JP7652829B2 - ムスカリンm1受容体アゴニストとしての二環式アザ化合物 - Google Patents
ムスカリンm1受容体アゴニストとしての二環式アザ化合物 Download PDFInfo
- Publication number
- JP7652829B2 JP7652829B2 JP2023060030A JP2023060030A JP7652829B2 JP 7652829 B2 JP7652829 B2 JP 7652829B2 JP 2023060030 A JP2023060030 A JP 2023060030A JP 2023060030 A JP2023060030 A JP 2023060030A JP 7652829 B2 JP7652829 B2 JP 7652829B2
- Authority
- JP
- Japan
- Prior art keywords
- mmol
- carboxylate
- reaction mixture
- tert
- stirred
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images



Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Epidemiology (AREA)
- Rheumatology (AREA)
- Addiction (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicinal Preparation (AREA)
- Indole Compounds (AREA)
Description
リンM1/M4受容体媒介性疾患の処置に有用である化合物に関する。また、本化合物を
含有する医薬組成物、及び本化合物に治療的使用も提供する。
方で神経伝達物質アセチルコリンの作用を媒介するGタンパク質共役受容体スーパーファ
ミリーのメンバーである。M1~M5の5種のmAChRサブタイプがクローニングされ
ている。M1 mAChRは主に皮質、海馬、線条体及び視床のシナプス後膜に発現し;
M2 mAChRは主に脳幹及び視床に位置するが、皮質、海馬及び線条体にも位置し、
そこでそれらはコリン作動性シナプス終末に存在する(Langmeadら、2008
Br J Pharmacol)。しかし、M2 mAChRは、心臓組織上(そこでそ
れらは心臓の迷走神経の神経支配を媒介する)及び平滑筋及び外分泌腺にも周辺的に発現
する。M3 mAChRは、CNSにおいて比較的低レベルで発現するが、平滑筋組織及
び、汗腺及び唾液腺などの腺組織に広く発現する(Langmeadら、2008 Br
J Pharmacol)。
えで重要な役割を果たす。アルツハイマー病などの認知機能障害に関連する疾患には、前
脳基底部におけるコリン作動性ニューロンの脱落が伴う(Whitehouseら、19
82 Science)。臨床像の重要な要素としてまた認知機能障害を有する統合失調
症では、統合失調症の対象の前頭前野、海馬及び尾状核被殻でmAChR密度が低下する
(Deanら、2002 Mol Psychiatry)。さらに、動物モデルで、中
枢コリン作動性経路が遮断または損傷されると、深刻な認知障害が起こり、非選択的mA
ChRアンタゴニストが精神疾患患者において精神異常作用を誘導することが示されてい
る。コリン補充療法は、内因性アセチルコリンの分解を防止するためのアセチルコリンエ
ステラーゼ阻害剤の使用に主として基づいている。これらの化合物は、臨床において認知
機能低下に対して対症療法的に有効性を示しているが、胃腸管運動の異常、徐脈、悪心及
び嘔吐を含む、末梢のM2及びM3 mAChRの刺激に起因する用量制限有害事象を引
き起こす(http://www.drugs.com/pro/donepezil.
html;http://www.drugs.com/pro/rivastigmi
ne.html)。
M1 mAChRアゴニストを同定することを目標としたさらなる発見努力がなされてき
た。そのような努力の結果、キサノメリン、AF267B、サブコメリン、ミラメリン及
びセビメリンなどの化合物により例示される一連のアゴニストが同定された。これらの化
合物の多くは、齧歯類及び/または非ヒト霊長類の両方の認知の前臨床モデルで非常に効
果的であることが示されている。ミラメリンは、齧歯類の作業記憶及び空間記憶における
スコポラミン誘発性障害に対して有効性を示しており;サブコメリンは、マーモセットの
視覚弁別課題において有効性を示し、キサノメリンは、受動的回避パラダイムの認知パフ
ォーマンスにおいてmAChRアンタゴニスト誘発性障害を回復させた。
世界全体で2,660万人)であり、深刻な記憶喪失及び認知機能障害をもたらす。本疾
患の病因は複雑であるが、主にアミロイド-βペプチド(Aβ)からなるアミロイド斑の
凝集、及び過剰リン酸化タウタンパク質により形成される神経原線維のもつれという2つ
の特徴的な脳の病理を特徴とする。Aβの蓄積はADの進行の中心的特徴であると考えら
れ、したがって、ADの処置の多くの推定治療は現在、Aβ産生の阻害を標的としている
。Aβは、膜結合アミロイド前駆体タンパク質(APP)のタンパク質分解切断に由来す
る。APPは、2つの経路、すなわち非アミロイドジェニック経路及びアミロイドジェニ
ック経路によりプロセシングされる。γ-セクレターゼによるAPPの切断は両経路に共
通であるが、前者の場合、APPはα-セクレターゼにより切断されて可溶性のAPPα
が得られる。しかしながら、アミロイドジェニック経路では、APPはβ-セクレターゼ
により切断されて可溶性APPβ、さらにAβも得られる。インビトロ試験により、mA
ChRアゴニストは可溶性の非アミロイドジェニック経路へのAPPのプロセシングを促
進し得ることが示されている。インビボ試験により、mAChRアゴニストである、AF
267Bが、アルツハイマー病の異なる要素を持つモデルである3xTgADトランスジ
ェニックマウスにおいて疾患様病理を変化させることが示された(Caccamoら、2
006 Neuron)。mAChRアゴニストセビメリンは、アルツハイマー型患者に
おけるAβの脳脊髄液レベルをわずかだが有意に低下させることが示されており、したが
って疾患改変有効性の可能性を実証している(Nitschら、2000 Neurol
)。
病剤のようなプロファイルを示すことを示唆している。mAChRアゴニストである、キ
サノメリンは、多くのドーパミン媒介性行動、例えば、ラットにおけるアンフェタミン誘
発歩行運動、マウスにおけるアポモルフィン誘発よじ登り、片側6-OH-DA破壊ラッ
トにおけるドーパミンアゴニスト誘導回転運動及びサルのアンフェタミン誘発運動過多を
回復させる(EPS傾向を伴わない)。キサノメリンは、ラットにおいてA9ではなくA
10のドーパミン細胞の発火及び条件回避を阻害し、前頭前皮質及び側坐核においてc-
fos発現を誘導するが、線条体では誘導しないことも示されている。これらのデータは
すべて、非定型抗精神病剤のようなプロファイルを示唆する(Mirzaら、1999
CNS Drug Rev)。ムスカリン受容体はまた、嗜癖の神経生物学に関係がある
とされている。コカイン及び他の嗜癖物質の強化効果は、中脳辺縁系ドーパミン系により
媒介され、ここで、行動的及び神経科学的試験により、コリン作動性ムスカリン受容体サ
ブタイプがドーパミン作動性神経伝達の調節において重要な役割を担うことが示されてい
る。例えば、M(4)(-/-)マウスは、コカインへの暴露の結果として有意に増強さ
れた報酬駆動性行動を示した(Schmidtら、Psychopharmacolog
y(2011)Aug;216(3):367-78)。さらに、キサノメリンは、これ
らのモデルにおいてコカインの作用を遮断することが実証されている。
ン病、トゥレット症候群及び疾患を誘発する根底的な病原的要因としてドーパミン作動性
機能障害と関連する他の症候群などの運動障害に対する新規処置を表す可能性がある。
及び/または統合失調症の処置のための臨床開発の様々な段階に進んでいる。キサノメリ
ンの第II相臨床試験では、アルツハイマー病に関連する行動障害及び幻覚を含む様々な
認知症状ドメインに対するその有効性が実証された(Bodickら、1997 Arc
h Neurol)この化合物は、統合失調症患者の小規模な第II相試験でも評価され
、プラセボ対照と比較して陽性症状及び陰性症状を有意に減少させた(Shekharら
、2008 Am J Psych)。しかしながら、すべての臨床試験においてキサノ
メリン及び他の関連するmAChRアゴニストは、コリン作動性の有害事象、例えば悪心
、胃腸痛、便通異常(diahorrhea)、発汗(過剰発汗)、唾液分泌過多(唾液
分泌過剰)、失神及び徐脈に関して許容できない安全域を示した。
る種類、すなわち急性疼痛、炎症性疼痛及び神経因性疼痛に分けることができる。急性疼
痛は、組織損傷をもたらし得る刺激から生体を安全に守るうえで重要な保護機能を担って
いる;しかし、術後の疼痛の管理は必要である。炎症性疼痛は、組織損傷、自己免疫応答
及び病原体侵入を含む多くの理由で起こり得、ニューロンの炎症及び疼痛を引き起こす炎
症性メディエーター、例えば神経ペプチド及びプロスタグランジンの作用により引き起こ
される。神経因性疼痛は、非痛み刺激に対する異常な痛み感覚を伴う。神経因性疼痛は、
多くの異なる疾患/外傷、例えば脊髄損傷、多発性硬化症、糖尿病(糖尿病性ニューロパ
チー)、ウイルス感染症(例えばHIVまたは疱疹)と関連する。神経因性疼痛は、疾患
または化学療法の副作用の両方に起因して癌においても一般的である。ムスカリン受容体
の活性化は、脊髄及び脳内の疼痛の高次中枢における受容体の活性化を通して多くの疼痛
状態で鎮痛作用を示すことが示されている。アセチルコリンエステラーゼ阻害剤によるア
セチルコリンの内因性レベルの上昇、アゴニストまたはアロステリックモジュレーターに
よるムスカリン受容体の直接的活性化は、鎮痛活性を有することが示されている。一方で
、アンタゴニストまたはノックアウトマウスの使用によりムスカリン受容体を遮断すると
、痛覚感受性が高まる。疼痛におけるM1受容体の役割に関する証拠については、D.F
.Fiorino and M.Garcia-Guzman,2012により概説され
ている。
対する改善された選択性を示す少数の化合物が同定されている(Bridgesら、20
08 Bioorg Med Chem Lett;Johnsonら、2010 Bi
oorg Med Chem Lett;Budzikら、2010 ACS Med
Chem Lett)。M3 mAChRサブタイプに対する選択性のレベルが高まって
いるにもかかわらず、これらの化合物のいくつかは、このサブタイプ及びM2 mACh
Rサブタイプの両方で顕著なアゴニスト活性を保持している。本明細書において我々は、
意外にもM2及びM3受容体サブタイプよりM1及び/またはM4 mAChRに対して
高レベルの選択性を示す一連の化合物を記載する。
物を提供する。より具体的には、本発明は、M2及びM3受容体サブタイプと比較してM
1受容体及び/またはM4受容体に対して選択性を呈する化合物を提供する。
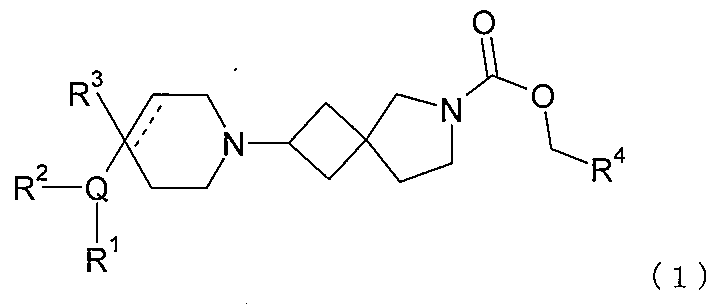
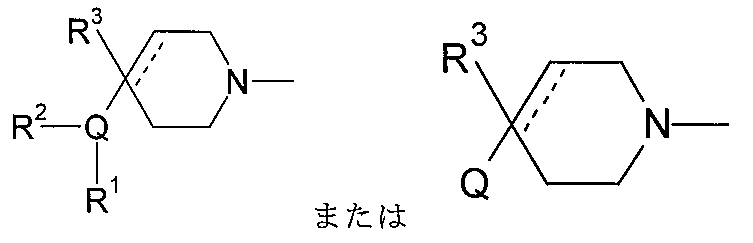
式中、Qは、N、O及びSから選択される1、2、3、または4個のヘテロ原子環員を
含有する5または6員の単環式複素環式の環であり;
R1は、水素;フッ素;塩素;臭素;シアノ;オキソ;ヒドロキシ;OR5;NR5R
6;COR5;COOR5;OCOR5;NR7COR5;CONR5R6;NR7CO
NR5R6;NR7COOR5;OCONR5R6;SR5;SOR5及びSO2R5;
1~6個のフッ素原子で任意選択的に置換されているC1-6非芳香族炭化水素基(1ま
たは2個の、しかし全部ではない、当該炭化水素基の炭素原子が、O、N及びSならびに
それらの酸化形態から選択されるヘテロ原子によって任意選択的に置き換えられてよい)
;及び、O、N及びSならびにそれらの酸化形態から選択される0、1、2または3個の
ヘテロ原子を含有する任意選択的に置換されている5または6員の環から選択され;
R2は、水素;フッ素;塩素;臭素;シアノ;ヒドロキシ;メトキシ;OR5;NR5
R6;COR5;COOR5;OCOR5;NR7COR5;CONR5R6;NR7C
ONR5R6;NR7COOR5;OCONR5R6;SR5;SOR5及びSO2R5
;C1-6非芳香族炭化水素基から選択されるか;またはR1及びR2は1つに接合して
6員の融合芳香族環を形成することができ;
R3は、水素;フッ素;シアノ;ヒドロキシ;アミノ;及び1~6個のフッ素原子で任
意選択的に置換されているC1-9非芳香族炭化水素基から選択され、1、2、または3
個の、しかし全部ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれら
の酸化形態から選択されるヘテロ原子によって任意選択的に置き換えられてよく;
R4は、水素、または1~6個のフッ素原子で任意選択的に置換されているC1-6非
芳香族炭化水素基であり、1または2個の、しかし全部ではない、当該炭化水素基の炭素
原子が、O、N及びSならびにそれらの酸化形態から選択されるヘテロ原子によって任意
選択的に置き換えられてよく;
R5、R6及びR7は、同じであるかまたは異なり、それぞれは、水素、1つまたは複
数のフッ素原子で任意選択的に置換されている非芳香族C1-4炭化水素基;または式C
H2N(Ra)COORbの基から独立して選択され;
Raは、水素及び非芳香族C1-4炭化水素基から選択され;
Rbは、フッ素;塩素;臭素;シアノ;ヒドロキシ;メトキシ;アミノ;またはシクロ
アルキル、ヘテロシクロアルキル、アリールまたはヘテロアリール基から選択される1つ
または複数の基で任意選択的に置換されている非芳香族C1-4炭化水素基であり;
点線は随意の第二の炭素-炭素結合を示すが、第二の炭素-炭素結合が存在するときに
はR3は存在しない。

式中、Qは、N、O及びSから選択される1、2、3または4個のヘテロ原子環員を含
有する5または6または7員の単環式複素環式の環であり;
R1は、水素;フッ素;塩素;臭素;シアノ;オキソ;ヒドロキシ;OR5;NR5R
6;COR5;COOR5;OCOR5;NR7COR5;CONR5R6;NR7CO
NR5R6;NR7COOR5;OCONR5R6;SR5;SOR5及びSO2R5;
1~6個のフッ素原子で任意選択的に置換されているC1-6非芳香族炭化水素基(ここ
で1または2個の、しかし全部ではない、当該炭化水素基の炭素原子が、O、N及びSな
らびにそれらの酸化形態から選択されるヘテロ原子によって任意選択的に置き換えられて
よい);及び、O、N及びSならびにそれらの酸化形態から選択される0、1、2または
3個のヘテロ原子を含有する任意選択的に置換されている5または6員の環から選択され
;
R2は、水素;フッ素;塩素;臭素;シアノ;ヒドロキシ;メトキシ;OR5;NR5
R6;COR5;COOR5;OCOR5;NR7COR5;CONR5R6;NR7C
ONR5R6;NR7COOR5;OCONR5R6;SR5;SOR5及びSO2R5
;C1-6非芳香族炭化水素基から選択されるか;またはR1及びR2は1つに接合して
6員の融合芳香族環を形成することができ;
R3は、水素;フッ素;シアノ;ヒドロキシ;アミノ;及び1~6個のフッ素原子で任
意選択的に置換されているC1-9非芳香族炭化水素基から選択され、1、2、または3
個の、しかし全部ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれら
の酸化形態から選択されるヘテロ原子によって任意選択的に置き換えられてよく;
R4は、水素、または1~6個のフッ素原子で任意選択的に置換されているC1-6非
芳香族炭化水素基であり、1または2個の、しかし全部ではない、当該炭化水素基の炭素
原子が、O、N及びSならびにそれらの酸化形態から選択されるヘテロ原子によって任意
選択的に置き換えられてよく;
R5、R6及びR7は、同じであるかまたは異なり、それぞれは、水素、1つまたは複
数のフッ素原子で任意選択的に置換されている非芳香族C1-4炭化水素基;または式C
H2N(Ra)COORbの基から独立して選択され;
Raは、水素及び非芳香族C1-4炭化水素基から選択され;
Rbは、フッ素;塩素;臭素;シアノ;ヒドロキシ;メトキシ;アミノ;またはシクロ
アルキル、ヘテロシクロアルキル、アリールまたはヘテロアリール基から選択される1つ
または複数の基で任意選択的に置換されている非芳香族C1-4炭化水素基であり;
点線は随意の第二の炭素-炭素結合を示すが、第二の炭素-炭素結合が存在するときに
はR3は存在しない。
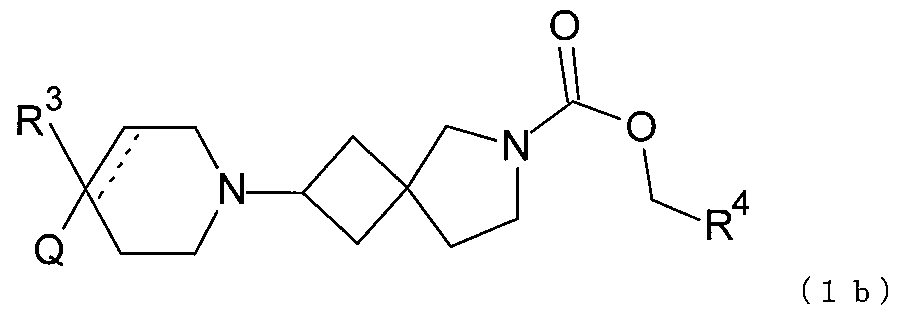
式中、Qは、N、O及びSから選択される1、2、3、または4個のヘテロ原子環員を
含有する任意選択的に置換されている5または6または7員の複素環であり;
R3は、水素;フッ素;シアノ;ヒドロキシ;アミノ;及び1~6個のフッ素原子で任
意選択的に置換されているC1-9非芳香族炭化水素基から選択され、1、2、または3
個の、しかし全部ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれら
の酸化形態から選択されるヘテロ原子によって任意選択的に置き換えられてよく;
R4は、水素、または1~6個のフッ素原子で任意選択的に置換されているC1-6非
芳香族炭化水素基であり、1または2個の、しかし全部ではない、当該炭化水素基の炭素
原子が、O、N及びSならびにそれらの酸化形態から選択されるヘテロ原子によって任意
選択的に置き換えられてよく;
点線は随意の第二の炭素-炭素結合を示すが、第二の炭素-炭素結合が存在するときに
はR3は存在しない。
80に定義される通りである。
個のさらなる環員を含有する芳香族複素環である、実施形態1.3に記載の化合物。
なる環員を含有する芳香族複素環である、実施形態1.4に記載の化合物。
5に記載の化合物。
環に連結している、実施形態1.1~1.6のいずれか1つに記載の化合物。
の窒素原子により隣接する6員環に連結している、実施形態1.1~1.6のいずれか1
つに記載の化合物。
-ピラゾリル、2-チアゾリル、2-オキサゾリル、トリアゾリル、テトラゾリル、チア
ジアゾリル、オキサジアゾリル、及びそれらの互変異性体から選択される、実施形態1.
1に記載の化合物。
体から選択される、実施形態1.6に記載の化合物。
に記載の化合物。
ontining)2-オキソ-3N(3-ピペリジン-2-オン)環である、実施形態
1.14に記載の化合物。
合物。
.16に記載の化合物。
記載の化合物。
、それは1つのR1及び/またはR2から選択されてよく、R1及びR2は同じであるか
または異なってよい。Qに対するさらなる置換基には、(L)-R10、(L)-R11
及び(L)-R12が含まれてよく、ここでLは、結合であるかまたはCH2基であり;
R10、R11及びR12は、水素;フッ素;塩素;臭素;シアノ;オキソ;ヒドロキシ
;OR15;NR15R16;COR15;CSR15;COOR15;COSR15;
OCOR15;NR17COR15;CONR15R16;CSNR15R16;NR1
7CONR15R16;R17COOR15;OCONR15R16;SR15;SOR
15及びSO2R15;1~6個のフッ素原子で任意選択的に置換されているC1-6非
芳香族炭化水素基(1または2個の、しかし全部ではない、当該炭化水素基の炭素原子が
、O、N及びSならびにそれらの酸化形態から選択されるヘテロ原子によって任意選択的
に置き換えられてよい);及び、O、N及びSならびにそれらの酸化形態から選択される
0、1、2または3個のヘテロ原子を含有する任意選択的に置換されている5または6員
の環から独立して選択され;
当該任意選択的に置換されている5または6員の環に対する随意の置換基は、水素;フッ
素;塩素;臭素;シアノ;オキソ;ヒドロキシ;OR5;NR5R6;COR5;COO
R5;OCOR5;NR7COR5;CONR5R6;NR7CONR5R6;NR7C
OOR5;OCONR5R6;SR5;SOR5及びSO2R5;及び1~6個のフッ素
原子で任意選択的に置換されているC1-6非芳香族炭化水素基(1または2個の、しか
し全部ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれらの酸化形態
から選択されるヘテロ原子によって任意選択的に置き換えられてよい)からなる基R8か
ら選択され;
R15、R16及びR17は同じであるかまたは異なり、または1つに接合して環を形成
してよく、それぞれは、水素、1つまたは複数のフッ素原子で任意選択的に置換されてい
る非芳香族C1-6炭化水素基(1または2個の、しかし全部ではない、当該炭化水素基
の炭素原子が、O、N及びSならびにそれらの酸化形態から選択されるヘテロ原子によっ
て任意選択的に置き換えられてよい);または式CH2N(Ra)COORbの基;また
は式(L)-R18の基(Lは、結合またはCH2基であり、R18はO、N及びSなら
びにそれらの酸化形態から選択される0、1、2または3個のヘテロ原子を含有する任意
選択的に置換されている5または6員の環である)から独立して選択され;
当該任意選択的に置換されている5または6員の環に対する随意の置換基は基R8から選
択される、実施形態1.1bに記載の化合物。
;NR5R6;COR5;COOR5;OCOR5;NR7COR5;CONR5R6;
NR7CONR5R6;NR7COOR5;OCONR5R6;SR5;SOR5及びS
O2R5;1~6個のフッ素原子で任意選択的に置換されているC1-6非芳香族炭化水
素基(1または2個の、しかし全部ではない、当該炭化水素基の炭素原子が、O、N及び
Sならびにそれらの酸化形態から選択されるヘテロ原子によって任意選択的に置き換えら
れてよい);及び、O、N及びSならびにそれらの酸化形態から選択される0、1、2ま
たは3個のヘテロ原子を含有する任意選択的に置換されている5または6員の環から選択
され、
当該任意選択的に置換されている5または6員の環に対する随意の置換基は、水素;フッ
素;塩素;臭素;シアノ;オキソ;ヒドロキシ;OR5;NR5R6;COR5;COO
R5;OCOR5;NR7COR5;CONR5R6;NR7CONR5R6;NR7C
OOR5;OCONR5R6;SR5;SOR5及びSO2R5;及び1~6個のフッ素
原子で任意選択的に置換されているC1-6非芳香族炭化水素基(1または2個の、しか
し全部ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれらの酸化形態
から選択されるヘテロ原子によって任意選択的に置き換えられてよい)からなる基R8か
ら選択される、実施形態1.1~1.19のいずれか1つに記載の化合物。
;NR5R6;COR5;COOR5;OCOR5;NR7COR5;CONR5R6;
NR7CONR5R6;NR7COOR5;OCONR5R6;SR5;SOR5及びS
O2R5;1~6個のフッ素原子で任意選択的に置換されているC1-5非芳香族炭化水
素基(1または2個の、しかし全部ではない、当該炭化水素基の炭素原子が、O、N及び
Sならびにそれらの酸化形態から選択されるヘテロ原子によって任意選択的に置き換えら
れてよい);及び、O、N及びSならびにそれらの酸化形態から選択される0、1、また
は2個のヘテロ原子を含有する任意選択的に置換されている5または6員の環から選択さ
れ;
当該任意選択的に置換されている5または6員の環に対する随意の置換基は、フッ素;塩
素;臭素;シアノ;オキソ;ヒドロキシ;OR5;NR5R6;COR5;COOR5;
OCOR5;NR7COR5;CONR5R6;NR7CONR5R6;NR7COOR
5;OCONR5R6;SR5;SOR5及びSO2R5;及び1~6個のフッ素原子で
任意選択的に置換されているC1-4非芳香族炭化水素基(1または2個の、しかし全部
ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれらの酸化形態から選
択されるヘテロ原子によって任意選択的に置き換えられてよい)からなる基R8から選択
される、実施形態1.20に記載の化合物。
;NR5R6;COR5;COOR5;OCOR5;NR7COR5;CONR5R6;
NR7CONR5R6;NR7COOR5;OCONR5R6;SR5;SOR5及びS
O2R5;1~6個のフッ素原子で任意選択的に置換されているC1-4非芳香族炭化水
素基(1または2個の、しかし全部ではない、当該炭化水素基の炭素原子が、O、N及び
Sならびにそれらの酸化形態から選択されるヘテロ原子によって任意選択的に置き換えら
れてよい);及び、O、N及びSならびにそれらの酸化形態から選択される0、1、また
は2個のヘテロ原子を含有する任意選択的に置換されている5または6員のアリールまた
はヘテロアリール環から選択され;
当該任意選択的に置換されている5または6員のアリールまたはヘテロアリール環に対す
る随意の置換基は、フッ素;塩素;臭素;シアノ;オキソ;ヒドロキシ;OR5;NR5
R6;COR5;COOR5;OCOR5;NR7COR5;CONR5R6;NR7C
ONR5R6;NR7COOR5;OCONR5R6;SR5;SOR5及びSO2R5
;及び1~6個のフッ素原子で任意選択的に置換されているC1-4非芳香族炭化水素基
(1または2個の、しかし全部ではない、当該炭化水素基の炭素原子が、O、N及びSな
らびにそれらの酸化形態から選択されるヘテロ原子によって任意選択的に置き換えられて
よい)からなる基R8から選択される、実施形態1.21に記載の化合物。
5R6;COR5;COOR5;OCOR5;NR7COR5;CONR5R6;NR7
CONR5R6;NR7COOR5;OCONR5R6;SO2R5;1~6個のフッ素
原子で任意選択的に置換されているC1-4非芳香族炭化水素基、(1または2個の、し
かし全部ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれらの酸化形
態から選択されるヘテロ原子によって任意選択的に置き換えられてよい);及び、O、N
及びSならびにそれらの酸化形態から選択される0、1、2または3個のヘテロ原子を含
有する任意選択的に置換されている5または6員の環から選択され、当該任意選択的に置
換されている5または6員のアリールまたはヘテロアリール環に対する随意の置換基は、
フッ素;塩素;臭素;シアノ;オキソ;ヒドロキシ;OR5;NR5R6;COR5;C
OOR5;OCOR5;NR7COR5;CONR5R6;NR7CONR5R6;NR
7COOR5;OCONR5R6;SR5;SOR5及びSO2R5;及び1~6個のフ
ッ素原子で任意選択的に置換されているC1-4非芳香族炭化水素基(1または2個の、
しかし全部ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれらの酸化
形態から選択されるヘテロ原子によって任意選択的に置き換えられてよい)からなる基R
8から選択される、実施形態1.1~1.19のいずれか1つに記載の化合物。
COR5;COOR5;OCOR5;NR7COR5;CONR5R6;NR7CONR
5R6;NR7COOR5;OCONR5R6;SO2R5;及び1~6個のフッ素原子
で任意選択的に置換されているC1-4非芳香族炭化水素基(1または2個の、しかし全
部ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれらの酸化形態から
選択されるヘテロ原子によって任意選択的に置き換えられてよい)から選択される、実施
形態1.23に記載の化合物。
COR5;COOR5;OCOR5;NR7COR5;CONR5R6;NR7CONR
5R6;NR7COOR5;SO2R5;及び1~6個のフッ素原子で任意選択的に置換
されているC1-4非芳香族炭化水素基から選択される、実施形態1.24に記載の化合
物。
及び1~6個のフッ素原子で任意選択的に置換されているC1-6非芳香族炭化水素基か
ら選択される、実施形態1.25に記載の化合物。
1~6個のフッ素原子で任意選択的に置換されているC1-4飽和非芳香族炭化水素基か
ら選択される、実施形態1.26に記載の化合物。
ル基から選択される、実施形態1.27に記載の化合物。
る、実施形態1.28に記載の化合物。
.29に記載の化合物。
3;CONHMe;CON(Me)2;COCF3;CO-シクロプロピル;CO-シク
ロブチル;CONHEt;COH;NH2;OMeである、実施形態1.20~1.30
に記載の化合物。
C1-6非芳香族炭化水素基から選択されるか;またはR1と接合して6員の融合芳香族
環を形成する、実施形態1.1~1.33のいずれか1つに記載の化合物。
水素基から選択される、実施形態1.34に記載の化合物。
れる、実施形態1.35に記載の化合物。
、実施形態1.36に記載の化合物。
記載の化合物。
。
はヘテロアリールであり得る、実施形態1.34に記載の化合物。
1.40のいずれか1つに記載の化合物。
~1.40のいずれか1つに記載の化合物。
素原子で任意選択的に置換されているC1-6非芳香族炭化水素基(1または2個の、し
かし全部ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれらの酸化形
態から選択されるヘテロ原子によって任意選択的に置き換えられてよい)から選択される
、実施形態1.42に記載の化合物。
素原子で任意選択的に置換されているC1-6非芳香族炭化水素基(1個の、しかし全部
ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれらの酸化形態から選
択されるヘテロ原子によって任意選択的に置き換えられてよい)から選択される、実施形
態1.43に記載の化合物。
びC1-4アルコキシから選択され、C1-4アルキル及びC1-4アルコキシはそれぞ
れ1~6個のフッ素原子で任意選択的に置換されている、実施形態1.44に記載の化合
物。
1.45に記載の化合物。
.47のいずれか1つに記載の化合物。
記載の化合物。
、実施形態1.49に記載の化合物。
る、実施形態1.50に記載の化合物。
。
れている非芳香族C1-4炭化水素基である;または式CH2N(Ra)COORbの基
である、先行実施形態のいずれか1つに記載の化合物。
1.54に記載の化合物。
1つに記載の化合物。
実施形態1.1~1.53のいずれか1つに記載の化合物。
または実施形態1.56に記載の化合物。
に記載の化合物。
実施形態1.59に記載の化合物。
のいずれか1つに記載の化合物。
1.62に記載の化合物。
1つに記載の化合物。
に記載の化合物。
実施形態1.65に記載の化合物。
のいずれか1つに記載の化合物。
1.67に記載の化合物。
1つに記載の化合物。
実施形態1.1~1.66のいずれか1つに記載の化合物。
または実施形態1.70に記載の化合物。
に記載の化合物。
実施形態1.72に記載の化合物。
、N及びSならびにそれらの酸化形態から選択される0、1または2または3個のヘテロ
原子を含有する芳香族環から選択される、先行実施形態のいずれか1つに記載の化合物。
、N及びSならびにそれらの酸化形態から選択される0、1または2または3個のヘテロ
原子を含有する非芳香族環から選択される、実施形態1.1~1.73のいずれか1つに
記載の化合物。
化合物。
化合物。
、1、2または3個の置換基R8で置換されている、先行実施形態のいずれか1つに記載
の化合物。
物。
R5R6;COR5;COOR5;OCOR5;NR7COR5;CONR5R6;SR
5;SOR5及びSO2R5;及び1~6個のフッ素原子で任意選択的に置換されている
C1-6非芳香族炭化水素基(1または2個の、しかし全部ではない、当該炭化水素基の
炭素原子が、O、N及びSならびにそれらの酸化形態から選択されるヘテロ原子によって
任意選択的に置き換えられてよい)から選択される、実施形態1.81、1.82、1.
83、1.85及び1.86のいずれか1つに記載の化合物。
R5;COOR5;OCOR5及びSO2R5;及び1~6個のフッ素原子で任意選択的
に置換されているC1-4非芳香族炭化水素基(1または2個の、しかし全部ではない、
当該炭化水素基の炭素原子が、O、N及びSならびにそれらの酸化形態から選択されるヘ
テロ原子によって任意選択的に置き換えられてよい)から選択される、実施形態1.87
に記載の化合物。
1~6個のフッ素原子で任意選択的に置換されているC1-4非芳香族炭化水素基から選
択される、実施形態1.88に記載の化合物。
アルキルから選択される、実施形態1.89に記載の化合物。



載の化合物。

6員の複素環式またはヘテロアリール(heteraryl)環であり、R4は実施形態
1.48~1.53のいずれか1つに定義される通りであり;
または式(2a)

たは7員の複素環式またはヘテロアリール(heteraryl)環であり、R4は実施
形態1.48~1.53のいずれか1つに定義される通りである、
に係る化合物。
複数の置換基、例えば、1、2または3つの置換基を有し、それは(L)-R10、(L
)-R11及び(L)-R12から選択され、ここでLは、結合であるかまたはCH2基
であり;R10、R11及びR12は、水素;フッ素;塩素;臭素;シアノ;オキソ;ヒ
ドロキシ;OR15;NR15R16;COR15;CSR15;COOR15;COS
R15;OCOR15;NR17COR15;CONR15R16;CSNR15R16
;NR17CONR15R16;R17COOR15;OCONR15R16;SR15
;SOR15及びSO2R15;1~6個のフッ素原子で任意選択的に置換されているC
1-6非芳香族炭化水素基(1または2個の、しかし全部ではない、当該炭化水素基の炭
素原子が、O、N及びSならびにそれらの酸化形態から選択されるヘテロ原子によって任
意選択的に置き換えられてよい);及びO、N及びSならびにそれらの酸化形態から選択
される0、1、2または3個のヘテロ原子を含有する任意選択的に置換されている5また
は6員の環から独立して選択され;
当該任意選択的に置換されている5または6員の環に対する随意の置換基は、水素;フッ
素;塩素;臭素;シアノ;オキソ;ヒドロキシ;OR5;NR5R6;COR5;COO
R5;OCOR5;NR7COR5;CONR5R6;NR7CONR5R6;NR7C
OOR5;OCONR5R6;SR5;SOR5及びSO2R5及び1~6個のフッ素原
子で任意選択的に置換されているC1-6非芳香族炭化水素基(1または2個の、しかし
全部ではない、当該炭化水素基の炭素原子が、O、N及びSならびにそれらの酸化形態か
ら選択されるヘテロ原子によって任意選択的に置き換えられてよい)からなる基R8から
選択され;
R15、R16及びR17は同じであるかまたは異なり、または1つに接合して環を形
成してよく、それぞれは、水素、1つまたは複数のフッ素原子で任意選択的に置換されて
いる非芳香族C1-6炭化水素基(1または2個の、しかし全部ではない、当該炭化水素
基の炭素原子が、O、N及びSならびにそれらの酸化形態から選択されるヘテロ原子によ
って任意選択的に置き換えられてよい);または式CH2N(Ra)COORbの基;ま
たは式(L)-R18の基から独立して選択され、ここでLは、結合またはCH2基であ
り、R18は、O、N及びSならびにそれらの酸化形態から選択される0、1、2または
3個のヘテロ原子を含有する任意選択的に置換されている5または6員の環であり;
当該任意選択的に置換されている5または6員の環に対する随意の置換基は、基R8か
ら選択される、当該化合物。

のいずれか1つに定義される通りであり、環Aは1または2個の窒素環員を含有する5員
の複素環式またはヘテロアリール環である、実施形態1.1~1.93に記載の化合物。
態に1.94記載の化合物。

0のいずれか1つに定義される通りである、実施形態1.96に記載の化合物。

0のいずれか1つに定義される通りである、実施形態1.98に記載の化合物。

0のいずれか1つに定義される通りである、実施形態1.98に記載の化合物。
4に記載の化合物。

0のいずれか1つに定義される通りである、実施形態1.101に記載の化合物。



0のいずれか1つに定義される通りである、実施形態1.101に記載の化合物。



員を含有する6員の単環式複素環式の環である、実施形態1.1に記載の化合物。


員を含有する7員の単環式複素環式の環である、実施形態1.1に記載の化合物。
1~4-20または5-1~5-2のいずれか1つに定義される通りである、実施形態1
.1に記載の化合物。
つに記載の化合物。
合物。
1.115に記載の化合物。
本出願では、別段の指示がない限り、以下の定義が適用される。
問題の疾患若しくは障害に罹患している、または罹患する恐れがある、または罹患する恐
れがある可能性がある対象に化合物が投与される、任意の形態の介入を説明するのに使用
される。ゆえに、「処置」という用語は、予防(preventative)(予防(p
rophylactic))処置、及び疾患または障害の測定可能または検出可能な症状
が示されている処置の両方を包含する。
効量」という用語は、所望の治療効果を生成するのに有効である、化合物の量を指す。例
えば、状態が疼痛である場合には、治療有効量は所望のレベルの疼痛緩和を提供するのに
十分な量である。疼痛緩和の所望のレベルは、例えば、疼痛の完全な除去または疼痛の重
症度における軽減であり得る。
おけるような)「非芳香族炭化水素基」という用語は、炭素原子及び水素原子からなり、
芳香族環を含有しない基を指す。炭化水素基は完全に飽和していてもよく、または1つま
たは複数の炭素-炭素二重結合若しくは炭素-炭素三重結合、または二重結合と三重結合
との混合物を含んでいてよい。炭化水素基は直鎖基または分岐鎖基であってよく、または
環式基からなっていても、環式基を含有していてもよい。ゆえに、非芳香族炭化水素とい
う用語には、アルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、
シクロアルキルアルキル、シクロアルケニルアルキルなどが含まれる。
ロアリール及び「シクロアルケニル」という用語は、別段の指示がない限り、それらの従
来の意味(例えば、IUPAC Gold Bookに定義される通り)で使用される。
炭素二重結合または三重結合を含有しない炭化水素基を指す。飽和炭化水素基は、それゆ
え、アルキル基、シクロアルキル基、シクロアルキルアルキル基、アルキルシクロアルキ
ル基またはアルキルシクロアルキルアルキル基であることができる。C1-4飽和炭化水
素基の例には、C1-4アルキル基、シクロプロピル、シクロブチル及びシクロプロピル
メチルが含まれる。
容される場合、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル及びシ
クロヘプチルなどの単環式シクロアルキル基と、二環式基及び三環式基との両方を含む。
二環式シクロアルキル基には、ビシクロヘプタン、ビシクロオクタン及びアダマンタンな
どの架橋環系が含まれる。
し全部ではない、非芳香族炭化水素基の炭素原子が、O、N及びSならびに(R1及びR
4の場合には)その酸化形態から選択されるヘテロ原子によって任意選択的に置き換えら
れてよい。炭素原子がヘテロ原子で置き換えられる場合、炭素と比べてヘテロ原子の原子
価が低いということは、置き換えられている炭素原子と結合していたであろう原子よりも
少ない原子がヘテロ原子と結合し得ることを意味することが理解されるだろう。ゆえに、
例えば、CH2基の炭素原子(原子価4)を酸素(原子価2)で置き換えることは、結果
として生じる分子が、2個少ない水素原子を含有し得ることを意味し、CH2基の炭素原
子(原子価4)を窒素(原子価3)で置き換えることは、結果として生じる分子が、1個
少ない水素原子を含有し得ることを意味するだろう。
はチオエーテル-CH2-S-CH2-を得るための酸素または硫黄による-CH2-C
H2-CH2-鎖中の炭素原子の置換、ニトリル(シアノ)基CH2-C≡Nを得るため
の窒素による基CH2-C≡C-H中の炭素原子の置換、ケトン-CH2-C(O)-C
H2-を得るためのC=Oによる基-CH2-CH2-CH2-中の炭素原子の置換、ス
ルホキシド-CH2-S(O)-CH2-またはスルホン-CH2-S(O)2-CH2
-を得るためのS=OまたはSO2による基-CH2-CH2-CH2-中の炭素原子の
置換、アミド-CH2-CH2-C(O)-NH-を得るためのC(O)NHによる-C
H2-CH2-CH2-鎖中の炭素原子の置換、アミン-CH2-NH-CH2-を得る
ための窒素による-CH2-CH2-CH2-鎖中の炭素原子の置換、及び、エステル(
またはカルボン酸)-CH2-CH2-C(O)-O-を得るためのC(O)Oによる-
CH2-CH2-CH2-鎖中の炭素原子の置換が挙げられる。そのような各置換の場合
、炭化水素基の少なくとも1つの炭素原子は残らなければならない。
式(1)、(1a)または(1b)の多くの化合物は、塩の形態、例えば酸付加塩、ま
たは、ある特定の場合にはカルボン酸塩、スルホン酸塩及びリン酸塩などの有機及び無機
塩基の塩で存在することができる。こうした塩はすべて、本発明の範囲内にあり、式(1
)、(1a)または(1b)の化合物に関しては、実施形態1.114~1.116に定
義されるような化合物の塩形態を含む。
Selection,and Use,P.Heinrich Stahl(編集),C
amille G.Wermuth(編集),ISBN:3-90639-026-8,
Hardcover,388頁,August 2002に記載される方法などの従来の
化学的方法により、塩基性または酸性部分を含有する親化合物から合成することができる
。一般に、そのような塩は、これらの化合物の遊離酸または塩基形態と適切な塩基または
酸とを、水中若しくは有機溶媒中、またはこの2つの混合液中で反応させることにより調
製することができ;一般には、エーテル、酢酸エチル、エタノール、イソプロパノールま
たはアセトニトリルなどの非水媒体が使用される。
両方で形成してよい。実施形態1.120に包含される酸付加塩の例として、酢酸、2,
2-ジクロロ酢酸、アジピン酸、アルギン酸、アスコルビン酸(例えば、L-アスコルビ
ン酸)、L-アスパラギン酸、ベンゼンスルホン酸、安息香酸、4-アセトアミド安息香
酸、ブタン酸、(+)ショウノウ酸、カンファー-スルホン酸、(+)-(1S)-カン
ファー-10-スルホン酸、カプリン酸、カプロン酸、カプリル酸、桂皮酸、クエン酸、
シクラミン酸、ドデシル硫酸、エタン-1,2-ジスルホン酸、エタンスルホン酸、2-
ヒドロキシエタンスルホン酸、ギ酸、フマル酸、ガラクタル酸、ゲンチシン酸、グルコヘ
プトン酸、D-グルコン酸、グルクロン酸(例えば、D-グルクロン酸)、グルタミン酸
(例えば、L-グルタミン酸)、α-オキソグルタル酸、グリコール酸、馬尿酸、ハロゲ
ン化水素酸(例えば、臭化水素酸、塩酸、ヨウ化水素酸)、イセチオン酸、乳酸(例えば
、(+)-L-乳酸、(±)-DL-乳酸)、ラクトビオン酸、マレイン酸、リンゴ酸、
(-)-L-リンゴ酸、マロン酸、(±)-DL-マンデル酸、メタンスルホン酸、ナフ
タレン-2-スルホン酸、ナフタレン-1,5-ジスルホン酸、1-ヒドロキシ-2-ナ
フトエ酸、ニコチン酸、硝酸、オレイン酸、オロト酸、シュウ酸、パルミチン酸、パモ酸
、リン酸、プロピオン酸、ピルビン酸、L-ピログルタミン酸、サリチル酸、4-アミノ
-サリチル酸、セバシン酸、ステアリン酸、コハク酸、硫酸、タンニン酸、(+)-L-
酒石酸、チオシアン酸、p-トルエンスルホン酸、ウンデシレン酸及び吉草酸からなる群
から選択される酸、ならびにアシル化アミノ酸及び陽イオン交換樹脂で形成されるモノ-
またはジ-塩が挙げられる。
、第四級アンモニウム塩を、例えば、当業者に周知の方法にしたがって、アルキル化剤と
の反応により形成し得る。そのような第四級アンモニウム化合物は式(1)、(1a)ま
たは(1b)の範囲内にある。
してよい。
され得る塩の例は、Bergeら、1977,「Pharmaceutically A
cceptable Salts,」J.Pharm.Sci.,Vol.66,pp.
1-19で論じられている。しかし、医薬的に許容されない塩を中間体形態として調製し
てもよく、その後それを医薬的に許容され得る塩に変換してよい。そのような医薬的に許
容されない塩形態は、例えば、本発明の化合物の精製または分離において有用である場合
があり、これも本発明の一部を形成する。
立体異性体は、同じ分子式及び結合原子の配列を有するが、空間におけるその原子の三
次元的配置においてのみ異なる異性体分子である。立体異性体は、例えば、幾何異性体ま
たは光学異性体であることができる。
幾何異性体の場合、異性は、炭素-炭素二重結合のまわりのシス及びトランス(Z及び
E)異性、またはアミド結合のまわりのシス及びトランス異性体、または(例えばオキシ
ムにおける)炭素窒素二重結合のまわりのsyn及びanti異性、または回転の束縛が
存在する結合のまわりの回転異性、またはシクロアルカン環などの環のまわりのシス及び
トランス異性のように二重結合のまわりの原子または基の配置が異なることによる。
1.116のいずれか1つに記載の化合物の幾何異性体を提供する。
式中の化合物が1つまたは複数のキラル中心を含有し、2つ以上の光学異性体の形態で
存在することができる場合、本化合物への言及は、文脈により別途要されない限り、その
全光学異性体形態(例えばエナンチオマー、エピマー及びジアステレオ異性体)を、個々
の光学異性体、または混合物(例えばラセミ混合物)若しくは2つ以上の光学異性体のい
ずれかとして含む。
する実施形態1.1~1.121のいずれか1つに記載の化合物を提供する。
び-異性体、またはd及びl異性体のように)、またはそれらは、Cahn、Ingol
d及びPrelogにより開発された「R及びS」命名法を用いてその絶対立体化学の点
から特徴付けされてもよい。Advanced Organic Chemistry
by Jerry March,第4版,John Wiley & Sons,New
York,1992,pages 109-114、さらにCahn,Ingold
& Prelog,Angew.Chem.Int.Ed.Engl.,1966,5,
385-415を参照されたい。光学異性体は、キラルクロマトグラフィー(キラル支持
体上でのクロマトグラフィー)を含むいくつかの技術により分離することができ、こうし
た技術は、当業者に周知である。キラルクロマトグラフィーに代わるものとして、(+)
-酒石酸、(-)-ピログルタミン酸、(-)-ジ-トルオイル-L-酒石酸、(+)-
マンデル酸、(-)-リンゴ酸及び(-)-カンファースルホン酸などのキラル酸でジア
ステレオ異性体の塩を形成し、優先晶出によりジアステレオ異性体を分離し、その後解塩
して遊離塩基の個別のエナンチオマーを得ることにより、光学異性体を分離することがで
きる。
ーの一方のエナンチオマーが、例えば生物活性の面で他方のエナンチオマーに対して利点
を呈することがある。ゆえに、ある特定の環境では、一対のエナンチオマーの一方のみ、
または複数のジアステレオ異性体の1つのみを治療剤として使用することが望ましくあり
得る。
キラル中心を有する、実施形態1.132に記載の化合物を含有する組成物であって、実
施形態1.108の化合物の少なくとも55%(例えば、少なくとも60%、65%、7
0%、75%、80%、85%、90%または95%)が単一の光学異性体(例えば、エ
ナンチオマーまたはジアステレオ異性体)として存在する、当該組成物を提供する。
または使用のための化合物)の総量の99%以上(例えば、実質的に全て)が単一の光学
異性体として存在する。
して存在する。
て存在する。
ゆえに、本発明は以下を提供する。
合物。
化合物。
実施形態1.1~1.138のいずれか1つに定義される本発明の化合物は、1つまた
は複数の同位体置換を含有してよく、特定の元素に関する言及は、その範囲内に元素の全
ての同位体を含む。例えば、水素に関する言及は、その範囲内に1H、2H(D)、及び
3H(T)を含む。同様に、炭素及び酸素に関する言及は、それらの範囲内にそれぞれ1
2C、13C及び14Cと16Oと18Oを含む。
囲内に同位体変種を含む。例えば、エチル基などのアルキル基に関する言及は、基内の1
つまたは複数の水素原子がジュウテリウム同位体またはトリチウム同位体の形態である、
例えば、全5個の水素原子がジュウテリウム同位体であるエチル基(パージュウテロエチ
ル基)のような変種も包含される。
0)では、実施形態1.1~1.138のいずれか1つの化合物は、放射性同位体を含有
しない。そのような化合物は治療的使用に好ましい。しかし、別の実施形態(実施形態1
.141)では、実施形態1.1~1.138のいずれか1つの化合物は、1つまたは複
数の放射性同位体を含有し得る。そのような放射性同位体を含有する化合物は、診断の場
面で有用であり得る。
実施形態1.1~1.141のいずれか1つに定義される式(1)、(1a)または(
1b)の化合物は、溶媒和物を形成し得る。好ましい溶媒和物は、本発明の化合物の固体
状態構造(例えば、結晶構造)に、非毒性の医薬的に許容され得る溶媒(以下、溶媒和性
溶媒と称する)の分子が取り込まれることにより形成される溶媒和物である。そのような
溶媒の例には、水、アルコール(例えば、エタノール、イソプロパノール及びブタノール
)及びジメチルスルホキシドが含まれる。溶媒和物は、本発明の化合物を溶媒または溶媒
和性溶媒を含有する溶媒の混合物で再結晶化することにより調製することができる。任意
の所与の例において、溶媒和物が形成されているか否かは、熱重量分析(TGE)、示差
走査熱量測定(DSC)及びX線結晶解析などの周知の標準的な技術を使用して化合物の
結晶を解析に供することにより決定することができる。溶媒和物は、化学量論的溶媒和物
または非化学量論的溶媒和物であり得る。特に好ましい溶媒和物は水和物であり、水和物
の例には、半水和物、一水和物及び二水和物が含まれる。
る。
載の化合物。
ては、Brynら、Solid-State Chemistry of Drugs,
第2版、米国ウェストラファイエットのSSCI,Incにより出版、1999,ISB
N 0-967-06710-3を参照されたい。
したがって、別の実施形態(実施形態1.153)では、本発明は、無水形態(例えば、
無水結晶形態)の、実施形態1.1~1.141のいずれか1つに定義される化合物を提
供する。
実施形態1.1~1.153のいずれか1つの化合物は、結晶または非結晶(例えば、
非晶質)状態で存在してよい。化合物が結晶状態で存在するか否かは、X線粉末回折(X
RPD)などの標準的な技術により容易に決定することができる。結晶及びその結晶構造
は、単結晶X線結晶解析、X線粉末回折(XRPD)、示差走査熱量測定(DSC)及び
赤外分光法、例えば、フーリエ変換赤外分光法(FTIR)を含むいくつかの技術を使用
して特徴付けることができる。水蒸気吸着重量測定試験、さらにXRPDにより、様々な
湿度条件下での結晶の挙動を解析することができる。化合物の結晶構造の決定は、本明細
書に記載の方法及びFundamentals of Crystallography
,C.Giacovazzo,H.L.Monaco,D.Viterbo,F.Sco
rdari,G.Gilli,G.Zanotti and M.Catti,(Int
ernational Union of Crystallography/Oxfo
rd University Press,1992 ISBN 0-19-85557
8-4(p/b),0-19-85579-2(h/b))に記載されているような方法
などの従来の方法に従い実施できるX線結晶解析により行うことができる。この技術は、
単結晶のX線回折の解析及び解釈を必然的に含む。非晶質固体では、結晶形態に通常存在
する三次元構造が存在せず、非晶質形態の互いの分子の相対位置は本質的にランダムであ
る。例えば、Hancockら、J.Pharm.Sci.(1997),86,1)を
参照されたい。
合物。
晶、または少なくとも60%結晶、または少なくとも70%結晶、または少なくとも80
%結晶、または少なくとも90%結晶、または少なくとも95%結晶、または少なくとも
98%結晶、または少なくとも99%結晶、または少なくとも99.5%結晶、または少
なくとも99.9%結晶、例えば、100%結晶である、実施形態1.1~1.153の
いずれか1つに記載の化合物。
化合物。
実施形態1.1~1.162のいずれか1つに定義される式(1)、(1a)または(
1b)の化合物は、プロドラッグの形態で存在してよい。「プロドラッグ」とは、例えば
、実施形態1.1~1.162のいずれか1つに定義される式(1)、(1a)または(
1b)の生物学的に活性な化合物にインビボで変換される任意の化合物を意味する。
る代謝的に不安定なエステル)である。代謝中、エステル基(-C(=O)OR)が切断
されて活性薬が得られる。そのようなエステルは、例えば、親化合物に存在する任意のヒ
ドロキシル基のエステル化により形成され得、適切な場合には、親化合物に存在する他の
任意の反応基を事前に保護し、その後必要があれば脱保護する。
なる化学反応の際に活性化合物になる化合物である(例えば、ADEPT、GDEPT、
LIDEPT等において)。例えば、プロドラッグは糖誘導体または他のグリコシドコン
ジュゲートであってよく、またはアミノ酸エステル誘導体であってよい。
1.170のいずれか1つに定義される化合物のプロドラッグであって、当該化合物がヒ
ドロキシル基またはアミノ基を形成するように生理条件下で変換可能である官能基を含有
する、当該プロドラッグを提供する。
実施形態1.1~1.170の式(1)、(1a)または(1b)には、実施形態1.
1~1.170の化合物の錯体(例えば、シクロデキストリンなどの化合物との包接錯体
若しくはクラスレート、または金属との錯体)も包含される。
レートの形態の、実施形態1.1~1.170のいずれか1つに記載の化合物を提供する
。
本発明の化合物は、ムスカリンM1受容体アゴニストとしての活性を有する。化合物の
ムスカリン活性は、下の実施例Aに記載のホスホ-ERK1/2アッセイを使用して決定
することができる。
1受容体に対して高度に選択的であることである。本発明の化合物は、M2及びM3受容
体サブタイプのアゴニストでない。例えば、本発明の化合物が、実施例Aに記載の機能ア
ッセイにおいてM1受容体に対して少なくとも6(好ましくは少なくとも6.5)のpE
C50値及び80を上回る(好ましくは95を上回る)Emax値を典型的に有するのに
対し、実施例Aの機能アッセイにおいてM2及びM3サブタイプに対して試験した時、そ
れらは5未満のpEC50値及び20%未満のEmax値を有し得る。
択的である。そのような化合物の例には、実施例1-6、1-9、1-21及び2-17
の化合物を含む。
の例には、実施例1-1~1-4及び1-8~1-10及び2-116の化合物を含む。
記載の化合物。
形態1.1~1.180のいずれか1つに記載の化合物。
てM1受容体に対して、6.0~8.1の範囲のpEC50及び少なくとも90のEma
xを有するムスカリンM1受容体アゴニストである、実施形態1.1~1.180のいず
れか1つに記載の化合物。
である、実施形態2.3に記載の化合物。
は実施形態2.4に記載の化合物。
てM4受容体に対して、6.0~9.0の範囲のpEC50及び少なくとも90のEma
xを有するムスカリンM4受容体アゴニストである、実施形態1.1~1.180のいず
れか1つに記載の化合物。
である、実施形態2.6に記載の化合物。
は実施形態2.7に記載の化合物。
て選択的である、実施形態2.3~2.8のいずれか1つに記載の化合物。
、実施形態2.9に記載の化合物。
、実施形態2.9に記載の化合物。
である、実施形態2.3~2.5のいずれか1つに記載の化合物。
である、実施形態2.6~2.8のいずれか1つに記載の化合物。
的である、実施形態2.3~2.8のいずれか1つに記載の化合物。
び50未満のEmaxを有する、実施形態2.3~2.14のいずれか1つに記載の化合
物。
0及び/または30未満のEmaxを有する、実施形態2.15に記載の化合物。
するための、実施形態1.1~1.180及び実施形態2.3~2.16のいずれか1つ
に記載の化合物。
は、アルツハイマー病、統合失調症及び他の精神病性障害、認知障害及びムスカリンM1
及び/またはM4受容体により媒介される他の疾患の処置において使用することができ、
様々な種類の疼痛の処置においても使用することができる。
~1.180のいずれか1つに記載の化合物。
頭型認知症、血管性認知症、レビー小体型認知症、初老期認知症、老年認知症、フリーデ
リヒ運動失調症、ダウン症候群、ハンチントン舞踏病、運動過剰症、躁病、トゥレット症
候群、アルツハイマー病、進行性球麻痺、注意、見当識、学習障害、記憶(すなわち記憶
障害、健忘、健忘障害、一過性全健忘症候群及び加齢に伴う記憶障害)及び言語機能を含
む認知機能の障害;脳卒中の結果としての認知機能障害、ハンチントン病、ピック病、エ
イズ関連認知症または、多発梗塞性認知症、アルコール性認知症、甲状腺機能(hypo
tiroidism)低下関連認知症などの他の認知症状態、及び小脳萎縮及び筋萎縮性
(amyotropic)側索硬化症などの他の変性障害に関連した認知症;認知機能低
下を引き起こし得る他の急性または亜急性状態、例えば、譫妄または鬱(仮性認知症状態
)外傷、頭部外傷、加齢に伴う認知機能低下、脳卒中、神経変性、薬剤誘発状態、神経毒
性薬、加齢に伴う認知機能障害、自閉症関連認知機能障害、ダウン症候群、認知障害関連
精神病、及び電撃治療後の関連する認知障害;ニコチン、大麻、アンフェタミン、コカイ
ンを含む薬物乱用または薬剤離脱に起因する認知障害、注意欠陥多動性障害(ADHD)
及び運動障害、例えば、パーキンソン病、神経遮断薬誘発パーキンソニズム及び遅発性ジ
スキネジア、統合失調症、統合失調症様疾患、精神病性鬱病、躁病、急性躁病、妄想障害
、幻覚障害及び妄想性障害、人格障害、強迫性障害、統合失調型障害、妄想性障害、悪性
腫瘍による精神病、代謝障害、内分泌疾患またはナルコレプシー、薬物乱用または薬剤離
脱に起因する精神病、双極性障害及び統合失調感情障害から選択される状態を含む、それ
から生じる、またはそれと関連する、実施形態2.18に従って使用するための化合物。
80のいずれか1つに記載の化合物。
いずれか1つに記載の化合物。
るヒト)において処置する方法であって、当該方法は、治療有効量の実施形態1.1~1
.180のいずれか1つに記載の化合物の投与を含む、当該方法。
またはそれと関連する、実施形態2.20に記載の方法。
2.23に記載の方法。
0のいずれか1つに記載の化合物の使用。
たはそれと関連する、実施形態2.26に記載の使用。
2.27に記載の使用。
叉神経痛、ヘルペス神経痛、全身の神経痛、内臓痛、変形性関節症の疼痛、帯状疱疹後神
経痛、糖尿病性ニューロパチー、根性痛、坐骨神経痛、背部痛、頭部または頸部の疼痛、
重度または難治性疼痛、侵害受容性疼痛、突出痛、術後の疼痛または癌性疼痛を処置する
またはそれらの重症度を軽減するための、実施形態1.1~1.180のいずれか1つに
記載の化合物。
叉神経痛、ヘルペス神経痛、全身の神経痛、内臓痛、変形性関節症痛、帯状疱疹後神経痛
、糖尿病性ニューロパチー、根性痛、坐骨神経痛、背部痛、頭部または頸部の疼痛、重度
または難治性疼痛、侵害受容性疼痛、突出痛、術後の疼痛または癌性疼痛を処置するまた
はそれらの重症度を軽減する方法であって、治療有効量の実施形態1.1~1.180の
いずれか1つに記載の化合物の投与を含む、当該方法。
候群を含むドライアイ及びドライマウスを処置するための、実施形態1.1~1.180
のいずれか1つに記載の化合物。
候群を含むドライアイ及びドライマウスを処置する方法であって、治療有効量の実施形態
1.1~1.180のいずれか1つに記載の化合物の投与を含む、当該方法。
叉神経痛、ヘルペス神経痛、全身の神経痛、内臓痛、変形性関節症の疼痛、帯状疱疹後神
経痛、糖尿病性ニューロパチー、根性痛、坐骨神経痛、背部痛、頭部または頸部の疼痛、
重度または難治性疼痛、侵害受容性疼痛、突出痛、術後の疼痛または癌性疼痛を処置する
またはそれらの重症度を軽減するため、または緑内障における眼圧の低下などの末梢性障
害を処置する及びシェーグレン症候群を含むドライアイ及びドライマウスを処置するため
の医薬品を製造するための、実施形態1.1~1.180のいずれか1つに記載の化合物
の使用。
0のいずれか1つに記載の化合物の使用。
誘発する根底的な病原的要因としてドーパミン作動性機能障害と関連する他の症候群など
の運動障害を処置するための、実施形態1.1~1.180のいずれか1つに記載の化合
物の使用。
式(1)、(1a)または(1b)の化合物は、当業者に周知である、及び本明細書に
記載の合成方法にしたがって調製することができる。
180のいずれか1つに定義される化合物を調製するためのプロセスであって、
(A)式(10)


は、実施形態1.1~1.180のいずれか1つに定義される;または
(B)式(12):

または
(C)式(10)
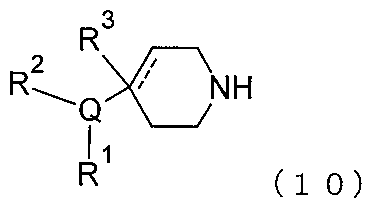

施形態1.1~1.180のいずれか1つに定義される;及び任意選択的に:
(D)式(1)、(1a)または(1b)のある化合物を式(1)、(1a)または(1
b)の別の化合物に変換することを含む、当該プロセスを提供する。
アミノ条件下で反応させる。還元的アミノ化反応は、酢酸を含有するジクロロメタンまた
はジクロロエタンなどの溶媒、ナトリウムトリアセトキシボロヒドリドなどの水素化ホウ
素還元剤を使用して周囲温度で典型的に実行される。
溶媒無し、またはテトラヒドロフラン、アセトニトリル若しくはジメチルアセトアミドな
どの好適な溶媒中のいずれかで、(例えば、約40℃から約70℃の温度への)穏やかな
加温を伴って典型的に実行される求核置換反応においてスルホン酸エステル(13、R=
メチル、トリフルオロメチルまたは4-メチルフェニル)と反応させる。
とができる。

、還元的アミノ化条件下で反応させる。還元的アミノ化反応は、酢酸を含有するジクロロ
メタンまたはジクロロエタンなどの溶媒中、塩化亜鉛と組み合わせたシアノ水素化ホウ素
ナトリウムまたはチタンイソプロポキシドと組み合わせた水素化トリアセトキシホウ素ナ
トリウムのいずれかの存在下で(例えば、約40℃から約70℃の温度への)穏やかな加
熱を伴って典型的に実行されて、中間体ピペリジン化合物(15)を得る。次いでこれを
酸(例えば、トリフルオロ酢酸を含むジクロロメタン)を用いた処理によりBoc基の除
去により脱保護して、化合物(12)を得る。
きる。

リウムを用いてアルコール(16)に還元する。次いで、アルコール(16)を、トリエ
チルアミンまたはN,N-ジイソプロピルエチルアミンなどの第三級アミンの存在下で、
ジクロロメタン中、対応する塩化スルホニルを使用してスルホン酸エステル(17、R=
メチル、トリフルオロメチルまたは4-メチルフェニル)として活性化させる。スルホン
酸エステル(17)をピペリジン複素環(10)と、無溶媒、つまり溶媒無し、またはテ
トラヒドロフラン、アセトニトリル若しくはジメチルアセトアミドなどの好適な溶媒中の
いずれかで、(例えば、約40℃から約70℃の温度への)穏やかな加温を伴って典型的
に実行される求核置換反応において反応させて、化合物(15)を得る。次いでこれを酸
(例えば、トリフルオロ酢酸を含むジクロロメタン)を用いた処理によりBoc基の除去
により脱保護して化合物(12)を得る。
保護された誘導体は、当業者に周知の方法により、式(1)、(1a)または(1b)の
別の化合物へと変換されることができる。ある官能基を別の官能基へと変換させるための
合成手法の例は、Advanced Organic Chemistry and O
rganic Syntheses(上記の参考文献を参照されたい)またはFiese
rs’ Reagents for Organic Synthesis,1-17巻
,John Wiley,Mary Fieserにより編集(ISBN:0-471-
58283-2)などの標準的な手引書に記載されている。これらの変換の例には、アミ
ド結合形成、尿素形成、カルバメート形成、アルキル化反応、N-アリール化反応及びC
-C結合カップリング反応が含まれる。
または複数の基を保護する必要があり得る。保護基の例、ならびに官能基を保護及び脱保
護する方法は、Protective Groups in Organic Synt
hesis(T.Greene and P.Wuts;第3版;John Wiley
and Sons,1999)に見出すことができる。
離及び精製されてよく、そのような方法の例には、再結晶技術及びクロマトグラフィー技
術、例えばカラムクロマトグラフィー(例えば、フラッシュクロマトグラフィー)及びH
PLCが含まれる。
活性化合物を単独投与することはできるが、医薬組成物(例えば、製剤)として提供す
る方が好ましい。
80のいずれか1つに定義される式(1)、(1a)または(1b)の少なくとも1種の
化合物を、少なくとも1種の医薬的に許容され得る賦形剤と共に含む、医薬組成物を提供
する。
体または半固体担体)、補助剤、希釈剤(例えば、充填剤または増量剤などの固体希釈剤
;及び溶媒及び共溶媒などの液体希釈剤)、造粒剤、バインダー、流れ助剤、コーティン
グ剤、放出制御剤(例えば、放出遅延(retarding or delaying)
ポリマーまたはワックス)、結合剤、崩壊剤、緩衝剤、滑沢剤、防腐剤、抗真菌剤及び抗
菌剤、抗酸化剤、緩衝剤、等張化剤、粘稠化剤、香味料、甘味料、色素、可塑剤、矯味剤
、安定剤または医薬組成物において従来的に使用される任意の他の賦形剤から選択するこ
とができる。
成物、及び/または剤形が、適切な医学的判断の範囲内で、過度の毒性、刺激、アレルギ
ー反応、または他の問題若しくは合併症を伴わずに、対象(例えば、ヒト対象)の組織と
の接触における使用に好適であり、妥当なベネフィット/リスク比と釣り合うことを意味
する。各賦形剤はまた、製剤の他の成分との適合性という意味で「許容可能」でなければ
ならない。
たがって製剤化されることができ、例えば、Remington’s Pharmace
utical Sciences,Mack Publishing Company,
Easton,PA,USAを参照されたい。
内または経皮投与に好適な任意の形態であることができる。
プセル剤(硬質または軟質シェル)、カプレット、丸剤、ロゼンジ剤、シロップ剤、溶液
剤、散剤、顆粒剤、エリキシル剤及び懸濁剤、舌下錠剤、ウエハース剤またはパッチ剤、
例えば、口腔内パッチが含まれる。
たは糖アルコール、例えば、ラクトース、スクロース、ソルビトールまたはマンニトール
;及び/または非糖系の希釈剤、例えば、炭酸ナトリウム、リン酸カルシウム、炭酸カル
シウム若しくはセルロースまたはその誘導体、例えば、微結晶性セルロース(MCC)、
メチルセルロース、エチルセルロース、ヒドロキシプロピルメチルセルロース、ならびに
デンプン、例えば、コーンスターチを含有することができる。錠剤は、結合剤及び造粒剤
、例えば、ポリビニルピロリドン、崩壊剤(例えば、膨潤性架橋ポリマー、例えば、架橋
カルボキシメチルセルロース)、滑沢剤(例えば、ステアレート)、防腐剤(例えば、パ
ラベン)、抗酸化剤(例えば、BHT)、緩衝剤(例えば、リン酸塩またはクエン酸塩緩
衝剤)、及び発泡剤、例えば、シトレート/ビカルボネート混合物のような標準的な成分
も含有してよい。こうした賦形剤は周知であり、ここで詳細に考察する必要はない。
時間にわたるまたは胃腸管の特定の領域で、制御様式で放出するように設計してもよい(
制御放出錠剤)。
性成分と、99%(w/w)~5%(w/w)の医薬的に許容され得る賦形剤(例えば、
上記で定義したような)またはこうした賦形剤の組み合わせを含む。好ましくは、組成物
は、約20%(w/w)~約90%(w/w)活性成分と80%(w/w)~10%の医
薬的に賦形剤または賦形剤の組み合わせとを含む。医薬組成物は、約1%~約95%、好
ましくは約20%~約90%の活性成分を含む。本発明に係る医薬組成物は、例えば、ア
ンプル、バイアル、坐剤、プレフィルドシリンジ、糖衣錠、散剤、錠剤またはカプセル剤
などの単位用量の形態であってよい。
流れ助剤及び/または0~99%(w/w)の充填剤/または増量剤を(薬物用量に応じ
て)含有してよい。それらは、0~10%(w/w)のポリマー結合剤、0~5%(w/
w)の抗酸化剤、0~5%(w/w)の色素も含有してよい。遅放性錠剤は、典型的には
、0~99%(w/w)の放出制御(例えば、遅延)ポリマーを(用量に応じて)さらに
含有するだろう。錠剤またはカプセル剤のフィルムコートは典型的には、0~10%(w
/w)のポリマー、0~3%(w/w)の色素、及び/または0~2%(w/w)の可塑
剤を含有する。
共溶媒及び/または0~99%(w/w)の注射用水(WFI)を(用量に応じて、及び
凍結乾燥されている場合)含有する。筋肉内デポー製剤用の製剤は0~99%(w/w)
の油も含有してよい。
者パック(patient pack)」として患者に提供してよい。
う。したがって、典型的には、所望の生物活性レベルを提供するのに十分な化合物を含有
するだろう。例えば、製剤は、1ナノグラム~2グラムの活性成分、例えば、1ナノグラ
ム~2ミリグラムの活性成分を含有してよい。これらの範囲内で、化合物の特定の部分範
囲は、0.1ミリグラム~2グラムの活性成分(より一般的には10ミリグラム~1グラ
ム、例えば、50ミリグラム~500ミリグラム)、または1マイクログラム~20ミリ
グラム(例えば、1マイクログラム~10ミリグラム、例えば、0.1ミリグラム~2ミ
リグラムの活性成分)である。
リグラム~1グラム、例えば、50ミリグラム~1グラム、例えば、100ミリグラム~
1グラムの活性化合物を含有してよい。
効果を達成するのに十分な量(有効量)で投与されるだろう。投与される化合物の正確な
量は、標準的な手順にしたがって担当の医師により決定されてよい。
るものではない。
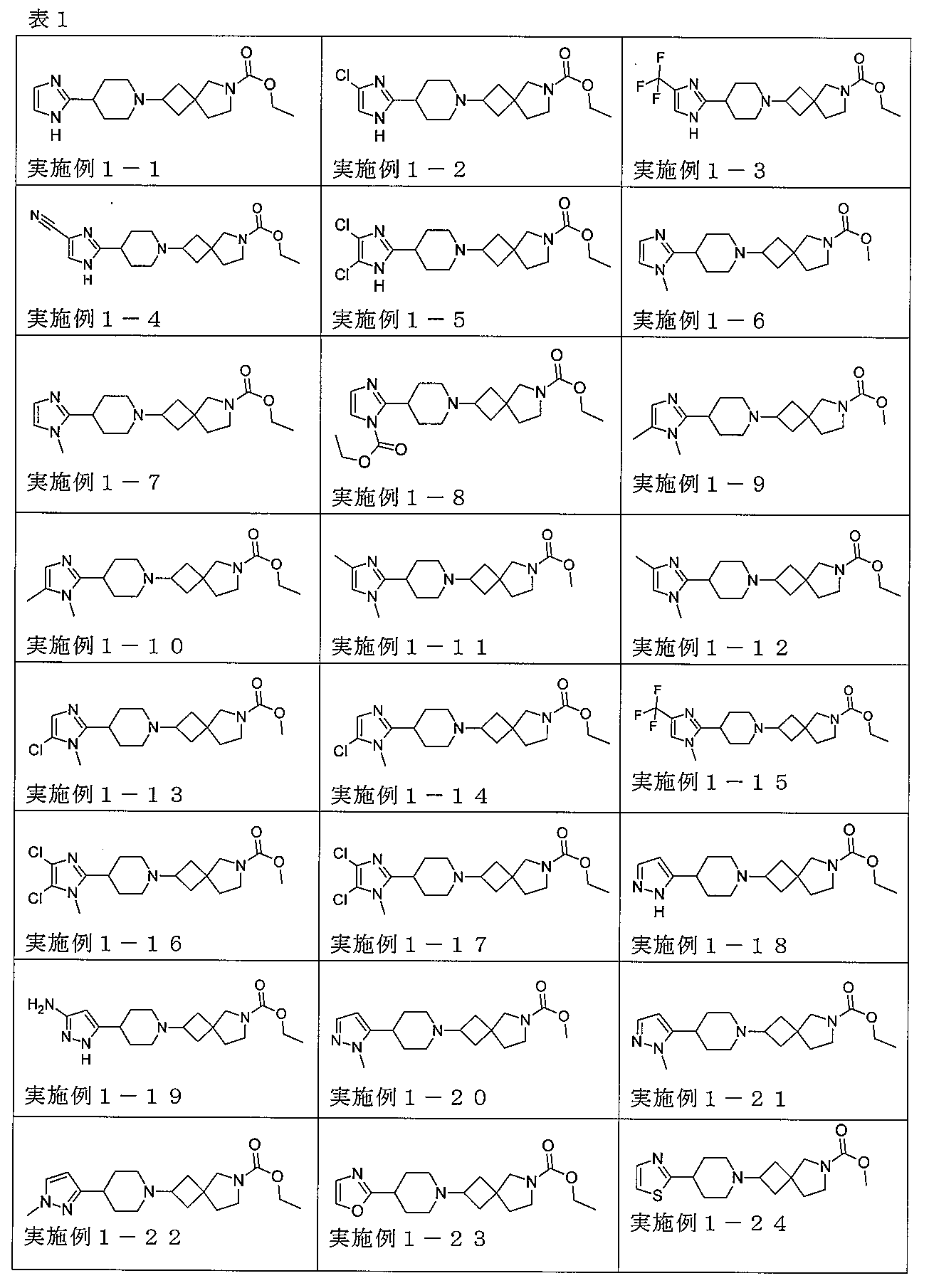
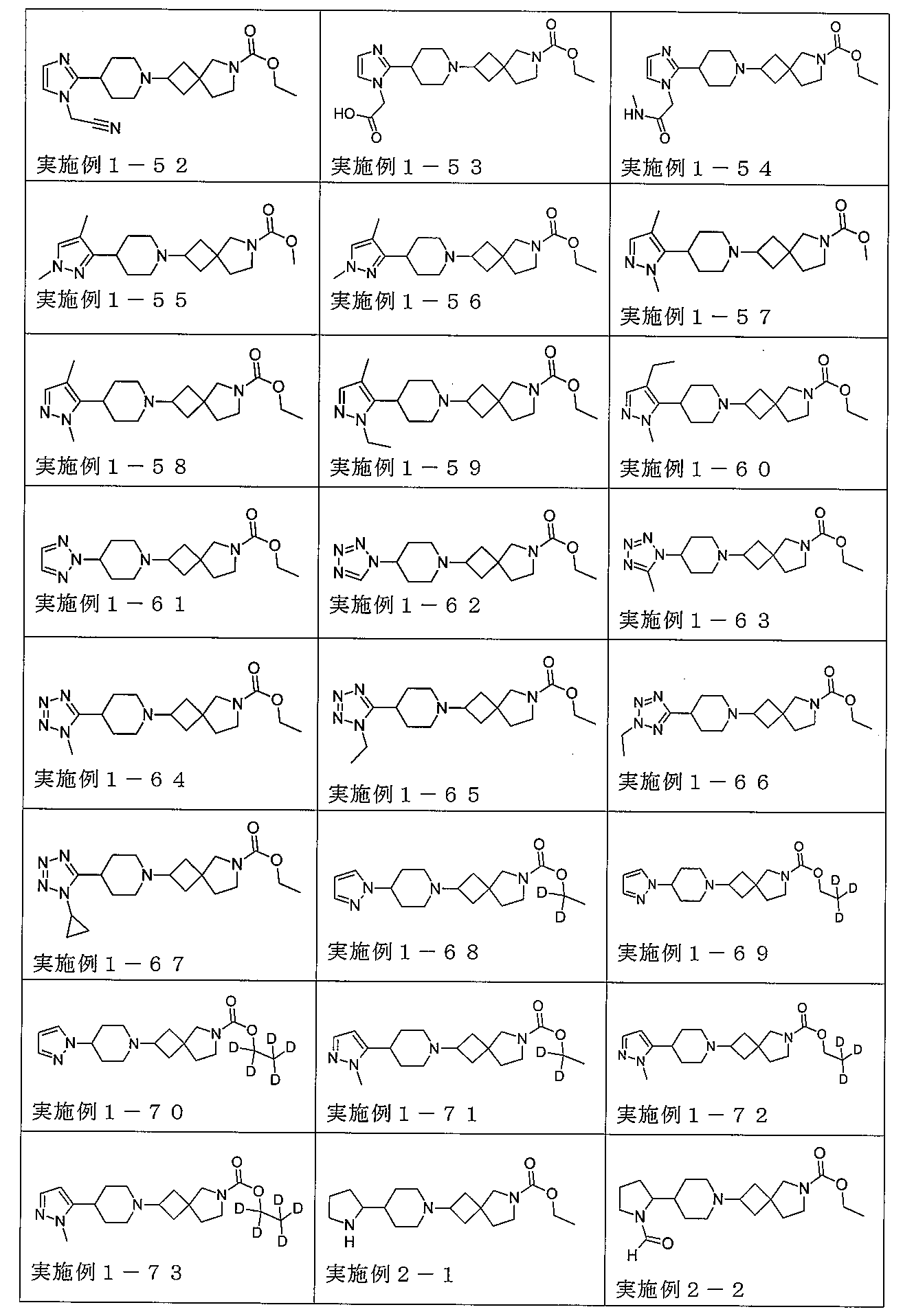
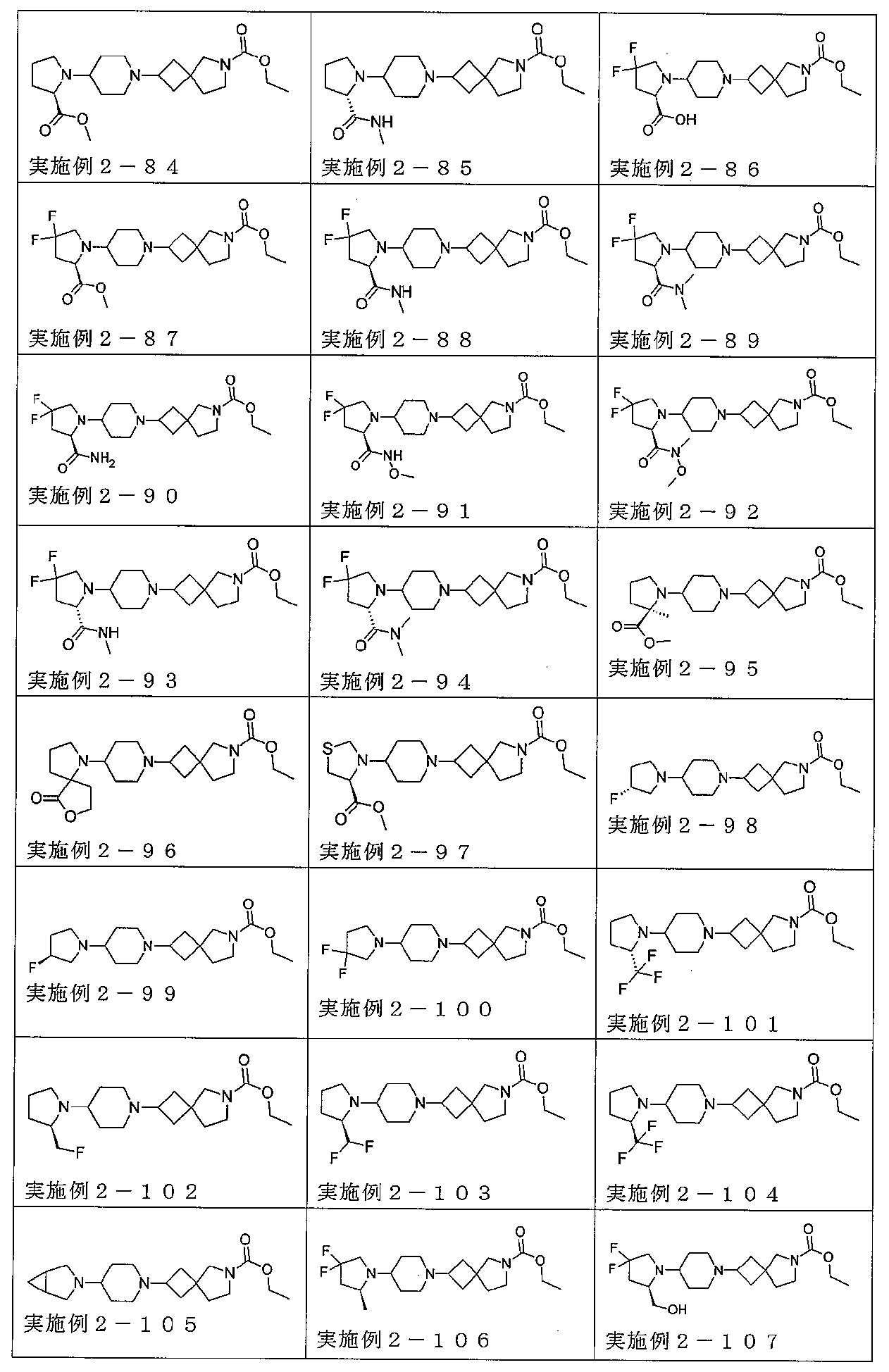
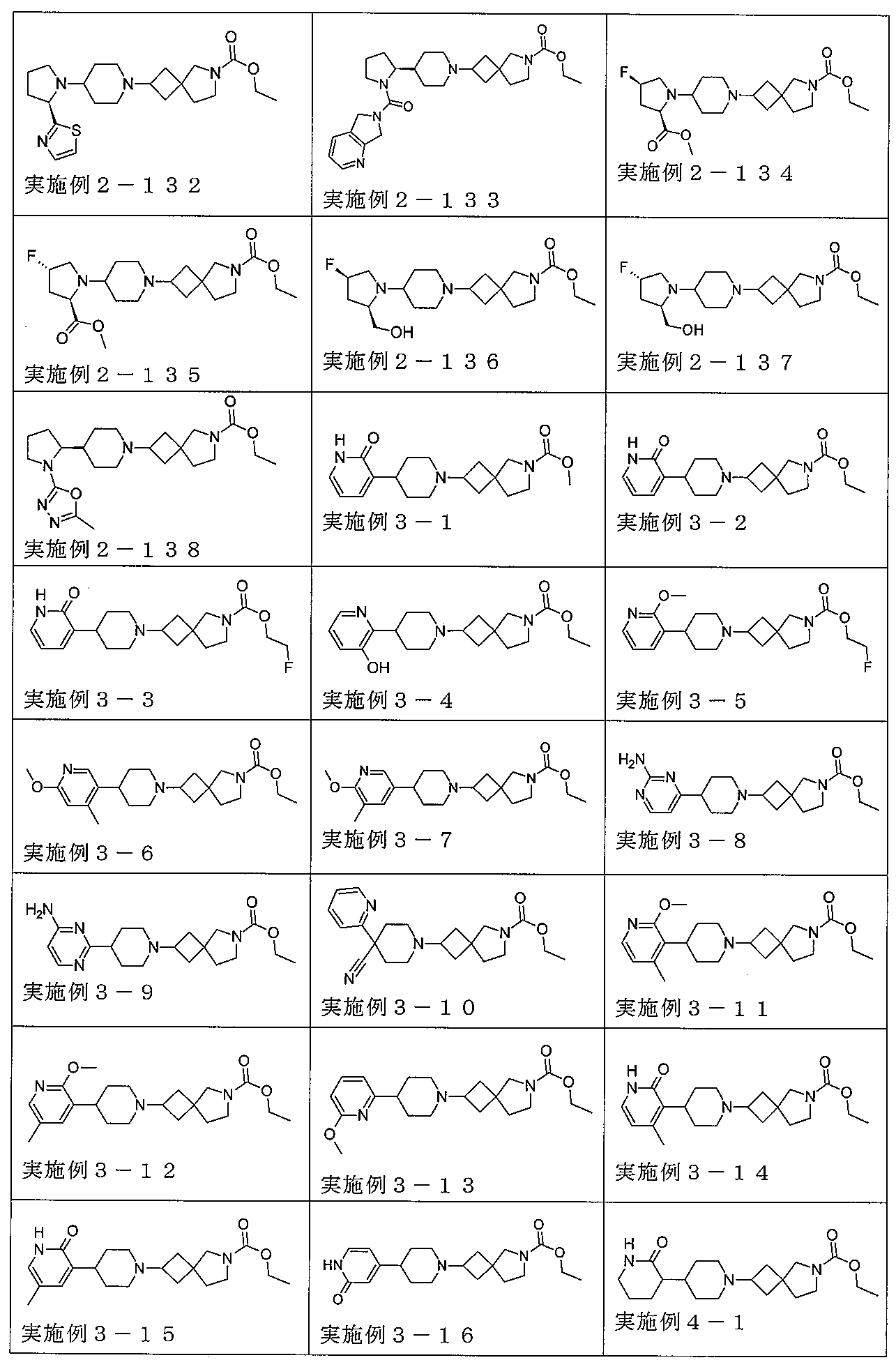
下記の表1に示す実施例1-1~5-2の化合物が調製された。そのNMR及びLCM
Sの特性及びそれらの調製に使用した方法を表3に記載する。







調製経路が含まれていない場合、当該中間体は市販されている。市販の試薬は、さらに
精製することなく利用した。室温(rt)は、約20~27℃を指す。1H NMRスペ
クトルは、Bruker製またはJeol製のいずれかの機器を用いて400MHzで記
録した。化学シフト値は、百万分率(ppm)単位、すなわち(δ:)値で表す。NM
Rシグナルの多重度には以下の略語を使用する:s=一重線、br=幅広、d=二重線、
t=三重線、q=四重線、quint=五重線、td=二重線の三重線、tt=三重線の
三重線、qd=二重線の四重線、ddd=二重線の二重線の二重線、ddt=三重線の二
重線の二重線、m=多重線。結合定数は、Hzで測定したJ値として記載する。NMR分
析及び質量分析の結果は、バックグランドピークを考慮して補正を行った。クロマトグラ
フィーとは、60~120メッシュシリカゲルを用いて行われ、窒素圧力(フラッシュク
ロマトグラフィー)条件下で実行されるカラムクロマトグラフィーを指す。反応をモニタ
リングするためのTLCとは、所定の移動相と固定相としてシリカゲルF254(Mer
ck)とを使用するTLC試験を指す。マイクロ波が媒介する反応は、Biotage
Initiatorマイクロ波反応器またはCEM Discoverマイクロ波反応器
にて行った。
用して次の条件下で実行された。
機器:Waters Alliance 2795、Waters 2996 PDA
検出器、Micromass ZQ;カラム:Waters X-Bridge C-1
8、2.5ミクロン、2.1×20mmまたはPhenomenex Gemini-N
X C-18、3ミクロン、2.0×30mm;勾配[時間(分)/溶媒C中D(%)]
:方法A:0.00/2、0.10/2、2.50/95、3.50/95、3.55/
2、4.00/2または方法B:0.00/2、0.10/2、8.40/95、9.4
0/95、9.50/2、10.00/2;溶媒:溶媒C=2.5L H2O+2.5m
Lアンモニア溶液;溶媒D=2.5L MeCN+135mL H2O+2.5mLアン
モニア溶液);注入量3μL;UV検出230~400nM;カラム温度45℃;流速1
.5mL/分。
機器:Agilent 1260 Infinity LCダイオードアレイ検出器、
Agilent 6120Bシングル四重極MS(API-ESソース);カラム:Ph
enomenex Gemini-NX C-18、3ミクロン、2.0×30mm;勾
配[時間(分)/溶媒A中B(%)]:方法:0.00/5、2.00/95、2.50
/95、2.60/5、3.00/5;溶媒:溶媒A=2.5L H2O+2.5mLの
(H2O中28%NH3);溶媒B=2.5L MeCN+129mL H2O+2.7
mLの(H2O中28%NH3);注入量0.5μL;UV検出190~400nM;カ
ラム温度40℃;流速1.5mL/分。
機器:HP1100 G1315A DAD、Micromass ZQ;カラム:W
aters X-Bridge C-18、2.5ミクロン、2.1×20mmまたはP
henomenex Gemini-NX C-18、3ミクロン、2.0×30mm;
勾配[時間(分)/溶媒C中D(%)]:方法D:0.00/2、0.10/2、2.5
0/95、3.50/95、3.55/2、4.00/2または方法E:0.00/2、
0.10/2、8.40/95、9.40/95、9.50/2、10.00/2;溶媒
:溶媒C=2.5L H2O+2.5mL H2O中28%アンモニア溶液;溶媒D=2
.5L MeCN+135mL H2O+2.5mL H2O中28%アンモニア溶液)
;注入量1μL;UV検出230~400nM;質量検出130~800AMU(+ve
及び-veエレクトロスプレー);カラム温度45℃;流速1.5mL/分。
機器:Waters Acquity H Class、フォトダイオードアレイ、S
Q検出器;カラム:BEH C18、1.7ミクロン、2.1×50mm;勾配[時間(
分)/溶媒A中B(%)]:0.00/5、0.40/5、0.8/35、1.20/5
5、2.50/100、3.30/100 4.00/5;溶媒:溶媒A=5mM酢酸ア
ンモニウム(mmmonium acetate)及びH2O中0.1%ギ酸;溶媒B=
MeCN中0.1%ギ酸;注入量2μL;UV検出200~400nM;質量検出100
~1200AMU(+veエレクトロスプレー);カラム温度は周囲温度;流速0.5m
L/分。
機器:Waters 2695、フォトダイオードアレイ、ZQ-2000検出器;カ
ラム:X-Bridge C18、5ミクロン、150×4.6mm;勾配[時間(分)
/溶媒A中B(%)]:0.00/10、5.00/90、7.00/100、11.0
0/100、11.01/10 12.00/10;溶媒:溶媒A=H2O中0.1%ア
ンモニア;溶媒B=MeCN中0.1%アンモニア;注入量10μL;UV検出200~
400nM;質量検出60~1000AMU(+veエレクトロスプレー);カラム温度
は周囲温度;流速1.0mL/分。
機器:Waters 2695、フォトダイオードアレイ、ZQ-2000検出器;カ
ラム:X-Bridge C18、5ミクロン、150×4.6mm;勾配[時間(分)
/溶媒A中B(%)]:0.00/100、7.00/50、9.00/0、11.00
/0、11.01/100、12.00/100;溶媒:溶媒A=H2O中0.1%アン
モニア;溶媒B=MeCN中0.1%アンモニア;注入量10μL;UV検出200~4
00nM;質量検出60~1000AMU(+veエレクトロスプレー);カラム温度は
周囲温度;流速1.0mL/分。
機器:Waters 2695、フォトダイオードアレイ、ZQ-2000検出器;カ
ラム:X-Bridge C18、3.5ミクロン、150×4.6mm;勾配[時間(
分)/溶媒A中B(%)]:0.00/5、5.00/90、5.80/95、10/9
5;溶媒:溶媒A=H2O中0.1%アンモニア;溶媒B=MeCN中0.1%アンモニ
ア;注入量10μL;UV検出200~400nM;質量検出60~1000AMU(+
veエレクトロスプレー);カラム温度は周囲温度;流速1.0mL/分。
機器:Waters 2695、フォトダイオードアレイ、ZQ-2000検出器;カ
ラム:X-Bridge C18、5ミクロン、150×4.6mm;勾配[時間(分)
/溶媒A中B(%)]:0.01/10、5.00/90、7.00/100、11.0
0/100、11.01/10、12.00/10;溶媒:溶媒A=H2O中20mM酢
酸アンモニウム;溶媒B=MeOH;注入量10μL;UV検出200~400nM;質
量検出60~1000AMU(+veエレクトロスプレー);カラム温度は周囲温度;流
速1.0mL/分。
機器:Waters 2695、フォトダイオードアレイ、ZQ-2000検出器;カ
ラム:X-Bridge C18、3.5ミクロン、50×4.6mm;勾配[時間(分
)/溶媒A中B(%)]:0.01/0、0.20/0、5.00/90、5.80/9
5、7.20/95、7.21/100、10.00/100;溶媒:溶媒A=H2O中
0.1%アンモニア;溶媒B=MeCN中0.1%アンモニア;注入量10μL;UV検
出200~400nM;質量検出60~1000AMU(+veエレクトロスプレー);
カラム温度は周囲温度;流速1.0mL/分。
機器:Waters Acquity UPLC、Waters 3100 PDA検
出器、SQD;カラム:Acquity BEH C-18、1.7ミクロン、2.1×
100mm;勾配[時間(分)/溶媒A中B(%)]:0.00/2、2.00/2、7
.00/50、8.50/80、9.50/2、10.0/2;溶媒:溶媒A=水中5m
M酢酸アンモニウム;溶媒B=アセトニトリル;注入量1μL;検出波長214nm;カ
ラム温度30℃;流速0.3mL/分。
機器:Agilent 1260 Infinity シリーズ UHPLC;ELS
D:Agilent 1260 infinity;カラム:Acquity C-18
、1.7ミクロン、2.1×50mm;勾配[時間(分)/溶媒A中B(%)]:0.0
0/10、1.00/10、2.00/15、4.50/55、6.00/90、8.0
0/90、9.00/10、10.00/10;溶媒:A=水中5mM酢酸アンモニウム
、B=アセトニトリル;注入量:1μL;ELSDによる検出;カラム温度:40℃;流
速:0.6mL/分。
機器:Waters Acquity UPLC、Waters 3100 PDA検
出器、SQD;カラム:Acquity BEH C-18、1.7ミクロン、2.1×
100mm;勾配[時間(分)/溶媒A中B(%)]:0.00/2、0.50/2、1
.50/20、4.00/92、5.00/92、5.50/50、6.00/2;溶媒
:溶媒A=水中5mM酢酸アンモニウム;溶媒B=アセトニトリル;注入量1μL;検出
波長214nm;カラム温度35℃;流速0.6mL/分。
機器:Waters Acquity UPLC、Waters 3100 PDA検
出器、SQD;カラム:Acquity HSS-T3、1.8ミクロン、2.1×10
0mm;勾配[時間(分)/溶媒A中B(%)]:0.00/10、1.00/10、2
.00/15、4.50/55、6.00/90、8.00/90、9.00/10、1
0.00/10;溶媒:溶媒A=水中0.1%トリフルオロ酢酸;溶媒B=アセトニトリ
ル;注入量1μL;検出波長214nm;カラム温度30℃;流速0.3mL/分。
AcOH=酢酸
CDI=1,1’-カルボニルジイミダゾール
d=日(数)
DAST=ジエチルアミノ硫黄トリフルオリド
DCE=ジクロロエタン
DCM=ジクロロメタン
DIPEA=ジイソプロピルエチルアミン
DIAD=ジイソプロピルアゾジカルボキシレート
DMF=ジメチルホルムアミド
DMP=デスマーチンペルヨージナン
DMSO=ジメチルスルホキシド
ES=エレクトロスプレーイオン化法
EtOAc=酢酸エチル
h=時間(数)
HATU=1-[ビス(ジメチルアミノ)メチレン]-1H-1,2,3-トリアゾロ[
4,5-b]ピリジウム3-オキシドヘキサフルオロフォスフェート
HPLC=高速液体クロマトグラフィー
LC=液体クロマトグラフィー
LiAlH4/LAH=水素化アルミニウムリチウム
MeCN=アセトニトリル
MeOH=メタノール
min=分(数)
MS=質量分析
Et3N=トリエチルアミン
NMR=核磁気共鳴
rt=室温
sat.=飽和
sol.=溶液
STAB=水素化トリアセトキシホウ素ナトリウム
THF=テトラヒドロフラン
TLC=薄層クロマトグラフィー
接頭辞n-、s-、i-、t-及びtert-は、それらの通常の意味:ノルマル、第二
級、イソ、及び第三級を有する。
中間体2、エチル2-オキソ-6-アザスピロ[3.4]オクタン-6-カルボキシレー
トを調製するための手順
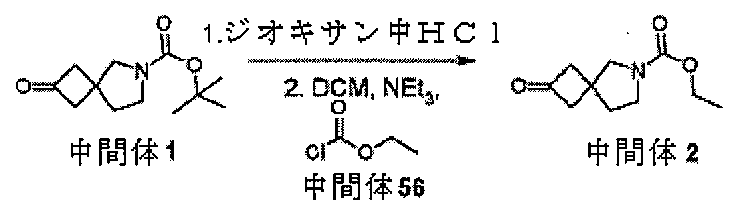
ol)を、塩化水素(4Mジオキサン溶液、50mL、210mmol)に少量ずつ加え
た。注意:発泡。24時間後、反応物を真空濃縮し、残存固体をEt3N(4.18mL
、30mmol)とDCM(66mL)の混合物中に溶解した。溶解が完了すると、溶液
を0℃まで直ちに冷却し、次いで、クロロギ酸エチル(1.57mL、16.5mmol
)を滴加した。18時間後、混合物をジクロロメタン(100mL)及びNaHCO3(
水溶液)(100mL)に注ぎ入れ、抽出した(2×100mL)。有機層を回収し、飽
和食塩水(20mL)で洗浄し、MgSO4で乾燥させ、次いで、蒸発後の残渣をカラム
クロマトグラフィー(順相、[Biotage SNAPカートリッジKP-sil 1
00g、40~63μm、60Å、50mL/分、勾配:DCM中0%~4%MeOH]
)によって精製して、中間体2、エチル2-オキソ-6-アザスピロ[3.4]オクタン
-6-カルボキシレートを、無色の油状物質(2.47g、83%)として得た。標記の
化合物に関するデータは表2にある。
トを調製するための手順

mmol)をジクロロメタン(5mL)中で塩化水素(4Mジオキサン溶液、45mL、
180mmol)に少量ずつ加えた。注意:発泡性。2時間後、反応物を真空濃縮し、1
.29gの残存固体をトリエチルアミン(2.23mL、16.0mmol)とジクロロ
メタン(10mL)の混合物中に溶解した。溶解が完了すると、溶液を0℃まで直ちに冷
却し、次いで、クロロギ酸メチル(0.68mL、8.83mmol)を滴加した。3時
間後、混合物をジクロロメタン(50mL)に注ぎ入れ、NaHCO3(水溶液)(2×
50mL)で洗浄し、DCM(50mL)で抽出した。有機層を合一し、飽和食塩水(5
0mL)で洗浄し、Biotageフェーズセパレータに通し、溶媒を真空除去し、残渣
をカラムクロマトグラフィー(順相、[Biotage SNAPカートリッジKP-s
il 50g、40~63μm、60Å、40mL/分、勾配:DCM中0%~10%M
eOH])によって精製して中間体3、メチル2-オキソ-6-アザスピロ[3.4]オ
クタン-6-カルボキシレートを橙色の油状物質(0.93g、66%)として得た。標
記の化合物に関するデータは表2にある。
ルボキシレートを調製するための手順

ート(5g、22.19mmol)を、1,4-ジオキサン中HCl溶液(25mL)中
で、10時間室温で攪拌した。反応混合物を真空濃縮し、アセトン(3×50mL)で研
和して、6-アザスピロ[3.4]オクタン-2-オン(2.77g、55.4%)を褐
色ゴム質として得た。残渣を、脱水DCM(20mL)中に溶解し、Et3N(0.7m
L、4.8mmol)を加えた。反応混合物を0℃まで冷却し、2-フルオロエチルカル
ボノクロリダート(0.45g、3.6mmol)を加えた。反応混合物を30℃で5時
間攪拌し、次いで、水(50mL)で希釈し、DCM(2×100mL)で抽出し、有機
層を合一し、乾燥させ(Na2SO4)、溶媒を真空除去した。残渣をカラムクロマトグ
ラフィー(順相、60~120メッシュシリカ、ヘキサン中0~10%EtOAc)によ
って精製して、中間体4、2-フルオロエチル2-オキソ-6-アザスピロ[3.4]オ
クタン-6-カルボキシレート(0.2g、38.8%)を褐色ゴム質として得た。標記
の化合物に関するデータは表2にある。
-イル)ピペリジントリフルオロ酢酸塩及び4-(4,5-ジクロロ-1-メチル-1H
-イミダゾール-2-イル)ピペリジントリフルオロ酢酸塩を調製するための手順

l)を、無水DCM(20mL)とEt3N(2.1mL、15.1mmol)の混合物
中に懸濁し、氷水浴にて冷却した。(BOC)2O(1.19g、5.45mmol)を
5分間かけて少量ずつ加え、混合物を室温まで加温し、48時間攪拌した。混合物をDC
Mで希釈し、飽和NaHCO3水溶液(×2)及び飽和食塩水(×1)で洗浄し、次いで
、フェーズセパレータに通し、真空濃縮して、tert-ブチル4-(1-メチル-1H
-イミダゾール-2-イル)ピペリジン-1-カルボキシレート(1.34g、定量)を
固体として得た。
LCMS(方法C):m/z 266(M+H)+(ES+)、1.43分時、UV活性
。
-カルボキシレート(0.250g、0.942mmol)を、MeCN(7.5mL)
に溶解し、NCS(0.314g、2.35mmol)で処理し、室温で一晩撹拌した。
反応混合物を、フラッシュシリカ(約15mL)で真空濃縮した。結果得られた粉末をカ
ラムクロマトグラフィー(順相、[Biotage SNAPカートリッジKP-sil
50g、40~63μm、60Å、40mL/分、勾配:15カラム容量にわたってD
CM中の2%~10%溶媒A、ここで、溶媒AはMeOH中10%の(7M NH3/M
eOH)である])によって精製して、tert-ブチル4-(5-クロロ-1-メチル
-1H-イミダゾール-2-イル)ピペリジン-1-カルボキシレート及びtert-ブ
チル4-(4,5-ジクロロ-1-メチル-1H-イミダゾール-2-イル)ピペリジン
-1-カルボキシレート及びスクシンイミド(0.495g)を含有する混合物を得た。
LCMS(方法C):モノクロロ:m/z 300/302(M+H)+(ES+)、1
.68分時、UV活性;ジクロロ:m/z 334/336/338(M+H)+(ES
+)、1.87分時、UV活性。モノクロロ;ジクロロの比率はLC-UVにより約16
:1。
ペリジン-1-カルボキシレート及びtert-ブチル4-(4,5-ジクロロ-1-メ
チル-1H-イミダゾール-2-イル)ピペリジン-1-カルボキシレート及びスクシン
イミド(0.495g)を含有する混合物をDCM(5mL)に溶解し、TFA(5mL
)で処理し、室温で6時間撹拌した。反応混合物を真空濃縮し、残渣をトルエンで共沸(
×2)して、スクシンイミドと混合された中間体20、4-(5-クロロ-1-メチル-
1H-イミダゾール-2-イル)ピペリジントリフルオロ酢酸塩及び中間体21、4-(
4,5-ジクロロ-1-メチル-1H-イミダゾール-2-イル)ピペリジントリフルオ
ロ酢酸塩の粗混合物を得た。定量的収率と想定。さらなる精製を行わずに使用された。標
記の化合物に関するデータは表2にある。
リジンジヒドロブロミド及び4-(4,5-ジクロロ-1H-イミダゾール-2-イル)
ピペリジンジヒドロブロミドを調製するための手順

.40g、1.79mmol)をMeCN(12mL)に溶解し、NCS(0.360g
、2.70mmol)で処理し、室温で5.5時間撹拌した。反応混合物をフラッシュシ
リカ(約10mL)で真空濃縮した。結果得られた粉末をカラムクロマトグラフィー(順
相、[Biotage SNAPカートリッジKP-sil 50g、40~63μm、
60Å、40mL/分、勾配:15カラム容量にわたってDCM中0%~5%溶媒A)、
次いで5カラム容量にわたってDCM中5%溶媒A、ここで、溶媒Aは、MeOH中10
%の(7M NH3/MeOH)である])によって精製して、両方ともスクシンイミド
と混合された、分離されたエチル4-(5-クロロ-1H-イミダゾール-2-イル)ピ
ペリジン-1-カルボキシレート及びエチル4-(4,5-ジクロロ-1H-イミダゾー
ル-2-イル)ピペリジン-1-カルボキシレートを得た。それぞれを、DCMに溶解し
て、H2O(×3)で洗浄し、フェーズセパレータに通して、真空濃縮してスクシンイミ
ドを除去した。
エチル4-(5-クロロ-1H-イミダゾール-2-イル)ピペリジン-1-カルボキシ
レート(0.12g、26%)、LCMS(方法C):m/z 258/260(M+H
)+(ES+)、1.34分時、UV活性。
エチル4-(4,5-ジクロロ-1H-イミダゾール-2-イル)ピペリジン-1-カ
ルボキシレート(0.24g、45%)、LCMS(方法C):m/z 292/294
/296(M+H)+(ES+)、1.24分時、UV活性。
シレート(0.12g、0.47mmol)をAcOH(2mL)に溶解し、48%HB
r水溶液(2mL)で処理し、約120℃で2時間加熱還流した。反応混合物を真空濃縮
し、残渣をトルエンで共沸(×1)し、真空濃縮して固体を得た。定量的収率の中間体2
2、4-(5-クロロ-1H-イミダゾール-2-イル)ピペリジンジヒドロブロミド塩
であると想定した。直ちに使用した。
ルボキシレート(0.24g、0.82mmol)をAcOH(2mL)に溶解し、48
%HBr水溶液(2mL)で処理し、約120℃で2時間加熱した。反応混合物を真空濃
縮し、残渣をトルエンで共沸(×1)し、真空濃縮して固体を得た。定量的収率の中間体
25、4-(4,5-ジクロロ-1H-イミダゾール-2-イル)ピペリジンジヒドロブ
ロミドであると想定した。直ちに使用した。標記の化合物に関するデータは表2にある。
-2-イル)ピペリジン-1-カルボキシレート及び中間体33、4-[4-(トリフル
オロメチル)-1H-イミダゾール-2-イル]ピペリジンを調製するための手順

mmol)をMeOH(10mL)に溶解し、その後7Mメタノール性アンモニアを0℃
に冷却して30分間加え、その後3,3-ジブロモ-1,1,1-トリフルオロプロパン
-2-オン(5.07g、18.5mmol)を少量ずつ加えた。結果得られた反応混合
物を25℃で2時間撹拌し、溶媒を真空除去し、残渣をH2O(80mL)とEtOAc
(50mL)に分配し、水層をEtOAc(2×50mL)で抽出し、有機層を合一し、
乾燥させ(Na2SO4)、溶媒を真空除去し、残渣をカラムクロマトグラフィー(DC
M中0.5%MeOHでの塩基性活性化アルミナ)によって精製して、中間体46、te
rt-ブチル4-(4-(トリフルオロメチル)-1H-イミダゾール-2-イル)ピペ
リジン-1-カルボキシレート(1.80g、60%)を白色固体として得た。
標記の化合物に関するデータは表2にある。
)ピペリジン-1-カルボキシレート(1g、3.13mmol)を1,4-ジオキサン
(5mL)に溶解し、その後1,4-ジオキサン中HCl(20mL、3M溶液)を滴加
した。結果得られた反応混合物を30℃で16時間撹拌し、溶媒を真空除去し、残渣をジ
エチルエーテル(3×5mL)で研和することによって精製して、中間体33、4-(4
-(トリフルオロメチル)-1H-イミダゾール-2-イル)ピペリジン塩酸塩(650
mg、95%)を白色固体として得た。標記の化合物に関するデータは表2にある。
-イル]ピペリジン塩酸塩を調製するための手順

)ピペリジン-1-カルボキシレート(200mg、0.63mmol)をTHF(5m
L)に溶解し、60%水素化ナトリウム(74mg、1.88mmol)を0℃で加えた
。反応混合物を0℃で10分間撹拌し、次いでヨウ化メチル(0.06mL、0.96m
mol)を加え、結果得られた反応混合物を25℃で2時間撹拌した。反応混合物をH2
O(60mL)とEtOAc(45mL)に分配し、水層をEtOAc(2×45mL)
でさらに抽出し、有機層を合一し、乾燥させ(Na2SO4)、溶媒を真空除去した。残
渣をカラムクロマトグラフィー(順相シリカ、メッシュサイズ:DCM中60~120、
0%~2.0%~3.5%MeOH)によって精製してtert-ブチル4-(1-メチ
ル-4-(トリフルオロメチル)-1H-イミダゾール-2-イル)ピペリジン-1-カ
ルボキシレート(190mg、91%)を黄色ゴム質として得た。
LCMS(方法F):m/z 334(M+H)+(ES+)、2.31分時、UV活性
ル-2-イル)ピペリジン-1-カルボキシレート(200mg、0.6mmol)を1
,4-ジオキサン(5mL)に溶解し、その後1,4-ジオキサン中HCl(20mL、
4M溶液)を滴加した。結果得られた反応混合物を30℃で16時間撹拌し、溶媒を真空
除去し、残渣をジエチルエーテル(3×5mL)で研和することにより精製して、中間体
37、4-[1-メチル-4-(トリフルオロメチル)-1H-イミダゾール-2-イル
]ピペリジン塩酸塩(140mg、86.8%)を白色固体として得た。標記の化合物に
関するデータは表2にある。
めの手順

15mL)に溶解し、Cs2CO3(7.4g、22.9mmol)を0℃で加えた。結
果得られた反応混合物を0~5℃で10分間撹拌し、次いでクロロギ酸ベンジル(3.2
g、19.1mmol)を滴加し、反応混合物を室温で18時間撹拌した。反応混合物を
、H2O(100mL)とEtOAc(200mL)に分配し、水層をEtOAc(2×
200mL)で抽出し、有機層を合一し、乾燥し(Na2SO4)、溶媒を真空除去した
。残渣をカラムクロマトグラフィー(順相シリカ、メッシュサイズ:60~120、ヘキ
サン中0%~10%EtOAc)によって精製して、1-ベンジル4-エチルピペリジン
-1,4-ジカルボキシレート(4.2g、76.4%)を黄色固体として得た。
LCMS(方法F):m/z 292(M+H)+(ES+)、2.35分時、UV活
性
43mmol)をEtOH(10mL)に溶解し、ヒドラジン一水和物(10mL、5.
41mmol)を加え、結果得られた反応混合物を90℃で一晩撹拌した。溶媒を真空除
去し、粗生成物をペンタン及びヘキサンで研和して、ベンジル4-(ヒドラジニルカルボ
ニル)ピペリジン-1-カルボキシレート(3.8g、95%)を白色固体として得た。
LCMS(方法H):m/z 278(M+H)+(ES+)、1.76分時、UV活
性
g、1.79mmol)をオルトギ酸トリエチル(8mL)に溶解し、次いでTFA(0
.1mL)を加えた。結果得られた反応混合物を80℃で一晩撹拌した。反応混合物をH
2O(50mL)とEtOAc(100mL)に分配し、水層をEtOAc(2×100
mL)で抽出し、有機層を合一し、乾燥(Na2SO4)させ、溶媒を真空除去した。残
渣を、カラムクロマトグラフィー(順相シリカ、メッシュサイズ:60~120、ヘキサ
ン中50%~60%EtOAc)によって精製して、ベンジル4-(1,3,4-オキサ
ジアゾール-2-イル)ピペリジン-1-カルボキシレート(0.19g、38%)を黄
色固体として得た。
LCMS(方法H):m/z 288(M+H)+(ES+)、2.03分時、UV活
性
シレート(0.15g、0.52mmol)をMeOH(10mL)に溶解した。乾燥1
0%パラジウム炭素触媒(20mg)を加え、反応混合物を、H2ガスを用いて室温でパ
ージした。反応混合物をH2雰囲気下で、室温で12時間攪拌した。反応塊を、セライト
を通して濾過し、溶媒を真空除去して、中間体43、4-(1,3,4-オキサジアゾー
ル-2-イル)ピペリジン(0.078g、99%)を無色のゴム質として得た。標記の
化合物に関するデータは表2にある。
を調製するための手順

)を90℃で24時間、加熱還流した。反応混合物を0℃まで冷却し、濾過した。残渣を
乾燥させて、(Z)-N-ヒドロキシエタンイミドアミド(2.1g、100%超)を白
色結晶固体として得た。
LCMS(方法H):m/z 74(M+H)+(ES+)、1.86分時、UV不活
性
チルピペリジン-4-カルボキシレート(1.17g、7.42mmol)をエタノール
(20mL)に溶解した。エタノール中21%ナトリウムエトキシド溶液(0.92mL
、13.4mmol)を滴加し、反応混合物を30分間室温で攪拌し、次いで100℃で
16時間撹拌した。溶媒を真空除去し、残渣をカラムクロマトグラフィー(順相シリカ、
DCM中0~12%メタノール)によって精製して、中間体44、4-(3-メチル-1
,2,4-オキサジアゾール-5-イル)ピペリジン(380mg、34%)を黄色ゴム
質として得た。標記の化合物に関するデータは表2にある。
製するための手順

し、水素化ナトリウム(4.7g、117.5mmol、油中60%)を室温で加えた。
反応混合物を室温で2時間撹拌し、2-(トリメチルシリル)エトキシメチルクロリド(
20.5g、123.38mmol)を反応混合物に室温で滴加し、加えた後、反応混合
物を室温で16時間撹拌した。反応混合物を氷冷水(1000mL)上へと注ぎ、EtO
Ac(500mL)で抽出し、水層をEtOAc(2×500mL)でさらに抽出し、有
機層を合一し、乾燥(Na2SO4)させ、溶媒を真空除去した。残渣をカラムクロマト
グラフィー(順相シリカ、DCM中0~1%メタノール)によって精製して、1-((2
-(トリメチルシリル)エトキシ)メチル)-1H-イミダゾール(16.2g、68.
6%)を淡緑色ゴム質として得た。
LCMS(方法F):m/z 199(M+H)+(ES+)、1.73分時、UV活
性
g、25.0mmol)をTHF(50mL)に溶解し、-78℃に冷却した。n-ブチ
ルリチウム(19.0mL、30mmol、ヘキサン中1.6M)を-78℃で滴加し、
次いで反応混合物を-78℃で1時間撹拌した。THF(10mL)中のtert-ブチ
ル4-オキソピペリジン-1-カルボキシレート(5.53g、27mmol)の溶液を
-78℃で反応混合物に滴加した。反応混合物を2時間にわたり室温まで放温した。反応
混合物を飽和NH4Cl溶液(100mL)でクエンチし、EtOAc(50mL)で抽
出し、水層をEtOAc(2×50mL)でさらに抽出し、有機層を合一し、乾燥(Na
2SO4)させ、溶媒を真空除去した。残渣をカラムクロマトグラフィー(順相シリカ、
ヘキサン中0~20%EtOAc)によって精製して、tert-ブチル4-ヒドロキシ
-4-(1-((2-(トリメチルシリル)エトキシ)メチル)-1H-イミダゾール-
2-イル)ピペリジン-1-カルボキシレート(8g、80.0%)を浅黄色ゴム質とし
て得た。
LCMS(方法F):m/z 398(M+H)+(ES+)、2.16分時、UV活
性
)メチル)-1H-イミダゾール-2-イル)ピペリジン-1-カルボキシレート(2.
0g、5.0mmol)を、1,4-ジオキサン中4MのHCl(20mL)に溶解し、
反応混合物を室温で10時間撹拌した。溶媒を真空除去し、残渣をアセトン(3×20m
L)で研和して、中間体47、4-(1H-イミダゾール-2-イル)ピペリジン-4-
オール(0.5g、60.2%)を褐色ゴム質として得た。標記の化合物に関するデータ
は表2にある。
調製するための手順

)メチル)-1H-イミダゾール-2-イル)ピペリジン-1-カルボキシレート(2.
0g、5.0mmol)をDMF(20mL)に溶解した。溶液をN2下で0℃まで冷却
し、NaH(0.24g、10.0mmol)を加えた。反応混合物を0℃で30分間撹
拌し、次いでヨウ化メチル(1.07g、7.5mmol)を加え、反応混合物を室温で
2時間撹拌した。反応混合物を、水(50mL)とEtOAc(50mL)に分配し、水
層(aqeous layer)をEtOAc(3×100mL)でさらに抽出し、有機
層を合一し、乾燥(Na2SO4)させ、溶媒を真空除去した。残渣をカラムクロマトグ
ラフィー(順相、60~120メッシュシリカ、ヘキサン中0~10%EtOAc)によ
って精製して、tert-ブチル4-メトキシ-4-(1-((2-(トリメチルシリル
)エトキシ)メチル)-1H-イミダゾール-2-イル)ピペリジン-1-カルボキシレ
ート(1.5g、75.0%)を黄色ゴム質として得た。
メチル)-1H-イミダゾール-2-イル)ピペリジン-1-カルボキシレート(1.5
g、3.6mmol)を1,4-ジオキサン中4MのHCl(20mL)に溶解し、反応
混合物を室温で10時間撹拌した。溶媒を真空除去し、残渣をアセトン(3×20mL)
で研和して、中間体48、4-(1H-イミダゾール-2-イル)-4-メトキシピペリ
ジン(0.5g、76.0%)を褐色固体として得た。標記の化合物に関するデータは表
2にある。
ル塩酸塩を調製するための手順

温で溶解し、反応混合物を-78℃まで窒素下で冷却し、ヘキサン中n-ブチルリチウム
(45.4mL、73.0mmol)をゆっくりと加えた。反応混合物を40℃まで穏や
かに加温し、4時間撹拌し、次いで-78℃まで冷却した。THF(100mL)中のt
ert-ブチル4-オキソピペリジン-1-カルボキシレート(14.56g、73.0
mmol)を加えた。反応混合物を40℃まで穏やかに加温し、10時間撹拌し、次いで
水(50mL)でクエンチした。反応混合物を、EtOAc(200mL)と水(150
mL)に分配し、水層をEtOAc(2×200mL)で抽出し、有機層を合一し、乾燥
(Na2SO4)させた。溶媒を真空除去し、残渣をメタノールで洗浄して、tert-
ブチル4-ヒドロキシ-4-(1-メチル-1H-イミダゾール-2-イル)ピペリジン
-1-カルボキシレート(14.0g、68.1%)を固体として得た。これを粗製のま
まその後の反応に使用した。
LCMS(方法F):m/z 282(M+H)+(ES+)、2.05分時、UV活
性
ペリジン-1-カルボキシレート(0.5g、1.7mmol)を1,4ジオキサン(3
0mL)に室温で溶解し、反応混合物を窒素下で0℃まで冷却し、ジオキサン中HCl(
15mL、4M溶液)をゆっくりと加えた。反応混合物を室温で6時間撹拌し、溶媒を真
空除去し、ペンタン(10mL)及びジエチルエーテル(10mL)から研和することに
よって残渣を精製して、中間体49、4-(1-メチル-1H-イミダゾール-2-イル
)ピペリジン-4-オール(0.2g、62.5%)を褐色固体として得た。標記の化合
物に関するデータは表2にある。
ジン塩酸塩を調製するための手順

)ピペリジン-1-カルボキシレート(3.0g、10.6mmol)をDMF(50m
L)に室温で溶解し、反応混合物を窒素下で0℃まで冷却し、NaH(0.64g、16
.0mmol、60%油中分散)を加えた。反応混合物を0℃で1時間撹拌し、次いでヨ
ウ化メチル(1.8g、128mmol)を滴加した。反応混合物を室温に加温し、10
時間撹拌し、次いで水(50mL)でクエンチした。反応混合物をEtOAc(3×20
0mL)で抽出し、有機層を合一し、乾燥(Na2SO4)させた。溶媒を真空除去し、
残渣をカラムクロマトグラフィー(順相、シリカ、60~120メッシュ、勾配:ヘキサ
ン中0%~50%EtOAc)によって精製して、tert-ブチル4-メトキシ-4-
(1-メチル-1H-イミダゾール-2-イル)ピペリジン-1-カルボキシレート(1
.3g、41.3%)を淡黄色固体として得た。
LCMS(方法F):m/z 296(M+H)+(ES+)、2.36分時、UV活性
ピペリジン-1-カルボキシレート(1.3g、3.3mmol)を1,4ジオキサン(
30mL)に室温で溶解し、反応混合物を窒素下で0℃まで冷却し、ジオキサン中HCl
(15mL、4M溶液)をゆっくりと加えた。反応混合物を室温で6時間撹拌し、溶媒を
真空除去し、ペンタン(10mL)及びジエチルエーテル(10mL)から研和すること
によって残渣を精製して、中間体50、4-メトキシ-4-(1-メチル-1H-イミダ
ゾール-2-イル)ピペリジン(0.80g、94.1%)を灰白色固体として得た。標
記の化合物に関するデータは表2にある。
-4-ヒドロキシピロリジン-2-イル]ピペリジン-1-カルボキシレートを調製する
ための手順
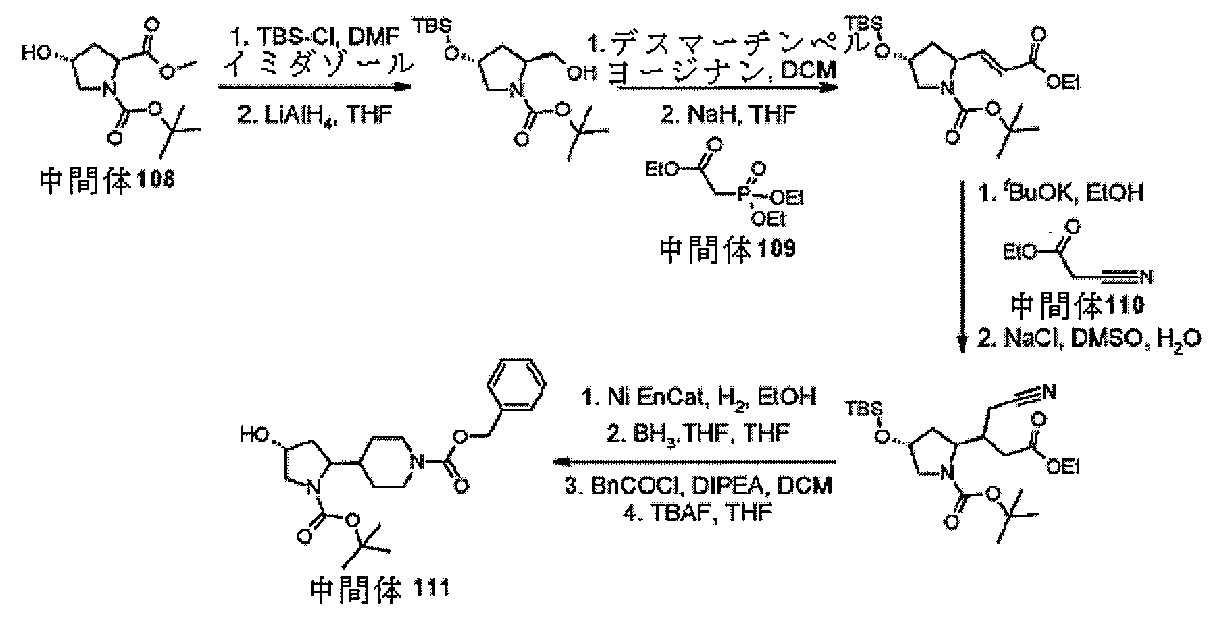
テル(25g、101.93mmol)及びイミダゾール(34.687g、509.5
mmol)をDMF(100mL)に溶解し、反応物を0℃まで冷却した。次いで、te
rt-ブチルジメチルシリルクロリドを加え(36.86g、244.56mmol)、
反応物を室温まで加温し、18時間撹拌した。揮発性物質をロータリーエバポレーターで
除去し、反応混合物(misture)をDCM(250mL)で希釈した。混合物をH
2O(2×250mL)で洗浄し、合一した水層をDCM(250mL)で洗浄し、合一
した有機層を飽和NH4Cl(水溶液)(250mL)及び飽和食塩水(250mL)で
洗浄し、Biotageフェーズセパレータに通した。揮発性物質を真空下で除去して、
(2S,4R)-1-Boc-4-tert-ブチルジメチルシリルエーテル-ピロリジ
ン-2-カルボン酸メチルエステル(35.812g、99%)を得た。
LCMS(方法D):m/z 260(M+H-Boc)+(ES+)、2.64分時、
UV不活性
ジン-2-カルボン酸メチルエステル(42.7g、118.76mmol)をTHF(
100mL)に溶解し、反応物を0℃まで冷却した。次いで、水素化アルミニウムリチウ
ムを加え(THF中120mLの1.0M溶液、120.0mmol)、反応物を0℃で
1時間撹拌した。反応物をH2O(4.5mL)、15%NaOH溶液(4.5mL)及
びH2O(13.5mL)でクエンチし、セライトプラグを通して濾過した。揮発性物質
を真空下で除去して、(2S,4R)-1-Boc-4-tert-ブチルジメチルシリ
ルエーテル-ピロリジン-2-ヒドロキシメチル(30.320g、77%)を得た。
LCMS(方法D):m/z 232(M+H-Boc)+(ES+)、2.00分時
、UV不活性
ジン-2-ヒドロキシメチル(10.050g、30.362mmol)をDCM(10
0mL)に溶解し、デスマーチンペルヨージナン(15.371g、36.253mmo
l)を加えた。反応物を室温で2時間撹拌し、次いで揮発性物質をロータリーエバポレー
ターで除去して、粗生成物をBiotage SNAPカラム(100g)へと直接ロー
ドして精製して(n-ヘキサン中10%~50%EtOA勾配)、(2S,4R)-1-
Boc-4-tert-ブチルジメチルシリルエーテル-ピロリジン-2-カルバルデヒ
ド(2.150g、22%)を得た。
38mmol)の懸濁液にホスホノ酢酸トリエチル(0.665mL、3.338mmo
l)を加えた。10分後に、THF(2mL)中の(2S,4R)-1-Boc-4-t
ert-ブチルジメチルシリルエーテル-ピロリジン-2-カルバルデヒド(1.002
g、3.034mmol)を加え、反応物を0℃で30分間撹拌した。揮発性物質をロー
タリーエバポレーターで除去し、反応混合物をDCM(20mL)で希釈した。混合物を
H2O(2×20mL)で洗浄し、合一した水層をDCM(20mL)で洗浄し、合一し
た有機層を飽和食塩水(20mL)で洗浄し、Biotageフェーズセパレータに通し
た。揮発性物質を真空下で除去し、粗混合物をBiotage SNAPカラム(100
g)に充填し、カラムクロマトグラフィー(ヘキサン中0~30%EtOAc)によって
精製して、tert-ブチル(2S,4R)-4-{[tert-ブチル(ジメチル)シ
リル]オキシ}-2-[(1E)-3-エトキシ-3-オキソプロパ-1-エン-1-イ
ル]ピロリジン-1-カルボキシレートを無色の油状物質(545mg、45%)として
得た。
LCMS(方法D):m/z 300(M+H-Boc)+(ES+)、2.85分時
、UV不活性。
ol)及びシアノ酢酸エチル(0.399mL、3.753mmol)の溶液に、ter
t-ブチル(2S,4R)-4-{[tert-ブチル(ジメチル)シリル]オキシ}-
2-[(1E)-3-エトキシ-3-オキソプロパ-1-エン-1-イル]ピロリジン-
1-カルボキシレート(500mg、1.251mmol)を加え、反応物を78℃で1
8時間撹拌した。AcOHを加え(0.200mL)、揮発性物質をロータリーエバポレ
ーターで除去した。反応混合物をDCM(50mL)で希釈し、H2O(2×50mL)
で洗浄し、合一した水層をDCM(50mL)で洗浄し、合一した有機層を飽和食塩水(
50mL)で洗浄し、Biotageフェーズセパレータに通した。揮発性物質を真空下
で除去し、粗混合物をBiotage SNAPカラム(100g)に充填し、カラムク
ロマトグラフィー(ヘキサン中0~30%EtOAc)によって精製して、ジエチル3-
[(4R)-1-(tert-ブトキシカルボニル)-4-{[tert-ブチル(ジメ
チル)シリル]オキシ}ピロリジン-2-イル]-2-シアノペンタンジオエート(cy
anopentanedioate)を黄色油状物質(567mg、89%)として得た
。
(0.040mL、2.204mmol)の溶液にジエチル3-[(4R)-1-(te
rt-ブトキシカルボニル)-4-{[tert-ブチル(ジメチル)シリル]オキシ}
ピロリジン-2-イル]-2-シアノペンタンジオエート(565mg、1.102mm
ol)を加え、反応物を145℃で2時間撹拌した。氷水を加え(50mL)、その後E
tOAc(50mL)を加え、有機層をH2O(2×50mL)で洗浄した。合一した有
機層を飽和食塩水(50mL)で洗浄し、Biotageフェーズセパレータに通した。
揮発性物質を真空下で除去し、粗混合物をBiotage SNAPカラム(50g)に
充填し、カラムクロマトグラフィー(ヘキサン中0~30%EtOAc)によって精製し
て、tert-ブチル(4R)-4-{[tert-ブチル(ジメチル)シリル]オキシ
}-2-(1-シアノ-4-エトキシ-4-オキソブタン-2-イル)ピロリジン-1-
カルボキシレートを黄色油状物質(351mg、72%)として得た。
LCMS(方法D):m/z 341(M+H-Boc)+(ES+)、2.77分時
、UV不活性
、EtOH(75mL)中のtert-ブチル(4R)-4-{[tert-ブチル(ジ
メチル)シリル]オキシ}-2-(1-シアノ-4-エトキシ-4-オキソブタン-2-
イル)ピロリジン-1-カルボキシレート(8.700g、19.7mmol)を加え、
反応物を78℃、水素バルーン雰囲気下で96時間撹拌した。反応混合物をセライトプラ
グで濾過し、揮発性物質を真空下で除去し、粗混合物をBiotage SNAPカラム
(340g)に充填し、カラムクロマトグラフィー(DCM中2.5~10%MeOH)
によって精製して、tert-ブチル(4R)-4-{[tert-ブチル(ジメチル)
シリル]オキシ}-2-(2-オキソピペリジン-4-イル)ピロリジン-1-カルボキ
シレートを黄色油状物質(4.665g、59%)として得た。
LCMS(方法D):m/z 399(M+H)+(ES+)、1.90分時、UV不
活性
-2-(2-オキソピペリジン-4-イル)ピロリジン-1-カルボキシレート(1.8
50g、4.648mmol)THF(30mL)の溶液に、ボラン:テトラヒドロフラ
ン(9.3mLのTHF中1.0M溶液、9.300mmol)を0℃で加え、反応物を
60℃で30分間撹拌した。反応物を室温まで冷却し、MeOH(10mL)でクエンチ
し、揮発性物質をロータリーエバポレーターで除去した。反応混合物をDCM(100m
L)で希釈し、1MのNaOH(水溶液)(2×100mL)で洗浄し、合一した水層を
DCM(100mL)で洗浄し、合一した有機層を飽和食塩水(250mL)で洗浄し、
Biotageフェーズセパレータに通した。揮発性物質を真空下で除去して、tert
-ブチル(2S,4R)-4-{[tert-ブチル(ジメチル)シリル]オキシ}-2
-(ピペリジン-4-イル)ピロリジン-1-カルボキシレートを黄色油状物質(1.8
30g、>99%)として得た。
LCMS(方法D):m/z 285(M+H-Boc)+(ES+)、3.00分時
、UV不活性
(ジメチル)シリル]オキシ}-2-(ピペリジン-4-イル)ピロリジン-1-カルボ
キシレート(1.830g、4.766mmol)の溶液に、ジイソプロピルエチルアミ
ン(1.814mL、10.484mmol)及びクロロギ酸ベンジル(0.816mL
、5.719mmol)を0℃で加えた。反応物を室温まで加温し、18時間撹拌した。
反応混合物をDCM(100mL)で希釈し、1MのNaOH(水溶液)(2×100m
L)で洗浄し、合一した水層をDCM(100mL)で洗浄し、合一した有機層を飽和食
塩水(250mL)で洗浄し、Biotageフェーズセパレータに通した。揮発性物質
を真空下で除去し、粗混合物をBiotage SNAPカラム(100g)に充填し、
カラムクロマトグラフィー(ヘキサン中10~40%EtOAc)によって精製して、ベ
ンジル4-[(2S,4R)-1-(tert-ブトキシカルボニル)-4-{[ter
t-ブチル(ジメチル)シリル]オキシ}ピロリジン-2-イル]ピペリジン-1-カル
ボキシレートを無色の油状物質(700mg、28%)として得た。
ボニル)-4-{[tert-ブチル(ジメチル)シリル]オキシ}ピロリジン-2-イ
ル]ピペリジン-1-カルボキシレート(0.780g、1.504mmol)の溶液に
、テトラブチルアンモニウムフルオリド(1.800mLの1.0M THF溶液、1.
800mmol)を加え、反応物を室温で1時間撹拌した。反応混合物をDCM(100
mL)で希釈し、H2O(2×100mL)で洗浄し、合一した水層をDCM(100m
L)で洗浄し、合一した有機層を飽和食塩水(250mL)で洗浄し、Biotageフ
ェーズセパレータに通した。揮発性物質を真空下で除去して、中間体111、ベンジル4
-[(2S,4R)-1-(tert-ブトキシカルボニル)-4-ヒドロキシピロリジ
ン-2-イル]ピペリジン-1-カルボキシレートを、無色の油状物質(500mg、8
2%)として得た。標記の化合物に関するデータは表2にある。
-イル)ピロリジン-1-カルボキシレートを調製するための手順

キシピロリジン-2-イル]ピペリジン-1-カルボキシレート(100mg、0.24
7mmol)をDCM(1mL)に-40℃で溶解し、DASTを加えた(0.049m
L、0.371mmol)。反応物を室温まで加温し、2時間撹拌した。反応混合物をD
CM(25mL)で希釈し、飽和NaHCO3(水溶液)(2×25mL)で洗浄し、合
一した水層をDCM(25mL)で洗浄し、合一した有機層を飽和食塩水(25mL)で
洗浄し、Biotageフェーズセパレータに通した。揮発性物質を真空下で除去して、
ベンジル4-[(2S,4S)-1-(tert-ブトキシカルボニル)-4-フルオロ
ピロリジン-2-イル]ピペリジン-1-カルボキシレート(0.090g、90%)を
得た。
LCMS(方法D):m/z 307(M+H-Boc)+(ES+)、2.31分時
、UV不活性
キシカルボニル)-4-フルオロピロリジン-2-イル]ピペリジン-1-カルボキシレ
ート(0.090g、0.221mmol)の溶液に、10%Pd/C(10mg)及び
1,4シクロヘキサジエン(0.147mL、1.530mmol)を加え、反応物を7
0℃で1時間撹拌した。反応物を、セライトプラグを通して濾過し、揮発性物質を真空下
で除去して、中間体112、tert-ブチル(2S,4S)-4-フルオロ-2-(ピ
ペリジン-4-イル)ピロリジン-1-カルボキシレート(55mg、92%)を得た。
標記の化合物に関するデータは表2にある。
-イル)ピロリジン-1-カルボキシレートを調製するための手順

℃で溶解し、DMSOを加えた(0.100mL)。5分後、-78℃で、ベンジル4-
[(2S,4R)-1-(tert-ブトキシカルボニル)-4-ヒドロキシピロリジン
-2-イル]ピペリジン-1-カルボキシレート(200mg、0.494mmol)を
DCM(2mL)中に加え、その後トリエチルアミン(0.345mL、2.47mmo
l)をさらに5分後に-78℃で加えた。反応物を室温まで加温し、30分間撹拌した。
反応混合物をDCM(25mL)で希釈し、飽和NaHCO3(水溶液)(2×25mL
)で洗浄し、合一した水層をDCM(25mL)で洗浄し、合一した有機層を飽和食塩水
(25mL)で洗浄し、Biotageフェーズセパレータに通した。揮発性物質を真空
下で除去して、ベンジル4-[(2S)-1-(tert-ブトキシカルボニル)-4-
オキソピロリジン-2-イル]ピペリジン-1-カルボキシレート(0.170g、85
%)を得た。
LCMS(方法D):m/z 303(M+H-Boc)+(ES+)、2.15分時
、UV不活性
ジン-2-イル]ピペリジン-1-カルボキシレート(170mg、0.422mmol
)をDCM(1mL)に-78℃で溶解し、DASTを加えた(0.167mL、1.2
67mmol)。反応物を室温まで加温し、18時間撹拌した。反応混合物をDCM(2
5mL)で希釈し、飽和NaHCO3(水溶液)(2×25mL)で洗浄し、合一した水
層をDCM(25mL)で洗浄し、合一した有機層を飽和食塩水(25mL)で洗浄し、
Biotageフェーズセパレータに通した。揮発性物質を真空下で除去して、ベンジル
4-[(2S)-1-(tert-ブトキシカルボニル)-4,4-ジフルオロピロリジ
ン-2-イル]ピペリジン-1-カルボキシレート(0.070g、39%)を得た。
LCMS(方法D):m/z 325(M+H-Boc)+(ES+)、2.41分時、
UV不活性
カルボニル)-4,4-ジフルオロピロリジン-2-イル]ピペリジン-1-カルボキシ
レート(0.067g、0.158mmol)の溶液に、10%Pd/C(10mg)及
び1,4シクロヘキサジエン(0.105mL、1.105mmol)を加え、反応物を
70℃で1時間撹拌した。反応物を、セライトプラグを通して濾過し、揮発性物質を真空
下で除去して、中間体113、tert-ブチル(2S)-4,4-ジフルオロ-2-(
ピペリジン-4-イル)ピロリジン-1-カルボキシレート(30mg、65%)を得た
。標記の化合物に関するデータは表2にある。
-1-カルボキシレートを調製するための手順

ロピロリジン-1,2-ジカルボキシレート(2g、7.5mmol)にTHF中の水素
化ホウ素リチウム溶液(2.0M、7.5mL、15mmol)を0℃で加え、反応物を
室温まで加温し、2時間撹拌した。反応物を、飽和NaHCO3水溶液を少量ずつ加える
ことによってクエンチし、ひとたび発泡が終わったら、混合物を濃縮してTHFを除去し
た。水性混合物を飽和NaHCO3水溶液とDCM(×2)に分配し、有機相をフェーズ
セパレータに通し、濃縮して、粗tert-ブチル(2R)-4,4-ジフルオロ-2-
(ヒドロキシメチル)ピロリジン-1-カルボキシレート(1.98g、100%超)を
油状物質として得た。
LCMS(方法C):m/z 260(M+Na)+(ES+)、1.09分時、UV
不活性。
(ヒドロキシメチル)ピロリジン-1-カルボキシレート(1g、4.2mmol)の溶
液及びトリエチルアミン(1.5mL、11mmol)にMsCl(0.42mL、5.
4mmol)を少量ずつ加えた。混合物を0℃で100分間撹拌し、次いで、氷冷飽和N
aHCO3水溶液と氷冷DCM(×2)に分配し、有機相をフェーズセパレータに通過さ
せ、濃縮して粗tert-ブチル(2R)-4,4-ジフルオロ-2-{[(メチルスル
ホニル)オキシ]メチル}ピロリジン-1-カルボキシレート(1.55g、100%超
)を油状物質として得た。
LCMS(方法C):m/z 338(M+Na)+(ES+)、1.28分時、UV不
活性。
{[(メチルスルホニル)オキシ]メチル}ピロリジン-1-カルボキシレート(1.5
5g、4.9mmol)の溶液にTHF(1.0M、9.8mL、9.8mmol)中の
LiBHEt3溶液を、10分間にわたって少量ずつ加えた。次いで、混合物を3日間撹
拌し、冷却浴を自然に終了させた。混合物を0℃にまで冷却し戻し、H2Oの添加により
クエンチし、次いで濃縮してTHFを除去した。水性混合物を飽和NaHCO3水溶液と
DCM(×2)に分配し、有機相をフェーズセパレータに通し、濃縮して、粗中間体12
5、tert-ブチル(2S)-4,4-ジフルオロ-2-メチルピロリジン-1-カル
ボキシレート(0.89g、82%)を油状物質として得た。標記の化合物に関するデー
タは表2にある。
ル)ピロリジン-1-カルボキシレートを調製するための手順

シレート(1.00g、4.111mmol)をDCM(10mL)に-78℃で溶解し
、DASTを加えた(1.629mL、12.332mmol)。反応物を室温まで加温
し、2時間撹拌した。反応混合物をDCM(100mL)で希釈し、飽和NaHCO3(
水溶液)(2×100mL)で洗浄し、合一した水層をDCM(100mL)で洗浄し、
合一した有機層を飽和食塩水(25mL)で洗浄し、Biotageフェーズセパレータ
に通した。溶媒(Sovent)を真空除去して、橙色の油状物質(0.957g、90
%)を得た。
ロリジン-1,2-ジカルボキシレート(500mg、1.885mmol)に、THF
(1.90mL、3.80mmol)中の2.0M溶液として水素化ホウ素リチウムを0
℃で加え、反応物を室温まで加温し、1時間撹拌した。溶媒を真空除去し、反応混合物を
DCM(50mL)で希釈し、飽和NaHCO3(水溶液)(2×50mL)で洗浄し、
合一した水層をDCM(50mL)で洗浄し、合一した有機層を飽和食塩水(50mL)
で洗浄し、Biotageフェーズセパレータに通した。揮発性物質を真空下で除去して
、中間体126、tert-ブチル(2R)-4,4-ジフルオロ-2-(ヒドロキシメ
チル)ピロリジン-1-カルボキシレート(452mg、92%)を得た。
を調製するための手順

00mmol)及び3-アミノプロパン-1-オール(0.330g、4.4mmol)
をCH2Cl2(20mL)中に室温で混合し、AcOH(0.68mL、12.0mm
ol)を加えて3時間撹拌した。STAB(2.34g、10.0mmol)を加えて、
反応混合物を窒素下、室温で一晩撹拌した。反応混合物をNaHCO3(飽和水溶液)(
40mL)の添加でクエンチしてCH2Cl2(4×45mL)で抽出し、合一した有機
層を飽和食塩水で洗浄し、次いでMgSO4で乾燥させ濾過した。溶媒を真空除去して、
粗tert-ブチル4-[(3-ヒドロキシプロピル)アミノ]ピペリジン-1-カルボ
キシレート(1.03g、4.00mmol)を得た。これを精製せずに使用した。
LCMS(方法B):m/z 259(M+H)+(ES+)、0.24分時、UV不活
性。
キシレート(1.03g、4.00mmol)、CDI(1.36g、8.4mmol)
及びDBU(0.24mL、1.60mmol)をTHF(40mL)に溶解し、混合物
を加熱還流し、72時間維持した。溶媒を真空除去し、残渣をカラムクロマトグラフィー
[Biotage SNAPカートリッジKP-sil 25g、40~63μm、60
Å、50mL/分、勾配:DCM中0%~10%MeOH])により精製して、tert
-ブチル4-(2-オキソ-1,3-オキサジナン-3-イル)ピペリジン-1-カルボ
キシレート(0.60g、53%)を無色の油状物質として得た。
LCMS(方法B):m/z 307(M+Na)+(ES+)、0.16分時、UV
不活性。
1-カルボキシレート(0.60g、2.11mmol)をCH2Cl2(21mL)に
溶解し、ジオキサン中4M塩化水素(2.64mL、10.5mmol)を加え、反応混
合物を室温で一晩撹拌した。沈殿物を濾過により回収し、CH2Cl2(2×20mL)
で洗浄し、乾燥させて、中間体132、3-(ピペリジン-4-イル)-1,3-オキサ
ジナン-2-オン塩酸塩(0.352g、76%)を無色の固体として得た。標記の化合
物に関するデータは表2にある。
4]オクタン-6-カルボキシレート塩酸塩を調製するための手順

985g、5.00mmol)及び1,4-ジオキサ-8-アザスピロ[4.5]デカン
(0.715g、5.00mmol)を室温でCH2Cl2(50mL)に混合し、Ac
OH(0.31mL、5.50mmol)を加え、3時間撹拌した。STAB(2.65
g、12.5mmol)を加え、反応混合物を窒素下、室温で一晩撹拌した。反応混合物
をNaHCO3(飽和水溶液)(40mL)の添加でクエンチし、CH2Cl2(4×4
5mL)で抽出し、合一した有機層を飽和食塩水で洗浄し、次いでMgSO4で乾燥させ
濾過した。溶媒を真空除去して、粗エチル2-(1,4-ジオキサ-8-アザスピロ[4
.5]デカ-8-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレートを
ジアステレオマーの混合物として得た。これをさらなる精製を行わずに用いた。
LCMS(方法D):m/z 325(M+H)+(ES+)、1.11分及び1.1
6分時、UV不活性。
アザスピロ[3.4]オクタン-6-カルボキシレート(1.62g、5.00mmol
)をTHF(10mL)に溶解し、水(10mL)及び濃塩酸(10mL)を加え、混合
物を室温で一晩撹拌した。溶媒を真空除去し、残渣をEt2Oから研和して、中間体15
1、エチル2-(4-オキソピペリジン-1-イル)-6-アザスピロ[3.4]オクタ
ン-6-カルボキシレート塩酸塩(1.30g、82%)を無色の固体として得た。標記
の化合物に関するデータは表2にある。
ための手順

(fritted glass tube)を通して窒素流を該液体へと15分間通過さ
せることによって脱気した。ベンジル4-(4,4,5,5-テトラメチル-1,3,2
-ジオキサボロラン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシ
レート(250mg、0.73mmol)、4-ブロモ-1,3-チアゾール(119m
g、0.73mmol)、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]ジク
ロロパラジウム(II)(32mg、0.044mmol)、脱気炭酸ナトリウム水溶液
(2M、1.1mL、2.2mmol)及び脱気1,4-ジオキサン(3mL)を、窒素
フラッシュした管内に配置し、密閉して圧力下、90℃で2.5時間加熱した。冷却した
反応混合物をH2Oで希釈し、EtOAcで抽出した。有機相をフェーズセパレータに通
過させ、フラッシュシリカ(15mL)で濃縮した。結果得られた粉末をカラムクロマト
グラフィー(順相、[Biotage SNAPカートリッジKP-sil 50g、4
0~63μm、60Å]、40mL/分、イソヘキサン中65%Et2O、アイソクラテ
ィック)によって精製して、ベンジル4-(1,3-チアゾール-4-イル)-3,6-
ジヒドロピリジン-1(2H)-カルボキシレート(173mg、79%)を得た。
LCMS(方法C):m/z 301(M+H)+(ES+)、1.46分時、UV活性
。
-ジヒドロピリジン-1(2H)-カルボキシレート(150mg、0.50mmol)
の溶液を10%パラジウム炭素触媒上、100バール圧、50℃、1mL/分の流速でH
-Cube装置を用いて、水素化した。溶液を濃縮して、ベンジル4-(1,3-チアゾ
ール-4-イル)ピペリジン-1-カルボキシレート(143mg、95%)を得た。
LCMS(方法A):m/z 303(M+H)+(ES+)、1.92分時、UV活性
。
アゾール-4-イル)ピペリジン-1-カルボキシレート(127mg、0.42mmo
l)の溶液を室温で一晩撹拌した。次いで、混合物を濃縮し、残渣をトルエンで共沸して
、中間体164、4-(1,3-チアゾール-4-イル)ピペリジン臭化水素酸(160
mg、100%超)を得た。標記の化合物に関するデータは表2にある。
3.3.1]ノナン-7-オンを調製するための手順

mmol)及びフェニルボロン酸(0.72g、6.0mmol)をトルエン(35mL
)に溶解し、120℃で16時間加熱還流した。反応混合物を濃縮して、粗(1R,5S
,7R)-3-フェニル-2,4-ジオキサ-3-ボラビシクロ[3.3.1]ノナン-
7-オール(1.43g、87%)を固体として得た。これを直ちに使用した。(1R,
5S,7R)-3-フェニル-2,4-ジオキサ-3-ボラビシクロ[3.3.1]ノナ
ン-7-オール(1.4g、6.4mmol)をDCM(50mL)に溶解した。酢酸ナ
トリウム(1.31g、16mmol)及びクロロクロム酸ピリジニウム(12.9g、
11mmol)を加え、反応混合物を16時間撹拌した。反応混合物を、セライトを通し
て濾過し、濾過物を濃縮して粗生成物を得た。これをDCM:ヘキサン(1:4)から再
結晶化して、中間体172、(1R,5S)-3-フェニル-2,4-ジオキサ-3-ボ
ラビシクロ[3.3.1]ノナン-7-オン(0.65g、38%)を固体として得た。
標記の化合物に関するデータは表2にある。
ン-1-イル]ピペリジントリフルオロ酢酸塩を調製するための手順

キシメチル)ピロリジン-1-カルボキシレート(150mg、0.63mmol)の溶
液を氷水にて冷却し、鉱油(30mg、0.75mmol)中の60%水素化ナトリウム
懸濁液で処理した。混合物を氷内で30分間撹拌し、次いで、室温で1.5時間撹拌し、
その後ヨウ化メチル(0.118mL、1.9mmol)を加え、室温で一晩撹拌した。
混合物を一滴のH2Oでクエンチし、次いで濃縮してTHFを除去した。残渣を、飽和N
aHCO3水溶液とDCM(×2)に分配し、有機相をフェーズセパレータに通過させ、
濃縮して、粗tert-ブチル(2R)-4,4-ジフルオロ-2-(メトキシメチル)
ピロリジン-1-カルボキシレート(110mg、69%)を油状物質として得た。
LCMS(方法C):m/z 274(M+Na)+(ES+)、1.35分時、UV不
活性。
フルオロ-2-(メトキシメチル)ピロリジン-1-カルボキシレート(110mg、0
.44mmol)の溶液を室温で40分間撹拌し、次いでトルエンで希釈して濃縮した。
残渣をトルエンで共沸(×2)して、粗(2R)-4,4-ジフルオロ-2-(メトキシ
メチル)ピロリジントリフルオロ酢酸塩を油状物質(172mg、100%超)として得
た。
LCMS(方法C):m/z 152(M+H)+(ES+)、0.73分時、UV不
活性。
酸塩(172mg、想定0.44mmol)をDMF(5mL)に溶解した。該溶液にD
IPEA(0.38mL、2.2mmol)、AcOH(0.038mL、0.66mm
ol)、tert-ブチル4-オキソピペリジン-1-カルボキシレート(0.087g
、0.44mmol)及びSTAB(0.278g、1.3mmol)をこの順に加えた
。混合物を室温で2日間撹拌し、次いで濃縮してDMFを除去した。残渣を飽和NaHC
O3水溶液とDCM(×2)に分配し、有機相をフェーズセパレータに通し、濃縮して、
粗tert-ブチル4-[(2R)-4,4-ジフルオロ-2-(メトキシメチル)ピロ
リジン-1-イル]ピペリジン-1-カルボキシレート(0.241g、100%超)を
油状物質として得た。
LCMS(方法C):m/z 335(M+H)+(ES+)、1.43分時、UV不
活性。
4-ジフルオロ-2-(メトキシメチル)ピロリジン-1-イル]ピペリジン-1-カル
ボキシレート(0.241g、想定0.44mmol)の溶液を室温で45分間撹拌し、
次いでトルエンで希釈し、濃縮した。残渣をトルエンで共沸(×2)して、粗中間体17
4、4-[(2R)-4,4-ジフルオロ-2-(メトキシメチル)ピロリジン-1-イ
ル]ピペリジントリフルオロ酢酸塩を油状物質として得た。
標記の化合物に関するデータは表2にある。
ロリジン-2-イル]エタノールトリフルオロ酢酸塩を調製するための手順

キシメチル)ピロリジン-1-カルボキシレート(150mg、0.63mmol)の溶
液を氷水にて冷却し、デスマーチンペルヨージナン(402mg、0.95mmol)で
処理した。冷却浴を除去して、混合物を室温で3時間撹拌した。飽和NaHCO3水溶液
(5mL)、飽和チオ硫酸ナトリウム水溶液(5mL)及びEtOAc(10mL)を加
え、混合物を30分間勢いよく撹拌した。相を分離し、水相をEtOAcで再抽出した。
合一した有機相をフェーズセパレータに通過させ、濃縮して、粗アルデヒドを得た。これ
をTHF(5mL)に直ちに溶解し、-78℃まで冷却し、エーテル(3M、0.42m
L、1.3mmol)中のメチルマグネシウムブロミドで処理した。混合物を冷却浴から
除去し、2.75時間撹拌し、次いで飽和NH4Cl水溶液の添加によりクエンチした。
混合物を濃縮してTHFを除去し、次いで、飽和NH4ClとDCM(×2)に分配した
。有機相をフェーズセパレータに通過させ、フラッシュシリカ(10mL)で濃縮した。
結果得られた粉末をカラムクロマトグラフィー(順相、[Biotage SNAPカー
トリッジKP-sil 25g、40~63μm、60Å]、30mL/分、イソヘキサ
ン中20~50%EtOAc、によって精製して、tert-ブチル(2R)-4,4-
ジフルオロ-2-(1-ヒドロキシエチル)ピロリジン-1-カルボキシレート(0.1
06g、67%)を油状物質として得た。
LCMS(方法C):m/z 152(M-BOC+H)+、196(M-tBu+H
)+(ES+)、1.24分時、UV不活性。
ルオロ-2-(1-ヒドロキシエチル)ピロリジン-1-カルボキシレート(102mg
、0.41mmol)の溶液を室温で30分間撹拌し、次いで、トルエンで希釈し、濃縮
した。残渣をトルエンで共沸して、粗1-[(2R)-4,4-ジフルオロピロリジン-
2-イル]エタノールトリフルオロ酢酸塩をゴム質として得た。直ちに使用した。
LCMS(方法C):m/z 152(M+H)+(ES+)、0.27分時、UV不活
性。
トリフルオロ酢酸塩(想定0.41mmol)をDMF(5mL)に溶解した。該溶液に
、DIPEA(0.38mL(mLm)、2.0mmol)、AcOH(0.035mL
、0.61mmol)、tert-ブチル4-オキソピペリジン-1-カルボキシレート
(0.081g、0.41mmol)及びSTAB(0.258g、1.2mmol)を
この順に加えた。混合物を室温で3日間撹拌し、次いで、濃縮してDMFを除去した。残
渣をトルエンで共沸し、MeOHに溶解し、フラッシュシリカ(5mL)で濃縮した。結
果得られた粉末をカラムクロマトグラフィー(順相、[Biotage SNAPカート
リッジKP-sil 25g、40~63μm、60Å]、30mL/分、DCM中0~
15%溶媒A、ここで、溶媒AはMeOH中10%の(7M NH3/MeOH)である
)によって精製して、tert-ブチル4-[(2R)-4,4-ジフルオロ-2-(1
-ヒドロキシエチル)ピロリジン-1-イル]ピペリジン-1-カルボキシレート(0.
301g、100%超)を油状物質として得た。
LCMS(方法C):m/z 335(M+H)+(ES+)、1.41分時、UV不
活性。
-ジフルオロ-2-(1-ヒドロキシエチル)ピロリジン-1-イル]ピペリジン-1-
カルボキシレート(0.301g、想定0.41mmol)の溶液を室温で30分間撹拌
し、次いでトルエンで希釈し、濃縮した。残渣をトルエンで共沸して、粗中間体179、
1-[(2R)-4,4-ジフルオロ-1-(ピペリジン-4-イル)ピロリジン-2-
イル]エタノールトリフルオロ酢酸塩を油状物質(0.553g、100%超)として得
た。標記の化合物に関するデータは表2にある。
の手順
リジン-1-カルボキシレート(0.80g、3.9mmol)及びアジ化ナトリウム(
1.52g、23mmol)を酢酸(50mL)に溶解した。結果得られた反応混合物を
100℃で6時間撹拌し、次いで室温まで冷却した。揮発性物質を濃縮によって除去し、
残渣をH2O(100mL)と酢酸エチル(150mL)に分配した。水層を酢酸エチル
(2×100mL)でさらに抽出し、合一した有機層を乾燥させ(Na2SO4)、溶媒
を濃縮により除去して、粗生成物を得た。これをジエチルエーテルで研和して、tert
-ブチル4-(1H-テトラゾール-1-イル)ピペリジン-1-カルボキシレート(0
.51g、9%)を固体として得た。
LCMS(方法F):m/z 254(M+H)+(ES+)、1.92分時、弱UV
活性。
レート(0.51g、2.0mmol)を1,4-ジオキサン(10mL)に溶解した。
1,4-ジオキサン(4M、5mL、20mmol)中のHClの溶液を滴加し、結果得
られた混合物を室温で16時間撹拌した。溶媒を濃縮により除去し、残渣をジエチルエー
テル(3×10mL)で研和することにより精製して、中間体215、4-(1H-テト
ラゾール-1-イル)ピペリジン塩酸塩(0.30g、97%)を固体として得た。標記
の化合物に関するデータは表2にある。
塩酸塩を調製するための手順

7mmol)及びシクロプロピルアミン(0.6mL、8.7mL)をDMF(45mL
)に溶解した。HATU(3.3g、8.7mmol)を室温で加え、その後DIPEA
(3.1mL、17mmol)を加えた。反応混合物を室温で3時間撹拌した。反応混合
物を、冷水(250mL)で希釈し、EtOAc(3×100mL)で抽出した。合一し
た有機層を乾燥させ(Na2SO4)、濃縮して粗生成物を得た。これをカラムクロマト
グラフィー(順相、中性シリカゲル、60~120メッシュ、ヘキサン中0~35%Et
OAC)によって濃縮して、tert-ブチル4-(シクロプロピルカルバモイル)ピペ
リジン-1-カルボキシレート(1.5g、64%)を固体として得た。
LCMS(方法F):m/z 269(M+H)+(ES+)、1.80分時、弱UV活
性。
ート(1.5g、5.6mmol)及びトリフェニルホスフィン(2.9g、11mmo
l)をTHF(160mL)に溶解した。DIAD(2.26g、11mmol)を室温
で15分かけて加えた。トリメチルシリルアジド(1.3g、11mmol)を加え、反
応混合物を室温で24時間撹拌した。反応混合物を水(250mL)で希釈し、EtOA
c(3×100mL)で抽出した。合一した有機層を乾燥させ(Na2SO4)、濃縮し
て粗生成物を得た。これをカラムクロマトグラフィー(順相、中性シリカゲル、60~1
20メッシュ、ヘキサン中0~30%EtOAC)により精製して、tert-ブチル4
-(1-シクロプロピル-1H-テトラゾール-5-イル)ピペリジン-1-カルボキシ
レート(400mg、24%)を固体として得た。
LCMS(方法F):m/z 294(M+H)+(ES+)、1.96分時、弱UV
活性。
ジン-1-カルボキシレート(400mg、1.4mmol)をジオキサン(5mL)に
溶解した。ジオキサン(4M、5mL、20mmol)中のHClの溶液を0℃で加え、
混合物を室温で5時間撹拌した。溶媒を濃縮により除去し、残渣をジエチルエーテル(1
0mL)で研和して、中間体218、4-(1-シクロプロピル-1H-テトラゾール-
5-イル)ピペリジン塩酸塩(260mg、98%)を固体として得た。標記の化合物に
関するデータは表2にある。
酢酸塩を調製するための手順

ピペリジン-1-カルボキシレート(0.221g、1.10mmol及びトリフェニル
ホスフィン(0.314g、1.20mmol)をTHF(5mL)に溶解し、次いで、
ジイソプロピルアゾジカルボキシレート(0.236mL、1.20mmol)で処理し
、室温で一晩撹拌した。反応混合物をフラッシュシリカ(5mL)で濃縮した。結果得ら
れた粉末をカラムクロマトグラフィー(順相、[Biotage SNAPカートリッジ
KP-sil 25g、40~63μm、60Å]、30mL/分、イソヘキサン中20
%~100%EtOAc)によって精製して、tert-ブチル4-(2,5-ジオキソ
ピロリジン-1-イル)ピペリジン-1-カルボキシレート(0.253g、90%)を
固体として得た。
LCMS(方法C):m/z 305(M+Na)+(ES+)、1.11分時、UV
不活性
ピロリジン-1-イル)ピペリジン-1-カルボキシレート(0.141g、0.50m
mol)の溶液を室温で30分間撹拌し、次いで、トルエンで希釈し、濃縮した。残渣を
トルエンで共沸して、中間体193、1-(ピペリジン-4-イル)ピロリジン-2,5
-ジオントリフルオロ酢酸塩をゴム質として得た。直ちに使用した。標記の化合物に関す
るデータは表2にある。
ロ[3.4]オクタン-6-カルボキシレートを調製するための手順
溶液に、n-ブチルリチウム(79.1mL、ヘキサン中2.5M、126mmol)を
-78℃でゆっくりと加えた。この温度で30分間撹拌した後、THF(40mL)中の
tert-ブチル4-オキソピペリジン-1-カルボキシレート(13.8g、69.6
mmol)をゆっくりと加えた。反応温度を徐々に室温まで持っていき、2時間撹拌した
。0℃に冷却した後、反応混合物を氷冷水(50mL)で注意深くクエンチした。揮発性
物質を除去した後、水層を酢酸エチル(3×50mL)で抽出した。有機層を合一し、飽
和食塩水で洗浄し、乾燥させ(Na2SO4)、濾過し、真空濃縮した。残渣をフラッシ
ュカラムクロマトグラフィー[順相、シリカゲル(100~200メッシュ)、勾配:ヘ
キサン中0%~30%酢酸エチル]によって精製して、tert-ブチル4-ヒドロキシ
-4-(ピリジン-2-イル)ピペリジン-1-カルボキシレート(11.4g、65%
)を黄色油状物質として得た。
1H-NMR(400 MHz; CDCl3) δ: 1.48(s, 9H), 1.56
- 1.63(m,2H), 1.90 - 2.0(m, 2H), 3.25 - 3.46(
m, 2H), 4.05- 4.22(m, 2H), 5.29(br.s., 1H), 7.
20 - 7.25(m, 1H), 7.32(d, J = 8.0, Hz, 1H), 7.73
(dt, J = 1.6,8.4 Hz, 1H), 8.53(d, J = 4.8 Hz, 1H
)。
イル)ピペリジン-1-カルボキシレート(11.4g、41.0mmol)の溶液に、
POCl3(5.7mL、61.5mmol)を加え、室温で20時間撹拌した。ピリジ
ンを真空除去した後、反応混合物をNaOH水溶液(10%、30mL)でクエンチし、
クロロホルム(2×30mL)で抽出した。有機層を合一し、乾燥させ(Na2SO4)
、濾過し、真空濃縮した。残渣をフラッシュカラムクロマトグラフィー[順相、シリカゲ
ル(100~200メッシュ)、勾配:ヘキサン中0%~30%酢酸エチル]によって精
製して、tert-ブチル3’,6’-ジヒドロ-[2,4’-ビピリジン]-1’(2
’H)-カルボキシレート(2.3g、21%)を黄色油状物質として得た。
1H-NMR(400 MHz; CDCl3) δ: 1.48(s, 9H), 2.61
- 2.70(m,2H), 3.60 - 3.70(m, 2H), 4.10 - 4.19
(m, 2H), 6.58- 6.62(m, 1H), 7.11 - 7.19(m, 1H)
, 7.36(d, J = 7.88Hz, 1H), 7.62 - 7.66(m, 1H),
8.55(d, J= 4.4 Hz, 1H)。
ビピリジン]-1’(2’H)-カルボキシレート(2.3g、8.84mmol)の溶
液に、PtO2(200mg、0.88mmol)を加え、反応混合物を室温で2日間H
2雰囲気下にて攪拌した。反応混合物を、セライトパッドを通して濾過し、MeOHで洗
浄し、真空濃縮した。残渣をフラッシュカラムクロマトグラフィー[順相、シリカゲル(
100~200メッシュ)、勾配:0.1%アンモニア水溶液を有するDCM中0%~1
5%MeOH]によって精製して、tert-ブチル[2,4’-ビピペリジン]-1’
-カルボキシレート(1.05g、44%)を無色の油状物質として得た。
1H-NMR(400 MHz; CDCl3) δ: 1.30 - 1.40(m,1H)
,1.48(s,9H),1.60 - 1.91(m,8H),2.45- 2.55(
m,2H),2.58 - 2.75(m,4H),3.24- 3.31(m,1H),
4.14 - 4.24(m,2H)。
シレート(1.05g、3.91mmol)の溶液に、THF(5mL)中のCbz-O
Su(975mg、3.91mmol)を加え、室温で2時間撹拌した。反応混合物を水
(20mL)でクエンチし、水層を酢酸エチル(2×20mL)で抽出した。有機層を合
一し、飽和食塩水で洗浄し、乾燥させ(Na2SO4)、濾過し、真空濃縮した。残渣を
フラッシュカラムクロマトグラフィー[順相、シリカゲル(100~200メッシュ)、
勾配:ヘキサン中0%~20%酢酸エチル]によって精製して、1-ベンジル1’-(t
ert-ブチル)[2,4’-ビピペリジン]-1,1’-ジカルボキシレート(840
mg、53%)を無色の油状物質として得た。
1H-NMR(400 MHz;CDCl3) δ: 1.44(s, 9H), 1.50
- 1.63(m,2H), 1.68 - 1.92(m, 4H), 1.95 - 2.14
(m, 2H), 2.55- 2.80(m, 2H), 2.81 - 2.95(m, 4H)
, 2.98 - 3.10(m,1H), 3.75 - 4.24(m, 3H), 5.11(
s, 2H), 7.34- 7.37(m, 5H)]。
ビピペリジン]-1,1’-ジカルボキシレート(840mg、2.1mmol)の溶液
に、ジオキサン中HCl(10mL、4M)を0℃でゆっくりと加え、反応混合物を室温
で2時間撹拌した。反応混合物を、飽和NaHCO3(20mL)でクエンチし、水層を
CH2Cl2(2×20mL)で抽出した。有機層を合一し、乾燥させ(Na2SO4)
、濾過し、真空濃縮した。残渣をフラッシュカラムクロマトグラフィー[順相、シリカゲ
ル(100~200メッシュ)、勾配:0.1%アンモニア水溶液を有するDCM中0%
~15%MeOH]により精製して、ベンジル[2,4’-ビピペリジン]-1-カルボ
キシレート(570mg、90%)を無色の粘着性の固体として得た。
1H-NMR(400 MHz;CDCl3) δ: 1.35 - 1.70(m, 6H
), 1.71 -1.98(m, 4H), 2.46 - 2.63(m, 2H), 2.68
- 2.73(m,1H), 3.03 - 3.18(m, 1H), 3.65 - 3.80
(m, 2H), 3.86- 4.16(m, 2H), 5.11(s, 2H), 7.34
- 7.37(m,5H)。
レート(520mg、1.72mmol)及びエチル2-オキソ-6-アザスピロ[3.
4]オクタン-6-カルボキシレート(305mg、1.55mmol)の溶液に、Ti
(OiPr)4(1.6mL、5.16mmol)を加え、0℃で40分間撹拌した。こ
の反応混合物にNaBH4(1.1g、5.16mmol)を加え、この温度で2時間撹
拌を続けた。反応混合物を水(20mL)でクエンチし、水層をCH2Cl2(2×20
mL)で抽出した。有機層を合一し、乾燥させ(Na2SO4)、濾過し、真空濃縮した
。残渣を分取HPLC(逆相、X BRIDGE、C-18、19×250mm、5μ、
勾配:0.1%NH4OHを含有する水中68%~90%ACN、214nm、室温:異
性体-1については7.45分及び異性体-2については8.37分によって精製して、
エチル2-オキソ-6-アザスピロ[3.4]オクタン-6-カルボキシレート異性体1
、(120mg、15%)及びエチル2-オキソ-6-アザスピロ[3.4]オクタン-
6-カルボキシレート異性体-2、(160mg、19%)を無色の粘着性の固体として
得た。
異性体-1:
LCMS(方法L):m/z 484(M+H)+(ES+)、5.70分時、UV活
性。
1H-NMR(400 MHz; CDCl3) δ: 1.10 - 1.32(m, 6H)
, 1.36 - 1.95(m,14H), 2.00 - 2.18(m, 2H), 2.52
- 3.05(m,4H), 3.20 - 3.45(m, 4H), 3.87 - 4.18
(m, 4H), 5.11(s,2H), 7.30 - 7.35(m, 5H)。
異性体-2:
LCMS(方法L):m/z 484(M+H)+(ES+)、5.81分時、UV活
性。
1H-NMR(400 MHz; CDCl3) δ: 1.10 - 1.32(m, 6H)
, 1.35 - 1.53(m,5H), 1.62 - 1.80(m, 5H), 1.81
- 1.97(m,4H), 2.00 - 2.18(m, 2H), 2.52 - 3.00
(m, 4H), 3.18- 3.52(m, 4H), 3.88 - 4.20(m, 4H)
, 5.11(s, 2H), 7.32- 7.37(m, 5H)。
-カルボキシレート(1.0g、2.06mmol)の異性体の混合物の溶液に、10%
パラジウム炭(320mg、50%湿潤)を加え、反応混合物を室温で16時間H2雰囲
気下にて攪拌した。反応混合物を、セライトパッドを通して濾過し、MeOHで洗浄し、
真空濃縮し、ペンタンで研和して、中間体229、エチル2-([2,4’-ビピペリジ
ン]-1’-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート(44
5mg、92%)を無色の液体として得た。標記の化合物に関するデータは表2にある。
H-イソインドール-2-カルボキシレートを調製するための手順
.0mmol)の溶液に、SOCl2(2.7mL、37.5mmol)を0℃でゆっく
りと加え、反応混合物を室温で16時間撹拌した。完了後、反応混合物を真空濃縮した。
残渣をジエチルエーテルで研和して、メチルイソインドリン-1-カルボキシレート塩酸
塩(4.9g、92%)を灰白色固体として得た。残渣をさらなる精製を行わずに次のス
テップに使用した。
1H-NMR(400 MHz; DMSO--d6) δ: 3.81(s, 3H), 4.
52 - 4.63(m, 2H), 5.70(s, 1H), 7.44 - 7.50(m, 4
H), 9.77(br.s.,2H)。
g、23.0mmol)の溶液に、Et3N(9.9mL、69.0mmol)及び(B
oc)2O(8.0mL、34.0mmol)を順次0℃で加えた。反応混合物を室温で
12時間撹拌した。反応混合物を水(20mL)でクエンチした。有機層を分離した後、
水層をCH2Cl2(3×15mL)で抽出した。有機層を合一し、飽和食塩水で洗浄し
、乾燥させ(Na2SO4)、濾過し、真空濃縮した。粗残渣をフラッシュカラムクロマ
トグラフィー[順相、シリカゲル(100~200メッシュ)、勾配:ヘキサン中10%
~30%酢酸エチル]によって精製して、2-(tert-ブチル)1-メチルイソイン
ドリン-1,2-ジカルボキシレート(6.5g、90%)を無色の液体として得た。
1H-NMR(400 MHz; CDCl3) δ: 1.52(s, 9H), 3.75(
s, 3H), 4.65- 4.85(m, 2H), 5.45(s, 1H), 7.25 -
7.43(m, 4H)。
-ジカルボキシレート(6.5g、23.0mmol)の溶液に、LAH(2M、11.
5mL、23.0mmol)を0℃でゆっくりと加え、30分間撹拌した。完了後、反応
混合物を飽和Na2SO4水溶液(20mL)でクエンチした。反応混合物を、セライト
パッドを通して濾過し、酢酸エチル(100mL)で洗浄し、乾燥させ(Na2SO4)
、真空濃縮して、tert-ブチル1-(ヒドロキシメチル)イソインドリン-2-カル
ボキシレート(5.2g、91%)を灰白色固体として得た。粗残渣をさらなる精製を行
わずに次のステップに使用した。
1H-NMR(400 MHz; CDCl3) δ: 1.52(s, 9H), 3.70
- 3.78(m,1H), 3.98 - 4.03(m, 1H), 4.60 - 4.69
(m, 1H), 4.70- 4.85(m, 2H), 5.22(br.s., 1H), 7
.25 - 7.40(m, 4H)。
-2-カルボキシレート(5.2g、20.0mmol)の溶液に、デスマーチンペルヨ
ージナン(27g、62.0mmol)を0℃で少量ずつ加え、室温で48時間撹拌した
。完了後、反応混合物を、セライトパッドを通して濾過し、ジエチルエーテル(3×20
mL)で洗浄した。濾過物を飽和NaHCO3水溶液、飽和食塩水で洗浄し、乾燥させ(
Na2SO4)、真空濃縮して、tert-ブチル1-ホルミルイソインドリン-2-カ
ルボキシレート(4.5g、88%)を褐色液体として得た。粗残渣をさらなる精製を行
わずに次のステップに使用した。
1H-NMR(400 MHz; CDCl3) δ: 1.48(s, 9H), 4.65
- 4.90(m,2H), 5.29 - 5.35(s, 1H), 7.25 - 7.35
(m, 4H), 9.51(s,1H)。
ル(3.3mL、18.2mmol)を-78℃で加えた。-78℃で1時間撹拌した後
、tert-ブチル1-ホルミルイソインドリン-2-カルボキシレート(4.5g、1
8.2mmol)をゆっくり加え、反応混合物を自然に0℃にした。完了後、反応混合物
を飽和NH4Cl水溶液(10mL)でクエンチし、水層を酢酸エチル(3×20mL)
で抽出した。有機層を合一し、乾燥させ(Na2SO4)、真空濃縮して、tert-ブ
チル(E)-1-(3-メトキシ-3-オキソプロパ-1-エン-1-イル)イソインド
リン-2-カルボキシレート(5.2g、92%)を褐色液体として得た。残渣をさらな
る精製を行わずに次のステップに使用した。
MS(ESI+ve):304
プロパ-1-エン-1-イル)イソインドリン-2-カルボキシレート(5.2g、17
.2mmol)の溶液に、Cs2CO3(11.1g、34.4mmol)を室温で少量
ずつ加えた。20分間撹拌した後、シアノ酢酸メチル(3.0mL、34.4mmol)
をゆっくりと加え、反応混合物を70℃で16時間撹拌した。完了後、反応混合物を、セ
ライトパッドを通して濾過し、ヘキサン(3×20mL)で徹底的に洗浄した。濾過物を
真空濃縮して、ジメチル3-(2-(tert-ブトキシカルボニル)イソインドリン-
1-イル)-2-シアノペンタンジオエート)(5.5g、粗(cr))を褐色の粘着性
の固体として得た。粗残渣をさらなる精製を行わずに次のステップに使用した。
MS(ESI+ve):403
インドリン-1-イル)-2-シアノペンタンジオエート)(1.6g、粗)の溶液に、
LiCl(500mg、11.7mmol)を加え、その後、水(0.1mL、触媒)を
加え、反応混合物を135℃で16時間撹拌した。完了後、反応混合物を水(20mL)
でクエンチし、水層をジエチルエーテル(3×20mL)で抽出した。有機層を合一し、
乾燥させ(Na2SO4)、真空濃縮して、tert-ブチル1-(1-シアノ-4-メ
トキシ-4-オキソブタン-2-イル)イソインドリン-2-カルボキシレート(1.5
g、粗)を褐色の半固体として得た。粗残渣をさらなる精製を行わずに次のステップに使
用した。
MS(ESI+ve):345
キソブタン-2-イル)イソインドリン-2-カルボキシレート(300mg、0.8m
mol)の溶液に、ラネーニッケル(0.30g、湿潤)を加え、反応混合物を50℃に
H2雰囲気下、50psiで2時間加熱した。次いで、反応温度を70℃まで上昇させ、
3時間撹拌した。完了後、反応混合物を、セライトパッドを通して濾過し、MeOH(2
5mL)で洗浄し、真空濃縮した。残渣をジエチルエーテル(30mL)で研和して、t
ert-ブチル1-(2-オキソピペリジン-4-イル)イソインドリン-2-カルボキ
シレート(0.21g、76%)を褐色固体として得た。
MS(ESI+ve):317
インドリン-2-カルボキシレート(210mg、0.60mmol)の溶液に、BH3
-DMS(0.5mL、6.60mmol)を0℃でゆっくりと加え、反応混合物を78
℃で8時間撹拌した。0℃で冷却した後、反応塊をメタノール(0.5mL)、その後、
水(1mL)でクエンチした。粗反応塊に、5%MeOH/DCM(30mL)を加え、
濾過した。濾過物を真空濃縮した。残渣をジエチルエーテル(20mL)で研和して、t
ert-ブチル1-(ピペリジン-4-イル)イソインドリン-2-カルボキシレート、
中間体243(200mg、99%)を褐色固体として得た。標記の化合物に関するデー
タは表2にある。
るための手順

2.4mmol)をCH2Cl2に溶解し、次いでDMAP(0.302g、2.4mm
ol)及びメタンスルホニルクロリド(0.284g、2.48mmol)を0℃で滴加
した。結果得られた反応混合物を室温で6時間撹拌し、次いで、H2O(70mL)とC
H2Cl2(70mL)に分配し、水層をCH2Cl2(2×70mL)でさらに抽出し
、有機層を合一し、乾燥させ(Na2SO4)、濾過し、溶媒を真空除去して、粗ter
t-ブチル4-((メチルスルホニル)オキシ)ピペリジン-1-カルボキシレート(0
.520g、75.0%)を白色固体として得た。これをさらなる精製を行わずに直接使
用した。
1H-NMR(400 MHz;DMSO) δ: 1.23(d, J = 9.38 H
z, 2H)1.54- 1.69(m, 4H)1.86 - 1.96(m, 2H)2.3
5(s, 1H)2.85- 3.00(m, 2H)3.18(d, J = 5.42 Hz,
5H)3.54 - 3.67(m, 4H)4.83(s, 1H)。
)に溶解し、NaH(0.037g、1.5mmol)を加え、0℃で30分間撹拌した
。tert-ブチル4-((メチルスルホニル)オキシ)ピペリジン-1-カルボキシレ
ート(0.400g、1.4mmol)を加え、150℃で1時間撹拌した。反応混合物
をH2O(50mL)とEtOAc(50mL)に分配し、水層をEtOAc(2×50
mL)でさらに抽出し、有機層を合一し、乾燥させ(Na2SO4)、濾過し、溶媒を真
空除去して、粗tert-ブチル4-(2H-1,2,3-トリアゾール-2-イル)ピ
ペリジン-1-カルボキシレート(0.350g、97.0%)を無色のゴム質として得
た。これをさらなる精製を行わずに直接使用した。
LCMS(方法F):m/z 253(M+H)+(ES+)、1.95分時、UV活性
。
-カルボキシレート(0.500g、1.9mmol)を1,4-ジオキサン(10mL
)に溶解し、その後、1,4-ジオキサン中HCl(5mL、4M)を滴加した。結果得
られた反応混合物を25℃で16時間撹拌し、溶媒を真空除去し、残渣をジエチルエーテ
ル(3×10mL)で研和することによって精製して、4-(2H-1,2,3-トリア
ゾール-2-イル)ピペリジン塩酸塩、中間体247、(0.290g、96.3%)を
淡白色固体として得た。標記の化合物に関するデータは表2にある。
調製するための手順

チル4-ブロモピペリジン-1-カルボキシレート(1.29g、4.8mmol)をD
MFに溶解した。K2CO3(1.64g、11.8mmol)を加え、結果得られた反
応混合物を100℃で6時間撹拌し、次いでH2O(100mL)と酢酸エチル(150
mL)に分配した。水層を酢酸エチル(2×100mL)でさらに抽出し、有機層を合一
し、乾燥させ(Na2SO4)、濾過し、溶媒を真空除去した。残渣をコンビフラッシュ
カラムクロマトグラフィー(順相、中性シリカゲル、60~120メッシュ、ヘキサン中
10~20%EtOAc)によって精製して、tert-ブチル4-(5-メチル-1H
-テトラゾール-1-イル)ピペリジン-1-カルボキシレート(0.280g、34.
6%)を白色固体として得た。
1H-NMR(400 MHz, DMSO) δ: 1.43(s, 9H), 1.73 -
1.88(m, 2H),2.01(br.s., 2H), 2.68 - 2.75(m,
3H), 2.88- 2.91(m, 2H), 4.03 - 4.10(m, 2H), 4.
60 - 4.70(m, 1H)。
-カルボキシレート(0.280g、1.04mmol)を1,4-ジオキサン(10m
L)に溶解し、その後1,4-ジオキサン中HCl(5mL、4M)を滴加した。結果得
られた反応混合物を25℃で16時間撹拌し、溶媒を真空除去し、残渣をジエチルエーテ
ル(3×10mL)で研和することによって精製して、4-(5-メチル-1H-テトラ
ゾール-1-イル)ピペリジン塩酸塩、中間体255(0.170g、97.6%)を白
色固体として得た。標記の化合物に関するデータは表2にある。
リジン-2-イル)プロパン-2-オール塩酸塩を得て調製するための手順

フルオロピロリジン-1,2-ジカルボキシレート(500mg、1.89mmol)の
溶液に、ジオキサン中HCl(4M、15mL)を0℃でゆっくりと加え、室温で3時間
撹拌した。反応混合物を真空濃縮して、残渣をヘキサン(10mL)で研和した。この残
渣を飽和NaHCO3水溶液(10mL)で塩基性化し、濃縮した。粗反応塊にCDM(
30mL)を加え、濾過した。濾過物を真空濃縮して、メチル(R)-4,4-ジフルオ
ロピロリジン-2-カルボキシレート(2,320mg、84%)を褐色液体として得た
。この粗残渣をさらなる精製を行わずに次のステップに使用した。
1H-NMR(400 MHz, CDCl3) δ:1.40 - 1.51(m, 1H),
2.58 - 2.84(m, 3H), 3.52 - 3.62(m, 1H), 3.84(
s, 3H), 4.40- 4.52(m, 1H)。
ボキシレート(200mg、1.21mmol)及びtert-ブチル4-オキソピペリ
ジン-1-カルボキシレート(240mg、1.21mmol)の溶液に、10%パラジ
ウム炭素(300mg、50%湿潤)を加え、反応混合物を、H2(1気圧)下、室温で
24時間撹拌した。完了後、反応混合物を、セライトパッドを通して濾過し、メタノール
で徹底的に洗浄し、真空濃縮して、tert-ブチル(R)-4-(4,4-ジフルオロ
-2-(メトキシカルボニル)ピロリジン-1-イル)ピペリジン-1-カルボキシレー
ト(400mg、95%)を無色の液体として得た。
1H-NMR(400 MHz, CDCl3) δ: 1.32 - 1.45(m, 1H
), 1.45(s,9H), 1.61 - 1.80(m, 4H), 2.39 - 2.49
(m, 1H), 2.50- 2.83(m, 2H), 3.19(s, 3H), 3.35
- 3.49(m,2H), 3.61 - 3.82(m, 3H), 3.94 - 4.05
(m, 1H)。
メトキシカルボニル)ピロリジン-1-イル)ピペリジン-1-カルボキシレート(37
5mg、1.07mmol)の溶液に、MeMgBr(3M、1.07mL、3.21m
mol)を0℃でゆっくりと加え、室温で4時間撹拌した。完了後、反応混合物を飽和N
H4Cl水溶液(10mL)でクエンチし、水層を酢酸エチル(3×10mL)で抽出し
た。有機層を合一し、乾燥させ(Na2SO4)、濾過し、真空濃縮した。粗残渣をフラ
ッシュカラムクロマトグラフィー[順相、シリカゲル(100~200メッシュ)、勾配
:ヘキサン中10%~30%酢酸エチル]によって精製して、tert-ブチル(R)-
4-(4,4-ジフルオロ-2-(2-ヒドロキシプロパン-2-イル)ピロリジン-1
-イル)ピペリジン-1-カルボキシレート(240mg、64%)を無色の液体として
得た。
1H-NMR(400 MHz, CDCl3) δ: 1.11(s, 3H), 1.21
(s, 3H), 1.21- 1.40(m, 2H), 1.46(s, 9H), 1.61
- 1.80(m,2H), 2.15 - 2.30(m, 3H), 2.50 - 2.83
(m, 3H), 3.02- 3.23(m, 2H), 4.09 - 4.30(m, 2H)
。O-H は観察されなかった。
シプロパン-2-イル)ピロリジン-1-イル)ピペリジン-1-カルボキシレート(2
40mg、0.69mmol)の溶液に、ジオキサン中HCl(4M、10mL)を0℃
でゆっくりと加え、室温で2時間撹拌した。反応混合物を真空濃縮し、残渣をヘキサン(
10mL)で研和した。粗反応塊にCH2Cl2(30mL)を加え、濾過した。濾過物
を真空濃縮して、(R)-2-(4,4-ジフルオロ-1-(ピペリジン-4-イル)ピ
ロリジン-2-イル)プロパン-2-オール塩酸塩、中間体258(150mg、87%
)を褐色液体として得た。標記の化合物に関するデータは表2にある。
1-カルボキシレートを調製するための手順

g、2.18mmol)を無水DCM(8mL)に溶解し、反応混合物を窒素下で0℃ま
で冷却した。1-エチル-3-(3-ジメチルアミノプロピル)-カルボジイミドHCl
(0.628g、3.275mmol)、ヒドロキシベンゾトリアゾール(0.334g
、2.183mmol)、N-メチルモルフォリン(1.104g、10.915mmo
l)及びジメチルアミン塩酸塩(0.356g、4.36mmol)を加え、反応混合物
を窒素下、室温で一晩撹拌した。反応混合物をDCM(20mL)で希釈し、飽和NaH
CO3(水溶液)(20mL)及び飽和NaCl(水溶液)(20mL)で洗浄した。有
機層をBiotageフェーズセパレータカートリッジに通し、溶媒を真空除去した。残
渣をカラムクロマトグラフィー(順相、[Biotage SNAPカートリッジKP-
sil 25g 40~63μm、60Å、25mL/分、勾配:0%~10%MeOH
/DCM])によって精製して、tert-ブチル(2R)-2-(ジメチルカルバモイ
ル)ピペリジン-1-カルボキシレート、中間体282、(0.241g、43%)を琥
珀色の油状物質として得た。標記の化合物に関するデータは表2にある。
ルボキシレートを調製するための手順

mmol)をDCM(8mL)に溶解し、窒素下で-78℃まで冷却した。N,N-ジエ
チルアミノ硫黄トリフルオリド(0.360g 2.24mmol)を反応混合物に滴加
し、反応混合物を窒素下、-78℃で4時間撹拌し、次いで、室温まで一晩加温した。反
応混合物を飽和NaHCO3(水溶液)(20mL)の添加によってクエンチし、DCM
(2×15mL)で抽出し、有機層を合一し、Biotageフェーズセパレータカート
リッジに通すことによって乾燥させ、溶媒を真空除去した。残渣をカラムクロマトグラフ
ィー(順相、[Biotage SNAPカートリッジKP-sil 10g 40~6
3μm、60Å、12mL/分、勾配:0%~4%MeOH/DCM])によって精製し
て、tert-ブチル(2R)-2-(フルオロメチル)ピロリジン-1-カルボキシレ
ート、中間体295、(0.104g、34%)を、琥珀色の油状物質として得た。標記
の化合物に関するデータは表2にある。
調製するための手順
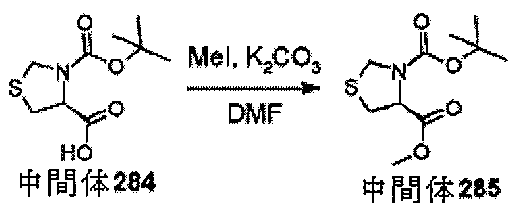
)を無水DMF(4mL)に溶解し、炭酸カリウム(2.372g、17.16mmol
)及びヨードメタン(0.730g、5.14mmol)を加えた。反応混合物を窒素下
、室温で一晩撹拌した。溶媒を真空除去し、残渣をEtOAc(40mL)に溶解し、水
(3×20mL)及び飽和NaCl(水溶液)(20mL)で洗浄し、乾燥させた(Mg
SO4)。溶媒を真空除去して、3-tert-ブチル-4-メチル(4S)-1,3-
チアゾリジン-3,4-ジカルボキシレート、中間体285、(0.812g、77%)
を淡黄色油状物質として得た。標記の化合物に関するデータは表2にある。
カルボキシレートを調製するための手順
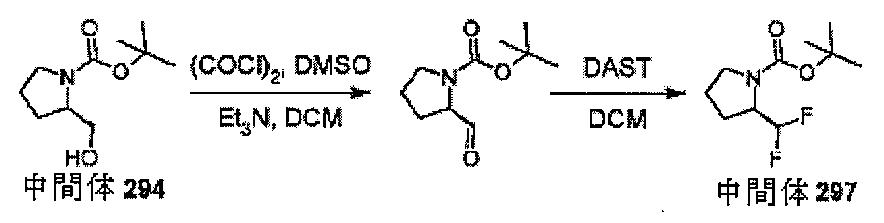
サリル(0.566g、2.93mmol)の溶液に窒素下、-78℃で滴加した。反応
混合物を窒素下、-78℃で15分間撹拌し、次いで無水DCM(4mL)中の(2R)
-(+)-1-Boc-2-ピロリジンメタノール(0.600g、2.98mmol)
の溶液を滴加した。反応混合物を窒素下、-78℃で15分間撹拌し、次いでEt3N(
1.06g、11.92mmol)を加え、反応混合物を窒素下、0℃で1時間撹拌した
。反応混合物を飽和NaHCO3(水溶液)(20mL)でクエンチし、DCM(2×2
0mL)で抽出し、有機層を合一し、Biotageフェーズセパレータカートリッジに
通すことによって乾燥させ、溶媒を真空除去した。残渣をカラムクロマトグラフィー(順
相、[Biotage SNAPカートリッジKP-sil 10g 40~63μm、
60Å、12mL/分、勾配:0%~4%MeOH/DCM])によって精製して、te
rt-ブチル(2R)-2-ホルミルピロリジン-1-カルボキシレート(0.435g
、73%)を得た。
35g、2.19mmol)を無水DCM(8mL)に溶解し、窒素下で-78℃まで冷
却した。N,N-ジエチルアミノ硫黄トリフルオリド(0.528g、3.28mmol
)を反応混合物に滴加し、反応混合物を窒素下、-78℃で3時間撹拌し、次いで、一晩
室温まで加温した。反応混合物を飽和NaHCO3(水溶液)(20mL)の添加により
クエンチし、DCM(2×15mL)で抽出し、有機層を合一し、Biotageフェー
ズセパレータカートリッジに通すことによって乾燥させ、溶媒を真空除去した。残渣をカ
ラムクロマトグラフィー(順相、[Biotage SNAPカートリッジKP-sil
10g 40~63μm、60Å、12mL/分、勾配:0%~4%MeOH/DCM
])によって精製して、tert-ブチル(2R)-2-(ジフルオロメチル)ピロリジ
ン-1-カルボキシレート、中間体297、(0.217g、45%)を琥珀色の油状物
質として得た。標記の化合物に関するデータは表2にある。
経路1
中間体30、5-(ピペリジン-4-イル)-1,2,4-チアジアゾールの調製によっ
て例示される鈴木反応、水素化及びBoc脱保護を介してピペリジンを調製するための典
型的な手順

ert-ブチル4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-
2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(200mg、
0.65mmol)及びCs2CO3(632mg、1.94mmol)をジオキサン:
水(10:2mL)に溶解した。反応混合物を30分間脱気し、その後PdCl2dpp
f(24mg、0.03mmol)を加え、次いで、90℃で16時間撹拌した。反応混
合物を、H2O(80mL)とEtOAc(50mL)に分配し、水層をEtOAc(2
×50mL)でさらに抽出し、有機層を合一し、乾燥させ(Na2SO4)、溶媒を真空
除去し、残渣をカラムクロマトグラフィー(順相シリカ、メッシュサイズ:60~120
、ヘキサン中16%~20%EtOAc)によって精製して、tert-ブチル4-(1
,2,4-チアジアゾール-5-イル)-3,6-ジヒドロピリジン-1(2H)-カル
ボキシレート(158mg、92.0%)を灰白色固体として得た。
LCMS(方法F):m/z 212(M+H-56)+(ES+)、2.37分時、U
V活性
ピリジン-1(2H)-カルボキシレート(200mg、0.74mmol)をMeOH
(15mL)に溶解し、10%Pd/C(20mg)を加えた。反応混合物をH2ガスで
パージし、25℃で8時間、H2圧下で攪拌した。反応混合物を、セライトを通して濾過
し、残渣をMeOHで洗浄し、溶媒を真空除去し、残渣をカラムクロマトグラフィー(順
相シリカ、メッシュサイズ:60~120、ヘキサン中20%~24%EtOAc)によ
って精製して、tert-ブチル4-(1,2,4-チアジアゾール-5-イル)ピペリ
ジン-1-カルボキシレート(150mg、74.6%)を暗緑色ゴム質として得た。
LCMS(方法F):m/z 214(M+H)+(ES+)、2.14分時、UV活性
ルボキシレート(150mg、0.56mmol)を1,4-ジオキサン(5mL)に溶
解し、ジオキサン中HCl(10mL、3.0M溶液)を滴加し、反応物を30℃で16
時間撹拌した。溶媒を真空除去し、残渣をジエチルエーテル(3×3mL)で研和するこ
とによって精製して、中間体30、5-(ピペリジン-4-イル)-1,2,4-チアジ
アゾール(102mg、89.5%)を暗緑色ゴム質として得た。標記の化合物に関する
データは表2にある。
中間体34、4-(1,5-ジメチル-1H-イミダゾール-2-イル)-1,2,3,
6-テトラヒドロピリジンを調製するための手順

ン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(2.0g
、6.55mmol)、2-ブロモ-1,5-ジメチル-1H-イミダゾール(1.13
g、6.45mmol)及びCsF(2.9g、1.85mmol)をDME:MeOH
(2:1、30mL)に溶解した。反応混合物を5分間脱気し、次いでPd(PPh3)
4(73mg、0.064mmol)を加え、結果得られた反応混合物を100℃で5時
間撹拌した。反応混合物を、H2O(100mL)とEtOAc(100mL)に分配し
、水層をEtOAc(2×100mL)でさらに抽出し、有機層を合一し、乾燥(Na2
SO4)させ、溶媒を真空除去した。残渣をカラムクロマトグラフィー(順相シリカ、メ
ッシュサイズ:60~120、ヘキサン中13%~17%酢酸エチル)によって精製して
、tert-ブチル4-(1,5-ジメチル-1H-イミダゾール-2-イル)-3,6
-ジヒドロピリジン-1(2H)-カルボキシレート(1g、55%)を黄色ゴム質とし
て得た。
LCMS(方法F):m/z 278(M+H)+(ES+)、1.70分時、UV活性
-ジヒドロピリジン-1(2H)-カルボキシレート(1.0g、3.61mmol)を
1,4-ジオキサン(20mL)に溶解し、その後、1,4-ジオキサン中HCl(20
mL、3M溶液)を滴加した。結果得られた反応混合物を30℃で16時間撹拌し、溶媒
を真空除去し、残渣をジエチルエーテル(3×5mL)で研和することによって精製して
、中間体34、4-(1,5-ジメチル-1H-イミダゾール-2-イル)-1,2,3
,6-テトラヒドロピリジン塩酸塩(0.5g、65%)を白色固体として得た。標記の
化合物に関するデータは表2にある。
中間体65、3-(ピペリジン-4-イル)ピリジン-2(1H)-オン塩酸塩の調製に
よって例示される鈴木反応、水素化及びBoc脱保護を介してピペリジンを調製するため
の典型的な手順
ン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(2.5g
、10.0mmol)、3-ヨード-2-メトキシピリジン(8.21g、26.0mm
ol)及びK2CO3(4.3g、31.8mmol)を1-4ジオキサン(10mL)
及び水(5mL)に溶解した。反応混合物を、N2を使用して15分間脱気し;Pd-1
32(0.376g、0.53mmol)を加え、反応混合物を80℃で2時間撹拌した
。反応混合物を水(50mL)で希釈し、EtOAc(2×100mL)で抽出し、有機
層を合一し、乾燥させ(Na2SO4)、溶媒を真空除去し、粗生成物をカラムクロマト
グラフィー(順相、60~120メッシュシリカ、ヘキサン中0~20%EtOAc)に
よって精製して、tert-ブチル2-メトキシ-3’,6’-ジヒドロ-[3,4’-
ビピリジン]-1’(2’H)-カルボキシレート(2.0g、69.0%)を灰白色固
体として得た。
LCMS(方法F):m/z 291(M+H)+(ES+)、2.39分時、UV活性
1’(2’H)-カルボキシレート(1.89g、6.51mmol)をMeOH(10
mL)に溶解し、10%Pd/C(0.2g)を加えた。反応混合物をH2ガスでパージ
し、室温で12時間、H2下で攪拌した。反応混合物を、セライトを通して濾過し、溶媒
を真空除去して、tert-ブチル4-(2-メトキシピリジン-3-イル)ピペリジン
-1-カルボキシレート(0.91g、47.9%)を無色のゴム質として得た。
LCMS(方法F):m/z 293(M+H)+(ES+)、2.50分時、UV活性
シレート(0.200g、0.6mmol)を1,4-ジオキサン(4.0mL)及び水
(2.0mL)に溶解し、濃塩酸を加え、反応混合物を100℃で10時間撹拌した。溶
媒を真空除去し、残渣をアセトン(3×10mL)で研和して、中間体65、3-(ピペ
リジン-4-イル)ピリジン-2(1H)-オン塩酸塩(0.100g、82.6%)を
褐色固体として得た。標記の化合物に関するデータは表2にある。
中間体66、2-メトキシ-3-(ピペリジン-4-イル)ピリジン塩酸塩の調製によっ
て例示される鈴木反応、水素化及びBoc脱保護を介してピペリジンを調製するための典
型的な手順

ン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(2.5g
、10.0mmol)、3-ヨード-2-メトキシピリジン(8.21g、26.0mm
ol)及びK2CO3(4.3g、31.8mmol)を1-4ジオキサン(10mL)
及び水(5mL)に溶解した。反応混合物を、N2を使用して15分間脱気し;Pd-1
32(0.376g、0.53mmol)を加え、反応混合物を80℃で2時間撹拌した
。反応混合物を水(50mL)で希釈し、EtOAc(2×100mL)で抽出し、有機
層を合一し、乾燥させ(Na2SO4)、溶媒を真空除去し、粗生成物をカラムクロマト
グラフィー(順相、60~120メッシュシリカ、ヘキサン中0~20%EtOAc)に
よって精製して、tert-ブチル2-メトキシ-3’,6’-ジヒドロ-[3,4’-
ビピリジン]-1’(2’H)-カルボキシレート(2.0g、69.0%)を灰白色固
体として得た。
LCMS(方法F):m/z 291(M+H)+(ES+)、2.39分時、UV活性
1’(2’H)-カルボキシレート(1.89g、6.51mmol)をMeOH(10
mL)に溶解し、10%Pd/C(0.2g)を加えた。反応混合物をH2ガスでパージ
し、室温で12時間、H2下で攪拌した。反応混合物を、セライトを通して濾過し、溶媒
を真空除去して、tert-ブチル4-(2-メトキシピリジン-3-イル)ピペリジン
-1-カルボキシレート(0.91g、47.9%)を無色のゴム質として得た。
LCMS(方法F):m/z 293(M+H)+(ES+)、2.50分時、UV活性
シレート(0.8g、2.7mmol)を、1,4-ジオキサン中HCl(4.0mL、
4.0M溶液)中で、室温で10時間撹拌した。溶媒を真空除去し、残渣をアセトン(3
×10mL)によって研和して、中間体66、2-メトキシ-3-(ピペリジン-4-イ
ル)ピリジン塩酸塩(0.135g、25.7%)を白色固体として得た。標記の化合物
に関するデータは表2にある。
中間体69、3,4’-ビピペリジン-2-オンの調製によって例示される水素化を介し
てピペリジンを調製するための典型的な手順

2.8mmol)をMeOH(10mL)に溶解し、PtO2(0.2g)を加えた。反
応混合物をH2ガスでパージし、室温で12時間、H2ガス下で攪拌した。反応混合物を
、セライトを通して濾過し、溶媒を真空除去して、中間体69、3,4’-ビピペリジン
-2-オン(0.4g、78.3%)を褐色ゴム質として得た。標記の化合物に関するデ
ータは表2にある。
中間体127、エチル2-{4-[(2S)-ピロリジン-2-イル]ピペリジン-1-
イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレートのジアステレオマー
の混合物の調製によって例示される還元的アミノ化及びBoc脱保護を介してピロリジン
を調製するための典型的な手順

レート(1.24g、6.29mmol)及びエチル2-オキソ-6-アザスピロ[3.
4]オクタン-6-カルボキシレート(1.60g、6.29mmol)をDMF(15
mL)に室温で溶解し、酢酸(0.54mL、9.44mmol)を加えた。反応混合物
を室温で3時間撹拌した。次いで、STAB(2.67g、12.6mmol)を加え、
反応混合物を窒素下、室温で一晩撹拌した。溶媒を真空除去し、残渣をカラムクロマトグ
ラフィー(順相、[Biotage SNAPカートリッジKP-sil 340g、4
0~63μm、60Å、80mL/分、勾配:DCM中、MeOH中の0%~10%7N
NH3])によって精製して、エチル2-{4-[(2S)-1-(tert-ブトキ
シカルボニル)ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.
4]オクタン-6-カルボキシレート(2.46g、90%)の異性体の分離不可能な混
合物を黄色固体として得た。
LCMS(方法D):m/z 436(M+H)+(ES+)、2.36分時、UV不活
性。
-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシ
レート(0.6g、1.4mmol)のジアステレオマーの混合物を1,4-ジオキサン
(10mL)に溶解し、1,4-ジオキサン中HCl(4M、15mL、60mmol)
で滴下処理した。結果得られた反応混合物を25℃で16時間撹拌し、溶媒を除去し、残
渣をジエチルエーテル(3×10mL)で研和することによって精製して、エチル2-{
4-[(2S)-ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3
.4]オクタン-6-カルボキシレートのジアステレオマーの混合物、中間体127を固
体(0.45g、97%)として得た。標記の化合物に関するデータは表2にある。
中間体137、1-(ピペリジン-4-イル)テトラヒドロピリミジン-2(1H)-オ
ンの調製によって例示される還元的アミノ化、Boc-脱保護、尿素形成、及び水素化分
解を介してピペリジンを調製するための典型的な手順

ol)及びtert-ブチル(3-アミノプロピル)カルバメート(0.766g、4.
4mmol)をCH2Cl2(20mL)に室温で混合し、AcOH(0.68mL、1
2.0mmol)を加え、3時間撹拌した。STAB(2.59g、12.0mmol)
を加え、反応混合物を窒素下、室温で一晩撹拌した。反応混合物をNaHCO3(飽和水
溶液)(40mL)の添加によりクエンチしてCH2Cl2(4×45mL)で抽出し、
合一した有機層を飽和食塩水で洗浄し、次いで、MgSO4で乾燥させ、濾過した。溶媒
を真空除去し、残渣をカラムクロマトグラフィー[Biotage SNAPカートリッ
ジKP-sil 25g、40~63μm、60Å、50mL/分、勾配:DCM中0%
~10%MeOH])によって精製して、ベンジル4-({3-[(tert-ブトキシ
カルボニル)アミノ]プロピル}アミノ)ピペリジン-1-カルボキシレート(1.54
g、98%)を無色の油状物質として得た。
LCMS(方法B):m/z 392(M+H)+(ES+)、1.73分時、UV活性
。
)ピペリジン-1-カルボキシレート(1.54g、3.92mmol)をCH2Cl2
(19.5mL)に溶解し、ジオキサン中4M塩化水素(4.90mL、19.6mmo
l)を加え、反応混合物を室温で一晩撹拌した。溶媒を真空除去し、残渣をCH2Cl2
(2×20mL)で洗浄し、乾燥させて、粗ベンジル4-[(3-アミノプロピル)アミ
ノ]ピペリジン-1-カルボキシレートジヒドロクロリド(1.41g、99%)を灰白
色固体として得た。
LCMS(方法B):m/z 292(M+H)+(ES+)、1.46分時、UV活
性。
ジヒドロクロリド(1.41g、3.88mmol)、CDI(0.778g、4.80
mmol)及びピリジン(0.24mL、12.0mmol)をTHF(39mL)に溶
解し、混合物を加熱還流し、18時間維持した。溶媒を真空除去し、残渣をカラムクロマ
トグラフィー[Biotage SNAPカートリッジKP-sil 50g、40~6
3μm、60Å、50mL/分、勾配:DCM中0%~10%MeOH])によって精製
して、ベンジル4-(2-オキソテトラヒドロピリミジン-1(2H)-イル)ピペリジ
ン-1-カルボキシレート(0.82g、65%)を無色の固体として得た。
LCMS(方法B):m/z 318(M+H)+(ES+)、2.62分時、UV活性
。
1-カルボキシレート(0.82g、2.59mmol)をEtOH(100mL)に溶
解し、50℃、1mL/分で40バールH2に設定したH-Cubeを使用して10%P
d/Cカートリッジに通した。溶出した溶液を真空濃縮して、中間体137、1-(ピペ
リジン-4-イル)テトラヒドロピリミジン-2(1H)-オン(0.470g、99%
)を無色の固体として得た。標記の化合物に関するデータは表2にある。
中間体139、(2S)-N-メチル-1-(ピペリジン-4-イル)ピロリジン-2-
カルボキサミドの調製によって例示される還元的アミノ化、及びBoc脱保護を介してピ
ペリジンを調製するための典型的な手順
NEt3(1.5mL、11.0mmol)、tert-ブチル4-オキソピペリジン-
1-カルボキシレート(0.38g、3.9mmol)及びZnCl2(0.15g、4
.5mmol)をMeOH(15mL)に窒素下で溶解し、50~60℃で1時間攪拌し
た。NaCNBH3(0.16g、0.67mmol)を0~10℃で少量ずつ加え、混
合物を室温で3時間撹拌した。反応混合物をEtOAc(2×100mL)と水(50m
L)に分配し、有機層を合一し、乾燥させ(Na2SO4)、濾過し、溶媒を真空除去し
、粗生成物をカラムクロマトグラフィー(順相シリカ、ヘキサン中0~20%EtOAc
)によって精製して、tert-ブチル(S)-4-(2-(メチルカルバモイル)ピロ
リジン-1-イル)ピペリジン-1-カルボキシレート(0.3g、25.0%)を淡褐
色液体として得た。
TLC観察:RF値:0.5(EA:Hex、5:5)。
LCMS(方法G):m/z 312(M+H)+(ES+)、1.61分時、UV不活
性。
ピペリジン-1-カルボキシレート(0.3g、0.96mmol)を、1,4-ジオキ
サン中HCl(5.00mL)溶液中で、室温で10時間攪拌した。反応混合物を、高真
空(high vaccum)下で濃縮し、アセトン(3×10mL)によって研和して
、中間体139、(S)-N-メチル-1-(ピペリジン-4-イル)ピロリジン-2-
カルボキサミドジヒドロクロリド(0.135g、67.16%)を無色の固体として得
た。標記の化合物に関するデータは表2にある。
中間体181、4-[2-(1H-ピラゾール-5-イル)ピロリジン-1-イル]ピペ
リジントリフルオロ酢酸塩の調製に例示される還元的アルキル化及び脱保護を介して4位
でN結合型環状アミンを有するピペリジンを調製するための一般的な手順

0.50mmol)をDMF(5mL)に溶解した。該溶液にDIPEA(0.435m
L、2.5mmol)、AcOH(0.043mL、0.75mmol)、tert-ブ
チル4-オキソピペリジン-1-カルボキシレート(0.100g、0.50mmol)
及びSTAB(0.318g、1.50mmol)をこの順に加えた。混合物を室温で2
日間撹拌し、次いで、濃縮してDMFを除去した。残渣を飽和NaHCO3水溶液とDC
M(×2)に分配し、有機相をフェーズセパレータに通し、濃縮して、粗tert-ブチ
ル4-[2-(1H-ピラゾール-5-イル)ピロリジン-1-イル]ピペリジン-1-
カルボキシレート(0.271g、100%超)を油状物質として得た。
LCMS(方法C):m/z 321(M+H)+(ES+)、1.18分時、UV活性
ラゾール-5-イル)ピロリジン-1-イル]ピペリジン-1-カルボキシレート(0.
271g、想定0.50mmol)の溶液を室温で110分間撹拌し、次いで、トルエン
で希釈し、濃縮した。残渣をトルエンで共沸して、粗中間体181、4-[2-(1H-
ピラゾール-5-イル)ピロリジン-1-イル]ピペリジントリフルオロ酢酸塩(0.5
98g、100%超)を油状物質として得た。直ちに使用した。標記の化合物に関するデ
ータは表2にある。
中間体184、5-メチル-1-(ピペリジン-4-イル)ピロリジン-2-オン酢酸塩
の調製によって例示されるピリジンへの銅触媒カップリング、その後の水素化を介してピ
ペリジンを含有するピロリジノンまたはオキサジアゾロンを調製するための一般的な手順

mmol)、4-ヨードピリジン(0.103g、0.50mmol)、(トランス)-
N,N’-ジメチルシクロヘキサン-1,2-ジアミン(0.016mL、0.10mm
ol)、CuI(0.019g、0.10mmol)及びK2CO3(0.209g、1
.5mmol)の混合物を窒素フラッシュしたガラス管に密閉し、150℃で一晩加熱し
ながら撹拌した。冷却した反応混合物をフラッシュシリカ(5mL)で濃縮した。結果得
られた粉末をカラムクロマトグラフィー(順相、[Biotage SNAPカートリッ
ジKP-sil 25g、40~63μm、60Å]、30mL/分、DCM中0~5%
溶媒A、ここで、溶媒AはMeOH中10%の(7M NH3/MeOH)である)によ
って精製して、5-メチル-1-(ピリジン-4-イル)ピロリジン-2-オン(0.0
88g、99%)を油状物質として得た。
LCMS(方法C):m/z 177(M+H)+(ES+)、0.69分時、UV活性
45mmol)をAcOH(8mL)に溶解し、H-Cubeを使用して、1mL/分の
流速、100℃、80バール圧で、10%Pt/C触媒で水素化した。次いで、該溶液を
濃縮し、残渣をトルエン(×2)で共沸して、粗中間体184、5-メチル-1-(ピペ
リジン-4-イル)ピロリジン-2-オン酢酸塩(0.166g、100%超)を油状物
質として得た。標記の化合物に関するデータは表2にある。
中間体199、(5R)-5-メチル-1-(ピペリジン-4-イル)ピロリジン-2-
オン酢酸塩、及び中間体200、(5R)-5-エチル-1-(ピペリジン-4-イル)
ピロリジン-2-オン酢酸塩の調製によって例示されるピリジンへの銅触媒カップリング
、その後の水素化を介してピペリジンを調製するための典型的な手順
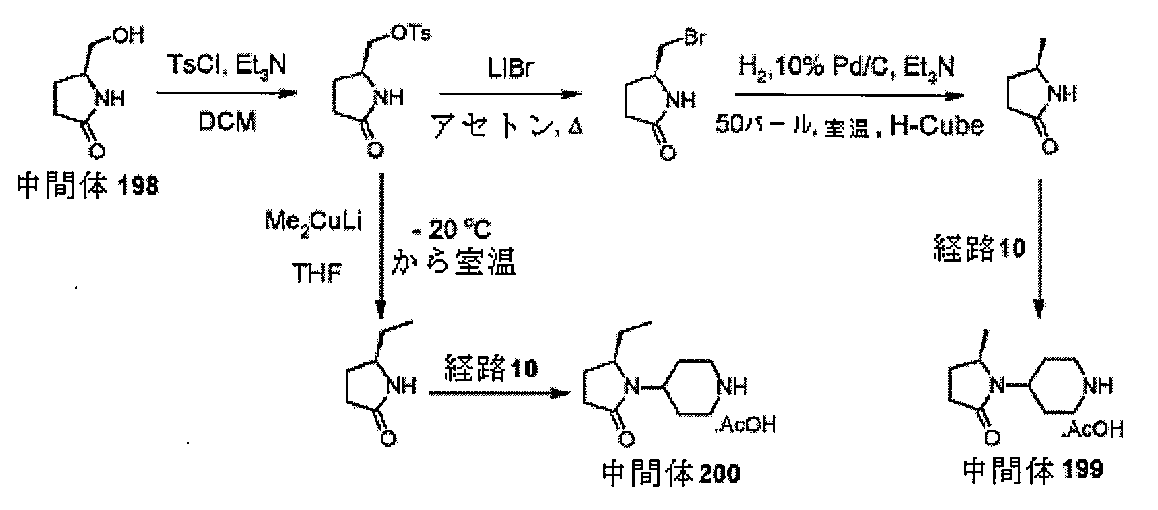
オン酢酸塩:
DCM(24mL)中の(5S)-5-(ヒドロキシメチル)ピロリジン-2-オン(
2.0g、17mmol)及び4-メチルベンゼンスルホニルクロリド(5.3g、28
mmol)の溶液に、トリエチルアミン(12mL、86mmol)を加えた。結果得ら
れた混合物を室温で一晩撹拌し、次いで濃縮した。残渣をDCMに溶解し、1MのHCl
水溶液(×3)及び飽和食塩水(×1)で洗浄し、次いで、フェーズセパレータに通し、
濃縮して褐色固体を得た。該固体をDCM/イソヘキサンから再結晶化して、黄褐色の固
体を得た。これを濾過により除去し、DCM/イソヘキサン混合物で洗浄し、空気乾燥さ
せて、[(2S)-5-オキソピロリジン-2-イル]メチル4-メチルベンゼンスルホ
ネート(3.13g、67%)を得た。
LCMS(方法C):m/z 270(M+H)+(ES+)、0.97分時、UV活性
チルベンゼンスルホネート(0.50g、1.9mmol)及び臭化リチウム(0.48
4g、5.6mmol)の混合物をN2下で一晩加熱還流し、次いで放冷した。溶媒を濃
縮により除去し、残渣をDCMとH2Oに分配し、相を分離した。水相をDCM(×3)
で抽出し、次いで、有機相をフェーズセパレータに通し、濃縮して、(5S)-5-(ブ
ロモメチル)ピロリジン-2-オン(0.284g、86%)をゴム質として得た。
LCMS(方法C):m/z 178/180(M+H)+(ES+)、0.37分時、
弱UV活性
の(5S)-5-(ブロモメチル)ピロリジン-2-オン(0.284g、1.6mmo
l)の溶液を、H-Cubeを使用して1mL/分の流速、室温、50バール圧で、10
%Pd/C触媒で水素化した。該溶液を濃縮して、粗(5R)-5-メチルピロリジン-
2-オン(0.445g、100%超)を粘着性の固体として得た。
LCMS(方法C):m/z 100(M+H)+(ES+)、0.34分時、弱UV活
性
を、経路10(中間体183とカップリング)にしたがって反応させて、粗中間体199
、(5R)-5-メチル-1-(ピペリジン-4-イル)ピロリジン-2-オン酢酸塩(
0.125g、46%)を油状物質として得た。標記の化合物に関するデータは表2にあ
る。
オン酢酸塩:
メチルリチウム(エーテル中1.5M、7.4mL、11mmol)をTHF(6mL
)中のヨウ化銅(1.06g、5.6mmol)の懸濁液に撹拌しながら素早く加えて、
氷水中N2下で予冷却した。淡褐色の溶液を氷水中で45分間撹拌し、次いで、-20℃
まで冷却した。THF(6mL)中の[(2S)-5-オキソピロリジン-2-イル]メ
チル4-メチルベンゼンスルホネート(0.50g、1.9mmol)の溶液を2分間か
けて少量ずつ加え、結果得られた溶液を-20℃で45分間撹拌し、次いで、氷水中で一
晩撹拌し、冷却浴をゆっくりと自然に終了させた。混合物をNH4Cl飽和水溶液(15
mL)でクエンチし、数時間撹拌した。2相混合物をエーテル(×3)で抽出し、有機相
を飽和食塩水で洗浄し、フェーズセパレータに通し、濃縮して、粗(5R)-5-エチル
ピロリジン-2-オン(0.124g、59%)を油状物質として得た。
LCMS(方法C):m/z 114(M+H)+(ES+)、0.50分時、弱UV活
性
経路10(中間体183とカップリング)にしたがって反応させて、粗中間体200、(
5R)-5-エチル-1-(ピペリジン-4-イル)ピロリジン-2-オン酢酸塩(0.
156g、72%)をゴム質として得た。標記の化合物に関するデータは表2にある。
中間体205、(4R)-4-メチル-3-(ピペリジン-4-イル)-1,3-オキサ
ゾリジン-2-オン酢酸塩の調製によって例示されるカルバメート形成、ピリジンへの銅
触媒カップリング、その後の水素化を介してピペリジンを調製するための典型的な手順
M(5mL)中の(2R)-2-アミノプロパン-1-オール(0.156mL、2.0
mmol)及びトリエチルアミン(0.56mL、4.0mmol)の溶液に1時間かけ
て少量ずつ加え、氷水中で予冷却した。混合物を氷水中でさらに2時間撹拌し、次いでエ
ーテル(6mL)を加えた。濃厚懸濁液を、焼結物を通して濾過し、該固体をさらなるエ
ーテル(6mL)で洗浄した。濾過物をフラッシュシリカ(5mL)で濃縮し、結果得ら
れた粉末をカラムクロマトグラフィー(順相、[Biotage SNAPカートリッジ
KP-sil 25g、40~63μm、60Å]、30mL/分、100%EtOAc
)によって精製して、(4R)-4-メチル-1,3-オキサゾリジン-2-オン(19
2mg、95%)を固体として得た。
LCMS(方法C):m/z 102(M+H)+(ES+)、0.14分時、UV不活
性
mol)を、経路10(中間体183とカップリング)にしたがって反応させて、粗中間
体205、(4R)-4-メチル-3-(ピペリジン-4-イル)-1,3-オキサゾリ
ジン-2-オン酢酸塩(0.343g、100%)を固体として得た。標記の化合物に関
するデータは表2にある。
中間体159、tert-ブチル4-[(2R)-2-(メトキシカルボニル)ピロリジ
ン-1-イル]ピペリジン-1-カルボキシレートの調製によって例示される還元的アミ
ノ化を介してピペリジンを調製するための典型的な手順
oc-4-ピペリジノン(0.24g、1.208mmol)をDMF(2mL)に室温
で溶解し、ジイソプロピルエチルアミン(0.209mL、1.208mmol)を加え
た。反応混合物を室温で3時間攪拌した。次いで、STAB(0.512g、2.416
mmol)を加え、反応混合物を窒素下、室温で一晩撹拌した。溶媒を真空除去し、残渣
をH2O(15mL)とEtOAc(25mL)に分配し、水層をEtOAc(2×25
mL)で抽出し、有機層を合一し、Na2SO4上で乾燥させ、溶媒を真空除去して、t
ert-ブチル4-[(2R)-2-(メトキシカルボニル)ピロリジン-1-イル]ピ
ペリジン-1-カルボキシレート、中間体159、を白色固体として得た(393mg、
>99%)。標記の化合物に関するデータは(ares)表2にある。
中間体271、tert-ブチル3,3-ジフルオロ-1,4’-ビピペリジン-1’-
カルボキシレートの調製によって例示される還元的アミノ化を介してピペリジンを調製す
るための典型的な手順

c-4-ピペリジノン(0.379g、1.90mmol)をDMF(8mL)に室温で
溶解し、ジイソプロピルエチルアミン(0.246g、1.90mmol)を加えた。反
応混合物を50℃、窒素下で2時間撹拌した。反応混合物を室温まで冷却し、次いで、氷
酢酸(0.114g、1.90mmol)及びSTAB(1.01g、4.76mmol
)を加え、反応混合物を50℃、窒素下で一晩撹拌した。水(2mL)を冷却した反応混
合物に加え、溶媒を真空除去した。残渣を飽和NaHCO3(水溶液)(10mL)で希
釈し、DCM(2×10mL)で抽出した。合一した有機層をBiotageフェーズセ
パレータカートリッジに通して乾燥させ、溶媒を真空除去した。残渣をカラムクロマトグ
ラフィー(順相、[Biotage SNAPカートリッジKP-sil 25g 40
~63μm、60Å、25mL/分、勾配:0%~10%MeOH/DCM])によって
精製して、tert-ブチル3,3-ジフルオロ-1,4’-ビピペリジン-1’-カル
ボキシレート、中間体271、(0.347g、60%)を琥珀色の油状物質として得た
。標記の化合物に関するデータは表2にある。
中間体195、4-(1-メチル-1H-テトラゾール-5-イル)ピペリジン塩酸塩の
調製によって例示されるテトラゾール形成、その後のアルキル化を介してピペリジンを調
製するための典型的な手順

ol)、アジ化ナトリウム(1.95g、30mmol)及び塩化アンモニウム(1.6
g、30mmol)をDMF(20mL)に溶解した。反応混合物を100℃で24時間
攪拌し、次いで、水(250mL)で希釈し、EtOAc(3×100mL)で抽出した
。合一した有機層を乾燥させ(Na2SO4)、濃縮して、粗生成物を得た。これをカラ
ムクロマトグラフィー(順相、中性シリカゲル、60~120メッシュ、DCM中0~5
%MeoH)によって精製して、tert-ブチル4-(1H-テトラゾール-5-イル
)ピペリジン-1-カルボキシレート(1.25g、50%)を固体として得た。
LCMS(方法F):m/z 198(M-tBu+H)+(ES+)、1.69分時、
UV不活性
レート(1.2g、4.7mmol)、ヨードメタン(2.0g、14mmol)及びC
s2CO3(9.6g、28mmol)を乾燥DMF(36mL)に溶解した。反応混合
物を100℃で2時間攪拌し、次いで、水(250mL)で希釈し、EtOAc(3×1
00mL)で抽出した。合一した有機層を乾燥させ(Na2SO4)、濃縮して、粗生成
物を得た。これをカラムクロマトグラフィー(順相、中性シリカゲル、60~120メッ
シュ、ヘキサン中0~35%EtOAc、次いで、ヘキサン中45~60%EtOAcに
よって精製して、2つの位置異性体を分離した。必要な位置異性体、tert-ブチル4
-(1-メチル-1H-テトラゾール-5-イル)ピペリジン-1-カルボキシレート(
0.160g、13%)、をカラムから2番目に溶出し、固体として得た。
LCMS(方法F):m/z 212(M-tBu+H)+(ES+)、1.79分時、
UV不活性
-カルボキシレート(0.160g、0.60mmol)をジオキサン(3mL)に溶解
した。ジオキサン中HCl(4M、3mL、12mmol)を0℃で加え、混合物を室温
で5時間撹拌した。溶媒を除去し、混合物をジエチルエーテル(5mL)で研和して、4
-(1-メチル-1H-テトラゾール-5-イル)ピペリジン塩酸塩、中間体195、(
0.130g、100%超)を固体として得た。標記の化合物に関するデータは表2にあ
る。
中間体223、(2R)-N-メチル-1,4’-ビピペリジン-2-カルボキサミドの
調製によって例示される還元的アミノ化、アミド形成及びBoc脱保護を介してピペリジ
ンを調製するための典型的な手順

-ブチル4-オキソピペリジン-1-カルボキシレート(2.31g、11.6mmol
)の溶液に、10%パラジウム炭(1g、50%湿潤)を加え、反応混合物を室温、H2
(1気圧)下で48時間撹拌した。反応混合物を、セライトベッドを通して濾過し、濾過
物を真空蒸発させた。この粗残渣をDCM(50mL)にて研和して、(R)-1’-(
tert-ブトキシカルボニル)-[1,4’-ビピペリジン]-2-カルボン酸(1.
2g、50%)を白色固体として得た。この粗残渣をさらなる精製を行わずに次のステッ
プに使用した。
1H-NMR(400 MHz; CDCl3) δ: 1.46(s, 9H), 1.50
- 1.59(m,1H), 1.75 - 1.91(m, 4H), 1.93 - 2.05
(m, 2H), 2.10- 2.19(m, 2H), 2.35 - 2.41(m, 1H)
, 2.51 - 2.69(m,3H), 3.41 - 3.49(m, 1H), 3.55
- 3.61(m,1H), 3.70 - 3.79(m, 1H), 4.25 - 4.36
(m, 2H)。
’-ビピペリジン]-2-カルボン酸(1.0g、3.20mmol)及びMeNH2(
THF中2M、3.2mL、6.41mmol)の溶液に、DIPEA(1.75mL、
9.60mmol)を0℃で加えた。10分間撹拌した後、1-プロパンホスホン酸無水
物[50%酢酸エチル溶液(4.07mL、6.41mmol)]を加え、室温で3時間
撹拌した。完了後、反応混合物を飽和NaHCO3水溶液でクエンチし、DCM(3×3
0mL)で抽出した。有機層を合一し、飽和食塩水で洗浄し、乾燥させ(Na2SO4)
、真空濃縮して、tert-ブチル(R)-2-(メチルカルバモイル)-[1,4’-
ビピペリジン]-1’-カルボキシレート(4、1g、97%)を無色のねばねばした液
体として得た。この粗残渣をさらなる精製を行わずに次のステップに使用した。
1H-NMR(400 MHz, DMSO) δ: 1.46(s, 9H),1.61 -
1.80(m, 4H),1.91 - 2.08(m, 4H), 2.25 - 2.33(m
, 2H), 2.61 - 2.71(m,4H), 2.82(d, J = 4.8 Hz, 3
H), 3.32 -3.45(m, 2H), 4.25 - 4.36(m, 2H), 6.8
5(br.s., 1H)。
[1,4’-ビピペリジン]-1’-カルボキシレート(700mg、2.15mmol
)の溶液に、ジオキサン中HCl(4M、10mL)を0℃でゆっくりと加え、室温で3
時間撹拌した。反応混合物を真空濃縮した。これを飽和NaHCO3水溶液(10mL)
で塩基性化し、濃縮した。粗反応塊に、5%MeOH/DCM(30mL)を加え、10
分間撹拌し、濾過した。濾過物を真空濃縮して、中間体223、(R)-N-メチル-[
1,4’-ビピペリジン]-2-カルボキサミド(400mg、83%)を褐色のねばね
ばした液体として得た。標記の化合物に関するデータは表2にある。
中間体250、4-エチル-5-ヨード-1-メチル-1H-ピラゾールの調製によって
例示されるザンドマイヤー反応を介してヨードピラゾールを調製するための典型的な手順

ジヨードメタン(9.0mL)に0~5℃、窒素雰囲気下で溶解し、その後亜硝酸イソア
ミルを滴加し、混合物を2時間80℃で、次いで2時間室温で攪拌した。反応混合物を、
H2O(100mL)とEtOAc(250mL)に分配し、水層をEtOAc(2×2
50mL)でさらに抽出し、合一した有機層を乾燥させ(Na2SO4)、濾過し、溶媒
を真空除去した。残渣をカラムクロマトグラフィー(順相、中性シリカゲル、60~12
0メッシュ、ヘキサン中30~50%酢酸エチル)によって精製して、4-エチル-5-
ヨード-1-メチル-1H-ピラゾール、中間体250(0.5g、53.23%)を淡
い黄色がかったゴム質として得た。標記の化合物に関するデータは表2にある。
中間体(Intermediate Intermediate)302、tert-ブ
チル(2R)-2-(ジフルオロメチル)ピロリジン-1-カルボキシレートの調製によ
って例示される脱保護、カルバメート形成、その後還元的アミノ化を介して活性化カルバ
メートを調製するための典型的な手順

17mol)をジオキサン中4MのHCl(25mL)に溶解し、窒素下、室温で一晩撹
拌した。溶媒を真空除去して、灰白色固体を得た。これをDCM(40mL)に懸濁し、
反応混合物を窒素下、0℃まで冷却した。Et3N(3.60g、0.036mol)及
び4-ニトロフェニルクロロギ酸(3.767g、0.0187mol)を加え、反応混
合物を室温で一晩撹拌した。反応混合物を飽和NaHCO3(水溶液)(30mL)でク
エンチし、DCM(3×20mL)で抽出した。有機層を合一し、Biotageフェー
ズセパレータカートリッジに通すことによって乾燥させ、溶媒を真空除去した。残渣をカ
ラムクロマトグラフィー(順相、[Biotage SNAPカートリッジKP-sil
50g 40~63μm、60Å、50mL/分、勾配:0%~6%MeOH/DCM
])によって精製して、4-ニトロフェニル2-オキソ-6-アザスピロ[3.4]オク
タン-6-カルボキシレートを黄色固体(1.40g、27%)として得た。
LCMS(方法C):m/z 291(M+H)+(ES+)、1.167分時
レート(0.700g、2.41mmol)をDMF(15mL)に溶解した。4-(1
H-ピラゾール-1-イル)ピペリジン(0.365g、2.41mmol)、氷酢酸(
0.144g、2.41mmol)及びSTAB(1.535g、7.24mmol)を
加え、反応混合物を窒素下、50℃で一晩撹拌した。反応混合物を水(2mL)でクエン
チし、溶媒を真空除去した。残渣をDCM(20mL)と飽和NaHCO3(水溶液)(
20mL)に分配し、水層をDCM(2×20mL)で抽出し、有機層を合一し、Bio
tageフェーズセパレータカートリッジに通すことによって乾燥させ、溶媒を真空除去
した。残渣をカラムクロマトグラフィー(順相、[Biotage SNAPカートリッ
ジKP-sil 25g 40~63μm、60Å、25mL/分、勾配:0%~10%
MeOH/DCM])によって精製して、4-ニトロフェニル2-[4-(1H-ピラゾ
ール-1-イル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カ
ルボキシレート、中間体302、(0.738g、72%)を得た。標記の化合物に関す
るデータは表2にある。
経路a
実施例1-1、エチル2-[4-(1H-イミダゾール-2-イル)ピペリジン-1-イ
ル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製によって例示さ
れる水素化トリアセトキシホウ素ナトリウム還元的アミノ化を介してピペリジンを調製す
るための典型的な手順

.1mmol)及びエチル2-オキソ-6-アザスピロ[3.4]オクタン-6-カルボ
キシレート(1.60g、7.1mmol)をDCM(60mL)に室温で溶解し、チタ
ンイソプロポキシド(2.31mL、7.81mmol)を加えた。反応混合物を室温で
1時間撹拌した。反応混合物を-5℃まで冷却し、次いで、STAB(3.01g、14
.2mmol)及び酢酸(350μL、4.26mmol)を加え、反応混合物を室温ま
で加温しながら、窒素下で一晩撹拌した。反応混合物をNaHCO3(飽和水溶液)(1
0mL)の添加によってクエンチし、DCMで希釈し、次いで、セライトパッドを通して
濾過した。層を分離し、水層をDCMで抽出した。合一したDCM層を飽和食塩水で洗浄
し、次いで、MgSO4で乾燥させた。溶媒を真空除去し、残渣をカラムクロマトグラフ
ィー(順相、[Biotage SNAPカートリッジKP-sil 50g、40~6
3μm、60Å、50mL/分、勾配:0.5%NEt3を伴うDCM中1%~10%M
eOH])によって精製して、エチル2-[4-(1H-イミダゾール-2-イル)ピペ
リジン]-6-アザスピロ[3.4]オクタン-6-カルボキシレート(2.645g、
98.3%)のジアステレオマーの分離不可能な混合物を白色固体として得た。分取HP
LCを使用して、ジアステレオマーを分離した。これには、Phenomenex Ge
mini-N C18カラム、150×21mmを使用し、18mL/分で28~38%
MeCN/H2Oで溶出し、218nmでモニターすることによって画分を収集した。そ
して、異性体1、エチル2-[4-(1H-イミダゾール-2-イル)ピペリジン]-6
-アザスピロ[3.4]オクタン-6-カルボキシレート(0.338g、14%)を無
色の固体として、異性体2、エチル2-[4-(1H-イミダゾール-2-イル)ピペリ
ジン]-6-アザスピロ[3.4]オクタン-6-カルボキシレート(0.369g、1
6%)を無色の固体として得た。異性体2に関するデータは表3にある。
実施例1-3、エチル2-(4-(4-(トリフルオロメチル)-1H-イミダゾール-
2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキ
シレートの調製によって例示されるシアノ水素化ホウ素ナトリウム及び塩化亜鉛還元的ア
ミノ化を介してピペリジンを調製するための典型的な手順

00mg、0.46mmol)、エチル2-オキソ-6-アザスピロ[3.4]オクタン
-6-カルボキシレート(89mg、0.46mmol)、ZnCl2(2mg、0.0
1mmol)及びトリエチルアミン(0.3mL、2.28mmol)をMeOH(5m
L)に溶解し、反応混合物を50℃で2時間撹拌した。反応混合物を0℃まで冷却し、N
aBH3CN(114mg、1.83mmol)を少量ずつ加えた。結果得られた反応混
合物を25℃で7時間撹拌し、溶媒を真空除去した。残渣をH2O(50mL)とEtO
Ac(35mL)に分配し、水層をEtOAc(2×35mL)で抽出し、有機層を合一
し、乾燥させ(Na2SO4)、溶媒を真空除去した。残渣を分取HPLC[逆相(X-
BRIDGE、C-18、250×19mm、5μm、18mL/分、勾配:MeCN/
水中28.0%(40.0分間)、100%(3.0分間)次いで28.0%(5.0分
間)、0.1%NH3]によって精製して、エチル2-(4-(4-(トリフルオロメチ
ル)-1H-イミダゾール-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.
4]オクタン-6-カルボキシレート、実施例1-3異性体1、(15mg、8.24%
)を黄色固体として、エチル2-(4-(4-(トリフルオロメチル)-1H-イミダゾ
ール-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カ
ルボキシレート、実施例1-3異性体2、(12mg、6.6%)を黄色固体として得た
。異性体2に関するデータは表3にある。
実施例1-4、エチル2-[4-(4-シアノ-1H-イミダゾール-2-イル)ピペリ
ジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製に
よって例示されるトリフルオロメチル置換イミダゾールをシアノ置換イミダゾールに変換
するための典型的な手順

ペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート(2
00mg、0.50mmol)をNH3溶液(20mL)に溶解し、60℃で8時間撹拌
した。溶媒を真空除去し、残渣をH2O(60mL)とEtOAc(40mL)に分配し
、水層をEtOAc(2×40mL)で抽出し、有機層を合一し、乾燥させた(Na2S
O4)。溶媒を真空除去及し、残渣を分取HPLC[逆相(DURASHELL、C-1
8、250×21.2mm、5μm、22mL/分、勾配:MeCN/水中25.0%(
30.0分間)、100%(3.0分間)、次いで25.0%(7.0分間)、0.1%
NH3]によって精製して、エチル2-(4-(4-シアノ-1H-イミダゾール-2-
イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレ
ート、実施例1-4異性体1、(26mg、14.6%)を黄色固体として、エチル2-
[4-(4-シアノ-1H-イミダゾール-2-イル)ピペリジン-1-イル]-6-ア
ザスピロ[3.4]オクタン-6-カルボキシレート、実施例1-4異性体2、(25m
g、14.06%)を黄色固体として得た。異性体2に関するデータは表3にある。
実施例1-7、エチル2-[4-(1-メチル-1H-イミダゾール-2-イル)ピペリ
ジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製に
よって例示される水素化トリアセトキシホウ素ナトリウム還元的アミノ化、Boc脱保護
及びエチルカルバメート形成を介してピペリジンを調製するための典型的な手順
1mmol)及び6-Boc-2-オキソ-6-アザスピロ[3,4]オクタン(0.2
73g、1.21mmol)をDCM(10mL)に室温で溶解し、チタンイソプロポキ
シド(0.4mL、2.42mmol)を加えた。反応混合物を室温で1時間撹拌した。
反応混合物を-5℃まで冷却し、次いで、STAB(0.513g、2.42mmol)
及び酢酸(27μL、480μmol)を加え、反応混合物を室温まで加温しながら、窒
素下で一晩撹拌した。反応混合物をNaHCO3(飽和水溶液)(10mL)の添加でク
エンチし、DCMで希釈し、次いでセライトパッドを通して濾過した。層を分離し、水層
をDCMで抽出した。合一したDCM層を飽和食塩水で洗浄し、次いで、MgSO4で乾
燥させた。溶媒を真空除去し、残渣をカラムクロマトグラフィー(順相、[Biotag
e SNAPカートリッジKP-sil 25g、40~63μm、60Å、50mL/
分、勾配:DCM中1%~10%MeOH])によって精製して、tert-ブチル2-
[4-(1-メチル-1H-イミダゾール-2-イル)ピペリジン]-6-アザスピロ[
3.4]オクタン-6-カルボキシレート(0.330g、72%)の異性体の分離不可
能な混合物を黄色ゴム質として得た。
LCMS(方法A):m/z 374(M+H)+(ES+)、1.68分時、UV不活
性。
ン]-6-アザスピロ[3.4]オクタン-6-カルボキシレート(0.326g、0.
87mmol)を、ジオキサン中4M塩化水素(1.2mL、5.2mmol)に溶解し
た。反応混合物を室温で18時間撹拌した。次いで、揮発性物質を真空除去し、残渣をD
CM(17mL)及びトリエチルアミン(0.49mL、3.49mmol)に溶解した
。クロロギ酸エチル(125μL、1.31mmol)を滴加し、該溶液を室温で18時
間撹拌した。次いで、混合物をNaHCO3(水溶液)(75mL)及びDCM(75m
L)に注ぎ入れ、抽出(2×75mL)し、合一したDCM抽出物を飽和食塩水(20m
L)で洗浄し、次いでMgSO4で乾燥させた。濃縮後、残渣をカラムクロマトグラフィ
ー(順相、[Biotage SNAPカートリッジKP-sil 25g、40~63
μm、60Å、50mL/分、勾配:DCM中1%~10%MeOH])によって精製し
て、エチル2-[4-(1-メチル-1H-イミダゾール-2-イル)ピペリジン]-6
-アザスピロ[3.4]オクタン-6-カルボキシレートを、褐色の油状物質として、ジ
アステレオマーの混合物(0.25g、83%)として提供した。分取HPLCを使用し
てジアステレオマーを分離した。これには、Phenomenex Gemini-N
C18カラム、150×21mmを使用し、18mL/分で38~48%MeCN/H2
Oで抽出し、218nmでモニターすることによって画分を収集した。そして、エチル2
-[4-(1-メチル-1H-イミダゾール-2-イル)ピペリジン]-6-アザスピロ
[3.4]オクタン-6-カルボキシレート、実施例1-7異性体1、(0.044g、
15%)を無色の油状物質として、エチル2-[4-(1-メチル-1H-イミダゾール
-2-イル)ピペリジン]-6-アザスピロ[3.4]オクタン-6-カルボキシレート
、実施例1-7異性体2、(0.031g、10%)を無色の油状物質として得た。異性
体2に関するデータは表3にある。
実施例1-9、メチル2-[4-(1,5-ジメチル-1H-イミダゾール-2-イル)
ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの
調製によって例示されるピペリジニルを含有する化合物を得るために、3,6-ジヒドロ
ピリジン-1(2H)-イルを含有する化合物を水素化するための典型的な手順
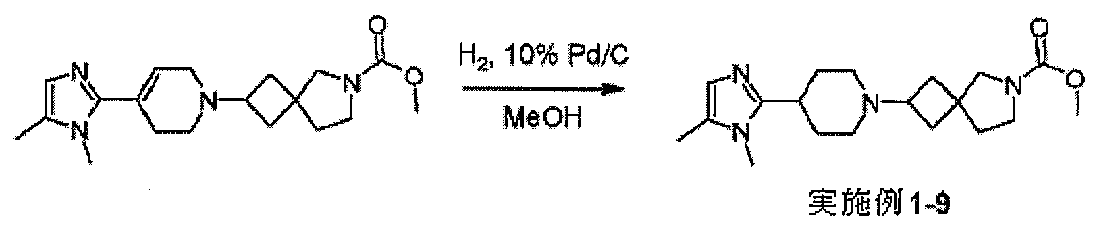
ヒドロピリジン-1(2H)-イル)-6-アザスピロ[3.4]オクタン-6-カルボ
キシレート(102mg、0.29mmol)[経路d及び中間体3及び34を介して合
成される]をMeOH(10mL)に溶解し、10%Pd/C(25mg)を加えた。反
応混合物をH2ガスでパージし、次いで、25℃で20時間、H2風船下で撹拌した。反
応混合物を、セライトを通して濾過し、MeOHで洗浄し、濾過物からの溶媒を真空除去
し、残渣を分取HPLC(X Bridge、C-18、150×30mm、5μm、4
0mL/分、勾配:アセトニトリル/水中30%(12.00分間)、100%(14.
00分間)、次いで、30%(14.01分間)、0.1%アンモニア]によって精製し
て、メチル2-[4-(1,5-ジメチル-1H-イミダゾール-2-イル)ピペリジン
-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例1-
9異性体1、(5.6mg、5.8%)を無色のゴム質として、メチル2-[4-(1,
5-ジメチル-1H-イミダゾール-2-イル)ピペリジン-1-イル]-6-アザスピ
ロ[3.4]オクタン-6-カルボキシレート、実施例1-9異性体2、(11.6mg
、11.7%)を無色のゴム質として得た。異性体2に関するデータは表3にある。
実施例1-36、エチル2-[4-(1H-1,2,4-トリアゾール-1-イル)ピペ
リジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製
によって例示される水素化トリアセトキシホウ素ナトリウム還元的アミノ化、Boc脱保
護及びエチルカルバメート形成を介してピペリジンを調製するための典型的な手順
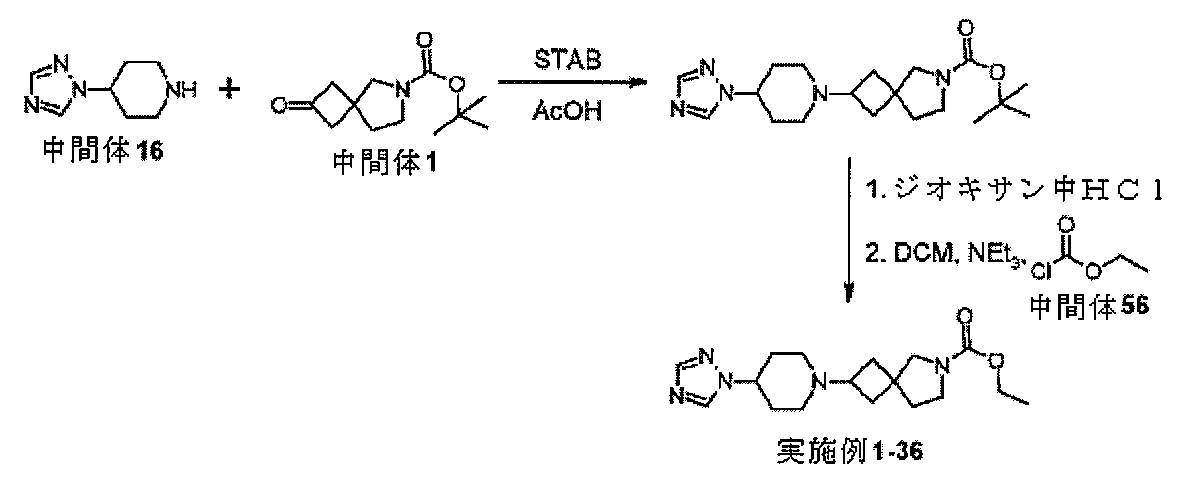
0mmol)及び6-Boc-2-オキソ-6-アザスピロ[3,4]オクタン(0.2
22g、1.05mmol)をDCM(10mL)にN2下、室温で溶解し、酢酸(0.
13mL、2.22mmol)を加えた。反応混合物を室温で2時間撹拌し、STAB(
0.53g、2.50mmol)を加え、反応混合物を室温で一晩撹拌した。反応混合物
をNaHCO3(飽和水溶液)(30mL)の添加でクエンチし、DCM(4×25mL
)で抽出し、合一したDCM層をBiotageフェーズセパレータに通した。溶媒を真
空除去して、tert-ブチル2-[4-(1H-1,2,4-トリアゾール-1-イル
)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレート
のジアステレオマーの粗混合物を得て、これを精製せずに使用した。
LCMS(方法C):m/z 362(M+H)+(ES+)、1.58分及び1.61
分時、UV不活性。
ジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレート(想定1
.0mmol)を、ジオキサン中4M塩化水素(1.2mL、5.2mmol)に溶解し
、反応混合物を室温で一晩撹拌した。揮発性物質を真空除去し、残渣をDCM(10mL
)に溶解し、NEt3(0.70mL、5.0mmol)を加えた。クロロギ酸エチル(
0.14mL、1.5mmol)を滴加し、溶液を室温で一晩撹拌した。混合物をNaH
CO3(水溶液)(40mL)に注ぎ入れ、DCM(4×40mL)で抽出し、合一した
DCM層をBiotageフェーズセパレータに通した。溶媒を真空除去し、残渣をカラ
ムクロマトグラフィー(順相、[Biotage SNAPカートリッジKP-sil
25g、40~63μm、60Å、40mL/分、勾配:DCM中0%~10%MeOH
)によって精製して、エチル2-[4-(1H-1,2,4-トリアゾール-1-イル)
ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの
ジアステレオマーの分離不可能な混合物を得た。分取HPLCを使用してジアステレオマ
ーを分離した。これには、Phenomenex Gemini-NX 5μm C18
110A Axiaカラム、100×30mmを使用し、30mL/分で14.4にわ
たり25~55%MeCN/溶媒Bで溶出し[ここで、溶媒BはH2O中0.2%の(2
8%NH3/H2O)である]、210nmでモニターすることによって画分を収集した
。そして、エチル2-[4-(1H-1,2,4-トリアゾール-1-イル)ピペリジン
-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例1-
36異性体1、(0.026g、8%)を無色の固体として、エチル2-[4-(1H-
1,2,4-トリアゾール-1-イル)ピペリジン-1-イル]-6-アザスピロ[3.
4]オクタン-6-カルボキシレート、実施例1-36異性体2、(0.026g、8%
)を無色の固体として得た。異性体2に関するデータは表3にある。
実施例1-51、エチル2-{4-[1-(2-メトキシエチル)-1H-イミダゾール
-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボ
キシレートの調製によって例示されるDMF中の水素化ナトリウムを使用してイミダゾー
ル含有化合物をアルキル化するための典型的な手順
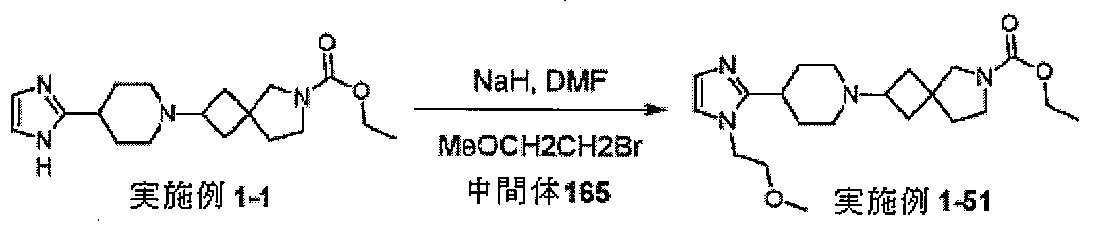
ザスピロ[3.4]オクタン-6-カルボキシレート(150mg、0.45mmol)
のジアステレオマーの混合物を無水DMF(3mL)に溶解し、水素化ナトリウムの60
%鉱油懸濁物(27mg、0.68mmol)で処理し、室温で2時間撹拌した。2-ブ
ロモエチルメチルエーテル(0.051mL、0.54mmol)を加え、混合物を室温
で一晩撹拌した。混合物を濃縮してDMFを除去した。残渣をMeOHに溶解し、フラッ
シュシリカ(5mL)で濃縮した。結果得られた粉末をカラムクロマトグラフィー(順相
、[Biotage SNAPカートリッジKP-sil 25g、40~63μm、6
0Å]、30mL/分、DCM中0~20%溶媒A、ここで、溶媒AはMeOH中10%
の(7M NH3/MeOH)である)によって精製して、エチル2-{4-[1-(2
-メトキシエチル)-1H-イミダゾール-2-イル]ピペリジン-1-イル}-6-ア
ザスピロ[3.4]オクタン-6-カルボキシレート(159mg、90%)のジアステ
レオマーの混合物を得た。この混合物をMeOHに溶解し、溶液を分取逆相HPLCによ
って精製した。これには、Phenomenex Gemini-NX 5μm C18
110A Axiaカラム、100×30mmを使用し、30mL/分で14.4分に
わたり15~45%MeCN/溶媒Bで溶出し[ここで、溶媒BはH2O中0.2%の(
28%NH3/H2O)である]、210nmでモニターすることによって画分を収集し
た。そして、エチル2-{4-[1-(2-メトキシエチル)-1H-イミダゾール-2
-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシ
レート、実施例1-51異性体1、(54mg、31%)及びエチル2-{4-[1-(
2-メトキシエチル)-1H-イミダゾール-2-イル]ピペリジン-1-イル}-6-
アザスピロ[3.4]オクタン-6-カルボキシレート、実施例1-51異性体2、(2
7mg、15%)を得た。
異性体2に関するデータは表3にある。
実施例1-52、エチル2-{4-[1-(シアノメチル)-1H-イミダゾール-2-
イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレ
ートの調製によって例示されるDMF中の炭酸カリウムを使用してイミダゾール含有化合
物をアルキル化するための典型的な手順

ザスピロ[3.4]オクタン-6-カルボキシレート(150mg、0.45mmol)
のジアステレオマーの混合物を無水DMF(3mL)に溶解した。炭酸カリウム(187
mg、1.4mmol)及びブロモアセトニトリル(0.114mL、1.6mmol)
を加え、混合物を室温で二晩撹拌した。混合物を濃縮してDMFを除去した。残渣をMe
OHに溶解し、フラッシュシリカ(5mL)で濃縮した。結果得られた粉末をカラムクロ
マトグラフィー(順相、[Biotage SNAPカートリッジKP-sil 25g
、40~63μm、60Å]、30mL/分、DCM中0~20%溶媒A、ここで、溶媒
AはMeOH中10%の(7M NH3/MeOH)である)によって精製して、エチル
2-{4-[1-(シアノメチル)-1H-イミダゾール-2-イル]ピペリジン-1-
イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート(91mg、54%
)のジアステレオマーの混合物を得た。この混合物をMeOHに溶解し、溶液を分取逆相
HPLCによって精製した。これには、Phenomenex Gemini-NX 5
μm C18 110A Axiaカラム、100×30mmを使用し、30mL/分で
14.4分間にわたって15~45%MeCN/溶媒Bで溶出し[ここで、溶媒BはH2
O中0.2%の(28%NH3/H2O)である]、210nmでモニターすることによ
って画分を収集して、エチル2-{4-[1-(シアノメチル)-1H-イミダゾール-
2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキ
シレート、実施例1-52異性体1、(8mg、5%)及びエチル2-{4-[1-(シ
アノメチル)-1H-イミダゾール-2-イル]ピペリジン-1-イル}-6-アザスピ
ロ[3.4]オクタン-6-カルボキシレート、実施例1-52異性体2、(5mg、3
%)を得た。異性体2に関するデータは表3にある。
実施例1-53、(2-{1-[6-(エトキシカルボニル)-6-アザスピロ[3.4
]オクト-2-イル]ピペリジン-4-イル}-1H-イミダゾール-1-イル)酢酸、
及び実施例1-54、エチル2-(4-{1-[2-(メチルアミノ)-2-オキソエチ
ル]-1H-イミダゾール-2-イル}ピペリジン-1-イル)-6-アザスピロ[3.
4]オクタン-6-カルボキシレートを調製するための手順
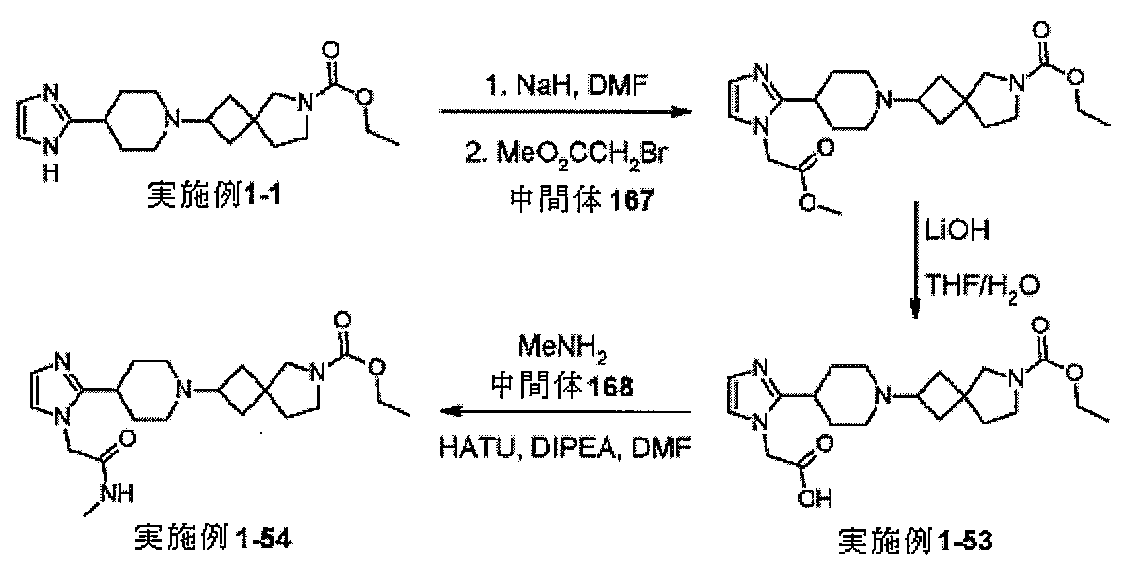
ザスピロ[3.4]オクタン-6-カルボキシレート(500mg、1.5mmol)の
ジアステレオマーの混合物を、DMF(10mL)中の水素化ナトリウムの60%鉱油分
散物(90mg、2.3mmol)及びブロモ酢酸メチル(0.171mL、1.8mm
ol)と、経路gの方法を使用して反応させて、エチル2-{4-[1-(2-メトキシ
-2-オキソエチル)-1H-イミダゾール-2-イル]ピペリジン-1-イル}-6-
アザスピロ[3.4]オクタン-6-カルボキシレート(393mg、65%)のジアス
テレオマーの混合物を得た。
LCMS(方法C):m/z 405(M+H)+(ES+)、1.12及び1.17分
時、弱UV活性。
2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキ
シレート(180mg、0.45mmol)のジアステレオマーの混合物を、THF(4
mL)及びH2O(1mL)中の水酸化リチウム一水和物(75mg、1.8mmol)
と室温で5日間撹拌した。混合物を濃縮してTHFを除去し、1MのHCl水溶液で酸性
化し、濃縮して、(2-{1-[6-(エトキシカルボニル)-6-アザスピロ[3.4
]オクト-2-イル]ピペリジン-4-イル}-1H-イミダゾール-1-イル)酢酸(
0.4g、100%超)のジアステレオマーの粗混合物を得た。おおよそ0.2gのこの
混合物をMeOHに溶解し、溶液を分取逆相HPLCによって精製した。これには、Ph
enomenex Gemini-NX 5μm C18 110A Axiaカラム、
100×30mmを使用し、30mL/分で14.4分間にわたって5~15%MeCN
/溶媒Bで溶出し[ここで、溶媒BはH2O中0.2%の(28%NH3/H2O)であ
る]、210nmでモニターすることによって画分を収集した。そして、(2-{1-[
6-(エトキシカルボニル)-6-アザスピロ[3.4]オクト-2-イル]ピペリジン
-4-イル}-1H-イミダゾール-1-イル)酢酸、実施例1-53異性体1、(30
mg、17%)及び(2-{1-[6-(エトキシカルボニル)-6-アザスピロ[3.
4]オクト-2-イル]ピペリジン-4-イル}-1H-イミダゾール-1-イル)酢酸
、実施例1-53異性体2、(22mg、13%)を得た。
異性体2に関するデータは表3にある。
-2-イル]ピペリジン-4-イル}-1H-イミダゾール-1-イル)酢酸のジアステ
レオマーの粗混合物、実施例1-53、(0.2g、想定0.22mmol)をDMF(
3mL)に溶解し、ジイソプロピルエチルアミン(0.155mL、0.89mmol)
とメタノール(2M、0.33mL、0.66mmol)中のメチルアミンの溶液とで処
理した。次いで、HATU(0.127g、0.33mmol)を加え、混合物を室温で
一晩撹拌した。混合物を濃縮してDMFを除去し、残渣をDCM及びMeOHの混合物に
溶解し、フラッシュシリカ(10mL)で濃縮した。結果得られた粉末をカラムクロマト
グラフィー(順相、[Biotage SNAPカートリッジKP-sil 25g、4
0~63μm、60Å]、30mL/分、DCM中0~20%溶媒A、ここで、溶媒Aは
MeOH中10%の(7M NH3/MeOH)である)によって精製して、エチル2-
(4-{1-[2-(メチルアミノ)-2-オキソエチル]-1H-イミダゾール-2-
イル}ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレ
ートのジアステレオマーの混合物を得た。この混合物をMeOHに溶解し、溶液を分取逆
相HPLCによって精製した。これには、Phenomenex Gemini-NX
5μm C18 110A Axiaカラム、100×30mmを使用し、30mL/分
で14.4分間にわたって15~45%MeCN/溶媒Bで溶出し[ここで、溶媒BはH
2O中0.2%の(28%NH3/H2O)である]、210nmでモニターすることに
よって画分を収集した。そして、エチル2-(4-{1-[2-(メチルアミノ)-2-
オキソエチル]-1H-イミダゾール-2-イル}ピペリジン-1-イル)-6-アザス
ピロ[3.4]オクタン-6-カルボキシレート、実施例1-54異性体1、(9mg、
4%)及びエチル2-(4-{1-[2-(メチルアミノ)-2-オキソエチル]-1H
-イミダゾール-2-イル}ピペリジン-1-イル)-6-アザスピロ[3.4]オクタ
ン-6-カルボキシレート、実施例1-54異性体2、(6mg、3%)を得た。異性体
2に関するデータは表3にある。
実施例1-70、エチル2-{4-[(2R)-4,4-ジフルオロ-2-(メチルカル
バモイル)ピロリジン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]
オクタン-6-カルボキシレートの調製によって例示されるカルバメート形成を介してピ
ペリジンを調製するための典型的な手順

]-6-アザスピロ[3.4]オクタン-6-カルボキシレート(0.125g、0.2
94mmol)を無水THF(4mL)に懸濁し、超音波処理して溶解を引き起こした。
60%鉱油懸濁物の水素化ナトリウム(0.026g、0.647mmol)を加え、反
応混合物を窒素下、室温で10分間撹拌した。エタノール-1,1-2,2,2-d5(
0.150g、2.94mmol)を加え、反応混合物を窒素下、室温で一晩撹拌した。
水(1mL)を反応混合物に加え、溶媒を真空除去した。残渣をDCM(20mL)と飽
和NaHCO3(水溶液)(10mL)に分配し、水層をDCM(2×10mL)で抽出
した。有機層を合一し、Biotageフェーズセパレータカートリッジに通すことによ
って乾燥させた。溶媒を真空除去し、残渣をカラムクロマトグラフィー(順相、[Bio
tage SNAPカートリッジKP-sil 10g 40~63μm、60Å、12
mL/分、勾配:0%~10%MeOH/DCM])によって精製した。残渣を分取逆相
HPLC(Phenomenex Gemini-NX 5μm C18 110A A
xiaカラム、100×30mm、30mL/分で14.4分間かけて20~50%Me
CN/溶媒Bで溶出し[ここで、溶媒BはH2O中0.2%の(28%NH3/H2O)
である]、210nmでモニターすることによって画分を収集する)によってさらに精製
して、(2H5)エチル2-[4-(1H-ピラゾール-1-イル)ピペリジン-1-イ
ル]-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例1-70異性
体1、(0.017g、17%)を白色固体として、(2H5)エチル2-[4-(1H
-ピラゾール-1-イル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン
-6-カルボキシレート、実施例1-70異性体2、(0.013g、13%)を白色固
体として得た。異性体2に関するデータは表3にある。
実施例2-2、エチル2-[4-(1-ホルミルピロリジン-2-イル)ピペリジン-1
-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製によって例
示されるホルムアミド形成を介してピペリジンを調製するための典型的な手順

時間撹拌し、次いで、反応物を0℃まで冷却し、THF(2mL)中のエチル2-(4-
(ピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン
-6-カルボキシレート塩酸塩(100mg、0.30mmol)のジアステレオマーの
混合物を滴加した。結果得られた反応混合物を60℃で8時間撹拌し、塩基性pHに調整
し、次いで、反応混合物をH2O(40mL)とEtOAc(25mL)に分配した。水
層をEtOAc(2×25mL)でさらに抽出し、有機層を合一し、Na2SO4で乾燥
させた。溶媒を真空除去し、残渣を分取HPLC(X Bridge、C-18、150
×30mm、5μm、40mL/分、勾配:アセトニトリル/水中30%(12.00分
間)、100%(14.00分間)、次いで、30%(14.01分間)、0.1%アン
モニア]によって精製して、エチル2-[4-(1-ホルミルピロリジン-2-イル)ピ
ペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実
施例2-2異性体1(14.6mg、13.0%)を黄色ゴム質として、エチル2-[4
-(1-ホルミルピロリジン-2-イル)ピペリジン-1-イル]-6-アザスピロ[3
.4]オクタン-6-カルボキシレート、実施例2-2異性体2(12.5mg、11.
1%)を黄色ゴム質として得た。異性体2に関するデータは表3にある。
実施例2-4、エチル2-{4-[1-(トリフルオロアセチル)ピロリジン-2-イル
]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート
の調製によって例示されるアミド形成を介してピペリジンを調製するための典型的な手順

[3.4]オクタン-6-カルボキシレート(50mg、0.15mmol)及びNEt
3(0.06mL、0.45mmol)をTHF(3mL)に室温で溶解した。エチル2
,2,2-トリフルオロ酢酸(0.03mg、0.22mmol)を滴加し、結果得られ
た反応混合物を室温で8時間撹拌した。反応混合物をH2O(40mL)とEtOAc(
25mL)に分配し、水層をEtOAc(2×25mL)でさらに抽出し、有機層を合一
し、Na2SO4で乾燥させた(dried ove)。溶媒を真空除去し、残渣を分取
HPLC(X Bridge、C-18、150×30mm、5μm、40mL/分、勾
配:アセトニトリル/水中30%(12.00分間)、100%(14.00分間)、次
いで30%(14.01分間)、0.1%アンモニア]によって精製して、エチル2-{
4-[1-(トリフルオロアセチル)ピロリジン-2-イル]ピペリジン-1-イル}-
6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-4異性体-1(
5.5mg、8.0%)を黄色ゴム質として、エチル2-{4-[1-(トリフルオロア
セチル)ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オ
クタン-6-カルボキシレート、実施例2-4異性体-2(6.2mg、9.7%)を黄
色ゴム質として得た。異性体2に関するデータは表3にある。
実施例2-17、エチル2-{4-[(2S)-1-(メチルカルバモイル)ピロリジン
-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボ
キシレートの調製によって例示されるアミド/カルバメート/尿素形成を介してピペリジ
ンを調製するための典型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(2.10g、5.65m
mol)のジアステレオマーの混合物をDCM(20mL)及びトリエチルアミン(1.
54mL、11.1mmol)に溶解した。メチルアミノホルミルクロリド(Methy
laminoformyl chloride)(620mg、6.63mmol)を加
え、溶液を室温で2時間撹拌した。次いで、混合物を1MのNaOH(水溶液)(50m
L)に注ぎ入れ、DCM(2×50mL)で抽出し、合一したDCM抽出物を飽和食塩水
(50mL)で洗浄し、次いで、Biotageフェーズセパレータに通し、真空濃縮し
て、エチル2-{4-[(2S)-1-(メチルカルバモイル)ピロリジン-2-イル]
ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレートを
黄色固体として、及びジアステレオマーの混合物(1.79g、82%)として提供した
。分取HPLCを使用してジアステレオマーを分離した。これには、Phenomene
x Gemini-NX C18カラム、100×30mmを使用し、18mL/分で、
H2O中25~35%MeCN/0.2%アンモニア(v/v)で溶出し、210nmで
モニターすることによって画分を収集した。そして、実施例2-17異性体1、エチル2
-{4-[(2S)-1-(メチルカルバモイル)ピロリジン-2-イル]ピペリジン-
1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート(0.78g、
36%)を無色の油状物質として、実施例2-17異性体2、エチル2-{4-[(2S
)-1-(メチルカルバモイル)ピロリジン-2-イル]ピペリジン-1-イル}-6-
アザスピロ[3.4]オクタン-6-カルボキシレート(0.67g、31%)を無色の
油状物質として得た。異性体2に関するデータは表3にある。
実施例2-19、エチル2-{4-[1-(エチルカルバモイル)ピロリジン-2-イル
]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート
の調製によって例示される尿素/カルバメート形成を介してピペリジンを調製するための
典型的な手順

[3.4]オクタン-6-カルボキシレート塩酸塩(100mg、0.30mmol)の
ジアステレオマーの混合物、ジエチルアミン(0.3mL、0.60mmol)及びNE
t3(0.1mL、0.90mmol)をDCE(5mL)に室温で溶解した。CDI(
145mg、0.60mmol)を加え、反応混合物を室温で15時間撹拌した。反応混
合物をH2O(40mL)とEtOAc(25mL)に分配し、水層をEtOAc(2×
25mL)でさらに抽出し、有機層を合一し、乾燥させ(Na2SO4)、溶媒を真空除
去し、残渣を分取HPLC[逆相HPLC(X-BRIDGE、C-18、250×19
mm、5μm、15mL/分、勾配:MeCN/水中30.0%~38.0%(25.0
分間)、100.0%(3.0分間)、次いで30.0%(2.0分間)、0.1%NH
3]によって精製して、エチル2-(4-(1-(エチルカルバモイル)ピロリジン-2
-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシ
レート、実施例2-19異性体-1(実施例219異性体-1)、(7.5mg、6.2
0%)を黄色ゴム質として、エチル2-(4-(1-(エチルカルバモイル)ピロリジン
-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボ
キシレート、実施例2-19異性体2、(8.1mg、6.60%)を黄色ゴム質として
得た。異性体2に関するデータは表3にある。
実施例2-22、エチル2-[4-(1-メチルピロリジン-2-イル)ピペリジン-1
-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートによって例示され
るアルキル化を介してピペリジンを調製するための典型的な手順

[3.4]オクタン-6-カルボキシレート塩酸塩(200mg、0.60mmol)の
ジアステレオマーの混合物及びホルムアルデヒド(40%溶液、1.01mL、3.60
mmol)をH2O(2mL)に25℃で溶解した。ギ酸(0.303mL、0.90m
mol)を滴加し、結果得られた混合物を70℃で14時間撹拌した。反応混合物をNa
HCO3溶液(5mL)でクエンチし、次いで反応混合物をH2O(50mL)とEtO
Ac(35mL)に分配した。水層をEtOAc(2×35mL)でさらに抽出し、有機
層を合一し、Na2SO4で乾燥させた。溶媒を真空除去し、残渣を分取HPLC[逆相
HPLC(X-Bridge、C-18、250×19.0mm、5μm、14mL/分
、勾配:MeCN/水中37%(28.0分間)、100%(4.0分間)、次いで37
%(3.0分間)、0.1%NH3]によって精製して、エチル2-(4-(1-メチル
ピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-
6-カルボキシレート、実施例2-22異性体1(12mg、5.80%)を黄色ゴム質
として、エチル2-(4-(1-メチルピロリジン-2-イル)ピペリジン-1-イル)
-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-22異性体2
(11mg、5.30%)を黄色ゴム質として得た。異性体2に関するデータは表3にあ
る。
実施例2-23、エチル2-{4-[1-(N-メチルグリシル)ピロリジン-2-イル
]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート
の調製によって例示されるアミド形成を介してピペリジンを調製するための典型的な手順

mol)をアセトニトリル(5mL)に溶解し、その後HATU(170mg、0.45
mmol)及びDIPEA(0.2mL、0.90mmol)を加えた。反応混合物を0
℃で10分間撹拌し、その後エチル2-(4-(ピロリジン-2-イル)ピペリジン-1
-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(100m
g、0.30mmol)のジアステレオマーの混合物を加え、結果得られた反応混合物を
25℃で3時間撹拌した。反応混合物をH2O(50mL)とEtOAc(35mL)に
分配し、水層をEtOAc(2×35mL)でさらに抽出し、有機層を合一し、Na2S
O4で乾燥させ、溶媒を真空除去した。最後に、残渣をカラムクロマトグラフィー(順相
、塩基性アルミナ(normal basic alumina)、活性、DCM中0.
5%~1.0%MeOH)によって精製して、エチル2-(4-(1-(N-((ベンジ
ルオキシ)カルボニル)-N-メチルグリシル)ピロリジン-2-イル)ピペリジン-1
-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート(130mg、8
0.74%)を褐色ゴム質として得た。エチル2-(4-(1-(N-((ベンジルオキ
シ)カルボニル)-N-メチルグリシル)ピロリジン-2-イル)ピペリジン-1-イル
)-6-アザスピロ[3.4]オクタン-6-カルボキシレート(130mg、0.24
mmol)をMeOH(10mL)に溶解し、その後、Pd/C(乾燥基準、13mg)
を加えた。次いで、反応物をH2ガスでパージし、結果得られた反応混合物を25℃で1
0時間撹拌した。反応混合物をセライトプラグに通して濾過し、メタノールで洗浄し、次
いで濾過物をNa2SO4で乾燥させ、溶媒を真空除去した。残渣を分取HPLC[逆相
HPLC(X-BRIDGE、C-18、250×19mm、5μm、15mL/分、勾
配:MeCN/水中20.0%~35.0%(30.0分間)、100.0%(3.0分
間)、次いで20.0%(2.0分間)、0.1%NH3]によって精製して、エチル2
-(4-(1-(メチルグリシル)ピロリジン-2-イル)ピペリジン-1-イル)-6
-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-23異性体1、(
9.0mg、9.27%)を黄色ゴム質として、エチル2-(4-(1-(エチルカルバ
モイル)ピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オ
クタン-6-カルボキシレート、実施例2-23異性体2、(8.0mg、8.50%)
を黄色ゴム質として得た。異性体2に関するデータは表3にある。
実施例2-27、エチル2-{4-[(2S)-1-(アゼチジン-1-イルカルボニル
)ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン
-6-カルボキシレートの調製によって例示される尿素形成を介してピペリジンを調製す
るための典型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(100mg、0.291
mmol)のジアステレオマーの混合物をDCM(5mL)及びジイソプロピルエチルア
ミン(0.099mL、0.58mmol)に溶解した。トリホスゲン(88mg、0.
291mmol)を0℃で加え、溶液を室温まで加温し1時間撹拌した。次いで、混合物
をDCM(50mL)で希釈し、H2O(70mL)で洗浄した。水層をDCM(2×5
0mL)で抽出し、合一したDCM抽出物をNa2SO4で乾燥させ、真空濃縮した。残
渣をDCM(5mL)に溶解し、アゼチジン(0.020mL、0.291mmol)及
びジイソプロピルエチルアミン(0.256mL、1.48mmol)を加えた。反応物
を室温で1時間撹拌した。次いで、混合物をDCM(50mL)で希釈し、H2O(70
mL)で洗浄した。水層をDCM(2×50mL)で抽出し、合一したDCM抽出物をN
a2SO4で乾燥させ、真空濃縮して、エチル2-{4-[(2S)-1-(アゼチジン
-1-イルカルボニル)ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピ
ロ[3.4]オクタン-6-カルボキシレートを黄色固体として、及びジアステレオマー
の混合物として提供した。残渣を分取HPLC[逆相HPLC(CHIRALPAK A
D-H、C-18、250×20mm、5μm、18.0mL/分、勾配:アセトニトリ
ル中0%~50%(15.0分間)、0.1%アンモニア及びH2O中0.1%アンモニ
アによって精製して、エチル(S)-2-(4-(1-(アゼチジン-1-カルボニル)
ピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-
6-カルボキシレート、実施例2-27異性体1(20mg、16.12%)を無色のゴ
ム質として、実施例2-27異性体2(20mg、16.12%)を無色のゴム質として
得た。異性体2に関するデータは表3にある。
実施例2-42、エチル2-(4-{(2S)-1-[エチル(プロパン-2-イル)カ
ルバモイル]ピロリジン-2-イル}ピペリジン-1-イル)-6-アザスピロ[3.4
]オクタン-6-カルボキシレート、及び実施例2-138、エチル2-{4-[(2S
)-1-(5-メチル-1,3,4-オキサジアゾール-2-イル)ピロリジン-2-イ
ル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレー
トの調製によって例示される尿素形成及び脱水を介してピペリジンを調製するための典型
的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(164mg、0.403
mmol)のジアステレオマーの混合物をDCM(2mL)及びジイソプロピルエチルア
ミン(0.209mL、1.21mmol)に溶解した。トリホスゲン(43mg、0.
145mmol)を0℃で加え、溶液を室温まで加温し、18時間撹拌した。この混合物
にカルバジン酸tert-ブチル(108mg、0.82mmol)及びジイソプロピル
エチルアミン(0.142mL、0.82mmol)を加え、反応物を室温で18時間撹
拌した。次いで、混合物をDCM(20mL)で希釈し、飽和NaHCO3(水溶液)(
2×20mL)で洗浄した。水層をDCM(20mL)で抽出し、合一したDCM抽出物
を飽和食塩水(50mL)で洗浄し、次いでBiotageフェーズセパレータに通し、
真空濃縮して、エチル2-{4-[(2S)-1-(ブチルカルバゾイル)ピロリジン-
2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキ
シレートを黄色油状物質として、及びジアステレオマーの混合物(192mg、97%)
として提供した。
LCMS(方法D):m/z 494(M+H)+(ES+)、1.83及び1.87分
時、UV不活性。
1mL)に溶解した。反応混合物を室温で1時間撹拌した。次いで、揮発性物質を真空除
去してから、反応混合物をDCM(2mL)及びジイソプロピルエチルアミン(0.14
2mL、0.82mmol)に再溶解した。塩化アセチル(0.031mL、0.428
mmol)を0℃で加え、溶液を室温まで加温し、2時間撹拌した。揮発性物質を真空除
去し、さらなる精製を行わずに次のステップに用いた。残渣をトルエン(2mL)及びジ
イソプロピルエチルアミン(0.135mL、0.78mmol)に溶解し、0℃まで冷
却した。塩化ホスホリルを加え(0.182mL、1.945mmol)、反応物を11
0℃で30分間撹拌してから、反応物を室温まで冷却し、氷水(20mL)でクエンチし
た。次いで、混合物をDCM(20mL)で希釈し、1MのNaOH(水溶液)(2×2
0mL)で洗浄した。水層をDCM(3×20mL)で抽出し、合一したDCM抽出物を
飽和食塩水(50mL)で洗浄し、次いでBiotageフェーズセパレータに通し、真
空濃縮した。残渣を分取HPLCによって精製した。これには、Phenomenex
Gemini-NX C18カラム、100×30mmを使用し、18mL/分でH2O
中25~45%MeCN/0.2%アンモニア(v/v)で溶出し、210nmでモニタ
ーすることによって画分を収集した。そして、実施例2-42異性体1、エチル2-(4
-{(2S)-1-[エチル(プロパン-2-イル)カルバモイル]ピロリジン-2-イ
ル}ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレー
ト(1.7mg、1%)を無色の油状物質として、実施例2-42異性体2、エチル2-
(4-{(2S)-1-[エチル(プロパン-2-イル)カルバモイル]ピロリジン-2
-イル}ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシ
レート(1.6mg、1%)を無色の油状物質として、実施例2-138異性体1、エチ
ル2-{4-[(2S)-1-(5-メチル-1,3,4-オキサジアゾール-2-イル
)ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン
-6-カルボキシレート(3.9mg、2.5%)を無色の油状物質として、実施例2-
138異性体2、エチル2-{4-[(2S)-1-(5-メチル-1,3,4-オキサ
ジアゾール-2-イル)ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピ
ロ[3.4]オクタン-6-カルボキシレート(3.0mg、2%)を無色の油状物質と
して得た。異性体2に関するデータは表3にある。
実施例2-47、エチル2-(4-{(2S)-1-[(2-フルオロエトキシ)カルボ
ニル]ピロリジン-2-イル}ピペリジン-1-イル)-6-アザスピロ[3.4]オク
タン-6-カルボキシレートの調製によって例示されるカルバメート形成を介してピペリ
ジンを調製するための典型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(0.15g、0.44m
mol)のジアステレオマーの混合物及びジイソプロピルエチルアミン(0.152mL
、0.89mmol)をDCM(5mL)に溶解し、次いで、反応混合物を0℃まで冷却
した。2-フルオロクロロギ酸エチル(0.062g、0.492mmol)を加え、結
果得られた反応混合物を25℃で16時間撹拌した。反応混合物をH2O(70mL)と
DCM(50mL)に分配し、水層をDCM(2×50mL)でさらに抽出し、有機層を
合一し、Na2SO4で乾燥させ、溶媒を真空除去した。残渣を分取HPLC[逆相HP
LC(CHIRALPAK AD-H、C-18、250×20mm、5μm、18.0
mL/分、勾配:アセトニトリル中0%~35%(52分間)、0.1%アンモニア及び
水中0.1%アンモニアによって精製して、エチル2-(4-{(2S)-1-[(2-
フルオロエトキシ)カルボニル]ピロリジン-2-イル}ピペリジン-1-イル)-6-
アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-47異性体-1(1
7mg、8.9%)を黄色ゴム質として、エチル2-(4-{(2S)-1-[(2-フ
ルオロエトキシ)カルボニル]ピロリジン-2-イル}ピペリジン-1-イル)-6-ア
ザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-47異性体-2(19
mg、10%)を黄色ゴム質として得た。異性体2に関するデータは表3にある。
実施例2-52、エチル2-{4-[(2S)-1-(ヒドロキシアセチル)ピロリジン
-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボ
キシレートの調製によって例示されるアミド形成を介してピペリジンを調製するための典
型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(0.2g、0.541m
mol)のジアステレオマーの混合物及びトリエチルアミン(0.152mL、1.1m
mol)をDCM(5mL)に溶解し、次いで反応混合物を0℃まで冷却し、塩化アセト
キシアセチル(0.080g、0.591mmol)を加えた。反応混合物を室温で2時
間撹拌した。揮発性物質を真空除去し、次いで、残渣をアセトニトリル(25mL)及び
20%NaOH水溶液(10mL)に溶解し、それを室温で1時間撹拌した。反応混合物
をH2O(70mL)とDCM(50mL)に分配し、水層をDCM(2×50mL)で
さらに抽出し、有機層を合一し、Na2SO4で乾燥させ、溶媒を真空除去し、残渣を分
取HPLC[逆相HPLC(CHIRALPAK AD-H、C-18、250×20m
m、5μm、18.0mL/分、勾配:0%~30%(35.0分間)、アセトニトリル
中0.1%アンモニア及び水中0.1%アンモニアによって精製して、エチル(S)-2
-(4-(1-(2-ヒドロキシアセチル)ピロリジン-2-イル)ピペリジン-1-イ
ル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-52異性
体1(8mg、8.3%)を無色のゴム質として、エチル(S)-2-(4-(1-(2
-ヒドロキシアセチル)ピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピ
ロ[3.4]オクタン-6-カルボキシレート、実施例2-52異性体2(12mg、1
2.24%)を無色のゴム質として得た。異性体2に関するデータは表3にある。
実施例2-53、エチル2-{4-[(2S)-1-(3,3,3-トリフルオロプロパ
ノイル)ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オ
クタン-6-カルボキシレートの調製によって例示されるアミド形成を介してピペリジン
を調製するための典型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(0.12g、0.36m
mol)のジアステレオマーの混合物及びジイソプロピルエチルアミン(0.123mL
、0.71mmol)をDCM(10mL)に溶解し、その後トリフルオロプロパニオン
酸(trifluoropropanioic acid)(0.045g、0.394
mmol)を加えた。反応混合物を0℃まで冷却し、プロピルホスホン酸無水物を加え(
0.140g、0.462mmol 50%EtOAc溶液)、結果得られた反応混合物
を25℃で2時間撹拌した。反応混合物をH2O(20mL)とDCM(50mL)に分
配し、水層をDCM(2×50mL)でさらに抽出し、有機層を合一し、Na2SO4で
乾燥させ、溶媒を真空除去した。残渣を分取HPLC[逆相HPLC(CHIRALPA
K AD-H、C-18、250×20mm、5μm、18.0mL/分、勾配:アセト
ニトリル中0%~30%(27.0分間)、0.1%アンモニア及び水中0.1%アンモ
ニアによって精製して、エチル(S)-2-(4-(1-(3,3,3-トリフルオロプ
ロパノイル)ピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4
]オクタン-6-カルボキシレート、実施例2-53異性体1(9mg、6.0%)を無
色のゴム質として、エチル(S)-2-(4-(1-(3,3,3-トリフルオロプロパ
ノイル)ピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オ
クタン-6-カルボキシレート、実施例2-53異性体2(9mg、6.0%)を無色の
ゴム質として得た。異性体2に関するデータは表3にある。
実施例2-58、エチル2-{4-[(2S)-1-プロパンチオイルピロリジン-2-
イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレ
ートの調製によって例示されるローソン試薬を介してチオアミドを調製するための典型的
な手順
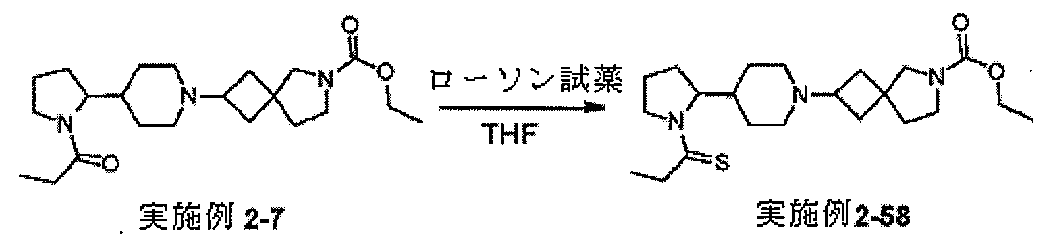
1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート(0.341g
、0.87mmol)をTHF(4mL)に溶解し、その後ローソン試薬(0.265g
、0.65mmol)を加えた。反応混合物を70℃で24時間撹拌した。揮発性物質を
真空除去し、反応混合物を1MのNaOH(水溶液)(50mL)とDCM(30mL)
に分配し、水層をDCM(2×50mL)でさらに抽出し、有機層を合一し、5%ピロ亜
硫酸ナトリウム(水溶液)で洗浄し、Na2SO4で乾燥させ、溶媒を真空除去した。残
渣を分取HPLCによって精製した。これには、Phenomenex Gemini-
NX C18カラム、100×30mmを使用し、18mL/分でH2O中25~45%
MeCN/0.2%アンモニア(v/v)で抽出し、210nmでモニターすることによ
って画分を収集した。そして、実施例2-58異性体1、エチル2-{4-[(2S)-
1-プロパンチオイルピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ
[3.4]オクタン-6-カルボキシレート(14.1mg、4%)を黄色油状物質とし
て、実施例2-58異性体2、エチル2-{4-[(2S)-1-プロパンチオイルピロ
リジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-
カルボキシレート(7.6mg、2%)を黄色油状物質として得た。異性体2に関するデ
ータは表3にある。
実施例2-61、エチル2-{4-[(2S)-1-エチルピロリジン-2-イル]ピペ
リジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製
によって例示されるアルキル化を介してピペリジンを調製するための典型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(0.100g、0.29
mmol)のジアステレオマーの混合物及び炭酸カリウム(0.123mg、0.89m
mol)をDMF(5mL)に溶解し、反応混合物を60℃で2時間撹拌した。次いで、
ヨードエタンを加え(0.049g、0.31mmol、反応混合物を100℃で62時
間撹拌した。反応混合物をH2O(70mL)とEtOAc(50mL)に分配し、水層
をEtOAc(2×50mL)でさらに抽出し、有機層を合一し、Na2SO4で乾燥さ
せ、溶媒を真空除去した。残渣を分取HPLC(X Bridge、C-18、150×
19mm、5μm、20mL/分、勾配:アセトニトリル/水中35%(0.01分間)
、100%(25.01分間)、次いで35%(30.00分間)、0.1%アンモニア
]によって精製して、エチル(S)-2-(4-(1-エチルピロリジン-2-イル)ピ
ペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実
施例2-61異性体1(43mg、38.8%)を無色のゴム質として、エチル(S)-
2-(4-(1-エチルピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピ
ロ[3.4]オクタン-6-カルボキシレート、実施例2-61異性体2(26mg、2
3.1%)を無色のゴム質として得た。異性体2に関するデータは表3にある。
実施例2-62、エチル2-(4-{(2S)-1-[3-(ピリジン-2-イル)プロ
パノイル]ピロリジン-2-イル}ピペリジン-1-イル)-6-アザスピロ[3.4]
オクタン-6-カルボキシレートによって例示されるアミド形成を介してピペリジンを調
製するための典型的な手順

の溶液に2-ピリジンプロピオン酸(106mg、0.704mmol)及びDMF(1
滴)を加えた。エチル2-{4-[(2S)-ピロリジン-2-イル]ピペリジン-1-
イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(262mg
、0.640mmol)のジアステレオマーの混合物をDCM(1mL)及びジイソプロ
ピルエチルアミン(0.355mL、2.049mmol)に溶解し、上記溶液に加え、
それを室温で2時間撹拌した。次いで、混合物を1MのNaOH(水溶液)(50mL)
に注ぎ入れ、DCM(2×50mL)で抽出し、合一したDCM抽出物を飽和食塩水(5
0mL)で洗浄し、次いでBiotageフェーズセパレータに通し、真空濃縮して、エ
チル2-(4-{(2S)-1-[3-(ピリジン-2-イル)プロパノイル]ピロリジ
ン-2-イル}ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カル
ボキシレートを黒色油状物質として、及びジアステレオマーの混合物(0.245g、8
2%)として提供した。分取HPLCを使用してジアステレオマーを分離した。これには
、Phenomenex Gemini-NX C18カラム、100×30mmを使用
し、18mL/分でH2O中25~45%MeCN/0.2%アンモニア(v/v)で溶
出し、210nmでモニターすることによって画分を収集した。そして、実施例2-62
異性体1、エチル2-(4-{(2S)-1-[3-(ピリジン-2-イル)プロパノイ
ル]ピロリジン-2-イル}ピペリジン-1-イル)-6-アザスピロ[3.4]オクタ
ン-6-カルボキシレート(0.042g、14%)を無色の油状物質として、実施例2
-62異性体2、エチル2-(4-{(2S)-1-[3-(ピリジン-2-イル)プロ
パノイル]ピロリジン-2-イル}ピペリジン-1-イル)-6-アザスピロ[3.4]
オクタン-6-カルボキシレート(0.030g、10%)を無色の油状物質として得た
。2に関するデータは表3にある。
実施例2-65、エチル2-{4-[(2S)-1-{N-[(ベンジルオキシ)カルボ
ニル]-β-アラニル}ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピ
ロ[3.4]オクタン-6-カルボキシレート、及び実施例2-66、エチル2-{4-
[(2S)-1-(β-アラニル)ピロリジン-2-イル]ピペリジン-1-イル}-6
-アザスピロ[3.4]オクタン-6-カルボキシレートの調製によって例示されるアミ
ド形成及びCBZ脱保護を介してピペリジンを調製するための典型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(100mg、0.298
mmol)のジアステレオマーの混合物及びDIPEA(0.102mL、0.597m
mol)をCH2Cl2(5mL)に溶解し、0℃まで冷却し、Z-β-アラ-OH(0
.066g、0.298mmol)を加え、その後プロパンホスホン酸無水物(0.12
3g、0.388mmol、50%酢酸エチル溶液)を加えた。結果得られた反応混合物
を25℃で3時間撹拌し、H2O(70mL)とCH2Cl2(50mL)に分配し、水
層をCH2Cl2(2×50mL)でさらに抽出した。合一した有機層を乾燥させ(Na
2SO4)、濾過し、溶媒を真空除去して、エチル2-{4-[(2S)-1-{N-[
(ベンジルオキシ)カルボニル]-β-アラニル}ピロリジン-2-イル]ピペリジン-
1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート(100mg、
71.4%)を無色のゴム質として得た。これを実施例2-66の合成のために精製を行
わずに直接使用した。
LCMS(方法I):m/z 541(M+H)+(ES+)4.38及び4.51分時
、UV不活性。
0×20mm、5μm、18.0mL/分、勾配:アセトニトリル中0%~50%(15
.0分間)、0.1%アンモニア及び水中0.1%アンモニアによって精製することがで
き、エチル2-{4-[(2S)-1-{N-[(ベンジルオキシ)カルボニル]-β-
アラニル}ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]
オクタン-6-カルボキシレート、実施例2-65異性体1(20mg、12.5%)を
無色のゴム質として、エチル2-{4-[(2S)-1-{N-[(ベンジルオキシ)カ
ルボニル]-β-アラニル}ピロリジン-2-イル]ピペリジン-1-イル}-6-アザ
スピロ[3.4]オクタン-6-カルボキシレート、実施例2-65異性体2(20mg
、12.5%)を無色のゴム質として得た。異性体2に関するデータは表3にある。
ラニル}ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オ
クタン-6-カルボキシレート(100mg、0.185mmol)のジアステレオマー
の混合物をTFA(2.0mL)に溶解した。結果得られた溶液を80℃で3時間撹拌し
、真空濃縮し、残渣を分取HPLC[逆相HPLC(CHIRALPAK AD-H、C
-18、250×19mm、5μm、13.0mL/分、勾配:アセトニトリル中0%~
30%(30.0分間)、0.1%アンモニア及び水中0.1%アンモニアによって精製
して、エチル(S)-2-(4-(1-(3-アミノプロパノイル)ピロリジン-2-イ
ル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレー
ト、実施例2-66異性体1(2mg、2.63%)を無色のゴム質として、エチル(S
)-2-(4-(1-(3-アミノプロパノイル)ピロリジン-2-イル)ピペリジン-
1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-6
6異性体2(3mg、4.0%)を無色のゴム質として得た。異性体2に関するデータは
表3にある。
実施例2-68、エチル2-{4-[(2S)-1-(2-フルオロエチル)ピロリジン
-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボ
キシレートの調製によって例示されるアルキル化を介してピペリジンを調製するための典
型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(100mg、0.27m
mol)のジアステレオマーの混合物をMeCN(5mL)に溶解し、CS2CO3(2
90mg、0.89mmol)を加え、その後2-ヨード-1-フルオロエタン(56m
gg、0.32mmol)を加え、反応混合物を50℃で16時間撹拌した。反応混合物
をH2O(70mL)と酢酸エチル(50mL)に分配し、水層を酢酸エチル(2×50
mL)でさらに抽出し、有機層を合一し、乾燥させ(Na2SO4)、濾過し、溶媒を真
空除去した。残渣を分取HPLC[逆相HPLC(CHIRALPAK AD-H、C-
18、250×19mm、5μm、15.0mL/分、勾配:アセトニトリル中0%~4
0%(19分間)、0.1%アンモニア及び水中0.1%アンモニア]によって精製して
、エチル(S)-2-(4-(1-(2-フルオロエチル)ピロリジン-2-イル)ピペ
リジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施
例2-68異性体1(25mg、25%)を黄色がかったゴム質として、エチル(S)-
2-(4-(1-(2-フルオロエチル)ピロリジン-2-イル)ピペリジン-1-イル
)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-68異性体
2(20mg、20.3%)を黄色がかったゴム質として得た。異性体2に関するデータ
は表3にある。
実施例2-69、エチル2-{4-[(2S)-1-(2,2,2-トリフルオロエチル
)ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン
-6-カルボキシレートの調製によって例示されるアルキル化を介してピペリジンを調製
するための典型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(0.100g、0.29
mmol)のジアステレオマーの混合物及びDIPEA(0.112g、0.87mmo
l)をTHF(5mL)に溶解し、60℃で2時間撹拌した。トリフルオロメタンスルホ
ン酸2,2,2-トリフルオロエチル(0.067g、0.29mmol)を0℃で滴加
し、結果得られた反応混合物を室温で24時間撹拌した。反応混合物をH2O(70mL
)とEtOAc(50mL)に分配し、水層をEtOAc(2×50mL)でさらに抽出
し、有機層を合一し、乾燥させ(Na2SO4)、濾過し、真空濃縮した。残渣を分取H
PLC(X Bridge、C-18、250×19mm、5μm、12mL/分、勾配
:アセトニトリル/水中45%(0.01分間)、100%(30.00分間)、次いで
45%(32.00分間)、0.1%アンモニアによって精製して、エチル(S)-2-
(4-(1-(2,2,2-トリフルオロエチル)ピロリジン-2-イル)ピペリジン-
1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-6
9異性体1(0.003g、2.4%)を無色のゴム質として、エチル(S)-2-(4
-(1-(2,2,2-トリフルオロエチル)ピロリジン-2-イル)ピペリジン-1-
イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-69異
性体2(0.002mg、1.6%)を無色のゴム質として得た。異性体2に関するデー
タは表3にある。
実施例2-70、エチル2-{4-[(2S)-1-(3,3,3-トリフルオロプロピ
ル)ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタ
ン-6-カルボキシレートの調製によって例示されるアルキル化を介してピペリジンを調
製するための典型的な手順
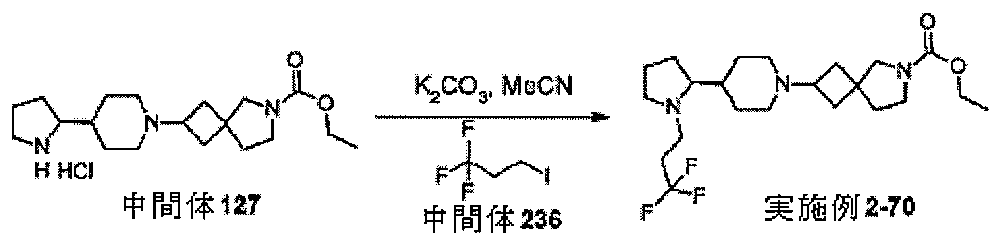
アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(0.100g、0.29
mmol)のジアステレオマーの混合物及びK2CO3(0.123g、0.89mmo
l)をMeCN(5mL)に溶解し、反応混合物を60℃で2時間撹拌した。1,1,1
-トリフルオロ-3-ヨードプロパン(0.066g、0.29mmol)を0℃で滴加
し、結果得られた混合物を室温で8時間撹拌した。反応混合物をH2O(70mL)とE
tOAc(50mL)に分配し、水層をEtOAc(2×50mL)でさらに抽出し、有
機層を合一し、乾燥させ(Na2SO4)、濾過し、溶媒を真空除去した。残渣を分取H
PLC(X Bridge、C-18、250×19mm、5μm、15mL/分、勾配
:アセトニトリル/水中60%(0.01分間)、100%(14.01分間)、次いで
60%(23.00分間)、0.1%アンモニアによって精製して、エチル(S)-2-
(4-(1-(3,3,3-トリフルオロプロピル)ピロリジン-2-イル)ピペリジン
-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-
70異性体1(0.005g、3.9%)を無色のゴム質として、エチル(S)-2-(
4-(1-(3,3,3-トリフルオロプロピル)ピロリジン-2-イル)ピペリジン-
1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-7
0異性体2(0.005mg、3.9%)を無色のゴム質として得た。異性体1及び異性
体2に関するデータは表3にある。
実施例2-72、エチル(S)-2-(4-(1-(2-メトキシ-2-オキソエチル)
ピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-
6-カルボキシレートの調製によって例示されるアルキル化を介してピペリジンを調製す
るための典型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(0.100g、0.29
mmol)のジアステレオマーの混合物及びDIPEA(0.14mL、0.87mmo
l)をMeCN(5mL)に溶解し、反応混合物を室温で1時間撹拌した。ブロモ酢酸メ
チル(0.044g、0.29mmol)を滴加し、結果得られた反応混合物を100℃
で3時間撹拌した。反応混合物をH2O(70mL)とEtOAc(50mL)に分配し
、水層をEtOAc(2×50mL)でさらに抽出し、有機層を合一し、乾燥させ(Na
2SO4)、濾過し、溶媒を真空除去した。残渣を分取HPLC(X Bridge、C
-18、250×19mm、5μm、15mL/分、勾配:アセトニトリル/水中48%
(0.01分間)、100%(11.1分間)、48%(48.00分間)、0.1%ア
ンモニア]によって精製して、エチル(S)-2-(4-(1-(2-メトキシ-2-オ
キソエチル)ピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4
]オクタン-6-カルボキシレート、実施例2-72異性体1(0.012g、9.9%
)を無色のゴム質として、エチル(S)-2-(4-(1-(2-メトキシ-2-オキソ
エチル)ピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オ
クタン-6-カルボキシレート、実施例2-72異性体2(0.013mg、10.7%
)を無色のゴム質として得た。異性体2に関するデータは表3にある。
実施例2-72、エチル2-(4-{(2S)-1-[2-(ジメチルアミノ)-2-オ
キソエチル]ピロリジン-2-イル}ピペリジン-1-イル)-6-アザスピロ[3.4
]オクタン-6-カルボキシレートの調製によって例示されるアルキル化を介してピペリ
ジンを調製するための典型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(0.100g、0.29
mmol)のジアステレオマーの混合物及びNEt3(0.087g、0.85mmol
)をジオキサン(5mL)に溶解し、反応混合物を60℃で30分間撹拌した。2-クロ
ロ-N,N-ジメチルアセトアミド(0.036g、0.29mmol)を0℃で滴加し
、結果得られた混合物を室温で12時間撹拌した。反応混合物をH2O(70mL)とE
tOAc(50mL)に分配し、水層をEtOAc(2×50mL)でさらに抽出し、有
機層を合一し、乾燥させ(Na2SO4)、濾過し、真空濃縮した。残渣を分取HPLC
(X Bridge、C-18、250×19mm、5μm、14mL/分、勾配:アセ
トニトリル/水中20%(0.01分間)、40%(36.00分間)、100%(44
.00分間)、次いで20%(52.00分間)、0.1%アンモニア]によって精製し
て、エチル(S)-2-(4-(1-(2-(ジメチルアミノ)-2-オキソエチル)ピ
ロリジン-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6
-カルボキシレート、実施例2-72異性体1(0.002g、1.6%)を黄色ゴム質
として、エチル(S)-2-(4-(1-(2-(ジメチルアミノ)-2-オキソエチル
)ピロリジン-2-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン
-6-カルボキシレート、実施例2-72異性体2(0.002mg、1.6%)を黄色
ゴム質として得た。異性体2に関するデータは表3にある。
実施例2-76、エチル2-{4-[2-(メチルカルバモイル)-2,3-ジヒドロ-
1H-イソインドール-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]
オクタン-6-カルボキシレートの調製によって例示される還元的アミノ化、Boc脱保
護及び尿素/アミド形成を介してピペリジンを調製するための典型的な手順

-2-カルボキシレート(135mg、0.45mmol)及びエチル3-オキソ-8-
アザビシクロ[3.2.1]オクタン-8-カルボキシレート(88mg、0.890m
mol)の溶液に、Ti(OiPr)4(0.40mL、1.34mmol)を0℃で加
え、反応混合物を1時間撹拌した。Na(OAc)3BH(283mg、1.34mmo
l)を反応混合物に少量ずつ加え、0℃で2時間撹拌した。完了後、反応混合物を飽和N
aHCO3水溶液でクエンチし、DCM(3×30mL)で抽出した。有機層を合一し、
飽和食塩水で洗浄し、乾燥させ(Na2SO4)、真空濃縮した。残渣をフラッシュカラ
ムクロマトグラフィー[順相、シリカゲル(100~200メッシュ)、勾配:DCM中
5%~10%メタノール]によって精製して、tert-ブチル1-(1-(6-(エト
キシカルボニル)-6-アザスピロ[3.4]オクタン-2-イル)ピペリジン-4-イ
ル)イソインドリン-2-カルボキシレート(35mg、75%)を無色の液体として得
た。
MS(ESI+ve):484
ルボニル)-6-アザスピロ[3.4]オクタン-2-イル)ピペリジン-4-イル)イ
ソインドリン-2-カルボキシレート(290mg、0.61mmol)の溶液に、ジオ
キサン中HCl(4M、5mL)を0℃でゆっくりと加え、混合物を室温で5時間撹拌し
た。溶媒を真空蒸発させた。固体残渣をジエチルエーテルで研和して、エチル2-(4-
(イソインドリン-1-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オク
タン-6-カルボキシレート塩酸塩(250mg、cr)を灰白色固体として得た。
MS(ESI+ve):384
1H-NMR(400 MHz; DMSO--d6) δ: 1.15(t, J = 6.9
Hz, 3H),1.16 - 1.26(m, 1H), 1.70 - 1.90(m, 5H
), 1.95 -2.28(m, 5H), 3.49 - 3.72(m, 4H), 3.60
- 3.72(m,4H), 3.98 - 4.15(m, 2H), 4.13(q, J =
6.9 Hz, 2H),4.45 - 4.59(m, 2H), 7.37 - 7.49(m,
5H), 9.54,10.19(2br.s., 2H)。
-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(240m
g、0.40mmol)の溶液に、DIPEA(0.43mL、2.38mmol)を0
℃で加えた。この反応混合物に、メチルカルバミン酸クロリド(methylcarba
mic chloride)(67mg、0.72mmol)を加え、室温で16時間撹
拌した。反応混合物を水(10mL)でクエンチし、水層をDCM(2×20mL)で抽
出した。有機層を合一し、乾燥させ(Na2SO4)、真空濃縮した。残渣をフラッシュ
カラムクロマトグラフィー[順相、シリカゲル(100~200メッシュ)、勾配:DC
M中5%~10%メタノール]によって精製して、エチル2-(4-(2-(メチルカル
バモイル)-イソインドリン-1-イル)ピペリジン-1-イル)-6-アザスピロ[3
.4]オクタン-6-カルボキシレートをジアステレオマーの混合物(130mg、48
%)として、ねばねばした液体として得た。
LCMS(方法M):m/z 441(M+H)+(ES+)、1.97及び1.99
分時、UV活性。
1H-NMR(400 MHz;DMSO-d6):δ: 1.15(t, J = 6.
9 Hz, 3H),1.16 - 1.26(m, 4 H), 1.49 - 1.90(m, 5
H), 1.91 -2.01(m, 2H), 2.62(d, J = 3.9 Hz, 3 H)
, 2.70 -2.90(m,2H), 3.09 - 3.25(m, 4H), 3.97(
q, J = 6.8Hz, 2H), 4.45 - 4.59(m, 2H), 5.01 - 5
.09(m, 1H),6.27(br.s., 1H), 7.22 - 7.32(m, 4H
)。
ピストンポンプ331及び332、171ダイオードアレイ検出器及びGX-271リキ
ッドハンドラーを含む、溶媒:水性=水+0.2%アンモニア(28%アンモニア溶液)
及び有機物=アセトニトリル、勾配:水中20~50%有機物溶液、流速:30mL/分
、カラム:Gemini-NX、C18、5μ、100×30mm)を使用したジアステ
レオマーの分離により、エチル2-(4-(2-(メチルカルバモイル)-イソインドリ
ン-1-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カル
ボキシレート、実施例2-76異性体1(8.99mg、12.3%)を無色のゴム質と
して、エチル2-(4-(2-(メチルカルバモイル)-イソインドリン-1-イル)ピ
ペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実
施例2-76異性体2(10.9mg、14.9%)を無色のゴム質として得た。異性体
1及び異性体2に関するデータは表3にある。
実施例2-77、エチル2-{4-[(2S)-1-フェニルピロリジン-2-イル]ピ
ペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調
製によって例示されるピロリジンをアリール化するための典型的な手順
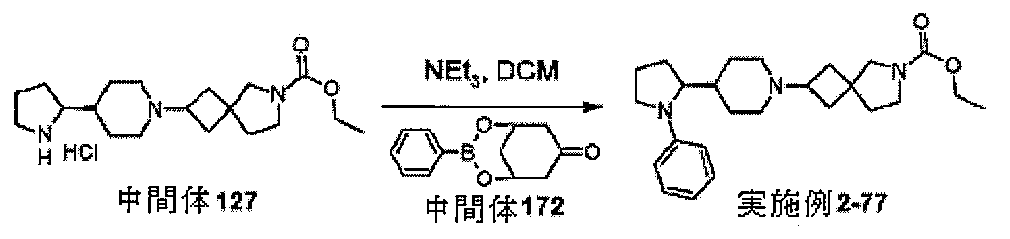
アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(0.100g、0.27
mmol)のジアステレオマーの混合物をDCM(5mL)に溶解し、トリエチルアミン
(54mg、0.54mmol)を加え、その後(1R,5S)-3-フェニル-2,4
-ジオキサ-3-ボラビシクロ[3.3.1]ノナン-7-オン(J.Luoら、Tet
rahedron Letters 54(2013),4505-4508を参照、5
8mg、0.53mmol)を加えた。反応混合物を室温で16時間撹拌し、次いで、H
2O(70mL)とDCM(100mL)に分配した。水層をDCM(2×50mL)で
さらに抽出し、有機層を合一し、乾燥させ(Na2SO4)、溶媒を除去し、残渣を分取
HPLC[逆相HPLC(CHIRALPAK AD-H、C-18、250×19mm
、5μm、15.0mL/分、勾配:アセトニトリル中0%~30%(21.0分間)、
0.1%アンモニア及び水中0.1%アンモニアによって精製して、エチル2-{4-[
(2S)-1-フェニルピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピ
ロ[3.4]オクタン-6-カルボキシレート、実施例2-77異性体1(10mg、1
3%)をゴム質として、エチル2-{4-[(2S)-1-フェニルピロリジン-2-イ
ル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレー
ト、実施例2-77異性体2(10mg、13%)をゴム質として得た。異性体2に関す
るデータは表3にある。
実施例2-78、メチル2-{4-[(2S)-1-(ピリジン-2-イル)ピロリジン
-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボ
キシレートの調製によって例示されるDMF中の炭酸セシウム及びヨウ化銅を使用して複
素環を有するピロリジン含有化合物をアリール化するための典型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(0.120g、0.37
mmol)のジアステレオマーの混合物、Cs2CO3(0.361g、1.1mmol
)及びCuI(0.105g、0.50mmol)をDMF(5mL)に溶解し、室温で
30分間撹拌した。次いで、2-ブロモピリジン(0.058g、0.37mmol)を
加え、結果得られた混合物を100℃で18時間撹拌した。反応混合物をH2O(70m
L)とEtOAc(50mL)に分配した。水層をEtOAc(2×50mL)でさらに
抽出した。有機層を合一し、乾燥させ(Na2SO4)、溶媒を濃縮によって除去し、残
渣を分取HPLC(X Bridge、C-18、150×19mm、5μm、13mL
/分、勾配:アセトニトリル/水中40%~100%(20分間)、0.1%アンモニア
]によって精製して、メチル2-{4-[(2S)-1-(ピリジン-2-イル)ピロリ
ジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カ
ルボキシレート、実施例2-78異性体1(0.011g、2%)をゴム質として、メチ
ル2-{4-[(2S)-1-(ピリジン-2-イル)ピロリジン-2-yl]ピペリジ
ン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2
-78異性体2(0.09mg、2%)をゴム質として得た。異性体2に関するデータは
表3にある。
実施例2-81、エチル2-{4-[(2S)-1-(ピリミジン-2-イル)ピロリジ
ン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カル
ボキシレートの調製によって例示されるエタノール中の炭酸ナトリウムを使用して複素環
を有するピロリジン含有化合物をアリール化するための典型的な手順

アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩(0.100g、0.29
mmol)のジアステレオマーの混合物及びNa2CO3(0.092g、0.87mm
ol)をエタノール(10mL)に溶解し、室温で30分間撹拌した。次いで、2-クロ
ロピリミジン(0.034g、0.29mmol)を0℃で加えた。結果得られた反応混
合物を80℃で6時間撹拌した。反応混合物を濃縮し、ジクロロメタンを加えた。混合物
を濾過し、濾過物を濃縮し、分取HPLC(X Bridge、C-18、150×19
mm、5μm、15mL/分、勾配:アセトニトリル/水中38%(0.01分間)、4
2%(15.00分間)、100%(19.00分間)、次いで38%(23.00分間
)、0.1%アンモニア]によって精製して、エチル2-{4-[(2S)-1-(ピリ
ミジン-2-イル)ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[
3.4]オクタン-6-カルボキシレート、実施例2-81異性体1(0.031g、2
5%)をゴム質として、エチル2-{4-[(2S)-1-(ピリミジン-2-イル)ピ
ロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6
-カルボキシレート、実施例2-81異性体2(0.017mg、14%)をゴム質とし
て得た。異性体2に関するデータは表3にある。
実施例2-82、エチル2-{4-[(2S)-1-(1,3-チアゾール-2-イル)
ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-
6-カルボキシレートの調製によって例示されるDMF中の炭酸セシウムを使用して複素
環を有するピロリジン含有化合物をアリール化するための典型的な手順
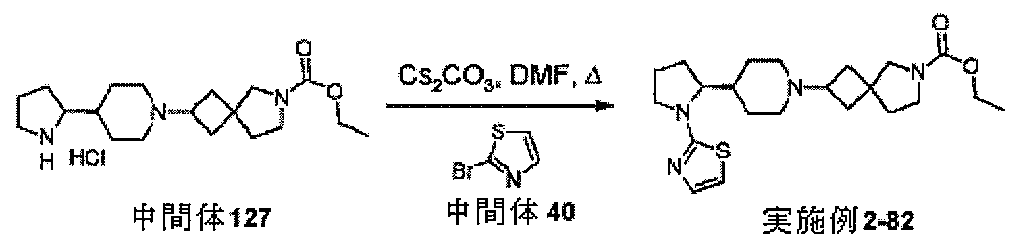
アザスピロ[3.4]オクタン-6-カルボキシレート塩酸塩のジアステレオマーの混合
物、中間体127、(100mg、0.27mmol)をDMF(5mL)に溶解し、C
S2CO3(260mg、0.81mmol)をそれに加え、その後2-ブロモチアゾー
ル(58mg、0.29mmol)を加えた。反応混合物を90℃で16時間撹拌した。
反応混合物をH2O(70mL)とEtOAc(50mL)に分配した。水層をEtOA
c(2×50mL)でさらに抽出した。有機層を合一し、乾燥させ(Na2SO4)、溶
媒を濃縮によって除去し、残渣を分取HPLC[逆相HPLC(CHIRALPAK A
D-H、C-18、250×19mm、5μm、15.0mL/分、勾配:アセトニトリ
ル中0%~30%(21.0分間)、0.1%アンモニア及び水中0.1%アンモニアに
よって精製して、エチル2-{4-[(2S)-1-(1,3-チアゾール-2-イル)
ピロリジン-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-
6-カルボキシレート、実施例2-82異性体1(20mg、18%)を無色のゴム質と
して、エチル2-{4-[(2S)-1-(1,3-チアゾール-2-イル)ピロリジン
-2-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボ
キシレート、実施例2-82異性体2(6mg、6%)を無色のゴム質として得た。異性
体1及び異性体2に関するデータは表3にある。
実施例2-84、エチル2-{4-[(2R)-2-(メトキシカルボニル)ピロリジン
-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボ
キシレートの調製によって例示される脱保護及び還元的アミノ化を介してピペリジンを調
製するための典型的な手順

]ピペリジン-1-カルボキシレート(0.396g、1.26mmol)をDCM(1
mL)に溶解し、その後ジオキサン中HCl(3mL、4.0M溶液)を滴加した。結果
得られた反応混合物を室温で1時間撹拌し、溶媒を真空除去し、残渣をさらなる精製を行
わずに次のステップに用いた。
mmol)及びエチル2-オキソ-6-アザスピロ[3.4]オクタン-6-カルボキシ
レート(0.266g、1.26mmol)をDMF(4mL)に室温で溶解し、DIP
EA(0.435mL、2.510mmol)を加えた。反応混合物を室温で3時間撹拌
した。次いで、STAB(0.533g、2.518mmol)を加え、反応混合物を窒
素下、室温で一晩撹拌した。溶媒を真空除去し、分取HPLCを使用してジアステレオマ
ーを分離した。これには、Phenomenex Gemini-NX C18カラム、
100×30mmを使用し、18mL/分でH2O中25~45%MeCN/0.2%ア
ンモニア(v/v)で溶出し、210nmでモニターすることによって画分を収集した。
そして、実施例2-84異性体1、エチル2-{4-[(2R)-2-(メトキシカルボ
ニル)ピロリジン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オク
タン-6-カルボキシレート(18.4mg、4%)を無色の油状物質として、実施例2
-84異性体2、エチル2-{4-[(2R)-2-(メトキシカルボニル)ピロリジン
-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボ
キシレート(13.9mg、3%)を無色の油状物質として得た。異性体2に関するデー
タは表3にある。
実施例2-85、エチル2-{4-[(2S)-2-(メチルカルバモイル)ピロリジン
-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボ
キシレートの調製によって例示される還元的アミノ化を介してピペリジンを調製するため
の典型的な手順

ジヒドロクロリド(0.2g、0.94mmol)、NEt3(0.75mL、5.0m
mol)、6-(エトキシカルボニル)-2-オキソ-6-アザスピロ[3.4]オクタ
ン-8-イリウム(0.188g、0.93mmol)及びZnCl2(30mg、0.
02mmol)をMeOH(15.00mL)に窒素下で溶解し、50~60℃で1時間
撹拌した。NaCNBH3(0.069g、1.0mmol)を0~10℃で少量ずつ加
え、反応混合物を室温で3時間撹拌した。反応混合物をEtOAc(2×50mL)と水
(30mL)に分配し、有機層を合一し、乾燥させ(Na2SO4)、濾過し、溶媒を真
空除去し、粗生成物を分取HPLC[逆相HPLC(X-Bridge PREP C1
8、250×19mm、5μm、15mL/分、勾配:アセトニトリル中30%~100
%(22分間)、次いで100%(2分間)、0.1%NH3によって精製して、実施例
2-85異性体1、エチル(S)-2-(4-(2-(メチルカルバモイル)ピロリジン
-1-イル)ピペリジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボ
キシレート(0.09g、24.32%)を白色固体として、実施例2-85異性体2、
エチル(S)-2-(4-(2-(メチルカルバモイル)ピロリジン-1-イル)ピペリ
ジン-1-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート(0.0
89g、24.10%)を白色固体として得た。異性体2に関するデータは表3にある。
実施例2-87、エチル2-{4-[(2R)-4,4-ジフルオロ-2-(メトキシカ
ルボニル)ピロリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキ
シレートの調製によって例示される脱保護及び水素化トリアセトキシホウ素ナトリウム還
元的アミノ化を介してピペリジンを調製するための典型的な手順
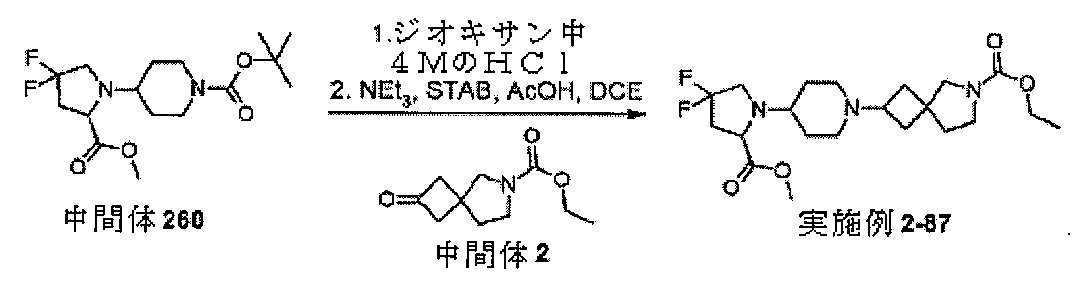
ピロリジン-1-イル]ピペリジン-1-カルボキシレート(0.36g、1.03mm
ol)をジオキサン中4.0MのHCl(10mL)に溶解し、反応混合物を室温で6時
間撹拌した。溶媒を真空除去し、残渣をさらなる精製を行わずに次のステップで使用した
。粗反応混合物及びエチル2-オキソ-6-アザスピロ[3.4]オクタン-6-カルボ
キシレート(0.243g、1.233mmol)をDCE(10mL)に室温で溶解し
、Et3N(0.249g、2.47mmol)を加えた。反応混合物を50℃窒素下で
2時間撹拌した。反応混合物を室温まで冷却し、氷酢酸(0.114g、1.90mmo
l)及びSTAB(0.784g、3.69mmol)を加え、反応混合物を50℃窒素
下で一晩撹拌した。水(2mL)を加えて反応混合物を冷却し、溶媒を真空除去した。残
渣をDCM(15mL)と飽和NaHCO3(水溶液)(15mL)に分配し、水層をD
CM(2×15mL)で洗浄した。有機層を合一し、Biotageフェーズセパレータ
カートリッジに通すことによって乾燥させた。溶媒を真空除去し、残渣をカラムクロマト
グラフィー(順相、[Biotage SNAPカートリッジKP-sil 10g 4
0~63μm、60Å、12mL/分、勾配:1%~10%MeOH/DCM])によっ
て精製した。残渣を分取逆相HPLC(Phenomenex Gemini-NX 5
μm C18 110A Axiaカラム、100×30mm、30mL/分で14.4
分間にわたって20~50%MeCN/溶媒Bで溶出し[ここで、溶媒BはH2O中0.
2%の(28%NH3/H2O)である]、210nmでモニターすることによって画分
を収集する)によってさらに精製して、エチル2-{4-[(2R)-4,4-ジフルオ
ロ-2-(メトキシカルボニル)ピロリジン-1-イル}-6-アザスピロ[3.4]オ
クタン-6-カルボキシレート、実施例2-87異性体1(0.020g、4.5%)、
及びエチル2-{4-[(2R)-4,4-ジフルオロ-2-(メトキシカルボニル)ピ
ロリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実
施例2-87異性体2(0.020g、4.5%)を得た。異性体2に関するデータは表
3にある。
実施例2-88、エチル2-{4-[(2R)-4,4-ジフルオロ-2-(メチルカル
バモイル)ピロリジン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]
オクタン-6-カルボキシレートの調製によって例示される脱保護及びアミド形成を介し
てピペリジンを調製するための典型的な手順

リジン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-
カルボキシレートのジアステレオマーの混合物、実施例2-87、(0.400g、0.
932mmol)をTHF(8mL)に溶解し、1MのLiOH(水溶液)(1.9mL
)を加え、反応混合物を室温で一晩撹拌した。反応混合物を2.0MのHCl溶液を使用
して中和し、溶媒を真空除去した。残渣をトルエンで共沸して、1-{1-[6-(エト
キシカルボニル)-6-アザスピロ[3.4]オクト-2-イル]ピペリジン-4-イル
}-4,4-ジフルオロ-D-プロリン(0.440g、100%)を黄色ガラスとして
得た。
LCMS(方法C):m/z 416(M+H)+(ES+)0.71分時、UV不活
性
ル]ピペリジン-4-イル}-4,4-ジフルオロ-D-プロリン(0.193g、0.
466mmol)を無水DMF(5mL)に溶解し、HATU(0.533g、1.39
8mmol)、THF(2.3mL、2.33mmol)中の2.0Mメチルアミン溶液
及びDIPEA(0.301g、2.33mmol)を加え、反応混合物を室温で一晩撹
拌した。溶媒を真空除去し、残渣をDCM(20mL)と飽和NaHCO3(水溶液)(
20mL)に分配し、水層をDCM(2×15mL)で抽出した。有機層を合一し、飽和
食塩水(20mL)で洗浄し、Biotageフェーズセパレータカートリッジに通すこ
とによって乾燥させた。溶媒を真空除去し、残渣をカラムクロマトグラフィー(順相、[
Biotage SNAPカートリッジKP-sil 10g 40~63μm、60Å
、12mL/分、勾配:0%~10%MeOH/DCM])によって精製した。残渣を分
取逆相HPLC(Phenomenex Gemini-NX 5μm C18 110
A Axiaカラム、100×30mm、30mL/分で14.4分間にわたって20~
50%MeCN/溶媒Bで溶出し[ここで、溶媒BはH2O中0.2%の(28%NH3
/H2O)である]、210nmでモニターすることによって画分を収集する)によって
さらに精製して、エチル2-{4-[(2R)-4,4-ジフルオロ-2-(メチルカル
バモイル)ピロリジン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]
オクタン-6-カルボキシレート、実施例2-88異性体1、(0.038g、18%)
を無色の油状物質として、エチル2-{4-[(2R)-4,4-ジフルオロ-2-(メ
チルカルバモイル)ピロリジン-1-イル]ピペリジン-1-イル}-6-アザスピロ[
3.4]オクタン-6-カルボキシレート、実施例2-88異性体2、(0.037g、
18%)を無色の油状物質として得た。異性体2に関するデータは表3にある。
実施例2-90、エチル2-{4-[(2R)-2-カルバモイル-4,4-ジフルオロ
ピロリジン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-
6-カルボキシレートの調製によって例示されるアミド形成を介してピペリジンを調製す
るための典型的な手順

ン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カル
ボキシレート(456mg、1.062mmol)のジアステレオマーの混合物をTHF
(1mL)及び28%NH3溶液(9mL)に60℃で混合し、反応物を18時間撹拌し
た。反応混合物を1MのHCl(水溶液)で中和し、DCM(25mL)で希釈し、H2
O(2×25mL)で洗浄し、合一した水層をDCM(25mL)で洗浄し、合一した有
機層を飽和食塩水(25mL)で洗浄し、Biotageフェーズセパレータに通した。
揮発性物質を真空下で除去して橙色の油状物質(0.290g、65%)を得た。分取H
PLCを使用してジアステレオマーを分離した。これには、Phenomenex Ge
mini-NX C18カラム、100×30mmを使用し、18mL/分でH2O中2
0~45%MeCN/0.2%アンモニアを含む(v/v)で溶出し、210nmでモニ
ターすることによって画分を収集した。そして、実施例2-90異性体1、エチル2-{
4-[(2R)-2-カルバモイル-4,4-ジフルオロピロリジン-1-イル]ピペリ
ジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート(16.
8mg、4%)を無色の油状物質として、実施例2-90異性体2、エチル2-{4-[
(2R)-2-カルバモイル-4,4-ジフルオロピロリジン-1-イル]ピペリジン-
1-イル}-6-アザスピロ[3.4]オクタン-6-カルボキシレート(12.4mg
、3%)を無色の油状物質として得た。異性体2に関するデータは表3にある。
実施例2-91、エチル2-{4-[(2R)-4,4-ジフルオロ-2-(メトキシカ
ルバモイル)ピロリジン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4
]オクタン-6-カルボキシレートの調製によって例示される、加水分解及びアミド形成
を介してピペリジンを調製するための典型的な手順
ン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-カル
ボキシレート(230mg、0.536mmol)のジアステレオマーの混合物をTHF
(6.5mL)及び1.0MのLiOH溶液(1.1mL、1.1mmol)に室温で溶
解し、反応物を18時間撹拌した。揮発性物質を真空下で除去し、化合物をさらなる精製
を行わずに通した。
ル]ピペリジン-4-イル}-4,4-ジフルオロ-D-プロリン(125mg、0.3
00mmol)をDMF(1mL)に溶解し、その後HATU(228mg、0.60m
mol)及びDIPEA(0.260mL、1.50mmol)を加えた。反応混合物を
0℃で10分間撹拌し、その後メトキシアミン塩酸塩(25mg、0.30mmol)を
加え、結果得られた反応混合物を室温で18時間撹拌した。反応混合物を真空濃縮し、次
いで、飽和NaHCO3(水溶液)(50mL)とDCM(50mL)に分配し、水層を
DCM(2×50mL)でさらに抽出し、有機層を合一し、飽和食塩水(50mL)で洗
浄し、Biotageフェーズセパレータに通した。揮発性物質を真空下で除去して、橙
色の油状物質(0.102g、77%)を得た。分取HPLCを使用してジアステレオマ
ーを分離した。これには、Phenomenex Gemini-NX C18カラム、
100×30mmを使用し、18mL/分でH2O中20~50%MeCN/0.2%ア
ンモニア(v/v)で溶出し、210nmでモニターすることによって画分を収集した。
そして、実施例2-91異性体1、エチル2-{4-[(2R)-4,4-ジフルオロ-
2-(メトキシカルバモイル)ピロリジン-1-イル]ピペリジン-1-イル}-6-ア
ザスピロ[3.4]オクタン-6-カルボキシレート(12.2mg、4%)を無色の油
状物質として、実施例2-91異性体2、エチル2-{4-[(2R)-4,4-ジフル
オロ-2-(メトキシカルバモイル)ピロリジン-1-イル]ピペリジン-1-イル}-
6-アザスピロ[3.4]オクタン-6-カルボキシレート(7.2mg、3%)を無色
の油状物質として得た。異性体2に関するデータは表3にある。
実施例2-111、エチル2-[4-(2-オキソピロリジン-1-イル)ピペリジン-
1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製によって
例示される脱保護及び水素化トリアセトキシホウ素ナトリウム還元的アミノ化を介してピ
ペリジンを調製するための典型的な手順

及びエチル2-オキソ-6-アザスピロ[3.4]オクタン-6-カルボキシレート(0
.212g、1.14mmol)をDMF(6mL)に室温で溶解し、反応混合物を40
℃窒素下で3時間撹拌した。反応混合物を室温まで冷却し、STAB(0.630g、2
.97mmol)及び氷酢酸(0.071g、1.189mmol)を加え、反応混合物
を40℃窒素下で一晩撹拌した。水(2mL)を冷却した反応混合物に加え、溶媒を真空
除去した。残渣をDCM(15mL)と飽和NaHCO3(水溶液)(15mL)に分配
し、水層をDCM(2×15mL)で洗浄した。有機層を合一し、Biotageフェー
ズセパレータカートリッジに通すことによって乾燥させた。溶媒を真空除去し、残渣をカ
ラムクロマトグラフィー(順相、[Biotage SNAPカートリッジKP-sil
10g 40~63μm、60Å、12mL/分、勾配:1%~10%MeOH/DC
M])によって精製した。残渣を分取逆相HPLC(Phenomenex Gemin
i-NX 5μm C18 110A Axiaカラム、100×30mm、30mL/
分で14.4分間にわたって20~35%MeCN/溶媒Bで溶出し[ここで、溶媒Bは
、H2O中0.2%の(28%NH3/H2O)である]、210nmでモニターするこ
とによって画分を収集する)によってさらに精製して、エチル2-[4-(2-オキソピ
ロリジン-1-イル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6
-カルボキシレート、実施例2-111異性体1、(0.008g、2%)を無色の油状
物質として、エチル2-[4-(2-オキソピロリジン-1-イル)ピペリジン-1-イ
ル]-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-111異
性体2、(0.009g、2%)を無色の油状物質として得た。異性体2に関するデータ
は表3にある。
ミン保護基で)保護された第二のアミン基を含有する場合。ひとたび、還元的アミノ化が
行われて標的のさらなる機能分化を可能にしたら、脱保護するための標準的な方法を使用
してこれらの保護基を除去することができる。
実施例2-124、エチル2-[4-(2-オキソ-1,2-ジヒドロピリジン-4-イ
ル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレー
トの調製によって例示される還元的アミノ化、Boc脱保護及び尿素形成を介してピペリ
ジンを調製するための典型的な手順

-6-カルボキシレート塩酸塩(0.316g、1.00mmol)及びtert-ブチ
ル(2-アミノエチル)カルバメート(0.320g、2.00mmol)をDCM(1
0mL)にN2下室温で溶解し、NEt3(0.15mL、1.10mmol)を加え、
反応混合物を室温で0.5時間撹拌した。酢酸(0.13mL、2.20mmol)を加
え、反応混合物を室温で2時間撹拌し、STAB(0.530g、2.50mmol)を
加え、反応混合物を一晩撹拌した。反応混合物をNaHCO3(飽和水溶液)(30mL
)の添加でクエンチし、DCM(4×25mL)で抽出し、合一したDCM層をBiot
ageフェーズセパレータに通し、真空濃縮して、粗エチル2-[4-({2-[(te
rt-ブトキシカルボニル)アミノ]エチル}アミノ)ピペリジン-1-イル]-6-ア
ザスピロ[3.4]オクタン-6-カルボキシレートをジアステレオマーの混合物として
得た。これをいかなるさらなる精製も行わずに使用した。
LCMS(方法D):m/z 425(M+H)+(ES+)、1.30及び1.35分
時、UV不活性。
ミノ)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレ
ート(0.424g、1.00mmol)をCH2Cl2(10mL)に溶解し、ジオキ
サン中4M塩化水素(1.25mL、5.0mmol)を加え、反応混合物を室温で一晩
撹拌した。揮発性物質を真空除去し、残渣をEtOH(10mL)に溶解し、NEt3(
1.40mL、10.0mmol)及びCDI(0.244g、1.50mmol)を加
え、混合物を加熱還流し一晩維持した。溶媒を真空除去し、残渣(resiude)をC
H2Cl2(20mL)と水(20mL)に分配し、水層をCH2Cl2(4×20mL
)で抽出した。合一した有機物を真空濃縮して、粗エチル2-[4-(2-オキソイミダ
ゾリジン-1-イル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6
-カルボキシレートをジアステレオマーの混合物として得た。分取HPLCを使用してジ
アステレオマーを分離した。これには、Phenomenex Gemini-N C1
8カラム、150×21mmを使用し、18mL/分で0.2%NH3/H2O中25~
45%MeCNで溶出し、210nmでモニターすることによって画分を収集した。そし
て、エチル2-[4-(2-オキソ-1,2-ジヒドロピリジン-4-イル)ピペリジン
-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例2-
124異性体1、(0.008g、2.3%)を無色の固体として、エチル2-[4-(
2-オキソ-1,2-ジヒドロピリジン-4-イル)ピペリジン-1-イル]-6-アザ
スピロ[3.4]オクタン-6-カルボキシレート、実施例2-124異性体2、(0.
008g、2.3%)を無色の固体として得た。異性体2に関するデータは表3にある。
実施例2-136、エチル2-{4-[(2R,4R)-4-フルオロ-2-(ヒドロキ
シメチル)ピロリジン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]
オクタン-6-カルボキシレートの調製によって例示されるエステル還元を介してピペリ
ジンを調製するための典型的な手順
リジン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3.4]オクタン-6-
カルボキシレート(0.140g、0.341mmol)のジアステレオマーの混合物を
無水THF(10mL)に溶解し、0℃まで窒素下で冷却した。THF(1.02mL、
1.023mmol)中の2.0M水素化ホウ素リチウム溶液を反応混合物に滴加し、次
いで反応混合物を室温まで一晩自然に温めた。反応混合物を飽和NaHCO3(水溶液)
(15mL)でクエンチし、次いでEtOAc(2×15mL)で抽出し、有機層を合一
し、乾燥させた(MgSO4)。溶媒を真空除去し、残渣をカラムクロマトグラフィー(
順相、[Biotage SNAPカートリッジKP-sil 10g 40~63μm
、60Å、12mL/分、勾配:0%~10%MeOH/DCM])によって精製した。
残渣を分取逆相HPLC(Phenomenex Gemini-NX 5μm C18
110A Axiaカラム、100×30mm、30mL/分で14.4分間にわたっ
て20~50%MeCN/溶媒Bで溶出し[ここで、溶媒Bは0.2%の(H2O中28
%NH3/H2O)である]、210nmでモニターすることによって画分を収集する)
によってさらに精製して、エチル2-{4-[(2R,4R)-4-フルオロ-2-(ヒ
ドロキシメチル)ピロリジン-1-イル]ピペリジン-1-イル}-6-アザスピロ[3
.4]オクタン-6-カルボキシレート、実施例2-136異性体1、(2.99mg、
0.23%)を白色固体として、エチル2-{4-[(2R,4R)-4-フルオロ-2
-(ヒドロキシメチル)ピロリジン-1-イル]ピペリジン-1-イル}-6-アザスピ
ロ[3.4]オクタン-6-カルボキシレート、実施例2-136異性体2、(3.10
mg、0.24%)を白色固体として得た。異性体2に関するデータは表3にある。
実施例3-4、エチル2-[4-(3-ヒドロキシピリジン-2-イル)ピペリジン-1
-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製によって例
示されるDMF中の水素化トリアセトキシホウ素ナトリウム還元的アミノ化を介してピペ
リジンを調製するための典型的な手順

0.8mmol)及びエチル2-オキソ-6-アザスピロ[3.4]オクタン-6-カル
ボキシレート(0.157g、0.8mmol)をDMF(8mL)に室温で混合した。
DIPEA(0.28mL、1.6mmol)及びAcOH(0.07mL、1.2mm
ol)を加え、その後STAB(0.34g、1.6mmol)を加えた。反応混合物を
窒素下、室温で一晩撹拌し、次いで少量のMeOHの添加でクエンチし、真空濃縮してす
べての溶媒を除去した。残渣をMeOH及びDCMの混合物に溶解し、フラッシュシリカ
(約10mL)で真空濃縮した。結果得られた粉末をカラムクロマトグラフィー(順相、
[Biotage SNAPカートリッジKP-sil 25g、40~63μm、60
Å、30mL/分、勾配:15カラム容量にわたってDCM中0%~15%溶媒A、ここ
で、溶媒AはMeOH中10%の(7M NH3/MeOH)である])によって精製し
て、ジアステレオマーの粗混合物を得た(0.258g)。この混合物をMeOHに溶解
し、少量の28%NH3/H2Oを加え(約0.1mL)、溶液を分取逆相HPLCによ
って精製した。これには、Phenomenex Gemini-NX 5μm C18
110A Axiaカラム、100×30mmを使用し、30mL/分で14.4分間
にわたって15~25%MeCN/溶媒Bで溶出し[ここで、溶媒BはH2O中0.2%
の(28%NH3/H2O)である]、230nmでモニターすることによって画分を収
集した。そしてエチル2-[4-(3-ヒドロキシピリジン-2-イル)ピペリジン-1
-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例3-4異
性体1、(0.034g、12%)、及びエチル2-[4-(3-ヒドロキシピリジン-
2-イル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキ
シレート、実施例3-4異性体2、(0.052g、18%)を得た。異性体2に関する
データは表3にある。
実施例3-10、エチル2-[4-シアノ-(ピリジン-2-イル)ピペリジン-1-イ
ル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製によって例示さ
れる水素化トリアセトキシホウ素ナトリウム還元的アミノ化を介してピペリジンを調製す
るための典型的な手順
1.0mmol)及びエチル2-オキソ-6-アザスピロ[3.4]オクタン-6-カル
ボキシレート(0.197g、1.0mmol)をDCM(10mL)にN2下室温で溶
解し、NEt3(0.15mL、1.1mmol)を加えた。反応混合物を室温で1時間
撹拌し、酢酸(0.13mL、2.2mmol)を加え、反応混合物を室温で3時間撹拌
した。STAB(0.636g、3.0mmol)を加え、反応混合物を一晩撹拌した。
反応混合物をNaHCO3(飽和水溶液)(30mL)の添加でクエンチし、DCM(4
×25mL)で抽出し、合一したDCM層をBiotageフェーズセパレータに通した
。溶媒を真空除去し、残渣をカラムクロマトグラフィー(順相、[Biotage SN
APカートリッジKP-sil 25g、40~63μm、60Å、40mL/分、勾配
:DCM中0%~10%MeOH)によって精製して、エチル2-[4-シアノ-(ピリ
ジン-2-イル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カ
ルボキシレートのジアステレオマーの分離不可能な混合物を得た。分取HPLCを使用し
てジアステレオマーを分離した。これには、Phenomenex Gemini-N
C18カラム、150×21mmを使用し、18mL/分で25~65%MeOH/H2
Oで溶出し、210nmでモニターすることによって画分を収集した。そして、エチル2
-[4-シアノ-(ピリジン-2-イル)ピペリジン-1-イル]-6-アザスピロ[3
.4]オクタン-6-カルボキシレート、実施例3-10異性体1、(0.012g、3
%)を無色の固体として、エチル2-[4-シアノ-(ピリジン-2-イル)ピペリジン
-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例3-
10異性体2、(0.014g、4%)を無色の固体として得た。両異性体に関するデー
タは表3にある。
実施例4-5、エチル2-(1-エチル-2-オキソ-3,4’-ビピペリジン-1’-
イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製によって例示
されるアルキル化を介してピペリジンを調製するための典型的な手順

ロ[3.4]オクタン-6-カルボキシレート、実施例4-1、(0.2g、0.55m
mol)、をDMF(3mL)に溶解し、0~5℃に冷却した。水素化ナトリウム(0.
080g、1.6mmol)及びヨードエタン(0.139g、0.8mmol)を加え
、反応混合物を室温で3時間撹拌した。反応物を水(50mL)でクエンチし、EtOA
c(3×30mL)で抽出し、合一した有機層を乾燥させ(Na2SO4)、濾過し、真
空濃縮して、粗エチル2-(1-エチル-2-オキソ-3,4’-ビピペリジン-1’-
イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレートをジアステレオマー
の混合物として得た。粗生成物を分取HPLC[X-Bridge C18(150×1
9mm、5μm、17mL/分、勾配:アセトニトリル中27%~100%(30分間)
、次いで100%(4分間)、0.1%NH3によって精製して、エチル2-(1-エチ
ル-2-オキソ-[3,4’-ビピペリジン]-1’-イル)-6-アザスピロ[3.4
]オクタン-6-カルボキシレート、実施例4-5異性体-1(0.011g、5.11
%)を無色のゴム質として、エチル2-(1-エチル-2-オキソ-[3,4’-ビピペ
リジン]-1’-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、
実施例4-5異性体-2(0.012g、5.80%)を無色のゴム質として得た。異性
体2に関するデータは表3にある。
実施例4-8、エチル2-[4-(2-オキソテトラヒドロピリミジン-1(2H)-イ
ル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレー
トの調製によって例示される水素化トリアセトキシホウ素ナトリウム還元的アミノ化を介
してピペリジンを調製するための典型的な手順
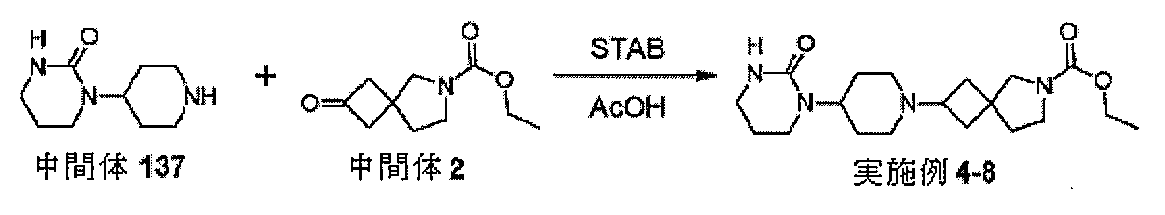
3g、1.0mmol)及びエチル2-オキソ-6-アザスピロ[3.4]オクタン-6
-カルボキシレート(0.197g、1.0mmol)をDCM(10mL)にN2下室
温で溶解し、酢酸(0.13mL、2.2mmol)を加え、反応混合物を室温で3時間
撹拌した。STAB(0.530g、2.5mmol)を加え、反応混合物を一晩撹拌し
た。反応混合物をNaHCO3(飽和水溶液)(30mL)の添加でクエンチし、DCM
(4×25mL)で抽出し、合一したDCM層をBiotageフェーズセパレータに通
した。溶媒を真空除去し、残渣をカラムクロマトグラフィー(順相、[Biotage
SNAPカートリッジKP-sil 25g、40~63μm、60Å、40mL/分、
勾配:DCM中0%~10%MeOH)によって精製して、エチル2-[4-(2-オキ
ソテトラヒドロピリミジン-1(2H)-イル)ピペリジン-1-イル]-6-アザスピ
ロ[3.4]オクタン-6-カルボキシレートのジアステレオマーの分離不可能な混合物
を得た。分取HPLCを使用してジアステレオマーを分離した。これには、Phenom
enex Gemini-N C18カラム、150×21mmを使用し、18mL/分
で0.2%NH3/H2O中15~30%MeCNで溶出し、210nmでモニターする
ことによって画分を収集した。そして、エチル2-[4-(2-オキソテトラヒドロピリ
ミジン-1(2H)-イル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタ
ン-6-カルボキシレート、実施例4-8異性体1、(0.028g、7.7%)を無色
の固体として、エチル2-[4-(2-オキソテトラヒドロピリミジン-1(2H)-イ
ル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレー
ト、実施例4-8異性体2、(0.025g、6.9%)を無色の固体として得た。
両異性体に関するデータは表3にある。
実施例4-13、エチル2-(3,3-ジフルオロ-1,4’-ビピペリジン-1’-イ
ル)-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製によって例示さ
れる脱保護及び水素化トリアセトキシホウ素ナトリウム還元的アミノ化を介してピペリジ
ンを調製するための典型的な手順

ート(0.347g、1.14mmol)をジオキサン中4.0MのHCl(5mL)に
溶解し、反応混合物を室温で一晩撹拌した。溶媒を真空除去し、残渣をさらなる精製を行
わずに次のステップで使用した。粗反応混合物及びエチル2-オキソ-6-アザスピロ[
3.4]オクタン-6-カルボキシレート(0.212g、1.14mmol)をDMF
(6mL)に室温で溶解し、DIPEA(0.295g、2.28mmol)を加えた。
反応混合物を50℃窒素下で2時間撹拌した。反応混合物を室温まで冷却し、氷酢酸(0
.068g、1.14mmol)及びSTAB(0.604g、2.85mmol)を加
え、反応混合物を50℃窒素下で一晩撹拌した。水(2mL)を冷却した反応混合物に加
え、溶媒を真空除去した。残渣をDCM(15mL)と飽和NaHCO3(水溶液)(1
5mL)に分配し、水層をDCM(2×15mL)で洗浄した。有機層を合一し、Bio
tageフェーズセパレータカートリッジに通すことによって乾燥させた。溶媒を真空除
去し、残渣をカラムクロマトグラフィー(順相、[Biotage SNAPカートリッ
ジKP-sil 10g 40~63μm、60Å、12mL/分、勾配:1%~10%
MeOH/DCM])によって精製した。残渣を分取逆相HPLC(Phenomene
x Gemini-NX 5μm C18 110A Axiaカラム、100×30m
m、30mL/分で14.4分間にわたって30~60%MeCN/溶媒Bで溶出し[こ
こで、溶媒BはH2O中0.2%の(28%NH3/H2O)である]、210nmでモ
ニターすることによって画分を収集する)によってさらに精製して、エチル2-(3,3
-ジフルオロ-1,4’-ビピペリジン-1’-イル)-6-アザスピロ[3.4]オク
タン-6-カルボキシレート、実施例4-13異性体1、(0.011g、2.6%)を
無色の油状物質として、エチル2-(3,3-ジフルオロ-1,4’-ビピペリジン-1
’-イル)-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例4-1
3異性体2、(0.005g、1.3%)を無色の油状物質として得た。異性体2に関す
るデータは表3にある。
実施例4-16、エチル2-[(2R)-2-(メチルカルバモイル)-1,4’-ビピ
ペリジン-1’-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの
調製によって例示される還元的アミノ化を介してピペリジンを調製するための典型的な手
順
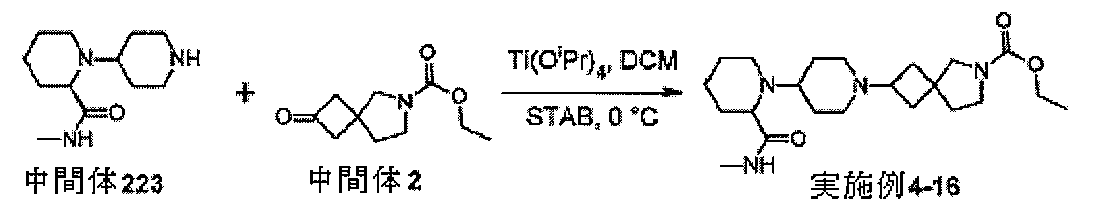
ルボキサミド(200mg、0.890mmol)及びエチル3-オキソ-8-アザビシ
クロ[3.2.1]オクタン-8-カルボキシレート(175mg、0.890mmol
)の溶液に、Ti(OiPr)4(0.80mL、2.67mmol)を0℃で加え、反
応混合物を1時間撹拌した。Na(OAc)3BH(562mg、2.67mmol)を
該反応混合物に少量ずつ加え、0℃で2時間撹拌した。完了後、反応混合物を飽和NaH
CO3水溶液でクエンチし、DCM(3×30mL)で抽出した。有機層を合一し、飽和
食塩水で洗浄し、乾燥させ(Na2SO4)、真空濃縮した。残渣を分取HPLC(逆相
、X BRIDGE、C-18、19×250mm、5μ、勾配:5mM NH4OAc
を含有する水中10%~90%ACN、によって精製して、25mg(7%)のエチル2
-[(2R)-2-(メチルカルバモイル)-1,4’-ビピペリジン-1’-イル]-
6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例4-16異性体-1
、及び25mg(7%)のエチル2-[(2R)-2-(メチルカルバモイル)-1,4
’-ビピペリジン-1’-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシ
レート、実施例4-16異性体-2を無色の半固体として得た。異性体2に関するデータ
は表3にある。
実施例5-1、エチル2-[4-(2-オキソアゼパン-1-イル)ピペリジン-1-イ
ル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートの調製によって例示さ
れるアルキル化、結晶化及び水素化トリアセトキシホウ素ナトリウム還元的アミノ化を介
してピペリジンを調製するための典型的な手順

ol)の溶液に、トリエチルアミン(0.153mL、1.1mmol)及び6-ブロモ
ヘキサノイルクロリド(0.168mL、1.098mmol)を加え、濁った懸濁液を
室温で2時間撹拌した。溶媒を真空除去し、残渣をH2O(15mL)とEtOAc(2
5mL)に分配し、水層をEtOAc(2×25mL)で抽出し、有機層を合一し、Na
2SO4で乾燥させ、溶媒を真空除去して、tert-ブチル4-[(6-ブロモヘキサ
ノイル)アミノ]ピペリジン-1-カルボキシレート(378mg、99%超)を橙色の
油状物質として得た。
キシレート(378mg、1.0mmol)をDMF(25mL)に溶解し、水素化ナト
リウムを加えた(48mg、1.2mmol)。反応混合物を80℃で1時間撹拌し、溶
媒を真空除去し、残渣をカラムクロマトグラフィー(順相、[Biotage SNAP
カートリッジKP-sil 10g、40~63μm、60Å、25mL/分、DCM中
1%~10%MeOH])によって精製して、tert-ブチル4-(2-オキソアゼパ
ン-1-イル)ピペリジン-1-カルボキシレート(178mg、60%)を得た。残渣
をDCM(1mL)に溶解し、その後、ジオキサン中HCl(3mL、4.0M溶液)を
滴加した。結果得られた反応混合物を室温で1時間撹拌し、溶媒を真空除去し、残渣をさ
らなる精製を行わずに次のステップに用いた。1-(ピペリジン-4-イル)アゼパン-
2-オン塩酸塩(0.182g、0.738mmol)及びエチル2-オキソ-6-アザ
スピロ[3.4]オクタン-6-カルボキシレート(0.155g、0.785mmol
)をDMF(2mL)に室温で溶解し、DIPEA(0.136mL、0.790mmo
l)を加えた。反応混合物を室温で3時間撹拌した。次いで、STAB(0.332g、
1.569mmol)を加え、反応混合物を窒素下、室温で一晩撹拌した。溶媒を真空除
去し、残渣を分取逆相HPLC(Phenomenex Gemini-NX 5μm
C18 110A Axiaカラム、100×30mm、30mL/分で14.4分間に
わたって25~45%MeCN/溶媒Bで溶出し[ここで、溶媒BはH2O中0.2%の
(28%NH3/H2O)である]、210nmでモニターすることによって画分を収集
する)によって精製して、エチル2-[4-(2-オキソアゼパン-1-イル)ピペリジ
ン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレート、実施例5
-1異性体1(6.2mg、2%)を無色の油状物質として、エチル2-[4-(2-オ
キソアゼパン-1-イル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン
-6-カルボキシレート、実施例5-1異性体2(3.9mg、1%)を無色の油状物質
として得た。異性体2に関するデータは表3にある。










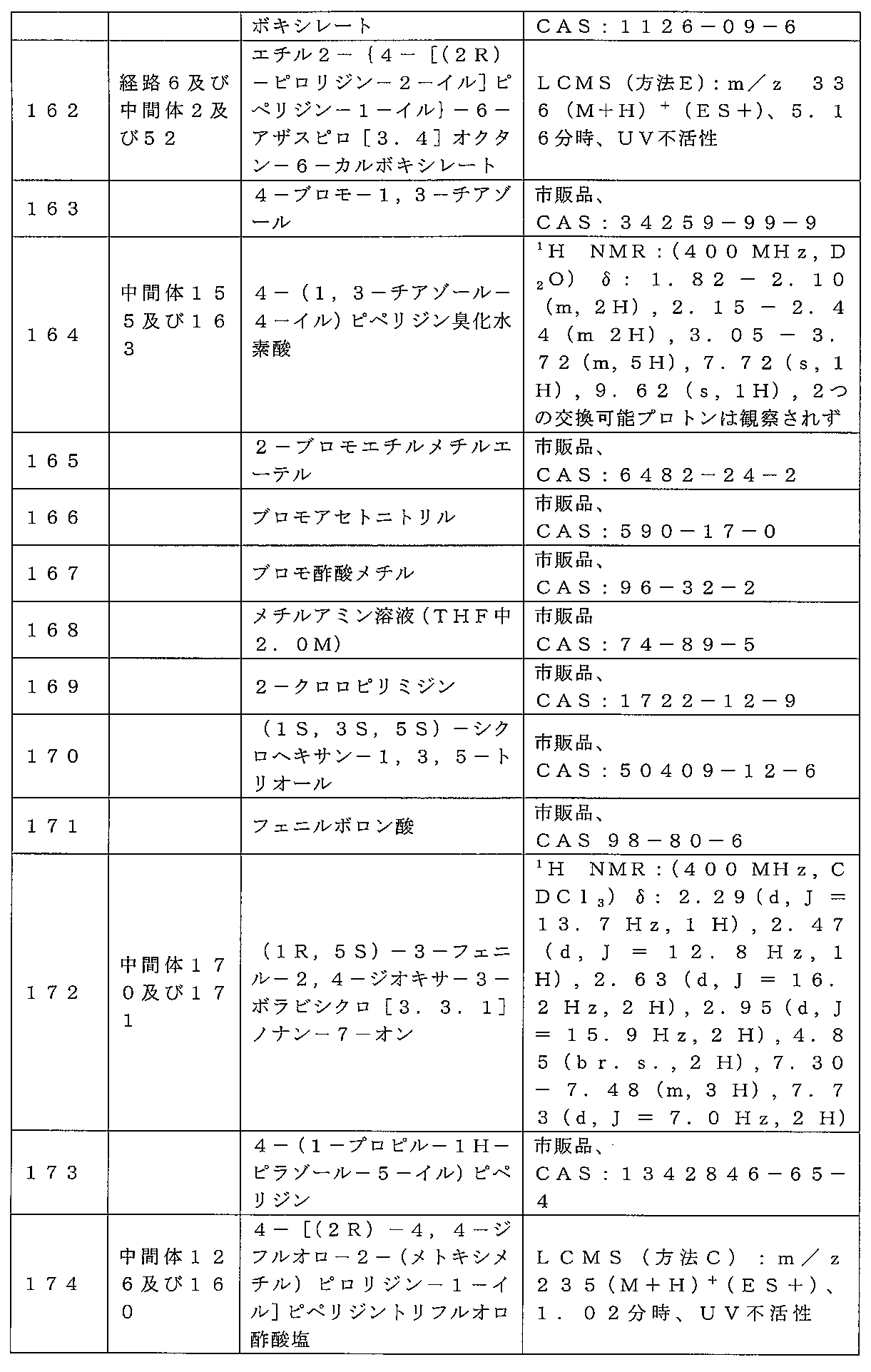

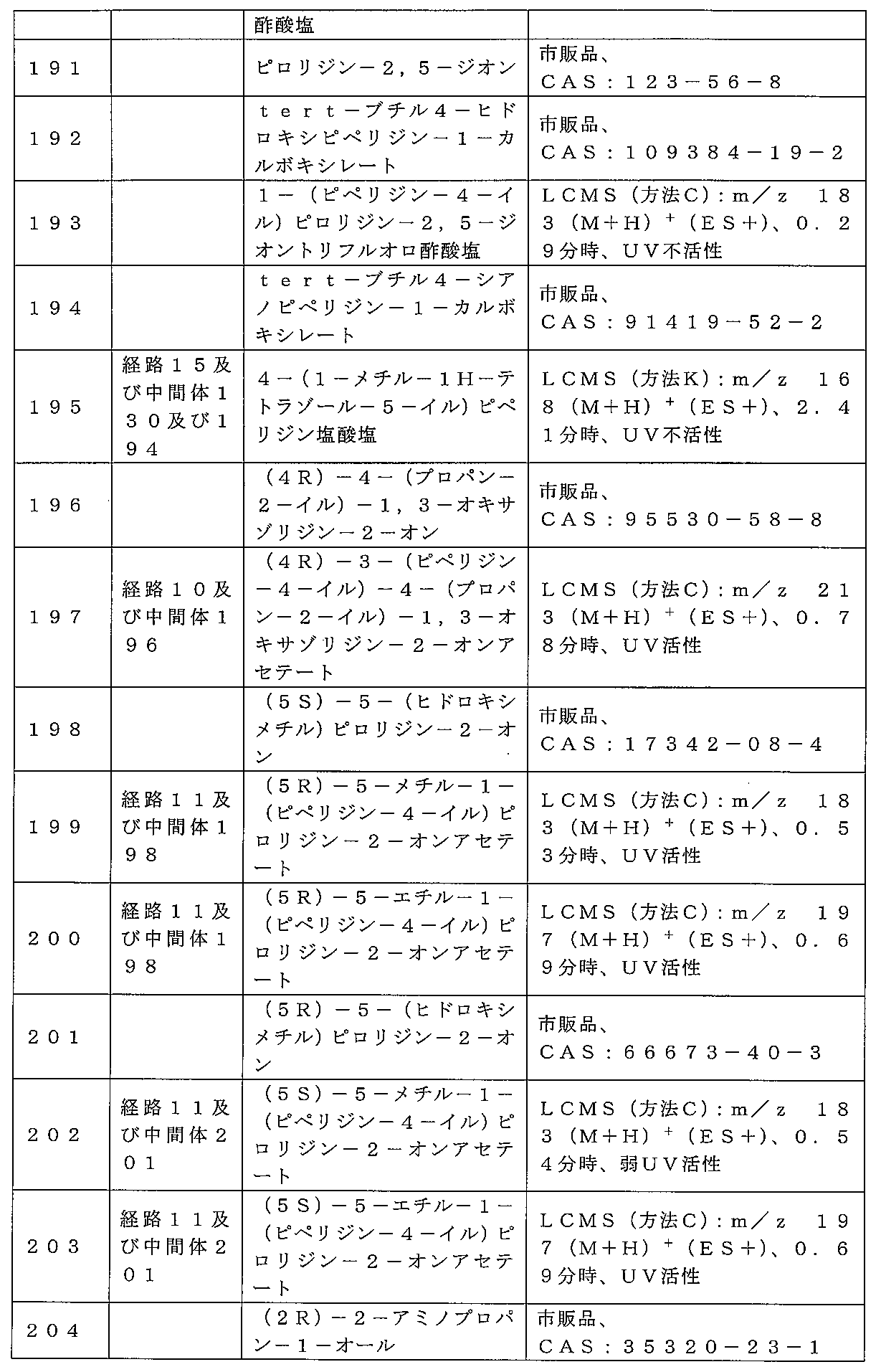


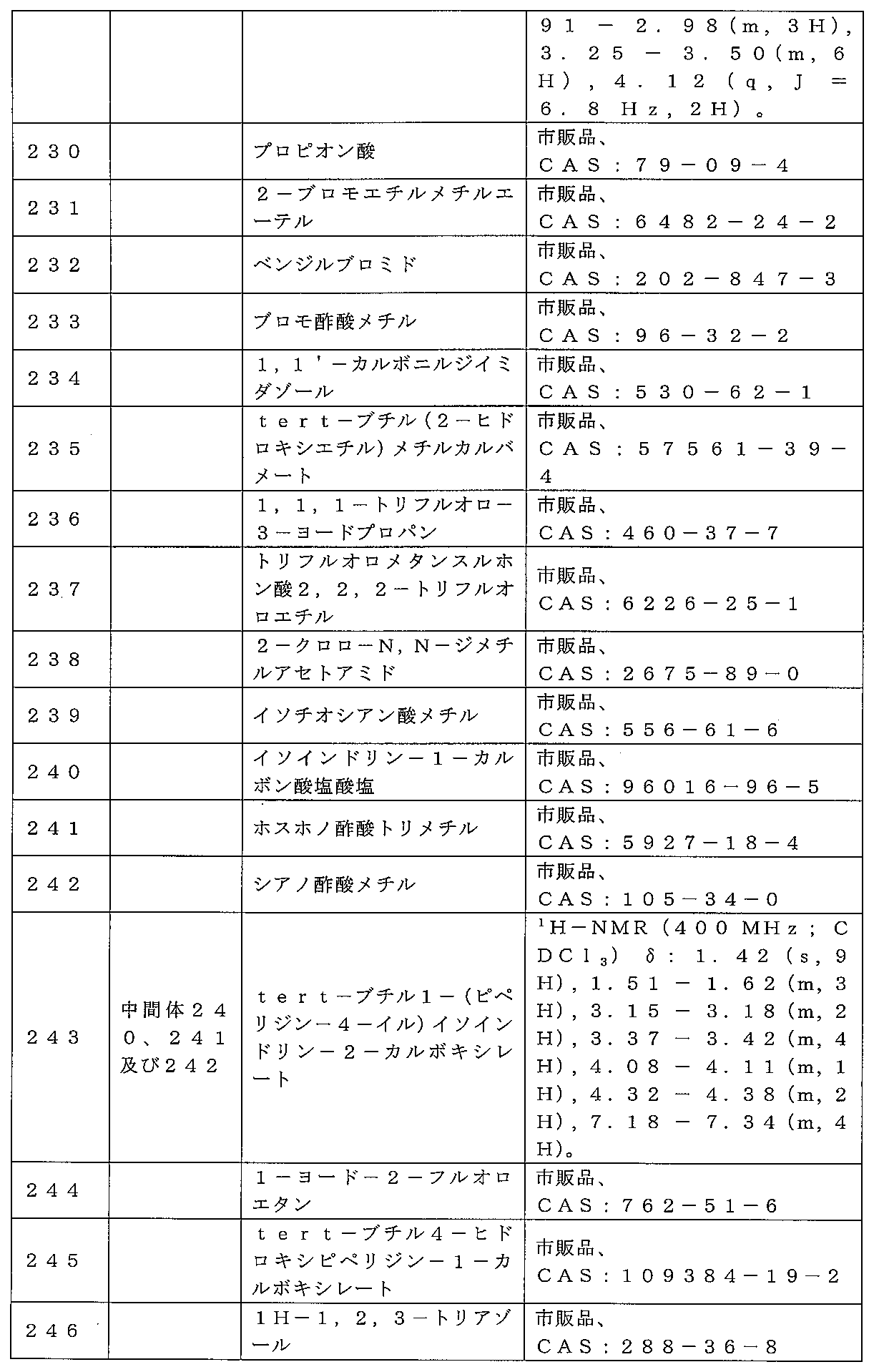





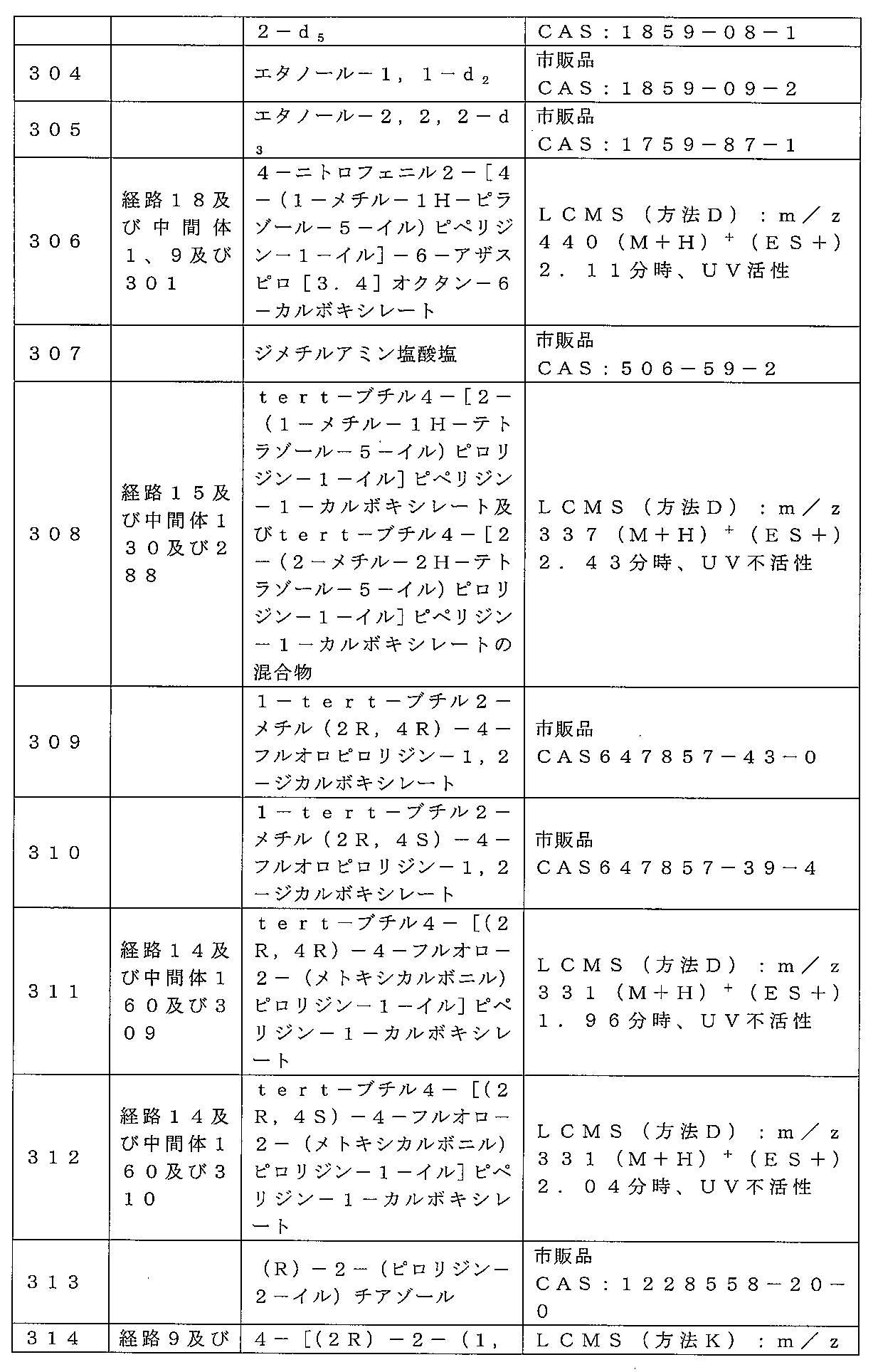




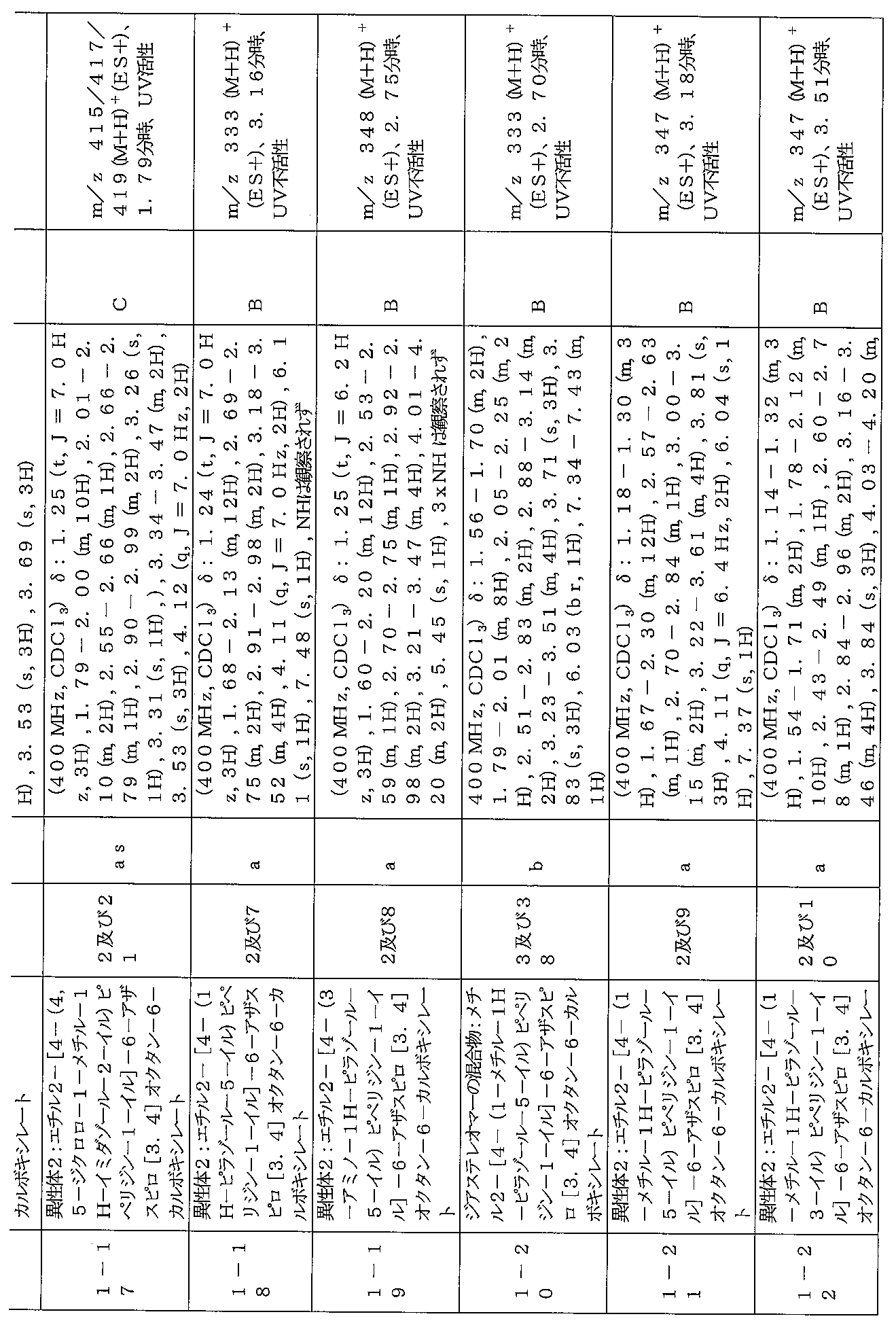
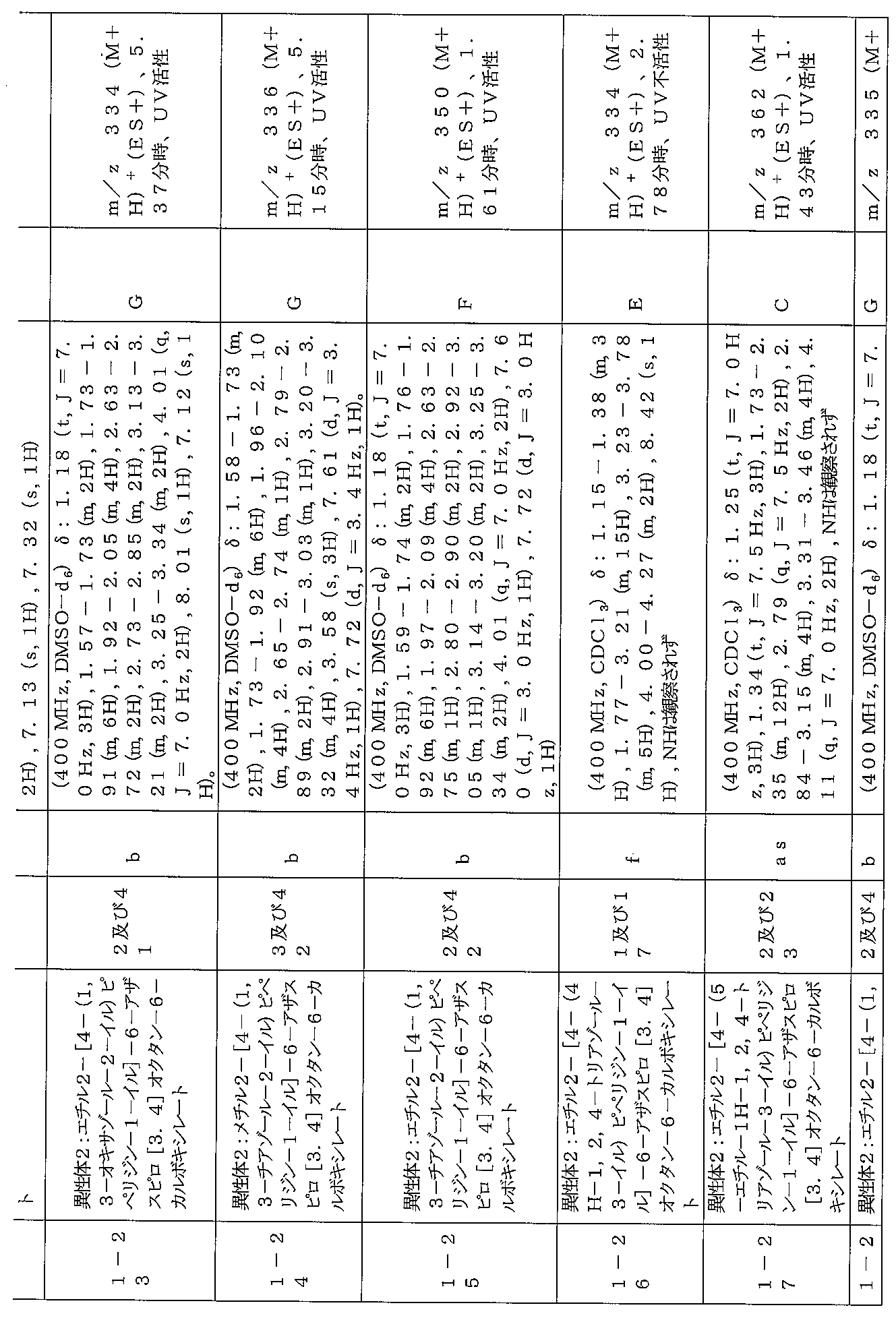


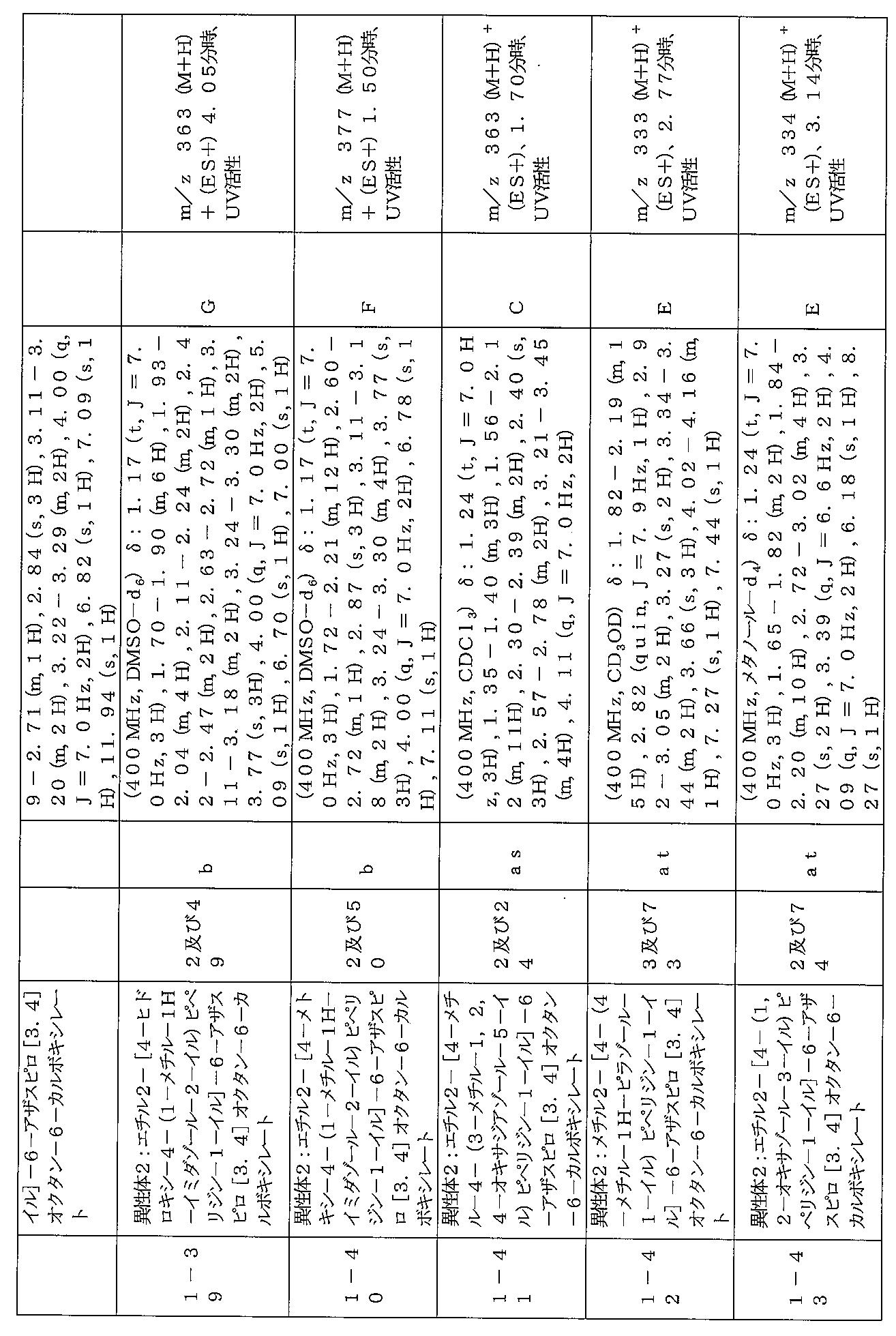







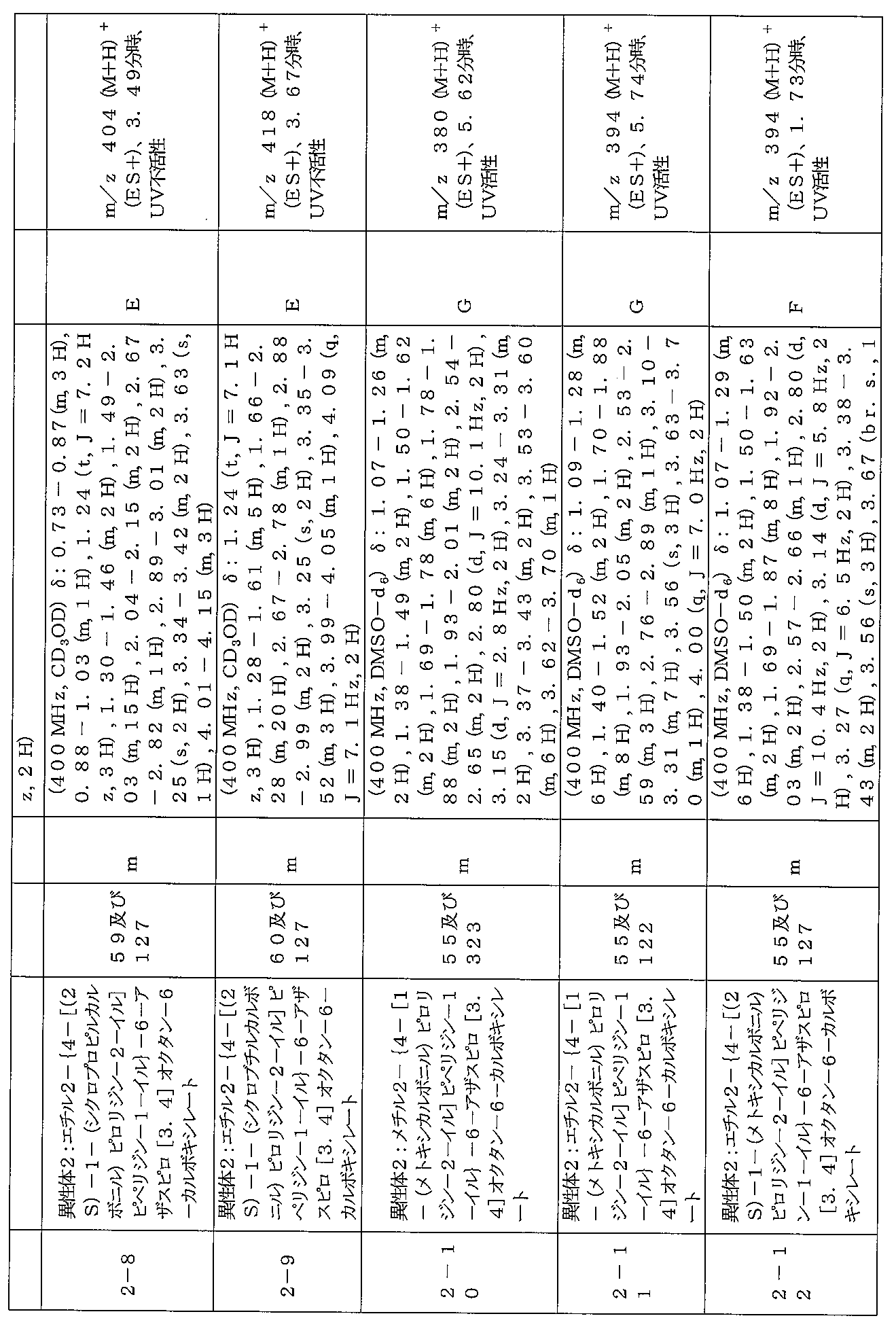











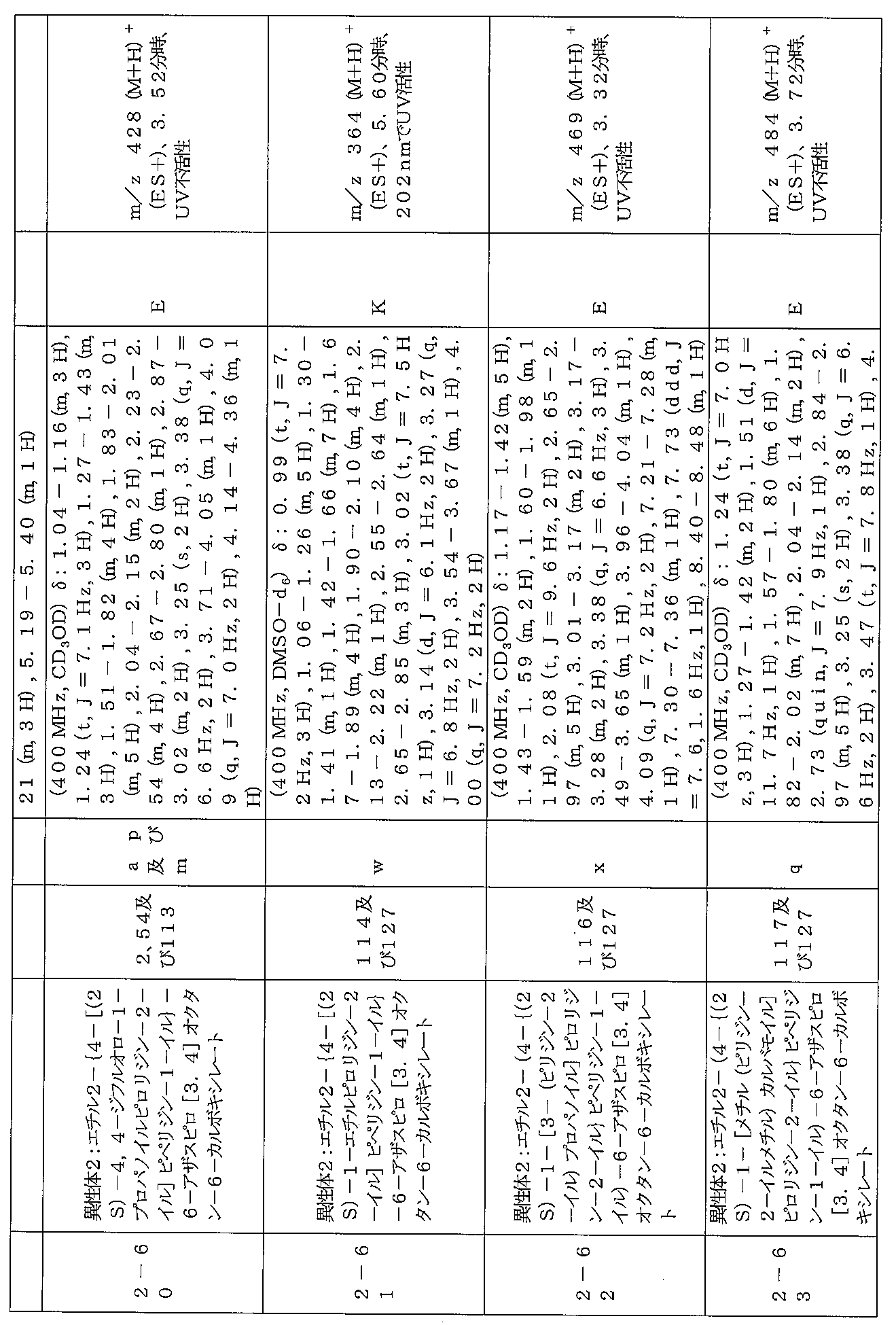

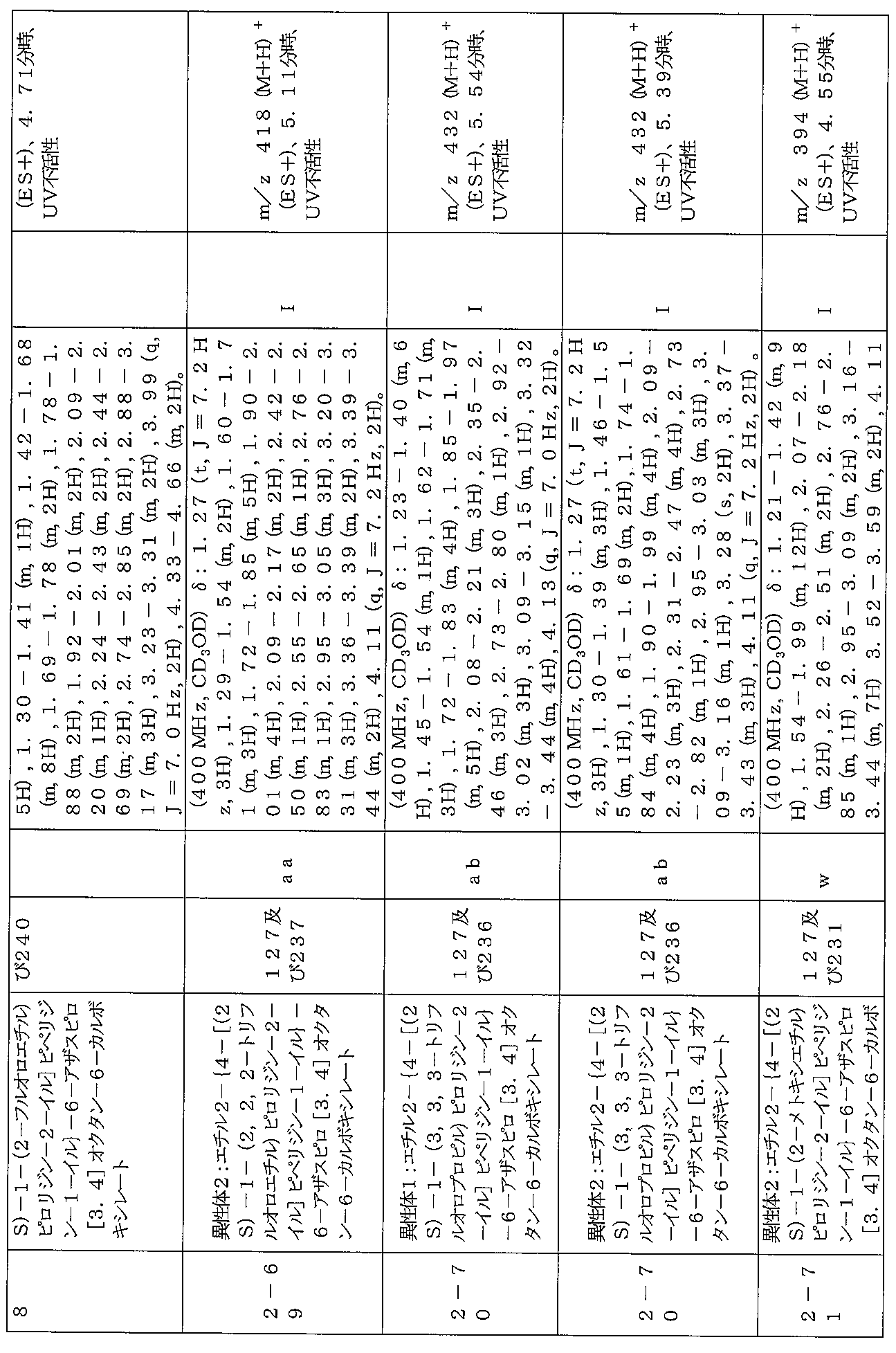
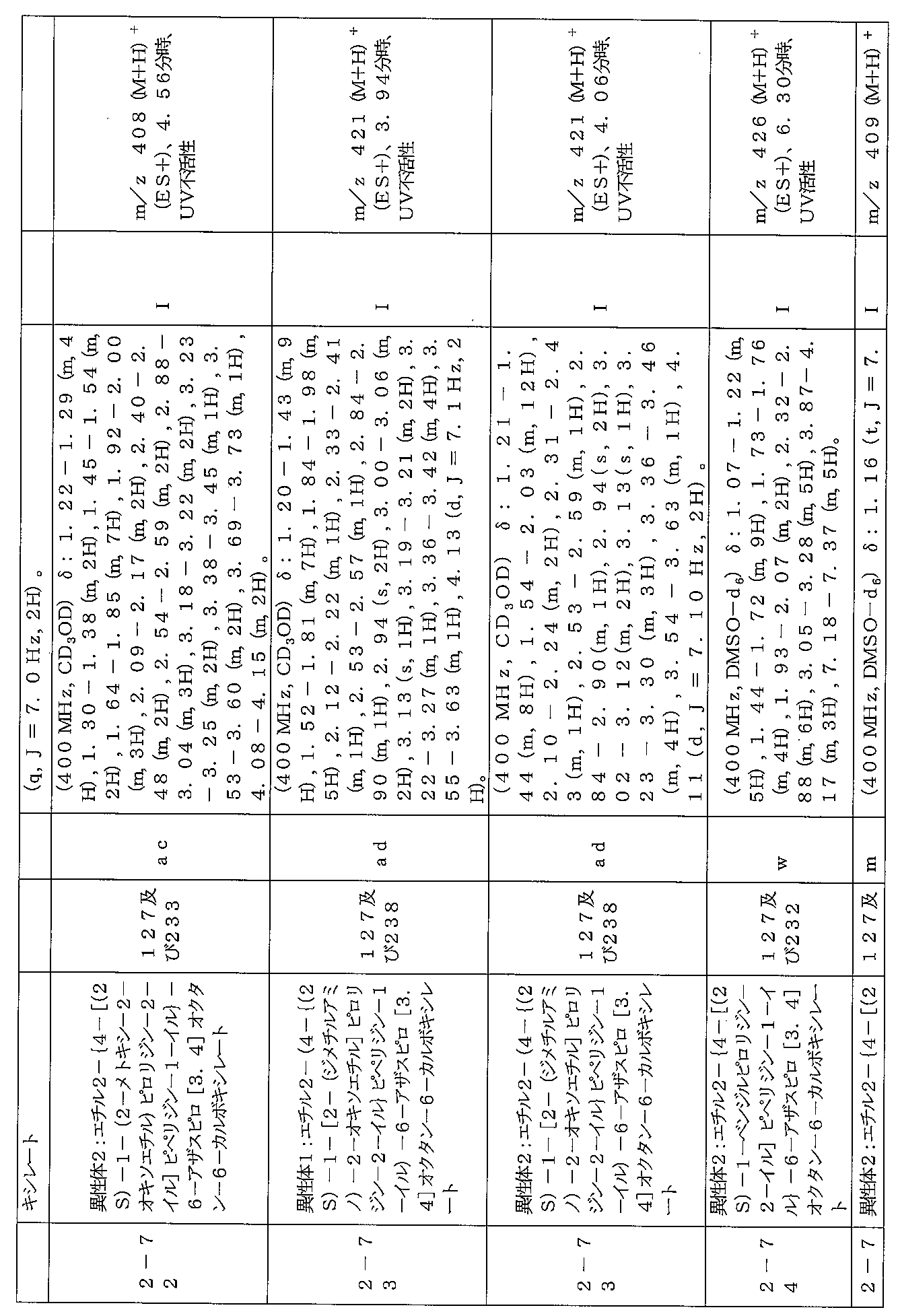




















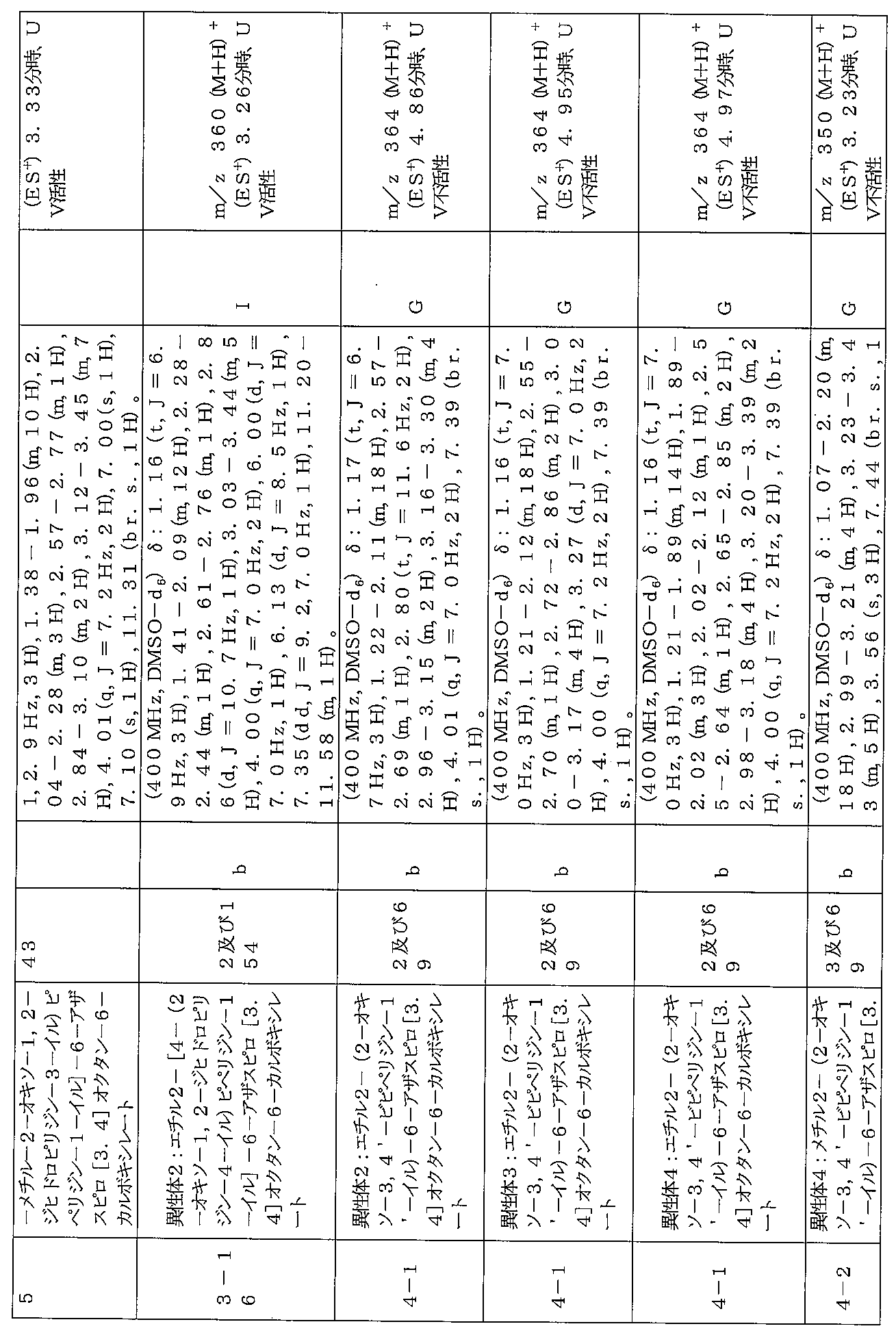

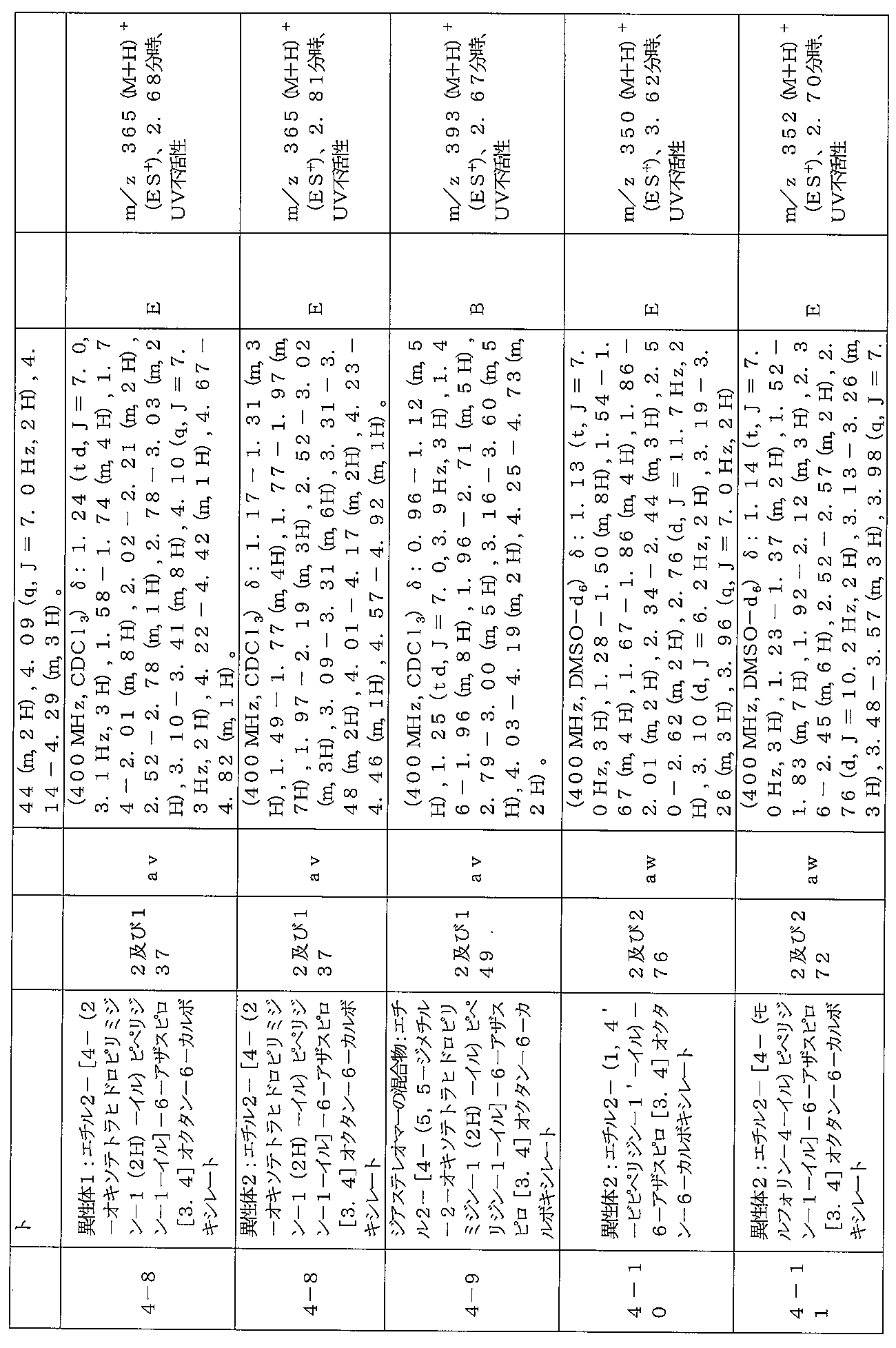



実施例A
ホスホ-ERK1/2アッセイ
Alphascreen Surefire ホスホ-ERK1/2アッセイ(Cro
uch & Osmond,Comb.Chem.High Throughput S
creen,2008)を用いて機能アッセイを行った。ERK1/2リン酸化は、Gq
/11及びGi/oタンパク質共役型受容体の両方の活性化の結果として下流で起きるた
め、M1、M3(Gq/11結合)及びM2、M4受容体(Gi/o結合)の評価には、
異なる受容体サブタイプに対する異なるアッセイ形式を使用するより非常に好適である。
ヒトムスカリンM1、M2、M3またはM4受容体を安定的に発現するCHO細胞を、M
EM-アルファ+10%透析FBS中、96ウェル組織培養プレートに蒔いた(25K/
ウェル)。ひとたび接着したら、細胞を一晩血清飢餓させた。アゴニスト刺激を5μLの
アゴニストを細胞に5分間(37℃)添加することにより行った。培地を除去し、50μ
Lの溶解緩衝液を加えた。15分後、4μLの試料を384ウェルプレートに移し、7μ
Lの検出混合物を加えた。プレートを暗所で穏やかに撹拌しながら2時間インキュベート
し、次いでPHERAstarプレートリーダーで読み取った。
した。
存在し、LCMS分析結果上のそれらの保持時間に基づいて割り当てた。大半の実施例で
は、異性体1は活性でない。活性異性体に関する分析データは表3に報告している。いく
つかの弱活性化合物に関するデータは表4に含まれて絶対立体化学の優先を強調する。

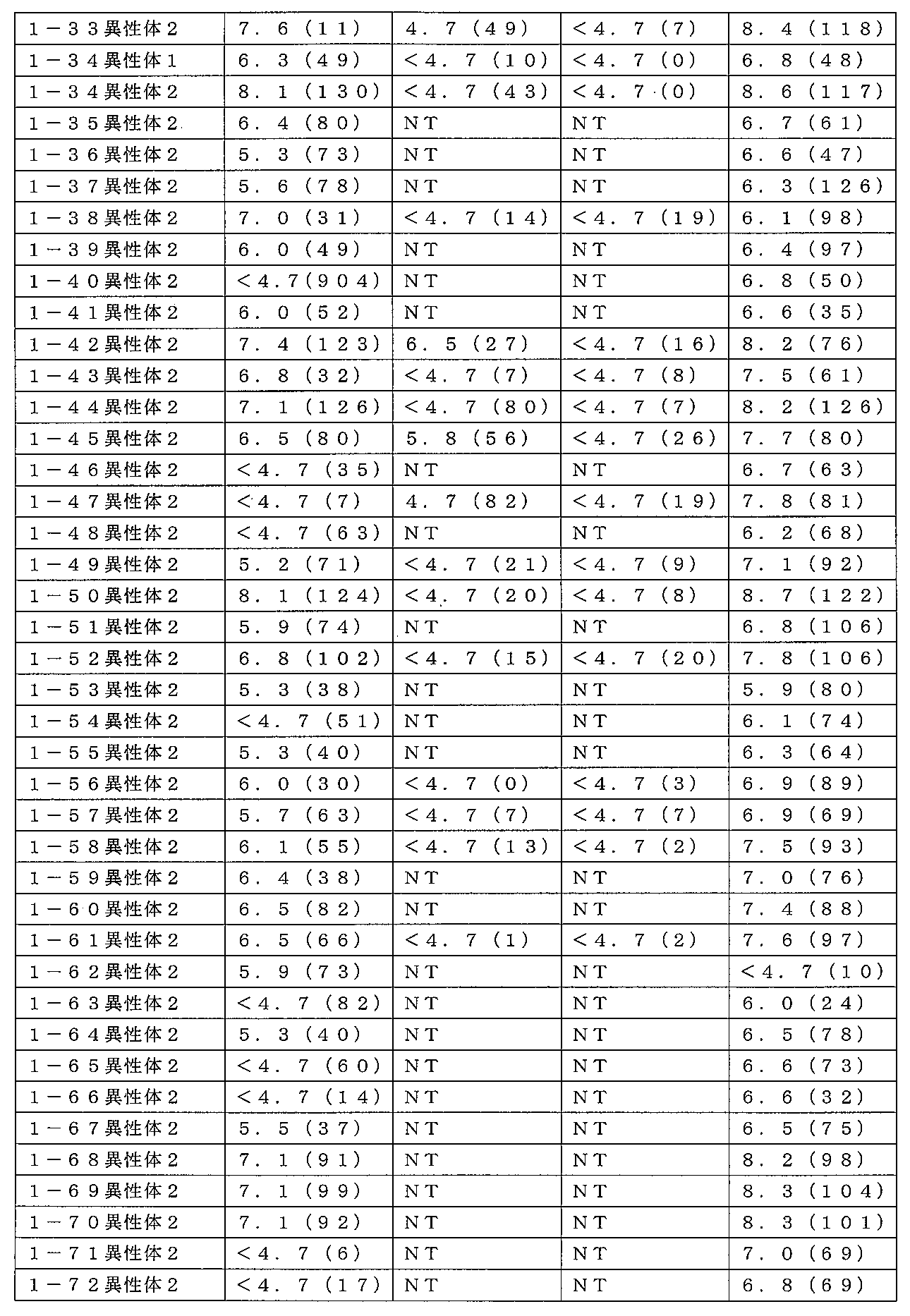



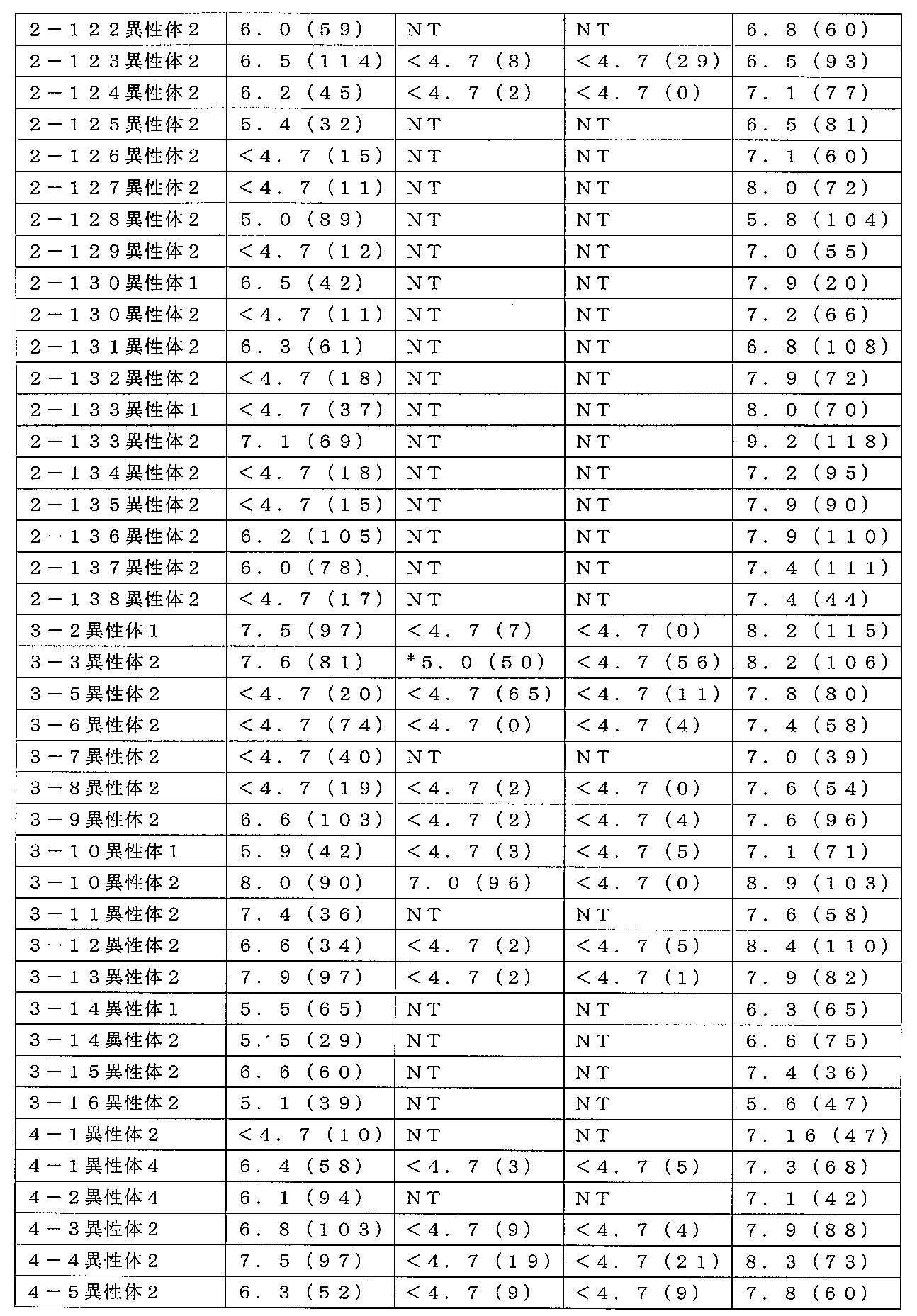

受動的回避
試験は、Foleyら、(2004)Neuropsychopharmacolog
yにより以前記載されたように実施した。受動的回避課題では訓練から6時間後にスコポ
ラミンを投与(1mg/kg、腹腔内)して動物に本パラダイムの健忘を起こさせた。強
制経口投与により訓練期間の90分前に3mg/kg、10mg/kg及び30mg/k
g(経口)の用量範囲の遊離塩基を投与して調査した。
で回復させ、おおよそのED50が約10mg/kg(経口)であることが見出された。
30mg/kgの作用は、陽性対照としたコリンエステラーゼ阻害剤ドネペジル(0.1
mg/kg、腹腔内)により生成された作用と同様であった(図1)。
ラットにおけるd-アンフェタミン誘発性運動亢進に及ぼす新規試験化合物及びキサノメ
リンの作用
本試験の目的は、ラットにおけるd-アンフェタミン誘発性運動亢進に及ぼす新規化合
物の作用を検査することである。統合失調症は、複合的な多機能疾患であり、単一の実験
手法によって完全に表すことはできない。抗精神病剤様行動を、d-アンフェタミンによ
り誘発される運動亢進(または自発運動の亢進)の阻害によりラットにおいて評価した。
この手法は、臨床的に適切であるドーパミン受容体アンタゴニストに感受性であり、それ
ゆえドーパミン作動性シグナル伝達に影響するムスカリンアゴニストと比較するのに適す
ると考えられる。d-アンフェタミン誘発性運動亢進を有意に低減させると事前に観察さ
れたある用量のキサノメリンを陽性対照として採用した。典型的に、統計学的解析には、
3元配置共分散分析またはロバスト回帰を必然的に含み、これは処置、日及びラックを因
子として、処置の前30分間の活性を共変数とし、その後に適切な多重比較試験を含んだ
。0.05未満のP値を統計学的に有意であるとみなし、それにしたがって全ての後続の
数字に記す。
2-17異性体2に関するデータを図2に示す。
医薬製剤
(i)錠剤製剤
式(1)、(1a)または(1b)の化合物を含有する錠剤組成物は、50mgの本化
合物を、希釈剤としての197mgのラクトース(BP)及び滑沢剤としての3mgのス
テアリン酸マグネシウムと混合し、既知の様式で圧縮して錠剤を形成することにより調製
する。
カプセル製剤は、100mgの式(1)、(1a)または(1b)の化合物を、100
mgのラクトース及び任意選択的に1重量%のステアリン酸マグネシウムと混合し、結果
得られた混合物を標準的な不透明硬質ゼラチンカプセルに充填することにより調製する。
前述の例は、本発明を説明するために示すものであり、本発明の範囲に何らかの限定を
加えるものと解釈してはならない。本発明の根底にある原理を逸脱することなく、上述し
、例に示した本発明の具体的な実施形態に多くの修正及び変更をなすことができることが
容易に明らかであるだろう。こうした修正及び変更の全ては、本出願に包含されると意図
される。
Claims (7)
-
またはエチル2-[4-(1-メチル-1H-ピラゾール-5-イル)ピペリジン-1-イル]-6-アザスピロ[3.4]オクタン-6-カルボキシレートによって表される化合物。 - 請求項1に記載の化合物の医薬的に許容され得る塩。
- 請求項1に記載の化合物または請求項2に記載の医薬的に許容され得る塩を含む医薬。
- 請求項1に記載の化合物または請求項2に記載の医薬的に許容され得る塩と、医薬的に許容され得る賦形剤とを含む医薬組成物。
- 請求項1に記載の化合物または請求項2に記載の医薬的に許容され得る塩を含む、統合失調症の処置のための医薬。
- 請求項1に記載の化合物または請求項2に記載の医薬的に許容され得る塩を含む、アルツハイマー病の処置のための医薬。
- 請求項1に記載の化合物または請求項2に記載の医薬的に許容され得る塩を含む、双極性障害の処置のための医薬。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023076025A JP7652833B2 (ja) | 2014-02-06 | 2023-05-02 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2023076024A JP7652832B2 (ja) | 2014-02-06 | 2023-05-02 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2025041135A JP2025085736A (ja) | 2014-02-06 | 2025-03-14 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB1402013.5 | 2014-02-06 | ||
| GBGB1402013.5A GB201402013D0 (en) | 2014-02-06 | 2014-02-06 | Pharmaceutical compounds |
| GBGB1416622.7A GB201416622D0 (en) | 2014-09-19 | 2014-09-19 | Pharmaceutical compounds |
| GB1416622.7 | 2014-09-19 | ||
| JP2020168341A JP2021006571A (ja) | 2014-02-06 | 2020-10-05 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020168341A Division JP2021006571A (ja) | 2014-02-06 | 2020-10-05 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
Related Child Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023076024A Division JP7652832B2 (ja) | 2014-02-06 | 2023-05-02 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2023076025A Division JP7652833B2 (ja) | 2014-02-06 | 2023-05-02 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2025041135A Division JP2025085736A (ja) | 2014-02-06 | 2025-03-14 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2023073504A JP2023073504A (ja) | 2023-05-25 |
| JP7652829B2 true JP7652829B2 (ja) | 2025-03-27 |
Family
ID=52465550
Family Applications (7)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016550483A Active JP6479029B2 (ja) | 2014-02-06 | 2015-02-06 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2019017086A Active JP6774513B2 (ja) | 2014-02-06 | 2019-02-01 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2020168341A Withdrawn JP2021006571A (ja) | 2014-02-06 | 2020-10-05 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2023060030A Active JP7652829B2 (ja) | 2014-02-06 | 2023-04-03 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2023076024A Active JP7652832B2 (ja) | 2014-02-06 | 2023-05-02 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2023076025A Active JP7652833B2 (ja) | 2014-02-06 | 2023-05-02 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2025041135A Pending JP2025085736A (ja) | 2014-02-06 | 2025-03-14 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
Family Applications Before (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016550483A Active JP6479029B2 (ja) | 2014-02-06 | 2015-02-06 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2019017086A Active JP6774513B2 (ja) | 2014-02-06 | 2019-02-01 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2020168341A Withdrawn JP2021006571A (ja) | 2014-02-06 | 2020-10-05 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023076024A Active JP7652832B2 (ja) | 2014-02-06 | 2023-05-02 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2023076025A Active JP7652833B2 (ja) | 2014-02-06 | 2023-05-02 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP2025041135A Pending JP2025085736A (ja) | 2014-02-06 | 2025-03-14 | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
Country Status (30)
| Country | Link |
|---|---|
| US (6) | US9670183B2 (ja) |
| EP (3) | EP4413985A3 (ja) |
| JP (7) | JP6479029B2 (ja) |
| KR (1) | KR102352388B1 (ja) |
| CN (2) | CN106458986B (ja) |
| AU (3) | AU2015213900B2 (ja) |
| BR (1) | BR112016017979B1 (ja) |
| CA (1) | CA2938169C (ja) |
| CL (1) | CL2016001983A1 (ja) |
| CY (1) | CY1120834T1 (ja) |
| DK (2) | DK3102568T3 (ja) |
| ES (2) | ES2688548T3 (ja) |
| FI (1) | FI3406609T3 (ja) |
| HR (2) | HRP20241145T1 (ja) |
| HU (1) | HUE068301T2 (ja) |
| IL (1) | IL247117B (ja) |
| LT (2) | LT3102568T (ja) |
| MX (2) | MX393637B (ja) |
| NZ (1) | NZ723103A (ja) |
| PH (1) | PH12016501570A1 (ja) |
| PL (2) | PL3406609T3 (ja) |
| PT (2) | PT3102568T (ja) |
| RS (2) | RS65894B1 (ja) |
| RU (1) | RU2685230C2 (ja) |
| SA (1) | SA516371614B1 (ja) |
| SG (1) | SG11201606269WA (ja) |
| SI (2) | SI3406609T1 (ja) |
| SM (1) | SMT202400446T1 (ja) |
| UA (1) | UA122121C2 (ja) |
| WO (1) | WO2015118342A1 (ja) |
Families Citing this family (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2012338581B2 (en) | 2011-11-18 | 2016-12-08 | Nxera Pharma Uk Limited | Muscarinic M1 receptor agonists |
| EP3421036B8 (en) | 2013-03-15 | 2020-12-30 | Verseon International Corporation | Multisubstituted aromatic compounds as serine protease inhibitors |
| HRP20241145T1 (hr) * | 2014-02-06 | 2024-11-22 | Nxera Pharma Uk Limited | Biciklički aza spojevi kao agonisti muskarinskih receptora |
| GB201404922D0 (en) * | 2014-03-19 | 2014-04-30 | Heptares Therapeutics Ltd | Pharmaceutical compounds |
| RU2017112739A (ru) | 2014-09-17 | 2018-10-17 | Версеон Корпорейшн | Пиразолил-замещенные пиридоновые соединения как ингибиторы сериновых протеаз |
| SG11201706411YA (en) | 2015-02-27 | 2017-09-28 | Verseon Corp | Substituted pyrazole compounds as serine protease inhibitors |
| GB201504675D0 (en) | 2015-03-19 | 2015-05-06 | Heptares Therapeutics Ltd | Pharmaceutical compounds |
| GB201513742D0 (en) * | 2015-08-03 | 2015-09-16 | Heptares Therapeutics Ltd | Muscarinic agonists |
| GB201513743D0 (en) * | 2015-08-03 | 2015-09-16 | Heptares Therapeutics Ltd | Muscarinic agonists |
| GB201513740D0 (en) * | 2015-08-03 | 2015-09-16 | Heptares Therapeutics Ltd | Muscarinic agonist |
| GB201519352D0 (en) * | 2015-11-02 | 2015-12-16 | Heptares Therapeutics Ltd | Pharmaceutical compounds |
| ES2789756T3 (es) | 2015-12-23 | 2020-10-26 | Merck Sharp & Dohme | Moduladores alostéricos de 6,7-dihidro-5H-pirrolo[3,4-b]piridin-5-ona del receptor de acetilcolina muscarínico M4 |
| WO2017107089A1 (en) | 2015-12-23 | 2017-06-29 | Merck Sharp & Dohme Corp. | 3- (1h-pyrazol-4-yl) pyridineallosteric modulators of the m4 muscarinic acetylcholine receptor |
| SG11201811712QA (en) | 2016-07-01 | 2019-01-30 | Pfizer | 5,7-dihydro-pyrrolo-pyridine derivatives for treating neurological and neurodegenerative diseases |
| GB201617454D0 (en) * | 2016-10-14 | 2016-11-30 | Heptares Therapeutics Limited | Pharmaceutical compounds |
| WO2018112842A1 (en) | 2016-12-22 | 2018-06-28 | Merck Sharp & Dohme Corp. | 6,6-fused heteroaryl piperidine ether allosteric modulators of m4 muscarinic acetylcholine receptor |
| WO2018112843A1 (en) | 2016-12-22 | 2018-06-28 | Merck Sharp & Dohme Corp. | Heteroaryl piperidine ether allosteric modulators of the m4 muscarinic acetylcholine receptor |
| GB201709652D0 (en) * | 2017-06-16 | 2017-08-02 | Heptares Therapeutics Ltd | Pharmaceutical compounds |
| TW201906838A (zh) | 2017-06-21 | 2019-02-16 | 日商第一三共股份有限公司 | Ep300/crebbp抑制劑 |
| WO2019000237A1 (en) | 2017-06-27 | 2019-01-03 | Merck Sharp & Dohme Corp. | ALLOSTERIC MODULATORS OF 3- (1H-PYRAZOL-4-YL) PYRIDINE FROM THE M4 ACETYLCHOLINE MUSCARINIC RECEPTOR |
| WO2019000238A1 (en) | 2017-06-27 | 2019-01-03 | Merck Sharp & Dohme Corp. | 5- (PYRIDIN-3-YL) OXAZOLE ALLOSTERIC MODULATORS OF M4 ACETYLCHOLINE MUSCARINIC RECEPTOR |
| WO2019000236A1 (en) | 2017-06-27 | 2019-01-03 | Merck Sharp & Dohme Corp. | ALLOSTERIC MODULATORS OF 3- (1H-PYRAZOL-4-YL) PYRIDINE FROM THE M4 ACETYLCHOLINE MUSCARINIC RECEPTOR |
| JP7166331B2 (ja) * | 2017-08-01 | 2022-11-07 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 中間体化合物及び方法 |
| TW201922758A (zh) | 2017-10-24 | 2019-06-16 | 美商歐樂根公司 | 烯胺及烯胺的非鏡像選擇性還原 |
| WO2019126559A1 (en) | 2017-12-20 | 2019-06-27 | Vanderbilt University | Antagonists of the muscarinic acetylcholine receptor m4 |
| WO2019183636A1 (en) | 2018-03-23 | 2019-09-26 | Pfizer Inc. | Piperazine azaspiro derivaves |
| GB201810239D0 (en) | 2018-06-22 | 2018-08-08 | Heptares Therapeutics Ltd | Pharmaceutical compounds |
| GB201810245D0 (en) * | 2018-06-22 | 2018-08-08 | Heptares Therapeutics Ltd | Pharmaceutical compounds |
| GB201819960D0 (en) | 2018-12-07 | 2019-01-23 | Heptares Therapeutics Ltd | Pharmaceutical compounds |
| GB201819961D0 (en) | 2018-12-07 | 2019-01-23 | Heptares Therapeutics Ltd | Pharmaceutical compounds |
| GB202020191D0 (en) | 2020-12-18 | 2021-02-03 | Heptares Therapeutics Ltd | Pharmaceutical compounds |
| PE20221453A1 (es) | 2019-10-09 | 2022-09-21 | Novartis Ag | Derivados de 2-azaspiro[3,4]octano como agonistas de m4 |
| AR120169A1 (es) * | 2019-10-09 | 2022-02-02 | Novartis Ag | Derivados de 2-azaespiro[3.4]octano como agonistas de m4 |
| EP4069367B1 (en) | 2019-12-06 | 2024-05-15 | Neurocrine Biosciences, Inc. | Muscarinic receptor 4 antagonists and methods of use |
| TWI889751B (zh) * | 2020-02-05 | 2025-07-11 | 美商紐羅克里生物科學有限公司 | 蕈毒受體4拮抗劑及使用方法 |
| US11912705B2 (en) * | 2020-08-20 | 2024-02-27 | The Corporation Of Mercer University | Cathinone derivatives, pharmaceutical formulations, and methods |
| EP4447953A1 (en) | 2021-12-13 | 2024-10-23 | Sage Therapeutics, Inc. | Combination of muscarinic receptor positive modulators and nmda positive allosteric modulators |
| US20250340531A1 (en) | 2022-08-04 | 2025-11-06 | Nxera Pharma Uk Limited | Crystalline forms and salts of a muscarinic receptor agonist |
| EP4587432A1 (en) * | 2022-09-16 | 2025-07-23 | Cerevel Therapeutics, LLC | M4 activators/modulators and uses thereof |
| CN120641409A (zh) * | 2023-01-12 | 2025-09-12 | 赛尔维治疗有限责任公司 | 作为m4活化剂/调节剂的螺衍生物及其用途 |
| CN120569376A (zh) | 2023-01-24 | 2025-08-29 | 纽罗克里生物科学有限公司 | 氘代化合物 |
| CN116496195A (zh) * | 2023-05-10 | 2023-07-28 | 南京优氟医药科技有限公司 | 一种(r)-4,4-二氟代吡咯烷-2-羧酸的制备方法 |
| WO2025030095A1 (en) | 2023-08-03 | 2025-02-06 | Neurocrine Biosciences, Inc. | Methods of synthesis |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6479029B2 (ja) | 2014-02-06 | 2019-03-06 | ヘプタレス セラピューティクス リミテッドHeptares Therapeutics Limited | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5852029A (en) | 1990-04-10 | 1998-12-22 | Israel Institute For Biological Research | Aza spiro compounds acting on the cholinergic system with muscarinic agonist activity |
| WO1999032489A1 (en) | 1997-12-23 | 1999-07-01 | Alcon Laboratories, Inc. | Muscarinic agents and use thereof to treat glaucoma, myopia and various other conditions |
| EP1386920A4 (en) * | 2001-04-20 | 2005-09-14 | Banyu Pharma Co Ltd | benzimidazolone derivatives |
| RU2006146985A (ru) * | 2004-05-28 | 2008-07-10 | Вертекс Фармасьютикалз Инкорпорейтед (Us) | Модуляторы мускариновых рецепторов |
| US7786308B2 (en) * | 2005-03-28 | 2010-08-31 | Vertex Pharmaceuticals Incorporated | Muscarinic modulators |
| WO2007076070A2 (en) | 2005-12-22 | 2007-07-05 | Vertex Pharmaceuticals Incorporated | Modulators of muscarinic receptors |
| WO2007100670A1 (en) * | 2006-02-22 | 2007-09-07 | Vertex Pharmaceuticals Incorporated | Spiro condensed piperidnes as modulators of muscarinic receptors |
| KR20080094964A (ko) | 2006-02-22 | 2008-10-27 | 버텍스 파마슈티칼스 인코포레이티드 | 무스카린성 수용체의 조절제 |
| JP2009529008A (ja) * | 2006-03-03 | 2009-08-13 | ノバルティス アクチエンゲゼルシャフト | N−ホルミルヒドロキシルアミン化合物 |
| GB0706188D0 (en) * | 2007-03-29 | 2007-05-09 | Glaxo Group Ltd | Compounds |
| US8119661B2 (en) * | 2007-09-11 | 2012-02-21 | Astrazeneca Ab | Piperidine derivatives and their use as muscarinic receptor modulators |
| WO2010049146A1 (en) | 2008-10-29 | 2010-05-06 | Grünenthal GmbH | Substituted spiroamines |
| FR2945531A1 (fr) | 2009-05-12 | 2010-11-19 | Sanofi Aventis | Derives de 7-aza-spiro°3,5!nonane-7-carboxylates, leur preparation et leur application en therapeutique |
| TW201211028A (en) * | 2010-08-10 | 2012-03-16 | Dainippon Sumitomo Pharma Co | Fused ring pyrrolidine derivatives |
| AU2012338581B2 (en) * | 2011-11-18 | 2016-12-08 | Nxera Pharma Uk Limited | Muscarinic M1 receptor agonists |
| CN104640851B (zh) * | 2012-09-18 | 2017-05-31 | 赫普泰雅治疗有限公司 | 作为毒蕈碱的m1受体激动剂的二环氮杂化合物 |
| CA2910873A1 (en) | 2013-04-30 | 2014-11-06 | Glaxosmithkline Intellectual Property (No.2) Limited | Enhancer of zeste homolog 2 inhibitors |
| PH12016501567B1 (en) | 2014-02-06 | 2023-12-06 | Riboscience Llc | 4`-difluoromethyl substituted nucleoside derivatives as inhibitors of influenza rna replication |
| US11078193B2 (en) | 2014-02-06 | 2021-08-03 | Janssen Sciences Ireland Uc | Sulphamoylpyrrolamide derivatives and the use thereof as medicaments for the treatment of hepatitis B |
| HUE062424T2 (hu) | 2014-09-19 | 2023-11-28 | Forma Therapeutics Inc | Piridin-2(1H)-on kinolinon származékok mint mutáns izocitrát dehidrogenáz inhibitorok |
| AU2015316796A1 (en) | 2014-09-19 | 2017-03-30 | Bayer Pharma Aktiengesellschaft | Benzyl substituted indazoles as Bub1 inhibitors |
| MX2017003621A (es) | 2014-09-19 | 2017-07-14 | Glaxosmithkline Ip Dev Ltd | Nuevos activadores de la guanilato ciclasa soluble y su uso. |
-
2015
- 2015-02-06 HR HRP20241145TT patent/HRP20241145T1/hr unknown
- 2015-02-06 RS RS20240960A patent/RS65894B1/sr unknown
- 2015-02-06 EP EP24177415.7A patent/EP4413985A3/en active Pending
- 2015-02-06 PT PT15703826T patent/PT3102568T/pt unknown
- 2015-02-06 AU AU2015213900A patent/AU2015213900B2/en active Active
- 2015-02-06 DK DK15703826.6T patent/DK3102568T3/en active
- 2015-02-06 PT PT181792581T patent/PT3406609T/pt unknown
- 2015-02-06 ES ES15703826.6T patent/ES2688548T3/es active Active
- 2015-02-06 UA UAA201609185A patent/UA122121C2/uk unknown
- 2015-02-06 PL PL18179258.1T patent/PL3406609T3/pl unknown
- 2015-02-06 JP JP2016550483A patent/JP6479029B2/ja active Active
- 2015-02-06 CA CA2938169A patent/CA2938169C/en active Active
- 2015-02-06 LT LTEP15703826.6T patent/LT3102568T/lt unknown
- 2015-02-06 MX MX2020001236A patent/MX393637B/es unknown
- 2015-02-06 SM SM20240446T patent/SMT202400446T1/it unknown
- 2015-02-06 BR BR112016017979-0A patent/BR112016017979B1/pt active IP Right Grant
- 2015-02-06 CN CN201580017993.6A patent/CN106458986B/zh active Active
- 2015-02-06 HR HRP20181499TT patent/HRP20181499T1/hr unknown
- 2015-02-06 SG SG11201606269WA patent/SG11201606269WA/en unknown
- 2015-02-06 NZ NZ723103A patent/NZ723103A/en unknown
- 2015-02-06 FI FIEP18179258.1T patent/FI3406609T3/fi active
- 2015-02-06 EP EP18179258.1A patent/EP3406609B1/en active Active
- 2015-02-06 HU HUE18179258A patent/HUE068301T2/hu unknown
- 2015-02-06 CN CN201910003375.6A patent/CN109851610B/zh active Active
- 2015-02-06 PL PL15703826T patent/PL3102568T3/pl unknown
- 2015-02-06 US US15/117,018 patent/US9670183B2/en active Active
- 2015-02-06 RS RS20181127A patent/RS57843B1/sr unknown
- 2015-02-06 EP EP15703826.6A patent/EP3102568B1/en active Active
- 2015-02-06 MX MX2016010322A patent/MX371529B/es active IP Right Grant
- 2015-02-06 SI SI201532034T patent/SI3406609T1/sl unknown
- 2015-02-06 RU RU2016133684A patent/RU2685230C2/ru active
- 2015-02-06 WO PCT/GB2015/050331 patent/WO2015118342A1/en not_active Ceased
- 2015-02-06 LT LTEP18179258.1T patent/LT3406609T/lt unknown
- 2015-02-06 SI SI201530399T patent/SI3102568T1/sl unknown
- 2015-02-06 ES ES18179258T patent/ES2986327T3/es active Active
- 2015-02-06 DK DK18179258.1T patent/DK3406609T3/da active
- 2015-02-06 KR KR1020167024572A patent/KR102352388B1/ko active Active
-
2016
- 2016-08-04 IL IL247117A patent/IL247117B/en active IP Right Grant
- 2016-08-04 SA SA516371614A patent/SA516371614B1/ar unknown
- 2016-08-05 CL CL2016001983A patent/CL2016001983A1/es unknown
- 2016-08-08 PH PH12016501570A patent/PH12016501570A1/en unknown
-
2017
- 2017-05-10 US US15/591,605 patent/US9926297B2/en active Active
-
2018
- 2018-02-22 US US15/902,537 patent/US10196380B2/en active Active
- 2018-09-27 CY CY181100996T patent/CY1120834T1/el unknown
- 2018-12-12 US US16/217,570 patent/US10385039B2/en active Active
-
2019
- 2019-02-01 JP JP2019017086A patent/JP6774513B2/ja active Active
- 2019-03-07 AU AU2019201579A patent/AU2019201579B2/en active Active
- 2019-07-01 US US16/459,179 patent/US10689368B2/en active Active
-
2020
- 2020-06-17 US US16/904,099 patent/US10961225B2/en active Active
- 2020-10-05 JP JP2020168341A patent/JP2021006571A/ja not_active Withdrawn
- 2020-11-26 AU AU2020277225A patent/AU2020277225A1/en not_active Abandoned
-
2023
- 2023-04-03 JP JP2023060030A patent/JP7652829B2/ja active Active
- 2023-05-02 JP JP2023076024A patent/JP7652832B2/ja active Active
- 2023-05-02 JP JP2023076025A patent/JP7652833B2/ja active Active
-
2025
- 2025-03-14 JP JP2025041135A patent/JP2025085736A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6479029B2 (ja) | 2014-02-06 | 2019-03-06 | ヘプタレス セラピューティクス リミテッドHeptares Therapeutics Limited | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
| JP6774513B2 (ja) | 2014-02-06 | 2020-10-28 | ヘプタレス セラピューティクス リミテッドHeptares Therapeutics Limited | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7652832B2 (ja) | ムスカリンm1受容体アゴニストとしての二環式アザ化合物 | |
| AU2016302047B2 (en) | Muscarinic agonists | |
| RU2811601C9 (ru) | Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | |
| RU2811601C1 (ru) | Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | |
| HK40114593A (en) | Pharmaceutical compounds | |
| HK40007500A (en) | Bicyclic aza compounds as muscarinic m1 receptor and/or m4 receptor agonists | |
| HK40007500B (zh) | 作为毒蕈碱m1和/或m4受体激动剂的双环氮杂化合物 | |
| HK1231473B (en) | Bicyclic aza compounds as muscarinic m1 receptor agonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230502 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20230502 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20240607 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20240906 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20241106 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20250307 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20250314 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7652829 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |