JP2017504601A - 免疫療法のためにマルチインプットシグナル感受性t細胞を操作する方法 - Google Patents
免疫療法のためにマルチインプットシグナル感受性t細胞を操作する方法 Download PDFInfo
- Publication number
- JP2017504601A JP2017504601A JP2016541194A JP2016541194A JP2017504601A JP 2017504601 A JP2017504601 A JP 2017504601A JP 2016541194 A JP2016541194 A JP 2016541194A JP 2016541194 A JP2016541194 A JP 2016541194A JP 2017504601 A JP2017504601 A JP 2017504601A
- Authority
- JP
- Japan
- Prior art keywords
- domain
- car
- cells
- cell
- seq
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000000034 method Methods 0.000 title claims abstract description 81
- 210000001744 T-lymphocyte Anatomy 0.000 title abstract description 92
- 238000009169 immunotherapy Methods 0.000 title abstract description 11
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 claims abstract description 283
- 210000004027 cell Anatomy 0.000 claims abstract description 217
- 239000003446 ligand Substances 0.000 claims abstract description 125
- 210000002865 immune cell Anatomy 0.000 claims abstract description 117
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 90
- 206010021143 Hypoxia Diseases 0.000 claims abstract description 78
- 230000007954 hypoxia Effects 0.000 claims abstract description 61
- 230000027455 binding Effects 0.000 claims abstract description 56
- 108020001756 ligand binding domains Proteins 0.000 claims abstract description 37
- 201000011510 cancer Diseases 0.000 claims abstract description 24
- 239000000427 antigen Substances 0.000 claims description 91
- 108091007433 antigens Proteins 0.000 claims description 91
- 102000036639 antigens Human genes 0.000 claims description 91
- 230000004913 activation Effects 0.000 claims description 76
- 230000003834 intracellular effect Effects 0.000 claims description 58
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 46
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 41
- 229910052760 oxygen Inorganic materials 0.000 claims description 37
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 35
- 239000001301 oxygen Substances 0.000 claims description 35
- 229920001184 polypeptide Polymers 0.000 claims description 34
- 210000004881 tumor cell Anatomy 0.000 claims description 31
- 230000000139 costimulatory effect Effects 0.000 claims description 30
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 claims description 19
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 claims description 19
- -1 LIGHT Proteins 0.000 claims description 15
- 230000005931 immune cell recruitment Effects 0.000 claims description 12
- 101000851370 Homo sapiens Tumor necrosis factor receptor superfamily member 9 Proteins 0.000 claims description 11
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 claims description 11
- 102100036856 Tumor necrosis factor receptor superfamily member 9 Human genes 0.000 claims description 11
- 230000001225 therapeutic effect Effects 0.000 claims description 11
- 230000003213 activating effect Effects 0.000 claims description 8
- 239000000203 mixture Substances 0.000 claims description 8
- 102100029360 Hematopoietic cell signal transducer Human genes 0.000 claims description 7
- 101000990188 Homo sapiens Hematopoietic cell signal transducer Proteins 0.000 claims description 7
- 108010028501 Hypoxia-Inducible Factor 1 Proteins 0.000 claims description 7
- 102000016878 Hypoxia-Inducible Factor 1 Human genes 0.000 claims description 7
- 230000001939 inductive effect Effects 0.000 claims description 7
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 claims description 5
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 claims description 5
- 101001103039 Homo sapiens Inactive tyrosine-protein kinase transmembrane receptor ROR1 Proteins 0.000 claims description 5
- 101001103036 Homo sapiens Nuclear receptor ROR-alpha Proteins 0.000 claims description 5
- 102100039615 Inactive tyrosine-protein kinase transmembrane receptor ROR1 Human genes 0.000 claims description 5
- 101001019455 Homo sapiens ICOS ligand Proteins 0.000 claims description 4
- 101000998120 Homo sapiens Interleukin-3 receptor subunit alpha Proteins 0.000 claims description 4
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 claims description 4
- 102100034980 ICOS ligand Human genes 0.000 claims description 4
- 102100033493 Interleukin-3 receptor subunit alpha Human genes 0.000 claims description 4
- 108010061593 Member 14 Tumor Necrosis Factor Receptors Proteins 0.000 claims description 4
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 claims description 4
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 claims description 4
- 102100028785 Tumor necrosis factor receptor superfamily member 14 Human genes 0.000 claims description 4
- 102100022156 Tumor necrosis factor receptor superfamily member 3 Human genes 0.000 claims description 4
- 102100022153 Tumor necrosis factor receptor superfamily member 4 Human genes 0.000 claims description 4
- 101710165473 Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 claims description 4
- 108010029697 CD40 Ligand Proteins 0.000 claims description 3
- 102100032937 CD40 ligand Human genes 0.000 claims description 3
- 108010087819 Fc receptors Proteins 0.000 claims description 3
- 102000009109 Fc receptors Human genes 0.000 claims description 3
- 102100034459 Hepatitis A virus cellular receptor 1 Human genes 0.000 claims description 3
- 101001068136 Homo sapiens Hepatitis A virus cellular receptor 1 Proteins 0.000 claims description 3
- 101001042104 Homo sapiens Inducible T-cell costimulator Proteins 0.000 claims description 3
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 claims description 3
- 101000884270 Homo sapiens Natural killer cell receptor 2B4 Proteins 0.000 claims description 3
- 101000831286 Homo sapiens Protein timeless homolog Proteins 0.000 claims description 3
- 101000752245 Homo sapiens Rho guanine nucleotide exchange factor 5 Proteins 0.000 claims description 3
- 101000633778 Homo sapiens SLAM family member 5 Proteins 0.000 claims description 3
- 101000633784 Homo sapiens SLAM family member 7 Proteins 0.000 claims description 3
- 101000934346 Homo sapiens T-cell surface antigen CD2 Proteins 0.000 claims description 3
- 101001018021 Homo sapiens T-lymphocyte surface antigen Ly-9 Proteins 0.000 claims description 3
- 101000809875 Homo sapiens TYRO protein tyrosine kinase-binding protein Proteins 0.000 claims description 3
- 101000801433 Homo sapiens Trophoblast glycoprotein Proteins 0.000 claims description 3
- 101000801234 Homo sapiens Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 claims description 3
- 101000679857 Homo sapiens Tumor necrosis factor receptor superfamily member 3 Proteins 0.000 claims description 3
- 102100021317 Inducible T-cell costimulator Human genes 0.000 claims description 3
- 102000017578 LAG3 Human genes 0.000 claims description 3
- 102100038082 Natural killer cell receptor 2B4 Human genes 0.000 claims description 3
- 102100029216 SLAM family member 5 Human genes 0.000 claims description 3
- 102000008115 Signaling Lymphocytic Activation Molecule Family Member 1 Human genes 0.000 claims description 3
- 108010074687 Signaling Lymphocytic Activation Molecule Family Member 1 Proteins 0.000 claims description 3
- 102100033447 T-lymphocyte surface antigen Ly-9 Human genes 0.000 claims description 3
- 102100038717 TYRO protein tyrosine kinase-binding protein Human genes 0.000 claims description 3
- 102100032990 Trem-like transcript 2 protein Human genes 0.000 claims description 3
- 101710149051 Trem-like transcript 2 protein Proteins 0.000 claims description 3
- 102100033579 Trophoblast glycoprotein Human genes 0.000 claims description 3
- 102100033728 Tumor necrosis factor receptor superfamily member 18 Human genes 0.000 claims description 3
- 101000971605 Homo sapiens Kita-kyushu lung cancer antigen 1 Proteins 0.000 claims description 2
- 101000951325 Homo sapiens Mitoferrin-1 Proteins 0.000 claims description 2
- 102100021533 Kita-kyushu lung cancer antigen 1 Human genes 0.000 claims description 2
- 102100037984 Mitoferrin-1 Human genes 0.000 claims description 2
- 230000002062 proliferating effect Effects 0.000 claims description 2
- 101001005269 Arabidopsis thaliana Ceramide synthase 1 LOH3 Proteins 0.000 claims 1
- 101001005312 Arabidopsis thaliana Ceramide synthase LOH1 Proteins 0.000 claims 1
- 101000668858 Spinacia oleracea 30S ribosomal protein S1, chloroplastic Proteins 0.000 claims 1
- 101000898746 Streptomyces clavuligerus Clavaminate synthase 1 Proteins 0.000 claims 1
- 108010087914 epidermal growth factor receptor VIII Proteins 0.000 claims 1
- 238000011282 treatment Methods 0.000 abstract description 17
- 230000009257 reactivity Effects 0.000 abstract description 4
- 108090000623 proteins and genes Proteins 0.000 description 143
- 102000004169 proteins and genes Human genes 0.000 description 92
- 235000018102 proteins Nutrition 0.000 description 90
- 108091006024 signal transducing proteins Proteins 0.000 description 50
- 102000034285 signal transducing proteins Human genes 0.000 description 50
- 102000035195 Peptidases Human genes 0.000 description 43
- 108091005804 Peptidases Proteins 0.000 description 43
- 239000004365 Protease Substances 0.000 description 42
- 239000013598 vector Substances 0.000 description 40
- 235000019419 proteases Nutrition 0.000 description 37
- 230000011664 signaling Effects 0.000 description 34
- 102100024965 Caspase recruitment domain-containing protein 11 Human genes 0.000 description 33
- 101000761179 Homo sapiens Caspase recruitment domain-containing protein 11 Proteins 0.000 description 31
- 102000018697 Membrane Proteins Human genes 0.000 description 31
- 108010052285 Membrane Proteins Proteins 0.000 description 31
- 230000019491 signal transduction Effects 0.000 description 28
- 230000014509 gene expression Effects 0.000 description 27
- 101001047681 Homo sapiens Tyrosine-protein kinase Lck Proteins 0.000 description 25
- 108091000080 Phosphotransferase Proteins 0.000 description 25
- 102100024036 Tyrosine-protein kinase Lck Human genes 0.000 description 25
- 102000020233 phosphotransferase Human genes 0.000 description 25
- 235000001014 amino acid Nutrition 0.000 description 23
- 230000017730 intein-mediated protein splicing Effects 0.000 description 23
- 230000003993 interaction Effects 0.000 description 23
- 230000000670 limiting effect Effects 0.000 description 23
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 21
- 108020004999 messenger RNA Proteins 0.000 description 21
- 150000007523 nucleic acids Chemical group 0.000 description 21
- 108091008874 T cell receptors Proteins 0.000 description 20
- 241000700605 Viruses Species 0.000 description 20
- 150000001413 amino acids Chemical class 0.000 description 20
- 102000005962 receptors Human genes 0.000 description 20
- 108020003175 receptors Proteins 0.000 description 20
- 230000000295 complement effect Effects 0.000 description 19
- 230000006870 function Effects 0.000 description 19
- 102000040430 polynucleotide Human genes 0.000 description 19
- 108091033319 polynucleotide Proteins 0.000 description 19
- 239000002157 polynucleotide Substances 0.000 description 19
- 239000012634 fragment Substances 0.000 description 18
- 230000002401 inhibitory effect Effects 0.000 description 18
- 230000007959 normoxia Effects 0.000 description 17
- 108091023040 Transcription factor Proteins 0.000 description 15
- 102000006495 integrins Human genes 0.000 description 15
- 108010044426 integrins Proteins 0.000 description 15
- 239000012528 membrane Substances 0.000 description 15
- 230000026731 phosphorylation Effects 0.000 description 15
- 238000006366 phosphorylation reaction Methods 0.000 description 15
- 230000001105 regulatory effect Effects 0.000 description 15
- 229920002477 rna polymer Polymers 0.000 description 15
- 230000006044 T cell activation Effects 0.000 description 14
- 102000040945 Transcription factor Human genes 0.000 description 14
- 125000003729 nucleotide group Chemical group 0.000 description 14
- 102100022875 Hypoxia-inducible factor 1-alpha Human genes 0.000 description 13
- 102100021125 Tyrosine-protein kinase ZAP-70 Human genes 0.000 description 13
- 150000001875 compounds Chemical class 0.000 description 13
- 238000000684 flow cytometry Methods 0.000 description 13
- 102000039446 nucleic acids Human genes 0.000 description 13
- 108020004707 nucleic acids Proteins 0.000 description 13
- 239000002773 nucleotide Substances 0.000 description 13
- 230000037361 pathway Effects 0.000 description 13
- 101710167800 Capsid assembly scaffolding protein Proteins 0.000 description 12
- 101000818543 Homo sapiens Tyrosine-protein kinase ZAP-70 Proteins 0.000 description 12
- 101710130420 Probable capsid assembly scaffolding protein Proteins 0.000 description 12
- 101710204410 Scaffold protein Proteins 0.000 description 12
- 210000004899 c-terminal region Anatomy 0.000 description 12
- 230000000694 effects Effects 0.000 description 12
- 108010055717 JNK Mitogen-Activated Protein Kinases Proteins 0.000 description 11
- 230000001086 cytosolic effect Effects 0.000 description 11
- 238000013461 design Methods 0.000 description 11
- 239000013612 plasmid Substances 0.000 description 11
- 239000000758 substrate Substances 0.000 description 11
- 210000001519 tissue Anatomy 0.000 description 11
- 101001046870 Homo sapiens Hypoxia-inducible factor 1-alpha Proteins 0.000 description 10
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 description 10
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 10
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 10
- 230000008685 targeting Effects 0.000 description 10
- 239000013603 viral vector Substances 0.000 description 10
- 108020004705 Codon Proteins 0.000 description 9
- 102000001301 EGF receptor Human genes 0.000 description 9
- 108060006698 EGF receptor Proteins 0.000 description 9
- 102000003945 NF-kappa B Human genes 0.000 description 9
- 231100000135 cytotoxicity Toxicity 0.000 description 9
- 230000003013 cytotoxicity Effects 0.000 description 9
- 239000012636 effector Substances 0.000 description 9
- 238000003752 polymerase chain reaction Methods 0.000 description 9
- 238000001890 transfection Methods 0.000 description 9
- 102000053602 DNA Human genes 0.000 description 8
- 108020004414 DNA Proteins 0.000 description 8
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 8
- 102000004190 Enzymes Human genes 0.000 description 8
- 108090000790 Enzymes Proteins 0.000 description 8
- 150000001540 azides Chemical class 0.000 description 8
- 238000003776 cleavage reaction Methods 0.000 description 8
- 229940088598 enzyme Drugs 0.000 description 8
- 230000006450 immune cell response Effects 0.000 description 8
- 239000003550 marker Substances 0.000 description 8
- 230000007017 scission Effects 0.000 description 8
- 238000013518 transcription Methods 0.000 description 8
- 230000035897 transcription Effects 0.000 description 8
- 230000003612 virological effect Effects 0.000 description 8
- 102000004127 Cytokines Human genes 0.000 description 7
- 108090000695 Cytokines Proteins 0.000 description 7
- 108700022150 Designed Ankyrin Repeat Proteins Proteins 0.000 description 7
- 108091028043 Nucleic acid sequence Proteins 0.000 description 7
- 108010076504 Protein Sorting Signals Proteins 0.000 description 7
- 238000003556 assay Methods 0.000 description 7
- 238000006471 dimerization reaction Methods 0.000 description 7
- 230000009977 dual effect Effects 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 239000002609 medium Substances 0.000 description 7
- 230000035772 mutation Effects 0.000 description 7
- 230000004044 response Effects 0.000 description 7
- 210000000130 stem cell Anatomy 0.000 description 7
- 230000000638 stimulation Effects 0.000 description 7
- 208000023275 Autoimmune disease Diseases 0.000 description 6
- 108700024832 B-Cell CLL-Lymphoma 10 Proteins 0.000 description 6
- 102000046016 B-Cell CLL-Lymphoma 10 Human genes 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- 108010057466 NF-kappa B Proteins 0.000 description 6
- 101710184528 Scaffolding protein Proteins 0.000 description 6
- 238000010459 TALEN Methods 0.000 description 6
- 108010043645 Transcription Activator-Like Effector Nucleases Proteins 0.000 description 6
- 208000036142 Viral infection Diseases 0.000 description 6
- 238000010586 diagram Methods 0.000 description 6
- 238000004520 electroporation Methods 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 230000001146 hypoxic effect Effects 0.000 description 6
- 210000004698 lymphocyte Anatomy 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 108020001580 protein domains Proteins 0.000 description 6
- 230000003248 secreting effect Effects 0.000 description 6
- 230000004083 survival effect Effects 0.000 description 6
- 210000000225 synapse Anatomy 0.000 description 6
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 6
- 230000009385 viral infection Effects 0.000 description 6
- 102000006306 Antigen Receptors Human genes 0.000 description 5
- 108010083359 Antigen Receptors Proteins 0.000 description 5
- 101150074953 BCL10 gene Proteins 0.000 description 5
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 5
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 5
- 241000713666 Lentivirus Species 0.000 description 5
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 5
- 108091034117 Oligonucleotide Proteins 0.000 description 5
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 5
- 230000033228 biological regulation Effects 0.000 description 5
- 230000008859 change Effects 0.000 description 5
- 230000001419 dependent effect Effects 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 230000007613 environmental effect Effects 0.000 description 5
- 239000003102 growth factor Substances 0.000 description 5
- 208000015181 infectious disease Diseases 0.000 description 5
- 239000003112 inhibitor Substances 0.000 description 5
- 238000001823 molecular biology technique Methods 0.000 description 5
- 150000003384 small molecules Chemical class 0.000 description 5
- 125000006850 spacer group Chemical group 0.000 description 5
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 4
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 4
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 4
- 108091007741 Chimeric antigen receptor T cells Proteins 0.000 description 4
- 108010001515 Galectin 4 Proteins 0.000 description 4
- 102100039556 Galectin-4 Human genes 0.000 description 4
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 4
- 102000004157 Hydrolases Human genes 0.000 description 4
- 108090000604 Hydrolases Proteins 0.000 description 4
- 206010025323 Lymphomas Diseases 0.000 description 4
- 102000001253 Protein Kinase Human genes 0.000 description 4
- 102000036646 Signalosomes Human genes 0.000 description 4
- 108091007411 Signalosomes Proteins 0.000 description 4
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 4
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 4
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 125000003275 alpha amino acid group Chemical group 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 238000007405 data analysis Methods 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 230000004927 fusion Effects 0.000 description 4
- 230000033444 hydroxylation Effects 0.000 description 4
- 238000005805 hydroxylation reaction Methods 0.000 description 4
- 239000000178 monomer Substances 0.000 description 4
- 230000004481 post-translational protein modification Effects 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- 108060006633 protein kinase Proteins 0.000 description 4
- 230000004850 protein–protein interaction Effects 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical group CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 4
- 238000011144 upstream manufacturing Methods 0.000 description 4
- 102100023635 Alpha-fetoprotein Human genes 0.000 description 3
- 101710145634 Antigen 1 Proteins 0.000 description 3
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 3
- 101150013553 CD40 gene Proteins 0.000 description 3
- 101100386910 Caenorhabditis elegans laf-1 gene Proteins 0.000 description 3
- 102000004631 Calcineurin Human genes 0.000 description 3
- 108010042955 Calcineurin Proteins 0.000 description 3
- 102000019034 Chemokines Human genes 0.000 description 3
- 108010012236 Chemokines Proteins 0.000 description 3
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 3
- 108010036949 Cyclosporine Proteins 0.000 description 3
- 230000004568 DNA-binding Effects 0.000 description 3
- 102100031780 Endonuclease Human genes 0.000 description 3
- 108020004202 Guanylate Kinase Proteins 0.000 description 3
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 3
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 description 3
- 108050009527 Hypoxia-inducible factor-1 alpha Proteins 0.000 description 3
- 101150113681 MALT1 gene Proteins 0.000 description 3
- 108700026676 Mucosa-Associated Lymphoid Tissue Lymphoma Translocation 1 Proteins 0.000 description 3
- 102100038732 Mucosa-associated lymphoid tissue lymphoma translocation protein 1 Human genes 0.000 description 3
- 108010014632 NF-kappa B kinase Proteins 0.000 description 3
- 102000015636 Oligopeptides Human genes 0.000 description 3
- 108010038807 Oligopeptides Proteins 0.000 description 3
- 238000012408 PCR amplification Methods 0.000 description 3
- 102000004079 Prolyl Hydroxylases Human genes 0.000 description 3
- 108010043005 Prolyl Hydroxylases Proteins 0.000 description 3
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 3
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 3
- 108091027981 Response element Proteins 0.000 description 3
- 206010039491 Sarcoma Diseases 0.000 description 3
- 101100204458 Schizosaccharomyces pombe (strain 972 / ATCC 24843) svf2 gene Proteins 0.000 description 3
- 241000713880 Spleen focus-forming virus Species 0.000 description 3
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 3
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 3
- 102100033726 Tumor necrosis factor receptor superfamily member 17 Human genes 0.000 description 3
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 3
- 108090000848 Ubiquitin Proteins 0.000 description 3
- 102000044159 Ubiquitin Human genes 0.000 description 3
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- 210000003719 b-lymphocyte Anatomy 0.000 description 3
- 239000011324 bead Substances 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 238000002512 chemotherapy Methods 0.000 description 3
- 229960001265 ciclosporin Drugs 0.000 description 3
- 229930182912 cyclosporin Natural products 0.000 description 3
- 230000001461 cytolytic effect Effects 0.000 description 3
- 210000000805 cytoplasm Anatomy 0.000 description 3
- 238000002784 cytotoxicity assay Methods 0.000 description 3
- 231100000263 cytotoxicity test Toxicity 0.000 description 3
- 210000004443 dendritic cell Anatomy 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000013604 expression vector Substances 0.000 description 3
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 3
- 230000002068 genetic effect Effects 0.000 description 3
- 102000006638 guanylate kinase Human genes 0.000 description 3
- 230000036039 immunity Effects 0.000 description 3
- 230000002779 inactivation Effects 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 230000004068 intracellular signaling Effects 0.000 description 3
- 238000002372 labelling Methods 0.000 description 3
- 208000032839 leukemia Diseases 0.000 description 3
- 238000007885 magnetic separation Methods 0.000 description 3
- 201000001441 melanoma Diseases 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 238000002703 mutagenesis Methods 0.000 description 3
- 231100000350 mutagenesis Toxicity 0.000 description 3
- 210000000822 natural killer cell Anatomy 0.000 description 3
- 239000008177 pharmaceutical agent Substances 0.000 description 3
- 230000034190 positive regulation of NF-kappaB transcription factor activity Effects 0.000 description 3
- 230000023603 positive regulation of transcription initiation, DNA-dependent Effects 0.000 description 3
- 230000016434 protein splicing Effects 0.000 description 3
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 3
- 230000006641 stabilisation Effects 0.000 description 3
- 238000011105 stabilization Methods 0.000 description 3
- 238000010186 staining Methods 0.000 description 3
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 3
- 230000002103 transcriptional effect Effects 0.000 description 3
- 238000010361 transduction Methods 0.000 description 3
- 230000026683 transduction Effects 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 238000011426 transformation method Methods 0.000 description 3
- 230000005945 translocation Effects 0.000 description 3
- 230000001960 triggered effect Effects 0.000 description 3
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 3
- 241000701161 unidentified adenovirus Species 0.000 description 3
- 241001430294 unidentified retrovirus Species 0.000 description 3
- 230000035899 viability Effects 0.000 description 3
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 102100026094 C-type lectin domain family 12 member A Human genes 0.000 description 2
- 101710188619 C-type lectin domain family 12 member A Proteins 0.000 description 2
- 102100038078 CD276 antigen Human genes 0.000 description 2
- 101710185679 CD276 antigen Proteins 0.000 description 2
- 102100032912 CD44 antigen Human genes 0.000 description 2
- 102100035793 CD83 antigen Human genes 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 description 2
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 2
- 101710205695 Caspase recruitment domain-containing protein 11 Proteins 0.000 description 2
- 102000016289 Cell Adhesion Molecules Human genes 0.000 description 2
- 108010067225 Cell Adhesion Molecules Proteins 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- 108010016626 Dipeptides Proteins 0.000 description 2
- 101100508533 Drosophila melanogaster IKKbeta gene Proteins 0.000 description 2
- 108010042407 Endonucleases Proteins 0.000 description 2
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 description 2
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 2
- 108010007457 Extracellular Signal-Regulated MAP Kinases Proteins 0.000 description 2
- 102000007665 Extracellular Signal-Regulated MAP Kinases Human genes 0.000 description 2
- 108091008794 FGF receptors Proteins 0.000 description 2
- 108010074860 Factor Xa Proteins 0.000 description 2
- 208000009329 Graft vs Host Disease Diseases 0.000 description 2
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 2
- 102100022057 Hepatocyte nuclear factor 1-alpha Human genes 0.000 description 2
- 102100022123 Hepatocyte nuclear factor 1-beta Human genes 0.000 description 2
- 108010077223 Homer Scaffolding Proteins Proteins 0.000 description 2
- 102000010029 Homer Scaffolding Proteins Human genes 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 2
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 description 2
- 101000856237 Homo sapiens Cancer/testis antigen 1 Proteins 0.000 description 2
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 2
- 101001045751 Homo sapiens Hepatocyte nuclear factor 1-alpha Proteins 0.000 description 2
- 101001045758 Homo sapiens Hepatocyte nuclear factor 1-beta Proteins 0.000 description 2
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 2
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 2
- 101000946860 Homo sapiens T-cell surface glycoprotein CD3 epsilon chain Proteins 0.000 description 2
- 101000801255 Homo sapiens Tumor necrosis factor receptor superfamily member 17 Proteins 0.000 description 2
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 2
- 101100268065 Homo sapiens ZAP70 gene Proteins 0.000 description 2
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 2
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 2
- 102000000588 Interleukin-2 Human genes 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 description 2
- 102100034872 Kallikrein-4 Human genes 0.000 description 2
- 102000003960 Ligases Human genes 0.000 description 2
- 108090000364 Ligases Proteins 0.000 description 2
- 239000007993 MOPS buffer Substances 0.000 description 2
- 108090000015 Mesothelin Proteins 0.000 description 2
- 102000003735 Mesothelin Human genes 0.000 description 2
- 108010082747 Mitogen-Activated Protein Kinase Phosphatases Proteins 0.000 description 2
- 102000004182 Mitogen-Activated Protein Kinase Phosphatases Human genes 0.000 description 2
- 102100034256 Mucin-1 Human genes 0.000 description 2
- 101100508818 Mus musculus Inpp5k gene Proteins 0.000 description 2
- 101150111783 NTRK1 gene Proteins 0.000 description 2
- 108091007494 Nucleic acid- binding domains Proteins 0.000 description 2
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 2
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 2
- 241000709664 Picornaviridae Species 0.000 description 2
- 108010014608 Proto-Oncogene Proteins c-kit Proteins 0.000 description 2
- 102000016971 Proto-Oncogene Proteins c-kit Human genes 0.000 description 2
- 101100366438 Rattus norvegicus Sphkap gene Proteins 0.000 description 2
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 2
- 241000242739 Renilla Species 0.000 description 2
- 108700008625 Reporter Genes Proteins 0.000 description 2
- 102000014400 SH2 domains Human genes 0.000 description 2
- 108050003452 SH2 domains Proteins 0.000 description 2
- 102000000551 Syk Kinase Human genes 0.000 description 2
- 108010016672 Syk Kinase Proteins 0.000 description 2
- 230000006052 T cell proliferation Effects 0.000 description 2
- 102100035794 T-cell surface glycoprotein CD3 epsilon chain Human genes 0.000 description 2
- 108010076818 TEV protease Proteins 0.000 description 2
- 108090000190 Thrombin Proteins 0.000 description 2
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 2
- 102000006275 Ubiquitin-Protein Ligases Human genes 0.000 description 2
- 108010083111 Ubiquitin-Protein Ligases Proteins 0.000 description 2
- 206010046865 Vaccinia virus infection Diseases 0.000 description 2
- 101150019975 ZAP70 gene Proteins 0.000 description 2
- 238000010317 ablation therapy Methods 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 230000008649 adaptation response Effects 0.000 description 2
- 239000011543 agarose gel Substances 0.000 description 2
- 230000000735 allogeneic effect Effects 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 238000009175 antibody therapy Methods 0.000 description 2
- 210000000612 antigen-presenting cell Anatomy 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 210000001185 bone marrow Anatomy 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229940112129 campath Drugs 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 238000002659 cell therapy Methods 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 230000005754 cellular signaling Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 230000002759 chromosomal effect Effects 0.000 description 2
- 230000006690 co-activation Effects 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 238000002716 delivery method Methods 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- 241001493065 dsRNA viruses Species 0.000 description 2
- 210000003162 effector t lymphocyte Anatomy 0.000 description 2
- 210000004700 fetal blood Anatomy 0.000 description 2
- 102000052178 fibroblast growth factor receptor activity proteins Human genes 0.000 description 2
- 102000034287 fluorescent proteins Human genes 0.000 description 2
- 108091006047 fluorescent proteins Proteins 0.000 description 2
- 238000001476 gene delivery Methods 0.000 description 2
- 208000024908 graft versus host disease Diseases 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 239000000833 heterodimer Substances 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- 230000028993 immune response Effects 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000001965 increasing effect Effects 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 108010024383 kallikrein 4 Proteins 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- 230000001483 mobilizing effect Effects 0.000 description 2
- 210000004877 mucosa Anatomy 0.000 description 2
- 238000006384 oligomerization reaction Methods 0.000 description 2
- 238000004806 packaging method and process Methods 0.000 description 2
- 230000010412 perfusion Effects 0.000 description 2
- 239000013600 plasmid vector Substances 0.000 description 2
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 2
- 210000004986 primary T-cell Anatomy 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 238000003157 protein complementation Methods 0.000 description 2
- 230000004853 protein function Effects 0.000 description 2
- 150000003212 purines Chemical class 0.000 description 2
- 150000003230 pyrimidines Chemical class 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 238000001959 radiotherapy Methods 0.000 description 2
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 2
- 230000007115 recruitment Effects 0.000 description 2
- 230000019908 regulation of T cell activation Effects 0.000 description 2
- 230000001718 repressive effect Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 210000003705 ribosome Anatomy 0.000 description 2
- 230000001235 sensitizing effect Effects 0.000 description 2
- 229960002930 sirolimus Drugs 0.000 description 2
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 2
- 108010087686 src-Family Kinases Proteins 0.000 description 2
- 102000009076 src-Family Kinases Human genes 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 229960004072 thrombin Drugs 0.000 description 2
- 229940113082 thymine Drugs 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- 208000007089 vaccinia Diseases 0.000 description 2
- FFILOTSTFMXQJC-QCFYAKGBSA-N (2r,4r,5s,6s)-2-[3-[(2s,3s,4r,6s)-6-[(2s,3r,4r,5s,6r)-5-[(2s,3r,4r,5r,6r)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-2-[(2r,3s,4r,5r,6r)-4,5-dihydroxy-2-(hydroxymethyl)-6-[(e)-3-hydroxy-2-(octadecanoylamino)octadec-4-enoxy]oxan-3-yl]oxy-3-hy Chemical compound O[C@@H]1[C@@H](O)[C@H](OCC(NC(=O)CCCCCCCCCCCCCCCCC)C(O)\C=C\CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@]2(O[C@@H]([C@@H](N)[C@H](O)C2)C(O)C(O)CO[C@]2(O[C@@H]([C@@H](N)[C@H](O)C2)C(O)C(O)CO)C(O)=O)C(O)=O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](CO)O1 FFILOTSTFMXQJC-QCFYAKGBSA-N 0.000 description 1
- BGFTWECWAICPDG-UHFFFAOYSA-N 2-[bis(4-chlorophenyl)methyl]-4-n-[3-[bis(4-chlorophenyl)methyl]-4-(dimethylamino)phenyl]-1-n,1-n-dimethylbenzene-1,4-diamine Chemical compound C1=C(C(C=2C=CC(Cl)=CC=2)C=2C=CC(Cl)=CC=2)C(N(C)C)=CC=C1NC(C=1)=CC=C(N(C)C)C=1C(C=1C=CC(Cl)=CC=1)C1=CC=C(Cl)C=C1 BGFTWECWAICPDG-UHFFFAOYSA-N 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- 102000002627 4-1BB Ligand Human genes 0.000 description 1
- 108010082808 4-1BB Ligand Proteins 0.000 description 1
- 108010013238 70-kDa Ribosomal Protein S6 Kinases Proteins 0.000 description 1
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical group N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 1
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 1
- 102100033793 ALK tyrosine kinase receptor Human genes 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 241000710929 Alphavirus Species 0.000 description 1
- 102100032187 Androgen receptor Human genes 0.000 description 1
- 108010049777 Ankyrins Proteins 0.000 description 1
- 102000008102 Ankyrins Human genes 0.000 description 1
- 101100366892 Anopheles gambiae Stat gene Proteins 0.000 description 1
- 241000710189 Aphthovirus Species 0.000 description 1
- 241000203069 Archaea Species 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 108010031480 Artificial Receptors Proteins 0.000 description 1
- 108090000448 Aryl Hydrocarbon Receptors Proteins 0.000 description 1
- 102100026792 Aryl hydrocarbon receptor Human genes 0.000 description 1
- 206010003445 Ascites Diseases 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 102100029822 B- and T-lymphocyte attenuator Human genes 0.000 description 1
- 108010008014 B-Cell Maturation Antigen Proteins 0.000 description 1
- 108010074708 B7-H1 Antigen Proteins 0.000 description 1
- 108010077805 Bacterial Proteins Proteins 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 102100021663 Baculoviral IAP repeat-containing protein 5 Human genes 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 102000003930 C-Type Lectins Human genes 0.000 description 1
- 108090000342 C-Type Lectins Proteins 0.000 description 1
- 102000002086 C-type lectin-like Human genes 0.000 description 1
- 108050009406 C-type lectin-like Proteins 0.000 description 1
- 102000002164 CARD domains Human genes 0.000 description 1
- 108050009503 CARD domains Proteins 0.000 description 1
- 102100027207 CD27 antigen Human genes 0.000 description 1
- 102100025221 CD70 antigen Human genes 0.000 description 1
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 1
- 229940045513 CTLA4 antagonist Drugs 0.000 description 1
- 101000741929 Caenorhabditis elegans Serine/threonine-protein phosphatase 2A catalytic subunit Proteins 0.000 description 1
- 102000004657 Calcium-Calmodulin-Dependent Protein Kinase Type 2 Human genes 0.000 description 1
- 108010003721 Calcium-Calmodulin-Dependent Protein Kinase Type 2 Proteins 0.000 description 1
- 101800001318 Capsid protein VP4 Proteins 0.000 description 1
- 102000013392 Carboxylesterase Human genes 0.000 description 1
- 108010051152 Carboxylesterase Proteins 0.000 description 1
- 102100025466 Carcinoembryonic antigen-related cell adhesion molecule 3 Human genes 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 102100034357 Casein kinase I isoform alpha Human genes 0.000 description 1
- 102000021350 Caspase recruitment domains Human genes 0.000 description 1
- 108091011189 Caspase recruitment domains Proteins 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 1
- 108010059480 Chondroitin Sulfate Proteoglycans Proteins 0.000 description 1
- 102000005598 Chondroitin Sulfate Proteoglycans Human genes 0.000 description 1
- VWFCHDSQECPREK-LURJTMIESA-N Cidofovir Chemical compound NC=1C=CN(C[C@@H](CO)OCP(O)(O)=O)C(=O)N=1 VWFCHDSQECPREK-LURJTMIESA-N 0.000 description 1
- 108020004638 Circular DNA Proteins 0.000 description 1
- 102100030953 Cleavage and polyadenylation specificity factor subunit 4 Human genes 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 241000711573 Coronaviridae Species 0.000 description 1
- 241000192700 Cyanobacteria Species 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- 108010037414 Cytoskeletal Proteins Proteins 0.000 description 1
- 102000010831 Cytoskeletal Proteins Human genes 0.000 description 1
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 1
- 241000450599 DNA viruses Species 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 102100024099 Disks large homolog 1 Human genes 0.000 description 1
- 102100037832 Docking protein 1 Human genes 0.000 description 1
- 102100037830 Docking protein 2 Human genes 0.000 description 1
- 101100366894 Drosophila melanogaster Stat92E gene Proteins 0.000 description 1
- 101150084967 EPCAM gene Proteins 0.000 description 1
- 108010036395 Endoglin Proteins 0.000 description 1
- 102100037241 Endoglin Human genes 0.000 description 1
- 108010013369 Enteropeptidase Proteins 0.000 description 1
- 102100029727 Enteropeptidase Human genes 0.000 description 1
- 108010092408 Eosinophil Peroxidase Proteins 0.000 description 1
- 108010055196 EphA2 Receptor Proteins 0.000 description 1
- 102100030340 Ephrin type-A receptor 2 Human genes 0.000 description 1
- 102100023721 Ephrin-B2 Human genes 0.000 description 1
- 108010044090 Ephrin-B2 Proteins 0.000 description 1
- 102000003951 Erythropoietin Human genes 0.000 description 1
- 102100031939 Erythropoietin Human genes 0.000 description 1
- 108090000394 Erythropoietin Proteins 0.000 description 1
- 108060002716 Exonuclease Proteins 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- 102000016359 Fibronectins Human genes 0.000 description 1
- 241000710831 Flavivirus Species 0.000 description 1
- 108050001931 Folate receptor alpha Proteins 0.000 description 1
- 102000010451 Folate receptor alpha Human genes 0.000 description 1
- 208000000666 Fowlpox Diseases 0.000 description 1
- 102100022086 GRB2-related adapter protein 2 Human genes 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- 108091052347 Glucose transporter family Proteins 0.000 description 1
- 102000042092 Glucose transporter family Human genes 0.000 description 1
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 1
- 102100031181 Glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 1
- 108060005986 Granzyme Proteins 0.000 description 1
- 102000001398 Granzyme Human genes 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- 102100033067 Growth factor receptor-bound protein 2 Human genes 0.000 description 1
- 102100028967 HLA class I histocompatibility antigen, alpha chain G Human genes 0.000 description 1
- 108010024164 HLA-G Antigens Proteins 0.000 description 1
- 102000016761 Haem oxygenases Human genes 0.000 description 1
- 108050006318 Haem oxygenases Proteins 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 102100022132 High affinity immunoglobulin epsilon receptor subunit gamma Human genes 0.000 description 1
- 108091010847 High affinity immunoglobulin epsilon receptor subunit gamma Proteins 0.000 description 1
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 1
- 102100025210 Histone-arginine methyltransferase CARM1 Human genes 0.000 description 1
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 1
- 101000779641 Homo sapiens ALK tyrosine kinase receptor Proteins 0.000 description 1
- 101000864344 Homo sapiens B- and T-lymphocyte attenuator Proteins 0.000 description 1
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 1
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 description 1
- 101000914337 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 3 Proteins 0.000 description 1
- 101000994700 Homo sapiens Casein kinase I isoform alpha Proteins 0.000 description 1
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 1
- 101000727105 Homo sapiens Cleavage and polyadenylation specificity factor subunit 4 Proteins 0.000 description 1
- 101001053984 Homo sapiens Disks large homolog 1 Proteins 0.000 description 1
- 101000951365 Homo sapiens Disks large-associated protein 5 Proteins 0.000 description 1
- 101000805172 Homo sapiens Docking protein 1 Proteins 0.000 description 1
- 101000805166 Homo sapiens Docking protein 2 Proteins 0.000 description 1
- 101001024566 Homo sapiens Ecto-ADP-ribosyltransferase 4 Proteins 0.000 description 1
- 101000900690 Homo sapiens GRB2-related adapter protein 2 Proteins 0.000 description 1
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 1
- 101000871017 Homo sapiens Growth factor receptor-bound protein 2 Proteins 0.000 description 1
- 101000599951 Homo sapiens Insulin-like growth factor I Proteins 0.000 description 1
- 101000994365 Homo sapiens Integrin alpha-6 Proteins 0.000 description 1
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 1
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 1
- 101000984189 Homo sapiens Leukocyte immunoglobulin-like receptor subfamily B member 2 Proteins 0.000 description 1
- 101000984186 Homo sapiens Leukocyte immunoglobulin-like receptor subfamily B member 4 Proteins 0.000 description 1
- 101001039199 Homo sapiens Low-density lipoprotein receptor-related protein 6 Proteins 0.000 description 1
- 101001023379 Homo sapiens Lysosome-associated membrane glycoprotein 1 Proteins 0.000 description 1
- 101001014223 Homo sapiens MAPK/MAK/MRK overlapping kinase Proteins 0.000 description 1
- 101001059991 Homo sapiens Mitogen-activated protein kinase kinase kinase kinase 1 Proteins 0.000 description 1
- 101001109503 Homo sapiens NKG2-C type II integral membrane protein Proteins 0.000 description 1
- 101001109501 Homo sapiens NKG2-D type II integral membrane protein Proteins 0.000 description 1
- 101000613490 Homo sapiens Paired box protein Pax-3 Proteins 0.000 description 1
- 101000616502 Homo sapiens Phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase 1 Proteins 0.000 description 1
- 101001136981 Homo sapiens Proteasome subunit beta type-9 Proteins 0.000 description 1
- 101000775749 Homo sapiens Proto-oncogene vav Proteins 0.000 description 1
- 101000884271 Homo sapiens Signal transducer CD24 Proteins 0.000 description 1
- 101000934341 Homo sapiens T-cell surface glycoprotein CD5 Proteins 0.000 description 1
- 101100207070 Homo sapiens TNFSF8 gene Proteins 0.000 description 1
- 101000655352 Homo sapiens Telomerase reverse transcriptase Proteins 0.000 description 1
- 101000597785 Homo sapiens Tumor necrosis factor receptor superfamily member 6B Proteins 0.000 description 1
- 101001135589 Homo sapiens Tyrosine-protein phosphatase non-receptor type 22 Proteins 0.000 description 1
- 101000617285 Homo sapiens Tyrosine-protein phosphatase non-receptor type 6 Proteins 0.000 description 1
- 102000009490 IgG Receptors Human genes 0.000 description 1
- 108010073807 IgG Receptors Proteins 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 101000668058 Infectious salmon anemia virus (isolate Atlantic salmon/Norway/810/9/99) RNA-directed RNA polymerase catalytic subunit Proteins 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 108090001117 Insulin-Like Growth Factor II Proteins 0.000 description 1
- 102000048143 Insulin-Like Growth Factor II Human genes 0.000 description 1
- 102100037852 Insulin-like growth factor I Human genes 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 108010061833 Integrases Proteins 0.000 description 1
- 102100032816 Integrin alpha-6 Human genes 0.000 description 1
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 1
- 102000003816 Interleukin-13 Human genes 0.000 description 1
- 108090000176 Interleukin-13 Proteins 0.000 description 1
- 102100020793 Interleukin-13 receptor subunit alpha-2 Human genes 0.000 description 1
- 101710112634 Interleukin-13 receptor subunit alpha-2 Proteins 0.000 description 1
- 108010038453 Interleukin-2 Receptors Proteins 0.000 description 1
- 102000010789 Interleukin-2 Receptors Human genes 0.000 description 1
- 108020003285 Isocitrate lyase Proteins 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 125000002842 L-seryl group Chemical group O=C([*])[C@](N([H])[H])([H])C([H])([H])O[H] 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- 101150028321 Lck gene Proteins 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 108010028275 Leukocyte Elastase Proteins 0.000 description 1
- 102100025583 Leukocyte immunoglobulin-like receptor subfamily B member 2 Human genes 0.000 description 1
- 102100025578 Leukocyte immunoglobulin-like receptor subfamily B member 4 Human genes 0.000 description 1
- 102100040704 Low-density lipoprotein receptor-related protein 6 Human genes 0.000 description 1
- 102100034709 Lymphocyte cytosolic protein 2 Human genes 0.000 description 1
- 101710195102 Lymphocyte cytosolic protein 2 Proteins 0.000 description 1
- 108010091221 Lymphotoxin beta Receptor Proteins 0.000 description 1
- 102100035133 Lysosome-associated membrane glycoprotein 1 Human genes 0.000 description 1
- 102000019149 MAP kinase activity proteins Human genes 0.000 description 1
- 108040008097 MAP kinase activity proteins Proteins 0.000 description 1
- 102100031520 MAPK/MAK/MRK overlapping kinase Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 102100030301 MHC class I polypeptide-related sequence A Human genes 0.000 description 1
- 241000701076 Macacine alphaherpesvirus 1 Species 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100028123 Macrophage colony-stimulating factor 1 Human genes 0.000 description 1
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 description 1
- 201000005505 Measles Diseases 0.000 description 1
- 102000012750 Membrane Glycoproteins Human genes 0.000 description 1
- 108010090054 Membrane Glycoproteins Proteins 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 102100028199 Mitogen-activated protein kinase kinase kinase kinase 1 Human genes 0.000 description 1
- 229930191564 Monensin Natural products 0.000 description 1
- GAOZTHIDHYLHMS-UHFFFAOYSA-N Monensin A Natural products O1C(CC)(C2C(CC(O2)C2C(CC(C)C(O)(CO)O2)C)C)CCC1C(O1)(C)CCC21CC(O)C(C)C(C(C)C(OC)C(C)C(O)=O)O2 GAOZTHIDHYLHMS-UHFFFAOYSA-N 0.000 description 1
- 101150097381 Mtor gene Proteins 0.000 description 1
- 108010063954 Mucins Proteins 0.000 description 1
- 101100180319 Mus musculus Itk gene Proteins 0.000 description 1
- 101100407308 Mus musculus Pdcd1lg2 gene Proteins 0.000 description 1
- 101100207071 Mus musculus Tnfsf8 gene Proteins 0.000 description 1
- 101100268066 Mus musculus Zap70 gene Proteins 0.000 description 1
- 108010018525 NFATC Transcription Factors Proteins 0.000 description 1
- 102000002673 NFATC Transcription Factors Human genes 0.000 description 1
- 108010001657 NK Cell Lectin-Like Receptor Subfamily K Proteins 0.000 description 1
- 102000000812 NK Cell Lectin-Like Receptor Subfamily K Human genes 0.000 description 1
- 102100022683 NKG2-C type II integral membrane protein Human genes 0.000 description 1
- 102100022680 NKG2-D type II integral membrane protein Human genes 0.000 description 1
- 102400000058 Neuregulin-1 Human genes 0.000 description 1
- 108090000556 Neuregulin-1 Proteins 0.000 description 1
- 102100033174 Neutrophil elastase Human genes 0.000 description 1
- 241000208125 Nicotiana Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- 102100029438 Nitric oxide synthase, inducible Human genes 0.000 description 1
- 101710089543 Nitric oxide synthase, inducible Proteins 0.000 description 1
- 241000714209 Norwalk virus Species 0.000 description 1
- 102000004473 OX40 Ligand Human genes 0.000 description 1
- 108010042215 OX40 Ligand Proteins 0.000 description 1
- 108700026244 Open Reading Frames Proteins 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 108010052444 Oxyrase Proteins 0.000 description 1
- 101150036732 PRKCQ gene Proteins 0.000 description 1
- 102100040891 Paired box protein Pax-3 Human genes 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- KHGNFPUMBJSZSM-UHFFFAOYSA-N Perforine Natural products COC1=C2CCC(O)C(CCC(C)(C)O)(OC)C2=NC2=C1C=CO2 KHGNFPUMBJSZSM-UHFFFAOYSA-N 0.000 description 1
- 102100021797 Phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase 1 Human genes 0.000 description 1
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 1
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 1
- 102000004422 Phospholipase C gamma Human genes 0.000 description 1
- 108010056751 Phospholipase C gamma Proteins 0.000 description 1
- 208000002151 Pleural effusion Diseases 0.000 description 1
- 108700030875 Programmed Cell Death 1 Ligand 2 Proteins 0.000 description 1
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 1
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 description 1
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 description 1
- 102100035764 Proteasome subunit beta type-9 Human genes 0.000 description 1
- 102100032190 Proto-oncogene vav Human genes 0.000 description 1
- 241000125945 Protoparvovirus Species 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical group C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 241000711798 Rabies lyssavirus Species 0.000 description 1
- 241000232299 Ralstonia Species 0.000 description 1
- 102000004278 Receptor Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000873 Receptor Protein-Tyrosine Kinases Proteins 0.000 description 1
- 241000702263 Reovirus sp. Species 0.000 description 1
- 108091081062 Repeated sequence (DNA) Proteins 0.000 description 1
- 241000712907 Retroviridae Species 0.000 description 1
- 102100027609 Rho-related GTP-binding protein RhoD Human genes 0.000 description 1
- 102000001332 SRC Human genes 0.000 description 1
- 108060006706 SRC Proteins 0.000 description 1
- 108091081021 Sense strand Proteins 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 102100023085 Serine/threonine-protein kinase mTOR Human genes 0.000 description 1
- 241000287219 Serinus canaria Species 0.000 description 1
- 102100038081 Signal transducer CD24 Human genes 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 1
- 102100037253 Solute carrier family 45 member 3 Human genes 0.000 description 1
- 102100038803 Somatotropin Human genes 0.000 description 1
- 241000713675 Spumavirus Species 0.000 description 1
- 101000677856 Stenotrophomonas maltophilia (strain K279a) Actin-binding protein Smlt3054 Proteins 0.000 description 1
- 108010002687 Survivin Proteins 0.000 description 1
- 230000005867 T cell response Effects 0.000 description 1
- 108010092262 T-Cell Antigen Receptors Proteins 0.000 description 1
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 description 1
- 102000006463 Talin Human genes 0.000 description 1
- 108010083809 Talin Proteins 0.000 description 1
- 108010017842 Telomerase Proteins 0.000 description 1
- 102000007000 Tenascin Human genes 0.000 description 1
- 108010008125 Tenascin Proteins 0.000 description 1
- 102000006601 Thymidine Kinase Human genes 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 108010034949 Thyroglobulin Proteins 0.000 description 1
- 102000009843 Thyroglobulin Human genes 0.000 description 1
- 108010073062 Transcription Activator-Like Effectors Proteins 0.000 description 1
- 102100023132 Transcription factor Jun Human genes 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- 101800000385 Transmembrane protein Proteins 0.000 description 1
- 102100032100 Tumor necrosis factor ligand superfamily member 8 Human genes 0.000 description 1
- 102100035284 Tumor necrosis factor receptor superfamily member 6B Human genes 0.000 description 1
- 102100039094 Tyrosinase Human genes 0.000 description 1
- 108060008724 Tyrosinase Proteins 0.000 description 1
- 102100033138 Tyrosine-protein phosphatase non-receptor type 22 Human genes 0.000 description 1
- 102100021657 Tyrosine-protein phosphatase non-receptor type 6 Human genes 0.000 description 1
- 108091023045 Untranslated Region Proteins 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 108020005202 Viral DNA Proteins 0.000 description 1
- 108020000999 Viral RNA Proteins 0.000 description 1
- 102000040856 WT1 Human genes 0.000 description 1
- 108700020467 WT1 Proteins 0.000 description 1
- 101150084041 WT1 gene Proteins 0.000 description 1
- 241000589634 Xanthomonas Species 0.000 description 1
- 108010046882 ZAP-70 Protein-Tyrosine Kinase Proteins 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000033289 adaptive immune response Effects 0.000 description 1
- 102000035181 adaptor proteins Human genes 0.000 description 1
- 108091005764 adaptor proteins Proteins 0.000 description 1
- 230000002730 additional effect Effects 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 102000019997 adhesion receptor Human genes 0.000 description 1
- 108010013985 adhesion receptor Proteins 0.000 description 1
- 210000004504 adult stem cell Anatomy 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 230000008860 allosteric change Effects 0.000 description 1
- 230000003281 allosteric effect Effects 0.000 description 1
- 229940125528 allosteric inhibitor Drugs 0.000 description 1
- 108010026331 alpha-Fetoproteins Proteins 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 108010080146 androgen receptors Proteins 0.000 description 1
- 230000002491 angiogenic effect Effects 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 101150010487 are gene Proteins 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N aspartic acid group Chemical group N[C@@H](CC(=O)O)C(=O)O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 230000001908 autoinhibitory effect Effects 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 201000000053 blastoma Diseases 0.000 description 1
- 238000010322 bone marrow transplantation Methods 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 238000010523 cascade reaction Methods 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000002458 cell surface marker Substances 0.000 description 1
- 230000010001 cellular homeostasis Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 208000011654 childhood malignant neoplasm Diseases 0.000 description 1
- 208000012191 childhood neoplasm Diseases 0.000 description 1
- 229960000724 cidofovir Drugs 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 108010030886 coactivator-associated arginine methyltransferase 1 Proteins 0.000 description 1
- 238000011284 combination treatment Methods 0.000 description 1
- 230000009137 competitive binding Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 102000003675 cytokine receptors Human genes 0.000 description 1
- 108010057085 cytokine receptors Proteins 0.000 description 1
- 210000005220 cytoplasmic tail Anatomy 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000012350 deep sequencing Methods 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 239000000412 dendrimer Substances 0.000 description 1
- 229920000736 dendritic polymer Polymers 0.000 description 1
- 230000030609 dephosphorylation Effects 0.000 description 1
- 238000006209 dephosphorylation reaction Methods 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 230000007783 downstream signaling Effects 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 239000002961 echo contrast media Substances 0.000 description 1
- 201000008184 embryoma Diseases 0.000 description 1
- 210000001671 embryonic stem cell Anatomy 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000010437 erythropoiesis Effects 0.000 description 1
- 229940105423 erythropoietin Drugs 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 102000013165 exonuclease Human genes 0.000 description 1
- 238000002710 external beam radiation therapy Methods 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 1
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 1
- 230000002414 glycolytic effect Effects 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 238000005734 heterodimerization reaction Methods 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 108091008039 hormone receptors Proteins 0.000 description 1
- 238000001794 hormone therapy Methods 0.000 description 1
- 229940084986 human chorionic gonadotropin Drugs 0.000 description 1
- 125000001165 hydrophobic group Chemical group 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 230000005965 immune activity Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 229940125721 immunosuppressive agent Drugs 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 210000004263 induced pluripotent stem cell Anatomy 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 230000015788 innate immune response Effects 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 230000008606 intracellular interaction Effects 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- PGHMRUGBZOYCAA-UHFFFAOYSA-N ionomycin Natural products O1C(CC(O)C(C)C(O)C(C)C=CCC(C)CC(C)C(O)=CC(=O)C(C)CC(C)CC(CCC(O)=O)C)CCC1(C)C1OC(C)(C(C)O)CC1 PGHMRUGBZOYCAA-UHFFFAOYSA-N 0.000 description 1
- PGHMRUGBZOYCAA-ADZNBVRBSA-N ionomycin Chemical compound O1[C@H](C[C@H](O)[C@H](C)[C@H](O)[C@H](C)/C=C/C[C@@H](C)C[C@@H](C)C(/O)=C/C(=O)[C@@H](C)C[C@@H](C)C[C@@H](CCC(O)=O)C)CC[C@@]1(C)[C@@H]1O[C@](C)([C@@H](C)O)CC1 PGHMRUGBZOYCAA-ADZNBVRBSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 229930027917 kanamycin Natural products 0.000 description 1
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 1
- 229960000318 kanamycin Drugs 0.000 description 1
- 229930182823 kanamycin A Natural products 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000002523 lectin Substances 0.000 description 1
- 230000023404 leukocyte cell-cell adhesion Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 208000019420 lymphoid neoplasm Diseases 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 239000010445 mica Substances 0.000 description 1
- 229910052618 mica group Inorganic materials 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 238000012737 microarray-based gene expression Methods 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 238000003032 molecular docking Methods 0.000 description 1
- 230000009456 molecular mechanism Effects 0.000 description 1
- 229960005358 monensin Drugs 0.000 description 1
- GAOZTHIDHYLHMS-KEOBGNEYSA-N monensin A Chemical compound C([C@@](O1)(C)[C@H]2CC[C@@](O2)(CC)[C@H]2[C@H](C[C@@H](O2)[C@@H]2[C@H](C[C@@H](C)[C@](O)(CO)O2)C)C)C[C@@]21C[C@H](O)[C@@H](C)[C@@H]([C@@H](C)[C@@H](OC)[C@H](C)C(O)=O)O2 GAOZTHIDHYLHMS-KEOBGNEYSA-N 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 238000012243 multiplex automated genomic engineering Methods 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 238000010606 normalization Methods 0.000 description 1
- 230000005937 nuclear translocation Effects 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 230000009437 off-target effect Effects 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 108010058266 p21-Activated Kinases Proteins 0.000 description 1
- 102000006271 p21-Activated Kinases Human genes 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 229930192851 perforin Natural products 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 1
- 230000008823 permeabilization Effects 0.000 description 1
- 101150079312 pgk1 gene Proteins 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- DCWXELXMIBXGTH-UHFFFAOYSA-N phosphotyrosine Chemical compound OC(=O)C(N)CC1=CC=C(OP(O)(O)=O)C=C1 DCWXELXMIBXGTH-UHFFFAOYSA-N 0.000 description 1
- 238000001126 phototherapy Methods 0.000 description 1
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 1
- 230000001242 postsynaptic effect Effects 0.000 description 1
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 108010079891 prostein Proteins 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 229940121649 protein inhibitor Drugs 0.000 description 1
- 239000012268 protein inhibitor Substances 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 230000004063 proteosomal degradation Effects 0.000 description 1
- 239000002213 purine nucleotide Substances 0.000 description 1
- 239000002719 pyrimidine nucleotide Substances 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 230000007420 reactivation Effects 0.000 description 1
- 239000007845 reactive nitrogen species Substances 0.000 description 1
- 239000003642 reactive oxygen metabolite Substances 0.000 description 1
- 108091008597 receptor serine/threonine kinases Proteins 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000022983 regulation of cell cycle Effects 0.000 description 1
- 210000003289 regulatory T cell Anatomy 0.000 description 1
- 230000007363 regulatory process Effects 0.000 description 1
- 230000003252 repetitive effect Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000020874 response to hypoxia Effects 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000005204 segregation Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 230000009919 sequestration Effects 0.000 description 1
- 230000009131 signaling function Effects 0.000 description 1
- 102000030938 small GTPase Human genes 0.000 description 1
- 108060007624 small GTPase Proteins 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 238000011272 standard treatment Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 208000003265 stomatitis Diseases 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000000946 synaptic effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 101150047061 tag-72 gene Proteins 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 229960002175 thyroglobulin Drugs 0.000 description 1
- 210000003014 totipotent stem cell Anatomy 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 108091006108 transcriptional coactivators Proteins 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 238000003146 transient transfection Methods 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- 230000004102 tricarboxylic acid cycle Effects 0.000 description 1
- 230000034512 ubiquitination Effects 0.000 description 1
- 238000010798 ubiquitination Methods 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 241000712461 unidentified influenza virus Species 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 230000024883 vasodilation Effects 0.000 description 1
- 108700026220 vif Genes Proteins 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Images
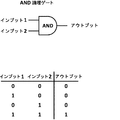
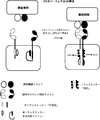
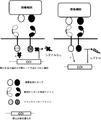
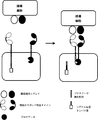

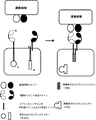
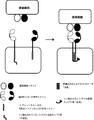
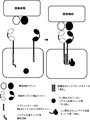

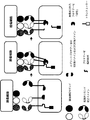
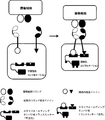


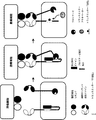
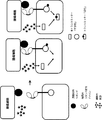
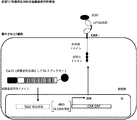
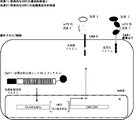














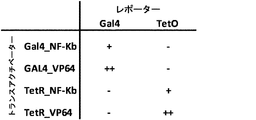
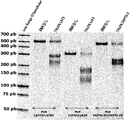





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/46—Hybrid immunoglobulins
- C07K16/468—Immunoglobulins having two or more different antigen binding sites, e.g. multifunctional antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/62—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising only variable region components
- C07K2317/622—Single chain antibody (scFv)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/03—Fusion polypeptide containing a localisation/targetting motif containing a transmembrane segment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/30—Non-immunoglobulin-derived peptide or protein having an immunoglobulin constant or Fc region, or a fragment thereof, attached thereto
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
- C12N2510/02—Cells for production
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Cell Biology (AREA)
- Toxicology (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description
本発明は、免疫療法のためにT細胞を操作する方法に関する。特に、前記T細胞は、インプットシグナルの組み合わせによって活性化されるように操作される。本発明は、リガンド結合ドメイン特性を利用して、選択された標的に免疫細胞の特異性および反応性を方向付け直すことができる新たな設計されたキメラ抗原受容体に関する。本発明はまた、治療的処置または予防的処置において使用するための、本方法によって得られた細胞、好ましくは、前記キメラ抗原受容体を含む細胞にも関する。
養子免疫治療は、エクスビボで作製された自己由来抗原特異的T細胞を導入することを伴い、ウイルス感染症および癌を処置する有望な戦略である。養子免疫治療に用いられるT細胞は、抗原特異的T細胞を増殖させることによって、または遺伝子工学によってT細胞を方向付け直すことによって作製することができる(Park, Rosenberg et al. 2011)。ウイルス抗原特異的T細胞の導入は、移植に関連するウイルス感染症と稀なウイルス関連新生物を処置するのに用いられる十分に確立した手順である。同様に、腫瘍特異的T細胞の単離および導入は黒色腫を処置することに成功したことが示されている。
(図2)ANDゲート:腫瘍抗原によって動かされる、受容体型チロシンキナーゼ(RTK)をベースとするキメラ抗原受容体の二量体化および活性化。腫瘍細胞表面に共存する2種類の腫瘍細胞リガンドが同時に存在すると、2種類のヘテロ二量体受容体型チロシンキナーゼベースキメラ抗原受容体が二量体化し、トランスリン酸化を介して活性化される。両CAR上では、トランスミッタードメインが自己抑制によって不活性状態に維持されている(例えば、キナーゼ活性部位は自己抑制ドメインによって隠されている)。腫瘍細胞表面に共存する2種類の腫瘍細胞リガンドが同時に存在すると、2種類のCARは二量体化して、キナーゼ自己抑制は解除され、非限定的な例としてトランスリン酸化または他の分子との相互作用を介してトランスミッタードメインは活性化される。
(図3)ANDゲート:前に不活化された遺伝子の補完:簡略化した例では、2つの異なるドメインを含む細胞質ドメインをもつ2つの異なるCARによって2つの異なる腫瘍細胞リガンドを認識することができる。同時に、T細胞のシグナル経路にある重要な遺伝子(GOI)のノックアウトが行われている。2種類のCARが共存し、その後に、腫瘍リガンド細胞が認識されると、第1のCARは、第2のCARによって媒介されるシグナルの伝達に必要なGOIの再活性化を可能にする因子を活性化することができる。
(図4)ANDゲート:プロテアーゼ系。2種類の腫瘍細胞リガンドが同時に存在すると、2種類のCARが活性化される。第1のCARの細胞内ドメインは、シグナル伝達タンパク質に連結されているプロテアーゼ標的配列を含む。第2のCARの細胞内ドメインはプロテアーゼを有する。それぞれのCARは独立して1種類の腫瘍リガンド細胞の存在によって活性化されず、活性化は、両腫瘍リガンド細胞の存在による2種類のCARの共存に由来する。2種類のCARが共存すると、標的配列プロテアーゼの切断とその後のシグナル伝達タンパク質の放出によって媒介される、2種類のCARの活性化が可能になる。
(図5)ANDゲート:スプリットタンパク質系。2種類の腫瘍細胞リガンドが同時に存在すると、2種類のCARが活性化される。第1のCARの細胞内ドメインは、「シグナル伝達ドメイン」の断片とインテインのCドメインまたはNドメインを含む。第2のCARの細胞内ドメインは、相補インテインドメインと相補シグナル伝達ドメイン断片を有する。それぞれのCARは独立して1種類の腫瘍リガンド細胞の存在によって活性化されず、活性化は、両腫瘍リガンド細胞の存在による2種類のCARの共存に由来する。2種類のCARが共存すると、T細胞の様々な活性化経路を開始することができる完全に活性な形のシグナル伝達タンパク質を再構成する完全活性スプリットインテインが再構成されることで2種類のCARの活性化が可能になる。シグナル伝達タンパク質の例は、ZAP70、SH2ドメイン、およびキナーゼドメインである。
(図6)ANDゲート:スプリットタンパク質系およびシグナル伝達タンパク質の放出。2種類の腫瘍細胞リガンドが同時に存在すると、2種類のCARが活性化される。第1のCARの細胞内ドメインは、「シグナル伝達タンパク質」のC末端不活性断片とインテインのCドメインまたはNドメインを含む。第2のCARの細胞内ドメインは、さらなるマルチドメインとホモ二量体化することができる二量体化ドメインを有する。このマルチドメインは、第2のインテインドメインとシグナル伝達タンパク質断片のNドメインによって構成される。それぞれのCARは独立して1種類の腫瘍リガンド細胞の存在によって活性化されず、活性化は、両腫瘍リガンド細胞の存在による2種類のCARの共存に由来する。2種類のCARが共存すると、細胞質に放出されてT細胞活性化を開始することができる完全に活性な形のシグナル伝達タンパク質を再構成する完全活性スプリットインテインが再構成されることで2種類のCARの活性化が可能になる。
(図7)ANDゲート:キナーゼをベースとするスプリットタンパク質系。2種類の腫瘍細胞リガンドが同時に存在すると、2種類のCARが活性化される。第1のCARの細胞内ドメインはシグナル伝達タンパク質結合領域とスプリットキナーゼのCドメインまたはNドメインを含む。第2のCARの細胞内ドメインは相補キナーゼドメインを有する。それぞれのCARは独立して1種類の腫瘍リガンド細胞の存在によって活性化されず、活性化は、両腫瘍リガンド細胞の存在による2種類のCARの共存に由来する。2種類のCARが共存すると、リン酸化することができる、従って、T細胞活性化を開始することができる完全活性キナーゼが再構成されることで2種類のCARの活性化が可能になる。スプリットキナーゼの例はLCKでもよい。
(図8)ANDゲート:キナーゼをベースとするスプリットタンパク質系のトランス活性化。2種類の腫瘍細胞リガンドが同時に存在すると、2種類のCARが活性化される。第1のCARの細胞内ドメインはシグナル伝達結合領域とスプリットキナーゼのCドメインまたはNドメインを含む。第2のCARの細胞内ドメインは相補キナーゼドメインを有する。それぞれのCARは独立して1種類の腫瘍リガンド細胞の存在によって活性化されず、活性化は、両腫瘍リガンド細胞の存在による2種類のCARの共存に由来する。2種類のCARが共存すると、シグナル伝達タンパク質結合領域上でのコンホメーション改変を引き起こすことができ、それによって、トランス活性化によって活性化することができるシグナル伝達タンパク質に結合することができる完全活性キナーゼが再構成されることで2種類のCARの活性化が可能になる。
(図9)ANDゲート:プロテアーゼをベースとするスプリット系およびシグナル伝達タンパク質の再局在化。2種類の腫瘍細胞リガンドが同時に存在すると、2種類のCARが活性化される。第1のCARの細胞内ドメインは、スプリットプロテアーゼのCドメインまたはNドメインと、プロテアーゼ標的配列と、シグナル伝達タンパク質を含む。第2のCARの細胞内ドメインは相補スプリットプロテアーゼドメインを有する。それぞれのCARは独立して1種類の腫瘍リガンド細胞の存在によって活性化されず、活性化は、両腫瘍リガンド細胞の存在による2種類のCARの共存に由来する。2種類のCARが共存すると、プロテアーゼ標的配列を切断し、シグナル伝達タンパク質を放出することができる完全活性プロテアーゼが再構成されることで2種類のCARの活性化が可能になる。
(図10)ANDゲート:3種類のCARを用いた、プロテアーゼをベースとするスプリット系。3種類の腫瘍細胞リガンドが同時に存在すると、CARが活性化される。第1のCARの細胞内ドメインはプロテアーゼ標的配列とシグナル伝達タンパク質を含む。第2のCARおよび第3のCARの細胞内ドメインは2種類の相補スプリットプロテアーゼドメインによって形成される。それぞれのCARは独立して1種類の腫瘍リガンド細胞の存在によって活性化されず、活性化は、3種類の腫瘍リガンド細胞の存在による3種類のCARの共存に由来する。3種類のCARが共存すると、プロテアーゼ標的配列を切断し、シグナル伝達タンパク質を放出することができる完全活性プロテアーゼが再構成されることで3種類のCARが活性化される。
(図11)ANDゲート:スキャフォールディングプロテアーゼをベースとする系。2種類の腫瘍細胞リガンドが同時に存在すると、2種類のCARが活性化される。第1のCARの細胞内ドメインは第1のタンパク質ドメインを含む。第2のCARの細胞内ドメインは第2のタンパク質ドメインを有する。それぞれのCARは独立して1種類の腫瘍リガンド細胞の存在によって活性化されず、活性化は、両腫瘍リガンド細胞の存在による2種類のCARの共存に由来する。2種類のCARが共存すると、タンパク質ドメイン1およびタンパク質ドメイン2が不活性スキャフォールディングタンパク質に結合することによって2種類のCARの活性化が可能になる。複合体が結合すると、活性型スキャフォールディングタンパク質が再構成され、T細胞を活性化することができる。スキャフォールディングタンパク質の例は、Carma1、SP76、hemITAM、DLG1、KSRである。
(図12)ANDゲート:操作されたヘテロ二量体ドメインの二重活性化に基づく。2種類の腫瘍細胞リガンドが同時に存在すると、2種類のCARが活性化される。第1のCARの細胞内ドメインは第1のトランスミッター結合ドメインを含む。第2のCARの細胞内ドメインは第2のトランスミッター結合ドメインを有する。それぞれのCarは独立して1種類の腫瘍リガンド細胞の存在によって活性化されず、活性化は、両腫瘍リガンド細胞の存在による2種類のCARの共存に由来する。2種類のCARが共存すると、2種類のトランスミッター結合ドメインの活性化(例えば、リン酸化および翻訳後修飾)が可能になり、これにより、T細胞を活性化することができるトランスミッターの動員を誘発することができる。
(図13)ANDゲート:自己抑制系:競合的結合が起こると活性化が誘導される。2種類の腫瘍細胞リガンドが同時に存在するとトランスミッターが活性化される。第1のCAR上では、トランスミッタードメインは自己抑制によって(例えば、「遮蔽(shielding)」タンパク質または抗体と相互作用することによって)不活性状態に維持されている。リガンド結合時に第2のCARが共存すると、遮蔽分子は、それ自身の、さらに高い親和性のドメインに移る(分子間移動)。次いで、「遮蔽されていない」トランスミッターは(例えば、翻訳後修飾または他の分子との相互作用によって)活性化することができる。
(図14)ANDゲート:自己抑制系:抑制ドメインの酵素的切断が起こると活性化が誘導される。2種類の腫瘍細胞リガンドが同時に存在するとトランスミッターが活性化される。第1のCAR上では、トランスミッタードメインは自己抑制によって(例えば、「遮蔽」タンパク質または抗体と相互作用することによって)不活性状態に維持されている。リガンド結合時に第2のCARが共存すると、プロテアーゼドメインが第1のcar上に存在するプロテアーゼ標的配列に接近し、従って、遮蔽分子は移動される。次いで、「遮蔽されていない」トランスミッターは(例えば、翻訳後修飾または他の分子との相互作用によって)活性化することができる。
(図15)ANDゲート:トランスミッタータンパク質の活性化を誘導するための受容体結合および外部刺激。CARとその腫瘍細胞リガンドとの結合と、操作されたT細胞の腫瘍細胞の細胞外刺激への曝露が同時に起こると、トランスミッターが活性化される。外部刺激は、代謝産物、低分子、ペプチド、低分子タンパク質(ケモカイン、サイトカイン)の濃度および物理的状態/化学的状態(pH、低酸素、酸化還元電位)の変化を包含する。
(図16)ANDゲート:1種類の腫瘍抗原の存在下での低酸素依存的活性化系。酸素枯渇環境で、操作されたT細胞と1種類の腫瘍細胞リガンドが同時に存在するとT細胞活性化が誘発される。このような論理ANDゲート活性化系を可能にするために、酸素誘導性の合成活性化経路を有するようにT細胞が操作される。このような合成経路は、操作された酸素濃度感受性転写因子(OxiTF)、第3の要素を発現させるOxiTF特異的合成プロモーター、キメラ抗原受容体(CARI)を含む3つの異なる要素から作られる。OxiTFは、操作されたT細胞の中で、腫瘍抗原に特異的なCARをコードする合成遺伝因子を活性化するように設計されている。固形腫瘍に遭遇したら、操作されたT細胞は酸素枯渇を検出し、CARIの産生を誘発する。CARIが細胞表面に露出したら腫瘍抗原の認識が可能になり、最終的に、CARIの中に存在する活性化ドメインおよび共刺激ドメインを介してT細胞の活性化および増殖が誘発される。
(図17)ANDゲート:2種類の腫瘍抗原の存在下での低酸素依存的活性化系。酸素枯渇環境で、操作されたT細胞と2種類の腫瘍細胞リガンドが同時に存在するとT細胞活性化が誘発される。このような論理ANDゲート活性化系を可能にするために、酸素誘導性の合成活性化経路を有するようにT細胞が操作される。このような合成経路は、操作された酸素濃度感受性転写因子(OxiTF)、第3の要素を発現させるOxiTF特異的合成プロモーター、腫瘍抗原IIに特異的なキメラ抗原受容体(CARII)を含む4つの異なる要素から作られる。この系は、腫瘍抗原Iに特異的な、構成的に発現しているCARIにある第4の要素を加えて完成する。OxiTFは、操作されたT細胞の中で、CARIIをコードする合成遺伝因子を活性化するように設計されている。固形腫瘍に遭遇したら、操作されたT細胞は酸素枯渇を検出し、CARIの産生を誘発する。既に存在するCARI-腫瘍抗原I複合体に加えて、CARIIとCARIが細胞表面に露出したら腫瘍抗原IIの認識が可能になる。最終的に、両方のCAR/腫瘍抗原複合体が同時に存在すると、CARIおよびIIの中に存在する活性化ドメインおよび共刺激ドメインを介してT細胞の活性化と増殖が誘発される。
(図18)ANDゲート:T細胞系活性化に適用されたANDゲート原理の図。CARの細胞外は、2つのコンホメーション(「活性」および「不活性」)で存在する2種類のリガンド結合ドメインを含む。2種類の腫瘍細胞リガンドの非存在下では、平衡は「不活性型」に強く向けられる。2種類のリガンド結合ドメインがそれぞれの腫瘍細胞リガンドに同時に結合する時にしか(2種類のインプット)、CARの細胞内ドメインへのプラスのシグナル(アウトプット)は誘発されない。
(図19)AND NOTゲート:一般的なスキーム。T細胞系活性化に適用されたAND NOTゲート原理の図。2種類の腫瘍細胞リガンドが同時に存在し、健常細胞リガンドが存在しない時にプラスのアウトプットが誘発される。インプット1およびインプット2は腫瘍細胞リガンドの存在に対応するのに対して、第3のインプットは健常細胞リガンドが存在しないことでなければならない。第1のCARおよび第2のCARは共刺激細胞質ドメインを有するのに対して、第3のCARは、健常細胞リガンドが認識されない場合に抑制作用がブロックされる抑制ドメインを有する。
(図20)T細胞シグナル伝達カスケードを抑制および刺激するための2のタイプのLCKの作製。第1のCARは、構成的に負に調節された形のLCK(-)の転写を刺激する抑制ドメインを用いて健常細胞の抗原を認識する。この第1のCARは、LCK(+)型の転写を活性化して、高レベルのT細胞活性化を生じる共刺激ドメインを含む第2のCARと対になっている。
(図21)CARを介したCARMA1タンパク質調節によるT細胞活性化の制御。抗原認識後のTCR刺激はCD28動員と結びつけられ、CD28動員によってPKCθ活性化が起こり、次に、PKCθ活性化によってCARMA1がリン酸化および活性化される。CARMA1は、T細胞受容体(TCR)シグナル伝達、一般的にはT細胞活性化のための重要なシグナロソームを構成する。CARMA1は様々なタンパク質を動員して、最終的に、2つの異なるシグナル伝達カスケード:NF-κBおよびc-jun N末端キナーゼ(JNK)を活性化することができる多タンパク質複合体を形成する。
(図22)HIF低酸素系の機能。酸素正常状態(高O2)では、HIFαは、酸素を検知するHIFα特異的プロリルヒドロキシラーゼ(PHD1-3)によって加水分解される。ヒドロキシル化によってHIFαポリユビキチン化が誘発され、HIFαはE3ユビキチンリガーゼによるプロテオソーム分解の標的とされる。低酸素(低O2)では、TCAサイクル中間体を介したヒドロキシル化の抑制、HIFαタンパク質の安定化、およびHIF転写活性の低下が起こる。
(図23)HIFαなどの酸素感受性ドメインを有する異なるキメラ抗原受容体(CAR)構造。左側には、単鎖CAR(scCAR)は、同じただ一つしかない鎖に、細胞外結合ドメイン(ここではscFv)と、酸素ドメイン(例えば、HIF1)と、活性化ドメインと同時活性化ドメインを有する。右側には、例示的なコンホメーションとして、α鎖がscFvと酸素ドメインを有し、β鎖が共刺激ドメインを有し、γ鎖が活性化ドメインを有する多鎖CAR(mcCAR)を示す。
(図24)(A)酸素正常状態または低酸素における鎖-HIF1(a.a.380-630)対α鎖WTの表面提示。低酸素では、HIF1を有するCAR T細胞の表面露出は(α-HIF1を有さない)対照CAR T細胞の表面露出と似ているが、酸素正常状態では表面露出はかなり少ない。このことは、CAR-αHIF1が良好に発現していることを示す。(B)低酸素から酸素正常状態に戻した後のα鎖-HIF1(a.a.380-630)対α鎖WTの表面提示。低酸素から酸素正常状態に戻すとCARαHIF1の発現は低下する。これは可逆的かつ動的な系である。すなわち、酸素正常状態では、CAR発現はα-HIF1およびα鎖ポリペプチドの分解によって一時的に抑制され、低酸素状態(すなわち、腫瘍環境)では、α-HIF1およびα鎖ポリペプチドは発現される。
(図25)(A)酸素正常状態または低酸素におけるα-HIF mcCAR対対照CARの表面検出。この実験では、さらに少ない全RNAが用いられ、得られた結果は図23の結果と同様である。(B)酸素正常状態における細胞傷害性の誘導。酸素正常状態では、対照多鎖CAR(α-HIF1を有さない)は多量の標的細胞死滅を示すのに対して、HIF-mcCARの場合、標的細胞は死滅しない。これらの細胞傷害性の結果と表面露出の結果を考慮すると、このことはHIF系がキメラ抗原受容体の中で完全に機能することを示している。
(図26)酸素正常状態または低酸素での様々なα鎖-HIF対WTα鎖の表面提示。(A)-EA-リンカーを有するHIF1-mcCAR(a.a.380-630)構築物;(B)HIF1-mcCAR(a.a.344-417);(C)HIF3-mcCAR(a.a.480-571);(D-E-F):(A-B-C)と同じであるが、低酸素から酸素正常状態に戻した。レンチウイルス送達によって、ここで得られた全ての結果から、HIF1系およびHIF3系はいずれも機能し、同じように振る舞うことが証明された。同様に、リンカーを用いて、またはリンカーを用いずにHIFタンパク質の様々な部分を使用できることが示された。
説明文:

(図28)両ゲート受容体の組成物の模式図。
(図29)両ゲートの成分の分子組み立て戦略の模式図。この図にはスペーサーが示されている。
(図30)7種類の膜タンパク質パートナーの表面発現。GG83、GG111、GG121、GG152、GG153、GG155、GG156、およびGG158を試験した。シグナル強度(++: 非常に強い、+: 強い)。
(図31)様々なトランスアクチベーターのmRNAトランスフェクションによる、レンチウイルスによって送達されたRQR8カセットの発現。これらの構築物はDNA結合ドメイン(TetOまたはGal4)と転写活性化ドメイン(VP64またはNF-κB)で構成され、トランスフェクトおよび試験される。得られたデータから、適切なトランスアクチベーターのmRNAトランスフェクションによって、レンチウイルスによって送達されたRQR8カセットが発現したことがはっきりと分かった。
(図32)設計されたTALENを用いた内因性遺伝子座における標的変異誘発を証明するT7エンドヌクレアーゼアッセイ。3つのパネルA、B、Cは全て、LAT、LCK、ZAP70、LFA、TRAT、またはCD28などのT細胞のシグナル伝達および/または機能に関与する酵素のノックアウト(KO)を示す。得られたデータから、設計されたTALENを用いた全標的遺伝子座における標的変異誘発レベルが高いことがはっきりと分かる。
(図33)ZAP70をノックアウトした後の脱顆粒実験。得られたデータから、WT T細胞と比べて、ノックアウト操作されたT細胞は濃い染色が減少したことがはっきりと分かる。
(図34)二重特異性CAR(biCAR)機能の模式図。二重特異性CAR(biCAR)は、2つの異なる標的細胞抗原に対して特異的親和性のあるscFvをもつ2種類のCAR(biCAR1およびbiCAR2)で構成される。これらのscFcの一方だけが特異的抗原に結合した時にCARは活性化されず、従って、細胞は死滅しない。両方のscFvが特異的抗原に結合した時にCARは活性化され、標的細胞は死滅する。
(表2)CARMA1リン酸化部位
養子T細胞療法において機能的応答を制御する能力は重要な問題である。このような治療方針では、T細胞は、高度に腫瘍特異的に標的細胞を認識する、表面に露出したキメラ抗原受容体(CAR)を発現させることによって操作される。しかしながら、潜在的な毒性オフターゲット作用を制御および最小化するにはマルチインプット系を設計することが非常に望ましい。
本発明者らは、論理ゲートに基づく複雑な2進法計算を含む、タスクを実行するための人工回路内の調節モジュールの合理的な組み合わせに基づいて、このような免疫細胞を操作する方法を開発した。「ゲート」という用語は、2種類以上のインプットシグナルに応答して特定の(予め決められた)アウトプットを生じる装置または分子機構を指すために用いられる。本発明によれば、論理ANDゲートとは、少なくとも2種類のインプットシグナルの組み合わせに起因する、異なるトランスミッタードメインの組み合わせと特定のタンパク質(シグナル伝達タンパク質)の活性化による免疫細胞活性化、特に、標的細胞に対するT細胞の細胞傷害性を指す。非限定的な例として、タンパク質機能を活性化するために、または抑制タンパク質を除去するために、それぞれのシグナルは一緒に働いてもよく、別々に働いてもよい。別の特定の態様において、前記インプットシグナルは、前のインプットシグナルに起因するアウトプットシグナルでもよい。特に、本発明は、免疫療法のために免疫細胞を操作する方法、特に、細胞を特異的に標的とするために免疫細胞を操作する方法に関し、本方法は、
(a)免疫細胞を準備する工程;
(b)インプットシグナルの組み合わせによって、前記免疫細胞を活性化する少なくとも2種類のトランスミッタードメインの組み合わせが誘導されるように、前記細胞が少なくとも2種類のインプットシグナルに対して感受性になるように前記免疫細胞を操作する工程であって、それぞれのトランスミッタードメインが単独では前記免疫細胞を活性化しない、工程
を含む。
特定の態様において、前記インプットシグナルは、前記操作された免疫細胞、特に、前記操作された免疫細胞の表面に発現しているキメラ抗原受容体によるリガンドの認識でもよい。
-前記特異的リガンドを認識することができる細胞外リガンド結合ドメイン;
-膜貫通ドメイン;
-少なくとも、活性化ドメインおよび同時活性化ドメインならびに酸素感受性ドメインを含む細胞内ドメイン
を含む。
トランスミッタータンパク質はまた、前に不活化された遺伝子を補完するか、または前に不活化された遺伝子を補完するように核内の遺伝子を活性化することもできる。従って、インプットシグナルが組み合わされた後に、2種類のトランスミッターシグナルが組み合わされると、不活化された遺伝子が補完され、従って、T細胞が活性化される。
本発明によるトランスミッタードメインは、プロテアーゼと、プロテアーゼ切断部位を介して膜アンカードメインに連結されているシグナル伝達タンパク質を含む基質タンパク質でもよい。2つのトランスミッタードメインの組み合わせによって免疫細胞が活性化される。実際には、プロテアーゼによって基質タンパク質が切断されるとシグナル伝達タンパク質が放出され、従って、免疫細胞が活性化される(図4)。前記膜アンカードメインは、基質タンパク質を細胞の膜に繋ぎ止める末端伸長部分でもよい。特定の態様において、前記基質タンパク質はキメラ抗原受容体の細胞内ドメインの一部である。前記プロテアーゼは、非限定的な例として、TEVプロテアーゼ、第Xa因子、トロンビン、操作されたウイルスポテアーゼ(potease)、エンテロキナーゼ、およびHRV3Cでもよい。
別の態様において、トランスミッタードメインはスプリットタンパク質である。この系は、機能分子が2つの非機能断片に切断されているタンパク質補完アッセイに基づいている。取り付けられているタンパク質間相互作用ドメインによって断片が再構成された時に、機能が回復する。タンパク質補完アッセイにおいて用いられる機能分子は活性酵素でもよく、シグナル伝達タンパク質でもよい。前記スプリットタンパク質は、非限定的な例として、スプリットキナーゼ、スプリットプロテアーゼ、およびスプリットインテインを包含する。
スキャフォールドタンパク質は多くの主要なシグナル伝達経路の重要な制御因子である。スキャフォールドタンパク質とは、シグナル伝達経路の複数のメンバーと相互作用および/または結合して、これらを繋ぎとめて複合体にすることができるタンパク質を意味する。本発明において、トランスミッタードメインは、スキャフォールドタンパク質を動員することができるシグナル伝達経路のメンバーでもよい。この組み立ては、スキャフォールド複合体内での成分の近さおよび有効濃度を増加することによってシグナル伝達の特異性および効率を高めて、免疫細胞を活性化することができるかもしれない。非限定的な例として、スキャフォールドタンパク質はプロテインキナーゼとその基質に結合し、それによって、特異的キナーゼリン酸化を確かなものにすることができる。または、前記スキャフォールドタンパク質はシグナル伝達メンバーのアロステリック変化をもたらすことができる。前記スキャフォールドタンパク質はシグナル伝達を調節してもよく、(複合体内で組織化される)経路成分が原形質膜などの特定の細胞領域に局在化するのを助けてもよく、シグナル伝達フィードバックを調整してもよく、活性化された分子を不活性化から保護してもよい。本発明による前記スキャフォールドタンパク質は、非限定的な例として、SYKチロシンキナーゼにあるようなSH2ドメイン、またはC型レクチンおよびhemITAM(図11)について述べたような様々なITAMドメイン(トランスミッタードメイン)を認識および結合することができるZAP70、または実施例2に記載のCARMA-1でもよい。
トランスミッタードメインはまた、ホモ二量体タンパク質またはヘテロ二量体タンパク質、特に、別のトランスミッタードメイン、例えば、別の受容体細胞内ドメインまたはサイトゾルタンパク質と二量化することができる受容体細胞内ドメインでもよい。これらのトランスミッタードメインが二量体化するとシグナルが下流に伝達される。タンパク質のホモ二量体化またはヘテロ二量体化が関与するシグナル伝達タンパク質の一例は、JAK/Statシグナル伝達経路が関与する前記のチロシンキナーゼ受容体でもよい。このような成分の活性化は、一般的に、リガンドを介した二量体化によって誘発される。ホモ二量体化する前記トランスミッタードメインは絶対ヘテロ二量体を形成するように操作されてもよい。特定の態様において、前記CARは、好ましくは、TrkA、c-Kit、FGFR、およびEGFR/Erbからなる群より選択される受容体型チロシンキナーゼの膜貫通ドメイン、任意で細胞内ドメインを含んでもよい。リガンドが認識されると受容体の二量体化が誘導され、従って、シグナル伝達タンパク質が活性化され、その結果、免疫細胞が活性化される(図12)。
トランスミッタードメインはまた自己抑制分子の非活性化型でもよい。自己抑制化合物は自己抑制された状態で存在してもよく、活性状態で存在してもよい。自己抑制された状態はタンパク質の触媒機能を混乱させるか、またはタンパク質が別のリガンドと相互作用する能力を混乱させる。自己抑制された状態は典型的にはキナーゼリン酸化の非存在下で起こる。このような自己抑制タンパク質の活性化は化合物のコンホメーション変化を伴ってもよい。このコンホメーション変化は別の化合物と相互作用した結果でもよい。前記抑制化合物はアロステリック抑制化合物でもよい。アロステリック抑制化合物は、自己抑制化合物の保存された(conserve)非活性化コンホメーション状態を保つように、自己抑制化合物と結合し、特異的な会合を形成する。自己抑制は、さらに高親和性の結合を有することができる(図13)、または、例えば、相互作用領域の共有結合修飾(例えば、脱リン酸/リン酸化)もしくはタンパク質分解を誘導することができる(図14)、別のトランスミッタードメインと相互作用することによって解放することができる。非限定的な例として、前記阻害剤は、クラスIおよびIIのp21活性化キナーゼ(pak)阻害剤、Rho活性化タンパク質阻害剤、自己抑制非受容体型セリン/スレオニンキナーゼ阻害剤、ホスファターゼ阻害剤、および自己抑制低分子GTPアーゼエフェクター阻害剤でもよい。
本発明の別の局面において、インプットシグナルは外部刺激でもよい(図15)。前記外部刺激は、非限定的な例として、操作されたT細胞の微小環境に腫瘍細胞が存在する時には低分子、ペプチド、低分子タンパク質、(ケモカイン、サイトカイン)、および物理化学的状態、例えば、pH、低酸素、酸化還元電位の変化を包含する。酸化還元調節要素は、非限定的な例として、酸素または窒素、例えば、活性酸素種または活性窒素種(NSおよびRNS)でもよい。
本発明の別の局面において、本発明者らはまた、一方のある特定のインプットシグナルだけが存在し、他方が存在しない時に特定の応答アウトプットが生じない論理ゲートに基づいて、このような免疫細胞を操作する方法も開発した(図19)。本発明によれば、免疫細胞の活性化、特に、標的細胞に対するT細胞の細胞傷害性は、いくつかのインプットシグナルの1つが認識された後に、特に、癌細胞上にあるリガンドが認識された後に誘導され、健常細胞上に存在するリガンドが認識された後に誘導されない。特に、本発明は、免疫療法のために免疫細胞を操作する方法、特に、細胞を特異的に標的とするために免疫細胞を操作する方法であって、
(a)免疫細胞を準備する工程;
(b)特定のインプットシグナルでしか前記免疫細胞応答の活性化が誘導されないように、前記細胞がいくつかのインプットシグナルのうちの少なくとも1つに対して感受性になるように前記免疫細胞を操作する工程
を含む、方法に関する。
前記の様々な方法は細胞表面にCARを発現させることを伴う。非限定的な例として、前記CARは、CARを細胞に導入することによって発現されてもよい。CARは、1つのプラスミドベクターによってコードされるトランスジーンとして導入されてもよい。前記プラスミドベクターはまた、前記ベクターを収容した細胞を特定および/または選択する選択マーカーも含有してよい。
本発明はまた、前記のように細胞外リガンド結合ドメインと、トランスミッタードメインを含む細胞内ドメインを含むキメラ抗原受容体に関する。特に、前記トランスミッタードメインは、プロテアーゼ、スプリットタンパク質、スキャフォールドタンパク質を動員するシグナル伝達経路のメンバー、二量体ドメインのうちの一方の単量体、自己抑制化合物からなる群より選択される。
細胞外にCD8ヒンジを含み、膜貫通ドメインとしてFcRαを含み、細胞内にHIF1αまたはHIF3αサブユニットと組み合わされたFcRαの一部を含む、α鎖;
細胞外ドメインおよび膜貫通ドメインとしてFcRβを含み、細胞内共刺激ドメインとしてΔITAM-41BBを含む、β鎖;
膜貫通ドメインとしてFcRγを含み、細胞内活性化ドメインとしてΔITAM-CD3ζを含む、γ鎖
を含む。
本発明はまた、細胞を操作する前記の方法によって入手しやすい、単離された細胞または細胞株にも関する。特に、前記単離された細胞は、前記の少なくとも1つのCARを含む。好ましい態様において、前記単離された細胞は、それぞれのCARが異なる細胞外リガンド結合ドメインを含むCAR集団を含む。特に、前記単離された細胞は、CARをコードする外因性ポリヌクレオチド配列を含む。
別の態様において、以前に述べられた様々な方法によって得られた単離された細胞または前記単離された細胞に由来する細胞株は医用薬剤として使用することができる。別の態様において、前記医用薬剤は、癌、自己免疫疾患、または感染症の処置を必要とする患者において癌、自己免疫疾患、または感染症を処置するのに使用することができる。別の態様において、本発明による前記単離された細胞または前記単離された細胞に由来する細胞株は、癌、ウイルス感染症、または自己免疫疾患の処置を必要とする患者において癌、ウイルス感染症、または自己免疫疾患を処置するための医用薬剤の製造において使用することができる。
(a)以前に述べられた方法のいずれか1つによって入手可能な免疫細胞を準備する工程;
(b)前記形質転換された免疫細胞を前記患者に投与する工程
の少なくとも1つを含む、それを必要とする患者を処置するための方法に依拠する。
ポリペプチド配列中のアミノ酸残基は一文字表記に従って本明細書において指定される。例えば、QはGlnまたはグルタミン残基を意味し、RはArgまたはアルギニン残基を意味し、DはAspまたはアスパラギン酸残基を意味する。
1. 細胞を特異的に標的とするために免疫細胞を操作する方法であって、
(a)免疫細胞を準備する工程;
(b)インプットシグナルの組み合わせによって、前記免疫細胞を活性化する少なくとも2種類のトランスミッタードメインの組み合わせが誘導されるように、前記細胞が少なくとも2種類のインプットシグナルに対して感受性になるように前記免疫細胞を操作する工程であって、それぞれのトランスミッタードメインが単独では前記免疫細胞を活性化しない、工程
を含む、方法。
2. 少なくとも1種類の前記インプットシグナルが、特異的リガンドを認識することができる細胞外リガンド結合ドメインと、任意でシグナル伝達タンパク質を介して、別のトランスミッタードメインと組み合わせて前記免疫細胞を活性化することができるトランスミッタードメインを含む細胞内ドメインを含むキメラ抗原受容体(CAR)を細胞表面に発現させることによって操作された前記免疫細胞による特異的リガンドの認識である、請求項1記載の方法。
3. 前記少なくとも2種類のインプットシグナルが、前記異なる特異的リガンドを認識することができる細胞外リガンド結合ドメインと、組み合わせて免疫細胞を活性化することができるトランスミッタードメインを含む細胞内ドメインを含むキメラ抗原受容体(CAR)を細胞表面に発現させることによって操作された前記免疫細胞による異なる特異的リガンドの認識である、請求項1記載の方法。
4. 前記インプットシグナルが、特定の低分子、ケモカイン、サイトカイン、物理化学的状態、低酸素の存在からなる群より選択される外部刺激である、請求項1〜3のいずれか一項記載の方法。
5. 外部刺激が低酸素であり、前記免疫細胞が、低酸素誘導性プロモーターの制御下で少なくとも1つのトランスミッタードメインを発現するように操作されている、請求項4記載の方法。
6. 外部刺激が低酸素であり、前記免疫細胞が、低酸素誘導性プロモーターの制御下でトランスミッタードメインを含む少なくとも1つのキメラ抗原受容体を発現するように操作されており、前記キメラ抗原受容体のリガンド認識が別のインプットシグナルである、請求項4記載の方法。
7.プロテアーゼ切断によって、前記免疫細胞を活性化することができるシグナル伝達タンパク質の放出が誘導されるように、前記トランスミッタードメインが、プロテアーゼと、プロテアーゼ切断部位を介して膜アンカードメインに連結されているシグナル伝達タンパク質を含む基質タンパク質である、請求項1〜6のいずれか一項記載の方法。
8. 前記プロテアーゼが、TEVプロテアーゼ、第Xa因子、トロンビン、操作されたウイルスプロテアーゼからなる群より選択される、請求項7記載の方法。
9. 前記トランスミッタードメインが、集合して、前記免疫細胞を活性化することができるシグナル伝達タンパク質を再構成するスプリットタンパク質である、請求項1〜8のいずれか一項記載の方法。
10. 前記スプリットタンパク質が、一緒に集合して、基質タンパク質を切断し、前記免疫細胞を活性化することができるシグナル伝達タンパク質を放出することができるプロテアーゼを再構成するスプリットプロテアーゼである、請求項9記載の方法。
11. 前記スプリットタンパク質が、一緒に集合して、前記免疫細胞を活性化することができるシグナル伝達タンパク質を活性化するキナーゼを再構成するスプリットキナーゼである、請求項9記載の方法。
12. 前記スプリットタンパク質が、一緒に集合して、インテイン配列を切り取り、前記免疫細胞を活性化することができる隣接するシグナル伝達タンパク質配列をペプチド結合でつなげるインテインを再構成するスプリットインテインである、請求項9記載の方法。
13. 前記トランスミッタードメインが、スキャフォールドタンパク質を動員することができるシグナル伝達経路のメンバーである、請求項1〜8のいずれか一項記載の方法。
14. 前記スキャフォールドタンパク質がSYKチロシンキナーゼまたはZAP70である、請求項13記載の方法。
15. 前記トランスミッタードメインが二量体タンパク質である、請求項1〜8のいずれか一項記載の方法。
16. 前記トランスミッタードメインが自己抑制化合物である、請求項1〜8のいずれか一項記載の方法。
17. 前記操作された免疫細胞が最初は遺伝子が不活化されており、前記トランスミッタードメインの組み合わせが、前記不活化された遺伝子を補完することができる、請求項1〜8のいずれか一項記載の方法。
18. 細胞外リガンド結合ドメインと、少なくとも1つのトランスミッタードメインを含む細胞内ドメインを含む、キメラ抗原受容体。
19. 前記トランスミッタードメインが、プロテアーゼ、スプリットタンパク質、二量体タンパク質、スキャフォールドタンパク質を動員することができるシグナル伝達経路のメンバー、および自己抑制化合物からなる群より選択される、請求項18記載のキメラ抗原受容体。
20. 請求項18または19記載のキメラ抗原受容体をコードする、ポリヌクレオチド。
21. 請求項18または19記載のキメラ抗原受容体を含む、単離された免疫細胞。
22. 請求項1〜17のいずれか一項記載の方法によって得られた、単離された免疫細胞。
23. 医用薬剤として使用するための、請求項21または22記載の単離された免疫細胞。
24. 癌、自己免疫状態、または病原体による感染症を処置するための、請求項21〜23のいずれか一項記載の単離された免疫細胞。
25. それを必要とする対象を処置する方法であって、
(a)請求項21または22記載の免疫細胞を準備する工程;
(b)前記免疫細胞を前記患者に投与する工程
を含む、方法。
スキャフォールドタンパク質であるカスパーゼ動員ドメイン含有膜関連グアニル酸キナーゼタンパク質-1(CARMA1)はMAGUKキナーゼファミリーのメンバーである(Roche, Ramadas et al. 2013)。CARMA1は、T細胞受容体(TCR)シグナル伝達、一般的にはT細胞活性化のための重要なシグナロソームを構成する。CARMA1の細胞内濃度はCARMA1活性の調節における重要な要素である。低濃度および中程度の濃度ではCARMA1シグナル伝達の強化が観察されているのに対して、高濃度では様々な成分が互いに離れて隔離されるために活性が減少することが報告されている(二相性の応答)。TCR結合後に、CARMA1は様々なタンパク質を動員して、最終的に、2つの異なるシグナル伝達カスケード: NF-κBおよびc-jun N末端キナーゼ(JNK)を活性化することができる多タンパク質複合体を形成する(Blonska and Lin 2009)。
第1の例では、本発明者は、(2種類のCARが共存した後に)再構成されたら、CARMA1タンパク質のリンカー領域上にあるセリン残基をリン酸化して、NF-κBおよびJNKに開始シグナルを送るスプリットタンパク質系として表2に列挙したキナーゼの1つを用いるつもりである。スプリットキナーゼの作製はチミジンキナーゼに関して言えば既に報告されており、成功を収めている(Massoud, Paulmurugan et al. 2010)。
CARMA1はカルボキシ末端MAGUK様特徴に加えて、機能上重要なCARDモチーフとコイルドコイルドメインを含有する。CARDモチーフは、2つのCARD含有結合パートナー間のホモタイプCARD/CARD相互作用、ことによると3つのCARD含有結合パートナー間のホモタイプCARD/CARD相互作用でも媒介することができるタンパク質間相互作用ドメインである。CARMA1のCARDは、アミノ末端CARDモチーフと、未知構造のSer/Thrリッチカルボキシル末端を含有するアダプターBcl10とのホモタイプ相互作用を媒介する。Bcl10は、JNKおよびNF-κBの活性化に必要なMALT1との複合体を構成的に形成する(図21を参照されたい)。ここでは、一方がカルボキシ末端MAGUK様特徴を有し、他方がCARDモチーフとコイルドコイルドメインを有する2種類のCARが共存した後に、完全に再構成されたCARMA1を得ることができる。CARMA1が再構成されるとBcl10およびMALT1が集合し、その結果として2つの内因性経路JNKおよびNF-κBが活性化される。
LCK(NCBI参照配列: NP_005347.3)は、TCR結合後に活性化される最初の分子の1つである(Borger, Filby et al. 2013; Borger, Zamoyska et al. 2013; Brownlie and Zamoyska 2013)。LCKはT細胞において構成的に活性化されており、TCRに会合しているCD3ζ鎖の低レベルリン酸化を維持している。TCRとペプチド-MHCが相互作用した後に、LCKはCD8細胞質ドメインに結合し、補助受容体CD8は、TCRに会合しているCD3ζ鎖の近くにLCKを動かす。LCKの標的は、TCRに会合しているCD3ζ鎖にあるが、CD3δ鎖、CD3ε鎖、およびZAP70にもあるITAM上のチロシン残基である。ZAP70のリン酸化は、そのキナーゼ活性を活性化するコンホメーション変化を促進して、LAT(T細胞を活性化するためのアダプター分子リンカー(adaptor molecule linker for activation T cells))をリン酸化する。次に、Latは複数の下流アダプターとシグナル伝達分子を動員する。
図式化したHIFα系の機能を図22に両状態(酸素正常状態および低酸素)で示した。図23には、異なるCAR構造(単鎖および多鎖)を示した。以下の実験では、多鎖CARコンホメーションを使用した。
全構築物が多鎖CAR(mcCAR)のα鎖(SEQ ID NO:2)、β鎖(SEQ ID NO:3)、およびγ鎖(SEQ ID NO:4)をコードするpCLS24707(SEQ ID NO:1)に由来した。低酸素誘導因子1-α(HIF1アクセッション番号Q16665)のアミノ酸380〜603をコードする配列(SEQ ID NO:5)を2つの部分に分けて新規合成し(GeneCust)、古典的な分子生物学技術を用いて、短い-GS-リンカー(SEQ ID NO:6)を用いてα鎖の下流にクローニングし、pCLS26580(SEQ ID NO:7)を得た。
Tリンパ球を、活性化して3〜6日後にAgilePulse MAX系(Harvard Apparatus)を用いたメッセンジャーRNAのエレクトロトランスファー(electrotransfer)によってトランスフェクトした。活性化ビーズを除去した後に、細胞をペレット化し、>28x106細胞/mlでcytoporation medium Tに再懸濁した。5x106個の細胞を27.5μgの全RNA(10μgのα鎖、7.5μgのβ鎖、および10μgのγ鎖)または32.5μgの全RNA(15μgのα鎖-HIF1、7.5μgのβ鎖、および10μgのγ鎖)と混合して0.4cmキュベットに入れた。エレクトロポレーションは2回の1200Vで0.1msのパルスと、それに続く4回の130Vで0.2msのパルスからなった。エレクトロポレーション後に、細胞を2mLの培養培地で希釈し、37℃/5%CO2(酸素正常状態と呼ぶ)または37℃、低O2濃度(低酸素と呼ぶ)で17時間インキュベートした。製造業者によって述べられたように、雰囲気発生システム(atmosphere generation system)(2.5L AnaeroJAR assembly, Anaerogen 2.5L, Anaerobic indicator BR0055 Oxoid)用いて低酸素状態を作り出した。低酸素状態からの細胞の画分を保存し、37℃/5%CO2(酸素正常状態)で4〜6時間インキュベートした。
α鎖を検出するための1回目の標識を、PBS SVF2%、EDTA 2mM、アジ化物 0.1%に溶解した抗Fab'2-ビオチン(ヤギ抗マウスIgG、Fab'2断片特異的, 115-066-072, Jackson Immunoresearch)を用いて4℃で20分間、行った後に、PBS SVF2% EDTA 2mM アジ化物 0.1%を用いて洗浄工程を行った。2回目の標識を、PBS SVF2% EDTA 2mM アジ化物 0.1%に溶解したストレプトアビジン-APCを用いて4℃で20分間、行った後に、 PBSで洗浄工程を行った。細胞生存率を、PBSに溶解したefluor450(ebioscience 65-0863-14)を用いて4℃で20分間モニタリングした後に、PBS SVF2% EDTA 2mM アジ化物 0.1%で洗浄工程を行った。フローサイトメトリーをMACSQUANT(Miltenyi Biotec)を用いて行い、データ分析をFlowJoソフトウェアを用いて行った。
実施例1のように、2μgの全RNA(0.94μgのα鎖、0.47μgのβ鎖、および0.62μgのγ鎖)を用いてT細胞のトランスフェクションを行った。実施例3に記載のように酸素正常状態および低酸素で表面検出を行った(図25A)。
α-HIF鎖、β鎖、γ鎖を、それぞれ、オリゴペアGAα鎖-F/GAα鎖-HIF-R、GAβ鎖-F/GAβ鎖-R、GAγ鎖-F/GAγ鎖-R(SEQ ID NO:15-20)を用いてPCR増幅した。3つの鎖を、Gibbson assembly protocol(New England Biolabs)を用いてレンチウイルスプラスミドの中でSFFVプロモーターの制御下で組み立てて、pCLS26949(SEQ ID NO:21)を得た。pCLS26949(SEQ ID NO:21)と、HIFドメインのないα鎖をコードするpCLS24707(SEQ ID NO:1)から、ウイルスベクターをGIGAウイルスベクター(Belgium)によって作製した。
レンチウイルス形質導入後に、細胞を37℃/5%CO2(酸素正常状態と呼ぶ)でインキュベートした。形質導入して3〜10日後に、操作されたT細胞を、37℃/5%CO2(酸素正常状態と呼ぶ)または37℃、低O2濃度(低酸素と呼ぶ)で様々な期間(1〜24時間)にわたってインキュベートした。製造業者によって述べられたように、雰囲気発生システム(2.5L AnaeroJAR assembly, Anaerogen 2.5L, Anaerobic indicator BR0055 Oxoid)もしくはOxyrase Enzyme System(EC-Oxyrase)のいずれか、または2つの方法の組み合わせを用いて低酸素状態を作り出した。CARの表面提示の検出は実施例3に記載のように行った。
操作されたT細胞の細胞溶解活性および特異性を、低酸素または酸素正常状態で、フローサイトメトリーをベースとする細胞傷害性アッセイを用いて評価した。このアッセイでは、CAR標的抗原を提示する標的細胞(標的+)と、CAR標的抗原を提示しない標的細胞(標的-)をCellTrace(商標)CFSEまたはCellTrace(商標)violetのいずれかで標識する。混合した標的細胞集団(1:1比)を、様々な比の操作されたエフェクターCAR T細胞(10:1〜1:1のエフェクター/標的比)と、X-Vivo-15培地に溶解した最終体積中、37℃で様々な期間(4時間〜24時間)にわたってコインキュベートした。
全構築物が多鎖CAR(mcCAR)のα鎖(SEQ ID NO:2)、β鎖(SEQ ID NO:3)、およびγ鎖(SEQ ID NO:4)をコードするpCLS24707(SEQ ID NO:1)に由来した。低酸素誘導因子1-α(HIF1アクセッション番号Q16665)のアミノ酸344〜417(SEQ ID NO:22)または530〜652(SEQ ID NO:23)をコードする配列を、実施例1のように新規合成した遺伝子(GeneCust)から組み立て、クローニングし、それぞれ、pCLS26959およびpCLS26960(SEQ ID NO:24〜25)を得た。
一般的な局面では、前記の系は、(2種類の標的抗原に結合することによって誘発される)共存が起こると相互作用し、トランスミッタータンパク質を放出する2種類の膜タンパク質パートナーで構成される(図27)。
第1の膜タンパク質パートナーは、(N末端からC末端にかけて)異なるブロック:(a)膜にアドレス指定(addressing)するためのシグナル配列および抗原特異的標的化領域(SEQ ID NO:32〜38)、(b)細胞外スペーサードメイン(いわゆるヒンジ)(SEQ ID NO:39〜41)、(c)膜貫通ドメイン(SEQ ID NO:42〜46)、ならびに(d)細胞内構造および/またはシグナル伝達リンカードメイン(SEQ ID NO:47〜70)、ならびに(e1)相互作用パートナードメインの1つ(SEQ ID NO:71〜77)で構成される(図28)。
組み立てられる膜タンパク質パートナーをコードする配列を、古典的な分子生物学技術を用いてプラスミドにT7プロモーター(SEQ ID NO:202)の制御下でサブクローニングする(NcoIおよびHindIII)。または、組み立てられる膜タンパク質パートナーをコードする配列を、古典的な分子生物学技術を用いて、T7プロモーター(SEQ ID NO:149〜151)を持ってくるオリゴヌクレオチドペアを用いてPCR増幅する。さらに、mcCARをベースとする膜タンパク質パートナーの場合、オリゴヌクレオチドβ鎖-F/β鎖-Rおよびγ鎖-F/γ鎖-R(SEQ ID NO:153〜156)を用いて、pCLS24707(SEQ ID NO:152)からβ鎖およびγ鎖を増幅する。
Tリンパ球を、活性化して3〜6日後にAgilePulse MAX系(Harvard Apparatus)を用いたメッセンジャーRNAのエレクトロトランスファーによってトランスフェクトする。活性化ビーズを除去した後に、細胞をペレット化し、>28x106細胞/mlでcytoporation medium Tに再懸濁する。5x106個の細胞を1〜30μgの全RNAと混合して0.4cmキュベットに入る。エレクトロポレーションは2回の1200Vで0.1msのパルスと、それに続く4回の130Vで0.2msのパルスからなった。エレクトロポレーション後に、細胞を2mLの培養培地で希釈し、37℃/5%CO2でインキュベートする。
膜タンパク質パートナーを検出するための1回目の標識を、PBS SVF2%、EDTA 2mM、アジ化物0.1%に溶解した抗Fab'2-ビオチン結合(ヤギ抗マウスIgG、Fab'2断片特異的, 115-066-072, Jackson Immunoresearch)を用いて4℃で20分間、行った後に、PBS SVF2% EDTA 2mM アジ化物 0.1%を用いて洗浄工程を行う。2回目の標識を、PBS SVF2% EDTA 2mM アジ化物 0.1%に溶解したストレプトアビジン-APCを用いて4℃で20分間、行った後に、 PBSで洗浄工程を行う。細胞生存率を、PBSに溶解したefluor450(ebioscience 65-0863-14)を用いて4℃で20分間モニタリングした後に、PBS SVF2% EDTA 2mM アジ化物 0.1%で洗浄工程を行う。フローサイトメトリーをMACSQUANT(Miltenyi Biotec)を用いて行い、データ分析をFlowJoソフトウェアを用いて行う。
二重膜タンパク質パートナー戦略の可能性を証明するために、レポーター遺伝子の発現に基づくリードアウトを構築する。これらのレポーター系は、最小CMVプロモーターの上流に配置された、いくつかのTetO(7x)オペレーター配列反復またはGal4(5x)オペレーター配列反復で構成される。これらにより、この人工プロモーターの下流に配置されたRQR8またはレニラ(renilla)レポーター遺伝子の発現が可能になり、pCLS26301、pCLS26303、pCLS27049、およびpCLS27050(SEQ ID NO:157〜160)が得られた。これらの構築物をレンチウイルス発現ベクターにクローニングする。市販のレンチウイルス発現系を用いて製造業者のプロトコールに従って、ウイルスベクターを調製する。
二重膜タンパク質パートナー戦略用のT細胞カスタムリードアウト系を作り出すために、TALEN(SEQ ID NO:175〜192)を用いて、TCR経路に関与するタンパク質をコードする遺伝子(SEQ ID NO:166〜174)をノックアウトする。実施例7に記載のように、mRNAの調製およびトランスフェクションを行う。T細胞内の内因性遺伝子座にあるTALEN活性を、従来のプロトコールを用いた酵素T7アッセイを用いてモニタリングする。得られたデータから、設計されたTALENを用いた、全標的遺伝子座における標的変異誘発レベルが高いことがはっきりと分かった(図32)。
実施例8に記載のリードアウト系にRQR8またはレニラ遺伝子の代わりに、KOタンパク質(例えば、ZAP70)をコードする遺伝子(SEQ ID NO:193)をクローニングする。または、リードアウト内のTetOまたはGal4オペレーター配列を取り替えるために、ヒト転写因子(例えば、HNF1BおよびHNF1A)(SEQ ID NO:195および196)の標的DNA配列(SEQ ID NO:247)をクローニングする。実施例7に記載の膜タンパク質パートナーの組み立てプロセスと適合するブロック(SEQ ID NO:197〜198)を作り出すために、これらのヒト転写因子(例えば、HNF1BおよびHNF1A)をコードするDNA配列を新規合成する。遺伝子(例えば、ZAP70)をノックアウトするのに使用するTALENの設計、レンチウイルスベクターの作製、mRNAの調製、T細胞へのリードアウトおよび膜タンパク質パートナーのトランスフェクションまたは形質導入を実施例7、8、および9に記載のように行う。(i)相互作用膜タンパク質パートナーが両方ともある場合、(ii)相互作用膜タンパク質パートナーが一方だけある場合、(iii)相互作用膜タンパク質パートナーがいずれも無い場合について、ノックアウトの補完を、抗原を提示する標的細胞の存在下で脱顆粒アッセイまたはフローベースの細胞傷害性アッセイを用いてモニタリングする。
biCARパートナーの組み立て
biCARパートナーは、(N末端からC末端にかけて)異なるブロック:(a)膜にアドレス指定するためのシグナル配列および抗原特異的標的化ドメイン、(b)細胞外スペーサードメイン(いわゆるヒンジ)、(c)膜貫通ドメイン、ならびに(d)細胞内活性化および/または共刺激ドメインで構成される(図23)。
biCARゲートを構成するbiCARパートナーを両方とも、レンチウイルスベクターとして個々に、またはライブラリーとして不死化ヒトT細胞(Jurkat)または初代T細胞に送達する。
[本発明1001]
少なくとも、
腫瘍細胞の表面に発現している特異的リガンドを認識することができる細胞外リガンド結合ドメイン;
膜貫通ドメイン;
少なくとも活性化ドメインを含む細胞内ドメイン;および
酸素感受性ポリペプチドドメイン
を含む、キメラ抗原受容体(CAR)。
[本発明1002]
酸素感受性ドメインが、HIF1αもしくはHIF3αの間で選択されるか、またはそれぞれSEQ ID NO:22、23、85もしくはSEQ ID NO:26、27と80%超、好ましくは90%、もしくはより好ましくは95%の同一性を有する、本発明1001のCAR。
[本発明1003]
細胞外ドメインがヒンジをさらに含む、本発明1001または1002のCAR。
[本発明1004]
ヒンジが、CD8a、IgG1、もしくはEpoR-D2より選択されるか、またはそれぞれSEQ ID NO:39、40、もしくは41と80%超、好ましくは90%、もしくはより好ましくは95%の同一性を有する、本発明1003のCAR。
[本発明1005]
細胞外結合ドメインが、EGFRvIII、CS1、CT83、GD3、MSCP、CD19、5T4、ROR1、CD123、またはCD33細胞標的抗原のエピトープに対して向けられるscFvを含む、本発明1001〜1004のいずれかのCAR。
[本発明1006]
細胞外結合ドメインが、SEQ ID NO:32、35、38;SEQ ID NO:33;SEQ ID NO:34;SEQ ID NO:36またはSEQ ID NO:37と80%超、好ましくは90%、またはより好ましくは95%の同一性を有するポリペプチドを含むscFvを含む、本発明1001〜1005のいずれかのCAR。
[本発明1007]
膜貫通ドメインが、CD8a、4-1BB、DAP10、CD28、またはFceRIαより選択され、それぞれSEQ ID NO:42、43、44、45、および46と80%超、好ましくは90%、またはより好ましくは95%の同一性を有する、本発明1001〜1006のいずれかのCAR。
[本発明1008]
細胞内ドメインが、
CD3ζ、FceRIg、CD28、4-1BB、OX40、DAP10、CD28、CD275、HVEM、LIGHT、CD40L、GITR、TIM1、SLAM、CD2、TLT-2、LAG3、DAP12、CD84、CD244、CD229、LTBR、およびCD278の中で選択されるか、またはそれぞれSEQ ID NO:47〜70と80%超、好ましくは90%、もしくはより好ましくは95%の同一性を有するリンカー
を含む、本発明1001〜1007のいずれかのCAR。
[本発明1009]
活性化ドメインがCD3ζである、本発明1001〜1008のいずれかのCAR。
[本発明1010]
共刺激ドメイン、例えば、4-1BBまたはCD28に由来する共刺激ドメインをさらに含む、本発明1001〜1009のいずれかのCAR。
[本発明1011]
単鎖CARである、本発明1001〜1010のいずれかのCAR。
[本発明1012]
多鎖CARである、本発明1001〜1010のいずれかのCAR。
[本発明1013]
多鎖CARが、
それぞれSEQ ID NO:7、3、および4と80%超の同一性、好ましくは90%の同一性、またはより好ましくは95%の同一性を有する、Fc受容体に由来するα鎖、β鎖、およびγ鎖の一部
を含む、本発明1012のCAR。
[本発明1014]
α鎖が、細胞外にCD8ヒンジ、膜貫通ドメインとしてFcRα、および細胞内にHIF1αまたはHIF3αサブユニットと組み合わされたFcRαの一部を含み、
β鎖が、細胞外ドメインおよび膜貫通ドメインとしてFcRβ、ならびに細胞内共刺激ドメインとしてΔITAM-41BBを含み、
γ鎖が、膜貫通ドメインとしてFcRγ、および細胞内活性化ドメインとしてΔITAM-CD3ζを含む、
本発明1013のCAR。
[本発明1015]
(a)免疫細胞を準備する工程;
(b)少なくとも2種類のインプットシグナルの組み合わせによって少なくとも第1のトランスミッタードメインと第2のトランスミッタードメインの組み合わせが誘導されるように、該免疫細胞が該少なくとも2種類のインプットシグナルに対して感受性になるように該免疫細胞を操作する工程であって、該組み合わせによって該免疫細胞の活性化シグナルが生じ、
第1のインプットシグナルが低酸素であり、
第2のインプットシグナルが、特異的リガンドを認識することができる細胞外リガンド結合ドメイン、および他方のトランスミッタードメインと組み合わせて該免疫細胞を活性化することができるトランスミッタードメインを含む細胞内ドメインを含む、免疫細胞の表面に発現しているキメラ抗原受容体(CAR)による特異的リガンドの認識によって生じ、
それぞれのトランスミッタードメインが単独で該免疫細胞を活性化しない、工程;ならびに
(c)該操作された免疫細胞を増殖させる工程
を含む、細胞を特異的に標的とするために免疫細胞を操作する方法。
[本発明1016]
低酸素誘導性プロモーターの制御下にあることにより、トランスミッタードメインが低酸素に対して感受性になる、本発明1015の方法。
[本発明1017]
低酸素状態に対する感受性が、α低酸素誘導因子1(HIF-Iα)またはα低酸素誘導因子3(HIF-3α)によって誘発される、本発明1015または1016の方法。
[本発明1018]
HIF-Iαポリペプチド配列が、SEQ ID NO:5もしくはSEQ ID NO:22〜23と80%超の同一性、好ましくは90%の同一性、もしくはより好ましくは95%の同一性を有するか、またはHIF-3αポリペプチド配列が、SEQ ID NO:26〜27と80%超の同一性、好ましくは90%の同一性、もしくはより好ましくは95%の同一性を有する、本発明1017の方法。
[本発明1019]
CARが本発明1001〜1014のいずれかのものである、本発明1015〜1018のいずれかの方法。
[本発明1020]
本発明1015〜1019のいずれかの方法によって入手可能な免疫細胞。
[本発明1021]
治療組成物として使用するための、本発明1001〜1008のいずれかによって入手可能な免疫細胞。
[本発明1022]
癌を処置するための、本発明1001〜1011のいずれかの単離された免疫細胞。
[本発明1023]
固形腫瘍を処置するための、本発明1012の単離された免疫細胞。
[本発明1024]
(a)本発明1020〜1023のいずれかの免疫細胞を準備する工程;
(b)該免疫細胞を患者に投与する工程
を含む、それを必要とする対象を処置するための方法。
Claims (24)
- 少なくとも、
腫瘍細胞の表面に発現している特異的リガンドを認識することができる細胞外リガンド結合ドメイン;
膜貫通ドメイン;
少なくとも活性化ドメインを含む細胞内ドメイン;および
酸素感受性ポリペプチドドメイン
を含む、キメラ抗原受容体(CAR)。 - 酸素感受性ドメインが、HIF1αもしくはHIF3αの間で選択されるか、またはそれぞれSEQ ID NO:22、23、85もしくはSEQ ID NO:26、27と80%超、好ましくは90%、もしくはより好ましくは95%の同一性を有する、請求項1記載のCAR。
- 細胞外ドメインがヒンジをさらに含む、請求項1または2記載のCAR。
- ヒンジが、CD8a、IgG1、もしくはEpoR-D2より選択されるか、またはそれぞれSEQ ID NO:39、40、もしくは41と80%超、好ましくは90%、もしくはより好ましくは95%の同一性を有する、請求項3記載のCAR。
- 細胞外結合ドメインが、EGFRvIII、CS1、CT83、GD3、MSCP、CD19、5T4、ROR1、CD123、またはCD33細胞標的抗原のエピトープに対して向けられるscFvを含む、請求項1〜4のいずれか一項記載のCAR。
- 細胞外結合ドメインが、SEQ ID NO:32、35、38;SEQ ID NO:33;SEQ ID NO:34;SEQ ID NO:36またはSEQ ID NO:37と80%超、好ましくは90%、またはより好ましくは95%の同一性を有するポリペプチドを含むscFvを含む、請求項1〜5のいずれか一項記載のCAR。
- 膜貫通ドメインが、CD8a、4-1BB、DAP10、CD28、またはFceRIαより選択され、それぞれSEQ ID NO:42、43、44、45、および46と80%超、好ましくは90%、またはより好ましくは95%の同一性を有する、請求項1〜6のいずれか一項記載のCAR。
- 細胞内ドメインが、
CD3ζ、FceRIg、CD28、4-1BB、OX40、DAP10、CD28、CD275、HVEM、LIGHT、CD40L、GITR、TIM1、SLAM、CD2、TLT-2、LAG3、DAP12、CD84、CD244、CD229、LTBR、およびCD278の中で選択されるか、またはそれぞれSEQ ID NO:47〜70と80%超、好ましくは90%、もしくはより好ましくは95%の同一性を有するリンカー
を含む、請求項1〜7のいずれか一項記載のCAR。 - 活性化ドメインがCD3ζである、請求項1〜8のいずれか一項記載のCAR。
- 共刺激ドメイン、例えば、4-1BBまたはCD28に由来する共刺激ドメインをさらに含む、請求項1〜9のいずれか一項記載のCAR。
- 単鎖CARである、請求項1〜10のいずれか一項記載のCAR。
- 多鎖CARである、請求項1〜10のいずれか一項記載のCAR。
- 多鎖CARが、
それぞれSEQ ID NO:7、3、および4と80%超の同一性、好ましくは90%の同一性、またはより好ましくは95%の同一性を有する、Fc受容体に由来するα鎖、β鎖、およびγ鎖の一部
を含む、請求項12記載のCAR。 - α鎖が、細胞外にCD8ヒンジ、膜貫通ドメインとしてFcRα、および細胞内にHIF1αまたはHIF3αサブユニットと組み合わされたFcRαの一部を含み、
β鎖が、細胞外ドメインおよび膜貫通ドメインとしてFcRβ、ならびに細胞内共刺激ドメインとしてΔITAM-41BBを含み、
γ鎖が、膜貫通ドメインとしてFcRγ、および細胞内活性化ドメインとしてΔITAM-CD3ζを含む、
請求項13記載のCAR。 - (a)免疫細胞を準備する工程;
(b)少なくとも2種類のインプットシグナルの組み合わせによって少なくとも第1のトランスミッタードメインと第2のトランスミッタードメインの組み合わせが誘導されるように、該免疫細胞が該少なくとも2種類のインプットシグナルに対して感受性になるように該免疫細胞を操作する工程であって、該組み合わせによって該免疫細胞の活性化シグナルが生じ、
第1のインプットシグナルが低酸素であり、
第2のインプットシグナルが、特異的リガンドを認識することができる細胞外リガンド結合ドメイン、および他方のトランスミッタードメインと組み合わせて該免疫細胞を活性化することができるトランスミッタードメインを含む細胞内ドメインを含む、免疫細胞の表面に発現しているキメラ抗原受容体(CAR)による特異的リガンドの認識によって生じ、
それぞれのトランスミッタードメインが単独で該免疫細胞を活性化しない、工程;ならびに
(c)該操作された免疫細胞を増殖させる工程
を含む、細胞を特異的に標的とするために免疫細胞を操作する方法。 - 低酸素誘導性プロモーターの制御下にあることにより、トランスミッタードメインが低酸素に対して感受性になる、請求項15記載の方法。
- 低酸素状態に対する感受性が、α低酸素誘導因子1(HIF-Iα)またはα低酸素誘導因子3(HIF-3α)によって誘発される、請求項15または16記載の方法。
- HIF-Iαポリペプチド配列が、SEQ ID NO:5もしくはSEQ ID NO:22〜23と80%超の同一性、好ましくは90%の同一性、もしくはより好ましくは95%の同一性を有するか、またはHIF-3αポリペプチド配列が、SEQ ID NO:26〜27と80%超の同一性、好ましくは90%の同一性、もしくはより好ましくは95%の同一性を有する、請求項17記載の方法。
- CARが請求項1〜14のいずれか一項記載のものである、請求項15〜18のいずれか一項記載の方法。
- 請求項15〜19のいずれか一項記載の方法によって入手可能な免疫細胞。
- 治療組成物として使用するための、請求項1〜8のいずれか一項によって入手可能な免疫細胞。
- 癌を処置するための、請求項1〜11のいずれか一項記載の単離された免疫細胞。
- 固形腫瘍を処置するための、請求項12記載の単離された免疫細胞。
- (a)請求項20〜23のいずれか一項記載の免疫細胞を準備する工程;
(b)該免疫細胞を患者に投与する工程
を含む、それを必要とする対象を処置するための方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DKPA201370806 | 2013-12-20 | ||
| DKPA201370806 | 2013-12-20 | ||
| PCT/EP2014/078876 WO2015092024A2 (en) | 2013-12-20 | 2014-12-19 | Method of engineering multi-input signal sensitive t cell for immunotherapy |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017504601A true JP2017504601A (ja) | 2017-02-09 |
| JP2017504601A5 JP2017504601A5 (ja) | 2018-01-18 |
Family
ID=49884853
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016541194A Pending JP2017504601A (ja) | 2013-12-20 | 2014-12-19 | 免疫療法のためにマルチインプットシグナル感受性t細胞を操作する方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10239948B2 (ja) |
| EP (2) | EP3808410A1 (ja) |
| JP (1) | JP2017504601A (ja) |
| AU (1) | AU2014368383B2 (ja) |
| CA (1) | CA2934436A1 (ja) |
| WO (1) | WO2015092024A2 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020512019A (ja) * | 2017-03-31 | 2020-04-23 | セレクティス ソシエテ アノニム | ユニバーサル抗cd22キメラ抗原受容体操作免疫細胞 |
| WO2020175366A1 (ja) * | 2019-02-25 | 2020-09-03 | 国立大学法人東京医科歯科大学 | キメラ抗原受容体 |
| WO2021131944A1 (ja) * | 2019-12-27 | 2021-07-01 | 国立大学法人神戸大学 | 癌遺伝子治療薬 |
| JP2022533806A (ja) * | 2018-11-06 | 2022-07-26 | イミュニティーバイオ、インコーポレイテッド | キメラ抗原受容体で修飾されたnk-92細胞 |
| US12109238B2 (en) | 2018-11-06 | 2024-10-08 | Immunitybio, Inc. | Chimeric antigen receptor-modified NK-92 cells |
Families Citing this family (75)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10548921B2 (en) | 2011-04-08 | 2020-02-04 | Baylor College Of Medicine | Reversing the effects of the tumor microenvironment using chimeric cytokine receptors |
| US10704021B2 (en) | 2012-03-15 | 2020-07-07 | Flodesign Sonics, Inc. | Acoustic perfusion devices |
| US10689609B2 (en) | 2012-03-15 | 2020-06-23 | Flodesign Sonics, Inc. | Acoustic bioreactor processes |
| US10967298B2 (en) | 2012-03-15 | 2021-04-06 | Flodesign Sonics, Inc. | Driver and control for variable impedence load |
| US9752113B2 (en) | 2012-03-15 | 2017-09-05 | Flodesign Sonics, Inc. | Acoustic perfusion devices |
| US10322949B2 (en) | 2012-03-15 | 2019-06-18 | Flodesign Sonics, Inc. | Transducer and reflector configurations for an acoustophoretic device |
| US9458450B2 (en) | 2012-03-15 | 2016-10-04 | Flodesign Sonics, Inc. | Acoustophoretic separation technology using multi-dimensional standing waves |
| US9950282B2 (en) | 2012-03-15 | 2018-04-24 | Flodesign Sonics, Inc. | Electronic configuration and control for acoustic standing wave generation |
| US9745548B2 (en) | 2012-03-15 | 2017-08-29 | Flodesign Sonics, Inc. | Acoustic perfusion devices |
| US10737953B2 (en) | 2012-04-20 | 2020-08-11 | Flodesign Sonics, Inc. | Acoustophoretic method for use in bioreactors |
| WO2014117121A1 (en) | 2013-01-28 | 2014-07-31 | St. Jude Children's Research Hospital, Inc. | A chimeric receptor with nkg2d specificity for use in cell therapy against cancer and infectious disease |
| US9745569B2 (en) | 2013-09-13 | 2017-08-29 | Flodesign Sonics, Inc. | System for generating high concentration factors for low cell density suspensions |
| WO2015105955A1 (en) | 2014-01-08 | 2015-07-16 | Flodesign Sonics, Inc. | Acoustophoresis device with dual acoustophoretic chamber |
| EP3119807B1 (en) * | 2014-03-19 | 2019-04-24 | Cellectis | Cd123 specific chimeric antigen receptors for cancer immunotherapy |
| CA2945620C (en) * | 2014-04-14 | 2022-12-06 | Cellectis | Bcma (cd269) specific chimeric antigen receptors for cancer immunotherapy |
| AU2015259877B2 (en) | 2014-05-15 | 2021-02-25 | National University Of Singapore | Modified natural killer cells and uses thereof |
| US9744483B2 (en) | 2014-07-02 | 2017-08-29 | Flodesign Sonics, Inc. | Large scale acoustic separation device |
| TWI751102B (zh) | 2014-08-28 | 2022-01-01 | 美商奇諾治療有限公司 | 對cd19具專一性之抗體及嵌合抗原受體 |
| IL292508B2 (en) | 2014-12-24 | 2023-08-01 | Autolus Ltd | A cell that simultaneously expresses a chimeric antigen receptor (car) of 19cd and 22cd |
| DK3250606T3 (da) | 2015-01-26 | 2021-01-04 | Cellectis | ANTI-CLL1-specifikke enkeltkædede kimære antigenreceptorer (SCCARS) til cancerimmunterapi |
| US11161907B2 (en) * | 2015-02-02 | 2021-11-02 | Novartis Ag | Car-expressing cells against multiple tumor antigens and uses thereof |
| US11377651B2 (en) | 2016-10-19 | 2022-07-05 | Flodesign Sonics, Inc. | Cell therapy processes utilizing acoustophoresis |
| US11021699B2 (en) | 2015-04-29 | 2021-06-01 | FioDesign Sonics, Inc. | Separation using angled acoustic waves |
| US11708572B2 (en) | 2015-04-29 | 2023-07-25 | Flodesign Sonics, Inc. | Acoustic cell separation techniques and processes |
| CN114634943A (zh) | 2015-05-18 | 2022-06-17 | T细胞受体治疗公司 | 使用融合蛋白对tcr重编程的组合物和方法 |
| MA42902A (fr) * | 2015-07-08 | 2018-05-16 | Univ Johns Hopkins | Lymphocytes infiltrant la moelle (mil) en tant que source de lymphocytes t pour une thérapie par des récepteurs chimériques d'un antigène (car) |
| US11474085B2 (en) | 2015-07-28 | 2022-10-18 | Flodesign Sonics, Inc. | Expanded bed affinity selection |
| US11459540B2 (en) | 2015-07-28 | 2022-10-04 | Flodesign Sonics, Inc. | Expanded bed affinity selection |
| HUE048939T2 (hu) | 2015-08-03 | 2020-09-28 | Engmab Sarl | Human B sejt érési antigén elleni monoklonális antitestek (BCMA) |
| EP3331920A4 (en) | 2015-08-07 | 2019-04-03 | Seattle Children's Hospital, dba Seattle Children's Research Institute | T CARRIER CARRIER CELLS FOR BISPECIFICATION OF SOLID TUMOR TARGETING |
| IL260532B2 (en) * | 2016-01-11 | 2023-12-01 | Univ Leland Stanford Junior | Systems containing chaperone proteins and their uses for controlling gene expression |
| BR112018013663A2 (pt) | 2016-01-11 | 2019-01-22 | Univ Leland Stanford Junior | proteínas quiméricas e métodos de imunoterapia |
| SG11201805872SA (en) | 2016-01-21 | 2018-08-30 | Pfizer | Chimeric antigen receptors targeting epidermal growth factor receptor variant iii |
| DK3405490T3 (da) | 2016-01-21 | 2022-01-10 | Pfizer | Mono- og bispecifikke antistoffer mod epidermal vækstfaktorreceptor variant iii og cd3 og anvendelser deraf |
| EP3202783A1 (en) * | 2016-02-02 | 2017-08-09 | Ecole Polytechnique Fédérale de Lausanne (EPFL) | Engineered antigen presenting cells and uses thereof |
| EP3420093B1 (en) * | 2016-02-26 | 2022-11-23 | Cellectis | Micelle based system nuclease encapsulation for in-vivo gene editing |
| CN107298715B (zh) * | 2016-04-15 | 2021-05-04 | 阿思科力(苏州)生物科技有限公司 | Slit2D2-嵌合抗原受体及其应用 |
| US11214789B2 (en) | 2016-05-03 | 2022-01-04 | Flodesign Sonics, Inc. | Concentration and washing of particles with acoustics |
| US11085035B2 (en) | 2016-05-03 | 2021-08-10 | Flodesign Sonics, Inc. | Therapeutic cell washing, concentration, and separation utilizing acoustophoresis |
| GB201609604D0 (en) * | 2016-06-01 | 2016-07-13 | Ucl Business Plc | Cell |
| WO2017217517A1 (ja) * | 2016-06-17 | 2017-12-21 | 国立大学法人大阪大学 | 腫瘍内静脈形成促進剤 |
| CA3032498A1 (en) | 2016-08-02 | 2018-02-08 | TCR2 Therapeutics Inc. | Compositions and methods for tcr reprogramming using fusion proteins |
| WO2018058432A1 (zh) * | 2016-09-28 | 2018-04-05 | 李华顺 | 一种多基因重组嵌合抗原受体分子及其应用 |
| JP7217970B2 (ja) | 2016-10-07 | 2023-02-06 | ティーシーアール2 セラピューティクス インク. | 融合タンパク質を用いてt細胞受容体をリプログラミングするための組成物及び方法 |
| WO2018073393A2 (en) | 2016-10-19 | 2018-04-26 | Cellectis | Tal-effector nuclease (talen) -modified allogenic cells suitable for therapy |
| KR20190127655A (ko) | 2016-10-19 | 2019-11-13 | 프로디자인 소닉스, 인크. | 음향학에 의한 친화성 세포 추출 |
| BR112019008426A2 (pt) | 2016-11-02 | 2019-09-03 | Engmab Sarl | anticorpo biespecífico contra bcma e cd3 e um fármaco imunológico para uso combinado no tratamento de mieloma múltiplo |
| CA3044593A1 (en) | 2016-11-22 | 2018-05-31 | TCR2 Therapeutics Inc. | Compositions and methods for tcr reprogramming using fusion proteins |
| US11408005B2 (en) | 2016-12-12 | 2022-08-09 | Seattle Children's Hospital | Chimeric transcription factor variants with augmented sensitivity to drug ligand induction of transgene expression in mammalian cells |
| CN117384929A (zh) | 2017-03-27 | 2024-01-12 | 新加坡国立大学 | 一种编码由细胞表达的嵌合受体的多核苷酸 |
| MX2019011570A (es) | 2017-03-27 | 2019-11-18 | Nat Univ Singapore | Lineas celulares estimuladoras para expansion y activacion ex vivo de celulas asesinas naturales. |
| US20200048618A1 (en) * | 2017-04-18 | 2020-02-13 | Autolus Limited | Cell |
| JP7264329B2 (ja) | 2017-05-17 | 2023-04-25 | シアトル チルドレンズ ホスピタル (ディービーエイ シアトル チルドレンズ リサーチ インスティテュート) | T細胞療法を向上させるための、哺乳動物t細胞の活性化により誘導可能な合成プロモーター(syn+pro)の作製 |
| WO2019002633A1 (en) | 2017-06-30 | 2019-01-03 | Cellectis | CELLULAR IMMUNOTHERAPY FOR REPETITIVE ADMINISTRATION |
| GB201711470D0 (en) * | 2017-07-17 | 2017-08-30 | Univ Oxford Innovation Ltd | Chimeric receptors |
| CN111164203A (zh) * | 2017-08-02 | 2020-05-15 | 奥托路斯有限公司 | 表达嵌合抗原受体或工程化tcr并包含选择性表达的核苷酸序列的细胞 |
| WO2019072824A1 (en) | 2017-10-09 | 2019-04-18 | Cellectis | IMPROVED ANTI-CD123 CAR IN UNIVERSAL MODIFIED IMMUNE T LYMPHOCYTES |
| KR102439221B1 (ko) | 2017-12-14 | 2022-09-01 | 프로디자인 소닉스, 인크. | 음향 트랜스듀서 구동기 및 제어기 |
| SG11202005609PA (en) * | 2017-12-15 | 2020-07-29 | Univ Leland Stanford Junior | Compositions and methods for inhibiting t cell exhaustion |
| US20210317181A1 (en) * | 2018-08-02 | 2021-10-14 | Asimov, Inc. | Universal chimeric receptors |
| CN112543651B (zh) * | 2018-08-07 | 2023-06-02 | 普渡研究基金会 | 使car t细胞恢复活力 |
| CN112912727A (zh) * | 2018-08-31 | 2021-06-04 | 因维克蒂斯股份有限公司 | 评估car功能性的方法 |
| JP2022506598A (ja) | 2018-11-01 | 2022-01-17 | ジュノー セラピューティクス インコーポレイテッド | Gタンパク質共役受容体クラスcグループ5メンバーd(gprc5d)に特異的なキメラ抗原受容体 |
| EP3773918A4 (en) | 2019-03-05 | 2022-01-05 | Nkarta, Inc. | CD19 DIRECTED CHIMERIC ANTIGEN RECEPTORS AND THEIR USES IN IMMUNOTHERAPY |
| JP2022529380A (ja) * | 2019-04-26 | 2022-06-21 | ザ ユニバーシティ オブ ノース カロライナ アット チャペル ヒル | キメラ抗原受容体構築物およびcar-t細胞におけるそれらの使用 |
| CN114945382A (zh) | 2019-11-26 | 2022-08-26 | 诺华股份有限公司 | Cd19和cd22嵌合抗原受体及其用途 |
| WO2022046755A1 (en) * | 2020-08-28 | 2022-03-03 | Innovative Cellular Therapeutics Holdings, Ltd. | Fusion protein enhancing cell therapy |
| TW202241935A (zh) | 2020-12-18 | 2022-11-01 | 美商世紀治療股份有限公司 | 具有可調適受體專一性之嵌合抗原受體系統 |
| IL311570A (en) | 2021-10-14 | 2024-05-01 | Arsenal Biosciences Inc | Immune cells with common expression chain and logic gate systems |
| EP4424714A1 (en) * | 2021-10-29 | 2024-09-04 | Shanghai Sinobay Biotechnology Co., Ltd. | Condition-controlled spliceable chimeric antigen receptor molecule and application thereof |
| WO2023087024A1 (en) * | 2021-11-15 | 2023-05-19 | The Board Of Trustees Of The Leland Stanford Junior University | Composition and method of valency controlled receptor systems for cell engineering and therapy |
| WO2024015383A1 (en) * | 2022-07-12 | 2024-01-18 | Northwestern University | Engineered hypoxia biosensors and methods of using the same |
| US20240041929A1 (en) | 2022-08-05 | 2024-02-08 | Juno Therapeutics, Inc. | Chimeric antigen receptors specific for gprc5d and bcma |
| WO2024040033A2 (en) * | 2022-08-15 | 2024-02-22 | The Regents Of The University Of California | Multi-chain synthetic receptors for simultaneous ligand‑induced transcriptional regulation and membrane‑proximal signal transduction |
| WO2024073775A2 (en) * | 2022-09-30 | 2024-04-04 | The Regents Of The University Of California | Compositions and methods for enhancing adoptive t cell therapeutics |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004524848A (ja) * | 2001-03-21 | 2004-08-19 | アイシス イノヴェイション リミテッド | アッセイ、方法および手段 |
| JP2008509209A (ja) * | 2004-08-09 | 2008-03-27 | キャンサー・リサーチ・テクノロジー・リミテッド | αケトグルタレート類および治療薬としてのその使用 |
| JP2009545310A (ja) * | 2006-07-31 | 2009-12-24 | ヴァスキュラー バイオジェニックス リミテッド | ポリペプチド、および、当該ポリペプチドをコードするポリヌクレオチド、および、虚血に関連する医学的状態の治療におけるそれらの使用 |
| WO2012099973A2 (en) * | 2011-01-18 | 2012-07-26 | The Trustees Of The University Of Pennsylvania | Compositions and methods for treating cancer |
| WO2013176915A1 (en) * | 2012-05-25 | 2013-11-28 | Roman Galetto | Methods for engineering allogeneic and immunosuppressive resistant t cell for immunotherapy |
| WO2014153270A1 (en) * | 2013-03-16 | 2014-09-25 | Novartis Ag | Treatment of cancer using humanized anti-cd19 chimeric antigen receptor |
| WO2014180306A1 (zh) * | 2013-05-08 | 2014-11-13 | 上海益杰生物技术有限公司 | 编码gpc3嵌合抗原受体蛋白的核酸及表达gpc3嵌合抗原受体蛋白的t淋巴细胞 |
| WO2014184143A1 (en) * | 2013-05-13 | 2014-11-20 | Cellectis | Cd19 specific chimeric antigen receptor and uses thereof |
| WO2014184744A1 (en) * | 2013-05-13 | 2014-11-20 | Cellectis | Methods for engineering highly active t cell for immunotherapy |
| WO2014184741A1 (en) * | 2013-05-13 | 2014-11-20 | Cellectis | Methods for engineering allogeneic and highly active t cell for immunotheraphy |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8611832D0 (en) | 1986-05-15 | 1986-06-25 | Holland I B | Polypeptide |
| US5037743A (en) | 1988-08-05 | 1991-08-06 | Zymogenetics, Inc. | BAR1 secretion signal |
| CA2597924C (en) * | 2005-02-15 | 2018-10-02 | Duke University | Anti-cd19 antibodies and uses in oncology |
| WO2009023386A2 (en) * | 2007-07-06 | 2009-02-19 | Trubion Pharmaceuticals, Inc. | Binding peptides having a c-terminally disposed specific binding domain |
| US8822647B2 (en) * | 2008-08-26 | 2014-09-02 | City Of Hope | Method and compositions using a chimeric antigen receptor for enhanced anti-tumor effector functioning of T cells |
| DK3214091T3 (en) * | 2010-12-09 | 2019-01-07 | Univ Pennsylvania | USE OF CHEMICAL ANTIGEN RECEPTOR MODIFIED T CELLS FOR TREATMENT OF CANCER |
| JP6177780B2 (ja) | 2011-09-28 | 2017-08-09 | エラ、ビオテック、ソシエダッド、アノニマEra Biotech, S.A. | スプリットインテインおよびその使用 |
| EP3594245A1 (en) | 2012-02-13 | 2020-01-15 | Seattle Children's Hospital d/b/a Seattle Children's Research Institute | Bispecific chimeric antigen receptors and therapeutic uses thereof |
| US20130280220A1 (en) | 2012-04-20 | 2013-10-24 | Nabil Ahmed | Chimeric antigen receptor for bispecific activation and targeting of t lymphocytes |
| CN103483452B (zh) * | 2012-06-12 | 2021-08-13 | 上海细胞治疗集团有限公司 | 双信号独立的嵌合抗原受体及其用途 |
| BR112015004522A2 (pt) * | 2012-09-04 | 2017-11-21 | Cellectis | receptor de antígeno quimérico multicadeia e usos destes |
| US9745368B2 (en) * | 2013-03-15 | 2017-08-29 | The Trustees Of The University Of Pennsylvania | Targeting cytotoxic cells with chimeric receptors for adoptive immunotherapy |
| CA2956667A1 (en) * | 2013-07-29 | 2015-02-05 | Bluebird Bio, Inc. | Multipartite signaling proteins and uses thereof |
-
2014
- 2014-12-19 JP JP2016541194A patent/JP2017504601A/ja active Pending
- 2014-12-19 CA CA2934436A patent/CA2934436A1/en not_active Abandoned
- 2014-12-19 WO PCT/EP2014/078876 patent/WO2015092024A2/en active Application Filing
- 2014-12-19 EP EP20188659.5A patent/EP3808410A1/en active Pending
- 2014-12-19 EP EP14830959.4A patent/EP3083691A2/en not_active Ceased
- 2014-12-19 US US15/106,783 patent/US10239948B2/en active Active
- 2014-12-19 AU AU2014368383A patent/AU2014368383B2/en not_active Ceased
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004524848A (ja) * | 2001-03-21 | 2004-08-19 | アイシス イノヴェイション リミテッド | アッセイ、方法および手段 |
| JP2008509209A (ja) * | 2004-08-09 | 2008-03-27 | キャンサー・リサーチ・テクノロジー・リミテッド | αケトグルタレート類および治療薬としてのその使用 |
| JP2009545310A (ja) * | 2006-07-31 | 2009-12-24 | ヴァスキュラー バイオジェニックス リミテッド | ポリペプチド、および、当該ポリペプチドをコードするポリヌクレオチド、および、虚血に関連する医学的状態の治療におけるそれらの使用 |
| WO2012099973A2 (en) * | 2011-01-18 | 2012-07-26 | The Trustees Of The University Of Pennsylvania | Compositions and methods for treating cancer |
| WO2013176915A1 (en) * | 2012-05-25 | 2013-11-28 | Roman Galetto | Methods for engineering allogeneic and immunosuppressive resistant t cell for immunotherapy |
| WO2013176916A1 (en) * | 2012-05-25 | 2013-11-28 | Roman Galetto | Use of pre t alpha or functional variant thereof for expanding tcr alpha deficient t cells |
| US20140134142A1 (en) * | 2012-05-25 | 2014-05-15 | Cellectis | Multi-Chain Chimeric Antigen Receptor and Uses Thereof |
| WO2014153270A1 (en) * | 2013-03-16 | 2014-09-25 | Novartis Ag | Treatment of cancer using humanized anti-cd19 chimeric antigen receptor |
| WO2014180306A1 (zh) * | 2013-05-08 | 2014-11-13 | 上海益杰生物技术有限公司 | 编码gpc3嵌合抗原受体蛋白的核酸及表达gpc3嵌合抗原受体蛋白的t淋巴细胞 |
| WO2014184143A1 (en) * | 2013-05-13 | 2014-11-20 | Cellectis | Cd19 specific chimeric antigen receptor and uses thereof |
| WO2014184744A1 (en) * | 2013-05-13 | 2014-11-20 | Cellectis | Methods for engineering highly active t cell for immunotherapy |
| WO2014184741A1 (en) * | 2013-05-13 | 2014-11-20 | Cellectis | Methods for engineering allogeneic and highly active t cell for immunotheraphy |
Non-Patent Citations (3)
| Title |
|---|
| ANG SONNY O; OLIVARES SIMON; DENIGER DREW C; LEE DEAN A; CHAMPLIN RICHARD E; COOPER LAURENCE J: "CONDITIONAL ACTIVATION OF T CELLS TO SPECIFICALLY TARGET C-MET UNDER HYPOXIA", MOLECULAR THERAPY, vol. VOL:17, NR:SUPPL.1, JPN5017001555, May 2009 (2009-05-01), GB, pages 25 - 26, ISSN: 0004286322 * |
| ANG SONNY O; OLIVARES SIMON; SHPALL ELIZABETH J; LEE DEAN A; CHAMPLIN RICHARD, ET AL.: "CONDITIONAL T-CELL ACTIVATION FOR TUMOR UNDER HYPOXIA", BLOOD, vol. VOL:112, NR:11, JPN5017001556, November 2008 (2008-11-01), US, pages 1331 - 1332, ISSN: 0004286323 * |
| PLOS ONE, vol. 8(2), JPN6019029963, 2013, pages 57695 - 1, ISSN: 0004286324 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020512019A (ja) * | 2017-03-31 | 2020-04-23 | セレクティス ソシエテ アノニム | ユニバーサル抗cd22キメラ抗原受容体操作免疫細胞 |
| JP7314115B2 (ja) | 2017-03-31 | 2023-07-25 | セレクティス ソシエテ アノニム | ユニバーサル抗cd22キメラ抗原受容体操作免疫細胞 |
| JP2022533806A (ja) * | 2018-11-06 | 2022-07-26 | イミュニティーバイオ、インコーポレイテッド | キメラ抗原受容体で修飾されたnk-92細胞 |
| JP7536649B2 (ja) | 2018-11-06 | 2024-08-20 | イミュニティーバイオ、インコーポレイテッド | キメラ抗原受容体で修飾されたnk-92細胞 |
| US12109238B2 (en) | 2018-11-06 | 2024-10-08 | Immunitybio, Inc. | Chimeric antigen receptor-modified NK-92 cells |
| WO2020175366A1 (ja) * | 2019-02-25 | 2020-09-03 | 国立大学法人東京医科歯科大学 | キメラ抗原受容体 |
| WO2021131944A1 (ja) * | 2019-12-27 | 2021-07-01 | 国立大学法人神戸大学 | 癌遺伝子治療薬 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20170073423A1 (en) | 2017-03-16 |
| EP3083691A2 (en) | 2016-10-26 |
| WO2015092024A3 (en) | 2015-08-13 |
| WO2015092024A2 (en) | 2015-06-25 |
| CA2934436A1 (en) | 2015-06-25 |
| EP3808410A1 (en) | 2021-04-21 |
| US10239948B2 (en) | 2019-03-26 |
| AU2014368383B2 (en) | 2020-01-16 |
| AU2014368383A1 (en) | 2016-06-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10239948B2 (en) | Method of engineering multi-input signal sensitive T cell for immunotherapy | |
| AU2021201967B2 (en) | Methods for engineering allogeneic and immunosuppressive resistant T cell for immunotherapy | |
| JP6998917B2 (ja) | 免疫療法のために同種異系かつ高活性のt細胞を操作するための方法 | |
| EP3341410B1 (en) | Chimeric antigen receptors with integrated controllable functions | |
| US11690873B2 (en) | Universal chimeric antigen receptor T cells specific for CD22 | |
| KR102170533B1 (ko) | 암 면역요법을 위한 cd33 특이적 키메라 항원 수용체들 | |
| EP2893004B1 (en) | Multi-chain chimeric antigen receptor and uses thereof | |
| US20170296623A1 (en) | INHIBITORY CHIMERIC ANTIGEN RECEPTOR (iCAR OR N-CAR) EXPRESSING NON-T CELL TRANSDUCTION DOMAIN | |
| TW201920250A (zh) | 用於以基因方式修飾且擴展淋巴球以及調節其活性之方法及組合物 | |
| CN110582299A (zh) | 高亲和力mage-a1特异性tcr及其用途 | |
| KR20160030103A (ko) | 면역요법을 위한 매우 활성인 t 세포를 조작하는 방법 | |
| RU2775453C2 (ru) | Сконструированные универсальные иммунные клетки с анти-cd22 химерным антигенным рецептором | |
| KR102726269B1 (ko) | 범용 항-cd22 키메라 항원 수용체 조작된 면역 세포 | |
| Zhu | Next Generation Receptors for the Enhanced Control of Cell-based Immunotherapies | |
| JP2020519267A (ja) | より安全な細胞免疫療法のためのプロテアーゼベースの切り替えキメラ抗原受容体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160818 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20161012 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171130 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20171130 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20181031 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20190115 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190507 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190805 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20191016 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20191219 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200205 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20200617 |