHFFFAOYSA-N cinnamic aldehyde Natural products O=CC=CC1=CC=CC=C1 KJPRLNWUNMBNBZ-UHFFFAOYSA-N 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- RWGFKTVRMDUZSP-UHFFFAOYSA-N cumene Chemical compound CC(C)C1=CC=CC=C1 RWGFKTVRMDUZSP-UHFFFAOYSA-N 0.000 description 2
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 2
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- VEUUMBGHMNQHGO-UHFFFAOYSA-N ethyl chloroacetate Chemical compound CCOC(=O)CCl VEUUMBGHMNQHGO-UHFFFAOYSA-N 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical compound C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- UXMYKOUNESIISI-UHFFFAOYSA-N n-[1-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperidin-4-yl]-2,3-dihydro-1,4-benzodioxine-6-sulfonamide Chemical compound O1CCOC2=CC(S(=O)(=O)NC3CCN(CC3)CC(O)C3=CC=CC4=NC=C(C=C43)OC)=CC=C21 UXMYKOUNESIISI-UHFFFAOYSA-N 0.000 description 2
- LKPFBGKZCCBZDK-UHFFFAOYSA-N n-hydroxypiperidine Chemical compound ON1CCCCC1 LKPFBGKZCCBZDK-UHFFFAOYSA-N 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 244000052769 pathogen Species 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- LWMPFIOTEAXAGV-UHFFFAOYSA-N piperidin-1-amine Chemical compound NN1CCCCC1 LWMPFIOTEAXAGV-UHFFFAOYSA-N 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- JPJALAQPGMAKDF-UHFFFAOYSA-N selenium dioxide Chemical compound O=[Se]=O JPJALAQPGMAKDF-UHFFFAOYSA-N 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000007901 soft capsule Substances 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- ROUYFJUVMYHXFJ-UHFFFAOYSA-N tert-butyl 4-oxopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=O)CC1 ROUYFJUVMYHXFJ-UHFFFAOYSA-N 0.000 description 2
- RQCNHUCCQJMSRG-UHFFFAOYSA-N tert-butyl piperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCCCC1 RQCNHUCCQJMSRG-UHFFFAOYSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- JJIHENIPUIXQDM-UHFFFAOYSA-N thiadiazole-5-carbaldehyde Chemical compound O=CC1=CN=NS1 JJIHENIPUIXQDM-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 229960003165 vancomycin Drugs 0.000 description 2
- MYPYJXKWCTUITO-UHFFFAOYSA-N vancomycin Natural products O1C(C(=C2)Cl)=CC=C2C(O)C(C(NC(C2=CC(O)=CC(O)=C2C=2C(O)=CC=C3C=2)C(O)=O)=O)NC(=O)C3NC(=O)C2NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(CC(C)C)NC)C(O)C(C=C3Cl)=CC=C3OC3=CC2=CC1=C3OC1OC(CO)C(O)C(O)C1OC1CC(C)(N)C(O)C(C)O1 MYPYJXKWCTUITO-UHFFFAOYSA-N 0.000 description 2
- MYPYJXKWCTUITO-LYRMYLQWSA-O vancomycin(1+) Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=C2C=C3C=C1OC1=CC=C(C=C1Cl)[C@@H](O)[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@H]3C(=O)N[C@H]1C(=O)N[C@H](C(N[C@@H](C3=CC(O)=CC(O)=C3C=3C(O)=CC=C1C=3)C([O-])=O)=O)[C@H](O)C1=CC=C(C(=C1)Cl)O2)=O)NC(=O)[C@@H](CC(C)C)[NH2+]C)[C@H]1C[C@](C)([NH3+])[C@H](O)[C@H](C)O1 MYPYJXKWCTUITO-LYRMYLQWSA-O 0.000 description 2
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 description 1
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 description 1
- 125000004916 (C1-C6) alkylcarbonyl group Chemical group 0.000 description 1
- ZVEJSCDZTXEUBM-HWKANZROSA-N (e)-3-pyridin-2-ylprop-2-enal Chemical compound O=C\C=C\C1=CC=CC=N1 ZVEJSCDZTXEUBM-HWKANZROSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- ATWLRNODAYAMQS-UHFFFAOYSA-N 1,1-dibromopropane Chemical compound CCC(Br)Br ATWLRNODAYAMQS-UHFFFAOYSA-N 0.000 description 1
- KEIFWROAQVVDBN-UHFFFAOYSA-N 1,2-dihydronaphthalene Chemical compound C1=CC=C2C=CCCC2=C1 KEIFWROAQVVDBN-UHFFFAOYSA-N 0.000 description 1
- VEFLKXRACNJHOV-UHFFFAOYSA-N 1,3-dibromopropane Chemical compound BrCCCBr VEFLKXRACNJHOV-UHFFFAOYSA-N 0.000 description 1
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 1
- KDHSQQUTHLMVCQ-UHFFFAOYSA-N 1-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-piperazin-1-ylethanol piperazine Chemical compound C1CNCCN1.C=1C=C2OCCOC2=CC=1C(O)CN1CCNCC1 KDHSQQUTHLMVCQ-UHFFFAOYSA-N 0.000 description 1
- HGVWMTAIIYNQSI-UHFFFAOYSA-N 1-(2,3-dihydro-1,4-benzodioxin-6-yl)ethanone Chemical compound O1CCOC2=CC(C(=O)C)=CC=C21 HGVWMTAIIYNQSI-UHFFFAOYSA-N 0.000 description 1
- VOJNAEKMXVNUQF-UHFFFAOYSA-N 1-(3-methoxyquinolin-5-yl)-2-(4-piperidin-4-ylpiperazin-1-yl)ethanol Chemical compound C12=CC(OC)=CN=C2C=CC=C1C(O)CN(CC1)CCN1C1CCNCC1 VOJNAEKMXVNUQF-UHFFFAOYSA-N 0.000 description 1
- FJWQELYSMDSWCM-UHFFFAOYSA-N 1-(3-methoxyquinolin-5-yl)-2-[4-(2-phenoxyethylamino)piperidin-1-yl]ethanol Chemical compound C12=CC(OC)=CN=C2C=CC=C1C(O)CN(CC1)CCC1NCCOC1=CC=CC=C1 FJWQELYSMDSWCM-UHFFFAOYSA-N 0.000 description 1
- PDGIOFDUZFPUBO-UHFFFAOYSA-N 1-(3-methoxyquinolin-5-yl)-2-[4-(3-phenylpropylamino)piperidin-1-yl]ethanol Chemical compound C12=CC(OC)=CN=C2C=CC=C1C(O)CN(CC1)CCC1NCCCC1=CC=CC=C1 PDGIOFDUZFPUBO-UHFFFAOYSA-N 0.000 description 1
- XNEYYKTXODAFFZ-UHFFFAOYSA-N 1-(3-methoxyquinolin-5-yl)-2-[4-(3-thiophen-2-ylsulfanylpropyl)piperazin-1-yl]ethanol Chemical compound C12=CC(OC)=CN=C2C=CC=C1C(O)CN(CC1)CCN1CCCSC1=CC=CS1 XNEYYKTXODAFFZ-UHFFFAOYSA-N 0.000 description 1
- NGXIKFXVDXOJHY-UHFFFAOYSA-N 1-(3-methoxyquinolin-5-yl)-2-[4-(naphthalen-2-ylmethylamino)piperidin-1-yl]ethanol Chemical compound C1=CC=CC2=CC(CNC3CCN(CC3)CC(O)C3=CC=CC4=NC=C(C=C43)OC)=CC=C21 NGXIKFXVDXOJHY-UHFFFAOYSA-N 0.000 description 1
- YADPPOFHPMCCHO-UHFFFAOYSA-N 1-(3-methoxyquinolin-5-yl)-2-[4-[(1-methylindol-2-yl)methylamino]piperidin-1-yl]ethanol Chemical compound C1=CC=C2N(C)C(CNC3CCN(CC3)CC(O)C3=CC=CC4=NC=C(C=C43)OC)=CC2=C1 YADPPOFHPMCCHO-UHFFFAOYSA-N 0.000 description 1
- CLUOHJOKZOUQNK-UHFFFAOYSA-N 1-(3-methoxyquinolin-5-yl)ethane-1,2-diol Chemical compound C1=CC=C(C(O)CO)C2=CC(OC)=CN=C21 CLUOHJOKZOUQNK-UHFFFAOYSA-N 0.000 description 1
- ROFTZKZVGWJCKM-UHFFFAOYSA-N 1-(3-thiophen-2-ylsulfanylpropyl)piperazine Chemical compound C1CNCCN1CCCSC1=CC=CS1 ROFTZKZVGWJCKM-UHFFFAOYSA-N 0.000 description 1
- MSYMXQLURAAOJQ-UHFFFAOYSA-N 1-[2-(1,3-benzodioxol-5-yl)ethyl]piperazine Chemical compound C=1C=C2OCOC2=CC=1CCN1CCNCC1 MSYMXQLURAAOJQ-UHFFFAOYSA-N 0.000 description 1
- QTMKEGCQTIHIMB-UHFFFAOYSA-N 1-benzyl-3-(2-tert-butylsilyloxypropan-2-yl)piperidin-4-ol Chemical compound C(C1=CC=CC=C1)N1CC(C(CC1)O)C(O[SiH2]C(C)(C)C)(C)C QTMKEGCQTIHIMB-UHFFFAOYSA-N 0.000 description 1
- BLKKQITUZVROHP-UHFFFAOYSA-N 1-benzyl-3-(hydroxymethyl)piperidin-4-ol Chemical compound C1CC(O)C(CO)CN1CC1=CC=CC=C1 BLKKQITUZVROHP-UHFFFAOYSA-N 0.000 description 1
- IQXXEPZFOOTTBA-UHFFFAOYSA-N 1-benzylpiperazine Chemical compound C=1C=CC=CC=1CN1CCNCC1 IQXXEPZFOOTTBA-UHFFFAOYSA-N 0.000 description 1
- MMZYCBHLNZVROM-UHFFFAOYSA-N 1-fluoro-2-methylbenzene Chemical compound CC1=CC=CC=C1F MMZYCBHLNZVROM-UHFFFAOYSA-N 0.000 description 1
- IBNGPIOSWCMJGG-UHFFFAOYSA-N 1-methylindole-2-carbaldehyde Chemical compound C1=CC=C2N(C)C(C=O)=CC2=C1 IBNGPIOSWCMJGG-UHFFFAOYSA-N 0.000 description 1
- 125000000453 2,2,2-trichloroethyl group Chemical group [H]C([H])(*)C(Cl)(Cl)Cl 0.000 description 1
- HGUFODBRKLSHSI-UHFFFAOYSA-N 2,3,7,8-tetrachloro-dibenzo-p-dioxin Chemical compound O1C2=CC(Cl)=C(Cl)C=C2OC2=C1C=C(Cl)C(Cl)=C2 HGUFODBRKLSHSI-UHFFFAOYSA-N 0.000 description 1
- HMBHAQMOBKLWRX-UHFFFAOYSA-N 2,3-dihydro-1,4-benzodioxine-3-carboxylic acid Chemical compound C1=CC=C2OC(C(=O)O)COC2=C1 HMBHAQMOBKLWRX-UHFFFAOYSA-N 0.000 description 1
- JWZQJTGQFHIRFQ-UHFFFAOYSA-N 2,3-dihydro-1,4-benzodioxine-6-carboxylic acid Chemical compound O1CCOC2=CC(C(=O)O)=CC=C21 JWZQJTGQFHIRFQ-UHFFFAOYSA-N 0.000 description 1
- JWHSRWUHRLYAPM-UHFFFAOYSA-N 2,3-dihydro-1,4-benzodioxine-6-sulfonyl chloride Chemical compound O1CCOC2=CC(S(=O)(=O)Cl)=CC=C21 JWHSRWUHRLYAPM-UHFFFAOYSA-N 0.000 description 1
- OMWBUWDHEZIMGQ-UHFFFAOYSA-N 2,3-dihydro-[1,4]dioxino[2,3-c]pyridine-7-carbaldehyde Chemical compound O1CCOC2=C1C=C(C=O)N=C2 OMWBUWDHEZIMGQ-UHFFFAOYSA-N 0.000 description 1
- NJYBIFYEWYWYAN-UHFFFAOYSA-N 2,4-difluorobenzoic acid Chemical compound OC(=O)C1=CC=C(F)C=C1F NJYBIFYEWYWYAN-UHFFFAOYSA-N 0.000 description 1
- OELSLLMORUCRJX-UHFFFAOYSA-N 2-(1,4-dioxan-2-yl)phenol Chemical compound OC1=CC=CC=C1C1OCCOC1 OELSLLMORUCRJX-UHFFFAOYSA-N 0.000 description 1
- MDKQNOCUQIBENI-UHFFFAOYSA-N 2-(3-bromopropylsulfanyl)thiophene Chemical compound BrCCCSC1=CC=CS1 MDKQNOCUQIBENI-UHFFFAOYSA-N 0.000 description 1
- OFFFCYNKZALCMH-UHFFFAOYSA-N 2-(4-amino-1-phenylmethoxycarbonylpiperidin-4-yl)acetic acid Chemical compound C1CC(N)(CC(O)=O)CCN1C(=O)OCC1=CC=CC=C1 OFFFCYNKZALCMH-UHFFFAOYSA-N 0.000 description 1
- RMVFJPUWIGGBIS-UHFFFAOYSA-N 2-(4-amino-3-fluoropiperidin-1-yl)-1-(3-methoxyquinolin-5-yl)ethanol Chemical compound C12=CC(OC)=CN=C2C=CC=C1C(O)CN1CCC(N)C(F)C1 RMVFJPUWIGGBIS-UHFFFAOYSA-N 0.000 description 1
- REBWSFKBXIVNRQ-UHFFFAOYSA-N 2-(furan-3-yl)furan Chemical group C1=COC(C2=COC=C2)=C1 REBWSFKBXIVNRQ-UHFFFAOYSA-N 0.000 description 1
- ZGEXYNBHYVEWKT-UHFFFAOYSA-N 2-(hydroxymethyl)-5-phenylmethoxypyran-4-one Chemical compound O1C(CO)=CC(=O)C(OCC=2C=CC=CC=2)=C1 ZGEXYNBHYVEWKT-UHFFFAOYSA-N 0.000 description 1
- ADDZHRRCUWNSCS-UHFFFAOYSA-N 2-Benzofurancarboxaldehyde Chemical compound C1=CC=C2OC(C=O)=CC2=C1 ADDZHRRCUWNSCS-UHFFFAOYSA-N 0.000 description 1
- NOZBALHPFKZOFE-UHFFFAOYSA-N 2-[4-(1-benzofuran-2-ylmethylamino)piperidin-1-yl]-1-(3-methoxyquinolin-5-yl)ethanol Chemical compound C1=CC=C2OC(CNC3CCN(CC3)CC(O)C3=CC=CC4=NC=C(C=C43)OC)=CC2=C1 NOZBALHPFKZOFE-UHFFFAOYSA-N 0.000 description 1
- NSGYNJVPKRNPLH-UHFFFAOYSA-N 2-[4-(2,1,3-benzothiadiazol-5-ylmethylamino)-3-fluoropiperidin-1-yl]-1-(3-methoxyquinolin-5-yl)ethanol Chemical compound C1=CC2=NSN=C2C=C1CNC(C(F)C1)CCN1CC(O)C1=CC=CC2=NC=C(OC)C=C21 NSGYNJVPKRNPLH-UHFFFAOYSA-N 0.000 description 1
- PGTDBVVVRRTHQG-UHFFFAOYSA-N 2-[[1-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperidin-4-yl]amino]-n-pyridin-2-ylacetamide Chemical compound C12=CC(OC)=CN=C2C=CC=C1C(O)CN(CC1)CCC1NCC(=O)NC1=CC=CC=N1 PGTDBVVVRRTHQG-UHFFFAOYSA-N 0.000 description 1
- LFIRHMDBLAELET-UHFFFAOYSA-N 2-fluoro-4-(2-methoxy-2-oxoethyl)sulfanyl-5-nitrobenzoic acid Chemical compound COC(=O)CSC1=CC(F)=C(C(O)=O)C=C1[N+]([O-])=O LFIRHMDBLAELET-UHFFFAOYSA-N 0.000 description 1
- 125000004198 2-fluorophenyl group Chemical group [H]C1=C([H])C(F)=C(*)C([H])=C1[H] 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- 125000004204 2-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 1
- XHNZTDLRVWGECR-UHFFFAOYSA-N 2-methyl-1-benzofuran-5-carbaldehyde Chemical compound O=CC1=CC=C2OC(C)=CC2=C1 XHNZTDLRVWGECR-UHFFFAOYSA-N 0.000 description 1
- DYFWPMHZVHAXTO-UHFFFAOYSA-N 2-oxo-3h-1,3-benzoxazole-5-carbaldehyde Chemical compound O=CC1=CC=C2OC(=O)NC2=C1 DYFWPMHZVHAXTO-UHFFFAOYSA-N 0.000 description 1
- XFFILAFLGDUMBF-UHFFFAOYSA-N 2-phenoxyacetaldehyde Chemical compound O=CCOC1=CC=CC=C1 XFFILAFLGDUMBF-UHFFFAOYSA-N 0.000 description 1
- WFCSWCVEJLETKA-UHFFFAOYSA-N 2-piperazin-1-ylethanol Chemical compound OCCN1CCNCC1 WFCSWCVEJLETKA-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- NZDHBOBUOPDISB-UHFFFAOYSA-N 2H-oxazine-6-sulfonic acid Chemical compound S(=O)(=O)(O)C=1ONC=CC=1 NZDHBOBUOPDISB-UHFFFAOYSA-N 0.000 description 1
- YHWMFDLNZGIJSD-UHFFFAOYSA-N 2h-1,4-oxazine Chemical compound C1OC=CN=C1 YHWMFDLNZGIJSD-UHFFFAOYSA-N 0.000 description 1
- IGDLCKRTTABTCE-UHFFFAOYSA-N 2h-oxazine-6-carbaldehyde Chemical compound O=CC1=CC=CNO1 IGDLCKRTTABTCE-UHFFFAOYSA-N 0.000 description 1
- CSUZMBGLKCRFKF-UHFFFAOYSA-N 2h-thiazine-6-carbaldehyde Chemical compound O=CC1=CC=CNS1 CSUZMBGLKCRFKF-UHFFFAOYSA-N 0.000 description 1
- VDIBWLYUICXIMH-UHFFFAOYSA-N 2h-thiazine-6-sulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CNS1 VDIBWLYUICXIMH-UHFFFAOYSA-N 0.000 description 1
- PCYGLFXKCBFGPC-UHFFFAOYSA-N 3,4-Dihydroxy hydroxymethyl benzene Natural products OCC1=CC=C(O)C(O)=C1 PCYGLFXKCBFGPC-UHFFFAOYSA-N 0.000 description 1
- LCSVYSVGXQQHSI-UHFFFAOYSA-N 3,4-dihydro-2h-1,5-benzodioxepine-7-carbaldehyde Chemical compound O1CCCOC2=CC(C=O)=CC=C21 LCSVYSVGXQQHSI-UHFFFAOYSA-N 0.000 description 1
- QXGFBAHKFOFROY-UHFFFAOYSA-N 3-(1,4-dioxan-2-yl)-1,3-benzoxazol-2-one Chemical compound O1C(COCC1)N1C(OC2=C1C=CC=C2)=O QXGFBAHKFOFROY-UHFFFAOYSA-N 0.000 description 1
- AYJNHDRREPUUCN-UHFFFAOYSA-N 3-(1,4-dioxan-2-yl)-2-nitrophenol Chemical compound O1C(COCC1)C=1C(=C(C=CC=1)O)[N+](=O)[O-] AYJNHDRREPUUCN-UHFFFAOYSA-N 0.000 description 1
- VZIRCHXYMBFNFD-HNQUOIGGSA-N 3-(2-Furanyl)-2-propenal Chemical compound O=C\C=C\C1=CC=CO1 VZIRCHXYMBFNFD-HNQUOIGGSA-N 0.000 description 1
- PFVMEXSUMYYVHK-UHFFFAOYSA-N 3-(2-tert-butylsilyloxypropan-2-yl)-N-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)piperidin-4-amine Chemical compound C(C)(C)(C)[SiH2]OC(C1CNCCC1NCC1=CC2=C(OCCO2)C=C1)(C)C PFVMEXSUMYYVHK-UHFFFAOYSA-N 0.000 description 1
- RVCWIRSZZWLYCQ-UHFFFAOYSA-N 3-(2-tert-butylsilyloxypropan-2-yl)piperidin-4-ol Chemical compound C(C)(C)(C)[SiH2]OC(C1CNCCC1O)(C)C RVCWIRSZZWLYCQ-UHFFFAOYSA-N 0.000 description 1
- JDCOGHHNKITDEW-UHFFFAOYSA-N 3-bromoquinoline;3,5-dibromoquinoline Chemical compound C1=CC=CC2=CC(Br)=CN=C21.C1=CC=C(Br)C2=CC(Br)=CN=C21 JDCOGHHNKITDEW-UHFFFAOYSA-N 0.000 description 1
- AUBBVPIQUDFRQI-UHFFFAOYSA-N 3-hydroxy-4-nitrobenzaldehyde Chemical compound OC1=CC(C=O)=CC=C1[N+]([O-])=O AUBBVPIQUDFRQI-UHFFFAOYSA-N 0.000 description 1
- MGEKTRDPBDREKY-UHFFFAOYSA-N 3-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)quinoline Chemical compound C12=CC(OC)=CN=C2C=CC=C1B1OC(C)(C)C(C)(C)O1 MGEKTRDPBDREKY-UHFFFAOYSA-N 0.000 description 1
- OIOCPCUFGCFOFZ-UHFFFAOYSA-N 3-methoxy-5-[2-[4-(2-thiophen-2-ylsulfanylethyl)piperazin-1-yl]ethoxy]quinoline Chemical compound C12=CC(OC)=CN=C2C=CC=C1OCCN(CC1)CCN1CCSC1=CC=CS1 OIOCPCUFGCFOFZ-UHFFFAOYSA-N 0.000 description 1
- SBXYWFIMBSFYBI-UHFFFAOYSA-N 3-methoxy-5-[2-[4-(naphthalen-2-ylmethyl)piperazin-1-yl]ethoxy]quinoline Chemical compound C1=CC=CC2=CC(CN3CCN(CC3)CCOC3=CC=CC4=NC=C(C=C43)OC)=CC=C21 SBXYWFIMBSFYBI-UHFFFAOYSA-N 0.000 description 1
- AWEZTTWEBOCJAV-UHFFFAOYSA-N 3-methoxyquinolin-5-ol Chemical compound C1=CC=C(O)C2=CC(OC)=CN=C21 AWEZTTWEBOCJAV-UHFFFAOYSA-N 0.000 description 1
- AQCRFDKOKZGTDE-UHFFFAOYSA-N 3-methoxyquinoline Chemical compound C1=CC=CC2=CC(OC)=CN=C21 AQCRFDKOKZGTDE-UHFFFAOYSA-N 0.000 description 1
- NBIVLYUEDPRLGI-UHFFFAOYSA-N 3-oxo-4h-1,4-benzothiazine-6-carboxylic acid Chemical compound S1CC(=O)NC2=CC(C(=O)O)=CC=C21 NBIVLYUEDPRLGI-UHFFFAOYSA-N 0.000 description 1
- FEXJVEZCIWUQMB-UHFFFAOYSA-N 3-oxo-4h-1,4-benzothiazine-6-sulfonyl chloride Chemical compound S1CC(=O)NC2=CC(S(=O)(=O)Cl)=CC=C21 FEXJVEZCIWUQMB-UHFFFAOYSA-N 0.000 description 1
- XOXXGNIBXYQQNS-UHFFFAOYSA-N 3-oxo-4h-pyrido[3,2-b][1,4]oxazine-6-carbaldehyde Chemical compound O1CC(=O)NC2=NC(C=O)=CC=C21 XOXXGNIBXYQQNS-UHFFFAOYSA-N 0.000 description 1
- VEPGNAIYGRUSIA-UHFFFAOYSA-N 3-oxo-4h-pyrido[3,2-b][1,4]thiazine-6-carbaldehyde Chemical compound S1CC(=O)NC2=NC(C=O)=CC=C21 VEPGNAIYGRUSIA-UHFFFAOYSA-N 0.000 description 1
- HETBKLHJEWXWBM-UHFFFAOYSA-N 4-chloro-3-nitrobenzaldehyde Chemical compound [O-][N+](=O)C1=CC(C=O)=CC=C1Cl HETBKLHJEWXWBM-UHFFFAOYSA-N 0.000 description 1
- YTHJCZRFJGXPTL-UHFFFAOYSA-N 4-hydroxy-3-nitrobenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1[N+]([O-])=O YTHJCZRFJGXPTL-UHFFFAOYSA-N 0.000 description 1
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- GSSBOYWRKTVVQX-UHFFFAOYSA-N 4-prop-2-ynoxybenzaldehyde Chemical compound O=CC1=CC=C(OCC#C)C=C1 GSSBOYWRKTVVQX-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- FBXGQDUVJBKEAJ-UHFFFAOYSA-N 4h-oxazin-3-one Chemical compound O=C1CC=CON1 FBXGQDUVJBKEAJ-UHFFFAOYSA-N 0.000 description 1
- IWJLZADTSIIYBX-UHFFFAOYSA-N 5-(benzyloxy)-2-(hydroxymethyl)-1,4-dihydropyridin-4-one Chemical compound N1C(CO)=CC(=O)C(OCC=2C=CC=CC=2)=C1 IWJLZADTSIIYBX-UHFFFAOYSA-N 0.000 description 1
- VNGBSMQCSIJMQI-UHFFFAOYSA-N 5-[[[1-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperidin-4-yl]amino]methyl]-3h-1,3-benzoxazol-2-one Chemical compound C1=C2OC(=O)NC2=CC(CNC2CCN(CC2)CC(O)C2=CC=CC3=NC=C(C=C32)OC)=C1 VNGBSMQCSIJMQI-UHFFFAOYSA-N 0.000 description 1
- AVEJOTKSCCWCQA-UHFFFAOYSA-N 5-ethenyl-3-methoxyquinoline Chemical compound C1=CC=C(C=C)C2=CC(OC)=CN=C21 AVEJOTKSCCWCQA-UHFFFAOYSA-N 0.000 description 1
- MWPCUSJFFDYNSY-UHFFFAOYSA-N 6-(1-hydroxy-2-piperazin-1-ylethyl)-4h-1,4-benzoxazin-3-one Chemical compound C=1C=C2OCC(=O)NC2=CC=1C(O)CN1CCNCC1 MWPCUSJFFDYNSY-UHFFFAOYSA-N 0.000 description 1
- YDYLPMPVYALQKQ-UHFFFAOYSA-N 6-(2-chloroacetyl)-4h-1,4-benzothiazin-3-one Chemical compound S1CC(=O)NC2=CC(C(=O)CCl)=CC=C21 YDYLPMPVYALQKQ-UHFFFAOYSA-N 0.000 description 1
- MIAHXWVABDHISZ-UHFFFAOYSA-N 6-(2-chloroacetyl)-4h-1,4-benzoxazin-3-one Chemical compound O1CC(=O)NC2=CC(C(=O)CCl)=CC=C21 MIAHXWVABDHISZ-UHFFFAOYSA-N 0.000 description 1
- QKCZJBYIJRYYFB-UHFFFAOYSA-N 6-(hydroxymethyl)-4h-pyrido[3,2-b][1,4]thiazin-3-one Chemical compound S1CC(=O)NC2=NC(CO)=CC=C21 QKCZJBYIJRYYFB-UHFFFAOYSA-N 0.000 description 1
- SBISQOVXKLBVBW-UHFFFAOYSA-N 6-[1-hydroxy-2-[4-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperazin-1-yl]ethyl]-4h-1,4-benzoxazin-3-one Chemical compound O1CC(=O)NC2=CC(C(O)CN3CCN(CC3)CC(O)C3=CC=CC4=NC=C(C=C43)OC)=CC=C21 SBISQOVXKLBVBW-UHFFFAOYSA-N 0.000 description 1
- OJJSLYBPMTVESL-UHFFFAOYSA-N 6-[4-[2-(3-methoxyquinolin-5-yl)oxyethyl]piperazine-1-carbonyl]-4h-1,4-benzoxazin-3-one Chemical compound O1CC(=O)NC2=CC(C(=O)N3CCN(CC3)CCOC3=CC=CC4=NC=C(C=C43)OC)=CC=C21 OJJSLYBPMTVESL-UHFFFAOYSA-N 0.000 description 1
- GTOPLAMCODSOKG-UHFFFAOYSA-N 6-[[4-[2-(3-methoxyquinolin-5-yl)oxyethyl]piperazin-1-yl]methyl]-4h-1,4-benzothiazin-3-one Chemical compound S1CC(=O)NC2=CC(CN3CCN(CC3)CCOC3=CC=CC4=NC=C(C=C43)OC)=CC=C21 GTOPLAMCODSOKG-UHFFFAOYSA-N 0.000 description 1
- JEUDSRPNHABCQA-UHFFFAOYSA-N 6-[[4-[2-(3-methoxyquinolin-5-yl)oxyethyl]piperazin-1-yl]methyl]-4h-1,4-benzoxazin-3-one Chemical compound O1CC(=O)NC2=CC(CN3CCN(CC3)CCOC3=CC=CC4=NC=C(C=C43)OC)=CC=C21 JEUDSRPNHABCQA-UHFFFAOYSA-N 0.000 description 1
- OJYZDRKPWNOYBK-UHFFFAOYSA-N 6-[[[1-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperidin-4-yl]amino]methyl]-4h-pyrido[3,2-b][1,4]thiazin-3-one Chemical compound S1CC(=O)NC2=NC(CNC3CCN(CC3)CC(O)C3=CC=CC4=NC=C(C=C43)OC)=CC=C21 OJYZDRKPWNOYBK-UHFFFAOYSA-N 0.000 description 1
- SSJGCILBSSEKLZ-UHFFFAOYSA-N 6-[[[3-fluoro-1-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperidin-4-yl]amino]methyl]-4h-1,4-benzothiazin-3-one Chemical compound S1CC(=O)NC2=CC(CNC3CCN(CC3F)CC(O)C3=CC=CC4=NC=C(C=C43)OC)=CC=C21 SSJGCILBSSEKLZ-UHFFFAOYSA-N 0.000 description 1
- BVDVUCLPNIHQIJ-UHFFFAOYSA-N 6-[[[4-(2-hydroxyethyl)-1-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperidin-4-yl]amino]methyl]-4h-1,4-benzoxazin-3-one Chemical compound O1CC(=O)NC2=CC(CNC3(CCO)CCN(CC3)CC(O)C3=CC=CC4=NC=C(C=C43)OC)=CC=C21 BVDVUCLPNIHQIJ-UHFFFAOYSA-N 0.000 description 1
- AFRIQPBBMQJEJG-UHFFFAOYSA-N 6-[[[4-(2-hydroxyethyl)piperidin-4-yl]amino]methyl]-4h-1,4-benzoxazin-3-one Chemical compound C=1C=C2OCC(=O)NC2=CC=1CNC1(CCO)CCNCC1 AFRIQPBBMQJEJG-UHFFFAOYSA-N 0.000 description 1
- LOMWKYHCHCZVCC-UHFFFAOYSA-N 6-acetamidopyridine-2-carboxylic acid Chemical compound CC(=O)NC1=CC=CC(C(O)=O)=N1 LOMWKYHCHCZVCC-UHFFFAOYSA-N 0.000 description 1
- UENBBJXGCWILBM-UHFFFAOYSA-N 6-methylpyridin-3-amine Chemical compound CC1=CC=C(N)C=N1 UENBBJXGCWILBM-UHFFFAOYSA-N 0.000 description 1
- GWVPOHCHOSLYSA-UHFFFAOYSA-N 7-(hydroxymethyl)-4h-1,4-benzoxazin-3-one Chemical compound N1C(=O)COC2=CC(CO)=CC=C21 GWVPOHCHOSLYSA-UHFFFAOYSA-N 0.000 description 1
- ISPVACVJFUIDPD-UHFFFAOYSA-N 7-chloro-1-cyclopropyl-6-fluoro-4-oxoquinoline-3-carboxylic acid Chemical compound C12=CC(Cl)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 ISPVACVJFUIDPD-UHFFFAOYSA-N 0.000 description 1
- AMKGKYQBASDDJB-UHFFFAOYSA-N 9$l^{2}-borabicyclo[3.3.1]nonane Chemical compound C1CCC2CCCC1[B]2 AMKGKYQBASDDJB-UHFFFAOYSA-N 0.000 description 1
- FEJUGLKDZJDVFY-UHFFFAOYSA-N 9-borabicyclo[3.3.1]nonane Substances C1CCC2CCCC1B2 FEJUGLKDZJDVFY-UHFFFAOYSA-N 0.000 description 1
- 241001232615 Acinetobacter baumannii ATCC 19606 = CIP 70.34 = JCM 6841 Species 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- RWRGEGWTWUJMII-UHFFFAOYSA-N C(C)(C)(C)[SiH2]OC(C1CN(CCC1NCC1=CC2=C(OCCO2)C=C1)CC(O)C1=C2C=C(C=NC2=CC=C1)OC)(C)C Chemical compound C(C)(C)(C)[SiH2]OC(C1CN(CCC1NCC1=CC2=C(OCCO2)C=C1)CC(O)C1=C2C=C(C=NC2=CC=C1)OC)(C)C RWRGEGWTWUJMII-UHFFFAOYSA-N 0.000 description 1
- HBFNPWDCUVZGET-UHFFFAOYSA-N C1CCNCC1.C=1C=C2OCC(=O)NC2=CC=1CNC1(CCO)CCNCC1 Chemical compound C1CCNCC1.C=1C=C2OCC(=O)NC2=CC=1CNC1(CCO)CCNCC1 HBFNPWDCUVZGET-UHFFFAOYSA-N 0.000 description 1
- GMVFWRGZBGOYAP-UHFFFAOYSA-N C1CCNCC1.CC(C)(C)[SiH2]OC(C)(C)C1CNCCC1NCC1=CC=C(OCC(=O)N2)C2=C1 Chemical compound C1CCNCC1.CC(C)(C)[SiH2]OC(C)(C)C1CNCCC1NCC1=CC=C(OCC(=O)N2)C2=C1 GMVFWRGZBGOYAP-UHFFFAOYSA-N 0.000 description 1
- 125000003320 C2-C6 alkenyloxy group Chemical group 0.000 description 1
- WJQPEPALENGIOE-UHFFFAOYSA-N CC(C)(C)[SiH2]OC(C)(C)C1CNCCC1NCC1=CC=C(OCC(=O)N2)C2=C1 Chemical compound CC(C)(C)[SiH2]OC(C)(C)C1CNCCC1NCC1=CC=C(OCC(=O)N2)C2=C1 WJQPEPALENGIOE-UHFFFAOYSA-N 0.000 description 1
- HXOZLTQYYYXMPP-UHFFFAOYSA-N COC1=CC2=C(C(CN(CC3)CCC3NC(C3=CC=CNO3)=O)O)C=CC=C2N=C1 Chemical compound COC1=CC2=C(C(CN(CC3)CCC3NC(C3=CC=CNO3)=O)O)C=CC=C2N=C1 HXOZLTQYYYXMPP-UHFFFAOYSA-N 0.000 description 1
- VEVFBNBBDQZZBA-UHFFFAOYSA-N COc1cc2c(C(CN(CCC3NCc4ccc5OCCOc5c4)CC3F)O)cccc2nc1 Chemical compound COc1cc2c(C(CN(CCC3NCc4ccc5OCCOc5c4)CC3F)O)cccc2nc1 VEVFBNBBDQZZBA-UHFFFAOYSA-N 0.000 description 1
- LZOVRZDBUFUPRS-UHFFFAOYSA-N COc1cnc(cccc2C(CN(CC3)CCC3NCc(cc(c(SC3)c4)NC3=O)c4F)O)c2c1 Chemical compound COc1cnc(cccc2C(CN(CC3)CCC3NCc(cc(c(SC3)c4)NC3=O)c4F)O)c2c1 LZOVRZDBUFUPRS-UHFFFAOYSA-N 0.000 description 1
- PPXXFFAJTWIDFU-UHFFFAOYSA-N COc1cnc(cccc2OCCN3CCN(Cc(cc4)nc(N5)c4SCC5=O)CC3)c2c1 Chemical compound COc1cnc(cccc2OCCN3CCN(Cc(cc4)nc(N5)c4SCC5=O)CC3)c2c1 PPXXFFAJTWIDFU-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 description 1
- 241000588697 Enterobacter cloacae Species 0.000 description 1
- 241000943303 Enterococcus faecalis ATCC 29212 Species 0.000 description 1
- 241000194031 Enterococcus faecium Species 0.000 description 1
- 241001360526 Escherichia coli ATCC 25922 Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 239000012448 Lithium borohydride Substances 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- RJQXTJLFIWVMTO-TYNCELHUSA-N Methicillin Chemical compound COC1=CC=CC(OC)=C1C(=O)N[C@@H]1C(=O)N2[C@@H](C(O)=O)C(C)(C)S[C@@H]21 RJQXTJLFIWVMTO-TYNCELHUSA-N 0.000 description 1
- 101100189356 Mus musculus Papolb gene Proteins 0.000 description 1
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 206010034133 Pathogen resistance Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 241001069992 Proteus mirabilis ATCC 29906 Species 0.000 description 1
- VOLMSPGWNYJHQQ-UHFFFAOYSA-N Pyranone Natural products CC1=C(O)C(=O)C(O)CO1 VOLMSPGWNYJHQQ-UHFFFAOYSA-N 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- 206010057190 Respiratory tract infections Diseases 0.000 description 1
- BXNCERKNSZFDFC-UHFFFAOYSA-N S1NC=CC=C1S(=O)(=O)O Chemical compound S1NC=CC=C1S(=O)(=O)O BXNCERKNSZFDFC-UHFFFAOYSA-N 0.000 description 1
- 241000295644 Staphylococcaceae Species 0.000 description 1
- 241000191963 Staphylococcus epidermidis Species 0.000 description 1
- 241000191984 Staphylococcus haemolyticus Species 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- SWEDAZLCYJDAGW-UHFFFAOYSA-N Thiophene-2-thiol Chemical compound SC1=CC=CS1 SWEDAZLCYJDAGW-UHFFFAOYSA-N 0.000 description 1
- 101710183280 Topoisomerase Proteins 0.000 description 1
- 235000018936 Vitellaria paradoxa Nutrition 0.000 description 1
- 241001135917 Vitellaria paradoxa Species 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 125000005354 acylalkyl group Chemical group 0.000 description 1
- 125000005041 acyloxyalkyl group Chemical group 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000003302 alkenyloxy group Chemical group 0.000 description 1
- 125000006193 alkinyl group Chemical group 0.000 description 1
- 125000005194 alkoxycarbonyloxy group Chemical group 0.000 description 1
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- HSMPSHPWCOOUJH-UHFFFAOYSA-N anilinyl Chemical group [NH]C1=CC=CC=C1 HSMPSHPWCOOUJH-UHFFFAOYSA-N 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 125000005018 aryl alkenyl group Chemical group 0.000 description 1
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 1
- 125000005015 aryl alkynyl group Chemical group 0.000 description 1
- 125000004350 aryl cycloalkyl group Chemical group 0.000 description 1
- 244000052616 bacterial pathogen Species 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- QNSAUKWWWIXHAN-UHFFFAOYSA-N benzyl 3-(2-tert-butylsilyloxypropan-2-yl)-4-hydroxypiperidine-1-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)N1CC(C(CC1)O)C(O[SiH2]C(C)(C)C)(C)C QNSAUKWWWIXHAN-UHFFFAOYSA-N 0.000 description 1
- NUWIXCNNEFUMDJ-UHFFFAOYSA-N benzyl 3-(2-tert-butylsilyloxypropan-2-yl)-4-oxopiperidine-1-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)N1CC(C(CC1)=O)C(O[SiH2]C(C)(C)C)(C)C NUWIXCNNEFUMDJ-UHFFFAOYSA-N 0.000 description 1
- WITCLTOOIPMTGJ-UHFFFAOYSA-N benzyl 4-(2-hydroxyethyl)piperazine-1-carboxylate Chemical compound C1CN(CCO)CCN1C(=O)OCC1=CC=CC=C1 WITCLTOOIPMTGJ-UHFFFAOYSA-N 0.000 description 1
- LNMUXMRJEMVUCL-UHFFFAOYSA-N benzyl 4-(2-methylsulfonyloxyethyl)piperazine-1-carboxylate Chemical compound C1CN(CCOS(=O)(=O)C)CCN1C(=O)OCC1=CC=CC=C1 LNMUXMRJEMVUCL-UHFFFAOYSA-N 0.000 description 1
- QFNKYCCDWGYGHM-UHFFFAOYSA-N benzyl 4-[2-(3-methoxyquinolin-5-yl)oxyethyl]piperazine-1-carboxylate Chemical compound C12=CC(OC)=CN=C2C=CC=C1OCCN(CC1)CCN1C(=O)OCC1=CC=CC=C1 QFNKYCCDWGYGHM-UHFFFAOYSA-N 0.000 description 1
- ZQADWZOIFPJNMA-UHFFFAOYSA-N benzyl 4-amino-3-(2-tert-butylsilyloxypropan-2-yl)piperidine-1-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)N1CC(C(CC1)N)C(O[SiH2]C(C)(C)C)(C)C ZQADWZOIFPJNMA-UHFFFAOYSA-N 0.000 description 1
- RUASIMJTCKBZDQ-UHFFFAOYSA-N benzyl 4-amino-4-(2-methoxy-2-oxoethyl)piperidine-1-carboxylate Chemical compound C1CC(CC(=O)OC)(N)CCN1C(=O)OCC1=CC=CC=C1 RUASIMJTCKBZDQ-UHFFFAOYSA-N 0.000 description 1
- VZOVOHRDLOYBJX-UHFFFAOYSA-N benzyl 4-oxopiperidine-1-carboxylate Chemical compound C1CC(=O)CCN1C(=O)OCC1=CC=CC=C1 VZOVOHRDLOYBJX-UHFFFAOYSA-N 0.000 description 1
- BZGZHDSMWXBMJR-UHFFFAOYSA-N benzyl N-[3-fluoro-1-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperidin-4-yl]carbamate Chemical compound C12=CC(OC)=CN=C2C=CC=C1C(O)CN(CC1F)CCC1NC(=O)OCC1=CC=CC=C1 BZGZHDSMWXBMJR-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- DWPWWTIOOLQURY-UHFFFAOYSA-N benzyl n-(3-fluoropiperidin-4-yl)carbamate Chemical compound FC1CNCCC1NC(=O)OCC1=CC=CC=C1 DWPWWTIOOLQURY-UHFFFAOYSA-N 0.000 description 1
- CTOUWUYDDUSBQE-UHFFFAOYSA-N benzyl piperazine-1-carboxylate Chemical compound C1CNCCN1C(=O)OCC1=CC=CC=C1 CTOUWUYDDUSBQE-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 229940075419 choline hydroxide Drugs 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- XLJMAIOERFSOGZ-UHFFFAOYSA-M cyanate Chemical compound [O-]C#N XLJMAIOERFSOGZ-UHFFFAOYSA-M 0.000 description 1
- 150000003997 cyclic ketones Chemical class 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- FWFSEYBSWVRWGL-UHFFFAOYSA-N cyclohex-2-enone Chemical compound O=C1CCCC=C1 FWFSEYBSWVRWGL-UHFFFAOYSA-N 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- SNRCKKQHDUIRIY-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloromethane;dichloropalladium;iron(2+) Chemical compound [Fe+2].ClCCl.Cl[Pd]Cl.C1=C[CH-]C(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.C1=C[CH-]C(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 SNRCKKQHDUIRIY-UHFFFAOYSA-L 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000004855 decalinyl group Chemical group C1(CCCC2CCCCC12)* 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- JGFBRKRYDCGYKD-UHFFFAOYSA-N dibutyl(oxo)tin Chemical compound CCCC[Sn](=O)CCCC JGFBRKRYDCGYKD-UHFFFAOYSA-N 0.000 description 1
- 125000006222 dimethylaminomethyl group Chemical group [H]C([H])([H])N(C([H])([H])[H])C([H])([H])* 0.000 description 1
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 150000002084 enol ethers Chemical class 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 1
- YPFMNHZRNXPYBG-UHFFFAOYSA-N ethyl 1-benzyl-4-oxopiperidin-1-ium-3-carboxylate;chloride Chemical compound Cl.C1CC(=O)C(C(=O)OCC)CN1CC1=CC=CC=C1 YPFMNHZRNXPYBG-UHFFFAOYSA-N 0.000 description 1
- HKGIEPXOWGJFCW-UHFFFAOYSA-N ethyl 2,4-difluoro-5-nitrobenzoate Chemical compound CCOC(=O)C1=CC([N+]([O-])=O)=C(F)C=C1F HKGIEPXOWGJFCW-UHFFFAOYSA-N 0.000 description 1
- ZLWAWDMYOMIDBX-UHFFFAOYSA-N ethyl 2-(5-formyl-2-nitrophenoxy)acetate Chemical compound CCOC(=O)COC1=CC(C=O)=CC=C1[N+]([O-])=O ZLWAWDMYOMIDBX-UHFFFAOYSA-N 0.000 description 1
- XGZFVYJLIMOTTF-UHFFFAOYSA-N ethyl 2H-thiazine-6-carboxylate Chemical compound C(C)OC(=O)C1=CC=CNS1 XGZFVYJLIMOTTF-UHFFFAOYSA-N 0.000 description 1
- 125000006534 ethyl amino methyl group Chemical group [H]N(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical compound C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 1
- RMBPEFMHABBEKP-UHFFFAOYSA-N fluorene Chemical compound C1=CC=C2C3=C[CH]C=CC3=CC2=C1 RMBPEFMHABBEKP-UHFFFAOYSA-N 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 244000000059 gram-positive pathogen Species 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 125000005349 heteroarylcycloalkyl group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- SXWRTZOXMUOJER-UHFFFAOYSA-N hydron;piperidin-4-one;chloride;hydrate Chemical compound O.Cl.O=C1CCNCC1 SXWRTZOXMUOJER-UHFFFAOYSA-N 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- QNXSIUBBGPHDDE-UHFFFAOYSA-N indan-1-one Chemical compound C1=CC=C2C(=O)CCC2=C1 QNXSIUBBGPHDDE-UHFFFAOYSA-N 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 239000008011 inorganic excipient Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 150000002527 isonitriles Chemical class 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- BEJNERDRQOWKJM-UHFFFAOYSA-N kojic acid Chemical compound OCC1=CC(=O)C(O)=CO1 BEJNERDRQOWKJM-UHFFFAOYSA-N 0.000 description 1
- 229960004705 kojic acid Drugs 0.000 description 1
- WZNJWVWKTVETCG-UHFFFAOYSA-N kojic acid Natural products OC(=O)C(N)CN1C=CC(=O)C(O)=C1 WZNJWVWKTVETCG-UHFFFAOYSA-N 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- AUHZEENZYGFFBQ-UHFFFAOYSA-N mesitylene Substances CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 description 1
- 125000001827 mesitylenyl group Chemical group [H]C1=C(C(*)=C(C([H])=C1C([H])([H])[H])C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- HNQIVZYLYMDVSB-UHFFFAOYSA-N methanesulfonimidic acid Chemical compound CS(N)(=O)=O HNQIVZYLYMDVSB-UHFFFAOYSA-N 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- JIQNWFBLYKVZFY-UHFFFAOYSA-N methoxycyclohexatriene Chemical compound COC1=C[C]=CC=C1 JIQNWFBLYKVZFY-UHFFFAOYSA-N 0.000 description 1
- JWERGOLFCYWFBY-UHFFFAOYSA-N methyl 2-(4-formyl-2-nitrophenyl)sulfanylacetate Chemical compound COC(=O)CSC1=CC=C(C=O)C=C1[N+]([O-])=O JWERGOLFCYWFBY-UHFFFAOYSA-N 0.000 description 1
- UIAHQILQYROVCF-UHFFFAOYSA-N methyl 3-oxo-4h-pyrido[3,2-b][1,4]thiazine-6-carboxylate Chemical compound S1CC(=O)NC2=NC(C(=O)OC)=CC=C21 UIAHQILQYROVCF-UHFFFAOYSA-N 0.000 description 1
- OQUCMNQHZFOMPC-UHFFFAOYSA-N methyl 6-amino-5-bromopyridine-2-carboxylate Chemical compound COC(=O)C1=CC=C(Br)C(N)=N1 OQUCMNQHZFOMPC-UHFFFAOYSA-N 0.000 description 1
- OHIHEJTUXNQOPM-UHFFFAOYSA-N methyl 6-aminopyridine-2-carboxylate Chemical compound COC(=O)C1=CC=CC(N)=N1 OHIHEJTUXNQOPM-UHFFFAOYSA-N 0.000 description 1
- LSEFCHWGJNHZNT-UHFFFAOYSA-M methyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(C)C1=CC=CC=C1 LSEFCHWGJNHZNT-UHFFFAOYSA-M 0.000 description 1
- 150000003956 methylamines Chemical class 0.000 description 1
- 229960003085 meticillin Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229940113083 morpholine Drugs 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- UEXYAZLLFZIXHN-UHFFFAOYSA-N n-(6-methylpyridin-2-yl)acetamide Chemical compound CC(=O)NC1=CC=CC(C)=N1 UEXYAZLLFZIXHN-UHFFFAOYSA-N 0.000 description 1
- OMOFUDLYVGGVSO-UHFFFAOYSA-N n-[1-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperidin-4-yl]-2,3-dihydro-1,4-benzodioxine-6-carboxamide Chemical compound O1CCOC2=CC(C(=O)NC3CCN(CC3)CC(O)C3=CC=CC4=NC=C(C=C43)OC)=CC=C21 OMOFUDLYVGGVSO-UHFFFAOYSA-N 0.000 description 1
- XTWCUKFKBWTZFO-UHFFFAOYSA-N n-[1-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperidin-4-yl]-3-oxo-4h-pyrido[3,2-b][1,4]thiazine-6-carboxamide Chemical compound S1CC(=O)NC2=NC(C(=O)NC3CCN(CC3)CC(O)C3=CC=CC4=NC=C(C=C43)OC)=CC=C21 XTWCUKFKBWTZFO-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 150000002825 nitriles Chemical group 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- JCXJVPUVTGWSNB-UHFFFAOYSA-N nitrogen dioxide Inorganic materials O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- NIHNNTQXNPWCJQ-UHFFFAOYSA-N o-biphenylenemethane Natural products C1=CC=C2CC3=CC=CC=C3C2=C1 NIHNNTQXNPWCJQ-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000008012 organic excipient Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 229920000166 polytrimethylene carbonate Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 229940074439 potassium sodium tartrate Drugs 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- UJEHWLFUEQHEEZ-UHFFFAOYSA-N quinoxaline-2-carbaldehyde Chemical compound C1=CC=CC2=NC(C=O)=CN=C21 UJEHWLFUEQHEEZ-UHFFFAOYSA-N 0.000 description 1
- 208000020029 respiratory tract infectious disease Diseases 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 239000012363 selectfluor Substances 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- DUOWOUTVIPTSKH-UHFFFAOYSA-N tert-butyl 3-fluoro-4-(phenylmethoxycarbonylamino)piperidine-1-carboxylate Chemical compound FC1CN(C(=O)OC(C)(C)C)CCC1NC(=O)OCC1=CC=CC=C1 DUOWOUTVIPTSKH-UHFFFAOYSA-N 0.000 description 1
- JZNWQLLPLOQGOI-UHFFFAOYSA-N tert-butyl 3-fluoro-4-oxopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=O)C(F)C1 JZNWQLLPLOQGOI-UHFFFAOYSA-N 0.000 description 1
- GGTHTGQXJVAKPM-UHFFFAOYSA-N tert-butyl 4-(3-thiophen-2-ylsulfanylpropyl)piperazine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCN1CCCSC1=CC=CS1 GGTHTGQXJVAKPM-UHFFFAOYSA-N 0.000 description 1
- RDSCDOZKNVEYOX-UHFFFAOYSA-N tert-butyl 4-(4-benzylpiperazin-1-yl)piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1N1CCN(CC=2C=CC=CC=2)CC1 RDSCDOZKNVEYOX-UHFFFAOYSA-N 0.000 description 1
- JXBLMDWEOQDVAC-UHFFFAOYSA-N tert-butyl 4-(benzylamino)-3-fluoropiperidine-1-carboxylate Chemical compound FC1CN(C(=O)OC(C)(C)C)CCC1NCC1=CC=CC=C1 JXBLMDWEOQDVAC-UHFFFAOYSA-N 0.000 description 1
- QAEMEZIMSSGJHN-UHFFFAOYSA-N tert-butyl 4-[4-[2-hydroxy-2-(3-methoxyquinolin-5-yl)ethyl]piperazin-1-yl]piperidine-1-carboxylate Chemical compound C12=CC(OC)=CN=C2C=CC=C1C(O)CN(CC1)CCN1C1CCN(C(=O)OC(C)(C)C)CC1 QAEMEZIMSSGJHN-UHFFFAOYSA-N 0.000 description 1
- ZQRYPCAUVKVMLZ-UHFFFAOYSA-N tert-butyl 4-amino-3-fluoropiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(N)C(F)C1 ZQRYPCAUVKVMLZ-UHFFFAOYSA-N 0.000 description 1
- XMGAKAOAPIZUJG-UHFFFAOYSA-N tert-butyl 4-piperazin-1-ylpiperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1N1CCNCC1 XMGAKAOAPIZUJG-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- CKXZPVPIDOJLLM-UHFFFAOYSA-N tert-butyl n-piperidin-4-ylcarbamate Chemical compound CC(C)(C)OC(=O)NC1CCNCC1 CKXZPVPIDOJLLM-UHFFFAOYSA-N 0.000 description 1
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 1
- VFJYIHQDILEQNR-UHFFFAOYSA-M trimethylsulfanium;iodide Chemical compound [I-].C[S+](C)C VFJYIHQDILEQNR-UHFFFAOYSA-M 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- 239000000052 vinegar Substances 0.000 description 1
- 235000021419 vinegar Nutrition 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the present application describes novel compounds having antibacterial activity. These compounds are, inter alia, as inhibitors of DNA gyrase and Topoisomerases (eg topoisomerase II and IV) of interest.
- the present invention relates to compounds of the general formula (I):
- R 1 is a hydrogen atom, a halogen atom, a hydroxy, an amino, a cyano, a nitro, a thiol, a
- Ci-C 6 alkyl a C 2 ⁇ C 6 alkenyl group, a C 2 -C 6 alkynyl, a heteroalkyl, an alkyloxy, a Heteroalkyloxy-, a cycloalkyloxy, a Alkylcycloalkyloxy-, a hetero - cycloalkyloxy or a heteroalkylcycloalkyloxy group;
- R 2 is a hydrogen atom, a halogen atom, a hydroxy, an amino, a cyano, C 1 -C 6 -alkyl, C 2 -C 6 -alkenyl, C 2 -C 6 -alkynyl or a heteroalkyl group;
- R 3 is a group of the following formula:
- U and V are independently nitrogen atoms or groups of the formula CH or CR 6 ;
- R 4 is independently halogen, hydroxy, amino, cyano, nitro or thiol, alkyl, alkenyl, alkynyl or heteroalkyl;
- n 0, 1 or 2;
- R 5 is an alkyl, alkenyl, alkynyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, alkylcycloalkyl, heteroalkyl, cycloalkyl, heterocycloalkyl, aralkyl or heteroaralkyl radical;
- the groups R 6 are independently a halogen atom, a hydroxy, alkyl, alkenyl, alkynyl or heteroalkyl group;
- R 7 is a hydrogen atom, a trifluoromethyl, Ci-C ⁇ alkyl, C 2 -C 6 alkenyl, Ci-C 6 alkoxycarbonyl, Ci-C ⁇ -alkylcarbonyl or a carbonylamino group wherein the amino group of carbonylamino optionally carbonyl- 6 alkenyloxy by a Ci-C ⁇ - alkoxycarbonyl, Ci-C ⁇ alkylcarbonyl, C2 ⁇ C, C 2 -C 6 -Alkenylcarbonyl-, dC 6 alkyl, C 2 -C 6 - alkenyl, and optionally further substituted by a Ci-C 6 alkyl or a C 2 -C 6 alkenyl group may be substituted;
- the radicals R 8 , R 9 , R 10 , R 12 and R 13 independently of one another represent a hydrogen atom, a halogen atom, an azide, a trifluoro- methyl, hydroxy, amino, Ci-C ⁇ -alkyloxy, Ci-C ⁇ alkyl thio, Ci-C 6 alkyl, C 2 -C 6 alkenyl, Ci-C ⁇ alkoxycarbonyl, C 2 -C 6 -Alkenyloxycarbonyl-, Ci-C 6 alkylsulfonyl, C 2 -C O - alkenylsulfonyl or a sulfonylamino group are wherein the amino group of the sulfonylamino group may be optionally substituted by a Ci-C ⁇ alkyl or phenyl;
- R 11 is a hydrogen atom, a trifluoromethyl, C 1 -C 6 -alkyl, C 2 -C 6 -alkenyl, C 1 -C 6 -alkoxycarbonyl, C 1 -C 6 -alkylcarbonyl or a carbonylamino group, where the amino group of the carbonylamino group is optionally carbonyl by a C C ⁇ - alkoxycarbonyl, Ci-C ⁇ -alkylcarbonyl, C 2 -C 6 alkenyloxy, C 2 -C 6 -Alkenylcarbonyl-, Ci-C ⁇ alkyl, C 2 -C O - alkenyl - And optionally further substituted by a Ci-C ö alkyl or a C 2 -C 6 alkenyl group may be substituted;
- alkyl refers to a saturated, straight or branched hydrocarbon group having 1 to 20 carbon atoms, preferably 1 to 12 carbon atoms, more preferably 1 to 6 carbon atoms, e.g. the methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, tert-butyl, n-pentyl, n-hexyl, 2, 2-dimethylbutyl or n-octyl group.
- alkenyl and alkynyl refer to at least partially unsaturated, straight-chain or branched hydrocarbon groups containing 2 to 20 Carbon atoms, preferably 2 to 12 carbon atoms, more preferably 2 to 6 carbon atoms, eg the ethenyl, allyl, acetylenyl, propargyl, isoprenyl or hex-2-enyl group.
- Alkenyl groups preferably have one or two (particularly preferably one) double bonds or alkinyl groups one or two (particularly preferably one) triple bonds.
- alkyl, alkenyl and alkynyl refer to groups in which one or more
- Hydrogen atoms are replaced by a halogen atom (preferably F or Cl), e.g. the 2, 2, 2-trichloroethyl or the trifluoromethyl group.
- a halogen atom preferably F or Cl
- e.g. the 2, 2, 2-trichloroethyl or the trifluoromethyl group e.g. the 2, 2, 2-trichloroethyl or the trifluoromethyl group.
- heteroalkyl refers to an alkyl, an alkenyl or an alkynyl group in which one or more (preferably 1, 2 or 3) carbon atoms are represented by an oxygen, nitrogen, phosphorus, boron, selenium, Silicon or sulfur atom are replaced (preferably oxygen, sulfur or nitrogen).
- heteroalkyl further refers to a carboxylic acid or a carboxylic acid derived group such as e.g. Acyl, acylalkyl, alkoxycarbonyl, acyloxy, acyloxyalkyl, carboxyalkylamide or alkoxycarbonyloxy.
- heteroalkyl groups are groups of the formulas R a -OY a , R a -SY a , R a -N (R b ) -Y a , R a -CO-Y a , R a -O-CO- Y a -, R a -CO-OY a -, R a -CO-N (R b) -Y a -, R a -N (R b) -CO-Y a -, R a -O-CO- N (R b ) -Y a -, R a -N (R b ) -CO-OY a -, R a -N (R b ) -CO-OY a -, R a -N (R b ) -CO-N (R c ) -Y a -, R a -O -CO-OY a -,
- R a is -O-CS-Y a -, R a is -CS-OY a -, R a is -CS-N (R b ) -Y a -, R a is -N (R b ) -CS-Y a -, R a -O-CS-N (R b ) -Y a -, R a -N (R b ) -CS-OY a -, R a -N (R b ) -CS-N (R c ) -Y a , R a -O-CS-OY a -, R a -S-CO-Y a -, R a -CO-SY a -, R a -CO-SY a -, R a -CO-SY a -, R a -S-CO-N (R b ) -Y a -, R a
- Alkynyl group and Y a is a direct bond, a Ci-C 6 - alkylene, a C 2 -C 6 alkenylene or a C 2 -C 6 - alkynylene, wherein each heteroalkyl group at least one carbon atom and one or more hydrogen atoms are replaced by Fluorine or chlorine atoms may be replaced.
- heteroalkyl groups are methoxy, trifluoromethoxy, ethoxy, n-propyloxy, isopropyloxy, tert-butyloxy, methoxymethyl, ethoxymethyl, -CH 2 CH 2 OH, -CH 2 OH, methoxyethyl, methylamines, ethylamino, dimethylamino, diethylamino, iso- Propylethylamino, methylaminomethyl, ethylaminomethyl, di-iso-propylaminoethyl, enol ether, dimethylaminomethyl, dimethylaminoethyl, acetyl, propionyl, butyryloxy, acetyloxy, methoxycarbonyl, ethoxycarbonyl, N-ethyl-N-methylcarbamoyl or N-methylcarbamoyl.
- heteroalkyl groups are nitrile, isonitrile, cyanate, thiocyanate, isocyanate, isothiocyanate and alkylnitrile groups.
- An example of a heteroalkylene group is a group of the formula -CH 2 CH (OH) -.
- cycloalkyl refers to a saturated or partially unsaturated (eg cycloalkenyl) cyclic Group which has one or more rings (preferably 1 or 2) and contains 3 to 14 ring carbon atoms, preferably 3 to 10 (in particular 3, 4, 5, 6 or 7) ring carbon atoms.
- the term cycloalkyl furthermore refers to groups in which one or more hydrogen atoms have been replaced by fluorine, chlorine, bromine or iodine atoms or OH, 0O, SH, SS, NH 2 , NHNH or NO 2 groups,
- cyclic ketones such as cyclohexanone, 2-cyclohexenone or cyclopentanone.
- Further concrete examples of cycloalkyl groups are the cyclopropyl, cyclobutyl,
- heterocycloalkyl refers to a cycloalkyl group as defined above in which one or more (preferably 1, 2 or 3) ring carbon atoms are represented by an oxygen, nitrogen, silicon, selenium, phosphorus or sulfur atom (preferably oxygen, sulfur or sulfur) Nitrogen) are replaced.
- a heterocycloalkyl group preferably has 1 or 2 rings with 3 to 10 (in particular 3, 4, 5, 6 or 7) ring atoms.
- heterocycloalkyl furthermore refers to groups in which one or more hydrogen atoms are replaced by fluorine,
- Examples are the piperidyl, piperazinyl, morpholinyl, urotropinyl, pyrrolidinyl, tetrahydrothiophenyl, tetrahydropyranyl, tetrahydrofuryl or 2-pyrazolinyl group as well as lactams, lactones, cyclic imides and cyclic anhydrides.
- alkylcycloalkyl refers to groups containing both cycloalkyl and alkyl, alkenyl or alkynyl groups according to the above definitions, for example alkylcycloalkyl, cycloalkylalkyl, alkylcycloalkenyl, alkenylcycloalkyl and alkynylcycloalkyl groups.
- an alkylcycloalkyl group contains a cycloalkyl group having one or two rings and containing 3 to 10 (especially 3, 4, 5, 6 or 7) ring carbon atoms and one or two alkyl, alkenyl or alkynyl groups having 1 or 2 to 6 carbon atoms.
- heteroalkylcycloalkyl refers to alkylcycloalkyl groups as defined above in which one or more (preferably 1, 2 or 3) carbon atoms are represented by an oxygen, nitrogen, silicon, selenium, phosphorus or sulfur atom (preferably oxygen, Sulfur or nitrogen) are replaced.
- a heteroalkylcycloalkyl group preferably has 1 or 2 rings having 3 to 10 (in particular 3, 4, 5, 6 or 7) ring atoms and one or two alkyl, alkenyl, alkynyl or heteroalkyl groups having 1 or 2 to 6 carbon atoms.
- Examples of such groups are alkylheterocycloalkyl, alkylheterocycloalkenyl, alkenyl-heterocycloalkyl, alkynylheterocycloalkyl, heteroalkylcycloalkyl, heteroalkylheterocycloalkyl and heteroalkyl-heterocyclic-alkenyl, where the cyclic groups are saturated or mono-, di- or tri-unsaturated.
- aryl or Ar refers to an aromatic group having one or more rings and containing 6 to 14 ring carbon atoms, preferably 6 to 10 (especially 6) ring carbon atoms.
- aryl (or Ar) further refers to groups in which one or several hydrogen atoms are replaced by fluorine, chlorine, bromine or iodine atoms or OH, SH, NH 2 , or NO 2 groups. Examples are the phenyl, naphthyl, biphenyl, 2-fluorophenyl, anilinyl, 3-nitrophenyl or 4-hydroxyphenyl group.
- heteroaryl refers to an aromatic group which has one or more rings and contains 5 to 14 ring atoms, preferably 5 to 10 (in particular 5 or 6) ring atoms and one or more (preferably 1, 2, 3 or 4) oxygen atoms. , Nitrogen, phosphorus or sulfur ring atoms (preferably O, S or N).
- heteroaryl furthermore refers to groups in which one or more hydrogen atoms have been replaced by fluorine, chlorine, bromine or iodine atoms or OH, SH, NH 2 or NO 2 groups.
- Examples are 4-pyridyl, 2-imidazolyl, 3-phenylpyrrolyl, thiazolyl, oxazolyl, triazolyl, tetrazolyl, isoxazolyl, indazolyl, indolyl, benzimidazolyl, pyridazinyl, quinolinyl, purinyl , Carbazolyl, acridinyl, pyrimidyl, 2,3'-bifuryl, 3-pyrazolyl and isoquinolinyl groups.
- aralkyl refers to groups containing both aryl and alkyl, alkenyl, alkynyl and / or cycloalkyl groups according to the above definitions, such as arylalkyl, arylalkenyl, arylalkynyl, arylcycloalkyl, arylcycloalkenyl, alkylarylcycloalkyl and alkylarylcycloalkenyl groups.
- aralkyls are toluene, xylene, mesitylene, styrene, benzyl chloride, o-fluorotoluene, 1H-indene, tetralin, dihydronaphthalene, indanone, phenylcyclopentyl, cumene, cyclohexylphenyl, fluorene and indane.
- an aralkyl group one or two aromatic ring systems (1 or 2 rings) having 6 to 10 carbon atoms and one or two alkyl, alkenyl and / or alkynyl groups having 1 or 2 to 6 carbon atoms and / or a cycloalkyl group having 5 or 6 ring carbon atoms.
- heteroaralkyl refers to an aralkyl group as defined above in which one or more (preferably 1, 2, 3 or 4) carbon atoms are represented by an oxygen, nitrogen, silicon, selenium, phosphorus,
- a heteroaralkyl group contains one or two aromatic ring systems (1 or 2 rings) having 5 or 6 to 10 carbon atoms and one or two alkyl, alkenyl and / or alkynyl groups having 1 or 2 to 6 carbon atoms and / or a cycloalkyl group 5 or 6 ring carbon atoms, wherein 1, 2, 3 or 4 of these carbon atoms are replaced by oxygen, sulfur or nitrogen atoms.
- Examples are arylheteroalkyl, arylheterocycloalkyl,
- This expression further refers to groups-substituted with unsubsti- Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, Ci-C6 - heteroalkyl, C 3 - are heteroaralkyl groups substituted - Ci 0 cycloalkyl, C 2 -C 9 heterocycloalkyl, Ce-Cio-aryl, Ci-C9 heteroaryl, C7-Ci2 aralkyl or C 2 -C n ,
- A is selected from the following groups: -CH (OH) CH 2 -, -CH 2 CH (OH) -, -OCH 2 CH 2 -, -CH 2 CH 2 O-, -CH 2 SO 2 -, -SO 2 CH 2 -, -CH 2 N (C 1 -C 4 -alkyl) -, -N (C 1 -C 4 -alkyl) CH 2 -, -CH 2 NH-, -NHCH 2 - , -CH 2 O-, -OCH 2 -, -CH 2 S-,
- R 1 is a cyano, a C 1 -C 4 -alkyloxy or a C 1 -C 4 -Heteroalkyloxy distr, wherein one or more hydrogen atoms of these groups may be replaced by fluorine atoms.
- R 1 is a methoxy group.
- R 2 is a hydrogen or H halo logging ratio.
- R 2 is a hydrogen, a chlorine or a fluorine atom.
- R 3 is selected from the following groups
- R 3 is particularly preferably selected from the following groups:
- the radicals R 4 are preferably in turn independently of one another a halogen atom, a hydroxy, a cyano, a Ci-C 4 - alkyl or a Ci-C4 heteroalkyl group (for example, a hydroxymethyl group).
- the radicals R 4 are particularly preferably independently of one another a fluorine or chlorine atom or a C 1 -C 4 -heteroalkyl group (for example a hydroxymethyl group).
- n is 0 or 1; more preferably 0.
- R 5 is a heteroalkylcycloalkyl or heteroaralkyl group.
- Heterocyclicalkylene group and Y is an aryl, heteroaryl, aralkyl, heteroaralkyl, cycloalkyl, heterocycloalkyl, alkylcycloalkyl, heteroalkylcycloalkyl, heteroarylheterocycloalkyl or an arylheterocycloalkyl group (in particular a heterocycloalkyl, a heteroaryl,
- Heteroalkylene group (in particular a C 1 -C 4 -Heteroalkylen- group) and Y 2 is an aryl, heteroaryl, aralkyl, heteroaralkyl, cycloalkyl, heterocycloalkyl, alkylcycloalkyl, Heteroalkylcycloalkyl-, Heteroarylhetero- cycloalkyl or a Arylheterocycloalkyl group (in particular a heterocycloalkyl, an aryl, a heteroaryl, aralkyl, heteroaralkyl, a heteroaryl) heterocycloalkyl or an arylheterocycloalkyl group).
- Y has one of the following structures:
- X 1 , X 2 and X 3 are independently nitrogen atoms or groups of the formula CR 20 , X 4 and X 5 are independently oxygen or sulfur atoms or groups of the formula NR 21 , o is 0, 1 or 2, R 14 , R 15 , R 16 , R 17 , R 19 and R 20 are independently hydrogen atoms, halogen atoms, hydroxy, Ci-C ⁇ -alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl or Ci -C ⁇ -heteroalkyl, and R 18 and R 21 are independently hydrogen, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl or C 6 heteroalkyl groups are.
- Y has one of the following structures:
- Y has one of the following structures:
- the radicals R 6 independently of one another are a halogen atom, a hydroxy, a C 1 -C 4 -alkyl or a C 1 -C 4 -heteroalkyl group (for example a hydroxyethyl group).
- the radicals R 6 are furthermore preferably, independently of one another, a fluorine or a chlorine atom or a hydroxy, a C 1 -C 4 -alkyloxy, a C 1 -C 4 -heteroalkyl (eg a hydroxyethyl group) or a C 3 -C 6 Dialkylamino- methyl group, wherein one or more hydrogen atoms of these groups may be replaced by fluorine atoms.
- the radicals R 6 are particularly preferably independently of one another a Ci-C 4 -Heteroalkyl distr (eg a hydroxyethyl group).
- R 2 , R 4a and R 6a are independently a hydrogen or halogen atom or a Ci-C 4 -Heteroalkyl distr (eg a hydroxymethyl or hydroxyethyl group) (in particular R 2 is a hydrogen atom and R 4a and R 6a is a hydrogen or Fluorine atom or a C ⁇ -C 4 heteroalkyl group).
- B and Y are as defined above.
- Particular preference is given to compounds of the formula (III):
- R 2 is a hydrogen or halogen atom (in particular, R 2 is a hydrogen atom).
- B and Y are as defined above.
- B is a bond or a group of the formula -CH 2 CH 2 -, -CH 2 CH 2 CH 2 - or -CH 2 CH (OH) -.
- R 2 is a hydrogen or halogen atom (in particular, R 2 is a hydrogen atom).
- B and Y are as defined above.
- Compounds of formulas (I) to (IV) may contain one or more chiral centers due to their substitution.
- the present invention therefore encompasses both all pure enantiomers and all pure diastereomers, as well as their mixtures in any mixing ratio.
- all cis / trans isomers of the compounds of the general formulas (I) to (IV) and mixtures thereof are also encompassed by the present invention.
- all tautomeric forms of the compounds of the formulas (I) to (IV) are encompassed by the present invention.
- compositions according to the present invention contain at least one compound of formulas (I) to (IV) as active ingredient and optionally excipients and / or adjuvants.
- Examples of pharmacologically acceptable salts of the compounds of the formulas (I) to (IV) are salts of physiologically acceptable mineral acids such as hydrochloric acid, sulfuric acid and phosphoric acid or salts of organic acids such as methanesulfonic acid, p-toluenesulfonic acid,
- pharmacologically acceptable salts of the compounds of formulas (I) to (IV) are alkali or alkaline earth salts such as sodium, potassium, lithium, calcium or magnesium salts, ammonium salts or salts of organic see bases such as methylamine, dimethylamine, triethylamine , Piperidine, ethylenediamine, lysine, choline hydroxide, meglumine, morpholine or arginine.
- Compounds of formula (I) to (IV) may be solvated, in particular hydrated.
- the hydration may occur, for example, during the manufacturing process or as a result of the hygroscopic nature of the initially anhydrous compounds of formulas (I) to (IV).
- the compounds of formulas (I) to (IV) may be present either as achiral compounds, diastereomeric mixtures, mixtures of enantiomers or as optically pure compounds.
- the prodrugs consist of a compound of formulas (I) to (IV) and at least one pharmacologically acceptable protecting group which is cleaved off under physiological conditions, eg an alkoxy, aralkyloxy, acyl or acyloxy group, e.g. a methoxy, ethoxy, benzyloxy, acetyl or acetyloxy group.
- compounds of formulas (I) to (IV) will be prepared using the known and acceptable modes, either alone or in combination with any of them administered to other therapeutic agents.
- Such therapeutically useful agents may be administered by one of the following routes: orally, eg as dragees, coated tablets, pills, semi-solids, soft or hard capsules, solutions, emulsions or suspensions; parenteral, eg as an injectable solution; rectally as suppositories; by inhalation, eg as
- the therapeutically useful product may be mixed with pharmacologically inert, inorganic or organic excipients, e.g. with lactose, sucrose, glucose, gelatin, malt, silica gel, starch or derivatives thereof, talc, stearic acid or theirs
- excipients such as vegetable oils, petroleum, animal or synthetic oils, wax, fat, polyols.
- excipients such as water, alcohols, aqueous saline, aqueous dextrose, polyols, glycerine, vegetable oils, petroleum, animal or synthetic oils may be used.
- excipients such as vegetable oils, petroleum, animal or synthetic oils, wax, fat and polyols.
- compressed gases that are suitable for this purpose, such as oxygen, nitrogen and carbon dioxide.
- the pharmaceutically acceptable agents may also contain additives for preservation, stabilization, emulsifiers, sweeteners, flavorings, salts for Change in osmotic pressure, buffer, coating additives and antioxidants.
- the compounds of the general formulas (I), (II), (III) and (IV) have improved properties as compared with the antibacterial compounds known in the art. Particularly noteworthy here are improved antibacterial activity, better solubility and improved PK properties.
- Combinations with other therapeutic agents may include other antimicrobial and antifungal agents.
- the dose of the biologically active compound of the present invention may vary within wide limits and may be adjusted to individual needs. In general, a dose of 10 mg to 4000 mg per day is suitable, with a preferred dose being 50 to 3000 mg per day. In appropriate cases, the dose may also be below or above the above values.
- the daily dose may be given as a single dose or in multiple doses. A typical single dose contains about 50 mg, 100 mg, 250 mg, 500 mg, 1 g or 2 g of the active ingredient. Examples
- tetrakis (triphenylphosphine) -palladium (0) (1155 g) was added to a solution of methoxyquinoline (Ib) (9.52 g) in dry 1, 2-dimethoxyethane (450 ml) at room temperature and stirred for 20 minutes. Then, potassium carbonate (5.57 g), water (120 ml) and 2,4,6-trivinylcycloboroxane-pyridine complex (3.85 g - O'Sheas Reagent - See J. Org. Ex., Vol. 67 (2002), 4968 -4971) was added and heated for 4 hours at 100 0 C.
- AD mix alpha (90.2 g) and methanesulfonic acid amide (7.6 g) were dissolved in water (280 ml) and tert-butanol (280 ml) at room temperature.
- the orange solution was cooled to 0 0 C, was added vinylquinoline (Ic) (14.4 g) and stirred for 2 days at 0-4 0 C.
- vinylquinoline (Ic) (14.4 g) and stirred for 2 days at 0-4 0 C.
- sodium pyrosulfite (108 g) was added at 0 ° C. and stirred for 30 minutes at this temperature. After warming to room temperature, it was extracted with ethyl acetate (5 x 150 ml). The combined organic phases were dried over sodium sulfate, filtered and concentrated.
- the crude product was purified by flash chromatography (silica gel, dichloromethane / methanol 29: 1, 4: 1) to afford the desired product (14.91 g).
- Epoxide (If) (689 mg) and 4- (tert-butoxycarbonyl amino) piperidine (686 mg) were dissolved in DMF (11 ml), treated with potassium carbonate (497 mg) and lithium perchlorate (364 mg) at 80 0 C. stirred for 2 days.
- the solution was concentrated, the residue dissolved in dichloromethane and washed with water and sat. Extracted sodium chloride solution.
- the organic phase was dried over magnesium sulfate, filtered and concentrated.
- the residue was purified by flash chromatography (silica gel, dichloromethane / methanol 97: 3) to afford the desired product (1.22 g).
- Boc-amine (Ig) (1.22 g) was dissolved in dichloromethane (23 ml), treated at 0 C with 0-5 Trifluoressigcicere (2.3 ml) and stirred at room temperature overnight. The solution was basified with 2N sodium hydroxide and the phases separated. The aqueous phase was extracted with dichloromethane. The combined organic phases were washed with sat. Washed sodium chloride solution, dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (silica gel, Dichloromethane / methanol 9: 1 + 1% ammonia) to afford the desired product (557 mg).
- ester (2e) (2.33 g) in dioxane (354 ml) and water (90 ml) was added dropwise over 2 hours 0.5N sodium hydroxide solution (24 ml) and then stirred at room temperature overnight. The solution was concentrated and the pH adjusted to 4 with 2N hydrochloric acid. The resulting solid was filtered off, washed with a little water and dried under vacuum overnight to yield the desired product (1.72 g).
- the compound was prepared as in Example Ik from aldehyde (2h) in 96% yield.
- 2,4-Difluorobenzoic acid (5.00 g) was dissolved in ethanol (50 ml) and bubbled through the solution for 20 minutes with HCl gas. The mixture was then refluxed for 5 hours, the solution was concentrated and the residue was dissolved in ether. The organic phase was washed with 1N sodium hydroxide solution and sat. Sodium chloride solution, dried over magnesium sulfate, filtered and concentrated to afford the desired product (3.8 g).
- Ethyl ester (3a) (3.8 g) was dissolved in fuming nitric acid (3 ml) and conc. Dissolved sulfuric acid (3 ml) at 0 0 C and stirred for 2.5 hours. Then it was washed with water (10 ml) diluted and extracted with dichloromethane (200 ml). The organic phase was washed with sat. Washed sodium chloride solution, dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (silica gel, hexane / ethyl acetate 6: 1) to afford the desired product (3.96 g).
- Nitrobenzoic acid (3b) (3.96 g) was dissolved in dichloromethane (75 ml), added with triethylamine (2.8 ml) and cooled to 0 ° C. After the addition of methyl thioglycolate (1.5 ml) was stirred at 0-5 0 C 3.5 hours and the solution stored overnight in the refrigerator. The solution was concentrated and the residue was purified by flash chromatography (silica gel, hexane / ethyl acetate 6: 1) to give the desired product (3.86 g).
- the compound was prepared as in Example Ik from aldehyde (3g) in 93% yield.
- the compound was prepared as in Example Ik from (2,3-dihydrobenzo [1,4] dioxin-6-carbaldehyde.
- AD mix beta was used to prepare the diol (Id).
- the compound was prepared from Boc-amine (7b) in 63% yield as in Example Ih.
- Example Ik The compound was prepared as in Example Ik from amine (7c) and aldehyde (Ij) in 86% yield.
- Example 8 7-Fluoro-6- ( ⁇ 1- [2-hydroxy-2- (3-methoxy-quinolin-5-yl) -ethyl] -piperidin-4-ylamino ⁇ -methyl) -4H-benzo [l , 4] thiazine-3-one (enantiomer 1)
- the compound was prepared from aldehyde (3g) in 78% yield as in Example 7d.
- the compound was prepared from aldehyde (4b) in 73% yield as in Example 7d.
- the compound was prepared from aldehyde (2h) in 83% yield as in Example 7d.
- Example 11 2- ⁇ 4- [(2,3-Dihydro- [l, 4] dioxino [2,3-c] pyridine-7-ylmethyl) -axnino] -piperidin-1-yl ⁇ -1- (3 -methoxy-quinolin-5-yl) -ethanol (enantiomer 1)
- the compound was prepared from aldehyde (5d) in 78% yield as in Example 7d.
- the compound was prepared as in Example 7d from 2,3-dihydro-benzo [1,4] dioxin-6-carbaldehyde.
- Example 14 2- ⁇ 4- [(2,3-Dihydro-benzo [l, 4] dioxin-6-ylmethyl) -amino] -piperidin-1-yl ⁇ -1- (3-methoxy-quinoline-5-) yl) ethanol (racemate)
- the compound was prepared from Boc-amine (14c) in 78% yield as in Example Ih.
- Example 16 1- (3-Methoxy-quinolin-5-yl) -2- ⁇ 4- [(2-methyl-benzofuran-5-ylin-ethyl) -amino] -piperidin-1-yl ⁇ -ethanol (racemate)
- PEG 300 (40 ml) was heated to 220 ° C. A solution of propynyl aldehyde (16a) (5g) in PEG 300 (10ml) was added and stirred at 220 ° C for 90 minutes. After cooling, the reaction mixture was poured onto ice (200 g) and extracted with dichloromethane / ether (1: 1, 2 x 300 ml). The combined organic phases were concentrated. The residue was removed by flash Chromatography (silica gel, hexane / ethyl acetate 9: 1) and gave the desired product (2 g).
- the compound was prepared from aldehyde (16b) as in Example 14e.
- Example 17 1- (3-Methoxy-quinolin-5-yl) -2- ⁇ 4- [(quinoxalin-2-ylmethyl) -amino] -piperidin-1-yl ⁇ -ethanol (racemate)
- Example 18 2- [4- ((E) -3-Furan-2-yl-allylamino) -piperidin-1-yl] -1- (3-methoxy-quinolin-5-yl) -ethanol (racemate)
- Example 14e The compound was prepared as in Example 14e from (E) -3-furan-2-yl-propenal.
- the compound was prepared from benzofuran-2-carbaldehyde as in Example 14e.
- Example 14e The compound was prepared as in Example 14e from phenoxyacetaldehyde (according to Syn. Lett., Vol. 11, 2004, p. 2010).
- the compound was prepared from aldehyde (2Id) as in Example 14e.
- Example 22 2- ⁇ 4- [(2,3-Dihydro- [l, 4] dioxino [2,3-c] pyridin-7-ylmethyl) -amino] -piperidin-1-yl ⁇ -1- (3 -methoxy-quinolin-5-yl) -ethanol (racemate)
- the compound was prepared from aldehyde (5d) as in Example 14e.
- the compound was prepared from aldehyde (23a) as in Example 14e.
- the compound was prepared from cinnamic aldehyde as in Example 14e.
- Example 25 1- (3-Methoxy-quinolin-5-yl) -2- ⁇ 4- [(1-methyl-1H-indol-2-ylmethyl) -amino] -piperidin-1-yl ⁇ -ethanol (racemate )
- the compound was prepared as in Example 14e from 1-methyl-1H-indole-2-carbaldehyde.
- the compound was prepared from 3-phenylpropionaldehyde as in Example 14e.
- Acetic acid 200 ml was added with iron powder (15.4 g) and heated to reflux. It was then filtered through Celite and washed with acetic acid. The filtrate was concentrated, the residue taken up in ethyl acetate and washed with sat. Washed sodium bicarbonate solution. The organic phase was dried over sodium sulfate, filtered and concentrated. The crude product was purified by flash chromatography (silica gel, hexane / ethyl acetate 1: 1) to afford the desired product (1.5 g).
- the compound was prepared from aldehyde (27c) as in Example 14e.
- Example 28 6- ( ⁇ 1- [2-hydroxy-2- (3-methoxy-quinolin-5-yl) ⁇ ethyl] piperidin-4-ylamino ⁇ -methyl) -4H-benzo [1,4] oxazin -3 ⁇ on (racemate)
- Example 14e The title compound was prepared as in Example 14e from 3-oxo-3,4-dihydro-2H-pyrido [3, 2-b] [1,4] oxazine-6-carbaldehyde (prepared according to WO2006 / 021448).
- Example 31 2- ⁇ 4- [(E) -3- (2,5-Difluoro-phenyl) -allylamino] -piperidin-1-yl ⁇ -1- (3-methoxy-quinolin-5-yl) -ethanol ( racemate)
- Example 14e The title compound was prepared as in Example 14e from (E) -3- (2,5-difluoro-phenyl) -propenal (prepared according to WO2004 / 087647).
- Example 32 1- (3-Methoxy-quinolin-5-yl) -2- ⁇ 4- [(naphthalen-2-ylmethyl) -amino] -piperidin-1-yl ⁇ -ethanol
- Example 40 2- ⁇ 4- [(2,3-Dihydro-benzo [l, 4] dioxin-6-ylmethyl) amino] -3-fluoro-piperidin-1-yl ⁇ -1- (3-methoxy) quinolin-5-yl) ethanol (enantiomer 1)
- the compound was prepared as in Example 7d from amine (40I) and 2, 3-dihydro-benzo [1,4] dioxine-6-carbaldehyde.
- the compound was prepared from aldehyde (Ij) as in Example 40j.
- the compound was prepared from cinnamic aldehyde as in Example 40j.
- the compound was prepared from aldehyde (2h) as in Example 40j.
- the compound was prepared from aldehyde (5d) as in Example 40j.
- Example 40j The compound was prepared as in Example 40j from benzo [l, 2,5] thiadiazole-5-carbaldehyde.
- the compound was prepared from aldehyde (4b) as in Example 40j.
- Example 48 2- [4- [(2,3-Dihydro-benzo [l, 4] dioxin-6-ylmethyl) -amino] -4- (2-hydroxy-ethyl) -piperidin-1-yl] -l - (3-methoxy-quinolin-5-yl) -ethanol (racemate)
- Dichloroethane (15 ml) was added with sodium triacetoxyborohydride (0.72 g) and stirred at room temperature for 16 hours. Then sat. Add bicarbonate solution (20 mL) and dichloromethane (50 mL), separate the layers and extract the aqueous layer with dichloromethane (50 mL). The combined organic phases were washed with sat. Washed sodium chloride solution, dried over sodium sulfate, filtered and concentrated. The residue was purified by flash chromatography (silica gel, ethyl acetate) to give the desired product (0.75 g).
- the piperidine (2- ⁇ 4- [(benzo [1,3-dioxol-5-ylmethyl) -amino] -piperidin-4-yl ⁇ -ethanol) was prepared analogously to steps 48c to 48e from benzo [1, 3] dioxole-5-carbaldehyde.
- the title compound was prepared as in Example 48f from epoxide (14b) and 2- ⁇ 4- [(benzo [1,3-dioxol-5-ylmethyl) -amino] -piperidin-4-yl ⁇ -ethanol.
- Example 50 6- ( ⁇ 4- (2-Hydroxyethyl) -1- [2-hydroxy-2- (3-methoxy-quinolin-5-yl) -ethyl] -piperidin-4-ylamino ⁇ -methyl) 4H-benzo [1,4] oxazin-3-one (racemate)
- the piperidine (6- ⁇ [4- (2-hydroxy-ethyl) -piperidin-4-ylamino] -methyl ⁇ -4 H -benzo [1,4] oxazin-3-one) was analogous to steps 48c to 48e Aldehyde (Ij) produced.
- the title compound was prepared as in Example 48f from epoxide (14b) and 6- ⁇ [4- (2-hydroxy-ethyl) -piperidin-4-ylamino] -methyl ⁇ -4H-benzo [1,4] oxazin-3-one produced .
- Example 51 2- ⁇ 4- [(2,3-Dihydro-benzo [l, 4] dioxin-6-ylmethyl) -amino] -3-hydroxymethyl-piperidin-1-yl ⁇ -l- (3-methoxy) quinolin-5-yl) ethanol (racemate)
- Lithium aluminum hydride (2.55 g) was added and stirred for 1 hour. Then water (2 ml), 3N sodium hydroxide solution (4 ml) and water (9 ml) were added, warmed to room temperature and added ether (150 ml). The solid was filtered off, the filtrate concentrated and afforded the desired product (5.33 g).
- Example 54 6- (1-Hydroxy-2- ⁇ 4- [2-hydroxy-2- (3-methoxy-quinolin-5-yl) -ethyl] -piperazin-1-yl ⁇ -ethyl) -4H-benzo [1,4] oxazin-3-one (racemate)
- the piperazine (6- (1-hydroxy-2-piperazin-1-yl-ethyl) -4H-benzo [l, 4] thiazine-3-one) was prepared analogously to steps 54a to 54b from 6- (2-chloro -acetyl) -4H-benzo [1, 4] thiazine-3-one.
- the piperazine (1- (2,3-dihydro-benzo [1,4] dioxin-6-yl) -2-piperazin-1-yl-ethanol) was prepared analogously to steps 54a to 54b from 2-chloro-1-one (2, 3-dihydro-benzo [1, 4] dioxin-6-yl) -ethanone produced.
- Example 54c The title compound was prepared as in Example 54c from epoxide (14b) and 1- (2,3-dihydro-benzo [1,4] dioxin-6-yl) -2-piperazin-1-yl-ethanol.
- Example Ik The compound was prepared as in Example Ik from 3-methoxy-5- (2-piperazin-1-yl-ethoxy) -quinoline (57f) and 2,3-dihydro-benzo [1, 4] dioxin-6-carbaldehyde in 66 % Yield produced.
- Example 60 6- ⁇ 4- [2- (3-Methoxy-quinolin-5-yloxy) -ethyl] -piperazin-1-ylmethyl ⁇ -4 H -pyrido [3, 2-b] [1,4] thiazine one 3-
- Example 65 6- ⁇ 4- [2- (3-Methoxy-quinolin-5-yloxy) ethyl] piperazine-1-carbonyl ⁇ -4 H -benzo [1,4] oxazin-3-one
- the reaction mixture was stirred for 1 hour at room temperature, then treated with 10% sodium sulfite solution (120 ml) and stirred for a further 30 minutes.
- the phases were separated and the aqueous phase extracted with ethyl acetate (2 x 100 ml).
- the combined organic phases were washed with sat. Washed sodium chloride solution, dried over sodium sulfate, filtered and concentrated.
- the residue was purified by flash chromatography (silica gel, ethyl acetate / hexane 2: 1) to afford the desired product (3.75 g).
- Example 70 2- [4- (2-Benzo [1,3] dioxol-5-yl-ethyl) -piperazin-1-yl] -1- (3-itethoxy-quinolin-5-yl) -ethanol (enantiomer 1)
- Example 71 1-Cyclopropyl-6-fluoro-7- ⁇ 4- [2-hydroxy-2- (3-methoxy-quinolin-5-yl) -ethyl] -piperazin-1-yl ⁇ -4-oxo-1 4-dihydro-quinoline-3-carboxylic acid
- Epoxide (14a) (50 mg) and ciprofloxacin (91.4 mg) were suspended in DMF (0.2 ml) and potassium carbonate (34.3 mg) added. The Letsgemsich was stirred overnight at 100 0 C. After cooling to room temperature, the reaction mixture was concentrated. The residue was purified by flash chromatography (silica gel, dichloromethane / methanol 9: 1). The concentrated fractions were recrystallized from ether / dichloromethane.
- N-tert-Boc-4-piperidinone (5.0 g) and 1-benzyl-piperazine (4.43 g) were dissolved in methanol (60 ml) and washed with
- Epoxide (14b) (0.39 g), piperazine (72b) (0.29 g), potassium carbonate (0.29 g) and lithium perchlorate (0.12 g) were dissolved in dry DMF (4 ml) and stirred at 100 ° C. under a nitrogen atmosphere for 24 hours , The mixture was on
- the maximum inhibitory concentration (MIC) ( ⁇ g / ml) of the examples against various organisms was determined: A. baumannii ATCC19606, E. cloacae ATCC23355, E. coli ATCC25922, K. pneumoniae ATCC27736, P. mirabilis ATCC29906, P. aeruginosa ATCC27853, p maltophilia ATCC13637, S. aureus ATCC43300, S. epidermidis ATCC14990, S. haemolyticus ATCC29970, E. faecalis ATCC29212 and E. faecium ATC19434.
- Examples 1-12, 14-15, 17-20, 22-26, 28-53 and 56-71 have an MIC of less than or equal to 4 ⁇ g / ml against at least two of the above organisms.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Quinoline Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Description

























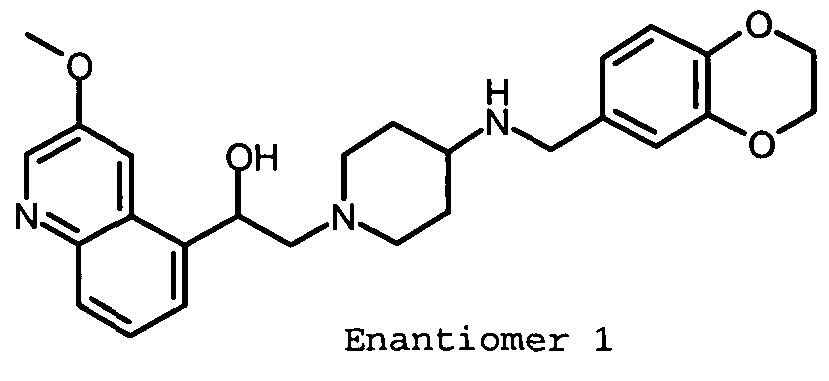





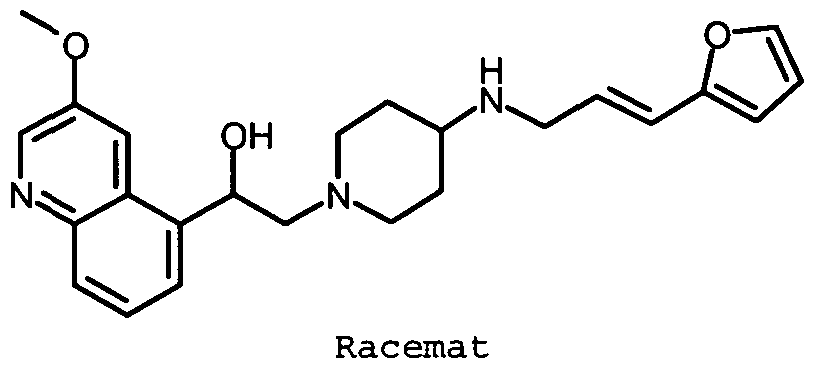














































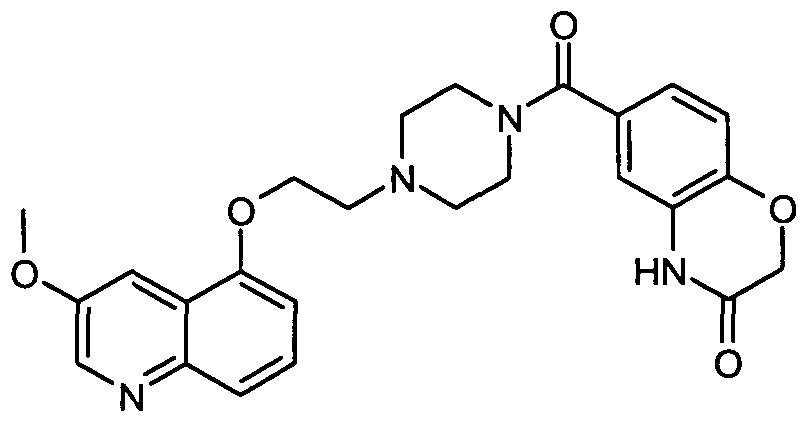







Claims








Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002625687A CA2625687A1 (en) | 2005-10-13 | 2006-10-13 | Antibacterial active 5-chinolin derivative |
| BRPI0617376-4A BRPI0617376A2 (pt) | 2005-10-13 | 2006-10-13 | derivados de 5-quinolina tendo uma atividade antibacteriana |
| JP2008534946A JP2009511530A (ja) | 2005-10-13 | 2006-10-13 | 抗菌活性を有する5−キノリン誘導体 |
| EP06806274A EP1943222A1 (de) | 2005-10-13 | 2006-10-13 | 5-chinolinderivate mit antibakterieller aktivität |
| US12/082,667 US20100324030A1 (en) | 2005-10-13 | 2008-04-11 | 5-Quinoline derivatives having an anti-bacterial activity |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102005049039.5 | 2005-10-13 | ||
| DE102005049039 | 2005-10-13 | ||
| DE102006028649A DE102006028649A1 (de) | 2006-06-22 | 2006-06-22 | Neue Verbindungen mit antibakterieller Aktivität |
| DE102006028649.9 | 2006-06-22 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/082,667 Continuation US20100324030A1 (en) | 2005-10-13 | 2008-04-11 | 5-Quinoline derivatives having an anti-bacterial activity |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007042325A1 true WO2007042325A1 (de) | 2007-04-19 |
Family
ID=37762231
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/009932 WO2007042325A1 (de) | 2005-10-13 | 2006-10-13 | 5-chinolinderivate mit antibakterieller aktivität |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20100324030A1 (de) |
| EP (1) | EP1943222A1 (de) |
| JP (1) | JP2009511530A (de) |
| KR (1) | KR20080064953A (de) |
| BR (1) | BRPI0617376A2 (de) |
| CA (1) | CA2625687A1 (de) |
| RU (1) | RU2008118422A (de) |
| WO (1) | WO2007042325A1 (de) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008139288A2 (en) * | 2007-05-09 | 2008-11-20 | Pfizer Inc. | Substituted heterocyclic derivatives and compositions and their pharmaceutical use as antibacterials |
| US7875715B2 (en) | 2005-06-16 | 2011-01-25 | Astrazeneca Ab | Compounds for the treatment of multi-drug resistant bacterial infections |
| US8012961B2 (en) | 2008-04-15 | 2011-09-06 | Actelion Pharmaceutical Ltd. | Tricyclic antibiotics |
| US10208052B1 (en) | 2017-03-20 | 2019-02-19 | Forma Therapeutics, Inc. | Compositions for activating pyruvate kinase |
| US10633336B2 (en) | 2014-12-19 | 2020-04-28 | The Broad Institute, Inc. | Dopamine D2 receptor ligands |
| US10675274B2 (en) | 2018-09-19 | 2020-06-09 | Forma Therapeutics, Inc. | Activating pyruvate kinase R |
| US10752588B2 (en) | 2014-12-19 | 2020-08-25 | The Broad Institute, Inc. | Dopamine D2 receptor ligands |
| US11001588B2 (en) | 2018-09-19 | 2021-05-11 | Forma Therapeutics, Inc. | Activating pyruvate kinase R and mutants thereof |
| US11820747B2 (en) | 2021-11-02 | 2023-11-21 | Flare Therapeutics Inc. | PPARG inverse agonists and uses thereof |
| US12054479B1 (en) | 2022-03-14 | 2024-08-06 | Slap Pharmaceuticals Llc | Multicyclic compounds |
| US12122778B2 (en) | 2019-09-19 | 2024-10-22 | Novo Nordisk Health Care Ag | Activating pyruvate kinase R |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100144717A1 (en) * | 2006-12-15 | 2010-06-10 | Janelle Comita-Prevoir | 2-quinolinone and 2-quinoxalinone-derivatives and their use as antibacterial agents |
| WO2016056581A1 (ja) * | 2014-10-08 | 2016-04-14 | 国立研究開発法人理化学研究所 | 植物成長促進剤及び植物成長促進方法 |
| MA41169A (fr) * | 2014-12-17 | 2017-10-24 | Acraf | Composés antibactériens à large spectre d'activité |
| TWI731420B (zh) * | 2018-09-27 | 2021-06-21 | 大陸商深圳微芯生物科技股份有限公司 | 具有吲哚胺-2,3-雙加氧酶抑制活性的喹啉衍生物及其製備方法、藥物組合物、聯合用藥物與用途 |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0363212A2 (de) * | 1988-10-06 | 1990-04-11 | MITSUI TOATSU CHEMICALS, Inc. | Heterozyklische Verbindungen und Arzneimittel, die die Wirkung von Antikrebsmitteln verstärken, welche diese als wirksame Bestandteile enthalten |
| US4918073A (en) * | 1986-01-09 | 1990-04-17 | Hoechst Aktiengesellschaft | Diarylalkyl-substituted alkylamines and medicaments containing them |
| WO1998050364A1 (en) * | 1997-05-03 | 1998-11-12 | Smithkline Beecham Plc | Tetrahydroisoquinoline derivatives as modulators of dopamine d3 receptors |
| WO2000021951A1 (en) * | 1998-10-08 | 2000-04-20 | Smithkline Beecham Plc | Tetrahydrobenzazepine derivatives useful as modulators of dopamine d3 receptors (antipsychotic agents) |
| EP1099692A1 (de) * | 1998-07-21 | 2001-05-16 | Eisai Co., Ltd. | N,n-substituierte cyclische aminderivate |
| WO2002034754A2 (en) * | 2000-10-26 | 2002-05-02 | Smithkline Beecham P.L.C. | Benzoxazinone derivatives, their preparation and use |
| WO2004002490A2 (en) * | 2002-06-26 | 2004-01-08 | Glaxo Group Limited | Piperidine compounds as antibacterials |
| WO2004089947A2 (de) * | 2003-04-08 | 2004-10-21 | Morphochem Ag | Neue verbindungen mit antibakterieller aktivität |
| WO2005016915A1 (en) * | 2003-08-14 | 2005-02-24 | Glaxo Group Limited | Piperidine/cyclohexane carboxamide derivatives for use as vanilloid receptor modulators |
| WO2005094251A2 (en) * | 2004-03-17 | 2005-10-13 | Glaxo Group Limited | M3muscarinic acetylcholine receptor antagonists |
| WO2005094834A1 (en) * | 2004-03-17 | 2005-10-13 | Glaxo Group Limited | Muscarinic acetylcholine receptor antagonists |
| US20050256103A1 (en) * | 2004-05-12 | 2005-11-17 | Eisai Co., Ltd. | Indole derivative having piperidine ring |
| WO2006021448A1 (en) * | 2004-08-25 | 2006-03-02 | Morphochem Aktiengesellschaft für kombinatorische Chemie | Novel compounds having an anti-bacterial activity |
| WO2006038172A1 (en) * | 2004-10-05 | 2006-04-13 | Actelion Pharmaceuticals Ltd | New piperidine antibiotics |
| WO2006099884A1 (en) * | 2005-03-24 | 2006-09-28 | Actelion Percurex Ag | Beta-aminoalcohol antibiotics |
| WO2007006926A2 (fr) * | 2005-07-11 | 2007-01-18 | Sanofi-Aventis | Nouveaux derives de 2,4-dianilinopyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk |
-
2006
- 2006-10-13 EP EP06806274A patent/EP1943222A1/de not_active Withdrawn
- 2006-10-13 KR KR1020087009733A patent/KR20080064953A/ko not_active Application Discontinuation
- 2006-10-13 CA CA002625687A patent/CA2625687A1/en not_active Abandoned
- 2006-10-13 BR BRPI0617376-4A patent/BRPI0617376A2/pt not_active IP Right Cessation
- 2006-10-13 JP JP2008534946A patent/JP2009511530A/ja not_active Withdrawn
- 2006-10-13 RU RU2008118422/04A patent/RU2008118422A/ru not_active IP Right Cessation
- 2006-10-13 WO PCT/EP2006/009932 patent/WO2007042325A1/de active Application Filing
-
2008
- 2008-04-11 US US12/082,667 patent/US20100324030A1/en not_active Abandoned
Patent Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4918073A (en) * | 1986-01-09 | 1990-04-17 | Hoechst Aktiengesellschaft | Diarylalkyl-substituted alkylamines and medicaments containing them |
| EP0363212A2 (de) * | 1988-10-06 | 1990-04-11 | MITSUI TOATSU CHEMICALS, Inc. | Heterozyklische Verbindungen und Arzneimittel, die die Wirkung von Antikrebsmitteln verstärken, welche diese als wirksame Bestandteile enthalten |
| WO1998050364A1 (en) * | 1997-05-03 | 1998-11-12 | Smithkline Beecham Plc | Tetrahydroisoquinoline derivatives as modulators of dopamine d3 receptors |
| EP1099692A1 (de) * | 1998-07-21 | 2001-05-16 | Eisai Co., Ltd. | N,n-substituierte cyclische aminderivate |
| WO2000021951A1 (en) * | 1998-10-08 | 2000-04-20 | Smithkline Beecham Plc | Tetrahydrobenzazepine derivatives useful as modulators of dopamine d3 receptors (antipsychotic agents) |
| WO2002034754A2 (en) * | 2000-10-26 | 2002-05-02 | Smithkline Beecham P.L.C. | Benzoxazinone derivatives, their preparation and use |
| WO2004002490A2 (en) * | 2002-06-26 | 2004-01-08 | Glaxo Group Limited | Piperidine compounds as antibacterials |
| WO2004089947A2 (de) * | 2003-04-08 | 2004-10-21 | Morphochem Ag | Neue verbindungen mit antibakterieller aktivität |
| WO2005016915A1 (en) * | 2003-08-14 | 2005-02-24 | Glaxo Group Limited | Piperidine/cyclohexane carboxamide derivatives for use as vanilloid receptor modulators |
| WO2005094251A2 (en) * | 2004-03-17 | 2005-10-13 | Glaxo Group Limited | M3muscarinic acetylcholine receptor antagonists |
| WO2005094834A1 (en) * | 2004-03-17 | 2005-10-13 | Glaxo Group Limited | Muscarinic acetylcholine receptor antagonists |
| US20050256103A1 (en) * | 2004-05-12 | 2005-11-17 | Eisai Co., Ltd. | Indole derivative having piperidine ring |
| WO2006021448A1 (en) * | 2004-08-25 | 2006-03-02 | Morphochem Aktiengesellschaft für kombinatorische Chemie | Novel compounds having an anti-bacterial activity |
| WO2006038172A1 (en) * | 2004-10-05 | 2006-04-13 | Actelion Pharmaceuticals Ltd | New piperidine antibiotics |
| WO2006099884A1 (en) * | 2005-03-24 | 2006-09-28 | Actelion Percurex Ag | Beta-aminoalcohol antibiotics |
| WO2007006926A2 (fr) * | 2005-07-11 | 2007-01-18 | Sanofi-Aventis | Nouveaux derives de 2,4-dianilinopyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk |
Non-Patent Citations (11)
| Title |
|---|
| ATKINSON, PETER J. ET AL: "3,4-Dihydro-2H-benzoxazinones are 5-HT1A receptor antagonists with potent 5-HT reuptake inhibitory activity", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS , 15(3), 737-741 CODEN: BMCLE8; ISSN: 0960-894X, 2005, XP002422250 * |
| BIOORGANIC & MEDICINAL CHEMISTRY LETTERS , 10(22), 2553-2555 CODEN: BMCLE8; ISSN: 0960-894X, 2000 * |
| CURRENT DRUG DISCOVERY TECHNOLOGIES , 2(2), 99-113 CODEN: CDDTAF; ISSN: 1570-1638, 2005 * |
| DATABASE CAPLUS [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; AUSTIN, NIGEL E. ET AL: "Novel 2,3,4,5-tetrahydro-1H-3-benzazepines with high affinity and selectivity for the dopamine D3 receptor", XP002422262, retrieved from STN Database accession no. 2000:844935 * |
| DATABASE CAPLUS [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; BALAKIN, KONSTANTIN V. ET AL: "Comprehensive computational assessment of ADME properties using mapping techniques", XP002422259, retrieved from STN Database accession no. 2005:607837 * |
| DATABASE CAPLUS [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; MACDONALD, GREGOR J. ET AL: "Design and Synthesis of trans-3-(2-(4-((3-(3-(5-Methyl-1,2,4- oxadiazolyl))-phenyl)carboxamido)cyclohexyl)ethyl)-7-methylsulfonyl- 2,3,4,5-tetrahydro-1H-3-benzazepine (SB-414796): A Potent and Selective Dopamine D3 Receptor Antagonist", XP002422261, retrieved from STN Database accession no. 2003:806660 * |
| DATABASE CAPLUS [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; SWANSON, DEVIN M. ET AL: "Identification and biological evaluation of 4-(3- trifluoromethylpyridin-2-yl)piperazine-1-carboxylic acid (5-trifluoromethylpyridin-2-yl)amide, a high affinity TRPV1 (VR1) vanilloid receptor antagonist", XP002422260, retrieved from STN Database accession no. 2004:967396 * |
| DATABASE CAPLUS [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; ZHU, TONG ET AL: "Polymer-Supported Synthesis of Pyridone-Focused Libraries as Inhibitors of Anaplastic Lymphoma Kinase", XP002422258, retrieved from STN Database accession no. 2006:333943 * |
| JOURNAL OF COMBINATORIAL CHEMISTRY , 8(3), 401-409 CODEN: JCCHFF; ISSN: 1520-4766, 2006 * |
| JOURNAL OF MEDICINAL CHEMISTRY , 46(23), 4952-4964 CODEN: JMCMAR; ISSN: 0022-2623, 2003 * |
| JOURNAL OF MEDICINAL CHEMISTRY , 48(6), 1857-1872 CODEN: JMCMAR; ISSN: 0022-2623, 2005 * |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7875715B2 (en) | 2005-06-16 | 2011-01-25 | Astrazeneca Ab | Compounds for the treatment of multi-drug resistant bacterial infections |
| WO2008139288A2 (en) * | 2007-05-09 | 2008-11-20 | Pfizer Inc. | Substituted heterocyclic derivatives and compositions and their pharmaceutical use as antibacterials |
| AU2008249745B2 (en) * | 2007-05-09 | 2012-01-12 | Pfizer Inc. | Substituted heterocyclic derivatives and compositions and their pharmaceutical use as antibacterials |
| EP2481735A1 (de) * | 2007-05-09 | 2012-08-01 | Pfizer Inc. | Substituierte heterocyclische Derivate und Zusammensetzungen und ihre pharmazeutische Verwendung als antibakterielle Mittel |
| WO2008139288A3 (en) * | 2007-05-09 | 2009-03-26 | Pfizer | Substituted heterocyclic derivatives and compositions and their pharmaceutical use as antibacterials |
| US8012961B2 (en) | 2008-04-15 | 2011-09-06 | Actelion Pharmaceutical Ltd. | Tricyclic antibiotics |
| US10752588B2 (en) | 2014-12-19 | 2020-08-25 | The Broad Institute, Inc. | Dopamine D2 receptor ligands |
| US11498896B2 (en) | 2014-12-19 | 2022-11-15 | The Broad Institute, Inc. | Dopamine D2 receptor ligands |
| US10633336B2 (en) | 2014-12-19 | 2020-04-28 | The Broad Institute, Inc. | Dopamine D2 receptor ligands |
| US12071440B2 (en) | 2017-03-20 | 2024-08-27 | Novo Nordisk Health Care Ag | Pyrrolopyrrole compositions as pyruvate kinase (PKR) activators |
| US10472371B2 (en) | 2017-03-20 | 2019-11-12 | Forma Therapeutics, Inc. | Compositions for activating pyruvate kinase |
| US10836771B2 (en) | 2017-03-20 | 2020-11-17 | Forma Therapeutics, Inc. | Compositions for activating pyruvate kinase |
| US10208052B1 (en) | 2017-03-20 | 2019-02-19 | Forma Therapeutics, Inc. | Compositions for activating pyruvate kinase |
| US11014927B2 (en) | 2017-03-20 | 2021-05-25 | Forma Therapeutics, Inc. | Pyrrolopyrrole compositions as pyruvate kinase (PKR) activators |
| US11649242B2 (en) | 2017-03-20 | 2023-05-16 | Forma Therapeutics, Inc. | Pyrrolopyrrole compositions as pyruvate kinase (PKR) activators |
| US11396513B2 (en) | 2017-03-20 | 2022-07-26 | Forma Therapeutics, Inc. | Compositions for activating pyruvate kinase |
| US11001588B2 (en) | 2018-09-19 | 2021-05-11 | Forma Therapeutics, Inc. | Activating pyruvate kinase R and mutants thereof |
| US11071725B2 (en) | 2018-09-19 | 2021-07-27 | Forma Therapeutics, Inc. | Activating pyruvate kinase R |
| US11844787B2 (en) | 2018-09-19 | 2023-12-19 | Novo Nordisk Health Care Ag | Activating pyruvate kinase R |
| US11980611B2 (en) | 2018-09-19 | 2024-05-14 | Novo Nordisk Health Care Ag | Treating sickle cell disease with a pyruvate kinase R activating compound |
| US12053458B2 (en) | 2018-09-19 | 2024-08-06 | Novo Nordisk Health Care Ag | Treating sickle cell disease with a pyruvate kinase R activating compound |
| US10675274B2 (en) | 2018-09-19 | 2020-06-09 | Forma Therapeutics, Inc. | Activating pyruvate kinase R |
| US12122778B2 (en) | 2019-09-19 | 2024-10-22 | Novo Nordisk Health Care Ag | Activating pyruvate kinase R |
| US11820747B2 (en) | 2021-11-02 | 2023-11-21 | Flare Therapeutics Inc. | PPARG inverse agonists and uses thereof |
| US12054479B1 (en) | 2022-03-14 | 2024-08-06 | Slap Pharmaceuticals Llc | Multicyclic compounds |
| US12128035B2 (en) | 2022-03-18 | 2024-10-29 | Novo Nordisk Health Care Ag | Activating pyruvate kinase R |
Also Published As
| Publication number | Publication date |
|---|---|
| BRPI0617376A2 (pt) | 2011-07-26 |
| KR20080064953A (ko) | 2008-07-10 |
| US20100324030A1 (en) | 2010-12-23 |
| CA2625687A1 (en) | 2007-04-19 |
| EP1943222A1 (de) | 2008-07-16 |
| RU2008118422A (ru) | 2009-11-20 |
| JP2009511530A (ja) | 2009-03-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007042325A1 (de) | 5-chinolinderivate mit antibakterieller aktivität | |
| RU2397982C2 (ru) | Новые соединения с антибактериальным действием | |
| WO2004035569A2 (de) | Neue verbindungen mit antibakterieller aktivität | |
| EP1497279B1 (de) | Substituierte indole und deren verwendung als 5ht-wiederaufnahme inhibitoren und als 5ht liganden | |
| DE602004007119T2 (de) | 4-(4-(heterocyclylalkoxy phenyl)-1-(heterocyclyl-carbonyl)piperidin derivate und verwandte verbindungen als histamin h3 antagonsiten zur behandlung von neurologischen erkrankungen wie alzheimer's | |
| DE69330823T2 (de) | Thiacyclische piperidinylderivate | |
| US20070244103A1 (en) | Novel Compounds Having an Anti-Bacterial Activity | |
| EP1392680A2 (de) | Selektive anthranylamidpyridinamide als vegfr-2 und vegfr-3 inhibitoren | |
| IL193176A (en) | Benzoyl-piperidine derivatives as 5ht2/d3 modulators, medicaments containing them and use thereof in the preparation of medicaments for therapy | |
| EP0530702A1 (de) | 1,2-Dihydro-2-oxopyridine als Angiotensin II-Antagonisten | |
| JPH08225569A (ja) | ベンジルピペリジン誘導体 | |
| DE60014143T2 (de) | Antimikrobielle 4-oxochinolizine mit 2-pyridongerüsten als teilstruktur | |
| CN116648245A (zh) | 四氢喹啉衍生物及其医药用途 | |
| JPH07505868A (ja) | 置換フェニルカルバメート類および尿素類 | |
| ZA200502862B (en) | Novel compounds with antibacterial activity | |
| CA3148365A1 (en) | Alpha-d-galactopyranoside derivatives | |
| DE60026581T2 (de) | Indolderivate zur behandlung von depression und angstzuständen | |
| DE19932314A1 (de) | Benzofuranderivate | |
| DE69613783T2 (de) | 4-(6-fluor-1,2-benzisoxazolyl)-1-piperidinyl-propoxy-chromen-4-on-derivate,ihre herstellung und ihre verwendung in der behandlung von psychosen, schizophrenie und angstzuständen | |
| DE10256405A1 (de) | Neue Verbindungen, die Topoisomerase IV inhibieren | |
| MX2008004780A (es) | Derivado activo 5-quinolina antibacteriano | |
| DE102006028649A1 (de) | Neue Verbindungen mit antibakterieller Aktivität | |
| WO2006072349A1 (de) | Thiophen-substituierte pyrazoline | |
| UA82518C2 (uk) | Сполуки азетидину, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі | |
| HU211665A9 (hu) | Az átmeneti oltalom az 1-19. igénypontokra vonatkozik. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680047155.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/004780 Country of ref document: MX Ref document number: 2625687 Country of ref document: CA Ref document number: 2008534946 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087009733 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006806274 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2328/CHENP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008118422 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006806274 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0617376 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080414 |