JP2016010771A - 複合半透膜 - Google Patents
複合半透膜 Download PDFInfo
- Publication number
- JP2016010771A JP2016010771A JP2014133606A JP2014133606A JP2016010771A JP 2016010771 A JP2016010771 A JP 2016010771A JP 2014133606 A JP2014133606 A JP 2014133606A JP 2014133606 A JP2014133606 A JP 2014133606A JP 2016010771 A JP2016010771 A JP 2016010771A
- Authority
- JP
- Japan
- Prior art keywords
- composite semipermeable
- semipermeable membrane
- membrane
- group
- functional layer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 CC*CC(C)C1[C@@]2(C)C(C)C3(CC3)CC12 Chemical compound CC*CC(C)C1[C@@]2(C)C(C)C3(CC3)CC12 0.000 description 2
Landscapes
- Separation Using Semi-Permeable Membranes (AREA)
- Coating Of Shaped Articles Made Of Macromolecular Substances (AREA)
- Polyamides (AREA)
- Polymerisation Methods In General (AREA)
Abstract
【課題】高いホウ素阻止性能を有し、重金属接触や薬液洗浄前後での膜性能変化の少ない複合半透膜を提供する。【解決手段】ポリアミド分離機能層が式(1)または(2)の構造を含む複合半透膜。【選択図】なし
Description
本発明は、液状混合物の選択的分離に有用な複合半透膜に関し、特に高いホウ素阻止性能を有し、重金属接触や薬液洗浄前後での膜性能変化の少ない複合半透膜に関する。
混合物の分離に関して、溶媒(例えば水)に溶解した物質(例えば塩類)を除くための技術には様々なものがあるが、近年、省エネルギーおよび省資源のためのプロセスとして膜分離法の利用が拡大している。膜分離法に使用される膜には、精密ろ過膜、限外ろ過膜、ナノろ過膜、逆浸透膜などがあり、これらの膜は、例えば海水、かん水、有害物を含んだ水などから飲料水を得る場合や、工業用超純水の製造、排水処理、有価物の回収などに用いられている(特許文献1、2)。
現在市販されている逆浸透膜およびナノろ過膜の大部分は複合半透膜であり、多孔性支持膜上にゲル層とポリマーを架橋した活性層を有するものと、多孔性支持膜上でモノマーを重縮合した活性層を有するものとの2種類がある。なかでも、多官能アミンと多官能酸ハロゲン化物との重縮合反応によって得られる架橋ポリアミドからなる分離機能層を多孔性支持膜上に被覆して得られる複合半透膜は、透過性や選択分離性の高い分離膜として広く用いられている。
選択分離すべきものとして、ホウ素があげられる。ホウ素は、人体及び動植物に対して神経障害の発症や成長阻害を引き起こすなどの毒性を持つが、海水に多く含まれていることから、海水淡水化においてホウ素除去は重要である。そこで、複合半透膜のホウ素除去性能を向上させる手段が種々提案されてきている(特許文献3、4)。特許文献3では、界面重合により製膜された複合半透膜を熱処理して性能向上させる方法が開示されている。特許文献4では、界面重合により製膜された複合半透膜を臭素含有遊離塩素水溶液に接触させる方法が開示されている。
また、複合半透膜の用途が広がるにつれ、重金属を多く含むなど様々な水質での長期的安定運転に耐えうる複合半透膜が望まれてきている。複合半透膜を使用し続けると、使用経過時間とともに膜表面に汚れが付着し、膜の造水量が低下するなどの膜性能劣化が起こる。そのため、ある期間運転後にアルカリや酸などによる薬液洗浄が必要となる。したがって、長期間にわたって安定な運転を継続するために、重金属などの微量成分による性能変化が起こりにくい複合半透膜や、アルカリや酸などの薬液洗浄前後での膜性能変化の少ない複合半透膜の開発が望まれている。
複合半透膜の耐アルカリ性を向上させるために、複合半透膜にpH9〜13の水素イオン濃度水溶液を接触させる方法(特許文献5)が開示されている。また、複合半透膜の耐酸性を向上させるために、複合半透膜に環状硫酸エステルを接触させる方法(特許文献6)が開示されている。
しかしこれらの膜でも、高いホウ素阻止性能、耐重金属性、耐アルカリ性を同時に満たすものはなく、様々な水質での長期運転における性能安定性は充分であるとはいえない。本発明は、高いホウ素阻止性能を有し、かつ重金属接触や薬液洗浄前後での膜性能変化の少ない複合半透膜を提供することを目的とする。
上記目的を達成するための本発明の複合半透膜は、以下のいずれかの構成をとる。
(1)微多孔性支持膜と、微多孔性支持膜上に設けられたポリアミド分離機能層とからなる複合半透膜であって、前記ポリアミド分離機能層が下記式(1)または(2)で示される構造を含むことを特徴とする複合半透膜。
(1)微多孔性支持膜と、微多孔性支持膜上に設けられたポリアミド分離機能層とからなる複合半透膜であって、前記ポリアミド分離機能層が下記式(1)または(2)で示される構造を含むことを特徴とする複合半透膜。
(2)前記ポリアミド分離機能層の黄色度が10.0以下であることを特徴とする前記に記載の複合半透膜。
(3)前記R1、R2が電子求引基を含むことを特徴とする前記(1)または(2)に記載の複合半透膜。
(4)前記R1、R2がシアノ基、カルボニル基、エステル結合のいずれかを含む有機基であることを特徴とする、前記(1)〜(3)のいずれかに記載の複合半透膜。
(5)微多孔性支持膜上に多官能第一級芳香族アミンと多官能酸ハロゲン化物との重縮合反応によるポリアミド層を形成する工程、該ポリアミド層が有する第一級芳香族アミノ基を、該アミノ基と反応してジアゾニウム塩またはその誘導体を生成する試薬に接触させ改質処理する工程、および、前記ポリアミド層を、下記式(3)を含む溶液に接触させ改質処理する工程とからなる複合半透膜の製造方法。
(3)前記R1、R2が電子求引基を含むことを特徴とする前記(1)または(2)に記載の複合半透膜。
(4)前記R1、R2がシアノ基、カルボニル基、エステル結合のいずれかを含む有機基であることを特徴とする、前記(1)〜(3)のいずれかに記載の複合半透膜。
(5)微多孔性支持膜上に多官能第一級芳香族アミンと多官能酸ハロゲン化物との重縮合反応によるポリアミド層を形成する工程、該ポリアミド層が有する第一級芳香族アミノ基を、該アミノ基と反応してジアゾニウム塩またはその誘導体を生成する試薬に接触させ改質処理する工程、および、前記ポリアミド層を、下記式(3)を含む溶液に接触させ改質処理する工程とからなる複合半透膜の製造方法。
(6)前記R3、R4がシアノ基、カルボニル基、エステル結合のいずれかを含む有機基であることを特徴とする前記(5)に記載の複合半透膜の製造方法。
本発明の複合半透膜は、高いホウ素阻止性能を有し、重金属接触や薬液洗浄前後での膜性能変化の少ない高性能な複合半透膜である。この膜を用いることで、特に、重金属を多く含む、あるいは水質が悪く洗浄頻度が多いかん水や海水を淡水化するにあたり長期間にわたって安定な運転の継続が期待される。
本発明の複合半透膜は、微多孔性支持膜と、微多孔性支持膜上に設けられたポリアミド分離機能層とからなる複合半透膜であって、前記ポリアミド分離機能層が下記式(1)または(2)で示される構造を含むことを特徴とする複合半透膜である。なお、本出願において式中の波線はポリアミド骨格を表す。
本発明において微多孔性支持膜とは、微多孔性支持膜として強度を付与するための基材と該基材上に設けられる多孔性支持体とを備えるものであり、実質的にイオン等の分離性能を有さず、実質的に分離性能を有する分離機能層に強度を与えるためのものである。微多孔性支持膜の孔のサイズや分布は特に限定されないが、例えば、均一で微細な孔、あるいは分離機能層が形成される側の表面からもう一方の面まで徐々に大きな微細孔をもち、かつ、分離機能層が形成される側の表面で微細孔の大きさが0.1nm以上100nm以下であるような微多孔性支持膜が好ましい。
微多孔性支持膜に使用する材料やその形状は特に限定されないが、微多孔性支持膜の基材としては、ポリエステル系重合体、ポリアミド系重合体、ポリオレフィン系重合体、あるいはこれらの混合物や共重合体等が挙げられる。中でも、機械的、熱的に安定性の高いポリエステルの布帛が特に好ましい。布帛の形態としては、長繊維不織布や短繊維不織布、さらには織編物を好ましく用いることができる。
基材上に形成される多孔性支持体の素材としては、ポリスルホンや酢酸セルロースやポリ塩化ビニル、あるいはそれらを混合したものが好ましく使用され、化学的、機械的、熱的に安定性の高いポリスルホンを使用するのが特に好ましい。
具体的には、次の化学式(4)に示す繰り返し単位からなるポリスルホンを用いると、多孔性支持体の孔径が制御しやすく、寸法安定性が高いため好ましい。
例えば、上記ポリスルホンのN,N−ジメチルホルムアミド(以降、DMFと記載)溶液を、密に織ったポリエステル布あるいは不織布の上に一定の厚さに注型し、それを水中で湿式凝固させることによって、表面の大部分が直径数10nm以下の微細な孔を有する微多孔性支持膜を得ることができる。
上記の微多孔性支持膜の厚みは、複合半透膜の強度およびそれをエレメントにしたときの充填密度に影響を与える。十分な機械的強度および充填密度を得るためには、微多孔性支持膜の厚みは、30〜300μmの範囲内にあることが好ましく、より好ましくは50〜250μmの範囲内である。また、多孔性支持体の厚みは、10〜200μmの範囲内にあることが好ましく、より好ましくは20〜100μmの範囲内である。
微多孔性支持膜の形態は、走査型電子顕微鏡や透過型電子顕微鏡、原子間力顕微鏡により観察できる。例えば走査型電子顕微鏡で微多孔性支持膜を構成する多孔性支持体を観察するのであれば、基材から多孔性支持体を剥がした後、この多孔性支持体を凍結割断法で切断して断面観察のサンプルとする。このサンプルに白金または白金−パラジウムまたは四塩化ルテニウム、好ましくは四塩化ルテニウムを薄くコーティングして3〜6kVの加速電圧で、高分解能電界放射型走査電子顕微鏡(UHR−FE−SEM)で観察する。高分解能電界放射型走査電子顕微鏡は、日立製S−900型電子顕微鏡などが使用できる。得られた電子顕微鏡写真から微多孔性支持膜を構成する多孔性支持体の膜厚や表面孔径を決定する。なお、本発明における厚みや孔径は平均値を意味するものである。
本発明に使用する微多孔性支持膜は、ミリポア社製”ミリポアフィルターVSWP”(商品名)や、東洋濾紙社製”ウルトラフィルターUK10”(商品名)のような各種市販材料から選択することもできるが、”オフィス・オブ・セイリーン・ウォーター・リサーチ・アンド・ディベロップメント・プログレス・レポート”No.359(1968)に記載された方法に従って製造することができる。
本発明において、ポリアミド分離機能層とは、ポリアミドからなる、ホウ素等を選択分離する機能を有する分離機能層である。分離機能層を構成するポリアミドは、多官能アミンと多官能酸ハロゲン化物との重縮合および改質処理により形成することができる。ここで、多官能アミンまたは多官能酸ハロゲン化物の少なくとも一方が3官能以上の化合物を含んでいることが好ましい。
ポリアミド分離機能層の厚みは、十分な分離性能および透過水量を得るために、通常0.01〜1μmの範囲内、好ましくは0.1〜0.5μmの範囲内である。
ここで、多官能アミンとは、一分子中に少なくとも2個の第一級アミノ基および/または第二級アミノ基を有し、そのアミノ基のうち少なくとも1つは第一級アミノ基であるアミンをいい、例えば、2個のアミノ基がオルト位やメタ位、パラ位のいずれかの位置関係でベンゼン環に結合したフェニレンジアミン、キシリレンジアミン、1,3,5−トリアミノベンゼン、1,2,4−トリアミノベンゼン、3,5−ジアミノ安息香酸、3−アミノベンジルアミン、4−アミノベンジルアミンなどの芳香族多官能アミン、エチレンジアミン、プロピレンジアミンなどの脂肪族アミン、1,2−ジアミノシクロヘキサン、1,4−ジアミノシクロヘキサン、4−アミノピペリジン、4−アミノエチルピペラジンなどの脂環式多官能アミン等を挙げることができる。中でも、膜の選択分離性や透過性、耐熱性を考慮すると、多官能アミンは一分子中に2〜4個の第一級アミノ基および/または第二級アミノ基を有する多官能芳香族アミンであることが好ましく、このような多官能芳香族アミンとしては、m−フェニレンジアミン、p−フェニレンジアミン、1,3,5−トリアミノベンゼンが好適に用いられる。中でも、入手の容易性や取り扱いのしやすさから、m−フェニレンジアミン(以下、m−PDAと記す)を用いることがより好ましい。これらの多官能アミンは、単独で用いても、2種以上を同時に用いてもよい。2種以上を同時に用いる場合、上記アミン同士を組み合わせてもよく、上記アミンと一分子中に少なくとも2個の第二級アミノ基を有するアミンを組み合わせてもよい。一分子中に少なくとも2個の第二級アミノ基を有するアミンとして、例えば、ピペラジン、1,3−ビスピペリジルプロパン等を挙げることができる。
多官能酸ハロゲン化物とは、一分子中に少なくとも2個のハロゲン化カルボニル基を有する酸ハロゲン化物をいう。例えば、3官能酸ハロゲン化物では、トリメシン酸クロリド、1,3,5−シクロヘキサントリカルボン酸トリクロリド、1,2,4−シクロブタントリカルボン酸トリクロリドなどを挙げることができ、2官能酸ハロゲン化物では、ビフェニルジカルボン酸ジクロリド、アゾベンゼンジカルボン酸ジクロリド、テレフタル酸クロリド、イソフタル酸クロリド、ナフタレンジカルボン酸クロリドなどの芳香族2官能酸ハロゲン化物、アジポイルクロリド、セバコイルクロリドなどの脂肪族2官能酸ハロゲン化物、シクロペンタンジカルボン酸ジクロリド、シクロヘキサンジカルボン酸ジクロリド、テトラヒドロフランジカルボン酸ジクロリドなどの脂環式2官能酸ハロゲン化物を挙げることができる。多官能アミンとの反応性を考慮すると、多官能酸ハロゲン化物は多官能酸塩化物であることが好ましく、また、膜の選択分離性、耐熱性を考慮すると、一分子中に2〜4個の塩化カルボニル基を有する多官能芳香族酸塩化物であることが好ましい。中でも、入手の容易性や取り扱いのしやすさの観点から、トリメシン酸クロリドを用いるとより好ましい。これらの多官能酸ハロゲン化物は、単独で用いても、2種以上を同時に用いてもよい。
また、上記架橋ポリアミド中に存在するアミノ基を、適宜選択した化学反応による改質処理によって、アゾ基やヒドラゾン基に変換することができる。これにより、アゾ基やヒドラゾン基を上記架橋ポリアミド中に導入することが可能である。例えば、ジアゾニウム塩生成を経由したアゾカップリング反応によりアミノ基をアゾ基に変換することができ、ジアゾニウム塩とカルバニオンの反応によりアミノ基をアゾ基あるいはその互変異性構造のヒドラゾン基に変換することができる。このとき、ジアゾニウム塩またはその誘導体の一部は、水と反応することにより、フェノール性水酸基へと変換される。これにより、フェノール性水酸基を上記架橋ポリアミド中に導入することも可能である。ここで、アゾカップリング反応する化合物として、電子密度の高い芳香族化合物である、芳香族アミン、ヘテロ芳香族アミン、フェノール、アルコキシベンゼン、アミドベンゼン、カルバニオンを生成してジアゾニウム塩と反応する化合物として、高い酸性度を示す炭化水素鎖を持つ化合物である、脂肪族ニトロ化合物、脂肪族ジシアノ化合物、β−ジケトン化合物、β−ケト酢酸化合物、ジカルボン酸化合物、複素環化合物などが挙げられる。これらの化合物が有する末端基により架橋ポリアミド中に種々の官能基が導入される。
本発明者らは鋭意検討を重ねた結果、ポリアミド分離機能層を有する複合半透膜において、一般式(1)あるいは(2)で示される構造を有する複合半透膜が、高いホウ素阻止性能を有し、同時に重金属接触や薬液洗浄への高い耐久性を具備することを見出し、本発明に至った。
本発明者らは鋭意検討を重ねた結果、ポリアミド分離機能層を有する複合半透膜において、一般式(1)あるいは(2)で示される構造を有する複合半透膜が、高いホウ素阻止性能を有し、同時に重金属接触や薬液洗浄への高い耐久性を具備することを見出し、本発明に至った。
本発明の複合半透膜は、微多孔性支持膜上にポリアミド分離機能層を有している。上記架橋ポリアミド中にアゾ基またはヒドラゾン基を含むことによって、ポリアミド機能層表面・内部の孔が緻密化する。緻密化することにより、複合半透膜の塩阻止性能およびホウ素除去率が向上する。
重金属イオンには一般的にπ電子系と相互作用しやすいものが多い。例えば、ポリアミド分離機能層のポリマー末端に電子密度の高い芳香環が存在すると、例えばマンガンや鉄、クロムなどの重金属イオンを多く含む水溶液に接触させた際、重金属イオンに対する配位が起こりやすく、化学構造や分子集合構造の変化の起点になりやすい。また、ポリアミド分離機能層のポリマーがフェノール性水酸基を有すると、アルカリ性条件や高溶質濃度条件下でイオン化して負電荷が発生し、そのクーロン反発により高次構造が変化するため膜性能の変化が起こることがある。
以上を勘案すると、上記ポリアミド分離機能層に一般式(1)あるいは(2)の構造を導入することで、アゾ基またはヒドラゾン基の含有割合を大きくし、末端基を脂肪族に置き換えて末端のπ電子密度を減少させることにより、分子内共役および電荷移動錯体形成を妨害し、好ましくはフェノール性水酸基の含有量を減少させることが可能となるため、高いホウ素除去性能と耐久性を両立できる。
ポリアミド分離機能層中の官能基の種類および官能基量は、例えば、固体NMR、X線光電子分光法(XPS)、赤外分光法(IR)、ラマン分光法を用いて分析することができる。また、ポリアミド機能層中の官能基の種類については、例えば、低分子でモデル化合物を合成し、NMR、赤外分光法(IR)、ラマン分光法を用いて分析することも可能である。
本発明において、ポリアミド分離機能層の黄色度は0以上10.0以下であることが好ましい。黄色度が10.0以下であると、十分な性能安定性と透水量を両立することができる。黄色度はより好ましくは、9.8以下である。黄色度が10.0より大きい場合は、π電子系の共役が大きく重金属と親和するため性能変化が起こりやすいか、あるいは、電子供与基や電子吸引基を有する末端基が多いためポリアミド分離機能層の孔を塞ぎ、透水量が低下する。
黄色度とは、日本工業規格JIS K7373:2006に規定されているポリマーの無色または白色から色相が黄方向に離れる度合いのことで、プラスの量として表される。
なお、ポリアミド分離機能層の黄色度は、カラーメーターにより測定できる。複合半透膜をポリアミド分離機能層面が下になるようにガラス板に乗せてから、微多孔性支持膜のみを溶解する溶媒にて微多孔性支持膜を溶解・除去し、ガラス板上に残る分離機能層試料の透過測定によって測定することができる。なお、複合半透膜をガラス板に乗せる際、後述の微多孔性支持膜を強化するための布帛は、あらかじめ剥離しておくことが好ましい。カラーメーターは、スガ試験器株式会社製SMカラーコンピュータSM−7などが使用できる。
ポリアミド分離機能層の黄色度は、ポリアミド分離機能層が有する芳香環に、電子供与基と電子吸引基を有する構造、および/または共役を延長する構造を持つことで大きくなる。電子供与基とは、例えば、ヒドロキシ基、アミノ基、アルコキシ基が挙げられる。電子吸引基とは、例えば、カルボキシ基、スルホン酸基、アルデヒド基、アシル基、アミノカルボニル基、アミノスルホニル基、シアノ基、ニトロ基、ニトロソ基が挙げられる。共役を延長する構造とは、例えば、多環芳香環、多環複素環、エテニレン基、エチニレン基、アゾ基、イミノ基、アリーレン基、ヘテロアリーレン基およびこれらの構造の組み合わせが挙げられる。これらの構造を持つことにより、ポリアミド分離機能層の黄色度は大きくなっていく。
ポリアミド分離機能層に上記構造を付与するためには、上記構造を持つ化合物をポリアミド分離機能層に担持させる方法、および/またはポリアミド分離機能層を化学的に処理し、上記構造を付与させる方法が挙げられる。長期に上記構造を保持させるためには、ポリアミド分離機能層を化学的に処理し、上記構造を付与させる方法が好ましい。
ポリアミド分離機能層を化学的に処理する方法としては、ポリアミド分離機能層が第一級アミノ基を有する複合半透膜を、第一級アミノ基と反応してジアゾニウム塩またはその誘導体を生成する試薬に接触させる方法が挙げられる。生成したジアゾニウム塩は、さらに、ジアゾニウム塩またはその誘導体と反応しうる試薬と接触させることにより、上記構造を形成する。上記ジアゾニウム塩またはその誘導体と反応する試薬のうち、接触により本発明の一般式(1)または(2)で示される構造を生成する試薬としては、高い酸性度を示す炭化水素鎖を持つ化合物が挙げられる。高い酸性度を示す炭化水素鎖を持つ化合物としては、式(3)の一般式で表される化合物を好適に用いることができる。R3はジアゾニウム塩またはその誘導体と接触して一般式(1)または(2)のR1またはR2のいずれかとなる。R3が反応してR1となった場合R4がR2となり、R3が反応してR2となった場合R4がR1となる。R3、R4、つまり、R1、R2は電子吸引基であることが好ましく、シアノ基、カルボニル基、エステル結合のいずれかを含む有機基であることが特に好ましい。この他にも、脂肪族ニトロ化合物、脂肪族ジシアノ化合物、β−ジケトン化合物、β−ケト酢酸化合物、ジカルボン酸化合物、複素環化合物等の一部を好適に用いることができる。上記化合物の具体的な例としては、例えば、ジニトロメタン、ニトロエタン、ニトロメタン、シアノ酢酸、シアノ酢酸メチル、シアノ酢酸エチル、シアノ酢酸アニライド、シアノアセチルクマロン、マロノニトリル、アセチル酢酸、アセチル酢酸メチル、アセチル酢酸エチル、アセチルアセトン、フェニルアセトニトリル、4−エチルフェニルアセトニトリル、3,4−ジメチルフェニルアセトニトリル、3−トリフルオロフェニルアセトニトリル、フェニル酢酸、フェニル酢酸メチル、フェニル酢酸エチル、4−クロロフェニル酢酸、4−クロロフェニル酢酸メチル、4−クロロフェニル酢酸エチル、4−エチルフェニル酢酸、4−エチルフェニル酢酸メチル、4−フェニル酢酸エチル、フェニルチオアセトニトリル、β−シアノフェニルプロピオニトリル、プロピオンアルデヒド、シクロヘキサノン、2−メチルシクロヘキサノン、ジメドン、アセト酢酸メチル、ピバロイル酢酸エチル、ベンゾイル酢酸メチル、アセト酢酸アニリド、ピバロイル酢酸アニリド、ベンゾイル酢酸アニリド、マロン酸、マロン酸ジメチル、マロン酸ジエチル、マロン酸ジアニリド、5−ピラゾロン、ピラゾロトリアゾール等が挙げられる。これらの化合物のうち脂肪族化合物は分離機能層中の末端構造の電子密度を小さく抑え、重金属イオンとの相互作用による構造の変化を抑制する効果が高いため、特に好適に用いられる。。
また、電子吸引基のpKaは13以下であることが好ましい。pKaが13以下であれば、高い酸性度を有するために開始反応であるプロトンの引き抜きが起こりやすく、反応が進行しやすくなり、競争反応であるジアゾニウム塩またはその誘導体の一部と水との反応によるフェノール性水酸基への変換割合も小さくなる。
次に、本発明の複合半透膜の製造方法について説明する。
本発明の複合半透膜を構成する分離機能層は、前記の多官能アミンを含有する水溶液と、多官能酸ハロゲン化物を含有する水と非混和性の有機溶媒溶液とを用い、微多孔性支持膜の表面で界面重縮合を行うことによりその骨格を形成する工程、形成されたポリアミド分離機能層を水溶液で洗浄する工程、該ポリアミド層が有する第一級アミノ基と反応してジアゾニウム塩またはその誘導体を生成する試薬に接触させる工程、ジアゾニウム塩またはその誘導体と反応する試薬と接触させる工程からなる製造方法により得ることができる。以下、本発明の複合半透膜を製造するための各製造工程を詳細に説明する。
複合半透膜における分離機能層は、例えば、前述の多官能アミンを含有する水溶液と、多官能酸ハロゲン化物を含有する、水と非混和性の有機溶媒溶液とを用い、微多孔性支持膜の表面で界面重縮合を行うことによりその骨格を形成できる。
ここで、多官能アミン水溶液における多官能アミンの濃度は0.1〜20重量%の範囲内であることが好ましく、より好ましくは0.5〜15重量%の範囲内である。多官能アミンの濃度がこの範囲であると十分な塩除去性能および透水性を得ることができる。多官能アミン水溶液には、多官能アミンと多官能酸ハロゲン化物との反応を妨害しないものであれば、界面活性剤や有機溶媒、アルカリ性化合物、酸化防止剤などが含まれていてもよい。界面活性剤は、微多孔性支持膜表面の濡れ性を向上させ、アミン水溶液と非極性溶媒との間の界面張力を減少させる効果がある。有機溶媒は界面重縮合反応の触媒として働くことがあり、添加することにより界面重宿合反応を効率よく行える場合がある。
界面重縮合を微多孔性支持膜上で行うために、まず、上述の多官能アミン水溶液を微多孔性支持膜に接触させる。接触は、微多孔性支持膜面上に均一にかつ連続的に行うことが好ましい。具体的には、例えば、多官能アミン水溶液を微多孔性支持膜にコーティングする方法や微多孔性支持膜を多官能アミン水溶液に浸漬する方法を挙げることができる。微多孔性支持膜と多官能アミン水溶液との接触時間は、1秒〜10分間の範囲内であることが好ましく、10秒〜3分間の範囲内であるとさらに好ましい。
多官能アミン水溶液を微多孔性支持膜に接触させた後は、膜上に液滴が残らないように十分に液切りする。十分に液切りすることで、膜形成後に液滴残存部分が膜欠点となって膜性能が低下することを防ぐことができる。液切りの方法としては、例えば、特開平2−78428号公報に記載されているように、多官能アミン水溶液接触後の微多孔性支持膜を垂直方向に把持して過剰の水溶液を自然流下させる方法や、エアーノズルから窒素などの気流を吹き付け、強制的に液切りする方法などを用いることができる。また、液切り後、膜面を乾燥させて水溶液の水分を一部除去することもできる。
次いで、多官能アミン水溶液接触後の微多孔性支持膜に、多官能酸ハロゲン化物を含む有機溶媒溶液を接触させ、界面重縮合により架橋ポリアミド分離機能層の骨格を形成させる。
有機溶媒溶液中の多官能酸ハロゲン化物の濃度は、0.01〜10重量%の範囲内であると好ましく、0.02〜2.0重量%の範囲内であるとさらに好ましい。0.01重量%以上とすることで十分な反応速度が得られ、また、10重量%以下とすることで副反応の発生を抑制することができるためである。さらに、この有機溶媒溶液にアシル化触媒を含有させると、界面重縮合が促進され、さらに好ましい。
有機溶媒は、水と非混和性であり、かつ多官能酸ハロゲン化物を溶解し、微多孔性支持膜を破壊しないものが望ましく、多官能アミン化合物および多官能酸ハロゲン化物に対して不活性であるものであればよい。好ましい例として、n−ヘキサン、n−オクタン、n−デカンなどの炭化水素化合物が挙げられる。
多官能酸ハロゲン化物の有機溶媒溶液の多官能アミン化合物水溶液相への接触の方法は、多官能アミン水溶液の微多孔性支持膜への被覆方法と同様に行えばよい。多官能酸ハロゲン化物の有機溶媒溶液を接触させて界面重縮合を行い、微多孔性支持膜上に架橋ポリアミドを含む分離機能層を形成したあとは、余剰の溶媒を液切りするとよい。液切りの方法は、例えば、膜を垂直方向に把持して過剰の有機溶媒を自然流下して除去する方法を用いることができる。この場合、垂直方向に把持する時間としては、1〜5分の間にあることが好ましく、1〜3分の間であるとより好ましい。短すぎると分離機能層が完全に形成せず、長すぎると有機溶媒が過乾燥となり欠点が発生しやすく、性能低下を起こしやすい。
さらに、微多孔性支持体上に分離機能層が形成された分離膜は、40〜100℃の範囲内、好ましくは60〜100℃の範囲内で、1〜10分間、より好ましくは2〜8分間熱水処理することで、複合半透膜の溶質阻止性能や透水性をより一層向上させることができる。
次に第一級アミノ基と反応してジアゾニウム塩またはその誘導体を生成する試薬に接触させる。接触させる第一級アミノ基と反応してジアゾニウム塩またはその誘導体を生成する試薬としては、亜硝酸およびその塩、ニトロシル化合物などの水溶液が挙げられる。亜硝酸やニトロシル化合物の水溶液は気体を発生して分解しやすいので、例えば、亜硝酸塩と酸性溶液との反応によって亜硝酸を逐次生成するのが好ましい。一般に、亜硝酸塩は水素イオンと反応して亜硝酸(HNO2)を生成するが、水溶液のpHが7以下、好ましくは5以下、さらに好ましくは4以下で効率よく生成する。中でも、取り扱いの簡便性から水溶液中で塩酸または硫酸と反応させた亜硝酸ナトリウムの水溶液が特に好ましい。
上記第一級アミノ基と反応してジアゾニウム塩またはその誘導体を生成する試薬中の亜硝酸や亜硝酸塩の濃度は、好ましくは0.01重量%以上1重量%以下の範囲であり、より好ましくは0.05重量%以上0.5重量%以下の範囲である。0.01重量%以上の濃度であれば十分な効果が得られ、濃度が1重量%以下であれば溶液の取扱いが容易である。
亜硝酸水溶液の温度は15℃〜45℃が好ましい。15℃以上であれば十分に反応が速く、45℃以下であれば亜硝酸の分解が起こり難いため取り扱いが容易である。
亜硝酸水溶液との接触時間は、ジアゾニウム塩および/またはその誘導体が生成する時間であればよく、高濃度では短時間で処理が可能であるが、低濃度であると長時間必要である。そのため、上記濃度の溶液では10分間以内であることが好ましく、3分間以内であることがさらに好ましい。また、接触させる方法は特に限定されず、該試薬の溶液を塗布しても、該試薬の溶液に該複合半透膜を浸漬してもよい。該試薬の溶媒は該試薬が溶解し、該複合半透膜が侵食されなければ、いかなる溶媒を用いてもかまわない。また、溶液には、第一級アミノ基と試薬との反応を妨害しないものであれば、界面活性剤や酸性化合物、アルカリ性化合物などが含まれていてもよい。
亜硝酸水溶液との接触時間は、ジアゾニウム塩および/またはその誘導体が生成する時間であればよく、高濃度では短時間で処理が可能であるが、低濃度であると長時間必要である。そのため、上記濃度の溶液では10分間以内であることが好ましく、3分間以内であることがさらに好ましい。また、接触させる方法は特に限定されず、該試薬の溶液を塗布しても、該試薬の溶液に該複合半透膜を浸漬してもよい。該試薬の溶媒は該試薬が溶解し、該複合半透膜が侵食されなければ、いかなる溶媒を用いてもかまわない。また、溶液には、第一級アミノ基と試薬との反応を妨害しないものであれば、界面活性剤や酸性化合物、アルカリ性化合物などが含まれていてもよい。
接触により生成したジアゾニウム塩またはその誘導体の一部は、微多孔性支持膜や分離機能層を形成する構造の芳香環、または分離機能層に保持した第一級アミノ基を有する化合物の芳香環とも反応し、アゾ基を形成する。それによりポリアミド機能層表面・内部の孔径が縮小し、ホウ素除去率の向上が望める。
次にジアゾニウム塩またはその誘導体と反応しうる試薬を含む溶液と接触させる。試薬は単一で用いてもよく、複数混合させて用いてもよく、異なる試薬に複数回接触させてもよい。これらのジアゾニウム塩またはその誘導体と反応する試薬を接触させる濃度と時間は、目的の効果を得るために適宜調節することができる。接触させる温度は10〜90℃が望ましい。10℃未満の時にはジアゾカップリング反応の速度が遅く、ジアゾニウム塩またはその誘導体の一部が水と反応することによりフェノール性水酸基量が増大するため、前述の理由で耐アルカリ性が低下し、望む効果が得られない。また90℃より高温ではポリマーの収縮がおこり透過水量が低下するため好ましくない。
本発明の複合半透膜は、プラスチックネットなどの原水流路材と、トリコットなどの透過水流路材と、必要に応じて耐圧性を高めるためのフィルムと共に、多数の孔を穿設した筒状の集水管の周りに巻回され、スパイラル型の複合半透膜エレメントとして好適に用いられる。さらに、このエレメントを直列または並列に接続して圧力容器に収納した複合半透膜モジュールとすることもできる。
また、上記の複合半透膜やそのエレメント、モジュールは、それらに原水を供給するポンプや、その原水を前処理する装置などと組み合わせて、流体分離装置を構成することができる。この分離装置を用いることにより、原水を飲料水などの透過水と膜を透過しなかった濃縮水とに分離して、目的にあった水を得ることができる。
流体分離装置の操作圧力は高い方が塩除去率は向上するが、運転に必要なエネルギーも増加すること、また、複合半透膜の耐久性を考慮すると、複合半透膜に被処理水を透過する際の操作圧力は、0.5MPa以上、10MPa以下が好ましい。供給水温度は、高くなると塩除去率が低下するが、低くなるにしたがい膜透過流束も減少するので、5℃以上、45℃以下が好ましい。
また、供給水pHは、高くなると海水などの高塩濃度の供給水の場合、マグネシウムなどのスケールが発生する恐れがあり、また、高pH運転による膜の劣化が懸念されるため、中性領域での運転が好ましい。
本発明に係る複合半透膜によって処理される原水としては、海水、かん水、排水等の500mg/L以上100g/L以下のTDS(Total Dissolved Solids:総溶解固形分)を含有する液状混合物が挙げられる。一般に、TDSは総溶解固形分量を指し、「質量÷体積」あるいは「重量比」で表される。定義によれば、0.45ミクロンのフィルターで濾過した溶液を39.5℃以上40.5℃以下の温度で蒸発させ残留物の重さから算出できるが、より簡便には実用塩分(S)から換算する。
なお、本発明の複合半透膜は、高い耐重金属性、耐アルカリ性を同時に満たすことを特徴とするが、耐薬品性の指標については、重金属イオンを含むpH12の水溶液への耐性を指標とするのが適当である。重金属イオンを多く含有する原水を用いて運転した場合膜面に重金属イオンが吸着していることが考えられ、アルカリ洗浄時に膜面に重金属イオンとアルカリが同時に接触することとなる。重金属イオンとして海水に比較的多く含まれるのは、マンガンイオン、鉄イオン、クロムイオンであり、その含有量は多くとも全価数合計でそれぞれ0.02〜0.2ppm程度である。また、アルカリ洗浄の薬剤としては、水酸化ナトリウム、水酸化カリウムなどが用いられ、一般的にpH12付近で洗浄する。そのため、重金属イオンとしてマンガンイオン、鉄イオン、クロムイオンをそれぞれ2ppmずつ混合したpH12のアルカリ水溶液接触前後での膜透過流束及びホウ素除去率の変化が小さいことを示せば、重金属やアルカリによって膜が劣化しにくいことが担保されるためである。
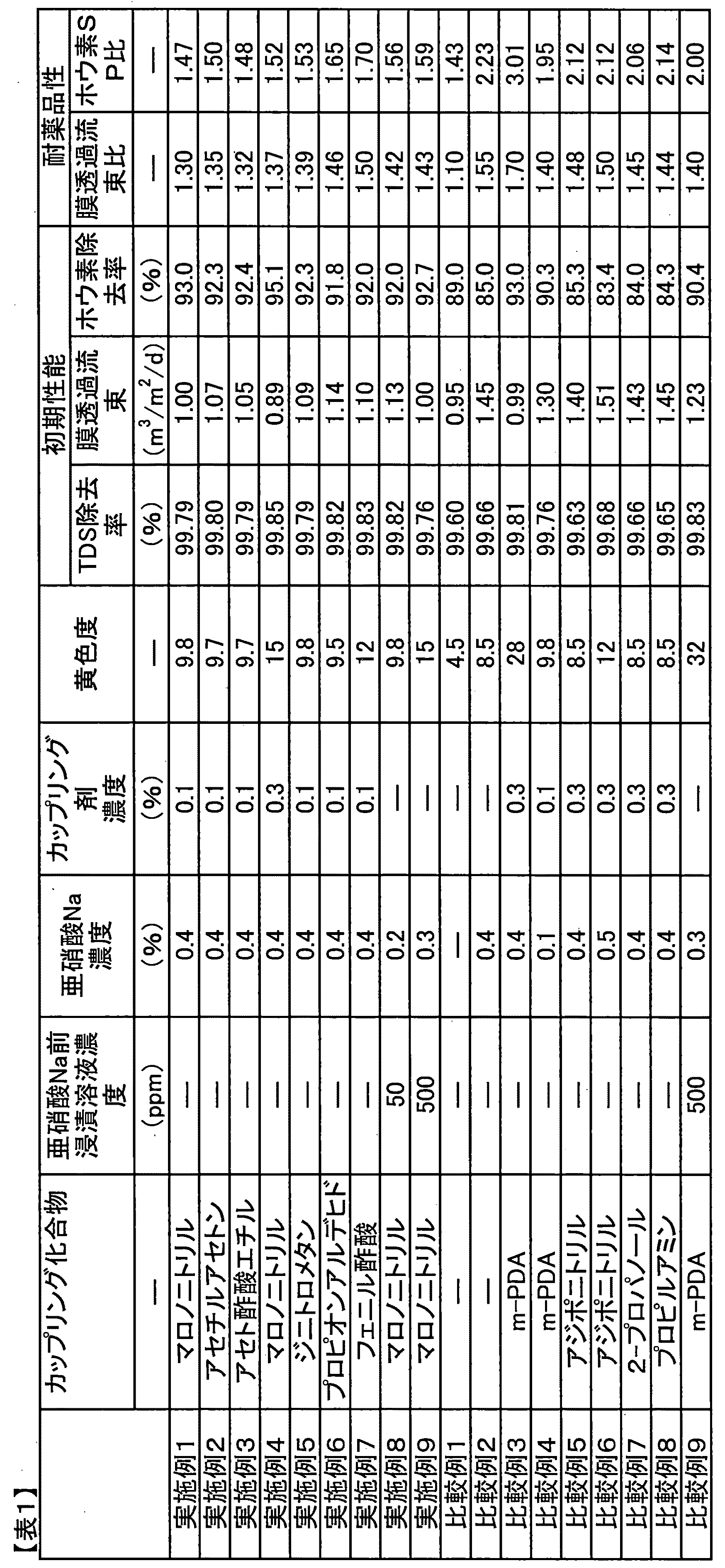
以下に実施例によって本発明をさらに詳細に説明するが、本発明はこれらの実施例によってなんら限定されるものではない。
比較例、実施例における分離機能層の黄色度は以下のように測定した。
(黄色度)
複合半透膜を室温で8時間乾燥したのち基材を剥離し、分離機能層面が下になるようにガラス板に乗せてから、ジクロロメタンを用いて微多孔性支持膜を溶解・除去した。ガラス板上に残った分離機能層の黄色度を、スガ試験器株式会社製SMカラーコンピュータSM−7により測定した。
複合半透膜を室温で8時間乾燥したのち基材を剥離し、分離機能層面が下になるようにガラス板に乗せてから、ジクロロメタンを用いて微多孔性支持膜を溶解・除去した。ガラス板上に残った分離機能層の黄色度を、スガ試験器株式会社製SMカラーコンピュータSM−7により測定した。
比較例、実施例における複合半透膜の各種特性は、複合半透膜に、温度25℃、pH6.5に調整した海水(TDS濃度約3.5%、ホウ素濃度約5ppm)を操作圧力5.5MPaで供給して膜ろ過処理を24時間行ない、その後の透過水、供給水の水質を測定することにより求めた。
(脱塩率(TDS除去率))
TDS除去率(%)=100×(1−(透過水中のTDS濃度/供給水中のTDS濃度))
(膜透過流束)
供給水(海水)の膜透過水量を、膜面1平方メートルあたり、1日あたりの透水量(立方メートル)でもって膜透過流束(m3/m2/日)を表した。
TDS除去率(%)=100×(1−(透過水中のTDS濃度/供給水中のTDS濃度))
(膜透過流束)
供給水(海水)の膜透過水量を、膜面1平方メートルあたり、1日あたりの透水量(立方メートル)でもって膜透過流束(m3/m2/日)を表した。
(ホウ素除去率)
供給水と透過水中のホウ素濃度を、日本工業規格JIS K0102:2013に規定の方法を用いて、ICP発光分析装置(日立製作所製 P−4010)で分析し、次の式から求めた。
ホウ素除去率(%)=100×(1−(透過水中のホウ素濃度/供給水中のホウ素濃度))
(耐薬品性)
過マンガン酸カリウム、塩化マンガン(II)四水和物、塩化鉄(II)四水和物、塩化鉄(III)六水和物、塩化クロム(III)六水和物、二クロム酸カリウムをそれぞれ1ppm含むpH12の水酸化ナトリウム水溶液を調製し、複合半透膜を室温で72時間浸漬した。耐薬品性は浸漬前後での膜透過流束比とホウ素SP比から求めた。
膜透過流束比=浸漬後の膜透過流束/浸漬前の膜透過流束
ホウ素SP比=(100−浸漬後のホウ素除去率)/(100−浸漬前のホウ素除去率)
なお、SPとはSubstance Peameation:物質透過の略である。
供給水と透過水中のホウ素濃度を、日本工業規格JIS K0102:2013に規定の方法を用いて、ICP発光分析装置(日立製作所製 P−4010)で分析し、次の式から求めた。
ホウ素除去率(%)=100×(1−(透過水中のホウ素濃度/供給水中のホウ素濃度))
(耐薬品性)
過マンガン酸カリウム、塩化マンガン(II)四水和物、塩化鉄(II)四水和物、塩化鉄(III)六水和物、塩化クロム(III)六水和物、二クロム酸カリウムをそれぞれ1ppm含むpH12の水酸化ナトリウム水溶液を調製し、複合半透膜を室温で72時間浸漬した。耐薬品性は浸漬前後での膜透過流束比とホウ素SP比から求めた。
膜透過流束比=浸漬後の膜透過流束/浸漬前の膜透過流束
ホウ素SP比=(100−浸漬後のホウ素除去率)/(100−浸漬前のホウ素除去率)
なお、SPとはSubstance Peameation:物質透過の略である。
(参考例1)
ポリスルホン18重量%のDMF溶液を、各溶媒および溶質の混合物を攪拌しながら90℃で2時間加熱保持することで調製した。長繊維からなるポリエステル不織布上にポリスルホン溶液を200μmの厚みで室温(25℃)でキャストし、ただちに純水中に浸漬して5分間静置することによって微多孔性支持膜(厚さ210〜215μm)を作製した。
ポリスルホン18重量%のDMF溶液を、各溶媒および溶質の混合物を攪拌しながら90℃で2時間加熱保持することで調製した。長繊維からなるポリエステル不織布上にポリスルホン溶液を200μmの厚みで室温(25℃)でキャストし、ただちに純水中に浸漬して5分間静置することによって微多孔性支持膜(厚さ210〜215μm)を作製した。
得られた微多孔性支持膜を、m−PDAの5.5重量%水溶液中に2分間浸漬し、該支持膜を垂直方向にゆっくりと引き上げ、エアーノズルから窒素を吹き付け微多孔性支持膜表面から余分な水溶液を取り除いた後、トリメシン酸クロリド0.165重量%を含む25℃のn−デカン溶液を表面が完全に濡れるように塗布して1分間静置した。次に、膜から余分な溶液を除去するために膜を1分間垂直に保持して液切りした後、90℃の熱水で2分間洗浄した。
(実施例1)
参考例1で得られた複合半透膜を、pH3.0、35℃に調整した、亜硝酸ナトリウム0.4重量%水溶液に40秒間浸漬した。亜硝酸ナトリウム水溶液のpHの調整は硫酸を用いて行った。次に、この複合半透膜を30℃のマロノニトリル0.1重量%水溶液に20秒間浸漬し、ジアゾカップリング反応を行った。複合半透膜をマロノニトリル水溶液から取り出した後、35℃の0.1重量%の亜硫酸ナトリウム水溶液に2分間浸漬した。このようにして得られた複合半透膜を評価したところ、TDS除去率、膜透過流束、ホウ素除去率はそれぞれ表1に示す値であった。また、複合半透膜の耐薬品性を評価したところ、薬品接触前後での膜透過流束比、ホウ素SP比は表1に示す通りであった。
参考例1で得られた複合半透膜を、pH3.0、35℃に調整した、亜硝酸ナトリウム0.4重量%水溶液に40秒間浸漬した。亜硝酸ナトリウム水溶液のpHの調整は硫酸を用いて行った。次に、この複合半透膜を30℃のマロノニトリル0.1重量%水溶液に20秒間浸漬し、ジアゾカップリング反応を行った。複合半透膜をマロノニトリル水溶液から取り出した後、35℃の0.1重量%の亜硫酸ナトリウム水溶液に2分間浸漬した。このようにして得られた複合半透膜を評価したところ、TDS除去率、膜透過流束、ホウ素除去率はそれぞれ表1に示す値であった。また、複合半透膜の耐薬品性を評価したところ、薬品接触前後での膜透過流束比、ホウ素SP比は表1に示す通りであった。
(実施例2〜7)
カップリング化合物、亜硝酸ナトリウム水溶液濃度、及びカップリング水溶液濃度を表1に記載した条件に変更した以外は、実施例1と同様にして複合半透膜を作製した。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
カップリング化合物、亜硝酸ナトリウム水溶液濃度、及びカップリング水溶液濃度を表1に記載した条件に変更した以外は、実施例1と同様にして複合半透膜を作製した。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
(実施例8)
参考例1で得られた複合半透膜を、25℃に調製したマロノニトリル50ppmの水溶液に120分間浸漬した後、硫酸によりpH3.0に調整した0.2重量%の亜硝酸ナトリウム水溶液により35℃で1分間処理した。複合半透膜を亜硝酸水溶液から取り除いた後、水洗し0.1重量%の亜硫酸ナトリウム水溶液に2分間浸漬した。このようにして得られた複合半透膜の初期性能及び耐薬品性を評価したところ、それぞれ表1に示す値であった。
参考例1で得られた複合半透膜を、25℃に調製したマロノニトリル50ppmの水溶液に120分間浸漬した後、硫酸によりpH3.0に調整した0.2重量%の亜硝酸ナトリウム水溶液により35℃で1分間処理した。複合半透膜を亜硝酸水溶液から取り除いた後、水洗し0.1重量%の亜硫酸ナトリウム水溶液に2分間浸漬した。このようにして得られた複合半透膜の初期性能及び耐薬品性を評価したところ、それぞれ表1に示す値であった。
(実施例9)
亜硝酸の濃度及び亜硝酸ナトリウム水溶液の濃度を表1に記載した条件に変更した以外は、実施例8と同様にして複合半透膜を作製した。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
亜硝酸の濃度及び亜硝酸ナトリウム水溶液の濃度を表1に記載した条件に変更した以外は、実施例8と同様にして複合半透膜を作製した。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
(比較例1)
表1に記載した条件を変更した以外は、実施例1と同様にして複合半透膜を作製した。すなわち、亜硝酸ナトリウム水溶液による処理及びジアゾカップリング反応を行っていない。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
表1に記載した条件を変更した以外は、実施例1と同様にして複合半透膜を作製した。すなわち、亜硝酸ナトリウム水溶液による処理及びジアゾカップリング反応を行っていない。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
(比較例2)
表1に記載した条件を変更した以外は、実施例1と同様にして複合半透膜を作製した。すなわち、亜硝酸ナトリウム水溶液による処理のみ行い、ジアゾカップリング反応を行っていない。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
表1に記載した条件を変更した以外は、実施例1と同様にして複合半透膜を作製した。すなわち、亜硝酸ナトリウム水溶液による処理のみ行い、ジアゾカップリング反応を行っていない。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
(比較例3〜8)
カップリング化合物、亜硝酸ナトリウム水溶液濃度、及びカップリング水溶液濃度を表1に記載した条件に変更した以外は、実施例1と同様にして複合半透膜を作製した。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
カップリング化合物、亜硝酸ナトリウム水溶液濃度、及びカップリング水溶液濃度を表1に記載した条件に変更した以外は、実施例1と同様にして複合半透膜を作製した。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
(比較例9)
表1に記載した条件を変更した以外は、実施例8と同様にして複合半透膜を作製した。すなわち、参考例1で得られた複合半透膜を、25℃に調製したm−PDA500ppmの水溶液に120分間浸漬した後、亜硝酸ナトリウム水溶液による処理をおこなった。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
表1に記載した条件を変更した以外は、実施例8と同様にして複合半透膜を作製した。すなわち、参考例1で得られた複合半透膜を、25℃に調製したm−PDA500ppmの水溶液に120分間浸漬した後、亜硝酸ナトリウム水溶液による処理をおこなった。得られた複合半透膜の初期性能及び耐薬品性を評価したところ、表1に示す性能を示した。
表から読み取れる通り、比較例1、2、5〜8では初期のホウ素阻止性能が実施例に比べ極端に低い。また、比較例3、4及び公知技術でおこなった比較例9に比して実施例では耐重金属性、耐アルカリ性が高い複合半透膜が得られることがわかる。
本発明の複合半透膜は、特に、かん水や海水の脱塩に好適に用いることができる。
Claims (6)
- 微多孔性支持膜と、微多孔性支持膜上に設けられたポリアミド分離機能層とからなる複合半透膜であって、前記ポリアミド分離機能層が下記式(1)または(2)で示される構造を含むことを特徴とする複合半透膜。
- 前記ポリアミド分離機能層の黄色度が10.0以下であることを特徴とする請求項1に記載の複合半透膜。
- 前記R1、R2が電子求引基を含むことを特徴とする請求項1または2に記載の複合半透膜。
- 前記R1、R2がシアノ基、カルボニル基、エステル結合のいずれかを含む有機基であることを特徴とする、請求項1〜3のいずれかに記載の複合半透膜。
- 微多孔性支持膜上に多官能第一級芳香族アミンと多官能酸ハロゲン化物との重縮合反応によるポリアミド層を形成する工程、該ポリアミド層が有する第一級芳香族アミノ基を、該アミノ基と反応してジアゾニウム塩またはその誘導体を生成する試薬に接触させ改質処理する工程、および、前記ポリアミド層を下記式(3)を含む溶液に接触させ改質処理する工程とからなる複合半透膜の製造方法。
- 前記R3、R4がシアノ基、カルボニル基、エステル結合のいずれかを含む有機基であることを特徴とする請求項5に記載の複合半透膜の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014133606A JP2016010771A (ja) | 2014-06-30 | 2014-06-30 | 複合半透膜 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014133606A JP2016010771A (ja) | 2014-06-30 | 2014-06-30 | 複合半透膜 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2016010771A true JP2016010771A (ja) | 2016-01-21 |
Family
ID=55227847
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014133606A Pending JP2016010771A (ja) | 2014-06-30 | 2014-06-30 | 複合半透膜 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2016010771A (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114749029A (zh) * | 2022-03-28 | 2022-07-15 | 浙江理工大学 | 一种聚酰胺复合反渗透膜修复方法 |
| CN115475540A (zh) * | 2022-09-29 | 2022-12-16 | 万华化学集团股份有限公司 | 一种聚酰胺复合膜及其制备方法和应用 |
-
2014
- 2014-06-30 JP JP2014133606A patent/JP2016010771A/ja active Pending
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114749029A (zh) * | 2022-03-28 | 2022-07-15 | 浙江理工大学 | 一种聚酰胺复合反渗透膜修复方法 |
| CN114749029B (zh) * | 2022-03-28 | 2023-01-03 | 浙江理工大学 | 一种聚酰胺复合反渗透膜修复方法 |
| CN115475540A (zh) * | 2022-09-29 | 2022-12-16 | 万华化学集团股份有限公司 | 一种聚酰胺复合膜及其制备方法和应用 |
| CN115475540B (zh) * | 2022-09-29 | 2024-05-03 | 万华化学集团股份有限公司 | 一种聚酰胺复合膜及其制备方法和应用 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6136266B2 (ja) | 複合半透膜 | |
| JP5741431B2 (ja) | 複合半透膜およびその製造方法 | |
| JP5895838B2 (ja) | 分離膜エレメントおよび複合半透膜の製造方法 | |
| JP5807547B2 (ja) | 半透膜およびその製造方法 | |
| JP6402627B2 (ja) | 複合半透膜 | |
| JP6295949B2 (ja) | 複合半透膜およびその製造方法 | |
| KR102293090B1 (ko) | 복합 반투막 | |
| JP2010094641A (ja) | 複合半透膜の処理方法 | |
| JP2013223861A (ja) | 複合半透膜 | |
| JP7342528B2 (ja) | 複合半透膜および複合半透膜の製造方法 | |
| JP5287353B2 (ja) | 複合半透膜 | |
| JP5177056B2 (ja) | 複合半透膜 | |
| JP2016010771A (ja) | 複合半透膜 | |
| JP5062136B2 (ja) | 複合半透膜の製造方法 | |
| JP2009262089A (ja) | 複合半透膜の製造方法 | |
| JP5120006B2 (ja) | 複合半透膜の製造方法 | |
| JP6511808B2 (ja) | 複合半透膜 | |
| JP5050335B2 (ja) | 複合半透膜の製造方法 | |
| WO2016136966A1 (ja) | 複合半透膜 | |
| JP2012143750A (ja) | 複合半透膜の製造方法 | |
| JP4872800B2 (ja) | 複合半透膜の処理方法及び塩処理済み複合半透膜の製造方法 | |
| JP2008260009A (ja) | 複合半透膜の製造方法 | |
| JP2009220023A (ja) | 複合半透膜の製造方法 | |
| JP5263104B2 (ja) | 複合半透膜の製造方法 |