JP5672016B2 - フッ素化合物の製造方法 - Google Patents
フッ素化合物の製造方法 Download PDFInfo
- Publication number
- JP5672016B2 JP5672016B2 JP2011006215A JP2011006215A JP5672016B2 JP 5672016 B2 JP5672016 B2 JP 5672016B2 JP 2011006215 A JP2011006215 A JP 2011006215A JP 2011006215 A JP2011006215 A JP 2011006215A JP 5672016 B2 JP5672016 B2 JP 5672016B2
- Authority
- JP
- Japan
- Prior art keywords
- alkali metal
- fluorine
- fluorine compound
- reaction
- solvent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 238000004519 manufacturing process Methods 0.000 title claims description 25
- 150000002222 fluorine compounds Chemical class 0.000 title claims description 23
- 229910001515 alkali metal fluoride Inorganic materials 0.000 claims description 23
- 239000002904 solvent Substances 0.000 claims description 19
- 239000013067 intermediate product Substances 0.000 claims description 16
- 229910052783 alkali metal Inorganic materials 0.000 claims description 15
- 229910052731 fluorine Inorganic materials 0.000 claims description 13
- 239000011737 fluorine Substances 0.000 claims description 13
- 150000002366 halogen compounds Chemical class 0.000 claims description 11
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 claims description 8
- 150000001340 alkali metals Chemical class 0.000 claims description 8
- 229910052794 bromium Inorganic materials 0.000 claims description 8
- 229910052801 chlorine Inorganic materials 0.000 claims description 8
- 229910052736 halogen Inorganic materials 0.000 claims description 8
- 150000002367 halogens Chemical class 0.000 claims description 8
- 229910052740 iodine Inorganic materials 0.000 claims description 8
- 230000002194 synthesizing effect Effects 0.000 claims description 7
- 229910052744 lithium Inorganic materials 0.000 claims description 6
- 229910052700 potassium Inorganic materials 0.000 claims description 6
- 229910052701 rubidium Inorganic materials 0.000 claims description 6
- 229910052708 sodium Inorganic materials 0.000 claims description 6
- 239000002798 polar solvent Substances 0.000 claims description 5
- 239000003586 protic polar solvent Substances 0.000 claims description 5
- 229910052792 caesium Inorganic materials 0.000 claims description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims 3
- 239000000460 chlorine Substances 0.000 description 36
- 238000006243 chemical reaction Methods 0.000 description 28
- 238000000034 method Methods 0.000 description 22
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- 239000000047 product Substances 0.000 description 13
- 150000003839 salts Chemical class 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 9
- 239000002994 raw material Substances 0.000 description 9
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 8
- 238000000806 fluorine-19 nuclear magnetic resonance spectrum Methods 0.000 description 7
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 6
- -1 imide salts Chemical class 0.000 description 6
- 238000001308 synthesis method Methods 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000006227 byproduct Substances 0.000 description 4
- 238000007796 conventional method Methods 0.000 description 4
- 125000001153 fluoro group Chemical group F* 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 230000000694 effects Effects 0.000 description 3
- 238000001228 spectrum Methods 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000003125 aqueous solvent Substances 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 238000003682 fluorination reaction Methods 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- KEQGZUUPPQEDPF-UHFFFAOYSA-N 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione Chemical compound CC1(C)N(Cl)C(=O)N(Cl)C1=O KEQGZUUPPQEDPF-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- HBBGRARXTFLTSG-UHFFFAOYSA-N Lithium ion Chemical compound [Li+] HBBGRARXTFLTSG-UHFFFAOYSA-N 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- XTHPWXDJESJLNJ-UHFFFAOYSA-N chlorosulfonic acid Substances OS(Cl)(=O)=O XTHPWXDJESJLNJ-UHFFFAOYSA-N 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000000151 deposition Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 229910001416 lithium ion Inorganic materials 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 239000011812 mixed powder Substances 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B21/00—Nitrogen; Compounds thereof
- C01B21/082—Compounds containing nitrogen and non-metals and optionally metals
- C01B21/086—Compounds containing nitrogen and non-metals and optionally metals containing one or more sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B21/00—Nitrogen; Compounds thereof
- C01B21/082—Compounds containing nitrogen and non-metals and optionally metals
- C01B21/087—Compounds containing nitrogen and non-metals and optionally metals containing one or more hydrogen atoms
- C01B21/093—Compounds containing nitrogen and non-metals and optionally metals containing one or more hydrogen atoms containing also one or more sulfur atoms
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/056—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes
- H01M10/0564—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes the electrolyte being constituted of organic materials only
- H01M10/0566—Liquid materials
- H01M10/0568—Liquid materials characterised by the solutes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- General Physics & Mathematics (AREA)
- Physics & Mathematics (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- Materials Engineering (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Secondary Cells (AREA)
Description
また、溶融塩電池の溶融塩として、KN(SO2F)2若しくはNaN(SO2F)2またはこれらの混合物が採用されている。溶融塩電池では、電池を作動させるために、溶融塩が溶解する温度にまで加熱することを要するが、KN(SO2F)2若しくはNaN(SO2F)2またはこれらの混合物は、従来の溶融塩と比べて溶融点が低いため、溶融塩電池の作動温度を低くする材料として注目されている。
(1)請求項1に記載の発明は、下記(1)式に示されるハロゲン化合物のハロゲン元素をフッ素に置換することにより下記(2)式に示されるフッ素化合物を合成するフッ素化合物の製造方法であって、前記ハロゲン化合物と、アルカリ金属Mのフッ化物であるアルカリ金属フッ化物MFとを無溶媒で反応させて中間生成物を生成し、その後、この中間生成物と前記アルカリ金属フッ化物MFとをプロトン性極性溶媒中で反応させることを要旨とする。
HN(SO2X1)(SO2X2) ・・・ (1)
MN(SO2F)2 ・・・ (2)
・X1およびX2は、それぞれ独立にCl、Br、Iのいずれかの元素を示す。
・アルカリ金属Mは、Li、Na、K、Rb、Csのいずれかを示す。
また、本発明では、極性溶媒としてプロトン性極性溶媒を用いる。
アルカリ金属フッ化物(MN(SO 2 X 1 )(SO 2 F))は、非プロトン性極性溶媒よりもプロトン性極性溶媒に多く溶解する。このため、この構成によれば、MN(SO 2 X 1 )(SO 2 F)とアルカリ金属フッ化物MFとの反応を促進することができる。
HN(SO2X)2 ・・・ (3)
MN(SO2X)(SO2F) ・・・ (4)
・XはCl、Br、Iのいずれかの元素を示す。
・アルカリ金属Mは、Li、Na、K、Rb、Csのいずれかを示す。
MN(SO2X)(SO2F) ・・・ (4)
MN(SO2F)2 ・・・ (5)
・XはCl、Br、Iのいずれかの元素を示す。
・アルカリ金属Mは、Li、Na、K、Rb、Csのいずれかを示す。
HN(SO2Cl)2は、従来の製造方法により生成する。次に、HN(SO2F)2を過剰の粉状のKFに滴下する。なお、KFに水分が含まれているとき、水とHN(SO2Cl)2とが反応して加水分解を起こすため、KFにHN(SO2Cl)2を滴下する前に、予めKFから水分を除去する。HN(SO2F)2とKFとの反応により、KN(SO2Cl)(SO2F)とHClが生成する。この反応は溶媒なしで行われるため、反応は2〜3分程度で完了する。なお、粉状のKFにHN(SO2F)2を滴下して形成されたもの、すなわちKN(SO2Cl)(SO2F)とKFとを含むものを中間生成物Aとする。
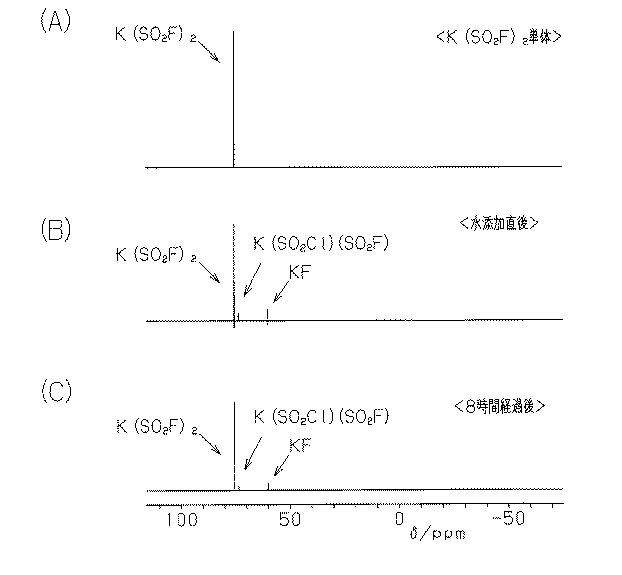
KN(SO2F)2の原料であるHN(SO2Cl)2の合成方法の一例を挙げる。なお、HN(SO2Cl)2の合成方法は以下に挙げる例に限定されない。
以下、KN(SO2F)2の合成方法を説明する。
第1工程では、粉体のKFを予め乾燥して、KFから水分を除去する。そして、予めHN(SO2F)2を温度37℃以上に加熱して液体とした上で、HN(SO2F)2をKFに滴下する。HN(SO2F)2の滴下量は、KF2.5〜3.0molに対してHN(SO2F)21.0molとされる。すなわち、KFは、HN(SO2F)2に対して過剰量とし、HN(SO2F)2の全てがKFと反応するように、両者の量が決められる。
図1(A)に示すように、KN(SO2F)2単体の19F−NMRスペクトルは、77δ/ppmに1本のピークを有す。
(1)本実施形態では、HN(SO2Cl)2をKFに滴下して中間生成物Aを生成し、その後、この中間生成物AとKFとを水溶媒中で反応させて、KN(SO2F)2を合成する。この方法によれば、従来の方法に比べて、短時間で、KN(SO2F)2を合成することができる。
なお、本発明の実施態様は上記実施形態にて示した態様に限られるものではなく、これを例えば以下に示すように変更して実施することもできる。また以下の各変形例は、上記各実施形態についてのみ適用されるものではなく、異なる変形例同士を互いに組み合わせて実施することもできる。
Claims (3)
- 下記(1)式に示されるハロゲン化合物のハロゲン元素をフッ素に置換することにより下記(2)式に示されるフッ素化合物を合成するフッ素化合物の製造方法であって、
前記ハロゲン化合物と、アルカリ金属Mのフッ化物であるアルカリ金属フッ化物MFとを無溶媒で反応させて中間生成物を生成し、その後、この中間生成物と前記アルカリ金属フッ化物MFとをプロトン性極性溶媒中で反応させる
ことを特徴とするフッ素化合物の製造方法。
HN(SO2X1)(SO2X2) ・・・ (1)
MN(SO2F)2 ・・・ (2)
X1およびX2は、それぞれ独立にCl、Br、Iのいずれかの元素を示す。
アルカリ金属Mは、Li、Na、K、Rb、Csのいずれかを示す。 - 請求項1に記載のフッ素化合物の製造方法において、
前記ハロゲン化合物と前記アルカリ金属フッ化物とを反応させる前に、前記アルカリ金属フッ化物から水分を除去する
ことを特徴とするフッ素化合物の製造方法。 - 下記(4)式に示されるハロゲン化合物のフッ素以外のハロゲン元素をフッ素に置換することにより下記(5)式に示されるフッ素化合物を合成するフッ素化合物の製造方法であって、
前記ハロゲン化合物とアルカリ金属Mのフッ化物であるアルカリ金属フッ化物MFとをプロトン性極性溶媒中で反応させる
ことを特徴とするフッ素化合物の製造方法。
MN(SO2X)(SO2F) ・・・ (4)
MN(SO2F)2 ・・・ (5)
XはCl、Br、Iのいずれかの元素を示す。
アルカリ金属Mは、Li、Na、K、Rb、Csのいずれかを示す。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011006215A JP5672016B2 (ja) | 2011-01-14 | 2011-01-14 | フッ素化合物の製造方法 |
| KR1020137017374A KR20140024841A (ko) | 2011-01-14 | 2012-01-13 | 불소 화합물의 제조 방법 |
| CN201280005069.2A CN103313933B (zh) | 2011-01-14 | 2012-01-13 | 制造氟化合物的方法 |
| US13/978,980 US20130294997A1 (en) | 2011-01-14 | 2012-01-13 | Method for producing fluorine compound |
| PCT/JP2012/050577 WO2012096371A1 (ja) | 2011-01-14 | 2012-01-13 | フッ素化合物の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011006215A JP5672016B2 (ja) | 2011-01-14 | 2011-01-14 | フッ素化合物の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012144412A JP2012144412A (ja) | 2012-08-02 |
| JP5672016B2 true JP5672016B2 (ja) | 2015-02-18 |
Family
ID=46507262
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011006215A Expired - Fee Related JP5672016B2 (ja) | 2011-01-14 | 2011-01-14 | フッ素化合物の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20130294997A1 (ja) |
| JP (1) | JP5672016B2 (ja) |
| KR (1) | KR20140024841A (ja) |
| CN (1) | CN103313933B (ja) |
| WO (1) | WO2012096371A1 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9650250B2 (en) | 2012-08-06 | 2017-05-16 | Nippon Soda Co., Ltd. | Method for producing bis(halosulfonyl)amine |
| CN105523530B (zh) * | 2014-10-23 | 2018-09-07 | 浙江蓝天环保高科技股份有限公司 | 一种双(氟磺酰)亚胺钾的制备方法 |
| JP2024525134A (ja) | 2021-06-10 | 2024-07-10 | スペシャルティ オペレーションズ フランス | ビス(フルオロスルホニル)イミドの塩を調製するための無溶媒方法 |
| EP4151592A1 (en) | 2021-09-15 | 2023-03-22 | Rhodia Operations | Solvent-free process for preparing a salt of bis(fluorosulfonyl)imide |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3623452B2 (ja) * | 2000-01-31 | 2005-02-23 | 森田化学工業株式会社 | スルホニルイミド化合物の製造方法 |
| FR2818972B1 (fr) * | 2000-12-29 | 2003-03-21 | Rhodia Chimie Sa | Procede de fluoration d'un compose halogene |
| JP2009504790A (ja) * | 2005-08-22 | 2009-02-05 | トランスファート プラス エスイーシー | スルホニルイミド及びその誘導体を調製するための方法 |
| JP4705476B2 (ja) * | 2006-01-10 | 2011-06-22 | 第一工業製薬株式会社 | フッ素化合物の製造方法 |
| JP4621783B2 (ja) * | 2008-03-31 | 2011-01-26 | 株式会社日本触媒 | フルオロスルホニルイミド類およびその製造方法 |
| KR101291903B1 (ko) * | 2008-07-23 | 2013-07-31 | 다이이치 고교 세이야쿠 가부시키가이샤 | 비스(플루오로설포닐)이미드 음이온 화합물의 제조 방법과 이온대화합물 |
| JP5471045B2 (ja) * | 2009-06-03 | 2014-04-16 | セントラル硝子株式会社 | イミド酸塩の製造方法 |
-
2011
- 2011-01-14 JP JP2011006215A patent/JP5672016B2/ja not_active Expired - Fee Related
-
2012
- 2012-01-13 WO PCT/JP2012/050577 patent/WO2012096371A1/ja not_active Ceased
- 2012-01-13 CN CN201280005069.2A patent/CN103313933B/zh not_active Expired - Fee Related
- 2012-01-13 US US13/978,980 patent/US20130294997A1/en not_active Abandoned
- 2012-01-13 KR KR1020137017374A patent/KR20140024841A/ko not_active Withdrawn
Also Published As
| Publication number | Publication date |
|---|---|
| KR20140024841A (ko) | 2014-03-03 |
| JP2012144412A (ja) | 2012-08-02 |
| WO2012096371A1 (ja) | 2012-07-19 |
| US20130294997A1 (en) | 2013-11-07 |
| CN103313933A (zh) | 2013-09-18 |
| CN103313933B (zh) | 2015-09-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5899789B2 (ja) | イミド塩の製造方法 | |
| CN104755418B (zh) | 用于制备含有氟磺酰基基团的酰亚胺盐的方法 | |
| CN103347811B (zh) | 氟磺酰亚胺铵盐的制造方法 | |
| JP6964595B2 (ja) | リチウムビス(フルオロスルホニル)イミドの新規の製造方法 | |
| JP6391081B2 (ja) | リチウムビス(フルオロスルホニル)イミドの生成方法 | |
| JP6495041B2 (ja) | ジハロリン酸アルカリ金属塩の製造方法およびジフルオロリン酸アルカリ金属塩の製造方法 | |
| JP4621783B2 (ja) | フルオロスルホニルイミド類およびその製造方法 | |
| US11420877B2 (en) | Lithium fluorosulfonate production method | |
| JP2017514779A (ja) | フルオロスルホニル基を含むイミドの調製 | |
| JP2004522681A (ja) | ハロスルホニル基、又はジハロホスホニル基を含む化合物をフッ素化するための方法 | |
| JP5672016B2 (ja) | フッ素化合物の製造方法 | |
| NL2020683B1 (en) | Production of lithium hexafluorophosphate | |
| JP6709686B2 (ja) | ビス(フルオロスルホニル)イミドアルカリ金属塩の製造方法 | |
| WO2019203095A1 (ja) | トリジオキシビフェニルシクロトリホスファゼンの製造方法 | |
| JP6921464B2 (ja) | リチウムビス(フルオロスルホニル)イミド塩の製造方法 | |
| KR20130021387A (ko) | 불소 함유 이미드 화합물의 제조 방법 | |
| JP2016504318A (ja) | 4−アミノ−5−フルオロ−3−クロロ−6−(置換)ピコリネートの調製方法 | |
| JP6779468B2 (ja) | ペンタフルオロスルファニルピリジン | |
| AU2020243365A1 (en) | Methods of preparing fluorinated alcohols | |
| JPWO2010001673A1 (ja) | フルオロプロピレンカーボネートの製造法 | |
| JP4642241B2 (ja) | N(cf3)2アニオンの生成およびその使用 | |
| JP2016509996A (ja) | イオン液体を製造する方法 | |
| JP2011037784A (ja) | ペルフルオロアルキルスルホンアミドの製造方法 | |
| JPH02240056A (ja) | メタンスルホニルフルオライドの製造方法 | |
| CN120641352A (zh) | 方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20130826 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140902 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140919 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20141125 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20141208 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5672016 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |