発明の詳細な説明
〔関連出願に対する相互参照〕
本願は、係属中の米国仮特許出願第61/044,330号(出願日:2008年4月11日)および係属中の米国仮特許出願第61/106,887号(出願日:2008年10月20日)に対する優先権を主張する。両方の出願の全体が、参照によって本明細書に援用される。
〔連邦政府に後援を受けた研究または開発に関する記載〕
本発明は、国立衛生研究所によって付与されたR01CA109025の下に、政府の支援を受けてなされた。政府は本発明に一定の権利を有している。
〔技術分野〕
本発明は医薬品化学の分野に属する。より詳細には、本発明は、ヘテロアリールによって置換されている、SmacのN末端配列の二環式のSmac模倣物に関する。SmacのN末端配列は、アポトーシスタンパク質の阻害因子の阻害剤として機能する。また、本発明は、アポトーシス性の細胞死の誘導を細胞に生じさせるか、または生じさせ易くするこれらの模倣物の使用に関する。
〔関連技術〕
浸襲性のがん細胞の表現型は、細胞内のシグナル伝達経路の調節解除を起こす遺伝的および後遺伝的な種々の変化の結果である(Ponder, Nature 411:336 (2001))。しかし、すべてのがん細胞に関する共通点は、アポトーシスプログラムを実施できないことであり、正常なアポトーシスの機序における欠損に起因して適切なアポトーシスが起こらないことが、がんの顕著な特徴である(Lowe et al., Carcinogenesis 21:485 (2000))。最近のがん療法(化学療法剤、放射線照射および免疫療法が挙げられる)は、がん細胞におけるアポトーシスを直接的に誘導することによって作用する。したがって、正常なアポトーシスの機序における欠損に起因してがん細胞がアポトーシスプログラムを実施できないことは、化学療法、放射線照射または免疫療法によって誘導されるアポトーシスに対する耐性の上昇としばしば関連付けられている。異なる系統に属するヒトのがんが有している、アポトーシスの異常に起因する現在の処置手順に対する本来または後天性の耐性は、現在のがん療法における重要な課題である(Lowe et al., Carcinogenesis 21:485 (2000); Nicholson, Nature 407:810 (2000))。したがって、新たな分子標的に特異的な抗がん療法を計画し、開発してがん患者の生存率および生活の質を向上させることに関する現在および今後の試みは、アポトーシスに対するがん細胞の耐性を特に目的にしている戦略を含んでいなければならない。この点について、がん細胞におけるアポトーシスを直接に阻害することに中心的な役割を果たしている非常に重要な負の調節因子の標的化は、新たな抗がん薬の設計のための非常に有望な治療戦略を意味している。
アポトーシスの中心的な負の調節因子の2つの部類が同定されている。調節因子の第1の部類は、2つの強力な抗アポトーシス分子、Bcl−2およびBcl−XLタンパク質によって例示されるようなタンパク質のBcl−2ファミリーである(Adams et al., Science 281:1322 (1998);Reed, Adv. Pharmacol. 41:501 (1997);Reed et al., J. Cell. Biochem. 60:23 (1996))。がん細胞の感受性を回復させ、がん細胞の耐性を克服するためにがんにおけるBcl−2およびBcl−XLを標的化する治療戦略は、広く調査されている(Adams et al., Science 281:1322 (1998); Reed, Adv. Pharmacol. 41:501 (1997); Reed et al., J. Cell. Biochem. 60:23 (1996))。いくつもの研究室が、Bcl−2およびBcl−XLの小分子阻害剤の設計に関心を寄せている。
アポトーシスの中心的な負の調節因子の第2の部類は、アポトーシスタンパク質阻害因子(IAPs)である(Deveraux et al., Genes Dev. 13:239 (1999); Salvesen et al., Nat. Rev. Mol. Cell. Biol. 3:401 (2002))。この部類としては、XIAP、cIAP−1、cIAP−2ML−IAP、HIAP、KIAP、TSIAP、NAIP、サバイビン、リビン、ILP−2、アポロンおよびBRUCEといったタンパク質が挙げられる。IAPタンパク質は、広範なアポトーシスの刺激(化学療法剤、放射線照射および免疫療法が挙げられる)によって誘導される、がん細胞におけるアポトーシスを強く抑制する。
X連鎖IAP(XIAP)は、すべてのIAPメンバーのうちで、アポトーシスを抑制する最も強力な阻害因子である(Holcik et al., Apoptosis 6:253 (2001); LaCasse et al., Oncogene 17:3247 (1998); Takahashi et al., J. Biol. Chem. 273:7787 (1998); Deveraux et al., Nature 388:300 (1997); Sun et al., Nature 401:818 (1999); Deveraux et al., EMBO J. 18:5242 (1999); Asselin et al., Cancer Res. 61:1862 (2001))。XIAPは、細胞死受容体媒介経路およびミトコンドリア媒介経路の両方において、アポトーシスの負の制御に重要な役割を果たす。XIAPは、酵素のカスパーゼファミリーの3つのメンバー、カスパーゼ−3,−7および−9と直接に結合し、強力に阻害することによって、内因性の強力なアポトーシス阻害因子として機能する(Takahashi et al., J. Biol. Chem. 273:7787 (1998); Deveraux et al., Nature 388:300 (1997); Sun et al., Nature 401:818 (1999); Deveraux et al., EMBO J. 18:5242 (1999); Asselin et al., Cancer Res. 61:1862 (2001); Riedl et al., Cell 104:791 (2001); Chai et al., Cell 104:769 (2001); Huang et al., Cell 104:781 (2001))。XIAPは、3つのバキュロウイルスのアポトーシスタンパク質阻害因子の繰返し(BIR)ドメイン、およびC末端のRINGフィンガーを含んでいる。第3のBIRドメイン(BIR3)は、ミトコンドリア経路における開始因子カスパーゼであるカスパーゼ−9を選択的に標的とし、BIR1とBIR2とのリンカー領域は、カスパーゼ−3およびカスパーゼ−7の両方を阻害する(Salvesen et al., Nat. Rev. Mol. Cell. Biol. 3:401 (2002))。XIAPとの結合が、3つのカスパーゼのすべての活性化を回避すると同時に、カスパーゼ−9との相互作用が、アポトーシスの阻害にもっとも重要であることが明らかになっている(Ekert et al., J. Cell Biol. 152:483 (2001); Srinivasula et al., Nature 410:112 (2001))。XIAPが、複数のシグナル伝達経路が集まる点である下流のエフェクターの段階においてアポトーシスを妨げるので、XIAPを標的化する戦略は、アポトーシスに対するがん細胞の耐性を克服するために特に有効であることが分かっている(Fulda et al., Nature Med. 8:808 (2002); Arnt et al., J. Biol. Chem. 277:44236 (2002))。
各タイプのがんにおけるXIAPの正確な役割は、完全に理解されている状態には程遠いが、多種類のがんにおいて、XIAPが非常に過剰発現されており、種々の現在の治療剤に対するがん細胞の耐性に重要な役割を果たし得ることを示す証拠が増えている(Holcik et al., Apoptosis 6:253 (2001); LaCasse et al., Oncogene 17:3247 (1998))。
XIAPタンパク質は、NCI60ヒトがん細胞株のほとんどにおいて発現されていることが見出された(Tamm et al., Clin. Cancer Res. 6:1796 (2000))。78人のこれまでに未処置の患者におけるがんのサンプルの分析によって、より低レベルのXIAPを有している患者が有意に長く生存したことが示された(Tamm et al., Clin. Cancer Res. 6:1796 (2000))。XIAPは、ヒトの悪性神経膠腫において発現されることが見出された(Wagenknecht et al., Cell Death Differ. 6:370 (1999); Fulda et al., Nature Med. 8:808 (2002))。XIAPは、ヒトの前立腺がん細胞において発現されることが見出され、ミトコンドリアが活性化状態にある前立腺がん細胞のリガンド媒介性のアポトーシスを誘導する、Apo2リガンド/腫瘍壊死因子と関連するアポトーシスを阻害する(McEleny et al., Prostate 51:133 (2002); Ng et al., Mol. Cancer Ther. 1:1051 (2002))。XIAPは、患者の非小細胞肺がん(NSCLC)において過剰発現され、NSCLCの病原性に関連している(Hofmann et al., J. Cancer Res. Clin. Oncol. 128:554 (2002))。XIAPの発現、およびシスプラチンを用いた処理によるXIAPの負の制御は、ヒトの卵巣がんのシスプラチン耐性に関連している(Li et al., Endocrinology 142:370 (2001); Cheng et al., Drug Resist. Update 5:131 (2002))。まとめると、これらのデータは、XIAPが現在の治療剤に対する種々のヒトのがんの耐性における重要な役割を果たし得ることを示唆している。
血管壁の完全な状態は、血管の恒常性および器官の機能にとって必須である。内皮細胞の生存およびアポトーシスの間の動的な釣合いは、血管の発達および病的な血管新生の間におけるこの完全な状態に寄与する。cIAP−1が、血管の発達の間における内皮細胞の生存および血管の恒常性の維持に必須であることについて示されている(Santoro et al., Nature Genetics 39:1397 (2007))。したがって、cIAP−1は、胚形成、再生および腫瘍形成の間における血管新生および血管の恒常性の制御に重要な役割を果たし得る。
アポトーシスは、単一の過程というよりは、ときに相互関連しており、細胞退行性変化を導く異なるシグナル伝達経路の多くと関連がある。アポトーシスの特定の形態に関連する経路が、多くの要因(例えば、当該過程を開始させる単一または複数の損傷)に依存している。他の要因としては、特定の受容体の活性化または過剰な活性化(例えば、腫瘍壊死因子α(TNFα)、腫瘍壊死因子と関連するアポトーシス誘導リガンド(Apo2L)またはFASリガンドによる“細胞死”受容体の活性化)が挙げられる。他の決定要因は関連する細胞種である。異なるシグナル伝達経路が、Fas受容体またはTNFα受容体の活性化後における、いわゆるタイプI細胞およびタイプII細胞に関して、示されているためである。
TRAIL(Apo2L)は、2つの前アポトーシスTRAIL受容体TRAIL−R1(またはDR4)(Pan et al., Science 276:111 (1997))、またはTRAIL−R2(KILLERまたはDR5)(Wu et al., Nat. Genet. 17:141-143 (1997); Pan et al., Science 277:815 (1997); Walczak et al., EMBO J. 16:5386 (1997))のいずれかと結合することによって、がん細胞におけるアポトーシスの選択的かつ強力な誘導因子であることが示されている。TRAILによる前アポトーシス性の細胞死受容体の活性化は、適合因子としての受容体FADDからなる、細胞死を誘導するシグナル伝達複合体(DISC)の形成を誘導する(Kischkel et al., Immunity 12:611 (2000); Kuang et al., J. Biol. Chem. 275:25065 (2000))。DISCが形成されると、カスパーゼ−8は、誘導された近接性によって自己プロセシングされ、活性化される(Medema et al., EMBO J. 16:2794 (1997); Muzio et al., J. Biol. Chem. 273:2926 (1998))。
TRAILは、有望ながん療法として多大な関心を集めている(French et al., Nat. Med. 5:146 (1999))。というのも、がん細胞を選択的に標的化すると同時に、通常の正常細胞がTRAILに対して耐性を示すためである(Ashkenazi et al., Science 281:1305 (1998); Walczak et al., Nat. Med. 5:157 (1999))。TRAILの全身性の投与は、異種移植された乳がんもしくは大腸がんの殺傷、およびマウスの生存の延長において、安全かつ有効であることが証明されている(Walczak et al., Nat. Med.5:157 (1999))。TRAILが多種類のがん細胞を特異的に殺傷し得るが、他の多くがTRAIL耐性を示す(Kim et al., Clin. Cancer Res. 6:335 (2000); Zhang et al., Cancer Res. 59:2747 (1999))。さらに、がん細胞は、TRAIL−R1またはTRAIL−R2のいずれかを特異的に認識する抗体(モノクローナルまたはポリクローナル)の使用によって殺傷される。
多くの機序が、TRAIL耐性を担う潜在的な要因として同定されている。当該機序は、多くの段階(受容体段階、ミトコンドリア段階、後ミトコンドリア段階およびDISC段階が挙げられる)に存在し得る。例えば、カスパーゼ−8発現の消失(Teitz et al., Nat. Med. 6:529 (2000); Griffith et al., J. Immunol. 161:2833 (1998))、または細胞性のFLICE阻害因子タンパク質(cFLIP)の高発現(Kim et al., Clin. Cancer Res. 6:335 (2000); Zhang et al., Cancer Res. 59:2747 1999; Kataoka et al., J. Immunol. 161:3936 (1998))が、TRAILに対して耐性のがん細胞を形成する。Yeh et al.は、cFLIPを欠損している胚性のマウスの線維芽細胞が、受容体媒介性のアポトーシスに対して特に高感受性であることを示している(Yeh et al., Immunity 12:533 (2000))。cFLIPの種々のスプライスバリアントが知られており、これらとしては、短いスプライスバリアントのcFLIP−Sおよびより長いスプライスバリアントのcFLIP−Lが挙げられる。cFLIPを欠損している胚性のマウスの線維芽細胞が、cFLIP−Sのレトロウイルスを用いた導入の結果として、TRAILによって誘導されるアポトーシスに耐性になることについて示されている(Bin et al., FEBS Lett. 510:37 (2002))。
TRAILは、腫瘍選択的な細胞死受容体の活性化(すなわち腫瘍細胞にアポトーシスを好ましく誘導するが、正常細胞には誘導しない)にとって潜在的に有望な候補物を代表している。しかし、多くのがん細胞は、上述のようなアポトーシスを誘導する薬剤に耐性である。結果として、当該薬剤を用いた処置は、治療効果を実現するために、放射線照射および/または細胞毒性の化学物質を用いた併用処置をしばしば必要とする。しかし、放射線照射および化学療法の両方が、重大な副作用を有しており、一般的には可能であれば避けられる。
したがって、周囲の正常な細胞の感受性を高めることなく、アポトーシスを誘導する選択的な薬剤(例えば、TRAILまたはTRAIL受容体抗体)に対して腫瘍細胞の感受性を選択的かつ効果的に高め得る薬剤に対する必要性がある。そのような薬剤は、受容体を媒介するアポトーシス性の制がん剤の使用と一般的に関連している薬剤耐性の低下または抑制、したがって、薬剤の有効性の向上および組み合わせ療法の必要性の排除にとって有用である。
近年、Smac/DIABLO(ミトコンドリア由来の二次的なカスパーゼの活性化因子)が、アポトーシス性の刺激に応じて、ミトコンドリアから細胞質基質に放出されるタンパク質として同定された(Budihardjo et al., Annu. Rev. Cell Dev. Biol. 15:269 (1999); Du et al., Cell 102:33 (2000))。Smacは、N末端のミトコンドリア標的化配列を有して合成されている。ミトコンドリア標的化配列は、成熟ポリペプチドへの成熟化の過程においてタンパク質分解性に除去される。Smacは、XIAPおよび他のIAPと直接に相互作用し、XIAPおよび他のIAPのカスパーゼとの結合を崩壊させ、カスパーゼの活性化を容易にすることが示された。
近年、Smacのタンパク質およびペプチドと複合体を形成するXIAPのBIR3ドメインの、高解像度の実験的な三次元(3D)構造が、決定されている(Sun et al., J. Biol. Chem. 275:36152 (2000); Wu et al., Nature 408:1008 (2000))。SmacのN末端の4ペプチド(Ala−Val−Pro−IleまたはAVPI(配列番号1))は、種々の水素結合相互作用およびファンデルワールス接触を介して、XIAPのBIR3ドメイン上にある表面の溝を認識する。また、BIR3およびカスパーゼ−9の間の相互作用が、BIR3ドメイン上にある表面の同じ溝に対して、カスパーゼ−9の小サブユニットのアミノ末端上の4残基(Ala−Thr−Pro−PheまたはATPF(配列番号2))を必要とすることが示されている。種々の最近の研究によって、Smacが、BIR3ドメインの表面にある同じ結合溝に関してカスパーゼ−9と競合することによって、カスパーゼ−9の触媒的な活性を促進することが十分に証明されている(Ekert et al., J. Cell Biol. 152:483 (2001); Srinivasula et al., Nature 410:112 (2001))。
ほとんどのタンパク質−タンパク質の相互作用とは異なり、Smac−XIAPの相互作用は、Smacタンパク質における4つのアミノ酸残基、およびXIAPのBIR3における十分に明確な表面の溝のみによって媒介されている。XIAPのBIR3に対するSmacのペプチドAVPI(配列番号1)のKd値(Kd=0.4μM)は、成熟Smacタンパク質(Kd=0.42μM)と実質的に同じである。十分に明確な表面のこの溝は、XIAPに対するSmacの結合を模倣する非ペプチド性の薬剤様の小分子の設計にとって理想的である。
細胞内輸送を容易にする輸送ペプチドに繋がれている、SmacのN末端の最初の4アミノ酸残基(AVPI(配列番号1))からなる細胞透過性のSmacペプチドは、細胞死受容体リガンドまたは細胞毒性薬剤によって誘導されるアポトーシスに対する、インビトロにおける種々の腫瘍細胞およびインビボにおける悪性の神経膠腫細胞の感受性を高めることが近年、示されている(Fulda et al., Nature Med. 8:808 (2002))。重要なことに、このSmacペプチドが、頭蓋内に異種移植された悪性の神経膠腫モデルにおけるApo2L/TRAILの抗腫瘍活性をインビボにおいて強く増強した。確立された腫瘍の完全な根絶およびマウスの生存は、SmacペプチドおよびApo2/TRAILとの組み合わせ処置によってのみ実現された。意義深いことに、Smacペプチドは、正常な脳組織に対する検出可能な毒性を有していない。
また、最近なされた独立した第2の研究によって、細胞内輸送を容易にする輸送ペプチドに繋がれている、SmacのN末端の最初の4〜8アミノ酸残基からなるペプチドが、MCF−7および他のヒト乳がん細胞株におけるアポトーシスの誘導、および種々の化学療法薬剤(パクリタキセル、エトポシド、SN−38およびドキソルビシンが挙げられる)の長期的な抗増殖性の作用を増強することが示された(Arnt et al., J. Biol. Chem. 277:44236 (2002))。この研究によって、XIAPおよびcIAP−1が細胞におけるこれらのペプチドにとっての主要な分子標的であると十分に示された。
第3の研究によって、ポリアルギニンに繋がれているN末端の最初の7残基が、非小細胞肺がんH460細胞におけるアポトソーム活性を回復させ、アポトーシス耐性を逆転させることが示された(Yang et al., Cancer Res. 63:831 (2003))。XIAPは、H460細胞におけるアポトソーム活性の欠損およびカスパーゼ活性の抑制を担っていることが示された。化学療法と組み合わせて使用される場合に、細胞透過性のSmacペプチドは、マウスにおけるわずかな毒性をともなって、インビボにおける腫瘍の成長を退行させた。まとめると、最近のこれらの独立した研究によって、強力かつ安定な細胞透過性のSmacの模倣物が、ヒトの乳がんおよび他の種類のがんの処置にとって優れた治療的な潜在性を有し得ると強く示唆された。
ペプチドに基づく阻害剤は、IAPの抗アポトーシス性の機能、および化学療法剤に対するがん細胞の応答におけるIAPの役割を解明するための有用なツールである。しかし、ペプチドに基づく阻害剤は、潜在的に有用な治療剤としての固有の制限を有している。これらの制限としては、細胞透過性に乏しいこと、およびインビボにおける安定性に乏しいことが挙げられる。実際に、Smacに基づくペプチド阻害剤を用いているこれらの3つの研究において、ペプチドは、それらを比較的に細胞透過性にするための輸送ペプチドと融合させる必要があった。
ペプチドに基づく阻害剤の固有の制限を克服するために、本発明は、二環式の骨格に芳香族複素環の置換を有しており、立体配置が制限されているSmac模倣物を提供する。
〔発明の概要〕
がん細胞またはそれらの支持細胞が遺伝的な障害またはアポトーシスの誘導要因(例えば、抗がん剤および放射線照射)に対する曝露に応じてアポトーシスを起こさないことは、がんの発症および進行の主な要因であると、一般的に受け入れられている。がん細胞またはそれらの支持細胞(例えば、腫瘍の脈管構造における新生血管細胞)におけるアポトーシスの誘導は、今日の市場または実践における有効ながん治療薬または放射線療法の実質的にすべてに関して、作用の共通する機序であると考えられている。細胞がアポトーシスを起こさないことの理由の1つは、IAPの増強された発現および蓄積である。
本発明は、がん、またはアポトーシスの調節不全と関連している他の過剰増殖性の障害もしくは疾患に罹っている動物の、IAPの(複数の)機能を阻害する治療有効量の(複数の)薬剤(例えば、小分子)に対する曝露が、異常細胞または支持細胞(生存し続けているこれらの細胞がIAPの過剰な活性または過剰発現に依存している)を完全に殺傷するか、および/または当該細胞を、がん治療薬または放射線療法の細胞死を誘導する活性に対して、より感受性の高い集団にさせることを意図している。本発明は、IAPの機能に依存してがん細胞におけるアポトーシスを誘導する単独療法として投与される場合か、または他の細胞死を誘導するがん治療薬もしくは放射線療法と時間的な関連を有して投与される場合に、IAPの阻害剤が、多種類のがんの処置に関してこれまでに対処されていない必要性を満たして、単独のがん治療薬もしくは放射線療法のみを用いて処置される動物における細胞の対応する割合と比べて、より多くの割合のがん細胞または支持細胞にアポトーシスプログラムを行わせ易くさせることを意図している。
また、本発明は、IAP(例えば、cIAP−c)の(複数の)機能を阻害する(複数の)薬剤(例えば、小分子)の治療有効量を用いた、内皮細胞に関連する疾患(例えば、腫瘍、血管新生、網膜症およびアテローム性動脈硬化症)に罹っている動物の処置が、血管新生を妨げ得るか、または阻害し得、異常な状態における血管の発達の間に血管の恒常性を崩壊させ得ることを意図している。本発明の化合物を用いて処置され得る特定の疾患としては、黄斑変性症、リューマチ性関節炎、乾癬、糖尿病性網膜症、未熟児網膜症、角膜移植片拒絶反応、血管新生緑内障、水晶体後繊維増殖症、皮膚潮紅、オスラー−ウェーバー症候群、心筋血管新生、プラーク血管新生、毛細血管拡張症、血友間接症、血管線維腫、創傷肉芽形成、腸管癒着症、強皮症および肥厚性瘢痕が挙げられる。
出願人は、二環式の骨格にヘテロアリール置換を有している特定のSmac模倣物が、がん細胞株において予想外のインビトロにおける有効性を示すことを見出している。したがって、本発明の化合物は、アポトーシス性の細胞死の誘導に対して応答性の広範な疾患の処置に有用であると予想される。
本発明の特定の実施形態において、本発明の化合物の治療有効量および一連の抗がん剤もしくは放射線照射を用いた動物の組み合わせ処置が、単独の当該化合物または抗がん剤/放射線照射を用いて処理された動物と比べて、そのような動物においてより大きな腫瘍の応答および臨床的な利益を生み出すと予想される。言い換えると、本発明の化合物が、IAPを発現するすべての細胞のアポトーシスの閾値を低下させると考えられるので、抗がん剤/放射線照射のアポトーシスを誘導する作用に応じてアポトーシスプログラムを首尾よく実施する細胞の割合が増加する。代替的に、本発明の化合物は、より少ない(したがってより毒性が低く、より許容可能な)用量の抗がん剤および/または線量の放射線線量を用いて、従来の用量の抗がん剤/線量の放射線と同じ腫瘍の応答/臨床的な利益を生じさせ得ると予想される。認可されているすべての抗がん剤および放射線処置に関する使用量が公知であるので、本発明は、本発明の化合物とそれらとの種々の組み合わせを意図している。また、本発明の化合物がIAPを阻害することによって少なくとも部分的に作用するので、治療有効量の当該化合物に対するがん細胞および支持細胞の曝露は、抗がん剤または放射線療法に応じてアポトーシスプログラムを実施する細胞の試みと一致させるために、時間を合わせられ得る。したがって、いくつかの実施形態において、特定の時間的な関連をともなう本発明の化合物の投与は、特に有効な治療方法を提供すると予想される。
本発明は、IAPタンパク質の活性の阻害、および特にアポトーシスの誘導に対する細胞の感受性の向上に有用なSmac模倣物に関する。一実施形態において、Smac模倣物は、式I:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、A2が存在しない場合にVがOであり;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキル、およびアラルキルからなる群から選択され;
Yが、CON(R1)、N(R1)CO、C(O)O、OC(O)、(CH2)1−3、任意に置換されているアリール、および任意に置換されているヘテロアリールからなる群から選択され、ここで、1つ以上のCH2基が、O、SまたはNR1によって置換され得;
Zが(CR2aR2b)rであり;
Dが(CR3aR3b)n−U−(CR4aR4b)mであり;
Uが、CR5aR5bおよびNR6からなる群から選択され;
Jが、(CR7aR7b)p−L−(CR8aR8b)qであり;
Tが、任意に置換されているヘテロアリールであり;
n、m、qおよびpが独立して、0−5からなる群から選択され;
rが、0−3であり;
R1が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R2a、R2b、R3a、R3b、R4a、R4b、R5a、R5b、R7a、R7b、R8aおよびR8bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロおよびCOR9からなる群から選択され;
Lが、O、S、NR1、NCOR9、CR7aR7b、C=O、C=SおよびC=NR1からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択される。
他の実施形態において、Smac模倣物は、式II:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1、A2、V、Z、W、X、Y、D、JおよびTは、式Iについて説明した意味と同じである。
他の実施形態において、Smac模倣物は、式III:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1、A2、V、Z、W、X、Y、UおよびTは、式Iについて説明した意味と同じであり、mは1または2である。
他の実施形態において、Smac模倣物は、式IV:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1、A2、XおよびUは、式Iについて説明した意味と同じであり、mは1または2である。
他の実施形態において、Smac模倣物は、式V:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1、A2、X、UおよびTは、式Iについて説明した意味と同じであり、mは1または2である。
他の実施形態において、Smac模倣物は、式VI:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXは、任意に置換されているアルキルであり、Tは、任意に置換されているヘテロアリールである。
他の実施形態において、Smac模倣物は、式VII:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXは、任意に置換されているアルキルであり、R9は、任意に置換されているアルキルまたはアラルキルであり、Tは、任意に置換されているヘテロアリールである。
他の実施形態において、Smac模倣物は、式I〜VIIの化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、Tは、
(ここで、Qが、O、SまたはNR12であり、R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールまたは任意に置換されているヘテロシクロであり、R10a、R10b、R11a、R11b、R11cおよびR11dが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドまたはスルホンアミドであり、Z1、Z2およびZ3が独立して、CR11eまたはNであり、ここで、Z1、Z2およびZ3の少なくとも1つがCR11eであり、Z1、Z2およびZ3の少なくとも1つがNであり、R11eが、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドまたはスルホンアミドである)
である。
他の実施形態において、Smac模倣物は、式I〜VIIの化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、Tは、
(ここで、Q、Z1、Z2、Z3、R10a、R10b、R11a、R11b、R11cおよびR11dが、上述のように説明した意味と同じである)
である。
他の実施形態において、Smac模倣物は、式VIII:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXが、任意に置換されているアルキルであり、mが1または2であり、Qが、O、SまたはNR12であり、R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールまたは任意に置換されているヘテロシクロであり、R10aおよびR10bが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択される。
他の実施形態において、Smac模倣物は、式IX:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXが、任意に置換されているアルキルであり、mが1または2であり、Qが、O、SまたはNR12であり、R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールまたは任意に置換されているヘテロシクロであり、R11a、R11b、R11cおよびR11dが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択される。
他の実施形態において、Smac模倣物は、式X:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXが、任意に置換されているアルキルであり、R9が、任意に置換されているアルキル、またはアラルキルであり、mが1または2であり、Qが、O、SまたはNR12であり、R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールまたは任意に置換されているヘテロシクロであり、R10aおよびR10bが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択される。
他の実施形態において、Smac模倣物は、式XI:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXが、任意に置換されているアルキルであり、R9が、任意に置換されているアルキル、またはアラルキルであり、mが1または2であり、Qが、O、SまたはNR12であり、R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールまたは任意に置換されているヘテロシクロであり、R11a、R11b、R11cおよびR11dが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択される。
本発明は、IAPタンパク質の阻害剤である、式I〜XIaによって表される化合物に関する。本発明は、細胞におけるアポトーシスの誘導、および血管形成の阻害のための本発明の化合物の使用に関する。上記化合物は、アポトーシス性の細胞死の誘導に応答する障害の処置、寛解または抑制に有用である。当該障害は、例えば、アポトーシスの調節不全によって特徴付けられる障害(がんなどの過剰増殖性の疾患)である。ある実施形態において、上記化合物は、がん療法に対する耐性によって特徴付けられるがん(例えば化学抵抗性、放射線抵抗性およびホルモン抵抗性などを示すがん)の処置、寛解または抑制に使用され得る。他の実施形態において、上記化合物は、それらを必要としている動物における血管形成を抑制または阻害する方法として使用され得る。本発明は、細胞にアポトーシスを誘導するか、またはアポトーシスの誘導因子に対する細胞の感受性を高めるために、治療有効量の式I〜XIaの化合物を含んでいる薬学的組成物を提供する。
本発明は、式Iの化合物、および当該化合物を動物に投与するための取扱説明書を含んでいるキットをさらに提供する。上記キットは、他の治療剤(例えば、抗がん剤またはアポトーシス調節剤)を必要に応じて含み得る。
また、本発明は、式XII:
の化合物を調製する方法であって、
(a)式XIII:
の化合物をアンモニアと縮合させて、式XIV:
の化合物を生成させる工程、
(b)式XIVの化合物を式XV:
の化合物に転化させる工程、
(c)式XVの化合物を式XVI:
(ここで、L2は離脱基である)
の化合物と縮合させて、式XVII:
の化合物を生成させる工程、
(b)式XVIIの化合物を環化させて式XIIの化合物を生成させる工程を包含している方法を提供する。
ここで、
R13が、N(H)P1、および、
からなる群から選択され;
P1が、アミン保護基であり;
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、A2が存在しない場合にVがOであり;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキルおよびアラルキルからなる群から選択され;
Yが、CON(R1)、N(R1)CO、C(O)O、OC(O)、(CH2)1−3、任意に置換されているアリール、および任意に置換されているヘテロアリールからなる群から選択され、ここで、1つ以上のCH2基が、O、SまたはNR6によって置換され得;
Zが、(CR2aR2b)rであり;
Uが、CR5aR5bおよびNR1からなる群から選択され;
mが、1または2であり;
rが、0−3であり;
R1が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R2a、R2b、R5aおよびR5bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールおよび任意に置換されているヘテロシクロからなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、およびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R10aおよびR10bが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択される。
また、本発明は、式XVIII:
の化合物を調製する方法であって、
(a)式XIII:
の化合物を、式XIX:
の化合物と縮合させて、式XX:
の化合物を生成させる工程、および、
(b)式XXの化合物を環化させて、式XVIIIの化合物を生成させる工程を包含している方法を提供する。
ここで、
R13が、N(H)P1および、
からなる群から選択され;
P1が、アミン保護基であり;
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、A2が存在しない場合にVがOであり;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキル、およびアラルキルからなる群から選択され;
Yが、CON(R1)、N(R1)CO、C(O)O、OC(O)、(CH2)1−3、任意に置換されているアリール、および任意に置換されているヘテロアリールからなる群から選択され、ここで、1つ以上のCH2基が、O、SまたはNR1によって置換され得;
Zが、(CR2aR2b)rであり;
Uが、CR5aR5bおよびNR6からなる群から選択され;
mが、1または2であり;
rが、0−3であり;
R1が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R2a、R2b、R5aおよびR5bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、およびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R11a、R11b、R11cおよびR11dが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択され;
R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択される。
また、本発明は、式XXI:
の化合物を調製する方法であって、
式XXIII:
の化合物を、式XXIV:
(ここで、L1は離脱基である)
の化合物と縮合させて、式XXIの化合物を生成させる工程を包含している方法を提供する。
ここで、
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、A2が存在しない場合にVがOであり;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキルおよびアラルキルからなる群から選択され;
Zが、(CR2aR2b)rであり;
Uが、CR5aR5bおよびNR6からなる群から選択され;
mが、1または2であり;
rが、0−3であり;
R2a、R2b、R5aおよびR5bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、およびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
Tが、任意に置換されているヘテロアリールである。
〔図面の簡単な説明〕
図1は、XIAP BIR3ドメインに対するSmac模倣物の競合的な結合の曲線を示しているグラフである。
図2は、cIAP1ドメインに対するSmac模倣物の競合的な結合の曲線を示しているグラフである。
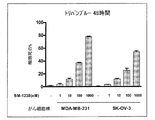
図3は、ヒト乳がんのMDA−MB−231細胞株およびヒト卵巣がんのSK−OV−3細胞株における、SM−1238による細胞死の誘導を示している棒グラフである。
〔発明の詳細な説明〕
本発明は、式I〜XIaによって表される、立体配置が制限されている化合物に関する。当該化合物は、Smac模倣物であり、IAPの阻害剤として機能する。式I〜XIaのSmac模倣物は、がん細胞株においてインビトロの強力な阻害剤活性を示す。本発明は、式I〜XIaの化合物の単独、またはアポトーシスの誘導剤との組み合わせと細胞を接触させることを包含している、アポトーシスの誘導剤に対する細胞の感受性を高める方法、ならびに細胞にアポトーシスを誘導する方法に関する。本発明は、アポトーシスの誘導に応答する動物の障害を処置、寛解または抑制する方法であって、式I〜XIaの化合物およびアポトーシス誘導剤を動物に投与することを包含している方法にさらに関する。当該障害としては、アポトーシスの調節不全によって特徴付けられる障害およびIAPの過剰発現によって特徴付けられる障害が挙げられる。本発明は、式I〜XIaの化合物を動物に投与することを包含している、それらを必要としている動物における血管形成を抑制または阻害する方法にさらに関する。
本明細書に使用されるとき、“IAP”という用語は、アポトーシスタンパク質の阻害剤のファミリーに属する任意の公知のメンバー(XIAP、cIAP−1、cIAP−2ML−IAP、HIAP、TSIAP、KIAP、NIAP、サバイビン、リビン、ILP−2、アポロンおよびBRUCEが挙げられるが、これらに限定されない)を指す。
本明細書に使用されるとき、“IAPの過剰発現”という用語は、IAPタンパク質をコードしているmRNAを通常のレベルにおいて発現しているか、または通常のレベルのIAPタンパク質を有している、病的ではない対応する類似の細胞と比べて、細胞内における(複数の)IAPタンパク質をコードしているmRNAの高いレベル(例えば、異常なレベル)、および/または(複数の)IAPタンパク質の高いレベルを指す。IAPタンパク質をコードしているmRNAの、細胞におけるレベル、またはIAPタンパク質のレベルを検出する方法としては、IAPタンパク質の抗体を用いたウエスタンブロッティング、免疫組織化学的方法、および核酸増幅法もしくは直接なRNAの検出方法が挙げられるが、これらに限定されない。当該細胞がIAPタンパク質を過剰発現していることを判定するために、細胞におけるIAPタンパク質の絶対的なレベルが重要であるように、当該細胞内における他の前アポトーシスシグナル伝達分子(例えば、前アポトーシス性のBcl−2ファミリーのタンパク質)に対するIAPタンパク質の相対的なレベルも重要である。これらの2つのバランスが、前アポトーシスシグナル伝達分子(IAPタンパク質のレベルではなく)が細胞にアポトーシスプログラムを実行させ、細胞を死に至らしめるに十分な場合に、当該細胞は、生存に関してIAPタンパク質に依存している。当該細胞において、阻害するために有効量のIAPタンパク質の阻害剤に対する曝露によって、十分に、細胞にアポトーシスプログラムを実行させ、細胞を死に至らしめる。したがって、“IAPの過剰発現”という用語はまた、前アポトーシスシグナルおよび抗アポトーシスシグナルの相対的なレベルに起因して、IAPタンパク質の機能を阻害する化合物の阻害に有効な量に応じたアポトーシスを起こす細胞を指す。
本明細書に使用されるときに、“抗がん剤(anticancer agent)”および“抗がん剤(anticancer drug)”は、がん(例えば動物における)などの過剰増殖性の疾患の処置に使用される、任意の治療剤(例えば、化学療法化合物および/または分子治療化合物)、放射線療法または外科的処置を指す。
本発明に使用されるときに、“プロドラッグ”という用語は、(例えば酵素的、生理学的、力学的、電磁気的に)活性な薬剤を放出するか、またはプロドラッグを活性な薬剤に変換するために、標的の生理学的システム内における(例えば、自発的または酵素的な)生体内変換を必要とする、親の“薬剤”分子の、薬理学的に不活性な誘導体を指す。プロドラッグは、安定性、毒性、特性の欠乏、または限られた生体適合性と関連する問題を克服するように設計されている。例示的なプロドラッグは、活性な薬剤分子そのものおよび化学的なマスキング基(例えば、薬剤の活性を可逆的に抑制する基)を含んでいる。いくつかの好ましいプロドラッグは、代謝条件下において切断可能な基を有している化合物の変種または誘導体である。例示的なプロドラッグは、それらが生理学的条件において加溶媒分解を受ける場合、または酵素的な分解もしくは他の生化学的な転換(例えば、リン酸化、水素化、脱水素化、糖付加)を受ける場合に、インビボまたはインビトロにおいて薬理的に活性になる。プロドラッグは、哺乳類の器官における可溶性、組織適合性または徐放性といった利点をしばしば示す。例えば、Bundgard, Design of Prodrugs, pp. 7-9, 21-24, Elsevier, Amsterdam (1985); and Silverman, The Organic Chemistry of Drug Design and Drug Action, pp. 352-401, Academic Press, San Diego, CA (1992)を参照すればよい。通常のプロドラッグとしては、酸の誘導体(例えば、親の酸および好適なアルコール(例えば、低級アルカノール)の反応によって調製されるエステル)、親の酸の化合物およびアミンの反応によって調製されるアミド、またはアクリル化された塩基性誘導体(例えば、低級アルキルアミド)を形成するために反応させた塩基性基が挙げられる。
本明細書に使用されるとき、“薬学的に受容可能な塩”という用語は、標的の動物(例えば、哺乳類)において生理学的に許容される、本発明の化合物の任意の塩(例えば、酸または塩基との反応によって得られる)を指す。本発明の化合物の塩は、有機または無機の酸および塩基から誘導され得る。酸の例としては、塩酸、臭化水素酸、硫酸、硝酸、過塩素酸、フマル酸、マレイン酸、リン酸、グリコール酸、乳酸、サリチル酸、コハク酸、トルエン−p−スルホン酸、酒石酸、酢酸、クエン酸、メタンスルホン酸、エタンスルホン酸、蟻酸、安息香酸、マロン酸、スルホン酸、ナフタレン−2−スルホン酸、およびベンゼンスルホン酸などが挙げられるが、これらに限定されない。それ自身は薬学的に受容可能ではないが、シュウ酸といった他の酸が、本発明の化合物を得るための中間体として有用な塩、および薬学的に受容可能なそれらの酸付加塩の調製に採用され得る。
塩基の例としては、アルカリ金属(例えば、ナトリウム)の水酸化物、アルカリ土類金属(例えばマグネシウム)の水酸化物、アンモニアおよびNW4 +(ここで、WはC1−4アルキルなどである)などが挙げられるが、これらに限定されない。
塩の例としては、酢酸塩、アジピン酸塩、アルギン酸塩、アスパラギン酸塩、安息香酸塩、ベンゼンスルホン酸塩、重硫酸塩、酪酸塩、クエン酸塩、ショウノウ酸塩、キャンホルスルホン酸塩、シクロペンタンプロピオン酸塩、ジグルコン酸塩、ドデシル硫酸塩、エタンスルホン酸塩、フマル酸塩、フルコヘプタン酸塩、グリセロリン酸塩、ヘミ硫酸塩、ヘプタン酸塩、ヘキサン酸塩、塩化物、臭化物、ヨウ化物、2−ヒドロキシ鉛丹スルホン酸塩、乳酸塩、マレイン酸塩、メシラート、メタンスルホン酸塩、2−ナフタレンスルホン酸塩、ニコチン酸塩、シュウ酸塩、パルモエート(palmoate)、ペクチン酸塩、過硫酸塩、フェニルプロピオン酸塩、ピクリン酸塩、ピバリン酸塩、プロピオン酸塩、コハク酸塩、酒石酸塩、チオシアン酸塩、トシレートおよびウンデカン酸塩などが挙げられるが、これらに限定されない。塩の他の例としては、好適なカチオン(例えば、Na+、NH4 +およびNW4 +(ここで、WはC1−4アルキルである))と化合された、本発明の化合物のアニオンなどが挙げられる。治療用途に関して、本発明の化合物の塩は、薬学的に受容可能であると考えられる。しかし、薬学的に受容可能ではない酸および塩基の塩はまた、例えば薬学的に受容可能な化合物の調製または精製における、用途を見出され得る。
本明細書に使用されるとき、“治療有効量”という用語は、障害の1つ以上の症状の寛解を生じるか、障害の進行を抑制するか、または障害の退行を引き起こすために十分な治療剤の量を指す。例えばがんの処置に関して、治療有効量は、少なくとも5%、好ましくは少なくとも10%、少なくとも15%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、少なくとも45%、少なくとも50%、少なくとも55%、少なくとも60%、少なくとも65%、少なくとも70%、少なくとも75%、少なくとも80%、少なくとも85%、少なくとも90%、少なくとも95%、または少なくとも100%だけ、腫瘍の成長の速度を低下させるか、腫瘍の重量を低下させるか、転移の数を低下させるか、腫瘍が進行に要する時間を延ばすか、または生存期間を延ばす治療剤の量を好ましく指す。
本明細書に使用されるとき、“感受性の向上”および“感受性を高めること”は、第1の薬剤(例えば、式Iの化合物)の投与を介して、動物または動物内の細胞を、第2の薬剤の生物学的な作用(例えば、細胞性機能の一側面(細胞分裂、細胞増殖、増殖、浸潤、血管形成またはアポトーシスが挙げられるが、これらに限定されない)の促進または妨害)に対して、より感受性または応答性の高い状態にすることを指す。標的細胞に対する感受性を高める第1の薬剤の作用は、第1の薬剤の非投与と第1の薬剤の投与との間に見られる、第2の薬剤の投与によって観察される差異であって、生物学的な目的の作用(例えば、細胞性機能の一側面(細胞分裂、細胞増殖、増殖、浸潤、血管形成またはアポトーシスが挙げられるが、これらに限定されない)の差異として測定され得る。感受性が向上された細胞の応答は、第1の薬剤の非投与時における応答を、少なくとも10%、少なくとも20%、少なくとも30%、少なくとも40%、少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも100%、少なくとも150%、少なくとも200%、少なくとも250%、少なくとも300%、少なくとも350%、少なくとも400%、少なくとも450%、または少なくとも500%だけ超えて増強され得る。
本明細書に使用されるとき、“アポトーシスの調節不全”という用語は、アポトーシスを介して細胞死に至る細胞の能力に関する任意の異常(例えば素因)を指す。アポトーシスの調節不全は、種々の障害と関連しているか、または当該障害によって誘導される。当該障害としては、例えば、自己免疫疾患(例えば、紅斑性狼蒼、リューマチ性の関節炎、移植片対宿主の疾患、重症筋無力症またはシェーグレン症候群)、過剰増殖性の疾患(例えば、腫瘍、B細胞リンパ腫またはT細胞リンパ腫)、ウイルス感染(例えば、ヘルペス、パピローマまたはHIV)、および他の障害(例えば、骨関節炎およびアテローム性動脈硬化症)が挙げられる。ここで留意されるべきことは、調節不全がウイルス感染によって誘導されているか、またはウイルス感染と関連している場合に、ウイルス感染は、調節不全が生じているときもしくは観察されるときに、検出され得るか、または検出され得ないことである。すなわち、ウイルスに誘導される調節不全は、ウイルス感染の症状の消失の後にさえ生じ得る。
本明細書に使用されるとき、“血管形成”という用語は、組織または器官における新たな血管の生成を意味する。本明細書に使用されるとき、“抗血管形成”という用語は、新たな血管の成長の抑制または縮小を指す。本発明の化合物を用いて処置され得る、血管形成と関連している疾患または障害としては、黄斑変性症、リューマチ性の関節炎、乾癬、糖尿病性網膜症、未熟児網膜症、角膜移植片拒絶反応、血管新生緑内障、水晶体後繊維増殖症、皮膚潮紅、オスラー−ウェーバー症候群、心筋血管新生、プラーク血管新生、毛細血管拡張症、血友間接症、血管線維腫、創傷肉芽形成、腸管癒着症、強皮症および肥厚性瘢痕が挙げられる。
本明細書に使用されるとき、“過剰増殖性の疾患”という用語は、動物における増殖細胞の局在化した集団が正常な増殖の通常の制限によって管理されていない任意の障害を指す。過剰増殖性の疾患の例としては、がん(例えば、腫瘍、新生物、およびリンパ腫など)または自己免疫の障害が挙げられるが、これらに限定されない。新生物は、浸潤または転移しない場合に良性であり、いずれかを生じる場合に悪性である。細胞の異常増殖は、構造または機能に重大な変化を生じることのない、組織または器官における細胞数の増加を含む、細胞増殖の一形態である。異形成は、完全に分化したある種類の細胞が分化した他の種類の細胞に置き換わる、制御された細胞成長の一形態である。他の実施形態において、過剰増殖性の疾患は、リューマチ性の関節炎、炎症性の腸疾患、骨関節炎、平滑筋腫、腺腫、脂肪腫、血管腫、線維腫、血管閉塞症、再狭窄、アテローム性動脈硬化症、前新生物性の障害(例えば、腺腫性の細胞異常増殖および前立腺における表皮内の新生物)、上皮内がん、口腔の毛髪状白斑または乾癬である。
活性化したリンパ球様細胞の病的な増殖は、自己免疫疾患または慢性の炎症性障害をしばしば生じる。本明細書に使用されるとき、“自己免疫疾患”は、生体が生体自身の分子、細胞または組織を認識する抗体または免疫細胞を生成している任意の障害を指す。自己免疫疾患の非限定的な例としては、自己免疫性溶血性貧血、自己免疫性肝炎、ベルジェ病もしくはIgA腎症、セリアック病、慢性の疲労症候群、クローン氏病、皮膚筋炎、線維筋肉痛、移植片対宿主疾患、グレイブズ病、橋本甲状腺炎、突発性血小板減少性紫斑病、扁平苔癬、多発性硬化症、重症筋無力症、乾癬、リューマチ熱、リュウマチ性関節炎、強皮症、シェーグレン症候群、全身性紅斑性狼瘡、1型糖尿病、潰瘍性大腸炎および白斑などが挙げられる。
本明細書に使用されるとき、“新生物性の疾患”は、良性(非がん性)または悪性(がん性)のいずれかである細胞の、任意の異常な成長を指す。
本明細書に使用されるとき、“抗新生物剤”という用語は、標的化された(例えば悪性の)新生物の増殖、成長または蔓延を遅らせる任意の化合物を指す。
本明細書に使用されるとき、“抑制する”、“抑制すること”および“抑制”という用語は、動物における病的な細胞(例えば、過剰増殖の細胞または新生物性の細胞)の発生を減らすことを指す。抑制は、例えば対象における病的な細胞の完全な非存在を達成し得る。また、抑制は、対象における病的な細胞の発生が本発明を用いない場合よりも減らされるように、部分的であり得る。
本明細書に使用されるとき、“アポトーシスの調節剤”という用語は、アポトーシスの調節(例えば阻害、減少、増強、促進)に関与する薬剤を指す。一実施形態において、アポトーシスの調整剤はアポトーシスの誘導剤である。本明細書に使用されるとき、“アポトーシスの誘導剤”という用語は、細胞(例えば、がん細胞)におけるアポトーシスを誘導して、アポトーシスプログラムを当該細胞に、より実行させ易くする薬剤を指す。一実施形態において、アポトーシスを誘導する薬剤は抗がん剤である。アポトーシスの誘導剤の例としては、デスドメインを含んでいるタンパク質が挙げられる。当該デスドメインとしては、Fas/CD95、TRAMP、TNF R1、DR1、DR2、DR3、DR4、DR5、DR6、FADDおよびRIPが挙げられるが、これらに限定されない。他のアポトーシスの調節剤の例としては、TNFα、Fasリガンド、Fas/CD95に対する抗体および他のTNFファミリー受容体、TRAIL(Apo2リガンドまたはApo2L/TRAILとしても知られている)、TRAIL−R1またはTRAIL−R2のアゴニスト(例えば、モノクローナルまたはポリクローナルのアゴニスト性の抗体)、Bcl−2、p53、BAX、BAD、Akt、CAD、PI3キナーゼ、PP1ならびにカスパーゼタンパク質が挙げられるが、これらに限定されない。調節剤としては、TNFファミリー受容体およびTNFファミリーリガンドのアゴニストおよびアンタゴニストが広く挙げられる。アポトーシスの調節剤は、可溶性であり得るか、または膜結合性(例えばリガンドまたは受容体)であり得る。好ましいアポトーシスの調節剤は、アポトーシスの誘導剤(例えば、TNFリガンドもしくはTNF関連リガンド、特にTRAMPリガンド、Fas/CD95リガンド、TNFR−1リガンドまたはTRAIL)である。
本発明のIAPの阻害剤は、一般式I:
を有しているSmac模倣物の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、A2が存在しない場合にVがOであり;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキル、およびアラルキルからなる群から選択され;
Yが、CON(R1)、N(R1)CO、C(O)O、OC(O)、(CH2)1−3、任意に置換されているアリール、および任意に置換されているヘテロアリールからなる群から選択され、ここで、1つ以上のCH2基が、O、SまたはNR1によって置換され得;
Zが(CR2aR2b)rであり;
Dが(CR3aR3b)n−U−(CR4aR4b)mであり;
Uが、CR5aR5bおよびNR6からなる群から選択され;
Jが、(CR7aR7b)p−L−(CR8aR8b)qであり;
Tが、任意に置換されているヘテロアリールであり;
n、m、qおよびpが独立して、0−5からなる群から選択され;
rが、0−3であり;
R1が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R2a、R2b、R3a、R3b、R4a、R4b、R5a、R5b、R7a、R7b、R8aおよびR8bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロおよびCOR9からなる群から選択され;
Lが、O、S、NR1、NCOR9、CR7aR7b、C=O、C=SおよびC=NR1からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択される。
他の実施形態において、Smac模倣物は、式Iの化合物であり、ここで、nは1であり、mは1または2であり、pは0であり、LはCH2であり、qは1である。他の実施形態において、Smac模倣物は式Iの化合物であり、ここで、R3a、R3b、R4a、R4b、R7a、R7b、R8aおよびR8bは水素である。他の実施形態において、Smac模倣物は式Iの化合物であり、ここで、YはCON(H)であり、WはCHであり、rは0であり、VはNである。他の実施形態において、Smac模倣物は式Iの化合物であり、ここで、YはCON(H)であり、WはCHであり、rは1であり、VはNであり、R2aは任意に置換されているアルキルであり、R2bは水素である。
特定の他の実施形態において、Smac模倣物は、式II:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1、A2、V、Z、W、X、Y、D、JおよびTは、式Iについて説明した意味と同じである。
一実施形態において、Smac模倣物は式IIの化合物であり、ここで、nは1であり、mは1または2であり、pは0であり、LはCH2であり、qは1である。他の実施形態において、Smac模倣物は式IIの化合物であり、ここで、R3a、R3b、R4a、R4b、R7a、R7b、R8aおよびR8bは水素である。他の実施形態において、Smac模倣物は式IIの化合物であり、ここで、YはCON(H)であり、WはCHであり、rは0であり、VはNである。他の実施形態において、Smac模倣物は式IIの化合物であり、ここで、YはCON(H)であり、WはCHであり、rは1であり、VはNであり、R2aは任意に置換されているアルキルであり、R2bは水素であり、Xは水素である。
特定の他の実施形態において、Smac模倣物は、式III:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1、A2、V、Z、W、X、Y、UおよびTは、式Iについて説明した意味と同じであり、mは1または2である。
他の実施形態において、Smac模倣物は式IIIの化合物であり、ここで、YはCON(H)であり、WはCHであり、rは1であり、VはNであり、R2aは任意に置換されているアルキルであり、R2bは水素であり、Xは水素である。一実施形態において、mは1である。他の実施形態において、mは2である。
特定の他の実施形態において、Smac模倣物は、式IV:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1、A2、Y、UおよびTは、式Iについて説明した意味と同じであり、mは1または2である。
他の実施形態において、Smac模倣物は式IVの化合物であり、ここで、mは1である。他の実施形態において、mは2である。
特定の他の実施形態において、Smac模倣物は、式V:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1、A2、V、Z、W、X、Y、UおよびTは、式Iについて説明した意味と同じであり、mは1または2である。
他の実施形態において、Smac模倣物は式Vの化合物であり、ここで、A1は任意に置換されているアルキルであり、A2は水素である。他の実施形態において、Xは任意に置換されているアルキルである。他の実施形態において、UはCH2である。他の実施形態において、UはNR6である。他の実施形態において、R6はCOR9である。他の実施形態において、R9は任意に置換されているアルキル、またはアラルキルである。他の実施形態において、R9は−CH2CH(CH3)2である。他の実施形態において、mは2である。他の実施形態において、mは1である。
特定の他の実施形態において、Smac模倣物は、式VI:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXは任意に置換されているアルキルであり、Tは任意に置換されているヘテロアリールである。
一実施形態において、A1およびXは独立して、任意に置換されているC1−4アルキルである。他の実施形態において、A1およびXは独立して、C1−4アルキルである。他の実施形態において、A1およびXは独立して、メチルおよびエチルからなる群から選択される。一実施形態において、A1およびXはメチルである。一実施形態において、A1はヒドロキシアルキルであり、Xは任意に置換されているアルキルである。他の実施形態において、A1はHOCH2CH2−である。
特定の他の実施形態において、Smac模倣物は、式VII:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXは任意に置換されているアルキルであり、R9は任意に置換されてアルキル、またはアラルキルであり、Tは任意に置換されているヘテロアリールである。
一実施形態において、A1およびXは独立して、任意に置換されているC1−4アルキルである。他の実施形態において、A1およびXは独立して、C1−4アルキルである。他の実施形態において、A1およびXは独立して、メチルおよびエチルからなる群から選択される。一実施形態において、A1およびXはメチルである。一実施形態において、A1はヒドロキシアルキルであり、Xは任意に置換されているアルキルである。他の実施形態において、A1はHOCH2CH2−である。
他の実施形態において、Smac模倣物は、式I〜VIIの化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、Tは、
である。ここで、Qは、O、SまたはNR12であり、R12は、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、または任意に置換されているヘテロシクロであり、R10a、R10b、R11a、R11b、R11cおよびR11dは独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択され、Z1、Z2およびZ3は独立して、CR11eまたはNであり、ここで、Z1、Z2およびZ3の少なくとも1つはCR11eであり、Z1、Z2およびZ3の少なくとも1つはNであり、R11eは、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択される。
一実施形態において、R10a、R10b、R11a、R11b、R11cおよびR11d少なくとも1つは、任意に置換されているフェニル、アラルキルまたは任意に置換されているアルキルである。一実施形態において、R12は水素または任意に置換されているアルキルである。一実施形態において、R12は水素である。一実施形態において、R10aは任意に置換されているフェニルである。一実施形態において、R11aは任意に置換されているフェニルである。
特定の他の実施形態において、Smac模倣物は、式I〜VIIの化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、Tは、
である。ここで、Q、Z1、Z2、Z3、R10a、R10b、R11a、R11b、R11cおよびR11dは上述の意味を有している。
特定の他の実施形態において、Smac模倣物は、式I〜VIIの化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、Tは、
である。ここで、Q、R10aおよびR10bは上述の意味を有している。
一実施形態において、R10aは、任意に置換されているアリール、アラルキルまたは任意に置換されているアルキルである。一実施形態において、R10bは水素である。他の実施形態において、R10aは任意に置換されているアリールであり、R10bは水素である。一実施形態において、QはSである。一実施形態において、QはNR12である。一実施形態において、R12は水素または任意に置換されているアルキルである。他の実施形態において、R12は水素である。
他の実施形態において、Smac模倣物は、式I〜VIIの化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、Tは、
である。ここで、Q、R11a、R11b、R11cおよびR11dは上述の意味を有している。
一実施形態において、R11aは、任意に置換されているアリール、アラルキルまたは任意に置換されているアルキルである。一実施形態において、R11b、R11cおよびR11dは水素である。他の実施形態において、R11aは任意に置換されているアリールであり、R11b、R11cおよびR11dは水素である。他の実施形態において、R11dは任意に置換されているアリールであり、R11a、R11bおよびR11cは水素である。一実施形態において、QはSである。一実施形態において、QはNR12である。一実施形態において、R12は水素または任意に置換されているアルキルである。他の実施形態において、R12は水素である。
他の実施形態において、Smac模倣物は、式I〜VIIの化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、Tは、
である。ここで、Q、Z1、Z2、Z3およびR11aは上述の意味を有している。
一実施形態において、R11aは任意に置換されているアリール、アラルキルまたは任意に置換されているアルキルであり、Z1、Z2およびZ3の少なくとも1つはNである。一実施形態において、R11aは任意に置換されているアリールである。一実施形態において、Z1はNであり、Z2およびZ3はCHである。一実施形態において、Z2はNであり、Z1およびZ3はCHである。一実施形態において、Z3はNであり、Z1およびZ2はCHである。一実施形態において、Z1およびZ3はNであり、Z2はCHである。一実施形態において、QはSである。一実施形態において、QはOである。一実施形態において、QはNR12である。一実施形態において、R12は水素または任意に置換されているアルキルである。他の実施形態において、R12は水素である。
特定の他の実施形態において、Smac模倣物は、式VIII:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXは、任意に置換されているアルキルであり、mは1または2であり、Qは、O、SまたはNR12であり、R12は、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールまたは任意に置換されているヘテロシクロであり、R10aおよびR10bは独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択される。
一実施形態において、mは2である。他の実施形態において、mは1である。一実施形態において、R10aは任意に置換されているアリールである。他の実施形態において、R10bは水素である。他の実施形態において、R10aは任意に置換されているアリールであり、R10bは水素である。一実施形態において、R9は水素または任意に置換されているアリールである。一実施形態において、QはSである。
特定の他の実施形態において、Smac模倣物は、式IX:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXは、任意に置換されているアルキルであり、mは1または2であり、Qは、O、SまたはNR12であり、R12は、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールまたは任意に置換されているヘテロシクロであり、R11a、R11b、R11cおよびR11dは独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択される。
一実施形態において、mは2である。他の実施形態において、mは1である。一実施形態において、R11aは任意に置換されているアリール、アラルキル、または任意に置換されているアルキルである。一実施形態において、R11b、R11cおよびR11dは水素である。他の実施形態において、R11aは任意に置換されているアリールであり、R11b、R11cおよびR11dは水素である。他の実施形態において、R11dは任意に置換されているアリールであり、R11a、R11bおよびR11cは水素である。一実施形態において、QはNR12である。一実施形態において、R12は水素または任意に置換されているアルキルである。他の実施形態において、R12は水素である。一実施形態において、QはSである。一実施形態において、QはOである。
特定の他の実施形態において、Smac模倣物は、式X:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXが、任意に置換されているアルキルであり、R9は、任意に置換されているアルキル、またはアラルキルであり、mは1または2であり、Qは、O、SまたはNR12であり、R12は、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールまたは任意に置換されているヘテロシクロであり、R10aおよびR10bは独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択される。
一実施形態において、mは2である。他の実施形態において、mは1である。一実施形態において、R10aは任意に置換されているアリールである。一実施形態において、R10bは水素である。他の実施形態において、R10aは任意に置換されているアリールであり、R10bは水素である。一実施形態において、R12は任意に水素または置換されているアルキルである。一実施形態において、R9は−CH2CH(CH3)2である。一実施形態において、QはSである。
特定の他の実施形態において、Smac模倣物は、式XI:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、A1およびXは、任意に置換されているアルキルであり、R9は、任意に置換されているアルキル、またはアラルキルであり、mは1または2であり、Qは、O、SまたはNR12であり、R12は、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールまたは任意に置換されているヘテロシクロであり、R11a、R11b、R11cおよびR11dは独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択される。
一実施形態において、mは2である。他の実施形態において、mは1である。一実施形態において、R11aは任意に置換されているアリール、アラルキル、または任意に置換されているアルキルである。一実施形態において、R11b、R11cおよびR11dは水素である。他の実施形態において、R11aは任意に置換されているアリールであり、R11b、R11cおよびR11dは水素である。他の実施形態において、R11dは任意に置換されているアリールであり、R11a、R11bおよびR11cは水素である。一実施形態において、QはNR12である。一実施形態において、R12は水素または任意に置換されているアルキルである。他の実施形態において、R12は水素である。一実施形態において、QはSである。一実施形態において、QはOである。一実施形態において、R9は−CH2CH(CH3)2である。
特定の他の実施形態において、Smac模倣物は、式Xの化合物であり、ここで、A1およびXはメチルであり、mは1であり、R9は任意に置換されているアルキル、またはアラルキルであり、R10aは任意に置換されているアリールであり、R10bは水素であり、QはSである。
特定の他の実施形態において、Smac模倣物は、式XIa:
の化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグであり、ここで、R9は任意に置換されているアルキル、またはアラルキルであり、R11aは任意に置換されているアリールである。
本明細書において、それ自身または他の基の一部によって使用されるとき、“アルキル”という用語は、1から18の炭素または示されている炭素数(例えば、C1−18は1から18の炭素を意味する)を有している、直鎖状または分枝鎖状の飽和脂肪族炭化水素を指す。他の実施形態において、アルキルはC1−6アルキルである。他の実施形態において、アルキルはC1−4アルキルである。例示的なアルキルとしては、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、sec-ブチル、イソブチル、tert-ブチル、n−ペンチル、n−ヘキシル、イソヘキシル、n−ヘプチル、4,4−ジメチルペンチル、n−オクチル、2,2,4−トリメチルペンチル、ノニルおよびデシルなどが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“任意に置換されているアルキル”という用語は、上述のように規定されるアルキルが、独立してヒドロキシ(すなわち−OH)、ニトロ(すなわち−NO2)、シアノ(すなわち−CN)、任意に置換されているシクロアルキル、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、アルコキシ、アリールオキシ、アラルキルオキシ、アルキルチオ、カルボキシアミドもしくはスルホンアミドから選択される置換基を1,2もしくは3つ用いて置換されているか、または置換されていないものを意味する。一実施形態において、任意に置換されているアルキルは置換基を2つ用いて置換されている、他の実施形態において、任意に置換されているアルキルは置換基を1つ用いて置換されている。他の実施形態において、置換基はヒドロキシ(すなわちヒドロキシアルキル)またはアミノ(すなわちアミノアルキル)から選択される。例示的な任意に置換されているアルキルとしては、−CH2OCH3、−CH2CH2NH2、−CH2CH2CN、CH2SO2CH3、ヒドロキシメチル、ヒドロキシエチルおよびヒドロキシプロピルなどが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“アルキレニル”という用語は、1つ,2つ,3つまたは4つの結合しているメチレン基を含んでいる2価のアルキルラジカルを指す。例示的なアルキレニル基としては、−(CH2)−、−(CH2)2−、−(CH2)3−および−(CH2)4−が挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“ハロアルキル”という用語は、1〜6のハロ置換基を有している、上述のように規定されるアルキルを指す。一実施形態において、ハロアルキルは、1つ,2つまたは3つのハロ置換基を有している。例示的なハロアルキル基としては、トリフルオロメチルおよび−CH2CH2Fなどが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“ヒドロキシアルキル”という用語は、1つ,2つまたは3つのヒドロキシ置換基を有している、上述のように規定されるアルキルを指す。一実施形態において、ヒドロキシアルキルは1つのヒドロキシ置換基を有している。例示的なヒドロキシアルキル基としては、ヒドロキシメチル、ヒドロキシエチルおよびヒドロキシプロピルなどが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“アラルキル”という用語は、任意に置換されているアリール置換基を1つ,2つまたは3つ有している、任意に置換されている上述のように規定されるアルキルを指す。一実施形態において、アラルキルは、任意に置換されているアリール置換基を2つ有している。他の実施形態において、アラルキルは、任意に置換されているアリール置換基を1つ有している。他の実施形態において、アラルキルはアリール(C1−6アルキル)である。他の実施形態において、アリール(C1−6アルキル)は、任意に置換されているアリール置換基を2つ有している。他の実施形態において、アリール(C1−6アルキル)は、任意に置換されているアリール置換基を1つ有している。例示的なアラルキル基としては、例えば、ベンジル、フェニルエチル、(4−フルオロフェニル)エチル、フェニルプロピル、ジフェニルメチル(すなわちPh2CH−)およびジフェニルエチル(すなわちPh2CHCH2−)が挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“シクロアルキル”という用語は、3から12の炭素原子(すなわちC3−12シクロアルキル)または示されている炭素数を有している1つから3つの環を含んでいる、飽和または部分的に不飽和の(1つまたは2つの二重結合を含んでいる)環状炭化水素基を指す。一実施形態において、シクロアルキルは1つの環を有している。他の実施形態において、シクロアルキルはC3−7シクロアルキルである。例示的なシクロアルキル基としては、シクロプロピル、シクロブチル、シクロペンチル。シクロヘキシル、シクロヘプチル、シクロオクチル、ノルボルニル、デカリンおよびアダマンチルが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“任意に置換されているシクロアルキル”という用語は、上述のように規定されるシクロアルキルが、1つ,2つまたは3つの置換基(それぞれが独立して、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、任意に置換されているアルキル、ハロアルキル、ヒドロキシアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、アルコキシ、アリールオキシ、アラルキルオキシ、アルキルチオ、カルボキシアミドまたはスルホンアミドから選択される)を用いて置換されているもの、または非置換であるものを意味する。また、“任意に置換されているシクロアルキル”は、上述のように規定されるシクロアルキルが任意に置換されているアリールに縮合され得るものを意味する。例示的な任意に置換されているシクロアルキルとしては、
本明細書において、それ自身または他の基の一部によって使用されるとき、“アルケニル”という用語は、炭素−炭素の二重結合を1つ,2つまたは3つ含んでいる、上述のように規定されるアルキル基を指す。一実施形態において、アルケニルは、炭素−炭素の二重結合を1つ有している。例示的なアルケニル基としては、−CH=CH2、−CH2CH=CH2、−CH2CH2CH=CH2、および−CH2CH2CH=CHCH3などが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“任意に置換されているアルケニル”という用語は、上述のように規定されるアルケニルが、1つ,2つまたは3つの置換基(それぞれが独立して、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、任意に置換されているアルキル、ハロアルキル、ヒドロキシアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、アルコキシ、アリールオキシ、アラルキルオキシ、アルキルチオ、カルボキシアミドまたはスルホンアミドから選択される)を用いて置換されているもの、または非置換であるものを意味する。例示的な任意に置換されているアルケニル基としては、−CH=CHPhおよび−CH2CH=CHPhなどが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“シクロアルケニル”という用語は、炭素−炭素の二重結合を1つ,2つまたは3つ含んでいる、上述のように規定されるシクロアルキル基を指す。一実施形態において、アルケニルは、炭素−炭素の二重結合を1つ有している。例示的なシクロアルケニル基としては、シクロペンテンおよびシクロヘキセンなどが挙げられ得る。
本明細書において、それ自身または他の基の一部によって使用されるとき、“任意に置換されているシクロアルケニル”という用語は、上述のように規定されるシクロアルケニルが、1つ,2つまたは3つの置換基(それぞれが独立して、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、任意に置換されているアルキル、ハロアルキル、ヒドロキシアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、アルコキシ、アリールオキシ、アラルキルオキシ、アルキルチオ、カルボキシアミドまたはスルホンアミドから選択される)を用いて置換されているもの、または非置換であるものを意味する。
本明細書において、それ自身または他の基の一部によって使用されるとき、“アルキニル”という用語は、炭素−炭素の三重結合を1つ〜3つ含んでいる上述のように規定されるアルキル基を指す。一実施形態において、アルキニルは、炭素−炭素の三重結合を1つ有している。例示的なアルキニル基としては、−C≡CH、−C≡CCH3、−CH2C≡CH、−CH2CH2C≡CHおよび−CH2CH2C≡CCH3が挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“任意に置換されているアルキニル”という用語は、上述のように規定されるアルキニルが、1つ,2つまたは3つの置換基(それぞれが独立して、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、任意に置換されているアルキル、ハロアルキル、ヒドロキシアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、アルコキシ、アリールオキシ、アラルキルオキシ、アルキルチオ、カルボキシアミドまたはスルホンアミドから選択される)を用いて置換されているもの、または非置換であるものを意味する。例示的な任意に置換されているアルキニルとしては、−C≡CPhおよび−CH2C≡CPhなどが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“アリール”という用語は、6〜14の炭素原子を有している単環式および二環式の芳香族環系(すなわちC6−14アリール)を指す。当該芳香族環系は、例えば、フェニル(Phと略される)、1−ナフチルおよび2−ナフチルなどである。
本明細書において、それ自身または他の基の一部によって使用されるとき、“任意に置換されているアリール”という用語は、上述のように規定されるアリールが、1つ,2つまたは3つの置換基(それぞれが独立して、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、任意に置換されているアルキル、ハロアルキル、ヒドロキシアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、アルコキシ、アリールオキシ、アラルキルオキシ、アルキルチオ、カルボキシアミドまたはスルホンアミドから選択される)を用いて置換されているもの、または非置換であるものを意味する。一実施形態において、任意に置換されているアリールは、任意に置換されているフェニルである。一実施形態において、任意に置換されているフェニルは、4つの置換基を有している。他の実施形態において、任意に置換されているフェニルは、3つの置換基を有している。他の実施形態において、任意に置換されているフェニルは、2つの置換基を有している。他の実施形態において、任意に置換されているフェニルは、1つの置換基を有している。例示的な任意に置換されているアリール基としては、2−メチルフェニル、2−メトキシフェニル、2−フルオロフェニル、2−クロロフェニル、2−ブロモフェニル、3−メチルフェニル、3−メトキシフェニル、3−フルオロフェニル、3−クロロフェニル、4−メチルフェニル、4−エチルフェニル、4−メトキシフェニル、4−フルオロフェニル、4−クロロフェニル、2,6−ジフルオロフェニル、2,6−ジクロロフェニル、2−メチルフェニル、3−メトキシフェニル、2−エチルフェニル、3−メトキシフェニル、3,4−ジメトキシフェニル、3,5−ジフルオロフェニル、3,5−ジメチルフェニルおよび3,5−ジメトキシフェニル、4−メチルフェニルなどが挙げられる。任意に置換されているアリールという用語は、任意に置換されている縮合されたシクロアルキルおよび任意に置換されている縮合されたヘテロシクロを有している基を包含することが意図されている。例としては、
本明細書において、それ自身または他の基の一部によって使用されるとき、“ヘテロアリール”という用語は、1つ,2つ,3つもしくは4つの異種原子(それぞれ独立して、酸素、窒素および硫黄からなる群から選択される)および5〜14の炭素原子を有している、単環式および二環式の芳香族環系(すなわちC5−14ヘテロアリール)を指す。一実施形態において、ヘテロアリールは3つの異種原子を有している。一実施形態において、ヘテロアリールは2つの異種原子を有している。一実施形態において、ヘテロアリールは1つの異種原子を有している。例示的なヘテロアリール基としては、1−ピロリル、2−ピロリル、3−ピロリル、2−イミダゾリル、4−イミダゾリル、ピラジニル、2−オキサゾリル、4−オキサゾリル、5−オキサゾリル、3−イソキサゾリル、4−イソキサゾリル、5−イソキサゾリル、2−チアゾリル、4−チアゾリル、5−チアゾリル、2−フリル、3−フリル、2−チエニル、3−チエニル、2−ピリジル、3−ピリジル、4−ピリジル、2−ピリミジル、4−ピリミジル、プリニル、2−ベンズイミダゾリル、4−ベンズイミダゾリル、5−ベンズイミダゾリル、2−ベンズチアゾリル、4−ベンズチアゾリル、5−ベンズチアゾリル、5−インドリル、3−インダゾリル、4−インダゾリル、5−インダゾリル、1−イソキノリル、5−イソキノリル、2−キノオキサリニル、5−キノオキサリニル、2−キノリル、3−キノリルおよび6−キノリルなどが挙げられる。ヘテロアリールという用語は、適切な窒素酸化物を包含することが意図されている。
本明細書において、それ自身または他の基の一部によって使用されるとき、“任意に置換されているヘテロアリール”という用語は、上述のように規定されるヘテロアリールが、1つ〜4つの置換基、典型的に1つまたは2つの置換基(それぞれが独立して、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、任意に置換されているアルキル、ハロアルキル、ヒドロキシアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、アルコキシ、アリールオキシ、アラルキルオキシ、アルキルチオ、カルボキシアミドまたはスルホンアミドから選択される)を用いて置換されているもの、または非置換であるものを意味する。一実施形態において、任意に置換されているヘテロアリールは1つの置換基を有している。他の実施形態において、上記置換基は、任意に置換されているアリール、アラルキル、または任意に置換されているアルキルである。他の実施形態において、上記置換基は、任意に置換されているフェニルである。利用可能な任意の炭素原子または窒素原子が置換され得る。例示的な任意に置換されているヘテロアリール基としては、
本明細書において、それ自身または他の基の一部によって使用されるとき、“ヘテロシクロ”は、部分的に不飽和(1つまたは2つの二重結合を含んでいる)または飽和の環状基を指す。当該環状基は、3から12の炭素原子ならびに1もしくは2の酸素原子、硫黄原子または窒素原子を有している1つから3つの環を含んでいる。ヘテロシクロは、必要に応じて、炭素原子または窒素原子を介して、分子の残りの部分と結合され得る。例示的なヘテロシクロ基としては、
本明細書において、それ自身または他の基の一部によって使用されるとき、“任意に置換されているヘテロシクロ”という用語は、上述のように規定されるヘテロシクロが、1つ〜4つの置換基、典型的に1つまたは2つの置換基(それぞれが独立して、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、任意に置換されているアルキル、ハロアルキル、ヒドロキシアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、アルコキシ、アリールオキシ、アラルキルオキシ、アルキルチオ、カルボキシアミド、スルホンアミド、−CORc、−SO2Rd、−N(Re)CORf、−N(Re)SO2Rgまたは−N(Re)C=N(Rh)−アミノから選択される)を用いて置換されているもの、または非置換であるものを意味する。置換は、任意に使用可能な炭素原子または窒素原子に存在し得る。例示的な置換されているヘテロシクロ基としては、
などが挙げられる。任意に置換されているヘテロシクロは、アリール基と縮合されて、上述のように規定される任意に置換されているアリールを提供し得る。
本明細書において、それ自身または他の基の一部によって使用されるとき、“アルコキシ”という用語は、末端の酸素原子に結合されている、ハロアルキル、任意に置換されているアルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニルまたは任意に置換されているアルキニルを指す。例示的なアルコキシ基としては、メトキシ、tert-ブトキシおよび−OCH2CH=CH2などが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“アリールオキシ”という用語は、末端の酸素原子に結合されている、任意に置換されているアリールを指す。例示的なアリールオキシとしては、フェノキシなどが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“アラルキルオキシ”という用語は、末端の酸素原子に結合されているアラルキルを指す。例示的なアラルキルオキシ基としては、ベンゾイルオキシなどが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“アルキルチオ”という用語は、末端の硫黄原子に結合されている、ハロアルキル、アラルキル、任意に置換されているアルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニルまたは任意に置換されているアルキニルを指す。例示的なアルキル基としては、−SCH3などが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“ハロ”または“ハロゲン”という用語は、フルオロ、クロロ、ブロモまたはヨードを指す。一実施形態において、ハロはフルオロまたはクロロである。
本明細書において、それ自身または他の基の一部によって使用されるとき、“アミノ”という用語は、式−NRaRbのラジカルを指す。ここで、RaおよびRbは独立して、水素、ハロアルキル、アラルキル、任意に置換されているアルキル、任意に置換されているシクロアルキル、任意に置換されているヘテロシクロ、任意に置換されているアリールまたは任意に置換されているヘテロアリールであり;RaおよびRbは、それらが結合している窒素原子とともに、任意に置換されている4員環から7員環のヘテロシクロを形成する。例示的なアミノ基としては、−NH2、−N(H)CH3、−N(CH3)2、N(H)CH2CH3、N(CH2CH3)および−N(H)CH2Phなどが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“カルボキシアミノ”という用語は、式−CO−アミノのラジカルを指す。例示的なカルボキシアミノ基としては、−CONH2−CON(H)CH3、−CON(H)Ph、−CON(H)CH2CH2Ph、−CON(CH3)2およびCON(H)CHPh2などが挙げられる。
本明細書において、それ自身または他の基の一部によって使用されるとき、“スルホンアミド”という用語は、式−SO2−アミノのラジカルを指す。例示的なスルホンアミド基としては、−SO2NH2、−SO2N(H)CH3および−SO2N(H)Phが挙げられる。
本明細書に使用されるとき、“約”という用語は記載されている数値の±10%の値を含んでいる。したがって、“約10”は9〜11を意味する。
本明細書に使用されるとき、“離脱基”という用語は、特定の反応において基質の残余部分または主要部分と見做される原子または基から引き離される原子または基を指す。アミドカップリング反応において、例示的な離脱基(すなわちL1によって表される離脱基)としては、−F、−Cl、−Br、−OH、−OC6F5およびO(CO)アルキルなどが挙げられ得る。一実施形態において、離脱基L1は−Clである。他の実施形態において、離脱基L1は−OHの活性化された形態(例えば、OBt、O−アシルイソ尿酸)である。活性化剤(例えば、ジシクロヘキシルカルボジイミド(DCC)、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド(EDC)、ベンゾトリアゾール−(1−イルオキシ)トリピロリジノホスホニウム ヘキサフルオロホスフェート(PyBop))が、アミドの形成に関するカルボン酸(すなわち離脱基は−OHである)を活性化するために採用され得る。当該活性化剤は、有機合成の当業者にとって周知である。また、他の添加剤(例えば、N−ヒドロキシベンゾトリアゾール(HOBt)またはN−ヒドロキシスクシニミド(HOSu))が加えられて、反応要素(例えば、速度、収量、純度、ラセミ化)を最適化し得る。求核置換反応(例えば、SN2反応)において、例示的な離脱基(すなわちL2によって表される離脱基)としては、−Cl、−Br、−I、−OSO2Me(メシレート)、−OSO2CF3(トリフレート)、−OSO2C6H5(ベシレート)および−OSO2CH3C6H4(トシレート)が挙げられる。一実施形態において、離脱基L2は−Clまたは−Brである。他の実施形態において、離脱基L2は−Brである。
本明細書に使用されるとき、“アミノ保護基”という用語は、分子の他の官能基または部分に対する反応が実施されている間に、アミノ官能性を遮断する(すなわち保護する)基を指す。当業者は、アミノ保護基の選択、結合および切断に精通しており、多くの保護基が当該技術において公知であり、1つまたは別の保護基の適合性が計画された合成手順の詳細に依存することを適切に理解する。これを主題とする論文(例えば、引用によってその開示事項が援用されるGreene and Wuts, "Protective Groups in Organic Synthesis," 3rd Ed., pp. 17-245 (J. Wiley & Sons, 1999))が、参考として利用可能である。好適なアミン保護基としては、カルボベンジルオキシ(Cbz)基、tert-ブチルオキシカルボニル(BOC)基、フルオレニルメチルオキシカルボニル(FMOC)基およびベンジル(Bn)基が挙げられる。
本明細書を通して、これらの基および置換基は好適な部分および化合物を提供するために選択される。
本発明の特定の化合物は、立体異性体(光学異性体が挙げられる)として存在し得る。本発明は、すべての立体異性体(個々の純粋な立体異性体調製物およびそれぞれの濃縮された調製物の両方、ならびに当業者にとって周知の方法にしたがって分離され得るそのような立体異性体のラセミ混合物および個々の光学異性体のラセミ混合物の両方)を包含している。
ある実施形態において、式Iの化合物は、
本発明の他の実施形態において、式Iの化合物は、
本発明の特定の実施形態において、式Iの化合物は、
または薬学的に受容可能なこれらの塩もしくはプロドラッグからなる群から選択される。
また、本発明は、式XII:
の化合物を調製する方法に関し、当該方法が、
(a)式XIII:
の化合物をアンモニアと縮合させて、式XIV:
の化合物を生成させる工程、
(b)式XIVの化合物を式XV:
の化合物に転化させる工程、
(c)式XVの化合物を式XVI:
(ここで、L2は離脱基である)
の化合物と縮合させて、式XVII:
の化合物を生成させる工程、ならびに、
(b)式XVIIの化合物を環化させて式XIIの化合物を生成させる工程を包含しており、
ここで、
R13が、N(H)P1および、
からなる群から選択され;
P1が、アミン保護基であり;
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、A2が存在しない場合にVがOであり;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキル、およびアラルキルからなる群から選択され;
Yが、CON(R1)、N(R1)CO、C(O)O、OC(O)、(CH2)1−3、任意に置換されているアリール、および任意に置換されているヘテロアリールからなる群から選択され、ここで、1つ以上のCH2基が、O、SまたはNR1によって置換され得;
Zが、(CR2aR2b)rであり;
Uが、CR5aR5bおよびNR6からなる群から選択され;
mが、1または2であり;
rが、0−3であり;
R1が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R2a、R2b、R5aおよびR5bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、およびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R10aおよびR10bが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択される。
一実施形態において、R13は−N(H)P1である。一実施形態において、P1はカルボベンジルオキシ、tert-ブチルオキシカルボニルおよびベンジルからなる群から選択される。一実施形態において、P1はカルボベンジルオキシおよびtert-ブチルオキシカルボニルからなる群から選択される。一実施形態において、P1はカルボベンジルオキシである。一実施形態において、P1はtert-ブチルオキシカルボニルである。一実施形態において、UはCH2である。一実施形態において、UはNCOR9である。他の実施形態において、R9は任意に置換されているアルキルである。一実施形態において、mは2である。一実施形態において、mは1である。
一実施形態において、L2は、−Cl、−Br、−I、−OSO2Me、−OSO2CF3、−OSO2C6H5、および−OSO2CH3C6H4からなる群から選択される。他の実施形態において、L2は−Clおよび−Brからなる群から選択される。他の実施形態において、L2は−Brである。一実施形態において、R10aは任意に置換されているフェニルである。一実施形態において、R10bは水素である。
一実施形態において、R13は、
である。一実施形態において、YはCON(H)であり、WはCHであり、rは0であり、VはNである。一実施形態において、UはCH2である。一実施形態において、UはNCOR9である。他の実施形態において、R9は任意に置換されているアルキルである。一実施形態において、mは2である。一実施形態において、mは1である。
一実施形態において、アンモニアは、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドおよびN−ヒドロキシベンゾトリアゾールの存在下において、約−20℃から約25℃、不活性な有機溶媒(例えば、アセトニトリル、テトラヒドロフラン、ジメチルホルムアミドなど)において、式XIIIの化合物と縮合される。一実施形態において、式XIVの化合物は、約−20℃から約45℃、不活性な有機溶媒(例えば、ジクロロメタンなど)において、P4S10を用いて式XVの化合物に転化される。一実施形態において、式XVの化合物と式XVIの化合物との縮合は、約0℃から約50℃、不活性な有機溶媒(例えば、アセトニトリル、テトラヒドロフラン、ジメチルホルムアミドなど)において実施される。一実施形態において、式XVIIの化合物は、メタノール、エタノール、プロパノール、イソプロパノールまたはブタノールにおいて環化される。一実施形態において、式XVIIの化合物の環化は、約25℃から約100℃、一実施形態において、約30℃において実施される。一実施形態において、式XVIIIの化合物の環化は、還流している溶液において実施される。一実施形態において、式XVIIIの化合物の環化は、還流しているエタノールにおいて実施される。
上述の反応のいずれかの進行は、当該分野において公知の分析法(例えば、TLC、LC、LC/MS、HPLC、NMRなど)によって監視される。式XIIの化合物および任意の(複数の)合成中間体(すなわち式XIV、式XVまたは式XVIIの化合物)は、当該分野において公知の手法(例えば、通常および逆相のカラムクロマトグラフィー(例えば、シリカゲル上におけるカラムクロマトグラフィーまたは逆相HPLC)、結晶化、抽出など)によって単離され得、精製され得る。このようにして単離された生成物は、純度が所望の水準に達するまで、さらなる精製(例えば、再結晶化)にかけられ得る。一実施形態において、式XIIの化合物は、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%またはそれ以上の純度を有している。
他の実施形態において、本発明は式XVIII:
の化合物を調製する方法に関し、当該方法は、
(a)式XIII:
の化合物を式XIX:
の化合物と縮合させて、式XX:
の化合物を生成させる工程、ならびに、
(b)式XXの化合物を環化させて、式XVIIIの化合物を生成させる工程を包含し、
ここで、
R13が、N(H)P1および、
からなる群から選択され;
P1が、アミン保護基であり;
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、A2が存在しない場合にVがOであり;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキル、およびアラルキルからなる群から選択され;
Yが、CON(R1)、N(R1)CO、C(O)O、OC(O)、(CH2)1−3、任意に置換されているアリール、および任意に置換されているヘテロアリールからなる群から選択され、ここで、1つ以上のCH2基が、O、SまたはNR1によって置換され得;
Zが、(CR2aR2b)rであり;
Uが、CR5aR5bおよびNR6からなる群から選択され;
mが、1または2であり;
rが、0−3であり;
R1が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R2a、R2b、R5aおよびR5bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールおよび任意に置換されているヘテロシクロからなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、およびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R11a、R11b、R11cおよびR11dが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択され;
R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択される。
一実施形態において、R11aは任意に置換されているフェニルである。一実施形態において、R11b、R11cおよびR11dは水素である。一実施形態において、R11aは任意に置換されているフェニルであり、R11b、R11cおよびR11dは水素である。一実施形態において、mは2である。一実施形態において、mは1である。一実施形態において、UはCH2である。一実施形態において、UはNCOR9である。他の実施形態において、R9は任意に置換されているアルキルである。一実施形態において、R12は水素である。
一実施形態において、R13は−N(H)P1である。一実施形態において、P1はカルボベンジルオキシ、tert-ブチルオキシカルボニルおよびベンジルからなる群から選択される。一実施形態において、P1はカルボベンジルオキシおよびtert−ブチルオキシカルボニルからなる群から選択される。一実施形態において、P1はカルボベンジルオキシである。一実施形態において、P1はtert−ブチルオキシカルボニルである。一実施形態において、UはCH2である。一実施形態において、UはNCOR9である。他の実施形態において、R9は任意に置換されているアルキルである。一実施形態において、mは2である。一実施形態において、mは1である。
一実施形態において、R13は、
である。一実施形態において、YはCON(H)であり、WはCHであり、rは0であり、VはNである。一実施形態において、UはCH2である。一実施形態において、UはNCOR9である。他の実施形態において、R9は任意に置換されているアルキルである。一実施形態において、mは2である。一実施形態において、mは1である。
一実施形態において、式XIIIの化合物は、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドおよびN−ヒドロキシベンゾトリアゾールの存在下において、約−20℃から約25℃、不活性な有機溶媒(例えば、アセトニトリル、テトラヒドロフラン、ジメチルホルムアミドなど)において、式XIXの化合物と縮合される。一実施形態において、式XXの化合物は、約25℃から約118℃の酢酸において環化される。他の実施形態において、式XXの化合物は、還流している酢酸において環化される。
上述の反応のいずれかの進行は、当該分野において公知の分析法(例えば、TLC、LC、LC/MS、HPLC、NMRなど)によって監視される。式XVIIIの化合物および任意の(複数の)合成中間体(すなわち式XIXの化合物)は、当該分野において公知の手法(例えば、通常および逆相のカラムクロマトグラフィー(例えば、シリカゲル上におけるカラムクロマトグラフィーまたは逆相HPLC)、結晶化、抽出など)によって単離され得、精製され得る。このようにして単離された生成物は、純度が所望の水準に達するまで、さらなる精製(例えば、再結晶化)にかけられ得る。一実施形態において、式XVIIIの化合物は、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%またはそれ以上の純度を有している。
また、本発明は、式XXI:
の化合物を調製する方法に関し、当該方法は、式XXIII:
の化合物を、式XXIV:
(ここでL1は離脱基である)
の化合物と縮合させて、式XXIの化合物を生成させる工程を包含し、
ここで、
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、A2が存在しない場合にVがOであり;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキルおよびアラルキルからなる群から選択され;
Zが、(CR2aR2b)rであり;
Uが、CR5aR5bおよびNR6からなる群から選択され;
mが、1または2であり;
rが、0−3であり;
R2a、R2b、R5aおよびR5bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、およびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロシクロからなる群から選択され;
Tが、任意に置換されているヘテロアリールである。
一実施形態において、WはCHであり、VはNである、一実施形態において、Tは、
からなる群から選択され、
ここで、
Qが、O、SおよびNR12からなる群から選択され;
R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールおよび任意に置換されているヘテロシクロからなる群から選択され;
R10a、R10b、R11a、R11b、R11cおよびR11dが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロ、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択される。一実施形態において、UはCH2である。一実施形態において、UはNCOR9である。一実施形態において、R9は任意に置換されているアルキルである。一実施形態において、mは2である。一実施形態において、mは1である。
一実施形態において、L1は−Clおよび−OHからなる群から選択される。一実施形態において、L1は−OHであり、反応は活性化剤の存在下において実施される。
一実施形態において、式XXIIIの化合物は、式XXII:
の化合物からP1を除去することによって調製され、ここで、P1は離脱基である。一実施形態において、P1は、カルボベンジルオキシ、tert−フルオレニルメチルオキシカルボニルまたはベンジルである。一実施形態において、P1はカルボベンジルオキシまたはtert−フルオレニルメチルオキシカルボニルである。一実施形態において、P1はtert−フルオレニルメチルオキシカルボニルである。
一実施形態において、縮合反応は、不活性な有機溶媒(例えば、アセトニトリル、ベンゼン、クロロホルム、1,2−ジクロロエタン、1,2−ジメトキシエタン、ジメチルホルムアミド、ジメチルスルホキシド、ジオキサン、ジクロロメタン、N−メチル−2−ピロリジノンまたはテトラヒドロフラン)において実施される。他の実施形態において、縮合反応はテトラヒドロフランにおいて実施される。他の実施形態において、縮合反応はジクロロメタンにおいて実施される。一実施形態において、縮合反応は約−20℃から約35℃において実施される。他の実施形態において、縮合反応は約25℃において実施される。一実施形態において、縮合反応は1時間から48時間かかって完了する。他の実施形態において、縮合反応は約12時間かかって完了する。
一実施形態において、L1は−Cl、−OHまたは−OBtである。一実施形態において、L1は−OHまたは−OBtである。他の実施形態において、縮合反応は、活性化剤の存在下において実施される。他の実施形態において、活性化剤は、ジシクロヘキシルカルボジイミド、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド、またはベンゾトリアゾール−(1−イルオキシ)トリピロリジノホスホニウム ヘキサフルオロホスフェートである。他の実施形態において、活性化剤は、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドである。他の実施形態において、縮合反応は、活性化剤、および反応要素(例えば、純度および収量)を最適化する添加剤の存在下において実施される。他の実施形態において、添加剤はN−ヒドロキシベンゾトリアゾールである。
式XXIIIの化合物と式XXIVの化合物との間における反応の進行は、当該分野において公知の分析法(例えば、TLC、LC、LC/MS、HPLC、NMRなど)によって監視される。式XXIの化合物は、当該分野において公知の手法(例えば、通常および逆相のカラムクロマトグラフィー(例えば、シリカゲル上におけるカラムクロマトグラフィーまたは逆相HPLC)、結晶化、抽出など)によって単離され得、精製され得る。このようにして単離された生成物は、純度が所望の水準に達するまで、さらなる精製(例えば、再結晶化)にかけられ得る。一実施形態において、式XXIの化合物は、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%またはそれ以上の純度を有している。
他の実施形態において、本発明は、式XXII:
を有している化合物に関し、
ここで、
Tが、任意に置換されているヘテロアリールであり;
mが、1または2であり;
Uが、CH2またはNR6であり;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロおよびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリールおよび任意に置換されているヘテロシクロからなる群から選択され;
P1が、アミン保護基である。
一実施形態において、P1はt−ブトキシカルボニルおよびベンジルオキシカルボニルからなる群から選択される。
他の実施形態において、本発明は、式XXV:
を有している化合物に関し、
ここで、
Tが、任意に置換されているヘテロアリールであり;
mが、1または2であり;
Uが、CH2またはNR6であり;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロシクロおよびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリールおよび任意に置換されているヘテロシクロからなる群から選択され;
Xが、水素、任意に置換されているアルキル、およびアラルキルからなる群から選択され;
A1が、水素および任意に置換されているアルキルからなる群から選択され;
P1が、アミン保護基である。
一実施形態において、P1はt−ブトキシカルボニルおよびベンジルオキシカルボニルからなる群から選択される。
一実施形態において、式XXVの化合物は、
本発明の化合物は、当業者に公知の方法を用いて調製され得る。より詳細には本発明の化合物は、実施例における例示的な反応によって示されているように調製され得る。
本発明の重要な側面は、アポトーシスの誘導シグナルに応答して、化学式I−XIaの化合物がアポトーシスを誘導すること、またアポトーシスの誘導を増強することである。したがって、これらの化合物がアポトーシス誘導因子に対する細胞(アポトーシス誘導因子に抵抗性である細胞が挙げられる)の感受性を向上させることが、意図されている。本発明のIAP阻害剤は、アポトーシスの誘導によって処置、寛解または抑制され得る任意の疾患において、アポトーシスの誘導に使用され得る。よって、本発明は、IAPタンパク質が過剰に発現している動物を対象とする組成物および方法を提供する。いくつかの実施形態において、細胞(例えばがん細胞)が、非病理学的なサンプル(例えば非がん性の細胞)と比べて、高いIAPタンパク質の発現レベルを示す。他の実施形態において、阻害に有効な量の化学式I−XIaの化合物に応答して、アポトーシスプログラムおよび細胞死を実行することによって、操作にしたがって、細胞が、IAPタンパク質の高い発現レベルを示していることを明らかにする。この応答は、生存に関するIAPタンパク質の機能に対する、当該細胞における依存性に少なくとも部分的に起因して、生じている。
他の実施形態において、本発明は、1つ以上のアポトーシス調節剤をともなって、アポトーシスと関連する状態を調節することに関する。アポトーシス調節剤の例としては、Fas/CD95、TRAMP、TNF RI、DR1、DR2、DR3、DR4、DR5、DR6、FADD、RIP、TNFα、Fasリガンド、TRAIL、TRAIL−R1またはTRAIL−R2に対する抗体、Bcl−2、p53、BAX、BAD、Akt、CAD、PI3キナーゼ、PP1、およびカスパーゼタンパク質が挙げられるが、これらに限定されない。また、アポトーシスの開始、決定および分解の過程に関与する他の薬剤が挙げられる。アポトーシス調節剤の例としては、薬剤の活性、存在、または濃度の変化によって、対象におけるアポトーシスを調節し得る薬剤が挙げられる。好ましいアポトーシス調節剤は、アポトーシスの誘導剤(例えば、TNFまたはTNFに関連するリガンド、より詳細にはTRAMPリガンド、Fas/CD95リガンド、TNFR−1リガンドまたはTRAIL)である。
いくつかの実施形態において、本発明に係る組成物および方法は、病的な細胞、病的な組織、病的な臓器、または動物における病的な状態および/または疾患状態を処置するために使用される。当該動物は、例えば、哺乳類の対象(ヒトおよび獣医学的な動物が挙げられるが、これらに限定されない)である。この点について、本発明に係る方法および組成物を用いる治療または抑制は、様々な疾患および病変に適している。これらの疾患および病状を例示する非限定的な例としては、乳癌、前立腺癌、リンパ腫、皮膚癌、膵(臓)癌、大腸癌、メラノーマ、悪性黒色腫、卵巣癌、脳腫瘍、原発性脳腫瘍、頭頸部癌、神経膠腫、膠芽腫、肝癌、膀胱癌、非小細胞肺癌、頭頸部癌、乳癌、卵巣癌、肺癌、小細胞肺癌、ウィルムス腫瘍、子宮癌、精巣癌、膀胱癌、膵臓癌、胃癌、大腸癌、前立腺癌、泌尿生殖器癌、甲状腺癌、食道癌、骨髄腫、多発性骨髄腫、副腎癌、腎細胞癌、子宮内膜癌、副腎皮質癌、悪性膵インスリノーマ、悪性カルチノイド腫瘍、絨毛癌、菌状息肉腫、悪性高カルシウム血症、子宮肥大症、白血病、急性リンパ性白血病、慢性リンパ性白血病、急性骨髄性白血病、慢性骨髄性白血病、慢性顆粒球性白血病、急性顆粒球性白血病、ヘアリー細胞白血病、神経芽細胞腫、横紋筋肉腫、カポジ肉腫、真性多血症、本態性血小板増加症、ホジキン病、非ホジキンリンパ腫、軟部組織肉腫、骨肉腫、原発性マクログロブリン血症および網膜芽細胞腫など、ならびにT細胞媒介性およびB細胞媒介性の自己免疫疾患;炎症性疾患;感染症(例えばピロリ菌感染と関連して、例えば抗潰瘍剤として)、増殖性疾患;エイズ;変性条件;血管疾患(例えば、主静脈瘤)などが挙げられるが、これらに限定されない。また、本発明の化合物は、プログラムされた細胞死またはアポトーシスの機序に欠陥がある疾患(例えば、多発性硬化症、喘息および関節硬化症など)の処置に有用であり得る。いくつかの実施形態において、処置されるがん細胞は転移性を示し得る。他の実施形態において、処置されるがん細胞は、抗がん剤に耐性であり得る。
いくつかの実施形態において、本発明に係る組成物および方法による処置に適した感染症としては、ウイルス、細菌、真菌、マイコプラズマおよびプリオンなどによって引き起こされる感染症が挙げられるが、これらに限定されない。
本発明に係るあるいくつかの実施形態は、化学式I−XIaの化合物および少なくとも1つの付加的な治療剤(化学療法抗悪性腫瘍剤、アポトーシス調節剤、抗菌剤、抗ウイルス剤、抗真菌薬および抗炎症剤が挙げられるが、これらに限定されない)の有効量を投与する方法、および/または治療手法(例えば、外科的処置および/または放射線療法)を提供する。
適切な抗がん剤の多くが、本発明に係る方法における使用について考慮される。実際に、本発明は、限定するわけではないが、様々な抗がん剤の投与を意図している。当該抗がん剤としては、アポトーシス誘導剤;ポリヌクレオチド(例えば、アンチセンス、リボザイム、siRNA);ポリペプチド(例えば、酵素および抗体);生物学的模倣物(例えば、ゴシポールまたはBH3模倣物);Bcl−2ファミリータンパク質(例えば、Bax)と結合(例えば、オリゴマー化または複合体化)する因子;アルカロイド;アルキル化剤;抗腫瘍性の抗生物質;代謝拮抗剤;ホルモン;白金の化合物;モノクローナル抗体またはポリクローナル抗体(例えば、抗がん剤、毒素またはディフェンシンと抱合されている抗体)、毒素;放射性核種;生物学的な反応の修飾剤(例えば、インターフェロン(例えばIFN−α)およびインターロイキン(例えばIL−2));養子免疫療法剤;造血成長因子;腫瘍細胞の分化を誘導する因子(例えば、全トランス型のレチノイン酸);遺伝子療法剤(アンチセンス療法の試薬およびヌクレオチドなど);腫瘍ワクチン;血管新生阻害剤;プロテアソーム阻害剤:NF−КB調節剤;抗CDK化合物;HDAC阻害阻などが挙げられる。開示されている上記化合物との同時投与に適した化学療法化合物および抗がん療法に関する多くの他の例が当業者に知られている。
特定の実施形態において、抗がん剤は、アポトーシスを誘導または刺激する因子を含んでいる。アポトーシスを誘導する因子としては、放射線(例えば、X線、ガンマ線、紫外線)、腫瘍壊死因子(TNF)と関連する因子(例えば、TNFファミリー受容体タンパク質、TNFファミリーリガンド、TRAIL、TRAIL−R1に対する抗体またはTRAIL−R2に対する抗体);キナーゼの阻害剤(例えば、上皮増殖因子受容体(EGFR)キナーゼの阻害剤、血管増殖因子受容体(VGFR)キナーゼの阻害剤、線維芽細胞増殖因子受容体(FGFR)キナーゼの阻害剤、血小板由来増殖因子受容体(PDGFR)キナーゼの阻害剤、およびBcr−Ablキナーゼの阻害剤(例えばGLEEVEC)など);アンチセンス分子;抗体(例えば、HERCEPTIN、RITUXAN、ZEVALINおよびAVASTIN);抗エストロゲン(例えば、ラロキシフェンおよびタモキシフェン);抗アンドロゲン(例えば、フルタミド、ビカルタミド、フィナステリド、アミノグルテチミド、ケトコナゾールおよびコルチコステロイド);シクロオキシゲナーゼ2(COX−2)阻害剤(例えば、セレコキシブ、メロキシカム、NS−398、および非ステロイド性抗炎症薬(NSAID));抗炎症薬(例えば、フェニルブタゾン、DECADRON、DELTASONE、デキサメタゾン、デキサメタゾン インテンゾル、DEXONE、HEXADROL、ヒドロキシクロロキン、METICORTEN、ORADEXON、ORASONE、オキシフェンブタゾン、PEDIAPRED、フェニルブタゾン、PLAQUENIL、プレドニゾロン、プレドニゾン、PRELONE、およびTANDEARIL);がんの化学療法剤(例えば、イリノテカン(CAMPTOSAR)、CPT−11、フルダラビン(FLUDARA)、ダカルバジン(DTIC)、デキサメタゾン、ミトキサントロン、MYLOTARG、VP−16、シスプラチン、カルボプラチン、オキサリプラチン、5−FU、ドキソルビシン、ゲムシタビン、ボルテゾミブ、ゲフィチニブ、ベバシズマブ、TAXOTERE、またはTAXOL);細胞内シグナル伝達分子;セラミドおよびサイトカイン;スタウロスポリンなどが挙げられるが、これらに限定されない。
さらなる他の実施形態において、本発明の組成物および方法は、式I−XIaの化合物、ならびにアルキル化剤、代謝拮抗剤および天然物(例えば、ハーブ、ならびに他の植物および/または動物に由来する化合物)から選択される少なくとも1つの抗増殖剤または抗腫瘍剤を提供する。
本発明に係る組成物および方法における使用に適しているアルキル化剤としては、(1)ナイトロジェンマスタード(例えば、メクロルエタミン、シクロホスファミド、イホスファミド、メルファラン(L−サルコリシン)、およびクロラムブシル);(2)エチレンイミンおよびメチルメラミン(例えば、ヘキサメチルメラミンおよびチオテパ);(3)アルキルスルホン酸塩(例えば、ブスルファン);(4)ニトロソウレア(例えば、カルムスチン(BCNU);ロムスチン(CCNU);セムスチン(メチル−CCNU);およびストレプトゾシン(ストレプトゾトシン));ならびに(5)トリアゼン(例えば、ダカルバジン(DTIC;ジメチルトリアゼノイミド−アゾールカルボキシアミド)が挙げられるが、これらに限定されない。
いくつかの実施形態において、本発明に係る組成物および方法における使用に適している代謝拮抗剤としては、(1)葉酸類似体(例えば、メトトレキサート(アメトプテリン);(2)ピリミジン類似体(例えば、フルオロウラシル(5−フルオロウラシル;5−FU)、フロクスウリジン(フルオロデ−オキシウリジン;FudR)、およびシタラビン(シトシンアラビノシド));(3)プリン類似体(例えば、メルカプトプリン(6−メルカプトプリン;6−MP)、チオグアニン(6−チオグアニン;TG)、およびペントスタチン(2’−デオキシコホルマイシン))が挙げられるが、これらに限定されない。
さらなる他の実施形態において、本発明の組成物および方法における使用に適している化学療法剤としては、(1)ビンカアルカロイド(例えば、ビンブラスチン(VLB)、ビンクリスチンなど);(2)エピポドフィロトキシン(例えば、エトポシド、テニポシド);(3)抗生物質(例えば、ダクチノマイシン(アクチノマイシンD)、ダウノルビシン(ダウノマイシン;ルビドマイシン)、ドキソルビシン、ブレオマイシン、プリカマイシン(ミスラマイシン)、およびマイトマイシン(マイトマイシンC));(4)酵素(例えば、L型アスパラギナーゼ);(5)生物学的反応の修飾因子(例えば、インターフェロンアルファ);(6)白金配位錯体(例えば、シスプラチン(シス−DDP)、およびカルボプラチン);(7)アントラセンディオン(例えば、ミトキサントロン);(8)置換ウレア(例えば、ヒドロキシウレア);(9)メチルヒドラジン誘導体(例えば、プロカルバジン(N−メチルヒドラジン;MIH));(10)副腎皮質抑制剤(例えば、ミトタン(o,p’−DDD)およびアミノグルテチミド);(11)副腎皮質ステロイド(例えばプレドニゾン);(12)プロゲスチン(例えば、カプロン酸ヒドロキシプロゲステロン、酢酸メドロキシプロゲステロン、および酢酸メゲストロール);(13)エストロゲン(例えば、ジエチルスチルベストロールおよびエチニルエストラジオール);(14)抗エストロゲン剤(例えば、タモキシフェン);(15)アンドロゲン(例えば、プロピオン酸テストステロンおよびフルオキシメステロン);(16)抗アンドロゲン剤(例えば、フルタミド);(17)性腺刺激ホルモン類似体(例えば、ロイプロリド)が挙げられるが、これらに限定されない。
がん療法において日常的に使用される任意の腫瘍の崩壊剤は、本発明の組成物および方法における用途が見出される。例えば、米国食品医薬品局(USFDA)は、米国における使用を認可された腫瘍の崩壊剤薬剤の処方を保持している。USFDAに対応する国際機関は、同様の処方を保持している。表1は、米国における使用を認可された例示的な抗腫瘍剤のリストを示している。当業者は、米国において認可されたすべての化学療法に対して要求される「製品ラベル」には、例示的な薬剤に関する、認可された適応症、用量の情報および毒性情報などについて記載されていることを適切に理解する。
抗がん剤としては、抗がん活性を有していることが同定されているが、現状において、米国食品医薬品局または他の対応機関によって認可されていないか、または新しい用途に関する調査を受けている化合物がさらに挙げられる。例としては、3−AP、12−O−テトラデカノイルホルボール−13−アセテート、17AAG、852A、ABI−007、ABR−217620、ABT−751、ADI−PEG20、AE−941、AG−013736、AGRO100、アラノシン、AMG706、抗体G250、抗新生物薬、AP23573、アパジクオン(apaziquone)、APC8015、アチプリモド、ATN−161、アトラセンタン、アザシチジン、BB−10901、BCX−1777、ベバシズマブ、BG00001、ビカルタミド、BMS247550、ボルテゾミブ、ブリオスタチン−1、ブセレリン、カルシトリオール、CCI−779、CDB−2914、セフィキシム、セツキシマブ、CG0070、シレンジタイド、クロファラビン、コンブレタスタチンA4リン酸、CP−675,206、CP−724,714、CpG7909、クルクミン、デシタビン、DENSPM、ドキセルカルシフェロール、E7070、E7389、エクチナサイジン743、エファプロキシラル、エフロルニチン、EKB−569、エンザスタウリン、エルロチニブ、エクシスリンド、フェンレチニド、フラボピリドール、フルダラビン、フルタミド、ホテムスチン、FR901228、G17DT、ガリキシマブ、ゲフィチニブ、ゲニステイン、グルホフファミド、GTI−2040、ヒストレリン、HKI−272、ホモハリントニン、HSPPC−96、hu14.18−インターロイキン−2の融合タンパク質、HuMax−CD4、イロプロスト、イミキモド、インフリキシマブ、インターロイキン−12、IPI−504、イロフルベン、イクサベピロン、ラパチニブ、レスタウルチニブ、ロイプロリド、LMB−9 抗毒素、ロナファーニブ、ルニリキシマブ、マホスファミド、MB07133、MDX−010、MLN2704、モノクローナル抗体3F8、モノクローナル抗体J591、モテキサフィン、MS−275、MVA−MUC1−IL2、ニルタミド、ニトロカンプトテシン、ノラトレキシド二塩酸塩、ノルバデックス、NS−9、O6−ベンジルグアニン、オブリメルセンナトリウム、ONYX−015、オレゴボマブ、OSI−774、パニツムマブ、パラプラチン、PD−0325901、ペメトレキセド、PHY906、ピオグリタゾン、ピルフェニドン、ピキサントロン、PS−341、PSC833、PXD101、ピラゾロアクリジン、R115777、RAD001、ランピルナーゼ、レベッカマイシン類似体、rhuアンジオスタチンタンパク質、rhuMab 2C4、ロシグリタゾン、ルビテカン(rubitecan)、S−1、S−8184、サトラプラチン(satraplatin)、SB−15992、SGN−0010、SGN−40、ソラフェニブ、SR31747A、ST1571、SU011248、ヒドロキサミン酸サブエロイルアニリド、スラミン、タラボスタット、タランパネル、タリキダル、テミシロキム、TGFa−PE38抗毒素、サリドマイド、チマルファシン、チピファルニブ、チラパザミン、TLK286、トラベクテジン、トリメトレキサートグルクロン酸、TroVax、UCN−1、バルプロ酸、ビンフルニン、VNP40101M、ボロシキシマブ、ボリノスタットと、VX−680、ZD1839、ZD6474、ジレウトン、およびゾスキダル三塩酸塩が挙げられるが、これらに限定されない。
一実施形態において、抗がん剤は、タキソテール、ゲムシタビン、ラパチニブ(タイケルブ(登録商標))およびエトポシドからなる群から選択される。
当業者は、抗がん剤および他の治療剤のさらなる詳細な説明に関する多くの有益な任意の手引書を任意に参照することができる。当該手引書としては、「the Physician's Desk Reference」、ならびにGoodmanおよびGilmanの「Pharmaceutical Basis of Therapeutics」(第10版、Hardman等編集、2002年)が挙げられるが、これらに限定されない。
本発明は、放射線療法を併用して、式I−XIaIの化合物を投与する方法を提供する。本発明は、治療線量の放射線を動物に照射するために使用される種類、量、または照射と投与との系によって限定されない。例えば、動物は、光子放射線療法、粒子ビーム放射線療法、他のタイプの放射線療法、およびそれらの組み合わせを受け得る。いくつかの実施形態において、放射線は、線形加速器を用いて動物に照射される。さらなる他の実施形態において、放射線は、ガンマナイフを用いて照射される。
放射線源は動物の外部または内部に存在し得る。体外からの照射療法は、最も一般的であり、例えば線形加速器を使用して、皮膚を透過させて、高エネルギーの放射線のビームを腫瘍部位に導くことに関する。放射線のビームは腫瘍部位に集中するが、通常の健康な組織の曝露を避けることは、ほぼ不可能である。しかし、外部からの放射線は、通常、動物によって十分に許容される。内部からの照射療法は、放射線源(例えば、ビーズ、ワイヤー、ペレット、カプセルおよび粒子)を体内の腫瘍部位またはその近傍に埋め込むこと(がん細胞を特異的に標的にする送達系の使用(例えば、がん細胞結合リガンドに接続されている粒子の使用)が挙げられる)に関する。このような包埋物は、処置の後に除去され得るが、または不活性な状態において体内に残され得る。内部からの照射療法の種類としては、近接照射療法、間質放射線療法、腔内療法および放射免疫療法などが挙げられる。
動物は、放射線増感剤(例えば、メトロニダゾール、ミソニダゾール、動脈内Budr、静脈内ヨードデオキシウリジン(IudR)、ニトロイミダゾール、5−置換−4−ニトロイミダゾール、2H−イソインドールジオン、[[(2−ブロモエチル)−アミノ]メチル]−ニトロ−1H−イミダゾール−1−エタノール、ニトロアニリン誘導体、DNA親和性の低酸素症選択的な細胞毒素、ハロゲン化DNAリガンド、1,2,4−ベンゾトリアジン酸化物、2−ニトロイミダゾール誘導体、含フッ素ニトロアゾール(nitroazole)誘導体、ベンズアミド、ニコチンアミド、アクリジンインターカレーター、5−チオトレトラゾール(thiotretrazole)誘導体、3−ニトロ−1,2,4−トリアゾール、4,5−ジニトロイミダゾール誘導体、水酸化テキサフィリン、シスプラチン、マイトマイシン、チリパザミン(tiripazamine)、ニトロソウレア、メルカプトプリン、メトトレキサート、フルオロウラシル、ブレオマイシン、ビンクリスチン、カルボプラチン、エピルビシン、ドキソルビシン、シクロホスファミド、ビンデシン、エトポシド、パクリタキセル、熱(温熱療法)など)、および放射線保護剤(例えば、システアミン、アミノアルキル二水素ホスホロチオエート、アミホスチン(WR2721)、IL−1、IL−6など)の投与を、必要に応じて受け得る。放射線増感剤は腫瘍細胞の殺傷度を高める。放射線保護剤は、放射線の有害な作用から、健康な組織を保護する。
許容不可能な負の副作用を生じることなく、放射線の照射量が患者に許容される範囲にある限り、任意の種類の放射線を動物に照射し得る。放射線療法の好適な種類としては、例えば、電離(電磁界)放射線療法(例えば、X線またはガンマ線)、または粒子ビーム照射療法(例えば、高い線エネルギーの照射)が挙げられる。電離放射線は、イオン化(すなわち、電子の獲得または放出)を起こすために十分なエネルギーを有している粒子または光子を含んでいる放射線と規定される(例えば、参照によってその全体が本明細書に援用される米国特許第5,770,581号に記載されている)。放射線の影響は、少なくとも部分的に医師によって制御され得る。標的細胞の暴露の最大化および低毒性を実現するために、分割照射法が好ましく採用される。
動物に照射される放射線の総線量は、好ましくは約0.01グレイ(Gy)から約100Gyである。より好ましくは、処置の全体を通して、約10Gyから約65Gy(例えば、約15Gy、20Gy、25Gy、30Gy、35Gy、40Gy、45Gy、50Gy、55Gyまたは60Gy)が照射される。いくつかの実施形態において、放射線の総線量を一日にわたって照射され得るが、理想的には、分割して数日にわたって総線量が照射される。放射線療法は、少なくとも約3日間、例えば、少なくとも5、7、10、14、17、21、25、28、32、35、38、42、46、52または56日(約1−8週間)にわたって施されることが望ましい。したがって、放射線の毎日の線量は、約1から5Gy(例えば、約1Gy、1.5Gy、1.8Gy、2Gy、2.5Gy、2.8Gy、3Gy、3.2Gy、3.5Gy、3.8Gy、4Gy、4.2Gy、または4.5Gy)、好ましくは1から2Gy(例えば、1.5から2Gy)になるだろう。放射線の毎日の線量は、標的細胞の崩壊を誘導するために十分な線量であるべきである。期間を延長する場合には、照射が毎日行われないことが好ましく、これによって、動物を休息させつつ、治療効果の実現を可能にする。例えば、処置の各週の間に、照射は、連続する5日間にわたって行われ、2日間には行われないことが好ましく、これによって一週につき2日間の休息が実現される。しかし、動物の応答性および起こり得る任意の副作用に応じて、照射は、1日/週、2日/週、3日/週、4日/週、5日/週、6日/週、または7日/週すべてに行われ得る。放射線療法は、治療期間の任意の時点において開始され得る。好ましくは、照射は、治療期間の1週目または2週目に開始され、残りの治療期間にわたって行われる。例えば、固形腫瘍を処置する6週間にわたる治療期間のうち、1−6週間または2−6週間にわたって放射線が照射される。また、5週間にわたる治療期間のうち、1−5週間または2−5週間にわたって放射線が照射され得る。しかし、これらの例示的な放射線療法の管理スケジュールは、本発明を限定することを意図していない。
また、抗菌療法剤は、本発明において治療剤として使用され得る。微生物の生体の機能を破壊し得るか、阻害し得るか、またはそうでなければ減弱させ得る任意の薬剤、ならびにこのような活性を示すと考えられる任意の薬剤が使用され得る。抗菌剤としては、単独にか、または組み合わせて使用される、天然または合成の抗生物質、抗体、阻害タンパク質(例えば、ディフェンシン)、アンチセンス核酸、および膜破壊剤などが挙げられる。実際には、抗生物質の任意の種類が使用され得、これらとしては、限定するわけではないが、抗菌剤、抗ウイルス剤および抗真菌剤などが挙げられる。
本発明のいくつかの実施形態において、式I−XIaの化合物および1つ以上の治療剤もしくは抗がん剤は、1つ以上の以下の条件において:異なる頻度において、異なる期間において、異なる濃度において、異なる投与経路によって、単一の組成物として、別個の組成物として、動物に投与される。いくつかの実施形態において、上記化合物は、治療剤または抗がん剤を投与する前(例えば、治療剤または抗がん剤の投与の、0.5、1、2、3、4、5、10、12もしくは18時間前、1、2、3、4、5もしくは6日前、または1、2、3もしくは4週間前)に投与される。いくつかの実施形態において、上記化合物は、治療剤または抗がん剤を投与した後(例えば、治療剤または抗がん剤の投与の、0.5、1、2、3、4、5、10もしくは18時間後、1、2、3、4、5もしくは6日後、または1、2、3もしくは4週間後)に投与される。いくつかの実施形態において、上記化合物および治療剤もしくは抗がん剤は、同時投与されるが、異なるスケジュールにおいて投与される。例えば、上記化合物は毎日投与され、一方で治療剤または抗がん剤は、1週間に1回、2週間に1回、3週間に1回または4週間に1回、投与される。他の実施形態において、上記化合物は1週間に1回、投与され、一方で治療剤または抗がん剤は、毎日、1週間に1回、2週間に1回、3週間に1回または4週間に1回、投与される。
本発明の範囲内の組成物は、意図される目的を達成するために有効な量の本発明の化合物が含まれている、すべての組成物を包含する。個体にとっての必要性が異なり、各成分の有効量の最適な範囲は、当業者の能力の範囲において決定される。典型的には、上記化合物は、アポトーシスの誘導に応答を示す疾患に関して処置される哺乳類の体重について、1日につき0.0025〜50mg/kgまたは薬学的に受容可能なそれらの塩の対応量の用量において、哺乳類(例えば、ヒト)に対して経口投与され得る。例えば、疾患の処置、寛解または抑制のために、約0.01〜約25mg/kgを経口投与する。筋肉内注射の用量は、経口投与の約半分が一般的である。例えば、適切な筋肉内への用量は、約0.0025〜約25mg/kg(例えば、約0.01〜約5mg/kg)である。
経口的な単位用量は、約0.01〜1000mg(例えば約0.1〜100mg)の上記化合物を含み得る。単位用量は、約0.1〜10mg(好都合には約0.25〜50mg)の上記化合物またはその溶媒和物をそれぞれが含んでいる1つ以上の錠剤またはカプセル剤として1日に1回以上にわたって投与され得る。
局所製剤において、上記化合物は、1gの担体につき約0.01〜100mgの濃度において存在し得る。一実施形態において、上記化合物は、約0.07〜1.0mg/ml、例えば約0.1〜0.5mg/ml、例えば約0.4mg/mlの濃度において存在している。
また、未加工の化学物質として上記化合物を投与することに加えて、本発明の上記化合物は、薬学的に許容可能な適切な担体を含有している薬学的な調製物の一部として投与され得る。当該担体は、上記調製物への上記化合物の加工を容易にする、薬学的に使用され得る賦形剤および助剤化合物から構成される。調製物(特に経口的もしくは局所的に投与され得、投与の好ましい種類のために使用され得る調製物(例えば、錠剤、糖衣錠、徐放トローチおよび徐放カプセル剤、口内洗浄剤およびうがい薬、ゲル、液体懸濁液、ヘアリンス、ヘアゲル、シャンプー)、および直腸に投与され得る調製物(例えば、坐剤)、ならびに点滴によってか、注射によってか、局所的にか、または経口的に投与されるために好適な溶液剤)は、約0.01から99パーセント、例えば、0.25から75パーセントの活性な(複数の)化合物を、賦形剤と共に含んでいることが好ましい。
本発明の薬学的組成物は、本発明の化合物の有益な効果を受け得る任意の動物に投与され得る。当該動物において最も重要な動物は、哺乳類(例えば、ヒト)であるが、本発明はそれに限定されることを意図されていない。その他の動物としては、家畜(ウシ、ヒツジ、ブタ、ウマ、イヌおよびネコなど)が挙げられる。
本発明の化合物および薬学的組成物は、意図される目的を達成する任意の手法によって投与され得る。例えば、非経口、皮下、静脈内、筋肉内、腹腔内、経皮、頬、髄腔内、頭蓋内、鼻腔内または局所経路によって、投与され得る。代わりにか、または同時に、経口経路によって投与され得る。用量は、受容者の年齢、受容者の健康、受容者の体重、現在の処置の種類、もしあれば処置の頻度、および所望される効果の性質に依存する。
本発明の薬学的調製物は、公知の手法(例えば、混合、造粒、糖衣錠製造、溶解または凍結乾燥の従来のプロセス)によって、製造され得る。したがって、経口用の薬学的調製物は、固体賦形剤と活性な化合物との混合によって入手され得る。経口用の薬学的調製物は、所望されるか、または必要とされる場合に錠剤または糖衣錠のコアを得るために、必要に応じて適切な補助剤を追加した後に、生じた混合物を粉砕すること、および顆粒の混合物を加工することによって入手され得る。
特に、適切な賦形剤は、充填剤(例えば、糖類(例えば、ラクトースまたはスクロース、マンニトールまたはソルビトール、セルロースの調製物)および/またはリン酸カルシウム(例えば、リン酸三カルシウムまたはリン酸水素カルシウム))および結合剤(例えばデンプン糊(例えば、トウモロコシデンプン、小麦デンプン、米デンプン、ジャガイモデンプンを用いたデンプン糊)、ゼラチン、トラガカント、メチルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロースナトリウム、および/またはポリビニルピロリドン)である。崩壊剤(例えば、上述のデンプン、およびカルボキシメチル−デンプン、架橋ポリビニルピロリドン、寒天またはアルギン酸もしくはそれらの塩(例えば、アルギン酸ナトリウム))が、必要に応じて加えられる。補助剤は、何よりも、流出調節剤および潤滑剤(例えば、シリカ、タルク、ステアリン酸もしくはその塩(例えば、ステアリン酸マグネシウムまたはステアリン酸カルシウム)、および/またはポリエチレングリコール)である。糖衣錠のコアは、必要に応じて、胃液に耐性を示す適切なコーティングが施されている。この目的のために、濃縮された糖の溶液が使用され得る。当該溶液は、アラビアゴム、タルク、ポリビニルピロリドン、ポリエチレングリコールおよび/または二酸化チタン、ラッカー溶液、ならびに適当な有機溶媒もしくは溶媒混合物を任意に含んでいる。胃液に耐性を示すコーティングを生成するために、適切なセルロース調製物(例えば、アセチルセルロースフタル酸またはヒドロキシプロピルメチルセルロースフタル酸)の溶液が用いられる。例えば、活性な化合物の用量の組み合わせを特徴づけるためにか、または識別するために、錠剤または糖衣錠コーティングに染料または顔料を添加し得る。
経口的に使用され得る他の薬学的調製物としては、ゼラチン製のプッシュフィットカプセル、ならびにゼラチンおよび可塑剤(例えば、グリセロールまたはソルビトール)からできている密閉ソフトカプセルが挙げられる。プッシュフィットカプセルは、充填剤(例えばラクトース)、結合剤(例えばデンプン)、および/または潤滑剤(例えばタルクまたはステアリン酸マグネシウム)、ならびに必要に応じて安定剤とともに混合され得る顆粒の形態として、活性な化合物を含み得る。ソフトカプセルにおいて、活性な化合物は、適切な液体(例えば、脂肪油または流動パラフィン)に溶解されるか、または懸濁されることが好ましい。さらに、安定剤が添加され得る。
直腸に使用され得る薬学的調製物としては、例えば、1つ以上の活性な化合物と坐剤の基剤との組み合わせからなる座剤が挙げられる。適切な坐剤の基剤は、例えば、天然のトリグリセリド、合成のトリグリセリドまたはパラフィン炭化水素である。それに加えて、活性な化合物と基剤との組み合わせからなるゼラチン直腸カプセルを使用することが可能である。考えられる基剤としては、例えば、液体トリグリセリド、ポリエチレングリコール、またはパラフィン炭化水素が挙げられる。
非経口投与のための適切な製剤としては、水溶性の形態である活性な化合物の水性溶液(例えば水溶性塩およびアルカリ溶液)が挙げられる。さらに、適切な油性の注射懸濁液として、活性な化合物の懸濁液を投与し得る。適切な親油性も溶媒または賦形剤としては、脂肪油(例えばゴマ油)、または合成脂肪酸エステル(例えば、オレイン酸エチルもしくはトリグリセリドもしくはポリエチレングリコール−400)が挙げられる。水性の注射懸濁液は、懸濁液の粘度を増加させる物質(例えば、カルボキシメチルセルロースナトリウム、ソルビトール、および/またはデキストラン)を含み得る。上記懸濁液は任意で安定剤を含み得る。
本発明の局所組成物は、適切な担体を選択することによって、オイル、クリーム、ローション、および軟膏などとして調合されることが好ましい。適切な担体としては、植物油または鉱物油、白色ワセリン(白色軟パラフィン)、分岐鎖状の脂肪または油、動物性脂肪、および高分子量のアルコール(C12を超える)が挙げられる。好ましい担体は、有効成分が溶解性を示す担体である。また、必要に応じて、色または香りを付与する薬剤と同様に、乳化剤、安定剤、保湿剤および酸化防止剤を含め得る。また、これらの局所製剤の中に経皮浸透の促進剤を用いることができる。このような促進剤の例は、米国特許第3989816号および米国特許第4444762号に見られ得る。
クリーム剤は、鉱物油、自己乳化蜜蝋および水の混合物から調合されることが好ましく、当該混合物には、少量のオイル(例えばアーモンド油)に溶解された活性成分が混合されている。このようなクリーム剤の典型的な例は、約40部の水、約20部の蜜蝋、約40部の鉱物油および約1部のアーモンド油を含んでいるクリーム剤である。
軟膏は、植物油(例えばアーモンド油)に活性成分が溶解されている溶液を、温めた軟パラフィンと混合し、この混合物を冷却することによって調製され得る。このような軟膏の典型的な例は、約30重量%のアーモンド油および約70重量%の白色軟パラフィンを含んでいる軟膏である。
ローション剤は、適切な高分子量のアルコール(例えばプロピレングリコールまたはポリエチレングリコール)に、活性成分を溶解させることによって好適に調製され得る。
ある側面において、本発明は、以下の特定の実施形態に描写される。
I.式I:
を有している化合物、または薬学的に受容可能なそれらの塩もしくはプロドラッグであって、
ここで、
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、A2が存在しない場合にVがOであり;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキル、およびアラルキルからなる群から選択され;
Yが、CON(R1)、N(R1)CO、C(O)O、OC(O)、(CH2)1−3、任意に置換されているアリール、および任意に置換されているヘテロアリールからなる群から選択され、ここで、1つ以上のCH2基が、O、SまたはNR1によって置換され得;
Zが(CR2aR2b)rであり;
Dが(CR3aR3b)n−U−(CR4aR4b)mであり;
Uが、CR5aR5bおよびNR6からなる群から選択され;
Jが、(CR7aR7b)p−L−(CR8aR8b)qであり;
Tが、任意に置換されているヘテロアリールであり;
n、m、qおよびpが独立して、0−5からなる群から選択され;
rが、0−3であり;
R1が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択され;
R2a、R2b、R3a、R3b、R4a、R4b、R5a、R5b、R7a、R7b、R8aおよびR8bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環およびCOR9からなる群から選択され;
Lが、O、S、NR1、NCOR9、CR7aR7b、C=O、C=SおよびC=NR1からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択される、化合物。
II.nが1であり、mが1または2であり、pは0であり、LがCR7aR7b、qが1、ならびに、R2a、R2b、R3a、R3b、R4a、R4b、R5a、R5b、R7a、R7b、R8aおよびR8bが水素である、Iの化合物。
III.YがCON(H)であり、WがCHであり、rが0であり、VがNである、IIの化合物。
IV.式II:
V.nが1であり、mが1または2であり、pが0であり、LがCH2であり、qが1であり、R3a、R3b、R4a、R4b、R7a、R7b、R8aおよびR8bが水素である、IVの化合物。
VI.YがCON(H)であり、WがCHであり、rが0であり、VがNである、Vの化合物。
VII.式V:
VIII.A1は任意に置換されているアルキルであり、A2は水素である、VIIの化合物。
IX.Xは任意に置換されているアルキルである、VIIの化合物。
X.UがCH2である、VIIの化合物。
XI.UがNR6である、VIIの化合物。
XII.R6がCOR9である、XIの化合物。
XIII.R9が任意に置換されているアルキルおよびアラルキルからなる群から選択される、XIIの化合物。
XIV.mは2である、VIIの化合物。
XV.mは1である、VIIの化合物。
XVI.A1が任意に置換されているアルキルであり、A2が水素であり、Xが任意に置換されているアルキルであり、UがNR6であり、R6がCOR9であり、R9が任意に置換されているアルキルおよびアラルキルからなる群から選択され、mが1である、VIIの化合物。
XVII.Tが、
からなる群から選択されるI−XVの化合物であって、
ここで、
Qが、O、SおよびNR12からなる群から選択され;
R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールおよび任意に置換されているヘテロ環からなる群から選択され;
R10a、R10b、R11a、R11b、R11cおよびR11dが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドなる群から選択され;
Z1、Z2およびZ3が独立して、CR11eおよびNからなる群から選択され、ここで、Z1、Z2およびZ3の少なくとも1つがCR11eであり、Z1、Z2およびZ3の少なくとも1つがNであり;
R11eが、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択される、化合物。
XVIII.Tが、
からなる群から選択される、XVIの化合物であって、
ここで、
Qが、O、SおよびNR12からなる群から選択され;
R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールおよび任意に置換されているヘテロ環からなる群から選択され;
R10a、R10b、R11a、R11b、R11cおよびR11dが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドなる群から選択される、化合物。
XIX.Tが、
XX.R10aが任意に置換されているアリールであり、R10bが水素である、XIXの化合物。
XXI.Tが、
XXII.R11aが、任意に置換されているアリール、アラルキルおよび任意に置換されているアルキルからなる群から選択され、R11b、R11c、およびR11dのそれぞれが水素である、XXIの化合物。
XXIII.R11dが、任意に置換されているアリール、アラルキルおよび任意に置換されているアルキルからなる群から選択され、R11a、R11b、およびR11cのそれぞれが水素である、XXIの化合物。
XXIV.式XIa:
を有している、XXIIの化合物であって、
ここで、R9が任意に置換されているアルキルまたはアラルキルであり、R11aが任意に置換されているアリールである、化合物。
XXV.以下のもの:
からなる群から選択される、Iの化合物または薬学的に受容可能なそれらの塩もしくはプロドラッグ。
XXVI.I−XXVのいずれか1つの化合物、および薬学的に受容可能な担体を含んでいる薬学的組成物。
XXVII.細胞におけるアポトーシスを誘導する方法であって、上記細胞をI−XXVのいずれか1つの化合物に接触させる工程を包含する方法。
XXVIII.アポトーシスの誘導因子に対して細胞を感受性にさせる方法であって、上記細胞をI−XXVのいずれか1つの化合物に接触させる工程を包含する方法。
XXIX.上記細胞をアポトーシスの誘導因子に接触させる工程をさらに包含する、XXXVIIIの方法。
XXX.アポトーシスの上記誘導因子が化学療法薬である、XXIXの方法。
XXXI.アポトーシスの上記誘導因子が放射線である、XXIXの方法。
XXXII.アポトーシスの上記誘導因子が、腫瘍壊死因子(TNF)、TNF関連リガンド、またはTRAIL−R1もしくはTRAIL−R2のアゴニストである、XXIXの方法。
XXXIII.上記TNF関連リガンドが、TRAMPリガンド、Fas/CD95リガンド、TNFR−1リガンドおよびTRAILからなる群から選択される、XXXIIの方法。
XXXIV.上記TFT関連リガンドがTRAILである、XIIIの方法。
XXXV.上記TRAIL−R1もしくはTRAIL−R2のアゴニストが抗体である、XXXIVの方法。
XXXVI.アポトーシスの誘導に対して反応性を有している、動物における疾患を処置、寛解または抑制する方法であって、I−XXVのいずれか1つの化合物の治療有効量を上記動物に投与する工程を包含する方法。
XXXVII.アポトーシスの上記誘導因子を投与する工程をさらに包含する、XXXVIの方法。
XXXVIII.アポトーシスの上記誘導因子が化学療法薬である、XXXVIIの方法。
XXXIX.アポトーシスの上記誘導因子が放射線である、XXXVIIIの方法。
XL.アポトーシスの上記誘導因子が、TNF、TNF関連リガンド、またはTRAIL−R1もしくはTRAIL−R2のアゴニストである、XXXVIIの方法。
XLI.上記TNF関連リガンドが、TRAMPリガンド、Fas/CD95リガンド、TNFR−1リガンドおよびTRAILからなる群から選択される、XLの方法。
XLII.上記TFT関連リガンドがTRAILである、XLIの方法。
XLIII.上記TRAIL−R1またはTRAIL−R2のアゴニストは、抗体である、XLIIの方法。
XLIV.アポトーシスの誘導に対して反応性を有している疾患が過剰増殖性疾患である、XXXVIの方法。
XLV.上記過剰増殖性疾患ががんである、KLIVの方法。
XLVI.アポトーシスの上記誘導因子の前に上記I−XXVの化合物を投与する、XXXVIの方法。
XLVII.アポトーシスの上記誘導因子の後に上記I−XXVの化合物を投与する、XXXVIの方法。
XLVIII.アポトーシスの上記誘導因子と同時に上記I−XXVの化合物を投与する、XXXVIの方法。
XLIX.動物における過剰増殖性疾患を処置、寛解または抑制する方法であって、I−XXVのいずれか1つの化合物の治療有効量を、上記動物に投与する工程を包含する方法。
L.抗がん剤を投与する工程をさらに包含する、XLIXの方法。
LI.上記抗がん剤がアポトーシスの誘導因子である、Lの方法。
LII.アポトーシスの上記誘導因子が化学療法薬である、LIの方法。
LIII.アポトーシスの上記誘導因子が放射線である、LIIの方法。
LIV.アポトーシスの上記誘導因子が、TNF、TNF関連リガンド、またはTRAIL−R1もしくはTRAIL−R2のアゴニストである、LIの方法。
LV.上記TNF関連リガンドが、TRAMPリガンド、Fas/CD95リガンド、TNFR−1リガンドおよびTRAILからなる群から選択される、LIVの方法。
LVI.上記TFT関連リガンドがTRAILである、LIVの方法。
LVII.上記TRAIL−R1またはTRAIL−R2のアゴニストは、抗体である、LIVの方法。
LVIII.上記過剰増殖性疾患ががんある、XLIXの方法。
LIX.上記抗がん剤の前に上記I−XXVの化合物を投与する、Lの方法。
LX.上記抗がん剤の後に上記I−XXVの化合物を投与する、Lの方法。
LXI.上記抗がん剤と同時に上記I−XXVの化合物を投与する、Lの方法。
LXII.上記抗がん剤が、タキソテール、ラパチニブおよびゲムシタビンから成る群から選択される、Lの方法。
LXIII.それらを必要とする動物における血管新生を抑制または阻害する方法であって、I−XXVのいずれか1つの化合物の治療有効量を、上記動物に投与する工程を包含する方法。
LXIV.上記動物が、黄斑変性症、関節リウマチ、乾癬、糖尿病性網膜症、未熟児網膜症、角膜移植片拒絶反応、血管新生緑内障、水晶体後線維増殖症、虹彩、オスラー-ウェバー症候群、心筋血管新生、プラーク血管新生、毛細血管拡張、血友病関節、血管線維腫、創傷造粒、腸の癒着、アテローム性動脈硬化症、強皮症、および肥厚性瘢痕からなる群から選択される疾患または障害にかかっている、LXIIIの方法。
LXV.上記I−XXVのいずれか1つの化合物、および当該化合物を動物に投与するための取扱説明書を含んでいる、キット。
LXVI.抗がん剤をさらに含んでいる、LXVのキット。
LXVII.上記抗がん剤がアポトーシスの誘導因子である、LXVIのキット。
LXVIII.アポトーシスの上記誘導因子が化学療法薬である、LXVIIのキット。
LXIX.アポトーシスの上記誘導因子が、TNF、TNF関連リガンド、またはTRAIL−R1もしくはTRAIL−R2のアゴニストである、LXVIIのキット。
LXX.上記TNF関連リガンドが、TRAMPリガンド、Fas/CD95リガンド、TNFR−1リガンドおよびTRAILからなる群から選択される、LXIXのキット。
LXXI.上記TFT関連リガンドがTRAILである、LXXのキット。
LXXII.上記TRAIL−R1またはTRAIL−R2のアゴニストは、抗体である、LXIXのキット。
LXXIII.上記取扱説明書が、過剰増殖性疾患にかかっている動物に上記化合物を投与するためのものである、LXVのキット。
LXXIV.上記過剰増殖性疾患ががんである、LXXIIIのキット。
LXXV.式XII:
(ここで、
R13が、N(H)P1および、
からなる群から選択され;
P1が、アミン保護基であり;
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、VがOである場合にA2が存在せず;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキルおよびアラルキルからなる群から選択され;
Yが、CON(R1)、N(R1)CO、C(O)O、OC(O)、(CH2)1−3、任意に置換されているアリール、および任意に置換されているヘテロアリールからなる群から選択され、ここで、1つ以上のCH2基が、O、SまたはNR6によって置換され得;
Zが、(CR2aR2b)rであり;
Uが、CR5aR5bおよびNR1からなる群から選択され;
mが、1または2であり;
rが、0−3であり;
R1が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択され;
R2a、R2b、R5aおよびR5bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールおよび任意に置換されているヘテロ環からなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環、およびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択され;
R10aおよびR10bが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択される)
の化合物を調製する方法であって、
(a)式XIII:
の化合物をアンモニアと縮合させて、式XIV:
の化合物を生成させる工程、
(b)式XIVの化合物を式XV:
の化合物に転化させる工程、
(c)式XVの化合物を式XVI:
(ここで、L2は離脱基である)
の化合物と縮合させて、式XVII:
を生成させる工程、ならびに、
(b)式XVIIの化合物を環化させて式XIIの化合物を生成させる工程を包含している、方法。
LXXVI.L2がClおよびBrからなる群から選択される、LXXVの方法。
LXXVII.L2がBrである、LXXVIの方法。
LXXVIII.R10aが任意に置換されているアリールである、LXXVIの方法。
LXXIX.mが1である、LXXVの方法。
LXXX.R13が−N(H)P1である、LXXVの方法。
LXXXI.P1がt−ブトキシカルボニルおよびベンジルオキシカルボニルからなる群から選択される、LXXXの方法。
LXXXII.R13が、
LXXXIII.YがCON(H)であり、WがCHであり、rが0であり、VがNである、LXXXIIの方法。
LXXXIV.UがNR6であり、R6がCOR9である、LXXVの方法。
LXXXV.式XVIII:
(ここで、
R13が、N(H)P1および、
からなる群から選択され;
P1が、アミン保護基であり;
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、VがOである場合にA2が存在せず;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキル、およびアラルキルからなる群から選択され;
Yが、CON(R1)、N(R1)CO、C(O)O、OC(O)、(CH2)1−3、任意に置換されているアリール、および任意に置換されているヘテロアリールからなる群から選択され、ここで、1つ以上のCH2基が、O、SまたはNR1によって置換され得;
Zが、(CR2aR2b)rであり;
Uが、CR5aR5bおよびNR6からなる群から選択され;
mが、1または2であり;
rが、0−3であり;
R1が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択され;
R2a、R2b、R5aおよびR5bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環、およびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択され;
R11a、R11b、R11cおよびR11dが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドからなる群から選択され;
R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択される)
の化合物を調製する方法であって、
(a)式XIII:
の化合物を式XIX:
の化合物と縮合させて式XX:
を生成させる工程、ならびに、
(b)式XXの化合物を環化させて式XVIIIの化合物を生成させる工程を包含している、方法。
LXXXVI.R12が水素である、LXXXVの方法。
LXXXVII.R11aが任意に置換されているフェニルである、LXXXVの方法。
LXXXVIII.mが1である、LXXXVの方法。
LXXXIX.R13が−N(H)P1である、LXXXVの方法。
XC.P1がt−ブトキシカルボニルおよびベンジルオキシカルボニルからなる群から選択される、LXXXIXの方法。
XCI.R13が、
XCII.YがCON(H)であり、WがCHであり、rが0であり、VがNである、XCIの方法。
XCIII.UがNR6であり、R6がCOR9である、LXXXVの方法。
XCIV.式XXI:
(ここで、
A1およびA2が独立して、水素および任意に置換されているアルキルからなる群から選択され、ここで、VがOである場合にA2が存在せず;
Vが、N、CHおよびOからなる群から選択され;
Wが、CHおよびNからなる群から選択され;
Xが、水素、任意に置換されているアルキルおよびアラルキルからなる群から選択され;
Zが、(CR2aR2b)rであり;
Uが、CR5aR5bおよびNR6からなる群から選択され;
mが、1または2であり;
rが、0−3であり;
R2a、R2b、R5aおよびR5bのそれぞれが独立して、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択され;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環、およびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択され;
Tが、任意に置換されているヘテロアリールである)
の化合物を調製する方法であって、
式XXIII:
の化合物を式XXIV:
(ここで、L1が離脱基である)
の化合物と反応させて、式XXIの化合物を生成させる工程を包含している、方法。
XCV.WがCHであり、VがNである、XCIVの方法。
XCVI.Tが、
からなる群から選択され、
ここで、
Qが、O、SおよびNR12からなる群から選択され;
R12が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリールおよび任意に置換されているヘテロ環からなる群から選択され;
R10a、R10b、R11a、R11b、R11cおよびR11dが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドなる群から選択される、XCIVの方法。
XCVII.UがCH2である、XCIVの方法。
XCVIII.UがNCOR9である、XCIVの方法。
XCIX.R9が任意に置換されているアルキルである、XCVIIIの方法。
C.L1が−Clおよび−OHからなる群から選択される、XCIVの方法。
CI.L1が−OHであり、活性化剤の存在下において縮合が実施される、Cの方法。
CII.上記XXIIIの式の化合物が、式XXIIの化合物:
(ここで、P1がアミン保護基である)
からP1を除去することによって調合される、XCIVの方法。
CIII.P1がt−ブトキシダルボニルおよびベンジルオキシカルボニルからなる群から選択される、CIIの方法。
CIV.式XXII:
を有している化合物であって、
ここで、
Tが、任意に置換されているヘテロアリールであり;
mが、1または2であり;
Uが、CH2またはNR6であり;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環、およびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、および任意に置換されているヘテロ環からなる群から選択され;
P1が、アミン保護基である、化合物。
CV.P1がt−ブトキシダルボニルおよびベンジルオキシカルボニルからなる群から選択される、CIVの化合物。
CVI.式XXV:
を有している化合物であって、
ここで、
Tが、任意に置換されているヘテロアリールであり;
mが、1または2であり;
Uが、CH2またはNR6であり;
R6が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環、およびCOR9からなる群から選択され;
R9が、水素、任意に置換されているアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリールおよび任意に置換されているヘテロ環からなる群から選択され;
Xが、水素、任意に置換されているアルキル、およびアラルキルからなる群から選択され;
A1が、水素および任意に置換されているアルキルからなる群から選択され;
P1が、アミン保護基である、化合物。
CVII.P1がt−ブトキシダルボニルおよびベンジルオキシカルボニルからなる群から選択される、CVIの化合物。
CVIII.以下の構造:
以下の実施例は例示的なものであり、本発明の方法および組成物を限定するものではない。臨床治療において通常に直面する様々な状態および要因の、当業者にとって明白な他の適切な変更および適合は、本発明の精神および範囲の内である。
(実施例1:共有結合的に制限されている(Covalently Constrained)Smac模倣物の合成)
一般的な手法:NMRスペクトルを陽子周波数300MHzにおいて得た。1H化学シフトを、Me4Si(0.00ppm)、CHCl3(7.26ppm)、CD2HOD(3.31ppm)またはDHO(4.79ppm)を内部標準として用いて記録している。13C化学シフトを、CDCl3(77.00ppm)、CD3OD(49.00ppm)、または1,4−ジオキサン(67.16ppm)を内部標準として用いて記録している。旋光度を室温において測定した。本発明の化合物は、逆層HPLC(水中0.1%のTFA、溶離液としてのアセトニトリル中0.1%のTFA)によって精製され、TFA塩として分離され得る。
一般的な手順A(カルボキシル酸とアミンとの縮合)
2つの上記基質のCH2Cl2溶液(副次的な基質について20mg/mL)に、EDC(アミノ基当たり1.1当量)、HOBt(アミノ基当たり1.1当量)、およびN,N−ジイソプロピルエチルアミン(アミノ基当たり4当量)を、攪拌しながら0℃において加えた。混合物を室温において8時間にわたって混合し、それから濃縮した。残余物をクロマトグラフィーによって精製して生成物を得た。
一般的な手順B(Bocの脱保護)
上記基質のメタノール溶液(20mg/mL)に、HClの1,4−ジオキサン溶液(4M、Boc当たり10〜20当量)を加えた。その溶液を室温において一晩にわたって混合し、それから凝縮して生成物を得た。
(実施例2:Smac模倣物中間体の合成)
立体配置的に制約されているSmac模倣物にとっての合成経路における中間体は、手順1〜7に記載の方法論を用いて合成され得る。
試薬および条件:(a)i.1,4−ジオキサン、メタノール中の1.4N HCl;ii.Boc−Dap(Z)−OH、EDC、HOBt、N,N−ジイソプロピルエチルアミン、CH2Cl2、2ステップの間52%;(b)O3、次に、PPh3、CH2Cl2、90%;(c)H2、10%Pd−C、i−PrOH、41%;(d)H2、10%Pd−C、i−PrOH;(e)NaBH(OAc)3、THF;(f)i.デス−マーチン−ペルヨージナン(Dess-Martin periodinane)、CH2Cl2;ii.H2、10%Pd−C、i−PrOH、2ステップの間50%;(h)H2、10%Pd−C、i−PrOH;(i)NaBH(OAc)3、THF。
中間体5および7の合成は手順1に示されている。化合物2は、報告されている方法((1) Zhang, J.; Xiong, C.; Wang, W.; Ying, J.; Hruby, V., J. Org. Lett., 2002, 4 (23), 4029-4032、(2) Polyak, F. and Lubell, W. D. J. Org, Chem. 1998, 63, 5937-5949、および(3)Tetrahedron Letters 2005, 46, 945-947参照)に従って、R型異性体を主要な生成物(比率約4:1)として有している2つのジアステレオマーの混合物として、ピログルタミン酸が1つから5つのステップにおいて調製され得る。2におけるBoc基の除去の後、N−α−(tert−ブトキシルカルボニル)−N−β−(ベンズオキキシルカルボニル)−L−ジアミノ−プロピオン酸(Boc−Dap(Z)−OH)との縮合によってアミド3が得られた。3におけるC−C二重結合のオゾン酸化によってアルデヒド4が得られた。4におけるCbz基の切断、生成されたアミンとアルデヒド基との分子内縮合、およびエナミンのその後の還元をワンポットにおいて実現し、長時間の反応において化合物5が生成じた。この代わりに、4のCbz基の脱保護、分子内環化、エナミン中間体の分離および還元によって5が生成される。この変換において化合物5のみが得られ、その異性体の検出可能な形成がなく、これらの条件において副次的な異性体からのアミノ酸アルデヒドが環化しないことを示唆している。
化合物2(540mg、2mmol)の20mLのメタノール溶液に、4規定のHClの4mlの1,4−ジオキサン溶液を加えた。この溶液を室温において一晩にわたって撹拌し、それから凝縮してアンモニウム塩が得られた。この塩と15mLのジクロロメタンとの混合物に、1.17g(2.4当量)のBoc−Dap(Z)−OH・DCHA、460mg(2.4mmol)のEDC、320g(2.4mmol)のHOBt、および3mLのN,N−ジイソプロピルエチルアミンを加えた。その混合物を室温において一晩にわたって攪拌し、それから凝縮した。残余物をクロマトグラフィーによって精製して、化合物3(YP−348)(580mg、59%)が得られた。1H NMR (300 MHz, CDCl3, TMS) (major isomer) δ 7.34-7.28 (m, 5H), 5.80-5.77 (m, 1H), 5.59 (m, 1H), 5.36-5.33 (d, J = 10.0 Hz, 2H), 5.19-5.01 (m, 4H), 4.67-4.62 (m, 1H), 4.47-4.44 (m, 1H), 3.76-3.74 (s, 1H), 3.74-3.71 (s, 2H), 2.32-2.30 (m, 1H), 2.16-2.12 (m, 1H), 1.99-1.95 (m, 2H), 1.42 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 172.4, 170.5, 156.5, 155.2, 136.4, 134.6, 133.8, 128.3, 127.9, 118.5, 117.1, 80.0, 66.6, 59.7, 58.2, 52.6, 43.4, 29.2, 28.1, 26.6。
化合物3(490mg、1mmol)の20mLのCH2Cl2溶液に、淡い青色になるまで、−78℃においてO3を通してバブリングした。15分間以上にわたってO3によってバブリングした後、空気によってバブリングして、過剰なO3を取り除いた。3mLのEt3Nを加えた後、その混合物を室温まで加熱し、1時間にわたって攪拌した。溶媒を蒸発させ、クロマトグラフィーによって残余物を精製してアルデヒド4(YP−367)(340mg、69%)が得られた。1H NMR (300 MHz, CDCl3, TMS) (major isomer) δ 9.78-9.67 (m, 1H), 7.53-7.32 (m, 5H), 5.44 (s, 1/2 H), 5.32 (s, 1/2 H), 5.15-5.06 (m, 2H), 4.64 (m, 1H), 4.40-4.39 (m, 1H), 3.78-3.76 (s, 3/2 H), 3.76-3.74 (s, 3/2H), 3.48-3.42 (m, 3H), 2.78-2.52 (m, 1H), 2.40-2.20 (m, 1H), 2.16 (m, 2H), 2.06-1.89 (m, 1H), 1.44-1.43 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 200.3, 199.5, 172.6, 172.2, 170.3, 156.5, 136.4, 128.4, 128.0, 66.7, 59.7, 59.1, 54.3, 52.4, 52.3, 48.4, 43.3, 29.6, 28.2, 21.0。
化合物4(290mg、0.6mmol)の20mLのイソプロパノール溶液に、0.2gの10%Pd/Cを加えた。その混合物をH2存在下の室温において一晩にわたって攪拌し、セライトに通して濾過し、凝縮した。残余物を無水THFに溶解させた。この溶液に、NaBH(OAc)3(380mg、1.8mmol)を加えた。この混合物を室温において一晩にわたって撹拌し、Na2SO4を用いて希釈し、塩水を用いて洗浄し、Na2SO4に通して乾燥させ、濃縮した。残余物をクロマトグラフィーによって精製して、化合物5(72mg、35%)が得られた。[α] 20 D - 30.2 (c = 1.7, CHCl3); 1H NMR (300 MHz, CDCl3, TMS) δ5.45 (brd, J = 8.0 Hz, 1H), 4.67 (m, 1H), 4.52 (t, J = 9.0 Hz, 1H), 4.23 (m, 1H), 3.74 (s, 3H), 3.20 (m, 2H), 2.94 (m, 1H), 2.74 (dd, J = 13.6, 10.9 Hz, 1), 2.35 (m, 1H), 2.14 (m, 1H), 1.99 (m, 1H), 1.86-1.74 (m, 3H), 1.66 (m, 1H), 1.43 (brs, 9H); 13C NMR (75 MHz, CDCl3, TMS) δ 173.42, 170.60, 155.16, 79.68, 59.46, 58.39, 54.92, 52.44, 46.72, 37.45, 32.15, 29.64, 28.29, 26.98。
9−BBNを用いて3におけるC−C二重結合を臭化水素化した後に、生じたボランのアルカリ酸化によって、アルコール6が得られた。デス−マーチン−ペルヨージナンを用いた6の酸化によって、2つのアルデヒドの混合物が得られた。この混合物を、化合物5にとっての手法と同じ手順において環化させて、化合物7が生じた。5と同様に、この変換において、単一の異性体のみが得られた。
化合物7に関する分析データ:[α] 20D - 23.2 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3, TMS) δ 5.23 (brd, J = 8.0 Hz, 1H), 4.79 (m, 1H), 4.65 (dd, J = 9.7, 8.2 Hz), 4.22 (m, 1H), 3.74 (s, 3H), 3.02-2.80 (m, 4H), 2.38-1.70 (m, 9H), 1.43 (brs, 9H); 13C NMR (75 MHz, CDCl3, TMS) δ 173.38, 171.59, 155.09, 79.68, 62.03, 59.82, 53.72, 53.15, 52.48, 50.09, 34.66, 34.55, 29.47, 28.31, 27.33。
YP−248Pに関する分析データ:1H NMRは、この化合物が2:1の比において2つの回転異性体を有していること示している。1H NMR (300 MHz, CDCl3, TMS) δ 7.47-7.44 (m, 1H), 7.38-7.32 (m, 4H), 5.65-5.62 (d, J = 8 Hz, 1H), 5.31-5.16 (m, 2H), 4.64-4.60 (m, 1H), 4.51-4.46 (t, J = 8 Hz, 1H), 4.24-4.23 (m, 1H), 4.23-4.21 (m, 1H), 3.75 (s, 1H), 3.73 (s, 2H), 3.66-3.63 (m, 1H), 3.63-3.61 (m, 1H), 3.61-3.31 (m, 1H), 2.36-2.34 (m, 1H), 2.11-1.76 (m, 6H), 1.44-1.45 (s, 9H)。
アミド中間体に関する分析データ:1H NMR (300 MHz, CDCl3, TMS) δ 5.79 (brd, J = 7.0 Hz, 1H), 4.50-4.35 (m, 2H), 4.05 (m, 1H), 3.98-3.85 (m, 2H), 3.70 (s, 3H), 3.32-3.04 (m, 2H), 2.54 (m, 1H), 2.40-2.26 (m, 2H), 2.25-1.60 (m, 6H), 1.39 (s, 9H), 0.98-0.89 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 173.12, 172.52, 168.85, 154.69, 79.80, 59.51, 56.11, 54.38, 53.51, 52.23, 46.18, 42.02, 32.51, 31.12, 28.12, 26.54, 25.81, 22.69, 22.40。
YP−237Pに関する分析データ:[α] 20 D -21.5° (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3, TMS) δ 3.71 (t, J = 6.5 Hz, 3H), 3.60 (dd, J = 9.0, 5.4 Hz, 1H), 3.11 (m, 1H), 2.05 (m, 1H), 1.95-1.63 (m, 3H), 1.46 (s, 9H), 1.25 (m, 1H), 0.89 (s, 9H), 0.05 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 174.5, 80.8, 61.5, 60.6, 57.5, 38.8, 31.8, 30.4, 28.0, 25.9, 18.2, -5.4; HRMS: calcd. m/z for [M+H]+330.2464; found 330.2466。
YP−238Pに関する分析データ:[α] 20 D-90.0° (c = 1.67, CHCl3); 1H NMRは、この化合物が1:1の比において2つの回転異性体を有していることを示している。1H NMR (300 MHz, CDCl3, TMS) δ 7.28 (m, 5H), 5.59 (m,1H), 5.35 (m, 1H), 5.20-5.05 (m, 2H), 4.85 (m, 1/2 H), 4.65 (m, 1/2 H), 4.46 (m, 1H), 4.35 (m, 1H), 3.80 (m, 1/2 H), 3.70-3.50 (m, 2H), 3.40 (m, 1H), 3.25 (m, 1/2 H), 2.32 (m, 1H), 2.20-1.50 (m, 4H), 1.46 (s, 4.5H), 1.44 (s, 4.5H), 1.43 (s, 4.5H), 1.41 (s, 4.5H); HRMS: calcd m/z 558.2791 for [M+Na]+; found 558.2794。
YP−239に関する分析データ:[α] 20 D-51.6° (c = 1.67, CHCl3); 1H NMRは、この化合物が2:1の比において2つの回転異性体を有していることを示している。1H NMR (300 MHz, CDCl3, TMS) δ 9.76 (s, 2/3 H), 9.71 (s, 1/3 H), 7.40-7.28 (m, 5H), 5.72-5.30 (m, 2H), 5.20-4.95 (m, 2H), 4.90-4.25 (m, 3H), 3.52-3.05 (m, 3H), 2.90-1.60 (m, 4H), 1.50-1.35 (m, 18H); HRMS: calcd m/z 556.2635 for [M+Na]+;found 556.2629。
YP−239Pに関する分析データ:[α] 20 D -8.4° (c = 0.65, CHCl3); 1H NMR (300 MHz, CDCl3, TMS) δ 5.49 (brd, J = 8.1 Hz, 1H), 4.70 (m, 1H), 4.41 (t, J = 9.3 Hz, 1H), 4.30 (m, 1H), 3.25-3.18 (m, 2H), 2.89 (m, 1H), 2.75 (dd, J = 13.5, 11.1 Hz, 1H), 2.34 (m, 1H), 2.18-1.60 (m, 6H), 1.49 (s, 9H), 1.44 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 171.8, 170.4, 155.2, 81.7, 79.5, 60.6, 58.5, 54.9, 52.3, 46.9, 37.5, 32.1, 28.3, 28.0, 27.0; HRMS: calcd m/z 406.2318 for [M+Na]+; found 406.2317。
化合物6は、文献(Duggan et al., Org. Biomol. Chem. 3:2287 (2005))に報告されている方法に従って、調製され得る(手順4)。アルケンの還元およびベンジルエステルの加水分解によって、酸7が得られた。
化学式Aによって表される化合物は、手順5に示される方法によって調製され得る。科学式Aにおいて、mは1〜2であり、R10aおよびR10bが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、または任意に置換されているヘテロ環であり、Uが、式Iについて上述されたような意味を有している。簡単に言えば、アンモニアと酸aの縮合によって、一級アミドbが生じる。一級アミドからチオアミドcへの選択的な変換は、室温において、CH2Cl2におけるP4S10との、bの反応によって実現され得る。cとの、dの反応によってeが得られる(ここで、L2が離脱基である)。L2は離脱基である。一実施形態において、dはα−ブロモケトンである。エタノールにおける還流によってeを環化させて、化学式Aのチオゾール(thiozole)が得られる。
化学式Bによって表される化合物は、手順6に記載されているように調製され得る。化学式Bにおいて、mが1〜2であり、R11a、R11b、R11cおよびR11dが独立して、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているシクロアルケニル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環、ハロ、ニトロ、シアノ、ヒドロキシ、アミノ、アルコキシ、アリールオキシ、アリールアルキルオキシ、アルキルチオ、カルボキシアミドおよびスルホンアミドであり;R12が、水素、任意に置換されているアルキル、ハロアルキル、アラルキル、任意に置換されているシクロアルキル、任意に置換されているアルケニル、任意に置換されているアルキニル、任意に置換されているアリール、任意に置換されているヘテロアリール、任意に置換されているヘテロ環であり、Uが、式Iについて上述されたような意味を有している。簡単に言えば、置換ジアミノベンゼンbと酸aとの縮合によってアミドcが得られる。AcOHにおける還流によってcを環化させて化学式Bの化合物が得られる。
化学式Cによって表される化合物は、手順7において示されたように調製され得る。化学式Cにおいて、mが1または2であり、A1、A2、Z、X、TおよびUは、式Iについて上述されたような意味を有している。簡単に言えば、aにおけるBoc保護基の除去によってアミンbが得られる。対応するBoc保護アミノ酸とbとの縮合によってアミドcが得られる。cにおけるBoc保護基の除去によってdが得られる。ハロゲン化アルキルを用いたdの置換によるA1基の導入、または対応するアルデヒドを用いたdの還元的アミノ化によって、化学式Cによって表されるSmac模倣物が得られる。
化学式Dによって表される化合物は、手順8に記載されているように調製され得る。化学式Dにおいて、mが1または2であり、TおよびR9は、式1について上述されたような意味を有している。簡単に言えば、カルボン酸R9CO2Hまたはカルボン酸塩化物R9COClとの、アミンaの反応によって、アミドbが得られる。bのBoc保護基の除去によって、化学式Dによって表される化合物が得られる。
試薬および条件:(a)3−ブロモ−1,2−ジアミン、EDC、HOBt、N,N−ジイソプロピルエチルアミン、CH2Cl2;(b)HOAc、70℃、10h;(c)アリールボロン酸、dba Pd、トリ−tert−ブチルホスフィン、K2CO3、メチレングリコールジメチルエーテル、還流、一晩;(d)i.1,4−ジオキサン中の4N HCl、メタノール;ii.L−N−Boc−N−メチル−アラニン、EDC、HOBt、N,N−ジイソプロピルエチルアミン、CH2Cl2;(e)i.H2、10%Pd−C、メタノール;ii.R9CO2H、EDC、HOBt、N,N−ジイソプロピルエチルアミン、CH2Cl2;iii.1,4−ジオキサン中の4規定のHCl、メタノール。
化学式Eによって表される化合物は、手順9に記載されているように調製され得る。化学式Eにおいて、mが1または2であり、R9は、式1について上述されたような意味を有している。簡単に言えば、3−ブロモ−1,2−ジアミンとの、酸1の反応によってアミド2が得られる。酢酸における2の環化によってベンズイミダゾール3が得られる。アリールボロン酸(例えば、2−,3−または4−フルオロフェニルボロン酸)との鈴木カップリングによって4が得られる。4のBoc脱保護、およびL−N−Boc−N−メチルアラニンとの反応によって5が得られる。Cbz脱保護、R9CO2Hとのカップリング、およびBoc脱保護によって化学式Eによって表される化合物が得られる。
(実施例3)
SM−1229に関する分析データ: 1H NMR (300 MHz, D2O) δ 7.65-7.50 (m, 2H), 7.21 (s, 1H), 7.18-6.96 (m, 3H), 5.20 (t, J = 6.86 Hz, 1H), 4.70 (m, 1H), 4.20 (m, 1H), 3.89 (m, 1H), 2.65 (s, 3H), 2.20-1.70 (m, 4H), 1.70-1.20 (m, 11H); 13C NMR (75 MHz, D2O) δ 173.18, 172.38, 169.49, 153.65, 133.89, 129.09, 128.52, 126.44, 114.79, 60.55, 60.26, 57.20, 50.95, 37.08, 33.03, 32.44, 31.37, 31.15, 25.28, 22.63, 15.72。
(実施例4)
SM−1235(主な配座異性体)に関する分析データ: 1H NMR (300 MHz, D2O) δ 7.72-7.65 (m, 2H), 7.48 (s, 1H), 7.40-7.20 (m, 3H), 5.21 (m, 1H), 5.02 (m, 1H), 4.32 (m, 1H), 3.98-3.20 (m, 5H), 2.55 (s, 3H), 2.35-1.70 (m, 9H), 1.42 (d, J = 7.2 Hz, 3H), 0.80-0.62 (m, 6H); 13C NMR (75 MHz, D2O) δ175.84, 173.60, 169.91, 154.59, 133.96, 129.42, 126.69, 114.57, 71.08, 61.57, 59.80, 57.28, 51.35, 46.62, 42.82, 34.30, 31.62, 22.25, 22.09, 15.61。
(実施例5)
SM−1237についての分析データ: 1H NMR (300 MHz, D2O) δ 7.45 (d, J = 7.5 Hz, 1H), 7.36-7.20 (m, 6H), 71.5 (d, J = 7.5 Hz, 1H), 5.13 (m, 1H), 4.72 (m, 1H), 4.33 (m, 1H), 3.80 (m, 1H), 2.51 (s, 3H), 2.45-2.10 (m, 3H), 1.92-1.37 (m, 8H), 1.36 (d, J = 7.2 Hz, 3H), 1.15 (m, 1H); 13C NMR (75 MHz, D2O) δ 173.85, 169.76, 153.02, 135.56, 131.66, 129.48, 129.18, 128.72, 128.50, 128.26, 127.10, 113.10, 60.97, 57.15, 55.36, 51.23, 36.59, 32.72, 32.34, 31.31, 29.58, 24.86, 22.80, 15.55。
(実施例6)
SM−1238(主な配座異性体)に関する分析データ: 1H NMR (300 MHz, D2O) δ7.70 (m, 1H), 7.60-7.40 (m, 7H), 5.40 (m, 1H), 5.15 (m, 1H), 4.55 (m, 1H), 4.03-3.86 (m, 2H), 3.62-3.55 (m, 2H), 3.20 (m, 1H), 2.62 (s, 3H), 2.60-2.20 (m, 4H), 2.15-1.70 (m, 3H), 1.50 (d, J = 7.2 Hz, 3H), 1.42 (m, 1H), 0.96 (m, 1H), 0.55 (d, J = 7.2 Hz, 3H), 0.36 (d, J = 7.2 Hz, 3H)。
(実施例7)
実施例7の化合物に関する分析データ: 1H NMR (300 MHz, CDCl3, TMS) δ7.70-7.35 (m, 6H), 7.32-7.15 (m, 2H), 6.88 (brs, 1H), 5.55 (m, 1H), 5.20 (m, 1H), 4.65 (brm, 1H), 4.42 (m, 1H), 4.15 (m, 1H), 3.25-3.08 (m, 2H), 2.85 (m, 1H), 2.75 (s, 3H), 2.70 (m, 1H), 2.52 (m, 1H), 2.35-1.75 (m, 5H), 1.45 (brs, 9H), 1.35 (d, J = 7.0 Hz, 3H); ESI MS (m/z) 561.3 (M+H)+。
(実施例8)
SM−1257(主な配座異性体)に関する分析データ: 1H NMR (300 MHz, D2O) δ 7.70-7.49 (m, 8H), 7.15-7.02 (m, 3H), 6.72-6.62 (m, 2H), 5.47 (m, 1H), 5.15 (m, 1H), 4.75 (m, 1H), 4.59 (m, 1H), 4.10-3.85 (m, 2H), 3.72-3.62 (m, 2H), 3.43 (m, 1H), 2.88 (m, 1H), 2.70 (s, 3H), 2.68-2.25 (m, 4H), 2.20-1.82 (m, 3H), 1.55 (d, J = 7.0 Hz, 3H); ESI MS (m/z) 579.3 (M+H)+。
(実施例9)
SM−1268(主な配座異性体)に関する分析データ: 1H NMR (300 MHz, D2O) δ 7.69-7.42 (m, 8H), 6.70-6.52 (m, 4H), 5.48 (m, 1H), 5.20 (m, 1H), 4.75 (m, 1H), 4.62 (m, 1H), 4.09-3.92 (m, 2H), 3.75-3.62 (m, 2H), 3.42 (m, 1H), 2.85 (m, 1H), 2.70 (s, 3H), 2.68-2.25 (m, 4H), 2.18-1.83 (m, 3H); ESI MS (m/z) 597.3 (M+H)+。
(実施例10)
SM−1270(主な配座異性体)に関する分析データ: 1H NMR (300 MHz, D2O) δ 7.82 (m, 1H), 7.70-6.97 (m, 7H), 7.20-7.09 (m, 3H), 6.73-6.65 (m, 2H), 5.49 (m, 1H), 5.23 (m, 1H), 4.75 (m, 1H), 4.53 (m, 1H), 4.10-3.95 (m, 2H), 3.68-3.58 (m, 2H), 3.32 (m, 1H), 2.72 (s, 3H), 2.72-2.55 (m, 2H), 2.52-1.95 (m, 6H), 1.80-1.62 (m, 2H), 1.55 (d, J = 7.0 Hz, 3H); ESI MS (m/z) 593.3 (M+H)+。
(実施例11)
SM−1271(主な配座異性体)に関する分析データ: 1H NMR (300 MHz, D2O) δ 7.64 (m, 1H), 7.45-7.20 (m, 6H), 7.05 (m, 1H), 6.65-6.50 (m, 2H), 6.50-6.39 (m, 2H), 5.42 (m, 1H), 5.20 (m, 1H), 4.85 (m, 1H), 4.55 (m, 1H), 4.10-3.90 (m, 2H), 3.65-3.45 (m, 2H), 3.25 (m, 1H), 2.68 (s, 3H), 2.65-2.02 (m, 7H), 1.95-1.70 (m, 1H), 1.55 (d, J = 7.0 Hz, 3H), 1.54 (m, 1H); ESI MS (m/z) 611.3 (M+H)+。
(実施例12)
SM−1306に関する分析データ: 1H NMR (300 MHz, D2O) δ 7.70-7.50 (m, 4H), 7.40-7.20 (m, 3H), 6.82-6.58 (m, 4H), 5.47 (m, 1H), 5.18 (m, 1H), 4.75-4.50 (m, 2H), 4.05-3.88 (m, 2H), 3.75-3.65 (m, 2H), 3.50-3.30 (m, 2H), 2.90 (m, 1H), 2.65 (m, 3H), 2.62-1.90 (m, 6H), 1.55 (d, J = 7.0 Hz, 3H); ESI MS (m/z) 615.3 (M+H)+。
(実施例13)
SM−1307に関する分析データ: 1H NMR (300 MHz, D2O) δ 7.40 (m, 1H), 7.25-7.15 (m, 3H), 7.05-5.85 (m, 3H), 6.60-6.35 (m, 4H), 5.38 (m, 1H), 5.15 (m, 1H), 4.75 (m, 1H), 4.50 (m, 1H), 4.05-3.80 (m, 2H), 3.70-3.50 (m, 2H), 3.50-3.20 (m, 2H), 2.70 (m, 1H), 2.63 (s, 3H), 2.60-1.70 (m, 6H), 1.55 (d, J = 7.0 Hz, 3H); ESI MS (m/z) 615.3 (M+H)+。
(実施例14)
SM−1308に関する分析データ: 1H NMR (300 MHz, D2O) δ 7.38 (m, 1H), 7.30-7.10 (m, 2H), 7.05-6.85 (m, 4H), 6.60-6.35 (m, 4H), 5.35 (m, 1H), 5.15 (m, 1H), 4.75 (m, 1H), 4.42 (m, 1H), 3.95 (m, 1H), 3.75 (m, 1H), 3.60-3.02 (m, 4H), 2.75-2.60 (m, 4), 2.58-1.60 (m, 6H), 4.05-3.80 (m, 2H), 3.70-3.50 (m, 2H), 3.50-3.20 (m, 2H), 2.70 (m, 1H), 2.63 (s, 3H), 2.60-1.70 (m, 6H), 1.55 (d, J = 7.0 Hz, 3H); ESI MS (m/z) 615.3 (M+H)+。
(実施例15)
SM−1316に関する分析データ: 1H NMR (300 MHz, D2O) δ 7.70 (m, 1H), 7.55-7.45 (m, 2H), 7.40 (m, 1H), 7.25-7.15 (m, 2H), 5.45 (m, 1H), 5.20 (m, 1H), 4.75 (m, 1H), 4.55 (m, 1H), 4.03-3.90 (m, 2H), 3.70-3.50 (m, 2H), 3.25 (m, 1H), 2.70 (s, 3H), 2.60-1.60 (m, 7H), 1.55 (d, J = 7.0 Hz, 3H); 1.50 (m, 1H), 1.02 (m, 1H), 0.55 (d, J = 7.2 Hz, 3H), 0.30 (d, J = 7.2 Hz, 3H); ESI MS (m/z) 563.3 (M+H)+。
(実施例16)
SM−1317に関する分析データ: 1H NMR (300 MHz, D2O) δ 7.55 (m, 1H), 7.50-7.30 (m, 3H), 7.22 (m, 1H), 7.18-7.05 (m, 2H), 6.98-6.90 (m, 3H), 6.85-6.50 (m, 2H), 5.38 (m, 1H), 5.10 (m, 1H), 4.80 (m, 1H), 4.50 (m, 1H), 4.02-3.80 (m, 2H), 3.75-3.50 (m, 2H), 3.45-3.20 (m, 2H), 2.78 (m, 1H), 2.68 (s, 3H), 2.58-1.75 (m, 6H), 1.55 (d, J = 7.0 Hz, 3H); ESI MS (m/z) 597.3 (M+H)+。
(実施例17)
XIAPに対する阻害剤の結合
蛍光偏光(FP)に基づく方法を用いて高感度かつ定量的なインビトロの結合アッセイを行い、XIAPタンパク質に対するSmac模倣物の結合親和性を決定した(Nikolovska-Coleska et al., Anal. Biochem. 332:261-73 (2004))。このアッセイについて、5−カルボキシフルオレセイン(5−Fam)が、変異Smacペプチド(AbuRPF−K−(5−FAM)−NH2(SM5Fと称する))のリジン側鎖に結合された。また、SM5Fよりも高い親和性を有している他の蛍光標識Smac模倣物(SM−F1と称する)を使用した。
Kd値を決定するために、ウルトラプレートリーダー(Ultra plate reader)(Tecan U.S., Research Triangle Park, NC)を用いて、マイクロフルオロ(Microfluor)2 96ウェルの黒い丸底プレート(thermo Scientific)における蛍光偏光度を測定した。SM−F1トレーサーの一例として、SM−F1(それぞれ2nMの、1nM、および1nMのXIAP−BIR3、cIAP1−BIR3、およびcIAP2−BIR3)および異なる濃度のタンパク質を各ウェルに対して、アッセイのバッファー(pH7.5の100mMのリン酸カリウム、100μg/mlウシのγ−グロブリン、0.02%のアジ化ナトリウム(Invitrogen社)、4%のDMSO)が125μlの最終容積になるまで加えた。プレートを混合し、穏やかに振とうさせながら室温において3時間にわたってインキュベートして、平衡状態を確保した。ミリ偏光ユニット(mP)の偏光度を、励起波長485nmおよび発光波長530nmにおいて測定した。それから、Graphpad Prism 5.0ソフトウェア(Graphpad Software, San Diego, CA)を用いて、タンパク質濃度に応じて増加するシグモイドの用量依存性のFPをフィッティングすることによって、平衡解離定数(Kd)を算出した。
組換えXIAP BIR3、cIAP−1 BIR3、cIAP−2 BIR3に対するSM5FのKd値は17.9nMであると測定された(Nikolovska-Coleska et al., Anal. Biochem. 332:261-73 (2004))。組換えXAIP−BIR3タンパク質に対するSM−F1のKd値は4.7nMであると測定された。
XIAP BIR3タンパク質に対するトレーサーとしてSM5Fを用いた競合的な結合実験において、試験化合物を、アッセイの緩衝液(100mMのリン酸カリウム、pH7.5;100μg/mlのウシのγグロブリン;0.02%のアジ化ナトリウム、Invitrogen社)において、XIAP BIR3タンパク質(30nM)およびSM5F(5nm)と共にインキュベートした。用量依存性の競合的な結合FP実験を、試験化合物の段階希釈液を用いて行った。各アッセイには、組換えXIAP BIR3タンパク質およびSM5Fを含んでいる結合ペプチドの対照(0%の阻害に相当)、ならびに遊離型のペプチドSM5Fのみを含んでいる遊離型ペプチドの対照(100%の阻害に相当)が含まれる。偏光度は、3時間のインキュベーション後、ウルトラリーダー(ULTRA READER)(Tecan U.S. Inc., Research Triangle Park, NC)を使用して結合が平衡に達したときに測定された。IC50値、すなわち、結合ペプチドの50%が置換されているときの阻害剤濃度は、非線形最小二乗解析を用いてプロットから決定される。曲線の適合化は、GRAPHPAD PRISMソフトフェア(GraphPad Software, Inc., San Diego, CA)を使用して実行される。
XIAP BIR3タンパク質に対するトレーサーとしてSM−F1を用いた競合的な結合実験では、試験化合物を、アッセイの緩衝液(100mMのリン酸カリウム、pH7.5;100μg/mlウシのγグロブリン;0.02%のアジ化ナトリウム、Invitrogen社)において、XIAP BIR3タンパク質(10nM)およびSM-1F(2nM)と共にインキュベートした。用量依存性の競合的な結合FPの実験を、試験化合物の段階希釈液を用いて行った。各アッセイには、組換えXIAP BIR3タンパク質およびSM−1Fを含んでいる結合ペプチドの対照(0%の阻害に相当)、ならびに遊離型のペプチドSM−1Fのみを含んでいる遊離型ペプチドの対照(100%の阻害に相当)が含まれる。偏光度を、3時間のインキュベーション後、ウルトラリーダー(Tecan U.S. Inc., Research Triangle Park, NC)を使用して結合が平衡に達したときに測定した。IC50値、すなわち、結合ペプチドの50%が置換されているときの阻害剤濃度は、非線形最小二乗解析を用いたプロットから決定される。曲線の適合化は、GRAPHPAD PRISMソフトフェア(GraphPad Software, Inc., San Diego, CA)を使用して実行される。
タンパク質/トレーサー複合体のみを含んでいる陰性対照(0%の阻害に相当)、および遊離型トレーサーのみを含んでいる陽性対照(100%の阻害に相当)が各アッセイ用プレートに入れられた。FP値は上述のように測定された。IC50値を、競合曲線の非線形回帰適合化によって測定した。競合的な阻害剤のKi値を、測定したIC50値、異なるタンパク質に対するトレーサーのKd値、ならびに競合アッセイにおけるタンパク質およびトレーサーの濃度に基づいて、前述の式(Nikolovska-Coleska et al., Anal. Biochem. 332:261-73 (2004))を用いて計算した。また、Ki値を、文献(Huang, X. J. Biomol. Screen. 8:34-38 (2003))で知られている一般的に使用される式を用いて算出した。
結合アッセイにおいて表2に示すようにSM5FまたはSM−1Fのいずれかをトレーサーとして用いて試験したとき、ならびに図1に示すようにトレーサーとしてSM5Fを用いて試験したとき、本発明に係るSmac模倣物は、XIAP BIR3タンパク質との強い結合親和性を示した。これらのデータによると、これらのSmac模倣物は、高い親和性をもってXIAPと結合することが分かる。
(実施例18)
他のIAPタンパク質に対する阻害剤の結合
他のIAPタンパク質(cIAP1およびcIAP2)に対する、立体的に制約されているSmac模倣物の結合能を試験するために、結合アッセイ条件を変更した。ヒスタグと融合させた組換えcIAP1 BIR3ドメイン(253残基から363残基)、cIAP2 BIR3ドメイン(238残基から349残基)を結合アッセイに使用した。他のIAPタンパク質の競合的な結合アッセイは、XIAP BIR3について説明したものと同様に実行される。組換えcIAP−1 BIR3およびcIAP−2 BIR3に対するSM5FのKd値は、それぞれ、4.1nM(Peng et al. J. Med. Chem.51: 8158-8162 (2008))、および6.6nM(Peng et al. J. Med. Chem.51: 8158-8162 (2008))であると測定された。組換えcIAP−BIR3、およびcIAP2−BIRsタンパク質に対するSM−F1のKd値は、それぞれ、1.1nMおよび2.3nMであると測定された。
cIAP−1 BIR3タンパク質に対するトレーサーとしてSM5Fを使用した競合的な結合実験では、試験化合物を、分析バッファー中で、cIAP−1 BIR3タンパク質(10nM)およびSM5F(2nM)と共にインキュベートした。cIAP−2 BIR3タンパク質に対するトレーサーとしてSM5Fを用いた競合的な結合実験では、試験化合物を、アッセイの緩衝液において、cIAP−2 BIR3タンパク質(25nM)およびSM5F(2nM)と共にインキュベートした。cIAP−1 BIR3およびcIAP−2 BIR3タンパク質のための他のすべての手順は、XIAP BIR3タンパク質競合実験に使用されるものと同じであった。
cIAP−1 BIR3タンパク質に対するトレーサーとしてSM−F1を用いた競合的な結合実験では、アッセイの緩衝液において、cIAP−1 BIR3タンパク質(3nM)およびSM−F1(1nM)と共に試験化合物をインキュベートした。cIAP−2 BIR3タンパク質に対するトレーサーとしてSM−F1を用いた競合的な結合実験では、アッセイの緩衝液において、cIAP−2 BIR3タンパク質(5nM)およびSM−F1(1nM)と共に試験化合物をインキュベートした。
トレーサーとしてSM5Fを用いると、図2に示すように、本発明のSmac模倣物は、cIAP1 BIR3タンパク質との強い結合親和性を示した。表3は、トレーサーとしてのSM5FまたはSM−F1のいずれかを使用した、cIAP1およびcIAP2タンパク質に対する本発明の化合物の結合親和性を示している。これらのデータによって、本発明の化合物がcIAP1およびcIAP2活性の強力な阻害剤として作用することが示唆された。
(実施例19)
立体的に制約されているSmac模倣物による細胞増殖の阻害
様々な癌細胞の成長に対する本発明の化合物の影響を試験した。96ウェルの平底細胞培養プレートに、試験化合物と共に細胞を各ウェルにつき3000の密度に播き、37℃、95%の空気および5%のCO2の環境において、4日間にわたって細胞をインキュベートした。異なる濃度の化合物を用いて処理した後の細胞増殖の阻害率を、WST−8キット(2−(2−メトキシ−4−ニトロフェニル)−3−(4−ニトロフェニル)−5−(2,4−ジスルフォフェニル)−2H−テトラゾリウムモノナトリウム塩(Dojindo Molecular Technologies, Inc., Gaithersburg, Maryland))を用いて測定した。10%の最終濃度のWST−8を各ウェルに加えた後、プレートを37℃において2〜3時間にわたってインキュベートした。ウルトラテカンリーダー(ULTRA Tecan Reader)(分子デバイス)を用いて450nmにおいてサンプルの吸光度を測定した。細胞増殖を50%抑制する試験化合物の濃度(IC50)を、試験化合物を用いて処理した細胞および未処理の細胞の吸光度を比較することによって算出した。
ヒト乳癌細胞株MDA−MB−231および卵巣癌細胞株SK−OV−3に対して試験したとき、本発明の化合物は、表4に示すように強い阻害活性を示した。これによれば、本発明の化合物が癌細胞の増殖の阻害剤であることが示された。
(実施例20)
細胞死の誘導
SM−1238による細胞死の誘導能を、乳がん細胞株MDA−MB−231および卵巣癌細胞株SK−OV−3において試験した(図3)。細胞を、SM−1238を用いて48時間にわたって処理し、トリパンブルー排除アッセイを用いて細胞の生存を判定した。SM−1238は濃度依存的に両方の細胞の細胞死を誘導した。
(実施例21)
ラットにおける経口投与後の薬物動態
雄のスピローグ−ダウリーラットにSM−1238(実施例6)を25mg/kgの用量において経口投与した後の、Cmax、Tmax、AUC(0−∞)、および半減期(T1/2)についての平均値±SD値は、それぞれ、831±135μg/L、2.0±1.7時間、7099±931μg/L*時間、および3.7±2.0時間であった(n=3)。
雄のスピローグ−ダウリーラットにSM−1268(実施例9)を25mg/kgの用量において経口投与した後において、Cmax、Tmax、AUC(0−∞)、および半減期(T1/2)についての平均値±SD値は、それぞれ、612±16μg/L、2.0±0.0時間、6489±965μg/L*時間、および4.2±1.0時間であった(n=3)。
雄のスピローグ−ダウリーラットにSM−1316(実施例15)を25mg/kgの用量において経口投与した後の、Cmax、Tmax、AUC(0−∞)、および半減期(T1/2)についての平均値±SD値は、それぞれ、1528±275μg/L、2.0±0.0時間、14304±1968μg/L*時間、および6.7±1.2時間であった(n=3)。
これらの実験は、本発明の化合物が経口的に生物適合性であることを示す。
本発明を十分に説明したが、本発明またはその任意の実施形態の範囲に影響を与えることなく、条件、処方、および他のパラメータについて広範かつ同等の範囲内で同様のことを行うことが可能であることは、当業者にとって理解されるだろう。前述した全ての特許、特許出願および刊行物は、参照によってその全体が本明細書に十分に援用される。
図1は、XIAP BIR3ドメインに対するSmac模倣物の競合的な結合の曲線を示しているグラフである。
図2は、cIAP1ドメインに対するSmac模倣物の競合的な結合の曲線を示しているグラフである。
図3は、ヒト乳がんのMDA−MB−231細胞株およびヒト卵巣がんのSK−OV−3細胞株における、SM−1238による細胞死の誘導を示している棒グラフである。