JP2017193584A - ヒドラジドを製造するための改善されたプロセス - Google Patents
ヒドラジドを製造するための改善されたプロセス Download PDFInfo
- Publication number
- JP2017193584A JP2017193584A JP2017150482A JP2017150482A JP2017193584A JP 2017193584 A JP2017193584 A JP 2017193584A JP 2017150482 A JP2017150482 A JP 2017150482A JP 2017150482 A JP2017150482 A JP 2017150482A JP 2017193584 A JP2017193584 A JP 2017193584A
- Authority
- JP
- Japan
- Prior art keywords
- hydrazide
- hydrazine
- reaction
- flask
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 CC(*)(SP)IC(NN)=O Chemical compound CC(*)(SP)IC(NN)=O 0.000 description 3
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/46—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having at least one of the nitrogen atoms, not being part of nitro or nitroso groups, further bound to other hetero atoms
- C07C323/48—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having at least one of the nitrogen atoms, not being part of nitro or nitroso groups, further bound to other hetero atoms to nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/14—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of sulfides
- C07C319/20—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of sulfides by reactions not involving the formation of sulfide groups
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C241/00—Preparation of compounds containing chains of nitrogen atoms singly-bound to each other, e.g. hydrazines, triazanes
- C07C241/04—Preparation of hydrazides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C243/00—Compounds containing chains of nitrogen atoms singly-bound to each other, e.g. hydrazines, triazanes
- C07C243/24—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids
- C07C243/26—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids with acylating carboxyl groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C243/28—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids with acylating carboxyl groups bound to hydrogen atoms or to acyclic carbon atoms to hydrogen atoms or to carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/02—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of thiols
- C07C319/12—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of thiols by reactions not involving the formation of mercapto groups
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Coating Apparatus (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description
例えば、本発明は以下の項目を提供する。
(項目1)
(a)ヒドラジンおよび不活性溶媒を含む攪拌された実質的に均一なスラリーを調製するステップと、
(b)該スラリーに塩化アシルを連続的に添加するステップと
を含む、ヒドラジンおよび塩化アシルからヒドラジドを調製する方法。
(項目2)
前記塩化アシルが、前記スラリーに実質的に1滴ずつ添加される、請求項1に記載の方法。
(項目3)
前記塩化アシルが、保護されたチオールをさらに含む、請求項1または2に記載の方法。
(項目4)
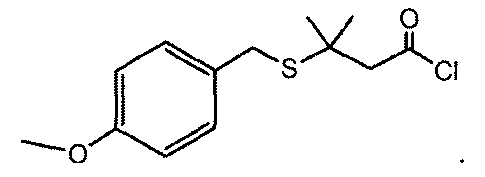
前記塩化アシルがベンジルチオエーテルを含む、請求項3に記載の方法。
(項目5)
前記塩化アシルが構造:
を有し、式中、Pはチオール保護基であり;
R1およびR2はそれぞれ、C1〜C5アルキルからなる群から選択され;
Lは、アルキレンリンカーである、
請求項1または2に記載の方法。
(項目6)
Lが−CH2−である、請求項5に記載の方法。
(項目7)
R1およびR2がそれぞれ独立してメチルである、請求項5または6に記載の方法。
(項目8)
Pが、フェニル環上で任意選択で置換されたベンジル基である、項目5、6、または7に記載の方法。
(項目9)
Pがp−メトキシベンジル基である、項目8に記載の方法。
(項目10)
前記塩化アシルが構造:
を有する、項目1または2に記載の方法。
(項目11)
前記不活性溶媒が塩化メチレンである、項目1から10のいずれか一項に記載の方法。
(項目12)
前記ヒドラジド生成物が構造:
を有し、式中、Pはチオール保護基であり;
R1およびR2はそれぞれ、C1〜C5アルキルからなる群から選択され;
Lは、アルキレンリンカーである、
項目1、2、または11に記載の方法。
(項目13)
Lが−CH2−である、項目12に記載の方法。
(項目14)
R1およびR2がそれぞれ独立してメチルである、項目12に記載の方法。
(項目15)
Pが、フェニル環上で任意選択で置換されたベンジル基である、項目12に記載の方法。
(項目16)
Pがp−メトキシベンジル基である、項目15に記載の方法。
(項目17)
所望のヒドラジドが、
であるか、またはその塩である、項目12に記載の方法。
(項目18)
前記ヒドラジドが、構造:
を有するビス−ヒドラジド副生成物を5%未満含有し、式中、RおよびR’は、任意選択で置換されたアルキル、ヘテロアルキル、またはヘテロアルカリール基である、
項目1から16のいずれか一項に記載の方法。
(項目19)
RおよびR’が、
であり、式中、Pはチオール保護基であり;
R1およびR2はそれぞれ、C1〜C5アルキルからなる群から選択され;
Lはアルキレンリンカーである、
項目18に記載の方法。
(項目20)
Lが−CH2−である、項目19に記載の方法。
(項目21)
R1およびR2がそれぞれ独立してメチルである、項目20に記載の方法。
(項目22)
Pが、フェニル環上で任意選択で置換されたベンジル基である、項目19に記載の方法。
(項目23)
Pがp−メトキシベンジル基である、項目22に記載の方法。
(項目24)
前記ビス−ヒドラジド副生成物が構造:
を有する、項目23に記載の方法。
(項目25)
酸塩化物溶液の前記連続的な添加が、約−68℃から約−75℃の反応温度を維持するように調節される、項目1から24のいずれか一項に記載の方法。
(項目26)
前記ヒドラジンスラリーが実質的に均一である、項目1から25のいずれか一項に記載の方法。
(項目27)
(a)攪拌された不活性溶媒を含む反応容器を、所望の低温に冷却するステップ、
(b)該反応容器に連続的な様式でヒドラジンを添加し、それによって、該ヒドラジンおよび該不活性溶媒を含む攪拌された実質的に均一なスラリーを調製するステップ、
(c)該スラリーに連続的な様式で酸塩化物を添加し、それによって、ヒドラジド結合を形成するステップ
を含む、ヒドラジド結合を含有する化合物を調製する方法。
(項目28)
前記不活性溶媒が塩化メチレンである、項目27に記載の方法。
(項目29)
(a)約−68℃から約−75℃の温度に不活性溶媒を冷却するステップと、
(b)不活性溶媒に溶解したヒドラジンを、該冷却した不活性溶媒に1滴ずつ添加するステップと
を含む、ヒドラジンスラリーを調製する方法。
(項目30)
前記不活性溶媒が塩化メチレンである、項目29に記載の方法。
(項目31)
前記ヒドラジンスラリーが、約270から約400rpmの速度で攪拌される、項目29または30に記載の方法。
(項目32)
項目16に記載の方法にしたがってヒドラジドを調製するステップを含む、3−メチル−3−メルカプトブタン酸ヒドラジドを調製する方法。
(項目33)
担体としてのモノクローナル抗体を含むカリケアマイシンのファミリーのメンバーの免疫複合体を調製する方法であって、該方法は、項目3に記載の方法にしたがって、アシル基がS保護メルカプト官能基を含有しているモノアシル化ヒドラジンを調製するステップと、該S保護メルカプト官能基の保護基を除去するステップと、得られたヒドラジドを、該免疫複合体を調製するために使用するステップとを含む、方法。
(項目34)
前記得られたヒドラジドが3−メチル−3−メルカプトブタン酸ヒドラジドである、項目33に記載の方法。
(項目35)
3−メチル−3−メルカプトブタン酸ヒドラジドがリンカーとして使用されて、ゲムツズマブオゾガマイシンまたはイノツズマブオゾガマイシンを作製する、項目34に記載の方法。
p−メトキシベンジルチオエーテル酸(1)の最初の調製
反応式Iを参照すると、熱電対、機械式攪拌子、N2入口を上部に備えた還流冷却器、および250mLの均圧添加漏斗を備えた5Lの丸底フラスコに、400g、465mL、4.70モルのピペリジンを投入した。3,3−ジメチルアクリル酸(215g、2.15モル)を少量ずつ、攪拌した5L反応フラスコに添加した。反応を、N2中で激しく攪拌した。反応温度は、添加中は35から40℃未満に維持した(注記:強力な発熱、即ち、ガス放出)。p−メトキシベンジルチオール(386g、323mL、2.32モル)を、(5L)反応フラスコに、15分にわたり均圧添加漏斗を介して投入した。混合物を、N2中で攪拌しながら82から88℃に加熱した。反応温度を、この範囲内で15分間維持した。注記:発熱。透明な黄色の混合物を、最短でも15時間、N2中で攪拌しながら92から95℃に加熱した。1mLのサンプルを、HPLC分析のために取り出した。反応は、3,3−ジメチルアクリル酸の3面積%未満が残ったときに終了したと見なした。反応を、加熱マントルを除去することによって70から75℃に冷却した。
p−メトキシベンジルチオエーテル酸塩化物(2)の最初の調製
反応式IIを参照すると、p−メトキシベンジルチオエーテル酸(400g、1.57モル)を、熱電対、機械式攪拌子、N2入口が上部にある還流冷却器、および0.5Lの均圧添加漏斗を備えた5Lの丸底フラスコに投入した。塩化メチレン(1600g、1212mL)を5L反応フラスコに投入した。透明な溶液を20から25℃に加熱した。塩化メチレン(300g)および塩化オキサリル(110g、78mL)を0.5L均圧添加漏斗に投入した。反応温度を20から30℃に維持しながら、塩化オキサリル/塩化メチレンの溶液350mLを、添加漏斗を介して添加した。透明な黄色の溶液を、バブリングが収まるまで最短でも30分間、20から25℃で攪拌した。塩化オキサリルの添加を繰り返した。反応温度を20から30℃に維持しながら、塩化オキサリル/塩化メチレンの溶液350mlを、均圧添加漏斗を介して反応フラスコに添加した(添加時間約45分)。反応混合物を、約32から38℃に加熱した。攪拌した溶液を、この温度範囲に最短でも1時間保持した。1mLのサンプルを、HPLC分析用に取り出した。反応は、メトキシベンジルチオエーテル酸出発材料の3面積%未満残存したときに、終了したと見なした。反応を、最短でも5分間、23から28℃に冷却した。溶液を、風袋計量済みの3L丸底フラスコに移した。反応フラスコを、100ml、132gの塩化メチレンで3Lのフラスコに濯いだ。反応溶液は、揮発物質が残存しなくなるまで33から36℃に設定された浴温度および25から28インチHgの圧力でロータリーエバポレータによって、真空中で濃縮した。最終重量は1367gであり、正味の重量は、p−メトキシベンジルチオエーテル酸塩化物が500.3gであった。留出物を廃棄した。
p−メトキシベンジルチオエーテルヒドラジド(3)の最初の調製
反応式IIIを参照すると、5Lのモートン型丸底フラスコは、熱電対、機械式攪拌子、N2入口が上部にある還流冷却器、および0.5L均圧添加漏斗を備えていた。p−メトキシベンジルチオエーテル酸塩化物を、500ml、660gの塩化メチレンに溶解した。溶液を、2Lのエルレンマイヤーフラスコに移した。500mlの塩化メチレンを添加して、全体積が1300mLの溶液を構成した。
p−メトキシベンジルチオエーテルヒドラジド(3)の修正された調製
副生成物であるビス−メトキシベンジルチオエーテルヒドラジド(6)のレベルを低下させるため、単離された生成物であるp−メトキシベンジルチオエーテルヒドラジド中のこの副生成物の形成に影響を及ぼす(1つまたは複数の)反応パラメータについて、調査した。実施例3の手順を繰り返した。−78℃で、ヒドラジン/CH2Cl2溶液は、非攪拌性の凍結混合物である。ゴム状の塊が反応フラスコに粘着し、それと共に攪拌翼は、空中で回転した。チオエーテル酸塩化物/CH2Cl2の溶液を、ヒドラジン/CH2Cl2(28%v/v)のこの凍結混合物に1滴ずつ添加し、それと共に温度を約−72℃に保持した。添加の終わり(温度は−72℃であった)のHPLC分析は、反応をほとんど示さなかった。これは、迅速な反応であるという予測に反していた。これは、反応における適正な混合が不十分であることに、起因していると考えられる。大部分が未反応の反応混合物を、そのままにして温めた。温度が約−50℃に達したとき、攪拌可能な不均一混合物が生じ、その後、素早い発熱が生じて、温度を即座に−28℃に押し上げ、反応の色が黄色からオフホワイトに変化した。これは、効果的ではない混合によって、ビス−ヒドラジドの発生を促す局所反応をもたらす可能性があるという仮説をもたらす。室温に温め、実施例3に示されるような反応の後処理によって、主な生成物(82%、HPLC面積%)としてビス−ヒドラジドを提供する。これは、典型的な望ましくない20%というレベルよりも、はるかに高い。添加されたチオエーテル酸塩化物(2)/CH2Cl2の溶液は、ヒドラジンの凍結した塊と効果的に混合しないと結論付けた。
p−メトキシベンジルチオエーテルヒドラジド(3)の調製に対する温度の影響
実施例5は、実施例4と同じ反応を繰り返したが、約−72℃ではなく0℃で実施した。チオエーテル酸塩化物/CH2Cl2の溶液を、ヒドラジン/CH2Cl2(28%v/v)の攪拌可能な均質溶液に1滴ずつ添加した。この場合、39%(HPLC面積%)のビス−ヒドラジドが形成された。これらの条件は、より低い温度および攪拌が、望ましくない副生成物の形成に影響を及ぼす因子であることを示唆している。実施例3.1および3.2の結果を、表3にまとめる。
p−メトキシベンジルチオエーテルヒドラジド(3)の調製に対するヒドラジン濃度の影響
次いで、低温でより低い濃度のヒドラジンを使用した影響について、試験をした。結果を、以下の表4にまとめる。−65から−72℃での、ヒドラジン/CH2Cl2の攪拌可能な不均一混合物は、28%(v/v)に対して5%および19%の濃度まで、ヒドラジン/CH2Cl2を希釈することによって調製した。ヒドラジン/CH2Cl2濃度を19%および5%として実施された表4の実験4.1および4.2は、それぞれ、チオエーテル酸塩化物と反応する攪拌可能な不均一混合物を提供し、それによって、ビス−ヒドラジド副生成物のレベルがそれぞれ3%および5%である所望の生成物をもたらした。少量のヒドラジンを使用して同じ反応を繰り返すことにより(5対10モル当量を使用した表4の実験4.3)、ビス−ヒドラジドは3%しか発生しなかった。ヒドラジンの典型的な量は、チオエーテル酸に対して5モル当量である。ヒドラジンの量を2倍にして10モル当量にした場合(表4:実験4.1および4.2)、最終生成物中のビス−ヒドラジドのレベルに著しい影響を与えなかった。
p−メトキシベンジルチオエーテルヒドラジド(3)の調製に対する温度の影響
生成物中のビス−ヒドラジドのレベルに対する温度の影響を、表5で試験した。チオエーテル酸塩化物を、ヒドラジン/CH2Cl2(19%v/v)の攪拌した混合物に−20および−72℃(表5:実験5.1および5.2)で添加した実験では、ビス−ヒドラジドがそれぞれ28%および4%のレベルで発生した。同様の結果が、実験5.3および5.4(表5)で観察された。上述の結果は、より低いレベル(3〜5%)のビス−ヒドラジドを得るのにより低い反応温度(約−70℃)が必要であることを示す。
p−メトキシベンジルチオエーテルヒドラジド(3)の調製に対するヒドラジン濃度の影響
表4の実験について試験すると、約−70℃の添加温度および19%または5%(v/v)のヒドラジン/CH2Cl2の濃度によって、同等の結果が得られたことが明らかにされた。この観察内容について、表6でさらに試験をした。チオエーテル酸塩化物が、19%、14%、および10%の濃度でヒドラジン/CH2Cl2の不均一混合物に添加された表6の実験6.1、6.2、および6.4では、ビス−ヒドラジドが、それぞれ6%、13%、および4%のレベルで発生した。反応体積、フラスコのサイズ、および攪拌速度は一定に保持した。結果は、19%のときに、反応が10%濃度での結果と同等であることを示した。ヒドラジン/CH2Cl2の濃度14%を、30gのスケール(表6の実験6.3)で繰り返した結果、わずか3%しかビス−ヒドラジドが混入していない所望の生成物が得られた。実験6.2におけるより高いレベルのビス−ヒドラジドは、素早く調節する前に反応温度を−57℃に急上昇させた、酸塩化物の初期の素早い添加が原因である。
p−メトキシベンジルチオエーテルヒドラジド(3)の調製に対する混合速度の影響
混合の影響について試験をした(表7)。実験7.2および7.1は、より速い混合(400rpm対200rpm)が、より少ないビス−ヒドラジド(22%対40%)を生成したことを示した。両方の実験における、通常レベルよりも高いビス−ヒドラジドは、表8の100mLフラスコ内の20mLの液体に比べて50mLフラスコ内のヒドラジン/CH2Cl2の初期液体レベル(36mL)を考慮すると、効果的でない混合によって引き起こされたと考えられる。これは、混合速度の他に、反応器の幾何形状および液体レベルを考慮しなければならないことを意味する。
p−メトキシベンジルチオエーテルヒドラジド(3)の調製に対するスケールアップの影響
ヒドラジン/CH2Cl2の濃度19%または5%(v/v)でp−メトキシベンジルチオエーテルヒドラジドを調製することにより、同等の結果が得られた。ヒドラジン/CH2Cl2濃度19%は、通常は−70℃で攪拌可能な混合物であるが、十分に混合されていない凍結混合物になる可能性があるという危険性がある。製造スケールのバッチ(400g)を5%のヒドラジン/CH2Cl2濃度で実行した場合、より大きな反応器(20Lモートン型)が必要になる。しかし、このプロセスをヒドラジン/CH2Cl2濃度14%で実行した場合、反応は、ガラス反応器、5Lモートン型丸底フラスコを使用して実施することができる。プロセスを、20gスケール(表8の実験8.1)で、ヒドラジン/CH2Cl2濃度14%v/vで実行した場合、単離した生成物p−メトキシベンジルチオエーテルヒドラジドには、4.4%(HPLC面積%)の副生成物であるビス−メトキシベンジルチオエーテルヒドラジドが混入していた。上述の条件(ヒドラジン/CH2Cl2濃度は14%であった)を、製造規模に拡大した条件で繰り返すことにより(表8の実験8.2)、4.2%(HPLC面積%)の副生成物であるビス−メトキシベンジルチオエーテルヒドラジドが混入しているp−メトキシベンジルチオエーテルヒドラジドが生成された。
1.ヒドラジン/CH2Cl2混合物へのメトキシベンジルチオ酸塩化物溶液の添加速度は、反応温度が−68から−75℃に維持されるように調節した。
p−メトキシベンジルチオエーエル酸中間体(1)の修正された調製
冷却器、N2入口、攪拌子、および温度プローブ/制御器を備えた5Lの反応フラスコを、設定した。ピペリジン(0.402kg)を、N2雰囲気中で容器に投入した。3,3−ジメチルアクリル酸(0.215kg)を、攪拌しながら少量ずつ添加し、その後、p−メトキシベンジルチオール(0.358g)を添加した。反応混合物を、最短でも15分間にわたって徐々に82から88℃に加熱し、その反応温度を、発熱が観察されるまで維持した。温度は、95℃を超えることがなかった。発熱が終了したら、加熱を92から98℃まで継続し、最短でも15時間維持した。
p−メトキシベンジルチオエーテル酸塩化物(2)の調製
冷却器、水スクラバー、温度プローブ、1L添加漏斗、N2入口、および攪拌子を備えた5L反応フラスコを、設定した。CH2Cl2(1.6kg)をN2雰囲気中で投入し、その後、p−メトキシベンジルチオエーテル酸(0.400kg)を攪拌しながら投入した。塩化オキサリル(0.220kg)およびCH2Cl2(0.600kg)の溶液を添加漏斗内で調製した。塩化オキサリル溶液の約半分を添加し、それと共に温度範囲を20から30℃に維持した(発熱!)。最短でも30分間攪拌した状態でCO2/COの発生が観察され、次いで温度を20から30℃の間に維持しながら、残りの塩化オキサリル溶液を添加した。気体の発生が弱まるまで(約30分)反応を攪拌し、次いで混合物を33から38℃に加熱した。この温度は、気体の発生が弱まるまで約60分間維持された。反応をサンプル採取し、HPLCを使用して、残りの酸の量を決定した。反応は、出発材料の量が5%以下であるときに終了したと判断した。反応が終了しなかった場合は、攪拌をさらに1時間、33から38℃で継続し、次いでサンプル採取し、再び試験をした。加熱マントルを除去し、反応混合物をそのまま20から30℃に冷却した。混合物を3Lの1つ口フラスコに移し、次いでCH2Cl2で濯いだ。バッチを、揮発物質のほとんどが除去されるまでロータリーエバポレータで濃縮した。
p−メトキシベンジルチオエーテルヒドラジド中間体(3)の調製
冷却器、N2入口、熱電対、攪拌子、および2Lの添加漏斗を備えた5Lの4つ口モートン型反応フラスコを、設定した。CH2Cl2(2.40kg)をN2雰囲気中で投入し、−75から−65℃に冷却した。無水ヒドラジン(0.252kg)を投入することにより、フラスコの側壁にヒドラジン結晶を形成することなく、ヒドラジン氷の均一なスラリーが得られた。チオエーテル酸塩化物溶液を添加漏斗に移し、必要に応じてCH2Cl2で濯ぐことにより、1.30Lの溶液体積が得られた。
チオール脱保護中間体(4)の調製
Dowex SRB OH陰イオン交換樹脂を、樹脂2.4kgを大型ブフナー漏斗に添加し、水(4回に分けて、それぞれ2.40kg)で洗浄し、次いでMeOH(4回に分けて、それぞれ1.92kg)で洗浄することによって調製した。樹脂をビーカー内で水で覆い、最短でも1時間浸漬し、次いで水を濾別した。樹脂を適切な貯蔵容器に移した。攪拌子、熱電対、N2入口、および250mLの添加漏斗を備えた5Lの反応フラスコを、設定した。トリフルオロ酢酸(2.80kg)をN2雰囲気中で充填し、5から10℃に冷却した。チオエーテルヒドラジド(0.380kg)を、温度を5から15℃の間に維持しながら少量ずつ添加した(発熱!)。溶液を、0から5℃に冷却した。
遊離塩基ヒドラジド(5)の調製
粗製の塩酸塩生成物(4)を、12Lの4つ口反応フラスコ内で水と混合し(20.2×Ckg)、攪拌することにより、溶液が得られた。溶液のpHを、6.5から7.5pH単位の範囲が実現するまで、処理済みの樹脂で調節した。バッチを約15分間攪拌し、次いでpHを再びチェックし、必要に応じて調節することにより、6.5から7.5pH単位の値が得られた。バッチを吸引濾過し、水で濯ぎ(3.00×Ckg)、次いで無水EtOH(2回に分け、それぞれ5.30kg×Ckg)で濯いだ。吸引濾過は、濾液の流れが本質的に停止するまで継続した。生成物の濾液を、風袋計量済みの3Lフラスコに移し、必要に応じて無水EtOHで濯いだ。バッチを、蒸留が本質的に停止するまでロータリーエバポレータで濃縮した。生成物の残渣を無水EtOH(1.58×Ckg)に再び溶解し、以前のように濃縮した。生成物の残渣を無水エーテル(2.57×Ckg)に再び溶解し、以前のように濃縮した。蒸留が本質的に停止したら、乾燥を高真空で継続し、蒸発を最短でも2時間継続させた。残渣の正味の重量は、定数Dであった。濃縮物を、CH2Cl2(66.3×Dkg)を使用して、攪拌子アセンブリ、温度プローブ、および窒素入口を備えた12L反応フラスコに移した。混合物を、15から30℃で、最短でも30分間で攪拌した。バッチを濾過し、濾液を第2の12L反応フラスコ内に収集した。第1のフラスコを、CH2Cl2(3.98×Dkg)を使用して、フィルタを通して第2のフラスコに濯いだ。バッチが入っている第2のフラスコは、攪拌子アセンブリ、温度プローブ、および窒素入口を備えていた。シリカゲル(0.700×Dkg)を攪拌しながら投入し、攪拌を約30分間継続する。バッチを吸引濾過し、シリカをCH2Cl2(2回に分け、それぞれ3.98×Dkg)で洗浄し、合わせた濾液を10L吸引フラスコに収集した。バッチをロータリーエバポレータで、1Lのフラスコに約30℃で濃縮した。10Lのフラスコを、CH2Cl2で必要に応じて、エバポレータに濯いだ。蒸留は、本質的に停止するまで継続した。ロータリーエバポレータを高真空源に切り替え、蒸発を35から40℃で約3時間継続した。
5ステッププロセスの全収率限界は、理論(0.105kg)の33%以上であり、最高収率と最低収率との間の差は、15%以下でなければならない。先のプロセスの限界は理論の33から43%であったが、ヒドラジド形成反応における塩化メチレンの増加によってもたらされた副生成物の形成の減少が、高い収率を引き起こすことになると予測される。確認用バッチでの実際の収率を、生産バッチの収率範囲を画定するのに使用することになる。
3−メチル−3−メルカプトブタン酸ヒドラジド、Cl−332258(DMHリンカー)の最終精製
塩化メチレン(1000mL、1325g)を、機械式攪拌子、N2入口、還流冷却器、および温度制御デバイスを備えた2Lの4つ口反応フラスコに投入した。DMHリンカー(20g)を反応フラスコに投入した。N2中、スラリーを、20±3℃で最短でも30分間攪拌した。得られたDMHリンカーの濁りのある溶液を、350mLの中間焼結ガラスブフナー漏斗に通して濾過した。濾液を、機械式攪拌子、N2入口、還流冷却器、および温度制御デバイスを備えた清浄な2Lの4つ口丸底フラスコに収集した。反応フラスコを、20mL、26.5gの塩化メチレンで清浄な反応フラスコに濯いだ。シリカゲル(20g)を、15〜25℃に温度を維持しながら反応フラスコ内の溶液に投入した。スラリーを、最短でも30分間、20±3℃でN2中で攪拌した。不均一混合物を、(350mL、中間)焼結ガラスブフナー漏斗に通して濾過した。濾液を、清浄な2Lの1つ口丸底フラスコ内に収集した。反応フラスコを、塩化メチレン(50mL、66.3g)でフィルターケーキに濯ぎ、濾液を1つ口フラスコ内に収集した。濾液を、ロータリーエバポレータ(浴=35±5℃)および機械式水流吸引器15〜30mmHgを使用して乾固濃縮し、その後、高真空(7〜10mmHg)にした。得られた白色固体を0〜5℃まで冷却し、7mmHgの高真空中で2時間乾燥した。n−ヘプタン(100mL、68.4g)を、硬質固体に投入し、均一な懸濁液が得られるまで室温で最短でも10分間攪拌した。
Claims (1)
- ヒドラジドを製造するための改善されたプロセスなど。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US93952907P | 2007-05-22 | 2007-05-22 | |
| US60/939,529 | 2007-05-22 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015218118A Division JP2016026219A (ja) | 2007-05-22 | 2015-11-06 | ヒドラジドを製造するための改善されたプロセス |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017193584A true JP2017193584A (ja) | 2017-10-26 |
Family
ID=39810161
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010509499A Active JP5697446B2 (ja) | 2007-05-22 | 2008-05-20 | ヒドラジドを製造するための改善されたプロセス |
| JP2013172956A Withdrawn JP2013234203A (ja) | 2007-05-22 | 2013-08-23 | ヒドラジドを製造するための改善されたプロセス |
| JP2015218118A Withdrawn JP2016026219A (ja) | 2007-05-22 | 2015-11-06 | ヒドラジドを製造するための改善されたプロセス |
| JP2017150482A Pending JP2017193584A (ja) | 2007-05-22 | 2017-08-03 | ヒドラジドを製造するための改善されたプロセス |
Family Applications Before (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010509499A Active JP5697446B2 (ja) | 2007-05-22 | 2008-05-20 | ヒドラジドを製造するための改善されたプロセス |
| JP2013172956A Withdrawn JP2013234203A (ja) | 2007-05-22 | 2013-08-23 | ヒドラジドを製造するための改善されたプロセス |
| JP2015218118A Withdrawn JP2016026219A (ja) | 2007-05-22 | 2015-11-06 | ヒドラジドを製造するための改善されたプロセス |
Country Status (24)
| Country | Link |
|---|---|
| US (5) | US8110705B2 (ja) |
| EP (2) | EP2155258B1 (ja) |
| JP (4) | JP5697446B2 (ja) |
| KR (1) | KR101547705B1 (ja) |
| CN (2) | CN101835494B (ja) |
| AR (1) | AR066674A1 (ja) |
| AU (1) | AU2008256905B2 (ja) |
| BR (1) | BRPI0811140B1 (ja) |
| CA (1) | CA2687852A1 (ja) |
| CL (1) | CL2008001490A1 (ja) |
| CY (2) | CY1119373T1 (ja) |
| DK (2) | DK2155258T3 (ja) |
| ES (2) | ES2686019T3 (ja) |
| HU (2) | HUE036704T2 (ja) |
| IL (1) | IL202242A (ja) |
| MX (1) | MX2009012587A (ja) |
| PA (1) | PA8781201A1 (ja) |
| PE (1) | PE20090305A1 (ja) |
| PL (2) | PL2155258T3 (ja) |
| PT (2) | PT2465541T (ja) |
| SI (2) | SI2465541T1 (ja) |
| TW (1) | TW200904784A (ja) |
| WO (1) | WO2008147765A1 (ja) |
| ZA (1) | ZA200908229B (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019069931A1 (ja) | 2017-10-03 | 2019-04-11 | 公立大学法人大阪府立大学 | 細胞培養容器、細胞の取得方法、および細胞の培養方法 |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101547705B1 (ko) * | 2007-05-22 | 2015-08-26 | 와이어쓰 엘엘씨 | 히드라지드의 개선된 제조 방법 |
| CN108047287B (zh) * | 2013-11-04 | 2021-04-27 | 辉瑞大药厂 | 用于合成加利车霉素衍生物的中间体和方法 |
| JP2016540826A (ja) | 2013-11-04 | 2016-12-28 | ファイザー・インク | 抗efna4抗体−薬物コンジュゲート |
| RU2708075C2 (ru) | 2014-04-30 | 2019-12-04 | Пфайзер Инк. | Конъюгаты анти-ртк7 антитело-лекарственное средство |
| WO2016105330A1 (en) * | 2014-12-22 | 2016-06-30 | Hewlett Packard Enterprise Development Lp | Response to an inoperative network device managed by a controller |
| WO2019018647A1 (en) | 2017-07-20 | 2019-01-24 | Pfizer Inc. | ANTI-GD3 ANTIBODIES AND CONJUGATES ANTIBODY-MEDICATION |
| CN109574870A (zh) * | 2018-12-25 | 2019-04-05 | 维思普新材料(苏州)有限公司 | 一种酰肼的连续制备方法 |
Family Cites Families (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US623230A (en) * | 1899-04-18 | Rotary ore-cooler | ||
| DE2608336A1 (de) | 1975-03-11 | 1976-09-30 | Sandoz Ag | Neue organische verbindungen, ihre herstellung und verwendung |
| US4291022A (en) * | 1975-03-11 | 1981-09-22 | Sandoz Ltd. | Organic compounds |
| US4671958A (en) | 1982-03-09 | 1987-06-09 | Cytogen Corporation | Antibody conjugates for the delivery of compounds to target sites |
| US4479953A (en) * | 1983-08-25 | 1984-10-30 | Merck & Co., Inc. | Pyrazine aldimine compounds as antimicrobial agents |
| US4970198A (en) | 1985-10-17 | 1990-11-13 | American Cyanamid Company | Antitumor antibiotics (LL-E33288 complex) |
| US4729781A (en) * | 1986-08-04 | 1988-03-08 | Sandoz Ltd. | 3,6-dichloro-2-methoxybenzohydroxamic acid derivatives and use as herbicidal agents |
| HUT48093A (en) * | 1986-08-04 | 1989-05-29 | Sandoz Ag | Herbicides comprising benzohydroxamic acid or benzoic acid hydrazide derivatives as active ingredient and process for producing benzohydroxamic acid or benzoic acid hydrazide derivatives |
| US5079233A (en) | 1987-01-30 | 1992-01-07 | American Cyanamid Company | N-acyl derivatives of the LL-E33288 antitumor antibiotics, composition and methods for using the same |
| US4939244A (en) | 1987-01-30 | 1990-07-03 | American Cyanamid Company | Pseudoaglycones of LL-E33288 antibiotics |
| US5053394A (en) * | 1988-09-21 | 1991-10-01 | American Cyanamid Company | Targeted forms of methyltrithio antitumor agents |
| US5770701A (en) | 1987-10-30 | 1998-06-23 | American Cyanamid Company | Process for preparing targeted forms of methyltrithio antitumor agents |
| JPH05222044A (ja) | 1991-10-25 | 1993-08-31 | Eastman Kodak Co | 2当量ピラゾロトリアゾールマゼンタカプラーの製造 |
| UA41298C2 (uk) * | 1991-11-22 | 2001-09-17 | Юніроял Кемікал Компані Інк. | Фенілгідразинове похідне, спосіб боротьби зі шкідниками сільськогосподарських культур та інсектоакарициднонематоцидна композиція |
| US5690904A (en) * | 1993-07-12 | 1997-11-25 | Amersham International Plc | Diagnostic radiopharmaceutical compounds (That) |
| US5443953A (en) | 1993-12-08 | 1995-08-22 | Immunomedics, Inc. | Preparation and use of immunoconjugates |
| US5403015A (en) * | 1993-12-09 | 1995-04-04 | Forte; Steven L. | Cards and methods for playing casino 21 or blackjack |
| WO1996006096A1 (en) * | 1994-08-23 | 1996-02-29 | Nissan Chemical Industries, Ltd. | Pyridine derivative |
| IT1292092B1 (it) | 1997-06-05 | 1999-01-25 | Geange Ltd | Impiego di derivati eterociclici aromatici azotati nel trattamento topico di affezioni di tessuti epiteliali |
| ATE371659T1 (de) * | 1998-10-16 | 2007-09-15 | Merck Sharp & Dohme | Pyrazolotriazinderivate als gaba-rezeptorliganden |
| GB0008696D0 (en) * | 2000-04-07 | 2000-05-31 | Merck Sharp & Dohme | Therapeutic agents |
| JP2005504014A (ja) | 2001-06-08 | 2005-02-10 | サイトビア インコーポレイテッド | カスパーゼの活性化因子およびアポトーシスの誘導因子としての置換された3−アリール−5−アリール−[1,2,4]−オキサジアゾール類および類似体、並びにその使用法 |
| SG187991A1 (en) * | 2002-05-02 | 2013-03-28 | Wyeth Corp | Calicheamicin derivative-carrier conjugates |
| US6803379B2 (en) * | 2002-06-04 | 2004-10-12 | Jose A. Fernandez-Pol | Pharmacological agents and methods of treatment that inactivate pathogenic prokaryotic and eukaryotic cells and viruses by attacking highly conserved domains in structural metalloprotein and metalloenzyme targets |
| CN100364531C (zh) * | 2002-12-18 | 2008-01-30 | 西托维亚公司 | 3,5-二取代-[1,2,4]-二唑及类似物和其用途 |
| KR100897642B1 (ko) | 2002-12-20 | 2009-05-14 | 글락소 그룹 리미티드 | 신경 장애 치료용 벤즈아제핀 유도체 |
| US7304161B2 (en) | 2003-02-10 | 2007-12-04 | Intrexon Corporation | Diaclhydrazine ligands for modulating the expression of exogenous genes in mammalian systems via an ecdysone receptor complex |
| EP1622615A4 (en) | 2003-05-13 | 2009-02-18 | Smithkline Beecham Corp | INHIBITORS OF THE INTEGRASE OF NAPHTHYRIDINE |
| TW200519094A (en) | 2003-07-02 | 2005-06-16 | Vertex Pharma | Pyrimidines useful as modulators of voltage-gated ion channels |
| MXPA06000276A (es) | 2003-07-02 | 2006-04-07 | Sugen Inc | Indolinona hidrazidas como inhibidores de c-met. |
| SE0302486D0 (sv) | 2003-09-18 | 2003-09-18 | Astrazeneca Ab | Novel compounds |
| JP2007532552A (ja) * | 2004-04-06 | 2007-11-15 | セマフォア ファーマシューティカルズ, インコーポレイテッド | Ptenインヒビター |
| TW200630106A (en) * | 2004-10-08 | 2006-09-01 | Wyeth Corp | Immunotherapy of autoimmune disorders |
| TWI404706B (zh) * | 2006-01-11 | 2013-08-11 | Actelion Pharmaceuticals Ltd | 新穎噻吩衍生物 |
| KR101547705B1 (ko) * | 2007-05-22 | 2015-08-26 | 와이어쓰 엘엘씨 | 히드라지드의 개선된 제조 방법 |
-
2008
- 2008-05-20 KR KR1020097026806A patent/KR101547705B1/ko active Active
- 2008-05-20 SI SI200831994T patent/SI2465541T1/sl unknown
- 2008-05-20 CA CA2687852A patent/CA2687852A1/en not_active Abandoned
- 2008-05-20 WO PCT/US2008/064213 patent/WO2008147765A1/en not_active Ceased
- 2008-05-20 MX MX2009012587A patent/MX2009012587A/es active IP Right Grant
- 2008-05-20 DK DK08755945.6T patent/DK2155258T3/en active
- 2008-05-20 EP EP08755945.6A patent/EP2155258B1/en active Active
- 2008-05-20 ES ES12159159.8T patent/ES2686019T3/es active Active
- 2008-05-20 CN CN2008800216526A patent/CN101835494B/zh active Active
- 2008-05-20 DK DK12159159.8T patent/DK2465541T3/en active
- 2008-05-20 HU HUE08755945A patent/HUE036704T2/hu unknown
- 2008-05-20 PE PE2008000869A patent/PE20090305A1/es not_active Application Discontinuation
- 2008-05-20 HU HUE12159159A patent/HUE039657T2/hu unknown
- 2008-05-20 AU AU2008256905A patent/AU2008256905B2/en active Active
- 2008-05-20 PT PT12159159T patent/PT2465541T/pt unknown
- 2008-05-20 PL PL08755945T patent/PL2155258T3/pl unknown
- 2008-05-20 PT PT87559456T patent/PT2155258T/pt unknown
- 2008-05-20 US US12/123,777 patent/US8110705B2/en active Active
- 2008-05-20 CN CN201110434577.XA patent/CN102558001B/zh active Active
- 2008-05-20 SI SI200831859T patent/SI2155258T1/sl unknown
- 2008-05-20 EP EP12159159.8A patent/EP2465541B1/en active Active
- 2008-05-20 BR BRPI0811140-5A patent/BRPI0811140B1/pt active IP Right Grant
- 2008-05-20 ES ES08755945.6T patent/ES2642888T3/es active Active
- 2008-05-20 PL PL12159159T patent/PL2465541T3/pl unknown
- 2008-05-20 JP JP2010509499A patent/JP5697446B2/ja active Active
- 2008-05-22 PA PA20088781201A patent/PA8781201A1/es unknown
- 2008-05-22 TW TW097118930A patent/TW200904784A/zh unknown
- 2008-05-22 AR ARP080102165A patent/AR066674A1/es unknown
- 2008-05-22 CL CL200801490A patent/CL2008001490A1/es unknown
-
2009
- 2009-11-19 IL IL202242A patent/IL202242A/en active IP Right Grant
- 2009-11-20 ZA ZA200908229A patent/ZA200908229B/xx unknown
-
2012
- 2012-01-27 US US13/360,220 patent/US8383857B2/en active Active
-
2013
- 2013-01-31 US US13/755,886 patent/US8853451B2/en active Active
- 2013-08-23 JP JP2013172956A patent/JP2013234203A/ja not_active Withdrawn
-
2014
- 2014-10-02 US US14/504,738 patent/US9227924B2/en active Active
-
2015
- 2015-11-06 JP JP2015218118A patent/JP2016026219A/ja not_active Withdrawn
- 2015-12-18 US US14/973,869 patent/US9738602B2/en active Active
-
2017
- 2017-08-03 JP JP2017150482A patent/JP2017193584A/ja active Pending
- 2017-09-20 CY CY20171100991T patent/CY1119373T1/el unknown
-
2018
- 2018-08-13 CY CY181100854T patent/CY1120592T1/el unknown
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019069931A1 (ja) | 2017-10-03 | 2019-04-11 | 公立大学法人大阪府立大学 | 細胞培養容器、細胞の取得方法、および細胞の培養方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2017193584A (ja) | ヒドラジドを製造するための改善されたプロセス | |
| JP7163439B2 (ja) | カリケアマイシン誘導体を合成するための中間体および方法 | |
| AU2016247232B2 (en) | Improved processes for making hydrazides | |
| AU2014208212B2 (en) | Improved processes for making hydrazides | |
| RU2484849C2 (ru) | Усовершенствованный способ получения гидразидов | |
| HK1172008B (en) | Improved processes for making hydrazides | |
| HK1147736B (en) | Improved processes for making hydrazides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20170803 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180226 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20180525 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20180725 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20180906 |