TWI637954B - 具有蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑活性之化合物 - Google Patents
具有蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑活性之化合物 Download PDFInfo
- Publication number
- TWI637954B TWI637954B TW102144561A TW102144561A TWI637954B TW I637954 B TWI637954 B TW I637954B TW 102144561 A TW102144561 A TW 102144561A TW 102144561 A TW102144561 A TW 102144561A TW I637954 B TWI637954 B TW I637954B
- Authority
- TW
- Taiwan
- Prior art keywords
- methyl
- compound
- phenyl
- formula
- group
- Prior art date
Links
- SBQUNTRLQMQGSF-UHFFFAOYSA-N Cc(cc1)c(C=CC(N2)=O)c2c1O Chemical compound Cc(cc1)c(C=CC(N2)=O)c2c1O SBQUNTRLQMQGSF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/439—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom the ring forming part of a bridged ring system, e.g. quinuclidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4709—Non-condensed quinolines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
- A61K9/0073—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/12—Aerosols; Foams
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M11/00—Sprayers or atomisers specially adapted for therapeutic purposes
- A61M11/005—Sprayers or atomisers specially adapted for therapeutic purposes using ultrasonics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M15/00—Inhalators
- A61M15/0028—Inhalators using prepacked dosages, one for each application, e.g. capsules to be perforated or broken-up
- A61M15/003—Inhalators using prepacked dosages, one for each application, e.g. capsules to be perforated or broken-up using capsules, e.g. to be perforated or broken-up
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M15/00—Inhalators
- A61M15/0028—Inhalators using prepacked dosages, one for each application, e.g. capsules to be perforated or broken-up
- A61M15/0045—Inhalators using prepacked dosages, one for each application, e.g. capsules to be perforated or broken-up using multiple prepacked dosages on a same carrier, e.g. blisters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M15/00—Inhalators
- A61M15/0065—Inhalators with dosage or measuring devices
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M15/00—Inhalators
- A61M15/009—Inhalators using medicine packages with incorporated spraying means, e.g. aerosol cans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M16/00—Devices for influencing the respiratory system of patients by gas treatment, e.g. mouth-to-mouth respiration; Tracheal tubes
- A61M16/10—Preparation of respiratory gases or vapours
- A61M16/14—Preparation of respiratory gases or vapours by mixing different fluids, one of them being in a liquid phase
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M2202/00—Special media to be introduced, removed or treated
- A61M2202/06—Solids
- A61M2202/064—Powder
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/38—Nitrogen atoms
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Pulmonology (AREA)
- Organic Chemistry (AREA)
- Anesthesiology (AREA)
- Biomedical Technology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Emergency Medicine (AREA)
- Dispersion Chemistry (AREA)
- Biophysics (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Otolaryngology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicinal Preparation (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
本發明係有關於用作為蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑二者之化合物、其製備方法、包含該等化合物之組成物、其治療用途及與其它醫藥上活性成分之組合。
Description
本發明係有關於通式I化合物,作為蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑二者、其製備方法、包含該等化合物之組成物、其治療用途及與其它醫藥上活性成分之組合。
肺部病症諸如氣喘及慢性阻塞性肺病(COPD)常見使用支氣管擴張劑治療。已知之一類支氣管擴張劑包含β2腎上腺素受體致效劑,諸如薩布它莫(salbutamol)、芬諾特羅(fenoterol)、福莫特羅(formoterol)及薩美特羅(salmeterol)。此等化合物通常藉由吸入投予。
另一類已知之支氣管擴張劑包含蕈毒鹼受體拮抗劑(抗膽鹼激性化合物),諸如伊帕托品(ipratropium)及堤歐托品(tiotropium)。此等化合物典型地也藉由吸入投予。
β2致效劑及蕈毒鹼受體拮抗劑二者的吸入配方乃治療氣喘及COPD的有價值藥劑,兩類藥劑由於能夠鬆弛縮窄的呼吸道故可提供症狀緩解。觀察得兩類藥劑的支氣管擴張劑效果乃加乘性,促成研究兩類藥劑的組合。於1975年,經由組合兩個成分諸如芬諾特羅及伊帕托品於單一噴霧劑顯示可達成有利效果。如此促成了溴化伊帕托品首先與芬諾特羅(Berodual,1980年問市)及其次與薩布它莫(Combivent,1994年問市)的固定劑量組合的發展。
更為晚近出現長效蕈毒鹼拮抗劑及長效β2致效劑二者,促成了此等藥劑之組合的發展。舉例言之,WO 00/69468揭示含有蕈毒鹼受體拮抗劑諸如溴化堤歐托品及β2腎上腺素受體致效劑,諸如反丁烯二酸福莫特羅及薩美特羅之醫藥組成物;及WO 2005/115467揭示一種組合包含β2致效劑及M3蕈毒鹼受體拮抗劑,其為3(R)-(2-羥基-2,2-二噻吩-2-基乙醯氧基)-1-(3-苯氧基丙基)-1-四價氮二環[2.2.2]辛烷之鹽。
固定劑量組合的發展之替代方案係為識別組合蕈毒鹼拮抗作用及β2致效作用兩種活性的分子。實際上,具有β2腎上腺素受體致效劑及蕈毒鹼受體拮抗劑兩種活性之化合物係被高度期望,原因在於此等雙功能化合物透過兩個獨立的作用機轉提供支氣管擴張作用,而具有單一分子藥力學。
此種化合物係描述於一些專利申請案中,諸如WO 2004/074246、WO 2004/074812、WO 2005/051946、WO 2006/023457、WO2006/023460、WO 2010/123766、WO 2011/048409及共同審查中之專利申請案PCT/EP2012/060795。
除了具有β2腎上腺素受體致效劑及蕈毒鹼受體拮抗劑二者活性之外,今日發現有些特定胺基甲酸酯衍生物具有對M3蕈毒鹼受體升高之親和力及長效支氣管擴張活性。
本發明係有關於通式I化合物,作為蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑二者、其製備方法、包含該等化合物之組成物、其治療用途及與其它醫藥上活性成分之組合,該等活性成分中例如為目前用於呼吸病症之治療者,其中包括β2致效劑、抗蕈毒鹼劑、有絲
分裂原活化蛋白質激酶(P38 MAP激酶)抑制劑、核因子κ-B激酶亞單位β(IKK2)抑制劑、人類嗜中性彈力蛋白酶(HNE)抑制劑、磷酸二酯酶4(PDE4)抑制劑、白三烯調節劑、非類固醇消炎藥(NSAID)、止咳劑、黏液調節劑、化痰劑、祛痰劑/黏液促動調節劑、胜肽化痰劑、抗生素、JAK抑制劑、SYK抑制劑、PI3Kδ或PI3Kγ抑制劑、皮質類固醇及M3拮抗劑/PDE4抑制劑(MAPI)。
更明確言之,本發明係有關於通式I化合物
其中Q為式Q1、Q2或Q3之基團
Z為H或OH;Y係選自Y’及Y1其為下式之二價基團
或
其中A1及A2獨立地為不存在或為(C1-C6)伸烷基;B為不存在或係選自於由伸芳基及伸雜芳基所組成之組群,選擇性地經以選自於鹵原子、(C1-C6)烷基、鹵(C1-C6)烷基及(C1-C6)烷氧基中之一或多個基團取代;C為不存在或為-OC(O)-或為下列基團C1-C3中之一者
其中R4為H或線性或分支(C1-C4)烷基;D為不存在或係選自於由伸芳基及伸雜芳基所組成之組群,選擇性地經以選自於一或多個(C1-C6)烷基取代;n及n’獨立地為0或1至3之整數;E為不存在或係選自-O-及-OC(O)-;G為伸芳基;R1及R2獨立地為H或芳基;R3為式J1或J2基團
其中R5為式K基團
其中p’為0或1,P為不存在或為CO,q為不存在或為1,及W為雜芳基;及其醫藥上可接受之鹽類或溶劑合物。
表示法「(C1-Cx)烷基」係指直鏈或分支鏈烷基其中碳原子數係為1至x。該等基團之實例為甲基、乙基、正丙基、異丙基、第三丁基、戊基、己基等。
表示法「鹵(C1-C6)烷基」係指經以一或多個鹵原子取代之直鏈或分支鏈烷基其中碳原子數係為1至6。
以類似方式,表示法「(C1-Cx)伸烷基」係指二價基團,諸如亞甲基、伸乙基、伸正丙基、伸異丙基、伸第三丁基、伸戊基、伸己基、伸辛基、伸壬基、伸癸基、伸十一烷基、伸十二烷基等。
表示法「(C1-C10)烷氧基」係指烷基-氧(例如烷氧)基,該烷基部分係如前文定義。該等基團之實例包含甲氧基、乙氧基、正丙氧基、異丙氧基、正丁氧基、異丁氧基、第二丁氧基、第三丁氧基、戊氧基、己氧基等。
表示法「芳基」係指含5至20較佳為5至15個環原子之一-、二-、或三環環系,及其中至少一個環為芳香族。
表示法「雜芳基」係指含5至20較佳為5至15個環原子之一-、二-、或三環環系,及其中至少一個環為芳香族及其中至少一個碳環原子為雜原子或雜芳香族基(例如N、NH、S或O)。
適合的芳基或雜芳基一環系之實例包括例如噻吩、苯、吡咯、吡唑、咪唑、異唑、唑、異噻唑、噻唑、吡啶、咪唑啶、呋喃基團等。
適合的芳基或雜芳基二環系之實例包括萘、伸聯苯基、嘌呤、喋啶、苯并三唑、喹啉、異喹啉、吲哚、異吲哚、苯并噻吩、二氫苯并二、二氫茚、二氫苯并二呯、苯并基團等。
適合的芳基或雜芳基三環系之實例包括茀基團以及前述雜芳基二環系之苯并稠合衍生物。
以類似方式,表示法「伸芳基」及「伸雜芳基」係指二價基團,諸如伸苯基、伸聯苯基及伸噻吩基。
每當鹼性胺基或第四銨基存在於式I化合物時,可存在有選自於下列之生理上可接受之陰離子:氯陰離子、溴陰離子、碘陰離子、三氟乙酸根、甲酸根、硫酸根、磷酸根、甲烷磺酸根、硝酸根、順丁烯二酸根、乙酸根、檸檬酸根、反丁烯二酸根、酒石酸根、草酸根、丁二酸根、苯甲酸根、對甲苯磺酸根、帕莫酸根(pamoate)及萘二磺酸根。同理,於酸性基諸如COOH基之存在下,也可存在有相對應的生理上陽離子鹽,例如包括鹼金屬離子或鹼土金屬離子。
顯然通式I化合物可含有非對稱中心。因此,本發明也包括光學立體異構物、非對映異構物、及其以任一種比例之混合物中之任一者。
更明確言之,取決於先前報告的提供給R1及R2之意義,
鏈接至R1、R2、G及-NH-基之碳原子可表示對掌中心。
於一具體例中,該組態為(S)。
於另一具體例中,此一對掌中心的絕對組態較佳為(R)。
於另一較佳具體例中,本發明描述的通式I化合物係呈非對映異構物之混合物存在。
熟諳技藝人士顯然易知通式I化合物其中R3為J1或J2
含有三個立體生成中心,如下以星號(*)指示。
如此表示式I結構式係以八個不同立體異構物加以特徵化。
須瞭解後述式I化合物之全部較佳組群或具體例可經必要修正彼此組合及應用)。
化合物之第一較佳組群為通式I化合物其中Q為式Q1、Q2或Q3基團
Z為H或OH;Y係選自Y’及Y1其為下式之二價基團
或
其中A1及A2獨立地為不存在或為(C1-C6)伸烷基;B為不存在或係選自於由伸芳基及伸雜芳基所組成之組群,選擇性地經以選自於鹵原子、(C1-C6)烷基、鹵(C1-C6)烷基及(C1-C6)烷氧基中之一或多個基團取代;C為不存在或為-OC(O)-或為下列基團C1-C3中之一者
其中R4為H或線性或分支(C1-C4)烷基;D為不存在或係選自於由伸芳基及伸雜芳基所組成之組群,選擇性地經以選自於一或多個(C1-C6)烷基取代;n及n’獨立地為0或1至3之整數;E為不存在或係選自-O-及-OC(O)-;G為伸芳基;R1、R2及R3係如前文定義。
於此第一組群中又更佳者為通式I化合物其中Q為Q1
Z為-OH,Y為Y’其為下式之二價基團
A1及A2獨立地為不存在或為(C1-C6)伸烷基;B為不存在或係選自於由伸芳基及伸雜芳基所組成之組群,選擇性地經以選自於鹵原子、(C1-C6)烷基、鹵(C1-C6)烷基及(C1-C6)烷氧基中之一或多個基團取代;C為不存在或為-OC(O)-或為下列基團C1-C3中之一者
其中R4為H或線性或分支(C1-C4)烷基;D為不存在或係選自於由伸芳基及伸雜芳基所組成之組群,選擇性地經以選自於一或多個(C1-C6)烷基取代;n及n’獨立地為0或1至3之整數;E為不存在或為-O-;及G為伸芳基。
於此第一組群中又更佳者為通式I化合物其中A1係選自於由亞甲基、伸乙基及伸丙基所組成之組群;A2係為不存在或係選自於由亞甲基、伸乙基及伸丙基所組成之組群;B係為不存在或係選自於由伸苯基、伸萘基、吡啶二基、呋喃二基、噻吩二基、及吡唑二基所組成之組群;C係為不存在或為-OC(O)-或為式C2基團
其中n為2;n’為1;D為不存在或為伸苯基;E為不存在或為-O-;及G為伸苯基。
較佳通式I化合物之第二組群為其中Q為Q1
Z為-OH;Y為Y1其係為下式之二價基團
A1為不存在或為(C1-C6)伸烷基;C係為不存在或為-OC(O)-或為C1
其中R4為H;B為不存在或係選自於由伸芳基及伸雜芳基所組成之組群,選擇性地經以選自於鹵原子、(C1-C6)烷基、鹵(C1-C6)烷基及(C1-C6)烷氧基中之一或多個基團取代;D為不存在或為伸芳基;n’為0或1至3之整數;E為不存在或為-O-;及G為伸芳基。
於此組群內部之又更佳者為通式I化合物其中A1為伸丁基;C為不存在或為-OC(O)-或為C1
其中R4為H;B為不存在或係選自於由吡唑二基、噻吩二基、吡啶
二基、呋喃二基、及唑二基所組成之組群,選擇性地經以選自於甲基之一或多個基團取代;D為不存在,n’為1,E為-O-,G為伸苯基及R3為式J1或J2基團
其中R5為式K基團
其中p’為0或1;P為不存在或為CO;q為不存在或為1,及W為雜芳基;及其醫藥上可接受之鹽類或溶劑合物。
本發明也提出單獨式I化合物或組合或混合一或多個醫藥上可接受之載劑及/或賦形劑的醫藥組成物。
本發明也提出式I化合物用於製備藥物之用途。
於又一態樣中,本發明提出式I化合物用於支氣管阻塞或發炎病,較佳地氣喘或慢性支氣管炎或慢性阻塞性肺病(COPD)之預防及/或治療之用途。
於又一態樣中,本發明提出式I化合物用於製造藥物之用途,該藥物係用於任何支氣管阻塞或發炎病,較佳地氣喘或慢性支氣管炎或慢性阻塞性肺病(COPD)之預防及/或治療。
本發明又提出任何支氣管阻塞或發炎病,較佳地氣喘或
慢性支氣管炎或慢性阻塞性肺病(COPD)之預防及/或治療之方法,該方法係包含對有需要之個體投予治療上有效量之通式I化合物。
本發明也提出適用於藉吸入投予之醫藥組成物。
吸入性製劑包括吸入性粉末劑、含推進劑之定量噴霧劑或不含推進劑之吸入性配方。
本發明也係有關於一種裝置其可為包含式I化合物之單劑或多劑乾粉吸入器、定量劑量吸入器及軟霧霧化器。
本發明也係有關於一種套組包含單獨式I化合物或組合或混合一或多個醫藥上可接受之載劑及/或賦形劑的醫藥組成物及一裝置其可為包含式I化合物之單劑或多劑乾粉吸入器、定量劑量吸入器及軟霧霧化器。
依據特定具體例,本發明提出如下報告之該等化合物:
本發明也提供包含本發明化合物本身或呈醫藥上可接受之鹽,及一或多個醫藥上可接受之載劑及/或賦形劑的醫藥組成物。
本發明化合物可作為唯一活性劑或組合其它醫藥上活性成分包括目前用於呼吸病症治療者投藥,例如β2致效劑、抗蕈毒鹼劑、有絲分裂原活化蛋白質激酶(P38 MAP激酶)抑制劑、核因子κ-B激酶亞單位β(IKK2)抑制劑、人類嗜中性彈力蛋白酶(HNE)抑制劑、磷酸二酯酶4(PDE4)抑制劑、白三烯調節劑、非類固醇消炎藥(NSAID)、止咳劑、黏液調節劑、化痰劑、祛痰劑/黏液促動調節劑、胜肽化痰劑、抗生素、JAK抑制劑、SYK抑制劑、PI3Kδ或PI3Kγ抑制劑、皮質類固醇及M3拮抗劑/PDE4抑制劑(MAPI)。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之β2致效劑的組合:卡莫特羅(carmoterol)、GSK-642444、英達卡特羅(indacaterol)、迷維特羅(milveterol)、亞弗莫特羅(arformoterol)、酒石酸亞弗莫特羅、福莫特羅(formoterol)、反丁烯二酸福莫特羅、薩美特羅(salmeterol)、羥萘甲酸薩美特羅、薩布特羅(salbutamol)、阿布特羅(albuterol)、雷瓦布特羅(levalbuterol)、特布塔林(terbutaline)、英達卡特羅(QAB-149)、AZD-3199、BI-1744-CL、LAS-100977、GSK159797、GSK59790、GSK159802、GSK642444、GSK678007、GSK96108、班布特羅(bambuterol)、異波特瑞諾(isoproterenol)、波卡特羅(procaterol)、克蘭布特羅(clenbuterol)、瑞波特羅(reproterol)、芬諾特羅(fenoterol)、比妥特羅(bitolterol)、波薩特羅(brodxatelor)及ASF-1020及其鹽類。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之抗蕈毒鹼劑的組合:雅葵帝寧
(aclidinium)、堤歐托品(tiotropium)、溴化堤歐托品(Spiriva®)、伊帕托品(ipratropium)、溴化伊帕托品、托斯品(trospium)、糖基吡咯酸酯(glycopyrrolate)、NVA237、LAS34273、GSK656398、GSK233705、GSK57319、LAS35201、OAT370及奧希托品(oxitropium)鹽類。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之PDE4抑制劑的組合:AN-2728、AN-2898、CBS-3595、阿沛米雷(apremilast)、ELB-353、KF-66490、K-34、LAS-37779、IBFB-211913、AWD-12-281、喜潘菲林(cipamfylline)、喜洛米雷(cilomilast)、羅芙米雷(roflumilast)、BAY19-8004及SCH-351591、AN-6415、英杜斯(indus)-82010,TPI-PD3、ELB-353、CC-11050、GSK-256066、歐格米雷(oglemilast)、OX-914、特妥米雷(tetomilast)、MEM-1414及RPL-554。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之P38 MAP激酶抑制劑的組合:塞瑪皮莫(semapimod)、塔瑪皮莫(talmapimod)、泊菲尼冬(pirfenidone)、PH-797804、GSK-725、GSK856553、GSK681323、米諾金(minokine)及洛瑪皮莫(losmapimod)及其鹽類。
於一較佳具體例中,本發明提供本發明化合物與IKK2抑制劑之組合。
本發明也提供本發明化合物與選自於下列所組成之組群的HNE抑制劑之組合:AAT、ADC-7828、亞利瓦(aeriva)、TAPI、AE-3763、KRP-109、AX-9657、POL-6014、AER-002、AGTC-0106、
瑞斯利瓦(respriva)、AZD-9668、哲麥拉(zemaira)、AAT IV、PGX-100、伊拉芬(elafin)、SPHD-400、波拉斯汀(prolastin)C及吸入性波拉斯汀。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之白三烯調節劑的組合:蒙特盧卡(montelukast)、哲佛盧卡(zafirlukast)及潘盧卡(pranlukast)。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之NSAID的組合:伊布波芬(ibuprofen)及奇托波芬(ketoprofen)。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之止咳劑的組合:可待因(codeine)及美沙芬(dextramorphan)。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之化痰劑的組合:N-乙醯基半胱胺酸及芙朵斯甜(fudostein)。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之袪痰劑/黏液促動調節劑的組合:安柏索(ambroxol)、高張溶液(例如食鹽水或甘露糖醇)及界面活性劑。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之胜肽化痰劑的組合:重組人類去氧核糖核酸I(dornase-alfa及rhDNase)及海理喜定(helicidin)。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之抗生素的組合:阿吉黴素
(azithromycin)、妥巴黴素(tobramycin)及雅崔歐南(aztreonam)。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之黏液調節劑的組合:INS-37217,蒂夸佛索(diquafosol)、希貝拿代(sibenadet)、CS-003、托內坦(talnetant)、DNK-333、MSI-1956及格菲提尼(gefitinib)。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之JAK抑制劑的組合:CP-690550及GLPG0634。
本發明也提供本發明化合物本身或呈醫藥上可接受之鹽與選自於由下列所組成之該組群之SYK抑制劑的組合:R406、R343及PRT062607。
本發明也提供式(I)化合物與選自於由下列所組成之組群之皮質類固醇的組合:德莎美沙松、芙提卡松(fluticasone)、糠酸芙提卡松、丙酸芙提卡松、沛尼索隆(prednisolone)、貝塔美沙松(betamethasone)、布迪索奈(budesonide)、摩米塔松(mometasone)、糠酸摩米塔松、崔安喜諾隆(triamcinolone)丙酮化物、喜雷索奈(ciclesonide)、TPI-1020、貝克美沙松(beclomethasone)、二丙酸貝克美沙松、沛尼松(prednisone)、迪法雜克(deflazacort)、氫皮質酮(hydrocortisone)、QAE-397及芙尼索萊(flunisolide)。
本發明化合物可使用如下概略方法及程序或藉使用熟諳技藝人士方便易得的其它資訊而從方便易得的起始物料製備。雖然本發明之特定具體例可於此處顯示或描述,但熟諳技藝人士將瞭解本
發明之全部具體例及態樣可使用此處描述之方法,或使用熟諳技藝人士已知之其它方法、反應物及起始物料製備。也須瞭解,除非另行陳述否則當給予典型或較佳製程條件(亦即反應溫度、時間、反應物之莫耳比、溶劑、壓力等)時,也可使用其它製程條件。雖然最佳反應條件可取決於所使用的特定反應物或溶劑,但此等條件也容易由熟諳技藝人士藉例行最佳化程序決定。
通式I化合物可依據如下合成反應式1及2製備。
通式VIII化合物表示一種化合物其中A1為經以側氧基取代之伸烷基,結果導致醛或酮被保護成環狀縮醛。環狀縮醛保護基(PG)可被去除而獲得通式XXIII化合物。
通式I化合物之合成除了前述方法外,可能要求潛在反應性官能基的保護。此種情況下,可相容保護基(PG)及其特定保護及脫保護方法之實例係說明於T.W.Green及P.Wutz之「Protecting groups in organic Synthesis」(Wiley-Interscience publication,1999)。通式I化合物例如可經由通式XVII化合物與通式XVIII化合物反應製備。此種還原胺化反應可遵照參考文獻中描述及技藝界已知之若干不同方案進行。舉例言之,可使用還原劑諸如NaBH4、NaCNBH3或NaBAcO3H,於溶劑諸如甲醇、乙醇、四氫呋喃(THF)或二氯甲烷(DCM)中進行。在添加還原劑之前獲得亞胺係有用的。反應於室溫順利進行1至12小時。
通式XVII之中間產物容易經由通式XIII化合物與通式XV化合物反應製備。反應於室溫或低溫於溶劑諸如DCM或吡啶順利進行1至16小時,結果獲得式XVI化合物,其於水性酸性溶液內溶液被脫保護而獲得通式XVII化合物(參考反應式1)。
通式XV化合物為市面上可得或可經由通式XIV醇與例如雙光氣於溶劑諸如DCM、THF或乙腈(ACN)於室溫或低溫反應0.5至12小時之時間,獲得通式XV化合物其中離去基LG為氯。另外,通式XIV醇可與例如甲醯基二咪唑(CDI)反應獲得相同中間產物其中LG為咪唑。具有其它LG的其它可能的中間產物可如參考文獻之說明製備。
通式XIII化合物可從通式XI化合物透過里特(Ritter)反應(乙腈及硫酸於室溫)接著於鹼性條件下進行中間產物乙醯胺的水解而
製備。
另外,通式XIII化合物可經由於氫氣環境或氫轉移條件下透過氫化還原疊氮化物式XII製備。反應於室溫或更高溫於醇發生及於1至12小時中止。另一種還原方法可為史陶丁哲(Staudinger)反應,涉及首先使用例如三苯基膦處理疊氮化物,接著使用水水解亞胺基磷烷中間產物。此種反應出現於室溫於水可相溶混溶劑例如THF。強力還原劑諸如LiAlH4於THF或醚於-40℃或低溫的使用容易許可式XIV轉換成為XIII。
疊氮化物XII係經由從式XI化合物藉與二苯基磷醯疊氮反應獲得。該反應係於高沸點諸如甲苯或二甲苯,於強鹼諸如但非限於1,8-二吖二環[5.4.0]十一碳-7-烯(DBU)於80℃至120℃範圍之溫度進行及於12至24小時反應完成。另外,式XI中間產物之羥基部分可轉化成適當離去基(LG),諸如甲烷磺醯基、甲苯磺醯基、或鹵素及然後與鹼性疊氮化物於極性溶劑諸如乙腈、DMF或N-甲基-2-吡咯啶酮(NMP)於室溫或更高溫反應。
通式XI中間產物可以數種不同方式製備。例如可從通式VII化合物其中E為-O-,與通式V醛反應製備,通式V醛具有適當羥基特徵,可方便地於標準光延反應(Mitsunobu)條件下反應。反應係於溶劑諸如THF或N-甲基-啉(NMM)於-10℃至室溫之溫度進行及於1至24小時完成。該反應係出現於偶氮二羧酸二乙酯(DEAD)或偶氮二羧酸二異丙酯(DIAD)及有機膦諸如但非限於三苯基膦之存在下。
通式VII之醇為市面上可得或可從式II化合物經由式VI化合物格利亞(Grignard)試劑之加成製備。反應正常係於質子惰性溶劑諸如醚或THF於室溫或低溫進行及於0.5至12小時完成。另外,可經由
通式II化合物,其中R2非為氫與還原劑諸如但非僅限於NaBH4還原製備,此種情況下結果獲得式VII化合物其中R1為氫。反應係於溶劑諸如甲醇、乙醇或THF內進行及於1至12小時時間完成。類似的合成方案可用於從通式IV化合物製備中間產物XI。
熟諳技藝人士顯然清楚通式VII或XI化合物之製備可透過反格利亞反應完成,其中式G-MgBr之格利亞試劑與式R1C(O)R2化合物於前述相同反應條件下反應。
通式IV化合物其中E為-O-,可遵照類似從式VII製備式XI化合物所述辦法而從通式II化合物製備。另外,通式IV化合物可經由通式II化合物與通式III化合物其中LG為離去基諸如甲苯磺酸根、甲烷磺酸根或鹵原子之烷化獲得。反應通常係於極性溶劑諸如乙腈或DMF進行,反應出現於鹼諸如鹼性碳酸鹽、重碳酸鹽或有機鹼存在下及於1至24小時不等時間完成。
式X化合物之製備可經由通式IX化合物或其類似物,其中溴係以碘或三氟甲烷磺酸根取代,與通式VIII化合物,其中n為2,於過渡金屬催化之交叉偶合反應條件下反應達成。末端烯VIII可於例如赫克(heck)反應條件下與IX反應,結果獲得伸烯基中間產物X,該產物容易利用典型雙鍵之催化氫化還原而獲得式IV化合物。如熟諳技藝人士已知,有大量方案、反應劑及催化劑可方便地用來達成期望的轉化。
另外,於鏈接基Y具有酯部分特徵的通式I化合物可經由於用於酯製備之縮合反應條件下,使用通式XXXVII化合物(其中A2係以OH官能化)處理通式XXXVI化合物製備。可能製備通式I化合物,其中C係等於C1,於始於羧酸及胺類製備醯胺之已知反應條件下,通式XXXVI化合物與化合物XXXVII(其中A2係經以-NR4取代)而製備通式I
化合物。
於本發明之另一具體例中,通式I化合物可從不同合成辦法製備,若反應係與式LG-A1-B-A2-C-D-CH2-(CH)2-E-H化合物進行,其中通式XXIV化合物與通式XXVI化合物於過渡金屬催化之交叉耦合反應條件下反應,接著還原雙鍵-(CH)2-,獲得式I化合物其中n=2或n=3。另外,可經由於前文對式II化合物與式III化合物反應所述條件下,經由式XXI化合物與式XXVII化合物反應製備。
式XXIV及XXI中間產物可經由於前文對式XVII與式XVIII化合物反應所述之還原胺化條件下,分別地始於式XXIII及XX化合物,經由使式XVIII化合物反應製備。另外,於前文針對經由化合物II與化合物III反應製備化合物IV所述之烷化條件下,分別使用式XXV及XXII化合物烷化化合物XVIII製備式XXIV及XXI化合物。
通式XVIII化合物可經由式XIX疊氮化物之單純還原獲得。該反應可利用於鈀催化劑存在下之催化氫化完成。反應係發生於極性溶劑諸如甲醇或乙醇,於氫氣環境下或於氫轉移條件下使用例如1,4-環己二烯或1-甲基1,4-環己二烯作為氫來源進行。反應於室溫(RT)進行。當於氫轉移條件下進行時可能要求較高溫。
疊氮化物XIX容易藉已知烷基溴與鹼性疊氮化物之親核取代而從XXIX製備。反應係於50℃至80℃溫度及於例如DMF或NMP之極性溶劑內進行,及可於鹼性碘化物之存在下加速進行。
於本發明之額外具體例中,式I化合物其中R3為J2或另一個第四銨鹽特徵基團可經由相對應式I第三胺前驅物,其中R3為J1與式XXVIII化合物反應製備。該反應係於室溫或更高溫於溶劑諸如DCM、乙腈、甲醇或AcOEt經1至24小時時間順利進行。
於本發明之另一個具體例中,通式XXI化合物可經由通式XXIX中間產物與通式XXX胺反應製備。此種反應為胺之常見烷化反應,其中離去基LG(通常為氯、溴或硫酸根)係藉親核基團類似胺XXX本身或於胺部分保護加以置換。通常於極性溶劑於高於室溫之溫度進行此種反應之數種方法係說明於參考文獻。類似反應可用於通式XXXVII化合物之製備。
熟諳技藝人士顯然易知通式I化合物其中R3為J1或J2含有三個立體產生中心,如下以符號星號(*)指示。如此表示式I結構式之特徵為具有八種不同的立體異構物。
於更方便之辦法中,於前述反應中各個單一立體異構物之合成可只使用對映異構物上純質的中間產物完成。
通式I化合物其中R3為J1或J2之製備必需對映異構純質醇為市面上可得。
單一對映異構純質通式XXIX化合物其中LG為溴之製備係說明於WO2005/080324、US2005-2222128、WO2004/032921、US2005/215590及WO2005/092861(由WO2007/107228引用)。通式XXXII之對映異構純質化合物可經由外消旋混合物之對掌層析分離獲
得,或始於通式XXXI之對映異構純質胺化合物獲得。式XXXI中間化合物含有鹼性基團,可經由使用對映異構純質羧酸鹽化外消旋混合物獲得的非對映異構物鹽,利用該鹽之結晶化獲得兩種對映異構物。廣用於此項目的之羧酸例如為扁桃酸、酒石酸及其衍生物。鹼XXXI溶解於適當極性溶劑,然後使用對映異構純質羧酸處理,造成兩種非對映異構物鹽中之一種沈澱。要求重複該程序數次以獲得期望的對映異構過量程度。
另外,式XXXI之胺可遵照例如參考文獻(Tetrahedron:Asymmetry 13(2002)303-310)所述辦法透過對映選擇性合成獲得,其中式II之醛,其中R2為H,首先使用對映異構純質第三丁基亞磺醯亞胺處理,及然後使用R2MgBr或R2Li(其中R2非為H)處理,接著中間產物水解,結果形成對映異構豐富式XXXI化合物,其可就此使用或進一步純化以提高對映異構物過量而使用。
通式XXXI之外消旋胺可以數種不同方式製備,例如經由添加羥基胺之通式II化合物,接著為所得肟中間產物之還原,該等反應可於技藝界已知之數種反應條件下進行。例如催化氫化或還原劑諸如LiAlH4或鋅於甲酸銨存在下的使用也是極為有效地將肟還原成為胺的方法。
可得的通式XXXI之胺容易於前述反應條件下進一步衍生。例如可於針對式II化合物使用式III化合物烷化所述條件下,使用經保護之式III醛處理,結果獲得通式XXXIII化合物。胺基之脫保護及式XV之反應以製備通式XVI化合物。
另外,通式I化合物可偶合通式XXXVI化合物與通式XXXVII化合物,獲得通式I化合物其中C為-OCO-或C1製備。此種酯或
醯胺可於熟諳技藝人士已知之不同反應條件下獲得。反應要求使用反應物活化酸XXXVI,該反應物諸如N,N’-二環己基甲二醯亞胺(DCC)、1-乙基-3-(3-二甲基胺基丙基)甲二醯亞胺(EDC)、2-(1H-苯并三唑-1-基)-1,1,3,3-四甲基脲鎓六氟磷酸鹽(HBTU)、(O-(7-吖苯并三唑-1-基)-N,N,N’,N’-四甲基脲鎓六氟磷酸鹽(HATU)或可轉換成為相對應之醯氯。活化酯可於DCM、吡啶或其它質子惰性溶劑內與式XXXVII化合物順利反應。
式XXXVI化合物可始於XXXII,透過式XXXIV化合物之烷化,脫保護及與式XV化合物之反應而予製備。此種轉化之反應條件係如前文說明且係描述於參考文獻。酸XXXVI係易於以如前文就與式XXXVII化合物反應所述方式而與式XXXVIII化合物反應,獲得式XVI化合物。
通式XXXVII化合物可經由通式XXIX化合物與式NH2-A1-B-A2-OH或NH2-A1-B-A2-NHR4之胺於針對通式XXIX化合物與通式XXX化合物反應所述之反應條件下反應製備。
針對全部前文之說明,通式I化合物之合成可遵照數種不同辦法進行。特別須注意必需反應條件強烈取決於鏈接基Y及Y1之本質及存在於鏈接基上的官能基。前文製備式I化合物其中C為-OC(O)-或C1之實施例係使熟諳技藝人士瞭解本發明之態樣。
用於本發明化合物特徵化之LCMS方法A、B、C及D說明如下:LCMS/HPLC方法
方法A(10cm_ESCI_FORMIC)
HPLC配置
溶劑:乙腈(遠UV級)含0.1%(V/V)甲酸
水(PureLab Option單元為高純度)含0.1%甲酸
管柱:-Phenomenex Luna 5μ C18(2),100×4.6mm。(Plus guard卡匣)
流速:-2毫升/分鐘
梯度:-A:水/甲酸B:MeCN/甲酸
典型注入2-7微升(濃度約0.2-1毫克/毫升)。
透過HP或Waters DAD之UV檢測
起始範圍(nm)210 結束範圍(nm)400 範圍間隔(nm)4.0。
其它波長軌跡係從DAD資料擷取。
選擇性ELS檢測係使用Polymer Labs ELS-1000。
MS檢測:Micromass ZQ,單一四極LC-MS或Quattro Micro LC-MS-MS。
分流器獲得約300微升/分鐘至質譜
MS資料之掃描範圍(m/z)
起始(m/z)100
結束(m/z)650或1500(需要時)
使用+ve/-ve切換
離子化為例行ESCI選項從單一回合獲得ESI及APCI二者資料。
典型ESI電壓及溫度為:來源120-150C 3.5KV毛細管 25V錐
典型APCI電壓及溫度為:來源140-160C 17uA電暈 25V錐
方法B(HPLC條件-15cm_Formic_Ascentis_HPLC_CH3CN)
HPLC配置
溶劑:-乙腈(遠UV級)含0.1%(V/V)甲酸
水(透過PureLab Ultra單元為高純度)含0.1%甲酸
管柱:-Supelco、Ascentis® Express C18或Hichrom Halo C18,2.7μm C18,150×4.6mm。
流速:-1毫升/分鐘
梯度:-A:水/甲酸 B:MeCN/甲酸
典型注入0.2-10微升
最大壓力設定值400巴。
儀器:Agilent 1100,雙泵浦,Agilent取樣器及Agilent DAD檢測器二極體陣列檢測:(300nm,帶寬200nm;參考450nm,帶寬100nm)
方法C(HPLC條件-10cm_Formic_ACE-AR_HPLC_CH3CN)
HPLC配置
溶劑:-乙腈(遠UV級)含0.1%(V/V)甲酸
水(透過PureLab Ultra單元為高純度)含0.1%甲酸
管柱:-Hichrom ACE 3 C18-AR混合模式管柱100×4.6mm。
流速:-1毫升/分鐘
梯度:-A:水/甲酸 B:MeCN/甲酸
典型注入0.2-10微升
最大壓力設定值400巴。
儀器:Agilent 1100,雙泵浦,Agilent取樣器及Agilent DAD檢測器二極體陣列檢測:(300nm,帶寬200nm;參考450nm,帶寬100nm)
方法D(HPLC條件-25cm_Acidic_Prodigy_HPLC)
HPLC配置
溶劑:-乙腈(遠UV級)含0.1%(V/V)甲酸
水(透過PureLab Option單元為高純度)含0.1%甲酸
管柱:-Phenominex Prodigy 5μ ODS 3,250×4.6mm。
流速;-1毫升/分鐘
梯度:-A:水/甲酸 B:MeCN/甲酸
典型注入2-7微升
儀器:Agilent 1100,雙泵浦,Agilent取樣器及Agilent DAD檢測器
本發明也提供式I化合物混合一或多種醫藥上可接受之載劑之醫藥組成物,例如Remington’s Pharmaceutical Sciences Handbook,XVII Ed.,Mack Pub.,N.Y.,U.S.A.所述。
本發明化合物之投予可依據病人的需求完成,例如經口、經鼻、經腸道外(皮下、靜脈、肌肉、胸內或藉輸注)、藉吸入、經直腸、經陰道、局部(topically)、局部(locally)、經皮、及經眼投予。
多種固體口服劑型可用以投予本發明化合物,包括諸如錠劑、粒狀膠囊劑、膠囊劑、橢圓錠、粒劑、菱形錠及散劑等固體劑型。本發明化合物可單獨投予或組合各種醫藥上可接受之載劑、稀釋劑(諸如蔗糖、甘露糖醇、乳糖、澱粉)及已知賦形劑投予,賦形劑包括懸浮劑、增溶劑、緩衝劑、黏結劑、崩散劑、保藏劑、著色劑、矯味劑、潤滑劑等。定時釋放膠囊劑、錠劑及膠漿劑也可優異地用以投予
本發明化合物。
各種液體口服劑型也可用以投予本發明化合物,包括水性及非水性溶液劑、乳液劑、懸浮液劑、糖漿劑及酏劑。此等劑型也可含有適當已知惰性稀釋劑諸如水及適當已知賦形劑,諸如保藏劑、濕潤劑、甜味劑、矯味劑及乳化及/或懸浮本發明化合物之作用劑。本發明化合物例如可呈等張性無菌溶液經靜脈注射。其它製劑亦屬可能。
經直腸投予本發明化合物之栓劑可經由混合該化合物與適當賦形劑製備,諸如可可脂、水楊酸酯類及聚乙二醇類。
也已知陰道投藥配方可呈乳膏劑、膠漿劑、糊劑、泡沫劑、或噴霧配方劑型,除了活性成分外,含有諸如適當載劑。
供局部投藥,醫藥組成物可呈適合投予皮膚、眼、耳或鼻的乳膏劑、軟膏劑、硬膏劑、洗劑、乳液劑、懸浮液劑、膠漿劑、溶液劑、糊劑、散劑、噴霧劑、及滴劑劑型。局部投藥也涉及透過諸如經皮貼片等手段的經皮投藥。
為了用於呼吸道疾病之治療,依據本發明化合物較佳係藉吸入投予。
吸入製劑包括吸入性粉末、含推進劑之定量噴霧劑或不含推進劑之吸入性配方。
用於呈乾粉投藥,可利用先前技藝已知之單劑或多劑吸入器。該種情況下,粉末可填充於明膠、塑膠或其它膠囊、卡匣或罩板泡胞包裝內或填充於貯器內。
稀釋劑或載劑通常為無毒且對本發明化合物為化學惰性例如乳糖或其它適合改良可呼吸分量的添加劑可添加至本發明之粉狀化合物。
含推進劑氣體諸如氫氟烷類之吸入噴霧劑可含有呈溶液形式或分散形式之本發明化合物。推進劑驅動之配方也可含有其它成分諸如助溶劑、安定劑及選擇性之其它賦形劑。
包含本發明化合物之不含推進劑之吸入性配方可呈於水性、醇性、或水醇性介質內之溶液或懸浮液劑型,可藉先前技術已知之噴射或超音波霧化器遞送或藉軟霧噴霧器諸如Respimat®遞送。
本發明化合物可作為唯一活性劑或可組合包括目前用於呼吸道病症之治療用的其它醫藥活性成分投予,該等活性成分包含β2致效劑、抗蕈毒鹼劑、有絲分裂原活化蛋白質激酶(P38 MAP激酶)抑制劑、核因子κ-B激酶亞單位β(IKK2)抑制劑、人類嗜中性彈力蛋白酶(HNE)抑制劑、磷酸二酯酶4(PDE4)抑制劑、白三烯調節劑、非類固醇消炎藥(NSAID)、止咳劑、黏液調節劑、化痰劑、祛痰劑/黏液促動調節劑、胜肽化痰劑、抗生素、JAK抑制劑、SYK抑制劑、PI3Kδ或PI3Kγ抑制劑、皮質類固醇及M3拮抗劑/PDE4抑制劑(MAPI)。
本發明化合物之劑量取決於多項因素包括欲治療特定疾病、症狀嚴重程度、投藥途徑、給藥間隔、所用的特定化合物、該化合物之功效、毒理學、及藥力學資料。
優異地,式I化合物例如可以0.001至1000毫克/日,較佳0.1至500毫克/日之劑量投予。
當式I化合物係藉吸入途徑投予時,較佳係以0.001至500毫克/日,較佳0.1至200毫克/日之劑量投予。
式I化合物可投予用於下列疾病之預防及/或治療:支氣管阻塞或發炎疾病諸如氣喘、慢性支氣管炎、慢性阻塞性肺病(COPD)、支氣管過度反應性、咳嗽、肺氣腫或鼻炎;泌尿病症諸如尿失禁、頻
尿、膀胱痙攣、慢性膀胱炎及膀胱過動症(OAB);胃腸道病症諸如腸躁症、痙攣性結腸炎、憩室炎、胃潰瘍、胃腸蠕動或胃酸分泌;口乾;散瞳、心搏過速;眼科手術;心血管病症諸如迷走神經誘發竇性心搏過緩。
現在將藉下列實施例進一步說明本發明。
用於通式(I)中化合物合成之中間化合物可透過後文說明之製備獲得。
(R)-5-(2-胺基-1-羥基乙基)-8-羥基喹啉-2(1H)-酮鹽酸鹽之製備
步驟1:8-(苄氧基)-5-(2-溴乙醯基)喹啉-2(1H)-酮
5-乙醯基-8-(苄氧基)喹啉-2(1H)-酮(19.4克(g),66.4毫莫耳(mmol))於無水THF(240毫升(mL))及無水甲醇(165mL)之懸浮液以1.5小時時間逐滴添加四-正-丁基三溴化銨(Bu4NBr3)(54.5g,113.0mmol)於無水THF(130mL)之溶液。未經加熱於減壓下濃縮之前,所得溶液於室溫攪拌隔夜。殘餘物再溶解於甲醇(200mL)。以冰冷卻添加飽和水性氯化銨溶液(390mL)。所得懸浮液經過濾及固體以水洗滌及於減壓下風乾。固體懸浮於DCM及甲醇(1:1v/v,100mL)90分鐘。藉過濾收集固體,以DCM洗滌及風乾獲得標題化合物(18.0g,73%)。
1H NMR(400MHz,DMSO-d6):δ 11.07(s,1 H);8.51(d,J=10.0Hz,1 H);7.94-7.83(m,1 H);7.60(d,J=7.5Hz,2 H);7.44-7.27(m,4 H);6.79-6.65(m,1 H);5.53-5.39(s,2 H);4.93(s,2 H)
步驟2:(R)-8-(苄氧基)-5-(2-溴-1-羥基乙基)喹啉-2(1H)-酮
8-(苄氧基)-5-(2-溴乙醯基)喹啉-2(1H)-酮(26.0g,69.9mmol)及(R)-3,3-二苯基-1-甲基四氫-3H-吡咯并[1,2-c][1,3,2]吖硼呃(21.3g,76.8mmol)與甲苯共沸蒸餾(3次)然後於氮氣氣氛下懸浮於無水THF(400mL)。懸浮液冷卻至-20℃(外溫)及藉注射泵浦以3小時時間
添加硼烷二甲基硫(BH3-Me2S)錯合物溶液(45.4mL,90.8mmol,2.0M於THF溶液)。添加完成後,反應混合物攪拌1小時,隨後以甲醇(25mL)淬熄。反應以20分鐘溫熱至室溫。混合物於減壓下濃縮及殘餘物懸浮於水性鹽酸(500毫升,1M溶液)及於室溫攪拌18小時。於此時間後,藉過濾收集固體及以水洗滌(3×100mL)。固體部分溶解於乙酸乙酯及回流2小時。其餘固體藉熱過濾去除,濾液經蒸發獲得標題化合物。收集自熱乙酸乙酯之固體再度部分溶解於乙酸乙酯及回流2小時,然後過濾獲得含純產物之濾液。此程序再重複四次。組合固體從乙酸乙酯及石油醚再結晶獲得標題化合物(20.0g,76%)。
1H NMR(400MHz,DMSO-d6):δ 10.68(s,1 H);8.19(d,J=9.9Hz,1 H);7.58(d,J=7.5Hz,2 H);7.41-7.36(m,2 H);7.34-7.29(m,1 H);7.23-7.19(m,2 H);6.57(d,J=9.8Hz,1 H);5.94(d,J=4.7Hz,1 H);5.31(s,2 H);5.25-5.19(m,1 H);3.71-3.58(m,2 H).
步驟3:(R)-8-(苄氧基)-5-(2-溴-1-((第三丁基二甲基矽烷基)氧基)乙基)喹啉-2(1H)-酮
2,6-二甲苯胺(6.9mL,59.5mmol)添加至(R)-8-(苄氧基)-5-(2-溴-1-羥基乙基)喹啉-2(1H)-酮(10.1g,27.0mmol)於DCM(100mL)於0℃之溶液。反應混合物攪拌5分鐘然後以15分鐘時間逐滴添加三氟甲烷磺酸第三丁基二甲基矽烷基酯(tBuMe2SiOtf)(13.0mL,56.8mmol)。混合物於0℃攪拌30分鐘,接著於室溫攪拌隔夜。於此時間後,
反應以飽和水性碳酸氫鈉溶液淬熄及以DCM(3次)萃取。組合有機萃取物經乾燥(硫酸鎂)、過濾及於減壓下濃縮。添加異己烷(500mL)至粗產物料,所得固體藉過濾收集。固體從乙酸乙酯及石油醚(40:60)再結晶獲得標題化合物(11.3g,85%)。
1H NMR(400MHz,CDCl3):δ 9.19(s,1 H);8.23(dd,J=9.9,4.4Hz,1 H);7.43(d,J=4.6Hz,5 H);7.17(dd,J=8.3,4.5Hz,1 H);7.03(dd,J=8.2,4.4Hz,1 H);6.71(dd,J=9.9,3.7Hz,1 H);5.18(d,J=4.5Hz,3 H);3.63-3.56(m,1 H);3.49(dd,J=10.4,4.8Hz,1 H);0.88(t,J=4.4Hz,9 H);0.14(d,J=4.4Hz,3 H);-0.11(d,J=4.4Hz,3 H).
步驟4:(R)-5-(2-疊氮基-1-((第三丁基二甲基矽烷基)氧基)乙基)-8-(苄氧基)喹啉-2(1H)-酮
(R)-8-(苄氧基)-5-(2-溴-1-((第三丁基二甲基矽烷基)氧基)乙基)喹啉-2(1H)-酮(10.0g,20.5mmol)溶解於二甲基甲醯胺(180mL)及水(20mL)。循序添加碘化鈉(3.39g,22.6mmol)及疊氮化鈉(1.47g,22.6mmol)。反應混合物於室溫攪拌至全部固體皆成為溶液。溶液於80℃加熱40小時然後冷卻至室溫及以乙酸乙酯(300mL)稀釋。混合物以水、食鹽水洗滌(2次)及有機萃取物經脫水(硫酸鎂)、過濾及於減壓下濃縮。粗產物殘餘物使用異己烷濕磨獲得期望化合物(8.16g,88%)。未經進一步純化及用於次一步驟。
1H NMR(400MHz,CDCl3):δ 9.19(s,1 H),8.18(d,J=9.9Hz,1 H),7.45-7.36(m,4 H),7.20(d,J=8.3Hz,1 H),7.04(d,J=8.3Hz,1 H),6.70(dd,J=9.9,2.2Hz,1 H),5.19-5.13(m,3 H),3.48(dd,J=12.7,8.1Hz,1 H),3.26(dd,J=12.7,3.8Hz,1 H),0.89(s,9 H),0.14(s,3 H),-0.11(s,3 H).
步驟5:(R)-5-(2-胺基-1-羥基乙基)-8-羥基喹啉-2(1H)-酮鹽酸鹽
(R)-5-(2-疊氮基-1-((第三丁基二甲基矽烷基)氧基)乙基)-8-(苄氧基)喹啉-2(1H)-酮(4.50g,10.0mmol)於乙醇(50mL)之溶液添加10%鈀/木炭(4.50g)接著添加1-甲基-1,4-環己二烯(11.0mL,97.9mmol)。反應溫熱至60℃及然後於60℃攪拌2小時。允許反應混合物冷卻及通過矽藻土墊過濾。濾餅以額外乙醇洗滌及濾液於減壓下蒸發。殘餘物從異丙醇蒸發(2次)及溶解於異丙醇(30mL)。添加HCl-二(4M,50mL,200mmol)及反應混合物於室溫攪拌18小時。所得懸浮液經過濾,濾餅以醚洗滌及固體於減壓下於P2O5存在下乾燥獲得標題化合物(1.65g,62%)。
1H NMR(400MHz,MeOD):δ 7.71(d,J=9.8Hz,1 H),6.57(d,J=8.2Hz,1 H),6.31(d,J=8.2Hz,1 H),6.02(dd,J=9.8,6.5Hz,1 H),4.58(dd,J=9.6,3.5Hz,1 H),2.47-2.31(m,2 H).
4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸2-(4-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)甲基)-2-甲基苯氧基)乙基酯(化合物1)
步驟1:N-((3-羥基苯基)(苯基)甲基)甲醯胺
3-羥基二苯甲酮(25g,126.1mmol)於甲醯胺(130mL,3.3mmol)加熱至180℃ 18小時。許可反應略微冷卻然後倒入冰冷水中及攪拌30分鐘,過濾及以水洗滌。固體於水(60mL)及乙醇(60mL)中攪拌及加熱至50℃ 1小時,然後許可其冷卻。固體經過濾及以水洗滌獲得標題化合物呈褐色固體(33.94g,118%)。
1H NMR(400MHz,CD3OD):δ 7.39-7.28(m,5 H);7.21-7.13(m,1 H);6.79(d,J=7.78Hz,1 H);6.73-6.68(m,2 H);5.45(s,1 H).
步驟2:3-(胺基(苯基)甲基)酚鹽酸鹽
甲醇(125mL)冷卻至0℃及逐滴添加乙醯氯(17.8mL)獲
得2M甲醇性鹽酸溶液。N-((3-羥基苯基)(苯基)甲基)甲醯胺於40℃與2M甲醇性鹽酸攪拌1.5小時。於減壓下去除溶劑,殘餘物再溶解於甲醇及於減壓下去除溶劑。此處理程序重複三次獲得標題化合物呈褐色固體(29.09g,97.9%)。
1H NMR(400MHz,DMSO-d6):δ 9.76(s,1 H);9.07(s,3 H);7.59-7.53(m,2 H);7.51-7.37(m,3 H);7.26(t,J=7.89Hz,1 H);6.99(d,J=7.75Hz,1 H);6.90(t,J=1.97Hz,1 H);6.81(dd,J=8.10,2.32Hz,1 H);5.58(d,J=5.82Hz,1 H).
步驟3:((3-羥基苯基)(苯基)甲基)胺基甲酸第三丁酯
3-(胺基(苯基)甲基)酚鹽酸鹽(29.09g,123.4mmol)於二氯甲烷(450mL)冷卻至0℃及緩慢添加二異丙基乙基胺(65.9mL,370.2mmol)及二碳酸二第三丁酯(59.2g,271.5mmol)。反應於0℃攪拌2小時,然後以16小時時間溫熱至室溫。去除溶劑及化合物經由二氧化矽柱塞純化,以0-20%乙酸乙酯於異己烷洗提獲得黑色油。此混合物於甲醇(300mL)內添加碳酸鉀(51g,370.2mmol)及於室溫攪拌16小時。懸浮液經過濾及濾液於減壓下蒸發,殘餘物再溶解於乙酸乙酯(370mL)。添加二氧化矽(73g)及懸浮液攪拌30分鐘,過濾,及濾餅又以乙酸乙酯洗滌。濾液蒸發至乾。深色固體殘餘物溶解於乙酸乙酯(200mL),添加木炭及懸浮液回流加熱1小時。懸浮液經矽藻土過濾及於減壓下去除溶劑。深色固體溶解於二氯甲烷及添加異己烷,然後蒸發去除溶劑(重複3次)獲得標題化合物呈黃色固體(34.81g,92%)。
1H NMR(400MHz,CDCl3):δ 7.36-7.16(m,6 H);6.80(d,J=7.79Hz,1 H);6.74-6.69(m,2 H);5.83(s,1 H);5.15(s,1 H);1.53-1.30(s,9 H).
步驟4:((3-羥基苯基)(苯基)甲基)胺基甲酸(S)-第三丁酯
得自步驟3之外消旋混合物藉SFC使用CHIRALPAK® AD 20μM 250×110毫米(mm)管柱使用正庚烷/2-丙醇/二乙基胺(60/40/0.1)作為洗提劑於25℃以570毫升/分鐘之流速純化。從54.1g粗產物獲得((3-羥基苯基)(苯基)甲基)胺基甲酸(S)-第三丁酯(Rt=8.5-8.6min,23.9g,99.2e.e.)。
步驟5:4-((3-(((第三-丁氧基甲醯基)胺基)(苯基)甲基)苯氧基)甲基)苯甲酸(S)-甲酯
((3-羥基苯基)(苯基)甲基)胺基甲酸(S)-第三丁酯(3.20g,10.7mmol),4-(溴甲基)苯甲酸甲酯(2.70g,11.8mmol)及碳酸鉀(2.20g,16.1mmol)於乙腈(54mL)之混合物於室溫攪拌16小時。反應混合物於減壓下濃縮,殘餘物分溶於乙酸乙酯及水。水相又以乙酸乙酯萃取及組合有機萃取物經組合,於無水硫酸鎂脫水,過濾及於減壓下蒸發去除
溶劑。殘餘物從乙酸乙酯及異己烷再結晶獲得標題化合物呈白色固體(3.25g,68%)。
1H NMR(400MHz,CDCl3):δ 8.04(d,J=8.2Hz,2 H);7.46(d,J=8.2Hz,2 H);7.34-7.20(m,6 H);6.90-6.81(m,3 H);5.87(s,1 H);5.13(s,1 H);5.07(s,2 H);3.92(s,3 H);1.44(s,9 H).
步驟6:4-((3-(胺基(苯基)甲基)苯氧基)甲基)苯甲酸(S)-甲酯鹽酸鹽
4-((3-(((第三-丁氧基甲醯基)胺基)(苯基)甲基)苯氧基)甲基)苯甲酸(S)-甲酯(3.21g,7.20mmol)於甲醇(36mL)之溶液添加鹽酸於二(4M,9.0mL,36mmol)。反應混合物於室溫攪拌16小時。於減壓下去除溶劑獲得標題化合物(2.65g,>95%)。
1H NMR(400MHz,CDCl3):δ 9.21(s,2 H);8.03(d,J=8.1Hz,2 H);7.64(d,J=8.1Hz,2 H);7.59(d,J=7.6Hz,2 H);7.49-7.34(m,5 H);7.17(d,J=7.7Hz,1 H);7.06(dd,J=8.3,2.4Hz,1 H);5.64(s,1 H);5.28(s,2 H);3.91(s,3 H).
步驟7:4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸甲酯
4-((3-(胺基(苯基)甲基)苯氧基)甲基)苯甲酸(S)-甲酯鹽酸鹽(12.0g,31.3mmol)於吡啶(100mL)於0℃經攪拌之溶液分成數份添
加(R)-啶-3-基氯代碳酸酯(8.50g,37.5mmol)。反應於0℃攪拌1小時然後許可溫熱至室溫16小時。加水至反應混合物及以乙酸乙酯萃取(3次)。組合萃取物以鹽水洗滌,脫水(硫酸鈉)、過濾及於減壓下蒸發去除溶劑。粗產物於KP-NH Biotage卡匣上使用0-20%甲醇於乙酸乙酯洗提藉層析術純化獲得標題化合物(10.3g,66%)。
步驟8:4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸
4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸甲酯(2.27g,4.50mmol)於THF(23mL)之經攪拌之溶液內添加氫氧化鋰水溶液(2.0M,9.30ml,18.0mmol)。混合物於室溫攪拌16小時。反應混合物之pH藉添加4M水性鹽酸調整至6。然後混合物以10%甲醇性乙酸乙酯萃取(2次)及組合有機萃取物於減壓下蒸發。然後殘餘物溶解於乙醇及再度於減壓下蒸發獲得標題化合物呈淺黃色固體(1.85g,84%)。
1H NMR(400MHz,DMSO-d6):δ 8.41(d,J=9.4Hz,1 H);7.99(d,J=7.9Hz,2 H);7.58(d,J=8.0Hz,2 H);7.42-7.26(m,6 H);7.09(s,1 H);7.02-6.91(m,2 H);5.87(d,J=9Hz,1 H);5.21(s,2 H);4.76(s,1 H);3.98-2.72(m,6 H);2.12-1.54(m,5 H).
步驟9:4-(2-羥基乙氧基)-3-甲基苯甲醛
4-羥基-3-甲基-苯甲醛(0.545g,4.00mmol)於DMF(10mL)
之經攪拌之溶液添加碳酸鉀(1.10g,7.97mmol)。反應混合物於室溫攪拌5分鐘及然後添加碳酸伸乙酯(0.705g,8.00mmol)於DMF(2mL)之溶液。所得混合物於80℃攪拌90小時。允許反應混合物冷卻及以乙酸乙酯及水稀釋。去除有機相,以食鹽水洗滌(2次),脫水(硫酸鎂),過濾及於減壓下蒸發去除溶劑獲得標題化合物(0.677g,94%)。
1H NMR(400MHz,CDCl3):δ 9.87(s,1 H);7.72-7.70(m,2 H);6.95-6.93(m,1 H);4.20-4.18(m,2 H);4.04-4.03(m,2 H);2.29(s,3 H);1.98(s,1 H).
步驟10:4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸2-(4-甲醯基-2-甲基苯氧基)乙基酯
4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸(0.778g,1.49mmol)於DMF(6mL)之經攪拌之溶液添加二異丙基乙基胺(0.649mL,1.79mmol)及HATU(0.679g,1.79mmol),混合物於室溫攪拌20分鐘。於所得溶液內添加4-(2-羥基乙氧基)-3-甲基苯甲醛(0.670g,3.72mmol)於DMF(2mL)之溶液。混合物於室溫攪拌18小時。混合物以乙酸乙酯稀釋,以10%水性碳酸鉀、食鹽水洗滌(2次),脫水(硫酸鎂),過濾及於減壓下蒸發去除溶劑。殘餘物載荷至SCX-2卡匣上及以乙腈(4管柱體積)洗提及然後以10%三乙基胺/乙腈(4管柱體積)洗提。
10%三乙基胺/乙腈洗提分藉TLC分析及組合含產物之洗提分及於減壓下蒸發。材料未經進一步純化即直接用於次一步驟。
步驟11:4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)
苯氧基)甲基)苯甲酸2-(4-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)甲基)-2-甲基苯氧基)乙基酯(化合物1)
(R)-5-(2-胺基-1-羥基乙基)-8-羥基喹啉-2(1H)-酮鹽酸鹽(0.211g,0.83mmol)於甲醇(6mL)之懸浮液添加三乙基胺(0.229mL,1.65mmol)。混合物攪拌10分鐘及然後添加4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸2-(4-甲醯基-2-甲基苯氧基)乙基酯(0.445g,0.69mmol)於甲醇(2mL)之溶液。混合物於室溫攪拌1小時。添加三乙醯氧基硼氫化鈉(0.292g,1.38mmol)接著添加乙酸(0.188mL,3.28mmol)及反應又持續18小時。反應混合物以水淬熄及於減壓下蒸發。殘餘物溶解於異丁醇及以水洗滌。有機相於減壓下蒸發及粗產物藉反相製備性HPLC純化獲得標題化合物(0.065g,11%)。
1H NMR(400MHz,DMSO-d6):δ 8.29-8.20(m,2 H);8.10(d,J=9.9Hz,1 H);7.97(d,J=8.0Hz,2 H);7.57(d,J=8.0Hz,2 H);7.32-7.18(m,6 H);7.17-7.01(m,4 H);6.98-6.85(m,4 H);6.47(d,J=9.9Hz,1 H);5.81(s,1 H);5.17(s,2 H);5.09(dd,J=7.9,4.7Hz,1 H);4.60(d,J=16.8Hz,3 H);4.31(d,J=5.0Hz,2 H);3.71(s,2 H);3.12(m,1 H);2.81-2.52(m,6 H);2.09(s,4 H);1.92(s,1 H);1.69-1.26(m,4 H).
下列化合物係偶合步驟9產生之必需醇與得自步驟8之酸以相同方式及將該產物用於隨後步驟製備。
製備必需醇之替代方法係藉3-氯-4-(2-羥基乙氧基)-5-甲氧基苯甲醛之合成突顯。
3-氯-4-(2-羥基乙氧基)-5-甲氧基苯甲醛之製備
氫化鈉(60%於礦油之懸浮液,0.24g,6.00mmol)於DMF(8mL)之懸浮液添加5-氯香草素(0.746g,4.00mol)於DMF(2mL)之溶液。反應混合物於室溫攪拌20分鐘及添加2-溴乙醇(0.42mL,5.93mmol)。反應混合物於50℃加熱90小時。反應混合物以乙酸乙酯稀釋及以水、食鹽水洗滌(2次),脫水(硫酸鎂),過濾及於減壓下蒸發去除溶劑。殘餘物藉急速管柱層析術純化,使用0-50%乙酸乙酯於異己烷洗提獲得標題化合物(0.413g,48%)。
1H NMR(400MHz,CDCl3):δ 9.87(s,1 H);7.53(d,J=1.6Hz,1 H);7.38(d,J=1.6Hz,1 H);4.29-4.27(m,2 H);3.96(s,3 H);3.90-3.85(m,2 H);2.77(t,d,J=6.4Hz,1 H).
下列化合物係以相同方式偶合步驟9所產生的必需醇與得自步驟8之酸,及使用該產物於隨後步驟製備。
3-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸4-(2-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)乙基)苄基酯(化合物18)
步驟1:4-(2-甲氧基乙烯基)苯甲酸甲酯
(甲氧基甲基)三苯基鏻氯化物(6.48g,20.0mmol)於THF(40mL)之以冰冷卻之懸浮液分成數份添加氫化鈉(60%於礦油之分散液,0.88g,22.0mmol)。反應混合物於此溫度攪拌10分鐘接著於室溫攪拌50分鐘。添加4-甲醯基苯甲酸甲酯(1.16g,10.0mmol)於THF(10mL)之溶液及混合物於室溫攪拌18小時。反應混合物以飽和水性碳酸氫鈉淬熄及以DCM萃取。有機相傾倒通過斥水性玻璃料及於減壓下蒸發去除溶液。殘餘物藉急速管柱層析術純化,使用0-10%乙酸乙酯於異己烷洗提獲得標題化合物(1.36g,71%)。
1H NMR(400MHz,CDCl3):異構物之混合物:δ 7.98-7.90(m,2 H);7.63-7.59(m,1 H);7.29-7.25(m,1 H);7.17(d,J=13.0Hz,0.5 H);6.25(d,J=7.0Hz,0.5 H);5.82(d,J=13.0Hz,0.5 H);5.31-5.25(m,0.5 H);3.90(s,3 H);3.83(s,1.5 H);3.72(s,1.5 H).
步驟2:4-(2,2-二甲氧基乙基)苯甲酸甲酯
4-(2-甲氧基乙烯基)苯甲酸甲酯(1.36g,7.08mmol)於甲醇(30mL)之溶液添加對-甲苯磺酸一水合物(pTSA)(0.135g,0.71mmol)及反應混合物回流加熱18小時。於減壓下蒸發去除溶劑。殘餘物分溶於DCM及10%水性碳酸鉀。有機相傾倒通過斥水性玻璃料及於減壓下蒸發去除溶劑。粗產物未經進一步純化即用於次一步驟。
步驟3:(4-(2,2-二甲氧基乙基)苯基)甲醇
4-(2,2-二甲氧基乙基)苯甲酸甲酯(推定7.08mmol)於THF(30mL)之冷(-78℃)溶液逐滴添加氫化鋰鋁溶液(2.0M於THF,3.50mL,7.00mmol)。許可反應混合物溫熱至室溫歷4小時。隨後混合物以水(0.266mL)、2M水性氫氧化鈉(0.266mL)及水(3×0.266mL)處理。混合物以乙酸乙酯稀釋及添加硫酸鎂。混合物於室溫攪拌1小時及然後通過矽藻土過濾。濾餅又以乙酸乙酯洗滌及濾液經組合。於減壓下蒸發去除溶劑及殘餘物藉急速管柱層析術純化,使用0-30%乙酸乙酯於異己烷洗提獲得標題化合物(0.854g,62%)。
1H NMR(400MHz,CDCl3):δ 7.32-7.30(m,2 H);7.26-7.23(m,2 H);4.68(d,J=5.6Hz,2 H);4.55-4.52(m,1 H);3.34(s,6 H);2.92(d,J=5.6Hz,1 H);1.28-1.24(m,1 H).
步驟4:3-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸
標題化合物係如實施例1步驟5至8所述以3-(溴甲基)苯甲酸甲酯置換步驟5之4-(溴甲基)苯甲酸甲酯製備。
步驟5:3-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸4-(2,2-二甲氧基乙基)苄基酯
標題化合物係如實施例1步驟10所述使用(4-(2,2-二甲氧基乙基)苯基)甲醇及3-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸分別置換4-(2-羥基乙氧基)-3-甲基苯甲醛及4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸甲酯鹽酸鹽製備。
步驟6:3-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸4-(2-側氧基乙基)苄基酯
3-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸4-(2,2-二甲氧基乙基)苄基酯(0.100g,0.15mmol)於丙酮(5mL)之經攪拌之溶液添加對甲苯磺酸一水合物(0.086g,0.45mmol)及混合物於室溫攪拌1小時。反應混合物以飽和碳酸氫鈉稀釋及以乙酸乙酯萃取。組合有機萃取物經脫水(硫酸鎂),過濾及於減壓下蒸發去除溶劑。殘餘物未經進一步純化即用於次一步驟。
步驟7:3-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯
氧基)甲基)苯甲酸4-(2-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)乙基)苄基酯(化合物18)
標題化合物係如實施例1步驟11使用3-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸4-(2-側氧基乙基)苄基酯置換4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸2-(4-甲醯基-2-甲基苯氧基)乙基酯製備。
1H NMR(400MHz,DMSO-d6):δ 8.28-8.19(m,3 H);8.17(d,J=9.9Hz,1 H);8.05(s,1 H);7.94(d,J=7.8Hz,1 H);7.72(d,J=7.7Hz,1 H);7.58-7.52(m,1 H);7.38(d,J=7.8Hz,2 H);7.31-7.17(m,7 H);7.10-7.02(m,2 H);6.96-6.86(m,3 H);6.51(d,J=9.9Hz,1 H);5.81(d,J=9.0Hz,1 H);5.33(s,2 H);5.16(s,2 H);5.07(dd,J=7.5,4.8Hz,1 H);4.57(s,1 H);3.10(s,1 H);2.86-2.65(m,8 H);2.50(m,4 H);1.90(s,1 H);1.79(s,1 H);1.46(t,J=49.4Hz,3 H).
下列化合物係以類似實施例2之方式偶合必需醇(如實施例2步驟1至3所述製備)至如實施例2步驟5所述之必需酸及隨後該產物用於實施例2步驟6及7製備。
4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸3-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)
胺基)甲基)苄基酯(化合物22)
步驟1:3-(1,3-二-2-基)苯甲酸甲酯
3-甲醯基苯甲酸甲酯(2.5g,15.2mmol),乙二醇(4.2mL,75mmol)及對甲苯磺酸一水合物(0.29g,1.52mmol)於甲苯(60mL)之混合物於丁及史塔克(Dean and Stark)條件下回流4小時。反應混合物以乙酸乙酯稀釋及依序以飽和碳酸氫鈉及鹽水洗滌。有機相經脫水(硫酸鎂),過濾及於減壓下蒸發去除溶劑獲得標題化合物(3.09g,98%)。
1H NMR(400MHz,DMSO-d6):δ 8.05-7.95(m,2 H),7.72(d,1 H),7.61-7.52(m,1 H),5.82(s,1 H),4.11-3.94(m,4 H),3.87(s,2 H).
步驟2:(3-(1,3-二-2-基)苯基)甲醇
標題化合物係如實施例2步驟3所述使用3-(1,3-二-2-基)苯甲酸甲酯置換4-(2,2-二甲氧基乙基)苯甲酸甲酯製備。
1H NMR(400MHz,DMSO-d6):δ 7.40(s,1 H),7.37-7.27(m,3 H),5.77-5.68(m,1 H),5.21(t,1 H),4.53-4.46(m,2 H),4.09-3.90(m,4 H).
步驟3:4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸3-(1,3-二-2-基)苄基酯
標題化合物係如實施例1步驟10所述以(3-(1,3-二-2-基)苯基)甲醇置換4-(2-羥基乙氧基)-3-甲基苄醛製備
步驟4:4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸3-甲醯基苄基酯
4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸3-(1,3-二-2-基)苄基酯(0.32g,0.57mmol)於THF(8mL)之經攪拌之溶液添加2M水性鹽酸(8mL)。反應混合物於室溫攪拌1小時。混合物以乙酸乙酯稀釋,及循序以飽和水性碳酸氫鈉及鹽水洗滌。有機萃取物經脫水(硫酸鎂),過濾及於減壓下蒸發去除溶劑。殘餘物未經進一步純化即用於次一步驟。
步驟5:4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸3-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)甲基)苄基酯(化合物22)
標題化合物係如實施例1步驟11所述以4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸3-甲醯基
苄基酯置換4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸2-(4-甲醯基-2-甲基苯氧基)乙基酯製備。
1H NMR(400MHz,DMSO-d6):δ 8.26(s,2 H);8.17(d,J=9.9Hz,1 H);8.04(d,J=8.0Hz,2 H);7.61(d,J=8.0Hz,2 H);7.48(s,1 H);7.41-7.22(m,9 H);7.13-7.07(m,2 H);7.01-6.90(m,3 H);6.51(d,J=9.9Hz,1 H);5.86(d,J=8.6Hz,1 H);5.39(s,2 H);5.23(s,2 H);5.12(dd,J=7.9,4.3Hz,1 H);4.63(s,1 H);3.85(s,2 H);3.17(d,J=14.5Hz,1 H);2.83-2.67(m,7 H);1.96(m,2 H);1.66(s,1 H);1.53(s,1 H);1.39(s,1 H).
下列化合物係以實施例3之類似方式,如實施例3步驟3所述偶合必需醇(如實施例3步驟1至2所述製備)至必需酸及隨後產物用於實施例3步驟4及5製備。
下列化合物係以實施例3之類似方式偶合必需醇(容後詳述)至如實施例3步驟3所述之必需酸及隨後所得產物用於實施例3步驟4及5製備。
必需醇之合成詳細說明如下:
化合物號碼32之必需醇之合成:3-(4-(二乙氧基甲基)苯基)丙-1-醇
步驟1:3-(4-(二乙氧基甲基)苯基)丙烯酸(E)-甲酯
4-(二乙氧基)苄醛(2.08g,10.0mmol)及(三苯基亞磷烷基)乙酸甲酯(3.68g,11.0mmol)於甲苯(30mL)之溶液回流加熱18小時。於減壓下蒸發去除溶劑。殘餘物藉急速管柱層析術純化使用0-15%乙酸乙酯於異己烷洗提獲得標題化合物(2.41g,91%)。
1H NMR(400MHz,CDCl3):δ 7.72(d,J=16Hz,1H);7.54-7.48(m,4 H);6.46(d,J=16Hz,1H);5.51(s,1 H);3.81(s,3 H);3.65-3.50(m,4 H);1.28-1.22(m,6 H).
步驟2:3-(4-(二乙氧基甲基)苯基)丙酸甲酯
1-甲基-1,4-環己二烯(10.0mL,89mmol)添加至3-(4-(二乙氧基甲基)苯基)丙烯酸(E)-甲酯(2.41g,9.13mmol)及10%鈀/碳(2.4g)於乙醇(40mL)之懸浮液。反應混合物於60℃加熱1.5小時。反應混合物通過矽藻土過濾及濾餅又以乙醇洗滌。於減壓下蒸發去除溶劑獲得標題化合物(2.15g,89%)。
1H NMR(400MHz,CDCl3):δ 7.40(m,2 H);7.20-7.18(m,2 H);5.46(s,1 H);3.73(s,3 H);3.70-3.48(m,4 H);2.97-2.92(m,2 H);2.69-2.60(m,2 H);1.26-1.23(m,6 H).
步驟3:3-(4-二乙氧基甲基)苯基)丙-1-醇
標題化合物係如實施例2步驟3所述使用3-(4-(二乙氧基甲基)苯基)丙酸甲酯置換4-(2,2-二甲氧基乙基)苯甲酸甲酯製備。
1H NMR(400MHz,CDCl3):δ 7.39(d,J=8.4Hz,2 H);7.20-7.18(m,2 H);5.47(s,1 H);3.69-3.49(m,6 H);2.73-2.69(m,2 H);1.90-1.85(m,2 H);1.28-1.24(m,6 H).
化合物33之必需醇之合成:2-(4-1,3-二-2-基)苯基)乙醇
步驟1:2-(4-甲醯基苯基)乙酸甲酯
乙醯氯(5mL)添加至4-(羥基甲基)苯基乙酸(5.78g,34.8mmol)於甲醇(200mL)之以冰冷卻之溶液內。允許反應混合物於此溫度攪拌42小時溫熱至室溫。於減壓下蒸發去除溶劑及殘餘物溶解於DCM(100mL)。添加二氧化錳(29.47g,339mmol)及所得懸浮液於室溫攪拌18小時。懸浮液通過矽藻土過濾及濾餅又以DCM洗滌。於減壓下蒸發去除溶劑,殘餘物藉急速管柱層析術純化,使用0-25%乙酸乙酯於異己烷
洗提獲得標題化合物(1.72g,28%)。
1H NMR(400MHz,CDCl3):δ 10.0(s,1 H);7.87-7.83(m,2 H);7.46(d,J=8Hz,2 H);3.69-3.65(m,5 H).
步驟2:2-(4-(1,3-二-2-基)苯基)乙醇
2-(4-甲醯基苯基)乙酸甲酯(1.72g,9.66mmol)溶解於乙二醇(2.3mL)及原甲酸三乙酯(1.8mL)。添加四丁基三溴化銨(0.048g)及反應於室溫攪拌1小時。又再添加四丁基三溴化銨(0.434g)及反應攪拌1小時。反應混合物以乙酸乙酯稀釋及以水及鹽水洗滌。有機相經脫水(硫酸鎂),過濾及於減壓下蒸發去除溶劑。殘餘物藉急速管柱層析術純化,以0-15%乙酸乙酯於異己烷洗提獲得不純的2-(4-(1,3-二-2-基)苯基)乙酸甲酯(0.84g)。材料溶解於THF(15mL)及冷卻至-78℃。逐滴添加氫化鋰鋁溶液(1.0M於THF,6.00mL,6.00mmol)。允許反應混合物以18小時溫熱至室溫。混合物依序以水(0.228mL)、2M水性氫氧化鈉(0.228mL)及水(3×0.228mL)處理。混合物以乙酸乙酯稀釋及添加硫酸鎂。混合物於室溫攪拌1小時,然後通過矽藻土過濾。濾餅又以乙酸乙酯洗滌及組合濾液。於減壓下蒸發去除溶劑,殘餘物藉急速管柱層析術純化,以0-40%乙酸乙酯於異己烷洗提獲得標題化合物(0.315g,17%)。
1H NMR(400MHz,CDCl3):7.44-7.42(m,2 H);7.26-7.24(m,2 H);5.80(s,1 H);4.17-4.00(m,4 H);3.88-3.83(m,2 H);2.89-2.87(m,2 H);1.39-1.34(m,1 H).
化合物號碼34之必需醇之合成:(1-(3-(1,3-二-2-基)丙基)-1H-吡唑-4-基)甲醇
步驟1:1-(3-(1,3-二-2-基)丙基)-1H-吡唑-4-羧酸乙酯
1H-吡唑-4-羧酸乙酯(2.0g,14.3mmol)於DMF(10毫升)之以冰冷卻經攪拌之溶液添加氫化鈉(60%於礦油之分散液,0.68g,17.1mmol)。許可反應混合物以20分鐘時間溫熱至室溫及添加2-(2-溴乙基)-1,3-二(2.84g,15.7mmol)。反應混合物於60℃加熱16小時。反應分溶於乙酸乙酯及鹽水。有機相又以鹽水洗滌,脫水(硫酸鎂),過濾及於減壓下蒸發去除溶劑。殘餘物藉急速管柱層析術純化,使用0-100%乙酸乙酯於異己烷洗提獲得標題化合物(2.14g,59%)。
1H NMR(400MHz,CDCl3):δ 7.91(d,J=4.3Hz,2 H);4.92-4.83(m,1 H);4.33-4.25(m,4 H);4.03-3.80(m,4 H);2.29-2.22(m,2 H);1.39-1.31(m,3 H).
步驟2:(1-(3-(1,3-二-2-基)丙基)-1H-吡唑-4-基)甲醇
標題化合物係如實施例2步驟2所述使用1-(3-(1,3-二 -2-基)丙基)-1H-吡唑-4-羧酸乙酯置換4-(2-甲氧基乙烯基)苯甲酸甲酯製備。
1H NMR(400MHz,CDCl3):δ 7.69(s,1 H);7.60-7.44(m,1 H);4.94-4.84(m,1 H);4.61-4.48(m,4 H);4.06-3.82(m,4 H);2.31-2.21(m,2 H);1.70-1.64(m,1 H).
化合物號碼35必需醇之合成:1-(2-羥基乙基)-1H-吡唑-4-羧醛
1H-吡唑-4-羧醛(0.50g,5.21mmol)、2-溴乙醇(1.30g,10.41mmol)及碳酸鉀(0.79g,5.73mmol)組合於微波小瓶內的乙腈(5mL)。微波小瓶於微波中於150℃加熱30分鐘。反應混合物經過濾,於減壓下蒸發去除溶劑。殘餘物藉急速管柱層析術純化,使用0-100%乙酸乙酯於異己烷洗提獲得標題化合物(0.50g,69%)。
1H NMR(400MHz,CDCl3):δ 9.86(s,1 H);8.02(s,1 H);7.99(s,1 H);4.33-4.25(m,2 H);4.05(t,J=4.8Hz,2 H).
1-甲基-5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)-1H-吡唑-3-羧酸4-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)丁基酯(化合物36)
步驟1:1-甲基-5-(((甲基磺醯基)氧基)甲基)-1H-吡唑-3-羧酸甲酯
5-(羥基甲基)-1-甲基-1H-吡唑-3-羧酸甲酯(0.60g,3.51mmol)及三乙基胺(1.22mL,8.77mmol)於DCM(10mL)之以冰冷卻之溶液添加甲烷磺醯氯(0.41mL,5.26mmol)。反應混合物於此溫度攪拌15
分鐘及去除冷卻劑。反應混合物於室溫攪拌1小時。反應混合物以DCM稀釋及以水及鹽水洗滌。有機相經脫水(硫酸鎂),過濾及於減壓下蒸發去除溶劑。粗產物未經進一步純化即用於次一步驟。
步驟2:5-((3-(((第三-丁氧基甲醯基)胺基)(苯基)甲基)苯氧基)甲基)-1-甲基-1H-吡唑-3-羧酸(S)-甲基酯
標題化合物係如實施例1步驟5所述使用1-甲基-5-(((甲基磺醯基)氧基)甲基)-1H-吡唑-3-羧酸甲酯置換4-(溴甲基)苯甲酸甲酯製備。
1H NMR(400MHz,CDCl3):δ 7.37-7.21(m,7 H);6.92(d,J=7.7Hz,1 H);6.87-6.81(m,3 H);5.88(s,1 H);5.25-4.98(m,1 H);5.00(s,2 H);4.05-3.91(m,3 H);3.92(s,3 H);1.44(s,9 H).
步驟3:1-甲基-5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)-1H-吡唑-3-羧酸甲酯
標題化合物係如實施例1步驟6所述使用5-((3-(((第三-丁氧基甲醯基)胺基)(苯基)甲基)苯氧基)甲基)-1-甲基-1H-吡唑-3-羧酸(S)-甲基酯置換4-((3-(((第三-丁氧基甲醯基)胺基)(苯基)甲基)苯氧基)甲基)苯甲酸甲酯及得自此步驟之產物用於實施例1步驟7製備。
步驟4:1-甲基-5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)
甲基)苯氧基)甲基)-1H-吡唑-3-羧酸
標題化合物係如實施例1步驟8所述以1-甲基-5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)-1H-吡唑-3-羧酸甲酯置換4-((3-(苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸甲酯製備。
步驟5:1-甲基-5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)-1H-吡唑-3-羧酸3-(1,3-二-2-基)丙基酯
標題化合物係如實施例1步驟10所述使用1-甲基-5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)-1H-吡唑-3-羧酸及3-(1,3-二-2-基)丙-1-醇分別置換4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸及4-(2-羥基乙氧基)-3-甲基苄醛製備。
步驟6:1-甲基-5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)-1H-吡唑-3-羧酸4-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)丁基酯(化合物36)
標題化合物係如實施例2步驟6及步驟7所述製備。
1H NMR(400MHz,DMSO-d6):δ 8.48-8.07(m,3 H);8.18(d,J=9.93Hz,1 H);7.33-7.19(m,6 H);7.13-7.04(m,2 H);7.00-6.90(m,3 H);6.88(s,1 H);6.51(d,J=9.86Hz,1 H);5.83(d,J=8.56Hz,1 H);5.32-5.03(m,3 H);4.60(s,2 H);4.26-4.19(m,2 H);4.10-3.66(m,3 H);3.20-3.08(m,1 H);2.93-2.54(m,9 H);2.06-1.28(m,8 H).
下列化合物係如實施例4所述使用步驟1之適合的烷化劑(以化合物36為例1-甲基-5-(((甲基磺醯基)氧基)甲基)-1H-吡唑-3-羧酸甲酯)用於步驟2及該產物用於實施例4之隨後各步驟製備。
((S)-(3-((3-((4-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)丁基)胺基甲醯基)-1-甲基-1H-吡唑-5-基)甲氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物39)
步驟1:((S)-(3-((3-((4,4-二乙氧基丁基)胺基甲醯基)-1-甲基-1H-吡
唑-5-基)甲氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯
甲基-5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)-1H-吡唑-3-羧酸(0.30g,0.61mmol)於DMF(6mL)之經攪拌之溶液添加三乙基胺(0.2mL,1.53mmol),4-胺基丁醛二乙基縮醛(0.20mL,1.22mmol),EDCI(0.18g,0.91mmol)及2-羥基吡啶-N-氧化物(0.09g,0.91mmol)。反應混合物於室溫攪拌90小時。反應混合物以乙酸乙酯稀釋及以飽和碳酸氫鈉、鹽水洗滌,脫水(硫酸鎂),過濾及於減壓下蒸發去除溶劑獲得粗產物目標化合物(0.22g,57%),其未經進一步純化即直接使用。
步驟2:((S)-(3-((3-((4-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)丁基)胺基甲醯基)-1-甲基-1H-吡唑-5-基)甲氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物39)
標題化合物係如實施例2步驟6及步驟7所述製備。
1H NMR(400MHz,DMSO-d6):δ 10.30(br s,1 H);8.48-8.07(m,3 H);8.21-8.10(m,2 H);7.34-7.19(m,6 H);7.15-7.04(m,2 H);7.00-6.91(m,2 H);6.75(s,1 H);6.53(d,J=9.86Hz,1 H);5.84(d,J=8.57Hz,1 H);5.31-5.10(m,3 H);4.61(s,1 H);3.88(s,3 H);3.31-2.97(m,3 H);2.95-2.56(m,7 H);1.94(s,1 H);1.82(s,1 H);1.73-1.16(m,9 H).
下列化合物係如實施例5所述使用適合的胺用於步驟1製備。
((S)-(3-((5-((4-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)丁基)胺基甲醯基)呋喃-2-基)甲氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物40)
步驟1:5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)呋喃-2-羧酸
標題化合物係如實施例4步驟2所述使用5-(溴甲基)呋喃-2-羧酸甲酯置換1-甲基-5-(((甲基磺醯基)氧基)甲基)-1H-吡唑-3-羧酸甲酯,接著藉實施例4步驟3之方法製備。粗產物未經進一步純化即用於次一步驟。
步驟2:((S)-(3-((5-((4,4-二乙氧基丁基)胺基甲醯基)呋喃-2-基)甲氧基)苯基)(苯基)甲基)-胺基甲酸(R)-啶-3-基酯
標題化合物係如實施例5步驟1所述使用5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)-甲基)呋喃-2-羧酸置
換1-甲基-5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)-1H-吡唑-3-羧酸製備。粗產物反應產物未經進一步純化即用於次一步驟。
步驟3:((S)-(3-((5-((4-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)丁基)胺基甲醯基)呋喃-2-基)甲氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物40)
標題化合物係如實施例5步驟2所述製備。
1H NMR(400MHz,DMSO-d6):δ 10.38(br s,1 H);8.46-8.39(m,1 H);8.33-8.21(m,3 H);8.18(d,J=9.93Hz,1 H);7.34-7.19(m,6 H);7.11(d,J=8.15Hz,1 H);7.05(t,J=3.66Hz,2 H);6.99-6.89(m,3 H);6.69(d,J=3.44Hz,1 H);6.54(d,J=9.86Hz,1 H);5.84(d,J=8.95Hz,1 H);5.20(dd,J=8.44,4.14Hz,1 H);5.08(s,2 H);4.60(s,1 H);3.28-3.01(m,3 H);2.88(d,J=8.88Hz,2 H);2.79(s,6 H);1.93(s,1 H);1.88-1.73(m,1 H);1.74-1.15(m,7 H).
下列化合物係如實施例6所述使用必需胺用在步驟2製備。
((S)-(3-((4-((4-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)丁基)胺基甲醯基)唑-2-基)甲氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物41)
步驟1:2-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)唑-4-羧酸
標題化合物係如實施例4步驟2所述使用2-(氯甲基)-1,3-唑-4-羧酸甲酯置換1-甲基-5-(((甲基磺醯基)氧基)甲基)-1H-吡唑-3-羧酸甲酯,接著為實施例4步驟3之方法製備。粗產物未經進一步純化即用於次一步驟。
步驟2:((S)-(3-((4-((4,4-二乙氧基丁基)胺基甲醯基)唑-2-基)甲氧基)苯基)(苯基)甲基)-胺基甲酸(R)-啶-3-基酯
標題化合物係如實施例5步驟1所述製備,使用2-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)-苯氧基)甲基)唑-4-羧酸置換1-甲基-5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)-胺基)甲基)苯氧基)甲基)-1H-吡唑-3-羧酸製備。粗產物反應產物未經進一步純化即
用於次一步驟。
步驟3:((S)-(3-((4-((4-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)丁基)胺基甲醯基)唑-2-基)甲氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯
標題化合物係如實施例5步驟2所述製備。
1H NMR(400MHz,DMSO-d6):δ 10.31(br s,1 H);8.61(s,1 H);8.37(t,J=6.01Hz,1 H);8.37-8.17(m,2 H);8.17(d,J=9.92Hz,1 H);7.33-7.19(m,6 H);7.10(d,J=8.15Hz,1 H);7.05-6.86(m,4 H);6.53(d,J=9.86Hz,1 H);5.83(d,J=8.65Hz,1 H);5.25-5.14(m,3 H);4.59(s,1 H);3.23(d,J=6.27Hz,2 H);3.21-3.06(m,1 H);2.86-2.52(m,8 H);1.92(s,1 H);1.80(s,1 H);1.71-1.16(m,8 H).
下列化合物係如實施例5所述以在步驟1使用適合的酸製備。
氯化(R)-3-((S)-(3-((5-((4-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基胺基)丁氧基)甲醯基)呋喃-2-基)甲氧基)苯基)(苯基)甲基胺基甲醯基氧基)-1-(2-側氧基-2-(噻吩-2-基)乙基)-1-四價氮二環[2.2.2]辛烷鹽酸鹽(化合物42)
步驟1:5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸鹽酸鹽
如實施例6步驟1所述製備的5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸進一步藉反相管柱
層析術純化使用洗提劑A:水/ACN 95/5+0.1% HCOOH及洗提劑B:ACN/水95/5+0.1% HCOOH之梯度洗提。5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸甲酸鹽(1:1)(8.36g,16.12mmol)分散於二(50mL)及然後於0℃添加4M HCl於二(16.12mL,64.5mmol)及混合物於室溫攪拌10分鐘直到完全溶解。前述溶液於激烈攪拌下逐滴添加於500mL乙醚中獲得白色沈澱,沈澱藉過濾收集。於真空中乾燥後分離5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸鹽酸鹽(8.2g,15.98mmol,99%產率)呈白色固體。
1H NMR(400MHz,DMSO-d 6)δ ppm 12.90-13.34(bs,1 H),10.10(bs,1 H),8.43(d,J=9.26Hz,1 H),7.13-7.42(m,7 H),7.04(m.,1 H),6.86-7.00(m,2 H),6.72(d,J=3.53Hz,1 H),5.83(d,J=9.26Hz,1 H),5.11(s,2 H),4.61-4.95(m,1 H),3.57-3.75(m,1 H),2.95-3.27(m,5 H),2.22(m,1 H),1.65-2.10(m,4 H)
步驟2:5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸4-(((R)-2-(8-(苄氧基)-2-側氧基-1,2-二氫喹啉-5-基)-2-(第三丁基二甲基矽烷基氧基)乙基)(第三丁氧基甲醯基)胺基)丁基酯
5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸鹽酸鹽(3.09g,6.03mmol)及2-(8-(苄氧基)-2-側氧基-1,2-二氫喹啉-5-基)-2-(第三丁基二甲基矽烷基氧基)乙基(4-羥基丁
基)胺基甲酸(R)-第三丁基酯(參考(R)-5-(2-胺基-1-羥基乙基)-8-羥基喹啉-2(1H)-酮鹽酸鹽之製備,步驟3)(3g,5.03mmol)溶解於15mL DMF,然後添加EDC(1.445g,7.54mmol)及混合物冷卻至0℃。DMAP(0.307g,2.51mmol)溶解於7mL DMF於0℃逐滴添加及混合物於0℃攪拌5分鐘,於室溫攪拌30分鐘及於50℃加熱3小時及然後於室溫放置隔夜。然後反應分溶於乙酸乙酯(300mL)及鹽水(600mL)。有機層以1:1混合物水/鹽水(2×300mL)洗兩次。有機相以鹽水(300mL)洗滌,以硫酸鈉脫水及蒸發獲得白色泡沫體。該泡沫體於300mL 95/5己烷/乙酸乙酯濕磨3小時及沈澱藉過濾收集。以己烷稀釋母液獲得第二收獲物。獲得5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸4-(((R)-2-(8-(苄氧基)-2-側氧基-1,2-二氫喹啉-5-基)-2-(第三丁基二甲基矽烷基氧基)乙基)(第三丁氧基甲醯基)胺基)丁基酯(4.3g得自兩次收獲物的組合,4.07mmol,81%產率)呈白色粉末。
1H NMR(400MHz,DMSO-d 6)δ ppm 10.64(bs,1 H),8.32(d,J=8.82Hz,2 H),7.57(d,J=7.50Hz,2 H),7.12-7.44(m,11 H),6.85-7.08(m,3 H),6.73(d,J=2.65Hz,1 H),6.47-6.64(m,1 H),5.82(d,J=8.82Hz,1 H),5.31(s,2 H),5.10(s,2 H),4.65-4.78(m,1 H),4.21(t,2 H),2.73-3.27(m,10 H),1.46-2.18(m,9 H),1.37(s,9 H),0.81(s,9 H),-0.01(s,3 H),-0.20(s,3 H)
步驟3:5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸4-(((R)-2-(8-(苄氧基)-2-側氧基-1,2-二氫喹啉-5-基)-2-(第三丁基二甲基矽烷基氧基)乙基)(第三丁氧基甲醯基)胺基)丁基酯甲酸鹽
5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸4-(((R)-2-(8-(苄氧基)-2-側氧基-1,2-二氫喹啉-5-基)-2-(第三丁基二甲基矽烷基氧基)乙基)(第三丁氧基甲醯基)胺基)丁基酯(2.1g,1.990mmol),10% Pd/C(0.106g,0.099mmol)及甲酸(0.092mL,2.388mmol)溶解於MeOH(44.2mL)及於室溫於氫氣氣球壓力下攪拌。2小時後反應完成及以矽藻土墊過濾及濾液於減壓下濃縮獲得無色黏稠油。油分散於Et2O/AcOEt及蒸發3次獲得重質白色泡沫體,其於己烷/Et2O/AcOEt濕磨獲得白色固體,白色固體藉過濾收集。母液經濃縮獲得0.44g白色泡沫體第二收獲物。兩次收獲物組合獲得5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸4-(第三丁氧基甲醯基((R)-2-(第三丁基二甲基矽烷基氧基)-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)丁基酯甲酸鹽(1.95g,1.928mmol,97%產率)呈白色非晶形固體。
1H NMR(400MHz,DMSO-d 6)δ ppm 10.38(br.s.,2 H),8.33(br.s.,2 H),8.14(s,1 H),7.16-7.40(m,7 H),6.84-7.06(m,5 H),6.73(m,1 H),6.41-6.60(m,1 H),5.82(d,J=9.26Hz,1 H),5.18-5.50(m,1 H),5.10(s,2 H),4.63-4.82(m,1 H),4.20(t,J=5.95Hz,2 H),2.71-3.22(m,10 H),1.44-2.16(m,9 H),1.44(s,9 H),0.80(s,9 H),-0.01(s,3 H),-0.19(s,3 H)
步驟4:5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸4-(第三丁氧基甲醯基((R)-2-(第三丁基二甲基矽烷
基氧基)-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)丁基酯氯化物鹽酸鹽。
5-((3-((S)-苯基(((R)-啶-3-基氧基)甲醯基胺基)甲基)苯氧基)甲基)呋喃-2-羧酸4-(第三丁氧基甲醯基((R)-2-(第三丁基二甲基矽烷基氧基)2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)丁基酯甲酸鹽(150mg,0.148mmol)溶解於500微升(uL)二及然後添加2-溴-1-(噻吩-2-基)乙酮(33.5mg,0.163mmol)於0.3mL二之溶液。2小時後添加第2當量烷化劑及混合物於室溫攪拌隔夜。為了推動反應完成,添加碳酸氫鈉(33mg,0.393mmol)及反應混合物於50℃加熱6小時。反應於室溫冷卻及然後添加Et2O。沈澱收集呈樹膠狀固體溶解於甲醇及蒸發獲得黃色殘餘物,殘餘物溶解於2-丙醇(1.8ml)。添加HCl於二(1.859ml,7.43mmol)及混合物於室溫攪拌隔夜。反應混合物以Et2O稀釋及藉過濾收集所形成的沈澱及藉反相管柱層析術純化,使用ACN:水(梯度從100%至40% ACN)獲得氯化(R)-3-((S)-(3-((5-((4-((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5基)乙基胺基)丁氧基)甲醯基)呋喃-2-基)甲氧基)苯基)(苯基)-甲基胺基甲醯基氧基)-1-(2-側氧基-2-(噻吩-2-基)乙基)-1-四價氮二環[2,2,2]辛烷鹽酸鹽(96mg,0.101mmol,68.1%
產率)呈淺黃色固體,該固體係利用EtOH及Et2O進行殘餘物之濕磨獲得。
1H NMR(400MHz,DMSO-d 6)δ ppm 9.87-10.55(bs,1 H),8.52(d,J=9.26Hz,1 H),8.33(s,2 H),8.11-8.20(m,2 H),8.05(d,J=3.53Hz,1 H),7.17-7.46(m,7 H),6.87-7.10(m,5 H),6.74(d,J=3.53Hz,1 H),6.48(d,J=9.70Hz,1 H),5.84(d,J=9.26Hz,1 H),4.93-5.23(m,6 H),4.23(t,J=6.39Hz,2 H),3.98-4.12(m,2 H),3.52-3.75(m,4 H),2.57-2.80(m,4 H),2.32(m,1 H),1.86-2.23(m,4 H),1.60-1.77(m,2 H),1.44-1.57(m,2 H)
((S)-(3-((4-((4-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)甲基)苯甲基)胺基甲醯基)苯甲基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物43)
步驟1:(4-(1,3-二-2-基)苯基)甲胺
4-氰基苄醛(1.31g,10.0mmol)及4-甲苯磺酸水合物(0.19g,1.0mmol)於甲苯(30mL)之經攪拌之溶液添加乙二醇(2.30mL,41.2mmol)。反應混合物於丁及史塔克條件下回流3小時。反應混合物以乙酸乙酯稀釋及以水、10%水性碳酸鉀溶液及鹽水洗滌。有機相經脫水(硫
酸鎂),過濾及於減壓下蒸發去除溶劑。殘餘物溶解於無水THF(10mL)及逐滴添加至氫化鋁鋰於THF之冰冷溶液(2M於THF,15.0mL,30.0mmol)。許可反應混合物溫熱至室溫,於攪拌下於此溫度維持18小時。依據添加水(1.2mL)、2M水性氫氧化鈉(1.2mL)及水(3.6mL)及隨後混合物於室溫攪拌30分鐘。添加乙酸乙酯及硫酸鎂及混合物又攪拌30分鐘。反應混合物經過濾及濾液於減壓下蒸發獲得標題化合物(1.56g,87%)。
1H NMR(400MHz,CDCl3):δ 7.46(m,2 H);7.34(d,J=8.4Hz,2 H);5.81(s,1 H);4.17-4.00(m,2 H);3.88(s,2 H);1.55(s,2 H).
步驟2:4-((3-(((第三丁氧基甲醯基)胺基)(苯基)甲基)苯氧基)甲基)苯甲酸(S)-甲基酯
((3-羥基苯基)(苯基)甲基)胺基甲酸(S)-第三丁基酯(3.20g,10.7mmol),4-(溴甲基)苯甲酸甲酯(2.70g,11.8mmol)及碳酸鉀(2.20g,16.1mmol)於乙腈(54mL)之混合物於室溫攪拌16小時。反應混合物於減壓下濃縮及殘餘物分溶於乙酸乙酯及水。水相又以乙酸乙酯萃取及組合有機萃取物,以無水硫酸鎂脫水,過濾及於減壓下蒸發去除溶劑。殘餘物從乙酸乙酯及異己烷再結晶獲得標題化合物呈白色固體(3.25g,68%)。
1H NMR(400MHz,CDCl3):δ 8.04(d,J=8.2Hz,2 H);7.46(d,J=8.2Hz,2 H);7.34-7.20(m,6 H);6.90-6.81(m,3 H);5.87(s,1 H);5.13(s,1 H);5.07(s,2 H);3.92(s,3 H);1.44(s,9 H).
步驟3:4-((3-(胺基(苯基)甲基)苯氧基)甲基)苯甲酸(S)-甲基酯鹽酸鹽
4-((3-(((第三丁氧基甲醯基)胺基)(苯基)甲基)苯氧基)甲基)苯甲酸甲基酯(3.21g,7.20mmol)於甲醇(36mL)之溶液添加氯化氫於二(4M,9.0mL,36mmol)。反應混合物於室溫攪拌16小時。於減壓下去除溶劑獲得標題化合物(2.65g,>95%)。
1H NMR(400MHz,CDCl3):δ 9.21(s,2 H);8.03(d,J=8.1Hz,2 H);7.64(d,J=8.1Hz,2 H);7.59(d,J=7.6Hz,2 H);7.49-7.34(m,5 H);7.17(d,J=7.7Hz,1 H);7.06(dd,J=8.3,2.4Hz,1 H);5.64(s,1 H);5.28(s,2 H);3.91(s,3 H).
步驟4:4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸甲酯
4-((3-(胺基(苯基)甲基)苯氧基)甲基)苯甲酸(S)-甲基酯鹽酸鹽(12.0g,31.3mmol)於吡啶(100mL)於0℃之經攪拌之溶液內分成數份添加(R)-啶-3-基氯代甲酸酯(8.50g,37.5mmol)。反應於0℃攪拌
1小時及然後讓其溫熱至室溫16小時。加水至反應混合物及以乙酸乙酯萃取(3次)。組合有機萃取物以鹽水洗滌,脫水(硫酸鈉),過濾及於減壓下蒸發去除溶劑。粗產物於KP-NH Biotage卡匣上藉層析術純化,以0-20%甲醇於乙酸乙酯洗提獲得標題化合物(10.3g,66%)。
步驟5:4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸
4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸甲酯(2.27g,4.50mmol)於THF(23mL)之經攪拌之溶液添加氫氧化鋰水溶液(2.0M,9.0ml,18.0mmol)。混合物於室溫攪拌16小時。反應混合物之pH藉添加4M水性鹽酸調整至6。然後混合物以10%甲醇性乙酸乙酯萃取(2次)及組合有機萃取物於減壓下蒸發。然後殘餘物溶解於乙醇及於減壓下再度蒸發獲得標題化合物呈淺黃色固體(1.85g,84%)。
1H NMR(400MHz,DMSO-d6):δ 8.41(d,J=9.4Hz,1 H);7.99(d,J=7.9Hz,2 H);7.58(d,J=8.0Hz,2 H);7.42-7.26(m,6 H);7.09(s,1 H);7.02-6.91(m,2 H);5.87(d,J=9Hz,1 H);5.21(s,2 H);4.76(s,1 H);3.98-2.72(m,6 H);2.12-1.54(m,5 H).
步驟6:((S)-(3-((4-((4-(1,3-二-2-基)苄基)胺基甲醯基)苄基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯
標題係如實施例5步驟1所述分別使用4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸及(4-(1,3-二-2-基)苯基)甲胺置換1-甲基-5-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)-1H-吡唑-3-羧酸及4-丁醛二乙基縮醛製備。
步驟7:((S)-(3-((4-((4-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)甲基)苯甲基)胺基甲醯基)苯甲基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物43)
標題化合物係如實施例3步驟4及步驟5使用((S)-(3-((4-((4-(1,3-二-2-基)苄基)胺基甲醯基)苄基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯置換4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸3-(1,3-二-2-基)苄基酯及該產物用於步驟5製備。
1H NMR(400MHz,DMSO-d6):δ 9.06-9.00(m,1 H);8.31-8.23(m,3 H);8.12(d,J=9.9Hz,1 H);7.90(d,J=8.1Hz,2 H);7.52(d,J=8.0Hz,2 H);7.32-7.17(m,10 H);7.05(t,J=4.2Hz,2 H);6.96-6.85(m,3 H);6.53-6.45(m,1 H);5.82(d,J=9.2Hz,1 H);5.18-5.06(m,3 H);4.62(s,2 H);4.47(d,J=5.9Hz,2 H);3.92-3.68(m,2 H);2.88-2.51(m,7 H);1.95(s,1 H);1.83(s,1 H);1.62(s,1 H);1.52(s,1 H);1.39(s,1 H).
下列化合物係如實施例9以必需醛用在步驟1製備。
下列化合物係如實施例9所述使用步驟2使用的必需鹵化物置換(4-溴甲基)苯甲酸甲酯及步驟6使用的必需胺置換(4-(1,3-二 -2-基)苯基)甲胺製備。
((S)-(3-((5’-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)甲基)-2’-甲氧基-[1,1’-聯苯]-4-基)甲氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物53)
步驟1:((S)-(3-((4-溴苄基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯
標題化合物係如實施例1步驟1至7使用4-溴苄基溴置換步驟5之4-(溴甲基)苯甲酸甲酯製備。
1H NMR(400MHz,DMSO-d6):δ 8.23(d,J=9.2Hz,1 H);7.59-7.56(m,2 H);7.40-7.38(m,2 H);7.32-7.22(m,6 H);7.01(s,1 H);6.95-6.86(m,2 H);5.83(d,J=9.2Hz,1 H);5.06(s,2 H);4.57-4.55(m,1 H);3.18(m,1 H);2.72-2.47(m,5 H);1.89(s,1 H);1.78(s,1 H);1.58(m,1 H);1.46(s,1 H);1.32(s,1 H).
步驟2:((S)-(3-((5’-甲醯基-2’-甲氧基-[1,1’-聯苯]-4-基)甲氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯
((S)-(3-((4-溴苄基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(0.30g,0.57mmol)及5-甲醯基-2-甲氧基苯基二羥硼酸(0.15g,0.86mmol)於甲苯/水(2mL/0.5mL)之溶液添加碳酸鈉(0.12g,1.14mmol)。氮氣冒泡通過反應混合物5分鐘,然後以肆(三苯基膦)鈀(0)(0.03g,0.03mmol)處理及反應混合物回流加熱2小時。反應混合物以乙酸乙酯稀釋及以水及鹽水洗滌。有機相經脫水(硫酸鎂,過濾及於減壓下蒸發去除溶劑。殘餘物載荷至SCX-2卡匣上及以乙腈(4管柱體積)及然後以10%三乙基胺/乙腈(4管柱體積)洗提。10%三乙基胺/乙腈洗提分藉TLC分析及含產物的洗提分經組合及於減壓下蒸發。材料未經進一步純化即用於次一步驟。
步驟3:((S)-(3-((5’-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)甲基)-2’-甲氧基-[1,1’-聯苯]-4-基)甲氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物53)
標題化合物係如實施例1步驟11所述使用((S)-(3-((5’-甲
醯基-2’-甲氧基-[1,1’-聯苯]-4-基)甲氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯置換4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸2-(4-甲醯基-2-甲基苯氧基)乙基酯製備。
1H NMR(400MHz,DMSO-d6):δ 8.32-8.23(m,3 H);8.11(d,J=9.9Hz,1 H);7.63-7.33(m,4 H);7.35-7.21(m,8 H);7.09-7.04(m,3 H);6.96-6.88(m,3 H);6.45(d,J=9.9Hz,1 H);5.84(d,J=9.1Hz,1 H);5.17-5.08(m,3 H);4.66-4.59(m,1 H);3.83(s,2 H);3.76(s,3 H);3.23-3.12(m,1 H);2.85-2.51(m,7 H);1.93(s,1 H);1.83(s,1 H);1.62(s,1 H);1.52(s,1 H);1.39(s,1 H).
下列化合物係如實施例10所述使用步驟2使用的必需二羥基硼酸置換5-甲醯基-2-甲氧基苯基二羥基硼酸製備。
((S)-(3-((4-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)甲基)苯甲基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物56)
步驟1:甲烷磺酸4-(1,3-二-2-基)苄基酯
(4-(1,3-二-2-基)苯基甲醇(如WO2012168359所述製備,1.2g,6.6mmol)及三乙基胺(2.8mL,20.0mmol)於DCM(40mL)之以冰冷卻經攪拌之溶液逐滴添加甲烷磺醯氯(0.77mL,10.0mmol)於DCM(10mL)之溶液。反應混合物於此溫度攪拌40分鐘及然後於室溫攪拌18小時。反應混合物以水洗滌及有機相經脫水(硫酸鎂)。濾液於減壓下蒸發獲得標題化合物,其未經進一步純化即立刻使用。
步驟2:((S)-(3-((4-(1,3-二-2-基)苄基)氧基)苯基)(苯基)甲基)胺基甲酸(R)啶-3-基酯
標題化合物係如實施例1步驟1至步驟7所述使用甲烷磺酸4-(1,3-二-2-基)苄基酯置換4-(溴甲基)苯甲酸甲酯製備。產物未經任何純化即直接用於次一步驟。
步驟3:((S)-(3-((4-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)甲基)苯甲基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物56)
標題化合物係如實施例3步驟4及步驟5所述使用((S)-(3-((4-(1,3-二-2-基)苄基)氧基)苯基)(苯基)甲基)胺基甲酸(R)啶-3-基酯置換4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸3-(1,3-二-2-基)苄基酯製備及該產物用於步驟5。
1H NMR(400MHz,DMSO-d6):δ 8.31-8.20(m,3 H);8.11(d,J=9.9Hz,1 H);7.41-7.27(m,8 H);7.26-7.20(m,2 H);7.12-7.00(m,2 H);6.94-6.83(m,3 H);6.48(d,J=9.9Hz,1 H);5.82(d,J=9.3Hz,1 H);5.10(dd,J=8.0,4.5Hz,1 H);5.04(s,2 H);4.63-4.62(m,1 H);3.81(s,2 H);3.27-3.08(m,1 H);2.82(br s,2 H);2.81-2.56(m,5 H);1.96(br s,1 H);1.90-1.84(m,1 H);1.70-1.60(m,1 H);1.63-1.46(m,1 H);1.44-1.38(m,1 H).
下列化合物係如實施例11所述使用步驟1必需醇置換(4-(1,3-二-2-基)苯基)甲醇製備。
((S)-(3-(2-((4-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)甲基)苯基)胺基)-2-側氧基乙氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物62)
步驟1:2-(3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)乙酸
標題化合物係如實施例1步驟1至步驟7所述使用溴乙酸甲酯置換步驟5的4-(溴甲基)苯甲酸甲酯製備。
1H NMR(400MHz,DMSO-d6):δ 8.32-8.24(m,1 H);7.34-7.12(m,6 H);6.89(s,1 H);6.84(d,J=7.6Hz,1 H);6.69(dd,J=8.2,2.5Hz,1 H);5.79(d,J=9.2Hz,1 H);4.72-4.70(m,1 H);4.37(s,2 H);3.30-3.29(m,1 H);2.93(s,2 H);2.89-2.69(m,4 H);2.05-2.03(m,1 H);1.92-1.88(m,1 H);1.70-1.50(m,3 H).
步驟2:((S)-(3-(2-((4-(羥基甲基)苯基)-胺基)-2-側氧基乙氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯
標題化合物係如實施例1步驟10所述使用2-(3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)乙酸置換4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)甲基)苯氧基)甲基)苯甲酸及4-胺基苄醇置換4-(2-羥基乙氧基)-3-甲基苄醛製備。
步驟3:((S)-(3-(2-((4-甲醯基苯基)胺基)-2-側氧基乙氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯
於1,4-二(10mL)之經攪拌之((S)-(3-(2-((4-羥基甲基)苯基)胺基)-2-側氧基乙氧基)苯基)(苯基)甲基)-胺基甲酸(R)-啶-3-基酯(0.68g,1.31mmol)溶液內添加氧化錳(IV)(0.57g,6.58mmol)。反應混合物於室溫攪拌60小時。懸浮液經過濾及濾餅以乙酸乙酯洗滌。濾液
於減壓下蒸發獲得粗產物標題化合物。此材料未經進一步純化即供使用。
步驟4:((S)-(3-(2-((4-((((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)甲基)苯基)胺基)-2-側氧基乙氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物62)
標題化合物係如實施例11步驟11所述製備。
1H NMR(400MHz,DMSO-d6):δ 10.50(s,1 H);10.23(s,1 H);9.63(s,1 H);9.05(s,1 H);8.48(d,J=9.2Hz,1 H);8.06(d,J=9.9Hz,1 H);7.66(d,J=8.2Hz,2 H);7.47(d,J=8.4Hz,2 H);7.36-7.17(m,7 H);7.12(d,J=8.2Hz,1 H);7.05-6.95(m,3 H);6.87(dd,J=8.3,2.5Hz,1 H);6.56(d,J=9.9Hz,1 H);6.18(s,1 H);5.86-5.81(m,1 H);5.32(d,J=9.6Hz,1 H);4.89-4.84(m,1 H);4.69(s,2 H);4.19(s,2 H);3.71-3.60(m,1 H);3.30-2.90(m,7 H);2.24(s,1 H);2.10-1.70(m,4 H).
下列化合物係如實施例12所述使用用於步驟2之必需胺置換4-胺基苄醇製備。
((S)-(3-((4-(2-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)乙基)-3-(三氟甲基)苯甲基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物66)
步驟1:((S)-(3-((4-溴-3-(三氟甲基)苄基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯
標題化合物係如實施例1步驟1至步驟7所述使用4-溴-3-三氟甲基-苄基溴置換步驟5之4-(溴甲基)苯甲酸甲酯製備。
1H NMR(400MHz,DMSO-d6):δ 8.22(d,J=9.6Hz,1 H);7.90(d,J=7.0Hz,2 H);7.65(d,J=8.3Hz,1 H);7.32-7.18(m,6 H);7.04(s,1 H);6.97(d,J=7.7Hz,1 H);6.90(dd,J=8.2,2.5Hz,1 H);5.83(d,J=9.5Hz,1 H);5.16(s,2 H);4.56(s,1 H);3.07(t,J=10.7Hz,1 H);2.79-2.54(m,5 H);1.89(s,1 H);1.78(s,1 H);1.57(s,1 H);1.46(s,1 H);1.32(s,1 H).
步驟2:((S)-(3-((4-((E)-2-乙氧基乙烯基)-3-(三氟甲基)苄基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯
((S)-(3-((4-溴-3-(三氟甲基)苄基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(0.295g,0.50mmol)及碳酸銫(0.494g,1.50mmol)之混合物添加已除氣的1,4-二/水(4/1,5mL)。此混合物以2-乙氧基伸乙基-1-二羥硼酸酯(0.152g,0.75mmol)處理及該混合物使用氮氣除氣。添加肆(三苯基膦)鈀(0)(0.029g,0.025mmol)及反應混合物回流3小時。反應混合物於減壓下蒸發及殘餘物分溶於乙酸乙酯及水。有機相以鹽水洗滌,脫水(硫酸鎂),過濾及於減壓下蒸發去除溶劑。粗產物藉層析術純化,以0-20%甲醇於乙酸乙酯洗提獲得標題化合物(0.258g,89%)。
1H NMR(400MHz,DMSO-d6):δ 8.23(d,J=9.5Hz,1 H);7.70(d,J=8.9Hz,2 H);7.67-7.55(m,1 H);7.34-7.20(m,6 H);7.05(s,1 H);6.97-6.85(m,2 H);5.98(dd,J=12.6,2.6Hz,1 H);5.82(d,J=9.5Hz,1 H);5.11(s,2 H);4.56(s,1 H);3.95(q,J=7.0Hz,2 H);3.07(t,J=10.8Hz,1 H);2.2.81-2.48(m,6 H);1.89(s,1 H);1.79(s,1 H);1.57(s,1 H);1.45(s,1 H);1.28(t,J=7.0Hz,4 H).
步驟3:((S)-(3-((4-(2-(((R)-2-羥基-2-(8-羥基-2-側氧基-1,2-二氫喹啉-5-基)乙基)胺基)乙基)-3-(三氟甲基)苯甲基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯(化合物66)
標題化合物係如實施例3步驟4及步驟5所述使用((S)-(3-((4-((E)-2-乙氧基乙烯基)-3-(三氟甲基)苄基)氧基)苯基)(苯基)甲基)胺基甲酸(R)-啶-3-基酯置換4-((3-((S)-苯基((((R)-啶-3-基氧基)甲醯基)胺基)-甲基)苯氧基)甲基)苯甲酸3-(1,3-二-2-基)苄基酯
及該產物用於步驟5製備。
1H NMR(400MHz,DMSO-d6):δ 10.51(s,2 H);9.67(s,1 H);8.94(br s,2 H);8.46(d,J=9.25Hz,1 H);8.18(d,J=9.95Hz,1 H);7.80(s,1 H);7.73(d,J=8.06Hz,1 H);7.55(d,J=8.00Hz,1 H);7.33-7.21(m,6 H);7.17(d,J=8.19Hz,1 H);7.05-6.97(m,3 H);6.91(dd,J=8.27,2.41Hz,1 H);6.59(m,2 H);6.24(s,1 H);5.84(d,J=9.09Hz,1 H);5.35(d,J=9.65Hz,1 H);5.15(s,2 H);4.88-4.83(m,1 H);3.72-3.60(m,1 H);3.27-3.11(m,10 H);2.23(br s,1 H);2.13-1.99(m,1 H);1.96-1.68(m,3 H).
下列化合物係如實施例13所述使用步驟1使用的必需苄基溴置換4-溴-3-三氟甲基-苄基溴製備。
(S)-3-(胺基(苯基)甲基)酚鹽酸鹽之大規模合成
步驟1:(3-甲氧基苯基)(苯基)甲酮
於五氯化磷(3763g,18.1mol)於7500mL苯之混合物內分成數份添加3-甲氧基苯甲酸(2500g,16.4mol)。混合物攪拌50分鐘直到變均質。醯氯之生成係藉TLC控制。完成後,混合物冷卻至10℃,反應器覆蓋鋁箔,及分成數份添加三氯化鋁(4820g,36.1mol)(內溫至多維持至30℃)。於室溫持續攪拌18小時。藉TLC(AcOEt:hex 1:9)監視反應。反應完成後,反應混合物傾倒入冰中及以AcOEt(7L)稀釋。然後有機層經分離及水層以AcOEt(2×10L,1×6L)萃取。組合有機層以水(5×3L)洗滌至pH約為6-7,以飽和水性碳酸氫鈉溶液(15L)洗滌,脫水(硫酸鈉),過濾及於減壓下蒸發去除溶劑獲得粗產物油。該產物藉減壓蒸餾(130-139℃,2毫巴)純化獲得標題化合物呈(2637g,76%)淺黃色油。
1H NMR(600MHz,CDCl3);δ 7.80(m,2 H);7.57(m,1 H);.7.46(m,2 H);7.32-7.37(m,3 H);7.12(m,1 H);3.83(s,3 H).
步驟2:(3-羥基苯基)(苯基)甲酮
1458g(6.9mol)(3-甲氧基苯基)(苯基)甲酮溶解於2090mL AcOH。於該溶液內,添加2320ml(20.6mol)48% HBr及混合物於90℃攪拌18小時。反應藉TLC(AcOEt:hex 1:9)監視。反應完成後,混合物冷卻至室溫及以攪拌傾倒入冰內。沈澱固體經過濾,以水洗滌及乾燥獲得標題化合物(1234g,91%)呈白色固體。
1H NMR(600MHz,CDCl3);δ 7.80(m,2 H);7.58(m,1 H);7.47(m,2 H);7.39(m,1 H);7.28-7.34(m,2 H);7.11(m,1 H);5.59(brs,1 H).
步驟3:3-(胺基(苯基)甲基)酚
(3-羥基苯基)(苯基)甲酮(400g,2mol)溶解於甲醇(4L)。羥基胺鹽酸鹽(168g,2.4mol)及乙酸鈉(331g,4mol)添加至所得溶液。混合物回流18小時。冷卻至室溫後,於減壓下蒸發去除溶劑,然後加水(3L)至殘餘物。產物以乙酸乙酯(3×3L)萃取。組合有機萃取物以飽和水性碳酸氫鈉、鹽水洗滌,脫水(硫酸鈉),過濾及於減壓下蒸發去除溶劑。粗產物殘餘物(1085g)未經進一步純化即用於次一步驟。
粗產物肟362g(基於分析287g,1.3mol純肟)溶解於乙醇(860mL)及25%水性氨(3000mL)。於此混合物內添加乙酸銨(52g,0.7mol),接著逐份添加鋅粉(440g,6.7mol)維持內溫不超過40℃。混合物未經加熱攪拌18小時,然後通過矽藻土墊過濾。濾餅以乙酸乙酯洗滌。濾液經收集及分離所形成的各層。水層以乙酸乙酯(5×5L)萃取。組合有機萃取物層以鹽水洗滌(2次)及於減壓下蒸發去除溶劑。產物於減壓
下乾燥(35℃,18小時)。
1H NMR(600MHz,DMSO-d6);δ 9.25(brs,1 H);7.36(m,2 H);7.25(m,2 H);7.15(m,1 H);7.03(m,1 H);6.79(m,2 H);6.54(m,1 H);4.98(s,1 H);2.17(brs,2 H).
步驟4:(S)-3-(胺基(苯基)甲基)酚(S)-扁桃酸鹽之結晶化
鹽形成:3-(胺基(苯基)甲基)酚(1081g,5.4mol)溶解於異丙醇(21.62L)及加熱至回流。於該混合物內逐滴添加S-扁桃酸(908g,6mol)於異丙醇(2160mL)之溶液。混合物回流1小時及然後任其冷卻至10℃(經18小時)。過濾所形成的沈澱,以冷異丙醇洗滌及於35℃於減壓下乾燥。
所得鹽於95%異丙醇回流1小時。讓混合物經18小時冷卻至10℃。固體經過濾,以冷異丙醇洗滌及於35℃於減壓烤爐內乾燥。結晶過程重複二次或多次直到藉對掌HPLC分析ee>98%。
步驟5:(S)-3-(胺基(苯基)甲基)酚鹽酸鹽
(S)-3-(胺基(苯基)甲基)酚(S)-扁桃酸鹽(1027g,2.9mol)懸浮於乙酸乙酯。逐滴添加碳酸氫鈉(737g,8.8mol)於水(11.05L)之溶液及該混合物於室溫攪拌18小時。混合物經分離及水層以乙酸乙酯(5×10L)萃取。組合有機萃取物經組合及於減壓下蒸發去除溶劑獲得464g(85%)胺呈淺黃色晶體。
胺(464g,2.3mol)懸浮於甲醇及逐滴添加4M HCl於AcOEt(3500mL,14mol)。混合物攪拌18小時及於減壓下蒸發去除溶劑。殘餘物以醚(2740mL)濕磨18小時。懸浮液經過濾,濾餅以醚洗滌及乾燥。
1H NMR(600MHz,DMSO-d6);δ 9.74(s,1 H);9.19(s,3 H);7.54(m,2 H);7.40(m,2 H);7.33(m,1 H);7.19(m,1 H);7.00(m,1 H);6.89(m,1 H);6.78(m,1 H);5.49(s,1H,CH).
得自Perkin Elmer之人類M3受體膜(15微克/孔)與0.52nM莨菪鹼甲基氯,[N-甲基-3H]含或不含試驗化合物,或與飽和濃度之顛茄鹼(5μM)一起培養以測定非專一性結合。檢定分析係於96孔聚丙烯孔板以250微升體積進行。使用的檢定分析緩衝液為50mM Tris-HCl,154mM NaCl(pH7.4)。DMSO之終檢定分析濃度為0.5%(v/v)。孔板經密封及於軌道振搖器上(慢速)於室溫培養2小時。使用過濾器歧管,於經使用0.5%聚伸乙基亞胺(v/v)前處理的96孔單一過濾器GF/C過濾板上獲得膜,以200微升檢定分析緩衝液洗四次。於添加50微閃爍(microscint)-0之前,板經乾燥,經密封,然後於Trilux Microbeta閃爍計數器上讀取。使用非線性曲線匹配程式,從競爭曲線決定IC50值。Ki值係藉陳氏及普索夫氏(Cheng and Prusoff)方程式從IC50值求出。
實施例中大部分化合物之Ki值係小於10nM。
得自Perkin Elmer之人類β2腎上腺素受體膜(7.5微克/孔)與含或不含試驗化合物之0.3nM 125-I Cyanopindolol或飽和濃度之s-propranolol(2μM)一起培養以測定非專一性結合。檢定分析係以200微升體積與96孔聚丙烯板進行。使用的檢定分析緩衝液為25mM HEPES,0.5% BSA(w/v),1mM EDTA,0.02%抗壞血酸(v/v),(pH7.4)。DMSO之終檢定分析濃度為0.5%(v/v)。孔板經密封及於軌道振搖器上(慢速)於室溫培養1小時。使用過濾器歧管,於經使用0.5%聚伸乙基亞胺(v/v)前處理的96孔單一過濾器GF/C過濾板上獲得膜,以含有10mM HEPES及500mM NaCl之200微升洗滌緩衝液洗六次。於添加50微閃爍-0之前板經乾燥,經密封,然後於Trilux Microbeta閃爍計數器上讀取。使用非線性曲線匹配程式,從競爭曲線決定IC50值。Ki值係藉陳氏及普索夫氏方程式從IC50值求出。實施例中大部分化合物之Ki值係小於10nM。
Claims (11)
- 一種通式I化合物,其中Q為式Q1之基團
 Z為OH;Y係選自Y’及Y1,其為下式之二價基團
Z為OH;Y係選自Y’及Y1,其為下式之二價基團 或
或 其中A1為伸丁基;A2為不存在或為(C1-C6)伸烷基;B係選自於由吡唑二基及噻吩二基所組成之組群,其各選擇性地經以一或多個甲基基團取代;C為-OC(O)-或為基團C1
其中A1為伸丁基;A2為不存在或為(C1-C6)伸烷基;B係選自於由吡唑二基及噻吩二基所組成之組群,其各選擇性地經以一或多個甲基基團取代;C為-OC(O)-或為基團C1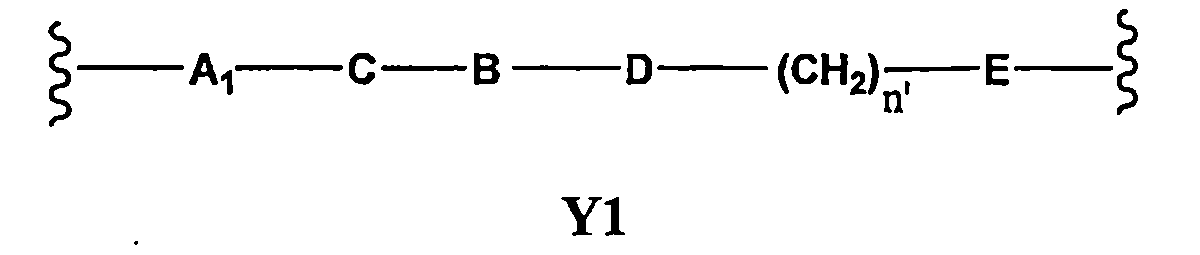 其中R4為H;D為不存在;n’為1;E為不存在或係-O-;G為伸苯基;R1為H;R2為苯基;R3為式J1或J2基團
其中R4為H;D為不存在;n’為1;E為不存在或係-O-;G為伸苯基;R1為H;R2為苯基;R3為式J1或J2基團 其中R5為式K基團
其中R5為式K基團 其中p’為0或1,P為不存在或為CO,q為不存在或為1,及W為雜芳基,X-為生理上可接受之陰離子,及其中該芳基係指含5至15個環原子之一、二、或三環環系,及其中至少一個環為芳香族,其中該雜芳基係指含5至15個環原子之一、二、或三環環系,及其中至少一個環為芳香族及其中至少一個碳環原子為選自N、S或O之雜原子,或其醫藥上可接受之鹽類。
其中p’為0或1,P為不存在或為CO,q為不存在或為1,及W為雜芳基,X-為生理上可接受之陰離子,及其中該芳基係指含5至15個環原子之一、二、或三環環系,及其中至少一個環為芳香族,其中該雜芳基係指含5至15個環原子之一、二、或三環環系,及其中至少一個環為芳香族及其中至少一個碳環原子為選自N、S或O之雜原子,或其醫藥上可接受之鹽類。
- 如申請專利範圍第1項之化合物,其中,Q為Q1Z為-OH,Y為Y’,其為下式之二價基團


- 如申請專利範圍第1或2項之化合物,其中,A2係為不存在或係選自於由亞甲基、伸乙基及伸丙基所組成之組群;B係選自於由噻吩二基、及吡唑二基所組成之組群。
- 如申請專利範圍第1項之化合物,其中,Q為Q1Z為-OH,Y為Y1,其係為下式之二價基團


- 一種醫藥組成物,其係包含申請專利範圍第1至4項中任一項定義之式I化合物及一或多個醫藥上可接受之載劑及/或賦形劑。
- 一種申請專利範圍第1至4項中任一項之式I化合物用於製造治療支氣管阻塞或發炎疾病之藥物之用途。
- 如申請專利範圍第6項之用途,其中,該支氣管阻塞或發炎疾病為氣喘或慢性支氣管炎或慢性阻塞性肺病(COPD)。
- 一種申請專利範圍第1至4項中任一項定義之式I化合物與一或多個活性成分之組合,該活性成分係選自於由下列所組成之組群:β2致效劑、抗蕈毒鹼劑、有絲分裂原活化蛋白質激酶(P38 MAP激酶)抑制劑、核因子κ-B激酶亞單位β(IKK2)抑制劑、人類嗜中性彈力蛋白酶(HNE)抑制劑、磷酸二酯酶4(PDE4)抑制劑、白三烯調節劑、非類固醇消炎藥(NSAID)、止咳劑、黏液調節劑、化痰劑、祛痰劑/黏液促動調節劑、胜肽化痰劑、抗生素、JAK抑制劑、SYK抑制劑、PI3Kδ或PI3Kγ抑制劑、皮質類固醇及M3拮抗劑/PDE4抑制劑(MAPI)。
- 如申請專利範圍第5項之醫藥組成物,其係藉吸入投予。
- 如申請專利範圍第9項之醫藥組成物,其係為吸入性粉末劑、含推進劑之定量噴霧劑或不含推進劑之吸入性配方。
- 一種包含申請專利範圍第9或10項之醫藥組成物之裝置,其可為單劑或多劑乾粉吸入器、定量劑量吸入器或軟霧霧化器。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12195891 | 2012-12-06 | ||
| ??12195891.2 | 2012-12-06 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW201427976A TW201427976A (zh) | 2014-07-16 |
| TWI637954B true TWI637954B (zh) | 2018-10-11 |
Family
ID=47323975
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW102144561A TWI637954B (zh) | 2012-12-06 | 2013-12-05 | 具有蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑活性之化合物 |
Country Status (38)
| Country | Link |
|---|---|
| US (2) | US8987299B2 (zh) |
| EP (2) | EP2928889B1 (zh) |
| JP (1) | JP6421989B2 (zh) |
| KR (2) | KR102156443B1 (zh) |
| CN (1) | CN104822678B (zh) |
| AR (1) | AR093825A1 (zh) |
| AU (1) | AU2013354043C1 (zh) |
| BR (1) | BR112015012730B1 (zh) |
| CA (1) | CA2893626C (zh) |
| CL (1) | CL2015001483A1 (zh) |
| CO (1) | CO7400870A2 (zh) |
| CY (2) | CY1120159T1 (zh) |
| DK (2) | DK2928889T3 (zh) |
| EA (1) | EA030552B1 (zh) |
| ES (2) | ES2670686T3 (zh) |
| GE (1) | GEP20186839B (zh) |
| HK (1) | HK1212989A1 (zh) |
| HR (2) | HRP20180915T1 (zh) |
| HU (2) | HUE037944T2 (zh) |
| IL (1) | IL239182B (zh) |
| LT (2) | LT3345904T (zh) |
| MA (1) | MA38147B1 (zh) |
| MX (1) | MX369311B (zh) |
| NZ (1) | NZ708738A (zh) |
| PE (1) | PE20151216A1 (zh) |
| PH (1) | PH12015501247A1 (zh) |
| PL (2) | PL2928889T3 (zh) |
| PT (2) | PT2928889T (zh) |
| RS (2) | RS57232B1 (zh) |
| SA (1) | SA515360514B1 (zh) |
| SG (2) | SG10201704032RA (zh) |
| SI (2) | SI2928889T1 (zh) |
| TN (1) | TN2015000244A1 (zh) |
| TR (1) | TR201808698T4 (zh) |
| TW (1) | TWI637954B (zh) |
| UA (1) | UA118181C2 (zh) |
| WO (1) | WO2014086924A1 (zh) |
| ZA (1) | ZA201503965B (zh) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ITRM20110083U1 (it) | 2010-05-13 | 2011-11-14 | De La Cruz Jose Antonio Freire | Piastra per la costruzione di carrelli per aeroplani |
| EP2386555A1 (en) | 2010-05-13 | 2011-11-16 | Almirall, S.A. | New cyclohexylamine derivatives having beta2 adrenergic agonist and m3 muscarinic antagonist activities |
| EP2592078A1 (en) | 2011-11-11 | 2013-05-15 | Almirall, S.A. | New cyclohexylamine derivatives having beta2 adrenergic agonist and M3 muscarinic antagonist activities |
| HUE037944T2 (hu) * | 2012-12-06 | 2018-09-28 | Chiesi Farm Spa | Muszkarin receptor antagonista és béta2 adrenerg receptor agonista akitvitással rendelkezõ vegyületek |
| TW201427977A (zh) * | 2012-12-06 | 2014-07-16 | Chiesi Farma Spa | 具有蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑活性之化合物 |
| BR112015013628A2 (pt) | 2012-12-18 | 2017-07-11 | Almirall Sa | derivados de carbamato de ciclo-hexila e quinuclidinila tendo atividades agonista adrenérgica de beta2 e antagonista muscarínica de m3 |
| TWI643853B (zh) | 2013-02-27 | 2018-12-11 | 阿爾米雷爾有限公司 | 同時具有β2腎上腺素受體促效劑和M3毒蕈鹼受體拮抗劑活性之2-氨基-1-羥乙基-8-羥基喹啉-2(1H)-酮衍生物之鹽類 |
| TWI641373B (zh) | 2013-07-25 | 2018-11-21 | 阿爾米雷爾有限公司 | 具有蕈毒鹼受體拮抗劑和β2腎上腺素受體促效劑二者之活性的2-胺基-1-羥乙基-8-羥基喹啉-2(1H)-酮衍生物之鹽 |
| TW201517906A (zh) | 2013-07-25 | 2015-05-16 | Almirall Sa | 含有maba化合物和皮質類固醇之組合 |
| TW201617343A (zh) | 2014-09-26 | 2016-05-16 | 阿爾米雷爾有限公司 | 具有β2腎上腺素促效劑及M3蕈毒拮抗劑活性之新穎雙環衍生物 |
| TWI703138B (zh) * | 2015-02-12 | 2020-09-01 | 義大利商吉斯藥品公司 | 具有蕈毒鹼受體拮抗劑及β2腎上腺素受體促效劑活性之化合物 |
| AR104828A1 (es) | 2015-06-01 | 2017-08-16 | Chiesi Farm Spa | COMPUESTOS CON ACTIVIDAD ANTAGONISTA DE LOS RECEPTORES MUSCARÍNICOS Y ACTIVIDAD AGONISTA DEL RECEPTOR b2 ADRENÉRGICO |
| ES2891073T3 (es) * | 2015-11-16 | 2022-01-26 | Chiesi Farm Spa | Un procedimiento para preparar una formulación en polvo seco que comprende un anticolinérgico, un corticoesteroide y un beta-adrenérgico |
| EP3383867B1 (en) | 2015-12-03 | 2021-04-14 | Chiesi Farmaceutici S.p.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| EP3484879B1 (en) | 2016-07-13 | 2020-12-30 | Chiesi Farmaceutici S.p.A. | Hydroxyquinolinone compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| CA2994290C (en) | 2017-11-06 | 2024-01-23 | Entech Solution As | Method and stimulation sleeve for well completion in a subterranean wellbore |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005115467A1 (en) * | 2004-05-31 | 2005-12-08 | Almirall Prodesfarma S.A. | Combinations comprising antimuscarinic agents and beta-adrenergic agonists |
| US20090181935A1 (en) * | 2008-01-15 | 2009-07-16 | Chiesi Farmaceutici S.P.A. | Compositions comprising an antimuscarinic and a long-acting beta-agonist |
| TW201302729A (zh) * | 2011-06-10 | 2013-01-16 | Chiesi Farma Spa | 具有蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑活性之化合物 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2222128A (en) | 1937-01-29 | 1940-11-19 | Pure Oil Co | Preparation and separation of aromatic hydrocarbons |
| DE19921693A1 (de) | 1999-05-12 | 2000-11-16 | Boehringer Ingelheim Pharma | Neuartige Arzneimittelkompositionen auf der Basis von anticholinergisch wirksamen Verbindungen und ß-Mimetika |
| CA2499314C (en) | 2002-10-11 | 2010-08-24 | Pfizer Inc. | Indole derivatives as beta-2 agonists |
| PE20040950A1 (es) | 2003-02-14 | 2005-01-01 | Theravance Inc | DERIVADOS DE BIFENILO COMO AGONISTAS DE LOS RECEPTORES ADRENERGICOS ß2 Y COMO ANTAGONISTAS DE LOS RECEPTORES MUSCARINICOS |
| JP4851937B2 (ja) | 2003-11-21 | 2012-01-11 | セラヴァンス, インコーポレーテッド | β2アドレナリン作動性受容体作動薬活性およびムスカリン受容体拮抗薬活性を有する化合物 |
| CA2553293C (en) | 2004-01-22 | 2010-12-14 | Pfizer Inc. | Sulfonamide derivatives for the treatment of diseases |
| WO2005092861A1 (en) | 2004-03-11 | 2005-10-06 | Pfizer Limited | Quinolinone derivatives pharmaceutical compositions containing them and their use |
| BRPI0507877A (pt) | 2004-03-23 | 2007-07-24 | Pfizer | derivados de formamida, composição farmacêutica, uso dos mesmos e combinação |
| US7538141B2 (en) | 2004-03-23 | 2009-05-26 | Alan Daniel Brown | Compounds for the treatment of diseases |
| US7528253B2 (en) | 2004-08-16 | 2009-05-05 | Theravance, Inc. | Compounds having β2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| WO2006023460A2 (en) | 2004-08-16 | 2006-03-02 | Theravance, Inc. | COMPOUNDS HAVING β2 ADRENERGIC RECEPTOR AGONIST AND MUSCARINIC RECEPTOR ANTAGONIST ACTIVITY |
| FR2898640B1 (fr) | 2006-03-20 | 2008-04-25 | Siemens Vdo Automotive Sas | Procede de transmission d'information relatif au fonctionnement d'un moteur a combustion interne |
| PL3210981T3 (pl) | 2009-04-23 | 2019-06-28 | Theravance Respiratory Company, Llc | Związki diamidowe wykazujące działanie jako antagonista receptora muskarynowego i agonista antagonista receptora beta 2 adrenergicznego |
| EP2426121A4 (en) * | 2009-04-30 | 2012-10-31 | Teijin Pharma Ltd | QUARTER AMMONIUM SALT CONNECTION |
| WO2011048409A1 (en) | 2009-10-20 | 2011-04-28 | Astrazeneca Ab | Cyclic amine derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| KR101906630B1 (ko) | 2011-06-10 | 2018-10-10 | 키에시 파르마슈티시 엣스. 피. 에이. | 무스카린 수용체 길항제 및 베타2 아드레날린 수용체 작용제 효능을 갖는 화합물 |
| HUE037944T2 (hu) * | 2012-12-06 | 2018-09-28 | Chiesi Farm Spa | Muszkarin receptor antagonista és béta2 adrenerg receptor agonista akitvitással rendelkezõ vegyületek |
| TW201427977A (zh) * | 2012-12-06 | 2014-07-16 | Chiesi Farma Spa | 具有蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑活性之化合物 |
-
2013
- 2013-12-05 HU HUE13811827A patent/HUE037944T2/hu unknown
- 2013-12-05 LT LTEP18156043.4T patent/LT3345904T/lt unknown
- 2013-12-05 MX MX2015006788A patent/MX369311B/es active IP Right Grant
- 2013-12-05 TW TW102144561A patent/TWI637954B/zh not_active IP Right Cessation
- 2013-12-05 RS RS20180529A patent/RS57232B1/sr unknown
- 2013-12-05 GE GEAP201313845A patent/GEP20186839B/en unknown
- 2013-12-05 LT LTEP13811827.8T patent/LT2928889T/lt unknown
- 2013-12-05 PL PL13811827T patent/PL2928889T3/pl unknown
- 2013-12-05 EA EA201590873A patent/EA030552B1/ru unknown
- 2013-12-05 ES ES13811827.8T patent/ES2670686T3/es active Active
- 2013-12-05 KR KR1020157014956A patent/KR102156443B1/ko active IP Right Grant
- 2013-12-05 DK DK13811827.8T patent/DK2928889T3/en active
- 2013-12-05 HU HUE18156043A patent/HUE051394T2/hu unknown
- 2013-12-05 CA CA2893626A patent/CA2893626C/en active Active
- 2013-12-05 AR ARP130104529A patent/AR093825A1/es unknown
- 2013-12-05 UA UAA201505478A patent/UA118181C2/uk unknown
- 2013-12-05 PT PT138118278T patent/PT2928889T/pt unknown
- 2013-12-05 PL PL18156043T patent/PL3345904T3/pl unknown
- 2013-12-05 MA MA38147A patent/MA38147B1/fr unknown
- 2013-12-05 DK DK18156043.4T patent/DK3345904T3/da active
- 2013-12-05 NZ NZ708738A patent/NZ708738A/en not_active IP Right Cessation
- 2013-12-05 JP JP2015546012A patent/JP6421989B2/ja active Active
- 2013-12-05 CN CN201380063365.2A patent/CN104822678B/zh active Active
- 2013-12-05 SG SG10201704032RA patent/SG10201704032RA/en unknown
- 2013-12-05 SI SI201330994T patent/SI2928889T1/en unknown
- 2013-12-05 ES ES18156043T patent/ES2816210T3/es active Active
- 2013-12-05 WO PCT/EP2013/075661 patent/WO2014086924A1/en active Application Filing
- 2013-12-05 EP EP13811827.8A patent/EP2928889B1/en active Active
- 2013-12-05 SI SI201331764T patent/SI3345904T1/sl unknown
- 2013-12-05 TR TR2018/08698T patent/TR201808698T4/tr unknown
- 2013-12-05 RS RS20201150A patent/RS60864B1/sr unknown
- 2013-12-05 EP EP18156043.4A patent/EP3345904B1/en active Active
- 2013-12-05 SG SG11201504318UA patent/SG11201504318UA/en unknown
- 2013-12-05 PE PE2015000738A patent/PE20151216A1/es active IP Right Grant
- 2013-12-05 AU AU2013354043A patent/AU2013354043C1/en active Active
- 2013-12-05 KR KR1020207006181A patent/KR102240459B1/ko active IP Right Grant
- 2013-12-05 PT PT181560434T patent/PT3345904T/pt unknown
- 2013-12-05 BR BR112015012730-4A patent/BR112015012730B1/pt active IP Right Grant
- 2013-12-06 US US14/098,735 patent/US8987299B2/en active Active
-
2015
- 2015-01-23 US US14/603,926 patent/US9371318B2/en active Active
- 2015-06-02 CL CL2015001483A patent/CL2015001483A1/es unknown
- 2015-06-03 TN TNP2015000244A patent/TN2015000244A1/fr unknown
- 2015-06-03 PH PH12015501247A patent/PH12015501247A1/en unknown
- 2015-06-03 SA SA515360514A patent/SA515360514B1/ar unknown
- 2015-06-03 ZA ZA2015/03965A patent/ZA201503965B/en unknown
- 2015-06-03 IL IL239182A patent/IL239182B/en active IP Right Grant
- 2015-06-03 CO CO15127118A patent/CO7400870A2/es unknown
-
2016
- 2016-01-29 HK HK16101057.0A patent/HK1212989A1/zh unknown
-
2018
- 2018-05-03 CY CY20181100469T patent/CY1120159T1/el unknown
- 2018-06-13 HR HRP20180915TT patent/HRP20180915T1/hr unknown
-
2020
- 2020-09-09 HR HRP20201435TT patent/HRP20201435T1/hr unknown
- 2020-09-28 CY CY20201100912T patent/CY1123365T1/el unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005115467A1 (en) * | 2004-05-31 | 2005-12-08 | Almirall Prodesfarma S.A. | Combinations comprising antimuscarinic agents and beta-adrenergic agonists |
| US20090181935A1 (en) * | 2008-01-15 | 2009-07-16 | Chiesi Farmaceutici S.P.A. | Compositions comprising an antimuscarinic and a long-acting beta-agonist |
| TW201302729A (zh) * | 2011-06-10 | 2013-01-16 | Chiesi Farma Spa | 具有蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑活性之化合物 |
Non-Patent Citations (1)
| Title |
|---|
| Hughes, Adam D., et al. "Discovery of muscarinic acetylcholine receptor antagonist and beta 2 adrenoceptor agonist (MABA) dual pharmacology molecules." Bioorganic & medicinal chemistry letters 21.5 (2011): 1354-1358. * |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI637954B (zh) | 具有蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑活性之化合物 | |
| TW201427977A (zh) | 具有蕈毒鹼受體拮抗劑及β2腎上腺素受體致效劑活性之化合物 | |
| AU2013354043A1 (en) | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity | |
| BR112013031493B1 (pt) | Composto, composição farmacêutica, uso de um composto, combinação de um composto, dispositivo e processo de preparação de um composto | |
| EP2928869B1 (en) | 1-phenyl-2-pyridinyl alkyl alcohol derivatives as phosphodiesterase inhibitors | |
| US9662323B2 (en) | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | Annulment or lapse of patent due to non-payment of fees |