JP2012504154A - シュードプロリンジペプチド - Google Patents
シュードプロリンジペプチド Download PDFInfo
- Publication number
- JP2012504154A JP2012504154A JP2011529526A JP2011529526A JP2012504154A JP 2012504154 A JP2012504154 A JP 2012504154A JP 2011529526 A JP2011529526 A JP 2011529526A JP 2011529526 A JP2011529526 A JP 2011529526A JP 2012504154 A JP2012504154 A JP 2012504154A
- Authority
- JP
- Japan
- Prior art keywords
- process according
- formula
- acid
- hydrogen
- water
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 108010016626 Dipeptides Proteins 0.000 title claims abstract description 15
- 238000000034 method Methods 0.000 claims abstract description 27
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 claims abstract description 21
- 125000006239 protecting group Chemical group 0.000 claims abstract description 16
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 13
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 13
- 239000001257 hydrogen Substances 0.000 claims abstract description 13
- 150000003862 amino acid derivatives Chemical class 0.000 claims abstract description 7
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims abstract description 7
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 claims abstract description 4
- 150000001370 alpha-amino acid derivatives Chemical class 0.000 claims abstract description 4
- 235000008206 alpha-amino acids Nutrition 0.000 claims abstract description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 4
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 54
- 239000000203 mixture Substances 0.000 claims description 23
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 20
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 17
- 229960001153 serine Drugs 0.000 claims description 16
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 claims description 15
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 14
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 claims description 13
- 239000004473 Threonine Substances 0.000 claims description 13
- 229960002898 threonine Drugs 0.000 claims description 13
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 claims description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 10
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 10
- 239000003960 organic solvent Substances 0.000 claims description 10
- 238000006243 chemical reaction Methods 0.000 claims description 9
- 150000001875 compounds Chemical class 0.000 claims description 9
- 239000011541 reaction mixture Substances 0.000 claims description 9
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 claims description 8
- 238000006798 ring closing metathesis reaction Methods 0.000 claims description 8
- 239000012190 activator Substances 0.000 claims description 7
- HEWZVZIVELJPQZ-UHFFFAOYSA-N 2,2-dimethoxypropane Chemical compound COC(C)(C)OC HEWZVZIVELJPQZ-UHFFFAOYSA-N 0.000 claims description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 6
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 claims description 6
- 239000003377 acid catalyst Substances 0.000 claims description 6
- 150000007529 inorganic bases Chemical class 0.000 claims description 6
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 5
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 claims description 4
- FUOOLUPWFVMBKG-UHFFFAOYSA-N 2-Aminoisobutyric acid Chemical compound CC(C)(N)C(O)=O FUOOLUPWFVMBKG-UHFFFAOYSA-N 0.000 claims description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 4
- 239000002253 acid Substances 0.000 claims description 4
- 229910052500 inorganic mineral Inorganic materials 0.000 claims description 4
- 239000011707 mineral Substances 0.000 claims description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 4
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 claims description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 claims description 3
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 claims description 3
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 claims description 3
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 claims description 3
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 claims description 3
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 claims description 3
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 claims description 3
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 claims description 3
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 claims description 3
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 claims description 3
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 claims description 3
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 claims description 3
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 claims description 3
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 claims description 3
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims description 3
- 239000004472 Lysine Substances 0.000 claims description 3
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 claims description 3
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 claims description 3
- 235000004279 alanine Nutrition 0.000 claims description 3
- 235000009582 asparagine Nutrition 0.000 claims description 3
- 229960001230 asparagine Drugs 0.000 claims description 3
- 235000003704 aspartic acid Nutrition 0.000 claims description 3
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 claims description 3
- 235000013922 glutamic acid Nutrition 0.000 claims description 3
- 239000004220 glutamic acid Substances 0.000 claims description 3
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 claims description 3
- 229960000310 isoleucine Drugs 0.000 claims description 3
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 claims description 3
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 claims description 3
- 150000003839 salts Chemical class 0.000 claims description 3
- 125000003944 tolyl group Chemical group 0.000 claims description 3
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 claims description 3
- 239000004474 valine Substances 0.000 claims description 3
- LJCZNYWLQZZIOS-UHFFFAOYSA-N 2,2,2-trichlorethoxycarbonyl chloride Chemical compound ClC(=O)OCC(Cl)(Cl)Cl LJCZNYWLQZZIOS-UHFFFAOYSA-N 0.000 claims description 2
- 239000004475 Arginine Substances 0.000 claims description 2
- 239000004471 Glycine Substances 0.000 claims description 2
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 claims description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 claims description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 claims description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 claims description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 claims description 2
- 239000003513 alkali Substances 0.000 claims description 2
- 239000008346 aqueous phase Substances 0.000 claims description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 claims description 2
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 claims description 2
- 150000004649 carbonic acid derivatives Chemical class 0.000 claims description 2
- 238000002425 crystallisation Methods 0.000 claims description 2
- 230000008025 crystallization Effects 0.000 claims description 2
- 235000018417 cysteine Nutrition 0.000 claims description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 claims description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 claims description 2
- 229930182817 methionine Natural products 0.000 claims description 2
- 239000012074 organic phase Substances 0.000 claims description 2
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 claims description 2
- 230000009466 transformation Effects 0.000 claims 2
- 229910001854 alkali hydroxide Inorganic materials 0.000 claims 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 claims 1
- 108090000765 processed proteins & peptides Proteins 0.000 abstract description 5
- 238000004519 manufacturing process Methods 0.000 abstract description 2
- 230000002441 reversible effect Effects 0.000 abstract description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 16
- 239000012044 organic layer Substances 0.000 description 15
- 239000010410 layer Substances 0.000 description 14
- 239000000243 solution Substances 0.000 description 13
- 239000007864 aqueous solution Substances 0.000 description 11
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 8
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 229910052808 lithium carbonate Inorganic materials 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 239000012455 biphasic mixture Substances 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- REITVGIIZHFVGU-IBGZPJMESA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-3-[(2-methylpropan-2-yl)oxy]propanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](COC(C)(C)C)C(O)=O)C3=CC=CC=C3C2=C1 REITVGIIZHFVGU-IBGZPJMESA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 235000010755 mineral Nutrition 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- -1 9H-fluoren-9-yl-methoxycarbonylamino Chemical group 0.000 description 2
- 238000010268 HPLC based assay Methods 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 150000001413 amino acids Chemical group 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 238000010533 azeotropic distillation Methods 0.000 description 2
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 2
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 239000003039 volatile agent Substances 0.000 description 2
- SKWOOIBKXDYTKR-VXKWHMMOSA-N (4s)-3-[(2s)-2-[9h-fluoren-9-yl(methoxycarbonyl)amino]-3-[(2-methylpropan-2-yl)oxy]propanoyl]-2,2-dimethyl-1,3-oxazolidine-4-carboxylic acid Chemical compound O=C([C@H](COC(C)(C)C)N(C(=O)OC)C1C2=CC=CC=C2C2=CC=CC=C21)N1[C@H](C(O)=O)COC1(C)C SKWOOIBKXDYTKR-VXKWHMMOSA-N 0.000 description 1
- 125000004066 1-hydroxyethyl group Chemical group [H]OC([H])([*])C([H])([H])[H] 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000010979 pH adjustment Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 235000011118 potassium hydroxide Nutrition 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000007614 solvation Methods 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/02—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length in solution
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/06—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/04—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D263/06—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by oxygen atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/0606—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing heteroatoms not provided for by C07K5/06086 - C07K5/06139, e.g. Ser, Met, Cys, Thr
- C07K5/06069—Ser-amino acid
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Analytical Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description
で示される化合物の新規な製造方法に関するものである。
[式中、R1、R2、R5、R6、R7及びR8は、上記PCT公報に定義されている]で示されるアンモニウム塩中間体を経由して達成される。
[式中、R1は、α−アミノ酸の側鎖であり、R2は、アミノ保護基であり、R3及びR4は、独立して水素(但し、R3及びR4の両方ともが水素ではない)又はC1−4−アルキルより選択され、R5は、水素又はメチルである]で示される化合物の製造方法であって、
a)式
[式中、R1及びR2は上記と同義である]
で示されるアミノ酸誘導体を、セリン又はトレオニンと共に、式
で示されるジペプチドに変換すること(それに関して活性化剤として水溶性カルボジイミドを使用する)、及び
b)酸触媒の存在下で、式
[式中、R3及びR4は、独立して水素又はC1−4−アルキルより選択されるが、但し、R3及びR4の両方ともが水素ではなく、R9a及びR9bは、独立してC1−4−アルキルである]で示される化合物と共に式IIIで示されるジペプチドの閉環を実施すること
を含む。
第1工程a)では、式
[式中、R1及びR2は、上記と同義である]
で示されるアミノ酸誘導体をセリン又はトレオニンと共に式
で示されるジペプチドに変換する(それに関して活性化剤として水溶性カルボジイミドを使用する)。
工程b)は、式
[式中、R3、R4、R9a及びR9bは、上記と同義である]
で示される化合物と共に式IIIで示されるジペプチドの閉環を酸触媒の存在下で、実施することを必要とする。
a)pHを7.0〜9.0の範囲、好ましくは7.5〜8.5の範囲に維持しながら反応混合物を水で抽出すること
b)pHを5.5〜6.0の範囲、好ましくは5.5〜5.7の範囲に維持しながら水相を水不混和性有機溶媒で抽出すること、
c)式Iで示される目的生成物を有機相から単離すること、及び場合により
d)式Iで示される目的生成物を有機溶媒中で結晶化させること
を含む手順を適用しながら実施することができる。
(S,S)−3−[3−tert−ブトキシ−2−(9H−フルオレン−9−イル−メトキシカルボニルアミノ)−プロピオニル]−2,2−ジメチル−オキサゾリジン−4−カルボン酸の合成
200mLのTHF中の16.1gのN−ヒドロキシスクシンイミド及び40.0gのFmoc−L−Ser(tBu)−OHの溶液を、80mLのDMF及び80mLのTHF中の26.0gの1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩の懸濁液に20℃で30〜60分以内に添加した。結果として得られた混合物を周囲温度で4時間撹拌し、次にそれを、240gの水中の8.75gの水酸化リチウム一水和物、6.1gの炭酸リチウム及び33.2gのL−セリンの予冷(−5℃)懸濁液に30〜45分以内に添加した。結果として得られた混合物を30分以内に室温まで温め、次に、この温度でもう1時間撹拌した。次に、その混合物を−5℃に冷却し、約150gの硫酸(20%水溶液)でpHを8.5から2.0〜2.5に調整した。その二相性混合物を室温まで温め、次に下の水層を分離した。その水層を200mLのトルエンで抽出した。合わせた有機層を150mLのトルエンで希釈し、次に150mLの水で5回洗浄した。トルエン及びTHFを用いた共沸蒸留により、有機層から水を除去した。無水(<0.05%)のトルエン/THF溶液(約500mL)を1.00gのメタンスルホン酸で処理した。その混合物に660mLのトルエン中の100gの2,2−ジメトキシプロパンの溶液を6〜10時間以内に添加した。全体の調合の間に、反応容積を一定(約600mL)に維持しながら揮発性物質を減圧下(80〜30mbar)で20〜28℃の温度で蒸発させて除いた。完全に添加した後に、その混合物を約500mLの最終容積まで濃縮し、次に1.35gのトリエチルアミンで処理した。水(50mL)を添加し、層を分離した。有機層を250gの重炭酸ナトリウム(5%水溶液)で処理した。その二相性混合物(pH約7.5)を35〜40℃に加温し、この温度で30〜45分間撹拌した。層を分離し、有機層を70gの重炭酸ナトリウム(5%水溶液)で3回抽出した。生成物を含有する水層を合わせたものを35〜40℃で360mLのトルエンで処理し、約50gの硫酸(20%水溶液)の滴加によりpHを5.5に調整した。水層を分離し、有機層を50gの水で2回洗浄した。結果として得られた有機層を周囲温度に冷却し、100mLの水で希釈した。数滴の硫酸(20%水溶液)の添加によりpHを4に調整した。下の水層を除去し、有機層を80gの水で2回洗浄した。有機層を乾燥するまで濃縮した。残渣を400mLのイソプロパノールで処理し、結果として得られた溶液を乾燥するまで濃縮した。残渣を90mLのイソプロパノール及び90mLのヘプタンで希釈し、その混合物を50℃に加温し、透明な溶液を実現した。400mLのヘプタンを3〜4時間以内に添加した。次に、その混合物を13〜16時間以内に−10℃に冷却し、結果として得られた懸濁液をこの温度で少なくとも4時間撹拌した。結晶を濾過して取り出し、80mLの予冷ヘプタンで洗浄し、40〜50℃/<30mbarで乾燥させて、無色結晶として31.6g(60%)の(S,S)−3−[3−tert−ブトキシ−2−(9H−フルオレン−9−イル−メトキシカルボニルアミノ)−プロピオニル]−2,2−ジメチル−オキサゾリジン−4−カルボン酸を得た(HPLCアッセイで99.0%(m/m))。
200mLのTHF中の16.1gのN−ヒドロキシスクシンイミド及び40.0gのFmoc−L−Ser(tBu)−OHの溶液を、80mLのDMF及び80mLのTHF中の26.0gの1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩の懸濁液に20℃で30〜60分以内に添加した。結果として得られた混合物を周囲温度で4時間撹拌し、次に、それを270gの水中の8.75g水酸化リチウム一水和物、6.1gの炭酸リチウム及び33.2gのL−セリンの予冷(−5℃)懸濁液に30〜60分以内に添加した。結果として得られた混合物を30分以内に室温まで温め、次に、この温度でもう1時間撹拌した。次に、その混合物を−5℃に冷却し、137gの硫酸(20%水溶液)でpHを2.5に調整した。その二相性混合物を室温まで温め、次に下の水層を分離した。その水層を210mLのトルエンで抽出した。合わせた有機層を100mLのトルエンで希釈し、次に130mLの水で5回洗浄した。トルエン及びTHFを用いた共沸蒸留により有機層から水を除去した。次に、結果として得られたトルエン/THF溶液(約550mL)を1.00gのメタンスルホン酸で処理した。次に、その混合物に1040mLのトルエン中の134gの2,2−ジメトキシプロパンの溶液を8〜10時間以内に添加した。全体の調合の間に、反応容積を一定(約600mL)に維持しながら揮発性物質を減圧下(80〜30mbar)で25〜32℃の温度で蒸発させて除いた。完全に添加した後に、その混合物を約500mLの最終容積まで濃縮した。反応混合物を250gの重炭酸ナトリウム(5%水溶液)で処理した。その二相性混合物を35〜40℃に加温し、この温度で15分間撹拌した。層を分離し、有機層を100gの重炭酸ナトリウム(5%水溶液)で抽出した。生成物を含有する水層を合わせたものをトルエン(150mL)で洗浄した。水層を300mLのトルエンで35〜40℃で処理し、約45gの硫酸(20%水溶液)の滴加によりpHを5.7に調整した。次に水層を分離し、有機層を80gの水で3回洗浄した。結果として得られた、生成物を含有する有機層を80mLの水で処理した。硫酸(20%水溶液)を数滴加えることによりpHを4に調整した。下の水層を除去し、有機層を80gの水で2回洗浄した。有機層を約170mLの残留容積まで濃縮した。その混合物を55〜60℃に加温し、イソプロパノール(15mL)を添加した。結果として得られた透明溶液を300mLのヘプタンで55〜60℃で2〜4時間以内に処理した。結果として得られた懸濁液を10時間以内に0℃に冷却し、この温度で3時間撹拌した。結晶を濾過して取り出し、100mLの予冷したヘプタンで洗浄し、50℃/<30mbarで乾燥させ、無色結晶として33.6g(63%)の(S,S)−3−[3−tert−ブトキシ−2−(9H−フルオレン−9−イル−メトキシカルボニルアミノ)−プロピオニル]−2,2−ジメチル−オキサゾリジン−4−カルボン酸を得た(HPLCアッセイで99.4%(m/m))。
Claims (20)
- 式
[式中、R1は、α−アミノ酸の側鎖であり、R2は、アミノ保護基であり、R3及びR4は、独立して水素(但し、R3及びR4の両方ともが水素ではない)又はC1−4−アルキルより選択され、R5は、水素又はメチルである]で示される化合物の製造方法であって、
a)式
[式中、R1及びR2は、上記と同義である]で示されるアミノ酸誘導体をセリン又はトレオニンと共に式
で示されるジペプチドに変換すること(それに関して活性化剤として水溶性カルボジイミドを使用する)、及び
b)式
[式中、R3及びR4は、独立して水素又はC1−4−アルキルより選択されるが、但し、R3及びR4の両方ともが水素ではなく、R9a及びR9bは、独立してC1−4−アルキルである]で示される化合物と共に式IIIで示されるジペプチドの閉環を酸触媒の存在下で実施すること
を含む方法。 - R1が、バリン、ロイシン、イソロイシン、メチオニン、フェニルアラニン、アスパラギン、グルタミン、グルタミン酸、ヒスチジン、リシン、アルギニン、アスパラギン酸、アラニン、セリン、トレオニン、チロシン、トリプトファン、システイン、グリシン、アミノイソ酪酸及びプロリンより選択される側鎖であることを特徴とする、請求項1記載の方法。
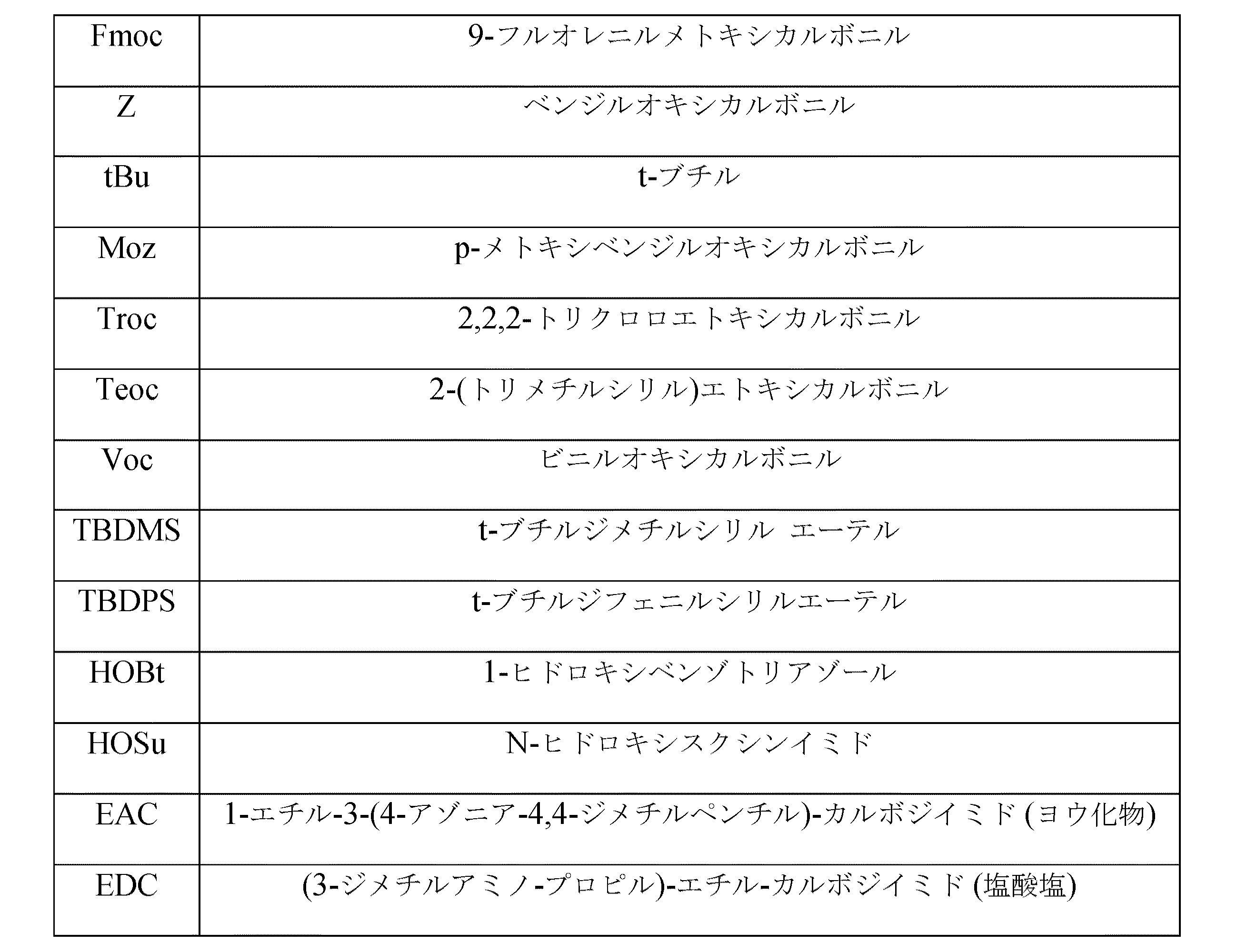
- R2が、Fmoc、Z、Moz、Troc、Teoc及びVocより選択されることを特徴とする、請求項1又は2記載の方法。
- 水溶性カルボジイミドが、EDC又はその塩であることを特徴とする、請求項1〜3記載の方法。
- EDC又はその塩が、HOSuと一緒に用いられることを特徴とする、請求項4記載の方法。
- セリン又はトレオニン:式IIで示されるアミノ酸誘導体の比が、1.5〜4.0:1の範囲より選択されることを特徴とする、請求項1〜5記載の方法。
- 工程a)における変換が、無機塩基の存在下で行われることを特徴とする、請求項1〜6記載の方法。
- 無機塩基が、アルカリ炭酸塩又はアルカリ水酸化物及びその混合物より選択されることを特徴とする、請求項7記載の方法。
- 工程a)における変換が、7.5〜9.5の範囲のpHで行われることを特徴とする、請求項1〜8記載の方法。
- 工程a)における変換が、−10℃〜25℃の範囲の温度で行われることを特徴とする、請求項1〜9記載の方法。
- 反応混合物が、工程a)における変換の後に鉱酸で酸性化されることを特徴とする、請求項1〜10記載の方法。
- 式IIIで示されるジペプチドが、単離されずに、方法工程b)において直接使用されることを特徴とする、請求項1〜11記載の方法。
- 工程b)において閉環に使用される式IVで示される化合物が、2,2−ジメトキシプロパンであることを特徴とする、請求項1〜12記載の方法。
- 2,2−ジメトキシプロパンが、反応混合物に連続的に添加され、発生したメタノールを、並行して連続的に留去することを特徴とする、請求項13記載の方法。
- 工程b)における閉環のための酸触媒が、メタンスルホン酸、(+)カンファー−10−スルホン酸、p−トルエンスルホン酸、p−トルエンスルホン酸ピリジニウムより選択されることを特徴とする、請求項1〜14記載の方法。
- 工程b)における閉環が、トルエン若しくはテトラヒドロフラン又はその混合物の存在下で実施されることを特徴とする、請求項1〜15記載の方法。
- 工程b)における閉環が、15℃〜35℃の範囲の温度で行われることを特徴とする、請求項1〜16記載の方法。
- a)pHを7.0〜9.0の範囲に維持しながら反応混合物を水で抽出すること;
b)pHを5.5〜6.0の範囲に維持しながら水相を水不混和性有機溶媒で抽出すること;
c)有機相から式Iで示される目的生成物を得ること、及び場合により
d)式Iで示される該目的生成物を有機溶媒中で結晶化させること
を含む処理手順により、式Iで示される目的化合物を得ることを特徴とする、請求項1〜17記載の方法。 - 水不混和性有機溶媒が、トルエンであることを特徴とする、請求項18記載の方法。
- 目的生成物を、トルエン、イソプロパノール及びヘプタンの混合物から結晶化させることを特徴とする、請求項18記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08165968 | 2008-10-07 | ||
| EP08165968.2 | 2008-10-07 | ||
| PCT/EP2009/062568 WO2010040660A1 (en) | 2008-10-07 | 2009-09-29 | Pseudoproline dipeptides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012504154A true JP2012504154A (ja) | 2012-02-16 |
| JP5389930B2 JP5389930B2 (ja) | 2014-01-15 |
Family
ID=41202596
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011529526A Expired - Fee Related JP5389930B2 (ja) | 2008-10-07 | 2009-09-29 | シュードプロリンジペプチド |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US8153815B2 (ja) |
| EP (1) | EP2344516B1 (ja) |
| JP (1) | JP5389930B2 (ja) |
| KR (1) | KR101268794B1 (ja) |
| CN (1) | CN102159587B (ja) |
| AU (1) | AU2009301209B2 (ja) |
| BR (1) | BRPI0919501A2 (ja) |
| CA (1) | CA2737675C (ja) |
| ES (1) | ES2410267T3 (ja) |
| IL (1) | IL210799A (ja) |
| MX (1) | MX2011002913A (ja) |
| WO (1) | WO2010040660A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2021512157A (ja) * | 2018-01-30 | 2021-05-13 | バッヘン・ホールディング・アクチエンゲゼルシャフト | グルカゴンペプチドの製造 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103524595A (zh) * | 2013-10-14 | 2014-01-22 | 苏州维泰生物技术有限公司 | 公斤级新方法合成伪二肽Fmoc-Gly-Thr(ψMe, Me pro)-OH的方法 |
| CN103897024A (zh) * | 2014-02-28 | 2014-07-02 | 苏州维泰生物技术有限公司 | 一种合成丙氨酸丝氨酸伪二肽模块的新方法 |
| CN106432468A (zh) * | 2016-11-03 | 2017-02-22 | 滨海吉尔多肽有限公司 | 一种制备艾塞那肽的固相合成方法 |
| CN107176970B (zh) * | 2017-07-06 | 2019-11-19 | 中国医药集团总公司四川抗菌素工业研究所 | 一种树脂催化法合成伪脯氨酸杂环肽的方法 |
| CN109836476A (zh) * | 2019-03-20 | 2019-06-04 | 吉尔生化(上海)有限公司 | 一种半胱氨酸假脯氨酸二肽的合成方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008000641A1 (en) * | 2006-06-28 | 2008-01-03 | F. Hoffmann-La Roche Ag | Pseudo proline dipeptides |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6790935B1 (en) * | 1999-02-05 | 2004-09-14 | Debiopharm S.A. | Cyclosporin derivatives and method for the production of said derivatives |
-
2009
- 2009-09-25 US US12/566,703 patent/US8153815B2/en not_active Expired - Fee Related
- 2009-09-29 ES ES09783513T patent/ES2410267T3/es active Active
- 2009-09-29 JP JP2011529526A patent/JP5389930B2/ja not_active Expired - Fee Related
- 2009-09-29 CA CA2737675A patent/CA2737675C/en not_active Expired - Fee Related
- 2009-09-29 WO PCT/EP2009/062568 patent/WO2010040660A1/en active Application Filing
- 2009-09-29 EP EP09783513A patent/EP2344516B1/en not_active Not-in-force
- 2009-09-29 MX MX2011002913A patent/MX2011002913A/es active IP Right Grant
- 2009-09-29 KR KR1020117007607A patent/KR101268794B1/ko not_active IP Right Cessation
- 2009-09-29 AU AU2009301209A patent/AU2009301209B2/en not_active Ceased
- 2009-09-29 CN CN2009801367246A patent/CN102159587B/zh not_active Expired - Fee Related
- 2009-09-29 BR BRPI0919501A patent/BRPI0919501A2/pt active Search and Examination
-
2011
- 2011-01-23 IL IL210799A patent/IL210799A/en not_active IP Right Cessation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008000641A1 (en) * | 2006-06-28 | 2008-01-03 | F. Hoffmann-La Roche Ag | Pseudo proline dipeptides |
Non-Patent Citations (1)
| Title |
|---|
| JPN6013019162; J. Am. Chem. Soc. vol.118, no.39, 1996, pp.9218-9227 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2021512157A (ja) * | 2018-01-30 | 2021-05-13 | バッヘン・ホールディング・アクチエンゲゼルシャフト | グルカゴンペプチドの製造 |
| JP7360397B2 (ja) | 2018-01-30 | 2023-10-12 | バッヘン・ホールディング・アクチエンゲゼルシャフト | グルカゴンペプチドの製造 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2344516A1 (en) | 2011-07-20 |
| KR20110061604A (ko) | 2011-06-09 |
| JP5389930B2 (ja) | 2014-01-15 |
| US20100087654A1 (en) | 2010-04-08 |
| CN102159587A (zh) | 2011-08-17 |
| AU2009301209A1 (en) | 2010-04-15 |
| CN102159587B (zh) | 2013-11-13 |
| KR101268794B1 (ko) | 2013-05-28 |
| CA2737675A1 (en) | 2010-04-15 |
| WO2010040660A1 (en) | 2010-04-15 |
| IL210799A0 (en) | 2011-04-28 |
| MX2011002913A (es) | 2011-04-12 |
| ES2410267T3 (es) | 2013-07-01 |
| IL210799A (en) | 2015-10-29 |
| CA2737675C (en) | 2016-02-09 |
| US8153815B2 (en) | 2012-04-10 |
| EP2344516B1 (en) | 2013-03-20 |
| AU2009301209B2 (en) | 2012-11-08 |
| BRPI0919501A2 (pt) | 2015-12-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA1329862C (en) | Aryloxy and arylacyloxy methyl ketones as thiol protease inhibitors | |
| JP5389930B2 (ja) | シュードプロリンジペプチド | |
| WO2001019849A1 (en) | A process for the preparation of h-tyr-d-ala-phe(f)-phe-nh¿2? | |
| FR2460291A1 (fr) | Nouveaux tripeptides agissant sur le systeme nerveux central et leur procede de preparation | |
| RU2715233C2 (ru) | Способ получения производных азотистого иприта | |
| JP5292289B2 (ja) | シュードプロリンジペプチド | |
| Katakai | Peptide synthesis using o-nitrophenylsulfenyl N-carboxy. alpha.-amino acid anhydrides | |
| HU213524B (en) | Reductive amination of an amino acid or its derivative by means of an alpha-ketoic acid or its derivative | |
| BE1005720A3 (fr) | Procede de synthese peptidique et nouveaux intermediaires de synthese. | |
| EP2139910B1 (fr) | Procede de synthese de peptides sans solvant | |
| CA2592969A1 (en) | New one-step synthesis of useful disubstituted amines | |
| US3891692A (en) | N-(cyclopropylalkoxycarbonyl)amino acids | |
| EP1123919A1 (en) | Process for producing peptidyl aldehydes | |
| KR20190092429A (ko) | 다이아제핀 유도체의 제조 방법 | |
| Naik et al. | Application of 2-(1h-benzotriazol-1-yl)-1, 1, 3, 3-tetramethyluronium tetrafluoroborate (tbtu) for the synthesis of acid azides | |
| JP2013095735A (ja) | フェノキシカルボニル基の脱離方法 | |
| WO2007097254A1 (ja) | 新規イソジペプチド | |
| JPH09255666A (ja) | ピペラジンアミド化合物及びピペラジンアミド誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130423 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130722 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130910 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20131009 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |