WO2006051808A1 - Hsp90ファミリー蛋白質阻害剤 - Google Patents
Hsp90ファミリー蛋白質阻害剤 Download PDFInfo
- Publication number
- WO2006051808A1 WO2006051808A1 PCT/JP2005/020519 JP2005020519W WO2006051808A1 WO 2006051808 A1 WO2006051808 A1 WO 2006051808A1 JP 2005020519 W JP2005020519 W JP 2005020519W WO 2006051808 A1 WO2006051808 A1 WO 2006051808A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- unsubstituted
- benzoic acid
- unsubstituted lower
- acceptable salt
- Prior art date
Links
- 102100034051 Heat shock protein HSP 90-alpha Human genes 0.000 title claims abstract description 97
- 101001016865 Homo sapiens Heat shock protein HSP 90-alpha Proteins 0.000 title claims abstract description 97
- 229940121649 protein inhibitor Drugs 0.000 title claims abstract description 40
- 239000012268 protein inhibitor Substances 0.000 title claims abstract description 40
- 150000003839 salts Chemical class 0.000 claims abstract description 107
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 82
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 57
- 239000000651 prodrug Substances 0.000 claims abstract description 46
- 229940002612 prodrug Drugs 0.000 claims abstract description 46
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims abstract description 43
- 125000003710 aryl alkyl group Chemical group 0.000 claims abstract description 42
- 239000004480 active ingredient Substances 0.000 claims abstract description 28
- 125000003435 aroyl group Chemical group 0.000 claims abstract description 25
- -1 nitro, formyl Chemical group 0.000 claims description 122
- 150000001875 compounds Chemical class 0.000 claims description 97
- IOHPVZBSOKLVMN-UHFFFAOYSA-N 2-(2-phenylethyl)benzoic acid Chemical compound OC(=O)C1=CC=CC=C1CCC1=CC=CC=C1 IOHPVZBSOKLVMN-UHFFFAOYSA-N 0.000 claims description 73
- 102000004169 proteins and genes Human genes 0.000 claims description 63
- 108090000623 proteins and genes Proteins 0.000 claims description 63
- 125000000623 heterocyclic group Chemical group 0.000 claims description 48
- 229910052736 halogen Inorganic materials 0.000 claims description 37
- 150000002367 halogens Chemical class 0.000 claims description 36
- 238000000034 method Methods 0.000 claims description 36
- 125000003118 aryl group Chemical group 0.000 claims description 33
- 125000005343 heterocyclic alkyl group Chemical group 0.000 claims description 32
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 30
- 125000003342 alkenyl group Chemical group 0.000 claims description 29
- 125000003545 alkoxy group Chemical group 0.000 claims description 24
- 125000000304 alkynyl group Chemical group 0.000 claims description 22
- 239000000126 substance Substances 0.000 claims description 20
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 19
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 18
- 239000003814 drug Substances 0.000 claims description 17
- 229910052757 nitrogen Inorganic materials 0.000 claims description 16
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 15
- 238000004519 manufacturing process Methods 0.000 claims description 15
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 14
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 14
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 claims description 12
- 125000003282 alkyl amino group Chemical group 0.000 claims description 10
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 claims description 10
- 201000010099 disease Diseases 0.000 claims description 10
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 10
- 230000002401 inhibitory effect Effects 0.000 claims description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 10
- 239000002246 antineoplastic agent Substances 0.000 claims description 9
- 125000001589 carboacyl group Chemical group 0.000 claims description 9
- 239000003795 chemical substances by application Substances 0.000 claims description 9
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 9
- 101710113864 Heat shock protein 90 Proteins 0.000 claims description 8
- 229940124597 therapeutic agent Drugs 0.000 claims description 8
- 125000004104 aryloxy group Chemical group 0.000 claims description 6
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 6
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 6
- 125000004391 aryl sulfonyl group Chemical group 0.000 claims description 5
- 230000000144 pharmacologic effect Effects 0.000 claims description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 3
- 201000011510 cancer Diseases 0.000 claims description 3
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 3
- 125000004208 3-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C([H])C(*)=C1[H] 0.000 claims description 2
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 claims description 2
- 230000005764 inhibitory process Effects 0.000 claims description 2
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 claims description 2
- 125000000449 nitro group Chemical class [O-][N+](*)=O 0.000 claims description 2
- 125000006503 p-nitrobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1[N+]([O-])=O)C([H])([H])* 0.000 claims description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims description 2
- 229920002554 vinyl polymer Polymers 0.000 claims description 2
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims 10
- XVTAQSGZOGYIEY-UHFFFAOYSA-N 3,4-dihydroisocoumarin Chemical class C1=CC=C2C(=O)OCCC2=C1 XVTAQSGZOGYIEY-UHFFFAOYSA-N 0.000 claims 1
- 125000005157 alkyl carboxy group Chemical group 0.000 claims 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 abstract description 2
- 229910052739 hydrogen Inorganic materials 0.000 abstract 3
- 239000001257 hydrogen Substances 0.000 abstract 3
- 125000005843 halogen group Chemical group 0.000 abstract 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 84
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 72
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 58
- 239000000243 solution Substances 0.000 description 55
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 54
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 51
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 49
- 235000018102 proteins Nutrition 0.000 description 46
- 239000000203 mixture Substances 0.000 description 45
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 39
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 32
- 239000002904 solvent Substances 0.000 description 32
- 238000005160 1H NMR spectroscopy Methods 0.000 description 31
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 31
- 239000011541 reaction mixture Substances 0.000 description 29
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 29
- 230000002829 reductive effect Effects 0.000 description 27
- 125000001424 substituent group Chemical group 0.000 description 24
- 238000006243 chemical reaction Methods 0.000 description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 21
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 20
- 229920006395 saturated elastomer Polymers 0.000 description 20
- 238000005481 NMR spectroscopy Methods 0.000 description 19
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 18
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 17
- 239000012044 organic layer Substances 0.000 description 17
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 15
- 239000011780 sodium chloride Substances 0.000 description 15
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 14
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 14
- 239000002253 acid Substances 0.000 description 14
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 14
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 239000012442 inert solvent Substances 0.000 description 12
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 11
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 11
- 238000012360 testing method Methods 0.000 description 11
- KJXAZGLVMAUYTK-UHFFFAOYSA-N 3-ethyl-4,6-bis(methoxymethoxy)-2-(2-methoxy-2-oxoethyl)benzoic acid Chemical compound CCC1=C(OCOC)C=C(OCOC)C(C(O)=O)=C1CC(=O)OC KJXAZGLVMAUYTK-UHFFFAOYSA-N 0.000 description 10
- 238000000668 atmospheric pressure chemical ionisation mass spectrometry Methods 0.000 description 10
- 238000012746 preparative thin layer chromatography Methods 0.000 description 10
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 8
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 8
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 8
- 125000004423 acyloxy group Chemical group 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 239000001569 carbon dioxide Substances 0.000 description 7
- 229910002092 carbon dioxide Inorganic materials 0.000 description 7
- SDVOWFYQPMNLBK-UHFFFAOYSA-N n-(methoxymethoxy)benzamide Chemical compound COCONC(=O)C1=CC=CC=C1 SDVOWFYQPMNLBK-UHFFFAOYSA-N 0.000 description 7
- VFYFMNCKPJDAPV-UHFFFAOYSA-N 2,2'-(5-oxo-1,3-dioxolan-4,4-diyl)diessigs Chemical compound C1N(C2)CN3CN1CN2C3.OC(=O)CC1(CC(O)=O)OCOC1=O VFYFMNCKPJDAPV-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 6
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 6
- 238000002835 absorbance Methods 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 6
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 6
- 230000027455 binding Effects 0.000 description 6
- 238000009739 binding Methods 0.000 description 6
- 239000012046 mixed solvent Substances 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 6
- 238000010898 silica gel chromatography Methods 0.000 description 6
- 125000004434 sulfur atom Chemical group 0.000 description 6
- YSTZNEUBFHSSMQ-UHFFFAOYSA-N 2,4-bis(methoxymethoxy)-6-(phenylmethoxymethyl)benzoic acid Chemical compound COCOC1=CC(OCOC)=CC(COCC=2C=CC=CC=2)=C1C(O)=O YSTZNEUBFHSSMQ-UHFFFAOYSA-N 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- 239000006180 TBST buffer Substances 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 239000001963 growth medium Substances 0.000 description 5
- 229910052751 metal Inorganic materials 0.000 description 5
- 239000002184 metal Chemical class 0.000 description 5
- 125000004430 oxygen atom Chemical group O* 0.000 description 5
- AECPBJMOGBFQDN-YMYQVXQQSA-N radicicol Chemical compound C1CCCC(=O)C[C@H]2[C@H](Cl)C(=O)CC(=O)[C@H]2C(=O)O[C@H](C)C[C@H]2O[C@@H]21 AECPBJMOGBFQDN-YMYQVXQQSA-N 0.000 description 5
- 229910052717 sulfur Inorganic materials 0.000 description 5
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 4
- ARNVPLFYBZBHPU-UHFFFAOYSA-N 2-(methoxymethoxy)benzoic acid Chemical compound COCOC1=CC=CC=C1C(O)=O ARNVPLFYBZBHPU-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 125000005041 acyloxyalkyl group Chemical group 0.000 description 4
- 125000006848 alicyclic heterocyclic group Chemical group 0.000 description 4
- 125000005205 alkoxycarbonyloxyalkyl group Chemical group 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 4
- 125000002619 bicyclic group Chemical group 0.000 description 4
- 230000010261 cell growth Effects 0.000 description 4
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 4
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- 150000004702 methyl esters Chemical class 0.000 description 4
- VYGYNVZNSSTDLJ-HKCOAVLJSA-N monorden Natural products CC1CC2OC2C=C/C=C/C(=O)CC3C(C(=CC(=C3Cl)O)O)C(=O)O1 VYGYNVZNSSTDLJ-HKCOAVLJSA-N 0.000 description 4
- 239000000825 pharmaceutical preparation Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 229930192524 radicicol Natural products 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 239000003656 tris buffered saline Substances 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 3
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 239000005711 Benzoic acid Substances 0.000 description 3
- 206010006187 Breast cancer Diseases 0.000 description 3
- 208000026310 Breast neoplasm Diseases 0.000 description 3
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 3
- 108010006519 Molecular Chaperones Proteins 0.000 description 3
- 102000005431 Molecular Chaperones Human genes 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 239000012190 activator Substances 0.000 description 3
- 125000005194 alkoxycarbonyloxy group Chemical group 0.000 description 3
- 125000000278 alkyl amino alkyl group Chemical group 0.000 description 3
- 125000004103 aminoalkyl group Chemical group 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 3
- 235000010233 benzoic acid Nutrition 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 229940125833 compound 23 Drugs 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- QTQAWLPCGQOSGP-GBTDJJJQSA-N geldanamycin Chemical compound N1C(=O)\C(C)=C/C=C\[C@@H](OC)[C@H](OC(N)=O)\C(C)=C/[C@@H](C)[C@@H](O)[C@H](OC)C[C@@H](C)CC2=C(OC)C(=O)C=C1C2=O QTQAWLPCGQOSGP-GBTDJJJQSA-N 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- OJURWUUOVGOHJZ-UHFFFAOYSA-N methyl 2-[(2-acetyloxyphenyl)methyl-[2-[(2-acetyloxyphenyl)methyl-(2-methoxy-2-oxoethyl)amino]ethyl]amino]acetate Chemical compound C=1C=CC=C(OC(C)=O)C=1CN(CC(=O)OC)CCN(CC(=O)OC)CC1=CC=CC=C1OC(C)=O OJURWUUOVGOHJZ-UHFFFAOYSA-N 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 239000002994 raw material Substances 0.000 description 3
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 description 3
- 229960002218 sodium chlorite Drugs 0.000 description 3
- 239000007790 solid phase Substances 0.000 description 3
- 125000003107 substituted aryl group Chemical group 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 2
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 2
- RNVFYQUEEMZKLR-UHFFFAOYSA-N 1-carbomethoxy-3,5-dihydroxybenzene Natural products COC(=O)C1=CC(O)=CC(O)=C1 RNVFYQUEEMZKLR-UHFFFAOYSA-N 0.000 description 2
- AXGMWZDGYAEJOY-UHFFFAOYSA-N 2,4-bis(methoxymethoxy)-6-(phenylmethoxymethyl)benzaldehyde Chemical compound COCOC1=CC(OCOC)=CC(COCC=2C=CC=CC=2)=C1C=O AXGMWZDGYAEJOY-UHFFFAOYSA-N 0.000 description 2
- FZFYFWFFCDXTLI-UHFFFAOYSA-N 2,4-bis(methoxymethoxy)-n-phenyl-6-(phenylmethoxymethyl)benzamide Chemical compound C=1C=CC=CC=1NC(=O)C=1C(OCOC)=CC(OCOC)=CC=1COCC1=CC=CC=C1 FZFYFWFFCDXTLI-UHFFFAOYSA-N 0.000 description 2
- MTCHCZWGQGCGNP-UHFFFAOYSA-N 2-(carboxymethyl)-3-ethyl-4,6-bis(methoxymethoxy)benzoic acid Chemical compound CCC1=C(OCOC)C=C(OCOC)C(C(O)=O)=C1CC(O)=O MTCHCZWGQGCGNP-UHFFFAOYSA-N 0.000 description 2
- RJJOOMXZXSWQOV-UHFFFAOYSA-N 2-bromo-1,5-bis(methoxymethoxy)-3-(phenylmethoxymethyl)benzene Chemical compound COCOC1=CC(OCOC)=CC(COCC=2C=CC=CC=2)=C1Br RJJOOMXZXSWQOV-UHFFFAOYSA-N 0.000 description 2
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 2
- 125000002927 2-methoxybenzyl group Chemical group [H]C1=C([H])C([H])=C(C(OC([H])([H])[H])=C1[H])C([H])([H])* 0.000 description 2
- BKOOMYPCSUNDGP-UHFFFAOYSA-N 2-methylbut-2-ene Chemical compound CC=C(C)C BKOOMYPCSUNDGP-UHFFFAOYSA-N 0.000 description 2
- RMWNVDVTMQWFLQ-UHFFFAOYSA-N 3-bromo-4,6-bis(methoxymethoxy)-2-(phenylmethoxymethyl)benzoic acid Chemical compound COCOC1=CC(OCOC)=C(C(O)=O)C(COCC=2C=CC=CC=2)=C1Br RMWNVDVTMQWFLQ-UHFFFAOYSA-N 0.000 description 2
- BYDRCRFKSUNNPO-UHFFFAOYSA-N 3-chloro-4,6-bis(methoxymethoxy)-2-(phenylmethoxymethyl)benzoic acid Chemical compound COCOC1=CC(OCOC)=C(C(O)=O)C(COCC=2C=CC=CC=2)=C1Cl BYDRCRFKSUNNPO-UHFFFAOYSA-N 0.000 description 2
- 125000006497 3-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C(OC([H])([H])[H])=C1[H])C([H])([H])* 0.000 description 2
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- XJUZRXYOEPSWMB-UHFFFAOYSA-N Chloromethyl methyl ether Chemical compound COCCl XJUZRXYOEPSWMB-UHFFFAOYSA-N 0.000 description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- RRSNDVCODIMOFX-MPKOGUQCSA-N Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O Chemical compound Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O RRSNDVCODIMOFX-MPKOGUQCSA-N 0.000 description 2
- JRZJKWGQFNTSRN-UHFFFAOYSA-N Geldanamycin Natural products C1C(C)CC(OC)C(O)C(C)C=C(C)C(OC(N)=O)C(OC)CCC=C(C)C(=O)NC2=CC(=O)C(OC)=C1C2=O JRZJKWGQFNTSRN-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 2
- BHHGXPLMPWCGHP-UHFFFAOYSA-N Phenethylamine Chemical compound NCCC1=CC=CC=C1 BHHGXPLMPWCGHP-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- MXZNUGFCDVAXLG-CHWSQXEVSA-N [(2S)-1-[(2R)-3-methyl-2-(pyridine-4-carbonylamino)butanoyl]pyrrolidin-2-yl]boronic acid Chemical compound CC(C)[C@@H](NC(=O)c1ccncc1)C(=O)N1CCC[C@@H]1B(O)O MXZNUGFCDVAXLG-CHWSQXEVSA-N 0.000 description 2
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 2
- QZORVGYVDDOQNT-UHFFFAOYSA-N [3,5-bis(methoxymethoxy)phenyl]methanol Chemical compound COCOC1=CC(CO)=CC(OCOC)=C1 QZORVGYVDDOQNT-UHFFFAOYSA-N 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 2
- 150000008041 alkali metal carbonates Chemical class 0.000 description 2
- 125000005236 alkanoylamino group Chemical group 0.000 description 2
- 125000005206 alkoxycarbonyloxymethyl group Chemical group 0.000 description 2
- 150000003973 alkyl amines Chemical class 0.000 description 2
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 230000006229 amino acid addition Effects 0.000 description 2
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- HOPRXXXSABQWAV-UHFFFAOYSA-N anhydrous collidine Natural products CC1=CC=NC(C)=C1C HOPRXXXSABQWAV-UHFFFAOYSA-N 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 229940061627 chloromethyl methyl ether Drugs 0.000 description 2
- UTBIMNXEDGNJFE-UHFFFAOYSA-N collidine Natural products CC1=CC=C(C)C(C)=N1 UTBIMNXEDGNJFE-UHFFFAOYSA-N 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940125810 compound 20 Drugs 0.000 description 2
- 229940126086 compound 21 Drugs 0.000 description 2
- 229940126208 compound 22 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- HPYNZHMRTTWQTB-UHFFFAOYSA-N dimethylpyridine Natural products CC1=CC=CN=C1C HPYNZHMRTTWQTB-UHFFFAOYSA-N 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 2
- 150000008282 halocarbons Chemical class 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- GJRQTCIYDGXPES-UHFFFAOYSA-N iso-butyl acetate Natural products CC(C)COC(C)=O GJRQTCIYDGXPES-UHFFFAOYSA-N 0.000 description 2
- FGKJLKRYENPLQH-UHFFFAOYSA-M isocaproate Chemical compound CC(C)CCC([O-])=O FGKJLKRYENPLQH-UHFFFAOYSA-M 0.000 description 2
- OQAGVSWESNCJJT-UHFFFAOYSA-N isovaleric acid methyl ester Natural products COC(=O)CC(C)C OQAGVSWESNCJJT-UHFFFAOYSA-N 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 2
- WGOPGODQLGJZGL-UHFFFAOYSA-N lithium;butane Chemical compound [Li+].CC[CH-]C WGOPGODQLGJZGL-UHFFFAOYSA-N 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 150000002736 metal compounds Chemical class 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- ZAVWSGIJJFAFER-UHFFFAOYSA-N methyl 2,4-bis(methoxymethoxy)-6-(phenylmethoxymethyl)benzoate Chemical compound COCOC1=CC(OCOC)=CC(COCC=2C=CC=CC=2)=C1C(=O)OC ZAVWSGIJJFAFER-UHFFFAOYSA-N 0.000 description 2
- KUZMDUMAEMKPJK-UHFFFAOYSA-N methyl 2-[2-(benzylcarbamoyl)-6-ethyl-3,5-bis(methoxymethoxy)phenyl]acetate Chemical compound C1=C(OCOC)C(CC)=C(CC(=O)OC)C(C(=O)NCC=2C=CC=CC=2)=C1OCOC KUZMDUMAEMKPJK-UHFFFAOYSA-N 0.000 description 2
- IISXFBLBSRVTIP-UHFFFAOYSA-N methyl 3,5-bis(methoxymethoxy)benzoate Chemical compound COCOC1=CC(OCOC)=CC(C(=O)OC)=C1 IISXFBLBSRVTIP-UHFFFAOYSA-N 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 2
- LGOPMBGZFRLHDT-UHFFFAOYSA-N n-benzyl-3-ethyl-2-(2-hydroxyethyl)-4,6-bis(methoxymethoxy)benzamide Chemical compound CCC1=C(OCOC)C=C(OCOC)C(C(=O)NCC=2C=CC=CC=2)=C1CCO LGOPMBGZFRLHDT-UHFFFAOYSA-N 0.000 description 2
- GVWISOJSERXQBM-UHFFFAOYSA-N n-methylpropan-1-amine Chemical compound CCCNC GVWISOJSERXQBM-UHFFFAOYSA-N 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 125000005882 oxadiazolinyl group Chemical group 0.000 description 2
- 125000005968 oxazolinyl group Chemical group 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 239000004014 plasticizer Substances 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- KMUONIBRACKNSN-UHFFFAOYSA-N potassium dichromate Chemical compound [K+].[K+].[O-][Cr](=O)(=O)O[Cr]([O-])(=O)=O KMUONIBRACKNSN-UHFFFAOYSA-N 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 125000000561 purinyl group Chemical class N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 150000003222 pyridines Chemical class 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- ATEBXHFBFRCZMA-VXTBVIBXSA-N rifabutin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC(=C2N3)C(=O)C=4C(O)=C5C)C)OC)C5=C1C=4C2=NC13CCN(CC(C)C)CC1 ATEBXHFBFRCZMA-VXTBVIBXSA-N 0.000 description 2
- 229960000885 rifabutin Drugs 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 2
- 235000019345 sodium thiosulphate Nutrition 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 2
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 2
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 2
- 125000005505 thiomorpholino group Chemical group 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- 229910052723 transition metal Inorganic materials 0.000 description 2
- 150000003624 transition metals Chemical class 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- PXJACNDVRNAFHD-UHFFFAOYSA-N (2-methoxyphenyl)methanamine Chemical compound COC1=CC=CC=C1CN PXJACNDVRNAFHD-UHFFFAOYSA-N 0.000 description 1
- GRRIMVWABNHKBX-UHFFFAOYSA-N (3-methoxyphenyl)methanamine Chemical compound COC1=CC=CC(CN)=C1 GRRIMVWABNHKBX-UHFFFAOYSA-N 0.000 description 1
- JPRPJUMQRZTTED-UHFFFAOYSA-N 1,3-dioxolanyl Chemical group [CH]1OCCO1 JPRPJUMQRZTTED-UHFFFAOYSA-N 0.000 description 1
- IDPURXSQCKYKIJ-UHFFFAOYSA-N 1-(4-methoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C=C1 IDPURXSQCKYKIJ-UHFFFAOYSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- YZUPZGFPHUVJKC-UHFFFAOYSA-N 1-bromo-2-methoxyethane Chemical compound COCCBr YZUPZGFPHUVJKC-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- KSTAYUMKBHPADG-UHFFFAOYSA-N 2,3-bis(methoxymethoxy)benzaldehyde Chemical compound COCOC1=CC=CC(C=O)=C1OCOC KSTAYUMKBHPADG-UHFFFAOYSA-N 0.000 description 1
- JYYPWNCZHOCLCV-UHFFFAOYSA-N 2,4-bis(methoxymethoxy)-n-(2-phenylethyl)-6-(phenylmethoxymethyl)benzamide Chemical compound C=1C=CC=CC=1CCNC(=O)C=1C(OCOC)=CC(OCOC)=CC=1COCC1=CC=CC=C1 JYYPWNCZHOCLCV-UHFFFAOYSA-N 0.000 description 1
- VDYDVRXKIROFLI-UHFFFAOYSA-N 2,4-dihydroxy-n-(2-phenylethyl)-6-(phenylmethoxymethyl)benzamide Chemical compound C=1C=CC=CC=1COCC1=CC(O)=CC(O)=C1C(=O)NCCC1=CC=CC=C1 VDYDVRXKIROFLI-UHFFFAOYSA-N 0.000 description 1
- WOXFMYVTSLAQMO-UHFFFAOYSA-N 2-Pyridinemethanamine Chemical compound NCC1=CC=CC=N1 WOXFMYVTSLAQMO-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- LBVWFGDWSVFGHI-UHFFFAOYSA-N 2-[2,4-dihydroxy-6-(3-hydroxyphenyl)phenyl]benzamide Chemical compound NC(=O)C1=CC=CC=C1C1=C(O)C=C(O)C=C1C1=CC=CC(O)=C1 LBVWFGDWSVFGHI-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- PTDNHYVEBIHJBK-UHFFFAOYSA-M 2-chloro-1,3-dimethylimidazol-1-ium;chloride Chemical compound [Cl-].CN1C=C[N+](C)=C1Cl PTDNHYVEBIHJBK-UHFFFAOYSA-M 0.000 description 1
- ISPYQTSUDJAMAB-UHFFFAOYSA-N 2-chlorophenol Chemical compound OC1=CC=CC=C1Cl ISPYQTSUDJAMAB-UHFFFAOYSA-N 0.000 description 1
- KOHBEDRJXKOYHL-UHFFFAOYSA-N 2-methoxy-n-methylethanamine Chemical compound CNCCOC KOHBEDRJXKOYHL-UHFFFAOYSA-N 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- KSSFGJUSMXZBDD-UHFFFAOYSA-N 3-oxo-4-pentenoic acid Chemical compound OC(=O)CC(=O)C=C KSSFGJUSMXZBDD-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- OQVYMXCRDHDTTH-UHFFFAOYSA-N 4-(diethoxyphosphorylmethyl)-2-[4-(diethoxyphosphorylmethyl)pyridin-2-yl]pyridine Chemical compound CCOP(=O)(OCC)CC1=CC=NC(C=2N=CC=C(CP(=O)(OCC)OCC)C=2)=C1 OQVYMXCRDHDTTH-UHFFFAOYSA-N 0.000 description 1
- 125000006306 4-iodophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1I 0.000 description 1
- DDFHBQSCUXNBSA-UHFFFAOYSA-N 5-(5-carboxythiophen-2-yl)thiophene-2-carboxylic acid Chemical compound S1C(C(=O)O)=CC=C1C1=CC=C(C(O)=O)S1 DDFHBQSCUXNBSA-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 241000726096 Aratinga Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- SLBDJHZFBKRMDP-UHFFFAOYSA-N COCOOC(=O)C1=CC=CC=C1 Chemical compound COCOOC(=O)C1=CC=CC=C1 SLBDJHZFBKRMDP-UHFFFAOYSA-N 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 102100038595 Estrogen receptor Human genes 0.000 description 1
- 229910052693 Europium Inorganic materials 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 102100026973 Heat shock protein 75 kDa, mitochondrial Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 1
- 101000619542 Homo sapiens E3 ubiquitin-protein ligase parkin Proteins 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 101710204707 Transforming growth factor-beta receptor-associated protein 1 Proteins 0.000 description 1
- DHANLYMXXSTEOI-UHFFFAOYSA-N [2-bromo-3,5-bis(methoxymethoxy)phenyl]methanol Chemical compound COCOC1=CC(CO)=C(Br)C(OCOC)=C1 DHANLYMXXSTEOI-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000005042 acyloxymethyl group Chemical group 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 125000005137 alkenylsulfonyl group Chemical group 0.000 description 1
- 125000000676 alkoxyimino group Chemical group 0.000 description 1
- 125000005115 alkyl carbamoyl group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical group 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 102000001307 androgen receptors Human genes 0.000 description 1
- 108010080146 androgen receptors Proteins 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 238000001815 biotherapy Methods 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 125000005708 carbonyloxy group Chemical group [*:2]OC([*:1])=O 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- PBGGNZZGJIKBMJ-UHFFFAOYSA-N di(propan-2-yl)azanide Chemical compound CC(C)[N-]C(C)C PBGGNZZGJIKBMJ-UHFFFAOYSA-N 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 125000000723 dihydrobenzofuranyl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- YUYGPCCUUFAHPR-UHFFFAOYSA-N dimethylamino propanoate Chemical compound CCC(=O)ON(C)C YUYGPCCUUFAHPR-UHFFFAOYSA-N 0.000 description 1
- QONGECDDDTYBGS-UHFFFAOYSA-N dimorpholin-4-ylmethanone Chemical compound C1COCCN1C(=O)N1CCOCC1 QONGECDDDTYBGS-UHFFFAOYSA-N 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 108010038795 estrogen receptors Proteins 0.000 description 1
- LUFGZDNJMKVIDY-UHFFFAOYSA-N ethyl 2-(benzoyloxymethyl)-4,6-dihydroxybenzoate Chemical compound CCOC(=O)C1=C(O)C=C(O)C=C1COC(=O)C1=CC=CC=C1 LUFGZDNJMKVIDY-UHFFFAOYSA-N 0.000 description 1
- KQWWVLVLVYYYDT-UHFFFAOYSA-N ethyl 3-oxohexanoate Chemical compound CCCC(=O)CC(=O)OCC KQWWVLVLVYYYDT-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- 125000000268 heptanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229940083761 high-ceiling diuretics pyrazolone derivative Drugs 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical group I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- LJJFWWCFMRNEPK-UHFFFAOYSA-N methyl 2,4-dihydroxy-6-(phenylmethoxymethyl)benzoate Chemical compound COC(=O)C1=C(O)C=C(O)C=C1COCC1=CC=CC=C1 LJJFWWCFMRNEPK-UHFFFAOYSA-N 0.000 description 1
- UBYWYGVADKPGNR-UHFFFAOYSA-N methyl 2-(methoxymethoxy)benzoate Chemical compound COCOC1=CC=CC=C1C(=O)OC UBYWYGVADKPGNR-UHFFFAOYSA-N 0.000 description 1
- DGYCBXQLEGUYSW-UHFFFAOYSA-N methyl 2-[2-ethyl-3,5-bis(methoxymethoxy)-6-(piperidine-1-carbonyl)phenyl]acetate Chemical compound CCC1=C(OCOC)C=C(OCOC)C(C(=O)N2CCCCC2)=C1CC(=O)OC DGYCBXQLEGUYSW-UHFFFAOYSA-N 0.000 description 1
- XMTHZANEURSBGV-UHFFFAOYSA-N methyl 2-[2-ethyl-3,5-dihydroxy-6-(piperidine-1-carbonyl)phenyl]acetate Chemical compound CCC1=C(O)C=C(O)C(C(=O)N2CCCCC2)=C1CC(=O)OC XMTHZANEURSBGV-UHFFFAOYSA-N 0.000 description 1
- XBJMSIODXYDODF-UHFFFAOYSA-N methyl 2-[2-ethyl-3,5-dihydroxy-6-(pyrrolidine-1-carbonyl)phenyl]acetate Chemical compound CCC1=C(O)C=C(O)C(C(=O)N2CCCC2)=C1CC(=O)OC XBJMSIODXYDODF-UHFFFAOYSA-N 0.000 description 1
- XTWLJQVUEGRUHV-UHFFFAOYSA-N methyl 2-[2-ethyl-6-[2-methoxyethyl(methyl)carbamoyl]-3,5-bis(methoxymethoxy)phenyl]acetate Chemical compound CCC1=C(OCOC)C=C(OCOC)C(C(=O)N(C)CCOC)=C1CC(=O)OC XTWLJQVUEGRUHV-UHFFFAOYSA-N 0.000 description 1
- IKLORODWISZOGR-UHFFFAOYSA-N methyl 3-bromo-4,6-dihydroxy-2-(phenylmethoxymethyl)benzoate Chemical compound COC(=O)C1=C(O)C=C(O)C(Br)=C1COCC1=CC=CC=C1 IKLORODWISZOGR-UHFFFAOYSA-N 0.000 description 1
- QYCIDWOOKULIEZ-UHFFFAOYSA-N methyl 3-ethyl-4,6-bis(methoxymethoxy)-2-(2-methoxy-2-oxoethyl)benzoate Chemical compound CCC1=C(OCOC)C=C(OCOC)C(C(=O)OC)=C1CC(=O)OC QYCIDWOOKULIEZ-UHFFFAOYSA-N 0.000 description 1
- HHPDDKOPEWTKJP-UHFFFAOYSA-N methyl 3-ethyl-4,6-dihydroxy-2-(2-methoxy-2-oxoethyl)benzoate Chemical compound CCC1=C(O)C=C(O)C(C(=O)OC)=C1CC(=O)OC HHPDDKOPEWTKJP-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 1
- 235000019799 monosodium phosphate Nutrition 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- FLCWYEUDIOQXEB-UHFFFAOYSA-N morpholin-4-yl(phenyl)methanone Chemical compound C=1C=CC=CC=1C(=O)N1CCOCC1 FLCWYEUDIOQXEB-UHFFFAOYSA-N 0.000 description 1
- 125000006518 morpholino carbonyl group Chemical group [H]C1([H])OC([H])([H])C([H])([H])N(C(*)=O)C1([H])[H] 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- LCANISONANLNKI-UHFFFAOYSA-N n-benzyl-2,4-dihydroxy-6-(phenylmethoxymethyl)benzamide Chemical compound C=1C=CC=CC=1COCC1=CC(O)=CC(O)=C1C(=O)NCC1=CC=CC=C1 LCANISONANLNKI-UHFFFAOYSA-N 0.000 description 1
- RIWRFSMVIUAEBX-UHFFFAOYSA-N n-methyl-1-phenylmethanamine Chemical compound CNCC1=CC=CC=C1 RIWRFSMVIUAEBX-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004923 naphthylmethyl group Chemical group C1(=CC=CC2=CC=CC=C12)C* 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 230000010807 negative regulation of binding Effects 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 125000005476 oxopyrrolidinyl group Chemical group 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000000636 p-nitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)[N+]([O-])=O 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 102000045222 parkin Human genes 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 229940083251 peripheral vasodilators purine derivative Drugs 0.000 description 1
- 229940117803 phenethylamine Drugs 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 125000006238 prop-1-en-1-yl group Chemical group [H]\C(*)=C(/[H])C([H])([H])[H] 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- JEXVQSWXXUJEMA-UHFFFAOYSA-N pyrazol-3-one Chemical class O=C1C=CN=N1 JEXVQSWXXUJEMA-UHFFFAOYSA-N 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- RSIJVJUOQBWMIM-UHFFFAOYSA-L sodium sulfate decahydrate Chemical compound O.O.O.O.O.O.O.O.O.O.[Na+].[Na+].[O-]S([O-])(=O)=O RSIJVJUOQBWMIM-UHFFFAOYSA-L 0.000 description 1
- JUJBNYBVVQSIOU-UHFFFAOYSA-M sodium;4-[2-(4-iodophenyl)-3-(4-nitrophenyl)tetrazol-2-ium-5-yl]benzene-1,3-disulfonate Chemical compound [Na+].C1=CC([N+](=O)[O-])=CC=C1N1[N+](C=2C=CC(I)=CC=2)=NC(C=2C(=CC(=CC=2)S([O-])(=O)=O)S([O-])(=O)=O)=N1 JUJBNYBVVQSIOU-UHFFFAOYSA-M 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-N sulfamic acid Chemical compound NS(O)(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-N 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- AYUNIORJHRXIBJ-TXHRRWQRSA-N tanespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCC=C)C(=O)C=C1C2=O AYUNIORJHRXIBJ-TXHRRWQRSA-N 0.000 description 1
- 229950007866 tanespimycin Drugs 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical compound C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 1
- FIQMHBFVRAXMOP-UHFFFAOYSA-N triphenylphosphane oxide Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=O)C1=CC=CC=C1 FIQMHBFVRAXMOP-UHFFFAOYSA-N 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/44—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring
- C07C235/58—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring with carbon atoms of carboxamide groups and singly-bound oxygen atoms, bound in ortho-position to carbon atoms of the same non-condensed six-membered aromatic ring
- C07C235/64—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring with carbon atoms of carboxamide groups and singly-bound oxygen atoms, bound in ortho-position to carbon atoms of the same non-condensed six-membered aromatic ring having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a six-membered aromatic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/44—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring
- C07C235/58—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring with carbon atoms of carboxamide groups and singly-bound oxygen atoms, bound in ortho-position to carbon atoms of the same non-condensed six-membered aromatic ring
- C07C235/60—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring with carbon atoms of carboxamide groups and singly-bound oxygen atoms, bound in ortho-position to carbon atoms of the same non-condensed six-membered aromatic ring having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/76—Esters of carboxylic acids having a carboxyl group bound to a carbon atom of a six-membered aromatic ring
- C07C69/84—Esters of carboxylic acids having a carboxyl group bound to a carbon atom of a six-membered aromatic ring of monocyclic hydroxy carboxylic acids, the hydroxy groups and the carboxyl groups of which are bound to carbon atoms of a six-membered aromatic ring
- C07C69/88—Esters of carboxylic acids having a carboxyl group bound to a carbon atom of a six-membered aromatic ring of monocyclic hydroxy carboxylic acids, the hydroxy groups and the carboxyl groups of which are bound to carbon atoms of a six-membered aromatic ring with esterified carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
- C07D213/40—Acylated substituent nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/192—Radicals derived from carboxylic acids from aromatic carboxylic acids
Definitions
- the present invention relates to a heat shock protein 90 (Hsp90) family protein inhibitor containing, for example, a benzoic acid derivative or a prodrug thereof or a pharmacologically acceptable salt thereof as an active ingredient.
- Hsp90 heat shock protein 90
- benzoic acid derivatives include 2-benzoyloxymethyl-4,6-dihydroxybenzoic acid ethyl ester (see Non-Patent Document 1), 3 -ethyl-4,6-dihydroxy-2-methoxycarbonylmethylbenzoic acid. Acid methyl esters (see Non-Patent Document 2), 3,5,3′-trihydroxybiphenyl-2-benzamide (see Non-Patent Document 3) and the like are known.
- Hsp90 heat shock protein 90 family proteins
- compounds that bind to heat shock protein 90 (Hsp90) family proteins include geldanamycin, harzomycin and other benzoquinone ansamycin antibiotics and Radicicol [Cell's Stress & Chaperones (Cell Stress & Chaperones), 3, 100-108 (1998), Journal 'Ob' Medicinal 'Chemistry (J. Med. Chem.), 42, 260-266 (1999)], and purine derivatives and Pyrazolone derivatives are known (WO 03/037860, W03 / 055860). All of these compounds have been reported to bind to Hsp90 family proteins and to exhibit pharmacological activities such as antitumor activity by inhibiting the functions of Hsp90 family proteins.
- a compound that binds to the Hsp90 family protein is considered to be useful as a therapeutic agent for a disease involving an Hsp90 family protein or a protein to which the Hsp90 family protein binds (Hsp90 client protein).
- Hsp90 family proteins Hsp90 protein, Hsp90 i3 protein, gnj94, hsp75 / TRAP1, etc. are known [Pharmacology & Therapeutics, 79, 129-168 (1998) , Molecular ⁇ ⁇ ⁇ Molecular End ocrinology, 13, 1435-1448 (1999) etc.].
- Non-Patent Document 1 "Journal 'Ob' the 'Chemical' Society, Parkin 'Transaction 1 (Journal of the Chemical Society, Perkin Transactions 1), 1979, No. 2, 529-532
- Non-Patent Document 2 "Tetrahedron Lett. J, 2000, 41st, p. 45 45-4547
- Non-Patent Document 3 "Tetrahedron Lett. J, 1968, 8th, p.491 9-4924
- An object of the present invention is to provide an Hsp90 family protein inhibitor containing, for example, a benzoic acid derivative or a prodrug thereof or a pharmacologically acceptable salt thereof as an active ingredient.
- the present invention relates to the following (1) to (49).
- n represents an integer from 0 to 0;
- R 1 is a hydrogen atom, hydroxy, carboxy-containing carboxy, nitro, formyl, halogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted cyclo Alkyl, substituted or unsubstituted Lower alkoxycarbonyl, substituted or unsubstituted aryl, substituted or unsubstituted lower alkanol, substituted or unsubstituted heterocyclic alkyl, substituted or unsubstituted aryl, substituted or unsubstituted aralkyl, substituted or unsubstituted arylolsulfonyl , Substituted or unsubstituted heterocyclic group, _CONR 7 R 8 (wherein R 7 and R 8 are the same or different, a hydrogen atom, substituted or unsubstitute
- R 2 is — NR 14 R 15 (wherein R 14 and R 15 are the same or different and each represents a hydrogen atom, a substituted or unsubstituted lower alkyl, a substituted or unsubstituted cycloalkyl, a substituted or unsubstituted lower alkanol, Represents substituted or unsubstituted aryl, substituted or unsubstituted heterocyclic group, substituted or unsubstituted aralkyl, substituted or unsubstituted heterocyclic alkyl or substituted or unsubstituted aroyl, or R 14 and R 15 are adjacent Together with the nitrogen atom to form a substituted or unsubstituted heterocyclic group) or —OR 16 (wherein R 16 is a substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted Is unsubstituted lower alkanol
- R 3 and R 5 are the same or different and each represents a hydrogen atom, force rubamoyl, sulfamoyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkanol, substituted or Unsubstituted cycloalkyl, substituted or unsubstituted lower alkylsulfonyl, substituted or unsubstituted arylsulfonyl, lower alkylaminocarbonyl, di-lower alkylaminocarbonyl, substituted or unsubstituted lower alkoxycarbonyl, heterocyclic carbonyl, Represents substituted or unsubstituted aralkyl or substituted or unsubstituted aroyl,
- R 4 and R 6 are the same or different and each represents a hydrogen atom, hydroxy, halogen, cyan nitro, formyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenino, substituted or unsubstituted lower Alkynyl, substituted or unsubstituted lower alkoxy, substituted or unsubstituted cycloalkyl, amino-substituted lower alkylami-substituted di-lower alkylami-substituted carboxy, substituted or unsubstituted lower alkoxycarbonyl, substituted or unsubstituted aryloxy, substituted or unsubstituted Represents a benzoyl represented by the following: aryl, substituted or unsubstituted heterocyclic group, substituted or unsubstituted lower alkanol, substituted or unsubstituted aroyl, substituted or unsubstitute
- R 1 is hydroxy, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted lower alkoxycarbonyl, _CONR 7 R 8 (wherein R 7 and R 8 are The Hsp90 family protein inhibitor according to any one of the above (1) to (4), which is synonymous) or —OR 13 (wherein R 13 is as defined above).
- R 1 is hydroxy, substituted or unsubstituted lower alkoxycarbonyl, one CONR 7 R 8 (wherein R 7 and R 8 are as defined above) or — ⁇ R 13 (wherein And R 13 is as defined above).
- the Hsp90 family protein inhibitor according to any one of (1) to (4) above.
- R 2 is — NR 14a R 15a (wherein R ′′ a and R 15a are the same or different and each represents a hydrogen atom, substituted or unsubstituted lower alkyl, substituted or unsubstituted aralkyl or substituted or Any one of the above (1) to (7), which represents an unsubstituted heterocyclic alkyl, or R 14a and R 15a together with the adjacent nitrogen atom form a substituted or unsubstituted heterocyclic group)
- R 3 and R 5 are the same or different and are a hydrogen atom, sulfamoyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkylsulfonyl, substituted or unsubstituted lower
- R M and R 5A are the same or different and are a hydrogen atom, sulfamoyl, substituted or unsubstituted lower alkyl (except methyl), substituted or unsubstituted lower alkenylsulfonyl, substituted Or an unsubstituted lower alkanoyl, a substituted or unsubstituted aralkyl (except for benzynole) or a substituted or unsubstituted aroyl; R represents a hydrogen atom, hydroxy or halogen,
- R 6A is a hydrogen atom, halogen, nitro-substituted nitro, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted lower alkoxy, substituted or unsubstituted Cycloalkyl, substituted or unsubstituted lower alkanol, amino lower alkylamidi dilower alkylamidi carboxy, substituted or unsubstituted lower alkoxycarbonyl, substituted or unsubstituted arylenoxy, substituted or unsubstituted aryl, substituted or Represents an unsubstituted heterocyclic group, a substituted or unsubstituted aralkyl, a substituted or unsubstituted heterocyclic alkyl or a substituted or unsubstituted aroyl;
- R 14 and R 15 are as defined above,
- nA represents an integer of 0 to 5
- R 1A is cyan carboxy, substituted or unsubstituted tert-butyl, substituted or unsubstituted isopropyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl Substituted or unsubstituted cycloalkyl, substituted or unsubstituted lower alkanol, substituted or unsubstituted lower alkoxycarbonyl, substituted or unsubstituted aryl (except 3-hydroxyphenyl), substituted or unsubstituted Aroyl, substituted or unsubstituted arylolsulfonyl, substituted or unsubstituted heterocyclic group or CONR 7 R 8 (wherein R 7 and R 8 are as defined above),
- R 1A is hydroxy, halogen, carboxy, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted Lower alkynyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted lower alkanol, substituted or unsubstituted lower alkoxycarbonyl, substituted or unsubstituted aryl, substituted or unsubstituted aryloyl, substituted or unsubstituted Heterocyclic alkyl, substituted or unsubstituted aralkyl, substituted or unsubstituted arylolsulfonyl, substituted or unsubstituted heterocyclic group, _ CONR 7 R 8 (wherein R 7 and R 8 are as defined above, respectively) ), _NRV ° (
- R 1A force S hydroxy, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted lower alkoxycarbonyl, -CONR ? R 8 (wherein R 7 and R 8 are each The benzoic acid derivative according to any one of the above (14) to (: 16) or pharmacologically, which is the same as defined above, or —OR 13 (wherein R 13 is as defined above). Acceptable salt.
- R 1A is hydroxy, substituted or unsubstituted lower alkoxycarbonyl, —CON R 7 R 8 (wherein R 7 and R 8 are as defined above) or —OR 13 ( In the formula, R 13 is as defined above, and the benzoic acid derivative or a pharmacologically acceptable salt thereof according to any one of the above (14) to (: 16).
- R 14 and R 15 are the same or different and are a hydrogen atom, a substituted or unsubstituted lower alkyl, a substituted or unsubstituted aralkyl or a substituted or unsubstituted heterocyclic alkyl, or R
- R 3A and R 5A are the same or different and each is a hydrogen atom, sulfamoyl, substituted or unsubstituted lower alkylsulfonyl, substituted or unsubstituted lower alkanol, or substituted or unsubstituted aroyl.
- R 3B and R 5B are the same or different and each represents a hydrogen atom, sulfamoyl, substituted or unsubstituted lower alkylsulfonyl, substituted or unsubstituted lower alkanol, or substituted or unsubstituted aroyl.
- R 4B represents a hydrogen atom, hydroxy or halogen
- R 6B is a hydrogen atom, halogen, nitro-substituted, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted lower alkoxy, substituted or unsubstituted Cycloalkyl, substituted or unsubstituted lower alkanol, amino lower alkylamidi dilower alkylami carboxy, substituted or unsubstituted lower alkoxycarbonyl, substituted or unsubstituted arylenoxy, substituted or unsubstituted aralkyl or substituted or Represents an unsubstituted heterocyclic alkynole,
- R 16B is substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkanol, substituted or unsubstituted aryl (except substituted phenyl), substituted or unsubstituted complex
- a cyclic group but substituted or unsubstituted 1- Represents substituted or unsubstituted aralkyl (except benzyl and 4-nitrobenzyl) or substituted or unsubstituted heterocyclic alkyl, except for oxoisochroman-6-yl),
- nB represents an integer of 0 to 5
- R 1B is cyan, carboxy, substituted or unsubstituted tert-butyl, substituted or unsubstituted isopropyl, substituted lower alkenyl (except substituted bulle), substituted or unsubstituted lower Alkynyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted lower alkanol (except pentanoyl), substituted or unsubstituted lower alkoxycarbonyl (except methoxycarbonyl), substituted or unsubstituted aroyl, substituted or Represents an unsubstituted heterocyclic alkyl, a substituted or unsubstituted aralkyl, a substituted or unsubstituted arylolsulfonyl, or —CONR 7 R 8 (wherein R 7 and R 8 are as defined above), ,
- nB is an integer of 1 to 5
- R 1B is cyan, substituted or unsubstituted lower alkynyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted lower alkoxycarbonyl, substituted or Unsubstituted aryl (except substituted or unsubstituted phenyl), substituted or unsubstituted aryl, substituted or unsubstituted heterocyclic alkyl, substituted or unsubstituted arylsulfonyl, substituted or unsubstituted heterocyclic A group (except substituted or unsubstituted 1,3-dioxorael and substituted or unsubstituted 4-oxochromen-2-yl), CONR 8 (wherein R 7 and R 8 are as defined above, respectively) there), in -NR3 ⁇ 4 10 (wherein, R 9 and R 1Q each have the same meanings as defined above)
- R 1B is benzyloxymethyl, phenyloxymethyl and ethoxymethyl
- R 1B is not methoxycarbonylmethyl, a benzoic acid derivative represented by
- a medicament comprising, as an active ingredient, the benzoic acid derivative according to any one of (14) to (33) or a prodrug thereof or a pharmacologically acceptable salt thereof.
- a medicament comprising the benzoic acid derivative or a pharmacologically acceptable salt thereof according to any one of (14) to (33) as an active ingredient.
- An Hsp90 family protein inhibitor comprising as an active ingredient the benzoic acid derivative according to any one of (14) to (33) or a prodrug thereof or a pharmacologically acceptable salt thereof.
- Hsp90 family protein inhibitor comprising, as an active ingredient, the benzoic acid derivative according to any one of (14) to (33) or a pharmacologically acceptable salt thereof.
- Hsp90 family protein or Hsp90 family protein containing the benzoic acid derivative or prodrug thereof or pharmacologically acceptable salt thereof according to any one of (14) to (33) as an active ingredient binds A therapeutic agent for diseases related to protein (Hsp90 client protein).
- Hsp90 family protein or Hsp90 family protein containing the benzoic acid derivative or pharmacologically acceptable salt thereof according to any of (14) to (33) as an active ingredient A therapeutic agent for diseases involving Hsp90 client protein.
- An antitumor agent comprising, as an active ingredient, the benzoic acid derivative according to any one of (14) to (33) or a prodrug thereof or a pharmacologically acceptable salt thereof.
- An antitumor agent comprising, as an active ingredient, the benzoic acid derivative according to any one of (14) to (33) or a pharmacologically acceptable salt thereof.
- An Hsp 90 family protein comprising administering an effective amount of the benzoic acid derivative according to any one of (14) to (33) or a prodrug thereof or a pharmacologically acceptable salt thereof. How to inhibit.
- a method for treating a malignant tumor comprising administering an effective amount of the benzoic acid derivative according to any one of (14) to (33) or a prodrug thereof or a pharmacologically acceptable salt thereof. .
- a benzoic acid derivative or a prodrug thereof or a pharmacologically acceptable salt thereof according to any one of (14) to (33) above for the production of a heat shock protein 90 (Hsp90) family protein inhibitor Use of salt.
- Hsp90 heat shock protein 90
- the present invention provides, for example, an Hsp90 family protein inhibitor containing, as an active ingredient, a benzoic acid derivative or a prodrug thereof or a pharmacologically acceptable salt thereof.
- the lower alkyl part of lower alkyl, lower alkoxy, lower alkoxycarbonyl, lower alkylsulfonyl, lower alkylaminocarbonyl, di-lower alkylaminocarbonyl, lower alkylamino and di-lower alkylamino includes, for example, linear or branched Examples thereof include alkyl having 1 to 8 carbon atoms, specifically methyl, ethyl, propyl, isopropyl, butyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, hexyl, heptyl, octyl and the like.
- the two lower alkyl moieties in di-lower alkylamino and di-lower alkylaminocarbonyl may be the same or different.
- Examples of the lower alkenyl include straight-chain or branched alkenyl having 2 to 8 carbon atoms, specifically bur, allyl, prop-1-en-1-yl, isopropenyl, crotinole, But-1-en-1-yl, But-2-en-1-yl, But-3-en-1-yl, Penta-2-en-1-yl, 3- Methylbut-1-ene-1-yl, 3-methylbut-2-ene-1-yl, penta-4-ene-1-yl, hex-2-en-1-yl Hexa-2-en-1-yl, Hepta-2-en-1-yl, Octa-2-en-1-yl and the like.
- Examples of the lower alkynyl include linear or branched alkynyl having 2 to 8 carbon atoms, specifically ethynyl, propiel, bucher, pentynyl, hexell, heptininor, otatur and the like.
- lower alkanoyl examples include straight-chain or branched alkanoinoles having 2 to 7 carbon atoms, specifically acetyl, propionyl, butyryl, isobutyryl, valeryl, isovaleryl, bivaloyl, hexanoyl, heptanoyl and the like.
- cycloalkyl examples include cycloalkyl having 3 to 8 carbon atoms, specifically cyclopropyl. Examples thereof include ropinole, cycloputinole, cyclopentinole, cyclohexinole, cycloheptinole, cyclooctyl and the like.
- aryl moiety of aryl, arylenoshonhonorile, aryloxy and aroyl includes, for example, monocyclic, bicyclic or tricyclic aryl having 6 to 14 carbon atoms, specifically vinyl, indul, naphthyl, Anthril and so on.
- aralkyl examples include aralkyl having 7 to 15 carbon atoms, specifically benzyl, phenethyl, benzhydryl, naphthylmethyl, and the like.
- heterocyclic group moiety of the heterocyclic group, heterocyclic alkyl, and heterocyclic carbonyl examples include aromatic heterocyclic groups and alicyclic heterocyclic groups.
- aromatic heterocyclic group examples include a 5-membered or 6-membered monocyclic aromatic heterocyclic group containing at least one atom selected from a nitrogen atom, an oxygen atom and a sulfur atom, and a 3- to 8-membered ring.
- aromatic heterocyclic groups examples include condensed bicyclic or tricyclic fused ring aromatic heterocyclic groups containing at least one atom selected from a nitrogen atom, an oxygen atom, and a sulfur atom.
- Specific examples include pyridyl, pyrajur, and pyrimigenol.
- Examples of the alicyclic heterocyclic group include a 5-membered or 6-membered monocyclic alicyclic heterocyclic group containing at least one atom selected from a nitrogen atom, an oxygen atom and a sulfur atom, A condensed alicyclic heterocyclic group containing at least one atom selected from a nitrogen atom, an oxygen atom and a sulfur atom, which is a bicyclic or tricyclic condensed 8-membered ring, is specifically mentioned.
- the heterocyclic group formed together with the adjacent nitrogen atom includes, for example, a 5-membered or 6-membered monocyclic heterocyclic group containing at least one nitrogen atom (the monocyclic heterocyclic group). May contain other nitrogen, oxygen or sulfur atoms), a bicyclic or tricyclic condensed ring-containing heterocyclic group containing 3 to 8 membered rings and containing at least one nitrogen atom (The condensed ring polycyclic group may contain other nitrogen atom, oxygen atom or sulfur atom).
- the alkylene part of the heterocyclic alkyl has the same meaning as that obtained by removing one hydrogen atom from the definition of the lower alkyl.
- Halogen means each atom of fluorine, chlorine, bromine and iodine.
- Substituent (A) in substituted lower alkyl, substituted tert-butyl, substituted isopropyl, substituted lower alkoxy, substituted lower alkoxycarbonyl, substituted lower alkylsulfonyl, substituted lower alkanol, substituted lower alkenyl, substituted lower alkynyl and substituted bule are the same or different and include, for example, a substituent having 1 to 3 substituents, specifically, hydroxy, cyan, nitro, carboxy, strong rubamoyl, amino, hydroxyimino, lower alkyloxy-imine halogen, substituted or non-substituted.
- the substitution position of the substituent is not particularly limited.
- the halogen, lower alkoxy, cycloalkyl, lower alkylol, lower alkoxycarbonyl, lower alkylaminocarbonyl, di-lower alkylaminocanorepinole, lower alkylamino and di-lower alkylamino exemplified in the substituent (A) are as follows: Each of the lower alkoxy moieties of the lower alkoxy imino is the same as the lower alkoxy, and the lower alkanol of the lower alkanoylamino is the same as the lower alkanoyl. It is synonymous.
- substituent (a) in the substituted lower alkoxy, substituted lower alkylaminocarbonyl, substituted dilower alkylaminocarbonyl, and substituted lower alkanoylamino exemplified in the examples of the substituent (A) is the same.
- substituents having 1 to 3 substituents are exemplified, specifically, hydroxy, halogen, lower alkoxy and the like, and halogen and lower alkoxy exemplified in the example of substituent (a) are as defined above. It is synonymous.
- Substituted cycloalkyl, substituted aryl, substituted phenyl, substituted arylsulfonyl, substituted aryloxy, substituted aralkyl, substituted aryl, substituted heterocyclic group, substituted 1,3-dioxolanyl, substituted 1_oxoisochroman-6- , Substituted 4-oxochromen-2_yl, substituted heterocyclic alkyl, and the substituted heterocyclic group formed together with the adjacent nitrogen atom (B) are the same or different, for example, substituted Specific examples of the substituent include 1 to 3, specifically, hydroxy, halogen, nitro, cyan, amino, carboxy, strength rubamoyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkoxy, aralkyloxy, lower Alkylsulfonyl, cycloalkyl, lower alkoxycarbonyl, heterocycle carbony
- Examples of the substituent in the substituted lower alkyl and substituted lower alkoxy exemplified in the substituent (B) include the same substituents as those exemplified in the above-mentioned substituent (a).
- Examples of the substituent (b) in the substituted heterocyclic group, substituted heterocyclic alkyl, and substituted aryl shown in the examples of the substituent (B) are the same or different, and examples thereof include a substituent having 1 to 3 substituents. Specific examples include hydroxy, cyanite halogen, lower alkyl, lower alkoxy, etc., and halogen, lower alkyl and lower alkyl exemplified in the substituent (b). Each class alkoxy is as defined above.
- compound (I) the compound represented by the general formula (I).
- compound (I) the compound represented by the general formula (I).
- prodrugs of compound (I) include compounds that are converted in vivo by various mechanisms such as hydrolysis in blood to yield compound (I) according to the present invention. Technologies well known to those skilled in the art [eg “Journal of 'Medicine' Chemistry”, 1997, No. 40, p. 2011-2016; “Drug“ Development ”Research (Drug Dev. Res.) J, 1995, 34th, p.220-230; "Advances in Drug Res. J, 1984, 13th, p.224 -331; Bundgaard, “Design of Prodrugs J, 1985, Elsevier Press, etc.”
- the prodrug of compound (I) includes lower alkyl, lower alkanoyloxyalkyl (for example, lower alkanoyloxy).
- lower alkanoyloxyalkyl for example, lower alkanoyloxy.
- N- (lower alkoxycarbonyl) aminoalkyl eg, N- (lower alkoxycarbonyl) aminomethyl, 1- [ ⁇ - (lower alkoxycarbonyl) amino) Etc.
- 3-phthalidyl 4-crotonolalatonyl, ⁇ -butyrate rataton-4-yl, di-lower alkylaminoalkyl, di-rubamoyl alkyl, di-lower alkyl-rubamoyl alkyl, piperidinoalkyl , Pyrrolidinoalkyl, morpholinoalkyl, and the like, and the like, and the like.
- examples of the prodrug of the compound (I) include lower alkanoyloxyalkyl, 1- (lower alkanoyloxy) ethyl, 1_methyl_1_ (lower alkanoyloxy) ethyl, lower alkoxycarbonyloxyalkyl, ⁇ - (lower alkoxycarbonyl) aminoalkyl, succinoyl, lower alkanol, hyamino-lower alkanol, and the like, Examples thereof include a compound in which a hydrogen atom is substituted.
- the prodrug of compound (I) is selected from lower alkylcarbonyl, lower alkoxycarbonyl, lower alkyl strength rubamoyl, di-lower alkyl strength rubamoyl and the like And a compound in which one or two hydrogen atoms of the amino are substituted.
- the lower alkyl part of carbonyloxymethyl, N-lower alkoxycarbonylaminomethyl, lower alkylcarbonyl, lower alkoxycarbonyl, lower alkylcarbamoyl, and di-lower alkyl rubamoyl has the same meaning as the lower alkyl, and di-lower alkylaminoalkyl.
- lower alkanoyloxyalkyl, lower alkanoyloxymethyl, 1- (lower alkanoyloxy) ethyl, 1-methyl-1- (lower alkanoyloxy) ethyl exemplified herein
- the lower alkanol part of the lower alkanoyl and the ⁇ -amino lower alkanoyl has the same meaning as the lower alkanol.
- lower alkanoyloxyalkyl, lower alkoxycarbonyloxyalkyl, (-(lower alkoxycarbonyl) aminoalkyl, di-lower alkylamino enoquinolole, rubamoyl alkyl, di-lower alkyl rubamoyl alkyl, piperi The definition is the same as that obtained by removing one hydrogen atom.
- the prodrugs of these compounds (I) can be prepared from compound (I) by, for example, Protective Groups in Organic Synthesis, third edition, Green ( TWGreene), John Wiley and 'Sons. Parakeet It can be carried out by the method described in John Wiley & Sons Inc. (1999) or a similar method.
- the pharmacologically acceptable salt of compound (I) or a prodrug thereof includes, for example, pharmaceutically acceptable acid addition salts, metal salts, ammonium salts, organic amine addition salts, amino acid addition salts, and the like. Include.
- Examples of the pharmacologically acceptable acid addition salt of Compound (I) or a prodrug thereof include inorganic acid salts such as hydrochloride, sulfate, nitrate, and phosphate, acetate, maleate, and fumarate.
- Organic salts such as acid salts and citrates are exemplified, and pharmacologically acceptable metal salts include alkali metal salts such as sodium salts and potassium salts, alkali salts such as magnesium salts and calcium salts. Examples thereof include earth metal salts, aluminum salts and zinc salts.
- Examples of pharmacologically acceptable ammonium salts include salts such as ammonium and tetramethylammonium.
- amine addition salts include addition salts such as monophorin and piperidine, and examples of pharmacologically acceptable amino acid addition salts include glycine and phenylalanine. Lysine, Asuparagin acid, glutamic acid for which addition salts.
- Inhibition of Hsp90 family protein means inhibition of binding between Hsp90 family protein and a protein to which Hsp90 family protein binds (Hsp90 client protein).
- Hsp90 family proteins include Hsp90 a protein, Hsp90 i3 protein, g ⁇ 94, hsp75 / TRAPl and the like.
- the protein to which the Hsp90 family protein binds may be any protein to which the Hsp90 family protein binds.
- EGFR Erb-B2, Bcr_Abl, src, raf_l, AKT, Flt_3, PLK, Weel, FAK, cMET, hTERT, HIF, mutation p53, estrogen receptor, androgen receptor ["Expert Opinion on Biological Therapy", 2002, 2nd, ⁇ ⁇ 3_24].
- Compound (I) and its intermediate can be obtained, for example, by the production method shown below.
- Compound (I) can be produced by the following steps.
- Compound (I) can be obtained by a condensation reaction between compound (Ila) and compound (III).
- compound (I) is reacted with compound (I) in a solvent in the presence of an activator such as 1-hydroxybenzotriazole, N-hydroxysuccinimide, and a condensing agent. Can be obtained.
- the reaction can be carried out by adding 1 to 20 equivalents of a base.
- 1 to 20 equivalents of the condensing agent, activator and compound ( ⁇ ) are used for each compound (Ila), and the reaction is carried out at a temperature between ⁇ 20 ° C. and the boiling point of the solvent used. It is performed for 24 minutes.
- Examples of the solvent include halogenated hydrocarbons such as dichloromethane and chloroform, esters such as methyl acetate, ethyl acetate, and isobutyl acetate, ethers such as jetyl ether, tetrahydrofuran, and 1,4-dioxane. , Aromatic hydrocarbons such as benzene and toluene, acetonitrile, ⁇ , ⁇ -dimethyl honolemamide, ⁇ -methylbiperidone, these Examples thereof include mixed solvents.
- halogenated hydrocarbons such as dichloromethane and chloroform
- esters such as methyl acetate, ethyl acetate, and isobutyl acetate
- ethers such as jetyl ether, tetrahydrofuran, and 1,4-dioxane.
- Aromatic hydrocarbons such as benzene and toluene, acetonitrile
- Examples of the condensing agent include dicyclohexylcarbodiimide, 1- [3- (dimethylamino) propyl] -3-ethylcarbopositimide hydrochloride, polymer bound-1- [3- (dimethylamino) propinole]- 3_ethyl carbodiimide, triphenylphosphine oxide 'trifluoromethanesulfonic acid anhydride, and the like.
- Examples of the base include alkylamines such as triethylamine, diisopropylethylamine, N-methylmorpholine, pyridines such as pyridine, lutidine, collidine, 4-dimethylaminopyridine, and alkali metals such as potassium carbonate and sodium bicarbonate. Examples include carbonates, alkali metal hydroxides such as potassium hydroxide, sodium hydroxide, and lithium hydroxide.
- the compound (Ila) is treated with an activator in advance and used in the reaction, or the carboxyl group of the compound (Ila) is converted into an acid chloride, an acid bromide, p-nitrophenoxycarbonyl, It can also be used in the reaction after being converted to a highly reactive group such as pentafluorophenoxycarbonyl, pentafluorothiooffenoxycarbonyl and the like.
- the starting I ⁇ product (III) is commercially or by known methods [for example, Nshi blanking to complement 'age 1 ⁇ force nick' Tofunsufome 1 ⁇ Shiyonzu two ⁇ (Comprehensive Organic Transfo rmations, second edition), written by RC Larock, John Wiley & Sons Inc. (1999), etc.] or a method according to them.
- the starting compound (Ila) is obtained by a known method (for example, J. Am. Chem. Soc, 93, 6708-6709 (1971)) or a method similar thereto. For example, it can be obtained by the following raw material production method 1.
- Compound (Ila) is compound (lib) in which Z is formyl, hydroxymethyl or methyl.
- Compound (Ilb-i) is added in an inert solvent in an amount of 1 to 20 equivalents such as sodium chlorite, potassium dichromate, excess It can be obtained by treating with an oxidizing agent such as potassium manganate.
- the inert solvent include dichloromethane, chloroform, tetrahydrofuran, N, N-dimethylformamide, tert-butyl alcohol, water, and mixed solvents thereof. The reaction is usually carried out at a temperature between 0 ° C. and 50 ° C. for 5 minutes to 24 hours.
- Compound (Ila) is compound (IIb_ii) in which Z is halogen among compounds (lib) in an inert solvent in an amount of 1 to 5 equivalents such as n-butyllithium, sec-butyllithium, tert-butyllithium, lithium It can be obtained by treating with an organic metal compound such as diisopropylamide or a metal such as magnesium or zinc and then treating with 1 to 100 equivalents of carbon dioxide.
- the inert solvent include tetrahydrofuran, jetyl ether, toluene, and a mixed solvent thereof. The reaction is usually carried out at a temperature between -78 ° C and 50 ° C for 5 minutes to 24 hours.
- the compound (Ila) is obtained by reacting the compound (Ilb-ii) with an acid such as formic acid, acetic acid, ammonium formate and the like in an inert solvent under 0.001 to 2 equivalents of a transition metal catalyst. It can also be obtained by treatment with 1 to 100 equivalents of carbon monoxide.
- the inert solvent include tetrahydrofuran, jetyl ether, toluene, and mixed solvents thereof.
- the transition metal catalyst include palladium chloride, tetrakis (triphenylphosphine) palladium, palladium acetate and the like. The reaction is usually carried out at a temperature between 0 ° C. and 200 ° C. for 5 minutes to 72 hours.
- Compound (Ila) is compound (I) in which R 2 is substituted or unsubstituted lower alkoxy or substituted or unsubstituted aralkyloxy in a mixed solvent of an inert solvent and water. It can also be obtained by treating with ⁇ 5 equivalents of a base such as sodium hydroxide, lithium hydroxide, potassium hydroxide.
- a base such as sodium hydroxide, lithium hydroxide, potassium hydroxide.
- the inert solvent include tetrahydrofuran, methanol, ethanol, dichloromethane and the like. The reaction is usually carried out at a temperature between 0 ° C. and 200 ° C. for 5 minutes to 24 hours.
- starting compound (lib) or a known method Commercially [e.g., Nshibu to completion 'O Ichiriki nick' Tofunsufo 1 to menu 1 ⁇ N'yonzu second piece (to omprehensive Organic Transformations, second edition), Rarokku ( RC Larock), John Wiley & Sons Inc. (1999) etc.] or a method according to them, and among the compounds (lib), Z is Compound (Ilb-iii) which is formyl can also be produced, for example, by the following steps.
- Compound (Ilb-iii) contains 1 to 5 equivalents of compound (lie) in an inert solvent such as n-butyllithium, sec-butyllithium, tert-butyllithium, lithium diisopropylamide and the like. It can be obtained by treating with an organic metal compound or a metal such as magnesium or zinc and then treating with a formylating agent such as ⁇ , ⁇ -dimethinorephonolemamide or ⁇ -formylmorpholine.
- the inert solvent include tetrahydrofuran, jetyl ether, toluene and the like. The reaction is usually carried out at a temperature between -78 ° C and 50 ° C for 5 minutes to 24 hours.
- starting compound (lie) is or methods known commercially [e.g., Nshibu to completion 'O ⁇ sword nick' Tofunsufo 1 to menu 1 ⁇ N'yonzu ⁇ 3 ⁇ 4 two pieces jx (Comprehensive Organic Transformatio ns, second edition) , By RC Larock, John Wiley & Sons Inc. (1999), etc.] or similar methods, for example, by the following process You can also
- Compound (lie) is obtained by combining compound (lid) with 1 to 2 equivalents of N-bromosuccinimide, N-chlorosuccinimide, chlorine, bromine, iodine, iodine and [bis (trifluoroacetoxy) in an inert solvent.
- Yodo Can be obtained by treating with a corresponding halogenating agent such as a combination of benzene.
- the inert solvent include dichloromethane, chlorophenol, carbon tetrachloride, ⁇ , ⁇ -dimethylformamide, and the like. The reaction is usually carried out at a temperature between 0 ° C and 50 ° C for 5 minutes to 24 hours.
- starting compound (lid) is, or known methods commercially [e.g., Nshibu to completion 'O ⁇ sword nick' Tofunsufo 1 to menu 1 ⁇ N'yonzu ⁇ 3 ⁇ 4 two pieces jx (Comprehensive Organic Transformatio ns, second edition), by RC Larock, John Wiley & Sons Inc. (1999), etc.] or similar methods.
- the compound (la) in which R 2 is _OR 16b (wherein R 16b represents a substituted or unsubstituted lower alkyl in the definition of R 16 ) is produced by the following method. You can also.
- R 1 R 3 to R 6 , R 16b and n are as defined above.
- Compound (la) can be obtained by alkylation reaction of compound (lla).
- compound (la) is compound (lla) in a solvent in an amount of 1 to 20 equivalents such as diazomethane, trimethylsilyldiazomethane, R 16b Y (wherein R 16b is as defined above, Y is It can be obtained by treating with a corresponding alkylating agent such as X. If necessary, the reaction can be carried out by adding 1 to 20 equivalents of a base. The reaction is usually carried out at a temperature between ⁇ 20 ° C. and the boiling point of the solvent for 5 minutes to 24 hours.
- Examples of the solvent include halogenated hydrocarbons such as dichloromethane and chloroform, esters such as methyl acetate, ethyl acetate, and isobutyl acetate, ethers such as jetyl ether, tetrahydrofuran, and 1,4-dioxane. , Aromatic hydrocarbons such as benzene, toluene, etc., acetonitrile, ⁇ , ⁇ -dimethenolehonolemamide, ⁇ -methinolepiperidone, hexane, and mixed solvents thereof.
- halogenated hydrocarbons such as dichloromethane and chloroform
- esters such as methyl acetate, ethyl acetate, and isobutyl acetate
- ethers such as jetyl ether, tetrahydrofuran, and 1,4-dioxane.
- Aromatic hydrocarbons such as benzene,
- R 16b when R 16b is methyl, a mixture of methanol or methanol and the solvent exemplified above can be used as the solvent.
- R 16b is ethyl, ethanol or ethanol and the solvent exemplified above as the solvent. It is also possible to use a mixture thereof.
- the base include alkylamines such as triethylamine, diisopropylethylamine, N-methylmorpholine, pyridines such as pyridine, lutidine, collidine, 4-dimethylaminopyridine, potassium carbonate, sodium bicarbonate, and the like.
- Alkali metal carbonates such as alkali metal carbonates, potassium hydroxide, sodium hydroxide and lithium hydroxide.
- each functional group in the compound (1), the raw material compound and the intermediate compound and the conversion of the functional group contained in the substituent may be carried out by a known method [for example, comprehensive 'Tomfosfo 1 ⁇ Me 1 ⁇ Nyons 2nd piece (RC Larock), John Wiley & Sons Inc. (1999) Etc.] or a method according to them.
- a compound (I) having a desired functional group at a desired position can be obtained by appropriately combining the above methods and the like.
- the intermediates and target compounds in each of the above production methods are subjected to, for example, filtration, extraction, washing, drying, concentration, recrystallization, various chromatography, etc., in an appropriate combination of separation and purification methods commonly used in organic synthetic chemistry. It can be isolated and purified. In addition, the intermediate can be subjected to the next reaction without any particular purification.
- compound (I) and its prodrugs may have stereoisomers such as geometric isomers and optical isomers. All these possible isomers and their isomers The mixture can be used for the Hsp90 family protein inhibitors of the present invention.
- Compound (I), a prodrug thereof, and a pharmacologically acceptable salt thereof may exist in the form of an adduct with water or various solvents. It can be used as a light Hsp90 family protein inhibitor.
- Hsp903 ⁇ 4a ⁇ ⁇ W ⁇ Human N-terminal recombinant Hsp90 protein (9-236 amino acid region) prepared according to the method described in Cell, SS, 239-250 (1997) was tris-buffered saline (TBS, pH 7. Dilute to 1 ⁇ g / mL in step 5), dispense 70 ⁇ L / well onto a 96-well ELISA assay plate manufactured by Grainer, and leave it at 4 ° C for 1 hour to solidify. Phased.
- test compound solution was prepared by diluting the test compound in 8 stages with a 10-fold dilution from the highest concentration of 0.1 mmol / L using TBST in a separate container.
- This test compound solution was added in an amount of 10 ⁇ L Zwell to an assembly plate in which TBST was dispensed in an amount of 90 ⁇ L / well, and allowed to stand at 24 ° C. for 1 hour.
- dimethyl sulfoxide is used at a final concentration of 0.1 / i L / wenore as a positive control for Atsy, and radicicol is used at a final concentration of 0.29 ⁇ / L as a negative control
- the test compound is used in the same plate as the test compound. The same operation was performed.
- the time-resolved fluorescence measurement value in the positive control is defined as 100% binding rate, the measurement value obtained in the negative control as 0% binding rate, and the binding rate in the wells to which the test compound is added is determined for each well. It calculated from the measured value in.
- the compounds 2-4, 9, 11-: 14, 17-: 19 and 23 inhibit the binding of piotylated radicicol to Hsp90 protein at a concentration of 10 ⁇ mol / L or less. It was further inhibited and shown to have binding activity to Hsp90 protein.
- geldanamycin, benzoquinone ansamycin antibiotics such as harpymycin, and radicicol are known as compounds that bind to Hsp90 family proteins [cells] 'Cell Stress & Chaperones,, 100-108 (1998), Journal' Ob 'Medical' Chemistry, 42, 260-266 (1999), etc.]. All of these compounds have been reported to bind to Hsp90 family proteins and exhibit pharmacological activities such as antitumor activity by inhibiting the functions of Hsp90 family proteins.
- geldanamycin derivatives [17-AAG, Invest.
- Compound (I) is considered useful as a therapeutic agent (for example, an antitumor agent) for diseases involving Hsp90 family protein or a protein to which Hsp90 family protein binds (Hsp90 client protein).
- Duplicated Dulbecco's modified Eagle's medium (DME M) containing 10% fetal bovine serum (FCS) in a 96-well microplate (manufactured by Nunk) with 2000 KPL-4 cells derived from human breast cancer per well ) was pre-cultured at 37 ° C for 24 hours in a 5% carbon dioxide incubator.
- a dimethyl sulfoxide (DMSO) solution of each test compound adjusted to 10 mmol / L, diluted in stages with the culture medium, was added to each well so that the total per well was 100 zL. Further, the cells were cultured at 37 ° C for 72 hours in a 5% carbon dioxide incubator.
- compounds 1 to 3, 9, 10, 14 to 19 and 23 have a GI value of 50 ⁇ 1 / ⁇ .
- the following shows cell growth inhibitory activity against human breast cancer-derived KPL-4 cells and is considered useful as an antitumor agent.
- compound (I) exhibits growth inhibitory activity against human chronic myeloid leukemia-derived K562 cells and can be confirmed to be useful as an antitumor agent.
- Compound (I) or a prodrug thereof or a pharmacologically acceptable salt thereof can be administered alone as it is, but it is usually desirable to provide it as various pharmaceutical preparations. These pharmaceutical preparations are used for animals and humans.
- the pharmaceutical preparation according to the present invention comprises compound (I) or a prodrug or pharmacologically acceptable salt thereof alone or as an active ingredient for any other treatment as an active ingredient. It can contain as a mixture of these.
- these pharmaceutical preparations are produced by any method well known in the technical field of pharmaceutics by mixing the active ingredient together with one or more pharmaceutically acceptable carriers.
- Examples of the dosage form include tablets and injections.
- Suitable for oral administration such as tablets, excipients such as lactose and mannitol, disintegrants such as starch, lubricants such as magnesium stearate, binders such as hydroxypropylcellulose, surface active agents such as fatty acid esters And a plasticizer such as glycerin.
- Formulations suitable for parenteral administration preferably contain active compounds that are isotonic with the blood of the recipient. Consists of a sterilized aqueous solution. For example, in the case of an injection, a solution for injection is prepared using a carrier made of a salt solution, a glucose solution, or a mixture of salt water and a glucose solution.
- these parenteral agents are also selected from excipients, disintegrating agents, lubricants, binders, surfactants and plasticizers exemplified as oral agents, diluents, preservatives, flavors and the like.
- One or more auxiliary ingredients can also be added.
- the dosage and frequency of administration of Compound (I) or a prodrug thereof or a pharmacologically acceptable salt thereof include the dosage form, patient age, body weight, nature or severity of symptoms to be treated, etc.
- 0.01 mg to 1 lg, preferably 0.05 to 50 mg is administered once or several times a day in the case of oral administration.
- parenteral administration such as intravenous administration
- 0.001 to 500 mg, preferably 0.01 to 100 mg per adult is administered once to several times a day.
- the dose and the number of doses vary depending on the various conditions described above.
- Step 1 2_ (Benzyloxymethyl) _4,6-bis (methoxymethoxy) benzoic acid (35 mg, 0.096 mmol) obtained in Step 6 of Example 22 was added to tetrahydrofuran (4 mL) and dichloromethane (1 mL ) And aniline (0.030 mL, 0.33 mmol), 1-hydroxybenzotriazole monohydrate (56 mg, 0.37 mmol) and 1_ [3- (dimethylamino) propyl] _3_ethyl carpositimide Hydrochloride (0.13 g, 0.68 mmol) was added, and the mixture was stirred at room temperature for 20 hr.
- Step 1 In the same manner as in Step 1 of Example 1, 2- (benzyloxymethyl) -4,6-bis (methoxymethoxy) benzoic acid (35 mg, 0.096 mmol) obtained in Step 6 of Example 22 Tetrahydrofuran (4 mL), dichloromethane (1 mL), benzylamine (0.032 mL, 0.29 mmol), 1-hydroxybenzotriazole monohydrate (60 mg, 0.39 mmol) and 1_ [3_ (dimethylamino) propiate.
- Step 2 N-Benzenore-2- (benzyloxymethyl) _4,6_bis (methoxymethoxy) benzamide (24 mg, 0.053 mmol) obtained in the same manner as in Step 2 of Example 1 From this, compound 2 (19 mg, 100%) was obtained using ethanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 In the same manner as in Step 1 of Example 1, 2- (benzyloxymethyl) -4,6-bis (methoxymethoxy) benzoic acid (32 mg, 0.88 mmol) obtained in Step 6 of Example 22 Tetrahydrofuran (4 mL), dichloromethane (1 mL), phenethylamine (0.030 mL, 0.24 mmol), 1-hydride Roxybenzotriazole monohydrate (68 mg, 0.44 mmol) and 1_ [3_ (dimethylamino) propyl] -3-ethylcarbodiimide hydrochloride (0.11 g, 0.57 mmol) were used to give 2_ (benzyloxymethyl) -4 , 6-Bis (methoxymethoxy) -N-phenethylbenzamide (27 mg, 69%) was obtained.
- Step 2 In the same manner as in Step 2 of Example 1, 2- (pendinoleoxymethyl) -4,6-bis (methoxymethoxy) -N-phenethylbenzamide (25 mg, 0.055 mmol) was used to obtain compound 3 (18 mg, 87%) using ethanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 2- (Benzyloxymethyl) -4,6-bis (methoxymethoxy) benzaldehyde (0.12 g, 0.33 mmol) obtained in Step 5 of Example 22 was dissolved in tetrahydrofuran (10 mL). To the obtained solution were added sodium chlorite (0.32 g, 3.6 mmol), amidosulfuric acid (0.69 g, 7.1 mmol) and distilled water (5 mL), and the mixture was stirred at room temperature for 3 hours. Water was added to the reaction mixture, and the mixture was extracted 3 times with chloroform.
- Step 2 2- (Benzyloxymethyl) -3-chloroguchi-4,6-bis (methoxymethoxy) obtained above
- B Dissolve benzoic acid (25 mg, 0.063 mmol) in methanol (2 mL), and add 2.0 mol / L trimethylsilyldiazomethane in hexane (0.30 mL, 0.60 mmol) to the resulting solution. Stir at room temperature for 1 hour. Acetic acid (0.050 mL) was added to the reaction mixture, and the solvent was evaporated under reduced pressure.
- Step 3 In the same manner as in Step 2 of Example 1, 2- (penzinoreoxymethyl) -3-chloro-4,6-bis (methoxymethoxy) benzoic acid methyl ester obtained above (4 mg) and concentrated hydrochloric acid (0.10 mL) were used to obtain compound 4 (10 mg, 77%).
- Step 1 In the same manner as in Step 1 of Example 1, 2- (benzyloxymethyl) _3_cloguchi-4,6_bis (methoxymethoxy) benzoic acid (37 mg) obtained in Step 1 of Example 4 , 0.093 mmol) from tetrahydrofuran (4 mL), dichloromethane (1 mL), benzylamine (0.033 mL, 0.30 mmol), 1-hydroxybenzotriazole monohydrate (62 mg, 0.40 mmol) and 1_ [3_ (Dimethylamino) propyl] -3_ethylcarbodiimide hydrochloride (94 mg, 0.49 mmol) was used, and N-benzyl-2- (benzyloxymethyl) -3-chloroguchi-4,6-bis (methoxymethoxy ) Benzamide (5.9 mg, 14%) was obtained.
- Step 1 In the same manner as in Step 1 of Example 1, 2- (benzyloxymethyl) -3-chloro-4,6-bis (methoxymethoxy) benzoic acid obtained in Step 1 of Example 4 (34 mg, 0.087 mmol), Tetrahydrofuran (4 mL), Dichloromethane (1 mL), 2_Methoxybenzylamine (0.039 mL, 0.30 mmol), 1-Hydroxybenzotriazole monohydrate (59 mg, 0.39 mmol) And 1_ [3_ (dimethylamino) propyl] -3-ethylcarbodiimide hydrochloride (0.11 g, 0.57 mmol) to give 2_ (benzyloxymethyl) -3-chloro-N- (2-methoxybenzyl) -4,6-bis (methoxymethoxy) benzamide (9.5 mg, 23%) was obtained.
- Step 2 In the same manner as in Step 2 of Example 1, 2- (pendinoleoxymethyl) -3-chloro-N- (2-methoxybenzyl) -4,6_ Compound 6 (5.8 mg, 6 8%) was obtained from bis (methoxymethoxy) benzamide (9.5 mg, 0.020 mmol) using ethanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 In the same manner as in Step 1 of Example 1, 2- (benzyloxymethyl) -3-chloro-4,6-bis (methoxymethoxy) benzoic acid obtained in Step 1 of Example 4 (33 mg, 0.084 mmol), tetrahydrofuran (4 mL), dichloromethane (1 mL), 3_methoxybenzylamine (0.038 mL, 0.3 0 mmol), 1-hydroxybenzotriazole monohydrate (68 mg, 0.44 mmol) and 1- [3_ (dimethylamino) propyl] -3_ethylcarbodiimide hydrochloride (98 mg, 0.51 mmol), 2_ (benzyloxymethyl) -3-black mouth _N- (3- Methoxybenzyl) -4,6_bis (methoxymethoxy) benzamide (9.0 mg, 23%) was obtained.
- Step 2 In the same manner as in Step 2 of Example 1, 2- (pendinoleoxymethyl) -3-chloro-N- (3-methoxybenzyl) -4,6- Compound 7 (5.1 mg, 63%) was obtained from bis (methoxymethoxy) benzamide (9.0 mg, 0.019 mmol) using ethanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 In the same manner as in Step 1 of Example 1, 2- (benzyloxymethyl) _3_cloguchi-4,6_bis (methoxymethoxy) benzoic acid (33 mg) obtained in Step 1 of Example 4 , 0.083 mmol), tetrahydrofuran (4 mL), dichloromethane (1 mL), 4_methoxybenzylamine (0.039 mL, 0.3 0 mmol), 1-hydroxybenzotriazole monohydrate (65 mg, 0.42 mmol) and 1_ [3_ (dimethylamino) propyl] -3-ethylcarbodiimide hydrochloride (0.11 g, 0.57 mmol), 2_ (benzyloxymethyl) -3-chloro-N- (4-methoxy Benzyl) -4,6-bis (methoxymethoxy) Benzamide (6.2 mg, 16%) was obtained.
- Step 2 In the same manner as in Step 2 of Example 1, 2- (pendinoleoxymethyl) -3-chloro-N- (4-methoxybenzyl) -4, 6- Compound 8 (3.7 mg, 6 7%) was obtained from bis (methoxymethoxy) benzamide (6.2 mg, 0.01 3 mmol) using ethanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 In the same manner as in Step 1 of Example 1, 2- (pendinoleoxymethyl) _3_cloguchi-4,6_bis (methoxymethoxy) benzoic acid obtained in Step 1 of Example 4 (51 mg, 0.13 mmol) to tetrahydrofuran (4 mL), dichloromethane (1 mL), 2-pyridylmethylamine (0.040 mL, 0.39 mmol), 1-hydroxybenzotriazole monohydrate (92 mg, 0.60 mmol) And 1_ [3_ (dimethylamino) propyl] -3_ethylcarbodiimide hydrochloride (0.13 g, 0.68 mmol), 2_ (benzyloxymethyl) -3-chloro-4,6-bis (methoxymethoxy) ) -N- (pyridin-2-ylmethinole) benzamide (21 mg, 33./o) was obtained.
- Step 2 In the same manner as in Step 2 of Example 1, 2- (pendinoleoxymethyl) -3-chloro-4,6-bis (methoxymethoxy) -N- (obtained above) Compound 9 (8.9 mg, 52%) was obtained from pyridine-2-ylmethyl) benzamide (21 mg, 0.043 mmol) using ethanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 2- (Benzyloxymethyl) -4,6-bis (methoxymethoxy) benzoic acid (60 mg, 0.17 mmol) obtained in Step 6 of Example 22 was replaced with N, N-dimethylformamide (10 N-bromosuccinimide (62 mg, 0.35 mmol) was added, and the mixture was stirred at room temperature for 2 hours. Water was added to the reaction mixture, and extracted three times with black mouth form. The organic layers were combined, washed successively with a saturated aqueous sodium thiosulfate solution and a saturated aqueous sodium chloride solution and dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure.
- Step 2_ (Benzyloxymethyl) _3_Bromo-4,6-bis (methoxymethoxy) benzoic acid (16 mg, 0.036 mmol) obtained above was dissolved in methanol (2 mL) Then, a 2.0 mol / L trimethylsilyldiazomethane in hexane solution (0.30 mL, 0.60 mmol) was added, and the mixture was stirred at room temperature for 1 hour. Acetic acid (0.050 mL) was added to the reaction mixture, and the solvent was evaporated under reduced pressure.
- Step 1 Compound 23 (2.0 g, 7.5 mmol) obtained in Reference Example 1 was dissolved in acetone (50 mL), and potassium carbonate (5.0 g, 36 mmol) and chloromethyl methyl ether ( 2.5 mL, 33 mmol) was added, and the mixture was stirred at 60 ° C for 4 hours. Water was added to the reaction mixture, and the mixture was extracted 3 times with chloroform. The organic layers were combined, washed with a saturated aqueous sodium chloride solution and dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure.
- Step 2 3-Ethyl-2- (methoxycarbonylmethyl) -4,6-bis (methoxymethoxy) benzoic acid methyl ester (1.1 g, 3.0 mmol) obtained above was dissolved in methanol (3 mL). The resulting solution was added with 4.0 mol / L lithium hydroxide aqueous solution (15 mL, 60 mmol), and stirred at 60 ° C for 30 hours. Water and 6.0 mol / L hydrochloric acid were added to the reaction mixture, and the pH value was adjusted to 3, followed by extraction three times with ethyl acetate.
- Step 3 2_ (Carboxymethyl) -3-ethyl-4,6_bis (methoxymethoxy) benzoic acid (0.38 g, 1.2 mmol) obtained above was dissolved in toluene (20 mL), Acetic anhydride (0.20 mL, 2.1 mmol) was added to the resulting solution, and the mixture was stirred at 100 ° C. for 4 hours. Water was added to the reaction mixture, and the mixture was extracted 3 times with diethyl ether. The organic layers were combined, washed with a saturated aqueous sodium chloride solution and dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure.
- Step 4 3_Ethyl-2- (methoxycarbonylmethyl) _4,6-bis (methoxymethoxy) benzoic acid (79 mg, 0.23 mmol) obtained above in the same manner as in Step 1 of Example 1. ) From tetrahydrofuran (4 mL), dichloromethane (1 mL), 50% aqueous dimethylamine (0.063 mL, 0.70 mmol), 1-hydroxybenzotriazole monohydrate (0.15 g, 0.98 mmol) and 1- [ Using 3- (dimethylamino) propyl] -3-ethylcarbodiimide hydrochloride (0.27 g, 1.4 mmol), [2_ (N, N-dimethylcarbamoyl) _6_ethyl-3,5-bis (methoxymethoxy) Methylesterol acetate (71 mg, 84%) was obtained.
- Step 5 In the same manner as in Step 2 of Example 1, [2- (N, N-dimethylcarbamoyl) -6-ethyl-3,5-bis (methoxymethoxy) was obtained as described above. ) Phenyl] acetic acid methyl ester (69 mg, 0.187 mmol) was used to give compound 11 (55 mg, 100%) using methanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 In the same manner as in Step 1 of Example 1, 3-ethyl-2- (methoxycarbonylmethyl) -4,6-bis (methoxymethoxy) benzoic acid obtained in Step 3 of Example 11 ( 79 mg, 0.23 mmol) , Tetrahydrofuran (4 mL), dichloromethane (1 mL), pyrrolidine (0.058 m, 0.70 mmol), 1-hydroxybenzotriazole monohydrate (0.15 g, 0.98 mmol) and 1- [3- (dimethylamino) [2-Ethyl-3,5-bis (methoxymethoxy) -6- (pyrrolidin-1-ylcarbonyl) phenyl] acetic acid methyl ester using propyl] -3-ethylcarbodiimide hydrochloride (0.27 g, 1.4 mmol) (70 mg, 77%) was obtained.
- Step 2 [2-Ethyl-3,5-bis (methoxymethoxy) -6- (pyrrolidine-1-ylcarbonyl) phenyl] acetic acid obtained in the same manner as in Step 2 of Example 1
- Compound 12 (56 mg, 100%) was obtained from the methyl ester (70 mg, 0.18 mmol) using methanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 In the same manner as in Step 1 of Example 1, 3-ethyl_2_ (methoxycarbonylmethyl) -4,6-bis (methoxymethoxy) benzoic acid (79 mg) obtained in Step 3 of Example 11 , 0.23 mmol) force, tetrahydrofuran (4 mL), dichloromethane (1 mL), N-methylbenzylamine (0.090 mL, 0.70 mmol), 1-hydroxybenzotriazole monohydrate (0.15 g, 0.98 mmol) And 1_ [3- (dimethylamino) propyl] -3_ethylcarbodiimide hydrochloride (0.27 g, 1.4 mmol) to give [2- (N-benzyl-N-methylcarbamoyl) _6_ethyl-3, 5- Bis (methoxymethoxy) phenyl] acetic acid methyl ester (61 mg, 60%) was obtained.
- Step 2 In the same manner as in Step 2 of Example 1, [2- (N-benzyl-N-methyl-powered rubamoyl) -6-ethyl-3,5-bis (methoxymethoxy) was obtained as described above. Compound 13 (48 mg, 99%) was obtained from) fenore] acetic acid methyl ester (60 mg, 0.14 mmol) using methanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 In the same manner as in Step 1 of Example 1, 3-ethyl-2- (methoxycarbonylmethyl) -4,6-bis (methoxymethoxy) benzoic acid obtained in Step 3 of Example 11 (79 mg, 0.23 mmol) force, tetrahydrofuran (4 mL), dichloromethane (1 mL), morpholine (0.061 mL, 0.70 mmol), 1-hydroxybenzotriazole monohydrate (0.15 g, 0.98 mmol) and 1- [3 [2-Ethyl-3,5-bis (methoxymethoxy) -6- (morpholinocarbonyl) phenyl] acetic acid using-(dimethylamino) propyl] -3_ethylcarbodiimide hydrochloride (0.27 g, 1.4 mmol) The methyl ester (82 mg, 86%) was obtained.
- Step 2 [2-Ethyl-3,5-bis (methoxymethoxy) -6- (morpholinocarboninole) phenol] obtained above in the same manner as in Step 2 of Example 1
- Compound 14 (50 mg, 80%) was obtained from acetic acid methyl ester (80 mg, 0.19 mmol) using methanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 In the same manner as in Step 1 of Example 1, 3-ethyl-2- (methoxycarbonylmethyl) -4,6-bis (methoxymethoxy) benzoic acid obtained in Step 3 of Example 11 ( 96 mg, 0.28 mmol) force, tetrahydrofuran (4 mL), dichloromethane (1 mL), N-methylpropylamine (0.070 mL, 0.68 mmol), 1-hydroxybenzotriazole monohydrate (0.15 g, 0.98 mmol) ) And 1_ [3- (dimethylamino) propyl] -3_ethylcarbodiimide hydrochloride (0.21 g, 1.1 mmol) were used to give [2-ethyl-3,5-bis (methoxymethoxy) -6- (N_ Methyl-N-propyl strength ruberamoyl) phenol] acetic acid methyl ester (61 mg, 55%) was obtained.
- Step 2 [2-Ethyl-3,5-bis (methoxymethoxy) -6- (N-methyl-N-propyl strength rubamoyl) obtained in the same manner as in Step 2 of Example 1
- Compound 15 (4 2 mg, 91%) was obtained from) phenyl] acetic acid methyl ester (60 mg, 0.15 mmol) using methanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 In the same manner as in Step 1 of Example 1, 3-ethyl_2_ (methoxycarbonylmethyl) -4,6-bis (methoxymethoxy) benzoic acid (96 mg, obtained in Step 3 of Example 11) 0.28 mmol) force, tetrahydrofuran (4 mL), dichloromethane (1 mL), 2_methoxy-N-methylethylamine (0.070 m, 0.65 mmol), 1_hydroxybenzotriazole monohydrate ( 0.15 g, 0.98 mmol) and 1- [3- (dimethylamino) propyl] -3-ethylcarbodiimide hydrochloride (0.25 g, 1.3 mmol) ) To give ⁇ 2-ethyl-6- [N- (2-methoxyethyl) -N-methylcarbamoyl] -3,5-bis (methoxymethoxy) phenyl ⁇ acetic acid methyl ester (0.10
- Step 2 In the same manner as in Step 2 of Example 1, ⁇ 2-ethyl-6- [N- (2-methoxychetyl) -N-methylcarbamoyl] -3, 5- Compound 16 (65 mg, 84%) was obtained from bis (methoxymethoxy) phenyl ⁇ methyl acetate (98 mg, 0.24 mmol) using methanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 3_Ethyl_2_ (methoxycarbonylmethyl) -4,6-bis (methoxymethoxy) benzoic acid (96 mg, obtained in Step 3 of Example 11 in the same manner as in Step 1 of Example 1 0.28 mmol) force, tetrahydrofuran (4 mL), dichloromethane (1 mL), piperidine (0.070 mL, 0.71 mmol), 1-hydroxybenzotriazole monohydrate (0.16 g, 1.0 mmol) and 1_ [3 -(Dimethylamino) propyl] -3_ethylcarbodiimide hydrochloride (0.27 g, 1.4 mmol) was used to produce [2-ethyl-3,5-bis (methoxymethoxy) -6- (piperidinocarbonyl) phenyl] Acetic acid methyl ester (57 mg, 49%) was obtained.
- Step 2 Methyl [2-ethyl-3,5-bis (methoxymethoxy) -6- (piperidinocarbonyl) phenyl] acetate obtained in the same manner as in Step 2 of Example 1
- Compound 17 (41 mg, 95%) was prepared from the ester (55 mg, 0.13 mmol) using methanol (4 mL) and concentrated hydrochloric acid (0.10 mL). Obtained.
- Step 1 In the same manner as in Step 1 of Example 1, 3-ethyl-2- (methoxycarbonylmethyl) -4,6-bis (methoxymethoxy) benzoic acid obtained in Step 3 of Example 11 (96 mg, 0.28 mmol) force, tetrahydrofuran (4 mL), dichloromethane (1 mL), N-methylbiperazine (0.070 mL, 0.6 3 mmol), 1_hydroxybenzotriazole monohydrate (0.17 g, 1.1 mmol) and 1_ Using [3_ (dimethylamino) propyl] -3-ethylcarbodiimide hydrochloride (0.26 g, 1.4 mmol), [2-ethyl-3,5-bis (methoxymethoxy) -6- (4-methylbiperazine-1 -Ylecarbonyl) phenol] acetic acid methyl ester (94 mg, 79%) was obtained.
- Step 1 [2-Ethyl-3,5-bis (methoxymethoxy) -6_ (4-methylpiperazine_1-ylcarbonyl) phenyl] obtained in the same manner as in Step 2 of Example 1
- Compound 18 (36 mg, 49%) was obtained from acetic acid methyl ester (55 mg, 0.13 mmol) using methanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- Step 1 In the same manner as in Step 1 of Example 1, 3-ethyl-2- (methoxycarbonylmethyl) -4,6-bis (methoxymethoxy) benzoic acid obtained in Step 3 of Example 11 ( 0.13 g, 0.29 mmol) , Tetrahydrofuran (4 mL), dichloromethane (1 mL), benzylamine (0.10 mL, 0.92 mm ol), 1-hydroxybenzotriazole monohydrate (0.25 g, 1.6 mmol) and 1- [3- (dimethylamino) Propyl] -3-ethylcarbodiimide hydrochloride (0.27 g, 1.4 mmol) was used to add [2_ (N_benzylcarbamoyl) -6-ethyl-3,5_bis (methoxymethoxy) phenyl] acetic acid methyl ester (0.11 g 84%).
- Step 2 [2- (N-Benzylcarbamoyl) _6_ethyl-3,5-bis (methoxymethoxy) phenyl] acetic acid methyl ester (46 mg, 0.11 mmol) obtained above was converted to tetrahydrofuran (5 mL) and cooled to 0 ° C. To the resulting solution was added 1.0 mol / L lithium aluminum hydride tetrahydrofuran solution (0.20 mL, 0.20 mmol), and the mixture was stirred at 0 ° C for 0.5 hr. A saturated aqueous sodium sulfate solution was added to the reaction mixture, and the mixture was extracted 3 times with ethyl acetate.
- Step 3 N-benzyl-3_ethyl _2_ (2-hydroxyethyl) -4,6_bis (methoxymethoxy) benzamide (31 Compound 19 (12 mg, 46%) was obtained using ethanol (4 mL) and concentrated hydrochloric acid (0.10 mL).
- 1H-NMR (270 MHz, CDCl) ⁇ (ppm): 1.09 (t, J 7.4 Hz, 3H), 2.39 (brs, 1H), 2.55 (q, J
- Step 1 [2-Ethyl-3,5-bis (methoxymethoxy) _6_ (morpholinocarbonyl) phenyl] acetic acid methyl ester (0.59 g, 1.4 mmol) obtained in Step 1 of Example 14 was added to tetrahydrofuran (8 mL) and cooled to 0 ° C. Lithium aluminum hydride (77 mg, 2.0 mmol) was added to the resulting solution, and the mixture was stirred at 0 ° C for 0.5 hr. To the reaction mixture were added anhydrous sodium sulfate and saturated aqueous sodium sulfate solution, and the mixture was stirred at room temperature for 2 hours and filtered.
- Step 2 3-Ethyl-2- (2-hydroxyethyl) -4,6-bis (methoxymethoxy) phenyl-morpholino-ketone (0.15 g, 0.39 mmol) obtained above was used. Dissolved in ⁇ , ⁇ -dimethylformamide (5 mL), 60% sodium hydride mineral oil dispersion (0.10 g, 2.5 mmol) was added to the resulting solution and stirred at room temperature for 1 hour. 2_Bromoethyl methyl ether (0.10 mL, 1.1 mmol) was added, and the mixture was stirred at 60 ° C for 12 hours. Water was added to the reaction mixture, and the mixture was extracted 3 times with black mouth form.
- Step 3 3-Ethyl-2- [2- (2-methoxyethoxy) ethyl] -4,6-bis (methoxymethoxy) obtained in the same manner as in Step 2 of Example 1
- Step 1 3,5-Dihydroxybenzoic acid methyl ester (11 g, 63 mmol) was dissolved in dichloromethane (0.10 L), and ⁇ , ⁇ -diisopropylethylamine (32 mL, 190 mmol) and chloromethyl methyl ether (14 mL, 190 mmol) were added, and the mixture was stirred at room temperature for 5 hours. Water was added to the reaction mixture, and the mixture was extracted 3 times with ethyl acetate. The organic layers were combined, washed with a saturated aqueous sodium chloride solution and dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure. The resulting residue was purified by silica gel column chromatography (black mouth form) to obtain 3,5-bis (methoxymethoxy) benzoic acid methyl ester (14 g, 87%).
- Step 2 3,5-Bis (methoxymethoxy) benzoic acid methyl ester obtained above (9.1 g, 36 mmol) was dissolved in jetyl ether (50 mL) and cooled to 0 ° C. . To the resulting solution was slowly added lithium aluminum hydride (2.0 g, 54 mmol), and the mixture was stirred at 0 ° C. for 2 hours. Sodium sulfate decahydrate (40 g) was added to the reaction mixture, and the mixture was further stirred at 0 ° C. for 1 hr. Reaction mixture And the obtained filtrate was concentrated under reduced pressure to obtain 3,5-bis (methoxymethoxy) phenyl methanol monole (7.6 g, 94%).
- Step 3 3,5-bis (methoxymethoxy) phenylmethanol (5.3 g, 23 mmol) obtained above was dissolved in N, N_dimethylformamide (20 mL), and the resulting solution To this was added N-bromosuccinimide (4.3 g, 24 mmol), and the mixture was stirred at room temperature for 5 hours. Water was added to the reaction mixture, and the mixture was extracted 3 times with chloroform. The organic layers were combined, washed with a saturated aqueous sodium chloride solution and dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure.
- Step 4 [2-Bromo-3,5-bis (methoxymethoxy) phenyl] methanol (3.5 g, 11 mmol) obtained above was dissolved in N, N-dimethylformamide (20 mL). And cooled to 0 ° C. To the resulting solution were added 60% sodium hydride mineral oil dispersion (0.59 g, 15 mmol) and benzyl bromide (2.0 mL, 17 mmol), and the mixture was stirred at room temperature for 1 hour. Methanol and water were added to the reaction mixture and extracted three times with black mouth form.
- Step 5 2_Bromo-1- (benzyloxymethyl) -3,5-bis (methoxymethoxy) benzene (1.6 g, 4.0 mmol) obtained above was converted into jetyl ether (25 mL). ) And dissolved in _78. Cool to C It was. To the resulting solution was added 1.52 mol / L n_butyllithium hexane solution (4.0 mL, 6.1 mmol), stirred at -78 ° C for 1 hour, then ⁇ , ⁇ -dimethylformamide (1.0 mL, 13 mmol) and stirred at room temperature for 3 hours. Methanol and water were added to the reaction mixture, and extracted three times with black mouth form.
- Step 6 2_ (Benzyloxymethyl) _4,6_bis (methoxymethoxy) benzaldehyde (0.80 g, 2.3 mmol) obtained above was dissolved in tert-butyl alcohol (20 mL), To the resulting solution was added 2-methyl-2-butene (10 mL, 94 mmol), sodium chlorite (2.0 g, 22 mmol), sodium dihydrogen phosphate (2.0 g, 17 mmol) and water (5 mL). ) And stirred at room temperature for 10 hours. The reaction mixture was extracted 3 times with black mouth form.
- Step 7 2- (Benzyloxymethyl) -4,6-bis (methoxymethoxy) benzoic acid (20 mg, 0.054 mmol) obtained above was dissolved in methanol (2 mL) to obtain To the resulting solution was added 2.0 mol / L trimethylsilyl diazomethane in hexane (0.30 mL, 0.60 mmol), and the mixture was stirred at room temperature for 1 hour. Acetic acid (0.050 mL) was added to the reaction mixture, and the solvent was evaporated under reduced pressure.
- Step 1 3-Oxohexanoic acid ethyl ester (20 mL, 0.13 mol) was dissolved in toluene (100 mL), and triethylamine (28 mL, 0.20 mol) and chlorotrimethylsilane (24 mL, 0.19 mol) was added, and the mixture was stirred at room temperature for 4 hours. Hexane (200 mL) and 0.5 mol / L aqueous sodium hydrogen carbonate solution were added to the reaction mixture, and the mixture was extracted 3 times with hexane.
- Step 2 3-Oxopentanenic acid dimethyl ester (11 mL, 75 mmol) was dissolved in dichloromethane (100 mL) and cooled to 0 ° C.
- the present invention provides, for example, an Hsp90 family protein inhibitor containing, as an active ingredient, a benzoic acid derivative, a prodrug thereof, or a pharmacologically acceptable salt thereof.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pyridine Compounds (AREA)
Abstract
Description


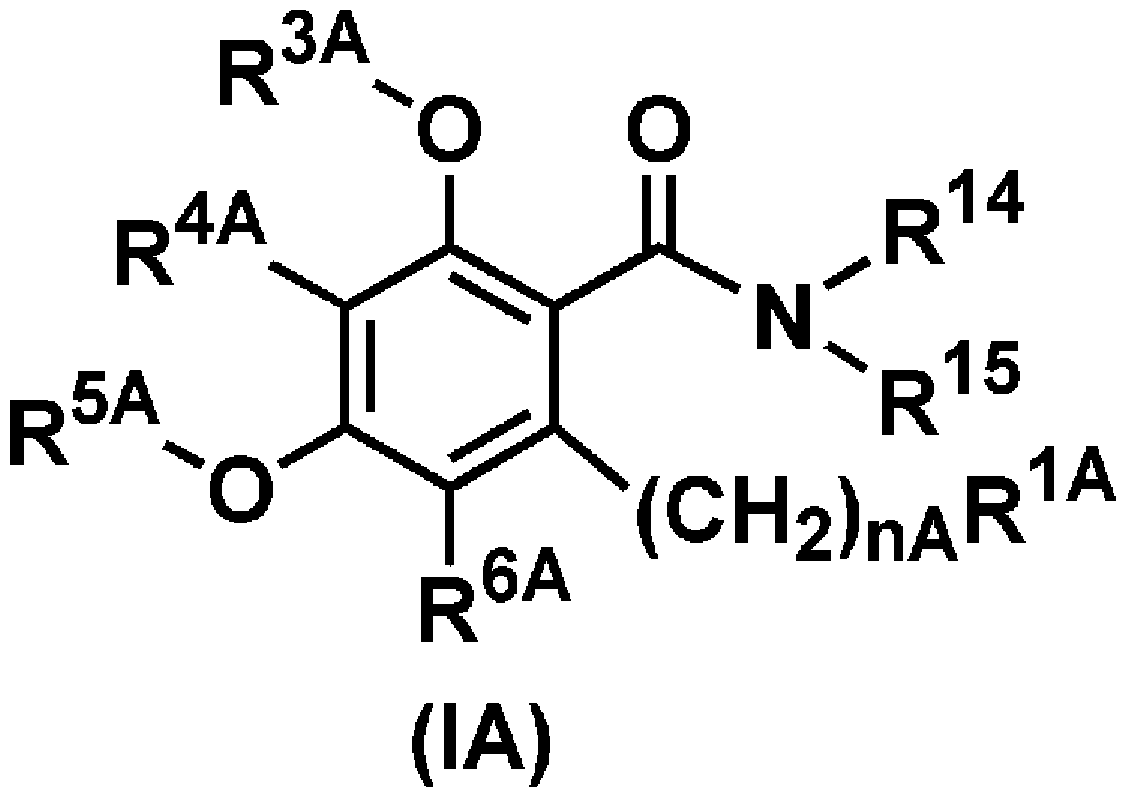











Claims






Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05806152A EP1813270A4 (en) | 2004-11-09 | 2005-11-09 | PROTEIN INHIBITORS OF THE HSP90 FAMILY |
| JP2006544912A JPWO2006051808A1 (ja) | 2004-11-09 | 2005-11-09 | Hsp90ファミリー蛋白質阻害剤 |
| US11/718,079 US7781485B2 (en) | 2004-11-09 | 2005-11-09 | Hsp90 family protein inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004324480 | 2004-11-09 | ||
| JP2004-324480 | 2004-11-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006051808A1 true WO2006051808A1 (ja) | 2006-05-18 |
Family
ID=36336485
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2005/020519 WO2006051808A1 (ja) | 2004-11-09 | 2005-11-09 | Hsp90ファミリー蛋白質阻害剤 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US7781485B2 (ja) |
| EP (1) | EP1813270A4 (ja) |
| JP (1) | JPWO2006051808A1 (ja) |
| WO (1) | WO2006051808A1 (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006109085A1 (en) | 2005-04-13 | 2006-10-19 | Astex Therapeutics Limited | Hydroxybenzamide derivatives and their use as inhibitors of hsp90 |
| WO2006117669A1 (en) * | 2005-05-03 | 2006-11-09 | Pfizer Inc. | Amide resorcinol compounds |
| WO2008044034A1 (en) * | 2006-10-12 | 2008-04-17 | Astex Therapeutics Limited | Hydrobenzamide derivatives as inhibitors of hsp90 |
| WO2008044027A2 (en) * | 2006-10-12 | 2008-04-17 | Astex Therapeutics Limited | Pharmaceutical compounds having hsp90 inhibitory or modulating activity |
| WO2008053319A1 (en) * | 2006-10-30 | 2008-05-08 | Pfizer Products Inc. | Amide resorcinol compounds |
| WO2009091921A1 (en) | 2008-01-15 | 2009-07-23 | Universite De Strasbourg | Synthesis of resorcylic acid lactones useful as therapeutic agents |
| US7754725B2 (en) | 2006-03-01 | 2010-07-13 | Astex Therapeutics Ltd. | Dihydroxyphenyl isoindolymethanones |
| US8883790B2 (en) | 2006-10-12 | 2014-11-11 | Astex Therapeutics Limited | Pharmaceutical combinations |
| US8916552B2 (en) | 2006-10-12 | 2014-12-23 | Astex Therapeutics Limited | Pharmaceutical combinations |
| US9730912B2 (en) | 2006-10-12 | 2017-08-15 | Astex Therapeutics Limited | Pharmaceutical compounds |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2073802A1 (en) | 2006-10-12 | 2009-07-01 | Astex Therapeutics Limited | Pharmaceutical combinations |
| GB0806527D0 (en) | 2008-04-11 | 2008-05-14 | Astex Therapeutics Ltd | Pharmaceutical compounds |
| US10464907B2 (en) | 2016-01-29 | 2019-11-05 | Industry Academic Cooperation Foundation Keimyung University | Compound having HSP90 inhibitory activity or pharmaceutically acceptable salt thereof, and medical use thereof |
| WO2019027203A1 (ko) * | 2017-08-02 | 2019-02-07 | 계명대학교 산학협력단 | Hsp90 억제 활성을 갖는 디히드록시페닐계 입체이성질체 및 이의 의학적 용도 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH03271261A (ja) * | 1990-03-22 | 1991-12-03 | Japan Tobacco Inc | 抗プロモーター作用を有する新規化合物およびその製造方法、並びにこれを含有する抗腫瘍薬 |
| JPH03271222A (ja) * | 1990-03-22 | 1991-12-03 | Japan Tobacco Inc | 抗プロモーター作用による抗腫瘍薬 |
| WO2001007429A1 (en) * | 1999-07-22 | 2001-02-01 | Mercian Corporation | Process for producing isocoumarins and intermediates for the same |
| WO2001044172A1 (en) * | 1999-12-15 | 2001-06-21 | Axys Pharmaceuticals, Inc. | Salicylamides as serine protease and factor xa inhibitors |
| WO2003103655A1 (ja) * | 2002-06-10 | 2003-12-18 | 株式会社医薬分子設計研究所 | 癌治療剤 |
| WO2004072051A1 (en) * | 2003-02-11 | 2004-08-26 | Vernalis (Cambridge) Limited | Isoxazole compounds as inhibitors of heat shock proteins |
| WO2005000778A1 (ja) * | 2003-06-27 | 2005-01-06 | Kyowa Hakko Kogyo Co., Ltd. | Hsp90ファミリー蛋白質阻害剤 |
| WO2005007151A1 (ja) * | 2003-07-16 | 2005-01-27 | Institute Of Medicinal Molecular Design. Inc. | 皮膚色素沈着の治療剤 |
| WO2005063222A1 (ja) * | 2003-12-26 | 2005-07-14 | Kyowa Hakko Kogyo Co., Ltd. | Hsp90ファミリー蛋白質阻害剤 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3271222B2 (ja) | 1994-02-22 | 2002-04-02 | ソニー株式会社 | 固体撮像装置 |
| US5832805A (en) | 1994-05-02 | 1998-11-10 | Ube Industries, Ltd. | Method and apparatus for controlling speed of hydraulic cylinder |
| JP2001097970A (ja) * | 1999-07-27 | 2001-04-10 | Mercian Corp | イソクマリン誘導体の製造方法 |
| US7208630B2 (en) * | 2004-10-27 | 2007-04-24 | University Of Kansas | Heat shock protein 90 inhibitors |
-
2005
- 2005-11-09 WO PCT/JP2005/020519 patent/WO2006051808A1/ja active Application Filing
- 2005-11-09 JP JP2006544912A patent/JPWO2006051808A1/ja active Pending
- 2005-11-09 EP EP05806152A patent/EP1813270A4/en not_active Withdrawn
- 2005-11-09 US US11/718,079 patent/US7781485B2/en not_active Expired - Fee Related
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH03271261A (ja) * | 1990-03-22 | 1991-12-03 | Japan Tobacco Inc | 抗プロモーター作用を有する新規化合物およびその製造方法、並びにこれを含有する抗腫瘍薬 |
| JPH03271222A (ja) * | 1990-03-22 | 1991-12-03 | Japan Tobacco Inc | 抗プロモーター作用による抗腫瘍薬 |
| WO2001007429A1 (en) * | 1999-07-22 | 2001-02-01 | Mercian Corporation | Process for producing isocoumarins and intermediates for the same |
| WO2001044172A1 (en) * | 1999-12-15 | 2001-06-21 | Axys Pharmaceuticals, Inc. | Salicylamides as serine protease and factor xa inhibitors |
| WO2003103655A1 (ja) * | 2002-06-10 | 2003-12-18 | 株式会社医薬分子設計研究所 | 癌治療剤 |
| WO2004072051A1 (en) * | 2003-02-11 | 2004-08-26 | Vernalis (Cambridge) Limited | Isoxazole compounds as inhibitors of heat shock proteins |
| WO2005000778A1 (ja) * | 2003-06-27 | 2005-01-06 | Kyowa Hakko Kogyo Co., Ltd. | Hsp90ファミリー蛋白質阻害剤 |
| WO2005007151A1 (ja) * | 2003-07-16 | 2005-01-27 | Institute Of Medicinal Molecular Design. Inc. | 皮膚色素沈着の治療剤 |
| WO2005063222A1 (ja) * | 2003-12-26 | 2005-07-14 | Kyowa Hakko Kogyo Co., Ltd. | Hsp90ファミリー蛋白質阻害剤 |
Non-Patent Citations (14)
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7700625B2 (en) | 2005-04-13 | 2010-04-20 | Astex Therapeutics Ltd. | Hydroxybenzamide derivatives and their use as inhibitors of Hsp90 |
| WO2006109075A2 (en) * | 2005-04-13 | 2006-10-19 | Astex Therapeutics Limited | Hydroxybenzamide derivatives and their use as inhibitors of hsp90 |
| WO2006109075A3 (en) * | 2005-04-13 | 2006-11-30 | Astex Therapeutics Ltd | Hydroxybenzamide derivatives and their use as inhibitors of hsp90 |
| US9914719B2 (en) | 2005-04-13 | 2018-03-13 | Astex Therapeutics Ltd. | Hydroxybenzamide derivatives and their use as inhibitors of HSP90 |
| US8816087B2 (en) | 2005-04-13 | 2014-08-26 | Astex Therapeutics Limited | Hydroxybenzamide derivatives and their use as inhibitors of Hsp90 |
| US8101648B2 (en) | 2005-04-13 | 2012-01-24 | Astex Therapeutics, Ltd. | Hydroxybenzamide derivatives and their use as inhibitors of HSP90 |
| EP2332909A1 (en) * | 2005-04-13 | 2011-06-15 | Astex Therapeutics Limited | Hydroxybenzamide derivatives and their use as inhibitors of HSP90 |
| JP2008537751A (ja) * | 2005-04-13 | 2008-09-25 | アステックス、セラピューティックス、リミテッド | 医薬化合物 |
| WO2006109085A1 (en) | 2005-04-13 | 2006-10-19 | Astex Therapeutics Limited | Hydroxybenzamide derivatives and their use as inhibitors of hsp90 |
| WO2006117669A1 (en) * | 2005-05-03 | 2006-11-09 | Pfizer Inc. | Amide resorcinol compounds |
| US7754725B2 (en) | 2006-03-01 | 2010-07-13 | Astex Therapeutics Ltd. | Dihydroxyphenyl isoindolymethanones |
| US8106057B2 (en) | 2006-03-01 | 2012-01-31 | Astex Therapeutics, Ltd. | Dihydroxyphenyl isoindolylmethanones |
| AU2007306104B2 (en) * | 2006-10-12 | 2014-11-06 | Astex Therapeutics Limited | Hydrobenzamide derivatives as inhibitors of Hsp90 |
| US8883790B2 (en) | 2006-10-12 | 2014-11-11 | Astex Therapeutics Limited | Pharmaceutical combinations |
| NO342242B1 (no) * | 2006-10-12 | 2018-04-30 | Astex Therapeutics Ltd | Hydrobenzamid derivater som inhibitorer av HSP90, fremgangsmåter for fremstilling, farmasøytiske sammensetninger, kombinasjoner og anvendelser. |
| WO2008044034A1 (en) * | 2006-10-12 | 2008-04-17 | Astex Therapeutics Limited | Hydrobenzamide derivatives as inhibitors of hsp90 |
| EP2546233A1 (en) * | 2006-10-12 | 2013-01-16 | Astex Therapeutics Limited | Hydrobenzamide derivatives as inhibitors of HSP90 |
| CN101848892B (zh) * | 2006-10-12 | 2013-08-21 | 阿斯特克斯治疗有限公司 | 作为hsp90抑制剂的氢化苯酰胺衍生物 |
| WO2008044027A3 (en) * | 2006-10-12 | 2008-06-19 | Astex Therapeutics Ltd | Pharmaceutical compounds having hsp90 inhibitory or modulating activity |
| JP2010505927A (ja) * | 2006-10-12 | 2010-02-25 | アステックス、セラピューティックス、リミテッド | 医薬化合物 |
| WO2008044027A2 (en) * | 2006-10-12 | 2008-04-17 | Astex Therapeutics Limited | Pharmaceutical compounds having hsp90 inhibitory or modulating activity |
| US8916552B2 (en) | 2006-10-12 | 2014-12-23 | Astex Therapeutics Limited | Pharmaceutical combinations |
| KR101571568B1 (ko) | 2006-10-12 | 2015-11-24 | 아스텍스 테라퓨틱스 리미티드 | Hsp90의 억제제인 하이드로벤즈아미드 유도체 |
| US9428439B2 (en) | 2006-10-12 | 2016-08-30 | Astex Therapeutics Ltd. | Hydrobenzamide derivatives as inhibitors of Hsp90 |
| US9730912B2 (en) | 2006-10-12 | 2017-08-15 | Astex Therapeutics Limited | Pharmaceutical compounds |
| WO2008053319A1 (en) * | 2006-10-30 | 2008-05-08 | Pfizer Products Inc. | Amide resorcinol compounds |
| WO2009091921A1 (en) | 2008-01-15 | 2009-07-23 | Universite De Strasbourg | Synthesis of resorcylic acid lactones useful as therapeutic agents |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1813270A1 (en) | 2007-08-01 |
| US7781485B2 (en) | 2010-08-24 |
| EP1813270A4 (en) | 2009-07-01 |
| JPWO2006051808A1 (ja) | 2008-05-29 |
| US20070265268A1 (en) | 2007-11-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006051808A1 (ja) | Hsp90ファミリー蛋白質阻害剤 | |
| JP6830671B2 (ja) | γ−ヒドロキシ酪酸(GHB)のプロドラッグ、その組成物および使用 | |
| CN106163521B (zh) | 有效的可溶性环氧化物水解酶抑制剂 | |
| JP4575294B2 (ja) | Hsp90ファミリー蛋白質阻害剤 | |
| RU2422446C2 (ru) | Производные спирохроманона в качестве ингибиторов ацетил коэнзим а карбоксилазы (асс) | |
| RU2523448C2 (ru) | Ингибиторы кинуренин-3-монооксигеназы | |
| KR101468449B1 (ko) | 신규한 페난스렌퀴논계 화합물 및 이를 포함하는대사증후군 질환의 치료 또는 예방용 약제 조성물 | |
| JP6763858B2 (ja) | Ras媒介性疾患を処置または予防する方法 | |
| US20220213083A1 (en) | Fxr small molecule agonist and preparation method therefor and use thereof | |
| JPWO2005063222A1 (ja) | Hsp90ファミリー蛋白質阻害剤 | |
| KR20200139702A (ko) | 칼페인 조정자 및 그 치료학적 용도 | |
| KR20200062243A (ko) | 그리세오풀빈 화합물과 의약 용도 | |
| US11572347B2 (en) | Orally available sEH/PDE4 dual inhibitors | |
| JP2009523711A (ja) | 新規環状置換フロピリミジン誘導体および心臓血管疾患を処置するためのその使用 | |
| CN108484598B (zh) | 吲哚嗪衍生物及其在医药上的应用 | |
| JP2003171381A (ja) | エントリー阻害剤 | |
| EP4223757A1 (en) | Fxr small-molecule agonist, and preparation method therefor and use thereof | |
| CA2685134A1 (en) | Use of cyclically substituted furopyrimidine derivatives for treating pulmonary arterial hypertonia | |
| JPH01283263A (ja) | 二置換ピロール類 | |
| JP2518497B2 (ja) | 抗脂血剤 | |
| JPWO2004024142A1 (ja) | Hsp90ファミリー蛋白質阻害剤 | |
| JPWO2004024141A1 (ja) | Hsp90ファミリー蛋白質阻害剤 | |
| RU2418786C2 (ru) | Энантиомерно чистые производные флавона для лечения пролиферативных нарушений и способы их получения | |
| JPH10512553A (ja) | 新規な化合物 | |
| KR20080088764A (ko) | 허혈성 질환의 예방 및 치료에 유효한 크로멘-4-온 화합물 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KN KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 11718079 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005806152 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006544912 Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005806152 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 11718079 Country of ref document: US |