JP4008024B2 - Gabaaレセプター複合体と相互作用させるための化合物 - Google Patents
Gabaaレセプター複合体と相互作用させるための化合物 Download PDFInfo
- Publication number
- JP4008024B2 JP4008024B2 JP50089295A JP50089295A JP4008024B2 JP 4008024 B2 JP4008024 B2 JP 4008024B2 JP 50089295 A JP50089295 A JP 50089295A JP 50089295 A JP50089295 A JP 50089295A JP 4008024 B2 JP4008024 B2 JP 4008024B2
- Authority
- JP
- Japan
- Prior art keywords
- pregnan
- hydroxy
- solution
- trifluoromethyl
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 150000001875 compounds Chemical class 0.000 title claims description 64
- 102000027484 GABAA receptors Human genes 0.000 title 1
- 108091008681 GABAA receptors Proteins 0.000 title 1
- -1 hydroxy, pyrid-4-ylthio group Chemical group 0.000 claims description 37
- 230000002829 reductive effect Effects 0.000 claims description 30
- 125000000217 alkyl group Chemical group 0.000 claims description 24
- FGMARUFVNCEQCL-SCYKMPJCSA-N 1-[(3r,5r,8r,9s,10s,13s,14s,17s)-3-ethynyl-3-hydroxy-10,13-dimethyl-1,2,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydrocyclopenta[a]phenanthren-17-yl]ethanone Chemical compound C([C@H]1CC2)[C@@](O)(C#C)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)C)[C@@]2(C)CC1 FGMARUFVNCEQCL-SCYKMPJCSA-N 0.000 claims description 23
- 229910052739 hydrogen Inorganic materials 0.000 claims description 20
- 239000001257 hydrogen Substances 0.000 claims description 19
- 159000000000 sodium salts Chemical class 0.000 claims description 14
- 125000003118 aryl group Chemical group 0.000 claims description 13
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 12
- 229910019142 PO4 Inorganic materials 0.000 claims description 10
- 239000010452 phosphate Substances 0.000 claims description 10
- FGMARUFVNCEQCL-NWWWSWJSSA-N 1-[(3r,5s,8r,9s,10s,13s,14s,17s)-3-ethynyl-3-hydroxy-10,13-dimethyl-1,2,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydrocyclopenta[a]phenanthren-17-yl]ethanone Chemical compound C([C@@H]1CC2)[C@@](O)(C#C)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)C)[C@@]2(C)CC1 FGMARUFVNCEQCL-NWWWSWJSSA-N 0.000 claims description 9
- 125000000304 alkynyl group Chemical group 0.000 claims description 9
- 125000005843 halogen group Chemical group 0.000 claims description 7
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 7
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 6
- 150000002431 hydrogen Chemical class 0.000 claims description 5
- WZUCWPNRRXLFPH-PLHHIASCSA-N 2-bromo-1-[(3r,5r,8r,9s,10s,13s,14s,17s)-3-ethynyl-3-hydroxy-10,13-dimethyl-1,2,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydrocyclopenta[a]phenanthren-17-yl]ethanone Chemical compound C1[C@@](O)(C#C)CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)C(=O)CBr)[C@@H]4[C@@H]3CC[C@@H]21 WZUCWPNRRXLFPH-PLHHIASCSA-N 0.000 claims description 3
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical class [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 claims description 3
- 125000000468 ketone group Chemical group 0.000 claims description 2
- 125000004950 trifluoroalkyl group Chemical group 0.000 claims 1
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 339
- 239000000243 solution Substances 0.000 description 265
- 239000000203 mixture Substances 0.000 description 221
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 190
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical group ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 157
- 239000007787 solid Substances 0.000 description 117
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 111
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 109
- AURFZBICLPNKBZ-UHFFFAOYSA-N Pregnanolone Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(=O)C)C1(C)CC2 AURFZBICLPNKBZ-UHFFFAOYSA-N 0.000 description 105
- 230000007958 sleep Effects 0.000 description 105
- AURFZBICLPNKBZ-YZRLXODZSA-N 3alpha-hydroxy-5beta-pregnan-20-one Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)C)[C@@]2(C)CC1 AURFZBICLPNKBZ-YZRLXODZSA-N 0.000 description 99
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 97
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 96
- 229950007402 eltanolone Drugs 0.000 description 92
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 90
- 238000006243 chemical reaction Methods 0.000 description 74
- CSNNHWWHGAXBCP-UHFFFAOYSA-L magnesium sulphate Substances [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 71
- 239000002904 solvent Substances 0.000 description 66
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 51
- 239000012044 organic layer Substances 0.000 description 47
- 239000000047 product Substances 0.000 description 46
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 44
- 239000012043 crude product Substances 0.000 description 44
- 235000019439 ethyl acetate Nutrition 0.000 description 44
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 40
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 39
- 238000009472 formulation Methods 0.000 description 38
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 36
- 229920006395 saturated elastomer Polymers 0.000 description 36
- 239000012267 brine Substances 0.000 description 35
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 35
- 150000003431 steroids Chemical class 0.000 description 35
- 238000010828 elution Methods 0.000 description 34
- 238000003756 stirring Methods 0.000 description 34
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 33
- 239000003814 drug Substances 0.000 description 30
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 30
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 29
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 28
- 229940079593 drug Drugs 0.000 description 28
- 239000010410 layer Substances 0.000 description 28
- 230000000694 effects Effects 0.000 description 26
- 238000003818 flash chromatography Methods 0.000 description 26
- 239000000741 silica gel Substances 0.000 description 25
- 229910002027 silica gel Inorganic materials 0.000 description 25
- 238000011282 treatment Methods 0.000 description 25
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 24
- ZAFYATHCZYHLPB-UHFFFAOYSA-N zolpidem Chemical compound N1=C2C=CC(C)=CN2C(CC(=O)N(C)C)=C1C1=CC=C(C)C=C1 ZAFYATHCZYHLPB-UHFFFAOYSA-N 0.000 description 23
- 229960001475 zolpidem Drugs 0.000 description 23
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 22
- 230000036760 body temperature Effects 0.000 description 22
- 230000036385 rapid eye movement (rem) sleep Effects 0.000 description 22
- 230000000147 hypnotic effect Effects 0.000 description 21
- JOFWLTCLBGQGBO-UHFFFAOYSA-N triazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1Cl JOFWLTCLBGQGBO-UHFFFAOYSA-N 0.000 description 21
- XMRPGKVKISIQBV-UHFFFAOYSA-N (+-)-5- Pregnane-3,20-dione Natural products C1CC2CC(=O)CCC2(C)C2C1C1CCC(C(=O)C)C1(C)CC2 XMRPGKVKISIQBV-UHFFFAOYSA-N 0.000 description 20
- 241000700159 Rattus Species 0.000 description 20
- 239000003153 chemical reaction reagent Substances 0.000 description 20
- 238000000034 method Methods 0.000 description 20
- 229960003386 triazolam Drugs 0.000 description 20
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 19
- 230000036470 plasma concentration Effects 0.000 description 19
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- 229910000027 potassium carbonate Inorganic materials 0.000 description 18
- 239000011541 reaction mixture Substances 0.000 description 18
- 239000007858 starting material Substances 0.000 description 17
- 238000001035 drying Methods 0.000 description 16
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 16
- 241000282414 Homo sapiens Species 0.000 description 15
- 239000012074 organic phase Substances 0.000 description 15
- 210000002966 serum Anatomy 0.000 description 15
- UIIMBOGNXHQVGW-UHFFFAOYSA-M sodium bicarbonate Substances [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 15
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 14
- 238000001816 cooling Methods 0.000 description 14
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 13
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 13
- 238000004587 chromatography analysis Methods 0.000 description 13
- 239000007788 liquid Substances 0.000 description 13
- 230000037023 motor activity Effects 0.000 description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- 239000002253 acid Substances 0.000 description 12
- 235000019270 ammonium chloride Nutrition 0.000 description 12
- 206010022437 insomnia Diseases 0.000 description 12
- 229920000858 Cyclodextrin Polymers 0.000 description 11
- 241000282412 Homo Species 0.000 description 11
- 239000003921 oil Substances 0.000 description 11
- 235000019198 oils Nutrition 0.000 description 11
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 11
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 11
- 239000001116 FEMA 4028 Substances 0.000 description 10
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 10
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 description 10
- 235000011175 beta-cyclodextrine Nutrition 0.000 description 10
- 229960004853 betadex Drugs 0.000 description 10
- 229910052794 bromium Inorganic materials 0.000 description 10
- 239000012141 concentrate Substances 0.000 description 10
- 235000008504 concentrate Nutrition 0.000 description 10
- 239000003480 eluent Substances 0.000 description 10
- 150000002148 esters Chemical class 0.000 description 10
- 239000000284 extract Substances 0.000 description 10
- 238000001914 filtration Methods 0.000 description 10
- 239000003326 hypnotic agent Substances 0.000 description 10
- 239000000651 prodrug Substances 0.000 description 10
- 229940002612 prodrug Drugs 0.000 description 10
- 229960000583 acetic acid Drugs 0.000 description 9
- 239000013078 crystal Substances 0.000 description 9
- 238000001647 drug administration Methods 0.000 description 9
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 9
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 9
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 9
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 9
- 238000001953 recrystallisation Methods 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- XMRPGKVKISIQBV-XWOJZHJZSA-N 5beta-pregnane-3,20-dione Chemical compound C([C@H]1CC2)C(=O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)C)[C@@]2(C)CC1 XMRPGKVKISIQBV-XWOJZHJZSA-N 0.000 description 8
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- 238000004440 column chromatography Methods 0.000 description 8
- 238000001704 evaporation Methods 0.000 description 8
- 230000008020 evaporation Effects 0.000 description 8
- 238000010898 silica gel chromatography Methods 0.000 description 8
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 7
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 7
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 7
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 7
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 7
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 7
- 230000008859 change Effects 0.000 description 7
- 239000007789 gas Substances 0.000 description 7
- 239000000377 silicon dioxide Substances 0.000 description 7
- 208000019116 sleep disease Diseases 0.000 description 7
- 239000011877 solvent mixture Substances 0.000 description 7
- RSRDWHPVTMQUGZ-OZIWPBGVSA-N 1-[(8r,9s,10s,13s,14s,17s)-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]ethanone Chemical class C1CC2CCCC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RSRDWHPVTMQUGZ-OZIWPBGVSA-N 0.000 description 6
- 239000005977 Ethylene Substances 0.000 description 6
- PGTVWKLGGCQMBR-FLBATMFCSA-N Ganaxolone Chemical compound C([C@@H]1CC2)[C@](C)(O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)C)[C@@]2(C)CC1 PGTVWKLGGCQMBR-FLBATMFCSA-N 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- 239000003054 catalyst Substances 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 102000005962 receptors Human genes 0.000 description 6
- 108020003175 receptors Proteins 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- 239000001384 succinic acid Substances 0.000 description 6
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 6
- XMRPGKVKISIQBV-BJMCWZGWSA-N 5alpha-pregnane-3,20-dione Chemical compound C([C@@H]1CC2)C(=O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)C)[C@@]2(C)CC1 XMRPGKVKISIQBV-BJMCWZGWSA-N 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 206010041349 Somnolence Diseases 0.000 description 5
- 125000003342 alkenyl group Chemical group 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 230000036765 blood level Effects 0.000 description 5
- 239000013058 crude material Substances 0.000 description 5
- 125000004122 cyclic group Chemical group 0.000 description 5
- 238000000354 decomposition reaction Methods 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 239000011777 magnesium Substances 0.000 description 5
- 239000012452 mother liquor Substances 0.000 description 5
- 150000002924 oxiranes Chemical class 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- 230000008667 sleep stage Effects 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 4
- HYQLYTIEHZUBGM-SIFBOAKMSA-N 2-(dimethylamino)-1-[(3R,5S,8R,9S,10S,13S,14S,17S)-3-hydroxy-3,10,13-trimethyl-1,2,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydrocyclopenta[a]phenanthren-17-yl]propan-1-one Chemical compound CC(C(=O)[C@H]1CC[C@@H]2[C@@]1(CC[C@H]3[C@H]2CC[C@@H]4[C@@]3(CC[C@@](C4)(C)O)C)C)N(C)C HYQLYTIEHZUBGM-SIFBOAKMSA-N 0.000 description 4
- IMKYPTWESUTRAJ-QNEXQYMPSA-N 2-hydroxy-1-[(3s,5s,8r,9s,10s,13s,14s,17s)-3,10,13-trimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]ethanone Chemical compound C1C[C@@H]2[C@@]3(C)CC[C@H](C)C[C@@H]3CC[C@H]2[C@@H]2CC[C@H](C(=O)CO)[C@]21C IMKYPTWESUTRAJ-QNEXQYMPSA-N 0.000 description 4
- VTBHBNXGFPTBJL-UHFFFAOYSA-N 4-tert-butyl-1-sulfanylidene-2,6,7-trioxa-1$l^{5}-phosphabicyclo[2.2.2]octane Chemical compound C1OP2(=S)OCC1(C(C)(C)C)CO2 VTBHBNXGFPTBJL-UHFFFAOYSA-N 0.000 description 4
- QGXBDMJGAMFCBF-HLUDHZFRSA-N 5α-Androsterone Chemical compound C1[C@H](O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC[C@H]21 QGXBDMJGAMFCBF-HLUDHZFRSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 4
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical group CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- 208000032140 Sleepiness Diseases 0.000 description 4
- FDCINQSOYQUNKB-UHFFFAOYSA-N UNPD98205 Natural products C1CC2C3(C)CCC(OC(=O)C)CC3CCC2C2CCC(=O)C21C FDCINQSOYQUNKB-UHFFFAOYSA-N 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 150000001336 alkenes Chemical class 0.000 description 4
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 239000012300 argon atmosphere Substances 0.000 description 4
- 210000004556 brain Anatomy 0.000 description 4
- AURFZBICLPNKBZ-SYBPFIFISA-N brexanolone Chemical compound C([C@@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)C)[C@@]2(C)CC1 AURFZBICLPNKBZ-SYBPFIFISA-N 0.000 description 4
- 230000000875 corresponding effect Effects 0.000 description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 4
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 4
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 238000000605 extraction Methods 0.000 description 4
- 230000001939 inductive effect Effects 0.000 description 4
- 150000002576 ketones Chemical class 0.000 description 4
- ACKFDYCQCBEDNU-UHFFFAOYSA-J lead(2+);tetraacetate Chemical compound [Pb+2].CC([O-])=O.CC([O-])=O.CC([O-])=O.CC([O-])=O ACKFDYCQCBEDNU-UHFFFAOYSA-J 0.000 description 4
- 239000003446 ligand Substances 0.000 description 4
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 4
- 239000004570 mortar (masonry) Substances 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 230000003647 oxidation Effects 0.000 description 4
- 238000007254 oxidation reaction Methods 0.000 description 4
- HFPZCAJZSCWRBC-UHFFFAOYSA-N p-cymene Chemical compound CC(C)C1=CC=C(C)C=C1 HFPZCAJZSCWRBC-UHFFFAOYSA-N 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 238000003127 radioimmunoassay Methods 0.000 description 4
- 230000001739 rebound effect Effects 0.000 description 4
- 230000033764 rhythmic process Effects 0.000 description 4
- 230000037321 sleepiness Effects 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 229940014800 succinic anhydride Drugs 0.000 description 4
- FKHIFSZMMVMEQY-UHFFFAOYSA-N talc Chemical compound [Mg+2].[O-][Si]([O-])=O FKHIFSZMMVMEQY-UHFFFAOYSA-N 0.000 description 4
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 4
- 125000004385 trihaloalkyl group Chemical group 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 235000013618 yogurt Nutrition 0.000 description 4
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 3
- NFERAWHWEBLQOA-PMFAJICFSA-N (3r,5s,8r,9s,10s,13s,14s,17s)-3-hydroxy-3,10,13-trimethyl-1,2,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydrocyclopenta[a]phenanthrene-17-carboxylic acid Chemical compound C1[C@](C)(O)CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)C(O)=O)[C@@H]4[C@@H]3CC[C@H]21 NFERAWHWEBLQOA-PMFAJICFSA-N 0.000 description 3
- UWTUEMKLYAGTNQ-OWOJBTEDSA-N (e)-1,2-dibromoethene Chemical group Br\C=C\Br UWTUEMKLYAGTNQ-OWOJBTEDSA-N 0.000 description 3
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- PPFHABGDIFKZDJ-WMCLBYQZSA-N CC(C(=O)[C@H]1CC[C@@H]2[C@@]1(CC[C@H]3[C@H]2CC[C@H]4[C@@]3(CC[C@H](C4)O)C)C)N(C)C Chemical compound CC(C(=O)[C@H]1CC[C@@H]2[C@@]1(CC[C@H]3[C@H]2CC[C@H]4[C@@]3(CC[C@H](C4)O)C)C)N(C)C PPFHABGDIFKZDJ-WMCLBYQZSA-N 0.000 description 3
- HEDRZPFGACZZDS-UHFFFAOYSA-N CHCl3 Substances ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 3
- QGXBDMJGAMFCBF-UHFFFAOYSA-N Etiocholanolone Natural products C1C(O)CCC2(C)C3CCC(C)(C(CC4)=O)C4C3CCC21 QGXBDMJGAMFCBF-UHFFFAOYSA-N 0.000 description 3
- 229910004373 HOAc Inorganic materials 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 3
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 3
- 125000003710 aryl alkyl group Chemical group 0.000 description 3
- 239000012148 binding buffer Substances 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 235000021152 breakfast Nutrition 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- HRLIOXLXPOHXTA-NSHDSACASA-N dexmedetomidine Chemical compound C1([C@@H](C)C=2C(=C(C)C=CC=2)C)=CN=C[N]1 HRLIOXLXPOHXTA-NSHDSACASA-N 0.000 description 3
- 229960004253 dexmedetomidine Drugs 0.000 description 3
- HDFFVHSMHLDSLO-UHFFFAOYSA-M dibenzyl phosphate Chemical compound C=1C=CC=CC=1COP(=O)([O-])OCC1=CC=CC=C1 HDFFVHSMHLDSLO-UHFFFAOYSA-M 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 230000000857 drug effect Effects 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 239000007943 implant Substances 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 229910052744 lithium Inorganic materials 0.000 description 3
- 235000019341 magnesium sulphate Nutrition 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- DVSDBMFJEQPWNO-UHFFFAOYSA-N methyllithium Chemical compound C[Li] DVSDBMFJEQPWNO-UHFFFAOYSA-N 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 239000002808 molecular sieve Substances 0.000 description 3
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000000186 progesterone Substances 0.000 description 3
- 229960003387 progesterone Drugs 0.000 description 3
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 230000001624 sedative effect Effects 0.000 description 3
- SQGYOTSLMSWVJD-UHFFFAOYSA-N silver(1+) nitrate Chemical compound [Ag+].[O-]N(=O)=O SQGYOTSLMSWVJD-UHFFFAOYSA-N 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 150000003462 sulfoxides Chemical class 0.000 description 3
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- YWYQTGBBEZQBGO-OQOYYRQQSA-N (3r,5r,8r,9s,10s,13s,14s,17s)-17-[(1r)-1-hydroxyethyl]-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-ol Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@H](O)C)[C@@]2(C)CC1 YWYQTGBBEZQBGO-OQOYYRQQSA-N 0.000 description 2
- VFBOQACQHXYULH-QWQRBHLCSA-N (5s,8r,9s,10s,13s,14s,17s)-10,13-dimethyl-17-(2-methyl-1,3-dioxolan-2-yl)-1,2,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydrocyclopenta[a]phenanthren-3-one Chemical compound CC1([C@H]2CC[C@H]3[C@H]4[C@@H]([C@]5(CCC(=O)C[C@@H]5CC4)C)CC[C@@]32C)OCCO1 VFBOQACQHXYULH-QWQRBHLCSA-N 0.000 description 2
- RTCGCDLZHDXORQ-UHFFFAOYSA-N 1,1,3-triphenylprop-2-ynylstannane Chemical compound C1(=CC=CC=C1)C(C#CC1=CC=CC=C1)(C1=CC=CC=C1)[SnH3] RTCGCDLZHDXORQ-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- BWZVCCNYKMEVEX-UHFFFAOYSA-N 2,4,6-Trimethylpyridine Chemical compound CC1=CC(C)=NC(C)=C1 BWZVCCNYKMEVEX-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- UOUIARGWRPHDBX-UHFFFAOYSA-N 3alpha-Hydroxy-5beta-oestran-17-on Natural products C1C(O)CCC2C3CCC(C)(C(CC4)=O)C4C3CCC21 UOUIARGWRPHDBX-UHFFFAOYSA-N 0.000 description 2
- QGXBDMJGAMFCBF-BNSUEQOYSA-N 3alpha-hydroxy-5beta-androstan-17-one Chemical compound C1[C@H](O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC[C@@H]21 QGXBDMJGAMFCBF-BNSUEQOYSA-N 0.000 description 2
- ALYNCZNDIQEVRV-UHFFFAOYSA-N 4-aminobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C=C1 ALYNCZNDIQEVRV-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 206010002091 Anaesthesia Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 208000019888 Circadian rhythm sleep disease Diseases 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- QXNVGIXVLWOKEQ-UHFFFAOYSA-N Disodium Chemical compound [Na][Na] QXNVGIXVLWOKEQ-UHFFFAOYSA-N 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 208000001456 Jet Lag Syndrome Diseases 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- OKIZCWYLBDKLSU-UHFFFAOYSA-M N,N,N-Trimethylmethanaminium chloride Chemical compound [Cl-].C[N+](C)(C)C OKIZCWYLBDKLSU-UHFFFAOYSA-M 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- YWYQTGBBEZQBGO-BERLURQNSA-N Pregnanediol Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](O)C)[C@@]2(C)CC1 YWYQTGBBEZQBGO-BERLURQNSA-N 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- 208000007271 Substance Withdrawal Syndrome Diseases 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 238000007239 Wittig reaction Methods 0.000 description 2
- 150000008065 acid anhydrides Chemical class 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 150000004703 alkoxides Chemical class 0.000 description 2
- 230000037005 anaesthesia Effects 0.000 description 2
- 230000003444 anaesthetic effect Effects 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 229940125717 barbiturate Drugs 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- FBPGAWABXWMRAR-UHFFFAOYSA-N benzenesulfinylmethylbenzene Chemical compound C=1C=CC=CC=1S(=O)CC1=CC=CC=C1 FBPGAWABXWMRAR-UHFFFAOYSA-N 0.000 description 2
- 229940049706 benzodiazepine Drugs 0.000 description 2
- 150000001557 benzodiazepines Chemical class 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 239000012455 biphasic mixture Substances 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- 229910000085 borane Inorganic materials 0.000 description 2
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical compound FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- RBHJBMIOOPYDBQ-UHFFFAOYSA-N carbon dioxide;propan-2-one Chemical compound O=C=O.CC(C)=O RBHJBMIOOPYDBQ-UHFFFAOYSA-N 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 230000001054 cortical effect Effects 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- QBKVEAIEIVKHGR-UHFFFAOYSA-M di(ethylidene)azanium;chloride Chemical compound [Cl-].CC=[N+]=CC QBKVEAIEIVKHGR-UHFFFAOYSA-M 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- 230000008034 disappearance Effects 0.000 description 2
- 229910000397 disodium phosphate Inorganic materials 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 230000035622 drinking Effects 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- JHYNXXDQQHTCHJ-UHFFFAOYSA-M ethyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(CC)C1=CC=CC=C1 JHYNXXDQQHTCHJ-UHFFFAOYSA-M 0.000 description 2
- 235000019197 fats Nutrition 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000003193 general anesthetic agent Substances 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 210000003128 head Anatomy 0.000 description 2
- 239000000017 hydrogel Substances 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000005286 illumination Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 239000002555 ionophore Substances 0.000 description 2
- 230000000236 ionophoric effect Effects 0.000 description 2
- 230000001788 irregular Effects 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 208000033915 jet lag type circadian rhythm sleep disease Diseases 0.000 description 2
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- RMGJCSHZTFKPNO-UHFFFAOYSA-M magnesium;ethene;bromide Chemical compound [Mg+2].[Br-].[CH-]=C RMGJCSHZTFKPNO-UHFFFAOYSA-M 0.000 description 2
- DQEUYIQDSMINEY-UHFFFAOYSA-M magnesium;prop-1-ene;bromide Chemical compound [Mg+2].[Br-].[CH2-]C=C DQEUYIQDSMINEY-UHFFFAOYSA-M 0.000 description 2
- YLERVAXAQFOFRI-UHFFFAOYSA-M magnesium;propa-1,2-diene;bromide Chemical compound [Mg+2].[Br-].[CH2-]C#C YLERVAXAQFOFRI-UHFFFAOYSA-M 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- LWJROJCJINYWOX-UHFFFAOYSA-L mercury dichloride Chemical compound Cl[Hg]Cl LWJROJCJINYWOX-UHFFFAOYSA-L 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 125000006682 monohaloalkyl group Chemical group 0.000 description 2
- 230000004973 motor coordination Effects 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 125000002524 organometallic group Chemical group 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- WLJVNTCWHIRURA-UHFFFAOYSA-N pimelic acid Chemical compound OC(=O)CCCCCC(O)=O WLJVNTCWHIRURA-UHFFFAOYSA-N 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 150000003128 pregnanes Chemical class 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 150000003146 progesterones Chemical class 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- 238000000159 protein binding assay Methods 0.000 description 2
- LEHBURLTIWGHEM-UHFFFAOYSA-N pyridinium chlorochromate Chemical compound [O-][Cr](Cl)(=O)=O.C1=CC=[NH+]C=C1 LEHBURLTIWGHEM-UHFFFAOYSA-N 0.000 description 2
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 230000004617 sleep duration Effects 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- 239000001488 sodium phosphate Substances 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 230000003595 spectral effect Effects 0.000 description 2
- 229910001220 stainless steel Inorganic materials 0.000 description 2
- 239000010935 stainless steel Substances 0.000 description 2
- 239000003270 steroid hormone Substances 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- DDFYFBUWEBINLX-UHFFFAOYSA-M tetramethylammonium bromide Chemical compound [Br-].C[N+](C)(C)C DDFYFBUWEBINLX-UHFFFAOYSA-M 0.000 description 2
- OSBSFAARYOCBHB-UHFFFAOYSA-N tetrapropylammonium Chemical compound CCC[N+](CCC)(CCC)CCC OSBSFAARYOCBHB-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- GKASDNZWUGIAMG-UHFFFAOYSA-N triethyl orthoformate Chemical compound CCOC(OCC)OCC GKASDNZWUGIAMG-UHFFFAOYSA-N 0.000 description 2
- MWKJTNBSKNUMFN-UHFFFAOYSA-N trifluoromethyltrimethylsilane Chemical compound C[Si](C)(C)C(F)(F)F MWKJTNBSKNUMFN-UHFFFAOYSA-N 0.000 description 2
- XMQSELBBYSAURN-UHFFFAOYSA-M triphenyl(propyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(CCC)C1=CC=CC=C1 XMQSELBBYSAURN-UHFFFAOYSA-M 0.000 description 2
- 238000005292 vacuum distillation Methods 0.000 description 2
- WJUFSDZVCOTFON-UHFFFAOYSA-N veratraldehyde Chemical compound COC1=CC=C(C=O)C=C1OC WJUFSDZVCOTFON-UHFFFAOYSA-N 0.000 description 2
- 238000010792 warming Methods 0.000 description 2
- LLYVBPVTNFOUPC-IZIDRRSZSA-N (1S,2S,5S,9S,10R,13S)-1,5-dimethyl-16,19-dioxapentacyclo[11.8.0.02,10.05,9.015,20]henicosan-6-one Chemical compound C1OC2C[C@@H]3CC[C@@H]4[C@H](CC[C@@]5(C(CC[C@H]54)=O)C)[C@]3(CC2OC1)C LLYVBPVTNFOUPC-IZIDRRSZSA-N 0.000 description 1
- BIJDHLPGEUAMQE-DISARSAHSA-N (3R,5S,8R,9S,10S,13S,14S,17S)-10,13-dimethyl-17-[2-[(E)-3-phenylprop-2-enyl]-1,3-dioxolan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-ol Chemical compound C1OC(C\C=C\C2=CC=CC=C2)([C@H]2CC[C@H]3[C@@H]4CC[C@H]5C[C@@H](CC[C@]5(C)[C@H]4CC[C@]23C)O)OC1 BIJDHLPGEUAMQE-DISARSAHSA-N 0.000 description 1
- GRYPSUHLFHWJNS-VCKBKBOKSA-N (3R,5S,8S,9S,10S,13R,14S,17S)-17-ethyl-10,13-dimethylspiro[1,2,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydrocyclopenta[a]phenanthrene-3,2'-oxirane] Chemical compound O1[C@@]2(C1)C[C@@H]1CC[C@H]3[C@@H]4CC[C@H](CC)[C@]4(CC[C@@H]3[C@]1(CC2)C)C GRYPSUHLFHWJNS-VCKBKBOKSA-N 0.000 description 1
- DVTDUTYENPZLFP-YBXIWKLVSA-N (3r,5s,8r,9s,10s,13s,14s,17s)-3-hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthrene-17-carbonitrile Chemical compound C1[C@H](O)CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)C#N)[C@@H]4[C@@H]3CC[C@H]21 DVTDUTYENPZLFP-YBXIWKLVSA-N 0.000 description 1
- YJDYCULVYZDESB-HQEMIIEJSA-N (8r,9s,10s,13s,14s)-10,13-dimethyl-1,2,3,4,5,6,7,8,9,11,12,14,15,16-tetradecahydrocyclopenta[a]phenanthren-17-one Chemical compound C1CCC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC21 YJDYCULVYZDESB-HQEMIIEJSA-N 0.000 description 1
- YBBNAGKQYPHZHZ-XFNFOBRPSA-N (8s,9s,10s,13r,14s,17r)-17-(2-bromoethyl)-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthrene Chemical class C1CCC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)CCBr)[C@@H]4[C@@H]3CCC21 YBBNAGKQYPHZHZ-XFNFOBRPSA-N 0.000 description 1
- UWTUEMKLYAGTNQ-UHFFFAOYSA-N 1,2-dibromoethene Chemical group BrC=CBr UWTUEMKLYAGTNQ-UHFFFAOYSA-N 0.000 description 1
- AUHZEENZYGFFBQ-UHFFFAOYSA-N 1,3,5-Me3C6H3 Natural products CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- JZJWCDQGIPQBAO-UHFFFAOYSA-N 1-(4-iodophenyl)ethanone Chemical compound CC(=O)C1=CC=C(I)C=C1 JZJWCDQGIPQBAO-UHFFFAOYSA-N 0.000 description 1
- YPEARZUTWVZHIZ-SRJHXTLLSA-N 1-[(8r,9s,10s,13s,14s,17s)-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]ethanol Chemical class C1CC2CCCC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(O)C)[C@@]1(C)CC2 YPEARZUTWVZHIZ-SRJHXTLLSA-N 0.000 description 1
- 125000004066 1-hydroxyethyl group Chemical group [H]OC([H])([*])C([H])([H])[H] 0.000 description 1
- BCFPLUJRJLEPGV-UHFFFAOYSA-N 1-methyl-1,4-dihydropyridin-1-ium-3-carboxylate Chemical compound CN1C=CCC(C(O)=O)=C1 BCFPLUJRJLEPGV-UHFFFAOYSA-N 0.000 description 1
- UUFQTNFCRMXOAE-UHFFFAOYSA-N 1-methylmethylene Chemical compound C[CH] UUFQTNFCRMXOAE-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- FHTDDANQIMVWKZ-UHFFFAOYSA-N 1h-pyridine-4-thione Chemical compound SC1=CC=NC=C1 FHTDDANQIMVWKZ-UHFFFAOYSA-N 0.000 description 1
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- YBCCBWKFKREPNU-DATPGIFZSA-N 2-bromo-1-[(3r,5r,8r,9s,10s,13s,14s,17s)-3-hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]ethanone Chemical compound C1[C@H](O)CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)C(=O)CBr)[C@@H]4[C@@H]3CC[C@@H]21 YBCCBWKFKREPNU-DATPGIFZSA-N 0.000 description 1
- MQHMRKCTPSYYCT-VQOUBXEKSA-N 2-bromo-1-[(3r,5s,8r,9s,10s,13s,14s,17s)-3-hydroxy-3,10,13-trimethyl-1,2,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydrocyclopenta[a]phenanthren-17-yl]ethanone Chemical compound C1[C@](C)(O)CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)C(=O)CBr)[C@@H]4[C@@H]3CC[C@H]21 MQHMRKCTPSYYCT-VQOUBXEKSA-N 0.000 description 1
- PTQSYCCQENSDIL-JIUSCHCVSA-N 2-bromo-1-[(8r,9s,10s,13s,14s,17s)-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]ethanone Chemical compound C1CCC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)C(=O)CBr)[C@@H]4[C@@H]3CCC21 PTQSYCCQENSDIL-JIUSCHCVSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- WPVLYXWAESTBRZ-UHFFFAOYSA-N 3-bromoprop-1-yne 1,1,3-triphenylprop-2-ynylstannane Chemical compound C(C#C)Br.C1(=CC=CC=C1)C(C#CC1=CC=CC=C1)(C1=CC=CC=C1)[SnH3] WPVLYXWAESTBRZ-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- BXRFQSNOROATLV-UHFFFAOYSA-N 4-nitrobenzaldehyde Chemical compound [O-][N+](=O)C1=CC=C(C=O)C=C1 BXRFQSNOROATLV-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 206010001497 Agitation Diseases 0.000 description 1
- 229910015900 BF3 Inorganic materials 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- BGXKFSTWOUXSDA-UHFFFAOYSA-N CI.C[S+](=O)(C)C Chemical compound CI.C[S+](=O)(C)C BGXKFSTWOUXSDA-UHFFFAOYSA-N 0.000 description 1
- GEEYLXLQQKCJTI-UHFFFAOYSA-N C[Si](C)(C)[N-][Si](C)(C)C.[Li+].P(=O)(OCC)(OCC)O Chemical compound C[Si](C)(C)[N-][Si](C)(C)C.[Li+].P(=O)(OCC)(OCC)O GEEYLXLQQKCJTI-UHFFFAOYSA-N 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 206010010904 Convulsion Diseases 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 206010049119 Emotional distress Diseases 0.000 description 1
- 102000005915 GABA Receptors Human genes 0.000 description 1
- 108010005551 GABA Receptors Proteins 0.000 description 1
- 102000004300 GABA-A Receptors Human genes 0.000 description 1
- 108090000839 GABA-A Receptors Proteins 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 238000003747 Grignard reaction Methods 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 229930194542 Keto Chemical group 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229910010082 LiAlH Inorganic materials 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- SKCPISXYQBYXQK-SOPGZXPBSA-N N1=CC=C(C=C1)SCC([C@H]1CC[C@H]2[C@@H]3CC[C@@H]4CCCC[C@]4(C)[C@H]3CC[C@]12C)=O Chemical compound N1=CC=C(C=C1)SCC([C@H]1CC[C@H]2[C@@H]3CC[C@@H]4CCCC[C@]4(C)[C@H]3CC[C@]12C)=O SKCPISXYQBYXQK-SOPGZXPBSA-N 0.000 description 1
- 229910003251 Na K Inorganic materials 0.000 description 1
- 229920002274 Nalgene Polymers 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- QGMRQYFBGABWDR-UHFFFAOYSA-M Pentobarbital sodium Chemical compound [Na+].CCCC(C)C1(CC)C(=O)NC(=O)[N-]C1=O QGMRQYFBGABWDR-UHFFFAOYSA-M 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 206010062519 Poor quality sleep Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 206010037660 Pyrexia Diseases 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 206010040981 Sleep attacks Diseases 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- GUGOEEXESWIERI-UHFFFAOYSA-N Terfenadine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GUGOEEXESWIERI-UHFFFAOYSA-N 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000005282 allenyl group Chemical group 0.000 description 1
- 229940125516 allosteric modulator Drugs 0.000 description 1
- 150000001371 alpha-amino acids Chemical class 0.000 description 1
- 235000008206 alpha-amino acids Nutrition 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229960004050 aminobenzoic acid Drugs 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 230000001387 anti-histamine Effects 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 230000037007 arousal Effects 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 230000003416 augmentation Effects 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- LKMCJXXOBRCATQ-UHFFFAOYSA-N benzylsulfanylbenzene Chemical compound C=1C=CC=CC=1CSC1=CC=CC=C1 LKMCJXXOBRCATQ-UHFFFAOYSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 125000005841 biaryl group Chemical group 0.000 description 1
- 150000005347 biaryls Chemical group 0.000 description 1
- 230000027455 binding Effects 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- IKWKJIWDLVYZIY-UHFFFAOYSA-M butyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(CCCC)C1=CC=CC=C1 IKWKJIWDLVYZIY-UHFFFAOYSA-M 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 238000003965 capillary gas chromatography Methods 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 210000003710 cerebral cortex Anatomy 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 230000002060 circadian Effects 0.000 description 1
- KFUSEUYYWQURPO-UPHRSURJSA-N cis-1,2-dichloroethene Chemical group Cl\C=C/Cl KFUSEUYYWQURPO-UPHRSURJSA-N 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- YNHIGQDRGKUECZ-UHFFFAOYSA-N dichloropalladium;triphenylphosphanium Chemical compound Cl[Pd]Cl.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-N 0.000 description 1
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 1
- LGTLXDJOAJDFLR-UHFFFAOYSA-N diethyl chlorophosphate Chemical compound CCOP(Cl)(=O)OCC LGTLXDJOAJDFLR-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000003651 drinking water Substances 0.000 description 1
- 235000020188 drinking water Nutrition 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- QGXBDMJGAMFCBF-LUJOEAJASA-N epiandrosterone Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC[C@H]21 QGXBDMJGAMFCBF-LUJOEAJASA-N 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 230000000763 evoking effect Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- NJVOZLGKTAPUTQ-UHFFFAOYSA-M fentin chloride Chemical compound C=1C=CC=CC=1[Sn](C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 NJVOZLGKTAPUTQ-UHFFFAOYSA-M 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 229960002598 fumaric acid Drugs 0.000 description 1
- GDSRMADSINPKSL-HSEONFRVSA-N gamma-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO GDSRMADSINPKSL-HSEONFRVSA-N 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- UHUSDOQQWJGJQS-UHFFFAOYSA-N glycerol 1,2-dioctadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(CO)OC(=O)CCCCCCCCCCCCCCCCC UHUSDOQQWJGJQS-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 229960004275 glycolic acid Drugs 0.000 description 1
- 229940027804 halcion Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000004970 halomethyl group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- KDCIHNCMPUBDKT-UHFFFAOYSA-N hexane;propan-2-one Chemical compound CC(C)=O.CCCCCC KDCIHNCMPUBDKT-UHFFFAOYSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 230000003284 homeostatic effect Effects 0.000 description 1
- 235000003642 hunger Nutrition 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 238000007327 hydrogenolysis reaction Methods 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 150000004694 iodide salts Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 210000004936 left thumb Anatomy 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 238000005567 liquid scintillation counting Methods 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- ARNWQMJQALNBBV-UHFFFAOYSA-N lithium carbide Chemical compound [Li+].[Li+].[C-]#[C-] ARNWQMJQALNBBV-UHFFFAOYSA-N 0.000 description 1
- GNSLYOGEBCFYDE-UHFFFAOYSA-N lithium;ethynylbenzene Chemical compound [Li+].[C-]#CC1=CC=CC=C1 GNSLYOGEBCFYDE-UHFFFAOYSA-N 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VXWPONVCMVLXBW-UHFFFAOYSA-M magnesium;carbanide;iodide Chemical compound [CH3-].[Mg+2].[I-] VXWPONVCMVLXBW-UHFFFAOYSA-M 0.000 description 1
- QGEFGPVWRJCFQP-UHFFFAOYSA-M magnesium;methanidylbenzene;bromide Chemical compound [Mg+2].[Br-].[CH2-]C1=CC=CC=C1 QGEFGPVWRJCFQP-UHFFFAOYSA-M 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229940098895 maleic acid Drugs 0.000 description 1
- 235000012054 meals Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- NOCGROPYCGRERZ-UHFFFAOYSA-M methoxymethyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(COC)C1=CC=CC=C1 NOCGROPYCGRERZ-UHFFFAOYSA-M 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- LSEFCHWGJNHZNT-UHFFFAOYSA-M methyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(C)C1=CC=CC=C1 LSEFCHWGJNHZNT-UHFFFAOYSA-M 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229960001207 micronized progesterone Drugs 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 230000008450 motivation Effects 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229940105631 nembutal Drugs 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000000820 nonprescription drug Substances 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 230000006911 nucleation Effects 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 125000000962 organic group Chemical group 0.000 description 1
- 238000006053 organic reaction Methods 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 230000036407 pain Effects 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- UEXCJVNBTNXOEH-UHFFFAOYSA-N phenyl acethylene Natural products C#CC1=CC=CC=C1 UEXCJVNBTNXOEH-UHFFFAOYSA-N 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- ANRQGKOBLBYXFM-UHFFFAOYSA-M phenylmagnesium bromide Chemical compound Br[Mg]C1=CC=CC=C1 ANRQGKOBLBYXFM-UHFFFAOYSA-M 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 229960004838 phosphoric acid Drugs 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical compound [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000006069 physical mixture Substances 0.000 description 1
- 229920000191 poly(N-vinyl pyrrolidone) Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229910052573 porcelain Inorganic materials 0.000 description 1
- NNFCIKHAZHQZJG-UHFFFAOYSA-N potassium cyanide Chemical compound [K+].N#[C-] NNFCIKHAZHQZJG-UHFFFAOYSA-N 0.000 description 1
- 238000009101 premedication Methods 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 239000000955 prescription drug Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000002250 progressing effect Effects 0.000 description 1
- 229940095574 propionic acid Drugs 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- OSFBJERFMQCEQY-UHFFFAOYSA-N propylidene Chemical compound [CH]CC OSFBJERFMQCEQY-UHFFFAOYSA-N 0.000 description 1
- 230000001698 pyrogenic effect Effects 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000004461 rapid eye movement Effects 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 230000001020 rhythmical effect Effects 0.000 description 1
- 210000004935 right thumb Anatomy 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 239000000932 sedative agent Substances 0.000 description 1
- 230000001953 sensory effect Effects 0.000 description 1
- 230000037152 sensory function Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000007873 sieving Methods 0.000 description 1
- 210000003625 skull Anatomy 0.000 description 1
- 201000002859 sleep apnea Diseases 0.000 description 1
- 208000022925 sleep disturbance Diseases 0.000 description 1
- 230000004622 sleep time Effects 0.000 description 1
- 230000037046 slow wave activity Effects 0.000 description 1
- 235000011888 snacks Nutrition 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 229940032330 sulfuric acid Drugs 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 1
- 230000001360 synchronised effect Effects 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical compound CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 150000003548 thiazolidines Chemical class 0.000 description 1
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 239000012485 toluene extract Substances 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- CFOAUYCPAUGDFF-UHFFFAOYSA-N tosmic Chemical compound CC1=CC=C(S(=O)(=O)C[N+]#[C-])C=C1 CFOAUYCPAUGDFF-UHFFFAOYSA-N 0.000 description 1
- 231100000440 toxicity profile Toxicity 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- YLGRTLMDMVAFNI-UHFFFAOYSA-N tributyl(prop-2-enyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)CC=C YLGRTLMDMVAFNI-UHFFFAOYSA-N 0.000 description 1
- 238000006692 trifluoromethylation reaction Methods 0.000 description 1
- BPLKQGGAXWRFOE-UHFFFAOYSA-M trimethylsulfoxonium iodide Chemical compound [I-].C[S+](C)(C)=O BPLKQGGAXWRFOE-UHFFFAOYSA-M 0.000 description 1
- AVWQQPYHYQKEIZ-UHFFFAOYSA-K trisodium;2-dodecylbenzenesulfonate;3-dodecylbenzenesulfonate;4-dodecylbenzenesulfonate Chemical compound [Na+].[Na+].[Na+].CCCCCCCCCCCCC1=CC=C(S([O-])(=O)=O)C=C1.CCCCCCCCCCCCC1=CC=CC(S([O-])(=O)=O)=C1.CCCCCCCCCCCCC1=CC=CC=C1S([O-])(=O)=O AVWQQPYHYQKEIZ-UHFFFAOYSA-K 0.000 description 1
- 229960003986 tuaminoheptane Drugs 0.000 description 1
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 238000009423 ventilation Methods 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000011345 viscous material Substances 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
Images
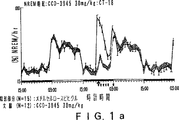
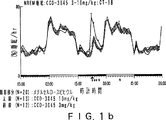

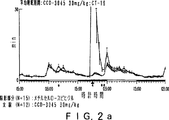
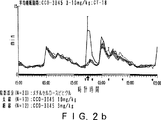


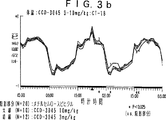



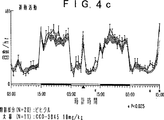

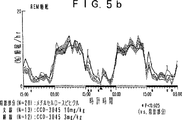

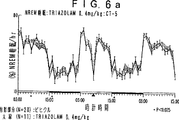

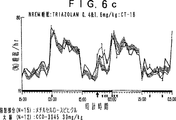

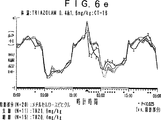
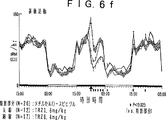

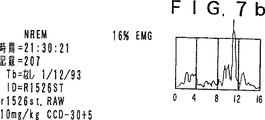


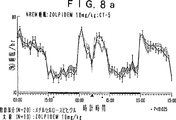




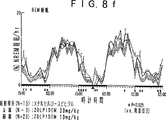
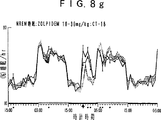
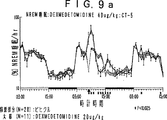






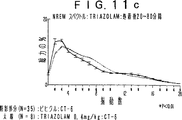


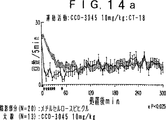

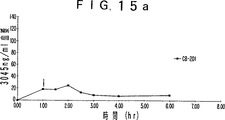
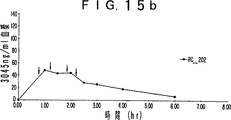
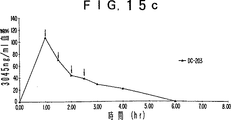
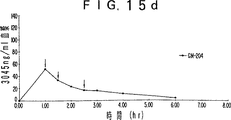
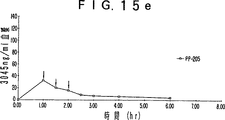
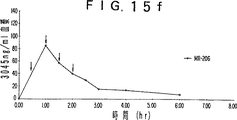
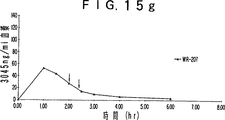
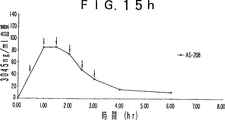

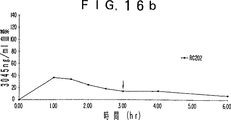
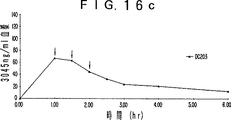

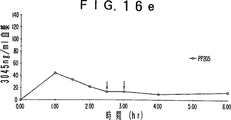
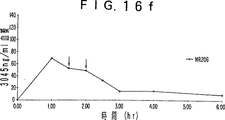


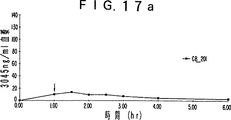


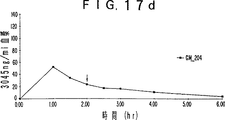
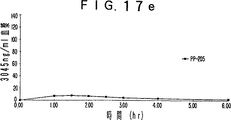
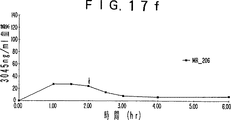

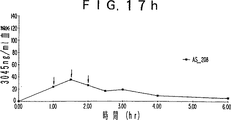


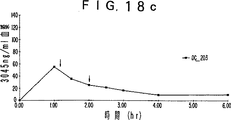
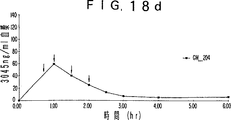
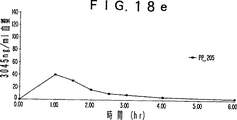
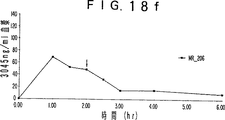

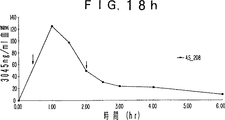




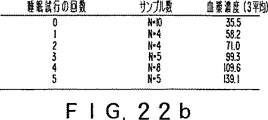



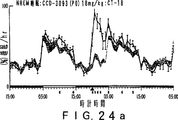




Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J43/003—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/565—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids not substituted in position 17 beta by a carbon atom, e.g. estrane, estradiol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/575—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of three or more carbon atoms, e.g. cholane, cholestane, ergosterol, sitosterol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/58—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids containing heterocyclic rings, e.g. danazol, stanozolol, pancuronium or digitogenin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J17/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, having an oxygen-containing hetero ring not condensed with the cyclopenta(a)hydrophenanthrene skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J31/00—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring
- C07J31/006—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring not covered by C07J31/003
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J33/00—Normal steroids having a sulfur-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J33/002—Normal steroids having a sulfur-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J41/00—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring
- C07J41/0033—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005
- C07J41/005—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005 the 17-beta position being substituted by an uninterrupted chain of only two carbon atoms, e.g. pregnane derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J41/00—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring
- C07J41/0033—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005
- C07J41/0094—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005 containing nitrile radicals, including thiocyanide radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J5/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond
- C07J5/0007—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond not substituted in position 17 alfa
- C07J5/0015—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond not substituted in position 17 alfa not substituted in position 16
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J51/00—Normal steroids with unmodified cyclopenta(a)hydrophenanthrene skeleton not provided for in groups C07J1/00 - C07J43/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J7/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms
- C07J7/0005—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21
- C07J7/001—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group
- C07J7/0015—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group not substituted in position 17 alfa
- C07J7/002—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group not substituted in position 17 alfa not substituted in position 16
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J7/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms
- C07J7/008—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms substituted in position 21
- C07J7/0085—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms substituted in position 21 by an halogen atom
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Anesthesiology (AREA)
- Toxicology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Steroid Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Description
この出願は1993年5月24日付で出願された同時係属出願第08/068,378号の一部継続であり;更にこれは1991年8月13日付で出願されて1993年8月3日付で特許第5,232,917号として発行された第745,216号の一部継続であり;更にこれは1990年5月10日付で出願されて1992年6月9日付で特許第5,120,723号として発行された出願第521,724号の一部継続であり;更にこれは1989年7月13日付で出願されて現在放棄されている出願第379,047号の一部継続であり;更にこれは1987年8月25日付で出願されて現在放棄されている第089,362号の一部継続である。しかも、この出願は1994年2月14日付で出願された第196,919号の一部継続であり;更にこれは1992年3月4日付で出願された第846,193号の一部継続であって、これは優先日が上記されている1993年8月3日付で発行された特許第5,232,917号の一部継続である。しかも、1994年2月14日付で出願された第196,919号は1993年8月2日付で出願された第101,497号の一部継続であり、更にこれは優先日が上記されている特許第5,232,917号の一部継続である。しかも、この出願は1994年2月14日付で出願された第196,972号の一部継続であり、更にこれは優先日が上記されている1993年8月2日付で出願された第101,497号の一部継続である。
背 景
本発明は、ヒトで睡眠を誘導するために、γ−アミノ酪酸レセプター−塩化物イオノホア複合体(“GABAAレセプター複合体”)と相互作用させる方法に関する。更に詳しくは、本発明は不眠症の治療におけるある3α−ヒドロキシ−5−還元ステロイドおよびその誘導体の用途に関する。
ステロイド、GABAAレセプター複合体および脳興奮性の関係は米国特許第5,120,723号および第5,232,917号明細書で既に記載されており、これらはそれら全ては引用することにより本明細書の開示の一部される。本発明者らは、このレセプター複合体のステロイド部位で作用する、神経作用ステロイドと称されるあるステロイドが、睡眠を誘導するため、特に不眠症を治療するために使用できることを発見した。例えば、本発明者らは、3α−ヒドロキシ−5β−プレグナン−20−オン(“プレグナノロン”)が経口投与されたときに睡眠を誘導できることを発見した。Arafatとその共同研究者らは、プロゲステロン代謝産物プレグナノロンおよび3α−ヒドロキシ−5α−プレグナン−20−オンとヒトでプロゲステロン投与後の睡眠との関係について明らかにした。Arafat,E.S.,Hargrove,J.T.,Maxson,W.G.,Desiderio,D.M.,Wentz,A.C.and Anderson,R.N.,"Sedative and Hypnotic Effects of Oral Administration of Micronized Progesterone May Be Mediated Through Its Metabolites",Am.J.Obstet.Gynecol.,159:1203-1209(1988)。プレグナノロンの経口活性は意外であり、Laszlo Gyermekはそれが"Pregnanolone: a Highly Potent,Naturally Occurring Hypnotic-Anesthetic Agent",Proc.Soc.Exp.Biol.Med.,1967,125:1058-1062において経口でほとんど不活性であることを見出していたからである。
しかしながら、初期の研究者らは、神経作用ステロイドがバルビツレート類のように作用して、睡眠を誘導するために用いられたとき麻酔の望ましくない副作用、低い治療インデックス、乱用可能性と、リバウンド不眠症のような有害副作用を表すことを示したため、プレグナノロンの経口活性処方を作る誘因は存在しなかった。Majewska,M.D.,Harrison,N.L.,Schwartz,R,D.,Barker,J.L.and Paul,S.M.,"Steroid Hormone Metabolites Are Barbiturate-Like Modulators of the GABA Receptor",Science,232:1004-1007,1986。
麻酔ステロイドのプロゲステロンは麻酔用量以下で投与されたときネコで睡眠を誘導することが、1967年以来知られている。Heuser,G.,"Induction of Anesthesia,Seizures and Sleep by Steroid Hormones",Anesthesiology,28:173-183(1967)。Heuserはプロゲステロンがネコで“自然”睡眠を起こすと結論付けたが、その理由はREM睡眠が薬物の投与中に抑制されず、しかもREM睡眠が薬物の投与中止で増加しなかったからである。重要なことに、Heuserはすべての麻酔ステロイドがこの“自然”睡眠を起こせるわけではないことを発見した。
The National Commission on Sleep Disorders Researchは、4千万のアメリカ人が不眠症、睡眠発作および睡眠無呼吸のような慢性睡眠障害にかかっているとみている。2〜3千万のアメリカ人が断続的睡眠関連問題を経験しているとみている。"Wake Up America: A National Sleep Alert",Vol.1,p.vi;Draft Report of the National Commission on Sleep Disorders Research,1992。不眠症および他の睡眠問題の大きな発生率は、正常な睡眠−覚醒パターンが情緒的苦痛、薬物、ジェットラグ、シフト作業、病気、そして痛みまたは空腹のような強い感覚刺激により遮断されうるとみている場合には、意外ではない。
睡眠障害および眠気のこの高発生率の社会的コストは重大である。これらのコストには生産性低下、知覚機能低下、アクシデント可能性増加、高い罹患および死亡リスクと、生活の質劣化がある(前掲)。このコミッションは、1990年だけで、睡眠障害および眠気のせいで、Exxon Valdezに基づくような睡眠関連事故による間接的コストを含めない直接的コストだけでも、米国において少くとも159億ドルを費やしている見積もった(前掲)。社会は切実に睡眠障害にとり安全で有効な治療法を要している。
現在、不眠症は様々な薬物で治療されているが、それらはすべてある欠点を有している。一般的治療は商品名Halcionで知られる薬物Triazolamの使用である。その欠点にはリバウンド不眠症と限定的な睡眠増加効力がある。リバウンド不眠症とは、薬物が消失するときにおける、あるいは薬物耐性または治療が停止されたときの禁断症状の結果としての、異常睡眠パターンの再発である。新規不眠症薬物Zolpidemもリバウンド不眠症を起こす傾向がある。
本発明による化合物は現行薬物より優れた特徴を有している。本発明は睡眠の速い開始、高い効能および効力と、最少のリバウンド不眠症を示す不眠症治療法を提供する。
本発明を十分に理解する上で役立てるため、睡眠の概要およびその研究の方法を下記に記載する。睡眠は2組の基準:1)電気生理学的、および2)挙動的基準を用いてしばしば規定される。電気生理学的基準には脳電図(EEG)、眼電図(EOG)および筋電図(EMG)がある。睡眠について研究する上で用いられる最も普通の基準は、脳の電気活動の記録であるEEGである。眠っている個体の皮質EEGを研究することにより、科学者は異なる電気的波形パターンの出現で示される2つの大きな睡眠段階があることをみつけた。これらの2段階はノンREM(NREM)およびREMと呼ばれる。NREM睡眠は慣例的に4つの段階に分けられ、EEGパターンは睡眠紡錘波、K複合波および高電圧徐波のような特徴的波形と同期して通常記載される。Kryger,M.H.,Roth,T.and Dement,W.C.,Principles and Practice of Sleep Medicine,p.3,1989(以下"PPSM"という)。REN睡眠は通常それより大きなEEG活動、筋肉アトニーおよび一過性の急速眼運動を示す(前掲)。
ヒトが睡眠のために目を閉じているとき、皮質電気リズムは8〜13Hzのリズムで同期している。これはα−リズムとして知られる。睡眠の第一段階で取られるEEGはそれより遅いリズム(4〜6Hz)および低い振幅を示す。Bowman,W.C.and Rand,M.J.,Textbook of Pharmacology,p6.24,2nd ed.,1980(以下、Bowman and Rand)。睡眠の第二段階では、1〜5Hzのより不規則で遅いEEG波がより大きな振幅で現れる。これらの不規則で遅い波は睡眠紡錘波と呼ばれるより速い波の出現で分散される。この第二段階の睡眠には、小さな鋭い波、その後1または2つのより大きな波、次いで約12Hzのより速い波からなるK複合波もある(前掲)。段階2は睡眠時間の45〜55%に相当する。PPSM,P.9。
段階3の睡眠は高電圧で特徴付けられ、遅いEEG波活動はこの段階でEEG活動の20〜50%に相当する。PPSM,p.7。段階4において、高電圧、徐波活動はEEGの>50%に相当し、K複合波は治験者の名前を呼ぶような刺激により喚起できない。PPSM,p.7;Bowman and Rand,p.6.24。
約1時間の睡眠後に、EEGパターンは同期していない4〜10Hzリズムの低振幅波までスピードアップする。これはREM睡眠であり、しばしばパラ睡眠と呼ばれ、その理由はEEGが覚醒の場合と似ているためであるが、それは非常に深い睡眠である。Bowman and Rand,P.6.24。REM睡眠の生物学的重要性はまだ不明である。しかしながら、REM睡眠の強いアルコール阻害は睡眠破壊と関係していた。Triazolamと他の不眠症薬物はREM睡眠を減少させるが、本発明による化合物はREM睡眠を最少で壊すにすぎないという利点を更に有している。
発明の要旨
本発明はヒトにおいて睡眠を誘導するための方法に関する。更に詳しくは、本発明は、不眠症を治療するための、GABAAレセプター複合体における最近特定された部位で作用するある3α−ヒドロキシル化−5−還元ステロイド誘導体およびそのプロドラッグの使用に関する。
本発明者らによる実験では、本発明を実施するときに用いられる化合物が睡眠の速い開始を行い、高い効能と効力を有することを明らかにした。それらは不眠症を治療するために用いられる既知催眠薬よりも少ないリバウンド不眠症および少ない体温への影響を示す。
【図面の簡単な説明】
本発明の上記および他の特徴、態様および利点は、以下の記載、添付された請求の範囲および添付図面からもっと良く理解されるようになるであろう。
図1Aおよび1Bは、日周期18時(CT−18)で各々投与された30mg/kgおよび3&10mg/kgのプレグナノロンに関する、ラットのNREM睡眠率/hr vs.時計時間のプロットである。
図1Cは、CT−5で投与された10mg/kgのプレグナノロンに関する、ラットのNREM睡眠率/hr vs.時計時間のプロットである。
図2Aおよび2Bは、CT−18で各々投与された30mg/kgおよび3&10mg/kgのプレグナノロンに関する、睡眠に費やすmin vs.時計時間のプロットである。
図2Cは、CT−5で投与された10mg/kgのプレグナノロンに関する、睡眠に費やすmin vs.時計時間のプロットである。
図3Aおよび3Bは、CT−18で各々投与された30mg/kgおよび3&10mg/kgのプレグナノロンに関する、体温の平均偏差vs.時計時間のプロットである。
図3Cは、CT−5で投与された10mg/kgのプレグナノロンに関する、体温の平均偏差vs.時計時間のプロットである。
図4Aおよび4Bは、CT−18で各々投与された30mg/kgおよび3&10mg/kgのプレグナノロンに関する、動いた回数/hr(運動活動)vs.時計時間のプロットである。
図4Cは、CT−5で投与された10mg/kgのプレグナノロンに関する、動いた回数/hr(運動活動)vs.時計時間のプロットである。
図5Aおよび5Bは、CT−18で各々投与された30mg/kgおよび3&10mg/kgのプレグナノロンに関する、REM睡眠率/hr vs.時計時間のプロットである。
図5Cは、CT−5で投与された10mg/kgのプレグナノロンに関する、REM睡眠率/hr vs.時計時間のプロットである。
図6A、6Bおよび6Cは、CT−5で投与された0.4mg/kgのTriazolam、CT−18で投与された0.4mg/kgのTriazolamと、CT−18で投与された0.4mg/kgおよび1.6mg/kgのTriazolamに各々関する、NREM睡眠率/hr vs.時計時間のプロットである。
図6Dは、CT−18で投与された0.4mg/kgおよび1.6mg/kgのTriazolamに関する、睡眠に費やすmin vs.時計時間のプロットである。
図6Eは、CT−18で投与された0.4mg/kgおよび1.6mg/kgのTriazolamに関する、体温の平均偏差vs.時計時間のプロットである。
図6Fは、CT−18で投与された0.4mg/kgおよび1.6mg/kgのTriazolamに関する、体動回数/hr(運動活動)vs.時計時間のプロットである。
図6Gは、CT−18で投与された0.4mg/kgおよび1.6mg/kgのTriazolamに関する、REM睡眠率/hr vs.時計時間のプロットである。
図7Aおよび7Bは10mg/kgのプレグナノロンに関するNREM睡眠中のEEGパターンについて示す。図7Bは図7Aの一部の拡大図である。
図7Cおよび7Dは30mg/kgのZolpidemに関するNREM睡眠中のEEGパターンについて示す。図7Dは図7Cの一部の拡大図である。
図8A、8Bおよび8Cは、CT−5およびCT−18で各々投与された10mg/kgのZolpidemと、CT−18で投与された30mg/kgのZolpidemに関する、NREM睡眠率/hr vs.時計時間のプロットである。
図8Cは、CT−18で投与された10mg/kgおよび30mg/kgのZolpidemに関する、睡眠に費やすmin vs.時計時間のプロットである。
図8Dは、CT−18で投与された10mg/kgおよび30mg/kgのZolpidemに関する、体温の平均偏差vs.時計時間のプロットである。
図8Eは、CT−18で投与された10mg/kgおよび30mg/kgのZolpidemに関する、体動回数/hr(運動活動)vs.時計時間のプロットである。
図8Fは、CT−18で投与された10mg/kgおよび30mg/kgのZolpidemに関する、REM睡眠率/hr vs.時計時間のプロットである。
図9Aおよび9Bは、CT−5およびCT−18で各々投与された40μg/kgのDexmedetomidineに関する、NREM睡眠率/hrvs.時計時間のプロットである。
図10A、10Bおよび10Cは、CT−5で各々投与されたプレグナノロン、ZolpidemおよびTriazolamに関する、総力率(NREM睡眠中の活動)vs.処置後minのプロットである。
図11A、11Bおよび11Cは、CT−5で各々投与されたプレグナノロン、ZolpidemおよびTriazolamに関する、総力率(NREM睡眠中の活動)vs.振動数のプロットである。
図12は様々な薬物で処置後最初の1時間における運動活動に関するベースラインからの変化を示した棒グラフである。運動活動は回数/hrで測定される。
図13は様々な薬物で処置後2〜3時間におけるREM睡眠の変化を示した棒グラフである。
図14Aおよび14Bは、CT−18で各々投与されたプレグナノロンおよびZolpidemに関する、5分間当たりで動いた回数vs.処置後minのプロットを示す。
図15A〜15Hは、5%プレグナノロン、94%ポリビニルピロリドンおよび1%ラウリル硫酸ナトリウムからなる処方1に関する、経時的なプレグナノロンのヒト血漿濃度のプロットである。矢印は睡眠が観察された時点を示す。
図16A〜16Hは、12.3%プレグナノロンおよび87.7%β−シクロデキストリンの包接複合体である処方2に関する、経時的なプレグナノロンのヒト血漿濃度のプロットである。矢印は睡眠が観察された時点を示す。
図17A〜17Hは、超臨界液体核形成によりナノサイズ化された99%プレグナノロンと1%ラウリル硫酸ナトリウムからなる処方3に関する、経時的なプレグナノロンのヒト血漿濃度のプロットである。矢印は睡眠が観察された時点を示す。
図18A〜18Hは、AKZO製の微粉砕99%プレグナノロンと1%ラウリル硫酸ナトリウムからなる処方4に関する、経時的なプレグナノロンのヒト血漿濃度のプロットである。矢印は睡眠が観察された時点を示す。
図19は4種すべての処方に関するプレグナノロンのヒト治験者血漿濃度vs.時間のプロットである。
図20は半時間間隔で2種の処方に関するプレグナノロンのヒト平均血漿濃度を示した表である。
図21は、2種のプレグナノロン処方について半時間間隔で、5分間ポリグラフ記録期間中における男性治験者の平均覚醒時間を示した表である。
図22Aは男性治験者による睡眠試行回数vs.プレグナノロンの血漿濃度についてプロットした図である。
図22Bは、試験日の男性治験者による睡眠試行回数がプレグナノロンの血漿濃度と関連していることを示した表である。
図23A〜Cは、3α−ヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−ホスフェート二ナトリウム塩、3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン、3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン、3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−ヘミサクシネートナトリウム塩、3α−ヒドロキシ−3β−エチニル−5α−プレグナン−20−オン、3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン 21−ヘミサクシネート、3α−ヒドロキシ−3β−トリフルオロメチル−19−ノル−5β−プレグナン−20−オン、3α−ヒドロキシ−21−(ピリジ−4−イルチオ)−5β−プレグナン−20−オン、3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノルプレグナン−20−オン 21−ホスフェート二ナトリウム塩またはコントロールをうけたラットで、NREM睡眠の平均変化、REMリバウンドの平均変化およびREM睡眠の平均変化について各々示した表である。
図24Aおよび24Bは、日周期18時(CT−18)で投与された18mg/kgの3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン(CCD−3093)に関する、ラットのNREM睡眠およびREM睡眠の各率/hr vs.時計時間のプロットである。
図25は、CT−18で投与された18mg/kgの3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オンに関する、睡眠に費やすmin vs.時計時間のプロットである。
図26Aおよび26Bは、CT−18で投与された18mg/kgの3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オンに関する、体温の平均偏差vs.時計時間および動いた回数/hr(運動活動)vs.時計時間のプロットである。
発明の具体的説明
本発明で用いられる化合物は、GABAAレセプター複合体と相互作用する3α−ヒドロキシル化−5−還元ステロイドである。これらの化合物には様々な3α−ヒドロキシル化−5−プレグナン−20−オン類、3α−ヒドロキシル化−5−アンドロスタン類および3α,21−プレグナンジオール−20−オン類および、プロドラッグと称されるそれらのケトン、エステル、エーテル、スルホン酸、硫酸、ホスホン酸、リン酸、オキシムおよびチアゾリジン誘導体がある。“プロドラッグ”という表現は直接の作用薬物、ここでは本発明で用いられる薬物の誘導体を表し、その誘導体はその薬物と比較して高いデリバリー特性および治療価を有し、患者体内で酵素的または化学的プロセスにより活性薬物に変換される。Notari,R.E.,"Theory and Practice of Prodrug Kinetics",Methods in Enzymology,112:309-323(1985);Bodor,N.,"Novel Approaches in Prodrug Design",Drugs of the Future,6(3):165-182(1981);Bundgaard,H.,"Design of Prodrugs: Bioreversible Derivatives for Various Functional Groups and Chemical Entities",Design of Prodrugs(H.Bundgaard,ed.),Elsevier,New York(1985)参照。本発明の方法で有用な合成誘導体の一部は真のプロドラッグではないことがあることに注意すべきであり、その理由は上記特徴に加えてそれらが固有の活性も有するからである。しかしながら、本目的から、それらもプロドラッグと称される。
本発明の方法で有用な3α−ヒドロキシプレグナン類および3α−ヒドロキシアンドロスタン類の中には、下記構造式Iを有する3α−ヒドロキシ−3β−R−5αまたは5β−19−R1−17β−R2−プレグナン類、および3α−ヒドロキシ−3β−R−5αまたは5β−19−R1−17β−R2−アンドロスタン類:
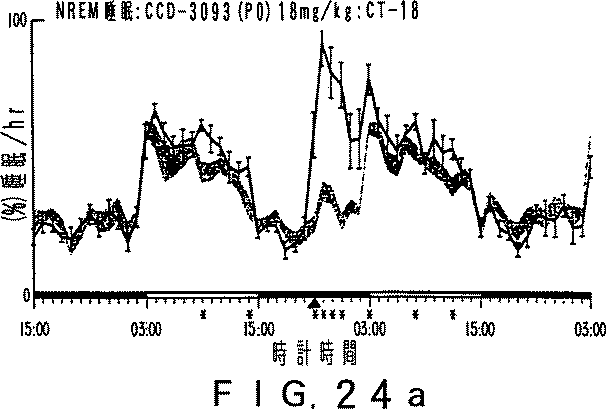
Rは水素、低級アルキル基、例えばメチルおよびメトキシメチル、低級アルキニル基、例えばエチニル、4−ヒドロキシペント−1−イニルおよび(4′−アセチルフェニル)エチニル、低級トリハロアルキル基、例えばトルフルオロメチル、低級モノハロアルキル基、例えばクロロメチル、低級アルケニル基、例えばエテニルおよび2−フェニルエテニル、アリール基、例えばフェニル、またはアラルキル基、例えばベンジルであり、
R1は水素またはメチルであり、
R2はメチレン(=CH2)、シアノ、ヒドロキシメチル、メトキシメチレン(=CHOCH3)、アセチル、2′−ヒドロキシアセチル、2′−ヒドロキシアセチルアセテート、1−ヒドロキシエチル、2′−ヒドロキシアセチルヘミサクシネート、2′−メトキシアセチル、ピリジ−4−イルチオアセチル、エチレン(=CHCH3)、プロピレン(=CHCH2CH3)、2′−ヒドロキシアセチルヘミサクシネートナトリウム塩、1−ヒドロキシブチル、1−ヒドロキシ−1−メチルエチル、1−ヒドロキシプロピル、1−プロピオニル、3−メトキシプロピオニル、エチニル、2′−メシルオキシアセチルまたは1′−(エチレンジオキシ)エチルである〕
および、それらの薬学上許容される3−エステル、20−エステル、21−エステル、3,20−ジエステルおよび3,21−ジエステルがあるが、但し3α−ヒドロキシ−5α−プレグナン類は21−ヒドロキシ置換基を有しない。
化合物の好ましい群にはRが水素でない場合を含み、その化合物は〔35S〕TBPS結合のアロステリックモジュレーターとして≦300nMのIC50値を有し、しかもその化合物は経口上活性である。
本発明の方法で使用できる化合物の他の好ましい群は、下記構造式IIを有する3α−ヒドロキシ−3β−R−5αまたは5β−19−R1−21−R2a−プレグナン−20−オン類:
Rは低級アルキル基、例えばメチル、低級アルキニル基、例えばエチニル、またはトリハロ(低級)アルキル基、例えばトルフルオロメチルであり、
R1は水素または低級アルキル基であり、
R2aは水素、ヒドロキシ、ピリジ−4−イルチオ基、ヘミサクシノイルオキシ基またはホスホリルオキシ基と、それらのナトリウム塩であるが、但し
(1)R1が水素であるとき、Rはトリハロ(低級)アルキル基であり、および
(2)R2aが水素またはピリジ−4−イルチオ基以外であるとき、Rはトリハロ(低級)アルキル基である〕
である。
式IIの化合物の好ましい群は、Rが低級アルキニル基またはトリハロ(低級アルキル)基である場合である。
式IIの化合物の他の好ましい群は、R2aがヘミサクシノイルオキシ基またはホスホリルオキシ基、あるいはそれらのナトリウム塩である場合である。
本発明の好ましい神経活性ステロイドには、3α−ヒドロキシ−5β−プレグナン−20−オン(プレグナノロン)(Diosynthより入手可)、3α−ヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−ホスフェート二ナトリウム塩、3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン、3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン、3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−ヘミサクシネートナトリウム塩、3α−ヒドロキシ−3β−エチニル−5α−プレグナン−20−オン、3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン 21−ヘミサクシネート、3α−ヒドロキシ−3β−トリフルオロメチル−19−ノル−5β−プレグナン−20−オン、3α−ヒドロキシ−21−(ピリジ−4−イルチオ)−5β−プレグナン−20−オン、および3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノルプレグナン−20−オン 21−ホスフェート二ナトリウム塩がある。
定 義
本発明に従いここで用いられる以下の用語は、特に断らないかぎり、下記の意味で定義される。
“アルキル”という用語は、炭素原子1〜10の直鎖、分岐鎖および環状基を含めた飽和脂肪族基に関する。アルキルには置換低級アルキルを含む。
“アルキニル”という用語は少くとも1つの炭素−炭素三重結合を含む不飽和基に関し、直鎖、分岐鎖および環状基を含む。アルキニルには置換低級アルキニルを含む。
“トリハロアルキル”という用語は、3つのハロゲン原子(F、Br、ClまたはI)で置換されたアルキル基に関する。好ましいトリハロアルキル基にはトリフルオロメチルおよび2′,2′,2′−トリフルオロエチルがある。
有機基または化合物に関してここで用いる“低級”という用語は、例えば8以内、好ましくは6以内、有利には1、2、3または4つの炭素原子を表す。
“アルケニル”という用語は少くとも1つの炭素−炭素二重結合を含む不飽和基に関し、直鎖、分岐鎖および環状基を含む。アルケニルとは2つの連続二重結合を有するアレニル基>C=C=C<を含めた意味である。アルケニルはアリールで置換されていてもよい。
“アリール”という用語は共役π電子系をもつ少くとも1つの環を有した芳香族基に関し、炭素環式アリールおよびビアリール基を含み、それらすべてが置換されていてもよい。
“炭素環式アリール”基とは、芳香族環の環原子が炭素原子である基である。
“ビアリール”という用語は、他のアリール基で置換されたアリール基を表す。
“置換低級アルキル”という用語は、アジド、シアノまたは低級アルコキシ基で置換された低級アルキル基に関する。
“アルコキシ”とは、酸素エーテル結合鎖で炭素原子に結合されたアルキル基に関する。
“モノハロアルキル”という用語は、1つのハロゲン原子(F、Br、ClまたはI)で置換されたアルキル基に関する。
“置換アリール”という用語は、低級アルキル、アリール、ヒドロキシ、低級アルコキシ、アミノ、ハロ、低級アシル、ニトロ、低級トリハロアルキル、シアノ、カルボキシおよび低級アシルオキシアセチルから独立して選択される1〜5つの置換基で置換されたアリール基に関する。
“アミノ”という用語は、RおよびR′が水素または低級アルキルから独立して選択される−NRR′に関するか、またはそれはモルホリノ基に関する。
“ハロ”という用語はハロゲン原子F、Br、ClおよびIに関する。
“アシル”という用語は、Rが低級アルキルである−C(O)Rに関する。
“カルボキシ”という用語は基−C(O)OHに関する。
“アシルオキシアセチル”という用語は、Rが低級アルキルである−C(O)CH2OC(O)Rに関する。
“置換低級アルキニル”という用語は、アリール、ヒドロキシ、ハロおよびケトからなる群より選択される1つの置換基で置換された低級アルキニル基に関する。
“アラルキル”という用語はアリール基で置換されたアルキル基に関する。適切なアラルキル基にはベンジルがあり、アリール上で置換されていてもよい。
“薬学上許容される”という用語は本発明で用いられるステロイドと有機または無機酸との組合せから誘導されるエステルまたは塩に関する。3、20および/または21位におけるヒドロキシ基の好ましいエステルは、それらの対応酸:酢酸、プロピオン酸、マレイン酸、フマル酸、アスコルビン酸、ピメリン酸、コハク酸、グルタル酸、ビスメチレンサリチル酸、メタンスルホン酸、エタンジスルホン酸、シュウ酸、酒石酸、サリチル酸、クエン酸、グルコン酸、イタコン酸、グリコール酸、p−アミノ安息香酸、アスパラギン酸、グルタミン酸、γ−アミノ酪酸、α−(2−ヒドロキシエチルアミノ)プロピオン酸、グリシンおよび他のα−アミノ酸、リン酸、硫酸、メタンスルホン酸、グルクロン酸と、1−メチル−1,4−ジヒドロニコチン酸により記載される場合である。特に好ましいのは酢酸、コハク酸およびリン酸のエステルである。
プロドラッグが薬物形に変換されるときエステルが開裂されるという事実により、3α位の置換基はヒドロキシルでもまたはエステルでもよいと考えられる。これらは開裂性エステルとしてここでは称される。
下記合成方法および例は、本発明で用いられる化合物の製造に関する。
合成方法
本発明による化合物は、いずれかの常法により、例えば"Steroid Reactions",Djerassi,Holden-Day,Inc.1963発行,San Franciscoまたは"Organic Reactions in Steroid Chemistry",Fried and Edwards,Van Nostrand-Reinhold Co.1972発行,New Yorkで記載されるような慣用的技術を用いて製造される。
本発明による化合物は、当業界で知られるいずれか適切な技術により製造しても、またはそれから誘導してもよい。
一般的方法
20−ヒドロキシプレグナン類は、慣用的な還元剤で20−ケトプレグナン類の還元により製造された。
21−ヘミサクシネート類は、対応21−ブロモプレグナン類を得るため最初に臭素分子で臭素化されたプレグナン−20−オン誘導体から製造された。次いでブロモ化合物はアミンの存在下でコハク酸のような様々な二酸と反応させ、21−ヒドロキシエステルを生じる。次いで二酸から得たエステルは慣用的手段でそれらのナトリウム塩に変換された。
そのエステルは上記化合物のヒドロキシル基と有機および無機酸、酸ハライド、酸無水物またはエステルとの当業界で周知の反応を用いて形成してもよい。
3β−置換基
ハロメチル
本発明による3β−モノハロメチル化合物は、不活性溶媒中ハライドイオン源、例えばトルエン中テトラメチルアンモニウムハライドと、好ましくは酢酸のようなプロトン源による3−スピロ−2′−オキシランステロイドの処理で製造される。
飽和または不飽和アルキル
他の3−置換ステロイドは、他の反応性官能基が必要に応じて保護された3−ケトステロイドへの有機金属試薬の付加により製造される。このため、3−アルキニル化合物は、有機金属試薬として不活性溶媒中でリチウムアセチリド、または1,2−ジブロモエチレンおよびブチルリチウムからその場で製造された試薬の使用により製造される。同様に、Rがアルケニル基である式Iの化合物は、3−ケトステロイドとビニルマグネシウムブロミドのようなビニル有機金属試薬との反応により製造してもよい。2−プロペニル基のような不飽和が反応の部位から除去された化合物も、アリルマグネシウムブロミドのような試薬で製造される。同様に、メチルマグネシウムヨージドのようなアルキルグリニャールの使用は3−アルキル化合物を生じる。
トリフルオロメチル
トリフルオロメチル基は、フッ化物イオンで触媒される3−ケトステロイドとトリメチルトリフルオロメチルシランとの反応により製造される。
21−酸素化化合物
21−メチルの四酢酸鉛酸化
このタイプの様々な化合物は、プレグナン−20−オンが四酢酸鉛で酸化されて21−アセトキシ誘導体を生じ、そのアセテートの加水分解で21−アルコールを生じる反応順序により製造される。次いで21−アルコールは酸無水物または酸クロリドのような適切なカルボン酸誘導体、あるいはヒドロキシル基の水素を代えることができる他の試薬、例えばメタンスルホニルクロリドでアシル化される。
C−21の臭素化
一方、プレグナン−20−オンはメタノール中において臭素のような臭素化剤で処理されて、21−ブロモプレグナン−20−オンを生じる。次いで臭素はコハク酸アニオンのような酸素求核剤と置き換えられて、他の21−置換プレグナン−20−オンを生じる。
プレグナン−17−エン
これらは、カリウムt−ブトキシドのような強塩基によるn−プロピルトリフェニルホスホニウムブロミドの処理から誘導されるイリドのようなWittig試薬と17−ケトステロイドとの反応により形成される。
3,20−ジオール
プレグナン−3,20−ジオールは、20−ヒドロキシを形成するために、プレグナン−17−エンの二重結合へのジボランのような水素化ホウ素の付加、その後例えば過酸化水素でこうして形成された有機ボランの酸化により形成される。一方、プレグナン−3,20−ジオールは20−オン基から20−ヒドロキシ基への還元により形成してもよい。適切な試薬は水素化ホウ素ナトリウムのような水素化試薬、またはn−プロパノール等の中におけるナトリウムのような溶解金属である。
例1
a.3α−ヒドロキシ−5β−プレグナン−20−オン 環状20−(1,2−エタンジイルケタール)
3α−ヒドロキシ−5β−プレグナン−20−オン(10.8g、34mmol)、エチレングリコール(45ml)およびトリエチルオルトホルメート(30ml)の混合液を室温で5分間攪拌した。次いでp−トルエンスルホン酸(pTSA)(200mg)を加え、攪拌を室温で1.5時間続けた。得られた濃厚ペーストを飽和NaHCO3溶液(250ml)中に注いだ。沈殿固体物を濾取し、冷水で十分に洗浄し、乾燥させた。この半乾燥生成物をCH2Cl2(350ml)に溶解し、無水K2CO3で乾燥させた。次いでケタールの溶液を濾過し、次の工程にそのまま用いた。
b.5β−プレグナン−3,20−ジオン 環状20−(1,2−エタンジイルケタール)
CH2Cl2中3α−ヒドロキシ−5β−プレグナン−20−オン 環状20−(1,2−エタンジイルケタール)の上記溶液をN2下で15分間にわたりN−メチルモルホリン−N−オキシド(8.8g、75mmol)および粉末4Åモレキュラーシーブ(58g)と共に攪拌した。次いで過ルテニウム酸テトラプロピルアンモニウム(400mg)を加え、攪拌を室温で2時間続けた。得られた暗緑色混合液をFlorisilの短カラムに通して、CH2Cl2で溶出させた。生成物を含有した(TLCによる)分画を合わせ、蒸発させた。次いで粗生成物をEtOAc:ヘキサン(1:1)の混合液から結晶化させ、長い棒状物として標題化合物(10.3g)を得た。
同様の方法を用いて、5α−プレグナン−3,20−ジオン 環状20−(1,2−エタンジイルケタール)を製造した。
c.3α−ヒドロキシ−3β−フェニルエチニル−5β−プレグナン−20−オン
乾燥THF(15ml)中5β−プレグナン−3,20−ジオン 環状20−(1,2−エタンジイルアセタール)(180mg、0.5mmol)の溶液を−70℃でリチウムフェニルアセチリド(THF中1M、1.5mmol、1.5ml)で処理した。混合液をこの温度で1時間、その後室温で2時間攪拌した後、それを2N HCl(1ml)で反応停止させた。溶媒を除去し、残渣をアセトン(20ml)に溶解した。1N HCl(5ml)を加えた後、溶液を室温で15時間攪拌した。溶媒を除去し、残渣をCH2Cl2で抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(300mg)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。トルエン:アセトン混合液(95:5)による溶出で、第一分画として3α−フェニルエチニル−3β−ヒドロキシ−5β−プレグナン−20−オン(70mg)を得た。更に、同溶媒混合液による溶出で、3β−フェニルエチニル−3α−ヒドロキシ−5β−プレグナン−20−オン(160mg)を得た。mp188−190℃
例2
a.3α−ヒドロキシ−3β−(2′−フェニルエチル)−5β−プレグナン−20−オン
3α−ヒドロキシ−3β−フェニルエチニル−5β−プレグナン−20−オン(44mg)の溶液をEtOAc(12ml)に溶解し、Pd/C(5%、12mg)を加え、混合液に400Kpa圧力で一夜にわたり室温で水素付加した。触媒の濾過、その後溶媒の蒸発により粗生成物を得、これをシリカゲルクロマトグラフィーにより精製して、純粋な標題化合物(33mg)を単離した。mp153−154℃;TLC Rf(ヘキサン:アセトン7:3)=0.4
例3
a.3β−(3′,4′−ジメトキシフェニルエチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン
乾燥THF(15ml)中2,2−ジブロモ−1−(3′,4′−ジメトキシフェニル)エテン(トリフェニルホスフィンの存在下で3,4−ジメトキシベンズアルデヒドと四臭化炭素とのWittig反応により製造)(966mg、3mmol)の溶液をN2下−78℃でn−BuLi(THF中2.5M、6mmol、2.4ml)で処理した。混合液をこの温度で2時間攪拌し、乾燥THF(10ml)中5β−プレグナン−3,20−ジオン 環状20−(1,2−エタンジイルアセタール)(720mg、2mmol)の溶液を30分間かけて滴下した。得られた混合液を−78℃で2時間攪拌した後、冷却浴を取除き、攪拌を室温で更に1時間続けた。次いでそれを−10℃で2N HCl溶液(1ml)で反応停止させた。溶媒を除去し、その後残渣をアセトン(25ml)に溶解した。2N HCl(10ml)を加えた後、溶液を室温で2時間攪拌した。飽和NaHCO3溶液を加えて、酸を中和させた。溶液を除去し、残渣をEtOAcで抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(1.2g)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。トルエン:アセトン混合液(96:4)による溶出でフェニルアセチレン化合物を得たが、これは特徴を明らかにしなかった。更に同溶媒による溶出で、第一分画として3α−(3′,4′−ジメトキシフェニルエチニル)−3β−ヒドロキシ−5β−プレグナン−20−オン(120mg)および第二分画として3β−(3′,4′−ジメトキシフェニルエチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン(430mg)を得た。mp82−88℃
同様の方法を用いて、3β−(4′−メトキシフェニルエチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン、3β−(4′−クロロフェニルエチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン、3β−(2′−メトキシフェニルエチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン、3β−(4′−ビフェニルエチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン、3β−(4′−ジメチルアミノフェニルエチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン、3β−(4′−シアノフェニルエチニル)−3α−ヒドロキシ−5β−プレグナン−20−オンを製造した。
例4
3β−(3′,4′−ジメトキシフェニルエチル)−3α−ヒドロキシ−5β−プレグナン−20−オン
Pd/C(5%、28mg)およびEtOAc(12ml)の混合液を水素下で10分間攪拌することにより水素で前飽和させた。次いでEtOAc(5ml)中3β−(3′,4′−ジメトキシフェニルエチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン(185mg)の溶液を加え、混合液に300Kpa圧力で一夜にわたり室温で水素付加した。触媒の濾過、その後溶媒の蒸発により粗生成物を得、これをシリカゲルクロマトグラフィー(ヘキサン:アセトン4:1)により精製して、純粋な標題化合物(135mg)を単離した。TLC Rf(ヘキサン:アセトン4:1)=0.14
例5
3β−(4′−ニトロフェニルエチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン
乾燥THF(20ml)中2,2−ジブロモ−1−(4−ニトロフェニル)エテン(トリフェニルホスフィンの存在下で4−ニトロベンズアルデヒドと四臭化炭素とのWittig反応により製造)(296mg、1mmol)の溶液をN2下−95℃でn−BuLi(THF中2.5M、2mmol、0.8ml)で処理した。混合液を−80〜−100℃で0.5時間攪拌し、その後乾燥THF(10ml)中5β−プレグナン−3,20−ジオン 環状20−(1,2−エタンジイルアセタール)(720mg、2mmol)の溶液を10分間かけて滴下した。得られた混合液を−80℃で1時間、その後0℃で更に1時間攪拌した後、それをNH4Cl溶液(3ml)で反応停止させた。溶媒を除去し、その後残渣をアセトン(25ml)に溶解した。2N HCl(10ml)を加えた後、溶液を室温で1時間攪拌した。飽和NaHCO3溶液を加えて、酸を中和させた。溶媒を除去し、残渣をCH2Cl2で抽出した。有機層を水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(400mg)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。トルエン:アセトン混合液(96:4)による溶出で褐色固体物として標題化合物(70mg)を得た。TLC Rf(トルエン:アセトン95:5)=0.18
例6
3α−ヒドロキシ−3β−フェニル−5β−プレグナン−20−オン
乾燥THF中5β−プレグナン−3,20−ジオン 環状20−(1,2−エタンジイルアセタール)(720mg、2mmol)の溶液を−70℃でフェニルマグネシウムブロミド(THF中3M、6mmol、2ml)で処理した。混合液をこの温度で3時間、その後室温で2時間攪拌した後、それを2N HCl(10ml)で反応停止させた。溶媒を除去し、残渣をアセトン(20ml)に溶解した。1N HCl(5ml)を加えた後、溶液を室温で15時間攪拌した。溶媒を除去し、残渣をCH2Cl2で抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(1.3g)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。トルエン:アセトン混合液(95:5)による溶出で、第一分画として3α−フェニル−3β−ヒドロキシ−5β−プレグナン−20−オン(420mg)を得た。更に、同溶媒混合液による溶出で、3β−フェニル−3α−ヒドロキシ−5β−プレグナン−20−オン(185mg)を得た。mp182−184℃
例7
3α−ヒドロキシ−3β−ベンジル−5β−プレグナン−20−オン
ベンジルマグネシウムブロミド(THF中2M、2mmol、1ml)の溶液をTHF(15ml)で希釈し、−60℃で乾燥THF(15ml)中5β−プレグナン−3,20−ジオン 環状20−(1,2−エタンジイルアセタール)(360mg、1mmol)の溶液で滴下処理した。混合液をこの温度で1時間、その後室温で15時間攪拌した後、それを2N HCl(10ml)で反応停止させた。溶媒を除去し、残渣をアセトン(20ml)に溶解した。1N HCl(5ml)を加えた後、溶液を室温で30分間攪拌した。それを2N NaOHで中和した。沈殿固体物を濾取し、水洗し、乾燥させて、3β−ヒドロキシ−3α−ベンジル−5β−プレグナン−20−オン(238mg)を得た。濾液をEtOAcで抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(160mg)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。トルエン:アセトン混合液(95:5)による溶出で、第一分画として3β−ヒドロキシ−3α−ベンジル−5β−プレグナン−20−オン(40mg)を得た。更に、同溶媒混合液による溶出で、3α−ヒドロキシ−3β−ベンジル−5β−プレグナン−20−オン(30mg)を得、無色棒状物としてヘキサン:CH2Cl2(4:1)から結晶化させた。mp133−141℃;TLC Rf(トルエン:アセトン9:1)=0.5
例8
3β−(1−ヘプチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン
乾燥THF(15ml)中1−ヘプチン(0.327ml、2.5mmol)の溶液を−78℃でn−BuLi(THF中2.5M、2.5mmol、1ml)で処理した。混合液をこの温度で1時間攪拌した後、乾燥THF(15ml)中5β−プレグナン−3,20−ジオン 環状20−(1,2−エタンジイルアセタール)(360mg、1mmol)の溶液を加え、混合液を−78℃で1時間攪拌した。次いでそれを2N HCl溶液(1ml)で反応停止させた。溶媒を除去し、その後残渣をアセトン(10ml)に溶解した。2N HCl(10ml)を加えた後、溶液を室温で1時間攪拌した。飽和NaHCO3溶液を加えて、酸を中和させた。溶媒を除去し、残渣をEtOAcで抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(400mg)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。トルエン:アセトン混合液(93:7)による溶出で、無色固体物として3β−(1−ヘプチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン(260mg)を得た。mp121−123℃
同様の方法を用いて、1−ヘプチン−6−オン 環状(1,2−エタンジイルアセタール)から3β−(1−ヘキシニル)−3α−ヒドロキシ−5β−プレグナン−20−オン、3β−(1−オクチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン、3α−ヒドロキシ−3β−(6−オキソ−1−ヘプチニル)−5β−プレグナン−20−オンを製造した。
例9
3β−〔3′(R/S)−ヒドロキシブチニル〕−3α−ヒドロキシ−5β−プレグナン−20−オン
乾燥THF(15ml)中3(R/S)−ヒドロキシブチン(0.470ml、6mmol)の溶液を−70℃でn−BuLi(THF中2.5M、12mmol、4ml)で処理した。混合液をこの温度で0.5時間攪拌した後、乾燥THF(30ml)中5β−プレグナン−3,20−ジオン 環状20−(1,2−エタンジイルアセタール)(1.08g、3mmol)の溶液を加え、混合液を−78℃で1時間攪拌した。冷却浴を取除き、混合液を室温で更に1.5時間攪拌した。次いでそれをNH4Cl溶液(3ml)で反応停止させた。溶媒を除去し、その後残渣をアセトン(10ml)に溶解した。2N HCl(5ml)を加えた後、溶液を室温で1時間攪拌した。飽和NaHCO3溶液を加えて、酸を中和させた。溶媒を除去し、残渣をEtOAcで抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(1.4g)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。トルエン:アセトン混合液(85:15)による溶出で、無色固体物として3β−〔3′(RS)−ヒドロキシブチニル〕−3α−ヒドロキシ−5β−プレグナン−20−オン(145mg)を得た。TLC Rf(トルエン:アセトン4:1)=0.24
同様の方法を用いて、3β−〔4′(R/S)−ヒドロキシペンチニル〕−3α−ヒドロキシ−5β−プレグナン−20−オンを製造した。
例10
3β−(4′−アセチルフェニルエチニル)−3α−ヒドロキシ−5α−プレグナン−20−オン
乾燥脱気ピロリジン(3ml)中4−ヨードアセトフェノン(95mg、0.39mmol)、3β−エチニル−3α−ヒドロキシ−5α−プレグナン−20−オン(106mg、0.3mmol)の溶液をアルゴン下室温で攪拌した。ビス(トリフェニルホスフィン)パラジウムクロリド(5mg)およびCuI(5mg)を加え、混合液を室温で15時間攪拌した。TLCでは出発物質の100%変換を示し、このため混合液にNH4Cl溶液(15ml)を加えて反応停止させ、EtOAcで抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(150mg)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。ヘキサン:アセトン混合液(4:1)による溶出で、無色固体物として3β−(4′−アセチルフェニルエチニル)−3α−ヒドロキシ−5α−プレグナン−20−オン(35mg)を得た。TLC Rf(ヘキサン:アセトン7:3)=0.4
同様の方法を用いて、3α−ヒドロキシ−3β−(4′−トリフルオロメチルフェニルエチニル)−5β−プレグナン−20−オン、3α−ヒドロキシ−3β−(4′−メチルフェニルエチニル)−5β−プレグナン−20−オン、3α−ヒドロキシ−3β−(4′−アセトキシアセチルフェニルエチニル)−5β−プレグナン−20−オン、3α−ヒドロキシ−3β−(ペンタフルオロフェニル)エチニル−5β−プレグナン−20−オン、3β−(2′−ヒドロキシフェニル)エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン、3β−(3′−ヒドロキシフェニル)エチニル−3α−ヒドロキシ−5β−プレグナン−20−オンおよび3α−ヒドロキシ−3β−(4′−カルボキシフェニルエチニル)−5β−プレグナン−20−オン、エチルエステルを製造した。
例11
3α−ヒドロキシ−3β−メトキシメチル−5α−プレグナン−20−オン
3(R)−エポキシ−5α−プレグナン−20−オンをTHF中カリウムt−ブトキシドのような強塩基の存在下で5α−プレグナン−3,20−ジオンとトリメチルスルホキソニウムメチルヨージドとの反応により製造した。
CH3OH(600ml)中3(R)−エポキシ−5α−プレグナン−20−オン(5.145g、15.57mmol)の溶液(未溶解固体粒子はほとんどなかった)に切断したばかりのNa(〜600mg)を加えた。注意:H2ガス発生。すべてのNaが反応した後、透明溶液が得られた。反応溶液を一夜攪拌した。TLC(3:1ヘキサン/アセトン)が主として出発物質を示したため、混合液を8時間(沸点近くまで)加熱し、その後室温で一夜おいた。TLCが2つの新たな低極性側スポット以外にもなお少量の出発物質を示した。したがって、混合液を更に8時間加熱したところ、未反応出発エポキシドはなかった。反応溶液が室温に戻った後、氷酢酸(3ml)を滴下した。次いで溶媒を減圧下で除去し、CH2Cl2および水を加えた。水層をCH2Cl2で逆抽出した。合わせた有機層を飽和NaHCO3溶液で洗浄し、(MgSO4)乾燥し、濾過し、減圧下で蒸発させて白色固体物(5.55g)を得たが、これはその1H NMRに基づくと3α−ヒドロキシ−3β−メトキシメチル−5α−プレグナン−20−オンおよびその17−エピマーの84:16混合物であることがわかった。加温1:1ヘキサン:アセトンからの再結晶化で、白色結晶として3α−ヒドロキシ−3β−メトキシメチル−5α−プレグナン−20−オン(3.270g、58%)のみを得た。mp163−4℃。予想通り、母液はほぼ等量で双方の17−エピマーを含有していた。
同様の方法を用いて、3α−ヒドロキシ−3β−エトキシメチル−5α−プレグナン−20−オン、3α−ヒドロキシ−3β−プロポキシメチル−5α−プレグナン−20−オンを製造した。
例12
3α−ヒドロキシ−3β−シアノメチル−5α−プレグナン−20−オン
シアン化カリウム(210mg、3.2mmol)および20,20−エチレンジオキシ−3R−スピロ−2′−オキシラン−5α−プレグナン−20−オン(1.0g、2.7mmol)をエタノール(175ml)中で2日間攪拌し、5時間還流し、その後一夜冷却させた。エーテル(100ml)および塩水(100ml)を反応混合液に加えた。分離した水相をエーテルで洗浄し、合わせた有機相を塩水で洗浄した。水性HCl(1M、6ml)およびTHF(15ml)を有機相に加え、溶液を一夜攪拌した。水性NaHCO3(20ml)、その後水(15ml)を加えた。分離した水相をエーテルで洗浄し、合わせた有機相をMgSO4で乾燥し、蒸発させた。白色固体残渣をフラッシュカラムクロマトグラフィー(40:20:1CH2Cl2/ヘキサン/アセトン)により精製して、白色固体物(493mg、52%)を得た。mp218.5−220℃
例13
3α−ヒドロキシ−3β−アジドメチル−5α−プレグナン−20−オン
乾燥DMF(Aldrich;50ml)中3(R)−エポキシ−5α−プレグナン−20−オン(1.012g、3.06mmol)の溶液にNaN3(226mg、3.48mmol)を加えたが、そのすべてが溶解したわけではなかった。次いで氷酢酸(1.05ml、17.5mmol)を加え、得られた混合液をヒートガンでわずかに加熱して、すべての固体物を溶解させた。混合液をしばらく攪拌して、透明な溶液を得た。TLC(100:1CH2Cl2/アセトンまたは3:1ヘキサン/アセトン)では、混合液を4時間攪拌した後で、出発物質の存在をなお示した。次いで反応混合液を35〜50℃で3日間攪拌しながら加熱した。TLCによると反応は完了していた。CH2Cl2および水を加えた。水層をCH2Cl2で逆抽出した。合わせた有機層を飽和NaHCO3で洗浄し、(MgSO4)乾燥し、濾過し、ロータリーエバポレーターで濃縮して、多量のDMFを含有した液体を得た。更に高真空下の蒸留による蒸発で固体物(1.09g)を得た。この固体物の1H NMRでは主に望ましい3α−ヒドロキシ−3β−アジドメチル−5α−プレグナン−20−オンの存在を示し、その17−エピマーの存在はほとんど示されなかった。溶出液としてCH2Cl2を用いたフラッシュカラムクロマトグラフィーによる精製で、白色固体物として3α−ヒドロキシ−3β−アジドメチル−5α−プレグナン−20−オン(834mg、73%)を得た。mp147−8℃
例14
3α,20α−ジヒドロキシ−5α−19−ノルプレグナン
THF(20ml)中3α−ヒドロキシ−5α−19−ノル−17(Z)−プレグネ−17(20)−エン(下記のようなWittig試薬でアンドロスタン−17−オンから製造)(232mg、0.81mmol)の溶液に0℃でジボランTHF錯体(THF中1M溶液、3.0ml)を滴下した。反応液を25℃に2.5時間加温した。次いで水酸化ナトリウムの溶液(2N、15ml)を0℃で非常にゆっくり加え、その後過酸化水素(30%、8ml)を加えた。次いで水を加え、THF層を分液漏斗で分離した。水層を酢酸エチル(2×40ml)で抽出した。有機溶液を炭酸カリウムで乾燥させ、溶媒を真空下で除去した。アセトンおよび塩化メチレンのヘキサン溶液(8:8:84)を用いたカラムクロマトグラフィーで、少量の純粋生成物(15mg)を得た。
例15
3α,21−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン,21−ヘミサクシネート
ピリジン12ml中3α,21−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン(600mg、1.72mmol)および無水コハク酸(388mg、3.88mmol)の混合液を室温で23時間攪拌した。反応液を真空下で濃縮し、残渣をEtOAcと水性HOAc溶液(pH4)に分配した。有機層を分離し、飽和NaCl溶液で洗浄し、濃縮した。粗製ヘミサクシネートをフラッシュクロマトグラフィー(3.5cm径カラム中シリカ12cm、45/55EtOAc/CH2Cl2400mlと50/50および60/40EtOAc/CH2Cl2各々200mlで溶出)により精製した。MeOH/水からの再結晶化で、白色固体物としてヘミサクシネート346mg(45%)を得た。
ヘミサクシネートのナトリウム塩は、その酸をメタノールに溶解して、1当量の炭酸水素ナトリウムの水溶液を加えることにより製造した。溶媒を真空下で除去し、残渣をエーテル/石油エーテルで摩砕して、白色固体物を得た。
例16
a.トリフェニルプロポ−2−イニルスタナン
臭化プロパルギル(トルエン中80w%溶液、1.7ml、11.43mmol)をMg(471mg、19.38mmol)および乾燥Et2O(25ml)の混合液に加えた。次いでHgCl2(少しの結晶)を加えた。混合液を〜5分間攪拌した。反応が生じないため、更にHgCl2(15mg)を加えた。得られた混合液を最初に室温で攪拌し、その後短時間加温した。混合液は混濁するようになり、反応が生じた。混合液を<20℃(冷水浴)で〜1時間攪拌すると、ほとんどのMgは反応した。未反応Mgの量は〜30分間にわたり未変化のままであった。そのため、臭化プロパルギル(トルエン中80w%溶液、0.4ml、2.69mmol)を加えた。混合液を<20℃で〜1時間攪拌した。その後、Et2O(50ml)中トリフェニルスズクロリド(95%、5.045g、12.43mmol)の懸濁液をカニューレでプロパルギルマグネシウムブロミドの上記溶液に滴下した。得られた混合液は白色固体物を含有しており、室温で〜3時間攪拌した。TLC(3:1ヘキサン:アセトン)では出発塩化スズの完全消費を示した。混合液に飽和水性NH4Cl(10ml)を加えて反応停止させた。白色固体物が生成した。有機層を白色固体物含有の水層から分離した。最後に、有機相を(MgSO4)乾燥し、濾過し、減圧下で蒸発させて、白色固体物(4.78g)を得た。加温ヘキサン(50ml)から固体物の再結晶化で、白色板状結晶としてトリフェニルプロピ−2−イニルスタナン(3.92g、81%)を得た。
b.3α−ヒドロキシ−3β−(ブタジ−2′,3′−エニル)−5α−プレグナン−20−オン(参考文献:Baldwin,J.E.,Adlington,R.M.,Basak,A.,J.Chem.Soc.,Chem.Commun.,1984,1284)
3β−ヨードメチル−3α−ヒドロキシ−5α−プレグナン−20−オン(234mg、0.51mmol)、トリフェニルプロピ−2−イニルスタナン(795mg、2.04mmol)、アゾビスイソブチロニトリル(18mg、0.11mmol)および乾燥ベンゼン(2ml)を含有した混合液を〜10分間脱気させた。次いで上記混合液を2時間還流した。TLC(100:1CH2Cl2/アセトン)で示されるように、未反応出発物質がなお一部(〜33%)存在し、その量は2時間にわたり未変化のままであった。TLCでは出発物質以外に2つのスポットを〜1:1比で示し、低極性側スポットは新しいスポットであり、他方は3β−メチル−3α−ヒドロキシ−5α−プレグナン−20−オンに相当した。反応混合液を室温まで冷却した。溶媒を除去した後、残渣をフラッシュクロマトグラフィーに付して、白色固体物として3α−ヒドロキシ−3β−(ブタジ−2′,3′−エニル)−5α−プレグナン−20−オン(20mg、11%)を得た。
例17
3α−ヒドロキシ−3β−(プロポ−2′−イニル)−5α−プレグナン−20−オン
Mg(268mg、11.02mmol)、臭化プロパルギル(80w%溶液、1.5ml、10.09mmol)および乾燥Et2O(15ml)の混合液にHgCl2(10mg)を加えた。得られた混合液を水浴でわずかに加温して、反応を開始させた。混合液は混濁するようになり、反応が生じた。混合液をすべてのMgが反応するまで(〜1時間)<20℃で攪拌した。
乾燥Et2O(20ml)および乾燥THF(10ml)中20,20−エチレンジオキシ−5α−プレグナン−3−オン(711mg、1.97mmol)の溶液を−78℃に冷却した。出発物質の一部は冷却時に沈殿した。その後、上記で製造されたプロパルギルマグネシウムブロミド溶液(〜0.6M、6.5ml)を−78℃で滴下した。得られた混合液を−78℃で〜1時間攪拌した。次いで混合液を1時間攪拌して、室温まで加温させた。1N HCl、p−トルエンスルホン酸一水和物およびアセトンを加えて、ケタール官能基を加水分解した。2相混合液を室温で一夜攪拌した。TLC(7:1ヘキサン:アセトン)では反応の完了を示した。Et2Oおよび水を加えた。水層をEt2Oで逆抽出した。合わせた有機層を飽和水性NaHCO3および塩水で洗浄し、(MgSO4)乾燥し、減圧下で蒸発させて、白色固体物(690mg)を得た。粗製固体物の1H NMRでは、それが望ましい3α−ヒドロキシ−3β−(プロピ−2′−イニル)−5α−プレグナン−20−オンおよびその3−エピマーの各々1:2混合物であることを示した。混合物を10:1ヘキサン:アセトンで2回フラッシュクロマトグラフィーに付して、白色固体物として3α−ヒドロキシ−3β−(プロピ−2′−イニル)−5α−プレグナン−20−オン(70mg、11%)を得た。mp220−225℃(分解)
例18
3α−ヒドロキシ−3β−(ブト−3′−エニル)−5α−プレグナン−20−オン(参考文献:Keck,G.E.,Yates,J.B.,J.Am.Chem.Soc.,1982,104,5829)
乾燥ベンゼン(3ml)に15分間Arを吹き込むことにより脱気させた。次いで3β−ヨードメチル−3α−ヒドロキシ−5α−プレグナン−20−オン(263mg、0.574mmol)、アリルトリブチルスズ(97%、0.5ml、1.56mmol)、1,2′−アゾビスイソブチロニトリル(20mg、0.122mmol)を連続的に加えた。得られた混合液を3.5時間還流した。TLC(100:1CH2Cl2/アセトン)では出発ヨージド誘導体の完全消費を示し、2つの新たなスポットの出現が〜1:1比でみられ、高極性側スポットは3β−メチル−3α−ヒドロキシ−5α−プレグナン−20−オンに相当した。反応混合液を室温まで冷却した。溶媒を除去した後、残渣をCH2Cl2と7:1ヘキサン:アセトンで2回フラッシュクロマトグラフィーに付した。3α−ヒドロキシ−3β−(ブテ−3′−エニル)−5α−プレグナン−20−オンを白色固体物として得た(75mg、35%)。mp140−141℃
例19
a.3α−ヒドロキシ−5β−プレグナン−20−オン,20−ケタール
3α−ヒドロキシ−5β−プレグナン−20−オン(10.8g、34mmol)、エチレングリコール(45ml)およびトリエチルオルトホルメート(30ml)の混合液を室温で5分間攪拌した。次いでp−トルエンスルホン酸(200mg)を加え、攪拌を室温で1.5時間続けた。得られた濃厚ペーストを飽和NaHCO3溶液(250ml)中に注いだ。沈殿固体物を濾取し、冷水で十分に洗浄し、乾燥させた。この半乾燥生成物をCH2Cl2(350ml)に溶解し、無水K2CO3で乾燥させた。次いでケタールの溶液を濾過し、次の工程でそのまま用いた。
b.5β−プレグナン−3,20−ジオン,20−ケタール
CH2Cl2中3α−ヒドロキシ−5β−プレグナン−20−オン,20−ケタールの上記溶液をN2下で15分間にわたりN−メチルモルホリン−N−オキシド(8.8g、75mmol)および粉末4Åモレキュラーシーブ(58g)と共に攪拌した。次いで過ルテニウム酸テトラプロピルアンモニウム(400mg)を加え、攪拌を室温で2時間続けた。得られた暗緑色混合液をFlorisilの短カラムに通して、CH2Cl2で溶出させた。生成物を含有した分画(TLC)を合わせ、蒸発させた。次いで粗生成物をEtOAc:Hx(1:1)の混合液から結晶化させ、長い棒状物として標題化合物(10.3g)を得た。
c.1,2−ジブロモエチレンからリチウム試薬の製造
N2ガスバブラー、温度計および滴下漏斗を装備した100ml3首フラスコに1,2−ジブロモエチレン(シス/トランス混合物、98%、Aldrich、0.164ml、2mmol、mw=186、d=2.246)を入れた。乾燥THF(15ml)を加え、溶液をドライアイス−アセトン浴で−78℃に冷却した。n−BuLi(THF中2.5M、1.6ml、4mmol)を10分間かけて滴下した。混合液をこの温度で1時間攪拌し、得られた試薬を次の工程に直ちに用いた。
d.3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン
−78℃で維持されたTHF中試薬の上記溶液をTHF(15ml)中5β−プレグナン−3,20−ジオン,20−ケタール(180mg、0.5mmol)の溶液で滴下処理した。温度を滴下中−70℃に維持した。攪拌をこの温度で15分間続けた(TLCで検査したところ100%変換)。冷却浴を取除き、得られた溶液に2N HCl(pH6)を加えて反応停止させた。溶媒を除去し、残渣をアセトン(10ml)に溶解した。2N HCl(4ml)を加えた後、溶液を環境温度で0.5時間攪拌した。混合液を希NaHCO3溶液で中和させた。沈殿固体物(158mg、93%)を濾取し、水洗し、乾燥させた。次いで粗生成物をEtOAcまたはアセトン−ヘキサンの混合液からの結晶化により精製して、標題化合物を得た。mp196−197℃;TLC−Rf0.45(ヘキサン:アセトン7:3)
e.3β−エチル−3α−ヒドロキシ−5β−プレグナン−20−オン
Pd/C(5%、24mg)およびMeOH(12ml)の混合液を水素下で10分間攪拌することにより水素で前飽和させた。次いでMeOH(5ml)中3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン(155mg)の溶液を加え、混合液に200Kpa圧力で1.5時間にわたり室温で水素付加した。触媒の濾過、その後溶媒の蒸発により標題化合物を得、これをヘキサン:アセトン(9:1)からの結晶化により精製した。収量105mg;mp115−117℃;TLC Rf(ヘキサン:アセトン7:3)=0.48
f.3β−エチル−3α−ヒドロキシ−5α−プレグナン−20−オン
アルゴン下で−60℃に冷却されたヨウ化第一銅570mg(3mmol)および乾燥エーテル10mlの混合液に1.5Mメチルリチウム4mlを加えた。反応混合液を0℃に加温し、無色溶液が得られるまで攪拌し、その後−60℃まで再冷却した。更に0.5〜1mlのメチルリチウム溶液を加えて、溶液を無色に保った。エーテル20ml中3R−スピロ−2′−オキシラン−5α−プレグナン−20−オン0.5g(1.5mmol)の冷却(0℃)溶液を上記反応混合液にゆっくり加え、その際に温度を−60℃に維持した。更に0.2mlのメチルリチウムを加え、反応混合液を−60℃で1時間攪拌した。次いでそれを飽和塩化アンモニウムの添加で反応停止させ、室温まで加温し、エーテルおよび塩化アンモニウム溶液で希釈した。有機相を分離した。水相をエーテルで抽出した。合わせた有機抽出液を乾燥し、濃縮して、粗生成物450mgを得た。5%アセトン/ヘキサンを用いたシリカゲルクロマトグラフィーで、356mgの生成物を得た。
例20
3α,20β−ジヒドロキシ−5β−プレグナン二ナトリウム,ビス(ヘミサクシネート)
乾燥ピリジン5ml中3α,20β−ジヒドロキシ−5β−プレグナン(Steraloids、250mg、0.78mmol)の懸濁液を無水コハク酸(200mg、2.0mmol)で処理した。混合液を100℃で合計10時間加熱した。更に6mmolの無水コハク酸を3回に分けて加え、同時に反応液を加熱した。暗色混合液を濃縮(0.05mmHg、30℃)して溶媒を除去し、その後90℃(0.05mmHg)で加熱して過剰の無水コハク酸を除去した。残渣をエーテル/ヘキサンから再結晶化して、主にコハク酸からなる固体物を得た。母液を濃縮し、カラムクロマトグラフィー(フラッシュシリカゲル、95/5/0.1CH2Cl2/MeOH/HOAcで溶出)に付して白色固体物を得、これをエーテル/ヘキサンから再結晶化した。ビス(ヘミサクシネート)、mp81−90℃に続いて、ビス(ナトリウム塩)を得た。
ビス(ヘミサクシネート)(100mg、0.192mmol)を少量のメタノールに溶解した。水0.6ml中NaHCO3(2eq、33mg、0.393mmol)の溶液を滴下した。3時間後に溶液を真空下で濃縮して、白色固体物を得た。
例21
a.3α−ヒドロキシ−3β−メチル−5α−アンドロスタン−17β−カルボン酸
臭素(2.0ml、38.8mmol)を0℃でNaOH(水30ml中3.746g)の水溶液に滴下した。その後、ジオキサン(250ml)および水(30ml)中3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オンの溶液を0℃で1時間かけて上記溶液に加えた。少し固体物を含有する得られた黄色混合液を室温まで加温した。しばらくして、すべての固体物が溶解した。反応溶液を一夜攪拌したところ、無色になった。次いでCH2Cl2(150ml)、水(100ml)および1N HCl(50ml)を加えた。塩水も加えて、2液相の分離を容易にさせた。水層を分離し、CH2Cl2(200ml)で逆抽出した。2つの有機相を合わせ、水洗し、(MgSO4)乾燥し、濾過し、減圧下で蒸発させて、白色固体物(3.851g)を得た。加温1:1ヘキサン:アセトン(500ml)からこの固体物の再結晶化で、白色固体物として3α−ヒドロキシ−3β−メチル−5α−アンドロスタン−17β−カルボン酸(1.838g、50%)を得た。mp222.5−224℃。母液の蒸発、その後加温メタノールからの再結晶化で、白色結晶として更に生成物(1.50g、40%)を得た。mp223−225℃
b.17β−(ヒドロキシメチル)−3β−メチル−5α−アンドロスタン−3α−オール
乾燥THF(30ml)中LiAlH4(95%、546mg、13.67mmol)の攪拌懸濁液に乾燥THF(50ml+洗浄用10ml)中3α−ヒドロキシ−3β−メチル−5α−アンドロスタン−17β−カルボン酸(1.633g、4.88mmol)の溶液を均圧漏斗から30分間かけて加えた。次いで得られた混合液を室温まで加温した。攪拌を一夜続けた。TLC(7:1ヘキサン:アセトン)では反応の完了を示した。反応混合液を0℃に冷却し、1N HCl(25ml)を滴下した。その後、Et2O(100ml)、水(25ml)および濃HCl(5ml)を加えた。水層はまだ完全に透明ではなかった。2つの相を分離した。濃HCl(15ml)を水層に加え、その後Et2O(100ml)で抽出した。2つの有機層を合わせ、飽和水性NaHCO3で洗浄し、(MgSO4)乾燥し、濾過し、減圧下で蒸発させ、白色固体物として17β−(ヒドロキシメチル)−3β−メチル−5α−アンドロスタン−3α−オール(1.605g)を得た。生成物の1H NMRでは、それが一部(4%)の未反応出発物質とTHF(4%)を含有していることを示した。
c.17β−ホルミル−3β−メチル−5α−アンドロスタン−3α−オール
乾燥CH2Cl2(90ml)中17β−(ヒドロキシメチル)−3β−メチル−5α−アンドロスタン−3α−オール(純度96%、1.58g、4.73mmol)の溶液に室温でクロロクロム酸ピリジニウム(98%、1.115g、5.07mmol)を加えた。得られた混合液を室温で7時間攪拌した。TLC(7:1ヘキサン:アセトン)では出発酸の完全消費を示した。次いで混合液を中性Florisilで濾過し、CH2Cl2(250ml)および5:1ヘキサン:アセトン(100ml)で連続洗浄した(濾液は生成物の存在についてTLCによりチェックした)。溶媒を除去して淡黄色固体物(1.390g)を得、これを溶出液としてCH2Cl2を用いてフラッシュクロマトグラフィーにより精製し、白色固体物として17β−ホルミル−3β−メチル−5α−アンドロスタン−3α−オール(1.081g、72%)を得た。mp156−166℃
d.17β−シアノ−3α−ヒドロキシ−5α−アンドロスタン
1,2−ジメトキシエタン(DME)135ml中3α−ヒドロキシ−5α−アンドロスタン−17−オン(1.511g、5.20mmol)の溶液にTHF中カリウムtert−ブトキシドの1M溶液(52ml、52mmol)を加えた。得られた混濁橙色溶液をDME30ml中P−トルエンスルホニルメチルイソシアニド2.06g(10.6mmol)の溶液で2.5時間処理した。更に2.5時間後、反応液をエーテル100mlで希釈し、水100mlを加えた。水層をエーテル(2×50ml)で抽出し、プールされた有機層を塩水溶液で洗浄し、(MgSO4)乾燥し、濃縮した。シリカゲルカラムクロマトグラフィー(溶出液として100%のCH2Cl2)によりやや不純な生成物860mgを得た。EtOAcからの再結晶化で純粋なニトリル300mg(19%)を得た。mp159−160℃
例22
3α−ヒドロキシ−5β−プレグン−17(20)−エン
エチルトリフェニルホスホニウムブロミド(Aldrich、1.606g、4.33mmol)および固体カリウムtert−ブトキシド(475mg、4.23mmol)から製造されたイリドに3α−ヒドロキシ−5β−アンドロスタン−17−オン(Steraloids、503mg、1.73mmol)を一度で加えた。赤橙色混合液を3.5時間還流下で加熱し、その後冷却させた。それを更に0℃に冷却し、その後氷冷飽和NH4Cl溶液/エーテル混合液に加えた。水層を分離し、エーテルで2回洗浄した。次いでエーテル層を飽和NaCl溶液で抽出し、(MgSO4)乾燥し、濃縮した。フラッシュクロマトグラフィー(4cm径カラム中シリカ17.5cm、2%アセトン/CH2Cl2400mlおよび2.5%アセトン/CH2Cl2200mlで溶出)により白色固体物としてアルケン530mg(100%)を得た。1H NMRによると、その化合物はZ−およびE−異性体の8:1混合物であった。
例23
3α−ヒドロキシ−3β−エテニル−5β−プレグナン−20−オン
乾燥THF(20ml)中5β−プレグナン−3,20−ジオン,20−ケタール(1.18g、3.3mmol)の溶液を−70℃でビニルマグネシウムブロミド(THF中1M、3.7mmol、3.7ml)で処理した。混合液をこの温度で5分間、その後室温で2.5時間攪拌した後、それを飽和NH4Cl溶液(10ml)で反応停止させた。溶媒を除去し、残渣をEtOAcで抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(1.2g)を得た。次いでこの粗生成物をアセトン(20ml)に溶解した。1N HCl(10ml)を加えた後、溶液を室温で15時間攪拌した。溶媒を除去し、残渣をCH2Cl2で抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(890mg)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。トルエン:アセトン混合液(94:6)による溶出で、第一分画として3α−エテニル−3β−ヒドロキシ−5β−プレグナン−20−オン(126mg)を得た。更に、同溶媒混合液による溶出で、3β−エテニル−3α−ヒドロキシ−5β−プレグナン−20−オン(189mg)を得た。mp113−116℃
例24
a.3α−ヒドロキシ−5α−アンドロスタン−17−オン
THF(50ml)中3β−ヒドロキシ−5α−アンドロスタン−17−オン(6g)およびアゾジカルボン酸ジエチル(5.04g)の溶液にトリフルオロ酢酸(3.3g)を加えたところ、混合液は黄色になった。次いでトリフェニルホスフィン(7.6g)を加えた。反応液は無色になり、発熱した。安息香酸ナトリウムを5分間後に加え、その後水(100ml)を加えた。混合液を塩化メチレン(3×80ml)で抽出し、有機層を硫酸マグネシウムで乾燥させた。溶液を真空下で除去し、粗生成物をメタノール(150ml)中水酸化カリウム(10%、10ml)で1時間かけて加水分解した。次いでほとんどのメタノールを真空下で除去し、残留物質を塩化メチレンと塩化アンモニウムに分配した。生成物(4.7g、78%)をクロマトグラフィー(CH2Cl2:アセトン=9:1)により精製した。
b.3α−ヒドロキシ−21−メチル−5α−プレグネ−17(Z)−エン
3α−ヒドロキシ−5α−アンドロスタン−17−オン(2g、6.9mmol)をTHF(20ml)中プロピルトリフェニルホスホニウムブロミド(13.3g)およびカリウムt−ブトキシド(3.9g)から製造されたWittig試薬に加えた。反応液を12時間還流し、25℃まで冷却した。次いで塩化アンモニウムの溶液(60ml)を加え、有機層を分液漏斗で分離した。水層を塩化メチレン(2×50ml)で抽出した。有機溶液を炭酸カリウムで乾燥させ、溶媒を真空下で除去した。粗生成物をクロマトグラフィー(アセトン:塩化メチレン:ヘキサン=1:2:7)により精製して、0.84gの生成物(39%)を得た。それはZおよびE異性体の混合物(Z:E=13:1)であった。
同様の操作を用いて、メチルトリフェニルホスホニウムブロミドから製造されたWittig試薬で3α−ヒドロキシ−17−メチレン−5α−アンドロスタンを製造した。
c.3α−t−ブチルジメチルシロキシ−21−メチル−5α−プレグン−17(Z)−エン
塩化メチレン(10ml)およびDMF(30ml)中3α−ヒドロキシ−21−メチル−5α−プレグネ−17(Z)−エン(0.84g、2.66mmol)、TBDMSCl(1.2g、8.0mmol)およびイミダゾール(0.91g、13.3mmol)の混合液を12時間攪拌し、その後塩化アンモニウムを加えた。次いでそれを塩化メチレン(3×40ml)で抽出し、塩水(50ml)で洗浄した。有機溶液を炭酸カリウムで乾燥させ、その後溶媒を除去したところ、まだ一部のDMFがあることがわかった。次いでそれをエーテルに再溶解し、塩水(2×50ml)で洗浄し、その後炭酸カリウムで乾燥させた。ヘキサンによるクロマトグラフィーで、純粋生成物1.14g(100%)を得た。
d.3α−t−ブチルジメチルシロキシ−20α−ヒドロキシ−21−メチル−5α−プレグナン
THF(30ml)中3α−t−ブチルジメチルシロキシ−21−メチル−5α−プレグネ−17(Z)−エン(1.14g、2.66mmol)の溶液に0℃でジボランTHF錯体(THF中1M溶液、5.3ml)を滴下した。反応液を25℃に1時間加温した。次いで水酸化ナトリウムの溶液(20%、10ml)を0℃で非常にゆっくり加え、その後過酸化水素(30%、10ml)を加えた。次いで水性塩化アンモニウムを加え、THF層を分液漏斗で分離した。水層をエーテル(2×40ml)で抽出した。有機溶液を炭酸カリウムで乾燥させ、溶媒を真空下で除去した。純粋生成物をカラムクロマトグラフィーにより得た(0.52g、44%)。
e.3α,20α−ジヒドロキシ−21−メチル−5α−プレグナン
HF(48%、5ml)およびCH3CN(30ml)を混合することで得た溶液を3α−t−ブチルジメチルシロキシ−20α−ヒドロキシ−21−メチル−5α−プレグナン(0.51g)含有のフラスコに加えたところ、白色沈殿物が出現した。反応液を1時間攪拌し、その後濾過した。白色固体物をエーテルで3回洗浄したところ、それは満足できる分析結果を示した(0.3g、79%)。mp227−231℃
例25
3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−19−ノルプレグナン−20−オン
トルエン(15ml)中3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノルプレグナン−20−オン(300mg、0.87mmol)の溶液にMeOH(1ml)およびBF3・OEt2(1.4ml、11.3mmol)を加えた。得られた混合液を0℃に冷却し、Pb(OAc)4(0.54g、1.21mmol)を加えた。反応液を攪拌下で25℃に45分間加温し、その後NaHCO3溶液(飽和、30ml)を加え、混合液を1時間攪拌した。それを水(50ml)含有の分液漏斗に注ぎ、エーテル(3×40ml)で抽出した。エーテル溶液を塩水(50ml)で洗浄し、その後K2CO3で乾燥させた。溶媒の除去で得られた粗生成物をMeOH(25ml)に溶解し、K2CO3(飽和、8ml)を加えた。反応液を5時間攪拌し、その後反応混合液を水(50ml)含有の分液漏斗に注いだ。次いでそれをCH2Cl2(3×30ml)で抽出した。合わせた抽出液をK2CO3で乾燥し、得られた粗製物質をクロマトグラフィーにより精製して、21−メトキシ副生成物(40mg)と共に本生成物(160mg)を得た。その生成物をヘキサン中10%アセトンからの再結晶化で更に精製し、白色固体物として純粋生成物88mg(28%)を得た。mp140−142℃
例26
a.3α−ヒドロキシ−3β−メチル−5β−19−ノルプレグン−(Z)17(20)−エン
THF(20ml)中5β−19−ノルプレグン−17(Z)−エン−3−オン(600mg、2.1mmol)の溶液にメチルマグネシウムブロミド(エーテル中3M、1 ml、3mmol)を加えた。反応液を25℃に加温した。塩化アンモニウム溶液(50ml)を加え、混合液をエーテル(3×30ml)で抽出した。クロマトグラフィー(ヘキサン中10%アセトン)により精製された生成物(577mg、91%)を白色ロウ状固体物として得た。
b.3α−ヒドロキシ−3β−メチル−5β−19−ノルプレグナン−20−オン
THF(30ml)中3α−ヒドロキシ−3β−メチル−5β−19−ノル−17(Z)−プレグネ−17(20)−エン(0.57g、1.9mmol)の溶液に25℃でBH3・THF溶液(THF中1M、3.8ml、3.8mmol)を加えた。反応は30分間で終了し、NaOH溶液(20%、8ml)を非常に慎重に加え、その後過酸化水素溶液(30%、8ml)を加えた。混合液を20分間攪拌し、その後NH4Cl溶液を加えた。混合液をCH2Cl2(3×30ml)で抽出し、合わせた抽出液をK2CO3で乾燥させた。溶媒の除去により粗製物質(760mg)を得、これをCH2Cl2(40ml)に溶解した。4−メチルモルホリン N−オキシド(0.6g、4.75mmol)および粉砕したばかりのモレキュラーシーブ(4Å、5g)を加え、得られた混合液を20分間攪拌した。過ルテニウム酸テトラブチルアンモニウム触媒(100mg)を加えた。酸化は40分間で完了し、反応混合液をFlorisilのパッドで濾過し、エーテルおよびCH2Cl2の2:1混合液(60ml)で洗浄した。溶媒の除去により得られた粗製物質(0.62mg)をクロマトグラフィー(ヘキサン中20%EtOAc)により精製し、白色固体物として生成物(53mg、9%)を得た。
例27
3β−エチニル−3α,20α−ジヒドロキシ−5β−プレグナンおよび3β−エチニル−3α,20β−ジヒドロキシ−5β−プレグナン
メタノール(20ml)中3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン(0.31g、0.91mmol)の溶液に水素化ホウ素ナトリウム(200mg、5.3mmol)を加え、それを25℃で1時間攪拌した。次いで塩化アンモニウム(50ml)溶液を加え、混合液をCH2Cl2(3×30ml)で抽出した。クロマトグラフィー(EtOAc:ヘキサン=3:7)により主生成物として3β−エチニル−3α,20β−ジヒドロキシ−5β−プレグナン(200mg、65%)を得た:mp221−223℃。副生成物3β−エチニル−3α,20α−ジヒドロキシ−5β−プレグナンを更に別のクロマトグラフィーにより精製した(ヘキサン中25〜30%酢酸エチル、16mg、5%):mp187−188℃
例28
a.3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン
メタノール45ml中3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン,21−アセテート(725mg、1.81mmol)の溶液に0℃で10%K2CO3水溶液(3.75ml)を加えた。室温で30分間攪拌した後、混合液を0℃に再冷却し、2N HOAc水溶液(1.8ml)を加えた。反応液をEtOAc/水混合液に加えた。水層をEtOAcで2回抽出し、プールされた有機層を塩水溶液で抽出し、(Na2SO4)乾燥し、濃縮した。フラッシュクロマトグラフィー(4cm径カラム中シリカ20cm、20%アセトン/ヘキサン2lで溶出)による精製で、白色固体物としてジオール582mg(90%)を得た。mp155.5−157℃
b.3β−エチニル−3α−ヒドロキシ−21−メトキシ−5β−プレグナン−20−オン
メタノール(4ml)およびトルエン(17ml)中3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン(503mg、1.47mmol)の溶液を氷/水浴で冷却し、ニートBF3−Et2O(2.9ml、3.35g、23.6mmol)とその後に固体四酢酸鉛1.0g(2.25mmol)を加えた。混合液を室温まで加温し、その後80分間攪拌した。次いで反応液を氷冷飽和NaHCO3溶液25mlに加えた。水層をEtOAc(2×10ml)で抽出し、合わせた有機層を飽和NaCl溶液で抽出し、(Na2SO4)乾燥し、濃縮した。2つのフラッシュカラム(シリカゲル、2.5%アセトン/CH2Cl2で溶出)により白色固体物として標題化合物148mg(27%)を得た。mp198.5−199.5℃
例29
(3R)−20,20−エチレンジオキシ−5α−プレグナン−スピロ−2′−オキシランの製造
乾燥THF125ml中トリメチルスルホキソニウムヨージド(Aldrich、8.79g、39.9mmol)および固体カリウムt−ブトキシド(Aldrich、95%、4.3g、36.6mmol)の混合液を還流下で1.5時間加熱した。反応液を室温まで冷却し、20,20−エチレンジオキシ−5α−プレグナン−3−オン(12.0g、33.3mmol)を固体として一度に加えた。得られた混合液を室温で20時間攪拌し、その後真空下で濃縮した。残渣をCH2Cl2に溶解し、水で抽出した。水層をCH2Cl2で逆抽出し、合わせた有機層を飽和NaCl溶液で洗浄し、(MgSO4)乾燥し、濃縮した。得られた白色固体物(12.0g、96%、mp160−165℃)を60/40アセトン/メタノールから再結晶化した。
例30
3α,20−ジヒドロキシ−3β,20−ジメチル−5α−プレグナン
5α−プレグナン−3,20−ジオン(1.5g、4.7mmol)を乾燥ベンゼン250mlに溶解し、3Mメチルマグネシウムブロミド15mlを加えた。反応混合液を室温で30分間攪拌した。過剰のメチルマグネシウムブロミドを0℃で硫酸100mlの滴下により分解させた。生成物溶液をジエチルエーテル250mlで抽出した。有機抽出液を合わせ、硫酸マグネシウムで乾燥し、蒸発させて、3β−メチルおよび3α−メチル異性体の60:40混合物を得た。生成物混合物をシリカゲルフラッシュクロマトグラフィーにより溶出液として70:30ヘキサン:酢酸エチルおよび1%メタノールを用いて分離させ、望ましい生成物0.476g(収率24%)を得た。
例31
3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン
MeOH(75ml)中3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン,21−アセテート(1.36g、3.06mmol)の溶液を0℃に冷却した。次いでK2CO3の溶液(10%水性、6.45ml、4.67mmol)を滴下した。0℃で1.5時間攪拌した後、酢酸の溶液(2N水性、2.5ml、5.0mmol)を滴下し、混合液を室温まで加温した。EtOAc、CH2Cl2および水(各々100ml)を加え、十分に混合した。有機相を分離し、水性NaHCO3およびNaCl溶液で洗浄し、MgSO4で乾燥し、真空下で蒸発させた。残渣をフラッシュカラムクロマトグラフィー(ヘキサン/EtOAc3:1)により精製して、白色固体物(973mg、79%)を得た。mp148−150℃
例32
a.3α−ヒドロキシ−5α−21−エチル−プレグン−17(Z)−エン
THF(10ml)中n−ブチルトリフェニルホスホニウムブロミド(3.9g、9.66mmol)およびカリウムt−ブトキシド(1.1g、9.66mmol)の混合液を30分間攪拌することにより製造されたWittig試薬に3α−ヒドロキシ−5α−アンドロスタン−17−オン(0.7g、2.4mmol)を加え、混合液を16時間加熱還流した。次いで反応を塩化アンモニウム溶液で停止させ、CH2Cl2で抽出した。純粋生成物(230mg、29%)をクロマトグラフィー(ヘキサン中10%アセトン)により得た。
b.3α−t−ブチルジメチルシロキシ−5α−21−エチル−プレグン−17(Z)−エン
t−ブチルジメチルシリルクロリド(0.32g、2.1mmol)、イミダゾール(0.24g、3.5mmol)および3α−ヒドロキシ−5α−21−エチル−プレグネ−17(Z)−エン(0.23g、0.7mmol)の混合液を25℃で12時間攪拌した。次いで塩化アンモニウム溶液を加え、抽出をエーテル(3×30ml)で行った。得られた抽出液を塩水(3×30ml)で洗浄した。次いで純粋生成物(0.27g、87%)をクロマトグラフィー(ヘキサン)により得た。
c.3α,20α−ジヒドロキシ−5α−21−エチルプレグナン
THF(20ml)中3α−t−ブチルジメチルシロキシ−5α−21−エチル−プレグネ−17(Z)−エン(0.27g、0.6mmol)の溶液に25℃でBH3・THF(THF中1M、1.2ml)を加えた。次いで反応液を1時間攪拌してから、水酸化ナトリウム(20%、5ml)を慎重に加えた。次いで過酸化水素溶液(30%、5ml)を加え、混合液を更に1時間攪拌した。次いで塩化アンモニウム溶液を加え、それをCH2Cl2で抽出した。20α−ヒドロキシ−3α−シリルエーテル化合物をクロマトグラフィー(ヘキサン中5%アセトン)により得た。この化合物60mgをアセトニトリル中フッ化水素酸で脱シリル化した。クロマトグラフィー(ヘキサン中20〜30%酢酸エチル)により純粋標題化合物11mgを得た(mp229−232℃、Rf=0.28、EtOAc:ヘキサン=3:7)。
例33
3β−フルオロメチル−3α−ヒドロキシ−5α−プレグナン−20−オン
n−Bu4NF・xH2O(7.873g)およびベンゼン(50ml)の混合液をDean-Starkトラップで一夜還流した。次いで透明溶液でない混合液を〜10mlまで濃縮(常圧)し、室温まで冷却した。乾燥ベンゼン(15ml+洗浄用5ml)中3(R)−5α−プレグナン−3−スピロ−2′−オキシラン−20−オン エチレンケタール(2.55g、6.81mmol)の溶液を二重末端針で上記濃縮溶液に加えた。得られた溶液を短絡蒸留装置で〜10mlに濃縮し、15分間還流した。濃縮反応溶液をTLCプレートにスポットすることが非常に困難であったため、乾燥ベンゼン(5ml)を加えた。TLC(100:1CH2Cl2/アセトンまたは3:1ヘキサン/アセトン)では2つの新たな低極性側スポット以外にも一部の未反応出発物質を示した。反応混合液を再び濃縮し、その後30分間還流した。TLC(混合液をベンゼンで希釈した後)では出発エポキシドのほぼ完全な消費を示した。前のように、混合液を濃縮し、しばらく還流した。この反応は混合液が高度に濃縮されたときだけ生じることにここでは注意しなければならない。混合液が室温に戻った(淡黄色固体物を生じた)後、エーテルおよび水を加えた。すべての固体物が溶解するわけではないため、CH2Cl2を加えた。水層をCH2Cl2で逆抽出した。合わせた有機抽出液を水(×2)で洗浄し、(Na2SO4)乾燥し、濾過し、減圧下で蒸発させて、白色固体物(3.33g)を得たが、これは1H NMRによると3β−フルオロメチル−3α−ヒドロキシ−5α−プレグナン−20−オン エチレンケタールおよび3α−フルオロ−3β−ヒドロキシメチル−5α−プレグナン−20−オン エチレンケタールの4:1混合物であることを示した。
ケタールを加水分解するために、アセトン(100ml)、水(5ml)およびp−TsOH・H2O(143mg、0.752mmol)を上記固体物に加えた。pHはやや酸性になるまでHClを加えることで調整した。混合液をヒートガンでしばらく加熱して透明溶液を得、これを室温で2時間攪拌した。混合液は混濁するようになるため、CH2Cl2を加えて透明溶液を得た。TLCでは反応の完了を示した。溶媒を減圧下で除去して白色固体物を得、これにCH2Cl2および水を加えた。水層をCH2Cl2で逆抽出した。合わせた有機層を飽和NaHCO3で洗浄し、(MgSO4)乾燥し、濾過し、減圧下で蒸発させて、白色結晶固体物(2.5g)を得た。溶出液としてCH2Cl2による粗生成物のフラッシュカラムクロマトグラフィーにより望ましい3β−フルオロメチル−3α−ヒドロキシ−5α−プレグナン−20−オン(1.41g、59%)を得た。mp201−3℃
例34
3α−ヒドロキシ−3β−メチル−5α−プレグン−17(20)−エン
乾燥THF8ml中5α−プレグン−17(20)−エン−3−オン(207mg、0.689mmol)の溶液に−30℃で(−78℃では、すべてのステロイドが溶解しない)エーテル中MeMgBrの3M溶液(Aldrich、0.34ml、1.02mmol)をシリンジで滴下した。−30℃で4.75時間攪拌した後、反応液を飽和NH4Cl溶液/エーテル混合液に加えた。水層をエーテル(3×30ml)で抽出し、有機層を合わせ、塩水溶液で洗浄し、(MgSO4)乾燥し、減圧下で濃縮した。粗製物質をCH2Cl2に溶解し、2cmカラム中32cmのフラッシュシリカゲルに加えた。カラムを未反応ケトン(8.7mg、4%)が溶出するまで100%CH2Cl2で溶出させた。各々100mlの2.5および5%アセトン/CH2Cl2による溶出で、38mg(18%)の3α−ヒドロキシ−3β−メチル−5α−プレグン−17(20)−エン(Rf0.35、100%CH2Cl2)および3β−ヒドロキシ−3α−メチル−5α−プレグン−17(20)−エン(Rf0.16、100%CH2Cl2)を得た。
例35
3α−ヒドロキシ−17Z−および17E−メトキシメチレン−5β−アンドロスタン
固体3α−ヒドロキシ−5β−アンドロスタン−17−オン(504mg、1.74mmol)をメトキシメチルトリフェニルホスホニウムブロミド(Aldrich、1.484g、4.33mmol)およびカリウムtert−ブトキシド(479mg、4.27mmol)から製造されたイリドに何回かに分けて加えた。この混合液を7時間還流し、その後0℃に冷却し、氷冷飽和NH4Cl溶液/エーテル混合液に加えた。水層をエーテルで2回抽出し、合わせた有機層を飽和NaCl溶液で洗浄し、(MgSO4)乾燥し、濃縮した。粗製アルケンをCH2Cl2に溶解し、3.5cm径カラム中13cmのフラッシュシリカに加えた。カラムを5%アセトン/CH2Cl2400mlおよび7.5%アセトン/CH2Cl2200mlで溶出させた。望ましいアルケンは回収された17−オン(220mg、44%)と共にZ−およびE−異性体の1:1混合物(158mg、28%)として単離した。アルケン異性体の混合物をフラッシュクロマトグラフィー(3.5cm径カラム中シリカ15cm、4%アセトン/トルエン500mlおよび5、6および7%アセトン/トルエン各々200mlで溶出)に再び付したが、うまく分離できなかった。プレパラティブHPLC(21.4mm径×25cm Microsorb5mカラム;溶媒系2.5%アセトン/CH2Cl2、10ml/min;RI検出)によりZ−異性体、mP150−152℃(高極性側)およびE−異性体、mp151−153℃(低極性側)を分離した。
例36
3α,20β−ジヒドロキシ−3β−メチル−5α−プレグナンおよび3α,20α−ジヒドロキシ−3β−メチル−5α−プレグナン
参考文献:House,H.O.,Mnller.H,C.,Pitt,C.G.,Wickam,P.P.,J.Org.Chem.,1963,28,2407
切断したばかりのナトリウム(250mg、10.87mmol)および沸騰トルエン(5ml)の混合液にイソプロピルアルコール(3ml、39.18mmol)およびトルエン(4ml+洗浄用2ml)中3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン(340mg、1.022mmol)の溶液を30分間かけて滴下した。得られた混合液を2.5時間還流したが、そのときのTLC(3:1ヘキサン:アセトン)では反応の完了を示し、出発ケトンよりも低いRfを有してほぼ同様の強度を有する2つの新たなスポットを現した。反応溶液を室温に戻し、その後氷浴で冷却した。1N HCl(10ml)を滴下した。混合液をCH2Cl2(〜50ml)で希釈した後、水層を分離し、CH2Cl2(×2)で逆抽出した。合わせた有機層を飽和NaHCO3で洗浄し、(MgSO4)乾燥し、濾過し、減圧下で蒸発させて、白色固体物(354mg)を得た。これら2生成物の精製は溶出液として15:1および10:1ヘキサン/アセトンを用いたフラッシュクロマトグラフィーにより行った。
初期分画の蒸発により、白色固体物として3α,20β−ジヒドロキシ−3β−メチル−5α−プレグナン(80mg)を得た。1H NMRおよびキャピラリーGCを用いて、純度を調べた。
更に溶出により3α,20α−ジヒドロキシ−3β−メチル−5α−プレグナンおよび3α,20β−ジヒドロキシ−3β−メチル−5α−プレグナンの9:1混合物(GCに基づく)(合計248mg)を得た。この混合物を加温エタノール(〜10ml)からの再結晶化に付したが、結晶は得られなかった。次に加温5:1ヘキサン/アセトン(〜50ml)で試みたところ、白色結晶(110mg)を得た。GCによると、これらの結晶は96%の純度を有するだけであった。最後に、純粋な3α,20α−ジヒドロキシ−3β−メチル−5α−プレグナン(80mg)を更に加温エタノールからの再結晶化で得ることができた。
例37
3α−ヒドロキシ−3β−ヨードメチル−5α−プレグナン−20−オン
アセトン(3ml)中3(R)−スピロ−2′−オキシラン−5α−プレグナン−20−オン(40mg、0.12mmol)の溶液にNaI(39mg、0.26mmol)を加えた。得られた溶液を室温で〜4時間攪拌した。TLC(3:1ヘキサン/アセトン)では新たな遅移動スポットの出現と共にほんの少量の出発物質の消費を示し、これら2スポットの相対強度は経時的に変化せず、出発物質とヨードメチル生成物(アルコキシドとして)との平衡の存在を示唆した。その後酢酸(〜0.02ml)を上記反応溶液に加えた。混合液を室温で一夜攪拌した。そのときTLCでは出発エポキシドの完全消失を示した。反応混合液は少量の白色結晶(酢酸ナトリウム)を含有していた。溶媒を減圧下で除去し、その後エーテルおよび水を加えた。水相を分離し、エーテルで逆抽出した。合わせたエーテル溶液を水および塩水で連続洗浄し、(MgSO4)乾燥し、濾過し、減圧下で蒸発させて、白色固体物として3α−ヒドロキシ−3β−ヨードメチル−5α−プレグナン−20−オン(51mg、92%)を得た。
例38
a.3,3−エチレンジオキシ−5β−プレグン−17(20)−エン
5β,3−エチレンジオキシ−アンドロスタン−17−オン(6g)をTHF(15ml)中エチルトリフェニルホスホニウムブロミド(15g)およびカリウムt−ブトキシド(4.5g)から製造されたWittig試薬に加えた。反応液を2時間還流し、25℃まで冷却した。次いで塩化メチレン(80ml)および塩化アンモニウム(60ml)の溶液を加え、有機層を分液漏斗で分離した。水層を塩化メチレン(2×50ml)で抽出した。有機溶液を炭酸カリウムで乾燥させ、溶媒を真空下で除去した。ほとんどのホスホロキシドはヘキサンで洗浄することにより除去した。得られた生成物(6g)をアセトン(100ml)に溶解し、塩酸(2N、10ml)を加えた。塩基性後処理とその後に塩化メチレン抽出により得られた粗生成物(5.5g)をクロマトグラフィーにより精製して、4.5gの生成物(83%)を得た。純粋な立体異性体をヘキサンからの反復再結晶化により得た。
b.3α−ヒドロキシ−3β−トリフルオロメチル−5β−プレグン−(Z)17(20)−エン
THF(15ml)中5β−プレグン−(Z)−17(20)−エン−3−オン(950mg、3.17mmol)の溶液に0℃でトリフルオロメチルトリメチルシラン(THF中0.5M溶液、9.5ml)を加えた。溶液は徐々に褐色になり、反応は30分間で終了した。水(30ml)を加え、有機層を集めた。水層をエーテル(3×50ml)で抽出し、有機溶液を炭酸カリウムで乾燥させた。純粋生成物(680mg、58%)をカラムクロマトグラフィーにより溶出液として酢酸エチルおよびヘキサン(1:9)を用いて単離した。
例39
a.3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン,21−アセテート
トルエン35ml中3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン(1.00g、2.92mmol)の懸濁液をメタノール2mlで処理した。得られた溶液を氷/水浴で冷却し、ニートBF3−Et2O(Aldrich、5.8ml、5.02g、35.4mmol)を加えた。固体四酢酸鉛1.0g(Aldrich、1.96g、4.42mmol)を数回に分けて加えた。淡紫色溶液が最初に生じ、攪拌を0℃で続けたとき淡褐色になった。混合液を室温で3時間攪拌し、その後0℃に再冷却した。冷反応液を飽和NaHCO3溶液、水および砕氷混合液52mlに加えた。得られた混合液をEtOAc(2×75ml)で抽出した。合わせた有機層を飽和NaCl溶液で抽出し、(Na2SO4)乾燥し、濃縮した。粗生成物をカラムクロマトグラフィー(5cm径カラム中フラッシュシリカゲル25cm、20%アセトン/ヘキサン31で溶出)により精製して、アセテート749mg(64%)を得た。mp196−198℃
b.3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン,21−ヘミサクシネート
アセトン50ml中21−ブロモ−3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン(1.5g、3.56mmol)の攪拌溶液にコハク酸(2.1g、17.8mmol)およびトリエチルアミン(3ml、21.4mmol)を加えた。反応液を3.5時間加熱還流し、その後室温まで冷却してから、酢酸エチルと希水性HClに分配した。有機層を飽和NaClで洗浄し、Na2SO4で乾燥し、真空下で濃縮して、粗製固体物を得た。固体物をジクロロメタンで摩砕して、固体物としてコハク酸を除去し、液体を真空下で濃縮して、泡状物(1.36g)を得た。3cmカラム中シリカゲル6inでフラッシュクロマトグラフィーにより、1:3→3:1酢酸エチル:ヘキサンで勾配を徐々に調整して溶出させて50ml分画を集め、標題化合物830mg(51%)を得た。
c.3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン,21−ヘミサクシネートナトリウム
ヘミサクシネート633mg、mp62−68℃をメタノールに溶解し、NaHCO3116mg(0.253mmol)の水溶液を加えた。3.5時間攪拌した後、溶媒を減圧下で除去し、残渣をエーテル/ヘキサン混合液で摩砕した。得られた淡黄色固体物616mgは>20mg/mlで水溶性であった。
例40
3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン,21−アセテート
無水アルゴン雰囲気下で、3α−ヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン(1.94g、5.02mmol)をトルエン(86ml)およびMeOH(5.2ml)に溶解した。三フッ化ホウ素エーテル錯体(10.4ml、84.3mmol)をシリンジで加えた。次いで四酢酸鉛(2.89g、6.51mmol)を加えた。混合液を70分間攪拌し、水中に注ぎ、CH2Cl2で3回抽出した。有機相を合わせ、水性NaHCO3およびNaCl溶液で洗浄し、MgSO4で乾燥し、真空下で蒸発させ、淡黄色固体物(2.18g)を得た。この固体物をフラッシュカラムクロマトグラフィー(CH2Cl2/EtOAc150:1およびヘキサン/EtOAc4:1)により精製して、白色固体物(1.54g、69%)を得た。mp167−168.5℃
例41
3β−クロロメチル−3α−ヒドロキシ−5α−プレグナン−20−オン
3(R)−スピロ−2′−オキシラン−5α−プレグナン−20−オン(46mg、0.14mmol)、テトラメチルアンモニウムクロリド(25mg、0.23mmol)および乾燥DMF(Aldrich、5ml)の混合液を〜130℃で加熱した。透明溶液をしばらくして得た。TLC(7:1ヘキサン/アセトン)では、出発物質以外にも、出発エポキシドより遅く移動する新たなスポットを示した。2時間後に反応の進行はもうなかった。更にテトラメチルアンモニウムクロリド(〜25mg)を反応混合液に加えた。得られた混合液を130〜150℃で1時間攪拌した。すべての固体物は溶解しなかった。TLCに変化はなかった。おそらく、クロロメチル生成物(アルコキシドとして)と出発エポキシドとの平衡があるのだろう。次いで混合液がまだ非常に熱いうちに酢酸(0.1ml、1.75mmol)を反応混合液に加えた。混合液中に存在するすべての固体粒子が溶解し、透明溶液が生じた。この溶液を〜100℃で2時間加熱したところ、TLCは反応の完了を示し、1つだけのスポットを現した。溶媒を真空蒸留により除去した。得られた固体物にCH2Cl2および水を加えた。水層をCH2Cl2で逆抽出した。合わせた有機層を(MgSO4)乾燥し、濾過し、減圧下で蒸発させて、固体物(46mg)を得た。この固体物の1H NMRでは望ましい位置異性体(regioisomer)(即ち、3β−クロロメチル−3α−ヒドロキシ−5α−プレグナン−20−オン)のみを示した。最後に、溶出液として9:1ヘキサン/アセトンを用いたフラッシュクロマトグラフィーにより白色固体物として標題化合物(35mg、68%)を得た。mp209−10℃(分解)
例42
a.3β−(3′−ブロモ−1−プロピニル)−3α−ヒドロキシ−5β−プレグナン−20−オン
乾燥THF(7ml)中臭化プロパルギル(トルエン中80%溶液、0.4ml、3.6mmol)の溶液を−78℃でn−BuLi(THF中2.5M、3mmol、1.2ml)で処理した。混合液をこの温度で10分間攪拌した後、5β−プレグナン−3,20−ジオン,20−ケタール(360mg、1mmol)の溶液を加え、混合液を−78℃で2時間攪拌した。次いでそれを飽和NH4Cl溶液(1ml)で反応停止させた。溶媒を除去し、その後残渣をアセトン(40ml)に溶解した。2N HCl(10ml)を加えた後、溶液を室温で3.5時間攪拌した。飽和NaHCO3溶液を加えて、酸を中和させた。溶媒を除去し、残渣をCH2Cl2で抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(360mg)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。ヘキサン:アセトン混合液(90:10)による溶出で、第一分画として3α−(3′−ブロモ−1−プロピニル)−3β−ヒドロキシ−5β−プレグナン−20−オン(50mg)を得た。更に同溶媒混合液による溶出で3β−(3′−ブロモ−1−プロピニル)−3α−ヒドロキシ−5β−プレグナン−20−オン(181mg)を得た。
例43
3β−ブロモメチル−3α−ヒドロキシ−5α−プレグナン−20−オン
3(R)−スピロ−2′−オキシラン−5α−プレグナン−20−オン(362mg、1.10mmol)、テトラメチルアンモニウムブロミド(322mg、2.16mmol)、酢酸(1.0ml、17.5mmol)および乾燥DMF(Aldrich、30ml)の混合液を〜130℃で加熱した。混合液中にはまだ一部の未溶解テトラメチルアンモニウムブロミドが存在し、110〜120℃で攪拌した。110〜120℃で30分間加熱した後のTLC(3:1ヘキサン/アセトン)では、出発物質以外にも、新たな高極性側スポットを示した。主生成物スポットより遅く移動する他の2つの非常に淡いスポットもあり、これら2つの非常に淡いスポットの強度は経時的に増加した。110〜120℃で3時間後、TLCでは経時的に量が変化しない未反応出発物質に相当するスポット以外にも3つのほぼ等しい強度のスポットを示した。溶媒を真空蒸留により除去した。得られた固体物にCH2Cl2、水および塩水(エマルジョン形成を防ぐため)を加えた。有機層を飽和NaHCO3で洗浄し、(MgSO4)乾燥し、濾過し、減圧下で蒸発させて、固体物(380mg)を得た。この固体物の1H NMRでは3種の生成物、即ち3β−ブロモメチル−3α−ヒドロキシ−5α−プレグナン−20−オン、3−ヒドロキシメチル−5α−プレグネ−2−エン−20−オンおよび3β−(アセトキシメチル)−3α−ヒドロキシ−5α−プレグナン−20−オンと未反応出発物質の存在を示した。最後に、溶出液として9:1ヘキサン/アセトンを用いたフラッシュクロマトグラフィーにより、白色固体物として純粋な3β−ブロモメチル−3α−ヒドロキシ−5α−プレグナン−20−オン(98mg、22%)を得た。mp196−97℃(分解)
例44
a.3α−ヒドロキシ−19−ノル−5α−17−メトキシメチレンアンドロスタン
THF(20ml)中19−ノル−5α−17−メトキシメチレンアンドロスタン−3−オン(200mg、0.65mmol)の溶液に−78℃でシリンジによりK−selectride(THF中1M)を加え、反応液をこの温度で4.5時間攪拌した。次いで混合液を塩化アンモニウム溶液(飽和、20ml)および塩化メチレン(40ml)含有の分液漏斗中に注いだ。水層を塩化メチレン(2×15ml)で抽出した。有機溶液を炭酸カリウムで乾燥し、溶媒を真空下で除去した。カラムクロマトグラフィー(ヘキサン中4%アセトン、4%塩化メチレン)により生成物130mg(64%)を得た。
b.3α−ヒドロキシ−19−ノル−5α−アンドロスタン−17β−カルボキサルデヒド
THF(10ml)中3α−ヒドロキシ−19−ノル−5α−17−メトキシメチレンアンドロスタン(130mg、0.43mmol)の溶液に25℃で塩酸の溶液(1N、1ml)を加え、反応液を6時間攪拌した。次いで炭酸水素ナトリウム溶液(NaHCO3、飽和、20ml)および塩化メチレン(30ml)を加えた。有機層を分離し、水層を塩化メチレン(2×20ml)で抽出した。有機溶液を炭酸カリウムで乾燥し、溶媒を真空下で除去した。純粋生成物(80mg、65%)をカラムクロマトグラフィーにより溶出液としてアセトンおよびヘキサン(1:4)を用いて単離した。
例45
3α,21−ジヒドロキシ−3β−メチル−5α−プレグナン−20−オン,21−リン酸二ナトリウム
THF10ml中21−ブロモ−3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン(1.0g、2.43mmol)の溶液に室温で攪拌しながらリン酸水素ジベンジル(2.1g、7.3mmol)およびトリエチルアミン(1.085ml、7.53mmol)を加えた。次いで反応液を4.5時間加熱還流し、その後室温まで冷却した。次いでジクロロメタン(25ml)を加え、溶液を分液漏斗に移し、1N HCl、飽和水性NaHCO3で洗浄し、MgSO4で乾燥し、真空下で濃縮して、粗製油状物としてジベンジルホスフェート(1.205g)を得た。ジベンジルホスフェート(790mg、1.3mmol)を数滴の硫酸と共に2:1EtOH:THF(30ml)に溶解し、5%Pd/C(180mg、20重量%)を加え、反応がTLCで終了するまで室温で50psi(約3.5kg/cm2)のH2に付した。触媒を濾去し、濾液を濃縮した。残渣を4:1MeOH:水(10ml)に溶解し、1M NaOHでpH11まで滴定した(溶液は混濁した)。溶液を易濾過性固体物が沈殿するまでアセトンで処理してから、混合液を0℃に冷却し、濾過して、粗製固体物260mgを単離した。固体物を水20mlに溶解して混濁溶液を形成し、それを濾過し、その後濃縮して、白色固体物として標題化合物220mgを得た。
例46
a.3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン,21−ヘミサクシネート
コハク酸(20g、170mmol)およびトリエチルアミン(19ml、140mmol)をアセトン(200ml)中粗製21−ブロモ−3α−ヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン(5.36g、10.9mmol)の溶液に加えた。混合液を1.75時間還流した。残渣を氷水(350ml)に注いだ。次いで更に水(100ml)を加えた。混合液を攪拌し、その後CH2Cl2(200ml、200mlおよび100ml)で3回抽出した。合わせた有機相をMgSO4で乾燥し、真空下で蒸発させて半固体残渣(5.71g)を得、これをフラッシュカラムクロマトグラフィー(CH2Cl2/MeOH、勾配90:1〜20:1)により精製して、白色固体物(2工程で3.63g、66%)を得た。mp:分解(広範囲)
b.3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン,21−ヘミサクシネート,ナトリウム塩
3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン,21−ヘミサクシネート(859mg、1.71mmol)をMeOH(35ml)に溶解した。次いで水(10ml)中NaHCO3(143mg、1.70mmol)を滴下した。更にMeOH(10ml)を加えた。2.5時間攪拌した後、溶媒を蒸発させた。トルエンを加え、真空下で蒸発させた。残渣をエーテル/ヘキサン、その後ヘキサンで処理して、白色固体物を得た。MeOHを加え、除去した。ヘプタンを加え、真空下で除去した。残渣をヘキサンで処理し、真空下で乾燥させて、白色固体物(878mg、98%)を得た。
例47
a.21−(N,N−ジメチルアミノ)メチル−3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン
アセトニトリル(15ml)中3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン(3.32g、10mmol)およびN,N−ジメチルメチレンアンモニウムクロリド(1.6g、17mmol)の溶液を2.5時間還流した。冷却した後、溶媒を除去し、残渣を飽和NaHCO3溶液の添加で塩基性化した。次いで混合液をCH2Cl2で抽出し、塩水で洗浄し、無水MgSO4で乾燥し、蒸発させて粗生成物を得た。この生成物をアセトンから結晶化し、無色棒状物として21−(N,N−ジメチルアミノ)メチル−3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン(550mg)を得た。
母液を蒸発乾固させ、残渣をシリカゲルクロマトグラフィーに付した。ヘキサン:アセトン(2:3、1:4)による溶出で、無色固体物として21−(N,N−ジメチルアミノ)メチル−3α−ヒドロキシ−3β−メチル−21−メチレン−5α−プレグナン−20−オン(200mg)を得た。更にアセトンによる溶出で、無色固体物として21−(N,N−ジメチルアミノ)メチル−3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン(1g)を更に得た。
b.3α−ヒドロキシ−3β−メチル−21−メチレン−5α−プレグナン−20−オン
21−(N,N−ジメチルアミノ)メチル−3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン(500mg)およびMeI(4ml)の混合液を室温で1.5時間攪拌した。CH2Cl24mlを加え、攪拌を更に15時間続けた。MeIおよびCH2Cl2を除去し、残渣をCH2Cl2(20ml)に溶解し、飽和NaHCO3溶液(15ml)で処理した。室温で2.5時間攪拌した後、有機層を分離し、塩水で洗浄し、無水MgSO4で乾燥し、蒸発させた。残渣(500mg)をシリカゲルクロマトグラフィーに付した。CH2Cl2:アセトン(95:5)による溶出で3α−ヒドロキシ−3β−メチル−21−メチレン−5α−プレグナン−20−オン(300mg)を得た。
c.3α−ヒドロキシ−3β,21−ジメチル−5α−プレグナン−20−オン
EtOAc(30ml)中3α−ヒドロキシ−3β−メチル−21−メチレン−5α−プレグナン−20−オン(250mg)の溶液にPd/C(27mg、5%)の存在下室温で1時間(2.5atmのH2)水素付加した。濾過、その後溶媒の除去後に、無色固体物として標題化合物(198mg)を得た。mp190−192℃
例48
a.21−(N,N−ジメチルアミノ)メチル−3α−ヒドロキシ−5β−プレグナン−20−オン
アセトニトリル(3ml)中3α−ヒドロキシ−5β−プレグナン−20−オン(318mg、1mmol)およびN,N−ジメチルメチレンアンモニウムクロリド(100mg、1.1mmol)の溶液を2時間還流した。冷却した後、溶媒を除去し、残渣を2N HCl 5mlの添加で酸性化した。次いで混合液をEtOAcで抽出した。水層を分離し、2N NaOHで中和した。沈殿固体物をCH2Cl2で抽出した。有機層を分離し、塩水で洗浄し、無水MgSO4で乾燥し、蒸発させて、無色固体物として粗製21−(N,N−ジメチルアミノ)メチル−3α−ヒドロキシ−5β−プレグナン−20−オン(200mg)を得、これを次の工程でそのまま用いた。
b.3α−ヒドロキシ−21−メチレン−5β−プレグナン−20−オンおよび3α−ヒドロキシ−21−メトキシメチル−5β−プレグナン−20−オン
21−(N,N−ジメチルアミノ)メチル−3α−ヒドロキシ−5β−プレグナン−20−オン(550mg)、CH2Cl2(2ml)およびMeI(4ml)の混合液を室温で1時間攪拌した。MeIおよびCH2Cl2を除去し、残渣をMeOH中で72時間還流した。冷却した後、混合液を氷水で処理し、EtOAcで抽出した。有機層を分離し、塩水で洗浄し、無水MgSO4で乾燥し、蒸発させた。残渣(500mg)をシリカゲルクロマトグラフィーに付した。CH2Cl2:アセトン(95:5)による溶出で3α−ヒドロキシ−21−メチレン−5β−プレグナン−20−オン(150mg)を得た。更に同溶媒による溶出で無色固体物として3α−ヒドロキシ−21−メトキシメチル−5β−プレグナン−20−オン(120mg)を得た。
例49
a.3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン,21−ホスフェート二ナトリウム
エタノール(20ml)中3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン,21−ジベンジルホスフェート(712mg、1.07mmol)および活性炭担持パラジウム(5%Pd、180mg)を(Parr装置で)水素の50psi雰囲気下で2時間おいた。炭素担持パラジウムを濾去し、濾液を蒸発させて、油状物を得た。この油状物をメタノール:水4:1(10ml)に溶解した。1.00M NaOH(1.97ml)を滴下して、pHを11(±1)にした。少量の固体物を濾去し、濾液からほとんどの溶媒を蒸発により除去した。残留物(ほとんど水)を凍結乾燥により除去して、白色固体物(494mg、88%)を得た。mP224℃分解。1H NMRあり
b.3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン,21−ジベンジルホスフェート
3α,21−ジヒドロキシ−3β−トリフルオロメチル−21−ブロモ−5β−プレグナン−20−オン(1.20g、2.58mmol)、ジベンジルホスフェート(2.15g、7.74mmol)およびトリエチルアミン(1.08g、7.74mmol)をTHF(10ml)中で攪拌し、その後3時間加熱還流した。室温まで冷却した後、EtOAcおよび0.5M HClを加えた。有機相を分離し、水性NaHCO3およびNaClで洗浄し、MgSO4で乾燥し、真空下で蒸発させて、褐色油状物を得た。この油状物をフラッシュカラムクロマトグラフィー(ヘキサン/アセトン4:1)により精製して、透明油状物(712mg、42%)を得た。
c.3α−ヒドロキシ−3β−トリフルオロメチル−21−ブロモ−5β−プレグナン−20−オン
臭化水素酸(48%、2滴)をメタノール(100ml)中3α−ヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン(4.20g、10.9mmol)の攪拌溶液に加えた。次いでメタノール(60ml)中臭素(2.17g、13.6mmol)の溶液を10分間かけて滴下した。30分間攪拌した後、固体NaHCO3を(すべての臭化水素酸が中和されるまで)加え、溶媒を蒸発させた。残渣をCH2Cl2と水に分配した。有機相を分離し、塩水で洗浄し、MgSO4で乾燥し、真空下で蒸発させて、黄色半固体残渣(5.36g)を得、これを更に精製せずに用いた。
d.3α−ヒドロキシ−3β−トリフルオロ−5β−プレグナン−20−オン
(参考文献:Krishnamurti,R.,Bellew,D.R.,Surya,Prakash,G.K.,J.Org.Chem.,56,984(1991))
乾燥THF(3ml)中5β−プレグナン−3,20−ジオン,20−エチレンケタール(60mg、0.166mmol)の溶液にTHF中0.5M F3CSi(CH3)3(0.5ml、0.25mmol)を加えた。得られた無色溶液を0℃に冷却し、n−Bu4NFxH2O(少量の結晶)を加えた。冷却浴を取除き、混合液を室温まで加温した。5α−プレグナン−3,20−ジオン,20−エチレンケタールに関する同様の反応とは異なり、上記反応混合液は黄色にならず、ガス発生がなかった。更にTHF中0.5M F3CSi(CH3)3(0.5ml、0.25mmol)を加えた。得られた混合液を室温で数分間攪拌した。TLC(3:1ヘキサン:アセトン)ではRf1に近い新たなスポットを示したが、一部の未反応出発物質がまだ存在していた。したがって、更にTHF中0.5M F3CSi(CH3)3(0.5ml、0.25mmol)を加えた。混合液を再び室温でしばらく攪拌した。未反応出発物質はみられなかった。1N HCl(〜3ml)を加え、得られた2相混合液を室温で一夜攪拌した。トリフルオロメチル化の結果として形成されたスポットは完全に消失し、2つの新たな低極性側スポットが存在して、低い方が主生成物であった。次いで混合液をエーテルおよび水で希釈した。水層を分離し、エーテルで抽出した。合わせた有機層を飽和NaHCO3および塩水で洗浄し、(MgSO4)乾燥し、濾過し、減圧下で蒸発させて、白色結晶(泡状)固体物を得た。2種のエピマーをフラッシュクロマトグラフィーにより15:1ヘキサン:アセトンで分離した。初期分画の蒸発により副異性体を得た。更にカラムの溶出で標題化合物50mgを得た。
例50
a.3β−トリフルオロメチル−3α−ヒドロキシ−21−ブロモ−5β−19−ノルプレグナン−20−オン
メタノール(10ml)中3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノルプレグナン−20−オン(0.65g、1.75mmol)の溶液を含有したフラスコに、褐色を維持するような速度でこの色が続くまで、メタノール(10ml)中臭素の溶液(0.15ml)を滴下した。次いで水(50ml)を加え、混合液をCH2Cl2(3×40ml)で抽出した。合わせた抽出液をK2CO3で乾燥した。溶媒の除去により泡状白色固体物として生成物(0.805g)を得た。
b.3β−トリフルオロメチル−3α−ヒドロキシ−5β−19−ノルプレグナン−20−オン,21−ジベンジルホスフェート
THF(10ml)中3β−トリフルオロメチル−3α−ヒドロキシ−21−ブロモ−5β−19−ノルプレグナン−20−オン(0.805g)の溶液にジベンジルホスフェート(1.48g、5.32mmol)およびトリエチルアミン(538mg、5.32mmol)を加え、混合液を1.5時間加熱還流した。HCl(0.5N、40ml)を加え、それをCH2Cl2(3×40ml)で抽出した。合わせた抽出液をNa2SO4で乾燥した。溶媒の除去により粗製物質を得、これをクロマトグラフィーにより精製して、生成物733mg(65%)を得た。
c.3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノルプレグナン−20−オン,21−リン酸二ナトリウム
エタノール(20ml)中3β−トリフルオロメチル−3α−ヒドロキシ−5β−19−ノルプレグナン−20−オン,21−ジベンジルホスフェート(733mg)およびPd/C(5%、200mg)の混合液をParr水素付加機に入れた。水素付加分解を約50psi下で30分間以内に行い、得られた混合液を#5濾紙で濾過して、触媒を除去した。溶媒を除去して、白色泡状固体物(494mg)を得た。この固体物をメタノール(10ml)に溶解し、水(8ml)中NaHCO3(190mg)の溶液を0℃で加えた。次いでメタノールを真空下で除去し、得られた水溶液を凍結乾燥して、生成物(530mg、92%)を得た。
例51
3α−ヒドロキシ−21−(ピリジ−4−イルチオ)−5β−プレグナン−20−オン
乾燥THF30ml中21−ブロモ−3α−ヒドロキシ−5β−プレグナン−20−オン(2.8g、7.05mmol)および4−メルカプトピリジン(940mg、8.46mmol)の溶液に25℃でトリエチルアミン(1ml)を滴下し、混合液を60℃に加熱し、1.5時間攪拌した。次いで溶液を酢酸エチルと1:1飽和NaHCO3および水に分配した。次いで有機層を飽和NaClで洗浄し、MgSO4で乾燥し、真空下で濃縮して、粗製固体物3.19gを得た。5.5cmカラム中シリカゲル6inでフラッシュクロマトグラフィーにより、10〜15%アセトン:CH2Cl2で溶出させて50ml分画を集め、黄色固体物として標題化合物2.246g(75%)を得た。mp192−4℃
例52
a.3β−エチニル−3α−ヒドロキシ−5α−プレグナン−20−オン,20−ケタール
ガス導入口、温度計およびコンデンサーを装備した250ml3首フラスコにリチウムアセチリド−EDA錯体(2.75g、90%、27.5mmol)を入れた。乾燥ベンゼン(60ml)を加え、アセチレンガスを適度な速度で混合液に吹き込んだ。次いで混合液を油浴で50〜55℃に加熱し、5α−プレグナン−3,20−ジオン,20−ケタール(9g、25mmol)で少しずつ処理した。攪拌をこの温度で5時間、その後室温で更に17時間続けた。得られた懸濁液を10℃に冷却し、飽和NaCl溶液(5ml)で処理した。溶媒を除去し、水を残渣に加えた。非水溶性生成物を濾取し、水洗し、真空下で乾燥させた。次いでこの粗生成物をEtOAcから結晶化して、3β−エチニル−3β−ヒドロキシ−5α−プレグナン−20−オン,20−ケタール(3.35g)を得た。母液を蒸発乾固させ、残渣をシリカゲルカラムクロマトグラフィーにより精製した。トルエン:アセトン混合液(92:8)による溶出で、未反応出発ケトン(1.3g)、その後に第二分画として3β−エチニル−3α−ヒドロキシ−5α−プレグナン−20−オン,20−ケタール(1.3g)を得た。更に同溶媒混合物による溶出でより高極性の3α−エチニル−3β−ヒドロキシ−5α−プレグナン−20−オン,20−ケタール(270mg)を得た。
b.3β−エチニル−3α−ヒドロキシ−5α−プレグナン−20−オン
3β−エチニル−3α−ヒドロキシ−5α−プレグナン−20−オン,20−ケタール(550mg)をアセトン(20ml)および2N HCl(10ml)の混合液に溶解し、混合液を室温で15時間攪拌した。溶媒を除去し、残渣をCH2Cl2で抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥した後、溶液を濾過し、蒸発させて、粗生成物(414mg)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルカラムに注いだ。トルエン:アセトン混合液(92:8)による溶出で標題化合物(280mg)を得た。mp175−177℃
c.3α−エチニル−3β−ニトロオキシ−5α−プレグナン−20−オン
CHCl3(45ml)中3α−エチニル−3β−ヒドロキシ−5α−プレグナン−20−オン,20−ケタール(2.15g)の溶液を−20℃に冷却し、無水酢酸(20ml)で処理した。次いで発煙硝酸(4ml)を加え、混合液をこの温度で45分間攪拌した。−5℃まで加温した後、黄色溶液を2N NaOH(70ml)および水(150ml)の混合液に注いで、pH3〜4の溶液を得、その後これをCHCl3で抽出し、水、飽和NaHCO3溶液、塩水で洗浄し、(MgSO4)乾燥し、蒸発させて、粘稠物質として標題化合物(3g)を得、これを次の工程でそのまま用いた。
d.3β−エチニル−3α−ヒドロキシ−5α−プレグナン−20−オン
上記工程からの粗製3α−エチニル−3β−ニトロオキシ−5α−プレグナン−20−オン(3g)をTHFおよび水(30ml、1:1)の混合液に溶解した。AgNO3(516mg)を加えた。室温で15時間攪拌した後、溶媒を除去し、残渣をCH2Cl2で抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥した後、溶液を濾過し、蒸発させて、粗生成物(2g)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルカラムに注いだ。トルエン:アセトン混合液(93:7)による溶出で、第一分画として3β−エチニル−3α−ヒドロキシ−5α−プレグナン−20−オン(550mg)を得た。
例53
a.1,2−ジブロモエチレンからリチウム試薬の製造
N2ガスバブラー、温度計および滴下漏斗を装備した250ml3首フラスコに1,2−ジブロモエチレン(シス/トランス混合物、98%、Aldrich、2.1ml、26mmol、mw=186、d=2.246)を入れた。乾燥THF(40ml)を加え、溶液をドライアイス−アセトン浴で−78℃に冷却した。n−BuLi(THF中2.5M、20ml、50mmol)を35分間かけて滴下した。混合液をこの温度で40分間攪拌し、得られた試薬を次の工程に直ちに用いた。
b.3β−エチニル−3α−ヒドロキシ−5α−プレグナン−20−オン,20−ケタール
−78℃に維持されたTHF中の上記試薬溶液をTHF(50ml)中5α−プレグナン−3,20−ジオン,20−ケタール(4.68g、13mmol)の溶液で1時間にわたり滴下処理した。温度は添加中−70℃以下に維持した。攪拌をこの温度で15分間続けた(TLCによる検査では100%変換)。冷却浴を取除き、得られた溶液に2N HClを加えて反応停止させた(pH6〜7)。溶媒を除去し、残渣をクロロホルムで抽出し、有機層を分離し、水洗し、無水MgSO4で乾燥させた。溶媒の除去により、標題生成物(3.9g)を対応3β−ヒドロキシエピマーとのエピマー混合物(85:15)として得た。
例54
3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノルプレグナン−20−オン
THF(80ml)中3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノルプレグネ−17(20)−エン(2.6g、7.3mmol)の溶液に25℃でジボランTHF錯体(THF中1M溶液、22ml)を滴下した。反応は1時間で完了し、その後水酸化ナトリウムの溶液(20%、50ml)を0℃で非常にゆっくり加え、その後過酸化水素(30%、30ml)を加えた。次いで水を加え、THF層を分液漏斗により分離した。水層を塩化メチレン(2×40ml)で抽出した。有機溶液を炭酸カリウムで乾燥させ、溶媒を真空下で除去した。速いカラム(ヘキサン:アセトン=1:1)で生成物1.8gを得、これをPCC酸化に付した(PCC、2.1g、9.6mmol;酢酸ナトリウム、0.8g、9.6mmol)。純粋生成物(700mg、26%)をカラムクロマトグラフィーにより溶出液として酢酸エチルおよびヘキサン(15:85)を用いて精製した。mp151.5−153.0℃
例55
a.ベンジルフェニルスルホキシドの合成
CH2Cl225ml中ベンジルフェニルスルフイド(Aldrich、1.068g、5.33mmol)の溶液に−78℃でCH2Cl210ml中m−クロロ過安息香酸(Aldrich、50〜60%;60%のとき760mg、2.64mmol)の溶液をゆっくり加えた。室温まで加温して一夜攪拌した後、溶液を飽和NaHCO3溶液20mlに加えた。水層を分離し、CH2Cl2(2×10ml)で抽出した。次いでプールされた有機層を(MgSO4)乾燥し、濃縮した。残渣をフラッシュカラムクロマトグラフィー(シリカゲル、10%アセトン/ヘキサンおよび15%アセトン/ヘキサン)に付して、白色固体物としてスルホキシド(632mg、55%)を得た。mp123−126℃
b.20,20−エチレンジオキシ−3α−ヒドロキシ−3β−〔〔2−(フェニルスルフィニル)−2−フェニル〕エチル〕−5α−プレグナン
乾燥THF2ml中ジイソプロピルアミン(Aldrich、CaH2から蒸留したばかり;0.5ml、361mg、3.57mmol)の溶液を−10℃に冷却し、シリンジで滴下されるヘキサン中n−BuLiの1.6M溶液(Aldrich、1.0ml、1.6mmol)で処理した。10分間後に反応液を−75℃に冷却し、乾燥THF5ml中ベンジルフェニルスルホキシド(347mg、1.60mmol)の溶液をシリンジで30分間かけて滴下した。得られた深黄色溶液に固体20,20−エチレンジオキシ−3(R)−5α−プレグナン−3−スピロ−2′−オキシラン297mg(0.79mmol)を加えた。反応液を室温まで加温し、その後50℃に加温した。5時間後に反応液を室温まで冷却し、氷冷水30mlに加えた。得られた混合液をEtOAc(3×20ml)で抽出した。合わせたEtOAc層を飽和NaCl溶液で逆抽出し、(Na2SO4)乾燥し、濃縮した。残渣をフラッシュクロマトグラフィー(シリカゲル、100%CH2Cl2〜20%アセトン/CH2Cl2の勾配)により精製して、2種ジアステレオマーの混合物としてスルホキシド(405mg、86%)を得た。この混合物を脱離工程に用いた。
c.3α−ヒドロキシ−3β−〔2−(E)−フェニルエテニル〕−5α−プレグナン−20−オン
2,4,6−コリジン(CaH2から蒸留)0.2ml含有p−イソプロピルトルエン1.5ml中スルホキシド(200mg、0.339mmol)の懸濁液を油浴中135℃で60分間加熱した。室温まで冷却した後、溶液を一夜攪拌した。白色沈殿物が生成し、これを単離して、p−イソプロピルトルエン(3×1ml)で洗浄した。1H NMRおよびTLCによると、沈殿物(69mg、44%)は望ましい20,20−エチレンジオキシ−3α−ヒドロキシ−2(E)−フェニルエテニル−5α−プレグナンであった。アセトン中ケタールの溶液を0℃で1M HClで処理し、冷却下で1時間攪拌した。反応液をEtOAc/水混合液中に注ぎ、有機層を水および飽和NaCl溶液で洗浄した。(Na2SO4)乾燥した後、溶媒を真空下で除去し、残渣をフラッシュクロマトグラフィー(シリカゲル、1%アセトン/CH2Cl2で溶出)により精製した。
例56
3α−ヒドロキシ−3β−(2′−プロペニル)−5β−プレグナン−20−オン
乾燥THF(20ml)中5β−プレグナン−3,20−ジオン,20−ケタール(180mg、0.5mmol)の溶液を−70℃でアリルマグネシウムブロミド(THF中2M、1mmol、0.5ml)で処理した。混合液をこの温度で5分間、その後室温で1.5時間攪拌した後、2N HCl(1ml)で反応停止させた。溶媒を除去し、残渣をアセトン(15ml)に溶解した。Dowex-40樹脂(2g)を加えた後、溶液を室温で20分間攪拌した。樹脂を濾去し、溶媒を除去した。残渣をCH2Cl2で抽出した。有機層を水、希NaHCO3溶液、水および塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(250mg)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。トルエン:アセトン混合液(95:5)による溶出で、第一分画として3α−(2′−プロペニル)−3β−ヒドロキシ−5β−プレグナン−20−オン(45mg)を得た。更に同溶媒混合液による溶出で3β−(2′−プロペニル)−3α−ヒドロキシ−5β−プレグナン−20−オン(85mg)を得た。mp119−120℃
例57
a.3α−ヒドロキシ−17b−エチニル−5α−アンドロスタン
3α−tert−ブチルジメチルシリルオキシ−17b−エチニル−5α−アンドロスタン(100mg、0.24mmol)をアセトニトリル/THF(30ml、2:1v/v)に溶解し、0℃に冷却した。無水フッ化水素酸(48%、2ml、60mmol)を滴下し、反応溶液を室温まで加温した。2時間後に反応溶液を水中に注ぎ、CH2Cl2で抽出した。有機相を水性炭酸水素ナトリウムで洗浄し、MgSO4で乾燥し、真空下で蒸発させて白色固体物(69mg)を得た。この固体物をフラッシュカラムクロマトグラフィー(ヘキサン/アセトン12:1)により精製して、白色固体物(59mg、82%)を得た。mp158−160℃
b.3α−tert−ブチルジメチルシリルオキシ−17b−エチニル−5α−アンドロスタン
乾燥アルゴン雰囲気下でジイソプロピルアミン(125ml、0.95mmol、CaH2から蒸留)をTHF(2ml)に溶解し、溶液を0℃に冷却した。n−ブチルリチウム(ヘキサン中2.5M、380ml、0.95mmol)を滴下した。この温度で5分間攪拌した後、溶液を−78℃に冷却した。3α−tert−ブチルジメチルシリルオキシ−5α−プレグナン−20−オン,20−エノールジエチルホスフェート(164mg、0.288mmol)をTHF(1ml)に溶解し、この溶液を既に製造されたリチウムジイソプロピルアミドの溶液(まだ−78℃)に滴下した。反応混合液を室温まで2.5時間かけて加温した。水(1ml)を加え、その後混合液をエーテル75mlで希釈した。有機相を1M HCl、水および水性炭酸水素ナトリウムで洗浄し、MgSO4で乾燥し、真空下で蒸発させて半固体残渣(100mg、83%)を得、これを更に精製せずに用いた。
c.3α−tert−ブチルジメチルシリルオキシ−5α−プレグナン−20−オン,20−エノールジエチルホスフェート
乾燥アルゴン雰囲気下でリチウムビス(トリメチルシリル)アミド(THF中1.0M溶液、1.02ml、1.02mmol)をTHF(1ml)で希釈した。橙色溶液を−78℃に冷却した。3α−tert−ブチルジメチルシリルオキシ−5α−プレグナン−20−オン(400mg、0.92mmol)をTHF(1ml)に溶解し、既に製造されたリチウムビス(トリメチルシリル)アミドの溶液(まだ−78℃)に滴下した。この温度で45分間攪拌した後、ジエチルクロロホスフェート(140ml、0.97mmol)を滴下し、得られた溶液を室温まで加温した。水(1ml)、その後エーテルを加えた。有機相を1N HCl、水および水性炭酸水素ナトリウムで洗浄し、MgSO4で乾燥し、真空下で蒸発させて黄色油状物(394mg)を得、これをフラッシュカラムクロマトグラフィー(ヘキサン/アセトン10:1)により精製して、透明油状物(164mg、31%)を得た。
d.3α−tert−ブチルジメチルシリルオキシ−5α−プレグナン−20−オン
乾燥アルゴン雰囲気下で3α−ヒドロキシ−5α−プレグナン−20−オン(1.1g、3.5mmol)を無水DMF(17ml)に溶解した。イミダゾール(0.94g、13.8mmol)を加え、混合液をすべてが溶解するまで攪拌した。次いでtert−ブチルジメチルシリルクロリド(1.04g、6.91mmol)を加え、反応混合液を一夜攪拌した。エーテル(約50ml)を加え、有機相を1N HCl、水および水性炭酸水素ナトリウムで洗浄し、MgSO4で乾燥し、真空下で蒸発させて白色固体物(1.93g)を得た。この固体物をフラッシュカラムクロマトグラフィー(100%CH2Cl2)により精製して、白色固体物(1.16g、78%)を得た。
例58
3α,21−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン,21−メシレート
ピリジン(1ml)中3α,21−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン(77mg、0.22mmol)の溶液に0℃でメタンスルホニルクロリド29ml(42mg、0.37mmol)を加えた。冷却下で2時間40分攪拌した後、反応液を水中に注ぎ、得られた沈殿物を集めた。フラッシュクロマトグラフィー(20mm径カラム中シリカ6″、0〜5%EtOAc/ヘキサンの勾配で溶出)により白色固体物としてメシレート54mg(57%)を得た。
例59
21−ブロモ−3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン
メタノール125ml中3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン(2.5g、7.3mmol)の攪拌溶液に0℃で数滴のHBrを加え、その後メタノール25ml中臭素の溶液(1.28g、8.0mmol)をゆっくり滴下した。反応液を25℃にゆっくり加温し、完全脱色したら、溶液を少量のNaHCO3含有の氷水に加えた。沈殿物を濾過し、ジクロロメタンに溶解し、MgSO4で乾燥し、真空下で濃縮して、粗製泡状物(2.837g)を得た。5.5cmカラム中シリカゲル6inでフラッシュクロマトグラフィーにより、1%アセトン:CH2Cl2で溶出させて50ml分画を集め、泡状物として標題化合物1.96g(64%)を得た。
例60
3β−クロロエチニル−3α−ヒドロキシ−5β−プレグナン−20−オン
乾燥THF(7ml)中シス−1,2−ジクロロエチレン(194mg、0.16ml、2mmol)の溶液をN2下−10℃でn−BuLi(THF中2.5M、4mmol、1.4ml)で処理した。混合液を−30℃で20分間、その後−5℃で10分間攪拌した。それを−30℃に再冷却し、乾燥THF(10ml)中5β−プレグナン−3,20−ジオン,20−ケタール(360mg、1mmol)の溶液を10分間かけて滴下した。冷却浴を取除き、混合液を室温で0.5時間攪拌した。NH4Cl溶液(3ml)を加えて、反応を停止させた。溶媒を除去し、その後残渣をアセトン(25ml)に溶解した。2N HCl(10ml)を加えた後、溶液を室温で1時間攪拌した。飽和NaHCO3溶液を加えて、酸を中和させた。溶媒を除去し、残渣をCH2Cl2で抽出した。有機層を水、その後塩水で洗浄した。無水MgSO4で乾燥させた後、溶液を濾過し、蒸発させて、粗生成物(427mg)を得た。次いでこの粗生成物を少量のCH2Cl2に溶解し、シリカゲルのカラムに注いだ。トルエン:アセトン混合液(95:5)による溶出で無色固体物として標題化合物(130mg)を得た。
例61
3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン
無水1,2−ジメトキシエタン(DME)(5ml)中3(R)−5α−プレグナン−3−スピロ−2′−オキシラン−20−オン(101mg、0.305mmol)およびNaI(115mg、0.767mmol)の溶液(淡黄色)に室温でn−Bu3SnH(0.22ml、0.238g、0.818mmol)を加えた。反応溶液は無色になった。次いでアゾビスイソブチルニトリル(AIBN)(10mg、0.061mmol)を加えた。得られた溶液を窒素雰囲気下で21時間還流したところ、TLC(3:1ヘキサン:アセトン)では反応の完了を示した。反応をメタノールで停止させ、混合液を室温でしばらく攪拌した。溶媒を真空下で除去して油状物を得たが、これはどちらにも溶解しなかった。CH2Cl2の添加で溶液を得、これを水、1N HClおよび飽和NaHCO3で洗浄した。有機層を(MgSO4)乾燥し、濾過し、減圧下で濃縮して、白色固体物を得た。勾配フラッシュクロマトグラフィー(ヘキサン、7:1ヘキサン/アセトン、5:1ヘキサン/アセトン)による精製で標題化合物(93mg、92%)を得た。
上記化合物が個別のジアステレオマーに分離できるジアステレオマーの混合物として存在しているかもしれないことは、当業者にとり明らかであろう。ジアステレオマーの分割は、好ましくはガスまたは液体クロマトグラフィーあるいは天然源からの単離により行える。他で指摘されないかぎり、前記のような本発明による化合物に関する明細書およびクレームでの言及は、分離されていようとまたはその混合物であろうと、すべての異性体を含めた意味である。
異性体を分離してみると、望ましい薬理活性はジアステレオマーの1つで優れていることが多い。ここで開示されるように、これら化合物の生物活性は高度の立体特異性を示す。
TBPS結合アッセイをGee,K.W.,Lawrence,L.J.and Yamamura,H.I.,"Modulation of the Chloride Ionophore by Benzodiazepine Receptor Ligands Influence of Gamma-Aminobutyric Acid and Ligand Efficacy",Molecular Pharmacology,30:218,1986で記載された操作に従い行った。雄性Sprague-Dawleyラット(200〜250g)を二酸化炭素で麻酔し、断頭した。ラットの脳を取出し、大脳皮質を切り刻んで、10倍容量の0.32Mスクロース中でホモゲナイズした。組織を1000×gで10分間遠心し、得られた上澄を9000gで20分間遠心した。得られたペレットを10倍容量の200mMNaCl/50mMリン酸Na−K pH7.4緩衝液(結合用緩衝液)に再懸濁し、その後9000×gで10分間遠心した。この洗浄操作を2回繰返し、最終P2ペレットを結合用緩衝液に再懸濁した。結合アッセイでは、〔35S〕TBPS(2nM)をP2膜、5μM GABAおよび、1)全結合量を測定するために無添加;2)非特異的結合を測定するために2μM未標識TPBS;3)濃度1nM〜10μM範囲内の神経ステロイドと共に、結合用緩衝液中で同時インキュベートした。22℃で90分間のインキュベート後に、懸濁液を濾過し、結合放射能を液体シンチレーションカウントにより調べた。TBPS結合の50%阻害を示す神経ステロイドの濃度であるIC50を、MicrosoftのExcel Solverで非線形曲線フィッティングルーチンを用いて計算した。低いIC50ほど、GABAAレセプター−塩化物チャンネル複合体のステロイド部位と高い親和性を示す。表1は本発明で用いられた代表的ステロイド類のIC50値を示している。
本発明の医薬組成物は、被治療体、動物またはヒトで望ましい薬学的活性を示すために十分な無毒性量で、本発明による活性化合物またはこのような化合物の混合物を無毒性製薬キャリアと配合することにより、慣用的な単位剤形で製造される。好ましくは、組成物は活性成分約5〜約250mg/投薬単位から選択される活性だが無毒性の量で活性成分を含有している。本発明による組成物および方法は不眠症を防止して、睡眠を生じさせる。
用いられる製薬キャリアには、例えば固体、液体または経時放出物質がある(例えば、Remington's Pharmaceutical Sciences,14th Edition,1970参照)。代表的固体キャリアはラクトース、白土、スクロース、タルク、ゼラチン、寒天、ペクチン、アラビアガム、ステアリン酸マグネシウム、ステアリン酸、微結晶セルロース、ポリマーヒドロゲルなどである。典型的な液体キャリアはプロピレングリコール、β−シクロデキストリンの水溶液、シロップ、ピーナツ油、オリーブ油および類似エマルジョンである。同様に、キャリアまたは希釈物にはグリセロールモノステアレートまたはグリセロールジステアレート単独、あるいはワックス、マイクロカプセル、微小球、リポソームまたはヒドロゲルと一緒にしたような、当業界に周知の遅延物質がある。
様々な薬剤形態が使用できる。このため、固体キャリアを用いるとき、製剤はプレインミル化され、微粉砕化され、油中にあるか、打錠され、微粉砕粉末またはペレット形で硬ゼラチンまたは腸溶性カプセル中に入れられ、あるいはトローチ、ロゼンジまたは坐剤の形をとる。本発明による化合物が直腸投与用坐剤の形で投与されるとき、化合物はカカオ脂およびポリエチレングリコール、または室温で固体だが直腸温度で液体の他の適切な無刺激物質のような物質と混合される。液体キャリアを用いるとき、製剤はアンプルのような液体、あるいは水性または非水性液体懸濁液の形をとる。液体剤形は薬学上許容される保存剤なども通常含有する。加えて、非経口投与、鼻スプレー、舌下および経口腔投与と、経時的放出皮膚パッチも本発明による化合物の局所投与用に適した薬剤形態である。
好ましい処方は経口投与である。本発明者らは、水に不溶性であり、したがって経口投与後にほとんど吸収されない傾向がある神経活性ステロイドの活性を特定の処方が有意に変えうることを発見した。例えば、プレグナノロンは経口で不活性であると考えられた。L.Gyermek,"Pregnanolone:a Highly Potent,Naturally Occurring Hypnotic-Anesthetic Agent",Proc.Soc.Exp.Biol.Med.,125:1058-1062,1967
本発明者らは、体が経口投与後に睡眠を誘導するために十分な量で胃腸管から吸収できるプレグナノロンの様々な処方を発見した。薬物のバイオアベイラビリティーは投与前および後に血清血液レベルを測定することにより定量される。この定量方法は、その全体を引用することにより本明細書の開示の一部とする、Robert H.Purdy et al.,"Radioimmunoassay of 3α-hydroxy-5α-pregnan-20-one in rat and human plasma",Steroids,55:290-296(1990)で記載されている。通常、血液サンプルが採取された後、血清が分離され、その後凍結される。血清は内在ステロイドを含めた競合物質を除去するためクロマトグラフィーにかけられる。プレグナノロンは、それが8cm Radial-Pak-5-μmシリカカラム(Waters Associates)でHPLC技術を用いて3ml/minの流速でジクロロメタン中0.2%エタノール(95%)の溶媒系を用いてクロマトグラフィーにかけられたとき、19.0分間の保持時間を有する。次いでラジオイムノアッセイがクロマトグラフィーにかけた物質の血清血液レベルを定量するためにランされる。
プレグナノロンがある処方で経口投与されたときヒトで睡眠を誘導する上で経口活性であることを証明するため、本発明者らは18〜35才の健常男性ボランティア8例を用いて試験を行った。研究は、治験者が薬物投与前および直後に隔離される、非盲検単一用量クロスオーバー研究であった。
ベースライン病歴、実験価値、薬物およびアルコールスクリーンを各治験者で行った。研究中、治験者は1日目から薬物投与後48時間日まで医療施設に留めた。病院では、カフェイン、アルコールまたは他の薬物の摂取を治験者に禁止した。
一夜絶食後、規定高脂肪朝食を各治験者に与え、15分以内で食べ終えさせた。0900GMTに始め、治験者にプロトコールで規定されたランダム化に従いプレグナノロンを投与した。プレグナノロン(500mg、顆粒粉末)を4処方のうち1つを経口投与した。処方1はプレグナノロン(微粉砕、AKZO,Arnheim,The Netherlandsから入手)(5%)、ポリビニルポビドン(ポリビニルピロリドンまたはPDP)(Luviskol K-30,BASF,Germany製)(94%)およびラウリル硫酸ナトリウム(SLS)(Nomeco,Copenhagen,Denmark製)(1%)を有していた。処方2はプレグナノロン(12.3%)およびβ−シクロデキストリン(シクロデキストリン1モル当たりH2O7モル、Sigma,St.Louis製)(87.7%)を含有していた。処方3はプレグナノロン(99%、ナノサイズ化)およびラウリル硫酸ナトリウム(1%)を含有していた。処方4はプレグナノロン(99%)およびラウリル硫酸ナトリウム(1%)を含有していた。研究で用いたプレグナノロンは、ナノサイズ化(サブミクロン粒度)した処方3を除き、すべての場合で均一に微粉砕化した(AKZOから入手)。薬物投与直後の1時間以外は、随意に水を与えた。食事は研究中適時にとるよう標準化させた。
処方1はすべての成分をエタノールに溶解することにより作った。混合液を攪拌し、ほとんどのエタノールが蒸発するまで加熱した。生成物を真空下で24時間かけて完全に乾燥させ、乳鉢ですりつぶした。
処方2は水50ml中プレグナノロン1.23g、シクロデキストリン6.77gを37℃で数時間振盪することにより作った。混合液を濾過し、真空下で24時間乾燥させ、乳鉢ですりつぶした。
処方3は高温および高圧(700bar以内)で二酸化炭素に諸成分を溶解させることにより作った。圧力を放出したところ、粒子が沈殿し、少量のサブミクロン粒子を生じた。
処方4は、微粉砕プレグナノロンをラウリル硫酸ナトリウムと乾燥混合し、その後篩にかけることにより作った。
プレグナノロンレベル分析用の血清サンプルを薬物投与前および直後に得て、24時間続けた。血清を分離し、凍結し、ラジオイムノアッセイで分析するまで貯蔵した。
研究中に得たサンプルはラジオイムノアッセイを用いてプレグナノロンに関し分析し、結果は適切な非区画薬物動態パラメーターを用いて分析した。相対的バイオアベイラビリティー評価はプレグナノロンの観察された血清濃度に基づき処方間で行った。
薬物投与に対する臨床応答の評価は、医療施設スタッフおよび訓練された観察者により行われた観察を通して得た。睡眠の開始および見掛け深さに関する観察は4時間の処置期間中記録し、その後血清プレグナノロン濃度と相関させた。睡眠の継続時間は、治験者が生命徴候および血液サンプルを得るプロセスでしばしば妨害されたため、調べなかった。
各個人は、3週間の試験中、薬物がなくなり排出された日(drug-free wash-out)により分けられた4種の処方を受けた。重度または予期せぬ有害作用はいずれの処置群でもみられなかった。体温は薬物投与前と薬物投与後0.5、1.0、1.5、2.5、3、4、8および12時間目に詳しくモニターおよび記録した。体温の低下はどの処方の研究でいずれの治験者でも観察されなかった。
約1時間後に、眠気および/または睡眠の徴候が観察された。β−シクロデキストリン中プレグナノロン(処方2)を受けたボランティアでは、8例中7例の治験者が薬物投与後<1時間から3時間までの範囲内で眠り始めることが観察された。睡眠の継続時間は、治験者が血液採取および生命徴候のため30分間毎に目覚めたため、調べることが不可能であった。主観的評価では軽い〜目覚め困難の範囲内で睡眠の深さを示した。結果は図16A〜16Hでまとめられ、そこでは睡眠の観察が記録された時点を矢印が示している。1例の治験者だけはβ−シクロデキストリン中で処方されたプレグナノロンを受けた後の研究期間中目覚めたままであることが観察された。図16A〜16Hはこの処方に関する血漿血液レベルも示している。ボランティア8例のピーク血漿レベルは約40〜120ng/mlの範囲であった。表2はこの処方の平均ピーク濃度が61.7ng/mlであったことを示す。
微粉砕プレグナノロンと1%ラウリル硫酸ナトリウムからなる処方4は、約40〜110ng/ml範囲のピーク血漿レベルを示した。表2は微粉砕処方の平均ピーク濃度が59.4ng/mlであったことを示す。
処方3は10〜35ng/ml範囲のピーク血漿レベルを示した。表2はナノサイズ化処方の平均ピーク濃度が22.0ng/mlであったことを示す。
相対的吸収またはバイオアベイラビリティーの評価は、ナノサイズ化処方を除くすべてが吸収の速度および程度の双方に関して同等の吸収性を有することを証明している。血清サンプルを各治験者について48時間の間隔にわたり分析した。図19は4種の異なる処方について観察された平均血清レベルを示している。3種の処方、PVP、βCDおよび微粉砕粉末+SLSはすべて同等の平均血清濃度−時間プロフィルを有していたが、ナノサイズ化プレグナノロン処方はかなり低いレベルを有して、そのピークは他の約35%であった。平均血清レベルを調べることに加えて、吸収のピークおよび程度の相対評価は、ピーク濃度と血清濃度−時間(AUC)プロフィル下の面積を調べることにより行える。表1はそれらの結果について示す。
図15A〜H、16A〜H、17A〜Hおよび18A〜Hは各治験者に関する血清濃度vs.時間のプロットである。鎮静効果が観察された各時点で、矢印がちょうどその箇所に示されている。観察されるように、血清濃度が高いほど、通常30ng/ml以上で、鎮静効果を観察できる。
もう1つのヒト研究では、血漿濃度および睡眠傾向を1回分のプレグナノロンの経口投与後2時間にわたり若い健康な男性ボランティア18例で測定した。2種の異なる処方を試験した。処方Iでは賦形剤としてβ−シクロデキストリンを用いた。処方IIでは賦形剤としてラウリル硫酸ナトリウムを用いた。β−シクロデキストリンおよびラウリル硫酸ナトリウム双方は公知の製薬賦形剤であり、化合物の毒性プロフィルに影響せずにプレグナノロンの吸収を高めるために選択された。
処方Iは1000mlエルレンマイヤーフラスコ内でプレグナノロン15.0gおよびβ−シクロデキストリン82.57gを蒸留水610mlに懸濁することにより製造されたβ−シクロデキストリン包接複合体であった。懸濁液を磁気棒スターラーにより37℃で36時間攪拌した。次いで懸濁液を0.22μmフィルター(Millipore、タイプGV)で濾過して、そこに複合体を集めた。得られた複合体を乾燥カップボード中50℃で12時間乾燥させ、その後塊を磁器乳鉢で粉砕し、顆粒を300μm篩に通した。顆粒を真空下P2O5により50℃で24時間乾燥させた。この処方物をプレグナノロン150mg含有の薬包にパッケージ化した。
処方IIは、125μm篩に通したプレグナノロン49.50gとラウリル硫酸ナトリウム0.50gを乳鉢および乳棒で30分間混合することにより製造された、プレグナノロンおよびラウリル硫酸ナトリウムの99:1物理的混合物であった。次いで混合物を300μm篩に通した。この処方物を250mgプレグナノロンカプセルおよび500mgプレグナノロン薬包としてパッケージ化した。
候補治験者は、病歴およびベースライン臨床実験価値を決めるために初回投薬の21日前に調べた。治験者には研究前または中にどんな処方薬または非処方薬をとっているか話さなかった。各治験者は2週間以内に2回参加した。連続した1回分処置は1週間隔で離した。
試験治験者は一夜絶食し、投薬の3時間前に研究センターに出向いた。軽食を投薬の2時間前に与えた。標準FDA承認高脂肪朝食を投薬の20分前に治験者に与え、投薬の5分前に終わらせた。朝食後、治験者に液体ヨーグルトで2つのうち一方の処方を与えた。薬包の内容物を液体ヨーグルト100mlに加え、20〜30秒間攪拌した。次いで治験者はそのヨーグルトを飲んだ。更に30mlのヨーグルトをカップ内の残りと混合して、全部の投与を確実に行わせた。これを飲んだ後、カップの側面および底をかきとり、治験者に与えた。液体は投薬後最初の1時間飲ませなかった。治験者は摂取後最初の1時間座るかまたは立ったままでいなければならなかった。
平均血漿濃度は、β−シクロデキストリン処方の場合、投薬後1時間で82.08±57.18ng/mlのピーク値に達した。平均血漿濃度は1.5時間で79.33±49.71ng/ml、2時間で74.21±37.21ng/mlであった。
ラウリル硫酸ナトリウム処方ではもっと後にそのピーク平均血漿濃度に達した。平均値は投薬後1.5時間で64.06±54.91ng/ml、2時間で66.63±49.60ng/mlであった。これらの統計は図20で示されている。
治験者はEEG、EOGおよびEMGでモニターしたが、これらは暗室で目を閉じて10分間記録した。記録は反応時間条件下で5分間行った。治験者は、彼等の左または右親指でボタンを押すことにより、ランダムな順序および間隔で与えられる2つの異なる調子のうち1つにできるだけ速く応答するように教えられていた。治験者を中断せずに5分間記録した。
図21で示されるように、覚醒時間は投薬後減少した。睡眠試行回数も調べた。睡眠試行とは、5分記録期間内で覚醒から何らかの睡眠段階への移行として規定した。各研究日に、治験者は5回のポリグラフ記録期間があった。したがって睡眠試行の最大回数は0〜5と様々である。5は睡眠が5分間投薬前記録期間中であっても観察されたことを示す。睡眠試行の回数を図22Aで最大血漿濃度(Cmax)に対してプロットした。低血漿濃度は通常少ない睡眠試行と符号し、高血漿濃度は睡眠試行の多い回数と符号した。図22Bは異なる回数の睡眠試行毎に3平均血漿濃度レベルを示す。
これらヒト研究からのデータは、(1)プレグナノロンが経口投与で適切な血中レベルを示すように処方でき、および(2)ボランティアでみられるプレグナノロンの血中レベルが眠気および/または睡眠の観察と相関していたことを示している。
本発明で用いられた化合物の薬理性質を明らかにするために、本発明者らは脳EEG、運動活動および体温に関するプレグナノロン、3α−ヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−ホスフェート二ナトリウム塩、3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン、3α−ヒドロキシ−3β−メチル−5α−プレグナン−20−オン、3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン21−ヘミサクシネートナトリウム塩、3α−ヒドロキシ−3β−エチニル−5α−プレグナン−20−オン、3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン21−ヘミサクシネート、3α−ヒドロキシ−3β−トリフルオロメチル−19−ノル−5β−プレグナン−20−オン、3α−ヒドロキシ−21(ピリジ−4−イルチオ)−5β−プレグナン−20−オン、3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノルプレグナン−20−オン 21−ホスフェート二ナトリウム塩の効果が測定されるインビボ実験をラットで行った。ラットはヒト睡眠の適切なモデルであるが、その理由はヒトで催眠薬であるすべての化合物がラットで催眠効果を有し、逆もまた同じだからである。Edgar,D.M.,"Drug Discovery and Evaluation Using the Advanced Technology Sleep-Wake Bioassay Laboratory at Atandard University",p.4,December 1992。加えて、ヒト睡眠用のラットモデルはNIHでピア−レビュー(peer-review)委員会により承認されていた。同上、p5。更に、ラット睡眠およびヒト睡眠は基本的に同じである。同上、p4。例えば、NREM vs.REM睡眠に費やされる時間の割合はラットおよびヒト双方で約4:1である。同上、p5。
ラットの準備、モニタリングおよびデータの分析は下記のように行った。成熟雄性Wistarラット(外科処置時275〜350g、Charles River Laboratories)を麻酔し(Nembutal、60mg/kg)、断続的EEGおよびEMG記録を行える頭蓋インプラントで外科的に処置した。体温および運動活動は腹部に外科的に設置された小型発信機(Minimitter)でモニターした。頭蓋インプラントはEEG記録用のステンレススチールネジ〔2個の前頭部(+3.9AP型ブレグマ、±2.0ML)および2個の後頭部(−6.4AP、±5.5ML)〕からなっていた。2本のテフロンコートスチールワイヤをEMG記録用に項部台形筋肉下に置いた。すべてのリード線を外科処置前に小型連結器にはんだ付けし、Glutarex(3M Co.)で化学的に滅菌した。インプラントアセンブリーを歯科用アクリルで頭蓋骨に固定した。最低3週間を回復に見込んだ。
各ラットを注文設計ステンレススチールキャビネットの分かれた換気区画内に位置するそれ自体の個別記録ケージで永続的に飼育した。個別記録ケージである各Nalgeneマイクロアイソレーターケージは、フィルター−トップ・ライザー(filter-top riser)および回転整流器で上げた。食物および水は随意に入手できた。24時間明暗サイクル(12時間オン、12時間オフ)をケージから5cmの4ワット蛍光灯を用いて研究中維持した。動物を処置前後双方の3日間平静にした。実際には、動物を週末中平静にした。処置を月曜朝または火曜朝に行い、動物を再び金曜まで平静にし、それからケージをきれいにして、データをコンピューターからコピーした。
コンピューターは同時にラット48匹から遠隔測定でEEG、EMG、体温および非特異的運動活動(LMA)と飲水活動についてモニターした。コンピューターは挙動依存性前後関係ルールを含めたパターンマッチアルゴリズムを用いて10秒毎に覚醒状態を分類した。飲水およびLMAは10秒毎に記録し、体温は1分毎に記録した。
夜行動物としてラットは明かりが消えているとき最も活発であり、ヒトのように、活動優勢期間の最後に催眠効果を最も受けやすい。したがって、処置時間は重要である。ラット活動優勢期間のピークであるCT−18で薬物を投与すれば、(3:00時計時間または日周期時間CT−0)に照明下で眠るラットの自然な性質は薬物の催眠活性を遮蔽できない。REM睡眠の傾向が最大であるCT−5で薬物を投与すれば、REM睡眠に対する薬物効果はより正確に測定できる。活動優勢期間は、明かりが図でCT−12または15:00時計時間に消されたときに始まる。
化合物は、ステロイドの物理的性質に応じて、無菌0.25%メチルセルロース、20〜50%ヒドロキシプロピルシクロデキストリンまたは水に懸濁した。これらのビヒクルは現在まで試験されたすべての催眠薬に向いたバイオアベイラビリティーを示す。薬物およびコントロールは、CT−18研究では暗赤色照明下でまたはCT−5研究では標準光下で、各々1mg/kgまたは10mg/kgの容量で腹腔内注射または経口投与した。
対抗コントロール群N=20は60匹の利用できるコントロール候補のプールから選択した。これらの対抗コントロール群は下記のように作った。可変要素(NREM、REM、LMA、体温および睡眠期間)の各々について、時間平均値の曲線を注射前24時間にわたり候補毎に調べた。曲線が処置群の平均曲線と最も良く(最小二乗法で)合う候補20匹を選択した。したがって、対抗ペア操作は用いなかったが、かなり匹敵する処置前ベースラインデータを有した並行処置群を用いた。
図1A、1Bおよび1Cは、CT−5またはCT−18で投与されたプレグナノロンが、それが投与された時間とは無関係に、NREM睡眠を促進する上で有効であることを照明している。双方の時間で、速やかな開始と高い効力がある。CT−5の方がラットは横になっていて、睡眠を誘導することが困難である。これらの結果は、シフト作業およびジェットラグで生じるような、睡眠が通常より早く誘導されねばならないときに、本発明による神経活性ステロイドが望ましい催眠剤であることを示している。更に、催眠薬を処方する医者は患者の睡眠負荷を確信できないため、患者の睡眠負荷とは無関係に予測できる催眠作用のある本ステロイドのような催眠薬を有することが有利である。
図1A〜Cはプレグナノロンがリバウンド作用を欠くことも示している。リバウンド作用は、処置の催眠作用がコントロールレベルに戻った後におけるNREM睡眠の減少として規定される。プレグナノロンはCT−5またはCT−18だとどの用量でもNREM睡眠のリバウンド減少を生じなかった。図24Aで示されるように、3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オンもCT−18でNREM睡眠のリバウンド減少を生じなかった。TriazolamはCT−18で投与されたときNREM睡眠のリバウンド減少を示した。図6Bは右手側で星印(“*”)を示しているが、これはNREM睡眠で統計上有意な減少を示す。
プレグナノロンは、CT−5およびCT−18双方で有効であり、リバウンド覚醒を生じないという点で、催眠剤の中では独特である。我々の研究ではZolpidemがリバウンド不眠症を生じなかったことを示すが、図8Aで示されるようにCT−5でNREM睡眠を有効に誘導することもできなかった。この特徴は、リバウンド不眠症を生じないプレグナノロンまたは他の神経活性ステロイドが最も重要な制限である耐性、禁断症状およびリバウンド不眠症を現行BZレセプターリガンドと比較して臨床的に少なく生じることを、おそらく示唆している。
図2A、2B、2Cおよび25は、プレグナノロンの投与および3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オンの投与が非断続的NREM睡眠の期間または“睡眠期間長さ”に強い増加を起こすことを証明している。睡眠期間長さは夜間周期的に目覚めるヒト傾向とパラレルであることから興味あり、それはヒトで睡眠の回復価値を決める上で重要なファクターであることが示された。睡眠のこの強い関連性は、年齢関連不眠症の治療にとり大きな可能性を有している。
図3A、3Bおよび3Cは、プレグナノロンの筋肉内投与がヒトで発熱作用を起こすことを見つけたAttallah Kappasらの開示とは対照的に、プレグナノロンが体温に影響しない傾向を有することを示している。"Fever-Producing Steroids of Endogenous Origin in Man",105:68/701-68/708,A.M.A.Archives of Internal Medicine,1960。注射前に何時間も、体温は近シヌソイドリズムで変動することに注意せよ。取扱いおよび注射で体温に短時間の増加を起こす。図26Aも、3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オンが体温に最小の作用しか有しないことを示している。TriazolamおよびZolpidemは図6Eおよび8Dで各々示されるように体温に影響を与える。体温作用の欠如は、プレグナノロンおよび本発明による神経活性ステロイドのNREM誘導メカニズムが恒常性コントロール機能に望ましくない副作用を示さないことを示唆している。
プレグナノロンは、図4A〜C、6Fおよび8Eで示されるように、TriazolamおよびZolpidemと比較して、30mg/kgのとき経時的に運動活動(LMA)の適度に小さな減少を生じ、10mg/kgのときには減少を生じない。3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オンも、図26Bで示されるように、経時的にLMAで適度に小さな減少を生じた。TriazolamおよびZolpidemは双方とも、図12の棒グラフで示されるように、CT−5で投与されたときビヒクルコントロールレベル以下にLMAを減少させたが、それにもかかわらずTriazolamはCT−5で催眠効果を有しなかった。図14Aおよび14Bは、ラットが眠っているプレグナノロンおよびZolpidemの有効期間中に、それらが動いていないことを証明している。しかしながら、Zolpidemは催眠作用が消失した後であってもLMAを抑制し続ける。運動協調不能は比較的減少するようであり、それは年長者で用いられる催眠薬にとり貴重な特色である。
図5A〜5Cおよび24Bは、プレグナノロンおよび3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オンが各々、確かにREM睡眠を減少させるベンゾジアゼピン類のような他の催眠薬と比較して、REM睡眠に対する阻害作用を欠くことを示している。図6Fおよび8Fは、CT−18で投与されたTriazolamおよびZolpidemが各々投与後最初の数時間にREM睡眠を減少させたことを示している。例えば、図13の棒グラフは、Zolpidemがプレグナノロンよりも有意に少なくNREM睡眠を誘導した用量でCT−5に投与したときREN睡眠を妨げることを証明している。
プレグナノロン、3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オンおよび本発明による他の神経活性ステロイドは、それらが有効なNREM促進効果とわずかなREM妨害性とを合せ持っているという点で独特である。アルコールおよびバルビツレート類のようなヒトにおけるREM睡眠の強い阻害は、薬物効果が消失してREMリバウンドが起こるような睡眠破壊と関連していた。REM睡眠の生物学的機能は未知であるが、本発明による神経活性ステロイドがREMをわずかにしか妨げず、強く睡眠を誘導するという発見は、好ましい属性である。
プレグナノロンはNREM睡眠中に高振動数、高振幅EEGパターンを示した。このEEGパターンは図7A〜Bで再現されている。逆に、深い正常NREM睡眠は通常高振動数、高振幅EEG波を有する。EEGスペクトルプロフィルに関するプレグナノロン効果はCT−5およびCT−18処置双方の後で非常に類似していた。このEEGパターンは図7C〜Dで示されるように他の催眠薬とは異なる。
図10Aで示されるように、プレグナノロンはNREM EEGデルタ活性(力)に特徴的な効果を有していた。NREM睡眠の“深さ”は0.1〜4.0Hzの低(デルタ)振動数で生じるEEG活性の割合として定量される。NREM睡眠であると決められたEEG活性の各10秒間毎に、EEGデルタ力を測定し、20分間隔で平均化し、その後ビヒクルコントロールと比較した。図10Aは、プレグナノロンが投与後少くとも5時間にわたり低デルタ範囲でデルタ力のパーセンテージを減少させたことを示している。逆に、図10Cおよび10Bは、TriazolamおよびZolpidemが各々処置後少くとも1時間にわたりデルタ範囲でEEG力のパーセンテージを増加させたことを示している。
プレグナノロンはピーク薬物効果期間中にEEGで振動数の特徴的分布を生じることもわかった。薬物の“EEGスペクトルプロフィル”を調べる目的は、振動数が処置後かなり増加または減少したかを調べるためである。図11Aは、プレグナノロンが低い振動数(0.1〜8Hz)から高い振動数(≧10Hz)に力をシフトさせたことを示している。逆に、ZolpidemおよびTriazolamは低いデルタ振動数範囲でEEG力のパーセンテージを増加させた。各々、図11Bおよび11C参照。
プレグナノロンの場合に用いたのと同様の実験法を用いて、本発明の様々な構造的修正をうけた9種の他の神経活性ステロイドをラットで試験した。9種すべての合成神経活性ステロイドはプレグナノロンと類似したプロフィルを生じた。REM、NREM、リバウンド不眠症、運動活動、体温および力スペクトルに対する効果はプレグナノロンおよび3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オンについて上記された場合と類似していた。これらの実験におけるREM、リバウンド不眠症およびNREM睡眠の変化は、各々図23A〜Cでまとめられている。プレグナノロンのように、9種すべてのステロイドはわずかなREM妨害を示しただけにすぎず、強いNREM促進効果を生じた(図23A&C)。これらのステロイドはリバウンド作用をほとんどまたは全く生じなかったことにも注意せよ。図23B参照。
要約すると、プレグナノロンと9種の他の合成神経活性ステロイドはNREM睡眠を強く増加させたが、温度降下をおこさず、REM睡眠を少し妨げるだけであり、催眠作用が消失した後運動活動を有意には減少させず、リバウンド覚醒も続かなかった。逆に、ZolpidemおよびTriazolamは催眠作用が消失した後に体温および運動活動を減少させた。ZolpidemおよびDexmedetomidine(眠気を起こすことが知られた薬物のクラスに属する抗ヒスタミン剤)はREM睡眠を強く妨げた。Dexmedetomidineは強いリバウンド作用を生じた。一緒にすると、これらの結果は、本発明による神経活性ステロイドがBZレセプターリガンドよりも特異的な催眠作用様式を有し、それにより他の重要な機能(即ち、体温と、おそらく運動協調性または動機)の障害を少くすることを示している。逆に、ベンゾジアゼピン類は催眠作用が消失した後にLMAを損ない、これは睡眠誘導薬物療法で望ましくない特徴である。
本発明により睡眠を誘導する方法は、睡眠を生じさせるために十分な無毒性量で、製薬キャリアと共に前記のように組成物として通常製造された本発明による化合物を、誘導睡眠の必要性のある被治療体に投与することからなる。正確な用量は活性化合物5〜250mgである。
好ましい態様が記載および説明されてきたが、様々な置換および修正を本発明の範囲から逸脱せずにそれに加えることができる。したがって、本発明が制限ではなく説明のために記載されていると理解するべきである。
Claims (6)
- 3βで置換された、下記構造式IIを有する3α−ヒドロキシプレグナン−20−オンもしくは19−ノルプレグナン−20−オン、またはそのナトリウム塩:
〔上記式中:
Rは、単一のアリール基、ヒドロキシ基、ハロ基もしくはケト基で置換されていてもよい低級アルキニル基、または低級トリフルオロアルキル基であり、
R1は水素または低級アルキル基であり、
R2aは水素、ヒドロキシ、ピリド−4−イルチオ基、ヘミサクシノイルオキシ基またはホスホリルオキシ基であり、但し、
(1)R1が水素であるとき、Rはトリフルオロ(低級)アルキル基であり、
(2)R2aが水素またはピリジ−4−イルチオ基以外であるとき、Rはトリフルオロ(低級)アルキル基である〕。 - Rが、トリフルオロメチルおよび2’,2’,2’−トリフルオロエチルからなる群より選択される、請求項1に記載の化合物。
- R2aが、ヘミサクシノイルオキシ基、ホスホリルオキシ基およびそれらのナトリウム塩からなる群より選択される、請求項1に記載の化合物。
- 化合物が、3α−ヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−ホスフェート二ナトリウム塩、3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン、3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−ヘミサクシネートナトリウム塩、3α−ヒドロキシ−3β−エチニル−5α−プレグナン−20−オン、3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン 21−ヘミサクシネート、3α−ヒドロキシ−3β−トリフルオロメチル−19−ノル−5β−プレグナン−20−オン、および3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノルプレグナン−20−オン 21−ホスフェート二ナトリウム塩からなる群より選択される、請求項1に記載の化合物。
- 以下の化合物からなる群より選択される、3α−ヒドロキシ−5−還元ステロイド化合物:
3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノル−プレグナン−20−オン;
3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノル−プレグナン−20−オン−21−ホスフェート二ナトリウム塩;
3α−ヒドロキシ−3β−トリフルオロメチル−21−ブロモ−5β−プレグナン−20−オン;
3α−ヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン;
3α−ヒドロキシ−3β−トリフルオロメチル−5β−プレグナ−(Z)17(20)−エン;
3α−ヒドロキシ−3β−トリフルオロメチル−19−ノル−5α−プレグナン−20−オン;
3α−ヒドロキシ−3β−トリフルオロメチル−19−ノル−5β−プレグナン−20−オン;
3α−ヒドロキシ−3β−トリフルオロメチル−19−ノル−5β−プレグナ−17(20)−エン;
3α−ヒドロキシ−21−メトキシ−3β−トリフルオロメチル−5β−19−ノル−プレグナン−20−オン;
3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−アセテート;
3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−ヘミサクシネートナトリウム塩;
3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−ヘミサクシネート;
3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−ジベンジルホスフェート;
3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン;
3α,21−ジヒドロキシ−3β−トリフルオロメチル−5β−プレグナン−20−オン 21−ホスフェート二ナトリウム塩;
3α,21−ジヒドロキシ−3β−トリフルオロメチル−19−ノル−5β−プレグナン−20−オン;
3β−トリフルオロメチル−3α−ヒドロキシ−21−ブロモ−5β−19−ノル−プレグナン−20−オン;および
3α−ヒドロキシ−3β−トリフルオロメチル−5β−19−ノル−プレグナン−20−オン 21−ジベンジルホスフェート。 - 以下の化合物からなる群より選択される、3α−ヒドロキシ−5−還元ステロイド化合物:
3β−(3’−ブロモ−1−プロピニル)−3α−ヒドロキシ−5β−プレグナン−20−オン;
3α−ヒドロキシ−3β−(2’−プロピニル)−5α−プレグナン−20−オン;
3α−ヒドロキシ−3β−(6−オキソ−1−ヘプチニル)−5β−プレグナン−20−オン;
3α−ヒドロキシ−3β−(プロピ−2’−イニル)−5α−プレグナン−20−オン;
3α−ヒドロキシ−3β−フェニルエチニル−5β−プレグナン−20−オン;
3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン 21−ヘミサクシネート;
3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン;
3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン 21−アセテート;
21−ブロモ−3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン;
3β−(1−ヘプチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン;
3β−(1−ヘキシニル)−3α−ヒドロキシ−5β−プレグナン−20−オン;
3β−(1−オクチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン;
3β−(3’−ブロモ−1−プロピニル)−3α−ヒドロキシ−5β−プレグナン−20−オン;
3β−(5’−シアノペンチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン;
3β−(クロロエチニル)−3α−ヒドロキシ−5β−プレグナン−20−オン;
3β−クロロエチニル−3α−ヒドロキシ−5β−プレグナン−20−オン;
3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン 20−ケタール;
3β−エチニル−3α−ヒドロキシ−5α−プレグナン−20−オン;
3β−エチニル−3α−ヒドロキシ−5β−プレグナン−20−オン;
3β−エチニル−3α−ヒドロキシ−21−メトキシ−5β−プレグナン−20−オン;
3β−エチニル−3α−ヒドロキシ−5β−19−ノル−プレグナ−17(Z)−エン;
3β−エチニル−3α−ヒドロキシ−19−ノル−5β−プレグナン−20−オン;
3β−エチニル−3α,20α−ジヒドロキシ−5β−プレグナン;
3β−エチニル−3α,20β−ジヒドロキシ−5β−プレグナン;および
3α,21−ジヒドロキシ−3β−エチニル−5β−プレグナン−20−オン 21−ヘミサクシネートナトリウム塩。
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US6837893A | 1993-05-24 | 1993-05-24 | |
| US08/068,378 | 1993-05-24 | ||
| US19691994A | 1994-02-14 | 1994-02-14 | |
| US19697294A | 1994-02-14 | 1994-02-14 | |
| US08/196,919 | 1994-02-14 | ||
| US08/196,972 | 1994-02-14 | ||
| US24627594A | 1994-05-19 | 1994-05-19 | |
| US08/246,275 | 1994-05-19 | ||
| PCT/US1994/005820 WO1994027608A1 (en) | 1993-05-24 | 1994-05-24 | Methods and compositions for inducing sleep |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005006594A Division JP4541166B2 (ja) | 1993-05-24 | 2005-01-13 | 睡眠を誘導するための方法および組成物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH08511771A JPH08511771A (ja) | 1996-12-10 |
| JP4008024B2 true JP4008024B2 (ja) | 2007-11-14 |
Family
ID=27490706
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50089295A Expired - Lifetime JP4008024B2 (ja) | 1993-05-24 | 1994-05-23 | Gabaaレセプター複合体と相互作用させるための化合物 |
| JP2005006594A Expired - Lifetime JP4541166B2 (ja) | 1993-05-24 | 2005-01-13 | 睡眠を誘導するための方法および組成物 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005006594A Expired - Lifetime JP4541166B2 (ja) | 1993-05-24 | 2005-01-13 | 睡眠を誘導するための方法および組成物 |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP0701444B1 (ja) |
| JP (2) | JP4008024B2 (ja) |
| AU (1) | AU698834B2 (ja) |
| DE (1) | DE69435286D1 (ja) |
| WO (1) | WO1994027608A1 (ja) |
Families Citing this family (77)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5939545A (en) | 1994-02-14 | 1999-08-17 | Cocensys, Inc. | Method, compositions, and compounds for allosteric modulation of the gaba receptor by members of the androstane and pregnane series |
| PT752860E (pt) * | 1994-02-14 | 2000-12-29 | Euro Celtique Sa | Androstanos e pregnanos para modulacao alosterica do receptor de gaba |
| US5994333A (en) * | 1994-08-04 | 1999-11-30 | Pherin Corporation | Pregnane and cholane steroids as neurochemical initiators of change in human hypothalamic function and related pharmaceutical compositions and methods |
| CN1171114A (zh) * | 1994-11-23 | 1998-01-21 | 科斯赛斯公司 | 用于γ-氨基丁酸受体的变构调节的雄甾烷和孕甾烷组系 |
| CA2223996A1 (en) * | 1995-06-06 | 1996-12-19 | Cocensys, Inc. | Neuroactive steroids of the androstane and pregnane series |
| US6780853B1 (en) | 1995-06-06 | 2004-08-24 | Euro-Celtique S.A. | Neuroactive steroids of the androstane and pregnane series |
| US5888996A (en) * | 1995-07-26 | 1999-03-30 | Trustees Of Boston University | Inhibition of NMDA receptor activity and modulation of glutamate-mediated synaptic activity |
| WO1997003677A1 (en) * | 1995-07-24 | 1997-02-06 | Trustees Of Boston University | Inhibition of nmda receptor activity by pregnenolone sulfate derivatives |
| AUPN498095A0 (en) * | 1995-08-23 | 1995-09-14 | Goodchild, Colin Stanley | Compounds and compositions |
| US5922699A (en) * | 1996-06-07 | 1999-07-13 | Pherin Corporation | 19-nor-cholane steroids as neurochemical initiators of change in human hypothalamic function |
| US6787530B1 (en) | 1996-08-23 | 2004-09-07 | Monash University | Use of pregnane-diones as analgesic agents |
| GR1003861B (el) * | 2000-12-29 | 2002-04-11 | Νεα νευροστεροειδη που αλληλεπιδρουν με τον υποδοχεα gabaa. | |
| EP2316468A1 (en) | 2002-02-22 | 2011-05-04 | Shire LLC | Delivery system and methods for protecting and administering dextroamphetamine |
| WO2005105822A2 (en) * | 2004-04-23 | 2005-11-10 | Euro-Celtique S.A. | 3-alpha-hydroxy 21-n- heteroaryl-pregnane derivatives for modulation of brain excitability and a process for the production thereof |
| WO2006010085A1 (en) * | 2004-07-09 | 2006-01-26 | Roxro Pharma, Inc. | Use of neurosteroids to treat neuropathic pain |
| EP2275095A3 (en) | 2005-08-26 | 2011-08-17 | Braincells, Inc. | Neurogenesis by muscarinic receptor modulation |
| EP2258359A3 (en) | 2005-08-26 | 2011-04-06 | Braincells, Inc. | Neurogenesis by muscarinic receptor modulation with sabcomelin |
| CA2625153A1 (en) | 2005-10-21 | 2007-04-26 | Braincells, Inc. | Modulation of neurogenesis by pde inhibition |
| CA2625210A1 (en) | 2005-10-31 | 2007-05-10 | Braincells, Inc. | Gaba receptor mediated modulation of neurogenesis |
| US20100216734A1 (en) | 2006-03-08 | 2010-08-26 | Braincells, Inc. | Modulation of neurogenesis by nootropic agents |
| JP2009536669A (ja) | 2006-05-09 | 2009-10-15 | ブレインセルス,インコーポレイティド | アンジオテンシン調節による神経新生 |
| US20100184806A1 (en) | 2006-09-19 | 2010-07-22 | Braincells, Inc. | Modulation of neurogenesis by ppar agents |
| EP2097437B1 (en) * | 2006-11-21 | 2015-05-27 | Umecrine Cognition AB | The use of pregnane and androstane steroids for the manufacture of a pharmaceutical composition for the treatment of cns disorders |
| JOP20180077A1 (ar) | 2007-06-19 | 2019-01-30 | Kythera Biopharmaceuticals Inc | تركيبات وطرق لحمض صفراوي تخليقي |
| US20080318870A1 (en) | 2007-06-19 | 2008-12-25 | Kythera Biopharmaceuticals, Inc. | Synthetic bile acid compositions and methods |
| US8242294B2 (en) | 2007-06-19 | 2012-08-14 | Kythera Biopharmaceuticals, Inc. | Synthetic bile acid compositions and methods |
| GB2460350B (en) * | 2008-04-25 | 2011-04-06 | Kythera Biopharmaceuticals Inc | Intermediates for the preparation of bile acids |
| US8555875B2 (en) | 2008-12-23 | 2013-10-15 | Map Pharmaceuticals, Inc. | Inhalation devices and related methods for administration of sedative hypnotic compounds |
| WO2010099217A1 (en) | 2009-02-25 | 2010-09-02 | Braincells, Inc. | Modulation of neurogenesis using d-cycloserine combinations |
| NZ597940A (en) * | 2009-08-13 | 2013-03-28 | Marinus Pharmaceuticals Inc | Method for making 3alpha-hydroxy, 3beta- methyl-5alpha-pregnan-20-one (ganaxolone) |
| WO2012021133A1 (en) | 2010-08-12 | 2012-02-16 | Kythera Biopharmaceuticals, Inc. | Synthetic bile acid compositions and methods |
| CN107936076B (zh) * | 2011-09-08 | 2021-10-15 | 萨奇治疗股份有限公司 | 神经活性类固醇、组合物、及其用途 |
| CN113234114A (zh) * | 2011-10-14 | 2021-08-10 | 萨奇治疗股份有限公司 | 3,3-二取代的19-去甲孕甾烷化合物、组合物、及其用途 |
| WO2013112605A2 (en) | 2012-01-23 | 2013-08-01 | Sage Therapeutics, Inc. | Neuroactive steroid formulations and methods of treating cns disorders |
| HUE056838T2 (hu) | 2012-08-21 | 2022-04-28 | Sage Therapeutics Inc | Allopregnanolon makacs epileptikus állapot kezelésére |
| WO2014085668A1 (en) | 2012-11-30 | 2014-06-05 | The Regents Of The University Of California | Anticonvulsant activity of steroids |
| MX362544B (es) | 2013-03-13 | 2019-01-24 | Sage Therapeutics Inc | Esteroides neuroactivos y metodos para su uso. |
| US20160068563A1 (en) | 2013-04-17 | 2016-03-10 | Boyd L. Harrison | 19-nor neuroactive steroids and methods of use thereof |
| SI2986624T1 (sl) * | 2013-04-17 | 2020-10-30 | Sage Therapeutics, Inc. | 19-nor nevroaktivni steroidi za metode zdravljenja |
| US9725481B2 (en) | 2013-04-17 | 2017-08-08 | Sage Therapeutics, Inc. | 19-nor C3, 3-disubstituted C21-C-bound heteroaryl steroids and methods of use thereof |
| TR201901745T4 (tr) | 2013-04-17 | 2019-03-21 | Sage Therapeutics Inc | 19-nor C3,3-disübstitüe edilmiş C21- n-pirazolil steroidleri ve bunların kullanım yöntemleri. |
| PL3021852T3 (pl) * | 2013-07-19 | 2021-07-05 | Sage Therapeutics, Inc. | Neuroaktywne steroidy, kompozycje i ich zastosowania |
| PT3035940T (pt) | 2013-08-23 | 2019-01-28 | Sage Therapeutics Inc | Esteroides neuroativos, composições e seus usos |
| US20160339040A1 (en) | 2014-01-29 | 2016-11-24 | Umecrine Cognition Ab | Steroid compound for use in the treatment of hepatic encephalopathy |
| US10246482B2 (en) | 2014-06-18 | 2019-04-02 | Sage Therapeutics, Inc. | Neuroactive steroids, compositions, and uses thereof |
| EP4306114A1 (en) | 2014-06-18 | 2024-01-17 | Sage Therapeutics, Inc. | Oxysterols and methods of use thereof |
| EP3188734A4 (en) | 2014-09-02 | 2018-01-10 | The Texas A&M University System | Method of treating organophosphate intoxication |
| JOP20200195A1 (ar) | 2014-09-08 | 2017-06-16 | Sage Therapeutics Inc | سترويدات وتركيبات نشطة عصبياً، واستخداماتها |
| PT3206493T (pt) | 2014-10-16 | 2020-08-03 | Sage Therapeutics Inc | Composições e métodos para tratamento de transtornos do snc |
| KR20230170816A (ko) | 2014-10-16 | 2023-12-19 | 세이지 테라퓨틱스, 인크. | Cns 장애의 치료를 위한 조성물 및 방법 |
| JP6893173B2 (ja) | 2014-11-27 | 2021-06-23 | セージ セラピューティクス, インコーポレイテッド | Cns障害を処置するための組成物および方法 |
| AR103384A1 (es) | 2015-01-12 | 2017-05-03 | Umecrine Cognition Ab | 3a-ETINILO, 3b-HIDROXI, 5a-PREGNAN-20-OXIMA COMO MODULADOR DE GABA |
| HUE054092T2 (hu) | 2015-01-26 | 2021-08-30 | Sage Therapeutics Inc | Központi idegrendszeri zavarok kezelésére szolgáló kompozíciók és eljárások |
| US10329320B2 (en) | 2015-02-20 | 2019-06-25 | Sage Therapeutics, Inc. | Neuroactive steroids, compositions, and uses thereof |
| PL3319612T3 (pl) | 2015-07-06 | 2021-12-20 | Sage Therapeutics, Inc. | Oksysterole i sposoby ich stosowania |
| HK1255500A1 (zh) | 2015-07-06 | 2019-08-16 | Sage Therapeutics, Inc. | 氧固醇及其使用方法 |
| EP3828194B1 (en) | 2015-07-06 | 2025-04-16 | Sage Therapeutics, Inc. | Oxysterols and methods of use thereof |
| RU2766155C2 (ru) | 2016-03-08 | 2022-02-08 | Сейдж Терапьютикс, Инк. | Нейроактивные стероиды, их композиции и применения |
| US20190330259A1 (en) | 2016-04-01 | 2019-10-31 | Sage Therapeutics, Inc. | Oxysterols and methods of use thereof |
| US10752653B2 (en) | 2016-05-06 | 2020-08-25 | Sage Therapeutics, Inc. | Oxysterols and methods of use thereof |
| US10278977B2 (en) | 2016-06-06 | 2019-05-07 | Umecrine Cognition Ab | Methods for treating hypersomnolence |
| MD3481846T2 (ro) | 2016-07-07 | 2021-11-30 | Sage Therapeutics Inc | 24-Hidroxisteroli 11-substituiți pentru utilizare în tratamentul stărilor legate de NMDA |
| CA3030420C (en) | 2016-07-11 | 2025-11-18 | Sage Therapeutics, Inc. | C7, C12 AND C16 SUBSTITUTED NEUROACTIVE STEROIDS AND ASSOCIATED METHODS OF USE |
| NZ790226A (en) | 2016-07-11 | 2025-08-29 | Sage Therapeutics Inc | C17, C20, and C21 substituted neuroactive steroids and their methods of use |
| CN121085986A (zh) | 2016-09-30 | 2025-12-09 | 萨奇治疗股份有限公司 | C7取代的氧固醇及其作为nmda调节剂的方法 |
| TW202444381A (zh) | 2016-10-18 | 2024-11-16 | 美商賽吉醫療公司 | 氧固醇(oxysterol)及其使用方法 |
| KR102521573B1 (ko) | 2016-10-18 | 2023-04-14 | 세이지 테라퓨틱스, 인크. | 옥시스테롤 및 그의 사용 방법 |
| WO2019075362A1 (en) * | 2017-10-12 | 2019-04-18 | Sage Therapeutics, Inc. | METHOD OF TREATING CENTRAL NERVOUS SYSTEM DISORDERS WITH NEUROSTEROIDS AND GABAERGIC COMPOUNDS |
| US11718642B2 (en) | 2018-01-12 | 2023-08-08 | Sage Therapeutics, Inc. | Compositions and methods for treating CNS disorders |
| WO2019209850A1 (en) * | 2018-04-23 | 2019-10-31 | Reddy Doodipala Samba | Neurosteroid compounds and methods for their preparation and use in treating central nervous system disorders |
| GB201821066D0 (en) * | 2018-12-21 | 2019-02-06 | Univ Oxford Innovation Ltd | Sleep modulation |
| CN111410673A (zh) * | 2019-01-08 | 2020-07-14 | 成都康弘药业集团股份有限公司 | 甾体类化合物、用途及其制备方法 |
| CN114206900A (zh) | 2019-05-31 | 2022-03-18 | 萨奇治疗股份有限公司 | 神经活性类固醇及其组合物 |
| JP2022552788A (ja) * | 2019-09-30 | 2022-12-20 | アテネン セラピューティクス, インコーポレイテッド | Gabaaレセプターのサブタイプを優先的に増強する組成物およびその使用方法 |
| WO2021188778A2 (en) * | 2020-03-18 | 2021-09-23 | Sage Therapeutics, Inc. | Neuroactive steroids and their methods of use |
| WO2023060067A1 (en) | 2021-10-04 | 2023-04-13 | Marinus Pharmaceuticals, Inc. | Amorphous solid dispersion ganaxolone formulation |
| CN117801047B (zh) * | 2023-01-19 | 2024-12-20 | 北京华睿鼎信科技有限公司 | 神经甾体衍生物及其用途 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3274179A (en) * | 1964-03-19 | 1966-09-20 | Merck & Co Inc | 3-haloethynyl-3-hydroxy derivatives of the androstane-17-one and pregnane-20-one series |
| DE2162555A1 (de) * | 1970-12-17 | 1972-06-22 | Glaxo Laboratories Ltd., Greenford, Middlesex (Grossbritannien) | Anästhetisch wirkende Steroidverbindungen |
| US5232917A (en) * | 1987-08-25 | 1993-08-03 | University Of Southern California | Methods, compositions, and compounds for allosteric modulation of the GABA receptor by members of the androstane and pregnane series |
| US5120723A (en) * | 1987-08-25 | 1992-06-09 | University Of Southern California | Method, compositions, and compounds for modulating brain excitability |
| AU609927B2 (en) * | 1987-08-25 | 1991-05-09 | Kelvin W. Gee | Compositions and methods for alleviating stress, anxiety and seizure activity |
| US5208227A (en) * | 1987-08-25 | 1993-05-04 | University Of Southern California | Method, compositions, and compounds for modulating brain excitability |
| CA2055308A1 (en) * | 1990-11-29 | 1992-05-30 | Roman Amrein | Imidazobenzodiazepine for the treatment of sleep disorders |
| JPH06510999A (ja) * | 1991-09-13 | 1994-12-08 | コセンシス・インコーポレイテッド | ステロイド結合部位を有する新規GABA↓aレセプター |
-
1994
- 1994-05-23 EP EP94918659A patent/EP0701444B1/en not_active Expired - Lifetime
- 1994-05-23 AU AU69883/94A patent/AU698834B2/en not_active Ceased
- 1994-05-23 JP JP50089295A patent/JP4008024B2/ja not_active Expired - Lifetime
- 1994-05-23 DE DE69435286T patent/DE69435286D1/de not_active Expired - Lifetime
- 1994-05-24 WO PCT/US1994/005820 patent/WO1994027608A1/en not_active Ceased
-
2005
- 2005-01-13 JP JP2005006594A patent/JP4541166B2/ja not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| JP4541166B2 (ja) | 2010-09-08 |
| EP0701444B1 (en) | 2010-04-07 |
| DE69435286D1 (de) | 2010-05-20 |
| JP2005179370A (ja) | 2005-07-07 |
| EP0701444A1 (en) | 1996-03-20 |
| JPH08511771A (ja) | 1996-12-10 |
| AU698834B2 (en) | 1998-11-12 |
| AU6988394A (en) | 1994-12-20 |
| WO1994027608A1 (en) | 1994-12-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4008024B2 (ja) | Gabaaレセプター複合体と相互作用させるための化合物 | |
| WO1994027608A9 (en) | Methods and compositions for inducing sleep | |
| CA2223397C (en) | Novel nor-pregnanes for inducing hypothalamic effects | |
| US5563131A (en) | Pregnane steroids as neurochemical initiators of change in human hypothalamic function and related pharmaceutical compositions and methods | |
| JPS5872600A (ja) | 新規シアノステロイド | |
| JP2002517405A (ja) | アゴニストまたはアンタゴニストホルモン特性を有する17β−ニトロ−11β−アリルステロイドとその誘導体 | |
| US6331534B1 (en) | Steroids as neurochemical stimulators of the VNO to alleviate pain | |
| CA2250309C (en) | Steroids as neurochemical initiators of change in human blood levels of lh or fsh | |
| EP1525215B1 (de) | Progesteronrezeptormodulatoren mit erhöhter antigonadotroper aktivität für die weibliche fertilitätskontrolle und hormonersatztherapie | |
| MASUDA | URINARY KETOSTEROID EXCRETION PATTERNS IN CONGENITAL ADRENAL HYPERPLASIAF | |
| NZ257218A (en) | Androstane steroid-containing medicament for nasal administration | |
| EP0715517B1 (en) | Estrene steroids as neurochemical initiators of change in human hypothalamic function and related pharmaceutical compositions | |
| JP4450435B2 (ja) | Pmsおよび不安の症状を緩和する、vnoにおける神経化学刺激物質としてのステロイド | |
| HU190746B (en) | Process for producing 6-alpha-methyl-corticoides and pharmaceutical compositions containing them | |
| WO1998003207A9 (en) | Steroids as neurochemical stimulators of the vno to alleviate symptoms of pms and anxiety | |
| CA2163748C (en) | Methods and compositions for inducing sleep | |
| CZ92697A3 (en) | Novel estrenes intended for inducing hypothalamus effects | |
| CZ32997A3 (en) | Pregnanes and cholanes as neurochemical initiators of function changes | |
| EP1032266A1 (en) | Use of delta5-androstene-3beta-ol-7,17-dione in the treatment of arthritis | |
| JPS629600B2 (ja) | ||
| NZ330570A (en) | the use of estrene steroids as neurochemical initiators of change in human hypothalamic funtion | |
| MXPA98007951A (en) | Steroids as neurochemical initiators of change in the levels in human blood of luteinizing hormone or stimulating hormone of folicu | |
| MXPA99000885A (en) | Steroids as neurochemical stimulants of the vomeronasal organ for mitigating symptoms of the pre-menstrual syndrome and ansie | |
| MXPA97009693A (en) | Novedosos nor-pregnanos, to induce hipotalami effects |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20040713 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20041013 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20041129 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050113 |
|
| A72 | Notification of change in name of applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A721 Effective date: 20050322 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20060530 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20060817 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20061002 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20061129 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20070313 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070607 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20070731 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20070829 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100907 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100907 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110907 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110907 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120907 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120907 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130907 Year of fee payment: 6 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |