JP5485237B2 - スピロケタール誘導体の結晶およびその製造方法 - Google Patents
スピロケタール誘導体の結晶およびその製造方法 Download PDFInfo
- Publication number
- JP5485237B2 JP5485237B2 JP2011193271A JP2011193271A JP5485237B2 JP 5485237 B2 JP5485237 B2 JP 5485237B2 JP 2011193271 A JP2011193271 A JP 2011193271A JP 2011193271 A JP2011193271 A JP 2011193271A JP 5485237 B2 JP5485237 B2 JP 5485237B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- formula
- crystal
- added
- water
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CCC(CCCC(**)CCC1)C1C1(OC)OC(CO)C(*)C(*)C1** Chemical compound CCC(CCCC(**)CCC1)C1C1(OC)OC(CO)C(*)C(*)C1** 0.000 description 5
- VWVKUNOPTJGDOB-BDHVOXNPSA-N CCc1ccc(Cc2cc([C@]([C@@H]([C@H]3O)O)(OC4)O[C@H](CO)[C@H]3O)c4cc2)cc1 Chemical compound CCc1ccc(Cc2cc([C@]([C@@H]([C@H]3O)O)(OC4)O[C@H](CO)[C@H]3O)c4cc2)cc1 VWVKUNOPTJGDOB-BDHVOXNPSA-N 0.000 description 3
- DLYCINLLALMWOI-AVGHQTNXSA-N CCc1ccc(CC(C2)=CC=C(CO3)C2[C@@]3([C@@H]([C@H]2O)O)O[C@H](CO)[C@H]2O)cc1 Chemical compound CCc1ccc(CC(C2)=CC=C(CO3)C2[C@@]3([C@@H]([C@H]2O)O)O[C@H](CO)[C@H]2O)cc1 DLYCINLLALMWOI-AVGHQTNXSA-N 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/04—Carbocyclic radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/01—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing oxygen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H13/00—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids
- C07H13/02—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids by carboxylic acids
- C07H13/04—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids by carboxylic acids having the esterifying carboxyl radicals attached to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H23/00—Compounds containing boron, silicon, or a metal, e.g. chelates, vitamin B12
Description
さらに、特許文献1は式(A)で示される化合物の製造方法を開示しており、スキーム3(特許文献1、第24頁)には、ジブロモベンゼン誘導体をアルキルリチウム試薬と作用させた後、ラクトンとカップリングし、さらにスズ化合物に変換した後に、パラジウム触媒存在下カップリング反応を行い、目的の化合物を得る方法が記載されている。
本発明の目的は、医薬品の活性成分として使用するスピロケタール誘導体の工業的製造に適した効率的かつ簡便な製造方法、および有用な合成中間体を提供すること、および医薬品あるいは医薬品原料として、保存安定性、製剤時の取り扱い易さなどの点で優れた特性を有する結晶を提供することである。
R1およびR2は、それぞれ独立に、1以上のRaにより置換されていてもよいC1−10アルキル、1以上のRaにより置換されていてもよいC3−10シクロアルキル、1以上のRaにより置換されていてもよいC2−10アルケニル、1以上のRaにより置換されていてもよいC3−10シクロアルケニル、1以上のRaにより置換されていてもよいC2−10アルキニル、1以上のRaにより置換されていてもよいアリール、1以上のRaにより置換されていてもよい飽和、部分不飽和、または不飽和のへテロシクリル、シアノ、ハロゲン原子、ニトロ、メルカプト、−OR3、−NR4R5、−S(O)pR6、−S(O)qNR7R8、−C(=O)R35、−CR36=NOR37、−C(=O)OR9、−C(=O)NR10R11、および−SiR12R13R14から選択され;nが2以上の場合、R1はそれぞれ同一であっても、異なっていてもよく;mが2以上の場合、R2はそれぞれ同一であっても、異なっていてもよく;または、隣接する炭素原子上に存在する2つのR1は、それらが結合する炭素原子と一緒になって、ベンゼン環に縮合する炭素環またはヘテロ環を形成してもよく;隣接する炭素原子上に存在する2つのR2は、それらが結合する炭素原子と一緒になって、ベンゼン環に縮合する炭素環またはヘテロ環を形成してもよく;
pは、0〜2から選択される整数であり;qは、1および2から選択される整数であり;
R3は、水素原子、C1−10アルキル、C3−10シクロアルキル、C2−10アルケニル、C3−10シクロアルケニル、C2−10アルキニル、アリール、ヘテロアリール、−SiR12R13R14、または−C(=O)R15であり;
R4およびR5は、それぞれ独立に、水素原子、ヒドロキシ、C1−10アルキル、C3−10シクロアルキル、C1−10アルコキシ、アリール、ヘテロアリール、−SiR12R13R14、および−C(=O)R15から選択され;
R6は、C1−10アルキル、C3−10シクロアルキル、アリール、またはヘテロアリールであり、ただし、pが0の場合、R6はさらに−SiR12R13R14、または−C(=O)R15であってもよく;
R7、R8、R10およびR11は、それぞれ独立に、水素原子、C1−10アルキル、C3−10シクロアルキル、アリール、ヘテロアリール、−SiR12R13R14、および−C(=O)R15から選択され;
R9は、水素原子、C1−10アルキル、C3−10シクロアルキル、アリール、ヘテロアリール、または−SiR12R13R14であり;
Raは、それぞれ独立に、C3−10シクロアルキル、C2−10アルケニル、C3−10シクロアルケニル、C2−10アルキニル、アリール、ヘテロアリール、ヒドロキシ、ハロゲン原子、−NR21R22、−OR38、−SR26、−S(O)2R27、−SiR23R24R25、カルボキシ、−C(O)NR28R29、−C(=O)R30、−CR31=NOR32、シアノ、および−S(O)rNR33R34から選択され;
rは、1および2から選択される整数であり;
R12、R13、R14、R23、R24、およびR25は、それぞれ独立に、C1−10アルキル、およびアリールから選択され;
R15およびR30は、それぞれ独立に、水素原子、C1−10アルキル、C3−10シクロアルキル、C1−10アルコキシ、C1−10アルキルアミノ、ジ(C1−10アルキル)アミノ、C1−10アルキルチオ、アリール、およびヘテロアリールから選択され;
R21、R22、R28、R29、R33およびR34は、それぞれ独立に、水素原子、ヒドロキシ、C1−10アルキル、C3−10シクロアルキル、C1−10アルコキシ、アリール、ヘテロアリール、−SiR23R24R25、および−C(=O)R30から選択され;
R26は、水素原子、C1−10アルキル、C1−10アルコキシ、C3−10シクロアルキルオキシ、アリールオキシ、C3−10シクロアルキル、アリール、ヘテロアリール、−C(=O)R30、または−SiR23R24R25であり;
R27は、ヒドロキシ、C1−10アルキル、C3−10シクロアルキル、アリール、ヘテロアリール、−SiR23R24R25、または−C(=O)R30であり;
R31は、水素原子、C1−10アルキル、またはC3−10シクロアルキルであり;
R32は、水素原子、C1−10アルキル、C3−10シクロアルキル、アリール、ヘテロアリール、−SiR23R24R25、または−C(=O)R30であり;
R35は、水素原子、C1−10アルキル、C3−10シクロアルキル、C2−10アルケニル、C3−10シクロアルケニル、C2−10アルキニル、C1−10アルキルチオ、アリール、またはヘテロアリールであり;
R36は、水素原子、C1−10アルキル、C3−10シクロアルキル、C2−10アルケニル、C3−10シクロアルケニル、またはC2−10アルキニルであり;
R37は、水素原子、C1−10アルキル、C3−10シクロアルキル、C2−10アルケニル、C3−10シクロアルケニル、アリール、ヘテロアリール、−SiR12R13R14、または−C(=O)R15であり;
R38は、C1−10アルキル、C3−10シクロアルキル、C2−10アルケニル、C3−10シクロアルケニル、C2−10アルキニル、C1−10アルキルチオ、アリール、ヘテロアリール、−SiR23R24R25、または−C(=O)R30である]
の化合物を製造する方法であって;
工程a)式(II):
P1は、金属イオン、水素原子またはヒドロキシ基の保護基であり;
R41は、R1として既に定義した基であり、ただし当該基は1以上の保護基を有していてもよく;nは既に定義したとおりである]
の化合物を有機金属試薬で処理し、その後、式(III):
を反応させて、式(IVa):
Xは、金属イオン、または水素原子である]
の化合物を得る工程;
工程b)式(IVb):
P6は、金属イオン、水素原子またはヒドロキシ基の保護基である]
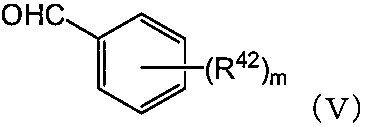
の化合物を、有機金属試薬で処理し、その後、式(V):
の化合物と反応させる工程;
を含み、さらに、上記工程中、および/またはその前後の任意の段階において、保護基を導入する工程、および/または保護基を除去する工程を含んでいてもよい、前記製造方法が提供される。
工程c)式(VI):
の化合物を、以下の2段階
段階(1):P1が水素原子である式(VI)の化合物を、酸性条件下で処理する工程(但し、P1が保護基である場合は、当該処理前の脱保護工程をさらに含む);および、
段階(2):還元反応により、工程b)の反応により生じたヒドロキシ基を除去する工程;
(但し、いずれの段階を先に行ってもよい)に付し、式(VII):
の化合物を得る工程をさらに含む。
用語「C1−10アルキル」は、炭素数1〜10の直鎖状または分岐鎖状のアルキル基を意味し、例えば、メチル、エチル、n−プロピル、i−プロピル、n−ブチル、s−ブチル、i−ブチル、t−ブチル、n−ペンチル、3−メチルブチル、2−メチルブチル、1−メチルブチル、1−エチルプロピル、n−ヘキシル、4−メチルペンチル、3−メチルペンチル、2−メチルペンチル、1−メチルペンチル、3−エチルブチル、2−エチルブチル、シクロプロピルメチル、およびシクロヘキシルメチルなどが含まれる。C1−10アルキルには、さらに、直鎖状または分岐鎖状のC1−6アルキル、およびC1−4アルキルが含まれる。
ここで、R51は、それぞれ独立に、1以上のR56により置換されていてもよいアリール、1以上のアリールにより置換されていてもよいC1−10アルコキシ、C1−10アルキルチオ、およびアリールセレニルから選択され;
R52は、それぞれ独立に、C1−10アルコキシから選択され;
R53およびR55は、それぞれ独立に、C1−10アルキル、およびアリールから選択され;
R54は、水素原子、C1−10アルキル、1以上のC1−10アルコキシにより置換されていてもよいアリール、ヘテロアリール、1以上のR57により置換されていてもよいアミノ、1以上のアリールにより置換されていてもよいC1−10アルコキシ、または1以上のニトロにより置換されていてもよいアリールオキシであり;
R56は、それぞれ独立に、C1−10アルキル、C1−10アルコキシ、アリール、およびヘテロアリールから選択され;
R57は、それぞれ独立に、C1−10アルキル、およびアリールから選択される。
本発明において定義されるR1およびR2は、特に限定はされないが、例えば、それぞれ独立に、1以上のRaにより置換されていてもよいC1−10アルキル、1以上のRaにより置換されていてもよいC3−10シクロアルキル、1以上のRaにより置換されていてもよいC2−10アルケニル、1以上のRaにより置換されていてもよいC3−10シクロアルケニル、1以上のRaにより置換されていてもよいC2−10アルキニル、1以上のRaにより置換されていてもよいアリール、1以上のRaにより置換されていてもよい飽和、部分不飽和、または不飽和のへテロシクリル、および−SiR12R13R14から選択される。より好ましくは、R1およびR2は、それぞれ独立に、C1−6アルキル、C3−6シクロアルキル、アリール、および−SiR12R13R14から選択される。本発明において、nまたはmが0の場合、ベンゼン環上にR1またはR2はそれぞれ存在しない。本発明の1つの態様において、nが0であり、mが0または1であり、R2がC1−4アルキルである。
P1、P6、およびXにおいて定義される金属イオンとは、アルコキシドイオンのカウンターイオンとなる金属イオンを意味し、例えば、リチウムイオン、ナトリウムイオン、カリウムイオン、セシウムイオン、マグネシウムイオンなどのアルカリ金属イオン、アルカリ土類金属イオンなどが挙げられ、他の金属と錯体を形成していてもよい。当該金属イオンには、例えば、本発明で使用する有機金属試薬をヒドロキシ基に作用させて生じる金属イオン(たとえば、リチウムイオン)なども含まれる。
工程a)で用いる式(III)の化合物において、P2、P3、P4、およびP5の例としては、例えば、C1−6アルコキシC1−6アルキル(例えば、メトキシメチル、エトキシメチル、1−メトキシエチル、1−メトキシ−1−メチルエチルなど)、アリールメチルオキシC1−6アルキル(例えば、ベンジルオキシメチルなど)、テトラヒドロピラニル、基−Si(R53)3(例えば、トリメチルシリル、トリエチルシリル、t−ブチルジメチルシリル、イソプロピルジメチルシリル、t−ブチルジフェニルシリルなど)、アラルキル(例えば、ベンジル、4−メトキシベンジル、トリフェニルメチルなど)、基−B(OR55)2、C1−6アルキルカルボニル(例えば、アセチル、プロピオニル、ピバロイルなど)、C1−6アルコキシカルボニル(例えば、メトキシカルボニル、イソプロピルオキシカルボニル、t−ブトキシカルボニル、ベンジルオキシカルボニルなど)、t−ブチルなどの保護基が挙げられる。また、P4およびP5は一緒になって、2つのヒドロキシ基を保護して環を形成する2価の基(例えば、−CH2−、−CH(CH3)−、−C(CH3)2−、−CHPh−など)であってもよい。
当該有機金属試薬は、例えば、−100〜30℃、好ましくは−90〜−10℃、特に−90〜−70℃の温度下で系中に少しずつ(例えば、滴下して)加えることができる。有機金属試薬の添加後、適当な温度下、例えば、−100〜30℃、好ましくは−90〜−10℃、特に−90〜−70℃で、一定時間、例えば、0.1〜5時間、好ましくは0.5〜2時間、反応を撹拌してもよい。
工程c)の段階(2)の還元反応は、適当な溶媒、例えば、テトラヒドロフラン(THF)、メチルテトラヒドロフラン、1,2−ジメトキシエタン、メタノール、エタノール、イソプロパノール、酢酸エステル(例えば、酢酸エチル、酢酸メチル、酢酸イソプロピルなど)、アセトン、水、または2以上の前記溶媒を含む混合溶媒中で、適当な温度下、例えば、−80〜80℃、好ましくは−30〜70℃、特に−20〜60℃で行うことができる。反応時間は適宜設定することができるが、例えば、0.5〜24時間、好ましくは5〜15時間程度である。当該還元反応は、式(VI)の化合物の2つのベンゼン環を連結する炭素原子上のヒドロキシ基を除去するために適当な還元剤および/または触媒を使用するものであれば特に限定されず、例えば、水素雰囲気下での金属触媒(例えば、炭素担持パラジウム、白金、均一系パラジウム錯体、均一系ルテニウム錯体、均一系ロジウム錯体);ルイス酸と組み合わせたヒドリド型還元剤(例えば、塩化アルミニウム−水素化ホウ素ナトリウム、トリフルオロ酢酸−トリエチルシラン)などを使用することができる。
工程d)式(I)の化合物を、式(X):
の化合物に変換する工程;
工程e)式(X)の化合物を結晶化し、再結晶により精製する工程;
工程f)式(X)の化合物から保護基を除去し、高純度の式(I)の化合物を得る工程;
を含む、前記製造方法が提供される。
の化合物が提供される。当該化合物は、式(I)の化合物の合成中間体として有用である。
の化合物が提供される。当該化合物もまた、式(I)の化合物の合成中間体として有用である。
NMRは、核磁気共鳴装置 JNM−ECP−500(JEOL製)、またはJNM−ECP−400(JEOL製)を用いて測定した。質量分析は、質量分析装置LCT Premier XE(Waters製)を用いて測定した。分取高速液体クロマトグラフィーは、ジーエルサイエンス分取システムを用いた。高速液体クロマトグラフィーは、Agilent 1100(Agilent製)を用いた。水分測定は、KF分析装置 型式KF−100(微量水分測定装置 三菱化学社製)を用いた。なお、生成物を精製なしで次の工程に使用する場合は、生成物の一部を取るか、または別途同じ手法で調製した生成物を適宜精製し、その後にNMRを測定した。
工程1:3,4,5−トリス(トリメチルシリルオキシ)−6−トリメチルシリルオキシメチル−テトラヒドロピラン−2−オンの合成
1H−NMR(500MHz,CDCl3)δ:−0.47(4.8H,s)、−0.40(4.2H,s)、−0.003〜0.004(5H,m)、0.07−0.08(13H,m)、0.15−0.17(18H,m)、1.200および1.202(3H,eacht,J=8.0Hz)、1.393および1.399(3H,each s)、1.44(3H,s)、2.61(2H,q,J=8.0Hz)、3.221および3.223(3H,eachs)、3.43(1H,t,J=8.5Hz)、3.54(1H,dd,J=8.5,3.0Hz)、3.61−3.66(1H,m)、3.80−3.85(3H,m)、4.56および4.58(1H,each d,J=12.4Hz)、4.92および4.93(1H,each d,J=12.4Hz)、5.80および5.82(1H,each d,J=3.0Hz)、7.14(2H,d,J=8.0Hz)、7.28−7.35(3H,m)、7.50−7.57(2H,m)。
ジアステレオマー3および4の混合物:
1H−NMR(500MHz,トルエン−d8,80℃)δ:−0.25(4H,s)、−0.22(5H,s)、0.13(5H,s)、0.16(4H,s)、0.211および0.214(9H,each s)、0.25(9H,s)、0.29(9H,s)、1.21(3H,t,J=7.5Hz)、1.43(3H,s)、1.45(3H,s)、2.49(2H,q,J=7.5Hz)、3.192および3.194(3H,each s)、3.91−4.04(4H,m)、4.33−4.39(2H,m)、4.93(1H,d,J=14.5Hz)、5.10−5.17(1H,m)、5.64および5.66(1H,each s)、7.03(2H,d,J=8.0Hz)、7.28−7.35(3H,m)、7.59−7.64(1H,m)、7.87−7.89(1H,m)。
工程4:1,1−アンヒドロ−1−C−[5−(4−エチルフェニル)ヒドロキシメチル−2−(ヒドロキシメチル)フェニル]−β−D−グルコピラノースの合成
工程5:1,1−アンヒドロ−1−C−[5−(4−エチルフェニル)メチル−2−(ヒドロキシメチル)フェニル]−β−D−グルコピラノース(粗生成物)の合成
高速液体クロマトグラフィーの測定条件:
カラム:Cadenza CD−C18 50mm P/NCD032
移動相:A液:H2O,B液:MeCN
グラジェント操作:B液:5%から100%(6分間)、100%(2分間)
流速:毎分1.0mL
温度:35.0℃
検出波長:210nm
工程6:1,1−アンヒドロ−1−C−[5−(4−エチルフェニル)メチル−2−(ヒドロキシメチル)フェニル]−2,3,4,6−テトラ−O−メトキシカルボニル−β−D−グルコピラノースの合成
なお、工程6とは別に、種結晶を用いなかったことを除き工程6と同様の方法を行ったところ、表題の化合物を結晶として得た。
高速液体クロマトグラフィー測定条件:
カラム:Capcell pack ODS UG−120(4.6mmI.D.×150mm,3μm,資生堂製)
移動相:A液:H2O、B液:MeCN
移動相の送液:A液およびB液の混合比を次のように変えて濃度勾配制御した。
温度:25.0℃
検出波長:220nm
水分量の測定方法:
分析法:電量滴定法
KF分析装置:微量水分測定装置 三菱化学社製 型式KF−100
陽極液:アクアミクロンAX(三菱化学製)
陰極液:アクアミクロンCXU(三菱化学製)
[実施例2]2,3,4,5−テトラキス(トリメチルシリルオキシ)−6−トリメチルシリルオキシメチル−2−(5−ブロモ−2−(1−メトキシ−1−メチルエトキシメチル)フェニル)テトラヒドロピランの合成
1H−NMR(500MHz,CDCl3)δ:−0.30(9H,s)、0.095(9H,s)、0.099(9H,s)、0.16(9H,s)、0.17(9H,s)、1.41(3H,s)、1.43(3H,s)、3.20(3H,s)、3.37−3.44(2H,m)、3.62(1H,dd,J=10.5,7.5Hz)、3.81−3.89(3H,m)、4.62(1H,d,J=13.2Hz)、4.81(1H,d,J=13.2Hz)、7.38(1H,dd,J=8.8,2.5Hz)、7.46(1H,d,J=8.8Hz)、7.70(1H,d,J=2.5Hz)。
ジアステレオマー6:
1H−NMR(500MHz,トルエン−d8,80℃)δ:−0.16(9H,s)、0.18(9H,s)、0.22(9H,s)、0.23(9H,s)、0.29(9H,s)、1.405(3H,s)、1.412(3H,s)、3.16(3H,s)、3.87(1H,dd,J=10.5,4.3Hz)、3.98(1H,dd,J=4.3,1.5Hz)、4.02(1H,dd,J=10.5,2.5Hz)、4.14(1H,s)、4.26(1H,brs)、4.38(1H,brs)、4.90−4.96(2H,m)、7.34(1H,dd,J=8.5,1.5Hz)、7.70(1H,d,J=8.5Hz)、7.97(1H,s,brs)。
[実施例3]1,1−アンヒドロ−1−C−[5−(4−エチルフェニル)メチル−2−(ヒドロキシメチル)フェニル]−2,3,4,6−テトラ−O−ピバロイル−β−D−グルコピラノースの合成
[実施例4]3,4,5−トリス(トリメチルシリルオキシ)−6−トリメチルシリルオキシメチル−テトラヒドロピラン−2−オンの合成
カラム:YMC−Pack ODS−AM 4.6mm I.D.×150mm、3μm (ワイエムシィ)
移動相:A液:2mM AcONH4/H2O, B液:50%(v/v)MeCN/MeOH
グラジェント操作:B液:50%から95%(15分間)、95%ホールド(15分間)、95%から100%(5分間)100%(15分間)
流速:1.0ml/分
カラム温度:40℃
検出波長:200nm
[実施例5]1,1−アンヒドロ−1−C−[5−(4−エチルフェニル)メチル−2−(ヒドロキシメチル)フェニル]−β−D−グルコピラノースの合成
工程1:2,3,4,5−テトラキス(トリメチルシリルオキシ)−6−トリメチルシリルオキシメチル−2−(5−(4−エチルフェニル)ヒドロキシメチル−2−(1−メトキシ−1−メチルエトキシメチル)フェニル)テトラヒドロピランの合成
カラム:Ascentis Express C18,3.0mm I.D.×100mm, 2.7μm(Supelco)
移動相:A液:2mM AcONH4水溶液、B液:MeCN
グラジェント操作:B液:30%から98%(25分間)、98%(5分間)
流速:毎分1.0mL
温度:40℃
検出波長:210nm
工程2:1,1−アンヒドロ−1−C−[5−(4−エチルフェニル)ヒドロキシメチル−2−(ヒドロキシメチル)フェニル]−β−D−グルコピラノースの合成
カラム:Atlantis dC18,4.6mm I.D.×75mm,3μm(Waters)
移動相:A液:H2O,B液:MeCN
グラジェント操作:B液:2%から20%(3分間)、20%から28%(5分間)、28%(12分間)、28%から100%(7分間)、100%(8分間)
流速:毎分1.2mL
温度:35℃
検出条件:210nm
工程4:1,1−アンヒドロ−1−C−[5−(4−エチルフェニル)メチル−2−(ヒドロキシメチル)フェニル]−2,3,4,6−テトラ−O−メトキシカルボニル−β−D−グルコピラノースの合成
水分量の測定方法:
分析法:電量滴定法
KF分析装置:微量水分測定装置 三菱化学社製 型式KF−100
陽極液:アクアミクロンAX(三菱化学製)
陰極液:アクアミクロンCXU(三菱化学製)
実施例5工程5にて使用した種結晶は、以下の方法により得た結晶の一部を使用した。
カラム:ZORBAX Eclipse XDB−C18,4.6mm I.D. x 50mm,1.8μm(Agilent)
移動相:A液:H2O,B液:MeOH
グラジェント操作:B液:40%から60%(11.5分間)、60%から80%(7分間)、80%から95%(4分間)、95%(5分間)
流速:毎分1.0ml
温度:50℃
検出波長:220nm
[実施例6]2,4−ジブロモ−1−(1−メトキシ−1−メチルエトキシメチル)ベンゼンのハロゲン金属交換反応
以下の条件(条件1〜4)において2,4−ジブロモ−1−(1−メトキシ−1−メチルエトキシメチル)ベンゼンのハロゲン金属交換反応を行い、反応の位置選択性を生成物の1H−NMR分析により確認した。
1H−NMR(CDCl3)δ:1.41(6H,s)、3.24(3H,s)、4.43(2H,s)、7.21−7.24(2H,m)、7.44−7.47(2H,m)。
1H−NMR(CDCl3)δ:1.46(6H,s)、3.24(3H,s)、4.55(2H,s)、7.10−7.14(1H,m)、7.29−7.33(1H,m)、7.51−7.55(2H,m)。
1,1−アンヒドロ−1−C−[5−(4−エチルフェニル)メチル−2−(ヒドロキシメチル)フェニル]−2,3,4,6−テトラ−O−メトキシカルボニル−β−D−グルコピラノース(湿性粉末88.2g)の1,2−ジメトキシエタン(285ml)溶液に、水酸化ナトリウム水溶液(5M、285ml)を室温にて加えて、同温で1時間攪拌した。この溶液に硫酸水溶液(1M、713ml)加えた後、さらに水(100ml)を加え、酢酸エチル(500ml)で2回抽出した。合わせた有機層を飽和塩化ナトリウム水溶液(1000ml)で洗浄し、さらに無水硫酸ナトリウム(250g)で乾燥後、減圧下で溶媒をおよそ半量留去し、析出した生成物を結晶性粉末として得た(10.3g)。得られた結晶性粉末の一部(4mg)をジメチルスルホキシド(0.02ml)に溶解させ、当該溶液を−20℃で2日間凍結乾燥し、ジメチルスルホキシドを除去した。残渣に水(0.02ml)を加えた後、ごく少量の上記の結晶性粉末を種結晶として加え、室温にて10日間振とう攪拌(100rpm、TAITEC社製DOUBLE SHAKER NR−3)し、表題の化合物を結晶として得た。得られた結晶の粉末X線回折を測定し、ピークが試験例5で測定した1水和物の回折パターンと同じ回折角(2θ)に観測されたことにより、当該結晶が1水和物であることを確認した。
1,1−アンヒドロ−1−C−[5−(4−エチルフェニル)メチル−2−(ヒドロキシメチル)フェニル]−β−D−グルコピラノースの1水和物結晶(200mg)と酢酸ナトリウム(40mg)をメタノール(1ml)に80℃にて溶解させ、室温に冷却した後、イソプロパノール(2ml)を加えた。減圧下に溶媒(約2ml)を留去した後、標題の共結晶の種結晶を加え、室温にて一夜撹拌し、析出した結晶をろ取し、イソプロパノール(4ml)で洗浄後、乾燥し、標題の共結晶(融点約162℃)を得た。共結晶の1H−NMR[(CD3)2SO]分析より、表題の化合物のエチル基のCH3のプロトン(δ1.12−1.16(3H,t))のピークと酢酸ナトリウムのCH3(δ1.56(3H,s))の積分比から存在比を算出したところ1:1の共結晶であった。
分析法:示差走査熱量測定(DSC)
装置:DSC6200R エスアイアイ・ナノテクノロジー社製
走査速度:10℃/分
走査範囲:30〜210℃
試料量:3〜4mg
[実施例9]1,1−アンヒドロ−1−C−[5−(4−エチルフェニル)メチル−2−(ヒドロキシメチル)フェニル]−β−D−グルコピラノース酢酸カリウム共結晶の調製
1,1−アンヒドロ−1−C−[5−(4−エチルフェニル)メチル−2−(ヒドロキシメチル)フェニル]−β−D−グルコピラノースの1水和物結晶(200mg)と酢酸カリウム(48mg)をメタノール(1ml)に80℃にて溶解させ、室温に冷却した後、イソプロパノール(2ml)を加えた。減圧下に溶媒(約2ml)を留去した後、標題の共結晶の種結晶を加え、室温にて一夜撹拌し、析出した結晶をろ取し、イソプロパノール(4ml)で洗浄後、乾燥し、標題の共結晶(融点約176℃)を得た。共結晶の1H−NMR[(CD3)2SO]分析より、表題の化合物のエチル基のCH3のプロトン(δ1.13−1.16(3H,t))のピークと酢酸カリウムのCH3(δ1.53(3H,s))の積分比から存在比を算出したところ1:1の共結晶であった。
[試験例1]1水和物結晶の水分吸着等温線の測定
式(XI):
本発明の式(XI)の化合物の1水和物結晶および当該化合物のアモルファスを用い、保存安定性試験を行った。式(XI)の化合物の1水和物結晶は、実施例1の工程7に記載の方法に準じて製造した。式(XI)の化合物のアモルファスは以下の方法で製造した。当該化合物の1水和結晶(15g)をホットステージ上で加熱し、融解後、調湿デシケーター(25℃/dry)内で室温にて放冷し、固化したものを乳鉢で粉砕することにより得たものを試料として使用した。各試料を25℃および40℃に設定した恒温槽に保存し、1ヶ月後、3ヶ月後および6ヶ月後に試料の純度を確認した。
使用機器:2695 Separations Module(Waters製)、2487 Dual λ Absorbance Detector(Waters製)または996 Photodiode Array Detector(Waters製)
カラム:YMC-Pack ODS-AM AM-302-3、内径4.6 mm×長さ15 cm、粒子径3μm(YMC製)
溶出液:A液=メタノール、B液=水
グラジェント操作:A液:55%(15分間)、A液55から100%(10分間)、100%(5分間)
流量:1.0mL/分
検出波長:220nm
サンプルクーラー温度:5℃
面積測定範囲:溶液注入後30分間
結果を表2に示す。25℃および40℃のいずれにおいても、アモルファスの不純物総量は経時的に増加したのに対し、25℃および40℃のいずれにおいても1水和物結晶の不純物総量は6ヶ月間ほぼ一定であった。
本発明の式(XI)の化合物の酢酸ナトリウム共結晶を用い、保存安定性試験を試験例2の方法に準じて行った。式(XI)の化合物の酢酸ナトリウム共結晶は、実施例5に記載の方法に準じて製造した。試料を25℃および40℃に設定した恒温槽に保存し、1ヶ月後、および3ヶ月後に試料の純度を確認した。得られた測定結果を試験例2で得られたアモルファスの結果と比較した。
本発明の式(XI)の化合物の酢酸カリウム共結晶を用い、保存安定性試験を試験例2の方法に準じて行った。式(XI)の化合物の酢酸カリウム共結晶は、実施例6に記載の方法に準じて製造した。試料を25℃および40℃に設定した恒温槽に保存し、1ヶ月後、および3ヶ月後に試料の純度を確認した。得られた測定結果を試験例2で得られたアモルファスの結果と比較した。
式(XI)の化合物の1水和物結晶、アモルファス、酢酸ナトリウム共結晶および酢酸カリウム共結晶の粉末X線回折を測定した。測定条件を以下に示す。
測定装置:RINT1100(Rigaku社製)
対陰極:Cu
管電圧:40kV
管電流:40mA
走査速度:2.000°/分
サンプリング幅:0.020°
発散スリット:1°
散乱スリット:1°
受光スリット:0.15mm
走査範囲:3〜35°
1水和物結晶の測定条件(測定条件2)
測定装置:X’Pert−Pro MPD(PANalytical社製)
対陰極:Cu
管電圧:45kV
管電流:40mA
走査方式:連続
ステップ幅:0.017
走査軸:2θ
ステップあたりのサンプリング時間:30秒
走査範囲:2〜35°
アモルファスの測定条件
測定装置:RINT1100(Rigaku社製)
対陰極:Cu
管電圧:40kV
管電流:20mA
走査速度:2.000°/分
サンプリング幅:0.020°
発散スリット:1°
散乱スリット:1°
受光スリット:0.15mm
走査範囲:2〜35°
酢酸ナトリウム共結晶および酢酸カリウム共結晶の測定条件
測定装置:X’Pert MPD(PANalytical社製)
対陰極:Cu
管電圧:45kV
管電流:40mA
走査速度:1.000°/分
サンプリング幅:0.050°
発散スリット:0.25°
散乱スリット:0.25°
受光スリット:0.2mm
走査範囲:3〜35°
1水和結晶の結果を図2に示す。3.5°、6.9°、10.4°、13.8°、16.0°、17.2°、18.4°、20.8°、21.4°、および24.4°付近の回折角(2θ)にピークが観測された。酢酸ナトリウム共結晶の結果を図4に示す。4.9°、8.7°、9.3°、11.9°、12.9°、14.7°、16.0°、17.1°、17.7°、19.6°、21.6°、および22.0°付近の回折角(2θ)にピークが観測された。酢酸カリウム共結晶の結果を図5に示す。5.0°、10.0°、10.4°、12.4°、14.5°、15.1°、19.0°、20.1°、21.4°、および25.2°付近の回折角(2θ)にピークが観測された。
(1)高純度の上記式(I)の化合物の製造方法であって;
工程d)式(I)の化合物を、式(X):
の化合物に変換する工程;
工程e)式(X)の化合物を結晶化し、再結晶により精製する工程;
工程f)式(X)の化合物から保護基を除去し、高純度の式(I)の化合物を得る工程;
を含む、前記製造方法。
(5)P7が、C1−6アルキルカルボニル、C1−6アルコキシカルボニル、−SiR23R24R25から選択され、R23、R24、およびR25は、本明細書に定義したとおりである、上記(1)〜(4)のいずれかに記載の製造方法。
(7)式(I)において、nが0であり、mが0または1であり、R2がC1−4アルキルである、上記(1)〜(6)のいずれかに記載の製造方法。
(8)式(XI):
(9)1水和物である、上記(8)に記載の式(XI)の化合物の結晶。
(10)粉末X線回折パターンにおいて、3.5°、6.9°、および13.8°付近の回折角(2θ)にピークを有する、上記(8)または(9)に記載の式(XI)の化合物の結晶。
(14)アセトン:水の容積比が1:3.5から1:7である、アセトンおよび水の混合溶媒を使用する、上記(13)に記載の式(XI)の化合物の結晶。
(16)粉末X線回折パターンにおいて、4.9°、14.7°、16.0°、17.1°、および19.6°付近の回折角(2θ)にピークを有する、上記(8)または(15)に記載の式(XI)の化合物の結晶。
(20)粉末X線回折パターンにおいて、5.0°、15.1°、19.0°、20.1°および25.2°付近の回折角(2θ)にピークを有する、上記(8)または(19)に記載の式(XI)の化合物の結晶。
Claims (16)
- 式(XI):
- 粉末X線回折パターンにおいて、3.5°、6.9°、および13.8°付近の回折角(2θ)にピークを有する、請求項1に記載の式(XI)の化合物の結晶。
- 粉末X線回折パターンにおいて、3.5°、6.9°、13.8°、16.0°、17.2°、および18.4°付近の回折角(2θ)にピークを有する、請求項1または2に記載の式(XI)の化合物の結晶。
- 粉末X線回折パターンにおいて、3.5°、6.9°、10.4°、13.8°、16.0°、17.2°、18.4°、20.8°、21.4°、および24.4°付近の回折角(2θ)にピークを有する、請求項1〜3のいずれか1項に記載の式(XI)の化合物の結晶。
- アセトンおよび水の混合溶媒からの結晶化により得られる、請求項1〜4のいずれか1項に記載の式(XI)の化合物の結晶。
- アセトン:水の容積比が1:3.5から1:7である、アセトンおよび水の混合溶媒を使用する、請求項5に記載の式(XI)の化合物の結晶。
- 式(XI):
- 粉末X線回折パターンにおいて、4.9°、14.7°、16.0°、17.1°、および19.6°付近の回折角(2θ)にピークを有する、請求項7に記載の式(XI)の化合物の結晶。
- 粉末X線回折パターンにおいて、4.9°、8.7°、9.3°、11.9°、12.9°、14.7°、16.0°、17.1°、17.7°、19.6°、21.6°、および22.0°付近の回折角(2θ)にピークを有する、請求項7または8に記載の式(XI)の化合物の結晶。
- メタノールおよびイソプロパノールの混合溶媒からの結晶化により得られる、請求項7〜9のいずれか1項に記載の式(XI)の化合物の結晶。
- 式(XI):
- 粉末X線回折パターンにおいて、5.0°、15.1°、19.0°、20.1°および25.2°付近の回折角(2θ)にピークを有する、請求項11に記載の式(XI)の化合物の結晶。
- 粉末X線回折パターンにおいて、5.0°、10.0°、10.4°、12.4°、14.5°、15.1°、19.0°、20.1°、21.4°、および25.2°付近の回折角(2θ)にピークを有する、請求項11または12に記載の式(XI)の化合物の結晶。
- メタノールおよびイソプロパノールの混合溶媒からの結晶化により得られる、請求項1、11〜13のいずれかに記載の式(XI)の化合物の結晶。
- 高純度の請求項1〜14のいずれか1項に記載した式(XI):
工程d)式(XI)の化合物を、式(Xc):
の化合物に変換する工程;
工程e)式(Xc)の化合物を結晶化し、再結晶により精製する工程;
工程f)式(Xc)の化合物から保護基を除去し、高純度の式(XI)の化合物を得て、該化合物を結晶化する工程;
を含む、前記製造方法。 - P7が、t−ブチルカルボニルおよびメトキシカルボニルから選択される、請求項15に記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011193271A JP5485237B2 (ja) | 2008-06-20 | 2011-09-05 | スピロケタール誘導体の結晶およびその製造方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008162073 | 2008-06-20 | ||
| JP2008162073 | 2008-06-20 | ||
| JP2011193271A JP5485237B2 (ja) | 2008-06-20 | 2011-09-05 | スピロケタール誘導体の結晶およびその製造方法 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010517979A Division JP4823385B2 (ja) | 2008-06-20 | 2009-06-19 | スピロケタール誘導体の結晶およびその製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012001554A JP2012001554A (ja) | 2012-01-05 |
| JP5485237B2 true JP5485237B2 (ja) | 2014-05-07 |
Family
ID=41434188
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010517979A Active JP4823385B2 (ja) | 2008-06-20 | 2009-06-19 | スピロケタール誘導体の結晶およびその製造方法 |
| JP2011193271A Active JP5485237B2 (ja) | 2008-06-20 | 2011-09-05 | スピロケタール誘導体の結晶およびその製造方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010517979A Active JP4823385B2 (ja) | 2008-06-20 | 2009-06-19 | スピロケタール誘導体の結晶およびその製造方法 |
Country Status (25)
| Country | Link |
|---|---|
| US (2) | US8569520B2 (ja) |
| EP (3) | EP3170834B8 (ja) |
| JP (2) | JP4823385B2 (ja) |
| KR (1) | KR101856784B1 (ja) |
| CN (1) | CN102046645B (ja) |
| AR (1) | AR072211A1 (ja) |
| AU (1) | AU2009261129A1 (ja) |
| CA (1) | CA2727923A1 (ja) |
| CL (1) | CL2010001485A1 (ja) |
| CO (1) | CO6351798A2 (ja) |
| CR (1) | CR20110033A (ja) |
| EC (1) | ECSP11010773A (ja) |
| ES (1) | ES2907611T3 (ja) |
| HK (1) | HK1154589A1 (ja) |
| IL (1) | IL210098A0 (ja) |
| MA (1) | MA32530B1 (ja) |
| MX (1) | MX2010014023A (ja) |
| NZ (1) | NZ589961A (ja) |
| PE (1) | PE20110115A1 (ja) |
| PL (1) | PL3170834T3 (ja) |
| RU (1) | RU2011101972A (ja) |
| SG (1) | SG193795A1 (ja) |
| TW (1) | TW201011043A (ja) |
| WO (1) | WO2009154276A1 (ja) |
| ZA (1) | ZA201100016B (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102656177B (zh) * | 2009-12-18 | 2016-01-27 | 中外制药株式会社 | 螺酮缩醇衍生物的制备方法 |
| WO2012115249A1 (ja) * | 2011-02-25 | 2012-08-30 | 中外製薬株式会社 | スピロケタール誘導体の結晶 |
| CA2851142A1 (en) * | 2011-10-06 | 2013-04-11 | Bayer Intellectual Property Gmbh | Heterocyclylpyri (mi) dinylpyrazole as fungicidals |
| TWI774159B (zh) * | 2013-12-27 | 2022-08-11 | 日商中外製藥股份有限公司 | 含有托格列淨(Tofogliflozin)之固體製劑及其製造方法 |
| CN106188190B (zh) * | 2016-07-28 | 2020-08-25 | 迪嘉药业集团有限公司 | 一种托格列净一水合物的制备方法 |
| WO2018073154A1 (en) | 2016-10-19 | 2018-04-26 | Boehringer Ingelheim International Gmbh | Combinations comprising an ssao/vap-1 inhibitor and a sglt2 inhibitor, uses thereof |
| CN108640958B (zh) * | 2017-05-05 | 2021-03-30 | 镇江圣安医药有限公司 | 异苯并呋喃衍生物、其药物组合物和制剂以及用途 |
| JP2021520394A (ja) | 2018-04-17 | 2021-08-19 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 医薬組成物、処置方法及びその使用 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL63968A (en) | 1980-10-01 | 1985-10-31 | Glaxo Group Ltd | Form 2 ranitidine hydrochloride,its preparation and pharmaceutical compositions containing it |
| HU196775B (en) | 1986-08-05 | 1989-01-30 | Richter Gedeon Vegyeszet | Process for production of morfologically unique new modifications of famotidin and medical compositions containing such substances |
| PT98344B (pt) | 1991-07-17 | 1999-04-30 | Lneti Lab Nac De Eng Tec Ind D | Processo para a obtencao de espirocetais labdanicos do tipo ambar cinzento |
| GB9426103D0 (en) | 1994-12-23 | 1995-02-22 | Merck Sharp & Dohme | Therapeutic agents |
| JP2000044534A (ja) | 1998-07-28 | 2000-02-15 | Mitsui Chemicals Inc | N保護基アミノ酸の製造法 |
| CA2399089C (en) | 2000-02-02 | 2010-05-11 | Banyu Pharmaceutical Co., Ltd. | Process for exchanging functional groups by halogen-metal exchange reaction |
| JP3849024B2 (ja) | 2003-03-26 | 2006-11-22 | 国立大学法人 東京大学 | 有機亜鉛錯体およびその製造方法 |
| TW200637869A (en) | 2005-01-28 | 2006-11-01 | Chugai Pharmaceutical Co Ltd | The spiroketal derivatives and the use as therapeutical agent for diabetes of the same |
| WO2007070984A1 (en) | 2005-12-23 | 2007-06-28 | Ecobiotics Limited | Spiroketals |
| WO2007140191A2 (en) * | 2006-05-23 | 2007-12-06 | Theracos, Inc. | Glucose transport inhibitors and methods of use |
| TWI403516B (zh) | 2006-07-27 | 2013-08-01 | Chugai Pharmaceutical Co Ltd | To replace spirocyclic alcohol derivatives, and its use as a therapeutic agent for diabetes |
| JP5791983B2 (ja) | 2011-07-07 | 2015-10-07 | 三井化学株式会社 | 樹脂組成物、これを用いたポリイミド金属積層体、及び電子回路用基板 |
-
2009
- 2009-06-18 TW TW098120460A patent/TW201011043A/zh unknown
- 2009-06-19 US US13/000,208 patent/US8569520B2/en active Active
- 2009-06-19 CN CN200980119081.4A patent/CN102046645B/zh active Active
- 2009-06-19 KR KR1020117000778A patent/KR101856784B1/ko active IP Right Grant
- 2009-06-19 SG SG2013061510A patent/SG193795A1/en unknown
- 2009-06-19 NZ NZ589961A patent/NZ589961A/xx not_active IP Right Cessation
- 2009-06-19 CA CA2727923A patent/CA2727923A1/en not_active Abandoned
- 2009-06-19 EP EP16202285.9A patent/EP3170834B8/en active Active
- 2009-06-19 AU AU2009261129A patent/AU2009261129A1/en not_active Abandoned
- 2009-06-19 WO PCT/JP2009/061226 patent/WO2009154276A1/ja active Application Filing
- 2009-06-19 PE PE2010001159A patent/PE20110115A1/es not_active Application Discontinuation
- 2009-06-19 EP EP22155547.7A patent/EP4036100A1/en not_active Withdrawn
- 2009-06-19 PL PL16202285T patent/PL3170834T3/pl unknown
- 2009-06-19 ES ES16202285T patent/ES2907611T3/es active Active
- 2009-06-19 RU RU2011101972/04A patent/RU2011101972A/ru not_active Application Discontinuation
- 2009-06-19 JP JP2010517979A patent/JP4823385B2/ja active Active
- 2009-06-19 EP EP09766724.0A patent/EP2308886B1/en active Active
- 2009-06-19 MX MX2010014023A patent/MX2010014023A/es not_active Application Discontinuation
- 2009-06-19 AR ARP090102252A patent/AR072211A1/es not_active Application Discontinuation
-
2010
- 2010-12-19 IL IL210098A patent/IL210098A0/en unknown
- 2010-12-20 MA MA33442A patent/MA32530B1/fr unknown
- 2010-12-20 CL CL2010001485A patent/CL2010001485A1/es unknown
-
2011
- 2011-01-03 ZA ZA2011/00016A patent/ZA201100016B/en unknown
- 2011-01-19 CR CR20110033A patent/CR20110033A/es not_active Application Discontinuation
- 2011-01-20 EC EC2011010773A patent/ECSP11010773A/es unknown
- 2011-01-20 CO CO11005627A patent/CO6351798A2/es not_active Application Discontinuation
- 2011-08-17 HK HK11108698.5A patent/HK1154589A1/xx unknown
- 2011-09-05 JP JP2011193271A patent/JP5485237B2/ja active Active
-
2013
- 2013-09-24 US US14/034,880 patent/US9163051B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5485237B2 (ja) | スピロケタール誘導体の結晶およびその製造方法 | |
| EP3663292B1 (en) | Diphenylmethane derivative in crystalline form | |
| EP2533640B1 (en) | Processes of synthesizing dihydropyridophthalazinone derivatives | |
| EP2277878B1 (en) | Process for production of ethynylthymidine compound using 5-methyluridine as starting raw material | |
| CN102875537A (zh) | 一种新的抗血栓药物的制备方法 | |
| Popsavin et al. | Synthesis and biological evaluation of new pyrazole-and tetrazole-related C-nucleosides with modified sugar moieties | |
| EP3567028B1 (en) | Biphenyl compound as ccr2/ccr5 receptor antagonist | |
| CN101302207A (zh) | 3-o-烷基-5,6-o-(1-甲基乙叉基)-l-抗坏血酸的制造方法以及5,6-o-(1-甲基乙叉基)-l-抗坏血酸的制造方法 | |
| EP4272836A2 (en) | Cd73 inhibitor, preparation method therefor and application thereof | |
| CN101805339B (zh) | 一种制备恩替卡韦化合物的方法 | |
| JP5711669B2 (ja) | スピロケタール誘導体の製造方法 | |
| CN117677616A (zh) | 制备血红蛋白调节剂的方法 | |
| JP5192807B2 (ja) | シュードウリジン保護体の安定結晶 | |
| CN110963955A (zh) | 一种单氟代螺环化合物的合成方法及其中间体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130514 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130710 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140121 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140219 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5485237 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |