JP5416089B2 - ジアゾ二環式smac模倣物およびその使用 - Google Patents
ジアゾ二環式smac模倣物およびその使用 Download PDFInfo
- Publication number
- JP5416089B2 JP5416089B2 JP2010503269A JP2010503269A JP5416089B2 JP 5416089 B2 JP5416089 B2 JP 5416089B2 JP 2010503269 A JP2010503269 A JP 2010503269A JP 2010503269 A JP2010503269 A JP 2010503269A JP 5416089 B2 JP5416089 B2 JP 5416089B2
- Authority
- JP
- Japan
- Prior art keywords
- optionally substituted
- compound
- group
- apoptosis
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 102100033189 Diablo IAP-binding mitochondrial protein Human genes 0.000 title description 24
- 101150082208 DIABLO gene Proteins 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims description 241
- 206010028980 Neoplasm Diseases 0.000 claims description 97
- 201000011510 cancer Diseases 0.000 claims description 74
- 239000002246 antineoplastic agent Substances 0.000 claims description 60
- 150000003839 salts Chemical class 0.000 claims description 52
- 241001465754 Metazoa Species 0.000 claims description 49
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 42
- 208000026310 Breast neoplasm Diseases 0.000 claims description 39
- 239000003814 drug Substances 0.000 claims description 39
- 206010006187 Breast cancer Diseases 0.000 claims description 38
- 208000035475 disorder Diseases 0.000 claims description 19
- 230000002401 inhibitory effect Effects 0.000 claims description 15
- 230000006882 induction of apoptosis Effects 0.000 claims description 14
- 206010060862 Prostate cancer Diseases 0.000 claims description 13
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 12
- 239000000556 agonist Substances 0.000 claims description 12
- 206010033128 Ovarian cancer Diseases 0.000 claims description 11
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 10
- 230000033115 angiogenesis Effects 0.000 claims description 10
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 9
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 8
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 8
- 201000002528 pancreatic cancer Diseases 0.000 claims description 8
- 239000008194 pharmaceutical composition Substances 0.000 claims description 6
- 201000001441 melanoma Diseases 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 4
- 208000032839 leukemia Diseases 0.000 claims description 4
- 206010009944 Colon cancer Diseases 0.000 claims description 3
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 3
- 208000029742 colonic neoplasm Diseases 0.000 claims description 3
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 2
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 2
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims description 2
- 208000034578 Multiple myelomas Diseases 0.000 claims description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 2
- 208000006265 Renal cell carcinoma Diseases 0.000 claims description 2
- 206010039491 Sarcoma Diseases 0.000 claims description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 201000007270 liver cancer Diseases 0.000 claims description 2
- 208000014018 liver neoplasm Diseases 0.000 claims description 2
- 108040000066 TRAIL receptor activity proteins Proteins 0.000 claims 4
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims 1
- 210000004027 cell Anatomy 0.000 description 208
- LSXUTRRVVSPWDZ-MKKUMYSQSA-N (5s,8s,10ar)-n-benzhydryl-5-[[(2s)-2-(methylamino)propanoyl]amino]-3-(3-methylbutanoyl)-6-oxo-1,2,4,5,8,9,10,10a-octahydropyrrolo[1,2-a][1,5]diazocine-8-carboxamide Chemical compound O=C([C@@H]1CC[C@@H]2CCN(C[C@@H](C(N21)=O)NC(=O)[C@H](C)NC)C(=O)CC(C)C)NC(C=1C=CC=CC=1)C1=CC=CC=C1 LSXUTRRVVSPWDZ-MKKUMYSQSA-N 0.000 description 141
- 230000006907 apoptotic process Effects 0.000 description 112
- 238000000034 method Methods 0.000 description 90
- 125000001072 heteroaryl group Chemical group 0.000 description 82
- 125000000547 substituted alkyl group Chemical group 0.000 description 81
- -1 HIAP Proteins 0.000 description 80
- 229910052739 hydrogen Inorganic materials 0.000 description 66
- 125000003118 aryl group Chemical group 0.000 description 59
- 125000000217 alkyl group Chemical group 0.000 description 58
- 239000001257 hydrogen Substances 0.000 description 57
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 54
- 108090000623 proteins and genes Proteins 0.000 description 50
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 49
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 49
- 102000004169 proteins and genes Human genes 0.000 description 49
- 108700012411 TNFSF10 Proteins 0.000 description 46
- 125000001188 haloalkyl group Chemical group 0.000 description 45
- 102100024598 Tumor necrosis factor ligand superfamily member 10 Human genes 0.000 description 44
- 238000006482 condensation reaction Methods 0.000 description 44
- 229910052760 oxygen Inorganic materials 0.000 description 43
- 101100369992 Homo sapiens TNFSF10 gene Proteins 0.000 description 42
- 150000001717 carbocyclic compounds Chemical class 0.000 description 42
- 239000003795 chemical substances by application Substances 0.000 description 42
- 229910052717 sulfur Inorganic materials 0.000 description 42
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 40
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 40
- 238000004458 analytical method Methods 0.000 description 39
- 230000005855 radiation Effects 0.000 description 39
- 125000003107 substituted aryl group Chemical group 0.000 description 39
- 230000000694 effects Effects 0.000 description 37
- 229940002612 prodrug Drugs 0.000 description 36
- 239000000651 prodrug Substances 0.000 description 36
- 108090000765 processed proteins & peptides Proteins 0.000 description 35
- 229940075439 smac mimetic Drugs 0.000 description 35
- 239000000411 inducer Substances 0.000 description 34
- 238000011282 treatment Methods 0.000 description 34
- 239000003446 ligand Substances 0.000 description 33
- 230000005764 inhibitory process Effects 0.000 description 32
- 230000000259 anti-tumor effect Effects 0.000 description 31
- 239000000203 mixture Substances 0.000 description 31
- 150000001420 substituted heterocyclic compounds Chemical class 0.000 description 30
- 229910052799 carbon Inorganic materials 0.000 description 29
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 27
- 230000010261 cell growth Effects 0.000 description 27
- 230000004663 cell proliferation Effects 0.000 description 27
- 238000006243 chemical reaction Methods 0.000 description 27
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 26
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 25
- 150000002431 hydrogen Chemical class 0.000 description 25
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 24
- 238000001959 radiotherapy Methods 0.000 description 24
- 125000001424 substituent group Chemical group 0.000 description 24
- 230000027455 binding Effects 0.000 description 23
- 201000010099 disease Diseases 0.000 description 23
- 239000003112 inhibitor Substances 0.000 description 23
- 101710101225 Diablo IAP-binding mitochondrial protein Proteins 0.000 description 22
- 239000012190 activator Substances 0.000 description 22
- 125000005843 halogen group Chemical group 0.000 description 22
- 229910052720 vanadium Inorganic materials 0.000 description 22
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 21
- 238000003556 assay Methods 0.000 description 19
- 229940063683 taxotere Drugs 0.000 description 19
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 17
- 238000009833 condensation Methods 0.000 description 17
- 230000005494 condensation Effects 0.000 description 17
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 16
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 16
- 101000610604 Homo sapiens Tumor necrosis factor receptor superfamily member 10B Proteins 0.000 description 16
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 16
- 102100040112 Tumor necrosis factor receptor superfamily member 10B Human genes 0.000 description 16
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 16
- 230000003463 hyperproliferative effect Effects 0.000 description 16
- 238000000338 in vitro Methods 0.000 description 16
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 16
- 125000006239 protecting group Chemical group 0.000 description 16
- 230000009467 reduction Effects 0.000 description 16
- 238000006722 reduction reaction Methods 0.000 description 16
- 239000000243 solution Substances 0.000 description 16
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 16
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 101000610605 Homo sapiens Tumor necrosis factor receptor superfamily member 10A Proteins 0.000 description 14
- 102100031988 Tumor necrosis factor ligand superfamily member 6 Human genes 0.000 description 14
- 102100040113 Tumor necrosis factor receptor superfamily member 10A Human genes 0.000 description 14
- 229940079593 drug Drugs 0.000 description 14
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 description 14
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 14
- 230000001225 therapeutic effect Effects 0.000 description 14
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 13
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 13
- 239000000460 chlorine Substances 0.000 description 13
- 230000001939 inductive effect Effects 0.000 description 13
- 238000002360 preparation method Methods 0.000 description 13
- 230000004044 response Effects 0.000 description 13
- 229940094060 tykerb Drugs 0.000 description 13
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 12
- 239000002253 acid Substances 0.000 description 12
- 239000000654 additive Substances 0.000 description 12
- 229960005420 etoposide Drugs 0.000 description 12
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 12
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 12
- 229960005277 gemcitabine Drugs 0.000 description 12
- 102000005962 receptors Human genes 0.000 description 12
- 108020003175 receptors Proteins 0.000 description 12
- 229940124597 therapeutic agent Drugs 0.000 description 12
- 102100027522 Baculoviral IAP repeat-containing protein 7 Human genes 0.000 description 11
- 229910052770 Uranium Inorganic materials 0.000 description 11
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 11
- 239000003960 organic solvent Substances 0.000 description 11
- 102000011727 Caspases Human genes 0.000 description 10
- 108010076667 Caspases Proteins 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- 101000936083 Homo sapiens Baculoviral IAP repeat-containing protein 7 Proteins 0.000 description 10
- 102100022203 Tumor necrosis factor receptor superfamily member 25 Human genes 0.000 description 10
- 125000006242 amine protecting group Chemical group 0.000 description 10
- 238000004440 column chromatography Methods 0.000 description 10
- 229910052757 nitrogen Inorganic materials 0.000 description 10
- 108091007065 BIRCs Proteins 0.000 description 9
- 108700003785 Baculoviral IAP Repeat-Containing 3 Proteins 0.000 description 9
- 102000051819 Baculoviral IAP Repeat-Containing 3 Human genes 0.000 description 9
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 9
- 150000001412 amines Chemical class 0.000 description 9
- 230000003389 potentiating effect Effects 0.000 description 9
- 102000004196 processed proteins & peptides Human genes 0.000 description 9
- 230000004083 survival effect Effects 0.000 description 9
- 101710088341 Dermatopontin Proteins 0.000 description 8
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 8
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 8
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 8
- 108050002568 Tumor necrosis factor ligand superfamily member 6 Proteins 0.000 description 8
- 230000004913 activation Effects 0.000 description 8
- 150000001408 amides Chemical class 0.000 description 8
- 125000003277 amino group Chemical group 0.000 description 8
- 230000001640 apoptogenic effect Effects 0.000 description 8
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 8
- 239000002775 capsule Substances 0.000 description 8
- 229940127089 cytotoxic agent Drugs 0.000 description 8
- 238000002875 fluorescence polarization Methods 0.000 description 8
- 238000005984 hydrogenation reaction Methods 0.000 description 8
- 239000000543 intermediate Substances 0.000 description 8
- 230000001404 mediated effect Effects 0.000 description 8
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 8
- 229960001156 mitoxantrone Drugs 0.000 description 8
- 238000011580 nude mouse model Methods 0.000 description 8
- 238000012360 testing method Methods 0.000 description 8
- 210000001519 tissue Anatomy 0.000 description 8
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 7
- 108090000566 Caspase-9 Proteins 0.000 description 7
- 102000004039 Caspase-9 Human genes 0.000 description 7
- 102000055031 Inhibitor of Apoptosis Proteins Human genes 0.000 description 7
- 125000005466 alkylenyl group Chemical group 0.000 description 7
- 229940076005 apoptosis modulator Drugs 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 125000000753 cycloalkyl group Chemical group 0.000 description 7
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 7
- 230000008482 dysregulation Effects 0.000 description 7
- 230000006698 induction Effects 0.000 description 7
- 230000003647 oxidation Effects 0.000 description 7
- 238000007254 oxidation reaction Methods 0.000 description 7
- 230000001575 pathological effect Effects 0.000 description 7
- 230000008569 process Effects 0.000 description 7
- 238000000159 protein binding assay Methods 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- FEJUGLKDZJDVFY-UHFFFAOYSA-N 9-borabicyclo(3.3.1)nonane Chemical compound C1CCC2CCCC1B2 FEJUGLKDZJDVFY-UHFFFAOYSA-N 0.000 description 6
- 208000023275 Autoimmune disease Diseases 0.000 description 6
- 108010039471 Fas Ligand Protein Proteins 0.000 description 6
- 241000124008 Mammalia Species 0.000 description 6
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 6
- 206010029113 Neovascularisation Diseases 0.000 description 6
- 201000004681 Psoriasis Diseases 0.000 description 6
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 6
- 101150009046 Tnfrsf1a gene Proteins 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 150000001336 alkenes Chemical class 0.000 description 6
- 230000030833 cell death Effects 0.000 description 6
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 6
- 229960003668 docetaxel Drugs 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 230000014509 gene expression Effects 0.000 description 6
- 230000012010 growth Effects 0.000 description 6
- 125000001475 halogen functional group Chemical group 0.000 description 6
- 125000004404 heteroalkyl group Chemical group 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 229940043355 kinase inhibitor Drugs 0.000 description 6
- 230000003211 malignant effect Effects 0.000 description 6
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 6
- 230000002265 prevention Effects 0.000 description 6
- 230000000861 pro-apoptotic effect Effects 0.000 description 6
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 238000004007 reversed phase HPLC Methods 0.000 description 6
- 230000002441 reversible effect Effects 0.000 description 6
- 206010039073 rheumatoid arthritis Diseases 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 210000004881 tumor cell Anatomy 0.000 description 6
- 125000006702 (C1-C18) alkyl group Chemical group 0.000 description 5
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 5
- 0 CC([C@@](CC1)*2[C@@]1C(OC)=O)/C=*/C[C@](*CC*)C2=O Chemical compound CC([C@@](CC1)*2[C@@]1C(OC)=O)/C=*/C[C@](*CC*)C2=O 0.000 description 5
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 5
- 229920002472 Starch Polymers 0.000 description 5
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 5
- 230000003213 activating effect Effects 0.000 description 5
- 230000000996 additive effect Effects 0.000 description 5
- 125000003710 aryl alkyl group Chemical group 0.000 description 5
- 125000006367 bivalent amino carbonyl group Chemical group [H]N([*:1])C([*:2])=O 0.000 description 5
- 239000012830 cancer therapeutic Substances 0.000 description 5
- 229960004316 cisplatin Drugs 0.000 description 5
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 5
- 238000002425 crystallisation Methods 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 229960002949 fluorouracil Drugs 0.000 description 5
- 150000002391 heterocyclic compounds Chemical class 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 238000006197 hydroboration reaction Methods 0.000 description 5
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 5
- 230000001965 increasing effect Effects 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 230000002438 mitochondrial effect Effects 0.000 description 5
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 5
- 230000002018 overexpression Effects 0.000 description 5
- 239000012188 paraffin wax Substances 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 5
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 5
- 238000001953 recrystallisation Methods 0.000 description 5
- BTIHMVBBUGXLCJ-OAHLLOKOSA-N seliciclib Chemical compound C=12N=CN(C(C)C)C2=NC(N[C@@H](CO)CC)=NC=1NCC1=CC=CC=C1 BTIHMVBBUGXLCJ-OAHLLOKOSA-N 0.000 description 5
- 230000001235 sensitizing effect Effects 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 5
- 230000002792 vascular Effects 0.000 description 5
- 235000019489 Almond oil Nutrition 0.000 description 4
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 4
- 201000001320 Atherosclerosis Diseases 0.000 description 4
- 102100027515 Baculoviral IAP repeat-containing protein 6 Human genes 0.000 description 4
- 125000005915 C6-C14 aryl group Chemical group 0.000 description 4
- 102100025752 CASP8 and FADD-like apoptosis regulator Human genes 0.000 description 4
- 102000004091 Caspase-8 Human genes 0.000 description 4
- 108090000538 Caspase-8 Proteins 0.000 description 4
- 101100044298 Drosophila melanogaster fand gene Proteins 0.000 description 4
- 101150064015 FAS gene Proteins 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- 206010018338 Glioma Diseases 0.000 description 4
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 4
- 101000914211 Homo sapiens CASP8 and FADD-like apoptosis regulator Proteins 0.000 description 4
- 101000611023 Homo sapiens Tumor necrosis factor receptor superfamily member 6 Proteins 0.000 description 4
- 241000699666 Mus <mouse, genus> Species 0.000 description 4
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- 101100335198 Pneumocystis carinii fol1 gene Proteins 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- 102100040403 Tumor necrosis factor receptor superfamily member 6 Human genes 0.000 description 4
- 208000036142 Viral infection Diseases 0.000 description 4
- 208000027418 Wounds and injury Diseases 0.000 description 4
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 239000008168 almond oil Substances 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 230000000692 anti-sense effect Effects 0.000 description 4
- 229940034982 antineoplastic agent Drugs 0.000 description 4
- 229940041181 antineoplastic drug Drugs 0.000 description 4
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 150000001735 carboxylic acids Chemical class 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 229940125898 compound 5 Drugs 0.000 description 4
- 238000013461 design Methods 0.000 description 4
- 229960004679 doxorubicin Drugs 0.000 description 4
- 239000008298 dragée Substances 0.000 description 4
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 4
- 229960002584 gefitinib Drugs 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 230000013632 homeostatic process Effects 0.000 description 4
- 238000009169 immunotherapy Methods 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 238000011275 oncology therapy Methods 0.000 description 4
- 210000000056 organ Anatomy 0.000 description 4
- 229960001592 paclitaxel Drugs 0.000 description 4
- 239000000825 pharmaceutical preparation Substances 0.000 description 4
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 150000003384 small molecules Chemical class 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- 230000008685 targeting Effects 0.000 description 4
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 4
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 4
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 4
- 231100000419 toxicity Toxicity 0.000 description 4
- 230000001988 toxicity Effects 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 4
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 4
- 230000009385 viral infection Effects 0.000 description 4
- QKMSMVGTLTVHLK-LBPRGKRZSA-N (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-(phenylmethoxycarbonylamino)propanoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CNC(=O)OCC1=CC=CC=C1 QKMSMVGTLTVHLK-LBPRGKRZSA-N 0.000 description 3
- 206010000050 Abdominal adhesions Diseases 0.000 description 3
- 108090000672 Annexin A5 Proteins 0.000 description 3
- 102000004121 Annexin A5 Human genes 0.000 description 3
- 241000283690 Bos taurus Species 0.000 description 3
- 201000009030 Carcinoma Diseases 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- 108010049207 Death Domain Receptors Proteins 0.000 description 3
- 102000009058 Death Domain Receptors Human genes 0.000 description 3
- 206010012689 Diabetic retinopathy Diseases 0.000 description 3
- 101710156605 Diablo homolog, mitochondrial Proteins 0.000 description 3
- XXPXYPLPSDPERN-UHFFFAOYSA-N Ecteinascidin 743 Natural products COc1cc2C(NCCc2cc1O)C(=O)OCC3N4C(O)C5Cc6cc(C)c(OC)c(O)c6C(C4C(S)c7c(OC(=O)C)c(C)c8OCOc8c37)N5C XXPXYPLPSDPERN-UHFFFAOYSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 102100026693 FAS-associated death domain protein Human genes 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- 208000010412 Glaucoma Diseases 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 101000911074 Homo sapiens FAS-associated death domain protein Proteins 0.000 description 3
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 3
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 229930012538 Paclitaxel Natural products 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 108010090931 Proto-Oncogene Proteins c-bcl-2 Proteins 0.000 description 3
- 102000013535 Proto-Oncogene Proteins c-bcl-2 Human genes 0.000 description 3
- 206010038933 Retinopathy of prematurity Diseases 0.000 description 3
- 206010039710 Scleroderma Diseases 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 108010000449 TNF-Related Apoptosis-Inducing Ligand Receptors Proteins 0.000 description 3
- 102000002259 TNF-Related Apoptosis-Inducing Ligand Receptors Human genes 0.000 description 3
- 206010043189 Telangiectasia Diseases 0.000 description 3
- 206010052779 Transplant rejections Diseases 0.000 description 3
- 208000013058 Weber syndrome Diseases 0.000 description 3
- 206010052428 Wound Diseases 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 125000004423 acyloxy group Chemical group 0.000 description 3
- 150000001350 alkyl halides Chemical class 0.000 description 3
- 125000004414 alkyl thio group Chemical group 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- 230000002424 anti-apoptotic effect Effects 0.000 description 3
- 230000000340 anti-metabolite Effects 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 229940121375 antifungal agent Drugs 0.000 description 3
- 239000003429 antifungal agent Substances 0.000 description 3
- 229940100197 antimetabolite Drugs 0.000 description 3
- 239000002256 antimetabolite Substances 0.000 description 3
- 230000009925 apoptotic mechanism Effects 0.000 description 3
- 108700000711 bcl-X Proteins 0.000 description 3
- 102000055104 bcl-X Human genes 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 3
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 3
- 229960001467 bortezomib Drugs 0.000 description 3
- 201000007293 brain stem infarction Diseases 0.000 description 3
- 125000002837 carbocyclic group Chemical group 0.000 description 3
- 229960004562 carboplatin Drugs 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 230000000973 chemotherapeutic effect Effects 0.000 description 3
- 238000002512 chemotherapy Methods 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 3
- 230000007547 defect Effects 0.000 description 3
- 230000002950 deficient Effects 0.000 description 3
- 231100000673 dose–response relationship Toxicity 0.000 description 3
- 210000002889 endothelial cell Anatomy 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 210000002950 fibroblast Anatomy 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 3
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 3
- 229960002074 flutamide Drugs 0.000 description 3
- 238000005469 granulation Methods 0.000 description 3
- 230000003179 granulation Effects 0.000 description 3
- 229940088597 hormone Drugs 0.000 description 3
- 239000005556 hormone Substances 0.000 description 3
- 206010020718 hyperplasia Diseases 0.000 description 3
- 230000001969 hypertrophic effect Effects 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 230000009545 invasion Effects 0.000 description 3
- 229960004891 lapatinib Drugs 0.000 description 3
- 208000002780 macular degeneration Diseases 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 108020004999 messenger RNA Proteins 0.000 description 3
- 229960000485 methotrexate Drugs 0.000 description 3
- 239000002480 mineral oil Substances 0.000 description 3
- 229960004857 mitomycin Drugs 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 230000002107 myocardial effect Effects 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 201000003142 neovascular glaucoma Diseases 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 238000005457 optimization Methods 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 3
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 3
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 3
- 150000003138 primary alcohols Chemical class 0.000 description 3
- 238000012342 propidium iodide staining Methods 0.000 description 3
- 231100000241 scar Toxicity 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 230000008093 supporting effect Effects 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- 208000009056 telangiectasis Diseases 0.000 description 3
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 3
- 125000001544 thienyl group Chemical group 0.000 description 3
- 229950009158 tipifarnib Drugs 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- PKVRCIRHQMSYJX-AIFWHQITSA-N trabectedin Chemical compound C([C@@]1(C(OC2)=O)NCCC3=C1C=C(C(=C3)O)OC)S[C@@H]1C3=C(OC(C)=O)C(C)=C4OCOC4=C3[C@H]2N2[C@@H](O)[C@H](CC=3C4=C(O)C(OC)=C(C)C=3)N(C)[C@H]4[C@@H]21 PKVRCIRHQMSYJX-AIFWHQITSA-N 0.000 description 3
- 229960000977 trabectedin Drugs 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- 150000003626 triacylglycerols Chemical class 0.000 description 3
- 230000006459 vascular development Effects 0.000 description 3
- 238000001262 western blot Methods 0.000 description 3
- VLHQXRIIQSTJCQ-LURJTMIESA-N (2s)-2-[methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]propanoic acid Chemical compound OC(=O)[C@H](C)N(C)C(=O)OC(C)(C)C VLHQXRIIQSTJCQ-LURJTMIESA-N 0.000 description 2
- YBYZHVZWINISJO-XPSCAIQQSA-N (5s,8s,10ar)-n-benzhydryl-5-[[(2s)-2-(methylamino)propanoyl]amino]-6-oxo-3-(2-phenylacetyl)-1,2,4,5,8,9,10,10a-octahydropyrrolo[1,2-a][1,5]diazocine-8-carboxamide Chemical compound O=C([C@@H]1CC[C@@H]2CCN(C[C@@H](C(N21)=O)NC(=O)[C@H](C)NC)C(=O)CC=1C=CC=CC=1)NC(C=1C=CC=CC=1)C1=CC=CC=C1 YBYZHVZWINISJO-XPSCAIQQSA-N 0.000 description 2
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- BKTRENAPTCBBFA-UHFFFAOYSA-N 4-[2-(4-hydroxy-3-phenylphenyl)propan-2-yl]-2-phenylphenol Chemical compound C=1C=C(O)C(C=2C=CC=CC=2)=CC=1C(C)(C)C(C=1)=CC=C(O)C=1C1=CC=CC=C1 BKTRENAPTCBBFA-UHFFFAOYSA-N 0.000 description 2
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical group CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 229940088872 Apoptosis inhibitor Drugs 0.000 description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 2
- 102100021676 Baculoviral IAP repeat-containing protein 1 Human genes 0.000 description 2
- 102100021663 Baculoviral IAP repeat-containing protein 5 Human genes 0.000 description 2
- 102100027517 Baculoviral IAP repeat-containing protein 8 Human genes 0.000 description 2
- 101710178104 Baculoviral IAP repeat-containing protein 8 Proteins 0.000 description 2
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 2
- 206010005003 Bladder cancer Diseases 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- 208000003174 Brain Neoplasms Diseases 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- FVLVBPDQNARYJU-XAHDHGMMSA-N C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O Chemical compound C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O FVLVBPDQNARYJU-XAHDHGMMSA-N 0.000 description 2
- 208000005623 Carcinogenesis Diseases 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- 108090000397 Caspase 3 Proteins 0.000 description 2
- 108090000567 Caspase 7 Proteins 0.000 description 2
- 102100029855 Caspase-3 Human genes 0.000 description 2
- 102100038902 Caspase-7 Human genes 0.000 description 2
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 2
- 208000032544 Cicatrix Diseases 0.000 description 2
- 101710082464 Cis-aconitate decarboxylase Proteins 0.000 description 2
- 206010010099 Combined immunodeficiency Diseases 0.000 description 2
- 208000011231 Crohn disease Diseases 0.000 description 2
- 108010037462 Cyclooxygenase 2 Proteins 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- 101710147299 DNA fragmentation factor subunit beta Proteins 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- 102000000541 Defensins Human genes 0.000 description 2
- 108010002069 Defensins Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 102000001301 EGF receptor Human genes 0.000 description 2
- 108060006698 EGF receptor Proteins 0.000 description 2
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000679921 Homo sapiens Tumor necrosis factor receptor superfamily member 21 Proteins 0.000 description 2
- 101000679903 Homo sapiens Tumor necrosis factor receptor superfamily member 25 Proteins 0.000 description 2
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 2
- 102000006992 Interferon-alpha Human genes 0.000 description 2
- 108010047761 Interferon-alpha Proteins 0.000 description 2
- 241000713321 Intracisternal A-particles Species 0.000 description 2
- 102100027670 Islet amyloid polypeptide Human genes 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 108010000817 Leuprolide Proteins 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 2
- 206010025323 Lymphomas Diseases 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- 229930192392 Mitomycin Natural products 0.000 description 2
- 241000699660 Mus musculus Species 0.000 description 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 2
- 108010006696 Neuronal Apoptosis-Inhibitory Protein Proteins 0.000 description 2
- 108700020796 Oncogene Proteins 0.000 description 2
- 108091008606 PDGF receptors Proteins 0.000 description 2
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 2
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 235000014676 Phragmites communis Nutrition 0.000 description 2
- 102000011653 Platelet-Derived Growth Factor Receptors Human genes 0.000 description 2
- 101710145525 Probable cinnamyl alcohol dehydrogenase Proteins 0.000 description 2
- 102100038280 Prostaglandin G/H synthase 2 Human genes 0.000 description 2
- 108091008611 Protein Kinase B Proteins 0.000 description 2
- 102100022419 RPA-interacting protein Human genes 0.000 description 2
- 108090000829 Ribosome Inactivating Proteins Proteins 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- 208000034189 Sclerosis Diseases 0.000 description 2
- 208000021386 Sjogren Syndrome Diseases 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 108010002687 Survivin Proteins 0.000 description 2
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 102100022205 Tumor necrosis factor receptor superfamily member 21 Human genes 0.000 description 2
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 2
- 241000700605 Viruses Species 0.000 description 2
- 108700031544 X-Linked Inhibitor of Apoptosis Proteins 0.000 description 2
- 102000050257 X-Linked Inhibitor of Apoptosis Human genes 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- JKOQGQFVAUAYPM-UHFFFAOYSA-N amifostine Chemical compound NCCCNCCSP(O)(O)=O JKOQGQFVAUAYPM-UHFFFAOYSA-N 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 230000002280 anti-androgenic effect Effects 0.000 description 2
- 230000001093 anti-cancer Effects 0.000 description 2
- 229940124650 anti-cancer therapies Drugs 0.000 description 2
- 229940046836 anti-estrogen Drugs 0.000 description 2
- 230000001833 anti-estrogenic effect Effects 0.000 description 2
- 229940124599 anti-inflammatory drug Drugs 0.000 description 2
- 230000001147 anti-toxic effect Effects 0.000 description 2
- 239000000051 antiandrogen Substances 0.000 description 2
- 229940030495 antiandrogen sex hormone and modulator of the genital system Drugs 0.000 description 2
- 238000011319 anticancer therapy Methods 0.000 description 2
- 239000004599 antimicrobial Substances 0.000 description 2
- 239000003443 antiviral agent Substances 0.000 description 2
- 239000000158 apoptosis inhibitor Substances 0.000 description 2
- 210000001367 artery Anatomy 0.000 description 2
- 125000000732 arylene group Chemical group 0.000 description 2
- 239000012131 assay buffer Substances 0.000 description 2
- 230000003143 atherosclerotic effect Effects 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 229960002756 azacitidine Drugs 0.000 description 2
- 150000001540 azides Chemical class 0.000 description 2
- 235000013871 bee wax Nutrition 0.000 description 2
- 239000012166 beeswax Substances 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 229960000397 bevacizumab Drugs 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 125000002529 biphenylenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C12)* 0.000 description 2
- 229960001561 bleomycin Drugs 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 210000004204 blood vessel Anatomy 0.000 description 2
- 229910000085 borane Inorganic materials 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- MJQUEDHRCUIRLF-TVIXENOKSA-N bryostatin 1 Chemical compound C([C@@H]1CC(/[C@@H]([C@@](C(C)(C)/C=C/2)(O)O1)OC(=O)/C=C/C=C/CCC)=C\C(=O)OC)[C@H]([C@@H](C)O)OC(=O)C[C@H](O)C[C@@H](O1)C[C@H](OC(C)=O)C(C)(C)[C@]1(O)C[C@@H]1C\C(=C\C(=O)OC)C[C@H]\2O1 MJQUEDHRCUIRLF-TVIXENOKSA-N 0.000 description 2
- 229960005539 bryostatin 1 Drugs 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 2
- 230000036952 cancer formation Effects 0.000 description 2
- 208000035269 cancer or benign tumor Diseases 0.000 description 2
- 125000001589 carboacyl group Chemical group 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 231100000504 carcinogenesis Toxicity 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 238000011284 combination treatment Methods 0.000 description 2
- 230000009137 competitive binding Effects 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- VFLDPWHFBUODDF-FCXRPNKRSA-N curcumin Chemical compound C1=C(O)C(OC)=CC(\C=C\C(=O)CC(=O)\C=C\C=2C=C(OC)C(O)=CC=2)=C1 VFLDPWHFBUODDF-FCXRPNKRSA-N 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 229960003901 dacarbazine Drugs 0.000 description 2
- 229960000640 dactinomycin Drugs 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 238000009510 drug design Methods 0.000 description 2
- 239000012039 electrophile Substances 0.000 description 2
- 150000002081 enamines Chemical class 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 229960001433 erlotinib Drugs 0.000 description 2
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- 239000000328 estrogen antagonist Substances 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 239000003925 fat Substances 0.000 description 2
- 239000010685 fatty oil Substances 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 108010074605 gamma-Globulins Proteins 0.000 description 2
- 210000004051 gastric juice Anatomy 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- QBKSWRVVCFFDOT-UHFFFAOYSA-N gossypol Chemical compound CC(C)C1=C(O)C(O)=C(C=O)C2=C(O)C(C=3C(O)=C4C(C=O)=C(O)C(O)=C(C4=CC=3C)C(C)C)=C(C)C=C21 QBKSWRVVCFFDOT-UHFFFAOYSA-N 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 208000029824 high grade glioma Diseases 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 2
- XXSMGPRMXLTPCZ-UHFFFAOYSA-N hydroxychloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CCO)CC)=CC=NC2=C1 XXSMGPRMXLTPCZ-UHFFFAOYSA-N 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 230000036039 immunity Effects 0.000 description 2
- 239000000367 immunologic factor Substances 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 230000004968 inflammatory condition Effects 0.000 description 2
- 108091006086 inhibitor proteins Proteins 0.000 description 2
- 239000003999 initiator Substances 0.000 description 2
- 238000007917 intracranial administration Methods 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 2
- FABUFPQFXZVHFB-CFWQTKTJSA-N ixabepilone Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@H](C)C(=O)C(C)(C)[C@H](O)CC(=O)N1)O)C)=C\C1=CSC(C)=N1 FABUFPQFXZVHFB-CFWQTKTJSA-N 0.000 description 2
- 229960002014 ixabepilone Drugs 0.000 description 2
- 230000002147 killing effect Effects 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 230000003902 lesion Effects 0.000 description 2
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 2
- 229960004338 leuprorelin Drugs 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000006210 lotion Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 201000011614 malignant glioma Diseases 0.000 description 2
- 229960004961 mechlorethamine Drugs 0.000 description 2
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 230000001394 metastastic effect Effects 0.000 description 2
- 206010061289 metastatic neoplasm Diseases 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 230000003278 mimic effect Effects 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical class CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 2
- 239000002324 mouth wash Substances 0.000 description 2
- 206010028417 myasthenia gravis Diseases 0.000 description 2
- 230000006654 negative regulation of apoptotic process Effects 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 238000003305 oral gavage Methods 0.000 description 2
- 201000008482 osteoarthritis Diseases 0.000 description 2
- 229940127084 other anti-cancer agent Drugs 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 230000008506 pathogenesis Effects 0.000 description 2
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 2
- VYMDGNCVAMGZFE-UHFFFAOYSA-N phenylbutazonum Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 VYMDGNCVAMGZFE-UHFFFAOYSA-N 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 230000010287 polarization Effects 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 2
- 229960004618 prednisone Drugs 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 210000005267 prostate cell Anatomy 0.000 description 2
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 230000003537 radioprotector Effects 0.000 description 2
- 238000006268 reductive amination reaction Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- VHXNKPBCCMUMSW-FQEVSTJZSA-N rubitecan Chemical compound C1=CC([N+]([O-])=O)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VHXNKPBCCMUMSW-FQEVSTJZSA-N 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 230000037387 scars Effects 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 229960003440 semustine Drugs 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 238000013207 serial dilution Methods 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- VSIVTUIKYVGDCX-UHFFFAOYSA-M sodium;4-[2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)tetrazol-2-ium-5-yl]benzene-1,3-disulfonate Chemical compound [Na+].COC1=CC([N+]([O-])=O)=CC=C1[N+]1=NC(C=2C(=CC(=CC=2)S([O-])(=O)=O)S([O-])(=O)=O)=NN1C1=CC=C([N+]([O-])=O)C=C1 VSIVTUIKYVGDCX-UHFFFAOYSA-M 0.000 description 2
- 229960001052 streptozocin Drugs 0.000 description 2
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000002511 suppository base Substances 0.000 description 2
- 238000011477 surgical intervention Methods 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 229960001603 tamoxifen Drugs 0.000 description 2
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 2
- 229960000235 temsirolimus Drugs 0.000 description 2
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 229960003087 tioguanine Drugs 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 239000012049 topical pharmaceutical composition Substances 0.000 description 2
- 239000003053 toxin Substances 0.000 description 2
- 231100000765 toxin Toxicity 0.000 description 2
- 108700012359 toxins Proteins 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical class [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- 229960000237 vorinostat Drugs 0.000 description 2
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- 230000004580 weight loss Effects 0.000 description 2
- MLBZLJCMHFCTQM-UHFFFAOYSA-N (2-methylphenyl)-diphenylphosphane Chemical compound CC1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 MLBZLJCMHFCTQM-UHFFFAOYSA-N 0.000 description 1
- HBUBKKRHXORPQB-FJFJXFQQSA-N (2R,3S,4S,5R)-2-(6-amino-2-fluoro-9-purinyl)-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O HBUBKKRHXORPQB-FJFJXFQQSA-N 0.000 description 1
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- MMHDBUJXLOFTLC-WOYTXXSLSA-N (2s)-2-[[(2r)-2-[[(2s)-2-[[(2s)-2-[[(2s)-1-acetylpyrrolidine-2-carbonyl]amino]-3-(1h-imidazol-5-yl)propanoyl]amino]-3-hydroxypropanoyl]amino]-3-sulfanylpropanoyl]amino]butanediamide Chemical compound CC(=O)N1CCC[C@H]1C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(N)=O)CC1=CN=CN1 MMHDBUJXLOFTLC-WOYTXXSLSA-N 0.000 description 1
- MHFUWOIXNMZFIW-WNQIDUERSA-N (2s)-2-hydroxypropanoic acid;n-[4-[4-(4-methylpiperazin-1-yl)-6-[(5-methyl-1h-pyrazol-3-yl)amino]pyrimidin-2-yl]sulfanylphenyl]cyclopropanecarboxamide Chemical compound C[C@H](O)C(O)=O.C1CN(C)CCN1C1=CC(NC2=NNC(C)=C2)=NC(SC=2C=CC(NC(=O)C3CC3)=CC=2)=N1 MHFUWOIXNMZFIW-WNQIDUERSA-N 0.000 description 1
- PSVUJBVBCOISSP-SPFKKGSWSA-N (2s,3r,4s,5s,6r)-2-bis(2-chloroethylamino)phosphoryloxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound OC[C@H]1O[C@@H](OP(=O)(NCCCl)NCCCl)[C@H](O)[C@@H](O)[C@@H]1O PSVUJBVBCOISSP-SPFKKGSWSA-N 0.000 description 1
- VEEGZPWAAPPXRB-BJMVGYQFSA-N (3e)-3-(1h-imidazol-5-ylmethylidene)-1h-indol-2-one Chemical compound O=C1NC2=CC=CC=C2\C1=C/C1=CN=CN1 VEEGZPWAAPPXRB-BJMVGYQFSA-N 0.000 description 1
- SZVCBOBNBHXZKC-MZRLSVQCSA-N (5s,8s,10ar)-5-[[(2s)-2-(methylamino)propanoyl]amino]-3-(3-methylbutanoyl)-6-oxo-n-[(2-phenylphenyl)methyl]-1,2,4,5,8,9,10,10a-octahydropyrrolo[1,2-a][1,5]diazocine-8-carboxamide Chemical compound O=C([C@@H]1CC[C@@H]2CCN(C[C@@H](C(N21)=O)NC(=O)[C@H](C)NC)C(=O)CC(C)C)NCC1=CC=CC=C1C1=CC=CC=C1 SZVCBOBNBHXZKC-MZRLSVQCSA-N 0.000 description 1
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- ZGNLFUXWZJGETL-YUSKDDKASA-N (Z)-[(2S)-2-amino-2-carboxyethyl]-hydroxyimino-oxidoazanium Chemical compound N[C@@H](C\[N+]([O-])=N\O)C(O)=O ZGNLFUXWZJGETL-YUSKDDKASA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- YHYKLKNNBYLTQY-UHFFFAOYSA-N 1,1-diphenylhydrazine Chemical compound C=1C=CC=CC=1N(N)C1=CC=CC=C1 YHYKLKNNBYLTQY-UHFFFAOYSA-N 0.000 description 1
- ABJFBJGGLJVMAQ-UHFFFAOYSA-N 1,4-dihydroquinoxaline-2,3-dione Chemical compound C1=CC=C2NC(=O)C(=O)NC2=C1 ABJFBJGGLJVMAQ-UHFFFAOYSA-N 0.000 description 1
- PVCULFYROUOVGJ-UHFFFAOYSA-N 1-[2-chloroethyl(methylsulfonyl)amino]-3-methyl-1-methylsulfonylurea Chemical compound CNC(=O)N(S(C)(=O)=O)N(S(C)(=O)=O)CCCl PVCULFYROUOVGJ-UHFFFAOYSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- BNZPFJBUHRBLNB-UHFFFAOYSA-N 1-oxido-1,2,4-benzotriazin-1-ium Chemical compound C1=CC=C2[N+]([O-])=NC=NC2=C1 BNZPFJBUHRBLNB-UHFFFAOYSA-N 0.000 description 1
- FXEDIXLHKQINFP-UHFFFAOYSA-N 12-O-tetradecanoylphorbol-13-acetate Natural products CCCCCCCCCCCCCC(=O)OC1CC2(O)C(C=C(CO)CC3(O)C2C=C(C)C3=O)C4C(C)(C)C14OC(=O)C FXEDIXLHKQINFP-UHFFFAOYSA-N 0.000 description 1
- BFPYWIDHMRZLRN-UHFFFAOYSA-N 17alpha-ethynyl estradiol Natural products OC1=CC=C2C3CCC(C)(C(CC4)(O)C#C)C4C3CCC2=C1 BFPYWIDHMRZLRN-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 1
- FOMKFBXHAKRALK-UHFFFAOYSA-N 2-[4-[(2-bromoethylamino)methyl]-2-nitroimidazol-1-yl]ethanol Chemical compound OCCN1C=C(CNCCBr)N=C1[N+]([O-])=O FOMKFBXHAKRALK-UHFFFAOYSA-N 0.000 description 1
- NYWSLZMTZNODJM-MCGDBQAWSA-N 2-[5-[(4e,20e)-35-butyl-19-[(2s,3s,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-10,12,14,16,18,22,26,30,34-nonahydroxy-3,5,21,33-tetramethyl-36-oxo-1-oxacyclohexatriaconta-4,20-dien-2-yl]-4-hydroxyhexyl]guanidine Chemical compound OC1CC(O)CC(O)CC(O)CC(O)CCCC\C(C)=C\C(C)C(C(C)C(O)CCCN=C(N)N)OC(=O)C(CCCC)C(O)C(C)CCC(O)CCCC(O)CCCC(O)\C(C)=C\C1O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 NYWSLZMTZNODJM-MCGDBQAWSA-N 0.000 description 1
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 1
- JZPGURKWXUBQLP-UHFFFAOYSA-N 2-[[2-(2-methylpropylsulfanyl)-1,3-benzothiazol-6-yl]iminomethyl]phenol Chemical compound CC(C)CSC1=NC2=C(S1)C=C(C=C2)N=CC3=CC=CC=C3O JZPGURKWXUBQLP-UHFFFAOYSA-N 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- TUMCWFMHZOUPDA-UHFFFAOYSA-N 2-ethylsulfanyl-1,3-benzothiazol-6-amine Chemical compound C1=C(N)C=C2SC(SCC)=NC2=C1 TUMCWFMHZOUPDA-UHFFFAOYSA-N 0.000 description 1
- HZLCGUXUOFWCCN-UHFFFAOYSA-N 2-hydroxynonadecane-1,2,3-tricarboxylic acid Chemical compound CCCCCCCCCCCCCCCCC(C(O)=O)C(O)(C(O)=O)CC(O)=O HZLCGUXUOFWCCN-UHFFFAOYSA-N 0.000 description 1
- CTRPRMNBTVRDFH-UHFFFAOYSA-N 2-n-methyl-1,3,5-triazine-2,4,6-triamine Chemical compound CNC1=NC(N)=NC(N)=N1 CTRPRMNBTVRDFH-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- FTBBGQKRYUTLMP-UHFFFAOYSA-N 2-nitro-1h-pyrrole Chemical class [O-][N+](=O)C1=CC=CN1 FTBBGQKRYUTLMP-UHFFFAOYSA-N 0.000 description 1
- YZEUHQHUFTYLPH-UHFFFAOYSA-N 2-nitroimidazole Chemical compound [O-][N+](=O)C1=NC=CN1 YZEUHQHUFTYLPH-UHFFFAOYSA-N 0.000 description 1
- 150000004959 2-nitroimidazoles Chemical class 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- KWYLVDGOCQSPDM-UHFFFAOYSA-N 3,7-dihydropurine-6-thione Chemical compound SC1=NC=NC2=C1NC=N2.S=C1N=CNC2=C1NC=N2 KWYLVDGOCQSPDM-UHFFFAOYSA-N 0.000 description 1
- AXRCEOKUDYDWLF-UHFFFAOYSA-N 3-(1-methyl-3-indolyl)-4-[1-[1-(2-pyridinylmethyl)-4-piperidinyl]-3-indolyl]pyrrole-2,5-dione Chemical compound C12=CC=CC=C2N(C)C=C1C(C(NC1=O)=O)=C1C(C1=CC=CC=C11)=CN1C(CC1)CCN1CC1=CC=CC=N1 AXRCEOKUDYDWLF-UHFFFAOYSA-N 0.000 description 1
- XMTQQYYKAHVGBJ-UHFFFAOYSA-N 3-(3,4-DICHLOROPHENYL)-1,1-DIMETHYLUREA Chemical compound CN(C)C(=O)NC1=CC=C(Cl)C(Cl)=C1 XMTQQYYKAHVGBJ-UHFFFAOYSA-N 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- CCRNCEKMSVYFLU-UHFFFAOYSA-N 4,5-dinitro-1h-imidazole Chemical class [O-][N+](=O)C=1N=CNC=1[N+]([O-])=O CCRNCEKMSVYFLU-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- AKJHMTWEGVYYSE-AIRMAKDCSA-N 4-HPR Chemical compound C=1C=C(O)C=CC=1NC(=O)/C=C(\C)/C=C/C=C(C)C=CC1=C(C)CCCC1(C)C AKJHMTWEGVYYSE-AIRMAKDCSA-N 0.000 description 1
- 125000004042 4-aminobutyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])N([H])[H] 0.000 description 1
- CNPURSDMOWDNOQ-UHFFFAOYSA-N 4-methoxy-7h-pyrrolo[2,3-d]pyrimidin-2-amine Chemical group COC1=NC(N)=NC2=C1C=CN2 CNPURSDMOWDNOQ-UHFFFAOYSA-N 0.000 description 1
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 1
- NJYVEMPWNAYQQN-UHFFFAOYSA-N 5-carboxyfluorescein Chemical compound C12=CC=C(O)C=C2OC2=CC(O)=CC=C2C21OC(=O)C1=CC(C(=O)O)=CC=C21 NJYVEMPWNAYQQN-UHFFFAOYSA-N 0.000 description 1
- KUEFXPHXHHANKS-UHFFFAOYSA-N 5-nitro-1h-1,2,4-triazole Chemical compound [O-][N+](=O)C1=NC=NN1 KUEFXPHXHHANKS-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- FJHBVJOVLFPMQE-QFIPXVFZSA-N 7-Ethyl-10-Hydroxy-Camptothecin Chemical compound C1=C(O)C=C2C(CC)=C(CN3C(C4=C([C@@](C(=O)OC4)(O)CC)C=C33)=O)C3=NC2=C1 FJHBVJOVLFPMQE-QFIPXVFZSA-N 0.000 description 1
- FOHUMFIQHBSPGD-UHFFFAOYSA-N 7-aminoisochromen-1-one Chemical compound C1=COC(=O)C2=CC(N)=CC=C21 FOHUMFIQHBSPGD-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- BUROJSBIWGDYCN-GAUTUEMISA-N AP 23573 Chemical compound C1C[C@@H](OP(C)(C)=O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 BUROJSBIWGDYCN-GAUTUEMISA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 208000003200 Adenoma Diseases 0.000 description 1
- 206010001233 Adenoma benign Diseases 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 108010012934 Albumin-Bound Paclitaxel Proteins 0.000 description 1
- 208000003120 Angiofibroma Diseases 0.000 description 1
- 102000010565 Apoptosis Regulatory Proteins Human genes 0.000 description 1
- 108010063104 Apoptosis Regulatory Proteins Proteins 0.000 description 1
- 108010089941 Apoptosomes Proteins 0.000 description 1
- 101100111638 Arabidopsis thaliana BIR2 gene Proteins 0.000 description 1
- 101100025412 Arabidopsis thaliana XI-A gene Proteins 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010003827 Autoimmune hepatitis Diseases 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 101710177963 Baculoviral IAP repeat-containing protein 7 Proteins 0.000 description 1
- 208000023328 Basedow disease Diseases 0.000 description 1
- 102000051485 Bcl-2 family Human genes 0.000 description 1
- 108700038897 Bcl-2 family Proteins 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 108010037003 Buserelin Proteins 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- UBTVVCSVUMFKAR-JCJIYJPFSA-N CC(C)CC(N(CC[C@@H](CC1)N2[C@@H]1C(NC(c1ccc(C)cc1)c(cc1)ccc1F)=O)C[C@H](CNC(C(C)C)=O)C2=O)=O Chemical compound CC(C)CC(N(CC[C@@H](CC1)N2[C@@H]1C(NC(c1ccc(C)cc1)c(cc1)ccc1F)=O)C[C@H](CNC(C(C)C)=O)C2=O)=O UBTVVCSVUMFKAR-JCJIYJPFSA-N 0.000 description 1
- JRPFGUVEGUMVRX-LQBCTSRJSA-N CC(C)CCCC(N(CC[C@@H](CC[C@H]1C(NC(c2ccccc2)c2ccccc2)=O)N1C1=O)C[C@@H]1NC([C@H](C)NC)=O)=O Chemical compound CC(C)CCCC(N(CC[C@@H](CC[C@H]1C(NC(c2ccccc2)c2ccccc2)=O)N1C1=O)C[C@@H]1NC([C@H](C)NC)=O)=O JRPFGUVEGUMVRX-LQBCTSRJSA-N 0.000 description 1
- FQSPQHRWQMGKNA-MQWKRIRWSA-N COC([C@H]1NC(CC=C)CC1)=O Chemical compound COC([C@H]1NC(CC=C)CC1)=O FQSPQHRWQMGKNA-MQWKRIRWSA-N 0.000 description 1
- OYFYMHHWDKFDNK-JBKTXMORSA-N C[C@@H](C(N[C@@H](CN(CC[C@@H](CC1)N2[C@@H]1C(NC(c1ccccc1)c1ccccc1)=O)C(C[C@H]1COCC1)=O)C2=O)=O)NC Chemical compound C[C@@H](C(N[C@@H](CN(CC[C@@H](CC1)N2[C@@H]1C(NC(c1ccccc1)c1ccccc1)=O)C(C[C@H]1COCC1)=O)C2=O)=O)NC OYFYMHHWDKFDNK-JBKTXMORSA-N 0.000 description 1
- 102100024436 Caldesmon Human genes 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 208000009458 Carcinoma in Situ Diseases 0.000 description 1
- 208000017897 Carcinoma of esophagus Diseases 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- 206010008874 Chronic Fatigue Syndrome Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 208000015943 Coeliac disease Diseases 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 229940126062 Compound A Drugs 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 101710112752 Cytotoxin Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- 102000010170 Death domains Human genes 0.000 description 1
- 108050001718 Death domains Proteins 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 101100175182 Drosophila melanogaster ppl gene Proteins 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 206010014733 Endometrial cancer Diseases 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 208000032027 Essential Thrombocythemia Diseases 0.000 description 1
- BFPYWIDHMRZLRN-SLHNCBLASA-N Ethinyl estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 BFPYWIDHMRZLRN-SLHNCBLASA-N 0.000 description 1
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 208000001640 Fibromyalgia Diseases 0.000 description 1
- 241000956293 Fulda Species 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 102100021239 G protein-activated inward rectifier potassium channel 2 Human genes 0.000 description 1
- 101710158550 G protein-activated inward rectifier potassium channel 2 Proteins 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 208000009329 Graft vs Host Disease Diseases 0.000 description 1
- 208000015023 Graves' disease Diseases 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000005794 Hairy Leukoplakia Diseases 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 208000035186 Hemolytic Autoimmune Anemia Diseases 0.000 description 1
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101000871228 Homo sapiens Diablo IAP-binding mitochondrial protein Proteins 0.000 description 1
- 101000804865 Homo sapiens E3 ubiquitin-protein ligase XIAP Proteins 0.000 description 1
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical group NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 1
- DOMWKUIIPQCAJU-LJHIYBGHSA-N Hydroxyprogesterone caproate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)CCCCC)[C@@]1(C)CC2 DOMWKUIIPQCAJU-LJHIYBGHSA-N 0.000 description 1
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 1
- 208000037147 Hypercalcaemia Diseases 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- 206010021245 Idiopathic thrombocytopenic purpura Diseases 0.000 description 1
- 208000010159 IgA glomerulonephritis Diseases 0.000 description 1
- 206010021263 IgA nephropathy Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 108010065805 Interleukin-12 Proteins 0.000 description 1
- 102000013462 Interleukin-12 Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 102100039064 Interleukin-3 Human genes 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- MLFKVJCWGUZWNV-UHFFFAOYSA-N L-alanosine Natural products OC(=O)C(N)CN(O)N=O MLFKVJCWGUZWNV-UHFFFAOYSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- UIARLYUEJFELEN-LROUJFHJSA-N LSM-1231 Chemical compound C12=C3N4C5=CC=CC=C5C3=C3C(=O)NCC3=C2C2=CC=CC=C2N1[C@]1(C)[C@](CO)(O)C[C@H]4O1 UIARLYUEJFELEN-LROUJFHJSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 206010024612 Lipoma Diseases 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- ZRVUJXDFFKFLMG-UHFFFAOYSA-N Meloxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=NC=C(C)S1 ZRVUJXDFFKFLMG-UHFFFAOYSA-N 0.000 description 1
- 206010054949 Metaplasia Diseases 0.000 description 1
- 101100519481 Mus musculus Ppl gene Proteins 0.000 description 1
- 241000204031 Mycoplasma Species 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- 125000000729 N-terminal amino-acid group Chemical group 0.000 description 1
- KTDZCOWXCWUPEO-UHFFFAOYSA-N NS-398 Chemical compound CS(=O)(=O)NC1=CC=C([N+]([O-])=O)C=C1OC1CCCCC1 KTDZCOWXCWUPEO-UHFFFAOYSA-N 0.000 description 1
- 206010061309 Neoplasm progression Diseases 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 150000007930 O-acyl isoureas Chemical class 0.000 description 1
- 229960005524 O6-benzylguanine Drugs 0.000 description 1
- KRWMERLEINMZFT-UHFFFAOYSA-N O6-benzylguanine Chemical compound C=12NC=NC2=NC(N)=NC=1OCC1=CC=CC=C1 KRWMERLEINMZFT-UHFFFAOYSA-N 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- SUDAHWBOROXANE-VIFPVBQESA-N PD 0325901-Cl Chemical compound OC[C@H](O)CONC(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F SUDAHWBOROXANE-VIFPVBQESA-N 0.000 description 1
- 239000009820 PHY 906 Substances 0.000 description 1
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 229920002230 Pectic acid Polymers 0.000 description 1
- 102000009658 Peptidylprolyl Isomerase Human genes 0.000 description 1
- 108010020062 Peptidylprolyl Isomerase Proteins 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 102000029797 Prion Human genes 0.000 description 1
- 108091000054 Prion Proteins 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 206010037211 Psychomotor hyperactivity Diseases 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 201000000582 Retinoblastoma Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- YJDYDFNKCBANTM-QCWCSKBGSA-N SDZ PSC 833 Chemical compound C\C=C\C[C@@H](C)C(=O)[C@@H]1N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C(=O)[C@H](C(C)C)NC1=O YJDYDFNKCBANTM-QCWCSKBGSA-N 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- ZSJLQEPLLKMAKR-UHFFFAOYSA-N Streptozotocin Natural products O=NN(C)C(=O)NC1C(O)OC(CO)C(O)C1O ZSJLQEPLLKMAKR-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 108700011582 TER 286 Proteins 0.000 description 1
- LGGHDPFKSSRQNS-UHFFFAOYSA-N Tariquidar Chemical compound C1=CC=CC2=CC(C(=O)NC3=CC(OC)=C(OC)C=C3C(=O)NC3=CC=C(C=C3)CCN3CCC=4C=C(C(=CC=4C3)OC)OC)=CN=C21 LGGHDPFKSSRQNS-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- PDMMFKSKQVNJMI-BLQWBTBKSA-N Testosterone propionate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](OC(=O)CC)[C@@]1(C)CC2 PDMMFKSKQVNJMI-BLQWBTBKSA-N 0.000 description 1
- QHOPXUFELLHKAS-UHFFFAOYSA-N Thespesin Natural products CC(C)c1c(O)c(O)c2C(O)Oc3c(c(C)cc1c23)-c1c2OC(O)c3c(O)c(O)c(C(C)C)c(cc1C)c23 QHOPXUFELLHKAS-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- 206010043540 Thromboangiitis obliterans Diseases 0.000 description 1
- 208000031981 Thrombocytopenic Idiopathic Purpura Diseases 0.000 description 1
- 108010078233 Thymalfasin Proteins 0.000 description 1
- 208000033781 Thyroid carcinoma Diseases 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 1
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 206010053648 Vascular occlusion Diseases 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 206010047642 Vitiligo Diseases 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- 208000008383 Wilms tumor Diseases 0.000 description 1
- 210000001766 X chromosome Anatomy 0.000 description 1
- SMEGJBVQLJJKKX-HOTMZDKISA-N [(2R,3S,4S,5R,6R)-5-acetyloxy-3,4,6-trihydroxyoxan-2-yl]methyl acetate Chemical compound CC(=O)OC[C@@H]1[C@H]([C@@H]([C@H]([C@@H](O1)O)OC(=O)C)O)O SMEGJBVQLJJKKX-HOTMZDKISA-N 0.000 description 1
- DFYPFJSPLUVPFJ-QJEDTDQSSA-N [(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-2-[[[(2R,3S,5R)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[5-(2-amino-6-oxo-1H-purin-9-yl)-2-(hydroxymethyl)oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphoryl]oxymethyl]oxolan-3-yl] [(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methyl hydrogen phosphate Chemical compound Cc1cn([C@H]2C[C@H](OP(O)(=O)OC[C@H]3O[C@H](C[C@@H]3OP(O)(=O)OC[C@H]3O[C@H](C[C@@H]3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)[C@@H](COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)O[C@H]3C[C@@H](O[C@@H]3COP(O)(=O)OC3CC(OC3CO)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)O2)c(=O)[nH]c1=O DFYPFJSPLUVPFJ-QJEDTDQSSA-N 0.000 description 1
- FKCMADOPPWWGNZ-YUMQZZPRSA-N [(2r)-1-[(2s)-2-amino-3-methylbutanoyl]pyrrolidin-2-yl]boronic acid Chemical compound CC(C)[C@H](N)C(=O)N1CCC[C@H]1B(O)O FKCMADOPPWWGNZ-YUMQZZPRSA-N 0.000 description 1
- XMYKNCNAZKMVQN-NYYWCZLTSA-N [(e)-(3-aminopyridin-2-yl)methylideneamino]thiourea Chemical compound NC(=S)N\N=C\C1=NC=CC=C1N XMYKNCNAZKMVQN-NYYWCZLTSA-N 0.000 description 1
- DFPAKSUCGFBDDF-ZQBYOMGUSA-N [14c]-nicotinamide Chemical compound N[14C](=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-ZQBYOMGUSA-N 0.000 description 1
- IBXPAFBDJCXCDW-MHFPCNPESA-A [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].Cc1cn([C@H]2C[C@H](O)[C@@H](COP([S-])(=O)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3CO)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O Chemical compound [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].Cc1cn([C@H]2C[C@H](O)[C@@H](COP([S-])(=O)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3CO)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O IBXPAFBDJCXCDW-MHFPCNPESA-A 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 230000035508 accumulation Effects 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- FXAGBTBXSJBNMD-UHFFFAOYSA-N acetic acid;2-hydroxypropane-1,2,3-tricarboxylic acid Chemical compound CC(O)=O.OC(=O)CC(O)(C(O)=O)CC(O)=O FXAGBTBXSJBNMD-UHFFFAOYSA-N 0.000 description 1
- 108010011755 acetyl-prolyl-histidyl-seryl-cysteinyl-asparaginamide Proteins 0.000 description 1
- 229940081735 acetylcellulose Drugs 0.000 description 1
- 208000017733 acquired polycythemia vera Diseases 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 201000005179 adrenal carcinoma Diseases 0.000 description 1
- 208000020990 adrenal cortex carcinoma Diseases 0.000 description 1
- 201000005188 adrenal gland cancer Diseases 0.000 description 1
- 208000007128 adrenocortical carcinoma Diseases 0.000 description 1
- 230000001780 adrenocortical effect Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 229940040563 agaric acid Drugs 0.000 description 1
- 230000001270 agonistic effect Effects 0.000 description 1
- 229950005033 alanosine Drugs 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 239000012670 alkaline solution Substances 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-BKBMJHBISA-N alpha-D-galacturonic acid Chemical compound O[C@H]1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-BKBMJHBISA-N 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229950010817 alvocidib Drugs 0.000 description 1
- BIIVYFLTOXDAOV-YVEFUNNKSA-N alvocidib Chemical compound O[C@@H]1CN(C)CC[C@@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O BIIVYFLTOXDAOV-YVEFUNNKSA-N 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 238000010640 amide synthesis reaction Methods 0.000 description 1
- 229960001097 amifostine Drugs 0.000 description 1
- 238000005576 amination reaction Methods 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 1
- 229960003437 aminoglutethimide Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 229940030486 androgens Drugs 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 230000002491 angiogenic effect Effects 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 125000004653 anthracenylene group Chemical group 0.000 description 1
- 230000003527 anti-angiogenesis Effects 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 229940045687 antimetabolites folic acid analogs Drugs 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- 229940045985 antineoplastic platinum compound Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000003699 antiulcer agent Substances 0.000 description 1
- 238000003782 apoptosis assay Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000005110 aryl thio group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 229910052789 astatine Inorganic materials 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 229950003462 atiprimod Drugs 0.000 description 1
- SERHTTSLBVGRBY-UHFFFAOYSA-N atiprimod Chemical compound C1CC(CCC)(CCC)CCC11CN(CCCN(CC)CC)CC1 SERHTTSLBVGRBY-UHFFFAOYSA-N 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 201000000448 autoimmune hemolytic anemia Diseases 0.000 description 1
- 201000003710 autoimmune thrombocytopenic purpura Diseases 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 229960003005 axitinib Drugs 0.000 description 1
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 description 1
- 125000003828 azulenyl group Chemical group 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 102000055102 bcl-2-Associated X Human genes 0.000 description 1
- 108700000707 bcl-2-Associated X Proteins 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 229960003094 belinostat Drugs 0.000 description 1
- NCNRHFGMJRPRSK-MDZDMXLPSA-N belinostat Chemical compound ONC(=O)\C=C\C1=CC=CC(S(=O)(=O)NC=2C=CC=CC=2)=C1 NCNRHFGMJRPRSK-MDZDMXLPSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000036983 biotransformation Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 201000001531 bladder carcinoma Diseases 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 238000002725 brachytherapy Methods 0.000 description 1
- 210000005013 brain tissue Anatomy 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- CUWODFFVMXJOKD-UVLQAERKSA-N buserelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](COC(C)(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 CUWODFFVMXJOKD-UVLQAERKSA-N 0.000 description 1
- 229960002719 buserelin Drugs 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- DNSISZSEWVHGLH-UHFFFAOYSA-N butanamide Chemical group CCCC(N)=O DNSISZSEWVHGLH-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 229960005084 calcitriol Drugs 0.000 description 1
- GMRQFYUYWCNGIN-NKMMMXOESA-N calcitriol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](CCCC(C)(C)O)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C GMRQFYUYWCNGIN-NKMMMXOESA-N 0.000 description 1
- 235000020964 calcitriol Nutrition 0.000 description 1
- 239000011612 calcitriol Substances 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 229940088954 camptosar Drugs 0.000 description 1
- 230000009702 cancer cell proliferation Effects 0.000 description 1
- 239000003560 cancer drug Substances 0.000 description 1
- OJLHWPALWODJPQ-QNWVGRARSA-N canfosfamide Chemical compound ClCCN(CCCl)P(=O)(N(CCCl)CCCl)OCCS(=O)(=O)C[C@H](NC(=O)CC[C@H](N)C(O)=O)C(=O)N[C@@H](C(O)=O)C1=CC=CC=C1 OJLHWPALWODJPQ-QNWVGRARSA-N 0.000 description 1
- 229950000772 canfosfamide Drugs 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 208000002458 carcinoid tumor Diseases 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- OKBVVJOGVLARMR-QSWIMTSFSA-N cefixime Chemical compound S1C(N)=NC(C(=N\OCC(O)=O)\C(=O)N[C@@H]2C(N3C(=C(C=C)CS[C@@H]32)C(O)=O)=O)=C1 OKBVVJOGVLARMR-QSWIMTSFSA-N 0.000 description 1
- 229960002129 cefixime Drugs 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000022534 cell killing Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 229940106189 ceramide Drugs 0.000 description 1
- 150000001783 ceramides Chemical class 0.000 description 1
- 208000019065 cervical carcinoma Diseases 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- HZCWPKGYTCJSEB-UHFFFAOYSA-N chembl118841 Chemical compound C12=CC(OC)=CC=C2NC2=C([N+]([O-])=O)C=CC3=C2C1=NN3CCCN(C)C HZCWPKGYTCJSEB-UHFFFAOYSA-N 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 229940044683 chemotherapy drug Drugs 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 125000004773 chlorofluoromethyl group Chemical group [H]C(F)(Cl)* 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 229950009003 cilengitide Drugs 0.000 description 1
- AMLYAMJWYAIXIA-VWNVYAMZSA-N cilengitide Chemical compound N1C(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](C(C)C)N(C)C(=O)[C@H]1CC1=CC=CC=C1 AMLYAMJWYAIXIA-VWNVYAMZSA-N 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 238000011281 clinical therapy Methods 0.000 description 1
- WDDPHFBMKLOVOX-AYQXTPAHSA-N clofarabine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1F WDDPHFBMKLOVOX-AYQXTPAHSA-N 0.000 description 1
- 229960000928 clofarabine Drugs 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- WDOGQTQEKVLZIJ-WAYWQWQTSA-N combretastatin a-4 phosphate Chemical compound C1=C(OP(O)(O)=O)C(OC)=CC=C1\C=C/C1=CC(OC)=C(OC)C(OC)=C1 WDOGQTQEKVLZIJ-WAYWQWQTSA-N 0.000 description 1
- 238000012875 competitive assay Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 229940109262 curcumin Drugs 0.000 description 1
- 239000004148 curcumin Substances 0.000 description 1
- 235000012754 curcumin Nutrition 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- BALGDZWGNCXXES-UHFFFAOYSA-N cyclopentane;propanoic acid Chemical compound CCC(O)=O.C1CCCC1 BALGDZWGNCXXES-UHFFFAOYSA-N 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- UFULAYFCSOUIOV-UHFFFAOYSA-N cysteamine Chemical compound NCCS UFULAYFCSOUIOV-UHFFFAOYSA-N 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 210000000172 cytosol Anatomy 0.000 description 1
- 231100000599 cytotoxic agent Toxicity 0.000 description 1
- 239000002619 cytotoxin Substances 0.000 description 1
- 229940026692 decadron Drugs 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 230000003412 degenerative effect Effects 0.000 description 1
- 238000006356 dehydrogenation reaction Methods 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 229940027008 deltasone Drugs 0.000 description 1
- 230000003831 deregulation Effects 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 1
- 229960000452 diethylstilbestrol Drugs 0.000 description 1
- VFLDPWHFBUODDF-UHFFFAOYSA-N diferuloylmethane Natural products C1=C(O)C(OC)=CC(C=CC(=O)CC(=O)C=CC=2C=C(OC)C(O)=CC=2)=C1 VFLDPWHFBUODDF-UHFFFAOYSA-N 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- MGHPNCMVUAKAIE-UHFFFAOYSA-N diphenylmethanamine Chemical compound C=1C=CC=CC=1C(N)C1=CC=CC=C1 MGHPNCMVUAKAIE-UHFFFAOYSA-N 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- HKXBNHCUPKIYDM-CGMHZMFXSA-N doxercalciferol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)/C=C/[C@H](C)C(C)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C HKXBNHCUPKIYDM-CGMHZMFXSA-N 0.000 description 1
- 229960000413 doxercalciferol Drugs 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- BNFRJXLZYUTIII-UHFFFAOYSA-N efaproxiral Chemical compound CC1=CC(C)=CC(NC(=O)CC=2C=CC(OC(C)(C)C(O)=O)=CC=2)=C1 BNFRJXLZYUTIII-UHFFFAOYSA-N 0.000 description 1
- 229960000925 efaproxiral Drugs 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000009462 endogenous apoptosis Effects 0.000 description 1
- 201000003914 endometrial carcinoma Diseases 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 1
- 229950002189 enzastaurin Drugs 0.000 description 1
- 230000007515 enzymatic degradation Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000008995 epigenetic change Effects 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- UFNVPOGXISZXJD-XJPMSQCNSA-N eribulin Chemical compound C([C@H]1CC[C@@H]2O[C@@H]3[C@H]4O[C@H]5C[C@](O[C@H]4[C@H]2O1)(O[C@@H]53)CC[C@@H]1O[C@H](C(C1)=C)CC1)C(=O)C[C@@H]2[C@@H](OC)[C@@H](C[C@H](O)CN)O[C@H]2C[C@@H]2C(=C)[C@H](C)C[C@H]1O2 UFNVPOGXISZXJD-XJPMSQCNSA-N 0.000 description 1
- 229960003649 eribulin Drugs 0.000 description 1
- 201000005619 esophageal carcinoma Diseases 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- 229960002568 ethinylestradiol Drugs 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 229960005167 everolimus Drugs 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 229950000484 exisulind Drugs 0.000 description 1
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 description 1
- 238000011347 external beam therapy Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 229950003662 fenretinide Drugs 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 206010016629 fibroma Diseases 0.000 description 1
- DBEPLOCGEIEOCV-WSBQPABSSA-N finasteride Chemical compound N([C@@H]1CC2)C(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)NC(C)(C)C)[C@@]2(C)CC1 DBEPLOCGEIEOCV-WSBQPABSSA-N 0.000 description 1
- 229960004039 finasteride Drugs 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 125000005567 fluorenylene group Chemical group 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 229960001751 fluoxymesterone Drugs 0.000 description 1
- YLRFCQOZQXIBAB-RBZZARIASA-N fluoxymesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)C[C@@H]2O YLRFCQOZQXIBAB-RBZZARIASA-N 0.000 description 1
- 150000002224 folic acids Chemical class 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 229950011423 forodesine Drugs 0.000 description 1
- 229960004783 fotemustine Drugs 0.000 description 1
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 229950001109 galiximab Drugs 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 208000010749 gastric carcinoma Diseases 0.000 description 1
- 229960003297 gemtuzumab ozogamicin Drugs 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 229940080856 gleevec Drugs 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229950011595 glufosfamide Drugs 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 230000002710 gonadal effect Effects 0.000 description 1
- 229930000755 gossypol Natural products 0.000 description 1
- 229950005277 gossypol Drugs 0.000 description 1
- 208000024908 graft versus host disease Diseases 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 230000009931 harmful effect Effects 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 201000003911 head and neck carcinoma Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 201000011066 hemangioma Diseases 0.000 description 1
- 208000031209 hemophilic arthropathy Diseases 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 235000008216 herbs Nutrition 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 125000004475 heteroaralkyl group Chemical group 0.000 description 1
- 125000005549 heteroarylene group Chemical group 0.000 description 1
- 125000005553 heteroaryloxy group Chemical group 0.000 description 1
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- ALBYIUDWACNRRB-UHFFFAOYSA-N hexanamide Chemical group CCCCCC(N)=O ALBYIUDWACNRRB-UHFFFAOYSA-N 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- HHXHVIJIIXKSOE-QILQGKCVSA-N histrelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC(N=C1)=CN1CC1=CC=CC=C1 HHXHVIJIIXKSOE-QILQGKCVSA-N 0.000 description 1
- 229960002193 histrelin Drugs 0.000 description 1
- 108700020746 histrelin Proteins 0.000 description 1
- HYFHYPWGAURHIV-UHFFFAOYSA-N homoharringtonine Natural products C1=C2CCN3CCCC43C=C(OC)C(OC(=O)C(O)(CCCC(C)(C)O)CC(=O)OC)C4C2=CC2=C1OCO2 HYFHYPWGAURHIV-UHFFFAOYSA-N 0.000 description 1
- 102000052732 human XIAP Human genes 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229960001330 hydroxycarbamide Drugs 0.000 description 1
- 229960004171 hydroxychloroquine Drugs 0.000 description 1
- 229950000801 hydroxyprogesterone caproate Drugs 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 1
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 1
- 230000000148 hypercalcaemia Effects 0.000 description 1
- 208000030915 hypercalcemia disease Diseases 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 1
- 229960004716 idoxuridine Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- HIFJCPQKFCZDDL-ACWOEMLNSA-N iloprost Chemical compound C1\C(=C/CCCC(O)=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)C(C)CC#CC)[C@H](O)C[C@@H]21 HIFJCPQKFCZDDL-ACWOEMLNSA-N 0.000 description 1
- 229960002240 iloprost Drugs 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- DOUYETYNHWVLEO-UHFFFAOYSA-N imiquimod Chemical compound C1=CC=CC2=C3N(CC(C)C)C=NC3=C(N)N=C21 DOUYETYNHWVLEO-UHFFFAOYSA-N 0.000 description 1
- 229960002751 imiquimod Drugs 0.000 description 1
- IWKXDMQDITUYRK-KUBHLMPHSA-N immucillin H Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)N[C@H]1C1=CNC2=C1N=CNC2=O IWKXDMQDITUYRK-KUBHLMPHSA-N 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 238000012151 immunohistochemical method Methods 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 201000004933 in situ carcinoma Diseases 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- SETFNECMODOHTO-UHFFFAOYSA-N indisulam Chemical compound C1=CC(S(=O)(=O)N)=CC=C1S(=O)(=O)NC1=CC=CC2=C1NC=C2Cl SETFNECMODOHTO-UHFFFAOYSA-N 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 229960000598 infliximab Drugs 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 239000000138 intercalating agent Substances 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229940117681 interleukin-12 Drugs 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 230000031146 intracellular signal transduction Effects 0.000 description 1
- 230000004068 intracellular signaling Effects 0.000 description 1
- 238000003402 intramolecular cyclocondensation reaction Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 230000005865 ionizing radiation Effects 0.000 description 1
- 229960005386 ipilimumab Drugs 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 229950005254 irofulven Drugs 0.000 description 1
- NICJCIQSJJKZAH-AWEZNQCLSA-N irofulven Chemical compound O=C([C@@]1(O)C)C2=CC(C)=C(CO)C2=C(C)C21CC2 NICJCIQSJJKZAH-AWEZNQCLSA-N 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- IQPQWNKOIGAROB-UHFFFAOYSA-N isocyanate Chemical compound [N-]=C=O IQPQWNKOIGAROB-UHFFFAOYSA-N 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229960004125 ketoconazole Drugs 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 201000010260 leiomyoma Diseases 0.000 description 1
- 229950001845 lestaurtinib Drugs 0.000 description 1
- 201000011486 lichen planus Diseases 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- DHMTURDWPRKSOA-RUZDIDTESA-N lonafarnib Chemical compound C1CN(C(=O)N)CCC1CC(=O)N1CCC([C@@H]2C3=C(Br)C=C(Cl)C=C3CCC3=CC(Br)=CN=C32)CC1 DHMTURDWPRKSOA-RUZDIDTESA-N 0.000 description 1
- 229950001750 lonafarnib Drugs 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 201000005296 lung carcinoma Diseases 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 201000000564 macroglobulinemia Diseases 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000012067 mathematical method Methods 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 229960002985 medroxyprogesterone acetate Drugs 0.000 description 1
- PSGAAPLEWMOORI-PEINSRQWSA-N medroxyprogesterone acetate Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 PSGAAPLEWMOORI-PEINSRQWSA-N 0.000 description 1
- 229960004296 megestrol acetate Drugs 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- 229960001929 meloxicam Drugs 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960003151 mercaptamine Drugs 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 238000003328 mesylation reaction Methods 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000015689 metaplastic ossification Effects 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- SIVLENRHVVVPKJ-UHFFFAOYSA-N methyl 4-chloro-3-[(2-methoxy-7-oxo-8h-pyrido[2,3-d]pyrimidine-6-carbonyl)amino]benzoate Chemical compound COC(=O)C1=CC=C(Cl)C(NC(=O)C=2C(NC3=NC(OC)=NC=C3C=2)=O)=C1 SIVLENRHVVVPKJ-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- VAOCPAMSLUNLGC-UHFFFAOYSA-N metronidazole Chemical compound CC1=NC=C([N+]([O-])=O)N1CCO VAOCPAMSLUNLGC-UHFFFAOYSA-N 0.000 description 1
- 229960000282 metronidazole Drugs 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- OBBCSXFCDPPXOL-UHFFFAOYSA-N misonidazole Chemical compound COCC(O)CN1C=CN=C1[N+]([O-])=O OBBCSXFCDPPXOL-UHFFFAOYSA-N 0.000 description 1
- 229950010514 misonidazole Drugs 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 230000006667 mitochondrial pathway Effects 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000009126 molecular therapy Methods 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- JFOHFDSMPQIOES-UHFFFAOYSA-N motexafin Chemical compound C1=NC2=CC(OCCOCCOCCOC)=C(OCCOCCOCCOC)C=C2N=CC(C(=C2CCCO)C)=NC2=CC(C(CC)=C2CC)=NC2=CC2=C(CCCO)C(C)=C1N2 JFOHFDSMPQIOES-UHFFFAOYSA-N 0.000 description 1
- 229950011637 motexafin Drugs 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 208000029766 myalgic encephalomeyelitis/chronic fatigue syndrome Diseases 0.000 description 1
- 201000005962 mycosis fungoides Diseases 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- ONDPWWDPQDCQNJ-UHFFFAOYSA-N n-(3,3-dimethyl-1,2-dihydroindol-6-yl)-2-(pyridin-4-ylmethylamino)pyridine-3-carboxamide;phosphoric acid Chemical compound OP(O)(O)=O.OP(O)(O)=O.C=1C=C2C(C)(C)CNC2=CC=1NC(=O)C1=CC=CN=C1NCC1=CC=NC=C1 ONDPWWDPQDCQNJ-UHFFFAOYSA-N 0.000 description 1
- KWQWWUXRGIIBAS-UHFFFAOYSA-N n-[2-(4-hydroxyanilino)pyridin-3-yl]-4-methoxybenzenesulfonamide;hydrochloride Chemical compound Cl.C1=CC(OC)=CC=C1S(=O)(=O)NC1=CC=CN=C1NC1=CC=C(O)C=C1 KWQWWUXRGIIBAS-UHFFFAOYSA-N 0.000 description 1
- YZOQZEXYFLXNKA-UHFFFAOYSA-N n-[4-(4-amino-2-ethylimidazo[4,5-c]quinolin-1-yl)butyl]methanesulfonamide Chemical compound C1=CC=CC2=C(N(C(CC)=N3)CCCCNS(C)(=O)=O)C3=C(N)N=C21 YZOQZEXYFLXNKA-UHFFFAOYSA-N 0.000 description 1
- RPWGTQRQPVPFKR-YDALLXLXSA-N n-cyclohexylcyclohexanamine;(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-(phenylmethoxycarbonylamino)propanoic acid Chemical compound C1CCCCC1NC1CCCCC1.CC(C)(C)OC(=O)N[C@H](C(O)=O)CNC(=O)OCC1=CC=CC=C1 RPWGTQRQPVPFKR-YDALLXLXSA-N 0.000 description 1
- UMJJGDUYVQCBMC-UHFFFAOYSA-N n-ethyl-n'-[3-[3-(ethylamino)propylamino]propyl]propane-1,3-diamine Chemical compound CCNCCCNCCCNCCCNCC UMJJGDUYVQCBMC-UHFFFAOYSA-N 0.000 description 1
- OHDXDNUPVVYWOV-UHFFFAOYSA-N n-methyl-1-(2-naphthalen-1-ylsulfanylphenyl)methanamine Chemical compound CNCC1=CC=CC=C1SC1=CC=CC2=CC=CC=C12 OHDXDNUPVVYWOV-UHFFFAOYSA-N 0.000 description 1
- VBEGHXKAFSLLGE-UHFFFAOYSA-N n-phenylnitramide Chemical class [O-][N+](=O)NC1=CC=CC=C1 VBEGHXKAFSLLGE-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004957 naphthylene group Chemical group 0.000 description 1
- 125000004923 naphthylmethyl group Chemical group C1(=CC=CC2=CC=CC=C12)C* 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 229950009793 naptumomab estafenatox Drugs 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 210000005170 neoplastic cell Anatomy 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 201000008026 nephroblastoma Diseases 0.000 description 1
- 229950008835 neratinib Drugs 0.000 description 1
- JWNPDZNEKVCWMY-VQHVLOKHSA-N neratinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 JWNPDZNEKVCWMY-VQHVLOKHSA-N 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229960002653 nilutamide Drugs 0.000 description 1
- XWXYUMMDTVBTOU-UHFFFAOYSA-N nilutamide Chemical compound O=C1C(C)(C)NC(=O)N1C1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 XWXYUMMDTVBTOU-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- OSTGTTZJOCZWJG-UHFFFAOYSA-N nitrosourea Chemical compound NC(=O)N=NO OSTGTTZJOCZWJG-UHFFFAOYSA-N 0.000 description 1
- 229940085033 nolvadex Drugs 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 229960002230 omacetaxine mepesuccinate Drugs 0.000 description 1
- HYFHYPWGAURHIV-JFIAXGOJSA-N omacetaxine mepesuccinate Chemical compound C1=C2CCN3CCC[C@]43C=C(OC)[C@@H](OC(=O)[C@@](O)(CCCC(C)(C)O)CC(=O)OC)[C@H]4C2=CC2=C1OCO2 HYFHYPWGAURHIV-JFIAXGOJSA-N 0.000 description 1
- 230000000174 oncolytic effect Effects 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 206010030979 oral hairy leukoplakia Diseases 0.000 description 1
- 239000008183 oral pharmaceutical preparation Substances 0.000 description 1
- 229950007283 oregovomab Drugs 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000003791 organic solvent mixture Substances 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- HFHZKZSRXITVMK-UHFFFAOYSA-N oxyphenbutazone Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=C(O)C=C1 HFHZKZSRXITVMK-UHFFFAOYSA-N 0.000 description 1
- 238000006385 ozonation reaction Methods 0.000 description 1
- 208000021255 pancreatic insulinoma Diseases 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 208000003154 papilloma Diseases 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000009589 pathological growth Effects 0.000 description 1
- 229940097097 pediapred Drugs 0.000 description 1
- WVUNYSQLFKLYNI-AATRIKPKSA-N pelitinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC1=CC=C(F)C(Cl)=C1 WVUNYSQLFKLYNI-AATRIKPKSA-N 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- IPWFJLQDVFKJDU-UHFFFAOYSA-N pentanamide Chemical group CCCCC(N)=O IPWFJLQDVFKJDU-UHFFFAOYSA-N 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- 125000005327 perimidinyl group Chemical group N1C(=NC2=CC=CC3=CC=CC1=C23)* 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 229960002087 pertuzumab Drugs 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 125000001792 phenanthrenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C=CC12)* 0.000 description 1
- 125000005560 phenanthrenylene group Chemical group 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical compound CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- 229960003424 phenylacetic acid Drugs 0.000 description 1
- 239000003279 phenylacetic acid Substances 0.000 description 1
- 229960002895 phenylbutazone Drugs 0.000 description 1
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 1
- PHEDXBVPIONUQT-RGYGYFBISA-N phorbol 13-acetate 12-myristate Chemical compound C([C@]1(O)C(=O)C(C)=C[C@H]1[C@@]1(O)[C@H](C)[C@H]2OC(=O)CCCCCCCCCCCCC)C(CO)=C[C@H]1[C@H]1[C@]2(OC(C)=O)C1(C)C PHEDXBVPIONUQT-RGYGYFBISA-N 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- ISWRGOKTTBVCFA-UHFFFAOYSA-N pirfenidone Chemical compound C1=C(C)C=CC(=O)N1C1=CC=CC=C1 ISWRGOKTTBVCFA-UHFFFAOYSA-N 0.000 description 1
- 229960003073 pirfenidone Drugs 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 229960004403 pixantrone Drugs 0.000 description 1
- PEZPMAYDXJQYRV-UHFFFAOYSA-N pixantrone Chemical compound O=C1C2=CN=CC=C2C(=O)C2=C1C(NCCN)=CC=C2NCCN PEZPMAYDXJQYRV-UHFFFAOYSA-N 0.000 description 1
- 229940072689 plaquenil Drugs 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 150000003058 platinum compounds Chemical class 0.000 description 1
- 229920000724 poly(L-arginine) polymer Polymers 0.000 description 1
- 108010011110 polyarginine Proteins 0.000 description 1
- 208000037244 polycythemia vera Diseases 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229940116317 potato starch Drugs 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- VJZLQIPZNBPASX-OJJGEMKLSA-L prednisolone sodium phosphate Chemical compound [Na+].[Na+].O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)COP([O-])([O-])=O)[C@@H]4[C@@H]3CCC2=C1 VJZLQIPZNBPASX-OJJGEMKLSA-L 0.000 description 1
- 229940096111 prelone Drugs 0.000 description 1
- 230000001855 preneoplastic effect Effects 0.000 description 1
- 208000030266 primary brain neoplasm Diseases 0.000 description 1
- 229950010664 primycin Drugs 0.000 description 1
- NYWSLZMTZNODJM-SDUQVVOESA-N primycin Natural products CCCC[C@H]1[C@H](O)[C@H](C)CC[C@@H](O)CCC[C@@H](O)CCC[C@@H](O)C(=C[C@H](O[C@H]2O[C@H](CO)[C@@H](O)[C@@H]2O)[C@@H](O)C[C@H](O)C[C@@H](O)C[C@H](O)C[C@H](O)CCCCC(=C[C@@H](C)[C@@H](OC1=O)[C@H](C)[C@H](O)CCCNC(=N)N)C)C NYWSLZMTZNODJM-SDUQVVOESA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 239000000583 progesterone congener Substances 0.000 description 1
- 230000005522 programmed cell death Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- QLNJFJADRCOGBJ-UHFFFAOYSA-N propionamide Chemical group CCC(N)=O QLNJFJADRCOGBJ-UHFFFAOYSA-N 0.000 description 1
- 229940080818 propionamide Drugs 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 201000001514 prostate carcinoma Diseases 0.000 description 1
- 208000021046 prostate intraepithelial neoplasia Diseases 0.000 description 1
- 229940121649 protein inhibitor Drugs 0.000 description 1
- 239000012268 protein inhibitor Substances 0.000 description 1
- 230000004850 protein–protein interaction Effects 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- NYJWYCAHJRGKMI-UHFFFAOYSA-N pyrido[1,2-a]pyrimidin-4-one Chemical compound C1=CC=CN2C(=O)C=CN=C21 NYJWYCAHJRGKMI-UHFFFAOYSA-N 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 230000006340 racemization Effects 0.000 description 1
- 239000000700 radioactive tracer Substances 0.000 description 1
- 238000011363 radioimmunotherapy Methods 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- 229950007649 ranpirnase Drugs 0.000 description 1
- 108010061338 ranpirnase Proteins 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- QEHOIJJIZXRMAN-QZQSLCQPSA-N rebeccamycin Chemical class O[C@@H]1[C@@H](O)[C@H](OC)[C@@H](CO)O[C@H]1N1C2=C3NC4=C(Cl)C=CC=C4C3=C3C(=O)NC(=O)C3=C2C2=CC=CC(Cl)=C21 QEHOIJJIZXRMAN-QZQSLCQPSA-N 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 239000003488 releasing hormone Substances 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- OAKGNIRUXAZDQF-TXHRRWQRSA-N retaspimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(O)C1=CC(O)=C2NCC=C OAKGNIRUXAZDQF-TXHRRWQRSA-N 0.000 description 1
- 229930002330 retinoic acid Natural products 0.000 description 1
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 1
- 201000003068 rheumatic fever Diseases 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 229940100486 rice starch Drugs 0.000 description 1
- 229960001302 ridaforolimus Drugs 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- OHRURASPPZQGQM-GCCNXGTGSA-N romidepsin Chemical compound O1C(=O)[C@H](C(C)C)NC(=O)C(=C/C)/NC(=O)[C@H]2CSSCC\C=C\[C@@H]1CC(=O)N[C@H](C(C)C)C(=O)N2 OHRURASPPZQGQM-GCCNXGTGSA-N 0.000 description 1
- 229960003452 romidepsin Drugs 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229950009213 rubitecan Drugs 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229960005399 satraplatin Drugs 0.000 description 1
- 190014017285 satraplatin Chemical compound 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000002453 shampoo Substances 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 238000003797 solvolysis reaction Methods 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 1
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 201000000498 stomach carcinoma Diseases 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- MVGSNCBCUWPVDA-MFOYZWKCSA-N sulindac sulfone Chemical compound CC1=C(CC(O)=O)C2=CC(F)=CC=C2\C1=C/C1=CC=C(S(C)(=O)=O)C=C1 MVGSNCBCUWPVDA-MFOYZWKCSA-N 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- FIAFUQMPZJWCLV-UHFFFAOYSA-N suramin Chemical compound OS(=O)(=O)C1=CC(S(O)(=O)=O)=C2C(NC(=O)C3=CC=C(C(=C3)NC(=O)C=3C=C(NC(=O)NC=4C=C(C=CC=4)C(=O)NC=4C(=CC=C(C=4)C(=O)NC=4C5=C(C=C(C=C5C(=CC=4)S(O)(=O)=O)S(O)(=O)=O)S(O)(=O)=O)C)C=CC=3)C)=CC=C(S(O)(=O)=O)C2=C1 FIAFUQMPZJWCLV-UHFFFAOYSA-N 0.000 description 1
- 229960005314 suramin Drugs 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 229950010637 talabostat Drugs 0.000 description 1
- 108010009573 talabostat Proteins 0.000 description 1
- AYUNIORJHRXIBJ-TXHRRWQRSA-N tanespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCC=C)C(=O)C=C1C2=O AYUNIORJHRXIBJ-TXHRRWQRSA-N 0.000 description 1
- 229950007866 tanespimycin Drugs 0.000 description 1
- 229950005890 tariquidar Drugs 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- QFJCIRLUMZQUOT-UHFFFAOYSA-N temsirolimus Natural products C1CC(O)C(OC)CC1CC(C)C1OC(=O)C2CCCCN2C(=O)C(=O)C(O)(O2)C(C)CCC2CC(OC)C(C)=CC=CC=CC(C)CC(C)C(=O)C(OC)C(O)C(C)=CC(C)C(=O)C1 QFJCIRLUMZQUOT-UHFFFAOYSA-N 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000002381 testicular Effects 0.000 description 1
- 229960001712 testosterone propionate Drugs 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 229960003433 thalidomide Drugs 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- NZVYCXVTEHPMHE-ZSUJOUNUSA-N thymalfasin Chemical compound CC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O NZVYCXVTEHPMHE-ZSUJOUNUSA-N 0.000 description 1
- 229960004231 thymalfasin Drugs 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 208000013077 thyroid gland carcinoma Diseases 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 231100000048 toxicity data Toxicity 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 150000004654 triazenes Chemical class 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 229940078499 tricalcium phosphate Drugs 0.000 description 1
- 235000019731 tricalcium phosphate Nutrition 0.000 description 1
- 229910000391 tricalcium phosphate Inorganic materials 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 230000005740 tumor formation Effects 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 208000010570 urinary bladder carcinoma Diseases 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 1
- 229960000604 valproic acid Drugs 0.000 description 1
- 229950010938 valspodar Drugs 0.000 description 1
- 108010082372 valspodar Proteins 0.000 description 1
- 229960000241 vandetanib Drugs 0.000 description 1
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 1
- 208000019553 vascular disease Diseases 0.000 description 1
- 230000006444 vascular growth Effects 0.000 description 1
- 208000021331 vascular occlusion disease Diseases 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- HHJUWIANJFBDHT-KOTLKJBCSA-N vindesine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(N)=O)N4C)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 HHJUWIANJFBDHT-KOTLKJBCSA-N 0.000 description 1
- NMDYYWFGPIMTKO-HBVLKOHWSA-N vinflunine Chemical compound C([C@@](C1=C(C2=CC=CC=C2N1)C1)(C2=C(OC)C=C3N(C)[C@@H]4[C@@]5(C3=C2)CCN2CC=C[C@]([C@@H]52)([C@H]([C@]4(O)C(=O)OC)OC(C)=O)CC)C(=O)OC)[C@H]2C[C@@H](C(C)(F)F)CN1C2 NMDYYWFGPIMTKO-HBVLKOHWSA-N 0.000 description 1
- 229960000922 vinflunine Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229940100445 wheat starch Drugs 0.000 description 1
- 239000003871 white petrolatum Substances 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
- 229960005332 zileuton Drugs 0.000 description 1
- MWLSOWXNZPKENC-SSDOTTSWSA-N zileuton Chemical compound C1=CC=C2SC([C@H](N(O)C(N)=O)C)=CC2=C1 MWLSOWXNZPKENC-SSDOTTSWSA-N 0.000 description 1
- ZPFVQKPWGDRLHL-ZLYBXYBFSA-N zosuquidar trihydrochloride Chemical compound Cl.Cl.Cl.C([C@H](COC=1C2=CC=CN=C2C=CC=1)O)N(CC1)CCN1C1C2=CC=CC=C2[C@H]2C(F)(F)[C@H]2C2=CC=CC=C12 ZPFVQKPWGDRLHL-ZLYBXYBFSA-N 0.000 description 1
- 125000004933 β-carbolinyl group Chemical group C1(=NC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
Images







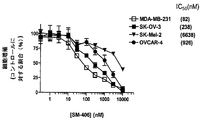




















Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5011—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics for testing antineoplastic activity
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Molecular Biology (AREA)
- Food Science & Technology (AREA)
- Physics & Mathematics (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Analytical Chemistry (AREA)
- Toxicology (AREA)
- Heart & Thoracic Surgery (AREA)
- Tropical Medicine & Parasitology (AREA)
- Biotechnology (AREA)
- Cell Biology (AREA)
- Microbiology (AREA)
- Cardiology (AREA)
- Vascular Medicine (AREA)
- Pain & Pain Management (AREA)
- Oncology (AREA)
- Dermatology (AREA)
- Ophthalmology & Optometry (AREA)
- Rheumatology (AREA)
- Epidemiology (AREA)
Description
本発明は、医薬化学の分野にある。特に、本発明は、アポトーシス阻害タンパク質のインヒビターとして作用するSmacのN末端配列の立体配座的に制限された模倣物に関する。本発明はまた、誘導するかまたはアポトーシス細胞死の誘導に対して細胞を感作するためのこれらの模倣物の使用に関する。
攻撃的な癌細胞表現型は、細胞内シグナル伝達経路の調節解除へ至る種々の遺伝的および後成的変化の結果である(Ponder, Nature 411:336 (2001))。しかし、全ての癌細胞の共通点は、それらがアポトーシスプログラムを実行しないことであり、正常なアポトーシス機構の欠陥に起因する適切なアポトーシスの欠如は、癌の特徴である(Lowe et al., Carcinogenesis 21:485 (2000))。化学療法剤、放射線、および免疫療法を含む、大抵の現在の癌療法は、癌細胞においてアポトーシスを間接的に誘導することによって機能する。従って、正常なアポトーシス機構の欠陥に起因して癌細胞がアポトーシスプログラムを実行できないことは、しばしば、化学療法、放射線、または免疫療法で誘導されるアポトーシスに対する耐性の増加と関連する。アポトーシス欠陥に起因する現在の治療プロトコルに対する、種々の起源のヒト癌の初期または獲得耐性は、現在の癌療法において主要な問題である(Lowe et al., Carcinogenesis 21:485 (2000);Nicholson, Nature 407:810 (2000))。従って、新規の分子標的特異的抗癌療法を設計および開発し癌患者の生存および生活の質を改善しようとする現在および未来の試みは、アポトーシスに対する癌細胞耐性を特異的に標的化する戦略を含まなければならない。この点で、癌細胞においてアポトーシスを直接阻害することにおいて中心的な役割を果たす重要な負の調節因子を標的化することは、新規の抗癌剤設計についての非常に有望な治療戦略を示す。
チャート1

A1およびA2は、独立して、水素および置換されていてもよいアルキルからなる群より選択され、ここでVがOである場合にはA2は存在せず;
Vは、N、CHおよびOからなる群より選択され;
Wは、CHおよびNからなる群より選択され;
Xは、水素および置換されていてもよいC1-3アルキルからなる群より選択され;
Yは、CONH、NHCO、C(O)O、OC(O)、1つまたは複数のCH2基がO、S、またはNR1によって置き換えられ得る(CH2)1-3、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Zは、(CR1R2)rであり;
Dは、(CR1R2)n-NR5-(CR3R4)mであり;
Jは、置換されていてもよいアルキレニルおよび(CR1R2)p-R6-(CR3R4)qからなる群より選択され;
Tは、C=O、C=S、C=NR1、S、S=O、SO2、O、NR1、CR1R2、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Uは、H、NR1R2、N(R1)COR7、OR1、SR1、置換されていてもよいアルキル、置換されていてもよいアリールおよびヘテロアリールからなる群より選択され;
n、m、pおよびqは、独立して、0〜5からなる群より選択され;
rは、0〜3であり;
R1、R2、R3およびR4は、各々、独立して、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
R5は、水素、置換されていてもよいアルキル、置換されていてもよいヘテロアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、置換されていてもよいヘテロアリールおよびCOR7からなる群より選択され;
R6は、O、S、NR1、CR1R2、C=O、C=SおよびC=NR1からなる群より選択され;かつ
R7は、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択される。
A1およびA2は、独立して、水素および置換されていてもよいアルキルからなる群より選択され、ここでVがOである場合にはA2は存在せず;
Vは、N、CHおよびOからなる群より選択され;
Wは、CHおよびNからなる群より選択され;
Xは、置換されていてもよいC1-3アルキルであり;
Yは、CONH、C(O)O、1つまたは複数のCH2基がO、S、またはNR1によって置き換えられ得る(CH2)1-3、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Dは、(CR1R2)n-NR5-(CR3R4)mであり;
Jは、置換されていてもよいアルキレニルおよび(CR1R2)p-R6-(CR3R4)qからなる群より選択され;
Tは、C=O、C=S、C=NR1、S、O、NR1、CR1R2、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Uは、H、NR1R2、OR1、SR1、置換されていてもよいアルキル、および置換されていてもよいアリールからなる群より選択され;
n、m、pおよびqは、独立して、0〜5より選択され;
R1、R2、R3およびR4は、各々、独立して、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
R5は、水素、置換されていてもよいアルキル、置換されていてもよいヘテロアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、置換されていてもよいヘテロアリールおよびCOR7からなる群より選択され;
R6は、O、S、NR1、CR1R2、C=O、C=SおよびC=NR1からなる群より選択され;
R7は、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択される。
A1およびA2は、独立して、水素および置換されていてもよいアルキルからなる群より選択され、ここでVがOである場合にはA2は存在せず;
Vは、N、CHおよびOからなる群より選択され;
Wは、CHおよびNからなる群より選択され;
Xは、水素および置換されていてもよいC1-3アルキルからなる群より選択され;
Yは、CONH、NHCO、C(O)O、OC(O)、1つまたは複数のCH2基がO、S、またはNR1によって置き換えられ得る(CH2)1-3、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Zは、(CR1R2)rであり;
Tは、C=O、C=S、C=NR1、S、S=O、SO2、O、NR1、CR1R2、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Uは、H、NR1R2、N(R1)COR7、OR1、SR1、置換されていてもよいアルキル、置換されていてもよいアリールおよびヘテロアリールからなる群より選択され;
mは、1または2であり;
rは、0〜3であり;
R1、R2、R3およびR4は、各々、独立して、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;かつ
R7は、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択される;
式XIIの化合物
と、
R7CO-L
式中、
R7は、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;かつ
Lは脱離基である;
とを縮合させ、式XIの化合物を形成させる段階。
a)式XXの化合物
とを縮合させ、式XXIIの化合物
b)式XXIIのアルケンをアルデヒドへ変換し、式XXIIIの化合物
c)式XXIIIの化合物のP2を除去し、式XXIVの化合物
d)式XXIVの化合物のC=N二重結合を還元し、式XIXの化合物を得る段階。
[請求項1001]
下記式Iを有する化合物またはその薬学的に許容される塩もしくはプロドラック:
A 1 およびA 2 は、独立して、水素および置換されていてもよいアルキルからなる群より選択され、ここでVがOである場合にはA 2 は存在せず;
Vは、N、CHおよびOからなる群より選択され;
Wは、CHおよびNからなる群より選択され;
Xは、水素および置換されていてもよいC 1-3 アルキルからなる群より選択され;
Yは、CONH、NHCO、C(O)O、OC(O)、1つまたは複数のCH 2 基がO、S、またはNR 1 によって置き換えられ得る(CH 2 ) 1-3 、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Zは、(CR 1 R 2 ) r であり;
Dは、(CR 1 R 2 ) n -NR 5 -(CR 3 R 4 ) m であり;
Jは、置換されていてもよいアルキレニルおよび(CR 1 R 2 ) p -R 6 -(CR 3 R 4 ) q からなる群より選択され;
Tは、C=O、C=S、C=NR 1 、S、S=O、SO 2 、O、NR 1 、CR 1 R 2 、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Uは、H、NR 1 R 2 、N(R 1 )COR 7 、OR 1 、SR 1 、置換されていてもよいアルキル、置換されていてもよいアリールおよびヘテロアリールからなる群より選択され;
n、m、pおよびqは、独立して、0〜5からなる群より選択され;
rは、0〜3であり;
R 1 、R 2 、R 3 およびR 4 は、各々、独立して、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
R 5 は、水素、置換されていてもよいアルキル、置換されていてもよいヘテロアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、置換されていてもよいヘテロアリールおよびCOR 7 からなる群より選択され;
R 6 は、O、S、NR 1 、CR 1 R 2 、C=O、C=SおよびC=NR 1 からなる群より選択され;かつ
R 7 は、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択される。
[請求項1002]
下記式IIを有する化合物またはその薬学的に許容される塩もしくはプロドラック:
A 1 およびA 2 は、独立して、水素および置換されていてもよいアルキルからなる群より選択され、ここでVがOである場合にはA 2 は存在せず;
Vは、N、CHおよびOからなる群より選択され;
Wは、CHおよびNからなる群より選択され;
Xは、置換されていてもよいC 1-3 アルキルであり;
Yは、CONH、C(O)O、1つまたは複数のCH 2 基がO、S、またはNR 1 によって置き換えられ得る(CH 2 ) 1-3 、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Dは、(CR 1 R 2 ) n -NR 5 -(CR 3 R 4 ) m であり;
Jは、置換されていてもよいアルキレニルおよび(CR 1 R 2 ) p -R 6 -(CR 3 R 4 ) q からなる群より選択され;
Tは、C=O、C=S、C=NR 1 、S、O、NR 1 、CR 1 R 2 、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Uは、H、NR 1 R 2 、OR 1 、SR 1 、置換されていてもよいアルキル、および置換されていてもよいアリールからなる群より選択され;
n、m、pおよびqは、独立して、0〜5より選択され;
R 1 、R 2 、R 3 およびR 4 は、各々、独立して、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
R 5 は、水素、置換されていてもよいアルキル、置換されていてもよいヘテロアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、置換されていてもよいヘテロアリールおよびCOR 7 からなる群より選択され;
R 6 は、O、S、NR 1 、CR 1 R 2 、C=O、C=SおよびC=NR 1 からなる群より選択され;
R 7 は、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
但し、任意の置換基は、カルボン酸、1つまたは複数のアルキル、ハロ、ヒドロキシルまたはハロアルキル基で置換されていてもよいシクロアルキル、またはアリール基で置換されていてもよいアリールを含まず、置換されていてもよいヘテロ環式基はオキソピペリジニルを含まない。
[請求項1003]
nおよびmが、n+mが3または4となるように0〜4より独立して選択される、請求項1002記載の化合物。
[請求項1004]
pおよびqが、p+qが1となるように0および1より独立して選択される、請求項1002記載の化合物。
[請求項1005]
nおよびmが、n+mが3または4となるように0〜4より独立して選択され、かつpおよびqが、p+qが1となるように0および1より独立して選択される、請求項1002記載の化合物。
[請求項1006]
TがC=Oである、請求項1005記載の化合物。
[請求項1007]
UがNR 1 R 2 である、請求項1005記載の化合物。
[請求項1008]
R 6 がCH 2 である、請求項1005記載の化合物。
[請求項1009]
YがCONHであり、WがCHであり、かつVがNである、請求項1005記載の化合物。
[請求項1010]
TがC=Oであり、UがNR 1 R 2 であり、R 6 がCH 2 であり、YがCONHであり、WがCHであり、かつVがNである、請求項1002記載の化合物。
[請求項1011]
下記式IIIを有する、請求項1002記載の化合物:
[請求項1012]
下記式IVを有する、請求項1001記載の化合物:
[請求項1013]
下記式Vを有する、請求項1012記載の化合物:
[請求項1014]
下記式VIを有する、請求項1013記載の化合物:
[請求項1015]
TがC=Oであり、UがNR 1 R 2 であり、かつR 5 がCOR 7 である、請求項1014記載の化合物。
[請求項1016]
A 1 が置換されていてもよいアルキルであり、かつA 2 が水素である、請求項1015記載の化合物。
[請求項1017]
R 1 が、置換されていてもよい炭素環式化合物および置換されていてもよいアルキルからなる群より選択される、請求項1015記載の化合物。
[請求項1018]
R 1 が置換されていてもよいアルキルであり、ここで、任意の置換基が、1つまたは複数の低級アルキル、ハロ、ハロアルキルまたはヘテロアリール基で置換されていてもよいアリールであり、かつR 2 が水素である、請求項1017記載の化合物。
[請求項1019]
R 7 が置換されていてもよいアルキルである、請求項1015記載の化合物。
[請求項1020]
以下からなる群より選択される、請求項1001記載の化合物またはその薬学的に許容される塩もしくはプロドラック:
[請求項1021]
以下からなる群より選択される、請求項1002記載の化合物またはその遊離塩基または別のその薬学的に許容される塩:
[請求項1022]
下記式の化合物またはその薬学的に許容される塩もしくはプロドラック:
[請求項1023]
請求項1001〜1021のいずれか一項記載の化合物と薬学的に許容される担体とを含む薬学的組成物。
[請求項1024]
請求項1022記載の化合物と薬学的に許容される担体とを含む薬学的組成物。
[請求項1025]
細胞を請求項1001〜1022のいずれか一項記載の化合物と接触させる段階を含む、細胞においてアポトーシスを誘導する方法。
[請求項1026]
細胞を請求項1001〜1022のいずれか一項記載の化合物と接触させる段階を含む、細胞をアポトーシスの誘導因子に対して感受性にする方法。
[請求項1027]
細胞をアポトーシスの誘導因子と接触させる段階をさらに含む、請求項1026記載の方法。
[請求項1028]
アポトーシスの誘導因子が化学療法剤である、請求項1027記載の方法。
[請求項1029]
アポトーシスの誘導因子が放射線である、請求項1027記載の方法。
[請求項1030]
アポトーシスの誘導因子が、腫瘍壊死因子(TNF)、TNF関連リガンド、またはTRAIL-R1もしくはTRAIL-R2のアゴニストである、請求項1027記載の方法。
[請求項1031]
TNF関連リガンドが、TRAMPリガンド、Fas/CD95リガンド、TNFR-1リガンド、およびTRAILからなる群より選択される、請求項1030記載の方法。
[請求項1032]
TNF関連リガンドがTRAILである、請求項1031記載の方法。
[請求項1033]
TRIAL-R1またはTRAIL-R2のアゴニストが抗体である、請求項1032記載の方法。
[請求項1034]
治療有効量の請求項1001〜1022のいずれか一項記載の化合物を動物へ投与する段階を含む、動物におけるアポトーシスの誘導に応答性である障害を治療、改善、または予防する方法。
[請求項1035]
アポトーシスの誘導因子を投与する段階をさらに含む、請求項1034記載の方法。
[請求項1036]
アポトーシスの誘導因子が化学療法剤である、請求項1035記載の方法。
[請求項1037]
アポトーシスの誘導因子が放射線である、請求項1035記載の方法。
[請求項1038]
アポトーシスの誘導因子が、TNF、TNF関連リガンド、またはTRAIL-R1もしくはTRAIL-R2のアゴニストである、請求項1035記載の方法。
[請求項1039]
TNF関連リガンドが、TRAMPリガンド、Fas/CD95リガンド、TNFR-1リガンド、およびTRAILからなる群より選択される、請求項1038記載の方法。
[請求項1040]
TNF関連リガンドがTRAILである、請求項1039記載の方法。
[請求項1041]
TRAIL-R1もしくはTRAIL-R2のアゴニストが抗体である、請求項1038記載の方法。
[請求項1042]
アポトーシスの誘導に応答性である障害が、過剰増殖性疾患である、請求項1034記載の方法。
[請求項1043]
過剰増殖性疾患が癌である、請求項1042記載の方法。
[請求項1044]
請求項1001〜1020記載の化合物をアポトーシスの誘導因子の前に投与する、請求項1035記載の方法。
[請求項1045]
請求項1001〜1022記載の化合物をアポトーシスの誘導因子の後に投与する、請求項1035記載の方法。
[請求項1046]
請求項1001〜1022記載の化合物をアポトーシスの誘導因子と同時に投与する、請求項1035記載の方法。
[請求項1047]
治療有効量の請求項1001〜1022のいずれか一項記載の化合物を動物へ投与する段階を含む、動物における過剰増殖性疾患を治療、改善、または予防する方法。
[請求項1048]
抗癌剤を投与する段階をさらに含む、請求項1047記載の方法。
[請求項1049]
抗癌剤がアポトーシスの誘導因子である、請求項1048記載の方法。
[請求項1050]
アポトーシスの誘導因子が化学療法剤である、請求項1049記載の方法。
[請求項1051]
アポトーシスの誘導因子が放射線である、請求項1049記載の方法。
[請求項1052]
アポトーシスの誘導因子が、TNF、TNF関連リガンド、またはTRAIL-R1もしくはTRAIL-R2のアゴニストである、請求項1049記載の方法。
[請求項1053]
TNF関連リガンドが、TRAMPリガンド、Fas/CD95リガンド、TNFR-1リガンド、およびTRAILからなる群より選択される、請求項1052記載の方法。
[請求項1054]
TNF関連リガンドがTRAILである、請求項1052記載の方法。
[請求項1055]
TRAIL-R1もしくはTRAIL-R2のアゴニストが抗体である、請求項1052記載の方法。
[請求項1056]
過剰増殖性疾患が癌である、請求項1047記載の方法。
[請求項1057]
請求項1001〜1022記載の化合物を抗癌剤の前に投与する、請求項1048記載の方法。
[請求項1058]
請求項1001〜1022記載の化合物を抗癌剤の後に投与する、請求項1048記載の方法。
[請求項1059]
請求項1001〜1022記載の化合物を抗癌剤と同時に投与する、請求項1048記載の方法。
[請求項1060]
抗癌剤が、タキソテル、ラパチニブおよびゲムシタビンからなる群より選択される、請求項1048記載の方法。
[請求項1061]
治療有効量の請求項1001〜1022のいずれか一項記載の化合物を動物へ投与する段階を含む、その必要がある動物において血管新生を予防または阻害する方法。
[請求項1062]
動物が、黄斑変性、関節リウマチ、乾癬、糖尿病性網膜症、未熟児網膜症、角膜移植拒絶、血管新生緑内障、水晶体後線維増殖症、ルベオーシス、オスラー−ウェーバー症候群、心筋血管新生、プラーク新生血管形成、毛細血管拡張症、血友病性関節症(hemophiliac joints)、血管線維腫、創傷肉芽形成、腸管癒着症、アテローム性動脈硬化症、強皮症、および肥厚性瘢痕からなる群より選択される疾患または障害を有する、請求項1061記載の方法。
[請求項1063]
請求項1001〜1022のいずれか一項記載の化合物と該化合物を動物へ投与するための使用説明書とを含む、キット。
[請求項1064]
抗癌剤をさらに含む、請求項1063記載のキット。
[請求項1065]
抗癌剤がアポトーシスの誘導因子である、請求項1064記載のキット。
[請求項1066]
アポトーシスの誘導因子が化学療法剤である、請求項1065記載のキット。
[請求項1067]
アポトーシスの誘導因子が、TNF、TNF関連リガンド、またはTRAIL-R1もしくはTRAIL-R2のアゴニストである、請求項1065記載のキット。
[請求項1068]
TNF関連リガンドが、TRAMPリガンド、Fas/CD95リガンド、TNFR-1リガンド、およびTRAILからなる群より選択される、請求項1067記載のキット。
[請求項1069]
TNF関連リガンドがTRAILである、請求項1068記載のキット。
[請求項1070]
TRAIL-R1もしくはTRAIL-R2のアゴニストが抗体である、請求項1067記載のキット。
[請求項1071]
使用説明書が、過剰増殖性疾患を有する動物へ化合物を投与するためのものである、請求項1064記載のキット。
[請求項1072]
過剰増殖性疾患が癌である、請求項1071記載のキット。
[請求項1073]
以下の段階を含む、下記式XIの化合物の製造方法:
A 1 およびA 2 は、独立して、水素および置換されていてもよいアルキルからなる群より選択され、ここでVがOである場合にはA 2 は存在せず;
Vは、N、CHおよびOからなる群より選択され;
Wは、CHおよびNからなる群より選択され;
Xは、水素および置換されていてもよいC 1-3 アルキルからなる群より選択され;
Yは、CONH、NHCO、C(O)O、OC(O)、1つまたは複数のCH 2 基がO、S、またはNR 1 によって置き換えられ得る(CH 2 ) 1-3 、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Zは、(CR 1 R 2 ) r であり;
Tは、C=O、C=S、C=NR 1 、S、S=O、SO 2 、O、NR 1 、CR 1 R 2 、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Uは、H、NR 1 R 2 、N(R 1 )COR 7 、OR 1 、SR 1 、置換されていてもよいアルキル、および置換されていてもよいアリールからなる群より選択され;
mは、1または2であり;
rは、0〜3であり;
R 1 、R 2 、R 3 およびR 4 は、各々、独立して、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;かつ
R 7 は、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択される;
下記式XIIの化合物
と、
R 7 CO-L
式中、
R 7 は、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;かつ
Lは脱離基である;
とを縮合させ、式XIの化合物を形成させる段階。
[請求項1074]
VがNであり、rが0であり、WがCHであり、YがCONHであり、TがC=Oであり、かつUがNR 1 R 2 である、請求項1073記載の方法。
[請求項1075]
LがClおよびOHからなる群より選択される、請求項1073記載の方法。
[請求項1076]
LがOHであり、かつ縮合が活性化剤の存在下で行われる、請求項1075記載の方法。
[請求項1077]
以下の段階を含む、下記式XIXの化合物の製造方法:
a)下記式XXの化合物
下記式XXIの化合物
とを縮合させ、下記式XXIIの化合物
b)式XXIIのアルケンをアルデヒドへ変換し、下記式XXIIIの化合物
c)式XXIIIの化合物のP 2 を除去し、下記式XXIVの化合物
d)式XXIVの化合物のC=N二重結合を還元し、式XIXの化合物を得る段階。
[請求項1078]
P 1 がtert-ブチルオキシカルボニルである、請求項1077記載の方法。
[請求項1079]
P 2 がカルボベンジルオキシである、請求項1078記載の方法。
[請求項1080]
式XXIVの化合物が、C=N二重結合の還元の前に単離される、請求項1077記載の方法。
[請求項1081]
還元がNaBH(OAc) 3 を用いて行われる、請求項1080記載の方法。
本発明は、Smacの模倣物でありかつIAPのインヒビターとして機能する、式I〜Xによって表される立体配座的に制限された化合物に関する。これらの化合物は、アポトーシスの誘導因子に対して細胞を感作し、そして、場合によっては、それら自体が、IAPを阻害することによってアポトーシスを誘導する。従って、本発明は、細胞を式I〜Xの化合物と単独でまたはアポトーシスの誘導因子と組み合わせて接触させることを含む、アポトーシスの誘導因子に対して細胞を感作する方法および細胞においてアポトーシスを誘導する方法に関する。本発明は、さらに、式I〜Xの化合物およびアポトーシスの誘導因子を動物へ投与することを含む、アポトーシスの誘導に応答性である動物における障害を治療、改善、または予防する方法に関する。このような障害としては、アポトーシスの調節異常を特徴とするもの、およびIAPの過剰発現を特徴とするものが挙げられる。本発明は、さらに、式I〜Xの化合物を動物へ投与することを含む、その必要がある動物において血管新生を予防または阻害する方法に関する。
A1およびA2は、独立して、水素および置換されていてもよいアルキルからなる群より選択され、ここでVがOである場合にはA2は存在せず;
Vは、N、CHおよびOからなる群より選択され;
Wは、CHおよびNからなる群より選択され;
Xは、水素および置換されていてもよいC1-3アルキルからなる群より選択され;
Yは、CONH、NHCO、C(O)O、OC(O)、1つまたは複数のCH2基がO、S、またはNR1によって置き換えられ得る(CH2)1-3、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Zは、(CR1R2)rであり;
Dは、(CR1R2)n-NR5-(CR3R4)mであり;
Jは、置換されていてもよいアルキレニルおよび(CR1R2)p-R6-(CR3R4)qからなる群より選択され;
Tは、C=O、C=S、C=NR1、S、S=O、SO2、O、NR1、CR1R2、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Uは、H、NR1R2、N(R1)COR7、OR1、SR1、置換されていてもよいアルキル、置換されていてもよいアリールおよびヘテロアリールからなる群より選択され;
n、m、pおよびqは、独立して、0〜5からなる群より選択され;
rは、0〜3であり;
R1、R2、R3およびR4は、各々、独立して、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
R5は、水素、置換されていてもよいアルキル、置換されていてもよいヘテロアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、置換されていてもよいヘテロアリールおよびCOR7からなる群より選択され;
R6は、O、S、NR1、CR1R2、C=O、C=SおよびC=NR1からなる群より選択され;
R7は、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択される。
A1およびA2は、独立して、水素および置換されていてもよいアルキルからなる群より選択され、ここでVがOである場合にはA2は存在せず;
Vは、N、CHおよびOからなる群より選択され;
Wは、CHおよびNからなる群より選択され;
Xは、置換されていてもよいC1-3アルキルであり;
Yは、CONH、C(O)O、1つまたは複数のCH2基がO、S、またはNR1によって置き換えられ得る(CH2)1-3、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Dは、(CR1R2)n-NR5-(CR3R4)mであり;
Jは、置換されていてもよいアルキレニルおよび(CR1R2)p-R6-(CR3R4)qからなる群より選択され;
Tは、C=O、C=S、C=NR1、S、O、NR1、CR1R2、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
Uは、H、NR1R2、OR1、SR1、置換されていてもよいアルキル、および置換されていてもよいアリールからなる群より選択され;
n、m、pおよびqは、独立して、0〜5より選択され;
R1、R2、R3およびR4は、各々、独立して、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択され;
R5は、水素、置換されていてもよいアルキル、置換されていてもよいヘテロアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、置換されていてもよいヘテロアリールおよびCOR7からなる群より選択され;
R6は、O、S、NR1、CR1R2、C=O、C=SおよびC=NR1からなる群より選択され;
R7は、水素、置換されていてもよいアルキル、置換されていてもよい炭素環式化合物、置換されていてもよいヘテロ環式化合物、置換されていてもよいアリール、および置換されていてもよいヘテロアリールからなる群より選択される。
a)式XXの化合物
とを縮合させ、式XXIIの化合物
b)式XXIIのアルケンをアルデヒドへ変換し、式XXIIIの化合物
c)式XXIIIの化合物のP2アミン保護基を除去し、式XXIVの化合物
d)式XXIVの化合物のC=N二重結合を還元し、式XIXの化合物を得る段階
を含む、方法に関する。
共有結合的に制限されたSmac模倣物の合成
一般方法:NMRスペクトルを、300 MHzのプロトン周波数で得た。1H化学シフトは、内部標準としてのMe4Si(0.00 ppm)、CHCl3(7.26 ppm)、CD2HOD(3.31 ppm)、またはDHO(4.79 ppm)で報告される。13C化学シフトは、内部標準としてのCDCl3 (77.00 ppm)、CD3OD(49.00 ppm)、または1,4-ジオキサン(67.16 ppm)で報告される。旋光度を室温で測定した。本発明の化合物は逆相HPLC(溶出液として水中の0.1% TFAとアセトニトリル中の0.1% TFA)により精製され、TFA塩として単離されてもよい。
CH2Cl2中の2つの基質の溶液(より少ない基質(substrate)について20 mg/mL)へ、撹拌しながら0℃で、EDC(1アミノ基当たり1.1 eq)、HOBt(1アミノ基当たり1.1 eq)およびN,N-ジイソプロピルエチルアミン(1アミノ基当たり4 eq)を添加した。混合物を室温で8時間撹拌し、次いで濃縮した。残渣をクロマトグラフィーによって精製し、生成物が得られた。
メタノール中の前記基質の溶液(20 mg/mL)へ、1,4-ジオキサン中のHClの溶液(4 M, 10-20 eq/Boc)を添加した。溶液を室温で一晩撹拌し、次いで濃縮し、生成物が得られた。
Smac模倣物中間体の合成
立体配座的に制限されたSmac模倣物についての合成経路における中間体は、スキーム1〜7に記載される方法を使用して合成され得る。
中間体5および7の合成をスキーム1に示す。化合物2は、主生成物としてR型異性体を含む2つのジアステレオマーの混合物(比率は約4:1である)として、報告された方法((1) Zhang, J.; Xiong, C.; Wang, W.; Ying, J.; Hruby, V., J. Org. Lett., 2002, 4 (23), 4029-4032、(2) Polyak, F. and Lubell, W. D. J. Org, Chem. 1998, 63, 5937-5949、および(3) Tetrahedron Letters 2005, 46, 945-947を参照のこと)に従って、ピログルタミン酸1から5段階で作製され得る。2のBoc基の除去、続いてのN-α-(tert-ブトキシルカルボニル)-N-β-(ベンゾキシルカルボニル)-L-ジアミノ-プロピオン酸(Boc-Dap(Z)-OH)との縮合によって、アミド3が得られた。3のC-C二重結合のオゾン酸化によって、アルデヒド4が得られた。4のCbz基の切断、得られるアミンとアルデヒド基との分子内縮合、続いてのエナミンの還元をワンポットで行い、延長された反応時間下で化合物5が得られる。または、4のCBz基の脱保護、分子内環化、エナミン中間体の単離、および還元によって、5を得た。この変換において、化合物5のみが得られ、その異性体の検出可能な形成は無く、このことは、少ない方の異性体由来のアミノアルデヒドはこれらの条件下で環化しないことを示唆している。
化合物7についての分析データ:
7とフェニル酢酸との縮合、続いてのメチルエステルの加水分解によって、酸が得られ、これをアミノジフェニルアミンと縮合させ、アミドが得られた。このアミドのBoc保護基の除去によって、アンモニウム塩が得られた。この塩とN-Boc-N-メチル-アラニンとの縮合、続いてのBoc保護基の除去によって、SH-207が得られた。
XIAPへのインヒビターの結合
IAPタンパク質への立体配座的に制限されたSmac模倣物の結合能力を試験するために、蛍光偏光(FP)に基づく方法を使用する高感度かつ定量的なインビトロ結合アッセイを開発し、そしてXIAPタンパク質へのSmac模倣物の結合を測定するために使用した(Nikolovska-Coleska et al., Anal Biochem. 332:261-73 (2004))。このアッセイのために、5-カルボキシフルオレセイン(5-Fam)を、変異Smacペプチドのリジン側鎖へカップリングした;AbuRPF-K-(5-Fam)-NH2(SM5Fと呼ぶ)。XIAP BIR3タンパク質へのSM5Fペプチドの結合のKd値は、17.92 nMであると測定され、このことは、このペプチドがXIAPタンパク質の表面ポケットへ高親和性で結合することを示している。Hisタグへ融合されたヒトXIAPの組換えXIAP BIR3タンパク質(残基241-356)は安定かつ可溶性であり、そしてFPに基づく結合アッセイのために使用した。
他のIAPタンパク質へのインヒビターの結合
他のIAPタンパク質(ML-IAP、cIAPl、およびcIAP2)への立体配座的に制限されたSmac模倣物の結合能力を試験するために、結合アッセイ条件を開発および最適化した。His-tagへ融合された、組換えcIAPl BIR3ドメイン(残基253-363)、cIAP2 BIR3ドメイン(残基238-349)、およびML-IAP BIR3ドメイン(残基63-179)を、結合アッセイにおいて使用した。他のIAPタンパク質についての競合結合アッセイを、XIAP BIR3について記載したものと同様に行った。簡単に記載すると、同一の高親和性トレーサーSM5F Smac-Fluペプチドを使用し、これは、それぞれ4.1 nM、6.6 nM、および160 nMの高親和性でcIAPl、cIAP2、およびML-IAPに結合する。以下のタンパク質濃度およびトレーサーを競合アッセイにおいて使用した:10 nM cIAPlおよび2 nM SM5F、25 nM cIAP2および2 nM SM5F、300 nM ML-IAPおよび5 nM SM5F。
立体配座的に制限されたSmac模倣物による細胞増殖阻害
種々の癌細胞株の増殖に対する本発明の化合物の効果を試験した。細胞を、試験化合物と共に、3000細胞/ウエルの密度で、96ウエル平底細胞培養プレート中に接種し、37℃で95%空気および5%CO2の雰囲気中において4日間インキュベートした。種々の濃度の化合物での処理後の細胞増殖阻害の割合を、WST-8キット(2-(2-メトキシ-4-ニトロフェニル)-3-(4-ニトロフェニル)-5-(2,4ジスルホフェニル)-2H-テトラゾリウム一ナトリウム塩;Dojindo Molecular Technologies, Inc., Gaithersburg, Maryland)を使用して測定した。WST-8を10%の最終濃度で各ウエルへ添加し、次いでプレートを37℃で2〜3時間インキュベートした。サンプルの吸光度を、ULTRA Tecan Reader(Molecular Device)を使用して450 nmで測定した。細胞増殖を50%阻害した試験化合物の濃度(IC50)を、未処理細胞および試験化合物で処理した細胞における吸光度を比較することによって計算した。
YP-337による細胞死の誘導
細胞死を誘導するYP-337の能力を、乳癌MDA-MB-231および卵巣癌SK-OV-3細胞株において試験した(表4)。細胞を48時間YP-337で処理し、細胞生存能力を、トリパンブルー排除アッセイを使用して測定した。
乳癌異種移植モデルにおけるSM-406(AT-406)の抗腫瘍活性
抗腫瘍剤として作用するSM-406(AT-406)の能力を、MDA-MB-231乳癌異種移植(ヌードマウス)モデルにおいて試験した。SM-406(AT-406)を、2週間、1週間当たりqd×5投与した。図1に示されるように、SM-406(AT-406)は、SM-406(AT-406)30、100および200 mg/kgの用量で、抗腫瘍活性を示した。11%までの用量依存性体重減少が、100および200 mg/kgで観察された。
前立腺癌異種移植モデルにおけるSM-406(AT-406)およびタキソテルの抗腫瘍活性
単独でおよびタキソテルと組み合わせて抗腫瘍剤として作用するSM-406(AT-406)の能力を、PC-3前立腺癌異種移植(ヌードマウス)モデルにおいて試験した。2週間、SM-406(AT-406)を1週間当たりqd×5、タキソテルを週1回投与した。図2に示されるように、SM-406(AT-406)は、100および200 mg/kgの用量で抗腫瘍活性を示した。さらに、SM-406(AT-406)(100 mg/kg)は、タキソテル(8 mg/kg)と組み合わせて、増強された抗腫瘍活性を示した。
前立腺癌異種移植モデルにおけるSM-406(AT-406)およびTykerb(登録商標)の抗腫瘍活性
単独でおよびTykerb(登録商標)と組み合わせて抗腫瘍剤として作用するSM-406(AT-406)の能力を、PC-3前立腺癌異種移植(ヌードマウス)モデルにおいて試験した。2週間、SM-406(AT-406)を1週間当たりqd×5、Tykerb(登録商標)を1週間当たりbid×5投与した。図3に示されるように、SM-406(AT-406)は、100および200 mg/kgの用量で抗腫瘍活性を示した。さらに、SM-406(AT-406)(100 mg/kg)は、Tykerb(登録商標)(90 mg/kg)と組み合わせて、増強された抗腫瘍活性を示した。
乳癌異種移植モデルにおけるSM-406(AT-406)およびタキソテルの抗腫瘍活性
単独でおよびタキソテルと組み合わせて抗腫瘍剤として作用するSM-406(AT-406)の能力を、2LMP乳癌異種移植モデルにおいて試験した。2週間、SM-406(AT-406)を1週間当たりqd×5、タキソテルを週1回投与した。図4に示されるように、SM-406(AT-406)は、100 mg/kgの用量で抗腫瘍活性を示した。さらに、SM-406(AT-406)(100 mg/kg)は、タキソテル(12 mg/kg)と組み合わせて、抗腫瘍活性を示した。
乳癌異種移植モデルにおけるSM-406(AT-406)およびTykerb(登録商標)の抗腫瘍活性
単独でおよびTykerb(登録商標)と組み合わせて抗腫瘍剤として作用するSM-406(AT-406)の能力を、2LMP乳癌異種移植モデルにおいて試験した。2週間、SM-406(AT-406)を1週間当たりqd×5、Tykerb(登録商標)をqd投与した。図5に示されるように、SM-406(AT-406)は、30、100および200 mg/kgの用量で抗腫瘍活性を示した。さらに、SM-406(AT-406)(30、100および200 mg/kg)は、Tykerb(登録商標)(90 mg/kg)と組み合わせて、増強された抗腫瘍活性を示した。
膵臓癌異種移植モデルにおけるSM-406(AT-406)の抗腫瘍活性
抗腫瘍剤として作用するSM-406(AT-406)の能力を、Panc-1膵臓癌異種移植モデルにおいて試験した。SM-406(AT-406)を、1週間当たりqd×5投与した。図6に示されるように、SM-406(AT-406)は、30および100 mg/kgの用量で抗腫瘍活性を示した。
乳癌異種移植モデルにおけるSM-406(AT-406)の用量スケジュール最適化
図7に示されるように、MDA-MB-231乳癌異種移植(ヌードマウス)モデルにおけるSM-406(AT-406)投薬スケジュールの最適化のための研究を、SM-406(AT-406)を200 mg/kg使用して行った。1週間当たりqd×5、第1〜5日、1週間当たりqd×3、第1〜3日、または週1回での投与によって、抗腫瘍活性が生じた。体重減少は、他の投薬スケジュールと比較して週1回投薬においてより少なかった(<5%)。
癌細胞株におけるSM-406のインビトロ活性
単一剤として細胞増殖を阻害するSM-406の能力を、MDA-MB-231乳癌細胞株、SK-OV-3およびOVCAR-4卵巣癌細胞株、ならびにSK-Mel-2メラノーマ癌細胞株において試験した。細胞を4日間処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図8に示されるように、SM-406は、MDA-MB-231、SK-OV-3、SK-Mel-2およびOVCAR-4についてそれぞれ、82 nM、238 nM、6638 nMおよび926 nMのIC50値で、4つ全ての癌細胞を阻害した。
癌細胞株におけるSM-406のインビトロ活性
単一剤として細胞増殖を阻害するSM-406の能力を、HL-60およびSR白血病細胞株において試験した。細胞を4日間処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図9に示されるように、SM-406は、それぞれ58 nMおよび7290 nMのIC50値で、両方の細胞株を阻害した。
癌細胞株におけるSM-406のインビトロ活性
単一剤として細胞増殖を阻害するSM-406の能力を、BT-549、MDA-MB-415、SUM-159、MDA-MB-468、MDA-MB-453および2LMPヒト乳癌細胞株において試験した。細胞を4日間処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図10に示されるように、SM-406は、6つ全ての細胞株において細胞増殖を阻害した。
MDA-MB-436細胞株におけるTNFαと組み合わせてのSM-406のインビトロ活性
TNFαと組み合わせてのSM-406、および単一剤としてのSM-428の、細胞増殖を阻害する能力を、ヒト乳癌細胞株MDA-MB-436において試験した。細胞を、4日間、単独もしくは種々の濃度のSM-406と組み合わせてのTNFαで、またはSM-428で、処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図11に示されるように、TNFαと組み合わせてSM-406は、TNFα単独での処理と比較して、増強された細胞増殖阻害を示した。
SUM-159細胞株におけるTNFαと組み合わせてのSM-406のインビトロ活性
TNFαと組み合わせてのSM-406、および単一剤としてのSM-428の、細胞増殖を阻害する能力を、ヒト乳癌細胞株SUM-159において試験した。細胞を、4日間、単独でもしくは種々の濃度のSM-406と組み合わせてTNFαで、またはSM-428で、処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図12に示されるように、TNFαと組み合わせてSM-406は、TNFα単独での処理と比較して、増強された細胞増殖阻害を示した。
Panc-1細胞株におけるTRAILと組み合わせてのSM-406のインビトロ活性
細胞増殖を阻害するTRAILと組み合わせてのSM-406の能力を、ヒト膵細胞株Panc-1において試験した。細胞を、4日間、単独でまたは種々の濃度のSM-406と組み合わせてTRAILで処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図13に示されるように、TRAILと組み合わせてSM-406は、TRAIL単独での処理と比較して、増強された細胞増殖阻害を示した。
MDA-231(2LMP)細胞株におけるTRAILと組み合わせてのSM-406のインビトロ活性
細胞増殖を阻害するTRAILと組み合わせてのSM-406の能力を、ヒト乳癌細胞株MDA-MB-231(2LMP)において試験した。細胞を、4日間、単独でまたは種々の濃度のSM-406と組み合わせてTRAILで処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図14に示されるように、TRAILと組み合わせてSM-406は、TRAIL単独での処理と比較して、増強された細胞増殖阻害を示した。
Panc-1細胞株におけるゲムシタビンと組み合わせてのSM-406のインビトロ活性
細胞増殖を阻害するゲムシタビンと組み合わせてのSM-406の能力を、ヒト膵細胞株Panc-1において試験した。細胞を、4日間、単独でまたは種々の濃度のSM-406と組み合わせてゲムシタビンで処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図15に示されるように、ゲムシタビンと組み合わせてSM-406は、ゲムシタビン単独での処理と比較して、増強された細胞増殖阻害を示した。
Panc-1細胞株におけるミトザントロンと組み合わせてのSM-406のインビトロ活性
細胞増殖を阻害するミトザントロンと組み合わせてのSM-406の能力を、ヒト膵細胞株Panc-1において試験した。細胞を、4日間、単独でまたは種々の濃度のSM-406と組み合わせてミトザントロンで処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図16に示されるように、ミトザントロンと組み合わせてSM-406は、ミトザントロン単独での処理と比較して、増強された細胞増殖阻害を示した。
PC3細胞株におけるロスコビチンと組み合わせてのSM-406のインビトロ活性
細胞増殖を阻害するロスコビチン(Ros)と組み合わせてのSM-406の能力を、ヒト前立腺細胞株PC3において試験した。細胞を、4日間、単独でまたは種々の濃度のロスコビチンと組み合わせてSM-406で処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図17に示されるように、ロスコビチンと組み合わせてSM-406は、細胞増殖を阻害した。
MDA-MB-453細胞株におけるタキソテルと組み合わせてのSM-406のインビトロ活性
細胞増殖を阻害するタキソテル(TXT)と組み合わせてのSM-406の能力を、ヒト乳癌細胞株MDA-MB-453において試験した。細胞を、4日間、単独でまたは種々の濃度のSM-406と組み合わせてTXTで処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図18に示されるように、タキソテルと組み合わせてSM-406は、タキソテル単独での処理と比較して、増強された細胞増殖阻害を示した。
Panc-1細胞株におけるVP-16と組み合わせてのSM-406のインビトロ活性
細胞増殖を阻害するVP-16と組み合わせてのSM-406の能力を、ヒト膵細胞株Panc-1において試験した。細胞を、4日間、単独でまたは種々の濃度のSM-406と組み合わせてVP-16で処理した。細胞増殖を、WSTに基づくアッセイを使用して測定した。図19に示されるように、VP-16と組み合わせてSM-406は、VP-16単独での処理と比較して、増強された細胞増殖阻害を示した。
MDA-MB-231細胞におけるcIAP-1タンパク質に対するSM-406の効果
SM-406およびSM-428の効果を、MDA-MB-231癌細胞においてcIAP-1タンパク質に対して測定した。細胞を、種々の濃度および時点で、SM-406またはSM-428で処理した。タンパク質レベルをウエスタンブロッティングによって分析した。図20に示されるように、SM-406はcIAP-1タンパク質レベルを低下させた。
SK-OV-3細胞におけるcIAP-1およびcIAP-2タンパク質に対するSM-406の効果
SM-406およびSM-428の効果を、SK-OV-3癌細胞においてcIAP-1およびcIAP-2タンパク質に対して測定した。細胞を、種々の濃度および時点で、SM-406またはSM-428で処理した。タンパク質レベルをウエスタンブロッティングによって分析した。図21に示されるように、SM-406は、cIAP-1およびcIAP-2タンパク質レベルを低下させた。
MDA-MB-231細胞におけるSM-406によるアポトーシスの誘導
SM-406またはSM-428によるアポトーシスの誘導を、MDA-MB-231乳癌細胞において測定した。細胞を種々の時間SM-406またはSM-428で処理し、アポトーシスを、フローサイトメーターを使用してFITCアネキシンV/ヨウ化プロピジウム染色によって分析した。図22に示されるように、3μMのSM-406は、MDA-MB-231乳癌細胞においてアポトーシスを誘導した。
SK-OV-3細胞におけるSM-406によるアポトーシスの誘導
SM-406またはSM-428によるアポトーシスの誘導を、SK-OV-3卵巣癌細胞において測定した。細胞を種々の濃度のSM-406またはSM-428で処理した。アポトーシスを、フローサイトメーターを使用してFITCアネキシンV/ヨウ化プロピジウム染色によって分析した。図23に示されるように、0.1、0.3、1および3μMのSM-406は、SK-OV-3卵巣癌細胞においてアポトーシスを誘導した。
Panc-1細胞におけるSM-406によるアポトーシスの誘導
SM-406またはSM-428によるアポトーシスの誘導を、Panc-1膵臓癌細胞において測定した。細胞を種々の濃度のSM-406またはSM-428で処理した。アポトーシスを、フローサイトメーターを使用してFITCアネキシンV/ヨウ化プロピジウム染色によって分析した。図24に示されるように、0.3、1および3μMのSM-406は、panc-1膵臓癌細胞においてアポトーシスを誘導した。
MDA-MB-231異種移植モデルにおけるSM-406の抗腫瘍活性
抗腫瘍剤として作用するSM-406の能力を、重篤複合免疫不全(SCID)マウスにおけるヒト乳癌のMDA-MB-231異種移植モデルにおいて試験した。腫瘍が150 mm3の平均サイズへ増殖した際に、処置を開始した。SM-406を、経口強制飼養によって、2週間の間、週5日、毎日与えた。タキソテルを、2週間の間、週1回、静脈内に(i.v.)与えた。図25に示されるように、SM-406は、30および100 mg/kg POで抗腫瘍活性を示した。
PC-3異種移植モデルにおけるSM-406の抗腫瘍活性
抗腫瘍剤として作用するSM-406の能力を、ヌードマウスにおけるヒト前立腺癌のPC-3異種移植モデルにおいて試験した。腫瘍が100 mm3の平均サイズへ増殖した際に、処置を開始した。SM-406を、経口強制飼養によって第15〜25日の2週間の間および第43〜53日のさらに2週間の間、週5日、毎日与えた。タキソテルを、3週間の間週1回、第16、23および44日に、静脈内に(i.v.)与えた。図26に示されるように、SM-406は、単独でまたはタキソテルと組み合わせて、抗腫瘍活性を示した。
2LMP異種移植モデルにおけるSM-406の抗腫瘍活性
抗腫瘍剤として作用するSM-406の能力を、ヌードマウスにおけるヒト乳癌の2LMP異種移植モデルにおいて試験した。SM-406を、第16〜26日の間、腹腔内(intrapentoneal)(i.p.)または経口(p.o.)のいずれで、10日間毎日与えた。タキソテルを、2週間の間、週1回、静脈内(i.v.)に与えた。瘍壊死因子(TNF)関連アポトーシス誘導リガンド(TRAIL)は、第16〜26日の10日間、1日1回、i.p.であった。図27に示されるように、SM-406は、単独でまたはTRAILと組み合わせて、抗腫瘍活性を示した。
IAPタンパク質のBIR3ドメインへのSM-406の結合
IAPタンパク質の4つのメンバーのBIR3ドメインへのSM-406およびSM-428の競合結合曲線を、蛍光偏光に基づく結合アッセイを使用して測定した。試験したIAPタンパク質は以下を含む:(A)XIAP;(B)cIAPl:(C)cIAP2;(D)ML-IAP。図28に示されるように、SM-406は、4つ全てのIAPタンパク質へ結合する。
Claims (11)
- 以下からなる群より選択される化合物またはその薬学的に許容される塩:
。 - 請求項1記載の化合物またはその薬学的に許容される塩と薬学的に許容される担体とを含む薬学的組成物。
- 請求項1記載の化合物またはその薬学的に許容される塩を含む、動物におけるアポトーシスの誘導に応答性である障害を治療、改善、または予防するための医薬。
- 請求項1記載の化合物またはその薬学的に許容される塩を含む、動物における癌を治療、改善、または予防するための医薬。
- さらに抗癌剤と併用投与される、請求項4記載の医薬。
- さらにTRAIL−R1(腫瘍壊死因子関連アポトーシス誘導リガンド−受容体1)もしくはTRAIL−R2(腫瘍壊死因子関連アポトーシス誘導リガンド−受容体2)のアゴニストと併用投与される、請求項4記載の医薬。
- TRAIL−R1(腫瘍壊死因子関連アポトーシス誘導リガンド−受容体1)もしくはTRAIL−R2(腫瘍壊死因子関連アポトーシス誘導リガンド−受容体2)のアゴニストが抗体である、請求項6記載の医薬。
- 前記癌が、乳癌、卵巣癌、前立腺癌、膵臓癌、白血病、黒色腫、結腸癌、肝臓癌、多発性骨髄腫、腎細胞癌腫、急性リンパ性白血病、慢性リンパ性白血病、急性骨髄性白血病、および軟部肉腫からなる群から選ばれる、請求項4〜7のいずれかに記載の医薬。
- 医薬が、動物において血管新生を予防または阻害するための、請求項4〜7のいずれかに記載の医薬。
- 請求項1記載の化合物またはその薬学的に許容される塩と、該化合物またはその薬学的に許容される塩を動物へ投与するための使用説明書とを含む、キット。
- 下記式の化合物またはその薬学的に許容される塩:
。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US92334807P | 2007-04-13 | 2007-04-13 | |
| US60/923,348 | 2007-04-13 | ||
| PCT/US2008/060215 WO2008128171A2 (en) | 2007-04-13 | 2008-04-14 | Diazo bicyclic smac mimetics and the uses thereof |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2010523722A JP2010523722A (ja) | 2010-07-15 |
| JP2010523722A5 JP2010523722A5 (ja) | 2012-04-19 |
| JP5416089B2 true JP5416089B2 (ja) | 2014-02-12 |
Family
ID=39864640
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010503269A Active JP5416089B2 (ja) | 2007-04-13 | 2008-04-14 | ジアゾ二環式smac模倣物およびその使用 |
Country Status (23)
| Country | Link |
|---|---|
| US (2) | US8278293B2 (ja) |
| EP (1) | EP2139490B1 (ja) |
| JP (1) | JP5416089B2 (ja) |
| KR (1) | KR101081685B1 (ja) |
| CN (1) | CN101686981B (ja) |
| AU (1) | AU2008240119B2 (ja) |
| BR (1) | BRPI0810522B8 (ja) |
| CA (1) | CA2683318C (ja) |
| CY (1) | CY1115735T1 (ja) |
| DK (1) | DK2139490T3 (ja) |
| EA (1) | EA017797B1 (ja) |
| ES (1) | ES2504216T3 (ja) |
| HK (1) | HK1141456A1 (ja) |
| HR (1) | HRP20140885T1 (ja) |
| HU (1) | HUE024142T2 (ja) |
| IL (1) | IL201474A (ja) |
| MX (1) | MX2009011069A (ja) |
| NZ (1) | NZ580468A (ja) |
| PL (1) | PL2139490T3 (ja) |
| PT (1) | PT2139490E (ja) |
| SI (1) | SI2139490T1 (ja) |
| WO (1) | WO2008128171A2 (ja) |
| ZA (1) | ZA200907055B (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7296180B2 (ja) | 2019-07-15 | 2023-06-22 | パブリクノエ アクツィオネルノエ オブシチェストヴォ“ノヴォシビルスキー ザヴォッド シムコンシントラトフ” | 原子炉用の燃料アセンブリの製造方法 |
Families Citing this family (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7772177B2 (en) | 2005-05-18 | 2010-08-10 | Aegera Therapeutics, Inc. | BIR domain binding compounds |
| CA2564872C (en) | 2005-10-25 | 2010-12-21 | Aegera Therapeutics Inc. | Iap bir domain binding compounds |
| TWI504597B (zh) | 2006-03-16 | 2015-10-21 | Pharmascience Inc | 結合於細胞凋亡抑制蛋白(iap)之桿狀病毒iap重複序列(bir)區域之化合物 |
| US8278293B2 (en) * | 2007-04-13 | 2012-10-02 | The Regents Of The University Of Michigan | Diazo bicyclic Smac mimetics and the uses thereof |
| US8815927B2 (en) | 2009-10-23 | 2014-08-26 | The Regents Of The University Of Michigan | Bivalent diazo bicyclic Smac mimetics and the uses thereof |
| WO2011057099A2 (en) | 2009-11-05 | 2011-05-12 | The Uab Research Foundation | Treating basal-like genotype cancers |
| SG182724A1 (en) | 2010-02-12 | 2012-08-30 | Pharmascience Inc | Iap bir domain binding compounds |
| WO2012052758A1 (en) | 2010-10-22 | 2012-04-26 | Astrazeneca Ab | Response biomarkers for iap antagonists in human cancers |
| GB201106817D0 (en) * | 2011-04-21 | 2011-06-01 | Astex Therapeutics Ltd | New compound |
| WO2014031487A1 (en) * | 2012-08-23 | 2014-02-27 | The Regentis Of The University Of Michigan | Bivalent inhibitors of iap proteins and therapeutic methods using the same |
| US9546174B2 (en) | 2012-11-30 | 2017-01-17 | Sanford-Burnham Medical Research Institute | Inhibitor of apoptosis protein (IAP) antagonists |
| GB201311888D0 (en) | 2013-07-03 | 2013-08-14 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| GB201311891D0 (en) | 2013-07-03 | 2013-08-14 | Glaxosmithkline Ip Dev Ltd | Novel compound |
| AU2013399606B2 (en) | 2013-09-09 | 2016-09-22 | Halliburton Energy Services, Inc. | Activation of set-delayed cement compositions by retarder exchange |
| CA2974651A1 (en) | 2014-01-24 | 2015-07-30 | Children's Hospital Of Eastern Ontario Research Institute Inc. | Smc combination therapy for the treatment of cancer |
| EP3151920A4 (en) | 2014-06-04 | 2017-12-27 | Sanford-Burnham Medical Research Institute | Use of inhibitor of apoptosis protein (iap) antagonists in hiv therapy |
| WO2016079527A1 (en) | 2014-11-19 | 2016-05-26 | Tetralogic Birinapant Uk Ltd | Combination therapy |
| WO2016097773A1 (en) | 2014-12-19 | 2016-06-23 | Children's Cancer Institute | Therapeutic iap antagonists for treating proliferative disorders |
| KR20200052995A (ko) | 2015-01-20 | 2020-05-15 | 아비나스 오퍼레이션스, 인코포레이티드 | 안드로겐 수용체의 표적화된 분해를 위한 화합물 및 방법 |
| US20170327469A1 (en) | 2015-01-20 | 2017-11-16 | Arvinas, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| GB201506872D0 (en) * | 2015-04-22 | 2015-06-03 | Ge Oil & Gas Uk Ltd | Novel compounds |
| GB201506871D0 (en) | 2015-04-22 | 2015-06-03 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| US20180147202A1 (en) | 2015-06-05 | 2018-05-31 | Arvinas, Inc. | TANK-BINDING KINASE-1 PROTACs AND ASSOCIATED METHODS OF USE |
| US10772962B2 (en) | 2015-08-19 | 2020-09-15 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of bromodomain-containing proteins |
| JP2019514878A (ja) | 2016-04-20 | 2019-06-06 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | Ripk2阻害剤を含むコンジュゲート |
| GB201610147D0 (en) | 2016-06-10 | 2016-07-27 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| KR102570992B1 (ko) | 2016-11-01 | 2023-08-28 | 아비나스 오퍼레이션스, 인코포레이티드 | 타우(Tau)-단백질 표적화 프로탁(PROTAC) 및 관련 사용 방법 |
| KR102173464B1 (ko) | 2016-12-01 | 2020-11-04 | 아비나스 오퍼레이션스, 인코포레이티드 | 에스트로겐 수용체 분해제로서의 테트라히드로나프탈렌 및 테트라히드로이소퀴놀린 유도체 |
| EP3559002A4 (en) | 2016-12-23 | 2021-02-17 | Arvinas Operations, Inc. | CHEMERICAL MOLECULES TARGETING EGFR PROTEOLYSIS AND RELATED METHODS OF USE |
| CN110741004B (zh) | 2016-12-23 | 2023-10-17 | 阿尔维纳斯运营股份有限公司 | 用于迅速加速性纤维肉瘤多肽的靶向降解的化合物和方法 |
| EP3559006A4 (en) | 2016-12-23 | 2021-03-03 | Arvinas Operations, Inc. | COMPOUNDS AND METHODS FOR TARGETED DEGRADATION OF FETAL LIVER KINASE POLYPEPTIDES |
| US11173211B2 (en) | 2016-12-23 | 2021-11-16 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of rapidly accelerated Fibrosarcoma polypeptides |
| US11191741B2 (en) | 2016-12-24 | 2021-12-07 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of enhancer of zeste homolog 2 polypeptide |
| EP3573977A4 (en) | 2017-01-26 | 2020-12-23 | Arvinas Operations, Inc. | EESTROGEN RECEPTOR PROTEOLYSIS MODULATORS AND RELATED METHOD OF USE |
| BR112020007046A2 (pt) | 2017-10-19 | 2020-11-17 | Debiopharm International S.A. | produto de combinação para o tratamento do câncer |
| US11065231B2 (en) | 2017-11-17 | 2021-07-20 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of interleukin-1 receptor- associated kinase 4 polypeptides |
| WO2019177902A1 (en) | 2018-03-10 | 2019-09-19 | Yale University | Modulators of btk proteolysis and methods of use |
| JP2021521112A (ja) | 2018-04-04 | 2021-08-26 | アルビナス・オペレーションズ・インコーポレイテッドArvinas Operations, Inc. | タンパク質分解の調節因子および関連する使用方法 |
| US20200038513A1 (en) | 2018-07-26 | 2020-02-06 | Arvinas Operations, Inc. | Modulators of fak proteolysis and associated methods of use |
| EP3831811A4 (en) | 2018-07-31 | 2022-04-20 | Fimecs, Inc. | HETEROCYCLIC COMPOUND |
| WO2020041331A1 (en) | 2018-08-20 | 2020-02-27 | Arvinas Operations, Inc. | Proteolysis targeting chimeric (protac) compound with e3 ubiquitin ligase binding activity and targeting alpha-synuclein protein for treating neurodegenerative diseases |
| CN113164775A (zh) | 2018-09-07 | 2021-07-23 | 阿尔维纳斯运营股份有限公司 | 用于迅速加速性纤维肉瘤多肽的靶向降解的多环化合物和方法 |
| CN113453678A (zh) | 2018-11-26 | 2021-09-28 | 德彪药业国际股份公司 | Hiv感染的联合治疗 |
| US10870663B2 (en) | 2018-11-30 | 2020-12-22 | Glaxosmithkline Intellectual Property Development Limited | Compounds useful in HIV therapy |
| WO2020148447A1 (en) | 2019-01-17 | 2020-07-23 | Debiopharm International S.A. | Combination product for the treatment of cancer |
| US20220153722A1 (en) | 2019-04-04 | 2022-05-19 | Dana-Farber Cancer Institute, Inc. | Cdk2/5 degraders and uses thereof |
| CN110028508B (zh) * | 2019-05-16 | 2021-05-28 | 南京华威医药科技集团有限公司 | 一种抗肿瘤的重氮双环类细胞凋亡蛋白抑制剂 |
| JP2022540935A (ja) | 2019-07-17 | 2022-09-20 | アルビナス・オペレーションズ・インコーポレイテッド | タウタンパク質標的化化合物および関連する使用方法 |
| JPWO2021020585A1 (ja) | 2019-07-31 | 2021-02-04 | ||
| BR112022005624A2 (pt) | 2019-09-25 | 2022-07-12 | Debiopharm Int Sa | Regimes de dosagem para tratamento de pacientes com carcinoma de células escamosas localmente avançado |
| US20230056604A1 (en) * | 2020-01-20 | 2023-02-23 | Astrazeneca Ab | Epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of cancer |
| WO2024022275A1 (zh) * | 2022-07-29 | 2024-02-01 | 苏州科睿思制药有限公司 | Xevinapant的晶型及其制备方法和用途 |
| US11957759B1 (en) | 2022-09-07 | 2024-04-16 | Arvinas Operations, Inc. | Rapidly accelerated fibrosarcoma (RAF) degrading compounds and associated methods of use |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3989816A (en) | 1975-06-19 | 1976-11-02 | Nelson Research & Development Company | Vehicle composition containing 1-substituted azacycloheptan-2-ones |
| US4444762A (en) | 1980-04-04 | 1984-04-24 | Nelson Research & Development Company | Vehicle composition containing 1-substituted azacyclopentan-2-ones |
| CA2462200A1 (en) * | 2001-08-10 | 2003-02-20 | Palatin Technologies, Inc. | Peptidomimetics of biologically active metallopeptides |
| AU2005206929B2 (en) * | 2004-01-16 | 2008-01-24 | The Regents Of The University Of Michigan | Conformationally constrained Smac mimetics and the uses thereof |
| US7674787B2 (en) * | 2004-07-09 | 2010-03-09 | The Regents Of The University Of Michigan | Conformationally constrained Smac mimetics and the uses thereof |
| WO2006010119A2 (en) | 2004-07-09 | 2006-01-26 | Michael Norris | Cargo retaining device for use in vehicle cargo storage areas |
| PT2019671E (pt) * | 2006-05-05 | 2014-12-18 | Univ Michigan | Intermediários para a preparação de miméticos de smac bivalentes |
| US8202902B2 (en) * | 2006-05-05 | 2012-06-19 | The Regents Of The University Of Michigan | Bivalent SMAC mimetics and the uses thereof |
| US8278293B2 (en) * | 2007-04-13 | 2012-10-02 | The Regents Of The University Of Michigan | Diazo bicyclic Smac mimetics and the uses thereof |
| JP5887134B2 (ja) * | 2008-04-11 | 2016-03-16 | ザ リージェンツ オブ ザ ユニバーシティ オブ ミシガン | ヘテロアリールによって置換されている二環式のSmacの模倣物 |
| US8625740B2 (en) * | 2011-04-14 | 2014-01-07 | Morpho Detection, Inc. | System and method for correcting X-ray diffraction profiles |
-
2008
- 2008-04-14 US US12/159,249 patent/US8278293B2/en active Active
- 2008-04-14 BR BRPI0810522A patent/BRPI0810522B8/pt active IP Right Grant
- 2008-04-14 CA CA2683318A patent/CA2683318C/en active Active
- 2008-04-14 SI SI200831285T patent/SI2139490T1/sl unknown
- 2008-04-14 EA EA200901400A patent/EA017797B1/ru unknown
- 2008-04-14 AU AU2008240119A patent/AU2008240119B2/en active Active
- 2008-04-14 EP EP08745750.3A patent/EP2139490B1/en active Active
- 2008-04-14 HU HUE08745750A patent/HUE024142T2/en unknown
- 2008-04-14 MX MX2009011069A patent/MX2009011069A/es active IP Right Grant
- 2008-04-14 JP JP2010503269A patent/JP5416089B2/ja active Active
- 2008-04-14 PL PL08745750T patent/PL2139490T3/pl unknown
- 2008-04-14 ES ES08745750.3T patent/ES2504216T3/es active Active
- 2008-04-14 WO PCT/US2008/060215 patent/WO2008128171A2/en active Application Filing
- 2008-04-14 PT PT87457503T patent/PT2139490E/pt unknown
- 2008-04-14 DK DK08745750.3T patent/DK2139490T3/da active
- 2008-04-14 KR KR1020097023384A patent/KR101081685B1/ko active IP Right Grant
- 2008-04-14 NZ NZ580468A patent/NZ580468A/en unknown
- 2008-04-14 CN CN2008800186484A patent/CN101686981B/zh active Active
-
2009
- 2009-10-09 ZA ZA2009/07055A patent/ZA200907055B/en unknown
- 2009-10-12 IL IL201474A patent/IL201474A/en active IP Right Grant
-
2010
- 2010-08-20 HK HK10107967.2A patent/HK1141456A1/xx unknown
-
2012
- 2012-07-25 US US13/557,804 patent/US8664212B2/en active Active
-
2014
- 2014-09-16 HR HRP20140885AT patent/HRP20140885T1/hr unknown
- 2014-09-29 CY CY20141100792T patent/CY1115735T1/el unknown
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7296180B2 (ja) | 2019-07-15 | 2023-06-22 | パブリクノエ アクツィオネルノエ オブシチェストヴォ“ノヴォシビルスキー ザヴォッド シムコンシントラトフ” | 原子炉用の燃料アセンブリの製造方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5416089B2 (ja) | ジアゾ二環式smac模倣物およびその使用 | |
| JP5230610B2 (ja) | 二価smac模倣物およびその使用 | |
| JP5638023B2 (ja) | Mdm2の小分子阻害剤およびその使用 | |
| WO2006010118A2 (en) | Conformationally constrained smac mimetics and the uses thereof | |
| JP2007522116A (ja) | コンホメーションが制約されたSmac模倣物およびその使用 | |
| WO2005069888A2 (en) | Smac peptidomimetics and the uses thereof | |
| WO2009126947A2 (en) | Heteroaryl-substituted bicyclic smac mimetics and the uses thereof | |
| KR100887045B1 (ko) | 구조적으로 강제된 smac 유사물 및 이들의 용도 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110405 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110405 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120228 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130415 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20130418 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130709 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130731 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130919 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20131024 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20131114 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5416089 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |