JP7607642B2 - Kras阻害剤である複素環式中間体に適用される合成方法 - Google Patents
Kras阻害剤である複素環式中間体に適用される合成方法 Download PDFInfo
- Publication number
- JP7607642B2 JP7607642B2 JP2022513928A JP2022513928A JP7607642B2 JP 7607642 B2 JP7607642 B2 JP 7607642B2 JP 2022513928 A JP2022513928 A JP 2022513928A JP 2022513928 A JP2022513928 A JP 2022513928A JP 7607642 B2 JP7607642 B2 JP 7607642B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- formula
- reaction
- organic solvent
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229940124785 KRAS inhibitor Drugs 0.000 title description 6
- 239000000543 intermediate Substances 0.000 title description 4
- 125000000623 heterocyclic group Chemical group 0.000 title description 2
- 238000010189 synthetic method Methods 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims description 69
- 238000000034 method Methods 0.000 claims description 49
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical group CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 37
- 239000003960 organic solvent Substances 0.000 claims description 37
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 36
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 28
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 27
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 27
- 239000000203 mixture Substances 0.000 claims description 24
- 238000006482 condensation reaction Methods 0.000 claims description 22
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 21
- CRWJEUDFKNYSBX-UHFFFAOYSA-N sodium;hypobromite Chemical compound [Na+].Br[O-] CRWJEUDFKNYSBX-UHFFFAOYSA-N 0.000 claims description 17
- 239000000243 solution Substances 0.000 claims description 17
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims description 15
- 239000011541 reaction mixture Substances 0.000 claims description 14
- 238000006460 hydrolysis reaction Methods 0.000 claims description 13
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 12
- 238000007363 ring formation reaction Methods 0.000 claims description 12
- 238000005577 Kumada cross-coupling reaction Methods 0.000 claims description 11
- 239000012074 organic phase Substances 0.000 claims description 11
- 238000010626 work up procedure Methods 0.000 claims description 11
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 10
- 229910021529 ammonia Inorganic materials 0.000 claims description 10
- 239000003054 catalyst Substances 0.000 claims description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 claims description 9
- UJKUJXVZLUTEAZ-UHFFFAOYSA-N n,n-dimethylformamide;dimethyl sulfate Chemical compound CN(C)C=O.COS(=O)(=O)OC UJKUJXVZLUTEAZ-UHFFFAOYSA-N 0.000 claims description 9
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 8
- 239000007818 Grignard reagent Substances 0.000 claims description 8
- -1 isopropyl Grignard reagent Chemical class 0.000 claims description 8
- 238000000746 purification Methods 0.000 claims description 8
- 238000006462 rearrangement reaction Methods 0.000 claims description 8
- 230000008569 process Effects 0.000 claims description 7
- 238000010898 silica gel chromatography Methods 0.000 claims description 7
- 239000002904 solvent Substances 0.000 claims description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 claims description 6
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 claims description 6
- 239000005695 Ammonium acetate Substances 0.000 claims description 6
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 claims description 6
- 239000002253 acid Substances 0.000 claims description 6
- 229940043376 ammonium acetate Drugs 0.000 claims description 6
- 235000019257 ammonium acetate Nutrition 0.000 claims description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 6
- 238000000605 extraction Methods 0.000 claims description 6
- 229910052742 iron Inorganic materials 0.000 claims description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 5
- 238000007167 Hofmann rearrangement reaction Methods 0.000 claims description 4
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 claims description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 claims description 4
- 239000008346 aqueous phase Substances 0.000 claims description 4
- 239000007864 aqueous solution Substances 0.000 claims description 4
- 239000003849 aromatic solvent Substances 0.000 claims description 4
- 230000007062 hydrolysis Effects 0.000 claims description 4
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 claims description 4
- 238000003756 stirring Methods 0.000 claims description 4
- 235000019270 ammonium chloride Nutrition 0.000 claims description 3
- 239000000706 filtrate Substances 0.000 claims description 3
- 238000001914 filtration Methods 0.000 claims description 3
- 230000003301 hydrolyzing effect Effects 0.000 claims description 3
- 238000005650 intramolecular substitution reaction Methods 0.000 claims description 3
- 239000000376 reactant Substances 0.000 claims description 3
- 229920006395 saturated elastomer Polymers 0.000 claims description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 claims description 3
- 125000003277 amino group Chemical group 0.000 claims description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 claims description 2
- 238000001035 drying Methods 0.000 claims description 2
- 125000001033 ether group Chemical group 0.000 claims description 2
- JBTWLSYIZRCDFO-UHFFFAOYSA-N ethyl methyl carbonate Chemical compound CCOC(=O)OC JBTWLSYIZRCDFO-UHFFFAOYSA-N 0.000 claims description 2
- 239000012065 filter cake Substances 0.000 claims description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 claims description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 claims description 2
- 238000005406 washing Methods 0.000 claims 4
- 238000000926 separation method Methods 0.000 claims 3
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 claims 2
- 125000003368 amide group Chemical group 0.000 claims 1
- LZKLAOYSENRNKR-LNTINUHCSA-N iron;(z)-4-oxoniumylidenepent-2-en-2-olate Chemical group [Fe].C\C(O)=C\C(C)=O.C\C(O)=C\C(C)=O.C\C(O)=C\C(C)=O LZKLAOYSENRNKR-LNTINUHCSA-N 0.000 claims 1
- 150000003839 salts Chemical class 0.000 claims 1
- 239000000126 substance Substances 0.000 claims 1
- 238000006243 chemical reaction Methods 0.000 description 41
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 24
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 description 20
- HTSGKJQDMSTCGS-UHFFFAOYSA-N 1,4-bis(4-chlorophenyl)-2-(4-methylphenyl)sulfonylbutane-1,4-dione Chemical compound C1=CC(C)=CC=C1S(=O)(=O)C(C(=O)C=1C=CC(Cl)=CC=1)CC(=O)C1=CC=C(Cl)C=C1 HTSGKJQDMSTCGS-UHFFFAOYSA-N 0.000 description 17
- 235000019439 ethyl acetate Nutrition 0.000 description 14
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 11
- 238000003786 synthesis reaction Methods 0.000 description 11
- FCSKOFQQCWLGMV-UHFFFAOYSA-N 5-{5-[2-chloro-4-(4,5-dihydro-1,3-oxazol-2-yl)phenoxy]pentyl}-3-methylisoxazole Chemical compound O1N=C(C)C=C1CCCCCOC1=CC=C(C=2OCCN=2)C=C1Cl FCSKOFQQCWLGMV-UHFFFAOYSA-N 0.000 description 9
- FKLJPTJMIBLJAV-UHFFFAOYSA-N Compound IV Chemical compound O1N=C(C)C=C1CCCCCCCOC1=CC=C(C=2OCCN=2)C=C1 FKLJPTJMIBLJAV-UHFFFAOYSA-N 0.000 description 9
- 238000005481 NMR spectroscopy Methods 0.000 description 9
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- 238000004440 column chromatography Methods 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 7
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 230000035484 reaction time Effects 0.000 description 6
- 238000001308 synthesis method Methods 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 238000004809 thin layer chromatography Methods 0.000 description 6
- 238000005859 coupling reaction Methods 0.000 description 4
- 239000003480 eluent Substances 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 238000006880 cross-coupling reaction Methods 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- AQBLLJNPHDIAPN-LNTINUHCSA-K iron(3+);(z)-4-oxopent-2-en-2-olate Chemical group [Fe+3].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O AQBLLJNPHDIAPN-LNTINUHCSA-K 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- NXQKSXLFSAEQCZ-SFHVURJKSA-N sotorasib Chemical compound FC1=CC2=C(N(C(N=C2N2[C@H](CN(CC2)C(C=C)=O)C)=O)C=2C(=NC=CC=2C)C(C)C)N=C1C1=C(C=CC=C1O)F NXQKSXLFSAEQCZ-SFHVURJKSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- VXPOAPWLMVAUCK-UHFFFAOYSA-N 4-methyl-2-propan-2-ylpyridin-3-amine Chemical compound C(C)(C)C1=NC=CC(=C1N)C VXPOAPWLMVAUCK-UHFFFAOYSA-N 0.000 description 1
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 1
- 102100030708 GTPase KRas Human genes 0.000 description 1
- 101000584612 Homo sapiens GTPase KRas Proteins 0.000 description 1
- 206010069755 K-ras gene mutation Diseases 0.000 description 1
- 102000008300 Mutant Proteins Human genes 0.000 description 1
- 108010021466 Mutant Proteins Proteins 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 238000006114 decarboxylation reaction Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 150000002611 lead compounds Chemical class 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- RYEXTBOQKFUPOE-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].CC[CH2-] RYEXTBOQKFUPOE-UHFFFAOYSA-M 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 239000012450 pharmaceutical intermediate Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 238000013341 scale-up Methods 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/06—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom containing only hydrogen and carbon atoms in addition to the ring nitrogen atom
- C07D213/127—Preparation from compounds containing pyridine rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C243/00—Compounds containing chains of nitrogen atoms singly-bound to each other, e.g. hydrazines, triazanes
- C07C243/24—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids
- C07C243/26—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids with acylating carboxyl groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C243/30—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids with acylating carboxyl groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of an unsaturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/01—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms
- C07C255/06—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms of an acyclic and unsaturated carbon skeleton
- C07C255/07—Mononitriles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/01—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms
- C07C255/30—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms containing cyano groups and singly-bound nitrogen atoms, not being further bound to other hetero atoms, bound to the same unsaturated acyclic carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/06—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom containing only hydrogen and carbon atoms in addition to the ring nitrogen atom
- C07D213/08—Preparation by ring-closure
- C07D213/09—Preparation by ring-closure involving the use of ammonia, amines, amine salts, or nitriles
- C07D213/12—Preparation by ring-closure involving the use of ammonia, amines, amine salts, or nitriles from unsaturated compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
- C07D213/82—Amides; Imides in position 3
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pyridine Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
本願は、2019年9月11日に出願した中国特許出願第2019108588762号の優先権を主張する。本願は、上記中国特許出願の全文を参照する。


式V:
式VI:


式V:
式VI:


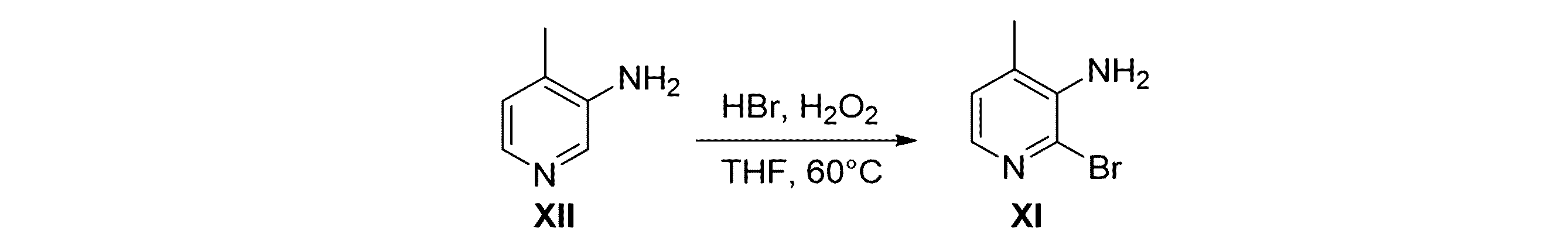
化合物IIはホフマン転位反応条件下で反応して化合物Iを生成する。ホフマン転位反応条件はNaOBrと水との混合系を含む。

酸は濃硫酸であり、例えば98%濃硫酸である。
酸と化合物IIIとの質量比は2.25:1である。
加水分解温度は105℃である。

有機溶媒は、テトラヒドロフラン等のエーテル溶媒である。
有機溶媒の量は、反応に影響を与えない限り、制限する必要はない。例えば、化合物IVの有機溶媒に対する質量対体積比は、0.03~0.04g/mLである。
N-メチル-ピロリドンと化合物IVとの質量比は9.5:1である。
鉄触媒は、鉄(III)アセチルアセトネートである。鉄触媒と化合物IVとの質量比は1:(2~2.2)である。
イソプロピルグリニャール試薬はイソプロピルマグネシウムクロリドである。イソプロピルグリニャール試薬は、溶液形態で使用することができる。溶液の溶媒は、エーテル溶媒等の有機溶媒である。例えば、1.0~2.5mol/Lテトラヒドロフラン溶液である。
イソプロピルグリニャール試薬と化合物IVとの体積対質量比は、(2.6~4.4):1である。
熊田カップリングの反応温度は0~10℃である。

有機溶媒は、メタノール等のアルコール溶媒である。
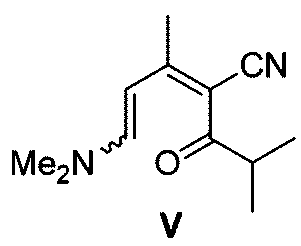
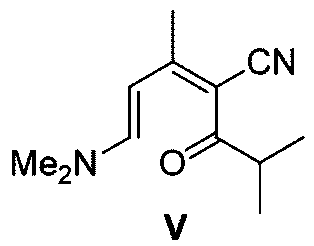
有機溶媒の量は、反応に影響を与えない限り、制限する必要はない。例えば、化合物Vと有機溶媒との質量対体積比は、0.03~0.04g/mLである。
アンモニア源は、アンモニア、酢酸アンモニウム、塩化アンモニウムであることができ;例えば、アンモニア源が酢酸アンモニウムである場合、酢酸アンモニウムと化合物Vとの質量比は、(3~4):1であり、例えば、3.67:1である。
環化反応の反応温度は0~50℃であり、例えば20~30℃である。

工程(1)における有機溶媒の量は、反応に影響を与えない限り、制限する必要はない。例えば、化合物VIIと有機溶媒との質量対体積比は、0.10~0.20g/mLであることができる。
工程(1)において、アルカリ性酸化アルミニウムと化合物VIIとの質量比は、(2~4):1であり、例えば2.3:1である。
工程(1)における縮合反応の反応温度は、0~50℃、例えば20~30℃である。
工程(2)において、N,N-ジメチルホルムアミドジメチルサルフェート縮合物(DMF-DMS)と化合物VIIとの質量比は、(2-4):1であり、例えば3:1である。
工程(2)において、無水酢酸と化合物VIIとの質量比は、(0.1~0.4):1であり、例えば0.18:1である。
工程(2)において、トリエチルアミンと化合物VIIとの体積対質量比は、1~2mL/gであり、例えば1.5mL/gである。
工程(2)における縮合反応の反応温度は、0~50℃、例えば0~10℃から20~30℃である。


1.原材料が容易に入手でき、安価である。
2.操作が単純であり、プロセスがスケールアップ及び工業生産に容易である。
3.より安全で環境にやさしい。

1H NMR(400MHz,DMSO-d6)δ8.63(d,J=5.1Hz,1H),7.35(dd,J=5.0,0.8Hz,1H),3.41(m,J=6.8Hz,1H),2.50(s,J=0.6Hz,3H),1.27(d,J=6.8Hz,6H)。

1H NMR(400MHz,DMSO-d6)δ8.38(d,J=4.9Hz,1H),7.90(s,1H),7.63(s,1H),7.08(d,J=4.7Hz,1H),3.14(m,1H),2.26(s,3H),1.20(d,J=6.8Hz,6H)。

1H NMR(400MHz,DMSO-d6)δ7.67(d,J=4.7Hz,1H),6.78(d,J=4.6,0.8Hz,1H),4.72(s,2H),3.19(m,1H),2.08(s,3H),1.15(d,J=6.7Hz,6H)。

1H NMR(400MHz,DMSO-d6)δ7.96(d,J=12.8Hz,1H),7.55(d,J=12.8Hz,1H),3.31(d,J=2.2Hz,1H),3.25(s,3H),3.13(m,1H),2.97(s,3H),2.24(s,3H),1.01(dd,J=6.8,3.6Hz,7H)。

1H NMR(400MHz,DMSO-d6)δ8.63(d,J=5.1Hz,1H),7.35(dd,J=5.0,0.8Hz,1H),3.41(m,1H),2.50(s,J=0.6Hz,3H),1.27(d,J=6.8Hz,6H)。
Claims (23)
- 式IIの化合物から式Iの化合物を調製する方法であって、ホフマン転位反応により、前記式IIの化合物からアミド基をアミノ基に変換して前記式Iの化合物を作製すること:

- 前記ホフマン転位反応の条件が、NaOBrと水との混合系を含む、請求項1に記載の方法。
- 前記NaOBrが、NaOBrの水溶液の形態である、請求項2に記載の方法。
- 前記NaOBrは、NaOHと水との混合物にBr2を加えて前記NaOBrの水溶液を得る工程によって調製され;NaOHとBr2との質量比を0.9:1とし;前記NaOHと水の混合物中におけるNaOHと水との質量比を0.3:1とし;前記式IIの化合物とBr2との質量比を1:1とし;前記式IIの化合物とNaOBrとのモル比は、(0.8~1.2):1である
請求項2に記載の方法。 - 前記NaOBrと水との混合系は、NaOBrを前記式IIの化合物と水との混合物に添加する工程で得られ;前記添加は滴下であり;前記添加は-10℃~10℃の温度で行われ;前記式IIの化合物と水との混合物において、前記式IIの化合物と前記水との質量比は、0.5:1である
請求項2に記載の方法。 - 前記混合系において、水の総量と前記式IIの化合物との質量比は7:1である、請求項2に記載の方法。
- 前記ホフマン転位反応は10~100℃の温度で行われる、請求項1に記載の方法。
- 前記方法は、ワークアップのプロセスをさらに含み、前記ワークアップのプロセスは、前記ホフマン転位反応が完了し反応混合物をもたらした後、抽出のために前記反応混合物に有機溶媒を添加し有機相を得て、前記有機相を洗浄、濃縮、分離、及び精製する工程を含み;前記有機溶媒は酢酸エチルであり;前記洗浄は、洗浄溶媒として飽和ブラインを用いて行われ;前記分離及び精製はシリカゲルカラムクロマトグラフィーで行われる
請求項1に記載の方法。 - 式IIIの化合物を酸で加水分解して前記式IIの化合物を得る工程:

- 前記酸が硫酸であり、前記酸と前記式IIIの化合物との質量比は2.25:1であり、及び、前記加水分解は、105℃で行われる、請求項9に記載の方法。
- 前記加水分解後、前記加水分解で得られた加水分解反応混合物に水を添加してpHを10~11に調整し;濾過し、乾燥して前記式IIの化合物を得る工程をさらに含み;50%水酸化ナトリウム水溶液を添加することによって前記pHを調整し;前記式IIの化合物を前記ホフマン転位反応に直接使用する
請求項9に記載の方法。 - N-メチルピロリドン及び鉄触媒の存在下で、有機溶媒中で式IVの化合物とイソプロピルグリニャール試薬との熊田カップリング反応を行い、前記式IIIの化合物を得る工程:

- 前記有機溶媒はエーテル溶媒であり;前記式IVの化合物と前記有機溶媒との質量-体積比は0.03~0.04g/mLであり;前記N-メチルピロリドンと前記式IVの化合物との体積対質量比は9.5:1であり;前記鉄触媒は鉄トリアセチルアセトナートであり;前記イソプロピルグリニャール試薬はエーテル溶媒中のイソプロピルマグネシウムクロリド溶液であり;前記イソプロピルグリニャール試薬と前記式IVの化合物との体積対質量比は、(2.6~4.4):1であり;及び、前記熊田カップリング反応は0~10℃で行われる
請求項12に記載の方法。 - 前記熊田カップリング反応の完了後、前記熊田カップリング反応から得られる熊田カップリング反応混合物に、クエン酸水溶液、飽和重炭酸ナトリウム水溶液、及び有機溶媒を順に添加して有機相を得て;前記有機相を抽出、濃縮、分離、及び精製して前記式IIIの化合物を得ることをさらに含み;抽出のための前記有機溶媒は、エステル溶媒であり;前記分離及び精製は、シリカゲルカラムクロマトグラフィーで行われる
請求項9に記載の方法。 - 式Vの化合物に環化反応を施して前記式IIIの化合物を得ること:

- 前記環化反応が、前記式Vの化合物とアンモニア源とを反応させ、次いで分子内置換反応を行って前記式IIIの化合物を形成することを含む、請求項15に記載の方法。
- 前記アンモニア源は、アンモニアガス、酢酸アンモニウム、及び塩化アンモニウムからなる群から選択される、請求項16に記載の方法。
- 式VIの化合物:

- 前記縮合反応は、反応物としてN,N-ジメチルホルムアミドジメチルアセタール(DMF-DMA)またはN,N-ジメチルホルムアミドジメチルサルフェート付加物(DMF-DMS)の存在下で行われる、請求項18に記載の方法。
- 式VIIの化合物にアセトンとの前記縮合反応を施して式VIの化合物を得ること:

- 工程(1):有機溶媒中、塩基性アルミナの存在下で、式VIIの化合物をアセトンと縮合反応させて式VIの化合物を得ること;
工程(2):有機溶媒中、無水酢酸及びトリエチルアミンの存在下で、前記式VIの化合物をN,N-ジメチルホルムアミドジメチルサルフェート縮合物と縮合反応させて、前記式Vの化合物を得ること:

をさらに含む、請求項15に記載の方法。 - 工程(1)における前記有機溶媒が芳香族溶媒であり;
及び/または、工程(1)において、前記式VIIの化合物と前記有機溶媒との質量-体積比は、0.10~0.20g/mLであり;
及び/または、工程(1)において、前記塩基性アルミナと前記式VIIの化合物との質量比は、(2~4):1であり;
及び/又は、工程(1)において、前記縮合反応の温度は0~50℃であり;
及び/または、工程(1)はまた、ワークアッププロセスを含み、前記ワークアッププロセスは、前記縮合反応が完了したとき、濾過し、前記濾過したケークを前記有機溶媒で洗浄し、得られた濾液を合わせて工程(2)で直接使用する工程を含み;
及び/または、工程(2)において、前記有機溶媒は芳香族溶媒であり;
及び/または、工程(2)において、前記N,N-ジメチルホルムアミドジメチルサルフェート縮合物と前記式VIIの化合物との質量比は、(2-4):1であり;
及び/または、工程(2)において、前記無水酢酸と前記式VIIの化合物との質量比は、(0.1~0.4):1であり;
及び/または、工程(2)において、前記トリエチルアミンと前記式VIIの化合物との質量対体積比は、1~2mL/gであり;
及び/または、工程(2)において、前記縮合反応の温度は0~50℃であり;
及び/または、工程(2)は、工程(1)で得られた前記式VIの化合物に無水酢酸及びトリエチルアミンを順次加えて、物質の混合物中でN,N-ジメチルホルムアミドジメチルサルフェートと縮合させ、前記縮合反応を行う工程であり;無水酢酸を添加する温度は0~10℃であり;トリエチルアミンを添加する温度は20~30℃であり;
及び/または、工程(2)はまた、ワークアッププロセスを含み、前記ワークアッププロセスは、縮合反応が完了したとき、水を添加して撹拌し、層を分離し、有機溶媒で水相を抽出し、前記有機相を合わせて濃縮し、分離し、精製して、前記式Vの化合物を得る工程を含み;抽出のための前記有機溶媒は、ジクロロメタンであることができ;前記分離及び精製は、シリカゲルカラムクロマトグラフィーであることができる
請求項17に記載の方法。 - 式II:

式V:

式VI:

それらの塩
を有する化合物であって、
式V中、ジメチルアミノ基に結合する二重結合は、Z、E、またはZとEとの混合物の形態である、化合物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201910858876 | 2019-09-11 | ||
| CN201910858876.2 | 2019-09-11 | ||
| PCT/CN2020/114560 WO2021047603A1 (zh) | 2019-09-11 | 2020-09-10 | 一种应用于kras抑制剂类药物杂环中间体的合成方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2022547007A JP2022547007A (ja) | 2022-11-10 |
| JP7607642B2 true JP7607642B2 (ja) | 2024-12-27 |
Family
ID=74866574
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022513928A Active JP7607642B2 (ja) | 2019-09-11 | 2020-09-10 | Kras阻害剤である複素環式中間体に適用される合成方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20220259150A1 (ja) |
| EP (1) | EP4029853A4 (ja) |
| JP (1) | JP7607642B2 (ja) |
| CN (1) | CN112479993A (ja) |
| WO (1) | WO2021047603A1 (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12473281B2 (en) | 2019-11-14 | 2025-11-18 | Amgen Inc. | Synthesis of KRAS G12C inhibitor compound |
| CN113666865B (zh) * | 2021-09-07 | 2023-05-26 | 杭州科耀医药科技有限公司 | 一种2-异丙基-3-氨基-4-甲基吡啶的合成方法 |
| MX2024010828A (es) | 2022-03-07 | 2024-09-17 | Amgen Inc | Procedimiento para preparar 4-metil-2-propan-2-il-piridin-3-carbon itrilo. |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019031476A (ja) | 2017-05-22 | 2019-02-28 | アムジエン・インコーポレーテツド | Kras g12c阻害剤及びその使用方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK0551459T3 (da) * | 1991-06-11 | 1997-04-07 | Boehringer Ingelheim Pharma | Fremgangsmåde til fremstilling af 3-amino-2-chlor-4-alkylpyridiner |
| CA2671336A1 (en) * | 2006-12-14 | 2008-06-19 | Novartis Ag | Epothilone analogues modified at positions c12-c13 as anticancer drugs |
| CN101965334A (zh) * | 2007-10-24 | 2011-02-02 | 比艾尔-坡特拉有限公司 | 新的前体 |
| EP2807157A1 (en) * | 2012-01-27 | 2014-12-03 | Novartis AG | 5-membered heteroarylcarboxamide derivatives as plasma kallikrein inhibitors |
| WO2016118586A1 (en) * | 2015-01-20 | 2016-07-28 | Virginia Commonwealth University | Lowcost, high yield synthesis of nevirapine |
| CN110546149B (zh) * | 2017-04-21 | 2022-11-11 | 深圳信立泰药业股份有限公司 | 作为pcsk9抑制剂的哌啶类化合物 |
| US12473281B2 (en) * | 2019-11-14 | 2025-11-18 | Amgen Inc. | Synthesis of KRAS G12C inhibitor compound |
-
2020
- 2020-09-10 EP EP20863253.9A patent/EP4029853A4/en active Pending
- 2020-09-10 US US17/627,892 patent/US20220259150A1/en active Pending
- 2020-09-10 WO PCT/CN2020/114560 patent/WO2021047603A1/zh not_active Ceased
- 2020-09-10 CN CN202010948760.0A patent/CN112479993A/zh active Pending
- 2020-09-10 JP JP2022513928A patent/JP7607642B2/ja active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019031476A (ja) | 2017-05-22 | 2019-02-28 | アムジエン・インコーポレーテツド | Kras g12c阻害剤及びその使用方法 |
Non-Patent Citations (1)
| Title |
|---|
| Sakamoto, Takao et al.,Condensed heteroaromatic ring systems. VI. Synthesis of indoles and pyrrolopyridines from o-nitroarylacetylenes,Chemical & Pharmaceutical Bulletin,1986年,34(6),2362-8 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2022547007A (ja) | 2022-11-10 |
| US20220259150A1 (en) | 2022-08-18 |
| EP4029853A1 (en) | 2022-07-20 |
| CN112479993A (zh) | 2021-03-12 |
| WO2021047603A1 (zh) | 2021-03-18 |
| EP4029853A4 (en) | 2023-03-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7607642B2 (ja) | Kras阻害剤である複素環式中間体に適用される合成方法 | |
| AU2018383864B2 (en) | Method for synthesis of Roxadustat and intermediate compounds thereof | |
| JP2023532317A (ja) | カンプトテシン誘導体を合成するための中間体及びその製造方法並びに用途 | |
| EP4212509B1 (en) | Method for preparing water-soluble magnolol derivative and honokiol derivative, methods for preparing intermediates of water-soluble magnolol derivative and honokiol derivative, and related monohydroxy protected intermediate | |
| CN108424388A (zh) | 一种慢性贫血药物的制备方法 | |
| CN111646922B (zh) | 一种2-(4-溴-2-氰基-6-氟苯基)乙酸的合成方法 | |
| CN114805314A (zh) | 一种恩赛特韦的合成方法 | |
| JPH02180868A (ja) | 3,5‐ジメチル‐4‐メトキシピリジン誘導体の製造方法ならびにそのための新規な中間生成物 | |
| CN111704573B (zh) | 一种雷贝拉唑氯化物及其中间体的制备方法 | |
| CN112300073B (zh) | 一种异喹啉衍生物的制备方法 | |
| RU2198176C2 (ru) | Способы получения производных азостероидов, производное азостероид-имидазолида, способ его получения | |
| CN112500337B (zh) | 3-溴-6-氯吡啶甲酰胺的合成方法 | |
| CN107641080B (zh) | 一种含螺环结构的二氢萘酮类衍生物及其制备方法 | |
| CN108358866A (zh) | 一种非布司他中间体的制备方法及其在制备非布司他中的应用 | |
| CN111100042B (zh) | 一种2-甲氧基-5-磺酰胺基苯甲酸的制备方法 | |
| CN113354573A (zh) | 一种可规模化生产α,α,α-三联吡啶的方法 | |
| AU2020233169A1 (en) | Process for preparing 4-amino-5-methylpyridone | |
| CN115477635B (zh) | 一种拉司米地坦的制备方法 | |
| JP2003212861A (ja) | ピリミジニルアルコール誘導体の製造方法及びその合成中間体 | |
| CN105017219B (zh) | 一种p53‑MDM2结合抑制剂二羟基异喹啉衍生物的合成方法 | |
| JP7751914B2 (ja) | B型肝炎ウイルスヌクレオカプシド阻害剤の製造方法 | |
| CN112679431A (zh) | 一种制备异喹啉酮类化合物的方法 | |
| CN110790657B (zh) | 7-甲基胡桃醌的合成方法 | |
| CN107602439B (zh) | 一种制备海洋生物碱Baculiferin-L中间体的合成方法 | |
| WO2024103319A1 (zh) | 一种合成(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3,1,0]己基-2-羧酸甲酯盐酸盐的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220518 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20230814 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20240711 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20240806 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20240926 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20241210 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20241217 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7607642 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |