JP6603529B2 - 再プログラム化多能性細胞の作製 - Google Patents
再プログラム化多能性細胞の作製 Download PDFInfo
- Publication number
- JP6603529B2 JP6603529B2 JP2015188541A JP2015188541A JP6603529B2 JP 6603529 B2 JP6603529 B2 JP 6603529B2 JP 2015188541 A JP2015188541 A JP 2015188541A JP 2015188541 A JP2015188541 A JP 2015188541A JP 6603529 B2 JP6603529 B2 JP 6603529B2
- Authority
- JP
- Japan
- Prior art keywords
- cells
- cell
- stro
- oct4
- pluripotent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0607—Non-embryonic pluripotent stem cells, e.g. MASC
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0696—Artificially induced pluripotent stem cells, e.g. iPS
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/5073—Stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/602—Sox-2
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/603—Oct-3/4
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/604—Klf-4
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/605—Nanog
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/606—Transcription factors c-Myc
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/608—Lin28
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/13—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells
- C12N2506/1346—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells
- C12N2506/1353—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells from bone marrow mesenchymal stem cells (BM-MSC)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/13—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells
- C12N2506/1346—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells
- C12N2506/1361—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells from dental pulp or dental follicle stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/13—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells
- C12N2506/1346—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells
- C12N2506/1384—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells from adipose-derived stem cells [ADSC], from adipose stromal stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/15011—Lentivirus, not HIV, e.g. FIV, SIV
- C12N2740/15041—Use of virus, viral particle or viral elements as a vector
- C12N2740/15043—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- General Health & Medical Sciences (AREA)
- Cell Biology (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Developmental Biology & Embryology (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Physics & Mathematics (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Transplantation (AREA)
- Medicinal Chemistry (AREA)
- Tropical Medicine & Parasitology (AREA)
- Food Science & Technology (AREA)
- Virology (AREA)
- Pathology (AREA)
- Biophysics (AREA)
- General Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Toxicology (AREA)
- Analytical Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
Description
なわち中胚葉、内胚葉および外肺葉すべての細胞に分化できると信じられている(Eva
ns & Kaufman、Nature 292:154〜156(1981))。ヒ
トES細胞およびそれに由来する細胞は、パーキンソン病、脊髄損傷および糖尿病などの
疾患の宿主の治療用に現在評価されている。しかし、ヒトES細胞はヒトの胚から得られ
るという事実が、多数の非常に議論の余地がある倫理規定を提起し、多くの国において、
これらの細胞の誘導が法律によって禁止されている。さらに、ES細胞およびそれに由来
する細胞は、それが由来する対象から抗原を発現するので、不適合な(例えば同じHLA
型(複数可)を発現しない)対象に投与された場合、それらの細胞が拒絶される危険性が
ある。したがって、科学者らはES細胞の現在の作製方法を避けるための技術的解決法を
探求した。これらの解決法を達成するための1つの望ましい方法は、出生後の個体、例え
ば、治療される対象もしくは関連する対象または他の適合する対象の体細胞から直接多能
性細胞を作製することであった。
ステップ(Wilmutら、Nature 385:810〜813(1997年))ま
たはES細胞と融合させるステップ(Cowanら、Science 309:1369
〜1373(2005年))を含み、未受精卵およびES細胞は体細胞において全能性ま
たは多能性をもたらす因子を含有することを示している。これらの方法に付随する問題は
卵子および/または胚の破壊の必要性を含み、このことは一部の国において倫理規定を提
起し得る。
Cell 95:379〜391(1998年))、Sox2(Avilionら、Ge
nes Dev.17:126〜140(2003年))およびNanog(Chamb
ersら、Cell 113:643〜655(2003年))を含むいくつかの転写因
子は、ES細胞の多能性の維持に関与しており、しかし、ES細胞の特定だけでは十分で
ない。
4、Sox2、c−MycおよびKlf4)を、マウスのES細胞培養に適切な条件下で
培養されたマウスのES細胞および成体マウスの線維芽細胞に導入した。どちらかの細胞
型に形質導入後、著者らは、マウスES細胞の形態および成長特性を示し、マウスES細
胞のマーカー遺伝子を発現した、人工多能性幹(iPS)細胞を得た(Takahash
i&Yamanaka、Cell 126:663〜676(2006年))。ヌードマ
ウスへのiPS細胞の皮下移植は、3種すべての胚葉に由来するさまざまな組織を含有す
る腫瘍をもたらした。胚盤胞への注射後、iPS細胞はマウスの胚発生に寄与した。これ
らのデータは、レトロウイルス形質導入を使用して、わずか数種の確定された因子を加え
ることによって、多能性細胞が直接マウス線維芽細胞培養物から作製され得ることを実証
している。しかし、この技術は、再プログラミングの率の低さ(処置された細胞の1%よ
りかなり低い)ならびにゲノムへの組み込みおよび発がん遺伝子c−MycおよびKlf
4の連続発現の必要性を含む、いくつかの重要な不利益に非常に悩まされる。これらの遺
伝子の発現は、細胞およびこの細胞に由来する細胞のレシピエントにおいて腫瘍の発生を
もたらし得る。この点で、おそらくこれらの発がん遺伝子の連続発現の結果として、これ
らの方法を用いて作製されたiPS細胞を使用して作製されたキメラマウスは腫瘍を発生
する。結果的に、この分野の調査の主要な目的は、これらの因子をコードする核酸のゲノ
ムへの組み込みを必要としないか、またはこれらの再プログラミング因子もしくは他の再
プログラミング因子の発現の数もしくは期間を最小化するかのいずれかの再プログラミン
グの方法を開発することである。
の可能性が最も高いため、iPS細胞に基づく研究の大部分は線維芽細胞を使用するが、
繊維芽細胞以外のさまざまな細胞集団がマウスにおけるiPS細胞の誘導に使用されてき
た。これらの研究からの重要な観察は、選択された体細胞の型が、iPS細胞作製の効率
および再プログラミングのレベルに有意な効果を有することである。この点において、神
経幹細胞、胃細胞および肝細胞などのいくつかの細胞型は、線維芽細胞と比較して比較的
高い効率で再プログラムすると思われる。しかし、ヒト由来のこれらの細胞の単離は、組
織採取の侵襲性および/または利用可能なドナー試料が限られていることにより困難また
は実現不可能である。いくつかのより入手しやすい細胞型(例えば、筋肉細胞または分化
した造血細胞)はiPS研究の基盤として使用されているが、再プログラミングの成功は
限られている。
能な最適な細胞型の特定の必要がある。効率的な再プログラミング特性を有するこのよう
な細胞型の特定は、ゲノムへの組み込みを必要とせず、かつ/または再プログラミング因
子の数を最小化もしくは減少させる再プログラミング方法の使用を容易にすると思われる
。このことは、腫瘍性形質転換の危険性が減少した、より安全な多能性iPS細胞集団を
もたらすであろう。再プログラミングの効率が高く、胚組織に頼らずに得られるこのよう
な細胞型は、存在がすでに企図されている適用である多能性ES細胞への使用に適してい
ると思われる。
を作製するために適切な供給源ではないという従来の見識にもかかわらず、さまざまな供
給源由来の細胞を使用してiPS細胞の作製を試みた。驚くべきことに、本発明者は、S
tro−1+多分化能細胞またはその子孫細胞(特に、脂肪組織または歯髄組織由来のも
の)が、高い効率、例えば線維芽細胞より高い効率を有するiPS細胞作製のための有用
な供給源であったことを発見した。
プログラムし、iPS細胞を作製するために、外から(exogenously)加えられることが普
通は必要とされる内因性因子、例えばKlf4および/またはc−mycを発現すること
も決定した。このような内因性(endocgenous)遺伝子の発現は、これらのタンパク質を
高レベルで導入することなく、またはこれらの非内因性型タンパク質を全く導入すること
なくiPS細胞を作製することを可能にできる。c−mycの内因性発現は、この発がん
遺伝子の構成的発現および/または強度の発現を必要としないので、c−mycの内因性
発現は、腫瘍形成の危険性が減少したiPS細胞の作製もまた容易にすると思われる。
−1+多分化能細胞および/またはその子孫細胞を、細胞を再プログラムするために十分
な条件下で1種または複数の能力決定因子(potency-determining factors)に曝露する
ステップおよび曝露された細胞を培養し、再プログラム化細胞を得るステップを含む。こ
の方法は、同様に、人工多能性幹(iPS)細胞を作製する方法に適用される。
o−1+多分化能細胞および/またはその子孫細胞が濃縮された細胞の集団を、細胞を再
プログラムするために十分な条件下で1種または複数の能力決定因子に曝露するステップ
を含む。
意の組織であってよい。好ましくは、Stro−1+多分化能細胞の供給源は脂肪組織ま
たは歯髄組織である。Stro−1+細胞の別の供給源は骨髄である。
能力決定因子に曝露する前の脂肪組織、歯髄組織、骨髄または他の部位から濃縮される。
胞および/またはその子孫細胞よりも広い分化能を有する、すなわち、Stro−1+多
分化能細胞および/またはその子孫細胞よりも広範囲の細胞系列(cell lineage)および
/または細胞型に分化できる、再プログラム化細胞を得るステップを含む。
Nanog、Lin28、c−Myc、bFGF、SCF、TERT、SV40ラージT
抗原、HPV16E6、HPV16E7、Bmil、Fbx15、Eras、ECAT1
5−2、Tcl1、β−カテニン、ECAT1、ESG1、Dnmt3L、ECAT8、
Gdf3、Sox15、ECAT15−1、Fthl17、Sal14、Rex1、UT
F1、Stella、Stat3、FoxD3、ZNF206、Mybl2、DPP A
2、Otx2およびGrb2または1種もしくは複数の前記因子と同一もしくは類似の活
性を有する化合物、例えばその活性断片または小分子からなる群から個別にまたは集約的
に選択される因子を含む。別の例示的な能力決定因子は、例えば本明細書に記載された、
化学物質、ペプチド、siRNA、shRNAまたはマイクロRNAである。例えば、1
種または複数の能力決定因子は、
(i)Oct4、
(ii)Oct4とSox2との組み合わせ、
(iii)Oct4、Sox2と、NanogおよびLin28のうちの少なくとも1つ
との組み合わせ、
(iv)Oct4、Klf4およびc−Mycの組み合わせ、
(v)Oct4、Sox2およびKlf4の組み合わせ、
(vi)Oct4、Sox2、Klf4およびc−Mycの組み合わせ、
(vii)Oct4、Sox2、NanogおよびLin28の組み合わせ、
(viii)Oct4、Sox2、Klf4、c−Myc、NanogおよびLin28
の組み合わせ、ならびに
(ix)(i)から(x)のうちのいずれか1つと、さらに化学物質、ペプチド、siR
NA、shRNAまたはマイクロRNAとの組み合わせ
からなる群から個別にまたは集約的に選択される。
胞は、出生後の対象から得られる。この実施形態によれば、本方法は、対照からStro
−1+多分化能細胞および/または子孫細胞を得る、または単離するステップをさらに含
み得る。
物対象は、限定するものではないが、ヒト、霊長類、家畜(例えば、ヒツジ、ウシ、ウマ
、ロバ、ブタ)、コンパニオン・アニマル(例えば、イヌ、ネコ)、実験動物(例えば、
マウス、ウサギ、ラット、モルモット、ハムスター)、捕らわれた野生動物(例えば、キ
ツネ、シカ)を含む。好ましくは、哺乳動物はヒトまたは霊長類である。最も好ましい哺
乳動物はヒトである。
胞を1種または複数の能力決定因子に曝露するステップは、プロモーターに作動可能に連
結された1種または複数の能力決定因子をコードする配列を含む核酸をStro−1+多
分化能細胞および/またはその子孫細胞に導入するステップを含む。核酸をコードする複
数の能力因子は互いに異なっていても、または単一の核酸において、例えば、別々のプロ
モーターにそれぞれが連結した複数の核酸、もしくは単一のプロモーターにそれぞれが連
結した複数の核酸を含む単一の発現ベクター、例えばマルチシストロン性ベクター中にあ
ってもよい。好ましくは、核酸はベクター、より好ましくはウイルスベクター、例えばレ
トロウイルスベクターまたはアデノウイルスベクター内に含有される。
細胞のゲノム中に組み込まれない。例えば、核酸(複数可)は、細胞(複数可)内で1つ
または複数のエピソームとして残存し、および/または最終的に細胞(複数可)から排出
される。
ましくは多能性細胞の作製をもたらす。一例において、再プログラム化細胞は、(i)O
ct−4、SSEA3、SSEA4、Tra−1−60およびTra−1−81からなる
群から選択される細胞マーカーを発現し、(ii)多能性細胞に特徴的な形態を示し、(
iii)免疫不全動物に導入されると奇形腫を形成する。
団、または所望の細胞型が濃縮された細胞集団に分化させるステップをさらに含む。本発
明の方法は、所望の細胞型を単離、濃縮または選択するステップをさらに含み得る。この
ような細胞は、例えば本明細書に記載された療法またはスクリーニングにおいて有用であ
る。または、例えば多能性細胞が病態を患っている対象から作製された場合、このような
分化細胞は疾患状態または病態の研究に有用である。
って作製された細胞の有効量を、薬学的に許容される担体または賦形剤を有する医薬組成
物に製剤化するステップを含む。
製された細胞またはその集団または再プログラム化細胞が濃縮された集団を提供する。同
様に、本発明の例示形態は、任意の実施形態に従って本明細書に記載の細胞または集団か
ら分化した細胞または細胞の集団を提供する。
る核酸を含む、Stro−1+多分化能細胞および/またはその子孫細胞を提供する。こ
のような細胞は、再プログラム化細胞の作製に有用である。
は、薬物療法、例えば疾患または障害を治療または予防する方法において有用であり、こ
の方法は、この細胞またはその集団を、それを必要とする対象に投与するステップを含む
。
は、スクリーニングにもまた有用である。例えば、本発明は、疾患または障害の治療また
は予防において有用な化合物をスクリーニングする方法を提供し、この方法は、本発明に
よる細胞または集団を前記化合物に曝露するステップを含む。
の方法は、
i)本発明に従って作製された多能性細胞またはその集団を被験化合物と接触させ、それ
から分化した細胞の量を決定するステップ、
ii)この化合物の不在下で、本発明に従って作製された多能性細胞またはその集団から
分化した細胞の量を決定するステップ
を含み、(ii)と比較して(i)において分化細胞の量が増加することが、化合物が多
能性細胞の分化を導くことを示す。
含む。このようにして、特定の細胞系列または細胞型への分化を導く化合物が決定される
。
方法をさらに提供することが、当業者には明らかであると思われる。
方法は、
(i)任意の実施形態に従って本明細書に記載の方法を実施して、病態を患う対象から多
能性細胞またはその集団を作製するステップ、および
(ii)細胞または集団を被験化合物と接触させ、病態の1種または複数の症状に対する
その効果を決定するステップ
を含み、病態の症状を改善または緩和する化合物が病態の治療に有用である。
(a)多能性細胞またはその集団を病態に侵された細胞に分化させるステップ、および
(b)(a)の細胞を被験化合物と接触させ、病態の1種または複数の症状に対するその
効果を決定するステップ
を含み、病態の症状を改善または緩和する化合物が病態の治療に有用である。
だけではなく、対象が、既存の治療化合物/予防化合物を用いた治療に応答する可能性が
あるかどうかを特定するためにも有用である。
たはその子孫細胞から成熟および分化および由来した1種または複数の標的組織に曝露し
た場合、化合物の任意の特定の毒性効果を特定するためにもさらに有用である。
本明細書を通して、特に明示しない限り、または文脈上別の解釈を要する場合を除き、
単一のステップ、組成物、ステップの群または組成物の群への言及は、それらのステップ
、組成物、ステップの群または組成物の群の1つまたは複数(すなわち、1つ以上)を包
含すると解釈されるべきである。
に変更すべきところは変更して適用される。
入れ可能であることが明らかであろう。本発明が、このような変更および改良をすべて含
むことは理解されるべきである。本発明は、本明細書に言及された、または示されたすべ
てのステップ、特徴、組成物および化合物、ならびに前記ステップまたは特徴のあらゆる
組み合わせまたは任意の2つ以上を個別にまたは集約的にさらに含む。
これらは単なる例示を目的とする意図である。機能的に同等である生成物、組成物および
方法が本発明の範囲内であることは、本明細書に記載のように明らかである。
技術、溶液中のペプチド合成、固相ペプチド合成および免疫学の従来技術を使用して、必
要以上の実験をせずに実施される。このような手順は、例えば、Sambrook、Fr
itsch & Maniatis、Molecular Cloning:A Lab
oratory Manual、Cold Spring Harbor Labora
tories、New York、第2版(1989年)、Vols I、IIおよびD
I全体;DNA Cloning:A Practical Approach、Vol
s.IおよびII(D.N.Glover編、1985年)、IRL Press、Ox
ford、テキスト全体;Oligonucleotide Synthesis:A
Practical Approach(M.J.Gait編、1984年)IRL P
ress、Oxford、テキスト全体および特にテキスト中のGaitによる1〜22
ページ;Atkinsonら、35〜81ページ;Sproatら、83〜115;およ
びWuら、135〜151ページの論文;4.Nucleic Acid Hybrid
ization:A Practical Approach(B.D.Hames &
S.J.Higgins編、1985年)IRL Press、Oxford、テキス
ト全体;Immobilized Cells and Enzymes:A Prac
tical Approach(1986年)IRL Press、Oxford、テキ
スト全体;Perbal,B.、A Practical Guide to Mole
cular Cloning(1984年);Methods In Enzymolo
gy(S.ColowickおよびN.Kaplan編、Academic Press
,Inc.)、シリーズ全体;J.F.Ramalho Ortigao、「The C
hemistry of Peptide Synthesis」:Knowledge
database of Access to Virtual Laborator
y website (Interactiva、Germany);Sakakiba
ra,D.、Teichman,J.、Lien,E.Land Fenichel,R
.L.(1976年).Biochem.Biophys.Res.Commun.73
336〜342ページ;Merrifield,R.B.(1963年).J.Am.
Chem.Soc.85、2149〜2154年;Barany,G.およびMerri
field,R.B.(1979年)、The Peptides(Gross,E.お
よびMeienhofer,J.編)vol.2、1〜284ページ、Academic
Press、New York.12.Wuunsch,E.編(1974年)Syn
these von Peptiden in Houben−Weyls Metod
en der Organischen Chemie(Muler,E.編)、vol
.15、第4版、パート1および2、Thieme,Stuttgart;Bodans
zky,M.(1984年)Principles of Peptide Synth
esis、Springer−Verlag、Heidelberg;Bodanszk
y,M.& Bodanszky,A.(1984年)The Practice of
Peptide Synthesis、Springer−Verlag、Heide
lberg;Bodanszky,M.(1985年)Int.J.Peptide P
rotein Res.25、449〜474ページ;Handbook of Exp
erimental Immunology、I〜IV巻(D. M.WeirおよびC
.C.Blackwell編、1986年、Blackwell Scientific
Publications);ならびにAnimal Cell Culture:
Practical Approach、第3版(John R.W.Masters編
、2000年)、ISBN0199637970、テキスト全体に記載されている。
「集約的」(collectively)は、本発明が、任意の数または組み合わせの列挙されたタ
ンパク質もしくはマーカーまたはタンパク質もしくはマーカーの群を包含することを意味
し、しかも、このような数または組み合わせのタンパク質もしくはマーカーまたはタンパ
ク質もしくはマーカーの群が本明細書に具体的に記載されていないにもかかわらず、添付
の特許請求の範囲が、任意の他の組み合わせのタンパク質もしくはマーカーまたはタンパ
ク質もしくはマーカーの群から単独におよび分割可能にこのような組み合わせまたは小型
の組み合わせ(sub-combinations)を定義し得ることを意味する。
」という用語または「含む(comprises)」もしくは「含むこと(compri
sing)」などの変形は、述べられたステップもしくは要素もしくは整数(integer)
またはステップもしくは要素もしくは整数の群を包含するが、他の任意のステップもしく
は要素もしくは整数またはステップもしくは要素もしくは整数の群を排除しないことを暗
示すると理解されるべきであろう。
の供給源から得ることができるが、必ずしも直接その供給源からである必要はないことを
示すと解釈されるべきである。
態と比較して、および/または細胞を投与されない対象と比較して、生理的過程もしくは
疾患状態を改善するため、または対象における疾患状態の発症を予防するための、再プロ
グラム化細胞またはそれから分化した細胞および/またはその子孫細胞の十分量を意味す
ると解釈されるべきである。
、再プログラム化細胞または多能性細胞の数またはパーセントが天然の細胞集団における
数またはパーセントを超えることを意味すると解釈されるべきである。例えば、再プログ
ラム化細胞または多能性細胞が濃縮された集団は、少なくとも約0.02%の前記細胞ま
たは少なくとも約0.05%の前記細胞または少なくとも約0.1%の前記細胞または少
なくとも約0.2%の前記細胞または少なくとも約0.5%の前記細胞または少なくとも
約0.5%の前記細胞または少なくとも約0.8%の前記細胞または少なくとも約1%の
前記細胞または少なくとも約2%の前記細胞または少なくとも約3%の前記細胞または少
なくとも約4%の前記細胞または少なくとも約5%の前記細胞または少なくとも約10%
の前記細胞または少なくとも約15%の前記細胞または少なくとも約20%の前記細胞ま
たは少なくとも約25%の前記細胞または少なくとも約30%の前記細胞または少なくと
も約40%の前記細胞または少なくとも約50%の前記細胞または少なくとも約60%の
前記細胞または少なくとも約70%の前記細胞または少なくとも約80%の前記細胞また
は少なくとも約85%の前記細胞または少なくとも約90%の前記細胞または少なくとも
約95%の前記細胞または少なくとも約97%の前記細胞または少なくとも約98%の前
記細胞または少なくとも約99%の前記細胞で構成されている。
xposing)」は、能力決定因子が細胞に生物学的効果を発揮するほど、細胞を能力
決定因子の十分近くに運ぶ任意の過程を意味すると解釈されるべきである。この用語は、
限定するものではないが、細胞をこの因子と接触させるステップおよび/または細胞をこ
の因子をコードする核酸と接触させるステップおよび/または細胞においてこの因子を発
現させるステップを含むと理解されるべきである。
しくはマーカーの群を単独に包含することを意味し、しかも、個別のタンパク質もしくは
マーカーまたはタンパク質もしくはマーカーの群が本明細書に単独で記載されていないと
しても、添付の特許請求の範囲が、このようなタンパク質もしくはマーカーまたはタンパ
ク質もしくはマーカーの群を、互いに単独におよび分割可能に定義し得ることを意味する
。
体細胞(例えば、Stro−1+多分化能細胞またはその子孫細胞)と実質的に、遺伝学
的に同一であり、ES細胞などの能力のより高い細胞と同様の特徴を示す細胞を指す。i
PS細胞は、ES細胞と類似の形態学的特性(すなわち、円形、大型の核小体およびわず
かな細胞質)および成長特性(すなわち、倍加時間;ES細胞は約17から18時間の倍
加時間を有する)を示す。さらに、iPS細胞は、多能性細胞特異的マーカー(例えば、
Oct−4、SSEA−3、SSEA−4、Tra−1−60、Tra−1−81、しか
しSSEA−Iは発現しない)を発現することが好ましい。しかし、iPS細胞は、胚に
直接由来するものではなく、少なくともそれらが多能性になるまで、選択された能力決定
因子の1つまたは複数のコピーを一時的にまたは安定して発現できる。本明細書において
使用する場合、「胚に直接由来しない」は、iPS細胞を作製するための出発細胞型が、
出生後の個体から得た非多能性Stro−1+多分化能(mutilpotential)細胞またはそ
の非多能性子孫であることを意味する。
3種の胚葉(中胚葉、内胚葉および外胚葉)の1種または2種または3種、好ましくは1
種または2種の胚葉の複数の異なる型の細胞に分化できることを意味すると解釈されるべ
きである。
種の胚葉、すなわち内胚葉、外胚葉および中胚葉のそれぞれの細胞に分化できることを意
味すると解釈されるべきである。多能性細胞は、さまざまな多能性細胞特異的マーカー(
例えば、以下の多能性細胞特異的マーカー:SSEA−3、SSEA−4、TRA−1−
60またはTRA1−81の1種または複数)を発現し、未分化細胞に特徴的な細胞形態
(すなわち、コンパクトなコロニー、核細胞質比の高さおよび顕著な核小体)を有し、S
CIDマウスなどの免疫不全動物に導入された場合、奇形腫を形成する。奇形腫は、通常
3種の胚葉すべての特徴を示す細胞または組織を含有する。当業者は、当分野において一
般的に使用される技術を使用することによってこれらの特徴を評価でき、例えば、Tho
msonら、Science 282:1145〜1147(1998年)を参照された
い。多能性細胞は、細胞培養における増殖、および多分化能特性を示す、系列が限定され
たさまざまな細胞集団への分化の両方ができる。多分化能体細胞は、多能性細胞と比較し
てより分化しているが、最終分化はしていない。したがって、多能性細胞は、多分化能細
胞より高い能力を有している。
酸、それらの機能的断片などの因子およびコードされた因子、例えばタンパク質もしくは
その機能的断片、または体細胞の能力を増加し、それにより多分化能、多能性または全能
になるために使用される小分子もしくは抗体を指す。能力決定因子は、場合により、再プ
ログラム化細胞中に一時的にだけ存在し得る、または核酸の場合、再プログラム化細胞の
ゲノム中に転写的に活性または不活性な状態で維持され得る。同様に、核酸の能力決定因
子は、再プログラム化細胞中に複数のコピーが存在でき、能力決定因子が細胞のゲノム中
に組み込まれ得る場合、染色体外または両方に存在し得る。例示的能力決定因子は、Oc
t4(例示的ヌクレオチドおよびアミノ酸配列は、Genbank受託番号BC1174
35.1またはNCBI受託番号NM002701で提示される)、Sox2(例示的ヌ
クレオチドおよびアミノ酸配列は、NCBI受託番号NM_003106.2で提示され
る)、Klf4(例示的ヌクレオチドおよびアミノ酸配列は、NCBI受託番号NM_0
04235.4で提示される)、Nanog(例示的ヌクレオチドおよびアミノ酸配列は
、NCBI受託番号NM_024865.2で提示される)、Lin28(例示的ヌクレ
オチドおよびアミノ酸配列は、NCBI受託番号NM_024674.4で提示される)
、c−Myc(例示的ヌクレオチドおよびアミノ酸配列は、Genbank受託番号L1
6785.1)、bFGF、SCF、TERT、SV40ラージT抗原、HPV16E6
、HPV16E7、Bmil、Fbx15、Eras、ECAT15−2、Tcl1、β
−カテニン、ECAT1、ESG1、Dnmt3L、ECAT8、Gdf3、Sox15
、ECAT15−1、Fthl17、Sal14、Rex1(例示的配列は、NCBI受
託番号NM174900で提示される)、UTF1(例示的配列は、NCBI受託番号N
M003577で提示される)、Stella(例示的配列は、NCBI受託番号NM1
99286で提示される)、Stat3、FoxD3(例示的配列は、NCBI受託番号
NM012183で提示される)、ZNF206、Mybl2、DPP A2、Otx2
およびGrb2を含む。本明細書に提供されたすべての受託番号は、2009年2月20
日現在のものである。当業者は、本明細書に記載の他の能力決定因子の構造を、例えばN
CBIまたはGenBankなどのデータベースを使用して容易に決定できるであろう。
前記因子と同じまたは同様の活性を有する化合物もまた含まれる。このような化合物は、
再プログラミングを強化または誘導できる抗体および小分子、例えば、ヒストンデアセチ
ラーゼ阻害剤またはDNAメチラーゼまたはその阻害剤を含む。当業者は、適切な化合物
を、例えば、1種または複数の能力決定因子が除外された本明細書に記載の方法および評
価された被験化合物のパネルを使用して、ならびに/または細胞および/もしくはMar
koulakiら、Nature Biotechnology 27、169〜171
ページ(2009年)に記載の方法を使用して決定できるであろう。
の能力を意味すると解釈されるべきである。したがって、優れた能力を有する細胞は、能
力のより低い細胞より多くの細胞型に分化できる。
複数の検出可能な症状を予防する、またはその発症を阻害する、または発症を遅延させる
ために十分な量の再プログラム化細胞またはそれから分化した細胞および/またはその子
孫細胞を意味すると解釈されるべきである。
と(preventing)」または「予防(prevention)」という用語は、
予防有効量の細胞を投与し、臨床症状の少なくとも1種の症状の発生を停止する、妨げる
、または遅延させるまたは減少させることを意味すると解釈されるべきである。
および/または多分化能/多能性/全能細胞に転換させ、したがってそれが由来した細胞
より優れた能力の可能性を有する過程を指す。好ましくは、再プログラム化細胞は多分化
能、多能性または全能であり、より好ましくは多能性である。「再プログラム化」という
用語は、体細胞を多分化能/多能性/全能にさせるために脱分化されている体細胞を指す
。
化能細胞コロニーを形成できる非造血性STRO−1+および/またはTNAP+の前駆
細胞を意味すると解釈されるべきである。好ましいSTRO−1+多分化能細胞は、本明
細書においてより詳細に考察される。
意の対象、好ましくは哺乳動物を意味すると解釈されるべきである。例示的対象は、限定
するものではないが、ヒト、霊長類、家畜(例えば、ヒツジ、ウシ、ウマ、ロバ、ブタ)
、コンパニオン・アニマル(例えば、イヌ、ネコ)、実験動物(例えば、マウス、ウサギ
、ラット、モルモット、ハムスター)、捕らわれた野生動物(例えば、キツネ、シカ)を
含む。好ましくは、哺乳動物はヒトまたは霊長類である。最も好ましい哺乳動物はヒトで
ある。
れぞれの細胞および胚外組織に分化できることを意味すると解釈されるべきである。
症状を減少する、または阻害するために十分な量の再プログラム化細胞またはそれから分
化した細胞および/またはその子孫細胞を意味すると解釈されるべきである。
tment)」または「治療すること(treating)」という用語は、治療有効量
の細胞を投与し、臨床症状の少なくとも1種の症状を減少させる、または阻害することを
意味すると理解されるべきである
STRO−1+多分化能細胞は、骨髄、血液、歯髄細胞、脂肪組織、皮膚、脾臓、膵臓
、脳、腎臓、肝臓、心臓、網膜、脳、毛嚢、腸、肺、リンパ節、胸腺、骨、靭帯、腱、骨
格筋、真皮および骨膜において発見された細胞であり、中胚葉および/または内胚葉およ
び/または外胚葉などの生殖細胞系に分化できる。好ましくは、STRO−1+細胞は骨
髄、歯髄または脂肪組織由来であり、より好ましくは歯髄または脂肪組織由来である。し
たがって、STRO−1+多分化能細胞は、限定するものではないが、脂肪組織、骨組織
、軟骨組織、弾性組織、筋肉組織および線維性結合組織を含む多数の細胞型に分化できる
。これらの細胞が進入する特定の分化系列決定および分化経路は、機序的な影響ならびに
/または内因性生物活性因子、例えば成長因子、サイトカインおよび/もしくは宿主組織
によって確立された局所的微小環境条件からのさまざまな影響に依存する。したがって、
STRO−1+多分化能細胞は、分裂し、時がたてば不可逆的に分化し表現型細胞をもた
らすであろう、幹細胞または前駆体細胞のいずれかである娘細胞をもたらす非造血性前駆
細胞である。
べき対象または関連する対象または関連のない対象(同じ種または異なる種のどちらでも
)から得られた試料から濃縮される。このような濃縮は、ex vivoまたはin v
itroで実施され得る。「濃縮された(enriched)」、「濃縮(enrich
ment)」という用語またはそれらの変形は、本明細書において、未処理の集団と比較
した場合、1種の特定の細胞型の割合または多数の特定の細胞型の割合が増加した細胞の
集団を表すために使用される。
−1+、THY−1+、STRO−2+、CD45+、CD146+、3G5+またはそ
れらの任意の組み合わせからなる群から個別にまたは集約的に選択される、1種または複
数のマーカーを発現する。
bri)である。好ましくは、STRO−1bright細胞は、さらにTNAP+、V
CAM−1+、THY−1+、STRO−2+および/またはCD146+の1つまたは
複数である。
に血管周囲の間葉前駆体細胞である。
において使用される蛍光または他のマーカーの強度に関連する場合、マーカーが細胞表面
に存在する程度に依存して、低レベル(loもしくはdim)または高レベル(brig
ht、bri)のいずれかでそのマーカーを発現できる。lo(もしくはdimもしくは
dull)とbriとの区別は、選別された特定の細胞集団に関して使用されるマーカー
との関連で理解されるであろう。所与のマーカーに対して「陰性」であると称される細胞
は、その細胞が必ずしも完全に不在ではない。この用語は、その細胞によって発現される
マーカーが比較的非常に低いレベルであることを意味し、検出可能に標識された場合、非
常に低いシグナルを発生するまたはバックグラウンドのレベルを上回って検出できないこ
とを意味する。
た場合、比較的高いシグナルを発生する細胞表面のマーカーを指す。理論に縛られること
を好むわけではないが、「bright」細胞が、試料中の他の細胞より多くの標的マー
カータンパク質(例えば、STRO−1により認識される抗原)を発現することが提唱さ
れる。例えば、STRO−1bri細胞は、FITC標識STRO−1抗体を用いて標識
された場合、蛍光標識細胞分取(FACS)分析による決定で非bright細胞(ST
RO−1dull/dim)より多くの蛍光シグナルを発生する。好ましくは、「bri
ght」細胞は出発試料中に含有される最も明るく標識された骨髄単核細胞の少なくとも
約0.1%を構成する。他の実施形態において、「bright」細胞は出発試料中に含
有される最も明るく標識された骨髄単核細胞の少なくとも約0.1%、少なくとも約0.
5%、少なくとも約1%、少なくとも約1.5%または少なくとも約2%を構成する。好
ましい実施形態において、STRO−1bright細胞は、「バックグラウンド」、す
なわちSTRO−1−である細胞と比較してSTRO−1の表面発現の2log量の発現
増加を有する。比較すると、STRO−1dimおよび/またはSTRO−1inter
mediate細胞は、2log量未満の発現増加、通常約1logまたは「バックグラ
ウンド」より低いSTRO−1の表面発現を有する。
スファターゼのすべてのアイソフォームを包含することを意図する。例えばこの用語は、
肝臓アイソフォーム(LAP)、骨アイソフォーム(BAP)および腎臓アイソフォーム
(KAP)を包含する。好ましい実施形態において、TNAPはBAPである。特に好ま
しい実施形態において、本明細書において使用する場合、TNAPは、ブダペスト条約の
規定の下に、2005年12月19日に受託番号PTA−7282でATCCに寄託され
た、ハイブリドーマ細胞系によって作製されたSTRO−3抗体に結合できる分子を指す
。
FU−Fを生じさせることができる。
が好ましい。多分化能細胞が決定(committed)され得る系列の限定されない例は、骨前
駆体細胞;胆管上皮細胞および肝細胞に関して多分化能である肝細胞前駆細胞;乏突起膠
細胞および星状細胞に進行するグリア細胞前駆体を発生できる神経制限細胞;ニューロン
に進行するニューロン前駆体;心筋および心筋細胞、グルコース反応性、インスリン分泌
膵臓β細胞系に関する前駆体を含む。他の系列は、限定するものではないが、象牙芽細胞
、象牙質産生細胞および軟骨細胞ならびに以下:細胞網膜色素上皮細胞、線維芽細胞、ケ
ラチノサイトなどの皮膚細胞、樹状細胞、毛嚢細胞、尿細管上皮細胞、平滑筋細胞および
骨格筋細胞、精巣前駆細胞、血管内皮細胞、腱、靭帯、軟骨、含脂肪細胞、線維芽細胞、
骨髄基質、心筋、平滑筋、骨格筋、周皮細胞、血管、上皮、グリア、ニューロン、星状細
胞および乏突起膠細胞の各細胞の前駆体細胞を含む。
生できない。
施形態に従って本明細書に記載の方法に使用する前に、標準的技術を使用してin vi
troで培養される。このような細胞またはそれらから分化した細胞は、自己または同種
の組成物において対象に投与するために有用である。別の実施形態において、確立された
ヒト細胞系の1種または複数の細胞が使用される。本発明の別の有用な実施形態において
、非ヒト動物(または対象がヒトではない場合、別の種由来)の細胞が使用される。
培養に関して使用される場合、「培地」という用語は、細胞周囲の環境構成要素を含む。
培地は、固体、液体、気体または相と材料の混合物であってよい。培地は、液体成長培地
および細胞成長を維持しない液体培地を含む。培地は、ゼラチン培地、例えば寒天、アガ
ロース、ゼラチンおよびコラーゲン基質をさらに含む。例示的ガス状培地は、ペトリ皿ま
たは他の固体もしくは半固体支持体上で成長する細胞が曝露されるガス相を含む。「培地
」という用語は、たとえ細胞に接触していなくても、細胞培養において使用するための材
料を指す。言い換えれば、細胞培養のために調製された栄養豊富な液体は培地である。水
または他の液体と混合した場合細胞培養に適切になる粉末混合物は、「粉末培地」と呼ぶ
ことができる。
標識された磁気ビーズを使用して骨髄からTNAP+STRO−1+多分化能細胞を単離
し、次いで培養して単離細胞を増殖させる(expand)ことによって得られる(適切な培養
条件の例に関してはGronthosら、Blood 85:929〜940ページ、1
995年を参照されたい)。
継代後)は、TNAP−、CC9−、HLAクラスI+、HLAクラスII−、CD14
−、CD19−、CD3−、CD11a−c−、CD31−、CD86−、CD34−お
よび/またはCD80−であり得る。しかし、本明細書に記載の培養条件と異なる培養条
件下では、さまざまなマーカーの発現は変動し得る可能性がある。さらに、これらの表現
型の細胞は増殖させた細胞集団において優勢であり得るが、この表現型(複数可)を有さ
ない細胞の少数の集団が存在することを意味するものではない(例えば、増殖させた細胞
は、少ない割合でCC9−であり得る。)。好ましい一実施形態において、増殖させた細
胞は、さまざまな細胞型に分化する能力を未だに有している。
1、ICAM−1、PECAM−1、P−セレクチン、L−セレクチン、3G5、CD4
9a/CD49b/CD29、CD49c/CD29、CD49d/CD29、CD90
、CD29、CD18、CD61、インテグリンβ 6−19、トロンボモジュリン、C
D10、CD13、SCF、PDGF−R、EGF−R、IGF1−R、NGF−R、F
GF−R、レプチン−R(STRO−2=レプチン−R)、RANKL、STRO−1b
rightおよびCD146またはこれらのマーカーの任意の組み合わせからなる群から
選択される、1種または複数のマーカーを集約的に、または個別に発現し得る。
よび/または記載された、多分化能性増殖STRO−1+多分化能細胞子孫(Multi
potential Expanded STRO−1+ Multipotentia
l cells Progeny)(MEMP)である。子孫が由来し得るSTRO−1
+多分化能細胞が濃縮された集団を調製する方法は、WO01/04268およびWO2
004/085630に記載される。in vitroの関連において、STRO−1+
多分化能細胞は、完全に純粋な調製品として存在することは稀であり、一般的には組織特
異的分化系列決定細胞(tissue specific committed cel
ls)(TSCC)である他の細胞と一緒に存在すると思われる。WO01/04268
は、約0.1%から90%の純度レベルで骨髄からこのような細胞を収穫するステップに
ついて言及している。子孫が由来する多分化能(mutilpotential)細胞を含む集団は、組
織供給源から直接収穫でき、またはex vivoですでに増殖させた集団であってもよ
い。
、20、30、40、50、60、70、80または95%を含む、収穫され、増殖しな
い実質的に精製されたSTRO−1+多分化能細胞の集団から得ることができる。このレ
ベルは、例えば、TNAP、STRO−1bright、3G5+、VCAM−1、TH
Y−1、CD146およびSTRO−2からなる群から個別にまたは集約的に選択される
、少なくとも1種のマーカーに陽性である細胞を選択することによって達成され得る。
ホスファターゼ(ALP)に対して陰性であるという点から、新鮮に収穫されたSTRO
−1+多分化能細胞とは識別され得る。対照的に、新鮮に単離されたSTRO−1+多分
化能細胞は、STRO−1briおよびALPの両方に対して陽性である。本発明の好ま
しい実施形態において、投与された細胞の少なくとも15%、20%、30%、40%、
50%、60%、70%、80%、90%または95%はSTRO−1bri、ALP−
の表現型を有する。さらなる好ましい実施形態において、MEMPは、マーカーのKi6
7、CD44および/またはCD49c/CD29、VLA−3、α3β1の1種または
複数に対して陽性である。その上さらなる好ましい実施形態において、MEMPはTER
T活性を示さない、および/またはマーカーのCD18に対して陰性である。
085630において示される任意の1種もしくは複数の組織型、すなわち骨髄、歯髄細
胞、脂肪組織および皮膚に由来し、または、おそらくより広範囲には、脂肪組織、歯、歯
髄、皮膚、肝臓、腎臓、心臓、網膜、脳、毛嚢、腸、肺、脾臓、リンパ節、胸腺、膵臓、
骨、靭帯、骨髄、腱および骨格筋に由来し得る。
のさまざまな方法によってもたらされ得るが、好ましい方法は、結合剤(例えば、抗体ま
たはその抗原結合断片)と関与するマーカーとの結合、その後の、高レベルの結合または
低レベルの結合または結合を示さないかのいずれかである、結合を示す細胞の分離に頼る
と理解されると思われる。最も都合の良い結合剤は、抗体または抗体に基づく分子であり
、好ましくは、これらの後者の薬剤の特異性のためモノクローナル抗体であり、またはモ
ノクローナル抗体に基づく。抗体は、両方のステップに使用できるが、他の薬剤もまた使
用でき、したがって、これらのマーカーに対するリガンドもまた、これらのマーカーを担
持する細胞またはそれを欠いた細胞の濃縮に用いることができる。
くは回収される分画の生存能力の保有を最大にする。異なる効率のさまざまな技術を用い
、比較的粗い分離を得ることができる。用いる特定の技術は、分離の効率、関連する細胞
毒性、実施の簡便さおよび速さならびに高性能の機器および/または技術的熟練の必要性
に依存すると思われる。分離の手順は、限定するものではないが、抗体コーティング磁気
ビーズを使用する磁気分離、アフィニティクロマトグラフィーおよび固体マトリックスに
結合した抗体を用いた「パニング」を含み得る。正確な分離を提供する技術は、限定する
ものではないが、FACSを含む。FACSを実施する方法は、当業者には明らかである
と思われる。
−1に対するモノクローナル抗体は、R&D Systems、USAから市販されてい
る)、ATCCまたは他の寄託組織から利用でき、および/または当分野において認識さ
れている技術を使用して作製できる。
現を認識する磁気活性化細胞分取法(MACS)を利用する固体相選別ステップである第
1のステップを含むことが好ましい。所望であれば、次いで、特許明細書WO01/14
268に記載の高レベルの前駆体細胞発現をもたらす第2の選別ステップを続けることが
できる。この第2の選別ステップは、複数のマーカーの使用を含み得る。
の前に、細胞供給源の収穫をさらに含み得る。したがって、組織は外科的に摘出される。
次いで、供給源組織を含む細胞は、いわゆる単一細胞懸濁液に分離されるであろう。この
分離は、物理的および/または酵素的方法によって達成され得る。
能細胞集団は、MEMPを得るための任意の適切な手段によって培養または増殖させ得る
。
ギ、げっ歯類、鳥類および魚類の細胞を含む任意の非ヒト動物種由来の細胞を使用して実
行できる。本発明を実施できる霊長類細胞は、限定するものではないが、チンパンジー、
ヒヒ、カニクイザルならびに他の任意の新世界ザルおよび旧世界ザル(New or Old World
monkeys)の細胞を含む。本発明を実施できる有蹄動物細胞は、限定するものではないが
、ウシ、ブタ、ヒツジ、ヤギ、ウマ、バッファローおよびバイソンの細胞を含む。本発明
を実施できるげっ歯類細胞は、限定するものではないが、マウス、ラット、モルモット、
ハムスターおよびスナネズミの細胞を含む。本発明を実施できるウサギ種の例は、家畜化
されたウサギ、ジャックラビット、野ウサギ、ワタオウサギ、カンジキウサギおよびナキ
ウサギを含む。ニワトリ(Gallus gallus)は、本発明を実施できる鳥類の例である。
できる。真核細胞および特に哺乳動物細胞の保管および保存の方法およびプロトコルは当
分野において公知である(例えば、Pollard,J.W.およびWalker,J.
M.(1997年)Basic Cell Culture Protocols、第2
版、Humana Press、Totowa、N.J.;Freshney,R.I.
(2000年)Culture of Animal Cells、第4版、Wiley
−Liss、Hoboken、N.J.を参照されたい)。単離された幹細胞、例えば間
葉幹細胞/前駆細胞またはその子孫の生物活性を維持する任意の方法は、本発明に関連し
て利用され得る。好ましい一実施形態において、細胞は、凍結保存を使用して維持および
保管される。
一実施形態において、STRO−1+多分化能細胞および/またはその子孫細胞は、例
えば、能力決定因子またはその複数を発現するように遺伝子改変される。
いて発現される核酸は、細胞、好ましくは多能性(plurlipotent)細胞における発現を誘
導するためのプロモーターに作動可能に連結される。例えば、核酸は、対象のさまざまな
細胞において作動可能であるプロモーター、例えば、ウイルスプロモーター、例えばCM
Vプロモーター(例えばCMV−IEプロモーター)またはSV−40プロモーターまた
は延長因子プロモーターまたは誘導プロモーター、例えばtet誘導プロモーターに作動
可能に連結される。さらなる適切なプロモーターは当分野において公知であり、本発明の
本実施形態に、必要な変更を加えて適用すると解釈されるべきである。本発明は、単一の
プロモーター、例えばOct4およびSox2から複数の能力決定因子の発現を可能にす
る、マルチシストロン性ベクターの使用をさらに包含する。このようなベクターは、一般
的に、さまざまな能力決定因子を個々にコードする2つの核酸を分離する配列内リボソー
ム進入部位(IRES)を含む。
れるべきであり、転写の開始に必要とされるTATAボックスまたはイニシエーターエレ
メントを含むゲノム遺伝子の転写制御配列を、例えば、発生刺激および/または外部刺激
に対する応答において、または組織特異的様式で遺伝子発現を改変するさらなる制御エレ
メント(すなわち、上流活性化配列、転写因子結合部位、エンハンサーおよびサイレンサ
ー)と一緒に含む、またはそれを伴わずに含む。本文脈において、「プロモーター」とい
う用語は、組み換え、合成もしくは融合分子または作動可能に連結された核酸の発現をも
たらす、活性化する、または強化する、かつ好ましくはペプチドまたはタンパク質をコー
ドする誘導体を表すためにもまた使用される。好ましいプロモーターは、1種または複数
の特異的制御エレメントのさらなるコピーを含有し、前記核酸分子の発現をさらに強化す
る、ならびに/または空間的発現および/もしくは時間的発現を改変することができる。
場合、核酸は、プロモーターと一緒に、またはプロモーターに「作動可能に連結される」
(すなわち、プロモーターの制御管理下にある)。プロモーターは、一般的に核酸の発現
5’(上流)に位置付けられ、プロモーターが核酸の発現を調節する。異種プロモーター
/核酸の組み合わせを構築するために、プロモーターは、そのプロモーターと遺伝子との
間の距離とおよそ同じ距離で遺伝子転写開始部位から離れて位置付けられ、プロモーター
がその自然の設定、すなわちプロモーターが由来する遺伝子において調節することが、一
般的に好ましい。当分野において公知であるように、この距離のいくつかの変更はプロモ
ーター機能を失うことなく調整され得る。同様に、その調節下で配置される異種核酸に対
する制御配列エレメントの好ましい位置付けは、その自然の設定、すなわちそれが由来す
る遺伝子において要素を位置付けることによって画定される。この場合も先と同様に、当
分野において公知であるように、この距離のいくつかの変更がまた起こり得る。
「発現構築体」という用語は、細胞において作動可能に連結される核酸に発現をもたらす
能力を有する核酸を指す。本発明の文脈において、発現構築体は、プラスミド、バクテリ
オファージ、ファージミド、コスミド、ウイルスのサブゲノムもしくはゲノム断片または
異種DNAに発現をもたらすことのできる他の核酸であってもよく、それを含んでいても
よいことが理解されよう。発現構築体は、細胞のゲノム中に組み込まれても、エピソーム
として残存してもよい。
であり得る。このような発現構築体は、例えば発現構築体がゲノムに組み込まれていない
再プログラム化細胞の作製に有用である。さらに、これらの発現構築体は多くの場合細胞
分裂の間に失われるので、組み換え発現構築体を含まない再プログラム化細胞の作製を可
能にする(例えば、Okitaら、Science、322:949〜53ページ、20
08年を参照されたい)。
われ、例えば、Ausubelら、(Current Protocols in Mo
lecular Biology.Wiley Interscience、ISBN
047 150338、1987年)またはSambrookら(Molecular
Cloning:Molecular Cloning:A Laboratory M
anual、Cold Spring Harbor Laboratories、Ne
w York、第3版、2001年)に記載されている。例えば、発現構築体の個々の要
素は、適切な鋳型核酸から、例えばPCRを使用して増幅され、その後、適切な発現構築
体、例えばプラスミドまたはファージミドなどの内部にクローン化される。
は本明細書に記載される。例えば、哺乳動物細胞中の本発明の方法に適切な発現ベクター
は、例えば、Invitrogenにより供給されるpcDNAベクタースイート(vect
or suite)のベクター、pCIベクタースイート(Promega)のベクター、pCM
Vベクタースイート(Clontech)のベクター、pMベクター(Clontech
)、pSIベクター(Promega)、VP16ベクター(Clontech)または
pcDNAベクタースイート(Invitrogen)のベクターである。
rogen Corporation、ClontechまたはPromegaを承知し
ているであろう。
ある。所与の生物に使用される技術は、公知の成功する技術に依存する。組み換えDNA
を細胞に導入する方法は、リン酸カルシウム沈殿(GrahamおよびVan Der
Eb、Virology、52:456〜467ページ、1973年;ChenおよびO
kayama、Mol.Cell Biol.、7:2745〜2752ページ、198
7年;Rippeら、Mol.Cell Biol.、10:689〜695ページ、1
990年)、DEAE−デキストラン(Gopal、Mol.Cell Biol.、5
:1188〜1190ページ、1985年)、エレクトロポレーション(Tur−Kas
paら、Mol.Cell Biol.、6:716〜718ページ、1986年;Po
tterら、Proc.Natl Acad.Sci.USA、81:7161〜716
5ページ、1984年)、直接微量注入、DNA負荷リポソーム(Nicolauおよび
Sene、Biochim.Biophys.Acta、721:185〜190ページ
、1982年;Fraleyら、Proc.Natl Acad.Sci.USA、76
:3348〜3352ページ、1979年)、細胞の超音波処理(Fechheimer
ら、Proc.Natl Acad.Sci.USA、84:8463〜8467ページ
、1987年)、高速微粒子銃を使用する遺伝子ボンバードメント(Yangら、Pro
c.Natl Acad.Sci USA、87:9568〜9572ページ、1990
年)、レセプター媒介トランスフェクション(WuおよびWu、J.Biol.Chem
.、262:4429〜4432ページ、19877年;WuおよびWu、Bioche
m.、27:887〜892ページ、1988年)を含む。他の実施形態において、核酸
の細胞への移入は、ナノキャップ(nanocaps)(例えばナノ粒子CaP04)、コロイド
金、ナノ粒子合成ポリマーおよび/またはリポソームと一緒に製剤化することによって達
成され得る。
体が細胞のゲノムに組み込まれていない限り、例えば上記、またはOkitaら、200
8年、上記に記載の方法を使用してトランスフェクトされる。好ましくは、プラスミドは
、前期細胞が再プログラムされるまで、前期細胞に繰り返しトランスフェクトされる。こ
の様式において、細胞のゲノムに組み込まれた異種DNAを有さない細胞が作製され、治
療的観点からこのことはより魅力的である。
分野において公知であり、市販されている。核酸の送達およびその核酸を宿主細胞のゲノ
ムに組み込まれるための従来のウイルスに基づく系は、例えば、レトロウイルスベクター
、レンチウイルスベクターまたはアデノ随伴ウイルスベクターを含む。または、アデノウ
イルスベクターはエピソーム性のままである核酸を宿主細胞に導入し、例えば、そのゲノ
ムに組み込まれた異種DNAを含まない再プログラム化細胞を作製するために有用である
。ウイルスベクターは、標的細胞および組織における有効かつ用途の広い遺伝子移入方法
である。さらに、高効率の形質導入が、多数のさまざまな細胞型および標的組織において
観察されている。例示的ウイルスベクターを以下に述べる。
一例において、本発明に有用なウイルス遺伝子送達系は、アデノウイルス由来のベクタ
ーを利用する。36kBの線状および二本鎖DNAウイルスであるアデノウイルスの遺伝
子構成の知見は、8kBまでの外来配列を有する大きなアデノウイルスDNA片の置換を
可能にする。アデノウイルスDNAは、遺伝毒性の可能性なしにエピソーム性様式で複製
できるので、アデノウイルスDNAの宿主細胞への感染は染色体への組み込みをもたらさ
ない。さらに、アデノウイルスは構造的に安定であり、ゲノムの再構成が、広大な増幅後
に検出されている。アデノウイルスは、その細胞周期の段階にかかわらず、事実上すべて
の上皮細胞に感染できる。組み換えアデノウイルスは、分裂細胞および非分裂細胞の両方
を形質導入できる。非分裂細胞を有効に形質導入する能力は、アデノウイルスを、遺伝子
を筋肉細胞または脂肪細胞に移入するための優れた候補にさせる。
および高い感染性のため、遺伝子移入ベクターとして特に適切である。ウイルスゲノムの
両方の末端は、100〜200塩基対(bp)の逆位末端反復(ITR)を含有し、これ
らはウイルスDNAの複製およびパッケージングに必要なシスエレメントである。
て分裂するさまざまな転写ユニットを含有する。E1領域(E1AおよびE1B)は、ウ
イルスゲノムおよび少数の細胞遺伝子の転写制御に関与するタンパク質をコードする。E
2領域(E2AおよびE2B)の発現は、ウイルスDNA複製のためのタンパク質の合成
をもたらす。これらのタンパク質は、DNAの複製、後期遺伝子発現および宿主細胞の遮
断に関与する(Renan(1990年)Radiotherap.Oncol.19:
197)。ウイルスカプシドタンパク質の大部分を含む後期遺伝子の生成物は、主要な後
期プロモーター(MLP)によって生じる単一の一次転写産物の重要なプロセシングの後
にのみ発現される。MLP(16.8μに位置する)は、感染後期において特に有効であ
り、このプロモーターから生じたすべてのmRNAは、それを翻訳のための例示的mRN
Aにさせる5’トリパータイトリーダー(TL)配列を保有する。
きるが、正常なウイルスの溶菌ライフサイクルにおいて複製するその能力に関しては不活
性化される(例えば、Berknerら、(1988年)BioTechniques
6:616ページ;Rosenfeldら、(1991年)Science 252:4
31〜434ページおよびRosenfeldら、(1992年)Cell 68:14
3〜155ページを参照されたい)。アデノウイルスAd株5 dl324型またはアデ
ノウイルスの他の株(例えば、Ad2、Ad3、Ad7など)に由来する適切なアデノウ
イルスベクター−は、当業者には公知である。
でき、気道上皮(上に引用されたRosenfeldら、(1992年))、内皮細胞(
Lemarchandら、(1992年)PNAS USA 89:6482〜6486
ページ)、肝細胞(HerzおよびGerard、(1993年)PNAS USA 9
0:2812〜2816ページ)および筋肉細胞(Quantinら、(1992年)P
NAS USA 89:2581〜2584ページ;Ragotら、(1993年)Na
ture 361:647ページ)を含む広範囲の細胞型を感染させるために使用され得
る、特定の状況において有利であり得る。
影響を及ぼすように改変され得る。
ムの担持能力は大きい(最大8キロベースまで)(Berknerら、上記;Haj−A
hmandおよびGraham(1986年)J.Virol.57:267ページ)。
現在使用され、したがって本発明によって支持される、大部分の複製欠損アデノウイルス
ベクターは、ウイルスのE1およびE3遺伝子の全部または一部が削除されているが、ア
デノウイルスの遺伝物質の80%程度を保有している(例えば、Jonesら、(197
9年)Cell16:683ページ;Berknerら、上記およびGrahamら、M
ethods in Molecular Biology、E.J.Murray編(
Humana、Clifton、NJ、1991年)7巻、109〜127ページを参照
されたい)。本発明の挿入ポリヌクレオチドの発現は、例えば、E1Aプロモーター、主
要後期プロモーター(MLP)および関連リーダー配列、ウイルスのE3プロモーターま
たは外から加えられたプロモーター配列の調節下であり得る。
き欠損であってもよい。アデノウイルスは、42種の公知の異なる血清型またはサブグル
ープA〜Fのいずれであってもよい。サブグループCのアデノウイルス5型は、本明細書
に記載の方法および組成物に従った使用のための、条件付き複製欠損アデノウイルスベク
ターを得るための例示的出発物質である。アデノウイルス5型は、大量の生化学的情報お
よび遺伝的情報が公知であるヒトアデノウイルスであり、アデノウイルスをベクターとし
て用いる大部分の構築に歴史的に使用されてきたからである。上記のように、本発明に従
った典型的なベクターは複製欠損であり、アデノウイルスE1領域を有さないであろう。
したがって、対象となる核酸を、E1をコードする配列が除去された場所に導入するため
に最も都合がよいと思われる。しかし、アデノウイルス配列内の領域中のポリヌクレオチ
ドの挿入位置は、本発明にとって重要ではない。例えば、Karlssonら、(198
6年)によって以前に記載されたように、E3置換ベクター中の欠失E3領域の代わりに
、またはE4を欠失したヘルパー細胞系またはヘルパーウイルス補体のE4領域にも挿入
され得る。
ージング細胞系」とも称されるこのヘルパー細胞系は、Frank Graham(Gr
ahamら、(1987年)J.Gen.Virol.36:59〜72ページおよびG
raham(1977年)J.General Virology 68:937〜94
0)によって開発され、E1AおよびE1Bをトランスで供給する。しかし、ヘルパー細
胞系は、ヒト細胞、例えばヒト胎児の腎臓細胞、筋肉細胞、造血細胞または他のヒト胎児
の間葉細胞もしくは上皮細胞にもまた由来し得る。または、ヘルパー細胞は、ヒトアデノ
ウイルスに対して許容である他の哺乳動物種の細胞にも由来し得る。このような細胞は、
例えば、ベロ細胞または他のサル胎児の間葉細胞もしくは上皮細胞を含む。
する、および/または限定された細胞型においてのみ所望のヌクレオチド配列を発現する
ことができる。例えば、ウイルスは、例えば米国特許第5,698,443号に記載のよ
うに、標的宿主細胞によって特異的に制御される転写開始領域の転写調節下で遺伝子を含
み得る。したがって、複製可能なアデノウイルスからの発現は、複製に必要なタンパク質
の合成を制御する細胞特異的応答エレメント、例えば、E1AまたはE1Bを挿入するこ
とによって、特定の細胞に制限され得る。
び哺乳動物のトランスフェクトへのその使用のための方法および材料を含む、本発明の実
施に有用であると思われるアデノウイルス技術に関するさらなる詳細なガイダンスに関し
ては、Wilsonら、WO94/28938、WO96/13597およびWO96/
2628ならびにそれらの引用された参考文献を参照されたい。
ある実施形態において、レトロウイルスベクターは、本明細書に記載の方法および組成
物に従って使用され得る。このようなウイルスは、再プログラム化細胞の作製にすでに使
用されているがStro−1+細胞においては使用されていない。レトロウイルスは、逆
転写の過程によって感染細胞においてそれらのRNAを二本鎖DNAに転換する能力を特
徴とする一本鎖RNAウイルスの群である(Coffin(1990年)「Retrov
iriae and their Replication」In Fields、Kn
ipe編Virology.New York:Raven Press)。次いで、得
られたDNAはプロウイルスとして細胞染色体に安定に組み込まれ、ウイルスタンパク質
を調節合成する。組み込まれると、レシピエント細胞およびその子孫においてウイルス遺
伝子配列が保有される。レトロウイルスゲノムは、それぞれカプシドタンパク質ポリメラ
ーゼ酵素およびエンベロープ成分をコードする3種の遺伝子、gag、polおよびen
vを含有する。gag遺伝子の上流で発見された配列はpsiと呼ばれ、ビリオンにゲノ
ムをパッケージングするためのシグナルとして機能する。2つの長い末端反復(LTR)
配列は、ウイルスゲノムの5’末端および3’末端に存在する。これらは、強いプロモー
ター配列およびエンハンサー配列を含有し、宿主細胞ゲノムへの組み込みにもまた必要と
される(Coffin(1990年)、上記)。
特定のウイルス配列に代わって、対象となる核酸がウイルスゲノムに挿入される。ビリオ
ンを作製するために、gag、polおよびenv遺伝子を含有するが、LTRおよびp
si成分を含有しないパッケージング細胞系が構築される(Mannら、(1983年)
Cell 33:153ページ)。本発明の核酸を含有する組み換えプラスミドと、レト
ロウイルスのLTRおよびpsi配列とを一緒に、(例えばリン酸カルシウム沈殿によっ
て)この細胞系に導入する場合、psi配列は組み換えプラスミドのRNA転写産物をウ
イルス粒子にパッケージング可能にし、その後培養培地に分泌する(Nicolasおよ
びRubenstein(1988年)「Retroviral Vectors」、R
odriguezおよびDenhardt編 Vectors:A Survey of
Molecular Cloning Vectors and their Use
s.Stoneham、ButterworthおよびTemin(1986年)「Re
trovirus Vectors for Gene Transfer:Effic
ient Integration into and Expression of
Exogenous DNA in Vertebrate Cell Genome」
、Kucherlapati編 Gene Transfer.New York:Pl
enum Press;Mannら、1983年、上記)。その後、組み換えレトロウイ
ルスを含有する培地が回収され、場合により濃縮され、遺伝子移入に使用される。レトロ
ウイルスベクターは、広範囲の細胞型に感染できる。
呼ばれる)の開発は、遺伝子療法のためのレトロウイルスの有用性を増加させ、欠損レト
ロウイルスは遺伝子療法目的のための遺伝子移入への使用を特徴とする(再考察として、
Miller,A.D.(1990年)Blood 76:271ページを参照されたい
)。したがって、レトロウイルスコーディング配列(gag、pol、env)の一部が
、ペプチドまたは本発明の類似体、例えばレトロウイルスに複製欠損をもたらす転写活性
化因子をコードする核酸によって置換されている組み換えレトロウイルスが構築され得る
。その後、複製欠損レトロウイルスはビリオンにパッケージングされ、このビリオンは、
標準的技術によってヘルパーウイルスの使用を介して標的細胞を感染させるために使用で
きる。組み換えレトロウイルスの作製およびこのようなウイルスをin vitroまた
はin vivoで細胞に感染させるためのプロトコルは、Current Proto
cols in Molecular Biology、Ausubel,F.M.ら(
編)Greene Publishing Associates、(1989年)、S
ections9.10〜9.14および他の標準的実験室マニュアルにおいて見出すこ
とができる。
は当業者には公知である。例示的レトロウイルスベクターは、pSR MSVtkNeo
(Mullerら、(1991年)Mol.Cell Biol.11:1785ページ
およびpSR MSV(XbaI)(Sawyersら、(1995年)J.Exp.M
ed.181:307ページ)およびそれらの誘導体である。例えば、これらのベクター
両方中の特有のBamHI部位は、ClacksonらによるWO96/41865に記
載のように、ベクターをBamHIを用いて消化し、Klenowを用いて埋め、それぞ
れ、pSMTN2およびpSMTX2を作製するために再接合することによって除去でき
る。同種指向性および両種指向性両方のレトロウイルス系を調製するための適切なパッケ
ージングウイルス系の例は、CripおよびCreを含む。
網膜細胞、内皮細胞、リンパ球、筋芽細胞、肝細胞、骨髄細胞を含む多数の異なる細胞型
にin vitroおよび/またはin vivoで導入するために使用されてきた(例
えば、Federico(1999年)Curr.Opin.Biotechnol.1
0:448ページ;Eglitisら、(1985年)Science 230:139
5〜1398ページ;DanosおよびMulligan、(1988年)PNAS U
SA 85:6460〜6464ページ;Wilsonら、(1988年)PNAS U
SA 85:3014〜3018ページ;Armentanoら、(1990年)PNA
S USA 87:6141〜6145ページ;Huberら、(1991年)PNAS
USA 88:8039〜8043ページ;Ferryら、(1991年)PNAS
USA 88:8377〜8381ページ;Chowdhuryら、(1991年)Sc
ience 254:1802〜1805ページ;Kayら、(1992年)Human
Gene Therapy 3:641〜647ページ;Daiら、(1992年)P
NAS USA 89:10892〜10895ページ;Hwuら、(1993年)J.
Immunol.150:4104〜4115ページ;米国特許第4,868,116号
;米国特許第4,980,286号;PCT出願WO89/07136;PCT出願WO
89/02468;PCT出願WO89/05345;およびPCT出願WO92/07
573による再考察を参照されたい)。
ターの感染範囲を、ウイルス粒子の表面上のウイルスパッケージングタンパク質を改変す
ることによって、制限できることが示されている(例えば、PCT公開WO93/252
34、WO94/06920およびWO94/11524を参照されたい)。例えば、レ
トロウイルスベクターの感染範囲の改変のための戦略は、細胞表面抗原に特異的な抗体と
、ウイルスenvタンパク質とのカップリング(Rouxら、(1989年)PNAS
USA 86:9079〜9083ページ;Julanら、(1992年)J.Gen
Virol 73:3251〜3255ページ;およびGoudら、(1983年)Vi
rology 163:251〜254ページ);または細胞表面リガンドと、ウイルス
envタンパク質とのカップリング(Nedaら、(1991年)J.Biol.Che
m.266:14143〜14146ページ)を含む。カップリングは、タンパク質また
は他の種類との化学的架橋(例えば、envタンパク質をアシアロ糖タンパク質に転換す
るためのラクトース)の形態で、および融合タンパク質の作製(例えば、一本鎖抗体/e
nv融合タンパク質)によって可能である。
本発明の核酸の送達に有用な例示的ウイルスベクター系はアデノ随伴ウイルス(AAV
)である。ヒトアデノウイルスは、受容体媒介エンドサイト−シスによって細胞に進入す
る二本鎖DNAウイルスである。これらのウイルスは成長および操作しやすく、in v
ivoおよびin vitroで広い宿主範囲を示すので、これらのウイルスは、遺伝子
移入に非常に適していると考えられている。アデノウイルスは、感染休止および標的細胞
の複製ができ、宿主ゲノム中に組み込まれるのではなく染色体外で生存できる。AAVは
、ディペンドウイルス属に属するヘルパー依存性DNAパルボウイルスである。AAVは
、病理が公知ではなく、有効な複製および生産的なライフサイクルのために、別のウイル
ス、例えば、アデノウイルス、ワクチニアまたはヘルペスウイルスによって提供されるさ
らなるヘルパー機能がなくては複製できない。
て潜伏状態を確立する。ヘルパーウイルスによるその後の感染は、その後、感染性のウイ
ルスの子孫を産生するために複製できる、組み込まれたコピーを救出する。野生型AAV
ウイルスとアデノウイルスまたはヘルペスウイルスのいずれかに由来するヘルパー機能と
の組み合わせは、複製可能な組み換えAVV(rAVV)を発生する。この系の1つの有
利性は、その相対的な安全性である(再考察のために、Xiaoら、(1997年)Ex
p.Neurol.144:113〜124ページを参照されたい。
る(BernsおよびBohenzky、(1987年)Advances in Vi
rus Research(Academic Press、Inc.)32:243〜
307ページ)。このゲノムは、各末端に、DNA複製の起点として、またウイルスのパ
ッケージングシグナルとしてcisに機能する逆位末端反復(ITR)を含む。ゲノムの
内部非反復部分は、それぞれAAVrepおよびcap領域として知られる、2つの大き
なオープンリーディングフレームを含む。これらの領域は、複製およびビリオンのパッケ
ージングに関与するウイルスタンパク質をコードする。AAVゲノムの詳細な説明のため
に、例えば、Muzyczka,N.(1992年)Current Topics i
n Microbiol. and Immunol.158:97〜129ページを参
照されたい。
込まれ得る。外来DNAの空間は約4.7kbに制限され、これは、本発明のペプチドま
たは類似体をコードする核酸の組み込みに十分である。Tratschinら、(198
5年)Mol.Cell.Biol.5:3251〜3260ページに記載のようなAA
Vベクターは、DNAを細胞に導入するために使用できる。さまざまな核酸が、AAVベ
クターを使用して異なる細胞型に導入されてきた(例えば、Hermonatら、(19
84年)PNAS USA 81:6466〜6470ページ;Tratschinら、
(1985年)Mol.Cell.Biol.4:2072〜2081ページ;Wond
isfordら、(1988年)Mol.Endocrinol.2:32〜39ページ
;Tratschinら、(1984年)J.Virol.51:611〜619ページ
;およびFlotteら、(1993年)J.Biol.Chem.268:3781〜
3790ページを参照されたい)。
例えば、Barlett,J.S.ら、(1996年)、Protocols for
Gene Transfer in Neuroscience; Towards G
ene Therapy of Neurological Disorders、11
5〜127ページに記載されている。
、または制限酵素を用いる所望のヌクレオチド配列の除去、その後の末端の平滑化、適切
なDNAリンカーのライゲーション、制限消化およびITRとの間の部位へのライゲーシ
ョンによるかのどちらかで所望のヌクレオチド配列のサブクローニングに使用され得る、
制限部位に隣接する145ヌクレオチドのAAV逆位末端配列(ITR)を含む。
増殖および精製ならびに細胞および哺乳動物のトランスフェクトへのその使用のための方
法および材料を含む、本発明の実施に有用であると思われるAAV技術に関するさらなる
詳細なガイダンスに関しては、例えば、Carterら、米国特許第4,797,368
号(1989年1月10日);Muzyczkaら、米国特許第5,139,941号(
1992年8月18日);Lebkowskiら、米国特許第5,173,414号(1
992年12月22日);Srivastava,米国特許第5,252,479号(1
993年10月12日);Lebkowskiら、米国特許第5,354,678号(1
994年、10月11日);Shenkら、米国特許第5,436,146号(1995
年7月25日);Chatterjeeら、米国特許第5,454,935号(1995
年12月12日)、Carterら、WO93/24641(1993年12月9日公開
)およびNatsoulis、米国特許第5,622,856号(1997年4月22日
)を参照されたい。
核酸の送達に使用され得る他のウイルスベクター系は、例えばヘルペスウイルス、例え
ば単純ヘルペスウイルス(Wooら、IJ St特許第5,631,236号、1997
年5月20日発行およびNeurovexによるWO00/08191)ワクチニアウイ
ルス(Ridgeway(1988年)Ridgeway、「Mammalian ex
pression vectors」、Rodriguez R L、Denhardt
D T編 Vectors:A survey of molecular clon
ing vectors and their uses.Stoneham:Butt
erworth,;BaichwalおよびSugden(1986年)「Vector
s for gene transfer derived from animal
DNA viruses:Transient and stable express
ion of transferred genes」、Kucherlapati R
編 Gene transfer.New York:Plenum Press;Co
uparら、(1988年)Gene、68:1〜10ページ)およびいくつかのRNA
ウイルスに由来し得る。例示的ウイルスは、例えば、アルファウイルス、ポックスウイル
ス、ワクチニアウイルス、ポリオウイルスなどを含む。それらは、さまざまな哺乳動物細
胞にとって魅力的な特徴をいくつかの提示する(Friedmann(1989年)Sc
ience、244:1275〜1281ページ;Ridgeway、1988年、上記
;BaichwalおよびSugden、1986年、上記、;Couparら、198
8年;Horwichら、(1990年)J.Virol.、64:642〜650ペー
ジ)。
胞への直接トランスフェクション
「自己切断型2Aペプチド」を組み込んだ、ポリシストロン性発現カセットからのすべ
ての遺伝的能力決定因子の同時発現は、「リボソームスキッピング」を起こし、単一プロ
モーターから個々の因子が同程度に発現できる(Sommerら、Stem Cells
.27:543〜549ページ、2008年;Careyら、Proc.Natl.Ac
ad.Sci.USA 106:157〜162ページ、2009年)。因子のペアを分
離する配列内リボソーム進入配列(IRES)を用いて、感染細胞はすべての能力決定因
子またはそのサブセットを発現できる。Careyら、(2009年、上記)は、IRE
S技術を用いずに、自己切断型2Aペプチドによって分離されたドキシサイクリン誘導因
子を構築した。
用して送達される。DNAトランスポゾンは、特異的「トランスポザーゼ」酵素によって
切り出され、ゲノムの至る所に再組み込みされる、転移と呼ばれる現象の遺伝要素である
。piggyBacは、TTAA配列を抱える転写DNAユニット中に優先的に挿入され
る、同義遺伝子ペイロードを抱えることができる、このようなトランスポゾンの1つであ
る。個別またはポリシストロン性のドキシサイクリン誘導構築体の誘導は、トランスポザ
ーゼを介した組み込みおよびその後の切り出しによって、ネズミおよびヒト線維芽細胞に
送達され、四倍体胚補完法による妊娠中期胚への貢献を含む、多能性のすべての特質を示
すiPS細胞を発生する(Woltjenら、Nature.458:766〜770ペ
ージ、2009年)。
する一時的Creリコンビナーゼの感染を介して切り出され得る(Kajiら、Natu
re.458:771〜775ページ、2009年)。
a)化学的取組み
一例において、能力決定因子は、ヒストンメチルトランスフェラーゼG9aの阻害剤で
あり、Oct4の代わりに、またはOct4に対する補完(例えば、Oct4の発現レベ
ルを減少させる)として使用される。G9aの化学的阻害は、例えばEnzo Life
sciences社によるBIX−01294(BIX)を用いて達成でき、ヒストン3
、リジン9のメチル化(H3K9me)により媒介されるOct4発現に対する拮抗作用
を軽減しており、いくつかの細胞において、iPS細胞の誘導のためにウイルスにより送
達されたOct4が完全に置換され得るShiら、Cell Stem Cell.2:
525〜528ページ、2008年)。
る。このようなshRNAは、Oct4プロモーターの脱メチル化およびOct4発現の
部分的再活性化をもたらすことが示されている(Maら、Stem Cells.26:
2131〜2141ページ、2008年)。
scienceによるBayk8644)は、G9aアンタゴニスト(例えば、BIX)
と組み合わせて使用され、Sox2およびcMycの代わりをする、または補完する(S
hiら、Cell Stem Cell.3:568〜574ページ、2008年)。
ら9日および数日の間(例えば5日間)連続する、体細胞周期の進行に関与するMEKの
化学的阻害(例えば、Cayman ChemicalによるPD0325901を使用
する)は、Oct4の発現が高い再プログラム化iPSコロニーの成長の強化をもたらす
(Shiら、2008年、上記)。
における内因性cMycの発現のβ−カテニン媒介誘導を刺激でき、再プログラミング効
率の劇的な改善をもたらす(Marsonら、2008年)。
リン/トレオニンホスファターゼ2A(PP2A)の強力な阻害剤である。PP2Aはc
Myc中の特定のセリン残基を脱リン酸化し、ユビキチンにより制御された迅速な分解の
ためにそれを標的とし、さらにKlf4の増加を誘発し、順に、cMyc遺伝子発現の上
方制御を誘発するcMycプロモーター中のOA応答性エレメントと結合する。EIFα
の抑制を介した、翻訳に関するOAのさらなる阻害効果は、mRNA転写産物の初期蓄積
およびその後のOA離脱に関するボーラス量の翻訳タンパク質の送達をもたらし得る。
レトロウイルス発現MEFがOct4選択可能なiPS細胞を発生するための、Klf4
と広範囲のタンパク質キナーゼ阻害剤であるケンパウロンとの置換は、生殖細胞系コンピ
テントキメラに寄与できる(Lyssiotisら、Proc.Natl.Acad.S
ci.USA 106:8912〜8917ページ、2009年)。
ある。iPS細胞に対する、ネズミ線維芽細胞のiPS再プログラミング効率が100倍
改善されることが、ヒストン脱アセチル化酵素活性の化学的阻害を介して観察されている
(Huangfuら、Nat Biotech.26:795〜797ページ、2008
年;Huangfuら、Nat Biotech.26:1269〜1275ページ、2
008年)。例えば、バルプロ酸は、遺伝的再プログラミングの再プログラミング効率を
改善する。
低分子化合物の使用と同様に、タンパク質の送達は、その可逆性のため、iPS細胞作
製のための魅力的な取組みである。
なるように、タンパク質の形質導入ドメインに接合される。タンパク質の形質導入ドメイ
ンは当分野において公知であり、例えばポリアルギニン、HIV Tat基本的ドメイン
、アンテナペディア(antannapedia)を含む(例えば、Jonesら、Br J Pha
rmacol.、145:1093〜102ページ、2005年に記載されている)。例
えば、能力決定因子に連結されたポリアルギニン標的化配列を組み込む大腸菌(E .coli
)により発現された組み換えタンパク質は、MEFをiPS細胞に転換できる。
okら、Molecular Cloning:A Laboratory Manua
l、Cold Spring Harbour Laboratory Press(1
989年)またはAusubelら(編)、Current Protocols in
Molecular Biology、Greene Pub.Associates
and Wiley−Interscience(1988年、現在までのすべての改
定を含む)に記載されている。
一例において、能力決定因子は、内因性遺伝子の発現をサイレンシングまたは減少させ
る核酸に基づく化合物である。
主に配列特異的様式で標的転写産物との結合を介して多数の生物学的過程を制御する、一
本鎖、非コーディングRNAである。miRNAは、まず一次転写産物として発現され、
次いでRNA誘導サイレンシング複合体(RISC)と複合する活性miRNAを放出す
るめに切断され、翻訳の抑制を開始する。0日目のmiR−294のトランスフェクショ
ンおよび6日目のレトロウイルス感染後、cMycを75%の効率まで置換できる(Ju
dsonら、Nat.Biotech.27:459〜461ページ、2009年)。
ログラム化へ超える援助ができ、再プログラミング効率は4倍に増加する(Mikkel
senら、Nature.454:49〜55ページ、2008年)。同様に、G9aの
短鎖ヘアピンRNA(shRNA)によるノックダウン、着床後の胚におけるOct4の
脱活性化に関与するヒストンメチルトランスフェラーゼは、vivoでのOct4プロモ
ーターの脱メチル化および部分的再活性化をもたらす(上記を参照されたい)。Oct4
/Sox2/Klf4感染と呼応したp53 siRNAの成人包皮線維芽細胞への添加
は、単独またはさらなる処置と組み合わせて、効率を増加する(Zhaoら、Cell
Stem Cell.3:475〜479ページ、2008年)。
ンチセンスポリヌクレオチド、リボザイム、PNA、干渉RNA、siRNA、短鎖ヘア
ピンRNA、マイクロRNAを含む。
「アンチセンスポリヌクレオチド」という用語は、任意の実施形態において本明細書に
記載のポリペプチドをコードする特定のmRNA分子の少なくとも一部に相補的であり、
mRNA翻訳などの転写後事象に干渉できるDNAもしくはRNAまたはそれらの組み合
わせを意味すると解釈されるべきである。アンチセンス法の使用は当分野において公知で
ある(例えば、HartmannおよびEndres、Manual of Antis
ense Methodology、Kluwer(1999年)を参照されたい)。
ハイブリダイズするであろう。アンチセンスポリヌクレオチドは構造遺伝子に一致する配
列または遺伝子発現またはスプライシングの調節をもたらす配列を含む。例えば、アンチ
センスポリヌクレオチドは、本発明の遺伝子の標的コーディング領域または5’非翻訳領
域(UTR)もしくは3’−UTRまたはこれらの組み合わせに一致し得る。アンチセン
スポリヌクレオチドは、イントロン配列と一部相補的であり得、これは転写の間または転
写後、好ましくは標的遺伝子のエクソン配列だけにスプライシングされ得る。アンチセン
ス配列の長さは、標的核酸またはそれをコードする構造遺伝子の、少なくとも19の連続
するヌクレオチド、好ましくは少なくとも50ヌクレオチドおよびより好ましくは少なく
とも100、200、500または1000ヌクレオチドでなくてはならない。全遺伝子
転写産物に相補的な完全長配列を使用できる。この長さは、最も好ましくは100〜20
00ヌクレオチドである。標的転写産物に対するアンチセンス配列の同一性の程度は、少
なくとも90%およびより好ましくは95〜100%でなくてはならない。
「触媒ポリヌクレオチド/核酸」という用語は、異なる基質を特異的に認識し、この基
質の化学的改変を触媒するDNA分子もしくはDNA含有分子(「デオキシリボザイム」
または「DNAザイム」としても当分野において公知である)またはRNAもしくはRN
A含有分子(「リボザイム」または「RNAザイム」としても公知である)を指す。触媒
核酸中の核酸塩基は、A、C、G、T(RNAに関してはU)の塩基であり得る。
チセンス配列(本明細書において「触媒ドメイン」とも称される)を含有する。本発明に
特に有用であるリボザイムの型は、ハンマーヘッドリボザイムおよびヘアピンリボザイム
である。
RNA干渉(RNAi)は、特定のタンパク質の作製を特異的に阻害するために有用で
ある。理論に縛られることを好むわけではないが、Waterhouseら(1998年
)は、dsRNA(二本鎖RNA)がタンパク質作製を減少させるために使用され得る機
序のためのモデルを提供している。この技術は、対象となる遺伝子のmRNAと本質的に
同一である配列またはその一部を含有するdsRNA分子の存在に頼る。好都合なことに
、dsRNAは組み換えベクターまたは宿主細胞において単一プロモーターから作製され
得、センス配列およびアンチセンス配列は、センス配列とアンチセンス配列とをハイブリ
ダイズできる関連のない配列に隣接し、ループ構造を形成する関連のない配列とともにd
sRNA分子を形成する。本発明にとって適切なdsRNA分子の設計および作製は、当
業者の能力の十分範囲内であり、特にWO99/32619、WO99/53050、W
O99/49029およびWO01/34815を考慮されたい。
19の連続するヌクレオチド、好ましくは少なくとも30または50ヌクレオチドおよび
より好ましくは少なくとも100、200、500または1000ヌクレオチドでなくて
はならない。全遺伝子転写産物に一致する完全長配列を使用できる。この長さは、最も好
ましくは100〜2000ヌクレオチドである。標的転写産物に対するセンス配列および
アンチセンス配列の同一性の程度は、少なくとも85%、好ましくは少なくとも90%お
よびより好ましくは95〜100%でなくてはならない。
連続するヌクレオチドと同一であるヌクレオチド配列を含む。好ましくは、siRNA配
列ジヌクレオチドAAで始まり、約30〜70%(好ましくは30〜60%、より好まし
くは40〜60%およびより好ましくは約45%〜55%)のGC含有量を含み、例えば
標準的BLAST検索によって決定して、導入される哺乳動物ゲノム中の標的以外の任意
のヌクレオチド配列に対して高い割合の同一性を有さない。
多能性細胞および/または再プログラム化細胞および/または再プログラミングを受け
た細胞は、多能性細胞の成長を支えるのに使用される任意の培地において培養され得る。
典型的な培養培地は、限定するものではないが、TeSR(商標)(StemCell
Technologies, Inc.; Vancouver、Canada)、mT
eSR(商標)(StemCell Technologies,Inc.)およびSt
emLine(登録商標)無血清培地(Sigma;St.Louis、MO)などの合
成培地、ならびにマウス胎児線維芽細胞(MEF)馴化培地などの馴化培地を含む。さら
なる培地は、塩基性培地、例えば、KoSR(Invitrogen Corporat
ion)を補充したDMEMまたはDMEM−F12を含む。または、Silvaら、P
LOS Biology、6:e253、2008年は、例えば、MAPKシグナル伝達
およびグリコーゲン合成酵素キナーゼ−3のシグナル伝達の阻害剤ならびに白血病阻止因
子(LIF)を含む、再プログラム化細胞の作製および未分化状態の再プログラム化細胞
の維持に有用な培地を記載している。本明細書において使用する場合、「規定培地」は、
単に生物化学的構成成分だけからなる生物化学的に規定された製剤を指す。規定培地はま
た、既知の化学組成を有する構成成分だけを含み得る。規定培地は、既知の供給源に由来
する構成成分をさらに含み得る。本明細書において使用する場合、「馴化培地」は、この
培地において培養された細胞由来の可溶性因子をさらに補充した成長培地を指す。または
、細胞は培養培地においてMEF上で維持されてもよい。
。細胞培養条件は、本発明の細胞のために大幅に変動し得るが、いくつかの実施形態にお
いて、細胞は、細胞成長に適切な、例えば空気中に5%O2、10%CO2、85%N2
を含む、または10%CO2を含む環境において維持される。
atrigel(商標)、ラミニン、コラーゲン、Culturex(登録商標)などの
上または内部で培養される。他の実施形態において、細胞は、細胞外マトリックスの存在
下で培養され得る。このようなマトリックスの存在下で細胞を増殖させる適切な手順は、
例えば米国特許第7,297,539号に記載されている。
下記の方法は、例えば、本明細書に記載のマーカーまたは当分野において公知のマーカ
ーを検出することによる、Stro−1+細胞および/または再プログラム化細胞/多能
性細胞の単離または濃縮にとって有用である。
任意の磁気を利用する細胞分取法、例えばDynabeads(登録商標)である。従来
のMACS手順は、Miltenyiら(Cytometry11:231〜238ペー
ジ、1990年)に記載されている。この手順において、細胞は抗体または細胞表面マー
カーもしくはタンパク質と結合する他の化合物に結合された磁気ビーズで標識され、細胞
は、常磁性分離カラムを通される、または磁場の別の形態に曝露される。分離カラムは強
力な磁石内に配置され、その結果、カラム内に磁場が発生する。磁気的に標識された細胞
は、カラム内に捕捉され、通過しない。捕捉された細胞は、その後カラムから溶出される
。
を発現する細胞を分離するためのMACSを使用して濃縮され得る。試料はタンパク質と
結合する免疫磁性ビーズと一緒にインキュベートされる。インキュベーション後、試料は
洗浄され、再懸濁され、磁場を通過させられ、免疫磁性ビーズに結合された細胞が取り除
かれ、ビーズに結合された細胞が回収される。これらの技術は陰性選択、例えば、望まし
くないマーカー、すなわち望ましくない細胞を発現する細胞の除去に同様に適用できる。
このような方法は、細胞集団を、望ましくない細胞型(複数可)において検出可能なレベ
ルで発現された細胞表面マーカーと結合する化合物で標識された磁性粒子と接触させるス
テップを含む。インキュベーション後、試料は洗浄され、再懸濁され、磁場を通過させら
れ、免疫磁性ビーズに結合された細胞が取り除かれる。ビーズに結合された細胞が回収さ
れる。望ましくない細胞型(複数可)を失った残存細胞が、その後回収される。
表面に固定され、細胞集団をそれと接触させる。未結合細胞を除去するための洗浄後、こ
の化合物と結合した細胞は回収、例えば溶出され得、その結果化合物が結合するタンパク
質を発現する細胞が単離または濃縮される。または所望であれば、化合物と結合しない細
胞を回収できる。
たは濃縮される。FACSは、粒子の蛍光特性に基づく、細胞を含む粒子を分離するため
の公知の方法であり、例えばKamarch,Methods Enzymol,151
:150〜165ページ、1987年に記載されている。一般的に、この方法は、細胞集
団を、1種または複数のタンパク質または細胞表面マーカーと結合できる化合物と接触さ
せるステップを含み、異なるマーカーと結合する化合物は、さまざまな蛍光部分、例えば
、蛍光色素分子を用いて標識される。細胞は、液体の狭く、速い流れの中心に同調される
。この流れは、それらの直径に関連して細胞間を分離するように構成される。振動機序は
、細胞の流れを引き起こし、個別の液滴に分ける。この系は、液滴中に複数の細胞が存在
する可能性が低いように調節される。流れが液滴に分かれる直前に、流れは、個々の細胞
の対象となる蛍光特徴、例えば、標識化合物が結合されているかどうかが測定される蛍光
測定ステーションを通過する。帯電リングは、流れが液滴に分かれるポイントに配置され
る。蛍光強度測定の直前に基づく電荷はリング上に配置され、液滴は流れから分離するの
で、対立する電荷は液滴上に捕捉されている。帯電した液滴は、その後液滴を迂回させる
静電偏向方式を介して、それらの電荷に基づいて容器に落とされる、例えば、標識化合物
が細胞に結合されている場合1つの容器に、そうでない場合は別の容器に落とされる。い
くつかの系において、電荷は、流れに直接印加され、分かれる液滴は流れと同じ符号の電
荷を保有する。この流れは、その後、液滴の分離後中性に戻る。
本発明の再プログラム化細胞または多能性細胞は、さまざまな商業的および治療的に重
要な組織型の集団を調製するために使用できる。一般に、このことは、細胞を所望の数に
増殖させることによって達成される。その後、任意のさまざまな分化戦略に従って分化が
引き起こされる。例えば、神経系列の細胞の高度に濃縮された集団は、細胞を、1種もし
くは複数のニュートロフィン(ニュートロフィン3または脳由来の神経栄養因子など)、
1種もしくは複数のマイトジェン(上皮成長因子、bFGF、PDGF、IGF1および
エリスロポエチンなど)または1種もしくは複数のビタミン(レチノイン酸、アスコルビ
ン酸など)を含有する培養培地に変えることによって作製できる。または、多分化能神経
幹細胞は、胚様体段階を介して作製でき、bFGFを含有する化学的規定培地において維
持できる。培養された細胞は、場合により、それらが神経前駆体細胞マーカー、例えば、
ネスチン、Musashi、ビメンチン、A2B5、nurr1またはNCAMを発現す
るかどうかに基づいて分離される。このような方法を使用して、(成熟ニューロンを含む
)神経細胞および(星状細胞および乏突起膠細胞を含む)グリア細胞の両方を作製する能
力を有する神経前駆細胞/幹細胞を得ることができる。または、分化細胞集団を形成する
能力を有する複製ニューロン前駆体を得ることができる。
セチラーゼ阻害剤の存在下で幹細胞を培養することによって再プログラム化細胞または多
能性細胞から分化され得る。培養された細胞は、場合により、EGF、インスリンまたは
FGFなどの肝細胞成熟因子と一緒に同時にまたは連続して培養される。
生的周期的収縮活性を有する細胞を作製するために使用され得る。この方法における分化
は、(5−アザ−デオキシ−シチジンなどの)DNAのメチル化をもたらすヌクレオチド
類似体、成長因子および骨形態形成タンパク質によって容易になる。細胞は、密度に基づ
く細胞分離によってさらに濃縮され得、クレアチン、カルニチンおよびタウリンを含有す
る培地において維持され得る。
F−ベータ受容体に対するリガンド、またはヒトビタミンD受容体に対するリガンドを含
有する培地において、間葉細胞または軟骨形成細胞への分化を対象とし得る。培地は、デ
キサメタゾン、アスコルビン酸−2−リン酸ならびにカルシウムおよびリン酸塩の供給源
をさらに含み得る。好ましい実施形態において、誘導細胞は、骨芽細胞系列の細胞の表現
型特徴を有する。
は、それを必要とするヒト患者において組織再構成または再生のために使用することもで
きる。この細胞は、それを意図される組織部位に接合し、機能的に欠損した領域を再構成
または再生できる様式で投与される。例えば、神経前駆体細胞は、治療される疾患に従っ
て、中枢神経系の実質部位または髄腔内部位に直接移植できる。神経細胞移植の有効性は
、McDonaldら、((1999年)Nat.Med.、5巻:1410ページ)お
よびKimら、((2002年)Nature、418巻:50ページ)に記載されるよ
うに、激しく損傷した脊髄に関してラットモデルにおいて評価され得る。移植の成功によ
り、2〜5週間後に病変に存在する、星状細胞、乏突起膠細胞および/またはニューロン
に分化し病変の終わりから脊髄に沿って遊走する、移植由来細胞および歩き方、協調およ
び体重支持における改善が示される。
有用性を実証している。
療をしなければ左心室壁組織の約55%が瘢痕組織になる心臓の低温傷害(cryoin
jury)に関する動物モデルにおいて評価され得る(Liら、(1996年)、Ann
.Thorac.Surg.、62巻:654ページ;Sakaiら、(1999年)、
Ann.Thorac.Surg.、8巻:2074ページ;Sakaiら、(1999
年)、J.Thorac.Cardiovasc.Surg.、118巻:715ページ
)。治療の成功により、瘢痕範囲が減少し、瘢痕の拡大が制限され、収縮、拡張および発
生圧力によって決定される心機能が改善されると思われる(Kehatら、(2004年
))。心外傷は、例えば、左下行前動脈の遠位部において塞栓コイルを使用しても(Wa
tanabeら、(1998年)、Cell Transplant.、7巻:239ペ
ージ)、または左冠動脈前下行枝の結紮(Minら、(2002年)、J.Appl.P
hysiol.、92巻:288ページ)によってもまたモデル化できる。治療の有効性
は、組織構造および心機能により評価し得る。本発明に具体化される心筋細胞調製品は、
心筋の再生のための療法および心機能不足の治療に使用され得る。
細胞および肝細胞前駆体を投与することによって回復できる。これらの分化細胞は、肝損
傷の修復能力に関して動物モデルにおいて評価され得る。このような一例は、D−ガラク
トサミンの腹腔内注射によって引き起こされる損傷である(Dabevaら、(1993
年)、Am.J.Pathol.、143巻:1606ページ)。治療の有効性は、肝細
胞マーカーに関する免疫組織化学的染色、細管構造が成長組織において形成されるかどう
かの顕微鏡的決定および肝臓特異的タンパク質の合成を回復するための治療能力によって
決定され得る。肝細胞は、直接投与によって、または例えば劇症肝不全の後に対象の肝組
織がそれ自体を再生する間に一時的に肝機能を提供する生物補助(bioassist)デバイス
の一部として、療法に使用され得る。
本発明の一例において、再プログラム化細胞または多能性細胞および/またはそれから
分化した細胞は、組成物の形態で投与され、またはこのような組成物に製剤化される。好
ましくは、このような組成物は、薬学的に許容される担体および/または賦形剤を含む。
活性を容易にするために当分野において従来から使用される組成物を指す(例えば、Re
mington’s Pharmaceutical Sciences、16版、Ma
c Publishing Company(1980年を参照されたい)。担体は、活
性化合物の任意の望ましくない副作用を減少することもできる。適切な担体は安定である
、例えば担体中の他の原料と反応できない。一例において、担体は、治療に用いた投与量
および濃度で、レシピエントにおいて有意な局所的または全身性の有害な影響を起こさな
い。
食塩水、水性デキストロース、ラクトース、リンガー液、緩衝化溶液、ヒアルロン酸およ
びグリコールが、(等張の場合)特に溶液のために好ましい液体担体である。適切な医薬
担体および賦形剤は、デンプン、セルロース、ブドウ糖、乳糖、ショ糖、ゼラチン、麦芽
、米、小麦粉、白亜、シリカゲル、ステアリン酸マグネシウム、ステアリン酸ナトリウム
、グリセロールモノステアレート(glycerol monostearate)、塩化ナトリウム、グリセ
ロール、プロピレングリコール、水、エタノールなどを含む。
る。好ましくは、このような培地組成物は投与された対象において任意の有害な影響を誘
導しない。
しくは有利な影響を発揮する細胞の能力に有害に影響を及ぼさない。
を適切なpHで維持し、その結果生物活性を発揮する、例えば、担体または賦形剤はリン
酸緩衝生理食塩水(PBS)である。PBSは細胞および因子との相互作用が最小であり
、細胞および因子の迅速な放出を可能にし、このような場合本発明の組成物は、血流また
は組織または組織周囲の領域もしくは組織に隣接する領域に、例えば、注射により直接適
用するための液体として作製できるので、PBSは魅力的な担体および賦形剤を代表する
。
エント適合性であり、レシピエントに対して害がない生成物に分解する足場内に組み込む
、または埋め込むこともできる。これらの足場は、レシピエント対象に移植された細胞に
支持および保護を提供する。天然および/または合成の生分解性足場はこのような足場の
例である。
するものではないが、生物学的に分解可能な足場である。天然の生分解性足場は、コラー
ゲン、フィブロネクチンおよびラミニンの足場を含む。細胞移植の足場に適切な合成材料
は、大規模な細胞成長および細胞機能を支持できなければならない。このような足場は、
さらに再吸収され得る。適切な足場は、例えば、Vacantiら、J.Ped.Sur
g.23:3〜9ページ 1988年;Cimaら、Biotechnol.Bioen
g.38:145ページ1991年;Vacantiら、Plast.Reconstr
.Surg.88:753〜9ページ 1991年により記載されたポリグリコール酸足
場またはポリ酸無水物、ポリオルトエステルおよびポリ乳酸などの合成ポリマーを含む。
の)ゲル足場において投与され得る。
発明の組成物と併せて投与され得る細胞は、限定するものではないが、他の多分化能細胞
もしくは多能性細胞または幹細胞または骨髄細胞を含む。異なる型の細胞は、投与直前ま
たは少し前に本発明の組成物と混合され得る、または投与前の期間一緒に共培養され得る
。
組成物は、約1×105の再プログラム化細胞または多能性細胞および/またはそれから
分化した細胞/kgから約1×107の再プログラム化細胞または多能性細胞および/ま
たはそれから分化した細胞/kg、あるいは、約1×106再プログラム化細胞または多
能性細胞および/またはそれから分化した細胞/kgから約5×106の再プログラム化
細胞または多能性細胞および/またはそれから分化した細胞/kgを含む。投与される細
胞の正確な量は、患者の年齢、体重および性別を含むさまざまな因子および治療される障
害の程度および重症度に依存する。
胞によって分泌される因子は循環に進入できるチャンバー内に含有される。この様式にお
いて、細胞が、対象の循環内に因子を分泌可能にすることによって可溶性因子は対象に投
与され得る。このようなチャンバーは、可溶性因子の局所レベルを増加するために対象中
の部位に同様に埋め込まれ得る。
者を免疫抑制する必要はなく、または望ましいとも思われない。したがって、同種間また
は異種間の移植であっても、再プログラム化細胞または多能性細胞および/またはそれか
ら分化した細胞は、ある場合においては容認され得る。
望ましいまたは適切であると思われる。このことは、免疫抑制剤の全身性または局所的使
用を介して達成され得る、または細胞をカプセル化デバイスにおいて送達することによっ
て達成され得る。細胞は、細胞および治療因子によって必要とされる栄養および酸素に対
して浸透性であるカプセルにカプセル化され得るが、細胞は、免疫液性の因子および細胞
に対して未だ不浸透性である。好ましくは、カプセルの材料は低アレルギー性であり、標
的組織中に容易かつ安定に位置し、埋め込み構造に追加の保護を提供する。移植細胞に対
する免疫反応を減少させる、または除去するこれらおよび他の手段は、当分野において公
知である。別の方法として、細胞は、その免疫原性を減少するように遺伝子改変され得る
。
本発明は、Stro−1+多分化能細胞および/またはその子孫細胞の再プログラミン
グを誘導または強化する化合物を特定または単離する方法をさらに提供し、前記方法は、
Stro−1+多分化能細胞および/またはその子孫細胞と化合物とを、一時の間、およ
び化合物が再プログラミングを誘導または強化する場合に再プログラミングが起こるため
に十分な条件下で接触させるステップ、および細胞が再プログラム化されるかどうかを決
定するステップを含む。
(i)Stro−1+多分化能細胞および/またはその子孫細胞が濃縮された集団と化
合物とを、一時の間、および化合物が再プログラミングを誘導または強化する場合に再プ
ログラミングが起こるために十分な条件下で接触させ、再プログラム化細胞の数を決定す
るステップ;ならびに
(ii)化合物と接触させられたことのないStro−1+多分化能細胞および/また
はその子孫細胞が濃縮された集団中の再プログラム化細胞の数を決定するステップ
を含み、
(ii)と比較して(i)において再プログラム化細胞の数が増加することが、化合物が
Stro−1+多分化能細胞および/またはその子孫細胞の再プログラミングを誘導また
は強化したことを示す。
の存在下で実施される。
強化する化合物を特定または単離する方法をさらに提供し、前記方法は、任意の実施形態
に従って本明細書に記載の方法を実施することによって作製された再プログラム化細胞ま
たは多能性細胞を接触させるステップ、および細胞が所望の細胞型に分化しているかどう
かを決定するステップを含む。
(i)任意の実施形態に従って本明細書に記載の方法を実施することによって作製され
た再プログラム化細胞または多能性細胞が濃縮された集団と化合物とを、一時の間、およ
び細胞の分化が起こるために十分な条件下で接触させ、所望の細胞型の細胞数を決定する
ステップ、ならびに
(ii)同じ条件であるが化合物が不在の下で、任意の実施形態に従って本明細書に記
載の方法を実施することによって作製された再プログラム化細胞または多能性細胞を培養
することによって作製された所望の細胞型の数を決定するステップ
を含み、
(ii)と比較して(i)において所望の細胞型の細胞数が増加することが、化合物が所
望の細胞型への分化を誘導または強化したことを示す。
し、本方法は、
(i)任意の実施形態に従って本明細書に記載の方法を実施し、病態を患う対象から多
能性細胞またはその集団を作製するステップ、
(ii)細胞または集団を被験化合物と接触させ、病態の1種または複数の症状に関す
るその効果を決定するステップ
を含み、
病態の症状を改善または緩和する化合物が病態の治療に有用である。
(a)多能性細胞またはその集団を、病態の影響を受ける細胞に分化させるステップ、
および
(b)(a)細胞と被験化合物を接触させ、病態の1種または複数の症状に関するその
効果を決定するステップ
を含み、病態の症状を改善または緩和する化合物が病態の治療に有用である。
有用ではなく、さらに、対象が既存の治療用/予防用化合物を用いた治療に応答する可能
性があるかないかを特定するためにも有用である。
の症状に基づく、細胞を分化させる適切な方法を承知していると思われる。例えば、病態
は、嚢胞性線維症であり、症状は肺細胞からの分泌であるか、または神経変性病態(例え
ば、アルツハイマー病またはハンチントン病)および症状は神経変性またはプラーク/細
胞内凝集体の形成であるか、または心臓の病態および症状は心筋細胞の収縮である。
毒性の危険性が減少した治療用化合物を決定する方法をさらに企図する。例えば、本発明
は、
(i)任意の実施形態に従って本明細書に記載の方法を実施し、多能性細胞またはその
集団を作製するステップ、
(ii)多能性細胞またはその集団を、1種または複数の特定の系列の細胞および/ま
たは組織に分化させるステップ、
(iii)この細胞と被験化合物とを接触させるステップ、ならびに
(iv)細胞の生存能力および/または増殖に対する化合物の効果を決定するステップ
を含み、
細胞または細胞集団の有意な割合を殺さない、または増殖を減少させない化合物が、毒性
が減少していると考えられる方法を提供する。
おいて公知であり、ターミナル・デオキシヌクレオチジル・トランスフェラーゼ媒介ビオ
チン化UTPニック末端標識(TUNEL)アッセイ、トリパンブルー色素排除アッセイ
、MTTアッセイ、チミジン取り込みアッセイまたはBrdU取り込みアッセイを含む。
を特定および/または単離するさまざまな方法を包含することを、前述より承知している
と思われる。スクリーニングに適切な化合物は、例えば、抗体、ペプチドまたは小分子を
含む。
がって、スクリーニング方法は、
(i)場合により、化合物の構造を決定するステップ、ならびに
(ii)化合物または化合物の名称もしくは構造を、例えば文書の形態、機械による読
み込み可能な形態またはコンピューターに読み込み可能な形態などで提供するステップ
によりさらに改変される。
いままでに試験されていない化合物については、化合物の構造の決定は黙示的(implicit
)である。当業者は、スクリーニングの実施時に、化合物の名称および/または構造を承
知していると思われるからである。
製するための任意の化学的または組み換えの合成手段、または任意の人または手段により
今までに合成されている化合物の供給を含むと解釈されるべきである。これは、化合物の
単離を明らかに含む。
細書に記載のスクリーニングにより決定されたその使用に関する指示と一緒に提供される
。
(i)場合により、化合物の構造を決定するステップ、
(ii)場合により、化合物の名称もしくは構造を、例えば文書の形態、機械による読
み込み可能な形態またはコンピューターに読み込み可能な形態などで提供するステップ、
ならびに
(iii)化合物を提供するステップ
によりさらに改変され得る。
ば、本明細書に記載のスクリーニングにより決定されたその使用に関する指示と一緒に提
供される。
本発明は以下を提供する。
[1]
再プログラム化細胞を作製する方法であって、Stro−1 + 多分化能細胞および/またはその子孫細胞を、細胞を再プログラムするために十分な条件下で1種または複数の能力決定因子に曝露するステップを含む方法。
[2]
再プログラム化細胞を作製する方法であって、Stro−1 + 多分化能細胞および/またはその子孫細胞が濃縮された細胞の集団を、細胞を再プログラムするために十分な条件下で1種または複数の能力決定因子に曝露するステップを含む方法。
[3]
Stro−1 + 多分化能細胞および/もしくはその子孫細胞、またはStro−1 + 多分化能細胞および/もしくはその子孫細胞が濃縮された細胞集団が脂肪組織、歯髄組織または骨髄に由来するものである、請求項1または2に記載の方法。
[4]
再プログラム化細胞を作製するために、曝露された細胞を培養するステップをさらに含む、請求項1または2に記載の方法。
[5]
再プログラム化細胞を単離するステップをさらに含む、請求項1または2に記載の方法。
[6]
Stro−1 + 多分化能細胞および/またはその子孫細胞よりも広い分化能を有する再プログラム化細胞を得るために、曝露された細胞を培養するステップを含む、請求項1から5のいずれか一項に記載の方法。
[7]
1種または複数の能力決定因子が、Oct4、Sox2、Klf4、Nanog、Lin28、c−Myc、bFGF、SCF、TERT、SV40ラージT抗原、HPV16E6、HPV16E7、Bmil、Fbx15、Eras、ECAT15−2、Tcl1、β−カテニン、ECAT1、ESG1、Dnmt3L、ECAT8、Gdf3、Sox15、ECAT15−1、Fthl17、Sal14、Rex1、UTF1、Stella、Stat3、およびGrb2または1種もしくは複数の前記因子と同一もしくは類似の活性を有する化合物からなる群から個別にまたは集約的に選択される、請求項1から6のいずれか一項に記載の方法。
[8]
1種または複数の能力決定因子が化学物質、ペプチド、siRNA、短鎖ヘアピンRN
AまたはマイクロRNAである、請求項1から7のいずれか一項に記載の方法。
[9]
1種または複数の能力決定因子が、
(i)Oct4、
(ii)Oct4とSox2との組み合わせ、
(iii)Oct4、Sox2と、NanogおよびLin28のうちの少なくとも1つとの組み合わせ、
(iv)Oct4、Klf4およびc−Mycの組み合わせ、
(v)Oct4、Sox2およびKlf4の組み合わせ、
(vi)Oct4、Sox2、Klf4およびc−Mycの組み合わせ、
(vii)Oct4、Sox2、NanogおよびLin28の組み合わせ、
(viii)Oct4、Sox2、Klf4、c−Myc、NanogおよびLin28の組み合わせ、ならびに
(ix)(i)から(x)のうちのいずれか1つと、さらに化学物質、ペプチド、siRNA、shRNAまたはマイクロRNAとの組み合わせ
からなる群から個別にまたは集約的に選択される、請求項1から7のいずれか一項に記載の方法。
[10]
Stro−1 + 多分化能細胞および/またはその子孫細胞が、出生後の哺乳動物から得られる、請求項1から9のいずれか一項に記載の方法。
[11]
哺乳動物がヒトである、請求項10に記載の方法。
[12]
Stro−1 + 多分化能細胞および/またはその子孫細胞を1種または複数の能力決定因子に曝露するステップが、プロモーターに作動可能に連結された1種または複数の能力決定因子をコードする配列を含む核酸をStro−1 + 多分化能細胞および/またはその子孫細胞に導入するステップを含む、請求項1または2に記載の方法。
[13]
プロモーターに作動可能に連結した異なる能力決定因子をコードする配列をそれぞれが含む複数の核酸を投与するステップを含む、請求項12に記載の方法。
[14]
核酸が1種または複数のベクターの内部にある、請求項12または13に記載の方法。
[15]
ベクターがウイルスベクターである、請求項14に記載の方法。
[16]
核酸が、Stro−1 + 多分化能細胞および/またはその子孫細胞のゲノム中に組み込まれない、請求項12に記載の方法。
[17]
再プログラム化細胞が多能性である、請求項1または2に記載の方法。
[18]
再プログラム化細胞が、(i)Oct−4、Nanog、SSEA3、SSEA4、Tra−1−60およびTra−1−81からなる群から選択される細胞マーカーを発現し、(ii)多能性細胞に特徴的な形態を示し、および/または(iii)免疫不全動物に
導入されると奇形腫を形成する、請求項1または2に記載の方法。
[19]
請求項1から18のいずれか一項に記載の方法を実施することによって作製される細胞。
[20]
請求項1から18のいずれか一項に記載の方法を実施することによって作製される、多能性細胞が濃縮された細胞集団。
[21]
多能性細胞が集団の少なくとも60%を占める、請求項20に記載の細胞の濃縮集団。
[22]
請求項18から21のいずれか一項に記載の細胞または集団から分化した細胞または細胞集団。
[23]
異種プロモーターに作動可能に連結された能力決定因子をコードする核酸を含む、Stro−1 + 多分化能細胞および/またはその子孫細胞。
[24]
医薬における請求項19から23のいずれか一項に記載の細胞または集団の使用。
[25]
疾患または障害を治療または予防する方法であって、請求項19から23のいずれか一項に記載の細胞または集団を、それを必要とする対象に投与するステップを含む方法。
[26]
疾患または障害の治療または予防において有用な化合物をスクリーニングする方法であって、請求項19から23のいずれか一項に記載の細胞または集団を前記化合物に曝露するステップを含む方法。
1.1 Stro−1+多分化能前駆細胞を濃縮した集団
Stro−1+多分化能前駆細胞は、骨髄、脂肪組織および歯髄組織を含むさまざまな
組織から得られる。異なる部位に由来するStro−1+多分化能前駆細胞の再プログラ
ミング効率を比較するために、本質的にGronthosおよびZannettino
Methods Mol Biol.449:45〜57ページ(2008年)に記載の
ように、これらの組織それぞれに由来する細胞を、STRO3 mAbを使用する免疫選
択によってStro−1Bright細胞を濃縮し、その後、培養により増殖し、Pro
FreezeTM−CDM(Lonza、USA)において凍結保存する。異なる免疫選
択法を使用して、同じ部位に由来するStro−1+多分化能前駆細胞の再プログラミン
グ効率を比較するために、同じドナー由来の対を成す骨髄試料を、STRO3またはST
RO1mAbのいずれかを使用する免疫選択によってStro−1Bright細胞を濃
縮し、培養により増殖し、ProFreezeTM−CDM(Lonza、USA)にお
いて凍結保存する。すべての研究に関して、第4継代の細胞を解凍し、即時使用のために
ビヒクルにおいて構成する。
導入遺伝子発現レンチウイルスベクターを、293FT細胞系(Invitrogen
)において作製する。293Tは、形質転換された293胎児腎細胞に由来する、成長が
速く、高度に移入可能なクローンの変異体であり、より高いウイルス価に寄与するパッケ
ージングタンパク質の高レベル発現のために、ラージT抗原を含有する。常法としての維
持および増殖のために、これらの細胞を、293FT培地(DMEM/10%FBS、2
mMのL−グルタミンおよび0.1mMのMEM非必須アミノ酸)において500μg/
mlのジェネテシンの存在下で培養する。パッケージングのために、293FT細胞を、
トリプシン処理によって収集する。遠心分離によりトリプシンを除去後、これらの細胞を
、ジェネテシンを含まない293FT培地において、T75フラスコにアリコートに分け
る(15×106細胞/フラスコおよび6フラスコ/構築物)。
、細胞をアリコートに分けた直後にSuperfect(登録商標)トランスフェクショ
ン試薬(Qiagen)を用いて実施する。翌日、トランスフェクション混合物を含有す
る培養培地を、1mMのピルビン酸ナトリウム(8ml/フラスコ)を補充した新鮮な2
93FT培地に取り換える。レンチウイルス含有上清を形質導入の約48から72時間後
に収集する。293FT細胞の残渣(debris)を、4℃において15分間の遠心分離によ
って上清から除去する。レンチウイルスを濃縮するために、0.4μMの酢酸セルロース
(CA)膜(Cornington、115ml 低タンパク質結合)を通して上清をろ
過し、70mlの滅菌ボトル(Beckman、カタログ番号355622、45Tiロ
ーターだけのためのポリカーボネート)において、40℃において2.5時間超遠心分離
にかける。上清の除去後、PBS(各構築物に関して約300μl)を加え、40℃にお
いて8から14時間、または室温において2時間、遠心管を振動させることによってペレ
ットを再懸濁する。残存する細胞残渣を遠心分離により除去し、再懸濁したレンチウイル
スをアリコートに分け、−80℃において保管した。レンチウイルスは、1種または複数
の能力決定因子をコードする配列を担持する。
1種または複数の能力決定因子(例えば、Oct4;またはOct4とSox2の組み
合わせ;またはOct4、Sox2と、NanogおよびLin28のうちの少なくとも
1つとの組み合わせ;またはOct4、Klf4およびc−Mycの組み合わせ;または
Oct4、Sox2およびKlf4の組み合わせ;またはOCT4、Sox2、Klf4
およびc−Mycの組み合わせ;またはOct4、Sox2、NanogおよびLin2
8の組み合わせ;またはOct4、Sox2、Klf4、c−Myc、Nanogおよび
Lin28の組み合わせ)をコードするレンチウイルスを、最終濃度約6μg/ml(S
igma)でポリブレン担体を加えた後、細胞培養液に加える。
に培養する。必要に応じて、形質導入の3日後に薬剤選択を開始する。
トリプシン処理(0.05%のトリプシン/0.5mMのEDTA、Invitroge
n)により解離させ、2%のパラホルムアルデヒド中で室温において20分間固定する。
細胞を、40μmのメッシュを通してろ過し、FACS緩衝液(2%のFBSおよび0.
1%のアジ化ナトリウムを含有するPBS)に再懸濁する。懸濁液において成長した細胞
を、1mMのEDTAおよび1%の正常マウス血清(Sigma)を補充したFACS緩
衝液において染色した。細胞内ミエロペルオキシダーゼ(MPO)染色を、Fix &
Perm(登録商標)試薬(Caltag Laboratories;Burling
ame、CA)を使用して実施する。5×105細胞を含有する約100μlの細胞懸濁
液を、個々の標識に使用する。(必要に応じて)一次抗体および二次抗体の両方のインキ
ュベーションを室温において約30分間実施する。対照試料を、アイソタイプ一致対照抗
体を用いて染色する。洗浄後、細胞を約300〜500μlのFACS緩衝液に再懸濁し
、FACSCaliburフローサイトメーター(BDIS;San Jose、CA)
において、Cell Quest(商標)acquisitionおよび分析ソフトウェ
ア(BDIS)を使用して分析する。合計で20,000事象(event)が得られる。検
出されたマーカーは、SSEA−3、SSEA−3、SSEA−4、Tra−1−60、
Tra−1−81、CD29、Tra−1−85、CD56、CD73、CD105、C
D31またはCD34から選択される。
presence or valproic acid)で維持する。
葉の分化誘導体を生じるための発生能を有することを実証する。
2.1 材料および方法
Stro−1+多分化能細胞を濃縮した集団
Stro−1+多分化能前駆細胞が濃縮された細胞集団を、骨髄、脂肪組織および歯髄
組織から得た。異なる部位に由来するStro−1+多分化能前駆細胞の再プログラミン
グ効率を比較するために、本質的にGronthosおよびZannettino Me
thods Mol Biol.449:45〜57ページ(2008年)に記載のよう
に、これらの組織それぞれに由来する細胞を、STRO3 mAbを使用する免疫選択に
よってStro−1Bright細胞を濃縮し、その後、培養により増殖し、ProFr
eezeTM−CDM(Lonza、USA)において凍結保存した。異なる免疫選択法
を使用して、同じ部位に由来するStro−1+多分化能前駆細胞の再プログラミング効
率を比較するために、同じドナー由来の対を成す骨髄試料を、STRO3またはSTRO
1mAbのいずれかを使用する免疫選択によってStro−1Bright細胞を濃縮し
、培養により増殖し、ProFreezeTM−CDM(Lonza、USA)において
凍結保存した。すべての研究に関して、第4継代の細胞を解凍し、即時使用のためにビヒ
クルにおいて構成した。
ウイルスパッケージング細胞である、プラチナ−A(Plat−A)細胞を、Cell
Biolabs,Inc.Detroit 551から得、線維芽細胞は実験のための
陽性対照であった。
OCT4、SOX2、KLF4およびcMYCのヒトcDNAを含有する、モロニーに
基づくレトロウイルスベクター(pMX)を、Addgeneから得た。9μgの各プラ
スミドを、Fugene6(Roche)を使用して、ウイルスパッケージングPlat
−A細胞にトランスフェクトした。ウイルス含有上清を、トランスフェクトの48時間後
及び72時間後に収集し、孔径0.45μmのフィルターを通してろ過し、4μg/ml
のポリブレン(Sigma)を補充した。標的細胞を、感染24時間前に1×103から
5×103細胞/cm2の密度で播種した。4種の転写因子のレトロウイルス上清を等量
で混合し、二重感染を24時間および48時間において標的細胞に加えた。感染細胞のた
めの培養培地を、感染後4日目にhES細胞培地に変えた。細胞は培地において維持し、
培地は、3週間まで、または細胞が集密に達するまで毎日新しくした。
iPS細胞系を確立するために、感染後約3週間目にhES細胞様コロニー形態に基づ
いてiPS細胞のコロニーを取り上げた。取り上げたコロニーを、hES細胞培地におい
て新鮮な、有糸分裂的に不活性化されたMEF上で増殖した。
ヒトiPS細胞を、20%のFBS(Hyclone)、1mMのL−Glutama
x、0.1mMの非必須アミノ酸、0.1mMのβ−メルカプトエタノール、1%のIT
Sおよび10ng/mlのbFGF(すべてInvitrogenから)を補充したDM
EMにおいて培養した。ヒトiPS細胞を、培養培地で毎日再生(refresh)した。機械
的解離を、29Gの針を付けた1mlのインスリン注射器を使用して、iPS細胞コロニ
ーをより小さい細胞の塊に解体することによって実施した。感染後8から10日後に、新
鮮な有糸分裂的に不活性化したMEF上にコロニーを移した。iPSコロニーを、7から
10日ごとに継代し、その後機械的解離を実施した。
細胞のトランスフェクション効率を推定するために、上記と同じ方法を使用して、pM
Xs−GFPレトロウイルスベクターをPlat−A細胞にさらにトランスフェクトした
。GFP cDNAを含有するpMXレトロウイルスは、追加の細胞であった。GFPを
発現する細胞の数を、フローサイトメトリーによって感染48時間後に評価した。細胞を
、0.25%のトリプシン−EDTA(Invitrogen)を用いて5分間解離させ
、フローサイトメーター(MoFLO)を使用して分析した。
脂肪細胞、歯髄細胞およびMPCを、上記の「Retroviral Product
ion and iPS Cell Generation」に記載の4種の因子に感染
させた。細胞を、17日間維持し、毎日培地を変えた。次いで細胞を、トリプシンを用い
て解離させ、4%のパラホルムアルデヒドを用いて固定した。細胞を、2.5%(w/v
)のスキムミルクパウダー、PBS中2%(v/v)のヤギ血清を用いてブロックした。
細胞を、一次抗体を用いて4℃において一晩標識し、次いで、一次抗体(マウス抗Oct
4、抗Nanogまたは抗SSEA4)を用いて4℃において一晩標識し、その後、ヤギ
抗マウスAlexa488二次抗体を用いて室温において1時間標識した。試料を、上記
のFACSによって分析し、陽性に染色された細胞の数を決定した。
すべてのStro−1+多分化能前駆細胞が濃縮された集団は、それが歯髄、脂肪組織
または骨髄から供給されたかどうかにかかわらず、また、STRO−3またはSTRO−
1mAbsを用いて免疫選択されたかどうかにかかわらず、再プログラムでき、iPS細
胞系を作製できる。
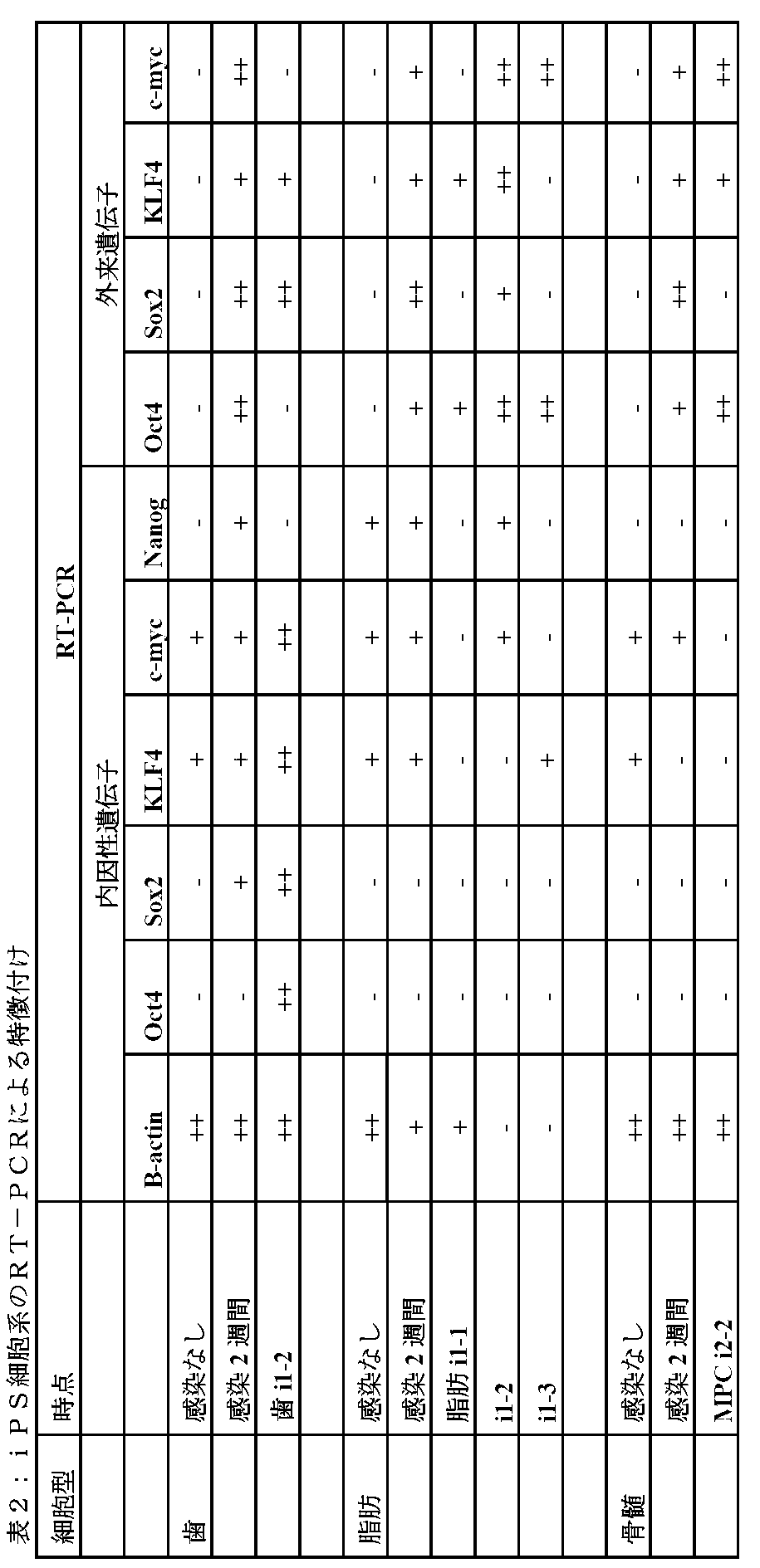
は同様であり、71.2〜98.3%の範囲であった。プレート当たりに成長するiPS
コロニーの数を測定する研究の結果は、D551胎児線維芽細胞に由来する系と比較して
、歯髄および脂肪組織を起源とするStro−1+多分化能前駆細胞が濃縮された細胞集
団に由来する系に関する有意により高い推定iPS細胞コロニーの形成を示す。さらに、
歯髄および脂肪組織を起源とするStro−1+多分化能前駆細胞が濃縮された細胞集団
に由来する系に関するiPS細胞のコロニー形成は、骨髄由来のStro−1+多分化能
前駆細胞に由来する系より有意に高かった。歯髄および脂肪組織(それぞれ56および2
2)に関する平均コロニー数(/播種された50,000細胞)は、D551線維芽細胞
(7)または骨髄(0.5)に関して観察されるより優位に高かった(p<0.05、カ
イ二乗検定)。
率の増加が、外からトランスフェクトされた遺伝子または誘導性外来遺伝子の持続発現と
関係があったかどうかを調べるために、本発明者らは、次にすべての培養されたトランス
フェクト細胞集団においてOct4およびNanogの発現を測定した。表1に示すよう
に、トランスフェクトされたD551線維芽細胞は、最も高いレベルのOct4の持続発
現(69%)を明示し、歯髄または脂肪組織を起源とするトランスフェクトされたStr
o−1+多分化能前駆細胞は中間レベルのOct4発現(それぞれ57%および56%)
を有し、トランスフェクトされた骨髄Stro−1+多分化能前駆細胞は最も低いレベル
のOct4発現(36%)を有した。
された集団は、Nanogを発現する細胞の合計数がトランスフェクトされたD551線
維芽細胞(28%)より低い(それぞれ、5%および10%)。骨髄Stro−1+多分
化能前駆細胞に関しては、対を成す同じ骨髄試料からSTRO−3またはSTRO−1m
Abを用いて免疫選択されたかどうかにかかわらず、同様のパターンのOct4およびN
anogの発現が一貫して見られる(Oct4に関してはそれぞれ30%および37%な
らびにNanogに関してはそれぞれ17%および21%)。
いiPS様コロニーと関連し得ることを示す以前に公開されたデータ(Chanら、Na
ture Biotech.27(11):1033〜1037ページ(2009年))
と併せて、歯髄または脂肪組織を起源とするトランスフェクトされたStro−1+多分
化能前駆細胞が、D551線維芽細胞より低いNanog発現を有するが、有意に高いi
PSコロニー数を有するという本発明者らの発見は、Stro−1+多分化能前駆細胞が
、能力因子への曝露後の決定的なiPSコロニー形成へ進行するためのより許容状態の細
胞型を代表することを示す。
1)トランスフェクション後の同様のOct4発現にもかかわらず、歯髄および脂肪組
織を起源とするStro−1+多分化能前駆細胞が濃縮された細胞集団は、iPS細胞コ
ロニーを、対照の線維芽細胞より、より効率的にかつより多数作製し、
2)トランスフェクション後にNanog発現の誘導が低下したにもかかわらず、歯髄
および脂肪組織を起源とするStro−1+多分化能前駆細胞が濃縮された細胞集団は、
決定的なiPS細胞コロニーを、対照の線維芽細胞より、より効率的にかつより多数作製
し、
3)歯髄および脂肪組織を起源とするStro−1+多分化能前駆細胞が濃縮された細
胞集団は、骨髄由来のStro−1+多分化能前駆細胞が濃縮された細胞集団より、iP
S細胞コロニーをより効率的に作製したので、組織の起源は、iPS細胞コロニーの形成
の効率に影響を及ぼし得る、
ことを示す。
歯髄由来のiPS細胞コロニーは、Oct4、Nanog、SSEA4、TRA1−6
0およびTRA1−81を発現することが免疫蛍光により示された。これらの細胞は、ア
ルカリホスファターゼを発現することがさらに示された。
れ、アルカリホスファターゼを発現することが示された。
遺伝子発現研究の結果を表2に提示する。
骨髄から得られたかどうかにかかわらず、すべてのStro−1+多分化能前駆細胞濃縮
細胞集団が、感染前にKLF4およびc−mycを内因性に発現した。理論および作用機
序に縛られることを好むものではないが、Stro−1+多分化能前駆細胞によるこれら
の能力因子の内因性発現は、Stro−1+多分化能前駆細胞を使用したiPS作製の、
線維芽細胞より高い効率を一部説明できる。しかし、さらなる因子は、歯髄又は脂肪組織
を起源とするStro−1+多分化能前駆細胞において観察されたiPS系を作製する効
率が、骨髄を起源とするものより高いことを説明できる。
ycおよびKlf4を発現した。外因性Oct4およびc−mycは、第3継代および第
8継代で発現停止されたが、Sox2およびKlf4は発現されたままである。
mycは発現停止されないままであった。
におい発現停止された唯一の外因性遺伝子はSox2であった。
Claims (12)
- 再プログラム化細胞を作製する方法であって、Stro−1bright間葉前駆体細胞および/またはその培養増殖により得られる多分化能子孫細胞が濃縮されたヒト細胞集団を、細胞を再プログラムするために十分な条件下で1種または複数の能力決定因子に曝露するステップを含み、前記細胞集団が脂肪組織または歯髄組織に由来する、方法。
- 再プログラム化細胞を単離するステップをさらに含む、請求項1に記載の方法。
- 1種または複数の能力決定因子が、Oct4、Sox2、Klf4、Nanog、Lin28、c−Myc、bFGF、SCF、TERT、SV40ラージT抗原、HPV16E6、HPV16E7、Bmil、Fbx15、Eras、ECAT15−2、Tcl1、β−カテニン、ECAT1、ESG1、Dnmt3L、ECAT8、Gdf3、Sox15、ECAT15−1、Fthl17、Sal14、Rex1、UTF1、Stella、Stat3、およびGrb2または1種もしくは複数の前記因子と同一もしくは類似の活性を有する化合物からなる群から個別にまたは集約的に選択される、請求項1または2に記載の方法。
- 1種または複数の能力決定因子が化学物質、ペプチド、siRNA、短鎖ヘアピンRNAまたはマイクロRNAを含む、請求項1から3のいずれか一項に記載の方法。
- 1種または複数の能力決定因子が、
(i)Oct4、
(ii)Oct4とSox2との組み合わせ、
(iii)Oct4、Sox2と、NanogおよびLin28のうちの少なくとも1つとの組み合わせ、
(iv)Oct4、Klf4およびc−Mycの組み合わせ、
(v)Oct4、Sox2およびKlf4の組み合わせ、
(vi)Oct4、Sox2、Klf4およびc−Mycの組み合わせ、
(vii)Oct4、Sox2、NanogおよびLin28の組み合わせ、
(viii)Oct4、Sox2、Klf4、c−Myc、NanogおよびLin28の組み合わせ、ならびに
(ix)(i)から(x)のうちのいずれか1つと、さらに化学物質、ペプチド、siRNA、shRNAまたはマイクロRNAとの組み合わせからなる群から個別にまたは集約的に選択される、請求項1から4のいずれか一項に記載の方法。 - 前記細胞集団が、出生後のヒトから得られる、請求項1から5のいずれか一項に記載の方法。
- 前記細胞集団を1種または複数の能力決定因子に曝露するステップが、プロモーターに作動可能に連結された1種または複数の能力決定因子をコードする配列を含む核酸をStro−1bright間葉前駆体細胞および/またはその多能性子孫細胞に導入するステップを含む、請求項1に記載の方法。
- プロモーターに作動可能に連結した異なる能力決定因子をコードする配列をそれぞれが含む複数の核酸を導入するステップを含む、請求項7に記載の方法。
- 核酸が1種または複数のベクターの内部にある、請求項7または8に記載の方法。
- ベクターがウイルスベクターである、請求項9に記載の方法。
- 核酸が、Stro−1bright間葉前駆体細胞および/またはその多能性子孫細胞のゲノム中に組み込まれない、請求項7に記載の方法。
- 多能性細胞が集団の少なくとも20%を占める、請求項1〜11のいずれか一項に記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US21064809P | 2009-03-20 | 2009-03-20 | |
| US61/210,648 | 2009-03-20 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012500009A Division JP6342115B2 (ja) | 2009-03-20 | 2010-03-22 | 再プログラム化多能性細胞の作製 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017235872A Division JP6708617B2 (ja) | 2009-03-20 | 2017-12-08 | 再プログラム化多能性細胞の作製 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2016073274A JP2016073274A (ja) | 2016-05-12 |
| JP6603529B2 true JP6603529B2 (ja) | 2019-11-06 |
Family
ID=42739058
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012500009A Active JP6342115B2 (ja) | 2009-03-20 | 2010-03-22 | 再プログラム化多能性細胞の作製 |
| JP2015188541A Active JP6603529B2 (ja) | 2009-03-20 | 2015-09-25 | 再プログラム化多能性細胞の作製 |
| JP2017235872A Active JP6708617B2 (ja) | 2009-03-20 | 2017-12-08 | 再プログラム化多能性細胞の作製 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012500009A Active JP6342115B2 (ja) | 2009-03-20 | 2010-03-22 | 再プログラム化多能性細胞の作製 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017235872A Active JP6708617B2 (ja) | 2009-03-20 | 2017-12-08 | 再プログラム化多能性細胞の作製 |
Country Status (10)
| Country | Link |
|---|---|
| US (3) | US9487756B2 (ja) |
| EP (2) | EP3293257B1 (ja) |
| JP (3) | JP6342115B2 (ja) |
| KR (1) | KR101764437B1 (ja) |
| CN (1) | CN102405280B (ja) |
| AU (1) | AU2010225469B2 (ja) |
| CA (1) | CA2755870C (ja) |
| ES (1) | ES2900277T3 (ja) |
| SG (1) | SG174426A1 (ja) |
| WO (1) | WO2010105311A1 (ja) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103562376B (zh) * | 2011-04-08 | 2017-12-29 | 国家医疗保健研究所 | 复壮细胞的方法 |
| WO2013147082A1 (ja) * | 2012-03-28 | 2013-10-03 | 株式会社クオリーメン | 不死化幹細胞及びその産生物を有効成分とする医薬組成物並びに医薬製剤 |
| EP2912164A4 (en) * | 2012-11-02 | 2016-08-31 | Lonza Walkersville | MICRO-RNAS AND CELL PROGRAMMING |
| KR101498156B1 (ko) * | 2013-04-16 | 2015-03-04 | 한국생명공학연구원 | 기형종 형성을 통한 유도만능줄기세포 유도용 체세포의 무한 재생산 방법 |
| WO2016076929A1 (en) * | 2014-11-13 | 2016-05-19 | University Of Iowa Research Foundation | Methods to generate epithelial cells |
| FR3059009B1 (fr) * | 2016-11-23 | 2018-12-07 | Universite de Bordeaux | Microcompartiment cellulaire et procedes de preparation |
| CN107151656A (zh) * | 2017-05-10 | 2017-09-12 | 中国科学院广州生物医药与健康研究院 | 一种提高体细胞向神经细胞转分化效率的方法 |
| CN108441517A (zh) * | 2018-03-28 | 2018-08-24 | 长春博邦企业管理咨询有限公司 | 一种人诱导多能干细胞的制备方法 |
| FR3081168B1 (fr) | 2018-05-21 | 2022-03-11 | Univ Bordeaux | Systeme de culture cellulaire en bioreacteur |
| FR3122883A1 (fr) | 2021-05-11 | 2022-11-18 | Treefrog Therapeutics | Microcompartiments cellulaires comprenant des cellules dont l’intégrité génomique est maintenue après amplification et procédé de préparation |
| FR3124193B3 (fr) | 2021-06-16 | 2023-09-22 | Treefrog Therapeutics | Microcompartiments cellulaires de grande taille comprenant plusieurs cystes |
| WO2022263601A1 (fr) | 2021-06-16 | 2022-12-22 | Treefrog Therapeutics | Microcompartiments cellulaires de grande taille comprenant plusieurs cystes |
| CN116333969A (zh) * | 2023-02-01 | 2023-06-27 | 宁波荣安生物药业有限公司 | 一种狂犬病毒用Vero细胞培养方法 |
Family Cites Families (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4797368A (en) | 1985-03-15 | 1989-01-10 | The United States Of America As Represented By The Department Of Health And Human Services | Adeno-associated virus as eukaryotic expression vector |
| US4980286A (en) | 1985-07-05 | 1990-12-25 | Whitehead Institute For Biomedical Research | In vivo introduction and expression of foreign genetic material in epithelial cells |
| JP2703893B2 (ja) | 1985-07-05 | 1998-01-26 | ホワイトヘッド・インスティテュ−ト・フォ−・バイオメディカル・リサ−チ | 外来遺伝子物質を発現する上皮細胞 |
| US5139941A (en) | 1985-10-31 | 1992-08-18 | University Of Florida Research Foundation, Inc. | AAV transduction vectors |
| EP0633318A1 (en) | 1987-09-11 | 1995-01-11 | Whitehead Institute For Biomedical Research | Transduced fibroblasts and uses therefor |
| WO1989005345A1 (en) | 1987-12-11 | 1989-06-15 | Whitehead Institute For Biomedical Research | Genetic modification of endothelial cells |
| EP0400047B1 (en) | 1988-02-05 | 1997-04-23 | Whitehead Institute For Biomedical Research | Modified hepatocytes and uses therefor |
| US5585362A (en) | 1989-08-22 | 1996-12-17 | The Regents Of The University Of Michigan | Adenovirus vectors for gene therapy |
| US5436146A (en) | 1989-09-07 | 1995-07-25 | The Trustees Of Princeton University | Helper-free stocks of recombinant adeno-associated virus vectors |
| US5173414A (en) | 1990-10-30 | 1992-12-22 | Applied Immune Sciences, Inc. | Production of recombinant adeno-associated virus vectors |
| EP0556345B2 (en) | 1990-10-31 | 2005-10-12 | Cell Genesys, Inc. | Retroviral vectors useful for gene therapy |
| US5252479A (en) | 1991-11-08 | 1993-10-12 | Research Corporation Technologies, Inc. | Safe vector for gene therapy |
| US5587308A (en) | 1992-06-02 | 1996-12-24 | The United States Of America As Represented By The Department Of Health & Human Services | Modified adeno-associated virus vector capable of expression from a novel promoter |
| JPH07507689A (ja) | 1992-06-08 | 1995-08-31 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 特定組織のターゲティング方法及び組成物 |
| ATE245703T1 (de) | 1992-09-22 | 2003-08-15 | Biofocus Discovery Ltd | Rekombinante viren, die an ihrer äusseren oberfläche ein nichtvirales polypeptid präsentieren |
| ATE263252T1 (de) | 1992-11-09 | 2004-04-15 | Us Health | Erzielbare vektorenpartikel |
| IT1264669B1 (it) | 1993-07-05 | 1996-10-04 | Comer Spa | Reattore per la rimozione di impurita' da un liquido |
| US5631236A (en) | 1993-08-26 | 1997-05-20 | Baylor College Of Medicine | Gene therapy for solid tumors, using a DNA sequence encoding HSV-Tk or VZV-Tk |
| US5698443A (en) | 1995-06-27 | 1997-12-16 | Calydon, Inc. | Tissue specific viral vectors |
| CA2203809C (en) | 1994-10-28 | 2008-06-03 | James M. Wilson | Recombinant adenovirus and methods of use thereof |
| US7410773B2 (en) * | 1995-02-02 | 2008-08-12 | Ghazi Jaswinder Dhoot | Method of preparing an undifferentiated cell |
| PT811073E (pt) | 1995-02-24 | 2007-03-30 | Univ Pennsylvania | Utilização de moduladores imunitários para preparar medicamentos que inibem as respostas imunitárias contra vírus recombinantes co-administrados |
| WO1996041865A1 (en) | 1995-06-07 | 1996-12-27 | Ariad Gene Therapeutics, Inc. | Rapamcycin-based regulation of biological events |
| US5622856A (en) | 1995-08-03 | 1997-04-22 | Avigen | High efficiency helper system for AAV vector production |
| US6506559B1 (en) | 1997-12-23 | 2003-01-14 | Carnegie Institute Of Washington | Genetic inhibition by double-stranded RNA |
| AU743316C (en) | 1998-03-20 | 2005-09-15 | Commonwealth Scientific And Industrial Research Organisation | Control of gene expression |
| CA2325344C (en) | 1998-04-08 | 2017-12-05 | Commonwealth Scientific And Industrial Research Organisation | Methods and means for obtaining modified phenotypes |
| GB9816781D0 (en) | 1998-07-31 | 1998-09-30 | Univ London | Herpes virus vectors for dendritic cells |
| US7410798B2 (en) | 2001-01-10 | 2008-08-12 | Geron Corporation | Culture system for rapid expansion of human embryonic stem cells |
| AUPQ147799A0 (en) | 1999-07-07 | 1999-07-29 | Medvet Science Pty. Ltd. | Mesenchymal precursor cell |
| US7670628B2 (en) * | 1999-07-07 | 2010-03-02 | Angioblast Systems, Inc. | Mesenchymal precursor cell |
| AU2003901668A0 (en) | 2003-03-28 | 2003-05-01 | Medvet Science Pty. Ltd. | Non-haemopoietic precursor cells |
| DE19939781C2 (de) | 1999-08-21 | 2003-06-18 | Schott Glas | Skulltiegel für das Erschmelzen oder das Läutern von anorganischen Substanzen, insbesondere von Gläsern und Glaskeramiken |
| US6326193B1 (en) | 1999-11-05 | 2001-12-04 | Cambria Biosciences, Llc | Insect control agent |
| US20030021771A1 (en) * | 2001-07-06 | 2003-01-30 | Chunhui Xu | Osteoblast precursors from human embryonic stem cells |
| AUPR703601A0 (en) * | 2001-08-15 | 2001-09-06 | Peter Maccallum Cancer Institute, The | Identification and isolation of somatic stem cells and uses thereof |
| WO2003070922A1 (en) * | 2002-02-19 | 2003-08-28 | Medipost Co., Ltd. | Isolation and culture-expansion methods of mesenchymal stem/progenitor cells from umbilical cord blood, and differentiation method of umbilical cord blood-derived mesenchymal stem/progenitor cells into various mesenchymal tissues |
| JP4572299B2 (ja) * | 2003-08-18 | 2010-11-04 | 財団法人ヒューマンサイエンス振興財団 | 改良されたsiRNA分子およびこれを用いた遺伝子発現の抑制法 |
| EP2348105B1 (en) | 2004-09-24 | 2018-10-24 | Mesoblast, Inc. | Multipotential expanded mesenchymal precursor cell progeny (MEMP) and uses thereof |
| US8192988B2 (en) * | 2004-10-22 | 2012-06-05 | University Of Central Florida Research Foundation, Inc. | Methods for increasing potency of adult mesenchymal stem cells |
| US20060134784A1 (en) * | 2004-11-30 | 2006-06-22 | Basch Ross S | Methods and compositions for the growth and maintenance of stem cells |
| US20060182724A1 (en) * | 2005-02-15 | 2006-08-17 | Riordan Neil H | Method for expansion of stem cells |
| JP5098028B2 (ja) | 2005-12-13 | 2012-12-12 | 国立大学法人京都大学 | 核初期化因子 |
| GB0619454D0 (en) * | 2006-10-02 | 2006-11-08 | Fujitsu Ltd | Communication systems |
| US8241621B2 (en) * | 2006-12-18 | 2012-08-14 | Medistem Laboratories | Stem cell mediated treg activation/expansion for therapeutic immune modulation |
| JP5813321B2 (ja) * | 2007-03-23 | 2015-11-17 | ウィスコンシン アラムニ リサーチ ファンデーション | 体細胞の再プログラム化 |
| CN105861443A (zh) * | 2007-04-07 | 2016-08-17 | 怀特黑德生物医学研究所 | 体细胞重编程 |
| JP2008307007A (ja) * | 2007-06-15 | 2008-12-25 | Bayer Schering Pharma Ag | 出生後のヒト組織由来未分化幹細胞から誘導したヒト多能性幹細胞 |
| WO2009046377A2 (en) * | 2007-10-04 | 2009-04-09 | Medistem Laboratories, Inc. | Compositions and methods of stem cell therapy for autism |
| DE102007051966A1 (de) * | 2007-10-31 | 2009-05-07 | Adc Automotive Distance Control Systems Gmbh | Verfahren und Vorrichtung zur Erkennung des Verlaufs einer Fahrspur |
| AU2008297024B2 (en) * | 2007-10-31 | 2014-08-28 | Kyoto University | Nuclear reprogramming method |
| WO2009061442A1 (en) * | 2007-11-06 | 2009-05-14 | Children's Medical Center Corporation | Method to produce induced pluripotent stem (ips) cells form non-embryonic human cells |
| US20090191171A1 (en) * | 2008-01-18 | 2009-07-30 | Yupo Ma | Reprogramming of Differentiated Progenitor or Somatic Cells Using Homologous Recombination |
| WO2009096049A1 (ja) * | 2008-02-01 | 2009-08-06 | Kyoto University | 人工多能性幹細胞由来分化細胞 |
| EP2250252A2 (en) * | 2008-02-11 | 2010-11-17 | Cambridge Enterprise Limited | Improved reprogramming of mammalian cells, and the cells obtained |
| EP2090649A1 (en) * | 2008-02-13 | 2009-08-19 | Fondazione Telethon | Method for reprogramming differentiated cells |
| US20110014164A1 (en) * | 2008-02-15 | 2011-01-20 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| CN101550406B (zh) * | 2008-04-03 | 2016-02-10 | 北京大学 | 制备多潜能干细胞的方法,试剂盒及用途 |
| KR101661940B1 (ko) | 2008-05-02 | 2016-10-04 | 고쿠리츠 다이가쿠 호진 교토 다이가쿠 | 핵 초기화 방법 |
| EP2128245A1 (en) * | 2008-05-27 | 2009-12-02 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Generation of induced pluripotent stem (iPS) cells |
| KR101372752B1 (ko) * | 2008-07-31 | 2014-03-14 | 고꾸리츠 다이가꾸호오징 기후다이가꾸 | 유도된 다능성 줄기 세포의 효율적인 확립 방법 |
| US20110263001A1 (en) * | 2008-11-14 | 2011-10-27 | Life Technologies Corporation | Compositions and Methods for Engineering Cells |
-
2010
- 2010-03-22 KR KR1020117023985A patent/KR101764437B1/ko active Active
- 2010-03-22 CA CA2755870A patent/CA2755870C/en active Active
- 2010-03-22 WO PCT/AU2010/000329 patent/WO2010105311A1/en not_active Ceased
- 2010-03-22 CN CN201080012939.XA patent/CN102405280B/zh active Active
- 2010-03-22 AU AU2010225469A patent/AU2010225469B2/en active Active
- 2010-03-22 EP EP17196545.2A patent/EP3293257B1/en active Active
- 2010-03-22 JP JP2012500009A patent/JP6342115B2/ja active Active
- 2010-03-22 ES ES17196545T patent/ES2900277T3/es active Active
- 2010-03-22 US US13/257,233 patent/US9487756B2/en active Active
- 2010-03-22 SG SG2011066743A patent/SG174426A1/en unknown
- 2010-03-22 EP EP10753012.3A patent/EP2408904B1/en active Active
-
2015
- 2015-09-25 JP JP2015188541A patent/JP6603529B2/ja active Active
-
2016
- 2016-10-20 US US15/299,088 patent/US20170037377A1/en not_active Abandoned
-
2017
- 2017-12-08 JP JP2017235872A patent/JP6708617B2/ja active Active
-
2025
- 2025-04-11 US US19/177,484 patent/US20250257328A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| KR20120000079A (ko) | 2012-01-03 |
| JP2018068307A (ja) | 2018-05-10 |
| CN102405280A (zh) | 2012-04-04 |
| JP6342115B2 (ja) | 2018-06-13 |
| CA2755870A1 (en) | 2010-09-23 |
| US20120121548A1 (en) | 2012-05-17 |
| EP2408904A1 (en) | 2012-01-25 |
| AU2010225469A1 (en) | 2011-10-06 |
| CN102405280B (zh) | 2015-03-04 |
| US9487756B2 (en) | 2016-11-08 |
| WO2010105311A1 (en) | 2010-09-23 |
| EP3293257B1 (en) | 2021-08-11 |
| JP2012520660A (ja) | 2012-09-10 |
| US20250257328A1 (en) | 2025-08-14 |
| US20170037377A1 (en) | 2017-02-09 |
| HK1252414A1 (en) | 2019-05-24 |
| EP2408904B1 (en) | 2017-10-18 |
| EP2408904A4 (en) | 2013-02-27 |
| CA2755870C (en) | 2019-04-09 |
| JP2016073274A (ja) | 2016-05-12 |
| ES2900277T3 (es) | 2022-03-16 |
| AU2010225469B2 (en) | 2016-05-05 |
| KR101764437B1 (ko) | 2017-08-02 |
| EP3293257A1 (en) | 2018-03-14 |
| JP6708617B2 (ja) | 2020-06-10 |
| SG174426A1 (en) | 2011-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6603529B2 (ja) | 再プログラム化多能性細胞の作製 | |
| WO2010042800A1 (en) | Methods of reprogramming somatic cells and methods of use for such cells | |
| CN104508131A (zh) | 通过合成的自我复制的RNA形成人iPS细胞 | |
| Wang et al. | Reprogramming efficiency and quality of induced Pluripotent Stem Cells (iPSCs) generated from muscle-derived fibroblasts of mdx mice at different ages | |
| WO2022235586A1 (en) | Methods of generating mature corneal endothelial cells | |
| US10190097B2 (en) | Method and composition for inducing human pluripotent stem cells | |
| Kim et al. | Generation of human myogenic progenitors from pluripotent stem cells for in vivo regeneration | |
| Wang et al. | Reprogramming of mouse renal tubular epithelial cells to induced pluripotent stem cells | |
| JP2015506168A (ja) | ヒト腎臓由来細胞から作製した人工多能性幹細胞 | |
| US20250197803A1 (en) | A method of differentiating an induced pluripotent stem cell into a retinal pigment epithelial cell, a retinal pigment epithelial cell and methods of using the retinal pigment epithelial cell | |
| US20210340495A1 (en) | Method for inducing and differentiating pluripotent stem cells and uses thereof | |
| TWI769410B (zh) | 新穎誘導性多能幹細胞(ipscs)及其應用 | |
| HK1252414B (en) | Production of reprogrammed pluripotent cells | |
| Henning | Identification and characterisation of a novel, multi-potent, skeletal muscle-derived stem cell with broad developmental plasticity | |
| Zhang | iPSC-Derived MSCs that Are Genetically Engineered for Systemic Bone Augmentation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20151026 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20151104 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160920 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20161213 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20170213 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170321 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20170808 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171208 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20180119 |
|
| A912 | Re-examination (zenchi) completed and case transferred to appeal board |
Free format text: JAPANESE INTERMEDIATE CODE: A912 Effective date: 20180216 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190723 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20191011 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6603529 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |