JP6529592B2 - 酢酸の製造方法 - Google Patents
酢酸の製造方法 Download PDFInfo
- Publication number
- JP6529592B2 JP6529592B2 JP2017536033A JP2017536033A JP6529592B2 JP 6529592 B2 JP6529592 B2 JP 6529592B2 JP 2017536033 A JP2017536033 A JP 2017536033A JP 2017536033 A JP2017536033 A JP 2017536033A JP 6529592 B2 JP6529592 B2 JP 6529592B2
- Authority
- JP
- Japan
- Prior art keywords
- acetic acid
- stream
- concentration
- mass ppm
- less
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 title claims description 1226
- 238000004519 manufacturing process Methods 0.000 title claims description 70
- 238000004821 distillation Methods 0.000 claims description 252
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 claims description 214
- MLUCVPSAIODCQM-NSCUHMNNSA-N crotonaldehyde Chemical compound C\C=C\C=O MLUCVPSAIODCQM-NSCUHMNNSA-N 0.000 claims description 181
- MLUCVPSAIODCQM-UHFFFAOYSA-N crotonaldehyde Natural products CC=CC=O MLUCVPSAIODCQM-UHFFFAOYSA-N 0.000 claims description 181
- 238000006243 chemical reaction Methods 0.000 claims description 151
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 135
- 238000010992 reflux Methods 0.000 claims description 104
- 239000012074 organic phase Substances 0.000 claims description 91
- 239000008346 aqueous phase Substances 0.000 claims description 84
- 238000009835 boiling Methods 0.000 claims description 84
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 84
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 claims description 77
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 claims description 77
- 239000011541 reaction mixture Substances 0.000 claims description 77
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 claims description 76
- 238000000034 method Methods 0.000 claims description 62
- IQGZCSXWIRBTRW-ZZXKWVIFSA-N (2E)-2-ethyl-2-butenal Chemical compound CC\C(=C/C)C=O IQGZCSXWIRBTRW-ZZXKWVIFSA-N 0.000 claims description 49
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 claims description 47
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 claims description 47
- 238000001704 evaporation Methods 0.000 claims description 47
- 230000008020 evaporation Effects 0.000 claims description 46
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims description 45
- 239000003054 catalyst Substances 0.000 claims description 41
- 229910052739 hydrogen Inorganic materials 0.000 claims description 33
- 239000001257 hydrogen Substances 0.000 claims description 33
- 238000000926 separation method Methods 0.000 claims description 30
- 229910052751 metal Inorganic materials 0.000 claims description 28
- 239000002184 metal Substances 0.000 claims description 28
- 239000012071 phase Substances 0.000 claims description 27
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 claims description 26
- 229910002091 carbon monoxide Inorganic materials 0.000 claims description 26
- 238000005810 carbonylation reaction Methods 0.000 claims description 26
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 24
- 238000004064 recycling Methods 0.000 claims description 21
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 claims description 19
- 239000007791 liquid phase Substances 0.000 claims description 12
- 239000010948 rhodium Substances 0.000 claims description 7
- 239000002253 acid Substances 0.000 claims description 5
- 229910052703 rhodium Inorganic materials 0.000 claims description 5
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical group [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 claims description 5
- 239000007788 liquid Substances 0.000 description 58
- 239000000047 product Substances 0.000 description 48
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 45
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 44
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 38
- 238000000605 extraction Methods 0.000 description 34
- 229910000043 hydrogen iodide Inorganic materials 0.000 description 34
- 239000007789 gas Substances 0.000 description 32
- 239000012286 potassium permanganate Substances 0.000 description 30
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 24
- 230000018044 dehydration Effects 0.000 description 23
- 238000006297 dehydration reaction Methods 0.000 description 23
- 239000003456 ion exchange resin Substances 0.000 description 23
- 229920003303 ion-exchange polymer Polymers 0.000 description 23
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 22
- 235000019253 formic acid Nutrition 0.000 description 22
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 20
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 20
- 239000002904 solvent Substances 0.000 description 20
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 16
- 239000000203 mixture Substances 0.000 description 16
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- 230000007797 corrosion Effects 0.000 description 14
- 238000005260 corrosion Methods 0.000 description 14
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 14
- 239000000243 solution Substances 0.000 description 13
- 241000122205 Chamaeleonidae Species 0.000 description 12
- 238000000895 extractive distillation Methods 0.000 description 12
- 230000000052 comparative effect Effects 0.000 description 11
- 230000007423 decrease Effects 0.000 description 11
- 235000019260 propionic acid Nutrition 0.000 description 10
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 10
- 150000002431 hydrogen Chemical class 0.000 description 9
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 9
- 239000001569 carbon dioxide Substances 0.000 description 8
- 229910002092 carbon dioxide Inorganic materials 0.000 description 8
- 230000005493 condensed matter Effects 0.000 description 8
- 238000001816 cooling Methods 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- 238000001179 sorption measurement Methods 0.000 description 8
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 7
- 238000010586 diagram Methods 0.000 description 7
- 150000002739 metals Chemical class 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 239000001301 oxygen Substances 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 6
- 150000001351 alkyl iodides Chemical class 0.000 description 6
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 5
- 229910052741 iridium Inorganic materials 0.000 description 5
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 5
- HSZCZNFXUDYRKD-UHFFFAOYSA-M lithium iodide Chemical compound [Li+].[I-] HSZCZNFXUDYRKD-UHFFFAOYSA-M 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- SKIDNYUZJPMKFC-UHFFFAOYSA-N 1-iododecane Chemical compound CCCCCCCCCCI SKIDNYUZJPMKFC-UHFFFAOYSA-N 0.000 description 4
- ANOOTOPTCJRUPK-UHFFFAOYSA-N 1-iodohexane Chemical compound CCCCCCI ANOOTOPTCJRUPK-UHFFFAOYSA-N 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- KMGBZBJJOKUPIA-UHFFFAOYSA-N butyl iodide Chemical compound CCCCI KMGBZBJJOKUPIA-UHFFFAOYSA-N 0.000 description 4
- 239000006227 byproduct Substances 0.000 description 4
- 230000006315 carbonylation Effects 0.000 description 4
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 4
- PVWOIHVRPOBWPI-UHFFFAOYSA-N n-propyl iodide Chemical compound CCCI PVWOIHVRPOBWPI-UHFFFAOYSA-N 0.000 description 4
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- 239000002250 absorbent Substances 0.000 description 3
- 230000002745 absorbent Effects 0.000 description 3
- 150000001242 acetic acid derivatives Chemical class 0.000 description 3
- 238000005882 aldol condensation reaction Methods 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 239000003426 co-catalyst Substances 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- -1 for example Substances 0.000 description 3
- 239000012535 impurity Substances 0.000 description 3
- 239000011630 iodine Substances 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 229910021645 metal ion Inorganic materials 0.000 description 3
- 229910052759 nickel Inorganic materials 0.000 description 3
- 235000011056 potassium acetate Nutrition 0.000 description 3
- 239000002994 raw material Substances 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- 241000246358 Thymus Species 0.000 description 2
- 235000007303 Thymus vulgaris Nutrition 0.000 description 2
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 2
- ZCHPKWUIAASXPV-UHFFFAOYSA-N acetic acid;methanol Chemical compound OC.CC(O)=O ZCHPKWUIAASXPV-UHFFFAOYSA-N 0.000 description 2
- 239000000956 alloy Substances 0.000 description 2
- 229910045601 alloy Inorganic materials 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000003990 capacitor Substances 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 239000012429 reaction media Substances 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- 239000012465 retentate Substances 0.000 description 2
- 239000001585 thymus vulgaris Substances 0.000 description 2
- 229910052726 zirconium Inorganic materials 0.000 description 2
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- 241001448862 Croton Species 0.000 description 1
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- AVMNFQHJOOYCAP-UHFFFAOYSA-N acetic acid;propanoic acid Chemical compound CC(O)=O.CCC(O)=O AVMNFQHJOOYCAP-UHFFFAOYSA-N 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910001516 alkali metal iodide Inorganic materials 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 239000003729 cation exchange resin Substances 0.000 description 1
- 239000012295 chemical reaction liquid Substances 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 239000011651 chromium Substances 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 150000004696 coordination complex Chemical class 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000005474 detonation Methods 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 239000012527 feed solution Substances 0.000 description 1
- CQEYYJGAYNLKBF-UHFFFAOYSA-N formic acid;propanoic acid Chemical compound OC=O.CCC(O)=O CQEYYJGAYNLKBF-UHFFFAOYSA-N 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-M iodide Chemical compound [I-] XMBWDFGMSWQBCA-UHFFFAOYSA-M 0.000 description 1
- 229940006461 iodide ion Drugs 0.000 description 1
- 150000004694 iodide salts Chemical group 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- ZDGGJQMSELMHLK-UHFFFAOYSA-N m-Trifluoromethylhippuric acid Chemical compound OC(=O)CNC(=O)C1=CC=CC(C(F)(F)F)=C1 ZDGGJQMSELMHLK-UHFFFAOYSA-N 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 229910001511 metal iodide Inorganic materials 0.000 description 1
- ACYINGGNKOBDCT-UHFFFAOYSA-N methanol;methoxymethane Chemical compound OC.COC ACYINGGNKOBDCT-UHFFFAOYSA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000011733 molybdenum Substances 0.000 description 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 1
- 150000002908 osmium compounds Chemical class 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N phosphonic acid group Chemical group P(O)(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 150000003304 ruthenium compounds Chemical class 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 125000000542 sulfonic acid group Chemical group 0.000 description 1
- 239000012808 vapor phase Substances 0.000 description 1
- 238000003809 water extraction Methods 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/10—Preparation of carboxylic acids or their salts, halides or anhydrides by reaction with carbon monoxide
- C07C51/12—Preparation of carboxylic acids or their salts, halides or anhydrides by reaction with carbon monoxide on an oxygen-containing group in organic compounds, e.g. alcohols
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/43—Separation; Purification; Stabilisation; Use of additives by change of the physical state, e.g. crystallisation
- C07C51/44—Separation; Purification; Stabilisation; Use of additives by change of the physical state, e.g. crystallisation by distillation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C53/00—Saturated compounds having only one carboxyl group bound to an acyclic carbon atom or hydrogen
- C07C53/08—Acetic acid
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Crystallography & Structural Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
前記カルボニル化反応工程で得られた反応混合物を蒸発槽において蒸気流と残液流とに分離する蒸発工程と、
前記蒸気流を第1蒸留塔によりヨウ化メチル及びアセトアルデヒドから選択された少なくとも一種の低沸成分に富む第1オーバーヘッド流と酢酸に富む第1酢酸流とに分離するとともに、前記第1オーバーヘッド流を凝縮、分液させて水相と有機相とを得る脱低沸工程と、
前記第1オーバーヘッド流を凝縮させた水相及び/又は有機相の少なくとも一部を反応槽にリサイクルする第1オーバーヘッド流リサイクル工程と、
を備えた酢酸の製造方法であって、
第1酢酸流におけるクロトンアルデヒド濃度を2.2質量ppm以下に制御することを特徴とする酢酸の製造方法(以下、「第1の酢酸の製造方法」と称する場合がある)を提供する。
前記カルボニル化反応工程で得られた反応混合物を蒸発槽において蒸気流と残液流とに分離する蒸発工程と、
前記蒸気流を第1蒸留塔によりヨウ化メチル及びアセトアルデヒドから選択された少なくとも一種の低沸成分に富む第1オーバーヘッド流と酢酸に富む第1酢酸流とに分離するとともに、前記第1オーバーヘッド流を凝縮、分液させて水相と有機相とを得る脱低沸工程と、
前記第1酢酸流を第2蒸留塔により水に富む第2オーバーヘッド流と第1酢酸流よりも酢酸に富む第2酢酸流とに分離する脱水工程と、
前記第1オーバーヘッド流を凝縮させた水相及び/又は有機相の少なくとも一部、及び/又は、前記第2オーバーヘッド流の一部を反応槽にリサイクルするオーバーヘッド流リサイクル工程と、
を備えた酢酸の製造方法であって、
第1酢酸流におけるクロトンアルデヒド濃度を2.2質量ppm以下に制御する、及び/又は、第2蒸留塔の還流比を0.32以上に制御することを特徴とする酢酸の製造方法(以下、「第2の酢酸の製造方法」と称する場合がある)を提供する。
CH3OH + CO → CH3COOH (1)
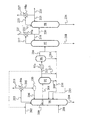
メタノール法酢酸パイロットプラントにおいて以下の実験を行った(図1参照)。
反応槽[全圧2.8MPa(絶対圧)、一酸化炭素分圧1.4MPa(絶対圧)、水素分圧0.02MPa(絶対圧)、反応温度187℃]で得られた反応混合液[組成:ヨウ化メチル(MeI)7.9%、酢酸メチル(MA)2.1%、水(H2O)2.5%、ロジウム錯体910ppm(Rh換算)、ヨウ化リチウム(LiI)14.1%、プロピオン酸110ppm、ギ酸30ppm、アセトアルデヒド(AD)410ppm、クロトンアルデヒド(CR)1.2ppm、2−エチルクロトンアルデヒド(2ECR)1.2ppm、酢酸ブチル(BA)9.9ppm、残り酢酸(但し、微量の不純物を含む)]400部を蒸発槽に仕込み、25%蒸発させた。蒸発槽の蒸気[組成:ヨウ化メチル28.1%、酢酸メチル4.9%、水1.9%、プロピオン酸73ppm、ギ酸85ppm、アセトアルデヒド1500ppm、クロトンアルデヒド2.5ppm、2−エチルクロトンアルデヒド0.09ppm、酢酸ブチル6.5ppm、残り酢酸(但し、微量の不純物を含む)]100部を脱低沸塔[実段数20段、仕込位置下から2段、塔頂圧250kPa(絶対圧)、塔頂温度140℃]に仕込み、塔頂蒸気を凝縮させ、水相と有機相とに分離後、有機相の一部(11部)を脱アセトアルデヒド塔[実段数80段、仕込位置下から11段、塔頂圧280kPa(絶対圧)、塔頂温度52℃]に送り、アセトアルデヒドを分離し系外に除去し、アセトアルデヒド除去後の有機相は反応系にリサイクルした。有機相の残り(41部)は、直接反応系にリサイクルした。水相の一部を脱低沸塔に還流(リサイクル)し、残りを留出液として1.5部を反応系にリサイクルした。水相の還流量/留出量を還流比とし、還流比を2とした。脱低沸塔の缶出から3部を抜取り、反応系にリサイクルした。脱低沸塔の中間部(下から4段)からサイドカット(SC)流として65部を抜取り、脱水塔[実段数50段、仕込位置下から34段、塔頂圧295kPa(絶対圧)、塔頂温度150℃]に仕込んだ。脱水塔の塔頂凝縮液の一部を脱水塔に還流(リサイクル)し、残りを留出液として19部を反応系にリサイクルした。脱水塔の還流比(還流量/留出量)を0.3とした。その結果、脱水塔の缶出液から製品酢酸46部を得た。製品酢酸中のクロトンアルデヒド含有量は2.2ppm、2−エチルクロトンアルデヒド含有量は0.08ppm、酢酸ブチル含有量は13ppmであった。製品酢酸の過マンガン酸タイム(カメレオンタイム)を測定したところ5分であった。結果を表1に示す。
反応槽の水素分圧を0.07MPaとした以外は比較例1と同様の実験を行った。結果を表1に示す。
脱低沸塔塔頂凝縮液の有機相の脱アセトアルデヒド塔への供給量を21部とした以外は比較例1と同様の実験を行った。なお、この変更により、反応混合液組成、蒸発槽の蒸気組成は変化した。結果を表1に示す。
脱低沸塔の還流比を5、脱水塔の還流比を0.5とした以外は比較例1と同様の実験を行った。なお、この変更により、反応混合液組成、蒸発槽の蒸気組成は変化した。結果を表1に示す。
脱低沸塔の還流比を5、脱水塔の還流比を0.5とし、脱低沸塔塔頂凝縮液の有機相の脱アセトアルデヒド塔への供給量を21部とした以外は比較例1と同様の実験を行った。なお、この変更により、反応混合液組成、蒸発槽の蒸気組成は変化した。結果を表1に示す。
脱低沸塔の還流比を10、脱水塔の還流比を5とし、脱低沸塔塔頂凝縮液の有機相の脱アセトアルデヒド塔への供給量を21部とした以外は比較例1と同様の実験を行った。なお、この変更により、反応混合液組成、蒸発槽の蒸気組成は変化した。結果を表1に示す。
脱低沸塔の還流比を15、脱水塔の還流比を10とし、脱低沸塔塔頂凝縮液の有機相の脱アセトアルデヒド塔への供給量を21部とした以外は比較例1と同様の実験を行った。なお、この変更により、反応混合液組成、蒸発槽の蒸気組成は変化した。結果を表1に示す。
脱低沸塔の還流比を20、脱水塔の還流比を20とし、脱低沸塔塔頂凝縮液の有機相の脱アセトアルデヒド塔への供給量を21部とした以外は比較例1と同様の実験を行った。なお、この変更により、反応混合液組成、蒸発槽の蒸気組成は変化した。結果を表1に示す。

比較例1と実施例1の対比より、反応槽の水素分圧を高くすると、製品カメレオンタイムが上昇することが分かる。これは、反応槽の水素分圧が高いと、クロトンアルデヒド(CR)が水素添加される量が増加するため、脱低沸塔仕込液中のCR濃度、脱低沸搭サイドカット液(第1酢酸流)中のCR濃度が低下し、その結果として脱水塔で得られる第2酢酸流中のCR濃度が低下し、製品カメレオンタイムが上昇するためと考えられる。
[1]金属触媒及びヨウ化メチルを含む触媒系、並びに、酢酸、酢酸メチル、水の存在下、メタノールと一酸化炭素とを反応槽で反応させて酢酸を生成させるカルボニル化反応工程と、
前記カルボニル化反応工程で得られた反応混合物を蒸発槽において蒸気流と残液流とに分離する蒸発工程と、
前記蒸気流を第1蒸留塔によりヨウ化メチル及びアセトアルデヒドから選択された少なくとも一種の低沸成分に富む第1オーバーヘッド流と酢酸に富む第1酢酸流とに分離するとともに、前記第1オーバーヘッド流を凝縮、分液させて水相と有機相とを得る脱低沸工程と、
前記第1オーバーヘッド流を凝縮させた水相及び/又は有機相の少なくとも一部を反応槽にリサイクルする第1オーバーヘッド流リサイクル工程と、
を備えた酢酸の製造方法であって、
第1酢酸流におけるクロトンアルデヒド濃度を2.2質量ppm以下に制御することを特徴とする酢酸の製造方法。
[2]金属触媒及びヨウ化メチルを含む触媒系、並びに、酢酸、酢酸メチル、水の存在下、メタノールと一酸化炭素とを反応槽で反応させて酢酸を生成させるカルボニル化反応工程と、
前記カルボニル化反応工程で得られた反応混合物を蒸発槽において蒸気流と残液流とに分離する蒸発工程と、
前記蒸気流を第1蒸留塔によりヨウ化メチル及びアセトアルデヒドから選択された少なくとも一種の低沸成分に富む第1オーバーヘッド流と酢酸に富む第1酢酸流とに分離するとともに、前記第1オーバーヘッド流を凝縮、分液させて水相と有機相とを得る脱低沸工程と、
前記第1酢酸流を第2蒸留塔により水に富む第2オーバーヘッド流と第1酢酸流よりも酢酸に富む第2酢酸流とに分離する脱水工程と、
前記第1オーバーヘッド流を凝縮させた水相及び/又は有機相の少なくとも一部、及び/又は、前記第2オーバーヘッド流の一部を反応槽にリサイクルするオーバーヘッド流リサイクル工程と、
を備えた酢酸の製造方法であって、
第1酢酸流におけるクロトンアルデヒド濃度を2.2質量ppm以下に制御する、及び/又は、第2蒸留塔の還流比を0.32以上に制御することを特徴とする酢酸の製造方法。
[3]第2酢酸流におけるクロトンアルデヒド濃度が2.0質量ppm以下(好ましくは1.8質量ppm以下、より好ましくは1.5質量ppm以下、さらに好ましくは1.2質量ppm以下、特に好ましくは0.7質量ppm以下、とりわけ0.5質量ppm以下)である[2]記載の酢酸の製造方法。
[4]第2酢酸流における2−エチルクロトンアルデヒド濃度が3.0質量ppm以下(好ましくは1.8質量ppm以下、より好ましくは1.5質量ppm以下、さらに好ましくは1.2質量ppm以下、特に好ましくは0.7質量ppm以下、とりわけ0.5質量ppm以下)である[2]又は[3]記載の酢酸の製造方法。
[5]第2酢酸流におけるクロトンアルデヒド濃度CCR(質量ppm)と2−エチルクロトンアルデヒド濃度CECR(質量ppm)との比(CCR/CECR)が35以下(好ましくは25以下、より好ましくは20以下、さらに好ましくは15以下)である[2]〜[4]のいずれか1つに記載の酢酸の製造方法。
[6]第2酢酸流における酢酸ブチル濃度が15質量ppm以下(好ましくは12質量ppm以下、より好ましくは10質量ppm以下、さらに好ましくは8質量ppm以下)である[2]〜[5]のいずれか1つに記載の酢酸の製造方法。
[7]第2酢酸流におけるクロトンアルデヒド濃度CCR(質量ppm)と酢酸ブチル濃度CBA(質量ppm)との比(CCR/CBA)が2.0以下(好ましくは1.5以下、より好ましくは1.0以下、さらに好ましくは0.6以下)である[2]〜[6]のいずれか1つに記載の酢酸の製造方法。
[8]第2蒸留塔の還流比を0.35以上(好ましくは0.4以上、より好ましくは1以上、さらに好ましくは2以上)に制御する[2]〜[7]のいずれか1つに記載の酢酸の製造方法。
[9]第2蒸留塔の還流比の上限が3000(好ましくは1000、より好ましくは100、さらに好ましくは10程度)である[2]〜[8]のいずれか1つに記載の酢酸の製造方法。
[10]触媒系がさらにイオン性ヨウ化物を含む[1]〜[9]のいずれか1つに記載の酢酸の製造方法。
[11] さらに、前記第1オーバーヘッド流を凝縮させた水相及び/又は有機相の少なくとも一部を蒸留してアセトアルデヒドを分離除去するためのアセトアルデヒド分離除去工程を有する[1]〜[10]のいずれか1つに記載の酢酸の製造方法。
[12]前記水相及び/又は前記有機相の少なくとも一部からアセトアルデヒドを分離除去した後の残液の少なくとも一部を反応槽にリサイクルする[11]記載の酢酸の製造方法。
[13]第1蒸留塔の運転条件につき、当該第1蒸留塔に水相のみを還流させる場合は水相の還流比を2以上(好ましくは3以上、より好ましくは4以上、さらに好ましくは8以上、特に好ましくは10以上)とし、有機相のみを還流させる場合は有機相の還流比を1以上(好ましくは1.5以上、より好ましくは2以上、さらに好ましくは4以上、特に好ましくは5以上)とし、水相及び有機相をともに還流させる場合は水相及び有機相の総和の還流比を1.5以上(好ましくは2.3以上、より好ましくは3以上、さらに好ましくは6以上、特に好ましくは7.5以上)とする[1]〜[12]のいずれか1つに記載の酢酸の製造方法。
[14] 反応槽の水素分圧が0.01MPa(絶対圧)以上(好ましくは0.015MPa(絶対圧)以上、より好ましくは0.02MPa(絶対圧)以上、さらに好ましくは0.04MPa(絶対圧)以上、特に好ましくは0.06MPa(絶対圧)以上、とりわけ0.07MPa(絶対圧)以上)である[1]〜[13]のいずれか1つに記載の酢酸の製造方法。
[15] 反応槽の反応混合液中のアセトアルデヒド濃度が500質量ppm以下(好ましくは450質量ppm以下、より好ましくは400質量ppm以下、さらに好ましくは350質量ppm以下、特に好ましくは300質量ppm以下、とりわけ250質量ppm以下)である[1]〜[14]のいずれか1つに記載の酢酸の製造方法。
[16]第1酢酸流における2−エチルクロトンアルデヒド濃度が3.0質量ppm以下(好ましくは2.0質量ppm以下、より好ましくは1.0質量ppm以下、さらに好ましくは0.8質量ppm以下、特に好ましくは0.5質量ppm以下)である[1]〜[15]のいずれか1つに記載の酢酸の製造方法。
[17]第1酢酸流におけるクロトンアルデヒド濃度CCR(質量ppm)と2−エチルクロトンアルデヒド濃度CECR(質量ppm)との比(CCR/CECR)が35以下(好ましくは25以下、より好ましくは20以下、さらに好ましくは15以下)である[1]〜[16]のいずれか1つに記載の酢酸の製造方法。
[18]第1酢酸流における酢酸ブチル濃度が15質量ppm以下(好ましくは12質量ppm以下、より好ましくは10質量ppm以下、さらに好ましくは8質量ppm以下)である[1]〜[17]のいずれか1つに記載の酢酸の製造方法。
[19]第1酢酸流におけるクロトンアルデヒド濃度CCR(質量ppm)と酢酸ブチル濃度CBA(質量ppm)との比(CCR/CBA)が2.0以下(好ましくは1.5以下、より好ましくは1.0以下、さらに好ましくは0.6以下)である[1]〜[18]のいずれか1つに記載の酢酸の製造方法。
[20]第1酢酸流中のクロトンアルデヒド濃度を2.0質量ppm以下(好ましくは1.8質量ppm以下、より好ましくは1.5質量ppm以下、さらに好ましくは1.2質量ppm以下、特に好ましくは1.0質量ppm以下、とりわけ0.8質量ppm以下、なかんずく0.5質量ppm以下)に制御する[1]〜[19]のいずれか1つに記載の酢酸の製造方法。
[21] 反応槽の水素分圧が0.5MPa(絶対圧)以下(好ましくは0.2MPa(絶対圧)以下)である[1]〜[20]のいずれか1つに記載の酢酸の製造方法。
[22]第1蒸留塔の還流比が0.5以上である[1]〜[21]のいずれか1つに記載の酢酸の製造方法。
[23]第1蒸留塔の還流比の上限が3000(好ましくは1000、より好ましくは100、さらに好ましくは30)である[1]〜[22]のいずれか1つに記載の酢酸の製造方法。
[24]反応槽の反応混合液中のクロトンアルデヒド濃度が5質量ppm以下(好ましくは3質量ppm以下、より好ましくは2質量ppm以下)である[1]〜[23]のいずれか1つに記載の酢酸の製造方法。
[25]反応槽の反応混合液中の2−エチルクロトンアルデヒド濃度が5質量ppm以下(好ましくは3質量ppm以下、より好ましくは2質量ppm以下)である[1]〜[24]のいずれか1つに記載の酢酸の製造方法。
[26]反応槽の反応混合液中の酢酸ブチル濃度が0.1〜15質量ppm(好ましくは1〜12質量ppm、より好ましくは2〜9質量ppm)である[1]〜[25]のいずれか1つに記載の酢酸の製造方法。
[27]蒸気流中のクロトンアルデヒド濃度が0〜5質量ppm(好ましくは0.1〜3質量ppm、より好ましくは0.2〜2質量ppm)である[1]〜[26]のいずれか1つに記載の酢酸の製造方法。
[28]蒸気流中の2−エチルクロトンアルデヒド濃度が0〜3質量ppm(好ましくは0.02〜2質量ppm、より好ましくは0.03〜0.8質量ppm)である[1]〜[27]のいずれか1つに記載の酢酸の製造方法。
[29]蒸気流中の酢酸ブチル濃度が0.1〜13質量ppm(好ましくは0.2〜12質量ppm、より好ましくは0.3〜9質量ppm)である[1]〜[28]のいずれか1つに記載の酢酸の製造方法。
2 蒸発槽
3,5,6 蒸留塔
4 デカンタ
7 イオン交換樹脂塔
8 スクラバーシステム
9 アセトアルデヒド分離除去システム
16 反応混合物供給ライン
17 蒸気流排出ライン
18,19 残液流リサイクルライン
54 一酸化炭素含有ガス導入ライン
55,56 水酸化カリウム導入ライン
57 触媒循環ポンプ
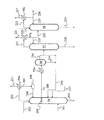
91 蒸留塔(第1脱アセトアルデヒド塔)
92 抽出塔
93 蒸留塔(第2脱アセトアルデヒド塔)
94 蒸留塔(抽出蒸留塔)
95 デカンタ
96 デカンタ
97 蒸留塔(脱アセトアルデヒド塔)
98 蒸留塔(抽出蒸留塔)
99 デカンタ
200 チムニートレイ
Claims (15)
- 金属触媒及びヨウ化メチルを含む触媒系、並びに、酢酸、酢酸メチル、水の存在下、メタノールと一酸化炭素とを反応槽で反応させて酢酸を生成させるカルボニル化反応工程と、
前記カルボニル化反応工程で得られた反応混合物を蒸発槽において蒸気流と残液流とに分離する蒸発工程と、
前記蒸気流を第1蒸留塔によりヨウ化メチル及びアセトアルデヒドから選択された少なくとも一種の低沸成分に富む第1オーバーヘッド流と酢酸に富む第1酢酸流とに分離するとともに、前記第1オーバーヘッド流を凝縮、分液させて水相と有機相とを得る脱低沸工程と、
前記第1オーバーヘッド流を凝縮させた水相及び/又は有機相の少なくとも一部を反応槽にリサイクルする第1オーバーヘッド流リサイクル工程と、
前記第1オーバーヘッド流を凝縮させた水相及び/又は有機相の少なくとも一部を蒸留してアセトアルデヒドを分離除去するためのアセトアルデヒド分離除去工程と、
を備えた酢酸の製造方法であって、
反応槽の反応混合物中の水の濃度は反応混合物の液相全体に対して1〜6質量%であり、
(a)下記の(i)、(ii)及び(iii)
(i)反応槽の水素分圧が0.02MPa(絶対圧)以上である
(ii)第1蒸留塔の運転条件につき、当該第1蒸留塔に水相のみを還流させる場合は水相の還流比を2以上とし、有機相のみを還流させる場合は有機相の還流比を1以上とし、水相及び有機相をともに還流させる場合は水相及び有機相の総和の還流比を1.5以上とする
(iii)反応槽の反応混合液中のアセトアルデヒド濃度が410質量ppm以下である
の3条件のうち少なくとも(i)の条件を満たし、且つ
(b)第1酢酸流におけるクロトンアルデヒド濃度を1.8質量ppm以下に制御することを特徴とする酢酸の製造方法。 - 金属触媒及びヨウ化メチルを含む触媒系、並びに、酢酸、酢酸メチル、水の存在下、メタノールと一酸化炭素とを反応槽で反応させて酢酸を生成させるカルボニル化反応工程と、
前記カルボニル化反応工程で得られた反応混合物を蒸発槽において蒸気流と残液流とに分離する蒸発工程と、
前記蒸気流を第1蒸留塔によりヨウ化メチル及びアセトアルデヒドから選択された少なくとも一種の低沸成分に富む第1オーバーヘッド流と酢酸に富む第1酢酸流とに分離するとともに、前記第1オーバーヘッド流を凝縮、分液させて水相と有機相とを得る脱低沸工程と、
前記第1酢酸流を第2蒸留塔により水に富む第2オーバーヘッド流と第1酢酸流よりも酢酸に富む第2酢酸流とに分離する脱水工程と、
前記第1オーバーヘッド流を凝縮させた水相及び/又は有機相の少なくとも一部、及び/又は、前記第2オーバーヘッド流の一部を反応槽にリサイクルするオーバーヘッド流リサイクル工程と、
前記第1オーバーヘッド流を凝縮させた水相及び/又は有機相の少なくとも一部を蒸留してアセトアルデヒドを分離除去するためのアセトアルデヒド分離除去工程と、
を備えた酢酸の製造方法であって、
反応槽の反応混合物中の水の濃度は反応混合物の液相全体に対して1〜6質量%であり、
(I)(a)下記の(i)、(ii)及び(iii)
(i)反応槽の水素分圧が0.02MPa(絶対圧)以上である
(ii)第1蒸留塔の運転条件につき、当該第1蒸留塔に水相のみを還流させる場合は水相の還流比を2以上とし、有機相のみを還流させる場合は有機相の還流比を1以上とし、水相及び有機相をともに還流させる場合は水相及び有機相の総和の還流比を1.5以上とする
(iii)反応槽の反応混合液中のアセトアルデヒド濃度が410質量ppm以下である
の3条件のうち少なくとも(i)の条件を満たし、且つ
(b)第1酢酸流におけるクロトンアルデヒド濃度を1.8質量ppm以下に制御する、
及び/又は、
(II)第2蒸留塔の還流比を0.32以上に制御する
ことを特徴とする酢酸の製造方法。 - 第2酢酸流におけるクロトンアルデヒド濃度が2.0質量ppm以下である請求項2記載の酢酸の製造方法。
- 第2酢酸流における2−エチルクロトンアルデヒド濃度が3.0質量ppm以下である請求項2又は3記載の酢酸の製造方法。
- 第2酢酸流におけるクロトンアルデヒド濃度CCR(質量ppm)と2−エチルクロトンアルデヒド濃度CECR(質量ppm)との比(CCR/CECR)が35以下である請求項2〜4のいずれか1項に記載の酢酸の製造方法。
- 第2酢酸流における酢酸ブチル濃度が15質量ppm以下である請求項2〜5のいずれか1項に記載の酢酸の製造方法。
- 第2酢酸流におけるクロトンアルデヒド濃度CCR(質量ppm)と酢酸ブチル濃度CBA(質量ppm)との比(CCR/CBA)が2.0以下である請求項2〜6のいずれか1項に記載の酢酸の製造方法。
- 反応槽の反応混合物中の水の濃度は反応混合物の液相全体に対して1〜4質量%である請求項1〜7のいずれか1項に記載の酢酸の製造方法。
- 金属触媒がロジウム触媒である請求項1〜8のいずれか1項に記載の酢酸の製造方法。
- 触媒系がさらにイオン性ヨウ化物を含む請求項1〜9のいずれか1項に記載の酢酸の製造方法。
- 前記水相及び/又は前記有機相の少なくとも一部からアセトアルデヒドを分離除去した後の残液の少なくとも一部を反応槽にリサイクルする請求項1〜10のいずれか1項に記載の酢酸の製造方法。
- 第1酢酸流における2−エチルクロトンアルデヒド濃度が3.0質量ppm以下である請求項1〜11のいずれか1項に記載の酢酸の製造方法。
- 第1酢酸流におけるクロトンアルデヒド濃度CCR(質量ppm)と2−エチルクロトンアルデヒド濃度CECR(質量ppm)との比(CCR/CECR)が35以下である請求項1〜12のいずれか1項に記載の酢酸の製造方法。
- 第1酢酸流における酢酸ブチル濃度が15質量ppm以下である請求項1〜13のいずれか1項に記載の酢酸の製造方法。
- 第1酢酸流におけるクロトンアルデヒド濃度CCR(質量ppm)と酢酸ブチル濃度CBA(質量ppm)との比(CCR/CBA)が2.0以下である請求項1〜14のいずれか1項に記載の酢酸の製造方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017044342 | 2017-03-08 | ||
| JP2017044342 | 2017-03-08 | ||
| PCT/JP2017/019577 WO2018163449A1 (ja) | 2017-03-08 | 2017-05-25 | 酢酸の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2018163449A1 JPWO2018163449A1 (ja) | 2019-03-22 |
| JP6529592B2 true JP6529592B2 (ja) | 2019-06-12 |
Family
ID=63447425
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017536033A Active JP6529592B2 (ja) | 2017-03-08 | 2017-05-25 | 酢酸の製造方法 |
Country Status (13)
| Country | Link |
|---|---|
| EP (1) | EP3401302B1 (ja) |
| JP (1) | JP6529592B2 (ja) |
| KR (1) | KR102257566B1 (ja) |
| CN (1) | CN110248921B (ja) |
| AR (1) | AR111120A1 (ja) |
| BR (1) | BR112019017835A2 (ja) |
| ES (1) | ES2816173T3 (ja) |
| MX (1) | MX2019010645A (ja) |
| MY (1) | MY186618A (ja) |
| PH (1) | PH12019550172A1 (ja) |
| SG (1) | SG11201907732PA (ja) |
| TW (1) | TWI705052B (ja) |
| WO (1) | WO2018163449A1 (ja) |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW283702B (ja) * | 1993-07-08 | 1996-08-21 | Daicel Chem | |
| JP3244350B2 (ja) | 1993-07-08 | 2002-01-07 | ダイセル化学工業株式会社 | 高純度酢酸の製造方法 |
| JPH07133249A (ja) * | 1993-09-17 | 1995-05-23 | Daicel Chem Ind Ltd | 高純度酢酸の製造方法 |
| EP0645362A1 (en) * | 1993-09-17 | 1995-03-29 | Daicel Chemical Industries, Ltd. | Process for producing highly purified acetic acid |
| SG44317A1 (en) * | 1994-06-15 | 1997-12-19 | Daicel Chem | Process for producing high purity acetic acid |
| JP3306227B2 (ja) * | 1994-07-06 | 2002-07-24 | ダイセル化学工業株式会社 | 酢酸および/または無水酢酸の製造法 |
| JP3332594B2 (ja) * | 1994-08-12 | 2002-10-07 | ダイセル化学工業株式会社 | 酢酸の精製方法 |
| JP3927237B2 (ja) * | 1995-04-27 | 2007-06-06 | ダイセル化学工業株式会社 | 酢酸の製造法 |
| IN192600B (ja) | 1996-10-18 | 2004-05-08 | Hoechst Celanese Corp | |
| GB9816385D0 (en) * | 1998-07-29 | 1998-09-23 | Bp Chem Int Ltd | Process |
| US6303813B1 (en) * | 1999-08-31 | 2001-10-16 | Celanese International Corporation | Rhodium/inorganic iodide catalyst system for methanol carbonylation process with improved impurity profile |
| JP4526381B2 (ja) * | 2004-12-27 | 2010-08-18 | ダイセル化学工業株式会社 | 酢酸の製造方法 |
| EP2628720B1 (en) * | 2010-10-06 | 2018-11-07 | Daicel Corporation | Acetic acid production method |
| SG190940A1 (en) * | 2010-12-15 | 2013-07-31 | Daicel Corp | Process for producing acetic acid |
| TWI547477B (zh) * | 2012-03-14 | 2016-09-01 | 大賽璐股份有限公司 | 醋酸之製造方法 |
| US9193657B2 (en) * | 2012-08-17 | 2015-11-24 | Celanese International Corporation | Catalyst stability in carbonylation processes |
| MX356188B (es) * | 2012-12-21 | 2018-05-16 | Daicel Corp | Procedimiento para producir acido acetico. |
| CN110845295A (zh) * | 2014-08-14 | 2020-02-28 | 科慕埃弗西有限公司 | 通过脱氟化氢来制备E-1,3,3,3-四氟丙烯(HFC-1234ze)的方法 |
| CN106715379B (zh) * | 2014-10-02 | 2020-05-19 | 国际人造丝公司 | 用于生产乙酸的方法 |
| JP6034478B2 (ja) * | 2015-01-30 | 2016-11-30 | セラニーズ・インターナショナル・コーポレーション | 酢酸の製造方法 |
-
2017
- 2017-05-25 MX MX2019010645A patent/MX2019010645A/es active IP Right Grant
- 2017-05-25 CN CN201780075561.XA patent/CN110248921B/zh active Active
- 2017-05-25 WO PCT/JP2017/019577 patent/WO2018163449A1/ja active Application Filing
- 2017-05-25 KR KR1020197028985A patent/KR102257566B1/ko active IP Right Grant
- 2017-05-25 JP JP2017536033A patent/JP6529592B2/ja active Active
- 2017-05-25 EP EP17739174.5A patent/EP3401302B1/en active Active
- 2017-05-25 ES ES17739174T patent/ES2816173T3/es active Active
- 2017-05-25 BR BR112019017835A patent/BR112019017835A2/pt not_active Application Discontinuation
- 2017-05-25 SG SG11201907732PA patent/SG11201907732PA/en unknown
- 2017-05-25 MY MYPI2019004851A patent/MY186618A/en unknown
- 2017-06-16 TW TW106120105A patent/TWI705052B/zh active
-
2018
- 2018-03-06 AR ARP180100512A patent/AR111120A1/es unknown
-
2019
- 2019-09-06 PH PH12019550172A patent/PH12019550172A1/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| ES2816173T3 (es) | 2021-03-31 |
| KR20190120815A (ko) | 2019-10-24 |
| TWI705052B (zh) | 2020-09-21 |
| MX2019010645A (es) | 2019-10-15 |
| SG11201907732PA (en) | 2019-09-27 |
| AR111120A1 (es) | 2019-06-05 |
| EP3401302A4 (en) | 2019-04-03 |
| BR112019017835A2 (pt) | 2020-04-14 |
| KR102257566B1 (ko) | 2021-05-31 |
| TW201840520A (zh) | 2018-11-16 |
| PH12019550172A1 (en) | 2020-06-01 |
| JPWO2018163449A1 (ja) | 2019-03-22 |
| EP3401302B1 (en) | 2020-07-22 |
| MY186618A (en) | 2021-07-30 |
| WO2018163449A1 (ja) | 2018-09-13 |
| CN110248921B (zh) | 2022-12-09 |
| EP3401302A1 (en) | 2018-11-14 |
| CN110248921A (zh) | 2019-09-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6481041B2 (ja) | 酢酸の製造方法 | |
| JP6626988B1 (ja) | 酢酸の製造方法 | |
| US10428005B2 (en) | Method for producing acetic acid | |
| JP6481042B2 (ja) | 酢酸の製造方法 | |
| JP6693959B2 (ja) | 酢酸の製造方法 | |
| JP6529592B2 (ja) | 酢酸の製造方法 | |
| JP6481040B2 (ja) | 酢酸の製造方法 | |
| US10308581B2 (en) | Method for producing acetic acid | |
| JP6481043B1 (ja) | 酢酸の製造方法 | |
| JP6588658B1 (ja) | 酢酸の製造方法 | |
| US10550058B2 (en) | Method for producing acetic acid | |
| JP6626987B1 (ja) | 酢酸の製造方法 | |
| JP6569019B1 (ja) | 酢酸の製造方法 | |
| JPWO2019230007A1 (ja) | 酢酸の製造方法 | |
| JPWO2018163448A1 (ja) | 酢酸の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20170725 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180710 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180910 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20180910 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190212 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190318 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190507 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190514 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6529592 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |