JP5587784B2 - 縮合複素環誘導体及びその医薬用途 - Google Patents
縮合複素環誘導体及びその医薬用途 Download PDFInfo
- Publication number
- JP5587784B2 JP5587784B2 JP2010533902A JP2010533902A JP5587784B2 JP 5587784 B2 JP5587784 B2 JP 5587784B2 JP 2010533902 A JP2010533902 A JP 2010533902A JP 2010533902 A JP2010533902 A JP 2010533902A JP 5587784 B2 JP5587784 B2 JP 5587784B2
- Authority
- JP
- Japan
- Prior art keywords
- alkyl
- ring
- alkoxy
- mono
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 125000000623 heterocyclic group Chemical group 0.000 title claims description 58
- 125000000217 alkyl group Chemical group 0.000 claims description 114
- LEHOTFFKMJEONL-UHFFFAOYSA-N Uric Acid Chemical compound N1C(=O)NC(=O)C2=C1NC(=O)N2 LEHOTFFKMJEONL-UHFFFAOYSA-N 0.000 claims description 71
- TVWHNULVHGKJHS-UHFFFAOYSA-N Uric acid Natural products N1C(=O)NC(=O)C2NC(=O)NC21 TVWHNULVHGKJHS-UHFFFAOYSA-N 0.000 claims description 68
- -1 amino, carboxy Chemical group 0.000 claims description 68
- 229940116269 uric acid Drugs 0.000 claims description 68
- 125000003545 alkoxy group Chemical group 0.000 claims description 63
- 150000003839 salts Chemical class 0.000 claims description 50
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 claims description 39
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 37
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 35
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 29
- 201000001431 Hyperuricemia Diseases 0.000 claims description 29
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 28
- 125000005115 alkyl carbamoyl group Chemical group 0.000 claims description 24
- 239000003814 drug Substances 0.000 claims description 24
- 238000004519 manufacturing process Methods 0.000 claims description 24
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 22
- 239000008194 pharmaceutical composition Substances 0.000 claims description 18
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 claims description 17
- 125000001153 fluoro group Chemical group F* 0.000 claims description 17
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 15
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 15
- 201000010099 disease Diseases 0.000 claims description 15
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 15
- 229940079593 drug Drugs 0.000 claims description 15
- 229910052731 fluorine Inorganic materials 0.000 claims description 14
- 125000002252 acyl group Chemical group 0.000 claims description 13
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 13
- 125000005843 halogen group Chemical group 0.000 claims description 12
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 11
- 125000004432 carbon atom Chemical group C* 0.000 claims description 11
- 125000006624 (C1-C6) alkoxycarbonylamino group Chemical group 0.000 claims description 10
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 10
- 201000005569 Gout Diseases 0.000 claims description 10
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 10
- 230000002265 prevention Effects 0.000 claims description 10
- 239000004480 active ingredient Substances 0.000 claims description 9
- 239000003112 inhibitor Substances 0.000 claims description 9
- 229910052757 nitrogen Inorganic materials 0.000 claims description 9
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 9
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 8
- 229910052799 carbon Inorganic materials 0.000 claims description 8
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 8
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 7
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 7
- 125000001424 substituent group Chemical group 0.000 claims description 7
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Chemical group C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 6
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 claims description 5
- 125000004845 (C1-C6) alkylsulfonylamino group Chemical group 0.000 claims description 5
- 125000004755 (C2-C7) acylamino group Chemical group 0.000 claims description 5
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 5
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 5
- 206010007027 Calculus urinary Diseases 0.000 claims description 5
- 125000003282 alkyl amino group Chemical group 0.000 claims description 5
- 125000005153 alkyl sulfamoyl group Chemical group 0.000 claims description 5
- 206010003246 arthritis Diseases 0.000 claims description 5
- 125000005223 heteroarylcarbonyl group Chemical group 0.000 claims description 5
- 208000017169 kidney disease Diseases 0.000 claims description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 5
- 229930192474 thiophene Chemical group 0.000 claims description 5
- 208000008281 urolithiasis Diseases 0.000 claims description 5
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical group C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 claims description 4
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical group C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 claims description 4
- 125000004429 atom Chemical group 0.000 claims description 4
- 125000005553 heteroaryloxy group Chemical group 0.000 claims description 4
- 125000004366 heterocycloalkenyl group Chemical group 0.000 claims description 4
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims description 3
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical group C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 3
- 229940123769 Xanthine oxidase inhibitor Drugs 0.000 claims description 3
- 125000005194 alkoxycarbonyloxy group Chemical group 0.000 claims description 3
- 125000005241 heteroarylamino group Chemical group 0.000 claims description 3
- 239000003064 xanthine oxidase inhibitor Substances 0.000 claims description 3
- 125000000081 (C5-C8) cycloalkenyl group Chemical group 0.000 claims description 2
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical group C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 claims description 2
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Chemical group C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 claims description 2
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical group C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 claims description 2
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical group C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 claims description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 2
- 230000001939 inductive effect Effects 0.000 claims 13
- 230000006698 induction Effects 0.000 claims 4
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 159
- 150000001875 compounds Chemical class 0.000 description 108
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 92
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 74
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 72
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 66
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 63
- 239000000203 mixture Substances 0.000 description 53
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 50
- 239000011541 reaction mixture Substances 0.000 description 46
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 42
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 37
- 239000000243 solution Substances 0.000 description 34
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 33
- 235000019341 magnesium sulphate Nutrition 0.000 description 33
- 239000012044 organic layer Substances 0.000 description 32
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 30
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 29
- 238000006243 chemical reaction Methods 0.000 description 28
- 239000000651 prodrug Substances 0.000 description 28
- 229940002612 prodrug Drugs 0.000 description 28
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 27
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 27
- 238000010898 silica gel chromatography Methods 0.000 description 23
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 21
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- 239000012442 inert solvent Substances 0.000 description 21
- 230000002829 reductive effect Effects 0.000 description 21
- 239000012156 elution solvent Substances 0.000 description 20
- 238000000034 method Methods 0.000 description 19
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 19
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 18
- 101000821903 Homo sapiens Solute carrier family 22 member 12 Proteins 0.000 description 18
- 230000029142 excretion Effects 0.000 description 18
- 239000002904 solvent Substances 0.000 description 18
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 16
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 238000012360 testing method Methods 0.000 description 15
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 14
- 102100021495 Solute carrier family 22 member 12 Human genes 0.000 description 14
- 238000001816 cooling Methods 0.000 description 14
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 14
- 230000002401 inhibitory effect Effects 0.000 description 14
- 239000002994 raw material Substances 0.000 description 14
- 239000011259 mixed solution Substances 0.000 description 13
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 12
- 230000035484 reaction time Effects 0.000 description 12
- 238000010992 reflux Methods 0.000 description 12
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 11
- 210000004027 cell Anatomy 0.000 description 11
- 238000001914 filtration Methods 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 10
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 9
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 9
- 108010093894 Xanthine oxidase Proteins 0.000 description 9
- 102100033220 Xanthine oxidase Human genes 0.000 description 9
- 125000003277 amino group Chemical group 0.000 description 9
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 9
- 229910000024 caesium carbonate Inorganic materials 0.000 description 9
- JYVHOGDBFNJNMR-UHFFFAOYSA-N hexane;hydrate Chemical compound O.CCCCCC JYVHOGDBFNJNMR-UHFFFAOYSA-N 0.000 description 9
- 239000012046 mixed solvent Substances 0.000 description 9
- 210000002966 serum Anatomy 0.000 description 9
- 229940124597 therapeutic agent Drugs 0.000 description 9
- 239000008096 xylene Substances 0.000 description 9
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 8
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 210000003734 kidney Anatomy 0.000 description 8
- 239000010410 layer Substances 0.000 description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 7
- OFCNXPDARWKPPY-UHFFFAOYSA-N allopurinol Chemical compound OC1=NC=NC2=C1C=NN2 OFCNXPDARWKPPY-UHFFFAOYSA-N 0.000 description 7
- 229960003459 allopurinol Drugs 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 238000007796 conventional method Methods 0.000 description 7
- 229910052763 palladium Inorganic materials 0.000 description 7
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 7
- 230000003449 preventive effect Effects 0.000 description 7
- 239000007858 starting material Substances 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 6
- 101100221606 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) COS7 gene Proteins 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- 230000002159 abnormal effect Effects 0.000 description 6
- 239000003054 catalyst Substances 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 6
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 6
- 229910052740 iodine Inorganic materials 0.000 description 6
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 6
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 6
- 239000002953 phosphate buffered saline Substances 0.000 description 6
- 235000011056 potassium acetate Nutrition 0.000 description 6
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 5
- 241000282412 Homo Species 0.000 description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 5
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 5
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 5
- 229910052801 chlorine Inorganic materials 0.000 description 5
- 125000001309 chloro group Chemical group Cl* 0.000 description 5
- 239000013604 expression vector Substances 0.000 description 5
- 102000056457 human SLC22A12 Human genes 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 239000012312 sodium hydride Substances 0.000 description 5
- 229910000104 sodium hydride Inorganic materials 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 4
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000012131 assay buffer Substances 0.000 description 4
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 4
- 239000012024 dehydrating agents Substances 0.000 description 4
- 238000010511 deprotection reaction Methods 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 4
- 230000001737 promoting effect Effects 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 4
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 4
- 230000002485 urinary effect Effects 0.000 description 4
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 3
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 3
- QEKCOTPRXZPSPZ-UHFFFAOYSA-N 5-bromo-1-methyl-7-nitroindole Chemical compound BrC1=CC([N+]([O-])=O)=C2N(C)C=CC2=C1 QEKCOTPRXZPSPZ-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 3
- 208000009911 Urinary Calculi Diseases 0.000 description 3
- 229960002529 benzbromarone Drugs 0.000 description 3
- WHQCHUCQKNIQEC-UHFFFAOYSA-N benzbromarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(Br)=C(O)C(Br)=C1 WHQCHUCQKNIQEC-UHFFFAOYSA-N 0.000 description 3
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 3
- CFBIOWPDDZPIDP-UHFFFAOYSA-N ethyl 2-bromo-4-methyl-1,3-thiazole-5-carboxylate Chemical compound CCOC(=O)C=1SC(Br)=NC=1C CFBIOWPDDZPIDP-UHFFFAOYSA-N 0.000 description 3
- IGRLNCOFYMWKBU-UHFFFAOYSA-N ethyl 2-chloropyridine-4-carboxylate Chemical compound CCOC(=O)C1=CC=NC(Cl)=C1 IGRLNCOFYMWKBU-UHFFFAOYSA-N 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 3
- 125000004043 oxo group Chemical group O=* 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 239000001632 sodium acetate Substances 0.000 description 3
- 235000017281 sodium acetate Nutrition 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 125000004434 sulfur atom Chemical group 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 125000005951 trifluoromethanesulfonyloxy group Chemical group 0.000 description 3
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 3
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 2
- IANQTJSKSUMEQM-UHFFFAOYSA-N 1-benzofuran Chemical compound C1=CC=C2OC=CC2=C1 IANQTJSKSUMEQM-UHFFFAOYSA-N 0.000 description 2
- AJUZODBSVAJVQQ-UHFFFAOYSA-N 1-methyl-7-nitro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indole Chemical compound C=1C([N+]([O-])=O)=C2N(C)C=CC2=CC=1B1OC(C)(C)C(C)(C)O1 AJUZODBSVAJVQQ-UHFFFAOYSA-N 0.000 description 2
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 2
- CKLDYHWZYWTBKQ-UHFFFAOYSA-N 2-(7-cyano-1-benzothiophen-5-yl)-4-methyl-1,3-thiazole-5-carboxylic acid Chemical compound S1C(C(O)=O)=C(C)N=C1C1=CC(C#N)=C(SC=C2)C2=C1 CKLDYHWZYWTBKQ-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- YGQCHVOXQFVYAB-UHFFFAOYSA-N 4-methyl-2-(1-methyl-7-nitroindol-5-yl)-1,3-thiazole-5-carboxylic acid Chemical compound S1C(C(O)=O)=C(C)N=C1C1=CC([N+]([O-])=O)=C(N(C)C=C2)C2=C1 YGQCHVOXQFVYAB-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- CABHADOSBTYWMQ-UHFFFAOYSA-N 5-bromo-7-nitro-1-benzofuran Chemical compound [O-][N+](=O)C1=CC(Br)=CC2=C1OC=C2 CABHADOSBTYWMQ-UHFFFAOYSA-N 0.000 description 2
- VULLKSFMGADJIO-UHFFFAOYSA-N 7-hydroxy-1-benzothiophene-5-carboxamide Chemical compound NC(=O)C1=CC(O)=C2SC=CC2=C1 VULLKSFMGADJIO-UHFFFAOYSA-N 0.000 description 2
- NEGUYZPTEJBCNN-UHFFFAOYSA-N 7-hydroxy-1-benzothiophene-5-carboxylic acid Chemical compound OC(=O)C1=CC(O)=C2SC=CC2=C1 NEGUYZPTEJBCNN-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- 108010078791 Carrier Proteins Proteins 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- 206010021131 Hypouricaemia Diseases 0.000 description 2
- 239000012097 Lipofectamine 2000 Substances 0.000 description 2
- 206010067125 Liver injury Diseases 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 102100030935 Solute carrier family 2, facilitated glucose transporter member 9 Human genes 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 238000006161 Suzuki-Miyaura coupling reaction Methods 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 108010092464 Urate Oxidase Proteins 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 150000008065 acid anhydrides Chemical class 0.000 description 2
- 239000012445 acidic reagent Substances 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 125000004949 alkyl amino carbonyl amino group Chemical group 0.000 description 2
- POJWUDADGALRAB-UHFFFAOYSA-N allantoin Chemical compound NC(=O)NC1NC(=O)NC1=O POJWUDADGALRAB-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- RDULEYWUGKOCMR-UHFFFAOYSA-N ethyl 2-chloro-3-oxobutanoate Chemical compound CCOC(=O)C(Cl)C(C)=O RDULEYWUGKOCMR-UHFFFAOYSA-N 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 231100000234 hepatic damage Toxicity 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 2
- 239000004310 lactic acid Substances 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 230000008818 liver damage Effects 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N naphthalene-acid Natural products C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 125000002524 organometallic group Chemical group 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- HXNFUBHNUDHIGC-UHFFFAOYSA-N oxypurinol Chemical compound O=C1NC(=O)N=C2NNC=C21 HXNFUBHNUDHIGC-UHFFFAOYSA-N 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 239000011698 potassium fluoride Substances 0.000 description 2
- 235000003270 potassium fluoride Nutrition 0.000 description 2
- 229960003081 probenecid Drugs 0.000 description 2
- DBABZHXKTCFAPX-UHFFFAOYSA-N probenecid Chemical compound CCCN(CCC)S(=O)(=O)C1=CC=C(C(O)=O)C=C1 DBABZHXKTCFAPX-UHFFFAOYSA-N 0.000 description 2
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000004742 propyloxycarbonyl group Chemical group 0.000 description 2
- NIPZZXUFJPQHNH-UHFFFAOYSA-N pyrazine-2-carboxylic acid Chemical compound OC(=O)C1=CN=CC=N1 NIPZZXUFJPQHNH-UHFFFAOYSA-N 0.000 description 2
- 230000009103 reabsorption Effects 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 229940126585 therapeutic drug Drugs 0.000 description 2
- YUKQRDCYNOVPGJ-UHFFFAOYSA-N thioacetamide Chemical compound CC(N)=S YUKQRDCYNOVPGJ-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N thioacetamide Natural products CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- NHDIQVFFNDKAQU-UHFFFAOYSA-N tripropan-2-yl borate Chemical compound CC(C)OB(OC(C)C)OC(C)C NHDIQVFFNDKAQU-UHFFFAOYSA-N 0.000 description 2
- 108010078530 urate transporter Proteins 0.000 description 2
- 229940075420 xanthine Drugs 0.000 description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- YJTKZCDBKVTVBY-UHFFFAOYSA-N 1,3-Diphenylbenzene Chemical group C1=CC=CC=C1C1=CC=CC(C=2C=CC=CC=2)=C1 YJTKZCDBKVTVBY-UHFFFAOYSA-N 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- SIXUTAQIEXGSDL-UHFFFAOYSA-N 1,3-dimethyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indole-7-carbonitrile Chemical compound Cc1cn(C)c2c(cc(cc12)B1OC(C)(C)C(C)(C)O1)C#N SIXUTAQIEXGSDL-UHFFFAOYSA-N 0.000 description 1
- YZVFSQQHQPPKNX-UHFFFAOYSA-N 1,3-thiazole-5-carboxylic acid Chemical compound OC(=O)C1=CN=CS1 YZVFSQQHQPPKNX-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- DDITYYPIADDMFF-UHFFFAOYSA-N 1-benzothiophen-3-amine;hydrochloride Chemical compound Cl.C1=CC=C2C(N)=CSC2=C1 DDITYYPIADDMFF-UHFFFAOYSA-N 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- AEQDJSLRWYMAQI-UHFFFAOYSA-N 2,3,9,10-tetramethoxy-6,8,13,13a-tetrahydro-5H-isoquinolino[2,1-b]isoquinoline Chemical compound C1CN2CC(C(=C(OC)C=C3)OC)=C3CC2C2=C1C=C(OC)C(OC)=C2 AEQDJSLRWYMAQI-UHFFFAOYSA-N 0.000 description 1
- IROGYIJRTPJAKF-UHFFFAOYSA-N 2-(1-methyl-7-nitroindol-5-yl)pyridine-4-carboxylic acid Chemical compound C=1C([N+]([O-])=O)=C2N(C)C=CC2=CC=1C1=CC(C(O)=O)=CC=N1 IROGYIJRTPJAKF-UHFFFAOYSA-N 0.000 description 1
- YQXSNUBLWXKBKH-UHFFFAOYSA-N 2-(7-cyano-1,2-dimethylindol-5-yl)-4-methyl-1,3-thiazole-5-carboxylic acid Chemical compound C=1C(C#N)=C2N(C)C(C)=CC2=CC=1C1=NC(C)=C(C(O)=O)S1 YQXSNUBLWXKBKH-UHFFFAOYSA-N 0.000 description 1
- HDGLAXYKCCHAIK-UHFFFAOYSA-N 2-(7-cyano-1-methylindol-5-yl)pyridine-4-carboxylic acid Chemical compound C=1C(C#N)=C2N(C)C=CC2=CC=1C1=CC(C(O)=O)=CC=N1 HDGLAXYKCCHAIK-UHFFFAOYSA-N 0.000 description 1
- YNLLJZXBTDPZEY-UHFFFAOYSA-N 2-(7-cyano-1h-indol-5-yl)pyridine-4-carboxylic acid Chemical compound OC(=O)C1=CC=NC(C=2C=C3C=CNC3=C(C#N)C=2)=C1 YNLLJZXBTDPZEY-UHFFFAOYSA-N 0.000 description 1
- GNCIAJYDXNXDRL-UHFFFAOYSA-N 2-(7-nitro-1-benzofuran-5-yl)pyridine-4-carboxylic acid Chemical compound OC(=O)C1=CC=NC(C=2C=C3C=COC3=C([N+]([O-])=O)C=2)=C1 GNCIAJYDXNXDRL-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- WLZCMGXAEXFAQA-UHFFFAOYSA-N 2-amino-3,5-dibromobenzonitrile Chemical compound NC1=C(Br)C=C(Br)C=C1C#N WLZCMGXAEXFAQA-UHFFFAOYSA-N 0.000 description 1
- VCBFAUSKNYAKJZ-UHFFFAOYSA-N 2-chloro-3-oxobutanoic acid Chemical class CC(=O)C(Cl)C(O)=O VCBFAUSKNYAKJZ-UHFFFAOYSA-N 0.000 description 1
- FVJPIFWJQUIPNO-UHFFFAOYSA-N 2-methylpropan-2-ol;oxolane Chemical compound CC(C)(C)O.C1CCOC1 FVJPIFWJQUIPNO-UHFFFAOYSA-N 0.000 description 1
- YNKRJGZPPAUPLQ-UHFFFAOYSA-N 2-methylsulfonyl-1h-imidazole Chemical compound CS(=O)(=O)C1=NC=CN1 YNKRJGZPPAUPLQ-UHFFFAOYSA-N 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- LZPWAYBEOJRFAX-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2$l^{2}-dioxaborolane Chemical compound CC1(C)O[B]OC1(C)C LZPWAYBEOJRFAX-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- GOYIODDQHINSAD-UHFFFAOYSA-N 5-bromo-1,3-dimethylindole-7-carbonitrile Chemical compound C1=C(Br)C=C2C(C)=CN(C)C2=C1C#N GOYIODDQHINSAD-UHFFFAOYSA-N 0.000 description 1
- DOYSDLJKECVNQO-UHFFFAOYSA-N 5-bromo-1-(2-methoxyethyl)-7-nitroindole Chemical compound BrC1=CC([N+]([O-])=O)=C2N(CCOC)C=CC2=C1 DOYSDLJKECVNQO-UHFFFAOYSA-N 0.000 description 1
- YGQNFONSYKGXNB-UHFFFAOYSA-N 5-bromo-1-(2-methylpropyl)-7-nitroindole Chemical compound BrC1=CC([N+]([O-])=O)=C2N(CC(C)C)C=CC2=C1 YGQNFONSYKGXNB-UHFFFAOYSA-N 0.000 description 1
- GVUFNQJVUFPHGD-UHFFFAOYSA-N 5-bromo-1-ethyl-3-methylindole-7-carbonitrile Chemical compound BrC1=CC(C#N)=C2N(CC)C=C(C)C2=C1 GVUFNQJVUFPHGD-UHFFFAOYSA-N 0.000 description 1
- YEYPSUQQZNDKDE-UHFFFAOYSA-N 5-bromo-2-hydroxy-3-nitrobenzaldehyde Chemical compound OC1=C(C=O)C=C(Br)C=C1[N+]([O-])=O YEYPSUQQZNDKDE-UHFFFAOYSA-N 0.000 description 1
- WSVZUNHHRDHIIP-UHFFFAOYSA-N 5-bromo-3-methyl-1-(2-methylpropyl)indole-7-carbonitrile Chemical compound BrC1=CC(C#N)=C2N(CC(C)C)C=C(C)C2=C1 WSVZUNHHRDHIIP-UHFFFAOYSA-N 0.000 description 1
- FJLCJKGJADCBES-UHFFFAOYSA-N 5-bromo-3-methyl-1-propan-2-ylindole-7-carbonitrile Chemical compound BrC1=CC(C#N)=C2N(C(C)C)C=C(C)C2=C1 FJLCJKGJADCBES-UHFFFAOYSA-N 0.000 description 1
- BIZYDCLBAGQWJE-UHFFFAOYSA-N 5-bromo-3-methyl-1-propylindole-7-carbonitrile Chemical compound BrC1=CC(C#N)=C2N(CCC)C=C(C)C2=C1 BIZYDCLBAGQWJE-UHFFFAOYSA-N 0.000 description 1
- KMQQSKDTKMRIBU-UHFFFAOYSA-N 5-bromo-3-methyl-1h-indole-7-carbonitrile Chemical compound C1=C(Br)C=C2C(C)=CNC2=C1C#N KMQQSKDTKMRIBU-UHFFFAOYSA-N 0.000 description 1
- MCDXSWCYMHBYDU-UHFFFAOYSA-N 5-bromo-7-nitro-1-benzofuran-2-carboxylic acid Chemical compound BrC1=CC([N+]([O-])=O)=C2OC(C(=O)O)=CC2=C1 MCDXSWCYMHBYDU-UHFFFAOYSA-N 0.000 description 1
- WERRDKWLHFDNTO-UHFFFAOYSA-N 5-bromo-7-nitro-1h-indole Chemical compound [O-][N+](=O)C1=CC(Br)=CC2=C1NC=C2 WERRDKWLHFDNTO-UHFFFAOYSA-N 0.000 description 1
- PZIQPEMTOGYRRJ-UHFFFAOYSA-N 7-hydroxy-1-benzothiophene-5-carbonitrile Chemical compound OC1=CC(C#N)=CC2=C1SC=C2 PZIQPEMTOGYRRJ-UHFFFAOYSA-N 0.000 description 1
- 208000009304 Acute Kidney Injury Diseases 0.000 description 1
- POJWUDADGALRAB-PVQJCKRUSA-N Allantoin Natural products NC(=O)N[C@@H]1NC(=O)NC1=O POJWUDADGALRAB-PVQJCKRUSA-N 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- KOTSLJITDVEVRJ-UHFFFAOYSA-N CC1=C(SC(=N1)C2=CC(=C3C(=C2)C=CN3)Br)C(=O)O Chemical compound CC1=C(SC(=N1)C2=CC(=C3C(=C2)C=CN3)Br)C(=O)O KOTSLJITDVEVRJ-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 239000005973 Carvone Substances 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 206010012455 Dermatitis exfoliative Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- MUXOBHXGJLMRAB-UHFFFAOYSA-N Dimethyl succinate Chemical compound COC(=O)CCC(=O)OC MUXOBHXGJLMRAB-UHFFFAOYSA-N 0.000 description 1
- 208000018522 Gastrointestinal disease Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 238000006130 Horner-Wadsworth-Emmons olefination reaction Methods 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 208000034624 Leukocytoclastic Cutaneous Vasculitis Diseases 0.000 description 1
- 208000032514 Leukocytoclastic vasculitis Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 208000001940 Massive Hepatic Necrosis Diseases 0.000 description 1
- YXOLAZRVSSWPPT-UHFFFAOYSA-N Morin Chemical compound OC1=CC(O)=CC=C1C1=C(O)C(=O)C2=C(O)C=C(O)C=C2O1 YXOLAZRVSSWPPT-UHFFFAOYSA-N 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- IIBOGKHTXBPGEI-UHFFFAOYSA-N N-benzylformamide Chemical compound O=CNCC1=CC=CC=C1 IIBOGKHTXBPGEI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 108091068479 OAT family Proteins 0.000 description 1
- 208000005374 Poisoning Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- HLCFGWHYROZGBI-JJKGCWMISA-M Potassium gluconate Chemical compound [K+].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O HLCFGWHYROZGBI-JJKGCWMISA-M 0.000 description 1
- TVQZAMVBTVNYLA-UHFFFAOYSA-N Pranoprofen Chemical compound C1=CC=C2CC3=CC(C(C(O)=O)C)=CC=C3OC2=N1 TVQZAMVBTVNYLA-UHFFFAOYSA-N 0.000 description 1
- 208000033626 Renal failure acute Diseases 0.000 description 1
- 241000219061 Rheum Species 0.000 description 1
- 108091006745 SLC22A12 Proteins 0.000 description 1
- 206010042033 Stevens-Johnson syndrome Diseases 0.000 description 1
- 231100000168 Stevens-Johnson syndrome Toxicity 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 241000269370 Xenopus <genus> Species 0.000 description 1
- CVNMBKFJYRAHPO-UHFFFAOYSA-N [chloro(methyl)phosphoryl]methane Chemical compound CP(C)(Cl)=O CVNMBKFJYRAHPO-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 201000011040 acute kidney failure Diseases 0.000 description 1
- 208000012998 acute renal failure Diseases 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000003113 alkalizing effect Effects 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 229960000458 allantoin Drugs 0.000 description 1
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 150000001510 aspartic acids Chemical class 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical class N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- NWNPYJBRVCYRMG-UHFFFAOYSA-N benzyl 2,3-dihydroindole-1-carboxylate Chemical compound C1CC2=CC=CC=C2N1C(=O)OCC1=CC=CC=C1 NWNPYJBRVCYRMG-UHFFFAOYSA-N 0.000 description 1
- AGHRMUGIUJNCSE-UHFFFAOYSA-N benzyl 5-(4-ethoxycarbonylpyridin-2-yl)-2,3-dihydroindole-1-carboxylate Chemical compound CCOC(=O)C1=CC=NC(C=2C=C3CCN(C3=CC=2)C(=O)OCC=2C=CC=CC=2)=C1 AGHRMUGIUJNCSE-UHFFFAOYSA-N 0.000 description 1
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 1
- 150000005347 biaryls Chemical group 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 239000004227 calcium gluconate Substances 0.000 description 1
- 235000013927 calcium gluconate Nutrition 0.000 description 1
- 229960004494 calcium gluconate Drugs 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- NEEHYRZPVYRGPP-UHFFFAOYSA-L calcium;2,3,4,5,6-pentahydroxyhexanoate Chemical compound [Ca+2].OCC(O)C(O)C(O)C(O)C([O-])=O.OCC(O)C(O)C(O)C(O)C([O-])=O NEEHYRZPVYRGPP-UHFFFAOYSA-L 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 230000006315 carbonylation Effects 0.000 description 1
- 238000005810 carbonylation reaction Methods 0.000 description 1
- ZDQWVKDDJDIVAL-UHFFFAOYSA-N catecholborane Chemical compound C1=CC=C2O[B]OC2=C1 ZDQWVKDDJDIVAL-UHFFFAOYSA-N 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229960001338 colchicine Drugs 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- IGARGHRYKHJQSM-UHFFFAOYSA-N cyclohexylbenzene Chemical compound C1CCCCC1C1=CC=CC=C1 IGARGHRYKHJQSM-UHFFFAOYSA-N 0.000 description 1
- 125000002933 cyclohexyloxy group Chemical group C1(CCCCC1)O* 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- LCSNDSFWVKMJCT-UHFFFAOYSA-N dicyclohexyl-(2-phenylphenyl)phosphane Chemical group C1CCCCC1P(C=1C(=CC=CC=1)C=1C=CC=CC=1)C1CCCCC1 LCSNDSFWVKMJCT-UHFFFAOYSA-N 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- FNJVDWXUKLTFFL-UHFFFAOYSA-N diethyl 2-bromopropanedioate Chemical compound CCOC(=O)C(Br)C(=O)OCC FNJVDWXUKLTFFL-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- CIPMPQGRFNDLAP-UHFFFAOYSA-N ethyl 1,3-thiazole-5-carboxylate Chemical compound CCOC(=O)C1=CN=CS1 CIPMPQGRFNDLAP-UHFFFAOYSA-N 0.000 description 1
- FOTGKPSJRVSZAN-UHFFFAOYSA-N ethyl 2-(7-cyano-1,2-dimethylindol-5-yl)-4-methyl-1,3-thiazole-5-carboxylate Chemical compound CC1=C(C(=O)OCC)SC(C=2C=C3C=C(C)N(C)C3=C(C#N)C=2)=N1 FOTGKPSJRVSZAN-UHFFFAOYSA-N 0.000 description 1
- SRBKLTQHZSDMLP-UHFFFAOYSA-N ethyl 2-(7-hydroxy-1-benzothiophen-5-yl)-4-methyl-1,3-thiazole-5-carboxylate Chemical compound CC1=C(C(=O)OCC)SC(C=2C=C3C=CSC3=C(O)C=2)=N1 SRBKLTQHZSDMLP-UHFFFAOYSA-N 0.000 description 1
- DWXKSCKBUSAOKS-UHFFFAOYSA-N ethyl 2-chloro-3-oxopropanoate Chemical compound CCOC(=O)C(Cl)C=O DWXKSCKBUSAOKS-UHFFFAOYSA-N 0.000 description 1
- ORCQTMZHDQSNOJ-UHFFFAOYSA-N ethyl 2-methyl-1,3-thiazole-5-carboxylate Chemical compound CCOC(=O)C1=CN=C(C)S1 ORCQTMZHDQSNOJ-UHFFFAOYSA-N 0.000 description 1
- ZAIZUIZXYBYBJF-UHFFFAOYSA-N ethyl 5-bromo-7-nitro-1-benzofuran-2-carboxylate Chemical compound BrC1=CC([N+]([O-])=O)=C2OC(C(=O)OCC)=CC2=C1 ZAIZUIZXYBYBJF-UHFFFAOYSA-N 0.000 description 1
- JPAQEUHUFZDVDQ-UHFFFAOYSA-N ethyl 8-iodoquinoline-6-carboxylate Chemical compound N1=CC=CC2=CC(C(=O)OCC)=CC(I)=C21 JPAQEUHUFZDVDQ-UHFFFAOYSA-N 0.000 description 1
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 1
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 208000004526 exfoliative dermatitis Diseases 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- ZPAKPRAICRBAOD-UHFFFAOYSA-N fenbufen Chemical compound C1=CC(C(=O)CCC(=O)O)=CC=C1C1=CC=CC=C1 ZPAKPRAICRBAOD-UHFFFAOYSA-N 0.000 description 1
- 229960001395 fenbufen Drugs 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000010575 fractional recrystallization Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 201000006362 hypersensitivity vasculitis Diseases 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000000654 isopropylidene group Chemical group C(C)(C)=* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- DKYWVDODHFEZIM-UHFFFAOYSA-N ketoprofen Chemical compound OC(=O)C(C)C1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 DKYWVDODHFEZIM-UHFFFAOYSA-N 0.000 description 1
- 229960000991 ketoprofen Drugs 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 1
- WGOPGODQLGJZGL-UHFFFAOYSA-N lithium;butane Chemical compound [Li+].CC[CH-]C WGOPGODQLGJZGL-UHFFFAOYSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 230000005976 liver dysfunction Effects 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- LVKCSZQWLOVUGB-UHFFFAOYSA-M magnesium;propane;bromide Chemical compound [Mg+2].[Br-].C[CH-]C LVKCSZQWLOVUGB-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- OHZZTXYKLXZFSZ-UHFFFAOYSA-I manganese(3+) 5,10,15-tris(1-methylpyridin-1-ium-4-yl)-20-(1-methylpyridin-4-ylidene)porphyrin-22-ide pentachloride Chemical compound [Cl-].[Cl-].[Cl-].[Cl-].[Cl-].[Mn+3].C1=CN(C)C=CC1=C1C(C=C2)=NC2=C(C=2C=C[N+](C)=CC=2)C([N-]2)=CC=C2C(C=2C=C[N+](C)=CC=2)=C(C=C2)N=C2C(C=2C=C[N+](C)=CC=2)=C2N=C1C=C2 OHZZTXYKLXZFSZ-UHFFFAOYSA-I 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000037353 metabolic pathway Effects 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- IAUBUELRSHDINQ-UHFFFAOYSA-N methyl 7-acetyloxy-1-benzothiophene-5-carboxylate Chemical compound COC(=O)C1=CC(OC(C)=O)=C2SC=CC2=C1 IAUBUELRSHDINQ-UHFFFAOYSA-N 0.000 description 1
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 210000000110 microvilli Anatomy 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- UXOUKMQIEVGVLY-UHFFFAOYSA-N morin Natural products OC1=CC(O)=CC(C2=C(C(=O)C3=C(O)C=C(O)C=C3O2)O)=C1 UXOUKMQIEVGVLY-UHFFFAOYSA-N 0.000 description 1
- 235000007708 morin Nutrition 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-M naproxen(1-) Chemical compound C1=C([C@H](C)C([O-])=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-M 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 1
- 210000000287 oocyte Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002891 organic anions Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- OFPXSFXSNFPTHF-UHFFFAOYSA-N oxaprozin Chemical compound O1C(CCC(=O)O)=NC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 OFPXSFXSNFPTHF-UHFFFAOYSA-N 0.000 description 1
- 229960002739 oxaprozin Drugs 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- PBDBXAQKXCXZCJ-UHFFFAOYSA-L palladium(2+);2,2,2-trifluoroacetate Chemical compound [Pd+2].[O-]C(=O)C(F)(F)F.[O-]C(=O)C(F)(F)F PBDBXAQKXCXZCJ-UHFFFAOYSA-L 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- ANRQGKOBLBYXFM-UHFFFAOYSA-M phenylmagnesium bromide Chemical compound Br[Mg]C1=CC=CC=C1 ANRQGKOBLBYXFM-UHFFFAOYSA-M 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 125000000612 phthaloyl group Chemical group C(C=1C(C(=O)*)=CC=CC1)(=O)* 0.000 description 1
- 231100000572 poisoning Toxicity 0.000 description 1
- 230000000607 poisoning effect Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229960003975 potassium Drugs 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 239000001508 potassium citrate Substances 0.000 description 1
- 229960002635 potassium citrate Drugs 0.000 description 1
- QEEAPRPFLLJWCF-UHFFFAOYSA-K potassium citrate (anhydrous) Chemical compound [K+].[K+].[K+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O QEEAPRPFLLJWCF-UHFFFAOYSA-K 0.000 description 1
- 235000011082 potassium citrates Nutrition 0.000 description 1
- NNFCIKHAZHQZJG-UHFFFAOYSA-N potassium cyanide Chemical compound [K+].N#[C-] NNFCIKHAZHQZJG-UHFFFAOYSA-N 0.000 description 1
- 239000000276 potassium ferrocyanide Substances 0.000 description 1
- 239000004224 potassium gluconate Substances 0.000 description 1
- 235000013926 potassium gluconate Nutrition 0.000 description 1
- 229960003189 potassium gluconate Drugs 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 229960003101 pranoprofen Drugs 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 210000000512 proximal kidney tubule Anatomy 0.000 description 1
- 230000004144 purine metabolism Effects 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 210000005084 renal tissue Anatomy 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- 239000000176 sodium gluconate Substances 0.000 description 1
- 235000012207 sodium gluconate Nutrition 0.000 description 1
- 229940005574 sodium gluconate Drugs 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 235000011044 succinic acid Nutrition 0.000 description 1
- 229960005137 succinic acid Drugs 0.000 description 1
- 239000007940 sugar coated tablet Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- LZNWYQJJBLGYLT-UHFFFAOYSA-N tenoxicam Chemical compound OC=1C=2SC=CC=2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 LZNWYQJJBLGYLT-UHFFFAOYSA-N 0.000 description 1
- 229960002871 tenoxicam Drugs 0.000 description 1
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- XOGGUFAVLNCTRS-UHFFFAOYSA-N tetrapotassium;iron(2+);hexacyanide Chemical compound [K+].[K+].[K+].[K+].[Fe+2].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-] XOGGUFAVLNCTRS-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 125000005505 thiomorpholino group Chemical group 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000010474 transient expression Effects 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- LGQXXHMEBUOXRP-UHFFFAOYSA-N tributyl borate Chemical compound CCCCOB(OCCCC)OCCCC LGQXXHMEBUOXRP-UHFFFAOYSA-N 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- AJSTXXYNEIHPMD-UHFFFAOYSA-N triethyl borate Chemical compound CCOB(OCC)OCC AJSTXXYNEIHPMD-UHFFFAOYSA-N 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- WRECIMRULFAWHA-UHFFFAOYSA-N trimethyl borate Chemical compound COB(OC)OC WRECIMRULFAWHA-UHFFFAOYSA-N 0.000 description 1
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- COIOYMYWGDAQPM-UHFFFAOYSA-N tris(2-methylphenyl)phosphane Chemical compound CC1=CC=CC=C1P(C=1C(=CC=CC=1)C)C1=CC=CC=C1C COIOYMYWGDAQPM-UHFFFAOYSA-N 0.000 description 1
- YZYKZHPNRDIPFA-UHFFFAOYSA-N tris(trimethylsilyl) borate Chemical compound C[Si](C)(C)OB(O[Si](C)(C)C)O[Si](C)(C)C YZYKZHPNRDIPFA-UHFFFAOYSA-N 0.000 description 1
- 229940005267 urate oxidase Drugs 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/443—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/04—Drugs for disorders of the urinary system for urolithiasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Urology & Nephrology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Immunology (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Description
尿酸は食事由来及び内因性に産生されたプリン体より、最終的にキサンチンがキサンチンオキシダーゼによる酸化を受けて産生する。アロプリノールは、キサンチンオキシダーゼ阻害剤として開発され、医療現場で用いられている唯一の尿酸生成抑制薬である。しかしながら、アロプリノールは、高尿酸血症及びこれに起因する各種疾患への有効性が報告されている反面、中毒症候群(過敏性血管炎)、スティーブンス・ジョンソン症候群、剥脱性皮膚炎、再生不良性貧血、肝機能障害などの重篤な副作用も報告されている(例えば、非特許文献2参照)。この原因のひとつとして、アロプリノールが核酸類似構造を有し、ピリミジン代謝経路を阻害することが指摘されている(例えば、非特許文献3参照)。
〔1〕 式(I)
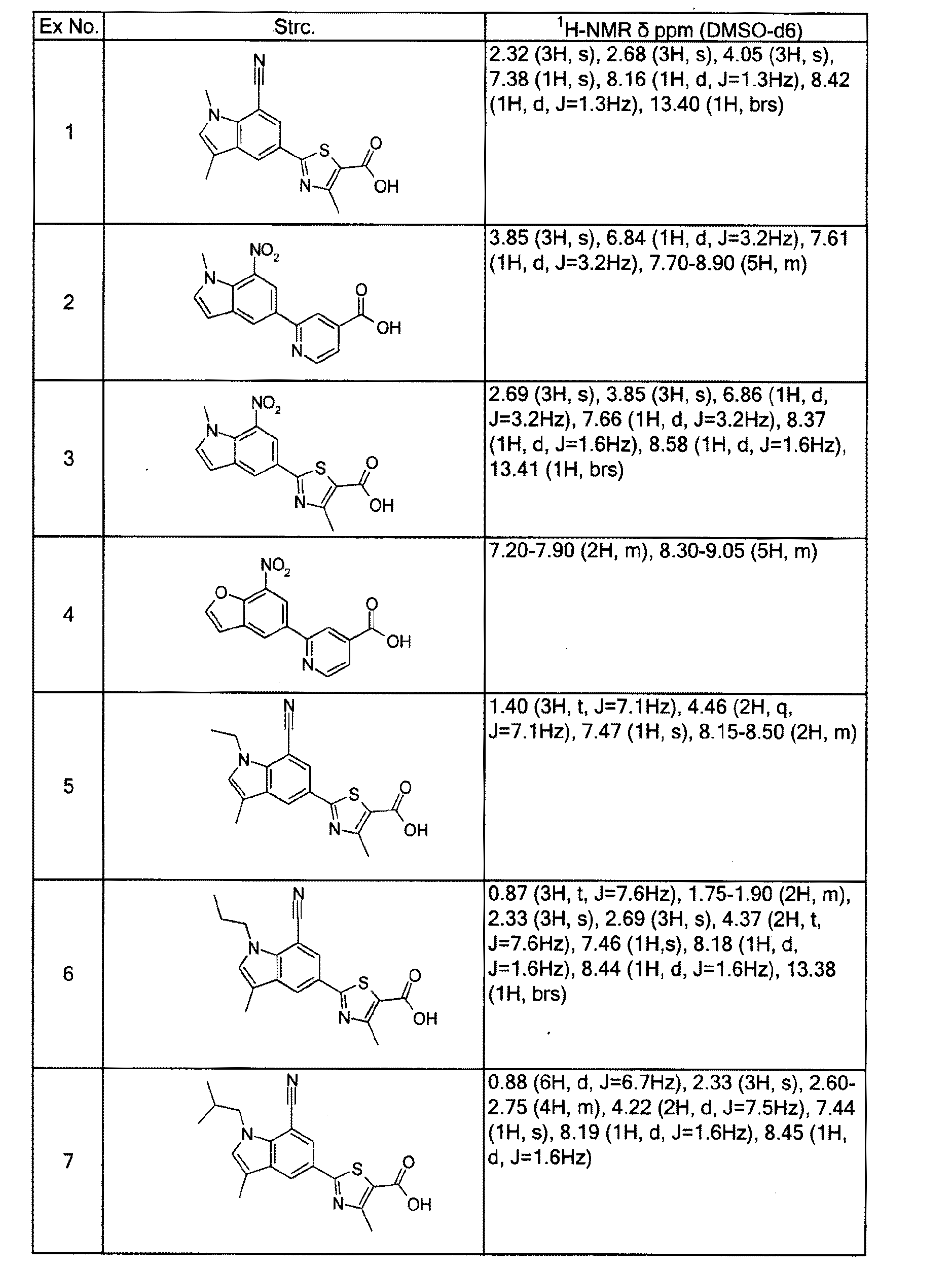
Tはトリフルオロメチル、ニトロ又はシアノ;
環Qは5又は6員環ヘテロアリール;
X1及びX2は独立して、CH又はN;
環UはC6アリール又は5又は6員環ヘテロアリール;
mは0〜2の整数;
nは0〜3の整数;
R1は水酸基、ハロゲン原子、アミノ又はC1−6アルキル(mが2であるとき、2つのR1は異なっていてもよい);
R2は、
(i)環Q内の炭素原子に結合している場合は、(1)〜(11):
(1)ハロゲン原子;
(2)水酸基;
(3)シアノ;
(4)ニトロ;
(5)カルボキシ;
(6)カルバモイル;
(7)アミノ;
(8)それぞれ独立して、置換基群αから選択される任意の(好ましくは1〜3個の)基を有していてもよいC1−6アルキル、C2−6アルケニル又はC1−6アルコキシ;
(9)それぞれ独立して、フッ素原子、水酸基及びアミノから選択される任意の基を1〜3個有していてもよいC2−6アルキニル、C1−6アルキルスルホニル、モノ(ジ)C1−6アルキルスルファモイル、C2−7アシル、C1−6アルコキシカルボニル、C1−6アルコキシカルボニルオキシ、モノ(ジ)C1−6アルキルアミノ、モノ(ジ)C1−6アルコキシC1−6アルキルアミノ、C1−6アルコキシC1−6アルキル(C1−6アルキル)アミノ、C2−7アシルアミノ、C1−6アルコキシカルボニルアミノ、C1−6アルコキシカルボニル(C1−6アルキル)アミノ、モノ(ジ)C1−6アルキルカルバモイル、モノ(ジ)C1−6アルコキシC1−6アルキルカルバモイル、C1−6アルコキシC1−6アルキル(C1−6アルキル)カルバモイル、モノ(ジ)C1−6アルキルアミノカルボニルアミノ、C1−6アルキルスルホニルアミノ又はC1−6アルキルチオ;
(10)それぞれ独立して、フッ素原子、水酸基、アミノ、オキソ、C1−6アルキル、C1−6アルコキシ、C1−6アルコキシC1−6アルキル、カルボキシ、C2−7アシル、C1−6アルコキシカルボニル、モノ(ジ)C1−6アルキルアミノ、カルバモイル、モノ(ジ)C1−6アルキルカルバモイル、モノ(ジ)C1−6アルコキシC1−6アルキルカルバモイル及びC1−6アルコキシC1−6アルキル(C1−6アルキル)カルバモイルから選択される任意の基を1〜3個有していてもよいC3−8シクロアルキル、3〜8員環ヘテロシクロアルキル、C5−8シクロアルケニル又は5〜8員環ヘテロシクロアルケニル;
(11)それぞれ独立して、ハロゲン原子、水酸基、アミノ、シアノ、ニトロ、C1−6アルキル、C1−6アルコキシ、C1−6アルコキシC1−6アルキル、カルボキシ、C2−7アシル、C1−6アルコキシカルボニル、カルバモイル、モノ(ジ)C1−6アルキルカルバモイル、モノ(ジ)C1−6アルキルアミノ、モノ(ジ)C1−6アルコキシC1−6アルキルアミノ、C1−6アルコキシC1−6アルキル(C1−6アルキル)アミノ、C1−6アルコキシカルボニルアミノ、モノ(ジ)C1−6アルコキシC1−6アルキルカルバモイル及びC1−6アルコキシC1−6アルキル(C1−6アルキル)カルバモイルから選択される任意の基を1〜3個有していてもよいC6アリール、C6アリールオキシ、C6アリールカルボニル、5又は6員環ヘテロアリール、5又は6員環ヘテロアリールオキシ、5又は6員環ヘテロアリールカルボニル、C6アリールアミノ、C6アリール(C1−6アルキル)アミノ、5又は6員環ヘテロアリールアミノ又は5又は6員環ヘテロアリール(C1−6アルキル)アミノ;の何れかであり、
(ii) 環Qの窒素原子に結合している場合は、(12)〜(15):
(12)それぞれ独立して、置換基群αから選択される任意の(好ましくは1〜3個の)基を有していてもよいC1−6アルキル又はC2−6アルケニル;
(13)それぞれ独立して、フッ素原子、水酸基及びアミノから選択される任意の基を1〜3個有していてもよいC2−6アルキニル、C1−6アルキルスルホニル、モノ(ジ)C1−6アルキルスルファモイル、C2−7アシル、C1−6アルコキシカルボニル又はモノ(ジ)C1−6アルキルカルバモイル;
(14)それぞれ独立して、フッ素原子、水酸基、アミノ、オキソ、C1−6アルキル、C1−6アルコキシ、C1−6アルコキシC1−6アルキル、カルボキシ、C2−7アシル、C1−6アルコキシカルボニル、カルバモイル、モノ(ジ)C1−6アルキルカルバモイル、モノ(ジ)C1−6アルコキシC1−6アルキルカルバモイル及びC1−6アルコキシC1−6アルキル(C1−6アルキル)カルバモイルから選択される任意の基を1〜3個有していてもよいC3−8シクロアルキル又は3〜8員環ヘテロシクロアルキル;
(15)それぞれ独立して、ハロゲン原子、水酸基、アミノ、シアノ、ニトロ、C1−6アルキル、C1−6アルコキシ、C1−6アルコキシC1−6アルキル、カルボキシ、C2−7アシル、C1−6アルコキシカルボニル、カルバモイル、モノ(ジ)C1−6アルキルカルバモイル、モノ(ジ)C1−6アルキルアミノ、モノ(ジ)C1−6アルコキシC1−6アルキルアミノ、C1−6アルコキシC1−6アルキル(C1−6アルキル)アミノ、C1−6アルコキシカルボニルアミノ、モノ(ジ)C1−6アルコキシC1−6アルキルカルバモイル及びC1−6アルコキシC1−6アルキル(C1−6アルキル)カルバモイルから選択される任意の基を1〜3個有していてもよいC6アリール、5又は6員環ヘテロアリール、C6アリールカルボニル又は5又は6員環ヘテロアリールカルボニル;の何れかであり;
(nが2又は3であるとき、R2はそれぞれ異なっていてもよく、更に、2つのR2が、環Q内の隣り合った原子に結合して存在し、それぞれC1−6アルコキシを有していてもよいC1−6アルキルである場合は、結合する環Q内の原子と共に、5〜8員環を形成してもよい。);
置換基群αは、フッ素原子;水酸基;アミノ;カルボキシ;それぞれフッ素原子、水酸基及びアミノから選択される任意の基を1〜3個有していてもよいC1−6アルコキシ、モノ(ジ)C1−6アルキルアミノ、モノ(ジ)C1−6アルコキシC1−6アルキルアミノ、C1−6アルコキシC1−6アルキル(C1−6アルキル)アミノ、C1−6アルコキシカルボニルアミノ、C2−7アシル、C1−6アルコキシカルボニル、モノ(ジ)C1−6アルキルカルバモイル、モノ(ジ)C1−6アルコキシC1−6アルキルカルバモイル、C1−6アルコキシC1−6アルキル(C1−6アルキル)カルバモイル、C1−6アルキルスルホニルアミノ、C2−7アシルアミノ及びC1−6アルコキシカルボニルアミノ;それぞれ独立して、フッ素原子、水酸基、アミノ、オキソ、C1−6アルキル、C1−6アルコキシ、C1−6アルコキシC1−6アルキル、カルボキシ、C2−7アシル、C1−6アルコキシカルボニル、カルバモイル、モノ(ジ)C1−6アルキルカルバモイル、モノ(ジ)C1−6アルコキシC1−6アルキルカルバモイル及びC1−6アルコキシC1−6アルキル(C1−6アルキル)カルバモイルから選択される任意の基を1〜3個有していてもよいC3−8シクロアルキル及び3〜8員環ヘテロシクロアルキル;及び、それぞれ独立して、ハロゲン原子、水酸基、アミノ、シアノ、ニトロ、C1−6アルキル、C1−6アルコキシ、C1−6アルコキシC1−6アルキル、カルボキシ、C2−7アシル、C1−6アルコキシカルボニル、カルバモイル、モノ(ジ)C1−6アルキルカルバモイル、モノ(ジ)C1−6アルキルアミノ、モノ(ジ)C1−6アルコキシC1−6アルキルアミノ、C1−6アルコキシC1−6アルキル(C1−6アルキル)アミノ、C1−6アルコキシカルボニルアミノ、モノ(ジ)C1−6アルコキシC1−6アルキルカルバモイル及びC1−6アルコキシC1−6アルキル(C1−6アルキル)カルバモイルから選択される任意の基を1〜3個有していてもよいC6アリール及び5又は6員環ヘテロアリールである〕で表される縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔3〕 X1がCHである前記〔1〕又は〔2〕記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔4〕 X2がCHである前記〔1〕〜〔3〕の何れかに記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔5〕 環Qがピリジン、ピリミジン、ピラジン、チアゾール、イミダゾール、ピラゾール、オキサゾール、イソチアゾール、イソオキサゾール、チオフェン、フラン又はピロール環である前記〔1〕〜〔4〕の何れかに記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔6〕 環Qがピリジン、チオフェン又はピロール環である前記〔5〕記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔7〕 環Uがベンゼン、ピリジン、チアゾール、ピラゾール又はチオフェン環である前記〔1〕〜〔6〕の何れかに記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔8〕 mが0であるか、又はmが1であり、かつ、環Uが下記式:
(式中、R1aは水酸基;アミノ;又はC1−6アルキルであり、Aは縮合環との結合を、Bはカルボキシとの結合をそれぞれ表す)で表される何れかの環である、前記〔7〕記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔10〕 環Uがチアゾール環である前記〔8〕又は〔9〕記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔11〕 mが0であるか、又はmが1;かつR1aが水酸基であり、かつ環Uがピリジン環である前記〔9〕記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔12〕 R1aがメチルである前記〔10〕記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔13〕 R1aが水酸基である前記〔11〕記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔14〕 nが0であるか、又はnが1〜3であり、かつR2が環Qの炭素原子に結合するハロゲン原子;水酸基;もしくはそれぞれフッ素原子、水酸基及びアミノから選択される任意の基を1〜3個有していてもよいC1−6アルキル、C1−6アルコキシ、C1−6アルコキシC1−6アルキルもしくはC1−6アルコキシC1−6アルコキシ;又は環Qの窒素原子に結合する、それぞれフッ素原子、水酸基及びアミノから選択される任意の基を1〜3個有していてもよいC1−6アルキルもしくはC1−6アルコキシC1−6アルキルである、前記〔1〕〜〔13〕の何れかに記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔15〕 nが0であるか、又はnが1〜3であり、かつR2が環Qの炭素原子に結合するハロゲン原子;水酸基;もしくはフッ素原子を1〜3個有していてもよいC1−6アルキル;又は環Qの窒素原子に結合する、それぞれフッ素原子を1〜3個有していてもよいC1−6アルキルもしくはC1−6アルコキシC1−6アルキルである、前記〔14〕記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔16〕 キサンチンオキシダーゼ阻害薬である、前記〔1〕〜〔15〕の何れかに記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩;
〔17〕 前記〔1〕〜〔15〕の何れかに記載の縮合複素環誘導体もしくはそのプロドラッグ又はその薬理学的に許容される塩を有効成分として含有する医薬組成物;
〔18〕 高尿酸血症、痛風結節、痛風関節炎、高尿酸血症による腎障害及び尿路結石から選択される疾患の予防又は治療用である、前記〔17〕記載の医薬組成物;
〔19〕 高尿酸血症の予防又は治療用である、前記〔18〕記載の医薬組成物;
〔20〕 血漿尿酸値低下薬である、前記〔17〕記載の医薬組成物;
〔21〕 尿酸生成抑制薬である、前記〔17〕記載の医薬組成物;等に関するものである。
「C1−6アルキル」とは、炭素数1〜6の直鎖状又は分岐状のアルキル基をいい、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル、tert−ブチル等が挙げられる。
「C1−6アルキレン」とは、上記C1−6アルキルから派生する2価の基をいう。
「C2−6アルケニル」とは、炭素数2〜6の直鎖状又は分岐状のアルケニル基をいい、ビニル、アリル、1−プロペニル、イソプロペニル等が挙げられる。
「C2−6アルキニル」とは、炭素数2〜6の直鎖状又は分岐状のアルキニル基をいい、エチニル、2−プロピニル等が挙げられる。
「C1−6アルコキシ」とは、炭素数1〜6の直鎖状又は分岐状のアルコキシ基をいい、メトキシ、エトキシ、プロポキシ、イソプロポキシ等が挙げられる。
「C1−6アルコキシカルボニル」とは、(C1−6アルコキシ)−C(O)−で表される基をいい、メトキシカルボニル、エトキシカルボニル、プロポキシカルボニル、イソプロポキシカルボニル等が挙げられる。
「C1−6アルコキシカルボニルオキシ」とは、(C1−6アルコキシ)−C(O)O−で表される基をいう。
「C1−6アルコキシC1−6アルキル」とは、上記C1−6アルコキシで置換された上記C1−6アルキルをいう。
「C1−6アルキルスルホニル」とは、(C1−6アルキル)−SO2−で表される基をいい、メチルスルホニル、エチルスルホニル等が挙げられる。
「C1−6アルキルスルホニルアミノ」とは、(C1−6アルキル)−SO2NH−で表される基をいい、メチルスルホニルアミノ、エチルスルホニルアミノ等が挙げられる。
「モノ(ジ)C1−6アルキルスルファモイル」とは、上記C1−6アルキルでモノ又はジ置換されたスルファモイル基をいう。
「C2−7アシル」とは、(C1−6アルキル)−C(O)−で表される基をいい、アセチル、プロピオニル、ブチリル、イソブチリル、ピバロイル等が挙げられる。
「C1−6アルキルチオ」とは、(C1−6アルキル)−S−で表される基をいう。
「C2−7アシルアミノ」とは、(C1−6アルキル)−C(O)NH−で表される基をいう。
「C1−6アルコキシカルボニルアミノ」とは、上記C1−6アルコキシカルボニルで置換されたアミノ基をいい、「C1−6アルコキシカルボニル(C1−6アルキル)アミノ」とは、上記C1−6アルコキシカルボニルと上記C1−6アルキルで置換されたアミノ基をいう。
「モノ(ジ)C1−6アルキルアミノカルボニルアミノ」とは、(モノ(ジ)C1−6アルキルアミノ)−C(O)NH−で表される基をいう。
「モノ(ジ)C1−6アルキルカルバモイル」とは、上記C1−6アルキルでモノ又はジ置換されたカルバモイル基をいい、「モノ(ジ)C1−6アルコキシC1−6アルキルカルバモイル」とは、上記C1−6アルコキシC1−6アルキルでモノ又はジ置換されたカルバモイル基をいい、「C1−6アルコキシC1−6アルキル(C1−6アルキル)カルバモイル」とは、上記C1−6アルコキシC1−6アルキルと上記C1−6アルキルで置換されたカルバモイル基をいう。
ジ置換の場合、それぞれの置換基は異なっていてもよい。
「C5−8シクロアルケニル」とは、5〜8員環のシクロアルケニル基をいい、シクロプロペニル、シクロブテニル、シクロペンテニル等が挙げられる。
「3〜8員環ヘテロシクロアルキル」とは、酸素原子、硫黄原子及び窒素原子から選択されるヘテロ原子を1〜2個環内に含む3〜8員環のヘテロシクロアルキル基をいい、アジリジノ、アゼチジノ、モルホリノ、2−モルホリニル、チオモルホリノ、1−ピロリジニル、ピペリジノ、4−ピペリジル、1−ピペラジニル、1−ピロリル、テトラヒドロフリル、テトラヒドロピラニル等が挙げられる。
「5〜8員環ヘテロシクロアルケニル」とは、酸素原子、硫黄原子及び窒素原子から選択されるヘテロ原子を1〜2個環内に含む5〜8員環のヘテロシクロアルケニル基をいい、2,3−ジヒドロフリル、2,5−ジヒドロフリル、3,4−ジヒドロ−2H−ピラン等が挙げられる。
「C6アリールオキシ」とは、(C6アリール)−O−で表される基をいい、フェニルオキシ等が挙げられる。
「C6アリールカルボニル」とは、(C6アリール)−C(O)−で表される基をいい、ベンゾイル等が挙げられる。
「C6アリールアミノ」とは、(C6アリール)−NH−で表される基をいう。
「C6アリール(C1−6アルキル)アミノ」とは、上記C6アリールと上記C1−6アルキルで置換されたアミノ基をいう。
「5又は6員環ヘテロアリールオキシ」とは、(5又は6員環ヘテロアリール)−O−で表される基をいう。
「5又は6員環ヘテロアリールカルボニル」とは、(5又は6員環ヘテロアリール)−C(O)−で表される基をいう。
「5又は6員環ヘテロアリールアミノ」とは、(5又は6員環ヘテロアリール)−NH−で表される基をいう。
「5又は6員環ヘテロアリール(C1−6アルキル)アミノ」とは、上記へテロアリールと上記C1−6アルキルで置換されたアミノ基をいう。
化合物(2)と化合物(3)とを、不活性溶媒中、塩基及びパラジウム触媒存在下、鈴木−宮浦カップリングを行い、必要に応じて脱保護を行うことにより、本発明の縮合複素環誘導体(I)を製造することもできる。不活性溶媒としては、ベンゼン、トルエン、キシレン、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン、ジクロロメタン、1,2−ジクロロエタン、クロロホルム、メタノール、エタノール、2−プロパノール、ブタノール、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、水、これらの混合溶媒等が挙げられる。塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、ナトリウムエトキシド、ナトリウムメトキシド、フッ化カリウム、フッ化セシウム、トリエチルアミン、N,N−ジイソプロピルエチルアミン、ピリジン、2,6−ルチジン、1,8−ジアザビシクロ[5,4,0]−7−ウンデセン等が挙げられる。パラジウム触媒としては、テトラキス(トリフェニルホスフィン)パラジウム、ジクロロビス(トリフェニルホスフィン)パラジウム、塩化パラジウム−1,1’ビス(ジフェニルホスフィノ)フェロセン等が挙げられる。反応温度は通常0℃から還流温度であり、反応時間は使用する原料物質や溶媒、反応温度などにより異なるが、通常30分〜7日間である。
化合物(4)と化合物(5)とを、工程1の方法と同様に、鈴木−宮浦カップリングを行い、必要に応じて脱保護を行うことにより、本発明の縮合複素環誘導体(Ia)を製造することもできる。
上記工程2で用いられる化合物(4)は、対応する化合物(2a)を不活性溶媒中、塩基及びパラジウム触媒存在下、リガンドの存在下又は非存在下、対応するボロン酸試薬と反応させることにより、製造することもできる。不活性溶媒としては、ベンゼン、トルエン、キシレン、N,N−ジメチルホルムアミド、1,2−ジメトキシエタン、1,4−ジオキサン、テトラヒドロフラン、ジメチルスルホキシド、N−メチルピロリドン、これらの混合溶媒等が挙げられる。塩基としては、トリエチルアミン、N,N−ジイソプロピルエチルアミン、ピリジン、2,6−ルチジン、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、酢酸カリウム、酢酸ナトリウム等が挙げられる。パラジウム触媒としては、酢酸パラジウム、ジクロロビス(トリフェニルホスフィン)パラジウム、塩化パラジウム−1,1’ビス(ジフェニルホスフィノ)フェロセン等が挙げられる。リガンドとしては、ビス(ジフェニルホスフィノ)フェロセン、トリシクロヘキシルホスフィン、2−(ジシクロヘキシルホスフィノ)ビフェニル等が挙げられる。ボロン酸試薬としては、ピナコールボラン、カテコールボラン、ビス(ピナコラート)ジボロン等が挙げられる。反応温度は通常0℃から還流温度であり、反応時間は使用する原料物質や溶媒、反応温度などにより異なるが、通常30分〜7日間である。
化合物(6)を、不活性溶媒中、塩基の存在下又は非存在下、パラジウム触媒存在下又は非存在下、シアノ化試薬と反応を行い、必要に応じて脱保護を行うことにより、本発明の縮合複素環誘導体(Ib)を製造することもできる。不活性溶媒としては、ベンゼン、トルエン、キシレン、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン、メタノール、エタノール、2−プロパノール、ブタノール、エチレングリコール、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシド、ヘキサメチルホスホルアミド、水、これらの混合溶媒等が挙げられる。塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、ナトリウムエトキシド、ナトリウムメトキシド、フッ化カリウム、フッ化セシウム、トリエチルアミン、N,N−ジイソプロピルエチルアミン、ピリジン、2,6−ルチジン、1,8−ジアザビシクロ[5,4,0]−7−ウンデセン等が挙げられる。パラジウム触媒としては、テトラキス(トリフェニルホスフィン)パラジウム、ジクロロビス(トリフェニルホスフィン)パラジウム、塩化パラジウム−1,1’ビス(ジフェニルホスフィノ)フェロセン、酢酸パラジウム、トリフルオロ酢酸パラジウム等が挙げられる。シアノ化試薬としては、シアン化銅、シアン化ナトリウム、シアン化カリウム、シアン化亜鉛、シアン化トリメチルシリル、カリウムフェロシアニド等が挙げられる。反応温度は通常0℃から還流温度であり、反応時間は使用する原料物質や溶媒、反応温度などにより異なるが、通常30分〜7日間である。
化合物(7)を不活性溶媒中、酸性条件下、チオアセトアミドと反応させることにより、化合物(8)を製造することもできる。不活性溶媒としては、テトラヒドロフラン、ジメトキシエタン、1,4−ジオキサン、N,N−ジメチルホルムアミド、N−メチルピロリドン、ベンゼン、トルエン、キシレン、これらの混合溶媒等が挙げられる。反応温度は通常0℃から還流温度であり、反応時間は使用する原料物質や溶媒、反応温度などにより異なるが、通常30分〜7日間である。
化合物(8)を不活性溶媒中、2−クロロ−3−オキソ酪酸誘導体と反応させ、必要に応じて脱保護を行うことにより、化合物(6a)を製造することもできる。不活性溶媒としては、メタノール、エタノール、n−ブタノール、イソプロパノール、N,N−ジメチルホルムアミド、テトラヒドロフラン、ベンゼン、トルエン等が挙げられる。反応温度は通常0℃から還流温度であり、反応時間は使用する原料物質や溶媒、反応温度などにより異なるが、通常30分〜7日間である。
化合物(9)を不活性溶媒中、塩基存在下、1−エチル−4−tert−ブチル−2−ジエチルホスホノスクシナートなどのHorner−Wadsworth−Emmons試薬と縮合した後、脱保護を行うか、又はコハク酸ジエステルと縮合することにより、化合物(10)を製造することもできる。不活性溶媒としては、ベンゼン、トルエン、キシレン、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン、N,N−ジメチルホルムアミド、N−メチルピロリドン、メタノール、エタノール、これらの混合溶媒等が挙げられる。塩基としては水素化ナトリウム、カリウムtert−ブトキシド、ナトリウムメトキシド、ナトリウムエトキシド等が挙げられる。反応温度は通常0℃から還流温度であり、反応時間は使用する原料物質や溶媒、反応温度などにより異なるが、通常30分〜7日間である。
化合物(10)を適当な溶媒中、脱水剤の存在下又は酸無水物溶媒中、塩基の存在下又は非存在下、反応を行い、必要に応じて加水分解を行うことにより、化合物(11)を製造することもできる。適当な溶媒としては、例えば、酢酸、硫酸、リン酸、テトラヒドロフラン、N,N−ジメチルホルムアミド、N−メチルピロリドン、ベンゼン、トルエン、キシレン、1,4−ジオキサン、水、これらの混合溶媒等が挙げられる。脱水剤としては、無水酢酸、トリフルオロ酢酸無水物、クロロギ酸メチル、クロロギ酸エチル等が挙げられる。酸無水物溶媒としては、無水酢酸、トリフルオロ酢酸無水物等が挙げられる。塩基としては、酢酸ナトリウム、酢酸カリウム等が挙げられる。反応温度は通常室温から還流温度であり、反応時間は使用する原料物質や溶媒、反応温度などにより異なるが、通常30分〜7日間である。加水分解に使われる不活性溶媒としては、メタノール、エタノール、イソプロパノール、テトラヒドロフラン、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメトキシエタン、水、これらの混合溶媒が挙げられる。加水分解に用いられる塩基としては、水酸化ナトリウム、水酸化カリウム、水酸化リチウムが挙げられる。反応温度は通常室温から還流温度であり、反応時間は使用する原料物質や溶媒、反応温度などにより異なるが、通常30分〜7日間である。
(ステップ1)
化合物(11)とアンモニアを不活性溶媒中、縮合剤の存在下、塩基の存在下又は非存在下、必要に応じて1−ヒドロキシベンゾトリアゾール等の添加剤を用いて、アミド化することにより、ナフタレンアミド化合物を製造することができる。不活性溶媒としては、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン、ベンゼン、トルエン、キシレン、ジクロロメタン、1,2−ジクロロエタン、クロロホルム又はこれらの混合溶媒等が挙げられる。縮合剤としては、無水酢酸、塩化チオニル、オギザリルクロリド、N,N’−カルボニルジイミダゾール、N,N’−ジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミド、N−エチル−N’−3−ジメチルアミノプロピルカルボジイミド及びその塩酸塩、ジフェニルホスホリルアジド等が挙げられる。塩基としてはトリエチルアミン、N,N−ジイソプロピルエチルアミン、ピリジン、2,6−ルチジン等が挙げられる。反応温度は通常0℃から還流温度であり、反応時間は使用する原料物質や溶媒、反応温度などにより異なるが、通常30分〜7日間である。
(ステップ2)
ステップ1で得られたナフタレンアミド化合物を不活性溶媒中、脱水剤の存在下、処理することにより、化合物(12)を得ることができる。不活性溶媒としては、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン、N,N−ジメチルホルムアミド、アセトニトリル、ベンゼン、トルエン、キシレン、ジクロロメタン、1,2−ジクロロエタン、クロロホルム、これらの混合物等が挙げられる。脱水剤としては、無水酢酸、塩化チオニル、オキシ塩化リン、メタンスルホニルイミダゾール、p−トルエンスルホニルクロリド、N,N’−ジシクロヘキシルカルボジイミド、五塩化ニリン、トリホスゲン等が挙げられる。反応温度は通常0℃から還流温度であり、反応時間は使用する原料物質や溶媒、反応温度などにより異なるが、通常30分〜7日間である。
化合物(12)を前記工程5及び工程6と同様の方法で反応させることにより、化合物(13)を得ることもできる。
化合物(13)を不活性溶媒中、塩基存在下、トリフルオロメタンスルホン酸無水物と反応させることにより、化合物(6b)を製造することもできる。不活性溶媒としては、ジクロロメタン、ジクロロエタン、クロロホルム、酢酸エチル、テトラヒドロフラン、N,N−ジメチルホルムアミド、N−メチルピロリドン、ベンゼン、トルエン、キシレン、1,4−ジオキサン等が挙げられる。塩基としては、トリエチルアミン、N,N−ジイソプロピルエチルアミン、ピリジン、2,6−ルチジン、1,8−ジアザビシクロ[5,4,0]−7−ウンデセン等が挙げられる。反応温度は通常0℃から還流温度であり、反応時間は使用する原料物質や溶媒、反応温度などにより異なるが、通常30分〜7日間である。
カルボキシ基において使用されるプロドラッグを構成する基としては、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、tert−ブチル等のC1−6アルキル;ピバロイルオキシメチル、アセチルオキシメチル、1−(ピバロイルオキシ)エチル、1−(アセチルオキシ)エチル等のC1−6アルキル−COO−C1−6アルキレン;エチルオキシカルボニルオキシメチル、1−(エチルオキシカルボニルオキシ)エチル、イソプロピルオキシカルボニルオキシメチル、1−(イソプロピルオキシカルボニルオキシ)エチル、tert−ブチルオキシカルボニルオキシメチル、1−(tert−ブチルオキシカルボニルオキシ)エチル等のC1−6アルキル−OCOO−C1−6アルキレン;シクロヘキシルオキシカルボニルメチル、1−(シクロヘキシルオキシカルボニル)エチル等のC3−8シクロアルキル−OCOO−C1−6アルキレン基等を挙げることができる。
5−ブロモ−1,3−ジメチル−1H−インドール−7−カルボニトリル
2−アミノ−3,5−ジブロモベンゾニトリル(1.0g)のテトラヒドロフラン(10mL)溶液に氷冷下、1mol/Lのカリウム tert−ブトキシド テトラヒドロフラン溶液(4.5mL)、臭化アリル(0.53g)を加え、室温にて16時間撹拌した。反応混合物を水に注ぎ酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン)で精製し、2−アリルアミノ−3,5−ジブロモベンゾニトリル(0.66g)を得た。
この化合物(0.23g)のアセトニトリル(10mL)溶液に酢酸パラジウム(0.016g)、トリ−O−トリルホスファン(0.067g)、トリエチルアミン(0.29g)を室温にて加え、85℃にて2時間撹拌した。反応混合物に、水、酢酸エチルを加え、セライトろ過し、2層を分離した。有機層を1規定塩酸、水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン)で精製し、5−ブロモ−3−メチル−1H−インドール−7−カルボニトリル(0.1g)を得た。
得られた化合物(0.05g)のジメチルホルムアミド(1mL)溶液に、氷冷下、ヨウ化メチル(0.090g)、水素化ナトリウム(0.015g)を加え、室温にて16時間撹拌した。反応混合物を水に注ぎ酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン)で精製し、標記化合物(0.035g)を得た。
5−ブロモ−1−メチル−7−ニトロ−1H−インドール
5−ブロモ−7−ニトロ−1H−インドール(0.2g)のジメチルホルムアミド(4mL)溶液に、氷冷下、ヨウ化メチル(0.42g)、水素化ナトリウム(0.060g)を加え、室温にて16時間撹拌した。反応混合物を水に注ぎ酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン)で精製し、標記化合物(0.11g)を得た。
5−ブロモ−7−ニトロベンゾフラン
5−ブロモ−2−ヒドロキシ−3−ニトロベンズアルデヒド(1.0g)のトルエン(10mL)溶液に2−ブロモマロン酸ジエチル(1.1g)、炭酸カリウム(0.84g)、テトラブチルアンモニウム ブロミド(0.13g)を室温にて加え、20時間還流した。反応混合物を水に注ぎ酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン)で精製し、5−ブロモ−7−ニトロベンゾフラン−2−カルボン酸エチル(0.68g)を得た。
この化合物(0.72g)のテトラヒドロフラン(12mL)、エタノール(4mL)、水(4mL)の混合溶液に水酸化リチウム一水和物(0.19g)を室温にて加え、同温にて16時間撹拌した。反応混合物に1規定塩酸、水を加え、析出した固体をろ取し、水、n−ヘキサンで洗浄後、減圧下50℃にて乾燥し、5−ブロモ−7−ニトロベンゾフラン−2−カルボン酸(0.53g)を得た。
得られた化合物(0.3g)にキノリン(3mL)、銅(0.073g)を室温にて加え、200℃にて3時間撹拌した。反応混合物に1規定塩酸、水を加え、酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン)で精製し、標記化合物(0.13g)を得た。
2−(7−ブロモ−1H−インドール−5−イル)イソニコチン酸エチル
5−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)−2,3−ジヒドロインドール−1−カルボン酸ベンジル(3.0g)のジメチルホルムアミド(40mL)、水(4mL)の混合溶液に室温にて、2−クロロイソニコチン酸エチル(1.5g)、テトラキス(トリフェニルホスフィン)パラジウム(0.46g)、炭酸セシウム(3.9g)を加え、80℃にて3時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/ヘキサン)で精製し、5−(4−エトキシカルボニルピリジン−2−イル)−2,3−ジヒドロインドール−1−カルボン酸ベンジル(1.4g)を得た。
この化合物(1.4g)のエタノール(20mL)、酢酸エチル(10mL)の混合溶液に室温にて、パラジウムカーボン(0.17g)、濃塩酸(0.57mL)を加え、水素雰囲気下、65℃にて3時間撹拌した。反応混合物に水、酢酸エチルを加え、セライトろ過し、2層を分離した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮し、2−(2,3−ジヒドロ−1H−インドール−5−イル)イソニコチン酸エチル
(0.91g)を得た。
得られた化合物(0.80g)の酢酸(16mL)溶液に氷冷下、臭素(0.57g)を加え、室温にて1時間撹拌した。反応混合物に1規定チオ硫酸ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮し、2−(7−ブロモ−2,3−ジヒドロ−1H−インドール−5−イル)イソニコチン酸エチル(0.92g)を得た。
この化合物(0.92g)のジクロロメタン(10mL)溶液に室温にて、二酸化マンガン(4.6g)を加え、室温にて4時間撹拌した。反応混合物をセライトろ過し、濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/ヘキサン)で精製し、標記化合物(0.39g)を得た。
5−ブロモ−1−エチル−3−メチル−1H−インドール−7−カルボニトリル、
5−ブロモ−3−メチル−1−プロピル−1H−インドール−7−カルボニトリル、
5−ブロモ−1−イソブチル−3−メチル−1H−インドール−7−カルボニトリル、
5−ブロモ−1−イソプロピル−3−メチル−1H−インドール−7−カルボニトリル
参考例1と同様の方法により、対応する原料を用いて標記化合物を合成した。
5−ブロモ−1−イソブチル−7−ニトロ−1H−インドール、
5−ブロモ−1−(2−メトキシエチル)−7−ニトロ−1H−インドール
参考例2と同様の方法により、対応する原料を用いて標記化合物を合成した。
2−(7−ブロモ−1H−インドール−5−イル)−4−メチルチアゾール−5−カルボン酸エチル
参考例4と同様の方法により、2−クロロイソニコチン酸エチルの代わりに2−ブロモ−4−メチルチアゾール−5−カルボン酸エチルを用いて標記化合物を合成した。
7−アセトキシベンゾ[b]チオフェン−5−カルボン酸メチル
チオフェン−3−カルバルデヒド(3.00g)、コハク酸ジメチル(4.69g)のメタノール(52mL)溶液に、ナトリウムメトキシド(28%メタノール溶液、6.2mL)を室温にて加え、一晩還流した。室温まで放冷後、反応混合物に水を注ぎ、ジエチルエーテルで抽出した。水層に1mol/L塩酸を加え酸性とし、ジエチルエーテルで抽出した。有機層を水、飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後濃縮した。残渣に無水酢酸(30mL)、酢酸ナトリウム(2.19g)を加え、一晩還流した。室温に放冷後、反応混合物にエタノール(50mL)を加え、濃縮した。残渣に2mol/L塩酸を加え、エーテルで抽出した。有機層を水、飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/ヘキサン=30/70〜100/0)にて精製し、標記化合物(1.17g)を得た。
7−ヒドロキシベンゾ[b]チオフェン−5−カルボン酸
7−アセトキシベンゾ[b]チオフェン−5−カルボン酸メチル(1.17g)のテトラヒドロフラン(20mL)、エタノール(7mL)及び水(7mL)の溶液に水酸化リチウム・1水和物(1.96g)を加え、室温にて一晩撹拌した。反応混合物に1mol/L塩酸及び水を加え、析出した固体をろ取し、水及びn−ヘキサンで洗浄後、減圧下50℃にて乾燥して、標記化合物(0.35g)を得た。
7−ヒドロキシベンゾ[b]チオフェン−5−カルボン酸アミド
7−ヒドロキシベンゾ[b]チオフェン−5−カルボン酸(0.35g)のテトラヒドロフラン(6mL)溶液に、1,1−カルボニルジイミダゾール(0.88g)を氷冷下加え、室温にて2時間撹拌した。反応混合物に28%アンモニア水溶液(3mL)を氷冷下加え、室温にて2時間撹拌した。反応混合物を減圧下濃縮し、残渣に1mol/L塩酸を加え、室温にて15分間撹拌した。析出した固体をろ取し、1mol/L塩酸、水で洗浄後乾燥して、標記化合物(0.17g)を得た。
7−ヒドロキシベンゾ[b]チオフェン−5−カルボニトリル
7−ヒドロキシベンゾ[b]チオフェン−5−カルボン酸アミド(0.17g)のジクロロメタン(5mL)溶液に、トリエチルアミン(0.71g)、トリフルオロ酢酸無水物(0.92g)を氷冷下加え、室温にて一晩撹拌した。反応混合物にメタノールを加え、減圧下濃縮した。残渣に水を加え、酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/ヘキサン=30/70〜100/0)にて精製し、標記化合物(0.10g)を得た。
7−ヒドロキシベンゾ[b]チオフェン−5−カルボチオ酸アミド
7−ヒドロキシベンゾ[b]チオフェン−5−カルボニトリル(0.10g)のN,N−ジメチルホルムアミド(2mL)及び4mol/L HCl 1,4−ジオキサン(2mL)の溶液に、チオアセトアミド(0.62g)を室温にて加え、80℃にて一晩撹拌した。反応混合物に水を注ぎ、ジエチルエーテルで抽出した。有機層を水で洗浄し、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/ヘキサン)にて精製し、標記化合物(0.08g)を得た。
2−(7−ヒドロキシベンゾ[b]チオフェン−5−イル)−4−メチルチアゾール−5−カルボン酸エチル
7−ヒドロキシベンゾ[b]チオフェン−5−カルボチオ酸アミド(0.08g)のエタノール(2mL)溶液に、2−クロロ−アセト酢酸エチル(0.20g)を室温にて加え、75℃にて一晩撹拌した。反応混合物を室温まで放冷後、析出した固体をろ取し、エタノールで洗浄した。減圧下50℃にて乾燥して、標記化合物(0.09g)を得た。
5−ブロモ−1,2−ジメチル−2,3−ジヒドロ−1H−インドール
5−ブロモ−2−メチル−2,3−ジヒドロ−1H−インドール(1.90g)のジメチルホルムアミド(20mL)溶液に、ヨードメタン(2.10g)及び水素化ナトリウム(0.5g)を氷冷下加え、50℃にて4時間撹拌した。反応混合物に水を注ぎ、酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/ヘキサン)にて精製し、標記化合物(1.60g)を得た。
5−ブロモ−1,2−ジメチル−2,3−ジヒドロ−1H−インドール−7−カルバルデヒド
5−ブロモ−1,2−ジメチル−2,3−ジヒドロ−1H−インドール(1.30g)のジメチルホルムアミド(10mL)溶液に塩化ホスホリル(1.3g)を氷冷下加え、80℃にて一晩撹拌した。放冷後、反応混合物に2mol/L水酸化ナトリウム水溶液を加え、室温にて30分間撹拌した。2mol/L塩酸で中和し、酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/ヘキサン)にて精製し、標記化合物(0.75g)を得た。
5−ブロモ−1,2−ジメチル−2,3−ジヒドロ−1H−インドール−7−カルボニトリル
5−ブロモ−1,2−ジメチル−2,3−ジヒドロ−1H−インドール−7−カルバルデヒド(0.75g)のテトラヒドロフラン(10mL)溶液にヒドロキシルアミン塩酸塩(0.24g)及びピリジン(0.93g)を室温にて加え、60℃にて5時間撹拌した。室温まで放冷後、無水酢酸(0.60g)を加え、60℃にて一晩撹拌した。反応混合物に水を注ぎ、酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/ヘキサン)にて精製し、標記化合物(0.54g)を得た。
5−ブロモキノリン−7−カルボン酸エチル
3−アミノ−5−ブロモ−安息香酸メチル(2.76g)、グリセロール(5.53g)、硫酸(75%水溶液、24mL)、3−ニトロベンゼンスルホン酸ナトリウム塩(5.41g)の混合物を、100℃にて3時間撹拌し、さらに140℃にて2時間撹拌した。反応混合物を60℃に冷却し、エタノールを加え、同温にて一晩撹拌した。反応混合物を室温に放冷し、溶媒を減圧下濃縮した。残渣に水を加え、さらにアンモニア水をゆっくりと加え、溶液をアルカリ性とし、酢酸エチルで抽出した。有機層を、水、飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後、濃縮した。残渣をシリカゲルカラムクロマトグラフィーにて精製し、標記化合物を得た。
8−ヨードキノリン−6−カルボン酸エチル
参考例21と同様の方法により、対応する原料を用いて標記化合物を合成した。
2−(5−ブロモキノリン−7−イル)−4−メチルチアゾール−5−カルボン酸エチル
2−(8−ヨードキノリン−6−イル)−4−メチルチアゾール−5−カルボン酸エチル
参考例17と同様の方法により、対応する原料を用いて標記化合物を合成した。
2−(5−ブロモキノリン−7−イル)チアゾール−5−カルボン酸エチル
参考例17と同様の方法により、2−クロロ−アセト酢酸エチルの代わりに2−クロロ−3−オキソプロピオン酸エチルを用いて標記化合物を合成した。
2−イソキノリン−7−イル−4−メチルチアゾール−5−カルボン酸エチル
7−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)イソキノリン(1.46g)、2−ブロモ−4−メチルチアゾール−5−カルボン酸エチル(1.43g)、テトラキス(トリフェニルフォスフィン)パラジウム(0.33g)、炭酸セシウム(2.80g)、N, N−ジメチルホルムアミド(11.5mL)、水(2.3mL)の混合物を、80℃にて一晩撹拌した。室温に放冷後、水、酢酸エチルを加え、5分間撹拌し、セライトろ過した。ろ液を分液し、有機層を飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン=0〜60%)にて精製し、続いてアミノプロピルシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン)にて精製し、標記化合物(0.53g)を得た。
2−(5−ブロモイソキノリン−7−イル)−4−メチルチアゾール−5−カルボン酸エチル
2−イソキノリン−7−イル−4−メチルチアゾール−5−カルボン酸エチル(0.49g)の硫酸(3.2mL)溶液に、N−ブロモスクシンイミド(0.29g)を氷冷下にて加え、室温にて一晩撹拌した。更にN−ブロモスクシンイミド(0.29g)を氷冷下にて加え、室温にて一晩撹拌した。反応混合物を氷中に注ぎ、飽和重曹水でPH8とした。酢酸エチルを加え抽出した。有機層を飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン=0〜40〜60%)にて精製し、標記化合物(0.094g)を得た。
2−(7−シアノ−1,3−ジメチル−1H−インドール−5−イル)−4−メチルチアゾール−5−カルボン酸
5−ブロモ−1,3−ジメチル−1H−インドール−7−カルボニトリル(0.035g)のジメチルホルムアミド(1mL)溶液にビス(ピナコラート)ジボロン(0.039g)、酢酸パラジウム(0.0016g)、酢酸カリウム(0.042g)を室温にて加え、80℃にて3時間撹拌した。反応混合物に水、酢酸エチルを加え、セライトろ過し、2層を分離した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮し、1,3−ジメチル−5−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)−1H−インドール−7−カルボニトリルを得た。
この化合物のジメチルホルムアミド(1mL)、水(0.1mL)の混合溶液に2−ブロモ−4−メチルチアゾール−5−カルボン酸エチル(0.042g)、テトラキス(トリフェニルホスフィン)パラジウム(0.0081g)、炭酸セシウム(0.069g)を室温にて加え、80℃にて4時間撹拌した。反応混合物を水に注ぎ酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン)で精製し、2−(7−シアノ−1,3−ジメチル−1H−インドール−5−イル)−4−メチルチアゾール−5−カルボン酸エチル(0.021g)を得た。
得られた化合物(0.021g)のテトラヒドロフラン(1mL)、エタノール(0.3mL)、水(0.3mL)の混合溶液に水酸化リチウム一水和物(0.013g)を室温にて加え、同温にて16時間撹拌した。反応混合物に1規定塩酸、水を加え、析出した固体をろ取し、水、n−ヘキサンで洗浄後、減圧下50℃にて乾燥し、標記化合物(0.013g)を得た。
2−(1−メチル−7−ニトロ−1H−インドール−5−イル)イソニコチン酸
5−ブロモ−1−メチル−7−ニトロ−1H−インドール(0.11g)のジメチルホルムアミド(2mL)溶液にビス(ピナコラート)ジボロン(0.123g)、酢酸パラジウム(0.005g)、酢酸カリウム(0.13g)を室温にて加え、80℃にて2時間撹拌した。反応混合物に水、酢酸エチルを加え、セライトろ過し、2層を分離した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮し、1−メチル−7−ニトロ−5−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)−1H−インドールを得た。
この化合物のジメチルホルムアミド(2mL)、水(0.2mL)の混合溶液に2−クロロイソニコチン酸エチル(0.099g)、テトラキス(トリフェニルホスフィン)パラジウム(0.026g)、炭酸セシウム(0.22g)を室温にて加え、80℃にて2時間撹拌した。反応混合物を水に注ぎ酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン)で精製し、2−(1−メチル−7−ニトロ−1H−インドール−5−イル)イソニコチン酸エチル(0.055g)を得た。
得られた化合物(0.055g)のテトラヒドロフラン(2.4mL)、エタノール(0.8mL)、水(0.8mL)の混合溶液に水酸化リチウム一水和物(0.036g)を室温にて加え、同温にて16時間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。得られた水層に1規定塩酸を加え、酢酸エチルで抽出した。有機層を硫酸マグネシウムで乾燥後、濃縮した。残渣を酢酸エチルで洗浄後、減圧下50℃にて乾燥し、標記化合物(0.017g)を得た。
4−メチル−2−(1−メチル−7−ニトロ−1H−インドール−5−イル)チアゾール−5−カルボン酸
5−ブロモ−1−メチル−7−ニトロ−1H−インドール(0.16g)のジメチルホルムアミド(3mL)溶液にビス(ピナコラート)ジボロン(0.17g)、酢酸パラジウム(0.007g)、酢酸カリウム(0.18g)を室温にて加え、80℃にて2時間撹拌した。反応混合物に水、酢酸エチルを加え、セライトろ過し、2層を分離した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮し、1−メチル−7−ニトロ−5−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)−1H−インドールを得た。
この化合物のジメチルホルムアミド(3mL)、水(0.3mL)の混合溶液に2−ブロモ−4−メチルチアゾール−5−カルボン酸エチル(0.18g)、テトラキス(トリフェニルホスフィン)パラジウム(0.035g)、炭酸セシウム(0.30g)を室温にて加え、80℃にて3時間撹拌した。反応混合物を水に注ぎ酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン)で精製し、4−メチル−2−(1−メチル−7−ニトロ−1H−インドール−5−イル)チアゾール−5−カルボン酸エチル(0.075g)を得た。
得られた化合物(0.075g)のテトラヒドロフラン(3mL)、エタノール(1mL)、水(1mL)の混合溶液に水酸化リチウム一水和物(0.046g)を室温にて加え、同温にて16時間撹拌した。反応混合物に1規定塩酸、水を加え、析出した固体をろ取し、水、n−ヘキサンで洗浄後、減圧下50℃にて乾燥し、標記化合物(0.051g)を得た。
2−(7−ニトロベンゾフラン−5−イル)イソニコチン酸
5−ブロモ−7−ニトロベンゾフラン(0.07g)のジメチルホルムアミド(2mL)溶液にビス(ピナコラート)ジボロン(0.081g)、酢酸パラジウム(0.003g)、酢酸カリウム(0.085g)を室温にて加え、80℃にて2時間撹拌した。反応混合物に水、酢酸エチルを加え、セライトろ過し、2層を分離した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮し、7−ニトロ−5−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)ベンゾフランを得た。
この化合物のジメチルホルムアミド(1mL)、水(0.1mL)の混合溶液に2−クロロイソニコチン酸エチル(0.062g)、テトラキス(トリフェニルホスフィン)パラジウム(0.016g)、炭酸セシウム(0.14g)を室温にて加え、80℃にて2時間撹拌した。反応混合物を水に注ぎ酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/n−ヘキサン)で精製し、2−(7−ニトロベンゾフラン−5−イル)イソニコチン酸エチル(0.02g)を得た。
得られた化合物の(0.02g)のテトラヒドロフラン(1mL)、エタノール(0.3mL)、水(0.3mL)の混合溶液に水酸化リチウム一水和物(0.013g)を室温にて加え、同温にて16時間撹拌した。反応混合物に1規定塩酸、水を加え、析出した固体をろ取し、水、メタノール、n−ヘキサンで洗浄後、減圧下50℃にて乾燥し、標記化合物(0.002g)を得た。
実施例1と同様の方法により、対応する原料を用いて、実施例5〜8の化合物を合成した。
実施例2と同様の方法により、対応する原料を用いて、実施例9〜10の化合物を合成した。
2−(7−シアノ−1H−インドール−5−イル)イソニコチン酸
2−(7−ブロモ−1H−インドール−5−イル)イソニコチン酸エチル(0.36g)の1−メチル1−2−ピロリドン(5mL)溶液にシアン化亜鉛(0.13g)、テトラキス(トリフェニルホスフィン)パラジウム(0.051g)を加え、マイクロウェーブ反応装置を用い、150℃にて1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:酢酸エチル/ヘキサン)で精製し、2−(7−シアノ−1H−インドール−5−イル)イソニコチン酸エチル(0.096g)を得た。
得られた化合物(0.020g)のテトラヒドロフラン(3mL)、エタノール(1mL)の混合溶液に1規定水酸化リチウム水溶液(1mL)を加え、室温にて16時間撹拌した。反応混合物に1規定塩酸(1mL)、水を加え、析出した固体をろ取し、水、n−ヘキサンで洗浄後、減圧下50℃にて乾燥し、標記化合物(0.007g)を得た。
2−(7−シアノ−1−メチル−1H−インドール−5−イル)イソニコチン酸
2−(7−シアノ−1H−インドール−5−イル)イソニコチン酸エチル(0.04g)のジメチルホルムアミド(2mL)溶液に氷冷下、水素化ナトリウム(0.007g)、ヨウ化メチル(0.03g)を加え、室温にて4時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し硫酸マグネシウムで乾燥後濃縮し、2−(7−シアノ−1H−インドール−5−イル)イソニコチン酸エチル(0.048g)を得た。
得られた化合物(0.048g)のテトラヒドロフラン(3mL)、エタノール(1mL)の混合溶液に1規定水酸化リチウム水溶液(1mL)を加え、室温にて16時間撹拌した。反応混合物に1規定塩酸(1mL)、水を加え、析出した固体をろ取し、水、n−ヘキサンで洗浄後、減圧下50℃にて乾燥し、標記化合物(0.032g)を得た。
実施例11と同様の方法により、2−(7−ブロモ−1H−インドール−5−イル)イソニコチン酸エチルの代わりに2−(7−ブロモ−1H−インドール−5−イル)−4−メチルチアゾール−5−カルボン酸エチルを用いて、実施例13の化合物を合成した。
実施例12と同様の方法により、対応する原料を用いて、実施例14の化合物を合成した。
2−(7−シアノベンゾ[b]チオフェン−5−イル)−4−メチルチアゾール−5−カルボン酸
2−(7−ヒドロキシベンゾ[b]チオフェン−5−イル)−4−メチルチアゾール−5−カルボン酸エチル(0.08g)のジクロロメタン(2mL)溶液に、ピリジン(0.06g)及びトリフルオロメタンスルホン酸無水物(0.14g)を氷冷下加え、室温にて1時間撹拌した。反応混合物に1mol/L塩酸を加え、ジクロロメタンで抽出し、有機層を水、飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後、濃縮し、4−メチル−2−(7−トリフルオロメタンスルホニルオキシベンゾ[b]チオフェン−5−イル)チアゾール−5−カルボン酸エチル(0.09g)を得た。得られた化合物のN−メチルピロリドン(2mL)溶液にシアン化亜鉛(0.11g)及びテトラキス(トリフェニルフォスフィン)パラジウム(0.06g)を室温にて加え、マイクロウェーブ反応装置(Biotage社)を用いて150℃にて1時間撹拌した。反応混合物に水を注ぎ、酢酸エチルで抽出し、有機層を水、飽和食塩水で洗浄した。硫酸マグネシウムで乾燥後濃縮し、残渣をジエチルエーテルで洗い、減圧下50℃にて乾燥して、2−(7−シアノベンゾ[b]チオフェン−5−イル)−4−メチルチアゾール−5−カルボン酸エチル(0.05g)を得た。2−(7−ヒドロキシベンゾ[b]チオフェン−5−イル)−4−メチルチアゾール−5−カルボン酸エチル(0.05g)のテトラヒドロフラン(1mL)、エタノール(0.18mL)の溶液に、1mol/L水酸化リチウム水溶液(0.18mL)を室温にて加え、室温にて4時間撹拌した。反応混合物に1mol/L塩酸及び水を加え、析出した固体をろ取し、水及びn−ヘキサンで洗浄後、減圧下50℃にて乾燥して、標記化合物(0.01g)を得た。
実施例15と同様の方法により、対応する原料を用いて標記化合物を合成した。
2−(7−シアノ−1,2−ジメチル−1H−インドール−5−イル)−4−メチルチアゾール−5−カルボン酸
2−(7−シアノ−1,2−ジメチル−2,3−ジヒドロ−1H−インドール−5−イル)−4−メチルチアゾール−5−カルボン酸エチル(0.16g)のジクロロメタン(4mL)溶液に二酸化マンガン(0.81g)を加え、室温にて10時間撹拌した。反応混合物をセライトろ過し、ろ液を濃縮し、2−(7−シアノ−1,2−ジメチル−1H−インドール−5−イル)−4−メチルチアゾール−5−カルボン酸エチル(0.15g)を得た。得られた化合物のテトラヒドロフラン(5mL)、エタノール(2mL)の溶液に、1mol/L水酸化リチウム水溶液(2mL)を室温にて加え、室温にて一晩撹拌した。反応混合物に1mol/L塩酸及び水を加え、析出した固体をろ取し、水及びn−ヘキサンで洗浄後、減圧下50℃にて乾燥して、標記化合物(0.10g)を得た。
実施例11と同様の方法により、対応する原料を用いて標記化合物を合成した。
キサンチンオキシダーゼ阻害活性
(1)試験化合物の調製
試験化合物をDMSO(和光純薬社製)にて40mMの濃度になるように溶解した後、リン酸緩衝生理食塩水(PBS)を用いて希釈して目的の濃度になるように調製した。
キサンチンオキシダーゼ(ウシミルク由来、シグマ社製)をリン酸緩衝生理食塩水(PBS)で0.02units/mLに調製し、96穴プレートに50μL/穴ずつ加えた。更にPBSを用いて希釈した試験化合物を50μL/穴ずつ加えた。PBSを用いて調製した200μMのキサンチン(和光純薬社製)を100μL/穴で加え、室温で10分間反応させた。290nmの条件下において、マイクロプレートリーダースペクトラマックスプラス384(モレキュラーデバイス社製)を用いて吸光度を測定した。キサンチンを加えない条件下での吸光度を0%とし、試験化合物を加えていない対照を100%として、50%抑制する試験化合物の濃度(IC50)を算出した(表5)。表中、Ex.Noは実施例番号を示す。
ヒトURAT1発現細胞を用いた尿酸輸送阻害活性
(1)ヒトURAT1一過的発現細胞の調製
ヒトURAT1完全長cDNA(NCBI Accession No.NM_144585)を発現ベクターpcDNA3.1(インビトロジェン社製)にサブクローニングを行った。ヒトURAT1発現ベクターをCOS7細胞(RIKEN CELL BANK RCB0539)にリポフェクトアミン2000(インビトロジェン社製)を用いて導入した。COS7細胞はコラーゲンコート24ウエルプレート(日本ベクトン・ディッキンソン社製)に90〜95%confluentになるよう播種し、10%ウシ胎児血清(三光純薬社製)含有D−MEM培地(インビトロジェン社製)を用いて、2時間、37℃、5%CO2条件下にて培養を行った。1穴あたり2μLのリポフェクトアミン2000を50μLのOPTI−MEM(インビトロジェン社製)で希釈し、室温で7分間静置した(以下Lipo2000−OPTIとする)。1穴あたり0.8μgヒトURAT1発現ベクターを50μLのOPTI−MEM(インビトロジェン社製)で希釈し、Lipo2000−OPTIに加えて穏やかに混和し、室温にて25分間放置した後、1穴あたり100μLずつCOS7細胞に添加した。更にCOS7細胞は、37℃、5%CO2条件下において2日間培養し、取り込み阻害活性の測定に供した。
試験化合物をDMSO(和光純薬社製)にて10mMの濃度になるように溶解した後、前処置用緩衝液(125mMグルコン酸ナトリウム、4.8mMグルコン酸カリウム、1.2mMリン酸2水素カリウム、1.2mM硫酸マグネシウム、1.3mMグルコン酸カルシウム、5.6mMグルコース、25mMヘペス、pH7.4)を用いて希釈して目的の2倍の濃度になるように調製した。試験化合物を含まない前処置用緩衝液を対照として用いた。更に14Cで標識された尿酸(American Radiolabeled Chemicals,Inc.社製)を含む前処置用緩衝液を試験化合物及び対照に等量加えて、最終的に20μMの尿酸を含むアッセイ緩衝液を作製した。
全ての試験は37℃のホットプレート上において実施した。前処置用緩衝液及びアッセイ緩衝液は37℃にて加温した後に用いた。プレートから培地を除去し、700μLの前処置用緩衝液を加えて、10分間プレインキュベーションした。同一操作を繰り返した後、前処置用緩衝液を取り除き、アッセイ緩衝液を400μL/穴で添加し、5分間取り込み反応を行った。反応終了後、直ちにアッセイ緩衝液を除去し、氷冷した前処置用緩衝液を1.2mL/穴ずつ加えて、細胞を2回洗浄した。0.2N水酸化ナトリウムを300μL/穴で添加して、細胞を溶解した。細胞溶解液はピコプレート(パーキンエルマー社製)に移し、マイクロシンチ40(パーキンエルマー社製)を600μL/穴で添加し、混合した後、液体シンチレーションカウンター(パーキンエルマー社製)にて放射活性を測定した。またURAT1発現ベクターを導入していないCOS7細胞も対照と同様の条件下において、放射活性を測定した。なお、試験化合物の阻害率は以下の式により算出した。その結果、実施例2の化合物は100μMの濃度において80%以上の阻害を示した。
阻害率(%)=[1−(B−C)/(A−C)]X 100
A:対照の放射活性
B:試験化合物を加えた場合の放射活性
C:URAT1発現ベクターを導入していないCOS7細胞の放射活性
Claims (21)
- 式
Tはトリフルオロメチル、ニトロ又はシアノ;
環Qは5又は6員環ヘテロアリール;
X1及びX2は独立して、CH又はN;
環UはC6アリール又は5又は6員環ヘテロアリール;
mは0〜2の整数;
nは0〜3の整数;
R1は水酸基、ハロゲン原子、アミノ又はC1−6アルキル(mが2であるとき、2つのR1は異なっていてもよい);
R2は、
(i)環Q内の炭素原子に結合している場合は、(1)〜(11):
(1)ハロゲン原子;
(2)水酸基;
(3)シアノ;
(4)ニトロ;
(5)カルボキシ;
(6)カルバモイル;
(7)アミノ;
(8)それぞれ独立して、置換基群αから選択される任意の基を有していてもよいC1−6アルキル、C2−6アルケニル又はC1−6アルコキシ;
(9)C2−6アルキニル、C1−6アルキルスルホニル、モノ(ジ)C1−6アルキルスルファモイル、C2−7アシル、C1−6アルコキシカルボニル、C1−6アルコキシカルボニルオキシ、モノ(ジ)C1−6アルキルアミノ、モノ(ジ)C1−6アルコキシC1−6アルキルアミノ、C1−6アルコキシC1−6アルキル(C1−6アルキル)アミノ、C2−7アシルアミノ、C1−6アルコキシカルボニルアミノ、C1−6アルコキシカルボニル(C1−6アルキル)アミノ、モノ(ジ)C1−6アルキルカルバモイル、モノ(ジ)C1−6アルコキシC1−6アルキルカルバモイル、C1−6アルコキシC1−6アルキル(C1−6アルキル)カルバモイル、モノ(ジ)C1−6アルキルアミノカルボニルアミノ、C1−6アルキルスルホニルアミノ又はC1−6アルキルチオ;
(10)C3−8シクロアルキル、3〜8員環ヘテロシクロアルキル、C5−8シクロアルケニル又は5〜8員環ヘテロシクロアルケニル;
(11)C6アリール、C6アリールオキシ、C6アリールカルボニル、5又は6員環ヘテロアリール、5又は6員環ヘテロアリールオキシ、5又は6員環ヘテロアリールカルボニル、C6アリールアミノ、C6アリール(C1−6アルキル)アミノ、5又は6員環ヘテロアリールアミノ又は5又は6員環ヘテロアリール(C1−6アルキル)アミノ;の何れかであり、
(ii) 環Qの窒素原子に結合している場合は、(12)〜(15):
(12)それぞれ独立して、置換基群αから選択される任意の基を有していてもよいC1−6アルキル又はC2−6アルケニル;
(13)C2−6アルキニル、C1−6アルキルスルホニル、モノ(ジ)C1−6アルキルスルファモイル、C2−7アシル、C1−6アルコキシカルボニル又はモノ(ジ)C1−6アルキルカルバモイル;
(14)C3−8シクロアルキル又は3〜8員環ヘテロシクロアルキル;
(15)C6アリール、5又は6員環ヘテロアリール、C6アリールカルボニル又は5又は6員環ヘテロアリールカルボニル;の何れかであり;
(nが2又は3であるとき、R2はそれぞれ異なっていてもよく、更に、2つのR2が、環Q内の隣り合った原子に結合して存在し、それぞれC1−6アルコキシを有していてもよいC1−6アルキルである場合は、結合する環Q内の原子と共に、5〜8員環を形成してもよい。);
置換基群αは、フッ素原子、水酸基、アミノ、カルボキシ、C1−6アルコキシ、モノ(ジ)C1−6アルキルアミノ、モノ(ジ)C1−6アルコキシC1−6アルキルアミノ、C1−6アルコキシC1−6アルキル(C1−6アルキル)アミノ、C1−6アルコキシカルボニルアミノ、C2−7アシル、C1−6アルコキシカルボニル、モノ(ジ)C1−6アルキルカルバモイル、モノ(ジ)C1−6アルコキシC1−6アルキルカルバモイル、C1−6アルコキシC1−6アルキル(C1−6アルキル)カルバモイル、C1−6アルキルスルホニルアミノ、C2−7アシルアミノ、C1−6アルコキシカルボニルアミノ、C3−8シクロアルキル、3〜8員環ヘテロシクロアルキル、C6アリール及び5又は6員環ヘテロアリールである〕で表される縮合複素環誘導体又はその薬理学的に許容される塩。 - Tがシアノである請求項1記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- X1がCHである請求項1又は2記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- X2がCHである請求項1〜3の何れかに記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- 環Qがピリジン、ピリミジン、ピラジン、チアゾール、イミダゾール、ピラゾール、オキサゾール、イソチアゾール、イソオキサゾール、チオフェン、フラン又はピロール環である請求項1〜4の何れかに記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- 環Qがピリジン、チオフェン又はピロール環である請求項5記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- 環Uがベンゼン、ピリジン、チアゾール、ピラゾール又はチオフェン環である請求項1〜6の何れかに記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- mが0であるか、又はmが1であり、かつ、環Uが下記式:
- mが0であるか、又はmが1であり、かつR1aが水酸基又はC1−6アルキルである、請求項8記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- 環Uがチアゾール環である請求項8又は9記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- mが0であるか、又はmが1;かつR1aが水酸基であり、かつ環Uがピリジン環である請求項9記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- R1aがメチルである、請求項10記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- R1aが水酸基である、請求項11記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- nが0であるか、又はnが1〜3であり、かつR2が環Qの炭素原子に結合するハロゲン原子;水酸基;それぞれフッ素原子、水酸基及びアミノから選択される任意の基を1〜3個有していてもよいC1−6アルキル又はC1−6アルコキシ;C1−6アルコキシC1−6アルキル;もしくはC1−6アルコキシC1−6アルコキシ;又は環Qの窒素原子に結合する、フッ素原子、水酸基及びアミノから選択される任意の基を1〜3個有していてもよいC1−6アルキル;もしくはC1−6アルコキシC1−6アルキルである、請求項1〜13の何れかに記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- nが0であるか、又はnが1〜3であり、かつR2が環Qの炭素原子に結合するハロゲン原子;水酸基;もしくはフッ素原子を1〜3個有していてもよいC1−6アルキル;又は環Qの窒素原子に結合する、フッ素原子を1〜3個有していてもよいC1−6アルキル;もしくはC1−6アルコキシC1−6アルキルである、請求項14記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- キサンチンオキシダーゼ阻害薬である、請求項1〜15の何れかに記載の縮合複素環誘導体又はその薬理学的に許容される塩。
- 請求項1〜15の何れかに記載の縮合複素環誘導体又はその薬理学的に許容される塩を有効成分として含有する医薬組成物。
- 高尿酸血症、痛風結節、痛風関節炎、高尿酸血症による腎障害及び尿路結石から選択される疾患の予防又は治療用である、請求項17記載の医薬組成物。
- 高尿酸血症の予防又は治療用である、請求項18記載の医薬組成物。
- 血漿尿酸値低下薬である、請求項17記載の医薬組成物。
- 尿酸生成抑制薬である、請求項17記載の医薬組成物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010533902A JP5587784B2 (ja) | 2008-10-15 | 2009-10-14 | 縮合複素環誘導体及びその医薬用途 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008266089 | 2008-10-15 | ||
| JP2008266089 | 2008-10-15 | ||
| JP2010533902A JP5587784B2 (ja) | 2008-10-15 | 2009-10-14 | 縮合複素環誘導体及びその医薬用途 |
| PCT/JP2009/067751 WO2010044404A1 (ja) | 2008-10-15 | 2009-10-14 | 縮合複素環誘導体及びその医薬用途 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2010044404A1 JPWO2010044404A1 (ja) | 2012-03-15 |
| JP5587784B2 true JP5587784B2 (ja) | 2014-09-10 |
Family
ID=42106567
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010533902A Expired - Fee Related JP5587784B2 (ja) | 2008-10-15 | 2009-10-14 | 縮合複素環誘導体及びその医薬用途 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20110201815A1 (ja) |
| EP (1) | EP2338887A4 (ja) |
| JP (1) | JP5587784B2 (ja) |
| CA (1) | CA2739058A1 (ja) |
| WO (1) | WO2010044404A1 (ja) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI423962B (zh) * | 2009-10-07 | 2014-01-21 | Lg Life Sciences Ltd | 有效作為黃嘌呤氧化酶抑制劑之新穎化合物、其製備方法及含該化合物之醫藥組成物 |
| SG2014012926A (en) * | 2011-08-30 | 2014-06-27 | Chdi Foundation Inc | Kynurenine-3-monooxygenase inhibitors, pharmaceutical compositions, and methods of use thereof |
| CN103980267B (zh) * | 2013-02-08 | 2018-02-06 | 镇江新元素医药科技有限公司 | 一类新型黄嘌呤氧化酶抑制剂的化合物及其药物组合物 |
| CN106061944A (zh) * | 2014-01-24 | 2016-10-26 | 普渡制药公司 | 吡啶类和嘧啶类物质及其用途 |
| US10258621B2 (en) | 2014-07-17 | 2019-04-16 | Chdi Foundation, Inc. | Methods and compositions for treating HIV-related disorders |
| CN106478619B (zh) * | 2015-08-29 | 2019-09-24 | 江苏新元素医药科技有限公司 | 一类黄嘌呤氧化酶抑制剂及其应用 |
| WO2017036404A1 (en) * | 2015-09-02 | 2017-03-09 | Sunshine Lake Pharma Co., Ltd. | Carboxy substituted (hetero) aromatic ring derivatives and preparation method and uses thereof |
| CN105541801B (zh) * | 2016-01-18 | 2018-07-17 | 常州大学 | Ezh2甲基转移酶抑制剂gsk126的合成方法 |
| AU2018354780A1 (en) | 2017-10-25 | 2020-04-09 | Bayer Pharma Aktiengesellschaft | Process for preparing benzothiophen-2yl boronate |
| CN111943957B (zh) * | 2019-05-17 | 2023-01-06 | 中国医学科学院药物研究所 | 喹啉甲酰胺类化合物及其制备方法和用途 |
| CA3218011A1 (en) * | 2021-04-29 | 2022-11-10 | Jiangsu Atom Bioscience And Pharmaceutical Co., Ltd. | A xanthine oxidase inhibitor |
| WO2023208108A1 (zh) * | 2022-04-27 | 2023-11-02 | 江苏新元素医药科技有限公司 | 可用于降尿酸的化合物 |
| TW202342455A (zh) * | 2022-04-27 | 2023-11-01 | 大陸商江蘇新元素醫藥科技有限公司 | 可用於痛風的化合物 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1398800A (en) * | 1972-05-05 | 1975-06-25 | Merck & Co Inc | Trisubstituted imidazoles |
| WO2006100095A1 (de) * | 2005-03-24 | 2006-09-28 | Curacyte Discovery Gmbh | Substituierte carbonsäureamide, verfahren zu ihrer herstellung und ihre verwendung als tnf-alpha-freisetzungsinhibitoren |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3914246A (en) * | 1972-05-05 | 1975-10-21 | Merck & Co Inc | Tri-substituted imidazoles |
| JP2000001431A (ja) | 1998-06-15 | 2000-01-07 | Kotobuki Seiyaku Kk | 尿酸排泄剤 |
| WO2005121132A1 (ja) | 2004-06-11 | 2005-12-22 | Shionogi & Co., Ltd. | 抗hcv作用を有する縮合ヘテロ環化合物 |
| EP1773840B1 (de) * | 2004-07-23 | 2010-01-20 | The Medicines Company (Leipzig) GmbH | Substituierte pyrido[3',2':4,5]thieno[3,2-d]pyrimidine und pyrido[3',2':4,5]furo[3,2-d]-pyrimidine zur verwendung als inhibitoren der pda-4 und/oder tnf-alpha freisetzung |
| RU2008118156A (ru) * | 2005-10-07 | 2009-11-20 | Астеллас Фарма Инк. (Jp) | Производное триарилкарбоновой кислоты |
-
2009
- 2009-10-14 CA CA2739058A patent/CA2739058A1/en not_active Abandoned
- 2009-10-14 US US13/124,336 patent/US20110201815A1/en not_active Abandoned
- 2009-10-14 EP EP09820582A patent/EP2338887A4/en not_active Withdrawn
- 2009-10-14 JP JP2010533902A patent/JP5587784B2/ja not_active Expired - Fee Related
- 2009-10-14 WO PCT/JP2009/067751 patent/WO2010044404A1/ja active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1398800A (en) * | 1972-05-05 | 1975-06-25 | Merck & Co Inc | Trisubstituted imidazoles |
| WO2006100095A1 (de) * | 2005-03-24 | 2006-09-28 | Curacyte Discovery Gmbh | Substituierte carbonsäureamide, verfahren zu ihrer herstellung und ihre verwendung als tnf-alpha-freisetzungsinhibitoren |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2739058A1 (en) | 2010-04-22 |
| EP2338887A4 (en) | 2012-05-09 |
| WO2010044404A1 (ja) | 2010-04-22 |
| EP2338887A1 (en) | 2011-06-29 |
| JPWO2010044404A1 (ja) | 2012-03-15 |
| US20110201815A1 (en) | 2011-08-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5587784B2 (ja) | 縮合複素環誘導体及びその医薬用途 | |
| JP5638513B2 (ja) | インドリジン誘導体及びその医薬用途 | |
| JP5906191B2 (ja) | (アザ)インドリジン誘導体及びその医薬用途 | |
| JP5330989B2 (ja) | (アザ)インドール誘導体及びその医薬用途 | |
| CA2682393C (en) | 5-membered heterocyclic derivative and use thereof for medical purposes | |
| JP5314037B2 (ja) | 縮合環誘導体及びその医薬用途 | |
| JPWO2010044403A1 (ja) | 5員環ヘテロアリール誘導体及びその医薬用途 | |
| JP5931744B2 (ja) | アセチレン誘導体及びその医薬用途 | |
| JP5563985B2 (ja) | フェニルイソニコチン酸誘導体及びその医薬用途 | |
| JP6023713B2 (ja) | 縮合へテロ環誘導体及びその医薬用途 | |
| JP5580204B2 (ja) | ビアリールイソニコチン酸誘導体及びその医薬用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20120912 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140304 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140408 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140722 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140724 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5587784 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |