JP5118968B2 - リピッドa類縁体の製造方法 - Google Patents
リピッドa類縁体の製造方法 Download PDFInfo
- Publication number
- JP5118968B2 JP5118968B2 JP2007533245A JP2007533245A JP5118968B2 JP 5118968 B2 JP5118968 B2 JP 5118968B2 JP 2007533245 A JP2007533245 A JP 2007533245A JP 2007533245 A JP2007533245 A JP 2007533245A JP 5118968 B2 JP5118968 B2 JP 5118968B2
- Authority
- JP
- Japan
- Prior art keywords
- formula
- compound represented
- solvent
- acid
- iii
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 CCCCCCCC(CC*C([C@@](C(COC)O*1O)OP(OCC=C)(OCC=C)=O)[C@]1NC(CCCCCCCCCC=CCCCCCC)=O)OC Chemical compound CCCCCCCC(CC*C([C@@](C(COC)O*1O)OP(OCC=C)(OCC=C)=O)[C@]1NC(CCCCCCCCCC=CCCCCCC)=O)OC 0.000 description 1
Landscapes
- Saccharide Compounds (AREA)
Description
2本のアシル型側鎖を予め導入後、2つの糖類を結合する特許文献3記載の製法はトータル工程数の減少、特に糖類の結合後における工程改善の面で優れているが、毒性を有する試薬の大量使用、爆発性試薬の使用、製造過程における操作性、再現性等に課題を有し、またジクロロメタンの使用も回避されていない。
したがって、環境に優しく、安全性、操作性、再現性に優れたE5564の製造方法が求められている。
<6> 上記<4>又は<5>において、有機スルホン酸は、メタンスルホン酸又はエタンスルホン酸であるのがよい。
<7> 上記<4>〜<6>のいずれかにおいて、第1の溶媒は、トルエン−ヘプタン混合溶媒であるのがよい。
<12> 上記<8>〜<11>のいずれかにおいて、水の含量が混合溶媒中、1〜10%(容量/容量比)であるのがよい。
<13> 上記<1>〜<12>のいずれかにおいて、求核剤が環状有機酸エステル類または環状ケトン類であるのがよい。
<15> 上記<1>〜<14>のいずれかにおいて、パラジウム触媒がテトラキス(トリフェニルホスフィン)パラジウムであるのがよい。
<16> 上記<15>において、テトラキス(トリフェニルホスフィン)パラジウムが酢酸パラジウムとトリフェニルホスフィンから系内で生成されるのがよい。
<A9> 上記<A5>〜<A8>のいずれかにおいて、水の含量が混合溶媒中、1〜10%(容量/容量比)であるのがよい。
<A10> 上記<A1>〜<A9>のいずれかにおいて、求核剤が環状有機酸エステル類または環状ケトン類であるのがよい。
<A12> 上記<A1>〜<A11>のいずれかにおいて、パラジウム触媒がテトラキス(トリフェニルホスフィン)パラジウムであるのがよい。
<A13> 上記<A12>において、テトラキス(トリフェニルホスフィン)パラジウムが酢酸パラジウムとトリフェニルホスフィンから系内で生成されるのがよい。
DDP: ジアリル N,N−ジイソプロピルホスホラミデート;
Py: ピリジン;及び
TFA: トリフルオロ酢酸。
式(I)の化合物は、以下の製造法により、製造することができる。
以下に実施例を挙げて本発明を更に具体的に説明するが、もとより本発明はこれらの実施例のみに限定されるものではない。
得られた粗体 289.1gにアセトニトリル 1065mLを加え、20℃で5分間攪拌後、4時間で0℃まで冷却し、さらに4時間攪拌した。析出した結晶を濾取し、減圧下室温にて終夜乾燥し、228.6g相当の標題化合物を得た。
得られた粗体にメタノール 1920mLを加え、不溶物をセライト濾過した。不溶物とセライトをメタノールで洗浄した。さらに、メタノール 1400mLを溶液に加えた後、17℃に冷却し水 375mLを滴下した。その後−20℃に冷却し45分間撹拌した後、濾過した。濾過物を事前に0℃に冷却した90%含水メタノール 400mLで洗浄、そのままヌッチェ上で減圧乾燥し 427.2gの湿体を得た。
10Lの4径フラスコに湿体 427.2gを入れ、メタノール 2400mLを加え溶解した。10℃に冷却後、水 180mLを滴下した。滴下終了後0℃に冷却し、50分間撹拌した後、濾過した。濾過物を事前に0℃に冷却した90%含水メタノール 400mLで洗浄後、35℃で減圧乾燥することにより標題化合物 199.5g(含量:92.2%)を得た。収率92.6%
このメタノール溶液3908.2mLを減圧下濃縮し、残渣440.9gを得た。残渣にアセトン450mLを加え、減圧下濃縮後、再びアセトン450mLを加え濃縮した。残渣を終夜冷蔵保存後、アセトン1800mLを加え40℃に加温し、1.5時間攪拌した。これを空冷し30℃以下で1.5時間攪拌後濾取した。濾過物をアセトン 750mLで洗浄後、取り出した固体を35〜40℃で減圧乾燥し、粗体として標題化合物のフリーリン酸体104.48g(含量74.2%)を定量的に得た。
得られたフリーリン酸体の粗体を0.1N水酸化ナトリウム水溶液で処理して、標題化合物を得た。
Claims (15)
- 第1の芳香族炭化水素溶媒中、ピリジン−トリフルオロ酢酸存在下、式(VII)で表される化合物にジアリル N,N−ジイソプロピルホスホラミデートおよび酸化剤を順次反応させて、式(VIII)で表される化合物を得、次いで求核剤の存在下、式(VIII)で表される化合物とパラジウム触媒とを反応させた後、ナトリウム源で処理して、式(I)で表される化合物を製造する方法。
- 式(VI)で表される化合物の1−プロペニル基を選択的に脱保護して、式(VII)で表される化合物を得、次いで、第1の芳香族炭化水素溶媒中、ピリジン−トリフルオロ酢酸存在下、式(VII)で表される化合物にジアリル N,N−ジイソプロピルホスホラミデートおよび酸化剤を順次反応させて、式(VIII)で表される化合物を得、次いで求核剤の存在下、式(VIII)で表される化合物とパラジウム触媒とを反応させた後、ナトリウム源で処理して、式(I)で表される化合物を製造する方法。
- 炭化水素類溶媒及び/又は第2の芳香族炭化水素溶媒を含んでなる第1の溶媒中、有機スルホン酸存在下、式(IV)で表される化合物と式(V)で表される化合物とを反応させて、式(VI)で表される化合物を得、次いで式(VI)で表される化合物の1−プロペニル基を選択的に脱保護して、式(VII)で表される化合物を得、次いで、第1の芳香族炭化水素溶媒中、ピリジン−トリフルオロ酢酸存在下、式(VII)で表される化合物にジアリル N,N−ジイソプロピルホスホラミデートおよび酸化剤を順次反応させて、式(VIII)で表される化合物を得、次いで求核剤の存在下、式(VIII)で表される化合物とパラジウム触媒とを反応させた後、ナトリウム源で処理して、式(I)で表される化合物を製造する方法。
- 前記第1の芳香族炭化水素溶媒がトルエン溶媒である請求項1〜3のいずれか1項記載の方法。
- 前記有機スルホン酸は、メタンスルホン酸又はエタンスルホン酸である請求項3又は4記載の方法。
- 前記第1の溶媒は、トルエン−ヘプタン混合溶媒である請求項3〜5のいずれか1項記載の方法。
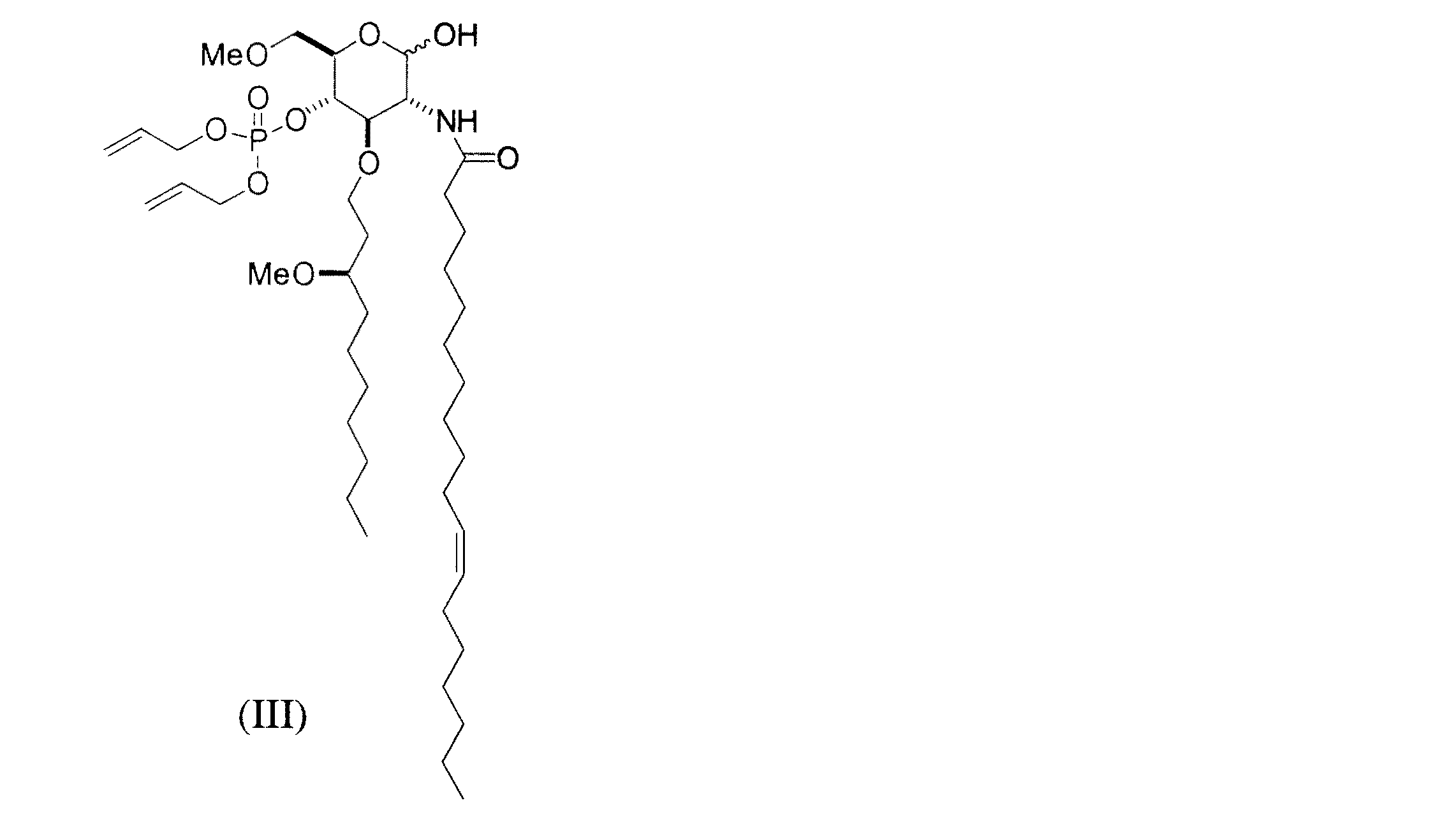
- 前記式(IV)で表される化合物は、酢酸エステル系溶媒と水との混合溶媒中、炭酸カリウム存在下、式(III)で表される化合物と、該式(III)で表される化合物1当量に対して1ないし10当量のトリクロロアセトニトリルとを反応させることにより得る請求項3〜6のいずれか1項記載の方法。
- 前記式(IV)で表される化合物は、式(X)で表される化合物の1−プロペニル基を選択的に脱保護して、式(III)で表される化合物を得、次いで酢酸エステル系溶媒と水との混合溶媒中、炭酸カリウム存在下、式(III)で表される化合物と、該式(III)で表される化合物1当量に対して1ないし10当量のトリクロロアセトニトリルとを反応させることにより得る請求項3〜7のいずれか1項記載の方法。
- 前記式(IV)で表される化合物は、ピリジン−トリフルオロ酢酸存在下、式(IX)で表される化合物にジアリル N,N−ジイソプロピルホスホラミデートおよび酸化剤を順次反応させて、式(X)で表される化合物を得、次いで式(X)で表される化合物の1−プロペニル基を選択的に脱保護して、式(III)で表される化合物を得、次いで酢酸エステル系溶媒と水との混合溶媒中、炭酸カリウム存在下、式(III)で表される化合物と、該式(III)で表される化合物1当量に対して1ないし10当量のトリクロロアセトニトリルとを反応させることにより得る請求項3〜8のいずれか1項記載の方法。
- 前記酢酸エステル系溶媒が酢酸メチルである請求項7〜9のいずれか1項記載の方法。
- 前記水の含量が混合溶媒中、1〜10%(容量/容量比)である請求項7〜10のいずれか1項記載の製造方法。
- 前記求核剤が環状有機酸エステル類または環状ケトン類である請求項1〜11のいずれか1項記載の方法。
- 前記求核剤がメルドラム酸またはジメドンである請求項1〜12のいずれか1項記載の方法。
- 前記パラジウム触媒がテトラキス(トリフェニルホスフィン)パラジウムである請求項1〜13のいずれか1項記載の方法。
- 前記テトラキス(トリフェニルホスフィン)パラジウムが酢酸パラジウムとトリフェニルホスフィンから系内で生成される請求項14記載の方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007533245A JP5118968B2 (ja) | 2005-08-31 | 2006-08-29 | リピッドa類縁体の製造方法 |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US71243105P | 2005-08-31 | 2005-08-31 | |
| US60/712,431 | 2005-08-31 | ||
| JP2005253044 | 2005-09-01 | ||
| JP2005253044 | 2005-09-01 | ||
| PCT/JP2006/316941 WO2007026675A1 (ja) | 2005-08-31 | 2006-08-29 | リピッドa類縁体の製造方法 |
| JP2007533245A JP5118968B2 (ja) | 2005-08-31 | 2006-08-29 | リピッドa類縁体の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2007026675A1 JPWO2007026675A1 (ja) | 2009-03-05 |
| JP5118968B2 true JP5118968B2 (ja) | 2013-01-16 |
Family
ID=47693090
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007533245A Expired - Fee Related JP5118968B2 (ja) | 2005-08-31 | 2006-08-29 | リピッドa類縁体の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5118968B2 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997011708A1 (en) * | 1995-09-29 | 1997-04-03 | Eisai Research Institute | Method for treating alcoholic liver disease |
| JPH11506793A (ja) * | 1995-06-05 | 1999-06-15 | エーザイ株式会社 | エンドトキセミアの治療及び防止に有用な置換されたリポ糖類 |
| WO2004074303A2 (en) * | 2003-02-20 | 2004-09-02 | Eisai Co, Ltd. | Reagents and methods for preparing lps antagonist b1287 and stereoisomers thereof |
-
2006
- 2006-08-29 JP JP2007533245A patent/JP5118968B2/ja not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11506793A (ja) * | 1995-06-05 | 1999-06-15 | エーザイ株式会社 | エンドトキセミアの治療及び防止に有用な置換されたリポ糖類 |
| WO1997011708A1 (en) * | 1995-09-29 | 1997-04-03 | Eisai Research Institute | Method for treating alcoholic liver disease |
| WO2004074303A2 (en) * | 2003-02-20 | 2004-09-02 | Eisai Co, Ltd. | Reagents and methods for preparing lps antagonist b1287 and stereoisomers thereof |
Non-Patent Citations (2)
| Title |
|---|
| JPN7012002200; Bioorganic & Medicinal Chemistry 10(4), 2002, pp.1129-36 * |
| JPN7012002201; Canadian Journal of Chemistry 80(8), 2002, pp.973-82 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2007026675A1 (ja) | 2009-03-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2417144B1 (en) | Synthesis of 2'-o-fucosyllactose | |
| Ying et al. | General methods for the synthesis of glycopyranosyluronic acid azides | |
| US7388083B2 (en) | Epimerization of 4′-C bond and modification of 14-CH3-(CO)-fragment in anthracyclin antibiotics | |
| CN101454333B (zh) | 乳糖胺衍生物 | |
| Dolhem et al. | Synthesis of glyco-1-ynitols via 1, 1-dibromo-1-alkenes from partially and unprotected aldoses | |
| Bianchi et al. | Neo-glycoconjugates: stereoselective synthesis of α-glycosyl amides via Staudinger ligation reactions | |
| JPH051092A (ja) | ヌクレオシド誘導体とその製造方法 | |
| JP5118968B2 (ja) | リピッドa類縁体の製造方法 | |
| IL189801A (en) | Process for production of lipid a analogue | |
| KR0139021B1 (ko) | 1-엔(n)-에틸시소마이신의 제조방법 | |
| CN101541820A (zh) | 二糖类化合物的钠盐及其制造方法以及其使用 | |
| Cai et al. | Efficient synthesis of d-xylo and d-ribo-phytosphingosines from methyl 2-amino-2-deoxy-β-d-hexopyranosides | |
| CN101238140A (zh) | 脂质a类似物的制造方法 | |
| JP4534024B2 (ja) | 化合物の分離用担体および化合物の分離方法 | |
| JP4691101B2 (ja) | 1−α−ハロ−2,2−ジフルオロ−2−デオキシ−D−リボフラノース誘導体及びその製造方法 | |
| JP2000226397A (ja) | リピドa中間体、その製法、リピドa及びその誘導体の製法 | |
| JP5478612B2 (ja) | コルヒチンおよびチオコルヒチンのグリコシド化の方法 | |
| RU2304583C1 (ru) | Способ синтеза ди- и триаминохлоринов | |
| JPH1059994A (ja) | シアル酸誘導体の製造方法 | |
| Kanaujiya et al. | N-nitrosamine directed stereoselective O-glycosylation reactions of 2-amino thioglycosides with NIS-TfOH | |
| JP5244608B2 (ja) | 二糖類化合物のナトリウム塩及びその製造方法並びにその使用 | |
| WO1997043295A1 (fr) | Derives de d-pentofuranose et leur procede de preparation | |
| TWI744448B (zh) | 甘草酸及半乳糖醛酸甘草酸之製造方法與該製造方法中所使用之中間體 | |
| Evans et al. | Synthesis and immunostimulatory activity of two α-S-galactosyl phenyl-capped ceramides | |
| JP2020158489A (ja) | 二環性ウロソン酸誘導体の製造方法およびウロソン酸誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090819 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120612 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120807 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121002 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121022 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5118968 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151026 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |