JP4515911B2 - アナンダミド加水分解の遮断による不安の調節 - Google Patents
アナンダミド加水分解の遮断による不安の調節 Download PDFInfo
- Publication number
- JP4515911B2 JP4515911B2 JP2004543510A JP2004543510A JP4515911B2 JP 4515911 B2 JP4515911 B2 JP 4515911B2 JP 2004543510 A JP2004543510 A JP 2004543510A JP 2004543510 A JP2004543510 A JP 2004543510A JP 4515911 B2 JP4515911 B2 JP 4515911B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- substituted
- faah
- unsubstituted
- compounds
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 208000019901 Anxiety disease Diseases 0.000 title claims description 99
- 230000036506 anxiety Effects 0.000 title claims description 64
- LGEQQWMQCRIYKG-DOFZRALJSA-N anandamide Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(=O)NCCO LGEQQWMQCRIYKG-DOFZRALJSA-N 0.000 title description 76
- LGEQQWMQCRIYKG-UHFFFAOYSA-N arachidonic acid ethanolamide Natural products CCCCCC=CCC=CCC=CCC=CCCCC(=O)NCCO LGEQQWMQCRIYKG-UHFFFAOYSA-N 0.000 title description 50
- 230000007062 hydrolysis Effects 0.000 title description 9
- 238000006460 hydrolysis reaction Methods 0.000 title description 9
- 230000000903 blocking effect Effects 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims description 304
- 125000000217 alkyl group Chemical group 0.000 claims description 86
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 57
- 238000011282 treatment Methods 0.000 claims description 49
- 239000003814 drug Substances 0.000 claims description 45
- 125000001424 substituent group Chemical group 0.000 claims description 39
- 229910052739 hydrogen Inorganic materials 0.000 claims description 37
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 27
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 26
- 235000019789 appetite Nutrition 0.000 claims description 23
- 230000036528 appetite Effects 0.000 claims description 23
- 208000002193 Pain Diseases 0.000 claims description 22
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 22
- 239000008194 pharmaceutical composition Substances 0.000 claims description 22
- 241000124008 Mammalia Species 0.000 claims description 19
- 230000036407 pain Effects 0.000 claims description 19
- 125000003545 alkoxy group Chemical group 0.000 claims description 15
- 125000005843 halogen group Chemical group 0.000 claims description 15
- 239000001257 hydrogen Substances 0.000 claims description 15
- 239000003937 drug carrier Substances 0.000 claims description 14
- 235000010290 biphenyl Nutrition 0.000 claims description 12
- 239000004305 biphenyl Substances 0.000 claims description 12
- 125000003917 carbamoyl group Chemical class [H]N([H])C(*)=O 0.000 claims description 12
- 229910052736 halogen Inorganic materials 0.000 claims description 12
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 11
- 208000027559 Appetite disease Diseases 0.000 claims description 10
- 208000019454 Feeding and Eating disease Diseases 0.000 claims description 10
- 238000004519 manufacturing process Methods 0.000 claims description 10
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 10
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 10
- 150000003839 salts Chemical class 0.000 claims description 9
- 150000002367 halogens Chemical class 0.000 claims description 8
- 125000001153 fluoro group Chemical group F* 0.000 claims description 5
- 125000004417 unsaturated alkyl group Chemical group 0.000 claims description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 3
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 claims description 2
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 claims 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 claims 1
- 108010046094 fatty-acid amide hydrolase Proteins 0.000 description 126
- 102100029111 Fatty-acid amide hydrolase 1 Human genes 0.000 description 123
- -1 biphenylyl Chemical group 0.000 description 115
- 238000000034 method Methods 0.000 description 102
- 239000003940 fatty acid amidase inhibitor Substances 0.000 description 92
- 230000000694 effects Effects 0.000 description 74
- 239000003112 inhibitor Substances 0.000 description 68
- 125000003118 aryl group Chemical group 0.000 description 61
- 238000012360 testing method Methods 0.000 description 59
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 51
- 239000000203 mixture Substances 0.000 description 43
- 241000700159 Rattus Species 0.000 description 41
- 125000001183 hydrocarbyl group Chemical group 0.000 description 41
- 239000000126 substance Substances 0.000 description 37
- 241001465754 Metazoa Species 0.000 description 35
- 125000000304 alkynyl group Chemical group 0.000 description 35
- 229940079593 drug Drugs 0.000 description 35
- 125000003342 alkenyl group Chemical group 0.000 description 34
- 125000004404 heteroalkyl group Chemical group 0.000 description 31
- 208000035475 disorder Diseases 0.000 description 29
- 102000004190 Enzymes Human genes 0.000 description 28
- 108090000790 Enzymes Proteins 0.000 description 28
- 230000027455 binding Effects 0.000 description 28
- 229940088598 enzyme Drugs 0.000 description 28
- 125000001072 heteroaryl group Chemical group 0.000 description 28
- 102000009132 CB1 Cannabinoid Receptor Human genes 0.000 description 27
- 108010073366 CB1 Cannabinoid Receptor Proteins 0.000 description 27
- 150000004665 fatty acids Chemical class 0.000 description 27
- 125000005842 heteroatom Chemical group 0.000 description 27
- 230000002401 inhibitory effect Effects 0.000 description 27
- 235000014113 dietary fatty acids Nutrition 0.000 description 26
- 239000000194 fatty acid Substances 0.000 description 26
- 229930195729 fatty acid Natural products 0.000 description 26
- 230000005764 inhibitory process Effects 0.000 description 26
- 229910052760 oxygen Inorganic materials 0.000 description 25
- 208000024891 symptom Diseases 0.000 description 25
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 24
- 210000004556 brain Anatomy 0.000 description 23
- 230000000949 anxiolytic effect Effects 0.000 description 22
- 201000010099 disease Diseases 0.000 description 22
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 21
- 229930003827 cannabinoid Natural products 0.000 description 21
- 239000003557 cannabinoid Substances 0.000 description 21
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 21
- 125000006850 spacer group Chemical group 0.000 description 21
- 241000282414 Homo sapiens Species 0.000 description 20
- 238000012216 screening Methods 0.000 description 20
- BOWVQLFMWHZBEF-KTKRTIGZSA-N oleoyl ethanolamide Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)NCCO BOWVQLFMWHZBEF-KTKRTIGZSA-N 0.000 description 19
- 230000002209 hydrophobic effect Effects 0.000 description 18
- 125000004433 nitrogen atom Chemical group N* 0.000 description 18
- 239000000243 solution Substances 0.000 description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- 239000004480 active ingredient Substances 0.000 description 17
- 239000012528 membrane Substances 0.000 description 17
- 238000002360 preparation method Methods 0.000 description 17
- 125000003710 aryl alkyl group Chemical group 0.000 description 16
- 230000006399 behavior Effects 0.000 description 15
- 210000002569 neuron Anatomy 0.000 description 15
- JZCPYUJPEARBJL-UHFFFAOYSA-N rimonabant Chemical compound CC=1C(C(=O)NN2CCCCC2)=NN(C=2C(=CC(Cl)=CC=2)Cl)C=1C1=CC=C(Cl)C=C1 JZCPYUJPEARBJL-UHFFFAOYSA-N 0.000 description 15
- 229960003015 rimonabant Drugs 0.000 description 15
- KGKDDSYRBQOMLE-UHFFFAOYSA-N (3-phenylphenyl) n-cyclohexylcarbamate Chemical compound C=1C=CC(C=2C=CC=CC=2)=CC=1OC(=O)NC1CCCCC1 KGKDDSYRBQOMLE-UHFFFAOYSA-N 0.000 description 14
- 241000699670 Mus sp. Species 0.000 description 14
- 239000000935 antidepressant agent Substances 0.000 description 14
- 229940005513 antidepressants Drugs 0.000 description 14
- 239000002249 anxiolytic agent Substances 0.000 description 14
- 206010015037 epilepsy Diseases 0.000 description 14
- 125000001188 haloalkyl group Chemical group 0.000 description 14
- 230000001965 increasing effect Effects 0.000 description 14
- 230000004044 response Effects 0.000 description 14
- 102100033639 Acetylcholinesterase Human genes 0.000 description 13
- 108010022752 Acetylcholinesterase Proteins 0.000 description 13
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 13
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 13
- 229930006000 Sucrose Natural products 0.000 description 13
- 229940022698 acetylcholinesterase Drugs 0.000 description 13
- 150000003857 carboxamides Chemical class 0.000 description 13
- 239000005720 sucrose Substances 0.000 description 13
- 208000020401 Depressive disease Diseases 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 125000002252 acyl group Chemical group 0.000 description 12
- 125000004103 aminoalkyl group Chemical group 0.000 description 12
- 238000010171 animal model Methods 0.000 description 12
- 238000003556 assay Methods 0.000 description 12
- 229960003638 dopamine Drugs 0.000 description 12
- 235000013305 food Nutrition 0.000 description 12
- 238000001727 in vivo Methods 0.000 description 12
- 229910052757 nitrogen Inorganic materials 0.000 description 12
- 230000001225 therapeutic effect Effects 0.000 description 12
- RCRCTBLIHCHWDZ-UHFFFAOYSA-N 2-Arachidonoyl Glycerol Chemical compound CCCCCC=CCC=CCC=CCC=CCCCC(=O)OC(CO)CO RCRCTBLIHCHWDZ-UHFFFAOYSA-N 0.000 description 11
- 230000001430 anti-depressive effect Effects 0.000 description 11
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- 230000008859 change Effects 0.000 description 11
- 230000002829 reductive effect Effects 0.000 description 11
- 201000000980 schizophrenia Diseases 0.000 description 11
- 229940124530 sulfonamide Drugs 0.000 description 11
- 150000003456 sulfonamides Chemical class 0.000 description 11
- 241000282412 Homo Species 0.000 description 10
- 102000005398 Monoacylglycerol Lipase Human genes 0.000 description 10
- 108020002334 Monoacylglycerol lipase Proteins 0.000 description 10
- 241000699666 Mus <mouse, genus> Species 0.000 description 10
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 10
- 230000003502 anti-nociceptive effect Effects 0.000 description 10
- 125000004104 aryloxy group Chemical group 0.000 description 10
- 210000004027 cell Anatomy 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 10
- 206010022437 insomnia Diseases 0.000 description 10
- 208000020016 psychiatric disease Diseases 0.000 description 10
- 230000007958 sleep Effects 0.000 description 10
- 125000003435 aroyl group Chemical group 0.000 description 9
- 125000004429 atom Chemical group 0.000 description 9
- 230000003197 catalytic effect Effects 0.000 description 9
- 230000007423 decrease Effects 0.000 description 9
- 235000019441 ethanol Nutrition 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 125000001624 naphthyl group Chemical group 0.000 description 9
- 125000003107 substituted aryl group Chemical group 0.000 description 9
- 229910052717 sulfur Inorganic materials 0.000 description 9
- 239000003826 tablet Substances 0.000 description 9
- 231100000027 toxicology Toxicity 0.000 description 9
- 125000006657 (C1-C10) hydrocarbyl group Chemical group 0.000 description 8
- 102000009135 CB2 Cannabinoid Receptor Human genes 0.000 description 8
- 108010073376 CB2 Cannabinoid Receptor Proteins 0.000 description 8
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 8
- 208000010412 Glaucoma Diseases 0.000 description 8
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 8
- 230000009471 action Effects 0.000 description 8
- 210000000577 adipose tissue Anatomy 0.000 description 8
- 238000009472 formulation Methods 0.000 description 8
- 230000036541 health Effects 0.000 description 8
- 125000000623 heterocyclic group Chemical group 0.000 description 8
- 238000001990 intravenous administration Methods 0.000 description 8
- 239000003446 ligand Substances 0.000 description 8
- 230000003389 potentiating effect Effects 0.000 description 8
- 239000007787 solid Substances 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- 238000004617 QSAR study Methods 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 230000009286 beneficial effect Effects 0.000 description 7
- 229940049706 benzodiazepine Drugs 0.000 description 7
- 230000001684 chronic effect Effects 0.000 description 7
- 238000013461 design Methods 0.000 description 7
- 239000002552 dosage form Substances 0.000 description 7
- 230000002996 emotional effect Effects 0.000 description 7
- 235000012631 food intake Nutrition 0.000 description 7
- 150000005363 heterobiaryls Chemical group 0.000 description 7
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 238000007912 intraperitoneal administration Methods 0.000 description 7
- 150000002632 lipids Chemical class 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 108010070612 neurotoxic esterase Proteins 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 230000009467 reduction Effects 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- 238000002604 ultrasonography Methods 0.000 description 7
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 229940124802 CB1 antagonist Drugs 0.000 description 6
- 102000018208 Cannabinoid Receptor Human genes 0.000 description 6
- 108050007331 Cannabinoid receptor Proteins 0.000 description 6
- 206010010904 Convulsion Diseases 0.000 description 6
- 239000005977 Ethylene Substances 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 208000034189 Sclerosis Diseases 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- ROFVXGGUISEHAM-UHFFFAOYSA-N URB597 Chemical compound NC(=O)C1=CC=CC(C=2C=C(OC(=O)NC3CCCCC3)C=CC=2)=C1 ROFVXGGUISEHAM-UHFFFAOYSA-N 0.000 description 6
- 239000013543 active substance Substances 0.000 description 6
- 125000002947 alkylene group Chemical group 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 239000001961 anticonvulsive agent Substances 0.000 description 6
- 125000004097 arachidonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])/C([H])=C([H])\C([H])([H])/C([H])=C([H])\C([H])([H])/C([H])=C([H])\C([H])([H])/C([H])=C([H])\C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 6
- 150000001557 benzodiazepines Chemical class 0.000 description 6
- 150000001720 carbohydrates Chemical class 0.000 description 6
- 235000014633 carbohydrates Nutrition 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 230000037406 food intake Effects 0.000 description 6
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 6
- 230000001939 inductive effect Effects 0.000 description 6
- 230000003993 interaction Effects 0.000 description 6
- 238000007918 intramuscular administration Methods 0.000 description 6
- 125000005647 linker group Chemical group 0.000 description 6
- 208000024714 major depressive disease Diseases 0.000 description 6
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 6
- 239000003755 preservative agent Substances 0.000 description 6
- 230000000069 prophylactic effect Effects 0.000 description 6
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 238000007920 subcutaneous administration Methods 0.000 description 6
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 6
- 239000003981 vehicle Substances 0.000 description 6
- XYUDDOIIYRPZLT-UHFFFAOYSA-N (4-phenylmethoxyphenyl) n-(4-fluorophenyl)carbamate Chemical compound C1=CC(F)=CC=C1NC(=O)OC(C=C1)=CC=C1OCC1=CC=CC=C1 XYUDDOIIYRPZLT-UHFFFAOYSA-N 0.000 description 5
- BQRRGDDGUKOPJO-UHFFFAOYSA-N 3-(3-hydroxyphenyl)benzamide Chemical compound NC(=O)C1=CC=CC(C=2C=C(O)C=CC=2)=C1 BQRRGDDGUKOPJO-UHFFFAOYSA-N 0.000 description 5
- 241000282472 Canis lupus familiaris Species 0.000 description 5
- 241000282326 Felis catus Species 0.000 description 5
- 102000004300 GABA-A Receptors Human genes 0.000 description 5
- 108090000839 GABA-A Receptors Proteins 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 238000010162 Tukey test Methods 0.000 description 5
- 230000001154 acute effect Effects 0.000 description 5
- 239000000556 agonist Substances 0.000 description 5
- 230000001197 anandamide transport Effects 0.000 description 5
- 230000003556 anti-epileptic effect Effects 0.000 description 5
- 229940005530 anxiolytics Drugs 0.000 description 5
- BNBSCAZCQDLUDU-DOFZRALJSA-N arachidonoyl amine Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(N)=O BNBSCAZCQDLUDU-DOFZRALJSA-N 0.000 description 5
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 5
- 230000004071 biological effect Effects 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 230000015556 catabolic process Effects 0.000 description 5
- 210000003169 central nervous system Anatomy 0.000 description 5
- 230000002490 cerebral effect Effects 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 238000006731 degradation reaction Methods 0.000 description 5
- 230000001419 dependent effect Effects 0.000 description 5
- 235000019439 ethyl acetate Nutrition 0.000 description 5
- 125000004474 heteroalkylene group Chemical group 0.000 description 5
- 125000001841 imino group Chemical group [H]N=* 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 5
- 208000019906 panic disease Diseases 0.000 description 5
- 230000000144 pharmacologic effect Effects 0.000 description 5
- 239000000523 sample Substances 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 230000035882 stress Effects 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical group C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- YJTKZCDBKVTVBY-UHFFFAOYSA-N 1,3-Diphenylbenzene Chemical group C1=CC=CC=C1C1=CC=CC(C=2C=CC=CC=2)=C1 YJTKZCDBKVTVBY-UHFFFAOYSA-N 0.000 description 4
- ODJFDWIECLJWSR-UHFFFAOYSA-N 3-bromobenzamide Chemical compound NC(=O)C1=CC=CC(Br)=C1 ODJFDWIECLJWSR-UHFFFAOYSA-N 0.000 description 4
- 208000008811 Agoraphobia Diseases 0.000 description 4
- 208000020925 Bipolar disease Diseases 0.000 description 4
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 4
- 241000277305 Electrophorus electricus Species 0.000 description 4
- OHCQJHSOBUTRHG-KGGHGJDLSA-N FORSKOLIN Chemical compound O=C([C@@]12O)C[C@](C)(C=C)O[C@]1(C)[C@@H](OC(=O)C)[C@@H](O)[C@@H]1[C@]2(C)[C@@H](O)CCC1(C)C OHCQJHSOBUTRHG-KGGHGJDLSA-N 0.000 description 4
- 229940122746 Fatty acid amide hydrolase inhibitor Drugs 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- 208000012902 Nervous system disease Diseases 0.000 description 4
- 208000025966 Neurological disease Diseases 0.000 description 4
- 208000008589 Obesity Diseases 0.000 description 4
- 206010033307 Overweight Diseases 0.000 description 4
- 206010034912 Phobia Diseases 0.000 description 4
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 4
- 238000000540 analysis of variance Methods 0.000 description 4
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 4
- 150000007860 aryl ester derivatives Chemical class 0.000 description 4
- 230000036760 body temperature Effects 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 238000002648 combination therapy Methods 0.000 description 4
- 230000000875 corresponding effect Effects 0.000 description 4
- 210000003618 cortical neuron Anatomy 0.000 description 4
- CYQFCXCEBYINGO-IAGOWNOFSA-N delta1-THC Chemical compound C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@@H]21 CYQFCXCEBYINGO-IAGOWNOFSA-N 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 231100000673 dose–response relationship Toxicity 0.000 description 4
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 239000002621 endocannabinoid Substances 0.000 description 4
- 125000002541 furyl group Chemical group 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 238000013537 high throughput screening Methods 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 230000004410 intraocular pressure Effects 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 238000003032 molecular docking Methods 0.000 description 4
- 230000000926 neurological effect Effects 0.000 description 4
- 235000020824 obesity Nutrition 0.000 description 4
- FATBGEAMYMYZAF-KTKRTIGZSA-N oleamide Chemical compound CCCCCCCC\C=C/CCCCCCCC(N)=O FATBGEAMYMYZAF-KTKRTIGZSA-N 0.000 description 4
- FATBGEAMYMYZAF-UHFFFAOYSA-N oleicacidamide-heptaglycolether Natural products CCCCCCCCC=CCCCCCCCC(N)=O FATBGEAMYMYZAF-UHFFFAOYSA-N 0.000 description 4
- 238000005457 optimization Methods 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 229950007031 palmidrol Drugs 0.000 description 4
- HXYVTAGFYLMHSO-UHFFFAOYSA-N palmitoyl ethanolamide Chemical compound CCCCCCCCCCCCCCCC(=O)NCCO HXYVTAGFYLMHSO-UHFFFAOYSA-N 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- 208000019899 phobic disease Diseases 0.000 description 4
- 239000006187 pill Substances 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 238000003825 pressing Methods 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 238000012545 processing Methods 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 229940044601 receptor agonist Drugs 0.000 description 4
- 239000000018 receptor agonist Substances 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- 210000004989 spleen cell Anatomy 0.000 description 4
- HWXVDEAMULKKDC-UHFFFAOYSA-N (4-phenylmethoxyphenyl) n-butylcarbamate Chemical compound C1=CC(OC(=O)NCCCC)=CC=C1OCC1=CC=CC=C1 HWXVDEAMULKKDC-UHFFFAOYSA-N 0.000 description 3
- XOHZBPMRSYEDHU-UHFFFAOYSA-N 3-(3-methoxyphenyl)benzamide Chemical compound COC1=CC=CC(C=2C=C(C=CC=2)C(N)=O)=C1 XOHZBPMRSYEDHU-UHFFFAOYSA-N 0.000 description 3
- STXAVEHFKAXGOX-UHFFFAOYSA-N 3-bromobenzonitrile Chemical compound BrC1=CC=CC(C#N)=C1 STXAVEHFKAXGOX-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 206010002869 Anxiety symptoms Diseases 0.000 description 3
- 206010003571 Astrocytoma Diseases 0.000 description 3
- 208000032841 Bulimia Diseases 0.000 description 3
- 206010006550 Bulimia nervosa Diseases 0.000 description 3
- 108010053652 Butyrylcholinesterase Proteins 0.000 description 3
- 0 C1C2C3=C*3CC12 Chemical compound C1C2C3=C*3CC12 0.000 description 3
- 102100033868 Cannabinoid receptor 1 Human genes 0.000 description 3
- 101710187010 Cannabinoid receptor 1 Proteins 0.000 description 3
- 102100036214 Cannabinoid receptor 2 Human genes 0.000 description 3
- 101710187022 Cannabinoid receptor 2 Proteins 0.000 description 3
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical group NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 3
- 102100032404 Cholinesterase Human genes 0.000 description 3
- 229920002261 Corn starch Polymers 0.000 description 3
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 3
- 206010012335 Dependence Diseases 0.000 description 3
- 206010054089 Depressive symptom Diseases 0.000 description 3
- 102000015554 Dopamine receptor Human genes 0.000 description 3
- 108050004812 Dopamine receptor Proteins 0.000 description 3
- 208000011688 Generalised anxiety disease Diseases 0.000 description 3
- 102000004157 Hydrolases Human genes 0.000 description 3
- 108090000604 Hydrolases Proteins 0.000 description 3
- 208000021384 Obsessive-Compulsive disease Diseases 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 229940124639 Selective inhibitor Drugs 0.000 description 3
- 208000031674 Traumatic Acute Stress disease Diseases 0.000 description 3
- 238000009825 accumulation Methods 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 230000006978 adaptation Effects 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 150000005347 biaryls Chemical group 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 125000002837 carbocyclic group Chemical group 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 239000007795 chemical reaction product Substances 0.000 description 3
- 239000008120 corn starch Substances 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- KQWGXHWJMSMDJJ-UHFFFAOYSA-N cyclohexyl isocyanate Chemical compound O=C=NC1CCCCC1 KQWGXHWJMSMDJJ-UHFFFAOYSA-N 0.000 description 3
- 230000002354 daily effect Effects 0.000 description 3
- 239000007884 disintegrant Substances 0.000 description 3
- 230000006397 emotional response Effects 0.000 description 3
- 230000007613 environmental effect Effects 0.000 description 3
- 239000000796 flavoring agent Substances 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 3
- 208000029364 generalized anxiety disease Diseases 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 230000002631 hypothermal effect Effects 0.000 description 3
- 229960004801 imipramine Drugs 0.000 description 3
- BCGWQEUPMDMJNV-UHFFFAOYSA-N imipramine Chemical compound C1CC2=CC=CC=C2N(CCCN(C)C)C2=CC=CC=C21 BCGWQEUPMDMJNV-UHFFFAOYSA-N 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 239000007928 intraperitoneal injection Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 244000005700 microbiome Species 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 230000036651 mood Effects 0.000 description 3
- 230000037023 motor activity Effects 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 238000001543 one-way ANOVA Methods 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 230000035945 sensitivity Effects 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 125000004434 sulfur atom Chemical group 0.000 description 3
- 230000009897 systematic effect Effects 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- HQVHOQAKMCMIIM-UHFFFAOYSA-N win 55,212-2 Chemical compound C=12N3C(C)=C(C(=O)C=4C5=CC=CC=C5C=CC=4)C2=CC=CC=1OCC3CN1CCOCC1 HQVHOQAKMCMIIM-UHFFFAOYSA-N 0.000 description 3
- LMTWQLGAIBXCTE-UHFFFAOYSA-N (2-phenylphenyl) carbamate Chemical class NC(=O)OC1=CC=CC=C1C1=CC=CC=C1 LMTWQLGAIBXCTE-UHFFFAOYSA-N 0.000 description 2
- DSVGFKBFFICWLZ-UHFFFAOYSA-N 1-fluoro-4-isocyanatobenzene Chemical compound FC1=CC=C(N=C=O)C=C1 DSVGFKBFFICWLZ-UHFFFAOYSA-N 0.000 description 2
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 2
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 2
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 2
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 2
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 2
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 2
- DPZMVZIQRMVBBW-UHFFFAOYSA-N 5-Phenyl-1-pentanol Chemical compound OCCCCCC1=CC=CC=C1 DPZMVZIQRMVBBW-UHFFFAOYSA-N 0.000 description 2
- IGGTVXYIQRVVOP-UHFFFAOYSA-N 5-phenylpentyl n-cyclohexylcarbamate Chemical compound C=1C=CC=CC=1CCCCCOC(=O)NC1CCCCC1 IGGTVXYIQRVVOP-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 241000218236 Cannabis Species 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 101800001982 Cholecystokinin Proteins 0.000 description 2
- 102100025841 Cholecystokinin Human genes 0.000 description 2
- 206010011971 Decreased interest Diseases 0.000 description 2
- 206010012239 Delusion Diseases 0.000 description 2
- SUZLHDUTVMZSEV-UHFFFAOYSA-N Deoxycoleonol Natural products C12C(=O)CC(C)(C=C)OC2(C)C(OC(=O)C)C(O)C2C1(C)C(O)CCC2(C)C SUZLHDUTVMZSEV-UHFFFAOYSA-N 0.000 description 2
- 206010012374 Depressed mood Diseases 0.000 description 2
- 206010016275 Fear Diseases 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 206010024264 Lethargy Diseases 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- 240000007472 Leucaena leucocephala Species 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 241000700157 Rattus norvegicus Species 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- HQVHOQAKMCMIIM-HXUWFJFHSA-N WIN 55212-2 Chemical compound C([C@@H]1COC=2C=CC=C3C(C(=O)C=4C5=CC=CC=C5C=CC=4)=C(N1C3=2)C)N1CCOCC1 HQVHOQAKMCMIIM-HXUWFJFHSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 208000005298 acute pain Diseases 0.000 description 2
- 208000026345 acute stress disease Diseases 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- 239000012790 adhesive layer Substances 0.000 description 2
- 238000013019 agitation Methods 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 210000004727 amygdala Anatomy 0.000 description 2
- 239000002269 analeptic agent Substances 0.000 description 2
- 230000001773 anti-convulsant effect Effects 0.000 description 2
- 229960003965 antiepileptics Drugs 0.000 description 2
- 230000007529 anxiety like behavior Effects 0.000 description 2
- 235000021407 appetite control Nutrition 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 229940114079 arachidonic acid Drugs 0.000 description 2
- 235000021342 arachidonic acid Nutrition 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000002238 attenuated effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 230000005540 biological transmission Effects 0.000 description 2
- UBXYXCRCOKCZIT-UHFFFAOYSA-N biphenyl-3-ol Chemical compound OC1=CC=CC(C=2C=CC=CC=2)=C1 UBXYXCRCOKCZIT-UHFFFAOYSA-N 0.000 description 2
- 125000000319 biphenyl-4-yl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 2
- 208000028683 bipolar I disease Diseases 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 239000003555 cannabinoid 1 receptor antagonist Substances 0.000 description 2
- 229960005286 carbaryl Drugs 0.000 description 2
- CVXBEEMKQHEXEN-UHFFFAOYSA-N carbaryl Chemical compound C1=CC=C2C(OC(=O)NC)=CC=CC2=C1 CVXBEEMKQHEXEN-UHFFFAOYSA-N 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 2
- 229940107137 cholecystokinin Drugs 0.000 description 2
- 238000011260 co-administration Methods 0.000 description 2
- ZPUCINDJVBIVPJ-LJISPDSOSA-N cocaine Chemical compound O([C@H]1C[C@@H]2CC[C@@H](N2C)[C@H]1C(=O)OC)C(=O)C1=CC=CC=C1 ZPUCINDJVBIVPJ-LJISPDSOSA-N 0.000 description 2
- OHCQJHSOBUTRHG-UHFFFAOYSA-N colforsin Natural products OC12C(=O)CC(C)(C=C)OC1(C)C(OC(=O)C)C(O)C1C2(C)C(O)CCC1(C)C OHCQJHSOBUTRHG-UHFFFAOYSA-N 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 230000002596 correlated effect Effects 0.000 description 2
- 235000019788 craving Nutrition 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 206010061428 decreased appetite Diseases 0.000 description 2
- 231100000868 delusion Toxicity 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 2
- 229960003529 diazepam Drugs 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 230000005593 dissociations Effects 0.000 description 2
- 239000012153 distilled water Substances 0.000 description 2
- 238000009509 drug development Methods 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 208000024732 dysthymic disease Diseases 0.000 description 2
- 201000003104 endogenous depression Diseases 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- 238000007824 enzymatic assay Methods 0.000 description 2
- 239000002532 enzyme inhibitor Substances 0.000 description 2
- 229940125532 enzyme inhibitor Drugs 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 238000005194 fractionation Methods 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 230000005660 hydrophilic surface Effects 0.000 description 2
- 230000002779 inactivation Effects 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 230000002045 lasting effect Effects 0.000 description 2
- 238000000670 ligand binding assay Methods 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- KNPNGMXRGITFLE-UHFFFAOYSA-N methylperoxy(phenyl)borinic acid Chemical compound COOB(O)C1=CC=CC=C1 KNPNGMXRGITFLE-UHFFFAOYSA-N 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- 230000003278 mimic effect Effects 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000000302 molecular modelling Methods 0.000 description 2
- VYQNWZOUAUKGHI-UHFFFAOYSA-N monobenzone Chemical compound C1=CC(O)=CC=C1OCC1=CC=CC=C1 VYQNWZOUAUKGHI-UHFFFAOYSA-N 0.000 description 2
- 125000004572 morpholin-3-yl group Chemical group N1C(COCC1)* 0.000 description 2
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 2
- 125000002757 morpholinyl group Chemical group 0.000 description 2
- HNHVTXYLRVGMHD-UHFFFAOYSA-N n-butyl isocyanate Chemical compound CCCCN=C=O HNHVTXYLRVGMHD-UHFFFAOYSA-N 0.000 description 2
- 230000003040 nociceptive effect Effects 0.000 description 2
- 230000009871 nonspecific binding Effects 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 239000006186 oral dosage form Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 230000008058 pain sensation Effects 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000002085 persistent effect Effects 0.000 description 2
- 239000002831 pharmacologic agent Substances 0.000 description 2
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 229920001592 potato starch Polymers 0.000 description 2
- 244000062645 predators Species 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 238000001525 receptor binding assay Methods 0.000 description 2
- 238000000611 regression analysis Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- IZTQOLKUZKXIRV-YRVFCXMDSA-N sincalide Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](N)CC(O)=O)C1=CC=C(OS(O)(=O)=O)C=C1 IZTQOLKUZKXIRV-YRVFCXMDSA-N 0.000 description 2
- 229960001922 sodium perborate Drugs 0.000 description 2
- YKLJGMBLPUQQOI-UHFFFAOYSA-M sodium;oxidooxy(oxo)borane Chemical compound [Na+].[O-]OB=O YKLJGMBLPUQQOI-UHFFFAOYSA-M 0.000 description 2
- 201000001716 specific phobia Diseases 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000012258 stirred mixture Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 208000011117 substance-related disease Diseases 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- 125000005309 thioalkoxy group Chemical group 0.000 description 2
- 230000001256 tonic effect Effects 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 239000011800 void material Substances 0.000 description 2
- 230000009184 walking Effects 0.000 description 2
- 238000005303 weighing Methods 0.000 description 2
- 235000019786 weight gain Nutrition 0.000 description 2
- 230000004584 weight gain Effects 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 1
- SNICXCGAKADSCV-JTQLQIEISA-N (-)-Nicotine Chemical compound CN1CCC[C@H]1C1=CC=CN=C1 SNICXCGAKADSCV-JTQLQIEISA-N 0.000 description 1
- QMGHHBHPDDAGGO-IIWOMYBWSA-N (2S,4R)-1-[(2S)-2-[[2-[3-[4-[3-[4-[[5-bromo-4-[3-[cyclobutanecarbonyl(methyl)amino]propylamino]pyrimidin-2-yl]amino]phenoxy]propoxy]butoxy]propoxy]acetyl]amino]-3,3-dimethylbutanoyl]-4-hydroxy-N-[[4-(4-methyl-1,3-thiazol-5-yl)phenyl]methyl]pyrrolidine-2-carboxamide Chemical compound CN(CCCNC1=NC(NC2=CC=C(OCCCOCCCCOCCCOCC(=O)N[C@H](C(=O)N3C[C@H](O)C[C@H]3C(=O)NCC3=CC=C(C=C3)C3=C(C)N=CS3)C(C)(C)C)C=C2)=NC=C1Br)C(=O)C1CCC1 QMGHHBHPDDAGGO-IIWOMYBWSA-N 0.000 description 1
- NLLGFYPSWCMUIV-UHFFFAOYSA-N (3-methoxyphenyl)boronic acid Chemical compound COC1=CC=CC(B(O)O)=C1 NLLGFYPSWCMUIV-UHFFFAOYSA-N 0.000 description 1
- BBRKGHBKCGBRNK-UHFFFAOYSA-N (4-fluorophenyl) n-butylcarbamate Chemical compound CCCCNC(=O)OC1=CC=C(F)C=C1 BBRKGHBKCGBRNK-UHFFFAOYSA-N 0.000 description 1
- FOGUWHRMZYPNRG-UHFFFAOYSA-N (4-methoxyphenyl) 4-butoxycarbonyloxybenzoate Chemical compound C1=CC(OC(=O)OCCCC)=CC=C1C(=O)OC1=CC=C(OC)C=C1 FOGUWHRMZYPNRG-UHFFFAOYSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- FHUDAMLDXFJHJE-UHFFFAOYSA-N 1,1,1-trifluoropropan-2-one Chemical compound CC(=O)C(F)(F)F FHUDAMLDXFJHJE-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- 125000000196 1,4-pentadienyl group Chemical group [H]C([*])=C([H])C([H])([H])C([H])=C([H])[H] 0.000 description 1
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- RCRCTBLIHCHWDZ-DOFZRALJSA-N 2-arachidonoylglycerol Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(=O)OC(CO)CO RCRCTBLIHCHWDZ-DOFZRALJSA-N 0.000 description 1
- 125000004174 2-benzimidazolyl group Chemical group [H]N1C(*)=NC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical group [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 208000036864 Attention deficit/hyperactivity disease Diseases 0.000 description 1
- 206010063659 Aversion Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 229940122226 Benzodiazepine receptor agonist Drugs 0.000 description 1
- ROFVEXUMMXZLPA-UHFFFAOYSA-N Bipyridyl Chemical group N1=CC=CC=C1C1=CC=CC=N1 ROFVEXUMMXZLPA-UHFFFAOYSA-N 0.000 description 1
- 208000031872 Body Remains Diseases 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 208000036632 Brain mass Diseases 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229940123158 Cannabinoid CB1 receptor antagonist Drugs 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- 102000003914 Cholinesterases Human genes 0.000 description 1
- 108090000322 Cholinesterases Proteins 0.000 description 1
- 208000019888 Circadian rhythm sleep disease Diseases 0.000 description 1
- 206010010144 Completed suicide Diseases 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 206010011469 Crying Diseases 0.000 description 1
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- 208000007590 Disorders of Excessive Somnolence Diseases 0.000 description 1
- 229940121891 Dopamine receptor antagonist Drugs 0.000 description 1
- 206010013654 Drug abuse Diseases 0.000 description 1
- 238000001061 Dunnett's test Methods 0.000 description 1
- 206010013954 Dysphoria Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 241001539473 Euphoria Species 0.000 description 1
- 206010015535 Euphoric mood Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 208000004454 Hyperalgesia Diseases 0.000 description 1
- 208000035154 Hyperesthesia Diseases 0.000 description 1
- 206010020710 Hyperphagia Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 238000012404 In vitro experiment Methods 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 208000001456 Jet Lag Syndrome Diseases 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- KWKZCGMJGHHOKJ-ZKWNWVNESA-N Methyl Arachidonyl Fluorophosphonate Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCCP(F)(=O)OC KWKZCGMJGHHOKJ-ZKWNWVNESA-N 0.000 description 1
- 208000007101 Muscle Cramp Diseases 0.000 description 1
- HOKKHZGPKSLGJE-GSVOUGTGSA-N N-Methyl-D-aspartic acid Chemical compound CN[C@@H](C(O)=O)CC(O)=O HOKKHZGPKSLGJE-GSVOUGTGSA-N 0.000 description 1
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 1
- 108010084810 Neurotransmitter Transport Proteins Proteins 0.000 description 1
- 102000005665 Neurotransmitter Transport Proteins Human genes 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- 102000019315 Nicotinic acetylcholine receptors Human genes 0.000 description 1
- 108050006807 Nicotinic acetylcholine receptors Proteins 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 229910003849 O-Si Inorganic materials 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 241000283283 Orcinus orca Species 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 229910003872 O—Si Inorganic materials 0.000 description 1
- 206010033664 Panic attack Diseases 0.000 description 1
- CWRVKFFCRWGWCS-UHFFFAOYSA-N Pentrazole Chemical compound C1CCCCC2=NN=NN21 CWRVKFFCRWGWCS-UHFFFAOYSA-N 0.000 description 1
- 229920005439 Perspex® Polymers 0.000 description 1
- PIJVFDBKTWXHHD-UHFFFAOYSA-N Physostigmine Natural products C12=CC(OC(=O)NC)=CC=C2N(C)C2C1(C)CCN2C PIJVFDBKTWXHHD-UHFFFAOYSA-N 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 206010039897 Sedation Diseases 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 208000005392 Spasm Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 208000013200 Stress disease Diseases 0.000 description 1
- 238000000692 Student's t-test Methods 0.000 description 1
- 208000007271 Substance Withdrawal Syndrome Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 206010042458 Suicidal ideation Diseases 0.000 description 1
- 206010042464 Suicide attempt Diseases 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 206010043169 Tearfulness Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 206010044565 Tremor Diseases 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- IVOMOUWHDPKRLL-UHFFFAOYSA-N UNPD107823 Natural products O1C2COP(O)(=O)OC2C(O)C1N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-UHFFFAOYSA-N 0.000 description 1
- BWVSKWYOFNUYPK-DOFZRALJSA-N [(5Z,8Z,11Z,14Z)-icosa-5,8,11,14-tetraenyl]phosphonic acid Chemical compound CCCCC/C=C\C/C=C\C/C=C\C/C=C\CCCCP(=O)(O)O BWVSKWYOFNUYPK-DOFZRALJSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 102000030621 adenylate cyclase Human genes 0.000 description 1
- 108060000200 adenylate cyclase Proteins 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000001800 adrenalinergic effect Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000008484 agonism Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 125000005237 alkyleneamino group Chemical group 0.000 description 1
- 125000005238 alkylenediamino group Chemical group 0.000 description 1
- 125000005530 alkylenedioxy group Chemical group 0.000 description 1
- 125000005529 alkyleneoxy group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- IJBZOOZRAXHERC-DOFZRALJSA-N am404 Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(=O)NC1=CC=C(O)C=C1 IJBZOOZRAXHERC-DOFZRALJSA-N 0.000 description 1
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 1
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 1
- 230000036592 analgesia Effects 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 230000001539 anorectic effect Effects 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 230000000049 anti-anxiety effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000000561 anti-psychotic effect Effects 0.000 description 1
- 230000002932 anti-schizophrenic effect Effects 0.000 description 1
- 229940053200 antiepileptics fatty acid derivative Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000000164 antipsychotic agent Substances 0.000 description 1
- 239000002948 appetite stimulant Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 125000002886 arachidonoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])/C([H])=C([H])\C([H])([H])/C([H])=C([H])\C([H])([H])/C([H])=C([H])\C([H])([H])/C([H])=C([H])\C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 230000035045 associative learning Effects 0.000 description 1
- 208000015802 attention deficit-hyperactivity disease Diseases 0.000 description 1
- 210000003050 axon Anatomy 0.000 description 1
- 239000000022 bacteriostatic agent Substances 0.000 description 1
- 210000004227 basal ganglia Anatomy 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 239000000759 benzodiazepine receptor stimulating agent Substances 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 238000010256 biochemical assay Methods 0.000 description 1
- 230000003851 biochemical process Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229960002495 buspirone Drugs 0.000 description 1
- QWCRAEMEVRGPNT-UHFFFAOYSA-N buspirone Chemical compound C1C(=O)N(CCCCN2CCN(CC2)C=2N=CC=CN=2)C(=O)CC21CCCC2 QWCRAEMEVRGPNT-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 230000001593 cAMP accumulation Effects 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 239000003554 cannabinoid 1 receptor agonist Substances 0.000 description 1
- 229940065144 cannabinoids Drugs 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 238000012569 chemometric method Methods 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 239000007958 cherry flavor Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- WORJEOGGNQDSOE-UHFFFAOYSA-N chloroform;methanol Chemical compound OC.ClC(Cl)Cl WORJEOGGNQDSOE-UHFFFAOYSA-N 0.000 description 1
- 229940048961 cholinesterase Drugs 0.000 description 1
- 239000000544 cholinesterase inhibitor Substances 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229960003920 cocaine Drugs 0.000 description 1
- 238000009225 cognitive behavioral therapy Methods 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 230000001143 conditioned effect Effects 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000003179 convulsant agent Substances 0.000 description 1
- 230000036461 convulsion Effects 0.000 description 1
- 230000002920 convulsive effect Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229940125808 covalent inhibitor Drugs 0.000 description 1
- 229940095074 cyclic amp Drugs 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 210000000172 cytosol Anatomy 0.000 description 1
- 230000009849 deactivation Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000003001 depressive effect Effects 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 239000010432 diamond Substances 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 235000021061 dietary behavior Nutrition 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 238000003113 dilution method Methods 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 239000003210 dopamine receptor blocking agent Substances 0.000 description 1
- 239000003651 drinking water Substances 0.000 description 1
- 235000020188 drinking water Nutrition 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 235000006694 eating habits Nutrition 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 210000002257 embryonic structure Anatomy 0.000 description 1
- 230000010482 emotional regulation Effects 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 150000002085 enols Chemical group 0.000 description 1
- 229960004756 ethanol Drugs 0.000 description 1
- 150000002169 ethanolamines Chemical class 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 230000021824 exploration behavior Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 230000008921 facial expression Effects 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000010685 fatty oil Substances 0.000 description 1
- 230000035558 fertility Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 210000005153 frontal cortex Anatomy 0.000 description 1
- 238000011990 functional testing Methods 0.000 description 1
- 230000003371 gabaergic effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000004518 granules dosage form Substances 0.000 description 1
- 210000004884 grey matter Anatomy 0.000 description 1
- 210000004326 gyrus cinguli Anatomy 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 1
- PXKAHSXZPDMGBF-UHFFFAOYSA-N hexyl n-cyclohexylcarbamate Chemical compound CCCCCCOC(=O)NC1CCCCC1 PXKAHSXZPDMGBF-UHFFFAOYSA-N 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 150000001469 hydantoins Chemical class 0.000 description 1
- 239000004093 hydrolase inhibitor Substances 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 206010020765 hypersomnia Diseases 0.000 description 1
- 230000000147 hypnotic effect Effects 0.000 description 1
- 238000007654 immersion Methods 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 239000005414 inactive ingredient Substances 0.000 description 1
- 206010021654 increased appetite Diseases 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 210000001153 interneuron Anatomy 0.000 description 1
- 230000010039 intracellular degradation Effects 0.000 description 1
- 229940125425 inverse agonist Drugs 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 208000033915 jet lag type circadian rhythm sleep disease Diseases 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 238000012933 kinetic analysis Methods 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 238000005567 liquid scintillation counting Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000006742 locomotor activity Effects 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 206010025482 malaise Diseases 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000028161 membrane depolarization Effects 0.000 description 1
- 230000003340 mental effect Effects 0.000 description 1
- 210000001259 mesencephalon Anatomy 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 229960001785 mirtazapine Drugs 0.000 description 1
- RONZAEMNMFQXRA-UHFFFAOYSA-N mirtazapine Chemical compound C1C2=CC=CN=C2N2CCN(C)CC2C2=CC=CC=C21 RONZAEMNMFQXRA-UHFFFAOYSA-N 0.000 description 1
- 230000000116 mitigating effect Effects 0.000 description 1
- 238000000329 molecular dynamics simulation Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 125000006682 monohaloalkyl group Chemical group 0.000 description 1
- 230000002969 morbid Effects 0.000 description 1
- 125000004312 morpholin-2-yl group Chemical group [H]N1C([H])([H])C([H])([H])OC([H])(*)C1([H])[H] 0.000 description 1
- 230000008450 motivation Effects 0.000 description 1
- 230000003551 muscarinic effect Effects 0.000 description 1
- ZZHGIUCYKGFIPV-UHFFFAOYSA-M n-butylcarbamate Chemical compound CCCCNC([O-])=O ZZHGIUCYKGFIPV-UHFFFAOYSA-M 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 208000004296 neuralgia Diseases 0.000 description 1
- 230000001722 neurochemical effect Effects 0.000 description 1
- 230000006764 neuronal dysfunction Effects 0.000 description 1
- 208000021722 neuropathic pain Diseases 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229960002715 nicotine Drugs 0.000 description 1
- SNICXCGAKADSCV-UHFFFAOYSA-N nicotine Natural products CN1CCCC1C1=CC=CN=C1 SNICXCGAKADSCV-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 229940127240 opiate Drugs 0.000 description 1
- 239000007968 orange flavor Substances 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 235000020830 overeating Nutrition 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 1
- 229960005152 pentetrazol Drugs 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000008447 perception Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- PIJVFDBKTWXHHD-HIFRSBDPSA-N physostigmine Chemical compound C12=CC(OC(=O)NC)=CC=C2N(C)[C@@H]2[C@@]1(C)CCN2C PIJVFDBKTWXHHD-HIFRSBDPSA-N 0.000 description 1
- 229960001697 physostigmine Drugs 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 1
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 125000006684 polyhaloalkyl group Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 208000028173 post-traumatic stress disease Diseases 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 210000004129 prosencephalon Anatomy 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 238000001671 psychotherapy Methods 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000005344 pyridylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 239000002287 radioligand Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000009183 running Effects 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 238000009738 saturating Methods 0.000 description 1
- 230000000698 schizophrenic effect Effects 0.000 description 1
- 230000036280 sedation Effects 0.000 description 1
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 description 1
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 230000003997 social interaction Effects 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 230000002739 subcortical effect Effects 0.000 description 1
- 201000009032 substance abuse Diseases 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000009182 swimming Effects 0.000 description 1
- 230000015883 synaptic transmission, dopaminergic Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 238000012353 t test Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 230000008448 thought Effects 0.000 description 1
- 230000036962 time dependent Effects 0.000 description 1
- KJAMZCVTJDTESW-UHFFFAOYSA-N tiracizine Chemical compound C1CC2=CC=CC=C2N(C(=O)CN(C)C)C2=CC(NC(=O)OCC)=CC=C21 KJAMZCVTJDTESW-UHFFFAOYSA-N 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- 208000037816 tissue injury Diseases 0.000 description 1
- 238000012876 topography Methods 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 238000013271 transdermal drug delivery Methods 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 238000011680 zucker rat Methods 0.000 description 1
Images





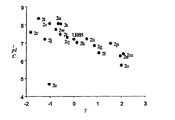


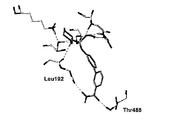
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/22—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms
- C07C311/29—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms having the sulfur atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/40—Esters of carbamic acids having oxygen atoms of carbamate groups bound to carbon atoms of six-membered aromatic rings
- C07C271/56—Esters of carbamic acids having oxygen atoms of carbamate groups bound to carbon atoms of six-membered aromatic rings with the nitrogen atom of at least one of the carbamate groups bound to a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/46—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups containing any of the groups, X being a hetero atom, Y being any atom, e.g. acylureas
- C07C275/48—Y being a hydrogen or a carbon atom
- C07C275/54—Y being a carbon atom of a six-membered aromatic ring, e.g. benzoylureas
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/04—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to the ring carbon atoms
- C07D215/06—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to the ring carbon atoms having only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/78—Benzo [b] furans; Hydrogenated benzo [b] furans
- C07D307/79—Benzo [b] furans; Hydrogenated benzo [b] furans with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Ophthalmology & Optometry (AREA)
- Psychiatry (AREA)
- Child & Adolescent Psychology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Diabetes (AREA)
- Anesthesiology (AREA)
- Epidemiology (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Indole Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Furan Compounds (AREA)
Description
本研究は、部分的に、米国国立衛生研究所が授与した認可番号DA12413、DA12447、およびDA12653の下、政府の支援で行われた。政府は、本発明について、一定の権利を有する。
本願は、2002年10月7日付けで出願された米国特許仮出願第60/417,008号の恩典を主張するものである。本願は同様に、2003年3月27日付けで出願された米国特許出願第10/112,509号および2003年8月15日付けで出願された米国特許出願第10/642,462号に関する。これらのそれぞれの開示は、その全体が組み入れられる。
不安は、病理学的には恐怖の一種(counterpart)であり、気分、思考、行動、および生理機能の障害を伴うことが多い。恐怖は通常、環境中の脅威の認識により引き起こされるが、不安障害は通常、環境からの脅威により刺激されたのではない恐怖または環境からの脅威に極めて不釣り合いな恐怖に伴って生じる。
本発明により、脂肪酸アミドヒドロラーゼ(FAAH)を阻害するための新規化合物ならびにFAAH阻害剤を被検体に投与することによる、不安もしくは疼痛、および他の神経性の、睡眠、または心理的障害の処置方法、睡眠を誘発するための方法、緑内障を処置するための方法、および食欲を制御するかまたは食欲障害を処置するための方法が提供される。その局面の一つとして、本発明により、不安および抑うつを処置するのに有用なFAAH阻害剤の使用が開示される。その局面の別のものとして、本発明により、下記式のFAAH阻害化合物が提供される:

からなる群より選択される芳香族部分またはアルキル部分または親油性部分である。
ある局面として、本発明により、脂肪酸アミドヒドロラーゼ(FAAH)、つまり内在性カンナビノイドのアナンダミドの細胞内分解に関与する酵素の新規阻害剤が提供される。本発明者らは、驚いたことに、FAAHをインビボにおいて低いIC50で阻害する化合物を発見した。本発明による典型的な化合物は、強力で、選択的で、全身活性なFAAH阻害剤とすることができる。FAAH阻害剤は、睡眠の誘発、不眠症の処置、および疼痛の緩和のような種々の目的に有用とすることができる。本発明により同様に、FAAH阻害剤の投与による不安の処置方法が提供される。臨床上用いられる抗不安薬と同様に、そのような阻害剤は、驚いたことに、高架式のゼロの迷路試験でベンゾジアゼピン様の特性を示し、ラットで、孤立による超音波発生を抑制する。さらに、それらの阻害剤は、急性疼痛のモデルで生体防御(疼痛回避)行動を低下する。これらの作用は、脳のアナンダミドの量が増加する(しかし、他の内在性カンナビノイドである2-アラキドノイルグリセロールの量は増加しない)ことに伴って生じており、CB1カンナビノイド受容体の遮断により阻止または拮抗される。この結果は、アナンダミドが情動状態の調節に関与することを示しており、FAAH阻害を抗不安療法に対する革新的な機構的研究法として暗示している。
本明細書では、単数形「一つの(a)」、「一つの(an)」、および「その(the)」には、文脈により他に明記されていない場合、複数対象が含まれることに留意されたい。
処置が施されるべきヒトを含めた哺乳類(例えば、ラット、マウス、ネコ、イヌ)を含む、任意の動物が含まれる。
本発明の化合物は、一つまたは複数の不斉中心を含むことができ、および従って、ラセミ化合物およびラセミ混合物、単一の鏡像異性体、偏左右異性体混合物ならびに個々の偏左右異性体として生じることができる。本発明には、本発明の化合物のそのような異性体の全てが包含されるものと意図される。
本発明により、次式の脂肪酸アミドヒドロラーゼ阻害剤が提供される:
からなる群より選択される芳香族部分またはアルキル部分または親油性部分である。Z1が-C(R6)=C(R7)-または-N=C(R6)-であり、およびpが0である場合、Z2が一員となっている芳香環は、Yに対してメタまたはパラ位にあることが好ましい。その位置は、メタ位であることがより好ましい。
からなる群より選択される。Yは、結合としてもよくまたは-O-、-S-、-N(R5)-、-S(R5)-、-C1〜C4アルキレン、(Z)-エチレンもしくは(E)-エチレン、および炭素原子3〜6個のシクロアルキルからなる群より選択されてもよく; 各Raおよび各Rbは、H、アルキル、ヘテロアルキル、アルケニル、アルキニル、シクロアルキル、アリール、置換アリール、アリールアルキル、置換アリールアルキル、ケトアルキル、ヒドロキシアルキル、アミノアルキル、-CH2-NR3R4、アルコキシ、アリールオキシ、アリールアルキルオキシ、ハロ、ハロアルキル、シアノ、ヒドロキシ、ニトロ、アミノ、-NR3R4、-SR5、カルボキサミド、-CONR3R4、-O-カルボキサミド、-O-CO-NR3R4、スルホンアミド、および-SO2NR3R4からなる群より独立して選択され、ここでR3およびR4は、H、アルキル、アルケニル、アルキニル、シクロアルキル、ヒドロキシアルキルおよびイミノ-メチルアミノより選択され、ならびに任意でR3およびR4は、これらが結合するN原子と共に5〜7員環を形成し; ならびにR1およびR2は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換シクロアルキル、および置換または非置換シクロヘテロアルキルからなる群より独立して選択され、および任意で、R1およびR2は、これらが結合するN原子と共に、置換または非置換環を形成してもよい。
。
本発明の化合物は、単純な化学反応を利用して、市販の出発物質で作り出すことができる。カルバメートは当技術分野においてよく知られている。以下の手順は典型的な合成経路であって、これは本発明を説明するが、限定するものではないことが意図される。当業者であれば、他の変形、変更、および代替を認識するであろう。
FAAH阻害活性を目的とした化合物のインビトロ・スクリーニング方法は、当業者によく知られている。そのような方法は、Quistand et al. in Toxicology and Applied Pharmacology 179: 57-63 (2002); Quistand et al. in Toxicology and Applied Pharmacology 173, 48-55 (2001); Boger et al., Proc. Natl. Acad. Sci. U.S.A. 97, 5044-49 (2000)に教示されている。
当業者であれば、ACHEまたはNTEに対する阻害効果を目的に物質をスクリーニングする方法を知っているものと思われる。例えば、Quistand et al. in Toxicology and Applied Pharmacology 179, 57-63 (2002); およびQuistand et al. in Toxicology and Applied Pharmacology 173, 48-55 (2001)を参照されたい。
本発明による化合物を同定するために、種々の方法を使用して、カンナビノイドCB1受容体結合活性をスクリーニングすることができる。種々のそのような方法は、U.S. Patent No.5,747,524およびU.S. Patent No.6,017,919に教示されている。
CB2受容体結合の研究方法は、当業者によく知られている。例えば、ヒトカンナビノイドCB2受容体との結合は、Showalter, et al., J. Pharmacol Exp Ther. 278(3), 989-99 (1996)の手順を用い、例えば、2002年2月28日に公開されたU.S. Patent Application No. 20020026050に教示されるように若干修正を加えて評価することができる。これらのそれぞれが参照として本明細書に組み入れられる。
最近になって、新たな化合物リードの創出の補助となるコンビナトリアル化学ライブラリの使用に注目が集まっている。コンビナトリアル化学ライブラリは、化学合成または生物合成のいずれかにより、試薬のような多数の化学的「基礎単位」を組み合わせることによって作製される多様な化合物群である。例えば、ポリペプチドライブラリのような直鎖状のコンビナトリアル化学ライブラリは、アミノ酸と呼ばれる一連の化学的基礎単位をあらゆる可能な方法で所定の化合物の長さ(すなわち、ポリペプチド化合物中のアミノ酸の数)に組み合わせることにより形成される。何百万もの化合物を化学的基礎単位のそのようなコンビナトリアル混合によって合成することができる。例えば、ある業界解説者によれば、100種類の可換性の化学的基礎単位の体系的なコンビナトリアル混合により、結果的に1億種類の四量体化合物または100億種類の五量体化合物の理論的合成にいたると観測されている(Gallop et al., J. Med. Chem. 37(9), 1233(1994))。
本明細書に記述した化合物のアッセイは、高速大量処理スクリーニングに従順である。従って、好ましいアッセイでは、FAAHとの阻害剤の結合、またはオレオイルエタノールアミドもしくはアナンダミドのような基質の加水分解により産生された反応産物(例えば、脂肪酸アミドまたはエタノールアミン)の放出が検出される。基質を標識して、放出された反応産物の検出を容易にすることができる。特定の反応産物の存在、非存在または定量化を目的とした高速大量処理アッセイは、当業者によく知られている。このように、例えば、U.S. Patent 5,559,410には、タンパク質に対する高速大量処理スクリーニング法が開示されており、ならびにU.S. Patent 5,576,220および5,541,061には、リガンド/抗体の結合に対する高速大量処理スクリーニング法が開示されている。
当業者であれば、化合物の抗不安作用を評価するのに利用できる多くの動物モデルが存在することを理解しているものと思われる。薬理学的に実証された不安に関する二つの動物モデルは、高架式のゼロの迷路試験、および孤立による超音波発生試験である。ゼロの迷路は、2つの開放四分円および2つの閉鎖四分円を有する高架式の環状形状のプラットホームからなり、その環境を探索する動物の本能と開放的な空間(ここでは、その動物は、捕食者により攻撃される可能性がある)のその恐怖との間の葛藤に基づいている(Bickerdike, M.J. et al., Eur. J. Pharmacol., 271, 403-411 (1994); Shepherd, J.K. et al., Psychopharmacology, 116, 56-64 (1994))。ベンゾジアゼピンのような、臨床上用いられる抗不安薬により、開放区画で過ごす時間の割合、および開放区画への侵入の数が増す。
抑うつの動物モデルが同様に、当業者によく知られている。例えば、抑うつの処置における本発明の化合物の効果は、慢性の軽いストレスによるラット無快感症モデルで試験することができる。このモデルは、慢性の軽いストレスにより褒美、例えば、ショ糖の消費に対する感受性の漸減が引き起こされるという観測結果、およびこの減少は、抗うつ剤を用いた慢性処置により用量依存的に食い止められるという観測結果に基づいている。この方法は以前に報告されており、この試験に関するさらなる情報は、Willner, Paul, Psychopharmacology, 1997, 134, 319-329から明らかになる。
当業者が試験化合物の抗けいれん活性を研究するのに、動物モデルを利用できる。例えば、そのような試験を実施するための方法を記述している、U.S. Patent No. 6,309,406およびU.S. Patent No. 6,326,156を参照されたい。さらに、化合物をてんかんまたは他のけいれん症状を患うヒトに投与して、けいれんの頻度または重症度または発症に対する効果を臨床的に評価することができる。
FAAHの阻害により、試験動物で睡眠が誘発されることが報告されている(U.S. Patent No. 6,096,784)。睡眠誘発化合物の研究方法は、当業者によく知られている。特に、試験用のFAAH阻害化合物が睡眠を誘発するかまたは不眠症を処置する能力を試験するための方法が同様に、U.S. Patent No. 6,096,784およびU.S. Patent No. 6,271,015に開示されている。最も明らかなのは、化合物を試験動物(例えば、ラットまたはマウス)またはヒトに投与することができ、眠って(例えば、眼を閉じた、運動静止状態)過ごしたその後の時間を監視することができる。同様に、WO 98/24396を参照されたい。
強硬症を誘発するFAAH阻害剤のスクリーニング方法が同様に、当業者によく知られている。Quistand et al. in Toxicology and Applied Pharmacology 173: 48-55 (2001)を参照されたい。Cravatt et al. Proc. Natl. Acad. Sci. U.S.A. 98: 9371-9376 (2001)を参照されたい。
抗侵害作用を目的としたFAAH阻害剤のスクリーニング方法は、当業者によく知られている。例えば、マウスホットプレート試験およびマウスホルマリン試験において、試験化合物を被検動物に投与して、熱によるまたは化学作用による組織傷害に対する侵害受容反応を測定することができる。同様に、抗侵害活性のスクリーニング方法を教示している、U.S. Patent No. 6,326,156を参照されたい。Cravatt et al. Proc. Natl. Acad. Sci. U.S.A. 98: 9371-9376 (2001)を参照されたい。
本発明の化合物を動物に投与して、それらが食糧摂取量および体重、体脂肪、食欲、食糧探索行動に影響を及ぼすかどうか、または調節因子による脂肪酸酸化を調節するかどうかを決定することができる。そのような試験の実施方法は、当業者に知られている。例えば、U.S. Patent Application No. 60/336,289(これは本出願と同一の譲受人に譲渡されおよびその全体が参照として本明細書に組み入れられる)を参照されたい。
理論に結び付けるわけではないが、CNSにおける過剰なドーパミン伝達が統合失調症および他の精神障害の一因となり得ると考えられている。全ての統合失調症患者の約3分の1で、明らかなドーパミン伝達物質および/または受容体の増加が現れる。この異常があからさまには現れない他の人でもやはり、ドーパミン受容体の薬理的遮断により、症候の改善が示される。これらのドーパミン受容体アンタゴニストは、最終的に、脱分極遮断およびドーパミン受容体拮抗作用により、ドーパミン濃度の総体的な減少をもたらす。従って、神経回路の機能異常(この多くで、ドーパミンが活性化に直接的および/または間接的役割を果たす)は、統合失調症の症候に関与しているように思われる。上記に示されているように、脳の皮質下領域におけるドーパミン受容体の遮断により、統合失調症の症候が実質的に軽減される。これらの領域におけるドーパミン産生の全般的な減少により、これらの疾患に苦しむ患者に同様の緩和が供与される。カンナビノイドは、CNSにおいてドーパミン活性を調節することが見出されている。
緑内障の処置に関して、眼の眼圧の測定方法は、医術のなかでは日常業務であり、使われるヒトまたは動物被検体で容易かつ安全に行うことができる。被検体の眼圧に対するFAAH阻害剤の効果は、化合物を眼に直接適用して、その後数時間にわたってまたは翌日にわたって眼圧を監視することにより、容易に評価することができる。もう一方の眼を対照として利用することができる。あるいは、FAAH阻害剤を全身投与することができ、別の媒体で処置した被検体を対照として利用することができる。
使用方法
不安および不安関連障害
いくつかの態様として、式IおよびIIの化合物を含む、FAAH阻害化合物、およびその薬学的組成物ならびにそれらの投与方法は、不安および不安障害または症状を処置するうえで有用である。この化合物および組成物は、例えば、不安、臨床的不安、パニック障害、広場恐怖症、全般性不安障害、特定の恐怖症、対人恐怖症、強迫性障害、急性ストレス障害、および心的外傷後ストレス障害; ならびに不安に起因した特徴を伴う適応障害、一般的健康状態に起因する不安障害、物質誘発性不安障害、および残りの部類の特定不能の不安障害を処置するうえで有用である。処置は予防的または治療的とすることができる。処置はヒト被検体に施すことができる。この化合物は、他の形では不眠症のような障害または症状に対するまたは痛みの軽減に対するどの薬学的介入も必要としない他の健常人に使用してもよい。
いくつかの態様として、式IおよびIIのFAAH阻害化合物、その薬学的組成物およびそれらの投与方法は、抑うつおよび抑うつ障害または症状を処置するうえで有用である。この化合物および組成物は、例えば、主要な抑うつ障害(単極性うつ病)、気分変調性障害(慢性で、軽度のうつ病)、および双極性障害(躁うつ病)を処置するうえで有用である。抑うつは、臨床うつまたは亜臨床うつとすることができる。処置は予防的または治療的とすることができる。処置はヒト被検体に施すことができる。この化合物は、他の形では不眠症のような疾患に対するまたは痛みの軽減に対するどの薬学的介入も必要としない他の健常人に使用してもよい。
いくつかの態様として、FAAH阻害化合物、その薬学的組成物およびそれらの投与方法は、てんかんおよびけいれん障害または発作を処置するうえで有用である。処置は予防的または治療的とすることができる。処置はヒト被検体に施すことができる。この化合物は、他の形では不眠症のような疾患に対するまたは痛みの軽減に対するどの薬学的介入も必要としない他の健常人に使用してもよい。
いくつかの態様として、本発明により、食欲を低下させるための、体脂肪を低下させるためのならびに哺乳類で肥満または体重超過を処置するかまたは予防するためのおよびこれらの健康状態と関連する疾患を予防するかまたは処置するためのFAAH阻害化合物の使用方法および薬学的組成物が提供される。本発明のある局面として、式Iおよび式IIによる阻害剤を含む、FAAH阻害剤を哺乳類に投与する段階による、食欲、体脂肪または体重を低下させるための方法、もしくは肥満または体重超過を処置するかまたは予防するための方法、または食物摂取量を低下させるか、もしくは哺乳類における食欲障害を処置するための方法が提供される。他の態様として、この阻害剤は、オレオイルエタノールアミド(OEA)もしくは他の脂肪酸アルカノールアミド化合物、またはオレオイルエタノールアミドもしくは脂肪酸アルカノールアミド化合物の同族体もしくは類似体(これらは食欲または食物消費を低下させ且つFAAHによる加水分解を受ける)との併用療法で投与される。
いくつかの態様として、式Iまたは式IIによるFAAH阻害化合物、その薬学的組成物およびそれらの投与方法は、統合失調症およびドーパミン関連障害を処置するうえで有用である。処置は予防的または治療的とすることができる。処置はヒト被検体に施すことができる。この化合物は、他の形では不眠症または痛覚過敏のような疾患に対するどの薬学的介入も必要としない他の健常人に使用してもよい。いくつかの態様として、この化合物は、発作性障害以外の他の疾患または障害に対する薬学的介入を必要としない他の健常人に使用してもよい。いくつかの態様として、被検体は他の形では、FAAH阻害剤を必要としない。
いくつかの態様として、式IおよびIIの化合物を投与して、哺乳類の被検体で睡眠を誘発するかまたは促進することができる。処置は予防的または治療的とすることができる。処置はヒト被検体に施すことができる。本発明の化合物および組成物は、不眠症の重症度または頻度または度合いを軽減するためだけに投与してもよい。
いくつかの態様として、式IおよびIIの化合物を投与して、被検体の疼痛を軽減することができる。処置は予防的または治療的とすることができる。処置はヒト被検体に施すことができる。本発明の化合物および組成物は、疼痛の重症度または頻度または度合いを軽減するためだけに投与してもよい。処置は、他の鎮痛剤または抗炎症剤との併用療法のなかで施してもよい。
いくつかの態様として、FAAH阻害剤を投与して、緑内障を処置するかもしくは予防することまたは眼内の眼圧を低下させることができる。いくつかの態様として、その化合物は全身投与されてもよい。他の態様として、FAAH阻害剤は眼の表面に直接適用される(例えば、目薬により)。
本発明の他の局面により、本発明の化合物と薬学的に許容される担体とを含む薬学的組成物が提供される。
本発明の化合物は同様に、非経口投与されてもよい。これらの活性化合物の液剤または懸濁剤をヒドロキシプロピルセルロースのような界面活性剤と適用に混合した水のなかで調製することができる。分散剤を同様に、油中のグリセロール、液状ポリエチレングリコールおよびその混合物のなかで調製することができる。通常の保存および使用条件下では、これらの製剤には、微生物の増殖を阻止する防腐剤が含まれる。
実施例1: 被検体
本発明者らは、雄および雌のWistarラット(200〜350 g)ならびに雄のSwissマウス(20 g)を使用した。全ての手順は、実験動物の取り扱いおよび利用に対する米国国立衛生研究所のガイドライン、ならびにイタリア衛生省のそのガイドライン(D. L. 116/92)を満たした。本発明者らは、18日齢のWistarラットの胚から皮質ニューロンの初代培養を調製して、それらを報告されている(Stella, N. et al., Eur. J. Pharmacol., 425, 189-196 (2001))ように維持した; 本発明者らは、American Type Culture Collection (Manassas, VA)からヒト星状細胞腫細胞を購入した。
アナンダミドおよび関連脂質は、実験室で合成された(Giuffrida, A. et al., Anal. Biochem., 280, 87-93 (2000))。SR141716A(リモナバント)は、米国国立衛生研究所の化学合成計画の一環としてRBI(Natick, MA)により提供された; AM404はTocris(Avonmouth, UK)から入手し、および他の薬物はSigma (St. Louis, MO)から入手した。阻害剤の調製に必要な全ての化学物質は、Aldrichから入手した。
n-ブチルカルバミン酸4-ベンジルオキシフェニルエステル(UCM532) (4)および4-フルオロフェニルカルバミン酸4-ベンジルオキシフェニルエステル(8)は、還流トルエン中で触媒量のトリエチルアミンを用い、それぞれ、n-ブチルイソシアネート、および4-フルオロフェニルイソシアネートによる4-ベンジルオキシフェノールの処理により得た。同様に、シクロヘキシルカルバミン酸ビフェニル-3-イルエステル(5)、シクロヘキシルカルバミン酸5-フェニルペンチルエステル(7)、およびシクロヘキシルカルバミン酸3'-カルバモイルビフェニル-3-イルエステル(UCM597) (6)は、シクロヘキシルイソシアネートをそれぞれ、3-フェニルフェノール、5-フェニルペンタン-1-オール、および3'-ヒドロキシビフェニル-3-カルボン酸アミドと反応させることにより合成した。後者の反応物質は、次のように調製した: 3-ブロモベンゾニトリルと過ホウ酸ナトリウムとの反応により得られた、3-ブロモ安息香酸アミドを、メトキシフェニルボロン酸と結合させて、3'-メトキシビフェニル-3-カルボン酸アミドを得て、これをBBr3で加水分解して、所望の3'-ヒドロキシビフェニル-3-カルボン酸アミドを生成させた。
3-ブロモベンゾニトリル(0.91 g、5 mmol)のジオキサン(19 mL)溶液に、NaBO3・4H2O(2.12 g、13.78 mmol)およびH2O(19 mL)を加えた。この混合液を80℃で16時間(h)撹拌し、冷却し、H2Oを加えて、CH2Cl2で抽出した。合わせた有機層をNa2SO4で乾燥し、そして蒸発させた。カラムクロマトグラフィー(ヘキサン/EtOAc 2:8)による残渣の精製および再結晶により、所望の生成物を無色の平たい小塊として得た。収率80%(EtOH)。Mp(融点): 156〜7℃(文献値156 ℃) (Pearson, D.E. et al., J. Org. Chem., 28: 3147-3149, (1963)。MS(EI): m/z 199(M+); 183(100%)。
3-ブロモ安息香酸アミド(0.76 g; 3.8 mmol)およびトルエン(25 mL)の撹拌混合液に、Pd(PPh3)4 (0.180 g; 0.16 mmol)、Na2CO3(2.543 g; 24 mmol)のH2O(10 mL)溶液、および3-メトキシフェニルボロン酸のEtOH(10mL)溶液を加えた。この混合液を激しく撹拌しながら1 h還流し、冷却し、そして水層をAcOEtで抽出した。合わせた有機層をNa2SO4で乾燥し、そして濃縮した。カラムクロマトグラフィー(シクロヘキサン/EtOAc 1:1その後4:6)による残渣の精製および再結晶により、所望の生成物(0.64 g)を白色固体として得た。収率74%。Mp: 138〜40℃(EtOH)。MS(EI): m/z 227 (M+、100%)。
3'-メトキシビフェニル-3-カルボン酸アミド(0.57 g; 2.5 mmol)の乾燥CH2Cl2 (28 mL)の撹拌・冷却溶液に、N2雰囲気下で、BBr3のCH2Cl2 (6.4 mL) 1 M溶液を加えた。この混合液を室温で1 h撹拌し、2N Na2CO3でクエンチし、そしてAcOEtで抽出した。合わせた有機層を塩水で洗浄し、Na2SO4で乾燥し、そして濃縮した。カラムクロマトグラフィー(シクロヘキサン/EtOAc 2:8)による残渣の精製により、所望の生成物を無定形固体として得た。収率91%。Mp: 148〜51℃(i-Pr2Oで温浸後)。MS(EI): m/z 213 (M+、100%)。
3'-ヒドロキシビフェニル-3-カルボン酸アミド(0.43 g、2 mmol)のトルエン(12 mL)の撹拌混合液に、Et3N(0.012 g、0.016 mL、0.12 mmol)、およびシクロヘキシルイソシアネート(0.28 g、0.28 mL、2.2 mmol)を加えた。20 h還流後、混合液を冷却し、そして濃縮した。カラムクロマトグラフィー(シクロヘキサン/EtOAc 4:6)による残渣の精製および再結晶により、5aを白色固体として得た。未反応量の4a(0.07 g、17%)も回収された。収率: 33%。Mp: 178℃(EtOH)(密閉キャピラーチューブ)。MS(EI): m/z 213(100%)。
立体配座解析、エネルギー最小化および化合物の重ね合わせを含む、分子モデリングの計算は、MMFF94s力場を使用したSybyl 6.8ソフトウェア(Tripos)を用いて行った。
本発明者らは、ラット脳のホモジネートから細胞分画を調製し、基質として、それぞれ、アナンダミド[エタノールアミン-3H] (American Radiolabeled Chemicals, ARC (St. Louis MO), 60 Ci/mmol)および2-モノ-オレオイル-グリセロール-[グリセロール-1,2,3-3H] (ARC, St. Louis MO, 20 Ci/mmol)を用い、膜FAAH活性および細胞質ゾルMGL活性をアッセイした(Dinh, T. Proc. Natl. Acad. Sci. U.S.A. (2002))。本発明者らは、ヒト星状細胞腫細胞で[3H]アナンダミド輸送アッセイ(Piomelli, D. et al., Proc. Natl. Acad. Sci. U. S. A., 96, 5802-5807 (1999)); リガンドとして[3H]WIN-55212-2(NEN-Dupont, Boston, MA, 40-60 Ci/mmol)を用い、それぞれ、ラット小脳膜およびCB2を過剰発現しているチャイニーズハムスター卵巣細胞(Receptor Biology-Perkin Elmer, Wellesley, MA)でCB1およびCB2結合アッセイ((Devane, W.A. et al. Science, 258, 1946-1949 (1992)); 精製酵素(電気ウナギアセチルコリンエステラーゼタイプV-Sおよびウマ血清コリンエステラーゼ; どちらもSigma, St. Louis, MOから入手)を用い、製造元の使用説明書に従って、市販のキット(Sigma, St. Louis, MO)でコリンエステラーゼアッセイを行った。ラット皮質ニューロンでアナンダミド輸送および加水分解を測定するため、本発明者らは、[3H]アナンダミドに4分間暴露する前に、細胞を適当な濃度のFAAH阻害剤と37℃で10分間プレインキュベートした。ある実験の場合、本発明者らは、0.1%ウシ血清アルブミン(タイプV、無脂肪酸、Sigma, St. Louis, MO)を含有する冷Tris-Krebs緩衝液で反応を停止させて、トリプシンEDTA処理により細胞を除去し、そしてクロロホルム/メタノール(体積比1:1)で細胞脂質を抽出した。本発明者らは、抽出液の有機層中の代謝されていない[3H]アナンダミドと、水層中の代謝された[3H]アナンダミド([3H]エタノールアミンとして)を測定した。他の実験の場合、ニューロンを[3H]アナンダミドに4分間暴露した後、本発明者らは、培地を交換し、細胞を洗浄し、そして培地に放出された[3H]アナンダミドを上記のように測定した。
本発明者らは、メタノール-クロロホルム混合液を使って組織から脂質を抽出して、それらをシリカゲルクロマトグラフィーにより分画した(Giuffrida, A. et al., Anal. Biochem., 280, 87-93 (2000))。アナンダミドおよび他の脂肪酸誘導体は、同位体希釈法を用いて、HPLC/MSにより定量化した(Giuffrida, A. et al., Anal. Biochem., 280, 87-93 (2000))。
本発明者らは、全ての化合物を生理食塩水/Tween 80/ポリエチレングリコール(90/5/5)に溶解して、試験の直前にそれらを腹腔内注射により投与した。本発明者らは、デジタル体温計(モデルBAT-12 Physitemp Instruments社, Clifton, NJ)に接続された直腸プローブ(タイプT, Copper-Costantan Thermocouple-Physitemp Instruments社, Clifton, NJ)で体温を; ならびにTsengおよびCraft (Tseng, A.H. et al., Eur. J. Pharmacol., 430, 41-47 (2001))らが報告した手順を利用して強硬症を測定した。
本発明者らは、UCM597をDMSO/生理食塩水(7/3)に溶解して、試験の45分前にそれを腹腔内注射により投与した。本発明者らは、自動システム(Scipro社, New York)を利用して、自由摂食ラットの食物摂取量を記録した。ラットは、試験前の3日間、試験かごに順応させた。各試験は、暗期の開始時に始めて、24時間続けた。
本発明者らは、FAAH阻害剤をポリエチレングリコール/水(1/1)におよびリモナバントを生理食塩水に溶解した。ホルマリンおよびホットプレートアッセイをマウスで、報告されているように行った(Beltramo, M. et al., FEBS Lett., 403, 263-267 (1997))。
本発明者らは、FAAH阻害剤およびリモナバントをジメチルスルホキシド(DMSO)/生理食塩水(それぞれ、7/3および9/1)に溶解した; 本発明者らは、腹腔内注射により試験の30分前にFAAH阻害剤をおよびUCM532の30分前にリモナバントを投与した。高架式のゼロの迷路は、地面からの高度65 cmに持ち上げられた黒色のPerspex製環状プラットホーム(直径105 cm、幅 10 cm)であって、4つの四分円に等しく分割されたそのプラットホームから成っていた(Bickerdike, M.J. et al., Eur. J. Pharmacol., 271, 403-411 (1994); Shepherd, J.K. et al., Psychopharmacology, 116, 56-64 (1994))。2つの対抗する四分円は、プラットホームの内縁および外縁が黒い壁(高さ27 cm)で囲まれ、その一方、その他の2つは、浅い縁(高さ1 cm)で取り囲まれているだけであった。この装置は、均一の暗赤色灯(40〜60ルクス)で照らされた。本発明者らは、閉鎖四分円にラットを入れて、セッションの間にその迷路を徹底的に清掃しつつ、それらを5分周期でビデオ録画した。ラットは、その四肢が四分円内にある場合、開放四分円にいると考えられた。結果は、開放四分円にいる時間/全時間の時間百分率(開放時間百分率)として表される。結果は、一方向ANOVAにより解析し、続けてTukeyの検定を行った。本発明者らは、コンピュータにオンライン接続されたOpto-Varimexかご(Columbus Instruments, Columbus, OH)の中で各ラットの運動活性を記録し、そして20-Wの白色光で照らされた音波減衰室に入れた。歩行活動で過ごした時間の量は、別記のとおりAuto-Trackソフトウェア(Columbus Instruments, Columbus, OH)を用いて解析した(Ali, M.M. et al., Neurosci. Lett., 284, 57-60 (2000); Wedzony, K. et al., Neuropsychopharmacology, 23, 547-559 (2000))。セッションの期間は、10日齢の成体ラットの場合には20分とし、10日齢の乳子の場合には60秒とした。本発明者らは、全体の一方向ANOVAにより解析し、続けて個々のグループ間の比較についてTukeyの検定を行った。本発明者らは、Cuomo et al. (Cuomo, V. et al., Neuropharmacology, 26, 701-705 (1987))により報告された手順に従って、音波減衰室に入れた10日齢の乳子の超音波発生を記録した。試験は900〜1400 hで行い、15 s続けた。薬物をベースライン値の収集(15 s)後に投与して、乳子を薬物投与から30分後、先と同様に試験した。データは、ベースラインからの変化百分率として表し、全体の一方向ANOVAにより解析し、続けて個々のグループ間の比較についてTukeyの検定を行った。
その特異な触媒機構(Patricelli, M.P. et al., Biochemistry, 38, 9804-9812 (1999))にもかかわらず、FAAHは、活性化カルボニルを有する化合物を含む種々のセリンヒドロラーゼ阻害剤により遮断される(Boger, D.L. et al. Proc. Natl. Acad. Sci. U. S. A., 97, 5044-5049 (2000))。従って、本発明者らは、抗コリンエステラーゼ薬カルバリル(表1、化合物1)のようなカルバミン酸のエステルがラット脳膜のFAAH活性を阻害できるかどうか調べた。1は効果がなかったが、その位置異性体2は、FAAHを弱く阻害し(半最大阻害濃度IC50 = 18.6 ± 0.7 μM; 平均±SEM、n = 3)、これはN-メチル置換基をシクロヘキシル基で置換することにより増強された(3、IC50 = 324 ± 31 nM)。アリールエステル4(そのベンジルオキシフェニル基は、2のナフチル部分が伸長された生物学的等価性の異型と見なすことができる)は、3に等しい効力(IC50 = 396 ± 63 nM)でFAAHを阻害した。4の立体配座解析により、O-CH2結合周囲のねじれ角が主に異なり、アンチまたはゴーシュ配座の置換基を有する、接近可能な配座異性体のファミリーが明らかとなった(データは示していない)。後者の配座は、ナフチル誘導体3の形状に一層よく似ていたので、本発明者らは、それらがFAAHの活性部位との4の相互作用に関与している可能性があるという仮説を立てた。この仮説の試験から、ビフェニル誘導体5(IC50 = 63 ± 9 nM)をデザインするに至り、その誘導体を末端フェニル基の系統的修飾によりさらに最適化することで、結果的に強力な阻害剤6(IC50 = 4.6 ± 1.6 nM)が得られた(表1)。
UCM532およびUCM597は、FAAHを阻害したが、しかしながら3つのさらなるセリンヒドロラーゼ、つまり電気ウナギアセチルコリンエステラーゼ、ウマ血漿ブチリルコリンエステラーゼ、およびラット脳モノグリセリドリパーゼ(MGL)に影響を及ぼさなかった(表2)。MGL阻害の欠如は、2-アラキドノイルグリセロール(2-AG) (Dinh, T. Proc. Natl. Acad. Sci. U. S. A. (2002))、脳に存在する他の内在性カンナビノイド(Mechoulam, R. et al. Biochem. Pharmacol., 50, 83-90 (1995); Sugiura, T. et al., Biochem. Biophys. Res. Commun., 215, 89-97 (1995); Stella, N. et al., Nature, 388, 773-778 (1997))の生物学的不活性化において、提唱されているこの酵素の役割という観点から特に注目に値する。さらに、UCM532およびUCM597は、ヒト星状細胞腫細胞におけるアナンダミド輸送に対し、またはCB1およびCB2受容体との高親和性リガンドの結合に対し効果がなかった(表2)。さらには、UCM532 (10 μM)は、21種の受容体のパネル、イオンチャネルおよび神経伝達物質輸送体(これらには、アデノシンA1、A2AおよびA2B; アドレナリン作動性α1A、α2A、β1およびβ2; ドーパミンD1およびD2; グルタメートN-メチル-(D)-アスパルテート; γ-アミノ-酪酸(GABA)Aアゴニスト部位; ヒスタミンH1; オピエートμ; ムスカリン性M2; ならびに脳ニコチン性受容体が含まれていた)と有意に相互作用しなかった(データは示していない)。FAAHに対するこの高選択性は、本発明者らが、生きた動物でUCM532およびUCM597の効果を調べることの後押しとなった。
UCM532またはUCM597(但しその不活性類似体7ではない)の腹腔内(i.p.)注射により、脳FAAH活性の著しい、用量依存的な阻害がもたらされた(図2a)。6回の
実験で、半最大阻害は、0.60 ± 0.09 mg kg-1 UCM532および0.150 ± 0.007 mg kg-1 UCM597に到達した。最大用量のUCM597(0.3 mg kg-1, i.p.)の注射後、FAAH阻害は、開始が早く(15 分未満)、持続的(6時間を超える)であり(図2b)、アナンダミド(図2c)およびFAAHの基質である他の脂肪酸エタノールアミドの脳含有量の有意な上昇が付随して起こった(注射後2 hのpmol/g組織では; オレオイルエタノールアミド: 媒体、137.0 ± 14.3; UCM597 0.3 mg kg-1、725.3 ± 28.6; パルミトイルエタノールアミド: 媒体、259.1 ± 15.0; UCM597、1324 ± 395; n = 8〜15)。FAAH活性および脂肪酸エタノールアミド量の類似変化が、種々の末梢組織でも測定された(データは示していない)。本発明者らのインビトロ実験で示されたMGL阻害の欠如(表2)に一致して、UCM597は、2-AGの脳含有量を変化させなかった(図2d)。
UCM532およびUCM597は、脳のアナンダミド量を増加させたが、それらは明白に、外来性アナンダミドによりもたらされた薬理反応の範囲を模倣していなかった。FAAH活性を最大限に遮断したUCM532(10 mg kg-1、i.p.)またはUCM597(0.3 mg kg-1、i.p.)の全身用量により、CB1受容体活性化の3つの典型的な徴候である(Chaperon, F. et al., Crit. Rev. Neurobiol., 13, 243-281 (1999))、強硬症(強直性不動)、低体温症または過食症(食物摂取量の増加)が引き起こされなかった(データは示していない)。しかしながら、これらの化合物は、急性疼痛の2つのモデルで中等度の抗侵害作用を及ぼした。有害な熱刺激に対する動物の反応を測定する、マウスホットプレート試験の場合、UCM597は、0.5 mg kg-1の用量(図3a)で反応潜時を引き延ばしたが、低い用量(0.1 mg kg-1; データは示していない)では引き延ばさなかった。さらに、化学物質による組織傷害に対する侵害防御反応を測定する、マウスホルマリン試験の場合、UCM597(0.5 mg kg-1)は、早期の疼痛行動を軽減したが、後期には変化がほとんどまたは全くなかった(図3b)。どちらの効果もCB1アンタゴニストSR1417161A(リモナバント) (0.2 mg kg-1, 静脈内, i.v.)により抑止され(図3aおよびb)、効果は低かったが、UCM532により模倣された(データは示していない)。本発明者らの結果は、FAAH-/-変異マウスで得られた結果(Cravatt B.F. et al., Proc. Natl. Acad. Sci. U. S. A., 98, 9371-9376 (2001))を裏付けており、FAAH活性の急性阻害によって、結果的に中等度のCB1媒介性抗侵害受容が引き起こされるが、しかし低体温症または強硬症は引き起こされないことが示唆される。
FAAH阻害により増幅される可能性があるアナンダミドの他の内因性作用を同定するため、本発明者らは、三つの理由で、感情反応の調節に目を向けた。第一に、CB1受容体は、不安および恐怖の制御に関与する、扁桃体のような、脳領域において高いレベルで発現される(Herkenham, M. et al., Proc. Natl. Acad. Sci. U. S.A., 87, 1932-1936 (1990); Glass, M. et al., Neuroscience, 77, 299-318 (1997); Katona, I. et al., J. Neurosci., 21, 9506-9518 (2001))。第二に、カンナビノイド薬の急性投与により、齧歯類(Chaperon, F. et al., Crit. Rev. Neurobiol., 13, 243-281 (1999))およびヒト(Hall, W. et al., Lancet, 352, 1611-1616 (1998); Robson, P. Br. J. Psychiatry, 178, 107-115 (2001))において著しい感情反応が引き起こされる。第三に、CB1アンタゴニストのリモナバントがラットで不安様行動を誘発することから、内在性カンナビノイドにより媒介される内因性の抗不安緊張状態の存在が示唆される(Rodriguez de Fonseca, F. et al., Science, 276, 2050-2054 (1997); C. Arevalo, R. et al., Pharmacol. Biochem. Behav., 70, 123-131(2001))。
式Iおよび式IIの脂肪酸アミドヒドロラーゼ阻害剤は、FAAHの細胞内酵素活性を標的化することにより、アナンダミドの不活性化を阻止する新しいクラスの薬剤に相当する。このクラスのうちで最も強力な一員である、UCM597は、脳膜の場合にはIC50値 4 nMでおよび完全なニューロンの場合には0.5 nMで、ならびにラットに全身投与後にはID50値0.15 mg kg-1でFAAH活性を阻害した。この化合物は、FAAHに対する選択性が、カンナビノイド受容体(選択指数: > 25,000)およびMGL、内在性カンナビノイドエステル、2-AGの非活性化に関与する酵素(選択指数: > 7,500)を含む、他のカンナビノイド関連標的よりもずっと高かった。そのような顕著な標的の識別は、インビボでのカンナビミメティック作用の欠如と一致していた。このように、FAAH活性をほとんど無効とし且つ実質的に脳のアナンダミド量を上昇させた用量で、UCM597およびその類似体UCM532は、齧歯類におけるカンナビノイド中毒の三つの主要な症候(Chaperon, F. et al., Crit. Rev. Neurobiol., 13, 243-281 (1999))、つまり強硬症を惹起せず、体温を低下させずまたは摂食を刺激しなかった。
本実施例は、式Iまたは式IIによる50種類を超える化合物のFAAH阻害IC50値を提供する。これらの化合物の試験結果を表4に示す。
本実施例は、O-アリールN-アルキルカルバミン酸アリールエステルのさらに詳細な3D-QSAR解析を示し、次に、FAAHの三次元構造にそれを関連付ける。
pIC50 = -0.49(±0.07)π + 7.26 (±0.09) (1)
n = 18 (URB524, 3g〜w) r2 = 0.74 s = 0.37 F = 46.0 q2 = 0.66 SDEP = 0.40
pIC = -0.52(±0.12)π - 1.20(±0.14)π2 + 7.33(±0.14) (2)
n = 7 (URB524, 3a〜f) r2 = 0.95 s = 0.22 F = 37.8 q2 = 0.80 SDEP = 0.33
pIC50 = -0.046(±0.009) MR + 0.80(±0.18) HB + 7.29(±0.17) (3)
n = 18 (URB524, 3g〜w) r2 = 0.76 s = 0.37 F = 23.2 q2 = 0.68 SDEP= 0.39
Claims (22)
- 下記式の化合物またはその薬学的に許容される塩:
(式中
XはOであり;
QはOであり;
Rは、置換または非置換メタビフェニルであり、ここで前記置換基は、ハロゲン、-OR'、=O、=NR'、=N-OR'、-NR'R''、-SR'、-OC(O)R'、-C(O)R'、-CO 2 R'、-CONR'R''、-OC(O)NR'R''、-NR''C(O)R'、-NR'-C(O)NR''R'''、-NR''C(O) 2 R'、-NR-C(NR'R''R''')=NR''''、-NR-C(NR'R'')=NR'''、-S(O)R'、-S(O) 2 R'、-S(O) 2 NR'R''、-NRSO 2 R'、-CNおよび-NO 2 、-R'、-N 3 、-CH(Ph) 2 、フルオロ(C 1 〜C 4 )アルコキシ、ならびにフルオロ(C 1 〜C 4 )アルキルからなる群より選択され、かつ、前記R'、R''、R'''およびR''''は、水素、および非置換飽和(C 1 〜C 8 )アルキルより独立して選択され;
R1 は非置換飽和シクロアルキルであり;
R2はHである。) - R 1 がシクロペンチルまたはシクロヘキシルである、請求項1記載の化合物。
- R 1 がシクロヘキシルである、請求項2記載の化合物。
- ビフェニルが、ハロゲン、非置換飽和アルキル、アミノ、アミド、またはトリフルオロメチルで置換されている、請求項3記載の化合物。
- ビフェニルが非置換である、請求項3記載の化合物。
- 化合物が下記式を有する、請求項1記載の化合物:
(式中、
Ra 1 およびRa 2 は、H、ハロゲン、非置換不飽和アルキル、フェニル、フェノキシ、トリフルオロメチル、アミノ、およびカルボキサミドからなる群より独立して選択され;および
RbはH、非置換飽和アルキル、およびカルボキサミドか選択される。) - Ra 1 およびRa 2 が、ハロゲン、非置換不飽和アルキル、トリフルオロメチル、アミノ、およびカルボキサミドからなる群より独立して選択される、請求項6記載の化合物。
- Rbが、メチル、アミノ、およびカルボキサミドからなる群より選択される、請求項7記載の化合物。
- Ra 1 、Ra 2 、およびRbが、下記a)〜m)で示される基の組み合わせのいずれかである、請求項6記載の化合物:
a) Ra 1 がフェニル、Ra 2 がH、およびRbがH;
b) Ra 1 がフェノキシ、Ra 2 がH、およびRbがH;
c) Ra 1 がH、Ra 2 がメチル、およびRbがH;
d) Ra 1 がH、Ra 2 がトリフルオロメチル、およびRbがH;
e) Ra 1 がH、Ra 2 がアミノ、およびRbがH;
f) Ra 1 がメチル、Ra 2 がH、およびRbがH;
g) Ra 1 がH、Ra 2 がフルオロ、およびRbがH;
h) Ra 1 がトリフルオロメチル、Ra 2 がH、およびRbがH;
i) Ra 1 がアミノ、Ra 2 がH、およびRbがH;
j) Ra 1 がカルボキサミド、Ra 2 がH、およびRbがH;
k) Ra 1 がフルオロ、Ra 2 がH、およびRbがH;
l) Ra 1 がH、Ra 2 がH、およびRbがメチル;または
m) Ra 1 がH、Ra 2 がH、およびRbがカルボキサミド。 - 薬学的に許容される担体、および請求項1〜9のいずれか一項記載の化合物を含む薬学的組成物。
- 哺乳動物における不安を治療するための薬剤の製造における、請求項1〜9のいずれか一項記載の化合物の使用。
- 哺乳動物における疼痛を治療するための薬剤の製造における、請求項1〜9のいずれか一項記載の化合物の使用。
- 哺乳動物における抑鬱を治療するための薬剤の製造における、請求項1〜9のいずれか一項記載の化合物の使用。
- 哺乳動物における食欲障害の治療または食欲の制御のための薬剤の製造における、請求項1〜9のいずれか一項記載の化合物の使用。
- 下記式の化合物またはその薬学的に許容される塩:
(式中、
XはOであり;
QはOであり;
Rは置換または非置換メタビフェニルであり、ここで前記置換基はハロゲン、非置換不飽和アルキル、トリフルオロメチル、アミノ、およびカルボキサミドからなる群より選択され;
R 1 はシクロヘキシルであり;
R 2 はHである。) - ビフェニルの置換基が、ハロゲン、メチル、トリフルオロメチル、アミノ、およびカルボキサミドからなる群より選択される、請求項15記載の化合物。
- 化合物が、N-シクロヘキシル3'-カルボキサミド ビフェニル-3-イル カルバメート、またはその薬学的に許容される塩である、請求項15記載の化合物。
- 薬学的に許容される担体、および請求項15〜17のいずれか一項記載の化合物を含む薬学的組成物。
- 哺乳動物における不安を治療するための薬剤の製造における、請求項15〜17のいずれか一項記載の化合物の使用。
- 哺乳動物における疼痛を治療するための薬剤の製造における、請求項15〜17のいずれか一項記載の化合物の使用。
- 哺乳動物における抑鬱を治療するための薬剤の製造における、請求項15〜17のいずれか一項記載の化合物の使用。
- 哺乳動物における食欲障害の治療または食欲の制御のための薬剤の製造における、請求項15〜17のいずれか一項記載の化合物の使用。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US41700802P | 2002-10-07 | 2002-10-07 | |
| PCT/US2003/031844 WO2004033422A2 (en) | 2002-10-07 | 2003-10-07 | Modulation of anxiety through blockade of anandamide hydrolysis |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2006511484A JP2006511484A (ja) | 2006-04-06 |
| JP2006511484A5 JP2006511484A5 (ja) | 2006-11-24 |
| JP4515911B2 true JP4515911B2 (ja) | 2010-08-04 |
Family
ID=32093946
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004543510A Expired - Fee Related JP4515911B2 (ja) | 2002-10-07 | 2003-10-07 | アナンダミド加水分解の遮断による不安の調節 |
Country Status (11)
| Country | Link |
|---|---|
| US (3) | US7176201B2 (ja) |
| EP (1) | EP1558591B1 (ja) |
| JP (1) | JP4515911B2 (ja) |
| KR (1) | KR20050088992A (ja) |
| CN (1) | CN100408556C (ja) |
| AU (1) | AU2003279877B2 (ja) |
| CA (1) | CA2501506C (ja) |
| EA (1) | EA010267B1 (ja) |
| MX (1) | MXPA05003715A (ja) |
| UA (1) | UA80841C2 (ja) |
| WO (1) | WO2004033422A2 (ja) |
Families Citing this family (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9923738D0 (en) * | 1999-10-07 | 1999-12-08 | Nestle Sa | Nutritional composition |
| US20050054730A1 (en) * | 2001-03-27 | 2005-03-10 | The Regents Of The University Of California | Compounds, compositions and treatment of oleoylethanolamide-like modulators of PPARalpha |
| CA2442330A1 (en) * | 2001-03-29 | 2002-10-10 | Emory University | Acute pharmacologic augmentation of psychotherapy with enhancers of learning or conditioning |
| WO2004034968A2 (en) * | 2002-08-20 | 2004-04-29 | The Regents Of The University Of California | Combination therapy for controlling appetites |
| KR20050088992A (ko) * | 2002-10-07 | 2005-09-07 | 더 리전트 오브 더 유니버시티 오브 캘리포니아 | 아난드아미드 가수분해 차단을 통한 불안감의 조절 |
| WO2004089296A2 (en) * | 2003-04-03 | 2004-10-21 | The Regents Of The University Of California | Improved inhibitors for the soluble epoxide hydrolase |
| CA2542547A1 (en) * | 2003-10-16 | 2005-05-26 | Daniele Piomelli | Dietary and other compositions, compounds, and methods for reducing body fat, controlling appetite, and modulating fatty acid metabolism |
| FR2865205B1 (fr) * | 2004-01-16 | 2006-02-24 | Sanofi Synthelabo | Derives de type aryloxyalkylcarbamates, leur preparation et leur application en therapeutique |
| FR2866886B1 (fr) * | 2004-02-26 | 2007-08-31 | Sanofi Synthelabo | Derives d'aryl-et d'heteroaryl-akylcarbamates, leur preparation et leur application en therapeutique |
| EP1765311A4 (en) | 2004-03-16 | 2009-04-29 | Univ California | REDUCTION OF NEPHROPATHY WITH INHIBITORS OF SOLUBLE EPOXY HYDROLASE AND EPOXYEICOSANOIDS |
| EP1761480B1 (en) * | 2004-06-23 | 2009-02-18 | F. Hoffmann-La Roche AG | Mao-b inhibitors |
| US20060084659A1 (en) * | 2004-10-19 | 2006-04-20 | Michael Davis | Augmentation of psychotherapy with cannabinoid reuptake inhibitors |
| NZ554555A (en) | 2004-10-20 | 2011-09-30 | Univ California | Cyclohexyl-urea derivatives as improved inhibitors for the soluble epoxide hydrolase |
| KR100693528B1 (ko) * | 2004-10-29 | 2007-03-14 | 주식회사 팬택 | 전원 지연 인가 기능을 가지는 무선통신 단말기 |
| EP1824821A2 (en) * | 2004-11-23 | 2007-08-29 | PTC Therapeutics, Inc. | Carbazole, carboline and indole derivatives useful in the inhibition of vegf production |
| ES2433290T3 (es) * | 2005-02-17 | 2013-12-10 | Astellas Pharma Inc. | Derivados de piperazina para el tratamiento de la incontinencia urinaria y el dolor |
| WO2006116773A2 (en) * | 2005-04-28 | 2006-11-02 | The Regents Of The University Of California | Methods, compositions, and compounds for modulation of monoacylglycerol lipase, pain, and stress-related disorders |
| US20090111778A1 (en) * | 2005-11-18 | 2009-04-30 | Richard Apodaca | 2-Keto-Oxazoles as Modulators of Fatty Acid Amide Hydrolase |
| US20070155707A1 (en) * | 2005-12-29 | 2007-07-05 | Kadmus Pharmaceuticals, Inc. | Ionizable inhibitors of fatty acid amide hydrolase |
| CA2635232A1 (en) * | 2005-12-29 | 2007-07-12 | N.V. Organon | Inhibitors of fatty acid amide hydrolase |
| WO2007106706A1 (en) * | 2006-03-10 | 2007-09-20 | Boehringer Ingelheim International Gmbh | Cyclic urea compounds as soluble epoxide hydrolase inhibitors effective for the treatment of cardiovascular disorders |
| JP2009538358A (ja) * | 2006-05-26 | 2009-11-05 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | 脂肪酸アミド加水分解酵素のオキサゾリルピペリジン・モジュレーター |
| EP2054055A2 (en) * | 2006-08-18 | 2009-05-06 | N.V. Organon | Combination faah inhibitor and analgesic, anti-inflammatory or anti-pyretic agent |
| US7888394B2 (en) | 2006-08-21 | 2011-02-15 | N.V. Organon | Synthesis, polymorphs, and pharmaceutical formulation of FAAH inhibitors |
| JPWO2008023720A1 (ja) | 2006-08-23 | 2010-01-14 | アステラス製薬株式会社 | ウレア化合物又はその塩 |
| US20080089845A1 (en) * | 2006-09-07 | 2008-04-17 | N.V. Organon | Methods for determining effective doses of fatty acid amide hydrolase inhibitors in vivo |
| US20090099240A1 (en) * | 2006-10-02 | 2009-04-16 | N.V. Organon | Methods for treating energy metabolism disorders by inhibiting fatty acid amide hydrolase activity |
| EP2096915A1 (en) * | 2006-11-20 | 2009-09-09 | N.V. Organon | Metabolically-stabilized inhibitors of fatty acid amide hydrolase |
| WO2008100977A2 (en) * | 2007-02-14 | 2008-08-21 | N.V. Organon | Carbamates therapeutic release agents as amidase inhibitors |
| DE102007022007A1 (de) * | 2007-05-08 | 2008-11-13 | Schebo Biotech Ag | Neuartike Pharmazeutika, Verfahren zu ihrer Herstellung und ihre Verwendung in der Prophylaxe und Therapie von ZNS-Erkrankungen und Diabetes |
| AU2008257154B2 (en) | 2007-05-25 | 2014-03-06 | The Scripps Research Institute | Tetracyclic inhibitors of fatty acid amide hydrolase |
| AU2008260496A1 (en) * | 2007-05-31 | 2008-12-11 | The Scripps Research Institute | Tricyclic inhibitors of fatty acid amide hydrolase |
| US8207226B1 (en) | 2008-06-03 | 2012-06-26 | Alcon Research, Ltd. | Use of FAAH antagonists for treating dry eye and ocular pain |
| TWI434842B (zh) | 2008-07-14 | 2014-04-21 | Astellas Pharma Inc | Azole compounds |
| KR20110091508A (ko) | 2008-11-06 | 2011-08-11 | 아스텔라스세이야쿠 가부시키가이샤 | 카르바메이트 화합물 또는 그의 염 |
| FR2938537B1 (fr) * | 2008-11-14 | 2012-10-26 | Sanofi Aventis | Derives de carbamates d'alkyl-heterocycles, leur preparation et leur application en therapeutique. |
| RU2553451C2 (ru) * | 2008-12-24 | 2015-06-20 | Биал-Портела Энд Ка, С.А. | Фармацевтические соединения |
| FR2941696B1 (fr) * | 2009-02-05 | 2011-04-15 | Sanofi Aventis | Derives d'azaspiranyl-alkylcarbamates d'heterocycles a 5 chainons, leur preparation et leur application en therapeutique |
| AU2010293429B2 (en) | 2009-09-09 | 2015-12-17 | Sumitomo Dainippon Pharma Co., Ltd. | 8-oxodihydropurine derivative |
| US20130150346A1 (en) | 2010-01-08 | 2013-06-13 | Quest Ventures Ltd. | Use of FAAH Inhibitors for Treating Parkinson's Disease and Restless Legs Syndrome |
| FR2955580A1 (fr) * | 2010-01-28 | 2011-07-29 | Sanofi Aventis | Derives de carbamate d'alkyl-heterocycles, leur preparation et leur application en therapeutique |
| RU2012135525A (ru) * | 2010-01-20 | 2014-02-27 | Санофи | Производные алкилгетероциклических карбаматов, их получение и применение в терапии |
| WO2011123719A2 (en) | 2010-03-31 | 2011-10-06 | Ironwood Pharmaceuticals, Inc. | Use of faah inhibitors for treating abdominal, visceral and pelvic pain |
| EP2389922A1 (en) | 2010-05-25 | 2011-11-30 | Symrise AG | Cyclohexyl carbamate compounds as anti-ageing actives |
| EP2593078B1 (en) | 2010-05-25 | 2016-11-30 | Symrise AG | Cyclohexyl carbamate compounds as active anti-cellulite ingredients |
| CN103025310B (zh) | 2010-05-25 | 2016-01-27 | 西姆莱斯有限公司 | 作为皮肤和/或毛发美白活性物的氨基甲酸环己酯化合物 |
| RU2583435C2 (ru) * | 2010-07-28 | 2016-05-10 | Те Риджентс Оф Те Юниверсити Оф Калифорния | Ингибиторы faah периферически-ограниченного действия |
| ES2404881T3 (es) * | 2010-09-28 | 2013-05-29 | Gruppo Cimbali S.P.A. | Varilla mejorada de formación automática de espuma de leche |
| CN104203906B (zh) * | 2011-08-19 | 2018-03-13 | 加利福尼亚大学董事会 | 间位取代的联苯基外周限制性faah抑制剂 |
| WO2014023325A1 (en) * | 2012-08-06 | 2014-02-13 | Fondazione Istituto Italiano Di Tecnologia | Multitarget faah and cox inhibitors and therapeutical uses thereof |
| ITMI20131180A1 (it) | 2013-07-15 | 2015-01-16 | Fond Istituto Italiano Di Tecnologia | O-alchil triazolil carbammati come inibitori di idrolasi delle ammidi degli acidi grassi (faah) |
| JP6390874B2 (ja) * | 2013-07-18 | 2018-09-19 | フォンダツィオーネ・イスティテュート・イタリアーノ・ディ・テクノロジア | フェニルカルバメート並びに脂肪酸アミドヒドロラーゼ(faah)酵素の阻害剤及びd3ドーパミン受容体(d3dr)の調節剤としてのその使用 |
| AR100387A1 (es) * | 2014-02-18 | 2016-10-05 | Basf Se | Copolímeros que comprenden etileno, ésteres de vinilo y ésteres de ácido (met)acrílico, sus formulaciones y usos como depresor del punto de fluidez, inhibidor de cera y potenciador de flujo para petróleos crudos |
| WO2015157313A1 (en) | 2014-04-07 | 2015-10-15 | The Regents Of The University Of California | Inhibitors of fatty acid amide hydrolase (faah) enzyme with improved oral bioavailability and their use as medicaments |
| EP3303289A4 (en) * | 2015-06-05 | 2018-11-14 | Dana-Farber Cancer Institute, Inc. | Compounds and methods for treating cancer |
| KR20180093898A (ko) | 2015-12-11 | 2018-08-22 | 닛산 가가쿠 가부시키가이샤 | 오일겔화제 |
| US10220061B1 (en) | 2017-09-26 | 2019-03-05 | Cynthia Denapoli | Method of reducing stress and anxiety in equines |
| US10414721B1 (en) | 2018-06-04 | 2019-09-17 | University Of Bern | Inhibitor of endocannabinoid cellular reuptake |
Family Cites Families (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL190462A (ja) * | 1953-09-02 | |||
| US3084096A (en) * | 1955-08-29 | 1963-04-02 | Union Carbide Corp | Indanyl n-methyl carbamate |
| US3275512A (en) | 1962-11-27 | 1966-09-27 | Upjohn Co | Process for treating inflammation with biphenylcarbamates |
| US3632631A (en) | 1967-09-08 | 1972-01-04 | Ethyl Corp | Sterically hindered bisphenyl carbamates |
| US3880908A (en) | 1972-01-24 | 1975-04-29 | Standard Oil Co | Urethane derivatives of polymethyl biphenyl useful as fungicides |
| DE2943480A1 (de) * | 1979-10-27 | 1981-05-07 | Bayer Ag, 5090 Leverkusen | Verfahren zur herstellung von n,o-disubstituierten urethanen, sowie ihre verwendung als ausgangsmaterial zur herstellung von isocyanaten |
| HU197831B (en) | 1984-12-20 | 1989-06-28 | Chinoin Gyogyszer Es Vegyeszet | Insecticide compositions containing alkenyl-oxy-phenyl-carbamat derivatives as active components and process for producing the active components |
| IL74497A (en) * | 1985-03-05 | 1990-02-09 | Proterra Ag | Pharmaceutical compositions containing phenyl carbamate derivatives and certain phenyl carbamate derivatives |
| JPS63303661A (ja) | 1987-01-22 | 1988-12-12 | Ishikawajima Harima Heavy Ind Co Ltd | 注湯装置 |
| US4987233A (en) * | 1988-06-15 | 1991-01-22 | Eli Lilly And Company | Process for preparing herbicidal ureas and insecticidal carbamates and carbamate derivatives |
| IL95994A0 (en) * | 1989-11-15 | 1991-07-18 | American Home Prod | Carbamate esters and pharmaceutical compositions containing them |
| US5112859A (en) * | 1990-09-14 | 1992-05-12 | American Home Products Corporation | Biphenyl amide cholesterol ester hydrolase inhibitors |
| US5476944A (en) | 1991-11-18 | 1995-12-19 | G. D. Searle & Co. | Derivatives of cyclic phenolic thioethers |
| US5541061A (en) | 1992-04-29 | 1996-07-30 | Affymax Technologies N.V. | Methods for screening factorial chemical libraries |
| US5288514A (en) | 1992-09-14 | 1994-02-22 | The Regents Of The University Of California | Solid phase and combinatorial synthesis of benzodiazepine compounds on a solid support |
| DE69314148T2 (de) | 1992-12-17 | 1998-01-15 | Valeo Systemes Essuyage | Stromversorgungsverfahren und -vorrichtung für einen elektrischen Motor zum Antrieb eines Scheibenwischers eines Kraftfahrzeugs |
| US5576220A (en) | 1993-02-19 | 1996-11-19 | Arris Pharmaceutical Corporation | Thin film HPMP matrix systems and methods for constructing and displaying ligands |
| US5519134A (en) | 1994-01-11 | 1996-05-21 | Isis Pharmaceuticals, Inc. | Pyrrolidine-containing monomers and oligomers |
| US5525735A (en) | 1994-06-22 | 1996-06-11 | Affymax Technologies Nv | Methods for synthesizing diverse collections of pyrrolidine compounds |
| US5549974A (en) | 1994-06-23 | 1996-08-27 | Affymax Technologies Nv | Methods for the solid phase synthesis of thiazolidinones, metathiazanones, and derivatives thereof |
| IT1271679B (it) * | 1994-07-18 | 1997-06-04 | Mediolanum Farmaceutici Srl | Derivati del fenilcarbammato atti all'impiego come anticolinesterasici |
| DE4430600A1 (de) * | 1994-08-22 | 1996-02-29 | Hoechst Schering Agrevo Gmbh | Biphenyl-Derivate |
| JPH0892167A (ja) * | 1994-09-21 | 1996-04-09 | Idemitsu Kosan Co Ltd | 芳香族炭酸ジエステルの製造方法 |
| US5585492A (en) * | 1994-10-11 | 1996-12-17 | G. D. Searle & Co. | LTA4 Hydrolase inhibitors |
| US6271015B1 (en) | 1995-06-12 | 2001-08-07 | The Scripps Research Institute | Fatty-acid amide hydrolase |
| CA2206192A1 (en) | 1996-06-13 | 1997-12-13 | F. Hoffmann-La Roche Ag | Modulation of lc132 (opioid-like) receptor function |
| US5856537A (en) | 1996-06-26 | 1999-01-05 | The Scripps Research Institute | Inhibitors of oleamide hydrolase |
| US6531506B1 (en) * | 1996-08-13 | 2003-03-11 | Regents Of The University Of California | Inhibitors of epoxide hydrolases for the treatment of hypertension |
| US6150415A (en) | 1996-08-13 | 2000-11-21 | The Regents Of The University Of California | Epoxide hydrolase complexes and methods therewith |
| NZ335419A (en) * | 1996-11-19 | 2000-05-26 | Icn Pharmaceuticals | multivalent quaternary ammonium salts of a cholinergic agent such as pyridostigmine |
| US5925672A (en) | 1996-12-06 | 1999-07-20 | Neurosciences Research Foundation, Inc. | Methods of treating mental diseases, inflammation and pain |
| TW492882B (en) | 1997-11-28 | 2002-07-01 | Caleb Pharmaceuticals Inc | Cholinergic antagonist plaster composition |
| US7897598B2 (en) | 1998-06-09 | 2011-03-01 | Alexandros Makriyannis | Inhibitors of the anandamide transporter |
| US6251931B1 (en) * | 1998-11-24 | 2001-06-26 | The Scripps Research Institute | Inhibitors of gap junction communication |
| US6414002B1 (en) | 1999-09-22 | 2002-07-02 | Bristol-Myers Squibb Company | Substituted acid derivatives useful as antidiabetic and antiobesity agents and method |
| US6359010B1 (en) | 1999-11-23 | 2002-03-19 | Thomas D. Geracioti, Jr. | Methods of treating anxiety and mood disorders with oleamide |
| US6261595B1 (en) | 2000-02-29 | 2001-07-17 | Zars, Inc. | Transdermal drug patch with attached pocket for controlled heating device |
| AU2001278479B2 (en) | 2000-07-19 | 2007-06-21 | F. Hoffmann-La Roche Ag | Pyrrolidine derivatives as metalloprotease inhibitors |
| FR2812876B1 (fr) * | 2000-08-08 | 2002-09-27 | Galderma Res & Dev | Nouveaux composes bi-aromatiques activateurs des recepteurs de type ppar-gamma et leur utilisation dans des compositions cosmetiques ou pharmaceutiques |
| DE60135230D1 (de) * | 2000-08-16 | 2008-09-18 | Hoffmann La Roche | Aminocyclohexan-derivate |
| ES2275808T3 (es) * | 2001-02-06 | 2007-06-16 | Pfizer Products Inc. | Composiciones farmaceuticas para el tratamiento de trastornos del snc y otros trastornos. |
| IL158300A0 (en) * | 2001-04-27 | 2004-05-12 | Bristol Myers Squibb Co | Bisarylimidazol derivatives and pharmaceutical compositions containing the same |
| IL162270A0 (en) | 2001-12-14 | 2005-11-20 | Novo Nordisk As | Compounds and uses thereof for decreasing activityof hormone-sensitive lipase |
| US6908939B2 (en) | 2001-12-21 | 2005-06-21 | Galderma Research & Development S.N.C. | Biaromatic ligand activators of PPARγ receptors |
| JP2005516986A (ja) * | 2002-02-08 | 2005-06-09 | ブリストル−マイヤーズ スクイブ カンパニー | (オキシム)カルバモイル脂肪酸アミド加水分解酵素インヒビター |
| FR2843964B1 (fr) * | 2002-08-29 | 2004-10-01 | Sanofi Synthelabo | Derives de dioxane-2-alkylcarbamates, leur preparation et leur application en therapeutique |
| KR20050088992A (ko) | 2002-10-07 | 2005-09-07 | 더 리전트 오브 더 유니버시티 오브 캘리포니아 | 아난드아미드 가수분해 차단을 통한 불안감의 조절 |
-
2003
- 2003-10-07 KR KR1020057005995A patent/KR20050088992A/ko not_active Application Discontinuation
- 2003-10-07 CN CNB2003801052130A patent/CN100408556C/zh not_active Expired - Fee Related
- 2003-10-07 US US10/681,858 patent/US7176201B2/en not_active Expired - Lifetime
- 2003-10-07 CA CA2501506A patent/CA2501506C/en not_active Expired - Fee Related
- 2003-10-07 AU AU2003279877A patent/AU2003279877B2/en not_active Ceased
- 2003-10-07 UA UAA200504339A patent/UA80841C2/xx unknown
- 2003-10-07 MX MXPA05003715A patent/MXPA05003715A/es unknown
- 2003-10-07 WO PCT/US2003/031844 patent/WO2004033422A2/en active Application Filing
- 2003-10-07 EP EP03773201.3A patent/EP1558591B1/en not_active Expired - Lifetime
- 2003-10-07 EA EA200500615A patent/EA010267B1/ru not_active IP Right Cessation
- 2003-10-07 JP JP2004543510A patent/JP4515911B2/ja not_active Expired - Fee Related
-
2006
- 2006-07-28 US US11/496,051 patent/US8003693B2/en active Active
-
2011
- 2011-08-16 US US13/211,219 patent/US20120010283A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| UA80841C2 (en) | 2007-11-12 |
| EA200500615A1 (ru) | 2005-10-27 |
| WO2004033422A3 (en) | 2004-07-29 |
| KR20050088992A (ko) | 2005-09-07 |
| US20040127518A1 (en) | 2004-07-01 |
| US8003693B2 (en) | 2011-08-23 |
| AU2003279877A1 (en) | 2004-05-04 |
| US20120010283A1 (en) | 2012-01-12 |
| EP1558591A4 (en) | 2007-09-12 |
| US7176201B2 (en) | 2007-02-13 |
| WO2004033422A2 (en) | 2004-04-22 |
| US20090048337A1 (en) | 2009-02-19 |
| MXPA05003715A (es) | 2005-09-30 |
| EP1558591A2 (en) | 2005-08-03 |
| CA2501506C (en) | 2014-02-11 |
| CN1729171A (zh) | 2006-02-01 |
| CN100408556C (zh) | 2008-08-06 |
| CA2501506A1 (en) | 2004-04-22 |
| EA010267B1 (ru) | 2008-06-30 |
| JP2006511484A (ja) | 2006-04-06 |
| EP1558591B1 (en) | 2014-05-07 |
| AU2003279877B2 (en) | 2010-05-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4515911B2 (ja) | アナンダミド加水分解の遮断による不安の調節 | |
| US11684598B2 (en) | Use of cannabinoids in the treatment of epilepsy | |
| WO2023283386A2 (en) | Safer psychoactive compositions | |
| WO2008024139A2 (en) | Inhibitors of fatty acid amide hydrolase | |
| AU2012299060A1 (en) | Meta-substituted biphenyl peripherally restricted FAAH inhibitors | |
| KR20080091132A (ko) | 지방산 아미드 가수분해 효소의 억제제 | |
| US20230181572A1 (en) | Carbamoyl cyclohexane derivatives for treating autism spectrum disorder | |
| JP2010120916A (ja) | 体内生理活性モノアミンの減少に関連して発症する精神・神経疾患の改善または治療剤 | |
| EP3952872B1 (en) | Carbamoyl cyclohexane derivatives for treating autism spectrum disorder | |
| US12043605B2 (en) | Deuterated empathogens | |
| CA3218884A1 (en) | Therapeutic aminoindane compounds and compositions | |
| US20240287016A1 (en) | Fluorinated Empathogens | |
| WO2024130044A1 (en) | Treatment of psychological factors affecting other medical conditions (pfaomc) | |
| OA20885A (en) | Carbamoyl cyclohexane derivatives for treating autism spectrum disorder. | |
| NZ622480B2 (en) | Meta-substituted biphenyl peripherally restricted faah inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20061006 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20061006 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100120 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20100120 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100326 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100428 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100513 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130521 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |