JP5265384B2 - 乳癌、結腸直腸癌、膵臓癌、卵巣癌及び肺癌の治療のための7−エチル−10−ヒドロキシカンプトセシンのマルチアーム・ポリマー複合体 - Google Patents
乳癌、結腸直腸癌、膵臓癌、卵巣癌及び肺癌の治療のための7−エチル−10−ヒドロキシカンプトセシンのマルチアーム・ポリマー複合体 Download PDFInfo
- Publication number
- JP5265384B2 JP5265384B2 JP2008554430A JP2008554430A JP5265384B2 JP 5265384 B2 JP5265384 B2 JP 5265384B2 JP 2008554430 A JP2008554430 A JP 2008554430A JP 2008554430 A JP2008554430 A JP 2008554430A JP 5265384 B2 JP5265384 B2 JP 5265384B2
- Authority
- JP
- Japan
- Prior art keywords
- alkyl
- acid
- group
- cycloalkyl
- substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 CCC(C(C*1C(CC2CCOC(C)=*)=O)=*C1=C[C@@]2OC=C)=C(C=C(C(C)=C)*(C)=C)*=* Chemical compound CCC(C(C*1C(CC2CCOC(C)=*)=O)=*C1=C[C@@]2OC=C)=C(C=C(C(C)=C)*(C)=C)*=* 0.000 description 4
- JEQIFRUVEJLHII-QFIPXVFZSA-N CC[C@](C(C=C1N2Cc3c(CC)c(cc(cc4)O)c4nc13)=C(CO1)C2=O)(C1=O)O[IH]I Chemical compound CC[C@](C(C=C1N2Cc3c(CC)c(cc(cc4)O)c4nc13)=C(CO1)C2=O)(C1=O)O[IH]I JEQIFRUVEJLHII-QFIPXVFZSA-N 0.000 description 1
- YXFVVABEGXRONW-UHFFFAOYSA-N Cc1ccccc1 Chemical compound Cc1ccccc1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 1
Images











Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/14—Ortho-condensed systems
- C07D491/147—Ortho-condensed systems the condensed system containing one ring with oxygen as ring hetero atom and two rings with nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Description

R1、R2、R3 及びR4 は、独立して、OHまたは(L)m−Dであり、
Lは、2官能性のリンカーであり、
Dは、例えば、
mは0または正の整数であり、
nは正の整数であり、
R1、R2、R3 及びR4 は、すべてがOHではないことを条件とする。
ここで、
R1、R2、R3 及びR4 は、独立して OHまたは(L)m−Dであり、
Lは二官能性のリンカーであり、
Dは、例えば、
mは0または正の整数であり、約1〜約10が好ましく、1がさらに好ましく、
nは正の整数であり、約28〜約341が好ましく、約227がさらに好ましく、
R1、R2、R3 及びR4 のすべてがOHではないことを条件とする。
カンプトセシンは、中国に自生しているカンレンボクの樹木、及びインドに自生しているクサミズキの樹木から産生される、水不溶性の細胞毒性アルカロイドである。カンプトセシンならびに関連する化合物及び類似体はまた、抗がん剤または抗腫瘍剤として有望であることが知られており、実験動物でのインビトロ及びインビボにおいて、これらの活性を発現することが示されている。
(a)R9及びR10は、独立して、水素、C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ−C1-6アルキルであるか、あるいは、
(b)R9は、水素、C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ−C1-6アルキルであって差し支えなく、R10は、−COR11であって差し支えなく、ここでR11は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ、C1-6アルコキシ−C1-6アルキルであるか、または
(c)R9及びR10は、それらに付加する窒素原子と一緒に、O、SまたはNR12 基を含んでいてもよい飽和3〜7員の複素環を形成し、ここでR12は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、アリール、あるいは、C1-6アルキル、ハロゲン、ニトロ、アミノ、C1-6アルキルアミノ、ペルハロ−C1-6アルキル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ、C1-6アルコキシ−C1-6アルキル及び−COR13からなる群より選択される1またはそれ以上の基で置換されたアリールであって、ここで、R13は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、C1-6アルコキシ、アリール、及び、C1-6アルキル、ペルハロ−C1-6アルキル、ヒドロキシ−C1-6アルキル、またはC1-6アルコキシ−C1-6アルキル基の1つ以上の基で置換されたアリール、であり、
R110〜R111は、それぞれ独立して、水素、ハロ、アシル、アルキル(例えば、C1-6アルキル)、置換アルキル、アルコキシ(例えば、C1-6アルコキシ)、置換アルコキシ、アルケニル、アルキニル、シクロアルキル、ヒドロキシル、シアノ、ニトロ、アジド、アミド、ヒドラジン、アミノ、置換アミノ(例えば、モノアルキルアミノ及びジアルキルアミノ)、ヒドロキシカルボニル、アルコキシカルボニル、アルキルカルボニルオキシ、アルキルカルボニルアミノ、カルバモイルオキシ、アリールスルホニルオキシ、アルキルスルホニルオキシ、−C(R117)=N−(O)j−R118、ここで、R117はH、アルキル、アルケニル、シクロアルキルまたはアリールであり、jは0または1であり、R118は、H、アルキル、アルケニル、シクロアルキル、または複素環である、及びR119C(O)O−、ここで、R119は、ハロゲン、アミノ、置換アミノ、複素環、置換複素環、または R120−O−(CH2)k−であって、ここでkは1〜10の整数であり、R120は、アルキル、フェニル、置換フェニル、シクロアルキル、置換シクロアルキル、複素環、または置換複素環である、からなる群より選択されるか、あるいは、
R7がR110と一緒に、またはR110がR111と一緒に、置換または非置換のメチレンジオキシ、エチレンジオキシ、またはエチレンオキシを形成し、
R112は、HまたはOR'であり、ここで、R'はアルキル、アルケニル、シクロアルキル、ハロアルキル、またはヒドロキシアルキルである。
本発明のある好ましい態様では、Lはアミノ酸残基である。既知の天然に存在するL-アミノ酸のいずれかから選択されるアミノ酸としては、いくつかの例を挙げるとすれば、アラニン、バリン、ロイシン、イソロイシン、グリシン、セリン、スレオニン、メチオニン、システイン、フェニルアラニン、チロシン、トリプトファン、アスパラギン酸、グルタミン酸、リジン、アルギニン、ヒスチジン、プロリン、及び/またはこれらの組合せが挙げられる。別の態様では、Lはペプチド残基であってもよい。ペプチドは、例えば、約2〜約10アミノ酸残基の長さであって差し支えない。
−[C(O)]vNR21(CR22R23O)t−、
−[C(O)]vNR21(CR22R23O)t(CR24R25)yO−、
−[C(O)]vNR21(CR22R23O)t(CR24R25)y−、
−[C(O)]vNR21(CR22R23)tO−、
−[C(O)]vNR21(CR22R23)t(CR24CR25O)yNR26−、
−[C(O)]vO(CR22R23)tNR26−、
−[C(O)]vO(CR22R23)tO−、
−[C(O)]vNR21(CR22R23)tNR26−、
−[C(O)]vNR21(CR22R23)t(CR24CR25O)y−、
−[C(O)]vNR21(CR22R23O)t(CR24CR25)yNR26−、
R21〜R26は、独立して、水素、C1-6アルキル、C2-6アルケニル、C2-6アルキニル、C3-19分岐鎖アルキル、C3-8 シクロアルキル、C1-6置換アルキル、C2-6置換アルケニル、C2-6置換アルキニル、C3-8置換シクロアルキル、アリール、置換アリール、アラルキル、C1-6ヘテロアルキル、置換C1-6ヘテロアルキル、C1-6アルコキシ、フェノキシ、C1-6へテロアルコキシからなる群より選択され、
R27は、水素、C1-6アルキル、C2-6アルケニル、C2-6アルキニル、C3-19分岐鎖アルキル、C3-8 シクロアルキル、C1-6置換アルキル、C2-6置換アルケニル、C2-6置換アルキニル、C3-8置換シクロアルキル、アリール、置換アリール、アラルキル、C1-6ヘテロアルキル、置換C1-6ヘテロアルキル、C1-6アルコキシ、フェノキシ、C1-6へテロアルコキシ、NO2、ハロアルキル及びハロゲンからなる群より選択され、
t及びyは、独立して、約1〜約4の正の整数から選択され、
vは0または1である。
マルチアームPEGは、参照することによりその開示を援用する、NOF社のDrug Delivery System catalog, Ver. 8, April 2006に記載されている。ある特に好ましいマルチアームPEGは、以下の構造式:
ここで、nは正の整数である。
一般に、本発明のプロドラッグは、1当量以上の活性化マルチアーム・ポリマーを、例えば活性部位当たり1当量以上のカンプトセシン−アミノ酸複合体と、該複合体がポリマーのカルボン酸と反応し、結合を形成するためのアミノ基を効率的に生じさせるのに十分な条件下で、反応させることによって調製される。
1)利用可能な20−ヒドロキシル基を含む、1当量のカンプトセシンまたはカンプトセシン類似体と、利用可能なカルボン酸基を含む1当量以上の二官能性リンカーとを提供し、
2)その2つの反応物質を反応させて、1,(3−ジメチルアミノプロピル)3−エチルカルボジイミド(EDC)(または1,3−ジイソプロピルカルボジイミド(DIPC)、任意の適切なジアルキルカルボジイミド、向山試薬(例えば2−ハロ−1−アルキル−ピリジニウムハライド)、またはプロパンホスホン酸環状無水物(PPACA)など)等のカップリング剤及びDMAPなどの適切な塩基の存在下、DCM(またはDMF、クロロホルム、トルエン、またはこれらの混合物)などの不活性溶媒中でカンプトセシン−二官能性リンカー中間体を形成し、
3)アミン基を有している、活性部位当たり1当量以上(実施例では2当量)の得られた中間体と、PEG−酸などの1当量の活性化ポリマーを、DCM(またはDMF、クロロホルム、トルエン、またはこれらの混合物)などの不活性溶媒中で、1,(3−ジメチルアミノプロピル)3−エチルカルボジイミド(EDC)、PPAC(または1,3−ジイソプロピルカルボジイミド(DIPC)、任意の適切なジアルキルカルボジイミド、向山試薬(例えば2−ハロ−1−アルキル−ピリジニウムハライド)、またはプロパンホスホン酸環状無水物(PPACA)など)等のカップリング剤、及び、例えばシグマ・ケミカル社などの商業的供給源から入手可能であるか、あるいは公知の技術を利用して合成された、DMAPなどの適切な塩基の存在下、0℃〜22℃までの温度で反応させる、
各工程を有していてもよい。
ここで、ポリマーの4つのアームすべてが、グリシンを介して、7−エチル−10−ヒドロキシカンプトセシンと共役している。本発明のこの態様に従って調製された化合物のHPLC分析は、平均して4つの7−エチル−10−ヒドロキシカンプトセシン分子が、1つのPEG分子に共役していることを示す(4重量%)。
本発明のポリマー複合体を含む医薬組成物は、例えば、さまざまな周知の混合、溶解、顆粒化、粉末化(levigating)、乳化、カプセル化、封入化、または凍結乾燥化する方法など、当技術分野で周知の方法によって製造されて差し支えない。活性化合物を薬剤的に使用可能な製剤に加工する工程を容易にする、補助剤及び賦形剤を含む1種類以上の生理的に許容できる担体と共に、組成物を製剤化してもよい。適切な製剤は、選択する投与経路に応じて決まる。本発明の多くの態様では、非経口の経路が好ましい。
治療に有効な量とは、カンプトセシンまたは関連する類似体、すなわち7−エチル−10−ヒドロキシカンプトセシン感受性の状態を予防、軽減、または改善するのに効果的な化合物の量のことをいう。治療に有効な量の決定は、特に、本明細書の開示を考慮すれば、十分に、当業者が対応できる範囲内にある。
上記を考慮して、化学式(I)で表される本発明の化合物を、それを必要としている患者に有効量で投与することを含む、哺乳動物を治療する方法も提供する。ここでDは生物活性部分、すなわちカンプトセシン類似体である。本発明のある好ましい態様では、Dは7−エチル−10−ヒドロキシカンプトセシン残基である。
40k4アーム−PEG−OH(12.5g、1当量)を220mlのトルエンと共に共沸蒸留し、35mlのトルエン/水を除去した。溶液を30℃まで冷却し、t−ブタノール中の1.0Mカリウム−t−ブトキシド(3.75ml、3当量×4=12当量)を加えた。混合物を30℃で30分間攪拌し、次に、ブロモ酢酸t−ブチル(0.975g、4当量×4=16当量)を加えた。反応を30℃で1時間保持した後、25度まで冷却した。150mlのエーテルをゆっくり加えて生成物を沈殿させた。得られた懸濁液を17℃まで冷却し、17℃で0.5時間置いた。粗生成物を濾過し、湿ったケーキをエーテルで2回洗浄した(2×125ml)。単離された湿ったケーキを50mlのDCMに溶解し、生成物を350mlのエーテルで沈殿させ、濾過した。湿ったケーキをエーテルで2回洗浄した(2×125ml)。生成物を40℃で真空乾燥させた(収率=98%、12.25g)。13C NMR(75.4 MHz, CDCl3):δ27.71, 68.48-70.71 (PEG), 80.94, 168.97。
40k4アーム−PEG−t−ブチルエステル(化合物2、12g)を120mlのDCMに溶解し、60mlのTFAを加えた。混合物を室温で3時間攪拌し、次に、溶媒を35℃で真空除去した。得られた油状残渣を37.5mlのDCMに溶解させた。粗生成物を375mlのエーテルで沈殿させた。湿ったケーキを30mlの0.5%NaHCO3に溶解させた。生成物をDCMで2回抽出した(2×150ml)。合わせた有機層を2.5gのMgSO4で乾燥させた。溶媒を室温で真空除去した。得られた残渣を37.5mlのDCMに溶解し、生成物を300mlのエーテルで沈殿させ、濾過した。湿ったケーキをエーテルで2回洗浄した(2×125ml)。生成物を40℃で真空乾燥させた(収率=90%、10.75g)。13C NMR(75.4 MHz, CDCl3):δ67.93 − 71.6 (PEG), 170.83。
100mlの無水DCM中の7−エチル−10−ヒドロキシカンプトセシン(化合物4、2.0g、5.10mmol、1当量)の懸濁液にEt3N(4.3ml、30.58mmol、6当量)及びTBDPSCl(7.8ml、30.58mmol、6当量)を加えた。反応混合物を一晩加熱還流した後、0.2NのHCl溶液(2×50ml)、飽和NaHCO3溶液(100ml)、及び塩水(100ml)で洗浄した。有機層をMgSO4で乾燥し、濾過し、真空下で蒸発濃縮させた。残渣を無水DCMに溶解し、ヘキサンを加えて沈殿させた。DCM/ヘキサンで沈殿を繰り返し、過剰のTBDPSClを除去した。固形物を濾過し、真空乾燥させて、2.09gの生成物を得た(収率65%)。1H NMR(300 MHz, CDCl3):δ0.90 (3 H, t, J = 7.6 Hz), 1.01 (3 H, t, J = 7.3 Hz), 1.17 (9H, s), 1.83-1.92 (2H, m), 2.64 (2H, q, 6.9 Hz), 3.89 (1 H, s, OH), 5.11 (2H, s), 5.27 (1H, d, J = 16.1 Hz), 5.72 (1H, d, J = 16.4 Hz), 7.07 (2 H, d, J = 2.63 Hz), 7.36-7.49 (7 H, m), 7.58 (1 H, s), 7.75-7.79 (4H, m), 8.05 (1 H, d, J = 9.4 Hz)。13C NMR(75.4 MHz, CDCl3):δ7.82, 13.28, 19.52, 22.86, 26.48, 31.52, 49.23, 66.25, 72.69, 97.25, 110.09, 117.57, 125.67, 126.57, 127.65, 127.81, 130.02, 131.69, 131.97, 135.26, 143.51, 145.05, 147.12, 149.55, 149.92, 154.73, 157.43, 173.72。
100mlの無水DCM中のTBDPS−(10)−(7−エチル−10−ヒドロキシカンプトセシン)(化合物5、3.78g、5.99mmol、1当量)及びBoc−Gly−OH(1.57g、8.99mmol、1.5当量)の0℃の溶液に、EDC(1.72g、8.99mmol、1.5当量)及びDMAP(329mg、2.69mmol、0.45当量)を加えた。反応混合物を0℃で、出発物質がHPLCで完全に消えるまで攪拌した(およそ1時間45分)。有機層を0.5%NaHCO3溶液(2×50ml)、水(1×50ml)、0.1NのHCl溶液(2×50ml)、及び塩水(1×50ml)で洗浄し、MgSO4で乾燥させた。濾過及び真空下で蒸発濃縮後、4.94gの粗生成物を得た(定量的収率)。粗固形物をさらに精製することなく、次の反応に使用した。1H NMR(300 MHz, CDCl3):δ0.89 (3 H, t, J = 7.6 Hz), 0.96 (3 H, t, J = 7.5 Hz), 1.18 (9H, s), 1.40 (9H, s), 2.07-2.29 (3H, m), 2.64 (2H, q, 7.5 Hz), 4.01-4.22 (2H, m), 5.00 (1 H, br s), 5.01 (2H, s), 5.37 (1H, d, J = 17.0 Hz), 5.66 (1H, d, J = 17.0 Hz), 7.08 (1 H, d, J = 2.34 Hz), 7.16 (1H, s), 7.37-7.50 (7 H, m), 7.77 (4H, d, J = 7.6 Hz), 8.05 (1 H, d, J = 9.4 Hz)。13C NMR(75.4 MHz, CDCl3):δ7.52, 13.30, 19.50, 22.86, 26.45, 28.21, 31.64, 42.28, 49.14, 67.00, 76.65, 79.96, 95.31, 110.13, 118.98, 125.75, 126.45, 127.68, 127.81, 130.03, 131.54, 131.92, 135.25, 143.65, 144.91, 145.19, 147.08, 149.27, 154.75, 155.14, 157.10, 166.98, 169.17。
5mlの無水ジオキサン中のTBDPS−(10)−(7−エチル−10−ヒドロキシカンプトセシン)−(20)−Gly−Boc(化合物6、1g、1.27mmol)にジオキサン中の4M HCl溶液5mlを加えた。反応混合物を室温で、出発物質がHPLCで完全に消えるまで攪拌した(1時間)。反応混合物に50mlのエチルエーテルを加え、得られた固形物を濾過した。固形物を50mlのDCMに溶かし、塩水で洗浄した(飽和NaHCO3溶液を加えてpHを2.5に合わせた)。有機層をMgSO4で乾燥し、真空下で蒸発濃縮した。残渣を5mlのDCMに溶かし、50mlのエチルエーテルを加えて沈殿させた。濾過により、770mg(収率84%)の最終生成物を得た。1H NMR(300 MHz, CDCl3):δ0.84 (3 H, t, J = 7.6 Hz), 1.05 (3 H, t, J = 7.3 Hz), 1.16 (9H, s), 2.15-2.30 (3H, m), 2.59 (2H, q, 7.6 Hz), 4.16 (1H, d, J = 17.9 Hz), 4.26 (1H, d, J = 17.9 Hz), 5.13 (2H, s), 5.46 (1H, d, J = 17.0 Hz), 5.60 (1H, d, J = 17.0 Hz), 7.11 (1 H, d, J = 2.34 Hz), 7.30 (1H, s), 7.40-7.51 (6 H, m), 7.56 (1H, dd, J = 2.34, 9.4 Hz), 7.77 (4H, dd, J = 7.6, 1.6 Hz), 7.98 (1 H, d, J = 9.1 Hz)。13C NMR(75.4 MHz, CDCl3):δ8.09, 13.72, 20.26, 23.61, 26.94, 31.83, 41.01, 50.71, 67.62, 79.51, 97.03, 111.65, 119.69, 127.13, 128.97, 128.99, 129.11, 131.43, 131.96, 133.00, 133.03,136.51, 145.62, 145.81, 147.24, 148.29, 150.58, 156.27, 158.68, 167.81, 168.34。
14mlの無水DCM中の40k4アーム−PEGCOOH(化合物3、1.4g、0.036mmol、1当量)の溶液にTBDPS−(10)−(7−エチル−10−ヒドロキシカンプトセシン)−(20)−Gly・HCl(化合物7、207mg、0.29mmol、活性部位当たり2.0当量)、DMAP(175mg、1.44mmol、10当量)及びPPAC(0.85mlのEtOAc中の50%溶液、1.44mmol、10当量)を加えた。反応混合物を室温で一晩攪拌した後、真空蒸発させた。得られた残渣をDCMに溶かし、生成物をエーテルで沈殿させて濾過した。残渣をDMF/IPAで再結晶化し、生成物(1.25g)を得た。13C NMR(75.4 MHz, CDCl3):δ7.45, 13.20, 19.39, 22.73, 26.42, 31.67, 40.21, 49.01, 66.83, 95.16, 110.02, 118.83, 125.58, 126.40, 127.53, 127.73, 129.96, 131.49, 131.76, 131.82, 135.12, 143.51, 144.78, 145.13, 146.95, 149.21, 154.61, 156.92, 166.70, 168.46, 170.30。
化合物40k4アーム−PEG−Gly−(20)−(7−エチル−10−ヒドロキシカンプトセシン)−(10)−TBDPS(化合物8、1.25g)に、THF及び0.05MのHCl溶液の1:1混合液(12.5ml)中のTBAF(122mg、0.46mmol、4当量)溶液を加えた。反応混合物を室温で4時間攪拌した後、DCMで2回抽出した。合わせた有機相をMgSO4で乾燥し、濾過し、真空下で蒸発濃縮した。残渣を7mlのDMFに溶かし、37mlのIPAで沈殿させた。固形物を濾過し、IPAで洗浄した。DMF/IPAで沈殿を繰り返した。最後に、残渣を2.5mlのDCMに溶かし、25mlのエーテルを加えて沈殿させた。固形物を濾過し、40℃の真空オーブンで一晩乾燥させた(860mg)。13C NMR(75.4 MHz, CDCl3):δ7.48, 13.52, 22.91, 31.67, 40.22, 49.12, 66.95, 94.82, 105.03, 118.68, 122.54, 126.37, 128.20, 131.36, 142.92, 144.20, 144.98, 147.25, 148.29, 156.44, 156.98, 166.82, 168.49, 170.39。このNMRデータには、PEG−COOHの痕跡がなく、このことは、すべてのCOOHが反応したことを示唆している。蛍光検出によって測定された結合数(loading)は、ポリマーの4本の分岐鎖のそれぞれに7−エチル−10−ヒドロキシカンプトセシンがすべて満たされた状態と一致する、3.9であると判明した。この実験のさらに大規模での反復実施においても、同一の結果を得た。
250mlの無水DCM中の7−エチル−10−ヒドロキシカンプトセシン(化合物4、2.45g、1当量)の懸濁液に、N2下、室温で、二炭酸ジ-t-ブチル(Di-tert-butyl dicarbonate)(1.764g、1.3当量)及び無水ピリジン(15.2ml、30当量)を加えた。懸濁液を室温で一晩攪拌した。濁った溶液を、セライト(10g)を通して濾過し、濾液を0.5NのHClで3回(3×150ml)、及びNaHCO3飽和溶液(1×150ml)で洗浄した。溶液をMgSO4(1.25g)で乾燥させた。溶媒を30℃で真空除去した。生成物を40℃で真空乾燥した(収率=82%、2.525g)。13C NMR(75.4 MHz, CDCl3):δ173.53, 157.38, 151.60, 151.28, 150.02, 149.70, 147.00, 146.50, 145.15, 131.83, 127.19, 127.13, 124.98, 118.53, 113.88, 98.06, 84.26, 72.80, 66.18, 49.33, 31.62, 27.73, 23.17, 13.98, 7.90。
無水CH2Cl2(20ml)中のBoc−(10)−(7−エチル−10−ヒドロキシカンプトセシン)(化合物10、0.85g、1.71mmol)及びBsmoc−Ala(0.68g、2.3mmol)の溶液に、0℃で、EDC(0.51g、2.67mmol)及びDMAP(0.065g、0.53mmol)を加えた。混合物をN2下、0℃で45分間攪拌した後、室温まで温めた。反応の完了をHPLCで確認し、反応混合物を1%NaHCO3(2×50ml)、水(50ml)及び0.1NのHCl(2×50ml)で洗浄した。有機相を無水MgSO4で乾燥させ、濾過した。溶媒を減圧除去した。得られた固形物を40℃以下で一晩真空乾燥し、収率95%で1.28gの生成物を得た。13C NMR(75.4 MHz, CDCl3):δ171.16,166.83, 157.16, 154.78, 151.59, 151.33, 149.82, 147.17, 146.68, 145.35, 145.15, 139.08, 136.88, 133.60, 131.83, 130.45, 130.40, 130.33, 127.40, 127.08, 125.32, 125.14, 121.38, 120.01, 114.17, 95.90, 84.38, 77.19, 76.64, 67.10, 56.66, 53.45, 49.96, 49.34, 31.7, 27.76, 17.94, 14.02, 7.53。ESI-MS, 786.20[M+H]+。
無水CH2Cl2(200ml)中のBoc−(10)−(7−エチル−10−ヒドロキシカンプトセシン)−(20)−Ala−Bsmoc(化合物11、4.2g、5.35mmol)及び4−ピペリジノピペリジン(1.17g、6.96mmol)の溶液を室温で5時間、攪拌した。次にこの混合物を0.1NのHCl(2×40ml)で洗浄後、有機層を無水MgSO4で乾燥した。この溶液を濾過し、溶媒を減圧蒸留によって除去し、HPLCによる純度93%、2.8gの生成物を得た。この生成物をさらに、エーテル(3×20ml)によって倍散(trituration)した後、酢酸エチル(4×20ml)によって倍散して精製し、純度97%、1.52g(2.70mmol)を得た。13C NMR(75.4 MHz, CDCl3):δ168.39, 166.63, 156.98, 151.20, 151.15, 149.69, 146.67, 146.56, 145.37, 144.53, 131.66, 127.13, 124.99, 119.80, 113.82, 96.15, 84.21, 77.67, 67.16, 49.48, 49.06, 31.56, 27.74, 23.14, 15.98, 13.98, 7.57。
無水CH2Cl2(100ml)に、Boc−(10)−(7−エチル−10−ヒドロキシカンプトセシン)−(20)−Ala(化合物12、1.50g、2.5mmol)及び4アームPEG−COOH(化合物3、10.01g、1.0mmol)を室温で加えた。溶液を0℃まで冷却した後、EDC(0.29g、1.5mmol)及びDMAP(0.30g、2.5mmol)を加えた。混合液をN2下、0℃で1時間攪拌した。その後、室温に一晩保った。溶媒を減圧下で蒸発させた。残渣を40mlのDCMに溶かし、エーテル(300ml)で粗生成物を沈殿させた。濾過によって得られた湿った固形物を、DMF/IPA(60/240ml)の混合液に65℃で溶解させた。溶液を2〜3時間のうちに室温まで冷却し、生成物を沈殿させた。次に、固形物を濾過し、エーテルで洗浄した(2×200ml)。湿ったケーキを40℃以下で一晩真空乾燥し、8.5gの生成物を得た。
無水CH2Cl2中の30%TFA溶液(130ml)に、40k4アーム−PEG−Ala−(20)−(7−エチル−10−ヒドロキシカンプトセシン)−(10)−Boc(化合物13、7.98g)を室温で加えた。混合物を、3時間、または出発物質の消失がHPLCで確認されるまで、攪拌した。溶媒を35℃、真空下で、可能な限り除去した。残渣を50mlのDCMに溶かし、エーテル(350ml)で粗生成物を沈殿させ、濾過した。湿った固形物をDMF/IPA(50/200ml)の混合液に65℃で溶解させた。溶液を2〜3時間のうちに室温まで冷却し、生成物を沈殿させた。次に、固形物を濾過し、エーテルで洗浄した(2×200ml)。湿ったケーキを40℃以下で一晩真空乾燥し、6.7gの生成物を得た。13C NMR(75.4 MHz, CDCl3):δ170.75, 169.30, 166.65, 157.00, 156.31, 148.36, 147.19, 145.03, 144.29, 143.00, 131.49, 128.26, 126.42, 122.47, 118.79, 105.10, 94.57, 78.08, 77.81, 77.20, 71.15, 70.88, 70.71, 70.33, 70.28, 70.06, 69.93, 69.57, 66.90, 49.14, 47.14, 31.53, 22.95, 17.78, 13.52, 7.46。
無水CH2Cl2(50ml)中のBoc−(10)−7−エチル−10−ヒドロキシカンプトセシン(化合物10、2.73g、5.53mmol)及びBsmoc−Met(3.19g、8.59mmol)の溶液に、EDC(1.64g、0.59mmol)及びDMAP(0.21g、1.72mmol)を0℃で加えた。混合液をN2下、0℃で45分間、攪拌した後、室温まで温めた。反応の完了をHPLCで確認し、反応混合物を1%NaHCO3(2×100ml)、水(100ml)及び0.1NのHCl(2×100ml)で洗浄した。有機相を無水MgSO4で乾燥させ、濾過した。溶媒を減圧除去した。得られた固形物を40℃以下で一晩真空乾燥し、収率88%、4.2gの生成物を得た。13C NMR(75.4 MHz, CDCl3):δ170.3, 166.8, 157.1, 155.2, 151.4, 151.2, 149.7, 147.0, 146.6, 145.3, 145.1, 138.9, 136.6, 133.5, 131.7, 130.5, 130.3, 130.2, 127.3, 127.0, 125.3, 125.1, 121.2, 119.8, 114.1, 96.1, 84.3, 76.7, 67.0, 56.7, 53.5, 53.4, 49.3, 31.6, 31.0, 29.7, 27.7, 23.1, 15.4, 13.9, 7.4; ESI-MS, 846.24[M+H]+。
無水CH2Cl2(200ml)中のBoc−(10)−(7−エチル−10−ヒドロキシカンプトセシン)−(20)−Met−Bsmoc(化合物15、4.1g、4.85mmol)及び4−ピペリジノピペリジン(1.06g、6.31mmol)の溶液を、室温で5時間、攪拌した。次に、この混合液を0.1NのHCl(2×40ml)で洗浄した後、有機層を無水MgSO4で乾燥した。この溶液を濾過後、溶媒を減圧蒸留によって除去し、HPLCによる純度97%で2.8gの生成物を得た。この生成物をさらに、エーテル(3×20ml)によって倍散した後、酢酸エチル(4×20ml)によって倍散して精製し、純度97%で1.54gを得た。13C NMR(75.4 MHz, CDCl3):δ167.2, 166.5, 156.9, 151.12, 150.9, 149.8, 146.3, 145.9, 145.8, 144.9, 131.3, 127.2, 127.0, 125.1, 119.6, 113.8, 96.7, 84.3, 78.2, 67.0, 60.4, 52.2, 49.4, 31.4, 29.6, 29.1, 27.7, 23.2, 15.1, 13.9, 7.7。
無水CH2Cl2(80ml)溶液に、Boc−(10)−(7−エチル−10−ヒドロキシカンプトセシン)−(20)−Met(化合物16、1.48g、2.25mmol)及び4アーム−PEG−COOH(化合物3、9.0g、0.9mmol)を室温で加えた。溶液を0℃まで冷却した後、EDC(0.26g、1.35mmol)及びDMAP(0.27g、2.25mmol)を加えた。混合液をN2下、0℃で1時間、攪拌した。その後、室温で一晩保持した。反応混合物を70mlのCH2Cl2で希釈し、30mlの0.1N HCl/1M NaCl水溶液で抽出した。有機層をMgSO4で乾燥し、溶媒を減圧蒸発させた。残渣を40mlのCH2Cl2に溶かし、エーテル(300ml)で粗生成物を沈殿させた。濾過によって得られた湿った固形物を65℃で270mlのDMF/IPAに溶解した。溶液を2〜3時間のうちに室温まで冷却し、生成物を沈殿させた。次に、固形物を濾過し、エーテルで洗浄した(2×400ml)。DMF/IPA中での上記結晶化の手順を繰り返した。湿ったケーキを40℃で一晩、真空乾燥し、7.0gの生成物を得た。13C NMR(75.4 MHz, CDCl3):δ169.8, 169.6, 166.5, 156.9, 151.2, 151.1, 149.9, 147.0, 146.6, 145.0, 131.7, 127,1, 126.8, 124.9, 119.7, 113.8, 95.5, 84.1, 70.1, 69.9, 66.9, 50.7, 49.2, 31.5, 31.2, 29.6, 27.6, 23.1, 15.3, 13.9, 7.5。
無水CH2Cl2(100ml)の30%TFA溶液に、硫化ジメチル(2.5ml)及び4アーム−PEG−Met−(20)−(7−エチル−10−ヒドロキシカンプトセシン)−(10)−Boc(化合物17、6.0g)を室温で加えた。混合物を、3時間、または出発物質の消失がHPLCで確認されるまで、攪拌した。溶媒を35℃、真空下で、可能な限り除去した。残渣を50mlのCH2Cl2に溶かし、エーテル(350ml)で粗生成物を沈殿させ、濾過した。湿った固形物をDMF/IPA(60/300ml)の混合液に65℃で溶解させた。溶液を2〜3時間のうちに室温まで冷却し、生成物を沈殿させた。次に、固形物を濾過し、エーテルで洗浄した(2×200ml)。湿ったケーキを40℃以下で一晩真空乾燥し、5.1gの生成物を得た。13C NMR(75.4 MHz, CDCl3):δ169.7, 166.6, 157.0, 156.3, 148.4, 147.3, 145.0, 144.4, 142.9, 131.5, 128.3, 126.4, 122.5, 118.7, 105.2, 94.7, 78.1, 67.0, 50.7, 49.2, 31.6, 31.3, 29.7, 23.0, 15.3, 13.5, 7.5;7−エチル−10−ヒドロキシカンプトセシンのPEGに対する割合:2.1重量%。
75mlのDCM中のBoc−(10)−(7−エチル−10−ヒドロキシカンプトセシン)(化合物10、750mg、1.52mmol)の溶液に、Boc−Sar−OH(432mg、2.287mmol)を加え、0℃まで冷却した。DMAP(432mg、2.287mmol)及びEDC(837mg、0.686mmol)を加え、反応混合物を0℃〜室温にて1.5時間、攪拌した。次に反応混合物を0.5%NaHCO3(2×75ml)、水(2×75ml)、最後に0.1NのHCl(1×75ml)で洗浄した。ジクロロメタン層を無水MgSO4で乾燥させ、溶媒を真空蒸発させ、乾燥した。収量=0.900mg(89%)。構造をNMRにて確認した。
Boc−(10)−(7−エチル−10−ヒドロキシカンプトセシン)−(20)−Sar−Boc(化合物19、900mg、1.357mmol)を、4mlのTFA及び16mlのDCMの溶液に加え、室温で1時間攪拌した。反応混合物を30℃でトルエンと共に蒸発させた。残渣を10mlのCHCl3に溶かし、エチルエーテルで沈殿させた。生成物を濾過し、乾燥させた。収量700mg(1.055mmol、78%)。13C NMR(67.8 MHz, CDCl3):δ168.26, 167.07, 158.84, 158.71, 148.82, 147.94, 147.22, 146.34, 144.04, 131.18, 130.08, 128.97, 124.46, 119.78, 106.02, 97.23, 79.84, 79.34, 66.87, 50.84, 49.86, 31.81, 23.94, 15.47, 13.84, 8.08。
無水DMF(30ml)中の7−エチル−10−ヒドロキシカンプトセシン−(20)−Sar・TFA(化合物20、2.17g、3.75mmol、1当量)の溶液を200mlの無水DCMで希釈した。Et3N(2.4ml、17.40mmol、4.5当量)を加えた後、TBDMSCl(2.04g、13.53mmol、3.5当量)を加えた。HPLCが出発物質の消失を示すまで(およそ1時間)、反応混合物を室温で攪拌した。有機層を0.5%NaHCO3で2回、水で1回、塩水で飽和させた0.1N HCl溶液で2回洗浄した後、MgSO4で乾燥させた。濾過し、溶媒を真空蒸発させた後、得られた油をDCMに溶解させた。エーテルを加えて得た固形物を、細または中程度のブフナー漏斗を使用して濾過した(2.00g、収率87%)。固形物のHPLCは、純度96%を示した。1H NMR及び13C NMRによって構造を確認した。1H NMR(300 MHz, CD3OD): δ0.23 (6H, s), 0.96 (9H, s), 0.98 (3 H, t, J = 7.3 Hz), 1.30 (3 H, t, J = 7.6 Hz), 2.13-2.18 (2H, m), 2.67 (3H, s), 3.11 (2 H, q, J = 7.6 Hz), 4.10 (1H, d, J = 17.6 Hz), 4.22 (1H, d, J = 17.6 Hz), 5.23 (2 H, s), 5.40 (1 H, d, J = 16.7 Hz), 5.55 (1H, d, J = 16.7 Hz), 7.32 (1H, s), 7.38-7.43 (2H, m), 8.00 (1H, d, J = 9.1 Hz)。13C NMR(75.4 MHz, CD3OD):δ-4.14, 8.01, 14.10, 19.30, 23.98, 26.16, 31.78, 33.52, 49.46, 50.95, 67.66, 79.80, 97.41, 111.96, 119.99, 127.75, 129.28, 129.67, 131.57, 145.24, 146.86, 147.16, 148.02, 150.34, 156.69, 158.72, 167.02, 168.27。
150mlの無水DCM中の40k4アーム−PEG−COOH(化合物3、10g、0.25mmol、1当量)の溶液に、20mlの無水DMF中のTBDMS−(10)−(7−エチル−10−ヒドロキシカンプトセシン)−Sar・HCl(化合物21、1.53g、2.5mmol、2.5当量)の溶液を加え、混合液を0℃まで冷却した。この溶液にEDC(767mg、4mmol、4当量)及びDMAP(367mg、3mmol、3当量)を加え、反応混合物を室温までゆっくりと温め、室温で一晩攪拌した。次に、反応混合物を真空蒸発させ、残渣を最小量のDCMに溶解させた。エーテルを加えて、固形物を形成させ、これを真空下で濾過した。残渣を30mlの無水CH3CNに溶かし、600mlのIPAを加えて沈殿させた。固形物を濾過し、IPA及びエーテルで洗浄し、生成物(9.5g)を得た。NMRで構造を確認した。
方法A.40k4アーム−PEG−Sar−(20)−(7−エチル−10−ヒドロキシカンプトセシン)−(10)−TBDMS(化合物22)を、H2O中のTFA50%混合液(200ml)に溶解させた。反応混合物を室温で10時間攪拌した後、100mlのH2Oで希釈し、DCM(2×300ml)で抽出した。合わせた有機相をH2O(2×100ml)で洗浄し、MgSO4で乾燥し、濾過し、真空蒸発させた。残渣を100mlの無水DMFに溶かし、ヒートガン(heat gun)で徐々に温め、400mlのDMFをゆっくり加えて沈殿させた。固形物を濾過し、IPA中の20%DMF及びエーテルで洗浄した。固形物をDCMに溶かし、エーテルで沈殿させた(6.8g)。構造をNMRで確認した。
実施例22.毒性データ
ヌードマウスを使用して、7−エチル−10−ヒドロキシカンプトセシンと共役した4アームPEGの最大耐用量(MTD)を試験した。マウスは、死亡率及び病気の徴候について14日間モニタされ、体重の減少が治療前体重の>20%の時点で屠殺した。
表2において、細胞毒性(IC50の結果を、各化合物についてμMで示す)は、インビトロにおける、各化合物の抗腫瘍作用の指標を提供する。この試験を、4アームPEG化された7−エチル−10−ヒドロキシカンプトセシン複合体のPEG化の効果の測定に用いた。PEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)、7−エチル−10−ヒドロキシカンプトセシン及びCPT−11のインビトロにおける細胞毒性を、MTS試験を用いて測定した。細胞を、37℃で72時間、薬剤で培養した。培養の後、MTS染料を加え、着色した生成物(ホルマザン)の形成を490nmで測定した。
ヌードマウスで増殖したヒトの皮下乳癌(MX−1)に対する4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)複合体(化合物9)の有効性を次のように測定した。少なくとも1週間の順応期間の後、供与マウスから、ヌードマウスの左側腹部(left auxiliary flank region)の単一の皮下部位に腫瘍小片を移植することにより、腫瘍を生着させた。腫瘍移植部位を週に2回観察し、1回は触診した。各マウスの腫瘍容積を、測径器で二次元測定し、公式:腫瘍容積=(長さ×幅2)を利用して計算することによって、決定した。腫瘍が、約100mm3の平均量に達した時点で、マウスを、非治療対照群、本明細書の実施例7に従って調製した4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)群、ここに参照することにより本願に援用されるConoverら(Anti-Cancer Drug Design, 1999, 14:499-506)の記載に従って調製したPEG−Ala−CPT群、及びCPT−11群からなる実験群に割付した。薬剤は、20mg/kgマウス体重の単回投与として、尾静脈を介した静脈注射によって投与された。複合体の用量は、7−エチル−10−ヒドロキシカンプトセシンに相当する量のことを指す。試験開始時及び単回投与後31日目に、マウスの体重及び腫瘍の大きさを測定した。
ヌードマウスにおけるヒトの乳癌(MX−1)の増殖に対する、一連の4アームPEG−アミノ酸(誘導体)−(7−エチル−10−ヒドロキシカンプトセシン)複合体の有効性を測定した。マウスを、本明細書の記載に従ってそれぞれ調製した、4アームPEG−Ala−(7−エチル−10−ヒドロキシカンプトセシン)、4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)、4アームPEG−Met−(7−エチル−10−ヒドロキシカンプトセシン)、4アームPEG−Sar−(7−エチル−10−ヒドロキシカンプトセシン)、CPT−11または生理食塩水対照の各群に割り付けた。マウスに、6日間連続して、5mg/kg/日を投与した(2日毎[q×2d]×6回)。マウス体重及び腫瘍の大きさを初回投与後及びその後の投与31日目までモニタした。
小(約100mm3)または大(約450mm3)のヒト乳房腫瘍MX−1を生着させたヌードマウスにおいて、4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)の抗腫瘍効果を、評価した。単回投与及び複数回投与の投与計画の両方につて、マウスにおける抗腫瘍治療活性を測定した。小さい乳房腫瘍を異種移植されたマウスでは、4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)の単回投与(20mg/kg)での治療で、TGIが100%に至った。4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)の複数回投与(20mg/kg、q2d×5)での治療の結果もまた、TGI100%に至った。CPT−11の単回投与(20mg/kg)では、腫瘍の増殖は抑制されず、CPT−11の複数回投与(20mg/kg、q2d×5)では、31日目の時点でTGI44%の結果となった。
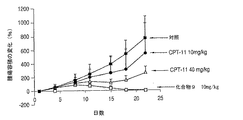
この実施例では、30mg/kgの4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)またはCPT−11を単回投与で静脈注射した後、4、8、11、15、18、及び22日目に、HT−29結腸直腸腫瘍細胞を異種移植したマウスの腫瘍の縮小を測定した。CPT−11は、80mg/kgの量でも投与した。結果を図7に記載する。4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)複合体は、顕著な抗腫瘍活性を示した。生理食塩水による対照群では、抑制は見られず、生存率0%であった。結果は、4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)が、結腸直腸癌の治療において、CPT−11と比較して著しく効果があることを示している。
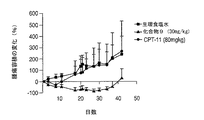
10mg/kg/用量の4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)またはCPT−11を5日間静脈注射した後、4、8、11、15、18、及び22日目に、HT−29結腸直腸腫瘍細胞を異種移植したマウスの腫瘍の縮小を測定した。CPT−11は、40mg/kg/用量を5日間の投与も行った。結果を図8に記載する。複数回投与でも、同様の結果が観察された。さらには、4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)の複数回投与では、CPT−11と比較して、顕著な治療効果を示した。結果は、4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)の複数回投与が、結腸直腸癌の治療において、CPT−11と比較して著しく効果があることを示している。MX−1腫瘍を異種移植されたマウスに見られたように、4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)またはCPT−11での治療では、治療期間中にマウスの体重は顕著には減少しなかった。
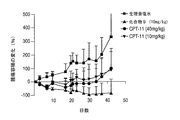
この実施例では、30mg/kgの4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)またはCPT−11を単回投与で静脈注射した後、4、8、11、15、18、及び22日目に、MiaPaCa−2膵臓腫瘍細胞を異種移植したマウスの腫瘍の縮小を測定した。CPT−11は、80mg/kgの量でも投与した。結果を図9に記載する。4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)複合体は、顕著な抗腫瘍活性を示した。生理食塩水による対照群及びCPT−11群では、抑制は見られなかった。結果は、4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)が、膵臓癌の治療において、CPT−11と比較して著しく効果があることを示している。4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)の単回投与での治療では、71%のTGIの結果となったのに対し、CPT−11の単回投与の注射での治療では、TGIは0%であった。
10mg/kg/用量の4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)またはCPT−11を5日間静脈注射した後、4、8、11、15、18、及び22日目に、MiaPaCa−2膵臓腫瘍細胞を異種移植したマウスの腫瘍の縮小を測定した。結果を図10に記載する。4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)の複数回投与では、CPT−11と比較して、顕著な治療効果を示した。結果は、4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)の複数回投与が、膵臓癌の治療において、CPT−11と比較して著しく効果があることを示している。4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)の複数回投与での治療では、95%のTGIの結果となったのに対し、CPT−11では、TGIは34%であった。
PEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)複合体のインビトロにおける代謝を、ラット肝細胞で観察した。PEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)をラット肝細胞と共に、37℃、pH7.5で2時間培養した。図11に示すように、7−エチル−10−ヒドロキシカンプトセシン及び7−エチル−10−ヒドロキシカンプトセシン−グルクロニド(7−エチル−10−ヒドロキシカンプトセシン−G)が主要な代謝産物として確認され、これは、インビボにおける7−エチル−10−ヒドロキシカンプトセシンの既知の代謝経路と一致している。
表6に、4種類のPEG−(7−エチル−10−ヒドロキシカンプトセシン)複合体の生理食塩水における溶解度を示す。4種類のPEG−(7−エチル−10−ヒドロキシカンプトセシン)複合体のすべてにおいて、7−エチル−10−ヒドロキシカンプトセシンで最大4mg/mlに相当する、良好な溶解性を示した。ヒト血漿中では、7−エチル−10−ヒドロキシカンプトセシンは、22〜52分の倍加時間でPEG複合体から着実に放出され、その放出は、次の実施例33に記載するように、pH及び濃度依存性のように見えた。
先の研究に基づいて、20−OH位のアシル化によって、活性化された閉環構造中に、ラクトン環を保護する。UV系のHPLC法を用いて、ラット及びヒト血漿における水溶液中の安定性及び加水分解の特性をモニタした。4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)複合体を、各試料と共に、室温で5分間、培養した。
腫瘍を有しないBalb/Cマウスに、4アームPEG−Gly−(7−エチル−10−ヒドロキシカンプトセシン)複合体を20mg/kgの単回投与によって注射した。さまざまな時点において、マウスを屠殺し、HPLCで、原形のままの複合体及び放出された7−エチル−10−ヒドロキシカンプトセシンについて、血漿を分析した。非コンパートメント解析(WinNonlin)を用いて、薬物動態学的分析を行った。詳細を表7に記す。
Claims (25)
- 下記式(I)の化合物:
R1、R2、R3及びR4は、独立して、OHまたは(L)m−Dであり、
Lは、アミノ酸、あるいは2−アミノアジピン酸、3−アミノアジピン酸、β−アラニン、β−アミノプロピオン酸、2−アミノ酪酸、4−アミノ酪酸、ピペリジン酸、6−アミノカプロン酸、2−アミノヘプタン酸、2−アミノイソ酪酸、3−アミノイソ酪酸、2−アミノピメリン酸、2,4−ジアミノ酪酸、デスモシン、2,2−ジアミノピメリン酸、2,3−ジアミノプロピオン酸、N−エチルグリシン、N−エチルアスパラギン、3−ヒドロキシプロリン、4−ヒドロキシプロリン、イソデスモシン、アロイソロイシン、N−メチルグリシン、サルコシン、N−メチルイソロイシン、6−N−メチルリジン、N−メチルバリン、ノルバリン、ノルロイシン及びオルニチンからなる群より選択されるアミノ酸誘導体の残基よりなる群から選択される二官能性のリンカーであり、
Dは、カンプトセシン、または下記式(II)を有するカンプトセシン類似体であり、
R7は、NO2、NH2、N3、水素、ハロゲン、F、Cl、Br、I、COOH、OH、O−C1-8アルキル、SH、S−C1-3アルキル、CN、CH2NH2、NH−C1-3アルキル、CH2−NH−C1-3アルキル、N(C1-3アルキル)2、CH2N(C1-3アルキル)2、O−、NH−及びS−CH2CH2N(CH2CH2OH)2、O−、NH−及びS−CH2CH2CH2N(CH2CH2OH)2、O−、NH−及びS−CH2CH2N(CH2CH2CH2OH)2、O−、NH−及びS−CH2CH2CH2N(CH2CH2CH2OH)2、O−、NH−及びS−CH2CH2N(C1-3アルキル)2、O−、NH−及びS−CH2CH2CH2N(C1-3アルキル)2、CHO及びC1-3アルキルからなる群より選択され、
R8は、H、C1-8アルキル及びCH2NR9R10からなる群より選択され、
ここで、
R9は、H、C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、及びC1-6アルコキシ−C1-6アルキルからなる群より選択され、
R10は、H、C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ−C1-6アルキル、及びCOR11 からなる群より選択され、
ここで、
R11 は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ、及びC1-6アルコキシ−C1-6アルキルからなる群より選択されるか、あるいは、
R9、R10、及びB環の窒素が、O、SまたはNR12を含む、飽和3〜7員の複素環を形成し、
ここで、
R12は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、アリール、ならびにC1-6アルキル、ハロゲン、ニトロ、アミノ、C1-6アルキルアミノ、ペルハロ−C1-6アルキル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ、C1-6アルコキシ−C1-6アルキル及びCOR13からなる群より選択される1つ以上の基で置換されたアリール、からなる群より選択され、ここでR13は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、C1-6アルコキシ、アリール、ならびに、C1-6アルキル、ペルハロ−C1-6アルキル、ヒドロキシ−C1-6アルキル、及びC1-6アルコキシ−C1-6アルキルのうちの1つ以上の基で置換されたアリール、からなる群より選択され、
R110 及びR111は、それぞれ独立して、水素、ハロ、アシル、アルキル、置換アルキル、アルコキシ、置換アルコキシ、アルケニル、アルキニル、シクロアルキル、ヒドロキシル、シアノ、ニトロ、アジド、アミド、ヒドラジン、アミノ、置換アミノ、ヒドロキシカルボニル、アルコキシカルボニル、アルキルカルボニルオキシ、アルキルカルボニルアミノ、カルバモイルオキシ、アリールスルホニルオキシ、アルキルスルホニルオキシ、−C(R117)=N−(O)j−R118 (ここで、R117は、H、アルキル、アルケニル、シクロアルキル、またはアリールであり、jは0または1であり、R118は、H、アルキル、アルケニル、シクロアルキル、または複素環である)、R119C(O)O−(ここで、 R119は、ハロゲン、アミノ、置換アミノ、複素環、置換複素環、またはR120−O−(CH2)k−(ここで、kは1〜10の整数であり、R120は、アルキル、フェニル、置換フェニル、シクロアルキル、置換シクロアルキル、複素環、または置換複素環である)である)からなる群より選択されるか、または
R7がR110と一緒に、またはR110がR111と一緒に、置換または非置換のメチレンジオキシ、エチレンジオキシ、またはエチレンオキシを形成し、
R112はHまたはOR'であり、ここで、R'は、アルキル、アルケニル、シクロアルキル、ハロアルキル、またはヒドロキシアルキルであり、
mは1であり、
前記Lは、前記アミノ酸またはアミノ酸誘導体のアミンおよびカルボン酸部分がそれぞれ、該化合物のポリマー部分のカルボキシル基、および前記カンプトセシンまたはカンプトセシン類似体の20−ヒドロキシル基と結合を形成して、Dとポリマー部分との間を連結しており、
nは、正の整数であり、
R1、R2、R3及びR4のすべてがOHではないことを条件とする。 - 前記カンプトセシン類似体が、
- Lが、グリシン、アラニン、メチオニンまたはサルコシン残基であることを特徴とする請求項1記載の化合物。
- Lがグリシン残基であることを特徴とする請求項1記載の化合物。
- nが28〜341であり、それによって該化合物のポリマー部分の総分子量が5,000〜60,000の範囲になることを特徴とする請求項1記載の化合物。
- nが114〜227であり、それによって該化合物のポリマー部分の総分子量が20,000〜40,000の範囲になることを特徴とする請求項1記載の化合物。
- nが227であり、それによって該化合物のポリマー部分の総分子量が40,000になることを特徴とする請求項1記載の化合物。
- R1、R2、R3及びR4が、(L)m−Dであることを特徴とする請求項1記載の化合物。
- 下記の化合物よりなる群から選択されることを特徴とする請求項1記載の化合物。
- 下記式で表されることを特徴とする請求項1記載の化合物。
- 薬剤として使用するための請求項1記載の化合物。
- Dが、7−エチル−10−ヒドロキシカンプトセシン残基であることを特徴とする請求項11記載の化合物。
- 癌を治療するための薬剤の調製における、有効量の請求項1記載の化合物の使用。
- 前記癌が、充実性腫瘍、リンパ腫、肺癌、小細胞肺癌、急性リンパ性白血病(ALL)、乳癌、結腸直腸癌、膵臓癌、膠芽腫、卵巣癌、および胃癌からなる群より選択されることを特徴とする請求項13記載の使用。
- 前記癌が転移性であることを特徴とする請求項13記載の使用。
- 前記化合物が、該化合物中に含まれる7−エチル−10−ヒドロキシカンプトセシンの重量で表して、1mg/kg/週〜100mg/kg/週の量で投与されることを特徴とする、請求項13記載の使用。
- 前記化合物が、該化合物中に含まれる7−エチル−10−ヒドロキシカンプトセシンの重量で表して、2mg/kg/週〜60mg/kg/週の量で投与されることを特徴とする請求項13記載の使用。
- 前記癌が乳癌であり、かつ前記化合物が下記式により表されることを特徴とする、請求項13記載の使用。
- 前記化合物が、該化合物中に含まれる7−エチル−10−ヒドロキシカンプトセシンの重量で表して、5〜20mg/kg/用量で投与されることを特徴とする、請求項18記載の使用。
- 前記癌が結腸直腸癌であり、かつ前記化合物が下記式により表されることを特徴とする、請求項13記載の使用。
- 前記化合物が、該化合物中に含まれる7−エチル−10−ヒドロキシカンプトセシンの重量で表して、10〜30mg/kg/用量で投与されることを特徴とする、請求項20記載の使用。
- 前記癌が膵臓癌であり、かつ前記化合物が下記式により表されることを特徴とする、請求項13記載の使用。
- 前記化合物が、該化合物中に含まれる7−エチル−10−ヒドロキシカンプトセシンの重量で表して、10〜30mg/kg/用量で投与されることを特徴とする、請求項22記載の使用。
- マルチアーム高分子プロドラッグの調製方法であって、
(a)利用可能な20−ヒドロキシル基を有する1当量のカンプトセシンまたはカンプトセシン類似体と、利用可能なカルボン酸基を有する1当量以上の二官能性リンカーとを提供し、
(b)利用可能なアミン基を有するカンプトセシン−二官能性リンカー中間体を形成するのに有効な条件下で、前記2つの反応物質を反応させ、
(c)活性部位当たり1当量以上の得られた中間体と、1当量の下記式
R1、R2、R3及びR4は、独立してOHまたは(L)m−Dであり、
Lは、アミノ酸、あるいは2−アミノアジピン酸、3−アミノアジピン酸、β−アラニン、β−アミノプロピオン酸、2−アミノ酪酸、4−アミノ酪酸、ピペリジン酸、6−アミノカプロン酸、2−アミノヘプタン酸、2−アミノイソ酪酸、3−アミノイソ酪酸、2−アミノピメリン酸、2,4−ジアミノ酪酸、デスモシン、2,2−ジアミノピメリン酸、2,3−ジアミノプロピオン酸、N−エチルグリシン、N−エチルアスパラギン、3−ヒドロキシプロリン、4−ヒドロキシプロリン、イソデスモシン、アロイソロイシン、N−メチルグリシン、サルコシン、N−メチルイソロイシン、6−N−メチルリジン、N−メチルバリン、ノルバリン、ノルロイシン及びオルニチンからなる群より選択されるアミノ酸誘導体の残基よりなる群から選択される二官能性のリンカーであり、
Dは、カンプトセシン、または下記式(II)を有するカンプトセシン類似体であり、
R7は、NO2、NH2、N3、水素、ハロゲン、F、Cl、Br、I、COOH、OH、O−C1-8アルキル、SH、S−C1-3アルキル、CN、CH2NH2、NH−C1-3アルキル、CH2−NH−C1-3アルキル、N(C1-3アルキル)2、CH2N(C1-3アルキル)2、O−、NH−及びS−CH2CH2N(CH2CH2OH)2、O−、NH−及びS−CH2CH2CH2N(CH2CH2OH)2、O−、NH−及びS−CH2CH2N(CH2CH2CH2OH)2、O−、NH−及びS−CH2CH2CH2N(CH2CH2CH2OH)2、O−、NH−及びS−CH2CH2N(C1-3アルキル)2、O−、NH−及びS−CH2CH2CH2N(C1-3アルキル)2、CHO及びC1-3アルキルからなる群より選択され、
R8は、H、C1-8アルキル及びCH2NR9R10からなる群より選択され、
ここで、
R9は、H、C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、及びC1-6アルコキシ−C1-6アルキルからなる群より選択され、
R10は、H、C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ−C1-6アルキル、及びCOR11 からなる群より選択され、
ここで、
R11 は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ、及びC1-6アルコキシ−C1-6アルキルからなる群より選択されるか、あるいは、
R9、R10、及びB環の窒素が、O、SまたはNR12を含む、飽和3〜7員の複素環を形成し、
ここで、
R12は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、アリール、ならびにC1-6アルキル、ハロゲン、ニトロ、アミノ、C1-6アルキルアミノ、ペルハロ−C1-6アルキル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ、C1-6アルコキシ−C1-6アルキル及びCOR13からなる群より選択される1つ以上の基で置換されたアリール、からなる群より選択され、ここでR13は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、C1-6アルコキシ、アリール、ならびに、C1-6アルキル、ペルハロ−C1-6アルキル、ヒドロキシ−C1-6アルキル、及びC1-6アルコキシ−C1-6アルキルのうちの1つ以上の基で置換されたアリール、からなる群より選択され、
R110 及びR111は、それぞれ独立して、水素、ハロ、アシル、アルキル、置換アルキル、アルコキシ、置換アルコキシ、アルケニル、アルキニル、シクロアルキル、ヒドロキシル、シアノ、ニトロ、アジド、アミド、ヒドラジン、アミノ、置換アミノ、ヒドロキシカルボニル、アルコキシカルボニル、アルキルカルボニルオキシ、アルキルカルボニルアミノ、カルバモイルオキシ、アリールスルホニルオキシ、アルキルスルホニルオキシ、−C(R117)=N−(O)j−R118 (ここで、R117は、H、アルキル、アルケニル、シクロアルキル、またはアリールであり、jは0または1であり、R118は、H、アルキル、アルケニル、シクロアルキル、または複素環である)、R119C(O)O−(ここで、 R119は、ハロゲン、アミノ、置換アミノ、複素環、置換複素環、またはR120−O−(CH2)k−(ここで、kは1〜10の整数であり、R120は、アルキル、フェニル、置換フェニル、シクロアルキル、置換シクロアルキル、複素環、または置換複素環である)である)からなる群より選択されるか、または
R7がR110と一緒に、またはR110がR111と一緒に、置換または非置換のメチレンジオキシ、エチレンジオキシ、またはエチレンオキシを形成し、
R112はHまたはOR'であり、ここで、R'は、アルキル、アルケニル、シクロアルキル、ハロアルキル、またはヒドロキシアルキルであり、
mは1であり、
前記Lは、前記アミノ酸またはアミノ酸誘導体のアミンおよびカルボン酸部分がそれぞれ、マルチアーム高分子プロドラッグのポリマー部分のカルボキシル基、および前記カンプトセシンまたはカンプトセシン類似体の20−ヒドロキシル基と結合を形成して、Dとポリマー部分との間を連結しており、
nは、正の整数であり、
R1、R2、R3及びR4のすべてがOHではないことを条件とする。 - マルチアーム高分子プロドラッグの調製方法であって、
(a)利用可能な20−ヒドロキシル基を有する1当量のカンプトセシンまたはカンプトセシン類似体を、利用可能なカルボン酸基を有する1当量以上の二官能性リンカーと、利用可能なアミン基を有するカンプトセシン−二官能性リンカー中間体を形成するのに有効な条件下で反応させ、
(b)活性部位当たり1当量以上の、工程(a)から得られた中間体を、1当量の下記式
R1、R2、R3及びR4は、独立してOHまたは(L)m−Dであり、
Lは、アミノ酸、あるいは2−アミノアジピン酸、3−アミノアジピン酸、β−アラニン、β−アミノプロピオン酸、2−アミノ酪酸、4−アミノ酪酸、ピペリジン酸、6−アミノカプロン酸、2−アミノヘプタン酸、2−アミノイソ酪酸、3−アミノイソ酪酸、2−アミノピメリン酸、2,4−ジアミノ酪酸、デスモシン、2,2−ジアミノピメリン酸、2,3−ジアミノプロピオン酸、N−エチルグリシン、N−エチルアスパラギン、3−ヒドロキシプロリン、4−ヒドロキシプロリン、イソデスモシン、アロイソロイシン、N−メチルグリシン、サルコシン、N−メチルイソロイシン、6−N−メチルリジン、N−メチルバリン、ノルバリン、ノルロイシン及びオルニチンからなる群より選択されるアミノ酸誘導体の残基よりなる群から選択される二官能性のリンカーであり、
Dは、カンプトセシン、または下記式(II)を有するカンプトセシン類似体であり、
R7は、NO2、NH2、N3、水素、ハロゲン、F、Cl、Br、I、COOH、OH、O−C1-8アルキル、SH、S−C1-3アルキル、CN、CH2NH2、NH−C1-3アルキル、CH2−NH−C1-3アルキル、N(C1-3アルキル)2、CH2N(C1-3アルキル)2、O−、NH−及びS−CH2CH2N(CH2CH2OH)2、O−、NH−及びS−CH2CH2CH2N(CH2CH2OH)2、O−、NH−及びS−CH2CH2N(CH2CH2CH2OH)2、O−、NH−及びS−CH2CH2CH2N(CH2CH2CH2OH)2、O−、NH−及びS−CH2CH2N(C1-3アルキル)2、O−、NH−及びS−CH2CH2CH2N(C1-3アルキル)2、CHO及びC1-3アルキルからなる群より選択され、
R8は、H、C1-8アルキル及びCH2NR9R10からなる群より選択され、
ここで、
R9は、H、C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、及びC1-6アルコキシ−C1-6アルキルからなる群より選択され、
R10は、H、C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ−C1-6アルキル、及びCOR11 からなる群より選択され、
ここで、
R11 は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、C3-7シクロアルキル、C3-7シクロアルキル−C1-6アルキル、C2-6アルケニル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ、及びC1-6アルコキシ−C1-6アルキルからなる群より選択されるか、あるいは、
R9、R10、及びB環の窒素が、O、SまたはNR12を含む飽和3〜7員の複素環を形成し、
ここで、
R12は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、アリール、ならびにC1-6アルキル、ハロゲン、ニトロ、アミノ、C1-6アルキルアミノ、ペルハロ−C1-6アルキル、ヒドロキシ−C1-6アルキル、C1-6アルコキシ、C1-6アルコキシ−C1-6アルキル及びCOR13からなる群より選択される1つ以上の基で置換されたアリール、からなる群より選択され、ここでR13は、水素、C1-6アルキル、ペルハロ−C1-6アルキル、C1-6アルコキシ、アリール、ならびに、C1-6アルキル、ペルハロ−C1-6アルキル、ヒドロキシ−C1-6アルキル、及びC1-6アルコキシ−C1-6アルキルのうちの1つ以上の基で置換されたアリール、からなる群より選択され、
R110 及びR111は、それぞれ独立して、水素、ハロ、アシル、アルキル、置換アルキル、アルコキシ、置換アルコキシ、アルケニル、アルキニル、シクロアルキル、ヒドロキシル、シアノ、ニトロ、アジド、アミド、ヒドラジン、アミノ、置換アミノ、ヒドロキシカルボニル、アルコキシカルボニル、アルキルカルボニルオキシ、アルキルカルボニルアミノ、カルバモイルオキシ、アリールスルホニルオキシ、アルキルスルホニルオキシ、−C(R117)=N−(O)j−R118 (ここで、R117は、H、アルキル、アルケニル、シクロアルキル、またはアリールであり、jは0または1であり、R118は、H、アルキル、アルケニル、シクロアルキル、または複素環である)、R119C(O)O−(ここで、 R119は、ハロゲン、アミノ、置換アミノ、複素環、置換複素環、またはR120−O−(CH2)k−(ここで、kは1〜10の整数であり、R120は、アルキル、フェニル、置換フェニル、シクロアルキル、置換シクロアルキル、複素環、または置換複素環である)である)からなる群より選択されるか、または
R7がR110と一緒に、またはR110がR111と一緒に、置換または非置換のメチレンジオキシ、エチレンジオキシ、またはエチレンオキシを形成し、
R112はHまたはOR'であり、ここで、R'は、アルキル、アルケニル、シクロアルキル、ハロアルキル、またはヒドロキシアルキルであり、
mは1であり、
前記Lは、前記アミノ酸またはアミノ酸誘導体のアミンおよびカルボン酸部分がそれぞれ、マルチアーム高分子プロドラッグのポリマー部分のカルボキシル基、および前記カンプトセシンまたはカンプトセシン類似体の20−ヒドロキシル基と結合を形成して、Dとポリマー部分との間を連結しており、
nは、正の整数であり、
R1、R2、R3及びR4のすべてがOHではないことを条件とする。
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US77246406P | 2006-02-09 | 2006-02-09 | |
| US60/772,464 | 2006-02-09 | ||
| US80439106P | 2006-06-09 | 2006-06-09 | |
| US60/804,391 | 2006-06-09 | ||
| US84493806P | 2006-09-15 | 2006-09-15 | |
| US60/844,938 | 2006-09-15 | ||
| US86451606P | 2006-11-06 | 2006-11-06 | |
| US60/864,516 | 2006-11-06 | ||
| PCT/US2007/003808 WO2007092646A2 (en) | 2006-02-09 | 2007-02-09 | Multi-arm polymeric conjugates of 7-ethyl-10-hydroxycamptothecin for treatment of breast, colorectal, pancreatic, ovarian and lung cancers |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2009526076A JP2009526076A (ja) | 2009-07-16 |
| JP2009526076A5 JP2009526076A5 (ja) | 2010-03-25 |
| JP5265384B2 true JP5265384B2 (ja) | 2013-08-14 |
Family
ID=38345853
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008554430A Expired - Fee Related JP5265384B2 (ja) | 2006-02-09 | 2007-02-09 | 乳癌、結腸直腸癌、膵臓癌、卵巣癌及び肺癌の治療のための7−エチル−10−ヒドロキシカンプトセシンのマルチアーム・ポリマー複合体 |
Country Status (11)
| Country | Link |
|---|---|
| EP (1) | EP1996208B1 (ja) |
| JP (1) | JP5265384B2 (ja) |
| KR (1) | KR20080091829A (ja) |
| CN (1) | CN101420963B (ja) |
| AU (1) | AU2007212201B2 (ja) |
| CA (1) | CA2640013A1 (ja) |
| ES (1) | ES2436109T3 (ja) |
| IL (1) | IL193164A0 (ja) |
| MX (1) | MX2008010303A (ja) |
| TW (1) | TWI426905B (ja) |
| WO (1) | WO2007092646A2 (ja) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7462627B2 (en) * | 2006-02-09 | 2008-12-09 | Enzon Pharmaceuticals, Inc. | Multi-arm polymeric conjugates of 7-ethyl-10-hydroxycamptothecin for treatment of breast, colorectal, pancreatic, ovarian and lung cancers |
| US7671067B2 (en) | 2006-02-09 | 2010-03-02 | Enzon Pharmaceuticals, Inc. | Treatment of non-hodgkin's lymphomas with multi-arm polymeric conjugates of 7-ethyl-10-hydroxycamtothecin |
| RU2009133793A (ru) * | 2007-02-09 | 2011-03-20 | Энзон Фармасьютикалз, Инк. (Us) | Лечение резистентных или невосприимчивых форм рака конъюгатами 7-этил-10-гидроксикампотецина с множеством ответвлений цепи |
| WO2010025337A1 (en) * | 2008-08-29 | 2010-03-04 | Enzon Pharmaceuticals, Inc. | Method of treating ras associated cancer |
| BRPI0919842A8 (pt) * | 2008-10-21 | 2015-09-22 | Enzon Pharmaceuticals Inc | tratamento de neuroblastioma com conjugados poliméricos de múltiplos braços de 7-etil-10-hidroxicamptotecina |
| KR20120044925A (ko) * | 2009-04-17 | 2012-05-08 | 엔즌 파마슈티칼스, 인코포레이티드 | 7-에틸-10-하이드록시캄토테신의 다중-가지 고분자 복합체를 이용한 신생혈관형성 억제 방법 |
| WO2013063286A1 (en) | 2011-10-25 | 2013-05-02 | Nektar Therapeutics | Treatment of patients suffering from cancer |
| US10132810B2 (en) | 2012-11-28 | 2018-11-20 | Nektar Therapeutics | Method for assessing and predicting efficacy of breast cancer treatment with a long-acting topoisomerase I inhibitor |
| EP3004888A1 (en) | 2013-05-31 | 2016-04-13 | Nektar Therapeutics | Method for predicting and evaluating responsiveness to cancer treatment with dna-damaging chemotherapeutic agents |
| CN104650342B (zh) * | 2013-11-18 | 2018-07-10 | 江苏豪森药业集团有限公司 | 多支链聚合药物前体及其应用 |
| CA2934552C (en) | 2014-01-14 | 2021-03-30 | Nektar Therapeutics | Combination-based treatment method |
| CN110448533B (zh) * | 2014-12-05 | 2022-03-29 | 天津键凯科技有限公司 | 一种聚乙二醇修饰的喜树碱类衍生物的药物组合物及其制备方法 |
| CN106831853B (zh) * | 2017-02-15 | 2019-02-22 | 浙江海正药业股份有限公司 | 7-乙基-10-o-叔丁基二苯基硅基喜树碱-20-o-甘氨酸盐酸盐的制备工艺 |
| CN108727584B (zh) * | 2017-04-21 | 2021-01-05 | 博瑞生物医药(苏州)股份有限公司 | 抗癌偶联物 |
| CN108727583B (zh) * | 2017-04-21 | 2022-03-22 | 高瑞耀业(北京)科技有限公司 | 多臂靶向抗癌偶联物 |
| CN108164419B (zh) * | 2017-12-25 | 2021-03-16 | 武汉大学 | 单分散聚乙二醇单甲醚修饰的丙泊酚前药的制备与应用 |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5880131A (en) * | 1993-10-20 | 1999-03-09 | Enzon, Inc. | High molecular weight polymer-based prodrugs |
| ES2267149T3 (es) * | 1996-08-30 | 2007-03-01 | University Of Hawaii | Nuevos derivados de criptoficina como agentes antineoplasicos. |
| US20040009229A1 (en) * | 2000-01-05 | 2004-01-15 | Unger Evan Charles | Stabilized nanoparticle formulations of camptotheca derivatives |
| US6548488B2 (en) * | 2000-03-17 | 2003-04-15 | Aventis Pharma S.A. | Composition comprising camptothecin or a camptothecin derivative and an alkylating agent for the treatment of cancer |
| US7132423B2 (en) * | 2001-09-21 | 2006-11-07 | Reddy Us Therapeutics, Inc. | Methods and compositions of novel triazine compounds |
| JP2006514064A (ja) * | 2003-01-16 | 2006-04-27 | テクノ マート シーオー.,エルティーディー. | ポルフィリン誘導体 |
| DE10307787A1 (de) * | 2003-02-24 | 2004-09-02 | Ktb Tumorforschungsgesellschaft Mbh | Protein-bindende Camptothecin-Derivate |
| AU2004228668B2 (en) * | 2003-04-03 | 2011-10-27 | Park Funding, Llc | PI-3 kinase inhibitor prodrugs |
| MXPA05010747A (es) * | 2003-04-11 | 2006-05-25 | Ptc Therapeutics Inc | Compuestos de acido 1,2,4-oxadiazolbenzoico y su uso para la supresion sin sentido y el tratamiento de enfermedades. |
| TWI334868B (en) * | 2003-06-03 | 2010-12-21 | Nippon Kayaku Kk | [1,2,4] triazoro [1,5-a] pyrimidine-2-ylurea derivative and use thereof |
| US20040247624A1 (en) * | 2003-06-05 | 2004-12-09 | Unger Evan Charles | Methods of making pharmaceutical formulations for the delivery of drugs having low aqueous solubility |
| LT1675622T (lt) * | 2003-09-17 | 2017-09-11 | Nektar Therapeutics | Daugiašakio polimero provaistai |
| WO2005068424A1 (en) * | 2004-01-20 | 2005-07-28 | Cell Therapeutics Europe S.R.L. | Indolinone derivatives as receptor tyrosine kinase ihibitors |
| TW200530236A (en) * | 2004-02-23 | 2005-09-16 | Chugai Pharmaceutical Co Ltd | Heteroaryl phenylurea |
| ITRM20040239A1 (it) * | 2004-05-13 | 2004-08-13 | Sigma Tau Ind Farmaceuti | Derivati ciclopeptidici ad attivita' antintegrine. |
-
2007
- 2007-02-09 JP JP2008554430A patent/JP5265384B2/ja not_active Expired - Fee Related
- 2007-02-09 AU AU2007212201A patent/AU2007212201B2/en not_active Ceased
- 2007-02-09 EP EP07763709.8A patent/EP1996208B1/en not_active Not-in-force
- 2007-02-09 MX MX2008010303A patent/MX2008010303A/es unknown
- 2007-02-09 ES ES07763709T patent/ES2436109T3/es active Active
- 2007-02-09 CN CN2007800127903A patent/CN101420963B/zh not_active Expired - Fee Related
- 2007-02-09 KR KR1020087021163A patent/KR20080091829A/ko not_active Application Discontinuation
- 2007-02-09 CA CA002640013A patent/CA2640013A1/en not_active Abandoned
- 2007-02-09 TW TW096104772A patent/TWI426905B/zh not_active IP Right Cessation
- 2007-02-09 WO PCT/US2007/003808 patent/WO2007092646A2/en active Application Filing
-
2008
- 2008-07-31 IL IL193164A patent/IL193164A0/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009526076A (ja) | 2009-07-16 |
| TWI426905B (zh) | 2014-02-21 |
| MX2008010303A (es) | 2009-03-05 |
| KR20080091829A (ko) | 2008-10-14 |
| AU2007212201B2 (en) | 2012-09-13 |
| AU2007212201A1 (en) | 2007-08-16 |
| WO2007092646A3 (en) | 2008-10-16 |
| EP1996208A2 (en) | 2008-12-03 |
| CA2640013A1 (en) | 2007-08-16 |
| ES2436109T3 (es) | 2013-12-27 |
| TW200810757A (en) | 2008-03-01 |
| CN101420963A (zh) | 2009-04-29 |
| CN101420963B (zh) | 2012-09-05 |
| EP1996208A4 (en) | 2011-04-06 |
| IL193164A0 (en) | 2009-08-03 |
| WO2007092646A2 (en) | 2007-08-16 |
| EP1996208B1 (en) | 2013-09-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5265384B2 (ja) | 乳癌、結腸直腸癌、膵臓癌、卵巣癌及び肺癌の治療のための7−エチル−10−ヒドロキシカンプトセシンのマルチアーム・ポリマー複合体 | |
| US7462627B2 (en) | Multi-arm polymeric conjugates of 7-ethyl-10-hydroxycamptothecin for treatment of breast, colorectal, pancreatic, ovarian and lung cancers | |
| EP2341774B1 (en) | Treatment of neuroblastoma with multi-arm polymeric conjugates of 7-ethyl-10-hydroxycamptothecin | |
| JP2005532429A (ja) | カンプトテシン誘導体およびそのポリマーコンジュゲート | |
| KR20090108082A (ko) | 7-에틸-10-하이드록시캄토테신 다분지형 고분자 접합체를 이용한 내성 또는 불응성 암의 치료방법 | |
| US8048891B2 (en) | Treatment of non-hodgkin's lymphomas with multi-arm polymeric conjugates of 7-ethyl-10-hydroxycamptothecin | |
| TW201010732A (en) | Method of treating RAS associated cancer | |
| JP2009539879A (ja) | インデノイソキノリン放出性ポリマー複合体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100205 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100205 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100806 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120911 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20121210 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130205 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130214 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130402 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130501 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |