WO1994004488A1 - Nouveaux composes poly-iodes, procede de preparation, produit de contraste les contenant - Google Patents
Nouveaux composes poly-iodes, procede de preparation, produit de contraste les contenant Download PDFInfo
- Publication number
- WO1994004488A1 WO1994004488A1 PCT/FR1993/000824 FR9300824W WO9404488A1 WO 1994004488 A1 WO1994004488 A1 WO 1994004488A1 FR 9300824 W FR9300824 W FR 9300824W WO 9404488 A1 WO9404488 A1 WO 9404488A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- represent
- formula
- groups
- compounds
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 86
- 238000002360 preparation method Methods 0.000 title abstract description 50
- 238000000034 method Methods 0.000 title abstract description 12
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 14
- 239000000460 chlorine Substances 0.000 claims description 38
- 229910052740 iodine Inorganic materials 0.000 claims description 19
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 claims description 16
- 239000011630 iodine Substances 0.000 claims description 15
- 125000003118 aryl group Chemical group 0.000 claims description 14
- 238000005917 acylation reaction Methods 0.000 claims description 11
- 150000001412 amines Chemical class 0.000 claims description 11
- 125000003277 amino group Chemical group 0.000 claims description 11
- 230000010933 acylation Effects 0.000 claims description 10
- 239000002904 solvent Substances 0.000 claims description 9
- 238000005804 alkylation reaction Methods 0.000 claims description 8
- 229910052794 bromium Inorganic materials 0.000 claims description 8
- 229910052801 chlorine Inorganic materials 0.000 claims description 8
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 8
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 claims description 7
- 230000029936 alkylation Effects 0.000 claims description 7
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 6
- 125000003368 amide group Chemical group 0.000 claims description 6
- 239000003054 catalyst Substances 0.000 claims description 6
- 239000003153 chemical reaction reagent Substances 0.000 claims description 6
- 238000010511 deprotection reaction Methods 0.000 claims description 6
- 238000005660 chlorination reaction Methods 0.000 claims description 5
- 230000008878 coupling Effects 0.000 claims description 5
- 238000010168 coupling process Methods 0.000 claims description 5
- 238000005859 coupling reaction Methods 0.000 claims description 5
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 4
- 150000001555 benzenes Chemical class 0.000 claims description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 4
- 125000006239 protecting group Chemical group 0.000 claims description 4
- 125000000217 alkyl group Chemical group 0.000 claims description 3
- 230000009435 amidation Effects 0.000 claims description 3
- 238000007112 amidation reaction Methods 0.000 claims description 3
- 239000007864 aqueous solution Substances 0.000 claims description 3
- 125000001424 substituent group Chemical group 0.000 claims description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 2
- 229910052751 metal Inorganic materials 0.000 claims description 2
- 239000002184 metal Substances 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 claims 2
- 125000003545 alkoxy group Chemical group 0.000 claims 1
- 238000002601 radiography Methods 0.000 abstract description 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 96
- 239000000243 solution Substances 0.000 description 48
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 30
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 28
- 238000001704 evaporation Methods 0.000 description 21
- 230000008020 evaporation Effects 0.000 description 21
- 239000000047 product Substances 0.000 description 18
- -1 amine alcohols Chemical class 0.000 description 17
- 239000013078 crystal Substances 0.000 description 17
- 239000002244 precipitate Substances 0.000 description 13
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 238000001914 filtration Methods 0.000 description 12
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 11
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 11
- 239000011347 resin Substances 0.000 description 11
- 229920005989 resin Polymers 0.000 description 11
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 10
- 239000000203 mixture Substances 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 238000000746 purification Methods 0.000 description 9
- 238000006722 reduction reaction Methods 0.000 description 9
- 238000003756 stirring Methods 0.000 description 8
- MHABMANUFPZXEB-UHFFFAOYSA-N O-demethyl-aloesaponarin I Natural products O=C1C2=CC=CC(O)=C2C(=O)C2=C1C=C(O)C(C(O)=O)=C2C MHABMANUFPZXEB-UHFFFAOYSA-N 0.000 description 7
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 7
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 7
- 238000010992 reflux Methods 0.000 description 7
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 6
- 229910004298 SiO 2 Inorganic materials 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 229910052739 hydrogen Inorganic materials 0.000 description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 238000005115 demineralization Methods 0.000 description 5
- 230000002328 demineralizing effect Effects 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 239000000377 silicon dioxide Substances 0.000 description 5
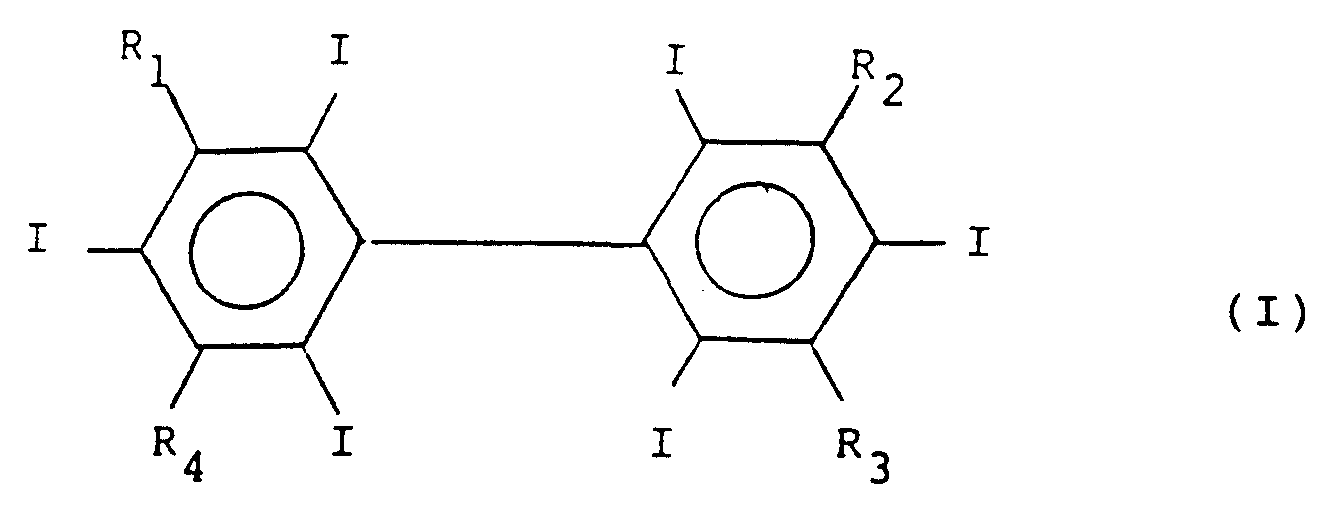
- SBMAZEDZVNSPTP-UHFFFAOYSA-N 1,3,5-triiodo-2-(2,4,6-triiodophenyl)benzene Chemical group IC1=CC(I)=CC(I)=C1C1=C(I)C=C(I)C=C1I SBMAZEDZVNSPTP-UHFFFAOYSA-N 0.000 description 4
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 239000006071 cream Substances 0.000 description 4
- 239000012429 reaction media Substances 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 3
- 239000004305 biphenyl Substances 0.000 description 3
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 3
- 239000007795 chemical reaction product Substances 0.000 description 3
- 239000002872 contrast media Substances 0.000 description 3
- 229910052802 copper Inorganic materials 0.000 description 3
- 239000010949 copper Substances 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- ILWRPSCZWQJDMK-UHFFFAOYSA-N triethylazanium;chloride Chemical compound Cl.CCN(CC)CC ILWRPSCZWQJDMK-UHFFFAOYSA-N 0.000 description 3
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 2
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical compound CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 2
- ZGCHLAJIRWDGFE-UHFFFAOYSA-N 1-aminopropane-1,1-diol Chemical compound CCC(N)(O)O ZGCHLAJIRWDGFE-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- GFGURBBHVJTXDU-UHFFFAOYSA-N 3-iodo-5-nitrobenzoic acid Chemical compound OC(=O)C1=CC(I)=CC([N+]([O-])=O)=C1 GFGURBBHVJTXDU-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- YTBSYETUWUMLBZ-UHFFFAOYSA-N D-Erythrose Natural products OCC(O)C(O)C=O YTBSYETUWUMLBZ-UHFFFAOYSA-N 0.000 description 2
- 206010056474 Erythrosis Diseases 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- GTTSNKDQDACYLV-UHFFFAOYSA-N Trihydroxybutane Chemical group CCCC(O)(O)O GTTSNKDQDACYLV-UHFFFAOYSA-N 0.000 description 2
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- IAQRGUVFOMOMEM-UHFFFAOYSA-N butene Natural products CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 2
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 2
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 230000020477 pH reduction Effects 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- ALHZEIINTQJLOT-UHFFFAOYSA-N (1-chloro-1-oxopropan-2-yl) acetate Chemical compound ClC(=O)C(C)OC(C)=O ALHZEIINTQJLOT-UHFFFAOYSA-N 0.000 description 1
- OZCRKDNRAAKDAN-HNQUOIGGSA-N (e)-but-1-ene-1,4-diol Chemical compound OCC\C=C\O OZCRKDNRAAKDAN-HNQUOIGGSA-N 0.000 description 1
- KRBIHOANUQUSRV-UHFFFAOYSA-N 1-(oxiran-2-yl)ethane-1,2-diol Chemical compound OCC(O)C1CO1 KRBIHOANUQUSRV-UHFFFAOYSA-N 0.000 description 1
- 239000005631 2,4-Dichlorophenoxyacetic acid Substances 0.000 description 1
- KJJPLEZQSCZCKE-UHFFFAOYSA-N 2-aminopropane-1,3-diol Chemical compound OCC(N)CO KJJPLEZQSCZCKE-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- BIRALNDXXLEWHV-UHFFFAOYSA-N 2-propan-2-yl-1,3-dioxane-5-carbonyl chloride Chemical compound CC(C)C1OCC(C(Cl)=O)CO1 BIRALNDXXLEWHV-UHFFFAOYSA-N 0.000 description 1
- YSNYXNGOASZBJN-UHFFFAOYSA-N 3-(2,3-dihydroxypropanoylamino)-5-[3-(2,3-dihydroxypropanoylamino)-5-[2-hydroxyethyl(2,3,4-trihydroxybutyl)carbamoyl]-2,4,6-triiodophenyl]-N-(2-hydroxyethyl)-2,4,6-triiodo-N-(2,3,4-trihydroxybutyl)benzamide Chemical group OC(C(=O)NC=1C(=C(C(=C(C=1I)C(N(CC(C(CO)O)O)CCO)=O)I)C1=C(C(=C(C(=C1I)C(N(CCO)CC(C(CO)O)O)=O)I)NC(C(CO)O)=O)I)I)CO YSNYXNGOASZBJN-UHFFFAOYSA-N 0.000 description 1
- GNFTZDOKVXKIBK-UHFFFAOYSA-N 3-(2-methoxyethoxy)benzohydrazide Chemical compound COCCOC1=CC=CC(C(=O)NN)=C1 GNFTZDOKVXKIBK-UHFFFAOYSA-N 0.000 description 1
- XVJSFQJMJQNVGO-UHFFFAOYSA-N 3-(3-carboxy-5-nitrophenyl)-5-nitrobenzoic acid Chemical group [O-][N+](=O)C1=CC(C(=O)O)=CC(C=2C=C(C=C(C=2)C(O)=O)[N+]([O-])=O)=C1 XVJSFQJMJQNVGO-UHFFFAOYSA-N 0.000 description 1
- MKCMCUCPJQAJKH-UHFFFAOYSA-N 3-[3-carbonochloridoyl-5-(diacetylamino)-2,4,6-triiodophenyl]-5-(diacetylamino)-2,4,6-triiodobenzoyl chloride Chemical group CC(=O)N(C(C)=O)C1=C(I)C(C(Cl)=O)=C(I)C(C=2C(=C(C(Cl)=O)C(I)=C(N(C(C)=O)C(C)=O)C=2I)I)=C1I MKCMCUCPJQAJKH-UHFFFAOYSA-N 0.000 description 1
- VUEHQMGMXMKHGT-UHFFFAOYSA-N 3-[3-carboxy-5-(diacetylamino)-2,4,6-triiodophenyl]-5-(diacetylamino)-2,4,6-triiodobenzoic acid Chemical group CC(=O)N(C(C)=O)C1=C(I)C(C(O)=O)=C(I)C(C=2C(=C(C(O)=O)C(I)=C(N(C(C)=O)C(C)=O)C=2I)I)=C1I VUEHQMGMXMKHGT-UHFFFAOYSA-N 0.000 description 1
- VEMYGEPVPLWHRR-UHFFFAOYSA-N 3-[[3-hydroxy-2-(hydroxymethyl)propanoyl]amino]-5-[3-[[3-hydroxy-2-(hydroxymethyl)propanoyl]amino]-2,4,6-triiodo-5-[methyl(2,3,4-trihydroxybutyl)carbamoyl]phenyl]-2,4,6-triiodo-N-methyl-N-(2,3,4-trihydroxybutyl)benzamide Chemical group OCC(C(=O)NC=1C(=C(C(=C(C=1I)C(N(CC(C(CO)O)O)C)=O)I)C1=C(C(=C(C(=C1I)C(N(C)CC(C(CO)O)O)=O)I)NC(C(CO)CO)=O)I)I)CO VEMYGEPVPLWHRR-UHFFFAOYSA-N 0.000 description 1
- LZFOCJBJYWXIQA-UHFFFAOYSA-N 3-acetyloxy-2-oxobutanoic acid Chemical compound CC(=O)OC(C)C(=O)C(O)=O LZFOCJBJYWXIQA-UHFFFAOYSA-N 0.000 description 1
- MFZWEPYJMNTNRS-UHFFFAOYSA-N 3-amino-5-(3-amino-5-carboxy-2,4,6-triiodophenyl)-2,4,6-triiodobenzoic acid Chemical group NC1=C(I)C(C(O)=O)=C(I)C(C=2C(=C(C(O)=O)C(I)=C(N)C=2I)I)=C1I MFZWEPYJMNTNRS-UHFFFAOYSA-N 0.000 description 1
- RNMWRTFLXBCPHP-UHFFFAOYSA-N 3-amino-5-(3-amino-5-carboxyphenyl)benzoic acid Chemical group NC1=CC(C(O)=O)=CC(C=2C=C(C=C(N)C=2)C(O)=O)=C1 RNMWRTFLXBCPHP-UHFFFAOYSA-N 0.000 description 1
- WAZWTTNGHMDUJD-UHFFFAOYSA-N 3-amino-5-[3-amino-5-[2,3-dihydroxypropyl(2,3,4-trihydroxybutyl)carbamoyl]-2,4,6-triiodophenyl]-N-(2,3-dihydroxypropyl)-2,4,6-triiodo-N-(2,3,4-trihydroxybutyl)benzamide Chemical group NC1=C(I)C(C(=O)N(CC(O)CO)CC(O)C(O)CO)=C(I)C(C=2C(=C(C(=O)N(CC(O)CO)CC(O)C(O)CO)C(I)=C(N)C=2I)I)=C1I WAZWTTNGHMDUJD-UHFFFAOYSA-N 0.000 description 1
- SHEBBMVCBRQVQX-UHFFFAOYSA-N 3-amino-5-[3-amino-5-[2-hydroxyethyl(2,3,4-trihydroxybutyl)carbamoyl]-2,4,6-triiodophenyl]-n-(2-hydroxyethyl)-2,4,6-triiodo-n-(2,3,4-trihydroxybutyl)benzamide Chemical group NC1=C(I)C(C(=O)N(CCO)CC(O)C(O)CO)=C(I)C(C=2C(=C(C(=O)N(CCO)CC(O)C(O)CO)C(I)=C(N)C=2I)I)=C1I SHEBBMVCBRQVQX-UHFFFAOYSA-N 0.000 description 1
- AFPHTEQTJZKQAQ-UHFFFAOYSA-N 3-nitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1 AFPHTEQTJZKQAQ-UHFFFAOYSA-N 0.000 description 1
- ABQCBCTXEZLEQC-UHFFFAOYSA-N 4-(2,3-dihydroxypropylamino)butane-1,2,3-triol Chemical compound OCC(O)CNCC(O)C(O)CO ABQCBCTXEZLEQC-UHFFFAOYSA-N 0.000 description 1
- PAHOOYLOBYJWTP-UHFFFAOYSA-N 4-(2-hydroxyethylamino)butane-1,2,3-triol Chemical compound OCCNCC(O)C(O)CO PAHOOYLOBYJWTP-UHFFFAOYSA-N 0.000 description 1
- QGRSTVXBRQIIPF-UHFFFAOYSA-N 4-(methylamino)butane-1,2,3-triol Chemical compound CNCC(O)C(O)CO QGRSTVXBRQIIPF-UHFFFAOYSA-N 0.000 description 1
- NUAPDTFBENBPBC-UHFFFAOYSA-N 4-[benzyl(2,3,4-trihydroxybutyl)amino]butane-1,2,3-triol Chemical compound OCC(O)C(O)CN(CC(O)C(O)CO)CC1=CC=CC=C1 NUAPDTFBENBPBC-UHFFFAOYSA-N 0.000 description 1
- WIFPJDJJFUSIFP-UHFFFAOYSA-N 4-aminobutane-1,2,3-triol Chemical compound NCC(O)C(O)CO WIFPJDJJFUSIFP-UHFFFAOYSA-N 0.000 description 1
- LLKFNPUXQZHIAE-UHFFFAOYSA-N 5-(3-aminopropyl)-8-bromo-3-methyl-2h-pyrazolo[4,3-c]quinolin-4-one Chemical compound O=C1N(CCCN)C2=CC=C(Br)C=C2C2=C1C(C)=NN2 LLKFNPUXQZHIAE-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- ALDVGIXMUCONHA-UHFFFAOYSA-N C(N)(=O)C=1C(=C(C(=CC=1I)I)C1=C(C=C(C=C1I)I)I)I Chemical group C(N)(=O)C=1C(=C(C(=CC=1I)I)C1=C(C=C(C=C1I)I)I)I ALDVGIXMUCONHA-UHFFFAOYSA-N 0.000 description 1
- FGUUSXIOTUKUDN-IBGZPJMESA-N C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 Chemical compound C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 FGUUSXIOTUKUDN-IBGZPJMESA-N 0.000 description 1
- ZTSCVRSXWWAKAD-UHFFFAOYSA-N CC(=O)OCCN(CC(C(COC(=O)C)OC(=O)C)OC(=O)C)C(=O)C1=C(C(=C(C(=C1I)C2=C(C(=C(C(=C2I)N)I)C(=O)N(CCOC(=O)C)CC(C(COC(=O)C)OC(=O)C)OC(=O)C)I)I)N)I Chemical group CC(=O)OCCN(CC(C(COC(=O)C)OC(=O)C)OC(=O)C)C(=O)C1=C(C(=C(C(=C1I)C2=C(C(=C(C(=C2I)N)I)C(=O)N(CCOC(=O)C)CC(C(COC(=O)C)OC(=O)C)OC(=O)C)I)I)N)I ZTSCVRSXWWAKAD-UHFFFAOYSA-N 0.000 description 1
- YTBSYETUWUMLBZ-IUYQGCFVSA-N D-erythrose Chemical compound OC[C@@H](O)[C@@H](O)C=O YTBSYETUWUMLBZ-IUYQGCFVSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- WNQYRJJSKBXWMN-UHFFFAOYSA-N N-(2,3-dihydroxypropyl)-3-[3-[2,3-dihydroxypropyl(2,3,4-trihydroxybutyl)carbamoyl]-5-nitrophenyl]-5-nitro-N-(2,3,4-trihydroxybutyl)benzamide Chemical group [N+](=O)([O-])C=1C=C(C=C(C=1)C(N(CC(C(CO)O)O)CC(CO)O)=O)C1=CC(=CC(=C1)C(N(CC(CO)O)CC(C(CO)O)O)=O)[N+](=O)[O-] WNQYRJJSKBXWMN-UHFFFAOYSA-N 0.000 description 1
- SNRRFRFNBFLFFZ-UHFFFAOYSA-N N-(2-hydroxyethyl)-3-[3-[2-hydroxyethyl(2,3,4-trihydroxybutyl)carbamoyl]-2,4,6-triiodophenyl]-2,4,6-triiodo-N-(2,3,4-trihydroxybutyl)benzamide Chemical group OCCN(C(=O)C=1C(=CC(=C(C=1I)C1=C(C=C(C(=C1I)C(N(CCO)CC(C(CO)O)O)=O)I)I)I)I)CC(C(CO)O)O SNRRFRFNBFLFFZ-UHFFFAOYSA-N 0.000 description 1
- MHHVXFHRRCSEGT-UHFFFAOYSA-N N-methyl-3-[3-[methyl(2,3,4-trihydroxybutyl)carbamoyl]-5-nitrophenyl]-5-nitro-N-(2,3,4-trihydroxybutyl)benzamide Chemical group [N+](=O)([O-])C=1C=C(C=C(C=1)C(N(CC(C(CO)O)O)C)=O)C1=CC(=CC(=C1)C(N(C)CC(C(CO)O)O)=O)[N+](=O)[O-] MHHVXFHRRCSEGT-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- LFZYTHQYPVCNAQ-UHFFFAOYSA-N [2,3-diacetyloxy-4-[[3-amino-5-[3-amino-2,4,6-triiodo-5-[methyl(2,3,4-triacetyloxybutyl)carbamoyl]phenyl]-2,4,6-triiodobenzoyl]-methylamino]butyl] acetate Chemical group CC(=O)OCC(OC(C)=O)C(OC(C)=O)CN(C)C(=O)C1=C(I)C(N)=C(I)C(C=2C(=C(C(=O)N(C)CC(OC(C)=O)C(COC(C)=O)OC(C)=O)C(I)=C(N)C=2I)I)=C1I LFZYTHQYPVCNAQ-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 238000010531 catalytic reduction reaction Methods 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 229940039231 contrast media Drugs 0.000 description 1
- 238000006264 debenzylation reaction Methods 0.000 description 1
- 238000006735 epoxidation reaction Methods 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- CGNYRXBURZLBKG-UHFFFAOYSA-N methyl 3-(3-methoxycarbonyl-5-nitrophenyl)-5-nitrobenzoate Chemical group [O-][N+](=O)C1=CC(C(=O)OC)=CC(C=2C=C(C=C(C=2)C(=O)OC)[N+]([O-])=O)=C1 CGNYRXBURZLBKG-UHFFFAOYSA-N 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- DJQIYGIGFCBHDK-UHFFFAOYSA-N n-(2,3-dihydroxypropyl)-3-[3-[2,3-dihydroxypropyl(2,3,4-trihydroxybutyl)carbamoyl]-5-(2-hydroxypropanoylamino)-2,4,6-triiodophenyl]-5-(2-hydroxypropanoylamino)-2,4,6-triiodo-n-(2,3,4-trihydroxybutyl)benzamide Chemical group CC(O)C(=O)NC1=C(I)C(C(=O)N(CC(O)CO)CC(O)C(O)CO)=C(I)C(C=2C(=C(C(=O)N(CC(O)CO)CC(O)C(O)CO)C(I)=C(NC(=O)C(C)O)C=2I)I)=C1I DJQIYGIGFCBHDK-UHFFFAOYSA-N 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- VLZLOWPYUQHHCG-UHFFFAOYSA-N nitromethylbenzene Chemical compound [O-][N+](=O)CC1=CC=CC=C1 VLZLOWPYUQHHCG-UHFFFAOYSA-N 0.000 description 1
- 239000003690 nonionic contrast media Substances 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 210000002330 subarachnoid space Anatomy 0.000 description 1
- UEUXEKPTXMALOB-UHFFFAOYSA-J tetrasodium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O UEUXEKPTXMALOB-UHFFFAOYSA-J 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/28—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton
- C07C237/46—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton having carbon atoms of carboxamide groups, amino groups and at least three atoms of bromine or iodine, bound to carbon atoms of the same non-condensed six-membered aromatic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/04—X-ray contrast preparations
- A61K49/0433—X-ray contrast preparations containing an organic halogenated X-ray contrast-enhancing agent
Abstract
Description











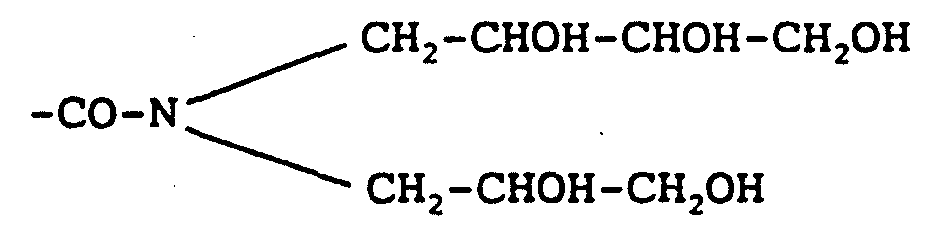






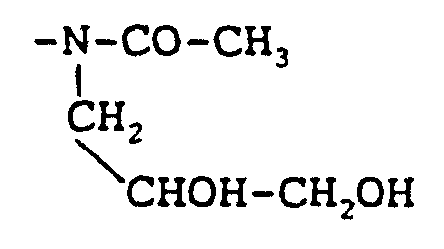












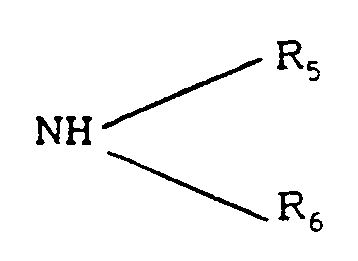


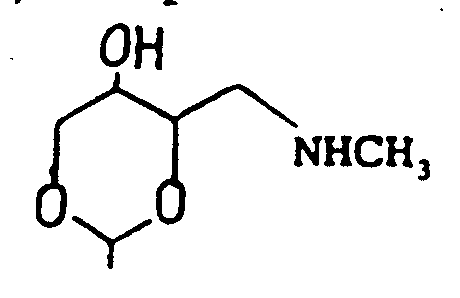









Claims








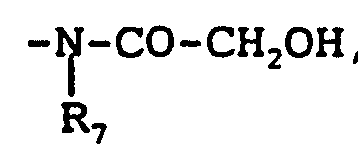
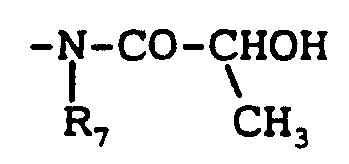


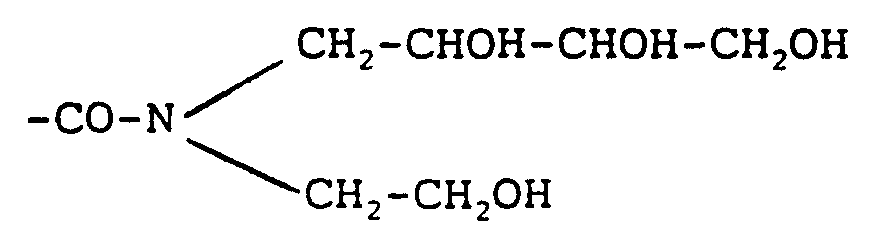





















Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU49644/93A AU4964493A (en) | 1992-08-25 | 1993-08-24 | Novel poly-iodated compounds, method of preparation and contrast product containing same |
| US08/387,721 US5618977A (en) | 1992-08-25 | 1993-08-24 | Polyiodinated compounds, process of preparation and contrast agent containing them |
| JP6505969A JPH08500355A (ja) | 1992-08-25 | 1993-08-24 | 新規ポリヨウ素化化合物、その製法およびそれらを含有する造影剤 |
| CA002142986A CA2142986A1 (fr) | 1992-08-25 | 1993-08-24 | Nouveaux composes polyiodes; methode de preparation et agent de contraste a base de ces composes |
| EP93919397A EP0656884A1 (fr) | 1992-08-25 | 1993-08-24 | Nouveaux composes poly-iodes, procede de preparation, produit de contraste les contenant |
| NO950665A NO950665L (no) | 1992-08-25 | 1995-02-22 | Nye polyjoderte forbindelser, fremgangsmåte for deres fremstilling og kontrastmidler inneholdende disse |
| FI950874A FI950874A (fi) | 1992-08-25 | 1995-02-24 | Uudet polyjoditetut yhdisteet, menetelmä niiden valmistamiseksi, niitä sisältävä varjoaine |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9210270A FR2695125B1 (fr) | 1992-08-25 | 1992-08-25 | Nouveaux composés poly-iodés, procédé de préparation, produit de contraste les contenant. |
| FR92/10270 | 1992-08-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994004488A1 true WO1994004488A1 (fr) | 1994-03-03 |
Family
ID=9433015
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR1993/000824 WO1994004488A1 (fr) | 1992-08-25 | 1993-08-24 | Nouveaux composes poly-iodes, procede de preparation, produit de contraste les contenant |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US5618977A (fr) |
| EP (1) | EP0656884A1 (fr) |
| JP (1) | JPH08500355A (fr) |
| CN (1) | CN1090572A (fr) |
| AU (1) | AU4964493A (fr) |
| CA (1) | CA2142986A1 (fr) |
| FI (1) | FI950874A (fr) |
| FR (1) | FR2695125B1 (fr) |
| HU (1) | HUT70943A (fr) |
| IL (1) | IL106768A0 (fr) |
| NO (1) | NO950665L (fr) |
| PL (1) | PL307587A1 (fr) |
| TR (1) | TR26907A (fr) |
| WO (1) | WO1994004488A1 (fr) |
| ZA (1) | ZA936188B (fr) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0714879A1 (fr) * | 1994-12-01 | 1996-06-05 | BRACCO S.p.A. | Dérivés biphényles iodés et leur utilisation diagnostique |
| US5958375A (en) * | 1994-09-23 | 1999-09-28 | Nycomed Imaging As | Urea-linked, iodinated bis phenyl compounds for X-ray contrast media |
| US8926945B2 (en) | 2005-10-07 | 2015-01-06 | Guerbet | Compounds comprising a biological target recognizing part, coupled to a signal part capable of complexing gallium |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9419203D0 (en) * | 1994-09-23 | 1994-11-09 | Nycomed Innovation Ab | Contrast media |
| US6310243B1 (en) | 1994-09-23 | 2001-10-30 | Nycomed Imaging As | Iodinated x-ray contrast media |
| US6265610B1 (en) * | 1999-01-12 | 2001-07-24 | The University Of North Carolina At Chapel Hill | Contrast media for angiography |
| WO2017165841A1 (fr) | 2016-03-25 | 2017-09-28 | Nanoprobes, Inc. | Particules à base d'iode |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3429949A1 (de) * | 1984-08-10 | 1986-02-20 | Schering AG, 1000 Berlin und 4709 Bergkamen | Neue nicht -ionische 2,4,6-trijod-isophthalsaeure-bis-amide, verfahren zu ihrer herstellung und ihre verwendung als roentgenkontrastmittel |
| EP0308364A2 (fr) * | 1987-09-17 | 1989-03-22 | Schering Aktiengesellschaft | Acides bis(3,5-dicarbamoyl-2-4-6-triiodanilide) ou carboxyliques, leur procédé de préparation ainsi que les agents de contraste aux rayons X qui les contiennent |
| EP0357467A1 (fr) * | 1988-06-02 | 1990-03-07 | Guerbet S.A. | Nouveaux composés triiodobenzéniques non ioniques iodés et produits de contraste les contenant |
-
1992
- 1992-08-25 FR FR9210270A patent/FR2695125B1/fr not_active Expired - Fee Related
-
1993
- 1993-08-23 IL IL106768A patent/IL106768A0/xx unknown
- 1993-08-24 PL PL93307587A patent/PL307587A1/xx unknown
- 1993-08-24 US US08/387,721 patent/US5618977A/en not_active Expired - Lifetime
- 1993-08-24 HU HU9500578A patent/HUT70943A/hu unknown
- 1993-08-24 ZA ZA936188A patent/ZA936188B/xx unknown
- 1993-08-24 JP JP6505969A patent/JPH08500355A/ja active Pending
- 1993-08-24 AU AU49644/93A patent/AU4964493A/en not_active Abandoned
- 1993-08-24 EP EP93919397A patent/EP0656884A1/fr not_active Withdrawn
- 1993-08-24 CA CA002142986A patent/CA2142986A1/fr not_active Abandoned
- 1993-08-24 WO PCT/FR1993/000824 patent/WO1994004488A1/fr not_active Application Discontinuation
- 1993-08-25 TR TR00731/93A patent/TR26907A/xx unknown
- 1993-08-25 CN CN93118233A patent/CN1090572A/zh active Pending
-
1995
- 1995-02-22 NO NO950665A patent/NO950665L/no unknown
- 1995-02-24 FI FI950874A patent/FI950874A/fi unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3429949A1 (de) * | 1984-08-10 | 1986-02-20 | Schering AG, 1000 Berlin und 4709 Bergkamen | Neue nicht -ionische 2,4,6-trijod-isophthalsaeure-bis-amide, verfahren zu ihrer herstellung und ihre verwendung als roentgenkontrastmittel |
| EP0308364A2 (fr) * | 1987-09-17 | 1989-03-22 | Schering Aktiengesellschaft | Acides bis(3,5-dicarbamoyl-2-4-6-triiodanilide) ou carboxyliques, leur procédé de préparation ainsi que les agents de contraste aux rayons X qui les contiennent |
| EP0357467A1 (fr) * | 1988-06-02 | 1990-03-07 | Guerbet S.A. | Nouveaux composés triiodobenzéniques non ioniques iodés et produits de contraste les contenant |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5958375A (en) * | 1994-09-23 | 1999-09-28 | Nycomed Imaging As | Urea-linked, iodinated bis phenyl compounds for X-ray contrast media |
| EP0714879A1 (fr) * | 1994-12-01 | 1996-06-05 | BRACCO S.p.A. | Dérivés biphényles iodés et leur utilisation diagnostique |
| US8926945B2 (en) | 2005-10-07 | 2015-01-06 | Guerbet | Compounds comprising a biological target recognizing part, coupled to a signal part capable of complexing gallium |
Also Published As
| Publication number | Publication date |
|---|---|
| AU4964493A (en) | 1994-03-15 |
| TR26907A (tr) | 1994-08-22 |
| FI950874A0 (fi) | 1995-02-24 |
| ZA936188B (en) | 1994-06-24 |
| JPH08500355A (ja) | 1996-01-16 |
| FI950874A (fi) | 1995-02-24 |
| EP0656884A1 (fr) | 1995-06-14 |
| IL106768A0 (en) | 1993-12-08 |
| US5618977A (en) | 1997-04-08 |
| HUT70943A (en) | 1995-11-28 |
| CN1090572A (zh) | 1994-08-10 |
| NO950665D0 (no) | 1995-02-22 |
| HU9500578D0 (en) | 1995-04-28 |
| FR2695125B1 (fr) | 1994-12-23 |
| CA2142986A1 (fr) | 1994-03-03 |
| NO950665L (no) | 1995-04-24 |
| PL307587A1 (en) | 1995-05-29 |
| FR2695125A1 (fr) | 1994-03-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0501875B1 (fr) | Nouveaux composés poly-iodés, procédé de préparation, produit de contraste les contenant | |
| EP0558395B1 (fr) | Composés utilisables dans des produits de contraste pour la radiographie | |
| BE897566A (fr) | Derives d'acides w-amines, leur preparation et utilisation ainsi que les compositions contenant ces derives. | |
| WO1994004488A1 (fr) | Nouveaux composes poly-iodes, procede de preparation, produit de contraste les contenant | |
| EP0461958B1 (fr) | Dérivés de 2-(aminoalkyl)-5-(arylalkyl)-1,3-dioxanes, leur préparation et leur application en thérapeutique | |
| EP0357467B1 (fr) | Nouveaux composés triiodobenzéniques non ioniques iodés et produits de contraste les contenant | |
| EP0675105B1 (fr) | Composés polyiodés, procédé de préparation et composition de diagnostic | |
| LU81676A1 (fr) | Nouveaux derives d'aurones,leur procede de preparation et leur utilisation comme produits pharmaceutiques | |
| EP0177414A1 (fr) | Composés triaminobenzéniques iodés, leur procédé de préparation et leur application dans des produits de contraste | |
| EP0118347A1 (fr) | Composés non-ioniques à structure benzénique iodée ou bromée et produits opacifiants en contenant | |
| CH615414A5 (fr) | ||
| EP0327455A1 (fr) | Ethers alkyliques ou benzyliques du phénol, leurs procédés de préparation et leur application en thérapeutique | |
| EP0437144B1 (fr) | Composés non ioniques iodés, leur procédé de préparation et produits de contraste en contenant | |
| WO1988008417A1 (fr) | Composes diamino benzeniques iodes, leur procede de preparation et produits de contraste les contenant | |
| EP0008973B1 (fr) | Procédé de fabrication des acides phénoxylactiques, de leurs dérivés et produits obtenus | |
| FR2693723A1 (fr) | Dérivés de la N-(phényléthyl-beta-ol) amine ainsi que leur procédé de préparation. | |
| CA1044697A (fr) | Procede de preparation de nouveaux azabicycloalcanes disubstitues | |
| FR2698355A1 (fr) | Nouveaux dérivés de 1-phénylbicyclo[2.2.2] octane, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent. | |
| FR2643077A1 (fr) | Nouveaux composes non ioniques iodes et produits de contraste les contenant | |
| CH475246A (fr) | Procédé de préparation d'un naphtalène substitué | |
| CH637378A5 (fr) | Benzamides cycloalkylalkyliques. | |
| EP0097072A1 (fr) | Nouveaux dérivés de benzylsulfinylacétamide, utilisation en thérapeutique et procédé de préparation | |
| BE504403A (fr) | ||
| FR2624857A1 (fr) | Derives de nicotinoyl-piperazine, procede de preparation et utilisation en therapeutique | |
| BE519971A (fr) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BB BG BR BY CA CZ FI HU JP KP KR KZ LK MG MN MW NO NZ PL RO RU SD SK UA US VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1993919397 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2142986 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 950874 Country of ref document: FI |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993919397 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08387721 Country of ref document: US |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1993919397 Country of ref document: EP |