KR20150113166A - 상호작용 성분을 이용하여 생체분자를 확인하기 위한 방법, 시스템, 및 소프트웨어 - Google Patents
상호작용 성분을 이용하여 생체분자를 확인하기 위한 방법, 시스템, 및 소프트웨어 Download PDFInfo
- Publication number
- KR20150113166A KR20150113166A KR1020157023727A KR20157023727A KR20150113166A KR 20150113166 A KR20150113166 A KR 20150113166A KR 1020157023727 A KR1020157023727 A KR 1020157023727A KR 20157023727 A KR20157023727 A KR 20157023727A KR 20150113166 A KR20150113166 A KR 20150113166A
- Authority
- KR
- South Korea
- Prior art keywords
- model
- sequence
- activity
- new
- interaction
- Prior art date
Links
Images















Classifications
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B35/00—ICT specially adapted for in silico combinatorial libraries of nucleic acids, proteins or peptides
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B35/00—ICT specially adapted for in silico combinatorial libraries of nucleic acids, proteins or peptides
- G16B35/20—Screening of libraries
-
- G06F19/701—
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1058—Directional evolution of libraries, e.g. evolution of libraries is achieved by mutagenesis and screening or selection of mixed population of organisms
-
- C40B50/02—
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
-
- G06F19/12—
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B10/00—ICT specially adapted for evolutionary bioinformatics, e.g. phylogenetic tree construction or analysis
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B15/00—ICT specially adapted for analysing two-dimensional or three-dimensional molecular structures, e.g. structural or functional relations or structure alignment
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/20—Allele or variant detection, e.g. single nucleotide polymorphism [SNP] detection
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/50—Mutagenesis
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B35/00—ICT specially adapted for in silico combinatorial libraries of nucleic acids, proteins or peptides
- G16B35/10—Design of libraries
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B5/00—ICT specially adapted for modelling or simulations in systems biology, e.g. gene-regulatory networks, protein interaction networks or metabolic networks
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B5/00—ICT specially adapted for modelling or simulations in systems biology, e.g. gene-regulatory networks, protein interaction networks or metabolic networks
- G16B5/20—Probabilistic models
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16C—COMPUTATIONAL CHEMISTRY; CHEMOINFORMATICS; COMPUTATIONAL MATERIALS SCIENCE
- G16C10/00—Computational theoretical chemistry, i.e. ICT specially adapted for theoretical aspects of quantum chemistry, molecular mechanics, molecular dynamics or the like
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16C—COMPUTATIONAL CHEMISTRY; CHEMOINFORMATICS; COMPUTATIONAL MATERIALS SCIENCE
- G16C20/00—Chemoinformatics, i.e. ICT specially adapted for the handling of physicochemical or structural data of chemical particles, elements, compounds or mixtures
- G16C20/30—Prediction of properties of chemical compounds, compositions or mixtures
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16C—COMPUTATIONAL CHEMISTRY; CHEMOINFORMATICS; COMPUTATIONAL MATERIALS SCIENCE
- G16C20/00—Chemoinformatics, i.e. ICT specially adapted for the handling of physicochemical or structural data of chemical particles, elements, compounds or mixtures
- G16C20/50—Molecular design, e.g. of drugs
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16C—COMPUTATIONAL CHEMISTRY; CHEMOINFORMATICS; COMPUTATIONAL MATERIALS SCIENCE
- G16C20/00—Chemoinformatics, i.e. ICT specially adapted for the handling of physicochemical or structural data of chemical particles, elements, compounds or mixtures
- G16C20/60—In silico combinatorial chemistry
Abstract
본 발명은 생물학적으로 관련된 데이터 공간을 빠르고 효율적으로 검색하는 방법을 제공한다. 더욱 구체적으로, 본 발명은 복합 생체분자 라이브러리, 또는 상기 라이브러리 세트로부터 원하는 특성을 가지거나, 상기 특성을 획득하는 데 가장 적합한 생체분자를 확인하는 방법을 제공한다. 본 발명은 또한 단계적 가산 또는 감산 기법, 베이지안 회귀, 앙상블 회귀 및 다른 방법을 포함하나, 이에 한정되지 않는, 서열 활성 관계를 모델링하는 방법을 제공한다. 본 발명은 본원에서 제공하는 방법을 수행하기 위한 디지털 시스템 및 소프트웨어를 추가로 제공한다.
Description
관련 출원에 대한 상호 참조
본 출원은 35 U.S.C. § 119(e)하에 2013년 1월 31일 출원된 미국 가특허 출원 번호 제61/759,276호(발명의 명칭: METHODS, SYSTEMS, AND SOFTWARE FOR IDENTIFYING BIO-MOLECULES WITH INTERACTING COMPONENTS), 및 2013년 3월 15일 출원된 미국 가특허 출원 번호 제61/799,377호(발명의 명칭: METHODS, SYSTEMS, AND SOFTWARE FOR IDENTIFYING BIO-MOLECULES USING MODELS OF MULTIPLICATIVE FORM)에 대한 이점을 주장하며, 상기 출원들은 모든 목적을 위해 그 전문이 본원에서 참조로 포함된다.
배경
본 개시내용은 분자 생물학, 분자 진화, 생물 정보학, 및 디지털 시스템 분야에 관한 것이다. 더욱 구체적으로, 본 개시내용은 전산적으로 생체분자의 활성을 예측하고/거나, 유도 진화(directed evolution)를 유도하는 방법에 관한 것이다. 디지털 시스템을 비롯한 시스템, 및 상기 방법을 실행하는 시스템 소프트웨어 또한 제공한다. 본 개시내용의 방법은 단백질의 최적화에서 산업적 및 치료학적 용도에 대한 유용성을 가진다.
검색가능한 서열 공간을 구성하는 가능한 분자의 조합상 급격한 증가 이외에는 다른 이유가 없었다면, 단백질 디자인은 어려운 과정이 되는 것으로 장기간 알려져 왔다. 단백질의 서열 공간은 방대하며, 현재 당업계에 공지된 방법을 사용하여 철저하게 조사하는 것은 불가능하다. 이와 같이 복잡하기 때문에, 우수한 단백질을 디자인하기 위해서는 다수의 근사 방법이 사용되어 왔다; 그중에서도 주된 방법은 유도 진화 방법이다. 오늘날, 단백질의 유도 진화는 흔히 반복적으로 실행되는 다양한 고처리량 스크리닝 재조합 포맷에 의해 지배된다.
동시에, 서열 활성 공간에 대한 조사를 위한 것으로 다양한 전산 기법이 제안되어 왔다. 각각의 전산 기법이 특정 상황하에서는 장점을 가지지만, 기능성 단백질을 확인하기 위하여 서열 공간을 효율적으로 검색하는 새로운 방법이 고도로 바람직할 것이다.
본 개시내용은 비1차 항, 특히, 서열 중 2개 이상의 서브유닛 사이의 상호작용을 나타내는 항을 이용하는 서열-활성 모델을 생성하고 사용하는 기법을 나타낸다. 서열-활성 모델은 다양한 생물학적 서열의 함수로서 생물학적 분자의 활성, 특징, 또는 특성을 기술한다. 상기 비1차 항은, 각각이 상호작용에 참여하는 서브유닛의 존재(또는 부재)를 나타내는 것인 2개 이상의 변수의 곱셈을 포함하는 "외적" 항일 수 있다. 일부 실시양태는 서열의 활성을 가장 잘 기술하는 비1차 항을 선택하는 기법을 포함한다. 흔히 서브유닛 사이의 진정한 상호작용보다 훨씬 더 가능성이 큰 비1차 상호작용 항이 존재한다는 것에 주의한다. 그러므로, 과대적합을 피하기 위해, 전형적으로는 단지 제한된 개수의 비1차 항이 고려되며, 사용되는 비1차 항은, 두드러지게 활성에 영향을 주는 상호작용을 반영하여야 한다.
본 개시내용의 한 측면은 (a) 복수 개의 생물학적 분자에 대한 서열 및 활성 데이터를 입수하는 단계; (b) 서열 및 활성 데이터로부터 베이스 모델을 제조하는 단계로서, 여기서, 베이스 모델은 서열의 서브유닛의 존재 또는 부재의 함수로서 활성을 예측하는 것인 단계; (c) 하나 이상의 신규 상호작용 항을 베이스 모델에 가하거나 베이스 모델로부터 감함으로써 하나 이상의 신규 모델을 제조하는 단계로서, 여기서, 신규 상호작용 항은 2개 이상의 상호작용 서브유닛 사이의 상호작용을 나타내는 것인 단계; (d) 서브유닛의 존재 또는 부재의 함수로서 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력을 측정하는 단계; 및 (e) (d)에서 측정된 바와 같은 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력에 기초하고, 신규 상호작용 항을 가하는 것에 대한 편향(bias)을 이용하여, 신규 상호작용 항을 베이스 모델에 가할지 또는 베이스 모델로부터 감할지 여부를 결정하는 단계를 포함하는, 원하는 활성이 증진된 생물학적 분자를 확인하는 것을 보조할 수 있는 서열-활성 모델을 제조하는 방법을 제공한다. 이어서, 유도된 모델은 다양한 적용에서, 예컨대, 원하는 생물학적 활성 및 특성을 가지는 단백질을 확인하기 위한 단백질 라이브러리의 유도 진화에서 사용될 수 있다.
일부 실시양태에서, 본 방법을 통해, 업데이트된 모델을 생성하기 위해서는 신규 상호작용 항을 베이스 모델에 가하여야 한다고 결정된 경우, 본 방법은 업데이트된 모델을 추가로 개선시킬 수 있는 추가의 상호작용 항을 검색하는 추가 단계를 추가로 포함한다. 구체적으로, 본 방법은 (f) 베이스 모델 대신 업데이트된 모델을 이용하여 (c)를 반복하고, (c)에서 가하거나 감한 것과는 다른 상호작용 항을 가하거나 감하는 단계; 및 (g) 베이스 모델 대신 업데이트된 모델을 이용하여 (d) 및 (e)를 반복하는 단계를 포함한다. 일부 실시양태에서, 본 방법은 (h) 추가의 업데이트된 모델을 이용하여 (f) 및 (g)를 반복하는 단계를 추가로 포함한다. 다양한 실시양태에서, 서열은 유전자들, 유전자, 핵산 서열, 단백질, 다당류 등을 상호작용 시키기 위한 전체 게놈, 전체 염색체, 염색체 세그먼트, 유전자 서열의 집합일 수 있다. 하나 이상의 실시양태에서, 서열의 서브유닛은 염색체, 염색체 세그먼트, 일배체형, 유전자, 뉴클레오티드, 코돈, 돌연변이, 아미노산, 탄수화물(단량체, 이량체, 삼량체, 또는 올리고머) 등일 수 있다.
상기 실시양태와 일관된 하나 이상의 구현에서, 단백질 변이체 라이브러리 중 변형시키고자 하는 아미노산 잔기를 확인하는 방법을 제공한다. 상기 실시양태에서, 복수 개의 생물학적 분자가 단백질 변이체 라이브러리의 트레이닝 세트를 구성한다. 단백질 변이체 라이브러리는 다양한 공급원으로부터의 단백질을 포함할 수 있다. 일례에서, 구성원으로는 자연적으로 발생된 단백질, 예컨대, 단일 유전자 패밀리의 구성원에 의해 코딩된 것을 포함한다. 또 다른 일례에서, 서열은 재조합 기반 다양성 생성 메커니즘을 사용함으로써 수득된 단백질을 포함한다. 예를 들어, 상기 목적으로 하나 이상의 자연적으로 발생된 모체 단백질 모두 또는 그의 일부를 코딩하는 핵산 상에서 DNA 단편화 매개 재조합, 합성 올리고뉴클레오티드 매개 재조합 또는 그의 조합이 수행될 수 있다. 추가의 또 다른 일례에서, 구성원은 체계적으로 다양화된 서열을 확인하는 실험 디자인(DOE: design of experiment) 프로토콜을 실행함으로써 얻는다.
일부 실시양태에서, 하나 이상의 상호작용 항은 한 상호작용 잔기의 존재를 나타내는 한 변수와, 또 다른 상호작용 잔기의 존재를 나타내는 또 다른 변수의 곱을 포함하는 외적 항이다. 서열-활성 모델의 형태는 하나 이상의 외적 항 및 하나 이상의 1차 항의 합일 수 있으며, 여기서, 각각의 1차 항은 단백질 변이체 라이브러리의 트레이닝 세트 중의 가변 잔기의 효과를 나타낸다. 하나 이상의 외적 항은 복원이 없는, 항의 단계적 가산 또는 감산을 포함하는 다양한 기법에 의해 잠재적인 외적 항의 군으로부터 선택될 수 있다.
하나 이상의 실시양태에서, 베이지안(Bayesian) 회귀 기법을 사용하여 외적 항을 포함하는 모델을 주어진 데이터에 적합화(fit)시키고, 여기서, 선험적 지식이 모델의 사후 확률 분포를 결정하는 데 사용된다.
하나 이상의 실시양태에서, 각각 하나 이상의 상이한 상호작용 항을 포함하는 것인 2개 이상의 신규 모델이 생성된다. 본 방법은 추가로 2개 이상의 신규 모델에 기반하여 앙상블 모델을 제조하는 것을 포함한다. 앙상블 모델은 2개 이상의 신규 모델로부터의 상호작용 항을 포함한다. 앙상블 모델은 관심의 대상이 되는 활성을 예측할 수 있는 2개 이상의 신규 모델의 능력에 따라 상호작용 항에 가중치를 부여한다.
서열-활성 모델은 다수의 상이한 기법에 의해 트레이닝 세트로부터 생성될 수 있다. 특정 실시양태에서, 모델은 회귀 모델, 예컨대, 부분 최소 제곱 모델, 베이지안 회귀 모델, 또는 주성분 회귀 모델이다. 또 다른 실시양태에서, 모델은 신경망이다.
고정 또는 변이를 위한 잔기를 확인하기 위해 서열-활성 모델을 이용하는 것은 다수의 상이한 가능한 분석 기법 중 임의의 것을 포함할 수 있다. 일부 경우에서, "참조 서열"은 변이를 정의하는 데 사용된다. 상기 서열은 모델에 의해 원하는 활성을 최고값(또는 최고값들 중 하나)으로 가지는 것으로 예측되는 것일 수 있다. 또 다른 경우에서, 참조 서열은 원래의 단백질 변이체 라이브러리의 구성원의 것일 수 있다. 본 방법은 참조 서열로부터 변이를 수행하기 위한 부분서열을 선택할 수 있다. 추가로 또는 별법으로, 서열-활성 모델은 원하는 활성에 미치는 영향 순서대로 잔기 위치(또는 특이적 위치의 특이적 잔기)를 순위화한다.
본 방법의 한 목표는 신규 단백질 변이체 라이브러리를 생성하고자 하는 것일 수 있다. 상기 프로세스의 일부로서, 본 방법은 상기 신규 라이브러리를 생성하는 데 사용하고자 하는 서열을 확인할 수 있다. 상기 서열은 상기 (e), (g), 또는 (h)에서 확인된 잔기에 변이를 포함하거나, 또는 이어서 상기 변이를 도입하는 데 사용되는 전구체이다. 서열은 단백질 변이체의 신규 라이브러리를 생성하는 돌연변이 유발법 또는 재조합 기반 다양성 생성 메커니즘을 수행함으로써 변형될 수 있다. 이는 유도 진화 방법의 일부를 형성할 수 있다. 신규 라이브러리는 또한 신규 서열-활성 모델을 개발하는 데 사용될 수 있다. 신규 단백질 변이체 라이브러리는 특정 활성, 예컨대, 안정성, 촉매 활성, 치료학적 활성, 병원체 또는 독소에 대한 저항성, 독성 등에 미치는 효과를 평가하기 위해 분석된다.
일부 실시양태에서, 본 방법은 제조를 위해 신규 단백질 변이체 라이브러리의 하나 이상의 구성원을 선택하는 것을 포함한다. 이어서, 이들 구성원들 중 하나 이상을 합성하고/거나, 발현 시스템에서 발현시킬 수 있다. 구체적인 실시양태에서, 본 방법은 하기 방식: (i) 그로부터 신규 단백질 변이체 라이브러리의 선택된 구성원이 발현될 수 있는 발현 시스템을 제공하는 방식; 및 (ii) 신규 단백질 변이체 라이브러리의 선택된 구성원을 발현하는 방식으로 계속 진행된다.
일부 실시양태에서, 본 방법은 아미노산 서열을 사용한다기 보다는 뉴클레오티드 서열을 사용하여 모델을 생성하고, 활성을 예측한다. 뉴클레오티드 군, 예컨대 코돈 중의 변이가 뉴클레오티드 서열에 의해 코딩되는 펩티드의 활성에 영향을 미친다. 일부 실시양태에서, 모델은 펩티드를 발현하는 데 사용되는 숙주에 따라 (같은 아미노산을 코딩하는 다른 코돈과 비교하여) 우선적으로 발현되는 코돈에 대한 편형을 제공할 수 있다.
본 개시내용의 또 다른 측면은 상기 기술된 방법 및 소프트웨어 시스템을 실행하기 위한 프로그램 명령어 및/또는 데이터 배열이 제공되어 있는 기계 판독가능한 매체를 포함하는 장치 및 컴퓨터 프로그램 제품에 관한 것이다. 빈번하게, 프로그램 명령어는 특정 방법 연산을 실행하기 위한 코드로서 제공된다. 본 개시내용의 특징을 수행하는 데 사용될 경우, 데이터는 데이터 구조, 데이터베이스 테이블, 데이터 객체, 또는 특수 정보의 다른 적절한 배열로서 제공될 수 있다. 본원에 기술된 방법 또는 시스템 중 임의의 것은 전체적으로 또는 부분적으로 임의의 적합한 기계 판독가능한 매체 상에 제공된 상기 프로그램 명령어 및/또는 데이터로서 제시될 수 있다.
하기 및 다른 특징은 하기 도면과 함께 하기의 상세한 설명에서 더욱 상세하게 기술된다.
도 1은 서열-활성 모델을 제조하기 위한 일반적인 단계적 방법을 도시한 것이다.
도 2는 하나 이상의 단백질 변이체 라이브러리 세대를 생성하기 위한 연산 순서를 도시한 순서도이며, 여기서, 연산은 서열-활성 모델, 예컨대, 도 1에서 얻은 것들 중 하나를 사용하여 단백질 변이체 라이브러리 세대를 유도한다. 생성된 변이체 라이브러리는 하나 이상의 신규 서열-활성 모델을 제조하기 위한 서열 및 활성 데이터를 제공할 수 있으며, 유도된 진화의 모델링-탐색 루프를 형성할 수 있다.
도 3a-3h는 특정의 선형 및 비선형 모델의 예측 능력을 비교하는 일례를 보여주는 그래프이다.
도 4a-4b는 서열-활성 모델을 제조하기 위한 단계적 가산 및 감산 방법을 실행하는 프로세스에 대한 순서도를 도시한 것이다. 도 4a는 모델을 제조하기 위한 단계적 가산 방법의 구체적인 일례를 도시한 것이고, 도 4b는 모델을 제조하기 위한 단계적 가산 방법의 구체적인 일례를 도시한 것이다.
도 5는 한 실시양태에 따른 서열 변이체의 유도 진화에서 베이지안 회귀를 실행하는 프로세스에 대한 순서도를 도시한 것이다.
도 6은 한 실시양태에 따른 서열 변이체의 유도 진화에서 앙상블 회귀를 실행하는 프로세스에 대한 순서도를 도시한 것이다.
도 7은 한 실시양태에 따라 단백질 변이체 라이브러리를 생성하는 부트스트랩 p 값 방법을 도시한 순서도이다.
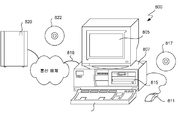
도 8은 예시적인 디지털 장치의 개략도이다.
도 2는 하나 이상의 단백질 변이체 라이브러리 세대를 생성하기 위한 연산 순서를 도시한 순서도이며, 여기서, 연산은 서열-활성 모델, 예컨대, 도 1에서 얻은 것들 중 하나를 사용하여 단백질 변이체 라이브러리 세대를 유도한다. 생성된 변이체 라이브러리는 하나 이상의 신규 서열-활성 모델을 제조하기 위한 서열 및 활성 데이터를 제공할 수 있으며, 유도된 진화의 모델링-탐색 루프를 형성할 수 있다.
도 3a-3h는 특정의 선형 및 비선형 모델의 예측 능력을 비교하는 일례를 보여주는 그래프이다.
도 4a-4b는 서열-활성 모델을 제조하기 위한 단계적 가산 및 감산 방법을 실행하는 프로세스에 대한 순서도를 도시한 것이다. 도 4a는 모델을 제조하기 위한 단계적 가산 방법의 구체적인 일례를 도시한 것이고, 도 4b는 모델을 제조하기 위한 단계적 가산 방법의 구체적인 일례를 도시한 것이다.
도 5는 한 실시양태에 따른 서열 변이체의 유도 진화에서 베이지안 회귀를 실행하는 프로세스에 대한 순서도를 도시한 것이다.
도 6은 한 실시양태에 따른 서열 변이체의 유도 진화에서 앙상블 회귀를 실행하는 프로세스에 대한 순서도를 도시한 것이다.
도 7은 한 실시양태에 따라 단백질 변이체 라이브러리를 생성하는 부트스트랩 p 값 방법을 도시한 순서도이다.
도 8은 예시적인 디지털 장치의 개략도이다.
상세한 설명
I. 정의
본원에서 달리 정의되지 한, 본원에서 사용되는 모든 기술 용어 및 과학 용어는 본 분야의 숙련가가 일반적으로 이해하는 바와 동일한 의미를 가진다. 본원에 포함된 용어를 포함하는 각종 과학 사전은 당업자에게 주지되어 있으며, 이용가능하다. 본원에 기술된 것과 유사하거나, 또는 등가인 임의의 방법 및 물질은 본원에 개시된 실시양태를 실시하는 데 사용될 수 있다는 것을 알 수 있다.
바로 다음에서 정의되는 용어들은 전체적으로 명세서를 참조함으로써 더욱 충분하게 이해된다. 정의는 단지 특정 실시양태를 기술하고, 본 명세서에 기술된 복작합 개념의 이해를 돕기 위한 것이다. 정의는 본 개시내용의 전체 범주를 한정하고자 하는 것이 아니다. 구체적으로, 기술된 특정 서열, 조성물, 알고리즘, 시스템, 방법론, 프로토콜 및 시약은 이러한 것들이 당업자에 의해 사용되는 상황에 따라 달라질 수 있는 바, 본 개시내용은 기술된 특정 서열, 조성물, 알고리즘, 시스템, 방법론, 프로토콜 및 시약으로 제한하는 것이 아님을 이해하여야 한다.
본 명세서 및 첨부된 청구범위에서 사용되는 바, 내용상 및 맥락상 달리 명확하게 명시되지 않는 한, "하나"("a," "an") 및 "그"라는 단수 형태는 복수의 지시 대상을 포함한다. 따라서, 예를 들어, "한 장치"라고 언급하는 것은 2개 이상의 상기 장치의 조합 등을 포함한다.
달리 명시되지 않는 한, "또는"이라는 접속사는 그의 정확한 의미로, 대안으로 특징을 선택하는 것(A 또는 B, 여기서, A를 선택하는 것은 B와 상호 배타적인 경우), 및 공동으로 특징을 선택하는 것(A 또는 B, 여기서, A 및 B, 둘 모두를 선택하는 경우), 둘 모두를 포함하는, 불(Boolean) 논리 연산자로서 사용되는 것으로 한다. 본 명세서 중 일부에서, "및/또는"이라는 용어는 같은 용도로 사용되며, 이는 "또는"이 상호 배타적인 대안에 관하여 사용되는 것을 암시하는 것으로 해석되지 않아야 한다.
"생체분자" 또는 "생물학적 분자"란 일반적으로 생물학적 유기체에서 발견되는 분자를 의미한다. 일부 실시양태에서, 생물학적 분자는 다중 서브유닛을 가지는 중합체성 생물학적 거대분자(즉, "생체중합체")를 포함한다. 전형적인 생체분자로는 자연적으로 발생된 중합체와 일부 구조상의 특징들을 공유하는 분자, 예컨대, RNA, RNA 유사체, DNA, DNA 유사체, 폴리펩티드, 폴리펩티드 유사체, 펩티드 핵산(PNA: peptide nucleic acid), RNA 및 DNA의 조합(예컨대, 키메라플라스트) 등을 비롯한, (뉴클레오티드 서브유닛으로부터 형성된) RNA, (뉴클레오티드 서브유닛으로부터 형성된) DNA, 및 (아미노산 서브유닛으로부터 형성된) 펩티드 또는 폴리펩티드를 포함하나, 이에 한정되지 않는다. 예컨대, 지질, 탄수화물, 또는 하나 이상의 유전적으로 코딩가능한 분자에 의해 제조된 다른 유기 분자(예컨대, 하나 이상의 효소 또는 효소 경로) 등을 포함하나, 이에 한정되지 않는, 임의의 적합한 생물학적 분자가 본 발명에서 사용될 수 있다는 것을 알 수 있는 바, 생체분자를 임의의 특정 분자로 한정하고자 하지 않는다.
"폴리뉴클레오티드" 및 "핵산"이라는 용어는 단일 또는 이중 가닥 형태의 데옥시리보뉴클레오티드 또는 리보뉴클레오티드 및 그의 중합체(예컨대, 올리고뉴클레오티드, 폴리뉴클레오티드 등)를 의미한다. 상기 용어는 단일, 이중 또는 삼중 가닥 DNA, 게놈 DNA, cDNA, RNA, DNA-RNA 하이브리드, 퓨린 및 피리미딘 염기, 및/또는 다른 천연, 화학적으로 또는 생화학적으로 변형된, 비천연 또는 유도체화된 뉴클레오티드 염기를 포함하는 중합체를 포함하나, 이에 한정되지 않는다. 하기는 폴리뉴클레오티드의 비제한적인 일례이다: 유전자, 유전자 단편, 염색체 단편, EST, 엑손, 인트론, mRNA, tRNA, rRNA, 리보자임, cDNA, 재조합 폴리뉴클레오티드, 분지형 폴리뉴클레오티드, 플라스미드, 벡터, 임의 서열의 단리된 DNA, 임의 서열의 단리된 RNA, 핵산 프로브, 및 프라이머. 일부 실시양태에서, 폴리뉴클레오티드는 변형된 뉴클레오티드, 예컨대, 메틸화된 뉴클레오티드 및 뉴클레오티드 유사체, 우라실, 다른 당 및 연결 기, 예컨대, 플루오로리보스 및 티오에이트, 및/또는 뉴클레오티드 분지를 포함한다. 일부 대체 실시양태에서, 뉴클레오티드의 서열은 비뉴클레오티드 성분에 의해 중단된다.
특별히 제한되지 않는 한, 본 용어는 참조 핵산과 결합 특성이 유사하고, 자연적으로 발생된 뉴클레오티드와 유사한 방식으로 대사되는 천연 뉴클레오티드의 공지된 유사체를 포함하는 핵산을 포함한다. 달리 명시되지 않는 한, 특정 핵산 서열은 또한 명확하게 명시된 서열 뿐만 아니라, 암시적으로 그의 보존적으로 변형된 변이체(예컨대, 축퇴성 코돈 치환) 및 보존적 서열을 포함한다. 구체적으로, 축퇴성 코돈 치환은 하나 이상의 선택된(또는 모든) 코돈의 3번째 위치가 혼합 염기 및/또는 데옥시이노신 잔기로 치환된 것인 서열을 생성함으로써 달성될 수 있다(문헌 [Batzer et al. (1991) Nucleic Acid Res. 19:5081]; [Ohtsuka et al. (1985) J. Biol. Chem . 260:2605-2608]; [Rossolini et al. (1994) Mol . Cell. Probes 8:91-98]). 핵산이라는 용어는 예컨대, 올리고뉴클레오티드, 폴리뉴클레오티드, cDNA, 및 mRNA와 상호교환적으로 사용된다.
"단백질," "폴리펩티드" 및 "펩티드"라는 용어는 상호교환적으로 사용되며, 이는 길이 또는 번역 후 변형(예컨대, 당화, 인산화, 지질화, 미리스틸화, 유비퀴틴화 등)에 상관없이, 아미드 결합에 의해 공유적으로 연결된 2개 이상의 아미노산으로 이루어진 중합체를 의미한다. 일부 경우에서, 중합체는 약 30개 이상의 아미노산 잔기, 및 일반적으로, 약 50개 이상의 아미노산 잔기를 가진다. 더욱 전형적으로, 중합체는 약 100개 이상의 아미노산 잔기를 함유한다. 본 용어는 통상 전장의 단백질 또는 펩티드의 단편으로 간주되는 조성물을 포함한다. D- 및 L-아미노산, 및 D- 및 L-아미노산의 혼합물도 본 정의에 포함된다. 본원에 기술된 폴리펩티드는 유전적으로 코딩된 아미노산으로 한정되지 않는다. 실제로, 유전적으로 코딩된 아미노산 이외에도, 본원에 기술된 폴리펩티드는 전체적으로 또는 부분적으로 자연적으로 발생된 및/또는 합성 비코딩된 아미노산으로 구성될 수 있다. 일부 실시양태에서, 폴리펩티드는 기능적 활성(예컨대, 촉매 활성)은 여전히 유지하면서, 전장의 모체 폴리펩티드의 아미노산 서열과 비교하였을 때, 아미노산 부가 또는 결실(예컨대, 갭) 또는 치환을 포함하는, 전장의 선조 또는 모체 폴리펩티드의 일부이다.
본원에서 사용되는 바, "셀룰라제"라는 용어는 셀룰로스(β-1,4-글루칸 또는 β-D-글루코시드 결합)를 더 짧은 셀룰로스 쇄, 올리고당, 셀로비오스 및/또는 글루코스로 가수분해시킬 수 있는 효소의 카테고리를 의미한다. 일부 실시양태에서, "셀룰라제"라는 용어는 베타-글루코시다제, 엔도글루카나제, 셀로비오하이드롤라제, 셀로비오스 데하이드로게나제, 엔도크실라나제, 베타-크실로시다제, 아라비노푸라노시다제, 알파 글루쿠로니다제, 아세틸크실란 에스터라제, 페룰로일 에스터라제, 및/또는 알파 글루쿠로닐 에스터라제를 포함한다. 일부 실시양태에서, "셀룰라제"라는 용어는 엔도크실라나제, 베타-크실로시다제, 아라비노푸라노시다제, 알파 글루쿠로니다제, 아세틸크실란 에스터라제, 페룰로일 에스터라제, 및 알파 글루쿠로닐 에스터라제를 포함하나, 이에 한정되지 않는, 헤미셀룰로스 가수분해 효소를 포함한다. "셀룰라제 생산 진균 세포"는 하나 이상의 셀룰로스 가수분해 효소를 발현하고, 분비하는 진균 세포이다. 일부 실시양태에서, 셀룰라제 생산 진균 세포는 셀룰로스 가수분해 효소의 혼합물을 발현하고, 분비한다. "셀룰로스 분해성," "셀룰로스 가수분해," "셀룰로스 분해," 및 유사 용어는 효소, 예컨대, 엔도글루카나제 및 셀로비오하이드롤라제(후자의 것은 또한 "엑소글루카나제"로도 지칭된다)가 시너지 작용에 의해 셀룰로스를 가용성 이당류 또는 올리고당, 예컨대, 셀로비오스로 분해한 후, 이어서, 이를 베타-글루코시다제에 의해 글루코스로 가수분해시킨다는 것을 의미하는 것이다. 일부 실시양태에서, 셀룰라제는 β-글루코시다제(BGL), 1형 셀로비오하이드롤라제(CBH1), 2형 셀로비오하이드롤라제(CBH2), 글리코시드 하이드롤라제 61(GH61), 및/또는 엔도글루카나제(EG)로부터 선택되는 재조합 셀룰라제이다. 일부 실시양태에서, 셀룰라제는 β-글루코시다제(BGL), 1형 셀로비오하이드롤라제(CBH1), 2형 셀로비오하이드롤라제(CBH2), 글리코시드 하이드롤라제 61(GH61), 및/또는 엔도글루카나제(EG)로부터 선택되는 재조합 마이셀리오프토라(Myceliophthora) 셀룰라제이다. 일부 추가의 실시양태에서, 셀룰라제는 EG1b, EG2, EG3, EG4, EG5, EG6, CBH1a, CBH1b, CBH2a, CBH2b, GH61a, 및/또는 BGL로부터 선택되는 재조합 셀룰라제이다.
본원에서 사용되는 바, "서열"이라는 용어는 전체 게놈, 전체 염색체, 염색체 세그먼트, 상호작용 유전자에 대한 유전자 서열의 집합, 유전자, 핵산 서열, 단백질, 다당류 등을 포함하나, 이에 한정되지 않는, 임의의 생물학적 서열의 순서 및 아이덴티티를 의미한다. 일부 맥락에서, 서열은 단백질(즉, 단백질 서열 또는 단백질 문자열) 중 아미노산 잔기의 순서 및 아이덴티티를, 또는 핵산(즉, 핵산 서열 또는 핵산 문자열) 중 뉴클레오티드의 순서 및 아이덴티티를 의미한다. 서열은 문자열로 표시될 수 있다. "핵산 서열"은 핵산을 포함하는 뉴클레오티드의 순서 및 아이덴티티를 의미한다. "단백질 서열"은 단백질 또는 펩티드를 포함하는 아미노산의 순서 및 아이덴티티를 의미한다.
"코돈"이란 유전자 코드의 일부이고, 단백질 중 특정 아미노산을 명시하거나, 또는 단백질 합성을 개시 또는 종결하는 3개의 연속된 뉴클레오티드로 이루어진 특이적 서열을 의미한다.
"천연 서열" 또는 "야생형 서열"이란 자연적으로 발생된 공급원으로부터 단리된 폴리뉴클레오티드 또는 폴리펩티드를 의미한다. 천연 폴리펩티드 의 재조합 형태 또는 천연 형태와 동일한 서열을 가지는 폴리뉴클레오티드가 "천연 서열"에 포함된다.
"유전자"라는 용어는 광범위하게 사용되며, 이는 생물학적 기능과 관련된 의 임의의 세그먼트를 의미한다. 따라서, 유전자는 코딩 서열, 및 그의 발현에 필요한 조절 서열을 포함한다. 유전자는 또한 임의적으로 예를 들어, 다른 단백질에 대한 인식 서열을 형성하는 비발현 핵산 세그먼트를 포함한다. 유전자는 관심의 대상이 되는 공급원으로부터의 클로닝, 또는 공지된 또는 예측된 서열 정보로부터의 합성을 비롯한, 다양한 공급원으로부터 수득될 수 있고, 원하는 파라미터를 가지도록 디자인된 서열을 포함할 수 있다.
"모티프"는 생물학적 분자내 또는 그 중의 서브유닛의 패턴을 의미한다. 예를 들어, "모티프"라는 용어는 비코딩 생물학적 분자의 서브유닛의 패턴, 또는 생물학적 분자의 코딩된 구현체의 서브유닛의 패턴과 관하여 사용될 수 있다.
"염색체"라는 용어는 다수의 유전자를 포함하는 꼬인 DNA, 조절 요소, 및 다른 뉴클레오티드 서열을 단일 조각을 포함하는, 세포에서 발견되는 DNA 및 회합된 단백질의 조직화된 구조에 관하여 사용된다. 본 용어는 또한 상기 구조의 DNA 서열에 관하여 사용된다.
"스크리닝"이란 하나 이상의 생체분자의 하나 이상의 특성을 측정하는 프로세스를 의미한다. 예를 들어, 전형적인 스크리닝 프로세스는 하나 이상의 라이브러리의 하나 이상의 구성원의 하나 이상의 특성을 측정하는 것을 포함한다. "발현 시스템"은 유전자 또는 다른 핵산에 의해 코딩되는 단백질 또는 펩티드를 발현하기 위한 시스템이다.
"숙주 세포" 또는 "재조합 숙주 세포"는 하나 이상의 재조합 핵산 분자를 포함하는 세포를 의미한다. 따라서, 예를 들어, 일부 실시양태에서, 재조합 숙주 세포는 천연 형태(즉, 비재조합)의 세포 내에서는 발견되지 않는 유전자를 발현한다.
"유도 진화," "유도된 진화," 또는 "인공 진화"란 인공 선택, 재조합 또는 다른 조작에 의해 나 이상의 생체분자 서열(또는 상기 서열을 나타내는 문자열)을 인공적으로 변이시키는 시험관내 또는 생체내 프로세스를 의미한다. 일부 실시양태에서, 유도 진화는, (1) 개체 변종이 존재하고, (2) 일부 변종은 유전성이고, (3) 그 중 일부 변종의 적합도는 상이한 것인 재생 집단에서 발생한다. 재생 성공은 미리 결정된 특성, 예컨대, 유익한 특성에 대한 선택 결과에 의해 결정된다. 재생 집단은 예컨대, 물리적 집단 또는 컴퓨터 시스템에서의 가상 집단일 수 있다.
특정 실시양태에서, 유도 진화 방법은 모체 단백질 변이체 라이브러리의 변이체를 코딩하는 유전자를 재조합함으로써 단백질 변이체 라이브러리를 생성한다. 본 방법은 모체 단백질 변이체 라이브러리의 단백질을 코딩하는 서열 또는 부분서열을 포함하는 올리고뉴클레오티드를 사용할 수 있다. 모체 변이체 라이브러리의 올리고뉴클레오티드 중 일부는 밀접한 관계를 가지며, 오직 다른 변이체와의 재조합에 의해 가변되도록 선택되는 대체 아미노산에 대한 코돈 선택에 있어서만 상이할 수 있다. 본 방법은 원하는 결과를 달성할 때까지 1회 또는 다회 사이클 동안 수행될 수 있다. 다회 사이클이 사용되는 경우, 각각은 허용가능한 성능을 가지는 어떤 변이체가 후속 재조합 사이클에서 사용될지를 확인하는 스크리닝 단계를 포함한다.
"셔플링" 및 "유전자 셔플링"은 일련의 쇄 연장 사이클을 통해 모체 폴리뉴클레오티드의 단편의 집합을 재조합함으로써 다양서을 도입하기 위한 유도 진화 방법을 의미한다. 특정 실시양태에서, 쇄 연장 사이클 중 1회 이상은 자가 프라이밍이다; 즉, 단편 그 자체 이외의 다른 프라이머는 첨가되지 않고 수행된다. 각각의 사이클은 하이브리드화를 통한 단일 가닥 단편 어닐링, 이어서, 쇄 연장을 통한 어닐링된 단편의 신장, 및 변성을 포함한다. 셔플링 과정 동안에 걸쳐 성장 핵산 가닥은 전형적으로는, 종종 "주형 교환"으로도 지칭되는 프로세스에서 다중의 상이한 어닐링 파트너에 노출된다. 본원에서 사용되는 바, "주형 교환"이란 제1 핵산으로부터의 제1 핵산 도메인을 제2 핵산으로부터의 제2 핵산 도메인과 교환할 수 있는 능력(즉, 제1 및 제2 핵산은 셔플링 방법에서 주형으로서의 역할을 한다)을 의미한다.
주형 교환으로는 빈번하게, 상이한 기원을 가지는 단편 사이의 교차 도입의 결과인 키메라 서열이 생산된다. 교차는 어닐링, 연장, 및 변성으로 이루어진 다회 사이클 동안의 주형 교환형 재조합을 통해 형성된다. 따라서, 셔플링을 통해서는 전형적으로 변이체 폴리뉴클레오티드 서열이 제조된다. 일부 실시양태에서, 변이체 서열은 변이체의 "라이브러리"를 포함한다. 상기 라이브러리의 일부 실시양태에서, 변이체는 모체 폴리뉴클레오티드 중 2개 이상의 것으로부터의 서열 세그먼트를 포함한다.
2개 이상의 모체 폴리뉴클레오티드가 사용될 때, 개별 모체 폴리뉴클레오티드는 상이한 모체로부터의 단편이 셔플링 사이클에서 사용되는 어닐링 조건하에서 하이브리드화하는 데 충분한 정도로 상동성을 띤다. 일부 실시양태에서, 셔플링을 통해 상동성이 상대적으로 제한된 모체 폴리뉴클레오티드의 재조합이 이루어질 수 있다. 대개, 개별 모체 폴리뉴클레오티드는 관심의 대상이 되는, 독특한 및/또는 고유의 도메인 및/또는 다른 서열 특징을 가진다. 독특한 서열 특징을 가지는 모체 폴리뉴클레오티드를 사용할 때, 셔플링을 통해서 고도로 다양한 변이체 폴리뉴클레오티드가 제조될 수 있다.
각종 셔플링 기법이 당업계에 공지되어 있다. 예컨대, 미국 특허 번호 제6,917,882호, 제7,776,598호, 제8,029,988호, 제7,024,312호, 및 제7,795,030호(이들 특허는 모두 그 전문이 본원에서 참조로 포함된다)를 참조할 수 있다.
"단편"은 뉴클레오티드 또는 아미노산 서열의 임의의 일부분이다. 단편은 폴리펩티드 또는 폴리뉴클레오티드 서열을 절단하는 것을 포함하나, 이에 한정되지 않는, 당업계에 공지된 임의의 적합한 방법을 사용하여 제조될 수 있다. 일부 실시양태에서, 단편은 폴리뉴클레오티드를 절단하는 뉴클레아제를 사용함으로써 제조된다. 일부 추가의 실시양태에서, 단편은 화학적 및/또는 생물학적 합성 기법을 사용하여 생성된다. 일부 실시양태에서, 단편은 상보적인 핵산(들)의 부분적인 쇄 신장을 사용하여 생성된, 하나 이상의 모체 서열의 부분서열을 포함한다.
"모체 폴리펩티드," "모체 폴리뉴클레오티드," "모체 핵산," 및 "모체"란 일반적으로 야생형 폴리펩티드, 야생형 폴리뉴클레오티드, 또는 다양성 생성 방법, 예컨대, 유도 진화에서 출발점으로서 사용되는 변이체를 의미하는 것으로 사용된다. 일부 실시양태에서, 모체 그 자체가 셔플링 또는 다른 다양성 생성 방법을 통해 제조된다. 일부 실시양태에서, 유도 진화에서 사용되는 돌연변이체는 모체 폴리펩티드와 직접적인 관련이 있다. 일부 실시양태에서, 모체 폴리펩티드는 극한의 온도, pH 및/또는 용매 조건에 노출되었을 때에도 안정적이고, 셔플링을 위한 변이체를 생성하기 위한 기반으로서의 역할을 할 수 있다. 일부 실시양태에서, 모체 폴리펩티드는 극한의 온도, pH 및/또는 용매 조건에 안정적이지 않으며, 모체 폴리펩티드는 진화되어 강력한 변이체가 제조된다.
"모체 핵산"이 모체 폴리펩티드를 코딩한다.
본원에서 사용되는 바, "돌연변이체," "변이체," 및 "변이체 서열"이란 일부 측면에서 표준 또는 참조 서열과 다른 생물학적 서열을 의미한다. 상기 차이는 "돌연변이"로 지칭될 수 있다. 일부 실시양태에서, 돌연변이체는 하나 이상의 치환, 삽입, 교차, 결실, 및/또는 다른 유전적 연산에 의해 변경된 아미노산(즉, 폴리펩티드) 또는 폴리뉴클레오티드 서열이다. 본 개시내용의 목적을 위해, 돌연변이체 및 변이체는 그가 생성되는 특정 방법으로 제한되지 않는다. 일부 실시양태에서, 돌연변이체 또는 변이체 서열은 모체 서열과 비교하여 증가된, 감소된, 또는 실질적으로 유사한 활성 또는 특성을 가진다. 일부 실시양태에서, 변이체 폴리펩티드는 야생형 폴리펩티드 (예컨대, 모체 폴리펩티드)의 아미노산 서열과 비교하여 돌연변이화된 하나 이상의 아미노산 잔기를 포함한다. 일부 실시양태에서, 복수개를 구성하는 변이체 폴리펩티드 중 폴리펩티드의 하나 이상의 아미노산 잔기는 모체 폴리펩티드와 비교하여 일정하게 유지되거나, 비변이체이거나, 또는 돌연변이화되지 않는다. 일부 실시양태에서, 모체 폴리펩티드는 안정성, 활성 또는 다른 특성이 개선된 변이체를 생성하기 위한 기반으로서 사용된다.
"돌연변이 유발법"은 돌연변이를 표준 또는 참조 서열, 예컨대, 모체 핵산 또는 모체 폴리펩티드로 도입하는 프로세스이다.
"라이브러리" 또는 "집단"은 2개 이상의 상이한 분자, 문자열, 및/또는 모델, 예컨대, 핵산 서열(예컨대, 유전자, 올리고뉴클레오티드 등) 또는 그로부터의 발현 생성물(예컨대, 효소 또는 다른 단백질)로 이루어진 집한을 의미한다. 라이브러리 또는 집단은 일반적으로 다수의 상이한 분자를 포함한다. 예를 들어, 라이브러리 또는 집단은 전형적으로 약 10개 이상의 상이한 분자를 포함한다. 거대 라이브러리는 전형적으로 약 100개 이상의 상이한 분자, 더욱 전형적으로, 약 1,000개 이상의 상이한 분자를 포함한다. 일부 적용을 위해, 라이브러리는 적어도 약 10,000개 이상의 상이한 분자를 포함한다. 특정 실시양태에서, 라이브러리는 유도 진화 방법에 의해 제조된 다수의 변이체 또는 키메라 핵산 또는 단백질을 포함한다.
각각의 두 핵산으로부터의 서열이 자손 핵산에 조합되어 있을 때, 두 핵산은 "재조합된" 것이다. 핵산 둘 모두가 재조합에 대한 기질일 경우, 두 서열은 "직접적으로" 재조합된 것이다.
"선택"이란 하나 이상의 생체분자가 관심의 대상이 되는 하나 이상의 특성을 가지는 것으로 확인되는 프로세스를 의미한다. 따라서, 예를 들어, 하나 이상의 라이브러리 구성원의 하나 이상의 특성을 측정하기 위한 라이브러리를 스크리닝할 수 있다. 라이브러리 구성원 중 하나 이상의 것이 관심의 대상이 되는 특성을 가지는 것으로 확인되었다면, 이는 선택된다. 선택은 라이브러리 구성원의 단리를 포함할 수 있지만, 이는 반드시 필요한 것은 아니다. 추가로, 선택 및 스크리닝은 동시 진행될 수 있고, 대개는 동시 진행된다.
"종속 변수"는 출력값 또는 효과를 나타내거나, 또는 그가 효과인지 여부를 알아보기 위해 검정된다. "독립 변수"는 입력값 또는 원인을 나타내거나, 또는 그가 원인인지 여부를 알아보기 위해 테스트된다. 종속 변수는 그가 독립 변수가 달라짐에 따라 달라지는지 여부, 및 얼마만큼 달라지는지를 알아보기 위해 연구될 수 있다.
단순 확률론적 선형 모델에서,
y
i
= a +
bx
i
+
e
i
여기서, y i 항은 i번째 종속 변수 값이고, x i 는 i번째 독립 변수 값이다. e i 항은 "오차"로도 알려져 있으며, 이는 독립 변수에 의해 설명되지 않는 종속 변수의 가변성을 포함한다.
독립 변수는 "예측 변수," "회귀변수," "통제 변수," "조작 변수," "설명 변수," 또는 "입력 변수"로도 알려져 있다.
"직교(orthogonal)/직교성(orthogonality)"이란 모델에서의 다른 독립 변수 또는 다른 관계로 수정되지 않는 독립 변수를 의미한다.
"서열-활성 모델"이란 한편으로는 생물학적 분자의 활성, 특징, 또는 특성과, 다른 한편으로는 각종 생물학적 서열 사이의 관계를 기술하는 임의의 수학적 모델을 의미한다.
"코딩된 문자열"이라는 용어는 생물학적 분자에 관한 서열/구조상의 정보를 보존하는 상기 분자의 구현체를 의미한다. 일부 실시양태에서, 코딩된 문자열은 변이체의 라이브러리 중의 서열 돌연변이에 대한 정보를 포함한다. 생체분자에 대한 활성 정보와 함께, 생체분자의 코딩된 문자열은 서열-활성 모델에 대한 트레이닝 세트로서 사용될 수 있다. 생체분자의 비서열 특성은 저장될 수 있거나, 또는 다르게는 생체분자에 대한 코딩된 문자열과 관련시켜 생각할 수 있다.
"참조 서열"은 그로부터 서열 변이가 이루어지는 서열이다. 일부 경우에서, "참조 서열"은 변이를 정의하는 데 사용된다. 상기 서열은 최고값(또는 최고값들 중 하나)의 원하는 활성을 가지는 모델에 의해 예측되는 것일 수 있다. 또 다른 경우에서, 참조 서열은 원래의 단백질 변이체 라이브러리의 구성원의 것일 수 있다. 특정 실시양태에서, 참조 서열은 모체 단백질 또는 핵산의 서열이다.
"트레이닝 세트"는 하나 이상의 모델의 적합화의 대상이 되고, 상기 모델이 그에 기반하는 것인 서열 활성 데이터 또는 관찰값 세트를 의미한다. 예를 들어, 단백질 서열-활성 모델의 경우, 트레이닝 세트는 초기 또는 개선된 단백질 변이체 라이브러리에 대한 잔기 서열을 포함한다. 전형적으로, 상기 데이터는 라이브러리 중 각각의 단백질에 대한 활성 값과 함께 완전한 또는 부분적인 잔기 서열 정보를 포함한다. 일부 경우에서, 다중의 활성 유형(예컨대, 속도 상수 데이터 및 열적 안정성 데이터)이 트레이닝 세트에서 함께 제공된다. 활성은 종종 유익한 특성이다.
"관찰값(observation)"이라는 용어는 모델, 예컨대, 서열-활성 모델을 생성하기 위해 트레이닝 세트에서 사용될 수 있는 단백질 또는 다른 생물학적 엔티티에 대한 정보이다. "관찰값"이라는 용어는 단백질 변이체를 비롯한, 서열 분석되고, 어세이(assay)된 임의의 생물학적 분자를 의미할 수 있다. 특정 실시양태에서, 각각의 관찰값은 라이브러리 중의 변이체에 대한 활성 값 및 관련 서열이다. 일반적으로, 서열-활성 모델을 생성하는 데 사용되는 관찰값이 많으면 많을수록, 상기 서열-활성 모델의 예측력은 더욱더 우수해진다.
본원에서 사용되는 바, "유익한 특성"이라는 용어는 단백질 또는 상기 단백질과 관련된 물질의 조성물 또는 프로세스에 일부 이점을 부여하는 표현형 또는 다른 확인가능한 특징을 의미하는 것으로 한다. 유익한 특성의 예로는 모체 단백질과 비교하였을 때, 변이체 단백질의 촉매 특성, 결합 특성, 극한의 온도, pH 등에 노출되었을 때의 안정성, 자극에 대한 감도, 억제 등의 증가 또는 감소를 포함한다. 다른 유익한 특성으로는 특정 자극에 대한 반응으로 변경된 프로파일을 포함할 수 있다. 유익한 특성에 대한 추가의 예는 하기에 기술된다. 유익한 특성의 값이 서열-활성 모델을 위한 트레이닝 세트에서 사용되는 관찰값에서의 활성 값으로서 사용될 수 있다.
"다음 세대 서열 분석" 또는 "고처리량 서열 분석"은 서열 분석 프로세스를 병렬화하여 동시 한꺼번에 수천 또는 수백만개의 서열이 제조되는 서열 분석 기법이다. 적합한 다음 세대 서열 분석 방법의 예로는 단일 분자 실시간 서열 분석(예컨대, 퍼시픽 바이오사이언시스(Pacific Biosciences: 미국 캘리포니아주 멘로 파크)), 이온 반도체 서열 분석(예컨대, 이온 토렌트(Ion Torrent: 미국 캘리포니아주 사우쓰 샌프란스시코)), 파이로시퀀싱(예컨대, 454, 미국 코네티컷주 브랜퍼드), 결찰에 의한 서열 분석(예컨대, 라이프 테크놀러지즈(Life Technologies: 미국 캘리포니아주 칼즈배드)의 SOLid 서열 분석), 합성 및 가역성 종결인자에 의한 서열 분석(예컨대, 일루미나(Illumina: 미국 캘리포니아주 샌디에고)), 핵산 영상화 기술, 예컨대, 투과 전자 현미경법 등을 포함하나, 이에 한정되지 않는다. 예시적인 기법에 관한 추가 설명은 본 개시내용의 상세한 설명에서 기술한다.
"예측력"이란 다양한 조건하에서 데이터에 대한 종속 변수의 값을 정확하게 예측할 수 있는 모델의 능력을 의미한다. 예를 들어, 서열-활성 모델의 예측력이란 서열 정보로부터 활성을 예측할 수 있는 모델의 능력을 의미한다.
"교차 검증"이란 관심의 대상이 되는 값(즉, 종속 변수의 값)을 예측할 수 있는 모델의 능력의 일반화 가능성은 검증하는 방법을 의미한다. 본 방법은 한 데이터 세트를 사용하여 모델을 제조하고, 상이한 데이터 세트를 사용하여 모델 오류를 검정한다. 제1 데이터 세트는 트레이닝 세트로서 간주되고, 제2 데이터 세트는 검증 세트이다.
"체계적 분산"이란 상이한 조합으로 달라지는 항목 또는 항목 세트의 상이한 디스크립터를 의미한다.
"체계적으로 가변된 데이터"란 항목 또는 항목 세트의 상이한 디스크립터로부터 생성, 도출, 또는 얻은 데이터가 상이한 조합으로 달라지는 것을 의미한다. 다수의 상이한 디스크립터는 동시에, 그러나, 상이한 조합으로 달라질 수 있다. 예를 들어, 아미노산의 조합이 바뀐 폴리펩티드로부터 수집된 활성 데이터는 체계적으로 가변된 데이터이다.
"체계적으로 가변된 서열"이라는 용어는 각각의 잔기가 다중 컨텍스트로 관찰되는 서열 세트를 의미한다. 원칙적으로, 체계적인 변이 수준은 서열이 서로로부터 직교성을 띠는 정도(즉, 평균과 비교하여 최대로 상이한 정도)에 의해 정량화될 수 있다.
"토글링"이라는 용어는 다중 아미노산 잔기 유형을 최적화된 라이브러리 중 단백질 변이체의 서열내 특정 위치 내로 도입하는 것을 의미한다.
"회귀" 및 "회귀 분석"이라는 용어는 독립 변수들 중에서 어느 것이 종속 변수와 관련이 있는지를 이해하고, 이들의 관련 형태를 탐색하는 데 사용되는 기법을 의미한다. 제한된 환경하에서, 회귀 분석은 독립 변수와 종속 변수 사이의 인과 관계를 추론하는 데 사용될 수 있다. 회귀 분석은 변수들 사이의 관계를 추정하기 위한 통계학적 기법이다. 회귀 분석은 종속 변수와 하나 이상의 독립 변수 사이의 관계에 초점이 맞춰질 때, 수개의 변수를 모델링하고 분석하기 위한 다수의 기법들을 포함한다. 더욱 구체적으로, 회귀 분석은, 다른 독립 변수들은 고정된 상태로 유지되면서, 독립 변수 중 어느 하나가 가변될 때, 종속 변수의 전형적인 값은 어떻게 달라지는지에 관한 이해를 돕는다. 회귀 기법은 서열 및 활성 정보를 포함할 수 있는 다중의 관찰값을 포함하는 트레이닝 세트로부터 서열-활성 모델을 생성하는 데 사용될 수 있다.
부분 최소 제곱 또는 PLS(Partial Least Squares)는 예측 변수(예컨대, 활성) 및 관찰 가능한 변수(예컨대, 서열)를 새 공간으로 투영하여 선형 회귀 모델을 찾는 계열의 방법이다. PLS는 또한 잠재 구조에의 투영으로도 알려져 있다. X(독립 변수) 및 Y(종속 변수) 데이터, 둘 모두 새 공간으로 투영된다. PLS는 두 행렬(X 및 Y) 사이의 기본 관계를 찾는 데 사용된다. 잠재 변수 접근법은 X 및 Y 공간에서 공분산 구조를 모델링하는 데 사용된다. PLS 모델은 Y 공간에서 최대 다차원 분산 방향을 설명하는 X 공간에서의 다차원 방향을 찾고자 할 것이다. PLS 회귀는 특히 예측 인자 행렬이 관찰값보다 더 많은 변수를 가질 때, X 값 사이에 다중공선성이 존재할 때 적합하다.
"디스크립터(descriptor)"란 항목을 기술하거나, 또는 확인해 주는 역할을 하는 것을 의미한다. 예를 들어, 문자열에서 문자는 문자열로 표시된 폴리펩티드 중의 아미노산의 디스크립터일 수 있다.
회귀 모델에서, 종속 변수는 항의 합에 의해 독립 변수에 연관된다. 각각의 항은 독립 변수와 관련 회귀 계수의 곱을 포함한다. 순(purely) 선형 회귀 모델의 경우, 회귀 계수는 하기 식에서 β로 제시된다:
y i = β 1 x i 1 + ... + β p x ip + ε i = x i T β + ε i
여기서, y i 는 종속 변수이고, x i 는 독립 변수이고, ε i 는 오차 변수이고, T는 벡터 x i 와 β의 내적인 전치 행렬을 의미한다.
"주성분 회귀"(PCR: Principal Component Regression)란 회귀 계수를 추정할 때, 주성분 분석을 사용하는 회귀 분석을 의미한다. 독립 변수에 대해 직접 종속 변수를 회귀하는 대신 PCR에서는 독립 변수의 주성분을 사용한다. PCR은 전형적으로 오직 회귀에서 주성분의 서브세트만을 사용한다.
"주성분 분석"(PCA: Principal Component Analysis)이란 가능하게 상관 변수의 관찰값 세트를 주성분으로 불리는 선형의 보정되지 않은 변수 값의 세트로 전환시키는 직교성 변환을 사용하는 수학적 절차를 의미한다. 주성분의 개수는 원래 변수의 개수보다 적거나, 또는 그와 동일하다. 상기 변환은 제1 주성분이 최대의 가능한 분산을 가지고(즉, 가능한 한 많은 데이터 가변성을 나타내고), 결국 각각의 후속 성분은 이전 성분에 직교성이 되도록(즉, 그 성분으로 보정되지 않도록) 하는 제약하에서 최고의 가능한 분산을 가지는 방식으로 정의된다.
"신경망"은 전산에의 연결주의 접근법을 사용하여 정보를 프로세싱하는 처리 요소 또는 "뉴런"의 상호 연결된 군을 포함하는 모델이다. 신경망은 입력값과 출력값 사이의 복합적인 관계를 모델링하거나, 데이터 패턴을 찾는 데 사용된다. 대부분의 신경망은 비선형, 분산, 병렬 방식으로 데이터를 처리한다. 대부분의 경우, 신경망은 학습 단계 동안 그의 구조를 바꾸는 적응계이다. 다양한 단위가 배정되는 서브태스크에 대한 명확한 묘사가 존재한다기보다 기능은 처리 요소에 의해 집합적으로 병렬로 실행된다.
일반적으로, 신경망은 처리 요소와 요소 파라미터 사이의 연결에 의해 결정되는 복잡한 전역적인 거동을 나타내는 간단한 처리 요소의 네트워크를 포함한다. 신경망은 네트워크에서 연결 강도를 변경시켜 원하는 신호 흐름을 생성하도록 디자인된 알고리즘과 함께 사용된다. 강도는 트레이닝 또는 학습 동안에 변경된다.
"랜덤 포레스트(random forest)"란 각각의 트리가 독립적으로 샘플링되고, 포레스트 중의 모든 트리에 대하여 같은 분포를 가지는 랜덤 벡터의 값에 의존하도록 하는 분류 트리 예측 인자의 조합을 의미한다. 랜덤 포레스트는 의사 결정 트리의 각각의 스플릿에 무작위로 선택된 특징을 가지는 프루닝되지 않은(un-pruned) 의사 결정 트리 학습자의 배깅으로 구성된 학습 앙상블이다. 랜덤 포레스트는 다수의 분류 트리로 성장하며, 이들 분류 트리는 각각 가장 인기가 많은 부류에 투표한다. 이어서, 랜덤 포레스트는 포레스트 중의 모든 트리 예측 인자로부터 투표로 가장 많은 인기를 얻은 부류를 취함으로써 변수를 분류한다.
불확실한 양 p의 "선험적 확률 분포," 또는 "선험적"은 관심의 대상이 되는 데이터(예컨대, 단백질 서열의 트레이닝 세트)를 고려하기 이전에 p에 대한 불확실성을 나타내는 확률 분포이다. 비공지된 양은 파라미터, 계수, 변수, 잠재 변수 등(예컨대, 다중 회귀 모델에서의 계수)일 수 있다.
불확실한 양 p의 "사후 확률 분포," 또는 "사후"는 관심의 대상이 되는 데이터를 고려한 이후에 p에 대한 불확실성을 나타내는 확률 분포이다.
"베이지안 선형 회귀"란 통계학적 분석이 베이지안 추측 맥락에서 착수되는 선형 회귀 접근법을 의미한다. 모델의 파라미터의 선험적 확률 분포 함수를 비롯한, 선형 회귀 모델에 대한 사전 신뢰는 베이스의 정리(Bayes theorem)에 따라 데이터의 가능도 함수와 조합되고, 이로써 파라미터에 대한 사후 확률 분포를 얻게 된다.
"과대적합(overfitting)"이란 근본적인 관계 대신 랜덤한 오차 또는 노이즈를 기술할 때 발생하는 상태를 의미한다. 과대적합은 일반적으로 모델이 과도하게 복잡할 때, 예컨대, 관찰값에 비하여 상대적으로 너무 많은 파라미터를 가질 때 발생한다. 데이터에서 작은 변동을 과장할 수 있는 바, 과대적합화된 모델은 일반적으로 부진한 예측 수행 능력을 가지게 될 것이다. 일부 실시양태에서, 하나 이상의 독립 변수(IV: independent variable)와 종속 변수(DV: dependent variable) 사이의 관계를 기술하는 데 수학적 모델이 사용된다. 모델은 DV = (IV)의 대수식으로 기재될 수 있다. "대수식"은 변수, 계수, 상수, 및 연산 기호, 예컨대, 플러스(+) 및 마이너스(-) 기호를 포함할 수 있다. 4x2 + 3xy + 7y + 5는 이변량 대수식이다.
일부 실시양태에서, 대수식 또는 수학적 모델의 "항"은 (+) 또는 (-) 기호로 이격되어 있는 요소이다. 이와 관련하여, 상기 예는 4개의 항, 4x2, 3xy, 7y, 및 5를 가진다. 항은 변수 및 계수(4x, 3xy, 및 7y), 또는 상수(5)로 구성될 수 있다. 대수식에서, 변수는 시스템의 상태 변화를 나타내는 다양한 값을 취할 수 있다. 예를 들어, 주행 차량의 속도를 나타내는 연속 변수 또는 아미노산 유형을 나타내는 다중의 불연속 값을 가지는 이산 변수일 수 있다. 변수는 엔티티의 존재 또는 부재, 예컨대, 특이적 위치의 특이적 유형의 잔기의 존재 또는 부재를 나타내는 비트 값 변수일 수 있다. 상기 대수식에서, 변수는 x 및 y이다.
일부 실시양태에서, 식의 "항"은 다른 기호에 의해, 예컨대, 곱셈 기호에 의해 경계가 표시되는 상기 식의 요소일 수 있다.
"계수"란 종속 변수, 또는 종속 변수를 포함하는 식으로 곱셈 처리된 스칼라 값을 의미한다. 상기 예에서, "계수"는 대수식 중 항의 수치 부분이다. 4x2 + 3xy + 7y + 5에서, 첫번째 항의 계수는 4이다. 두번째 항의 계수는 3이다. 세번째 항의 계수는 7이다. 항이 단 하나의 변수로만 구성될 경우, 그의 계수는 1이다.
"상수"는 대수식에 오직 수치만을 포함하는 항이다. 즉, 변수가 없는 항이다. 식 4x2 + 3xy + 7y + 5에서, 상수항은 "5"이다.
"1차 항"은 차수가 1인 항, 또는 거듭제곱이 1인 단일 변수이다. 상기 예에서, 7y 항은 그의 차수가 1이기 때문에(y1 또는 간단하게 y) 1차 항이다. 대조적으로, 4x2 항은 x의 차수가 2이기 때문에 2차 항이고, 3xy는 x 및 y 각각의 차수가 1이고, 그의 곱으로 차수는 2가 되기 때문에 이변량 2차 항이다.
본 본 명세서 중 일부에서, "1차 항" 및 "비상호작용 항"은 상호교환적으로 사용되며, 이는 단일 독립 변수와 관련 계수의 곱을 포함하며, 여기서, 단일 IV는 단일 잔기의 존재/부재를 나타내는 것인, 회귀 모델의 항을 의미한다.
일부 실시양태에서, "비1차 항," "외적 항," 및 "상호작용 항"은 상기 항들이 2개 이상의 독립 변수와 관련 계수의 곱을 포함하는 회귀 모델의 항을 의미할 때에는 본 개시내용에서 상호교환적으로 사용된다. 더욱 일반적으로, "비1차 항"은 예컨대, 독립 변수의 멱함수 또는 지수 함수와 같이, 차수가 1보다 크거나, 또는 작은 항을 명시하는 데 사용된다. 비1차 항의 일부 예로는 xy, x2, x⅓, xy, 및 ex를 포함한다. 따라서, 본 명세서 중 일부에서, "비1차 항"은 두 독립 변수의 곱을 포함하는 항보다 더 넓은 의미의 항을 의미한다.
일부 실시양태에서, 상호작용 항은, 각각의 IV가 특정 위치의 특정 유형의 잔기의 존재를 나타내는 것인, 2개 이상의 IV의 비선형 함수, 예컨대, 2개 이상의 IV의 곱함수, 멱함수 또는 지수 함수를 포함하는 항으로서 실행될 수 있다. 예를 들어, y = ax 1 + bx 2 + cx 1 x 2 에서, 변수 x 1 및 x 2 는 하나의 특정 위치의 두 특정 잔기의 존재/부재를 나타낼 수 있고, cx 1 x 2 는 두 특정 잔기의 상호작용의 효과를 나타내는 상호작용 항이다. 다른 실시양태에서, 상호작용 항은 2개 이상의 잔기의 상호작용을 나타내는 단일 IV를 포함하는 항으로서 실행될 수 있다. 예를 들어, y = ax 1 + bx 2 + cz에서, 변수 x 1 및 x 2 는 특정 위치의 두 특정 잔기의 존재/부재를 나타낼 수 있고, cz는 두 특정 잔기의 상호작용의 효과를 나타내는 상호작용 항이다. 이에 대한 마지막 일례에서, 상호작용 항 cz는 외적 항이 아니다. 비록 기술상으로는 cz가 1차 항이기는 하지만, 선형, 비상호작용 항인 ax 1 및 bx 2 와 혼돈하지 않도록 하기 위해 본원에서는 그렇게 표지화하지 않는다. 본 개시내용에서 사용되는 바, "선형 모델"이라는 용어는 오직 1차 항만을 포함하는 모델을 의미한다. 그에 반해, "비선형 모델"이라는 용어는 1차 항 및 비1차 항, 둘 모두를 포함하는 모델을 의미한다. 일부 실시양태에서, 비선형 모델은 외적 항으로서 실행되는 상호작용 항을 포함한다.
더욱 일반적으로, 선형 모델 또는 선형 시스템은 중첩 원리 및 차수가 1인 동차성을 충족시킨다. 중첩 원리란, 모든 선형 시스템의 경우, 주어진 위치 및 시간에서 2개 이상의 자극에 의해 유발되는 순 반응은 각각의 자극에 의해 개별적으로 유발되었을 반응의 총합이라는 것을 언급한다. 이는 또한 가성성으로도 알려져 있다. 입력값 A가 반응 X를 일으키고, 입력값 B가 반응 Y를 일으킬 경우, 이때, 입력 (A + B)는 반응 (X + Y)를 일으킨다. 차수가 1인 동차성이라는 것은 그의 출력값 또는 종속 변수(DV: dependent variable)는 그의 입력값 또는 독립 변수에 따라 그에 비례하여 변하는 임의의 모델을 의미한다. 역으로, "비선형 모델"은 중첩 원리 및 차수가 1인 동차성을 충족시키지 않는 모델이다.
"상호작용 서브유닛"은 서열의 모델링된 활성에 대하여 시너지 효과를 가지는 서열의 2개 이상의 서브유닛을 의미하는 것이며, 여기서, 시너지 효과는 모델링된 활성에 미치는 서브유닛의 개별 효과와는 별개의 것이고, 상이한 것이다.
"베이스 모델"이라는 용어는 모델을 개선시키는 프로세스 초반에 제공되는 서열-활성 모델과 관련하여 사용된다.
"업데이트된 모델"이라는 용어는 베이스 모델 및/또는 그의 유도 기점이 되는 또 다른 모델과 비교하였을 때 예측력이 개선된 것인, 베이스 모델로부터 직접 또는 간접적으로 유도되는 서열-활성 모델과 관련하여 사용된다.
모델의 "가능도 함수" 또는 "가능도"는 통계학적 모델의 파라미터의 함수이다. 일부 관찰값이 주어졌을 때, 파라미터 값 세트의 가능도는 상기 파라미터 값이 주어졌을 때, 관찰값의 확률과 같으며, 즉, L(θ\x) = P (x\θ)이다.
"몬테카를로 시뮬레이션(Monte Carlo simulation)"은 다수의 무작위 샘플링에 의존하여 실제 현상을 모의하는 수치 결과를 얻는 시뮬레이션이다. 예를 들어, 간격 (0,1]로부터 다수의 의사 무작위 균일 변수를 끌어내고, 0.50 이하의 값을 헤드로서 할당하고, 0.50 초과의 값을 테일로서 할당하는 것이 반복적인 동전 던지기 행동의 몬테카를로 시뮬레이션이다.
"메트로폴리스(Metropolis) 알고리즘" 또는 "메트로폴리스-해스팅스(Metropolis-Hastings) 알고리즘"은 직접 샘플링하기 어려운 확률 분포로부터 일련의 무작위 샘플을 수득하기 위한 마르코프 연쇄 몬테카를로(MCMC: Markov chain Monte Carlo) 방법이다. 이러한 일련의 샘플링은 분포를 모의하는 데(즉, 히스토그램을 생성하는 데), 또는 적분(예컨대, 예상치)을 계산하는 데 사용될 수 있다. 메트로폴리스-해스팅 및 다른 MCMC 알고리즘은 일반적으로 다차원 분포로부터 샘플링하는 데, 특히, 고차원일 때에 사용된다. 메트로폴리스-해스팅 알고리즘의 목적은 원하는 분포 P(x)에 따라 상태 x를 점근적으로 생성하는 것이고, 이를 이행하기 위해 확률 과정을 사용한다. 상기 알고리즘의 개념은 독특한 분포 P(x)로 점근적으로 수렴되도록 확률 과정을 조건화하는 것이다.
"마르코프 연쇄"는 마르코프 특성을 가지는 일련의 확률 변수 X 1 , X 2 , X 3 ...이다. 다시 말해, 현재 상태가 주어졌을 때, 미래 및 과거 상태는 독립적이다. 형식적으로는,
X i 의 가능한 값은 소위 연쇄의 상태 공간이라 불리는 추가 집합 S를 형성한다. "마르코프 연쇄" 시스템은 유한 또는 가산 개수의 가능상 상태 사이의 한 상태로부터 또 다른 상태로 변환되는 수학적 시스템이다. 이는 일반적으로 무기억으로 특징화되는 무작위 과정이며: 다음 상태는 선행된 일련의 이벤트가 아닌, 오직 현재 상태에만 의존한다.
"아카이케 정보 기준"(AIC: Akaike Information Criterion)은 통계학적 모델의 상대적인 적합도의 척도이고, 이는 흔히 유한 모델 집합 사이에서 모델을 선택하기 위한 기준으로서 사용된다. AIC는 사실상, 주어진 모델이 현실을 기술하는 데 사용될 때 정보 누락의 상대적인 척도를 제공하는 정보 엔트로피 개념에 근거를 두고 있다. 모델 구성에서 편향과 분산 사이의, 또는 대략적으로 말하면, 모델의 정확도와 복잡도 사이의 트레이드오프를 기술한다고 볼 수 있다. AIC는 AIC = - 2log e L + 2k(여기서, L은 함수의 최대 가능도이고, k는 추정하고자 하는 모델의 자유 파라미터 개수이다)로 계산될 수 있다.
"베이지안 정보 기준"은 유한 모델 집합 사이에서 모델을 선택하기 위한 기준이고, AIC와 밀접한 관계를 가진다. BIC는 BIC = - 2log e L + klog e (n)(여기서, n은 데이터 관찰값의 개수이다)로 계산될 수 있다. 관찰값의 개수가 증가함에 따라, BIC가 흔히 AIC보다 더 심하게 추가 개수의 자유 파라미터에 벌점을 부가한다.
"유전 알고리즘"은 진화 과정을 모방한 과정이다. 유전 알고리즘(GA: Genetic Algorithm)은 특징 규명이 완전하게 이루어지지 못하였거나, 너무 복잡하여 완전하게 특징 규명될 수 없지만, 그에 대한 일부 분석적 평가는 이용가능한 문제의 해법을 찾는 데 있어서 매우 다양한 분야에서 사용된다. 즉, GA는 해답의 상대적인 값(또는 적어도 또 다른 것과 비교할 때 잠재적인 한 해답의 상대적인 값)에 대한 일부 정량가능한 척도에 의해 평가될 수 있는 문제의 해법을 찾는 데 사용된다. 본 개시내용과 관련하여, 유전 알고리즘은 전형적으로 문자열이 하나 이상의 생물학적 분자(예컨대, 핵산, 단백질 등)에 상응하는 경우에, 컴퓨터에서 문자열을 선택하거나, 조작하기 위한 과정이다.
"유전 연산"(또는 "GO: genetic operation")이라는 용어는 임의 유형의 문자열로 이루어진 임의 집단에서의(따라서, 상기 문자열에 의해 코딩된 물리적 객체의 임의의 물리적 특정에서의) 모든 변화는 논리 대수 함수의 유한 집합의 무작위 및/또는 미리 결정된 응용의 결과로서 기술될 수 있는 경우의, 생물학적 및/또는 전산학적 유전 연산을 의미한다. GO의 예로는 증식, 교차, 재조합, 돌연변이, 결찰, 단편화 등을 포함하나, 이에 한정되지 않는다.
"앙상블 모델"은 그의 항이 모델 군의 항들 모두를 포함하는 모델로서, 여기서, 상기 항의 앙상블 모델의 계수는 군의 개별 모델의 상응하는 항의 가중치가 부여된 계수에 기초하는 것인 모델이다. 계수에 가중치를 부여하는 것은 개별 모델의 예측력 및/또는 적합도에 기초한다.
II. 개선된 단백질 변이체 라이브러리 생성
단백질 서열을 탐색하는 유도된 진화 접근법에서, 단백질 변이체 생성을 유도하기 위해 서열-활성 모델이 사용된다. 본 개시내용의 한 측면은 단백질 라이브러리에 기초하고, 신규 및 개선된 단백질 라이브러리를 검색하는 데 사용될 수 있는 서열-활성 모델을 제조하는 다양한 방법을 제공한다. 본 섹션에서는 먼저 신규 및 개선된 단백질을 검색하기 위한 프로세스에 관한 개요를 제공한 후, 출발 라이브러리를 선택하고, 서열-활성 모델을 구축하고, 상기 모델을 이용하여 신규 단백질의 탐색을 유도하는 것과 관련된 문제에 관하여 추가로 상세하게 설명한다.
본 개시내용은 아미노산 잔기 서열 및 단백질 활성을 포함하는 예시적인 일례를 제공하지만, 본원에 기술된 접근법은 또한 다른 생물학적 서열 및 활성에 대해서도 실행될 수 있다는 것을 이해하여야 한다. 예를 들어, 다양한 실시양태에서, 서열은 전체 게놈, 전체 염색체, 염색체 세그먼트, 상호작용 유전자에 대한 유전자 서열의 집합, 유전자, 핵산 서열, 단백질, 다당류 등일 수 있다. 하나 이상의 실시양태에서, 서열의 서브유닛은 염색체, 염색체 세그먼트, 일배체형, 유전자, 뉴클레오티드, 코돈, 돌연변이, 아미노산, 단량체, 이량체, 삼량체, 또는 올리고머 탄수화물 등일 수 있다.
전형적으로, 특정 회차(round)의 서열 유도 진화 초반에 서열 분석되고, 어세이된 단백질 변이체의 트레이닝 세트가 얻어진다. 주어진 회차의 유도 진화를 통해, 모체 펩티드 또는 상기 회차의 유도 진화 초반에 사용된 펩티드와 하나 이상의 돌연변이가 다른 다수의 변이체 단백질이 생성된다. 한 회차의 유도 진화 동안에 생성되는 변이체 펩티드는 활성에 대해 어세이된다. 추가의 1회차 이상의 유도 진화에서의 사용을 위해 원하는 활성 및/또는 모체 펩티드(들)와 비교하여 개선된 활성을 가지는 상기 펩티드가 선택된다.
서열 분석되고, 어세이된 단백질 변이체 또한 서열-활성 모델을 생성하는 데 사용될 수 있다. 전형적으로, 상기 단백질 변이체가 실제로 서열 분석되었다면, 이는 서열-활성 모델에서 사용된다. 각각의 서열 분석되고, 어세이된 단백질 변이체는 "관찰값"으로 지칭된다. 일반적으로, 서열-활성 모델을 생성하는 데 사용되는 관찰값이 많으면 많을수록, 상기 서열-활성 모델의 예측력은 더욱더 우수해진다.
다음 세대 대량 병렬 서열 분석 기술이 도래할 때까지는 임의 회차의 유도 진화에서 생성되는 10 내지 30개 초과의 변이체 펩티드를 경제적으로 서열 분석하기는 어려웠다. 이제 다음 세대 서열 분석의 적용으로 한 회차의 유도 진화에서 생성된 다수의 더 많은 변이체 단백질이 서열 분석될 수 있다. 그 결과, 서열-활성 모델을 생성하는 데 훨씬 더 큰 트레이닝 세트 데이터 풀이 사용될 수 있다. 이제 서열-활성 모델은, 한 회차로부터 상위의 실행 펩티드 뿐만 아니라, 추가 회차의 유도 진화를 위해서는 관심의 대상이 되지는 못하지만, 그의 서열 활성 정보는 더욱 강건한 서열-활성 모델을 생성하는 데 적용될 수 있는 것인 일부 펩티드를 포함하는 트레이닝 세트를 사용하여 생성될 수 있다.
일부 실시양태에서, 일반적으로 임의 서열의 활성을 예측할 수 있는 능력이 우수한 서열-활성 모델을 생성하는 것이 바람직할 수 있다. 예측력은 예측 정확도 뿐만 아니라, 모델이 활성을 정확하게 예측하는 일관성을 특징으로 할 수 있다. 추가로, 모델은 광범위한 서열 공간에 걸쳐 활성을 정확하게 예측할 수 있는 그의 능력을 특징으로 할 수 있다. 예를 들어, 예측력은 펩티드의 주어진 검정 및/또는 검증에 대하여 계산된 활성과 실제 활성 사이의 잔차를 특징으로 할 수 있다. 일반화된 예측력이 높은 모델일수록 상이한 검증 데이터 세트 간의 오차는 더 작고, 더욱 일관된 경향이 있다. 검정 데이터 세트로 과대적합화된 모델은 하기 실시예에 제시된 바와 같이, 검증 데이터에 대하여 일관성이 더 큰 및 더 적은 오차를 수득하는 경향이 있다. 본 개시내용의 한 측면은 상이한 데이터 세트 간에 예측력이 높은 모델을 효율적으로 찾는 방법을 제공한다.
A. 개선된 단백질
변이체를
검색하기 위한 프로세스에 관한 개요
본원에 기술된 서열-활성 모델을 이용하여, 초기 변이체 라이브러리 중 유도 진화가 이루어지게 되는 하나 이상의 모체 "유전자"를 확인하는 것을 지원할 수 있다. 한 회차의 진화가 수행된 후, 신규의 관찰값 세트를 제공하는 신규 변이체 라이브러리를 확인한 후, 신규 또는 리파이닝된 서열-활성 모델을 제조하기 위한 데이터로서 피드백될 수 있다. 신규 관찰값에 기초하여 서열-활성 모델을 제조하는 것과 서열-활성 모델에 기초하여 유도 진화를 수행하는 것을 교대로 수행하는 본 프로세스는 반복적인 모델링-탐색 루프를 형성할 수 있으며, 이는 원하는 단백질 및 라이브러리가 수득될 때까지 반복될 수 있다.
서열-활성 모델과 변이체 라이브리 사이의 피드백 루프 때문에, 더욱 우수한 모델 및 더욱 우수한 변이체 라이브러리는 활성이 개선된 단백질을 탐색하는 데 있어 서로에 의존한다. 그러므로, 모델링 도메인 및/또는 서열 분석 도메인에서 병목 및 개선이 두 도메인 모두에 영향을 줄 수 있다. 본 발명의 일부 실시양태에서, 모델링 기법에 기인한 모델링 효율의 개선은 서열 탐색을 유도하는 데 있어 더욱 우수한 모델을 제공한다. 일부 실시양태에서, 다음 세대 서열 분석 기술은 시험관내에서 서열 분석 속도를 개선시키는 데 뿐만 아니라, 교차 검증 데이터를 제공하여 인실리코 전산 모델을 개선시키는 데에도 사용된다.
본 발명의 일부 실시양태에서, 유용한 서열-활성 모델은 강건한 수학적 모델링 기법 및 다수의 "관찰값"을 필요로 한다. 이러한 관찰값은 모델에 대하여 트레이닝 세트로 제공되는 데이터이다. 구체적으로, 각각의 관찰값은 라이브러리 중 변이체에 대한 활성 값 및 관련 서열이다. 역사적으로, 서열 분석은 큰 트레이닝 세트의 개발에서 제한된 단계였고, 결과적으로 강건한 서열-활성 모델이 증가하게 되었다. 현재 일반적으로 사용되는 방법에서, 아마도 수백개의 변이체를 포함하는 변이체 라이브러리가 생성된다. 그러나, 실제로는 상기 변이체 중 단지 소부분만이 서열 분석된다. 전형적인 유도 진화 회차에서는 실제로 최고 활성을 가지는 단지 약 10 내지 30개의 변이체만이 서열 분석된다. 상대적으로 활성이 낮은 일부 변이체를 비롯한, 라이브러리 중 훨씬 더 큰 부분의 변이체가 서열 분석되는 것이 이상적이다. 다음 세대 서열 분석 도구는 서열 분석 속도를 크게 개선시켰으며, 이를 통해 트레이닝 세트 중 저활성 및 고활성 변이체를 포함할 수 있게 된다. 일부 실시양태에서, 활성 수준이 광범위한 변이체를 포함함에 따라 수행능이 더욱 우수하고/거나, 더욱 광범위한 서열 및 활성 공간에 걸쳐 활성을 예측하는 데 있어 더욱 우수한 모델이 제조된다.
본원에서 언급되는 일부 선형 서열 활성 회귀 모델은 관심의 대상이 되는 임의의 활성을 예측하는 독립 변수로서 개별 잔기를 포함한다. 선형 서열 활성 회귀 모델은 2개 이상의 잔기 사이의 상호작용을 나타내는 항을 포함하지 않는다. 두 잔기 사이의 상호작용이 활성에 대하여 시너지 효과를 가지는 경우, 선형 모델은 두 상호작용 잔기와 관련된, 인위적으로 폭등된 계수 값을 제공할 수 있다. 그 결과, 상기 모델을 이용하여 연구하는 당사자는 상대적으로 높은 계수 값에 의해 제안되는 바와 같이, 단순히 잔기를 치환함으로써 생성된 펩티드의 활성은 예상보다 더 높을 것이라는 잘못된 결론을 내릴 수 있다. 이는 연구원이 선형 모델을 이용하는 것으로부터 잔기 치환과 관련된 활성 증가는 주로 치환의 또 다른 치환과의 상호작용의 결과라는 것을 이해하지 못하기 때문이다. 연구원이 이러한 상호작용의 중요성을 이해한다면, 이때 상기 연구원은 두 치환 모두를 동시에 수행할 수 있고, 선형 모델에 의해 제안되는 활성 증가를 달성할 수 있다.
두 잔기가 상호작용하여 비선형 방식으로 활성을 억제한다면, 선형 모델은, 잔기가 순전히 서로로부터 분리되는 데 고려되는 경우에 적절한 것으로 보이는 것보다 더 낮은 값은 상기 잔기와 관련된 계수가 원인이 된다고 본다. 다시 말해, 상호작용 잔기에 대하여 치환 중 다른 나머지 하나를 제외한, 하나만 수행함으로써 선형 모델에 의해 제안되는 것보다 더 큰 활성 결과를 얻게 될 것이다.
잔기-잔기 상호작용이 활성에 대하여 강력한 영향을 미치는 경우에는 선형 모델이 부적절할 수 있기 때문에, 잔기 사이의 상호작용을 나타내는 비1차 상호작용 항을 포함하는 비선형 모델은 대개 활성을 정확하게 예측하는 데 필요하다. 그러나, 비1차 항을 사용하는 모델은 전산 및 실험상의 도전 과제를 제기한다. 가장 특히, 모델을 개발/이용하는 데 매우 많은 잠재적 상호작용 항이 고려되며, 이는 상당량의 전산을 필요로 한다. 훨씬 더 큰 한계는 상당수의 잔기-잔기 상호작용 항을 이용하여 모델을 생성하는 데 필요한 잠재적인 관찰값 개수이다. 추가로, 특정 개수의 이용가능한 관찰값을 고려해 볼 때, 모델 생성 기법은 데이터를 과대적합화시키는 경향이 있을 수 있다. 이러한 도전 과제를 처리하기 위해서는 서열-활성 모델에서 제공된 상호작용 항을 주의깊게 선택하고, 제한하는 것이 다수의 모델을 개발하는 데 있어 중요한 고려 사항이 된다.
도 1은 서열-활성 모델을 제조하는 프로세스의 실행을 보여주는 순서도를 제시한 것이다. 도시된 바와 같이, 프로세스 (100)은 변이체 유전자에 대한 서열 및 활성 데이터("관찰값")를 제공하는 블록 (103)으로 시작된다. 서열 데이터는 초기 또는 개선된 단백질 변이체 라이브러리를 위해 예를 들어, 잔기 서열을 포함하는 트레이닝 세트로부터 취할 수 있다. 전형적으로, 이들 데이터는 라이브러리 중의 각각의 단백질에 대한 활성값과 함께, 완전한 또는 부분적인 잔기 서열 정보를 포함한다. 일부 경우에서, 다중 유형의 활성(예컨대, 속도 상수 데이터 및 열적 안정성 데이터)이 트레이닝 세트에 함께 제공된다. 원하는 결과에 의해 결정되는 바와 같이, 다른 데이터 소스도 또한 고려될 수 있다. 일부 적합한 데이터 소스로는 구축 중인 서열-활성 모델과 관련된 특정 펩티드에 관한 정보를 기술하는 문헌상의 참고 문헌을 포함하나, 이에 한정되지 않는다. 추가의 정보 공급원으로는 같은 프로젝트에서의 이전 회차 또는 상이한 회차의 유도 진화를 포함하나, 이에 한정되지 않는다. 실제로, (본원에서 제공된 방법을 포함하나, 이에 한정되지 않는 임의의 적합한 방법을 사용하여) 이전 회차의 유도 진화로부터 유도된 정보가 후속하여 제조되는 라이브러리, 변이체 등을 개발하는 데 사용될 수 있다는 것을 알 수 있는 것으로 한다.
다수의 실시양태에서, 단백질 변이체 라이브러리의 개별 구성원들은 광범위한 서열 및 활성을 나타낸다. 이는 광범위한 영역의 서열 공간에 걸쳐 적용가능한 서열-활성 모델을 생성하는 것을 용이하게 한다. 상기와 같이 다양한 라이브러리를 생성하는 기법으로는 본원에 기술된 바와 같은, 단백질 서열의 체계적인 변이 및 유도 진화 기법을 포함하나, 이에 한정되지 않는다. 그러나, 일부 대체 실시양태에서, 특정 유전자 패밀리 중의 유전자 서열 (예컨대, 다중의 종 또는 유기체에서 발견되는 특정 키나제)로부터 모델을 생성하는 것이 바람직할 수 있다. 패밀리의 모든 구성원 간에 다수의 잔기가 동일하기 때문에, 모델은 단지 다른 잔기만을 기술한다. 따라서, 일부 실시양태에서, 가능한 모든 변이체로 이루어진 세트와 비교하여 상대적으로 작은 트레이닝 세트에 기초한 통계학적 모델은 로컬 센스로 유효하다. 즉, 모델은 오직 주어진 변이체의 주어진 관찰값에 대해서만 유효하다. 일부 실시양태에서, 일부 모델에서는 고려 중인 모델 시스템(들)의 능력 및/또는 요구를 넘어선다는 것이 인지되고 있는 바와 같이, 본 목적은 전역적인 적합도 기능을 찾고자 하는 것이 아니다.
활성 데이터는 관심의 대상이 되는 활성/활성들의 크기를 측정할 수 있도록 적절하게 디자인된 어세이 및/또는 스크린을 포함하나, 이에 한정되지 않는 당업계에 공지된 임의의 적합한 수단을 사용하여 수득될 수 있다. 상기 기법은 주지되어 있고, 본 발명에 필수적인 것은 아니다. 적절한 어세이 또는 스크린을 디자인하는 원리는 당업계에 광범위하게 이해되고 있고, 주지되어 있다. 단백질 서열을 수득하는 기법 또한 주지되어 있으며, 본 발명에 중요한 것은 아니다. 언급한 바와 같이, 다음 세대 서열 분석 기술이 사용될 수 있다. 본원에 기술된 실시양태와 함께 사용되는 활성은 단백질 안정성(예컨대, 열적 안정성)일 수 있다. 그러나, 다수의 중요한 실시양태는 다른 활성, 예컨대, 촉매 활성, 병원체 및/또는 독소에 대한 저항, 독성 등을 고려한다. 실제로, 당업계에 공지된 임의의 적합한 방법이 본 발명에서 사용될 수 있는 바, 본 발명을 임의의 특정 어세이/스크리닝 방법(들), 및/또는 서열 분석 방법(들)로 한정하고자 하지 않는다.
트레이닝 세트 데이터를 생성 또는 획득한 후, 프로세스는 상기 데이터를 사용하여 서열 정보의 함수로서 활성을 예측하는 베이스 서열-활성 모델을 생성한다. 블록 (105)를 참조할 수 있다. 상기 모델은 특정 단백질에 대한 서열 정보가 제공될 때 그 단백질의 상대적인 활성을 예측하는 식, 알고리즘, 또는 다른 도구이다. 다시 말해, 단백질 서열 정보는 입력값이고, 활성 예측은 출력값이다. 일부 실시양태에서, 베이스 모델은 임의의 상호작용 항을 포함하지 않는다. 이 경우, 베이스 모델은 "선형 모델"로서 기술될 수 있다. 다른 실시양태에서, 베이스 모델은 모든 이용가능한 상호작용 항을 포함하며, 이 경우, 베이스 모델은 비선형 모델 또는 상호작용 모델로서 기술될 수 있다.
많은 실시양태의 경우, 베이스 모델은 활성에 대한 각종 잔기의 기여도를 순위화할 수 있다. 모두가 기계 학습의 범주하에 포함되는 것인, 상기 모델을 생성하는 방법(예컨대, 부분 최소 제곱 회귀(PLS), 주성분 회귀(PCR), 및 다중 선형 회귀(MLR: multiple linear regression), 베이지안 선형 회귀)은 독립 변수 포맷(서열 정보), 종속 변수 포맷(활성), 및 모델 그 자체 형태(예컨대, 선형 1차식)와 함께 하기에서 논의된다.
베이스 서열-활성 모델 생성 후, 프로세스는 이용가능한 상호작용 항의 풀로부터의 상호작용 항을 반복적으로 베이스 모델에 가하거나 그로부터 감산하고, 생성된 신규 모델을 베이스 모델에 대한 개선에 대해 평가하여 최종 모델을 제조한다. 블록 (107)을 참조할 수 있다. 베이스 모델이 모든 이용가능한 상호작용 항을 포함할 경우, 프로세스는 단계적으로 상기 항을 제거한다.
신규 모델을 평가할 때, 본 개시내용의 방법은 모델이 주어진 데이터 세트에 대해 나타내는 분산 뿐만 아니라, 신규 데이터를 예측할 수 있는 모델의 능력도 고려한다. 일부 실시양태에서, 상기 모델 선택 접근법은 모델이 주어진 데이터 세트에 대하여 과대적합화되지 못하도록 하기 위해 계수/파라미터가 더 적은 등가의 모델보다 계수/파라미터가 더 많은 모델에는 벌점을 부가한다. 선택 방법의 예로는 아카이케 정보 기준(AIC) 및 베이지안 정보 기준(BIC: Bayesian Information Criterion), 및 그의 변형을 포함하나, 이에 한정되지 않는다.
베이스 모델보다 누진적으로 더 많은 상호작용 항(및 관련 계수)을 포함하는 희귀 모델에서와 같이, 일련의 내포된 모델에서는 심지어 추가의 계수가 스퓨리어스(spurious)한 경우에도, 더욱 복잡한 모델은 추가의 자유도를 누릴 수 있기 때문에, 더욱 복잡한 모델이 더 단순한 모델과 동등하게 우수하거나, 또는 그보다 더 우수한 정도로 적합하다. 본 개시내용의 특정 실시양태는 적합도 증가가 스퓨리어스한 파라미터에 대한 대가에 의한 오프셋보다 더 큰 정도로 더욱 복잡한 모델에 벌점을 부가하는 모델 선택 방법을 사용한다.
블록(105) 및 (107)에서의 연산에 따라 서열-활성 모델을 생성하는 예시적인 알고리즘을 하기에 제시한다. 상기 기법으로는 모델에 추가의 상호작용 항을 포함하는 것에 대해 편향하는 단계적 기법을 포함하나, 이에 한정되지 않는다. 그러나, 본 개시내용을 이러한 구체적인 일례로 한정하고자 하지 않는다.
한 측면에서, 본 개시내용은 원하는 활성에 영향을 주는 생물학적 분자를 확인하는 것을 보조할 수 있는 서열-활성 모델을 제조하는 방법을 제공한다. 일부 실시양태에서, 본 방법은 (a) 복수 개의 생물학적 분자에 대한 서열 및 활성 데이터를 입수하는 단계; (b) 서열 및 활성 데이터로부터 베이스 모델을 제조하는 단계로서, 여기서, 베이스 모델은 서열의 서브유닛의 존재 또는 부재의 함수로서 활성을 예측하는 것인 단계; (c) 하나 이상의 신규 상호작용 항을 베이스 모델에 가하거나 또는 베이스 모델로부터 감함으로써 하나 이상의 신규 모델을 제조하는 단계로서, 여기서, 신규 상호작용 항은 2개 이상의 상호작용 서브유닛 사이의 상호작용을 나타내는 것인 단계; (d) 서브유닛의 존재 또는 부재의 함수로서 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력을 측정하는 단계; 및 (e) (d)에서 측정된 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력에 기초하고, 신규 상호작용 항을 가하는 것에 대한 편향을 이용하여, 베이스 모델에, 또는 베이스 모델로부터 신규 상호작용 항을 가할지 또는 감할지 여부를 결정하는 단계를 포함한다. 이어서, 유도된 모델은 다양한 적용에서, 예컨대, 원하는 생물학적 활성 및 특성을 가지는 단백질을 확인하기 위한 단백질 라이브러리의 유도 진화에서 사용될 수 있다.
일부 실시양태에서, 일부 실시양태에서, 본 방법을 통해, 업데이트된 모델을 생성하기 위해서는 신규 상호작용 항을 베이스 모델에 가하여야 한다고 결정된 경우, 본 방법은 또한 (f) 베이스 모델 대신 업데이트된 모델을 이용하여 (c)를 반복하고, (c)에서 가하거나 감한 것과는 다른 상호작용 항을 가하거나 감하는 단계; 및 (g) 베이스 모델 대신 업데이트된 모델을 이용하여 (d) 및 (e)를 반복하는 단계를 포함한다. 일부 실시양태에서, 본 방법은 (h) 추가의 업데이트된 모델을 이용하여 (f) 및 (g)를 반복하는 단계를 추가로 포함한다.
트레이닝 세트를 위한 관찰값을 선택하고, 서열-활성 모델을 생성하기 위한 수학적 기법을 선택한 후, 베이스 모델이 생성된다. 베이스 모델은 전형적으로는 그의 예측 능력을 고려하지 않고 생성된다. 이는 본원에 기술된 바와 같이, 이용가능한 관찰값(즉, 관찰값 세트)으로부터 베이스 모델을 생성하기 위한 정의된 방법에 따라 간단하게 생성된다. 상기 언급한 바와 같이, 일부 실시양태에서, 모델은 단백질을 기술하지만, 서열 모델은 다양한 서열을 기술할 수 있다. 모델이 단백질을 기술하는 경우, 베이스 모델은 간단하게 트레이닝 세트를 생성하는 데 사용되는 펩티드의 집합에 존재하는 각각의 돌연변이에 대하여 단일 항을 가지는 선형 모델이다. 상기 실시양태에서, 베이스 모델은 펩티드 중의 잔기 사이의 상호작용을 나타내는 어떤 항도 포함하지 않는다. 일부 실시양태에서, 베이스 모델은 관찰값 세트에 존재하는 각각의 모든 돌연변이에 대한 개별 항을 포함하지 않는다.
대안적 접근법에서, 베이스 모델은 분리시 각각의 돌연변이를 기술하는 항을 포함할 뿐만 아니라, 잠재적인 상호작용 잔기들 모두에 대한 항도 포함한다. 극단적인 경우에, 주목받는 돌연변이들 사이의 모두 가능한 상호작용도 베이스 모델에서 사용된다. 베이스 모델은 돌연변이 사이의 각각의 모든 쌍별 상호작용에 대한 항 뿐만 아니라, 각각의 모든 가능한 3개 잔기의 상호작용 뿐만 아니라, 모든 가능한 4개 잔기의 상호작용에 대한 항 등도 포함한다. 일부 실시양태는 쌍별 상호작용만, 또는 쌍별 상호작용 및 3원 상호작용을 포함한다. 3원 상호작용은 활성에 영향을 미치는, 3개의 독특한 서브유닛들 사이의 상호작용이다.
베이스 모델로서 단순 선형 모델을 이용하는 하나 이상의 실시양태에서, 모델을 개선시키기 위한 후속 노력으로는 독특한 상호작용을 나타내는 신규 항을 가하는 것을 포함한다. 베이스 모델이 1차 항 및 비1차 항을 포함하는 대체 실시양태에서, 모델을 개선시키기 위한 후속 노력으로는 비1차, 상호작용 항 중 일부를 선택적으로 제거하는 것을 포함한다.
본 발명의 하나 이상의 실시양태에서, 베이스 모델을 개선시키기 위한 프로세스는 생성된 모델이 모델의 정질을 충분히 개선시키는지 여부를 결정할 때, 베이스 모델로부터 상호작용 항을 반복적으로 가하거나 감산하는 것을 포함한다. 매회 반복시, 현재 모델의 예측력을 측정하고, 또 다른 모델, 예컨대, 베이스 모델 또는 업데이트된 모델과 비교한다.
예측력의 척도가 다른 데이터 세트로 일반화될 수 있는 모델의 능력을 이미 고려하고 있는 실시양태에서, 상기 척도만이 단독으로 후보 모델이 선택되어야 하는지 여부를 결정할 수 있다. 예를 들어, 척도, 예컨대, AIC 또는 BIC는 모델 가능도(또는 잔류 오차) 및 파라미터 개수, 둘 모두를 고려한다. 모델의 "가능도 함수" 또는 "가능도"는 통계학적 모델의 파라미터의 함수이다. 일부 관찰된 결과가 주어졌을 때, 파라미터 값 세트의 가능도는 상기 파라미터 값이 주어졌을 때, 상기 관찰된 결과의 확률과 같으며, 즉, L(θ\x) = P(x\θ)이다. 모델 가능도를 계산하는 것에 관한 일례는 하기 섹션에서 기술한다. 더 적은 파라미터를 가지는 모델과 같이, 더 많은 파라미터를 가지는 모델이 같은 양의 데이터 분산을 포착한다면, 척도, 예컨대, AIC 및 BIC는 더 많은 파라미터를 가지는 모델에 대해 편향된다. 예측력의 척도가 오직 잔류 오차만을 고려할 경우, 현재 진행 중인 반복과 관련된 변화를 가장 최근 업데이트된 현재 모델로 도입해야 할지 여부를 결정하기 위해서는 잔류 오차의 개선 규모가 고려되어야 한다. 이는 개선 규모를 역치와 비교함으로써 달성될 수 있다. 그 규모가 역치보다 작을 경우, 현재 진행 중인 반복에서 고려 중에 있는 변화는 수락되지 않는다. 별법으로, 개선 규모가 역치를 초과할 경우, 고려 중에 있는 변화는 업데이트된 모델로 도입되고, 업데이트된 모델은 최적 신규 모델로서의 역할을 하게 되며, 남은 반복은 계속해서 진행된다.
특정 실시양태에서, 각각의 반복은 고려 중에 있는 현 최적 모델로부터 단일의 상호작용 항을 가하거나 감하는 것을 고려한다. 가법 모델의 경우, 즉, 베이스 모델이 오직 1차 항만을 포함하는 경우, 모든 이용가능한 상호작용 항의 풀이 고려될 수 있다. 이들 상호작용 항들은 각각 프로세스가 완료되고, 최종 최적 모델이 수득될 때까지 계속해서 고려된다.
일부 경우에서, 본 프로세스가 효과적으로 수렴되었고, 및 추가 개선이 가능성이 없다고 결정되었을 때, 풀 중의 모든 이용가능한 상호작용 항이 고려되기 이전에 모델 생성 프로세스는 종결된다.
도 2는 프로세스에서 단백질 서열 및 활성 공간을 탐색하기 위한 목적으로 신규 단백질 변이체 라이브러리의 생성을 유도하기 위해 모델이 어떻게 반복적으로 사용될 수 있는지를 도시한 것이다((200) 참조). 최종 모델 생성 후, 최종 모델은 활성에 영향을 주는 것으로 예측되는 다중의 잔기 위치(예컨대, 35번 위치) 또는 특이적 잔기 값(예컨대, 35번 위치의 글루타민)을 확인하는 데 사용된다. 블록 (207)을 참조할 수 있다. 상기 위치를 확인하는 것 이외에도, 모델은 원하는 활성(활성들?)에 대한 그의 기여도에 기초하여 잔기 위치 또는 잔기 값을 "순위화"하는 데 사용될 수 있다. 예를 들어, 모델은 35번 위치의 글루타민이 활성에 대하여 가장 현저한 양성 효과를 가지며, 208번 위치의 페닐알라닌이 활성에 대하여 두번째로 가장 현저한 양성 효과를 가진다는 것 등을 예측할 수 있다. 하기 기술되는 한 구체적인 접근법에서, PLS 또는 PCR 회귀 계수는 특이적 잔기의 중요도를 순위화하는 데 사용된다. 또 다른 구체적인 접근법에서, PLS 로드 행렬은 특이적 잔기 위치의 중요도를 순위화하는 데 사용된다.
프로세스가 활성에 영향을 주는 잔기를 확인한 후, 잔기 중 일부는 블록 (209)에 명시된 바와 같이 변이를 위해 선택된다(도 2). 이는 서열 공간 탐색을 위해 수행된다. 잔기는 다수의 상이한 선택 프로토콜 중 임의의 것을 사용하여 선택되며, 상기 프로토콜 중 일부는 하기에 기술한다. 한 예시적인 일례에서, 활성에 가장 유익한 영향을 주는 것으로 예측되는 특이적 잔기는 보존된다(즉, 변이되지 않는다). 그러나, 영향을 덜 주는 것으로 예측되는 특정 개수의 다른 잔기가 변이를 위해 선택된다. 또 다른 예시적인 일례에서, 활성에 가능 큰 영향을 주는 것으로 밝혀진 잔기 위치는, 단, 오직 그 위치가 트레이닝 세트의 고성능 구성원에서 다른 것으로 나타난 경우에만 변이를 위해 선택된다. 예를 들어, 모델을 통해 잔기 위치 197번이 활성에 가능 큰 영향을 주는 것으로 예측되되, 단, 고활성을 가지는 단백질 모두 또는 그 대부분이 상기 위치에 류신을 가진다면, 197번 위치는 본 접근법에서의 변이를 위해서는 선택되지 않을 것이다. 다시 말해, 다음 세대 라이브러리 중 모든 단백질 또는 그 대부분은 197번 위치에 류신을 가질 것이다. 그러나, 일부 "우수한" 단백질이 상기 위치에 발린을 가지고, 다른 것들은 류신을 가진다면, 그때는 프로세스는 상기 위치의 아미노산을 가변시키기 위해 선택하게 될 것이다. 일부 경우에서, 2개 이상의 상호작용 잔기의 조합이 활성에 가장 큰 영향을 미친다는 것을 알게 될 것이다. 그러나, 일부 전략법에서, 이들 잔기는 공변이된다.
변이에 대한 잔기를 확인한 후, 본 방법은 이어서 명시된 잔기 변이를 가지는 신규 변이체 라이브러리를 생성한다. 블록 (211)을 참조할 수 있다(도 2). 본 목적을 위해 다양한 방법론이 이용될 수 있다. 한 일례에서, 신규 변이체 라이브러리를 생성하는 데 시험관내 또는 생체내 재조합 기반 다양성 생성 메커니즘이 수행된다. 상기 방법은 모체 변이체 라이브러리의 단백질을 코딩하는 서열 또는 부분서열을 포함하는 올리고뉴클레오티드를 사용할 수 있다. 올리고뉴클레오티드 중 일부는 밀접한 관련을 가질 것이며, (209)에서 변이를 위해 선택된 대체 아미노산의 경우 코돈 선택에서만 차이가 날 것이다. 재조합 기반 다양성 생성 메커니즘은 1회 또는 다회 사이클로 수행될 수 있다. 다회 사이클이 사용될 경우, 매회 사이클은 어느 변이체가 후속 재조합 사이클에서 사용되는 데 허용되는 성능을 가지는지를 확인하기 위한 스크리닝 단계를 포함한다. 이것이 유도 진화의 한 형태이다. 그러나, 임의의 적합한 방법/기법이 본 발명에서 사용될 수 있다는 것을 알 수 있는 바, 본 발명을 재조합 기반 다양성 생성 방법 중 임의의 구체적인 방법으로 한정하고자 하지 않는다.
추가의 예시적인 일례에서, "참조" 단백질 서열이 선택되고, 도 2의 (209)에서 선택되는 잔기는 변이체 라이브러리의 개별 구성원의 확인을 위해 "토글링"된다. 그렇게 확인된 신규 단백질은 신규 라이브러리를 생성하는 데 적절한 기법에 의해 합성된다. 한 일례에서, 참조 서열은 PLS 또는 PCR 모델에 의해 예측되는, 트레이닝 세트의 최적 구성원 또는 "최적" 서열일 수 있다.
또 다른 예시적인 일례에서, 한 회차의 유도 진화에서 변이를 위한 잔기는 단일 모체 서열에서 선택된다. 모체는 이전 회차의 유도 진화로부터 생성된 모델을 이용하여, 또는 어세이 성능이 최적 라이브러리 구성원을 확인하는 데이터를 사용하여 확인될 수 있다. 다음 회차의 유도 진화를 위한 올리고뉴클레오티드는 현 회차 동안 서열-활성 모델로부터 알고리즘 방식으로 예측된 하나 이상의 돌연변이를 포함하는 선택된 모체의 골격의 일부를 포함하는 것으로 정의될 수 있다. 이러한 올리고뉴클레오티드는 합성 방법을 포함하나, 이에 한정되지 않는 임의의 적합한 수단을 사용하여 제조될 수 있다.
신규 라이브러리 생성 후, 블록 (213)에 명시되어 있는 바와 같이 (도 2), 활성에 대해 스크리닝된다. 이상적으로, 신규 라이브러리는 이전 라이브러리에서 관찰되었던 것보다 우수한 활성을 가지는 하나 이상의 구성원을 제공한다. 그러나, 그러한 장점이 없는 경우에도, 신규 라이브러리는 유익한 정보를 제공할 수 있다. 그의 구성원은 (209)(도 2)에서 선택된 변이의 효과의 원인이 되며,는 개선된 모델을 생성하는 데 사용될 수 있고, 이로써, (209)(도 2)에서 선택된 변이의 효과를 나타내고, 이로써, 더욱 광범위한 영역의 서열 공간에 걸쳐 활성을 정확하게 예측하는 개선된 모델을 생성하는 데 사용될 수 있다. 추가로, 라이브러리는 (예컨대, 활성에서) 국소 최대에서 전역 최대로의 서열 공간 상의 추이를 나타낼 수 있다.
일부 실시양태에서, 프로세스 (200)(도 2)의 목표에 의존하여, 각각의 것이 트레이닝 세트의 신규 구성원을 제공하는 것인, 일련의 신규 단백질 변이체 라이브러리를 생성하는 것이 바람직할 수 있다. 이어서, 업데이트된 트레이닝 세트는 개선된 모델을 생성하는 데 사용된다. 개선된 모델을 달성하기 위해, 추가의 또 다른 단백질 변이체 라이브러리가 생성되어야 하는지 여부를 결정하는, 블록 (215)에 제시된 바와 같은 결정 연산을 포함하는 프로세스 (200)이 제시되어 있다. 결정하는 데 다양한 기준이 사용될 수 있다. 결정 기준의 예로는 지금까지 생성된 단백질 변이체 라이브러리 개수, 현 라이브러리로부터의 상위 단백질의 활성, 원하는 활성 규모, 및 최근의 신규 라이브러리에서 관찰된 개선 수준을 포함하나, 이에 한정되지 않는다.
프로세스가 계속해서 신규 라이브러리를 사용한다고 가정할 때, 프로세스는 현 단백질 변이체 라이브러리에 대해 얻은 서열 및 활성 데이터로부터 신규 서열-활성 모델이 생성되는, 블록 (100)의 연산으로 복귀한다(도 2). 다시 말해, 현 단백질 변이체 라이브러리에 대한 서열 및 활성 데이터는 신규 모델을 위한 트레이닝 세트의 일부로서의 역할을 한다(또는 전체 트레이닝 세트로서의 역할을 할 수 있다). 이후, 상기 기술된 바와 같이, 단, 신규 모델을 이용하여 블록 (207), (209), (211), (213), 및 (215)의 연산(도 2)이 실행된다.
본 방법의 종점에 도달한 것으로 결정되었을 때, 도 2에 도시된 사이클은 종료되고, 더 이상 신규 라이브러리는 생성되지 않는다. 상기 시점에서, 프로세스는 간단하게 종료되거나, 또는 일부 실시양태에서, 라이브러리 중 하나 이상의 것으로부터의 하나 이상의 서열(들)이 개발 및/또는 제조를 위해 선택된다. 블록 (217)을 참조할 수 있다.
B. 관찰값 생성
단백질 변이체 라이브러리는 라이브러리 중 구성원 간에 서로 상이한 하나 이상의 잔기를 가지는 다중 단백질로 이루어진 군이다. 이러한 라이브러리는 본원에 기술된 방법 및/또는 당업계에 공지된 임의의 적합한 수단을 사용하여 생성될 수 있다. 이러한 라이브러리는 본 발명의 다양한 실시양태에 따라 서열-활성 모델을 생성하는 데 사용되는 트레이닝 세트에 데이터를 제공하는 데 사용될 수 있다는 것을 알 수 있다. 단백질 변이체 라이브러리에 포함된 단백질의 개수는 대개 적용 및 그의 생성과 관련된 비용에 의존한다. 본 발명은 본 발명의 방법에 사용되는 단백질 라이브러리 중의 단백질의 개수를 임의의 특정 개수로 한정하고자 하지 않는다. 추가로 본 발명은 임의의 특정 단백질 변이체 라이브러리 또는 라이브러리들로 한정하고자 하지 않는다.
일례에서, 단백질 변이체 라이브러리는 단일 유전자 패밀리에 의해 코딩될 수 있는 하나 이상의 자연적으로 발생된 단백질로부터 생성된다. 공지된 단백질 또는 신규한 합성 단백질의 재조합을 포함하나, 이에 한정되지 않는 다른 출발점이 사용될 수 있다. 라이브러리는 다양한 기법에 의해 상기 시드 또는 출발 단백질로부터 생성될 수 있다. 한 경우에서, 라이브러리는 문헌 [Stemmer (1994) Proceedings of the National Academy of Sciences, USA, 10747-10751] 및 WO 95/22625(상기 두 문헌 모두 본원에서 참조로 포함된다)에 기술된 바와 같은 DNA 단편화 매개 재조합에 의해, 문헌 [Ness et al. (2002) Nature Biotechnology 20:1251-1255 및 WO 00/42561(상기 두 문헌 모두 본원에서 참조로 포함된다)에 기술된 바와 같은 합성 올리고뉴클레오티드 매개 재조합에 의해, 또는 하나 이상의 모체 단백질의 일부 또는 그 모두를 코딩하는 핵산에 의해 생성된다. 상기 방법들의 조합 또한 사용될 수 있으며(예컨대, DNA 단편 및 합성 올리고뉴클레오티드의 재조합), 그뿐만 아니라, 예를 들어, WO97/20078 및 WO98/27230(상기 두 문헌 모두 본원에서 참조로 포함된다)에 기술된 바와 같은 다른 재조합 기반 방법도 사용될 수 있다. 단백질 변이체 라이브러리를 생성하는 사용되는 임의의 적합한 방법은 본 발명에서 사용될 수 있다는 것을 알 수 있다. 실제로, 본 발명을 변이체 라이브러리를 생성하는 임의의 특정 방법으로 한정하고자 하지 않는다.
일부 실시양태에서, 단일 "출발" 서열(이는 "선조" 서열일 수 있다)은 모델링 프로세스에 사용되는 돌연변이 군을 정의하고자 하는 목적으로 사용될 수 있다. 일부 실시양태에서, 출발 서열 중 하나 이상의 것은 야생형 서열이다.
특정 실시양태에서, 돌연변이는 (a) 문헌상에서 기질 특이성, 선택성, 안정성, 또는 다른 유익한 특성에 영향을 주는 것으로서 확인된 것이고/거나, (b) 전산적으로 단백질 폴딩 패턴(예컨대, 단백질의 내부 잔기를 패킹하는 것), 리간드 결합, 서브유닛 상호작용, 다중의 다양한 동족체 간의 패밀리 셔플링 등을 개선시키는 것으로 예측되는 것이다. 별법으로, 돌연변이는 출발 서열 내로 물리적으로 도입될 수 있고, 발현 생성물은 유익한 특정에 대하여 스크리닝될 수 있다. 비록 임의의 적합한 방법도 사용될 수는 있지만, 부위 지정 돌연변이 유발법이 돌연변이를 도입하는 데 있어 유용한 기법 중 한 일례이다. 따라서, 별법으로 또는 추가로, 돌연변이체는 유전자 합성, 포화 무작위 돌연변이 유발법, 잔기의 반합성 조합 라이브러리, 유도 진화, 반복적 서열 재조합("RSR: recursive sequence recombination")(예컨대, 미국 특허 출원 번호 제2006/0223143호(그의 전문이 본원에 참조로 포함된다) 참조), 유전자 셔플링, 오류 유발 PCR, 및/또는 임의의 다른 적합한 방법에 의해 제공될 수 있다. 적합한 포화 돌연변이 유발 방법의 한 일례는 미국 공개 특허 출원 번호 제20100093560호(그의 전문이 본원에 참조로 포함된다)에 기술되어 있다.
출발 서열이 야생형 단백질의 아미노산 서열과 동일할 필요는 없다. 그러나, 일부 실시양태에서, 출발 서열은 야생형 단백질의 서열이다. 일부 실시양태에서, 출발 서열은 야생형 단백질에 존재하지 않는 돌연변이를 포함한다. 일부 실시양태에서, 출발 서열은 예컨대, 단백질 패밀리와 같이, 공통된 특성을 가지는 단백질 군으로부터 유래된 컨센서스 서열이다.
모체 서열의 공급원으로서의 역할을 할 수 있는 효소 패밀리 또는 부류에 관한 비제한적 대표적인 목록으로는 하기: 옥시도리덕타제(E.C.1); 트랜스퍼라제(E.C.2); 하이드롤리아제(E.C.3); 리아제(E.C.4); 이소머라제(E.C.5) 및 리가제(E.C.6)를 포함하나, 이에 한정되지 않는다. 더욱 구체적이되, 비제한적인 옥시도리덕타제의 서브군으로는 데하이드로게나제(예컨대, 알콜 데하이드로게나제(카보닐 리덕타제), 크실루로스 리덕타제, 알데히드 리덕타제, 파르네솔 데하이드로게나제, 락테이트 데하이드로게나제, 아라비노스 데하이드로게나제, 글루코스 데하이드로게나제, 프럭토스 데하이드로게나제, 크실로스 리덕타제 및 숙시네이트 데하이드로게나제), 옥시다제(예컨대, 글루코스 옥시다제, 헥소스 옥시다제, 갈락토스 옥시다제 및 릭카제), 모노아민 옥시다제, 리폭시게나제, 퍼옥시다제, 알데히드 데하이드로게나제, 리덕타제, 장쇄 아실-[아실-캐리어-단백질] 리덕타제, 아실-CoA 데하이드로게나제, 에네-리덕타제, 신타제(예컨대, 글루타메이트 신타제), 니트레이트 리덕타제, 모노 및 디옥시게나제, 및 카탈라제를 포함한다. 더욱 구체적이되, 비제한적인 트랜스퍼라제의 서브군으로는 메틸, 아미디노, 및 카복실 트랜스퍼라제, 트랜스케톨라제, 트랜스알돌라제, 아실트랜스퍼라제, 글리코실트랜스퍼라제, 트랜스아미나제, 트랜스글루타미나제 및 폴리머라제를 포함한다. 더욱 구체적이되, 비제한적인 하이드롤라제의 서브군으로는 에스테르 하이드롤라제, 펩티다제, 글리코실라제, 아밀라제, 셀룰라제, 헤미셀룰라제, 크실라나제, 키티나제, 글루코시다제, 글루카나제, 글루코아밀라제, 아실라제, 갈락토시다제, 플루라나제, 피타제, 락타제, 아라비노시다제, 뉴클레오시다제, 니트릴라제, 포스파타제, 리파제, 포스포리파제, 프로테아제, ATP아제, 및 데할로게나제를 포함한다. 더욱 구체적이되, 비제한적인 리아제의 서브군으로는 데카복실라제, 알돌라제, 하이드라타제, 데하이드라타제(예컨대, 카보닉 안하이드라제), 신타제(예컨대, 이소프렌, 피넨 및 파르네센 신타제), 펙티나제(예컨대, 펙틴 리아제) 및 할로하이드린 데하이드로게나제를 포함한다. 더욱 구체적이되, 비제한적인 이소머라제의 서브군으로는 라세마제, 에피머라제, 이소머라제(예컨대, 크실로스, 아라비노스, 리보스, 글루코스, 갈락토스 및 만노스 이소머라제), 토토머라제, 및 뮤타제(예컨대, 아실 전달 뮤타제, 포스포뮤타제, 및 아미노뮤타제)를 포함한다. 더욱 구체적이되, 비제한적인 리가제의 서브군으로는 에스테르 신타제를 포함한다. 모체 서열의 공급원으로서 사용될 수 있는 다른 효소 패밀리 또는 부류로는 트랜스아미나제, 프로테아제, 키나제, 및 신타제를 포함한다. 본 개시내용의 가능한 효소에 관한 특정의 구체적인 측면을 예시하였지만, 상기 목록은 배타적인 것으로 간주되지 않으며, 본 개시내용을 한정하거나, 본 개시내용의 범주를 제한하는 것은 아니다.
일부 경우에서, 본원에 기술된 방법에서 유용한 후보 효소는 예를 들어, 거울상이성질체 선택적 반응, 예컨대, 거울상이성질체 선택적 환원 반응을 촉매화시킬 수 있다. 상기 효소는 예를 들어, 제약 화합물의 합성에 유용한 중간체를 제조하는 데 사용될 수 있다.
일부 실시양태에서, 후보 효소는 엔도크실라나제(EC 3.2.1.8); β-크실로시다제(EC 3.2.1.37); 알파 L-아라비노푸라노시다제(EC 3.2.1.55); 알파 글루쿠로니다제(EC 3.2.1.139); 아세틸크실란에스터라제(EC 3.1.1.72); 페룰로일 에스터라제(EC 3.1.1.73); 쿠마로일 에스터라제(EC 3.1.1.73); 알파 갈락토시다제(EC 3.2.1.22); 베타-갈락토시다제(EC 3.2.1.23); 베타-만나나제(EC 3.2.1.78); 베타-만노시다제(EC 3.2.1.25); 엔도-폴리갈락투로나제(EC 3.2.1.15); 펙틴 메틸 에스터라제(EC 3.1.1.11); 엔도-갈락타나제(EC 3.2.1.89); 펙틴 아세틸 에스터라제(EC 3.1.1.6); 엔도-펙틴 리아제(EC 4.2.2.10); 펙테이트 리아제(EC 4.2.2.2); 알파 람노시다제(EC 3.2.1.40); 엑소-폴리-알파 갈락투로노시다제(EC 3.2.1.82); 1,4-알파 갈락투로니다제(EC 3.2.1.67); 엑소폴리갈락투로네이트 리아제(EC 4.2.2.9); 람노갈락투로난 엔도리아제(EC 4.2.2.B3); 람노갈락투로난 아세틸에스터라제(EC 3.2.1.B11); 람노갈락투로난 갈락투로노하이드롤라제(EC 3.2.1.B11); 엔도-아라비나나제(EC 3.2.1.99); 락카제(EC 1.10.3.2); 망간 의존성 퍼옥시다제(EC 1.10.3.2); 아밀라제(EC 3.2.1.1), 글루코아밀라제(EC 3.2.1.3), 프로테아제, 리파제, 및 리그닌 퍼옥시다제(EC 1.11.1.14)로부터 선택된다. 1, 2, 3, 4, 5개 또는 5개 초과의 효소로 이루어진 임의의 조합이 본 발명의 조성물에 사용될 수 있다는 것을 알 수 있다.
본 발명의 하나 이상의 실시양태에서, 단일 출발 서열은 라이브러리 생성을 위해 다양한 방식으로 변형된다. 일부 실시양태에서, 라이브러리는 출발 서열의 개별 잔기를 체계적으로 가변시킴으로써 생성된다. 한 예시적인 일례에서, 체계적으로 가변된 서열을 확인하는 데 실험 디자인(DOE) 방법이 사용된다. 또 다른 일례에서, 어느 정도 수준의 체계적인 변이를 도입하는 데 "실습 실험실(wet lab)" 방법, 예컨대, 올리고뉴클레오티드 매개 재조합이 사용된다. 임의의 적합한 방법이 사용될 수 있다는 것을 알 수 있는 바, 본 발명을 체계적으로 가변된 서열을 생성하는 임의의 특정 방법으로 한정하고자 하지 않는다.
본원에서 사용되는 바, "체계적으로 가변된 서열"이라는 용어는 각각의 잔기가 다중 컨텍스트로 관찰되는 서열 세트를 의미한다. 원칙적으로, 체계적인 변이 수준은 서열이 서로로부터 직교성을 띠는 정도(즉, 평균과 비교하여 최대로 상이한 정도)에 의해 정량화될 수 있다. 일부 실시양태에서, 프로세스는 최대의 직교성을 띠는 서열을 가지는 것에 의존하지 않는다. 그러나, 모델의 정질은 테스트되는 서열 공간의 직교성에 직접적으로 관련하여 개선될 것이다. 간단한 예시적인 일례에서, 펩티드 서열은 2개의 잔기 위치를 확인함으로써 체계적으로 가변되며, 이들은 각각 2개의 상이한 아미노산 중 하나를 가질 수 있다. 최대로 다양한 라이브러리는 4개의 가능한 서열 모두를 포함한다. 상기 최대의 체계적인 변이는 예컨대, 2N에 의해(각각의 N 잔기 위치에 2가지 옵션이 있는 경우), 변수 위치의 개수에 따라 지수적으로 증가한다. 그러나, 최대의 체계적인 변이가 요구되는 것은 아니라는 것을 당업계의 숙련가는 쉽게 이해할 수 있을 것이다. 체계적인 변이는 서열 공간을 잘 샘플링하는 검정을 위한 상대적으로 소규모의 서열 세트를 확인하는 메커니즘을 제공한다.
체계적으로 가변된 서열을 가지는 단백질 변이체는 당업계의 숙련가에게 주지된 기법을 사용하여 다수의 방식으로 수득될 수 있다. 명시된 바와 같이, 적합한 방법으로는 하나 이상의 "모체" 폴리뉴클레오티드 서열에 기초하여 변이체를 생성하는 재조합 기반 방법을 포함하나, 이에 한정되지 않는다. 폴리뉴클레오티드 서열은 예를 들어, 재조합시키고자 하는 폴리뉴클레오티드를 DN아제로 분해시킨 후, 핵산을 결찰시키고/거나, PCR 재조립을 수행하는 것과 같은 다양한 기법을 사용하여 재조합될 수 있다. 이러한 방법은 예를 들어, 문헌 [Stemmer (1994) Proceedings of the National Academy of Sciences USA, 91:10747-10751], 미국 특허 번호 제5,605,793호(발명의 명칭: "Methods for In Vitro Recombination"), 미국 특허 번호 제5,811,238호(발명의 명칭: "Methods for Generating Polynucleotides having Desired Characteristics by Iterative Selection and Recombination"), 미국 특허 번호 제5,830,721호(발명의 명칭: "DNA Mutagenesis by Random Fragmentation and Reassembly"), 미국 특허 번호 제5,834,252호(발명의 명칭: "End Complementary Polymerase Reaction"), 미국 특허 번호 제5,837,458호(발명의 명칭: "Methods and Compositions for Cellular and Metabolic Engineering"), W098/42832(발명의 명칭: "Recombination of Polynucleotide Sequences Using Random or Defined Primers"), WO 98/27230(발명의 명칭: "Methods and Compositions for Polypeptide Engineering"), WO 99/29902(발명의 명칭: "Method for Creating Polynucleotide and Polypeptide Sequences") 등(상기 문헌은 모두 본원에서 참조로 포함된다)에 기술된 것과 같은 방법을 포함하나, 이에 한정되지 않는다.
합성 재조합 방법 또한 특히, 체계적인 변이를 가지는 단백질 변이체 라이브러리를 생성하는 데 매우 적합하다. 합성 재조합 방법에서, 재조합시키고자 하는 복수 개의 유전자를 전체로서 코딩하는 복수 개의 올리고뉴클레오티드가 합성된다. 일부 실시양태에서, 올리고뉴클레오티드는 전체로서 상동성인 모체 유전자로부터 유도된 서열을 코딩한다. 예를 들어, 서열 정렬 프로그램, 예컨대, BLAST를 사용하여 관심의 대상이 되는 상동성 유전자를 정렬한다(예컨대, 문헌 [Atschul, et al., Journal of Molecular Biology, 215:403-410 (1990)] 참조). 상동체 사이의 아미노산 변이에 상응하는 뉴클레오티드가 주목된다. 이러한 변이는 임의적으로는 모체 서열의 공변이 분석, 모체 서열에 대한 기능 정보, 모체 서열 사이의 보존적 또는 비보존적 변이 선별, 또는 다른 적합한 기준에 기초하여 전체 가능한 변이의 서브세트로 추가로 제한된다. 예를 들어, 모체 서열의 공변이 분석, 모체 서열에 대한 기능 정보, 모체 서열 사이의 보존적 또는 비보존적 변이 선별, 또는 변이에 대한 위치의 자명한 내성에 의해 확인된 위치에서의 추가의 아미노산 다양성을 코딩하도록 변이는 임의적으로 추가로 증가된다. 본 결과는 모체 유전자 서열로부터 유도된 컨센서스 아미노산 서열 을 코딩하는 축퇴성 유전자 서열이며, 여기서, 위치의 축퇴성 뉴클레오티드는 아미노산 변이를 코딩한다. 축퇴성 유전자에 존재하는 다양성으 조립하는 데 필요하는 뉴클레오티드를 함유하는 올리고뉴클레오티드가 디자인된다. 이러한 접근법에 대한 상세한 설명은 예를 들어, 문헌 [Ness et al. (2002), Nature Biotechnology, 20:1251-1255], WO 00/42561("Oligonucleotide Mediated Nucleic Acid Recombination"), WO00/42560("Methods for Making Character Strings, Polynucleotides and Polypeptides having Desired Characteristics"), WO 01/75767("In Silico Cross-Over Site Selection"), 및 WO 01/64864("Single-Stranded Nucleic Acid Template-Mediated Recombination and Nucleic Acid Fragment Isolation")(상기 문헌은 각각 본원에서 참조로 포함된다)에서 살펴볼 수 있다. 확인된 폴리뉴클레오티드 변이체 서열은 시험관내 또는 생체내에서 전사 및 번역되어 단백질 변이체 서열의 세트 또는 라이브러리를 생성할 수 있다.
체계적으로 가변된 서열의 세트는 또한 데이터 세트 증의 서열을 정의하는 실험 디자인(DOE) 방법을 사용하여 선험적으로 디자인될 수 있다. DOE 방법에 대한 설명은 문헌 [Diamond, W.J. (2001) Practical Experiment Designs: for Engineers and Scientists, John Wiley & Sons] 및 ["Practical Experimental Design for Engineers and Scientists" by William J Drummond (1981) Van Nostrand Reinhold Co New York], ["Statistics for experimenters" George E.P. Box, William G Hunter and J. Stuart Hunter (1978) John Wiley and Sons, New York], 또는 예컨대, itl.nist.gov/div898/handbook/의 월드 와이드 웹에서 살펴볼 수 있다. 관련된 수학적 계산을 실행하는 데 이용가능한 전산 패키지가 수개 존재하며, 이는 스타티스틱스 툴박스(Statistics Toolbox)(MATLAB®), JMP®, STATISTICA®, 및 STAT-EASE® DESIGN EXPERT®를 포함한다. 결과는 본 발명의 서열-활성 모델을 구축하는 데 적합한 서열의 체계적으로 가변되고, 직교성의 분산된 데이터 세트이다. DOE 기반 데이터 세트 또한 당업계에 공지된 바와 같이 플레킷-버만(Plackett-Burman) 또는 부분 요인 디자인(Fractional Factorial Design)을 이용하여 쉽게 생성될 수 있다.
공학 및 화학에서, 부분 요인 디자인은 전체 요인 디자인과 비교하여 더 소수의 실험을 정의하는 데 사용된다. 상기 방법에서, 인자는 2개 이상의 수준 사이에서 가변된다(즉, "토글링된다"). 최적화 기법을 사용하여 선택된 실험이 확실하게 인자 공간 분산을 나타내는 데 있어 최대로 유익하도록 한다. 같은 디자인 접근법(예컨대, 부분 요인, D-최적 디자인)은 주어진 개수의 위치가 2개 이상의 잔기 사이에서 토글링되는 더욱 적은 개수의 서열을 구성하기 위해 단백질을 조작하는 데 적용될 수 있다. 일부 실시양태에서, 이러한 서열 세트는 해당 단백질 서열 공간에 존재하는 체계적 분산을 최적으로 기술한다.
단백질 조작에 적용되는 DOE 접근법에 대한 예시적인 예는 하기 연산을 포함한다:
1) 본원에 기술된 원리에 기초하여(예컨대, 모체 서열 중의 존재, 보존 수준 등) 토글링 위치를 확인하는 연산;
2) 인자 번호(즉, 변수 위치), 수준 개수(즉, 각각의 위치에서의 선택 사항), 및 실행 실험 개수를 정의함으로써 통상 이용가능한 통계학적 소프트웨어 패키지 중 하나를 이용하여 DOE 실험을 생성하여 출력 행렬을 제공하는 연산. (각 위치의 잔기 선택 사항을 나타내는 1 및 0으로 구성된) 출력 행렬의 정보 내용은 실행 실험 개수에 직접적으로 의존한다(전형적으로 많을수록 더 우수하다);
3) 출력 행렬을 이용하여 1 및 0을 다시 각각의 위치의 특이적 잔기 선택 사항으로 코딩하는 단백질 정렬을 구성하는 연산.
4) 단백질 정렬에 제시된 단백질을 코딩하는 유전자를 합성하는 연산.
5) 관련 어세이(들)로 합성된 유전자에 의해 코딩되는 단백질을 테스트하는 연산.
6) 테스트된 유전자/단백질에 기초하여 모델을 구축하는 연산.
7) 본원에 기술된 단계를 따라 수행함으로써 중요 위치를 확인하고, 적합도가 개선된 하나 이상의 후속 라이브러리를 구축하는 연산.
예시적인 일례에서, 20개의 위치에서 기능적으로 최고인 아미노산 잔기가 결정되는 단백질(예컨대, 각각의 위치에서 이용가능한 2개의 가능한 아미노산이 존재한다)을 조사한다. 본 일례에서, 분해 IV 요인 디자인이 적합할 것이다. 분해 IV 디자인은 중복시키는 2 인자 효과도 없이, 모든 단일 변수의 효과를 설명할 수 있는 디자인으로서 정의된다. 디자인은 220개(~100만개)의 가능한 서열의 전체 다양성을 포괄하는 40개의 특이적 아미노산 서열로 이루어진 세트를 명시할 것이다. 이어서, 임의의 표준 유전자 합성 프로토콜을 사용하여 상기 서열이 생성되고, 이들 클론의 기능 및 적합도가 측정된다.
상기 접근법에 대한 대안은 일부 또는 모든 이용가능한 서열(예컨대, 진뱅크(GENBANK)® 데이터베이스 및 다른 공개 공급원)을 사용하여 단백질 변이체 라이브러리를 제공하는 것이다. 이러한 접근법은 관심의 대상이 되는 서열 공간 영역을 지시해 준다.
C. 서열 분석 방법
역사적으로, 서열 분석은 큰 트레이닝 세트 개발에 있어 제한하는 단계였으며, 그 결과, 강건한 서열-활성 모델은 증가하였다. 서열 변이체에 고비용이 소비되고, 장시간이 소요되기 때문에 관찰값의 개수는 수천개의 변이체로 제한되었다. 다음 세대 서열 분석 도구는 비용을 크게 축소시켰고, 서열 분석 속도 및 부피를 크게 증가시켰으며, 이로써 트레이닝 세트 중에 저활성 및 고활성 변이체, 둘 모두를 포함할 수 있게 되었다.
다음 세대 서열 분석 도구는 1회에 걸쳐 다수의 염기쌍(예컨대, 약 1,000,000,000개 이상의 염기쌍)을 저렴하게 서열 분석할 수 있다. 이러한 능력은 전형적으로 길이가 단지 수 킬로염기쌍인 변이체 단백질을 1회에 걸쳐 서열 분석할 때 사용될 수 있다. 흔히 다음 세대 서열 분석 도구는 다수의 소형 서열보다는 단일의 대형 게놈(예컨대, 인간 게놈)을 1회에 걸쳐 서열 분석하는 데 최적화된 것이다. 다수의 관찰값을 동시에 서열 분석하기 위하여 다음 세대 서열 분석 도구의 잠재능을 실행시키기 위해서는 1회에 걸쳐 서열 분석되는 각각의 관찰값의 기원은 독특하게 확인되어야 한다. 일부 실시양태에서, 바코딩된 서열은 1회차 동안 다음 세대 서열 분석기에 공급된 각각의 모든 단편에서 사용된다. 일례에서, 바코드는 특정 플레이트 상의 특정 웰(예컨대, 96 웰 플레이트)을 독특하게 확인한다. 이러한 실시양태 중 일부에서, 각각의 플레이트의 각각의 웰은 단일의 독특한 변이체를 함유한다. 각각의 변이체, 또는 더욱 구체적으로, 각각의 변이체의 각각의 단편을 바코딩함으로써, 다중의 상이한 변이체의 유전자 서열을 1회에 걸쳐 서열 분석할 수 있고 확인할 수 있다. 프로세스에서, 바코드가 동일한 모든 단편 판독치를 확인하고, 변이체에 대한 서열의 길이를 확인하는 알고리즘에 의해 함께 프로세싱한다.
일부 실시양태에서, 주어진 웰 중의 변이체로의 세포로부터의 DNA를 추출한 후, 단편화시킨다. 이어서, 단편을 바코딩하여 상기 변이체와 관련된, 적어도 웰, 및 때때로 웰 및 플레이트를 확인한다. 이어서, 생성된 단편을 크기 선별하여 다음 세대 서열 분석기에 적절한 길이의 서열을 제조한다. 한 예시적인 일례에서, 판독 길이는 약 200개의 염기쌍 길이다. 일부 실시양태에서, 먼저 플레이트의 다양한 웰로부터의 DNA 단편을 풀링한 후까지는 플레이트 바코드를 적용시키지 않는다. 이어서, 풀링된 DNA를 바코딩하여 플레이트를 확인한다. 일부 실시양태에서, 단편이 어떤 웰로부터 유도되었는지와는 상관없이, 각각의 단편은 동일한 플레이트 바코드를 가질 것이다. 그러나, 일부 대체 실시양태에서, 단편은 상이한 바코드를 가진다. 추가로, 웰 및 플레이트 바코드는 주어진 웰로부터 추출된 DNA를 확인하기 위해 적용될 수 있다.
하나 이상의 실시양태에서, 서열 데이터는 예를 들어, 제1 세대 서열 분석 방법으로 간주되는 것인 생어(Sanger) 서열 분석 또는 맥삼-길버트(Maxam-Gilbert) 서열 분석을 비롯한 벌크 서열 분석 방법을 사용하여 수득될 수 있다. 표지화된 디데옥시 쇄 종결인자를 사용하는 것을 포함하는 생어 서열 분석은 당업계에 주지되어 있으며; 예컨대, 문헌 [Sanger et al., Proceedings of the National Academy of Sciences of the United States of America 74, 5463-5467 (1997)]을 참조할 수 있다. 핵산 샘플 분획에 대해 다중의 부분 화학적 분해 반응을 수행한 후, 단편을 검출하고 분석하고 서열 추론해내는 것을 포함하는 맥삼-길버트 서열 분석 또한 당업계에 주지되어 있다; 예컨대, 문헌 [Maxam et al., Proceedings of the National Academy of Sciences of the United States of America 74, 560-564 (1977)]을 참조할 수 있다. 또 다른 벌크 서열 분석 방법은 샘플의 서열이 예컨대, 마이크로어레이 또는 유전자 칩 상의 복수 개의 서열에 대한 그의 하이브리드화 특성에 기초하여 도출되는 것인, 하이브리드화에 의한 서열 분석이다; 예컨대, 문헌 [Drmanac, et al, Nature Biotechnology 16, 54-58 (1998)]을 참조할 수 있다.
하나 이상의 실시양태에서, 서열 데이터는 다음 세대 서열 분석 방법을 사용하여 얻는다. 다음 세대 서열 분석은 또한 "고처리량 서열 분석"으로도 지칭된다. 상기 기법은 서열 분석 프로세스을 병행하여 수천 또는 수백만 개의 서열을 한번에 제조한다. 적합한 다음 세대 서열 분석 방법의 예로는 단일 분자 실시간 서열 분석(예컨대, 퍼시픽 바이오사이언시스: 미국 캘리포니아주 멘로 파크), 이온 반도체 서열 분석(예컨대, 이온 토렌트: 미국 캘리포니아주 사우쓰 샌프란시스코), 파이로시퀀싱(예컨대, 454, 미국 코네티컷 브래드포드), 결찰에 의한 서열 분석(예컨대, 라이프 테크놀러지즈(Life Technologies: 미국 캘리포니아주 칼즈배드)의 SOLid 서열 분석), 합성 및 가역성 종결인자에 의한 서열 분석(예컨대, 일루미나: 미국 캘리포니아주 샌디에고), 핵산 영상화 기술, 예컨대, 투과 전자 현미경법 등을 포함하나, 이에 한정되지 않는다,
일반적으로, 다음 세대 서열 분석 방법은 전형적으로 개별 DNA 분자를 증폭시키는 시험관내 클로닝 단계를 사용한다. 에멀젼 PCR(emPCR: Emulsion PCR)은 오일상 내의 수성 소적 중의 프라이머로 코팅된 비드와 함께 개별 DNA 분자를 단리시킨다. PCR을 통해 비드 상의 프라이머에 결합하는 DNA 분자 카피가 제조되고, 이후 추후 서열 분석을 위해 고정화시킨다. emPCR은 (Marguilis) 등에 의한(454 라이프 사이언시즈(454 Life Sciences: 미국 코네티컷 브래드포드), (Shendure 및 Porreca 등에 의한(이는 또한 "폴로니(polony) 서열 분석"으로도 알려져 있다) 방법, 및 SOLiD 서열 분석(어플라이드 바이오시스템즈 인코포레이티드(Applied Biosystems Inc.: 미국 캘리포니아주 포스터 시티)에서 사용된다. 문헌 [M. Margulies, et al. (2005) "Genome sequencing in microfabricated high-density picolitre reactors" Nature 437: 376-380]; [J. Shendure, et al. (2005) "Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome" Science 309 (5741): 1728-1732]를 참조할 수 있다. 시험관내 클론 증폭 또한 고체 표면에 부착된 프라이머 상에서 단편이 증폭되는, "브릿지 PCR"에 의해 수행될 수 있다. (Braslavsky) 등은 DNA 분자를 표면에 직접 고정시키면서, 상기 증폭 단계는 생략한, 단일 분자 방법(헬리코스 바이오사이언시스 코포레이션(Helicos Biosciences Corp.: 미국 매사추세츠 케임브리지)에 의해 상업화됨)를 개발하였다(문헌 [I. Braslavsky, et al. (2003) "Sequence information can be obtained from single DNA molecules" Proceedings of the National Academy of Sciences of the United States of America 100: 3960-3964]).
물리적으로 표면에 결합된 DNA 분자는 동시에 서열 분석될 수 있다. "합성에 의한 서열 분석"에서, 상보적 가닥은 염료 종결 전기영동 서열 분석과 같이 DNA 폴리머라제를 사용하여 주형 가닥의 서열에 기초하여 구축된다. 가역적 종결인자 방법(일루미나(미국 캘리포니아주 샌디에고) 및 헬리코스 바이오사이언시스 코포레이션(미국 매사추세츠 케임브리지)에 의해 상업화됨)은 가역성 버전의 염료 종결인자를 사용하여 뉴클레오티드를 한번에 하나씩 부가하고, 또 다른 뉴클레오티드가 중합화될 수 있도록 차단기를 반복적으로 제거함으로써 실시간으로 각각의 위치의 형광을 검출한다. "파이로시퀀싱" 또한 DNA 중합화를 사용하여 뉴클레오티드를 한번에 하나씩 부가하고, 부착된 피로포스페이트의 유리에 의해 방출된 빛을 통해 주어진 위치에 부가된 뉴클레오티드를 검출하고, 그 개수를 정량화한다(454 라이프 사이언시즈(미국 코네티컷 브래드포드)에 의해 상업화됨). 문헌 [M. Ronaghi, et al. (1996). "Real-time DNA sequencing using detection of pyrophosphate release" Analytical Biochemistry 242: 84-89]를 참조할 수 있다.
다음 세대 서열 분석 방법의 구체적인 예는 하기에 추가로 상세하게 기술된다. 본 발명의 하나 이상의 실행은 본 발명의 원리로부터 벗어남 없이 하기 서열 분석 방법 중 하나 이상의 것을 사용할 수 있다.
단일 분자 실시간 서열 분석(SMRT: single molecule real time sequencing로도 알려져 있다)은 퍼시픽 바이오사이언시스에 의해 개발된 합성 기술에 의한 병행 단일 분자 DNA 서열 분석이다. 단일 분자 실시간 서열 분석은 제로 모드 도파관(ZMW: zero-mode waveguide)을 사용하였다. 단일 DNA 폴리머라제 효소는 ZMW 바닥에 고정되고, 여기서, DNA 단일 분자는 주형으로서 사용된다. ZMW는 DNA 폴리머라제에 의해 도입되는 DNA(이는 또한 염기로도 알려져 있다)의 단일 뉴클레오티드만을 관찰하는 데 충분한 정도로 작은 조사된 관찰 부피를 생성하는 구조물이다. 4개의 DNA 염기 각각이 4개의 상이한 형광성 염료 중 하나에 부착된다. DNA 폴리머라제에 의해 뉴클레오티드가 도입되었을 때, 형광성 태그는 절단되고, 그의 형광이 더 이상은 관찰될 수 없는 ZMW의 관찰 영역 밖으로 확산된다. 검출기는 뉴클레오티드 도입의 형광성 신호를 검출하고, 염료의 상응하는 형광성에 따라 염기는 결정된다(base call).
적용가능한 또 다른 단일 분자 서열 분석 기술은 (예컨대, 문헌 [Harris T.D. et al., Science 320: 106-109 [2008]]에 기술되어 있는 바와 같이) 헬리코스 트루 싱글 몰레큘 시퀀싱(tSMS: Helicos True Single Molecule Sequencing) 기술이다. tSMS 기법에서, DNA 샘플을 대략 100 내지 200개의 뉴클레오티드로 이루어진 가닥으로 절단하고, 폴리A 서열을 각각의 DNA 가닥의 3' 단부에 부가한다. 각각의 가닥을 형광으로 표지화된 아데노신 뉴클레오티드의 부가에 의해 표지화한다. 이어서, DNA 가닥을, 유세포 표면에 고정화되어 있는 수백만 개의 올리고 T 포획 부위를 포함하는 유세포에 하이브리드화시킨다. 특정 실시양태에서, 주형의 밀도는 약 1억개의 주형/㎠일 수 있다. 이어서, 유세포를 장치, 예컨대, 헬리스코프(HeliScope)™ 서열 분석기에 로딩하고, 레이저를 유세포의 표면에 조사하여 각각의 주형의 위치를 밝혀낸다. CCD 카메라는 유세포 표면상의 주형의 위치를 지도화할 수 있다. 이어서, 주형 형광성 표지를 절단하고, 세척해 낸다. DNA 폴리머라제 및 형광으로 표지화된 뉴클레오티드를 도입함으로써 서열 분석 반응을 시작한다. 올리고 T 핵산이 프라이머로서의 역할을 한다. 폴리머라제는 표지화된 뉴클레오티드를 주형 지정된 방식으로 프라이머에 도입한다. 폴리머라제 및 도입되지 않은 뉴클레오티드를 제거한다. 형광으로 표지화된 뉴클레오티드가 지정된 방식으로 도입되어 있는 주형을 유세포 표면을 영상화함으로써 식별한다. 영상화한 후, 절단 단계를 통해 형광성 표지를 제거하고, 원하는 판독 길이에 도달할 때까지 다른 형광으로 표지화된 뉴클레오티드를 이용하여 프로세스를 반복한다. 각각의 뉴클레오티드 부가 단계를 이용하여 서열 정보를 수집한다. 단일 분자 서열 분석 기술에 의한 전체 게놈 서열 분석은 서열 분석 라이브러리 제조에서 PCR 기반 증폭을 제외시키거나, 또는 전형적으로 배제하고, 본 방법을 통해서는 샘플 카피를 측정하기보다는 샘플을 직접적으로 측정할 수 있다.
이온 반도체 서열 분석은 DNA의 중합화 동안 유리되는 수소 이온의 검출에 기초하여 DNA 서열 분석 방법이다. 이는 "합성에 의한 서열 분석" 방법으로서, 그 동안 상보적 가닥은 주형 가닥의 서열에 기초하여 구축된다. 서열 분석하고자 하는 주형 DNA 가닥을 함유하는 마이크로웰에 단일 종의 데옥시리보뉴클레오티드 트리포스페이트(dNTP: deoxyribonucleotide triphosphate)를 가득 채운다. 도입된 dNTP이 선도 주형 뉴클레오티드에 상보적일 경우, 성장하는 상보적 가닥 내로 도입된다. 이는 반응이 발생하였음을 나타내는 ISFET 이온 센서를 촉발시키는 수소 이온을 유리시킨다. 주형 서열에 동종중합체 반복부가 존재할 경우, 다중 dNTP 분자는 단일 사이클에서 도입될 것이다. 이로써 상응하는 개수의 수소가 방출되고, 비례하여 더 높은 전기 신호가 발생하게 된다. 이러한 기술은, 변형된 뉴클레오티드 또는 광학이 사용되지 않는다는 점에서 다른 서열 분석 기술과는 상이하다. 이온 반도체 서열 분석은 또한 이온 토렌트 서열 분석, pH 매개 서열 분석, 실리콘 서열 분석, 또는 반도체 서열 분석으로도 지칭될 수 있다.
파이로시퀀싱에서, 중합화 반응에 의해 유리되는 피로포스페이트 이온은 ATP 술푸릴라제에 의해 아데노신 5' 포스포술페이트와 반응하여 ATP를 제조하고; 이어서, ATP는 루시퍼라제에 의해 루시페린의 옥시루시페린 + 빛으로의 전환을 구동시킨다. 형광은 일시적이기 때문에, 본 방법에서는 형광을 제거하는 분리 단계는 필요 없다. 한번에 한 유형의 데옥시리보뉴클레오티드 트리포스페이트(dNTP)가 부가되고, 서열 정보는 dNTP가 반응 부위에서 유의적인 신호를 생성하는 것에 따라 식별된다. 상업적으로 이용가능한 로슈(Roche) GS FLX 장치는 본 방법을 사용하여 서열을 획득한다. 상기 기법 및 그의 적용은 예를 들어, 문헌 [Ronaghi et al., Analytical Biochemistry 242, 84-89 (1996)] 및 [Margulies et al., Nature 437, 376-380 (2005)]([Nature 441, 120 (2006)]의 정오표)에서 상세하게 논의되고 있다. 상업적으로 이용가능한 파이로시퀀싱 기술은 (예컨대, 문헌 [Margulies, M. et al. Nature 437:376-380 [2005]]에 기술되어 있는 것과 같은) 454 서열 분석(로슈)이다.
결찰 서열 분석에서, 오버행이 있는 부분적으로 이중 가닥 올리고뉴클레오티드를 오버행을 가진, 서열 분석되는 핵산에 연결하는 데 리가제 효소가 사용되며; 결찰이 이루어지도록 하기 위해서는 오버행은 상보적이어야 한다. 부분적으로 이중 가닥 올리고뉴클레오티드 중 염기는 부분적으로 이중 가닥 올리고뉴클레오티드 및/또는 부분적으로 이중 가닥 올리고뉴클레오티드의 또 다른 부분에 하이브리드화된 제2 올리고뉴클레오티드에 컨쥬게이트된 형광단에 따라 확인될 수 있다. 형광 데이터를 획득한 후, (부분적으로 이중 가닥 올리고뉴클레오티드에 포함되어 있던) 그의 인식 부위로부터 고정된 거리에 있는 부위에서 절단하는 예컨대, II형 제한 효소, 예를 들어, Bbvl에 의해 결찰된 복합체를 결찰 부위의 상류쪽에서 절단한다. 이러한 절단 반응은 이전 오버행의 상류쪽으로 바로 옆에 있는 신규 오버행을 노출시키고, 프로세스는 반복된다. 상기 기법 및 그의 적용은 예를 들어,문헌 [Brenner et al., Nature Biotechnology 18, 630-634 (2000)]에서 상세하게 논의된다. 일부 실시양태에서, 결찰 서열 분석은 환형 핵산 분자의 회전환 증폭 생성물을 수득하고, 결찰 서열 분석을 위한 주형으로서 회전환 증폭 생성물을 사용함으로써 본 발명의 방법에 적합화된다.
결찰 서열 분석 기술의 상업적으로 이용가능한 예는 SOLiD™ 기술(어플라이드 바이오시스템즈(Applied Biosystems))이다. 결찰에 의한 SOLiD™ 서열 분석에서, 게놈 DNA를 단편으로 전단하고, 어댑터를 단편의 5' 및 3' 단부에 부착시켜 단편 라이브러리를 생성한다. 별법으로, 어댑터를 단편의 5' 및 3' 단부에 결찰시키고, 단편을 환형화하고, 환형화된 단편을 분해하여 내부 어댑터를 생성하고, 어댑터를 생성된 단편의 5' 및 3' 단부에 부착시켜 짝을 이루어 쌍으로 형성된 라이브러리를 생성함으로써 내부 어댑터를 도입시킬 수 있다. 이어서, 비드, 프라이머, 주형, 및 PCR 성분을 함유하는 마이크로반응기에서 클론 비드 집단을 제조한다. PCR 후, 주형을 변성시키고, 비드를 강화시켜 주형 연장된 비드를 분리시킨다. 선택된 비드 상의 주형을 3' 변형시켜 유리 슬라이드에 결합할 수 있도록 만든다. 순차적인 하이브리드화, 및 특이적 형광단에 의해 확인되는 중앙 결정된 염기(또는 염기쌍)와 부분적 무작위 올리고뉴클레오티드의 결찰에 의해 서열을 결정할 수 있다. 색상을 기록하고, 결찰된 올리고뉴클레오티드를 절단하고, 제거하고, 이어서, 프로세스를 반복한다.
가역성 종결인자 서열 분석에서, 차단기의 존재에 기인하여 가역성 쇄 종결인자인 것인 형광성 염료로 표지화된 뉴클레오티드 유사체를 단일 염기 연장 반응에서 도입한다. 염기의 아이덴티티는 형광단에 따라 측정되고; 다시 말해, 각각의 염기는 상이한 형광단과 쌍을 형성한다. 형광/서열 데이터를 획득한 후, 형광단 및 차단기를 화학적으로 제거하고, 다음 염기의 서열 정보를 획득할 때까지 사이클을 반복하다. 일루미나 GA 장치는 상기 방법에 의해 작동한다. 상기 기법 및 그의 적용은 예를 들어, 문헌 [Ruparel et al., Proceedings of the National Academy of Sciences of the United States of America 102, 5932-5937 (2005)], 및 [Harris et al., Science 320, 106-109 (2008)]에서 상세하게 기술된다.
가역성 종결인자 서열 분석 방법의 상업적으로 이용가능한 예는 (예컨대, 문헌 [Bentley et al., Nature 6:53-59 [2009]]에 기술되어 있는 바와 같은) 일루미나의 합서에 의한 서열 분석 및 가역성 종결인자 기반 분석이다. 일루미나의 서열 분석 기술은 올리고뉴클레오티드 앵커가 결합되는 평면형인, 광학적으로 투명한 표면에의 단편화된 게놈 DNA의 부착에 의존한다. 주형 DNA는 단부를 수복시켜 5' 인산화된 블런트 단부를 생성하고, 클레노우(Klenow) 단편의 폴리머라제 활성을 사용하여 단일 A 염기를 블런트 인산화된 DNA 단편의 3' 단부에 부가한다. 이러한 부가를 통해 올리고뉴클레오티드 어댑터에의 결찰을 위한 DNA 단편이 제조되며, 이는 결찰율을 증가시키기 위해 그의 3' 단부에 단일 T 염기로 이루어진 오버행을 가진다. 어댑터 올리고뉴클레오티드는 유세포 앵커에 상보적이다. 제한 희석 조건하에서, 어댑터 변형된 단일 가닥 주형 DNA를 유세포에 부가하고, 하이브리드화에 의해 앵커에 고정화시킨다. 부착된 DNA 단편을 연장시키고, 브릿지 증폭시켜 각각 ~1,000개의 같은 주형의 카피를 함유하는, 수억개의 클러스터를 가지는 초고밀도 서열 분석 유세포를 생성한다. 제거가능한 형광성 염료와 함께 가역성 종결인자를 아용하는 합성에 의한 강건한 4색 DNA 서열 분석 기술을 사용하여 주형을 서열 분석한다. 레이저 여기 및 내부 전반사 광학을 이용하여 고밀도 형광 검출을 달성한다. 약 20-40 bp, 예컨대 36 bp의 짧은 서열 리드(reads)를 반복 차폐된 참조 게놈에 대해 정렬하고, 특수 개발된 데이터 분석 파이프라인 소프트웨어를 이용하여 짧은 서열 리드의 참조 게놈에 대한 독특한 지도화를 확인한다. 비반복 차폐된 참조 게놈 또한 사용될 수 있다. 반복 차폐된 또는 비반복 차폐된 참조 게놈이 사용되는지 여부와는 상관없이, 오직 참조 게놈에 대하여 독특하게 지도화된 리드만이 계수된다. 제2 리드 완료 후, 주형은 계내에서 재생됨으로써 단편의 반대쪽 단부로부터 제2 리드가 이루어질 수 있다. 따라서, DNA 단편에 대한 단일 단부 또는 쌍을 이룬 단부의 서열 분석이 사용될 수 있다. 샘플 중에 존재하는 DNA 단편의 부분적인 서열 분석이 수행되고, 길이가 미리 결정된, 약 36 bp의 서열 태그를 포함하는 리드가 공지된 참조 게놈에 대해 지도화되고 계수된다.
나노포어 서열 분석에서, 단일 가닥 핵산 분자는 예컨대, 전기영동 구동력을 사용하여 포어를 통해 트레딩되고, 서열은 단일 가닥 핵산 분자가 포어를 통해 통과함에 따라 수득되는 데이터를 분석함으로써 도출된다. 데이터는 이온 전류 데이터일 수 있고, 여기서, 각각의 염기는 예컨대, 포어를 통해 통과하는 전류를 상이하게 식별가능한 정도로 부분적으로 차단함으로써 전류를 변경시킨다.
또 다른 예시적인, 그러나, 비제한적인 실시양태에서, 본원에 기술된 본 방법은 투과 전자 현미경법(TEM: transmission electron microscopy)을 사용하여 서열 정보를 수득하는 것을 포함한다. 본 방법은 중원자 마커로 선택적으로 표지화된 고분자량(150 kb 이상) DNA를 단일 원자 해상도 투과 전자 현미경으로 영상화하고, 이들 분자를 초박 필름상에 과조밀(가닥 사이 3 nm) 평행 어레이로 염기 사이의 이격 거리는 일관되게 하여 배열하는 것을 포함한다. 전자 현미경을 사용하여 필름 상의 분자를 영상화하여 중원자 마커의 위치를 측정하고, DNA로부터 베이스 서열 정보를 추출한다. 본 방법은 PCR 특허 공개 WO 2009/046445에 추가로 기술되어 있다.
또 다른 예시적인, 그러나, 비제한적인 실시양태에서, 본원에 기술된 본 방법은 제3 세대 서열 분석을 사용하여 서열 정보를 수득하는 것을 포함한다. 제3 세대 서열 분석에서, 다수의 작은 (~50 nm) 홀이 있는, 알루미늄 코팅을 포함하는 슬라이드가 제로 모드 도파관으로서 사용된다(예컨대, 문헌 [Levene et al., Science 299, 682-686 (2003)] 참조). 알루미늄 표면은 폴리포스포네이트 화학법, 예컨대, 폴리비닐포스포네이트 화학법에 의해 DNA 폴리머라제의 부착으로부터 보호된다(예컨대, 문헌 [Korlach et al., Proceedings of the National Academy of Sciences of the United States of America 105, 1176-1181 (2008)] 참조). 이로써 DNA 폴리머라제 분자는 알루미늄 코팅의 홀 중 노출된 실리카에 우선적으로 부착된다. 이러한 구성으로 사용되는 소산파 현상은 형광 배경을 감소시킬 수 있으며, 이로써 보다 고농도의 형광으로 표지화된 dNTP가 사용될 수 있다. 형광단은 dNTP의 말단 포스페이트에 부착되고, 이로써 형광은 dNTP 도입시에 방출되지만, 형광단은 새로 도입된 뉴클레오티드에 부착된 상태로 남아있지 않으며, 이는 복합체가 또 다른 회차의 도입을 위해 즉시 사용될 준비가 되어 있음을 의미한다. 이 방법에 의해, 알루미늄 코팅의 홀에 존재하는 개별 프라이머-주형 복합체 내로의 dNTP 도입이 검출될 수 있다. 예컨대, 문헌 [Eid et al., Science 323, 133-138 (2009)]를 참조할 수 있다.
D. 서열-활성 모델 생성
상기 언급된 바와 같이, 본원의 실시양태와 함께 사용되는 서열-활성 모델은 단백질 활성에 대한 단백질 서열 정보에 관한 것이다. 모델에 의해 사용되는 단백질 서열 정보는 많은 형태를 취할 수 있다. 일부 실시양태에서, 단백질 중 아미노산 잔기의 완전한 서열이다(예컨대, HGPVFSTGGA...). 그러나, 일부 실시양태에서, 완전한 아미노산 서열은 불필요하다. 예를 들어, 일부 실시양태에서, 특정 연구 노력으로 가변시키고자 하는 잔기만을 제공하기만 하면 충분하다. 후기 연구 단계를 포함하는 일부 실시양태에서, 다수의 잔기가 고정되고, 서열 공간 중 단지 제한된 영역만이 탐색 상태로 유지된다. 상기와 같은 상황 중 일부에서, 입력값으로서 오직, 탐색이 계속 진행되는 단백질 영역 중의 상기 잔기 확인만을 필요로 하는 서열-활성 모델을 제공하는 것이 편리하다. 일부 추가의 실시양태에서, 모델은 잔기 위치의 잔기의 정확한 아이덴티티를 알아야 하는 것을 요하지 않는다. 상기 일부 실시양태에서, 특정 잔기 위치의 아미노산을 특징화하는 하나 이상의 물리적 또는 화학적 특성(들)을 확인한다. 한 예시적인 일례에서, 모델은 벌크, 소수성, 산성 등에 의해 잔기 위치를 명시할 것을 필요로 한다. 추가로, 일부 모델에서, 상기 특성의 조합이 사용된다. 실제로, 모델이 서열 정보, 활성 정보 및/또는 다른 물리적 특성(예컨대, 소수성 등)의 다양한 구성에서 사용될 수 있다는 것을 알 수 있는 바, 본 발명을 임의의 특정 접근법으로 한정하고자 하지 않는다.
따라서, 서열-활성 모델의 형태는, 그가 원하는 바에 따라 서열 정보에 기초하여 단백질의 상대적인 활성에 가까운 근사값을 정확하게 구하기 위한 비히클을 제공하는 한, 광범위하게 달라질 수 있다. 일부 실시양태에서, 모델은 일반적으로 활성을 종속 변수로서, 및/또는 서열/잔기 값을 독립 변수로서 처리한다. 수학적/논리적 형태의 모델에 대한 예로는 다양한 차수의 선형 및 비선형 수학식, 신경망, 분류 및 회귀 트리/그래프, 클러스터링 접근법, 반복 분할, 지지 벡터 기계 등을 포함한다. 한 실시양태에서, 모델 형태는 계수 및 잔기 값의 곱을 합산하는 선형 추가형 모델이다. 또 다른 실시양태에서, 모델 형태는 (잔기 사이의 상호작용 항을 나타내는) 특정 잔기 외적을 비롯한, 다양한 서열/잔기 항의 비선형 곱이다. 실제로, 본원에 예시된 바와 같이, 임의의 적합한 포맷이 사용될 수 있다는 것을 알 수 있는 바, 개시된 실시양태는 임의의 특정 포맷으로 한정하고자 하지 않는다.
일부 실시양태에서, 모델은 활성과 서열 사이의 수학적/논리적 관계를 제공하는 활성 대 서열 정보의 트레이닝 세트로부터 개발된다. 상기 관계는 전형적으로 신규 서열의 활성 또는 잔기가 관심의 대상이 되는 활성에 미치는 영향을 예측하기 위해 사용 이전에 검증된다.
모델을 생성하는 데 다양한 기법이 이용가능하고, 본 발명에 사용될 수 있다는 것을 알 수 있다. 일부 실시양태에서, 기법은 모델의 최적화 또는 모델 오류의 최소화를 포함한다. 구체적인 예로는 부분 최소 제곱, 앙상블 회귀, 랜덤 포레스트, 각종의 다른 회귀 기법 뿐만 아니라, 신경망 기법, 반복 분할, 지지 벡터 기계 기법, CART(분류 및 회귀 트리: classification and regression tree) 등을 포함한다. 일반적으로, 기법은 활성에 유의적인 영향을 미치는 잔기와 그렇지 못한 잔기를 구별할 수 있는 모델을 생성하여야 한다. 일부 실시양태에서, 모델은 개별 잔기 또는 잔기 위치가 활성에 미치는 영향에 기초하여 그를 순위화한다. 당업계에 공지된 임의의 적합한 방법이 본 발명에서 사용될 수 있다는 것을 알 수 있는 바, 본 발명을 모델을 생성하는 임의의 구체적인 기법으로 한정하고자 하지 않는다.
일부 실시양태에서, 모델은 트레이닝 세트에서 독립 변수 및 종속 변수의 공변동을 확인하는 회귀 기법에 의해 생성된다. 다양한 회귀 기법이 공지되어 있고, 널리 사용된다. 예로는 다중 선형 회귀(MLR), 주성분 회귀(PCR) 및 부분 최소 제곱 회귀(PLS)를 포함한다. 일부 실시양태에서, 모델은 앙상블 회귀 및 랜덤 포레스트를 포함하나, 이에 한정되지 않는 다중 구성 요소를 포함하는 기법을 사용하여 생성된다. 상기 방법 및 임의의 다른 적합한 방법이 본 발명에 사용될 수 있다는 것을 알 수 있다. 본 발명을 임의의 특정 기법으로 한정하고자 하지 않는다.
MLR이 상기 기법들 중 가장 기본적인 것이다. 이는 트레이닝 세트의 구성원에 대한 한 세트의 계수 방정식을 간단하게 풀이하는 데 사용된다. 각각의 방정식은 특정 위치에의 특정 잔기의 존재 또는 부재(즉, 독립 변수)와 트레이닝 세트 구성원의 활성(즉, 종속 변수)에 관한 것이다. 트레이닝 세트 중 잔기 옵션의 개수에 따라, 상기 방정식의 계수는 매우 커질 수 있다.
MLR과 같이, PLS 및 PCR은 활성 활성을 잔기 값과 관련시키는 방정식으로부터 모델을 생성한다. 그러나, 상기 기법은 상이한 방식으로 이를 수행한다. 먼저 독립 변수의 개수를 감소시키는 좌표 변환을 실행한다. 이어서, 변환된 변수에 대한 회귀를 실행한다. MLR에서, 잠재적으로 매우 많은 독립 변수가 존재하며: 각각의 잔기 위치에 대해 2개 이상의 것이 트레이닝 세트 내에서 달라질 수 있다. 관심의 대상이 되는 단백질 및 펩티드가 흔히 매우 많고, 트레이닝 세트가 다수의 상이한 서열을 제공할 수 있다고 가정할 때, 독립 변수의 개수는 빠르게 매우 많아질 수 있다. 데이터 세트에서 가장 많은 변이를 제공하는 것에 주력하기 위해 변수의 개수를 감소시킴으로써, PLS 및 PCR은 일반적으로 더 적은 개수의 샘플을 필요로 하고, 모델을 생성하는 데 관여하는 단계를 간소화한다.
PCR은 원시 독립 변수(즉, 잔기 값)의 좌표 변환에 의해 얻은 상대적으로 적은 개수의 잠재 변수에 대해 실제 회귀가 수행된다는 점에서 PLS 회귀와 유사하다. PLS와 PCR 사이의 차이는 PCR에서 잠재 변수는 독립 변수(즉, 잔기 값) 사이의 공변동을 최대화시킴으로써 구성된다는 점이다. PLS 회귀에서, 잠재 변수는 독립 변수와 종속 변수(즉, 활성 값) 사이의 공변동을 최대화시키는 방식으로 구성된다. 부분 최소 제곱 회귀는 문헌 [Hand, D.J., et al. (2001) Principles of Data Mining (Adaptive Computation and Machine Learning), Boston, MA, MIT Press], 및 [Geladi, et al. (1986) "Partial Least-Squares Regression: a Tutorial," Analvtica Chimica Acta, 198:1-17]에 기술되어 있다. 상기 두 문헌 모두 모든 목적을 위해 본원에서 참조로 포함된다.
PCR 및 PLS에서, 회귀 분석의 직접적인 결과는 가중화된 잠재 변수의 함수인 활성에 대한 식이다. 상기 식은 잠재 변수를 다시 원래의 독립 변수로 역 전환시키는 좌표 변환을 실행함으로써 원래의 독립 변수의 함수로서 활성에 대한 식으로 변환될 수 있다.
본질적으로, PCR 및 PLS, 둘 모두는 먼저 트레이닝 세트에 포함된 정보의 차원수를 감소시킨 후, 변환되어 신규 독립 변수를 산출하였지만, 원래의 종속 변수 값은 보존하는 것인 변환된 데이터 세트에 대한 회귀 분석을 실행한다. 데이터 세트의 변환된 버전을 통해서는 오직 회귀 분석을 실행하기 위한 상대적으로 소수의 방정식만을 얻을 수 있다. 차원수를 감소시키는 것을 실행하지 않는 프로토콜에서, 그에 대한 변이가 존재할 수 있는 각각의 개별 잔기가 고려되어야 한다. 이는 계수의 매우 큰 세트일 수 있다(예컨대, 2원 상호작용의 경우, 2N 계수, 여기서, N은 트레이닝 세트에서 달라질 수 있는 잔기 위치의 개수이다). 전형적인 주성분 분석에서, 단지 3, 4, 5, 6개의 주성분만이 사용된다.
트레이닝 데이터를 적합화할 수 있는 기계 학습 기법의 능력은 흔히 "모델 피트"로 지칭되고, 회귀 기법, 예컨대, MLR, PCR 및 PLS에서, 모델 피트는 전형적으로 측정값과 예측값 사이의 제곱 차의 합에 의해 측정된다. 주어진 트레이닝 세트의 경우, 최적의 모델 피트는 MLR을 사용하여 획득될 것이며, PCR 및 PLS를 사용한 경우에는 흔히 성능이 더 떨어지는 모델 피트(측정치와 예측치 사이의 제곱 오차의 합이 더 높은 것)를 획득하게 될 것이다. 그러나, 잠재 변수 회귀 기법, 예컨대, PCR 및 PLS를 사용하였을 때의 주된 장점은 상기 모델의 예측 능력에 있다. 제곱 오차의 합이 매우 작은 모델 피트를 수득하는 것은 결코 모델이 트레이닝 세트에서 볼 수 없었던 신규 샘플을 정확하게 예측할 수 있을 것이라는 것을 보장하지는 못한다-사실상, 흔히 그 반대의 경우로, 특히, 다수의 변수 및 오직 소수의 관찰값(즉, 샘플)이 존재하는 경우에 그러하다. 따라서, 잠재 변수 회귀 기법(예컨대, PCR, PLS)은 흔히 트레이닝 데이터에 성능이 더 떨어지는 모델 피트를 가짐과 동시에, 일반적으로는 더욱 강건하고, 트레이닝 세트 이외의 신규 샘플을 더욱 정확하게 예측할 수 있다.
본 개시내용에 따라 모델을 생성하는 데 사용될 수 있는 또 다른 부류의 도구는 지지 벡터 기계(SVM: support vector machine)이다. 상기 수학적 도구는 입력값으로서 활성에 기초하여 2개 이상의 군으로 분류된 서열의 트레이닝 세트를 취한다. 지지 벡터 기계는 트레이닝 세트의 "활성" 및 "불활성" 구성원을 분리하는 하이퍼플레인 인터페이스에 얼마나 가까운지에 따라 다르게 의존하여 트레이닝 세트의 상이한 구성원에 가중치를 부여함으로써 작동한다. 상기 기법에서 과학자는 먼저 어느 트레이닝 세트 구성원을 "활성" 군에 배치하여야 할지, 및 어느 트레이닝 세트 구성원을 "불활성" 군에 배치하여야 할지를 결정하여야 한다. 일부 실시양태에서, 이는 트레이닝 세트의 "활성" 구성원과 "불활성" 구성원 사이의 경계로서의 역할을 하는 활성 수준에 대한 적절한 수치값을 선택함으로써 달성된다. 이러한 분류로부터, 지지 벡터 기계는 트레이닝 세트 중 활성 군과 불활성 군의 구성원의 서열을 정의하는 개별 독립 변수에 대한 계수 값을 제공할 수 있는 벡터 W를 생성한다. 상기 계수는 본원 다른 곳에 기술된 바와 같이 개별 잔기를 "순위화"하는 데 사용될 수 있다. 기법은 하이퍼플레인의 맞은편 상의 가장 가까이 있는 트레이닝 세트 구성원 사이의 거리를 최대화시키기는 하이퍼플레인을 확인하는 데 사용된다. 또 다른 실시양태에서, 지지 벡터 회귀 모델링을 수행한다. 이 경우, 종속 변수는 연속 활성 값의 벡터이다. 지지 벡터 회귀 모델은, 개별 잔기를 순위화하는 데 사용될 수 있는 계수 벡터, W를 생성한다.
SVM은 다수의 연구에서 큰 데이터 세트를 검토하는 데 사용되어 왔으며, DNA 마이크로어레이와 함께 널리 사용되어 왔음을 알 수 있다. 그의 잠재적인 강도로는 샘플을 서로로부터 분리하는 인자를 (가중치를 부여함으로써) 정밀하게 구별할 수 있는 능력을 포함한다. SVM이 어느 잔기가 기능에 기여하는지를 정확하게 알아낼 수 있을 정도로 이는 잔기를 순위화하는 데 있어 특히 유용한 도구가 될 수 있다. SVM은 문헌 [S. Gunn (1998) "Support Vector Machines for Classification and Regressions," Technical Report, Faculty of Engineering and Applied Science, Department of Electronics and Computer Science, University of Southampton](이는 모든 목적을 위해 본원에서 참조로 포함된다)에 기술되어 있다.
본 발명의 일부 실시양태에서, 모델을 생성하는 데 사용될 수 있는 또 다른 부류의 도구는 랜덤 입력값을 사용하는 분류 트리의 앙상블에 기초하는 분류 및 회귀이며, 그의 일례로는 랜덤 포레스트가 있다. 문헌 [Breiman (2001). "Random Forests," Machine Learning 45 (1): 5-32]를 참조할 수 있다. 랜덤 포레스트는 각각의 트리가 독립적으로 샘플링되고, 포레스트 중의 모든 트리에 대하여 같은 분포를 가지는 랜덤 벡터의 값에 의존하도록 하는 트리 예측 인자의 조합이다. 랜덤 포레스트는 의사 결정 트리의 각각의 스플릿에 무작위로 선택된 특징을 가지는 프루닝되지 않은 의사 결정 트리 학습자의 배깅으로 구성된 학습 앙상블이다. 포레스트 중 트리의 개수가 많아짐에 따라 포레스트에 대한 일반화 오류는 한계점으로 수렴된다.
랜덤 포레스트는 하기 방식으로 구성될 수 있다:
1) 트레이닝 세트 중 경우의 수가 N일 경우, 원래의 데이터로부터 무작위로, 단, 교체하면서 N가지의 경우를 샘플링한다. 상기 샘플은 트리를 증가시키기 위한 트레이닝 세트가 될 것이다.
2) M개의 입력 독립 변수가 존재할 경우, 트리 중 각각의 노드에서 M개 중 m개의 변수가 무작위로 선택되고, 이들 m에서 최상의 스플릿을 사용하여 노드를 스플릿할 수 있도록 개수 m<<M으로 지정된다. m 값은 포레스트가 증가하는 동안 일정하게 유지된다.
3) 일부 실행에서, 각각의 트리는 가능한 가장 큰 정도로까지 증가한다. 프루닝은 없다.
4) 이어서, 다수의 트리가 생성된다(k = 1, ..., K(일반적으로, K ≥ 100).
5) 다수의 트리 생성 후, 트리는 모두 관심의 대상이 되는 변수의 분류에 투표한다. 예를 들어, 트리는 각각 활성의 최종 예측 또는 특정 돌연변이에 기여할 수 있다.
6) 이어서, 랜덤 포레스트는 포레스트 중 트리 예측 인자 모두로부터 가장 많은 인기의 투표를 받은 부류를 취함으로써 x(예컨대, 돌연변이 서열 또는 다른 독립 변수)를 분류한다.
포레스트 오류율은 포레스트 중 임의의 두 트리 사이의 상관관계에 의존한다. 상관관계를 증가시키면, 포레스트 오류율은 증가하게 된다. 포레스트 오류율은 포레스트 중 각각의 개별 트리의 강도에 의존한다. 오류율이 낮은 트리는 강한 분류기이다. 개별 트리의 강도를 증가시키면, 포레스트 오류율은 감소하게 된다. m을 감소시키면, 상관관계 및 강도, 둘 모두는 감소하게 된다. m을 증가시키면, 상기 둘 모두는 증가하게 된다. m의 "최적" 범위는 대략적이며 - 일반적으로 매우 광범위하다.
랜덤 포레스트 기법은 카테고리 변수 뿐만 아니라, 회귀 모델에서 연속 변수에 대해 사용될 수 있다. 본 발명의 일부 실시양태에서, 랜덤 포레스트 모델은 SVM 및 신경망 모델과 유사한 예측력을 가지지만, 다른 이유들 중에서도 교차 검증이 모델링 프로세스로 구축되고, 교차 검증을 위한 별도의 프로세스는 필요하지 않기 때문에 계산 효율은 더 높은 경향이 있다.
i) 선형 모델
본 개시내용은 비선형 모델에 관한 것이지만, 서열 대 활성의 선형 모델 맥락하에서 더욱 쉽게 이해될 수 있다. 추가로, 일부 실시양태에서, 선형 모델은 비선형 모델을 생성하기 위한 단계적 프로세스에서 "베이스" 모델로서 사용된다. 일반적으로, 활성 대 서열의 하기 형식을 가진다:
<방정식 1>

상기 선형식에서, y는 예측 반응이고, cij 및 xij는 각각 서열 중 i번 위치에서의 정선된 잔기를 나타내는 데 사용되는 회귀 계수 및 비트 값 또는 더미 변수이다. 단백질 변이체 라이브러리의 서열에는 N개의 잔기 위치가 존재하고, 이들은 각각 하나 이상의 잔기에 의해 점유될 수 있다. 임의의 주어진 위치에서, M개의 별도의 잔기 유형에 걸쳐 j = 1로 존재할 수 있다. 상기 모델은 모든 위치의 잔기 사이의 선형(가산) 관계를 추정한다. 방정식 1의 확장 버전은 하기와 같다:
명시된 바와 같이, 활성 및 서열 정보 형태의 데이터는 초기 단백질 변이체 라이브러리로부터 도출되고, 이는 모델의 회귀 계수를 측정하는 데 사용된다. 먼저, 단백질 변이체 서열의 정렬로부터 더미 변수를 확인한다. 아미노산 잔기 위치의 아미노산 잔기가 서열들과 다른 아미노산 잔기 위치를 단백질 변이체 서열로부터 확인한다. 이들 가변 잔기 위치들 중 일부 또는 모두에서의 아미노산 잔기 정보가 서열-활성 모델에 포함될 수 있다.
하기 표 I는 각각의 변이체 단백질에 상응하는 활성 값과 함께, 10개의 예시적인 변이체 단백질에 대한 가변 잔기 위치 형태의 서열 정보 및 잔기 유형을 포함한다. 이는 계수 모두에 대해서 풀 수 있을 정도로 충분한 방정식을 생성하는 데 필요한 더욱 많은 세트의 대표적인 구성원이다. 따라서, 예를 들어, 표 I의 예시적인 단백질 변이체 서열의 경우, 10번, 166번, 175번, 및 340번 위치는 가변 잔기 위치이고, 다른 위치 모두, 즉, 표에서 명시되지 않은 위치는 변이체 1-10 사이에 동일한 잔기를 포함한다. 상기 일례에서, 10개의 변이체는 야생형 골격 서열을 포함할 수 있거나, 포함하지 않을 수도 있다. 일부 실시양태에서, 야생형 골격 서열을 포함하는 모든 변이체의 데이터를 나타내도록 개발된 모델은 완벽한 다중공선성, 또는 더미 가변 트랩의 문제를 도입할 수 있다. 이러한 문제는 다양한 기법에 의해 처리될 수 있다. 일부 실시양태는 모델 개발로부터 야생형 골격 데이터를 배제시킬 수 있다. 일부 실시양태는 야생형 골격을 나타내는 계수를 드롭 아웃시킬 수 있다. 일부 실시양태는 기법, 예컨대, 다중공선성을 처리하는 PLS 회귀를 사용할 수 있다.
<표 I>

따라서, 방정식 1에 기초하여, 회귀 모델은 표 I의 체계적으로 가변된 라이브러리로부터 도출될 수 있고, 즉, 하기와 같다:
<방정식 2>

비트 값(x 더미 변수)은 지정된 아미노산 잔기의 존재 또는 부재를 반영하는 1 또는 0, 별법으로, 1 또는 -1, 또는 일부 다른 대용 표현 방식으로 나타낼 수 있다. 예를 들어, 1 또는 0 지정을 사용할 경우, x10Ala는 변이체 1의 경우에는 "1"이고, 변이체 2의 경우에는 "2"가 될 것이다. 1 또는 -1 지정을 사용할 경우, x10Ala는 변이체 1의 경우에는 "1"이고, 변이체 2의 경우에는 "-1"이 될 것이다. 따라서, 회귀 계수는 라이브러리 중의 모든 변이체에 대한 서열 활성 정보에 기초하여 회귀 방정식으로부터 도출될 수 있다. (x에 대한 1 또는 0 지정을 사용하였을 때) 변이체 1-10에 대한 상기 방정식의 예는 하기와 같다:

완전한 방정식 세트는 임의의 적합한 회귀 기법(예컨대, PCR, PLS, 또는 MLR)을 사용하여 쉽게 풀릴 수 있고, 이로써 관심의 대상이 되는 각각의 잔기 및 위치에 상응하는 회귀 계수 값을 측정할 수 있다. 상기 예에서, 회귀 계수의 상대적인 크기는 특정 위치의 특정 잔기가 활성에 기여하는 기여도의 상대적인 크기와 상관관계가 있다. 이어서, 회귀 계수를 순위화하거나, 또는 다르게는 분류하여 어느 잔기가 원하는 활성에 바람직하게 기여할 수 있는 가능성이 더 큰지를 측정할 수 있다. 하기 표 II는 표 I에 예시된 체계적으로 가변된 라이브러리에 상응하는 예시적인 회귀 계수 값을 제공한다:
<표 II>

순위화된 회귀 계수 목록을 사용하여 원하는 활성(즉, 적합도 개선)과 관련하여 최적화된 단백질 변이체의 신규 라이브러리를 구성할 수 있다. 이는 다양한 방식으로 수행될 수 있다. 한 실시양태에서, 관찰값이 가장 높은 계수를 가지는 아미노산 잔기를 유지함으로써 달성된다. 이는 회귀 모델에 의해 지시되는 바, 원하는 활성에 가장 크게 기여하는 잔기이다. 음의 디스크립터가 잔기를 확인하는 데 사용될 경우(예컨대, 류신의 경우, 1, 및 글리신의 경우, -1), 이는 계수의 절대치에 기초하여 잔기 위치를 순위화하는 것이 필요하게 된다. 상기와 같은 상황하에서는 전형적으로 각각의 잔기에 대해서는 오직 단일 계수만이 존재한다는 것에 주의한다. 계수 크기의 절대치를 통해 상응하는 잔기 위치가 순위화된다. 이어서, 그들 각각이 원하는 활성면에서 유해한지 또는 유익한지 여부를 측정하기 위해서는 개별 잔기의 부호(sign)를 고려하는 것이 필요하게 된다.
ii) 비선형 모델
비선형 모델링은 단백질 중 활성에 기여하는 잔기-잔기 상호작용을 나타내는 데 사용된다. N-K 랜드스케이프는 상기 문제를 기술한다. 파라미터 N은 관련 폴리펩티드 서열 집합에서 가변 잔기의 개수를 지칭한다. 파라미터 K는 상기 폴리펩티드들 중 임의의 것 내에 존재하는 개별 잔기 사이의 상호작용을 나타낸다. 상호작용은 보통 폴리펩티드의 1차, 2차, 또는 3차 구조와는 상관없이, 다양한 잔기 사이에 물리적으로 가까운 인접함의 결과이다. 상호작용은 직접 상호작용, 간접 상호작용, 물리화학적 상호작용, 폴리 중간체에 기인하는 상호작용, 번역 효과 등에 기인할 수 있다. 문헌 [Kauffman, S. and Levin, S. (1987), "Towards a general theory of adaptive walks on rugged landscapes", Journal of Theoretical Biology 128 (1) 11-45]을 참조할 수 있다.
파라미터 K는 값 K=1일 경우, 각각의 가변 잔기(예컨대, 그중 20개 존재)가 그의 서열 중 정확히 하나의 다른 잔기와 상호작용할 수 있도록 정의된다. 모든 잔기가 모든 다른 잔기의 효과와 물리적으로 및 화학적으로 독립된 경우, K 값은 0이다. K는 폴리펩티드의 구조에 의존하여 광범위한 범위의 상이한 값을 가질 수 있다는 것을 확실하다. 문제의 폴리펩티드 구조가 철저하게 풀린 경우, K 값은 추정될 수 있다. 그러나, 흔히 이는 상기 경우의 것이 아니다.
(상기 기술된 바와 같은) 폴리펩티드 활성의 전적으로 선형인 가법 모델은 2개 이상의 잔기 사이의 특이적인 상호작용을 나타내는 하나 이상의 비1차 상호작용 항을 포함함으로써 개선될 수 있다. 상기 제시된 모델 형태와 관련하여, 상기 항은 상호작용하여 활성에 대하여 유의적으로 긍정적인 또는 부정적인 영향을 미치는 2개 이상의 특정 잔기(이는 각각 서열에서 특정 위치와 관련되어 있다)를 나타내는 2개 이상의 더미 변수를 포함하는 "외적"으로 서술된다. 예를 들어, 외적 항은 식 c abXaXb를 가질 수 있고, 여기서, Xa는 서열 상의 특정 위치에 특정 잔기가 존재한다는 것을 나타내는 더미 변수이고, 변수 Xb는 폴리펩티드 서열 중 (제1 위치와 상호작용하는) 상이한 위치의 특정 잔기의 존재를 나타낸다. 모델의 예시적인 형태에 대한 상세한 설명은 하기에 제시한다.
외적 항에 표시된 모든 잔기의 존재(즉, 2개 이상의 특이적 유형의 잔기들 각각의 것이 구체적으로 확인된 위치에 존재)는 폴리펩티드의 전반적인 활성에 영향을 미친다. 영향은 다수의 방식으로 명시될 수 있다. 예를 들어, 개별 상호작용 잔기들이 각각 폴리펩티드 중에 단독으로 존재할 때, 이는 활성에 부정적인 영향을 미칠 수 있지만, 개별 상호작용 잔기들이 폴리펩티드에 존재할 때 전반적인 효과는 긍정적인 영향을 미치는 것이다. 그 반대는 다른 경우에서도 진정 그러할 수 있다. 추가로, 시너지 효과가 일어날 수 있으며, 이때, 개별 잔기는 각각 단독으로 활성에 대하여 상대적으로 제한된 영향을 주지만, 잔기 모두가 존재할 경우에 활성에 미치는 효과는 개별 잔기 모두의 누적 효과보다 더 크다.
일부 실시양태에서, 비선형 모델은 서열 중 상호작용 가변 잔기의 모든 가능한 조합에 대한 외적 항을 포함한다. 그러나, 실제로는 오직 가변 잔기의 서브세트만이 서로 상호작용하기 때문에, 비선형 모델이 물리적 실체를 나타내지는 못한다. 추가로, 모델을 생성하는 데 사용되는 특정 폴리펩티드를 나타내는 것이지만, 폴리펩티드 내의 실제 상호작용을 나타내는 것은 아닌 스퓨리어스 결과를 제공하는 모델을 생성하기 위한 "과대적합"을 일으킬 것이다. 물리적 실체를 나타내고, 과대적합을 회피하는 모델에 대한 외적 항의 정확한 개수는 K 값에 의해 좌우된다. 예를 들어, K=1일 경우, 외적 상호작용 항의 개수는 N과 같다.
일부 실시양태에서, 비선형 모델을 구성하는 데 있어서, 활성에 유의적인 영향을 미치는 진정한 구조적 상호작용을 나타내느 외적 상호작용 항을 확인하는 것이 중요하다. 이는, 항 추가가 더 이상 통계학상 유의적이지 않을 때까지, 후보 외적 항을 초기 1차 항 모델에만 한번에 하나씩 첨가하는 것인 정방향 추가, 및 모든 가능한 외적 항이 초기 모델에 제공되고, 한번에 하나씩 제거되는 것인 역방향 제거를 포함하나, 이에 한정되지 않는 다양한 방식으로 달성될 수 있다. 하기 제시되는 예시적인 일례는 유용한 비1차 상호작용 항을 확인하기 위해 단계적 가산 및 감산 기법을 사용하는 것을 포함한다.
일부 실시양태에서, 상기 상호작용 항을 포함하는 비선형 모델을 생성하기 위한 접근법은 상기 기술된 선형 모델을 생성하기 위한 접근법과 동일한 것이다. 다시 말해, 트레이닝 세트는 데이터를 모델에 "적합화"시키는 데 사용된다. 그러나, 하나 이상 비1차 항, 바람직하게, 상기 논의된 외적 항이 모델에 추가된다. 추가로, 상기 기술된 선형 모델과 같이, 생성된 비선형 모델은 폴리펩티드의 전반적인 활성에 대한 다양한 잔기의 중요도를 순위화하는 데 사용될 수 있다. 비선형 방정식에 의해 예측되는 바와 같이 가변 잔기의 최상의 조합을 확인하는 데 다양한 기법이 사용될 수 있다. 잔기를 순위화하는 접근법은 하기 기술된다. 일부 실시양태에서, 심지어 단 2개의 잔기에 의해 유발되는 상호작용으로만 제한될 때에도, 가변 잔기에 대하여 가능한 외적 항들이 아주 많이 사용된다. 더 많은 상호작용이 발생함에 따라, 비선형 모델에 대하여 고려되는 잠재적인 상호작용의 수는 기하급수적으로 성장한다. 모델이 3개 이상의 잔기를 포함하는 상호작용의 가능성을 포함할 경우, 잠재적인 항의 개수는 더욱더 빠르게 성장한다.
20개의 가변 잔기가 존재하고, K=1인(각 가변 잔기가 다른 가변 잔기와 상호작용한다고 가정하는 것인)인 간단한 예시적인 일례에서, 모델에 20개의 상호작용 항(외적)이 존재하여야 한다. 임의의 더 적은 개수의 상호작용 항이 존재한다면, (비록 상호작용 중 일부가 활성에 대하여 유의적인 영향을 미치지 않을 수도 있지만) 모델은 상호작용을 충분히 기술하지 못할 것이다. 반대로, 임의의 더 많은 개수의 상호작용 항이 존재한다면, 모델은 데이터 세트를 과대적합화할 수 있다. 본 일례에서, N*(N-1)/2 또는 190개의 가능한 상호작용 쌍이 존재한다. 대략 5.48 x 1026개의 가능한 조합이 존재하는 바, 서열 중 20개의 상호작용을 기술하는 20개의 독특한 쌍의 조합을 찾는 것이 전산학상 중요한 문제가 된다.
관련 외적 항을 확인하는 데 다수의 기법이 사용될 수 있다. 문제 크기 및 이용가능한 계산력에 의존하여, 모든 가능한 조합을 탐색함으로써 데이터를 최고로 적합화하는 한 모델을 확인할 수 있다. 그러나, 흔히 문제는 전산상 부담이 된다. 따라서, 일부 실시양태에서, 효율적인 검색 알고리즘 또는 근사치가 사용된다. 본원에서 명시되는 바와 같이, 한 적합한 검색 기법은 단계적 기법이다. 그러나, 본 발명을 관련 외적 항을 확인하는 데 있어 임의의 특정 방법으로 한정하고자 하지 않는다.
예시적인 일례는 서열 정보로부터 활성을 예측하는 모델에 비선형 외적 항을 도입하는 값을 보여주는 하기 표 III에 제시되어 있다. 본 일례는 서열 중 각각의 가변 위치에는 단 2개의 잔기 옵션이 존재한다고 가정하는 비선형 모델이다. 본 일례에서, 단백질 서열은 각각 +1 및 -1을 사용함으로써, 선택 A 또는 선택 B에 상응하는 더미 변수를 사용하여 코딩 서열로 배정된다. 모델은 각각의 잔기 선택을 지정하는 데 사용되는 수치 값 중 어느 것이 임의로 선택되는지에 대해 영향을 받지 않는다. 표 III의 제1행에 제시된 가변 위치는 단백질 서열의 실제 서열 위치를 나타내는 것은 아니다. 대신, 잔기 선택 A 및 잔기 선택 B에 대하여 표 III의 제2 및 제3행에 제시된 2가지 옵션 중 하나로 달라질 수 있는 단백질 서열 중 임의의 10개의 가상 위치를 나타내는 임의 표지이다.
<표 III>

이러한 코딩 체계를 이용할 때, 단백질 서열을 활성과 연관시키는 데 사용되는 선형 모델은 하기와 같이 작성될 수 있다:
<방정식 3>
여기서, y는 반응(활성)이고, c n 은 위치 n에서의 잔기 선택에 대한 회귀 계수이고, x는 위치 n에서의 잔기 선택을 코딩하는 더비 변수(+1/-1)이고, c 0 은 반응의 평균값이다. 이러한 형태의 모델은 가변 잔기 사이에 상호작용이 없다고(즉, 각각의 잔기 선택이 단백질의 전체 적합도에 대하여 독립적으로 기여한다고) 가정한다.
비선형 모델은 잔기 사이의 상호작용을 나타내는 특정 개수의 (아직 결정되지 않은) 외적 항을 포함한다:
<방정식 4>
여기서, 변수는 방정식 (3)의 것과 동일하지만, 이 경우, 비1차 항이 존재하며, 예컨대, c 1,2 는 변수 위치 1과 2 사이의 상호작용에 대한 회귀 계수이다.
선형 모델 및 비선형 모델의 성능을 평가하기 위해, NK 랜드스케이프로 알려져 있는 합성 데이터 소스가 사용되었다(문헌 [Kauffman and Levin, 1987]). 상기 언급된 바와 같이, N은 모의 단백질 중 가변 위치의 개수이고, K는 잔기 사이의 상위성 커플링이다. 추가로, 합성 데이터 세트는 인실리코로 생성되었다.
이러한 데이터 세트를 사용하여 N=20개의 가변 위치 및 K=1(반복을 위해, K=1인 경우, 각각의 가변 위치는 다른 가변 위치에 기능적으로 커플링된다)과 함께 S=40개의 합성 샘플을 포함하는 초기 트레이닝 세트를 생성하였다(반복을 위해, K=1인 경우, 각각의 가변 위치는 다른 가변 위치에 기능적으로 커플링된다). 무작위화된 단백질을 생성할 때, 각각의 가변 위치는 더미 변수 +1 또는 -1을 포함하는 것에 대하여 동일한 확률을 가졌다. 합성 트레이닝 세트의 각각의 구성원에 대한 (외적으로 표현되는) 잔기-잔기 상호작용 및 실제 활성이 공지되어 있다. 검증에서의 사용을 위해 또 다른 V=100개의 샘플이 생성되었다. 또한, 검증 세트의 각각의 구성원에 대한 잔기-잔기 상호작용 및 활성이 공지되었다.
트레이닝 세트를 사용하여 선형 모델 및 비선형 모델, 둘 모두를 구성하였다. 일부 비선형 모델은 외적 항을 선택함으로써 생성되었고, 다른 비선형 모델은 상기 항을 선택하지 않고 생성되었다. 도 3a-f에 대한 모델은 유전 알고리즘 모델링 방법을 사용하여 생성된 반면, 도 g-h에 대한 모델은 단계적 모델링 방법을 사용하여 생성되었다. 비록 단지 1차 항만을 가지는 모델과 비교하여 1차 및 비1차 항, 둘 모두를 가지는 모델이 가지는 정량적 이점이 유전 알고리즘 모델링 방법과 단계적 모델링 방법 사이에는 차이가 있기는 하지만, 그 결과는 모델링 방법과는 상관없이, 비1차 항을 가지는 모델이 가지는 일반화할 수 있는 이점을 나타낸다. 실제로, 임의의 적합한 모델링 방법이 본 발명에 사용될 수 있다는 것을 알 수 있는 바, 본 발명을 임의의 특정 방법으로 한정하고자 하지 않는다.
상기 기술된 바와 같이 트레이닝 세트 크기가 S=40인 경우, 선형 모델은 측정값과 예측값을 충분히 잘 연관시킬 수 있었지만, 트레이닝 세트에 제시되지 않은 데이터에 대해 검증되는 경우에는 보다 약한 상관 관계를 보였다(도 3a 참조). 제시된 바와 같이, 검은색으로 표시된(dark) 데이터 포인트는 선형 모델에 의해 이루어진 예측에 대한 40개의 트레이닝 데이터 포인트의 관찰된 활성을 나타낸다. 흰색으로 표시된(light) 데이터 포인트는 40개의 트레이닝 샘플로부터 구성되고, 어느 것도 원래의 트레이닝 세트에서는 제시되지 않았던 검증 샘플 V를 예측하는 데 사용된 동일 모델에 의해 이루어진 예측을 나타낸다. 검증 세트는 트레이닝 세트와는 대조적으로, 모델의 진정한 예측 능력을 나타내는 우수한 척도를 제공하는 데, 이는 특히 하기 기술되는 비선형 경우에서는 모델 과대적합 문제에 직면하게 될 수 있다.
선형 모델을 이용하여 비선형 적합도 랜드스케이프를 모델링하였다는 것을 고려해 볼 때, 상기 기술된 S=40개인 트레이닝 세트에 대한 결과는 주목할만하다. 이 경우, 선형 모델은 기껏해야 주어진 잔기의 선택을 위한 적합도에 대한 평균 기여도를 포착할 수 있다. 충분한 개수의 평균 기여도가 조합하여 고려되었다고 가정해 볼 때, 선형 모델은 실제 측정된 반응을 대략적으로 예측한다. 선형 모델에 대한 검증 결과는, 트레이닝 크기가 S=100개로 증가되었을 때 약간 더 우수하였다(도 3b 참조). 상대적으로 단순한 모델이 데이터를 과소적합화하는 경향은 "편향"으로 알려져 있다.
단지 S=40개의 샘플만을 사용하여 비선형 모델을 트레이닝시켰을 때, 트레이닝 세트 구성원과의 상관관계는 탁월하였다(도 3c 참조). 불행하게도, 이러한 예시적인 일례에서, 상기 모델은 검증 세트에서 측정값과 그의 제한된 상관관계로 입증된 바와 같이, 트레이닝 세트 밖으로 제한된 예측력을 제공하였다. 다수의 잠재적인 변수(가능하게는 210개)를 가지고, 적절한 외적 항의 확인을 용이하게 하는 제한된 트레이닝 데이터를 포함하는 상기 비선형 모델은 본질적으로는 단지 트레이닝된 데이터 세트를 기억할 수 있을 뿐이었다. 고도로 복잡한 모델이 데이터를 과대적합화하는 이러한 경향은 "분산"으로 알려져 있다. 편향-분산 트레이드오프는 기계 학습에서의 근본적인 문제를 나타내고, 신규의 또는 특징이 규명되지 않은 기계 학습 문제를 다룰 경우, 이를 처리하기 위해서는 일부 검증 형태가 거의 항상 요구된다.
그러나, 도 3d에 제시된 바와 같이 더 큰 트레이닝 세트(S=100)를 사용하여 비선형 모델을 트레이닝시켰을 때, 비선형 모델은 트레이닝 예측, 및 더욱 중요하게는, 검증 예측, 둘 모두에 대해 매우 잘 실행하였다. 검증 예측은 충분히 정확하였고, 데이터 포인트 대부분은 트레이닝 세트를 플롯팅하는 데 사용되는 검은색으로 표시된 동그라미로 가려져 있다.
비교를 위해, 도 3e 및 도 3f는 주의하여 외적 항을 선택하지 않고 제조된 비선형 모델의 성능을 보여주는 것이다. 도 3c 및 3d의 모델과 달리, 모든 가능한 외적 항이 선택되었다(즉, N=20인 경우, 190개의 외적 항). 상기 도에 제시되어 있는 바와 같이, 검증 세트 활성을 예측할 수 있는 능력은 주의하여 외적 항을 선택함으로써 생성된 비선형 모델의 것과 비교하였을 때, 상대적으로 부족하다. 이와 같이 검증 데이터를 예측할 수 있는 능력이 부족한 것은 과대적합을 나타내는 것이다.
도 3g 및 3h는 각각 인실리코로 모의된 데이터에 대한 선형 모델 및 단계적, 비선형 모델의 잔차로 표시되는 예측력을 보여주는 것이다. 단계적 비선형 모델은 일반적으로는 상기 기술된 바와 같이, 및 더욱 구체적으로는 하기와 같이 실행되었다.
이들 모델을 검정하기 위해, 모의된 데이터를 생성하였다. 평균이 MN 이고, 표준 편차가 SD 인 정규 분포에 기초하여 난수 발생기 R 을 생성하였다. 이어서, 10개의 돌연변이로 이루어진 세트를 정의하였다. 이들 네이밍은 M1, M2...M10이었다(이러한 네이밍 방식은 임의적이다). 이 단계는 다양성 생성을 모의하는 것이다.
각 돌연변이는 단백질 서열 내의 주어진 위치에서의 아미노산 변화를 나타내었고, 각각의 위치는 다른 위치와는 독립적이다. 상기 각각의 돌연변이는 R ( MN = 0, SD = 0.2)에 기초하여 지정된 무작위 활성 값 A 를 가졌다. 상기 6개의 돌연변이를 선택하고, 함께 쌍을 이루게 하여 3쌍의 P를 만들었다. 이들 쌍은 돌연변이 사이의 상위성 상호작용을 나타내었다.
R ( MN = 0, SD = 0.2)에 기초하여 각각의 쌍 P 에 활성 값 AP 를 지정하였다. 각각의 변이체가 상기 정의된 돌연변이 M 의 난수를 포함하는 50개의 변이체로 이루어진 라이브러리 L 이 구성되었고 - 돌연변이의 난수는 R ( MN = 4, SD = 0.25)의 반올림된 절대값에 의해 정의되었다. 이 단계는 라이브러리 구성 및 서열 분석을 모의하는 것이다.
먼저 1.0(돌연변이 서열이 없는, 야생형의 정의된 활성)에 (두 돌연변이 모두가 존재한다면) 각각의 쌍별 돌연변이 PA 로부터의 활성 값을 가산한 후, 남은 단일 돌연변이의 값 ( A )을 가산하여 L 중 각각의 변이체의 활성을 계산하였다. 각각의 변이체에 대한 최종 값에 R ( MN = 0, SD = 0.005)로부터의 무작위 값을 가산함으로써 어세이 노이즈를 모의하였다. 이 단계는 변이체의 스크리닝을 모의하는 것이다.
최종 단계로부터의 데이터에 기초하여 선형 모델 LM을 구성하였다. 이 모델은 10개의 독립 변수/계수를 포함하였는데, 이들 각각은 M 중 한 돌연변이를 나타내는 것이다. 이어서, 상기 얻은 데이터 및 최소 제곱법 회귀를 사용하여 선형 모델을 적합화시켰다.
단계적 가산 방법을 이용하고, 베이스 모델은 LM 으로 하고, 선택 기준으로서 AIC를 이용하고, 오직 단일 돌연변이 및 쌍별 상호작용을 나타내는 계수만을 포함하는 모델을 선택함으로써 상기 얻은 데이터에 기초하여 모델 M M 을 선택하였다. 모델 선택 방법에 대한 추가의 상세한 설명에 대해 하기의 모델 선택 설명부를 참조할 수 있다. 최소 제곱법 회귀를 사용하여 AIC에 의해 선택된 최적 모델을 적합화시켰다.
선형 모델 및 비선형 모델의 예측 능력을 평가하기 위하여 상기 기술된 절차를 20회 반복하였다. 모델 예측은 모의된 데이터에 대하여 플롯팅하였고, 여기서, 도 3g는 선형 모델을 나타낸 것이고, 도 3h는 단계적 비선형 모델을 나타낸 것이다. 상기 모델을 이용하여 상기 기술된 단일 돌연변이의 값을 예측하였다. 관심의 대상이 되는 단 하나의 돌연변이만을 함유하는 변이체를 예측하기 상기 모델을 이용하고, 1.0(야생형)을 감함으로써 이러한 예측을 수행하였다. 도 3g 및 3h로부터 자명한 바, 비선형 모델이, 선형 경향은 더 크고, 잔차는 더 작은, 더욱 정확학 값을 예측한다.
iii) 모델 선택
일부 실시양태에서, 단계적 가산 또는 감산 방법은 비1차 상호작용 항을 가지는 모델을 제조하는 데 사용된다. 도 1의 블록 (107)에 제시된 연산을 실행함으로써 베이스 모델로부터 상호작용 항을 단계적으로 추가하거나 제거함으로써 상호작용 항을 포함하며, 예측력이 높은 최종을 제공한다. 도 4a는 베이스 모델에 상호작용 항을 가산하고, 신규 모델을 평가함으로써 최종의 최적 모델을 생성하여 도 1의 블록 (107)의 연산 실행의 순서도를 제공한다.
본 일례에서, 베이스 서열 모델은 상호작용 항을 포함하지 않는다. 본 방법은 먼저 현 서열 모델 및 최적 서열 모델을 베이스 서열 모델로 설정한다(블록 (409)). 본 방법은 서열 변이체에 대한 상호작용 항의 풀을 정의한다. 이들 상호작용 항은 임의 개수의, 2개 이상의 아미노산 잔기의 쌍별 또는 고차 상호작용을 포함할 수 있다. 블록 (411)을 참조할 수 있다. 비록 블록 (409)가 블록 (411) 앞에 존재하는 것으로 도시되어 있기는 하지만, 두 단계의 순서는 중요하지 않다. 일부 실시양태에서, 상호작용 항의 풀은 관심의 대상이 되는 모든 아미노산 잔기의 계승 조합을 포함한다. 일부 추가의 실시양태에서, 적어도 모든 쌍별 상호작용 항을 포함한다. 일부 추가의 실시양태에서, 쌍별 및 3원 상호작용 항을 포함한다.
베이스 모델을 생성한 후, 본 방법은 풀로부터 검정되지 않은 상호작용 항을 선택한다. 이어서, 본 방법은 선택된 상호작용 항을 현 서열 모델에 추가함으로써 신규 서열 모델을 생성한다. 블록 (413)을 참조할 수 있다. 이어서, 본 방법은 추가의 상호작용 항을 포함하는 것에 대한 편향을 포함하는 모델 선택 방법을 이용하여 신규 서열 모델의 예측력을 평가한다. 블록 (415)를 참조할 수 있다. 본 방법은 신규 서열 모델의 예측력이 최적 서열 모델의 것보다 큰지 그 여부를 측정한다. 결정 블록 (417)을 참조할 수 있다. 일례로서, 본 방법은 모델 선택 기준으로서 "가능도" 결정(예컨대, AIC)을 사용하는 기법을 사용할 수 있다. 상기 경우에서, AIC 값이 앞서 검정된 모델의 것보다 작은 모델만이 오직 보다 높은 예측력을 가지는 것으로 간주된다.
일부 실시양태에서, 선택 방법은 더 많은 파라미터를 가지는 모델에 대해 편향된다. 상기 선택 방법의 예로는 아카이케 정보 기준(AIC) 및 베이지안 정보 기준(BIC), 및 그의 변형을 포함하나, 이에 한정되지 않는다. 예를 들어, AIC는
AIC
= -
2log
e
L
+ 2k
(여기서, L은 데이터 세트가 주어졌을 때, 모델의 가능도이고, k는 모델의 자유 파라미터 개수이다)로 계산될 수 있다.
일부 실시양태에서, 데이터 세트가 주어졌을 때, 모델의 가능도는 최대 가능도 방법을 포함하나, 이에 한정되지 않는, 다양한 방법에 의해 계산될 수 있다. 예를 들어, 한 관찰값에 대하여 활성이 존재하거나, 또는 존재하는 2원 종속 변수의 경우, 모델의 가능도는

(여기서, n은 데이터 세트 중 데이터 포인트의 총 개수이고; a i 및 b i 는 i번째 조건을 포함하는 관찰된 시행 개수이고; p는 모델에 의해 예측된 바와 같이 관찰된 종속 변수의 확률이다)로서 계산될 수 있다.
베이스 모델보다 누진적으로 더 많은 상호작용 항(및 관련 계수)을 포함하는 회귀 모델에서와 같이, 일련의 내포된 모델을 포함하는 일부 실시양태에서, 심지어 추가의 계수가 스퓨리어스한 경우에도, 더욱 복잡한 모델은 추가의 자유도를 누릴 수 있기 때문에, 더욱 복잡한 모델이 더 단순한 모델과 동등하게 우수하거나, 또는 그보다 더 우수한 정도로 적합하다. 일부 실시양태에서, AIC는 적합도 증가가 스퓨리어스한 파라미터에 대한 대가에 의한 오프셋보다 더 큰 정도로 더욱 복잡한 모델에 벌점을 부가한다. 모델 선택에서, AIC 값이 작을수록 더 우수한 모델임을 나타낸다.
도 4a에 제시된 일례에서, 신규 서열 모델의 예측력이 최적 서열 모델의 것보다 클 경우, 이때, 본 방법은 신규 모델을 최적 모델로서 설정한다. 블록 (419)를 참조할 수 있다. 이어서, 본 방법은 검정되지 않은 임의의 추가의 상호작용 항이 풀에 남아있는지 여부를 체크한다. 결정 블록 (421)을 참조할 수 있다. 남아있을 경우, 프로세스는 블록 (413)으로 다시 되돌아감으로써 상호작용 풀에서 이용가능한, 이용가능한 상호작용 항들 모두를 검정하는 내부 루프를 형성한다. 내부 루프의 반복을 통해, 단일의 최적 상호작용 항을 찾을 수 있고, 모델에 추가할 수 있다.
모든 상호작용 항을 검정하고, 내부 루프가 종료된 후, 이전 최적 모델보다 더 큰 예측력을 가지는 모델이 존재한다고 가정한다면, 하나의 추가의 상호작용 항을 가지는 최적 모델이 확인된다. 결정 블록 (423)을 참조할 수 있다. 상기 실시양태에서, 본 방법은 현재 모델을 최적 모델로 설정하고, 최적 모델의 상호작용 항을 상호작용 항의 이용가능한 풀로부터 배제시킨다. 블록 (425)를 참조할 수 있다. 이어서, 본 방법은 블록 (413)으로 다시 루프를 형성한다. 이러한 외부 루프는 모델의 예측력을 개선시킬 수 있는 후속의 최적 상호작용 항을 검색한다. 상호작용 항이 발견되고 나면, 이전 최적 서열 모델의 것보다 더 큰 예측력을 가지는 신규 모델이 확인되지 않을 때까지 후속 최적 상호작용 항에 대한 검색은 외부 루프에서 계속 진행된다.
모델을 개선시키는 추가의 상호작용 항이 더 이상 발견되지 않을 때, 본 방법은 최적 모델을 최종 모델로서 설정한다. 블록 (427)을 참조할 수 있다. 서열 및 활성 데이터가 주어졌을 때, 최적 모델에 대한 검색이 종료된다. 이어서, 모델은 신규 서열의 활성을 예측하는 데 사용된다. 상기 예측은 추가 변이 및 검정을 위한 서열 선택을 유도할 수 있다.
특정 실시양태에서, 상호작용 항 풀 중 이용가능한 상호작용 항들은 각각 모델의 품질 또는 예측력에 대하여 잠재적으로 동등한 영향을 미치는 것으로 처리된다. 다시 말해, 실행시 풀 중 각각의 이용가능한 상호작용 항은 특정 반복 동안 고려의 대상이 되는 것으로 선택될 수 있는 가능성을 동등하게 가진다. 일부 실시양태에서, 이용가능한 상호작용 항은 무작위로, 또는 일부 임의 순서대로 선택된다. 일부 다른 실시양태에서, 상호작용 항은 일부 항이 주어진 반복 동안 고려의 대상이 되는 것으로 선택될 수 있는 가능성이 다른 것보다 더 크게 하는 방식으로 편향되거나, 가중화된다. 특정 실시양태에서, 편향 또는 가중화는 상호작용에 대한 물리적 또는 이론적 정보에 기초하여 적용될 수 있다. 예를 들어, 단백질의 두 특정 영역에서의 돌연변이는 물리적으로 서로 인접해 있고, 이로써 상호작용할 수 있는 가능성이 있는 것으로 공지될 수 있다. 이들 두 일반적인 영역 내의 잔기에 관한 상호작용 항은 모델을 개선시키는 반복적 프로세스 동안 선택을 위해 편향될 수 있다.
도 4a에 대한 것과 유사한 프로세스를 예시하는 의사 코드는 하기와 같다::

도 4b는 도 1의 블록 (107)에 제시된 연산의 실시양태를 보여주는 순서도를 제공하는 것이다. 본 프로세스에서, 상호작용 항을 상기 항의 풀로부터 상호작용 항을 모든 가능한 상호작용 항을 포함하는 베이스 모델로부터 제거하여 최종의 최적 모델을 생성한다.
본 실시양태에서, 베이스 서열 모델은 정의된 풀 내에 모든 상호작용 항을 포함한다. 본 방법은 먼저 프로세스 초반에 현 서열 모델 및 최적 서열 모델을 베이스 서열 모델과 동등한 것으로 설정한다(블록 (439)). 본 실시양태는 상호작용 항의 완전한 풀이 임의 개수의, 2개 이상의 아미노산 잔기의 쌍별 또는 고차 상호작용을 포함할 수 있다는 점에서 상기 기술된 최종 모델과 유사하다 . 일부 실시양태에서, 상호작용 항의 풀은 관심의 대상이 되는 모든 아미노산 잔기의 계승 조합을 포함한다.
베이스 모델 생성 후, 본 방법은 베이스 모델에 이미 포함된 항의 풀로부터 아직 검정되지 못한 상호작용 항을 선택한다. 이어서, 본 방법은 선택된 상호작용 항을 현 서열 모델로부터 제거함으로써 신규 서열 모델을 생성한다. 블록 (441)을 참조할 수 있다. 이어서, 본 방법은 추가의 상호작용 항에 대한 편향을 가지는 모델 선택 방법을 사용하여 신규 서열 모델의 예측력을 평가한다. 블록 (443)을 참조할 수 있다. 본 방법은 신규 서열 모델의 예측력이 최적 서열 모델의 것보다 큰지 그 여부를 평가한다. 블록 (445)에 제시된 결정 연산을 참조할 수 있다. 일부 실시양태에서, AIC가 모델 선택 기준으로서 사용되며, 이로써, AIC 값이 이전 검정된 모델보다 더 작은 모델의 경우, 이는 예측력은 더 높은 것으로 간주된다.
상기 예시적인 일례에서, 신규 서열 모델의 예측력이 최적 서열 모델의 것보다 클 경우, 이때 본 방법은 최적 모델로서 신규 모델을 설정한다. 블록 (447)을 참조할 수 있다. 이어서, 본 방법은 임의의 추가의 상호작용 항을 검정되지 않은 풀에 남겨둘지 여부(즉, 현 서열 모델로부터 감할지 여부)를 체크한다. 결정 블록 (449)를 참조할 수 있다. 검정되지 않은 임의의 항에 존재할 경우, 본 방법은 블록 (441)로 되돌아감으로써 내부 루프를 형성하여 상호작용 풀 중 이용가능한, 이용가능한 상호작용 항들 모두를 검정한다. 내부 루프의 반복을 통해, 단일의 단일의 상호작용 항을 확인한다. 이를 모델로부터 누락시키면, 모델은 가장 크게 개선된다(그리고, AIC가 모델의 예측력을 측정하는 데 사용된다면, AIC를 가장 큰 차이로 하락시킨다).
모든 상호작용 항을 검정하고, 내부 루프가 종료된 후, 이전 최적 모델보다 더 큰 예측력을 가지는 모델이 존재한다고 가정한다면, 하나의 더 적은 개수의 상호작용 항을 가지는 최적 모델이 확인된다. 결정 블록 (451)을 참조할 수 있다. 이 경우, 본 방법은 현재 모델을 최적 모델로서 설정한다. 블록 (453)을 참조할 수 있다. 이어서, 본 방법은 블록 (441)로 다시 루프를 형성한다. 이러한 외부 루프는 가장 큰 차이로 모델의 예측력을 개선시킬 수 있는 후속 상호작용 항을 검색한다. 그러한 상호작용 항이 발견되고 나면, 이전 최적 서열 모델의 것보다 더 큰 예측력을 가지는 신규 모델이 확인되지 않을 때까지 제거하고자 하는 후속 상호작용 항에 대한 검색은 외부 루프에서 계속 진행된다.
내부 루프가 완료되고, 모델을 개선시키는 제거하고자 하는 추가의 상호작용 항이 더 이상 발견되지 않을 때(즉, 블록 (451)에 제시된 결정 연산에서 "아니오'라고 답하였을 때), 본 방법은 최종 최적 모델을 최종 모델로서 설정한다. 블록 (455)를 참조할 수 있다. 서열 및 활성 데이터가 주어졌을 때, 최적 모델에 대한 검색이 종료된다.
iv) 대안적
모델링
옵션
상기 접근법에 대한 많은 추가의 변형도 본 개시내용의 범주 내에 포함된다. 실제로, 임의의 적합한 모델이 본 발명에 사용될 수 있다는 것을 알 수 있는 바, 본 발명을 임의의 특정 모델로 한정하고자 하지 않는다. 한 예시적인 일례로서, xij 변수는 아미노산 그 자체의 정확한 아이덴티티(류신 대 발린 대 프롤린...)라기 보다는 아미노산의 물리적 또는 화학적 특성을 나타내는 것이다. 상기 특성의 예로는 친유성, 벌크, 및 전자 특성(예컨대, 형식 전하, 부분 전하와 관련된 반 데르 발스 표면적 등)을 포함한다. 본 접근법을 실행하기 위해, 아미노산 잔기를 나타내는 xij 값은 그의 특성 또는 상기 특성으로부터 구성된 주성분으로 제시될 수 있다. 임의의 적합한 특성이 본 발명의 방법에서 사용될 수 있다는 것을 알 수 있는 바, 본 발명을 아미노산, 펩티드, 및/또는 폴리펩티드의 임의의 특정 특성으로 한정하고자 하지 않는다.
일부 추가의 실시양태에서, xij 변수는 아미노산 잔기라기보다는 뉴클레오티드를 나타낸다. 상기 실시양태에서, 본 목적은 단백질 변이체 라이브러리를 위해 단백질을 코딩하는 핵산 서열을 확인하는 것이다. 아미노산보다는 뉴클레오티드를 사용함으로써, 활성(예컨대, 특이적 활성) 이외의 다른 파라미터를 원하는 대로 최적화시킬 수 있다. 예를 들어, 특정 숙주 또는 벡터에서의 단백질 발현은 뉴클레오티드 서열의 함수일 수 있다. 2개의 상이한 뉴클레오티드 서열이 같은 아미노산 서열을 가지는 단백질을 코딩할 수 있지만, 뉴클레오티드 서열 중 하나가 단백질을 더 많은 양으로 생산할 수 있도록 할 수 있고/거나, 단백질은 더욱 큰 활성을 가진다. 아미노산 서열보다는 뉴클레오티드 서열을 사용함으로써, 본원에 기술된 본 방법은 개선된 유전자 발현 특성 및/또는 개선된 특성(예컨대, 특이적 활성, 안정성 등)을 보이는 미생물 균주를 최적화시키는 데 사용될 수 있다.
일부 실시양태에서, 뉴클레오티드 서열은 코돈 서열로서 제시된다. 일부 실시양태에서, 모델은 예측되는 활성이 뉴클레오티드 서열 중에 존재하는 다양한 코돈의 함수가 되도록 뉴클레오티드 서열의 원자 단위로서 코돈을 사용한다. 각각의 코돈은 전체 뉴클레오티드 서열 중 그의 위치와 함께 서열-활성 모델을 생성하기 위한 독립 변수로서의 역할을 한다. 일부 경우에서, 주어진 아미노산에 대하여 상이한 코돈은 주어진 유기체에서 다르게 발현된다는 것에 주의한다. 일부 실시양태에서, 각각의 유기체는 주어진 아미노산에 대해 선호 코돈, 또는 코돈 빈도의 분포를 가진다. 독립 변수로서 코돈을 사용함으로써, 본 실시양태는 상기 선호를 나타낸다. 따라서, 본 실시양태는 발현 변이체의 라이브러리를 생성하는 데 사용될 수 있다(예컨대, 여기서, "활성"은 특정 숙주 유기체의 유전자 발현 수준을 포함한다).
일부 실시양태에서, 본 방법은 하기 연산: (a) 단백질 변이체 라이브러리의 트레이닝 세트를 특징화하는 데이터를 입수하는 연산; (b) (a)에서 수득한 데이터에 기초하여 뉴클레오티드 유형 및 뉴클레오티드 서열 중의 상응하는 위치의 함수로서 활성을 예측하는 비선형 서열-활성 모델을 발생시키는 연산; (c) 서열-활성 모델을 이용하여 뉴클레오티드 서열 중의 위치 및/또는 뉴클레오티드 서열 중 특정 위치의 뉴클레오티드 유형을 원하는 활성에 미치는 영향 순서대로 순위화하는 연산; 및 (d) 원하는 활성을 개선시키기 위해 순위화를 사용하여 뉴클레오티드 서열 중의 가변시키고자 하거나, 또는 고정시키고자 하는 하나 이상의 뉴클레오티드를 확인하는 연산을 포함한다. 명시된 바와 같이, 일부 실시양태에서, 가변시키고자 하는 뉴클레오티드는 특이적 아미노산을 코딩한다.
일부 다른 실시양태에서, 본 방법은 특정 특성과 관련된 그의 중요도에 대해 잔기를 순위화하거나, 다르게는 특징화하는 상이한 기법의 용도를 포함한다. 선형 모델에 대하여 상기 기술된 바와 같이, 회귀 계수의 크기를 사용하여 잔기를 순위화하였다. 크기가 큰 계수를 가지는 잔기(예컨대, 166 Ile)를 고순위 잔기로 간주하였다. 특징화를 사용하여 단백질 변이체의 신규의 최적화된 라이브러리 생성에서 특정 잔기를 가변시킬지 여부를 결정하였다. 비선형 모델의 경우, 본원에 기술된 바와 같이, 감도 분석은 더욱 복잡하였다.
PLS 및 다른 기법은 특이적 잔기 또는 잔기 위치를 순위화하는 데 사용될 수 있는, 회귀 계수 크기 이외의 추가 정보를 제공한다. 기법, 예컨대, PLS 및 주성분 분석(PCA) 또는 PCR은 주성분 또는 잠재 벡터의 형태로 정보를 제공한다. 이는 본원에 개시된 본 발명의 실시양태와 함께 사용되는 다차원 데이터 세트, 예컨대, 단백질 서열 활성 공간을 통해 최대 변이의 방향 또는 벡터를 나타낸다. 이러한 잠재 벡터는 다양한 서열 크기의 함수이다; 즉, 트레이닝 세트를 구성하는 데 사용되는 변이체 라이브러리를 포함하는 단백질 서열을 포함하는 개별 잔기 또는 잔기 위치. 그러므로, 잠재 벡터는 트레이닝 세트 중 각각의 잔기 위치로부터의 기여도의 총합을 포함한다. 일부 위치는 벡터의 방향에 더욱 강하게 기여한다. 이는 상대적으로 큰 "로드," 즉, 벡터를 기술하는 데 사용되는 계수에 의해 드러난다. 간단한 예시적인 일례로서, 트레이닝 세트는 트리펩티드로 구성될 수 있다. 이러한 일례에서, 제1 잠재 벡터는 3개의 잔기 모두로부터의 기여도를 포함한다.
벡터 1 = a1(잔기 위치 1) + a2(잔기 위치 2) + a3(잔기 위치 3)
계수, a1, a2, 및 a3은 로드이다. 이는 데이터세트에서 변이에 대한 상응하는 잔기 위치의 중요도를 반영하기 때문에, 이는 상기 기술된 바와 같이, "토글링" 결정을 목적으로 개별 잔기 위치의 중요도를 순위화하는 데 사용될 수 있다. 회귀 계수와 같은 로드는 각각의 토글링된 위치에서의 잔기를 순위화하는 데 사용될 수 있다. 다양한 파라미터가 이들 로드의 중요도를 기술한다. 일부 실시양태는 로드 행렬을 사용하기 위해 예컨대, 투영에서 변수 중요도(VIP: Variable Importance in Projection)와 같은 방법을 사용한다. 로드 행렬은 트레이닝 세트로부터 취한 다중 잠재 벡터에 대한 로드로 구성된다. PLS 투영법을 위한 변수 중요도에서, 변수(예컨대, 잔기 위치)의 중요도는 VIP를 계산함으로써 전산화된다. 주어진 PLS 크기에 대해, a, (VIN)ak 2는 상기 PLS 크기에 의한 y(종속 변수, 예컨대, 특정 기능)의 설명된 가변성(%)을 변수의 PLS 가중치 제곱(wak)2에 곱한 값과 같다. 모든 PLS 크기(성분)에 대하여 (VIN)ak 2의 총합을 구한다. 이어서, 총합을 PLS 모델에서 설명된 y의 전체 가변성(%)으로 나누고, 모델 중 변수의 개수를 곱하여 VIP를 계산한다. VIP가 1보다 큰 변수가 특정 함수(y)와의 상관 관계에 대하여 가장 관련성을 가지며, 따라서, 토글링 결정을 위한 최고 순위를 차지하게 된다.
다수의 실시양태에서, 본 발명은 조합 라이브러리 중 돌연변이가 관심의 대상이 되는 서열 활성에 미치는 효과를 확인하기 위한 일반적인 선형 회귀 방법을 이용한다. 대안적 모델링 옵션 및 기법, 예컨대, 베이지안 회귀, 앙상블 회귀, 부트스트랩핑이 상기 언급된 본 방법과 함께 조합하여, 또는 그를 대신하여 사용될 수 있다. 실제로, 임의의 적합한 방법(들)은 본 발명에 사용될 수 있다는 것을 알 수 있는 바, 본 발명을 임의의 특이적인 모델링 옵션 및/또는 기법으로 한정하고자 하지 않는다.
베이지안 선형 회귀
본 발명의 일부 실시양태에서, 베이지안 선형 회귀가 사용될 수 있다는 것을 알 수 있다. 본 방법은 통계학적 분석이 베이지안 추측의 맥락에서 착수되는 선형 회귀 접근법이다. 회귀 모델이 정규 분포를 가지는 오차를 가질 때, 및 특정 형태의 선험적 분포가 가정되는 경우, 모델의 파라미터의 사후 확률 분포는 베이지안 추측 기법을 사용하여 결정된다.
선형 회귀 모델의 최소 제곱법 해법은 분석 계산 방법, 예컨대, 무어-펜로즈 의사 역(Moore-Penrose pseudo inverse)을 이용하여 데이터의 가능도 함수에 기초하여 계수 벡터 및 모델 오차를 추정한다. 이는 모든 서열에 대한 서열 활성 관계를 나타내는 데이터의 관찰값이 충분히 존재한다고 가정하는 빈도주의적 접근법이다. 그러나, 샘플의 모든 관찰값은 거의 집단 구성원들 모두를 나타낼 정도로는 결코 충분하지 않다. 이는 특히 샘플(또는 트레이닝 세트) 크기가 제한되어 있을 때 문제가 된다. 베이지안 접근법에서, 샘플 데이터는 선험적 확률 분포 형태의 추가 정보로 보충된다. 파라미터에 대한 사전 신뢰는 베이스의 정리에 따라 데이터의 가능도 함수와 조합되고, 이로써 파라미터에 대한 사후 신뢰를 얻게 된다. 사전 신뢰는 이용가능한 선험적 관념인 정보와 도메인에 의존하는 상이한 함수 형태를 취할 수 있다.
예를 들어, 일부 실시양태에서, 베이지안 회귀는 모델 적합화 이전에 계수에 가중치를 가하기 위해 선험적 정보를 이용할 수 있다. 일부 실시양태에서, 이전 회차의 유도 진화, 예컨대, 모체 또는 참조 골격 및 이전 회차에서 사용된 돌연변이 중 적어도 일부를 사용하여 실행된 회차의 것으로부터 얻은 서열/활성 데이터는 선형 계수에 가중치를 가하는 데 사용될 수 있다. 추가로, 2개 이상의 돌연변이 사이의 상위성 관계의 예측은 비선형, 상호작용 계수에 가중치를 가하는 데 사용될 수 있다. 이 접근법의 주된 장점 중 하나는 모델 예측을 유도하는 선험적 정보를 포함한다는 점이다.
선험적 정보 공급처에 대한 한 예시적인 일례로는 참조 골격에의 다중 돌연변이들 각각에 대한 독립항 및 상호작용 항을 가지는 모델이 있다. 일부 실시양태에서, 데이터는 변이체당 하나의 돌연변이를 포함하는 변이체 집합으로부터 얻는다.
본 발명에서 사용될 수 있다는 것을 알 수 있는 선험적 정보에 관한 추가 예로는 특정 돌연변이의 역할 또는 돌연변이의 유형에 대한 직관적 또는 물리적 정보를 포함하나, 이에 한정되지 않는다. 공급처와 상관없이, 선험적 정보는 서열과 활성 사이의 관계에 관한 선입관으로서의 역할을 한다.
모델의 파라미터를 추정하기 위한 일부 실시양태에서, 베이지안 선형 회귀는 모델을 주어진 데이터에 대해 적합화하기 위해 몬테 카를로(Monte Carlo) 시뮬레이션, 예컨대, 깁스 샘플링(Gibbs sampling) 또는 메트로폴리스(Metropolis) 알고리즘을 이용한다. 깁스 샘플링은 직접적인 샘플링이 어려울 때, 대략, 명시된 다중분산 확률 분포로부터(즉, 2개 이상의 확률 변수의 결합 확률 분포로부터)의 것인 관찰값의 서열을 수득하기 위한 마르코프 연쇄 몬테 카를로(Markov chain Monte Carlo) 알고리즘이다.
도 5는 변이체 라이브러리의 유도된 진화에서의 베이지안 회귀 사용을 도시한 순서도이다. 각각의 회차의 서열 진화는 이전 회차로부터의 서열에 기초한 돌연변이를 포함하며, 이는 지식, 예컨대, 서열-활성 모델에 의해 유도될 수 있다. 예를 들어, 블록(501)에서와 같이 n회차 진화에는 변이체당 하나의 돌연변이가 존재한다. 블록 (503)에 제시된 바와 같은 다음 또는 n+1회차 진화는 현 회차이다. 각각의 변이체에 대하여 하나 이상의 신규 돌연변이가 존재하며, 이로써 변이체당 2개 이상의 돌연변이에 이르게 된다. 베이지안 회귀는 본 예시적인 일례의 현 회차에서 실행된다.
n+1회차의 서열 변이체가 신규 모델에 대한 데이터의 트레이닝 세트를 제공한다. 신규 모델은 블록(507)에 명시된 바와 같이, 개별 잔기에 대한 1차 항만을 포함하는 베이스 모델, 또는 모든 가능한 상호작용 항/계수를 포함하는 완전 모델을 포함할 수 있다. 신규 모델은 또한 상기에서 설명된 단계적 가산 또는 감산 기법을 비롯한, 다양한 기법에 의해 선택되는 모델을 포함할 수 있으며, 블록 (505)를 참조할 수 있다. 모델은 하기 논의되는 바와 같은 유전 알고리즘 또는 부트스트랩 기법을 사용하여 교대로 선택될 수 있다. 이러한 모델은 모두 현 데이터/n+1 회차의 트레이닝 세트 데이터로부터의 신규 데이터에 기초한다. 베이지안 추측 기법은 이러한 모델에 적용될 수 있고, 이로써, 모델은 현 데이터의 확률 함수 및 선험적 정보 분포, 둘 모두에 기초한다. 선험적 정보는 블록(501)에 의해 명시된 n 회차에서와 같이, 이전 회차의 서열 변이체의 데이터로부터 얻은 결과일 수 있다. 정보는 또한 블록(503)에 의해 명시된 바와 같이, 임의의 이전 회차의 진화로부터의 서열 활성 데이터, 또는 지식에 대한 다른 사전 직관로부터 얻은 결과일 수 있다. 블록(509)에 명시된 베이지안 회귀 모델은 현 데이터 및 선험적 정보에 의해 제공된 정보에 기초하여 활성을 예측하며, 이는 블록 (511)을 참조할 수 있다. 비록 도 5는 베이지안 회귀 기법을 n+1 회차에 적용시킨 것만을 도시하지만, 이는 다양한 단계에서도 적용될 수 있다. 임의의 적합한 방법이 본 발명에 사용될 수 있다는 것을 알 수 있는 바, 본 발명을 도 5에 제공된 특정 단계로 한정하고자 하지 않는다.
앙상블 회귀
일부 실시양태에서, 본 발명은 서열-활성 모델을 제조하는 데 앙상블 회귀 기법을 사용한다. 앙상블 회귀 모델은 수개의 회귀 모델에 기초한다. 각각의 모델의 예측은 특정 정보 기준(IC: information criterion)에 기초하여 가중되고, 앙상블 예측은 그를 포함하는 모든 모델의 예측의 가중 합이다. 일부 실시양태에서, 모델 발생은 1차 항 모두를 포함하는 베이스 모델에서부터 시작된다. 후속 모델은 상호작용 계수를 일부 또는 모든 가능한 조합으로 추가함으로써 구성된다. 일부 실시양태에서, 상호작용 계수는 단계적 프로세스로 추가된다. 각각의 모델은 데이터에 적합화되고, IC는 생성된다. 각각의 모델에 대한 가중치는 IC 그 자체, 또는 변환된 버전, 예컨대, 로그값, 부정값(negated value) 등일 수 있는 IC에 기초한다. 앙상블에서 각각의 모델의 예측값을 생성하고, 각각의 모델로부터 예측값의 가중 평균을 구하여 앙상블 예측값을 측정함으로써 관찰값에 대해 예측할 수 있다. 완전 앙상블은 모든 가능한 모델을 포함하지만, 그를 포함하는 모델의 개수에 대하여 또는 IC에 대하여 역치를 설정함하여 트리밍함으로써 실행 성능이 불량한 모델을 제거할 수 있다.
다양한 기법을 이용하여 앙상블의 구성 모델을 생성할 수 있다. 예를 들어, 일부 실시양태에서, 유전 알고리즘을 이용하여 구성 모델을 생성한다. 서열/활성 데이터를 사용하여, 각각은 그 자신의 계수 세트를 가지는 복수 개의 회귀 모델을 생성한다. 적합도 기준(예컨대, AIC 또는 BIC)에 따라 최적 모델이 선택된다. 상기 모델을 "메이팅"시켜 신규 하이브리드 모델을 생성하고, 이어서, 이를 적합도에 대해 평가하고, 그에 따라 선택한다. 일부 실시양태에서, 상기 프로세스는 다중 회차의 "전산적 진화" 동안 반복되고, 이로써 최적 모델의 앙상블이 생성된다. 별법으로, 일부 실시양태에서, 앙상블 구성 요소는 상기 기술된 바와 같이 단계적 회귀에 의해 생성되고, 앙상블을 형성하기 위해 최적 n 모델이 선택된다.
도 6은 본 발명의 실시양태에 따라 서열 변이체의 유도 진화에서 앙상블 회귀를 실행하는 프로세스에 대한 순서도를 제공하는 것이다. 본 실시양태에서, 앙상블 회귀 기법은 다중 회차의 서열 진화 중 임의 단계에서 적용될 수 있다. 예를 들어, n 회차에서 블록 (601)에 제시된 서열 변이체는 다양한 모델에 대한 데이터의 트레이닝 세트를 제공하여 블록 (603)에 명시된 바와 같은 모델 풀을 형성한다. 모델 풀 중의 모델은 유전 알고리즘 및/또는 단계적 선택에 의해 생성된 모델일 수 있다. 다른 실시양태에서, 모델 풀은 n 분할 교차 검증 모델 및/또는 부트스트랩핑 모델을 포함한다. 일부 실시양태에서, 다양한 모델 선택 기준, 예컨대, 예컨대, AIC 또는 BIC에 기초하여 예측력이 우수한 모델만이 풀에 들어간다.
별법으로, 또는 추가로, 일부 실시양태에서, 모델 선택에 의해 스크리닝되지 못한 모델 또한 모델 풀에 들어간다. 한 실시양태에서, 1차 항 및 비1차 항을 가지는 모든 모델이 모델 풀에 들어간다. 다수의 잔기 및 잔기 간의 훨씬 더 많은 요인 상호작용의 경우, 본 실시양태는 전산적으로 매우 집약적일 수 있다. 일부 대체 실시양태에서, 1차 항 및 쌍별 상호작용 항을 포함하는 모델만이 모델 풀에 들어간다. 모델 풀을 포함시키는 방법과는 상관없이, 앙상블 모델은 그의 구성 요소 항 모두를 포함한다. 모델 풀은 베이지안 모델을 포함하나, 이에 한정되지 않는 임의 개수의 모델을 포함할 수 있으며, 베이지안 모델의 경우, 선험적 정보가 앙상블 내로 도입될 수 있다.
일부 실시양태에서, 블록 (605)에 명시된 바와 같이, 앙상블은 풀 중의 각각의 모델의 계수의 가중 평균에 기초하여 서열 활성을 예측하고, 여기서, 가중치는 상응하는 모델의 예측력에 의해 결정된다.
일부 실시양태에서, 앙상블 회귀는 하기: (1) 공(empty) 앙상블을 제공하는 단계; (2) 군 크기 n이 1 이상인 것을 선택하는 단계; (3) 데이터 포인트를 크기 n의 군으로 분류하는 단계로서, 여기서, 데이터 포인트는 복원 없이 분류되는 것인 단계; (4) 앙상블 모델을 제조하여 개별 및 상호작용 계수를 예측하는 단계인 것인 작업 흐름을 사용한다. 일부 실시양태에서, 앙상블 모델을 제조하는 단계 (4)는 a) 각각의 군에 대한 데이터 포인트를 제거하고, 여기서, 남은 데이터가 트레이닝 세트를 형성하고, 제거된 데이터가 검증 세트를 형성하고; b) 단계적 회귀를 사용하여 훈력 세트를 적합화함으로써 모델을 제조하고; c) 모델의 예측력을 나타내는 것인 검증 세트를 사용하여 모델을 검정하고; d) 상기 기술된 바와 같이 앙상블 모델을 생성하는 데 사용되는 모델 풀에 모델을 추가하는 것을 추가로 포함한다.
부트스트랩 접근법
주어진 반복에서 고려 중에 있는 모델의 예측력의 특징을 규명하기 위한 다른 기법이 본 발명에서 사용될 수 있다는 것을 알 수 있다. 일부 실시양태에서, 이러한 기법은 교차 검증 또는 부트스트랩 기법을 포함한다. 일부 실시양태에서, 교차 검증은 모델을 생성하는 데 사용되는 관찰값 세트를 사용하지만, 모델의 강도를 평가하는 데에는 관찰값 중 일부를 배제시킨다. 일부 실시양태에서, 부트스트랩 기법은 복원 검정되는 샘플 세트를 사용하는 것을 포함한다. 일부 실시양태에서, 교차 검증 또는 부트스트랩핑에 의해 생성된 모델은 상기 기술된 바와 같은 앙상블 모델로 조합될 수 있다.
일부 추가의 실시양태에서, 본 방법은 잔기를 단순히 활성에의 그의 예측된 기여 크기에 의해서 뿐만 아니라, 상기 예측된 기여에 대한 신뢰에 의해서도 또한 순위화한다. 일부 경우에서, 연구원은 한 데이터 세트로부터 또 다른 세트로의 모델의 일반화 가능성에 대해 관심을 가지고 있다. 다시 말해, 연구원은 계수 또는 주성분의 값이 스퓨리어스한지 여부를 알고자 한다. 교차 검증 및 부트스트랩핑 기법은 모델이 다양한 데이터로 일반화 가능한지에 대한 신뢰 수준을 나타내는 척도를 제공한다.
일부 실시양태에서, 크기와 분포의 조합에 기초하여 순위화하는 통계학상 더욱 철저한 접근법이 사용된다. 이들 실시양태 중 일부에서, 크기가 높고, 분포가 조밀한, 이 둘 모두의 계수가 가장 높은 순위를 부여한다. 일부 경우에서, 또 다른 것보다 크기가 더 낮은 계수는 변동이 적기 때문에 더 높은 순위를 부여받을 수 있다. 따라서, 일부 실시양태는 크기 및 표준 편차 또는 분산, 이 둘 모두에 기초하여 아미노산 잔기 또는 뉴클레오티드를 순위화한다. 이를 달성하는 데 다양한 기법들이 사용될 수 있다. 실제로, 본 발명을 임의의 구체적인 순위화 기법으로 한정하고자 하지 않는다. 부트스트랩 p 값 접근법을 사용하는 한 실시양태는 하기에 기술된다.
부트스트랩 방법을 사용하는 방법에 관한 예시적인 일례는 도 7에 도시되어 있다. 도 7에 제시되어 있는 바와 같이, 본 방법 (725)는 원래의 데이터 세트 S를 제공하는 블록 (727)에서 시작된다. 일부 실시양태에서, 이는 상기 기술된 바와 같은 트레이닝 세트이다. 예를 들어, 일부 실시양태에서, 출발 서열의 개별 잔기들을 임의 방식으로(예컨대, 상기 기술된 바와 같이) 체계적으로 가변시킴으로써 생성된다. 방법 (725)에 의해 예시되는 것과 같은 경우에서, 데이터 세트 S는 분석에서 사용하기 위한 M개의 상이한 데이터 포인트(아미노산 또는 뉴클레오티드 서열로부터 수집된 활성 및 서열 정보)를 가진다.
데이터 세트 S로부터 다양한 부트스트랩 세트 B가 생성된다. 이들 세트들은 각각, 세트 S로부터 복원 추출함으로써 모두 원래의 세트 X로부터 취하여진 M개의 구성원으로 이루어진 신규 세트를 생성함으로써 얻는다. 블록 (729)를 참조할 수 있다. "복원(with replacement)" 조건은 원래의 세트 S에 변동을 일으킨다. 신규 부트스트랩 세트인 B는 종종 S로부터 복제 샘플을 포함할 것이다. 일부 경우에서, 부트스트랩 세트 B는 또한 원래 S에 포함되어 있는특정 샘플을 포함하지 않다.
예시적인 일례로서, 100개의 서열로 이루어진 세트 S를 제공한다. 부트스트랩 세트 B는 원래의 세트 S 중의 100개의 서열로부터 100개의 구성원 서열을 무작위로 선택함으로써 생성된다. 본 방법에서 사용되는 각각의 부트스트랩 세트 B는 100개의 서열을 포함한다. 따라서, 일부 서열은 1회 이상의 선택되고, 나머지 다른 것은 선택되지 않을 수도 있다. 이어서, 100개의 서열로 이루어진 세트 S로부터 생성된 부트스트랩 세트 B를 사용하여, 본 방법은 모델을 구축한다. 블록 (731)을 참조할 수 있다. 모델은 PLS, PCR, SVM, 단계적 회귀 등을 사용하여, 상기 기술된 바와 같이 구축될 수 있다. 실제로, 임의의 적합한 모델이 모델을 구축하는 데 사용될 수 있다는 것을 알 수 있도록 한다. 이러한 모델은 세트 B로부터의 다양한 샘플 중에서 발견되는 잔기 또는 뉴클레오티드에 대한 계수 또는 다른 순위 지표를 제공한다. 블록 (733)에 제시된 바와 같이, 후속 사용을 위해 이들 계수 또는 다른 지표를 기록한다.
이어서, 결정 블록 (735)에서, 본 방법은 또 다른 부트스트랩 세트가 생성되어야 하는지 여부를 결정한다. 생성되어야 할 경우, 본 방법은 신규 부트스트랩 세트 B가 상기 기술된 바와 같이 생성되는 블록 (729)로 돌아간다. 생성될 필요가 없는 경우, 본 방법은 하기 논의되는 블록 (737)로 진행된다. 블록 (735)에서의 결정은 계수 값의 분포를 평가하는 데 계수 값의 상이한 세트 몇개가 사용되는지에 달려있다. 정확한 통계치를 생성하기 위해서는 세트 B의 개수가 충분하여야 한다. 일부 실시양태에서, 100 내지 1,000개의 부트스트랩 세트가 제조되고, 분석된다. 이는 100 내지 1,000개가 방법 (725)의 블록 (729), (731), 및 (733)을 통과하는 것으로 나타낼 수 있다. 그러나, 원하는 분석을 위해 적합한 임의 개수가 사용될 수 있는 것을 알 수 있는 바, 본 발명을 임의의 특정 개수의 부트스트랩 세트로 한정하고자 하지 않는다.
충분한 개수의 부트스트랩 세트 B를 제조하고, 분석한 후에는 결정 (735)에서 부정의 답변을 얻게 된다. 명시된 바와 같이, 이어서, 본 방법은 블록 (737)로 진행된다. 여기서, 계수 값(예컨대, 100 내지 1,000개의 값, 각각의 부트스트랩 세트로부터 하나씩)을 사용하여 각각의 잔기 또는 뉴클레오티드(코돈 포함)에 대한 계수 평균 및 표준 편차(또는 모델에 의해 생성된 다른 지표)를 계산한다. 이 정보로부터, 본 방법은 t 통계치를 계산하고, 측정값이 0과 다른 신뢰 구간을 결정할 수 있다. t 통계치로부터, 신뢰 구간에 대한 p 값을 계산한다. 이러한 예시적인 경우에서, p 값이 작을수록, 측정된 회귀 계수가 0과 다르다는 것에 대한 신뢰는 더 커진다.
p 값은 단지 계수 또는 잔기의 중요성을 나타내는 다른 지표에서의 통계학적 변동을 설명할 수 있는 다수의 상이한 유형의 특징화들 중 하나일 뿐이라는 것에 주목한다. 예로는 회귀 계수에 대한 95% 신뢰 구간을 계산하고, 95% 신뢰 구간이 제로 라인과 교차하는 것으로 간주되는 임의의 회귀 계수는 배제시키는 것을 포함하나, 이에 한정되지 않는다. 기본적으로, 일부 실시양태에서, 표준 편차, 분산, 또는 데이터 분포의 다른 통계학상 관련된 척도를 설명하는 임의의 특징화가 사용될 수 있다. 일부 실시양태에서, 이러한 특징화 단계는 또한 계수의 크기를 설명한다.
일부 실시양태에서, 큰 표준 편차를 얻게 된다. 표준 편차가 큰 이유는 데이터 세트 측정이 불량하고/거나, 원래의 데이터 세트에 특정 잔기 또는 뉴클레오티드가 나타나는 것에는 한계가 있는 것을 포함하나, 이에 한정되지 않는 다양한 원인에 기인할 수 있다. 상기 중 후자의 경우에, 일부 부트스트랩 세트는 어떤 특정 잔기 또는 뉴클레오티드도 존재하지 않는 것을 포함할 것이다. 이 경우, 상기 잔기에 대한 계수 값은 0이 될 것이다. 다른 부트스트랩 세트는 잔기 또는 뉴클레오티드 중 적어도 일부는 존재하는 것을 포함하고, 상응하는 계수는 0이 아닌 값을 제공할 것이다. 그러나, 0인 값을 제공하는 세트는 계수의 표준 편차를 상대적으로 크게 만들 것이다. 이는 계수 값의 신뢰를 감소시키며, 순위를 하락시키게 된다. 그러나, 이는 포함된 잔기 또는 뉴클레오티드에 대한 데이터가 상대적으로 거의 없다면, 예상된다.
이어서, 블록 (739)에서, 본 방법은 최저 (최상) p 값에서부터 최고 (최악) p 값까지로 회귀 계수(또는 다른 지표)를 순위화한다. 절대값이 클수록, 더 많은 표준 편차가 0에서부터 떨어져 있다는 사실 때문에, 상기 순위화는 회귀 계수 그 자체의 절대값과 고도한 상관 관계가 있다고 볼 수 있다. 따라서, 표준 편차가 제공된 경우, 회귀 계수가 커짐에 따라 p 값은 작아진다. 그러나, 절대 순위가 항상 p 값 및 순 크기 방법, 둘 모두와 동일한 것은 아니며, 특히 세트 S를 시작하는 데 상대적으로 소수의 데이터 포인트가 이용가능한 경우에 그러하다.
마지막으로, 블록 (741)에 제시된 바와 같이, 본 방법은 블록 (739)의 연산에서 관찰된 순위에 기초하여 특정 잔기를 고정시키고, 토글링한다. 이는 다른 실시양태에 대해 상기 기술된 순위화 사용과 본질적으로 동일하다. 한 접근법에서, 본 방법은 최상의 잔기(이제, 최저 p 값을 가지는 것)를 고정시키고, 그 나머지 다른 잔기(최고 p 값을 가지는 것)는 토글링시킨다.
본 방법 (725)는 인실리코로 잘 실행되는 것으로 나타났다. 또한, 일부 실시양태에서, p 값 순위화 접근법은 물론 단일 또는 소수의 잔기도 처리한다: 부트스트랩 프로세스에서 대개 원래의 데이터 세트에서는 출현하지 않았던 잔기가 무작위로 선택될 가능성은 더 적기 때문에, p 값은 일반적으로 더 높을 것이다(더 나쁠 것이다). 심지어 그의 계수가 큰 경우에도, (표준 편차로 측정된) 그의 가변성은 꽤 높을 것이다. 일부 실시양태에서, 잘 나타나지 않는 잔기(즉, 충분한 빈도로 관찰되지 않거나, 또는 회귀 계수가 더 낮은 잔기)가 다음 회차의 라이브러리 디자인에서 토글링에 대해 우수한 후보가 될 수 있는 바, 이는 원하는 결과가 된다.
E. 모델 예측 서열을 변형시킴으로써 수행되는 최적화된 단백질
변이체
라이브러리 생성
본 발명의 목표 중 하나는 유도 진화를 통해 최적화된 단백질 변이체 라이브러리를 생성하는 것이다. 본 발명의 일부 실시양태는 생성된 서열-활성 모델을 이용하여 단백질 변이체의 유도 진화를 유도하는 방법을 제공한다. 상기 기술된 본 방법에 따라 제조되고, 리파이닝된 다양한 서열-활성 모델은 단백질 또는 생물학적 분자의 유도 진화를 유도하는 데 적합하다. 프로세스의 일부로서, 본 방법은 신규 단백질 변이체 라이브러리를 생성하는 데 사용되는 서열을 확인할 수 있다. 상기 서열은 상기 확인된 정의된 잔기 상에 변이를 포함하거나, 추후에 상기 변이를 도입하는 데 사용되는 전구체이다. 서열은 돌연변이 유발법 또는 재조합 기반 다양성 생성 메커니즘을 수행함으로써 변형될 수 있고, 이로써 단백질 변이체의 신규 라이브러리가 생성될 수 있다. 신규 라이브러리는 또한 신규 서열-활성 모델을 개발하는 데에도 사용될 수 있다.
일부 실시양태에서, 올리고뉴클레오티드 또는 핵산 서열의 제조는 핵산 합성기를 사용하여 올리고뉴클레오티드 또는 핵산 서열을 합성함으로써 달성된다. 본 발명의 일부 실시양태는 제조된 올리고뉴클레오티드 또는 단백질 서열을 유도 진화를 위한 빌딩 블록으로서 사용하여 한 회차의 유도 진화를 수행하는 것을 포함한다. 본 발명의 다양한 실시양태는 상기 빌딩 블록에 재조합 및/또는 돌연변이 유발법을 적용시켜 다양성을 생성할 수 있다.
한 구체적인 일례로서, 일부 실시양태는 재조합 기법을 올리고뉴클레오티드에 적용시킨다. 상기 실시양태에서, 본 방법은 서열-활성 모델의 항의 계수를 평가함으로써 한 회차의 유도 진화를 위한 하나 이상의 돌연변이를 선택하는 것을 포함한다. 돌연변이는 모델에 의해 예측되는 바와 같이, 단백질의 활성에 대한 그의 기여도에 기초하여 특이적 위치의 특이적 유형의 정의된 아미노산 또는 뉴클레오티드의 조합으로부터 선택된다. 일부 실시양태에서, 돌연변이 선택은 다른 계수보다 더 큰 것으로 결정된 하나 이상의 계수를 확인하는 단계, 및 그렇게 확인된 하나 이상의 계수에 의해 제시되는 정의된 위치의 정의된 아미노산 또는 뉴클레오티드를 선택하는 단계를 포함한다. 일부 실시양태에서, 서열-활성 모델에 따라 돌연변이를 선택한 후, 본 방법은 하나 이상의 돌연변이를 포함하거나, 또는 그를 코딩하는 복수 개의 올리고뉴클레오티드를 제조하는 단계, 및 제조된 올리고뉴클레오티드를 사용하여 한 회차의 유도 진화를 수행하는 단계를 포함한다. 일부 실시양태에서, 유도 진화 기법은 올리고뉴클레오티드를 조합 및/또는 재조합하는 것을 포함한다.
본 발명의 다른 실시양태는 재조합 기법을 단백질 서열에 적용시킨다. 일부 실시양태에서, 본 방법은 신규 단백질 또는 신규 핵산 서열을 확인하는 단계, 및 신규 단백질 또는 신규 핵산 서열에 의해 코딩된 단백질을 제조 및 어세이하는 단계를 포함한다. 일부 실시양태에서, 본 방법은 신규 단백질 또는 신규 핵산 서열에 의해 코딩된 단백질을 추가 유도 진화를 위한 출발점으로서 사용하는 단계를 추가로 포함한다. 일부 실시양태에서, 유도 진화 프로세스는 모델에 의해 원하는 수준의 활성을 가지는 것으로 예측된 단백질 서열을 단편화하고 재조합하는 단계를 포함한다.
일부 실시양태에서, 본 방법은 모델에 의해 중요한 것으로 예측된 개별 돌연변이에 기초하여 신규 단백질 또는 신규 핵산 서열을 확인하고/거나, 제조한다. 본 방법은 서열-활성 모델의 항의 계수를 평가하여 활성에 기여하는, 정의된 위치의 정의된 아미노산 또는 뉴클레오티드 중 하나 이상의 것을 확인함으로써 하나 이상의 돌연변이를 선택하는 단계; 상기 선택된 하나 이상의 돌연변이를 포함하는 신규 단백질 또는 신규 핵산 서열을 확인하는 단계, 및 신규 단백질 또는 신규 핵산 서열에 의해 코딩된 단백질을 제조 및 어세이하는 단계를 포함한다.
다른 실시양태에서, 본 방법은 개별 돌연변이 대신 전체 서열의 예측되는 활성에 기초하여 신규 단백질 또는 신규 핵산 서열을 확인하고/거나, 제조한다. 이러한 실시양태 중 일부에서, 본 방법은 다중 단백질 서열 또는 다중 아미노산 서열을 서열-활성 모델에 적용시키는 단계, 및 서열-활성 모델에 의해 예측되는, 다중 단백질 서열 또는 핵산 서열 각각에 대한 활성 값을 측정하는 단계를 포함한다. 본 방법은 서열-활성 모델에 의해 예측되는, 다중 서열에 대한 활성 값을 평가함으로써 상기 적용된 다중 단백질 서열 또는 다중 아미노산 서열로부터 신규 단백질 서열 또는 신규 핵산 서열을 선택하는 단계를 추가로 포함한다. 본 방법은 또한 신규 단백질 서열 또는 신규 핵산 서열에 의해 코딩된 단백질을 가지는 단백질을 제조하고 어세이하는 단계를 포함한다.
일부 실시양태에서, 간단하게 최상으로 예측된 단일 단백질을 합성하는 것 이외에도, 단백질 중 각각의 위치에서의 잔기 선택에서 최적 변이의 감도 분석에 기초하여 단백질의 조합 라이브러리를 생성한다. 본 실시양태에서, 예측된 단백질에 대하여 주어진 잔기 선택의 감도가 클수록, 예측되는 적합도 변화는 더 커질 것이다. 일부 실시양태에서, 이러한 감도는 최고에서부터 최저까지에 이르며, 감도 점수를 사용하여 (즉, 감도에 기초하여 상기 잔기를 도입함으로써) 후속 회차에서 조합 단백질 라이브러리를 생성한다. 일부 실시양태에서, 선형 모델이 사용될 때, 감도는 간단하게 모델에서 주어진 잔기 항과 관련된 계수의 크기를 고려함으로써 확인된다. 그러나, 비선형 모델의 경우에는 불가능하다. 대신, 비선형 모델을 이용하는 실시양태에서, 잔기 감도는, "최상으로" 예측된 서열에서 단일 잔기가 가변될 때, 모델을 이용하여 활성 변화를 계산함으로써 측정된다.
본 발명의 일부 실시양태는 단백질 서열 또는 핵산 서열 중 하나 이상의 위치를 선택하고, 그렇게 확인된 하나 이상의 위치에서 포화 돌연변이 유발법을 수행하는 것을 포함한다. 일부 실시양태에서, 위치는 서열-활성 모델의 항의 계수를 평가하여 활성에 기여하는, 정의된 위치의 정의된 아미노산 또는 뉴클레오티드 중 하나 이상의 것을 확인함으로써 선택된다. 따라서, 일부 실시양태에서, 한 회차의 유도 진화는 서열-활성 모델을 이용하여 선택되는 위치에서 단백질 서열에 대해 포화 돌연변이 유발법을 수행하는 것을 포함한다. 하나 이상의 상호작용 항을 포함하는 모델을 포함하는 일부 실시양태에서, 본 방법은 2개 이상의 상호작용 잔기에 동시에 돌연변이 유발법을 적용시키는 것을 포함한다.
일부 실시양태에서, 잔기는 순위화된 순서로 고려된다. 일부 실시양태에서, 고려 중에 있는 각각의 잔기에 대하여, 프로세스는 상기 잔기를 "토글링"할지 여부를 결정한다. "토글링"이라는 용어는 다중 아미노산 잔기 유형을 최적화된 라이브러리 중 단백질 변이체의 서열내 특정 위치 내로 도입하는 것을 의미한다. 예를 들어, 세린이 한 단백질 변이체 중의 166번 위치에 출현할 수 있는 반면, 페닐알라닌은 같은 라이브러리에서 또 다른 단백질 변이체 중의 166번 위치에 출현할 수 있다. 트레이닝 세트에서 단백질 변이체 서열 간에 차이가 나지 않는 아미노산 잔기는 전형적으로 최적화된 라이브러리에서 고정된 상태로 유지된다. 그러나, 최적화된 라이브러리에 항상 변이가 존재할 수 있는 것은 아니다.
일부 실시양태에서, 최적화된 단백질 변이체 라이브러리는 확인된 "고위" 회귀 계수 잔기는 모두 고정화되고, 나머지 보다 낮은 하위 회귀 계수 잔기는 토글링되도록 디자인된다. 상기 실시양태에 대한 이론적 설명은 '최상으로' 예측된 단백질 주변의 국소 공간이 검색되어야 한다는 점이다. 토글이 도입되는 출발점 "골격"은 모델에 의해 예측된 최상의 단백질, 및/또는 스크리닝된 라이브러리로부터 이미 '최적' 단백질인 것으로 검증된 것일 수 있다는 것에 주의한다. 실제로, 출발점 골격을 임의의 특정 단백질으로 한정하고자 하지 않는다.
대체 실시양태에서, 확인된 고위 회귀 계수 잔기 중 적어도 하나 이상이되, 단, 그들 모두는 아닌 것인 잔기가 최적화된 라이브러리에서 고정화되고, 나머지는 토글링된다. 일부 실시양태에서, 이러한 접근법은 한번에 너무 많은 변이를 도입함으로써 다른 아미노산 잔기의 컨텍스트를 급격하게 변이시키는 것을 원하지 않는 경우에 권고된다. 또한, 토글링을 위한 출발점은 모델에 의해 예측된 바와 같은 최적 잔기 세트, 현존 라이브러리로부터 최적 단백질인 것으로 검증된 것, 또는 잘 모델링되는 "평균" 클론일 수 있다. 후자의 경우, 앞서 샘플링으로부터 생략되었던 활성 힐(hill)에 대한 검색을 위해서는 더 큰 공간이 탐색되어야 하는 바, 더욱더 중요한 것으로 예측되는 잔기를 토글링하는 것이 바람직할 수도 있다. 이러한 유형의 라이브러리는 후속 회차에 대해 더 리파이닝된 영상을 생성하는 바, 전형적으로 조기 회차의 라이브러리 생성에 더 많이 관련되어 있다. 또한, 출발점 골격을 임의의 특정 단백질로 한정하고자 하지 않는다.
상기 실시양태의 일부 대안으로는 어느 잔기를 토글링할지를 결정하는 데 잔기의 중요도 (순위)을 사용하는 상이한 방법을 포함한다. 상기의 한 대체 실시양태에서, 보다 높은 고위 잔기 위치가 토글링을 위해 더욱 적극적으로 선호된다. 본 접그법에서 요구되는 정보로는 트레이닝 세트로부터의 최적 단백질의 서열, PLS 또는 PCR 예측 최적 서열, 및 PLS 또는 PCR 모델로부터의 잔기의 순위를 포함한다. "최적" 단백질은 데이터세트 중 실습 실험실에서 검증된 "최적" 클론(즉, 교차 검증에서 예측된 값에 비교적 가깝다는 점에서 여전히 잘 모델링하는 것인, 측정된 기능이 최고인 것인 클론)이다. 본 방법은 상기 단백질로부터의 각각의 잔기를 원하는 활성 값이 최고값인 "최상으로 예측된" 서열로부터의 상응하는 잔기와 비교하였다. 로드 또는 회귀 계수가 최고값인 잔기가 '최적' 클론에 존재하지 않을 경우, 본 방법은 상기 위치를 후속 라이브러리를 위한 토글 위치로서 도입한다. 잔기가 최적 클론에 존재할 경우, 본 방법은 상기 위치를 토글 위치로서 처리하지 않고, 계속해서 다음 위치로 이동하게 될 것이다. 프로세스는 충분한 크기의 라이브러리가 생성될 때까지, 연속하여 더 낮은 로드 값을 거쳐 이동하면서 각종 잔기에 대해 반복된다.
일부 실시양태에서, 유지되는 회귀 계수 잔기의 개수, 및 토글링되는 회귀 계수 잔기의 개수는 가변된다. 어느 잔기를 토글링할지 및 어느 잔기를 유지할지를 결정하는 것은 원하는 라이브러리 크기, 회귀 계수 사이의 차이 규모, 및 비선형성이 존재하는 것으로 판독되는 정도를 포함하나, 이에 한정되지 않는 다양한 인자에 기초한다. 계수가 작은(중립) 잔기를 유지시키면 후속 회차의 진화에서 중요한 비선형성을 밝혀낼 수 있다. 일부 실시양태에서, 최적화된 단백질 변이체 라이브러리는 약 2N개의 단백질 변이체를 포함하며, 여기서, N은 두 잔기 사이에 토글링되는 위치의 개수를 나타낸다. 또 다른 방식으로 언급하자면, 추가의 각각의 토글에 의해 추가되는 다양성은 라이브러리의 크기를 배가시킴에 따라 10개의 토글 위치는 ~1,000개의 클론(1,024)을, 13개의 토글 위치는 ~10,000개의 클론(8,192)을, 및 20개의 토글 위치는 ~1,000,000개의 클론(1,048,576)을 생산한다. 적절한 라이브러리 크기는 인자, 예컨대, 스크린 비용, 랜드스케이프의 견고성, 바람직한 공간 샘플링 비율(%) 등에 의존한다. 일부 경우에서, 비교적 많은 개수의 변이된 잔기는 과도하게 높은 비율의 클론이 비기능성인 라이브러리를 생성하는 것으로 밝혀졌다. 그러므로, 일부 실시양태에서, 토글링을 위한 잔기 개수의 범위는 약 2 내지 약 30개이고; 즉, 라이브러리 크기 범위는 약 4 내지 230~109개의 클론이다.
추가로, 후속 회차의 다양한 라이브러리 전략법이 동시에 이용되며, 여기서, 일부 전략법은 더욱 적극적이고(더 많은 "유익한" 잔기가 고정되고), 다른 나머지 전략법은 더욱 보존적이라는 것(더욱 철저하게 공간을 탐색하기 위한 목적으로 더 적은 "유익한" 잔기는 고정된다는 것)이 고려된다.
일부 실시양태에서, 대부분의 자연적으로 발생된 펩티드 또는 다르게는 계승적인 펩티드에 존재하는 기 또는 잔기 또는 "모티프"는 단백질의 기능성(예컨대, 활성, 안정성 등)에 중요할 수 있는 바, 그를 확인하고/거나, 보존한다. 예를 들어, 자연적으로 발생된 펩티드에서 3번 가변 위치의 Ile는 항상 11번 가변 위치의 Val과 커플링된다는 것을 알 수 있다. 그러므로, 한 실시양태에서, 상기 기를 보존하는 것이 임의의 토글링 전략법에서 요구된다. 다시 말해, 유일하게 허용되는 토글은 기준 단백질에서 특정 군을 보존하는 것 또는 활성 단백질에서도 또한 발견되는 상이한 군을 생성하는 것이다. 후자의 경우, 2개 이상의 잔기를 토글링하는 데 필요하다.
일부 추가의 실시양태에서, 현 최적화된 라이브러리 중 실습 실험실에서 검증된 '최적' 단백질(또는 소수의 최적 단백질들 중 하나)(즉, 여전히 잘 모델링하는 것인, 즉, 교차 검증에서 예측된 값에 비교적 가까운 것인, 측정된 기능이 최고인 단백질, 또는 최고인 소수의 단백질들 중 하나)이 다양한 변이가 도입되는 골격으로서의 역할을 한다. 또 다른 접근법에서, 잘 모델링할 수 없는 현 라이브러리 중 실습 실험실에서 검증된 '최적' 단백질(또는 소수의 최적 단백질들 중 하나)이 다양한 변이가 도입되는 골격으로서의 역할을 한다. 일부 다른 접근법에서, 서열-활성 모델에 의해 원하는 활성에 대해 최고값(또는 최고값들 중 하나)를 가지는 것으로 예측되는 서열이 골격으로서의 역할을 한다. 이러한 접근법에서, "다음 세대" 라이브러리(및 가능하게는 상응하는 모델)에 대한 데이터 세트는 최적 단백질 중 하나 또는 수개에서 잔기를 변이시킴으로써 얻는다. 한 실시양태에서, 이러한 변이는 골격 중 잔기의 체계적인 변이를 포함한다. 일부 경우에서, 변이는 다양한 돌연변이 유발법, 재조합 및/또는 부분서열 선택 기법을 포함한다. 이들은 각각 시험관내, 생체내, 및/또는 인실리코로 수행될 수 있다. 실제로, 임의의 적합한 포맷이 사용될 수 있다는 것을 알 수 있는 바, 본 발명을 임의의 특정 포맷으로 한정하고자 하지 않는다.
일부 실시양태에서, 선형 모델에 의해 예측되는 최적의 서열이 상기 기술된 바와 같은 검사에 의해 확인될 수 있지만, 비선형 모델의 경우에는 그러하지 않다. 특정 잔기가 1차 항 및 외적 항, 둘 모두에 출현하고, 다른 잔기의 다수의 가능한 조합과 관련하여 그가 활성에 미치는 전반적인 효과는 문제가 될 수 있다. 따라서, 비선형 모델의 경우 외적 항의 선택과 같이, 비선형 모델에 의해 예측되는 최적의 서열은 (전산 자원이 충분하다는 가정하에) 모델을 이용하여 모든 가능한 서열을 검정함으로써, 또는 검색 알고리즘, 예컨대, 단계적 알고리즘을 이용함으로써 확인될 수 있다.
일부 실시양태에서, 상기 기술된 바와 같이 확인된 컴퓨터 진화된 단백질에 포함되어 있는 정보는 신규 단백질을 합성하고, 그를 물리적 어세이로 테스트하는 데 사용된다. 실제 실습 실험실에서 측정된 적합도 기능에 관하여 인실리코로 정확하게 나타냄으로써 연구원들은 실험실에서 스크리닝되는 데 필요한 변이체 개수 및/또는 진화 사이클수를 감소시킬 수 있다. 일부 실시양태에서, 최적화된 단백질 변이체 라이브러리는 본원에 기술된 재조합 방법을 사용하여, 또는 별법으로, 유전자 합성 방법 후, 생체내 또는 시험관내 발현을 수행함으로써 생성된다. 일부 실시양태에서, 최적화된 단백질 변이체 라이브러리를 원하는 활성에 대하여 스크리닝한 후, 그를 서열 분석한다. 상기 도 1 및 2의 논의에 명시된 바와 같이, 본원에 기술된 방법을 사용하여 최적화된 단백질 변이체 라이브러리로부터의 활성 및 서열 정보를 이용함으로써 추가의 최적화된 라이브러리의 디자인의 기원이 될 수 있는 또 다른 서열-활성 모델을 생성할 수 있다. 한 실시양태에서, 이러한 신규 라이브러리로부터의 모든 단백질이 데이터세트의 일부로서 사용된다.
III. 디지털 장치 및 시스템
자명한 바, 본원에 기술된 실시양태는 하나 이상 컴퓨터 시스템에 저장되거나, 그를 통해 전달되는 명령 및/또는 데이터의 제어하에 작동하는 프로세스를 이용한다. 본원에 개시된 실시양태는 또한 이들 연산을 실행하는 장치에 관한 것이다. 일부 실시양태에서, 장치는 필요한 목적을 위해 특수 디자인되고/거나, 구축되거나, 또는 컴퓨터에 저장된 컴퓨터 프로그램 및/또는 데이터 구조에 의해 선택적으로 활성화되거나, 변경되는 범용 컴퓨터일 수 있다. 본 발명에 의해 제공되는 프로세스는 본질적으로 임의의 특정 컴퓨터 또는 다른 특정 장치와 관련이 있는 것은 아니다. 특히, 다양한 범용 기계는 본원의 교시에 따라 작성된 프로그램과 함께 사용된다는 것을 알 수 있다. 그러나, 일부 실시양태에서, 필요한 방법 연산을 실행하기 위해 특수 장치가 구축된다. 상기와 같은 다양한 기계를 위한 특정 구조에 관한 한 실시양태는 하기에 기술된다.
추가로, 본 발명의 특정 실시양태는 다양한 컴퓨터 실행 연산 수행을 위한 프로그램 명령어 및/또는 데이터(데이터 구조 포함)를 포함하는 컴퓨터 판독가능한 매체 또는 컴퓨터 프로그램 제품에 관한 것이다. 컴퓨터 판독가능한 매체의 예로는 자기 매체, 예컨대, 하드 디스크, 플로피 디스크, 자기 테이프; 광학 매체, 예컨대, CD-ROM 장치 및 홀로그램 장치; 광자기 매체; 반도체 메모리 장치; 및 프로그램 명령어를 저장하고, 실행하도록 특수 설정된 하드웨어 장치, 예컨대, 읽기 전용 메모리 장치(ROM: read-only memory) 및 랜덤 액세스 메모리(RAM: random access memory), 응용 주문형 집적 회로(ASIC: application-specific integrated circuit), 및 프로그램 가능 논리 소자(PLD: programmable logic device)를 포함하나, 이에 한정되지 않는다. 데이터 및 프로그램 명령어는 또한 반송파 또는 다른 수송 매체(예컨대, 광회선, 전기선, 및/또는 방송 전파) 상에서 구현될 수 있다. 실제로, 본 발명을 컴퓨터 실행 연산 수행을 위한 명령어 및/또는 데이터를 포함하는 임의의 특정 컴퓨터 판독가능한 매체 또는 임의의 다른 컴퓨터 프로그램 제품으로 한정하고자 하지 않는다.
프로그램 명령어의 예로는 예컨대, 컴파일러에 의해 작성된 로우 레벨 코드, 및 인터프리터를 사용하여 컴퓨터에 의해 실행될 수 있는 하이어 레벨 코드를 포함하는 파일을 포함하나, 이에 한정되지 않는다. 추가로, 프로그램 명령어로는 기계 코드, 원시 코드 및 본 발명에 따라 컴퓨팅 기계의 연산을 직접 또는 간접적으로 제어하는 임의의 다른 코드를 포함하나, 이에 한정되지 않는다. 코드는 입력, 출력, 계산, 조건부, 분기, 반복 루프 등을 명시할 수 있다.
한 예시적인 일례에서, 본원에 개시된 방법을 구현하는 코드는 적절하게 설정된 컴퓨팅 장치로 로딩되었을 때, 장치가 하나 이상의 문자열(들) 상에서 모의 유전 연산(GO)을 실행하도록 하는 논리 명령어 및/또는 데이터를 포함하는 고정 매체 또는 전달가능한 프로그램 구성 요소에서 구현된다. 도 8은 매체(817), 네트워크 포트(819), 사용자 입력 키보드(809), 사용자 입력(811), 또는 다른 입력 수단으로부터 명령어를 판독할 수 있는 논리 장치인 예시적인 디지털 장치(800)를 보여주는 것이다. 이하 장치(800)은 데이터 공간에서 통계학적 연산을 지시하기 위해, 예컨대, 하나 이상 데이터세트를 구축하기 위해(예컨대, 데이터 공간의 복수 개의 대표적인 구성원을 측정하기 위해) 상기 명령어를 사용할 수 있다. 개시된 실시양태를 구현할 수 있는 논리 장치의 한 유형으로는 CPU(807), 임의적 사용자 입력 장치 키보드(809), 및 GUI 위치 결정 장치(811) 뿐만 아니라, 주변 구성 요소, 예컨대, 디스크 드라이버(815) 및 모니터(805)(GO 변형된 문자열을 제시하고, 사용장 의해 상기 문자열의 서브세트의 간소화된 선택을 제공한다)를 포함하는 컴퓨터 시스템(800)에서와 같은 컴퓨터 시스템이 있다. 고정된 매체(817)은 임의적으로 전체 시스템을 프로그램화하는 데 사용되고, 이는 예컨대, 디스크형의 광학 또는 자기 매체 또는 다른 전자 메모리 저장 소자를 포함할 수 있다. 통신 포트(819)는 시스템을 프로그램화하는 데 사용되고, 이는 임의 유형의 통신 연결부를 나타낼 수 있다.
일부 실시양태에서, 본 개시내용은 하나 이상 프로세서; 시스템 메모리; 및 하나 이상의 프로세서에 의해 실행될 때, 컴퓨터 시스템이 생물학적 분자의 유도 진화를 수행하는 방법을 실행하도록 하는 컴퓨터 실행가능 명령어가 그에 저장되어 있는 하나 이상의 컴퓨터 판독가능한 저장 매체를 포함하는 컴퓨터 시스템을 제공한다. 일부 실시양태에서, 본 방법은 (a) 복수 개의 생물학적 분자에 대한 서열 및 활성 데이터를 입수하는 단계; (b) 서열 및 활성 데이터로부터 베이스 모델을 제조하는 단계로서, 여기서, 베이스 모델은 서열의 서브유닛의 존재 또는 부재의 함수로서 활성을 예측하는 것인 단계; (c) 베이스 모델에, 또는 그로부터 하나 이상의 신규 상호작용 항을 가하거나 제거함으로써 하나 이상의 신규 모델을 제조하는 단계로서, 여기서, 신규 상호작용 항은 2개 이상의 상호작용 서브유닛 사이의 상호작용을 나타내는 것인 단계; (d) 서브유닛의 존재 또는 부재의 함수로서 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력을 측정하는 단계; 및 (e) (d)에서 측정된 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력에 기초하고, 추가의 상호작용 항을 포함하는 것에 대한 편향을 이용하여, 베이스 모델에, 또는 베이스 모델로부터 신규 상호작용 항을 가할지 또는 감할지 여부를 결정하는 단계를 포함한다.
특정 실시양태는 또한 응용 주문형 집적 회로(ASIC), 및 프로그램 가능 논리 소자(PLD)의 회로망 내에서 구현될 수 있다. 상기 경우에서, 본 실시양태는 ASIC 또는 PLD를 생성하는 데 사용될 수 있는 컴퓨터 판독가능한 디스크립터 언어로 실행된다. 본 발명의 일부 실시양태는 다양한 다른 디지털 장치, 예컨대, PDA, 랩톱 컴퓨터 시스템, 디스플레이, 영상 편집 장치 등의 회로망 또는 논리 프로세서 내에서 실행된다.
일부 실시양태에서, 본 발명은 컴퓨터 시스템의 하나 이상의 프로세서에 의해 실행될 때, 컴퓨터 시스템이 원하는 활성에 영향을 주는 생물학적 분자를 확인하는 방법을 실행하도록 하는 컴퓨터 실행가능 명령어가 그에 저장되어 있는 하나 이상의 컴퓨터 판독가능한 저장 매체를 포함하는 컴퓨터 프로그램 제품에 관한 것이다. 상기 방법은 본원에 기술된 임의의 방법, 예컨대, 도면 및 유사 부호에 의해 포함되는 것일 수 있다. 일부 실시양태에서, 본 방법은 복수 개의 생물학적 분자에 대한 서열 및 활성 데이터를 입수하고, 서열 및 활성 데이터로부터 베이스 모델 및 개선된 모델을 제조한다. 일부 실시양태에서, 모델은 서열의 서브유닛의 존재 또는 부재의 함수로서 활성을 예측한다.
본 발명의 일부 실시양태에서, 컴퓨터 프로그램 제품에 의해 실행되는 본 방법은 베이스 모델에, 또는 베이스 모델로부터 하나 이상의 신규 상호작용 항을 가하거나 제거함으로써 하나 이상의 신규 모델을 제조하며, 여기서, 신규 상호작용 항은 2개 이상의 상호작용 서브유닛 사이의 상호작용을 나타낸다. 일부 실시양태에서, 본 방법은 서브유닛의 존재 또는 부재의 함수로서 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력을 측정한다. 본 방법은 또한 상기에서 측정된 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력에 기초하고, 추가의 상호작용 항을 포함하는 것에 대한 편향을 이용하여, 베이스 모델에, 또는 베이스 모델로부터 신규 상호작용 항을 가할지 또는 감할지 여부를 결정한다.
상기 내용은 명확하게 하고, 이해시키기 위한 목적으로 좀 더 상세하게 기술되었지만, 본 개시내용의 진정한 범주로부터 벗어남 없이 형태 및 세부 사항은 다양하게 변형될 수 있다는 것이 당업자에게는 본 개시내용의 판독으로부터 명백해질 것이다. 예를 들어, 상기 기술된 모든 기법 및 장치는 다양한 조합으로 사용될 수 있다. 본 출원에서 인용된 모든 공개 문헌, 특허, 특허 출원, 또는 다른 문헌은 마치 각각의 개별 공개 문헌, 특허, 특허 출원, 또는 다른 문헌이 개별적으로 모든 목적을 위해 참조로 포함된 것으로 명시된 바와 같은 정도로 모든 목적을 위해 그 전문이 참조로 포함된다.
SEQUENCE LISTING
<110> CODEXIS, INC.
COPE, GREGORY A
<120> METHODS, SYSTEMS, AND SOFTWARE FOR IDENTIFYING BIOMOLECULES WITH
INTERACTING COMPONENTS
<130> CDXSP018WO
<140> PCT/US2014/013666
<141> 2014-01-29
<150> 61/759,276
<151> 2013-01-31
<150> 61/799,377
<151> 2013-03-15
<160> 13
<170> PatentIn version 3.5
<210> 1
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 1
Ala Ser Gly Phe
1
<210> 2
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 2
Asp Phe Val Ala
1
<210> 3
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 3
Lys Leu Gly Ala
1
<210> 4
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 4
Asp Ile Val Phe
1
<210> 5
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 5
Ala Ile Val Ala
1
<210> 6
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 6
Asp Ser Gly Phe
1
<210> 7
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 7
Leu Phe Gly Phe
1
<210> 8
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 8
Ala Phe Val Ala
1
<210> 9
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 9
Leu Ser Gly Phe
1
<210> 10
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 10
Asp Leu Val Ala
1
<210> 11
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 11
Ile Leu Leu Met Gly Trp Lys Cys Ser Phe
1 5 10
<210> 12
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 12
Val Ala Ile Pro His Asn Arg Thr Ala Tyr
1 5 10
<210> 13
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic Peptide
<400> 13
Val Ala Leu Pro Gly Trp Lys Thr Ser Phe
1 5 10
Claims (13)
- (a) 복수 개의 생물학적 분자에 대한 서열 및 활성 데이터를 입수하는 단계;
(b) 서열 및 활성 데이터로부터 베이스 모델을 제조하는 단계로서, 여기서, 베이스 모델은 서열의 서브유닛의 존재 또는 부재의 함수로서 활성을 예측하는 것인 단계;
(c) 하나 이상의 신규 상호작용 항을 베이스 모델에 가하거나 베이스 모델로부터 감함으로써 하나 이상의 신규 모델을 제조하는 단계로서, 여기서, 신규 상호작용 항은 2개 이상의 상호작용 서브유닛 사이의 상호작용을 나타내는 것인 단계;
(d) 서브유닛의 존재 또는 부재의 함수로서 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력을 측정하는 단계; 및
(e) (d)에서 측정된, 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력에 기초하고, 추가의 상호작용 항을 포함하는 것에 대한 편향(bias)을 이용하여, 신규 상호작용 항을 베이스 모델에 가할지 또는 베이스 모델로부터 감할지 여부를 결정하는 단계
를 포함하는, 원하는 활성에 영향을 미치는 생물학적 분자를 확인하는 방법. - 제1항에 있어서, (c)에서 하나 이상의 신규 모델을 제조하는 단계가 선험적 정보를 이용하여 신규 모델의 사후 확률 분포를 결정하는 것을 포함하는 것인 방법.
- 제2항에 있어서, 베이스 모델 및/또는 신규 모델을 제조하는 단계가 깁스 샘플링(Gibbs sampling)을 이용하여 서열 및 활성 데이터에 모델을 적합화(fit)시키는 것을 포함하는 것인 방법.
- 제1항에 있어서, 하나 이상의 신규 모델이, 각각 상이한 상호작용 항을 포함하는 2개 이상의 신규 모델을 포함하는 것인 방법.
- 제4항에 있어서, 2개 이상의 신규 모델에 기초하여 앙상블 모델을 제조하는 단계를 추가로 포함하며, 여기서,
앙상블 모델은 2개 이상의 신규 모델로부터의 상호작용 항을 포함하고,
상호작용 항은, (d)에서 측정된, 활성을 예측할 수 있는 2개 이상의 신규 모델의 능력에 의해 가중화되는 것인 방법. - 제1항에 있어서, 신규 상호작용 항을 베이스 모델에 가하거나 베이스 모델로부터 감함으로써 업데이트된 모델을 생성하며,
(f) 베이스 모델 대신 업데이트된 모델을 이용하여 (c)를 반복하고, (c)에서 가하거나 감한 것과는 다른 상호작용 항을 가하거나 감하는 단계; 및
(g) 베이스 모델 대신 업데이트된 모델을 이용하여 (d) 및 (e)를 반복하는 단계
를 추가로 포함하는 방법. - 제6항에 있어서, (h) 추가의 업데이트된 모델을 이용하여 (f) 및 (g)를 반복하는 단계를 추가로 포함하는 방법.
- 제1항에 있어서, (d)에서 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력을 아카이케 정보 기준(Akaike Information Criterion) 또는 베이지안 정보 기준(Bayesian Information Criterion)에 의해 측정하는 것인 방법.
- 제1항에 있어서, 서열이 전체 게놈, 전체 염색체, 염색체 세그먼트, 상호작용 유전자에 대한 유전자 서열의 집합, 유전자, 또는 단백질인 것인 방법.
- 제1항에 있어서, 서브유닛이 염색체, 염색체 세그먼트, 일배체형, 유전자, 뉴클레오티드, 코돈, 돌연변이, 아미노산, 또는 잔기인 것인 방법.
- 제1항에 있어서, 복수 개의 생물학적 분자가 단백질 변이체 라이브러리의 트레이닝 세트를 구성하는 것인 방법.
- 컴퓨터 시스템의 하나 이상의 프로세서에 의해 실행될 때, 컴퓨터 시스템이 원하는 활성에 영향을 주는 생물학적 분자를 확인하는 방법을 구현하도록 하는 컴퓨터 실행가능 명령어가 저장되어 있는 하나 이상의 컴퓨터 판독가능한 비일시적인 저장 매체를 포함하는 컴퓨터 프로그램 제품으로서, 상기 방법은
(a) 복수 개의 생물학적 분자에 대한 서열 및 활성 데이터를 입수하는 단계;
(b) 서열 및 활성 데이터로부터 베이스 모델을 제조하는 단계로서, 여기서, 베이스 모델은 서열의 서브유닛의 존재 또는 부재의 함수로서 활성을 예측하는 것인 단계;
(c) 하나 이상의 신규 상호작용 항을 베이스 모델에 가하거나 베이스 모델로부터 감함으로써 하나 이상의 신규 모델을 제조하는 단계로서, 여기서, 신규 상호작용 항은 2개 이상의 상호작용 서브유닛 사이의 상호작용을 나타내는 것인 단계;
(d) 서브유닛의 존재 또는 부재의 함수로서 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력을 측정하는 단계; 및
(e) (d)에서 측정된, 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력에 기초하고, 추가의 상호작용 항을 포함하는 것에 대한 편향을 이용하여, 신규 상호작용 항을 베이스 모델에 가할지 또는 베이스 모델로부터 감할지 여부를 결정하는 단계
를 포함하는 것인 컴퓨터 프로그램 제품. - 하나 이상 프로세서;
시스템 메모리; 및
하나 이상의 프로세서에 의해 실행될 때, 컴퓨터 시스템이 생물학적 분자의 유도 진화를 수행하는 방법을 구현하도록 하는 컴퓨터 실행가능 명령어가 저장되어 있는 하나 이상의 컴퓨터 판독가능한 저장 매체
를 포함하는 컴퓨터 시스템으로서, 상기 방법은
(a) 복수 개의 생물학적 분자에 대한 서열 및 활성 데이터를 입수하는 단계;
(b) 서열 및 활성 데이터로부터 베이스 모델을 제조하는 단계로서, 여기서, 베이스 모델은 서열의 서브유닛의 존재 또는 부재의 함수로서 활성을 예측하는 것인 단계;
(c) 하나 이상의 신규 상호작용 항을 베이스 모델에 가하거나 베이스 모델로부터 감함으로써 하나 이상의 신규 모델을 제조하는 단계로서, 여기서, 신규 상호작용 항은 2개 이상의 상호작용 서브유닛 사이의 상호작용을 나타내는 것인 단계;
(d) 서브유닛의 존재 또는 부재의 함수로서 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력을 측정하는 단계; 및
(e) (d)에서 측정된, 활성을 예측할 수 있는 하나 이상의 신규 모델의 능력에 기초하고, 추가의 상호작용 항을 포함하는 것에 대한 편향을 이용하여, 신규 상호작용 항을 베이스 모델에 가할지 또는 베이스 모델로부터 감할지 여부를 결정하는 단계
를 포함하는 것인 컴퓨터 시스템.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020217000247A KR102490720B1 (ko) | 2013-01-31 | 2014-01-29 | 상호작용 성분을 이용하여 생체분자를 확인하기 위한 방법, 시스템, 및 소프트웨어 |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361759276P | 2013-01-31 | 2013-01-31 | |
| US61/759,276 | 2013-01-31 | ||
| US201361799377P | 2013-03-15 | 2013-03-15 | |
| US61/799,377 | 2013-03-15 | ||
| PCT/US2014/013666 WO2014120819A1 (en) | 2013-01-31 | 2014-01-29 | Methods, systems, and software for identifying bio-molecules with interacting components |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020217000247A Division KR102490720B1 (ko) | 2013-01-31 | 2014-01-29 | 상호작용 성분을 이용하여 생체분자를 확인하기 위한 방법, 시스템, 및 소프트웨어 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20150113166A true KR20150113166A (ko) | 2015-10-07 |
Family
ID=51223867
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020157023727A KR20150113166A (ko) | 2013-01-31 | 2014-01-29 | 상호작용 성분을 이용하여 생체분자를 확인하기 위한 방법, 시스템, 및 소프트웨어 |
| KR1020157023739A KR102215219B1 (ko) | 2013-01-31 | 2014-01-29 | 승법형 모델을 이용하여 생체분자를 확인하기 위한 방법, 시스템, 및 소프트웨어 |
| KR1020217000247A KR102490720B1 (ko) | 2013-01-31 | 2014-01-29 | 상호작용 성분을 이용하여 생체분자를 확인하기 위한 방법, 시스템, 및 소프트웨어 |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020157023739A KR102215219B1 (ko) | 2013-01-31 | 2014-01-29 | 승법형 모델을 이용하여 생체분자를 확인하기 위한 방법, 시스템, 및 소프트웨어 |
| KR1020217000247A KR102490720B1 (ko) | 2013-01-31 | 2014-01-29 | 상호작용 성분을 이용하여 생체분자를 확인하기 위한 방법, 시스템, 및 소프트웨어 |
Country Status (15)
| Country | Link |
|---|---|
| US (4) | US9665694B2 (ko) |
| EP (2) | EP2951579B1 (ko) |
| JP (4) | JP6433028B2 (ko) |
| KR (3) | KR20150113166A (ko) |
| CN (3) | CN109360608B (ko) |
| AU (2) | AU2014212430B2 (ko) |
| BR (2) | BR112015018449B1 (ko) |
| CA (2) | CA2898777C (ko) |
| DK (1) | DK2951754T3 (ko) |
| IL (2) | IL240055A0 (ko) |
| LT (1) | LT2951754T (ko) |
| NZ (2) | NZ710323A (ko) |
| RU (2) | RU2695146C2 (ko) |
| SG (2) | SG11201505969XA (ko) |
| WO (2) | WO2014120819A1 (ko) |
Families Citing this family (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6433028B2 (ja) | 2013-01-31 | 2018-12-05 | コデクシス, インコーポレイテッド | 乗法形式のモデルを使用して生体分子を同定する方法、システム、およびソフトウェア |
| SG11201508193TA (en) | 2013-04-17 | 2015-11-27 | Agency Science Tech & Res | Method for generating extended sequence reads |
| EP2911075A1 (de) * | 2014-02-25 | 2015-08-26 | LTS LOHMANN Therapie-Systeme AG | System zur Bestimmung einer Eignung eines Wirkstoffs für die transdermale oder transmukosale Applikation sowie entsprechendes Verfahren |
| WO2016085916A1 (en) | 2014-11-25 | 2016-06-02 | Codexis, Inc. | Engineered imine reductases and methods for the reductive amination of ketone and amine compounds |
| FI3237621T3 (fi) | 2014-12-22 | 2023-06-01 | Codexis Inc | Ihmisen alfa-galaktosidaasivariantteja |
| WO2016105579A1 (en) * | 2014-12-22 | 2016-06-30 | Board Of Regents Of The University Of Texas System | Systems and methods for processing sequence data for variant detection and analysis |
| EP3298524A4 (en) | 2015-05-22 | 2019-03-20 | CSTS Health Care Inc. | THERMODYNAMIC MEASUREMENTS RELATING TO PROTEIN-PROTEIN INTERACTION NETWORKS FOR THE TREATMENT OF CANCER |
| FR3047013A1 (fr) * | 2016-01-22 | 2017-07-28 | Univ Montpellier | Procede de classification d'un echantillon biologique. |
| WO2017190211A1 (en) * | 2016-05-04 | 2017-11-09 | Deep Genomics Incorporated | Methods and systems for producing an expanded training set for machine learning using biological sequences |
| US10184117B2 (en) | 2016-06-09 | 2019-01-22 | Codexis, Inc. | Biocatalysts and methods for hydroxylation of chemical compounds |
| JP2019517801A (ja) | 2016-06-15 | 2019-06-27 | コデクシス, インコーポレイテッド | 操作されたβ−グルコシダーゼおよびグルコシル化方法 |
| IL264686B1 (en) | 2016-08-26 | 2024-03-01 | Codexis Inc | Engineered imine reductases and methods for reversible amination of ketone and amine compounds |
| US10118610B2 (en) * | 2016-08-31 | 2018-11-06 | Ford Global Technologies, Llc | Autonomous vehicle using path prediction |
| KR102573324B1 (ko) | 2017-02-13 | 2023-08-30 | 코덱시스, 인코포레이티드 | 조작된 페닐알라닌 암모니아 리아제 폴리펩티드 |
| CN108693787A (zh) * | 2017-03-29 | 2018-10-23 | 株式会社东芝 | 模型构建系统以及模型构建方法 |
| AU2018292105A1 (en) | 2017-06-30 | 2019-12-19 | Codexis, Inc. | T7 RNA polymerase variants |
| US10738286B2 (en) | 2017-06-30 | 2020-08-11 | Codexis, Inc. | T7 RNA polymerase variants |
| CN107677997B (zh) * | 2017-09-28 | 2021-06-29 | 杭州电子科技大学 | 基于GLMB滤波和Gibbs采样的扩展目标跟踪方法 |
| EP3676846A1 (en) * | 2017-10-06 | 2020-07-08 | Grail, Inc. | Site-specific noise model for targeted sequencing |
| JP7258871B2 (ja) | 2017-10-17 | 2023-04-17 | インビタエ コーポレイション | 遺伝子及びゲノムの検査並びに分析におけるバリアント解釈の、監査可能な継続的な最適化のための分子エビデンスプラットフォーム |
| JP2021506252A (ja) | 2017-12-13 | 2021-02-22 | コデクシス, インコーポレイテッド | アミドカップリングのためのカルボキシエステラーゼポリペプチド |
| CN108281192B (zh) * | 2017-12-29 | 2022-03-22 | 一诺仪器(中国)有限公司 | 基于集成学习算法的人体成分预测方法及系统 |
| WO2020028039A1 (en) | 2018-07-30 | 2020-02-06 | Codexis, Inc. | Engineered glycosyltransferases and steviol glycoside glucosylation methods |
| US11398297B2 (en) * | 2018-10-11 | 2022-07-26 | Chun-Chieh Chang | Systems and methods for using machine learning and DNA sequencing to extract latent information for DNA, RNA and protein sequences |
| CA3115808A1 (en) * | 2018-10-11 | 2020-04-16 | Berkeley Lights, Inc. | Systems and methods for identification of optimized protein production and kits therefor |
| JP2022512847A (ja) | 2018-10-29 | 2022-02-07 | コデクシス, インコーポレイテッド | 操作されたdnaポリメラーゼバリアント |
| JP2022513199A (ja) | 2018-12-14 | 2022-02-07 | コデクシス, インコーポレイテッド | 操作されたチロシンアンモニアリアーゼ |
| AU2019403323A1 (en) | 2018-12-20 | 2021-07-01 | Codexis, Inc. | Human alpha-galactosidase variants |
| WO2020154386A1 (en) * | 2019-01-22 | 2020-07-30 | EMULATE, Inc. | High-content imaging of microfluidic devices |
| WO2020168286A1 (en) * | 2019-02-14 | 2020-08-20 | University Of Washington | Systems and methods for improved nanopore-based analysis of nucleic acids |
| CN109902389B (zh) * | 2019-03-01 | 2021-01-01 | 厦门大学 | 基于改进通用似然估计的不确定性有限元模型修正方法 |
| CN109979539B (zh) * | 2019-04-10 | 2020-10-02 | 电子科技大学 | 基因序列优化方法、装置及数据处理终端 |
| CA3138861A1 (en) | 2019-05-02 | 2020-12-10 | Board Of Regents, The University Of Texas System | System and method for increasing synthesized protein stability |
| JP7298284B2 (ja) * | 2019-05-09 | 2023-06-27 | 富士通株式会社 | 演算処理装置、演算処理プログラム、及び演算処理方法 |
| JP2022542751A (ja) * | 2019-06-07 | 2022-10-07 | ライカ マイクロシステムズ シーエムエス ゲゼルシャフト ミット ベシュレンクテル ハフツング | 生物学関連のデータを処理するためのシステムおよび方法、顕微鏡を制御するためのシステムおよび方法ならびに顕微鏡 |
| AU2020290510A1 (en) * | 2019-06-12 | 2022-02-03 | Quantum-Si Incorporated | Techniques for protein identification using machine learning and related systems and methods |
| JP7438693B2 (ja) | 2019-09-02 | 2024-02-27 | キヤノンメディカルシステムズ株式会社 | 診療支援装置 |
| US20210174909A1 (en) * | 2019-12-10 | 2021-06-10 | Homodeus, Inc. | Generative machine learning models for predicting functional protein sequences |
| US11188778B1 (en) * | 2020-05-05 | 2021-11-30 | Illumina, Inc. | Equalization-based image processing and spatial crosstalk attenuator |
| JP2023539632A (ja) | 2020-08-28 | 2023-09-15 | コデクシス, インコーポレイテッド | 操作されたプロテアーゼバリアント |
| JP2023539634A (ja) | 2020-08-28 | 2023-09-15 | コデクシス, インコーポレイテッド | 操作されたアミラーゼバリアント |
| US11361194B2 (en) | 2020-10-27 | 2022-06-14 | Illumina, Inc. | Systems and methods for per-cluster intensity correction and base calling |
| CA3204825A1 (en) | 2020-12-18 | 2022-06-23 | Codexis, Inc. | Engineered uridine phosphorylase variant enzymes |
| US11439159B2 (en) | 2021-03-22 | 2022-09-13 | Shiru, Inc. | System for identifying and developing individual naturally-occurring proteins as food ingredients by machine learning and database mining combined with empirical testing for a target food function |
| IL305924A (en) | 2021-04-02 | 2023-11-01 | Codexis Inc | Cyclic GMP-AMP synthase (CGAS) variant transgenic enzymes |
| EP4314262A1 (en) | 2021-04-02 | 2024-02-07 | Codexis, Inc. | Engineered guanylate kinase variant enzymes |
| CN117120600A (zh) | 2021-04-02 | 2023-11-24 | 科德克希思公司 | 工程化腺苷酸激酶变体酶 |
| CN117222735A (zh) | 2021-04-02 | 2023-12-12 | 科德克希思公司 | 工程化乙酸激酶变体酶 |
| US11455487B1 (en) | 2021-10-26 | 2022-09-27 | Illumina Software, Inc. | Intensity extraction and crosstalk attenuation using interpolation and adaptation for base calling |
| CN114913939B (zh) * | 2022-07-19 | 2022-11-15 | 北京科技大学 | 高通量平台和机器学习优化的药物组合设计方法及装置 |
| CN116884503B (zh) * | 2023-09-06 | 2023-12-26 | 北京齐碳科技有限公司 | 序列和后验矩阵的处理方法、装置和计算设备 |
Family Cites Families (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6117679A (en) | 1994-02-17 | 2000-09-12 | Maxygen, Inc. | Methods for generating polynucleotides having desired characteristics by iterative selection and recombination |
| US5834252A (en) | 1995-04-18 | 1998-11-10 | Glaxo Group Limited | End-complementary polymerase reaction |
| US5837458A (en) | 1994-02-17 | 1998-11-17 | Maxygen, Inc. | Methods and compositions for cellular and metabolic engineering |
| US20060257890A1 (en) | 1996-05-20 | 2006-11-16 | Maxygen, Inc. | Methods and compositions for cellular and metabolic engineering |
| US5605793A (en) | 1994-02-17 | 1997-02-25 | Affymax Technologies N.V. | Methods for in vitro recombination |
| US6335160B1 (en) | 1995-02-17 | 2002-01-01 | Maxygen, Inc. | Methods and compositions for polypeptide engineering |
| US6171820B1 (en) * | 1995-12-07 | 2001-01-09 | Diversa Corporation | Saturation mutagenesis in directed evolution |
| US6537776B1 (en) | 1999-06-14 | 2003-03-25 | Diversa Corporation | Synthetic ligation reassembly in directed evolution |
| US6153410A (en) | 1997-03-25 | 2000-11-28 | California Institute Of Technology | Recombination of polynucleotide sequences using random or defined primers |
| AU746786B2 (en) | 1997-12-08 | 2002-05-02 | California Institute Of Technology | Method for creating polynucleotide and polypeptide sequences |
| US20020048772A1 (en) | 2000-02-10 | 2002-04-25 | Dahiyat Bassil I. | Protein design automation for protein libraries |
| US7315786B2 (en) | 1998-10-16 | 2008-01-01 | Xencor | Protein design automation for protein libraries |
| US6917882B2 (en) | 1999-01-19 | 2005-07-12 | Maxygen, Inc. | Methods for making character strings, polynucleotides and polypeptides having desired characteristics |
| US6376246B1 (en) | 1999-02-05 | 2002-04-23 | Maxygen, Inc. | Oligonucleotide mediated nucleic acid recombination |
| US7024312B1 (en) | 1999-01-19 | 2006-04-04 | Maxygen, Inc. | Methods for making character strings, polynucleotides and polypeptides having desired characteristics |
| DK1072010T3 (da) | 1999-01-19 | 2010-06-21 | Maxygen Inc | Oligonukleotidmedieret nukleinsyrerekombination |
| US20070065838A1 (en) | 1999-01-19 | 2007-03-22 | Maxygen, Inc. | Oligonucleotide mediated nucleic acid recombination |
| AU780941B2 (en) * | 1999-11-22 | 2005-04-28 | Yissum Research Development Company Of The Hebrew University Of Jerusalem | System and method for searching a combinatorial space |
| US20010051855A1 (en) * | 2000-02-17 | 2001-12-13 | California Institute Of Technology | Computationally targeted evolutionary design |
| AU2001241939A1 (en) | 2000-02-28 | 2001-09-12 | Maxygen, Inc. | Single-stranded nucleic acid template-mediated recombination and nucleic acid fragment isolation |
| WO2001075767A2 (en) | 2000-03-30 | 2001-10-11 | Maxygen, Inc. | In silico cross-over site selection |
| US20030032059A1 (en) | 2000-05-23 | 2003-02-13 | Zhen-Gang Wang | Gene recombination and hybrid protein development |
| AU2001263411A1 (en) | 2000-05-23 | 2001-12-03 | California Institute Of Technology | Gene recombination and hybrid protein development |
| EP1325457A4 (en) | 2000-10-10 | 2007-10-24 | Genencor Int | LIBRARIES RICH IN INFORMATION |
| JP2004536819A (ja) * | 2001-06-11 | 2004-12-09 | トランジション・セラピューティックス・インコーポレーテッド | ウイルス性、増殖性および炎症性の疾患の治療のためにビタミンb12および治療薬を用いた複合治療 |
| EP2278509B1 (en) * | 2002-03-01 | 2014-11-19 | Codexis Mayflower Holdings, LLC | Methods, systems, and software for identifying functional biomolecules |
| US20050084907A1 (en) | 2002-03-01 | 2005-04-21 | Maxygen, Inc. | Methods, systems, and software for identifying functional biomolecules |
| US7747391B2 (en) | 2002-03-01 | 2010-06-29 | Maxygen, Inc. | Methods, systems, and software for identifying functional biomolecules |
| EP1498825A1 (en) | 2002-04-04 | 2005-01-19 | Ishihara Sangyo Kaisha, Ltd. | Apparatus and method for analyzing data |
| WO2003099999A2 (en) * | 2002-05-20 | 2003-12-04 | Abmaxis, Inc. | Generation and selection of protein library in silico |
| JP4042492B2 (ja) * | 2002-08-07 | 2008-02-06 | トヨタ自動車株式会社 | エンジン制御パラメータの適合方法及び適合システム |
| JP2004355174A (ja) * | 2003-05-28 | 2004-12-16 | Ishihara Sangyo Kaisha Ltd | データ解析方法及びそのシステム |
| KR20070007253A (ko) * | 2003-10-08 | 2007-01-15 | 이바이오사이언스 | 천연 면역글로불린 결합 물질 및 이의 제조 방법 및 사용방법 |
| US7529714B2 (en) * | 2004-12-14 | 2009-05-05 | International Business Machines Corporation | Cost management of software application portfolio |
| CN1763227A (zh) * | 2005-09-01 | 2006-04-26 | 南京工业大学 | 一种提高蛋白质生物学功能的方法 |
| CA2697193C (en) * | 2007-09-14 | 2017-06-06 | Adimab, Inc. | Rationally designed, synthetic antibody libraries and uses therefor |
| KR20100089060A (ko) | 2007-10-04 | 2010-08-11 | 할싸이언 몰레큘러 | 전자 현미경으로 핵산 중합체를 시퀀싱하는 방법 |
| US20090312196A1 (en) | 2008-06-13 | 2009-12-17 | Codexis, Inc. | Method of synthesizing polynucleotide variants |
| US8383346B2 (en) | 2008-06-13 | 2013-02-26 | Codexis, Inc. | Combined automated parallel synthesis of polynucleotide variants |
| CN101353372A (zh) * | 2008-08-04 | 2009-01-28 | 林峻 | 一种新型蛋白质分子定向进化方法 |
| CN102317473A (zh) * | 2008-12-11 | 2012-01-11 | 加利福尼亚太平洋生物科学股份有限公司 | 核酸模板的分类 |
| US8551096B2 (en) * | 2009-05-13 | 2013-10-08 | Boston Scientific Scimed, Inc. | Directional delivery of energy and bioactives |
| CN101792934A (zh) * | 2009-08-26 | 2010-08-04 | 青岛科技大学 | 一种基于组合原理和pcr建立超大容量基因文库的新方法 |
| JP5649424B2 (ja) * | 2010-02-03 | 2015-01-07 | 大和ハウス工業株式会社 | 防水シート診断方法および診断装置 |
| US20120115734A1 (en) * | 2010-11-04 | 2012-05-10 | Laura Potter | In silico prediction of high expression gene combinations and other combinations of biological components |
| JP4839416B1 (ja) | 2011-01-06 | 2011-12-21 | アクアエンタープライズ株式会社 | 移動過程予測システム、移動過程予測方法、移動過程予測装置及びコンピュータプログラム |
| US20120231447A1 (en) * | 2011-02-15 | 2012-09-13 | Howard Hughes Medical Institute | Surface Passivation Methods for Single Molecule Imaging of Biochemical Reactions |
| CN102206625A (zh) * | 2011-03-04 | 2011-10-05 | 东华大学 | 易错pcr/卡那霉素筛选系统定向进化蛋白质内含子 |
| US20130017540A1 (en) | 2011-06-07 | 2013-01-17 | Yun Yen | Identification of mutation types associated with acquired resistance and methods for using same |
| JP6433028B2 (ja) | 2013-01-31 | 2018-12-05 | コデクシス, インコーポレイテッド | 乗法形式のモデルを使用して生体分子を同定する方法、システム、およびソフトウェア |
-
2014
- 2014-01-29 JP JP2015556114A patent/JP6433028B2/ja active Active
- 2014-01-29 AU AU2014212430A patent/AU2014212430B2/en active Active
- 2014-01-29 KR KR1020157023727A patent/KR20150113166A/ko active Application Filing
- 2014-01-29 AU AU2014212432A patent/AU2014212432B2/en active Active
- 2014-01-29 WO PCT/US2014/013666 patent/WO2014120819A1/en active Application Filing
- 2014-01-29 EP EP14746406.9A patent/EP2951579B1/en active Active
- 2014-01-29 RU RU2015136776A patent/RU2695146C2/ru active
- 2014-01-29 KR KR1020157023739A patent/KR102215219B1/ko active IP Right Grant
- 2014-01-29 NZ NZ710323A patent/NZ710323A/en unknown
- 2014-01-29 CN CN201811237910.6A patent/CN109360608B/zh active Active
- 2014-01-29 CA CA2898777A patent/CA2898777C/en active Active
- 2014-01-29 BR BR112015018449-9A patent/BR112015018449B1/pt active IP Right Grant
- 2014-01-29 CN CN201480018421.5A patent/CN105144190B/zh active Active
- 2014-01-29 JP JP2015556112A patent/JP6377078B2/ja active Active
- 2014-01-29 CA CA2899859A patent/CA2899859C/en active Active
- 2014-01-29 DK DK14746677.5T patent/DK2951754T3/da active
- 2014-01-29 WO PCT/US2014/013668 patent/WO2014120821A1/en active Application Filing
- 2014-01-29 RU RU2015136780A patent/RU2664695C2/ru active
- 2014-01-29 KR KR1020217000247A patent/KR102490720B1/ko active IP Right Grant
- 2014-01-29 US US14/167,709 patent/US9665694B2/en active Active
- 2014-01-29 SG SG11201505969XA patent/SG11201505969XA/en unknown
- 2014-01-29 LT LTEPPCT/US2014/013666T patent/LT2951754T/lt unknown
- 2014-01-29 SG SG11201505977RA patent/SG11201505977RA/en unknown
- 2014-01-29 NZ NZ710299A patent/NZ710299A/en unknown
- 2014-01-29 US US14/167,713 patent/US9684771B2/en active Active
- 2014-01-29 BR BR112015018454-5A patent/BR112015018454B1/pt active IP Right Grant
- 2014-01-29 CN CN201480019389.2A patent/CN105074463B/zh active Active
- 2014-01-29 EP EP14746677.5A patent/EP2951754B1/en active Active
-
2015
- 2015-07-21 IL IL240055A patent/IL240055A0/en active IP Right Grant
- 2015-07-21 IL IL240056A patent/IL240056A0/en active IP Right Grant
-
2017
- 2017-04-04 US US15/479,222 patent/US20170204405A1/en not_active Abandoned
- 2017-04-04 US US15/479,220 patent/US20170211206A1/en not_active Abandoned
- 2017-07-20 JP JP2017140863A patent/JP2017189176A/ja not_active Withdrawn
-
2018
- 2018-07-13 JP JP2018133041A patent/JP6655670B2/ja active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6655670B2 (ja) | 乗法形式のモデルを使用して生体分子を同定する方法、システム、およびソフトウェア | |
| US11342046B2 (en) | Methods and systems for engineering biomolecules | |
| CN105765592A (zh) | 酶变体的自动筛选 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AMND | Amendment | ||
| E902 | Notification of reason for refusal | ||
| AMND | Amendment | ||
| E601 | Decision to refuse application | ||
| X091 | Application refused [patent] | ||
| AMND | Amendment | ||
| X601 | Decision of rejection after re-examination | ||
| A107 | Divisional application of patent | ||
| J201 | Request for trial against refusal decision | ||
| J301 | Trial decision |
Free format text: TRIAL NUMBER: 2021101000022; TRIAL DECISION FOR APPEAL AGAINST DECISION TO DECLINE REFUSAL REQUESTED 20210105 Effective date: 20210928 |