JP2012201603A - ジフェノール化合物の製造方法 - Google Patents
ジフェノール化合物の製造方法 Download PDFInfo
- Publication number
- JP2012201603A JP2012201603A JP2011065387A JP2011065387A JP2012201603A JP 2012201603 A JP2012201603 A JP 2012201603A JP 2011065387 A JP2011065387 A JP 2011065387A JP 2011065387 A JP2011065387 A JP 2011065387A JP 2012201603 A JP2012201603 A JP 2012201603A
- Authority
- JP
- Japan
- Prior art keywords
- compound
- acid
- reaction
- diphenol
- diphenol compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
【課題】式(2)
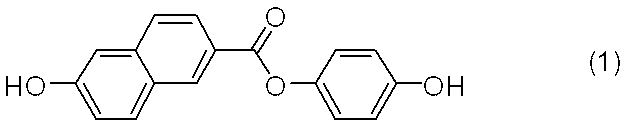
で示されるジエステル化合物の副生を抑制し、且つ、式(1)
で示されるジフェノール化合物の生成選択性が向上した、当該ジフェノール化合物の製造方法を提供すること。
【解決手段】硫酸及びリン酸の存在下、6−ヒドロキシ−2−ナフトエ酸とヒドロキノンとを脱水縮合させる工程を有することを特徴とする式(1)で示されるジフェノール化合物の製造方法、及び、更に、反応系内に二酸化ケイ素を主成分とする無機粉末を共存させることを特徴とする前記の製造方法等。
【選択図】なし
で示されるジエステル化合物の副生を抑制し、且つ、式(1)
で示されるジフェノール化合物の生成選択性が向上した、当該ジフェノール化合物の製造方法を提供すること。
【解決手段】硫酸及びリン酸の存在下、6−ヒドロキシ−2−ナフトエ酸とヒドロキノンとを脱水縮合させる工程を有することを特徴とする式(1)で示されるジフェノール化合物の製造方法、及び、更に、反応系内に二酸化ケイ素を主成分とする無機粉末を共存させることを特徴とする前記の製造方法等。
【選択図】なし
Description
本発明は、ジフェノール化合物の製造方法に関する。
ジフェノール化合物は、例えば、ポリエステル、ポリウレタン、ポリエーテル、エポキシ樹脂等の種々のポリマーの原料として知られている。
特許文献1には、式(1)
で示されるジフェノール化合物(以下、ジフェノール化合物(1)と記すことがある。)が、液晶性アクリル酸エステルモノマー及び優れた光学特性を示す液晶性ポリマーの原料となりうることが記載されている。
ジフェノール化合物(1)を製造する方法としては、例えば、特許文献1の段落番号「0131」には、原料である6−ヒドロキシ−2−ナフトエ酸とヒドロキノンとを、酸触媒として硫酸の存在下、脱水縮合される反応が具体的に記載される方法が知られている。
特許文献1には、式(1)
ジフェノール化合物(1)を製造する方法としては、例えば、特許文献1の段落番号「0131」には、原料である6−ヒドロキシ−2−ナフトエ酸とヒドロキノンとを、酸触媒として硫酸の存在下、脱水縮合される反応が具体的に記載される方法が知られている。
上記方法における脱水縮合反応では、原料の一つであるヒドロキノンが2つの水酸基を有しているため、式(2)
で示される、6−ヒドロキシ−2−ナフトエ酸が2分子縮合されたジエステル体であるジフェノール化合物(以下、ジエステル化合物(2)と記すことがある。)が副生する。このため、ジエステル化合物(2)の副生を抑制し、且つ、ジフェノール化合物(1)の生成選択性が向上した製造方法の開発が望まれていた。
本発明者らは、このような条件下鋭意検討した結果、本発明に至った。
即ち、本発明は、
1.硫酸及びリン酸の存在下、6−ヒドロキシ−2−ナフトエ酸とヒドロキノンとを脱水縮合させる工程を有することを特徴とする式(1)
で示されるジフェノール化合物の製造方法(以下、本発明製造方法と記すこともある。);
2.更に、反応系内に二酸化ケイ素を主成分とする無機粉末(以下、本無機粉末と記すこともある。)を共存させることを特徴とする前項1記載の製造方法;
3.前記無機粉末(即ち、本無機粉末)が、珪藻土からなる無機粉末であることを特徴とする前項2記載の製造方法;
等を提供するものである。
即ち、本発明は、
1.硫酸及びリン酸の存在下、6−ヒドロキシ−2−ナフトエ酸とヒドロキノンとを脱水縮合させる工程を有することを特徴とする式(1)
2.更に、反応系内に二酸化ケイ素を主成分とする無機粉末(以下、本無機粉末と記すこともある。)を共存させることを特徴とする前項1記載の製造方法;
3.前記無機粉末(即ち、本無機粉末)が、珪藻土からなる無機粉末であることを特徴とする前項2記載の製造方法;
等を提供するものである。
本発明により、ジエステル化合物(2)の副生を抑制し、且つ、ジフェノール化合物(1)の生成選択性が向上したジフェノール化合物(1)の製造方法を提供することが可能となる。
以下、本発明を詳細に説明する。
本発明製造は、式(1)
で示されるジフェノール化合物の製造方法であり、硫酸及びリン酸の存在下、6−ヒドロキシ−2−ナフトエ酸とヒドロキノンとを脱水縮合させる工程を有することを特徴とする。
本発明製造は、式(1)
本発明製造において用いられるヒドロキノンの使用量としては、6−ヒドロキシ−2−ナフトエ酸1モルに対して、例えば、1モル〜10モルの範囲を挙げることができる。好ましくは、1.2モル〜5モルの範囲が挙げられる。
本発明製造方法における脱水縮合反応(即ち、エステル化反応)は、通常、溶媒の存在下実施される。尚、溶媒は、反応中、還流させておいてもよい。
本発明製造において用いられる溶媒としては、当該反応を阻害しないものであれば特に制限はないが、例えば、ベンゼン、トルエン、キシレン、エチルベンゼン、メシチレン、クメン、シメン等の芳香族炭化水素溶媒;例えば、クロロベンゼン、ジクロロベンゼン、トリクロロベンゼン、クロロトルエン等の芳香族ハロゲン化炭化水素溶媒;例えば、ヘキサン、ペンタン、オクタン、ノナン、デカン、シクロヘキサン、メチルシクロヘキサン等の脂肪族炭化水素溶媒;例えば、クロロホルム、1,2−ジクロロエタン、1,1,1−トリクロロエタン、1,1,2,2−テトラクロロエタン、1−クロロブタン、1,2−ジブロモエタン等のハロゲン化炭化水素溶媒;これらの混合物;等を挙げることができる。好ましくは、例えば、芳香族炭化水素溶媒、芳香族ハロゲン化炭化水素溶媒等が挙げられる。中でも、より好ましくは、例えば、トルエン、キシレン、クロロベンゼン等を挙げることができる。尚、溶媒は、複数種類の溶媒を混合して用いてもよい。
本発明製造において用いられる溶媒の使用量としては、6−ヒドロキシ−2−ナフトエ酸1重量部に対して、例えば、1重量部〜50重量部の範囲を挙げることができる。
尚、反応の進行に伴って水が副生することから、副生する水は、反応中、反応系外へ除去することが好ましい。
本発明製造において用いられる溶媒としては、当該反応を阻害しないものであれば特に制限はないが、例えば、ベンゼン、トルエン、キシレン、エチルベンゼン、メシチレン、クメン、シメン等の芳香族炭化水素溶媒;例えば、クロロベンゼン、ジクロロベンゼン、トリクロロベンゼン、クロロトルエン等の芳香族ハロゲン化炭化水素溶媒;例えば、ヘキサン、ペンタン、オクタン、ノナン、デカン、シクロヘキサン、メチルシクロヘキサン等の脂肪族炭化水素溶媒;例えば、クロロホルム、1,2−ジクロロエタン、1,1,1−トリクロロエタン、1,1,2,2−テトラクロロエタン、1−クロロブタン、1,2−ジブロモエタン等のハロゲン化炭化水素溶媒;これらの混合物;等を挙げることができる。好ましくは、例えば、芳香族炭化水素溶媒、芳香族ハロゲン化炭化水素溶媒等が挙げられる。中でも、より好ましくは、例えば、トルエン、キシレン、クロロベンゼン等を挙げることができる。尚、溶媒は、複数種類の溶媒を混合して用いてもよい。
本発明製造において用いられる溶媒の使用量としては、6−ヒドロキシ−2−ナフトエ酸1重量部に対して、例えば、1重量部〜50重量部の範囲を挙げることができる。
尚、反応の進行に伴って水が副生することから、副生する水は、反応中、反応系外へ除去することが好ましい。
本発明製造方法における脱水縮合反応(即ち、エステル化反応)は、常圧条件下で実施してもよいし、加圧条件下で実施してもよいし、減圧条件下で実施してもよい。また、窒素ガス、アルゴンガス等の不活性ガスの雰囲気下で実施してもよい。
本発明製造方法における脱水縮合反応(即ち、エステル化反応)の反応温度としては、例えば、40℃〜250℃の範囲を挙げることができる。好ましくは、60℃〜200℃の範囲が挙げられる。より好ましくは、80℃〜180℃の範囲を挙げることができる。
本発明製造方法における脱水縮合反応(即ち、エステル化反応)の反応温度としては、例えば、40℃〜250℃の範囲を挙げることができる。好ましくは、60℃〜200℃の範囲が挙げられる。より好ましくは、80℃〜180℃の範囲を挙げることができる。
本発明製造において、反応時間を短縮するために、必要に応じて、二酸化ケイ素を主成分とする無機粉末(即ち、本無機粉末)を反応系内に追加的に共存させることができる。
本発明製造において用いられる本無機粉末としては、硫酸及びリン酸や原料と反応しないものがよく(尚、硫酸及びリン酸以外の他の酸触媒が反応系内に共存する場合には、当該他の酸触媒とも反応しないものがよい。)、例えば、珪藻土からなる無機粉末、パーライトからなる無機粉末、シリカゲルからなる無機粉末、これらの混合物からなる無機粉末等を挙げることができる。好ましくは、例えば、珪藻土からなる無機粉末(具体的には例えば、セライト(Celite 登録商標)と呼ばれる市販品を挙げることができる。)が挙げられる。尚、ここで「珪藻土からなる無機粉末」とは、珪藻土のみからなる無機粉末であってもよく、珪藻土以外に他の無機物等を含む無機粉末であってもよい。また、例えば、珪藻土を他の無機物等とともに焼成してなる無機粉末であってよい。
本発明製造において用いられる本無機粉末の使用量としては、酸触媒1重量部に対して、例えば、0.2重量部〜10重量部の範囲を挙げることができる。
本発明製造において用いられる本無機粉末としては、硫酸及びリン酸や原料と反応しないものがよく(尚、硫酸及びリン酸以外の他の酸触媒が反応系内に共存する場合には、当該他の酸触媒とも反応しないものがよい。)、例えば、珪藻土からなる無機粉末、パーライトからなる無機粉末、シリカゲルからなる無機粉末、これらの混合物からなる無機粉末等を挙げることができる。好ましくは、例えば、珪藻土からなる無機粉末(具体的には例えば、セライト(Celite 登録商標)と呼ばれる市販品を挙げることができる。)が挙げられる。尚、ここで「珪藻土からなる無機粉末」とは、珪藻土のみからなる無機粉末であってもよく、珪藻土以外に他の無機物等を含む無機粉末であってもよい。また、例えば、珪藻土を他の無機物等とともに焼成してなる無機粉末であってよい。
本発明製造において用いられる本無機粉末の使用量としては、酸触媒1重量部に対して、例えば、0.2重量部〜10重量部の範囲を挙げることができる。
本発明製造方法における脱水縮合反応(即ち、エステル化反応)は、酸触媒としての、硫酸及びリン酸の両者の存在下で実施する。
本発明製造において用いられる酸触媒(即ち、硫酸及びリン酸)の使用量としては、6−ヒドロキシ−2−ナフトエ酸1モルに対して、例えば、0.0001モル〜1モルの範囲を挙げることができる。好ましくは、0.001モル〜0.5モルの範囲が挙げられる。
本発明製造において、必要に応じて、前記酸触媒(即ち、硫酸及びリン酸)以外の「他の酸触媒」を共存させてもよい。当該「他の酸触媒」を共存させる場合には、前記酸触媒(即ち、硫酸及びリン酸)と「他の酸触媒」との合計の使用量として、6−ヒドロキシ−2−ナフトエ酸1モルに対して、例えば、0.0001モル〜1モルの範囲を挙げることができる。好ましくは、0.001モル〜0.5モルの範囲が挙げられる。
ここで「他の酸触媒」としては、例えば、塩酸、硫酸、リン酸、臭化水素酸、ヨウ化水素酸、ホウ酸等の無機酸;例えば、ベンゼンスルホン酸、p−トルエンスルホン酸、メタンスルホン酸等のスルホン酸;例えば、フェニルボロン酸、ペンタフルオロフェニルボロン酸等の有機ホウ酸化合物;例えば、三フッ化ホウ素、トリス(ペンタフルオロフェニル)ボラン等のルイス酸化合物;これらの混合物;等を挙げることができる。
本発明製造において用いられる酸触媒(即ち、硫酸及びリン酸)の使用量としては、6−ヒドロキシ−2−ナフトエ酸1モルに対して、例えば、0.0001モル〜1モルの範囲を挙げることができる。好ましくは、0.001モル〜0.5モルの範囲が挙げられる。
本発明製造において、必要に応じて、前記酸触媒(即ち、硫酸及びリン酸)以外の「他の酸触媒」を共存させてもよい。当該「他の酸触媒」を共存させる場合には、前記酸触媒(即ち、硫酸及びリン酸)と「他の酸触媒」との合計の使用量として、6−ヒドロキシ−2−ナフトエ酸1モルに対して、例えば、0.0001モル〜1モルの範囲を挙げることができる。好ましくは、0.001モル〜0.5モルの範囲が挙げられる。
ここで「他の酸触媒」としては、例えば、塩酸、硫酸、リン酸、臭化水素酸、ヨウ化水素酸、ホウ酸等の無機酸;例えば、ベンゼンスルホン酸、p−トルエンスルホン酸、メタンスルホン酸等のスルホン酸;例えば、フェニルボロン酸、ペンタフルオロフェニルボロン酸等の有機ホウ酸化合物;例えば、三フッ化ホウ素、トリス(ペンタフルオロフェニル)ボラン等のルイス酸化合物;これらの混合物;等を挙げることができる。
本発明製造方法における脱水縮合反応(即ち、エステル化反応)後、必要に応じて、得られた反応マスの中に存在する酸触媒を中和処理し、次いで得られた中和処理物を濃縮処理及び/又はろ過処理することにより、ジフェノール化合物(1)を粗結晶として得ることができる。更に、ジフェノール化合物(1)の粗結晶を溶媒に溶解させた後、得られた溶液をろ過処理することにより、本無機粉末が除去されたジフェノール化合物(1)の溶液として得ることもできる。
このようにして得られたジフェノール化合物(1)の粗結晶、及び、ジフェノール化合物(1)の溶液、並びに、本発明製造方法における脱水縮合反応により得られた反応マスは、例えば、再結晶等の精製手段を施すことにより、更に精製することもできる。
このようにして得られたジフェノール化合物(1)の粗結晶、及び、ジフェノール化合物(1)の溶液、並びに、本発明製造方法における脱水縮合反応により得られた反応マスは、例えば、再結晶等の精製手段を施すことにより、更に精製することもできる。
以下、実施例により本発明をさらに詳細に説明する。
実施例1
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.1g、ヒドロキノン29.3g、98%硫酸0.67g、85%リン酸0.65g、セライト(Celite 登録商標)545 1.33g及びトルエン150mLを仕込み、還流条件下で6時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去された。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=1.9:96.3:1.9(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液100gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水で3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール300gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量135gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水80gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液20gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶33.3g(純度:88.0%(LC面積百分率値)、見かけ収率:89.2%)を得た。
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.1g、ヒドロキノン29.3g、98%硫酸0.67g、85%リン酸0.65g、セライト(Celite 登録商標)545 1.33g及びトルエン150mLを仕込み、還流条件下で6時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去された。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=1.9:96.3:1.9(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液100gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水で3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール300gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量135gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水80gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液20gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶33.3g(純度:88.0%(LC面積百分率値)、見かけ収率:89.2%)を得た。
実施例2
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸300.0g、ヒドロキノン351.1g、98%硫酸8.0g、85%リン酸9.2g、セライト(Celite 登録商標)545 17.2g及びトルエン1800mLを仕込み、還流条件下で8時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=1.8:96.6:1.6(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液1200gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水500gで3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール3600gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量1611gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水960gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液250gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶408.7g(純度:95.2%(LC面積百分率値)、見かけ収率:91.5%)を得た。
1H−NMR(DMSO−d6,TMS基準,単位:ppm)
δ6.83(d,J=8.9Hz,2H),7.09(d,J=8.9Hz,2H),7.17〜7.26(m,2H),7.84(d,J=8.7Hz,1H),7.98(dd,J=8.7Hz,1.4Hz,1H),8.04(d,J=8.7Hz,1H),8.67(s,1H),9.49(s,br,1H),10.27(s,br,1H)
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸300.0g、ヒドロキノン351.1g、98%硫酸8.0g、85%リン酸9.2g、セライト(Celite 登録商標)545 17.2g及びトルエン1800mLを仕込み、還流条件下で8時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=1.8:96.6:1.6(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液1200gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水500gで3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール3600gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量1611gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水960gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液250gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶408.7g(純度:95.2%(LC面積百分率値)、見かけ収率:91.5%)を得た。
1H−NMR(DMSO−d6,TMS基準,単位:ppm)
δ6.83(d,J=8.9Hz,2H),7.09(d,J=8.9Hz,2H),7.17〜7.26(m,2H),7.84(d,J=8.7Hz,1H),7.98(dd,J=8.7Hz,1.4Hz,1H),8.04(d,J=8.7Hz,1H),8.67(s,1H),9.49(s,br,1H),10.27(s,br,1H)
実施例3
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.3g、98%硫酸0.67g、85%リン酸0.33g、セライト(Celite 登録商標)545 0.99g及びトルエン150mLを仕込み、還流条件下で6時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=2.3:95.3:2.3(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液100gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水で3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール300gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量135gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水80gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液20gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶34.8g(純度:93.7%(LC面積百分率値)、見かけ収率:93.3%)を得た。
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.3g、98%硫酸0.67g、85%リン酸0.33g、セライト(Celite 登録商標)545 0.99g及びトルエン150mLを仕込み、還流条件下で6時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=2.3:95.3:2.3(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液100gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水で3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール300gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量135gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水80gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液20gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶34.8g(純度:93.7%(LC面積百分率値)、見かけ収率:93.3%)を得た。
実施例4
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.3g、98%硫酸0.67g、85%リン酸1.30g、セライト(Celite 登録商標)545 1.97g及びトルエン150mLを仕込み、還流条件下で6時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=3.2:95.0:1.7(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液100gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水で3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール300gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量135gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水80gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液20gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶33.6g(純度:91.5%(LC面積百分率値)、見かけ収率:90.1%)を得た。
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.3g、98%硫酸0.67g、85%リン酸1.30g、セライト(Celite 登録商標)545 1.97g及びトルエン150mLを仕込み、還流条件下で6時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=3.2:95.0:1.7(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液100gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水で3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール300gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量135gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水80gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液20gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶33.6g(純度:91.5%(LC面積百分率値)、見かけ収率:90.1%)を得た。
実施例5
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.2g、98%硫酸0.67g、85%リン酸0.77g、セライト(Celite 登録商標)545 2.86g及びトルエン150mLを仕込み、還流条件下で6時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=2.3:95.5:2.2(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液100gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水で3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール300gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量135gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水80gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液20gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶34.3g(純度:92.9%(LC面積百分率値)、見かけ収率:92.1%)を得た。
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.2g、98%硫酸0.67g、85%リン酸0.77g、セライト(Celite 登録商標)545 2.86g及びトルエン150mLを仕込み、還流条件下で6時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=2.3:95.5:2.2(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液100gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水で3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール300gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量135gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水80gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液20gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶34.3g(純度:92.9%(LC面積百分率値)、見かけ収率:92.1%)を得た。
実施例6
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.1g、ヒドロキノン29.3g、98%硫酸0.67g、85%リン酸0.77g、セライト(Celite 登録商標)545 1.43g及びクロロベンゼン150mLを仕込み、内圧61kPa、還流条件下で8時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流中の内温は110℃であり、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=1.8:96.1:2.1(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液100gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水で3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール300gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量135gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水80gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液20gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶35.3g(純度:93.7%(LC面積百分率値)、見かけ収率:94.5%)を得た。
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.1g、ヒドロキノン29.3g、98%硫酸0.67g、85%リン酸0.77g、セライト(Celite 登録商標)545 1.43g及びクロロベンゼン150mLを仕込み、内圧61kPa、還流条件下で8時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流中の内温は110℃であり、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=1.8:96.1:2.1(LC面積比)であった。
次に、上記で得られた反応マスに、5重量%の炭酸水素ナトリウム水溶液100gを加えた後、得られた混合物を30分間撹拌した。次いで、得られた混合物から濾別により得られた固体成分をイオン交換水で3回洗浄した。
このようにして得られた固体成分(粗結晶)とメタノール300gとを混合した後、得られた混合物を50℃で撹拌した後、同温度のまま、前記混合物から不溶分を濾別除去することにより、ろ液を回収し、これを重量135gになるまで濃縮した。尚、濃縮途中で灰色結晶が析出した。
このようにして得られた濃縮残渣を60℃で保温しながら、イオン交換水80gを滴下した後、2時間保温した。次いで、これを室温まで冷却した後、得られた混合物から濾別により得られた固体成分を50重量%メタノール水溶液20gで3回洗浄した。
このようにして得られた固体成分(精製結晶)を真空乾燥することにより、ジフェノール化合物(1)の灰色結晶35.3g(純度:93.7%(LC面積百分率値)、見かけ収率:94.5%)を得た。
実施例7
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.2g、98%硫酸0.33g、85%リン酸0.38g及びトルエン150mLを仕込み、還流条件下で8時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=1.7:96.6:1.7(LC面積比)であった。
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.2g、98%硫酸0.33g、85%リン酸0.38g及びトルエン150mLを仕込み、還流条件下で8時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=1.7:96.6:1.7(LC面積比)であった。
実施例8
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.3g、98%硫酸0.67g、85%リン酸0.77g及びトルエン150mLを仕込み、還流条件下で7時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=2.4:96.3:1.3(LC面積比)であった。
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.3g、98%硫酸0.67g、85%リン酸0.77g及びトルエン150mLを仕込み、還流条件下で7時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=2.4:96.3:1.3(LC面積比)であった。
比較例1
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.2g、98%硫酸0.67g、セライト(Celite 登録商標)545 0.67g及びトルエン150mLを仕込み、還流条件下で6時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=3.3:93.8:2.9(LC面積比)であり、ジエステル化合物(副生物:ジエステル化合物(2))の生成量が多い結果となった。
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.2g、98%硫酸0.67g、セライト(Celite 登録商標)545 0.67g及びトルエン150mLを仕込み、還流条件下で6時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=3.3:93.8:2.9(LC面積比)であり、ジエステル化合物(副生物:ジエステル化合物(2))の生成量が多い結果となった。
比較例2
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.3g、98%硫酸0.67g、p−トルエンスルホン酸一水和物1.27g、セライト(Celite 登録商標)545 1.93g及びトルエン150mLを仕込み、還流条件下で4時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=1.7:94.6:3.7(LC面積比)であり、ジエステル化合物(副生物:ジエステル化合物(2))の生成量が多い結果となった。
温度計、ディーンスターク装置、冷却管及び撹拌装置を備えた四つ口フラスコに、6−ヒドロキシ−2−ナフトエ酸25.0g、ヒドロキノン29.3g、98%硫酸0.67g、p−トルエンスルホン酸一水和物1.27g、セライト(Celite 登録商標)545 1.93g及びトルエン150mLを仕込み、還流条件下で4時間反応させた。反応後、得られた反応マスを室温まで冷却した。尚、還流に伴い留出した水は、ディーンスターク装置により、反応容器から除去した。
反応終了時、得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比は、原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))=1.7:94.6:3.7(LC面積比)であり、ジエステル化合物(副生物:ジエステル化合物(2))の生成量が多い結果となった。
前述の実施例1〜実施例8、及び、比較例1〜比較例2で得られた「反応終了時に得られた反応マスの中の原料カルボン酸:モノエステル化合物(目的物:ジフェノール化合物(1)):ジエステル化合物(副生物:ジエステル化合物(2))に係る組成比」の結果を纏めて表1に示した。尚、ジフェノール化合物(1)の生成選択性として、目的物:ジフェノール化合物(1)/副生物:ジエステル化合物(2)の比を算出し、表1に併記した。
本発明により、ジエステル化合物(2)の副生を抑制し、且つ、ジフェノール化合物(1)の生成選択性が向上したジフェノール化合物(1)の製造方法を提供することが可能となる。
Claims (3)
- 硫酸及びリン酸の存在下、6−ヒドロキシ−2−ナフトエ酸とヒドロキノンとを脱水縮合させる工程を有することを特徴とする式(1)
で示されるジフェノール化合物の製造方法。 - 更に、反応系内に二酸化ケイ素を主成分とする無機粉末を共存させることを特徴とする請求項1記載の製造方法。
- 前記無機粉末が、珪藻土からなる無機粉末であることを特徴とする請求項2記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011065387A JP2012201603A (ja) | 2011-03-24 | 2011-03-24 | ジフェノール化合物の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011065387A JP2012201603A (ja) | 2011-03-24 | 2011-03-24 | ジフェノール化合物の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2012201603A true JP2012201603A (ja) | 2012-10-22 |
Family
ID=47182956
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011065387A Withdrawn JP2012201603A (ja) | 2011-03-24 | 2011-03-24 | ジフェノール化合物の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2012201603A (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7409965B2 (ja) | 2020-05-27 | 2024-01-09 | 上野製薬株式会社 | 4-ヒドロキシ安息香酸2’-ヒドロキシ-(1,1’-ビナフタレン)-2-イルおよびその製造方法 |
-
2011
- 2011-03-24 JP JP2011065387A patent/JP2012201603A/ja not_active Withdrawn
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7409965B2 (ja) | 2020-05-27 | 2024-01-09 | 上野製薬株式会社 | 4-ヒドロキシ安息香酸2’-ヒドロキシ-(1,1’-ビナフタレン)-2-イルおよびその製造方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6083901B2 (ja) | ビナフタレン化合物の製造方法 | |
| JP5958734B2 (ja) | 新規エポキシ化合物及びその製造方法 | |
| KR20150113005A (ko) | 신규 알릴 화합물 및 그 제조방법 | |
| EP3495342B1 (en) | Polymerizable triptycene derivative compound | |
| JPS6322540A (ja) | 4−ハイドロキシフエニル4−ハイドロキシベンゾエ−トの製法 | |
| JP2012201603A (ja) | ジフェノール化合物の製造方法 | |
| JP2015003875A (ja) | リン系化合物及びその製造方法 | |
| EP3162789B1 (en) | Production intermediate of polymerizable compound, method for producing same, composition and stabilization method | |
| JP6319805B2 (ja) | スピロビフルオレン骨格を有するジオール化合物およびその製造方法 | |
| JP2007001888A (ja) | 1,4−ジヒドロ−1,4−メタノアントラセン類の製造方法 | |
| JP6027910B2 (ja) | 触媒の製造方法、及び光学活性アンチ−1,2−ニトロアルカノール化合物の製造方法 | |
| JP5432605B2 (ja) | エステル基を有する芳香族カルボン酸二無水物の製造方法 | |
| JP2008162967A (ja) | 非対称型ビス(ターピリジン)化合物の合成方法 | |
| JP6190256B2 (ja) | 新規なビス(ヒドロキシフェニル)ベンゾオキサゾール化合物 | |
| JP6420106B2 (ja) | 環構造を有するジカルボン酸モノエステルの製造方法、ジカルボン酸モノエステルアミン塩、ジカルボン酸モノエステルを用いた液晶化合物の製造方法 | |
| JPWO2019117019A1 (ja) | ジオールの製造方法 | |
| JP2015131786A (ja) | 9,9’−スピロビフルオレン類の製造方法 | |
| Morikawa et al. | Sc (OTf) 3-catalyzed cyclooligomerization of 2, 4-dialkoxybenzyl alcohols. Formation of resorcin [n] arene peralkyl ethers | |
| CN114805122B (zh) | 一种酯化反应的方法 | |
| JP5603626B2 (ja) | 2−endo−6−exo−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの製造方法 | |
| RU2676767C2 (ru) | Политриазол и способ его получения | |
| CN114805121A (zh) | 一种酯化反应的方法 | |
| JP2021042154A (ja) | ビナフタレン骨格を有する化合物の製造方法 | |
| JPH08119939A (ja) | 高純度エーテル型ビスマレイミドの製造方法 | |
| JP2015105239A (ja) | N−ビニルカルバゾールの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A300 | Withdrawal of application because of no request for examination |
Free format text: JAPANESE INTERMEDIATE CODE: A300 Effective date: 20140603 |