JP7166399B2 - タピナロフを調製するためのプロセス - Google Patents
タピナロフを調製するためのプロセス Download PDFInfo
- Publication number
- JP7166399B2 JP7166399B2 JP2021113606A JP2021113606A JP7166399B2 JP 7166399 B2 JP7166399 B2 JP 7166399B2 JP 2021113606 A JP2021113606 A JP 2021113606A JP 2021113606 A JP2021113606 A JP 2021113606A JP 7166399 B2 JP7166399 B2 JP 7166399B2
- Authority
- JP
- Japan
- Prior art keywords
- formula
- compound
- salt
- iii
- solvate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C39/00—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a six-membered aromatic ring
- C07C39/02—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a six-membered aromatic ring monocyclic with no unsaturation outside the aromatic ring
- C07C39/08—Dihydroxy benzenes; Alkylated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/045—Hydroxy compounds, e.g. alcohols; Salts thereof, e.g. alcoholates
- A61K31/05—Phenols
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C37/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring
- C07C37/001—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring by modification in a side chain
- C07C37/002—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring by modification in a side chain by transformation of a functional group, e.g. oxo, carboxyl
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C37/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring
- C07C37/06—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring by conversion of non-aromatic six-membered rings or of such rings formed in situ into aromatic six-membered rings, e.g. by dehydrogenation
- C07C37/07—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring by conversion of non-aromatic six-membered rings or of such rings formed in situ into aromatic six-membered rings, e.g. by dehydrogenation with simultaneous reduction of C=O group in that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C39/00—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a six-membered aromatic ring
- C07C39/205—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a six-membered aromatic ring polycyclic, containing only six-membered aromatic rings as cyclic parts with unsaturation outside the rings
- C07C39/21—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a six-membered aromatic ring polycyclic, containing only six-membered aromatic rings as cyclic parts with unsaturation outside the rings with at least one hydroxy group on a non-condensed ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/51—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition
- C07C45/54—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition of compounds containing doubly bound oxygen atoms, e.g. esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/63—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by introduction of halogen; by substitution of halogen atoms by other halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C45/72—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction of compounds containing >C = O groups with the same or other compounds containing >C = O groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/20—Unsaturated compounds containing keto groups bound to acyclic carbon atoms
- C07C49/213—Unsaturated compounds containing keto groups bound to acyclic carbon atoms containing six-membered aromatic rings
- C07C49/217—Unsaturated compounds containing keto groups bound to acyclic carbon atoms containing six-membered aromatic rings having unsaturation outside the aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/657—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings
- C07C49/683—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings having unsaturation outside the aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/687—Unsaturated compounds containing a keto groups being part of a ring containing halogen
- C07C49/697—Unsaturated compounds containing a keto groups being part of a ring containing halogen containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C50/00—Quinones
- C07C50/24—Quinones containing halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/09—Preparation of carboxylic acids or their salts, halides or anhydrides from carboxylic acid esters or lactones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
- C07C51/377—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups
- C07C51/38—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups by decarboxylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/235—Saturated compounds containing more than one carboxyl group
- C07C59/347—Saturated compounds containing more than one carboxyl group containing keto groups
- C07C59/353—Saturated compounds containing more than one carboxyl group containing keto groups containing rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/76—Unsaturated compounds containing keto groups
- C07C59/84—Unsaturated compounds containing keto groups containing six membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/03—Preparation of carboxylic acid esters by reacting an ester group with a hydroxy group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/08—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides with the hydroxy or O-metal group of organic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/28—Preparation of carboxylic acid esters by modifying the hydroxylic moiety of the ester, such modification not being an introduction of an ester group
- C07C67/293—Preparation of carboxylic acid esters by modifying the hydroxylic moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/67—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of saturated acids
- C07C69/716—Esters of keto-carboxylic acids or aldehydo-carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Catalysts (AREA)
- Compounds Of Unknown Constitution (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
本出願は、2017年11月10日に提出された「PROCESS」と題された米国仮特許出願第62/584,192号の利益および優先権を主張し、その内容は参照によりその全体が本明細書に組み込まれる。
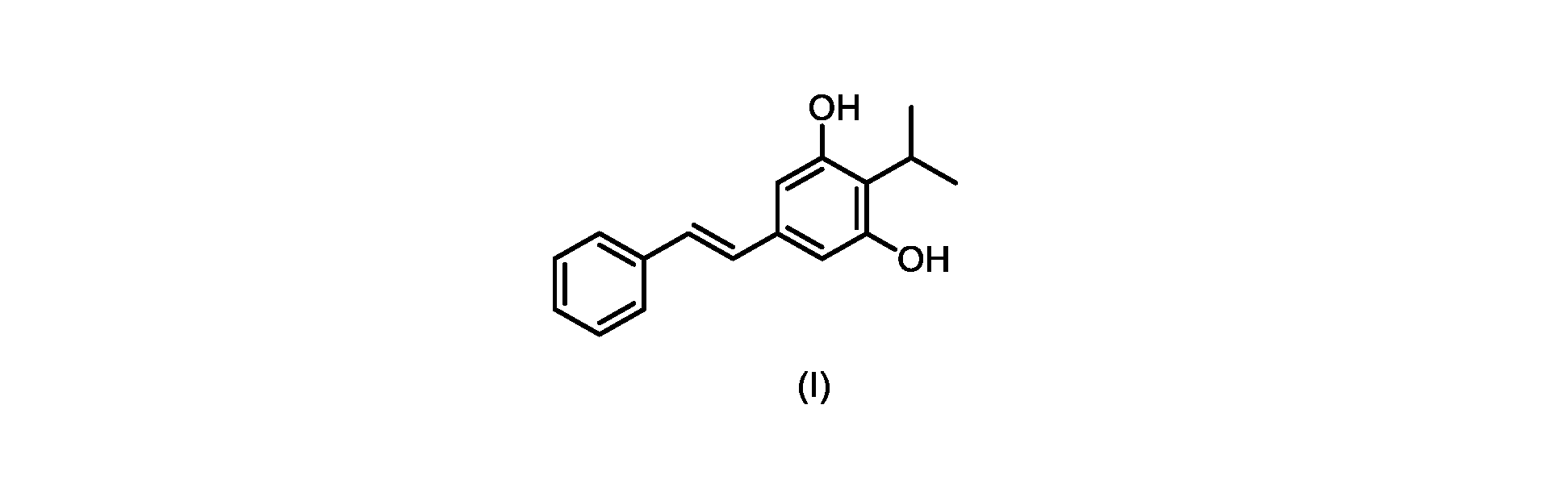
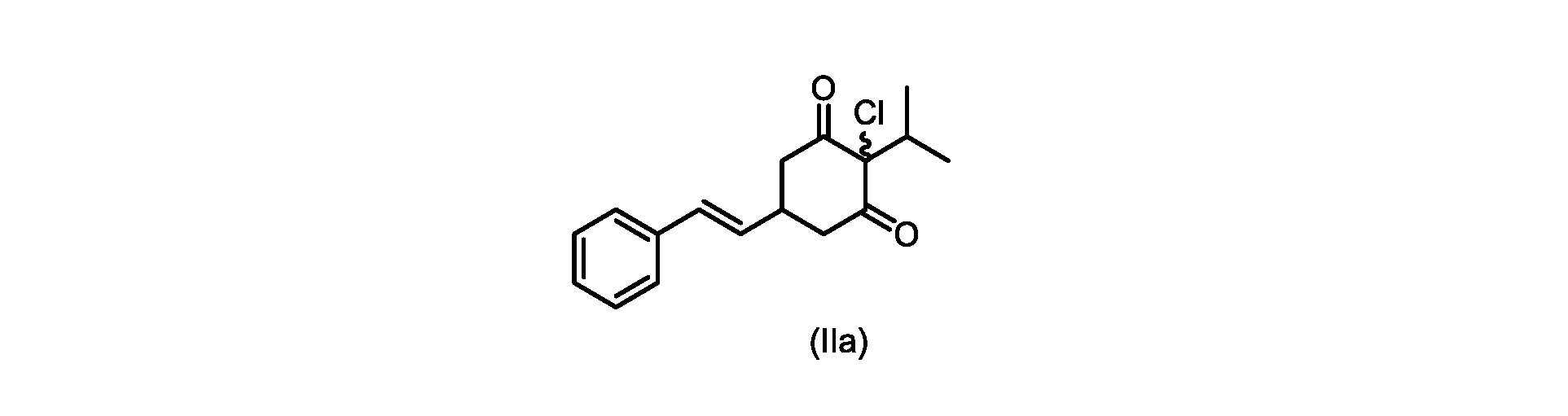
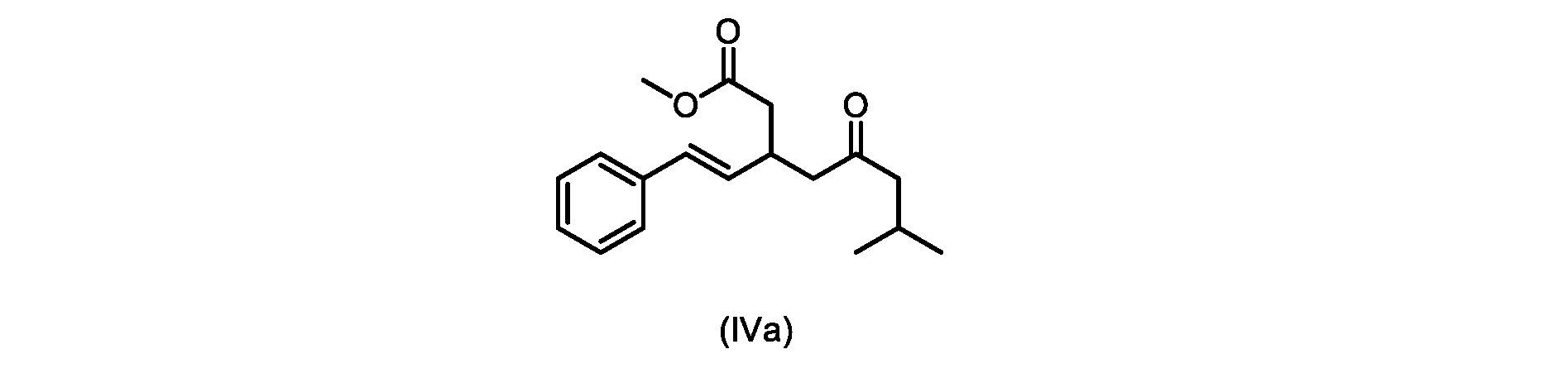
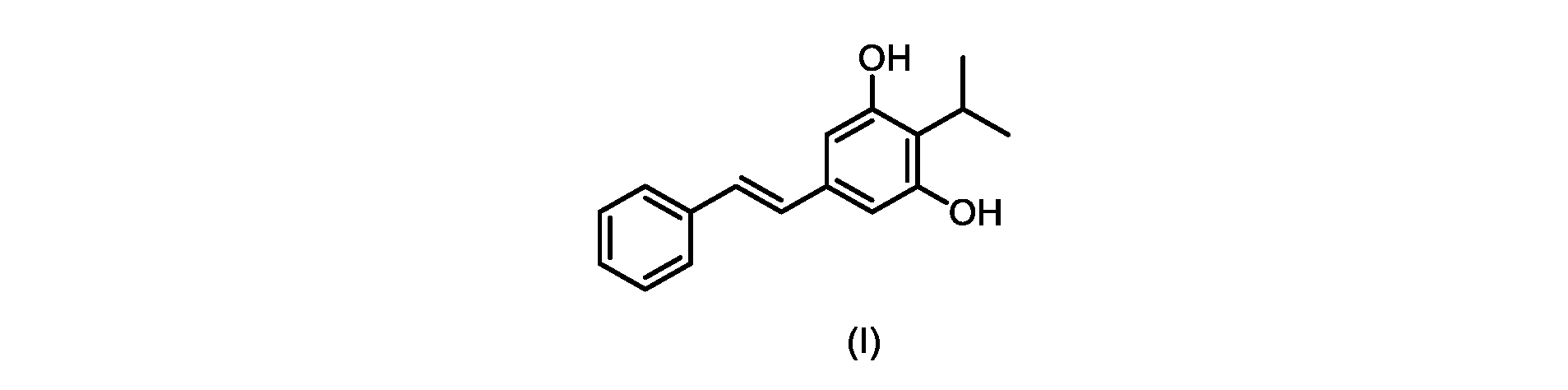
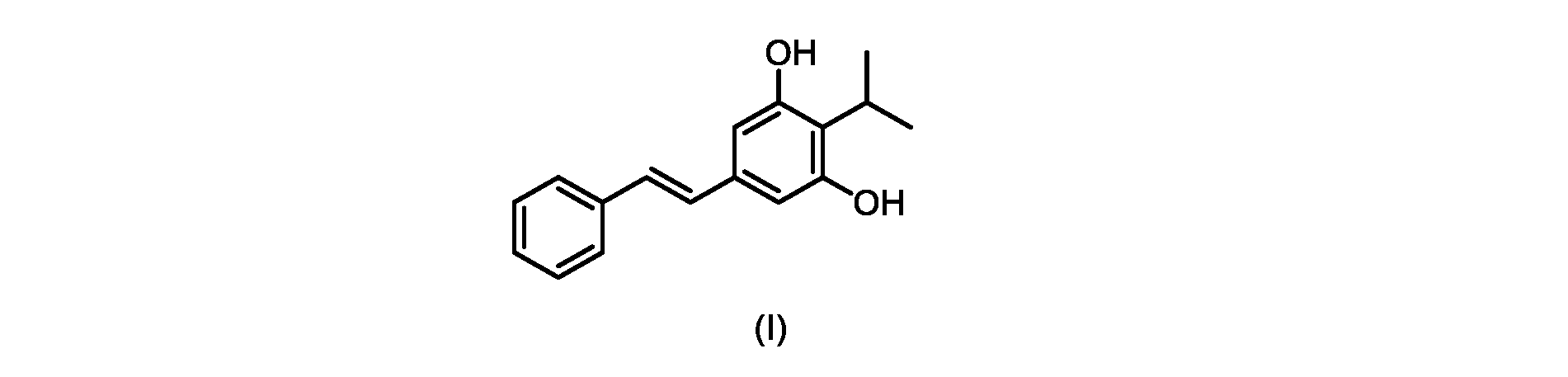
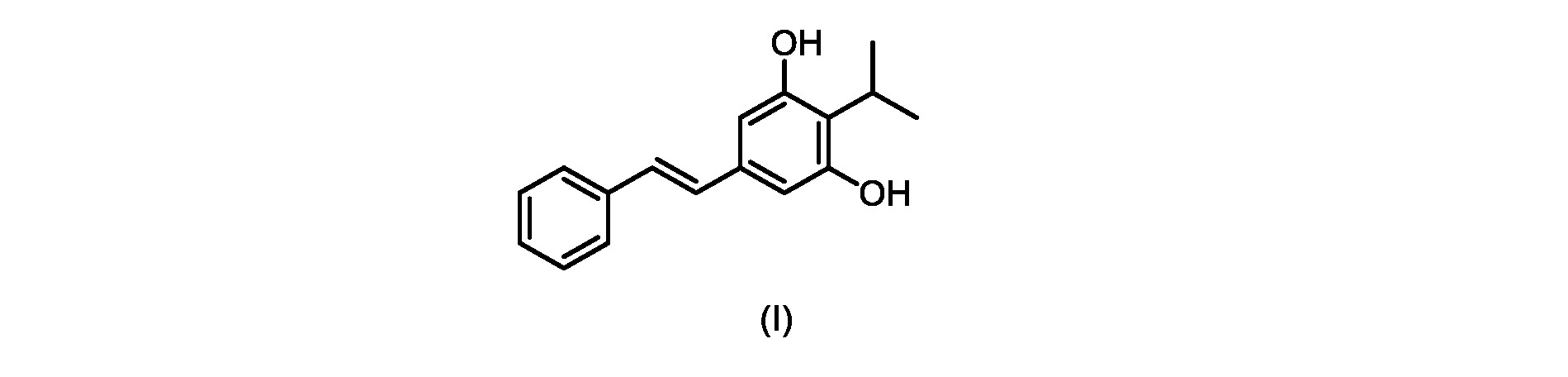
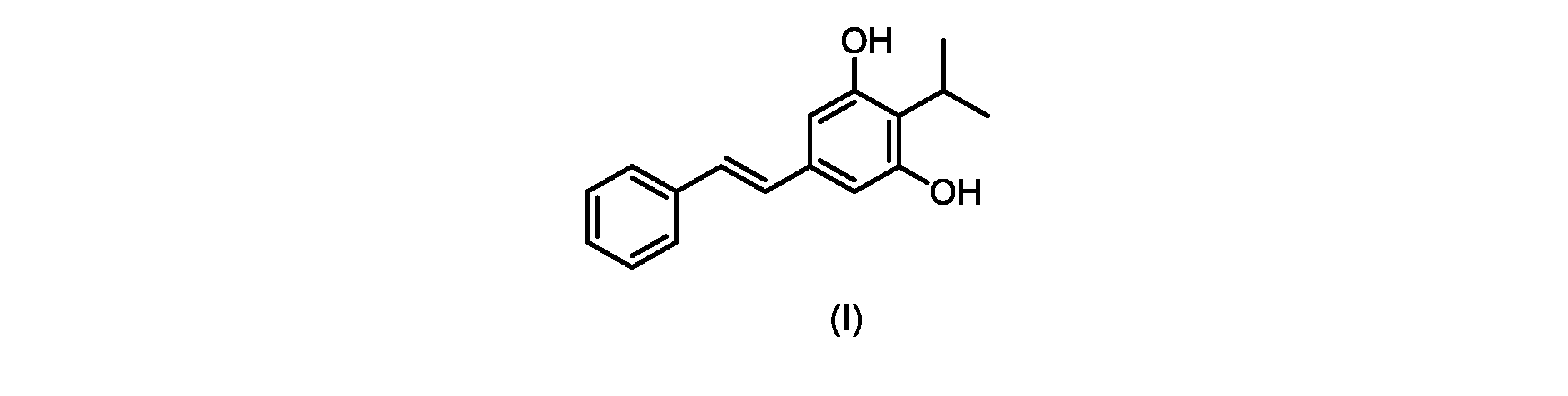
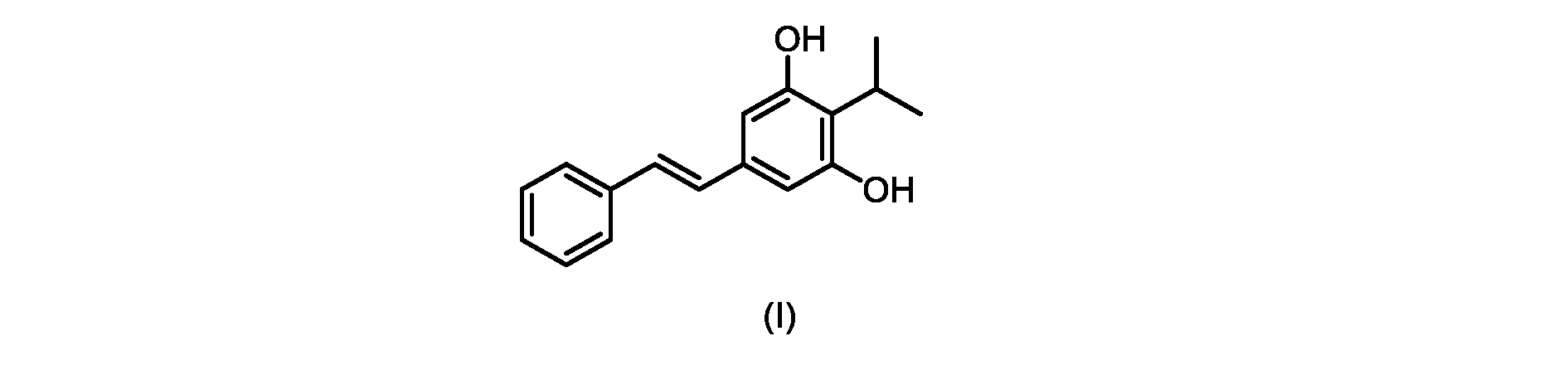
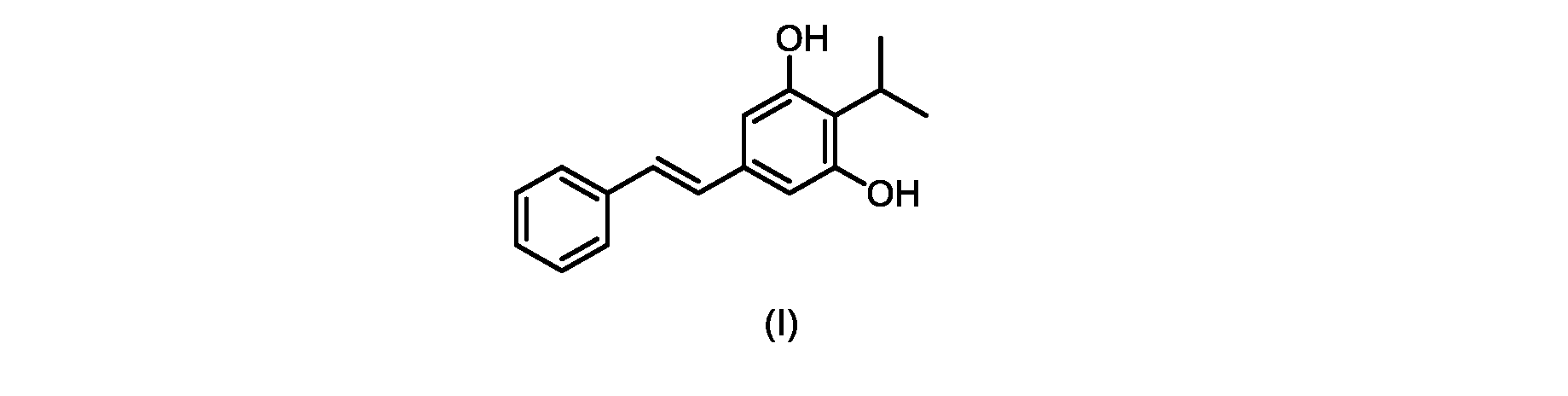
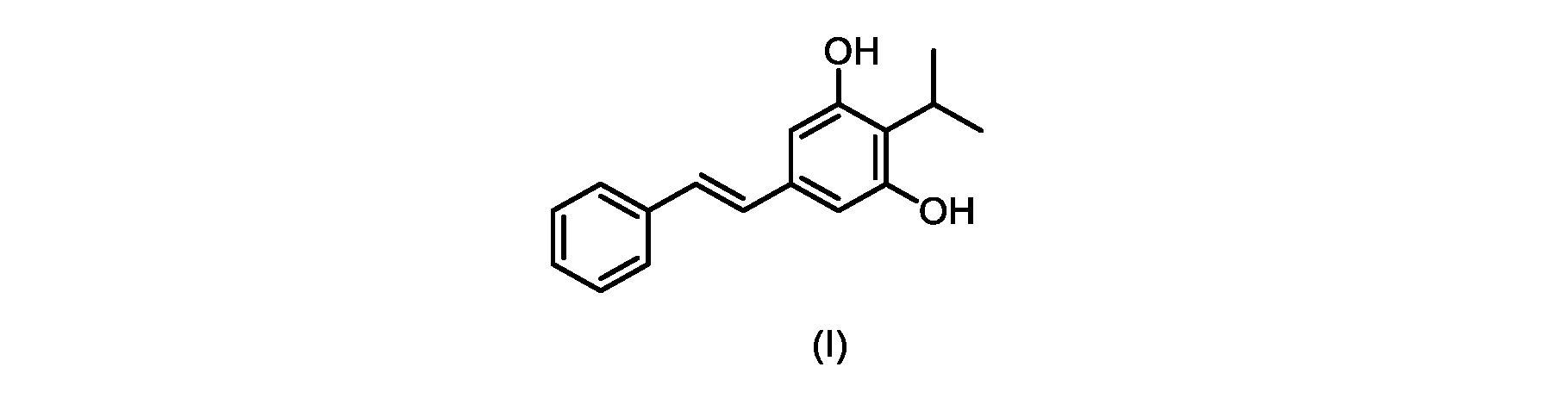
いくつかの実施形態は、式(I)の化合物またはその塩もしくは溶媒和物の調製のためのプロセスであって、
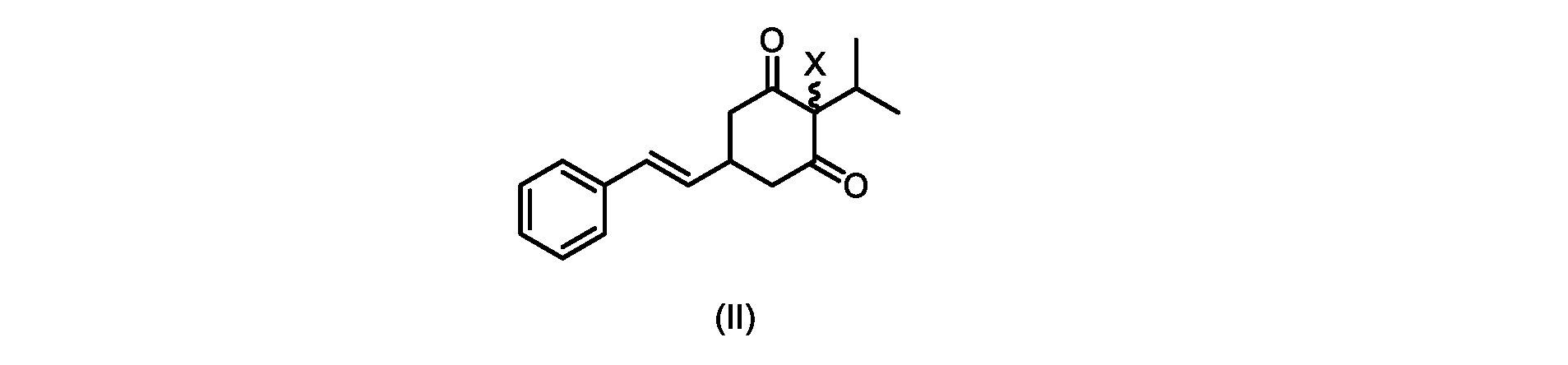
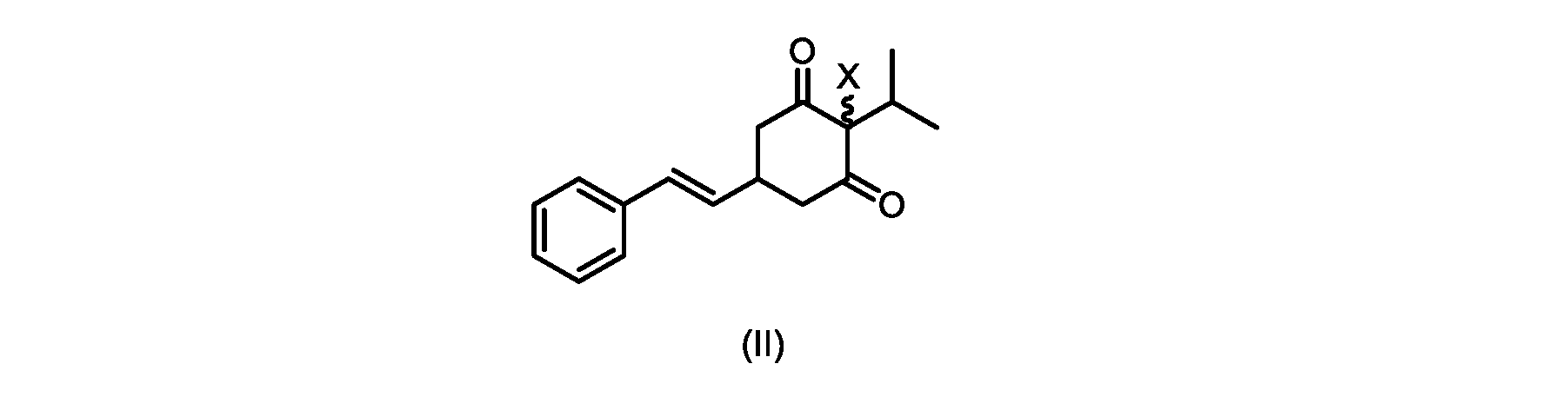
(a)が、式(II)の化合物またはその塩を形成するための
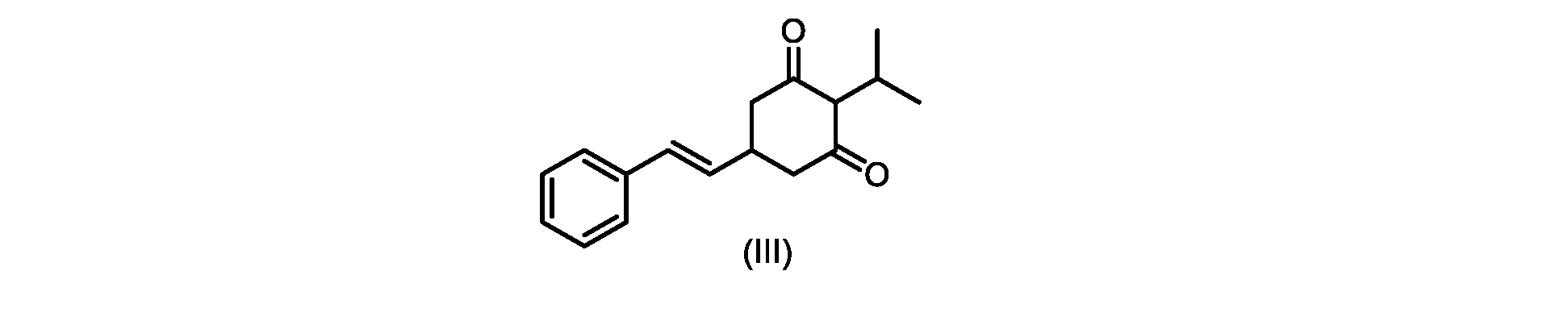
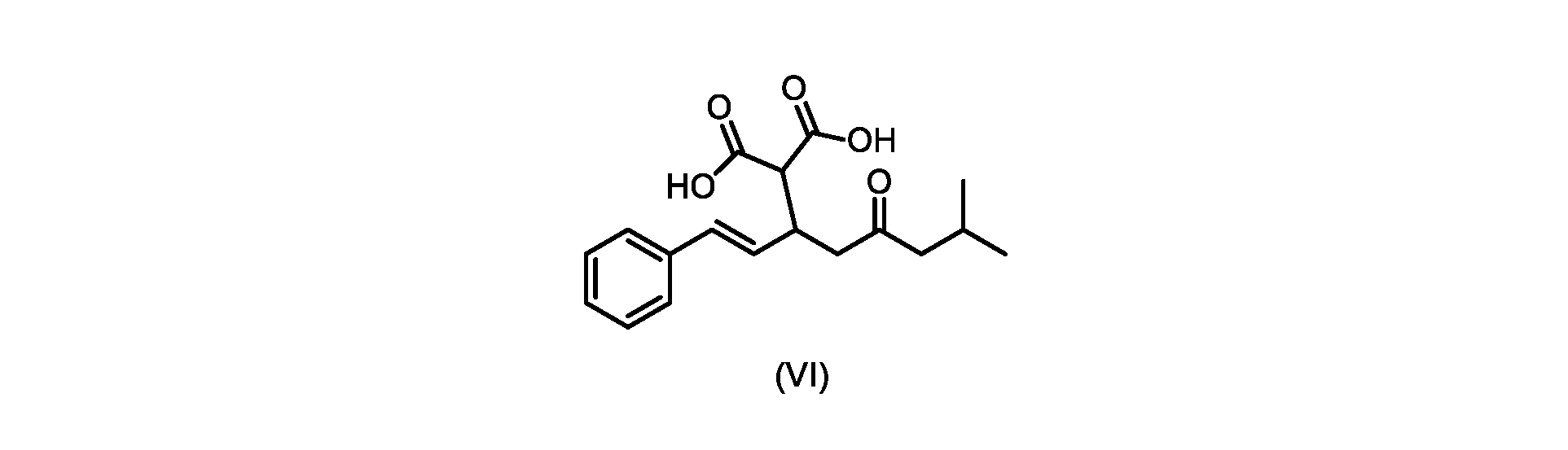
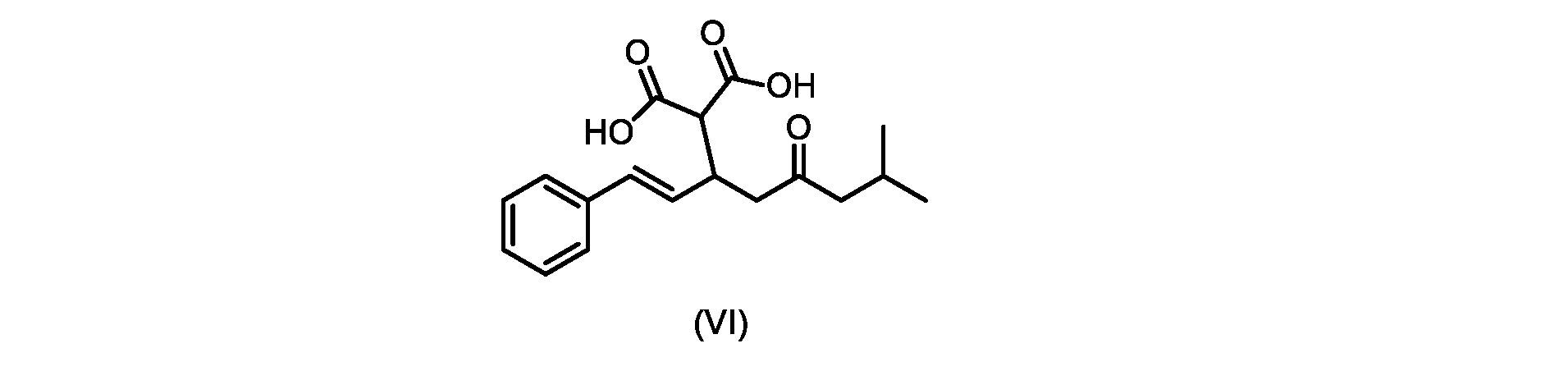
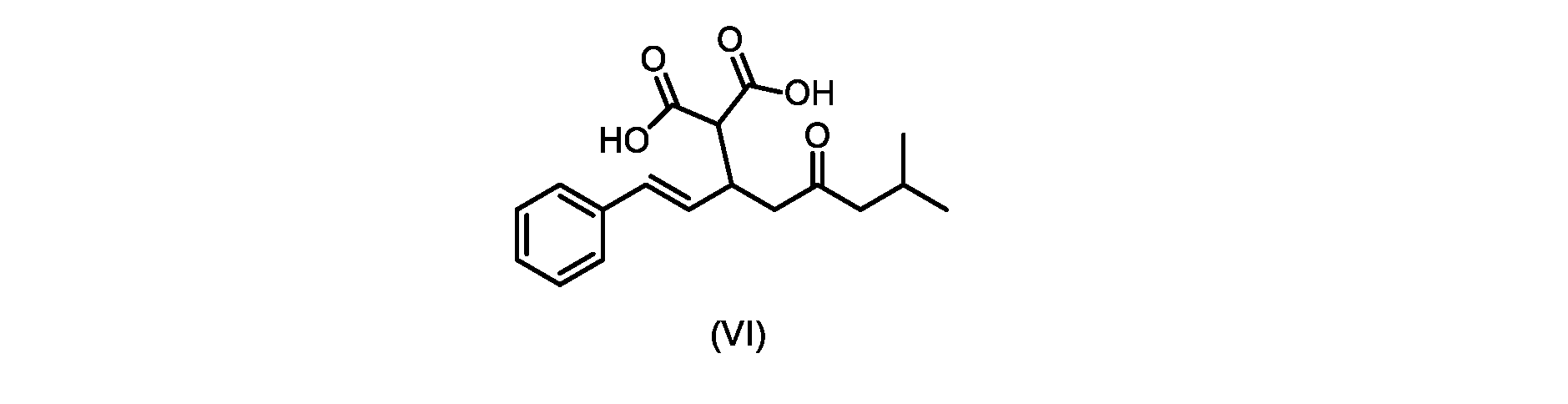
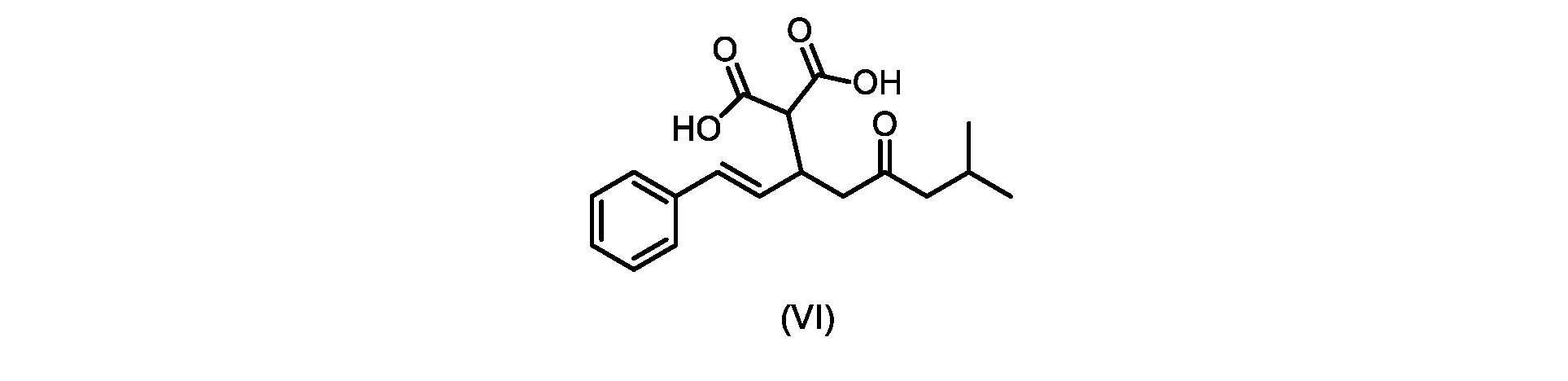
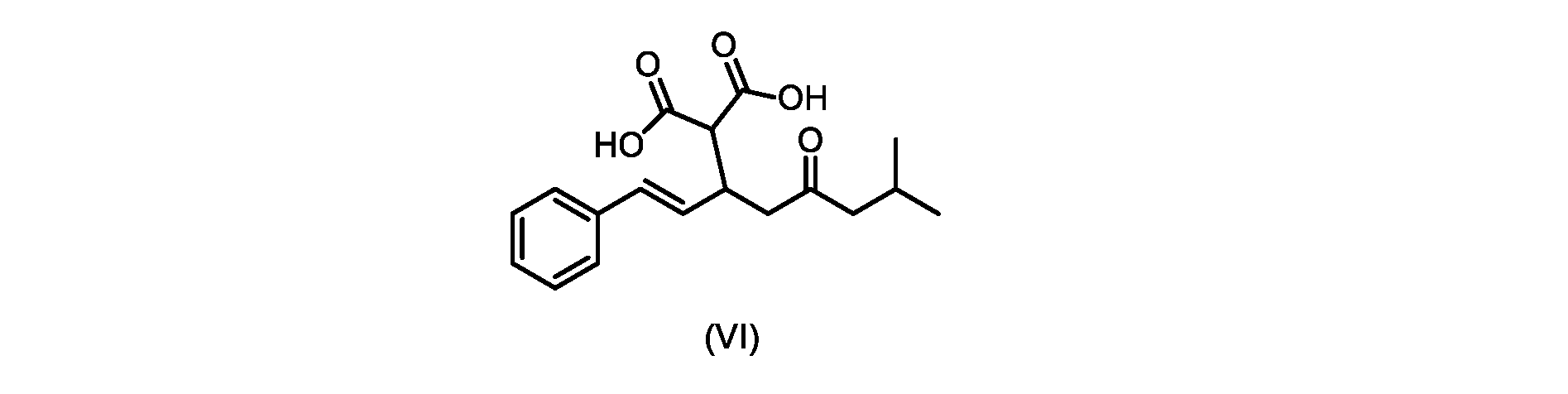
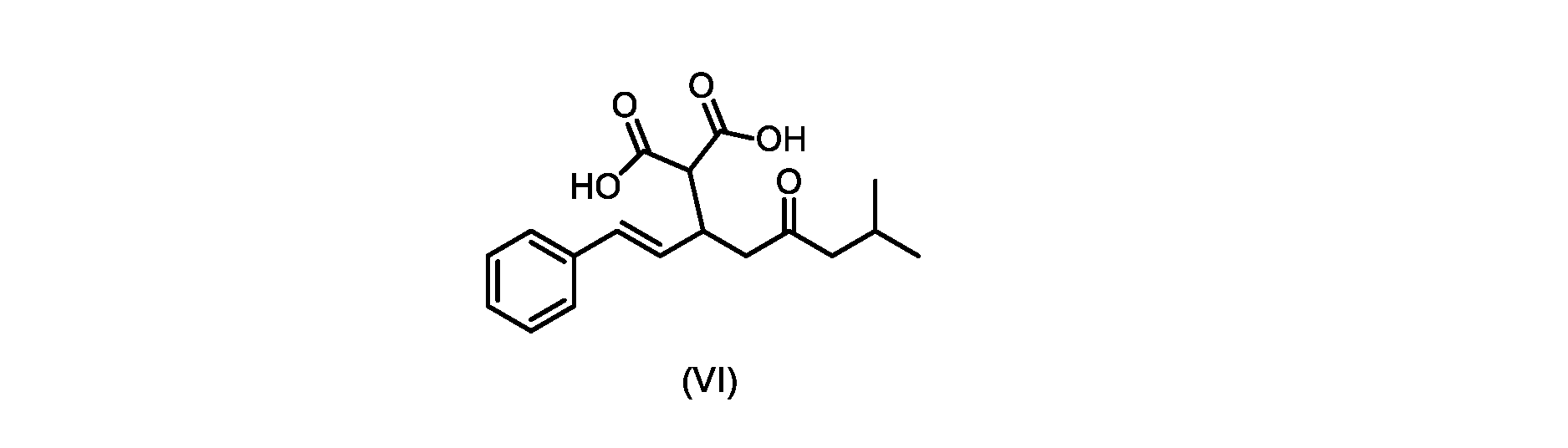
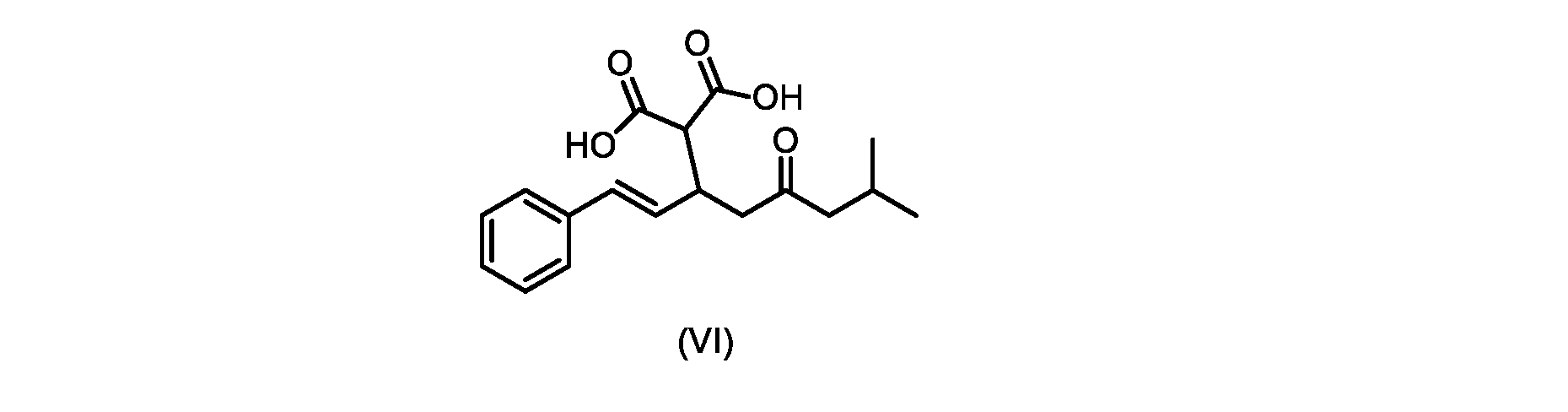
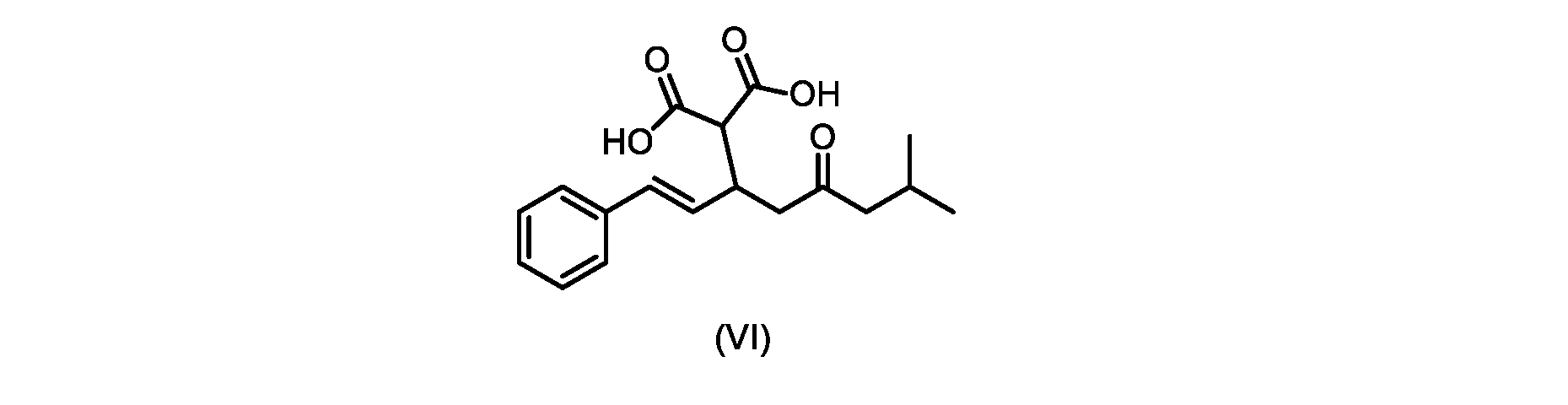
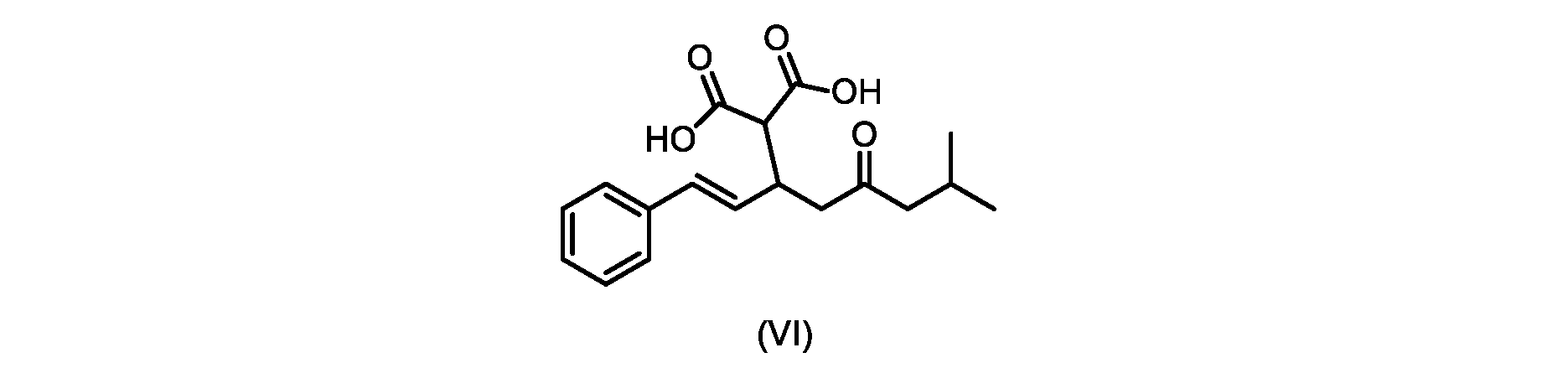
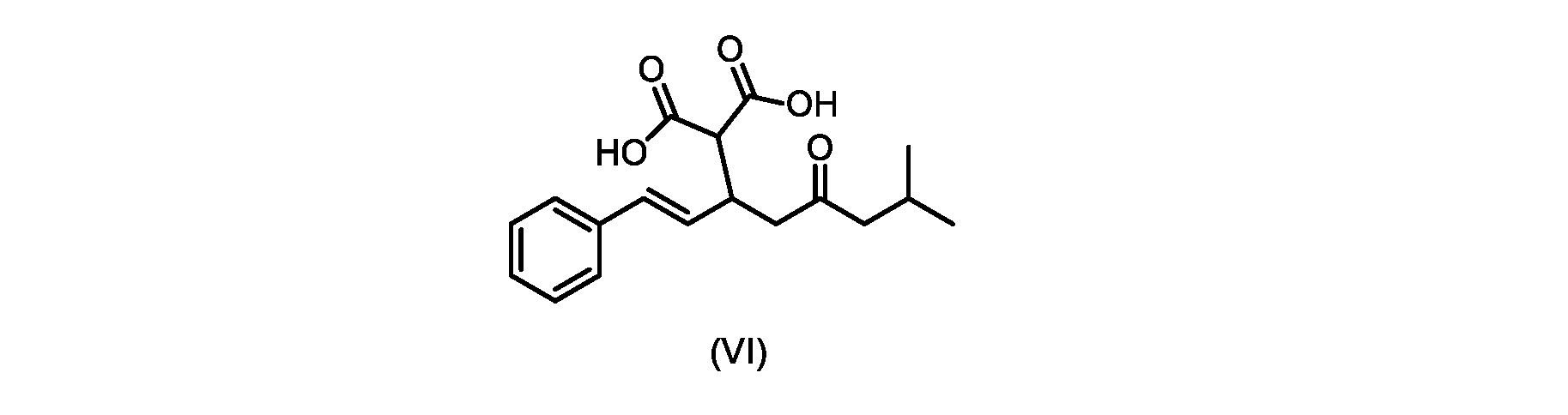
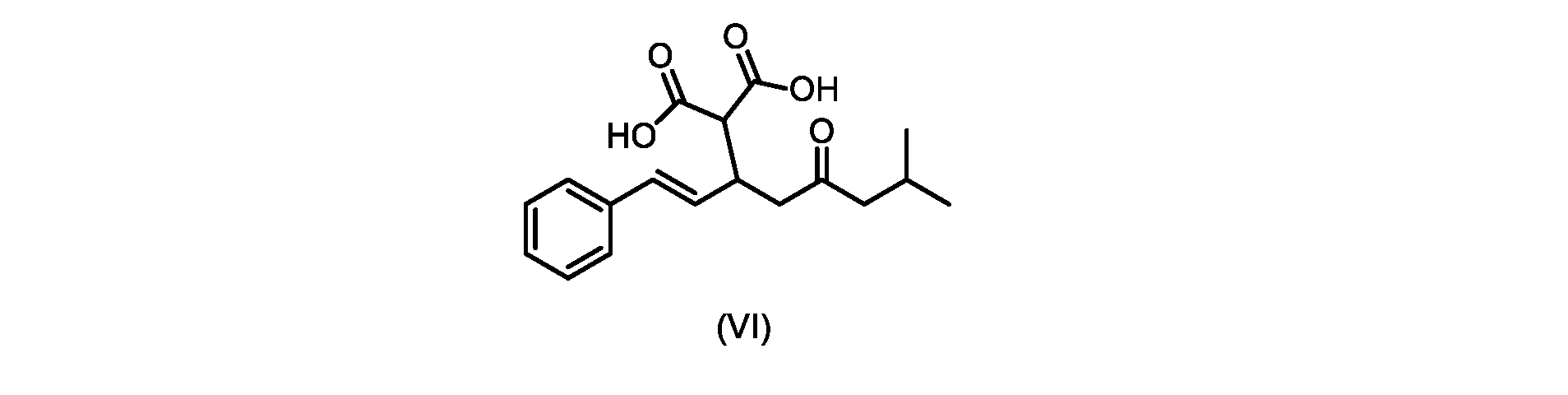
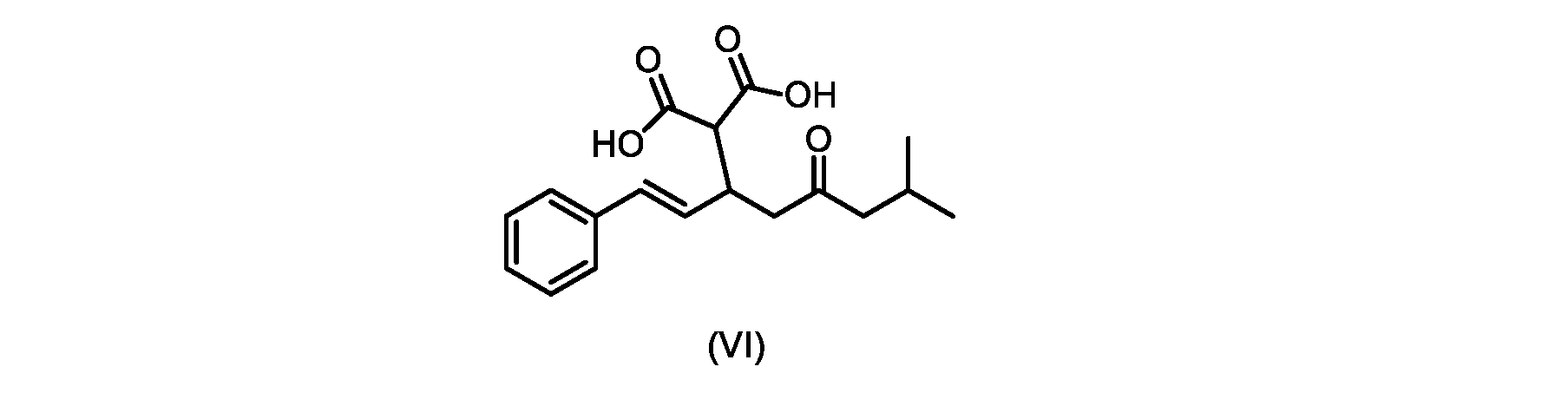
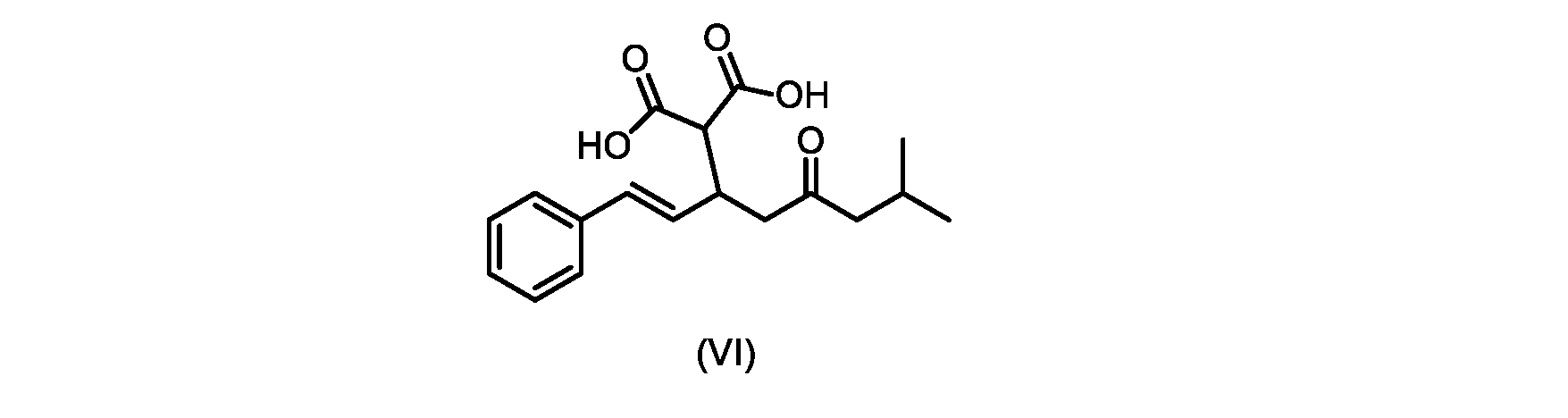
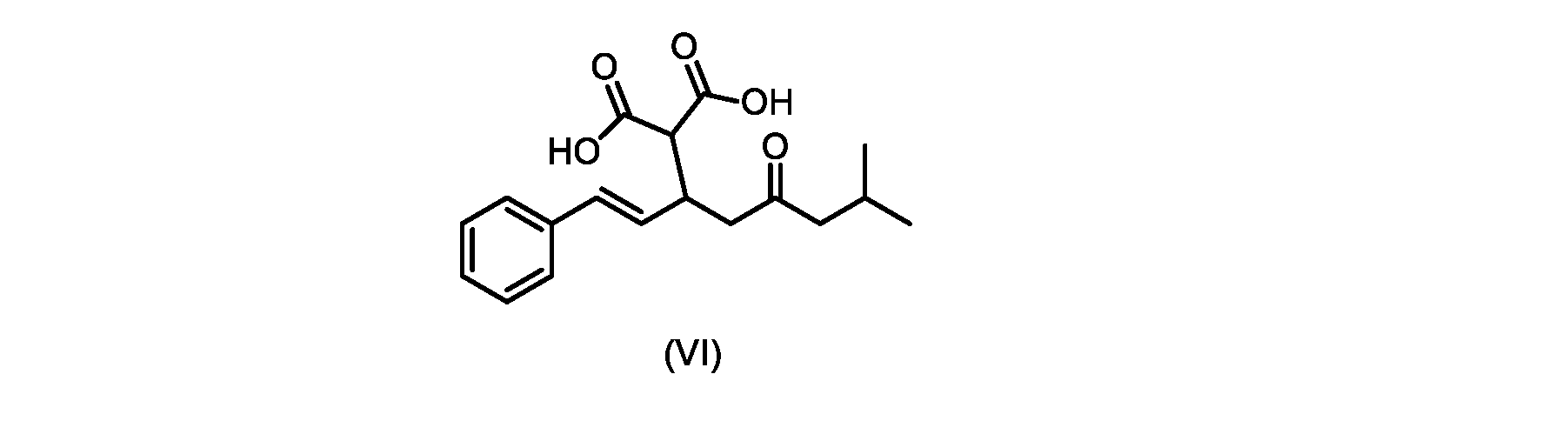
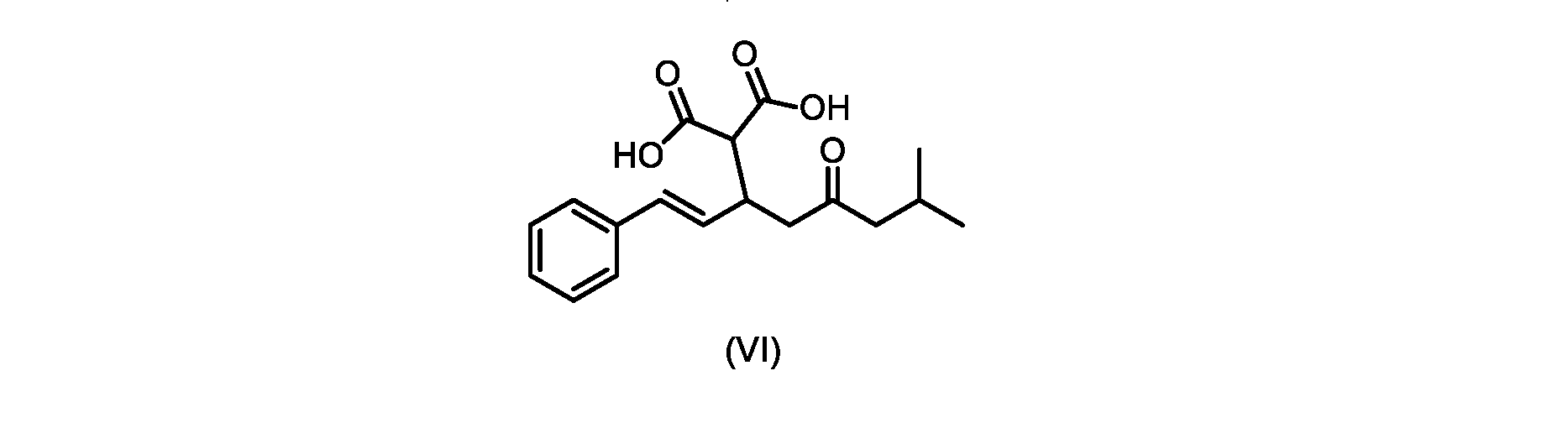
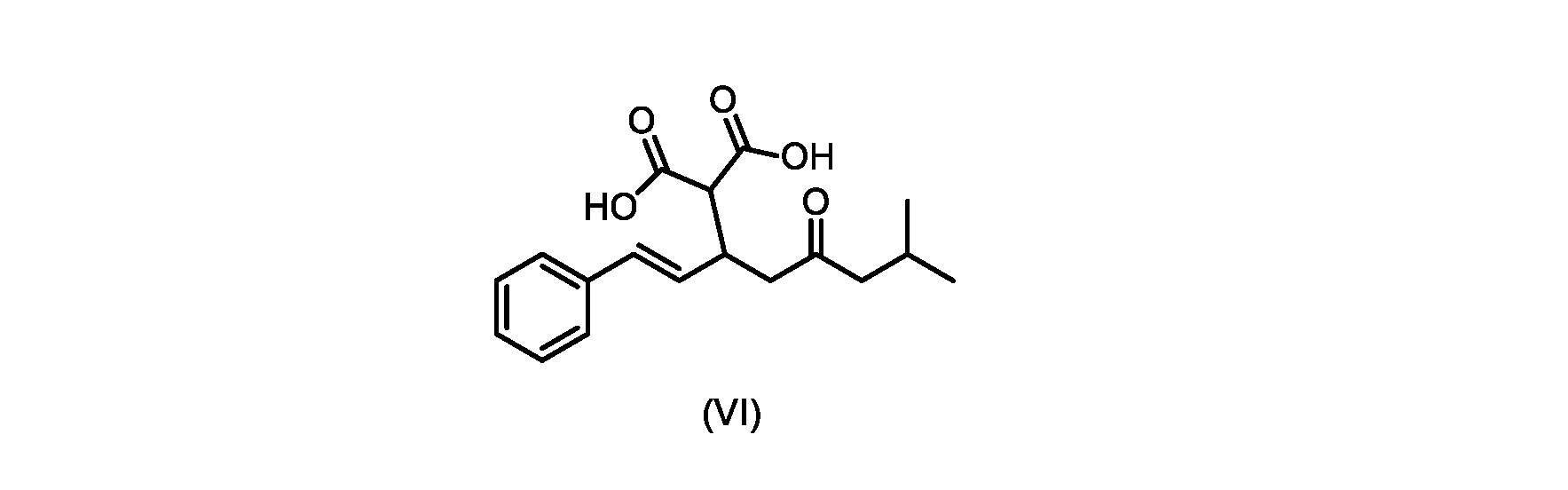
(b)が、式(VI)の化合物またはその塩の、

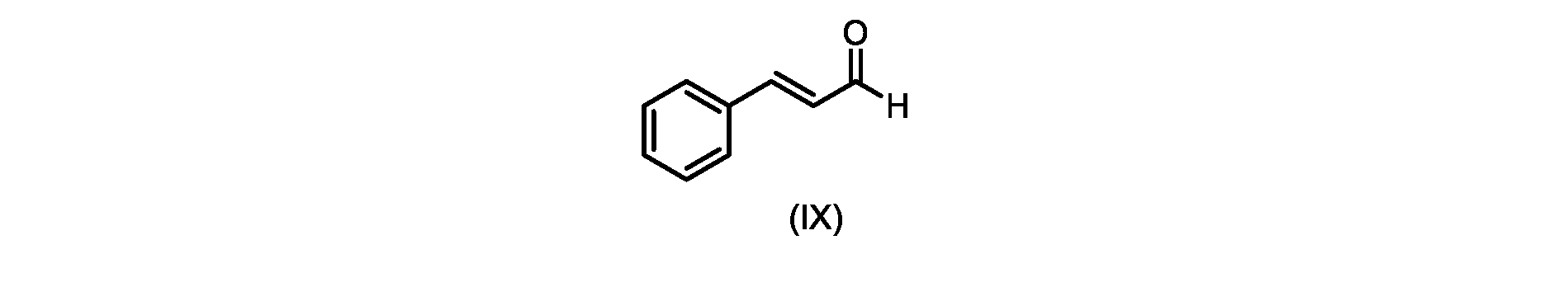
(c)が、式(IX)の化合物またはその塩の、

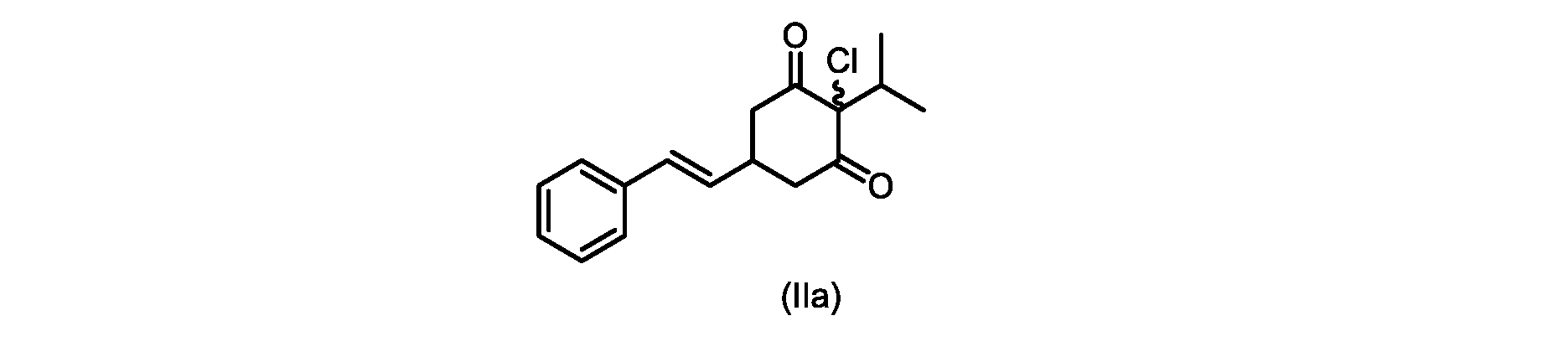
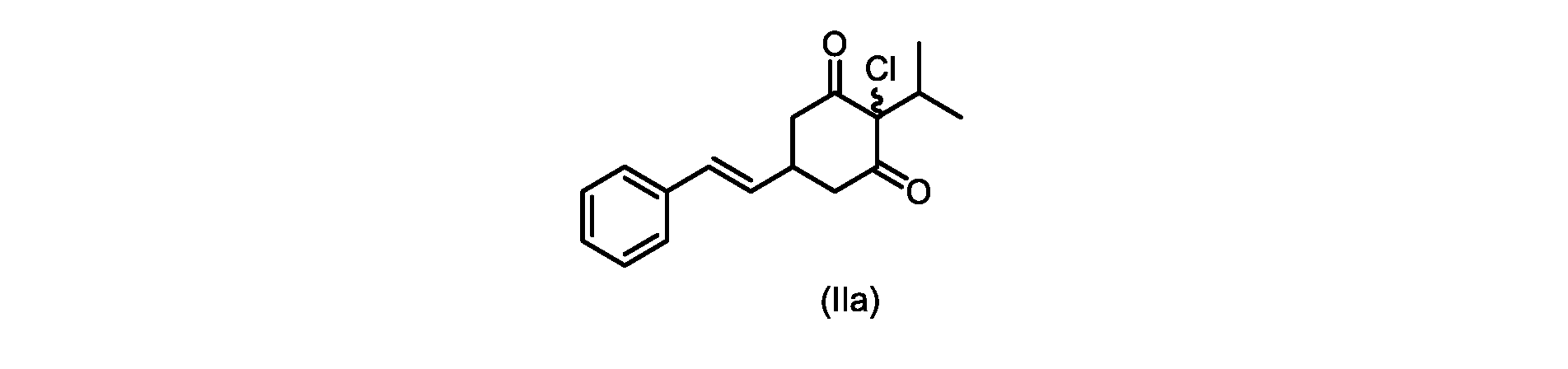
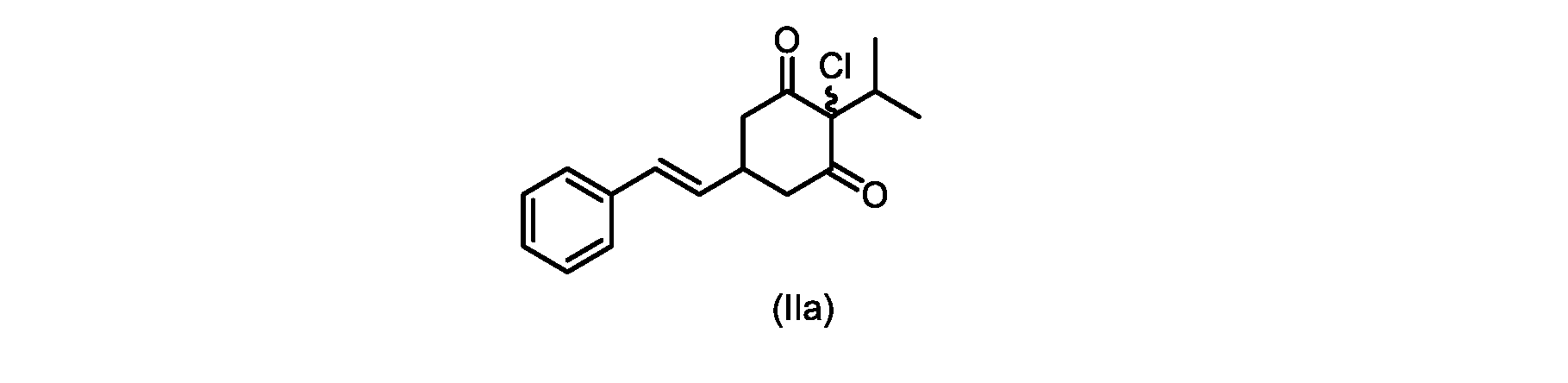
いくつかの実施形態では、Xは、Clである。
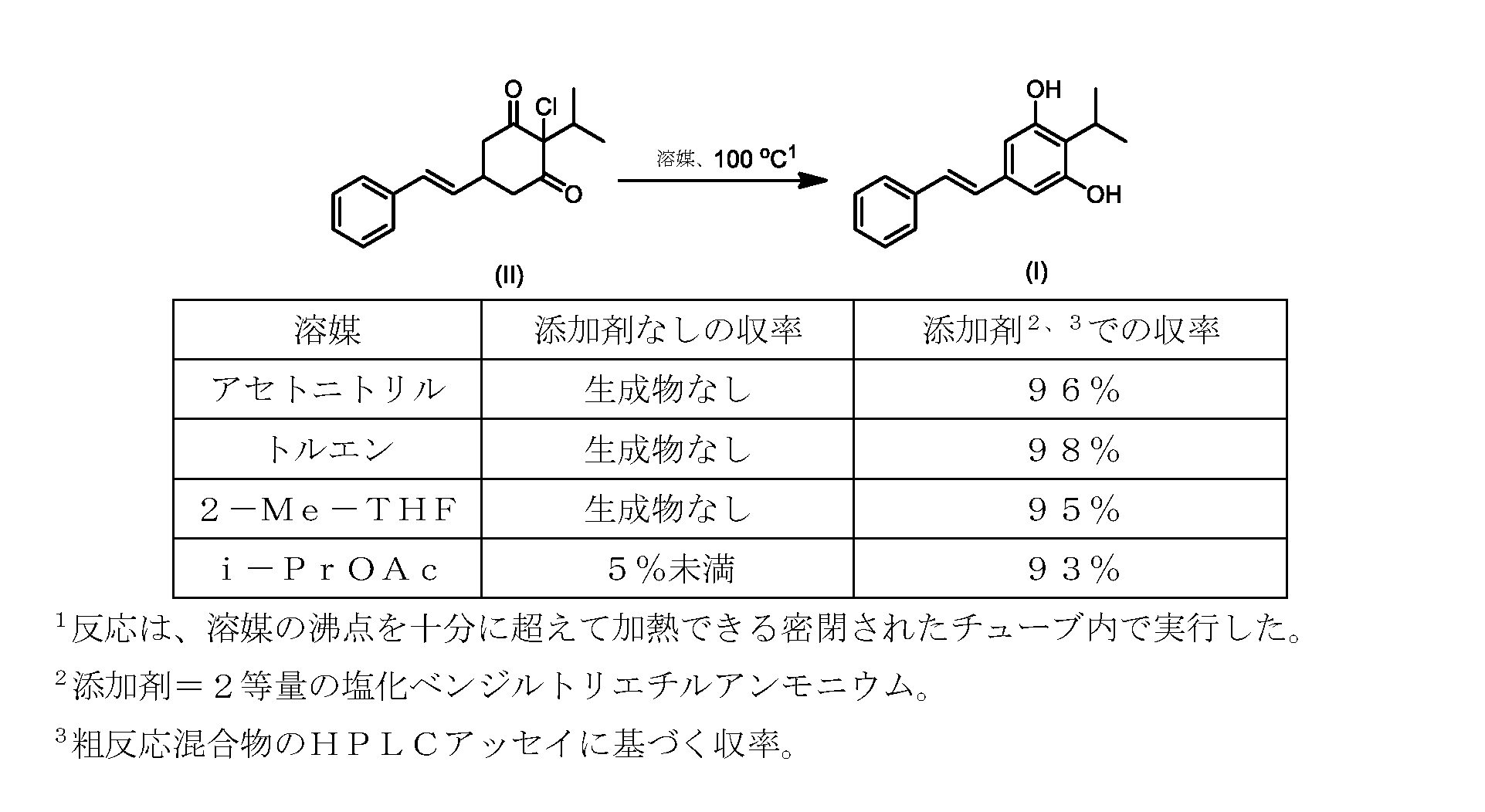
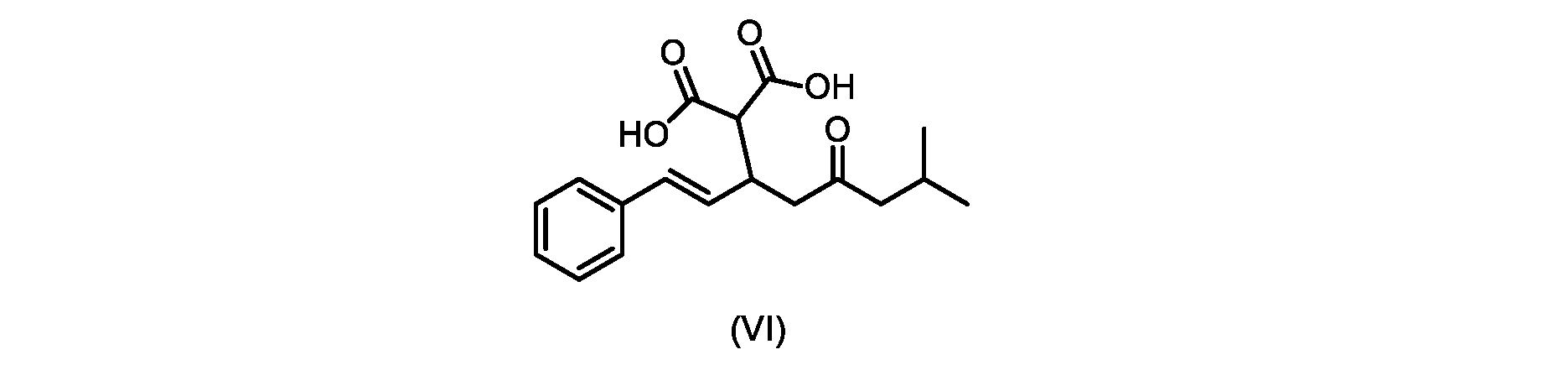
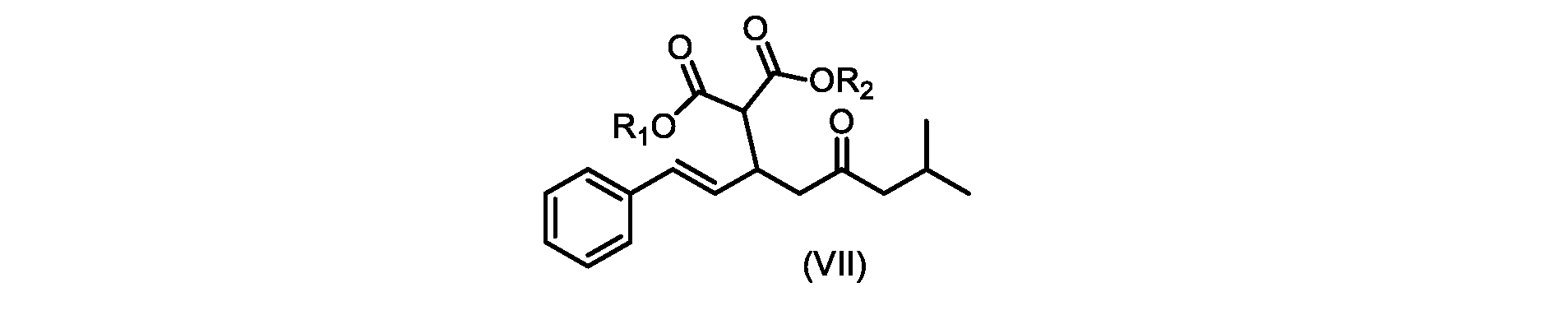
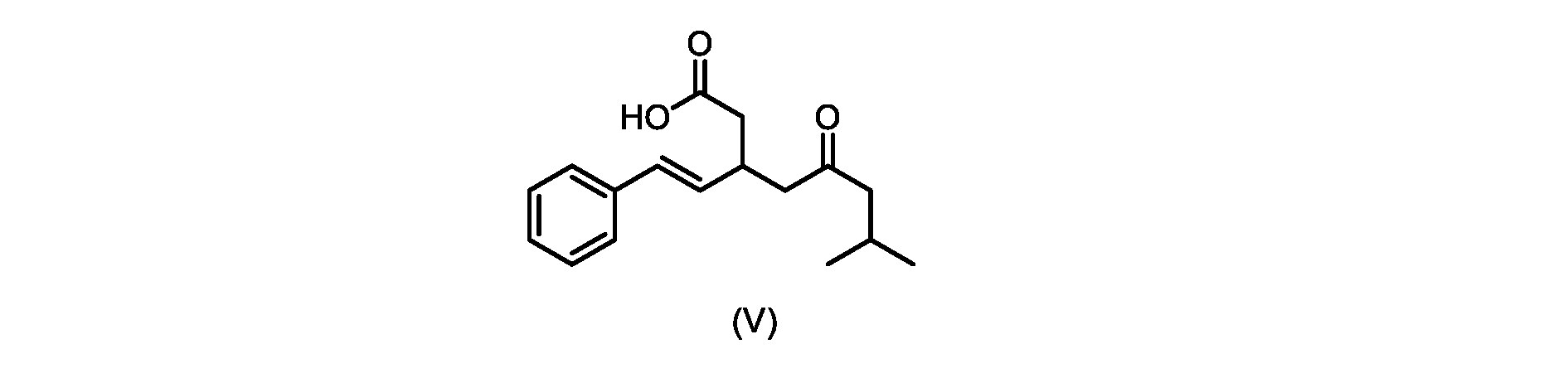
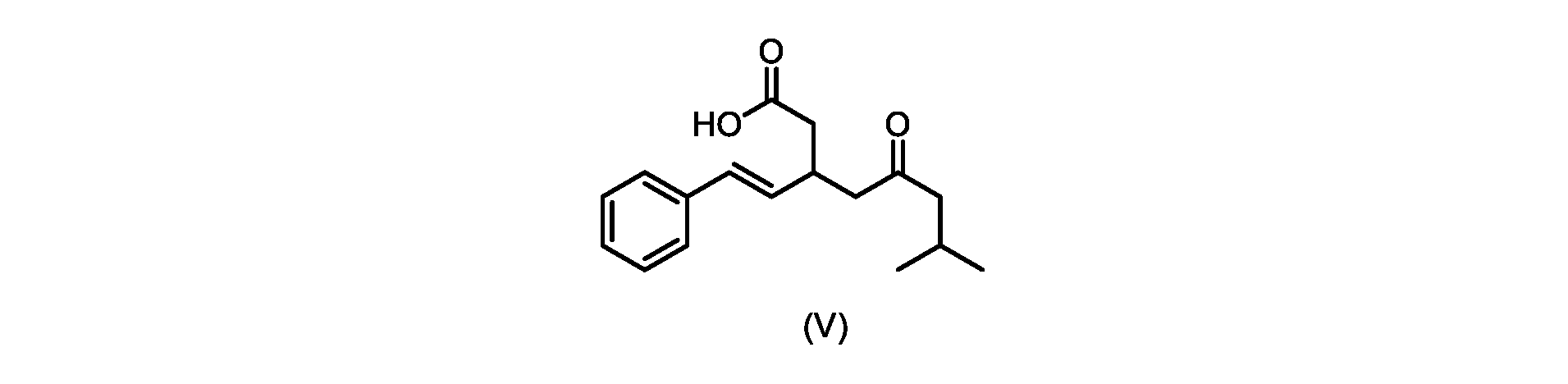
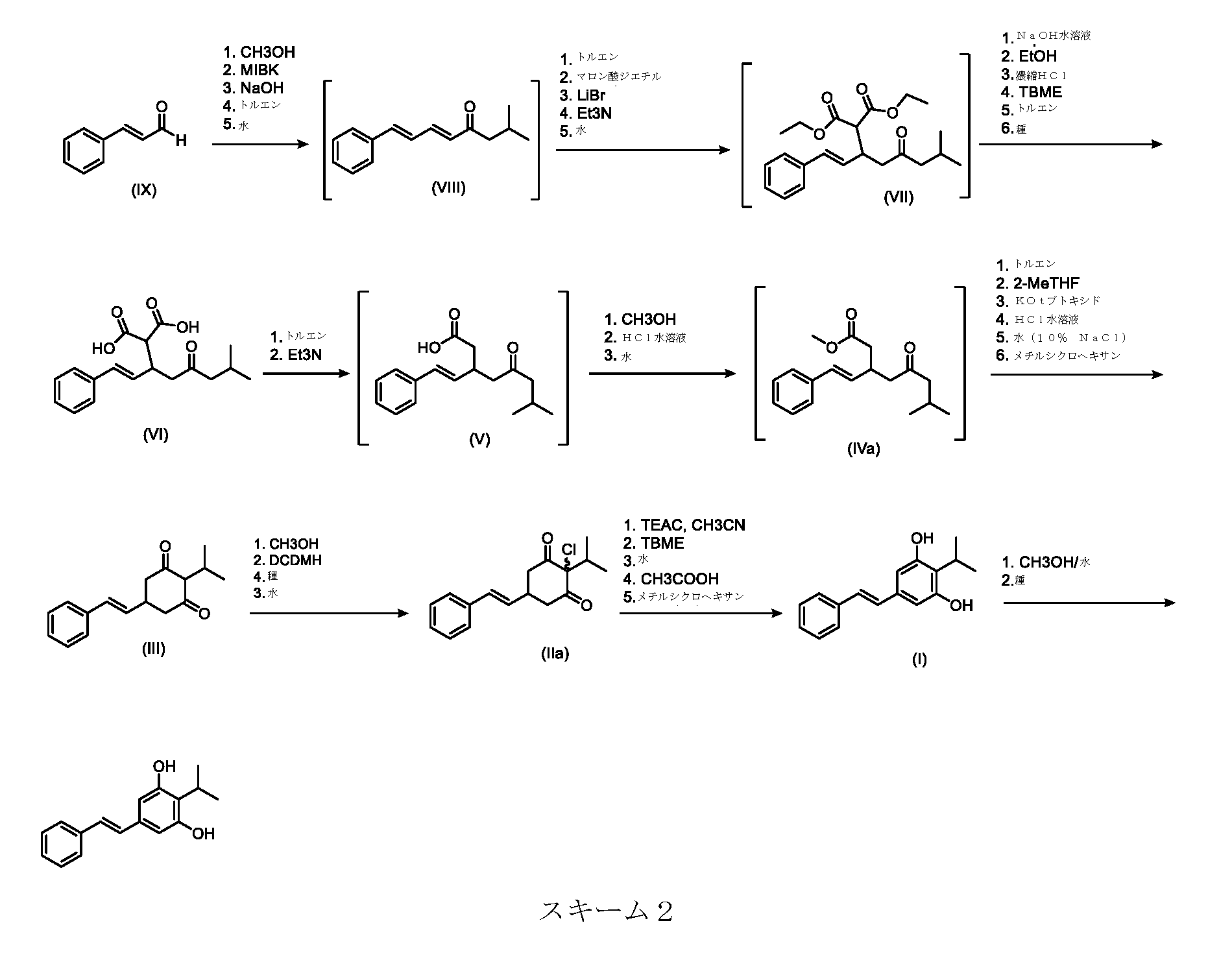
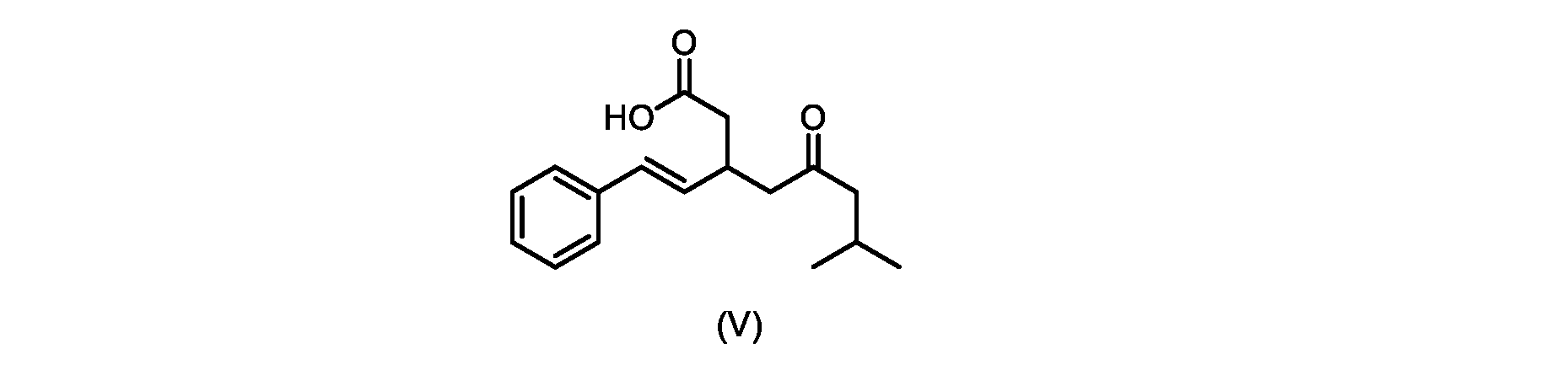
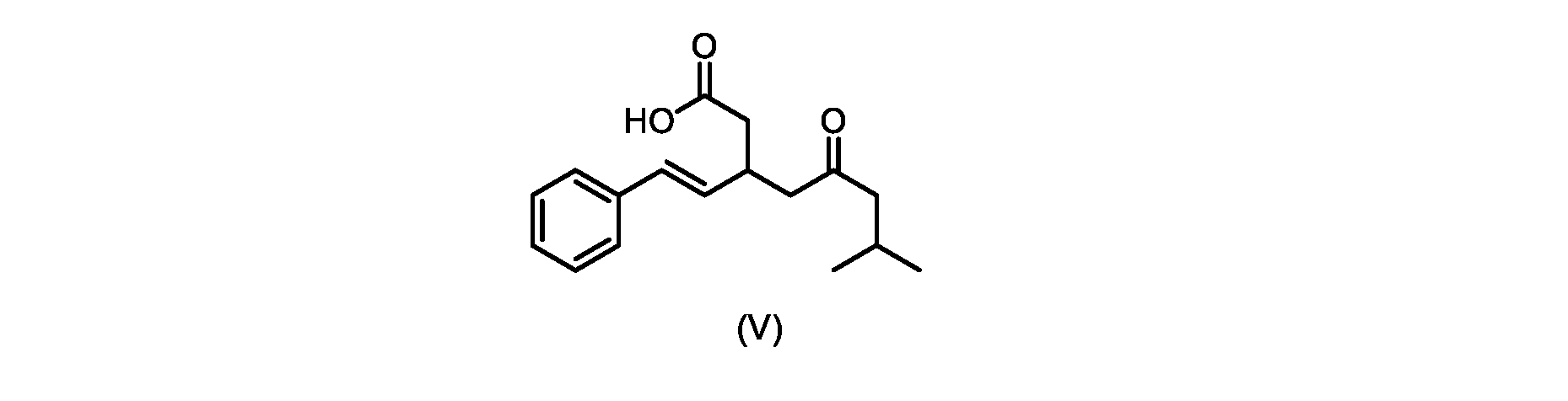
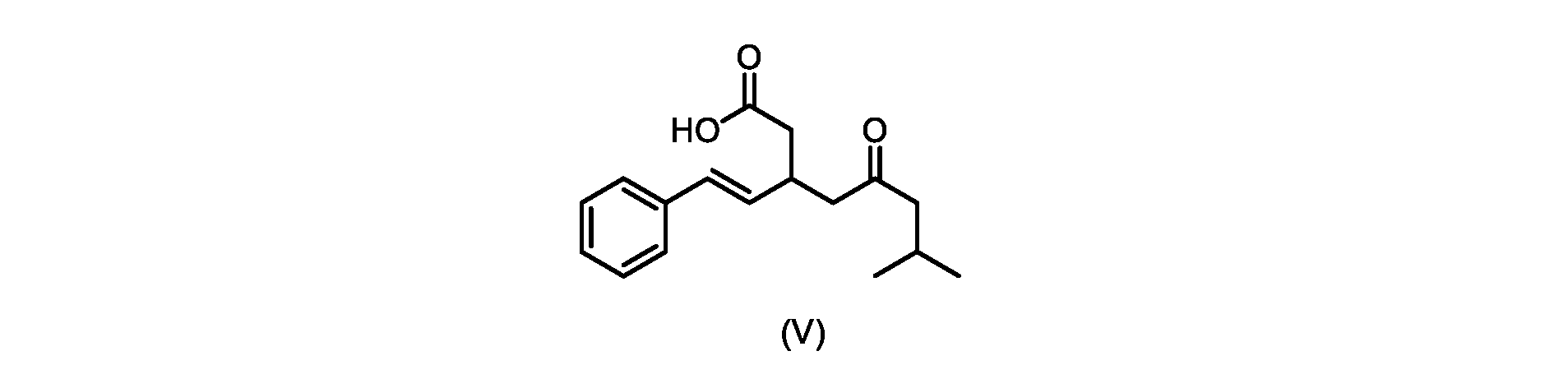
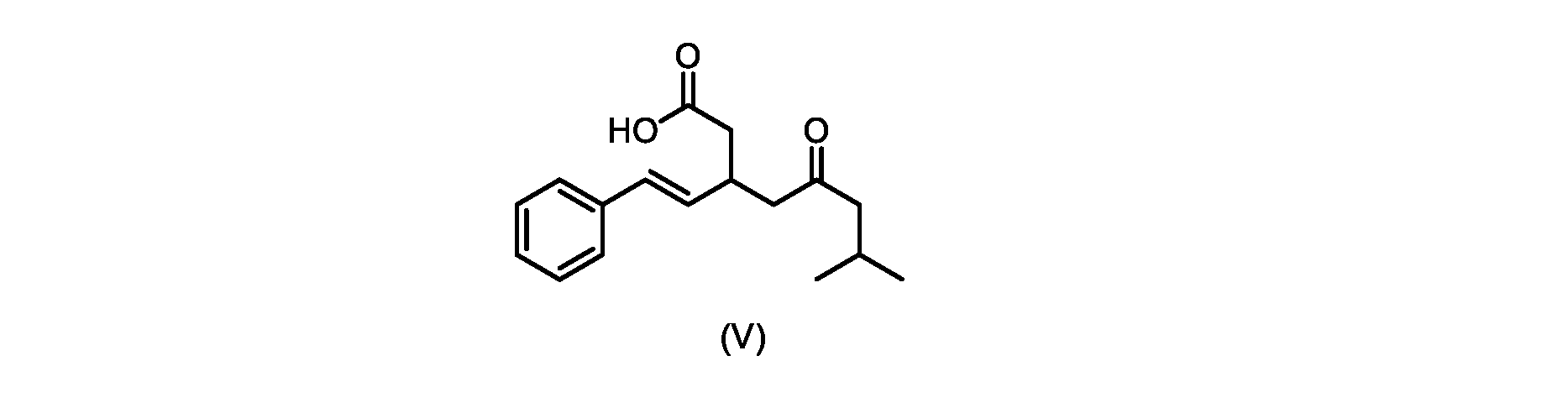
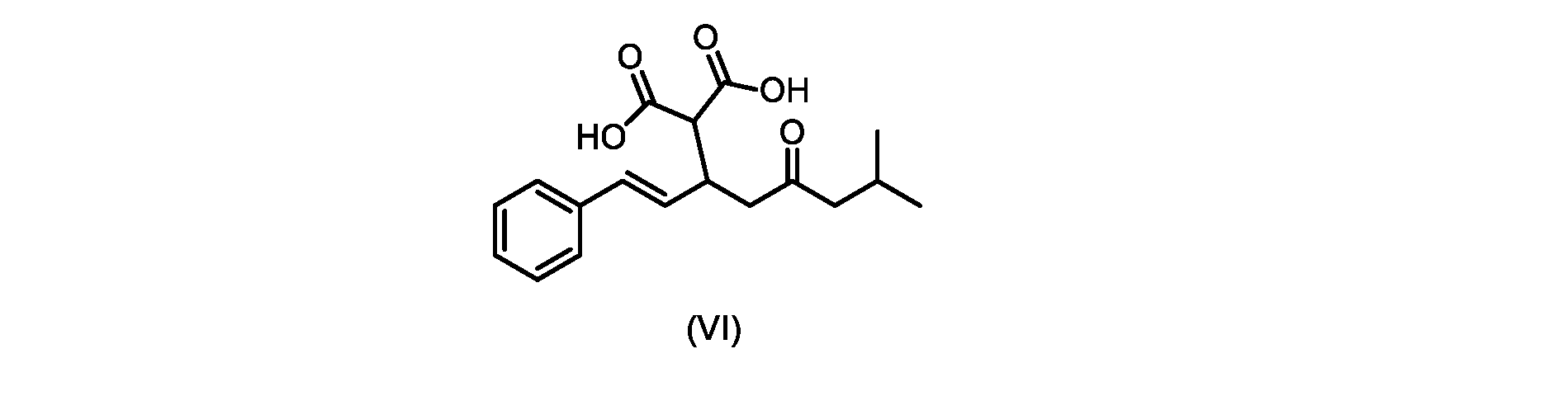
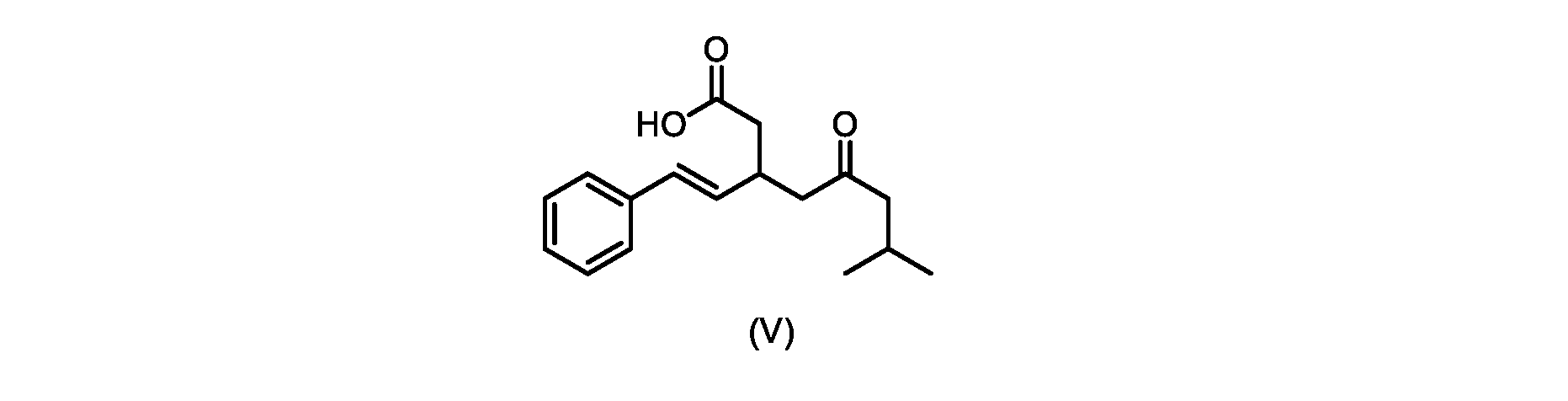
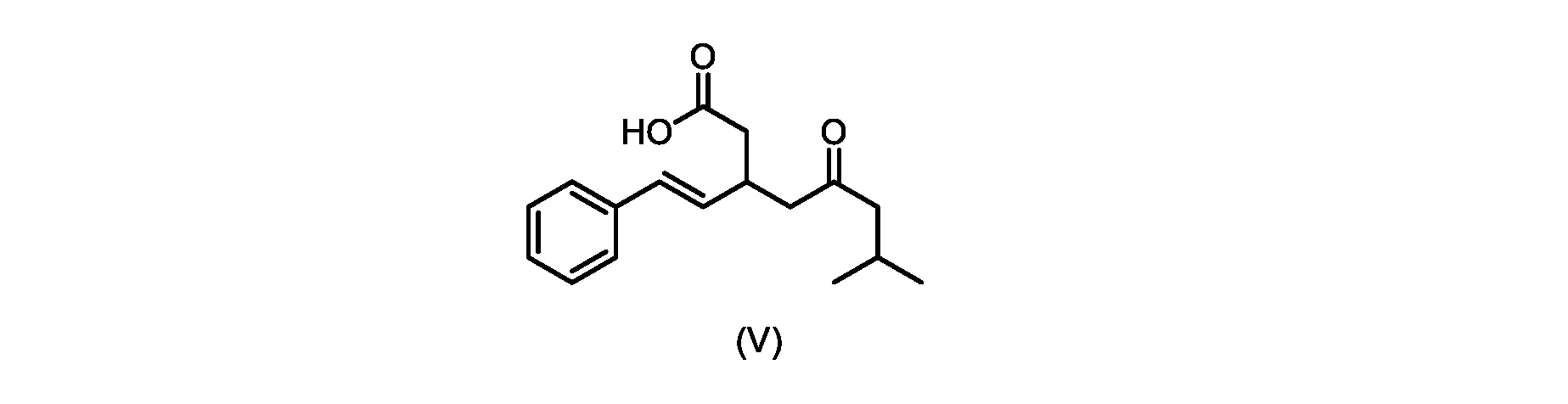
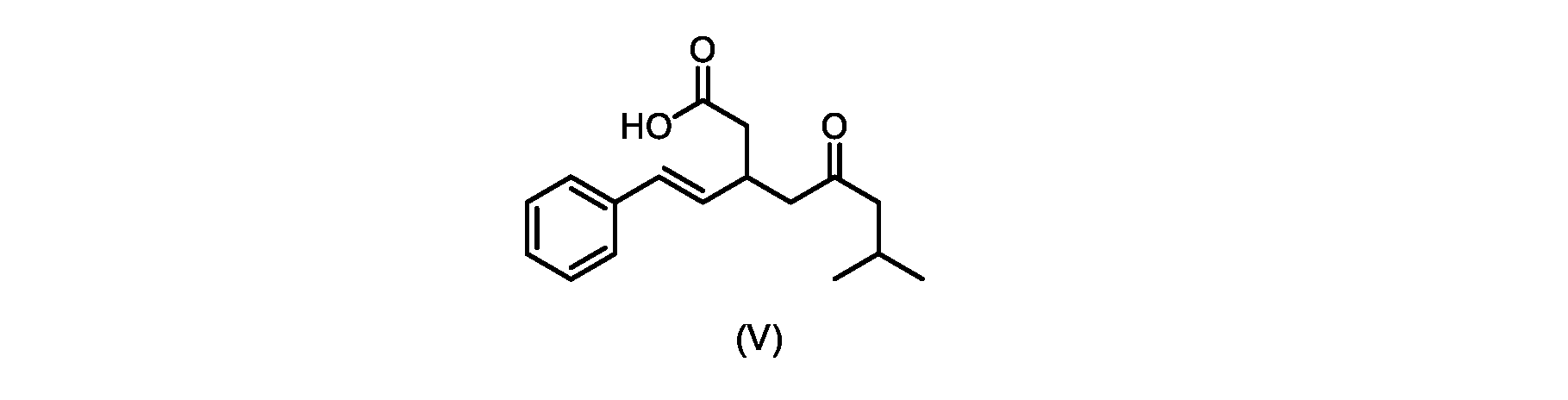
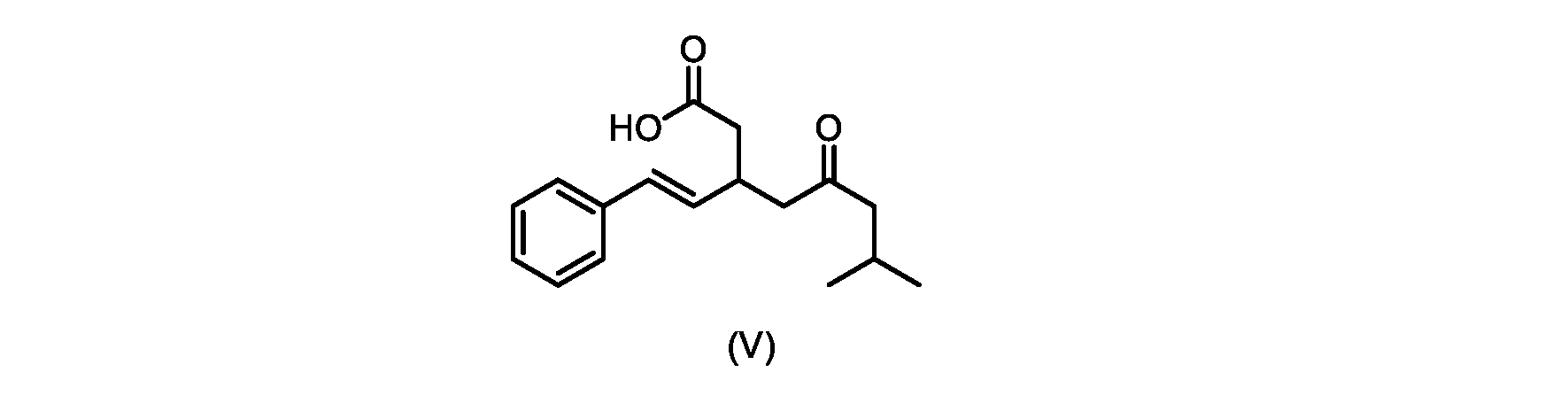
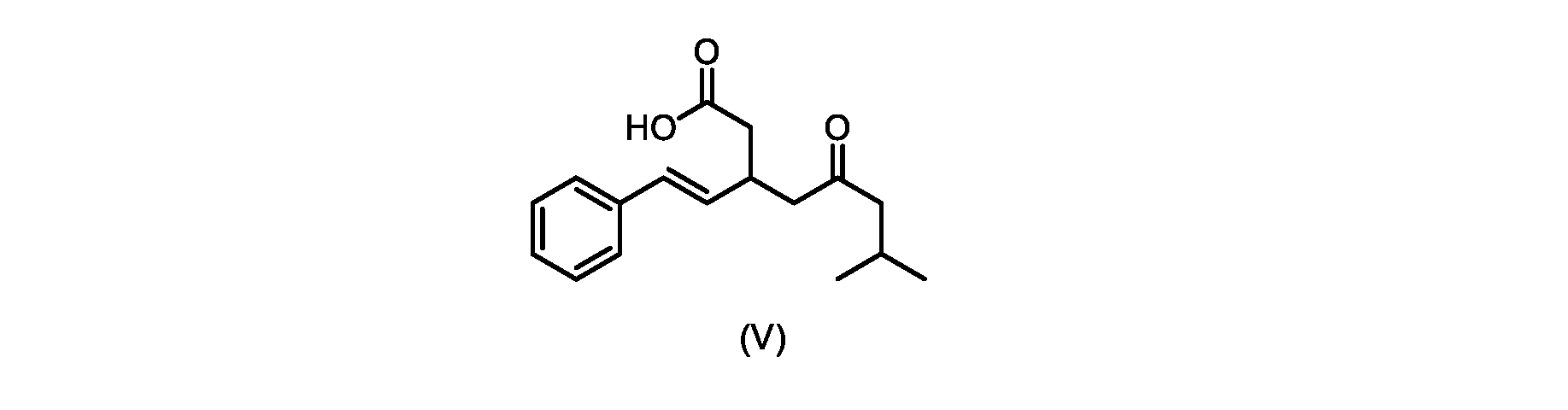
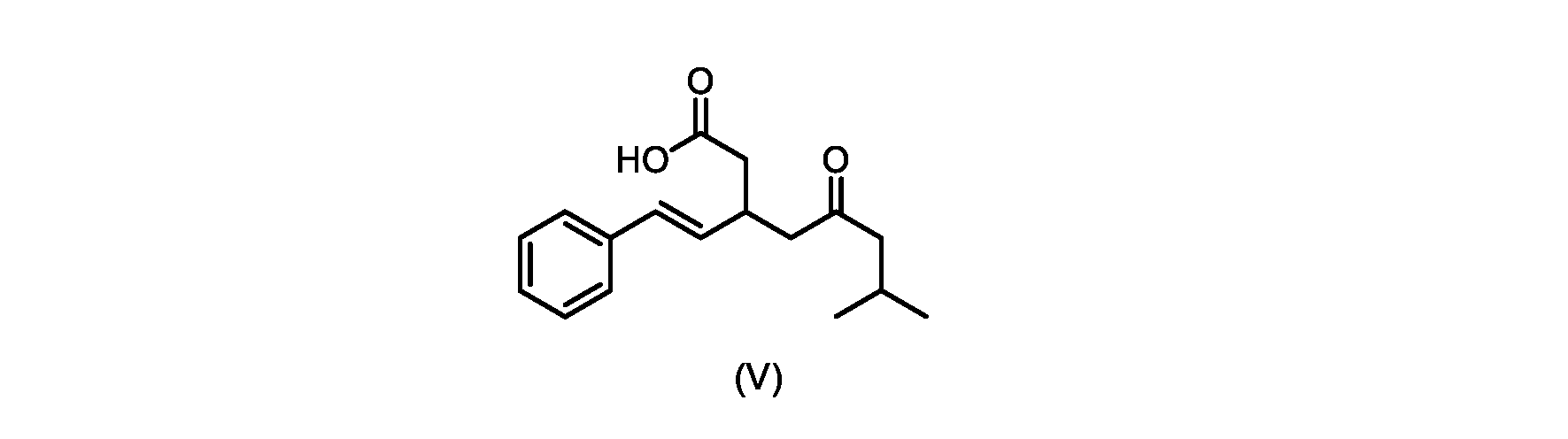
いくつかの実施形態では、式(VI)の化合物またはその塩の、式(III)の化合物またはその塩への変換は、式(V)の化合物またはその塩を形成するための式(VI)の化合物またはその塩の脱炭酸と、
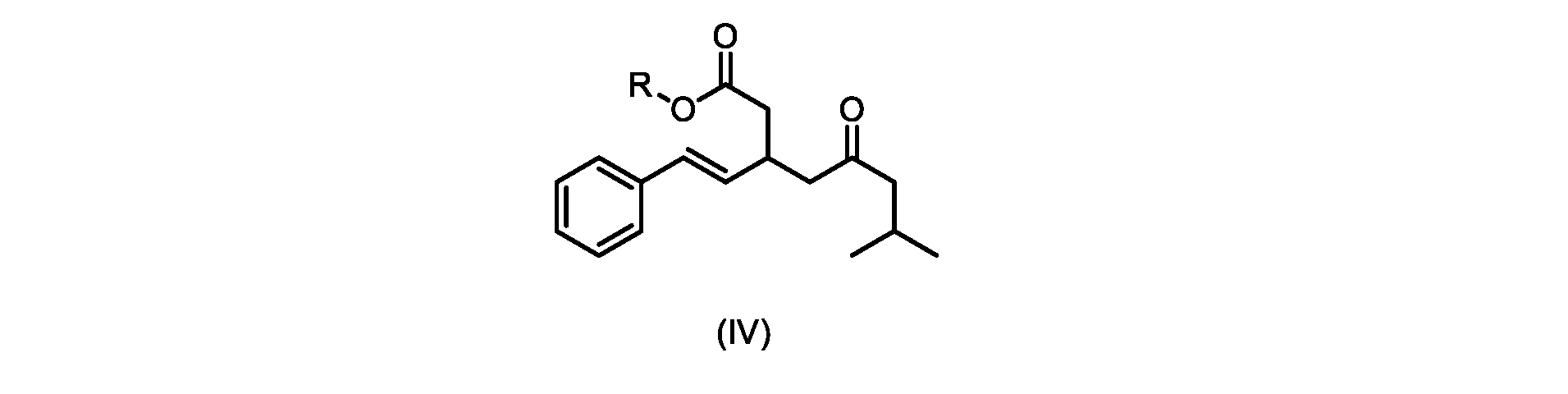
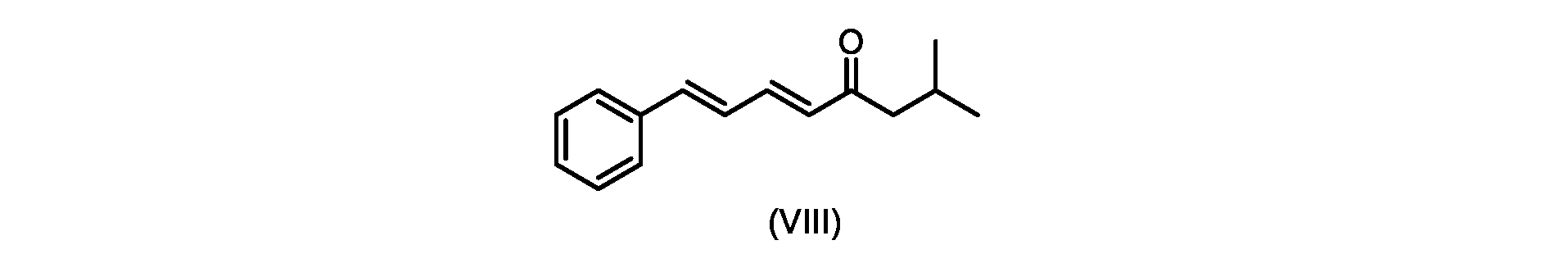
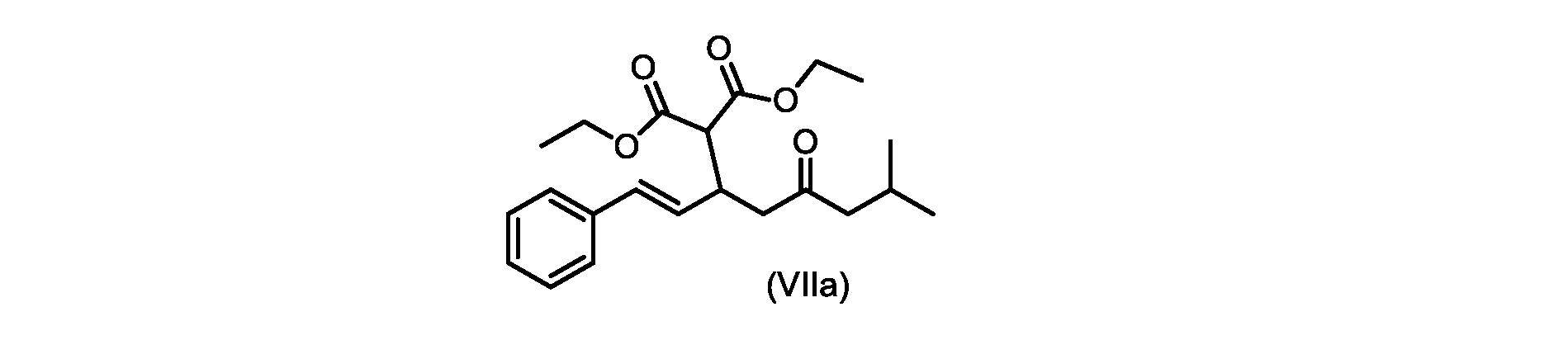
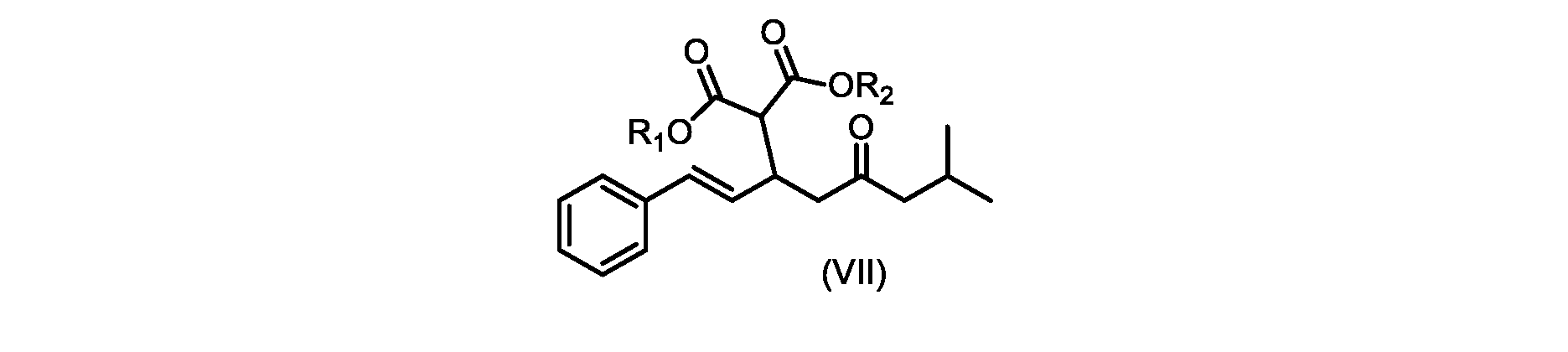
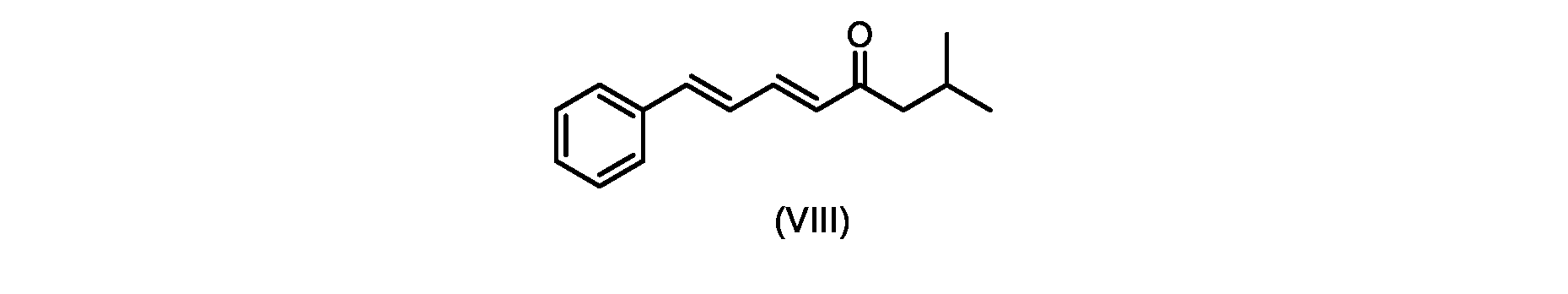
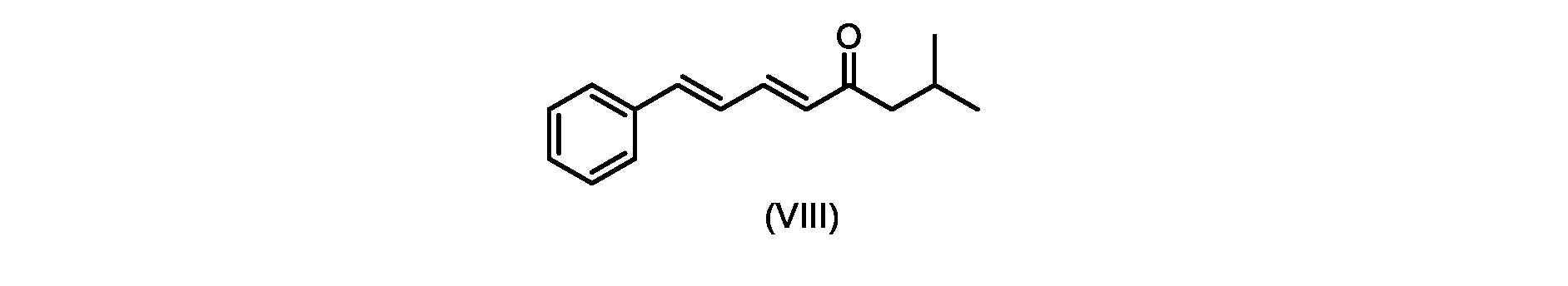
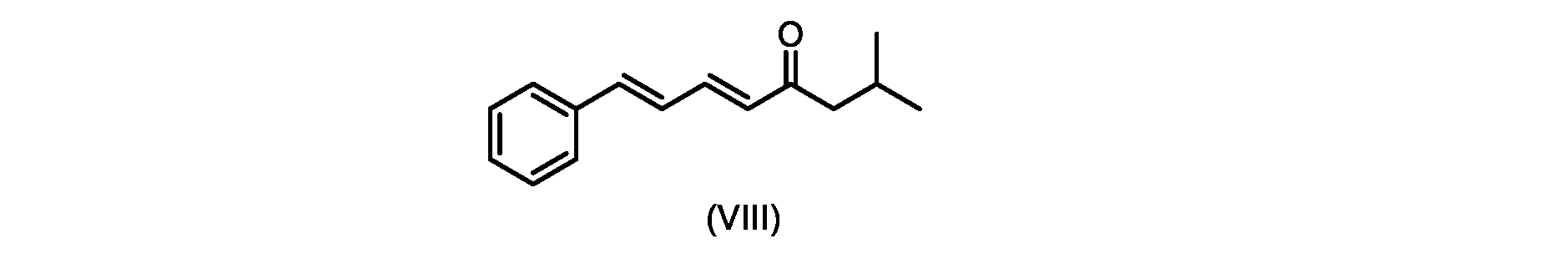
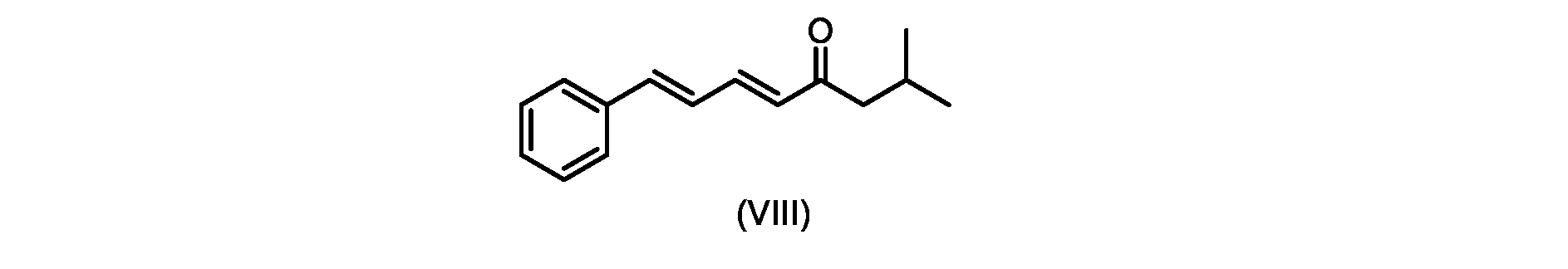
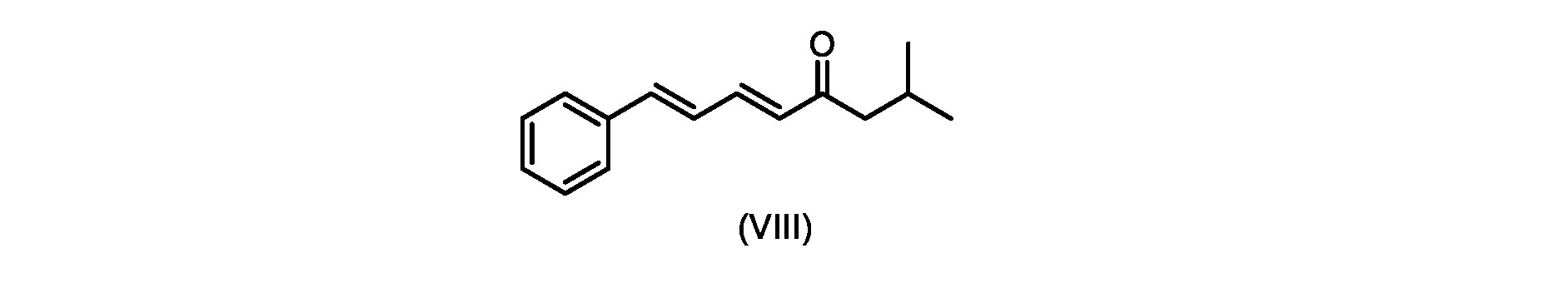
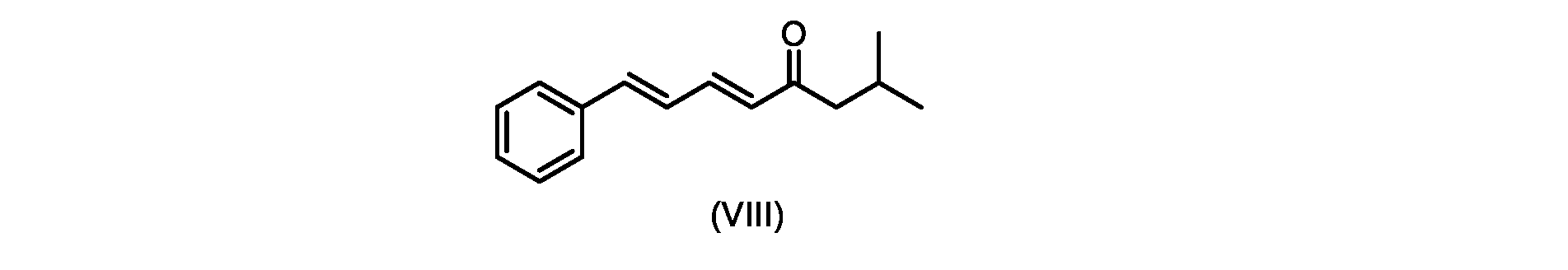
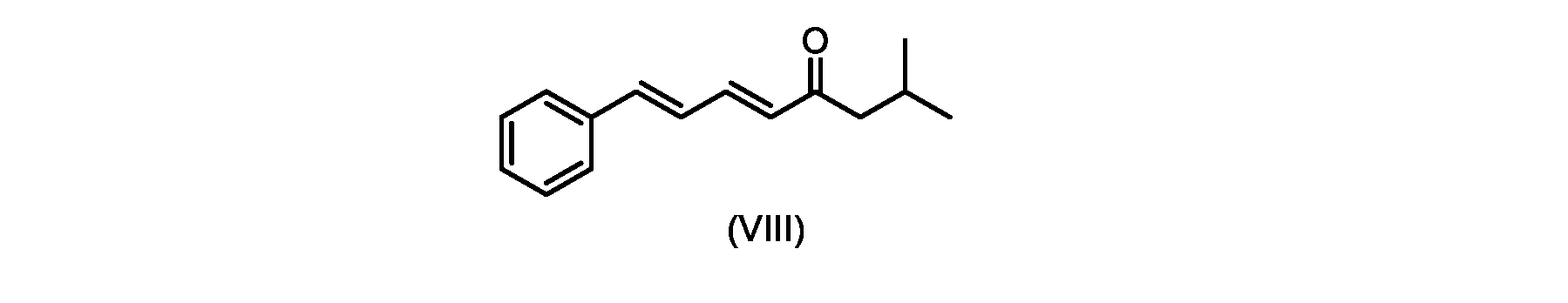
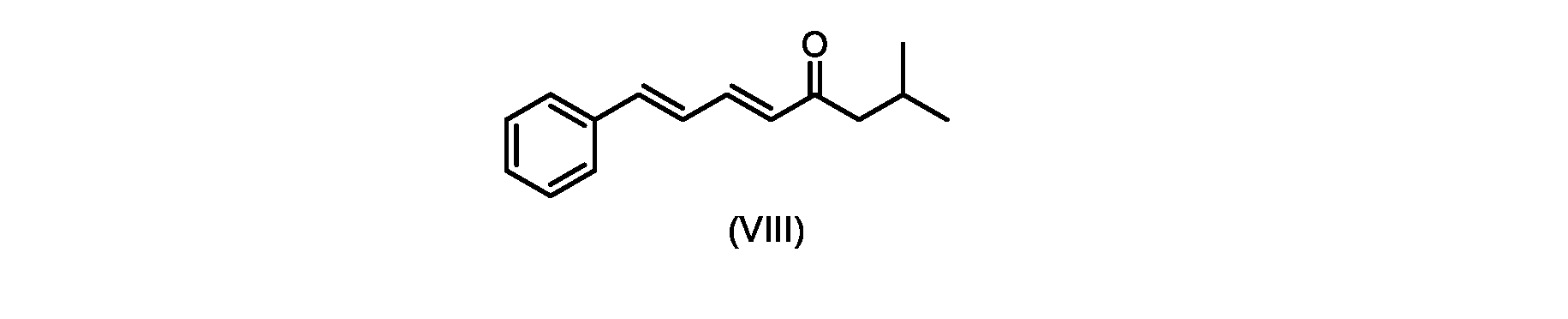
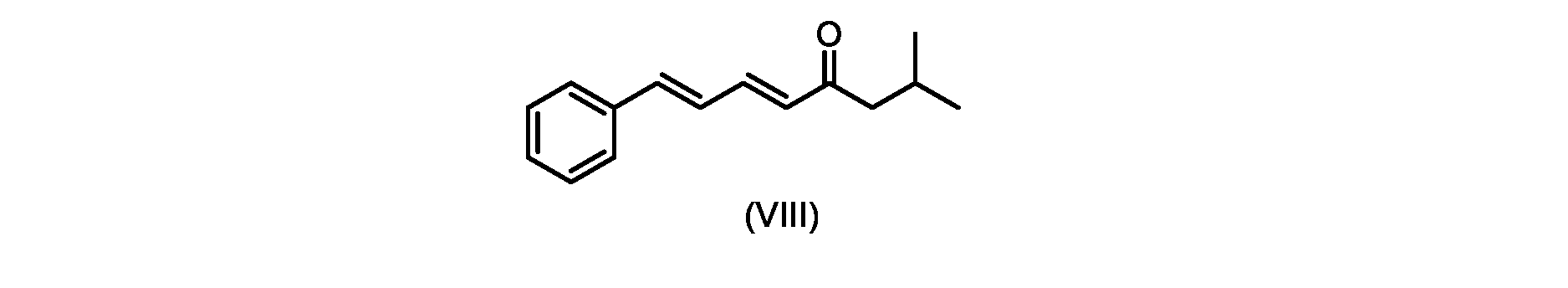
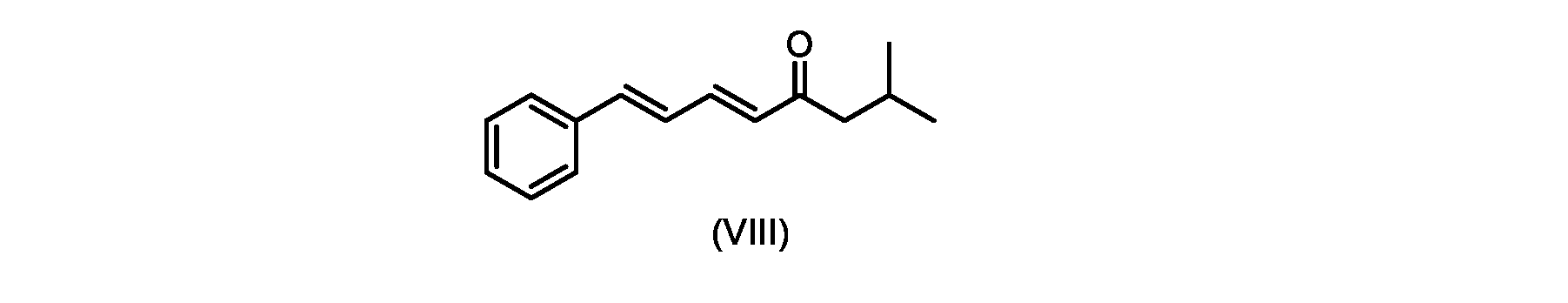
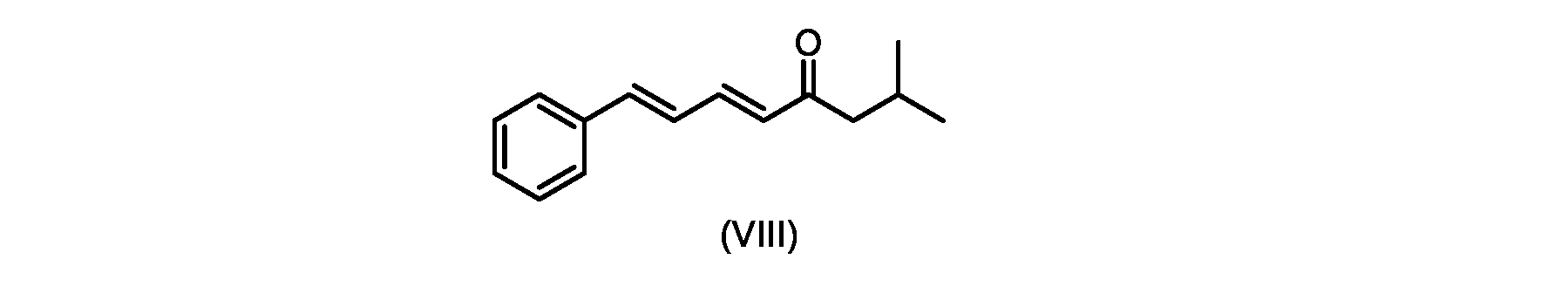
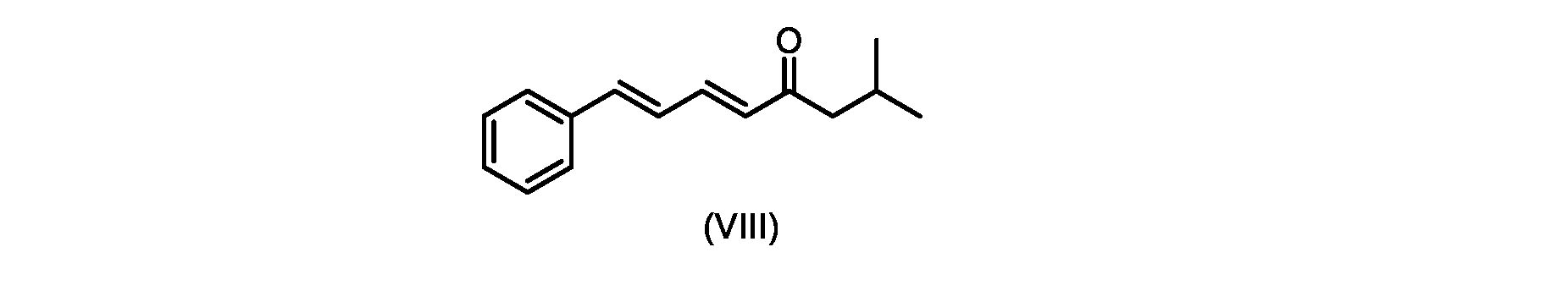
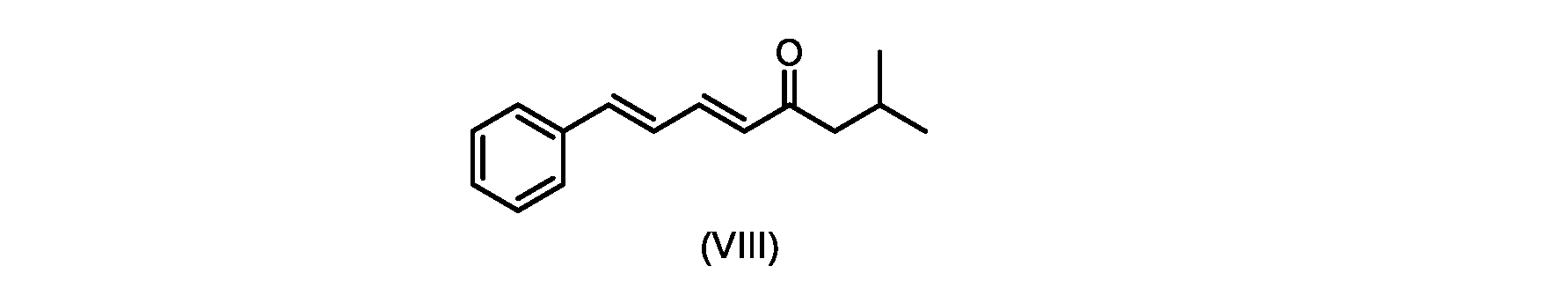
いくつかの実施形態では、式(IX)の化合物またはその塩の、式(VI)の化合物またはその塩への変換は、式(VIII)の化合物またはその塩を形成するための、式(IX)の化合物またはその塩の、メチルイソブチルケトンとの縮合と、
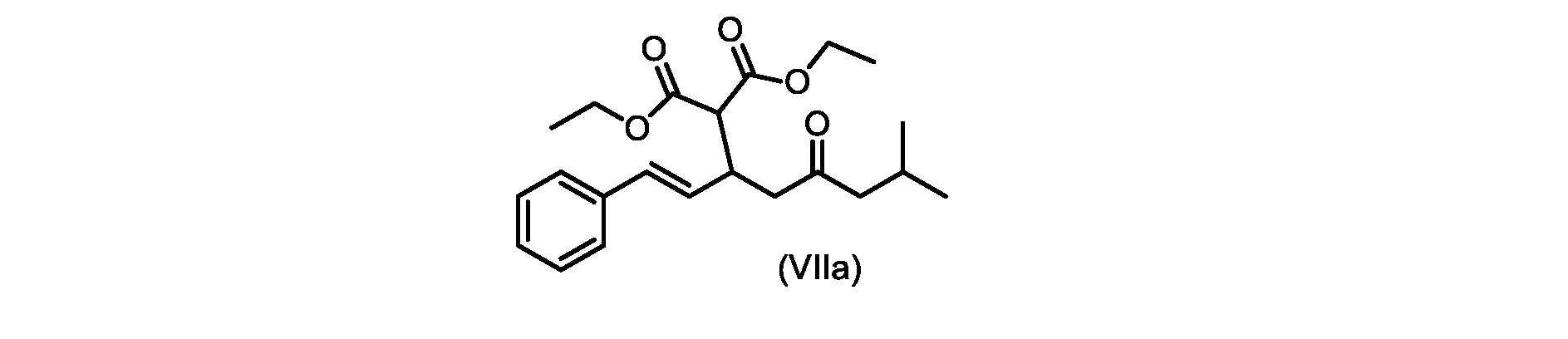
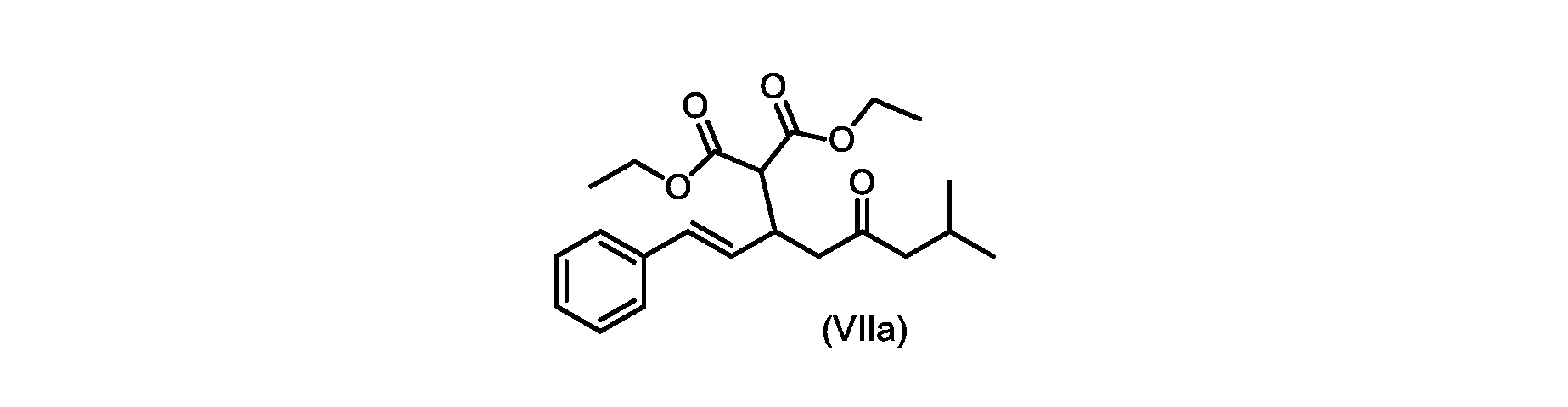
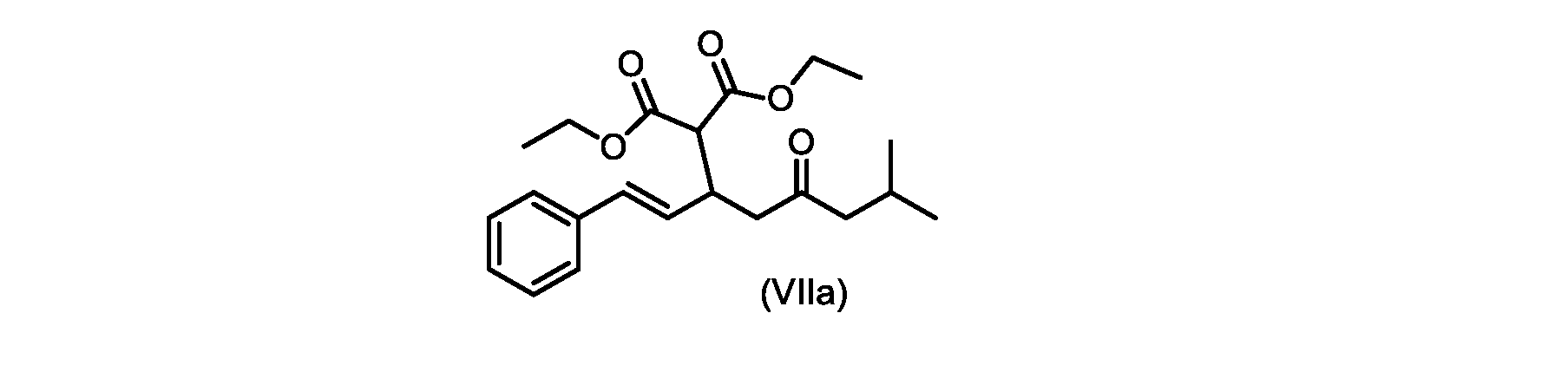
その後、式(VI)の化合物またはその塩を形成するための式(VII)の化合物またはその塩の加水分解と、を含む。
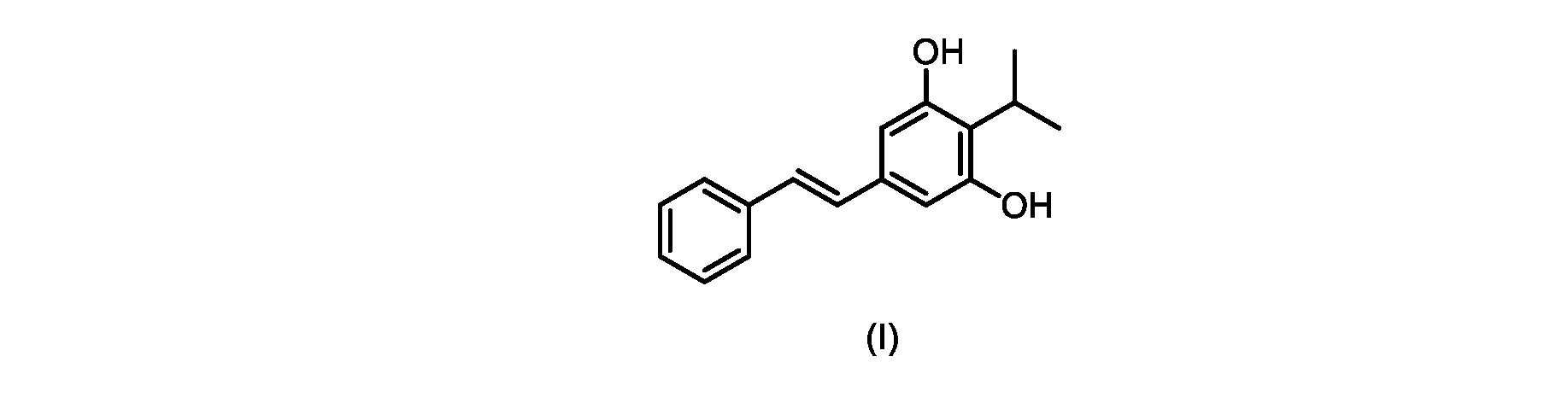
本明細書のいくつかの実施形態は、式(I)の化合物またはその塩もしくは溶媒和物を調製するためのプロセスであって、
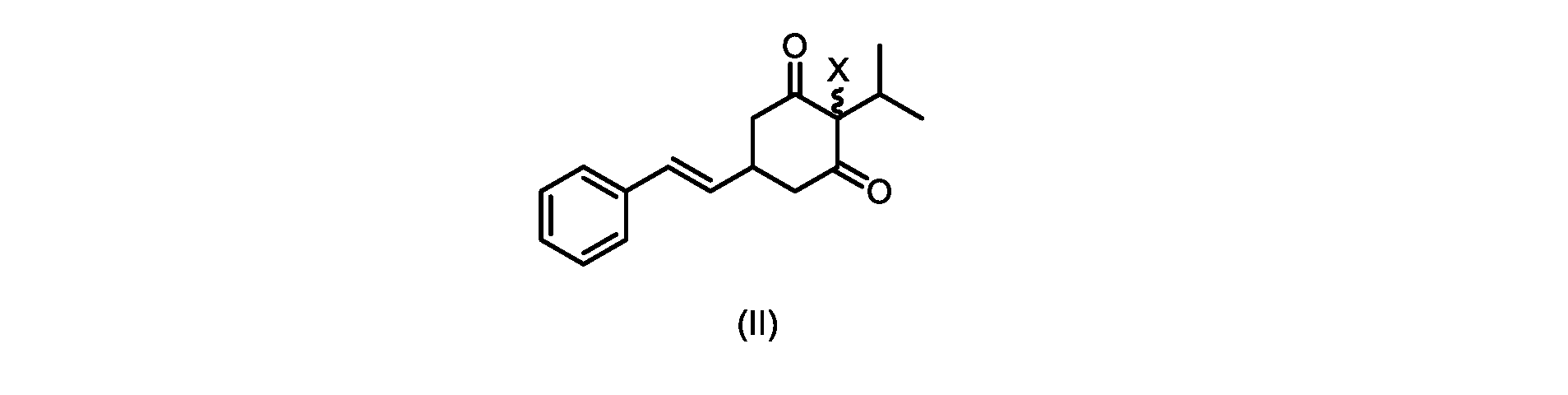
いくつかの実施形態では、式(I)の化合物またはその塩もしくは溶媒和物を調製するためのプロセスであって、
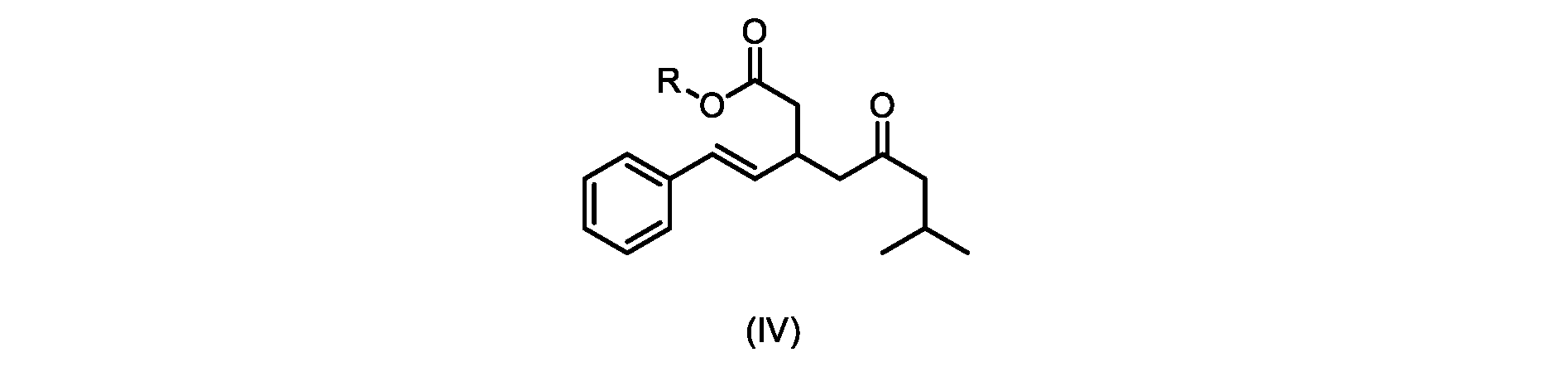
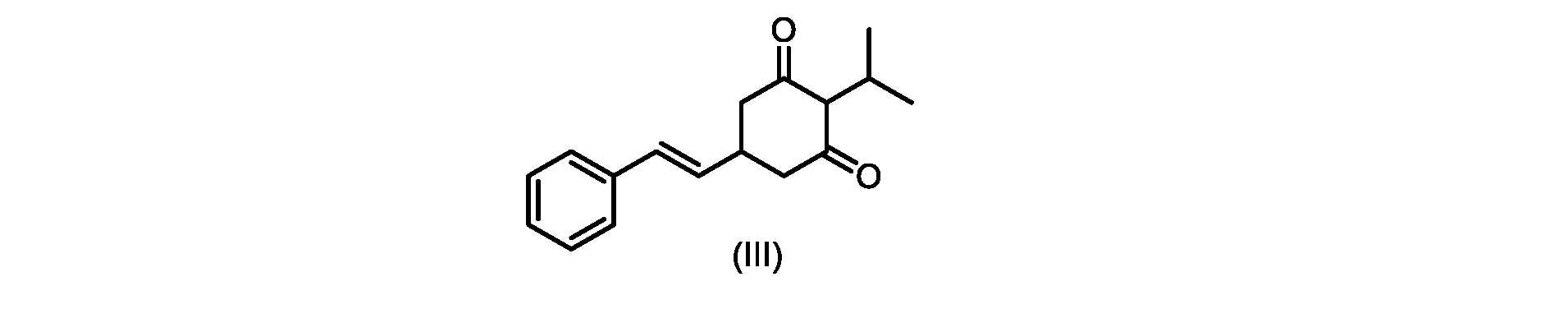
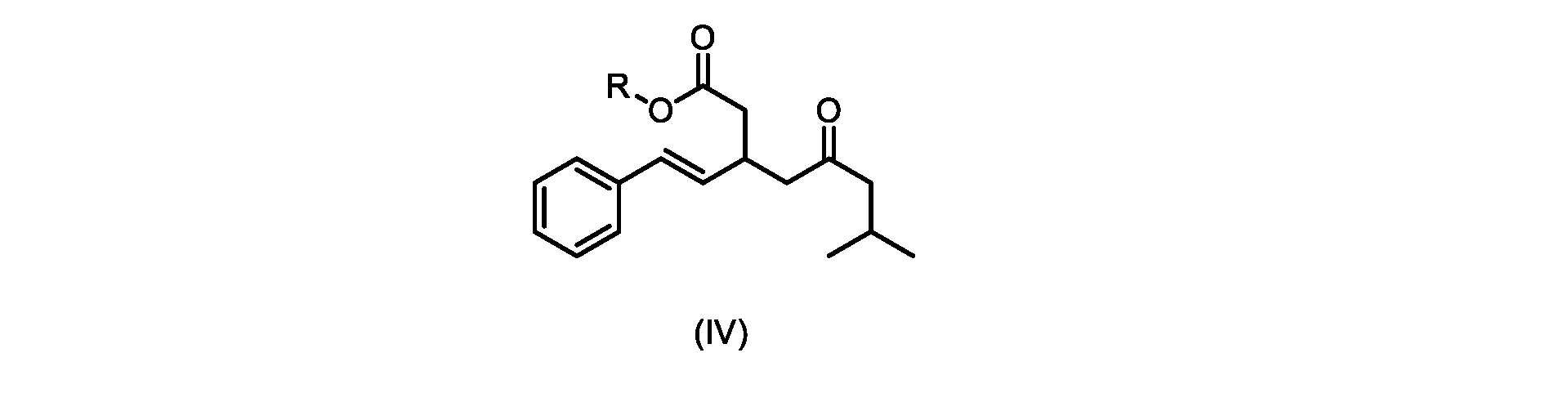
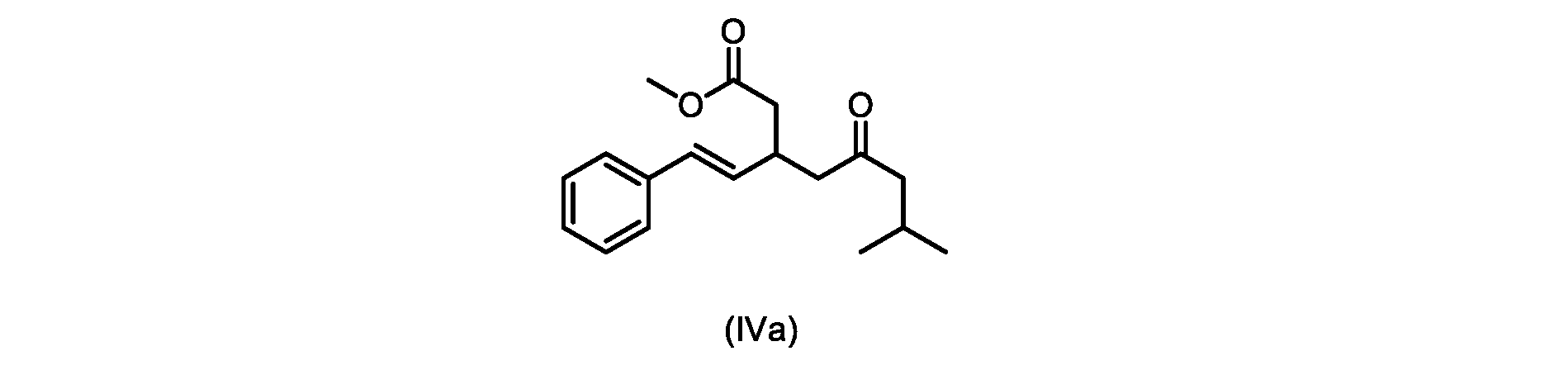
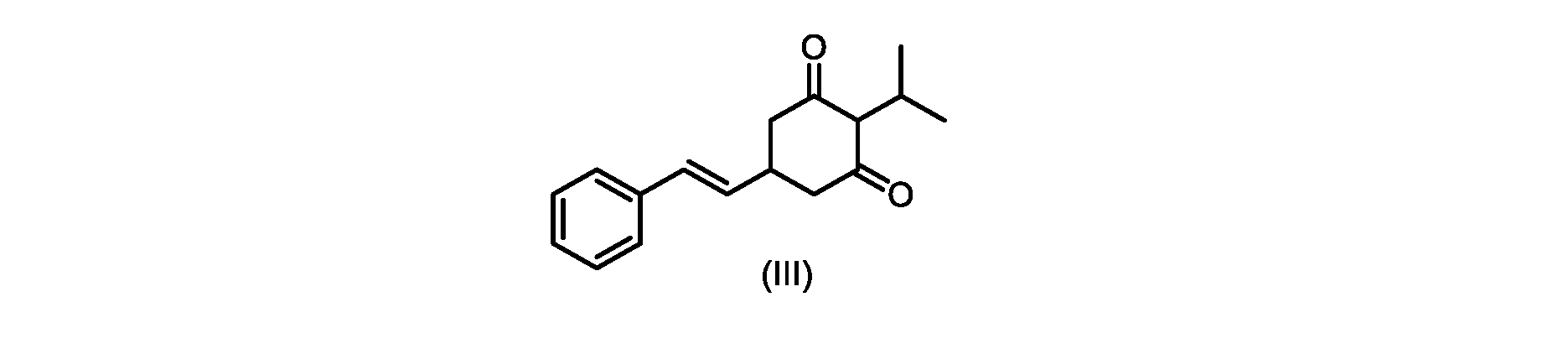
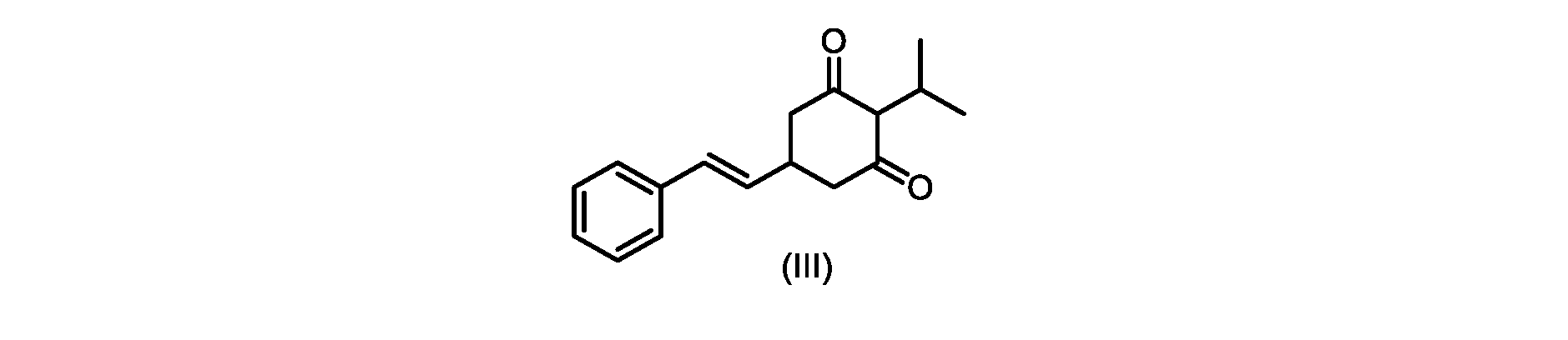
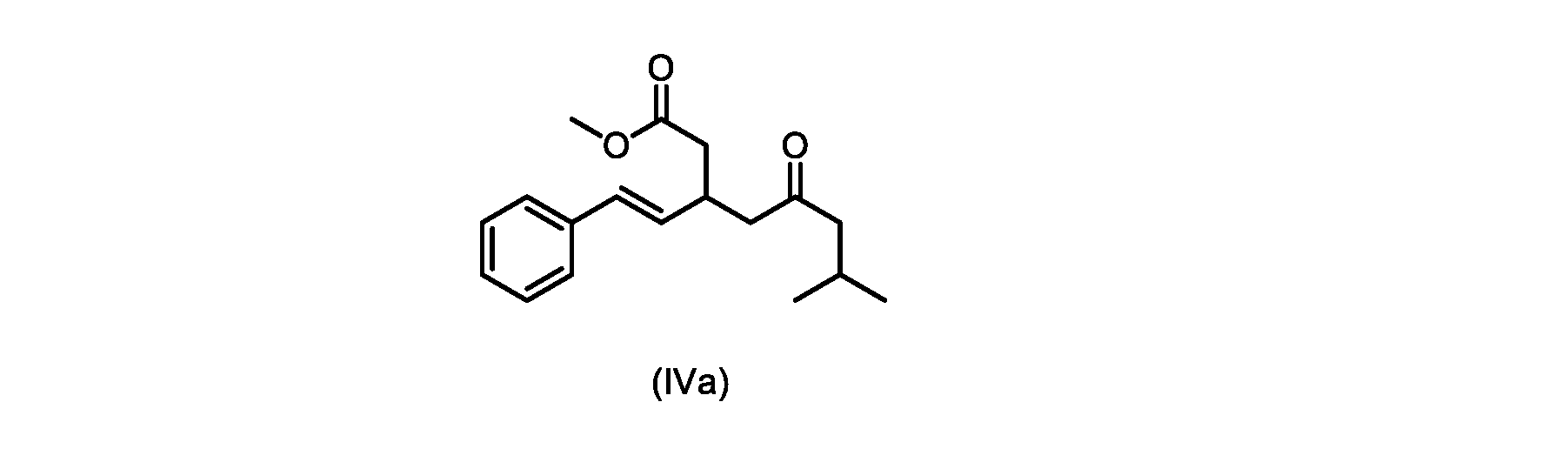
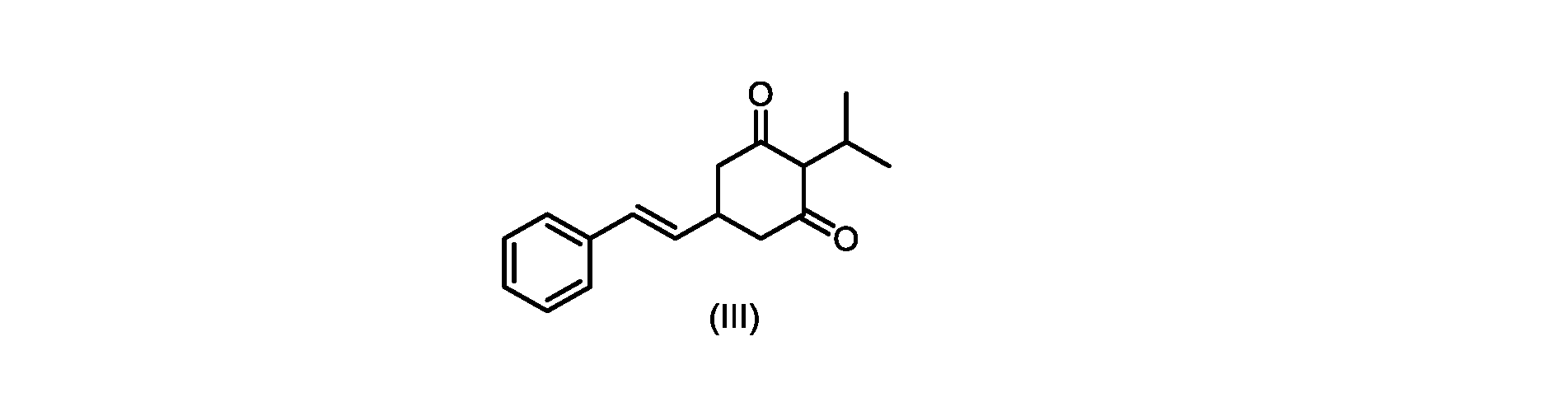
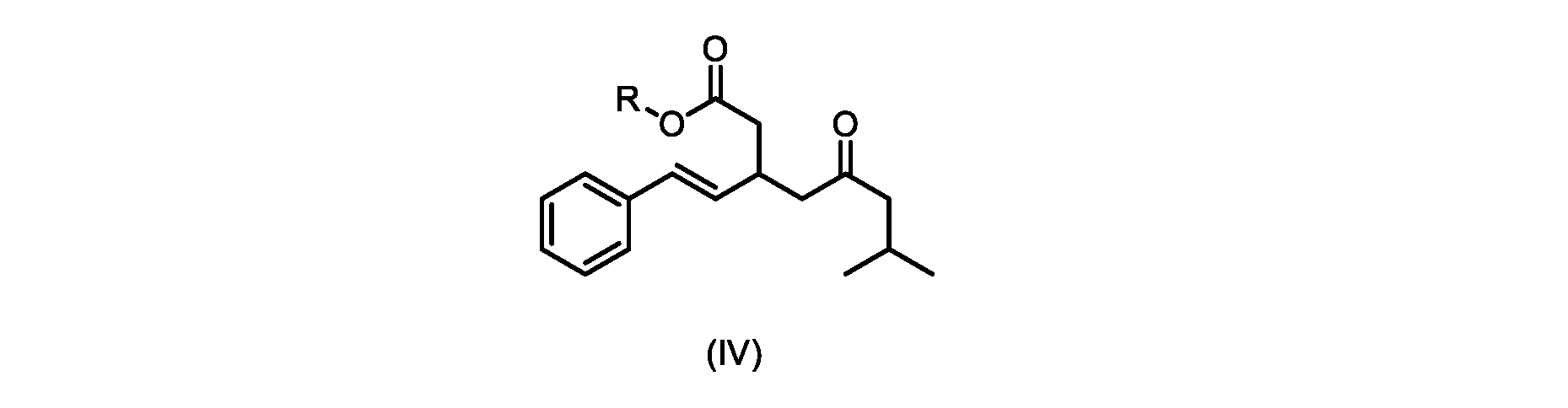
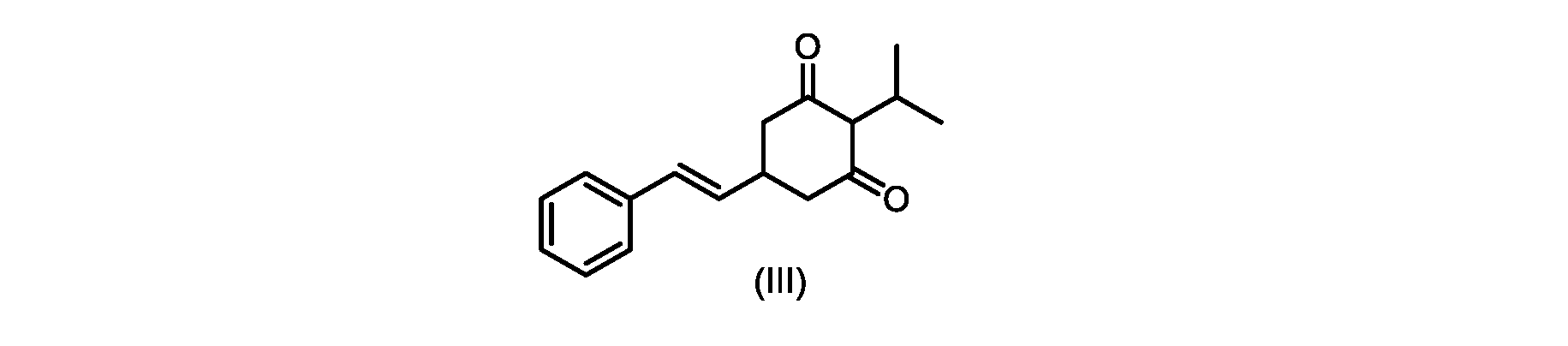
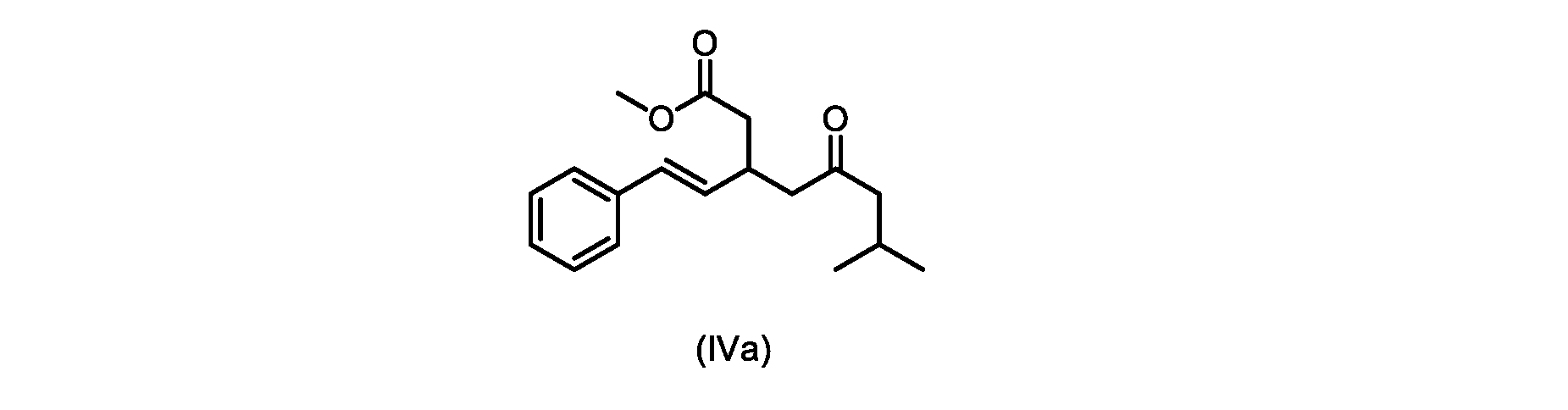
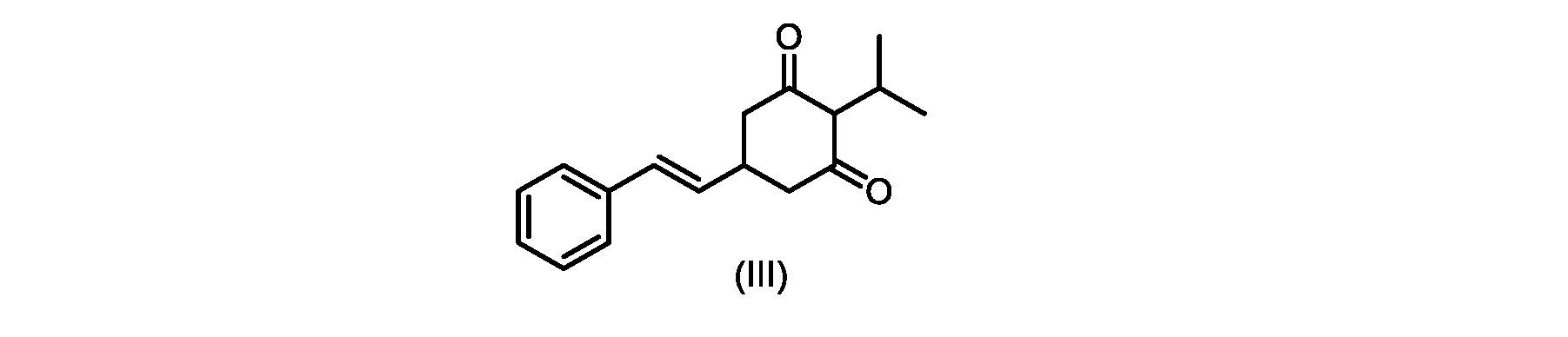
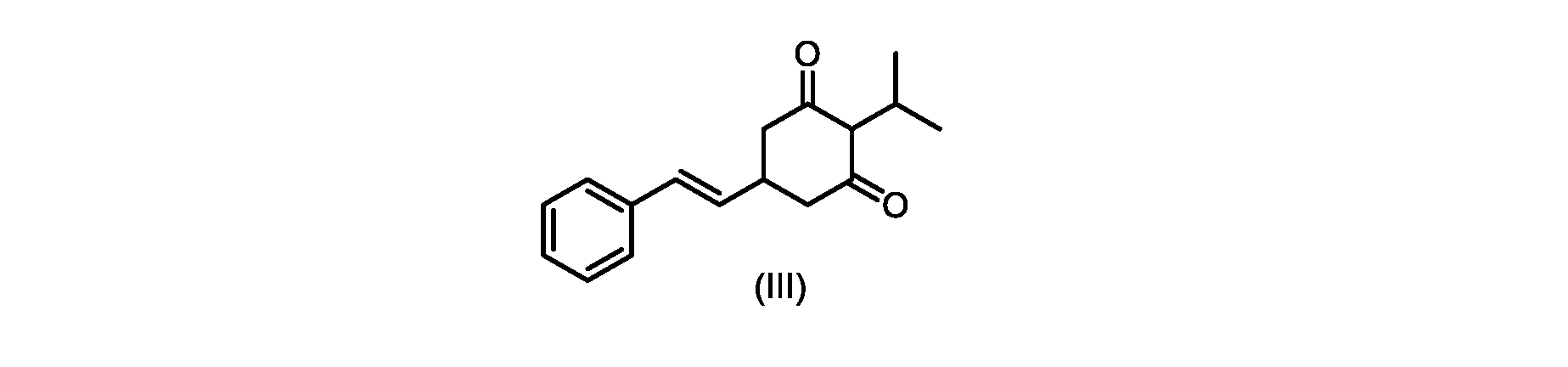
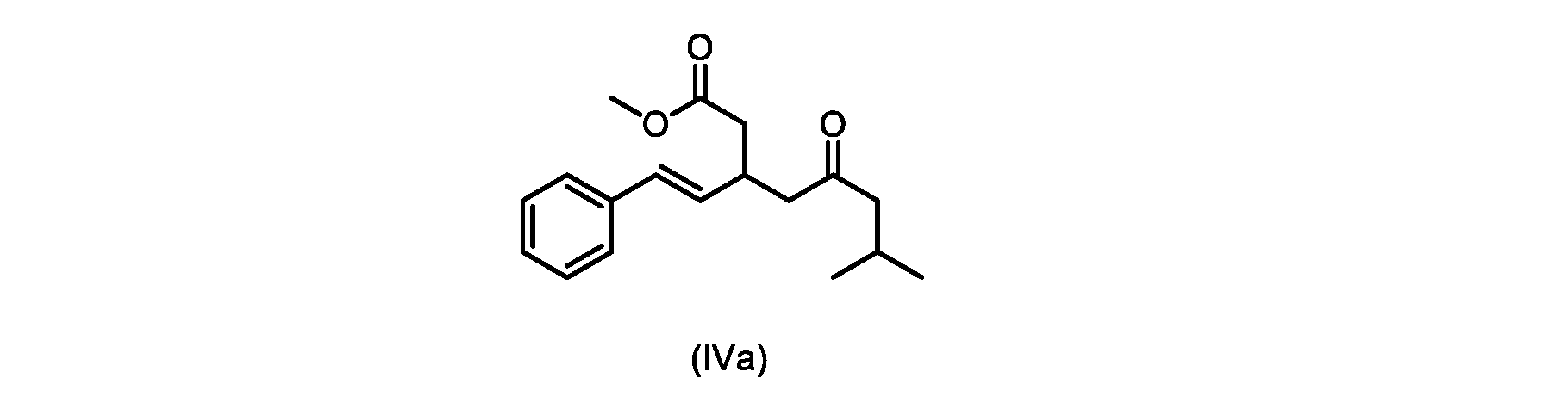
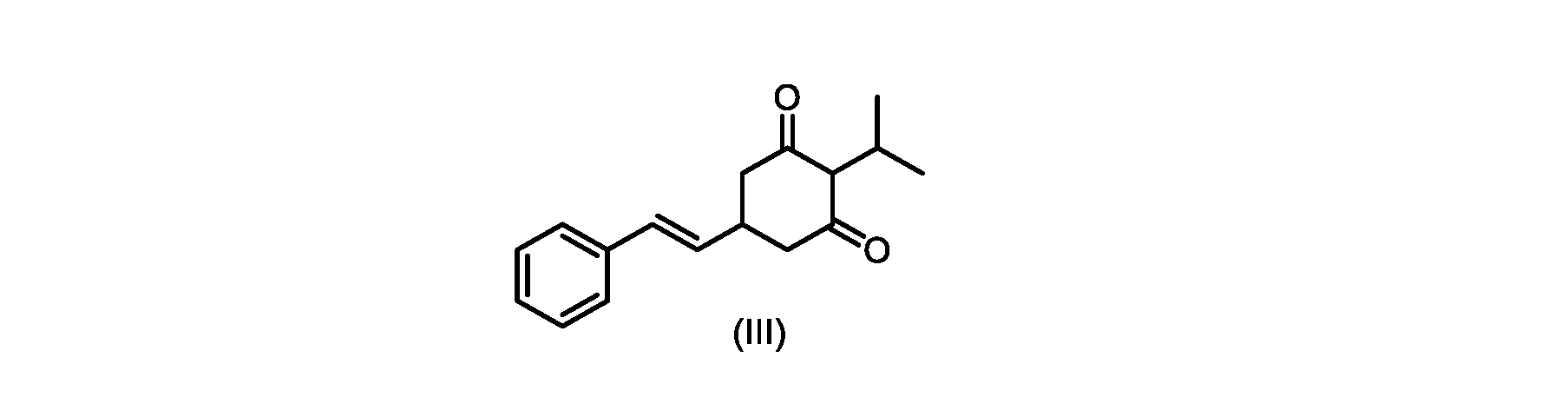
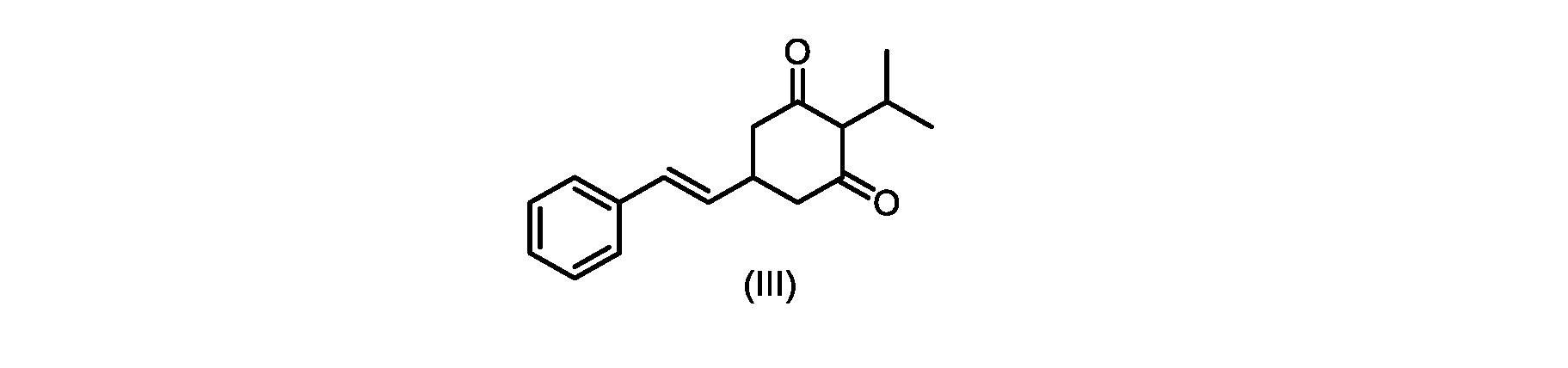
c)式(IV)の化合物またはその塩を環化して、式(III)の化合物またはその塩を形成することと、
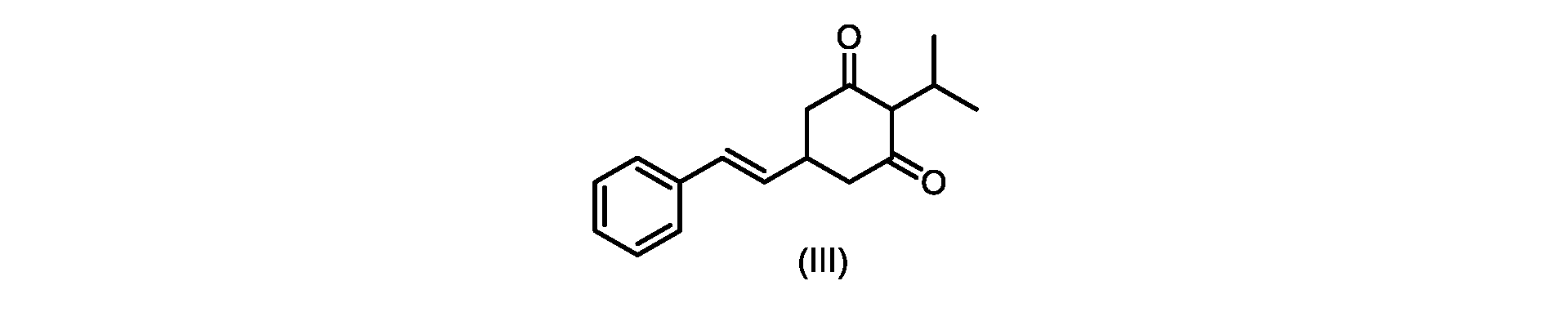
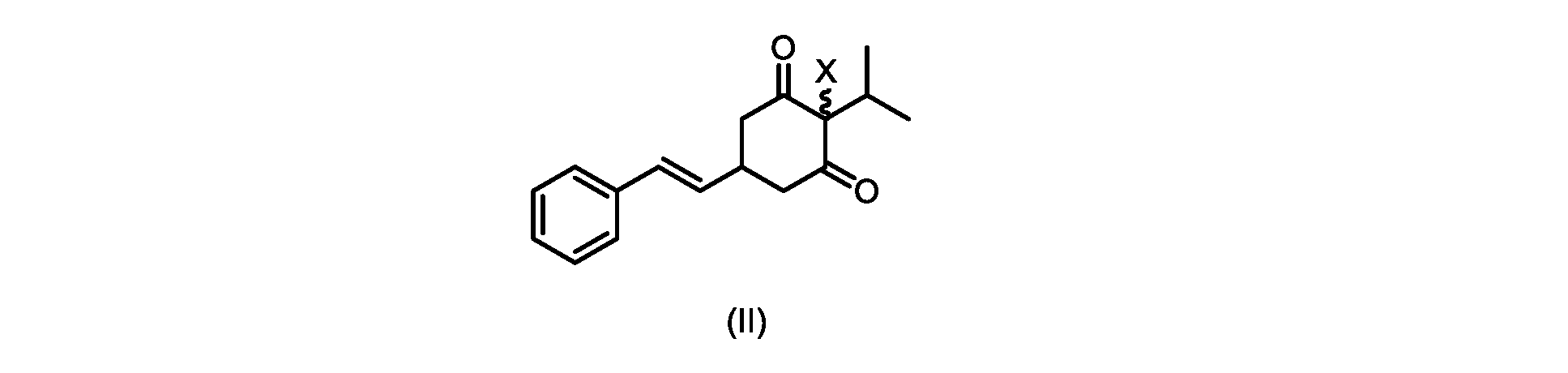
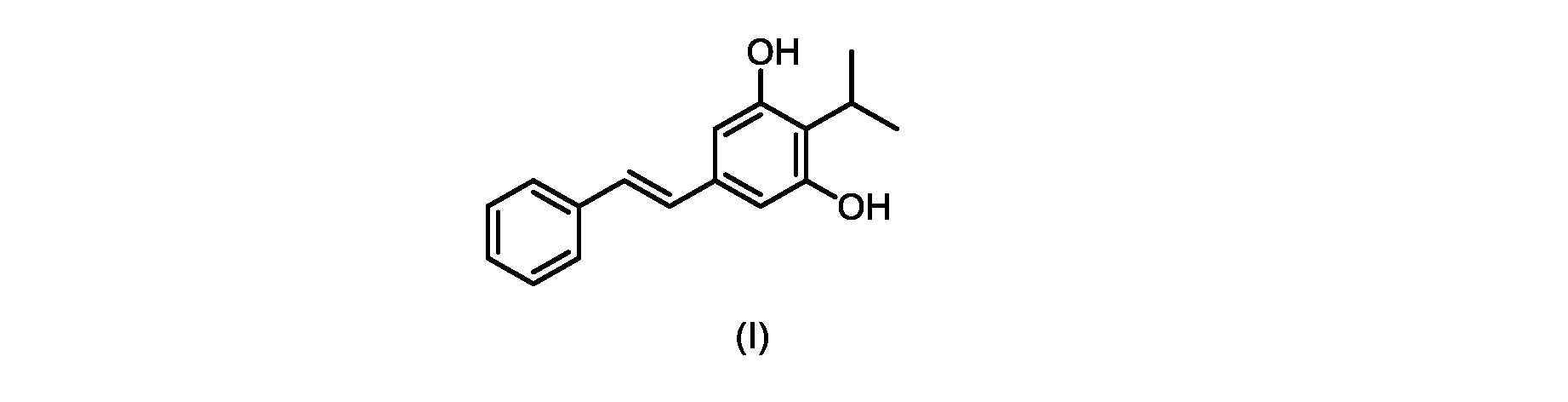
e)式(II)の化合物またはその塩を芳香族化して、式(I)の化合物またはその塩を形成することと、を含むプロセスが本明細書に記載される。
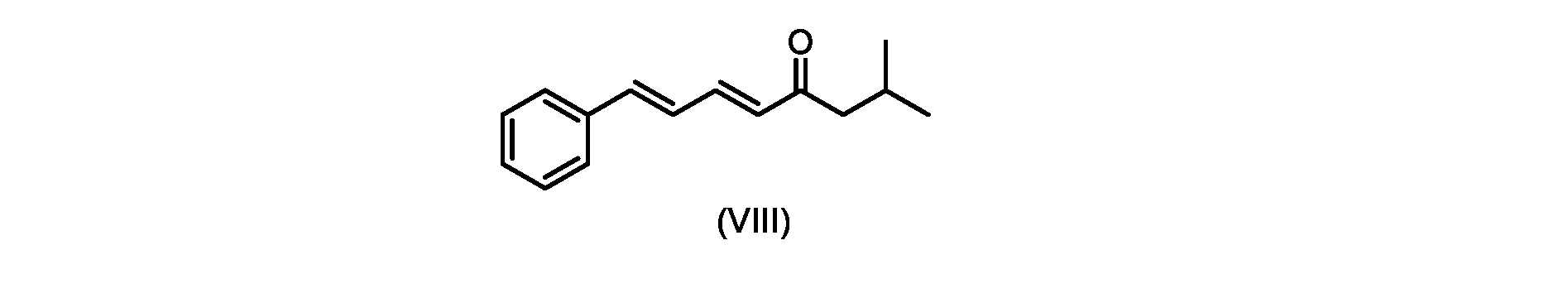
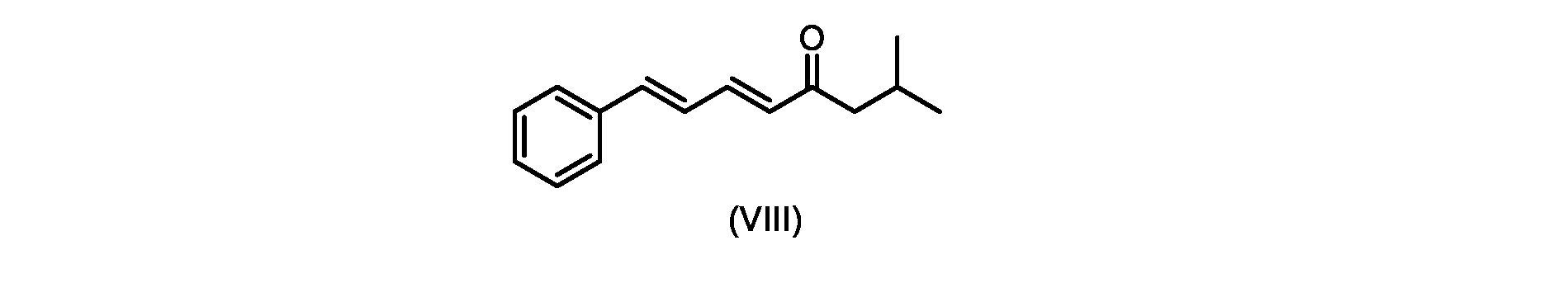
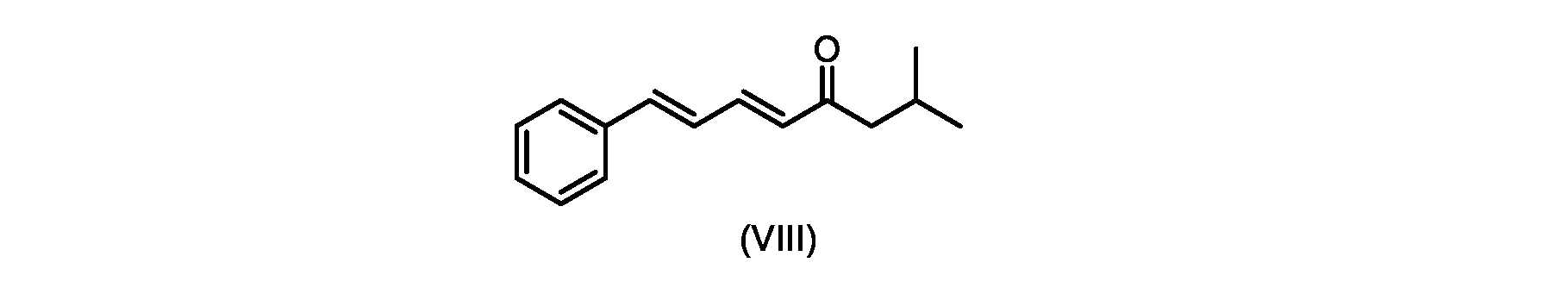
i.トランス-シンナムアルデヒドまたはその塩を、メチルイソブチルケトンと縮合して、式(VIII)の化合物またはその塩を形成することと、
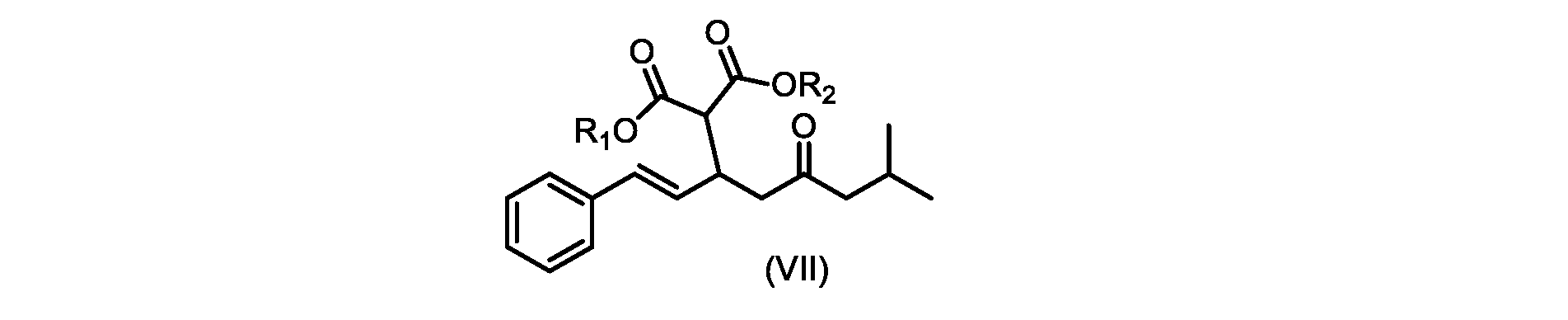
iii.式(VII)の化合物またはその塩を加水分解して、式(VI)の化合物またはその塩を形成することと、を含むプロセスによって調製される。
本明細書のいくつかの実施形態は、式(I)の化合物またはその塩もしくは溶媒和物を調製するためのプロセスであって、
i.メチルイソブチルケトンを、メタノール性水酸化ナトリウムの存在下でトランス-シンナムアルデヒドで処理して、式(VIII)の化合物を形成することと、
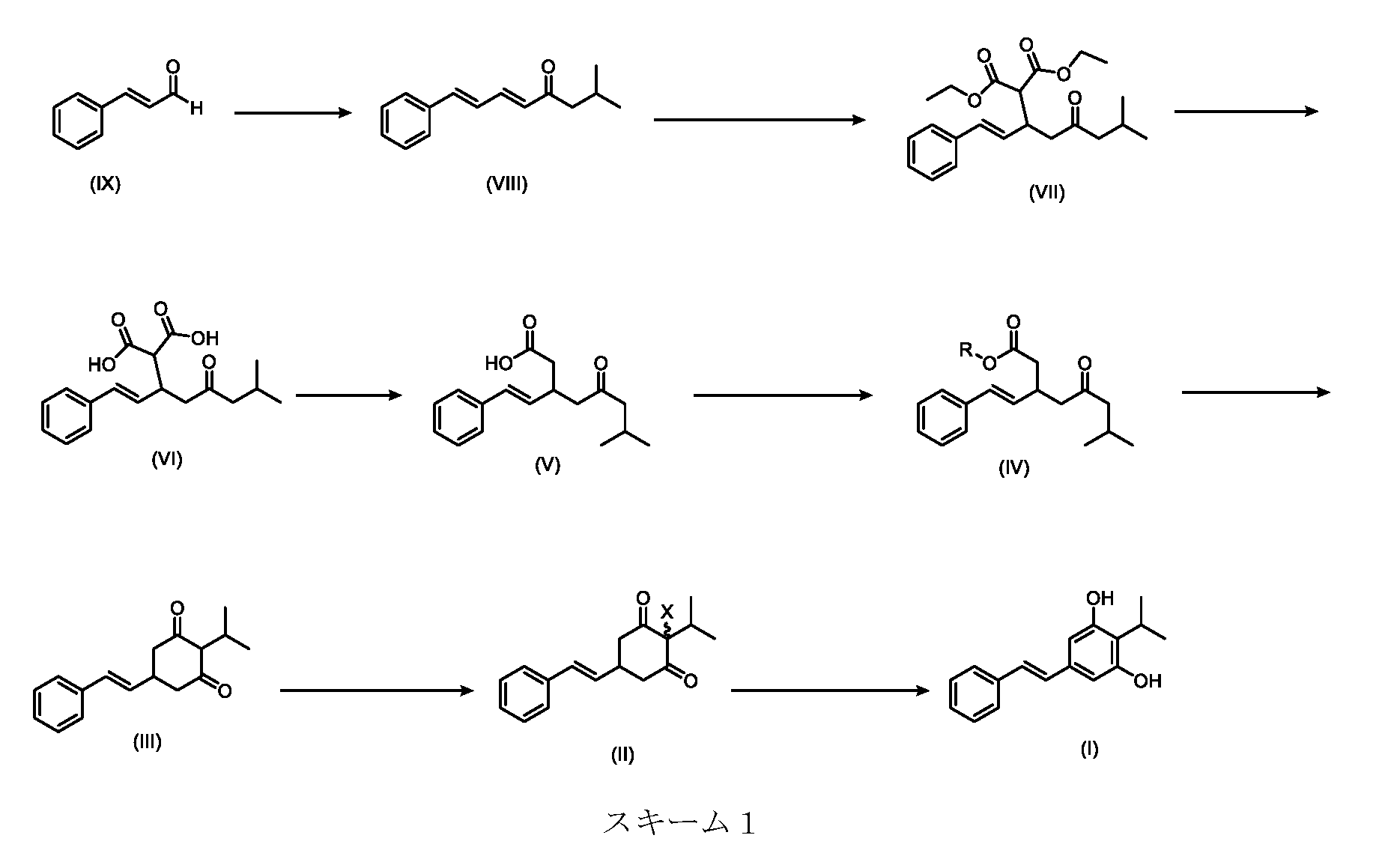
いくつかの実施形態は、下のスキーム2に示され、実施例で詳細に説明される本発明の特定の実施形態のプロセスを説明する。
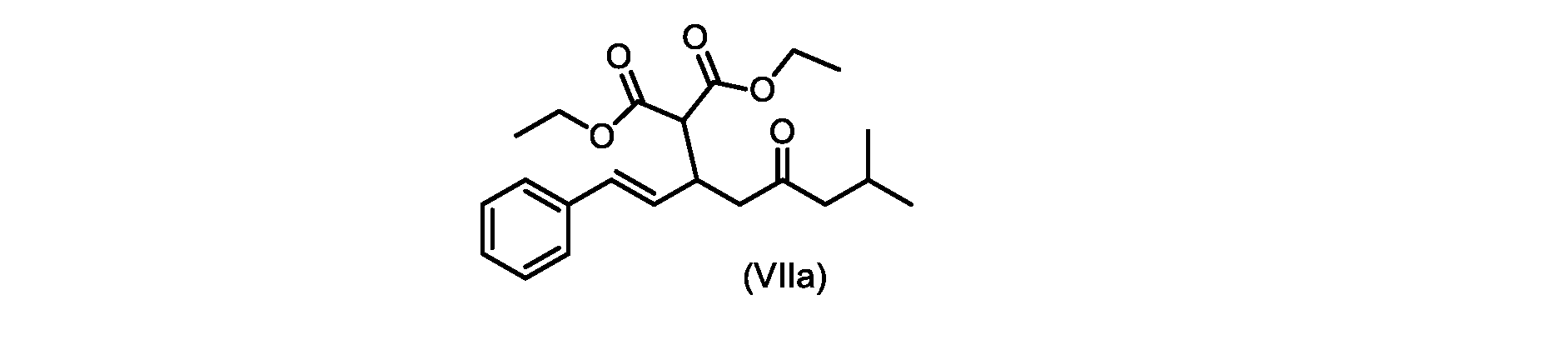
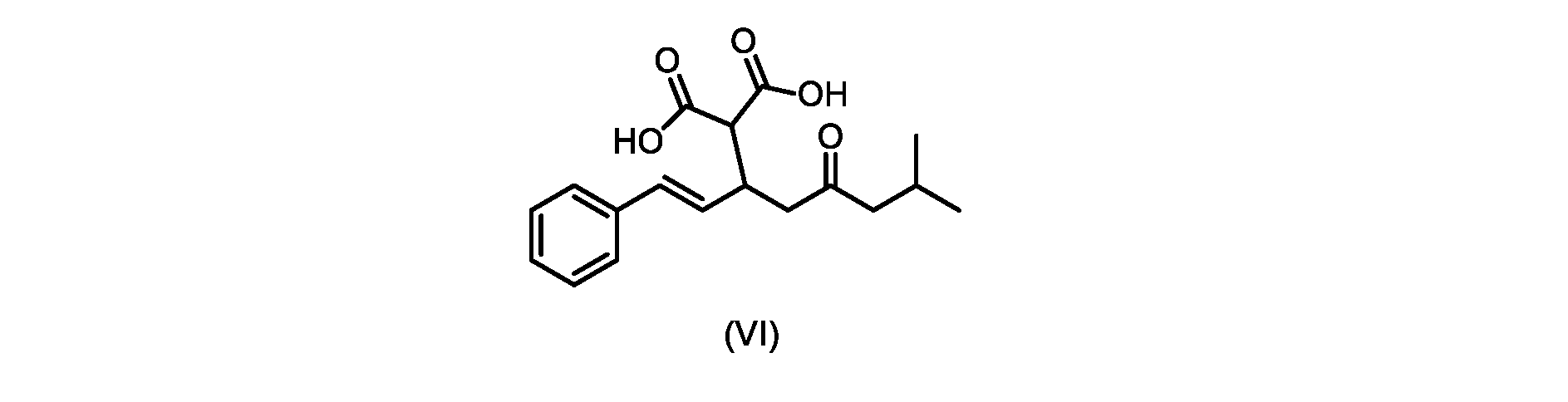
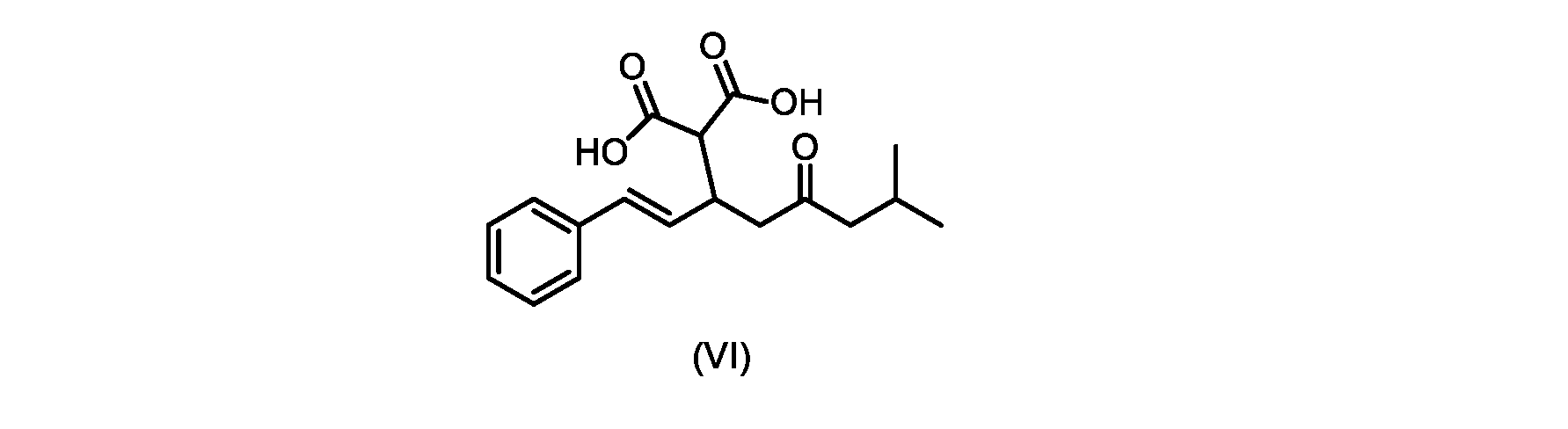
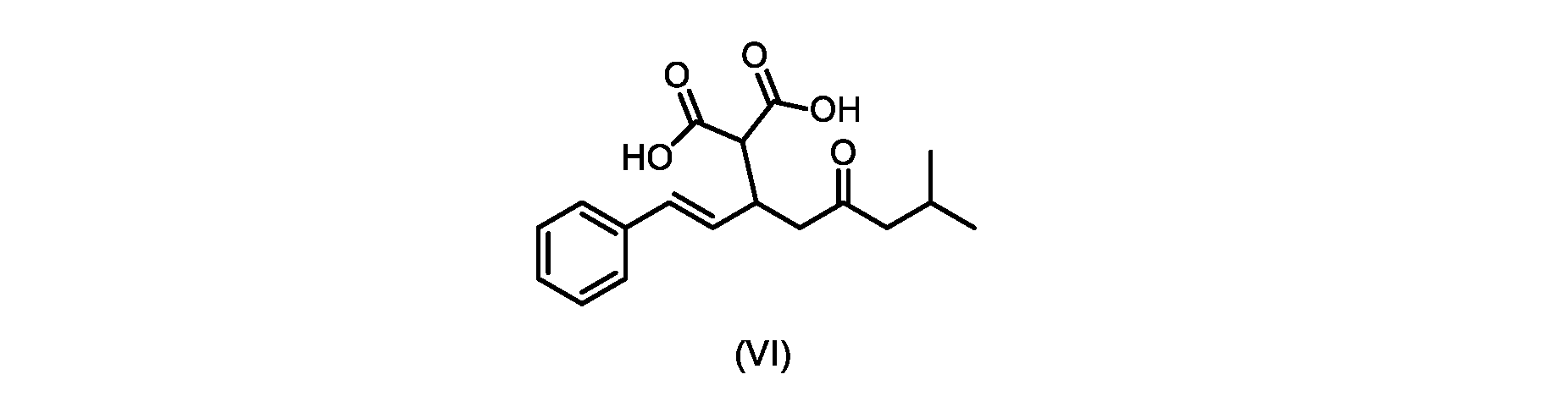
本明細書のいくつかの実施形態は、式(VI)の化合物またはその塩を調製するためのプロセスであって
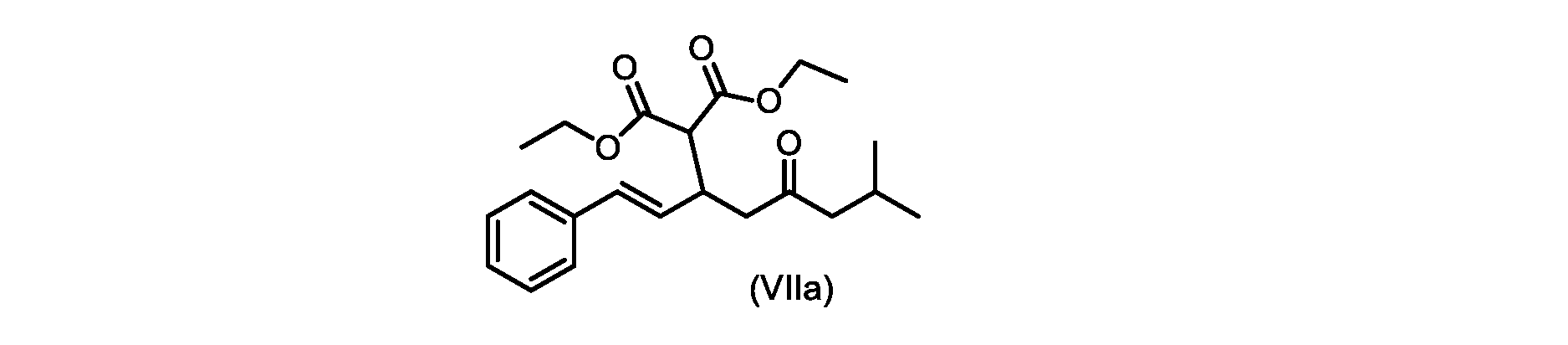
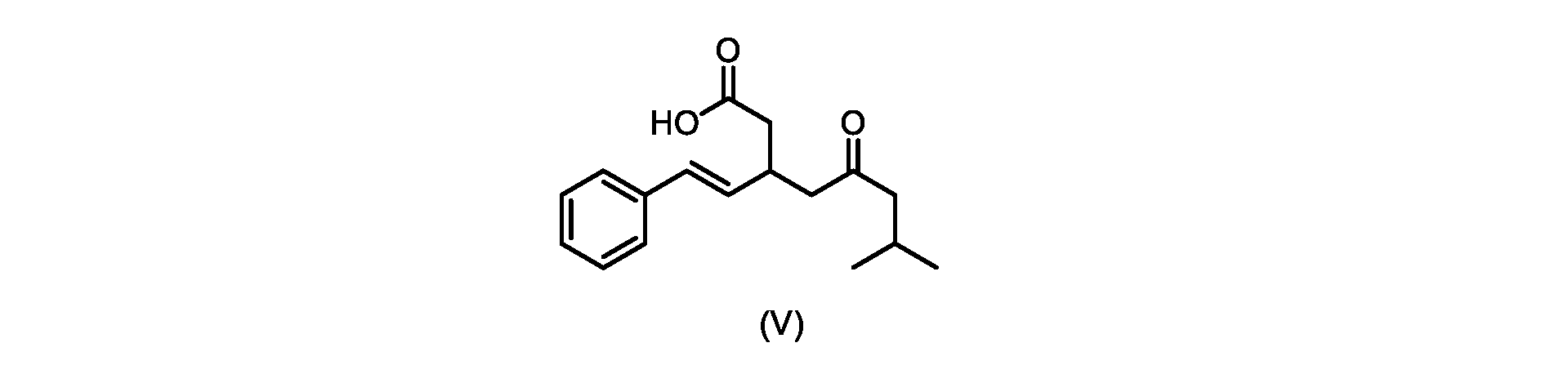
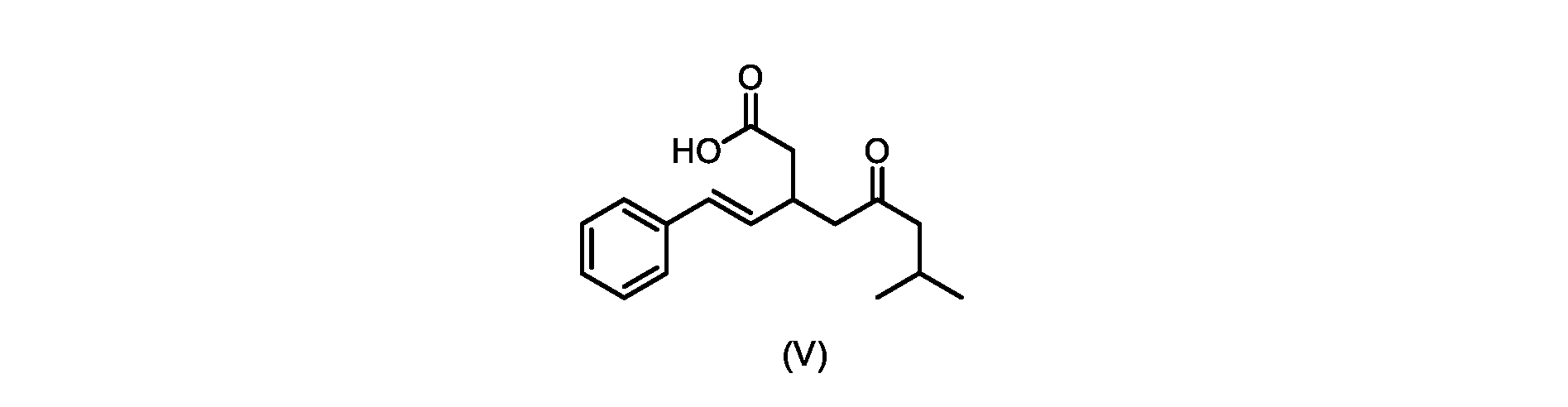
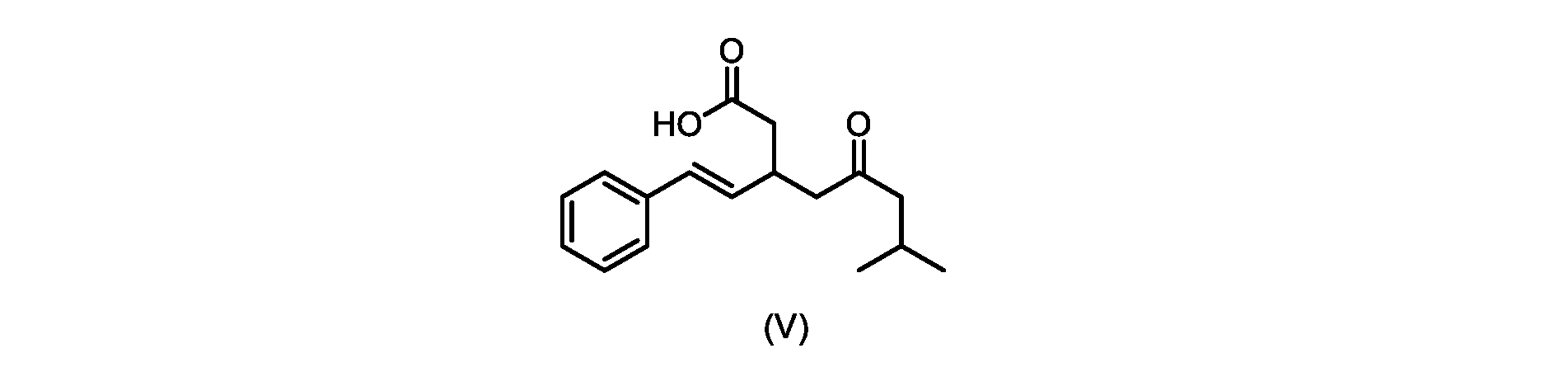
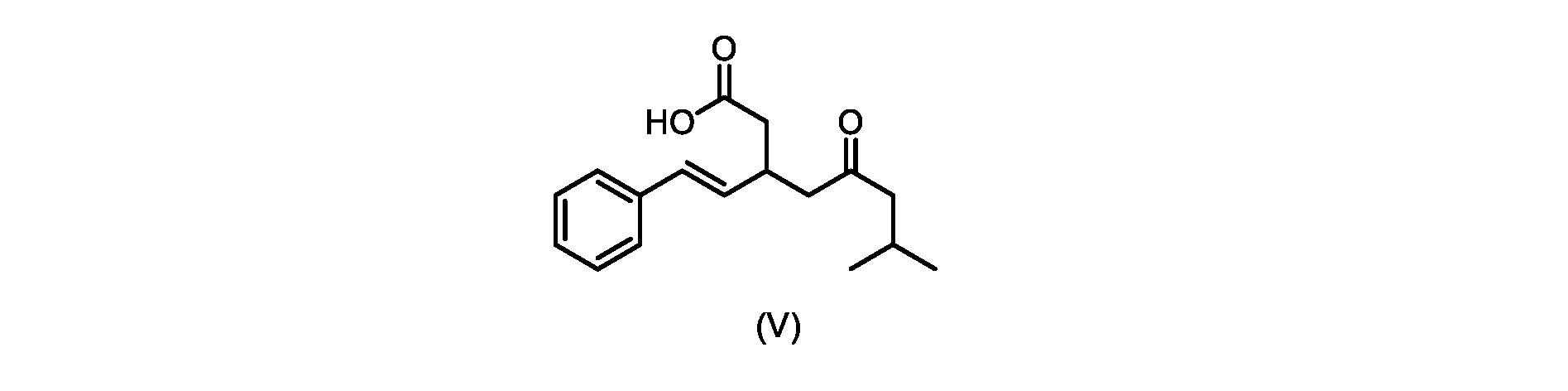
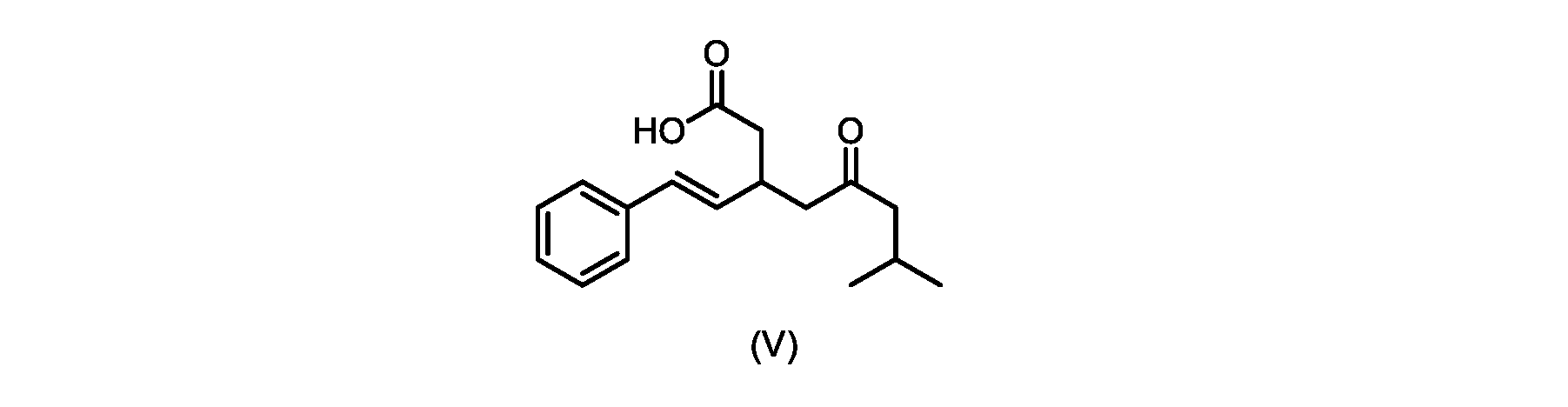
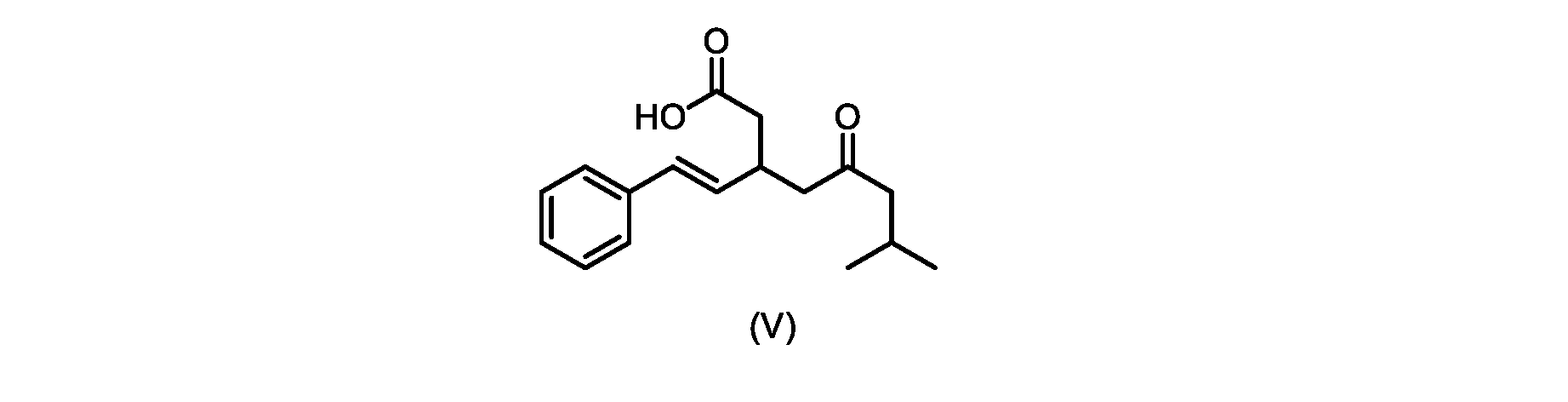
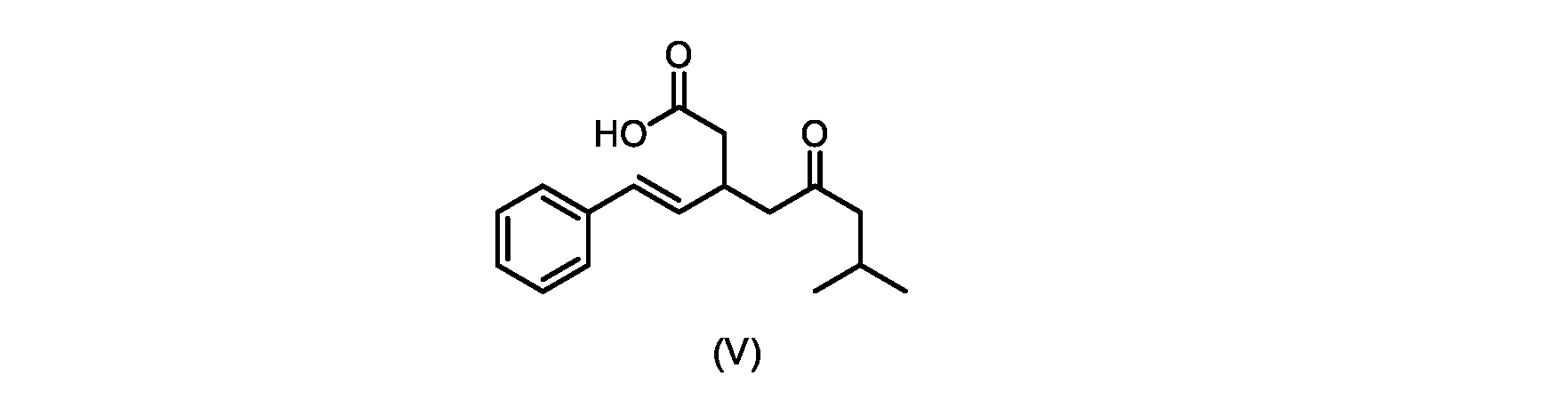
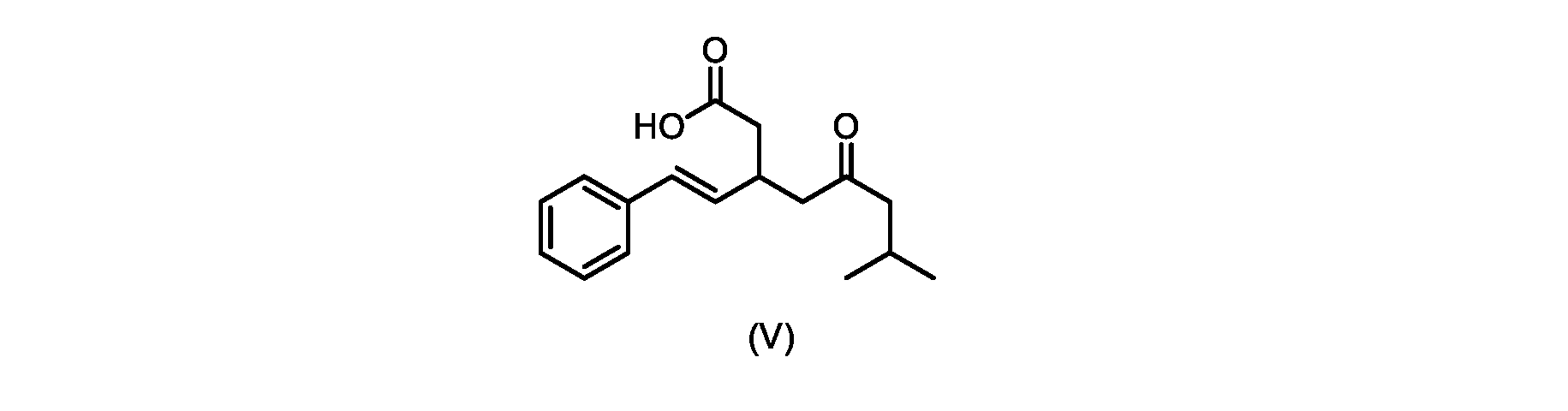
本明細書のいくつかの実施形態は、式(V)の化合物またはその塩を調製するためのプロセスであって、

a)トランス-シンナムアルデヒドまたはその塩を、メチルイソブチルケトンと縮合して、式(VIII)の化合物またはその塩を形成することと、
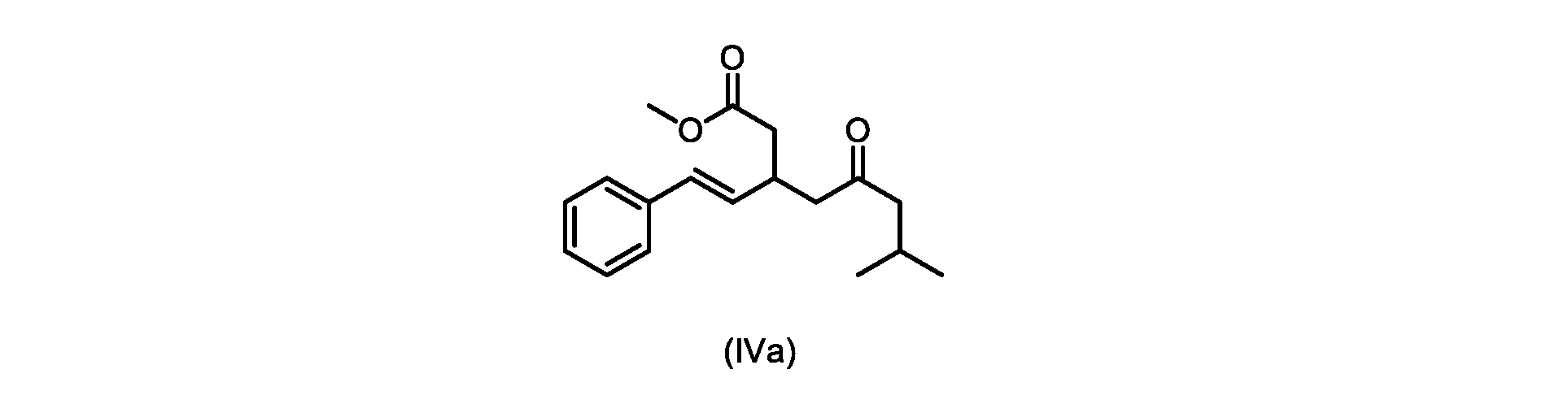
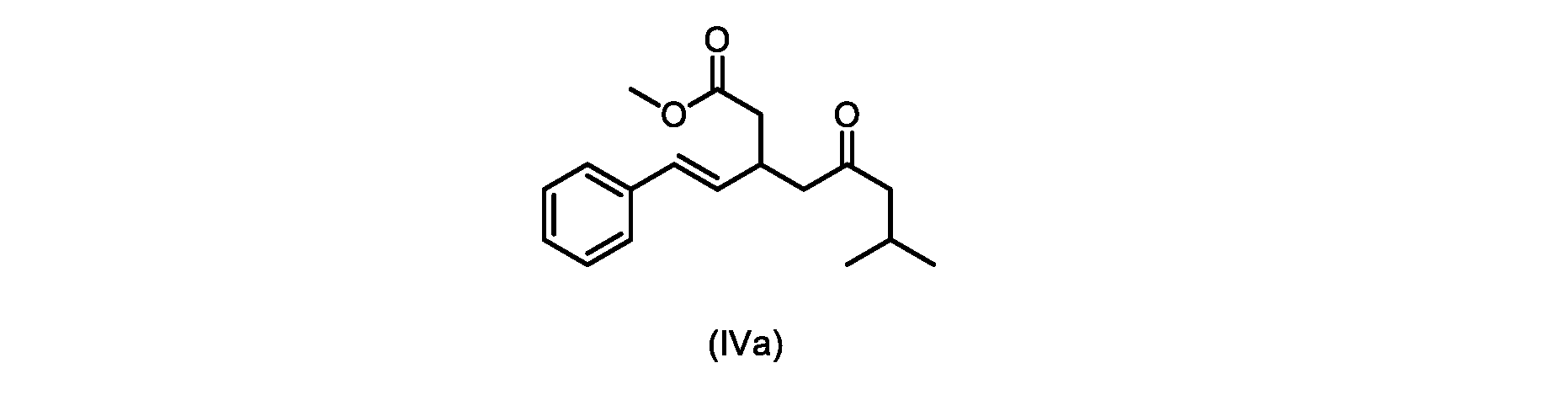
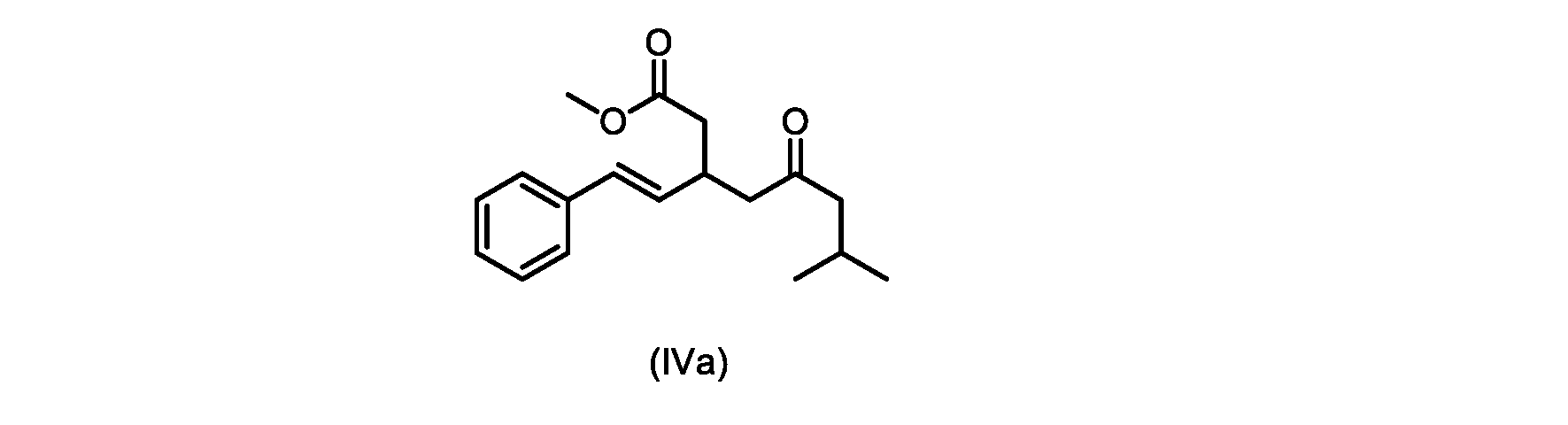
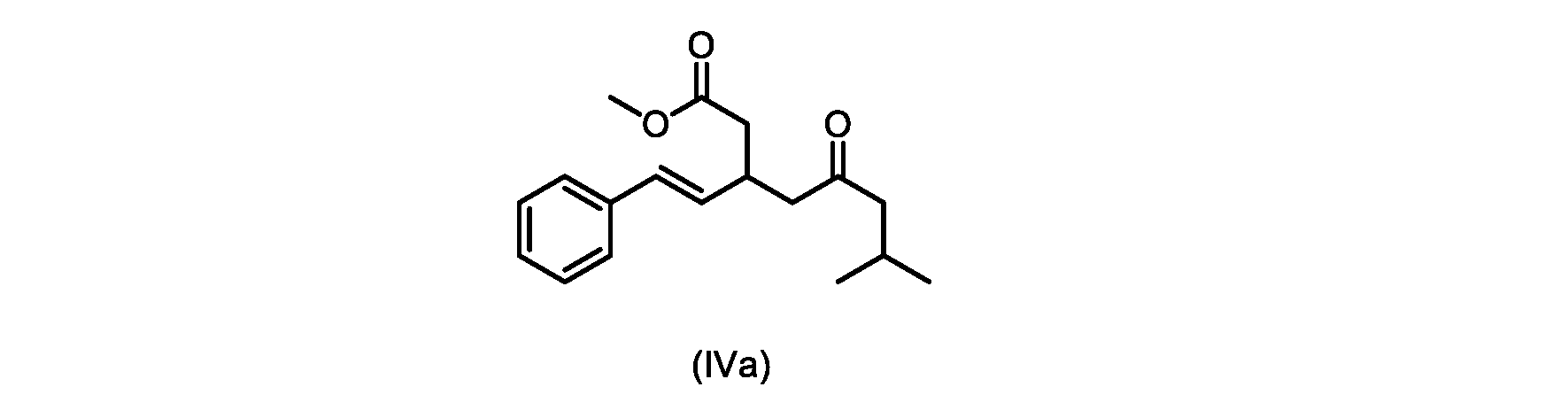
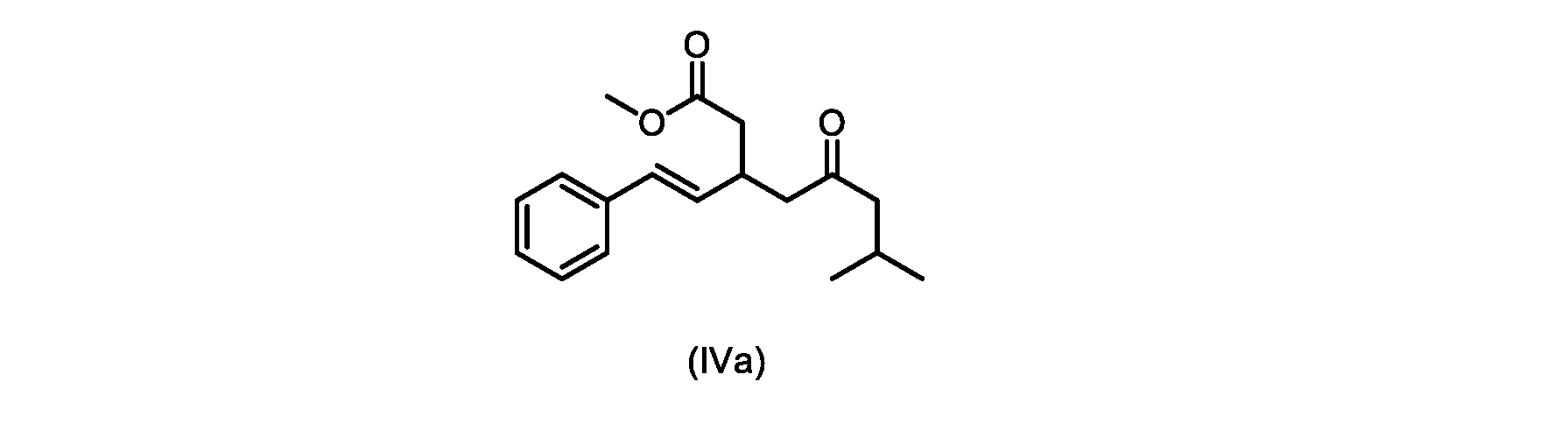
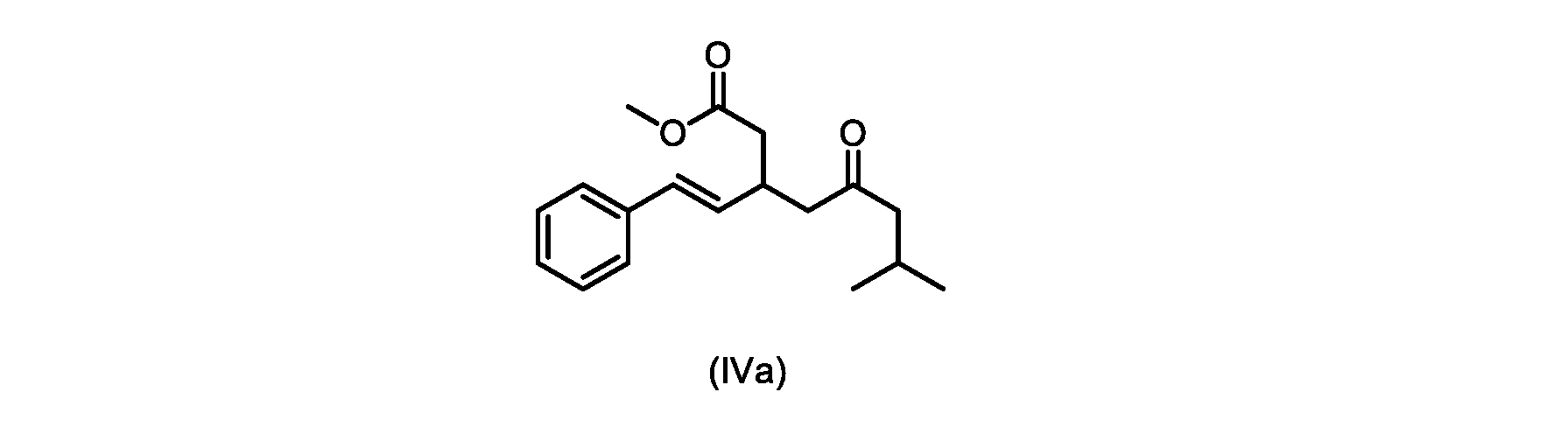
本明細書のいくつかの実施形態は、式(IVa)の化合物またはその塩を調製するためのプロセスであって、
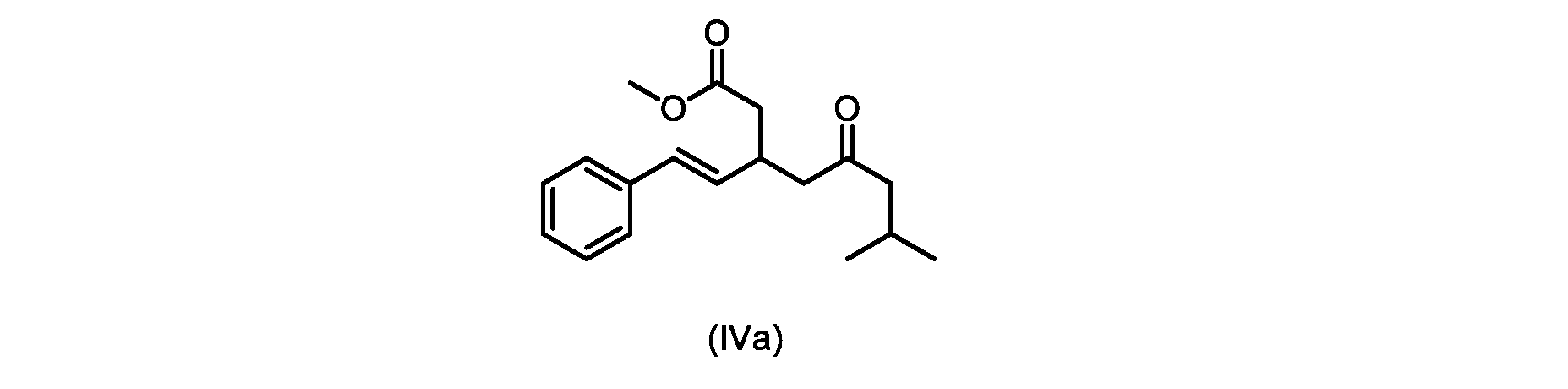
i.トランス-シンナムアルデヒドまたはその塩またはその塩を、メチルイソブチルケトンと縮合して、式(VIII)の化合物またはその塩を形成することと、
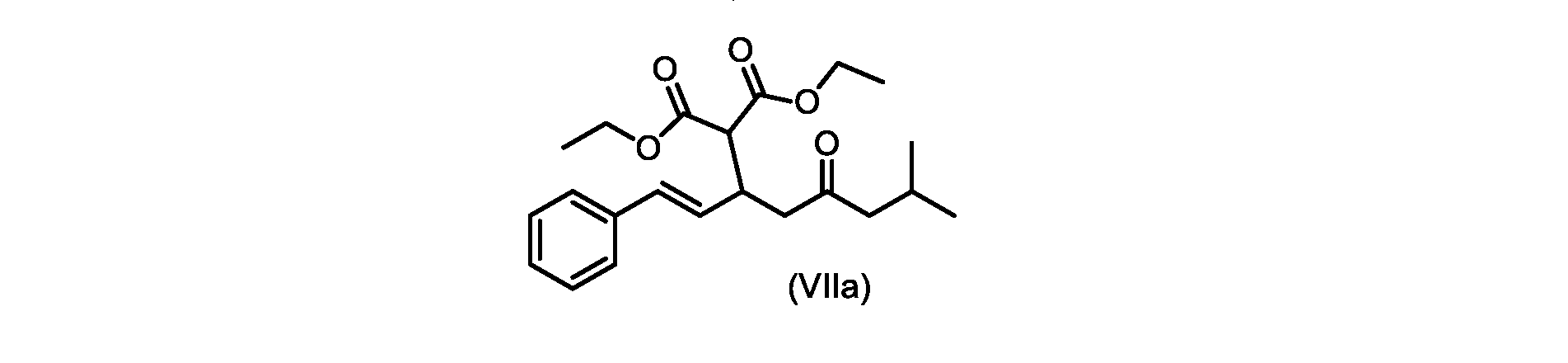
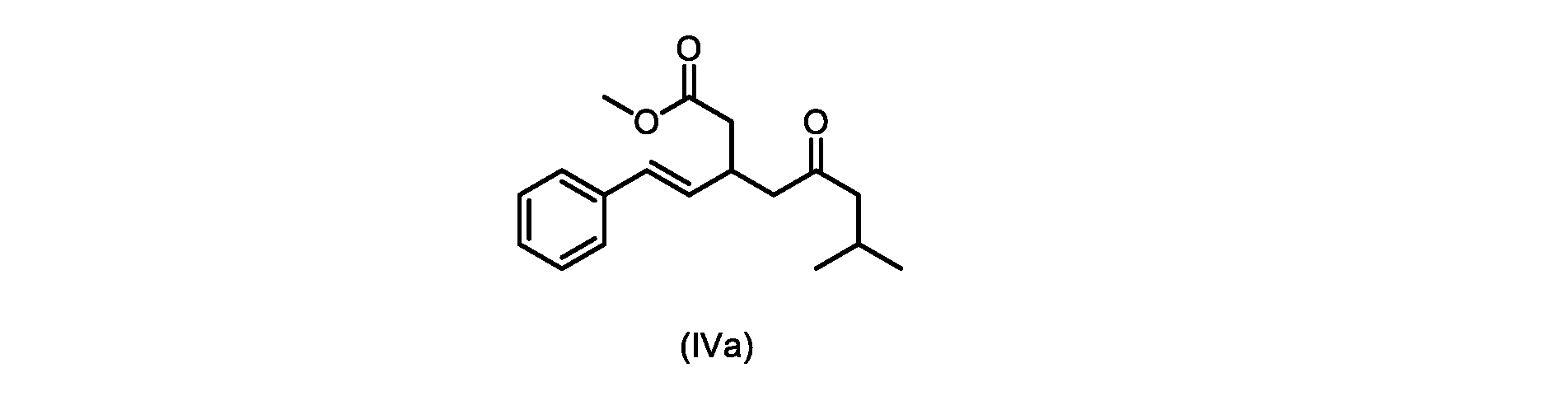
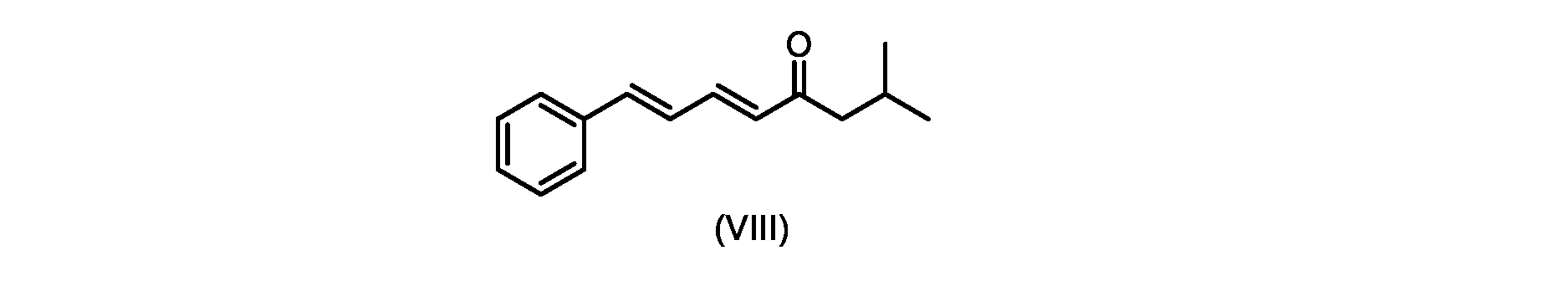
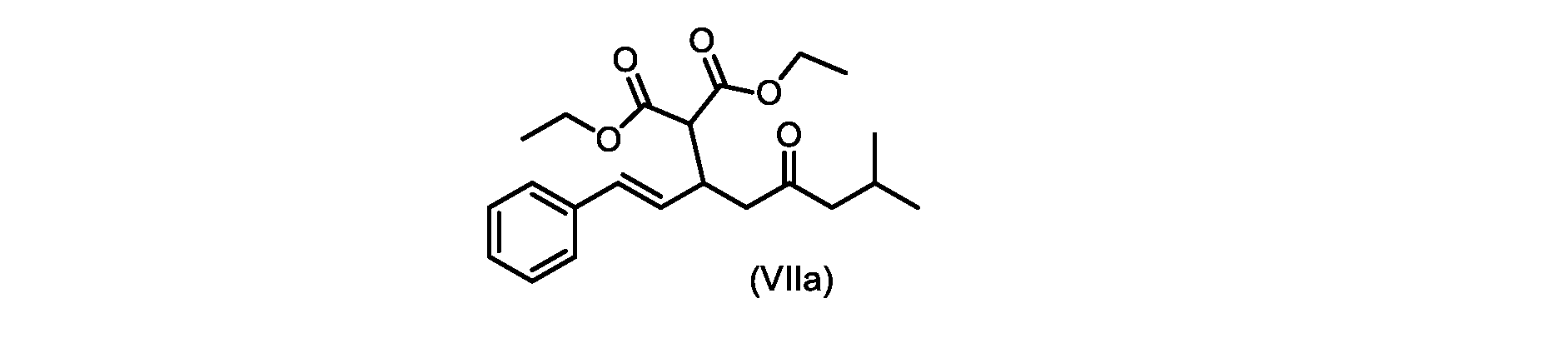
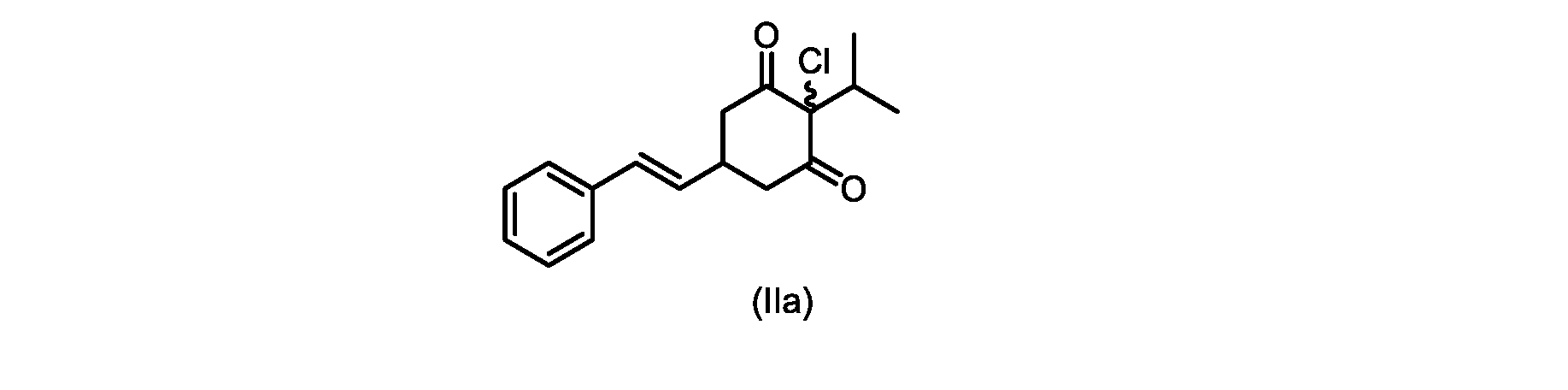
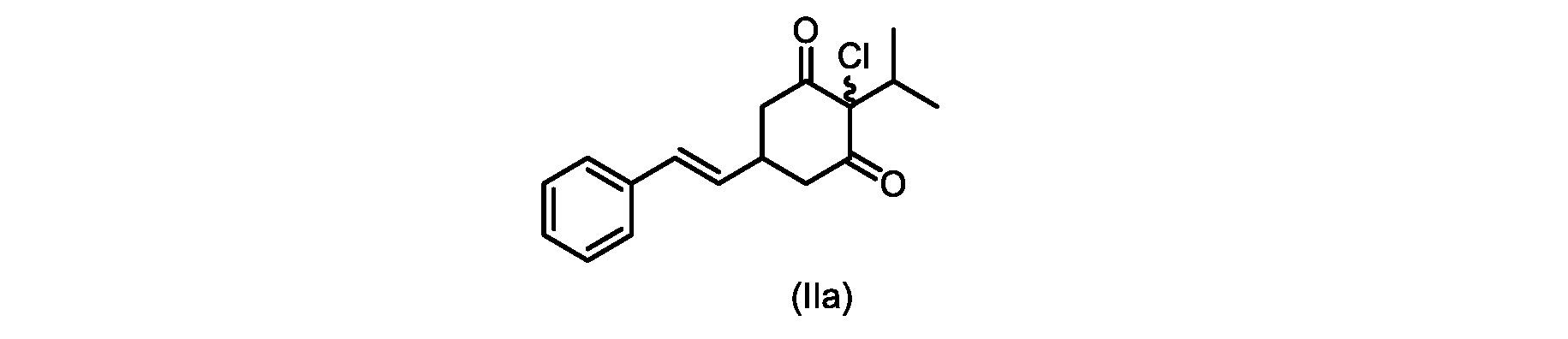
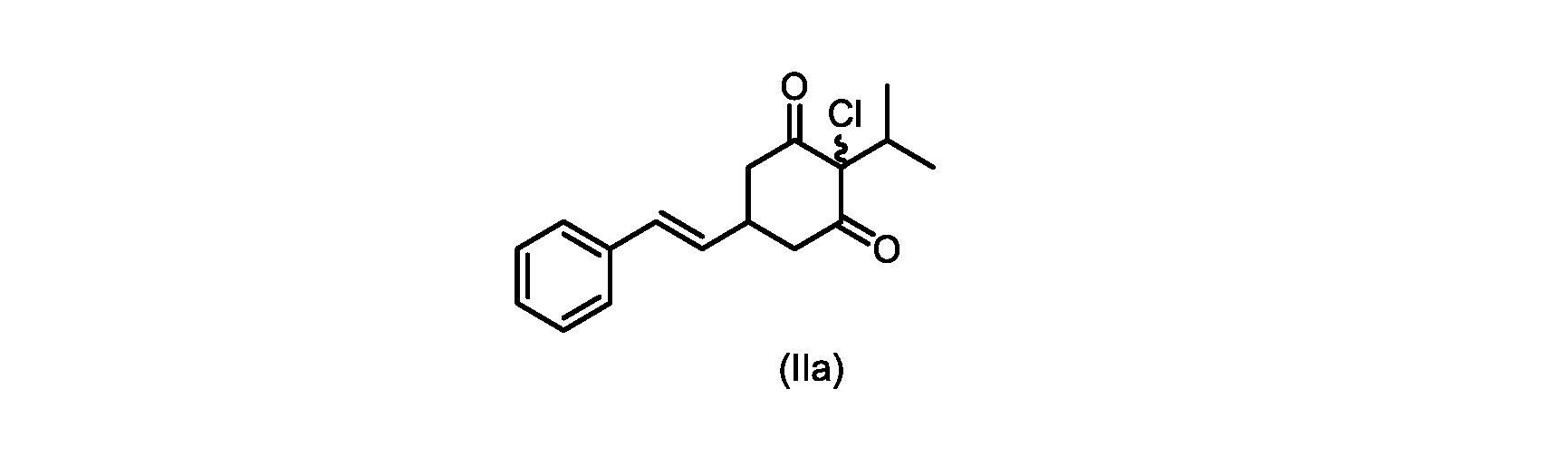
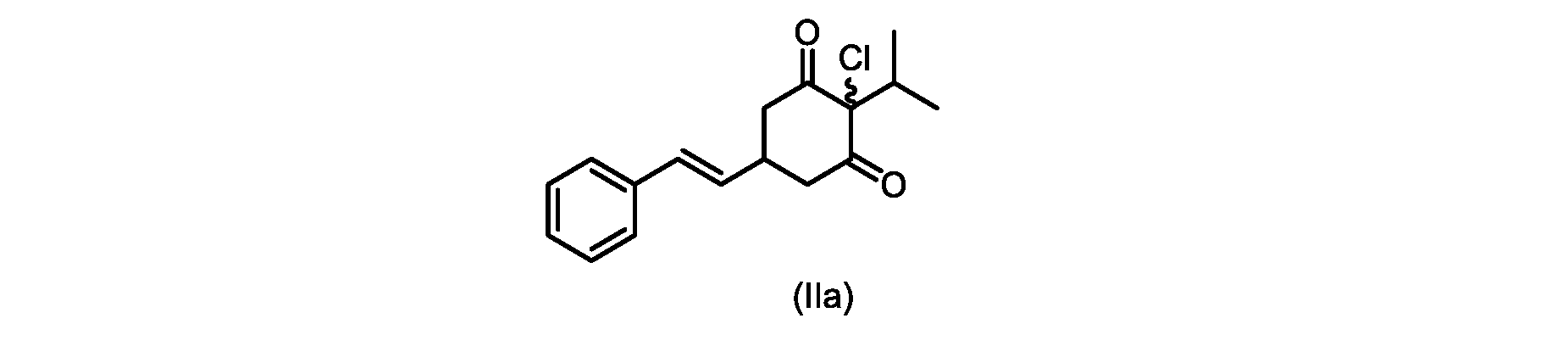
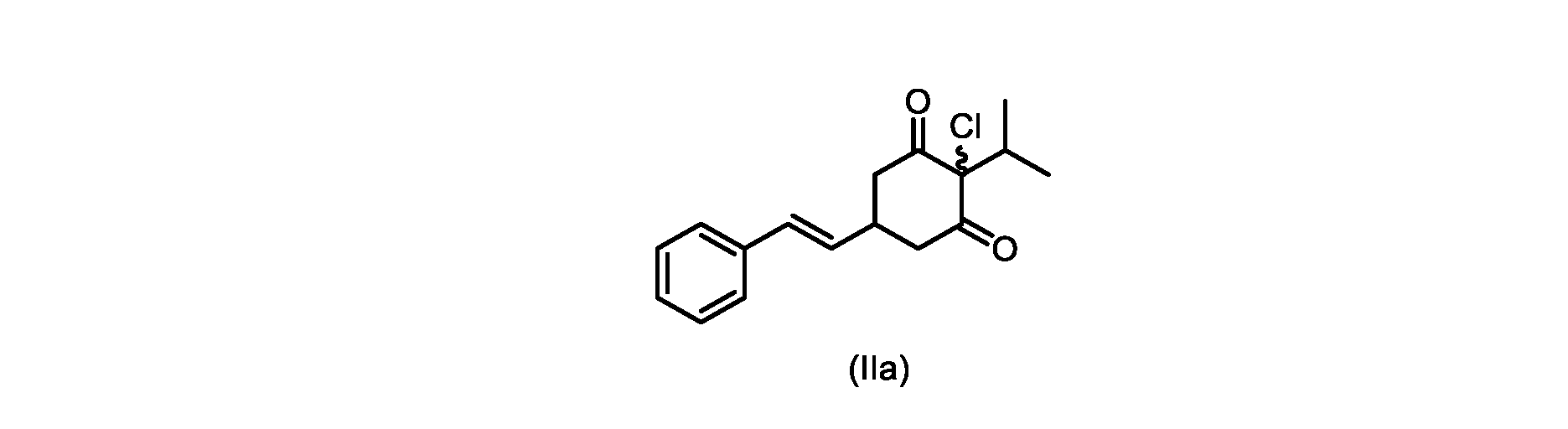
本明細書のいくつかの実施形態は、式(IIa)の化合物またはその塩を調製するためのプロセスであって、

i.トランス-シンナムアルデヒドまたはその塩を、メチルイソブチルケトンと縮合して、式(VIII)の化合物またはその塩を形成することと、

本明細書のいくつかの実施形態は、
c)式(IV)の化合物またはその塩を環化して、式(III)の化合物またはその塩を形成することと、
e)式(II)の化合物またはその塩を芳香族化して、式(I)の化合物またはその塩を形成することと、を含むプロセスによって調製される、式(I)の化合物またはその塩もしくは溶媒和物を説明する。
i.トランス-シンナムアルデヒドまたはその塩を、メチルイソブチルケトンと縮合して、式(VIII)の化合物またはその塩を形成することと、
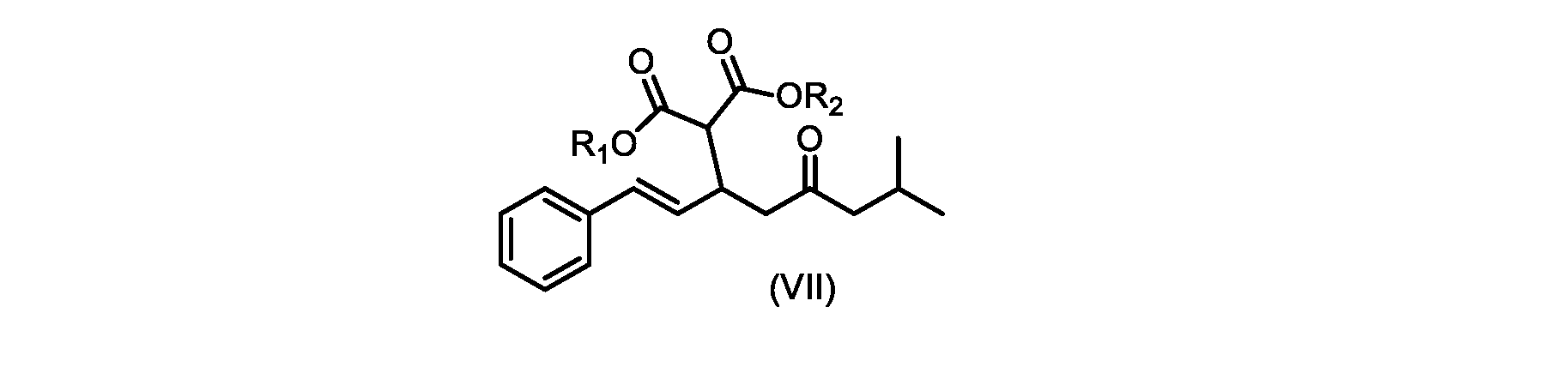
iii.式(VII)の前記化合物またはその塩を加水分解して、式(VI)の前記化合物またはその塩を形成することと、を含むプロセスによって調製される、式(I)の化合物またはその塩もしくは溶媒和物を説明する。
本明細書のいくつかの実施形態は、
i.トランス-シンナムアルデヒドまたはその塩を、メチルイソブチルケトンと縮合して、式(VIII)の化合物またはその塩を形成することと、
いくつかの実施形態は、
i.メチルイソブチルケトンを、メタノール性水酸化ナトリウムの存在下でトランス-シンナムアルデヒドで処理して、式(VIII)の化合物を形成することと、
本明細書のいくつかの実施形態は、
本明細書のいくつかの実施形態は、
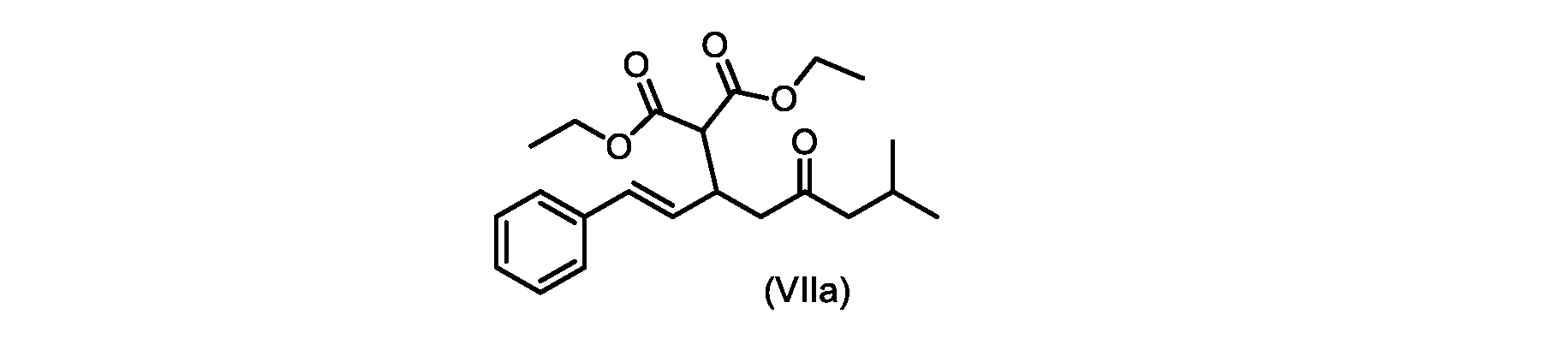
i.トランス-シンナムアルデヒドまたはその塩を、メチルイソブチルケトンと縮合して、式(VIII)の化合物またはその塩を形成することと、
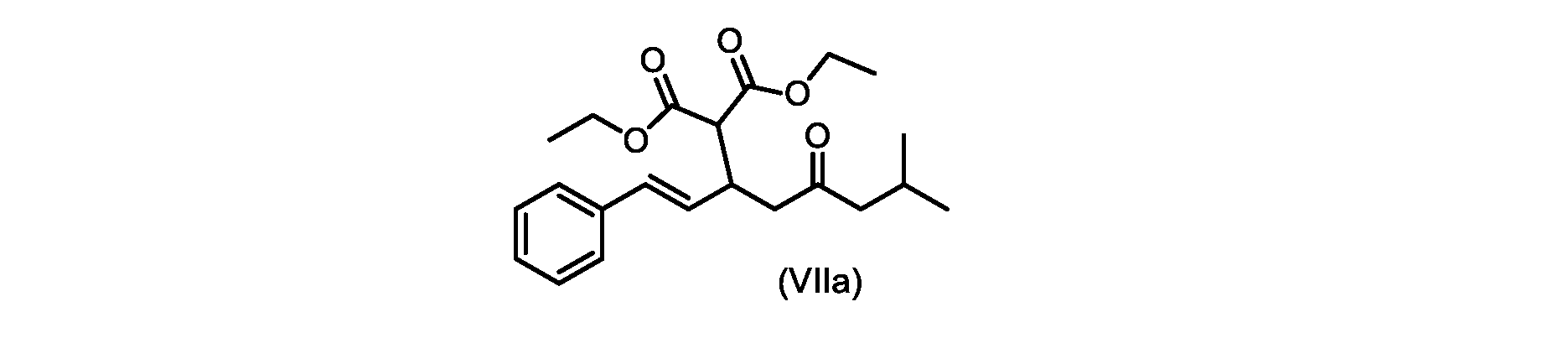
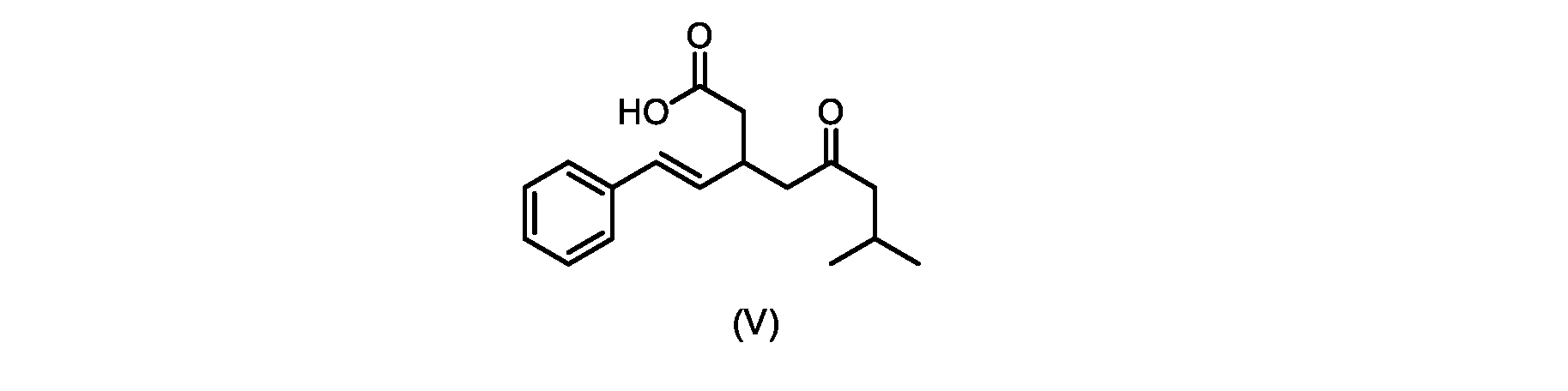
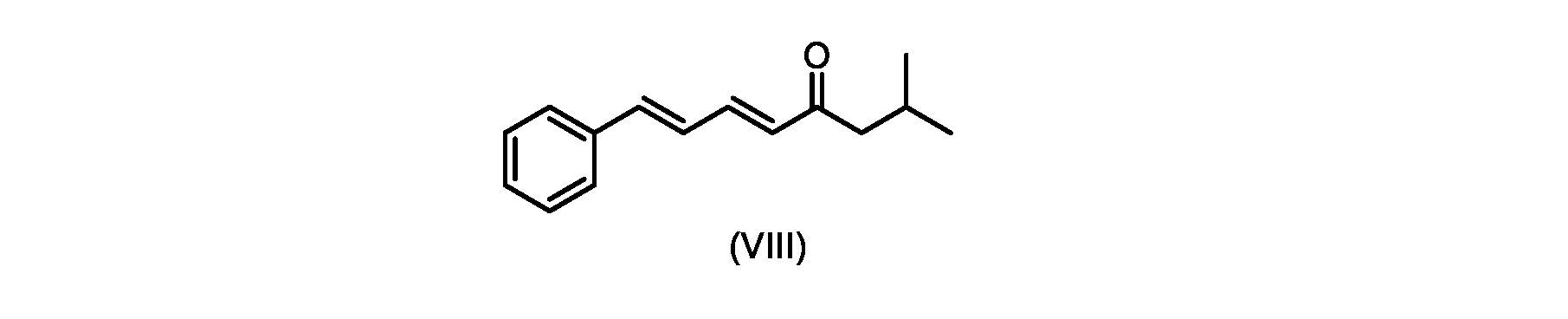
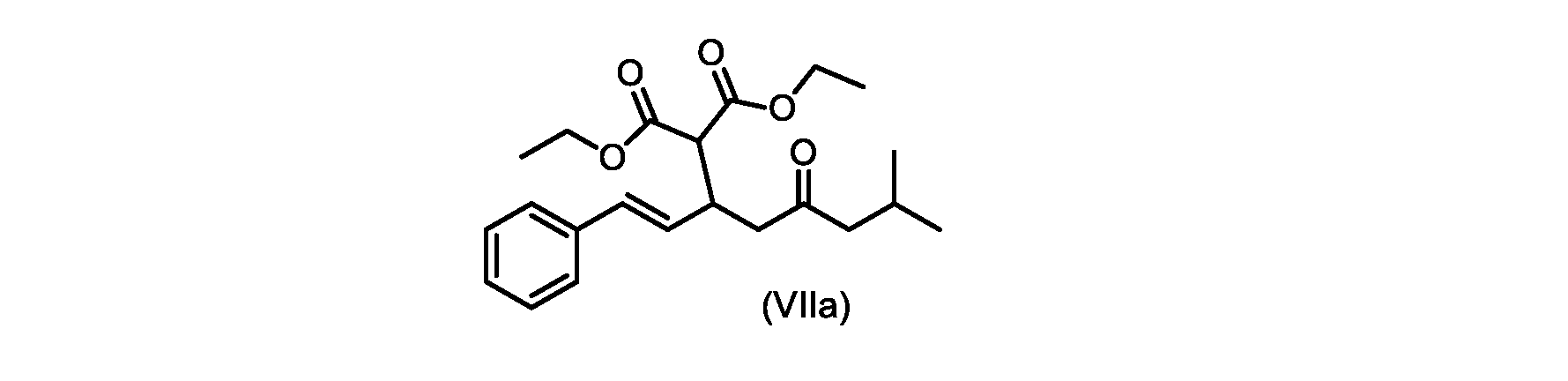
本明細書のいくつかの実施形態は、
i.トランス-シンナムアルデヒドまたはその塩を、メチルイソブチルケトンと縮合して、式(VIII)の化合物またはその塩を形成することと、

本明細書のいくつかの実施形態は、
i.トランス-シンナムアルデヒドまたはその塩を、メチルイソブチルケトンと縮合して、式(VIII)の化合物またはその塩を形成することと、

いくつかの実施形態は、実質的に図1に示されるようなX線粉末回折パターンを有する結晶性固体形態(形態1)の式(I)の化合物を説明する。いくつかの実施形態では、形態1の式(I)の化合物は、15.0、17.8、19.1、20.2、21.5、22.4、23.3、24.5、26.2、および27.9度(すべての値±0.1°2θ実験誤差)に特定のピークを有するX線粉末回折(XRPD)パターンを特徴とする。別の実施形態では、形態1の式(I)の化合物は、15.0、17.8、19.1、20.2、21.5、22.4、23.3、24.5、26.2、および27.9度(すべての値±0.1°2θ実験誤差)から選択される少なくとも9つ、または少なくとも8つ、または少なくとも7つ、または少なくとも6つ、または少なくとも5つ、または少なくとも4つ、または少なくとも3つの特定のピークを有するX線粉末回折(XRPD)パターンを特徴とする。いくつかの実施形態では、式(I)の化合物は、少なくとも80%が形態1である。いくつかの実施形態では、式(I)の化合物は、少なくとも85%が形態1である。いくつかの実施形態では、式(I)の化合物は、少なくとも90%が形態1である。いくつかの実施形態では、式(I)の化合物は、少なくとも95%が形態1である。いくつかの実施形態では、式(I)の化合物は、少なくとも99%が形態1である。いくつかの実施形態では、式(I)の化合物は、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、または約99%が形態1である。
本明細書の実施形態は、本明細書に記載の任意の実施形態に従って調製される式(I)の化合物またはその塩もしくは溶媒和物および薬学的に許容される賦形剤を含む薬学的組成物を説明する。
「C1~4アルキル」という用語は、少なくとも1個、多くとも4個の炭素原子を含有する直鎖または分岐鎖アルキルを意味する。本明細書で使用される「C1~4アルキル」の例には、メチル、エチル、n-プロピル、n-ブチル、イソブチル、イソプロピル、およびt-ブチルが含まれるが、これらに限定されない。

特に明記しない限り、プロトン核磁気共鳴スペクトル(1H NMR)は、25℃、400MHzで記録した。化学シフトは、テトラメチルシランからの100万分の1(ppm、δスケール)ダウンフィールドで表され、NMR溶媒中の残留プロトンを基準とする。データは次のように表される:化学シフト、多重度(s=一重項、d=二重項、sep=七重項、m=多重項および/または多重共鳴、br=広域)、積分、ヘルツ単位のカップリング定数、および割り当て。プロトンデカップリング炭素核磁気共鳴スペクトル(13C NMR)は、25℃、100MHzで記録した。化学シフトは、テトラメチルシランからの100万分の1(ppm、δスケール)ダウンフィールドで表され、溶媒の炭素共鳴を基準とする。高分解能質量分析(HRMS)は、Orbitrap質量分析計を使用して取得した。
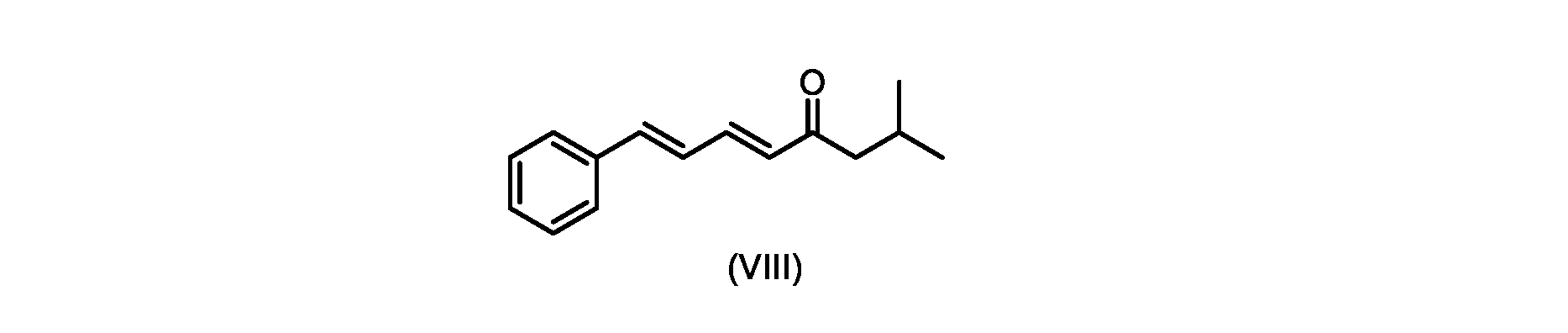
(5E,7E)-2-メチル-8-フェニルオクタ-5,7-ジエン-4-オン(式(VIII)の化合物)の合成

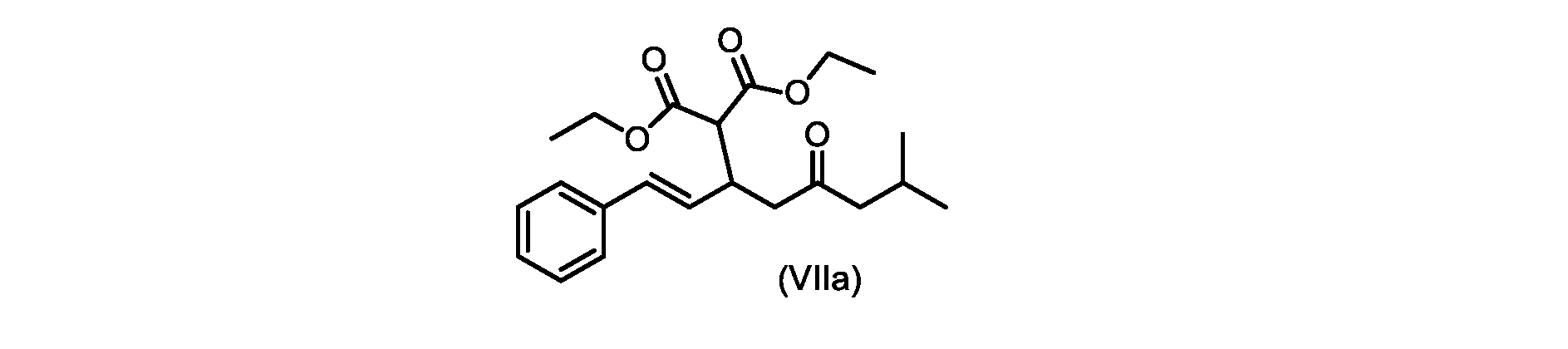
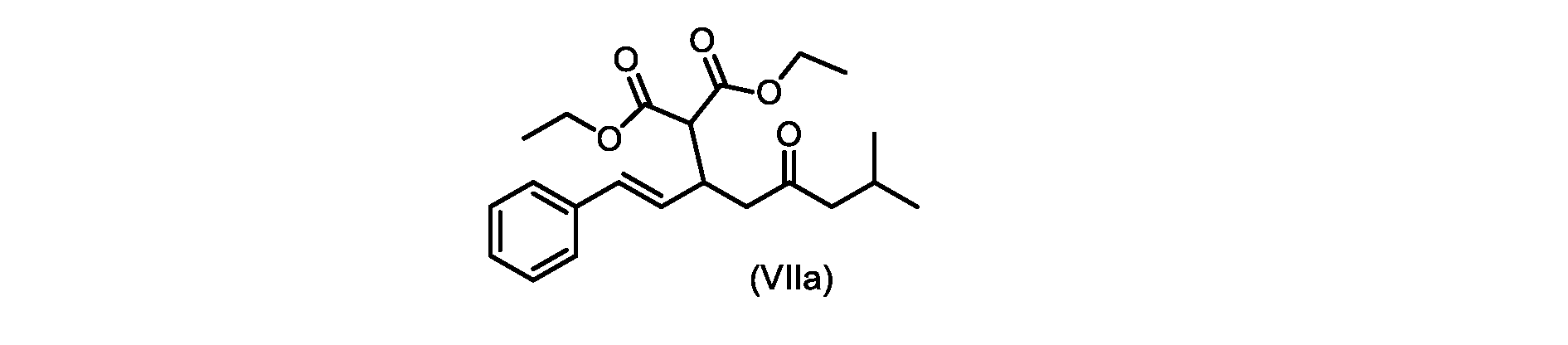
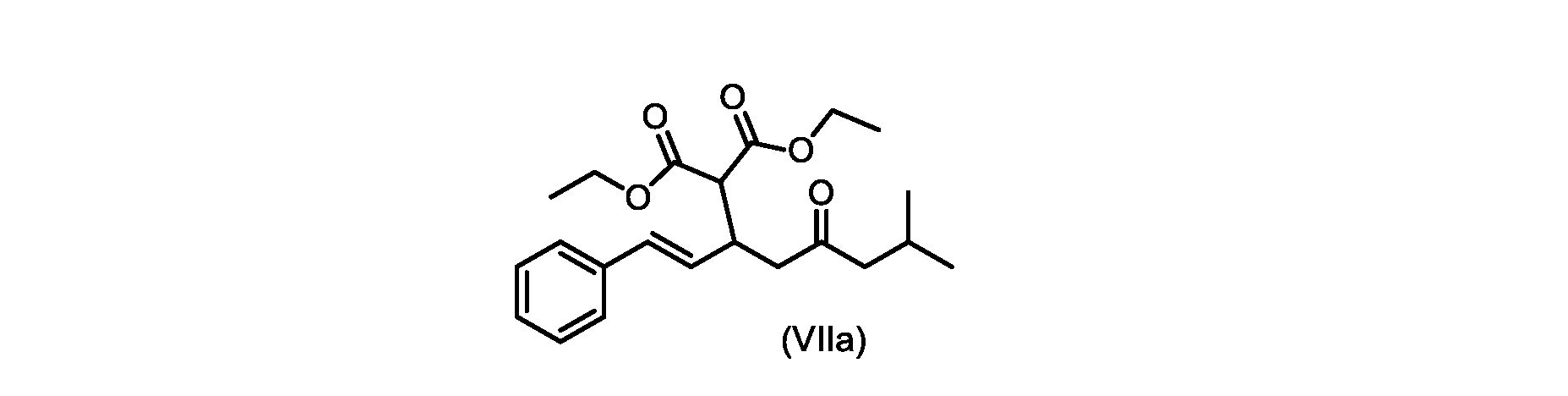
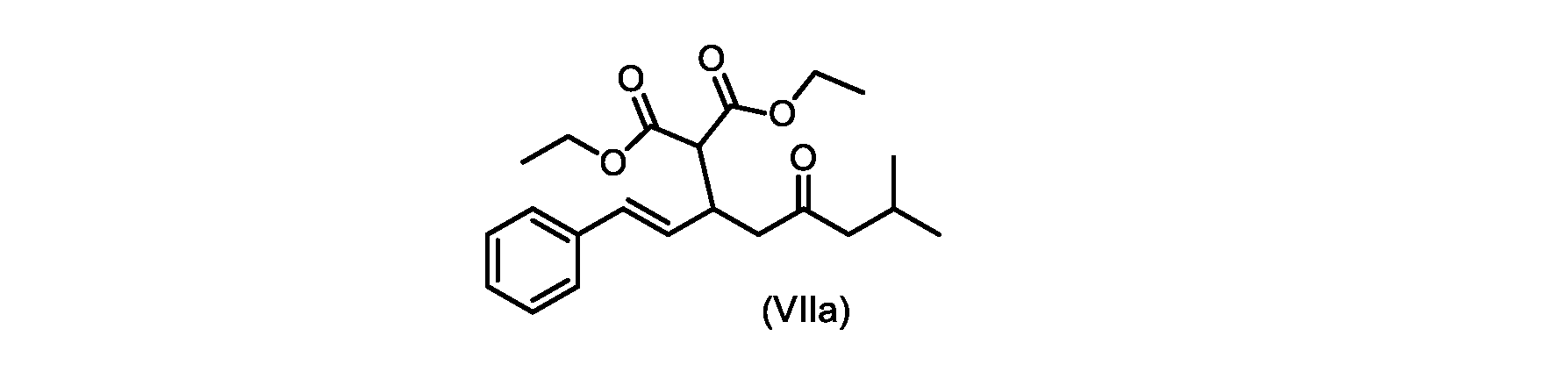
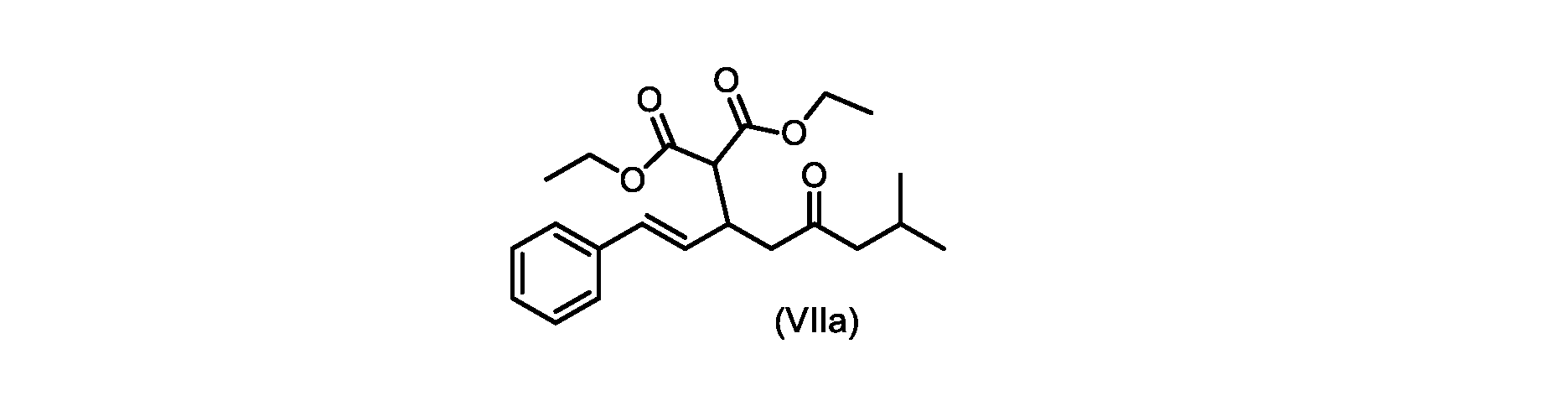
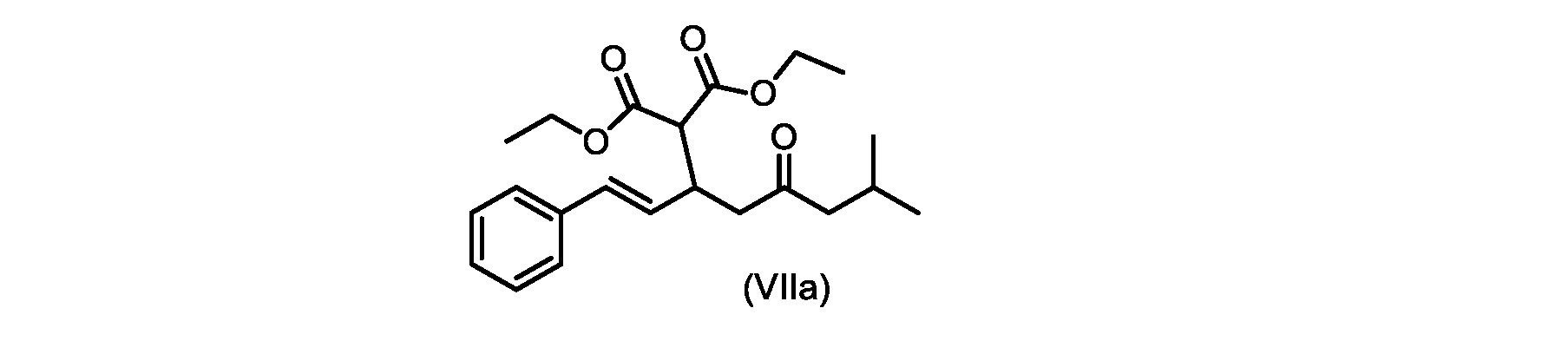
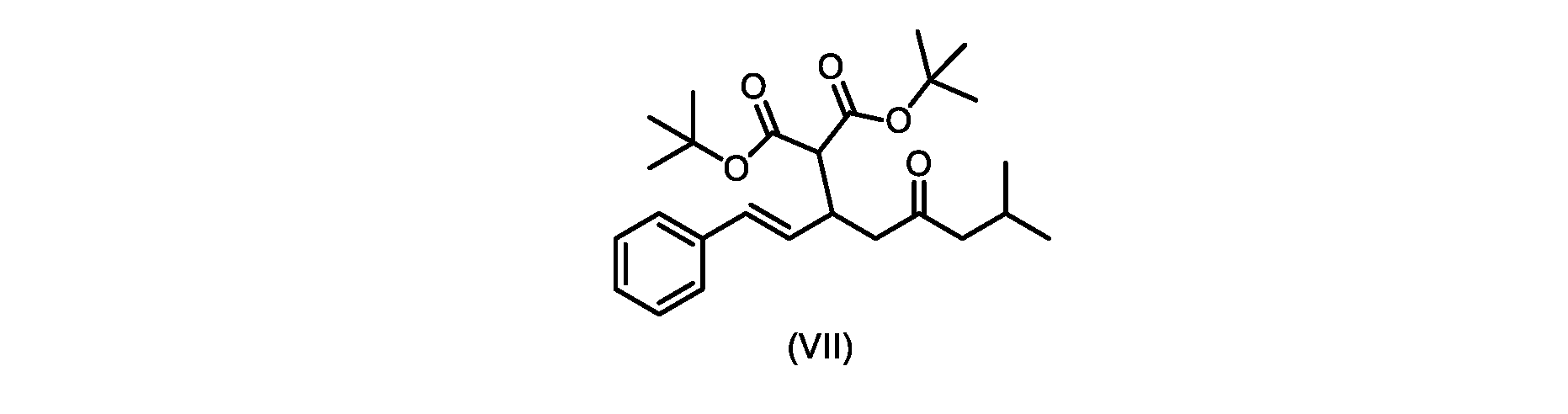
(E)-ジ-tert-ブチル2-(7-メチル-5-オキソ-1-フェニルオクタ-1-エン-3-イル)マロネート(R1+R2=tBuである式VIIの化合物)の合成
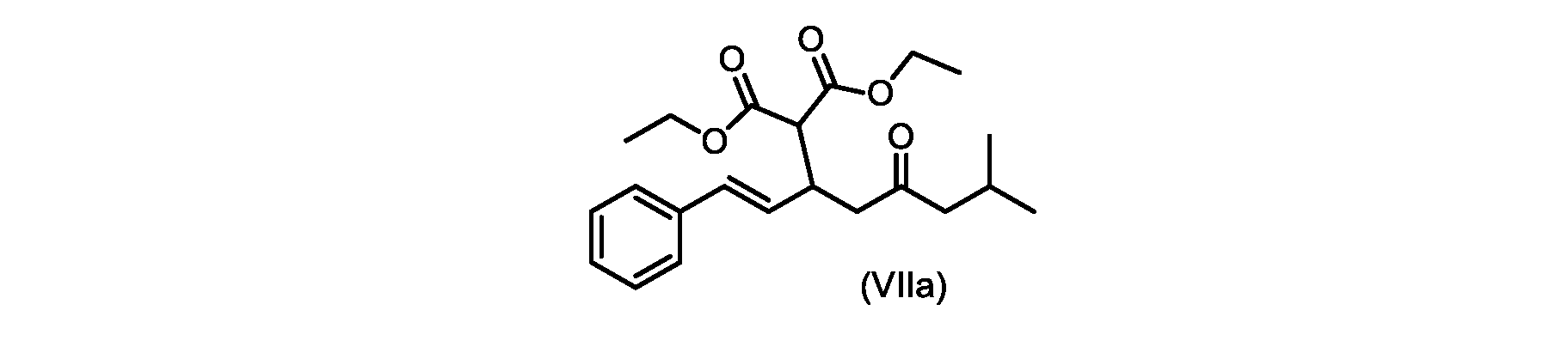
(E)-ジエチル2-(7-メチル-5-オキソ-1-フェニルオクタ-1-エン-3-イル)マロネート(式VIIaの化合物)の合成
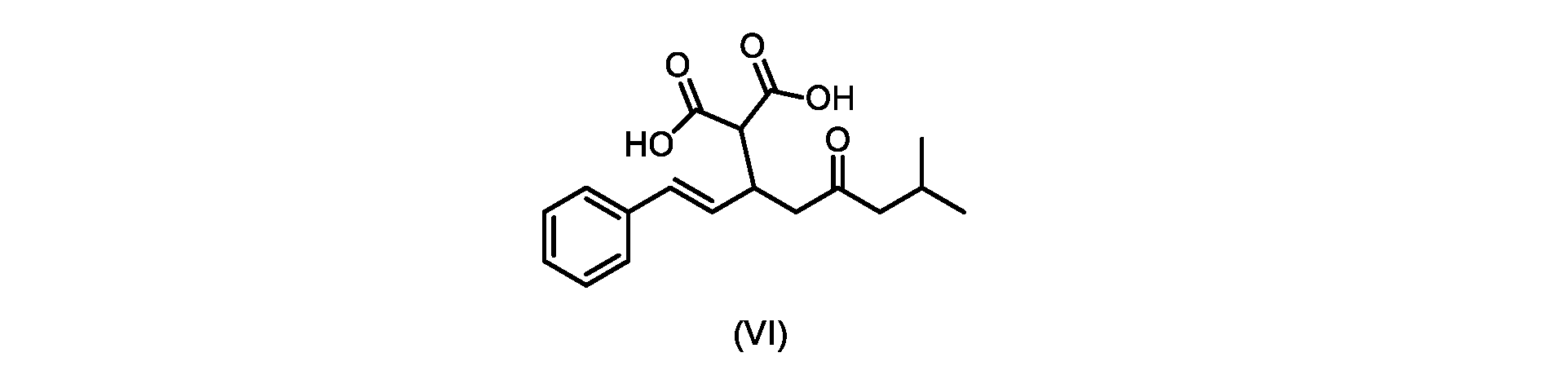
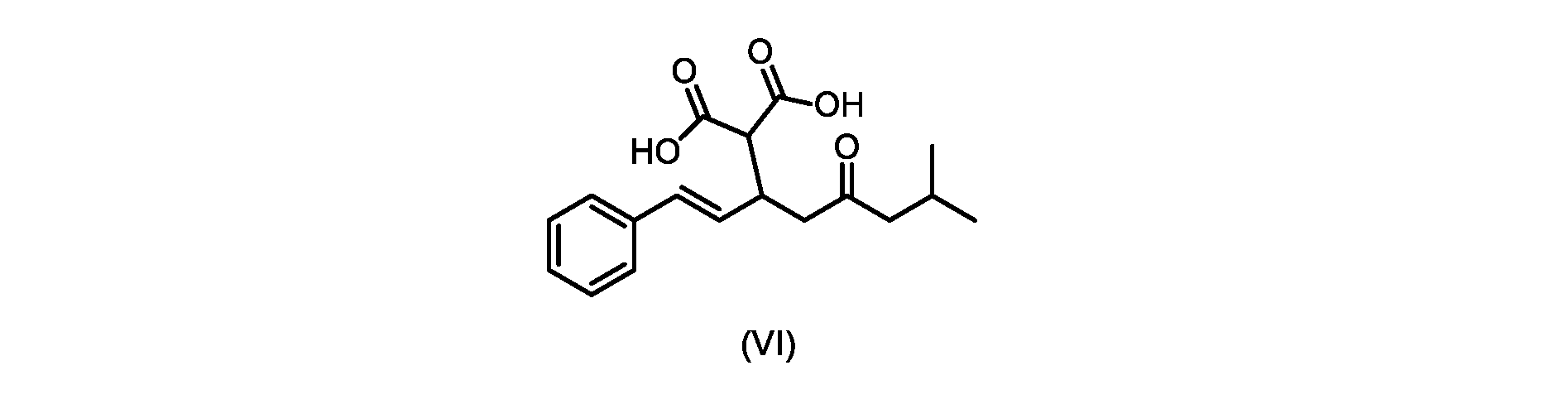
(E)-2-(7-メチル-5-オキソ-1-フェニルオクタ-1-エン-3-イル)マロン酸(式VIの化合物)の合成
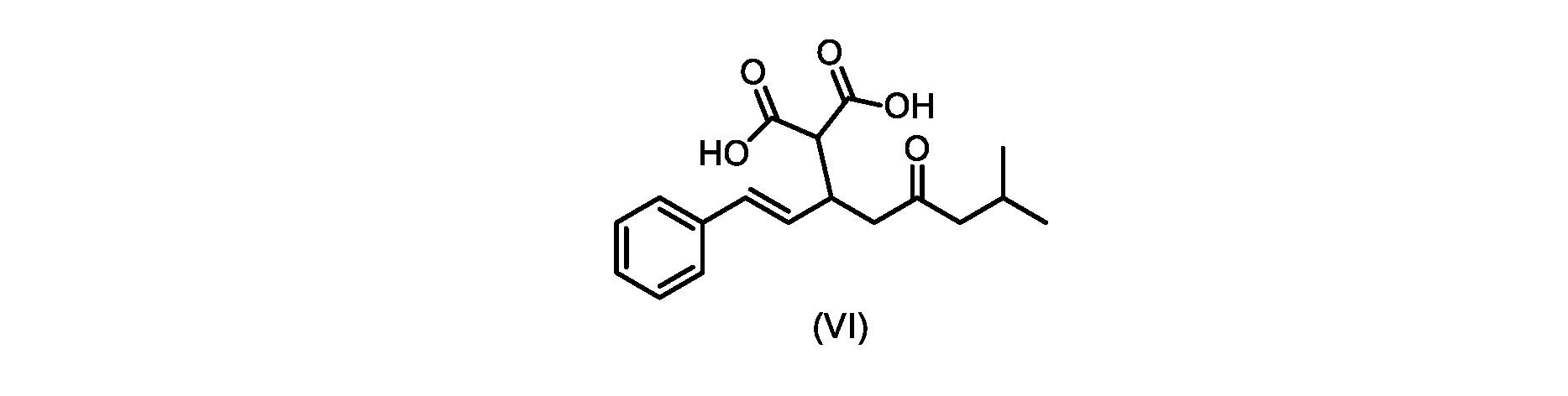
前のステップで得られた(E)-ジ-tert-ブチル2-(7-メチル-5-オキソ-1-フェニルオクタ-1-エン-3-イル)マロネート(式(VII)の化合物)のトルエン溶液を酢酸(50mL)で希釈し、酢酸(100mL)および濃塩酸水溶液(75mL)からなる撹拌溶液に60℃で1時間かけて滴加した。添加が完了したら、得られた溶液を60℃で4時間撹拌した。生成物混合物を30分かけて20℃に冷却し、水(200mL)を添加した。2層を20℃で激しく撹拌し、次いで静置した。次いで、水層を廃棄し、トルエン層を濃縮乾固させた。得られた油にトルエン(250mL)を添加し、溶液を撹拌しながら60℃に加熱した。次いで、温かいトルエン溶液を1時間かけて20℃までゆっくりと冷却し、その時点で(E)-2-(7-メチル-5-オキソ-1-フェニルオクタ-1-エン-3-イル)マロン酸(式(VI)の化合物)が溶液から沈殿した。次いで、固体を濾過し、ウェットケーキをトルエン(100mL)で洗浄した。次いで、ウェットケーキを真空下30℃で12時間乾燥させ、(E)-2-(7-メチル-5-オキソ-1-フェニルオクタ-1-エン-3-イル)マロン酸(式(VI)の化合物)を、白色の結晶性固体(37.3g、シンナムアルデヒド(式(IX)の化合物)から62%)として得た。
実施例3からの(E)-ジエチル2-(7-メチル-5-オキソ-1-フェニルオクタ-1-エン-3-イル)マロネート(約7.6モル、式(VIIa)の化合物)のトルエン溶液に、20~30℃で、6M水酸化ナトリウム水溶液(5.05L、4当量)、トルエン(0.5Lまたは0.5容量)、および200プルーフエタノール(2容量)を入れ、内容物をTJ=20℃で少なくとも4時間撹拌した。完了したら、温度を35~50℃に調整し、少なくとも30分間撹拌した後、撹拌を停止し、層を分離した。添加中は温度を10℃以下に維持しながら、水層を0~5℃に冷却し、濃縮塩酸でpHを0~1に調整した(2.9Lまたは4.7等量を必要とした)。所望のpHに達したら、容器にTBME(3L、3容量)を入れ、二相混合物を20~25℃に温めた。混合物を15~30分間撹拌し、層を分離した。有機層にトルエン(7L、7容量)および水(6L、6容量)を添加し、混合物を15~30分間撹拌した。層を分離し、トルエンを添加して、16~18容量(3L、3容量)の充填を達成した。混合物を真空蒸留を使用して約9~9.5容量まで蒸留し、温度を40~45℃に調整し、混合物に5gの化合物VIを播種した(理論的収率に対して0.2%w/w)。混合物を40~45℃で30~60分間撹拌した。核形成が観察されたら、混合物をトルエンで12容量に希釈し、40~45℃で少なくとも1時間保持した。スラリーを0.5℃/分で10~20℃に冷却し、10~20℃で少なくとも1時間保持した。固体を濾過によって単離し、フィルターケーキをトルエン(2×7容量)で洗浄した。固体を真空オーブンで25~35℃で一晩乾燥させて、(E)-2-(7-メチル-5-オキソ-1-フェニルオクタ-1-エン-3-イル)マロン酸(式(VI)の化合物)を得た。
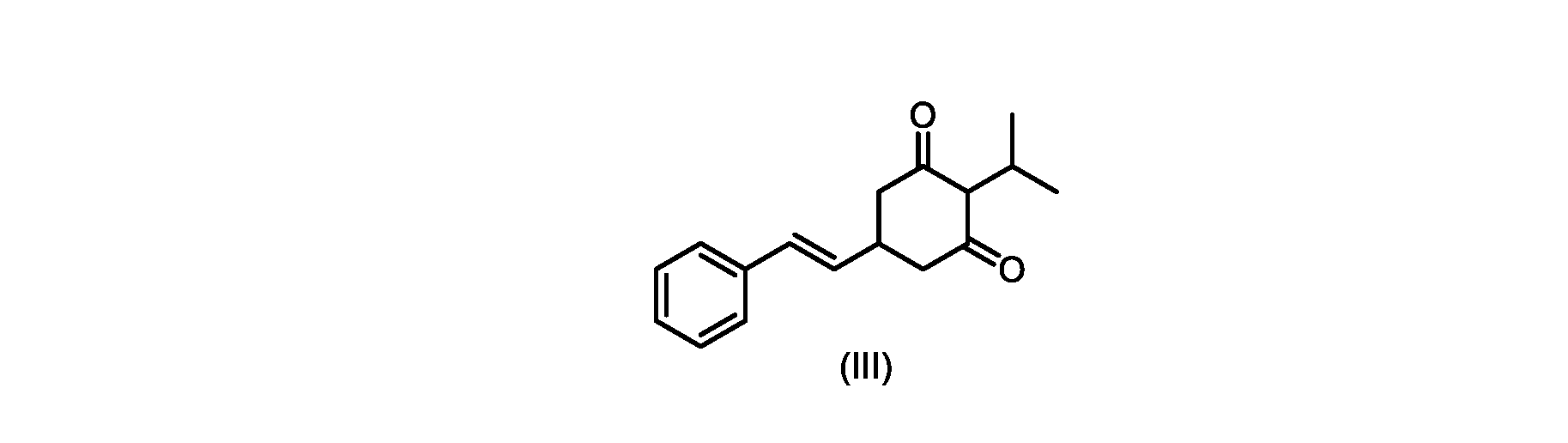
(E)-2-イソプロピル-5-スチリルシクロヘキサン-1,3-ジオン(式(III)の化合物)の合成
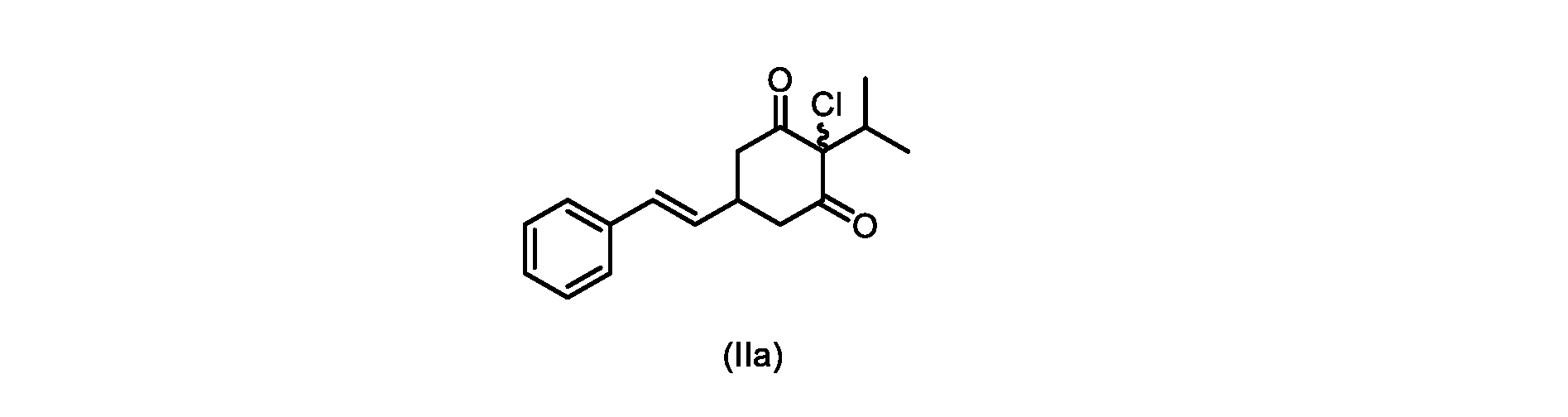
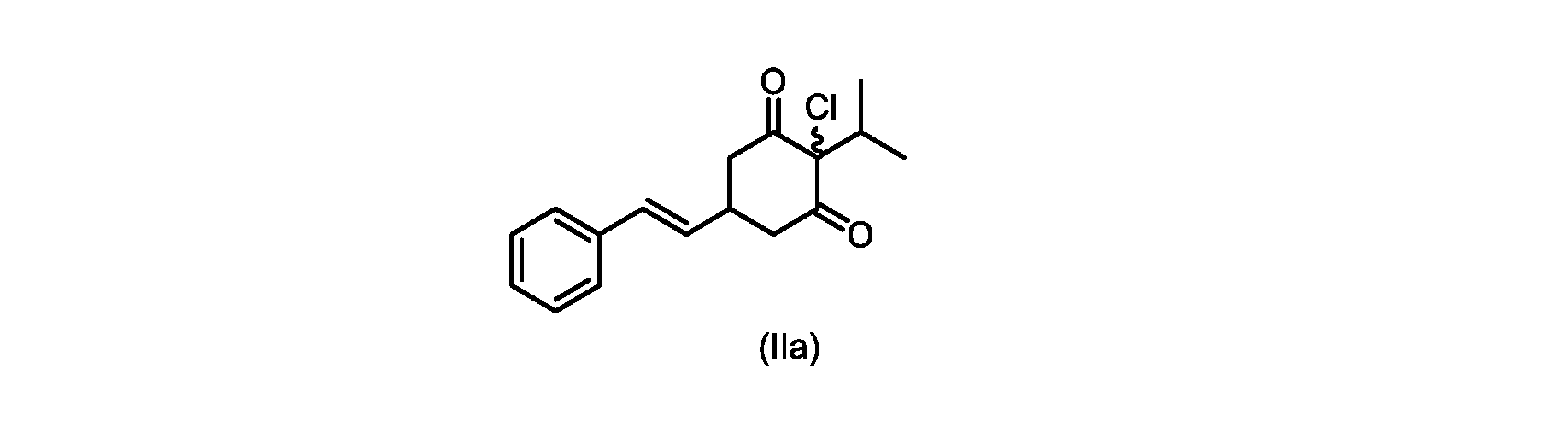
(E)-2-クロロ-2-イソプロピル-5-スチリルシクロヘキサン-1,3-ジオン(式(IIa)の化合物)の合成
1H NMR(400MHz,DMSO)δ7.41-7.39(m,2H),7.36-7.32(m,2H),7.23-7.22(m,1H),6.54(d,1H,J=16Hz),6.32(dd,1H,J=16,7.2Hz),3.20-3.14(m,3H),2.84-2.77(m,1H),2.73-2.72(m,1H),2.70-2.69(m,1H),0.82(d,6H,J=6.8Hz)。
1H NMR(400MHz,DMSO)δ7.33-7.28(m,4H),7.25-7.21(m,2H),6.35(dd,1H,J=16,2Hz),6.03(dd,1H,J=16,5.6Hz),3.48(dd,1H,J=14.8,6Hz),3.24-3.18(m,1H),3.10(sep,1H,J=6.4Hz),2.86(dd,1H,J=14.8,3.6Hz),0.83(d,6H,J=6.4Hz)。
(E)-2-イソプロピル-5-スチリルベンゼン-1,3-ジオール(式(I)の化合物)の合成
Claims (30)
- 式(I)の化合物またはその塩もしくは溶媒和物を調製するためのプロセスであって、
式(III)の化合物を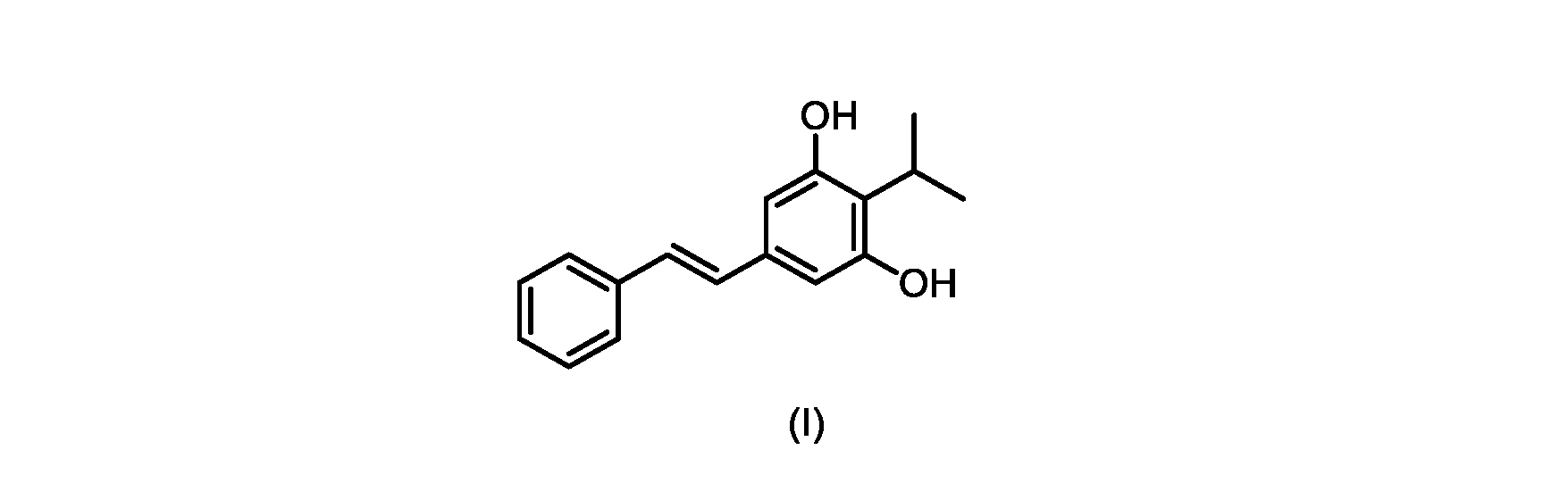
ハロゲン化剤でハロゲン化して、式(II)の化合物を得る工程と、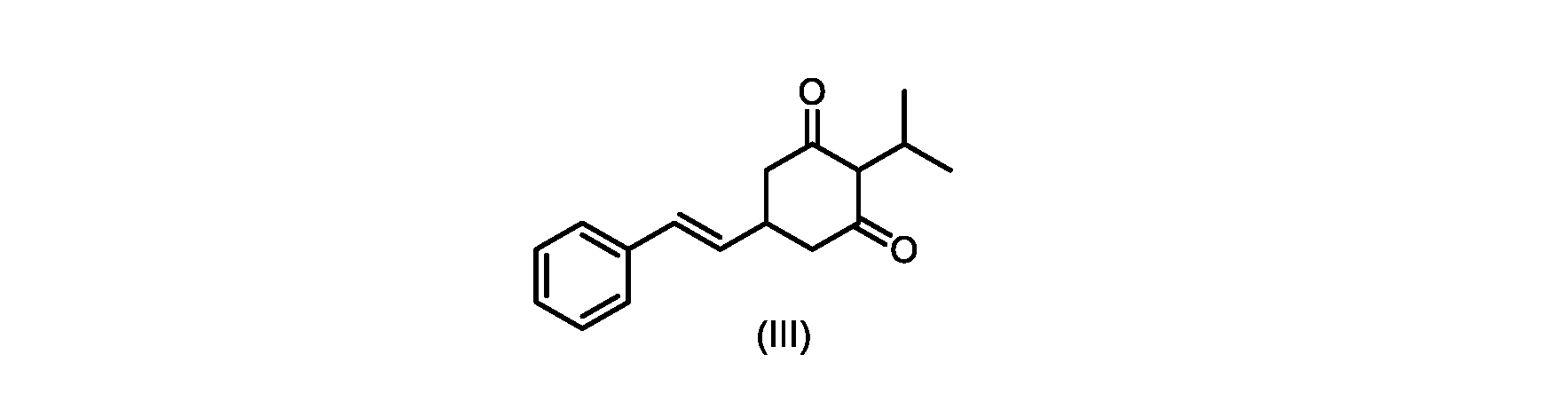
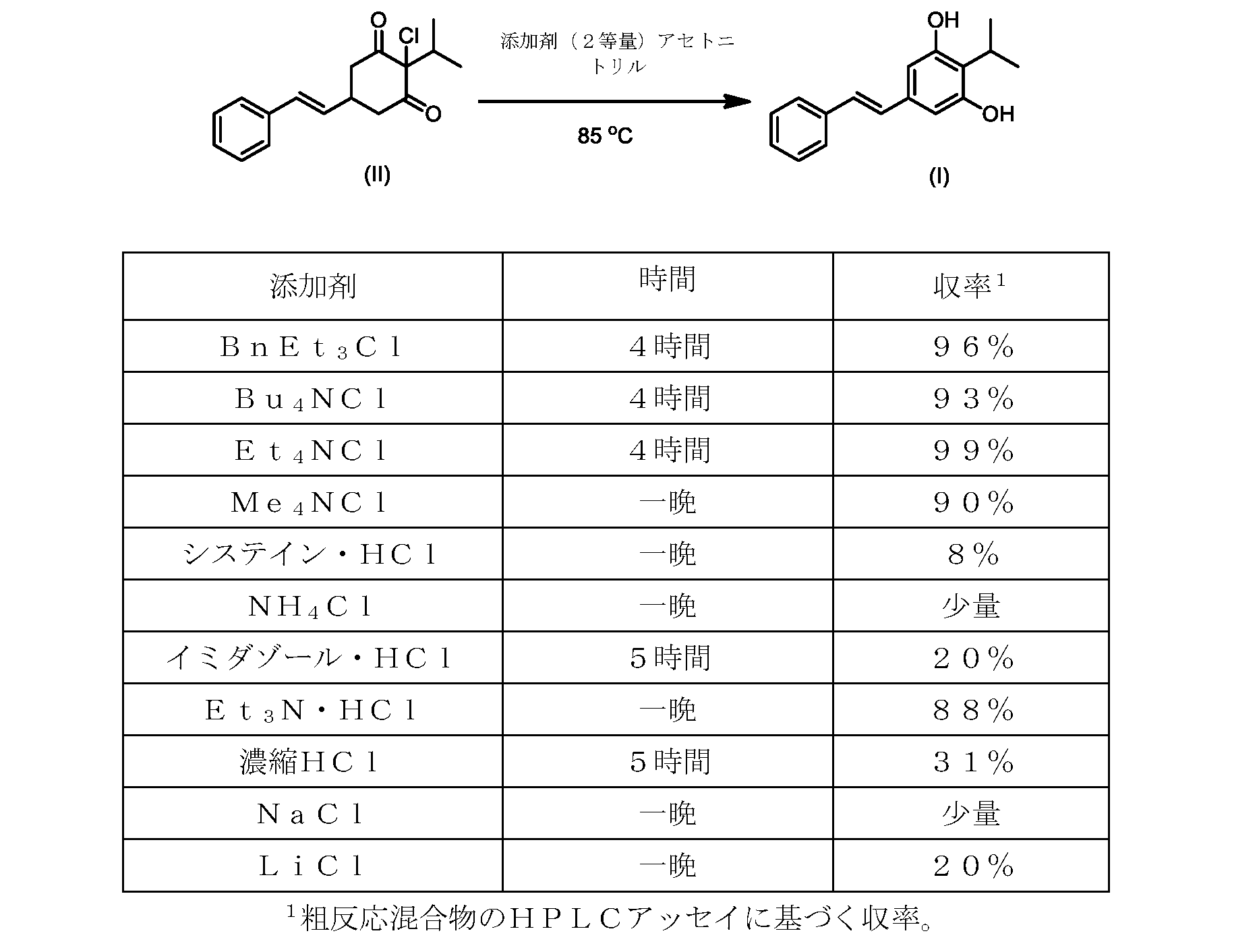
式(II)の前記化合物(式中、Xが、Cl、Br、またはIである)を溶媒中の第四級アンモニウム塩で芳香族化して、式(I)の前記化合物を得る工程と、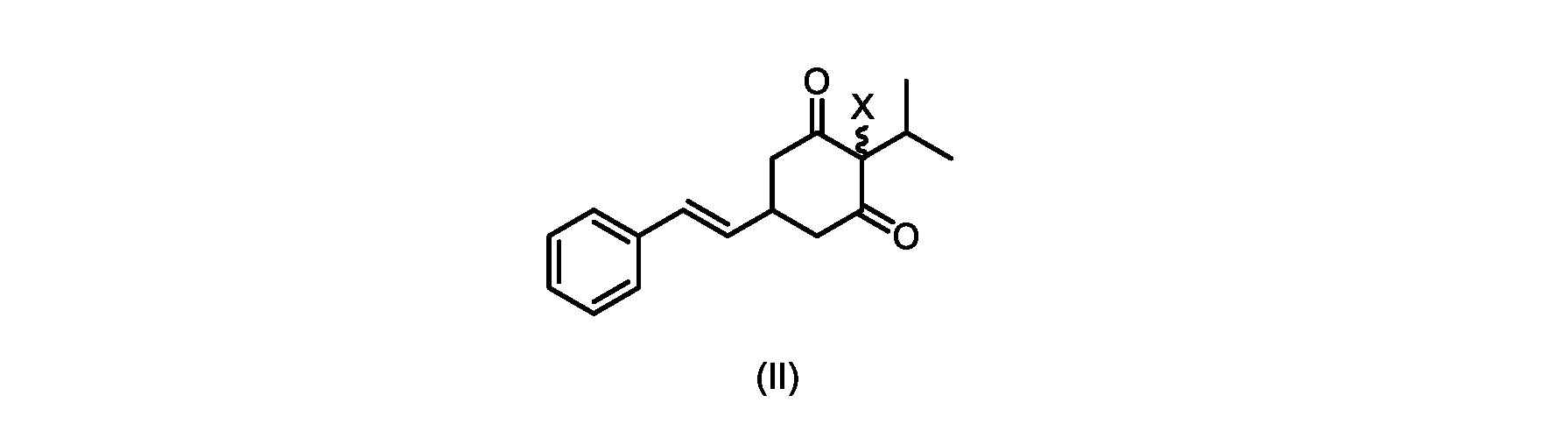
を有する、プロセス。 - 前記第四級アンモニウム塩が、第四級塩化アンモニウム塩である、請求項1に記載のプロセス。
- 前記第四級塩化アンモニウム塩が、塩化ベンジルトリエチルアンモニウム、塩化テトラブチルアンモニウム、塩化テトラエチルアンモニウム、および塩化テトラメチルアンモニウムからなる群から選択される、請求項2に記載のプロセス。
- 前記芳香族化が、アセトニトリル、トルエン、2-メチルテトラヒドロフラン、酢酸イソプロピル、アセトン、およびメチルイソブチルケトンからなる群から選択される溶媒中で実行される、請求項1~3のいずれか1つに記載のプロセス。
- Xが、Clである、請求項1~4のいずれか1つに記載のプロセス。
- 式(I)の前記化合物の酢酸溶媒和物の形成をさらに有する、請求項1~5のいずれか1つに記載のプロセス。
- 式(I)の前記化合物の結晶化による精製をさらに有する、請求項1~5のいずれか1つに記載のプロセス。
- 前記結晶化が、メタノールおよび水を使用して実行される、請求項7に記載のプロセス。
- 前記ハロゲン化剤が、1,3-ジクロロ-5,5-ジメチルヒダントイン、N-クロロスクシンイミド、およびトリクロロイソシアヌル酸からなる群から選択される、請求項5に記載のプロセス。
- 前記ハロゲン化がメタノール中で実行され、および、前記ハロゲン化剤が、1,3-ジクロロ-5,5-ジメチルヒダントインである、請求項5~9のいずれか1つに記載のプロセス。
- 式(IV)の化合物
(式中、Rが、C1~4アルキルである)を環化して、式(III)の前記化合物を得る工程をさらに有する、請求項1~10のいずれか1つに記載のプロセス。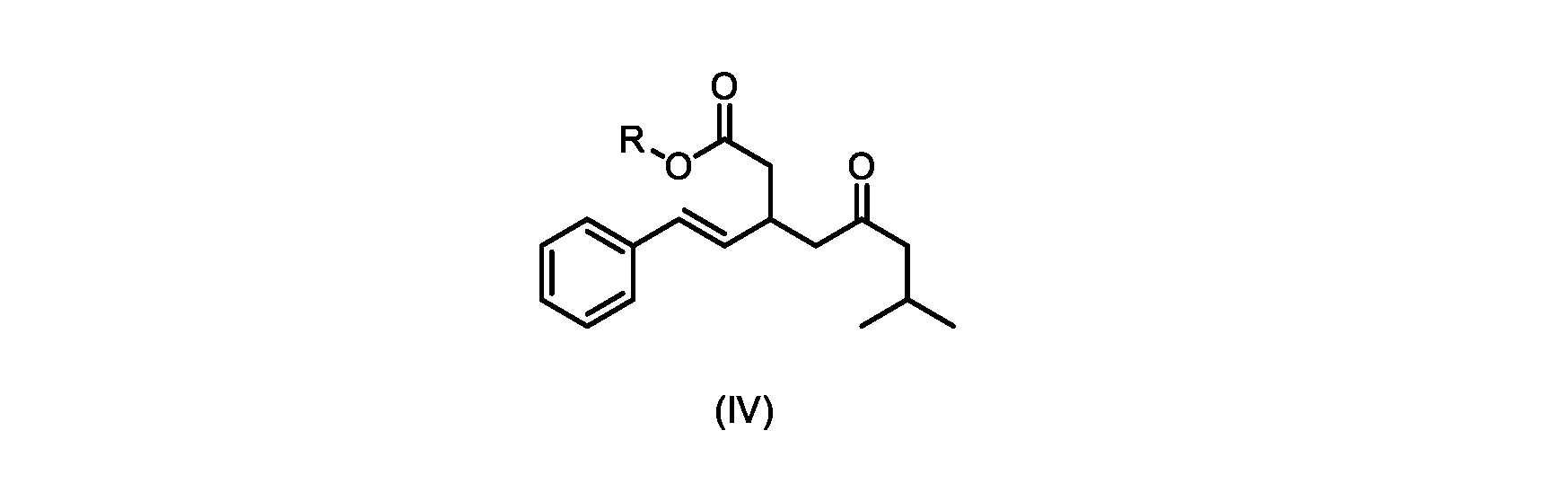
- Rが、メチル、エチル、プロピル、またはブチルからなる群から選択される、請求項11に記載のプロセス。
- Rが、メチル、またはt-ブチルである、請求項11または12に記載のプロセス。
- 前記環化が、2-メチルテトラヒドロフラン中で、式(IV)の前記化合物とカリウムtert-ブトキシドを接触させる工程を有するものである、請求項11~13のいずれか1つに記載のプロセス。
- 式(III)の前記化合物が酸性化され、メチルシクロヘキサンでの沈殿によって単離される、請求項11~14のいずれか1つに記載のプロセス。
- 式(V)の化合物またはその塩をエステル化して、
式(IV)の前記化合物を得る工程をさらに有する、請求項11~15のいずれか1つに記載のプロセス。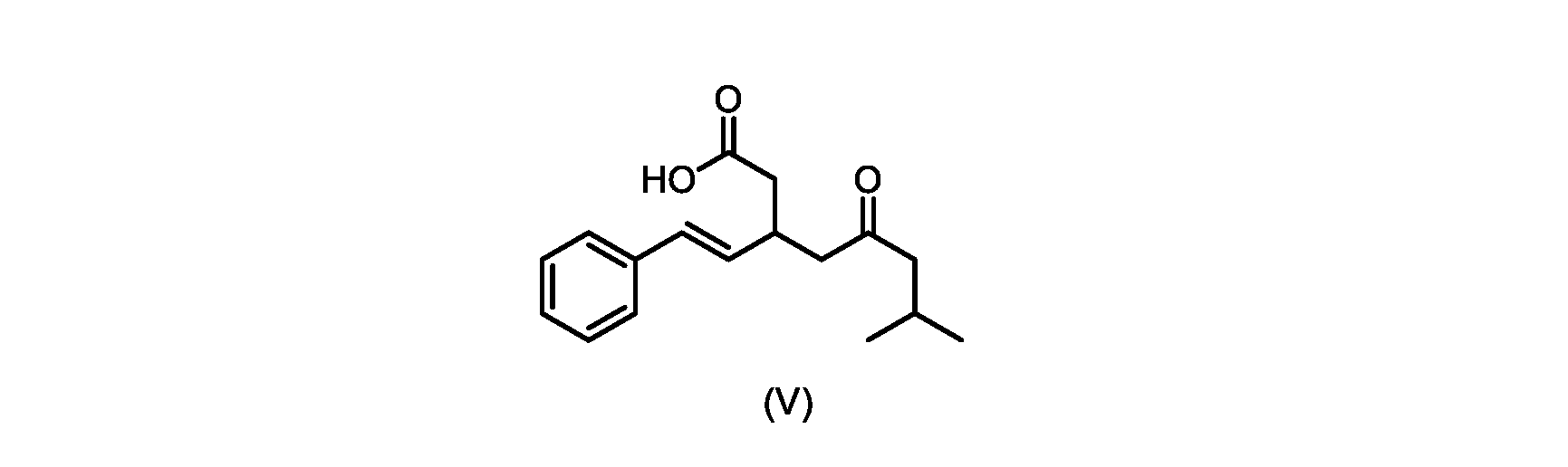
- 前記エステル化が、式(V)の前記化合物またはその塩を、メタノール中の塩酸水溶液と共に加熱する工程を有する、請求項16に記載のプロセス。
- 式(VI)の化合物を、
塩基の存在下で脱炭酸して、式(V)の前記化合物またはその塩を得る工程をさらに有する、請求項16または17に記載のプロセス。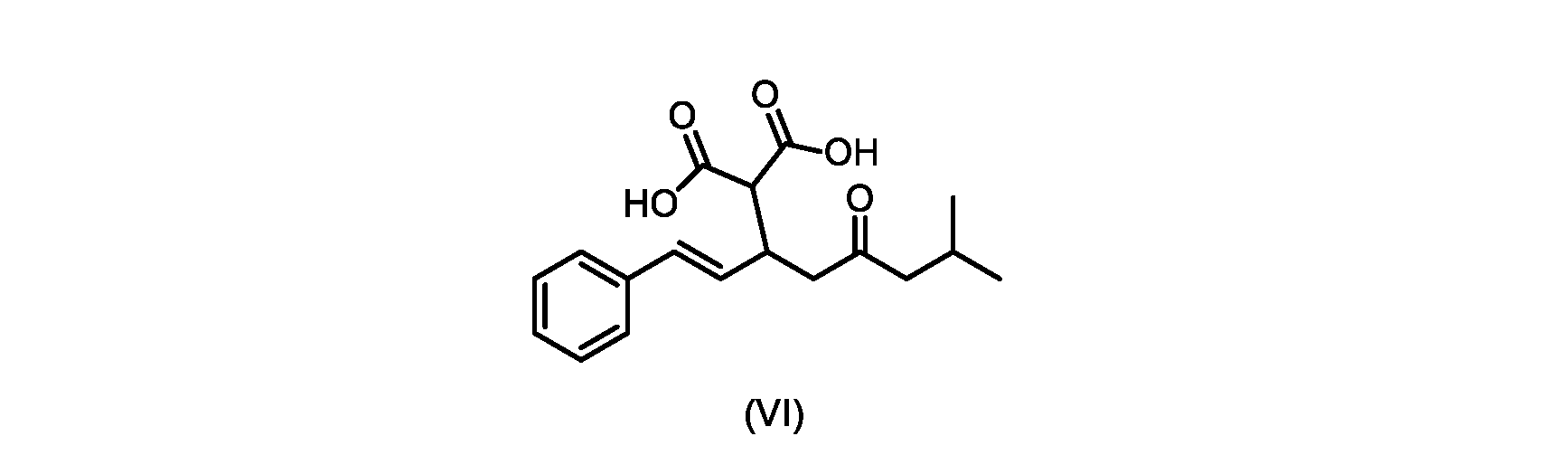
- 前記脱炭酸が、式(VI)の前記化合物を、トリエチルアミンの存在下で加熱する工程を有する、請求項18に記載のプロセス。
- 式(VII)の化合物
(式中、R1およびR2の各々が、独立して、C1~4アルキルである)を加水分解して、式(VI)の前記化合物を得る工程をさらに有する、請求項18または19に記載のプロセス。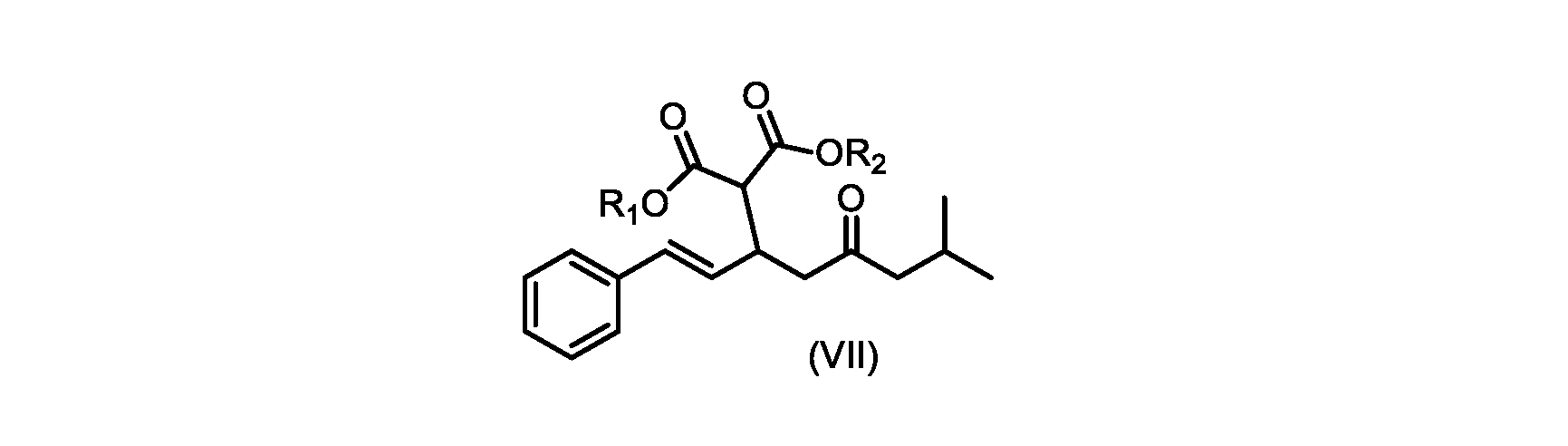
- R1およびR2の各々が、エチルである、請求項20に記載のプロセス。
- 前記加水分解が、式(VII)の前記化合物を、エタノール中の水酸化カリウムまたは水酸化ナトリウムで処理する工程を有する、請求項20または21に記載のプロセス。
- ジアルキルマロン酸エステルを、式(VIII)の化合物に添加して、
式(VII)の前記化合物を得る工程をさらに有する、請求項20~22のいずれか1つに記載のプロセス。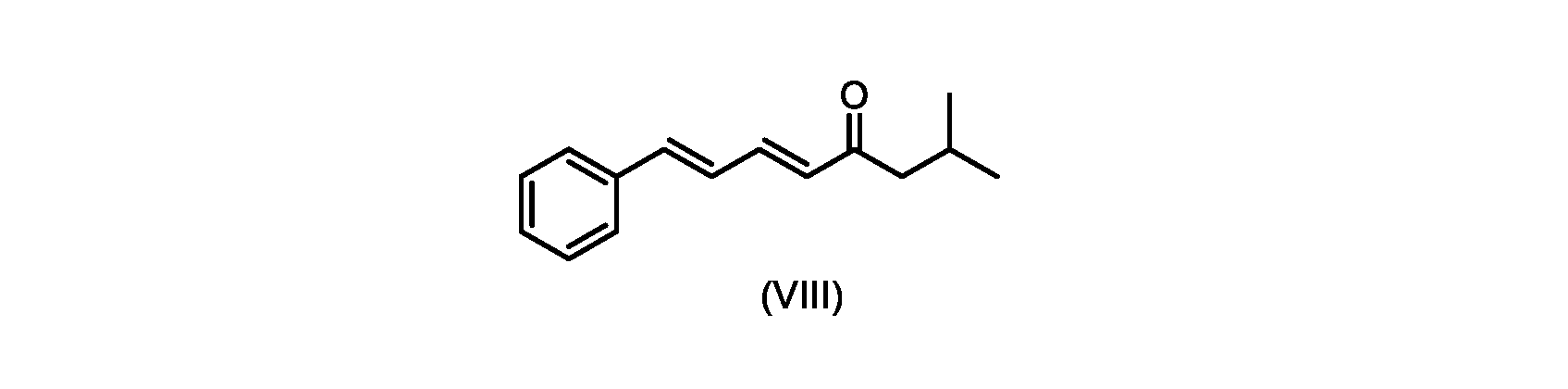
- 前記ジアルキルマロン酸エステルが、ジ-tert-ブチルマロネートまたはマロン酸ジエチルである、請求項23に記載のプロセス。
- 前記添加が、前記ジアルキルマロン酸エステルを、臭化リチウム/トリエチルアミンの存在下で式(VIII)の前記化合物と接触させる工程を有する、請求項23または24に記載のプロセス。
- トランス-シンナムアルデヒド(IX)を
メチルイソブチルケトンと縮合して、式(VIII)の前記化合物を形成する工程をさらに有する、請求項23~25のいずれか1つに記載のプロセス。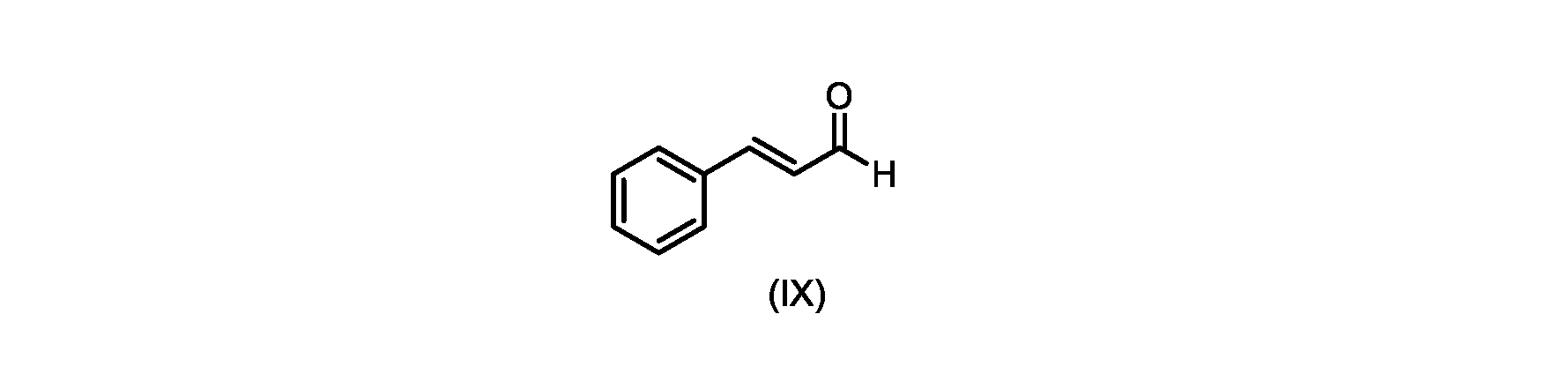
- 前記縮合が、前記メチルイソブチルケトンを、メタノール中の水酸化ナトリウムの存在下で前記トランス-シンナムアルデヒドで処理する工程を有する、請求項26に記載のプロセス。
- 式(I)の化合物またはその塩もしくは溶媒和物を調製するためのプロセスであって、
a)式(VI)の化合物を、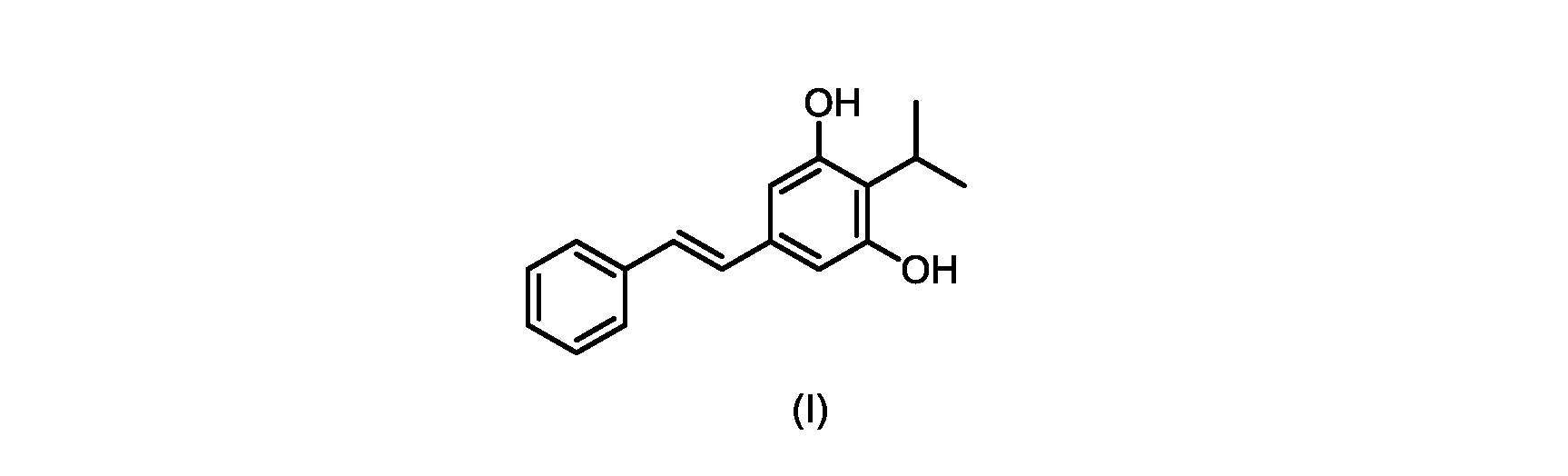
触媒トリエチルアミンと共に加熱して、式(V)の化合物を形成する工程と、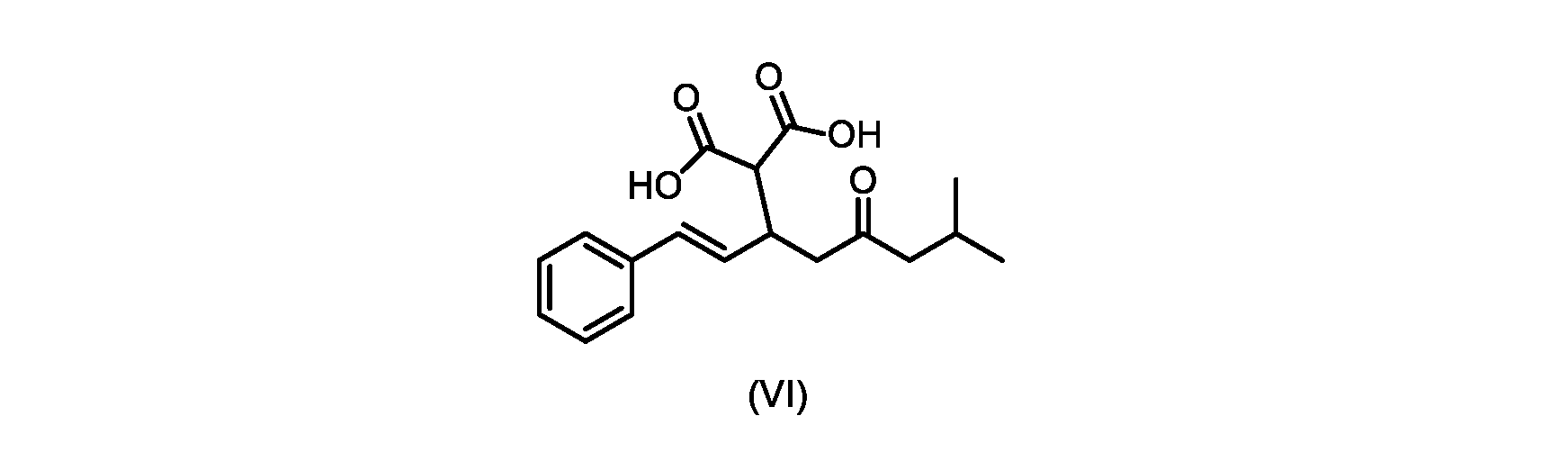
b)式(V)の前記化合物をメタノールおよび塩酸水溶液と共に加熱して、式(IVa)の化合物を形成する工程と、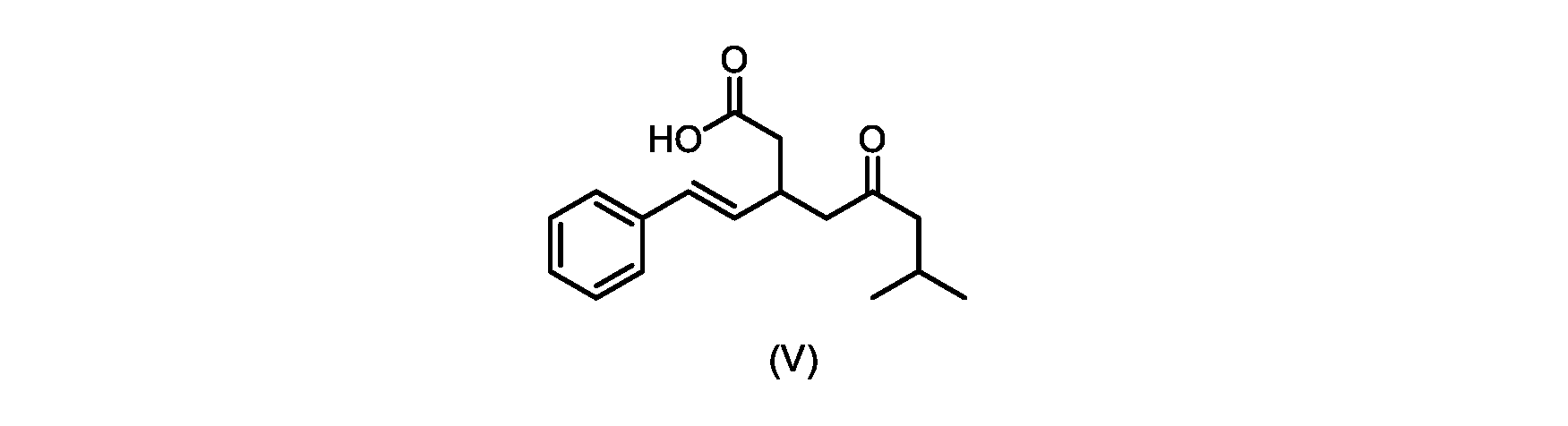
c)式(IVa)の前記化合物の冷却溶液を、カリウムtert-ブトキシドで処理して、式(III)の化合物を形成する工程と、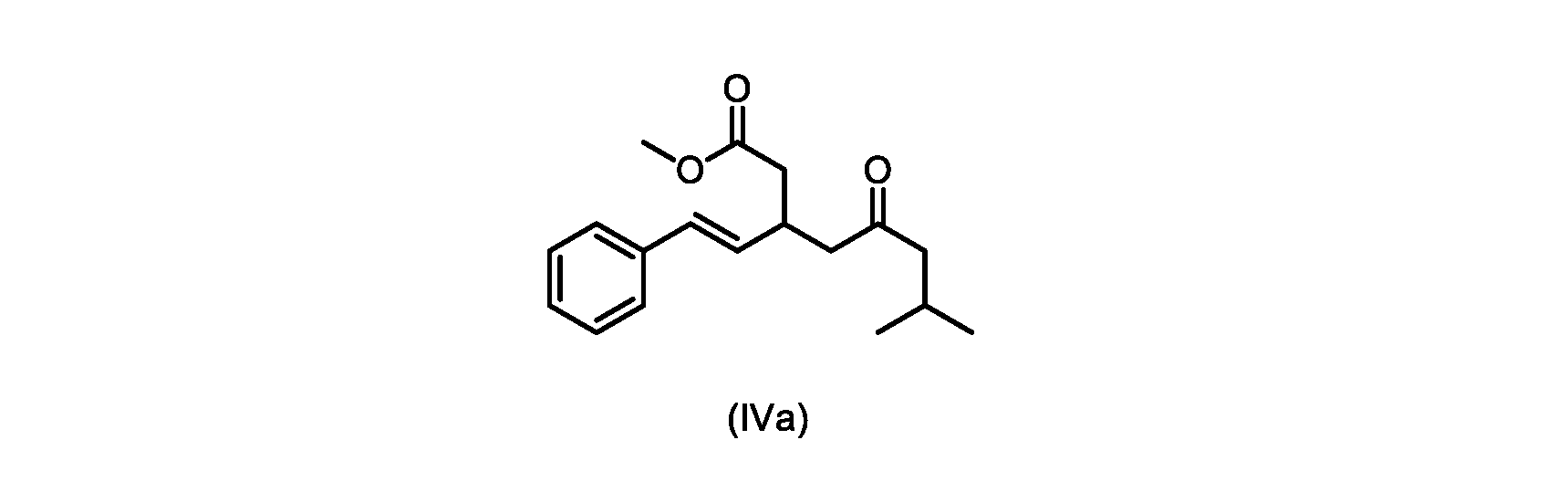
d)式(III)の前記化合物を、1,3-ジクロロ-5,5-ジメチルヒダントインと共に加熱して、式(IIa)の化合物を形成する工程と、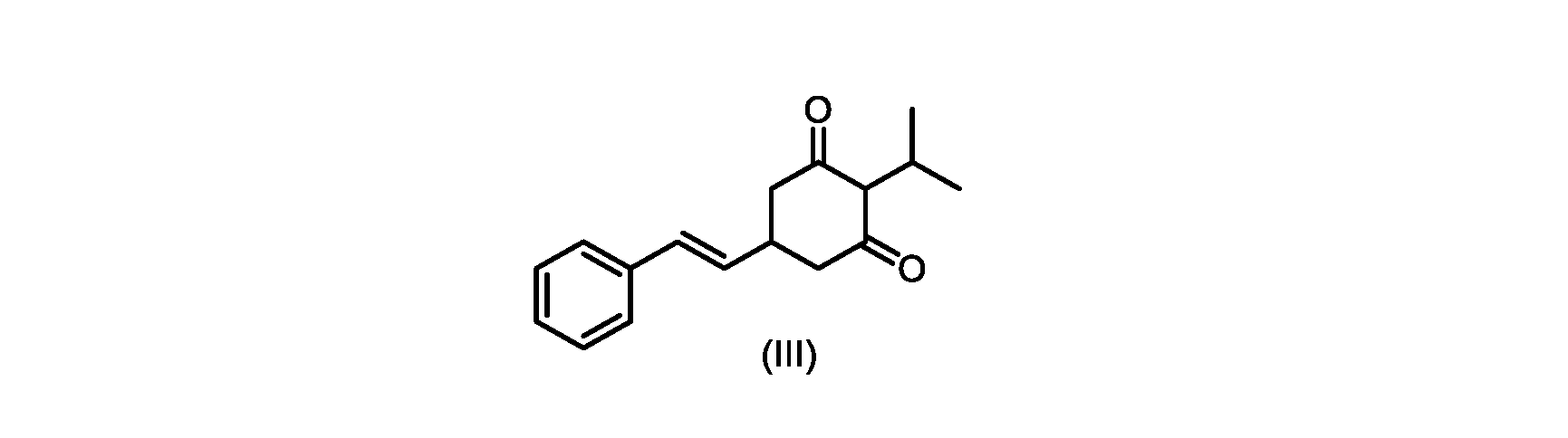
e)式(IIa)の前記化合物を、塩化テトラエチルアンモニウムと共に加熱して、式(I)の前記化合物を形成する工程と、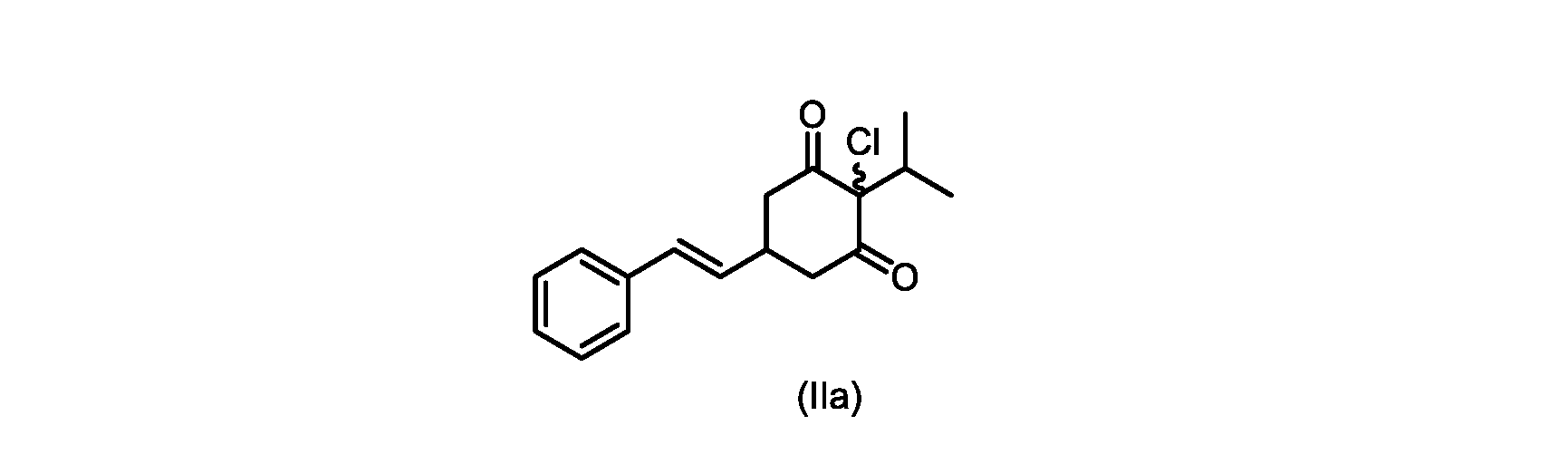
を有する、プロセス。 - f)結晶化によるステップe)からの式(I)の精製をさらに有する、請求項28に記載のプロセス。
- 式(VI)の前記化合物が、
i.メチルイソブチルケトンを、メタノール性水酸化ナトリウムの存在下でトランス-シンナムアルデヒドで処理して、式(VIII)の化合物を形成する工程と、
ii.式(VIII)の前記化合物を、臭化リチウムおよびトリエチルアミンの存在下でマロン酸ジエチルで処理して、式(VIIa)の化合物を形成する工程と、
iii.式(VIIa)の前記化合物を、水酸化ナトリウムおよびエタノールで加水分解して、式(VI)の前記化合物を得る工程と、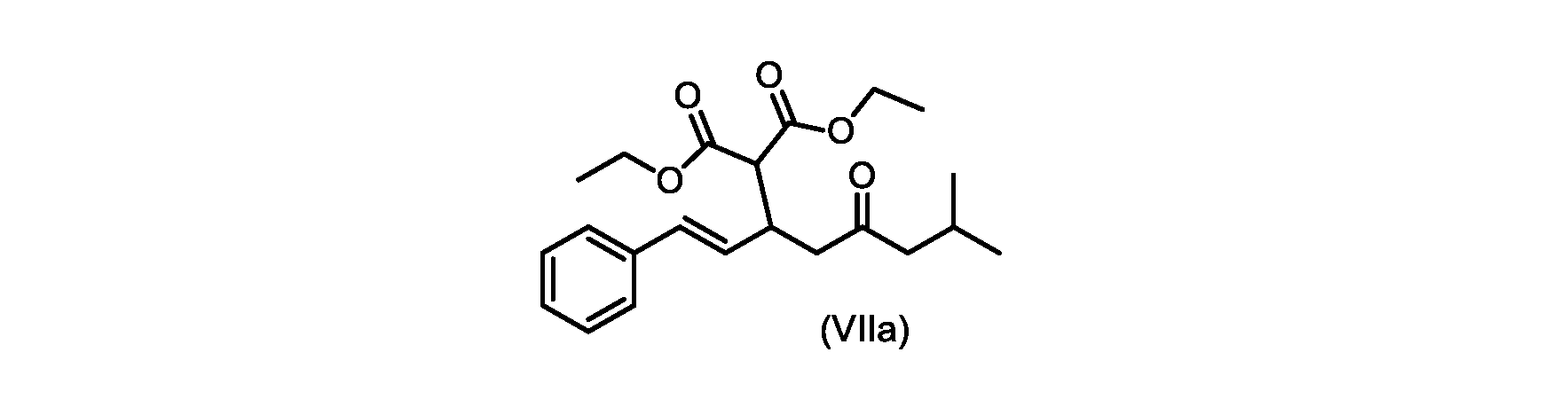
を有するプロセスによって調製される、請求項28または29に記載のプロセス。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022148508A JP2023002516A (ja) | 2017-11-10 | 2022-09-16 | タピナロフを調製するためのプロセス |
| JP2024022828A JP2024069233A (ja) | 2017-11-10 | 2024-02-19 | タピナロフを調製するためのプロセス |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762584192P | 2017-11-10 | 2017-11-10 | |
| US62/584,192 | 2017-11-10 | ||
| JP2020523700A JP6912664B2 (ja) | 2017-11-10 | 2018-11-13 | タピナロフを調製するためのプロセス |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020523700A Division JP6912664B2 (ja) | 2017-11-10 | 2018-11-13 | タピナロフを調製するためのプロセス |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022148508A Division JP2023002516A (ja) | 2017-11-10 | 2022-09-16 | タピナロフを調製するためのプロセス |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2021169482A JP2021169482A (ja) | 2021-10-28 |
| JP7166399B2 true JP7166399B2 (ja) | 2022-11-07 |
Family
ID=66431171
Family Applications (5)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020523700A Active JP6912664B2 (ja) | 2017-11-10 | 2018-11-13 | タピナロフを調製するためのプロセス |
| JP2020216295A Pending JP2021063100A (ja) | 2017-11-10 | 2020-12-25 | タピナロフを調製するためのプロセス |
| JP2021113606A Active JP7166399B2 (ja) | 2017-11-10 | 2021-07-08 | タピナロフを調製するためのプロセス |
| JP2022148508A Pending JP2023002516A (ja) | 2017-11-10 | 2022-09-16 | タピナロフを調製するためのプロセス |
| JP2024022828A Pending JP2024069233A (ja) | 2017-11-10 | 2024-02-19 | タピナロフを調製するためのプロセス |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020523700A Active JP6912664B2 (ja) | 2017-11-10 | 2018-11-13 | タピナロフを調製するためのプロセス |
| JP2020216295A Pending JP2021063100A (ja) | 2017-11-10 | 2020-12-25 | タピナロフを調製するためのプロセス |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022148508A Pending JP2023002516A (ja) | 2017-11-10 | 2022-09-16 | タピナロフを調製するためのプロセス |
| JP2024022828A Pending JP2024069233A (ja) | 2017-11-10 | 2024-02-19 | タピナロフを調製するためのプロセス |
Country Status (14)
| Country | Link |
|---|---|
| US (5) | US10647649B2 (ja) |
| EP (1) | EP3706725A4 (ja) |
| JP (5) | JP6912664B2 (ja) |
| KR (1) | KR102773538B1 (ja) |
| CN (1) | CN111511357B (ja) |
| AU (2) | AU2018365241B2 (ja) |
| BR (1) | BR112020009158A2 (ja) |
| CA (2) | CA3082115A1 (ja) |
| CL (3) | CL2020001226A1 (ja) |
| CO (1) | CO2020007018A2 (ja) |
| IL (2) | IL274439B2 (ja) |
| MX (2) | MX2020004785A (ja) |
| SG (1) | SG11202002576TA (ja) |
| WO (1) | WO2019094934A1 (ja) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT3297605T (pt) | 2015-05-21 | 2022-05-30 | Dermavant Sciences GmbH | Composições farmacêuticas tópicas |
| US11617724B2 (en) | 2015-05-21 | 2023-04-04 | Dermavant Sciences GmbH | Topical pharmaceutical compositions |
| SG11202002576TA (en) | 2017-11-10 | 2020-04-29 | Dermavant Sciences GmbH | Process for preparing tapinarof |
| US11497718B2 (en) | 2018-11-13 | 2022-11-15 | Dermavant Sciences GmbH | Use of tapinarof for the treatment of atopic dermatitis |
| IL297415A (en) * | 2020-04-22 | 2022-12-01 | Yissum Res Dev Co Of Hebrew Univ Jerusalem Ltd | Active phenolate salt complex |
| WO2021236709A1 (en) | 2020-05-19 | 2021-11-25 | Teva Pharmaceuticals International Gmbh | Solid state forms of tapinarof |
| WO2022109626A1 (en) * | 2020-11-23 | 2022-05-27 | Dermavant Sciences GmbH | Gel, ointment, and foam formulations of tapinarof and methods of use |
| CN115703700B (zh) * | 2021-08-03 | 2024-08-16 | 北京颖泰嘉和生物科技股份有限公司 | 1,3-环己二酮的制备方法 |
| WO2023067606A1 (en) * | 2021-10-21 | 2023-04-27 | Sol-Gel Technologies Ltd. | Crystalline polymorph of tapinarof |
| WO2025021674A2 (en) | 2023-07-21 | 2025-01-30 | Interquim, S.A. | Process for the preparation of tapinarof |
| WO2025031935A1 (en) | 2023-08-04 | 2025-02-13 | Sandoz Ag | Crystalline forms of tapinarof |
| CN121969598A (zh) * | 2023-08-24 | 2026-05-01 | 塔罗制药工业有限公司 | 制备本维莫德及相关化合物的方法 |
| IT202300023508A1 (it) | 2023-11-08 | 2025-05-08 | Procos Spa | Processo per la preparazione della forma polimorfa iii di tapinarof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003516400A (ja) | 1999-12-06 | 2003-05-13 | ウェリケム,バイオテック インコーポレーテッド | ヒドロキシルスチルベンならびに新規スチルベン誘導体および類似体による抗炎症治療および乾癬治療ならびにプロテインキナーゼ阻害 |
| CN103992212A (zh) | 2014-05-29 | 2014-08-20 | 河北科技大学 | 顺式苯烯莫德的合成方法及顺式苯烯莫德的应用 |
Family Cites Families (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1442295A (fr) | 1964-10-08 | 1966-06-17 | Smith Kline French Lab | Nouveaux dérivés de l'acide beta, beta-diphényl acrylique et leur procédé de préparation |
| JPS5111748A (en) | 1974-07-16 | 1976-01-30 | Taisho Pharmaceutical Co Ltd | Suchirubenjudotaino seizoho |
| JPS5721341A (en) * | 1980-07-15 | 1982-02-04 | Nippon Zeon Co Ltd | Preparation of substituted acetic ester |
| JPS58159410A (ja) | 1982-03-18 | 1983-09-21 | Mitsubishi Chem Ind Ltd | 抗炎症剤 |
| US4515883A (en) | 1983-04-14 | 1985-05-07 | Ricoh Company, Ltd. | Stilbene derivatives, distyryl derivatives and electrophotographic photoconductor comprising at least one of the derivatives |
| DE3580134D1 (de) | 1984-07-07 | 1990-11-22 | Shudo Koichi Prof Dr Chem | Benzoesaeurederivate. |
| DE68907095T2 (de) | 1988-03-21 | 1994-01-05 | Boehringer Ingelheim Pharma | Verbindungen zur Verhinderung der Biosynthese von der Lipoxygenase abgeleiteten Metaboliten der Arachidonsäure. |
| GB8915104D0 (en) | 1989-06-30 | 1989-08-23 | Beecham Group Plc | Chemical process |
| US5268517A (en) | 1989-07-14 | 1993-12-07 | Basf Aktiengesellschaft | Stereoselective preparation of Z-1,2-diarylallyl chlorides and the conversion thereof into azolylmethyloxiranes, and novel intermediates |
| GB9106177D0 (en) | 1991-03-22 | 1991-05-08 | Aston Molecules Ltd | Substituted diphenylethylenes and analogues or derivatives thereof |
| JPH06135948A (ja) | 1992-10-30 | 1994-05-17 | Taiho Yakuhin Kogyo Kk | スチレン誘導体又はその塩 |
| AU669279B2 (en) | 1993-03-10 | 1996-05-30 | Morinaga Milk Industry Company Limited | Stilbene derivative and stilbene analog derivative, and use thereof |
| JPH0753359A (ja) | 1993-06-11 | 1995-02-28 | Kao Corp | リポキシゲナーゼ阻害剤 |
| US5760277A (en) | 1995-06-08 | 1998-06-02 | Firmenich Sa | Process for the manufacture of unsaturated cycloaliphatic ketones |
| JPH1072330A (ja) | 1996-09-03 | 1998-03-17 | Kansai Kouso Kk | チロシナーゼ活性阻害剤及び化粧料 |
| FR2777183B1 (fr) | 1998-04-10 | 2001-03-02 | Oreal | Utilisation d'au moins un hydroxystilbene dans une composition destinee a favoriser la desquamation de la peau et composition le comprenant |
| US6624197B1 (en) | 1998-05-08 | 2003-09-23 | Calyx Therapeutics, Inc. | Diphenylethylene compounds |
| AU4084599A (en) | 1998-05-18 | 1999-12-06 | Oklahoma Medical Research Foundation | Resveratrol inhibition of myeloperoxidase |
| FR2785284B1 (fr) | 1998-11-02 | 2000-12-01 | Galderma Res & Dev | Analogues de la vitamine d |
| US20080255245A1 (en) * | 1999-12-06 | 2008-10-16 | Welichem Biotech Inc. | Anti-Inflammatory and Psoriasis Treatment and Protein Kinase Inhibition by Hydroxystilbenes and Novel Stilbene Derivatives and Analogues |
| US7321050B2 (en) | 1999-12-06 | 2008-01-22 | Welichem Biotech Inc. | Anti-inflammatory and psoriasis treatment and protein kinase inhibition by hydroxy stilbenes and novel stilbene derivatives and analogues |
| JP2001261585A (ja) | 2000-03-15 | 2001-09-26 | Chemiprokasei Kaisha Ltd | ハロゲン化ビニル化合物の合成法 |
| US6552085B2 (en) | 2000-08-16 | 2003-04-22 | Insmed Incorporated | Compositions containing hypoglycemically active stilbenoids |
| US6410596B1 (en) | 2000-08-16 | 2002-06-25 | Insmed Incorporated | Compositions containing hypoglycemically active stillbenoids |
| AU2002226225B2 (en) | 2001-01-18 | 2008-03-20 | Dermavant Sciences GmbH | Novel 1,2-diphenylethene derivatives for treatment of immune diseases |
| JP4425628B2 (ja) | 2001-07-23 | 2010-03-03 | ジョンソン・アンド・ジョンソン・コンシューマー・カンパニーズ・インコーポレイテッド | 細胞保護化合物、薬学的処方物および美容用処方物、ならびに方法 |
| AU2003271470B2 (en) | 2002-10-01 | 2010-03-18 | Welichem Biotech Inc. | Novel bioactive diphenyl ethene compounds and their therapeutic applications |
| WO2009108762A2 (en) | 2008-02-26 | 2009-09-03 | The Penn State Research Foundation | Methods and compositions for treatment of retinoid-responsive conditions |
| JP4426633B2 (ja) * | 2008-04-11 | 2010-03-03 | 花王株式会社 | 2−アルキル−2−シクロアルケン−1−オンの製造方法 |
| US8524782B2 (en) | 2008-07-23 | 2013-09-03 | Laurus Labs Private Limited | Key intermediate for the preparation of Stilbenes, solid forms of Pterostilbene, and methods for making the same |
| CN101531571B (zh) | 2009-04-17 | 2013-03-20 | 河北科技大学 | 六次甲基四胺氧化合成二苯乙烯类化合物的方法 |
| CN101564537B (zh) | 2009-06-08 | 2011-05-25 | 河北科技大学 | 3,5-二羟基-4-异丙基二苯乙烯-溴乙酸乙酯-聚乙二醇复合物及其合成方法 |
| CN101648851B (zh) | 2009-09-03 | 2012-10-10 | 河北科技大学 | (e)-3,5-二羟基-4-异丙基二苯乙烯的清洁制备方法 |
| CN103172497A (zh) * | 2011-12-23 | 2013-06-26 | 重庆市科学技术研究院 | 治疗牛皮癣新药苯烯莫德的工业化生产工艺 |
| CN104003848B (zh) * | 2013-02-25 | 2016-02-10 | 武汉诺安药业有限公司 | (e)-3,5-二羟基-4-异丙基二苯乙烯制备方法 |
| US10376475B2 (en) | 2014-12-12 | 2019-08-13 | Dermavant Sciences GmbH | Method of use |
| CN105884581A (zh) * | 2014-12-30 | 2016-08-24 | 上海复星医药产业发展有限公司 | 苯烯莫德的制备方法 |
| PT3297605T (pt) | 2015-05-21 | 2022-05-30 | Dermavant Sciences GmbH | Composições farmacêuticas tópicas |
| US11617724B2 (en) | 2015-05-21 | 2023-04-04 | Dermavant Sciences GmbH | Topical pharmaceutical compositions |
| CN111148729A (zh) | 2017-09-30 | 2020-05-12 | 北京文丰天济医药科技有限公司 | 苯烯莫德的晶型及其用途与制备方法 |
| SG11202002576TA (en) | 2017-11-10 | 2020-04-29 | Dermavant Sciences GmbH | Process for preparing tapinarof |
| WO2021236709A1 (en) | 2020-05-19 | 2021-11-25 | Teva Pharmaceuticals International Gmbh | Solid state forms of tapinarof |
-
2018
- 2018-11-13 SG SG11202002576TA patent/SG11202002576TA/en unknown
- 2018-11-13 AU AU2018365241A patent/AU2018365241B2/en active Active
- 2018-11-13 MX MX2020004785A patent/MX2020004785A/es unknown
- 2018-11-13 CA CA3082115A patent/CA3082115A1/en active Pending
- 2018-11-13 JP JP2020523700A patent/JP6912664B2/ja active Active
- 2018-11-13 US US16/189,268 patent/US10647649B2/en active Active
- 2018-11-13 KR KR1020207016222A patent/KR102773538B1/ko active Active
- 2018-11-13 CA CA3253169A patent/CA3253169A1/en active Pending
- 2018-11-13 EP EP18875722.3A patent/EP3706725A4/en active Pending
- 2018-11-13 BR BR112020009158-8A patent/BR112020009158A2/pt unknown
- 2018-11-13 IL IL274439A patent/IL274439B2/en unknown
- 2018-11-13 WO PCT/US2018/060749 patent/WO2019094934A1/en not_active Ceased
- 2018-11-13 IL IL304090A patent/IL304090B2/en unknown
- 2018-11-13 CN CN201880071224.8A patent/CN111511357B/zh active Active
-
2020
- 2020-04-15 US US16/849,346 patent/US10961175B2/en active Active
- 2020-05-08 CL CL2020001226A patent/CL2020001226A1/es unknown
- 2020-06-09 CO CONC2020/0007018A patent/CO2020007018A2/es unknown
- 2020-07-13 MX MX2022015106A patent/MX2022015106A/es unknown
- 2020-12-25 JP JP2020216295A patent/JP2021063100A/ja active Pending
-
2021
- 2021-02-12 US US17/174,566 patent/US11597692B2/en active Active
- 2021-07-08 JP JP2021113606A patent/JP7166399B2/ja active Active
-
2022
- 2022-09-16 JP JP2022148508A patent/JP2023002516A/ja active Pending
-
2023
- 2023-01-16 CL CL2023000154A patent/CL2023000154A1/es unknown
- 2023-02-03 US US18/164,309 patent/US12275696B2/en active Active
- 2023-12-04 AU AU2023274224A patent/AU2023274224B2/en active Active
-
2024
- 2024-02-19 JP JP2024022828A patent/JP2024069233A/ja active Pending
- 2024-12-16 CL CL2024003879A patent/CL2024003879A1/es unknown
-
2025
- 2025-03-12 US US19/077,700 patent/US20250270158A1/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003516400A (ja) | 1999-12-06 | 2003-05-13 | ウェリケム,バイオテック インコーポレーテッド | ヒドロキシルスチルベンならびに新規スチルベン誘導体および類似体による抗炎症治療および乾癬治療ならびにプロテインキナーゼ阻害 |
| CN103992212A (zh) | 2014-05-29 | 2014-08-20 | 河北科技大学 | 顺式苯烯莫德的合成方法及顺式苯烯莫德的应用 |
Non-Patent Citations (3)
| Title |
|---|
| European Journal of Organic Chemistry,2014年,Vol.2014, No.36,pp.8026-8028 |
| Journal of the Chemical Society,1958年,pp.911-912 |
| Tetrahedron,1973年,Vol.29, No.23,pp.3857-3859 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7166399B2 (ja) | タピナロフを調製するためのプロセス | |
| AU2009290849B2 (en) | Novel imidazolidine compounds as androgen receptor modulators | |
| KR840002306B1 (ko) | 치환된 옥시란카복실산의 제조방법 | |
| JPH0261950B2 (ja) | ||
| RU2279433C2 (ru) | Новое соединение для лечения импотенции | |
| RU2795144C2 (ru) | Способ получения тапинарофа | |
| JP2006519241A (ja) | 5−ht2c受容体活性を有する化合物およびその使用 | |
| EP0074121A1 (de) | 2,3,4,5-Tetrahydro-1-benzoxepin-3,5-dion-Derivate und Verfahren zu deren Herstellung | |
| RU2722717C1 (ru) | Способ получения N6-ди(перфторалкокси)фосфорилсиднониминов | |
| EA021299B1 (ru) | Способ получения 5-(2-аминопиримидин-4-ил)-2-арил-1н-пиррол-3-карбоксамидов | |
| RU2058979C1 (ru) | Производные алкенкарбоновой кислоты, или смеси их изомеров, или их индивидуальные изомеры, или соли, обладающие свойствами антагонистов лейкотриена, способы их получения, промежуточные для их получения, и фармацевтическая композиция на их основе | |
| US20170158637A1 (en) | Synthetic Processes of Carprofen | |
| ES2857128T3 (es) | Procedimiento para la síntesis de firocoxib | |
| EA027231B1 (ru) | Способ получения бисфосфоновых кислот |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20210721 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20220610 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220621 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220916 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20221004 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20221025 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7166399 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S201 | Request for registration of exclusive licence |
Free format text: JAPANESE INTERMEDIATE CODE: R314201 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |