JP3935861B2 - 新規蛋白質及びその製造方法 - Google Patents
新規蛋白質及びその製造方法 Download PDFInfo
- Publication number
- JP3935861B2 JP3935861B2 JP2003169309A JP2003169309A JP3935861B2 JP 3935861 B2 JP3935861 B2 JP 3935861B2 JP 2003169309 A JP2003169309 A JP 2003169309A JP 2003169309 A JP2003169309 A JP 2003169309A JP 3935861 B2 JP3935861 B2 JP 3935861B2
- Authority
- JP
- Japan
- Prior art keywords
- protein
- ocif
- obm
- activity
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images






























Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/715—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2875—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the NGF/TNF superfamily, e.g. CD70, CD95L, CD153, CD154
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/12—Drugs for disorders of the metabolism for electrolyte homeostasis
- A61P3/14—Drugs for disorders of the metabolism for electrolyte homeostasis for calcium homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/525—Tumour necrosis factor [TNF]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70578—NGF-receptor/TNF-receptor superfamily, e.g. CD27, CD30, CD40, CD95
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2878—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the NGF-receptor/TNF-receptor superfamily, e.g. CD27, CD30, CD40, CD95
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/566—Immunoassay; Biospecific binding assay; Materials therefor using specific carrier or receptor proteins as ligand binding reagents where possible specific carrier or receptor proteins are classified with their target compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/52—Assays involving cytokines
- G01N2333/525—Tumor necrosis factor [TNF]
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
- G01N2333/70578—NGF-receptor/TNF-receptor superfamily, e.g. CD27, CD30 CD40 or CD95
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/10—Musculoskeletal or connective tissue disorders
- G01N2800/108—Osteoporosis
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Physical Education & Sports Medicine (AREA)
- Gastroenterology & Hepatology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Toxicology (AREA)
- Zoology (AREA)
- Cell Biology (AREA)
- Rheumatology (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Urology & Nephrology (AREA)
- Physics & Mathematics (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Food Science & Technology (AREA)
- Endocrinology (AREA)
- Diabetes (AREA)
Description
【発明の属する技術分野】
本発明は、破骨細胞形成抑制因子(osteoclastogenesis inhibitory factor;以下、OCIFということがある)に結合する新規な蛋白質(OCIF結合分子;以下OBMということがある)、及びその製造方法に関する。
また本発明は、該蛋白質をコードするDNA、このDNAによりコードされるアミノ酸配列を有する蛋白質、該蛋白質を遺伝子工学的に製造する方法、及び該蛋白質を含有する医薬組成物に関する。
さらに本発明は、該蛋白質びそのDNAを用いた、該蛋白質の発現を調節する物質、該蛋白質の生物活性を阻害又は修飾する物質、あるいは該蛋白質と結合しその作用を伝達するレセプターのスクリーニング方法、これにより得られた物質、及び得られた物質を含有する医薬組成物に関する。
またさらに本発明は、該蛋白質に対する抗体、その製造方法、それを用いた該蛋白質の測定方法、及び抗体を含有する医薬に関する。
【0002】
【従来技術】
骨代謝は、骨形成を担当する骨芽細胞と骨吸収を担当する破骨細胞の、総合された活性に依存している。骨代謝異常は、骨形成と骨吸収の均衡が崩れることにより発生すると考えられている。骨代謝の異常を伴う疾患としては、骨粗鬆症、高カルシウム血症、骨ページェット病、腎性骨異栄養症、慢性関節リューマチ、及び変形性関節炎などが知られている。これらの骨代謝異常疾患の代表として骨粗鬆症が挙げられる。この疾患は、破骨細胞による骨吸収が骨芽細胞による骨形成を上まわることにより発生する疾患であり、骨の石灰質と骨基質とが等しく減少することを特徴とする。この疾患の発生メカニズムについては未だ完全には解明されていないが、骨の疼痛が発生し、骨の脆弱化による骨折が起こる疾患である。高齢人口の増加に伴い、骨折による寝たきり老人の原因となり、この疾患は社会問題にもなっており、その治療薬の開発が急務となっている。このような骨代謝異常による骨量減少症は、骨形成の促進、骨吸収の抑制、あるいはこれらのバランスの改善により治療できることが期待される。即ち、骨形成は、骨形成を担当する骨芽細胞の増殖、分化、機能を促進すること、破骨細胞前駆細胞からの破骨細胞への分化、成熟を抑制すること、あるいは破骨細胞の骨吸収活性などの機能を抑制することにより促進されると期待される。このような活性を有するホルモン、低分子物質、あるいは生理活性蛋白質について、現在精力的な探索及び開発研究が進められている。
【0003】
すでに、骨に関わる疾患の治療及び治療期間の短縮を図る医薬品として、カルシトニン製剤、活性型ビタミンD3製剤、エストラジオールを含有するホルモン製剤、イプリフラボン、ビタミンK2、ビスフォスフォネート系化合物などがあり、さらに、より副作用が少なく、有効性に優れた治療薬の開発を目指して活性型ビタミンD3誘導体、エストラジオール誘導体、第2世代または第3世代のビスフォスフォネート系化合物などの臨床試験が実施されている。
【0004】
しかし、これらの薬剤を用いた治療法は、その効果並びに治療結果において必ずしも満足出来るものではなく、より安全かつ有効性の高い新しい治療薬の開発が期待されている。また、骨代謝疾患の治療に使用されている薬剤の中には、その副作用により治療可能な疾患が限定されているものもある。更に現在、骨粗鬆症などの骨代謝疾患の治療には、複数の薬剤を同時に使用する多剤併用療法が主流になってきている。このような観点から、従来の医薬品とは異なった作用メカニズムを持ち、しかもより有効性が高くかつ副作用の少ない医薬品の開発が期待されている。
【0005】
前述したように、骨代謝を担当する細胞は骨芽細胞と破骨細胞である。これらの細胞は互いに密接に相互作用していることが知られており、この現象はカップリングと呼ばれている。即ち、破骨細胞の分化、成熟には骨芽細胞様ストローマ細胞が分泌するサイトカイン類、インターロイキン1(IL-1)、3(IL-3)、6(IL-6)、11(IL-11)、顆粒球マクロファージコロニー刺激因子(GM-CSF)、マクロファージコロニー刺激因子(M-CSF)、インターフェロンガンマー(IFN‐γ)、腫瘍壊死因子α(TNF‐α)、トランスフォーミング増殖因子β(TGF‐β)などが促進的又は抑制的に作用することが報告されている(非特許文献1; 非特許文献2;非特許文献3;非特許文献4)。骨芽細胞様ストローマ細胞は、未熟な破骨細胞前駆細胞や破骨細胞との細胞間接着により、それぞれ破骨細胞の分化、成熟や成熟破骨細胞による骨吸収などの機能発現に重要な役割を演じていることが知られている。この細胞間接着による破骨細胞形成に関与する因子として、骨芽細胞様ストローマ細胞の膜上に発現される破骨細胞分化誘導因子(osteoclast differentiation factor, ODF)(非特許文献5;非特許文献6)という分子が仮想されており、この仮説によれば破骨細胞の前駆細胞にODFのレセプターが存在している。
しかし、ODF及びこのレセプターとも未だ精製及び同定されておらず、それらの性質、作用機作、及び構造に関する報告もない。このように、破骨細胞の分化、成熟のメカニズムは未だ十分に解明されておらず、このメカニズムの解明は基礎医学領域に多大な貢献をするだけでなく、新しい作用メカニズムに基づく新規な骨代謝異常症治療薬の開発にも貢献することが期待される。
【0006】
本発明者らは、このような状況に鑑み鋭意探索を進めた結果、ヒト胎児肺線維芽細胞、IMR-90(ATCC CCL186)の培養液中に破骨細胞形成抑制因子(osteoclastogenesis inhibitory factor, OCIF)を見出した(特許文献1)。
さらに、本発明者らは、OCIFのDNAクローニング、動物細胞を用いた遺伝子組み換え型OCIFの生産、遺伝子組み換え型OCIFによるin vivo薬効(骨代謝改善効果)の確認などに成功した。OCIFは、従来の医薬品よりも有効性が高くかつ副作用が少ない、骨代謝異常に関連する疾患の予防及び治療剤として期待されている。
【0007】
【特許文献1】
WO96/26217号
【非特許文献1】
Raisz: Disorders of Bone and Mineral Metabolism, 287-311, 1992
【非特許文献2】
Suda et al.:Princeples of Bone Biology, 87-102, 1996
【非特許文献3】
Suda et al.: Endocrine Reviews, 4, 266-270, 1955
【非特許文献4】
Lacey et al.:Endocrinology, 186, 2369-2376, 1995
【非特許文献5】
Suda et al.:Endocrine Rev.13,66-80, 1992
【非特許文献6】
Suda et al.:Bone 17, 87S-91S, 1995
【0008】
【発明が解決しようとする課題】
本発明者らは破骨細胞形成抑制因子OCIFを用い、その結合蛋白質の存在を鋭意探索した結果、OCIF結合蛋白質が活性型ビタミンD3や副甲状腺ホルモン(parathyroid hormone, PTH)などの骨吸収促進因子の存在下で培養した骨芽細胞様ストローマ細胞上に特異的に発現することを見出した。さらに、このOCIF結合蛋白質の性質及びその生理的機能を調べたところ、該蛋白質は未熟な破骨細胞前駆細胞からの破骨細胞への分化、成熟に関与する、いわゆる破骨細胞分化、成熟を支持又は促進する因子としての生物活性を有していることを見出し、本発明を成すに至った。さらに、本発明蛋白質につき研究を重ねた結果、この新規な膜蛋白質は骨芽細胞様ストローマ細胞と脾臓細胞との共培養系において、骨芽細胞様ストローマ細胞による未熟な破骨細胞前駆細胞からの破骨細胞への分化、成熟を担う重要な蛋白質であることを明らかにした。本発明における破骨細胞の分化、成熟を支持又は促進する因子としての蛋白質の同定及びその単離精製の成功により、本発明蛋白質を利用した、生体の骨代謝機構に基づいた新規な骨代謝異常症治療薬のスクリーニングを可能にした。
【0009】
従って本発明は、破骨細胞形成抑制因子OCIFに結合する新規な蛋白質(OCIF結合分子;OBM)、及びその製造方法を提供することを課題とする。また本発明は、該蛋白質をコードするDNA、このDNAによりコードされるアミノ酸配列を有する蛋白質、該蛋白質を遺伝子工学的に製造する方法、及び該蛋白質を含有する医薬組成物を提供することを課題とする。また、該蛋白質を含有する骨代謝異常症予防及び/又は治療剤を提供することを課題とする。さらに本発明は、該蛋白質を及びそのDNAを用いた、該蛋白質の発現を調節する物質、該蛋白質の生物活性を阻害又は修飾する物質、あるいは該蛋白質と結合しその作用を伝達するレセプターのスクリーニング方法、これにより得られた物質、及び得られた物質を含有する医薬組成物を提供することを課題とする。またさらに本発明は、該蛋白質に対する抗体、その製造方法、それを用いた該蛋白質の測定方法、及び抗体を含有する医薬を提供することを課題とする。
【0010】
本発明蛋白質は、以下の物理化学的性質及び生物活性を示す。
即ち、
a.親和性:破骨細胞形成抑制因子(osteqclastogenesis inhibitory factor; OCIF)に特異的に結合し、高い親和性(細胞膜上での解離定数、Kd値が10-9M以下)を有する。
b.分子量:非還元条件下におけるSDS―ポリアクリルアミド電気泳動による分子量測定で、約30,000-40,000の分子量を示し、又、モノマータイプのOCIFとクロスリンクさせた場合の見かけ上の分子量は、約90,000-110,000を示す。
c.生物活性:活性型ビタミンD3及び副甲状腺ホルモン(PTH)などの、骨吸収促進因子の存在下でのマウス骨芽細胞様ストローマ細胞とマウス脾臓細胞共培養において、破骨細胞の分化、成熟を支持又は促進する活性を有する。
破骨細胞形成の代表的なインビトロ培養系として、活性型ビタミンD3あるいはPTH存在下でのマウス由来骨芽細胞様ストローマ細胞株・ST2とマウス脾臓細胞との共培養系がよく知られている。本発明蛋白質の発現細胞は活性型ビタミンD3存在あるいは非存在下でそれぞれ培養したマウス骨芽細胞様ストローマ細胞あるいはマウス脾臓細胞へのOCIFの結合性を試験することにより調べることができる。
【0011】
本発明蛋白質は、活性型ビタミンD3やPTHのような骨吸収促進因子の存在下で培養した骨芽細胞様ストローマ細胞上に特異的に誘導される蛋白質として特定される。又、活性型ビタミンD3存在下の上記共培養系にOCIFを添加することにより1から40ng/mlの範囲で用量依存的に破骨細胞の形成が阻害されること、活性型ビタミンD3存在下でST2細胞上に誘導される本発明蛋白質の発現の経時変化と破骨細胞形成の経時変化とがよく相関すること、又、ST2細胞に発現される本発明蛋白質の多寡とST2細胞による破骨細胞形成支持能の強弱が一致し、ST2細胞上の本発明蛋白質にOCIFが結合することにより破骨細胞形成が完全に抑制されることから、本発明蛋白質は破骨細胞の分化、成熟を支持又は促進する生物活性(作用)を有する蛋白質として特定される。
【0012】
本発明蛋白質のOCIFに対する親和性は、OCIFを標識し、その標識体の動物細胞膜表面への結合性を試験することによって評価することができる。OCIFは、放射性同位元素による標識や蛍光標識などのような通常の蛋白質標識法を用いることによって標識することができる。例えば、OCIFの放射性同位元素による標識としてはチロシン残基の125I標識が挙げられ、ヨードジェン法、クロラミンT法、及び酵素法などの標識法を利用することができる。このようにして得た標識OCIFを用いた、OCIFの動物細胞膜表面への結合性試験は常法に従い実施でき、又、結合性試験に用いる培地に標識OCIFの100倍から400倍濃度未標識OCIFを添加することにより、非特異的な結合量を測定することができる。OCIFの特異的結合量は、標識OCIFの総結合量から非特異的な結合量を差し引くことにより算出される。細胞膜上に発現された本発明蛋白質の親和性は標識OCIFの添加量を変えて試験を行い、特異的結合量をScatchard plotで解析することによって評価される。このようにして求められた、本発明蛋白質のOCIFに対する親和力は約100-500pMであり、本発明蛋白質はこのように破骨細胞形成抑制因子OCIFに高い親和性(細胞膜上での解離定数、Kd値が10-9M以下)を有する蛋白質として特定される。OBMの分子量は、ゲル濾過クロマトグラフィーやSDS-PAGE等を用いて測定される。より正確に分子量を測定するためにはSDS-PAGEを用いるのが好ましく、OBMは還元条件下で約40,000 (40,000±4,000)の分子量を有する蛋白質として特定される。
【0013】
本発明蛋白質は、マウス骨芽細胞様ストローマ細胞株ST2、マウス前脂肪細胞株PA6、その他のヒト骨芽細胞様細胞株、あるいはヒト、マウス、ラットなどの哺乳動物から採取し濃縮した骨芽細胞様細胞などから得ることができる。又、これらの細胞に本発明蛋白質を発現させるために必要な誘導物質としては、活性型ビタミンD3(Calcitriol)、副甲状腺ホルモン(PTH)、インターロイキン(IL)‐1、IL-6、IL-11、オンコスタチンM及び白血病細胞増殖抑制因子LIF)などの骨吸収促進因子が挙げられる。又、これらの誘導物質の添加濃度としては、活性型ビタミンD3やPTHでは、10-8M、IL-11やオンコスタチンMではそれぞれ10ng/ml及び1ng/ml、IL-6では20ng/mlのIL-6と500ng/mlのIL-6可溶性レセプターを用いるのが望ましい。好ましくは、マウス骨芽細胞様ストローマ細胞株ST2を用い、10-8M活性型ビタミンD3、10-7Mデキサメサゾン、及び10%牛胎児血清を添加したα‐MEM中で一週間以上培養してコンフルエントにした細胞を用いるとよい。このようにして培養した細胞はセルスクレイパーなどを用いて剥離させ、回収するとよい。又、回収した細胞は使用するまでの間‐80℃で保存しておくことができる。
【0014】
本発明蛋白質は、このようにして回収した細胞の膜画分から精製すると効率良く精製することができる。膜画分の調製は、細胞内器官の分画に利用される通常の方法に従って行うことかできる。膜画分の調製に使用する緩衝液には、好ましくは各種プロテアーゼ阻害剤を添加するとよい。添加するプロテアーゼ阻害剤としては、例えばPMSF、APMSF、EDTA、0‐フェナントロリン、ロイペプチン、ペプスタチンA、アプロチニン、大豆トリプシンインヒビターなどのセリンプロテアーゼ阻害剤、チオールプロテアーゼ阻害剤、及びメタロプロテアーゼ阻害剤などが挙げられる。細胞の破砕には、Daunceホモジナイザー、ポリトロンホモジナイザー、超音波破砕装置なとを用いることが出来る。破砕した細胞は0.5Mシュクロースを加えた緩衝液に懸濁させ、600×gで10分間遠心分離することにより、細胞核と未破砕の細胞を沈殿画分として分離することかできる。遠心分離で得た上清を150,000×gで90分間遠心分離することにより、沈殿画分として膜画分を得ることかできる。このようにして得た膜画分を各種の界面活性剤で処理することにより、細胞膜に存在する本発明蛋白質を効率良く可溶化し、抽出することかできる。可溶化に用いる界面活性剤としては、細胞膜蛋白質の可溶化に一般的に使用されている各種界面活性剤、例えばCHAPS(3‐[(3-cholamidopropyl)‐dimethylammonio-1-propanesulfonate)、Triton X-100、Nikkol、n‐オクチルグリコシドなどを用いることができる。好ましくは、0.5%CHAPSを添加して4℃で2時間攪拌し、本発明蛋白質を可溶化するのが良い。このようにして調製したサンプルを、150,000×gで60分間遠心分離し、その上清を可溶化膜画分として得ることかできる。
【0015】
このようにして得られた可溶化膜画分からの本発明蛋白質の精製は、OCIFを固定化したカラム、ゲル、樹脂などを用いて効率よく精製することかできる。固定化に使用するOCIFとしては、WO96/26217号公報記載の方法に従って、ヒト胎児肺線維芽細胞株IMR-90の培養液から単離したもの、あるいは遺伝子工学的手法により得られたもの(rOCIF)を使用することができる。これらのrOCIFは、ヒト、ラット、あるいはマウスのcDNAを常法に従って発現ベクターに組み込み、CHO細胞、BHK細胞、Namalwa細胞などの動物細胞あるいは昆虫細胞などで発現させ、精製することによって得ることができる。このようにして得られたOCIFは、分子量約60kDa(モノマー型)と120kDa(ダイマー型)の分子量を示すが、固定化に用いるOCIFとしては、好ましくはダイマー型OCIFを用いる。OCIFを固定化するためのゲル、樹脂としてはECHセファロース4B、EAHセファロース4B、チオプロピルセファロース6B、CNBr‐活性化セファロース4B、活性化CHセファロース4B、エポキシ活性化セファロース6B、活性化チオールセファロース4B(以上、ファルマシア社)、TSKgel AF‐エポキシトヨパール650、TSKgel AF‐アミノトヨパール650、TSKgel AF‐フォルミルトヨパール650、TSKgel AF‐カルボキシトヨパール650、TSKgel AF‐トレシルトヨパール650(以上、トーソー社)、アミノ-セルロファイン、カルボキシ-セルロファイン、FMP活性化セルロファイン、フォルミル-セルロファイン(以上、生化学工業社)、アフィゲル10, アフィゲル15, アフィプレップ10(以上、BioRad社)などを用いることができる。又、OCIFを固定化するためのカラムとしてはHiTrap NHS-activatedカラム(ファルマシア社)、TSKgel Tresyl-5PW(トーソー社)などを用いることができる。HiTrap NHS-activatedカラム(1ml,ファルマシア社)を用いたOCIFの固定化法として、具体的には以下の方法を挙げることができる。即ち、OCIF 13.0 mgを含む0.2M NaHCO3/0.5M NaCl(pH8.3)溶液1mlをカラムに添加し、室温で30分間カップリング反応させる。
【0016】
次いで、0.5Mエタノールアミン/0.5M NaCl(pH8.3)と0.1M酢酸/0.5M NaCl(pH4.0)を流し、再度0.5Mエタノールアミン/0.5M NaCl(pH8.3)に置き換え、室温で1時間放置し、過剰な活性基を不活性化する。その後、0.5Mエタノールアミン/0.5M NaCl(pH8.3)と0.1M酢酸/0.5M NaCl(pH4.0)で2度洗浄し、50mM Tris/1 M NaCl/0.1%CHAPS緩衝液(pH7.5)で置換することにより、OCIF固定化カラムを作製することができる。このようにして作製したOCIF固定化カラムやOCIF固定化ゲルあるいは樹脂などを用い、本発明蛋白質を効率よく精製することができる。本発明蛋白質の分解を抑制するために、精製に用いる緩衝液にも上記の各種プロテアーゼ阻害剤を添加するとよい。本発明蛋白質は、上記の可溶化膜画分をOCIF固定化カラムに負荷する、あるいはOCIF固定化ゲル、樹脂などと混ぜて攪拌することによって吸着させ、酸、各種蛋白質変性剤、カコジレートバッファーなどによってOCIF固定化カラム、ゲル、あるいは樹脂などから溶出することができる。好ましくは、本発明蛋白質の変性を最小限に抑えるため、酸を用いて溶出し直ちに中和するとよい。溶出に用いる酸性緩衝液としては、例えば0.1Mグリシン-塩酸緩衝液(pH3.0)、0.1Mグリシン-塩酸緩衝液(pH2.0)、及び0.1Mクエン酸ナトリウム緩衝液(pH2.0)などを用いることができる。
【0017】
このようにして精製した本発明蛋白質は、生物試料からの蛋白質の精製に汎用される通常の方法を用いて、本発明蛋白質の物理化学的性質を利用した各種の精製操作により更に精製することができる。本発明蛋白質溶液の濃縮には限外濾過、凍結乾燥及び塩析など、通常、蛋白質の精製過程で用いられる手法が挙げられる。
好ましくは、Centricon‐10(BioRad杜)などを用いた遠心による限外濾過を用いるのが良い。又、精製手段としては、イオン交換クロマトグラフィー、ゲル濾過クロマトグラフィー、疎水クロマトグラフィー、逆相クロマトグラフィー、調製用電気泳動などを用いた通常の蛋白質の精製に利用される各種の手法を組み合わせて用いることができる。より具体的には、Superose 12カラム(ファルマシア社)などを用いたゲル濾過クロマトグラフィーや逆相クロマトグラフィーを組み合わせて用いることにより、本発明蛋白質を純化することが可能である。又、精製過程中の本発明蛋白質の同定は、固定化OCIFとの結合活性あるいはOCIFとの結合物を抗OCIF抗体による免疫沈降後、SDS‐ポリアクリルアミド電気泳動(SDS-PAGE)で分析することにより検出できる。
このようにして得られた本発明蛋白質は、その活性より骨代謝異常症、例えば大理石病などの治療剤としての医薬、あるいは研究・診断用試薬として有用である。
【0018】
また本発明は、破骨細胞形成抑制因子OCIFに結合する新規な蛋白質(OCIF結合分子;OBM)をコードするDNA、そのDNAによりコードされるアミノ酸配列を有する蛋白質、それを用いてOCIFに特異的に結合する蛋白質を遺伝子工学的に製造する方法、及び該蛋白質を含有する骨代謝治療剤に関する。さらに本発明は、OBMの発現を調節する物質のスクリーニング方法、OBMと結合しその作用を阻害又は修飾する物質のスクリーニング方法、OBMと結合しOBMの作用を伝達するレセプターのスクリーニング方法、これらのスクリーニングの結果得られた物質を含有する医薬組成物に関する。
【0019】
本発明のDNAによりコードされる新規蛋白質OBMは、以下の物理化学的性質及び生物活性を示す。
即ち、
a.破骨細胞形成抑制因子(OCIF)に特異的に結合する。
b.還元条件下におけるSDS-PAGEによる分子量測定で、約40,000(±4,000)の分子量を示す。又、モノマータイプのOCIFとクロスリンクさせた場合の見かけ上の分子量は約90,000-110,000である。
c.破骨細胞の分化、成熟を支持、促進する活性を有する。
ものである。
【0020】
本発明のOCIF結合分子、OBMをコードするDNAを同定し、OBMの性質を評価するためのプローブとして使用するヒト破骨細胞形成抑制因子(OCIF)は、WO96/26217号に従ってヒト胎児性線維芽細胞株IMR-90の培養液から単離することができる。OBMのDNAの単離、同定には、遺伝子組換え型ヒトOCIF、遺伝子組換え型マウスOCIF、遺伝子組換え型ラットOCIF等を用いることもできる。これらの遺伝子組換え型OCIFはそれぞれのDNAを常法に従って発現ベクターに組み込み、CHO細胞、BHK細胞、Namalwa細胞等の動物細胞あるいは昆虫細胞などで発現させ、精製することによって得ることができる。
【0021】
目的の蛋白質をコードするcDNAを分離する(cDNAクローニング)方法としては、その蛋白質の部分アミノ酸配列を決定しそれに対応する塩基配列をもとにハイブリダイゼーション法により目的のcDNAを単離する方法の他に、蛋白質のアミノ酸配列が不明であっても発現ベクター中にcDNAライプラリーを構築し、それらを細胞中に導入し、目的の蛋白質の発現の有無をスクリーニングすることにより、目的のcDNAを単離する方法(発現クローニング法)がある(D'Andrea et al.: Cell 57, 277-285, 1989; Fukunaga et al.: Cell 61, 341-350, 1990)。
発現クローニング法では、宿主細胞として細菌、酵母、動物細胞などが目的に応じて使い分けられている。本発明のように動物細胞の膜表面に存在すると考えられる蛋白質をコードするcDNAをクローニングする際には、動物細胞を宿主とする場合が多い。又、DNAの導入効率が高く、導入されたDNAの発現効率の高い宿主が通常使用される。そのような特徴を持つ細胞のひとつが、本発明で用いたサル腎臓細胞COS-7細胞である。COS-7細胞ではSV40 large T antigenが発現しているため、SV40の複製開始点を持っプラスミドは細胞中で多コピーのエピソームとして存在するようになり、通常より高発現が期待できる。又、DNAを導入した時点から数日のうちに最高の発現レベルに到達するので迅速なスクリーニングに適している。この宿主細胞に高発現可能なプラスミドを組み合わせることによって、極めて高レベルの遺伝子発現が可能となる。プラスミド上で最も遺伝子の発現量に影響を与える因子はプロモーターであり、高発現用のプロモーターとしてSRαプロモーターやサイトメガロウィルス由来プロモーター等がよく使用される。
【0022】
発現クローニングにより膜蛋白質のcDNAをクローニングしようとする際のスクリーニング法として、バインディング法(binding法)、パニング法(panning法)、フィルムエマルジョン法などが考案されている。
本発明は、発現クローニング法とバインディング法を組み合わせることによって得たOCIFに特異的に結合する蛋白質(OBM)をコードするDNA及び発現蛋白質、及びDNA又は発現蛋白質を用いた生理活性物質のスクリーニングに関する。本発明のDNAがコードするOBMは、OCIFを標識し、その標識体の動物細胞膜表面への結合性を試験することによって検出することができる。OCIFの標識法としては、放射性同位元素による標識や蛍光標識等、一般的な蛋白質の標識法を用いることができる。例えば、OCIFの放射性同位元素による標識としてはチロシン残基の125I標識か挙げられ、具体的標識法としてヨードジェン法、クロラミンT法及び酵素法等がある。OCIFの動物細胞膜表面への結合性はこのようにして得た標識OCIFを用い、常法に従った方法で試験できる。又、結合性の試験に用いる培地に標識OCIFの100倍から400倍濃度の未標識OCIFを添加することにより、非特異的な結合量を測定できる。OCIFの特異的結合量は、標識OCIFの総結合量から非特異的な結合量を差し引くことにより算出される。
【0023】
本発明者らは、破骨細胞の分化に関与する因子はOCIFと相互作用するとの仮定のもと、遺伝子組み換え型OCIFを用い、OCIFが結合する蛋白質を分離するため、マウス骨芽細胞様ストローマ細胞株ST2のmRNAから作製した発現ライブラリーを以下に述べる方法でスクリーニングした。ST2mRNAをもとに合成したDNAを動物細胞用発現ベクターに挿入し、それらをサル腎臓細胞COS-7細胞に遺伝子導入した。125I標識したOCIFをプローブとして用いてCOS-7細胞上に発現した目的の蛋白質をスクリーニングした。その結果、OCIFと特異的に結合する蛋白質をコードするDNAを分離するに至り、このOCIF結合分子(OCIF結合分子;OBM)をコードするDNAの塩基配列を決定した。又、このDNAにコードされるOBMは細胞膜上でOCIFと強くしかも特異的に結合することを見出した。
【0024】
本発明でいう比較的温和な条件でのDNAハイブリダイゼーションとは、例えば常法に従いナイロンメンブレンにDNAをトランスファーし固定化した後、ハイブリダイゼーション用バッファー中で放射線標識したプローブのDNAと40〜70℃、2時間〜1晩程度ハイブリダイゼーションさせ、0.5×SSC (0.075M塩化ナトリウム及び0.0075Mクエン酸ナトリウム)で45℃10分洗浄する条件を言う。具体的には、常法に従い、ナイロンメンブレンにハイボンドN(アマシャム杜)を用い、DNAをトランスファーし固定化した後、Rapid Hybridization Buffer(アマシャム社)中で32P標識したプローブのDNAと65℃2時間ハイブリダイゼーションさせて、0.5×SSC (0.075M塩化ナトリウム及び0.0075Mクエン酸ナトリウム)で45℃10分洗浄する条件を言う。
【0025】
破骨細胞形成の代表的なインビトロ培養系として、活性型ビタミンD3あるいはPTHの存在下でのマウス由来骨芽細胞様ストローマ細胞株、ST2とマウス脾臓細胞との共培養系がよく知られている。本発明のOBMは活性型ビタミンD3やPTHのような骨吸収促進因子の存在下で培養した骨芽細胞様ストローマ細胞上に特異的に誘導される蛋白質として特定される。更に、活性型ビタミンD3あるいはPTH非存在下でもマウス脾臓細胞の培養系に本発明のDNAによりコードされる蛋白質を添加することによって破骨細胞の形成が促進されることから、本発明のDNAによりコードされるOBMは破骨細胞の分化、成熟に関与していると考えられる。
【0026】
本発明のDNAを発現ベクターに挿入してOBM発現プラスミドを作製し、これを各種の細胞又は菌株に導入して発現させることにより、組み換え型OBMを製造することができる。発現させる際の哺乳動物細胞の宿主としてCOS-7、CHO、Namalwaなど、又は細菌の宿主として大腸菌などを、用いることができる。その際、DNAの全長を用いて膜結合型蛋白質として発現させる、あるいは膜結合部位をコードする部分を除去することにより分泌型、可溶化型としても発現させることができる。このようにして製造された組み換え型OBMは、通常用いられる蛋白精製法、例えばOCIF固定化カラムを用いたアフィニティークロマトグラフィー一、イオン交換クロマトグラフィー、あるいはゲル濾過クロマトグラフィーなどを組み合わせることにより、効率よく精製することができる。このようにして得られた本発明蛋白質は、その活性より骨代謝異常症、例えば大理石病などの治療剤としての医薬、あるいは研究・診断用試薬として有用である。
【0027】
本発明のDNAにコードされる蛋白質OBMを利用して、(1)OBMの発現を調節する物質のスクリーニング、(2)OBMに特異的に結合し、OBMの生物活性を阻害、あるいは修飾する物質のスクリーニング、及び(3)破骨細胞前駆細胞に存在し、OBMの生物活性を伝達する蛋白質(OBMレセプター)のスクリーニング、更にはこのOBMレセプターを利用したアンタゴニストやアゴニストの開発が可能である。上述のOBMあるいはOBMレセプターを用いたコンビナトリアルケミストリーにおいて、アンタゴニストあるいはアゴニストの探索に必要なペプチドライブラリーは具体的に以下の方法で作製することができる。その一つに、スプリット法がある
(Lam et al.; Nature 354, 82-84, 1991)。この方法は、合成担体(ビーズ)にそれぞれのアミノ酸(ユニット)を結合させたものを各ユニットごと別々に合成する。この合成された担体を一度すべて混ぜ、次にユニットの数に等分し、次のユニットをまた各々結合させる。この操作をn回繰り返すことにより、担体にn個のユニットが結合したライブラリーが作製される。このように合成を行うと一つの担体群には、1種類の配列しか合成されないので、本発明の蛋白質を用いた上記のスクリーニング法で陽性を示した担体群を選びだし、そのアミノ酸配列を決定すれば、特異的に結合するペプチドを同定することができる。又、別の方法としてファージディスプレイ法を用いることもできる。この方法はランダムなペプチドをコードする合成遺伝子をファージで発現させるもので、上記合成ライブラリーに比べ、ライブラリー中の分子数を多くできるという利点があるものの、ファージが嫌う配列はライブラリーに存在し得ないことなどがあり、分子数当たりの多様性が低いという欠点がある。ファージディスプレイ法でも同様に、本発明の蛋白質を用いたスクリーニング系を利用し、それに特異的に結合するファージをパニングにより濃縮し、得られた特異的結合性を有するファージを大腸菌で増幅し、そのペプチドをコードする塩基配列を決定すればよい。更に、前記(2)及び(3)のスクリーニング系を使用し、ペプチドライブラリーからOBMあるいはOBMレセプターに特異的でかつ高親和性のペプチドをスクリーニングしたい場合、スクリーニング時にそれぞれOCIFやOBMを共存させ、それぞれの濃度を上げながら、陽性を示した担体あるいはファージをスクリーニングすることにより、特異的かつ極めて高親和性のペプチドを得ることができる。例をあげれば、すでに造血ホルモンであるエリスロポエチン(EPO)のレセプターを用い、多様性に富んだペプチドライブラリーからのEPO様活性を有する低分子ペプチドアゴニストのスクリーニング及びその立体構造解析、更にはその立体構造に基づいた有機合成展開によるEPO活性を有する低分子物質(アゴニスト)の創製に成功している(Nicholas et a1.: Science, 273, 458-463, 1996)。
【0028】
また本発明者らは、破骨細胞形成抑制因子OCIFを用い、その結合蛋白質が活性型ビタミンD3や副甲状腺ホルモン(parathyroid hormone, PTH)などの骨吸収因子の存在下で培養した骨芽細胞様ストローマ細胞株、ST2の細胞上に特異的に発現することをすでに見出した。さらに、該蛋白質は未熟な破骨細胞前駆細胞からの破骨細胞への分化、成熟に関与する、いわゆる破骨細胞分化、成熟を支持又は促進する因子としての生物活性を有していることを見出し、該蛋白質を精製することによりその物理化学的諸性質及びその生物活性を明らかにした。本発明者らは、本発明のDNAにより発現される遺伝子組み換え型蛋白質OBMと、すでに前記OCIFと特異的に結合する精製天然型蛋白質との異同を明らかにすべく、物理化学的性質及び生物活性を比較した。その結果、両蛋白質とも▲1▼膜結合蛋白質であり、特異的にOCIFと結合し、▲2▼SDS-PAGEでの分子量は約40,000、▲3▼モノマータイプのOCIFとクロスリンキングさせた場合の見かけの分子量は約90,000-110,000であり、物理化学的諸性質がよく一致していること、又、生物活性においても、両蛋白質は破骨細胞の分化、成熟を支持又は促進する活性を有していることから、両蛋白質は同一である可能性が示唆された。さらに、本発明DNAを用いて遺伝子工学的に発現させ、精製した蛋白質(遺伝子組み換え型OBM)で作製したウサギ抗OBMポリクローナル抗体は、前記の方法で得られる精製天然型蛋白質に交差性を有し、OBMとOCIFとの特異的結合を阻害するのと同様に、前記の精製天然型蛋白質とOCIFとの結合を特異的に阻害した。これらの結果から、本発明のDNAにより発現される遺伝子組み換え型蛋白質OBMは、前記OCIFに特異的に結合する天然型蛋白質と同一であることが明らかである。
【0029】
さらに本発明者らは、OCIFに特異的に結合し、天然型あるいは遺伝子組み換え型マウスOBMと同様にマウス脾臓細胞からの破骨細胞の分化、成熟を支持、促進する活性を有するヒト由来OCIF結合蛋白分子(以下ヒトOBMと称する)をコードする遺伝子(cDNA)を単離するため、前述のとおりマウスOBM cDNAから作製したプライマーを用い、ヒトリンパ節由来cDNAを鋳型としてpolymerase chain reaction(PCR)を行い、得られたヒトOBMc DNA断片をプローブとして上記cDNAライブラリーをスクリーニングした。その結果、OCIFと特異的に結合するヒト由来蛋白質をコードするcDNAを分離することに成功し、このヒト由来OCIF結合蛋白分子、即ちヒトOBMをコードするcDNAの塩基配列を決定した。このcDNAにコードされるヒトOBMは、マウスOBMと同様に細胞膜上でOCIFと強く、しかも特異的に結合する特性を有し、またマウス脾臓細胞からの破骨細胞の分化、成熟を支持、促進する生物活性を有することを見出した。
【0030】
即ち、本発明は破骨細胞形成抑制因子OCIFに結合する新規なヒト由来蛋白質であるヒトOBMをコードするDNA、そのDNAによりコードされるアミノ酸配列を有する蛋白質、そのDNAを用いてOCIFに特異的に結合する性質を有し、マウス脾臓細胞からの破骨細胞の分化、成熟を支持、促進する活性を有する蛋白質を遺伝子工学的に製造する方法、及び該蛋白質を含有する骨代謝異常症治療剤の提供、更にヒトOBMの発現を調節する物質のスクリーニング方法、ヒトOBMと結合しその作用を阻害又は修飾する物質のスクリーニング方法、ヒトOBMと結合しその作用を伝達するレセプターのスクリーニング方法及びこれらのスクリーニングの結果得られた物質を含有する医薬組成物を提供することを課題とする。
【0031】
本発明は、OCIFに特異的に結合する特性を有し、また破骨細胞の分化、成熟を支持、促進する生物活性を有する新規なヒト蛋白質であるヒトOBMをコードするDNA、そのDNAによりコードされるアミノ酸配列を有する蛋白質、そのDNAを用いてOCIFに特異的に結合する特性を有し、また破骨細胞の分化、成熟を支持、促進する生物活性を有する蛋白質を遺伝子工学的に製造する方法、及び該蛋白質を含有する骨代謝異常症治療剤に関する。更に、本発明はヒトOBMの発現を調節する物質のスクリーニング方法、ヒトOBMと結合しその作用を阻害又は修飾する物質のスクリーニング方法、ヒトOBMと結合しOBMの生物活性を伝達するレセプターのスクリーニング方法、及びこれらのスクリーニングの結果得られた物質を含有する医薬組成物、さらにヒト由来OCIF結合蛋白質に対する抗体及びその抗体を用いた骨代謝異常症の予防及び/又は治療薬に関する。
【0032】
本発明のDNAによりコードされる新規ヒト由来OCIF結合蛋白分子、即ちヒトOBMは、以下の物理化学的性質及び生物活性を示す。
即ち、
a.破骨細胞形成抑制因子(osteoclastogenesis inhibitory factor, OCIF)(WO96/26217号)に特異的に結合する。
b.還元条件下でのSDS-PAGEによる分子量測定で、約40,000(±5,000)の分子量を示す。又、モノマータイプのOCIFとクロスリンクさせた場合の見かけ上の分子量は約90,000‐110,000である。
c.破骨細胞の分化、成熟を支持、促進する生物活性を有する。
ものである。
【0033】
本発明のヒトOBMをコードするcDNAを分離同定するための、プローブとして使用するマウス由来OCIF結合蛋白質であるマウスOBM cDNAは、前述の方法に従って、マウス骨芽細胞様ストローマ細胞株、ST2のcDNAライブラリーから単離することができる。又、ヒトOBMc DNAを発現することにより得られる蛋白質の特性及びその生物活性を評価するために必要なヒト破骨細胞形成抑制因子OCIFは、WO96/26217号記載の方法に従って、ヒト線維芽細胞株IMR-90の培養液から単離、あるいはそのDNAを用いて遺伝子工学的に製造することができる。ヒトOBMの特性や生物活性の評価には、遺伝子組み換えヒトOCIF、遺伝子組み換えマウスOCIF、遺伝子組み換えラットOCIF等を用いることもできる。これらの遺伝子組み換え型OCIFは、それぞれのcDNAを常法に従って発現ベクターに組み込み、CHO細胞、BHK細胞、Namalwa細胞等の動物細胞あるいは昆虫細胞などで発現させ、精製することによって得ることができる。
【0034】
目的の蛋白質をコードするヒト型のcDNAを単離する(cDNAクローニング)方法としては、▲1▼その蛋白質を精製し、その部分アミノ酸配列を決定し、それに対応する塩基配列を有するDNAをプローブとして用い、ハイブリダイゼーション法により目的のcDNAを単離する方法、▲2▼目的の蛋白質のアミノ酸配列が不明であっても発現ベクター中にcDNAライブラリーを構築し、それらを細胞中に導入し、目的蛋白質の発現の有無をスクリーニングすることにより、目的のcDNAを単離する方法(発現クローニング法)、及び▲3▼ヒト由来の目的蛋白質と同じ特性及び生物活性を有するヒト以外の哺乳動物由来の蛋白質をコードする遺伝子、cDNAがクローニングしようとするヒト由来の目的蛋白質のcDNAと高いホモロジーを有すると想定し、前者のcDNAをプローブとし、ヒト細胞あるいは組織から構築したcDNAライブラリーからハイブリダイゼーション法やpolymerase chain reaction (PCR)法により目的のヒト蛋白質をコードするcDNAを単離する方法がある。
【0035】
ヒトOBM cDNAは、前記マウスOBM cDNAと高い相同性を有するとの想定のもと、ヒトOBM産生細胞あるいは組織は後者のcDNAをプローブとして、ノーザンハイブリダイゼーション法により調べることができる。ヒトOBM cDNAは、マウスOBM cDNAから作製したマウスOBMプライマーを用い、上記のようにして同定したヒトOBM産生組織、例えばヒトリンパ節等から構築したcDNAを鋳型として、PCRによりヒトOBM cDNA断片を得て、このヒトOBMc DNA断片をプローブとし、上記の様に同定したヒトOBM産生細胞あるいは組織のcDNAライブラリーをスクリーニングすることにより得ることができる。本発明は、このようにして得られたOCIFに特異的に結合する性質を有し、また破骨細胞の分化、成熟を支持、促進する生物活性を有するヒト由来蛋白質、即ちヒトOBMをコードするDNAに関する。本発明のDNAがコードするヒトOBMは、膜貫通領域を有する膜結合型蛋白質をコードすることから、OCIFを標識し、本発明のcDNAを発現した動物細胞表面へのOCIF標識体の結合によって検出することができる。OCIFの標識法としては、前述と同様、放射性同位元素による標識や蛍光標識等、一般的な蛋白質の標識法を用いることができる。
本発明のヒトOBM cDNAにより発現される蛋白質の分子量は、ゲル濾過クロマトグラフィーやSDS-PAGE等を用いて測定される。より正確に分子量を測定するためには、SDS-PAGEを用いるのが好ましく、ヒトOBMは、還元条件下で約40,000(40,000±5,000)の分子量を有する蛋白質として特定される。
【0036】
本発明でいう比較的温和な条件でのDNAハイブリダイゼーションとは、例えば常法に従いナイロンメンブランにDNAをトランスファーし、固定化した後、ハイブリダイゼーション用バッファー中で放射線標識したプローブのDNAと40-70℃、2時間から一夜程度ハイブリダイゼーションさせ、0.5×SSC(0.075M塩化ナトリウム及び0.0075Mクエン酸ナトリウム)で45℃、10分間洗浄する条件を言う。より具体的には、常法に従い、ナイロンメンブランにハイボンドN(アマシャム社)を用い、DNAをトランスファーし、固定化した後、Rapid Hybridization Buffer(アマシャム社)中で32P標識したプローブのDNAと65℃、2時間ハイブリダイゼーションさせて、上記の0.5xSSCで45℃、10分間洗浄する条件を言う。
【0037】
破骨細胞形成の代表的なインビトロ培養系として、活性型ビタミンD3あるいはPTHの存在下でのマウス由来骨芽細胞様ストローマ細胞株、ST2とマウス脾臓細胞との共培養系がよく知られている。このインビトロ培養系での破骨細胞形成には、骨芽細胞様ストローマ細胞と脾臓細胞間の接着による相互作用及び活性型ビタミンD3やPTH等の骨吸収促進因子の存在が不可欠である。このインビトロ培養系において、骨吸収促進因子非存在下で、しかも破骨細胞形成支持能のないサル腎臓細胞株であるCOS細胞に本発明のcDNAを発現させた遺伝子組み換えCOS紬胞株は、骨芽細胞様ストローマ細胞株、ST2と同様に脾臓細胞からの破骨細胞形成支持能を獲得した。また本発明のcDNAは膜結合型蛋白質をコードすることから、この膜結合部位をコードする部分を除去することにより分泌型、可溶型として発現させることもできる。骨吸収促進因子非存在下の上記インビトロ培養系に、この分泌型ヒトOBMを添加するだけで破骨細胞の形成が起こることも確認された。
これらの結果から、本発明のcDNAによりコードされるヒトOBMは破骨細胞の分化、成熟に関与する因子として特定される。
【0038】
本発明のcDNAを発現ベクタ一に挿入してヒトOBM発現プラスミドを作製し、これを各種の細胞または菌株に導入して発現させることにより、組み換え型ヒトOBMを製造することができる。発現させる際の哺乳動物細胞の宿主としてCOS-7、CHO、Namalwa細胞などを、また細菌の宿主として大腸菌等を用いることができる。その際、cDNAの全長を用いて膜結合型蛋白質として発現させることができるし、また膜結合部位をコードする部分を除去することにより分泌型、可溶化型としても発現させることができる。このようにして製造された組み換え型ヒトOBMは、通常用いられる蛋白精製法、例えばOCIF固定化カラムを用いたアフィニティクロマトグラフィー、イオン交換クロマトグラフィーやゲル濾過クロマトグラフィー等を組み合わせることにより効率良く精製できる。このようにして得られた本発明ヒトOBMは、その活性より骨代謝異常症、例えば大理石病などの治療剤としての医薬、あるいは研究・診断用試薬として有用である。
【0039】
本発明のcDNAにコードされる蛋白質、ヒトOBMを利用することにより、▲1▼ヒトOBMの発現を調節する物質のスクリーニング、▲2▼ヒトOBMに特異的に結合し、その生物活性を阻害あるいは修飾する物質のスクリーニング、及び▲3▼ヒト破骨細胞前駆細胞に存在し、ヒトOBMの生物活性を伝達するヒト蛋白質(ヒトOBMレセプター)のスクリーニング、さらにはこのヒトOBMレセプターを利用したアンタゴニストやアゴニストの開発が可能である。上述のヒトOBMあるいはヒトOBMレセプターを用いたコンビナトリアルケミストリーにおいて、アンタゴニストあるいはアゴニストの探索に必要なペプチドライブラリーはマウスOBMを用いたスクリーニングの方法と同様の方法で作製することができ、マウスOBMの代わりにヒトOBMを用いて、ペプチドライブラリーを同様にスクリーニングすることにより、特異的かつ極めて高親和性のペプチドを得ることができる。
【0040】
さらにまた、前述のようにOBMは有用性の高い蛋白質であるが、この蛋白質の測定を行うにはOBMを特異的に認識する抗体とその抗体を用いた酵素免疫測定法の構築が必須である。しかしながら、これまでにOBMの測定に有用な抗体は得られていない。さらに、OBMあるいはsOBMの生物活性を中和する抗OBM/sOBM抗体は、OBMあるいはsOBMの作用、即ち破骨細胞形成の促進作用を抑制することが想定され、骨代謝異常症の治療薬としての開発が期待されるが、このような抗体も未だ得られていない。
【0041】
上述の状況に鑑み本発明者らは鋭意研究の結果、破骨細胞形成抑制因子(OCIF)に特異的に結合する膜結合蛋白質(OCIF結合分子;OBM)及び膜結合部位を欠損した可溶性OBM(sOBM)の両抗原を認識する抗体(抗OBM/sOBM抗体)を見出すに至った。従って本発明は、破骨細胞形成抑制因子OCIFに特異的に結合する膜結合蛋白質OBM及び膜結合部位を欠損した可溶性OBM(sOBM)の両抗原を認識する抗体(抗OBM/sOBM抗体)、その製造方法、この抗体を用いたOBM及びsOBMの測定方法、さらにこの抗体を有効成分とする骨代謝異常症予防及び/又は治療剤を提供することを課題とする。
【0042】
本発明は、破骨細胞形成抑制因子(Osteoclastogenesis inhibitory factor; OCIF)に特異的に結合する膜結合蛋白質(OCIF binding molecule; OBM)及び膜結合部位を欠損した可溶性OBM(sOBM)の両抗原を認識する抗体(抗OBM/sOBM抗体)、その製造方法、この抗体を用いたOBM及びsOBMの測定方法、さらにこの抗体を有効成分とする医薬、特に骨代謝異常症予防及び/又は治療剤に関する。
本発明の抗体は、OBM及びsOBMの生物活性である破骨細胞形成促進活性を中和する活性を有し、以下の性質を持つ抗体から構成される。
即ち、
a)マウスOBM及びマウスsOBMの両抗原を認識するポリクローナル抗体(抗マウスOBM/sOBMポリクローナル抗体)
b)ヒトOBM及びヒトsOBMの両抗原を認識するポリクローナル抗体(抗ヒトOBM/sOBMポリクローナル抗体)
c)マウスOBM及びマウスsOBMの両抗原を認識するモノクローナル抗体(抗マウスOBM/sOBMモノクローナル抗体)
d)ヒトOBM及びヒトsOBMの両抗原を認識するモノクローナル抗体(抗ヒトOBM/sOBMモノクローナル抗体)
e)マウスOBM及びマウスsOBMの両抗原に交差性を有する抗ヒトOBM/sOBMモノクローナル抗体、から構成される。
【0043】
マウスOBM及びマウスsOBMの両抗原を認識するポリクローナル抗体(以下抗マウスOBM/sOBMポリクローナル抗体と称する)及びヒトOBM及びヒトsOBMの両抗原を認識するポリクローナル抗体(以下抗ヒトOBM/sOBMポリクローナル抗体と称する)は、以下の手段によって得ることができる。免疫用抗原としての精製マウスOBMは、前述の方法に従って得ることができる。即ち、マウス骨芽細胞様ストローマ細胞株ST-2を活性ビタミンD3処理し、その細胞膜上のOBMをOCIF固定化カラム及びゲル濾過クロマトグラフィーにより精製することにより、天然型マウスOBM(native OBM)を得ることができる。又、前述のマウスOBM cDNA(配列表配列番号15)又はヒトOBM cDNA(配列表配列番号12)を常法により発現ベクターに組み込み、CHO細胞、BHK細胞、Namalwa、COS-7細胞等の動物細胞、昆虫細胞、あるいは大腸菌などで発現させて、上記と同様の方法で精製することにより、遺伝子組み換えマウスOBM(配列表配列番号1)又はヒトOBM(配列表配列番号11)を得ることができ、これを免疫用抗原として用いても良い。この時、膜結合蛋白質であるマウスOBMあるいはヒトOBMを大量かつ高度に精製するには、多大の労力を要する。一方、膜結合型蛋白質であるOBMとその膜結合部位を欠損させて得られる可溶化蛋白質である可溶性OBM(sOBM)とは、前述のように破骨細胞の分化、成熟促進活性において差異がないことが認められている。従って、マウスsOBM及びヒトsOBMは発現及びその高度精製が比較的容易であることから、これら可溶化蛋白質であるsOBMを免疫用抗原として用いても良い。
【0044】
マウスsOBM(配列表配列番号16)及びヒトsOBM(配列表配列番号17)は、マウスsOBM cDNA(配列表配列番号18)又はヒトsOBM cDNA(配列表配列番号19)の5'上流側に他の分泌蛋白質由来の既知シグナル配列をコードする塩基配列を付加し、上記と同様に遺伝子工学的手法により発現ベクターに組み込み、各種動物細胞、昆虫細胞、あるいは大腸菌などを宿主として発現し、精製することにより、それぞれ得ることができる。このようにして得られた免疫用抗原を、リン酸塩緩衝生理食塩水(PBS)に溶解し、必要に応じ同容量のフロイント完全アジュバントと混合し乳化後、動物に約1週間間隔で皮下投与し、数回免疫する。抗体価を測定し、最高の抗体価に達した時点でブースター投与し、投与10日後に全採血を行う。得られた抗血清を硫酸アンモニウム分画沈澱し、グロブリン分画を陰イオン交換クロマトグラフィーにより精製するか、あるいは抗血清をバインディングバッファー(Biorad社)で2倍希釈し、その希釈抗血清をプロテインA又はプロテインGセファロースカラムクロマトグラフィーにより精製することにより、目的とする抗マウス又は抗ヒトOBM/sOBMポリクローナル型抗体を得ることができる。
【0045】
本発明のモノクローナル抗体は、以下の方法により得ることができる。即ち、モノクローナル抗体の作製に必要な免疫用抗原としては、ポリクローナル抗体の時と同様に天然型マウスOBM(native OBM)、遺伝子組み換えマウス型又はヒトOBM、遺伝子組み換えマウス型又はヒト型sOBMを用いることができる。それぞれの抗原により哺乳動物を免疫するか、あるいはインビトロ法により免疫したリンパ球細胞を骨髄腫細胞株(ミエローマ)などと融合させ、常法によりハイブリドーマを作製する。このハイブリドーマ培養液について、高度に精製されたそれぞれの抗原を用いて、ソリッドフェーズELISAによりそれぞれの抗原を認識する抗体生産ハイブリドーマを選択する。得られたハイブリドーマをクローニングし、樹立した安定な抗体産生ハイブリドーマをそれぞれ培養することにより、目的とする抗体が得られる。ハイブリドーマの作製に当たっては、哺乳動物を使用する場合、マウスやラットなどの小動物を使用するのが一般的である。免疫は、抗原を適当な溶媒、例えば生理食塩水などで適当な濃度に希釈し、この溶液を静脈内や腹腔内に投与し、これに必要に応じてフロイント完全アジュバントを併用投与し、動物に1〜2週間間隔で3〜4回程度投与する方法が一般的である。このようにして免疫された動物を最終免疫後3日目に解剖し、摘出した脾臓から得られた脾臓細胞を免疫細胞として使用する。免疫細胞と細胞融合するマウス由来のミエローマとしては、例えばp3/x63-Ag8、p3-U1、NS-1、MPC-11、SP-2/0、FO、P3x63 Ag8. 653及びS194などが挙げられる。又、ラット由来の細胞としては、R-210などの細胞株が挙げられる。ヒト型の抗体を生産する場合には、ヒトBリンパ球をインビトロで免疫し、ヒトミエローマ細胞やEBウイルスにより形質転換した細胞株と細胞融合させることにより、ヒト型の抗体を生産することができる。免疫された細胞とミエローマ細胞株との融合は公知の方法、例えばKoehlerとMilsteinらの方法(Koehler et al.: Nature 256, 495-497, 1975)が一般的に使用されるが、電気パルスを利用した電気パルス法などでも良い。免疫リンパ球とミエローマ細胞株は、通常行われている細胞数の比率に混合し、一般に使用される牛胎児血清(FCS)不含細胞培養用培地にポリエチレングリコールを添加して融合処理を行い、FCS添加HAT選択培地で培養を行い融合細胞(ハイブリドーマ)を選別する。
【0046】
次に、抗体を生産するハイブリドーマをELISA、プラーク法、オクタロニー法、凝集法など通常用いられる抗体の検出方法により選択し、安定なハイブリドーマを樹立する。このようにして樹立されたハイブリドーマは、通常の培養方法により継代培養可能であり、必要に応じて凍結保存できる。ハイプリドーマは常法により培養して、その培養液、または哺乳動物の腹腔内に移植することにより、腹水から抗体を回収することができる。培養液あるいは腹水中の抗体は、塩析法、イオン交換及びゲル濾過クロマトグラフィー、プロテインAまたはプロテインGアフィニティークロマトグラフィーなど通常用いられる方法により精製することができる。このような方法により、抗原としてsOBMを用いて得られたモノクローナル抗体のほとんど全ては、sOBMだけでなくOBMをも特異的に認識できる抗体(抗OBM/sOBMモノクローナル抗体と称する)である。これらの抗体は、OBM及びsOBMのそれぞれの測定に利用することができる。これらの抗体は放射性アイソトープや酵素ラベルすることにより、ラジオイムノアッセイ(RIA)やエンザイムイムノアッセイ(EIA)として知られている測定系に用いることにより、OBM量及びsOBM量を測定することができる。これらの測定系を用いることにより、血液、尿などの生体試料中のsOBM量を容易にかつ高感度で測定することができる。また、これらの抗体を用いることにより、組織や細胞表面に結合したOBM量をバインディングアッセイ等により容易にかつ高感度で測定することかできる。
【0047】
得られた抗体をヒトに対する医薬として用いる場合、抗原性の問題からヒト型抗ヒトOBM/sOBM抗体を作製することが望ましい。ヒト型抗ヒトOBM/sOBM抗体の作製は、次に述べるような手法により得ることができる。即ち、▲1▼ヒト末梢血あるいは脾臓から採取したヒトリンパ球をin vitroでIL-4存在下、抗原であるヒトOBMあるいはヒトsOBMで感作し、感作したヒトリンパ球をマウスとヒトとのヘテロハイブリドーマであるK6H6/B5(ATCC CRL1823)と細胞融合させることにより、目的の抗体産生ハイブリドーマをスクリーニングする。得られた抗体産生ハイブリドーマが生産する抗体は、ヒト型抗ヒトOBM/sOBMモノクローナル抗体である。これらの抗体の中からヒトOBM/sOBMの活性を中和する抗体を選別する。しかしながら、このようにヒトリンパ球をin vitroで感作する方法では、一般的に抗原に対して高親和性の抗体を得るのは困難である。
【0048】
従って、ヒトOBMおよびsOBMに高親和性のモノクローナル抗体を得るには、上記のようにして得られた低親和性ヒト型抗ヒトOBM/sOBMモノクローナル抗体を高親和化する必要がある。それには、上記のようにして得られ、中和抗体であるものの低親和性であるヒト型抗ヒトOBM/sOBMモノクローナル抗体のCDR領域(特にCDR-3)にランダム変異を導入し、これをファージで発現させてヒトOBM/sOBMを固相化したプレートを用いてファージディスプレイ法により、抗原であるヒトOBM/sOBMに強力に結合するファージを選択し、そのファージを大腸菌で増やし、その塩基配列から高親和性を有するCDRのアミノ酸配列を決定すればよい。このようにして得られたヒト型抗ヒトOBM/sOBMモノクローナル抗体をコードする遺伝子を一般的に使用されている哺乳動物細胞用発現ベクターに組み込んで、発現させることによりヒト型抗ヒトOBM/sOBMモノクローナル抗体が得られる。これらの中から、ヒトOBM/sOBMの生物活性を中和し、かつ高親和性である目的のヒト型抗ヒトOBM/sOBMモノクローナル抗体を選別することができる。また、▲2▼Balb/cマウスを用いて、本発明と同じように常法(Koehler et al.:Nature 256, 495497, 1975)によりマウス型抗ヒトOBM/sOBMモノクローナル抗体を作製し、ヒトOBM/sOBMの生物活性を中和し、かつ高親和性を有するモノクローナル抗体を選択する。この高親和性マウス型抗ヒトOBM/sOBMモノクローナル抗体のCDR‐領域(CDR-1,2および3)をヒトIgGのCDR領域に移植するCDR-grafting(Winter and Milstein: Nature 349, 293-299, 1991)の手法を駆使することによりヒト型化が可能である。上記に加え、さらに▲3▼ヒト末梢血リンパ球をSevere combined immune deficiency (SCID)マウスに移植し、この移植されたSCIDマウスはヒト型抗体を生産する(Mosier D.E.et al.: Nature 335, 256-259, 1988; Duchosal M.A. et al.: Nature 355, 258-262, 1992)ので、抗原としてヒトOBMあるいはsOBMを抗原として感作し、スクリーニングすることにより、ヒトOBM/sOBMに特異的なヒト型モノクローナル抗体を生産するリンパ球をそのマウスから採取することかできる。得られたリンパ球を前述のヒト型抗体の作製法▲1▼と同様に、マウスとヒトとのヘテロハイブリドーマであるK6H6/B5(ATCC CRL1823)と細胞融合させ、得られたハイブリドーマをスクリーニングすることにより、目的のヒト型モノクローナル抗体を産生するハイブリドーマを得ることができる。このようにして得られたハイブリドーマを培養することにより、目的のヒト型モノクローナル抗体を大量に製造でき、上述の方法と同様に精製することにより精製品を得ることができる。又、目的のヒト型モノクローナル抗体を産生するハイブリドーマからcDNAライブラリーを構築し、目的のヒト型モノクローナル抗体をコードする遺伝子(cDNA)をクローニングし、この遺伝子を遺伝子工学的手法により適当な発現ベクターに組み込み、各種動物細胞、昆虫細胞、あるいは大腸菌などを宿主として発現させることにより、遺伝子組み換えヒト型モノクローナル抗体を大量に製造することができる。
【0049】
得られた培養液から上述と同様の方法により精製することにより、精製されたヒト型モノクローナル抗体を大量に得ることができる。
さらに、上述の方法で得られた抗OBM/sOBMモノクローナル抗体の中から、OBM/sOBMの生物活性を中和する抗体を得ることかできる。これらOBM/sOBMの生物活性を中和する抗体は、生体内でのOBM/sOBMの生物作用、即ち破骨細胞形成促進作用をブロックすることから、医薬として、特に骨代謝異常症の予防及び/又は治療剤として期待される。抗OBM/sOBM抗体によるOBMあるいはsOBMの生物活性の中和活性は、in vitroでの破骨細胞形成系における破骨細胞形成の抑制活性で測定することができる。in vitroアッセイ系として、以下の3つの方法がある。即ち、破骨細胞形成(osteoclastogenesis)のin vitro培養系として、▲1▼活性型ビタミンD3とデキサメサゾンの存在下、マウス骨芽細胞様ストローマ細胞株、ST2細胞とマウス脾臓細胞との共培養系、▲2▼サル腎臓細胞株、COS-7細胞上にOBMを発現させ、ホルムアルデヒドにより固定化し、M-CSF存在下、その細胞上でマウス脾臓細胞を培養する共培養系、▲3▼遺伝子組み換えsOBM及びM-CSF存在下でマウス脾臓細胞を培養する系などが挙げられる。これら培養系に種々の濃度で抗OBM/sOBM抗体を添加し、破骨細胞形成に及ぼす影響を調べることにより、抗OBM/sOBM抗体による破骨細胞形成抑制活性を測定することができる。又、in vivoでの実験動物を利用した骨吸収抑制活性により抗OBM/sOBM抗体の破骨細胞形成抑制活性を評価することができる。即ち、破骨細胞形成が亢進している動物モデルとして、卵巣摘出モデルがある。この種の実験動物に抗OBM/sOBM抗体を投与し、骨吸収抑制活性(骨密度の増強活性)を測定することにより、抗OBM/sOBM抗体による破骨細胞形成抑制活性を測定することができる。
【0050】
このようにして得られたOBM/sOBMの生物活性を中和する抗体は、医薬として、特に骨代謝異常症の予防及び/又は治療を目的とした医薬組成物として、あるいはこのような疾患の免疫学的診断のための抗体として有用である。本発明抗体は、製剤化して経口的あるいは非経口的に投与することができる。本発明抗体を含む製剤は、OBM及び/又はsOBMを認識する抗体を有効成分として含有する医薬組成物として、ヒトあるいは動物に対し安全に投与されるものである。医薬組成物の形態としては、点滴を含む注射剤、坐剤、経鼻剤、舌下剤、経皮吸収剤などが挙げられる。モノクローナル抗体は高分子蛋白質であることから、バイアル瓶などのガラス容器や注射筒などへの吸着が著しい上に不安定であり、種々の物理化学的因子、例えば熱、pHおよび湿度等により容易に失活する。従って、安定な形で製剤化するために、安定化剤、pH調整剤、緩衝剤、可溶化剤、界面活性剤などを添加する。
【0051】
安定化剤としては、グリシン、アラニン等のアミノ酸類、デキストラン40およびマンノース等の糖類、ソルビトール、マンニトール、キシリトール等の糖アルコール等が挙げられ、またこれらの二種以上を併用してもよい。これらの安定化剤の添加量は、抗体の重量に対して0.01〜100倍、特に0.1〜10倍添加するのが好ましい。これら安定化剤を加えることにより、液状製剤あるいは凍結乾燥製剤の保存安定性を向上することができる。緩衝剤としては、例えばリン酸バッファー、クエン酸バッファー等が挙げられる。緩衝剤は、液状製剤あるいは凍結乾燥製剤の再溶解後の水溶液のpHを調整し、抗体の安定性、溶解性に寄与する。緩衝剤の添加量としては、例えば液状製剤あるいは凍結乾燥製剤を再溶解した後の水量に対し、1〜10mMとするのが好ましい。界面活性剤としては、好ましくはポリソルベート20、プルロニックF-68、ポリエチレングリコール等、特に好ましくはポリソルベート80が挙げられ、またこれらの2種以上を併用してもよい。抗体のような高分子蛋白質は容器の材質であるガラスや樹脂などに吸着しやすい。従って、界面活性剤を添加することによって、液状製剤あるいは凍結乾燥製剤の再溶解後の抗体の、容器への吸着を防止することができる。界面活性剤の添加量としは、液状製剤あるいは凍結乾燥製剤の再溶解後の水重量に対し0.001〜1.0%添加することが好ましい。以上のような安定化剤、緩衝剤、あるいは吸着防止剤を加えて本発明抗体の製剤を調製することができるが、特に医療用又は動物薬用注射剤として用いる場合は、浸透圧比として許容される浸透圧比は1〜2が好ましい。浸透圧比は、製剤化に際して塩化ナトリウムの増減により調製することができる。製剤中の抗体含量は、適用疾患、適用投与経路などに応じて適宜調整することができ、ヒトに対するヒト型化抗体の投与量は、抗体のヒトOBM/sOBMに対する親和性、即ちヒトOBM/sOBMに対する解離定数(Kd値)に依存し、親和性が高い(Kd値が低い)ほど、ヒトヘの投与量を少なく薬効を発現することができる。又、ヒト型抗体はヒト血中での半減期が約20日間と長いことから、例えばヒトに対して投与する際には、約0.1〜100mg/kgを1〜30日間に1回以上投与すれば良い。
【0052】
【実施例】
以下の実施例により本発明をより詳紬に説明するが、これらは単に例示するのみであり、本発明はこれらによって何ら限定されるものではない。
【実施例1】
本発明蛋白質の製造
(1)ST2 細胞の大量培養
マウス骨芽細胞様ストローマ細胞株ST2(RIKEN CELL BANK, RCBO224)は、10%牛胎児血清を含むα‐MEM培地を用いて培養した。付着細胞用225cm2Tフラスコでコンフルエントになるまで培養したST2細胞を、トリプシン処理してTフラスコから剥がし洗浄した後、5枚の225cm2Tフラスコに移し、10-8M活性型ビタミンD3(Calcitriol)、10-7Mデキサメサゾン、及び10%牛胎児血清を添加したα‐MEM培地各々60mlを加えてCO2インキュベーター中で7-10日間培養した。培養したST2細胞はセルスクレイパーを用いて回収し、使用するまで‐80℃で保存した。
【0053】
(2) 膜画分の調製と膜結合蛋白質の可溶化
225cm2のTフラスコ80枚を用いて培養した実施例1‐(1)記載のST2細胞(容量約12ml)に、プロテアーゼ阻害剤(2mM APMSFP, 2mM EDTA, 2mM o-phenanthroline, 1mM leupeptin, 1μg/ml pepstatin A及び100 units/ml aprotinin)を含む10mMトリス-塩酸緩衝液(pH7.2)を3倍容量(36ml)加えた。ボルテックスミキサーを用いて30秒間この細胞を激しく攪拌した後、氷中で10分間放置した。
ホモジナイザー(DOUNCE TISSUE GRINDER, A syrindge, WHEATON SCIENTIFIC社)を用い、細胞を破砕した。この細胞破砕液に、上記のプロテアーゼ阻害剤、0.5Mシュクロース、0.1M塩化カリウム、10mM塩化マグネシウム、及び2mM塩化カルシウムを加えた10mMトリスー塩酸緩衝液(pH7.2)の等量(48ml)を加え、攪拌した後、4℃、600×gで10分間遠心した。この遠心分離により、細胞核と未破砕の細胞を沈殿画分として分離した。遠心分離で得た上清を4℃、150,000×gで90分間遠心し、沈殿画分としてST2細胞の膜画分を得た。この膜画分に、上記のプロテアーゼ阻害剤、15mM塩化ナトリウム、及び0.1Mシュクロースを含む10mMトリス-塩酸緩衝液(pH7.2)8mlを加えた後、20% CHAPS (3‐[(3-cholamidopropyl)‐dimethylammonio]‐1-propanesulfonate、Sigma社)200μlを加え、4℃で2時間攪拌した。この液を4℃、150,000×gで60分間遠心し、その上清を可溶化膜画分として得た。
【0054】
【実施例2】
本発明蛋白質の精製
(1)OCIF 固定化アフィニティーカラムの調製
HiTrap NHS-activatedカラム(1ml,ファルマシア社)内のイソプロパノールを1mM塩酸で置換した後、WO96/26217号公報記載の方法で調製した遺伝子組み換え型OCIF 13.0mgを含む0.2M NaHCO3/0.5MNaCl(pH 8.3)溶液1mlをシリンジ(5ml,テルモ社)を用いカラムに添加した。室温で30分間カップリング反応させた後、過剰な活性基を不活性化する為、0.5Mエタノールアミン/0.5M NaCl(pH 8.3)と0.1M酢酸/0.5M NaCl(pH 4.0)を3mlずつ交互に3回流し、再度0.5Mエタノールアミン/0.5M NaCl(pH 8.3)に置き換え室温で1時間放置した。その後、0.5Mエタノールアミン/0.5M NaCl(pH 8.3)と0.lM 酢酸/0.5M NaCl(pH 4.0)で2度洗浄し、50mM Tris/1M NaCl/0.1%CHAPS緩衝液(pH 7.5)に置換した。
【0055】
(2)OCIF 固定化アフィニティーカラムによる本発明蛋白質の精製
OCIF結合蛋白質の精製は、特に断らない限り4℃で行った。前述のOCIF固定化アフィニティーカラムを、実施例1‐(2)記載のプロテアーゼ阻害剤、0.15M塩化ナトリウム、及び0.5% CHAPSを加えた10mMトリス-塩酸緩衝液(pH 7.2)で平衡化した。このカラムに、実施例1‐(2)項記載の可溶化膜画分約8mlを流速0.01ml/分で負荷した。このカラムに、上記のプロテアーゼ阻害剤、0.15M塩化ナトリウム、及び0.5%CHAPSを加えた10mMトリス-塩酸緩衝液(pH 7.2)を流速0.5ml/分で100分間流してカラムを洗浄した。次に、プロテアーゼ阻害剤、0.2M塩化ナトリウム、及び0.5%CHAPSを加えた0.1Mグリシン-塩酸緩衝液(pH 3.3)を流速0.1ml/分で50分間流し、カラムに吸着した蛋白質を溶出した。同様に、プロテアーゼ阻害剤、0.2M塩化ナトリウム、及ひ0.5%CHAPSを加えた0.1Mクエン酸ナトリウム緩衝液(pH 2.0)を流速0.1ml/分で50分間流し、カラムに吸着した蛋白質を溶出した。溶出液は0.5ml/フラクションにて分取した。分取した画分には、2Mトリス溶液を加え、直ちに中和した。各緩衝液で溶出したフラクション(溶出液量1.0-5.0ml)の各々をセントリコン-10(Centricon‐10, Amicon, USA)を用いて50から100μlに濃縮した。濃縮した各フラクションの一部を分取し、それぞれにOCIFを添加後、OCIFポリクローナル抗体で免疫沈殿させた。その沈殿画分をSDS化して後、SDS-PAGEにかけてOCIFに特異的な結合能を有する蛋白質のバンドが出現するフラクション(Fr. No.3-10)を本発明蛋白質画分とした。
【0056】
(3) ゲル濾過による本発明蛋白質の精製
実施例2‐(2)記載の方法で精製し、濃縮したOCIF結合蛋白質(0.lMグリシン-塩酸緩衝液、pH 3.3及び0.lMクエン酸ナトリウム緩衝液、pH 2.0溶出画分)を10mMTris-HCl, 0.5M NaCl, 0.5% CHAPS(pH 7.0)で平衡化したSuperose 12HR10/30カラム(ファルマシア杜、1.0x30cm)にかけ、平衡化緩衝液を移動相として用い流速0.5 ml/minで展開させ、0.5 mlずつの画分を集めた。上記と同様にして本発明蛋白質画分(Fr. No. 27-32)を同定し、各々のフラクションをCentricon-10 (Amicon)で濃縮した。
【0057】
(4) 逆相高速液体クロマトグラフィーによる精製
前述のゲル濾過で精製したOCIF結合蛋白質を0.1% トリフルオロ酢酸(TFA),30%アセトニトリルで平衡化したC4カラム(2.1x250mm、Vydac, USA)に負荷した。最初の50分間でアセトニトリル濃度を30%から55%に、次の10分間でアセトニトリル濃度を80%にする勾配、流速0.2ml/minで溶出を行い、溶出された蛋白質のピークを215nmで検出した。溶出された各ピークの蛋白質を分取し本発明蛋白質のピークを同定することにより、高度に精製された本発明蛋白質を得た。
【0058】
【実施例3】
精製された本発明蛋白質の SDS-PAGE
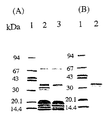
まず、活性型ビタミンD3存在下あるいは非存在下で培養したST2細胞から調製した膜可溶化画分を上記のようにOCIF固定化アフィニティーカラムで精製し、その精製標品をSDS-PAGEにかけた。図1(A)に示したように、活性型ビタミンD3存在下で培養したST2細胞からの精製標品のみに、約30,000-40,000のメジャーな蛋白質バンドが検出され、OCIF固定化アフィニティーカラムにより、OCIFに特異的に結合する蛋白質、即ち本発明蛋白質が選択的に濃縮、精製されることが明らかになった。しかしながら、本発明蛋白質以外にOCIF固定化カラムの担体やスペサーなどに非特異的に結合した数種の蛋白質バンドが両精製サンプル中に共通して検出された。これらの本発明蛋白質以外の蛋白質を上記のようにゲル濾過及びC4逆相クロマトグラフィーで除去し、得られた高度精製本発明蛋白質のSDS-PAGEを図1(B)に示した。高度精製本発明蛋白質は電気泳動的に均一であり、その分子量は約30,000-40,000であった。
【0059】
【実施例4】
OCIF の骨芽細胞への結合試験
(1) 125 I 標識 OCIF の調製
OCIFはヨードジェン(Iodogen)法により125標識した。即ち、2.5mg/ml Iodogen‐クロロホルム溶液20μlを1.5mlエッペンドルフチューブに移し、40℃でクロロホルムを蒸発させて、ヨードジェンコートしたチューブを調製した。このチューブを0.5Mリン酸ナトリウム緩衝液(Na-Pi,pH 7.0)400μlで3回洗浄した後、0.5M Na-Pi, pH 7.0, 5μlを加えた。このチューブにNa-125I溶液(Amersham社、NEZ-033H20)1.3μl(18.5 MBq)を加えた後、直ちに1mg/ml rOCIF溶液(モノマー型あるいはダイマー型)10μlを加え、ボルテックスミキサーで攪拌した後、室温で30秒間放置した。この溶液を、予め10mg/mlヨウ化カリウム、0.5M Na-Pi, pH 7.0溶液80μlと5%牛血清アルブミンのリン酸塩緩衝生理食塩水5μlを添加しておいたチューブに移し攪拌した。この溶液を0.25%牛血清アルブミンのリン酸塩緩衝生理食塩水溶液で平衡化しておいたスピンカラム(1ml, G-25 fine, ファルマシア社)に負荷し、2,000rpmで5分間遠心した。カラムから溶出された画分に0.25%牛血清アルブミンのリン酸塩緩衝生理食塩水溶液400μlを加え攪拌した後、2μlを取り、その放射能をガンマーカウンターで測定した。このようにして調製した125I標識OCIF溶液の放射化学純度は10%TCAにより沈殿する画分の放射能を測定することにより求めた。又、125I標識OCIF溶液のOCIF生物活性は、WO96/26217号公報記載の方法に従い測定した。
又、125I標識OCIF濃度は以下のようにELISAにより測定した。
【0060】
(2) 125 I標識 OCIF 濃度の ELISA による測定
WO96/26217号公報記載の抗OCIFウサギポリクローナル抗体を2μg/mlになるように溶解させた50mM NaHCO3(pH 9.6)100μlずつを96ウェルイムノプレート(MaxiSorpTM、Nunc社)の各ウェルに加えて4℃で一夜放置した。この液を吸い取った後、ブロックエース(雪印乳業)/リン酸塩緩衝生理食塩水溶液(25/75)300μlずつを各ウェルに加え、室温で2時間放置した。この液を吸い取った後、各ウェルを0.01 %ポリソルベート80を含むリン酸塩緩衝生理食塩水(P-PBS)で3回洗浄し、次いで125I標識OCIFサンプルあるいはOCIF標準品を添加したブロックエース/リン酸塩緩衝生理食塩水溶液(25/75)300μlずつを各ウェルに加え、室温で2時間放置した。この液を吸い取った後、P-PBS 200μlで各ウェルを6回洗浄した。パーオキシダーゼ標識したウサギ抗OCIFポリクローナル抗体を含むブロックエース(雪印乳業)/リン酸塩緩衝生理食塩水溶液(25/75)100μlずつを各ウェルに加え、室温で2時間放置した。この液を吸い取った後、P-PBS 200μlで各ウェルを6回洗浄した。TMB溶液(TMB Soluble Reagent, High Sensitivity, Scytek社)100μlずつを各ウェルに加え、室温で2-3分放置した後、停止液(Stopping Reagent, Scytek社)100μlずつを各ウェルに加えた。各ウェルの490nmにおける吸光度をマイクロプレートリーダーで測定した。OCIF標準品を用いて作製した検量線より、125I標識OCIFの濃度を求めた。
【0061】
(3)OCIF の骨芽細胞あるいは脾臓細胞への結合試験
マウス骨芽細胞様ストローマ細胞株ST2あるいは脾臓細胞をそれぞれ4×104 cell/ml及び2x106 cell/mlの濃度になるように、10-8M活性型ビタミンD3(Calcitriol)及び10-7Mデキサメサゾン添加あるいは無添加の10%牛胎児血清(FBS)を含むα‐MEM培地に懸濁させ、この培地1mlずつを24ウェルマイクロプレートに播種した。細胞をCO2インキュベーター中で4日間培養しα‐MEM培地で洗浄した後、上記の125I標識OCIF(モノマー型又はダイマー型)20ng/mlを加えた結合試験用培地(0.2%牛血清アルブミン、20mM Hepes緩衝液、0.2% NaN3を加えたα‐MEM培地)200μlを各ウェルに加えた。又、別のウェルには、8μg/ml rOCIF(400倍濃度)を更に添加した結合試験用培地を200μg/ml加え、非特異的吸着量測定に供した。CO2インキュベーター中で1時間培養した後、1 mlのリン酸塩緩衝生理食塩水 1 mlで3回洗浄した。この際、脾臓細胞は浮遊細胞であるため、24ウェルプレートを遠心分離しながら、各ウェルの細胞を洗浄した。洗浄後、0.1N NaOH溶液500μlを各ウェルに加え、室温に10分間放置することにより細胞を溶解させ、細胞に結合したRI量をガンマーカウンターで測定した。
125I標識OCIFは、培養した脾臓細胞には結合せず、図2に示したように、活性型ビタミンD3で培養した骨芽細胞様ストローマ細胞にのみ特異的に結合した。
このことから、本発明蛋白質は活性型ビタミンD3とデキサメサゾンにより骨芽細胞様ストローマ細胞上に誘導される膜結合蛋白質であることが明らかになった。
【0062】
【実施例5】
本発明蛋白質の生物活性
(1) 骨芽細胞様ストローマ細胞の破骨細胞形成支持能
骨芽細胞の破骨細胞形成支持能は、形成される破骨細胞の酒石酸耐性酸性フォスファターゼ活性(TRAP活性)を測定することによって評価した。即ち、ddyマウス(8-12週齢)の脾臓細胞(2×105 cells/100μl/well)とマウス骨芽細胞様ストローマ細胞ST2(5×103 cells/100μl/well)を10-8M活性型ビタミンD3、10-7Mデキサメサゾン、及ひ10%牛胎仔血清を加えたα‐MEM培地に懸濁させ、96ウェルプレートに播種した。CO2インキュベーター中で1週間培養した後、各ウェルをリン酸塩緩衝生理食塩水で洗浄し、更に100μlのエタノール/アセトン(1:1)を加えて、室温で1分間固定した。固定後、5.mM p-nitrophenol phosphateと10mM酒石酸ナトリウムを含む50mMクエン酸緩衝液、pH 4.5、100μlを各ウェルに加え、室温で15分間反応させた。反応後、0.1N NaOH溶液を各ウェルに添加し、405 nmの吸光度をマイクロプレートリーダーで測定した。RIKEN CELL BANKから購入した後の継代数約10代のST2細胞と継代数約40代のST2細胞の破骨細胞形成支持能を試験した結果を図3に示す。この結果から、継代数の多いST2細胞は破骨細胞形成支持能が高いことか明らかになった。
【0063】
(2) 活性型ビタミン D 3 及びデキサメサゾン存在下での培養における骨芽細胞様ストローマ細胞膜上の本発明蛋白質発現の経時変化と共培養系における破骨細胞形 成の経時変化
実施例4‐(3)と同様に、骨芽細胞様ストローマ細胞株ST2を活性型ビタミンD3及びデキサメサゾン存在下で7日間培養した。OCIF結合試験は、実験例4‐(1)に記載した125I標識OCIF(モノマータイプ)を用いて行った。非特異的結合は400倍濃度の非標識OCIFを用いて125I標識OCIFのST2細胞への結合を競合させることにより測定した。その結果、活性型ビタミンD3とデキサメサゾンにより、培養日数の経過と共に125I標識OCIFの特異的結合量が上昇した。即ち、図4及び図5に示したように本発明蛋白質は活性型ビタミンD3によりST2細胞の表面に培養日数と共に発現し、その発現は培養4日目に最大に達した。一方、マウス脾臓細胞とST2細胞の共培養を活性型ビタミンD3存在下で行うことにより、破骨細胞様の細胞が形成される。破骨細胞のマーカー酵素であるTRAP陽性の単核前破骨細胞様細胞は培養3、4日目には形成され、さらに分化、成熟したTRAP陽性の多核細胞は培養5、6日目に形成される。本発明蛋白質の発現の経時変化と破骨細胞形成のそれとは良く相関していることが明らかになった。
【0064】
(3) 共培養期間において、種々の培養期間のみ OCIF 処理した場合の破骨細胞形成抑制効果
本発明蛋白質が破骨細胞形成に関与する因子であることを更に明確にするために、前述の実施例5‐(2)における6日間の共培養期間において、種々の培養期間(各2日間、但し5日目のみ1日間)の細胞を、100ng/mlのOCIFで処理した。
その結果、図6に示したように、ST2細胞上に最も本発明蛋白質が発現される培養48-96時間目でのOCIF処理が最も効果的に破骨細胞の形成を抑制した。即ち、OCIFは本発明蛋白質を介してST2細胞に結合することにより、破骨細胞形成を抑制することが明らかになった。
以上の結果から、本発明蛋白質は活性型ビタミンD3とデキサメサゾンにより骨芽細胞様ストローマ細胞膜上に誘導され、破骨細胞の分化、成熟を支持又は促進する因子としての生物活性(作用)を有することが明らかになった。
【0065】
【実施例6】
125 I標識 OCIF と本発明蛋白質とのクロスリンキング試験
本発明蛋白質の存在をさらに確認するため、125I標識OCIFと本発明蛋白質とのクロスリンキングを行った。実施例4‐(3)と同様にマウス骨芽細胞様細胞株ST2を活性型ビタミンD3及びデキサメサゾン存在下及び非存在下で4日間培養した。
細胞をリン酸塩緩衝生理食塩水1mlで洗浄した後、上記の125I標識OCIF(モノマー型)25ng/ml、又はWO96/26217号公報にある配列表配列番号76記載の蛋白質を動物細胞で発現させ上記の方法で標識することにより得られた125I標識OCIF‐CDDl 40ng/mlを加えた結合試験用培地(0.2% 牛血清アルブミン、20mM Hepes緩衝液、0.2% NaN3、及び100μg/mlヘパリンを加えたα‐MEM培地)200μlを添加した。又、別のウェルには、400倍濃度のOCIFを更に添加した結合試験用培地を添加し、非特異的吸着試験に供した。CO2インキュベーター中で1時間培養した後、100μg/mlヘパリンを加えたリン酸塩緩衝生理食塩水1mlで3回洗浄した。これらのウェルに100μg/mlのクロスリンキング剤、DSS(Disuccinimidyl suberate、Pierce社)を溶解させたリン酸塩緩衝生理食塩水500μlを加え、0℃で10分間反応させた。
これらのウェルの細胞を0℃に冷却したリン酸塩緩衝生理食塩水1mlで2回洗った後、1% Triton X-100、2mM PMSF(phenylmethylsulfonyl fluoride)、10μM pepstatin、10μM leupeptin、10μM antipain、及び2mM EDTAを加えた20mM Hepes緩衝液100μlを各ウェルに加え室温に30分間放置して細胞を溶解させた。これらのサンプル15μlを常法により非還元条件下でSDS化した後、SDS‐ポリアクリルアミド電気泳動用ゲル(4-20%ポリアクリルアミドグラジェント、第一化学社)を用いて泳動させた。泳動後、ゲルを乾燥させ、BioMax MSフィルム(Kodak社)とBioMax MS増感スクリーン(Kodak社)を用いて‐80℃で24時間感光させた。感光させたフィルムを常法により現像した。
125I標識OCIF(モノマー型、60kDa)を用いた場合には分子量約90,000-10,000のクロスリンクされた蛋白質が検出された。又、125I標識OCIF-CDDl(31kDa)を用いた場合、図7に示すように約70-80kDa(平均78kDa)のクロスリンクされた蛋白質が検出された。
【0066】
【実施例7】
ST 細胞上に発現する本発明蛋白質の Scatchard Plot による解析
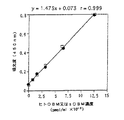
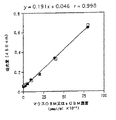
上記の125I標識OCIF(モノマー型)を1,000pMになるように加えた結合試験用培地(0.2% 牛血清アルブミン、20mM Hepes緩衝液、0.2% NaN3を加えたα‐MEM培地)を調製し、結合試験用培地を用い、1/2希釈倍率で段階的に希釈した。又、非特異的な結合を求めるため、これらの溶液に更に400倍濃度の未標識モノマー型OCIFを添加した溶液を調製した。調製したこれらの溶液200μlを10-8M活性型ビタミンD3(Calcitriol)と10-7Mデキサメサゾン存在下で4日間培養した上記のST2細胞(約10継代目)のウェルに加え、実施例4‐(3)と同様の方法で125I標識OCIFの結合を試験した。得られた結果を常法に従ってScatchard Plotし、OCIFとOCIF結合蛋白質の解離定数とST2細胞当たりのOCIF結合蛋白質の個数(サイト数)を求めた。
その結果、OCIFと本発明蛋白質の解離定数は280pM、ST2細胞当たりのOCIF結合蛋白質のサイト数は約33,000個/細胞であった。又、実施例5‐(1)で記載したように、約40継代培養したST2細胞は破骨細胞形成支持能が10継代培養したST2細胞よりも高かったことから、前者のST2細胞上に発現した本発明蛋白質のサイト数を測定したところ、サイト数が58,000個/細胞であり、明らかに10継代培養したST2細胞よりも多く、本発明蛋白質の発現量の多寡がST2細胞による破骨細胞形成支持能の強弱に繋がっていることが明らかになった。このことは、本発明蛋白質は破骨細胞の分化、成熟を支持又は促進する因子であることを示す。
【0067】
【実施例8】
OBMcDNA のクローニング
(1) マウス ST2 細胞からの RNA の抽出
マウス骨芽細胞様ストローマ細胞株ST2(RIKEN CELL BANK, RCBO224)は、10% 牛胎児血清を含むα‐MEM培地(ギブコBRL杜)を用いて培養した。付着細胞用225cm2T‐フラスコでコンフルエントになるまで培養したST2細胞を、トリプシン処理してT‐フラスコから剥がし洗浄した後、5枚の225cm2T‐フラスコに移し、10-8M活性化ビタミンD3(Calcitriol、和光純薬杜)、lO-7Mデキサメサゾン、及び10%牛胎児血清を添加したα-MEM培地を各々60mlずつ加えてCO2インキュベーター中で5日間培養した。培養したST2細胞からISOGEN(和光純薬社)を用いて総RNAを抽出した。総RNA約600μgからOligo(dT)‐celluloseカラム(5'‐3'Prime社)を用いてポリA+RNAを調製した。約8μgのポリA+RNAを得た。
【0068】
(2) 発現ライブラリーの構築
実施例8-(1)で得られたポリA+RNA 2μgからGreat Lengths cDNA Synthesis kit(Clontech社)を用い、その説明書に従い二本鎖cDNAを合成した。即ち、ポリ+RNA 2μgとOligo(dT)25(dN)プライマーを混合し蒸留水を加え最終容量を6.25μlとし、70℃で3分保温後、氷中で2分冷却した。この溶液に蒸留水2.2μl、5X First-strand buffer 2.5μl、100mM DTT (ジチオスレイトール)0.25μl、PRIME RNase Inhibitor (1U/ml) (5'‐3'Prime社) 0.5μl、5倍希釈した[α‐32P] dCTP(アマシャム杜、3000 Ci/mmol、2μCi/μl) 0.5μl、dNTP(各20mM) 0.65μl、MMLV (RNaseH‐)逆転写酵素1.25μl(250ユニット)を加え、42℃で90分保温した。さらに、蒸留水を62.25μl、5X second-strand buffer 20μl、dNTP (各20mM) 0.75μl、Second-strand enzyme cocktail 5μlを添加し、16℃で2時間保温した。この反応液にT4DNA polymerase 7.5ユニットを加え、16℃でさらに30分保温後0.2M EDTA 5μl添加して反応を停止し、フェノール・クロロフォルム処理後、エタノール沈殿を行った。この二本鎖cDNAの末端にEcoRI-Sal I-NotIリンカー(Clontech社)を付加し末端をリン酸化した。サイズフラクショネーション用カラムにより500bp以上のcDNAを分離し、エタノール沈殿を行った。沈殿したDNAを水に溶解し、あらかじめ制限酵素EcoRI(宝酒造社)切断及びCIAP(ウシ小腸アルカリフォスファターゼ、宝酒造社)処理して調製したpcDL-SR α296 (Molecular and Cellular Biology, Vol.8, pp466-472、1988)に挿入した。
【0069】
(3) OCIF との結合を指標とした発現ライブラリーのスクリーニング
実施例8-(2)で得られたDNAを用い大腸菌、XL2 BlueMRF’(東洋紡社)を形質転換し、1ウェルあたり約100コロニーとなるように細胞培養用24ウェルプラスティックプレートに調製したLカーベニシリン寒天培地(1%トリプトン、0.5%イーストエキス、1%NaCl、60μg/mlカーベニシリン、1.5%寒天)上に増殖させた。各ウェル中の形質転換株を3mlのTerrific Brothアンピシリン培地(1.2%トリプトン、2.4%イーストエキス、0.4%グリセロール、0.017M KH2PO4、0.072M K2HPO4, 100μg/mlアンピシリン)に懸濁し、37℃で一晩振盪培養した。遠心により集菌しQIAwell kit(QIAGEN社)を用いてプラスミドDNAを調製した。260nmにおける吸光度によりDNA含量を測定し、エタノール沈殿により濃縮し、200ng/μlとなるように蒸留水に溶解した。この様にしてそれぞれ約100個のコロニーから由来するDNAプールを500プール調製し、COS-7細胞(RIKEN CELL BANK,RCBO539)へのトランスフェクションに用いた。COS-7細胞を24ウェルプレートに8×104細胞/ウェルとなるように播種し、10% 牛胎児血清を含むDMEM培地を用いて、CO2インキュベーター中で37℃にて一晩培養した。翌日、培地を除いた後、無血清DMEM培地で細胞を洗浄した。トランスフェクション用試薬リポフェクトアミン(ギブコBRL社)添付のプロトコールに従い、予めOPTI-MEM培地 (ギブコBRL 社)を用いて希釈しておいた前記プラスミドDNAとリポフェクトアミン(ギブコBRL 社)を混合し、15分後この混合液を各ウェルの細胞に加えた。用いたDNA及びリポフェクタミンの量はそれぞれ1μg及び4μlとした。5時間後、培地を除去し、1mlの10% 牛胎児血清を含むDMEM培地(ギブコ社)を添加し、CO2 インキュベーター中(5%CO2)、37℃で2-3日培養した。上記の様にトランスフェクトし2-3日培養したCOS-7 細胞を無血清DMEM培地で洗浄した後、125I標識したOCIF 20ng/mlを加えた結合試験用培地(0.2%牛血清アルブミン、20mM Hepes緩衝液、0.1mg/ml heparin及び0.02% NaN3を加えた無血清DMEM培地) 200μlを各ウェルに加えた。CO2 インキュベーター中(5% CO2)で37℃ 1時間培養した後、細胞を0.1mg/ml heparinを含むリン酸塩緩衝生理食塩水500μlで2回洗浄した。洗浄後、0.1N NaOH溶液 500μlを各ウェルに加え、室温に10分間放置することにより細胞を溶解させ、各ウェル中の125Iの量をガンマーカウンター(パッカード社)で測定した。計500 プールをスクリーニングすることにより、OCIFと特異的に結合する蛋白質をコードするcDNAを含むDNAプール1っを分離した。さらに本発明のcDNAを含むDNAプールを細分化し、前記と同様のトランスフェクションとスクリーニング操作を繰り返すことにより、OCIFと結合する蛋白質をコードするcDNAを単離した。このcDNAを含むプラスミドをpOBM291と名付けた。このプラスミドを含む大腸菌は、pOBM291として通商産業省工業技術院生命工学工業技術研究所(現独立行政法人産業技術総合研究所 微生物寄託センター)に受託番号FERM BP-5953として平成9年5月23日に寄託されている。OCIFの125I標識ならびにELISAによる125I標識OCIFの定量法を以下に示す。OCIFはヨードジェン(Iodogen)法により125I標識した。2.5 mg/ml Iodogen-クロロホルム溶液20μlを1.5 mlエッペンドルフチューブに移し、40℃でクロロホルムを蒸発させて、ヨードジェンコートしたチューブを調製した。このチューブを0.5Mリン酸ナトリウム緩衝液(Na-Pi, pH 7.0)400μlで3回洗浄した後、0.5 M Na-Pi, pH 7.0, 5μlを加えた。このチューブにNa-125I溶液 (Amersham社、NEZ-033H20)1.3μl(18.5 MBq)を加えた後、直ちに1mg/mlOCIF溶液(モノマー型あるいはダイマー型) 10μlを加え、ボルテクスミキサーで攪拌した後、室温で30秒間放置した。
この溶液を、予め10mg/ml沃化カリウム、0.5M Na-Pi, pH7.0溶液80μlと 5% 牛血清アルブミンを含むリン酸塩緩衝生理食塩水(BSA-PBS)5μl を添加しておいたチューブに移し攪拌した。この溶液をBSA-PBSで平衝化しておいたスピンカラム(1ml, G-25 fine, ファルマシア社)に負荷し、2,000rpmで5分間遠心した。カラムから溶出された画分にBSA-PBSを400μl加え撹拌した後、2μlを取り、その放射能をガンマーカウンターで測定した。このようにして調製した 125I標識OCIF溶液の放射化学純度は 10% TCAにより沈殿する画分の放射能を測定することにより求めた。また、125I標識OCIF溶液のOCIF生物活性は、WO96/26217号公報記載の方法に従い測定した。また、125I標識OCIF濃度は、以下のようにELISAにより測定した。即ち、WO96/26217号公報記載のウサギ抗OCIFポリクローナル抗体を2μg/mlになるように溶解させた50mM NaHCO3, pH 9.6を100 μlずつ96ウェルイムノプレート(Nunc,MaxiSorpTM)の各ウェルに加えて4℃で一夜放置した。この液を吸い取った後、ブロックエース(雪印乳業)とリン酸塩緩衝生理食塩の混合水溶液 (混合比25:75)(B-PBS)を200μlずつ各ウェルに加え、室温で2時間放置した。この液を吸い取った後、各ウェルを0.01%Polysorbate 80を含むリン酸塩緩衝生理食塩水 (P-PBS)で3回洗浄し、次いで125I標識OCIFサンプルあるいはOCIF標準品を添加した B-PBSを100μlずつ各ウェルに加え、室温で2時間放置した。この液を吸い取った後、P-PBS200μlで各ウェルを6回洗浄した。パーオキシダーゼ標識したウサギ抗OCIFポリクローナル抗体をB-PBSで希釈した溶液を100μlずつ各ウェルに加え、室温で2時間放置した。この液を吸い取った後、P-PBS200μlで各ウェルを6回洗浄した。TMB 溶液 (TMB Soluble Reagent, High Sensitivity, Scytek社)を100μlずつ各ウェルに加え、室温で2-3分放置した後、停止液(Stopping Reagent, Scytek社)を100μlずつ各ウェルに加えた。各ウェルの450 nmにおける吸光度をマイクロプレートリーダーで測定した。OCIF標準品を用いて作製した検量線より125I標識OCIFの濃度を求めた。
【0070】
(4) OBM の全アミノ酸配列をコードする cDNA の塩基配列の決定
実施例8-(3)で得られたOBMcDNAの塩基配列を、タックダイデオキシターミネーターサイクルシークエンシングキット(パーキンエルマー社)を用いて決定した。
即ち、pOBM291を鋳型とし、直接挿入断片の塩基配列を決定した。また、pOBM291を制限酵素EcoRI で切断し得られた約1.0kb ならびに約0.7kb の断片をプラスミドpUC19(宝酒造社)のEcoRI部位に挿入し、それらの断片の塩基配列も決定した。用いたプライマーは、pcDL-SRα296 の挿入断片 DNAの塩基配列を決定するためのプライマーSRR2、プラスミドpUC19 の挿入断片 DNAの塩基配列を決定するためのプライマーml3PrimerM3、ml3PrimerRV(ともに宝酒造社)及びOBMcDNAの塩基配列に基づいて設計された合成プライマーOBM#8である。これらプライマーの配列を配列表配列番号3〜6に示す。
また、決定されたOBMcDNAの塩基配列を配列番号2に、その配列から推定されるアミノ酸配列を配列番号1にそれぞれ示す。
【0071】
〔実施例9〕
本発明cDNAにコードされる蛋白質の発現
プラスミドpOBM291 を6ウェルプレートの各ウェル中でCOS-7細胞にリポフェクトアミンを用いトランスフェクションし、10%牛胎児血清を含むDMEM培地中で2日間培養した。5%透析牛胎児血清を添加した、システイン・メチオニン不含DMEM (大日本製薬社) に培地交換し(800μl/well) 15分培養した後、Express Protein Labeling Mix (NEN社、10mCi/ml)を14μl添加した。4時間培養後、10% 牛胎児血清を含むDMEM培地を 200μl加え1時間培養した。細胞をPBSで2回洗浄後、1%TritonX-100、1% bovine hemoglobin、10μg/ml leupeptin、0.2 TIU/ml aprotinin、1mM PMSFを含むTSAバッファー (0.14M NaCl、0.025% NaN3を含む 10mMTris-HCl (pH 8.0))を0.5ml加え、氷上1時間静置した。ピペッティングにより細胞を破砕した後、4℃で3000Xg 10分間遠心分離し上清を得た。この上清100μlに200μlの希釈バッファー (0.1 % TritcmX-100、0.1% bovine hemoglobin、10μg/ml leupeptin、0.2TIU/ml aprotinin、1mM PMSFを含むTSAバッファー)を加え、protein A Sepharose(50μl)とともに4℃で1時間振とうさせた後、4℃で1500Xg にて1分間遠心分離し上清を回収することにより、protein A Sepharoseに非特異的に吸着する分画を除いた。この上清にOCIF(1μg)を加え、4℃で1時間振とうさせながらOBMとOCIFを結合させた後、抗OCIFポリクローナル抗体 (50μg)を加えて4℃で1時間振とうした。これにprotein A Sepharose(10μl)を加えさらに4℃で1時間振とうした。4℃で1500Xg にて1分間遠心分離し沈澱画分を回収した。4℃、1500X g で1分間の遠心分離による沈澱の洗浄を、希釈バッファーで2回、bovine hemoglobinを含まない希釈バッファーで2回、TSAバッファーで1回、50mM Tris-HCl(pH 6.5)で1回行った。洗浄後、沈澱に10 %βメルカプトエタノールを含むSDS バッファー(0.125 MTris-HCl、4%ドデシル硫酸ナトリウム、20% グリセロール、0.002%ブロモフェノールブルー、pH 6.8)を加え、100℃で5分加熱後 SDS-PAGE(12.5%ポリアクリルアミドゲル、第一化学社)を行った。常法によりゲルを固定した後、Amplify(アマシャム社)により、アイソトープのシグナルを増強した後、Bio Max MRフィルム (KODAK社) を用いて−80℃で感光させた。結果を図8に示す。
本発明cDNAにコードされる蛋白質の分子量は約40,000であることが示された。
【0072】
〔実施例10〕
本発明cDNAにコードされる蛋白質と OCIF との結合
プラスミドpOBM291 を24ウェルプレートの各ウェル中でCOS細胞にリポフェクトアミンを用いてトランスフェクトし、その細胞を2-3日培養したのち無血清DMEM培地で洗浄し、これに125I標識したOCIF 20ng/mlを加えた結合試験用培地 (0.2%牛血清アルブミン、20mM Hepes緩衝液、0.1mg/ml heparin、0.2% NaN3を加えた無血清DMEM培地)200 μl を加えた。また別のウェルには、125I標識したOCIF 20ng/mlに加えて8μg/mlの非標識OCIFを更に添加した結合試験用培地を200μl加えて実験を行った。CO2 インキュベーター中(5% CO2)で37℃にて1時間培養した後、細胞を0.1mg/mlのheparinを含むリン酸緩衝生理食塩水500μlで2回洗浄した。洗浄後、0.1N NaOH溶液500μlを各ウェルに加え、室温に10分間放置することにより細胞を溶解させ、ウェル中の125Iの量をガンマーカウンターで測定した。その結果、図9に示したようにプラスミドpOBM291 をトランスフェクトした細胞にのみ125I標識したOCIFが結合することが確認された。また、その結合は、400倍濃度のOCIFを添加することで著しく阻害されることが確認された。以上の結果から、プラスミドpOBM291上のcDNAにコードされる蛋白質、OBMは、COS-7細胞表面でOCIFと特異的に結合することが明らかとなった。
【0073】
〔実施例11〕
125 I標識した OCIF と本発明 cDNA にコードされる蛋白質とのクロスリンキング試験
本発明cDNAにコードされる蛋白質の性質をさらに詳しく解析するため、125I標識したモノマー型OCIFと本発明cDNAにコードされる蛋白質とのクロスリンキングを行った。実施例8-(3)に記載の方法に従ってプラスミドpOBM291をCOS-7 細胞にトランスフェクトした後、上記の 125I標識したOCIF (25ng/ml) を加えた結合試験用培地200μl を添加した。また、別のウェルには、125I標識したOCIFに加え400 倍濃度の非標識OCIFをさらに添加した結合試験用培地を加えた。CO2インキュベーター中(5% CO2)で37℃ 1時間培養した後、細胞を0.1mg/ml heparinを含むリン酸塩緩衝生理食塩水500 μlで2回洗浄した。この細胞に100μg/mlのクロスリンキング剤、DSS (Disuccinimidyl suberate, Pierce社) を溶解させたリン酸塩緩衝生理食塩水 500μl を加え、0℃で10 分間反応させた。これらのウェルの細胞を0 ℃に冷却したリン酸緩衝生理食塩水 1ml で2回洗った後、この細胞に1% Triton X-100(和光純薬社)、2mM PMSF (phenylmethylsulfonyl fluoride、シグマ社)、10μM pepstatin (和光純薬社)、10μM leupeptin(和光純薬社)、10μM antipain (和光純薬杜)、及び2mM EDTA (和光純薬社)を含む 20 mM Hepes緩衝液 100μl を加え、室温に30分間放置して細胞を溶解させた。これらのサンプル15μlを常法により還元条件下でSDS化した後、SDS-電気泳動用ゲル(4-20% ポリアクリルアミドグラジエント、第一化学社)を用いて泳動した。泳動後、ゲルを乾燥させ、BioMax MSフィルム(Kodak社) とBioMaxMS 増感スクリーン (Kodak社)を用いて−80℃で24時間感光させた。感光させたフィルムを常法により現像した。この結果、125I標識モノマー型OCIFと本発明cDNAによりコードされる蛋白質をクロスリンクさせることにより、図10に示すように分子量約90,000-110,000のバンドが検出された。
【0074】
〔実施例12〕
ノーザンブロットによる解析
付着細胞用25cm2T フラスコでコンフルエントになるまで培養したST2細胞を、トリプシン処理してT フラスコから剥がし洗浄した後、1枚の225cm2T フラスコに播種し、10- 8M 活性化ビタミンD3、10- 7M デキサメサゾン、及び10%牛胎児血清を添加したα-MEM培地各々60mlを加えてCO2インキュベーター中で4日間培養した。培養したST2細胞からISOGEN (和光純薬社) を用いて総 RNAを抽出した。また、活性化ビタミンD3とデキサメサゾン非存在下で培養したST2細胞からも上記の方法で総RNAを抽出した。各総RNA 20 μg (4.5μl)にそれぞれ2.0μlの 5X ゲル泳動緩衝液(0.2M モルホリノプロパンスルホン酸、pH 7.0、50mM 酢酸ナトリウム、5mM EDTA)、3.5 μlのホルムアルデヒド及び10.0μlのホルムアミドを加え55℃で15分保温後、電気泳動に供した。電気泳動用のゲルは1.0%アガロース、2.2M脱イオン化ホルムアルデヒド、40mM モルホリノプロパンスルホン酸 pH 7.0、10mM 酢酸ナトリウム、1mM EDTAの組成で調製した。
また、電気泳動は40 mM モルホリノプロパンスルホン酸、pH 7.0、10mM 酢酸ナトリウム、1mM EDTAの緩衝液中で行った。電気泳動後、RNAをナイロンメンブレンにトランスファーした。pOBM291を制限酵素EcoRI で切断して得られた、約1.0kb の DNA断片を、メガプライム DNAラベリングキット (アマシャム社)及びα-32P-dCTP(アマシャム社) を用いて標識したものをプローブとしてハイブリダイゼーションを行った。その結果、図11に示すように、ST2細胞では活性化ビタミンD3とデキサメサゾンの存在下で培養した場合に、本発明cDNAにコードされる蛋白質 (OBM)の遺伝子発現が強く誘導されていることが明らかとなった。
【0075】
〔実施例13〕
本発明 cDNA にコードされる蛋白質の破骨細胞形成支持能
実施例8-(3)に記載の方法でCOS細胞にpOBM291 をトランスフェクトした。3日後トリプシナイズした細胞をリン酸塩緩衝生理食塩水で一回遠心洗浄したのち、1%パラフォルムアルデヒドを含むPBS に細胞をサスペンドしながら室温で5分間固定し、PBSにて6回遠心洗浄した。10-8M 活性化ビタミンD3、10−7M デキサメサゾン、10% 牛胎児血清を含むα-MEM培地にて調製した1×106個/ml のマウス脾臓細胞及び4×104個/ml のST2 細胞を24ウェルプレートにそれぞれ 700μl、350μl添加したのちTCインサート (Nunc社)を各ウェルにセットした。上記培地にて段階希釈した固定化COS細胞(350μl)及びOCIF(50μl)をTCインサート中に添加して37℃で6日間培養した。この結果、OCIFによる破骨細胞形成阻害活性が、本発明cDNAにコードされる蛋白質により抑制されることが明らかとなった。
【0076】
〔実施例14〕
分泌型 OBM の発現
(1) 分泌型 OBM 発現プラスミドの構築
OBM HF (配列表配列番号7)とOBM XR (配列表配列番号8)をプライマー、pOBM291を鋳型としPCR反応を行った。この生成物を、アガロースゲル電気泳動で精製した後、制限酵素HindIII、EcoRIで切断し、さらにアガロースゲル電気泳動で精製した。この精製断片(0.6kb)を、pSec TagA(インビトローゲン社)のHindIII/EcoRV 断片 (5.2kb)と、OBM cDNAのEcoRI/PmaCI断片 (0.32kb)とともにライゲーションキットver.2(宝酒造社) を用いてライゲーションを行い、その反応生成物を用いて大腸菌 DH5αを形質転換した。得られたアンピシリン耐性株より、アルカリSDS 法によりプラスミドを精製し、制限酵素で切断することによりpSecTagA に0.6Kb と0.32kbの断片が挿入されたプラスミドを選び出した。
さらにこのプラスミドにつきダイターミネーターサイクルシークエンシングFSキット(パーキン‐エルマー社)を用いてシークエンスを行い、このプラスミドが分泌型OBMをコードする配列 (塩基配列:配列表配列番号2の338〜1355番、アミノ酸配列:配列表配列番号1の72〜316番)を持っていることを確認した。このプラスミドを制限酵素NheI、XhoIで切断した後、分泌型OBM cDNAに相当する断片(1.0kb)をアガロースゲル電気泳動で回収した。この断片をライゲーションキットを用いて発現ベクターpCEP4(インビトローゲン社)のNheI/XhoI断片(10.4kb)に挿入し、その反応生成物を用いて大腸菌DH5 αを形質転換した。得られたアンピシリン耐性株より、アルカリSDS法によりプラスミドを精製し、制限酵素で切断して解析することにより、目的の構造を持った分泌型OBM発現プラスミド(pCEP sOBM)を持つ大腸菌株を選び出した。pCEP sOBMを持つ大腸菌株を培養し、キアフィルタープラスミドミディキット(キアゲン社)を用いてpCEP sOBMを精製した。
【0077】
(2) 分泌型 OBM の発現
293-EBNA細胞を10%FCSを含むIMDM (IMDM-10%FCS)に懸濁し、コラーゲンコートした24ウェルプレート(住友べ一クライト社)に2x105 個/2ml/ウェルになるように播種し、一晩培養した。この細胞に1μgのpCEP sOBM 又はpCEP4をリポフェクトアミン(ギブコ社)4μlを用いて形質導入し、0.5ml の無血清IMDM又はIMDM-10 %FCS 中でさらに2日間培養し、その培養上清を回収した。分泌型OBMの培養上清中での発現は、以下のようにして確認した。培養上清に最終濃度0.1Mになるよう炭酸水素ナトリウムを加えた後、培養液を96ウェルプレートに添加し、4℃で一晩放置し、培養上清中のOBMを96ウェルプレートに固相化した。このプレートをPBS で4倍希釈したブロックエース (雪印乳業社) 溶液 (B-PBS)で室温に2時間放置しブロッキングした後、B-PBSで希釈した3-100ng/mlのOCIFを各ウェルに100μlずつ加え37℃で2時間放置した。0.05% Tween 20 を含むPBS(PBS-T)でプレートを洗浄した後、B-PBS で希釈した WO96/26217号記載のパーオキシダーゼ標識ウサギ抗OCIFポリクローナル抗体を各ウェルに100μlずつ添加し、37℃で2時間放置した。PBS-Tで各ウェルを6回洗浄した後、TMB溶液(TMB Soluble Reagent, High Sensitivity, Scytek社)を100μlずつ各ウェルに加え、室温で約10分放置した後、停止液(Stopping Reagent, Scytek社) を100μlずつ各ウェルに加えた。各ウェルの 450nmにおける吸光度をマイクロプレートリーダーで測定した。結果を図12に示す。pCEP sOBMを形質導入した細胞の培養上清を固相化したプレートでは、添加したOCIFの濃度に依存して450nmの吸収が増加したが、ベクターpCEP4を形質導入した細胞の培養上清を固定化した場合は吸収の増加はみられなかった。又、固定化に用いる培養上清の割合を5-90%の範囲で種々に変化させ、一定濃度のOCIF(50ng/ml)を添加した際の実験の結果を図13に示す。pCEP sOBMを形質導入した細胞の培養上清を固相化したプレートでは、培養上清の割合が高くなるほど450nmの吸収が増加したが、ベクターpCEP4を形質導入した細胞の培養上清を固定化したプレートでは、吸収の増加は見られなかった。以上の結果より、分泌型OBMが、pCEP sOBMを形質導入した細胞の培養上清中に生産されていることが確認された。
【0078】
〔実施例15〕
チオレドキシン‐ OBM 融合蛋白質 (Trx-OBM) の発現
(1) チオレドキシン‐ OBM 融合蛋白質 (Trx-OBM) 発現ベクターの構築
10μlの10X ExTaqバッファー(宝酒造社)、8 μl の10mMdNTP (宝酒造社)、77.5μl の滅菌蒸留水、2μl のpOBM291 水溶液(10ng/μl)、1μlのプライマーOBM3 (100pmol/μl、配列表配列番号9)、1μlのプライマーOBMSalR2(100pmol/μl、配列表配列番号10)、0.5μlのExTaq (5u/μl) (宝酒造社)を混合しマイクロ遠心管中でPCR反応を行った。95℃5分、50℃1秒、55℃1分、74℃1秒、72℃5分の反応後、96℃1分、50℃1秒、55℃1分、74℃1秒、72℃3分の反応を25サイクル行った。1%アガロースゲル電気泳動により、反応液全量から約750bpのDNA 断片をQIAEX II gel extraction kit(QIAGEN社)を用いて精製した。精製したDNA 断片全量を制限酵素SaII, EcoRI(宝酒造杜)で切断した後、1.5%アガロースゲル電気泳動を行い約160bp のDNA断片 (断片1)を精製し20μlの滅菌蒸留水に溶解した。同様に、4μg のpOBM291を制限酵素BamHI, EcoRI(宝酒造社)で切断し得られた約580bp のDNA断片 (断片2)、及び2μgのpTrXFus (InVitrogen社)を制限酵素BamHI,SalI(宝酒造社)で切断し得られた約3.6kbのDNA断片(断片3)をそれぞれ精製し、それぞれを20μlの滅菌蒸留水に溶解した。DNA断片の精製にはQIAEXII gel extraction kitを用いた。
断片1、断片2、断片3をDNAligation kit ver.2 (宝酒造社) を用い、16℃で2時間30分保温することにより結合させた。ライゲーション反応液を用い大腸菌GI724 株(インビトローゲン社)をThioFusion Expression System (インビトロ一ゲン社)添付のInstruction Manualに記載の方法で形質転換した。得られたアンピシリン耐性形質転換株の中から、制限酵素切断により得られるDNA断片地図の解析並びにDNA 配列の決定により、OBMcDNA 断片(塩基配列:配列表配列番号2の350〜1111、アミノ酸配列:配列表配列番号1の76〜316)がチオレドキシン遺伝子に同じ読み枠で結合しているプラスミドを持つ菌株を選び出した。得られた菌株をGI724/pTrxOBM25と名付けた。
【0079】
(2) 大腸菌での OBM の発現
GI724/pTrxOBM25株及びpTrxFus を持つGI724株 (GI724/pTrxFus)それぞれを2mlのRMG-Amp 培地(0.6 %Na2HPO4, 0.3 %KH2PO4, 0.05 %NaCl, 0.1 % NH4Cl 1.2%カザミノ酸(Difco社)、1%グリセロール、1mM MgCl2、100 μg/mlアンピシリン (Sigma社) pH7.4)で30℃で6時間振とう培養した。菌液0.5ml を50mlのInduction培地(0.6% Na2HPO4、0.3 %KH2PO4、0.05%NaCl、0.1% NH4Cl、0.2%カザミノ酸、0.5%グルコース、1mM MgCl2、100μg/mlアンピシリン、pH 7.4)に添加し、30℃で振とう培養した。OD600nmでの値が約0.5になった時点で最終濃度が0.1mg/mlになるようにL-トリプトファンを添加し、さらに30℃での振とう培養を6時間継続した。菌液を3000×gで遠心し菌体を集め、12.5mlのPBS (10mM リン酸バッファー、0.15M NaCl、pH7.4)に懸濁した。懸濁液を超音波発生装置 (Ultrasonics社)にかけ菌体を破砕し、7000×g で30分遠心して上清を集め可溶性蛋白質画分とした。この可溶性蛋白質画分液10μlずつを還元条件下でのSDS ポリアクリルアミド(10%)電気泳動に供した。この結果、GI724/pTrxOBM25の可溶性蛋白質画分液にはGI724/pTrxFus の可溶性蛋白質画分液には見られない分子量約40kDaのバンドが検出された(図14)。よって、チオレドキシンとOBMの融合蛋白質(Trx-OBM)が大腸菌中で発現していることが確認された。
【0080】
(3)Trx-OBM の OCIF との結合能
発現させたTrx-OBM がOCIFと結合することを以下の実験により確認した。即ち、96ウェルイムノプレート(Nunc社)の各ウェルに10mM炭酸水素ナトリウム水溶液で5000倍に希釈した抗チオレドキシン抗体(インビトローゲン社) を100μlずつ添加し4℃で一晩放置した。各ウェル中の液を捨てた後、ブロックエース(雪印乳業社)をPBS で2倍希釈した溶液 (BA-PBS)を各ウェルに200μlずつ添加し室温で1時間放置した。液を捨てた後、各ウェルにBA-PBSで段階希釈した上記GI724/pTrxOBM25 由来及びGI724/pTrxFus 由来可溶性蛋白質画分液を100μlずつ添加し室温で2時間放置した。PBS-T で各ウェルを3回洗浄した後、BA-PBSで希釈したOCIF (100ng/ml)を各ウェルに100μlずつ添加し室温で2時間放置した。PBS-Tで各ウェルを3回洗浄した後、BA-PBSで2000倍希釈したWO96/26217号記載のパーオキシダーゼ標識ウサギ抗OCIFポリクローナル抗体を各ウェルに100μlずつ添加し、2時間室温で放置した。PBS-Tで各ウェルを6回洗浄した後、TMB溶液(TMB Soluble Reagent, High Sensitivity, Scytek社)を100μlずつ各ウェルに加え、室温で約10分放置した後、停止液(Stopping Reagent, Scytek社)を100μlずつ各ウェルに加えた。各ウェルの 450nm における吸光度をマイクロプレートリーダーで測定した。結果を図15に示す。GI724/pTrxFus由来可溶性蛋白質画分液を添加したものでは添加しなかったものと比べて吸光度に差は認められなかったが、GI724/pTrxOBM25 由来可溶性蛋白質画分液を添加したものでは、その添加濃度の増加に依存して吸光度が上昇した。又、添加する可溶性画分液の希釈割合を一定(1%)にし、BA-PBSで段階希釈したOCIF(0-100ng/ml)を添加した場合の実験結果を図16に示す。GI724/pTrxFus 由来可溶性蛋白質画分液を用いたものではOCIF濃度に係わりなく吸光度は低いままであったが、GI724/pTrxOBM25由来可溶性蛋白質画分液を用いたものでは添加したOCIF濃度に依存して吸光度が上昇した。この結果より、GI724/pTrxOBM25 で生産されるTrx-OBMは、OCIFと結合する能力があることが確認された。
【0081】
(4)Trx-OBM を生産する大腸菌の大量培養
GI724/pTrxOBM25 をRMG-Amp 寒天培地 (0.6%Na2HPO4、0.3%KH2PO4、0.05%NaCl、0.1%NH4Cl、2%カザミノ酸、1%グリセロール、1mM MgCl2、100μg/mlアンピシリン、1.5%寒天、pH7.4) に白金耳で塗まつし、30℃で一晩培養した。菌を10mlのInduction培地に懸濁し、5mlずつを500mlのInduction培地のはいった2L用三角フラスコ計2本に添加し、30℃で振とう培養した。OD600nmでの値が約0.5 になった時点で最終濃度が0.1mg/mlになるようにL-トリプトファンを添加し、さらに30℃で振とう培養を6時間継続した。菌液を3000×gで20分間遠心分離して菌体を集め、菌体を160ml のPBSに懸濁した。懸濁液を超音波発生装置 (Ultrasonics 社)にかけ菌体を破砕し、7000×gで30分間遠心して上清を集め可溶性蛋白質画分とした。
【0082】
(5)OCIF 固定化アフィニティーカラムの調製
TSKgel AF-トレシルトヨパール650(東ソー社) 2gとWO96/26217号に記載の方法で調製した遺伝子組み換え型OCIF 35.0 mgを含む1.0 Mリン酸カリウム緩衝液(pH7.5) 40mlとを混合し4℃で一晩ゆっくり振とうしカップリング反応を行った。過剰な活性基を不活性化する為、遠心分離により上清を除去した後、40mlの0.1Mトリス塩酸緩衝液 (pH7.5)を沈殿した担体に加え、室温で1時間おだやかに振とうした。0.01% Polysorbate 80 と0.2M NaClを含む0.1M グリシン‐塩酸緩衝液 (pH3.3) 及び0.01% Polysorbate 80 と0.2M NaClを含む0.1 M クエン酸ナトリウム緩衝液 (pH2.0)を通液しカラムを洗浄した後、0.01% Polysorbate 80 を含む10mM リン酸ナトリウム緩衝液(pH 7.4)で2度洗浄し、平衡化した。
【0083】
(6) OCIF 固定化アフィニティーカラムによる Trx-OBM の精製
Trx-OBM の精製は、特に断らない限り4℃で行った。上記のOCIF固定化アフィニティー担体(10ml)と上記実施例15-(4)記載の可溶性蛋白質画分液 (120ml)とを混合後、50ml遠心管(4本)中で、ローターを用いて4℃で一晩おだやかに振とうした。この混合液中の担体をエコノカラム(バイオラッド社、内径1.5cm 長さ15cm)に充填した。0.01%Polysorbate 80 を含むPBS 300ml、0.01% Polysorbate 80 と2M NaClを含む10mMリン酸ナトリウム緩衝液(pH7.0)100ml、及び0.01% Polysorbate 80 と0.2M NaClを含む0.1 M グリシン‐塩酸緩衝液(pH3.3) 100ml を順次通液してカラムを洗浄した。次に、0.01%Polysorbate 80 と0.2M NaClを含む0.1 Mクエン酸ナトリウム緩衝液 (pH2.0) を通液し、カラムに吸着した蛋白質を溶出した。溶出液は5mlずつ分取した。分取した画分には、10%容量の2Mトリス溶液(pH8.0)を加え、直ちに中和した。溶出液各画分中のTrx-OBMの有無を上記実施例15-(3)記載の方法(OCIFとの結合能)により調べた。
Trx-OBMを含む画分を集めさらに精製した。
【0084】
(7) ゲル濾過による Trx-OBM の精製
上記実施例15-(6)のTrx-OBMフラクション約25mlを、Centriplus10 及びCentricon 10 (amicon杜)を用いて、約0.5mlに遠心濃縮した。このサンプルを、予め0.01 %Polysorbate 80 を含むPBSで平衡化したSuperose 12 HR 1O/30 カラム (1.0 x 30cm、ファルマシア社) にかけた。分離には、0.01% Polysorbate 80 を含む PBSを移動相として用い、流速 0.25ml/minで展開した。カラムからの溶出液は0.25 mlずつ分取した。分取した画分のTrx-OBMは、実施例15-(3)記載の方法及びPhast System (ファルマシア社)を用いたSDS-ポリアクリルアミド電気泳動(10-15%ポリアクリルアミドゲル、ファルマシア社)と銀染色で検出した。純化されたTrx-OBMを含むフラクション(Fr.20-23)を集め、Trx-OBMの蛋白質濃度を測定した。蛋白質濃度の測定は、ウシ血清アルブミンを標準品として用いDC-protein assay kit (バイオラッド社)で行った。
【0085】
〔実施例16〕
OBM の破骨細胞形成誘導活性
pOBM291及びpcDL-SRα296 をリポフェクトアミン(ギブコ社)を用いてCOS-7細胞にそれぞれトランスフェクトした。それぞれの細胞を10%FCSを含むDMEMで1日培養後トリプシン処理し、カバーグラス(15mm 丸形、マツナミ社)を敷いた24ウェルプレートに1ウェルあたり5×104個の細胞を播種し、さらに2日間培養した。培養プレートをPBSで1回洗浄したのち、1%パラフォルムアルデヒドを含むPBSを加えて室温で8分間インキュベートし細胞を固定した。細胞を固定したプレートをPBSにて6回洗浄したのち、10-8M 活性型ビタミンD3、10-7Mデキサメサゾン、10%牛胎児血清を含むα-MEMにて1X106個/mlに懸濁したマウス脾臓細胞を1ウェルあたり700 μl添加した。各ウェル上にMillicell PCF(ミリポア社)をセットし、上記培地にて4x104個/mlに懸濁したST2細胞をMillicell PCF中に 700μl 添加し37℃、6日間培養した。培養終了後、Millicell PCFを取り除き、プレートをPBS で1回洗浄後、アセトン‐エタノール溶液(50:50)で1分間細胞を固定した後、LEUKOCYTE ACID PHOSPHATASEキット (シグマ社)を用いて破骨細胞特異的マーカーである酒石酸耐性酸性フォスファターゼ活性(TRAP活性)を示す細胞のみを染色した。顕微鏡観察の結果、pcDL-SRα296をトランスフェクトしたCOS-7細胞を固定したウェル中にはTRAP活性を示す細胞は全く検出されなかったのに対し、pOBM291 をトランスフェクトした細胞を固定したウェル中には45±18個(平均±標準偏差、n=3)のTRAP陽性細胞が観察された。又、これらのTRAP陽性細胞に、カルシトニンが結合することも確認された。これより、OBMには破骨細胞形成を誘導する活性があることが明らかとなった。
【0086】
〔実施例17〕
Trx-OBM 及び分泌型 OBM による破骨細胞形成誘導活性
マウス脾臓細胞を10−8M活性型ビタミンD3、10−7M デキサメサゾン、10%牛胎児血清を含むα-MEMに2×106個/mlになるように懸濁し、この懸濁液を24ウェルプレートに1ウェルあたり350 μl添加した。精製したTrx-OBMを上記培地で希釈した溶液(40ng/ml)、pCEP sOBM 又はpCEP4 を形質導入した293-EBNA細胞をIMDM-10 %FCS で培養した培養上清を上記培地にて10倍希釈した溶液、又は上記培地のみをそれぞれ350μl 上記のウェルに添加した後、Millicell PCF(ミリポア社)を各ウェルにセットし、上記培地にて懸濁した4 ×104個/ml のST2細胞をMillicell PCF に600μl 添加した。6日間培養した後、Millicell PCFを取り除き、プレートをPBSで一回洗浄後、アセトン‐エタノール溶液(50:50)で1分間細胞を固定したのち、LEUKOCYTE ACID PHOSPHATASEキット (シグマ社)を用いて酒石酸耐性酸性フォスファターゼ活性 (TRAP活性)を示す細胞のみを染色した。顕微鏡観察の結果、Trx-OBMを添加しなかったウェル中にはTRAP活性を示す細胞は全く検出されなかったのに対し、Trx-OBMを添加したウェルには106± 21個(平均±標準偏差、n=3)のTRAP陽性細胞が観察された。同様にpCEP4で形質導入された293-EBNAの培養上清を添加したウェル中にはTRAP活性を示す細胞は全く検出されなかったのに対し、pCEP sOBM で形質導入された293-EBNAの培養上清を添加したウェルには120±31個(平均±標準偏差、n=3)のTRAP陽性細胞が観察された。又、これらのTRAP陽性細胞に、カルシトニンが結合することも確認された。これより、Trx-OBM及び分泌型OBMには、破骨細胞形成を誘導する活性があることが明らかとなった。
【0087】
〔実施例18〕
本発明の cDNA により発現される蛋白質 OBM と本発明の天然型の OCIF 結合蛋白質との同一性
(1) ウサギ抗 OBM ポリクローナル抗体の作製
雄性日本白色ウサギ (体重2.5-3.0 kg、北山ラベス社より入手)3羽に、実施例14-(6)及び(7)記載の方法により得られた精製OBM(チオレドキシン-OBM融合蛋白質) 200 μg/mlをフロイント完全アジュバント (DIFCO社)と等量混合してエマルジョンとしたものを、1回1mlずつ皮下免疫した。免疫は1週間間隔で合計6回行い、最終免疫後10日目に全採血を行った。分離した血清から抗体を以下の様に精製した。即ち、PBSで2倍希釈した抗血清に最終濃度が40% (w/v %)となるように硫酸アンモニウムを添加して4℃で1時間放置後、8000 x gで20分間遠心分離を行い、沈殿を得た。沈殿を少量のPBSに溶解し、PBS に対して4℃で透析した後、Protein G-Sepharose カラム (ファルマシア社製) に負荷した。PBS で洗浄後、0.1 M グリシン‐塩酸緩衝液(pH 3.0) で吸着した免疫グロブリンGを溶出し、直ちに1.5Mトリス‐塩酸緩衝液 (pH 8.7) で中性pHとした。溶出蛋白質画分をPBS に対して透析後、280nmにおける吸光度を測定し、その濃度を決定した(E1%13.5)。西洋ワサビパーオキシダーゼ標識した抗 OBM抗体は、マレイミド活性化パーオキシダーゼキット(ピアス社)を用いて作製した。即ち、1mgの精製抗体に80μgのN-スクシンイミド-S-アセチルチオ酢酸を添加し、室温で30分間反応させた。これに5mgのヒドロキシルアミンを添加して脱アセチル化した後、修飾された抗体をポリアクリルアミド脱塩カラムにて分画した。蛋白質画分を1mgのマレイミド活性化パーオキシダーゼと混合し、室温で1時間反応させ酵素標識抗体を得た。
【0088】
(2) ウサギ抗 OBM ポリクローナル抗体による本発明の cDNA により発現される蛋白質 (OBM) あるいは本発明の天然型の蛋白質と OCIF との特異的結合能の阻害
実施例15‐(6)及び(7)記載の方法で得られた精製OBM(チオレドキシン−OBM融合蛋白質)及び実施例2-(4)の天然型精製OCIF結合蛋白質をそれぞれ0.1 M 炭酸水素ナトリウムに2 μg/mlとなるように溶解し、それぞれの溶液100μlずつを96ウェルイムノプレート(Nunc社製)の各ウェルに加え、4℃で一夜放置した。各ウェルに50%濃度のブロックエースを200 μl ずつ加え、室温で1時間放置した。各ウェルを0.1 %ポリソルベート20を含むPBS(P20-PBS)で3回洗浄した後、P20-PBSで希釈した25%濃度のブロックエースにウサギ抗OBM抗体を200 μg/mlとなるように溶解し、その抗体溶液あるいは抗体を含まない25%濃度のブロックエース溶液を100μlずつ各ウェルに添加し、37℃、1時間インキュベートした。各ウェルをP20-PBSで3回洗浄した後、実施例8-(3)記載の125I標識したOCIF20ng/mlを加えた結合試験溶液(0.2%牛血清アルブミン、20mM Hepes、0.1mg/mlヘパリンを加えたP20-PBS)を100μlずつ各ウェルに添加した。又、別のウェルには、125I標識したOCIF 20ng/mlに加えて8μg/mlの非標識OCIFを含む結合試験溶液を100 μl 加えて実験を行った。そのイムノプレートを37℃、1時間インキュベートした後、各ウェルをP20-PBSで6回洗浄した。各ウェル中の125Iの量をガンマーカウンターで測定した。結果を図17に示す。図に示したように、本発明cDNAにより発現され、精製して得られるOBM及び本発明の天然型のOCIFに特異的に結合する蛋白質は、ウサギ抗OBMポリクローナル抗体で処理することによって、125I標識したOCIFとの結合は全く起こらなかった。一方、抗体処理しなかった両蛋白質には、125I標識OCIFが結合することが確認された。又、それらの結合は400倍濃度の非標識OCIF(8μg/ml)を添加することにより顕著に阻害されることから、両蛋白質のOCIFへの結合は特異的結合であることが明らかになった。以上の結果から、ウサギ抗OBMポリクローナル抗体は、本発明cDNAにより発現される蛋白質であるOBM及び本発明の天然型のOCIF結合蛋白質を共に認識し、両蛋白質とOCIFとの特異的結合を阻害することが明らかになった。
【0089】
〔実施例19〕
ヒト OBMcDNA のクローニング
(1) マウス OBM プライマーの調製
ヒトOBMcDNAのスクリーニングには、上記の実施例の方法に従い調製したマウスOBMプライマー、OBM#3 及びOBM#8 を用いた。それぞれの配列を配列表配列番号9及び6に示す。
【0090】
(2)PCR によるヒト OBMcDNA 断片の取得
ヒトリンパ節由来cDNAライブラリーとしてHuman Lymph Node Marathon ready cDNA(クロンテック社)を鋳型として、上記(1)で調製したマウスOBMcDNA プライマーを用いて、PCRを行いヒトOBMcDNA断片を取得した。
以下に用いたPCRの条件を示す。

【0091】
(3)PCR により増幅されたヒト OBMcDNA 断片の精製と塩基配列の決定
実施例19-(2)で得られたヒトOBMcDNA断片をアガロース電気泳動により分離し、さらにQIAEXゲルエクストラクションキット(キアゲン社)を用いて精製した。
この精製されたヒトOBMcDNA断片を鋳型として、再度上記マウスOBMcDNAプライマーを用いてPCRを行い、このヒトOBMcDNA 断片を大量に調製し、同様にQIAEXゲルエクストラクションキットにより精製した。精製されたヒトOBMcDNA断片の塩基配列を、OBM#3及び OBM#8 (配列表9及び6)をプライマーとして、タックダイデオキシターミネーターサイクルシークエンシングFSキット(Taq Dye Deoxy Terminator Cycle Sequencing FS kit;パーキンエルマー社)を用いて決定した。このヒトOBMcDNA 断片の塩基配列は、マウスOBMcDNAの相当する領域と比較した結果、80.7%の相同性を示した。
【0092】
(4) 約 690 bp のヒト OBMcDNA 断片をプローブとしたハイブリダイゼーション法による全長ヒト OBMcDNA のスクリーニング
実施例19-(3)で精製された約690 bpのヒトOBMcDNA断片をメガプライム DNAラベリングキット(アマシャム社)を用いて[α32P] dCTP で標識し、全長のヒトOBMcDNAをスクリーニングした。スクリーニングの対象としては、Human Lymph Node 5’-STRETCH PLUS cDNA ライブラリー (クロンテック社、USA)を用いた。同社のプロトコールに従い、組み換えファージを37℃で15分間大腸菌C600 Hflに感染させた後、その大腸菌を45℃に加温したLB寒天培地 (1%トリプトン、0.5%イーストエキス、1% NaCl、0.7 %寒天)に添加し、1.5%寒天を含むLB寒天培地プレート上に流し込んだ。37℃で一夜培養後、プラークの生じたプレート上にハイボンドN(アマシャム社)を約3分間密着させた。このフィルターを常法に従いアルカリ変性処理した後、中和し、2×SSC溶液に浸した後、UVクロスリンク(ストラタジーン社) によりDNAをフィルターに固定した。得られたフィルターをRapid-hyb バッファー (アマシャム社)に浸漬し、65℃で15分間前処理した後、熱変性した上記ヒトOBMcDNA断片(約690bp、5×105 cpm/ml)を添加した上記バッファーに移し替え、65℃で一夜ハイブリダイゼーションさせた。反応後、フィルターをそれぞれ0.1%SDSを含む2×SSC、1×SSC及び0.1×SSCで順次1回ずつ65℃で15分間洗浄した。得られたいくつかの陽性クローンをさらに2回スクリーニングを繰り返すことにより純化した。それらの中から、約2.2kbのインサートを持つクローンを選択し、以下の実験に用いた。この純化したファージをλhOBMと名付けた。純化したλhOBMからQIAGEN Lambdaキット(キアゲン社)のプロトコールに従い、約10μgのDNAを得た。このDNAを制限酵素SalIで消化した後、アガロース電気泳動により約2.2kbのhOBMインサートcDNAを分離した。QIAEXゲルエクストラクションキット(キアゲン社)を用いて精製したこのDNA断片を、あらかじめ制限酵素SalIで切断し、脱リン酸化しておいたプラスミドpUC19(MBI社)にDNA ライゲーションキット ver.2(宝酒造社)を用いて挿入した。得られたDNA断片を含むpUC19 で大腸菌DH 5α (ギブコBRL杜)を形質転換させた。得られた形質転換株はpUC19hOBM と名付け、これを増殖させ、約2.2kbのヒトOBMcDNA が挿入されたプラスミドを常法により精製した。
【0093】
(5) ヒト OBM の全アミノ酸配列をコードする cDNA の塩基配列の決定
実施例19-(4)で得られたヒトOBMcDNAの塩基配列を、タックダイデオキシターミネーターサイクルシークエンシングFSキット(パーキンエルマー社)を用いて決定した。即ち、pUC19hOBM を鋳型とし、挿入断片の塩基配列を決定した。プライマーとして、pUC19 の挿入断片 DNAの塩基配列を決定するためのプライマー、ml3PrimerM3及びml3 PrimerRV (ともに宝酒造社)、及びヒトOBMcDNA 断片(約690bp)の塩基配列に基づいて設計した合成プライマーヒト OBM#8を用いた。用いたプライマー、ml3PrimerM3及びml3PrimerRV の配列を、それぞれ配列表配列番号4及び5に示す。これにより決定された、ヒトOBMcDNA の塩基配列から推定されるヒトOBMのアミノ酸配列を配列表配列番号11、又、ヒトOBMcDNAの塩基配列を配列表配列番号12にそれぞれ示す。
得られたヒトOBMcDNA を含むプラスミド、pUC19hOBMで形質転換した大腸菌は、通商産業省工業技術院生命工学工業技術研究所(現独立行政法人産業技術総合研究所微生物寄託センター)に受託番号 FERM BP-6058として平成9年8月13日に寄託されている。
【0094】
〔実施例20〕
OCIF の 125 I標識ならびに ELISA による 125 I標識 OCIF の定量
OCIFはヨードジェン(Iodogen)法により125I標識した。2.5mg/ml Iodogen-クロロホルム溶液20μlを1.5mlエッペンドルフチューブに移し、40℃でクロロホルムを蒸発させて、ヨードジェンコートしたチューブを調製した。このチューブを0.5Mリン酸ナトリウム緩衝液(Na-Pi,pH7.0)400μlで3回洗浄した後、5μlの0.5M Na-Pi, pH 7.0を加えた。このチューブにNa-125I溶液(アマシャム社、NEZ-033H) 1.3 μl (18.5MBq)を加えた後、直ちに1mg/ml OCIF溶液(モノマー型あるいはダイマー型)10μlを加え、ボルテックスミキサーで攪拌した後、室温で30秒間放置した。この溶液を、予め10 mg/ml沃化カリウムを含む0.5MNa-Pi(pH7.0)溶液80μlと5%牛血清アルブミンを含むリン酸緩衝生理食塩水(BSA-PBS)5μlを添加しておいたチューブに移し撹伴した。この溶液をBSA-PBS で平衡化したスピンカラム(1ml, G-25 Sephadex fine, ファルマシア社)に負荷し、2,000rpmで5分間遠心した。カラムから溶出された画分にBSA-PBSを400μl加え攪拌した後、2μlを取り、その放射能をガンマーカウンターで測定した。このようにして調製した125I標識OCIF溶液の放射化学純度は10%トリクロロ酢酸(TCA)により沈殿する画分の放射能を測定することにより求めた。
125I標識OCIFのOCIF生物活性は、WO96/26217号記載の方法に従い測定した。又、125I標識OCIF濃度は、以下のようにELISA により測定した。即ち、WO96/26217号公報記載のウサギ抗OCIFポリクローナル抗体を2μg/mlになるように溶解した50mMNaHCO3, pH 9.6を100 μlずつ96ウェルイムノプレート(Nunc社、MaxiSorpTM)の各ウェルに加えて、4℃一夜放置した。この溶液を捨てた後、ブロックエース(雪印乳業社)とリン酸塩緩衝生理食塩水との混合水溶液(混合比25:75)(B-PBS)を200 μlずつ各ウェルに加え、室温で2時間放置した。この液を捨てた後、各ウェルを0.01% Polysorbate 80 を含むリン酸塩緩衝生理食塩水(P-PBS)で3回洗浄し、次いで125I標識OCIFサンプルあるいはOCIF標準品を添加したB-PBSを 100μlずつ各ウェルに加え、室温で2時間放置した。この液を捨てた後、P-PBS200 μlで各ウェルを6回洗浄した。パーオキシダーゼ標識したウサギ抗OCIFポリクローナル抗体をB-PBS で希釈した溶液を100μlずつ各ウェルに加え、室温で2時間放置した。この溶液を捨てた後、P-PBS200μlで各ウェルを6回洗浄した。TMB 溶液(TMB Soluble Reagent, High Sensitivity, Scytek社)を100μlずつ各ウェルに加え、室温で2〜3分間放置した後、停止液(Stopping Reagent, Scytek社)を100μlずつ各ウェルに加えた。各ウェルの450 nmにおける吸光度をマイクロプレートリーダーで測定した。OCIF標準品を用いて作成した検量線より125I標識OCIFの濃度を求めた。
【0095】
〔実施例21〕
本発明 cDNA にコードされる蛋白質の発現
(1) 動物細胞用 hOBM 発現ベクターの構築
pUChOBM を制限酵素SalIで切断し、約2.2 kbのDNA断片を1%アガロース電気泳動により精製し、DNAブランティングキット(宝酒造社)により末端を平滑化した(平滑化 hOBMcDNA 断片)。発現プラスミドpcDL-SRα296 (Molecular and Cellar Biology, Vol 8, pp466-472(1988))を制限酵素EcoRI で切断し、ブランティングキットにより末端を平滑化したものと、平滑化したhOBMcDNA 断片とをDNAライゲーションキットver.2により結合させた。ライゲーション反応液を用い大腸菌DHαを形質転換した。得られたアンピシリン耐性形質転換株から、制限酵素切断により得られるDNA断片地図の解析並びにDNA配列の決定により、SRαプロモーターによる転写方向に対してhOBMcDNA の転写方向が順向きに挿入されているプラスミド、phOBMを持つ菌株を選びだし、DH5α/phOBMと名付けた。
【0096】
(2)COS-7 細胞でのヒト OBM の発現
大腸菌、DH5 α/phOBMを培養し、キアフィルタープラスミドミディキット(キアゲン社)を用い、プラスミドphOBMを精製した。phOBMを6ウェルプレートの各ウェル中でCOS-7細胞にリポフェクトアミンを用いてトランスフェクションし、10%牛胎児血清を含むDMEM中で2日間培養した。5%透析牛胎児血清を添加した、システイン・メチオニン不含DMEM(大日本製薬社)に培地交換し(88μl/well)15分間培養した後、ExpressProtein Labelng Mix (NEN社、10 mCi/ml)を14μl添加した。4時間培養後、10%牛胎児血清を含むDMEMを200 μl 加え1時間培養した。細胞をPBSで2回洗浄後、1%TritonX-100、1%牛ヘモグロビン、10μg/mlロイペプチン(leupeptin)、0.2 TIU/mlアプロチニン(aprotinin)及び1mM PMSF を含むTSA バッファー(0.14M NaCl、0.025% NaN3を含む10mMトリス‐塩酸、pH 8.0)を0.5ml加え、氷上で1時間静置した。ピペッティングにより細胞を破砕した後、4℃、3,000×g、10分間遠心し上清を得た。この上清100μl に200 μlの希釈バッファー(0.1% TritonX-100、0.1%牛ヘモグロビン、10μg/mlロイペプチン、0.2TIU/mlアプロチニン及び1mM PMSF を含むTSA バッファー)を加え、Protein A Sepharose (50μl)とともに4℃で1時間振盪させた後、4℃、1,500×gで1分間遠心分離し上清を回収することにより: Protein A Sepharose に非特異的に吸着する蛋白質を除去した。この上清にOCIF (1μg)を加え、4℃で1時間振盪させながらヒトOBMとOCIFを結合させた後、ウサギ抗OCIFポリクローナル抗体(50μg)を加えて4℃で1時間振盪した。この溶液にProtein A Sepharose (10μl)を加え、更に4℃で1時間振盪した。4℃、1,500×gで1分間遠心分離し沈殿画分を回収した。4℃、1,500×g で1分間遠心分離して得られた沈殿を希釈バッファーで2回、牛ヘモグロビンを含まない希釈バッファーで2回、TSAバッファーで1回、50mMトリス‐塩酸(pH 6.5)で1回、それぞれ洗浄後、沈殿に10% βメルカプトエタノールを含むSDS バッファー(0.125M トリス‐塩酸、4%ドデシル硫酸ナトリウム、20%グリセロール、0.002%ブロモフェノールブルー、pH6.8) を加え、100℃で5分間加熱後、SDS-PAGE (12.5%ポリアクリルアミドゲル、第一化学社) にかけた。常法によりゲルを固定、乾燥させ、Amplify(アマシャム社)により、アイソトープのシグナルを増強した後、Bio Max MRフィルム(コダック社)を用いて‐80℃で感光させた。結果を図18に示す。この結果、本発明のcDNAにコードされる蛋白質の分子量は、約40,000であることが明らかとなった。
【0097】
〔実施例22〕
本発明 cDNA にコードされる蛋白質と OCIF との結合
実施例21-(2)と同様に精製されたphOBM を24ウェルプレートの各ウェル中でCOS-7細胞にリポフェクトアミンを用いてトランスフェクトし、その細胞を2〜3日間培養した後、無血清DMEMで洗浄し、これに125I標識したOCIF 20 ng/mlを加えた結合試験培地 (0.2%牛血清アルブミン、20mM Hepes緩衝液、0.1mg/mlヘパリン、0.2% NaN3 を加えた無血清DMEM)200μlを加えた。また別のウェルには、125I標識したOCIF 20 ng/mlに加えて更に8μg/mlの非標識OCIFを添加した結合試験培地を200 μl加えて実験を行った。CO2 インキュベーター中(5% CO2)、37℃で1時間培養した後、細胞を0.1mg/mlのヘパリンを含むリン酸塩緩衝生理食塩水500μlで2回洗浄した。洗浄後、0.1NNaOH 溶液 500μlを各ウェルに加え、室温に10分間放置することにより細胞を溶解させ、ウェル中の125Iの量をガンマーカウンターで測定した。この結果、図19に示したように、phOBMをトランスフェクトした細胞にのみ125I標識したOCIFが結合することが確認された。又、その結合は400 倍濃度の非標識 OCIF (8μg/ml)を添加することにより、顕著に阻害されることが確認された。以上の結果から、phOBM上のcDNAにコードされる蛋白質ヒトOBMは、COS-7 細胞表面でOCIFに特異的に結合することが明らかとなった。
【0098】
〔実施例23〕
125 I標識した OCIF と本発明 cDNA にコードされる蛋白質とのクロスリンキング試験
本発明cDNAにコードされる蛋白質の性質を更に詳しく解析するため、125I標識したモノマー型OCIFと本発明cDNAにコードされる蛋白質とのクロスリンキングを行った。即ち、実施例21-(1)及び(2)に記載した方法に従って、それぞれ発現ベクターをphOBMを構築し、COS-7 細胞にトランスフェクトした後、上記の125I標識したOCIF (25ng/ml)を加えた結合試験用培地を200 μlずつ各ウェルに添加した。又、別のウェルには、125I標識したOCIFに加え400 倍濃度の非標識OCIFを更に添加した結合試験用培地を加えた。CO2インキュベーター中(5% CO2)、37℃で1時間培養した後、細胞を0.1mg/ml のヘパリンを含むリン酸塩緩衝生理食塩水500μl で2回洗浄した。この細胞に100μg/mlのクロスリンキング剤、DSS (Disuccinimidyl suberate, Pierce社) を溶解させたリン酸塩緩衝生理食塩水 500μlを加え、0℃で10分間反応させた。これらのウェルの細胞を0℃に冷却したリン酸塩緩衝生理食塩水1mlで2回洗浄した後、この細胞に1%Triton X-100(和光純薬社)、2 mM PMSF(phenylmethylsulfonyl fluoride、シグマ社)、10μM ペプスタチン (和光純薬社)、10μMロイペプチン (和光純薬社)、10μM アンチパイン (antipain, 和光純薬社)及び2 mM EDTA (和光純薬社)を含む20 mM Hepes 緩衝液100μlを加え、室温に30分間放置して細胞を溶解させた。これらのサンプル15 μlを常法により還元条件下でSDS化した後、SDS−電気泳動用ゲル(4-20%ポリアクリルアミドグラジエント、第一化学社)を用いて泳動した。泳動後、ゲルを乾燥させ、Bio MAX MS フィルム(コダック社)とBioMax MS 増感スクリーン(コダック社)を用いて‐80℃で24時間感光させた。感光させたフィルムを常法により現像した。この結果、125I標識モノマー型OCIFと本発明cDNAによりコードされる蛋白質をクロスリンクさせることにより、図20に示すように、分子量約90,000‐110,000の蛋白質バンドが検出された。
【0099】
〔実施例24〕
分泌型ヒト OBM の発現
(1) 分泌型ヒト OBM 発現プラスミドの構築
ヒトOBM SF (配列表配列番号13) とマウスOBM #8(配列表配列番号6)をプライマー、pUC19hOBM を鋳型としてPCR 反応を行った。この生成物をアガロースゲル電気泳動で精製した後、制限酵素SPlI、HindIIIで切断し、さらにアガロースゲル電気泳動で精製して、0.27kbの断片を得た。ヒトOBM cDNAを制限酵素DraIで部分消化し、一ヶ所切断された断片をアガロース電気泳動で精製し、その精製断片をさらに制限酵素HindIIIで切断した。0.53kbのDraI/HindIII断片をアガロース電気泳動で精製し、この精製断片と上記のPCR生成物のSplI,HindIII 断片(0.27 kb)をpSec Tag A(インビトローゲン社)のSPlI/EcoRV 断片(5.2 kb)と共にライゲーションキットver.2(宝酒造社)を用いてライゲーションを行い、その反応生成物を用いて大腸菌DH5 αを形質転換した。得られたアンピシリン耐性株より、アルカリSDS 法によりプラスミドを精製し、制限酵素で切断することによって、pSec Tag A に0.27 kbと0.53 kbの断片が挿入されたプラスミドを選び出した。さらに、このプラスミドをタックダイデオキシターミネーターサイクルシークエンシングFSキット(パーキンエルマー社)を用いてシークエンスを行い、このプラスミドが分泌型ヒトOBMをコードする配列を持っていることを確認した。
このプラスミドを制限酵素NheI、XhoIで切断した後、分泌型ヒトOBM cDNAに相当する断片(0.8kb)をアガロースゲル電気泳動で回収した。この断片をライゲーションキットを用いて発現ベクターpCEP4(インビトローゲン社)のNheI/XhoI断片(10.4kb)に挿入し、その反応生成物を用いて大腸菌DH5αを形質転換した。得られたアンピシリン耐性株より、アルカリSDS法によりプラスミドを精製し、制限酵素で切断することにより、目的の構造を持った分泌型ヒトOBM発現プラスミド(pCEPshOBM)を持つ大腸菌株を選択した。pCEPshOBMを持つ大腸菌株を培養し、キアフィルタープラスミドミディキット(キアゲン社)を用いてpCEPshOBMを精製した。
【0100】
(2) 分泌型 OBM の発現
293-EBNA細胞を10%FCSを含むIMDM (IMDM-10% FCS) に懸濁し、コラーゲンコートした24ウェルプレート(住友べ一クライト社)に2×105細胞/2ml/ウェルになるように播種し、一夜培養した。この細胞に1μg のpCEPshOBM 及びpCEP4 のそれぞれをリポフェクトアミン(ギブコ社) 4μlを用いて形質導入し、0.5mlの無血清IMDMまたはIMDM-10 % FCS中で更に2日間培養し、その培養上清を回収した。分泌型ヒトOBMの培養上清中での発現は、以下のようにして確認した。即ち、培養上清に最終濃度が0.1Mになるように炭酸水素ナトリウムを加えて4℃で一夜放置し、培養上清中のヒトOBMを96ウェルプレートに固相化した。このプレートをPBSで4倍希釈したブロックエース(雪印乳業社) 溶液(B-PBS)で室温に2時間放置しブロッキングした後、B-PBSで希釈した3-100 ng/mlのOCIFを加えて37℃で2時間放置した。0.05%Polysorbate 20 を含むPBS (P-PBS)でプレートを洗浄した後、B-PBSで希釈したWO96/26217号記載のパーオキシダーゼ標識抗OCIF抗体を各ウェルに100μlずつ添加し、37℃で2時間放置した。P-PBSで各ウェルを6回洗浄した後、TMB溶液(TMB Soluble Reagent, High Sensitivity, Scytek社)を100μlずつ各ウェルに加え、室温で約10分間放置した後、停止液(Stopping Reagent. Scytek杜)を100μlずつ各ウェルに加えた。各ウェルの450 nmにおける吸光度をマイクロプレートリーダーで測定した。結果を図21に示す。pCEPshOBMを形質導入した細胞の培養上清を固相化したプレートでは、添加したOCIFの濃度に依存して450nmの吸光度が増加した。一方、ベクターのみのpCEP4を形質導入した細胞の培養上清を固相化した場合には、その吸光度の増加は見られなかった。又、固相化に用いる培養上清量の割合を5〜90%の範囲で種々変化させ、一定濃度のOCIF(50ng/ml)を添加した際の結果を図22に示す。pCEPshOBM を形質導入した細胞の培養上清を固相化したプレートでは、添加する培養上清量の増加につれて450nmにおける吸光度が増加したが、ベクターpCEP4を形質導入した細胞の培養上清を固相化したプレートでは、吸光度の増加は見られなかった。これらの結果により、分泌型ヒトOBM がpCEPshOBM を形質導入した細胞の培養上清中に発現していることが確認された。
【0101】
〔実施例25〕
チオレドキシン - ヒト OBM 融合蛋白質 (Trx-hOBM) の発現
(1) チオレドキシン - ヒト OBM 融合蛋白質 (Trx-hOBM) 発現ベクターの構築
10μlの10×ExTaqバッファ-(宝酒造社)、8μlの 10mM dNTP (宝酒造社)、77.5μlの滅菌蒸留水、2μlのpUC19hOBM 水溶液 (10ng/μl)、1μlのプライマーマウスOBM #3 (配列表配列番号9)(100pmol/μl)、1μlのプライマーhOBM SalR2 (配列表配列番号14)(100pmol/μl)、0.5 μlのExTaq (5u/μl)(宝酒造社)を混合しマイクロ遠心管中でPCR反応を行った。95℃5分、50℃1秒、55℃1分、74℃1秒、72℃5分の反応後、96℃1分、50℃1秒、55℃1分、74℃1秒、72℃3分の反応を25サイクル行った。反応液全量から約750bpのDNA断片を精製した。精製したDNA断片(全量)を制限酵素SalI(宝酒造社)及びBspHI(ニューイングランドバイラブズ杜)で切断した後、1%アガロース電気泳動を行い、約320bpのDNA断片(断片1)を精製し20μlの滅菌蒸留水に溶解した。同様に、実施例19-(3)記載のpUC19hOBM4μgを制限酵素BamHI, BspHI(宝酒造社)で切断して得られた約450bpのDNA断片(断片2)、及び2μgのpTrxFus(インビトロジェン社)を制限酵素BamHI, SalI(宝酒造社)で切断して得られた約3.6 kbのDNA断片(断片3)をそれぞれ精製し、それぞれを20μlの滅菌蒸留水に溶解した。DNA断片の精製にはQIAEXII gel extraction kitを用いた。断片1、断片2、及び断片3をDNA ligation kit ver.2 (宝酒造社)を用い、16℃で2時間30分保温することにより結合させた。ライゲーション反応液を用い大腸菌GI724株(インビトロジェン社)をThioFusion Expression System(インビトロジェン社)添付のInstruction Manualに記載の方法で形質転換した。得られたアンピシリン耐性形質転換株の中から、制限酵素切断により得られるDNA断片地図の解析並びにDNA配列の決定により、hOBM cDNA断片がチオレドキシン遺伝子に同じ読み枠で結合しているプラスミドを持っ菌株を選択した。得られた菌株をGI724/pTrxhOBMと名付けた。
【0102】
(2) 大腸菌での Trx-hOBM の発現
GI724/pTrxhOBM株及びpTrxFus を持つGI724株(GI724/pTrxFus)をそれぞれ2mlのRMG‐Amp培地(0.6% Na2HPO4、0.3% KH2PO4、0.05% NaCl、0.1% NH4Cl、2% カザミノ酸、1% グリセロール、1mM MgCl2、100 μg/mlアンピシリン、pH 7.4) 中、37℃で6時間振盪培養した。菌液 0.5ml を50ml のInduction培地(0.6% Na2HPO4、0.3% KH2PO4、0.05% NaCl、0.1% NH4Cl、0.2%カザミノ酸、0.5%グルコース、1mM MgCl2、100μg/mlアンピシリン、pH7.4)に添加し、30℃で振盪培養した。OD600nm における値が約0.5になった時点で、最終濃度が0.1mg/mlになるようにレトリプトファンを添加し、更に30℃で6時間振盪培養した。菌液を3,000×gで遠心し菌体を集め、12.5 ml のPBS に懸濁した。懸濁液を超音波発生装置(Ultrasonics社)にかけ菌体を破砕し、7,000 ×gで30分間遠心して上清を集めて可溶性蛋白質画分とした。これらの画分溶液10μlずつを還元条件下でのSDS-PAGE(10%ポリアクリルアミド)に供した。この結果、図23に示すように、GI724/pTrxOBM の可溶性蛋白質画分には、GI724/pTrxFus の可溶性蛋白質画分には見られない分子量約40,000の蛋白質のバンドが検出された。以上の結果から、チオレドキシンとヒトOBMの融合蛋白質(Trx-hOBM) が大腸菌中で発現していることが確認された。
【0103】
(3) Trx-hOBM の OCIF との結合能
発現させたTrx-hOBMがOCIFと結合することを、以下の実験により確認した。即ち、96ウェルイムノプレート(Nunc社)の各ウェルに10mM 炭酸水素ナトリウム水溶液で5000倍に希釈した抗チオレドキシン抗体(インビトロジェン社)を100μl ずつ添加し4℃で一夜放置した。各ウェル中の液を捨てた後、ブロックエース(雪印乳業社)をPBSで2倍希釈した溶液 (BA-PBS) を各ウェルに200 μlずつ添加し室温で1時間放置した。液を捨てた後、各ウェルをP-PBSで3回洗浄した。各ウェルにBA-PBSで段階希釈した上記GI724/pTrxhOBM 由来及び GI724/pTrxFus 由来可溶性蛋白質画分溶液を 100μl ずつ添加し室温で2時間放置した。P-PBSで各ウェルを3回洗浄した後、BA-PBSで希釈したOCIF(100ng/ml)を各ウェルに100μずつ添加し室温に2時間放置した。P-PBSで各ウェルを3回洗浄した後、BA-PBSで2000倍に希釈したWO96/26217号記載のパーオキシダーゼ標識した抗OCIF抗体を各ウェルに100μlずつ添加し、室温で2時間放置した。P-PBS で各ウェルを6回洗浄した後、TMB 溶液を 100μlずつ各ウェルに加え、室温で約10分間放置した後、停止液(Stopping Reagent)を 100μlずつ各ウェルに加えた。各ウェルの450 nmにおける吸光度をマイクロプレートリーダーで測定した。結果を図24に示す。 GI724/pTrxFus由来可溶性蛋白質画分溶液を添加したもの、添加しなかったものの間で吸光度には差異は見られなかったが、GI724/pTrxhOBM 由来可溶性蛋白質画分溶液を添加したものでは、その添加濃度が増加するにつれて吸光度が上昇した。又、添加する可溶性蛋白質画分溶液の希釈割合を一定(1%濃度)にし、BA-PBSで段階希釈したOCIF(0-100ng/ml)を添加した場合の実験結果を図25に示す。 GI724/pTrxFus 由来可溶性蛋白質画分溶液を用いたものでは、OCIFの濃度に係わりなく吸光度は低値であったが、GI724/pTrxhOBMに由来可溶性蛋白質画分溶液を添加したものでは、OCIF濃度が増加するにつれて吸光度が増加した。これらの結果から、GI724/pTrxhOBMで生産されるTrx-hOBMには、OCIFと結合する能力があることが確認された。
【0104】
(4)Trx-hOBM を生産する大腸菌の大量培養
GI724/pTrxhOBMをRMG-Amp 寒天培地(0.6%Na2HPO4、0.3% KH2PO4、0.05% NaCl、0.1% NH4Cl、2% カザミノ酸、1.5% 寒天、pH 7.4)に白金耳で塗抹し、30℃で一夜培養した。菌体を10ml のInduction培地に懸濁し、5mlずつを500 mlのInduction 培地の入った2 l用三角フラスコ計2本に添加し、30℃で振盪培養した。OD600nm での吸光度が約0.5になった時点で最終濃度が0.1 mg/mlとなるようにL‐トリプトファンを添加し、更に30℃で6時間振盪培養した。菌液を3,000×gで20分間遠心分離して菌体を集め、菌体を160 mlのPBS に懸濁した。懸濁液を超音波発生装置(Ultrasonics社)にかけ菌体を破砕し、7,000×g で30分間遠心分離して上清を集め可溶性蛋白質画分とした。
【0105】
(5) OCIF 固定化アフィニティーカラムの調製
TSKgel AF-トレシルトヨパール650 (東ソー社) 2gと、WO96/26217号公報記載の方法で調製した遺伝子組み換え型OCIF35.0 mgを含む1.0Mリン酸カリウム緩衝液 (pH 7.5) 40mlとを混合し、4℃で一夜緩やかに振盪することによりカップリング反応を行った。過剰な活性基を不活性化するため、遠心分離により上清を除去した後、40mlの0.1Mトリス‐塩酸緩衝液 (pH 7.5)を沈殿した担体に加え、室温で1時間穏やかに振盪した。0.01% Polysorbate 80 と0.2M NaCl を含む0.1Mグリシン‐塩酸緩衝液 (pH 3.3) 及び0.01% Polysorbate 80 と0.2M NaClを含む0.1M クエン酸ナトリウム緩衝液 (pH 2.0)を通液しカラムを洗浄した後、0.01%Polysorbate 80 を含む10 mM リン酸ナトリウム緩衝液 (pH7.4)で2回洗浄し、カラムを平衡化した。
【0106】
(6) OCIF 固定化アフィニティーカラムによる Trx-hOBM の精製
Trx-hOBMの精製は、特に断らない限り4℃で行った。上記のOCIF固定化アフィニティー担体(10ml) と上記実施例25-(4)記載の可溶性蛋白質画分溶液 (120ml)とを混合後、50ml遠心管 (4本)中で、ローテーターを用いて4℃で一夜穏やかに振盪した。この混合液中の担体をエコノカラム(内径1.5cm×長さ15cm、バイオラッド社)に充填した。0.01% Polysorbate 80 を含むPBS 300ml、0.01% Polysorbate 80 及び2.0MNaClを含む10 mM リン酸緩衝液 (pH 7.0) 100ml、0.01% Polysorbate 80 及び0.2M NaClを含む0.1M グリシン‐塩酸緩衝液(pH 3.3) 100mlを順次通液してカラムを洗浄した。次に、0.01% Polysorbate 80 及び0.2M NaClを含む0.1 M クエン酸ナトリウム緩衝液(pH 2.0)を通液し、カラムに吸着した蛋白質を溶出した。溶出液は5mlずつ分取した。分取した画分には、10%容量の2Mトリス溶液 (pH 8.0) を加えて直ちに中和した。溶出液各画分中のTrx-hOBMの有無を、実施例25-(3)記載の方法により調べた。又、Trx-hOBMを含む画分を集め、更に精製を進めた。
【0107】
(7) ゲル濾過による Trx-hOBM の精製
上記実施例25-(6)記載のTrx-hOBMフラクション約25mlをCentriplus10及びCentricon 10 (Amicon社)を用いて、約0.5mlに遠心濃縮した。この濃縮サンプルを予め0.01% Polysorbate 80 を含むPBSで平衡化したSuperose12 HR 10/30カラム(1.0×30cm、ファルマシア社)に負荷した。移動相として0.01% Polysorbate 80 を含むPBSを用い、流速0.25ml/minでカラムを展開した。カラムからの溶出液は0.25mlずつ分取した。分取した画分のTrx-hOBMは、実施例25-(3)記載の方法及びSDS-PAGEにより検出した。純化されたTrx-hOBMを含むフラクションを集め、Trx-hOBMの蛋白質濃度を測定した。蛋白質濃度は牛血清アルブミンを標準品として用い、DC-protein assay kit(バイオラッド社)で測定した。
【0108】
〔実施例26〕
hOBM の破骨細胞形成誘導活性
phOBM及びpcDL-SRα296をリポフェクトアミン(ギブコ社)を用いてCOS-7細胞にそれぞれトランスフェクトした。それぞれの細胞を10% FCSを含むDMEMで1日間培養後トリプシン処理し、カバーグラス(1mm丸形、マツナミ社)を敷いた24ウェルプレートに1ウェル当たり5×104個の細胞を播種し、さらに2日間培養した。培養プレートをPBSで1回洗浄した後、1%パラフォルムアルデヒドを含むPBSを加えて室温で8分間インキュベートし細胞をカバーグラス上に固定化した。細胞を固定化したプレートをPBSで6回洗浄した後、10-8M活性化ビタミンD3、10− 7Mデキサメサゾン、10%牛胎児血清を含むα-MEMにて1×106個/mlになるように懸濁したマウス脾臓細胞を1ウェル当たり700μlずつ添加した。各ウェル上にMillicell PCF(ミリポア社)をセットし、上記培地にて4×104個/mlに懸濁したST2細胞をMillicell PCF中に700μlずつ添加し、37℃で6日間培養した。培養終了後、Millicell PCFを取り除き、プレートをPBSで1回洗浄後、アセトン-エタノール溶液(50:50)で1分間細胞を固定した後、LEUKOCYTE ACID PHOSPHATASEキット(シグマ社)を用いて破骨細胞特異的マーカーである酒石酸耐性酸性フォスファターゼ活性(TRAP活性)を示す細胞のみを染色した。顕微鏡観察の結果、ヒトOBM cDNAを含まないベクターのみのpcDL-SRα296をトランスフェクトしたCOS-7細胞を固定化したウェル中にはTRAP活性を示す細胞は全く検出されなかったのに対し、phOBMをトランスフェクトした細胞を固定化したウェル中には65±18個(n=3、平均値±標準偏差)のTRAP陽性細胞が観察された。又、これらのTRAP陽性細胞は125I -標識サケカルシトニン(アマシャム社)と特異的結合を示したことから、カルシトニンレセプターを発現していることが確認された。これらの結果から、本発明cDNAによりコードされる蛋白質であるヒトOBMは破骨細胞形成を誘導する活性があることが明らかとなった。
【0109】
〔実施例27〕
Trx-hOBM 及び分泌型ヒト OBM による破骨細胞形成誘導活性
マウス脾臓細胞を10-8M活性型ビタミンD3、10− 7Mデキサメサゾン、10%牛胎児血清を含むα-MEMに2×106個/mlになるように懸濁し、24ウェルプレートに1ウェルあたり350μl添加した。精製したTrx-hOBMを上記培地で希釈した溶液(40ng/ml)、pCEPshOBMまたはpCEP4を形質導入した293-EBNA細胞をIMDM-10%FBSで培養した培養上清を上記培地にて10倍希釈した溶液または上記培地のみをそれぞれ350μl添加した後、Millicell PCF(ミリポア社)を各ウェルにセットし、上記培地にて懸濁した4×104個/mlのST2細胞をMillicell PCFに600μl添加した。6日間培養した後、MillicellPCFを取り除き、プレートをPBSで1回洗浄後、アセトン-エタノール溶液(50:50)で1分間細胞を固定した後、LEUKOCYTE ACID PHOSPHATASEキット(シグマ社)を用いて酒石酸耐性酸性フォスファターゼ活性(TRAP活性)を示す細胞のみを染色した。顕微鏡観察の結果、Trx-hOBMを添加しなかったウェル中にはTRAP活性を示す細胞は全く検出されなかったのに対し、Trx-hOBMを添加したウェルには115±19個(n=3、平均値±標準偏差)のTRAP陽性細胞が観察された。同様に、pCEP4で形質導入された293-EBNAの培養上清を添加したウェル中にはTRAP活性を示す細胞は全く検出されなかったのに対し、pCEPshOBMで形質導入された293-EBNAの培養上清を添加したウェルには125±23個(n=3、平均値±標準偏差)のTRAP陽性細胞が観察された。又、これらTRAP陽性細胞は125I標識したサケカルシトニン(アマシャム社)と特異的結合を示しことから、カルシトニンレセプターを発現していることが確認された。これらの結果から、Trx-hOBM及び分泌型OBMには、破骨細胞形成を誘導する活性があることが明らかとなった。
【0110】
〔実施例28〕
ポリクローナル抗体の作製
免疫抗原として用いるマウスsOBM又はヒトsOBMは、前記の方法に従って得た。即ち、マウスsOBM cDNA (マウスOBMのN末端から72番目までのアミノ酸を欠損させた膜結合部位を持たないマウスsOBM(配列表配列番号16)をコードするcDNA、配列表配列番号18)又はヒトOBM cDNA(ヒトOBMのN末端から71番目までのアミノ酸を欠損させた膜結合部位を持たないヒトsOBM(配列表配列番号17)をコードするcDNA、配列表配列番号19)を、イムノグロブリンのκ-チェーンのシグナルペプチドをコードする塩基配列を含む発現ベクターpSecTagA(インビトローゲン社)のHindIII/EcoRV断片(5.2kb)と、OBMcDNAのEcoRI/PmaCI断片(0.32kb)とともにライゲーションキットver.2(宝酒造社)を用いてライゲーションを行い、その反応生成物を用いて大腸菌DH5αを形質転換した。得られたアンピシリン耐性株からアルカリSDS法によりプラスミドを精製し、制限酵素で切断することによりpSec TagAに0.6Kbと0.32 Kbの断片が挿入されたプラスミドを選び出した。このプラスミドをダイターミネーターサイクルシークエンシングFSキット(パーキン-エルマー社)を用いてシークエンスを確認した結果、このプラスミドがマウス又はヒトsOBMをコードする配列を有することを確認した。このプラスミドを制限酵素NheI、XhoIで切断した後、分泌型OBM cDNAに相当する断片(1.0kb)をアガロースゲル電気泳動で回収した。この断片をライゲーションキットを用いて発現ベクターpCEP4(インビトローゲン社)のNheI/XhoI断片(10.4kb)に挿入し、その反応生成物を用いて大腸菌DH5αを形質転換した。得られたアンピシリン耐性株からアルカリSDS法によりプラスミドを精製し、制限酵素で切断して解析することにより、目的の構造を持った分泌型OBM発現プラスミド(pCEP sOBM)を持つ大腸菌株を選び出した。pCEP sOBMを持つ大腸菌株を培養し、キアフィルタープラスミドミディキット(キアゲン社)を用いてpCEP sOBMを精製した。次に、293-EBNA細胞を10%FCSを含むIMDM(IMDM-10%FCS)に懸濁し、コラーゲンコートした24ウェルプレート(住友べ一クライト社)に2×105個/2ml/ウェルになるように播種し、一夜培養した。この細胞に1μgのpCEP sOBM又はpCEP4をリポフェタミン(ギブコ社)4μlを用いて形質導入し、0.5mlの無血清IMDM又はIMDM-10%FCS中でさらに2日間培養し、その培養上清を回収した。遺伝子組み換えマウス可溶性OBM(msOBM)あるいはヒト可溶性OBM(hsOBM)の高生産細胞株を以下のようにスクリーニングした。msOBMあるいはhsOBMを含むと見られる培養上清に最終濃度が0.1Mになるように炭酸水素ナトリウムを添加後、それぞれの培養上清を100μlずつ96ウェルイムノプレート(Nunc社)の各ウェルに添加し、4℃で一夜放置して培養上清中のmsOBMあるいはhsOBMを各ウェルに固相化した。このプレートの各ウェルにPBSで4倍希釈したブロックエース(雪印乳業社)溶液(B-PBS)を200μl加え、室温で2時間放置した。0.1%polysprbate20を含むPBS(P-PBS)で3回洗浄後、B-PBSで段階的に希釈した遺伝子組み換えOCIF(rOCIF)溶液(3〜100ng/ml)を100μlずつ各ウェルに添加し、37℃で2時間放置した。そのプレートをPBSで3回洗浄した後、B-PBSで希釈したパーオキシダーゼ標識抗OCIFポリクローナル抗体(WO96/26217号)を100μlずつ各ウェルに添加し、37℃で2時間放置した。P-PBSで各ウェルを6回洗浄後、TMB溶液(TMB Soluble Agent, Highsensitivity, Scytek社)を100μlずつ各ウェルに加え、室温で約10分間放置した後、停止液(Stopping Reagent, Scytek社)を100μlずっ各ウェルに加えた。各ウェルの450nmにおける吸光度をマイクロプレートリーダーで測定した。msOBMあるいはhsOBMの生産細胞株の培養上清を固相化したプレートでは、OCIFの添加濃度に比例して、吸光度が顕著に増加した。msOBMあるいはhsOBMの生産細胞株について、その吸光度の増加の割合の高い生産株を、それぞれ高生産株として選択した。このように選択されたmsOBMあるいはhsOBM高生産293-EBNA細胞をそれぞれ5%FCSを含有するIMDMを培地として用い、T-フラスコ(T-225)を25枚用いて大量培養した。コンフルエントに達した後、新鮮な培地をT-225フラスコ1枚当たり100mlずつ加え、3〜4日間培養して、培養上清を回収した。この操作を4回繰り返して、msOBMあるいはhsOBMを含む培養上清をそれぞれ10リットル得た。実施例25-(6)及び(7)記載の方法に従いrOCIF固定化カラムを用いたアフィニティークロマトグラフィーおよびゲル濾過クロマトグラフィーにより精製することにより、これら培養上清からSDS-ポリアクリルアミド電気泳動的に均一(分子量32kDa)な精製msOBM及びhsOBMをそれぞれ約10mg及び12mg得た。このようにして得られた精製標品を、それぞれ免疫抗原として用いた。得られた各抗原を、リン酸塩緩衝生理食塩水(PBS)に200μg/mlになるように溶解し、等容量のフロイント完全アジュバントと混合し乳化後、日本白色ウサギ各3羽に約1週間間隔で1mlを皮下投与し免疫した。抗体価を測定し、抗体価が最高に到達した時点で、ブースター投与し、投与10日後に全採血した。抗血清をプロテインAセファロースクロマトグラフィー用バインディングバッファー(BioRad社)で2倍希釈し、同バッファーで平衡化したプロテインAカラムに負荷した。同バッファーでカラムを充分洗浄後、カラムに吸着した抗sOBM抗体をエルーションバッファー(BioRad社)あるいは0.1Mグリシン-HClバッファー、pH2.9-3.0で溶出した。抗体溶出液を直ちに中和する目的で、1.0MTris-HCl,pH8.0を少量含む試験管を用いて溶出溶液を分画した。抗体溶出液をPBSに対して4℃、一夜透析した。抗体溶液の蛋白量をウシIgGをスタンダードとしてLowry法により測定した。このようにして、本発明のポリクローナル抗体を含む精製イムノグロブリン(IgG)が、ウサギ抗血清1ml当たり約10mg得られた。
【0111】
〔実施例29〕
ポリクローナル抗体を用いた ELISA による OBM 及び sOBM の測定
実施例28で得られたウサギ抗ヒトsOBMポリクローナル抗体を、固相抗体及び酵素標識抗体としたサンドイッチELISAを構築した。酵素標識は、石川らの方法(Ishikawa et al:J. Immuno assay, Vol.4, 209-327, 1983)に従いパーオキシダーゼ(POD)標識した。実施例28で得られた抗ヒトsOBMポリクローナル抗体を2μg/mlとなるように0.1M NaHCO3溶液に溶解し、96-ウェルイムノプレート(Nunc社)の各ウェルに100μlずつ添加、室温で一夜放置した。次いで、各ウェルに200μlずつ50%ブロックエース(雪印乳業社)を加え、室温で1時間放置し、各ウェルを0.1%polysorbate20を含むPBS(洗浄バッファー)で3回洗浄した。
実施例26の方法と同様にヒトOBMを発現し実施例2の方法で精製することにより得られた精製ヒトOBM、及び実施例28で得られた精製ヒトsOBMをそれぞれ一次反応用バッファー(40%ブロックエース、0.1%polysorbate20を含む0.2MTris-HClバッファー、pH7.2)で段階希釈した溶液を、100μlずつ各ウェルに加えた。室温で2時間放置後、各ウェルを上記洗浄バッファーで3回洗浄した。
二次反応用バッファー(25%ブロックエース、0.1%polysorbate20を含む0.1MTris-HClバッファー、pH7.2)で1000倍希釈したPOD標識抗ヒトsOBMポリクローナル抗体を100μlずつ各ウェルに加え、室温で2時間放置した後、各ウェルを3回洗浄バッファーで洗浄した。各ウェルに酵素基質溶液(TMB, ScyTek社)を100μlずつ添加し、室温で10分間放置後、反応停止液(Stopping reagent, ScyTek社)を100μlずつ各ウェルに加えて酵素反応を停止させた。各ウェルの450nmにおける吸光度をマイクロプレートリーダーを用いて測定した。結果を図26に示す。ウサギ抗ヒトsOBMポリクローナル抗体を用いたサンドイッチELISAは、ヒトsOBM(分子量約32kDa)及びヒトOBM(分子量約40kDa)をほぼ等しく認識し、それぞれの測定感度は約12.5x10-3pmol/ml(ヒトOBMで約500pg/ml、ヒトsOBMで約400pg/ml)であった。実施例28で得られたウサギ抗マウスsOBMポリクローナル抗体を用いたELISAによるマウスsOBM及びマウスOBMの測定は上記と同様に実施でき、それぞれの測定感度も同程度で極めて微量のマウスsOBMあるいはマウスOBMを測定できることが明らかとなった。
以上のように、実施例28で得られた本発明抗ヒトsOBMポリクローナル抗体は、ヒトsOBM及びヒトOBMの両抗原を等しく認識することから、抗ヒトOBM/sOBMポリクローナル抗体と名付けた。又、実施例28で得られた抗マウスsOBMポリクローナル抗体は、同様にマウスsOBM及びマウス0BMの両抗原を等しく認識することから、抗マウスOBM/sOBMポリクローナル抗体と名付けた。
【0112】
〔実施例30〕
モノクローナル抗体の作製
免疫用抗原には、実施例28で得られた精製ヒトsOBMを用いた。精製ヒトsOBMを10μg/mlになるように生理食塩水に溶解した。このようにして調製したヒトsOBM溶液に、同容量のフロイント完全アジュバントを加え良く乳化した後、それぞれの抗原をBalb/cマウスー匹当たり腹腔内に200μlずつ1週間間隔で3回投与し、マウスを免疫した。次に、ヒトsOBMを5μg/ml含む生理食塩水に同容量のフロイント不完全アジュバントを添加し、充分乳化したものを上記Balb/cマウスー匹あたり200μlずつ、1週間間隔で4回追加免疫した。4回目の追加免疫の1週間後、ブースターとしてヒトsOBMを10μg/ml含む生理食塩水をBalb/cマウス1匹当たり100μlずつ静脈内に投与した。最終免疫後3日目に脾臓を摘出、脾臓細胞を分離し、マウスミエローマ細胞 P3x63-AG8.653とを公知の方法(Koehler,G. and Milstein, C., Nature,256, 495(1975))に従い細胞融合した。融合終了後、細胞懸濁液をヒポキサンチン、アミノプテリン及びチミヂンを含むHAT培地中で10日間培養した。ミエローマ細胞が死滅し、ハイブリドーマが出現した後、HAT培地からアミノプテリンを除いたHT培地に培地交換し、培養を継続した。
【0113】
〔実施例31〕
ハイブリドーマの選択及びクローニング
実施例30の細胞融合及び培養を開始して10日後にハイブリドーマの出現を認めたので、下記に記載した改良ソリッドフェーズELISAにより、ヒトsOBMを認識する高親和性抗体及びその抗体を産生するハイブリドーマの選択を行った。又、ヒトsOBMとマウスsOBMの両方を認識する抗OBMモノクローナル抗体を選択するために、ソリッドフェーズELISA用抗原として、ヒトsOBM以外に実施例27で得られたマウスsOBMも使用した。ヒトsOBM及びマウスsOBMをそれぞれ5μg/mlになるように0.1M炭酸水素ナトリウム溶液に溶解し、それぞれの抗原溶液を50μlずつ96-ウェルイムノプレート(Nunc社)の各ウェルに加え、4℃で一夜静置しそれぞれの抗原を固相化した。各ウェル中の抗原溶液を捨て、各ウェルに50%ブロックエース(雪印乳業社)を200μlずつ加え、室温で1時間放置してブロッキングした。各ウェルを0.1%polysorbate20を含むリン酸塩緩衝生理食塩水(PBS-P)で洗浄し、各ウェルに40μlの牛血清(ハイクローン社)を加えた。次いで、各ウェルに10μlのハイブリドーマ培養上清を加え、80%血清濃度下、室温で2時間反応させた。80%血清存在下でのソリッドフェーズELISAの目的は、高濃度の蛋白質及び血清由来の免疫反応妨害物質等の存在中でも微量のヒトsOBMあるいはマウスsOBMと結合できる抗体、即ちヒトsOBMあるいはマウスsOBMに対して高親和性の抗体生産ハイブリドーマを選択するためである。室温で2時間反応後、プレートをPBS-Pで洗浄し、25%ブロックエースを含む生理食塩水で5000分の1に希釈したパーオキシダーゼ標識抗マウスIgG(KPL社)を各ウェルに50μl加え、室温で2時間反応させた。プレートをPBS-Pで3回洗浄した後、各ウェルに50μlの酵素基質溶液(TMB, ScyTek社)を加え、室温で5分間反応させた。次いで、反応停止液(stopping reagent, ScyTek社)50μlを加えて酵素反応を停止させた。各ウェルの450nmにおける吸光度をマイクロプレートリーダー(イムノリーダーNJ2000、日本インターメッド社)を用いて測定し、ヒトsOBMあるいはマウスsOBMを認識する抗体生産ハイブリドーマを選択した。特に高い吸光度(OD450nm)を示すそれぞれの抗体生産ハイブリドーマを選択し、それらを限界希釈法により3〜5回クローニングを繰り返すことにより、安定な抗体生産ハイブリドーマを樹立した。得られた抗体生産株の中から、さらに抗体の生産性が高いハイブリドーマ株を選別した。
【0114】
〔実施例32〕
モノクローナル抗体の生産及び精製
実施例31で得られた抗体、即ち高親和性でヒトsOBMを認識する抗体生産ハィブリドーマ及びマウスsOBMにも交差性を有する抗体を産生するハイブリドーマをそれぞれ培養し、マウスー匹当たりそれぞれのハイブリドーマを1×106細胞ずつ、約1週間前にプリスタン(アルドリッチケミカル社)を投与しておいたBalb/c系マウスの腹腔内に移植した。約2〜3週間後、蓄積した腹水を採取し、本発明のヒトsOBMあるいはヒトsOBM及びマウスsOBMを認識するモノクローナル抗体を含む腹水を得た。実施例28に記載したプロテインAカラムによる抗OBM/sOBMポリクローナル抗体の精製法に準じて、プロテインAカラム(ファルマシア社)クロマトグラフィーにより腹水から精製モノクローナル抗体を得た。
【0115】
〔実施例33〕
モノクローナル抗体の抗原特異性
ヒトsOBMを特異的に認識するモノクローナル抗体並びにヒトsOBM及びマウスsOBMにも交差性を有するモノクローナル抗体について、ヒトsOBM、膜結合部位を有するintactなヒトOBM、マウスsOBM、及び膜結合部位を有するintactなマウスOBMを抗原とした時の抗原特異性について調べた。30数種類のモノクローナル抗体を得たが、それらのうち代表的なモノクローナル抗体にっいての結果を第1表に示す。この結果、ヒトsOBMを特異的に認識する抗ヒトsOBMモノクローナル抗体のほとんど全ては膜結合部位を有するintactなヒトOBMも認識するが、マウスsOBM及び膜結合部位を有するintactなマウスOBMは認識しないことが明らかとなった。一方、ヒトsOBM及びマウスsOBMも認識するモノクローナル抗体も得られたが、その数は極めて少なく、これらの抗体はヒトOBM並びにマウスOBMにも交差性を有していることがわかった。これらの結果は、ヒトOBMとマウスOBMには共通した抗原認識部位、即ちエピトープが存在していることを示すものである。ヒトsOBMを免疫抗原として作製した抗ヒトsOBMモノクローナル抗体は、膜結合型のintactな蛋白質であるヒトOBMも等しく認識することから、抗ヒトsOBMモノクローナル抗体を抗ヒトOBM/sOBMモノクローナル抗体と名付けた。
【0116】
【表1】
〔実施例34〕
モノクローナル抗体のクラス及びサブクラスの検定
本発明のモノクローナル抗体のクラス及びサブクラスを、イムノグロブリンクラス及びサブクラス分析キット(アマシャム社)を用いて検定した。検定は、キットに指示されているプロトコールに従い実施した。代表的なモノクローナル抗体についての結果を表2に示す。抗ヒトOBM/sOBMモノクローナル抗体の大多数はIgG1であり、その他IgG2a及びIgG2bも存在していた。又、いずれの抗体もライトチェーンはκ鎖であった。
【0118】
【表2】
〔実施例35〕
モノクローナル抗体の解離定数 (Kd 値 ) の測定
公知の方法(Betrand Friguet et al.:Journal of Immunological Methods, 77,305-319,1986)に従い、モノクローナル抗体の解離定数を測定した。即ち、実施例32で得られた精製抗体を5ng/mlに40%ブロックエース、0.1% polysorbate20を含む0.4M Tris-HCl pH7.4(1次バッファー)で希釈し、これに実施例28で得られた精製ヒト可溶性OBM(hsOBM)を6.25ng/ml〜10μg/mlになるように1次バッファーで希釈したものを等容量混ぜ、4℃で15時間静置することによりhsOBMとモノクローナル抗体を結合させた。15時間後、hsOBMと未結合の抗体をhsOBM(10μg/ml、100μl/ウェル)を固相化したソリッドフェーズELISAにて測定することにより、モノクローナル抗体のhsOBMに対する解離定数を算定した。又、hsOBMに対するモノクローナル抗体でありながら、マウス可溶性OBM(msOBM)にも交差する抗体のmsOBMに対する親和性についても、上記のhsOBMの代わりにmsOBMを使用することにより同様に測定した。特に、各抗原に高い親和性を示し、酵素免疫測定法やその他バインディングアッセイ等に有用な抗体についての結果を第3表に示す。
【0120】
【表3】
【0121】
〔実施例36〕
抗ヒト OBM/sOBM モノクローナル抗体を用いたサンドイッチ ELISA によるヒト OBM 及び sOBM の測定方法
実施例35で得られた2種類の高親和性モノクローナル抗体、H-OBml及びH-OBM4をそれぞれ固相抗体と酵素標識抗体として、サンドイッチELISAを構築した。
抗体の標識は、マレイミド活性化パーオキシダーゼキット(ピアス社)を用いて行った。H-OBml抗体を10μg/mlになるよう0.1M炭酸水素ナトリウム溶液に溶解し、100μlずつ96-ウェルイムノプレート(Nunc社)の各ウェルに加え、4℃で一夜静置し固相化した。各ウェルの溶液を捨て、50%濃度のブロックエース300μlを加え、室温で2時間静置しブロッキングした。ブロッキング後、プレートを0.1%polysorbate20を含むリン酸塩緩衝生理食塩水(PBS-P)で洗浄した。ヒト可溶性sOBM及びヒトOBMをそれぞれ40%ブロックエース(雪印乳業社)及び0.1%polysorbate20(和光純薬社)を含む0.4MTris-HC(pH7.4)(1次反応バッファー)に溶解、希釈し、種々の濃度の被験溶液を調製した。種々の濃度に調製したそれぞれの被験溶液を100μlずつ各ウェルに加え、室温で2時間反応させた。2時間後、プレートをPBS-Pで洗浄し、25%ブロックエース及び0.1%polysorbate20を含む0.2MTris-HCl(pH7.4)(2次反応バッファー)で希釈したPOD標識H-OBM4抗体を各ウェルに100μl加え、室温で2時間反応させた。
プレートをPBS-Pで洗浄した後、各ウェルに100μlの酵素基質溶液(TMB,ScyTek社)を加えて発色させた後、反応停止液(stopping reagent, ScyTek社)を100μlずつ各ウェルに加えて酵素反応を停止した。各ウェルの450nmにおける吸光度をマイクロプレートリーダーを用いて測定した。結果を図27に示す。
この結果、実施例35で得られた2種類の高親和性抗ヒトOBM/sOBMモノクローナル抗体、H-OBml及びH-OBM4を用いて構築したサンドイッチELISAは、ヒトOBM及びヒトsOBMを等しく認識し、その測定感度は約1.25-2.5×10-3pmol/ml(分子量約40kDaのヒトOBMでは約50-100pg/ml、分子量約32kDaのヒトsOBMでは約40-80pg/ml)で極めて微量のヒトOBM及びヒトsOBMを測定できることが明らかとなった。尚、これら2種類の抗ヒトOBM/sOBMモノクローナル抗体、H-OBml及びH-OBM4を生産するハイブリドーマをH-OBml及びH-OBM4と命名し、またマウスOBM及びマウスsOBMも認識し、破骨細胞形成抑制活性を発揮する抗ヒトOBM/sOBMモノクローナル抗体、H-OBM9を生産するハイブリドーマをH-OBM9と命名した。これらのハイブリドーマは、通商産業省工業技術院生命工学工業技術研究所(現独立行政法人産業技術総合研究所 微生物寄託センター)に受託番号FERM BP-6264 (H-OBml)、FERM BP-6265 (H-OBM4)、及びFERM BP-6266 (H-OBM9)として、平成9年11月5日にそれぞれ寄託されている。
【0122】
〔実施例37〕
マウス OBM 及びマウス sOBM を認識する抗ヒト OBM/sOBM モノクローナル抗体を用いたマウス OBM 及びマウス sOBM の測定
実施例33及び35で得られたマウスOBM及びマウスsOBMも認識する抗ヒトOBM/sOBMモノクローナル抗体、H-OBM9を固相抗体とし、実施例28で得られた抗マウスOBM/sOBMポリクローナル抗体を酵素標識抗体とするサンドイッチELISAを構築した。
マウスOBM及びマウスsOBMをそれぞれ実施例35と同様に一次反応用バッファーで段階的に希釈し、実施例36と同様の方法によりマウスOBM及びマウスsOBMを測定した。結果を図28に示す。この結果、本発明のマウスOBM及びマウスsOBMも認識する本発明の抗ヒトOBM/sOBMモノクローナル抗体、H-OBM9を使用することにより、マウスOBM及びマウスsOBMを同様に測定できることが確認された。実施例35の結果に示すように、この抗ヒトOBM/sOBMモノクローナル抗体、H-OBM9はマウスsOBMに対する解離定数が高い、即ちマウスsOBMに対する親和性が比較的低いことから、本ELISAによるマウスOBM(分子量約40kDa)及びマウスsOBM(分子量約32kDa)の測定感度は、約25×10-3pmol/ml (マウスOBMでは約1ng/ml、マウスsOBMでは約0.8ng/ml)程度であった。
【0123】
〔実施例38〕
抗 OBM/sOBM 抗体による破骨細胞形成抑制活性試験
マウス脾細胞とST2細胞(マウス骨髄由来間質細胞)とのco-cultureでosteoclast-like cell(OCL)が誘導されることが知られている(Endocrinology, 125, 1805-1813(1989))。そこで、OBM/sOBM抗体をこのco-cultureに添加した時、OCL誘導を抑制するか否かを検討した。このco-culture系においてはマウスOBMが発現しているので、実施例に使用する抗体はマウスOBMを認識するウサギ抗マウスOBM/sOBMポリクローナル抗体、及びヒトOBM及びマウスOBMの両抗原を認識するヒト抗OBM/sOBMモノクローナル抗体、H-OBM9を用いた。10%FCSを含むαMEMにて段階希釈したそれぞれのOBM抗体を24well plate(ヌンク社)に700μl/well、上記培地にて懸濁した雄マウス脾細胞(2×106/ml)を350μl/well添加した。次いでトリプシナイズしたST2細胞を4×10-8M VitaminD3及び4×10-7MDexamethazoneを含む上記培地に懸濁し(8×104cells/ml)350μl/well添加し、37℃で6日間培養した。培養プレートをPBSにて1回洗浄したのち、50%エタノール、50%アセトンの混合液にて室温で1分間固定した。プレートを風乾した後、LEUKOCYTE ACID PHOSPHATASEキット(シグマ社)のプロトコールに従い基質液を500μl/well添加し、37℃で55分間反応させた。この反応により破骨細胞特異的マーカーである酒石酸耐性酸性フォスファターゼ活性(TRAP活性)を示す細胞のみが染色される。プレートを蒸留水で1回洗浄した後風乾してTRAP陽性細胞数をカウントした。結果を表4に示す。この結果、ウサギ抗マウスOBM/sOBMポリクローナル抗体及びマウスOBMを認識する抗ヒトOBM/sOBMモノクローナル抗体、H-OBM9とも抗体濃度に依存してOCL誘導を阻害することがわかった。
これらの抗体は、破骨細胞形成因子、OCIF/OPGと同様に破骨細胞形成抑制活性を有し、骨代謝異常症治療薬として有望であることが明らかになった。
【0124】
【表4】
〔実施例39〕
Trx-OBM のヒト破骨細胞形成誘導活性
成人健常者より静脈採取した全血から、Histopaque(sigma社)を用い、添付のプロトコールに従いdensity gradient法によって単核細胞を調製した。この単核細胞を10-7Mデキサメサゾン、200ng/mlマクロファージコロニー刺激因子(ミドリ十字杜)、10%牛胎児血清及び実施例15で得られた。精製されたTrx-OBM(0-100ng/ml)を含むα-MEMに1.3×106/mlになるように懸濁し、この懸濁液を48ウェルプレートに1ウェルあたり300μl添加し3日間37℃で培養した。培養液を上記培地で交換後、さらに4日間37℃で培養した。培養した細胞を実施例5に示した方法で酒石酸耐性酸性フォスファターゼ活性(TRAP活性)を示す細胞を選択的に染色し、染色された多核細胞の数を顕微鏡観察により測定した。結果を図29に示す。この結果、Trx-OBMを添加しなかったウェル中にはTRAP活性を示す細胞はほとんど検出されなかったのに対し、Trx-OBMを添加すると濃度依存的にTRAP陽性多核細胞が出現した。又、これらのTRAP陽性多核細胞は破骨細胞のマーカーであるビトロネクチンレセプターも陽性であった。さらに、同様の培養を48ウェルプレートに静置した象牙質切片上で行うと、Trx-OBM存在下でのみ象牙質切片上に吸収窩が形成された。これより、Trx-OBMにはヒト破骨細胞形成を誘導する活性があることが明らかとなった。
【0126】
〔実施例40〕
抗 OBM/sOBM 抗体による骨吸収抑制活性
妊娠15日目のddYマウス(日本SLC社)に[45Ca]-CaCl2溶液(Amersham社)を1匹あたり25μCi皮下注射し、胎仔の骨を45Caでラベルした。翌日、マウスを屠殺し開腹して、子宮から胎仔を取り出した。胎仔より前肢を摘出して、皮膚及び筋肉を剥がし長管骨を摘出し、さらに軟骨を除去して長管骨の骨幹部のみにした。骨幹部は24穴プレートに入れた0.5mlの培養液(0.2%牛血清アルブミン(sigma社)を含むBGJb medium(GIBCO BRL社))に1本ずつ浮かべ、37℃, 5% CO2存在下で24時間培養した。前培養終了後、各種骨吸収因子(ビタミンD3、プロスタグランジンE2、副甲状腺ホルモン、インターロイキン1α)、及び正常ウサギIgG(100μg/ml;対照として)又は実施例28で得られたウサギ抗OBM/sOBMポリクローナル抗体を含む新しい培養液(0.5ml)に長管骨を移し、さらに72時間培養した。培養終了後、長管骨は0.5mlの5%トリクロロ酢酸水溶液(和光純薬社)に入れ、室温で3時間以上処理して脱灰した。培養液及びトリクロロ酢酸抽出液(共に0.5ml)にシンチレーター(AQUASOL-2、PACKARD社)5mlを加え、45Caの放射活性を測定し、骨吸収により培養液中に遊離した45Caの割合を算出した。結果を図30〜図33にそれぞれ示す。この結果、ビタミンD3 (10-8M)は骨吸収活性を上昇させたが、ウサギ抗OBM/sOBMポリクローナル抗体を加えることにより、濃度依存的にビタミンD3による骨吸収を抑制し、100μg/mlの濃度において完全に抑制した(図30)。又、プロスタグランジンE2 (10-6M)あるいは副甲状腺ホルモン(100ng/ml)は骨吸収活性を上昇させたが、ウサギ抗OBM/sOBMポリクローナル抗体の添加(l00μg/ml)により、プロスタグランジンE2及び副甲状腺ホルモンによる骨吸収をほぼ完全に抑制した(図31及び図32)。一方、陽性対照として用いた正常ウサギIgG(100μg/ml)は、プロスタグランジンE2及び副甲状腺ホルモンによる骨吸収に対して影響しなかった。又、インターロイキン1α(10ng/ml)によっても骨吸収が引き起こされたが、ウサギ抗OBM/sOBMポリクローナル抗体(100μg/ml)により有意に抑制された(図23)。これらの結果より、本発明抗体は骨吸収抑制物質として優れていることが明らかとなった。マウス抗ヒトOBM/sOBM抗体であるH-OBM9についても同様の実験を行った結果、ウサギ抗OBM/sOBMポリクローナル抗体とほぼ同等の骨吸収抑制活性があることが確認された。
【0127】
産業上の利用の可能性
本発明により、破骨細胞形成抑制因子(Osteoc1astogenesis inhibitory factor;OCIF)に結合する新規な蛋白質、その製造方法、この蛋白質を用いて該蛋白質の発現を調節する物質のスクリーニング方法、該蛋白質の作用を阻害又は修飾する物質のスクリーニング方法、該蛋白質と結合してその作用を伝達するレセプターのスクリーニング方法、及びこれらのスクリーニング方法により得られた物質を含有する医薬組成物、該蛋白質に対する抗体、及びその抗体を用いた骨代謝異常治療剤が提供される。
また、本発明により、破骨細胞形成抑制因子OCIFに結合する新規な蛋白質(OCIF結合分子)をコードするDNA、そのDNAによりコードされるアミノ酸配列を有する蛋白質、そのDNAを用いてOCIFに特異的に結合する蛋白質を遺伝子工学的に製造する方法、及び該蛋白質を含有する骨代謝治療剤が提供される。更に、OCIF結合分子の発現を調節するスクリーニング方法、OCIF結合分子と結合しその作用を阻害又は修飾する物質のスクリーニング方法、OCIF結合分子と結合しその作用を伝達するレセプターのスクリーニング方法、これらのスクリーニングの結果得られた物質を含有する医薬組成物が提供される。
さらに、破骨細胞形成抑制因子OCIFに結合する新規なヒト蛋白質(ヒト由来OCIF結合分子、ヒトOBM)をコードするDNA、そのDNAによりコードされるアミノ酸配列を有する蛋白質、そのDNAを用いてOCIFに特異的に結合する性質を有し、破骨細胞の分化、成熟を支持、促進する生物活性を有する蛋白質を遺伝子工学的に製造する方法、及び該蛋白質を含有する骨代謝異常症治療剤が提供される。OCIF結合分子の発現を調節する物質のスクリーニング方法、OCIF結合分子と結合し、その作用を阻害又は修飾する物質のスクリーニング方法、OCIF結合分子と結合し、その生物作用を伝達するレセプターのスクリーニング方法、及びこれらのスクリーニングの結果得られた物質を含有する医薬組成物、さらにヒト由来OCIF結合蛋白質に対する抗体及びその抗体を用いた骨代謝異常症の予防及び/又は治療薬が提供される。
またさらに、本発明により、OCIFに特異的に結合する膜結合分子(OCIF bindingmolecule;OBM)蛋白質及び膜結合部位を欠損した可溶性OBM(sOBM)の両抗原を認識する抗体(抗OBM/sOBM抗体)、その製造方法、この抗体を用いたOBM及びsOBMの測定方法、さらにこの抗体を有効成分とする骨代謝異常症予防及び/又は治療剤が提供される。
本発明により得られた蛋白質あるいは抗体は、医薬あるいは研究又は試験用試薬として有用である。
【0128】
【配列表フリーテキスト】
配列番号3:PCRプライマー SRR2
配列番号4:PCRプライマー ml3PrimerM3
配列番号5:PCRプライマー ml3PrimerRV
配列番号6:PCRプライマー OBM#8
配列番号7:PCRプライマー OBM HF
配列番号8:PCRプライマー OBM XR
配列番号9:PCRプライマー OBM #3
配列番号10:PCRプライマー OBM SalR2
配列番号13:PCRプライマー ヒトOBM SF
配列番号14:PCRプライマー ヒトOBM SalR2
【配列表】
【図面の簡単な説明】
【図1】本発明実施例3のマウスOBM蛋白質のSDS-PAGEの結果を示す。
〔符号の説明〕
(A):レーン1:分子量マーカー
レーン2:活性型ビタミンD3とデキサメサゾン存在下で培養したST2細胞からの部分精製標品(Gly-HCl(pH2.0)溶出画分)
レーン3:活性型ビタミンD3とデキサメサゾン非存在下で培養したST2細胞からの部分精製標品(Gly-HCl(pH2.0)溶出画分)
(B):レーン1:分子量マーカー
レーン2:逆相高速液体クロマトグラフィーで精製した本発明マウス
OBM蛋白質(実施例3)
【図2】実施例4の125Iで標識したOCIFの骨芽細胞様ストローマ細胞ST2への結合試験の結果を示す。
【図3】実施例5(1)の継代数の違う骨芽細胞様ストローマ細胞ST2による破骨細胞形成支持能を示す。
〔符号の説明〕
1:継代数約10代のST2細胞の破骨細胞形成支持能
2:継代数約40代のST2細胞の破骨細胞形成支持能
【図4】実施例5(2)の活性型ビタミンD3及びデキサメサゾン存在下での培養における骨芽細胞様ストロ一マ細胞膜上の本発明蛋白質発現の経時変化を示す。
【図5】実施例5(2)の共培養系における破骨細胞形成の経時変化を示す。
【図6】実施例5(3)の共培養期間において、種々の培養期間のみOCIF処理した場合の破骨細胞形成抑制効果を示す。
【図7】実施例6の1251標識OCIFと本発明蛋白質とのクロスリンキング試験の結果を示す。
〔符号の説明〕
レーン1:125I標識OCIF-CDDl
レーン2:125I標識OCIF-CDDlとST2細胞をクロスリンクさせたもの
レーン3:400倍濃度の未標識OCIFを添加してクロスリンクさせたもの
【図8】実施例9における、SDS-PAGEの結果を示す。
〔符号の説明〕
レーン1:pOBM291をトランスフェクトしたCOS-7細胞の蛋白質を、OCIF無添加で免疫沈降させたもの
レーン2: pOBM291をトランスフェクトしたCOS-7細胞の蛋白質を、OCIFを添加して免疫沈降させたもの
【図9】実施例10における、125Iで標識したOCIFの、pOBM291をトランスフェクトしたCOS-7細胞への結合試験の結果を示す。
〔符号の説明〕
レーン1及び2:125Iで標識したOCIFの、pOBM291をトランスフェクトしたCOS-7細胞への結合量
レーン3及び4:125Iで標識したOCIFの、pOBM291をトランスフェクしたCOS-7細胞への結合量(400倍濃度の未標識OCIF添加時)
【図10】実施例11における、125Iで標識したOCIFを用いたクロスリンク試験の結果を示す。
〔符号の説明〕
レーン1:125I標識OCIF
レーン2:125I標識OCIFと、pOBM291をトランスフェクトしたCOS-7細胞をクロスリンクさせたもの
レーン3:400倍濃度の未標識OCIF存在下て、125I標識OCIFと、pOBM291をトランスフェクトしたCOS-7細胞をクロスリンクさせたもの
【図11】実施例12における、ノーザンブロットの結果を示す。
〔符号の説明〕
レーン1:ビタミンD及びデキサメサゾン無添加で培養したST2細胞由来RNA
レーン2:ビタミンD及びデキサメサゾンを添加し培養したST2細胞由来RNA
【図12】実施例13‐(2)における、OCIF濃度を変えた時の培養上清中蛋白質のOCIF結合能を示す。
〔符号の説明〕
○:pCEP4
●:pCEP sOBM
【図13】実施例13‐(2)における、培養上清の割合を変えた時の培養上清中蛋白質のOCIF結合能を示す。
〔符号の説明〕
○:pCEP4
●:pCEP sOBM
【図14】実施例14-(2)における、大腸菌で発現させたチオレドキシンとマウスOBMの融合蛋白質のSDS-PAGEの結果を示す。
〔符号の説明〕
レーン1:分子量マーカー
レーン2:GI724/pTrxFus由来可溶性蛋白質画分
レーン3:GI724/pTrxOBM25由来可溶性蛋白質画分
【図15】実施例14‐(3)における、可溶性蛋白質画分の割合を変えた時の0CIF結合能を示す。
〔符号の説明〕
□:GI724/pTrxFus
○:GI724/pTrxOBM25
【図16】実施例14‐(3)における、OCIF濃度を変えた時の可溶性蛋白質画分(1%)のOCIF結合能を示す。
〔符号の説明〕
□:GI724/pTrxFus
○:GI724/pTrxOBM25
【図17】本発明のマウスOBM cDNAにより発現され精製して得られる蛋白質マウスOBM、及び天然型の精製OCIF結合蛋白質とOCIFとの特異的結合能の、ウサギ抗マウスOBM抗体による阻害結果を示す。
〔符号の説明〕
1:抗体で処理したcDNAにより発現され精製して得られる蛋白質、OBM+125I-OCIF
2:抗体で処理した天然型の蛋白質+125I‐OCIF
3:抗体未処理のcDNAにより発現されて得られる蛋白質、マウスOBM+125I‐OCIF
4:抗体未処理の天然型の蛋白質+125I‐OCIF
5:3+非標識OCIF(125I-OCIFの400倍量)
6:4+非標識OCIF(125I-OCIFの400倍量)
【図18】本発明のcDNAにより発現される蛋白質、ヒトOBMのSDS-PAGEの結果を示す。
〔符号の説明〕
レーン1:分子量マーカー
レーン2:本発明のcDNAを含む発現ベクター、phOBMをトランスフェクトしたCOS-7細胞の蛋白質をOCIF無添加でウサギ抗OCIFポリクローナル抗体で免疫沈降させたもの
レーン3:本発明cDNAを含む発現ベクター、phOBMをトランスフェクトしたCOS-7細胞の蛋白質をOCIF添加でウサギ抗OCIFポリクローナル抗体で免疫沈降させたもの
【図19】本発明cDNAを含む発現ベクター、phOBMをトランスフェクトしたCOS-7細胞へのOCIFの結合試験の結果を示す。
〔符号の説明〕
レーン1:phOBMをトランスフェクトしたCOS-7細胞に125I -OCIFを添加したもの
レーン2:phOBMをトランスフェクトしたCOS-7細胞に125I -OCIFを添加し、さらに400倍量の非標識OCIFを加えたもの
【図20】本発明cDNAでコードされる蛋白質、ヒトOBMと125I -OCIF(モノマー型)とのクロスリンキング試験の結果を示す。
〔符号の説明〕
レーン1: 125I-OCIF
レーン2: 125I‐OCIFとphOBMをトランスフェクトしたCOS-7細胞膜上の蛋白質とをクロスリンクさせたもの
レーン3:400倍濃度の非標識OCIF存在下で、125I -OCIFとphOBMをトランスフェクトしたCOS-7細胞膜上の蛋白質とをクロスリンクさせたもの
【図21】実施例23‐(2)における、OCIF濃度を変えた時の培養液上清中蛋白質(分泌型hOBM)のOCIF結合能を示す。
〔符号の説明〕
○:分泌型ヒトOBMをコードするcDNAを含まないベクターのみのpCEP4をトランスフェクトした293-EBNA細胞の培養液
●:分泌型ヒトOBMをコードするcDNAを含む発現ベクターのpCEPshOBMをトランスフェクした293-EBNA細胞の培養液
【図22】実施例23‐(2)における、OCIF濃度を一定にし、添加する培養液量を変えた時の培養液上清中蛋白質(分泌型ヒトOBM)のOCIF結合能を示す。
〔符号の説明〕
○:分泌型ヒトOBMをコードするcDNAを含まないベクターのみのpCEP4をトランスフェクトした293-EBNA細胞の培養液
●:分泌型ヒトOBMをコードするcDNAを含む発現ベクターのpCEPshOBMをトランスフェクトした293-EBNA細胞の培養液
【図23】大腸菌で発現させたチオレドキシンとヒトOBMの融合蛋白質のSDS-PAGEの結果を示す。
〔符号の説明〕
レーン1:分子量マーカー
レーン2:大腸菌GI724/pTrxFus由来可溶性蛋白質画分
レーン3:大腸菌GI724/pTrxhOBM由来可溶性蛋白質画分
【図24】実施例24‐(3)における、チオレドトキシンとヒトOBMの融合蛋白質を含む大腸菌由来可溶性蛋白質画分の添加割合を変えた時の融合蛋白質と、OCIFとの結合能を示す。
〔符号の説明〕
○:大腸菌GI724/pTrxFus由来可溶性蛋白質画分
●:大腸菌GI724/pTrxshOBM由来可溶性蛋白質画分
【図25】実施例24‐(3)における、OCIF濃度を変えた時の大腸菌由来可溶性蛋白質画分中のチオレドトキシンとヒトOBMの融合蛋白質と、OCIFとの結合能を示す。
〔符号の説明〕
○:大腸菌GI724/pTrxFus由来可溶性蛋白質画分
●:大腸菌GI724/pTrxshOBM由来可溶性蛋白質画分
【図26】本発明のウサギ抗ヒトOBM/sOBMポリクローナル抗体を用いたサンドイッチELISAによる、ヒトOBM及びsOBMの測定結果を示す。
〔符号の説明〕
口:ヒトOBM
●:ヒトsOBM
【図27】本発明の抗ヒトOBM/sOBMモノクローナル抗体を用いたサンドイッチELISAによる、ヒトOBM及びsOBMの測定結果を示す。
〔符号の説明〕
□:ヒトOBM
●:ヒトsOBM
【図28】本発明のマウスOBM及びsOBMにも交差性を有する抗ヒトOBM/sOBMモノクローナル抗体を用いたサンドイッチELISAによる、マウスOBM及びsOBMの測定結果を示す。
〔符号の説明〕
□:マウスOBM
●:マウスsOBM
【図29】チオレドトキシンとマウスOBMの融合蛋白質によるヒト破骨細胞様細胞形成の促進活性を示す。
【図30】ビタミンD3刺激による骨吸収活性の、抗OBM/sOBM抗体による抑制を示す。
【図31】プロスタグランジンE2(PGE2)刺激による骨吸収活性の、抗OBM/sOBM抗体による抑制を示す。
【図32】副甲状腺ホルモン(PTH)刺激による骨吸収活性の、抗OBM/sOBM抗体による抑制を示す。
【図33】インターロイキン1α(1L-1)刺激による骨吸収活性の、抗OBM/sOBM抗体による抑制を示す。
Claims (47)
- 下記の(a)乃至(e)に記載の性質を有する蛋白質に結合し、且つ100ng/ml以下の濃度において該蛋白質による破骨細胞形成活性を抑制する活性を示す抗体:
(a)破骨細胞形成抑制因子(osteoclastogenesis inhibitory factor:OCIF)に特異的に結合する;
(b)SDS−ポリアクリルアミド電気泳動により、約30,000−40,000の分子量を示す;
(c)モノマータイプのOCIFとクロスリンクさせた複合蛋白質が、SDS−ポリアクリルアミド電気泳動により、約90,000−110,000の分子量を示す;
(d)骨吸収促進因子存在下での骨芽細胞様ストローマ細胞と脾臓細胞共培養において、破骨細胞の分化、成熟を支持又は促進する活性を示す;
(e)骨吸収促進因子の存在下で培養した骨芽細胞様ストローマ細胞上に発現する。 - 10ng/ml以下の濃度において破骨細胞形成活性を抑制する活性を示す、請求項1記載の抗体。
- 配列表の配列番号11に記載のアミノ酸配列からなる蛋白質に結合し、且つ、100ng/ml以下の濃度において該蛋白質による破骨細胞形成活性を抑制する活性を示す抗体。
- 10ng/ml以下の濃度において破骨細胞形成活性を抑制する活性を示す、前記請求項3に記載の抗体。
- 骨吸収抑制活性を有する、請求項1乃至請求項4のいずれか一つに記載の抗体。
- モノクローナル抗体であることを特徴とする、請求項1乃至請求項5のいずれか一つに記載の抗体。
- 下記の(a)乃至(e)に記載の性質を有する蛋白質、又は、配列表の配列番号17に記載のアミノ酸配列からなる蛋白質との結合において、10-8M以下の解離定数を示すことを特徴とする、請求項6記載の抗体:
(a)破骨細胞形成抑制因子(osteoclastogenesis inhibitory factor:OCIF)に特異的に結合する;
(b)SDS−ポリアクリルアミド電気泳動により、約30,000−40,000の分子量を示す;
(c)モノマータイプのOCIFとクロスリンクさせた複合蛋白質が、SDS−ポリアクリルアミド電気泳動により、約90,000−110,000の分子量を示す;
(d)骨吸収促進因子存在下での骨芽細胞様ストローマ細胞と脾臓細胞共培養において、破骨細胞の分化、成熟を支持又は促進する活性を示す;
(e)骨吸収促進因子の存在下で培養した骨芽細胞様ストローマ細胞上に発現する。 - 10-10乃至10-8Mの解離定数を示す、請求項7記載の抗体。
- 10-10M以下の解離定数を示す、請求項7記載の抗体。
- 10-11乃至10-10Mの解離定数を示す、請求項9記載の抗体。
- クラスがIgG1であり、且つ軽鎖がκ鎖である、請求項6乃至請求項10のいずれか一つに記載の抗体。
- 以下の工程1)および2)を含むことからなる方法により得られる、下記の(a)乃至(e)に記載の性質を有する蛋白質、又は、配列表の配列番号11に記載のアミノ酸配列からなる蛋白質に結合し、且つ、100 ng/ml 以下の濃度において該蛋白質による破骨細胞形成活性を抑制する活性、及び、骨吸収抑制活性の両方又は何れか一方の活性を示す抗体:
(a)破骨細胞形成抑制因子(osteoclastogenesis inhibitory factor:OCIF)に特異的に結合する;
(b)SDS−ポリアクリルアミド電気泳動により、約30,000−40,000の分子量を示す;
(c)モノマータイプのOCIFとクロスリンクさせた複合蛋白質が、SDS−ポリアクリルアミド電気泳動により、約90,000−110,000の分子量を示す;
(d)骨吸収促進因子存在下での骨芽細胞様ストローマ細胞と脾臓細胞共培養において、破骨細胞の分化、成熟を支持又は促進する活性を示す;
(e)骨吸収促進因子の存在下で培養した骨芽細胞様ストローマ細胞上に発現する;
1)上記の(a)乃至(e)に記載の性質を有する蛋白質、又は、配列表の配列番号11に記載のアミノ酸配列からなる蛋白質に結合する抗体を作製する工程;
2)破骨細胞形成抑制活性及び/又は骨吸収抑制活性を示す抗体を選抜する工程。 - 配列表の配列番号1に記載のアミノ酸配列からなる蛋白質は認識しない、請求項1乃至12のいずれか一つに記載の抗体。
- 骨代謝異常を伴う疾患の治療に用いられる、請求項1乃至請求項13のいずれか一つに記載の抗体。
- 骨代謝異常を伴う疾患が、骨粗鬆症、高カルシウム血症、骨ページェット病、腎性骨異栄養症、慢性関節リューマチ及び変形性関節炎から選択される一つまたは複数である、請求項14記載の抗体。
- 骨代謝異常による骨量減少症の治療に用いられる、請求項1乃至請求項13のいずれか一つに記載の抗体。
- 配列表の配列番号17記載のアミノ酸配列からなる蛋白質を特異的に認識し、且つ、配列表の配列番号11記載のアミノ酸配列からなる蛋白質を認識するモノクローナル抗体。
- 配列表の配列番号16記載のアミノ酸配列からなる蛋白質は認識しない、請求項17記載のモノクローナル抗体。
- 配列表の配列番号1記載のアミノ酸配列からなる蛋白質は認識しない、請求項17又は18記載のモノクローナル抗体。
- 配列表の配列番号16記載のアミノ酸配列からなる蛋白質および配列表の配列番号1記載のアミノ酸配列からなる蛋白質にも交差性を有している、請求項17記載のモノクローナル抗体。
- 配列表の配列番号17に記載のアミノ酸配列からなる蛋白質との結合において、10-8M以下の解離定数を示す、請求項17乃至請求項20のいずれか一つに記載のモノクローナル抗体。
- 配列表の配列番号17に記載のアミノ酸配列からなる蛋白質との結合において、10-10乃至10-8Mの解離定数を示す、請求項21記載のモノクローナル抗体。
- 配列表の配列番号17に記載のアミノ酸配列からなる蛋白質との結合において、10-10M以下の解離定数を示す、請求項21記載のモノクローナル抗体。
- 配列表の配列番号17に記載のアミノ酸配列からなる蛋白質との結合において、10-11乃至10-10Mの解離定数を示す、請求項23記載のモノクローナル抗体。
- 100ng/ml以下の濃度において破骨細胞形成抑制活性を示す、請求項17乃至請求項24のいずれか一つに記載のモノクローナル抗体。
- 10ng/ml以下の濃度において破骨細胞形成活性を抑制する活性を示す、請求項25記載のモノクローナル抗体。
- 骨吸収抑制活性を有する、請求項17乃至請求項26のいずれか一つに記載のモノクローナル抗体。
- 以下の工程1)および2)を含むことからなる方法により得られる、配列表の配列番号11に記載のアミノ酸配列からなる蛋白質に結合し、且つ100 ng/ml 以下の濃度において該蛋白質による破骨細胞形成活性を抑制する活性、及び、骨吸収抑制活性の両方又は何れか一方の活性を示すモノクローナル抗体:
1)配列表の配列番号11に記載のアミノ酸配列からなる蛋白質に結合するモノクローナル抗体を作製する工程;
2)破骨細胞形成抑制活性及び/又は骨吸収抑制活性を示すモノクローナル抗体を選抜する工程。 - クラスがIgG1であり、且つ軽鎖がκ鎖である、請求項17乃至請求項28のいずれか一つに記載のモノクローナル抗体。
- 骨代謝異常を伴う疾患の治療に用いられる、請求項17乃至請求項29のいずれか一つに記載のモノクローナル抗体。
- 骨代謝異常を伴う疾患が、骨粗鬆症、高カルシウム血症、骨ページェット病、腎性骨異栄養症、慢性関節リューマチ及び変形性関節炎から選択される一つまたは複数である、請求項30記載のモノクローナル抗体。
- 骨代謝異常による骨量減少症の治療に用いられる、請求項17乃至請求項29のいずれか一つに記載のモノクローナル抗体。
- 配列表の配列番号11に記載のアミノ酸配列を有する蛋白質を認識するが、配列表の配列番号1に記載のアミノ酸配列を有する蛋白質は認識しないモノクローナル抗体。
- 配列表の配列番号17に記載のアミノ酸配列を有する蛋白質との結合において10-8以下の解離定数を示す、請求項33記載のモノクローナル抗体。
- 配列表の配列番号17に記載のアミノ酸配列を有する蛋白質との結合において10-10以下の解離定数を示す、請求項34記載のモノクローナル抗体。
- 下記の(a)乃至(e)に記載の性質を有する蛋白質に10-8M以下の解離定数をもって結合し、且つ100ng/ml以下の濃度において該蛋白質による破骨細胞形成活性を抑制する活性を示す抗体:
(a)破骨細胞形成抑制因子(osteoclastogenesis inhibitory factor:OCIF)に特異的に結合する;
(b)SDS−ポリアクリルアミド電気泳動により、約30,000−40,000の分子量を示す;
(c)モノマータイプのOCIFとクロスリンクさせた複合蛋白質が、SDS−ポリアクリルアミド電気泳動により、約90,000−110,000の分子量を示す;
(d)骨吸収促進因子存在下での骨芽細胞様ストローマ細胞と脾臓細胞共培養において、破骨細胞の分化、成熟を支持又は促進する活性を示す;
(e)骨吸収促進因子の存在下で培養した骨芽細胞様ストローマ細胞上に発現する。 - 10-10M以下の解離定数をもって結合する、請求項36記載の抗体。
- 10ng/ml以下の濃度において破骨細胞形成活性を抑制する活性を示す、請求項36又は37記載の抗体。
- 配列表の配列番号17に記載のアミノ酸配列からなる蛋白質に10-8M以下の解離定数をもって結合し、且つ、100ng/ml以下の濃度において破骨細胞形成抑制活性を示すモノクローナル抗体。
- 配列表の配列番号17に記載のアミノ酸配列からなる蛋白質に10-10M以下の解離定数をもって結合する、請求項39記載のモノクローナル抗体。
- 10ng/ml以下の濃度において破骨細胞形成活性を抑制する活性を示す、請求項39又は40記載のモノクローナル抗体。
- 骨吸収抑制活性を有する、請求項39乃至請求項41のいずれか一つに記載のモノクローナル抗体。
- 配列表の配列番号17に記載のアミノ酸配列からなる蛋白質に10-8M以下の解離定数をもって結合するモノクローナル抗体。
- 配列表の配列番号17に記載のアミノ酸配列からなる蛋白質に10-10M以下の解離定数をもって結合するモノクローナル抗体。
- 100ng/ml以下の濃度において破骨細胞形成抑制活性を示す、請求項43又は44記載のモノクローナル抗体。
- 10ng/ml以下の濃度において破骨細胞形成抑制活性を示す、請求項45記載のモノクローナル抗体。
- 骨吸収抑制活性を有する、請求項43乃至請求項48のいずれか一つに記載のモノクローナル抗体。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003169309A JP3935861B2 (ja) | 1997-04-15 | 2003-06-13 | 新規蛋白質及びその製造方法 |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP9780897 | 1997-04-15 | ||
| JP15143497 | 1997-06-09 | ||
| JP21789797 | 1997-08-12 | ||
| JP22480397 | 1997-08-21 | ||
| JP33224197 | 1997-12-02 | ||
| JP2003169309A JP3935861B2 (ja) | 1997-04-15 | 2003-06-13 | 新規蛋白質及びその製造方法 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP54374198A Division JP3523650B2 (ja) | 1997-04-15 | 1998-04-15 | 新規蛋白質及びその製造方法 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004381995A Division JP4230993B2 (ja) | 1997-04-15 | 2004-12-28 | 新規蛋白質及びその製造方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2004041195A JP2004041195A (ja) | 2004-02-12 |
| JP2004041195A5 JP2004041195A5 (ja) | 2005-08-04 |
| JP3935861B2 true JP3935861B2 (ja) | 2007-06-27 |
Family
ID=27525882
Family Applications (7)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP54374198A Expired - Lifetime JP3523650B2 (ja) | 1997-04-15 | 1998-04-15 | 新規蛋白質及びその製造方法 |
| JP2003169309A Expired - Lifetime JP3935861B2 (ja) | 1997-04-15 | 2003-06-13 | 新規蛋白質及びその製造方法 |
| JP2004381995A Expired - Lifetime JP4230993B2 (ja) | 1997-04-15 | 2004-12-28 | 新規蛋白質及びその製造方法 |
| JP2008191860A Expired - Lifetime JP4355357B2 (ja) | 1997-04-15 | 2008-07-25 | 新規蛋白質及びその製造方法 |
| JP2008260450A Expired - Lifetime JP4878616B2 (ja) | 1997-04-15 | 2008-10-07 | 新規蛋白質及びその製造方法 |
| JP2011195938A Expired - Lifetime JP5285751B2 (ja) | 1997-04-15 | 2011-09-08 | 新規蛋白質及びその製造方法 |
| JP2013050045A Expired - Lifetime JP5593407B2 (ja) | 1997-04-15 | 2013-03-13 | 新規蛋白質及びその製造方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP54374198A Expired - Lifetime JP3523650B2 (ja) | 1997-04-15 | 1998-04-15 | 新規蛋白質及びその製造方法 |
Family Applications After (5)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004381995A Expired - Lifetime JP4230993B2 (ja) | 1997-04-15 | 2004-12-28 | 新規蛋白質及びその製造方法 |
| JP2008191860A Expired - Lifetime JP4355357B2 (ja) | 1997-04-15 | 2008-07-25 | 新規蛋白質及びその製造方法 |
| JP2008260450A Expired - Lifetime JP4878616B2 (ja) | 1997-04-15 | 2008-10-07 | 新規蛋白質及びその製造方法 |
| JP2011195938A Expired - Lifetime JP5285751B2 (ja) | 1997-04-15 | 2011-09-08 | 新規蛋白質及びその製造方法 |
| JP2013050045A Expired - Lifetime JP5593407B2 (ja) | 1997-04-15 | 2013-03-13 | 新規蛋白質及びその製造方法 |
Country Status (21)
| Country | Link |
|---|---|
| US (7) | US7527790B2 (ja) |
| EP (3) | EP2336168A1 (ja) |
| JP (7) | JP3523650B2 (ja) |
| KR (1) | KR100496063B1 (ja) |
| CN (1) | CN1138786C (ja) |
| AT (1) | ATE328006T1 (ja) |
| AU (1) | AU735355B2 (ja) |
| CA (2) | CA2257247C (ja) |
| CY (1) | CY2010017I2 (ja) |
| DE (2) | DE69834699T3 (ja) |
| DK (1) | DK0911342T4 (ja) |
| ES (1) | ES2263204T5 (ja) |
| FR (1) | FR10C0048I2 (ja) |
| HU (3) | HU229476B1 (ja) |
| IL (1) | IL127115A (ja) |
| LU (1) | LU91759I2 (ja) |
| NO (2) | NO322632B1 (ja) |
| NZ (1) | NZ332995A (ja) |
| PT (1) | PT911342E (ja) |
| RU (1) | RU2238949C2 (ja) |
| WO (1) | WO1998046644A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013139465A (ja) * | 1997-04-15 | 2013-07-18 | Daiichi Sankyo Co Ltd | 新規蛋白質及びその製造方法 |
Families Citing this family (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL117175A (en) | 1995-02-20 | 2005-11-20 | Sankyo Co | Osteoclastogenesis inhibitory factor protein |
| US7632922B1 (en) * | 1995-12-22 | 2009-12-15 | Amgen, Inc. | Osteoprotegerin |
| US20050147611A1 (en) * | 1995-12-22 | 2005-07-07 | Amgen Inc. | Combination therapy for conditions leading to bone loss |
| EP1803816A3 (en) * | 1996-12-13 | 2007-09-26 | Schering Corporation | Mammalian cell surface antigens; related reagents |
| US6271349B1 (en) | 1996-12-23 | 2001-08-07 | Immunex Corporation | Receptor activator of NF-κB |
| US6242213B1 (en) | 1996-12-23 | 2001-06-05 | Immunex Corporation | Isolated DNA molecules encoding RANK-L |
| WO1999058674A2 (en) * | 1998-05-14 | 1999-11-18 | Immunex Corporation | Method of inhibiting osteoclast activity |
| EP0975754B2 (en) * | 1997-04-16 | 2016-01-06 | Amgen Inc., | Osteoprotegerin binding proteins and their receptors |
| US6316408B1 (en) * | 1997-04-16 | 2001-11-13 | Amgen Inc. | Methods of use for osetoprotegerin binding protein receptors |
| US5843678A (en) * | 1997-04-16 | 1998-12-01 | Amgen Inc. | Osteoprotegerin binding proteins |
| AU2011202547B2 (en) * | 1997-04-16 | 2014-08-28 | Amgen Inc. | Osteoprotegerin binding proteins and receptors |
| HUP0104126A3 (en) | 1998-10-28 | 2004-07-28 | Sankyo Company Ltd Chuo Ku | Remedies for bone metabolic errors |
| AUPQ314799A0 (en) * | 1999-09-29 | 1999-10-21 | University Of Western Australia, The | Bone cell factor |
| DK2105449T3 (da) † | 2000-02-23 | 2019-10-07 | Amgen Inc | Antagonistiske selektive bindemidler af osteoprotegerinbindende protein |
| US20030103978A1 (en) * | 2000-02-23 | 2003-06-05 | Amgen Inc. | Selective binding agents of osteoprotegerin binding protein |
| ATE366306T1 (de) | 2000-09-22 | 2007-07-15 | Immunex Corp | Screeningverfahren für agonisten und antagonisten des rezeptoraktivators von nf-kappa b |
| WO2002062990A1 (fr) * | 2001-02-07 | 2002-08-15 | Sankyo Company, Limited | Anticorps et utilisation de cet anticorps |
| WO2002079256A1 (fr) * | 2001-03-26 | 2002-10-10 | Sankyo Company, Limited | Anticorps et son utilisation |
| JP4921687B2 (ja) | 2001-04-03 | 2012-04-25 | ソシエテ・デ・プロデュイ・ネスレ・エス・アー | 乳中の破骨細胞分化抑制因子 |
| PH12012502440A1 (en) | 2001-06-26 | 2013-06-17 | Amgen Inc | Antibodies to opgl |
| WO2003086289A2 (en) | 2002-04-05 | 2003-10-23 | Amgen Inc. | Human anti-opgl neutralizing antibodies as selective opgl pathway inhibitors |
| US7462700B2 (en) * | 2002-12-10 | 2008-12-09 | Schering-Plough Animal Health Corporation | Canine RANKL and methods for preparing and using the same |
| US20050009097A1 (en) * | 2003-03-31 | 2005-01-13 | Better Marc D. | Human engineered antibodies to Ep-CAM |
| WO2005016111A2 (en) | 2003-08-08 | 2005-02-24 | Abgenix, Inc. | Antibodies directed to parathyroid hormone (pth) and uses thereof |
| US7318925B2 (en) | 2003-08-08 | 2008-01-15 | Amgen Fremont, Inc. | Methods of use for antibodies against parathyroid hormone |
| AR045563A1 (es) | 2003-09-10 | 2005-11-02 | Warner Lambert Co | Anticuerpos dirigidos a m-csf |
| DK2311873T3 (en) | 2004-01-07 | 2018-11-26 | Novartis Vaccines & Diagnostics Inc | M-CSF-SPECIFIC MONOCLONAL ANTIBODY AND APPLICATIONS THEREOF |
| TWI359026B (en) * | 2004-02-12 | 2012-03-01 | Sankyo Co | Pharmaceutical composition for the osteoclast rela |
| RU2324425C2 (ru) * | 2005-08-30 | 2008-05-20 | Государственное образовательное учреждение "Институт усовершенствования врачей" Министерства здравоохранения и социального развития | Способ профилактики рождения больных остеопетрозом детей |
| AR056806A1 (es) | 2005-11-14 | 2007-10-24 | Amgen Inc | Moleculas quimericas de anticuerpo rankl- pth/ pthrp |
| US8168181B2 (en) | 2006-02-13 | 2012-05-01 | Alethia Biotherapeutics, Inc. | Methods of impairing osteoclast differentiation using antibodies that bind siglec-15 |
| US20070190560A1 (en) * | 2006-02-13 | 2007-08-16 | Olga Ornatsky | Element-tagged olignucleotide gene expression analysis |
| EP2020445B1 (en) | 2006-05-12 | 2013-01-02 | Keio University | Detection of inflammatory disease and composition for prevention or treatment of inflammatory disease |
| WO2008044797A1 (fr) | 2006-10-11 | 2008-04-17 | Oriental Yeast Co., Ltd. | Animal modèle d'ostéopénie |
| WO2008044379A1 (fr) | 2006-10-11 | 2008-04-17 | Oriental Yeast Co., Ltd. | Modèle animal de perte osseuse |
| KR101235439B1 (ko) | 2006-10-11 | 2013-02-20 | 오리엔탈 이스트 컴파니 리미티드 | 가용형 rankl과 에피토프 태그의 융합 단백질을 포함하는 시약 |
| JP5191487B2 (ja) | 2007-06-05 | 2013-05-08 | オリエンタル酵母工業株式会社 | 新しい骨量増加薬 |
| JPWO2009048072A1 (ja) | 2007-10-11 | 2011-02-17 | 第一三共株式会社 | 破骨細胞関連蛋白質Siglec−15を標的とした抗体 |
| WO2010053610A2 (en) * | 2008-07-30 | 2010-05-14 | Emergent Biosolutions, Inc. | Stable anthrax vaccine formulations |
| WO2010038610A1 (ja) | 2008-09-30 | 2010-04-08 | オリエンタル酵母工業株式会社 | 新しい軟骨細胞増殖及び分化誘導剤 |
| US20120018338A1 (en) * | 2009-03-30 | 2012-01-26 | Hoffman-La Roche Inc. | Method for avoiding glass fogging |
| RU2539790C2 (ru) | 2009-04-09 | 2015-01-27 | Дайити Санкио Компани, Лимитед | АНТИТЕЛО ПРОТИВ Siglec-15 |
| WO2011017294A1 (en) * | 2009-08-07 | 2011-02-10 | Schering Corporation | Human anti-rankl antibodies |
| EP2433644A1 (en) | 2010-09-22 | 2012-03-28 | IMBA-Institut für Molekulare Biotechnologie GmbH | Breast cancer therapeutics |
| EP2434285A1 (en) | 2010-09-22 | 2012-03-28 | IMBA-Institut für Molekulare Biotechnologie GmbH | Breast cancer diagnostics |
| JP2013543382A (ja) | 2010-10-05 | 2013-12-05 | 第一三共株式会社 | 破骨細胞関連蛋白質Siglec−15を標的とした抗体 |
| WO2012050154A1 (ja) * | 2010-10-13 | 2012-04-19 | 学校法人 東京女子医科大学 | 抗vdac抗体を含有する破骨細胞形成抑制剤 |
| JPWO2012133914A1 (ja) | 2011-03-31 | 2014-07-28 | オリエンタル酵母工業株式会社 | Ranklアンタゴニストを含む癌免疫増強剤 |
| JP6346602B2 (ja) * | 2012-03-21 | 2018-06-20 | ダナ−ファーバー キャンサー インスティテュート, インコーポレイテッド | ヒトリンパ器官由来の抑制性ストローマ細胞の単離および使用 |
| MX2014011828A (es) | 2012-03-30 | 2014-12-10 | Daiichi Sankyo Co Ltd | Anticuerpo anti-siglec-15 con region determinante de complementariedad modificada. |
| US9493562B2 (en) | 2012-07-19 | 2016-11-15 | Alethia Biotherapeutics Inc. | Anti-Siglec-15 antibodies |
| CA2920376A1 (en) | 2013-08-07 | 2015-02-12 | Martin Blomberg JENSEN | Methods and composition comprising denosumab or an antibody or antigen binding domain, fragment or derivative thereof, immunoreactive with a rankl/opgbp peptide for use in the treatment, prevention or alleviation of male infertility |
| WO2015100344A1 (en) | 2013-12-27 | 2015-07-02 | Emergent Product Development Gaithersburg Inc. | Temperature stable vaccine formulations |
| US11002739B2 (en) * | 2016-09-13 | 2021-05-11 | E&S Healthcare Co., Ltd. | Monoclonal antibody specifically binding to thioredoxin 1, and use thereof |
| CN108410990B (zh) * | 2018-05-30 | 2021-07-06 | 中国医学科学院北京协和医院 | Igfbp3在制备诊断i型神经纤维瘤合并脊柱畸形病产品中的应用 |
| CN112521466B (zh) * | 2020-01-22 | 2022-06-17 | 天津科技大学 | 一种提高异源蛋白分泌量的信号肽突变体及其用途 |
| CN111333709B (zh) * | 2020-03-17 | 2023-09-08 | 吉林大学 | 旋毛虫肌幼虫期丝氨酸蛋白酶抑制剂的b细胞表位多肽、杂交瘤细胞株、单克隆抗体及应用 |
Family Cites Families (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4179337A (en) * | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| US4710457A (en) * | 1983-06-29 | 1987-12-01 | Sloan-Kettering Institute For Cancer Research | Monoclonal antibody for human hematopoietic glycoproteins and method |
| US4710473A (en) * | 1983-08-10 | 1987-12-01 | Amgen, Inc. | DNA plasmids |
| WO1986000922A1 (en) | 1984-07-30 | 1986-02-13 | The Salk Institute For Biological Studies | Retroviral gene transfer vectors |
| US4959314A (en) * | 1984-11-09 | 1990-09-25 | Cetus Corporation | Cysteine-depleted muteins of biologically active proteins |
| US5846534A (en) * | 1988-02-12 | 1998-12-08 | British Technology Group Limited | Antibodies to the antigen campath-1 |
| US6027729A (en) * | 1989-04-20 | 2000-02-22 | Chiron Corporation | NANBV Diagnostics and vaccines |
| JP2877509B2 (ja) | 1989-05-19 | 1999-03-31 | アムジエン・インコーポレーテツド | メタロプロテイナーゼ阻害剤 |
| EP0513334A4 (en) * | 1990-11-30 | 1993-08-04 | Celtrix Laboratories, Inc. | Use of a bone morphogenetic protein in synergistic combination with tgf--g(b) for bone repair |
| US5118667A (en) * | 1991-05-03 | 1992-06-02 | Celtrix Pharmaceuticals, Inc. | Bone growth factors and inhibitors of bone resorption for promoting bone formation |
| CA2068389A1 (en) | 1991-05-13 | 1992-11-14 | Masahiko Sato | Method for inhibiting bone resorption |
| MX9204303A (es) * | 1991-07-23 | 1993-11-01 | Rhone Poulenc Rorer Int | Factor regulador del crecimiento de osteoclasto. |
| IL99120A0 (en) | 1991-08-07 | 1992-07-15 | Yeda Res & Dev | Multimers of the soluble forms of tnf receptors,their preparation and pharmaceutical compositions containing them |
| CA2124967C (en) | 1991-12-17 | 2008-04-08 | Nils Lonberg | Transgenic non-human animals capable of producing heterologous antibodies |
| US5623053A (en) * | 1992-01-10 | 1997-04-22 | California Institute Of Technology | Soluble mammal-derived Fc receptor which binds at a pH ranging from about 5.5 to 6.5 and releases at a pH ranging from about 7.5 to 8.5 |
| US5447851B1 (en) * | 1992-04-02 | 1999-07-06 | Univ Texas System Board Of | Dna encoding a chimeric polypeptide comprising the extracellular domain of tnf receptor fused to igg vectors and host cells |
| JPH07509223A (ja) | 1992-04-30 | 1995-10-12 | アムジェン インコーポレイテッド | インターロイキン−1媒介疾患および腫瘍壊死因子媒介疾患の治療方法 |
| US5585479A (en) * | 1992-07-24 | 1996-12-17 | The United States Of America As Represented By The Secretary Of The Navy | Antisense oligonucleotides directed against human ELAM-I RNA |
| US5578569A (en) * | 1993-03-12 | 1996-11-26 | Tam; Cherk S. | Method of increasing bone growth |
| US5457035A (en) * | 1993-07-23 | 1995-10-10 | Immunex Corporation | Cytokine which is a ligand for OX40 |
| ATE202571T1 (de) | 1993-09-14 | 2001-07-15 | Merck & Co Inc | Humane protein-tyrosinphosphatase decodierende cdna |
| US6268212B1 (en) | 1993-10-18 | 2001-07-31 | Amgen Inc. | Tissue specific transgene expression |
| AU4440496A (en) | 1995-02-10 | 1996-08-22 | Smithkline Beecham Corporation | Use of src SH2 specific compounds to treat a bone resorption disease |
| IL117175A (en) * | 1995-02-20 | 2005-11-20 | Sankyo Co | Osteoclastogenesis inhibitory factor protein |
| WO1996028546A1 (en) | 1995-03-15 | 1996-09-19 | Human Genome Sciences, Inc. | Human tumor necrosis factor receptor |
| US20030166097A1 (en) * | 1995-03-15 | 2003-09-04 | Human Genome Sciences, Inc. | Human tumor necrosis factor receptor |
| US7094564B1 (en) * | 1995-03-15 | 2006-08-22 | Human Genome Sciences, Inc. | Human tumor necrosis factor receptor |
| AU5985996A (en) | 1995-06-07 | 1997-01-15 | Osteosa Inc. | Osteoclast growth regulatory factor |
| WO1997000318A1 (en) | 1995-06-07 | 1997-01-03 | Osteosa Inc. | Osteoclast growth regulatory factor |
| US5843901A (en) * | 1995-06-07 | 1998-12-01 | Advanced Research & Technology Institute | LHRH antagonist peptides |
| US6369027B1 (en) * | 1995-12-22 | 2002-04-09 | Amgen Inc. | Osteoprotegerin |
| US6613544B1 (en) * | 1995-12-22 | 2003-09-02 | Amgen Inc. | Osteoprotegerin |
| US5662948A (en) * | 1996-01-29 | 1997-09-02 | Chrysler Corporation | Adjustable and removable trimline inserts for a molding tool |
| US6750091B1 (en) * | 1996-03-01 | 2004-06-15 | Micron Technology | Diode formation method |
| JPH1057071A (ja) | 1996-08-19 | 1998-03-03 | Snow Brand Milk Prod Co Ltd | 新規dna及びそれを用いた蛋白質の製造方法 |
| EP1803816A3 (en) * | 1996-12-13 | 2007-09-26 | Schering Corporation | Mammalian cell surface antigens; related reagents |
| FR2757507B1 (fr) | 1996-12-20 | 1999-01-29 | Inst Francais Du Petrole | Procede de separation de paraxylene comprenant une adsorption avec injection d'eau et une cristallisation |
| AU5901598A (en) * | 1996-12-20 | 1998-07-17 | Board Of Regents, The University Of Texas System | Compositions and methods of use for osteoclast inhibitory factors |
| WO1999058674A2 (en) | 1998-05-14 | 1999-11-18 | Immunex Corporation | Method of inhibiting osteoclast activity |
| US6271349B1 (en) * | 1996-12-23 | 2001-08-07 | Immunex Corporation | Receptor activator of NF-κB |
| US6242213B1 (en) * | 1996-12-23 | 2001-06-05 | Immunex Corporation | Isolated DNA molecules encoding RANK-L |
| KR100496063B1 (ko) | 1997-04-15 | 2005-06-21 | 산쿄 가부시키가이샤 | 신규단백질 및 그 제조방법 |
| US6316408B1 (en) * | 1997-04-16 | 2001-11-13 | Amgen Inc. | Methods of use for osetoprotegerin binding protein receptors |
| US5843678A (en) * | 1997-04-16 | 1998-12-01 | Amgen Inc. | Osteoprotegerin binding proteins |
| EP0975754B2 (en) | 1997-04-16 | 2016-01-06 | Amgen Inc., | Osteoprotegerin binding proteins and their receptors |
| JP2002514079A (ja) | 1997-05-01 | 2002-05-14 | アムジエン・インコーポレーテツド | キメラopgポリペプチド |
| WO1998054201A1 (en) | 1997-05-29 | 1998-12-03 | Human Genome Sciences, Inc. | Human tumor necrosis factor receptor-like protein 8 |
| US6087555A (en) * | 1997-10-15 | 2000-07-11 | Amgen Inc. | Mice lacking expression of osteoprotegerin |
| US6790823B1 (en) | 1998-04-23 | 2004-09-14 | Amgen Inc. | Compositions and methods for the prevention and treatment of cardiovascular diseases |
| WO2001003719A2 (en) | 1999-07-09 | 2001-01-18 | Amgen Inc. | Combination therapy for conditions leading to bone loss |
| DE60034871T2 (de) | 1999-09-03 | 2008-01-17 | Amgen Inc., Thousand Oaks | Verfahren und zusammensetzungen zur prevention oder behandlung von krebs und von krebs assoziertem knochenverlust |
| US20030144187A1 (en) | 1999-09-03 | 2003-07-31 | Colin R. Dunstan | Opg fusion protein compositions and methods |
| US20030103978A1 (en) * | 2000-02-23 | 2003-06-05 | Amgen Inc. | Selective binding agents of osteoprotegerin binding protein |
| PH12012502440A1 (en) * | 2001-06-26 | 2013-06-17 | Amgen Inc | Antibodies to opgl |
| WO2003086289A2 (en) * | 2002-04-05 | 2003-10-23 | Amgen Inc. | Human anti-opgl neutralizing antibodies as selective opgl pathway inhibitors |
-
1998
- 1998-04-15 KR KR10-2003-7009671A patent/KR100496063B1/ko not_active Expired - Lifetime
- 1998-04-15 AT AT98914034T patent/ATE328006T1/de active
- 1998-04-15 CN CNB988004771A patent/CN1138786C/zh not_active Expired - Lifetime
- 1998-04-15 DE DE69834699T patent/DE69834699T3/de not_active Expired - Lifetime
- 1998-04-15 NZ NZ332995A patent/NZ332995A/xx not_active IP Right Cessation
- 1998-04-15 CA CA2257247A patent/CA2257247C/en not_active Expired - Lifetime
- 1998-04-15 AU AU68518/98A patent/AU735355B2/en not_active Expired
- 1998-04-15 JP JP54374198A patent/JP3523650B2/ja not_active Expired - Lifetime
- 1998-04-15 HU HU0000717A patent/HU229476B1/hu active Protection Beyond IP Right Term
- 1998-04-15 RU RU99100615/13A patent/RU2238949C2/ru active Protection Beyond IP Right Term
- 1998-04-15 EP EP20100186133 patent/EP2336168A1/en not_active Withdrawn
- 1998-04-15 PT PT98914034T patent/PT911342E/pt unknown
- 1998-04-15 ES ES98914034T patent/ES2263204T5/es not_active Expired - Lifetime
- 1998-04-15 IL IL127115A patent/IL127115A/en not_active IP Right Cessation
- 1998-04-15 HU HU0800628A patent/HU229841B1/hu unknown
- 1998-04-15 EP EP05017241.0A patent/EP1657255B1/en not_active Expired - Lifetime
- 1998-04-15 CA CA2781623A patent/CA2781623A1/en not_active Abandoned
- 1998-04-15 EP EP98914034.8A patent/EP0911342B2/en not_active Expired - Lifetime
- 1998-04-15 DK DK98914034.8T patent/DK0911342T4/da active
- 1998-04-15 WO PCT/JP1998/001728 patent/WO1998046644A1/ja not_active Ceased
- 1998-04-15 DE DE122010000049C patent/DE122010000049I1/de active Pending
- 1998-12-14 NO NO19985848A patent/NO322632B1/no not_active IP Right Cessation
-
2002
- 2002-06-11 US US10/167,182 patent/US7527790B2/en not_active Expired - Lifetime
-
2003
- 2003-06-13 JP JP2003169309A patent/JP3935861B2/ja not_active Expired - Lifetime
- 2003-06-13 US US10/460,623 patent/US7192718B2/en not_active Expired - Lifetime
-
2004
- 2004-05-27 US US10/854,300 patent/US7449185B2/en not_active Expired - Fee Related
- 2004-12-28 JP JP2004381995A patent/JP4230993B2/ja not_active Expired - Lifetime
-
2005
- 2005-05-24 US US11/135,521 patent/US20050208580A1/en not_active Abandoned
-
2006
- 2006-08-31 US US11/513,178 patent/US20080187540A9/en not_active Abandoned
-
2008
- 2008-07-25 JP JP2008191860A patent/JP4355357B2/ja not_active Expired - Lifetime
- 2008-10-07 JP JP2008260450A patent/JP4878616B2/ja not_active Expired - Lifetime
- 2008-12-19 US US12/340,327 patent/US20100136011A1/en not_active Abandoned
-
2010
- 2010-11-24 LU LU91759C patent/LU91759I2/fr unknown
- 2010-11-25 FR FR10C0048C patent/FR10C0048I2/fr active Active
- 2010-11-25 NO NO2010021C patent/NO2010021I2/no unknown
- 2010-11-30 CY CY2010017C patent/CY2010017I2/el unknown
-
2011
- 2011-09-08 JP JP2011195938A patent/JP5285751B2/ja not_active Expired - Lifetime
-
2012
- 2012-08-16 US US13/587,791 patent/US20120329671A1/en not_active Abandoned
-
2013
- 2013-03-13 JP JP2013050045A patent/JP5593407B2/ja not_active Expired - Lifetime
-
2014
- 2014-05-07 HU HUS1400027C patent/HUS1400027I1/hu unknown
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013139465A (ja) * | 1997-04-15 | 2013-07-18 | Daiichi Sankyo Co Ltd | 新規蛋白質及びその製造方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3935861B2 (ja) | 新規蛋白質及びその製造方法 | |
| JPWO1998046644A1 (ja) | 新規蛋白質及びその製造方法 | |
| AU2008200700C1 (en) | Novel Protein and Process for Producing The Same | |
| KR100436092B1 (ko) | 신규단백질및그제조방법 | |
| HK1159137A (en) | Novel protein and method for producing the protein | |
| AU2014250718A1 (en) | Novel Protein and Process for Producing The Same | |
| MXPA98010700A (en) | Novel protein and process for producing the same | |
| AU2012204072A1 (en) | Novel Protein and Process for Producing The Same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20030613 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20041228 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20050422 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050613 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20070314 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20070320 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 3935861 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100330 Year of fee payment: 3 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100330 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110330 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110330 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110330 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120330 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120330 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130330 Year of fee payment: 6 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130330 Year of fee payment: 6 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130330 Year of fee payment: 6 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130330 Year of fee payment: 6 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20230330 Year of fee payment: 16 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: R3D02 |
|
| EXPY | Cancellation because of completion of term |