JP4774150B2 - 分析物の電気化学的分析法 - Google Patents
分析物の電気化学的分析法 Download PDFInfo
- Publication number
- JP4774150B2 JP4774150B2 JP2000566678A JP2000566678A JP4774150B2 JP 4774150 B2 JP4774150 B2 JP 4774150B2 JP 2000566678 A JP2000566678 A JP 2000566678A JP 2000566678 A JP2000566678 A JP 2000566678A JP 4774150 B2 JP4774150 B2 JP 4774150B2
- Authority
- JP
- Japan
- Prior art keywords
- electrode
- detection electrode
- receptor
- solution
- analyte
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images









Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/543—Immunoassay; Biospecific binding assay; Materials therefor with an insoluble carrier for immobilising immunochemicals
- G01N33/54366—Apparatus specially adapted for solid-phase testing
- G01N33/54373—Apparatus specially adapted for solid-phase testing involving physiochemical end-point determination, e.g. wave-guides, FETS, gratings
- G01N33/5438—Electrodes
Landscapes
- Health & Medical Sciences (AREA)
- Immunology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Chemical & Material Sciences (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Cell Biology (AREA)
- Food Science & Technology (AREA)
- Medicinal Chemistry (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Description
本発明は、分析物の電気化学的検出法、および電気化学的検出法に用いる検出電極に関する。
バイオセンサーを用いる、生物学的液体中の分析物、例えば、種々の抗原、抗体、DNA分子などの電気化学的分析は、機器分析の最も有望で魅力的な方法の一つである。この分野における興味の持続と多数の刊行物は、この方法の多くの基本的利点、すなわち、高感受性、簡単さ、および比較的簡単で費用のかからない装置の使用により説明づけられる。
【0002】
分析物の存在に伴う化学信号を測定可能な電気信号に変換する、電導性ポリマーフィルム、例えばポリピロールまたはポリチオフェンの使用に基づくバイオセンサー用具の構築は当該分野で知られている([1]および[2]参照)。
EP−A−0193154には、電気化学的検出に用いる、ポリピロールまたはポリチオフェンフィルムでコートした電極が記載されている。試験すべき分析物と相補的なバイオレセプターは、重合後の電導性ポリマーフィルム表面上に吸着する。WO89/11649には、電気化学的アッセイに用いる重合(高分子)電極の別の製造方法が記載されている。この方法では、所望の結合特異性を有するバイオレセプター分子は、重合中に電導性ポリマーフィルムに組み込まれる。各該アッセイにEP−A−0193154およびWO89/11649に記載の方法を用いるには、試験すべき分析物と特異的に結合することができる固定化バイオレセプターを有する種々の分析物用電極を合成する必要がある。
【0003】
本出願人の公開された出願PCT/GB98/00548には、試験する分析物と特異的に結合する固定化バイオレセプター分子を含む電導性ポリマー層でさらにコートされた金属性電位差測定電極を含む電気化学的検出電極を用いる電気化学的分析の電位差測定法が記載されている。分析物の存在は、固定化バイオレセプターに分析物が結合した時の検出電極表面の荷電変化により示される。分析物検出法は、最初、固定(一定)pHのワーキング緩衝溶液に浸漬した測定用具で一緒に接続した検出電極と参照電極を含む電気化学的セルを組み立てることにより実施される。検出電極と参照電極間の電位差のベース値を記録し、次いで、検出電極および参照電極を、ワーキング緩衝液と同じpHの、分析物を含むと思われるより高いイオン強度の溶液と接触させ、再度電位差を記録する。検出電極および参照電極を最終的に不純物を含まないワーキング緩衝液に移し、再度電位を記録する。分析物存在下で一定pHにおける緩衝液のイオン強度の変化から生じる検出電極と参照電極間の電気差の変化は、分析物の濃度に比例する。
【0004】
参考文献[13、14、15、16]および[3]に示すように、バイオレセプターを含むポリマーフィルムを有する検出電極の、周囲溶液のイオン強度の段階的変化に対する反応(電位変化の量および速度)は(いわゆる「イオン−ステップ」法)、大部分、ポリマーフィルムの荷電により決定される。ポリマーフィルムが作られている物質を除き、ポリマーフィルムの荷電は、それと結合するレセプター分子の荷電により決定される。特異的分析物とのアフィニティ(親和性)反応の結果としてレセプターの荷電が変化するならば、検出電極の反応も検出電極と被検液との接触後に行われるイオン−ステップ法の結果変化するであろう。大多数の分析物が両向性(amphoteric)の性質を持つため、レセプターの荷電は溶液のpHに依存し、したがって、該荷電はイオン−ステップ法時の溶液のpHを一定に維持するのに非常に重要である。
【0005】
このように、検出電極と被検液の接触前および後に行われるイオン−ステップ法に対する検出電極の反応の変化を測定することに基づく、先に記載の方法では、被検液中の、検出電極と結合するレセプターに特異的な分析物の存在を検出することができる。理想的な場合では、膜中のレセプターの荷電の変動、従って検出電極反応の変化は、検出電極に結合したレセプターに特異的な分析物の、被検液中濃度に正比例する。しかしながら、実際の条件では、同じ分析物の荷電はかなり変動し得るため、定量的に矛盾した結果が生じる。さらに、レセプターの荷電の変化が常にアフィニティ反応を伴うわけではない。これは、通常、小さいかまたは非荷電の抗原を試験する時に生じる[14]。
まとめると、先行技術文献に記載の、電導性ポリマー電極の使用に基づく電気化学的検出法の欠点には、検出電極の製造法が産業化になじみにくく複雑であること、得られる検出電極の特性の矛盾、検出電極を性能を失わずに保存する能力に限界があることが含まれる。さらに、電気化学的検出のための先に記載のプロトコール、特に、PCT/GB98/00548に記載の方法は、小さい非荷電の分子、または等電点が検出電極表面に固定化されたレセプターの等電点に近い分子を検出するための使用は限られている。
【0006】
本発明は、小さい非荷電の分子を分析できることにより応用範囲が広く、上記方法に固有の欠点をほぼ持たず、厳密な定量結果をもたらし、該電極の製造法を産業的製造法によりなじむようにし、分析の生産性を増し、再現性を改善することにより得られる結果の信頼性を増大させる試料中の分析物の電気化学的分析法を提供する。
すなわち、最初の局面において、本発明は、アビジン、ストレプトアビジン、抗FITC抗体、および電導性ポリマーに固定化または吸着した、少なくとも1種類のレセプター分子と結合することができる分子からなる群から選ばれるアダプター分子を含む電導性ポリマーでコートされた電導性電極を含む、分析物の電気化学的分析法に用いる検出電極を提供する。
【0007】
検出電極を用いる電気化学的分析法に固有の重要な問題の1つは、検出電極上に固定されたレセプターの天然の性質を一定期間にわたり保持する問題である。この分野で比較的進歩が見られているのは、限られた数の酵素検出電極についてのみである[7]。文献[8、9、10]で知られた精製レセプターを用いる電気化学的検出電極の大部分については、その有効な貯蔵期限が全く記載されていない。固定化レセプターの天然の性質の保持は、抗体をレセプターとして用いる場合に特に重要であるが、これはそれらに固有の高度の構造的変動性があるためである。
これに対して、抗体や他の生体分子は濃縮溶液の形で保存すると非常に長期にわたってその有用な特性を保持することが知られており、したがって、実用的特性を失わずに検出電極を長期貯蔵する問題は、電気化学的検出法の使用前または実施中にレセプターを急速に固定化することにより克服することができよう。
【0008】
この問題は、電導性ポリマーに固定化または吸着するいわゆるアダプター分子を用いることにより本宣誓発明(本発明)において解決される。アダプター分子の目的は、試験する分析物に特異的なレセプター分子を検出電極表面と結合することである。以下に述べるように、アダプター分子を用いることにより、検出電極の製造工程を一時的に分けることができる。すなわち、固定化/吸着アダプター分子を含む電極を製造し、それらを長期間貯蔵し、次いで電気化学的分析前または分析中に電極上に特異的レセプターを固定するのに便利である。適切なアダプター分子を選ぶことにより、全範囲の種々のレセプター分子と結合することができるアダプター分子を含む「普遍的」検出電極を製造することも可能である。試験する分析物の特異性は、単に、適切な特異性のアダプター分子のレセプターとの結合により「普遍的」検出電極に付与される。したがって、もはや電気沈殿工程中に所望の特異性のレセプターを組み込む必要はない。
【0009】
タンパク質アビジンおよびストレプトアビジンは、アダプター分子として用いるのに好ましい。生卵から得られたタンパク質であるアビジンは、それぞれ補助因子ビオチン1分子と結合することができる1つの部位を有する、4つの同じペプチドサブユニットからなる。ビオチン(ビタミンH)はすべての生体中に極微量存在する酵素の補助因子であり、主としてタンパク質やポリペプチドに結合してみいだされる。アビジンまたはストレプトアビジン(ある種の細菌培養、例えば、Streptomyces abiationから単離されたアビジン)分子との結合反応を開始し、この反応中に実質的に解離しない(解離定数〜10-15Mol/l「ビオチン−アビジン」複合体を形成するビオチン分子の能力はよく知られている[11、12]。
【0010】
本発明の著者が行った研究は、電導性ポリマーフィルムに固定化したアビジンおよびストレプトアビジンは長期間にわたりその天然の特性を保持し(少なくとも1年、おそらくそれ以上)、この期間を通してビオチンコンジュゲートレセプターと結合させるのに用いることができることを示した。広範囲の種々の分子にビオチンをコンジュゲートさせることができる技術は当該分野でよく知られている。すなわち、固定化アビジンまたはストレプトアビジンを含む検出電極は、適切なビオチン化レセプターを結合させるだけで簡単に該分析物に対して特異的にすることができる。
アビジンおよびストレプトアビジンは好ましいアダプター分子であるが、別のアダプター分子、特に少なくとも1種類のレセプター分子と特異的に結合することができる分子を用いることも本発明の範囲内である。この別のアダプター分子群に含まれるのはプロテインA、プロテインG、およびレクチンである。これらの分子はすべて、少なくとも1種類のレセプター分子と結合する能力を共有し、このことはそれらがある種のレセプター分子の各メンバーに存在する共通結合部位モチーフと特異的に結合することができることを意味する(結合相互関係に関する解離定数は10-8Mol/l以下である)。例として、プロテインA(Staphylococcus aureusから単離するか、または組換えDNA技術により得られた42kDのポリペプチド)は、免疫グロブリン、特に、広範囲の哺乳動物種由来のIgGのFc領域と結合し、プロテインG(IgG FcレセプタータイプIII、Bjorck,L.およびKronvall,G., J.Immunol., 133, 969(1984)参照)も、広範囲の哺乳動物種由来のIgG分子のFc領域と結合する。レクチンは糖タンパク質や炭水化物に存在してよい糖部分と結合するタンパク質である。レクチンの各タイプは、該糖部分に対する特異性を持つため、ある範囲の糖タンパク質や正しい糖部分を保持する複雑な炭水化物と結合することができよう。
【0011】
さらなる態様において、抗FITC抗体をアダプター分子として用いることができる。この態様では、分析物に対する検出電極の特異性は、適切な特異性のFITC標識レセプターを抗FITC抗体と結合させることによりもたらすことができる。
電導性ポリマーフィルム中/上にアダプター分子を用いることによっても、アダプター分子による電導性ポリマーの遊離表面のブロッキングと結びついた、検出電極と接触中の被検溶液成分の非特異的相互作用を減らすことにより電気化学的分析中に得られる結果の信頼性がかなり改善される。アダプター分子の使用も、例えば、さらなる表面ブロッキング手順の必要性を除去することにより、検出電極の製造方法の技術的効率を増大させる。
本発明の電位差測定検出電極は、製造に費用がかからず、便宜上、1回の電気化学的検出実験や一連の検出実験に使用して捨てることを意図した、ディスポーザブル形式で製造することができる。さらに本発明は、電気化学的検出に必要な検出電極と参照電極の両方を含む電極アセンブリーを提供する。以下で述べるように、適切な参照電極には銀/塩化銀およびカロメル(塩化水銀)電極が含まれる。該電極アセンブリーは、検出電極と参照電極を接続するための電気的接点を備えた、安価な物質から製造された覆いまたはホルダーを含むディスポーザブルユニットとして提供するのが好都合である。
本発明の検出電極は、限定されるものではないが、抗原の二重抗体サンドイッチアッセイ、抗体の二重抗原サンドイッチアッセイ、抗原の競合アッセイ、抗体の競合アッセイ、ヒト抗体検出用の血清学的アッセイ(例えば、標識抗ヒト抗体を用いるRubella IgG抗体)、およびIgMアッセイ(例えば、IgM−Rubella抗体)を含む広範囲の電気化学的分析法に用いることができる。
【0012】
第二の局面において、本発明は、アビジン、ストレプトアビジン、および電導性ポリマーに固定化した少なくとも1種類のレセプター分子と結合することができる分子からなる群から選ばれるアダプター分子を含む電導性ポリマー層でコートされた電導性電極を含む、分析物の電気化学的検出法に用いる検出電極の製造方法であって、
a)電導性ポリマーのモノマー単位とアダプター分子を含む電気化学的重合溶液を調製し、
b)コートする電極を該電気化学的重合溶液に浸漬し、
c)該電極と電気化学的重合溶液間の周期的電位を適用して該溶液からポリマーを電気化学的に合成することにより電極をコートする(ここで、周期的電位は少なくとも1完全周期を適用する)工程を含む方法を提供する。
さらに本発明は、アビジン、ストレプトアビジン、および電導性ポリマーに吸着した少なくとも1種類のレセプター分子と結合することができる分子からなる群から選ばれるアダプター分子を含む電導性ポリマー層でコートされた電導性電極を含む、分析物の電気化学的検出法に用いる検出電極の製造方法であって、
a)電導性ポリマーのモノマー単位を含む電気化学的重合溶液を調製し、
b)コートする電極を該電気化学的重合溶液に浸漬し、
c)該電極と電気化学的重合溶液間の周期的電位を適用して該溶液からポリマーを電気化学的に合成することにより電極をコートし(ここで、周期的電位は少なくとも1完全周期で適用する)、
d)該アダプター分子が該電極をコーティングする電導性ポリマー上に吸着するようなアダプター分子を含む溶液と、該コートされた電極を接触させる工程を含む方法を提供する。
【0013】
本発明の方法によれば、電導性ポリマーフィルムは、モノマー溶液からの電気化学的合成により電導性電極表面に沈殿する。電導性電極は、水性媒質中で安定な金属または金属に準ずる電導性を有する標準的電位差測定用電極であることが好ましい。本明細書中の実施例に示すように、電導性ポリマーフィルムの電気的沈殿は、モノマー、極性溶媒、およびバックグラウンドの電解質を含む溶液を用いて行われる。ピロール、チオフェン、フラン、またはアニリンが好ましいモノマーである。極性溶媒として用いるには脱イオン水が好ましい。
当業者によく知られているように、電導性ポリマーは、該ポリマーの構造および/または電導特性を修飾するために、電気化学的合成段階で添加(dope)されることが多い。典型的ドーパント(dopant)アニオンは、重合工程中に組み込まれ、ポリマーの基本骨格(バックボーン)の正の荷電を中和する、サルフェート(SO4 2−)である。サルフェートはイオン交換により容易に放出されないので、ポリマー構造を維持するのを助ける。本発明において、電極の感度を増大させるには、該ポリマー周囲の溶液によるイオン交換について最大能力を有するドーパントアニオンを用いることが好ましい。これは、電気化学的重合溶液の調整時にバックグラウンドの電解質として、アニオンが大イオン半径を有する塩を用いることにより達成される。アニオンが大イオン半径を有する適切な塩には、ドデシル硫酸ナトリウムおよびデキストラン硫酸ナトリウムが含まれる。電気化学的重合溶液中のこれら塩の濃度は試験の種類に応じて0.005〜0.05Mの範囲内で変動する。
【0014】
多くの論文[4、5]が報告しているように、溶液に浸漬した電導性(導電性)ポリマーについてイオン平衡が達成される速度およびイオン交換が生じる容易さは、本質的に、電気沈殿段階で導入されるドーパントアニオンのサイズに依存し、ドーパントアニオンのイオン半径がより大きければ、より急速なイオン交換が生じ、より急速に平衡状態に達する。これは、溶液のイオン組成の変動に応じた「金属電極−電導性ポリマー」の電位が変化する速度と値に直接結びついている[6]。
電導性ポリマー膜は、いずれも検出電極表面へのレセプターの結合と、緩衝溶液組成の変動に対して検出電極を敏感にするのを助ける二重の機能を果たす。特に、電導性ポリマーの酸化還元組成に影響を及ぼす緩衝溶液の組成変化は、検出電極の定常状態における電位に、対応する変化を生じる。
【0015】
アダプター分子は、アダプター分子を電気化学的重合溶液に加えることにより電気化学的合成段階で電導性ポリマーフィルムに固定化されても、電気化学的重合後に電導性ポリマーフィルム表面に吸着してもよい。前者の場合は、アダプター分子の溶液を電気沈殿工程の直前に電気沈殿用溶液に加えてよい。完成した溶液の貯蔵時間が30分以内である場合、沈殿工程は最適に行われる。特定の試験の種類に応じて、溶液中のアダプター分子濃度は5.00〜100.00μg/mlの範囲で変動してよい。アダプター分子含有溶液からの電導性ポリマーの電気沈殿工程は本明細書中の実施例に記載している。電気沈殿工程が完結したら、得られた検出電極を、脱イオン水および0.01Mリン酸緩衝生理食塩溶液で連続してリンスし、次いで、試験の種類に応じて、微生物増殖阻害剤や殺菌剤(例えばゲンタマイシン)を含む特別な貯蔵用緩衝溶液に入れるか、室温で無塵空気中で乾燥してよい。
【0016】
電気沈殿工程完結後にアダプター分子が吸着されるべきである場合は、以下のプロトコールを用いてよい(本発明はこの特定の方法の使用に何ら限定されるものではないことを述べておくが)。検出電極を最初脱イオン水でリンスし、新たに調製した0.02M炭酸緩衝溶液に入れ、これを15−60分間保つ。次に、溶液を満たした容器に検出電極を浸漬するか、または検出電極表面に溶液を1滴のせることにより、検出電極を濃度1.00〜50.00μg/mlのアダプター分子を含む、新たに調製した0.02M炭酸緩衝溶液と接触させる。検出電極を、典型的には+4℃で1〜24時間、アダプター分子の溶液とインキュベートする。インキュベーション後、検出電極を脱イオン水でリンスし、0.1Mリン酸緩衝生理食塩水溶液中に1〜4時間置く。試験の種類に応じて、次いで検出電極を微生物増殖阻害剤または殺菌剤を含む特別な貯蔵緩衝液中に置くか、または室温で無塵空気で乾燥する。
【0017】
アダプター分子がアビジンまたはストレプトアビジンである場合は、検出電極を製造するための上記本発明方法は、所望により、さらにビオチンと結合した特異的レセプターが、ビオチン/アビジンまたはビオチン/ストレプトアビジン結合相互作用により電極の電導性ポリマーコーティングに固定化または吸着したアビジンまたはストレプトアビジン分子と結合するように、該ビオチン化レセプターを含む溶液と該コートされた電極を接触させる工程を含んでいてよい。
本発明の著者および他の研究者[12]がともに行った研究は、最適条件下でレセプターをビオチン化すると、ビオチン化していないものに比べて特性(アフィニティ、貯蔵品質など)が変化しないことを示した。
ビオチン化として当業者に知られた方法である、ビオチンと対応するレセプターのコンジュゲーション(結合)は、知られた手順の1つ(例えば[12]に記載のような)を用いて行うことができる。さらに、多くの既製品の、特異性の異なるビオチン化抗体が市販されている(例えば、抗ヒトIgGまたは抗ヒトIgAビオチン標識ヤギ抗体(Calbiochem-Novabiochem, USA)。
【0018】
ビオチン化レセプターを用いる大きな利点の1つは、検出電極と、結合したアビジンまたはストレプトアビジンと対応するビオチン化レセプターの間の反応を生じさせることにより検出電極の特異性を変化させることができることである。先に記載したように、アビジン/ストレプトアビジンが結合した検出電極は事実上「普遍的検出電極」であり、試験する所望の分子に対する特異性は適切なビオチン化レセプターの結合により付与される。試験する分析物に特異的な、アビジンまたはストレプトアビジンが結合した検出電極を製造するには、検出電極上に結合したアビジンまたはストレプトアビジンとビオチン化レセプター間で反応を行うが、このためには、溶液を満たした溶液に検出電極を浸漬するか、または検出電極表面に溶液を滴下することにより、検出電極を室温で後者の溶液と接触させる(溶液中のビオチン化レセプター濃度は一般に0.1〜100μg/mlであり、接触時間は3〜15分間である)。
レセプター分子は別の分子(分析物)と特異的に結合することができるあらゆる分子であり得る。適切なレセプターの種類には、モノクローナルおよびポリクローナル抗体、キメラ抗体、抗原認識能を保持している抗体断片(例えば、FabおよびFab2断片)、組換えタンパク質およびその断片、合成ペプチド、抗原、一本鎖DNA、RNAまたはPNA分子、ホルモン、ホルモンレセプター、酵素、化合物などが含まれる。
【0019】
上記のように、当該分野で知られた、電位差測定用検出電極を用いる電気化学的検出法は、小さい非荷電の抗原の検出に使用するには限界がある。この問題を克服し、厳密な定量結果を得るために、荷電標識と結合した第二レセプターまたは競合分子を用いてよい。
したがって、さらなる局面において、本発明は、試料中の分析物の電気化学的検出法であって、
a)試料中の検出すべき所望の分析物を結合することができるレセプターを固定化または吸着した電導性ポリマーコーティングを有する検出電極を得、
b)該所望の分析物が該固定化または吸着レセプターと結合するように試料を含む被検溶液に検出電極を浸漬し、
c)固定化または吸着レセプターと結合する部位から空間的に離れた部位で該分析物と結合することができる、荷電標識と結合した第二レセプターを含む溶液と検出電極を接触させ、
d)処理した検出電極と参照電極の両方を電解液中に浸漬したときの、それらの間の電位差をモニターし、
e)一定pHにおける電解液のイオン強度の変化に伴う検出電極と参照電極間の電位差をモニターする工程を含む方法を提供する。
上記方法のアフィニティ反応工程は、同業者によく知られた標準的サンドイッチ法に等しい。分析のサンドイッチ形式は、試験に用いるレセプターおよび標識第二レセプターが抗原上の空間的に異なる種々のエピトープと結合する抗体である多価抗原の検出に特に有用である。該サンドイッチ形式は、抗原が空間的に分離した2またはそれ以上の同じエピトープを保持する場合にも用い得る。この後者の場合、試験に用いるレセプターおよび標識第二レセプターは同じ特異性の抗体であってよい。
【0020】
競合アッセイ形式で電気化学的分析を行うことも本発明の範囲内である。したがって、本発明は、試料中の分析物の電気化学的検出法であって、
a)レセプター試料中の検出すべき所望の分析物と結合することができるレセプターを固定化または吸着した電導性ポリマーコーティングを有する検出電極を得、
b)該所望の分析物が該固定化または吸着レセプターと結合するように試料を含む被検溶液に検出電極を浸漬し、
c)該固定化または吸着レセプターと結合することができる、荷電標識と結合した競合分子を含む溶液と検出電極を接触させ、
d)電解液中に浸漬したときの、処理した検出電極と参照電極の間の電位差をモニターし、
e)一定pHにおける電解液のイオン強度の変化に伴う検出電極と参照電極間の電位差をモニターする工程を含む方法も提供する。
【0021】
この競合電気化学的アッセイにおいて、競合分子は、標識分析物、または固定化/吸着レセプター上の同じ分析物結合部位と結合することができる、標識された、分析物の構造的類似体であるかもしれない(図5Bおよび図7B参照)。低分子の分析物(例えば、本明細書中の実施例に記載のジゴキシン)の検出には、標識分析物を競合分子として用いるのが特に好ましい。あるいはまた、競合分子は固定化/吸着レセプターの異なる部位に結合してよい。例えば、固定化レセプターが抗体である場合は、抗免疫グロブリン抗体(好ましくはFab特異的)、または適切な特異性の抗イディオタイプ抗体であるかも知れない(図5Aおよび図7A参照)。
【0022】
本出願の図5A、5B、7A、および7Bを参照して当業者が容易に理解するであろうように、競合検出法は、通常、検出電極表面に過剰のレセプター部位があることに依存する。分析物と結合しないレセプターは競合分子と結合するのに利用されよう。レセプター部位の総数が一定のままであるとすると、結合競合分子の量は、存在する分析物の量と反比例するであろう。
分析物の濃度と結びついた化学的信号を測定可能な電気信号に変換するには、第二レセプターまたは競合分子と結合する荷電標識は以下の特性を有するあらゆる荷電標識であり得る。(i)パートd)の電解液のpHで正味の荷電を保持し、(ii)この荷電の大きさが一定pHでの電解液のイオン強度の変化に応じて変化する。
好ましい荷電標識は、高度に荷電されており、1静電単位(e)より大きいパート(d)の電解質のpHで正味の荷電を有する。適切な荷電標識には、金、フェロセン、およびラテックスミクロスフェアが含まれる。荷電標識の荷電の大きさは、工程(d)および(e)間のイオン強度のステップ変化が検出電極と参照電極間の検出可能な電位差の変化を生じるように検出電極上の電導性ポリマーコーティングの酸化還元組成に影響を与える。荷電標識は、レセプター/分析物/第二レセプター複合体(サンドイッチアッセイ)、またはレセプター/競合分子複合体(競合アッセイ)の形成において電導性ポリマーに極めて接近するのみである。すなわち、荷電標識を用いることにより、正しい定量結果を得ることができ、小さな非荷電の分析物を試験する能力に関して、試験可能な分析物の範囲がかなり広がる。
【0023】
ラテックスミクロスフェアは、第二レセプターまたは競合分子と結合した、好ましい荷電標識である。ラテックスミクロスフェアを用いるコンジュゲーションは、例えば[17]または[18]に記載の、知られた技術の1つ用いるか、またはラテックスミクロスフェアと抗体をコンジュゲーションさせるための特殊な市販キット、例えば、「Carbodiimide Kit for Carboxylated Beads」(Polysciences Inc., USA)を用い、製造業者が提供するプロトコールに従って行うことができよう。ある種の既製のラテックスコンジュゲートは、専門の製造業者(例えば、Polysciences Inc., USA)から市販されている。
荷電標識を用いる代わりに、分析物濃度に関連する化学信号を測定可能な電気信号に変換するために、酵素標識と結合した第二レセプターまたは競合分子を用いる上記方法と同等の電気化学的検出法を実施することもできる。
【0024】
したがって、本発明は、さらに試料中の分析物の電気化学的検出法であって、
a)レセプター試料中の検出すべき所望の分析物と結合することができるレセプターを固定化または吸着した電導性ポリマーコーティングを有する検出電極を得、
b)該分析物が該固定化または吸着レセプターと結合するように試料を含む被検溶液に検出電極を浸漬し、
c)固定化または吸着レセプターと結合する部位から空間的に離れた部位で該分析物と結合することができる、酵素と結合した第二レセプターを含む溶液と検出電極を接触させ、
d)処理した検出電極と参照電極の両方を電解液中に浸漬したときの、それらの間の電位差をモニターし、
e)該酵素の基質を含む電解液に曝露した後の検出電極と参照電極間の電位差をモニターする工程を含む方法を提供する。
【0025】
a)レセプター試料中の検出すべき所望の分析物を結合することができるレセプターを固定化または吸着した電導性ポリマーコーティングを有する検出電極を得、
b)該所望の分析物が該固定化または吸着レセプターに結合するように試料を含む被検溶液に検出電極を浸漬し、
c)該固定化または吸着レセプターと結合することができる、酵素と結合した競合分子を含む溶液と検出電極を接触させ、
d)処理した検出電極と参照電極の両方を電解液中に浸漬したときの、それらの間の電位差をモニターし、
e)該酵素の基質を含む電解液に曝露した後の検出電極と参照電極間の電位差をモニターする工程を含む、対応する競合電気化学的検出法も本発明の範囲内である。
【0026】
上記方法のある態様において、第二レセプターまたは競合分子と結合した酵素は、検出電極の電導性ポリマーコーティングの酸化還元組成に直接影響を及ぼす基質を、電導性ポリマーコーティングの酸化還元組成に対する検出可能な効果を持たない生成物に変換することができる。
別の態様において、第二レセプターまたは競合分子と結合した酵素は、検出電極の電導性ポリマーコーティングの酸化還元組成に対する検出可能な効果を持たない基質を、電導性ポリマーコーティングの酸化還元組成に直接または間接的に影響を及ぼすことができる生成物に変換することができる。そのような酵素の例はホースラディッシュパーオキシダーゼである。酵素反応の生成物が電導性ポリマーの酸化還元組成に間接的影響を及ぼし得る1つの方法は、電解液のpHを変化させることである(この態様において、電解液のpHは緩衝されない)。そのような生成物を生じる酵素の例はウレアーゼである。
さらなる態様において、第二レセプターまたは競合分子と結合した酵素は、検出電極の電導性ポリマーコーティングの酸化還元組成に対する検出可能な効果を持たない生成物を、第二酵素の基質である生成物に変換することができる(ここで、第二酵素の作用により電導性ポリマーの酸化還元組成に直接または間接的に影響を及ぼす第二生成物が生じる)。
【0027】
すべての態様において、コンジュゲート酵素は、レセプター/分析物/第二レセプター複合体(サンドイッチアッセイ)の形成、またはレセプター/競合分子複合体(競合アッセイ)の形成により電導性ポリマーに極めて接近する。
第二レセプターまたは競合分子と酵素標識のコンジュゲーションは当該分野で知られたあらゆる技術により行ってよい(例えば、[19]参照)。広く利用可能な、種々の酵素標識と種々の特異性のレセプターとのコンジュゲートの市販調製物も使用できる。
上記電気化学的検出法はすべて、別の分子(分析物)と特異的に結合することができるあらゆる種類のレセプターを用いて行うことができる。適切なレセプターには、モノクローナルおよびポリクローナル抗体、キメラ抗体、抗原認識能を保持する抗体断片(例えば、FabおよびFab2断片)、組換えタンパク質およびその断片、合成ペプチド、抗原、一本鎖DNA、RNAもしくはPNA分子、ホルモン、ホルモンレセプター、酵素、化合物などが含まれる。
第二レセプターまたは競合分子が荷電または酵素標識とコンジュゲートするかどうかに関わらず、本発明の検出法すべての感度は「連続(逐次)」形式のアフィニティ反応(すなわち、工程(a)〜(c))を行うことにより達成される。これは、試験する分析物が多価抗原である場合に特にそうである(すなわち、サンドイッチアッセイ)。該連続形式では、レセプターが結合した検出電極を、最初、分析物の存在を試験する試料を含む被検溶液と接触させる。本明細書で用いている用語「試料」には、その範囲内に、全血、血清、血漿、尿、リンパ液、能脊髄液、または精液、周囲(環境)の液体、食品および飲料品産業で使用または生産される物質、またはそれらのあらゆる希釈物もしくは抽出物が含まれる。試料には固体物質の溶液もしくは抽出物も含まれてよい。試験溶液用に用いる容器は、マイクロタイタープレートのウェル、微量遠心チューブ、または適切な大きさの他のあらゆる容器であってよい。試験溶液の容量は、検出電極の幾何学的大きさに応じて一般に5〜200μlであろう。検出電極と被検溶液の接触時間は、連続的に攪拌するか攪拌せずに、典型的には15〜40℃で3〜30分間である。
【0028】
被検溶液と接触後、検出電極を標識第二抗レセプターの溶液を含む溶液に移す。用いる容器と溶液の容量は検出電極と被検溶液の接触に用いるものと同様である。溶液中の標識第二レセプターまたは標識競合分子の濃度は、試験に必要な感度に応じて典型的には1〜100μg/mlである。連続的に攪拌するか攪拌せずに、15〜40℃で3〜30分間接触させる。
「連続」形式に代わるものとして、検出電極を適切な標識第二レセプターまたは標識競合分子を加えた被検溶液と約5〜60分間の接触時間で接触させることにより行程(b)および(c)を同時に行うことにより、実質的に全試験時間を減らし、試験手順を簡略化することができる。被検溶液に加える標識第二レセプターまたは標識競合分子の濃度は、その種類、特異的特性、および試験に必要な感度に応じて、典型的には1〜100μg/mlである。
得られる結果をゆがめることになる、試験する生物学的液体成分と検出電極の間に生じうる非特異的相互作用や、標識第二レセプターまたは標識競合分子の検出電極表面への非特異的吸着を排除するために、種々のブロッキング剤を標識第二レセプターまたは標識競合分子の溶液に加えてよい。適切なブロッキング剤には、ウシ血清アルブミン(0.5〜5重量%)、希正常ヒトまたは動物血清(5〜10容量%)、ゼラチン(10〜50容量%)などが含まれる。そうすることにより検出電極のあらゆる遊離表面が同時にブロッキングされ、標識第二レセプターまたは標識競合分子と検出電極の相互作用が達成される。
【0029】
本発明の検出法のすべてにおいて、固定化/吸着アダプター分子を含む電極を用いることができる。特に、ビオチン/アビジン、ビオチン/ストレプトアビジン、プロテインA/抗体、プロテインG/抗体、FITC/抗FITCまたはレクチン/糖結合相互作用によりレセプター分子を検出電極表面に結合させてよい。
アダプター分子を含む「普遍的」検出電極を用いることにより「ワン−ポット」形式で検出方法を実施することができる。この態様において、アフィニティ反応は、相互作用する分子間の接触を最大にし、感度を最大にし、試験時間を最小にする均質な溶液中で行われる。この場合、レセプター溶液と標識第二レセプターまたは標識競合分子の溶液は、同時または連続的に、単一反応溶液中の分析物を含む懸濁試料を含む被検溶液に加えられる。被検溶液中のレセプターおよび標識第二レセプターまたは標識競合分子の濃度は、典型的にはそれぞれ0.1〜100μg/mlおよび1−100μg/mlである。次に、結合反応が生じるように、被検溶液を連続的に混合するか混合せずに15〜40℃で5〜60分間インキュベートする。次いで、適切なアダプター分子を含む検出電極を、被検溶液を含む溶液に浸漬するか、または検出電極表面に被検溶液を1滴おくことにより被検溶液と接触させる。検出電極と被検溶液の接触時間は、典型的には15〜40℃で3〜30分間である。次に、検出電極に結合した分析物の量を、第二レセプターまたは競合分子が荷電標識または酵素標識のいずれで標識されているかに応じて、「イオン−ステップ」法を用いるかまたは適切な酵素基質を加えることにより測定する。
【0030】
アフィニティ反応行程が完結したら、電気化学的測定用セルを、測定器でつなげた検出電極および参照電極を電解質溶液(本明細書ではワーキング溶液とも呼ぶ)と接触させることにより組み立て、測定装置を用いて一定時間の参照電極に対する検出電極電位を記録する。市販の適切な大きさの参照電極や本発明を実施するために設計された電極も参照電極として用いてよい。測定器は、標準的電位差測定器またはポテンシオスタット(potentiostat)である。本発明を実施するために設計され、カスタムソフトウエアで制御されるPC適合電子測定器も用いることができる。
検出電極と参照電極を接続するための電気的接点を備え、ケーブルまたは他の手段で測定器と接続した特殊なホルダーにより検出電極および参照電極を測定器と好都合に繋ぐことができる。測定システムの全体の大きさを小型化することができる、測定器と一体になったホルダーも使用できよう。
リン酸生理食塩水、トリス−HCl、カーボネート、ビカーボネート、アセテート、ボレート(borate)などの水性緩衝溶液をワーキング溶液として用いる。電気化学的セル中のワーキング溶液の容量は、検出電極の幾何学的大きさに応じて典型的には50〜5000μlである。緩衝溶液の容器は、吸着特性が最小限の物質でできた、あらゆる適切な大きさの容器、例えば標準的マイクロタイタープレートであってよい。本発明の別の態様は、緩衝溶液をペリスタルティック(peristaltic)ポンプや他の手段で送り込むことができる、検出電極および参照電極の一体化ホルダーと連結して使用可能な低容量(<1cm3)のフロースルーセルを用いる変形(バリアント)である。
【0031】
参照電極電位に対する検出電極電位を、電位差測定装置またはポテンシオスタットに接続したチャートレコーダーを用いるか、またはPC適合電子計測器を用いる特別なプログラムにより一定時間記録する。後者の場合は、該プログラムでは、予め決定した時間間隔(典型的には3〜5秒毎に合計10〜100秒間)で参照電極電位に対する検出電極電位を評価し、座標「検出電極信号−時間」上に点の形で結果を表示する。参照電極電位に対する検出電極電位の記録を記録することにより、検出電極のバックグラウンド電位値V1を測定し、検出電極のバックグラウンド電位ドリフト(γ)を評価する(これは、最小二乗法を用いて得られる「検出電極信号−時間」曲線の直線化により計算される)。
第二レセプターまたは競合分子を荷電標識と結合させる場合は、一定pHの電解質溶液のイオン強度を変化させることにより(いわゆる「イオン−ステップ」法)検出電極に結合した分析物の量を評価する。
イオン−ステップ法では、検出電極および参照電極を備えたホルダーを最初のワーキング溶液からイオン強度が異なる同じ組成の第二ワーキング溶液に移すか、または検出電極および参照電極を浸漬したワーキング溶液にイオン強度の異なる(高いかまたは低い)緩衝溶液を直接加えることにより、電解質溶液のイオン強度を変更(上方または下方に)してよい。フロースルーセルを用いる場合は、異なるイオン強度の緩衝溶液を用いて該セルから最初のワーキング溶液を追い出すことにより電解質溶液のイオン強度を変更することができる。
【0032】
種々のイオン強度を有するワーキング溶液は、種々の濃度の塩、例えばKCl、Na2SO4などを加えることにより達成される(それらは、溶液に加えたときに完全に解離し、溶液のpHを偏らせないことから用いられる)。ワーキング溶液の塩濃度は0.01〜0.1Mである。
第二レセプターまたは競合分子を酵素と結合する場合は、「イオン−ステップ」法を行う必要はない。そのかわり、ワーキング溶液の組成はその酵素に適した基質を加えることにより変更される。このために、検出電極および参照電極を備えたホルダーを最初のワーキング溶液を含む容器からワーキング溶液および基質を含む容器に移すか、または基質溶液を検出電極および参照電極を浸漬した最初のワーキング溶液に直接加えることができる。フロースルーセルを用いる場合は、基質を含むワーキング溶液を用いてセルから最初のワーキング溶液を追い出すことによりワーキング溶液の組成を変更することができる。
【0033】
用いてよい基質には、ABTS({2,2’−アジノ−ビス−[3−エチルベンズチアゾリン−6−スルホン酸]})、TMB(3,3,5,5’−テトラエチルベンジジン)、DAB(3,3’ジアミノベンジジン)(ここで、酵素標識はパーオキシダーゼである)、尿素(ここで、酵素標識はウレアーゼである)、p−NPP(p−ニトロフェニルホスフェート)、BCIP(5−ブロモ−4−クロロ−3−インドリルホスフェート)(ここで、酵素標識はアルカリホスファターゼである)が含まれる。
【0034】
ワーキング溶液のイオン強度の段階的変化または酵素基質の添加に応じた参照電極電位に対する検出電極電位の変動を測定器を用いて一定時間記録する。さらに、電位差測定装置またはポテンシオスタットに接続したチャートレコーダーを用いるか、またはPC適合電子計測器を用いる特別なプログラムにより記録を行う。後者の場合には、該プログラムでは、予め決定した時間間隔(典型的には3〜5秒毎)で参照電極電位に対する検出電極電位を評価し、座標「検出電極信号−時間」上に点の形で結果を表示する。個々の試験の種類に応じて、参照電極電位に対する検出電極電位の変動を記録するの要する時間は30秒間から600秒間の間で変動する。
緩衝溶液のイオン強度が変化する場合は、参照電極電位に対する検出電極電位の変動について得られる曲線は、通常、放物線の形を取り、電導性ポリマーフィルムの総荷電(導電点)により調節される、緩衝溶液のイオン強度変化に対する検出電極の反応を示す。
【0035】
分析をサンドイッチアッセイとして行う場合、該ポリマーフィルムの総荷電の変動は、試験する分析物の量に正比例する。しかしながら、分析を競合アッセイとして行う場合、該ポリマーフィルムの総荷電の変動は、試験する分析物の量に反比例する。
該方法のこの段階が完結したら、参照電極電位に対する検出電極電位の最終値V2を測定する。次に、参照電極電位に対する検出電極電位の変化について以下の定量的特性を計算することができる。
1.参照電極電位に対する検出電極電位の変化について得られた曲線により示された面積(積分):
【数1】

2.検出電極のバックグラウンドと最終電位の差(ミリボルト):
δ=V2−V1
ワーキング溶液の組成またはイオン強度の変化に応じた検出電極電位変動の定量的特性に基づいて、被検溶液中の標的分析物の定量的含有量を測定する。
【0036】
上記のごとく得られたγ、S2、および/またはδの値を用い、被検溶液中の標的分析物量の測定に基づいて、ゼロラインのドリフトγを考慮する値を再計算し、S2 γおよび/またはδγ値を得ることができる。訂正値S2 γおよびδγを、「分析結果対標的分析物量」の較正曲線と比較してよい。当業者が容易にわかるであろうように、既知の標的分析物量を含む、一定範囲の被検溶液を用い、上記方法と同様の方法で較正曲線を構築するためのデータを得ることができる。
さらなる局面において、本発明は、試料中の分析物の電気化学的検出法であって、
(a)電導性ポリマーに固定化または吸着したアビジンまたはストレプトアビジンを含む電導性ポリマー層でコートした電導性電極を含む検出電極を得(ここで、該アビジンまたはストレプトアビジン分子は、ビオチン/アビジンまたはビオチン/ストレプトアビジン結合相互作用により、検出すべき分析物を結合することができるレセプター分子と結合する)、
(b)該所望の分析物が該固定化または吸着レセプター分子と結合するように試料を含む被検溶液と検出電極を接触させ、
(c)検出電極と参照電極の両方を電解液に浸漬したときの参照電極に対する検出電極電位をモニターし、
(d)一定pHでの電解液のイオン強度または組成の変化に伴う参照電極に対する検出電極電位差をモニターする工程を含む方法を提供する。
【0037】
本電気化学的検出法は、標的分析物のレセプターに対する結合が、別個の荷電または酵素標識を必要とせずに測定できるような十分大きな検出電極表面の荷電変化を生じる場合に有用である。特に、この方法は核酸の電気化学的検出に有用である。検出電極表面に結合した核酸プローブ(例えばオリゴヌクレオチド)に対する標的核酸のハイブリダイゼーションは、「イオン−ステップ」法により検出できるような十分大きな荷電変化により達成される。すなわち、荷電標識と結合した第二レセプターまたは標的分子を用いる必要はない。特異的核酸を含むと思われる物質(例えば生物学的液体)は、通常、検出法にかける前に増幅工程(例えばPCR)にかけてよい。したがって、増幅法にかけた試料について特異的核酸の電気化学的検出法を行うことも本発明の範囲内である。
さらに本発明は、以下の実施例(限定のためではない)およびそのおもな実施段階を示す添付図を参照して理解されよう。
【0038】
実施例で用いた試薬、物質、および装置は以下の通りであった。
試薬および物質:
ピロール(>98%)はMerckから購入し、使用前に減圧蒸留により2回精製し、次いで+4℃、N2下にて不透明容器中で貯蔵した。
以下の試薬はすべてSigma Chemical Co.(USA)から購入した:
水酸化カリウム(KOH)、ACS試薬)、水酸化ナトリウム(NaOH、ACS試薬)、アジ化ナトリウム(Sigma Ultra)、硫酸セリウム(IV)(ACS試薬)、塩化カリウム(Sigma Ultra)、トリス[ヒドロキシメチル]アミノメタン(Sigma Ultra)、ドデシル硫酸ナトリウム(SDS、>99%)、デキストラン硫酸ナトリウム(mol.wt.〜8000)、イソプロピルアルコール(ACS試薬)、アセトン、塩化水素酸(ACS試薬)、塩素酸(ACS試薬)、N,N−ジメチルホルムアミド(ACS試薬)、グリシン緩衝溶液0.2M)、「トリス緩衝生理食塩水錠」、「リン酸緩衝生理食塩水錠」、o-フェニレンジアミン・2塩酸錠」、ウシ血清アルブミン(RIA級、分画V)、凍結ウシ血清、ストレプトアビジン(Streptomyces abidinii由来)、NHS−d−ビオチン、ジメチルスルホシキド(DMSO、ACS試薬)、
ビオチン−X−X−NHS、ウシアルブミンに対するウサギポリクローナル抗体、マウスIgGに対するヤギポリクローナル抗体とウレアーゼのコンジュゲート、ジコキシン、マウスモノクローナル抗ジコキシンクローンDI−22ビオチンコンジュゲート、パーオキシダーゼ(100単位/mg固体)、ディスポーザブル透析バッグ(MWCO 1,000)、特記しない限り、リン酸緩衝生理食塩水溶液(PBS)はpH7.4を用いた。
【0039】
B型肝炎表面抗原に対するマウスモノクローナル抗体(HBsAg)(5.7mg/ml、0.01%アジ化ナトリウムを含むリン酸緩衝生理食塩水溶液中)、マウスIgGに対するヒツジポリクローナル抗体(10.0mg/ml、0.01%アジ化ナトリウムを含むリン酸緩衝生理食塩水溶液中)、およびHBsAgに対するマウスモノクローナル抗体とパーオキシダーゼのコンジュゲート(2.5mg/ml、0.01%アジ化ナトリウムを含むリン酸緩衝生理食塩水溶液中)は、Sorbent-Service Ltd.(Russia)から購入した。
凍結乾燥インスリン(ウシ、〜20IU/mg)、およびインスリンに対するモノクローナル抗体(1.2mg/ml、リン酸緩衝生理食塩水溶液中)はRussian Academy of Medical Sciences付属のCardiology Research Centreのメンバーより寄付。
ニックトランスレーションによりビオチン化した二本鎖DNAプローブ(長さ〜1kb)の凍結乾燥調製物[20]、ビオチン化DNAプローブと相補的な二本鎖DNAの凍結乾燥調製物、ビオチン化DNAプローブと相補的でない二本鎖DNAの凍結乾燥調製物、サケ精液由来DNAの凍結乾燥調製物、およびDNAハイブリダイゼーション用緩衝溶液は、すべてMinistry of Health付属のScientific Research Institute of Biophysicsのメンバーより寄付。
【0040】
Polybead(登録商標)Sulphate Microspheres (2.5% Solids-Latex、0.2μm)はPolysciences Inc.から購入した。
Lavsan(登録商標)フィルム(〜500μm厚)はVladimir Chemical Plant(Russia)から購入。
真空中に分散したクロミウム標的およびフォトレジスト<<FP−383>>は、NIIPIK Institute(Russia)から購入。
脱イオン水(試薬級、抵抗>18MΩ)は、Millipore Milli-RO/Milli-Q Systemを用いて得た。
プラチナ線、厚さ〜0.5mm。
B型肝炎表面抗原の第二英国ワーキング標準品(HbsAg濃度、0.50iU/ml)、North London Blood Transfusion Centre, UKより寄付。
HBsAg陽性ヒト血清試料、North London Blood Transfusion Centre, UKより寄付。
【0041】
装置:
UVN真空沈殿(deposition)ユニット(Russia)。
<<PNF−6Ts>>フォトレジスト適用ユニット(Russia)。
自家製フォト−テンプレート(WO96/02001の図1、図3または図4に示す電極デザインをもたらすフォト−テンプレートが適している)。
自家製Soxhlet装置。
EU18乾燥加熱キャビネット(Jouan,France)。
<<PI−50.1>>標準積分器(integrator)と2座標(twin-coordinate)レコーダー付きポテンシオスタット−ガルバノスタット(galbanstat)。
2チャンネルレコーダー2210(LKB-Pribori, Sweden)。
標準コンビネーション電極付きチェックメートpHメーター(Mettler, Switzerland)。
自家製Ag/AgClセミミクロ参照電極;直径〜2.5mm、飽和KCl溶液充填。
Ag/AgCl参照電極N°476370(Corning)。
増幅器とアナログ−デジタル変換器を含む、目的に合わせて設計したPC適合測定器;注文(カスタム)ソフトウエアで制御。
目的に合わせて設計した検出電極および参照電極ホルダー。
【0042】
(実施例)
実施例1.
HBsAg測定法:競合アッセイ;試料−希釈標本;受容体(レセプター)−ビオチン化したモノクローナルマウス抗−HBsAg;競合分子−標識化したヒツジポリクローナル抗−マウスIgG;電荷標識−ラテックス。
【0043】
1.1. 電極物質は、60×48mmのラブサン(Lavsan)フィルムから製造し、次いで熱イソプロパノール蒸気中で洗浄し、熱イソプロパノール蒸気中で乾燥した。マグネトロン析出によって、該物質にクロム層(0.05μm)を塗布した。遠心分離によって、そのクロム層にフォトレジストを塗布し、+80℃で20分間乾燥した。パターン化した感光基板を通した紫外線を用いて、フォトレジスト層を露光させた。フォトレジスト層をKOH溶液中で現像し、次いで+100℃で20分間乾燥した。硫酸第2セリウム中でエッチングすることによって、クロムパターンを得た。ジメチルホルムアミドを用いて、フォトレジストを除き、続いてイソプロパノール蒸気中ですすぎ、乾燥した。塩化金(III)溶液からガルバニ析出によって、クロムパターンに金層(0.50μm)を塗布し、続いてイソプロパノール中ですすぎ、乾燥した。
【0044】
1.2. その結果得られた電極を、2〜5%のKOH溶液中で30分間かけて2回洗浄し、次いで脱イオン水ですすぎ、アセトン中で5分間、2回洗浄し、次いで室温で20分間、風乾した。次いで、その電極をフッ素プラスチックホルダーに装着し、ソックスレット容器(Soxhlet vessel)中に置き、熱イソプロピルアルコール中で0.5〜2時間洗浄した。次いで、電極を完備したホルダーをソックスレット容器から取りだし、電極をイソプロピルアルコール蒸気中で乾燥した。これらの操作が完結後、その電極を封したガラス容器中に置いた。
【0045】
1.3. 塩化ナトリウム(8.0g)、トリス(ヒドロキシメチル)アミノメタン(12.2g)、塩化カリウム(0.2g)およびアジ化ナトリウム(0.1g)を脱イオン水(1L)に溶解することによって、検出電極用の保存液を調製した。
【0046】
1.4. HBsAgのマウスモノクローナル抗体を、以下の通りビオチン化した。
【0047】
20トリスで緩衝化した生理食塩水の錠剤およびアジ化ナトリウム(0.15g)を脱イオン水(1.50L)に溶解することによって、アジ化ナトリウムを含有する0.01M トリスで緩衝化した生理食塩水(pH 8.0)を調製した。
【0048】
NHS−d−ビオチン(1.0mg)をDMSO(1mL)に溶解した。
【0049】
購入したHBSAgのマウスモノクローナル抗体(5.7mg/mL、176μL)を、0.01M リン酸塩で緩衝化した生理食塩水(824μL)を含むミクロチューブに加えた。
【0050】
その得られた溶液に、NHS−d−ビオチンのDMSO溶液(50μL)を加えた。
【0051】
その混合物を含むミクロチューブをサーモミキサーに入れ、絶えず混合しながら、+22℃の温度で4時間保った。
【0052】
次いで、その混合物を、500倍の過剰量のアジ化ナトリウムを含有するトリスで緩衝化した生理食塩水(0.01M)を用いて4℃で終夜透析した。
【0053】
得られたビオチン化抗体の溶液を少量のアリコート(〜10μL)毎に分け、+4℃で保存した。
【0054】
1.5. マウスIgGのヒツジポリクローナル抗体を、以下の通りラテックスミクロフェアと接合した。
【0055】
0.2M グリシン緩衝液(500mL)を脱イオン水(500mL)に加え、得られた混合物に塩化ナトリウム(8.5g)およびアジ化ナトリウム(0.1g)を溶解し、0.1M NaOH溶液を用いてpHを8.2に調節することによって、アジ化ナトリウムを含有するグリシンで緩衝化した生理食塩水(pH 8.2)を調製した。
【0056】
購入したマウスIgGのヒツジポリクローナル抗体(10mg/mL、60μL)を、グリシンで緩衝化した生理食塩水(940μL)を含むミクロチューブに加えた(標識化する前に、ポリクローナル抗体をアフィニティー精製することが好ましい)。
【0057】
ポリベッド(登録商標)スルフェートミクロフェアの懸濁液(2.5%、200μL)を得られた溶液に加えた。
【0058】
その混合物を含むミクロチューブをサーモミキサーに入れ、絶えず混合しながら、37±1℃の温度で30分間保った。
【0059】
次いで、その混合物を含むミクロチューブを室温まで冷却し、それにウシ血清アルブミン(0.01g)を加えた。
【0060】
得られた標識化抗体の溶液を少量(〜5μL)のアリコート毎に分け、+4℃で保存した。
【0061】
1.6. 重合する前に、モノマー(例えば、ピロール)を標準水冷装置中で常圧下、135〜140℃で蒸留し、封した不透明な容器中でN2下、−20℃〜−5℃で保存した。電気化学的重合の溶液中でのモノマー濃度は、試験の種類に応じて0.3〜1.0Mの範囲内で変わる。
【0062】
本実施例において、ピロールの電気化学的重合用の溶液を、以下の通り調製した。
【0063】
新たに蒸留したピロール(2.5mL)およびSDS(0.02g)を脱イオン水(20.0mL)に溶解した。
【0064】
リン酸塩で緩衝化した生理食塩水の錠剤を脱イオン水(200mL)に溶解し、ストレプトアビジン(4.0mg)をPBS(2mL)に溶解した。
【0065】
ピロールおよびSDSの溶液に、ストレプトアビジンのPBS溶液(1mL)を加えた。
【0066】
最終的な溶液を回転式ミキサー中に入れ、10分間混合した。
【0067】
1.7. 電析プロセスは、3電極の電気化学的セル(例えば、作用電極、参照電極および補助(対)電極を含む)中で行われる。作用電極は、上記の通り製造した金属の電位差電極であり、補助電極は金または白金のワイヤーの1片であり、参照電極は銀/塩化銀電極である。
【0068】
析出は、作用電極に連続して掃引電圧をかけたポテンシオスタットを用いて行う。高分子フィルムの所望の厚さおよび性質に応じて、電位掃引下限、電位掃引上限、電圧掃引速度および掃引の周期数は変化し、典型的にはそれぞれ、−500mV〜+800mV、+1000mV〜+2000mV(Ag/AgCl参照電極に対する相対値)、25〜200mV/秒および3〜30である。
【0069】
本実施例において、以下の通り、電着によってストレプトアビジンが結合した電極上のポリピロールフィルムを得た。
【0070】
電気化学的重合の溶液(200μL)を、ミクロタイタープレートのウェルに入れた。
【0071】
ポテンシオスタットにつないだ電極、白金ワイヤーおよび半微量参照電極をそのウェルに浸した。
【0072】
参照電極に対する電位の周期的な掃引は、掃引速度を150mv/分で、+800〜+1800mVの範囲とした。
【0073】
ポリピロールフィルムの形成プロセスは、ポテンシオスタットの対応する出力につないだ対座標チャート記録計を用いる電圧−電流曲線について、およびポテンシオスタットの対応する出力につないだ積分器およびチャート記録計を用いる電極を通過する総電気量について追跡した。析出方法全般にわたって、最初に作用電極を通過する電気量と、次の周期のものとが15%より多く違わないことを確認するためにチェックを行った。
【0074】
ポリピロールフィルムが特定の厚さに達すれば(電位掃引周期数−8;電極を通過する総電気量−750mC)、そのプロセスを停止した。
【0075】
1.8. ストレプトアビジンが結合したポリピロールフィルムでコーティングされた検出電極をウェルから取り出し、脱イオン水、続いて0.01M リン酸塩で緩衝化した生理食塩水(pH 7.4)ですすぎ、保存液(300μL)を入れたミクロチューブ中に置き、このものを+4℃で保存した。
【0076】
1.9. 検出電極の必要な電気量を得るために、工程1.7から1.8を繰り返した。
【0077】
1.10. 別々のミクロチューブ中で、B型肝炎表面抗原用第2英国ワーキング標準液(Second British Working Standard for Hepatitis B Surface Antigen)の100、50、25および20μLに、ウシ血清をそれぞれ100、150、175および180μLを加えて、希釈倍数が2、4、8および16で、予め解凍したウシ血清中でB型肝炎表面抗原用第2英国ワーキング標準液を希釈することによって、HBsAg濃度が既知の試料群(200μL)を製造した。純粋なウシ血清は、HBsAg含有量が0である試料を得るために使用した。
【0078】
1.11. ビオチン化したHBsAgのマウスモノクローナル抗体の適当な力価溶液(0.2mL)を、0.01M リン酸塩で緩衝化した生理食塩水(19.8mL)に加え、回転式振とう器中で十分に撹拌し、次いでアリコート(200μL)毎にミクロチューブ中に分配した。
【0079】
1.12. 標識化した(ラテックス接合した)マウスIgGのヒツジポリクローナル抗体の適当な力価溶液(0.1mL)を、0.01M リン酸塩で緩衝化した生理食塩水(19.9mL)に加え、回転式ミキサー中で十分に混合し、次いでアリコート(200μL)毎にミクロチューブ中に分配した。
【0080】
1.13. 作用緩衝液N°1を以下の通り調製した。
【0081】
リン酸塩で緩衝化した生理食塩水の錠剤を、脱イオン水(200mL)に溶解した。
【0082】
ウシ血清アルブミン(2g)およびKCl(0.37g)を、得られた溶液に溶解した。
【0083】
1.14. リン酸塩で緩衝化した生理食塩水の錠剤を脱イオン水(200mL)に溶解することによって、作用緩衝液N°2を調製した。
【0084】
1.15. ストレプトアビジンが結合したポリピロールフィルムでコーティングされた検出電極の適当な数を保存緩衝液から取り出し、各々を、ビオチン化したHBsAgのマウスモノクローナル抗体溶液を含む1ミクロチューブ(工程1.11由来)中に置き、室温で10分間インキュベートした。
【0085】
1.16. 1.15の完結後、ビオチン化した抗体溶液を含むミクロチューブから検出電極を取り出し、各々を、HBsAg濃度が既知の試料を含む1ミクロチューブ(工程1.10由来)中に置き;次いで、検出電極を入れたミクロチューブをサーモミキサー中に置き、絶えず撹拌しながら、37±1℃の温度で15分間保った。
【0086】
1.17. 1.16の完結後、試料を含むミクロチューブから検出電極を取り出し、ラテックスを接合させたマウスIgGのヒツジポリクローナル抗体を含むミクロチューブ(工程1.12由来)中に置き、次いで回転振とう器に置き、絶えず撹拌しながら、室温で5分間保った。
【0087】
1.18. 1.17の完結後、そのミクロチューブから検出電極を取り出し、0.01M リン酸塩で緩衝化した生理食塩水中で3〜5秒間すすぎ、各々を作用緩衝液N°1で十分に満たしたミクロタイタープレート中に置いた。
【0088】
1.19. PCベースの測定装置につないだホルダーの電気接触子に、検出電極および参照電極をつなぎ、その検出電極および参照電極が溶液に浸るように、作用緩衝液N°1で十分に満たしたミクロタイタープレートの上方にそのホルダーを設置した。
【0089】
1.20. 特注のソフトウェアを始動させ、参照電極電位に対する検出電極電位(ミリボルト単位)を記録するために、30秒間使用した。
【0090】
1.21. 1.20の完結後、1,19に記載の方法と同様にして、作用緩衝液N°2で十分に満たしたミクロタイタープレートの上方に、そのホルダーを設置した。
【0091】
1.22. 参照電極電位に対する検出電極電位(ミリボルト単位)の変化を記録するために、特注のソフトウェアを100秒間、使用した。
【0092】
1.23. 特注のソフトウェアを用いて、バックグラウンドと検出電極の最終電位間の電位差δ(ミリボルト単位)を算出した。
【0093】
1.24. 1,19−1.21に記載の操作を、工程1.10で製造したHBsAgの濃度が既知の試料を用いて順番に繰り返した。
【0094】
1.25. 工程1.24で得られた結果に基づいて、曲線「試料中のHBsAg濃度に対するδ」(図10)をプロットするために、外注ソフトウェアを使用し、検出電極システムの絶対感度の閾値下限をこの曲線から決定した。
【0095】
実施例2
HBsAg測定法:競合アッセイ;試料−希釈標本;受容体−ビオチン化したモノクローナルマウス抗−HBsAg;競合分子−標識化したヒツジポリクローナル抗−マウスIgG;電荷標識−ラテックス。
【0096】
2.1. 1.1−1.5に記載の方法を行った。
【0097】
2.2. ピロールの電気化学的重合用の溶液を、以下の通り調製した。
【0098】
新たに蒸留したピロール(2.5mL)およびSDS(0.05g)を、脱イオン水(20.0mL)に溶解した。
【0099】
2.3. 以下の通り、ポリピロールフィルムを電着によって得た。
【0100】
電気化学的重合の溶液(200μL)をミクロタイタープレートのウェルに入れた。
【0101】
ポテンシオスタットにつないだ電極、白金ワイヤーおよび半微量参照電極をウェルに浸した。
【0102】
参照電極に対する電位の周期的な掃引は、掃引速度を100mV/秒で、+800〜+2200mVの範囲をかけた。
【0103】
ポリピロールフィルムの製造プロセスは、ポテンシオスタットの対応する出力につないだX−Yチャート記録計を用いる電圧−電極曲線について、およびポテンシオスタットの対応する出力につないだ積分器およびチャート記録計を用いる電極を通過する総電気量について追跡した。
【0104】
特定の厚さのポリピロールフィルムに達すれば(電位掃引周期数−6;電極を通過する総電気量−750mC)、そのプロセスを停止した。
【0105】
2.4. ポリピロールフィルムでコーティングされた検出電極をウェルから取り出し、脱イオン水、続いて0.01M リン酸塩で緩衝化した生理食塩水(pH 7.4)ですすぎ、保存液(300μL)を入れたミクロチューブ中に置き、これを+4℃で保存した。
【0106】
2.5. 検出電極の必要な電気量を得るために、2.1〜2.4に記載の方法を繰り返した。
【0107】
2.6. リン酸塩で緩衝化した生理食塩水の錠剤を脱イオン水(200mL)に溶解し、得られた溶液にストレプトアビジン(1.0mg)を溶解することによって、ストレプトアビジン溶液を調製した。
【0108】
2.7. ストレプトアビジンは、以下の通り検出電極を覆っているポリピロールフィルムの表面に結合した。
【0109】
ストレプトアビジン溶液は、アリコート(200μL)毎にミクロチューブ中に分配した。
【0110】
ポリピロールフィルムでコーティングされている検出電極を保存緩衝液から取り出し、各々を、ストレプトアビジン溶液を入れたミクロチューブ中に置き、+4℃で18時間インキュベートした。
【0111】
ストレプトアビジン溶液を含むミクロチューブから検出電極を取り出し、0.01M リン酸塩で緩衝化した生理食塩水で洗浄し、各々を保存緩衝液に入れ、次いで+4℃で保存した。
【0112】
2.8. HBsAg濃度が既知の試料群を、1.10に記載の通り調製した。
【0113】
2.9. ビオチン化したHBsAgのマウスモノクローナル抗体の適当な力価溶液(0.1mL)を、0.01M リン酸塩で緩衝化した生理食塩水(19.9mL)に加え、回転振とう機中で十分に混合し、次いでアリコート(200μL)毎にミクロチューブ中に分配した。
【0114】
2.10. 標識化した(ラテックス接合した)マウスIgGのヒツジポリクローナル抗体の適当な力価溶液(0.1ml)を0.01M リン酸塩で緩衝化した生理食塩水(19.9mL)に加え、回転式ミクサー中で十分に混合し、次いでアリコート(200μL)毎にミクロチューブ中に分配した。
【0115】
2.11. 作用緩衝液N°1は、1.13に記載の通り調製した。
【0116】
2.12. 作用緩衝液N°2は、1.14に記載の通り調製した。
【0117】
2.13. ストレプトアビジンが結合したポリピロールフィルムでコーティングされた検出電極を保存緩衝液から取り出し、各々を、ビオチン化したHBsAgのマウスモノクローナル抗体溶液を含むミクロチューブ(工程2.9由来)中に置き、室温で10分間インキュベートした。
【0118】
2.14. 2.13の完結後、ビオチン化抗体の溶液を含むミクロチューブから検出電極を取り出し、各々を、HBsAg濃度が既知の試料を含む1ミクロチューブ(工程2.8由来)中に置き、次いで、その検出電極を入れたミクロチューブをサーモミキサー中に置き、絶えず混合しながら、37±1℃で15分間保った。
【0119】
2.15. 2.14の完結後、試料を含むミクロチューブから検出電極を取り出し、ラテックスを接合させたマウスIgGのヒツジポリクローナル抗体溶液を含むミクロチューブ(工程2.10)中に置き、次いで回転式振とう機中に置き、絶えず混合しながら、室温で5分間保った。
【0120】
2.16. 2.15の完結後、ミクロチューブから検出電極を取り出し、0.01M リン酸塩で緩衝化した生理食塩水中で3〜5秒間すすぎ、各々を作用緩衝液N°1で十分に満たしたミクロタイタープレート中に置いた。
【0121】
2.17. PCベースの測定装置につないだホルダーの電気接触子に、検出電極および参照電極をつなぎ、その検出電極および参照電極が溶液に浸るように、作用緩衝液N°1で十分に満たしたミクロタイタープレートウェルの上方にそのホルダーを設置した。
【0122】
2.18. 特注のソフトウェアを始動させ、参照電極電位に対する検出電極電位(ミリボルト単位)を記録するのに、30秒間使用した。
【0123】
2.19. 2.18の完結後、2.17に記載と同様な方法で、作用緩衝液N°2で十分に満たしたミクロタイターウェルプレートの上方に、そのホルダーを設置した。
【0124】
2.20. 参照電極に対する検出電極電位の変化(ミリボルト単位)を記録するために、特注のソフトウェアを100秒間使用した。
【0125】
2.21. 特注のソフトウェアを用いて、バックグラウンドと検出電極の最終電位間の電位差(δ)(ミリボルト単位)を算出した。
【0126】
2.22. 工程2.8で製造した濃度が既知の試料を用いて、2.17〜2.10に記載の操作を順番に繰り返した。
【0127】
2.23. 工程2.22で得られた結果に基づいて、検量曲線「試料中のHBsAg濃度に対するδ」をプロットするために、外注ソフトウェアを使用した。
【0128】
2.24. 希釈倍数が10、100、1000、5000および10000の試料群を製造するために、HBsAg陽性血清の多数の試料(HBsAg濃度が未知のものを含む)を、予め解凍したウシ血清中で各々逐次希釈した。各希釈液のアリコート(200μL)を別々のミクロチューブに入れた。
【0129】
2.25. 各希釈液中のHBsAg濃度を測定するために、工程2.13〜2.21に記載の方法を用い、これらの結果および工程2.23で得られた検量曲線を、元の(希釈していない)血清試料中でのHBsAg濃度を算出するのに使用した(図11)。
【0130】
実施例3.
抗−アルブミン抗体測定法:競合アッセイ;試料−アルブミンに対するウサギポリクローナル抗体;受容体−ビオチン化したウシ血清アルブミン;競合分子−標識化したアルブミンのウサギポリクローナル抗体;電荷標識−ラテックス。
【0131】
3.1. 1.1〜1.3に記載の方法を行った。
【0132】
3.2. 1.4に記載の通り、ウシ血清アルブミン(BSA)をビオチン化した。得られたビオチン化BSA溶液を、少量のアリコート(〜10μL)毎に分け、+4℃で保存した。
【0133】
3.3. 1.5に記載の方法に従って、ウシアルブミンのウサギポリクローナル抗体をラテックスミクロフェアと接合した。得られた標識化抗体の溶液を少量のアリコート(〜5μL)毎に分け、+4℃で保存した。
【0134】
3.4. 2.2.に記載の通り、ピロールの電気化学的重合用の溶液を調製した。
【0135】
3.5. 2.3.に記載の通り、電着によってポリピロールフィルムを得た。
【0136】
3.6. ポロピロールフィルム中でコーティングされた検出電極をウェルから取り出し、脱イオン水、続いて0.01M リン酸塩で緩衝化した生理食塩水(pH 7.4)ですすぎ、保存液(300μL)を入れたミクロチューブ中に置き、これを+4℃で保存した。
【0137】
3.7. 検出電極の必要な電気量を得るために、3.1〜3.6に記載の方法を繰り返した。
【0138】
3.8. 2.6に記載の通り、ストレプトアビジン溶液を調製した。
【0139】
3.9. 2.7に記載の通り、ストレプトアビジンは、検出電極を覆っているポリピロールフィルムの表面に結合した。
【0140】
3.10. 濃度が既知で非標識のウシアルブミンのウサギポリクローナル抗体の試料群を、以下の通り調製した。
【0141】
購入したウシアルブミンのウサギポリクローナル抗体の試料を、500倍過剰量の0.01M リン酸塩で緩衝化した生理食塩水を用いて+4℃で終夜透析した。
【0142】
得られた0.01M リン酸塩で緩衝化した生理食塩水中のウシアルブミンのウサギポリクローナル抗体の溶液を、10,20、50、100、1000および5000の希釈倍数で0.01M リン酸塩で緩衝化した生理食塩水を用いて逐次希釈した。
【0143】
各々の希釈試料のアリコート(200μL)を、別々のミクルチューブに入れた。
【0144】
3.11. 適当に希釈したビオチン化BSAの溶液(0.8mL)を、0.01M リン酸塩で緩衝化した生理食塩水(19.2mL)に加え、回転振とう機中で十分に混合し、次いでアリコート(200μL)毎にミクロチューブ中に分配した。
【0145】
3.12. 標識化した(上記の通り、ラテックスで接合した)ウシアルブミンのウサギポリクローナル抗体の適当な力価溶液(0.1mL)を、0.01M リン酸塩で緩衝化した生理食塩水(19.9mL)に加え、回転式ミキサー中で十分に混合し、次いでアリコート(200μL)毎にミクロチューブ中に分配した。
【0146】
3.13. 作用緩衝液N°1は、以下の通り調製した。
【0147】
リン酸塩で緩衝化した生理食塩水の錠剤を、脱イオン水(200mL)に溶解した。
【0148】
得られた溶液に、KCl(0.37g)を溶解した。
【0149】
3.14. 1.14に記載の通り、作用緩衝液N°2を調製した。
【0150】
3.15. ストレプトアビジンが結合したポリピロールフィルムでコーティングされた検出電極を保存緩衝液から取り出し、各々を、ビオチン化BSAの溶液を含む1ミクロチューブ(工程3.11由来)中に置き、室温で25分間インキュベートした。
【0151】
3.16. 3.15の完結後、ビオチン化BSAの溶液を含むミクロチューブから検出電極を取り出し、各々を、濃度が既知で非標識のウシアルブミンのウサギポリクローナル抗体試料を含む1ミクロチューブ(工程3.10由来)中に置き、次いで検出電極を入れたミクロチューブをサーモミキサー中に置き、絶えず混合しながら、37±1℃の温度で25分間保った。
【0152】
3.17. 3.16の完結後、試料を含むミクロチューブから検出電極を取り出し、このものをラテックスを接合したウシアルブミンのウサギポリクローナル抗体溶液を含むミクロチューブ(工程3.12由来)に移し、次いで回転振とう機中に置き、絶えず混合しながら室温で10分間保った。
【0153】
3.18. 3.17の完結後、ミクロチューブから検出電極を取り出し、0.01M リン酸塩で緩衝化した生理食塩水中で3〜5秒間すすぎ、各々を、作用緩衝液N°1で十分に満たしたミクロタイタープレート中に置いた。
【0154】
3.19. PCベースの測定装置につないだホルダーの電気接触子に検出電極および参照電極をつなぎ、その検出電極および参照電極が溶液に浸るように、参照緩衝液N°1で十分に満たしたミクロタイタープレートの上方にそのホルダーを設置した。
【0155】
3.20. 特注のソフトウェアを始動させ、参照電位に対する検出電極電位(ミリボルト単位)を記録するために、100秒間使用した。
【0156】
3.21. 3.20の完結後、3.19に記載の方法と同様な方法で、作用緩衝液N°2で十分に満たしたミクロタイタープレートの上方に、ホルダーを設置した。
【0157】
3.22. 参照電極電位に対する検出電極電位(ミリボルト単位)の変化を記録するために、特注のソフトウェアを200秒間使用した。
【0158】
3.23. 特注のソフトウェアを用いて、作用電極電位に対する検出電極電位の変化についての曲線が示す領域(積分)S2を算出した。
【0159】
3.24. 工程3.10で製造した濃度が既知で非標識のウシアルブミンのウサギポリクローナル抗体の試料を用いて、3.19〜3.23に記載の操作を順番に繰り返した。
【0160】
3.25. 工程3.24で得られた結果に基づいて、検量線「試料中の標識化したウシアルブミンのウサギポリクローナル抗体の濃度に対するS2」をプロットするために、特注のソフトウェアを使用した。
【0161】
実施例4.
HBsAg測定法;サンドイッチアッセイ;試料−HBsAg濃度が既知である試料;受容体−ビオチン化したHBsAgのマウスモノクローナル抗体;標識化したHBsAgのマウスモノクローナル抗体;標識−ペルオキシダーゼ。
【0162】
4.1. 1.1〜1.4に記載の方法を行った。
【0163】
4.2. 2.2.に記載の通り、ピロールの電気化学的重合用の溶液を調製した。
【0164】
4.3. 2.3に記載の通り、電着によってポリピロールフィルムを得た。
【0165】
4.4. ポリピロールフィルムでコーティングされた検出電極をウェルから取り出し、脱イオン水、続いて0.01M リン酸塩で緩衝化した生理食塩水(pH 7.4)ですすぎ、保存液(300μL)を入れたミクロチューブ中に置き、このものを+4℃で保存した。
【0166】
4.5. 検出電極の必要な電気量を得るために、4.1〜4.4に記載の方法を繰り返した。
【0167】
4.6. ストレプトアビジン溶液を、2.6に記載の通り調製した。
【0168】
4.7. 2.7に記載の通り、ストレプトアビジンは、検出電極を覆っているポリピロールフィルムの表面に結合した。
【0169】
4.8. HBsAg濃度が既知である試料群を、1.10に記載の通り製造した。
【0170】
4.9. ビオチン化したHBsAgのマウスモノクローナル抗体の適当な力価溶液(0.2mL)を、0.01M リン酸塩で緩衝化した生理食塩水(19.8mL)に加え、回転振とう機中で十分に混合し、次いでアリコート(200μL)毎に、ミクロチューブ中に分配した。
【0171】
4.10. 予め解凍したウシ血清(5mL)を0.01M リン酸塩で緩衝化した生理食塩水(15mL)に加え、得られた溶液(19.6mL)に、購入したペルオキシダーゼを接合したHBsAgのマウスモノクローナル抗体の適当な力価溶液(0.4mL)を加え、次いで回転振とう機中で十分に混合し、アリコート(200μL)毎にミクロチューブ中に分配した。
【0172】
4.11. リン酸塩で緩衝化した生理食塩水の錠剤を脱イオン水(200mL)に溶解することによって、作用緩衝液N°1を調製した。
【0173】
4.12. 作用緩衝液N°2を、以下の通り調製した。
【0174】
o−フェニレンジアミン・二塩酸塩の錠剤および尿素過酸化水素化物/緩衝液の錠剤を、脱イオン水(20mL)に溶解した。
【0175】
得られた溶液(0.1mL)を、0.01M リン酸塩で緩衝化した生理食塩水(19.9mL)に加え、回転振とう機中で十分に混合し、次いで回転式ガラス容器中に置き、試験開始まで+4℃で保存した。
【0176】
4.13. ストレプトアビジンが結合したポリピロールフィルムでコーティングされた検出電極を保存緩衝液から取り出し、各々を、ビオチン化したHBsAgのマウスモノクローナル抗体溶液(200μL)を含むミクロチューブ(工程4.9由来)中に置き、室温で5分間インキュベートした。
【0177】
4.14. 4.13の完結後、ビオチン化抗体の溶液を含むミクロチューブから検出電極を取り出し、HBsAg濃度が既知の試料を含むミクロチューブ(工程4.8由来)中に置き、次いでその検出電極を入れたミクロチューブをサーモミキサー中に置き、絶えず混合しながら、37±1℃の温度で10分間保った。
【0178】
4.15. 4.14の完結後、試料を含むミクロチューブから検出電極を取り出し、ペルオキシダーゼを接合させたHBsAgのマウスモノクローナル抗体溶液を含むミクロチューブ(工程4.10由来)中に置き、次いでこれを回転振とう機中に入れ、絶えず混合しながら、室温で5分間保った。
【0179】
4.16. 4.15の完結後、ミクロチューブから検出電極を取り出し、0.01M リン酸塩で緩衝化した生理食塩水中で3〜5秒間すすぎ、各々を、作用緩衝液N°1で十分に満たしたミクロタイタープレート中に置いた。
【0180】
4.17. PCベースの測定装置につないだホルダーの電気接触子に検出電極および参照電極をつなぎ、その検出電極および参照電極が溶液に浸るように、作用緩衝液N°1で十分に満たしたミクロタイタープレートの上方に、ホルダーを設置した。
【0181】
4.18. 特注のソフトウェアを始動させ、参照電極電位に対する検出電極電位(単位ミクロボルト)を記録するために、50秒間使用した。
【0182】
4.19. 4.18の完結後、4.17に記載の方法と同様な方法で、作用緩衝液N°2で十分に満たしたミクロタイタープレートの上方に、ホルダーを設置した。
【0183】
4.20. 参照電極電位に対する検出電極電位(ミリボルト単位)の変化を記録するために、特注のソフトウェアを100秒間使用した。
【0184】
4.21. 特注のソフトウェアを用いて、バックグラウンドと検出電極の最終電位間の電位差(δ)(ミリボルト単位)を算出した。
【0185】
4.22. 工程4.8で製造したHBsAg濃度が既知の試料を用いて、4.17〜4.21に記載の操作を順番に繰り返した。
【0186】
4.23. 工程4.22で得られた結果に基づいて、検量曲線「試料中のHBsAg濃度に対するδ」をプロットするために、特注のソフトウェアを使用した。
【0187】
4.24. 2.24に記載の通り、希釈した血清試料群を製造した。
【0188】
4.25. 4.24で製造した各々の希釈試料中でのHBsAg濃度を測定するために、工程4.13〜4.21に記載の方法を使用し、これらの結果および工程4.23で得られた検量曲線を、元の(希釈していない)血清試料中でのHbsAg濃度を算出するために使用した。
【0189】
実施例5
HBsAg測定;サンドイッチ試験;試料−既知のHBsAg濃度を有する試料;受容体−HBsAgに対するビオチン化マウスモノクローナル抗体;HBsAgに対する標識化したマウスモノクローナル抗体;標識−パーオキシダーゼ;”逐次試験”
【0190】
5.1. 1.1−1.4に記載の方法を行った。
5.2. 2.2に記載のように、ピロールの電気化学的重合のための溶液を調製した。
5.3. 2.3に記載のように、ポリピロールのフイルムを電気化学的電析により形成した。
5.4. ポリピロールのフイルムを被覆した検出電極をウエルから除去し、脱イオン水、次に0.01Mリン酸緩衝生理食塩水溶液(pH7.4)で濯ぎ、300μlの貯蔵溶液を有するマイクロチューブに入れ、+4℃で貯蔵した。
5.5. 必要量の検出電極を得るため、5.1−5.4に記載の方法を繰り返した。
5.6. ストレプトアビジンの溶液を2.6に記載のように調製した。
5.7. ストレプトアビジンを2.6に記載のように検出電極を被覆するポリピロールのフイルムの表面に結合させた。
【0191】
5.8. 既知のHBsAg濃度を有する一連の試料を1.10に記載のように調製した。
5.9. HBsAgに対するビオチン化マウスモノクローナル抗体の溶液0.5mlを0.01Mリン酸塩で緩衝化した食塩溶液19.5mlに添加し、ロータリーシェイカーで十分攪拌し、200μlのアリコートでマイクロチューブに分配した。
【0192】
5.10. パーオキシダーゼで標識したHBsAgに対するマウスモノクローナル抗体の溶液1.7mlを、0.01Mリン酸塩で緩衝化した食塩溶液18.3mlに添加し、オービタルミキサーで十分攪拌し、200μlのアリコートでマイクロチューブに分配した。
5.11. ワーキング緩衝液NO1を4.11に記載のように調製した。
5.12. ワーキング緩衝液NO2を4.12に記載のように調製した。
5.13. 結合したストレプトアビジンを有するポリピロールのフイルムを被覆した検出電極を貯蔵緩衝剤溶液から除去し、それぞれをHBsAgに対するビオチン化マウスモノクローナル抗体(工程5.9からの)の溶液を有するマイクロチューブに入れ、5分間室温でインキュベートした。
5.14. 工程5.13と同時に、購入した、HBsAgに対するパーオキシダーゼで標識したマウスモノクローナル抗体の適切にタイターした(titered)溶液を既知のHBsAg濃度を有する各試料(工程5.8からの)に添加し、試料から最初に10μlを除去し;次にマイクロチューブ及び試料を37±1℃で5−10分間サーモミキサー中に置いた。
【0193】
5.15. 5.13−5.14を完了したら、検出電極をビオチン化抗体の溶液を含むマイクロチューブから除き、既知のHBsAg濃度を有する試料(工程5.14からの)を含むマイクロチューブ中に置き、次に37±1℃で15分間サーモミキサー中に保ち、連続的に攪拌した。
5.16. 5.16を完了したら、検出電極をマイクロチューブから取り除き、0.01Mリン酸塩で緩衝化した食塩溶液で3−5秒間濯ぎ、それぞれをワーキング緩衝液NO1を満たしたマイクロタイタープレートウエルに置いた。
5.17. 検出電極及び参照電極をPCをベースにする測定装置に結合したホルダーの電気接触に結合し、ホルダーをワーキング緩衝液NO1を満たしたマイクロタイタープレートウエル上に置き、検出電極及び参照電極が溶液中に浸るようにした。
【0194】
5.18. カスタムソフトウエアを開始させ、50秒にわたって参照電極電位に対する検出電極電位をミリボルトで記録するのに用いた。
5.19. 5.18を完了したら、5.17に記載したのと同様の方法で、ホルダーをワーキング緩衝液NO2を満たしたマイクロタイタープレートウエル上に置いた。
5.20. カスタムソフトウエアを用い、200秒にわたって参照電極電位に対する検出電極電位のミリボルトの変化を記録した。
【0195】
5.21. カスタムソフトウエアを用い、検出電極電位変化 対 参照電極電位の曲線により示される面積(積分)S2を計算した。
5.22. 5.17−5.21に記載の操作を、工程5.8で調製した既知のHBsAg濃度を有する試料を用いて順に繰り返した。
5.23. 工程5.22で得られた結果に基づいて、カスタムソフトウエアを用いて、キャリブレーション曲線「S2 対 試料中のHBsAg濃度」をプロットした。
5.24. 一連の希釈した血清試料を2.24に記載のように調製した。
5.25. 工程5.13−5.21に記載の方法を用いて、5.24で調製した希釈した各試料中のHBsAg濃度を測定し、これらの結果を工程5.23で得られたキャリブレーション(較正)曲線と共に用いて血清のもとの(希釈しない)試料中のHBsAg濃度を計算した。
【0196】
実施例6
HBsAg測定;サンドイッチ試験;試料−既知のHBsAg濃度を有する試料;受容体−HBsAgに対するビオチン化マウスモノクローナル抗体;HBsAgに対する標識化マウスモノクローナル抗体;標識−パーオキシダーゼ;”ワンポット試験”
【0197】
6.1. 1.1−1.4に記載の方法を行った。
6.2. 2.2に記載のように、ピロールの電気化学的重合のための溶液を調製した。
6.3. 2.3に記載のように、ポリピロールのフイルムを電気化学的電析により形成した。
6.4. ポリピロールのフイルムでコートした検出電極をウエルから除去し、脱イオン水、次に0.01Mリン酸緩衝生理食塩水溶液(pH7.4)で濯ぎ、300μlの貯蔵溶液を有するマイクロチューブに入れ、+4℃で貯蔵した。
6.5. 必要量の検出電極を得るため、6.1−6.4に記載の方法を繰り返した。
【0198】
6.6. ストレプトアビジンの溶液を2.6に記載のように調製した。
6.7. ストレプトアビジンを2.7に記載のように検出電極を被覆するポリピロールのフイルムの表面に結合させた。
6.8. 既知のHBsAg濃度を有する一連の試料を1.10に記載のように調製した。
6.9. HBsAgに対するビオチン化マウスモノクローナル抗体の適切にタイターした溶液2.5mlを、0.01Mリン酸塩で緩衝化した食塩溶液17.5mlに添加し、ロータリーシェイカーで十分攪拌し、次に200μlのアリコートでマイクロチューブに分配した。
6.10. HBsAgに対するパーオキシダーゼで標識したマウスモノクローナル抗体の溶液1.7mlを0.01Mリン酸塩で緩衝化した食塩溶液18.3mlに添加し、ロータリーシェカーで十分攪拌し、200μlのアリコートでマイクロチューブに分配した。
6.11. ワーキング緩衝剤NO1を4.11に記載のように調製した。
6.12. ワーキング緩衝剤NO2を4.12に記載のように調製した。
【0199】
6.13. HBsAgに対するビオチン化マウスモノクローナル抗体の溶液10μl(工程6.9からの)及びHBsAgに対するパーオキシダーゼで標識したマウスモノクローナル抗体10μlを既知のHBsAg濃度の各試料に添加し、先ずその試料から20μlを徐去し、;次にマイクロチューブ及び試料をサーモミキサーに置き、37±1℃で10分間インキュベートし、連続的に混合した。
6.14. 6.13が完了したら、結合したストレプトアビジンを有するポリピロールのフイルムを被覆した検出電極を貯蔵緩衝剤溶液から除去し、既知のHBsAg濃度を有する試料(工程6.13からの)を含むマイクロチューブに入れ、次に37 1℃で5分間サーモミキサー中に保ち、連続的に攪拌した。
6.15. 6.14が完了したら、検出電極をマイクロチューブから除き、0.01Mリン酸塩で緩衝化した食塩溶液で3−5秒間濯ぎ、それぞれをワーキング緩衝液NO1を満たしたマイクロタイタープレートウエルに置いた。
【0200】
6.16. 検出電極及び参照電極をPCをベースにする測定装置に結合したホルダーの電気接触に連結し、ホルダーをワーキング緩衝液NO1を満たしたマイクロタイタープレートウエル上に置き、検出電極及び参照電極が溶液中に浸るようにした。
6.17. カスタムソフトウエアを開始させ、100秒にわたって参照電極電位に対する検出電極電位をミリボルトで記録するのに用いた。
6.18. 6.17を完了したら、6.16に記載したのと類似の方法で、ホルダーをワーキング緩衝剤NO2を満たしたマイクロタイタープレートウエル上に置いた。
6.19. カスタムソフトウエアを用い、200秒にわたって参照電極電位に対する検出電極電位のミリボルトの変化を記録した。
【0201】
6.20. カスタムソフトウエアを用い、検出電極電位変化 対参照電極電位の曲線により示される面積(積分)S2を計算した。
6.21. 6.16−6.20に記載の操作を、工程6.8で調製した既知のHBsAg濃度を有する試料を用いて順に繰り返した。
6.22. 6.21で得られた結果に基づいて、カスタムソフトウエアを用いて、キャリブレーション曲線「S2−試料中のHBsAg濃度」をプロットした。
6.23. 一連の希釈した血清試料を2.24に記載のように調製した。
6.24. 工程6.13−6.21に記載の方法を用いて、6.23で調製した希釈した各試料中のHBsAg濃度を測定し、これらの結果を工程6.22で得られたキャリブレーション曲線と共に用いて血清のもとの(希釈しない)試料中のHBsAg濃度を計算した。
【0202】
実施例7
インスリン測定;競合試験;試料−既知のインスリン濃度を有する試料;受容体−インスリンに対するビオチン化マウスモノクローナル抗体;競合分子−標識ポリクローナルヤギ抗マウスIgG抗体;標識−ウレアーゼ”
【0203】
7.1. 1.1−1.3に記載の方法を行った。
7.2. インスリンに対するマウスモノクローナル抗体のビオチン化を1.4に記載のように行なった。インスリンに対するビオチン化マウスモノクローナル抗体の得られた溶液を少容量のアリコート(約10μl)に分け、+4℃で貯蔵した。
7.3. 2.2に記載のようにして、ピロールの電気化学的重合のための溶液を調製した。
7.4. 2.3に記載のようにして、ポリピロールのフイルムを電気化学的電析により形成した。
【0204】
7.5. ポリピロールのフイルムでコートした検出電極をウエルから除去し、脱イオン水、次に0.01Mリン酸緩衝生理食塩水溶液(pH7.4)で濯ぎ、300μlの貯蔵溶液を有するマイクロチューブに入れ、+4℃で貯蔵した。
7.6. 必要量の検出電極を得るため、7.4−7.5に記載の方法を繰り返した。
7.7. ストレプトアビジンの溶液を2.6に記載のように調製した。
7.8. ストレプトアビジンを2.7に記載のように検出電極を被覆するポリピロールのフイルムの表面に結合した。
【0205】
7.9. 既知のインスリン濃度を有する一連の試料を次のように調製した:
1.12gの塩化カリウム及び1.0gのウシ血清アルブミンを100mlの脱イオン水に溶解した;
100μgの凍結乾燥したインスリンを得られた溶液200μlに溶解した;
得られたインスリン溶液を、希釈倍率10、20、50、100、1000、及び10,000倍で、塩化カリウム及びウシ血清アルブミンを含む脱イオン水で希釈した;
希釈した各試料の200μlアリコートを別のマイクロチューブに入れた。
7.10. インスリンに対するビオチン化マウスモノクローナル抗体の適切にタイターした溶液2.5mlを0.01Mリン酸塩で緩衝化した食塩溶液19.2mlに添加し、ロータリーシェイカーで十分攪拌し、次にマイクロチューブに200μlアリコートした。
7.11. マウスIgGに対する購入したウレアーゼをコンジュゲートしたヤギポリクローナル抗体0.02mlを、0.01Mリン酸塩で緩衝化した食塩溶液19.98mlに添加し、ロータリーシェイカーで十分攪拌し、次にマイクロチューブに200μlアリコートした。
【0206】
7.12. ワーキング緩衝液NO1を、2.44gの塩化カリウムを200mlの脱イオン水に溶解することによって調製した。
7.13. ワーキング緩衝液NO2を、20mlのワーキング緩衝液NO1中に0.012gの尿素を溶解することによって調製した。
7.14. 結合したストレプトアビジンを有するポリピロールのフイルムでコートした検出電極を貯蔵溶液から除去し、インスリンに対するビオチン化マウスモノクローナル抗体の溶液(工程7.10からの)を有するマイクロチューブに入れ、10分間インキュベートした。
【0207】
7.15. 7.14が完了したら、検出電極をビオチン化抗体の溶液を有するマイクロチューブから除去し、既知のインスリン濃度の試料(工程7.9からの)を含むマイクロチューブ中に置き、37±1℃で15分間保持し、連続的に混合した。
7.16. 7.15が完了したら、検出電極を試料を含むマイクロチューブから除去し、マウスIgGに対するウレアーゼをコンジュゲートしたヤギポリクローナル抗体の溶液(工程7.11からの)を含むマイクロチューブ中に置き、次にロータリーシェーカーに置き、室温で10分間保持し、連続的に攪拌した。
7.17. 7.16が完了したら、検出電極をマイクロチューブから除去し、塩化カリウム及びウシ血清アルブミンを有する脱イオン水で3−5秒濯ぎ、それぞれをワーキング緩衝液NO1を満たしたマイクロタイタープレートウエルエルに置いた。
7.18. 検出電極及び参照電極をPCをベースにする測定装置に結合したホルダーの電気接触に連結し、ホルダーをワーキング緩衝液NO1を満たしたマイクロタイタープレートウエル上に置き、検出電極及び参照電極が溶液中に浸るようにした。
【0208】
7.19. カスタムソフトウエアを開始させ、200秒にわたって参照電極電位に対する検出電極電位をミリボルトで記録するのに用いた。
7.20. 7.19を完了したら、7.18に記載したのと類似の方法で、ホルダーをワーキング緩衝液NO2を満たしたマイクロタイタープレートウエル上に置いた。
7.21. カスタムソフトウエアを用い、400秒にわたって参照電極電位 対 検出電極電位のミリボルトの変化を記録した。
7.22. カスタムソフトウエアを用い、検出電極電位変化に対する参照電極電位の曲線により示される面積(積分)S2を計算した。
7.23. 7.18−7.22に記載の操作を、工程7.9で調製した既知のインスリン濃度を有する試料を用いて順に繰り返した。
7.24. 7.23で得られた結果に基づいて、カスタムソフトウエアを用いて、キャリブレーション曲線「S2 対 試料中のインスリン濃度」をプロットした。
【0209】
実施例8
核酸ハイブリダイゼーション
8.1. 1.1−1.3に記載の方法を行なった。
8.2. 2.2に記載のようにして、ピロールの電気化学的重合用の溶液を調製した。
8.3. 2.3に記載のようにして、ポリピロールのフイルムを電気化学的電析により形成した。
8.4. ポリピロールのフイルムでコートした検出電極をウエルから除去し、脱イオン水、次に0.01Mリン酸緩衝生理食塩水溶液(pH7.4)で濯ぎ、300μlの貯蔵溶液を有するマイクロチューブに入れ、+4℃で貯蔵した。
【0210】
8.5. 必要量の検出電極を得るため、8.3−8.4に記載の方法を繰り返した。
8.6. 200mlの脱イオン水にリン酸緩衝生理食塩水錠を溶解し、得られた溶液にストレプトアビジンを溶解することによってストレプトアビジンの溶液を調製した。
8.7. ストレプトアビジンを以下のようにして検出電極を被覆するポリピロールのフイルムの表面に結合させた:
ストレプトアビジン溶液をマイクロチューブに200μlのアリコートで分配した;
ポリピロールのフイルムで被覆した検出電極を貯蔵溶液から除去し、それぞれをストレプトアビジン溶液を含むマイクロチューブ中に置き、24時間+4℃でインキュベートした;
検出電極をストレプトアビジン溶液を含むマイクロチューブから除去し、0.01Mリン酸緩衝生理食塩水溶液、及び、0.01%アジ化ナトリウム、及び0.15M塩化カリウムを含む脱イオン水で2度濯ぎ、各検出電極を貯蔵溶液中に置き、+4℃で貯蔵した。
【0211】
8.8. ビオチン化1本鎖DNAプローブの溶液を以下のように調製した:
1mgのビオチン化1本鎖DNAプローブ(長さ約1kb)の凍結乾燥物を1mlの脱イオン水に溶解し、得られた溶液をマイクロチューブに入れた;
DNAプローブ溶液を含むマイクロチューブを水浴に置き、5−8分+100℃でインキュベートした;
DNAプローブ溶液を含むマイクロチューブを氷を有する容器中に移し、急速に0℃まで冷却した;
DNAプローブ溶液を含むマイクロチューブを次にフリーザーに移し、−20℃で貯蔵した。
8.9. ビオチン化DNAプローブに相補的な1本鎖DNAの溶液を以下のように調製した:
ビオチン化DNAプローブに相補的な2本鎖DNAの凍結乾燥物10mgを脱イオン水1mlに溶解し、得られた溶液をマイクロチューブに入れた;
DNA溶液を有するマイクロチューブを水浴中に置き、+100℃で5−8分インキュベートした;
DNA溶液を有するマイクロチューブを氷を含む容器に移し、0℃まで急速に冷却した;
DNA溶液を有するマイクロチューブを次にフリーザーに移し、−20℃で凍結保存した。
【0212】
8.10. ビオチン化DNAプローブに非相補的な1本鎖DNAの溶液を8.9に記載のように調製した。
8.11. ワーキング緩衝液NO1を、以下のように調製した:
リン酸緩衝生理食塩水錠剤を200mlの脱イオン水に溶解した;
2gのウシ血清アルブミン、0.37gの塩化カリウム、及び0.12gのクエン酸ナトリウムを得られた溶液に溶解した。
8.12. ワーキング緩衝液NO2を、以下のように調製した:
リン酸緩衝生理食塩水錠剤を200mlの脱イオン水に溶解した;
2gのウシ血清アルブミン、1gのデキストラン硫酸ナトリウムを得られた溶液に溶解した。
8.13. 結合したストレプトアビジンを有するポリピロールのフイルムで被覆した検出電極を、0.01%アジ化ナトリウム及び0.15M塩化カリウムを含む脱イオン水から除去し、前もって解凍し、室温まで暖めた1本鎖DNAプローブの溶液20μlを各検出電極の表面をワーキングに適用した。
8.14. 8.13が完了したら、検出電極をヒュミディティチャンバーに入れ、+44℃で60分インキュベートした。
【0213】
8.15. 8.14が完了したら、検出電極をヒュミディティチャンバーから出し、それぞれをDNAハイブリダイゼーション用の最初の緩衝剤溶液200μlを含むマイクロチューブに入れ、+4℃で短時間保持した。(DNAハイブリダイゼーション緩衝液は当業者に公知のいずれの標準的なハイブリダイゼーション緩衝液でもよい、Sambrook, J., Fritsch, E. F. and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edition, Cold Spring Harbor laboratory Press, Cold Spring Harbor, NY, USA 参照)
【0214】
8.16. ビオチン化DNAプローブに相補的な1本鎖DNAを含む試料を以下のように調製した:
サケの精子からの凍結乾燥したDNA10mgをDNAハイブリダイゼーション用の最初の緩衝剤溶液10mlに溶解した;
前もって解凍し、室温まで暖めたビオチン化DNAプローブに相補的な1本鎖DNAの溶液10μlを得られた溶液0.99mlに添加した;
得られた溶液をロータリーシェーカーで十分攪拌し、200μlのアリコートでマイクロチューブに分配した。
8.17. ビオチン化DNAプローブに非相補的な1本鎖DNAを含む試料を以下のように調製した:
サケの精子からの凍結乾燥したDNA10mgをDNAハイブリダイゼーション用の最初の緩衝剤溶液10mlに溶解した;
前もって解凍し、室温まで暖めたビオチン化DNAプローブに非相補的な1本鎖DNAの溶液100μlを得られた溶液0.9mlに添加した;
得られた溶液をロータリーシェーカーで十分攪拌し、200μlのアリコートでマイクロチューブに分配した。
8.18. 固定化ビオチン化1本鎖DNAプローブを有する検出電極の半分をDNAハイブリダイゼーション用の最初の緩衝剤溶液を含むマイクロチューブから除去し、ビオチン化DNAプローブに相補的なDNAを含む試料を有するマイクロチューブ中に置き;検出電極を含むマイクロチューブを次にサーモミキサーに置き、連続的に攪拌しながら+42℃で120分保った。
【0215】
8.19. 8.18が終わったら、検出電極をマイクロチューブから除去し、ワーキング緩衝液NO1で3−5秒濯ぎ、それぞれをワーキング緩衝液NO1を満たしたマイクロタイタープレートウエルエルのウエルに置いた。
8.20. 検出電極及び参照電極をPCをベースにする測定装置に連結したホルダーの電気接触に連結し、ホルダーをワーキング緩衝液NO1を満たしたマイクロタイタープレートウエル上に置き、検出電極及び参照電極が溶液中に浸るようにした。
8.21. カスタムソフトウエアを開始させ、200秒にわたって参照電極電位に対する検出電極電位をミリボルトで記録するのに用いた。
8.22. 8.21が完了したら、8.20に記載されたのと類似の方法で、ホルダーをワーキング緩衝剤NO2を満たしたマイクロタイタープレートウエル上に置いた。
【0216】
8.23. カスタムソフトウエアを用い、600秒にわたって参照電極電位に対する検出電極電位のミリボルトの変化を記録した。
8.24. カスタムソフトウエアを用い、バックグランド及び最終電位間のミリボルト差(δ)を計算した。
8.25. 8.20−8.24に記載の操作を、工程4.8で調製したビオチン化DNAプローブに相補的なDNAを含む試料を用いて順に繰り返した。
8.26. 固定化ビオチン化1本鎖DNAプローブを有する検出電極の他の半分をDNAハイブリダイゼーション用の最初の緩衝剤溶液を含むマイクロチューブから除去し、DNAプローブに非相補的なDNAを含む試料を有するマイクロチューブ中に置き;検出電極を含むマイクロチューブを次にサーモミキサーに置き、連続的に攪拌しながら+42℃で120分保った。
8.27. 8.19−8.25に記載の方法を、ビオチン化DNAプローブに非相補的なDNAを含む試料を用いて繰り返した。
8.28. 8.19−8.26で得られた結果に基づいて、カスタムソフトウエアを用い、ビオチン化DNAプローブに相補的な、及び非相補的なDNAの試料について得られたδ値の統計的な分布曲線をプロットした。
【0217】
実施例9
ジゴキシン測定;競合的試験;試料−既知のジゴキシン濃度を有する試料;受容体−ジゴキシンに対するビオチン化マウスモノクローナル抗体;競合分子−標識したジゴキシン;標識−パーオキシダーゼ。
【0218】
9.1. 1.1−1.4に記載の方法を行なった。
9.2. 2.2に記載のようにして、ピロールの電気化学的重合用の溶液を調製した。
9.3. 2.3に記載のようにして、ポリピロールのフイルムを電気化学的電析により形成した。
9.4. ポリピロールのフイルムでコートした検出電極をウエルから除去し、脱イオン水、次に0.01Mリン酸緩衝生理食塩水溶液(pH7.4)で濯ぎ、300μlの貯蔵溶液を有するマイクロチューブに入れ、+4℃で貯蔵した。
【0219】
9.5. 必要数の検出電極を得るため、9.1−9.4に記載の方法を繰り返した。
9.6. 2.6に記載のようにしてストレプトアビジンの溶液を調製した。
9.7. ストレプトアビジンを2.7に記載のようにして検出電極を被覆するポリピロールのフイルムの表面に結合させた。
9.8. 既知のジゴキシン濃度を有する一連の試料を以下のように調製した:
250mlのエタノールを750mlの脱イオン水に添加した;
250mgのジゴキシンを得られたエタノール溶液1000mlに溶解した;
リン酸緩衝生理食塩水錠剤及び10.0gのウシ血清アルブミンを200mlの脱イオン水に溶解した;
得られたジゴキシン溶液をウシ血清アルブミンを有するPBS溶液で250、2500、25000、50000、125000、250000及び500000倍の希釈倍率で順次希釈した;
希釈した試料のそれぞれの200μlアリコートを別々のマイクロチューブに入れた。
【0220】
9.9. ジゴキシンに対するビオチンをコンジュゲートしたマウスモノクローナル抗体の溶液20μlを0.01Mリン酸塩緩衝溶液(pH7.4)19.98mlに添加し、ロータリーシェイカーで十分混合し、次にマイクロチューブに200μlアリコートした。
9.10. ジゴキシンを前記したプロトコールに従ってパーオキシダーゼとコンジュゲートさせた。[21]を参照。パーオキシダーゼで標識したジゴキシンの得られた溶液(最終濃度約0.1mg/ml)を希釈割合10倍で0.01Mリン酸塩食塩緩衝液(pH7.4)で希釈し、少量(10μl)のアリコートに分割し、4℃で貯蔵した。
9.11. ワーキング緩衝液NO1を、4.11に記載のようにして調製した。
9.12. ワーキング緩衝液NO2を、4.12に記載のようにして調製した。
9.13. 結合したストレプトアビジンを有するポリピロールのフイルムで被覆した検出電極を貯蔵緩衝剤溶液から除去し、それぞれをジゴキシンに対するビオチンをコンジュゲートしたマウスモノクローナル抗体の溶液を含むマイクロチューブ中に置き、室温で10分間インキュベートした。
【0221】
9.14. 9.13と同時にパーオキシダーゼで標識したジゴキシン(工程9.10からの)の溶液2μlを既知ジゴキシン濃度を有する試料(工程9.8からの)のそれぞれに添加し、最初に試料から2μlを除去した。
9.15. 工程9.13及び9.14が完了したら、検出電極をパーオキシダーゼで標識した、及び標識しないジゴキシン(工程9.14からの)を含む試験管に移し;その試験管及び検出電極をサーモミキサーに置き、10分間37±1℃で保持し、連続的に攪拌した。
9.16. 工程9.15が完了したら、検出電極をマイクロチューブから除去し、0.01M PBSで3−5秒間濯ぎ、それぞれをワーキング緩衝液NO1を満たしたマイクロタイタープレートウエルに置いた。
【0222】
9.17. 検出電極及び参照電極をPCをベースにする測定装置に結合したホルダーの電気接触に連結し、ホルダーをワーキング緩衝液NO1を満たしたマイクロタイタープレートウエル上に置き、検出電極及び参照電極を溶液中に浸すようにした。
9.18. カスタムソフトウエアを開始させ、30秒にわたって参照電極電位に対する検出電極電位をミリボルトで記録するのに用いた。
9.19. 9.18を完了したら、9.17に記載されたのと類似の方法で、ホルダーをワーキング緩衝液NO2を満たしたマイクロタイタープレートウエル上に置いた。
9.20. カスタムソフトウエアを用い、100秒にわたって参照電極電位に対する検出電極電位のミリボルトの変化を記録した。
9.21. カスタムソフトウエアを用い、検出電極電位変化 対 参照電極電位の曲線により示される面積(積分)S2を計算した。
9.22. 工程9.8で製造した既知のジゴキシン濃度を有する試料を用いて9.17−9.21に記載の操作を順に繰り返した。
9.23. 工程9.22で得られた結果に基づいてカスタムソフトウエアを用いキャリブレーション曲線「S2 対 試料中ののジゴキシン濃度」をプロットした。
【0223】
文献
【表1】
【図面の簡単な説明】
【図1A】 ポリマー層に固定化したアビジンまたはストレプトアビジン分子(3)を含む電導性ポリマー層(2)でコートした電位差測定電極(1)を含む検出電極を模式的に示す図である。
【図1B】 ポリマー層上に吸着したアビジンまたはストレプトアビジン分子(3)を含む電導性ポリマー層(2)でコートした電位差測定電極(1)を含む検出電極を模式的に示す図である。
【図2A】 ポリマー層に固定化したアビジンまたはストレプトアビジンと、ビオチン(5)と結合した抗体(4)の結合反応により、検出電極を分析物(この場合、抗原は中〜高分子量の物質)に特異的にする方法を示す図である。
【図2B】 ポリマー層に固定化したアビジンまたはストレプトアビジンと、ビオチン(5)と結合した抗原(6)の結合反応により、検出電極を試験する分析物(この場合は抗体)に特異的にする方法を示す図である。
【図2C】 ポリマー層に固定化したアビジンまたはストレプトアビジンと、ビオチン(5)と結合したDNAプローブ(7)の結合反応により、検出電極を分析物(この場合はDNA分子)に特異的にする方法を示す図である。
【図3A】 検出電極に固定化したビオチン化抗体に特異的な抗原(6)を含む被検溶液と接触した検出電極の配置を示す図である。
【図3B】 検出電極に固定化したビオチン化抗原に特異的な抗体(4)を含む被検溶液と接触した検出電極の配置を示す図である。
【図3C】 検出電極に固定化したビオチン化DNAプローブに特異的なDNA分子(8)を含む被検溶液と接触した検出電極の配置を示す図である。
【図4A】 試験する抗原に特異的な、荷電標識(10)と結合した第二レセプター(9)の溶液と接触する検出電極の配置(例えば、自己抗体が荷電標識と結合した抗アイソタイプ抗体を用いて測定される血清学的アッセイ形式)を示す図である。
【図4B】 試験する抗原に特異的な、荷電標識(10)と結合した第二レセプター(11)の溶液と接触する検出電極の配置(サンドイッチアッセイ形式、この場合、第二レセプターは抗アイソタイプ抗体である)を示す図である。
【図5A】 ビオチン化抗体に特異的な、荷電標識(10)と結合した競合分子(12)の溶液と接触した検出電極の配置(競合アッセイ形式)を示す図である。
【図5B】 ビオチン化抗原に特異的な、荷電標識(10)と結合した競合分子(13)の溶液と接触した検出電極の配置(競合アッセイ形式)を示す図である。
【図6A】 試験する抗原に特異的な、酵素(14)と結合した第二レセプター(9)の溶液と接触した検出電極の配置(サンドイッチアッセイ形式)を示す図である。
【図6B】 試験する抗体に特異的な、酵素(14)と結合した第二レセプター(11)の溶液と接触した検出電極の配置(例えば、自己抗体が酵素と結合した抗アイソタイプ抗体を用いて測定される血清学的アッセイ形式)を示す図である。
【図7A】 ビオチン化抗体に特異的な、酵素(14)と結合した競合分子(12)の溶液と接触した検出電極の配置(競合アッセイ形式)を示す図である。
【図7B】 ビオチン化抗体に特異的な、酵素(14)と結合した競合分子(13)の溶液と接触した検出電極の配置(競合アッセイ形式)を示す図である。
【図8A】 ビオチン化レセプター溶液[図8A1]、被検溶液[図8A2]、および標識第二レセプター溶液[図8A3]と接触する検出電極の連続的配置、次いで、参照電極電位に対する検出電極電位の測定を含む「連続」形式の電気化学的分析工程を示す図である。
【図8B】 最初に、検出電極をビオチン化レセプター溶液と接触させ[図8B1]、次いで検出電極を、標識第二レセプター溶液を加えた被検溶液と接触させ[図8B2]、次いで参照電極電位に対する検出電極電位を測定する[図8B3]電気化学的分析工程を示す図である。
【図8C】 ビオチン化レセプターおよび標識第二レセプターの溶液を被検溶液に加え[図8C1]、次いで検出電極を被検溶液と接触させ[図8C2]、次いで参照電極電位に対する検出電極電位を測定する[図8C3] 電気化学的分析工程を示す図である。
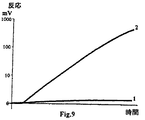
【図9】 検出電極をいかなる分析物も含まない被検溶液とインキュベーションした後(曲線1)、および検出電極をビオチン化レセプターに特異的な分析物を含む被検溶液とインキュベーションした後(曲線2)の、ワーキング溶液のイオン強度または組成の段階的変化に対する検出電極電位の変動曲線の典型的な形を示す図である(座標「ミリボルト−時間」)。
【図10】 血清試料中のHBsAg濃度対参照電極に対する測定された検出電極のバックグラウンド電位と最終電位の差(ミリボルト)の較正曲線を示す図である。
【図11】 HBsAg陽性血清の希釈対参照電極に対する測定された検出電極のバックグラウンド電気と最終電位の差(ミリボルト)の較正曲線を示す図である。
Claims (34)
- 試料中の分析物の電気化学的検出法であって、
a)試料中の検出すべき所望の分析物を結合することができるレセプターを固定化または吸着した導電性ポリマーコーティングを有する検出電極を得、
b)該所望の分析物が該固定化または吸着レセプターに結合するように試料を含む被検溶液と検出電極を接触させ、
c)固定化または吸着レセプターと結合する部位から空間的に離れた部位で該分析物と結合することができる、荷電標識と結合した第二レセプターを含む溶液と検出電極を接触させ、
d)処理した検出電極と参照電極の両方を電解液中に浸漬したときの、それらの間の電位差をモニターし、
e)一定pHにおける電解液のイオン強度の変化に伴う検出電極と参照電極間の電位差をモニターする工程を含む方法。 - 試料中の分析物の電気化学的検出法であって、
a)レセプター試料中の検出すべき所望の分析物と結合することができるレセプターを固定化または吸着した導電性ポリマーコーティングを有する検出電極を得、
b)該分析物が該固定化または吸着レセプターに結合するように試料を含む被検溶液と検出電極を接触させ、
c)該固定化または吸着レセプターと結合することができる、荷電標識と結合した競合分子を含む溶液と検出電極を接触させ、
d)処理した検出電極と参照電極の両方を電解液中に浸漬したときの、それらの間の電位差をモニターし、
e)一定pHにおける電解液のイオン強度の変化に伴う検出電極と参照電極間の電位差をモニターする工程を含む方法。 - 荷電標識が以下の特性を有する請求項1または2に記載の方法:
(i)パートd)の電解液のpHで正味の荷電を保持し、
(ii)この荷電の大きさが一定pHでの電解液のイオン強度の変化に応じて変化する。 - 荷電標識が3.33564 x 10-10クーロン(1静電単位)より大きい電解液のpHで正味の荷電を有する請求項1または2に記載の方法。
- 荷電標識がフェロセン(ferrocene)、ラテックスミクロスフェア、または金である請求項3または4記載の方法。
- 検出電極を、荷電標識と結合した第二レセプターまたは競合分子を添加した被検溶液と接触させることにより工程(b)および(c)を同時に行う請求項1〜5のいずれかに記載の方法。
- 試料中の分析物の電気化学的検出法であって、
a)レセプター試料中の検出すべき所望の分析物と結合することができるレセプターを固定化または吸着した導電性ポリマーコーティングを有する検出電極を得、
b)該分析物が該固定化または吸着レセプターと結合するように試料を含む被検溶液と検出電極を接触させ、
c)固定化または吸着レセプターと結合する部位から空間的に離れた部位で該分析物と結合することができる、酵素と結合した第二レセプターを含む溶液と検出電極を接触させ、
d)処理した検出電極と参照電極の両方を電解液中に浸漬したときの、それらの間の電位差をモニターし、
e)該酵素の基質を含む電解液に曝露した後の検出電極と参照電極間の電位差をモニターする工程を含む、
該酵素が、該酵素と基質の反応により生成される生成物が検出電極と参照電極間の電位差に影響を及ぼす酵素である方法。 - 試料中の分析物の電気化学的検出法であって、
a)レセプター試料中の検出すべき所望の分析物を結合することができるレセプターを固定化または吸着した導電性ポリマーコーティングを有する検出電極を得、
b)該所望の分析物が該固定化または吸着レセプターに結合するように試料を含む被検溶液と検出電極を接触させ、
c)該固定化または吸着レセプターと結合することができる、酵素と結合した競合分子を含む溶液と検出電極を接触させ、
d)処理した検出電極と参照電極の両方を電解液中に浸漬したときの、それらの間の電位差をモニターし、
e)該酵素の基質を含む電解液に曝露した後の検出電極と参照電極間の電位差をモニターする工程を含む、
該酵素が、該酵素と基質の反応により生成される生成物が検出電極と参照電極間の電位差に影響を及ぼす酵素である方法。 - 該酵素が、検出電極と参照電極間の電位差に対して検出可能な効果を持たない基質を、検出電極と参照電極間の電位差に直接または間接的に影響を及ぼすことができる生成物に変換することができる請求項7または8記載の方法。
- 該酵素がパーオキシダーゼである請求項9記載の方法。
- 該導電性ポリマー膜の酸化還元組成に間接的に影響を及ぼすことができる生成物がパート(e)の電解液のpHを変化させる請求項9記載の方法。
- 該酵素がウレアーゼである請求項11記載の方法。
- 該酵素が、検出電極と参照電極間の電位差に対する検出可能な効果を持たない基質を、第二酵素の基質である生成物に変換することができる請求項7または8記載の方法であって、第二酵素の作用により検出電極と参照電極間の電位差に直接または間接的に影響を及ぼす第二生成物が生じる方法。
- 該酵素が、検出電極と参照電極間の電位差に直接影響する基質を、検出電極と参照電極間の電位差に対する検出可能な効果を持たない生成物に変換することができる請求項7または8記載の方法。
- 該検出電極を、酵素標識と結合した第二レセプターまたは競合分子を加えた被検溶液と接触させることにより工程(b)および(c)が同時に行われる請求項7〜14のいずれかに記載の方法。
- レセプター分子が、細菌、植物もしくは動物細胞の断片、タンパク質(ハプテン)と結合した化合物、レクチン、糖タンパク質、または炭水化物、モノクローナル抗体、ポリクローナル抗体、抗体断片、抗体擬似物、キメラ抗体ウイルス溶解物、組換えタンパク質、合成ペプチド、ホルモン、ホルモンレセプター、一本鎖核酸、低分子である請求項1〜15のいずれかに記載の方法。
- 該検出電極の導電性ポリマーコーティングにドーパントアニオンが添加されている請求項1〜16のいずれかに記載の方法。
- 該ドーパントアニオンがドデシルサルフェートまたはデキストランサルフェートである請求項17記載の方法。
- 該検出電極がその導電性ポリマーコーティングに固定化されるか吸着されたアダプター分子を含み、検出すべき分析物と結合することができるレセプターが該アダプター分子と結合する請求項1〜18のいずれかに記載の方法。
- 導電性ポリマー層に固定化されるか吸着されたアダプター分子を有する検出電極を、荷電標識または酵素と結合した第二レセプターまたは競合分子およびレセプターを加えた被検溶液と接触させることにより検出電極とレセプターを接触させる工程とともに、工程(b)および(c)を同時に行う請求項19記載の方法。
- 検出すべき分析物と結合することができるレセプターがビオチン化されており、アダプター分子がアビジンまたはストレプトアビジンであり、該レセプターが、ビオチン/アビジンまたはビオチン/ストレプトアビジン結合相互作用により該アダプター分子と結合する、請求項19または20に記載の方法。
- 検出すべき分析物と結合することができるレセプターが、抗体であり、アダプター分子がプロテインAまたはプロテインGであり、該抗体がプロテインA/抗体またはプロテインG/抗体結合相互作用により該アダプター分子と結合する、請求項19または20に記載の方法。
- 検出すべき分析物と結合することができるレセプターが、糖部分を含んでおり、アダプター分子がレクチンであり、該レセプターがレクチン/糖結合相互作用により該アダプター分子と結合する、請求項19または20に記載の方法。
- 検出すべき分析物と結合することができるレセプターが、FITCで標識されており、アダプター分子が抗FITC抗体であり、該レセプターがFITC/抗FITC結合相互作用により該アダプター分子と結合する、請求項19または20に記載の方法。
- 全血、血清、リンパ液、尿、唾液、脳脊髄液、および精液からなる群から選ばれる生物学的液体を被検溶液として用いる請求項1〜24のいずれかに記載の方法。
- 少なくとも工程(d)および(e)が測定用フロースルーセル中で行われる請求項1〜25のいずれかに記載の方法。
- 導電性ポリマーコーティングに固定化されたアダプター分子を有する検出電極を得る工程が、
a)導電性ポリマーのモノマー単位とアダプター分子を含む電気化学的重合溶液を調製し、
b)導電性電極を該電気化学的重合溶液に浸漬し、
c)該電極と電気化学的重合溶液間の、少なくとも1完全周期で適用する周期的電位を適用して該溶液からポリマーを電気化学的に合成することにより電極をコートする工程、を含む方法を用いて該電極を製造することを含む、請求項19〜26のいずれかに記載の方法。 - 導電性ポリマーコーティングに吸着したアダプター分子を有する検出電極を得る工程が、
a)導電性ポリマーのモノマー単位を含む電気化学的重合溶液を調製し、
b)導電性電極を該電気化学的重合溶液に浸漬し、
c)該電極と電気化学的重合溶液間の、少なくとも1完全周期で適用する周期的電位を適用して該溶液からポリマーを電気化学的に合成することにより電極をコートし、
d)該アダプター分子が該電極をコーティングする導電性ポリマー上に吸着するようなアダプター分子を含む溶液と、該コートされた電極を接触させる工程を含む方法を用いて該電極を製造することを含む、請求項19〜26のいずれかに記載の方法。 - 導電性ポリマーのモノマー単位がアニリン、チオフェン、フラン、またはピロールである請求項27または28に記載の方法。
- 周期的電位がノコギリ歯の形をしている請求項27〜29のいずれかに記載の方法。
- 周期的電位が少なくとも2周期で適用される請求項27〜30のいずれかに記載の方法。
- 周期的電位が、参照電極に対して+2ボルトに等しいかまたはそれ以下の、電極に適用される最高値を有する請求項27〜31のいずれかに記載の方法。
- 試料中の分析物の電気化学的検出法であって、
(a)導電性ポリマーに固定化または吸着したアビジンまたはストレプトアビジン分子を含む導電性ポリマー層でコートした導電性電極を含む検出電極を得、ここで、該アビジンまたはストレプトアビジン分子は、ビオチン/アビジンまたはビオチン/ストレプトアビジン結合相互作用により、検出すべき分析物を結合することができるレセプター分子と結合する、
(b)該所望の分析物が該固定化または吸着レセプター分子と結合するように試料を含む被検溶液と検出電極を接触させ、
(c)検出電極と参照電極の両方を電解液に浸漬したときの参照電極に対する検出電極電位をモニターし、
(d)一定pHでの電解液のイオン強度または組成の変化に伴う参照電極に対する検出電極電位差をモニターする工程を含む方法。 - 検出すべき分析物が核酸であり、レセプター分子がオリゴヌクレオチドである請求項33記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU98116346/13A RU2161653C2 (ru) | 1998-08-24 | 1998-08-24 | Способ количественного электрохимического анализа биомолекул |
| RU98116346 | 1998-08-24 | ||
| PCT/GB1999/002785 WO2000011473A1 (en) | 1998-08-24 | 1999-08-24 | Method of electrochemical analysis of an analyte |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2002523746A JP2002523746A (ja) | 2002-07-30 |
| JP2002523746A5 JP2002523746A5 (ja) | 2010-12-02 |
| JP4774150B2 true JP4774150B2 (ja) | 2011-09-14 |
Family
ID=20210003
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000566678A Expired - Fee Related JP4774150B2 (ja) | 1998-08-24 | 1999-08-24 | 分析物の電気化学的分析法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US6770190B1 (ja) |
| EP (1) | EP1108213B1 (ja) |
| JP (1) | JP4774150B2 (ja) |
| CN (1) | CN1135390C (ja) |
| AT (1) | ATE446513T1 (ja) |
| AU (1) | AU5437999A (ja) |
| DE (1) | DE69941572D1 (ja) |
| GB (1) | GB2347223B (ja) |
| RU (1) | RU2161653C2 (ja) |
| WO (1) | WO2000011473A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7174065B2 (ja) | 2018-04-17 | 2022-11-17 | コリア リサーチ インスティテュート オブ ケミカル テクノロジー | マルチウェル電極基盤のバイオセンサー |
Families Citing this family (84)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2273270A1 (de) | 1998-08-28 | 2011-01-12 | febit holding GmbH | Verfahren und Vorrichtung zur Herstellung und/oder Analyse von biochemischen Reaktionsträgern |
| US7034660B2 (en) * | 1999-02-26 | 2006-04-25 | Sri International | Sensor devices for structural health monitoring |
| WO2001038873A2 (en) * | 1999-11-24 | 2001-05-31 | Biotronic Technologies, Inc. | Devices and methods for detecting analytes using electrosensor having capture reagent |
| DE10022750A1 (de) * | 2000-05-10 | 2001-11-22 | Wolfgang Schuhmann | Verfahren zur Immobilisierung von Erkennungskomponenten |
| DE10051396A1 (de) | 2000-10-17 | 2002-04-18 | Febit Ferrarius Biotech Gmbh | Verfahren und Vorrichtung zur integrierten Synthese und Analytbestimmung an einem Träger |
| US6670131B2 (en) * | 2000-11-30 | 2003-12-30 | Kabushiki Kaisha Toshiba | Nucleic acid detection method and apparatus, and vessel for detecting nucleic acid |
| JP4739579B2 (ja) * | 2001-06-25 | 2011-08-03 | 独立行政法人産業技術総合研究所 | 内分泌攪乱物質の高感度検出方法、及びこれを適用した内分泌攪乱物質の高感度検出装置 |
| JP4567333B2 (ja) * | 2001-08-24 | 2010-10-20 | センサーテック・リミテッド | 高感度電位差測定センサを製造する方法 |
| DE10204652B4 (de) * | 2002-02-05 | 2004-07-22 | Infineon Technologies Ag | Schaltkreis-Anordnung, elektrochemischer Sensor, Sensor-Anordnung und Verfahren zum Verarbeiten eines über eine Sensor-Elektrode bereitgestellten Stromsignals |
| GB0205455D0 (en) | 2002-03-07 | 2002-04-24 | Molecular Sensing Plc | Nucleic acid probes, their synthesis and use |
| US6987164B2 (en) * | 2002-03-09 | 2006-01-17 | Samsung Electronics Co., Ltd. | Electrically conductive polymer, sensor using the same, and method for detecting target molecule using the sensor |
| DE10211900A1 (de) * | 2002-03-18 | 2003-10-16 | Infineon Technologies Ag | Biosensor zum Erfassen von makromolekularen Biopolymeren und Verfahren zur Herstellung eines Biosensors zum Erfassen von makromolekularen Biopolymeren |
| GB2386950A (en) * | 2002-03-26 | 2003-10-01 | Sensor Tech Ltd | A sensing electrode for analysis/detection of an analyte in a test sample |
| DE10224567B4 (de) * | 2002-06-03 | 2014-10-23 | Boehringer Ingelheim Vetmedica Gmbh | Sensor-Anordnung und Verfahren zum Betreiben einer Sensor-Anordnung |
| DE10228260A1 (de) * | 2002-06-25 | 2004-01-22 | Bayer Ag | Methode und Vorrichtung zum impedimetrischen Nachweis eines oder mehrerer Analyten in einer Probe |
| FR2847581B1 (fr) * | 2002-11-21 | 2007-03-23 | Commissariat Energie Atomique | Procede de fixation d'une proteine sur un polymere a base de pyrrole et son utilisation pour la fabrication d'un capteur |
| CN1217192C (zh) | 2002-11-21 | 2005-08-31 | 北京博奥生物芯片有限责任公司 | 光电化学标记物和用其进行光电化学分析的方法、仪器与试剂盒 |
| CN100392385C (zh) * | 2002-12-03 | 2008-06-04 | 博奥生物有限公司 | 亲和反应的化学放大电化学检测方法及其试剂盒 |
| DE10319155B4 (de) * | 2003-04-29 | 2008-02-14 | Bruker Daltonik Gmbh | Elektrisch auslesbare Bindungen von Analytmolekülen an immobilisierten Sondenmolekülen |
| DE10324912A1 (de) | 2003-05-30 | 2005-01-05 | Siemens Ag | Verfahren zur Detektion von DNA-Punktmutationen (SNP-Analyse) sowie zugehörige Anordnung |
| KR100549227B1 (ko) * | 2003-09-06 | 2006-02-03 | 한국전자통신연구원 | 유기분자 소자의 제작 방법 |
| JP4497903B2 (ja) * | 2003-12-02 | 2010-07-07 | 財団法人大阪産業振興機構 | タンパク質チップおよびそれを用いたバイオセンサー |
| DE102004005711A1 (de) * | 2004-02-05 | 2006-05-11 | Siemens Ag | Biosensor zur Bestimmung eines Allergenes mit Betriebsverfahren |
| DE102004005710A1 (de) * | 2004-02-05 | 2006-05-04 | Siemens Ag | Biosensor und Verfahren zu dessen Betrieb |
| JP2006133137A (ja) * | 2004-11-08 | 2006-05-25 | Eiichi Tamiya | 被検物質の検出方法 |
| JP4744910B2 (ja) * | 2004-11-11 | 2011-08-10 | 株式会社堀場製作所 | バイオセンサ、マルチバイオセンサ及びこれを用いた多成分測定システム |
| GB2420180A (en) * | 2004-11-11 | 2006-05-17 | Sensor Tech Ltd | Method of electrochemical analysis of an analyte |
| JP4232108B2 (ja) | 2005-05-20 | 2009-03-04 | セイコーエプソン株式会社 | 標的物質の検出または定量方法、該方法に用いられる電極基板、装置、およびキット |
| US8117902B2 (en) * | 2005-11-03 | 2012-02-21 | University Of Massachusetts | Nanopatterned surfaces and related methods for selective adhesion, sensing and separation |
| US20070202561A1 (en) * | 2006-02-10 | 2007-08-30 | Becton Dickinson And Company | Electronic Detection Immunoassays that Utilize a Binder Support Medium |
| AU2007229320B2 (en) * | 2006-03-17 | 2013-01-10 | Newsouth Innovations Pty Limited | Electrochemical sensor |
| US20080103064A1 (en) * | 2006-06-13 | 2008-05-01 | Antara Biosciences Inc. | Microscale fluidic devices for electrochemical detection of biological molecules |
| CA2656203C (en) * | 2006-06-30 | 2014-07-22 | Chisso Corporation | Kit for detection/quantification of analyte, and method for detection/quantification of analyte |
| JP4936536B2 (ja) * | 2006-07-13 | 2012-05-23 | 国立大学法人富山大学 | 定量・定性分析方法 |
| EP2061572A1 (en) * | 2006-08-31 | 2009-05-27 | Technion Research & Development Foundation Ltd. | Controllable binding and dissociation of chemical entities and electrode devices therefore |
| WO2008054611A2 (en) * | 2006-10-04 | 2008-05-08 | President And Fellows Of Harvard College | Engineered conductive polymer films to mediate biochemical interactions |
| JP4751302B2 (ja) * | 2006-11-21 | 2011-08-17 | 株式会社日立製作所 | 電位差式センサ及び分析用素子 |
| WO2008112653A1 (en) * | 2007-03-09 | 2008-09-18 | Dxtech, Llc | Electrochemical detection system |
| US8197650B2 (en) * | 2007-06-07 | 2012-06-12 | Sensor Innovations, Inc. | Silicon electrochemical sensors |
| EP2210268A4 (en) | 2007-10-17 | 2012-02-15 | Ohmx Corp | IN THE BIOSENSORS USED NEW CHEMISTRY |
| CN101464424B (zh) * | 2007-12-18 | 2013-02-13 | 宁波大学 | 一种研究dna分子导电性的测试方法及其测试系统 |
| CN102854232B (zh) * | 2008-12-08 | 2015-12-02 | 拜尔健康护理有限责任公司 | 具有信号调节功能的生物传感器系统 |
| US8025789B2 (en) * | 2008-12-17 | 2011-09-27 | General Electric Company | Anionically-charged polymer detection method |
| WO2011034668A1 (en) | 2009-08-07 | 2011-03-24 | Ohmx Corporation | Enzyme triggered redox altering chemical elimination (e-trace) immunoassay |
| WO2011057347A1 (en) | 2009-11-12 | 2011-05-19 | Tgr Biosciences Pty Ltd | Analyte detection |
| GB2476057B (en) * | 2009-12-09 | 2012-05-30 | Schlumberger Holdings | Electro-chemical sensor |
| EP2390664B1 (de) * | 2010-05-25 | 2013-04-17 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Verfahren zur elektrochemischen Detektion von Bindungsreaktionen |
| WO2012012537A1 (en) | 2010-07-20 | 2012-01-26 | Ohmx Corporation | Novel chemistry used in biosensors |
| GB2500550A (en) | 2010-12-16 | 2013-09-25 | Sensor Innovations Inc | Electrochemical sensors |
| KR101274854B1 (ko) | 2010-12-23 | 2013-06-13 | 한남대학교 산학협력단 | 감염성 호흡기 질환 진단용 전기화학 dna 센서 및 이의 제조방법 |
| WO2012100078A1 (en) | 2011-01-19 | 2012-07-26 | Ohmx Corporation | Enzyme triggered redox altering chemical elimination (e-trace) immmunoassay |
| WO2012121229A1 (ja) * | 2011-03-08 | 2012-09-13 | 公立大学法人大阪府立大学 | 微生物検出用センサーおよびその製造方法 |
| EP3435082B8 (en) | 2011-03-31 | 2020-11-25 | Tgr Biosciences Pty Ltd | Detection of multiple analytes |
| RU2466680C1 (ru) | 2011-10-03 | 2012-11-20 | Общество С Ограниченной Ответственностью "Витацел" | Способ диагностики состояния кожи пациента (варианты) |
| JP6203183B2 (ja) | 2011-10-17 | 2017-09-27 | オームクス コーポレイション | 酵素誘発レドックス変化化学的脱離(e−trace)イムノアッセイを使用する、ヘモグロビンa1cのパーセンテージの単回直接検出 |
| JP2014532720A (ja) | 2011-11-04 | 2014-12-08 | オームクス コーポレイション | バイオセンサーにおいて使用される新規化学 |
| CA2860739A1 (en) | 2012-01-09 | 2013-07-18 | Ohmx Corporation | Enzyme cascade methods for e-trace assay signal amplification |
| JP2015529809A (ja) | 2012-07-27 | 2015-10-08 | オームクス コーポレイション | 酵素及び他の標的分析物の存在及び活性の切断前検出後の単分子層の電気測定 |
| EP2877592B1 (en) | 2012-07-27 | 2016-09-21 | Ohmx Corporation | Electronic measurements of monolayers following homogeneous reactions of their components |
| CN102875623B (zh) * | 2012-09-19 | 2016-01-20 | 华东理工大学 | 糖基蒽醌类化合物及其石墨烯传感器构建 |
| CN102901822A (zh) * | 2012-10-23 | 2013-01-30 | 扬州大学 | 聚合物自组装超微孔膜免疫组合传感器的制备方法 |
| WO2014118543A2 (en) * | 2013-01-30 | 2014-08-07 | Vantix Holdings Limited | Electrochemical total protein detection system |
| EP2972333B1 (en) | 2013-03-11 | 2018-09-19 | The University of Toledo | A biosensor device to target analytes in situ, in vivo, and/or in real time, and methods of making and using the same |
| US9594047B2 (en) * | 2013-04-26 | 2017-03-14 | Universiteit Antwerpen | Potentiometric sensors and method for measuring intermolecular interactions |
| DE102013211837A1 (de) * | 2013-06-21 | 2014-12-24 | Gilupi Gmbh | Katheter mit Detektionsvorrichtung zum Echtzeitnachweis eines Probenmaterials |
| JP5565783B1 (ja) * | 2013-08-08 | 2014-08-06 | 国立大学法人 東京大学 | バイオセンサ |
| JP5599012B1 (ja) * | 2014-06-23 | 2014-10-01 | 国立大学法人 東京大学 | 採取部及びバイオセンサ |
| WO2016043078A1 (ja) * | 2014-09-19 | 2016-03-24 | 国立大学法人 新潟大学 | 基質抗原同時検出バイオセンサ、電極、基質抗原同時検出方法、および、プログラム |
| EP3078965B1 (en) * | 2015-04-06 | 2018-04-04 | ARKRAY, Inc. | Biosensor comprising electrode for measuring hematocrit value |
| CN105353015B (zh) * | 2015-12-02 | 2019-04-12 | 厦门大学 | 一种聚二氧乙烯噻吩复合膜的制备方法及应用 |
| CN105606675B (zh) * | 2015-12-30 | 2018-09-18 | 湖南大学 | 用于检测铅的适配体传感器及其制备方法和应用 |
| US11202593B2 (en) | 2017-04-21 | 2021-12-21 | 2Pi-Sigma Corp. | Adjustable lancet and test cartridge for automated medical sample collection and testing |
| US11175303B2 (en) | 2017-04-21 | 2021-11-16 | 2Pi-Sigma Corp. | Automated medical sample collection and testing for providing blood coagulation indication |
| US10928411B2 (en) | 2017-04-21 | 2021-02-23 | 2Pi-Sigma Corporation | Automated medical sample collection and testing |
| WO2018195544A1 (en) * | 2017-04-21 | 2018-10-25 | 2Pi-Sigma Corporation | Automated medical sample collection, testing, and analysis |
| US10791972B2 (en) | 2017-04-21 | 2020-10-06 | 2Pi-Sigma Corporation | Fluid measurement for automated medical sample collection and testing |
| US11103163B2 (en) | 2017-04-21 | 2021-08-31 | 2Pi-Sigma Corporation | Test cartridge and lancet for automated medical sample collection and testing |
| CN106979966A (zh) * | 2017-05-09 | 2017-07-25 | 福建医科大学附属协和医院 | 一种检测纤维连接蛋白的电化学免疫传感器的制备方法 |
| JP7278147B2 (ja) * | 2018-05-22 | 2023-05-19 | アークレイ株式会社 | 新規バイオセンシング法 |
| BR112020023538A2 (pt) * | 2018-05-22 | 2021-05-04 | Katherine Konstantin Sizov | biossensor de receptor de etileno |
| CN111351938A (zh) * | 2018-12-20 | 2020-06-30 | 麦德龙生物株式会社 | 电激发标记分子的方法和绝缘膜涂覆的电极 |
| JP2022523691A (ja) * | 2019-01-30 | 2022-04-26 | アリゾナ・ボード・オブ・リージェンツ・オン・ビハーフ・オブ・アリゾナ・ステイト・ユニバーシティー | 生体電気回路、それを製造および使用するためのシステムおよび方法 |
| CN110376380B (zh) * | 2019-07-25 | 2020-07-24 | 华中科技大学 | 一种电化学酶联免疫传感器及其制备与检测抗原的应用 |
| CN114441614A (zh) * | 2021-12-30 | 2022-05-06 | 广州市赛特检测有限公司 | 一种电化学微生物快速检测仪及生物探针的修饰方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02179461A (ja) * | 1988-12-30 | 1990-07-12 | Chemo Sero Therapeut Res Inst | 酵素免疫センサ用膜 |
| WO1998035232A2 (en) * | 1997-02-06 | 1998-08-13 | The University Of North Carolina At Chapel Hill | Electrochemical detection of specific binding |
| WO1998037409A1 (en) * | 1997-02-20 | 1998-08-27 | Biosensor Technology Limited | Method of electrochemical detection of immunoactive macromolecules |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0697219B2 (ja) * | 1985-02-25 | 1994-11-30 | 財団法人化学及血清療法研究所 | 免疫センサー用電極及びその製造方法 |
| JPH0721478B2 (ja) * | 1986-03-31 | 1995-03-08 | 財団法人化学及血清療法研究所 | 免疫センサ−用作用膜 |
| WO1989011649A1 (en) * | 1988-05-16 | 1989-11-30 | Wollongong Uniadvice Limited | Antibody containing electrode |
| US5312762A (en) | 1989-03-13 | 1994-05-17 | Guiseppi Elie Anthony | Method of measuring an analyte by measuring electrical resistance of a polymer film reacting with the analyte |
| GB2276724A (en) * | 1993-03-31 | 1994-10-05 | Cambridge Life Sciences | Electrochemical detection of specific binding species |
-
1998
- 1998-08-24 RU RU98116346/13A patent/RU2161653C2/ru active
-
1999
- 1999-08-24 AU AU54379/99A patent/AU5437999A/en not_active Abandoned
- 1999-08-24 CN CNB998125474A patent/CN1135390C/zh not_active Expired - Lifetime
- 1999-08-24 JP JP2000566678A patent/JP4774150B2/ja not_active Expired - Fee Related
- 1999-08-24 EP EP99940398A patent/EP1108213B1/en not_active Expired - Lifetime
- 1999-08-24 WO PCT/GB1999/002785 patent/WO2000011473A1/en active Application Filing
- 1999-08-24 US US09/763,345 patent/US6770190B1/en not_active Expired - Lifetime
- 1999-08-24 GB GB0012057A patent/GB2347223B/en not_active Expired - Lifetime
- 1999-08-24 AT AT99940398T patent/ATE446513T1/de not_active IP Right Cessation
- 1999-08-24 DE DE69941572T patent/DE69941572D1/de not_active Expired - Lifetime
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02179461A (ja) * | 1988-12-30 | 1990-07-12 | Chemo Sero Therapeut Res Inst | 酵素免疫センサ用膜 |
| WO1998035232A2 (en) * | 1997-02-06 | 1998-08-13 | The University Of North Carolina At Chapel Hill | Electrochemical detection of specific binding |
| WO1998037409A1 (en) * | 1997-02-20 | 1998-08-27 | Biosensor Technology Limited | Method of electrochemical detection of immunoactive macromolecules |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7174065B2 (ja) | 2018-04-17 | 2022-11-17 | コリア リサーチ インスティテュート オブ ケミカル テクノロジー | マルチウェル電極基盤のバイオセンサー |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002523746A (ja) | 2002-07-30 |
| AU5437999A (en) | 2000-03-14 |
| ATE446513T1 (de) | 2009-11-15 |
| EP1108213A1 (en) | 2001-06-20 |
| EP1108213B1 (en) | 2009-10-21 |
| GB2347223B (en) | 2003-03-26 |
| CN1135390C (zh) | 2004-01-21 |
| GB2347223A (en) | 2000-08-30 |
| WO2000011473A1 (en) | 2000-03-02 |
| CN1325490A (zh) | 2001-12-05 |
| RU2161653C2 (ru) | 2001-01-10 |
| DE69941572D1 (de) | 2009-12-03 |
| GB0012057D0 (en) | 2000-07-12 |
| US6770190B1 (en) | 2004-08-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4774150B2 (ja) | 分析物の電気化学的分析法 | |
| Bakirhan et al. | Recent progress on the sensitive detection of cardiovascular disease markers by electrochemical-based biosensors | |
| JP4714745B2 (ja) | 検体の電気化学分析の改良方法 | |
| Darain et al. | Development of an immunosensor for the detection of vitellogenin using impedance spectroscopy | |
| Darain et al. | Disposable amperometric immunosensor system for rabbit IgG using a conducting polymer modified screen-printed electrode | |
| Dong et al. | Heterogeneous immunosensing using antigen and antibody monolayers on gold surfaces with electrochemical and scanning probe detection | |
| Çevik et al. | Construction of novel electrochemical immunosensor for detection of prostate specific antigen using ferrocene-PAMAM dendrimers | |
| Omidfar et al. | Development of urinary albumin immunosensor based on colloidal AuNP and PVA | |
| Wang et al. | A piezoelectric immunoagglutination assay for Toxoplasma gondii antibodies using gold nanoparticles | |
| Vikholm-Lundin et al. | Site-directed immobilisation of antibody fragments for detection of C-reactive protein | |
| Kang et al. | New architecture for reagentless, protein-based electrochemical biosensors | |
| WO2009032901A1 (en) | Biosensors and related methods | |
| Horak et al. | Polymer-modified microfluidic immunochip for enhanced electrochemical detection of troponin I | |
| WO2001038873A2 (en) | Devices and methods for detecting analytes using electrosensor having capture reagent | |
| US20060240492A1 (en) | Carbon nanotube based immunosensors and methods of making and using | |
| KR20190121247A (ko) | 멀티웰 전극 기반 바이오센서 | |
| Zhuo et al. | A tris (2, 2′-bipyridyl) cobalt (III)-bovine serum albumin composite membrane for biosensors | |
| Liu | Electrochemical detection of prostate-specific antigen based on gold colloids/alumina derived sol-gel film | |
| Zhou et al. | An amperometric immunosensor based on an electrochemically pretreated carbon–paraffin electrode for complement III (C3) assay | |
| Li et al. | Ultrasensitive multiplexed protein biomarker detection based on electrochemical tag incorporated polystyrene spheres as label | |
| Wang et al. | A reusable piezo-immunosensor with amplified sensitivity for ceruloplasmin based on plasma-polymerized film | |
| Ameur et al. | Impedimetric measurements on polarized functionalized platinum electrodes: application to direct immunosensing | |
| Liang et al. | Interdigitated conductometric immunosensor for determination of interleukin‐6 in humans based on dendrimer G4 and colloidal gold modified composite film | |
| Ni et al. | A one-step potentiometric immunoassay for plasma cardiac troponin I using an antibody-functionalized bis-MPA–COOH dendrimer as a competitor with improved sensitivity | |
| Ren et al. | Development of a new and simple method for the detection of histidine-tagged proteins based on thionine-chitosan/gold nanoparticles/horseradish peroxidase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060605 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060605 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20090723 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090818 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20091118 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20091126 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20091218 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20091228 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100115 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100122 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100216 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100615 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100915 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100924 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101014 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20101014 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110118 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110411 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20110531 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20110627 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140701 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |