WO2004012816A1 - Antibakterielle ester-makrozyklen - Google Patents
Antibakterielle ester-makrozyklen Download PDFInfo
- Publication number
- WO2004012816A1 WO2004012816A1 PCT/EP2003/007824 EP0307824W WO2004012816A1 WO 2004012816 A1 WO2004012816 A1 WO 2004012816A1 EP 0307824 W EP0307824 W EP 0307824W WO 2004012816 A1 WO2004012816 A1 WO 2004012816A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrogen
- alkyl
- substituents
- amino
- cycloalkyl
- Prior art date
Links
- 0 *C(*)(C(N(*)C(Cc1cc(-c(cc2)cc(CC3N)c2O*)ccc1O*)C(O*)=O)=O)N(*)C3=O Chemical compound *C(*)(C(N(*)C(Cc1cc(-c(cc2)cc(CC3N)c2O*)ccc1O*)C(O*)=O)=O)N(*)C3=O 0.000 description 5
- TVCKSKQJJDQDTI-UHFFFAOYSA-N CC(C)(C)OC(NC(CC(C[N+](OC)=[OH+])=O)C(OCc1ccccc1)=O)=O Chemical compound CC(C)(C)OC(NC(CC(C[N+](OC)=[OH+])=O)C(OCc1ccccc1)=O)=O TVCKSKQJJDQDTI-UHFFFAOYSA-N 0.000 description 1
- ONMWQAVYQVMVEQ-KGLIPLIRSA-N CC(C)(C)OC(N[C@@H](C[C@H](C[NH+]([O-])[O-])O)C(OCc1ccccc1)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](C[C@H](C[NH+]([O-])[O-])O)C(OCc1ccccc1)=O)=O ONMWQAVYQVMVEQ-KGLIPLIRSA-N 0.000 description 1
- LWFLLSLYPAGSTQ-DEOSSOPVSA-N CC(C)(C)OC(N[C@@H](Cc(cc(cc1)I)c1OCc1ccccc1)C(OCc1ccccc1)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](Cc(cc(cc1)I)c1OCc1ccccc1)C(OCc1ccccc1)=O)=O LWFLLSLYPAGSTQ-DEOSSOPVSA-N 0.000 description 1
- WPQAXSUSTBENDT-VABKMULXSA-N CC(C)COC([C@H](Cc(cc(cc1)-c(cc2C[C@@H](C(N[C@H]3CCCN)=O)N)ccc2O)c1O)N(C)C3=O)=O Chemical compound CC(C)COC([C@H](Cc(cc(cc1)-c(cc2C[C@@H](C(N[C@H]3CCCN)=O)N)ccc2O)c1O)N(C)C3=O)=O WPQAXSUSTBENDT-VABKMULXSA-N 0.000 description 1
- QQQPSGRFCXUIDM-UFYCRDLUSA-N CN([C@@H](Cc1cc(-c(cc2)cc(C[C@@H](C(N[C@H]3CCCN)=O)N)c2O)ccc1O)C(OC)=O)C3=O Chemical compound CN([C@@H](Cc1cc(-c(cc2)cc(C[C@@H](C(N[C@H]3CCCN)=O)N)c2O)ccc1O)C(OC)=O)C3=O QQQPSGRFCXUIDM-UFYCRDLUSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0812—Tripeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- the invention relates to antibacterial ester macrocycles and processes for their preparation and their use for the manufacture of medicaments for the treatment and / or prophylaxis of diseases, in particular bacterial infections.
- biphenomycin B (R 1 , R 2 is hydrogen, R 3 ', R 4 , R 7 , R 8 and R 9 is hydrogen, R 3 is 3- Amino-2-hydroxy-prop-1-yl and free carboxyl instead of an ester group) are described as having antibacterial activity. Partial steps of the synthesis of biphenomycin B are in
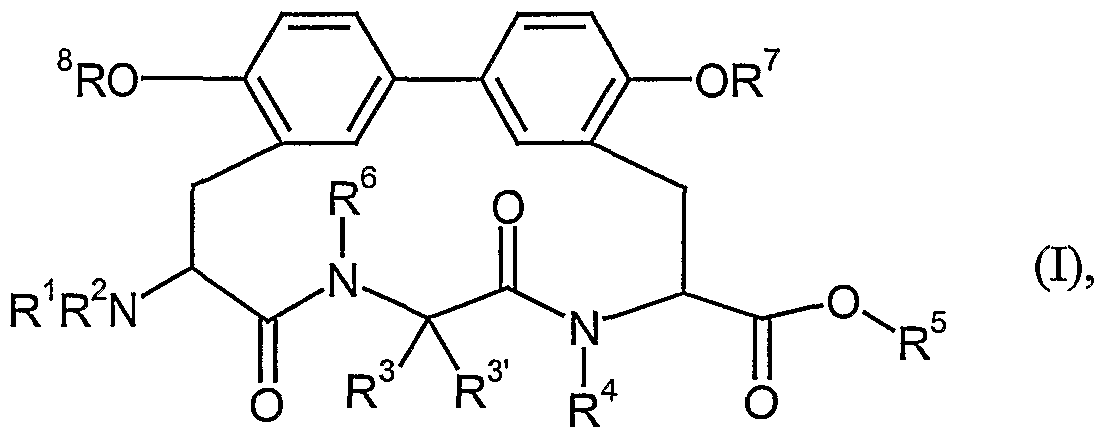
- the invention relates to compounds of the formula
- R 1 is hydrogen, alkyl, aryl, heteroaryl, heterocyclyl, alkylcarbonyl,
- R 1 can be substituted, apart from hydrogen, with 0, 1, 2 or 3 substituents R 1 "1 , the substituents R 1" 1 being selected independently of one another from the group consisting of halogen, alkyl, trifluoromethyl, trifluoromethoxy, nitro, Cyano, amino, AlJ ylarnino, dialkylamino, cycloalkyl, aryl, heteroaryl, heterocyclyl, hydroxy, alkoxy and carboxyl, R is hydrogen or alkyl,
- alkyl can be substituted with 0, 1, 2 or 3 substituents R 2 "1 , the substituents R 2" 1 being selected independently of one another from the group consisting of halogen, amino, alkylamino and dialkylamino,
- R 1 and R 2 together with the nitrogen atom to which they are attached form a heterocycle which can be substituted with 0, 1 or 2 substituents
- R 1 "2 where the substituents R 1" 2 are selected independently of one another from the group consisting of halogen, trifluoromethyl, amino, alkylamino, dialkylamino, cycloalkyl, aryl, heteroaryl, heterocyclyl, hydroxy, alkoxy, carboxyl, alkoxycarbonyl and aminocarbonyl,
- R 3 is hydrogen, alkyl or the side group of an amino acid, wherein
- Alkyl can be substituted with 0, 1, 2 or 3 substituents R " , the substituents R 3" 1 being selected independently of one another from the group consisting of trifluoromethyl, nitro, amino, alkylamino, dialkylamino, cycloalkyl, aryl, heteroaryl, Heterocyclyl, hydroxy, alkoxy,
- cycloalkyl, aryl, heteroaryl and heterocyclyl can be substituted with 0, 1 or 2 substituents R 3 "2 , the substituents R 3" 2 being selected independently of one another from the group consisting of halogen, alkyl, trifluoromethyl and amino,
- R 3 ' is hydrogen, -CC 6 - alkyl or C 3 -C 8 cycloalkyl
- R 4 is hydrogen, -CC 6 - alkyl or C 3 -C 8 cycloalkyl
- R 5 is alkyl, cycloalkyl, aryl, heteroaryl, heterocyclyl or a hydroxy-functional amino acid residue, where R 5 can be substituted with
- substituents R 5 "1 , the substituents R 5" 1 being selected independently of one another from the group consisting of halogen, alkyl, trifluoromethyl, trifluoromethoxy, cyano, amino, alkylamino, dialkylamino, cycloalkyl, aryl, heteroaryl , Heterocyclyl, hydroxy, alkoxy, carboxyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl and
- alkylamino and dialkylamino can be substituted with 0, 1 or 2 substituents R 5 "2 , the substituents R 5" 2 being selected independently of one another from the group consisting of hydroxyl, amino, alkoxy,
- R 6 is hydrogen, Ci-C 6 alkyl or C 3 -C 8 cycloalkyl
- R 7 is hydrogen, -C ⁇ alkyl, alkylcarbonyl or C 3 -C 8 cycloalkyl
- R is hydrogen or Ci-C ö alkyl
- Compounds according to the invention are the compounds of the formula (I) and their salts, solvates and solvates of the salts, the compounds of the formula (I 1 ) mentioned below and their salts, solvates and solvates of the salts and those of the formula (I) I) and or T) comprised compounds hereinafter referred to as exemplary embodiment (s) and their salts, solvates and solvates of the salts, insofar as they included the compounds mentioned below of formula (I) and / or (I ') are not already salts, solvates and solvates of the salts.
- preferred salts are physiologically acceptable salts of the compounds according to the invention.
- Physiologically acceptable salts of compounds (I) include acid addition salts of mineral acids, carboxylic acids and sulfonic acids, e.g. Salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, emansulfonic acid, toluenesulfonic acid, benzenesulfonic acid, naphthalenedisulfonic acid,
- Physiologically acceptable salts of the compounds (I) also include salts of conventional bases, such as, for example and preferably, alkali metal salts (e.g. sodium and potassium
- Potassium salts alkaline earth metal salts (eg calcium and magnesium salts) and ammonium salts, derived from ammonia or organic amines with 1 to 16 carbon atoms, such as, for example and preferably, emylamine, diemylarnine, triemyla in, ethyldiisopropylamine, monoethanolamine, the anolamine, triethanolamine, Dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, dihydroabietylamine, argfine, lysine, ylenm " amine and methylpiperidine.
- alkaline earth metal salts eg calcium and magnesium salts
- ammonium salts derived from ammonia or organic amines with 1 to 16 carbon atoms, such as, for example and preferably, emylamine, diemylarnine, triemyla in, ethyldiisopropyl
- solvates are those forms of the compounds which form a complex in the solid or liquid state by coordination with solvent molecules. Hydrates are a special form of solvate, in which coordination takes place with water. In the context of the present invention, unless otherwise specified, the substituents have the following meaning:
- Alkyl and the alkyl parts in substituents such as alkoxy, mono- and dialkylamino,
- Alkylsulfonyl include linear and branched alkyl, for example Ci-C 2 -, especially dC 6 - and -C-C 4 alkyl.
- -Cg- alkyl includes methyl, ethyl, n- and i-propyl, n-, i-, sec- and tert-butyl, n-pentyl, isopentyl, neopentyl and hexyl.
- C -C 4 alkyl includes methyl, ethyl, n- and i-propyl, n-, i-, sec- and tert-butyl.
- alkylcarbonyl preferably represents a straight-chain or branched alkyl radical having 1 to 6 or 1 to 4 carbon atoms. Examples and preferably mentioned are: methylcarbonyl, ethylcarbonyl, n-propylcarbonyl, isopropylcarbonyl and t-butylcarbonyl.
- Alkenyl comprises linear and branched C 2 -C 12 -, in particular C 2 -C 6 - and C 2 -C - alkenyl, such as vinyl, allyl, prop-1-en-1-yl, isopropenyl, but-1-enyl , But-2-enyl,
- Buta-1,2-dienyl and buta-1,3-dienyl are buta-1,2-dienyl and buta-1,3-dienyl.
- Alkynyl comprises linear and branched C 2 -C 12 -, in particular C 2 -C 6 - and C 2 -C 4 -
- Alkynyl e.g. Ethynyl, propargyl (2-propynyl), 1-propynyl, but-1-ynyl, but-2-ynyl.
- Cycloalkyl comprises polycyclic saturated hydrocarbon radicals with up to 14 carbon atoms, namely monocyclic C 3 -C 12 , preferably C 3 -C 8 alkyl, in particular C 3 -C 6 alkyl, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl , Cyclooctyl, Cyclononyl, and polycyclic alkyl, ie preferably bicyclic and tricyclic, optionally spirocyclic C 7 -C 1 alkyl, such as, for example, bicyclo [2.2.1] -hept-l-yl, bicyclo [2.2.1] -he ⁇ t -2-yl, bicyclo [2.2.1] -hept-7-yl, Bicyclo [2.2.2] oct-2-yl, bicyclo [3.2.1] oct-2-yl, bicyclo [3.2.2] non-2-y
- aryl stands for an aromatic radical having preferably 6 to 10 carbon atoms.
- Preferred aryl radicals are phenyl and naphthyl.
- alkoxy preferably represents a straight-chain or branched alkoxy radical, in particular having 1 to 6, 1 to 4 or 1 to 3 carbon atoms.
- a straight-chain or branched alkoxy radical having 1 to 3 carbon atoms is preferred. The following may be mentioned by way of example and preferably: methoxy, ethoxy, n-propoxy, isopropoxy, tert-butoxy, n-pentoxy and n-hexoxy.
- alkoxycarbonyl preferably represents a straight-chain or branched alkoxy radical having 1 to 6 or 1 to 4 carbon atoms, which is linked via a carbonyl group.
- a straight-chain or branched is preferred
- Alkoxycarbonylrest with 1 to 4 carbon atoms The following may be mentioned as examples and preferably: methoxycarbonyl, ethoxycarbonyl, n-propoxycarbonyl, isopropoxycarbonyl and tert-butoxycarbonyl.
- monoalkylamino (alkylarnino) stands for an amino
- Group with a straight-chain or branched alkyl substituent which preferably has 1 to 6, 1 to 4 or 1 to 2 carbon atoms.
- a straight-chain or branched monoall ⁇ lamino radical having 1 to 4 carbon atoms is preferred. Examples and preferably mentioned are: Me yla ino, emylamino, n-propylamino, isopropylamino, tert-butylamino, n-pentylamino and n-hexylamino.
- dialallamino stands for an amino group with two identical or different straight-chain or branched alkyl substituents, which preferably each have 1 to 6, 1 to 4 or 1 or 2 carbon atoms.
- Straight-chain or branched dialkylamino radicals each having 1, 2, 3 or 4 carbon atoms per alkyl substituent are preferred.
- NN-dimemylamino NN-diemylamino, N-emyl-N-me ylamino, N-methyl-Nn-propylamino, N-isopropyl-Nn-propylamino, Nt-butyl-N-memylamino, N-ethyl-Nn-pentylamino and Nn-hexyl-N-memylamino.
- Monoalkylaminocarbonyl (Al ⁇ laminocarbonyl) or dialkylaminocarbonyl is in
- Examples and preferably mentioned are: Memylaminocarbonyl, Emylaminocarbonyl, Isopropylaminocarbonyl, t-
- Bulylaminocarbonyl NN-Dime ylarninocarbonyl, NN-Diemylaminocarbonyl, N-Emyl-N-memylaminocarbonyl and N-t-Butyl-N-memylaminocarbonyl.
- arylaminocarbonyl stands for an aromatic radical with preferably 6 to 10 carbon atoms, which is linked via an aminocarbonyl group.
- Preferred radicals are phenylaminocarbonyl and ⁇ aphthylaminocarbonyl.
- alkylcarbonylamino represents an amino group with a straight-chain or branched alkanoyl substituent which preferably has 1 to 6, 1 to 4 or 1 to 2 carbon atoms and is linked via the carbonyl group.
- a monoacylarnino radical having 1 to 2 carbon atoms is preferred. Examples include and are preferably: formamido, acetamido, propionamido, n-butyramido and pivaloylamido.
- Heterocyclyl represents a mono- or polycyclic, heterocyclic radical with 4 to 10 ring atoms and up to 3, preferably 1 heteroatoms or hetero groups from the series ⁇ , O, S, SO, SO 2 . 4- to 8-membered, in particular 5- to 6-membered, heterocyclyl is preferred. Mono- or bicyclic heterocyclyl is preferred. Monocyclic heterocyclyl is particularly preferred.
- heterocyclyl residues can be saturated or be partially unsaturated. Saturated heterocyclyl residues are preferred.
- the heterocycly radicals can be bonded via a carbon atom or a hetero atom. 5- to 6-membered, monocyclic saturated heterocyclyl radicals having up to two heteroatoms from the O, N and S series are particularly preferred. Examples and, preferably, are: oxetan-3-yL pyrrolidin-2-yl, pyrrolidin-3- yl,
- a nitrogen heterocyclyl ring is a heterocycle which has only nitrogen atoms as heteroatoms.
- Heteroaryl stands for an aromatic, mono- or bicyclic radical with 5 to 10 ring atoms and up to 5 heteroatoms from the series S, O and / or N. 5- to 6-membered heteroaryls with up to 4 heteroatoms are preferred.
- the heteroaryl radical can be bonded via a carbon or heteroatom.
- alkoxycarbonylamino represents an amino group with a straight-chain or branched alkoxycarbonyl substituent which preferably has 1 to 6 or 1 to 4 carbon atoms in the alkoxy radical and is linked via the carbonyl group.
- An alkoxycarbonylamino radical having 1 to 4 carbon atoms is preferred. Examples that may be mentioned are: methoxycarbonyl-a ino, emoxycarbonylamino, n-propoxycarbonylamino and t-butoxycarbonylamino.
- Carbonyl represents a -C (O) group. Accordingly, arylcarbonyl, heterocyclocarbonyl and heteroarylcarbonyl on the carbonyl group are substituted with the corresponding radicals, ie aryl, heterocyclyl etc.
- Sulfonyl represents an -S (O) 2 group. Accordingly, alkylsulfonyl, arylsulfonyl, heterocyclylsulfonyl and heteroarylsulfonyl on the sulfonyl group are substituted with the corresponding radicals, ie alkyl, aryl etc.
- Aminosulfonyl represents an -S (O) 2 NH 2 group. Accordingly, alkylaminosulfonyl, dialkylaminosulfonyl, arylaminosulfonyl, heterocyclylaminosulfonyl and heteroarylaminosulfonyl on the amino group are substituted with the corresponding radicals, ie alkyl, aryl etc.
- Halogen in the context of the invention includes fluorine, chlorine, bromine and iodine.
- Fluorine or chlorine are preferred.
- the side group of an amino acid is understood to mean that organic residue of an amino acid molecule which is bonded to the ⁇ -carbon atom of the amino acid.
- the residues of naturally occurring ⁇ -amino acids in the L or in the D configuration are preferred, in particular naturally occurring ⁇ -amino acids in the natural L configuration.
- Indolyl methyl group (tryptophan), a benzyl group (phenylalanine), a
- Methylthioethyl group (Met onin), hydroxymethyl (serine), p-hydroxybenzyl
- Carbonylbound amino acid residue stands for an amino acid residue which is bonded via the carbonyl group of the arm ⁇ oleic acid function.
- ⁇ -amino acids in the L or D configuration in particular naturally occurring ⁇ -amino acids in the natural L configuration, for example glycine, L-alanine and L-proline.
- Hydroxyfunction-bound amino acid residue stands for an amino acid residue which is bound via a hyroxy function of the amino acid. These include, for example, serine (-OCH (NH 2 ) COOH) or threonine (-OCH (CH 3 ) CH (NH 2 ) COOH. Preferred are ⁇ -amino acids in the L or in the D configuration, in particular naturally occurring ⁇ Amino acids in the natural L configuration, for example serine or threonine.
- amino protective groups are understood to be those organic radicals with which amino groups are temporarily against
- Oxycarbonyl derivatives such as carbamates and in particular the following groups are preferred: benzyloxycarbonyl, 4-bromo-benzyloxycarbonyl, 2-chlorobenzyloxycarbonyl, 3-chlorobenzyloxycarbonyl, dichlorobenzyloxycarbonyl, 3,4-dimethoxybenzyloxycarbonyl, 3,5-dimethoxybenzyloxycarbonyl, 2,4-dimethoxy - benzyloxycarbonyl, 4-methoxybenzyloxycarbonyl, 4-nitrobenzyloxycarbonyl, 2-nitrobenzyloxycarbonyl, 2-nitto-4,5-dimethoxybenzyloxycarbonyl, 3,4,5-tri-methoxybenzyloxycarbonyl, methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, Isopropoxycarbonyl, butoxycarbonyl, isobutoxycarbonyl, tert-butoxycarbonyl, pentoxycarbonyl, is
- a symbol * on a bond means a chhality center.
- R 1 is hydrogen, alkyl, aryl, heteroaryl, heterocyclyl, alkylcarbonyl,
- R 1 can be substituted, apart from hydrogen, with 0, 1, 2 or 3 substituents R 1 "1 , the substituents R 1" 1 being selected independently of one another from the group consisting of halogen, alkyl, trifluoromethyl, trifluoromethoxy, nitro, cyano, Amino, alkylamino, dialkylamino, cycloalkyl, aryl, heteroaryl, heterocyclyl, hydroxy, alkoxy and carboxyl,
- R 2 is hydrogen or alkyl
- alkyl can be substituted with 0, 1, 2 or 3 substituents R 2 "1 , where the substituents R 2" 1 are independently selected from the
- R 1 and R 2 together with the nitrogen atom to which they are attached form a heterocycle which can be substituted by 0, 1 or 2 substituents R 1 "2 , the substituents R 1" 2 being selected independently of one another from the group consisting of halogen, trifluoromethyl, amino, allylamino, dialkylamino, cycloalkyl, aryl, heteroaryl, heterocyclyl, hydroxy, alkoxy, carboxyl, alkoxycarbonyl and aminocarbonyl,
- R 3 is hydrogen, alkyl or the side group of an amino acid, in which alkyl can be substituted with 0, 1, 2 or 3 substituents R " , the substituents R " being selected independently of one another from the group consisting of trifluoromethyl, nitro, amino, Al lamino, dialkylamino, cycloalkyl, aryl, heteroaryl, heterocyclyl, hydroxy, alkoxy,
- cycloalkyl, aryl, heteroaryl and heterocyclyl can be substituted with 0, 1 or 2 substituents R 3 "2 , the substituents R 3" 2 being selected independently of one another from the group consisting of halogen, alkyl, trifluoromethyl and amino,
- Heteroarylcarbonyl, heterocyclylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, arylaminocarbonyl, alkylsulfonyl, arylsulfonyl, heterocyclylsulfonyl or heteroarylsulfonyl may be substituted,
- R is hydrogen or -Ce-alkyl
- R 4 is hydrogen, Ci-C ⁇ -alkyl or C 3 -C 8 cycloalkyl
- R 5 is alkyl, cycloalkyl, aryl, heteroaryl, heterocyclyl or a hydroxy-functional amino acid residue, where R 5 can be substituted with 0, 1, 2 or 3 substituents R " , the substituents R " being selected independently of one another from the group consisting of halogen, alkyl, trifluoromethyl, trifluoromethoxy, cyano, amino, alkylamino, dialkylamino, cycloalkyl, aryl, heteroaryl, heterocyclyl, hydroxy, alkoxy, carboxyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl and dialkylaminocarbonyl,
- R is hydrogen, Ci-C ö alkyl or C 3 -C 8 cycloalkyl
- R 7 is hydrogen or -CC 6 alkyl
- R is hydrogen or Ci-C 6 alkyl.
- R 1 is hydrogen, alkyl, alkylcarbonyl, arylcarbonyl, heterocyclylcarbonyl, heteroarylcarbonyl, alkoxycarbonyl or a carbonyl-bonded
- R 1 can be substituted with 0, 1 or 2 substituents R 1 "1 , wherein the
- Substituents R 1 "1 are independently selected from the group consisting of halogen, trifluoromethyl, amino, alkylamino, dialkyl a ino, phenyl, 5- to 6-membered heteroaryl, 5- to 6-membered heterocyclyl, hydroxy and alkoxy,
- R 2 is hydrogen or methyl
- R 3 is equal to aminocarbonylmethyl, 3-aminopropyl, 2-hydroxy-3-aminopropyl, 3-guanidinopropyl, 2-aminocarbonylethyl, 2-hydroxycarbonylethyl, 4-amino-butyl, hydroxymethyl, 2-hydroxyethyl or 4-amino-3-hydroxybutane-l -yl is,
- R 3 is hydrogen
- R 4 is hydrogen or methyl
- R 5 is alkyl, C 3 -C 6 -cycloalkyl, phenyl, 5- to 6-membered heteroaryl, 5- to 6-membered heterocyclyl or a hychoxy-functional amino acid residue,
- R 5 is alkyl, C 3 -C 6 cycloalkyl or 5- to 6-membered heterocyclyl, this can be substituted with 0, 1 or 2 substituents R 5 "2 , where the substituents R 5" 2 are selected independently of one another from the group consisting of alkyl, trifluoromethyl, amino,
- R 5 is phenyl or 5- to 6-membered heteroaryl
- this can be substituted with 0, 1 or 2 substituents R " , the substituents R 5" 3 being selected independently of one another from the group consisting of Halogen, trifluoromethyl, amino, alkylamino, dialkylamino, C 3 -C 6 -cycloalkyl, 5- to 6-membered heteroaryl, 5- to 6-membered heterocyclyl, hydroxy, alkoxy, carboxyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl and dialkylaminocarbonyl,
- R 6 is hydrogen or methyl
- R 7 is hydrogen
- R is hydrogen
- R 1 is hydrogen, alkyl or alkylcarbonyl
- R 2 is hydrogen
- R 3 is alkyl or the side group of an amino acid, in which alkyl can be substituted with 0, 1, 2 or 3 substituents R 3- " 1, where the Substituents R 3 "1 are independently selected from the
- cycloalkyl, aryl, heteroaryl and heterocyclyl can be substituted with 0, 1 or 2 substituents R 3 "2 , the substituents R 3" 2 being selected independently of one another from the group consisting of halogen, alkyl, trifluoromethyl and amino,
- R 3 ' is hydrogen, Ci-C ⁇ - alkyl or C 3 -C 8 -cycloalkyl
- R 4 is hydrogen, -C ⁇ - alkyl or C 3 -C 8 cycloalkyl
- R 5 is alkyl, cycloalkyl, aryl, heteroaryl or heterocyclyl, where R 5 can be substituted with 0, 1, 2 or 3 substituents R 5 "1 , where the
- Substituents R 5 "1 are independently selected from the group consisting of halogen, alkyl, trifluoromethyl, trifluoromethoxy, cyano, amino, alkylamino, dialkylamino, cycloalkyl, aryl, heteroaryl, heterocyclyl, hydroxy, alkoxy, carboxyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl and dialkylaminocarbonyl,
- alkylamino and diallamino can be substituted with 0, 1 or 2 substituents R 5 "2 , the substituents R 5" 2 being selected independently of one another from the group consisting of hydroxyl, amino, alkoxy, alkylamino and dialkylamino, R 6 is hydrogen,
- R 7 is hydrogen, -CC 6 alkyl, alkylcarbonyl or C 3 -C 8 cycloalkyl
- R is hydrogen
- R 1 is hydrogen
- R 2 is hydrogen
- R 3 is alkyl or the side group of an amino acid, in which alkyl can be substituted with 0, 1, 2 or 3 substituents R 3 "1 , the substituents R 3" 1 being selected independently of one another from the group consisting of amino, al lamino , Dialkylamino, cycloalkyl, heteroaryl, heterocyclyl, hydroxy, alkoxy, carboxyl, alkoxycarbonyl,
- cycloalkyl, heteroaryl and heterocyclyl can be substituted with 0, 1 or 2 substituents R 3 "2 , the substituents R 3" 2 being selected independently of one another from the group consisting of alkyl and amino,
- R is hydrogen
- R 4 is hydrogen, CC alkyl or C 3 -C 8 cycloalkyl
- R 5 is alkyl, cycloalkyl, aryl, heteroaryl or heterocyclyl, where R 5 can be substituted with 0, 1, 2 or 3 substituents R 5 "1 , the substituents R 5" 1 being selected independently of one another from the group consisting of Alkyl, cyano, amino, alkylamino, dialkylamino,
- alkylamino and dialkylamino can be substituted with 0, 1 or 2
- Substituents R 5 "2 where the substituents R 5" 2 are selected independently of one another from the group consisting of hydroxyl, amino, alkoxy, alkylamino and dialkylamino,
- R 6 is hydrogen
- R 7 is hydrogen
- R is hydrogen
- R 1 is hydrogen
- R 2 is hydrogen
- R 3 is aminocarbonylmethyl, 3-aminoprop-l-yl, 2-hydroxy-3-aminoprop-l-yl, l-hydroxy-3-aminoprop-l-yl, 3-guanidinoprop-l-yl, 2-aminocarbonyl- is ethyl, 2-hydroxycarbonylethyl, 4-amino-but-l-yl, hydroxymethyl, 2-hydroxyethyl, 2-aminoethyl, 4-amino-3-hydroxybut-l-yl or (1-piperidin-3-yl) methyl,
- R is hydrogen
- R 4 is hydrogen, methyl, ethyl, isopropyl or cyclopropyl
- R 5 is alkyl or C 3 -C 6 cycloalkyl, where R 5 can be substituted with 0, 1, 2 or 3 substituents R 5 "1 , the substituents R 5" 1 being selected independently of one another from the group consisting of Alkyl, amino, alkylamino, dialkylamino, cycloalkyl, hydroxy, alkoxy, carboxyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl and dialkylaminocarbonyl,
- alkylamino and dialkylamino can be substituted with 0, 1 or 2
- R 6 is hydrogen
- R 7 is hydrogen
- R 8 is hydrogen
- R 1 is hydrogen
- R 2 is hydrogen
- R 3 is 3-amino -rop-l-yl or 2-hydroxy-3-aminoprop-l-yl
- R 3 is hydrogen
- R 4 is hydrogen or methyl
- R 5 is C 1 -C alkyl, where alkyl can be substituted with 0, 1 or 2 substituents independently selected from the group consisting of amino, hydroxy and carboxyl,
- R 6 is hydrogen
- R 7 is hydrogen
- R is hydrogen
- R 5 is alkyl or C 3 -C 6 cycloalkyl, where R 5 can be substituted by 0,
- substituents R 5 "1 , the substituents R 5" 1 being selected independently of one another from the group consisting of alkyl, amino, alkylamino, dialkylamino, cycloalkyl, hydroxy, alkoxy, carboxyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl and dialkylaminocarbonyl .
- alkylamino and dialkylamino can be substituted with 0, 1 or 2 substituents R 5 "2 , the substituents R 5" 2 being selected independently of one another from the group consisting of hydroxyl and amino.
- R 5 is C 1 -C 4 -alkyl, where alkyl can be substituted with 0, 1 or 2 substituents independently selected from the group consisting of amino, hydroxy and carboxyl.
- the invention further relates to a process for the preparation of the compounds of the formula (I) or their salts, compounds of the formula
- R 1 to R 4 and R 6 to R 8 have the meaning given above, where the
- R has the meaning given above, are implemented.
- reactive functionalities for example free ammo functions or hydroxy functions
- reactive functionalities for example free ammo functions or hydroxy functions
- compounds of the formula (H) are blocked by protective groups before the reaction of compounds of the formula (JJ) with compounds of the formula (TU).
- Acid-labile protecting groups on R 1 (or R 2 ) or as substituents in are preferred the radicals R 3 and R 3 ' , particularly preferred is Boc.
- Reactive functionalities in R 5 of compounds of the formula (UT) are already incorporated in the synthesis with protection.
- Preference is given to acid-labile protective groups (eg Boc) or hydrogen-ololytically cleavable protective groups (eg benzyl or benzyloxycarbonyl).
- the protective groups can be removed by deprotection reactions. This is done according to standard protective group chemistry. Deprotection reactions under acidic conditions are preferred.
- R> 2 in compounds of formula (I) is a selectively removable protective group, can be functionalized after deprotection (for example after hydrogenolysis in the case R 2 is Z) the exposed amino function (R> 2 is hydrogen) with the desired substituent R 2 become.
- carbodiimides such as e.g. NN'-diethyl, NN-dipropyl, NN'-diisopropyl (DIC) and NN'-dicyclohexylcarbodiimide, ⁇ - (3-dimethylaminoisopropyl) - N'-ethylcarbodiimide hydrochloride (EDC), N-cyclohexylcarbodiimide-N 'propyloxy methyl polystyrene (PS carbodiimide) or carbonyl compounds such as carbonyldimidazole are suitable. If appropriate, the activation takes place in the presence of 4-dimethylaminopyridine.
- carbodiimides such as e.g. NN'-diethyl, NN-dipropyl, NN'-diisopropyl (DIC) and NN'-dicyclohexylcarbodiimide, ⁇ - (3-dimethylaminoisoprop
- Inert organic solvents which do not change under the reaction conditions are suitable as solvents. These include halogenated hydrocarbons such as dichloromethane or trichloromethane, hydrocarbons such as benzene, toluene,
- Acetonitrile, tetrahydrofuran, dioxane or dimethylformamide It is also possible to use mixtures of the solvents.
- Anhydrous dichloromethane, dimethylformamide and acetonitrile are particularly preferred.
- Reactions with activation by EDC or DIC in absolute acetonitrile, dimethylformamide or dichloromethane at low temperature (-10 ° C.) in the presence of 4-dimethylamino ⁇ yridine are preferred.
- the invention further relates to an alternative process for the preparation of the compounds of the formula (I) or their salts, characterized in that compounds of the formula (II) can also be reacted with compounds of the formula (HI) with acid catalysis.
- the compounds of the formula (IT) are mixed with an excess of anhydrous alcohol HO-R 5 , if appropriate in the presence of an inert solvent, and at room temperature or up to
- the boiling point of the solution is mixed with an acid (preferably with a mineral acid) or acid-releasing reagents (e.g. thionyl chloride) and converted into compounds of the formula (I).
- an acid preferably with a mineral acid
- acid-releasing reagents e.g. thionyl chloride
- halogenated hydrocarbons such as dichloromethane or trichloromethane
- hydrocarbons such as benzene, toluene, tetahydrofuran and dioxane. It is also possible to use mixtures of the solvents.
- ester cleavage is preferably carried out with hydrogen in the presence of palladium on carbon.
- Organic solvents which do not change under the reaction conditions are suitable as solvents. These include halogenated hydrocarbons such as dichloromethane or trichloromethane, hydrocarbons such as tetrahydrofuran, dioxane, dimethylformamide, acetic acid, mixtures of acetic acid and water or alcohols (preferably methanol, ethanol and isopropanol), optionally in the presence of one or more acid equivalents. It is also possible to use mixtures of the solvents. Mixtures of acetic acid, water and ethanol or THF are particularly preferred.
- the ester cleavage is preferably carried out in the presence of palladium (0) catalysts by standard methods of protective group chemistry.
- Degassed (oxygen-free) organic solvents that do not change under the reaction conditions are suitable as solvents. These include halogenated hydrocarbons such as dichloromethane or trichloromethane, hydrocarbons such as tetrahydrofuran, dioxane and dimethylformamide, optionally in the presence of one or more acid equivalents.
- esters R 5 equal to benzyl, alkyl
- esters can also be split into the corresponding carboxylic acids by basic hydrolysis.
- Aqueous lithium or sodium hydroxide are preferably used as bases.
- Suitable solvents are organic solvents which are partially or unlimitedly miscible with water. These include alcohols (methanol and ethanol are preferred), tetrahydrofuran, dioxane and dimethylformamide. It is also possible to use mixtures of the solvents. Methanol, tetrahydrofuran and dimethylformamide are particularly preferred.
- the invention further relates to an alternative process for the preparation of the compounds of the formulas (I) and (Ia) or their salts, characterized in that compounds of the formula
- R> 1 - b, i-.s R have the meaning given above, where these are optionally present in activated form, are cyclized under peptide coupling conditions.
- R 1 to R 8 have the meaning given above
- R is an amine protecting group (preferably Boc),
- carbodiimides such as NN'-diethyl, NN'-dipropyl, NN'-diisopropyl, NN-dicyclohexylcarbodiimide, N- (3-dimethylammoisopropyl) -N'-ylcarbom " imide -
- EDC Hydrochloride
- PFP pentafluorophenol
- PS-carbodiimide N-cyclohexylcarbodiimide-N'-propyloxymethyl-polystyrene
- carbonyl compounds such as carbonyldiimidazole
- 1,2-oxazolium compounds such as 2-ethyl-5-phenyl-l, 2-oxazohum-3-sulfate or 2-tert-butyl-5-methyl-isoxazolium perchlorate
- acylamino compounds such as 2-ethoxy-l-ethoxycarbonyl-l, 2 -dihydroquinoline, or propanephosphonic anhydride, or isobutyl chloroformate, or bis (2-oxo-3-oxazolidinyl) phosphoryl chloride or benzotriazolyloxy-tri (dimethylammo) phosphom ⁇ x ⁇ hexafluorophosphate, or O- (benz
- N-tefra-methyluromum hexafluorophosphate HBTU
- 2- (2-oxo-l- (2H) -pyridyl) -l, l, 3,3-teframethylx ⁇ roniumtetrafluoroborat TPTU
- Bases are, for example, alkali carbonates, e.g. Sodium or potassium carbonate, or hydrogen carbonate, or preferably organic bases such as trialkylamines e.g. Triethylamine, N-methylmorpholine, N-methylpiperidine, 4-dimethylaminopyridine or diisopropylethylamine.
- alkali carbonates e.g. Sodium or potassium carbonate
- hydrogen carbonate e.g. Sodium or potassium carbonate
- organic bases e.g. Triethylamine, N-methylmorpholine, N-methylpiperidine, 4-dimethylaminopyridine or diisopropylethylamine.
- Inert organic solvents which do not change under the reaction conditions are suitable as solvents. These include halogenated hydrocarbons such as dichloromethane or trichloromethane, hydrocarbons such as benzene, toluene,
- Tetrahydrofuran, dioxane, dimethylformamide or acetonitrile It is also possible to use mixtures of the solvents. Dichloromethane and dimethylformamide are particularly preferred.
- the compounds of the formula (I) according to the invention can be prepared according to the following synthesis scheme.
- R 9 is a silyl protective group, in particular 2- (trimethylsilyl) ethyl, after the protective group on R 10 has been removed, be reacted with fluoride, in particular with tetrabutylammonium fluoride.
- solvents which do not change under the reaction conditions are suitable as solvents.
- solvents include halogenated hydrocarbons such as dichloromethane, hydrocarbons such as benzene, toluene, tetrahydrofuran, dioxane and dimethylformamide. It is also possible to use mixtures of the solvents.
- Preferred solvents are tetrahydrofuran and dimethylformamide.
- R 9 is a silyl protective group, in particular 2- (trimethylsilyl) ethyl,
- Carbodiimides such as e.g. NN'-diethyl-, NN'-dipropyl-, NN'-diisopropyl-, NN- dicyclohexylcarbodiimide, N- (3-dimethylaminoisopropyl) -N'-ethylcarbodiimide-
- EDC Hydrochloride
- PFP pentafluorophenol
- PS-carbodiimide N-cyclohexylcarbodiimide-N'-propyloxymethyl-polystyrene
- carbonyl compounds such as carbonyldiimidazole
- 1,2-oxazolium compounds such as 2-ethyl-5-phenyl- l, 2-oxazolium-3-sulfate or 2-tert-butyl-5-methyl-isoxazolium perchlorate
- acylamino compounds such as 2-ethoxy-l-ethoxycarbonyl-l, 2-dihydroquinoline, or propanephosphonic anhydride, or isobutyl- chloroformate, or bis (2-oxo-3-oxazolidinyl) phosphoryl chloride or benzo-1riazolyloxy-tri (dimethylanxmo) phosphomumhexafluorophosphate, or O-
- Bases are, for example, alkali carbonates, such as sodium or potassium carbonate, or hydrogen carbonate, or preferably organic bases, such as trial lamines, for example triethylamine, N-methylmorpholine, N-memylpiperidine, 4-dimethylaminopyridine or diisopropylethylamine.
- alkali carbonates such as sodium or potassium carbonate
- hydrogen carbonate or preferably organic bases, such as trial lamines, for example triethylamine, N-methylmorpholine, N-memylpiperidine, 4-dimethylaminopyridine or diisopropylethylamine.
- Inert organic solvents which do not change under the reaction conditions are suitable as solvents. These include halogenated hydrocarbons such as dichloromethane or trichloromethane, hydrocarbons such as benzene, toluene, acetonitrile, tetrahydrofuran, dioxane or dimethylformamide. It is also possible to use mixtures of the solvents. Anhydrous dichloromethane and dimethylformamide are particularly preferred.
- reaction in the presence of HATU and NN-diisopropylethylamine is particularly preferred.
- R> 9 is a silyl protective group
- R 11 is an amino protective group, in particular Boc,
- R ⁇ can be produced by deprotection at R ⁇ . This is done according to standard methods of protecting group chemistry, in the case of R 11 Boc preferably with hydrogen chloride in dioxane.
- R 4 , R 5 and R 7 have the meaning given above and
- R 11 is an amino protective group (preferably Boc),
- R 1 , R 2 and R s have the meaning given above and
- R 9 is a silyl protective group, in particular 2- (trimethylsilyl) ethyl,
- Catalysts and a base preferably in the presence of bis (diphenylphosphino) ferrocene palladium (TI) chloride and cesium carbonate
- Inert organic solvents which do not change under the reaction conditions are suitable as solvents. These include hydrocarbons such as benzene, toluene, tetrahydrofuran, dioxane, dimethylformamide and dimethyl sulfoxide. It is also possible to use mixtures of the solvents. Dimethylformamide and dimethyl sulfoxide are particularly preferred.
- R 4 , R 5 and R 7 have the meaning given above and
- R .11 is an amino protective group (preferably Boc),
- R 4 and R 7 have the meaning given above and
- R 11 is an amino protective group (preferably Boc),
- carbodiimides such as e.g. NN'-diethyl, NN'-dipropyl, NN'-diisopropyl, NN-
- EDC N-cyclohexylcarbodiimide-N'-propyloxymethyl-polystyrene
- PS-carbodiimide N-cyclohexylcarbodiimide-N'-propyloxymethyl-polystyrene
- carbonyl compounds such as carbonyldiimidazole are suitable.
- Do not change reaction conditions include halogenated hydrocarbons such as dichloromethane or trichloromethane, hydrocarbons such as benzene, toluene, acetonitrile, tetrahydrofuran, dioxane or dimethylformamide. It is also possible to use mixtures of the solvents. Anhydrous dichloromethane and acetonitrile are particularly preferred.
- R 9 -OH preferably 2-trimethylsilylethanol
- carbodiimides such as e.g. NN'-diethyl, NN'-dipropyl, NN'-diisopropyl, NN'-dicyclohexylcarbodiimide, N- (3-dimemylamoisopropyl) -N'-emylcarbodiimide hydrochloride (EDC) N-cyclohexylcarbodiine and N-propyloxymethyl polystyrene (PS-
- EDC N-dimemylamoisopropyl
- Carbodiimide or carbonyl compounds such as carbonyldiimidazole.
- Inert organic solvents which do not change under the reaction conditions are suitable as solvents. These include halogenated hydrocarbons such as dichloromethane or trichloromethane, hydrocarbons such as benzene, toluene, acetonitrile, tetrahydrofuran, dioxane or dimethylformamide. It is also possible to use mixtures of the solvents. Anhydrous dichloromethane and acetonitrile are particularly preferred.
- R 1 and R 8 have the meaning given above and
- R .13 is an amino protecting group, in particular Boc,
- R 13 deprotected in the first stage on R 13 . This is done according to standard methods of protecting group chemistry, in the case of R 13 equal to Boc, preferably with anhydrous hydrogen chloride in dioxane or with trifluoroacetic acid in dichloromethane in the presence of small amounts of water.
- R 13 equal to Boc
- amine can optionally be in the form of a salt, preferably hydrochloride or trifluoroacetate,
- R 2 has the meaning given above and X represents a leaving group, in the presence of a base in inert solvents, if appropriate in the presence of potassium iodide, preferably in a temperature range from 0 ° C. Room temperature until the solvents reflux at normal pressure.
- Mesylate, tosylate, succinate or halogen are preferred for X, chlorine, bromine or iodine being preferred for halogen.
- Bases are, for example, alkali carbonates, e.g. Sodium or potassium carbonate, or bicarbonate, or organic bases such as trialkylamines e.g. Triethylamine, N-methylpiperidine, 4-dimethylaminopyridine or diisopropylethylamine.
- alkali carbonates e.g. Sodium or potassium carbonate, or bicarbonate
- organic bases such as trialkylamines e.g. Triethylamine, N-methylpiperidine, 4-dimethylaminopyridine or diisopropylethylamine.
- Inert organic solvents which do not change under the reaction conditions are suitable as solvents. These include halogenated hydrocarbons such as dichloromethane or trichloromethane, hydrocarbons such as benzene, toluene, acetonitrile, tetrahydrofuran, dioxane, acetone or dimethylformamide. It is also possible to use mixtures of the solvents. Dimethylformamide and dichloromethane are particularly preferred.
- R 2 can optionally be a protective group (eg Z, ie benzyloxycarbonyl or aloe, ie
- the compounds of formula (Va) can be prepared by using compounds of formula (Va).
- R 4 , R 5 and R have the meaning given above and
- R 11 is an amino protective group (preferably Boc),
- R 1 , R 2 and R 8 have the meaning given above and
- R 9 is a silyl protective group, in particular 2- (trimethylsilyl) ethyl,
- the reaction known as the Suzuki reaction (Synlett 1992, 207-210; Chem. Rev. 1995, 95, 2457-2483), takes place in the presence of palladium Catalysts and a base, preferably in the presence of bis (diphenylphosphino) ferrocene palladium (II) chloride and cesium carbonate.
- solvents which do not change under the reaction conditions are suitable as solvents.
- hydrocarbons such as
- Benzene, toluene, tetxahydrofuran, dioxane, dimethylformamide and dimethyl sulfoxide It is also possible to use mixtures of the solvents. Dimethylformamide and dimethyl sulfoxide are particularly preferred.
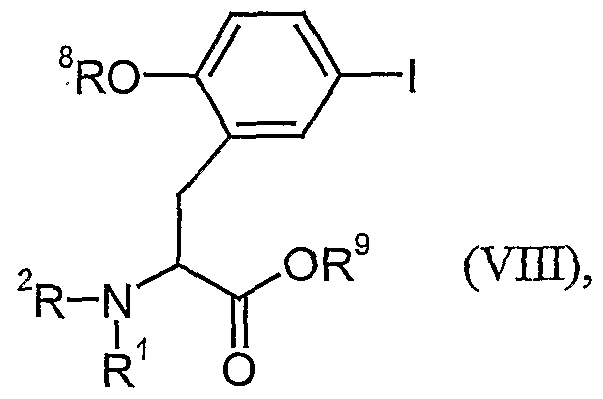
- the compounds of the formula (Villa) can be obtained from the compounds of the formula
- the enantiomerically pure compounds of the formulas (IX) and (IXb) are known or can be obtained from racemic precursors analogously to known processes such as crystallization with chiral amine bases or by chromatography on chiral, stationary phases.
- R 4 and R 7 and R 1 and R 8 have the meaning given above, R 11 and R 13 are an amino protecting group and
- R 12 is alkyl (particularly preferably ethyl),
- This reaction preferably takes place in a basic medium in a water-ethanol mixture.
- R 7 and R 8 have the meaning given above
- R> 4 - and A ⁇ R> l have the meaning given above, 1 1 1 ⁇
- R and R are an amino protecting group
- R 12 is alkyl (particularly preferably ethyl),
- This reaction preferably takes place with alkali alcoholate in lower aliphatic alcohols, especially with sodium ethylate in ethanol.
- R 7 and R 8 have the meaning given above
- reaction preferably takes place in toluene.
- X represents a leaving group, are reacted in inert solvents, if appropriate in the presence of a base, if appropriate in the presence of potassium iodide, preferably in a temperature range from room temperature to the reflux of the solvents at atmospheric pressure.
- a base if appropriate in the presence of potassium iodide
- X is preferred for X, and halogen is preferred
- Chlorine, bromine or iodine are preferred.
- Inert solvents are, for example, halogenated hydrocarbons such as methylene chloride, trichloromethane or 1,2-dichloroethane, ethers such as dioxane, tetrahydrofuran or 1,2-dimethoxyethane, or other solvents such as acetone, dimethylformamide,
- Dimethylformamide is preferred.
- Bases are, for example, alkali carbonates such as cesium carbonate, sodium or potassium carbonate, or sodium or potassium methoxide, or sodium or potassium ethanolate or potassium tert-butoxide, or amides such as sodium amide, lithium bis (trimethylsilyl) amide or lithium diisopropylamide, or organometallic compounds such as butyllithium or phenyllithium, tertiary amine bases such as triethylamine or diisopropylethylamine, or other bases such as sodium hydride, DBU, preferably potassium tert-butoxide, cesium carbonate, DBU, sodium hydride, potassium carbonate or sodium carbonate. Potassium carbonate is preferred.
- alkali carbonates such as cesium carbonate, sodium or potassium carbonate, or sodium or potassium methoxide, or sodium or potassium ethanolate or potassium tert-butoxide
- amides such as sodium amide, lithium bis (trimethylsilyl) amide or lithium di
- the compounds of the invention can because of their phaimacological
- Gram-positive cocci e.g. Staphylococci (Staph. Aureus, Staph. Epidermidis) and streptococci (Strept. Agalactiae, Strept. Faecalis, Strept. Pneumoniae, Strept. Pyogenes); gram-negative cocci (neisseria gonorrhoeae) as well as gram-negative rods such as enterobacteria, e.g. Escherichia coli, Haemophilus influenzae, Citrobacter (Citrob. Freundii, Citrob. Divernis), Salmonella and Shigella; further Klebsielle (Klebs. pneumoniae, Klebs.
- Pr. Vulgaris Providencia, Yersinia, and the genus Acinetobacter.
- the antibacterial spectrum also includes the genus Pseudomonas (Ps. aeruginosa, Ps. maltophilia) and strictly anaerobic bacteria such as Bacteroides fragilis, representatives of the genus Peptococcus, Peptostreptococcus and the genus Clostridium; also mycoplasma (M. pneumoniae, M. hominis, M. urealyticum) and mycobacteria, eg Mycobacterium tuberculosis.
- Pseudomonas Ps. aeruginosa, Ps. maltophilia
- strictly anaerobic bacteria such as Bacteroides fragilis, representatives of the genus Peptococcus, Peptostreptococcus and the genus Clostridium
- mycoplasma M. pneumoniae, M. homini
- Infectious diseases in humans such as B. septic infections, bone and joint infections, skin infections, postoperative wound infections, abscesses, phlegmon, wound infections, infected burns, burns, infections in the mouth area, infections after dental surgery, septic arthritis, mastitis, tonsillitis, genital infections and eye infections.
- bacterial infections can also be treated in other species. Examples include:
- Pig coli diarrhea, enterotoxemia, sepsis, dysentery, salmonellosis, metritis-mastitis-agalaktiae syndrome, mastitis;
- Ruminants (cattle, sheep, goats): diarrhea, sepsis, bronchopneumonia,
- Horse bronchopneumonia, foal paralysis, puerperal and postpuerperal infections, salmonellosis; Dog and cat: bronchopneumonia, diarrhea, dermatitis, otitis, urinary tract infections, prostatitis;
- Poultry (chicken, turkey, quail, pigeon, ornamental birds and others): mycoplasmosis, E. coh infections, chronic respiratory diseases, salmonellosis, pasteurellosis,
- Bacterial diseases in the rearing and keeping of farmed and ornamental fish can also be treated, the antibacterial spectrum extending beyond the previously mentioned pathogens to other pathogens such as e.g. Pasteurella, brucella,
- Campylobacter Listeria, Erysipelothris, Corynebacteria, Borellia, Treponema, Nocardia, Rikettsie, Yersinia.
- the present invention further relates to compounds of the general formula (I) for combating diseases, in particular bacterial diseases,
- the present invention further relates to a method for combating bacterial infections in humans and animals by administering an antibacterially effective amount of at least one compound of the formula (I).
- the present invention further relates to medicaments which contain at least one compound according to the invention, preferably together with one or more pharmacologically acceptable auxiliaries or excipients, and to their use for the purposes mentioned above.
- the active substance can act systemically and / or locally.
- it can be applied in a suitable manner, such as orally, parenterally, pulmonally, nasally, sublingual, lingual, buccal, rectal, transdermal, conjunctival, otic or as an implant.
- the active ingredient can be administered in suitable administration forms for these administration routes.
- Non-coated and coated tablets e.g. tablets coated with enteric coatings or film-coated tablets
- capsules coated tablets, granules, pellets, powder
- Emulsions, suspensions, solutions and aerosols Emulsions, suspensions, solutions and aerosols.
- Parenteral administration can be done by bypassing a resorption step (intravenous, intraarterial, intracardial, intraspinal or intralumbal) or by switching on absorption (intramuscular, subcutaneous, intracutaneous, percutaneous, or intraperitoneal).
- Suitable forms of application for parenteral administration include: Injection and auxiliary preparations in the form of solutions, suspensions, emulsions, lyophilisates and sterile powders.
- Inhalation drug forms e.g.
- the active compounds can be converted into the administration forms mentioned in a manner known per se. This is done using inert, non-toxic, pharmaceutically suitable excipients. These include carriers (e.g. microcrystalline cellulose), solvents (e.g. liquid polyethylene glycols), emulsifiers (e.g. sodium dodecyl sulfate), dispersants (e.g. polyvinyl pyrrolidone), synthetic and natural biopolymers (e.g. albumin), stabilizers (e.g. antioxidants such as ascorbic acid), dyes (e.g. inorganic pigments such as iron oxides) or taste and / or odor correctors.
- carriers e.g. microcrystalline cellulose
- solvents e.g. liquid polyethylene glycols
- emulsifiers e.g. sodium dodecyl sulfate
- dispersants e.g. polyvinyl pyrrolidone
- synthetic and natural biopolymers e.g
- Dilution ratios and concentration data for liquid / liquid solutions each relate to the volume.
- Method 1 column: Kromasil C18, LR temperature: 30 ° C; Flow: 0.75 ml / min; Eluent A: 0.01 M HClO 4 , eluent B: acetonitrile, gradient: ⁇ 0.5 min 98% A ⁇ 4.5 min 10% A ⁇ 6.5 min 10% A.
- Method 2 HPLC: column: Kromasil C18 60 * 2 mm, LR temperature: 30 ° C; Flow: 0.75 ml / min, eluent A: 0.01 MH 3 PO 4 , eluent B: acetonitrile, gradient: -> 0.5 min 90% A -> 4.5 min 10% A - »6.5 min 10% A.
- Method 5 Micromass Quattro LCZ instrument; Syrnmetry C18 column, 50 mm x 2.1 mm, 3.5 ⁇ m; Temperature: 40 ° C; Flow: 0.5 ml / min; Eluent A: acetonitrile + 0.1% formic acid, Eluent B: water + 0.1% formic acid, gradient:
- Method 8 HPLC: column: 250 * 4 mm, Kromasil 100, C-18, 5 ⁇ m; Temperature: 40 ° C; Flow: 1 ml / min; Eluent: acetonitrile 15% and 0.2% perchloric acid 85%; UV detection: 210 ⁇ m.
- Method 9 Instrument: Waters Alliance 2790 LC; Column: Symmetry C18, 50 mm x 2.1 mm, 3.5 ⁇ m; Eluent A: water + 0.1% formic acid, eluent B: acetonitrile + 0.1% formic acid; Gradient: 0.0 min 5% B -> 5.0 min 10% B ->
- Method 12 (LC-MS): TSQ 7000, Finnigan MAT, Bremen; Column: Inertsil ODS3 50 mm x 2.1 mm, 3 ⁇ m; Temperature: 25 ° C; Flow: 0.5 ml / min; Eluent A: water + 0.05% formic acid, eluent B: acetonitrile + 0.05% formic acid, gradient: 0.0 min 15% B ⁇ 15 min ⁇ 100% B ⁇ 30 min 100% B.
- Method 13 7 Tesla Apex II with external electrospray ion source, Bruker Daltronics; Column: X-terra C18 50 mm x 2.1 mm, 2.5 ⁇ m; Temperature: 25 ° C; Flow: 0.5 ml / min; Eluent A: water + 0.1% formic acid, eluent B: acetonitrile + 0.1% formic acid, gradient: 0.0 min 5% B - 13 min ⁇ 100% B ⁇ 15 min 100% B.
- Method 14 (HPLC): Column: X-Terra TM from Waters, RP 8 , 5 ⁇ m, 3.9x150 mm; Start: 95% A, 5% B; 12 min: 5% A, 95% B. Eluent A: water + 0.01% trifluoroacetic acid; Eluent B: acetonitrile + 0.01% trifluoroacetic acid; Flow: 1.2 ml / min.
- Method 15 Device type MS: Micromass ZQ; Device type HPLC: Waters Alliance 2795; Column: Merck Chromolith SpeedROD RP-18e 50x4.6mm; Eluent A: water + 500 ⁇ l 50% formic acid / 1; Eluent B: acetonitrile + 500 ⁇ l 50% formic acid / 1; Gradient: 0.0 min 10% B ⁇ 3.0 min 95% B ⁇ 4.0 min 95% B; Oven: 35 ° C; Flow: 0.0 min 1.0 ml / min - 3.0 min 3.0 ml / min ⁇ 4.0 min 3.0 ml / min; UV-
- Method 16 Device type MS: Micromass ZQ; Device type HPLC: Waters Alliance 2795; Column: Merck Chromolith SpeedROD RP-18e 50x4.6mm; Eluent A: water + 500 ⁇ l 50% formic acid / 1; Eluent B: acetonitrile + 500 ⁇ l 50%
- Method 18 Device type MS: Micromass ZQ; Device type HPLC: Waters Alliance 2795; Column: Merck Chromolith SpeedROD RP-18e 50x4.6mm; Eluent A: water + 500 ⁇ l 50% formic acid / 1; Eluent B: acetonitrile + 500 ⁇ l 50% formic acid / 1; Gradient: 0.0 min 10% B- »3.0 min 95% B- 4.0 min 95% B; Oven:
- Method 20 Device type MS: Micromass ZQ; Device type HPLC: HP 1100 Series; UV DAD; Column: Grom-Sil 120 ODS-4 HE 50x2 mm, 3.0 ⁇ m; Eluent A: water + 500 ⁇ l 50% formic acid / 1, eluent B: acetonitrile + 500 ⁇ l 50% formic acid / 1; Gradient: 0.0 min 0% B - »2.9 min 70% B - ⁇ 3.1 min 90% B - 4.5 min 90% B; Oven: 50 ° C; Flow: 0.8 ml / min; UV detection: 210 mn.
- Method 21 Device type MS: Micromass ZQ; Device type HPLC: Waters Alliance 2795; Column: Phenomenex Synergi 2 ⁇ Hydro-RP Mercury 20x4 mm; Eluent A: 1 1 water + 0.5 ml 50% formic acid, eluent B: 1 1 acetonitrile + 0.5 ml 50% formic acid; Gradient: 0.0 min 90% A (flow: 1 ml / min) -> 2.5 min 30% A (flow: 2 ml / min) -> 3.0 min 5% A (flow: 2 ml / min) - 4.5 min 5% A (flow:
- Method 22 Device type MS: Micromass ZQ; Device type HPLC: HP 1100 Series; UV DAD; Column: Grom-Sil 120 ODS-4 HE 50x2 mm, 3.0 ⁇ m; Eluent A: water + 500 ⁇ l 50% formic acid / 1, eluent B: acetonitrile + 500 ⁇ l 50%
- Method 24 Device type MS: Micromass ZQ; Device type HPLC: Waters Alliance 2790; Column: Grom-Sil 120 ODS-4 HE 50x2 mm, 3.0 ⁇ m; Eluent A: water + 500 ⁇ l 50% formic acid; Eluent B: acetonitrile + 500 ⁇ l 50% formic acid / 1; Gradient: 0.0 min 5% B- 2.0 min 40% B- 4.5 min 90% B ⁇ 5.5 min 90% B; Oven: 45 ° C; Flow: 0.0 min 0.75 ml / min ⁇ 4.5 min 0.75 ml 5.5 min- 5.5 min
- Method 25 Instrument: HP 1100 with DAD detection; Column: Kromasil RP-18, 60 mm x 2 mm, 3.5 ⁇ m; Eluent A: 5 ml HClO 4 / l water, eluent B: acetonitrile; Gradient: 0 min 2% B, 0.5 min 2% B, 4.5 min 90% B, 15 min 90% B; Flow:
- Example 6A [(+/-) - 3- (2-benzyloxy-5-iodophenyl) -2 (S) -tert-butoxycarbonylamino-propionic acid] is based on a chiral stationary silica gel phase, based on the selector from poly (N-methacryloyl-L-leucine-dicyclopropyl-methylamide), separated with a mixture of z ' -hexane / ethyl acetate as the eluent.
- the first eluted enantiomer (98.9% ee) is clockwise in dichloromethane
- Method B A solution of 6.99 g (11.06 mmol) of 2 (S) -benzyloxycarbonylamino-3- (2-benzyl-oxy-5-iodophenyl) -propionic acid- (2-trimethylsilyl) -ethyl ester (Example 11 A) and 6.50 g (11.06 mmol) 3- [2-benzyloxy-5- (4,4,5,5-tetramethyl- [l, 3,2] dioxaborolan-2- yl) -phenyl] -2 (S) -tert-butoxycarbonylamino-propionic acid benzyl ester (Example 8A) in 40 ml DMF is degassed by passing argon through (approx.
- Example 20A 5,17-bis-benzyloxy-14 (S) -benzyloxycarbonylamino-ll (S) - (3-benzyloxycarbonylamino-2 (R) -hydroxy-propyl) -10,13-dioxo-9,12-diaza-tricyclo [14.3.1.1 2 ' 6 ] henicosa-l (19), 2,4,6 (21), 16 (20), 17-hexaen-8 (S) - benzyl carboxylate
- Example 16A The preparation is carried out analogously to Example 16A from 0.47 g (0.51 mmol) of the compound from Example 15A and 0.19 g (0.51 mmol) of N ⁇ -Boc-N ⁇ -ZL-ornithine with 0.19 g (0.51 mmol) of HATU and 0.35 ml (1.65 mmol ) N, N-Diisopropylethylamine in 5.55 ml dry DMF.
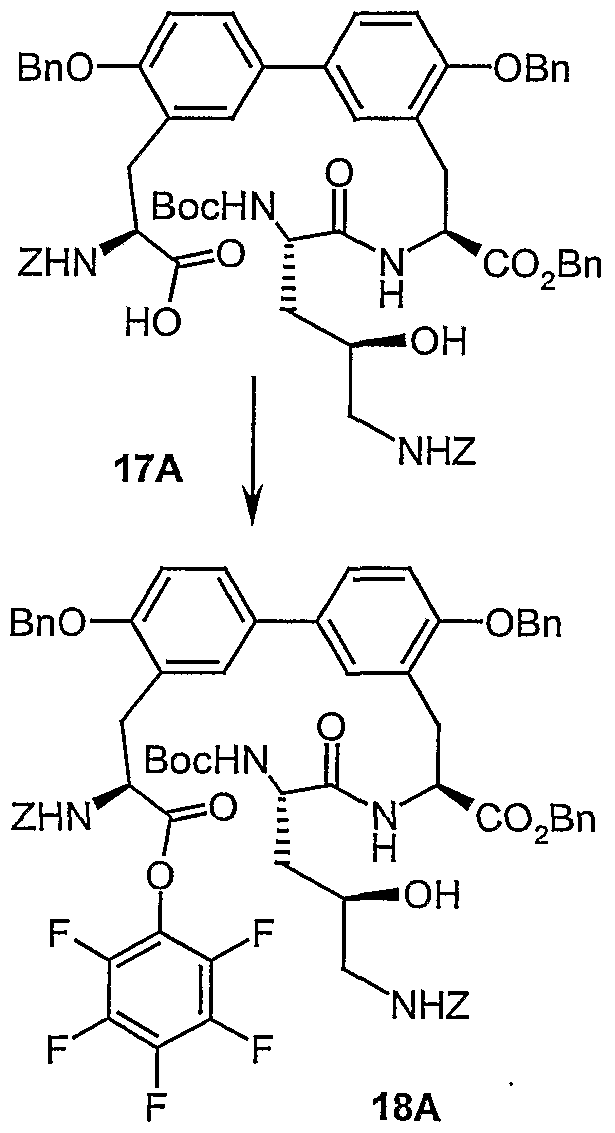
- Example 17A The preparation is carried out analogously to Example 17A from 0.82 g (0.68 mmol) of the compound from Example 23 A with 2 equiv. (1.3 ml) tetrabutylammonium fluoride (1 M in THF) in 30 ml dried DMF.
- Example 29 A (8S, 11S, 14S) -14 - [(tert-butoxycarbonyl) amino-ll- [3 - [(tert-butoxycarbonyl) - amino] propyl ⁇ -5,17-dihydroxy-10,13-dioxo -9,12-diazatricyclo [14.3.1.1 2 ' 6 ] -henicosa-l (20), 2 (21), 3,5,16,18-hexaen-8-carboxylic acid
- Example 8A Analogously to Example 8A, 380 mg (0.63 mmol) of the compound from Example 31 A are placed in a heated flask in 4 ml of DMF and, with stirring, at room temperature with 184.5 mg (0.73 mmol) of 4,4,4 ', 4', 5 3 5,5 ', 5'-octamethyl-2,2'-bi- 1,3,2-dioxaborolane, 186 mg (1.9 mmol) potassium acetate and 23.15 mg (0.03 mmol) bis (diphenylphosphino) ferrocene palladium (III) chloride added. The mixture is left to react at 80 ° C. for 4 h.
- Example 12A Metal B
- Example 12A Metal B
- 190 mg (0.32 mmol) of the compound from Example 32A 199.5 mg (0.32 mmol) of the compound from Example 11A, 195.5 mg (0.63 mmol) of cesium carbonate and 23.15 mg (0.03 mmol)
- Bis (diphenylphosphino) Fe ⁇ ocen-palladium ( ⁇ ) chloride in 1.5 ml DMF under an argon atmosphere.
- Example 15A The preparation is carried out analogously to Example 15A from 930 mg (0.95 mmol) of the compound from Example 33A and 22.14 ml of a 4 M dioxane / hydrogen chloride solution in 15 ml of dioxane.
- Example 16A The preparation is carried out analogously to Example 16A from 922 mg (1.01 mmol) of the compound from Example 34A, 0.5 g (1.01 mmol) of the compound from Example 14A, 421 mg (1.11 mmol) of HATU and 0.7 ml (518 mg; 3.27 mmol) of DIPEA in 4.2 ml DMF.
- Example 17A The preparation is carried out analogously to Example 17A from 360 mg (0.27 mmol) of the compound from Example 35A and 0.8 ml (3 equiv.) Of 1 M tetrabutylammonium fluoride solution (THF) in 20 ml of DMF. Yield: 159 mg (53% of theory)
- Example 37A 2 (S) - [Methyl- (5-benzyloxycarbonylamino) -2 (S) -tert-butoxycarbonylamino-4 (R) -hydroxy-pentanoyl] amino-3- [4,4 * -bis-benzyloxy-3 ' - (2 (S) -benzyloxycarbonyl-amino-2-pentafluorophenyloxycarbonyl-ethyl) -biphenyl-3-yl] -propionic acid benzyl ester
- Examples 42A to 48A listed in the following table are prepared from the corresponding starting materials:
- Example 52A (8S, 11S, 14S) -14 - [(tert-Butoxycarbonyl) amino] -ll- ⁇ 3 - [(tert-butoxycarbonyl) - amino] propyl ⁇ -5,17-dihydroxy-10,13-dioxo- 9,12-diazatricyclo [14.3.1.1 2,6 ] - henicosa-l (20), 2 (21), 3,5,16,18-hexaen-8-carboxylic acid
- Example 49A The preparation is carried out analogously to Example 49A from 20 mg (0.03 mmol) of the compound from Example 52A and 9.4 mg (0.06 mmol) of tert-butyl-2-hydroxyethyl carbonate
- Example 5 The preparation is carried out analogously to Example 5 from 1.2 mg of the compound from Example 40A with 0.3 ml of absolute methanol and 3 drops of 4M dioxane / hydrogen chloride solution.
- Example 5 The preparation is carried out analogously to Example 5 from 10 mg (0.02 mmol) of the compound from Example 19A and 1 ml of isopropanol with 10 drops of 4M dioxane / hydrogen chloride solution.
- cAMP 11.25 mg / ml
- 50 ⁇ l reaction mix 1 ⁇ l cAMP (11.25 mg / ml) per 50 ⁇ l reaction mix are added to the reaction mix of the in vitro transcription-translation test.
- the test batch is 105 ⁇ l, with 5 ⁇ l of the substance to be tested being presented in 5% DMSO.
- IC50 indicates the concentration of an inhibitor which leads to a 50% inhibition of the translation of Firefly luciferase.
- the plasmid pBESTluc (Promega Corporation, USA) is used to construct a reporter plasmid which can be used in an in vitro S. aureus transcription-translation assay.
- the CAPFor ' primer contains the capAl promoter, the ribosome binding site and the 5' region of the luciferase gene.
- PCR product can be isolated, which contains the Firefly luciferase gene with the fused capAl promoter. After restriction with Clal and Hindm, this is ligated into the vector pBESTluc, also digested with Clal and Hindlü. The resulting plasmid pla can be replicated in E. coli and used as a template in the S. aureus in vitro transcription-translation test.
- BHI medium Six liters of BHI medium are inoculated with a 250 ml overnight culture of a S. aureus strain and grown at 37 ° C to an OD600nm of 2-4. The cells are harvested by centrifugation and placed in 500 ml of cold buffer A
- lysostaphin 0.8 mg / ml
- buffer B 1.5 ml lysostaphin (0.8 mg / ml) in buffer B are placed in 3 pre-cooled centrifuge beakers and mixed with 33 ml of the cell suspension each. The samples are incubated for 45 to 60 min at 37 ° C with occasional shaking before adding 150 ⁇ l of a 0.5 M DTT solution. The lysed cells are centrifuged at 30,000 xg for 30 min at 4 ° C. After being taken up in buffer B, the cell pellet is centrifuged again under the same conditions and the collected supernatants are combined.
- the supernatants are centrifuged again under the same conditions and 0.25 volumes of buffer C (670 mM tris-acetate, pH 8.0, 20 mM Mg-acetate, 7 mM Na 3 -phosphoenolpyruvate, 7 ) are added to the upper 2/3 of the supernatant mM DTT, 5.5 mM ATP, 70 ⁇ M amino acids (complete from Promega), 75 ⁇ g pyruvate kinase (Sigma,
- the samples are incubated for 30 min at 37 ° C.
- the supernatants are overnight at 4 ° C against 2 1 dialysis buffer (10 mM Tris-acetate, pH 8.0, 14 mM Mg-acetate, 1 mM DTT, 60 mM K-acetate) with a buffer change in a dialysis tube with 3500 Da Exclusion dialyzed.
- the dialysate is concentrated to a protein concentration of about 10 mg / ml by the
- Dialysis tubing is covered with cold PEG 8000 powder (Sigma, Germany) at 4 ° C.
- the S30 extracts can be stored aliquoted at -70 ° C.
- Transcription-translation assay can be shown.
- the assay is based on cell-free transcription and translation of Firefly Luciferase using the reporter plasmid pla as a template and cell-free S30 extracts derived from S. aureus. The activity of the resulting luciferase can be demonstrated by luminescence measurement.
- the amount of S30 extract or plasmid pla to be used must be re-tested for each preparation in order to ensure an optimal concentration in the test. 3 ⁇ l of the substance to be tested dissolved in 5% DMSO are placed in an MTP. Then 10 ⁇ l of a suitably concentrated plasmid solution p1 are added.
- the IC 50 is the concentration of an inhibitor which leads to a 50% inhibition of the translation of Firefly luciferase.
- the minimum inhibitory concentration is the minimum concentration of an antibiotic with which a test germ is inhibited in its growth over 18-24 h.
- the inhibitor concentration can be determined using standard microbiological methods (see e.g. The National Committee for Clinical Laboratory Standards. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard-fifth edition. NCCLS document M7-A5 [ISBN 1-56238-394 -9]. NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania
- the MIC of the compounds according to the invention is determined in the liquid dilution test on a 96-microtiter plate scale.
- the bacterial germs were in a minimal medium (18.5 mM Na 2 HPO 4 , 5.7 mM KH 2 PO 4 , 9.3 mM NH 4 C1, 2.8 mM MgSO 4 , 17.1 mM NaCl, 0.033 ⁇ g / ml Thiamine hydrochloride, 1.2 ⁇ g / ml nicotinic acid, 0.003 ⁇ g / ml biotin, 1% glucose, 25 ⁇ g / ml of any proteinogenic amino acid with the exception of phenylalanine; [H.-P.
- the lowest substance concentration at which no visible bacterial growth occurred is defined as the MIC.
- the MIC values in ⁇ M of some compounds according to the invention compared to a number of test germs are listed in the table below as examples. The compounds show a graded antibacterial effect against most of the test germs.
- the suitability of the compounds according to the invention for the treatment of bacterial infections can be shown in various animal models.
- the animals are generally infected with a suitable virulent germ and then treated with the compound to be tested, which is present in a formulation adapted to the particular therapy model.
- the suitability of the compounds according to the invention for the treatment of bacterial infections in a sepsis model in mice after infection with S. aureus can be demonstrated.
- S. aureus 133 cells are grown overnight in BH broth (Oxoid, Germany). The overnight culture was diluted 1: 100 in fresh bra broth and turned up for 3 hours.
- the bacteria in the logarithmic growth phase are centrifuged off and washed twice with buffered, physiological saline solution.
- the animal model is set so that untreated animals die within 24 hours of infection.
- the compounds according to the invention can be converted into pharmaceutical preparations as follows:
- Example 2 100 mg of the compound from Example 2, 50 mg lactose (monohydrate), 50 mg corn starch (native), 10 mg polyvinylpyrolidone (PVP 25) (from BASF, Ludwigshafen, Germany) and 2 mg magnesium stearate.
- the mixture of active ingredient, lactose and starch is granulated with a 5% solution (m m) of the PVP in water.
- the granules are dried with the magnesium stearate for 5 min. mixed.
- This mixture is compressed with a conventional tablet press (tablet format see above).
- a pressing force of 15 kN is used as a guideline for the pressing.
- Rhodigel is suspended in ethanol, the active ingredient is added to the suspension. The water is added with stirring. The swelling of the Rhodigel is stopped for about 6 hours.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Communicable Diseases (AREA)
- Biophysics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description
































































































Claims



Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03766214A EP1526896A1 (de) | 2002-07-29 | 2003-07-18 | Antibakterielle ester-makrozyklen |
| AU2003246716A AU2003246716A1 (en) | 2002-07-29 | 2003-07-18 | Antibacterial ester macrocycles |
| US10/522,667 US20060258571A1 (en) | 2002-07-29 | 2003-07-18 | Antibacterial ester macrocycles |
| JP2004525241A JP2006502128A (ja) | 2002-07-29 | 2003-07-18 | 抗菌性エステル−マクロサイクル |
| CA002495479A CA2495479A1 (en) | 2002-07-29 | 2003-07-18 | Antibacterial ester macrocycles |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10234422A DE10234422A1 (de) | 2002-07-29 | 2002-07-29 | Antibakterielle Ester-Makrozyklen |
| DE10234422.1 | 2002-07-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004012816A1 true WO2004012816A1 (de) | 2004-02-12 |
Family
ID=30128472
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2003/007824 WO2004012816A1 (de) | 2002-07-29 | 2003-07-18 | Antibakterielle ester-makrozyklen |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20060258571A1 (de) |
| EP (1) | EP1526896A1 (de) |
| JP (1) | JP2006502128A (de) |
| AU (1) | AU2003246716A1 (de) |
| CA (1) | CA2495479A1 (de) |
| DE (1) | DE10234422A1 (de) |
| WO (1) | WO2004012816A1 (de) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005118613A2 (de) * | 2004-05-26 | 2005-12-15 | Aicuris Gmbh & Co.Kg | Antibakterielle amid-makrozyklen |
| WO2007006548A2 (de) * | 2005-07-14 | 2007-01-18 | Aicuris Gmbh & Co. Kg | Antibakterielle amid-makrozyklen vii |
| JP2008514561A (ja) * | 2004-09-24 | 2008-05-08 | アイキュリス・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング・ウント・コムパニー・コマンディットゲゼルシャフト | 抗菌性アミドマクロサイクルiv |
| US7655643B2 (en) | 2003-12-16 | 2010-02-02 | Aicuris Gmbh & Co. Kg | Antibacterial macrocycles with substituted biphenyl |
| EP2386547A1 (de) | 2005-12-29 | 2011-11-16 | Lexicon Pharmaceuticals, Inc. | Multizyklische Aminosäurederivate und Verwendungsverfahren dafür |
| US8129561B2 (en) | 2003-06-18 | 2012-03-06 | Tranzyme Pharma Inc. | Processes for intermediates for macrocyclic compounds |
| CN112262150A (zh) * | 2018-06-15 | 2021-01-22 | 横河电机株式会社 | 酰胺的制备方法 |
| US11505573B2 (en) | 2018-03-28 | 2022-11-22 | Hoffmann-La Roche Inc. | Peptide macrocycles against Acinetobacter baumannii |
| US11819532B2 (en) | 2018-04-23 | 2023-11-21 | Hoffmann-La Roche Inc. | Peptide macrocycles against Acinetobacter baumannii |
| US12012466B2 (en) | 2015-10-27 | 2024-06-18 | Hoffmann-La Roche Inc. | Peptide macrocycles against Acinetobacter baumannii |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10226921A1 (de) * | 2002-06-17 | 2003-12-24 | Bayer Ag | Antibakterielle Amid-Makrozyklen |
| DE102005014247A1 (de) * | 2005-03-30 | 2006-10-05 | Aicuris Gmbh & Co. Kg | Antibakterielle Amid-Makrozyklen VI |
| DE102005014245A1 (de) * | 2005-03-30 | 2006-10-05 | Aicuris Gmbh & Co. Kg | Antibakterielle Amid-Makrozyklen V |
| AU2019284745B2 (en) * | 2018-06-15 | 2023-05-11 | Yokogawa Electric Corporation | Method for producing amide |
| EP3904317A4 (de) * | 2018-12-25 | 2022-09-21 | Tokyo Institute of Technology | Verfahren zur herstellung von amid |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2801591A1 (fr) * | 1999-11-30 | 2001-06-01 | Aventis Pharma Sa | Derives macrocycliques de l'acide hydroxamique, leur preparation et les compositions qui les contiennent |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10358824A1 (de) * | 2003-12-16 | 2005-07-21 | Bayer Healthcare Ag | Antibakterielle Makrozyklen mit substituiertem Biphenyl |
-
2002
- 2002-07-29 DE DE10234422A patent/DE10234422A1/de not_active Withdrawn
-
2003
- 2003-07-18 JP JP2004525241A patent/JP2006502128A/ja not_active Withdrawn
- 2003-07-18 AU AU2003246716A patent/AU2003246716A1/en not_active Abandoned
- 2003-07-18 WO PCT/EP2003/007824 patent/WO2004012816A1/de active Application Filing
- 2003-07-18 US US10/522,667 patent/US20060258571A1/en not_active Abandoned
- 2003-07-18 EP EP03766214A patent/EP1526896A1/de not_active Withdrawn
- 2003-07-18 CA CA002495479A patent/CA2495479A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2801591A1 (fr) * | 1999-11-30 | 2001-06-01 | Aventis Pharma Sa | Derives macrocycliques de l'acide hydroxamique, leur preparation et les compositions qui les contiennent |
Non-Patent Citations (4)
| Title |
|---|
| BROWN, ALLAN G. ET AL: "Application of the Suzuki biphenyl synthesis to the natural products biphenomycin and vancomycin", JOURNAL OF THE CHEMICAL SOCIETY, PERKIN TRANSACTIONS 1: ORGANIC AND BIO-ORGANIC CHEMISTRY (1972-1999) (1992), (1), 123-30, XP009018532 * |
| CHANG, CONWAY C. ET AL: "LL-AF283 antibiotics, cyclic biphenyl peptides", JOURNAL OF ANTIBIOTICS (1991), 44(6), 674-7, XP009018534 * |
| SCHMIDT, ULRICH ET AL: "Amino acids and peptides. 84. Synthesis of biologically active cyclopeptides. 24. Total synthesis of the biphenomycins. III. Synthesis of biphenomycin B", SYNTHESIS (1992), (10), 1025-30, XP001155274 * |
| SCHMIDT, ULRICH ET AL: "Amino acids and peptides. 88. Synthesis of biologically active cyclopeptides. 26. Total synthesis of the biphenomycins. V. Synthesis of biphenomycin A", SYNTHESIS (1992), (12), 1248-54, XP001155271 * |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8497242B2 (en) | 2003-06-18 | 2013-07-30 | Tranzyme Pharma Inc. | Processes for intermediates for macrocyclic compounds |
| US10040751B2 (en) | 2003-06-18 | 2018-08-07 | Ocera Therapeutics, Inc. | Intermediates for macrocyclic compounds |
| US9181298B2 (en) | 2003-06-18 | 2015-11-10 | Ocera Therapeutics, Inc. | Intermediates for macrocyclic compounds |
| US8129561B2 (en) | 2003-06-18 | 2012-03-06 | Tranzyme Pharma Inc. | Processes for intermediates for macrocyclic compounds |
| US7655643B2 (en) | 2003-12-16 | 2010-02-02 | Aicuris Gmbh & Co. Kg | Antibacterial macrocycles with substituted biphenyl |
| WO2005118613A3 (de) * | 2004-05-26 | 2006-01-12 | Bayer Healthcare Ag | Antibakterielle amid-makrozyklen |
| WO2005118613A2 (de) * | 2004-05-26 | 2005-12-15 | Aicuris Gmbh & Co.Kg | Antibakterielle amid-makrozyklen |
| JP2008514561A (ja) * | 2004-09-24 | 2008-05-08 | アイキュリス・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング・ウント・コムパニー・コマンディットゲゼルシャフト | 抗菌性アミドマクロサイクルiv |
| WO2007006548A3 (de) * | 2005-07-14 | 2007-03-22 | Aicuris Gmbh & Co Kg | Antibakterielle amid-makrozyklen vii |
| WO2007006548A2 (de) * | 2005-07-14 | 2007-01-18 | Aicuris Gmbh & Co. Kg | Antibakterielle amid-makrozyklen vii |
| EP2386547A1 (de) | 2005-12-29 | 2011-11-16 | Lexicon Pharmaceuticals, Inc. | Multizyklische Aminosäurederivate und Verwendungsverfahren dafür |
| US12012466B2 (en) | 2015-10-27 | 2024-06-18 | Hoffmann-La Roche Inc. | Peptide macrocycles against Acinetobacter baumannii |
| US11505573B2 (en) | 2018-03-28 | 2022-11-22 | Hoffmann-La Roche Inc. | Peptide macrocycles against Acinetobacter baumannii |
| US11819532B2 (en) | 2018-04-23 | 2023-11-21 | Hoffmann-La Roche Inc. | Peptide macrocycles against Acinetobacter baumannii |
| CN112262150A (zh) * | 2018-06-15 | 2021-01-22 | 横河电机株式会社 | 酰胺的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2006502128A (ja) | 2006-01-19 |
| EP1526896A1 (de) | 2005-05-04 |
| CA2495479A1 (en) | 2004-02-12 |
| US20060258571A1 (en) | 2006-11-16 |
| DE10234422A1 (de) | 2004-02-12 |
| AU2003246716A1 (en) | 2004-02-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1515983B1 (de) | Antibakterielle amid-makrozyklen | |
| WO2004012816A1 (de) | Antibakterielle ester-makrozyklen | |
| EP1869068A1 (de) | Antibakterielle amid-makrozyklen v | |
| AU2015228898B2 (en) | Polymyxin derivatives and their use in combination therapy together with different antibiotics | |
| EP1797110B1 (de) | Antibakterielle amid-makrozyklen iv | |
| EP1697400B1 (de) | Antibakterielle makrozyklen mit substituiertem biphenyl | |
| EP1866291B1 (de) | Antibakterielle amid-makrozyklen vi | |
| WO2005118613A2 (de) | Antibakterielle amid-makrozyklen | |
| WO2005033129A1 (de) | Antibakterielle amid-makrozyklen | |
| WO2005100380A1 (de) | Antibakterielle amid-makrozyklen ii | |
| IL165630A (en) | Antibacterial amide macrocycles | |
| EP1907413A2 (de) | Antibakterielle amid-makrozyklen vii | |
| DE10243201A1 (de) | Alkyl- und Alkenylamide | |
| WO2004113290A1 (de) | Substituierte alkylamide | |
| EP1448521A1 (de) | Derivate von andrimid und moiramid b mit antibakteriellen eigenschaften |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003766214 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2495479 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004525241 Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003766214 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2003766214 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006258571 Country of ref document: US Ref document number: 10522667 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 10522667 Country of ref document: US |