JP7089636B2 - アンジオテンシンii2型受容体拮抗薬の塩形、結晶形及びその製造方法 - Google Patents
アンジオテンシンii2型受容体拮抗薬の塩形、結晶形及びその製造方法 Download PDFInfo
- Publication number
- JP7089636B2 JP7089636B2 JP2021507501A JP2021507501A JP7089636B2 JP 7089636 B2 JP7089636 B2 JP 7089636B2 JP 2021507501 A JP2021507501 A JP 2021507501A JP 2021507501 A JP2021507501 A JP 2021507501A JP 7089636 B2 JP7089636 B2 JP 7089636B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- solvent
- reagent
- formula
- crystalline form
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
- C07D217/26—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurology (AREA)
- Engineering & Computer Science (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description



式(I)化合物を混合溶剤に加えて溶解するステップ(a)と、
30~50℃で10~30時間撹拌するステップ(b)と、
ろ過後水洗し、30~50℃でろ過ケーキを15~25時間乾燥させるステップ(c)と、を含み、
前記混合溶剤は、アセトンと水の体積比が1:1.5~2.5である、式(I)化合物の結晶形Aの調製方法をさらに提供する。


式(I)化合物を溶剤に加えて、懸濁液を形成するステップ(a)と、
懸濁液を35~45℃で30~60時間撹拌するステップ(b)と、
遠心分離後、ろ過ケーキを8~16時間乾燥させるステップ(c)と、を含み、
前記溶剤は、メタノール、エタノール、アセトニトリルから選ばれるものであり、または、アセトンと水との体積比が3:2の混合溶剤である、式(I)化合物の結晶形Bの調製方法をさらに提供する。


溶剤Fは、n-ヘプタン、ジクロロメタン、テトラヒドロフラン、シクロヘキサン、及びジオキサンから選ばれ、
試薬Gは酸化銀、硫酸マグネシウム、硫酸カルシウム、及び硫酸ナトリウムから選ばれる。)

試薬Iは、水酸化リチウム一水和物及び水酸化ナトリウムから選ばれ、
溶剤Jはジクロロメタンから選ばれ、
触媒KはN,N-ジメチルホルムアミドから選ばれ、
試薬Lは塩化オクサリルから選ばれ、
溶剤Mはジクロロメタンから選ばれ、
試薬Nはピラゾールから選ばれ、
試薬OはN-メチルモルホリンから選ばれ、
溶剤PはN,N-ジメチルホルムアミド、ジメチルスルホキシド、ジクロロメタン、及びテトラヒドロフランから選ばれ、
試薬Qはテトラメチルグアニジン、1,8-ジアザビシクロウンデク-7-エン、トリエチルアミン、ジイソプロピルエチルアミン、及び2,6-ジメチルピリジンから選ばれる。)
本発明の化合物はインビトロで優れた生物学的活性を示し、そして複数の属において良好な薬物動態特性を示す。その結晶形は安定的であり、吸湿性が低い。
特に断らない限り、本明細書で使用される下記用語及び句は下記定義を含むことを意図する。1つの特定の句または用語は、特に定義されない場合、不明確または不明瞭であると理解できず、一般的な定義として理解すべきである。本明細書に商品名が記載されている場合、該商品名に対応する商品またはその活性成分を意味する。
r.t.は室温を表し、THFはテトラヒドロフランを表し、NMPはN-メチルピロリドンを表し、MeSO3Hはメタンスルホン酸を表し、DMEはエチレングリコールジメチルエーテルを表し、DCMはジクロロメタンを表し、Xphosは2-ジシクロヘキシルホスフィノ-2',4',6'-トリイソプロピルビフェニルを表し、EtOAcは酢酸エチルを表し、MeOHはメタノールを表し、acetoneはアセトンを表し、2-Me-THFは2-メチルテトラヒドロフランを表し、IPAはイソプロパノールを表し、HATUはO-(7-アザベンゾトリアゾール-1-イル)-N,N,N',N'-テトラメチルウロニウムヘキサフルオロホスファートを表す。
化合物は手動またはChemDraw(登録商標)ソフトウェアで命名され、市販化合物はサプライヤーのカタログ名を用いる。
本発明のX-線粉末回折(X-ray powder diffractometer、XRPD)方法
器具型番:Bruker D8advance X-線粉末回折計
テスト方法:XRPD検出に約10~20mgのサンプルが使用される。
詳細なXRPDパラメータは以下のとおりである。
X線管:Cu、kα(λ=1.54056Å)、
X線管電圧:40kV、X線管電流流:40mA
発散スリット:0.60mm
検出器スリット:10.50mm
散乱防止スリット:7.10mm
走査範囲:3~40degまたは4~40deg
ステップ幅:0.02deg
ステップ:0.12秒
サンプルトレイの回転数:15rpm
本発明の示差走査熱量分析(Differential Scanning Calorimeter、DSC)方法
器具型番号:TA Q2000示差走査熱量計
テスト方法:サンプル(~1mg)をDSC用アルミ鍋に入れてテストし、50mL/min、N2条件下、10℃/minの昇温速度で、サンプルを室温から250℃(または280℃)に加熱する。
本発明の熱重量分析(Thermal Gravimetric Analyzer、TGA)方法
器具型番:TA Q5000熱重量分析計
テスト方法:サンプル(2~5mg)をTGA用プラチナ鍋に入れてテストし、25mL/min、N2条件下、10℃/minの昇温速度で、サンプルを室温から30℃または重量が20%損失されるまで加熱する。
本発明の動的蒸気収着分析(Dynamic Vapor Sorption、DVS)方法
器具型番:SMS DVS Advantage動的蒸気収着装置
テスト条件:サンプル(10~15mg)をDVSサンプルトレイに入れてテストする。
詳細なDVSパラメータは以下のとおりである。
温度:25℃
平衡:dm/dt=0.01%/min(最短:10min、最長:180min)
乾燥:0%RH下、120min乾燥
RH(%)テスト勾配:10%
RH(%)テスト勾配範囲:0%~90%~0%
吸湿性の評価は以下のように分類される。


詳細なパラメータは以下のとおりである。


酸化銀(1.5g、6.6mmol)を化合物S-マンデル酸(500.0mg、3.3mmol)とブロモシクロペンタン(49.0g、328.6mmol)の混合液に加え、次に、20~25℃の条件下で撹拌しながら16時間反応させた。反応液をろ過し、ろ液を真空濃縮させて溶剤を除去し、得た粗製物をシリカクロマトグラフィーカラム(溶離液:酢酸エチル/石油エーテル0~10%)により分離して精製し、化合物S-A1-1を得た。1H NMR(400MHz,CDCl3):δ7.49-7.40(m,2H),7.38-7.28(m,3H),5.22-5.19(m,1H),4.88(s,1H),4.03-3.99(m,1H),1.89-1.64(m,10H),1.57-1.45(m,6H).MS m/z:311.1[M+Na]+.
化合物S-A1-1(340.0mg、1.2mmol)をテトラヒドロフラン(6.0mL)と水(3.0mL)の混合溶剤に溶解し、水酸化リチウム一水和物(283.0mg、11.8mmol)を加え、反応液を20~25℃で48時間撹拌した。反応液を1N塩酸でpH<3に調整した後、酢酸エチル(20mL×3)で抽出した。合併した有機相を飽和食塩水(50mL)で洗浄して、無水硫酸ナトリウムで乾燥させ、真空濃縮させて、得た粗製物をシリカクロマトグラフィーカラム(溶離液:0~37.5%石油エーテル/酢酸エチル)により分離して精製し、化合物S-A1を得た。1H NMR(400MHz,CDCl3):δ7.45-7.34(m,5H),4.93(s,1H),4.07-4.03(m,1H),1.78-1.69(m,6H),1.62-1.48(m,2H).
SFC:カラム:ChiralCel OJ-H(150mm×4.6mm、5um);移動相:A:CO2、B:エタノール[0.05%ジエチルアミン];B%:5%~40%5.5min、40%3min、5%1.5min;Rt=2.321min;95.6%ee.

窒素ガスの保護下、化合物C1-1(200.0g、1.31mol)を無水エタノール(1.50L)に溶解した。15℃で撹拌しながら無水炭酸カリウム(181.1g、1.31mol)及び臭化ベンジル(268.9g、1.57mol)を順次加え、次に反応液を100℃に加熱して15時間撹拌し続けた。反応液を室温に冷却させた後ろ過し、ろ液を真空濃縮させ、得た油状物を酢酸エチル(3.0L)で再溶解した後、2N水酸化ナトリウム水溶液(500mL×2)及び飽和食塩水(600mL×2)で順次洗浄し、無水硫酸ナトリウムで乾燥させてろ過して、真空濃縮させて粗製物を得た。粗製物を石油エーテルに分散させて1時間撹拌し、ろ過して化合物C1-2を得た。1H NMR(400MHz,CDCl3):δ10.25(s,1H),7.42-7.34(m,6H),7.21-7.12(m,2H),5.19(s,2H),3.96(s,3H).
窒素ガスの保護下、化合物C1-2(220.0g、908.08mmol)、2-ニトロ酢酸エチル(145.0g、1.09mol)、及びジエチルアミン塩酸塩(149.3g、1.36mol)の、無水トルエン(2.1L)中の混合溶液を130℃に加熱して15時間還流させ、反応により生成した水をDeane-Stark分水器で分離した。反応液を室温に冷却した後、真空濃縮させてトルエンを除去した。残留物をジクロロメタン(500mL)に再溶解した後、飽和食塩水(1000mL×2)で洗浄し、無水硫酸ナトリウムで乾燥させてろ過し、真空濃縮させて化合物C1-3を得て、この化合物を精製せずに次のステップの反応に用いた。
窒素ガスの保護下、上記ステップ2で得られた粗品化合物C1-3(430.0g、1.2mol)をイソプロパノール(2.2g、36.0mmol)及びクロロホルム(4.5L)に溶解し、混合液を0℃に冷却した後、撹拌しながら100~200メッシュのシリカゲル(1.8kg)に加え、次に、1.5時間かけてホウ水素化ナトリウム(201.1g、5.3mol)をバッチで加えた。反応液を15℃に昇温した後、撹拌しながら12時間反応させ続けた。酢酸(210mL)を緩やかに加えた後、15分間撹拌し続け、反応液をろ過し、ろ過ケーキをジクロロメタン(500mL)で洗浄した。合併したろ液を真空濃縮させて、得た残留物をシリカクロマトグラフィーカラム(溶離液:6%~10%石油エーテル/酢酸エチル)により分離して精製し、化合物C1-4を得た。1H NMR(400MHz,CDCl3):δ7.48-7.33(m,5H),7.02-6.97(m,1H),6.94-6.90(m,1H),6.64-6.62(dd,J=1.6,7.6Hz,1H),5.33-5.30(dd,J=6.0,9.2Hz,1H),5.19-5.05(m,2H),4.15-4.10(q,J=7.2Hz,2H),3.91(s,3H),3.44-3.31(m,2H),1.16-1.12(t,J=7.2Hz,3H).
15℃で、化合物C1-4(76.2g、212.04mmol)を酢酸(700mL)に溶解し、反応温度を60~65℃の間に維持しながら亜鉛粉(110.9g、1.70mol)を緩やかに加え、添加終了後、60℃で撹拌しながら2時間反応させ続けた。反応液を室温に冷却した後、ろ過し、ろ過ケーキを酢酸(300mL)で洗浄した。合併したろ液を真空濃縮させて、得た残留物をジクロロメタン(500mL)に再溶解し、飽和重炭酸ナトリウム水溶液(200mL×2)及び飽和食塩水(200mL×2)で洗浄し、無水硫酸ナトリウムで乾燥させてろ過し、真空濃縮させて粗製物C1-5を得て、この化合物を精製せずに次のステップに用いた。MS m/z:330.1[M+1]+.
15℃、窒素ガスの保護下、化合物C1-5(48.9g、149.4mmol)を2N塩酸溶液(500mL)に溶解し、その後、37%ホルムアルデヒド水溶液(36.4g、448.1mmol)を加えて、反応液を25時間撹拌した。ろ過し、ろ過ケーキを水(100mL)で洗浄し、真空乾燥させて、化合物C1の塩酸塩を得た。MS m/z:342.1[M+1]+.
化合物C1(40.0g、117.2mmol)をキラルカラムにより分離して、2つの異性体(-)-C1及び(+)-C1を得た。
(-)-C1:1H NMR(400MHz,CDCl3):δ7.40-7.38(m,2H),7.33-7.22(m,3H),6.73-6.71(m,2H),4.93-4.92(m,2H),4.17-4.15(q,J=7.2Hz,2H),4.10-3.93(m,2H),3.79(s,3H),3.62-3.58(m,1H),3.07-3.06(m,1H),2.77-2.65(m,1H),1.21(t,J=7.2Hz,3H).MS m/z:342.1[M+1]+.[α]=-23.4.
(+)-C1:1H NMR(400MHz,CDCl3):δ7.43-7.40(m,2H),7.33-7.22(m,3H),6.86(s,2H),5.06-4.95(q,J=11.2Hz,2H),4.54-4.50(m,1H),4.33-4.21(m,3H),4.07-4.05(m,1H),3.88(s,3H),3.34-3.25(m,1H),3.20-3.14(m,1H),1.30-1.26(t,J=7.2Hz,3H).MS m/z:342.1[M+1]+.[α]=+9.8.

アセトン(21.2L)を50Lの反応釜に加えて、撹拌を開始し、次に、出発原料D1-0(2.69kg)、炭酸カリウム(3.48kg)及び臭化ベンジル(3.39kg)を反応釜に順次加え、反応液を55~60℃で約18時間撹拌した。反応液を10~20℃に降温して、減圧して吸引濾過し、ろ過ケーキをアセトン(2L、1.5L)で洗浄した。ろ液をロータリーエバポレータに移し、外温40~45℃で減圧濃縮させ、得た粗品を酢酸エチル(26L)に溶解して、13L水で2回洗浄し(毎回6.5L)、13L飽和塩化ナトリウム水溶液で2回洗浄し(毎回6.5L)、有機相を無水硫酸ナトリウム(1.5kg)で乾燥させてろ過し、ろ液を外温40~45℃で減圧濃縮させ、別のバッチと合併して処理し、さらに、得た粗品を石油エーテル30Lに加え、外温0~5℃で21時間撹拌し、ろ過して、ろ過ケーキを石油エーテル4Lで2回洗浄し(毎回2L)、得たろ過ケーキをシリカゲルカラムクロマトグラフィー(溶離剤:0~10%酢酸エチル/石油エーテル)により分離して精製し、化合物D1-1を得た。
1H NMR(400MHz,CDCl3):δ10.25(s,1H),7.41-7.36(m,6H),7.17-7.07(m,2H),5.19(s,2H),3.95(s,3H)。
テトラヒドロフラン(5.0L)を50Lの反応釜に加え、撹拌を開始し、次に、出発原料D1-2(3.21kg)及びテトラメチルグアニジン(1.31kg)を反応釜に順次加え、降温して、内温を10℃以下に維持しながら原料A-1(2.3kg)のテトラヒドロフラン(4.8L)溶液を滴下し、反応液を20~30℃で約16時間撹拌した。反応液を外温35~40℃で減圧濃縮させ、得た粗品を酢酸エチル(20L)に溶解し、10%のクエン酸水溶液8L、飽和塩化ナトリウム水溶液10Lで順次2回洗浄し(毎回5L)、有機相を無水硫酸ナトリウム(1.0kg)で乾燥させてろ過し、ろ液を外温35~40℃で減圧濃縮させ、得た粗製物をシリカゲルカラムクロマトグラフィー(溶離剤:0~30%酢酸エチル/石油エーテル)により分離して精製し、化合物D1-3を得た。
1H NMR(400MHz,CDCl3):δ7.38-7.32(m,5H),7.08-7.03(m,2H),6.94-6.91(m,1H),4.98(s,2H),3.90(s,3H),3.84(s,3H),1.39(s,9H)。
ビス(1,5-シクロオクタジエン)ロジウム(I)トリフルオロメタンスルホナート(249.20mg)及び(+)-1,2-ビス[(2R,5R)-2,5-ジエチルホスフォラノ]ベンゼン(210.40mg)をメタノール(20mL)に溶解し、混合液を窒素ガスの保護下、15分間撹拌し、アルゴンガスの雰囲気下、化合物D1-3(200g)のメタノール(1L)溶液に加え、アルゴンガスを用いて3回置換し、水素ガスを用いて3回置換し、反応液を水素ガス(50psi)雰囲気、及び20~25℃の条件で18時間撹拌した。有機溶剤を減圧除去し、得た粗製物を別のバッチと合併し、シリカゲルでろ過して、化合物D1-4を得た。
1H NMR(400MHz,CDCl3):δ7.54-7.46(m,2H),7.41-7.35(m,3H),7.03-7.00(m,1H),6.90-6.86(m,1H),6.76-6.74(m,1H),5.35-5.32(m,1H),5.05(s,2H),4.49-4.44(m,1H),3.90(s,3H),3.62(s,3H),3.06-2.95(m,2H),1.39(s,9H)。
SFC:カラム:Lux Cellulose-2(150mm×4.6mm、3um);移動相:B:イソプロパノール[0.05%エチルアミン];B%:5%~40%5.5min、40%3min、5%1.5min;Rt=3.247min;97.8%ee.
水酸化リチウム一水和物(0.76kg)を水(16.0L)に溶解し、内温を15℃以下に維持しながら化合物A-4(3.69kg)のテトラヒドロフラン(10.6L)溶液を滴下し、反応液を10~20℃の条件下で18時間撹拌した。飽和クエン酸水溶液でpHを約5に調整し、有機溶剤を減圧除去し、得た粗製物を酢酸エチル16.0Lに加えて、分液し、有機相を10%のクエン酸水溶液(8L)で洗浄し、10%の塩化ナトリウム水溶液で3回洗浄し(毎回6.0L)、無水硫酸ナトリウム1.0kgで乾燥させてろ過し、有機溶剤を減圧除去し、化合物D1-5を得た。
1H NMR(400MHz,CDCl3):δ7.49-7.33(m,5H),7.03-7.00(m,1H),6.90-6.86(m,1H),6.76-6.74(m,1H),5.47-5.45(m,1H),5.11(s,2H),4.49-4.44(m,1H),3.90(s,3H),3.06-2.95(m,2H),1.39(s,9H)。
酢酸エチル(4.0L)を10L三口フラスコに加え、ドライアイス/エタノールで冷却し、塩化水素ガス(900.0g)を導入して、さらに酢酸エチル1Lを加えて希釈し、4M塩化水素酢酸エチル溶液として使用に備えた。化合物A-5(1.5kg)を酢酸エチル(10.0L)に溶解して、内温を10℃以下に維持しながら4Mの塩化水素酢酸エチル溶液を加え、反応液を5~15℃の条件下で2時間撹拌した。イソプロピルエーテル6.0Lを加えて、反応液を5~10℃の条件下で16時間撹拌し続けた。ろ過し、ろ過ケーキをイソプロピルエーテルで2回洗浄し(毎回1.2L)、化合物D1-6を得た。
1H NMR(400MHz,CD3OD):δ7.49-7.44(m,2H),7.37-7.33(m,3H),7.09-7.04(m,2H),6.83-6.80(m,1H),5.19-5.07(m,2H),4.18-4.14(m,1H),3.93(s,3H),3.33-3.28(m,1H),2.90-2.84(m,1H)。
化合物D1-6(2.24kg)を8個のバッチ(302.56g)に等分して遊離し、各バッチを水(4.8L)に加えて、炭酸ナトリウム(53.96g)の水(302.5mL)溶液を滴下し、反応液を0.5時間撹拌し、ろ過したろ過ケーキを合併して、水(6.2L)で1回洗浄した。遊離したろ過ケーキを水(22.0L)に加え、85%のリン酸(850.0mL)及び37%のホルムアルデヒド水溶液(900.0mL)を順次加え、反応液を55~65℃に昇温した条件下で16時間撹拌し続けた。酢酸ナトリウム(988.7g)の水(3.0L)溶液を滴下し、pHを約3に調整してろ過し、ろ過ケーキを水で4回洗浄し(毎回6.0L)、アセトン(12.0L)で1回洗浄し、真空乾燥させて、化合物D1を得た。
1H NMR(400MHz,CD3OD):δ7.49-7.30(m,5H),7.02-6.93(m,2H),5.04(s,2H),4.30-4.20(m,2H),3.89(s,3H),3.74-3.70(m,1H),3.54-3.48(m,1H),2.92-2.84(m,1H)。

化合物(-)-C1(155.00mg、454.00μmol)及び化合物S-A1(90.00mg、408.60μmol)をジクロロメタン(5.00mL)に溶解し、HATU(259.00mg、681.00μmol)及びジイソプロピルエチルアミン(118.00mg、912.54μmol、159.46μL)を順次加え、反応液を20~25℃で16時間撹拌し続けた。反応液を水15mLに注入して、分液し、水相をジクロロメタンで3回抽出し(20mL×3)、合併した有機相を飽和食塩水30mLで1回洗浄し、無水硫酸ナトリウムで乾燥させ、真空乾燥させて粗品を得た。シリカゲルカラムクロマトグラフィー(溶離剤:0-50%石油エーテル/酢酸エチル)により分離して精製し、化合物I-1を得た。1H NMR(400MHz,CDCl3)δ:7.50-7.20(m,10H),6.84(m,1H),6.66(d,J=8.0Hz,0.5H),6.43(d,J=12.0Hz,0.5H),5.50-5.48(m,0.5H),5.32(d,J=8.0Hz,1H),5.07-4.76(m,3H),4.60(d,J=16.0Hz,0.5H),4.51-4.45(m,1H),4.18-4.07(m,2H),3.84(d,J=12Hz,3H),3.66-3.61(m,0.5H),3.55-3.40(m,1H),3.17-3.11(m,0.5H),2.99-2.93(m,0.5H),2.74-2.68(m,0.5H),1.90-1.70(m,5H),1.60-1.57(m,3H),1.29-1.17(m,3H).MS m/z=544.4[M+H]+.
SFC:カラム:ChiralPak AD-3(150mm×4.6mm、3μm);移動相:A:CO2、B:イソプロパノール[0.05%ジエチルアミン];B%:5%~40%5.5min、40%3min、5%1.5min;Rt=5.034min;86.3%de.
化合物I-1(163.00mg、299.83μmol)をテトラヒドロフラン(3.00mL)に溶解し、水酸化リチウム(72.00mg、3.01mmol)の水(1.50mL)溶液を加え、反応液を15~20℃で48時間撹拌し続けた。反応液に1Mの塩酸水溶液を加えてpH<4に調整し、酢酸エチルで抽出し(15mL×3)、合併した有機相を飽和食塩水30mLで1回洗浄し、無水硫酸ナトリウムで乾燥させ、有機溶剤を減圧除去し、得た粗製物をシリカゲルカラムクロマトグラフィー(溶離剤:0-20%ジクロロメタン/メタノール)により分離して精製し、得た化合物を再度キラルカラムにより分離し、化合物Iを得た。1H NMR(400MHz,DMSO-d6):δ7.53-7.16(m,10H),7.00-6.80(m,1.5H),6.68(d,J=8.0Hz,0.5H),5.36(d,J=12.0Hz,1H),5.01-4.66(m,3H),4.41(d,J=24.0Hz,0.5 H),4.30(d,J=24Hz,0.5H),4.13-3.95(m,1H),3.79(s,3H),2.84-2.79(m,1H),2.68-2.64(m,1H),2.39-2.27(m,1H),1.80-1.38(m,8H).MS m/z:516.3[M+1]+.
SFC:カラム:Chiralpak AD-3(100mm×4.6mm、3μm);移動相:B:イソプロパノール[0.05%ジエチルアミン];B%:5%~40%4.5min、40%2.5min、5%1min;Rt=4.198min;100.0%de.

n-ヘプタン(8.0L)を50Lの反応釜に加え、撹拌を開始し、出発原料1-0(1.0kg)、ブロモシクロペンタン(3.4kg)、硫酸マグネシウム(1.0kg)、酸化銀(2.0kg)を反応釜に順次加え、反応液を20~30℃で約19時間撹拌し、硫酸マグネシウム(0.3kg)及び酸化銀(0.7kg)を補充して、反応液を20~30℃で撹拌しながら約46時間反応させ続けた。反応液をシリカゲル(100~200メッシュ、2.0kg)に通し、卓上式吸引濾過漏斗で減圧吸引濾過し、ろ過ケーキを12.0Lジクロロメタンで3回に分けて洗浄し、毎回ジクロロメタン4.0Lを加えた。ろ液をロータリーエバポレータに移し、外温35~40℃で減圧濃縮させ、化合物1-1と1-2の混合物を得て、この混合物をさらに精製せずに次のステップの反応に用いた。
1H NMR(400MHz,CDCl3):δ7.49-7.43(m,2H),7.40-7.29(m,3H),4.95(s,1H),4.04-3.96(m,1H),3.72(s,3H),1.81-1.69(m,6H),1.58-1.46(m,2H)。
テトラヒドロフラン(5.3L)を50L反応釜に加え、撹拌を開始し、化合物1-1と1-2の混合物(1.3kg)を加え、水酸化リチウム一水和物(0.3kg)の水(2.7L)溶液を加え、反応液を20~30℃で4時間撹拌し続けた。反応液にn-ヘプタン(10.5L)を加え、10分間撹拌して、分液し、水相を2M塩化水素水溶液でpH3~4に調整し、ジクロロメタン16.0Lで2回抽出し、毎回8.0Lとし、合併した有機相を無水硫酸ナトリウム(1.0kg)で乾燥させ、ろ過して、ろ液を外温35~40℃で減圧濃縮させ、化合物1-3を得た。
1H NMR(400MHz,CDCl3):δ7.41-7.29(m,5H),4.88(s,1H),4.00-3.94(m,1H),1.74-1.57(m,6H),1.52-1.43(m,2H)。
ジクロロメタン(12.0L)を50L反応釜に加え、化合物1-3(1.2kg)を加えて、撹拌を開始し、N,N-ジメチルホルムアミド(12.0g)を加えて、1.5時間内で塩化オクサリル(1.04kg)を滴下し、反応液を20~30℃で1時間撹拌した。反応液を外温35~40℃で減圧濃縮させ、得た粗品を使用に備えた。ジクロロメタン(8.0L)を50L反応釜に加え、窒素ガスを用いて2回置換し、撹拌を開始し、ピラゾール(0.4kg)及びN-メチルモルホリン(0.7kg)を順次加え、0~10℃に降温し、窒素ガスを用いて1回置換し、上記粗品をジクロロメタン(4.0L)に溶解して溶液を調製し、反応釜に緩やかに滴下し、滴下終了後、窒素ガスを用いて1回置換し、反応液を20~30℃に昇温し、約16時間撹拌し続けた。反応液を順次、1M硫酸水溶液10.0Lで2回洗浄し(毎回5.0L)、飽和重炭酸ナトリウム水溶液11.0Lで2回洗浄し(毎回5.5L)、水7.0Lで1回洗浄し、飽和塩化ナトリウム溶液8.0Lで1回洗浄し、有機相を無水硫酸ナトリウム(0.5kg)で乾燥させてろ過し、ろ液を外温35~40℃で減圧濃縮させて、粗品を得た。粗品をn-ヘキサン7.2Lに分散させ、反応液を65~75℃に昇温して2時間撹拌し、15~25℃に降温して16時間撹拌し続けた。ろ過し、ろ過ケーキをn-ヘキサンで2回洗浄し(毎回600.0mL)、ろ過ケーキを外温20~30℃で5時間減圧乾燥させ、化合物1-4を得た。
1H NMR(400MHz,CDCl3):δ8.24(d,J=2.8Hz,1H),7.73(s,1H),7.59(d,J=6.8Hz,2H),7.41-7.27(m,3H),6.43(dd,J=1.5,2.8Hz,1H),6.36(s,1H),4.21-3.98(m,1H),1.87-1.67(m,6H),1.57-1.43(m,2H)。
N,N-ジメチルホルムアミド(12.0L)を50L反応釜に加え、撹拌を開始し、化合物D-1(1220.3g)及びテトラメチルグアニジン(493.8g)を順次加え、反応液を15~25℃で1時間撹拌し、化合物1-4(1211.6g)を反応釜に加え、反応液を15~25℃で17時間撹拌し続けた。反応液を水12.0Lに投入し、2M塩酸水溶液でpHを3に調整し、酢酸エチル24.0Lで2回抽出し(毎回12.0L)、合併した有機相を水12.0Lで3回洗浄し(毎回4.0L)、飽和塩化ナトリウム水溶液(3.0L)で1回洗浄し、無水硫酸ナトリウム(500.0g)で乾燥させてろ過し、ろ液を35~40℃の条件下で減圧濃縮させて、粗品を得た。アセトン(4.0L)、水(8.0L)、及び粗品を50L反応釜に順次加え、反応液を35~45℃で20時間撹拌し、ろ過し、ろ過ケーキを水8.0Lで2回洗浄し(毎回4.0L)、40℃の条件下で真空乾燥箱によりろ過ケーキを21時間乾燥させ、式(I)化合物の結晶形Aを得た。
1H NMR(400MHz,DMSO-d6):δ12.68(brs,1H),7.48-7.30(m,10H),6.95-6.70(m,2H),5.38-5.21(m,1.5H),4.96-4.69(m,3.5H),4.42-4.32(m,1H),4.10-3.95(m,1H),3.80(s,3H),3.39-3.36(m,0.5H),3.24-3.19(m,0.5H),2.88-2.70(m,0.5H),2.48-2.42(m,0.5H),1.73-1.48(m,8H)。LCMS(ESI)m/z:516.0[M+1]+。
式(I)化合物の結晶形A約50mgを適量のメタノールに加えた。サンプル液を磁気撹拌機(40℃)にて2日間撹拌して遠心分離し、沈殿を得て、室温で溶液から溶剤を揮発させて結晶化させた。その後、真空乾燥箱に入れて室温で一晩乾燥させ、式(I)化合物の結晶形Bを得た。
1H NMR(400MHz,DMSO-d6):δ12.68(brs,1H),7.48-7.30(m,10H),6.95-6.70(m,2H),5.38-5.21(m,1.5H),4.96-4.69(m,3.5H),4.42-4.32(m,1H),4.10-3.95(m,1H),3.80(s,3H),3.39-3.36(m,0.5H),3.24-3.19(m,0.5H),2.88-2.70(m,0.5H),2.48-2.42(m,0.5H),1.73-1.48(m,8H)。
LCMS(ESI)m/z:516.0[M+1]+。
実験材料:
SMS DVS Advantage動的蒸気収着装置
実験方法:
式(I)化合物の結晶形B 10~15mgをDVSサンプルトレイに入れてテストした。
実験結果:
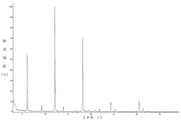
式(I)化合物の結晶形BのDVSパターンを図に示し、△W=0.05327%であった。
実験結論:
式(I)化合物の結晶形Bは、25℃、80%RHで、吸湿による重量増加が0.05327%であり、実質的には吸湿性無しであった。
『原薬及び製剤安定性試験の指導原則』(中国薬局方2015版四部通則9001)に準じて、高温(60℃、開放)、高湿(室温/相対湿度92.5%、開放)及び光照射(総照度1.2×106Lux・hr/近紫外線200w・hr/m2、開放)条件での式(I)化合物の結晶形Bの安定性を調べた。

試薬:
溶液及び緩衝液
緩衝液
50mM Tris
100mM NaCl
5mM MgCl2
0.1%BSA
プロテアーゼ阻害剤混合物、エチレンジアミン四酢酸不含-1錠(Roche#11873580001)(50mLの場合、1錠追加)
pH 7.4
1.化合物の調製
リガンドPD123319及びテスト化合物を参照して、DMSOを用いて750μMの母液を調製し、化合物ごとに8個の濃度勾配(最高濃度750uM、3倍希釈)に調製し、10ul/ウェルで384ウェルプレートのマザープレートに加えた。
SPA beadsを緩衝液で25mg/mlの母液とし、
同位体[125I]-Sar1-Ile8-Angiotensin IIに純水を加えて50uCi/mlの母液を調製した。
2.膜の製造
HEK-293細胞のhAT2を過剰発現させた細胞膜を緩衝液で2.5mg/mlとした。
3.具体的な操作
ECHOを用いてマザープレートから化合物200nlをテスト用384プレートの各ウェルに吸い取った。ZPEに等体積のDMSOを加えた。(テスト化合物の反応中の濃度について250倍希釈した)。
10μg/μlの磁気ビーズ、0.05μg/μlのAT2膜を含有する溶液を50ml調製し、シェーカーにて均一に混合した(100rpm、30min)。テストプレートには、最終的に1.25μg/ウェルのhAT2膜、250μg/ウェルの磁気ビーズが含まれている。
Multidrop Combiピペットを用いて、1ウェルあたり25μlで3.2の膜混合液を化合物のテストプレートに加えた。
50uCi/mlの同位体[125I]-Sar1-Ile8-Angiotensin II母液を用いて緩衝液で0.2nMの溶液とし、Multidrop Combiピペットを用いて0.2nMの125Iを、体積で1ウェルあたり25μlで化合物テストプレートに加えた。125I同位体の最終濃度を0.1nMとした。
製造されたテストプレートをシェーカーに置き、200rpmで、室温下一晩放置した。
遠心分離機を用いて1200rpmでテストプレートを1min遠心分離した。
遠心分離後のテストプレートについてMicrobetaにより読み取った。
実験結果は表4に示される。

測定対象化合物をDMSOに溶解して、10mmol/Lの原液を調製した。ピペット(Eppendorf Research社)を用いて溶出媒体980μLを2mLのスクリューキャップ付きガラス管形瓶に加えた。各試験化合物の原液20μL及びQCサンプルをpH 7.4の動力学的検出溶液に相当する緩衝溶液に加えた。試験化合物及びDMSO溶液のそれぞれの最終濃度は200μM及び2%であった。薬瓶にキャップを付けた。最大濃度の理論値は200μMであった。室温下、1分間あたり880回転の速度でこの混合物を24時間回転して振とうさせた。バイアルを1分間あたり13000回転で30分間遠心分離した。デジタルピペットを用いて上澄み液200μLを96ウェルプレートに加えた。高速液体クロマトグラフィーのスペクトルにより測定された試験化合物の溶解度、実験結果を表5に示した。

研究項目として、各アイソザイムの特異的プローブ基質を用いて、ヒト肝ミクロソームチトクロームP450アイソザイム(CYP1A2、CYP2C9、CYP2C19、CYP2D6、及びCYP3A4)に対する測定対象化合物の阻害性を評価した。
希釈済みの一連の濃度の測定対象化合物の作動液を、ヒト肝ミクロソーム、プローブ基質、及び循環系の補因子を含有するインキュベート系に加え、メタノールの含有量を最終的なインキュベート系の約1%(v/v)とした。測定対象化合物を含有せず溶剤を含有する対照を酵素活性対照(100%)とした。サンプル中の分析物の濃度は、液体クロマトグラフィー-タンデム質量分析(LC/MS/MS)方法によって測定する。サンプル(空白溶剤、陽性対照阻害剤または測定対象化合物)濃度の平均値を用いて計算した。SigmaPlot(V.11)を用いて測定対象化合物の平均百分率で示される活性を用いて、濃度について非線形回帰分析を行った。3パラメータまたは4パラメータ逆対数方程式を用いてIC50値を計算した。実験結果を表6に示した。

測定対象化合物のCaco-2細胞での双方向透過性を測定し、そして測定対象化合物が外部へ排出されて輸送されるか否かをテストした。
実験方法:
ストック液の調製
化合物をジメチルスルホキシド(DMSO)または他の適切な溶剤に溶解し、適切な濃度のストック液を調製した。
適切な内部標準(internal standard、IS)をアセトニトリル(acetonitrile、ACN)または他の適切な有機溶剤に溶解して停止液とし、具体的な情報は研究レポートに記載されている。
ナドロール(nadolol)、メトプロロール(metoprolol)、ジゴキシン(digoxin)、3-(ポタシオスルホオキシ)エストラ-1,3,5(10)-トリエン-17-オン(estrone3-sulfate potassium、E3S)、及びGF120918は、本研究においてそれぞれ低張性対照化合物、高張性対照化合物、P-糖タンパク質(P-gp)基質、乳癌耐性タンパク質(BCRP)基質、及び排出トランスポーター阻害剤とした。これらの化合物のストック液はDMSOを用いて調製され、≦-30℃に保存し、6ヵ月内で使用する。
投与液及び受容液の調製
本研究では、10mM HEPES(2-[4-(2-ヒドロキシエチル)-1-ピペラジニル]-エタンスルホン酸)を含有するHBSS(Hanks Balanced Salt Solution)を輸送緩衝液(pH7.40±0.05)とした。投与液及び受容液の調製方法を下記表7に示した。

37±1℃、5%CO2、及び飽和湿度の培養条件で、Caco-2細胞をMEM培地(Minimum Essential Media)で培養した。次に、1×105細胞/cm2の接種密度で細胞をCorning Transwell-96ウェルプレートに接種し、次に、細胞を二酸化炭素インキュベータに入れて21~28日間培養した後、輸送実験に用い、この間、4~5日間おきに培地を1回交換した。
化合物の投与濃度を2、10、及び100μMとし、10μM GF120918含有または不含の条件下で双方向に(A-B及びB-A方向)投与し、投与濃度ごとに3つの平行試験を行った。Digoxin及びE3Sは、テスト濃度をそれぞれ10、及び5μMとし、10μM GF120918含有または不含の条件で双方向に投与し、nadolol及びmetoprololは、いずれのテスト濃度も2μMであり、10μM GF120918不含の条件下で一方向(A-B方向)に投与し、3つの対照化合物についても3つの平行試験を行った。
サンプル収集情報を下記表8に示した。

Caco-2細胞の完全性はルシファーイエロー検出実験(Lucifer Yellow Rejection Assay)を用いてテストした。細胞プレートごとに6個の細胞ウェルをランダムに選択し、それぞれ100μMルシファーイエローを加え、ルシファーイエロー検出実験を輸送実験と同時に行った。120分間インキュベート後、ルシファーイエローサンプルを取り、425/528nm(励起/放射)スペクトルでサンプル中のルシファーイエローの相対蛍光強度(the relative fluorescence unit、RFU)を検出した。
サンプル中の測定対象化合物、対照化合物nadolol、metoprolol、digoxin及びE3Sの濃度はすべて液体クロマトグラフィー-タンデム質量分析(LC/MS/MS)方法によって測定した。分析物及び内部標準の保留時間、クロマトグラムの収集、及びクロマトグラムの積分は、ソフトウェアAnalyst(AB Sciex、Framingham、Massachusetts、USA)で処理され、実験結果は表9に示された。

Claims (15)
- X線粉末回折パターンにおいて、2θ角が3.52±0.20°、6.04±0.20°、18.21±0.20°に特徴的な回折ピークを有する、ことを特徴とする式(I)化合物の結晶形A。

- X線粉末回折パターンにおいて、2θ角が3.52±0.20°、6.04±0.20°、14.40±0.20°、15.11±0.20°、18.21±0.20°、18.46±0.20°、20.12±0.20°、24.13±0.20°に特徴的な回折ピークを有する、請求項1に記載の結晶形A。
- 示差走査熱量測定曲線において155.36℃±3℃に吸熱ピークの開始点を有する、請求項1または2に記載の結晶形A。
- 熱重量分析曲線において、100.00℃±3℃に0.1489%の重量損失に達する、請求項1または2に記載の結晶形A。
- 式(I)化合物を混合溶剤に加えて溶解するステップ(a)と、
30~50℃で10~30時間撹拌するステップ(b)と、
ろ過後、30~50℃でろ過ケーキを15~25時間乾燥させるステップ(c)と、を含み、
前記混合溶剤は、アセトンと水の体積比が1:1.5~2.5である、式(I)化合物の結晶形Aの調製方法。 - X線粉末回折パターンにおいて、2θ角が3.52±0.20°、6.08±0.20°、9.25±0.20°、12.12±0.20°、14.00±0.20°、18.19±0.20°、24.31±0.20°、30.50±0.20°に特徴的な回折ピークを有する、ことを特徴とする式(I)化合物の結晶形B。
- 示差走査熱量測定曲線において、150.95℃±3℃に吸熱ピークの開始点を有する、請求項6に記載の結晶形B。
- 熱重量分析曲線において、120.00℃±3℃に0.0558%の重量損失に達する、請求項6に記載の結晶形B。
- 式(I)化合物を溶剤に加えて、懸濁液を形成するステップ(a)と、
懸濁液を35~45℃で30~60時間撹拌するステップ(b)と、
遠心分離後、ろ過ケーキを8~16時間乾燥させるステップ(c)と、を含み、
前記溶剤は、メタノール、エタノール、アセトニトリルから選ばれるものであり、または、アセトンと水との体積比が3:2の混合溶剤である、式(I)化合物の結晶形Bの調製方法。 - 慢性疼痛治療薬の調製における請求項1~4のいずれか1項に記載の結晶形Aまたは請求項6~8のいずれか1項に記載の結晶形Bの使用。
- 以下のステップを含む、式(I)化合物の調製方法。


- 以下のステップを含む、請求項11に記載の調製方法。

- 化合物1Eと化合物1-0とのモル比は1.2~5:1であり、或いは、化合物1-4と化合物D1-1とのモル比は、1.1~1.5:1であり、或いは、化合物1-1、1-2、及び1-3を調製するステップでは、反応系の温度範囲が25±5℃に制御され、或いは、化合物1-4の調製には、ステップaとステップbを含み、ステップaでは、反応系の温度範囲が25±5℃に制御され、ステップbでは、反応系に材料を投入する際に、反応系の温度範囲が5±5℃に制御され、試薬投入の終了後、反応系の温度範囲が25±5℃に制御され、或いは、溶剤Fはn-ヘプタンから選ばれ、前記n-ヘプタンの体積と化合物1-0の質量との比が8.0~10.0:1であり、試薬Gは、酸化銀、硫酸マグネシウムから選ばれ、前記酸化銀、硫酸マグネシウムと化合物1-0とのモル比が1.0~5.0:1である、請求項12に記載の調製方法。
- 試薬Gは投入過程においてバッチで投入される、請求項11または12に記載の調製方法。
- 溶剤Hは、テトラヒドロフランと水から選ばれる混合物であり、前記テトラヒドロフランと水の体積比が1~2:1であり、試薬Iは水酸化リチウム一水和物であり、前記水酸化リチウム一水和物と化合物1-0とのモル比が1.0~2.0:1であり、溶剤Jと化合物1-3との質量比は10:1であり、触媒Kと化合物1-3とのモル比は0.002~0.004:1であり、試薬Lと化合物1-3とのモル比は1.2~2.0:1であり、溶剤Mと化合物1-3との質量比は6~10:1であり、試薬Nと化合物1-3とのモル比は1.0~1.5:1であり、試薬Oと化合物1-3とのモル比は1.0~1.5:1であり、溶剤PはN,N-ジメチルホルムアミドから選ばれ、前記N,N-ジメチルホルムアミドから選ばれる溶剤Pと化合物1-4との質量比が10:1であり、試薬Qはテトラメチルグアニジンから選ばれ、前記テトラメチルグアニジンから選ばれる試薬Qと化合物1-4とのモル比が1~1.2:1である、請求項12に記載の調製方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201811301892.3 | 2018-11-02 | ||
| CN201811301892 | 2018-11-02 | ||
| PCT/CN2019/115149 WO2020088677A1 (zh) | 2018-11-02 | 2019-11-01 | 一种血管紧张素ii受体2拮抗剂的盐型、晶型及其制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2021534143A JP2021534143A (ja) | 2021-12-09 |
| JP7089636B2 true JP7089636B2 (ja) | 2022-06-22 |
Family
ID=70461977
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021507501A Active JP7089636B2 (ja) | 2018-11-02 | 2019-11-01 | アンジオテンシンii2型受容体拮抗薬の塩形、結晶形及びその製造方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US11286239B2 (ja) |
| EP (1) | EP3819293B1 (ja) |
| JP (1) | JP7089636B2 (ja) |
| KR (1) | KR102619333B1 (ja) |
| CN (1) | CN112585120B (ja) |
| AU (1) | AU2019373178B2 (ja) |
| ES (1) | ES2944065T3 (ja) |
| FI (1) | FI3819293T3 (ja) |
| PT (1) | PT3819293T (ja) |
| WO (1) | WO2020088677A1 (ja) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011088504A1 (en) | 2010-01-19 | 2011-07-28 | Spinifex Pharmaceuticals Pty Ltd | Methods and compositions for improved nerve conduction velocity |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2018279669B8 (en) * | 2017-06-09 | 2020-10-15 | Shandong Danhong Pharmaceutical Co., Ltd. | Carboxylic acid derivative as at AT2R receptor antagonist |
-
2019
- 2019-11-01 JP JP2021507501A patent/JP7089636B2/ja active Active
- 2019-11-01 KR KR1020217006383A patent/KR102619333B1/ko active IP Right Grant
- 2019-11-01 EP EP19878016.5A patent/EP3819293B1/en active Active
- 2019-11-01 PT PT198780165T patent/PT3819293T/pt unknown
- 2019-11-01 CN CN201980050569.XA patent/CN112585120B/zh active Active
- 2019-11-01 FI FIEP19878016.5T patent/FI3819293T3/fi active
- 2019-11-01 AU AU2019373178A patent/AU2019373178B2/en active Active
- 2019-11-01 WO PCT/CN2019/115149 patent/WO2020088677A1/zh unknown
- 2019-11-01 ES ES19878016T patent/ES2944065T3/es active Active
-
2021
- 2021-02-18 US US17/179,382 patent/US11286239B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011088504A1 (en) | 2010-01-19 | 2011-07-28 | Spinifex Pharmaceuticals Pty Ltd | Methods and compositions for improved nerve conduction velocity |
| CN102821765A (zh) | 2010-01-19 | 2012-12-12 | 西芬克斯医药有限公司 | 改良神经传导速度的方法和组合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3819293A1 (en) | 2021-05-12 |
| ES2944065T3 (es) | 2023-06-19 |
| EP3819293A4 (en) | 2021-09-01 |
| US20210206724A1 (en) | 2021-07-08 |
| EP3819293B1 (en) | 2023-03-29 |
| FI3819293T3 (fi) | 2023-05-03 |
| US11286239B2 (en) | 2022-03-29 |
| AU2019373178B2 (en) | 2022-03-24 |
| KR20210039436A (ko) | 2021-04-09 |
| PT3819293T (pt) | 2023-04-19 |
| CN112585120A (zh) | 2021-03-30 |
| WO2020088677A1 (zh) | 2020-05-07 |
| AU2019373178A1 (en) | 2021-03-04 |
| CN112585120B (zh) | 2022-11-11 |
| JP2021534143A (ja) | 2021-12-09 |
| KR102619333B1 (ko) | 2023-12-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2386974T3 (es) | Formas cristalinas de la 4-metil-N-[3-(4-metil-imidazol-1-il)-5-trifluoro-metil-fenil]-3-(4-piridin-3-il-pirimidin-2-il-amino)-benzamida | |
| ES2746031T3 (es) | Sal de N-(5S,6S,9R)-5-amino-6-(2,3-difluorofenil)-6,7,8,9-tetrahidro-5H-ciclohepta[B]piridin-9-IL-4-(2-oxo-2,3-dihidro-1H-imidazo[4,5-B]piridin-1-il)piperidina-1-carboxilato | |
| UA123472C2 (uk) | N-[4-фтор-5-[[(2s,4s)-2-метил-4-[(5-метил-1,2,4-оксадіазол-3-іл)метокси]-1-піперидил]метил]тіазол-2-іл]ацетамід як інгібітор oga | |
| EP3315499B1 (en) | Crystal of (6S,9aS)-N-benzyl-8-({6-[3-(4-ethylpiperazin-1-yl)azetidin-1-yl]pyridin-2-yl}methyl)-6-(2-fluoro-4-hydroxybenzyl)-4,7-dioxo-2-(prop-2-en-1-yl)hexahydro-2H-pyrazino[2,1-c][1,2,4]triazine-1(6H)-carboxamide | |
| UA121138C2 (uk) | Нафтиридинові сполуки як інгібітори jak-кінази | |
| EA031030B1 (ru) | Ингибиторы фактора в комплемента на основе производных пиперидинилиндола и их применение | |
| CA3121289C (en) | Histone acetylase p300 inhibitor and use thereof | |
| AU2016366306B2 (en) | Fumagillol derivatives and polymorphs thereof | |
| JP6974618B2 (ja) | Fgfr及びvegfr阻害剤としての化合物の塩形態、結晶形およびその製造方法 | |
| JP7089636B2 (ja) | アンジオテンシンii2型受容体拮抗薬の塩形、結晶形及びその製造方法 | |
| CA3116141C (en) | Cycloalkane-1,3-diamine derivative | |
| EP3620454B1 (en) | Carboxylic acid derivative as at2r receptor antagonist | |
| CA3239813A1 (en) | Crystal forms of thienoimidazole compound and preparation method thereof | |
| WO2001002397A1 (fr) | Composes tricycliques presentant une jonction spiro | |
| US20220081446A1 (en) | Crystal form of 1,2,3-triazolo[1,5-a]pyrazines derivative and preparation method for crystal form | |
| EP3816167A1 (en) | Crystal of benzoxazole derivative | |
| US20210230188A1 (en) | Crystal form of tri-cycle compound and application thereof | |
| RU2787767C2 (ru) | Кристалл производного бензоксазола | |
| WO2024051771A1 (zh) | 一种五元并六元杂环化合物的晶型及其制备方法和应用 | |
| JP2022530812A (ja) | Wee1阻害剤化合物の結晶形及びその応用 | |
| WO2018209667A1 (zh) | 多环杂环化合物的晶型、其制备方法、应用及组合物 | |
| CN115433257A (zh) | 咪唑并哒嗪类双功能protac分子化合物及其制备和应用 | |
| Manetsch et al. | Photoactivatable probes and uses thereof | |
| CN117510490A (zh) | 一种稠合氮杂萘烷类衍生物的晶型及其制备方法 | |
| CN111484505A (zh) | 一种双环RORγ抑制剂的盐酸盐结晶型 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210210 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20210210 |
|
| RD01 | Notification of change of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7426 Effective date: 20210806 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220208 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220420 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20220607 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20220610 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7089636 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |