JP2009519041A - 核酸配列決定のためのプローブおよび使用方法 - Google Patents
核酸配列決定のためのプローブおよび使用方法 Download PDFInfo
- Publication number
- JP2009519041A JP2009519041A JP2008545768A JP2008545768A JP2009519041A JP 2009519041 A JP2009519041 A JP 2009519041A JP 2008545768 A JP2008545768 A JP 2008545768A JP 2008545768 A JP2008545768 A JP 2008545768A JP 2009519041 A JP2009519041 A JP 2009519041A
- Authority
- JP
- Japan
- Prior art keywords
- nucleic acid
- probe
- acid molecule
- molecular
- linker
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images












Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/536—Immunoassay; Biospecific binding assay; Materials therefor with immune complex formed in liquid phase
- G01N33/542—Immunoassay; Biospecific binding assay; Materials therefor with immune complex formed in liquid phase with steric inhibition or signal modification, e.g. fluorescent quenching
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6816—Hybridisation assays characterised by the detection means
- C12Q1/6818—Hybridisation assays characterised by the detection means involving interaction of two or more labels, e.g. resonant energy transfer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6869—Methods for sequencing
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T436/00—Chemistry: analytical and immunological testing
- Y10T436/14—Heterocyclic carbon compound [i.e., O, S, N, Se, Te, as only ring hetero atom]
- Y10T436/142222—Hetero-O [e.g., ascorbic acid, etc.]
- Y10T436/143333—Saccharide [e.g., DNA, etc.]
Abstract
核酸分子の配列決定のためのナノプローブおよびナノプローブを使用するための方法を提供する。特定の例において、該プローブは、重合剤と、標的核酸分子内の相補的ヌクレオチドと特異的に結合することによって、リンカーから分離することなく、核酸分子の鋳型鎖に可逆的に結合することができる化学的部分を有する、1つまたは複数の分子リンカーとを含む。標的核酸分子内の相補的ヌクレオチドとリンカー上の化学的部分との可逆的な結合は、相補的ヌクレオチドとリンカー上の化学的部分との対合を示す特性シグナルの放出によって示される。かかる化学的部分の1つの例は、非加水分解性ヌクレオチド類似体である。特定の例において、重合剤および化学的部分は、化学的部分の特定の型を特徴とする供与体フルオロフォア、および受容体フルオロフォアなどのタグに関連する。
Description
関連出願に対する相互参照
本出願は、2005年12月12日に双方とも出願された米国仮出願第60/749,729号および第60/749,858号に対する優先権を主張し、参照により本明細書に組み入れられる。
本出願は、2005年12月12日に双方とも出願された米国仮出願第60/749,729号および第60/749,858号に対する優先権を主張し、参照により本明細書に組み入れられる。
分野
本開示は、プローブ、ならびにDNAおよびRNAなどの核酸分子を配列決定するための方法に関し、研究および臨床的用途における疾病の診断のために使用することができる。
本開示は、プローブ、ならびにDNAおよびRNAなどの核酸分子を配列決定するための方法に関し、研究および臨床的用途における疾病の診断のために使用することができる。
背景
多数の方法が核酸分子を配列決定するために使用されてきた。従来のマクサムギルバート化学分解法は、DNAの化学特異的開裂に関与する(Maxam and Gilbert,Proc.Natl.Acad.Sci,USA 74:560,1977(非特許文献1))。本方法において、放射能標識化DNA分子を、4つの別個の反応混合物内でインキュベートし、それぞれは、特異的識別性(G、A+G、CまたはC+T)の1つまたは2つのヌクレオチドでDNAを部分的に開裂する。得られたDNAフラグメントは、4つの反応のそれぞれを、ゲルの別個のレーンに分けた、ポリアクリルアミドゲル電気泳動によって分離される。DNA配列は、ゲルの4つのレーン内でフラグメントの高分子分離を観察することによって、オートラジオグラフィーの後に決定される。
多数の方法が核酸分子を配列決定するために使用されてきた。従来のマクサムギルバート化学分解法は、DNAの化学特異的開裂に関与する(Maxam and Gilbert,Proc.Natl.Acad.Sci,USA 74:560,1977(非特許文献1))。本方法において、放射能標識化DNA分子を、4つの別個の反応混合物内でインキュベートし、それぞれは、特異的識別性(G、A+G、CまたはC+T)の1つまたは2つのヌクレオチドでDNAを部分的に開裂する。得られたDNAフラグメントは、4つの反応のそれぞれを、ゲルの別個のレーンに分けた、ポリアクリルアミドゲル電気泳動によって分離される。DNA配列は、ゲルの4つのレーン内でフラグメントの高分子分離を観察することによって、オートラジオグラフィーの後に決定される。
サンガージデオキシ法は、DNAポリメラーゼおよびデオキシヌクレオシド三リン酸およびジデオキシヌクレオシド三リン酸の混合物を用いた合成プライマーの酵素伸長によって、異なる長さのDNA分子の生成に関する(Sanger et al,Proc.Natl.Acad.Sci,USA74:5463,1977(非特許文献2))。反応は、ポリアクリルアミドゲル上の4つの平行なレーンに分離され、オートラジオグラフィーの後に配列が決定される。
蛍光ヌクレオチドを使用することにより、放射性ヌクレオチドの必要性がなくなり、DNA配列決定を自動化する手段を提供する(例えば、Ansorgeへの米国特許第5,124,247号(特許文献1)、Proberらへの米国特許第5,242,796号(特許文献2)、Proberらへの米国特許第5,306,618号(特許文献3)、Middendorfらへの米国特許第5,360,523号(非特許文献4)、Pettitへの米国特許第5,556,790号(特許文献5)、Smithへの米国特許第5,821,058号(特許文献6)を参照)。しかしながら、一般に、フルオロフォアを使用する方法は、ゲル電気泳動またはキャピラリー電気泳動を今もなお必要とし、従って緩慢であり巨視的である。
蛍光標識dNTPを使用するその他の潜在的障壁は、誰も蛍光標識DNA分子を完全には合成することができなかったことである。従って、相補的核酸鎖の合成を可能にする配列決定方法は、今もなお必要とされている。
概要
本開示は、核酸分子の配列決定に使用できる改良型プローブ、および該プローブを使用する方法を提供する。特定の例において、プローブは、1つまたは複数の遺伝子の転写レベルを決定するために使用することができる。例えば、プローブは、個別のRNAの転写を計数し、それによって細胞内で生成された数の推定を提供するために使用することができる。特定の例において、本明細書に開示するプローブおよび方法は、現在利用可能なマイクロアレイ技術の代替物として使用される。
本開示は、核酸分子の配列決定に使用できる改良型プローブ、および該プローブを使用する方法を提供する。特定の例において、プローブは、1つまたは複数の遺伝子の転写レベルを決定するために使用することができる。例えば、プローブは、個別のRNAの転写を計数し、それによって細胞内で生成された数の推定を提供するために使用することができる。特定の例において、本明細書に開示するプローブおよび方法は、現在利用可能なマイクロアレイ技術の代替物として使用される。
特定の例において、「Medusa」と称されるプローブは、1つまたは複数の化学的部分(非加水分解性ヌクレオチドなど)を重合剤に連結する(一部の例では、離間して)、重合剤に結合した1つ以上の(複数など)分子リンカーを有する重合剤を含む。化学的部分は、リンカーから分離することなく、標的核酸分子内の相補的ヌクレオチドに特異的に結合することによって、核酸分子の鋳型鎖に可逆的に結合することができる。開示する例において、可逆的な組み込みは、重合剤の活性部位で発生する。しかしながら、化学的部分は、成長する核酸鎖に永久に組み込むことができないことが理想的である。標的核酸分子内の相補的ヌクレオチドとリンカー上の化学的部分との特異的な結合は、相補的ヌクレオチドとリンカー上の化学的部分との対合を示す特性シグナルの放出によって示される。
重合剤は、配列決定される標的核酸分子に結合することができる活性部位を含み、一部の例において、標的核酸分子に相補的な核酸分子の合成を促進することができ、相補的核酸分子は、相補的ヌクレオチドが相補的核酸分子に組み込まれるように伸長する。重合剤は、線状鎖(相補的核酸分子など)を形成する化学反応で、モノマー分子(ヌクレオチドなど)を一緒に反応させることができる化合物を含む。例示的な重合剤には、DNAポリメラーゼ、RNAポリメラーゼ、および逆転写酵素が含まれるが、これらに限定されるものではない。特定の例において、ポリメラーゼは、GFP−ポリメラーゼである。重合剤の選択は、配列決定される核酸に依存しうる。例えば、標的核酸分子がDNAである場合は、重合剤は、DNAまたはRNAポリメラーゼであってもよいが、標的核酸分子がRNAである場合は、重合剤は、逆転写酵素であってもよい。
リンカーから分離することなく、標的核酸分子の鋳型鎖内で相補的ヌクレオチドと可逆的に結合することができる化学的部分は、非加水分解性ヌクレオチド類似体などのヌクレオチド類似体を含むことができる。かかる類似体は、標的核酸分子内の相補的ヌクレオチドと対になることができるが、伸長している相補的核酸鎖に永久に組み込まれることはない。非加水分解性ヌクレオチド類似体は、当技術分野では公知であり、非加水分解性であるα−β結合を有する非加水分解性ヌクレオチド三リン酸類似体などの、非加水分解性ヌクレオチド三リン酸類似体を含む。
特定の例において、プローブは、少なくとも4つの独立したリンカーを含み、それぞれは、標的核酸分子内の異なるヌクレオチドと特に対合することができるが、伸長している相補的核酸分子に永久に組み込むことができない、異なる化学的部分を有する。その他の例において、プローブは、分岐構造を形成するために結合される複数のリンカーを含み、それぞれの分岐は、標的核酸分子内の異なるヌクレオチドと特に対合することができるが、伸長している相補的核酸分子に永久に組み込まれることはできない、異なる化学的部分を有する。例えば、分岐構造は、1点でのみ重合剤に結合してもよい。
分子リンカーは、重合剤を核酸分子の鋳型(または標的)鎖に可逆的に結合することができる1つまたは複数の化学的部分に連結する。特定の例において、分子リンカーは、標的核酸分子の存在下で、重合剤と化学的部分との相互作用を可能にすると同時に、標的または鋳型核酸分子の非存在下で、重合剤と化学的部分との実質上の絡み合いを回避するように、重合剤および化学的部分を互いに十分な距離をおいて維持する。例えば、分子リンカーは、リンカーの絡み合いを抑制するために十分な距離をおいて重合剤の周囲で離間することができ、重合剤の活性部位に到達するほど十分に長い。一部の例において、分子リンカー(または少なくともその一部分)は、標的核酸分子の非存在下で、重合酵素と化学的部分との相互作用を減少させるように十分な剛性を有する。
分子リンカー(または、分子リンカーの一部分である分子ロッドなど、その一部)は、標的核酸分子の非存在下で、望ましくない相互作用を回避するために、重合剤と化学的部分とを十分な距離をおいて離間する柔軟性を考慮して十分な長さを有するが、例えば、重合剤が標的核酸分子に結合する場合、重合剤と化学的部分とが互いに、および標的核酸分子と相互に作用しうる十分な柔軟性を保持する。例えば、分子リンカーの少なくとも一部は、分子リンカーの少なくとも一部が、標的核酸分子の非存在下で、重合剤(重合剤に関連するタグなど)と化学的部分との相互作用を減少させるような十分な剛性および長さとなることを可能し、標的核酸分子の存在下で、重合剤と化学的部分との相互作用を可能にする持続長を有することができる。
特定の例において、分子リンカーの全長は、二本鎖または一本鎖核酸分子などのリンカーを構成する1つまたは複数の成分の持続長と異なる(それより長いまたは短いなど)。しかしながら、特定の例において、分子リンカーの全長は、標的核酸分子の存在下で、標的核酸分子と同様に、重合剤と化学的部分との有意な相互作用を可能にすると同時に、有意な相互作用が、標的核酸分子の非存在下で、重合剤と化学的部分との間に発生する長さを超えることはない。かかる相互作用は、当技術分野で公知の方法を用いて、例えば、重合剤が、供与体フルオロフォアを含み、1つまたは複数の化学的部分が、FRET対の対応する受容体フルオロフォアを含む場合、受容体発光蛍光を測定することによって測定することができる。その他の例において、重合剤は、標的の非存在下で、化学的部分からフォスターの半径(22から90Åのフォスターの半径など)の少なくとも2倍の距離で実質上維持される。
持続長(lp)は、線状鎖のための平均的局所立体構造であり、任意のセグメントによって画定される方向のすべての鎖セグメントの平均的延在の合計を反映する。従って、持続長は、ポリマー鎖の剛性または弾性の基準である。特定の例において、持続長は、実質的には、平均にして68.40度の屈曲で発生する周長距離を測定する屈曲度(従って、鎖の有効弾性)である。従って、持続長は、分子リンカーの組成物に応じて変化する。例えば、二本鎖DNA(dsDNA)分子のための持続長は、一本鎖DNA(ssDNA)分子およびポリエチレングリコール(PEG)のものと異なる。特定の例において、例えば、イオン強度が約0.2Mおよび温度が20℃で、dsDNAは、持続長が約400〜500Å(450〜500Åなど)であり、dsRNAは、持続長が約700〜750Åである。特定の例において、ssDNAは、持続長が約40Å(例えば20℃)である(Clossey and Carlon,Phys.Rev.E.Stat.Nonlin.Soft.Matter.Phys.68(6Ptl):061911,2003)。特定の例において、PEGは、持続長が約3.8Åである。
特定の例において、分子リンカーには、核酸のポリマーなどの線状ポリマー、アミノ酸、糖、PEG、またはそれらの組み合わせが含まれる。例えば、分子リンカーには、テザー(tether)、分子ロッド、またはそれらの組み合わせが含まれるが、これらに限定されるものではない。例えば、十分な剛性の分子リンカーは、分子ロッド、例えば、dsDNAから成る分子ロッドを含むことができる。一部の例において、十分な剛性の分子リンカーには、テザーによって連結される複数の分子ロッド、または分子ロッドによって連結される複数のテザーが含まれる。テザーの特定の一例は、ポリエチレングリコール(PEG)から成る(または一部の例においては、構成される)分子である。
標的核酸分子の非存在下で、重合剤と化学的部分が相互作用して反応を提供しないように、重合剤および化学的部分を、1つまたは複数の分子リンカーによって空間的に分離した方向に連結することができる。しかしながら、分子リンカーは、重合剤および化学的部分に、所定の条件下で、相互に作用するために互いに十分に接近させ、検出可能なシグナルなどの所定の反応、または標的核酸分子との相互作用を引き起こすことを可能にする。例えば、重合剤または化学的部分に関連するタグのうちの少なくとも1つは、供与体および受容体が、互い十分に接近する場合、供与体フルオロフォアタグによる受容体フルオロフォアタグの励起など、別のタグに十分に接近する場合に活性化されうる。
さらに、本開示は、標的核酸分子に結合することができ、相補的ヌクレオチドが相補的核酸分子に組み込まれるように伸長する相補的核酸分子の合成を促進することができる活性部位を含む重合剤を提供する。重合剤は、絡み合いを抑制するために、重合剤上で離間する1つまたは複数の分子リンカーをさらに含み、それぞれのリンカーは、標的核酸分子内の相補的ヌクレオチドに特異的に結合することによって、リンカーから分離することなく、核酸分子の鋳型鎖に可逆的に結合することができる、異なる化学的部分(非加水分解性ヌクレオチド類似体など)を有する。特定の例において、重合剤は、リンカーが有する化学的部分を識別するそれぞれの化学的部分に関連するタグをさらに含む。さらに、ポリメラーゼは、リンカーが有する化学的部分を識別する特性シグナルを放出する化学的部分に関連するタグと相互に作用するタグに関連することができる。
さらに、本開示は、例えば、標的核酸分子の核酸配列を決定するために、開示するナノプローブを使用する方法を提供する。特定の例において、該方法は、特定の標的分子が、試料内に存在するかを決定するために使用され、一部の例において、存在する標的核酸分子の量の定量化を含む。例えば、該方法は、1つまたは複数の核酸突然変異に関連する疾病を有する対象を診断するために、プローブを使用するために提供される。
配列決定は、インビトロまたはインサイチュー(例えば、顕微鏡用スライド上)およびインビボ(例えば、プローブを細胞内に導入し、mRNAが生成される時にその配列を観察することによって)で行うことができる。該方法は、いくつかの核酸を分子レベルで同時に配列決定することを可能にする。例えば、複数の配列決定反応は、実質上同時に行うことができ、複数の配列決定反応からのシグナルを検出し、核酸配列に変換することができる。
特定の例において、該方法は、オリゴヌクレオチドプライマー、および標的核酸分子内の相補的ヌクレオチドとの塩基対合に、伸長する核酸分子に組み込まれることができ、核酸分子の鋳型鎖に可逆的に結合するリンカーが有する化学的部分を置換することができる加水分解性ヌクレオチド(dATP、dCTP、dGTP、およびdTTPまたはATP、CTP、GTPおよびUTPなど)の混合物の存在下で、標的核酸分子を本明細書に開示するプローブに曝露する工程を含む。一連のシグナルの放出は検出され、シグナルは、相補的ヌクレオチドとのリンカー上の化学的部分の対合を示す複数の特性シグナルの放出を含む。一部の例において、一連のシグナルの放出は、核酸配列に変換される。
特定の例において、重合剤は、タグ(供与体フルオロフォアなど)に関連し、化学的部分(非加水分解性A、T/U、CまたはGヌクレオチド類似体など)のそれぞれの異なる型は、リンカーが有する特定の化学的部分を識別する一意的なタグに関連し、重合剤に関連するタグと、化学的部分に関連するタグとの相互作用は、相補的ヌクレオチドとリンカー上の化学的部分との対合を示す特性シグナルの放出を誘発する。特定の例において、タグは、重合剤または化学的部分に直接結合される。しかしながら、タグは、直接結合される必要はなく、代わりに、リンカー上の化学的部分が、相補的ヌクレオチドと対になる場合、特性シグナルの放出を生成するために、重合剤または化学的部分に十分に接近した分子リンカー上に見ることができる。
例えば、重合剤に関連するタグは、供与体フルオロフォアであってもよく、特定の化学的部分を識別するタグは、1つまたは複数の受容体フルオロフォアを含むことができ、重合剤と合成核酸分子に組み込むことができない化学的部分との相互作用は、供与体フルオロフォアによる受容体フルオロフォアの励起を可能にする供与体フルオロフォアに受容体フルオロフォアを接近させる。かかる例において、シグナルを検出する工程は、受容体フルオロフォアから放出された蛍光性シグナル(または供与体フルオロフォアからの減少された放出シグナル)を検出する工程を含むことができる。特定の例において、該方法は、受容体フルオロフォアではなく、供与体フルオロフォアを特に励起する電磁放射(レーザーなど)源によって、供与体フルオロフォアを励起する工程をさらに含む。あるいは、供与体フルオロフォアは、化学発光分子、例えば、エクオリンである。この例において、化学発光法による供与体フルオロフォアは、必然的に励起状態にあるので、供与体フルオロフォアは、電磁放射源による励起を必要としない。この励起は、標的核酸分子と対合する化学的部分に関連する受容体フルオロフォアを励起するのに十分な距離のみエネルギーを移動させることができる波長で、光を放出する供与体を含む。
特定の例において、プローブは、例えば、ポリメラーゼ成分を基板に結合するリンカー分子を介してアドレス可能な場所で基板に結合または固定される。例となるリンカーには、ストレプトアビジン−ビオチン、ヒスチジン−Ni、S−タグ−S−タンパク質、およびグルタチオン−グルタチオン−S−トランスフェラーゼ(GST)が含まれる。その他の例では、配列決定される標的核酸分子は、例えば、アドレス可能な場所で基板に結合または固定される。特定の例において、オリゴヌクレオチドプライマーは、例えば、5’末端で、基板に固定される。例えば、核酸分子は、5’末端、3’末端、または中間のどこかにより、基板に結合することができる。特定の例において、配列決定反応は、三次元ポリアクリルアミドゲルで行われ、配列決定に必要な試薬のすべては、ゲル内に存在する。
一部の例において、複数のプローブ、プライマー、または核酸分子は、所定のパターン、例えば、アドレス可能な場所で、基板に直接的または間接的に固定される。例えば、薬剤は、規則的なアレイに、またはスライド上に薬剤を含む液滴をマイクロピペット操作することによって、例えば、手動ピペット操作か、もしくは自動アレイヤーを使用し、エッチングされたチャネル内に蒸着することができる。かかる方法は、シグナルが、配列決定反応のそれぞれから検出される場合に、単一の基板上での同時の(または実質上同時の)配列決定を可能にする。一意的な放出シグナルは、例えば、電荷結合素子(CCD)カメラにより検出することができ、基板上の所定の位置から一連のシグナルを検出し、それらを核酸配列に変換することができる。一意的な放出シグナルは、コンピュータ可読媒体に保存することができる。
本開示の前述のおよびその他の特性および利点は、添付図面への参照によって進められる、以下のいくつかの例の詳細な説明からより明白となるであろう。
配列表
ヌクレオチド塩基の標準略語を使用して、添付の配列表に記載の核酸配列を示す。特定の例において、核酸配列の鎖を1つのみ示すが、相補的鎖は、示された鎖への任意の言及によって含まれると理解される(例えば、dsDNA分子ロッドの場合)。
ヌクレオチド塩基の標準略語を使用して、添付の配列表に記載の核酸配列を示す。特定の例において、核酸配列の鎖を1つのみ示すが、相補的鎖は、示された鎖への任意の言及によって含まれると理解される(例えば、dsDNA分子ロッドの場合)。
SEQ ID NO:1は、例示的な標的配列である。
SEQ ID NO:2は、SEQ ID NO:1の圧縮版である。
SEQ ID NO:3〜26は、図2Cに示すプローブを作成するために使用することができる配列である。
SEQ ID NO:27〜30は、SEQ ID NO:3、5、7、および9とそれぞれ置換することができる配列である。
SEQ ID NO:31〜38は、ヘアピンループを形成することができる配列である。
SEQ ID NO:39は、例示的な標的配列である。
SEQ ID NO:2は、SEQ ID NO:1の圧縮版である。
SEQ ID NO:3〜26は、図2Cに示すプローブを作成するために使用することができる配列である。
SEQ ID NO:27〜30は、SEQ ID NO:3、5、7、および9とそれぞれ置換することができる配列である。
SEQ ID NO:31〜38は、ヘアピンループを形成することができる配列である。
SEQ ID NO:39は、例示的な標的配列である。
一部の態様の詳細な説明
略語および用語
本開示をより適切に説明し、本開示を実践する上で当業者の指針となるように、用語および方法の説明を以下に提供する。本明細書で使用される限り、「含む」とは、「包含する」を意味し、単数形「1つの(aまたはan)」または「その(the)」は、文脈による別段の明確な指示がない限り、複数形の引例を含む。例えば、「1つの分子リンカー」に対する引例は、1つまたは複数のかかる分子リンカーを含み、「そのプローブ」に対する引例は、1つまたは複数のプローブおよび当業者には公知のその同等物などに対する引例を含む。用語「or」とは、文脈による別段の明確な指示がない限り、記載する代替要素の単一の要素、または2つ以上の要素の組み合わせを指す。例えば、句「1つのテザーまたは1つの分子ロッド」とは、1つまたは複数のテザー、1つまたは複数の分子ロッド、または1つまたは複数のテザーと1つまたは複数の分子ロッドの両方の組み合わせを指す。
略語および用語
本開示をより適切に説明し、本開示を実践する上で当業者の指針となるように、用語および方法の説明を以下に提供する。本明細書で使用される限り、「含む」とは、「包含する」を意味し、単数形「1つの(aまたはan)」または「その(the)」は、文脈による別段の明確な指示がない限り、複数形の引例を含む。例えば、「1つの分子リンカー」に対する引例は、1つまたは複数のかかる分子リンカーを含み、「そのプローブ」に対する引例は、1つまたは複数のプローブおよび当業者には公知のその同等物などに対する引例を含む。用語「or」とは、文脈による別段の明確な指示がない限り、記載する代替要素の単一の要素、または2つ以上の要素の組み合わせを指す。例えば、句「1つのテザーまたは1つの分子ロッド」とは、1つまたは複数のテザー、1つまたは複数の分子ロッド、または1つまたは複数のテザーと1つまたは複数の分子ロッドの両方の組み合わせを指す。
別段の説明がない限り、本明細書に使用されるすべての技術用語および科学用語は、本開示が属する当業者に一般に理解されるものと同様の意味を有する。本明細書に記載のものと類似または同等の方法および材料は、本開示の実践または検証において使用することができるが、適切な方法および材料を以下に記載する。材料、方法、および例は、一例にすぎず、限定することを意図しない。開示のその他の特性および利点は、以下の詳細な説明および特許請求の範囲から明白である。
Å オングストローム
dsDNA 二本鎖DNA
FRET 蛍光共鳴エネルギー移動
GFP 緑色蛍光タンパク質
LNA ロックト核酸
PEG ポリエチレングリコール
PNA ペプチド核酸
RT 逆転写酵素
ssDNA 一本鎖DNA
Å オングストローム
dsDNA 二本鎖DNA
FRET 蛍光共鳴エネルギー移動
GFP 緑色蛍光タンパク質
LNA ロックト核酸
PEG ポリエチレングリコール
PNA ペプチド核酸
RT 逆転写酵素
ssDNA 一本鎖DNA
受容体フルオロフォア:例えば、約400から900nmの範囲内(約500から800nmの範囲内など)で、供与体フルオロフォアからエネルギーを吸収する化合物。一般に、受容体フルオロフォアは、通常、供与体フルオロフォアの最大吸収波長よりも少なくとも10nm長い(少なくとも20nm長いなど)波長で光を吸収し、約400から900nmに及ぶ波長で最大限の蛍光放射を有する。供与体によって放射されるエネルギーが、受容体を励起することができるように、受容体フルオロフォアは、供与体フルオロフォアの放出と重なる励起スペクトルを有する。理想的には、受容体フルオロフォアは、開示するナノプローブに結合されることができる。
例示的受容体フルオロフォアには、ローダミンおよびその誘導体(N,N,N’,N’−テトラメチル−6−カルボキシローダミン(TAMRA)、6−カルボキシ−X−ローダミン(ROX)など)、フルオレセイン誘導体(5−カルボキシフルオレセイン(FAM)および2’7’−ジメトキシ−4’5’−ジクロロ−6−カルボキシフルオレセイン(JOE)など)、緑色蛍光タンパク質(GFP)、BODIPY(4,4−ジフルオロ−4−ボラ−3a,4a−ジアザ−s−インダセン)およびシアニン染料が含まれるが、これらに限定されるものではない。特定の例において、受容体フルオロフォアは、塩基などのヌクレオチド類似体、糖、またはヌクレオチドのリン酸(α、β、またはγ)に結合されうる。
特定の例において、受容体フルオロフォアは、Dabcyl、Glen ResearchからのBlack Hole Quenchers(商標)、Epoch BiosciencesからのEclipse(商標)Dark Quencher、Integrated DNA TechnologiesからのIowa Black(商標)などのダーククエンチャーである。かかる例において、供与体フルオロフォアに十分に接近する場合に受容体フルオロフォアからの放出シグナルの増加を検出する代わりに、クエンチャーに十分に接近する場合に、供与体フルオロフォアからの放出シグナルの減少を検出することができる。
活性部位:化学反応が発生するポリメラーゼの領域などの、酵素または抗体の触媒部位。活性部位は、基質との相互作用を可能にする空間的配置に1つまたは複数の残基または原子を含み、後者の反応を発生させる。
結合:複合体の形成など、2つ以上の分子間の連関。一般に、複合体内の分子の結合が強力であるほど、解離の速度は遅くなる。特異的結合とは、薬剤と標的との間の選択的結合を指す。
特異的結合の特定の例には、相補的核酸分子への1つの核酸分子のハイブリダイゼーション、タンパク質(ポリメラーゼなど)と標的タンパク質または核酸分子との連関が含まれるが、これらに限定されない。
特定の例において、十分な量、例えば、その結合の検出を可能にする十分な量のタンパク質が核酸分子への非共有化学結合を形成する場合は、タンパク質は、核酸分子に結合することが知られている。
一例において、その結合の検出を可能にする十分な量のオリゴヌクレオチド分子が、塩基対を形成する、または標的核酸分子にハイブリダイズされる場合は、オリゴヌクレオチド分子(プライマーなど)は、標的核酸分子に結合することが観察される。多くの場合、オリゴヌクレオチドとその標的核酸分子との間の結合は、50%のオリゴヌクレオチドが標的から融解する温度(Tm)に特徴付けられる。高温(Tm)は、低温(Tm)の複合体と比較してより強力または安定した複合体を意味する。
特定の例において、結合は、ナノプローブ上に存在する標識を検出することによって評価される。例えば、供与体と受容体フルオロフォアとの相互作用に続いて生成される蛍光性シグナルは、ナノプローブ上のヌクレオチド類似体と標的核酸分子内の相補的ヌクレオチドとの結合の指標として測定することができる。
化学的部分:分子の部分基または官能基。例は、標的核酸分子内の相補的ヌクレオチドに特異的に結合することによって、標的核酸分子の鋳型鎖に可逆的に結合することができる、ヌクレオチドなどの薬剤を含む。特定の例において、化学的部分は、分子リンカーを介してプローブに結合され、化学的部分が、標的核酸分子上の相補的ヌクレオチドに特異的に結合する場合、リンカーから分離しない。
化学的部分の特定の例には、非加水分解性ヌクレオチド類似体など、成長する相補的核酸鎖に組み込むことができないヌクレオチド類似体を含むが、これに限定されない。
cDNA(相補的DNA):内部、非コードセグメント(イントロン)、および転写を決定する調節配列を欠くDNAの一片。cDNAは、mRNAに相補的であり、逆転写酵素を使用して合成することができる。
相補的:二本鎖DNAまたはRNA鎖は、塩基対の2つの相補的鎖から成る。DNA/RNAに見られるそれぞれの塩基に対して1つの相補的塩基(A/T、およびC/Gなど)があるので、任意の単一鎖に対する相補的鎖を決定することができる。
検出:薬剤が存在するか、または存在しないかを決定すること。一部の例において、これは、定量化をさらに含むことができる。例えば、特定の例で開示するプローブを使用すると、例えば、化学的部分が、リンカーから分離されることなく、標的核酸分子内の相補的ヌクレオチドに結合するので、化学的部分の検出が可能になる。
巨視的な数の分子(少なくとも1023の分子など)が、同時に観察することができるように、検出は大量であってもよい。さらに、検出は、顕微鏡法およびバックグラウンドノイズを減少させる全反射などの技術を使用して、単一の分子からのシグナルの識別を含むことができる。個別の分子のスペクトルは、これらの技術によって取得することができる(Ha et al,Proc.Natl.Acad.Sci.USA.93:6264−8,1996)。
供与体フルオロフォア:エネルギーを受容体フルオロフォアへ移動し、それによって検出可能な蛍光性シグナルを生成することができるフルオロフォアまたは発光分子。一般に、供与体フルオロフォアは、約300から900nm、例えば、約350から800nmの範囲で吸収する化合物である。供与体フルオロフォアは、望ましい励起波長、例えば、約103M−1cm−1超で、強いモル吸光係数を有する。種々の化合物は、フルオレセイン(およびその誘導体)、ローダミン(およびその誘導体)、GFP、フィコエリトリン、BODIPY、DAPI(4’,6−ジアミジノ−2−フェニルインドール)、インド−1、クマリン、ダンシル、テルビウム(およびその誘導体)、およびシアニン染料を含む、供与体蛍光成分として用いることができる。特定の例において、供与体フルオロフォアは、エクオリンなどの化学発光分子である。
電磁放射:電界強度および磁場強度の同時周期的変動によって伝播され、電波、赤外線、可視光線、紫外線、X線およびガンマ線を含む一連の電磁波。特定の例において、電磁放射は、単色性、方向性、コヒーレンス、偏光および強度の性質を保有することができるレーザーによって放射される。レーザーからのエネルギーが、供与体フルオロフォアを励起するが、受容体フルオロフォアは励起しないように、レーザーは、特定の波長(または波長の比較的狭い範囲にわたり)で光を放射することができる。
放出シグナル:フルオロフォアが、励起波長で光を吸収した後、フルオロフォアから生成される特定の波長の光。
放出スペクトル:フルオロフォアの後に発生するエネルギースペクトルは、光の特定の波長によって励起される。それぞれのフルオロフォアは、特徴のある放出スペクトルを有する。一例において、個別のフルオロフォア(またはフルオロフォアの一意的な組み合わせ)は、ヌクレオチド類似体に関連し、フルオロフォアからの放出スペクトルは、異なるヌクレオチド類似体間を区別するための手段を提供する。
絡み合う:例えば、複雑に入り組んだ塊において、絡み合うこと。特定の例において、ナノプローブの絡み合いは、標的分子の存在下で、化学的部分(ヌクレオチド類似体など)が、標的核酸分子の相補的ヌクレオチドと相互に作用することを減少または阻止する。その他の特定の例において、ナノプローブの絡み合いは、化学的部分(ヌクレオチド類似体など)間、または化学的部分と重合剤間との望ましくない相互作用、例えば、標的核酸分子との相互作用を阻止する相互作用という結果になる。
励起または励起シグナル:フルオロフォアが、光の異なる(より長いなど)波長を放出する状態までフルオロフォアを励起することが必要な特定の波長の光。
フルオロフォア:光の定義済み波長など、特定の刺激に曝露することによって励起される場合、例えば、異なる波長で、光を放出する(蛍光を発する)化合物。
フルオロフォアは、発光化合物の比較的大きい部類の一部である。発光化合物は、冷光を発する光の特定の波長を必要とせず、エネルギーの化学源を使用する化学発光分子を含む。従って、化学発光分子を使用すると、レーザーなどの、外部の電磁放射源の必要性がなくなる。化学発光分子の例には、エクオリンが含まれるが、これに限定されるものではない(Tsien,1998,Ann.Rev.Biochem.67:509)。
本明細書に開示するナノプローブに使用することができる特定のフルオロフォアの例は、4−アセトアミド−4’−イソチオシアナトスチルベン−2,2’ジスルホン酸、アクリジンおよびアクリジンイソチオシアネートなどのアクリジンおよび誘導体、5−(2’−アミノエチル)アミノナフタレン−l−スルホン酸(EDANS)、4−アミノ−N−[3−ビニルスルホニル)フェニル]ナフタルイミド−3,5二硫酸塩(ルシファーイエローVS)、N−(4−アニリノ−l−ナフチル)マレイミド、アントラニルアミド、ブリリアントイエロー、クマリン、7−アミノ−4−メチルクマリン(AMC、クマリン120)などのクマリンおよび誘導体、7−アミノ−4−トリフルオロメチルコウルアリン(クマラン151);シアノシン;4’,6−ジアミニジノ−2−フェニルインドール(DAPI);5’,5”−ジブロモピロガロール−スルホンフタレイン(ブロモピロガロールレッド);7−ジエチルアミノ−3−(4’−イソチオシアナトフェニル)−4−メチルクマリン;ジエチレントリアミンペンタアセテート;4,4’−ジイソチオシアナトジヒドロ−スチルベン−2,2’−ジスルホン酸;4,4’−ジイソチオシアナトスチルベン−2,2’−ジスルホン酸;5−[ジメチルアミノ]ナフタレン−l−塩化スルホニル(DNS、ダンシルクロライド);4−ジメチルアミノフェニルアゾフェニル−4’−イソチオシアネート(DABITC);エオシンおよびエオシンイソチオシアネートなどのエオシンおよび誘導体;エリスロシンBおよびエリスロシンイソチオシアネートなどのエリスロシンおよび誘導体;エチジウム;5−カルボキシフルオレセイン(FAM)、5−(4,6−ジクロロトリアジン−2−イル)アミノフルオレセイン(DTAF)、2’7’−ジメトキシ−4’5’−ジクロロ−6−カルボキシフルオレセイン(JOE)、フルオレセイン、フルオレセインイソチオシアネート(FITC)、およびQFITC(XRITC)などのフルオレセインおよび誘導体;フルオレスカミン;IR144;IR1446;マラカイトグリーンイソチオシアネート;4−メチルウンベリフェロン;オルトクレゾールフタレイン;ニトロチロシン;パラローザニリン;フェノールレッド;B−フィコエリトリン;o−フタルジアルデヒド;ピレン、ピレンブチラートおよびスクシンイミジル1−ピレンブチラートなどのピレンおよび誘導体;リアクティブレッド4(チバクロン-RTM、ブリリアントレッド3B−A);6−カルボキシ−X−ローダミン(ROX)、6−カルボキシローダミン(R6G)、リサミンローダミンB塩化スルホニル、ローダミン(Rhod)、ローダミンB、ローダミン123、ローダミンXイソチオシアネート、スルホローダミンB、スルホローダミン101およびスルホローダミン101(テキサスレッド)の塩化スルホニル誘導体などのローダミンおよび誘導体;N,N,N’,N’−テトラメチル−6−カルボキシローダミン(TAMRA);テトラメチルローダミン;テトラメチルローダミンイソチオシアネート(TRITC);リボフラビン;ロゾール酸およびテルビウムキレート誘導体など、Nazarenkoらへの米国特許第5,866,366号に記載されている。
その他の適切なフルオロフォアには、約617nmで放出するチオール反応性ユーロピウムキレート(HeydukおよびHeyduk,Analyt.Biochem.248:216−27,1997;J.Biol.Chem.274:3315−22,1999)の他に、GFP、リサミン(商標)、ジエチルアミノクマリン、フルオレセインクロロトリアジニル、ナフトフルオレセイン、4,7−ジクロロローダミン、およびキサンテン(Leeらへの米国特許第5,800,996号に記載)、ならびにその誘導体が含まれる。一例において、フルオロフォアは、大きなストークシフトタンパク質(Kogure et al.,Nat.Biotech.24:577−81,2006を参照)である。例えば、Molecular Probes(Eugene,OR)より入手可能なものなど、当業者に公知のその他のフルオロフォアもまた使用することができる。
特定の例において、フルオロフォアは、供与体フルオロフォアまたは受容体フルオロフォアとして使用される。理想的には、フルオロフォアは、標的生体分子に相互に作用するナノプローブの能力を十分に妨害することなく、ナノプローブ要素に結合される能力を有し、光退色に対して安定しており、高量子効率を有する。複数の受容体フルオロフォアが、例えば、単一のナノプローブ上で、または一緒に使用される異なるナノプローブ上で使用される例では、1つのフルオロフォア(または2以上のフルオロフォアの組み合わせ)からの放出が、その他のフルオロフォア(または2つ以上のフルオロフォアの組み合わせ)と区別可能となるように、フルオロフォアは、区別可能な放出スペクトルを有するように有利に選択される。
本明細書に開示するフルオロフォアは、供与体フルオロフォアまたは受容体フルオロフォアとして使用することができる。特に有用なフルオロフォアは、ナノプローブ(例えば、ポリメラーゼ、分子リンカー、またはヌクレオチド類似体に)に結合される能力を有し、光退色に対して安定しており、高量子効率を有する。さらに、1つのフルオロフォア(Aに関連する1つのものなど)からの放出が、その他のヌクレオチド類似体(Tに関連する1つのものなど)に関連するフルオロフォアと区別可能となるように、ヌクレオチド類似体の異なる集まり(A、T/U、G、およびCに対応するものなど)に関連するフルオロフォアは、区別可能な放出スペクトルを有するように有利に選択される。
フォスター(または蛍光)共鳴エネルギー移動(FRET):励起フルオロフォア(供与体)が、低エネルギー光吸収分子(受容体)に励起状態エネルギーを移動する過程。このエネルギー移動は、無放射であり、主として供与体フルオロフォアと受容体フルオロフォアとの間の双極子−双極子相互作用による。このエネルギーは、距離、例えば、10〜100Åなど、限定される距離を超えて伝達することができる。FRET効率は、式中R0が、FRET効率が50%の距離である、1/(1+(R/R0)^6)に従い低下する。
FRET対:蛍光共鳴エネルギー移動(FRET)に携わることができるフルオロフォアの集まり(対など)。使用可能なFRET対の例を以下に記載する。しかしながら、当業者は、フルオロフォアの他の多数の組み合わせも使用できることを認識するであろう。
FAMは、波長が488nmの光によって最も効率的に励起され、500から650nmのスペクトルで光を放出し、放出極大525nmを有する。FAMは、JOE、TAMRA、およびROXとともに使用するのに適した供与体フルオロフォアである(そのすべては、励起極大514nmを有し、FAMを刺激する光によって有意に刺激されることはない)。
399nmで励起され、511nmで放出する、GFP突然変異体H9−40(Tsien,1998,Ann.Rev.Biochem.67:509)は、BODIPY、フルオレセイン、ローダミングリーンおよびオレゴングリーンとともに使用するのに適した供与体フルオロフォアの役割を果たすことができる。さらに、フルオロフォアテトラメチルローダミン、リサミン(商標)、テキサスレッド、およびナフトフルオレセインは、このGFP突然変異体とともに受容体フルオロフォアとして使用することができる。
フルオロフォア3−(ε−カルボキシ−ペンチル)−3’−エチル−5,5’−ジメチルオキサカルボシアニン(CYA)は、488nmで極大励起され、ひいては、受容体フルオロフォアと使用することができるローダミン誘導体(R6G、TAMRA5およびROXなど)のための供与体フルオロフォアの役割を果たすことができる(Hung et al,Analytical Biochemistry,243:15−27,1996参照)。しかしながら、CYAおよびFAMは双方とも、同じ波長(488nm)で極大励起されるので、適切なFRET対の例ではない。
FRET対の1つ特定の例は、GFP2およびYFPである。
フルオロフォアが、適切な供与体−受容体FRET対を作出する、当分野で公知の技術の分光測光法を使用し、当業者は、容易に決定することができる。さらに、Grantら(Biosens Bioelectron16:231−7,2001)は、本明細書に開示するナノプローブに使用することができるFRET対の特定の例を提供する。
融合タンパク質:本来は結合が見られない2つのアミノ酸配列を含むタンパク質。用語「GFP−ポリメラーゼ融合タンパク質」とは、第1のアミノ酸配列および第2のアミノ酸配列を含むタンパク質を指し、第1のアミノ酸配列は、GFP分子(突然変異体または野生型)であり、第2のアミノ酸配列は、ポリメラーゼである。同様に、用語「GFP−エクオリン融合タンパク質」とは、第1のアミノ酸配列および第2のアミノ酸配列を含むタンパク質を指し、第1のアミノ酸配列は、GFP分子(突然変異体または野生型)であり、第2のアミノ酸配列は、エクオリンである。GFP−エクオリン融合タンパク質は、Baubetら(Proc.Natl.Acad.Sci.USA97:7260−5,2000、参照により本明細書に組み入れられる)の方法を使用して生成することができる。
これらの融合タンパク質は、式X−Yによって表すことができ、式中、Xは、GFPなどのタグであり、Yは、ポリメラーゼなどの重合剤である。一部の例において、アミノ酸鎖は、融合タンパク質の第1および第2のドメインに連結するために使用することができる。
緑色蛍光タンパク質(GFP):Aequorea victoriaにおける蛍光放出源。本明細書で使用される限り、GFPは、例えば、参照により本明細書に組み入れられる、Tsien,1998,Ann.Rev.Biochem.67:509、およびTsienとHeimへの米国特許第5,777,079号および第5,625,048号に記載されるような、野生型タンパク質、およびスペクトルが移動するその突然変異体の双方を指す。特定の例において、GFPは、レーザーを使用して励起される。その他の例において、GFPは、エクオリン、例えば、GFP−エクオリン融合タンパク質を使用して励起される。
GFP−ポリメラーゼ:機能性GFP分子および機能性ポリメラーゼの両方を含む組換え型融合タンパク質。GFPは、ポリメラーゼのN−またはC−末端、またはポリメラーゼが、有意な重合活性を保持する限り(例えば、相補的核酸鎖の伸長を触媒する能力を保持する)、ポリメラーゼ内のどこにでも位置することができる。さらに、GFP−ポリメラーゼは、例えば、精製または基板への結合に役立つように、リンカー(リンカー−GFP−ポリメラーゼ)を含むことができる。さらに、例えば、LRETの使用が望ましい場合は、GFP−ポリメラーゼはまた、機能性エクオリン配列を含むことができる。
ハイブリダイゼーション:DNA、RNAの2つの鎖の相補的領域間、またはDNAとRNAとの間に塩基対を形成し、それによって二本鎖分子を形成すること。特定の度合いのストリンジェンシーをもたらすハイブリダイゼーション条件は、ハイブリダイゼーションの方法および組成物の性質ならびにハイブリダイズする核酸配列の長さに応じて変化することになる。一般に、ハイブリダイゼーションの温度およびハイブリダイゼーション液のイオン強度(Na+濃度など)は、ハイブリダイゼーションのストリンジェンシーを決定する。特定の度合いのストリンジェンシーに達するためのハイブリダイゼーション条件に関する計算は、Sambrook et al.,(1989)Molecular Cloning,second edition,Cold Spring Harbor Laboratory,Plainview,NY(chapters9and11)に論じられている。以下は、ハイブリダイゼーション条件の例示的な集まりであり、これらに限定されるものではない。
非常に高いストリンジェンシー(少なくとも90%の同一性を共有する配列を検出する)
ハイブリダイゼーション:5×SSCにて65℃で16時間
洗浄2回: 2×SSCにて室温(RT)で各15分
洗浄2回: 0.5×SSCにて65°Cで各20分
ハイブリダイゼーション:5×SSCにて65℃で16時間
洗浄2回: 2×SSCにて室温(RT)で各15分
洗浄2回: 0.5×SSCにて65°Cで各20分
高いストリンジェンシー(少なくとも80%の同一性を共有する配列を検出する)
ハイブリダイゼーション:5×〜6×SSCにて65℃〜70℃で16〜20時間
洗浄2回: 2×SSCにてRTで各5〜20分
洗浄2回: 1×SSCにて55℃〜70℃で各30分
ハイブリダイゼーション:5×〜6×SSCにて65℃〜70℃で16〜20時間
洗浄2回: 2×SSCにてRTで各5〜20分
洗浄2回: 1×SSCにて55℃〜70℃で各30分
低いストリンジェンシー(少なくとも50%の同一性を共有する配列を検出する)
ハイブリダイゼーション:6×SSCにてRTから55℃で16〜20時間
少なくとも2回洗浄: 2×〜3×SSCにてRTから55℃で各20〜30分
ハイブリダイゼーション:6×SSCにてRTから55℃で16〜20時間
少なくとも2回洗浄: 2×〜3×SSCにてRTから55℃で各20〜30分
20×SSCは、3.0M NaCl/0.3Mクエン酸三ナトリウムである。
リンカー:本開示のプローブの基板への結合など、1つの分子を別の分子に結合する構造であり、リンカーの一部分は、基板に機能的に連結され、リンカーの別の部分は、プローブに機能的に連結される。
リンカーの特定の1タイプは、重合剤を1つまたは複数の化学的部分(1つまたは複数のヌクレオチド類似体など)に結合することができる、テザー、ロッド、またはそれらの組み合わせなどの分子リンカーであり、リンカーの一部分は、重合剤に機能的に連結され、リンカーの別の部分は、1つまたは複数の化学的部分に機能的に連結される。
ロックト核酸(LNA(商標)):リボヌクレオシドが、2’−酸素原子と4’−炭素原子との間でメチレン単位に連結される二環式核酸。この連結は、ヌクレオチド類似体のリボフラノース環の柔軟性を制限し、剛性を有する二環式N型立体構造にロックされる。さらに、LNAは、A二本鎖の、より熱力学的に安定した形態から成る立体構造に適合するように、隣接した塩基を誘導する。
LNAオリゴヌクレオチドは、DNA合成装置を使用して、標準的なホスホラミダイトの化学的性質によって合成することができる。さらに、LNAは、その他の核酸類似体と同様にDNA、RNAと混合することができる。特例の例において、LNAは、分子リンカーの一部として含まれる。
発光共鳴エネルギー移動(LRET):供与体分子が、発光分子であるか、または例えばレーザーの代わりに、発光分子によって励起される、FRETと類似した過程。LRETを使用すると、バックグラウンドの蛍光を減少させることができる。特定の例において、化学発光分子は、外部の電磁放射源の必要性がなく、供与体フルオロフォア(GFPなど)を励起するために使用することができる。その他の例において、発光分子は、発光分子の励起された共鳴が、1つまたは複数の受容体フルオロフォアを励起する供与体である。
使用できる発光分子の例には、エクオリンおよびルシフェラーゼが含まれるが、これらに限定されない。470nmで頂点に達する、エクオリンからの生体発光は、供与体GFPフルオロフォアを励起するために使用することができる(Tsien,1998,Ann.Rev.Biochem.67:509;Baubet et al.,2000,Proc.Natl.Acad.Sci.U.S.A.,97:7260−5)。その後、GFPは、本明細書に開示する受容体フルオロフォアを励起する。この例において、エクオリンおよびGFPの両方とも、本開示のナノプローブに結合することができる。555nmで頂点に達する、ホタル(Photinus pyralis)のルシフェラーゼからの生体発光は、本明細書に開示する受容体フルオロフォアを励起することができる。この例において、ルシフェラーゼおよびGFPの両方とも、本開示のナノプローブに結合することができる。ルシフェラーゼが使用される一部の例において、受容体フルオロフォアの双極子は、ルシフェラーゼ光の偏光と整列する。その他の例において、多数のルシフェラーゼ分子は、ナノプローブに隣接してまたは周囲にさえ整列する。例えば、内部または表面上にルシフェラーゼの多くの分子を有する球体、デンドリマーまたはシートを作出することも可能である。
ナノプローブまたはプローブ:核酸分子を配列決定するために使用できる分子素子。特定の例において、ナノプローブまたはプローブは、受容体および供与体フルオロフォア対などの配列の検出を可能とする1つまたは複数のタグを含む。
核酸分子(または配列):限定されるものではないが、cDNA、mRNA、ゲノムDNA、および合成(化学的に合成されるなど)DNAまたはRNAを含む、デオキシリボヌクレオチドまたはリボヌクレオチドポリマー。核酸分子は、二本鎖(ds)または一本鎖(ss)であってもよい。一本鎖の場合、核酸分子は、センス鎖またはアンチセンス鎖であってもよい。核酸分子は、天然ヌクレオチド(A、T/U、C、およびGなど)を含むことができ、天然ヌクレオチドの類似体もまた含むことができる。ペプチド核酸(PNA)内など、ペプチド骨格に連結される塩基の集まりは、核酸分子の置換基として使用することができる。
標的核酸分子は、配列決定される核酸であり、当業者に公知の任意の方法(例えば、Ulmerへの米国特許第5,674,743号に記載)によって、精製した形で取得することができる。相補的核酸分子は、標的核酸分子に相補的であり、標的核酸分子を配列決定する場合、伸長される核酸鎖である。
ヌクレオチド:糖および1つまたは複数のリン酸基に連結される、ピリミジン、プリン、またはその合成類似体など、塩基を含むモノマー。ヌクレオチドは、ポリヌクレオチド内の1つのモノマーである。ヌクレオチド配列とは、ポリヌクレオチド内の塩基の配列を指す。
DNAの主なヌクレオチドは、デオキシアデノシン5’−三リン酸(dATPまたはA)、デオキシグアノシン5’−三リン酸(dGTPまたはG)、デオキシシチジン5’−三リン酸(dCTPまたはC)およびデオキシチミジン5’−三リン酸(dTTPまたはT)である。RNAの主なヌクレオチドは、アデノシン5’−三リン酸(ATPまたはA)、グアノシン5’−三リン酸(GTPまたはG)、シチジン5’−三リン酸(CTPまたはC)およびウリジン5’−三リン酸(UTPまたはU)である。
ヌクレオチド前駆体の選択は、配列決定される核酸に依存する。鋳型が、一本鎖DNA分子である場合は、デオキシリボヌクレオチド前駆体(dNTP)は、DNA指令のDNAポリメラーゼの存在下で使用される。あるいは、リボヌクレオチド前駆体(NTP)は、DNA指令のRNAポリメラーゼの存在下で使用される。しかしながら、配列決定される核酸がRNAである場合、dNTPおよびRNA指令のDNAポリメラーゼが使用される。
本明細書に開示するヌクレオチドは、例えば、Nazarenkoらへの米国特許第5,866,336号(参照により本明細書に組み入れられる)に記載されている、修飾塩基、修飾糖成分、修飾リン酸骨格を含有するヌクレオチドをさらに含む。しかしながら、かかる修飾形態は、成長する核酸鎖へのヌクレオチドの組み込み、または相補的核酸鎖へのヌクレオチドの結合を可能にさせる。本明細書に記載される修飾形態は、核酸合成の停止をもたらさない。
ヌクレオチドは、構造上の任意の位置で修飾することができる。例には、修飾ヌクレオチド5−フルオロウラシル、5−ブロモウラシル、5−クロロウラシル、5−ヨードウラシル、ヒポキサンチン、キサンチン、アセチルシトシン、5−(カルボキシヒドロキシルメチル)ウラシル、5−カルボキシメチルアミノメチル−2−チオウリジン、5−カルボキシメチルアミノメチルウラシル、ジヒドロウラシル、β−D−ガラクトシルケウオシン、イノシン、N−6−ソペンテニルアデニン、1−メチルグアニン、1−メチルイノシン、2,2−ジメチルグアニン、2−メチルアデニン、2−メチルグアニン、3−メチルシトシン、5−メチルシトシン、N6−アデニン、7−メチルグアニン、5−メチルアミノメチルウラシル、メトキシアミノメチル−2−チオウラシル、β−D−マンノシルケウオシン、5’−メトキシカルボキシメチルウラシル、5−メトキシウラシル、2−メチルチオ−N6−イソペンテニルアデニン、ウラシル−5−オキシ酢酸、シュードウラシル(pseudouracil)、ケウオシン、2−チオシトシン、5−メチル−2−チオウラシル、2−チオウラシル、4−チオウラシル、5−メチルウラシル、ウラシル−5−オキシ酢酸メチルエステル、ウラシル−S−オキシ酢酸、5−メチル−2−チオウラシル、3−(3−アミノ−3−N−2−カルボキシプロピル)ウラシル、および2、6−ジアミノプリンが含まれるが、これらに限定されるものではない。
構造上の任意の位置でヌクレオチドを修飾するために使用できる修飾糖成分の例には、アラビノース、2−フルオロアラビノース、キシロース、およびヘキソース、またはホスホロチオアート、ホスホロジチオアート、ホスホルアミドチオアート、ホスホロアミダート、ホスホロジアミダート、メチルホスホン酸塩、アルキルホスホトリエステル、もしくはホルムアセタールまたはその類似体などのリン酸骨格の修飾成分が含まれるが、これらに限定されるものではない。
ヌクレオチド類似体:天然塩基、糖、リン酸骨格、またはそれらの組み合わせの1つまたは複数の修飾を含むヌクレオチド。かかる修飾は、ヌクレオチドが成長する核酸鎖へ組み込まれることが不可能になる可能性をもたらす。特定の例には、非加水分解性ヌクレオチドが含まれる。非加水分解性ヌクレオチドには、αとβリン酸間の酸素が、窒素または炭素と置換されている、モノヌクレオチドおよびトリヌクレオチドが含まれる(Jena Bioscience)。HIV−I逆転写酵素は、窒素で置換されたαおよびβリン酸との間の酸素でdTTPを加水分解することはできない(Ma et al,J.Med.Chem.,35:1938−41,1992)。
ヌクレオチド類似体の「型」とは、検出される共通する特徴を共有のヌクレオチド類似体の集まりのうちの1つを指す。例えば、ヌクレオチド類似体の集まりは、A、T、CおよびG類似体(DNAに対して)またはA、U、CおよびG類似体(RNAに対して)の4つの型に分けることができる。この例において、ヌクレオチド類似体のそれぞれの型は、集まりの中でその他のヌクレオチド類似体から区別可能となるように(例えば、蛍光分光法またはその他の光学的手段によって)、1つまたは複数の受容体フルオロフォアなどの一意的なタグに関連付けられる。
「C」の代わりに使用できる例示的なヌクレオチド類似体は、G−クランプである(Glen Research)。G−クランプは、三環系アミノエチル−フェノキサジン2’−デオキシシチジン類似体(AP−dC)である。G−クランプは、ホスホラミダイトとして入手可能であり、従って、DNA構造に合成することができる。かかる類似体は、本明細書で提供されるナノプローブに使用することができる(例えば、図1Aに示すプローブの化学的部分22、24、26、28のうちの1つとして、または図1Bに示すdCTP22の代用となることができる)。
オリゴヌクレオチド:少なくとも6つのヌクレオチド、例えば、少なくとも9つ、少なくとも15、少なくとも18、少なくとも24、少なくとも30、少なくとも50、少なくとも100、少なくとも200、またはさらに少なくとも500のヌクレオチドの長さの直鎖ポリヌクレオチド(DNAまたはRNAなど)配列。オリゴヌクレオチドは、改変糖成分などの非天然部分、またはホスホロチオアート−オリゴデオキシヌクレオチドなどの糖間の連結を含むことができる。特定の例において、非天然部分を含むオリゴヌクレオチドは、RNAまたはDNAに結合し、ペプチド核酸(PNA)分子を含むことができる。
ORF(オープンリーディングフレーム):終止コドンが全くないアミノ酸をコードする一連のヌクレオチドトリプレット(コドン)。通常、これらの配列は、ペプチドに翻訳可能である。
対合:標的核酸分子上の相補的ヌクレオチドへの化学物質(ヌクレオチド類似体など)の結合など、対に結合する過程。特定の例において、対合は、共有結合の形成をもたらす。その他の例において、対合は、化学結合の形成をもたらすことはない。
ペプチド核酸(PNA):核酸塩基を有する中性ペプチド様骨格を含む、情報分子の部類であり、従来のオリゴヌクレオチドよりも高い親和性および特異性で、相補的RNAまたはDNAにハイブリダイズすることを可能にする。PNA分子の構造は、DNAと類似しており、デオキシリボースリン酸骨格は、ペプチドに見られるものと類似する骨格と置換されている。特定の例において、PNAは、ヌクレアーゼおよびプロテアーゼに耐性を示す。PNAは、フルオロフォア(例えば、受容体フルオロフォア)など、N(5)−末端で官能基を含むことができる。
持続長(lp):線状鎖のための平均的局所立体構造であり、任意のセグメントによって画定される方向のすべての鎖セグメントの平均的延在の合計を反映する。特定の例において、持続長は、実質的には、平均にして68.40度の屈曲で発生する周長距離を測定する屈曲度(従って、鎖の有効弾性)である。
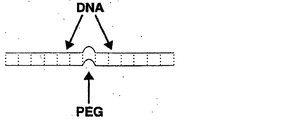
ポリエチレングリコール(PEG):エチレン化合物のポリマー、H(OCH2CH2)nOH。PEG付加は、PEG構造をその他の分子、例えば、標的または活性化可能な部分などの機能分子に付加する行為である。PEGは、水、メタノール、ベンゼン、ジクロロメタンに可溶性であり、ジエチルエーテルおよびヘキサンに不溶性である。
開示するナノプローブに使用可能なPEGの特定の例には、3〜5ユニットのスペーサー18などの1〜7ユニットのスペーサー18(Integrated DNA Technologies,Coralville,IA)、C3スペーサーホスホラミダイト(1〜10ユニットなど)、スペーサー9(1〜10ユニットなど)、PC(光開裂可能)スペーサー(1〜10ユニットなど)、(すべてIntegrated DNA Technologiesより入手可能)が含まれるが、これらに限定されるものではない。その他の例において、開示するナノプローブで使用できるPEGの長さは、PEGの1から40のモノマーが含まれるが、これらに限定されるものではない。
重合剤:線状鎖またはポリマー鎖の三次元網目構造を形成するために、化学反応内にモノマー分子(ヌクレオチドなど)を一緒に反応させることができる化合物。重合剤の特定の例は、ポリメラーゼ、すなわち核酸鋳型に相補的な核酸鎖を合成する酵素である。核酸分子を配列決定するために使用できるポリメラーゼの例には、大腸菌(E.coli)DNAポリメラーゼI、特に、3’から5’エキソヌクレアーゼ活性を有するクレノウフラグメント、Taqポリメラーゼ,逆転写酵素(HIV−I RTまたはLlレトロトランスポゾンの逆転写酵素など)、大腸菌RNAポリメラーゼ、および麦芽RNAポリメラーゼIIが含まれるが、これらに限定されるものではない。
ポリメラーゼの選択は、配列決定される核酸に依存する。鋳型が、一本鎖DNA分子である場合、DNA指令のDNAまたはRNAポリメラーゼを使用することができ、鋳型が、一本鎖RNA分子である場合、逆転写酵素(RNA指令のDNAポリメラーゼなど)を使用することができる。
プライマー:短い核酸分子、例えば、プライマーと標的核酸鎖との間にハイブリッドを形成するために、核酸ハイブリダイゼーションによって、相補的標的核酸分子にアニーリングすることができる、少なくとも9のヌクレオチドの配列。プライマーは、ポリメラーゼ酵素によって、標的核酸分子に沿って延在することができる。従って、例えば、プライマーが、ストリンジェントなハイブリダイゼーション条件下で、標的核酸分子をハイブリダイズするように、個別のプライマーは核酸配列決定のために使用することができ、ここでプライマーの配列は標的核酸分子に特異的である。
特定の例において、プライマーは、配列決定される標的核酸分子に相補的な少なくとも10の連続ヌクレオチドなど、長さが少なくとも10のヌクレオチドである。特異性を強化するために、配列決定される標的核酸分子に相補的な少なくとも12、少なくとも15、少なくとも20、または少なくとも30の連続ヌクレオチドを有するプライマーなど、より長いプライマーを用いることができる。プライマーを調製および使用する方法は、例えば、Sambrook et al(1989)Molecular Cloning:A Laboratory Manual,Cold Spring Harbor,New York;Ausubel et al.(1987)Current Protocols in Molecular Biology,Greene Publ.Assoc.&Wiley−Intersciencesに記載されている。
精製:精製という用語は、絶対的純度を意味せず、むしろ相対的な用語を意図する。従って、例えば、精製GFP−ポリメラーゼタンパク質調製物は、GFP−ポリメラーゼタンパク質が、細胞内の環境にあるタンパク質より純粋であるものである。特定の例において、GFP−ポリメラーゼタンパク質が、調製物の総タンパク質含有量の少なくとも50%を表すように、GFP−ポリメラーゼタンパク質の調製物は精製されるが、例えば、総タンパク質含有量の90%または98%でもよい。
量子ドット:安定的に蛍光を発し、均一の表面領域を保有し、生体分子(1つまたは複数のナノプローブなど)を結合させるように化学修飾することができる、設計された無機半導体結晶ナノ粒子略球状であるが、本開示のナノプローブに結合される量子ドットは、いずれの形状(球状、管状、錐体状、円錐形、または立方体など)であってもよいが、特に適切なナノ粒子は球状である。
一般に、量子ドットは、既に記載したように、相対的単分散度(例えば、調製物内量子ドット間で約10%未満変化する核の直径を有する)で調製することができる(Bawendi et al.,J.Am.Chem.Soc.115:8706,1993)。当技術分野で公知の量子ドットは、例えば、CdSe、CdS、およびCdTe(集合的に「CdX」と称される)から成る群から選択される核を有する。
組換え型:組換え型核酸分子またはタンパク質配列は、天然に存在しない配列を有するか、または2つの人工的組み合わせによって作出される配列、さもなければ配列の別個のセグメントを有するものである。特定の例において、この人工的組み合わせは、化学合成によって、または核酸もしくはタンパク質配列の単離セグメントの人為的操作、例えば、遺伝子工学技術によって達成される。特定の例において、dsDNAから成る分子ロッドは、組換え型分子である。
逆転写酵素:一般にRNAを使用するが、鋳型としてDNAを使用することができる鋳型特異的DNAポリメラーゼ。
標的核酸分子に可逆的に結合:可逆平衡の状態で存在する一時的結合。例えば、ポリメラーゼの活性部位で相補体へのヌクレオチドの一過性対合を含み、ヌクレオチドは、ポリメラーゼによって形成される核酸分子にヌクレオチドを共有結合的に組み込む化学反応(加水分解または共有結合形成など)を受けない。
RNAポリメラーゼ:DNA鋳型に相補的なリボヌクレオチド前駆体の重合を触媒する酵素。
ロッドまたは分子ロッド:分子リンカーの一部など、ナノプローブの一部の剛性を高めるために、ナノプローブの分子リンカー内に含むことができる構造。分子ロッドは、標的核酸分子の非存在下で、化学的部分、重合剤、またはそれらの組み合わせの相互作用を減少させるように十分な剛性を有する。さらに、分子ロッドは、標的生体分子の存在下で、化学的部分、重合剤、またはそれらの組み合わせが相互に作用することを可能にする長さである。
特定の例において、分子ロッドは、持続長よりも短い長さを有する分子リンカー内に存在し、それによって、標的生体分子の非存在下で、化学的部分、重合剤、またはそれらの組み合わせの相互作用を有意に減少させる。一例において、dsDNAから成る分子ロッドは、dsDNAの持続長、約150のヌクレオチドより短い、10〜140のヌクレオチドの長さを有する。
例示的な分子ロッドには、dsDNA分子、ペプチド核酸(PNA)、カーボンナノチューブ、ロックト核酸分子(LNA)、微小管、バクテリア、線形ウイルス粒子、ウイルス尾部繊維、またはその他のタンパク質構造(αヘリックスもしくはβバレルを含むタンパク質成分、またはロイシンジッパー構造などのその他のタンパク質構造など)が含まれるが、これらに限定されるものではない。分子ロッドは、DNAから構築される立方体または八面体など、三次元分子構造の一部であってもよい(例えば、Seeman,Sci.Am.290:64−9および72−5,2004を参照)。特定の例において、分子ロッドは、10〜150のヌクレオチド、10〜140のヌクレオチド、20〜100のヌクレオチド、20〜50のヌクレオチド、20〜40のヌクレオチド、30〜50のヌクレオチド、または約20、30、または40のヌクレオチドなど、少なくとも10のヌクレオチド、少なくとも35のヌクレオチド、または150のヌクレオチドまたはそれ未満のdsDNA分子である。
試料:核酸分子(例えば、ゲノムDNA、cDNA、RNA、またはmRNA)など、生体分子を含む試料など生物検体。例示的な試料は、末梢血(または血清などその一部)、尿、唾液、組織生検、頬のぬぐい液、外科標本、微細針吸引物、羊水穿刺試料、および剖検材料に存在するものなど、対象からの細胞または細胞溶解物を含むものである。
一連のシグナル:相補的ヌクレオチドとの化学的部分の対合を示す、標的核酸分子内の相補的ヌクレオチドと化学的部分(ヌクレオチド類似体など)との特異的結合により放出される、光またはスペクトルシグナルなどの電磁シグナルを含む放出シグナルの順次的連続である。特定の例において、一連のシグナルは、一連の受容体フルオロフォア放出シグナルであり、それぞれの一意的なシグナルは、特定の化学的部分に関連する。
シグナル:情報を提供する物理的性質に備わる検出可能な変化またはインパルス。開示する方法との関連において、例には、光、例えば、特定の量または波長の光などの電磁シグナルが含まれる。特定の例において、シグナルは、光の消失など、物理事象の消滅である。特性シグナルは、ナノプローブ上の特定のヌクレオチド類似体が、標的核酸分子内の相補的ヌクレオチドと特異的に結合する場合に期待される特定のシグナルである。例えば、特性シグナルは、ヌクレオチド類似体に結合される、または関連するフルオロフォアによって予想することができる、蛍光タグを付けられた非加水分解性ヌクレオチド類似体から放出される、結果として生じるシグナルであってもよい。
対象:ヒトならびにウシ、ブタ、ウマ、イヌ、ネコ、トリ、爬虫類および魚などの動物の対象を含む、多細胞性脊椎動物の生命のある生物。
基板:その他の分子(タンパク質または核酸分子など)を結合または内部に埋め込むことができる材料または表面。特定の例において、基板は、ガラスおよび石英を含む光透過性の生体適合性材料から作出される。例えば、基板は、長さ3cm×幅1cm×厚さ0.25cmのガラス顕微鏡用スライドであってもよい。さらにその他の例において、例えば、LRETが使用される場合、基板は不透明であってもよい。一例において、基板は、ゲルマトリクスである。特定の例において、基板は、放物線フローチャネルプロファイルを有するマイクロ流体デバイスである。
特定の例において、基板は、その他の分子を結合する前に処理される。例えば、ガラス顕微鏡用スライドを、水中で30分間超音波処理によって洗浄し、10%のNaOH中に30分間浸し、蒸留水ですすぎ、80℃で10分間乾燥または一晩風乾することができる。
タグ:例えば、分光測光法、フローサイトメトリー、または顕微鏡法によって検出することができる薬剤。例えば、1つまたは複数のタグは、ナノプローブに結合されることができ、それによって核酸分子の配列決定が可能になる。例示的なタグには、放射性同位元素、フルオロフォア、化学発光薬剤、電荷、酵素およびそれらの組み合わせが含まれる。
テザー:直接的または間接的に、1つまたは複数の化学物質(ヌクレオチド類似体など)を重合剤に連結するために、ナノプローブに含むことができる構造。例えば、1つまたは複数の分子ロッドと組み合わせて、1つまたは複数のテザーは、1つまたは複数の化学物質(ヌクレオチド類似体など)を重合剤に連結するために使用することができる。理想的には、テザーは、標的核酸分子の存在下で、化学物質(ヌクレオチド類似体など)、重合剤、タグ、またはそれらの組み合わせに相互作用させると同時に、テザーが、それ自体と、またはナノプローブのその他の要素とからまる可能性を減少させる長さである。
例示的なテザーには、PEGなどの水溶性長連鎖分子、ペプチド(少なくとも30のアミノ酸のペプチド、例えば、ヘリカーゼをヌクレアーゼに結合するRecBタンパク質の70アミノ酸長の柔軟性のあるテザーの少なくとも30の連続アミノ酸など(Singleton et al.,Nature432:187−93,2004))、糖鎖(2000〜14000の残基など)、脱塩基リン酸ジエステルスペーサー(IDT5’dSpacerなど)、炭水化物鎖(少なくとも10の糖分子)、およびポリカプロラクトン鎖(少なくとも10のモノマーなど)が含まれる。特定の例において、テザーは、PEG、例えば、約23−164Å長のPEGから成る。
標的核酸配列または分子:事前に選択した核酸分子、例えば、検出または配列が所望されるもの。核酸分子は、精製した形態である必要はない。その他の種々の生体分子もまた、標的核酸分子内に存在することができる。例えば、標的核酸分子は、細胞または生体試料(その他の核酸分子およびタンパク質を含むことができる)内に存在することができる。
形質転換:形質転換細胞は、分子生物学技術によって核酸分子を導入された細胞である。本明細書で使用される限り、形質転換という用語は、ウイルスベクターによるトランスフェクション、プラスミドベクターによる形質転換、ならびにエレクトロポレーション、リポフェクション、注入、およびパーティクルガン加速による裸のDNAの導入を含み、核酸分子をかかる細胞に導入することができるすべての技術を含む。特定の例において、細胞は、本明細書に開示するプローブにより形質転換される。
十分な条件下:所望の活性を可能にする任意の環境を画定するために使用される句。
例には、例えば、1つまたは複数の突然変異を含む標的核酸分子などの標的核酸分子が、試料内に存在するかどうか決定するために、試料内の標的核酸分子の配列決定を可能にするのに十分な条件下で、プローブを試料に接触させる工程が含まれる。
一意的な放出シグナル:特定のフルオロフォアのための放出スペクトルなど、特定の事象についての情報を伝達する放出シグナルであり、その他のシグナル(その他の放出スペクトルシグナルなど)と区別することができる。標的核酸分子上の相補的塩基との化学的部分の対合が、一意的なシグナルまたはシグナルの組み合わせ(異なる一意的な波長で放出するフルオロフォアなど)をもたらすように、開示する方法に関連する例は、1つまたは複数の個別のフルオロフォアまたはその他のタグを、それぞれの型の化学的部分(A、T/U、CまたはG非加水分解性ヌクレオチド類似体など)に関連させる工程を含む。それぞれの化学的部分は、例では、化学的部分に関連するタグに基づく一意的な放出シグナルを有する。このシグナルは、どの型の化学的部分(A、T/U、CまたはG非加水分解性ヌクレオチド類似体など)が、標的核酸内の相補的ヌクレオチドと対になったか決定するために使用することができ、これらのシグナルは、組み合わせて核酸配列を示す。
シグナルは、異なる波長のみでなく、一意的なスペクトルを形成する、種々の波長で異なる強度を特徴とすることができる。特に、波長が同じ集まりを有する2つのシグナルは、それらが、特定の波長でいくつかの異なる強度を有する場合に区別することができる。
一般戦略
標的核酸分子を配列決定するために使用できる、開示するナノプローブは、重合剤、および標的核酸分子内の相補的ヌクレオチドに結合することができる1つまたは複数の化学的部分を離間する1つまたは複数の分子リンカーを含む。一般に、化学的部分は、伸長している核酸分子に永久に組み込むことができない。リンカーは、標的核酸分子の非存在下で、重合剤と化学的部分とを所望の距離だけ離間して実質上維持するが、標的生体分子の存在下で、それらが実質上相互に作用しうる、長さおよび柔軟性の組み合わせを有する。さらに、分子リンカーは、分子リンカー同士の絡み合いを実質上回避し、リンカーの末端の化学的部分の絡み合いを実質上回避する。表1は、使用可能な重合剤および化学的部分のいくつかの組み合わせを例示する。さらに、重合剤および化学的部分に関連することができる例示的なタグを提供する。
標的核酸分子を配列決定するために使用できる、開示するナノプローブは、重合剤、および標的核酸分子内の相補的ヌクレオチドに結合することができる1つまたは複数の化学的部分を離間する1つまたは複数の分子リンカーを含む。一般に、化学的部分は、伸長している核酸分子に永久に組み込むことができない。リンカーは、標的核酸分子の非存在下で、重合剤と化学的部分とを所望の距離だけ離間して実質上維持するが、標的生体分子の存在下で、それらが実質上相互に作用しうる、長さおよび柔軟性の組み合わせを有する。さらに、分子リンカーは、分子リンカー同士の絡み合いを実質上回避し、リンカーの末端の化学的部分の絡み合いを実質上回避する。表1は、使用可能な重合剤および化学的部分のいくつかの組み合わせを例示する。さらに、重合剤および化学的部分に関連することができる例示的なタグを提供する。
(表1)例示的な重合剤/化学的部分/タグの組み合わせ

プローブは、最小距離の外側に重合剤と化学的部分との潜在的な相互作用を維持のみする必要がある。さらに、分子成分の場所が、統計的確率の観点のみから表すことができるので、相互作用の非存在は、絶対的ではなく、代わりに、重合剤と化学的部分との間(および化学的部分自体の間)の望ましくない相互作用を所望のレベルまで減少させる方法において、動的分子運動の制限を意味することが理解される。プライマーの存在下で、重合剤が標的核酸分子に結合すると、分子リンカーの柔軟性は、重合剤と化学的部分とが相互に作用するのに十分となる(例えば、非加水分解性ヌクレオチド類似体と、ポリメラーゼの活性部位との相互作用)。
核酸分子を配列決定するためのナノプローブ
本開示は、標的核酸分子を配列決定するためのナノプローブを提供する。特定の例において、開示するナノプローブは、インビトロ、エクスビボ、インサイチュー、またはインビボでも使用される。「Medusa」プローブと称されるプローブには、重合剤、および重合剤上で離間する1つまたは複数の分子リンカーが含まれ、リンカーは、標的核酸分子内の相補的ヌクレオチドと特異的に結合することによって、リンカーから分離することなく、核酸分子の鋳型鎖と可逆的に結合することができる化学的部分を有する。標的核酸分子内の相補的ヌクレオチドへのリンカー上の化学的部分の可逆的な組み込みは、標的核酸分子内の相補的ヌクレオチドとリンカー上の化学的部分との対合を示す、特性シグナルの放出(供与体フルオロフォア放出における減少、または受容体フルオロフォア放出における増加)によって示される。
本開示は、標的核酸分子を配列決定するためのナノプローブを提供する。特定の例において、開示するナノプローブは、インビトロ、エクスビボ、インサイチュー、またはインビボでも使用される。「Medusa」プローブと称されるプローブには、重合剤、および重合剤上で離間する1つまたは複数の分子リンカーが含まれ、リンカーは、標的核酸分子内の相補的ヌクレオチドと特異的に結合することによって、リンカーから分離することなく、核酸分子の鋳型鎖と可逆的に結合することができる化学的部分を有する。標的核酸分子内の相補的ヌクレオチドへのリンカー上の化学的部分の可逆的な組み込みは、標的核酸分子内の相補的ヌクレオチドとリンカー上の化学的部分との対合を示す、特性シグナルの放出(供与体フルオロフォア放出における減少、または受容体フルオロフォア放出における増加)によって示される。
ポリメラーゼなどの重合剤は、標的核酸分子に結合することができ、相補的ヌクレオチドが相補的核酸分子に組み込まれるように伸長する相補的核酸分子の合成を促進することができる活性部位を含む。特定の例において、相補的ヌクレオチドは、標的核酸分子に相補的な伸長している核酸分子に永久に組み込まれることができる加水分解性ヌクレオチドである。例えば、これは、標的核酸分子に可逆的に結合する場合があるが(例えば、相補的ヌクレオチドとの水素結合を形成することによって)、伸長している相補的核酸分子に永久に組み込むことができない、化学的部分(非加水分解性ヌクレオチド類似体、例えば、非加水分解性dNTPなど)と対照的である。
リンカーから分離することなく、核酸分子の鋳型鎖に可逆的に結合することができる化学的部分は、標的核酸分子に相補的な伸長している核酸分子に永久に組み込まれるようにならないものである。例えば、化学的部分は、標的核酸分子の鋳型鎖との1つまたは複数の非共有化学結合(水素結合など)を形成することがあるが、かかる結合は、伸長している鎖に共有結合的に組み込まれる加水分解性ヌクレオチド(ATP、GTP、CTP、UTP、dATP、dGTP、dCTP、dTTPなど)、相補的鎖の伸長を可能にするヌクレオチドとの化学的部分の置換を可能にする速度の可逆平衡の状態で存在する。化学的部分は、標的核酸分子内の相補的ヌクレオチドと特異的におよび可逆的に結合することができ、化学的部分に関連する受容体標識を、受容体標識を刺激する供与体標識に十分に接近させる。かかる対合は、対になる特定の化学的部分を特徴とするシグナルの放出をもたらすことができる。例えば、結合が、化学的部分の拡散に関係するので、異なる化学的部分のすべては、同一の速度で重合剤の活性部位に最初に結合することがある。しかしながら、異なる化学的部分の放出の動力学が、形成される結合の数および強度に関係するので、標的鎖内の曝露ヌクレオチドに相補的な化学的部分は、その他の化学的部分より強力に結合し、それより長い間活性部位に結合を保つことになる。この結合時間の増加は、対になる特定の化学的部分を特徴とするシグナルの生成および検出を可能にする。
かかる化学的部分の特定の例は、ヌクレオチド類似体、例えば、非加水分解性ヌクレオチド類似体である。化学的部分は、標的核酸鎖内の相補的ヌクレオチドと対になるために、重合剤の活性部位と相互に作用する化学的部分の能力、および伸長している核酸分子に可逆的に非共有結合する(通常、積み重ねによる)能力を実質上妨害しない任意の手段によって分子リンカーに結合することができる。例えば、化学的部分が、ヌクレオチド類似体である場合、塩基、糖、またはリン酸を介して、分子リンカーに結合することができる。特定の例において、化学的部分がモノヌクレオチドである場合、塩基または3’リボース炭素上の分子リンカーに結合することができる。
特定の例において、化学的部分を重合剤に結合するのに使用される分子リンカーは、複数の点で重合剤に結合される、複数の個別のリンカーを含む。例えば、それぞれが、標的核酸分子内の異なるヌクレオチドに特異的に結合することができる、異なる型の化学的部分を有する、少なくとも4つの独立した分子リンカーは、重合剤に結合することができる。特定の例において、それぞれが、標的核酸分子内の異なるヌクレオチドに特異的に結合することができる、特定の化学的部分に関連する、異なるフルオロフォアまたはフルオロフォアの組み合わせを有する、少なくとも8つの独立した分子リンカーは、重合剤に結合することができる。例えば、8つのリンカーのうちの2つは、dCTPを含むことができ、それぞれのdCTPが、検出可能な別個のシグナルを生成するように、それぞれのdCTPは、異なる受容体フルオロフォアまたはフルオロフォアの組み合わせに関連する。これは、塩基、この場合はGの連続が、より容易に区別されることを可能にする。しかしながらその他の例において、分子リンカーは、例えば自由回転を可能とし、それによってリンカーの末端上の化学的部分が、重合剤活性部位への平等なアクセスを有することを可能にする単一の共有結合によって、ある点で重合剤に結合される。例えば、分子リンカーは、1つの場所で重合剤に連結および結合することができる。特定の例において、分子リンカーは、その他の薬剤(リンカーなど)に連結および結合され、次いで重合剤に結合される。化学的部分を重合剤に結合するために使用される分子リンカーは、重合剤に結合される、分岐構造を形成する複数の分子リンカーを含むことができる。例えば、それぞれの分岐は、標的核酸分子内の異なるヌクレオチドと特異的に結合することができる異なる化学的部分を有することができる。特定の例において、分岐構造は、少なくとも4つの分岐を含み、それぞれの分岐は、異なる化学的部分を有する。一部の例において、分岐分子リンカーは、単一の点で重合剤に結合する。
特定の例において重合剤はタグに関連し、化学的部分のそれぞれは、リンカーが有する特定の化学的部分を識別するタグに関連し、重合剤に関連するタグと、化学的部分に関連するタグとの相互作用は、相補的ヌクレオチドとリンカー上の化学的部分との対合を示す特性シグナルの放出を誘発する。プローブの要素とのタグの関連は、該要素とのタグの直接的結合を含むことができる。例えば、重合剤は、GFP−ポリメラーゼ融合タンパク質であってもよい。同様に、化学的部分が、ヌクレオチド類似体である場合、タグは、ヌクレオチド類似体の塩基、糖、またはリン酸に結合することができる。理想的には、プローブの重合剤または化学的部分へのタグの直接的または間接的結合は、その要素の生物学的活性を有意に抑制しない。例えば、理想的には、重合剤へのタグの結合は、ポリメラーゼ活性を20%超低下させない。その他の例において、重合剤または化学的部分へのタグの関連は、その要素へのタグの直接的結合を必要としない。例えば、タグは、分子リンカー(例えば、テザー上またはロッド上)など、プローブのその他の部分上にあってもよい。かかる例において、タグは、重合剤の活性部位での化学的部分の検出、および化学的部分と相補的ヌクレオチドとの対合を可能とするように、重合剤または化学的部分へ十分に接近する。
特定の例において、重合剤に関連するタグは、それぞれの化学的部分に関連するタグにより供与体−受容体の対を形成し、それによって、供与体−受容体の対の相互作用は、特性シグナルの放出を刺激する。一部の例において、供与体は、受容体が特性シグナルを放出するために反応する刺激を放出するために、外部刺激(レーザーまたは化学発光分子など)を加えることによって刺激される。例えば、重合剤に関連するタグは、供与体フルオロフォアであってもよく、化学的部分に関連するタグのそれぞれは、特定の化学的部分に対して一意的な放出シグナルを放出する1つまたは複数の受容体フルオロフォアを含む。
特定の例において、分子リンカーは、ヌクレオチドなどの線状ポリマーを含む。例えば、分子リンカーは、テザー、ロッド、またはそれらの組み合わせを含むことができる。分子リンカーは、リンカーの絡み合いを抑制するために十分な距離をおいて重合剤の周囲で離間することができ、化学的部分が重合剤の活性部位に到達するほど十分に長い。さらに、分子リンカーは、標的核酸鎖の非存在下で、重合剤および化学的部分の実質上の絡み合いを回避するように、重合剤および化学的部分を互いから十分な距離をおいて離間して維持することができる。一部の例において、長さが少なくとも持続長と同じくらい長い分子ロッド、例えば、10〜150のヌクレオチドの長さを有するdsDNAロッドなど、分子リンカーの少なくとも一部(ロッド)は、標的核酸分子の非存在下で、重合剤と化学的部分との相互作用を減少させるのに十分な剛性を有する。特定の例において、分子ロッドは、20〜100のヌクレオチド、例えば、40のヌクレオチドなど、10から140のヌクレオチドのdsDNA配列である。特定の例において、分子ロッドは、120オングストローム(Å)長である。
例えば、分子リンカーの剛性を高めるために、分子リンカーは、分子ロッドによって連結される少なくとも2つのテザーなど、1つまたは複数の分子ロッドを含むことができる。二本鎖DNA分子など、分子ロッドの包含は、プローブの剛性を高めるために含まれてもよく、さらに官能基を分離することもできる。例えば、両端にテザーを有する分子ロッドを含む分子リンカーは、化学的部分および重合剤が、ブラウン運動によって移動する少なくとも2つの点を有することができる。すなわち、それぞれが、テザーによって可能となるように、ロッドの末端に対して、化学的部分の可能性のあるすべての場所を表す、少なくとも2つの雲球体があることになる。これらの球体は、ある程度まで交差する。標的核酸分子の非存在下で、ナノプローブは、実質上交差しない球体を有することができる。一部の例において、分子ロッドの2つの末端間の距離は、2つのテザーの長さの合計に満たない。特定の例において、分子ロッドは、標的核酸分子の非存在下で、ナノプローブ上の供与体および受容体フルオロフォア間のFRETを減少させるために使用される。
本明細書に開示する重合剤は、標的核酸分子に結合することができ、かつ相補的ヌクレオチドが相補的核酸分子に組み込まれるように伸長する相補的核酸分子の合成を促進することができる活性部位を含む。さらに、重合剤は、絡み合いを有意に回避するのに十分な距離をおいて、重合剤上で離間される1つまたは複数の分子リンカーを含む。それぞれのリンカーは、リンカーから分離することなく、標的核酸分子内の相補的ヌクレオチドに特異的に結合することによって、核酸分子の鋳型鎖に可逆的に結合することができる、異なる化学的部分(非加水分解性ヌクレオチド類似体など)を有する。重合剤は、例えば、重合剤に関連するタグ、およびリンカーが有する化学的部分を識別するそれぞれの化学的部分に関連するタグなどのタグをさらに含むことができる。ポリメラーゼに関連するタグは、リンカーが有する化学的部分を識別する特性シグナルを放出するために、化学的部分に関連するそれぞれのタグを相互に作用することができる。
上述のとおり、特定の例において、重合剤(例えば、重合剤に関連するタグ)および化学的部分が、標的生体分子の非存在下で、実質上相互に作用しないように、分子リンカーの長さは、重合剤および化学的部分を互いから十分に離間して維持するものである。例えば、標的分子の存在下または非存在下で、プローブの一部がプローブの1つまたは複数の他の部分と相互に作用するかどうか決定するための方法は、当技術分野では公知である。一例において、特定の長さの分子リンカーが適切であるかどうか決定するために、特定の分子リンカー長を有する本開示のプローブは、本明細書に開示する方法を使用して生成される。特定の例において、複数のプローブが生成され、それぞれは、異なる分子リンカー長を有する。使用するのに適切な分子リンカーの長さを識別するために、供与体フルオロフォアは、分子リンカーの一末端で重合剤に結合され、適切な受容体フルオロフォアは、分子リンカーのもう一方の末端で化学的部分に結合される。特定の例において、供与体および受容体は、FRET対である。分子リンカーの末端が、互いと相互に作用することができるかどうか決定するために、分子リンカーは、標的核酸分子および適切なプライマーの存在下または非存在下で溶液中に入れることができ、例えば分光測光法または蛍光顕微鏡検査法によって受容体放出蛍光が検出される。特定の例において、標的核酸分子およびプライマーが存在する場合にのみ有意な受容体放出蛍光(例えば、所定の閾値を超える)を生成し、標的核酸分子およびプライマーの非存在下では、バックグラウンドレベルの受容体放出蛍光しか生成しない分子リンカーの長さは、本開示のプローブで使用することができる。対照的に、特定の例において、標的核酸分子およびプライマーが存在する場合には有意な受容体放出蛍光を生成せず、または標的核酸分子およびプライマーの非存在下ではバックグラウンドを有意に超える受容体放出蛍光を生成する分子リンカーの長さは、本開示のプローブには使用されない。一部の実施例において、所望の結果を生み出す分子リンカーの長さは、使用する特定のFRET対に応じて変化することができる。例えば、GFP/フルオレセインFRET対がプローブの一部である場合に使用される分子リンカーの長さは、Alexa Fluor430/BODIPY630FRET対が、プローブの一部である場合に使用される分子リンカーの長さと異なる場合がある。
標的核酸分子の非存在下で、重合剤と化学的部分との相互作用を減少させるために、分子リンカー(またはその部分)は、(標的核酸分子の存在下または非存在下における、化学的部分自体の間での相互作用と同様に)標的核酸分子の非存在下で、分子リンカーが、重合剤(重合剤に関連するタグ)と化学的部分との相互作用を減少させるのに十分に剛性を有することができる持続長を有することができる。テザーなどの分子リンカーのその他の部分は、標的核酸分子の存在下で、重合剤(そのタグなど)と化学的部分との相互作用(または重合剤と標的核酸分子を有する化学的部分との相互作用)を可能にする。
長さの差異が、望ましくない重合剤と化学的部分との相互作用(または化学的部分自体の間の相互作用)を引き起こすのに不十分である限り、分子リンカーの全長は、分子リンカーの特定の要素に対する持続長と同一であっても異なっていてもよい。例えば、分子リンカーが、特定の持続長を有する分子ロッドを含む場合、分子リンカーは、その持続長より短くてもまたは長くてもよい。さらに、分子ロッド長それ自体は、分子ロッドを生成するために使用されるポリマーの持続長より短くてもまたは長くてもよい。特定の例において、分子リンカーは、分子ロッドを含み、ロッドの全長は、分子ロッドを含む分子の持続長より短い(分子ロッドを含む分子の持続長の0.l倍、0.5倍、または1倍など)。さらにその他の特定の例において、リンカーの長さは、その要素のうちのいずれか1つの持続長より長くてもまたは短くてもよい。例えば、分子ロッドを含む分子リンカーに対して、分子リンカーの全長はせいぜい、分子ロッドを含む分子の持続長より5倍短いまたは長い(分子ロッドを含む分子リンカーの持続長の1〜5倍、1〜4倍、または1〜3倍など)。一例において、分子ロッドは、dsDNA(持続長は400〜500Å)から成り、分子ロッドの長さは、400〜500Å(550〜700Åまたは550〜1000Åなど)より長い、または持続長(100〜350Åまたは200〜350Åなど)より短い。
当業者は、1つの持続長において、ロッドの遠末端は多くの場合、それでも元の方向と同じ方向(68.40度)に実質上向いており、この長さのロッド、従って柔軟性は、有用な機能的剛性をそれでも提供することができることを認識するであろう。持続長より長いロッドは、いくつかの用途において受け入れることができる、それ以上の柔軟性を提供する。その他の用途において、リンカーの比較的長い部分は、ブラウン運動、および結合できる標的分子の存在に応じて、部分が一体となる、またはならないようにさせる分子テザーの役割をするために十分に遠く離間するとはいえ、単一のリンカーは、均一の種類の単一の分子から成り(持続長より実質上長い、1000bpのdsDNAなど)、そのリンカーのある部分は、局所的に分子ロッドの役割をし、局所的にナノプローブ機能を提供できるほど十分に近い(例えば、40bp)。DNAのさらなる一部は、例えば、エンハンサー、活性化因子またはリプレッサーを供給するために「ループ」になることができるが、かかる状況は、局所転写因子が、互いに基本的に剛性の有する位置で、DNAに結合する場合に(例えば、GaIR binding sites of E.coli,Semsey et al.,GenesDev.18:1898−907,2004のように)発生する。かかるナノプローブ構造は可能であるが、一般に、本明細書に記載の構造は、持続長より実質上大きくない分子ロッドと、対応する持続長より実質上長い分子テザーとを明確に区別する。実際に見られるdsDNA転写制御機構とは異なり、一般に、本明細書に記載のナノプローブにおいて、分子ロッドおよびテザーは、例えば、PEGを有するdsDNAのように、実質上異なる持続長を有する異なる種類の分子を結合させることによって構築される。
持続長は、組成に応じて変化する。例えば、二本鎖DNA(dsDNA)分子の持続長は、一本鎖DNA(ssDNA)分子のものおよびポリエチレングリコール(PEG)と異なる。例えば、dsDNAは、持続長が400〜500Åである。特定の例において、ssDNAは、持続長が約40Åである。特定の例において、PEGは、持続長が約3.8±0.02Åである(Kienberger et al,Single Molecules1:123−8,2000)。
標的核酸分子の非存在下で、重合剤と化学的部分との相互作用を実質上回避するために(標的核酸分子の存在下または非存在下で、化学的部分自体の間の相互作用と同様に)、かつ標的生体分子の存在下で、重合剤と化学的部分との相互作用が可能となるように、リンカーの長さは、使用される特定の供与体および受容体フルオロフォアに対して、22から90Åの距離など、少なくとも、官能基を少なくともフォスターの半径だけ離間して維持するのに十分である。一部の例において、リンカーの長さは、10から1000Åの距離など、重合剤と化学的部分との電荷を分離するのに十分である。特定の例において、分子リンカーの全長は、10から300Å、10から200Å、20から200Å、20から187Å、20から150Å、60から120Å、または60から200Åなど、約10から500Åである。
分子リンカーの例には、テザー、分子ロッド、またはそれらの組み合わせが含まれるが、これらに限定されるものではない。例えば、分子リンカーは、テザーによって連結される複数の分子ロッド、または分子ロッドによって連結される複数のテザーを含むことができる。ナノプローブ10が、分子リンカー20によって連結され、離間される重合剤12および化学的部分28を含む、特定の一例を図1Aに示し、分子リンカー20は、テザー68、70によって連結される分子ロッド66から構成される。特定の例において、分子ロッドは、約100から200Å(約120〜140Åなど)であり、それぞれのテザーは、約23から187Å(約60Åなど)である。
重合剤および化学的部分は、互いに、また標的核酸分子と相互に作用することができ、検出可能なシグナルなど、所定の反応を提供する。重合剤および化学的部分が、標的分子の非存在下で、反応を提供するために相互に作用しないように、重合剤および化学的部分は、分子リンカーによって、空間的に分離した方向に維持することができる。しかしながら、分子リンカーは、重合剤および化学的部分に、所定の条件下で、相互に作用するために互いに十分に接近させ、検出可能なシグナルなどの所定の反応を引き起こすことを可能にする。
一例において、分子リンカーのそれぞれは、同じ長さであるか、またはほぼ同一の長さである。その他の例において、2つ以上の分子リンカーは、異なる長さである。多様な長さの分子リンカーを有するプローブは、化学的部分の結合頻度を制御するために使用することができる。例えば、AおよびT非加水分解性ヌクレオチド類似体が、比較的短い分子リンカーに結合されるが、CおよびG非加水分解性ヌクレオチド類似体が、比較的長い分子リンカーに結合される場合、AおよびTヌクレオチド類似体にとって、重合剤の活性部位に到達し、それによって、より強い放出シグナルを生成することはより容易となる。
重合剤
Medusaプローブは、重合剤を含む。重合剤は、線状鎖を形成する化学反応で、モノマー分子(ヌクレオチドなど)を一緒に反応させることができる化合物(酵素など)である。
Medusaプローブは、重合剤を含む。重合剤は、線状鎖を形成する化学反応で、モノマー分子(ヌクレオチドなど)を一緒に反応させることができる化合物(酵素など)である。
重合剤の特定の例には、DNAまたはRNAポリメラーゼなどのポリメラーゼ、およびリボソームが含まれる。ポリメラーゼの選択は、配列決定される核酸に依存する。例えば、鋳型が、一本鎖DNA分子である場合、DNA指令のDNAまたはRNAポリメラーゼを使用することができ、鋳型が、一本鎖RNA分子である場合、逆転写酵素(RNA指令のDNAポリメラーゼなど)を使用することができる。
限定されることがないポリメラーゼの特定の例には、大腸菌DNAポリメラーゼI(3’から5’エキソヌクレアーゼ活性を有するクレノウフラグメントなど)、Taqポリメラーゼ、逆転写酵素(HIV−I RT、例えば、Le Grice et al,J.Virol.62:2525:9,1988に開示される一本鎖HIV RTなど)、大腸菌RNAポリメラーゼ、および麦芽RNAポリメラーゼIIが含まれる。
一例において、ポリメラーゼは、DNAおよびRNAの両方のために配列を提供するために使用することができるHIV逆転写酵素であり、化学的部分は、αとβリン酸との間の酸素が、窒素と置換されるヌクレオチド類似体である。かかるヌクレオチド類似体は、標的核酸分子上の相補的ヌクレオチドと結合することができるが、伸長している相補的鎖に組み込むことができない。
一例において、ポリメラーゼは、修飾ポリメラーゼである。かかる修飾は、ポリメラーゼの生物学的活性を変化させる、例えば、基質の特異性、前進性、または精度を変化させるために使用することができる。例えば、DNAポリメラーゼの正確性は、突然変異によって向上させることができる(Wisniewski et al.,J.Biol.Chem.274:28175−84,1999)。例えば、かかるポリメラーゼは、折り畳まれていないクロマチン領域と同様に、二本鎖ゲノムDNAを配列決定するために使用することができる。その他の例において、DNAポリメラーゼの前進性は、ポリメラーゼドメインを非特異的dsDNA結合タンパク質(Sso7dなど)に共有結合的に連結することによって改善することができる(例えば、Wang et al.,Nucleic Acids Res.32(3):1197−207,2004参照)。その他の例において、HIV−1のRTは、酵素のヌクレオチド結合特異性を変化させるために、K65の位置に突然変異を含む。
本明細書に記載するように、特定の例において、重合剤は、供与体フルオロフォアなどのタグを含む。タグは、ポリメラーゼに直接的に結合される必要はないが(例えば、リンカーを介してポリメラーゼに結合される)、特定の例において、タグはポリメラーゼに結合される。例えば、GFP−ポリメラーゼ融合タンパク質は、標準的分子生物学的方法を使用して生成することができる(例えば、Liu et al,J.Biol.Chem.277:46712−9,2002;およびKratz et al.,Proc.Natl.Acad.Sci.USA,96:1915−20,1999参照)。
従って、ポリメラーゼは、タグの結合を可能にするために、修飾することができる(例えば、1つまたは複数のアミノ酸置換を含む)。例えば、1つまたは複数のアミノ酸は、タグの結合を可能にするために、当技術分野で公知の標準的方法(PCRを使用した部位特異的突然変異誘発法など)を使用してCysと置換することができる。理想的には、ポリメラーゼのアミノ酸配列を変化させることは、相補的核酸分子の合成を促進する能力など、ポリメラーゼの生物学的活性を有意に妨害しない。必要に応じて、溶媒に影響をうけやすいシステイン残基は、セリン残基と置換することができる。例えば、HIV RT p66サブユニットは、フルオロフォアAlexa488など、これらの位置でタグの結合を可能にするために、K287CまたはW24Cに突然変異させることができる(例えば、Rothwell et al.,Proc.Natl.Acad.Sci.USA.100:1655−60,2003およびKensch et al.,J.MoI.Biol.301:1029−39;2000参照)。
特定の例において、重合剤は、システイン287でRTに結合される供与体フルオロフォアを有する突然変異体HIV−1 RT(K287C)−Tus融合タンパク質である。
一例において、重合剤は、ストレプトアビジンを含む融合タンパク質である。かかる融合タンパク質は、標準的分子生物学的方法を使用して生成することができる。分子リンカーへのビオチンの結合は、1つまたは複数の分子リンカーをストレプトアビジン−ポリメラーゼ融合タンパク質に結合するために使用することができる。
一例において、ポリメラーゼは、供与体フルオロフォアおよび受容体フルオロフォアの両方を含む。両方のフルオロフォアを含むことによって、ポリメラーゼの立体構造内の運動または変化は、例えば、タイミングシグナルのようにモニタリングすることができる。特定の例において、供与体フルオロフォアは、ポリメラーゼの一面(「フィンガー」など)に結合され、受容体フルオロフォアは、ポリメラーゼのその他の面(「サム(thumb)」など)に結合される。ポリメラーゼが閉状態になるに従い、供与体および受容体フルオロフォアは、供与体が受容体を励起し、検出可能な受容体放出シグナルの生成(または検出可能な供与体放出シグナルの減少)をもたらすように、十分に接近させる。ポリメラーゼが開状態になるに従い、供与体が、十分に受容体を励起できず、検出可能な受容体放出シグナルの減少(または検出可能な供与体放出シグナルの増加)をもたらすように、供与体および受容体フルオロフォアは、十分に離間される。受容体からの放出がモニタリングされる場合、シグナルの増加、続く放出シグナルの減少(または供与体発光がモニタリングされる場合は、逆もまた同様)は、タイミングシグナルとして使用することができる。
特定の例において、供与体フルオロフォアは、EGFP(励起484nm;放出510nm)であり、受容体フルオロフォアは、EYFP(励起512nm;放出529nm)であり、かかる標識ポリメラーゼを含むプローブの480nmでの照射は、512nmでEYFPを励起する、510nmでのEGFPの放出をもたらす。
一例において、HTV RT p66サブユニットは、K287Cへの供与体フルオロフォアの結合、およびW24Cへの受容体フルオロフォアの結合(または逆もまた同様)を可能にするために、K287CまたはW24Cへ突然変異させる。これらの2つのアミノ酸残基は、それぞれ、ポリメラーゼのフィンガーおよびサム上にあり、従って、それらの間の距離は、ポリメラーゼの状態に応じて変化することができる。従って、かかるポリメラーゼは、タイミングシグナルを取得するために使用することができる。(24または287の位置の周囲のエピトープに対する抗体は、1つのフルオロフォアが結合されている一方で、他方の結合を阻止するために使用することができる。あるいは、その他の化学的性質は、供与体および受容体フルオロフォアを一意的に結合するために使用することも可能である。)
表2は、空間移動度が、通常のポリメラーゼ活性の間に最大限度まで変化するKlentaq 1 DNAポリメラーゼ上のアミノ酸を示す(Li et al,EMBOJ.17:7514−25,1998)。かかる残基は、供与体または受容体フルオロフォアが結合することができる残基の例である。例えば、TRP24は、フィンガー上にあり、LYS287は、HIV−1 RTのサム上にある。ポリメラーゼの開状態から閉状態へ移行の間、それらの距離は、約15Å変化する。
(表2)空間移動度が、Klentaq 1 DNAポリメラーゼで変化するアミノ酸
化学的部分
上述のように、分子リンカーを介してポリメラーゼに連結される化学的部分は、標的核酸分子内の曝露相補的ヌクレオチドと対合することによって、リンカーから分離されることなく、鋳型核酸分子に結合することができる薬剤である。例えば、化学的部分は、伸長している相補的鎖に永久に組み込まれることなく、検出可能なシグナルを生成するために十分な時間、ポリメラーゼ活性部位に入る。化学的部分は、活性部位との1つまたは複数の化学結合、伸長している相補的鎖との塩基対、および鋳型核酸との結合を形成することがあるが、かかる結合は、加水分解性ヌクレオチドによる化学的部分の置換を可能にするように可逆的であり、加水分解性ヌクレオチドは、伸長している相補的鎖に非可逆的に組み込まれるようになる。これは、重合剤を1つの塩基ごとに前に進める。化学的部分は、リンカーから除去されないため、プローブを再度使用することができる。
上述のように、分子リンカーを介してポリメラーゼに連結される化学的部分は、標的核酸分子内の曝露相補的ヌクレオチドと対合することによって、リンカーから分離されることなく、鋳型核酸分子に結合することができる薬剤である。例えば、化学的部分は、伸長している相補的鎖に永久に組み込まれることなく、検出可能なシグナルを生成するために十分な時間、ポリメラーゼ活性部位に入る。化学的部分は、活性部位との1つまたは複数の化学結合、伸長している相補的鎖との塩基対、および鋳型核酸との結合を形成することがあるが、かかる結合は、加水分解性ヌクレオチドによる化学的部分の置換を可能にするように可逆的であり、加水分解性ヌクレオチドは、伸長している相補的鎖に非可逆的に組み込まれるようになる。これは、重合剤を1つの塩基ごとに前に進める。化学的部分は、リンカーから除去されないため、プローブを再度使用することができる。
かかる化学的部分の特定の例には、非加水分解性ヌクレオチド類似体(例えば、非加水分解性ヌクレオチド三リン酸類似体)などの、ヌクレオチド類似体、例えば、モノヌクレオチドと同様に、BODIPY FL AMPPNP(B22356)およびBODIPY FL GMPPNP(B22355)などの、Molecular Probesから入手可能なものが含まれる。特定の例において、非加水分解性ヌクレオチド三リン酸類似体は、非加水分解性であるα−β結合を有する非加水分解性ヌクレオチド三リン酸類似体である。
非加水分解性ヌクレオチド類似体は、市販されている(例えば、Jena Bioscience(Jena,Germany)は、α,β−メチレン−ATP;α,β−N−dUTP;α,β−C−GTPなど4つすべてのdNTP塩基のための非加水分解性類似体を販売している)。さらに、αとβ−リン酸との間に−NH−または−CH2−基を含む4つすべてのdNTP類似体は、(例えば、Jena Bioscienceによって)商業生産が可能である。
特定の例において、本明細書に開示するナノプローブは、多数のヌクレオチド(加水分解性ヌクレオチドなど)を含み、それによって、内蔵型配列決定プローブを提供する。これにより、配列決定プローブが、配列内の限定された数の塩基を報告できるようになる。ヌクレオチドは、分子リンカー上に存在することができる。例えば、γ−リン酸上に最初から結合されているヌクレオチドが使用されると、配列決定される核酸分子に組み込まれ、従って、その分子リンカーは、配列決定反応にもはや加わることはない。
テザー
分子リンカーは、プローブに対する柔軟性を提供することができる、1つまたは複数のテザーを含むことができる。理想的には、テザーは、化学的部分の運動を可能にする、例えば、化学的部分が重合剤と、また標的核酸分子(標的核酸分子上の曝露塩基など)と相互に作用するのに十分な柔軟性がある。テザーの長さは、標的核酸分子の非存在下で、化学的部分と重合剤との相互作用を実質上回避し、標的核酸分子の存在下で、化学的部分と重合剤との相互作用が可能になるのに十分であるべきである。しかしながら、テザーは、理想的には、テザーまたは化学的部分の絡み合い(例えば、互いに、または重合剤と)を引き起こすほど長くはない。特定の例において、テザーは、水溶性であり、非毒性である。
分子リンカーは、プローブに対する柔軟性を提供することができる、1つまたは複数のテザーを含むことができる。理想的には、テザーは、化学的部分の運動を可能にする、例えば、化学的部分が重合剤と、また標的核酸分子(標的核酸分子上の曝露塩基など)と相互に作用するのに十分な柔軟性がある。テザーの長さは、標的核酸分子の非存在下で、化学的部分と重合剤との相互作用を実質上回避し、標的核酸分子の存在下で、化学的部分と重合剤との相互作用が可能になるのに十分であるべきである。しかしながら、テザーは、理想的には、テザーまたは化学的部分の絡み合い(例えば、互いに、または重合剤と)を引き起こすほど長くはない。特定の例において、テザーは、水溶性であり、非毒性である。
特定の例において、テザーの長さは、標的核酸分子の非存在下で、化学的部分を分離するのに十分長いが、ナノプローブまたは化学的部分のからまりを引き起こすほど長くはなく、化学的部分に標的核酸分子と、および重合剤との相互に作用させるには十分短い。
テザーとして使用することができる特定の材料の例には、一本鎖DNA分子、糖鎖、ペプチド(RecBタンパク質の2つの部分の間の連結部など)、およびポリエチレングリコール(PEG)または本明細書に開示する特性を有する任意のその他の柔軟性のあるポリマーが含まれるが、これらに限定されるものではない。特定の例において、テザーは、2つ以上のこれらの薬剤から構成される。特定の例において、テザーはPEGを含み、一部の例においては、PEGから成る。
特定の例において、テザーは、20〜200Å、23〜187Å、100〜140Å、または70〜94Å、例えば120Åなど、約10〜500Åである。一例において、テザーは、長さが187Å未満である。
特定の例において、テザーは、2〜4つまたは3〜4つのかかるスペーサーなど、長さが23.4Åである、18−原子PEGスペーサーの3から7つのユニットなどのPEGから成る。PEGは、非毒性で、柔軟性があり、親水性であり、またDNA合成の間にスペーサーとして挿入することができる(SyntheGen,Glen Research)。
一例において、テザーは、例えば、10〜30のヌクレオチド、10〜20のヌクレオチドなど、10〜40のヌクレオチド、例えば、10のヌクレオチド、20のヌクレオチド、または40のヌクレオチドの長さを有する、一本鎖DNA(ssDNA)分子である。特定の例において、ssDNAテザーは、その他の核酸鎖にアニーリングすることができ、それによって、柔軟性のあるテザーを剛性のある分子ロッドに変換する。理想的には、配列は、それ自体、官能基、または分析される試料内の核酸配列に特異的にハイブリダイズしないものである。
一例において、テザーは、糖鎖である(例えば、10〜75、10〜50、または20〜40のなど、10〜100の糖成分の長さを有する)。
一例において、テザーは、電荷付きテザーを含む分子リンカーの絡み合いを緩和するために、電荷(−COO−または−NH3 +など)を含む。
分子ロッド
分子リンカーは、標的核酸分子の非存在下で、化学的部分と重合剤との相互作用(または、例えば、分子リンカーの絡み合いに起因する、標的核酸分子の存在下または非存在下で、化学的部分自体の相互作用)を減少させるために、プローブに十分な剛性を提供することができる、1つまたは複数の分子ロッドを含むことができる。しかしながら、ロッドの長さは、標的核酸分子の存在下で、化学的部分と重合剤との相互作用を可能にするのに十分である。一部の例では、ナノプローブ内の分子ロッドの存在は、絡み合いの可能性を軽減し、重合剤の活性部位への化学的部分の結合の速度を増加することができる。
分子リンカーは、標的核酸分子の非存在下で、化学的部分と重合剤との相互作用(または、例えば、分子リンカーの絡み合いに起因する、標的核酸分子の存在下または非存在下で、化学的部分自体の相互作用)を減少させるために、プローブに十分な剛性を提供することができる、1つまたは複数の分子ロッドを含むことができる。しかしながら、ロッドの長さは、標的核酸分子の存在下で、化学的部分と重合剤との相互作用を可能にするのに十分である。一部の例では、ナノプローブ内の分子ロッドの存在は、絡み合いの可能性を軽減し、重合剤の活性部位への化学的部分の結合の速度を増加することができる。
開示するナノプローブは、少なくとも2つの分子ロッド、例えば1、2、3、4、5、6、7、8、9、または10の分子ロッドなど、1つまたは複数の分子ロッドを含むことができる。一例において、1つまたは複数の分子ロッドを使用すると、所要のテザーの長さを減少し、それによって、デバイスの費用および大きさを減少させる。
特定の例において、分子ロッドは、dsDNA配列である。dsDNAの長さは、標的核酸分子の存在下で、化学的部分と重合剤との相互作用を可能にするが、標的核酸分子の非存在下で、それらの相互作用を減少させるものである。ナノプローブが供与体および受容体フルオロフォアを含む場合、dsDNAの長さは、標的核酸分子の存在下で、フルオロフォアの相互作用を可能にするが、標的核酸分子の非存在下で、それらの相互作用を減少させるものである。特定の例において、dsDNA分子ロッドは、400〜500Åの持続長にほぼ相当する長さである。しかしながら、ロッドが、標的生体分子の非存在下で、官能基の相互作用を減少させ、分子リンカーの有意な絡み合いを引き起こさない限り、当業者は、より短いまたはより長い長さが使用できることを認識するであろう。特定の例において、dsDNA分子ロッドは、150から200のヌクレオチド、あるいは、10〜140のヌクレオチド、20〜140のヌクレオチド、20〜100のヌクレオチド、20〜50のヌクレオチド、30〜50のヌクレオチド、または例えば35、36、37、38、39、40、41、42、43、44、もしくは45のヌクレオチドなどの30〜40のヌクレオチドなど、10〜150のヌクレオチドである。特定の例において、dsDNA分子ロッドは、少なくとも20のヌクレオチドなど、少なくとも10のヌクレオチドである。特定の例において、分子ロッドは、40の塩基のdsDNAである。塩基は、厚さ3.38Åであり、従って、40の塩基対は、長さ135Åであり、典型的なFRETの距離よりも長い。特定の例において、dsDNAの配列は、NANEVプログラムを使用して選ばれる(Goodman et al.,BioTechniques,38:548−50,2005)。
その他の特定の例において、分子ロッドは、ペプチド骨格DNA(ペプチド核酸、PNA)またはロックト核酸(LNA)など、DNAの修飾体または変異体を含むDNA分子から構成される。特定の例において、かかるDNA変異体は、ヘリックス熱安定性およびヌクレアーゼへの耐性を変化させるために使用される。さらにその他の例において、分子ロッドは、カーボンナノチューブ(例えば、長さが100〜200Åであるナノチューブ)から構成される。さらにその他の例において、分子ロッドは、バクテリア、ウイルス粒子、またはウイルス尾部繊維を含む。
分岐分子リンカー
特定の例において、例えば、図1Cに示すように、分子リンカーは、分岐構造を形成する複数の部分から構成される。かかる構造は、本明細書に記載の合成DNAの一本鎖を適切にアニーリングすることによって作製することができる。
特定の例において、例えば、図1Cに示すように、分子リンカーは、分岐構造を形成する複数の部分から構成される。かかる構造は、本明細書に記載の合成DNAの一本鎖を適切にアニーリングすることによって作製することができる。
あるいは、5−Me−dC分岐ホスホラミダイトは、オリゴヌクレオチド合成の際に使用可能である。5−Me−dC分岐ホスホラミダイトが、鎖の中央に組み込まれるように、第1の鎖の合成を行うことができる。その後鎖の端部は覆われ、次いで5−Me−dC分岐は、障害物が除去され、次いで合成は、第2の分岐を開始する。これは、Y形状の分子を作製する。かかる2つの分子は、相補的配列を有するように合成できるので、4つのアームによって中心側面で単一ロッドを形成することができる。該アームにおける配列、およびこれらのアームに相補的なその他のオリゴヌクレオチドの設計を適切に選択することによって、図1Cに示すような構造を作製することができる。
タグ
特定の例において、本明細書に開示のナノプローブは、例えば、標的核酸分子の配列決定を可能にするために、検出可能な標識など、1つまたは複数のタグを含む。使用可能な例示的なタグには、フルオロフォア、化学発光薬剤、および電荷が含まれる。特定の例において、標的核酸分子がキャパシタに接近するに従い、電荷の変化が検出される。
特定の例において、本明細書に開示のナノプローブは、例えば、標的核酸分子の配列決定を可能にするために、検出可能な標識など、1つまたは複数のタグを含む。使用可能な例示的なタグには、フルオロフォア、化学発光薬剤、および電荷が含まれる。特定の例において、標的核酸分子がキャパシタに接近するに従い、電荷の変化が検出される。
特定の例において、ナノプローブは、受容体フルオロフォアおよび1つまたは複数の供与体フルオロフォアを含む。ナノプローブ上の単一供与体または単一受容体フルオロフォアのみを示す図において、当業者は、複数のフルオロフォアが、例えば、シグナルを増加させる、またはスペクトルの組み合わせを提供するために、ナノプローブ上に含まれることができることを理解するであろう。理想的には、受容体および供与体フルオロフォアは、標的生体分子の非存在下で、相互作用を減少させる位置でナノプローブに結合される(それによって、検出可能なシグナルを減少させる)。しかしながら、供与体フルオロフォアが、受容体フルオロフォアを励起し、受容体が、特徴のある波長を放出し、それによって検出可能なシグナルを生成するように、標的生体分子の存在下で、重合剤および化学的部分の互いとの、また標的核酸分子上の曝露塩基との相互作用は、受容体および供与体フルオロフォアが相互に作用することを可能にする。
特定の例において、供与体フルオロフォアは、大きなストークシフトを有する。これは、供与体励起光周波数によって、受容体フルオロフォアの励起を減少させる。さらに、適切な濾過は、励起波長を減少または除去することができ、検出される受容体からの放出スペクトルのみを残す。
特定の例において、供与体フルオロフォアは、緑色蛍光タンパク質(GFP)、またはその変異体であり、例えば、1つまたは複数のGFP分子は、標準的分子生物学的方法を使用して、重合剤(例えば、GFP−ポリメラーゼ融合タンパク質を生成するための)にクローン化することができる。その他の特定の例において、供与体フルオロフォアは、エクオリンなどの化学発光分子である。キレートランタニドは、明るく、大きなストークシフトの、鋭い放出スペクトルを有する非退色発光団を提供し、従って、供与体として使用することができる。供与体フルオロフォアとして化学発光分子を使用することにより、外部光源の必要性がなくなる。GFPと同様に、エクオリンなどの化学発光分子は、標準的分子生物学的方法を使用して(例えば、エクオリン−ポリメラーゼ融合タンパク質を生成するために)、重合剤にクローン化することができる。
重合酵素に連結される化学的部分のそれぞれの異なる型は、1つまたは複数のフルオロフォアなど、1つまたは複数の異なるタグに関連することができる。特定の例において、化学的部分のそれぞれの異なる型は、1つまたは複数の異なる受容体フルオロフォアに関連し、供与体フルオロフォア(または重合剤に関連する発光分子)によって励起することができる。
使用可能な受容体および供与体フルオロフォア対の特定の例には、BODIPYとともに使用するために適切な供与体フルオロフォアとしてのGFP突然変異体H9−40(Tsien,1998,Ann.Rev.Biochem.67:509)、フルオレセイン、ローダミングリーン、オレゴングリーン、テトラメチルローダミン、リサミン(商標)、受容体フルオロフォアとしてのテキサスレッドおよびナフトフルオレセイン、およびフルオレセインのための供与体フルオロフォアとしてのフルオロフォア3−(ε−カルボキシ−ペンチル)−3’−エチル−5、5’−ジメチルオキサカルボシアニン(CYA)、または受容体フルオロフォアとしてのローダミン誘導体(R6G、TAMRA、およびROXなど)が含まれるが、これらに限定されるものではない。受容体および供与体フルオロフォア対のその他の特定の例には、7−ジメチルアミノクマリン−4−酢酸(DMACA)およびフルオレセイン−5−イソチオシアネート(FITC);7−アミノ−4−メチル−3−酢酸クマリン(AMCA)およびフルオレセイン−5−イソチオシアネート(FITC);ならびにフルオレセイン−5−イソチオシアネート(FITC)およびテトラメチルローダミンイソチオシアネート(TRITC)が含まれるが、これらに限定されるものではない。
使用可能な蛍光タグの特定に例には、Alexa Fluorシリーズ(Molecular Probes,Eugene,OR)が含まれる。Alexa Fluor430は、430nmで吸収し、その高ストークシフトのために、540nmで遠方に放出し、従って、供与体フルオロフォアとして使用することができる。励起スペクトルは、Alexa Fluor430の540nm放出ピークに重なるので、Alexa Fluor430は、特定の例において、受容体フルオロフォアとしてAlexa Fluors546、555、568、594、647、およびBODIPY630とともに使用することができる。
さらに、供与体および受容体分子は、二分子蛍光相補性(BiFC)を使用して設計することができる(Hu et al.,Nat.Biotechnol.21:539−45,2003;Hu et al,Mol Cell.9:789−98,2002)。2つの部分的GFPフラグメントは、相補性、従って蛍光性を与えるために結合する。相補性は、ほんの瞬間だけを要するが、発色団の形成は、t1/2=300秒と長い時間を要する。従って、該方法は、FRETより遅くなる可能性がある。発色団は、永久に形成するので、長続きする結果を提供するために、ナノプローブ内で使用することができる。
一例において、タグは、クエンチャーである。例えば、クエンチャーが、受容体フルオロフォアと組み合わせて使用される場合は、減少した受容体シグナルは、検出可能なシグナルである。その他の例において、クエンチャーは、供与体フルオロフォアと組み合わせて使用され、減少された受容体シグナルは、検出可能なシグナルである。
量子ドット
一例において、本明細書に開示する1つまたは複数のMedusaプローブは、量子ドットと称される蛍光ナノ粒子に結合される。量子ドットに結合されるナノプローブは、1つまたは複数の対応する受容体フルオロフォア(例えば、化学的部分に関連する)を含むことができ、量子ドットまたはコーネル(Cornell)ドット(シリカをコーティングしたフルオロフォア)は、供与体フルオロフォアでありうる。特定の例において、ナノプローブは、直接またはリンカーを介して量子ドットに結合され、例えば、量子ドットをコーティングする抗体の例が挙げられる。
一例において、本明細書に開示する1つまたは複数のMedusaプローブは、量子ドットと称される蛍光ナノ粒子に結合される。量子ドットに結合されるナノプローブは、1つまたは複数の対応する受容体フルオロフォア(例えば、化学的部分に関連する)を含むことができ、量子ドットまたはコーネル(Cornell)ドット(シリカをコーティングしたフルオロフォア)は、供与体フルオロフォアでありうる。特定の例において、ナノプローブは、直接またはリンカーを介して量子ドットに結合され、例えば、量子ドットをコーティングする抗体の例が挙げられる。
量子ドットは、有意なFRETを回避するために、例えば、十分な長さの分子リンカーとともにつなぐことができる。その他の例において、量子ドットは、量子ドットの表面から有意な距離に化学的部分を保持する分子リンカー(4面体構造など)を使用して繋がれる。その後、ナノプローブ上の化学的部分が、標的核酸分子上の相補的な曝露塩基と対になる場合、化学的部分は、FRETによって検出される。
基板へのプローブの結合
特定の例において、本開示のプローブは、例えば、リンカーを介して、基板に結合される。従って、基板に結合されるプローブが、本開示によって提供される。
特定の例において、本開示のプローブは、例えば、リンカーを介して、基板に結合される。従って、基板に結合されるプローブが、本開示によって提供される。
特定の例において、プローブの重合剤は、ガラスまたはプラスチック材料などの基板に結合される。タンパク質を基板に結合する方法は、当技術分野で公知である。例えば、リンカーは、重合剤を基板に結合するために使用することができる。理想的には、リンカーは、重合剤の生物学的活性を有意に妨害しない。リンカーは、結合の共有結合または非共有手段であることができる。
一例において、リンカーは、互いに、ポリメラーゼ上の1つの分子(アフィニティータグなど)、基板上の別のものに対して高親和性を有する、分子の対である。かかる高親和性分子には、ストレプトアビジンとビオチン、ヒスチジンとニッケル(Ni)、ヒスチジンとSi、およびGSTとグルタチオンが含まれる。ポリメラーゼと基板が接触する場合、それらは、高親和性分子の相互作用のために、互いに結合する。例えば、ポリメラーゼは、6X His−タグ(例えば、標準的分子生物学的方法を使用して)を含むように設計され、その後、Siを含む表面に結合させることができる(例えば、Chaetal.,Proteomics4:1965−76,2004参照)。同様に、ポリメラーゼは、S−タグ、グルタチオン−S−トランスフェラーゼ(GST)、またはストレプトアビジンを含むように設計することができる。
その他の例において、リンカーは、非分岐鎖における2つ以上の炭素原子を有する、直鎖または分岐アミノ−またはメルカプト−炭化水素である。例には、アミノアルキル、アミノアルケニルおよびアミノアルキニル基が含まれる。あるいは、リンカーは、長さで10〜20の炭素のアルキル鎖であり、Si−C直接的結合により、またはエステル、Si−O−C、連結により結合される場合がある(参照により本明細書に組み入れられる、Footeへの米国特許第5,661,028号参照)。その他のリンカーは、Proberらへの米国特許第5,306,518号、コラム19;Wardらへの米国特許第4,711,955号、コラム8〜9;Mathiesらへの米国特許第5,707,804、コラム6〜7(参照によりすべて本明細書に組み入れられる)に記載されている。
配列決定のための例示的なナノプローブ
本開示は、1つまたは複数の標的核酸配列を配列決定するために使用できる、ナノプローブの複数の例を提供する。
本開示は、1つまたは複数の標的核酸配列を配列決定するために使用できる、ナノプローブの複数の例を提供する。
本開示のプローブの特定の一例を図1Aに示す。プローブ10は、重合剤12、および化学的部分22、24、26、28を重合剤12に結合する、4つの分子リンカー14、16、18、20を含む。化学的部分22、24、26、28は、所定の反応において、互いに、または重合剤12と相互作用することができ、分子リンカー14、16、18、20は、標的核酸の非存在下で、化学的部分22、24、26、28を、互いに、また重合剤12から十分に離間して離して維持する。特定の例において、化学的部分22、24、26、28は、化学的部分22、24、26、28の実質上の絡み合いを回避するために、互いに離間する。
一部の例において、プローブ10は、重合剤12に関連するタグ30、および化学的部分22、24、26、28に関連する異なるタグ32、34、36、38をそれぞれに含む。プローブ10は、重合剤12に直接結合されるタグ30、および化学的部分24、26、28に直接結合されるタグ34、36、38をそれぞれに示す。対照的に、タグ32は、化学的部分22に直接結合されないが、化学的部分22と標的核酸分子内のその相補的ヌクレオチドとが対になる際にシグナルを放出するために、化学的部分22に十分に接近しているリンカー14と結合する。同様に、タグ30は、リンカーを介して重合剤12に結合することも可能である。
分子リンカー14、16、18、20は、化学的部分22、24、26、28が、標的核酸分子の非存在下で、互いと、または重合剤12と実質上相互に作用しない長さおよび/または剛性を有する。しかしながら標的核酸分子の存在下において、分子リンカー14、16、18、20は、所定の反応において、標的核酸分子の存在下で化学的部分22、24、26、28が重合剤12と十分に相互に作用することを可能にする。例えば、化学的部分22、24、26、28上またはその付近のタグ32、34、36、38が、重合剤12のタグ30と(例えば、タグ30が、重合剤の活性部位に隣接する場合)、および標的核酸分子と相互に作用して、シグナル(光など)を生成できるように、重合剤12は、標的核酸分子の存在下で、標的およびその相補的プライマーに結合する。例えば、図1Bに示すように、プローブ39は、標的核酸分子40(プローブの一部ではない)、および相補的鎖が伸長するその相補的プライマー42(プローブの一部ではない)に結合することができる。図1Bに示すように、標的核酸分子40上の次のヌクレオチドは、「T」44であり、これは相補的化学的部分(本明細書ではモノヌクレオチド「A」26として示される)と特異的に対になることができる。分子リンカー18は、重合剤12の方へ屈曲するほど十分に長くかつ柔軟性があり、モノヌクレオチド「A」26が相補的鎖42と可逆的に相互に作用し、相補的ヌクレオチドT44と対になることを可能にする。しかしながら、「A」26は、相補的鎖42に永久に組み込まれることはない。代わりに、「A」26は、配列決定反応において(プローブの一部ではない、その他のヌクレオチドdCTP48、dGTP50、dTTP52とともに)存在し、伸長している相補的鎖42に組む込むことができる、加水分解性dATP46(プローブの一部ではない)と置換されることになる。標的核酸分子40上の「T」44との「A」26の対合は、重合剤12に関連するタグ30と、「A」26に関連するタグ36との相互作用に起因して、検出可能なシグナルを生成することになる。しかしながら、その他の化学的部分22、24、28は、標的核酸分子40内の「T」44に相補的でないので、検出可能なシグナルを生成するのに十分な時間、重合剤の活性部位に存在しない考えられる。
図1Aに示すプローブ10は、重合酵素12上の複数の点への複数の分子リンカー14、16、18、20の結合を示す例である。重合酵素上の単一点への複数の分子リンカーの結合を示す代替の例を図1Cに示す。プローブ60は、重合酵素12、および「ハブ」の役割をする分子ロッド62に結合される複数のテザー14、16、18、20から構成される分子リンカーを含む。分子リンカーは、「ハブ」62が自由に回転することによって重合酵素12への平等なアクセスを提供するリンカー64を介して、ある点で、重合酵素12に結合される。
図2A〜Dは、核酸分子を配列決定するために使用することができるMedusaナノプローブを示し、この図において、分子リンカーは、単一点で重合剤に結合される分岐構造である。しかしながら、当業者は、複数の分岐分子リンカーが単一の重合剤に結合可能であることを理解するであろう。
図2Aに示すプローブ100は、分岐構造を有する分子リンカー102を含む。分子リンカー102は、テザー106を介して単一点で重合剤104に結合される。分子リンカー102は、複数のテザー106、108、110、112、114、116、118、120、および複数の分子ロッド122、124、126、128を含む。テザー106、108、110、112、114、116、118、120は、プローブに柔軟性を提供し、分子ロッド122、124、126、128は、例えば、分子リンカー102の分岐の絡み合いを減少させるために剛性を提供する。非加水分解性A130、C132、G134、およびT136類似体として本明細書に示す化学的部分130、132、134、136は、テザー112、114、118、120の末端で分子リンカー102にそれぞれ結合する。例えば、4つの非加水分解性ヌクレオチド類似体130、132、134、136は、これらのγ−リン酸によって、アミノ末端ポリエチレングリコール(PEG)テザー112、114、118、120に結合させることができる。テザー112、114、118、120は、それぞれの断片の自由な回転を可能にするので、4つの化学的部分130、132、134、136のそれぞれは、重合剤104への平等なアクセスを有することができる。テザー112、114は、分子ロッド126に結合され、テザー118、120は、分子ロッド128に結合される。分子ロッド126、128は、別の分子ロッド124によって結合されるテザー110、116によって一緒に結合される。分子ロッド124はテザー108に結合し、テザー108は分子ロッド122に結合し、分子ロッド122はテザー106に結合し、テザー106は重合剤104に結合する。例えば、テザー106は、重合剤104上のシステインまたは異なる化学修飾された残基を介して重合剤104に結合されるアミノ末端を含むことができる。特定の例において、テザー106、108、110、112、114、116、118、120は、PEGから構成され、分子ロッド122、124、126、128は、dsDNAから構成される。必要に応じて、dsDNAは、プローブの適切な構造を確認するために使用することができる、図2Aに示すような制限部位(EcoRI、BamHI、PstIまたはHindIIIなど)を含むことができる。
さらに、プローブ100は、重合剤104に関連するタグ138、およびそれぞれの化学的部分130、132、134、136に関連する異なるタグ140、142、144、146をそれぞれに含む。プローブ100において、タグ138、140、142、144、146は、重合剤104または化学的部分130、132、134、136に直接的には結合されない。代わりに、タグ138、140、142、144、146は、分子リンカー106、112、114、118、120の一部に位置する。プローブ100は、標的核酸分子150にハイブリダイズされる、プライマー148に結合することが示されている。しかしながら、プライマー148および標的核酸分子150は、プローブ100の一部ではない。分子ロッド122、124、126、128は、例えば、標的核酸分子150の非存在下で、化学的部分130、132、134、136と重合剤104との相互作用を減少させるために、または標的核酸分子150の非存在下で、タグ138とタグ140、142、144、146との相互作用を減少させるために、剛性を提供することができる。さらに、相補的化学的部分が、重合剤104の結合ポケット内の標的核酸分子150に結合される場合、分子ロッド122、124、126、128は、3つの非相補的化学的部分の相互作用を減少させる。
プローブ100の変異体を図2Bに示す。さらに、プローブ200は、分岐構造を有する分子リンカー202を含むが、該分岐構造は、より対称的である。例えば、プローブ100において、化学的部分のうちの1つ(130など)が、重合剤104の活性部位に結合される場合、別の化学的部分のうちの1つ(132など)は、その他の化学的部分(134、136など)よりも近い。これは、例えば供与体および受容体フルオロフォアが、プローブ100に含まれる場合、バックグラウンドシグナルを増加させることがある。対照的に、プローブ200は、すべての化学的部分の、重合剤への同一のアクセスを可能にする。
分子リンカー202は、テザー206を介して単一点で重合剤204に結合される。分子リンカー202は、複数のテザー206、208、210、212、214、および複数の分子ロッド216、218、220、222、224を含む。テザー206、208、210、212、214は、プローブに柔軟性を提供し、分子ロッド216、218、220、222、224は、例えば、分子リンカー202の分岐の絡み合いを減少させるために、剛性を提供する。非加水分解性A226、C228、G230、およびT232類似体として本明細書に示す化学的部分226、228、230、232は、テザー208、210、212、214の末端で分子リンカー202にそれぞれ結合される。例えば、4つの非加水分解性ヌクレオチド類似体226、228、230、232は、これらのγ−リン酸塩によって、アミノ末端ポリエチレングリコール(PEG)テザー208、210、212、214に結合させることができる。テザー206、208、210、212、214は、それぞれのMedusa「アーム」の自由な回転を可能にするので、4つの化学的部分226、228、230、232のそれぞれは、重合剤204への平等なアクセスを有する。テザー208、210は、分子ロッド216、218にそれぞれ結合され、分子ロッド216、218は、分子ロッド220に結合される。特定の例において、分子ロッド216、218、222、224は、それぞれ20ヌクレオチドのdsDNAである。さらに、分子ロッド220は、テザー212および214にそれぞれ結合される、分子ロッド222、224に結合される。分子ロッド220は、重合剤204に結合される、テザー206に結合される。特定の例において、テザー206、208、210、212、214は、PEGから構成され、分子ロッド216、218、220、222、224は、dsDNAから構成される。必要に応じて、dsDNAは、プローブの適切な構造を確認するために使用することができる、図2Bに示すような制限部位(EcoRI、BamHI、PstIまたはHindIIIなど)を含むことができる。一例において、重合剤204は、分子ロッド220内にter DNA部位を含むことによって、分子リンカーに結合され、ここでは重合剤204は、ポリメラーゼ−Tus融合タンパク質である。ter DNA部位は、Tusタンパク質に特異的に結合することになる。これによって、テザー206の必要性がなくなる。
さらに、プローブ200は、重合剤204に関連するタグ234、およびそれぞれの化学的部分226、228、230、232に関連する異なるタグ236、238、240、242をそれぞれ含む。プローブ200において、タグ234、236、238、240、242は、重合剤204または化学的部分226、228、230、232に直接的には結合されない。代わりに、タグ234、236、238、240、242は、分子リンカー206、208、210、212、214の一部に位置する。プローブ200は、標的核酸分子244にハイブリダイズされる、プライマー242に結合することが示されている。しかしながら、プライマー242および標的核酸分子244は、プローブ200の一部ではない。分子ロッド216、218、220、222、224は、例えば、標的核酸分子244の非存在下で、化学的部分226、228、230、232と重合剤204との相互作用を減少させるために、または標的核酸分子244の非存在下で、タグ234とタグ236、238、240、242との相互作用を減少させるために、剛性を提供することができる。例えば、分子ロッド220は、標的核酸分子244の非存在下で、分子リンカー202の個別の分岐をタグ234から離れた状態で維持することができる。さらに、相補的化学的部分が、重合剤204の結合ポケット内の標的核酸分子244に結合される場合、分子ロッドは、3つの非相補的化学的部分の相互作用を減少させる。
プローブ100および200の変異体を図2Cに示す。さらに、プローブ300は、分岐構造を有する分子リンカー302を含む。プローブ300は、分子ロッド/テザー(DNA/PEG鎖など)につき1つのアミノ基のみを使用してプローブを構築するための、DNAハイブリダイゼーションの使用法を示す。分子リンカー302は、分子ロッド310によって結合される、テザー306、308を介して単一点で重合剤304に結合される。分子リンカー302は、複数のテザー306、308、312、314、316、318、320、322、324、326および複数の分子ロッド310、328、330、332、334、336、338、340、342、344を含む。テザー306、308、312、314、316、318、320、322、324、326は、プローブに柔軟性を提供し、分子ロッド310、328、330、332、334、336、338、340、342、344は、例えば、分子リンカー302の分岐の絡み合いを減少させるために剛性を提供する。非加水分解性A346、C348、G350、およびT352類似体として本明細書に示す化学的部分346、348、350、352は、テザー318、312、326、320の末端で分子リンカー302にそれぞれ結合される。例えば、4つの非加水分解性ヌクレオチド類似体346、348、350、352は、これらのγ−リン酸によって、アミノ末端ポリエチレングリコール(PEG)テザー318、312、326、320に結合させることができる。テザー314、316、322、324は、それぞれのセグメントの自由な回転を可能にするので、4つの化学的部分346、348、350、352のそれぞれは、重合剤304への平等なアクセスを有する。特定の例において、テザー306、308、312、314、316、318、320、322、324、326は、PEGから構成され、分子ロッド310、328、330、332、334、336、338、340、342、344は、dsDNAから構成される。必要に応じて、dsDNAは、プローブの適切な構造を確認するために使用することができる、図2Cに示すような制限部位(EcoRI、BamHI、PstIまたはHindIIIなど)を含むことができる。
さらに、プローブ300は、重合剤304に関連するタグ354、およびそれぞれの化学的部分346、348、350、352に関連する異なるタグ356、358、360、362をそれぞれ含む。プローブ300において、タグ354、356、358、360、362は、重合剤304または化学的部分346、348、350、352に直接的には結合されない。代わりに、タグ354、356、358、360、362は、分子リンカー310、334、328、344、338の一部に位置する。さらに、図2C(両方向矢印)は、例えば、退色によってシグナルの喪失を軽減するために、複数のタグが、いかにそれぞれの化学的部分(複数のフルオロフォアまたはデンドリマーなど)に関連できるかを示す。例えば分子ロッド334は、1つのタグのみを含むssDNAの代わりに、複数のタグ364を含むssDNAとのDNAハイブリダイゼーションによって形成することができる。プローブ300は、標的核酸分子368にハイブリダイズされる、プライマー366に結合することが示されている。しかしながら、プライマー366および標的核酸分子368は、プローブ300の一部ではない。分子ロッド310、328、330、332、334、336、338、340、342、344は、例えば、標的核酸分子368の非存在下で、化学的部分346、348、350、352と重合剤304との相互作用を減少させるために、または標的核酸分子368の非存在下で、タグ354とタグ356、358、360、362との相互作用を減少させるために、剛性を提供することができる。さらに、相補的化学的部分が、重合剤304の結合ポケット内の標的核酸分子368に結合される場合、分子ロッドは、3つの非相補的化学的部分の相互作用を減少させる。
プローブ300の変異体を図2Dに示す。プローブ400が分子リンカー402を含むことを除き、プローブ400は、プローブ300と同一であり、単一のタグの代わりに、複数の種類のタグは、それぞれの化学的部分346、348、350、352に関連する。例えば、化学的部分346は、タグ404、406に関連し、化学的部分348は、タグ404、408に関連し、化学的部分350は、タグ404、410に関連し、および化学的部分352は、タグ406、408、410に関連する。いくつかのタグを使用することによって、化学的部分と標的核酸鎖内のその相補的塩基とが対合すると放出されるシグナルに修正を加えることができる。例えば、タグが、受容体フルオロフォアである場合は、プローブ400は検出を可能にし、一部の例において、フルオロフォア退色の修正、または1つまたは複数の分子リンカー分岐の喪失を可能にする。プローブ400は、標的核酸分子368にハイブリダイズされる、プライマー366に結合することが示されている。しかしながら、プライマー366および標的核酸分子368は、プローブ400の一部ではない。
図1A〜図1Cおよび図2A〜図2Dに示す化学的部分がヌクレオチド類似体である例において、ヌクレオチド類似体は、糖の3’ヒドロキシルで、もしくはリン酸(α、β、またはγリン酸など)で、または重合剤の活性部位もしくは相補的塩基対合への特異的結合を妨害しないヌクレオチド上の任意の点で、塩基によって分子リンカーに結合することができる。図1A〜図1Cおよび図2A〜図2Dに示す化学的部分は、異なる化学的部分(図1A〜図1Cおよび図2A〜図2Dに示すように)であってもよく、または、同じ化学的部分(この場合、それぞれの型の化学的部分を有するナノプローブは、配列決定反応に含まれ得る)であってもよい。特定の例において、図1A〜図1Cおよび図2A〜図2Dに示す重合剤に関連するタグは、供与体フルオロフォアであり、それぞれの化学的部分に関連するタグは、受容体フルオロフォアを含む。例えば、複数の型の化学的部分が、同一のプローブ上にある場合は、それぞれの化学的部分は、一意的な受容体フルオロフォアまたはフルオロフォアの組み合わせに関連することができる。
ナノプローブの生成
多くの方法が開示されたナノプローブを生成するために利用可能である。例えば、タグを別の分子に結合させる方法は公知である。さらに、DNA−PEG構造を生成する方法は公知である。特定の方法が本明細書に提供されるが、本開示はこれらの方法に限定されない。
多くの方法が開示されたナノプローブを生成するために利用可能である。例えば、タグを別の分子に結合させる方法は公知である。さらに、DNA−PEG構造を生成する方法は公知である。特定の方法が本明細書に提供されるが、本開示はこれらの方法に限定されない。
DNA/PEGの合成および結合
分子リンカーが1つまたは複数のDNA分子ロッドおよび1つまたは複数のPEGテザーを含む例において、以下の方法を使用することができる。任意の所望の配列のDNAを、様々な市販のソース(Invitrogen、Synthegen、Sigmaなど)から入手することができる。DNAの配列は、NANEVプログラムを使用して生成することができ、これは「核酸ナノ構造の設計のための発展的方法」を使用する(Goodman et al.,BioTechniques,38:548−50,2005)。本プログラムは、所望の構造のみハイブリダイゼーションにより形成されるように、ナノプローブおけるDNA配列を設計するために使用することができる。特定の例において、PEGテザーは、標準フォスフォラミダイト「スペーサー」として分子リンカー内のどこにでも組み込まれる。DNA配列においてどこでもアミノ基を導入することも可能である。
分子リンカーが1つまたは複数のDNA分子ロッドおよび1つまたは複数のPEGテザーを含む例において、以下の方法を使用することができる。任意の所望の配列のDNAを、様々な市販のソース(Invitrogen、Synthegen、Sigmaなど)から入手することができる。DNAの配列は、NANEVプログラムを使用して生成することができ、これは「核酸ナノ構造の設計のための発展的方法」を使用する(Goodman et al.,BioTechniques,38:548−50,2005)。本プログラムは、所望の構造のみハイブリダイゼーションにより形成されるように、ナノプローブおけるDNA配列を設計するために使用することができる。特定の例において、PEGテザーは、標準フォスフォラミダイト「スペーサー」として分子リンカー内のどこにでも組み込まれる。DNA配列においてどこでもアミノ基を導入することも可能である。
DNA−DNAハイブリダイゼーションの適切な使用により、ナノプローブは、DNA/PEGリンカーにつき1つのアミノ基のみを使用して、構築することができる。これは、例えば、図1Aに示されるように、ナノプローブ上のフルオロフォアまたはタンパク質を結合させるためにアミノ基を使用することを可能にする。
一例において、DNA−PEG−NH2−dNTPは、ポリメラーゼが結合したビーズまたは別の基板を使用して、DNA−PEG−NH2から離れて精製される。DNA−PEG−NH2−dNTPは、ポリメラーゼに結合するが、DNA−PEG−NH2は結合しない。
化学的部分へのタグの結合
タグは、化学的部分に結合させることができる。例えば、化学的部分が、ヌクレオチド類似体である場合、タグは、塩基、糖、α、β、またはγリン酸に結合することができる。
タグは、化学的部分に結合させることができる。例えば、化学的部分が、ヌクレオチド類似体である場合、タグは、塩基、糖、α、β、またはγリン酸に結合することができる。
化学的部分への分子リンカーの結合
化学的部分は、分子リンカーに結合させることができる。例えば、化学的部分がヌクレオチド類似体である場合、分子リンカーは、塩基、糖(例えば、糖の3´ヒドロキシルで)、α、β、またはγリン酸に結合することができる。理想的には、そのような結合は、化学的部分の、重合剤の活性部位に結合する能力、または相補的ヌクレオチド塩基と対になる能力を阻害しない。化学的部分を分子リンカーに結合させる方法は当技術分野において公知であり、開示される方法は、特定の方法に限定されない。例えば、分子リンカーをγリン酸に結合させるために、5’アミノ変性剤 C6 TFAを利用することができる(Synthegen、Houston,TXおよびIDT、Coralville,IAより入手可能)。ヌクレオチド類似体をリンカーに結合させるために使用可能な別の方法は、米国特許第6,936,702号に開示される(参照により本明細書に組み入れられる)。
化学的部分は、分子リンカーに結合させることができる。例えば、化学的部分がヌクレオチド類似体である場合、分子リンカーは、塩基、糖(例えば、糖の3´ヒドロキシルで)、α、β、またはγリン酸に結合することができる。理想的には、そのような結合は、化学的部分の、重合剤の活性部位に結合する能力、または相補的ヌクレオチド塩基と対になる能力を阻害しない。化学的部分を分子リンカーに結合させる方法は当技術分野において公知であり、開示される方法は、特定の方法に限定されない。例えば、分子リンカーをγリン酸に結合させるために、5’アミノ変性剤 C6 TFAを利用することができる(Synthegen、Houston,TXおよびIDT、Coralville,IAより入手可能)。ヌクレオチド類似体をリンカーに結合させるために使用可能な別の方法は、米国特許第6,936,702号に開示される(参照により本明細書に組み入れられる)。
重合剤への分子リンカーの結合
一例において、1つまたは複数の分子リンカーは、ter DNA部位に結合させるためにTusタンパク質を使用して、重合剤に結合される。例えば、Tusタンパク質は、標準的なクローニング技術を使用して、重合剤に融合させることができる。分子リンカー部分は、ter DNA部位を含むことができ、これはTusタンパク質に特異的に結合する。Tus−ter結合切断定数は、10−13Mである(Neylon et al.,Microbiol.Mol.Biol.Rev.69:501−26,2005)。
一例において、1つまたは複数の分子リンカーは、ter DNA部位に結合させるためにTusタンパク質を使用して、重合剤に結合される。例えば、Tusタンパク質は、標準的なクローニング技術を使用して、重合剤に融合させることができる。分子リンカー部分は、ter DNA部位を含むことができ、これはTusタンパク質に特異的に結合する。Tus−ter結合切断定数は、10−13Mである(Neylon et al.,Microbiol.Mol.Biol.Rev.69:501−26,2005)。
タンパク質リンカー
特定の例において、分子リンカーは、ser−glyなどの柔軟性のあるタンパク質鎖から成る。例えば、分子リンカーを形成する柔軟性のあるループを有するように重合剤を拡張することができる。鎖は、ポリメラーゼを継続させるためにループして戻る。例えば、リシンまたはシステインなどのアミノ酸に結合することにより、化学的部分(dNTPなど)を酵素的に付加することができる。ポリメラーゼは、特定の分子リンカーを認識し、適切な塩基を結合する。例えば、結果が非加水分解性である場合、結合は通常のdNTP上にある。一例において、ポリメラーゼおよび柔軟性のある分子リンカーは、改変されたポリメラーゼに対する遺伝子を有するファージの表面上に発現する。特定のdNTPは、例えば、3’末端上でカラムなどの固体支持体に結合することができる。改変されたポリメラーゼを発現するファージは、dNTPへのポリメラーゼの結合を可能にする条件下で、固体支持体と接触する。これは、改変されたポリメラーゼを発現するファージの選択を可能にする。改変されたポリメラーゼは、GFP、YFP、RFP、CFPおよびエクオリンなどの供与体フルオロフォアをさらに含むことができる。
特定の例において、分子リンカーは、ser−glyなどの柔軟性のあるタンパク質鎖から成る。例えば、分子リンカーを形成する柔軟性のあるループを有するように重合剤を拡張することができる。鎖は、ポリメラーゼを継続させるためにループして戻る。例えば、リシンまたはシステインなどのアミノ酸に結合することにより、化学的部分(dNTPなど)を酵素的に付加することができる。ポリメラーゼは、特定の分子リンカーを認識し、適切な塩基を結合する。例えば、結果が非加水分解性である場合、結合は通常のdNTP上にある。一例において、ポリメラーゼおよび柔軟性のある分子リンカーは、改変されたポリメラーゼに対する遺伝子を有するファージの表面上に発現する。特定のdNTPは、例えば、3’末端上でカラムなどの固体支持体に結合することができる。改変されたポリメラーゼを発現するファージは、dNTPへのポリメラーゼの結合を可能にする条件下で、固体支持体と接触する。これは、改変されたポリメラーゼを発現するファージの選択を可能にする。改変されたポリメラーゼは、GFP、YFP、RFP、CFPおよびエクオリンなどの供与体フルオロフォアをさらに含むことができる。
標的核酸分子を配列決定する方法
本開示は、2つ以上の標的核酸分子などの標的核酸分子を同時に配列決定する方法を提供する。配列決定は、インビトロ、エクスビボ、インサイチュー(例えば、対象から取得された生体試料を使用して)、またはインビボ(例えば、細胞内で配列決定することにより)で実施することができる。特定の例において、標的核酸鎖は、疾病に関連する1つまたは複数の突然変異を含む。
本開示は、2つ以上の標的核酸分子などの標的核酸分子を同時に配列決定する方法を提供する。配列決定は、インビトロ、エクスビボ、インサイチュー(例えば、対象から取得された生体試料を使用して)、またはインビボ(例えば、細胞内で配列決定することにより)で実施することができる。特定の例において、標的核酸鎖は、疾病に関連する1つまたは複数の突然変異を含む。
特定の例において、標的核酸分子は、対象から取得される。例えば、標的核酸分子は、対象から取得した生体試料に存在することができる。別の例において、標的核酸分子は、対象内に存在し、鋳型核酸分子のオリゴヌクレオチドプライマーおよびプローブへの曝露は、対象の細胞へのオリゴヌクレオチドプライマーおよびプローブの導入を含む。
特定の例において、標的核酸分子の核酸配列を決定する方法は、オリゴヌクレオチドプライマーおよび加水分解性ヌクレオチドの混合物の存在下で標的核酸分子を本明細書に開示される1つまたは複数のプローブに曝露する段階を含む。加水分解性ヌクレオチドは、標的核酸分子内の相補的ヌクレオチドと対合することによって、伸長する核酸分子に組み込まれ、標的核酸分子上の相補的ヌクレオチドに可逆的に結合する化学的部分を置換することができる。加水分解性ヌクレオチドは、化学的部分に置換される場合、これは、1塩基先に重合剤を進める。化学的部分は、伸長する相補鎖に永久的には組み込まれないため、重合酵素の活性部位によって、最終的に拡散する。一連のシグナルの発光が検出され、特性シグナルの発光は、リンカー上の化学的部分とその相補的ヌクレオチドとの対合を示す。そのような特性シグナルはまた、どの加水分解性ヌクレオチドが、標的核酸分子に相補的である伸長する核酸分子に次に組み込まれるかを示すことができる。特定の例において、一連のシグナルの発光は、核酸配列に変換される。一部の例において、一連のシグナルの発光は、発光共鳴エネルギー移動(LRET)または蛍光共鳴エネルギー移動(FRET)によって生成される。特定の例において、一連のシグナルの発光は、電荷結合素子(CCD)カメラで検出され、核酸配列に変換される。一連のシグナルはまた、コンピュータ可読媒体に保存することもできる。化学的部分はリンカーから除去されないため、プローブを再度使用することができる。
従って、非標識加水分解性ヌクレオチドは、伸長する鎖に組み込まれる唯一のものであるため、本方法は、伸長する相補的核酸分子に蛍光標識ヌクレオチドを組み込む問題を解決する。得られる相補的核酸分子は、正常ヌクレオチドを含む。
特定の例において、重合剤はタグに関連し、化学的部分のそれぞれはまた、リンカーが有する特定の化学的部分を識別するタグに関連し、ここで、重合剤に関連するタグと、化学的部分に関連するタグとの相互作用は、化学的部分と相補的ヌクレオチドとの対合を示す特性シグナルの発生を誘発する。上記に記載されるように、タグは、重合剤および化学的部分に直接結合することができるか、または、分子リンカーに間接的に関連する、例えば、重合剤または化学的部分に十分に接近して分子リンカー上に存在することができるかのいずれかである。
一部の例において、重合剤に関連するタグは、供与体フルオロフォアを含み、特定の化学的部分を識別するタグは、1つまたは複数の受容体フルオロフォアを含む。重合剤と化学的部分との十分な相互作用、例えば、重合剤内の活性部位での化学的部分の存在、および標的核酸鎖上での化学的部分と相補的ヌクレオチドとの対合は、受容体フルオロフォアを供与体フルオロフォアの近傍に到達させ、供与体フルオロフォアによる受容体フルオロフォアの励起を可能にする。かかる例において、シグナルを検出する段階は、受容体フルオロフォアから放出された蛍光性シグナル(蛍光の増加など)を検出する段階、または供与体フルオロフォアから放出された蛍光性シグナル(蛍光の減少など)を検出する段階を含むことができる。特定の例において、供与体フルオロフォアはGFPであり、受容体フルオロフォアは、BODIPY、フルオレセイン、ローダミングリーン、オレゴングリーン、またはその誘導体である。別の特定の例において、供与体フルオロフォアは、Alexa Fluor 430であり、受容体フルオロフォアは、Alexa Fluor 546、568、594、および647である。
タグの1つが供与体フルオロフォアである一部の例において、本方法は、リンカー上の化学的部分とその相補的ヌクレオチドとの対合を示す特性シグナルを放出するように1つまたは複数の受容体フルオロフォアを刺激する励起シグナルを放出させるため、供与体フルオロフォアを励起する段階をさらに含む。例えば、供与体は、例えば、特定の波長の電磁放射を放出するレーザーによって提供される光、または狭い範囲内の波長の光のコヒーレントビームなどの電磁放射を使用して、励起することができる。その他の例において、供与体は、発光分子(エクオリンなど)によって励起される。一部の例において、供与体は継続的に励起される。しかしながら、すべての供与体フルオロフォアが外部源による励起を必要とするわけではない。例えば、化学発光による供与体分子は、外部源による励起を必要としない。理想的には、供与体フルオロフォアの励起源は、受容体フルオロフォアを著しく励起しない。
分子リンカー上の特定の化学的部分(非加水分解性A、C、G、またはTアナログなど)とその相補的ヌクレオチドとの対合を示す特性シグナルの発光は、核酸配列に変換することができる。例えば、各化学的部分がその相補的ヌクレオチドと対合し、顕微鏡の視野において放出される場合、一連の発光シグナルが取得される。例えば、発光シグナルは、顕微鏡対物レンズで収集することができ、化学的部分に関連するそれぞれのタグに対する完全な発光スペクトルは、分光光度計により生成される。完全な発光スペクトルは、顕微鏡の視野において、それぞれの化学的部分がその相補的ヌクレオチドと対合する場合、化学的部分に関連するそれぞれのタグに対して、CCDカメラなどの検出デバイスにより取得される。CCDカメラは、発光スペクトルを収集し、一連の電荷にスペクトルを変換する。シグナルスペクトルとそれぞれのクラスの化学的部分上のタグに対するスペクトルとの間の最小二乗適合などのアルゴリズムを使用して、顕微鏡の視野において、それぞれの核酸について一連の発光スペクトルを核酸配列に変換するために、それぞれの化学的部分の対合についての電荷をコンピュータにより記録することができる。
多くの異なるアルゴリズムは、発光スペクトルを核酸配列に変換するために使用することができるが、この特定の例は1つのアプローチを例示する。4つの蛍光スペクトル(Anm、Cnm、GnmおよびT/Unm)は、巨視的測定から生成される。試料から、未知のノイズスペクトル(Snm)が生成される。未知のスペクトルは、非加水分解性アナログの相対的比率を示す4つの重量、a、c、gおよびt/uのみを有する4つの既知のスペクトルの合計であると考えられる。従って、520nm〜523nmで、これは、5つの等式をもたらす。
A520*a+C520*c+G520*g+T520*t=S520
A521*a+C521*c+G521*g+T521*t=S521
A522*a+C522*c+G522*g+T522*t=S522
A523*a+C523*c+G523*g+T523*t=S523
A524*a+C524*c+G524*g+T524*t=S524
A520*a+C520*c+G520*g+T520*t=S520
A521*a+C521*c+G521*g+T521*t=S521
A522*a+C522*c+G522*g+T522*t=S522
A523*a+C523*c+G523*g+T523*t=S523
A524*a+C524*c+G524*g+T524*t=S524
例えば、A520などの既知の値を代入し、最小二乗線形回帰を使用することによって、未知の値a、c、g、およびt/uを求める。
この特定の例において、重合剤に関連する供与体フルオロフォアは、GFP H9−40であり、以下の通り、化学的部分は受容体フルオロフォアに関連する:非加水分解性Aは、BODIPYで標識され;非加水分解性Tは、フルオレセインで標識され;非加水分解性Cは、ローダミンで標識され;非加水分解性Gは、オレゴングリーンで標識される。別の例において、ポリメラーゼに関連する供与体フルオロフォアは、H9−40であり、以下の通り、化学的部分は受容体フルオロフォアに関連する:非加水分解性Aは、テトラメチルローダミンで標識され;非加水分解性T/Uは、ナフトフルオレセインで標識され;非加水分解性Cは、リサミンで標識され;非加水分解性Gは、テキサスレッドで標識される。それぞれの異なる型の化学的部分とその相補的塩基との対合を検出することができるように、それぞれの受容体フルオロフォアの発光スペクトルがモニタリングされ、それぞれのフルオロフォアのスペクトルは互いに区別することができる。
未知のタグの相対的比率を決定する過程は、「線形アンミキシング法」として公知である(Dickinson et al,Biotechniques 31:1272,1274−6,1278,2001)。ナノプローブの収集がヌクレオチド配列の混合を与えることができる一方、個別のナノプローブは、主に単一のヌクレオチド配列を与える。もし、ナノプローブの収集が、一定時期同期化され、または、同時にただ1つの加水分解性ヌクレオチドを付加することにより配列に沿ってそれらを進めることによって(フローセルにおけるように)、収集が実質的に単一の配列を報告する。配列における塩基の相対的比率は、配列ロゴ技術により示すことができる(Schneider and Stephens,Nucleic Acids Res.18:6097−100, 1990)。
特定の例において、配列決定反応の1つまたは複数の要素は、ガラス、プラスチック、または金属基板などの基板に結合する、または固定される。例えば、プローブ、標的核酸、またはプライマーは、基板に固定することができる。上記に記載されるように、プローブは、重合剤を介して基板に結合することができる。核酸分子(標的核酸またはプライマーなど)は、5’末端、3’末端または内部で、基板に結合することができる。
本方法は、実質的に同時に複数の配列決定反応を行う段階と、複数の配列決定反応から一連のシグナルを検出する段階とを含むことができる。例えば、複数の重合剤、鋳型核酸分子、またはオリゴヌクレオチドプライマーは、所定のパターンで基板に直接または間接的に固定することができる。一連のシグナルを検出する段階は、シグナルを、そのようなパターン内の所定の位置に対応する核酸分子に相関させる段階を含むことができる。重合剤、鋳型核酸分子、またはオリゴヌクレオチドプライマーは、基板上に液滴を微量ピペッティングすることにより、規則的なアレイにエッチングされたチャネル内で、所定のパターンで基板に固定することができる。
一部の例において、配列決定反応中に存在するヌクレオチドのレベルは、制御される。例えば、プローブは、付加された加水分解性ヌクレオチドの非存在下で、プライマーおよび標的核酸分子とともにインキュベートされる。生成したシグナルは、標的核酸分子上に露出したヌクレオチドを示し、従って、付加されるべき次のヌクレオチドを示す。そのようなヌクレオチドは、その後、付加され、1つまたは複数の位置だけ先にプローブを進める。ヌクレオチドは、除去され(例えば、洗浄により)、サイクルを反復する。
図1Bは、開示されたプローブが核酸分子を配列決定するためにどのように使用され得るかについて、特定の例を提供する。開示された任意のプローブは、プローブ39と置換することができることを当業者は理解するであろう。本方法は、非標識加水分解性ヌクレオチド46、48、50、52(dATP、dCTP、dGTP、およびdTTP;またはRNAポリメラーゼについてはNTPなど)の混合物の存在下で、標的核酸配列40をオリゴヌクレオチドプライマー42およびプローブ39(または、図1A〜Bおよび2A〜Dに示されるものなど、本明細書に示される他の任意のプローブ)に接触させる段階を含むことができる。配列決定される核酸分子40(ナノプローブの部分ではない)の非存在下で、検出可能なシグナルはほとんどない。重合剤12が、プライマー42において標的核酸配列40に結合する場合、塩基44(本明細書では、「T」)は、標的鎖40上に露出される。分子リンカー14、16、18、20は、ブラウン運動によって移動し、末端の化学的部分22、24、26、28を、重合剤12の活性部位における標的核酸分子40上に露出した塩基44に接近させ、対合させることができる。分子リンカー14、16、18、20の末端の化学的部分22、24、26、28は、塩基44に結合するために競争するが、4つの化学的部分の1つだけ(この場合、26)が、露出した塩基44に相補的である。不正確な対合が非相補的化学的部分(この場合、22、24、28)により生じる場合、その化学的部分(この場合、22、24、または28)は、迅速に離れる。しかしながら、正確な対合が生じる場合(本例において、相補的化学的部分26)、正確な化学的部分は、重合剤12の活性部位において相当な時間滞留し、相補的塩基44と対合する。この間、相補的化学的部分26上の対応するタグ36(受容体フルオロフォアなど)は、タグ30に対する重合剤12に関連するタグ30(供与体フルオロフォアなど)に十分に接近してタグ36と相互作用し、それによって、そのような化学的部分26に対して特性シグナル(受容体発光シグナルなど)を生成する。タグ32、34、36、38のそれぞれは、区別できる発光シグナルを生成する。すべての化学的部分22、24、26、28は、活性部位の内外に拡散するが、露出した塩基44に相補的である化学的部分26に関連するタグ36は、活性部位を最も長い時間占有するため、シグナルを独占し、適切に相補的塩基44と対合することができる。化学的部分22、24、26、28は、伸長する核酸鎖42に付加できないため、重合剤の活性部位における化学的部分が非標識加水分解性ヌクレオチド(この場合、46)に置換されない限り、鎖は伸長しない。従って、検出可能なシグナルは、伸長する相補鎖42に組み込まれるべき次の非標識加水分解性ヌクレオチド(この場合、46)を示す。適切な加水分解性ヌクレオチド46は、伸長する相補鎖42に最終的に組み込まれ、重合剤12は1つ先の位置に進む。これにより、標的核酸40上で次の相補的塩基が露出し、サイクルを反復する。変化する発光シグナルは、標的ヌクレオチド配列に対応する。加水分解性ヌクレオチド46が組み込まれるステッピング過程中、供与体シグナルのみが放出される。
特定の例において、重合剤12は、顕微鏡スライドなどの基板に(例えば、リンカーにより)結合される。基板は、顕微鏡のステージに取り付けることができる。特定の例において、配列決定反応は、水性の環境で生じ、例えば、ガラスのカバースリップで覆うことにより、乾燥を避けるために密閉することができる。かかる例において、配列決定される核酸40は、アニーリングしたオリゴヌクレオチドプライマー42を有し、固定された重合剤12により結合される。配列決定反応を開始するために、非標識加水分解性ヌクレオチド44、46、48、50の混合物は、ナノプローブとともに付加される。配列決定反応は、上記に記載されるように進行する。配列決定のためのすべての必要な成分が常に供給され、利用可能であるため、外部のポンプデバイスまたはリザーバーは必要としない。
従って、本方法は、シグナルが核酸分子の大集団を代表する電気泳動ゲル上のパターンなどの高分子事象をモニタリングすることにより配列決定する代わりに、分子レベルにおいて標的核酸分子上の相補的ヌクレオチドに可逆的に結合する化学的部分の対合をモニタリングすることにより核酸分子を配列決定することを可能にする。大きな視野と組み合わせた本方法を使用して、毎秒最大750塩基対の配列決定速度で、同時に、1000以上のDNA分子を配列決定することが可能であり、これは高速のDNAポリメラーゼの速度である(Watson et al,Molecular Biology of the Gene,4th Edition,The Benjamin/Cummings Publishing Co.,Inc.,Menlo Park,California,1987,page110)。コピー/配列決定されるそれぞれのDNA分子、およびそれに関連するプローブは、重合剤媒介反応が生じる視野の特定の部分に対応し得る。
配列決定速度の改変
特定の例において、核酸配列決定の速度は制御される。例えば、配列決定反応は、0〜30℃、例えば、4℃、または室温(20〜25℃など)の低温で実施され、配列決定速度を減少させることができる。これらの低温で、重合剤は、低温で適切に機能することができるものであってよい。この温度範囲は、より狭いスペクトル線、従って、より高い解読複雑性を可能にする。低温は、スペクトルを鋭くし、解読するためのさらに明確なスペクトルを可能にする。重合反応を阻害する可能性があるため、冷凍を避ける。別の例において、配列決定速度を減少させるために、配列決定反応を実施する環境は、20%のFicol70中の9.95cPの粘度などの粘性にさらされる。さらに、ポリメラーゼを遅延させる1つまたは複数の突然変異を含むポリメラーゼなどの突然変異体ポリメラーゼを、配列決定速度を減少させるために使用することができる。配列決定速度はまた、配列決定反応中に存在する遊離の加水分解性ヌクレオチド(dNTPなど)の濃度によって制御することもでき、遊離のdNTPの濃度の減少は、重合速度を減少させる。
特定の例において、核酸配列決定の速度は制御される。例えば、配列決定反応は、0〜30℃、例えば、4℃、または室温(20〜25℃など)の低温で実施され、配列決定速度を減少させることができる。これらの低温で、重合剤は、低温で適切に機能することができるものであってよい。この温度範囲は、より狭いスペクトル線、従って、より高い解読複雑性を可能にする。低温は、スペクトルを鋭くし、解読するためのさらに明確なスペクトルを可能にする。重合反応を阻害する可能性があるため、冷凍を避ける。別の例において、配列決定速度を減少させるために、配列決定反応を実施する環境は、20%のFicol70中の9.95cPの粘度などの粘性にさらされる。さらに、ポリメラーゼを遅延させる1つまたは複数の突然変異を含むポリメラーゼなどの突然変異体ポリメラーゼを、配列決定速度を減少させるために使用することができる。配列決定速度はまた、配列決定反応中に存在する遊離の加水分解性ヌクレオチド(dNTPなど)の濃度によって制御することもでき、遊離のdNTPの濃度の減少は、重合速度を減少させる。
圧縮配列
特定の例において開示される方法は、圧縮配列を提供する。つまり、標的配列がある列において2つ以上の同一塩基を含む場合、本方法は、2つ以上の同一塩基間の差異を検出しない可能性があり、それによって、圧縮配列を生成する。例えば、大腸菌の配列の初めの30塩基である
は、類似塩基の列を除去することにより、
と圧縮され、
を生じる。
特定の例において開示される方法は、圧縮配列を提供する。つまり、標的配列がある列において2つ以上の同一塩基を含む場合、本方法は、2つ以上の同一塩基間の差異を検出しない可能性があり、それによって、圧縮配列を生成する。例えば、大腸菌の配列の初めの30塩基である
そのような圧縮配列は一意的であるため、さらに使用可能である。例えば、大腸菌由来のRNAが配列決定され、上記の配列3を生じる場合、RNAの位置は、上記の配列3を全大腸菌ゲノムの圧縮されたバージョンと比較することにより決定することができる。全ヒトゲノムが配列決定される場合、本方法は、個別のmRNA分子を直接計数するために使用することができる。第1の工程は、全ヒトゲノム配列を圧縮することである。その後、NCBI Basic Local Alignment Search Tool(BLAST)、またはその他のプログラムを用い、本開示の配列決定方法により得られた結果を使用して、圧縮されたヒトゲノム配列を検索する。本方法は、ハイスループット分析に対する巨視的処理を必要とせず、遺伝子発現の研究のために極めて有用である。
しかしながら、完全な、非圧縮配列の検出を可能にする方法を提供する。例えば、正確な化学的部分(正確な塩基など)が活性部位にある場合のみ、重合剤は、活性部位に近づく。ポリメラーゼ部分の距離の変化は、例えば、FRETにより測定できる。このタイミングシグナルは、重合剤にタグを含むことによって生成することができ、タグの運動は検出される。例えば、重合剤は、供与体および受容体フルオロフォアの対を含むことができ、重合剤の開閉は、フルオロフォアの相互作用により検出可能なシグナル(供与体による受容体の励起、検出することができる得られた受容体発光)を生じる。タイミングシグナルの検出は、取り込みを示すので、2つの塩基が同一である場合であっても、2つの塩基が新生DNAポリマーに付加されていることが明らかである。
完全な、非圧縮配列を検出するために使用することができる別の方法は、フローセルのように、薬剤を付加および除去することができるシステムにおいて、配列決定反応を実施することである。例えば、プローブは、表面(ビーズなど)、および付加したプライマー/標的核酸分子に結合することができる。フローは、4つの加水分解性塩基(dNTPなど)のいずれも提供することができる。化学的部分は、標的核酸分子上の相補的塩基と対合するので、得られるシグナルは、次に付加する必要がある加水分解性塩基を示す。そのような加水分解性塩基は、配列決定反応に加えられ、プローブは、1つ先の塩基に進む。加水分解性塩基はその後除去され、過程は反復される。集団において、プローブは、次の不足塩基への指数関数的遷移を示し、停止する。従って、集団は、時間とともにモニタリングすることができる。付加される2つの塩基がある場合、遷移はより遅くなる。つまり、付加した塩基の数は、遷移の半減期を測定することにより決定することができる。
別の例において、本開示のプローブは、基板に結合され、標的核酸およびそのプライマーに結合される。そのような例において、プローブは、それぞれの化学的部分に関連する同一のタグ(同一の受容体フルオロフォア、または受容体フルオロフォアの組み合わせなど)を含む。重合剤はまた、タグ(供与体フルオロフォアなど)に関連する。非標識加水分解性ヌクレオチドは、例えば、同時に1つの型を付加することができる。非標識加水分解性ヌクレオチドの非存在下で、化学的部分は活性部位にとどまり、検出可能なシグナル(受容体フルオロフォア発光など)を生成する。その後、非標識加水分解性ヌクレオチドの1つが付加される。不正確な非標識加水分解性ヌクレオチドが付加される場合、これは化学的部分を置換せず、従って、検出可能なシグナルは有意に変化しない。対照的に、正確な非標識加水分解性ヌクレオチドが付加される場合、これは化学的部分を置換し、従って、検出可能なシグナルを有意に変化させる(例えば、受容体発光の減少または供与体発光の増加など)。回復のタイミングは、伸長する相補鎖に付加される非標識加水分解性ヌクレオチドの数に依存するため、2つの非標識加水分解性ヌクレオチドが付加される場合、1つの非標識加水分解性ヌクレオチドが付加される場合と比べ、検出可能なシグナルが回復するのに時間を要する。従って、4つの非標識加水分解性ヌクレオチドを通じて循環することにより、完全な配列を得ることができる。
完全な、非圧縮配列を検出するために使用することができるさらに別の方法は、ナノプローブ上の化学的部分上の受容体フルオロフォアとは異なる受容体フルオロフォアを特異的に活性化する供与体フルオロフォア(重合剤に結合された第2の供与体フルオロフォアなど)を有するナノプローブを使用することである。配列決定される核酸は、受容体フルオロフォア(第2の供与体フルオロフォアにより励起するものなど)を有する少なくとも1つのヌクレオチドにおいて標識される。ナノプローブは、配列決定される核酸分子に沿って進むため、ポリメラーゼ上の供与体と受容体標識された鋳型核酸分子との間の距離は、変化するシグナルを与える。従って、ナノプローブが2つの同一塩基上に進む場合、受容体から供与体へのシグナルが同時に変化するため、これらを区別することができる。
単一の塩基の進行は、物理的に決定することができる(Greenleaf and Block, Science,313:801,2006)。そのような方法は、本明細書に開示されるMedusaナノプローブと組み合わせることができる。DNAに沿って進むことは、3.4Å工程(0.34nm)により、および約34度の回転(360°あたり10.6bp)により検出することができる。回転は、配列決定される核酸(DNAなど)に結合された分子リンカー上に蛍光検出器を設置することによって、増幅することができる。DNAは回転するため、34°回転は、より長いアームで、より長い距離を移動する。アームが長いほど、変化が大きい。ポリメラーゼ(本明細書に提供されるMedusaシーケンサーなど)が移動するか、DNAが移動するかのどちらかである。(Greenleaf and Block, Science,313:801,2006の方法を使用して)移動するポリメラーゼを観察することができる。移動する標的核酸分子は、標的核酸分子を標識することにより(例えば、DNA結合GFP融合タンパク質を結合することにより)検出することができる。標識された標的核酸分子は回転するため、標識の回転は、それにより、ステッピングの検出を可能にする。標的核酸分子が回転する場合に偏光の角度が変わるように標識が標的核酸分子に密に結合する場合、これは、標的核酸分子に対して移動するポリメラーゼの検出を可能にする。回転は、それぞれの位置からの光子の数を変える。GFP−DNA結合タンパク質は、ポリメラーゼがそれを過ぎて、またはヌクレオソーム様物体を通じて読み込むために除去することができるように(T7 RNAポリメラーゼに対して観察されているように)、結合される。
標的核酸分子(標的DNA分子など)の運動を検出するために使用することができる別の方法は、標的核酸分子に沿って、あるいは標的核酸分子周辺のいずれかに結合するナノプローブを使用することである。結合する場合、偏光したFRETシグナルが生じる。分子リンカーは、柔軟性のある、または硬性であることができる。硬性分子リンカーは、DNAから離れて検出点を移動させることができ、レバー分子リンカーを増加させる。別の例は、「Molecular Beacon scissors DNA検出器」を使用することである。これは、2つのアームがDNAの対面上で標的DNA分子に結合する硬性交差型形状分子である。中央の柔軟性のあるヒンジは、リンカーが互いに近接する場合にFRETシグナルが生成され検出されるように、2つの分子リンカーをDNAに結合することを可能にする。このデバイスは、DNAなどの標的核酸分子に結合しない場合、有意な検出可能なFRETシグナルを生成しない。結合する場合、FRETシグナルは検出され、MedusaシーケンサーがDNAを移動する場合、これは変化する偏光シグナルを生成するDNA周辺を移動する。
使用することができるさらに別の方法は、DNAをエチジウムブロマイドなどのDNA挿入色素と接触させることである。双極子は、ポリメラーゼが通過する場合に回転する。あるいは、ピロロdCは、内部蛍光標識としてDNAに組み込むことができる。DNAが回転するにつれて、ピロロdCからのシグナルは、偏光角を吸収し放出する双極子が変化するため、変化する。この双極子軸の変化は、検出することができるシグナル(例えば、エチジウムブロマイドまたはピロロdC由来)の変化を生じる。
インビボ配列決定
特定の例において、1つまたは複数のMedusaプローブが細胞に導入され、それによって、生細胞内での配列決定を可能にする。例えば、開示されたプローブは、生成されたmRNAの配列を観察するために(例えば、プローブが重合剤として逆転写酵素を含む場合)使用することができる。特定の例において、本方法は、遺伝的疾病または癌を診断するために使用される。
特定の例において、1つまたは複数のMedusaプローブが細胞に導入され、それによって、生細胞内での配列決定を可能にする。例えば、開示されたプローブは、生成されたmRNAの配列を観察するために(例えば、プローブが重合剤として逆転写酵素を含む場合)使用することができる。特定の例において、本方法は、遺伝的疾病または癌を診断するために使用される。
細胞に薬剤を導入する方法は、当技術分野で公知である。一例において、1つまたは複数のMedusaプローブはリポソームに組み込まれ、これは当技術分野で公知の標準的方法を使用して細胞に導入される。別の例において、プローブは、例えば、Sokolら(Proc.Natl.Acad.Sci.USA95:11538−43,1998)の方法を使用して、細胞に直接注入される。特定の例において、細胞は、ヒトなどの哺乳類の対象から取得される。例えば、細胞は、血液サンプル(またはその画分)または頬スワブなど、対象の生体試料から取得することができる。
細胞に存在するヌクレオチドは、成長する相補鎖(dNTPなどの加水分解性ヌクレオチド)に組み込まれるものである。プライマーはまた、プローブと共に細胞に導入することもできる。特定の例において、プライマーは、例えば分子リンカーを介して、重合剤に結合される。特定の例において、プライマーの配列は、標的配列に基づいて決定される。例えば、特定の突然変異が標的配列に存在するか否かを決定することが目標である場合、プライマー配列は、Medusaプローブをそのような配列に誘導するために使用することができる。あるいは、プライマーが任意の配列に結合するように普遍的塩基から成ることができる(例えば、Berger et al. Nucleic Acids Res.28:2911−4,2000を参照)。
特定の例において、成熟RNA上の選択的スプライシング変異体が識別される。
実施例1
タグ付きポリメラーゼ
本実施例は、フルオロフォアまたは発光分子(例えば、供与体フルオロフォア)などの少なくとも1つのタグを含むポリメラーゼを生成するために使用できる方法を記載する。特定のフルオロフォアまたは発光分子を記載するが、その他のタグは類似方法を使用してポリメラーゼに結合することができることを当業者は理解するであろう。
タグ付きポリメラーゼ
本実施例は、フルオロフォアまたは発光分子(例えば、供与体フルオロフォア)などの少なくとも1つのタグを含むポリメラーゼを生成するために使用できる方法を記載する。特定のフルオロフォアまたは発光分子を記載するが、その他のタグは類似方法を使用してポリメラーゼに結合することができることを当業者は理解するであろう。
タグの一般的な結合
タグは、標準組換え技術を使用して重合剤に結合させることができる。一般に、タグは、重合剤のNまたはC末端に置かれる。しかしながら、タグはまた、重合剤内の任意のアミノ酸、例えば、タンパク質表面に露出したアミノ酸に結合させることもできる。理想的には、タグ結合は、重合剤の重合活性を著しく妨害しない。例えば、融合重合剤タンパク質は、伸長する核酸分子にヌクレオチドを組み込む能力をさらに有するであろう。
タグは、標準組換え技術を使用して重合剤に結合させることができる。一般に、タグは、重合剤のNまたはC末端に置かれる。しかしながら、タグはまた、重合剤内の任意のアミノ酸、例えば、タンパク質表面に露出したアミノ酸に結合させることもできる。理想的には、タグ結合は、重合剤の重合活性を著しく妨害しない。例えば、融合重合剤タンパク質は、伸長する核酸分子にヌクレオチドを組み込む能力をさらに有するであろう。
融合タンパク質の生成方法は、当業者に公知である(例えば、Sambrook et al,Molecular Cloning,A Laboratory Manual,Cold Spring Harbor Laboratory,Cold Spring Harbor,New York,Chapter17,1989を参照)。タグポリメラーゼ組換え融合タンパク質を調製するために、タグおよびポリメラーゼをコード化した配列を含むベクターを構成することができる。配列は、所望のタグポリメラーゼ組換え融合タンパク質を生成するために順序付けされる。本ベクターは、大腸菌などのバクテリアで発現され、タンパク質は精製される。精製法は、結合したタグに依存することができる。例えば、アフィニティータグはまた、重合剤に結合させる場合、細菌溶解物は、融合タンパク質にあるタグに対して高アフィニティーを有する樹脂を含むカラムに加えられる。溶解物を加え、タグ付き融合タンパク質を結合させた後、非結合タンパク質を流出し、続いて融合タンパク質を溶出する。
あるいは、タグを、化学的方法を使用して重合剤に結合させることができる。例えば、タグは、Cys残基に結合させようとする場合、タグは、マレイミドなどのチオール反応性化合物を使用して、重合剤に結合させることができる。
アフィニティータグ
一例において、アフィニティータグは、GFP−ポリメラーゼタンパク質などの重合剤に結合され、その精製およびその後の基板への結合に役立つ(実施例7を参照)。アフィニティータグの例には、ヒスチジン(His)、ストレプトアビジン、S−タグ、およびグルタチオン−S−トランスフェラーゼ(GST)を含む。当業者に公知の他のタグをまた使用することもできる。市販のベクターは、1つまたは複数のアフィニティータグを含む。これらのベクターは、直接使用することができ、または必要に応じて、タグをコード化した配列は、PCRを使用してベクターから増幅し、その後、上記重合剤を含むベクターなどの異なるベクターに連結することができる。
一例において、アフィニティータグは、GFP−ポリメラーゼタンパク質などの重合剤に結合され、その精製およびその後の基板への結合に役立つ(実施例7を参照)。アフィニティータグの例には、ヒスチジン(His)、ストレプトアビジン、S−タグ、およびグルタチオン−S−トランスフェラーゼ(GST)を含む。当業者に公知の他のタグをまた使用することもできる。市販のベクターは、1つまたは複数のアフィニティータグを含む。これらのベクターは、直接使用することができ、または必要に応じて、タグをコード化した配列は、PCRを使用してベクターから増幅し、その後、上記重合剤を含むベクターなどの異なるベクターに連結することができる。
6または10の連続したヒスチジン(His)残基部分は、金属イオンのための高アフィニティーを有する。His−6またはHis−10部分は、pETベクター(Novagen,Madison,WI)を使用して重合剤に結合させることができる。GFP−His(Park and Raines,Protein Sci.6:2344−9,1997)およびタンパク質−GFP−His組換えタンパク質の生成は、前述されている(Prescott et al.,FEBS Lett.411:97−101,1997)。Hisを含有する融合タンパク質は、Paborskyら(Anal.Biochem.,234:60−5,1996)に記載されるように精製することができる。手短に言えば、細胞溶解物から得られたタンパク質は、アフィニティークロマトグラフィーを使用してNi2+−NTA−アガロース(QIAGEN,Valencia,CA)に固定化される。非結合タンパク質を洗浄除去した後、例えば、8mMイミダゾール、50mM Tris HCl、pH7.5、150mM NaClを含む緩衝液を使用して、結合組換えタンパク質は、例えば100〜500mMの高濃度のイミダゾールを含む同一の緩衝液を使用して溶出される。
S−タグシステムは、膵臓リボ核酸分解酵素A由来のS−タンパク質と15アミノ酸S−タグペプチドの相互作用に基づく。S−タグ付きタンパク質の精製キットに加えて、S−タグ融合タンパク質を生成するためのいくつかのベクターは、Novagen(Madison,WI)から市販されている。例えば、ベクターpET29a−cおよびpET30a−cを使用することができる。S−タグ融合タンパク質は、S−タグ融合タンパク質を保持するS−タンパク質アガロースビーズを使用して、細胞溶解物をインキュベートすることにより精製される。非結合タンパク質を洗浄除去した後、融合タンパク質は、S−タグペプチドを残す部位特異的プロテアーゼを有するアガロースビーズの培養により放出される。
アフィニティータグストレプトアビジンは、非常に高いアフィニティーでD−ビオチンに結合する。ストレプトアビジン−融合タンパク質を生成するベクターおよびこれらのタンパク質を精製する方法は、SantoおよびCantorに記載されている(Biochem.Biophys.Res.Commun.176:571−7,1991、参照により本明細書に組み込まれる)。融合タンパク質を精製するために、細胞溶解物は、2−イミノビオチンアガロースカラムに加えられ(その他のビオチンを含有するカラムを使用してもよい)、非結合タンパク質を洗浄除去した後、融合タンパク質は、例えば、6M尿素、50mM酢酸アンモニウム(pH4.0)を使用して溶出される。
酵素グルタチオン−S−トランスフェラーゼ(GST)は、グルタチオンに対する高アフィニティーを有する。GST(pGEX)を含有するプラスミド発現ベクターは、Smithへの米国特許第5,654,176号およびSharrocks(Gene,138:105−8,1994)に開示される。pGEXベクターは、Amersham Pharmacia Biotech(Piscataway,NJ)から市販される。細胞溶解物は、グルタチオン−アガロースビーズでインキュベートされ、洗浄後、融合タンパク質は、例えば、5mM還元したグルタチオンを含有する50mM Tris−HCl(pH8.0)を使用して、溶出される。GST−重合剤の融合タンパク質の精製後、GST部分は、特定のタンパク質分解的切断により放出できる。GST−融合タンパク質は不溶性である場合、1%のTriton X−100、1%のTween20、10mM ジチオスレイトールまたは0.03%のNaDodSO4などのグルタチオン−アガロースに結合するために分離しない可溶化剤において、タンパク質が可溶化する場合、アフィニティークロマトグラフィーにより精製することができる。GST−融合タンパク質を可溶化するために使用される他の方法は、FrangioniおよびNeelにより記載される(Anal.Biochem.210:179−87,1993)。
組換えGFP−ポリメラーゼ
緑色蛍光タンパク質(GFP)は、GFPの中央にアミノ酸により形成された発色団を含む。野生型GFPは、508nmで発光を生成するために393nmまたは476nmで励起される。GFP突然変異体は、励起および発光スペクトルのどちらかを有する。GFP突然変異体H9−40は、398nmで単一吸収のみを有し、511nmで放出する。赤方移動GFP突然変異体RSGFP4(Delagrave et al.,Biotechnology13:151−4,1995)は、490nmで励起し、505nmで発光する。青方移動GFP突然変異体BFP5は、385nmで吸収し、450nmで放射する(Mitra et al.,Gene,173:13−7,1996)。
緑色蛍光タンパク質(GFP)は、GFPの中央にアミノ酸により形成された発色団を含む。野生型GFPは、508nmで発光を生成するために393nmまたは476nmで励起される。GFP突然変異体は、励起および発光スペクトルのどちらかを有する。GFP突然変異体H9−40は、398nmで単一吸収のみを有し、511nmで放出する。赤方移動GFP突然変異体RSGFP4(Delagrave et al.,Biotechnology13:151−4,1995)は、490nmで励起し、505nmで発光する。青方移動GFP突然変異体BFP5は、385nmで吸収し、450nmで放射する(Mitra et al.,Gene,173:13−7,1996)。
重合剤は、当業者に公知の組換え技術により、GFP−ポリメラーゼなどの融合タンパク質を生成するためにGFPに結合することができる。野生型または突然変異体GFP遺伝子配列およびポリメラーゼ配列を挿入することができる多重クローニング部位(MCS)を含むプラスミド(pGFPなど)は、Clontech(Palo Alto,CA)から市販されている。
手短に言えば、ポリメラーゼDNAおよびGFPプラスミドの双方は、センス方向において、GFPプラスミドのMCSにポリメラーゼの挿入を可能にする適切な制限酵素で消化される。得られたフラグメントは、連結され、大腸菌などのバクテリアで発現される。発現した組換えGFP−ポリメラーゼは、その後、当業者に公知の方法を使用して精製される。GFPは、ポリメラーゼのNまたはC末端で、またはその間のどこでも(露出したCys残基など)、置くことができる。理想的には、GFP−ポリメラーゼは、重合活性およびGFPの蛍光特性を保持する。
組換えGFP−エクオリン−ポリメラーゼ
組換えGFP−エクオリン−ポリメラーゼは、当業者に公知の方法、例えば、Baubetらにより開示された方法を使用して、生成することができる(Proc.Natl.Acad.Sci.USA97:7260−5,2000、参照により本明細書に組み入れられる)。
組換えGFP−エクオリン−ポリメラーゼは、当業者に公知の方法、例えば、Baubetらにより開示された方法を使用して、生成することができる(Proc.Natl.Acad.Sci.USA97:7260−5,2000、参照により本明細書に組み入れられる)。
手短に言えば、エクオリンcDNA(例えば、Genbank Accession No.L29571)、ポリメラーゼDNA、およびGFPプラスミドは、センス方向において、GFPプラスミドのMCSにエクオリンおよびポリメラーゼの挿入を可能にする適切な制限酵素で消化される。得られたフラグメントは、連結され、大腸菌などのバクテリアで発現される。発現した組換えGFP−エクオリン−ポリメラーゼは、その後、上記のように精製される。また、アフィニティータグを、加えることもできる。
GFP、エクオリン、およびポリメラーゼ配列の順序付けは、最適化することができる。配列決定のための最適特性を有することを決定するために、得られるGFP−エクオリン−ポリメラーゼを試験する。そのような特性は、タンパク質精製の容易さ、生成されるタンパク質量、放出した化学発光シグナル量、励起後放出した蛍光性シグナル量、GFPおよびエクオリンの蛍光特性の最小変化、およびポリメラーゼ活性量を含むことができる。
ポリメラーゼへのフルオロフォア結合
GFP−ポリメラーゼ融合タンパク質を生成する代替物として、その他のフルオロフォアを使用することができる。特定の例において、蛍光標識された重合剤は、高蛍光収率を有し、重合剤の生物学的活性を保持し、主として、核酸分子の相補的鎖を合成する能力を有する。重合剤は、従って、重合剤の機能を維持するために最大ではない蛍光収率を有する。反応染料でタンパク質を標識化する方法は、当業者には公知である。さらに、分子プローブ(Eugene,OR)などのそのような蛍光染料の製造者は、そのような反応を実施するために指示を提供する。1つまたは複数のフルオロフォアの重合剤への接合に続き、非接合染料は、例えば、ゲルろ過、透析またはその組み合わせにより除去することができる。
GFP−ポリメラーゼ融合タンパク質を生成する代替物として、その他のフルオロフォアを使用することができる。特定の例において、蛍光標識された重合剤は、高蛍光収率を有し、重合剤の生物学的活性を保持し、主として、核酸分子の相補的鎖を合成する能力を有する。重合剤は、従って、重合剤の機能を維持するために最大ではない蛍光収率を有する。反応染料でタンパク質を標識化する方法は、当業者には公知である。さらに、分子プローブ(Eugene,OR)などのそのような蛍光染料の製造者は、そのような反応を実施するために指示を提供する。1つまたは複数のフルオロフォアの重合剤への接合に続き、非接合染料は、例えば、ゲルろ過、透析またはその組み合わせにより除去することができる。
例えば、アミン反応性フルオロフォアは、重合剤に結合することができる。使用可能なアミン反応性プローブの例は、フルオレセイン、BODIPY、ローダミン、テキサスレッドおよびそれらの誘導体を含むがそれらに限定されない。そのような染料は、N末端で遊離アミンと同様に、ポリメラーゼ内のリジン残基に結合するであろう。アミン反応性フルオロフォアの反応は、通常pH7〜10の範囲のpH値で開始する。
あるいは、チオール反応性プローブは、蛍光標識されたポリメラーゼを生成するために使用することができる。タンパク質では、チオール基は、システイン残基に存在する。チオールを伴うフローライト反応は、化学的に安定したチオエステルを得るために、生理学的pH領域(pH6.5〜8.0)において、室温で、または室温以下で、通常迅速に行われる。使用可能なチオール反応性プローブの例には、フルオレセイン、BODIPY、クマリン、ローダミン、テキサスレッドおよびそれらの誘導体を含むがそれらに限定されない。
アルコール(セリン、トレオニン、およびチオシン残基)、カルボン酸およびグルタミンを含むタンパク質の他の官能基は、エーテルフルオロフォアをポリメラーゼに接合するために使用することができる。
ポリメラーゼに結合することができる別のフルオロフォアは、AllenおよびBenkovicにより記載されるように、4−[N−[(ヨードアセトキシ)エチル]−N−メチルアミノ]−7−ニトロベンズ−2−オキサ−l,3−ジアゾール(IANBD)である(Biochemistry,1989,28:9586)。
実施例2
タグ付き化学的部分
本実施例は、1つまたは複数のタグを非加水分解性ヌクレオチド類似体(例えば、dNMPまたはdNDP)などの化学的部分に結合する方法を記載する。さらに、当業者は、市販のタグ付き化学的部分は、本開示のプローブに使用することができることを認識するであろう。
タグ付き化学的部分
本実施例は、1つまたは複数のタグを非加水分解性ヌクレオチド類似体(例えば、dNMPまたはdNDP)などの化学的部分に結合する方法を記載する。さらに、当業者は、市販のタグ付き化学的部分は、本開示のプローブに使用することができることを認識するであろう。
1つまたは複数の受容体フルオロフォアなどのタグは、化学的部分のいずれの部分にも結合することができる。例えば、化学的部分がヌクレオチド類似体である場合、タグは、塩基、糖質、またはリン酸(α、β、またはγ−リン酸など)に直接的または間接的に結合することができる。理想的には、そのような結合は、鋳型鎖に可逆的結合し、標的核酸鎖において相補的ヌクレオチドと対にする化学的部分の能力を阻害しない。さらに、理想的には、タグは、検出可能なシグナルを提供するために化学的部分に結合することができる。
一例において、受容体フルオロフォアなどのタグは、リンカー分子により化学的部分に間接的に結合される。例えば、ストレプトアビジン連鎖を使用することができる。連鎖は、ペプチダーゼ感受性である場合があり、発光後、タグを放出させることができ、シグナルは、標的核酸鎖における化学的部分とその相補的塩基との間の対合の結果として検出される。Hobbsらへの米国特許第5,047,519号および第5,151,507号(参照により本明細書に組み入れられる)は、ヌクレオチドをフルオロフォアから単離するためにリンカーの使用を教示する。リンカーの例は、直鎖アルキレン、C1−C20を含み、任意的に、二重鎖結合、三重結合、アリール基またはN、OまたはSなどのヘテロ原子を含んでもよい。ジラジカル部分の置換基は、C1−C6アルキル、アリール、エステル、エーテル、アミン、アミドまたはクロロ基を含むことができる。
Jena BioScienceは、dGMPCPP、dAMPCPP、dCMPCPP、TMPCPP、dGMPNPP、dAMPNPP、dCMPNPP、およびTMPNPPなどの加水分解性ではない複数の異なるヌクレオチド類似体を提供し、αリン酸とβリン酸の間の酸素の代わりにCは、CH2基を示し、Nは、NH基を示す。
化学的部分のそれぞれ異なる型(dAMP、dCMP、dGMPおよびdTMPなど)に関連するタグが異なる供与体フルオロフォアの例において、理想的には、重合剤に供与体フルオロフォアを励起するために使用される周波数は、受容体フルオロフォアの励起スペクトルに有意に重複するとは言えない。しかしながら、各化学的部分は、供与体フルオロフォアからの発光が受容体フルオロフォアを励起するように、重合剤に結合される供与体フルオロフォアの発光スペクトルを重複する励起スペクトルを有する少なくとも1つの受容体フルオロフォアを保有しなければならない。さらに、化学的部分のそれぞれの型(dAMP、dCMP、dGMPおよびdTMPなど)は、それぞれの型(sdAMP、dCMP、dGMPおよびdTMPなど)がその他の型(dAMP、dCMP、dGMPおよびdTMPなど)と区別できる発光シグナル(発光スペクトルなど)を有するように、結合した一意的なタグ(または一意的な組み合わせのタグ)を有するであろう。故に、「A」に相補的である化学的部分は、「C」、「G」、または「T」に相補的である化学的部分と異なる発光シグナルを発し、「C」に相補的である化学的部分は、「T」、「G」、または「A」に相補的である化学的部分と異なる発光シグナルを発し、「G」に相補的である化学的部分は、「C」、「A」、または「T」に相補的である化学的部分と異なる発光シグナルを発し、および「T」に相補的である化学的部分は、「C」、「G」、または「A」に相補的である化学的部分と異なる発光シグナルを発するだろう。RNAの場合、本例において、Uは、Tの代わりに機能するだろう。
タグ付きの化学的部分が、標的核酸に相補的ヌクレオチドと組み合わせることができることを確実にするのに役立てるために、タグ付きの化学的部分は、例えば、タグが受容体フルオロフォアである場合、蛍光分光光度計を使用して試験することができる。
オリゴヌクレオチドが4つの塩基突出を持つdsDNAを形成するためにそれ自体にアニーリングするように、Tm値が高いヘアピンを含む5’フルオレセイン標識付きのオリゴヌクレオチドを合成する(Lyakhov et al,Nucl.Acid Res.29:4892−4900,2001)。ヘアピンオリゴヌクレオチドは、DNAの上の鎖および下の鎖の正確な等モル量を可能にし、それは、いずれのアニーリング調製工程を必要としない。ポリメラーゼは、突出部を埋めることができ、付加される次の塩基は、dTTPである。本オリゴヌクレオチドの一バージョンにおいて、3’末端は、Cy3に接合されるdTTPを使用して1つの塩基により延長することができるヒドロキシル末端を有する正常dCを有する。埋めることにより、フルオレセインは、Cy3に近く、FRETは、蛍光分光光度計を使用して2つのフルオロフォア間で測定される。これは、ポリメラーゼがDNAに結合し、与条件下で、重合化することができることを示す。ジデオキシC(IDT、Coralville,IA)で終わる別の5’フルオレセイン標識付きオリゴヌクレオチドが合成される。末端平滑化反応条件(Fill in reaction condition)は、第1オリゴヌクレオチドと同様に設定される。ポリメラーゼが存在する場合、FRETは、5’フルオレセインと、ジデオキシターミネーション化されたオリゴヌクレオチドに共有結合できないdTTP−Cy3との間で観測される。ポリメラーゼなしでは、FRETは観測されず、(ポリメラーゼが存在する場合)観測したFRETは、DNAヘアピン上のその相補塩基にCy3−dTTPを保持するポリメラーゼにより生じることを示す。これらの同一条件下で、dATP、dGTP、dCTPはFRETから離れ競合しないが、dTTPは競合し、不正確なものではなく、正確な次のヌクレオチドを結合させるためのポリメラーゼ−ヘアピン複合体による選択を示す。
実施例3
化学的部分へのリンカーの結合
本実施例は、図1に示すように、それぞれの分子リンカー14、16、18、20への化学的部分22、24、26、28の結合など、リンカー(PEGなど)を化学的部分(ヌクレオチド類似体など)に結合するために使用することができる方法を記載する。
化学的部分へのリンカーの結合
本実施例は、図1に示すように、それぞれの分子リンカー14、16、18、20への化学的部分22、24、26、28の結合など、リンカー(PEGなど)を化学的部分(ヌクレオチド類似体など)に結合するために使用することができる方法を記載する。
テザー末端は、アミノ基を含むことができ、ホスホラミダイト連結を介して非加水分解性のdNTPアナログのγ−リン酸に結合することができる。
DNA−PEG−DNA−NH2+PPNP−デオキシリボヌクレオチド→
DNA−PEG−DNA−NH−PPNP−デオキシリボヌクレオチド
式中PPNPは、α,β−P−O−P結合は、イミド基、P−N−Pにより置換されることを意味する。反応は、1M 1−エチル−3−(ジメチルアミノプロピル)カルボジイミド(EDC)、pH6.5、20°Cで10〜12時間、使用する(Chatterji et al.,Methods Enzymol.274:456−78,1996)。
DNA−PEG−DNA−NH2+PPNP−デオキシリボヌクレオチド→
DNA−PEG−DNA−NH−PPNP−デオキシリボヌクレオチド
式中PPNPは、α,β−P−O−P結合は、イミド基、P−N−Pにより置換されることを意味する。反応は、1M 1−エチル−3−(ジメチルアミノプロピル)カルボジイミド(EDC)、pH6.5、20°Cで10〜12時間、使用する(Chatterji et al.,Methods Enzymol.274:456−78,1996)。
さらに、Jena BioScienceは、アミノ基(γ−[(8−アミノオクチル)−イミド]−dUTP)で終わるγリン酸に結合されるテザーを有するdUTPを提供する。
実施例4
例示的分子リンカー
本実施例は、特異な屈曲を有する例示的な分子リンカーを記載する。これは、プローブが極めて特異な運動をしか有さないことを可能にし、絡み合いの可能性を取り除くことができる。
例示的分子リンカー
本実施例は、特異な屈曲を有する例示的な分子リンカーを記載する。これは、プローブが極めて特異な運動をしか有さないことを可能にし、絡み合いの可能性を取り除くことができる。
特定の実施例において、分子リンカーは、隆起を有するdsDNA、欠損した塩基、またはdsDNA内のニック(例えば、中央部付近または中央部で)を含む。別の実施例において、dsDNAは、両鎖における同一位置で短いPEGテザーを含む(図3)。このような分子リンカーは、分子リンカーの柔軟性および剛性をさらに制御するために使用することができる。例えば、屈曲は、重合剤の活性部位に化学的部分の先端を誘導するために分子リンカーの屈曲部を方向付けるための特定角度の範囲で設計することができる。特定の実施例において、dsDNAの3’および5’末端の双方は、回転を避けるためにポリメラーゼに結合される。
実施例5
核酸分子を配列決定するためのプローブ
本実施例は、標的核酸分子を配列決定するために使用することができる特定のプローブを記載する。特定のフルオロフォア、分子リンカー、およびポリメラーゼが記載されるが、これらへの変化は、本明細書の教示に基づいて行うことができることを当業者は理解するであろう。
核酸分子を配列決定するためのプローブ
本実施例は、標的核酸分子を配列決定するために使用することができる特定のプローブを記載する。特定のフルオロフォア、分子リンカー、およびポリメラーゼが記載されるが、これらへの変化は、本明細書の教示に基づいて行うことができることを当業者は理解するであろう。
設計は、図2Cに基づくが、ポリメラーゼへの分子リンカーの結合方法は、テザー306、dsDNA310、およびテザー308を除去することにより変更される。dsDNA336は、いかなる切断もなく、Tusタンパク質に対する結合部位(ter)を含む連続dsDNAにより置換される。Tusタンパク質は、HIV−1 RT(例えば、実施例1に記載される方法を使用して)に翻訳的に融合される。
DNA配列は、NANEVプログラムを使用して設計され、図2Cに示される制限酵素認識部位が唯一であることを確保するために確認された。さらに、MaeIII制限酵素認識部位は、ter配列に必然的に出現し、これは、プローブにおいて、唯一である。使用したTus配列は、9つの既知のTus部位からのコンセンサス塩基である。ある対のssDNA配列が互いに相補的であり、いくつかの配列が図2Cに示される制限酵素認識部位を含む制限を考慮し、NANEVプログラムが、構造を進化させるために使用された。
NANEVは、dsDNA鎖の名前に単一文字を使用する。小文字(a、c、g、t、e、h、b、p、m)は、大文字(A、C、G、T、E、H、B、P、M)で標識化された対応するssDNAにハイブリダイズされるssDNAのセグメントを示す。「ハブ」部分がそれらを切断する制限酵素により命名される(E、H、B、P、M)が、それぞれのdsDNAの分岐は、対応する非加水分解性塩基(A、C、G、T)により命名される。例えば、非加水分解性アデノシン346を有する分岐334に関しては、1つのオリゴヌクレオチドは、356−334−1aと称され、フルオロフォア356に結合される。356−334−1aは、332−316−334−318−346−11E−2Aと共にアニーリングする。
NANEVを使用して設計された14のdsDNA部分を表3に示す。
(表3)ナノプローブを生成するための配列
これらの14の成分のいくつかは、PEGにより接合され、10のオリゴヌクレオチドを生成するために適切なフルオロフォアに結合される。IDT(Coralville,IA)またはMidland(Midland,Texas)は商業的に10のオリゴヌクレオチドを以下のように合成することができる。
ここで[フルオロフォア356]は、Rhodamine Red(TM)−X(最大吸収:574nm、最大発光:594)「緑色」である。
ここで[フルオロフォア358]は、Cy3(TM)(最大吸収:550nm、最大発光:564)「青色」である。
ここで[フルオロフォア360]は、テキサスレッド(登録商標)−X(最大吸収:598nm、最大発光:617)「黄色」である。
ここで[フルオロフォア362]は、Cy5(商標)(最大吸収:648nm、最大発光:668)「赤色」である。
4つの非加水分解性のdNTPは、合成される(例えば、Jena Bioscienceにより):dGMPCPP、dAMPCPP、dCMPCPP、およびTMPCPPであり、ここでCはαとβリン酸との間で酸素の代わりにCH2基を示す。ここで留意すべきは、古い用語TMPCPPは、dTMPCPP、つまりデオキシリボ−TMPCPPを意味することである。(Jena Bioscienceはまた、dGMPNPP、dAMPNPP、dCMPNPP、およびTMPNPPを提供することができ、ここでNは、αとβリン酸の間の酸素の代わりのNH基を示す。ここで留意すべきは、古い用語TMPNPPは、dTMPNPP、つまりデオキシリボ−TMPNPPを意味することである。)
標識およびオリゴヌクレオチドの役割が逆であることを除き、Pierce Technical Resource TR0030.1「修正および標識オリゴヌクレオチド5’リン酸基」から得られる以下の反応プロトコルを使用して、それぞれの非加水分解性のdNTPを、対応するオリゴヌクレオチド分岐におけるアミノ基にそのγリン酸により共有結合させる。
1.10μlの反応緩衝液に非加水分解性のdNTPを溶解する。(Pierce社により推奨される反応緩衝液は、「EDTA入りリン酸緩衝生理食塩水(PBS):10mMリン酸ナトリウム、0.15M NaCl、10mM EDTA、pH7.2などの反応緩衝液である。意図した反応を妨害する、10mMより多いリン酸を含有するPBSの使用を避けなければならない。その他のアミンを含まないおよびカルボン酸を含まない緩衝液を代用することができるが、反応をクエンチする1級アミンを含むTrisを避けなければならない。」)
2.オリゴヌクレオチドを10μlの0.1Mイミダゾール、pH6中に溶解し、1mMの最終濃度にする。
3.1.25mg(6.52micromol)のEDC(1−エチル−3−[3−ジメチルアミノプロピル]カルボジイミド塩酸塩、Pierce Product No.22980)を量り、微小遠心管に入れる。
4.EDCを含む前記管に7.5μlの調製した非加水分解性のdNTPを加え、直ちに5μlのオリゴヌクレオチド/イミダゾール溶液を加える。
5.内容物が完全に溶解するまで前記管をボルテックスし、その後内容物を収集するために該管を少しの間、遠心分離する。
6.さらに20μlの0.1Mイミダゾール、pH6をさらに加える。
7.室温で一晩反応物をインキュベートする。
8.10%のポリアクリルアミドゲルに反応生成物から未反応のオリゴヌクレオチドを分離する。
2.オリゴヌクレオチドを10μlの0.1Mイミダゾール、pH6中に溶解し、1mMの最終濃度にする。
3.1.25mg(6.52micromol)のEDC(1−エチル−3−[3−ジメチルアミノプロピル]カルボジイミド塩酸塩、Pierce Product No.22980)を量り、微小遠心管に入れる。
4.EDCを含む前記管に7.5μlの調製した非加水分解性のdNTPを加え、直ちに5μlのオリゴヌクレオチド/イミダゾール溶液を加える。
5.内容物が完全に溶解するまで前記管をボルテックスし、その後内容物を収集するために該管を少しの間、遠心分離する。
6.さらに20μlの0.1Mイミダゾール、pH6をさらに加える。
7.室温で一晩反応物をインキュベートする。
8.10%のポリアクリルアミドゲルに反応生成物から未反応のオリゴヌクレオチドを分離する。
DNAポリメラーゼへの結合能力により生成物を精製することも可能である。初めに、サイズ排除カラムまたは透析を使用して未反応ヌクレオチドを除去する。次に、HIV−1 RTまたは結合した別のポリメラーゼ(例えば、ニッケルカラムへのヒスチジン−6タグ結合を有するHIV−1 RT)を有するカラムを作製する。鋳型DNAおよびアニーリングしたプライマーDNAを追加することができる。本ポリメラーゼカラムは、ヌクレオチドに結合したオリゴヌクレオチドを、テザーヌクレオチドを持たない未反応のオリゴヌクレオチドと比較して、遅延させなければならない。上記のカルボジイミド架橋結合反応の詳細は、ChatterjiおよびGopalで得られる(Methods Enzymol.274:456−78,1996)。本プロトコルは、それぞれのオリゴヌクレオチドについて別々に実施される。
これにより以下の構造が構築される。
次に、332−316−334−318−346−11E−2A(SEQ ID NO:23);340−322−338−320−352−14P−8T(SEQ ID NO:24);350−326−344−324−342−6G−13H(SEQ ID NO:25);348−312−328−314−330−4C−12B(SEQ ID NO:26);356−334−1a(SEQ ID NO:3);358−328−3c(SEQ ID NO:5);360−344−5g(SEQ ID NO:7);362−338−7t(SEQ ID NO:9);332−336−342−9eMh(SEQ ID NO:11);340−336−330−10pmb(SEQ ID NO:12)の10のオリゴヌクレオチドが、図2Cに示されるプローブの分子リンカー302を形成するため、一緒にハイブリダイズされる。分子リンカー302はその後ゲル電気泳動、排除カラムまたはショ糖密度勾配により精製される。得られた分子リンカー302の構造は、5つの制限酵素を単独でおよび組み合わせて用いて消化して、ポリアクリルアミドゲル上で生成物を観測することにより、試験することができる。
大腸菌由来のtus(終結利用因子)遺伝子は、pBAD33tusにクローン化された(Henderson et al,Mol.Genet.Genomics265:941−53,2001,Guzman et al,J.Bacterial.77:4121−30,1995)。HIV−1 RT p66サブユニットは、すべての溶媒利用しやすいシステイン残基はセリン残基に置換し(C38SおよびC280S)、287のリジンを唯一のシステインに置きかえる(K287C)(Kensch et al,J.Mol.Biol.301:1029−39,2000)。287の唯一のシステインは、ポリメラーゼの「サム(thumb)」にあり、ポリメラーゼの活性部位に近いが、DNA結合または活性部位を阻害しないように十分遠く離れている。Tusタンパク質はわずか2つのシステイン、CYS99およびCYS255、があり、双方は完全に埋められているため(PDB 1ECR Kamada et al.,Nature383:598−603,1996)、露出したシステインを避けるためにTusを遺伝子操作する必要はない。Tusは、突然変異HIV−1 RTを用いた翻訳による融合の際に、クローン化される。
HIV−1 RT(PDB entry 1RTD Huang et al.,Science282:1669−1675,1998)およびDNAに結合したTus結合(1ECR Kamada et al.,Nature383:598−603,1996)の三次元構造に見られるように、双方のタンパク質のNおよびC末端は、活性部位から十分距離を置いて表面にあるので、2つのタンパク質の融合は、それらの構造または機能を阻害しない。RecBタンパク質の2つの部分を結合する親水性ポリペプチド(PDB entry 1W36,Singleton et al.,Nature432:187−93,2004)は、TusをHIV−1 RTに接続してTus−HIV−1 RTを生成するために使用される。
当業者は、TusかHIV−1 RTタンパク質のどちらかが融合物のN末端に置かれ、それらは入れ替え可能であることを認識するであろう。当業者はまた、分離に役立てるために構造のいずれかの末端に6−ヒスチジンタグを置くことができることを認識するであろう。TusのN末端の6−ヒスチジンタグは、結合にほとんど影響がないが、HIV−1のC末端の6−ヒスチジンタグは、ポリメラーゼ活性に既知の影響を全く与えない。
供与体フルオロフォアは、マレイミド標識試薬フルオレセイン−5−マレイミド(Pierce、Rockford,IL製造者の使用説明書を使用)を使用することにより、Tus−HIV−1 RTにおいて唯一のシステインに結合する。この供与体フルオロフォアは、上記の4つの受容体の対のそれぞれとFRETの対を形成する。当業者はまた、相対シグナル強度を必要に応じて調整するために、対応するオリゴヌクレオチドに追加の受容体フルオロフォアを加えることができることも認識するであろう。当業者は、フルオロフォアの多くの他の組み合わせが可能であることを認識するであろう。
完成したプローブを生成するためにTus−HIV−1 RTタンパク質をコアに加える。プローブはその後ゲル電気泳動、排除カラムまたはショ糖密度勾配により精製される。最終プローブ構造は、5つの制限酵素で単独でおよび組合せにより消化し、ポリアクリルアミドゲルの生成物を観測することにより、確認することができる。
第2の実施例において、逆転写酵素は、そのエラー頻度を低減するためにF227Aの定方向突然変異誘発により改変される(Wisniewski et al.,J.Biol.Chem.274:28175−84,1999)。第3の実施例において、それが単鎖Fv(scFv)リンカー配列の場合と同様、TusとHIV−1 RTとの間の結合は、決定される。使用した古典的な配列は、(Gly4Ser)3であるが、ファージディスプレイ技術は、その他の組み合わせを得るために使用することができる(Tang et al.,J.Biol.Chem.271:15682−6,1996;Hennecke et al,Protein Eng.11:405−10,1998)。
当業者は、多くの他の組み合わせが本願開示のナノプローブに対して可能であることを認識するであろう。
実施例6
核酸分子を配列決定するためのコード化したプローブ
本実施例は、複数のフルオロフォアがそれぞれの非加水分解性のヌクレオチド類似体に関連する、実施例5に記載されるプローブの変異体を記載する。フルオロフォアの特定の組み合わせを記載するが、その他の組み合わせを使用することができることを当業者は理解するであろう。そのようなプローブは、それぞれの化学的部分に関連する受容体フルオロフォアの光退色の検出を可能にし、特定の実施例において、そのような光退色の修正を可能にする。
核酸分子を配列決定するためのコード化したプローブ
本実施例は、複数のフルオロフォアがそれぞれの非加水分解性のヌクレオチド類似体に関連する、実施例5に記載されるプローブの変異体を記載する。フルオロフォアの特定の組み合わせを記載するが、その他の組み合わせを使用することができることを当業者は理解するであろう。そのようなプローブは、それぞれの化学的部分に関連する受容体フルオロフォアの光退色の検出を可能にし、特定の実施例において、そのような光退色の修正を可能にする。
プローブの化学的部分のそれぞれが単一受容体フルオロフォアにのみ関連する場合、その受容体フルオロフォアは、退色により失い、受容体発光シグナルはその受容体フルオロフォアから検出されない。この光退色した受容体フルオロフォアからシグナルがない場合、シグナルの喪失が、光退色によるものであるか否か、または一続きの標的配列がその塩基を単に持たないか否かを識別することは困難である。それぞれの化学的部分に2つ以上のタグを関連させることにより、フルオロフォアの退色は、検出することができ、一部の実施例において修正されることができる。
図2Dに示されるプローブは、化学的部分346、348、350、352のそれぞれに関連する複数のタグ404、406、408、410を含む。複数の受容体フルオロフォアの含有は、退色により、または分子リンカー部分の喪失により、フルオロフォアの喪失の検出を可能にする。
図2Dでは、異なるタグ354、404、406、408、410は、Hammingエラー修正コードに類似のコードを形成する。これらのタグのいずれか1つが光退色により破壊される場合、得られるシグナルは、損傷を受けていないプローブにより生成されたものとは異なる。例えば、供与体354が破壊される場合、その後、供与体発光は、生成されず、他のフルオロフォアのどれも励起されない。そのようなプローブからの配列データは無視することができる。プローブの「A」アームは、2つの受容体フルオロフォア404および406を有する。相補的Tがポリメラーゼ結合部位の標的DNAにある場合、双方のフルオロフォアは、供与体354により励起される。これは、その他の3つのアームに比べて一意的なスペクトルを生成する。つまり、Aは、404、406に関連し、Cは、404、408に関連し、Gは、404、410に関連し、Tは、406、408、410に関連する。ロッド334のフルオロフォア404は、光退色化され、その後、Aアームは、406のスペクトルのみを含むシグナルを報告する。これは、損傷を受けていないAのシグナルおよびその他の3つのアームにより生成されるシグナルとは異なる。従って、記録コンピュータは、プローブが損傷され、終了しなければならないプローブから読み取るシグナルを決定することができる。Tアーム338は3つのフルオロフォアを有するため、フルオロフォア408および410の双方が同時に破壊される可能性が低い。従って、406からのスペクトルのみを含むシグナルは、標的ヌクレオチドへのAアームの結合によって生じる可能性が高い。従って、コンピュータは、読み込みを終了する前に配列の次の塩基が実際にはAであることを推定することができる。つまり、コンピュータは、エラー検出およびエラー修正を実施することができる。
ロッド334上のフルオロフォア406が破壊された場合、受信したシグナルは、スペクトル404のみで構成される。スペクトル404はまた、ロッド328上のフルオロフォア408を退色することにより、またはロッド344上のフルオロフォア410を退色することにより生成されることもできる。従って、シグナル406を発出したアームを、明確に決定することができない。この場合、DNA配列を読み込むコンピュータは、エラーが生じることを含むことができるが、エラーを修正することができないと結論づけることができる。この時、Medusaからの読み込みは終了する。
ロッド328または344上のフルオロフォアの破壊は、ロッド334上のフルオロフォアの破壊と類似の結果を生じる。これらの全ての場合、共通のフルオロフォア404が破壊される場合、あるエラーが検出され、そのエラーは、修正されることができる。404でないその他のフルオロフォア(つまり、ロッド334、328、344上のそれぞれフルオロフォア406、408、410)が破壊される場合、あるエラーが検出され、エラーは、修正されることができない。一度エラーが検出され、修正されると、第二のエラーが不明確な結果を生じるため、配列決定は、終了する。
ロッド338上の3つのフルオロフォア406、408、410のいずれかは、光退色され、その後残った2つのフルオロフォアはさらに、一意的なシグナルを生じる。例えば、フルオロフォア406が破壊され、その後Tアームが標的ヌクレオチド上のAに結合する場合、シグナルは、フルオロフォア408および410からのスペクトルで構成される。これらのスペクトルは単独で損傷していないプローブにより生成されないため、あるエラーが検出され、そのエラーは、修正されることができる。この場合、その他の3つのアームのように、第二のエラーが不明確な結果を生じるため、エラー修正後、配列決定は、終了される。
その他のコード化スキームは、本明細書に開示されるプローブに対して可能であり、4つ以上のフルオロフォアを有するスキームを含む。当業者は、種々のコード化スキームにより検出され、修正されることができるエラー数を予測することができる。しかしながら、図2Dに示されるものは、実施する4つのフルオロフォアのみ必要とする。図2Cに示されるような基本プローブは、十分な配列データを作製するために4つのフルオロフォアを使用するが、4つのフルオロフォアのみの冗長再配列は、エラーを検出し、修正するプローブの構築を可能にする。プローブ設計は、フルオロフォアの交換を可能にするため、図2Cに示されるように、ssDNA364とssDNA334を交換でき、基本非コード化のプローブが構築されると、図2Dに示されるようにコード化したプローブを作製することを可能にする。実施例5において、コード化プローブは、オリゴヌクレオチド356−334−1a(SEQ ID NO:3)、358−328−3c(SEQ ID NO:5)、360−344−5g(SEQ ID NO:7)、および362−338−7t(SEQ ID NO:9)が以下の4つのオリゴヌクレオチドにより置換される場合を除いて、構築される。
「青色」「赤色」
「青色」「黄色」
「青色」「緑色」
「赤色」「黄色」「緑色」
ここで[フルオロフォア404]はCy3(登録商標)(最大吸光度:550nm、最大発光:564)「青色」(図2Cのフルオロフォア358に対応)であり、[フルオロフォア406]は、Cy5(登録商標)(最大吸光度:648nm、最大発光:668)「赤色」(図2Cのフルオロフォア362に対応)であり、[フルオロフォア408]は、テキサスレッド(登録商標)−X(最大吸光度:598nm、最大発光:617)「黄色」(図2Cのフルオロフォア360に対応)であり、[フルオロフォア410]は、ローダミンレッド(商標)−X(最大吸光度:574nm、最大発光:594)「緑色」(図2Cのフルオロフォア356に対応)である。
ここで[フルオロフォア404]はCy3(登録商標)(最大吸光度:550nm、最大発光:564)「青色」(図2Cのフルオロフォア358に対応)であり、[フルオロフォア406]は、Cy5(登録商標)(最大吸光度:648nm、最大発光:668)「赤色」(図2Cのフルオロフォア362に対応)であり、[フルオロフォア408]は、テキサスレッド(登録商標)−X(最大吸光度:598nm、最大発光:617)「黄色」(図2Cのフルオロフォア360に対応)であり、[フルオロフォア410]は、ローダミンレッド(商標)−X(最大吸光度:574nm、最大発光:594)「緑色」(図2Cのフルオロフォア356に対応)である。
実施例7
基板へのプローブまたは核酸分子の結合
本実施例は、顕微鏡用スライドまたはゲルマトリクスなどの基板への実施例1において生成されたタグ標識された重合剤(例えば、実施例5および6のプローブ部分)、または核酸分子を結合するために使用することができる方法を記載する。配列決定反応中、配列決定される標的核酸分子、オリゴヌクレオチドプライマー、またはプローブは、基板(例えば、顕微鏡の視野において)に結合することができる。
基板へのプローブまたは核酸分子の結合
本実施例は、顕微鏡用スライドまたはゲルマトリクスなどの基板への実施例1において生成されたタグ標識された重合剤(例えば、実施例5および6のプローブ部分)、または核酸分子を結合するために使用することができる方法を記載する。配列決定反応中、配列決定される標的核酸分子、オリゴヌクレオチドプライマー、またはプローブは、基板(例えば、顕微鏡の視野において)に結合することができる。
核酸の結合
基板への核酸(例えば、配列決定される標的核酸分子またはオリゴヌクレオチドプライマー)を結合するいくつかの方法が使用できる。核酸分子は、5’または3’末端、または中間のどこかにより、基板に結合させることができる。例えば、5’ビオチン化プライマーを、合成し(Beaucage,Tetrahedron Letters22:1859−62,1981;Caruthers,Meth.Enzym.154:287−313,1987)、ストレプトアビジンをコーティングした基板表面に付着することができる(Hultman,Nucl.Acids Res.17:4937−46,1989)。別の例において、核酸分子は、Haらにより記載されるように、アミノプロピルシラン化された(APS)ガラス上で乾燥することができる(Proc.Natl.Acad.Sci.USA.93:6264−68,1996、参照により本明細書に組み入れられる)。リンカーを介してオリゴヌクレオチドプライマーを基板に結合する方法は、Cheesemanへの米国特許第5,302,509号に開示され、参照により本明細書に組み入れられる。
基板への核酸(例えば、配列決定される標的核酸分子またはオリゴヌクレオチドプライマー)を結合するいくつかの方法が使用できる。核酸分子は、5’または3’末端、または中間のどこかにより、基板に結合させることができる。例えば、5’ビオチン化プライマーを、合成し(Beaucage,Tetrahedron Letters22:1859−62,1981;Caruthers,Meth.Enzym.154:287−313,1987)、ストレプトアビジンをコーティングした基板表面に付着することができる(Hultman,Nucl.Acids Res.17:4937−46,1989)。別の例において、核酸分子は、Haらにより記載されるように、アミノプロピルシラン化された(APS)ガラス上で乾燥することができる(Proc.Natl.Acad.Sci.USA.93:6264−68,1996、参照により本明細書に組み入れられる)。リンカーを介してオリゴヌクレオチドプライマーを基板に結合する方法は、Cheesemanへの米国特許第5,302,509号に開示され、参照により本明細書に組み入れられる。
さらに他の例において、核酸分子は、活性シリル部分を核酸分子に結合することにより(例えば、Kumarらにより開示された方法を使用することにより(Nucleic Acids Res.28:e71,2000))、修飾されていない基板に交差結合する。手短に言えば、シランは、以下のように核酸に結合される。メルカプトシラン[(3−メルカプトプロピル)−トリメトキシシラン]を酢酸ナトリウム(30mM、pH4.3)またはクエン酸ナトリウム(30mM、pH4)などの反応緩衝液により5mMの保存液にまで希釈する。メルカプトシランによる5’−チオール−標識ヌクレオチドの結合に対して、1nmolのヌクレオチドを、10〜120分間、室温で20μlの同一の緩衝液中の5nmolのメルカプトシランと反応させる。反応混合物を、直接使用するか、またはガラスの顕微鏡用スライドなどの基板に固定化のために所望の濃度に反応緩衝液で希釈する。5’−アクリル−標識オリゴヌクレオチドは、同一工程を使用して、メルカプトシランに結合される。
5’−チオール−標識ヌクレオチドは、ヘテロ二官能性リンカーN−スクシンイミジル−3−(2−ピリジルジチオール)−プロピオネート(SPDP)またはスクシンイミジル−6−(ヨードアセチル−アミノ)−ヘキサノエート(SIAX)の存在下で、ジメチルスルホキシド(DMSO)中のアミノシラン[(3−アミノプロピル)−トリメトキシシラン]で結合される。ヌクレオチド(最終濃度5〜50μM)は、10μlのDMSO中で2.5nmolのアミノシラン(エタノール中で5mMの溶液から添加)および2.5nmolの二官能性試薬(DMSO中の5mMの原液から添加)と混合され、反応は、1〜2時間、室温で進行させた。
アクリル−標識オリゴヌクレオチド(50〜500pmol)は、10μlの30mM NaOAc、pH4.3中の25nmolのアクリルシラン(γ−メタクリルオキシプロピル−トリメトキシシラン)と混合させる。過硫酸アンモニウム(H2O中10%)およびN,N,N’,N’−テトラメチルエチレンジアミン(TEMED)は、それぞれ最終濃度0.5および2%まで添加され、混合物は、30分間、室温で反応させた。
抱合反応後、反応混合物は、シラン化した核酸と称され、基板へのスポッティングのために直接使用することができる。シラン化核酸を、スライドガラス上に手動で(120nl/スポット)または自動アレイヤー(Genetic Microsystem,Woburn.USA)(1nl/スポット)でスポットすることができる。水溶液中の核酸を、スライドガラス上にスポット後、15分間、室温で加湿チャンバ内に保持し、50℃で5分間乾燥させ、非共有結合核酸を除去するために沸騰水に30秒間浸し、ハイブリダイゼーション前に窒素で乾燥させることができる。DMSO中のヌクレオチドは、スライドガラス上にスポット後、室温で15分間放置し、50℃で10分間乾燥させる。これらのスライドは、DMSO(3×2分)、エタノール(3×2分)および沸騰水(2分)で連続的に洗浄され、その後の使用のために窒素で乾燥させる。
オリゴヌクレオチドプライマーなどの基板に結合させる核酸分子に相補的核酸分子をハイブリダイズするために、ハイブリダイズされた核酸分子は、0.1%のTween−20で5×SSC(750mM NaCl、125mMクエン酸ナトリウム、pH7)中の20nMと1μMとの間まで希釈される。ハイブリダイゼーションは、加湿器中のカバースリップ下で、37℃で30分間一晩行われる。非ハイブリダイズおよび非特異的核酸分子は、0.1%のTween−20を含む5×SSCで洗浄され(3×1分)、続いて0.1%のTween−20(2×15分)を含む1×SSCで洗浄することにより除去される。
試料核酸分子などのより長い核酸分子をハイブリダイズする場合、ハイブリダイゼーションを、0.1%のSDSおよび1μg/μlの酵母tRNAを有する3×SSC中で、65℃で4時間実施することができる。スライドを、その後室温で0.1%のSDSを含む1×SSC(3×2分)および0.1%のSDS(3×5分)を含む0.1×SSCで洗浄する。
洗浄後、スライドは、窒素ガスで乾燥させることができる。同一の基板上で反復したハイブリダイゼーションが望ましい場合、基板は、1分間、水中で煮沸され、次のハイブリダイゼーション反応に進む前に窒素ガスで乾燥させる。
3’末端で核酸を結合するために、末端トランスフェラーゼを、その分子を「テール」に使用することができる。
プローブの結合
本開示のプローブを、重合剤を介して基板に結合、または固定することができる。例えば、ストレプトアビジン−ポリメラーゼ融合タンパク質を生成することができ(例えば、実施例1に記載される方法を使用して)、その後例えば、MazzolaおよびFodorにより記載されるように、ビオチン化基板に添付することができる(Biophys.J.68:1653−60,1995)または、Itakuraらの(Biochem.Biophys.Res.Commun.196:1504−10,1993)。
本開示のプローブを、重合剤を介して基板に結合、または固定することができる。例えば、ストレプトアビジン−ポリメラーゼ融合タンパク質を生成することができ(例えば、実施例1に記載される方法を使用して)、その後例えば、MazzolaおよびFodorにより記載されるように、ビオチン化基板に添付することができる(Biophys.J.68:1653−60,1995)または、Itakuraらの(Biochem.Biophys.Res.Commun.196:1504−10,1993)。
重合剤を基板に結合する他の方法は、当業者には周知である。例えば、原子間力顕微鏡の顕微鏡チップは、基板の表面を化学変化させるために使用してもよい(Travis,Science268:30−1,1995)。あるいは、重合剤が6〜10の連続的なヒスチジン残基を含む場合、ニッケルコーティングした基板に結合する。例えば、Paborskyら(Anal.Biochem.234:60−5,1996)は、ニッケルをプラスチック基板に結合するための方法を記載する。手短に言えば、マイクロタイターポリスチレンプレートを満たすために、100μlのN,N−ビス[カルボキシメチル]リシン(BCML)を、それぞれのウェルに添加(0,1M NaPQ4中に10mM BCML、pH8)し、室温で一晩インキュベートする。プレートは、その後、200μlの0.05% Tweenで洗浄し、ブロッキングされ(50mM Tris HCl中の3% BSA、pH7.5、150mM NaCl、0.05% Tween)、一連の緩衝液で洗浄される。第1に、50mM Tris HCl、pH7.5、500mM イミダゾール、0.05% Tweenで、第2に、0.05% Tweenで、第3に、100mM EDTA、pH8.0で、最後に0.05% Tweenで洗浄される。次に、プレートを、20分間室温で、10mM NiSO4でインキュベートする。最後に、プレートを、0.05% Tween、その後50mM Tris HCl、500 mM NaCl、pH7.5で洗浄する。
基板へのプローブのランダム結合は、低濃度のプローブで十分でなければならない。視野においてシグナルを配列決定するための密充てんを可能にするために、プローブを、整理されたアレイにおいて、二次元の基板表面に配列することができる。プローブは、Mullerらにより記載されるようにマイクロメータの間隔で間隔をあけることができる(Science 268:272−3, 1995、参照により本明細書に組み入れられる)。さらに、幅約50μm、深さ約10〜20μmのチャネルのパターンを基板において形成し、Footeへの米国特許第5,661,028号に記載されるように、標準フォトリソグラフィー過程を使用し、続いて化学的ウェットエッチングを使用することができる(参照により本明細書に組み入れられる)。非常に小さいチャネルを、ナノリソグラフィー技法を使用して生成することができる。20nmにわたる穴またはチャンバの穴の密度が高い周期的配列は、Parkらの方法による窒化ケイ素でコーティングされた基板に製造される(Science,276:1401−4,1997、参照により本明細書に組み入れられる)。それぞれのチャンバにおいて、単一配列決定反応を実施する。プローブはまた、基板の表面上にプローブを含む液滴を微量ピペットにより規則的なアレイにおいて基板に結合することもできる。液滴を、例えば、ガラスのカバースリップで、蒸発を防止するために覆うことができる。
ゲルマトリクス中の包埋
プローブまたは核酸分子を二次元表面に結合するための代替案として、それらを三次元ゲルマトリクスに包理することができる。例えば、プローブ、核酸分子、またはその双方は、凝固できる液体マトリクスに添加され、その中に薬剤を捕捉する。この型のマトリクスの例は、例えば、Ni2+−NTA−アガロース(QIAGEN,Valencia,CA)などのアガロースおよびアクリルアミドを含む。
プローブまたは核酸分子を二次元表面に結合するための代替案として、それらを三次元ゲルマトリクスに包理することができる。例えば、プローブ、核酸分子、またはその双方は、凝固できる液体マトリクスに添加され、その中に薬剤を捕捉する。この型のマトリクスの例は、例えば、Ni2+−NTA−アガロース(QIAGEN,Valencia,CA)などのアガロースおよびアクリルアミドを含む。
実施例8
異なる長さのロッドを持つフルオロフォア間の距離の算定
上記に記載されるように、開示されたナノプローブは、例えば、テザー、例えば、PEGから構成されるテザーを含むロッドなどの分子ロッドを介して化学的部分に結合される重合剤を含むことができる(例えば、図1Aの分子リンカー14、16、18を参照)。しかしながら、それらを分離し続ける力がない、遊離型PEG鎖のそれぞれは、一点周辺の集団を形成するために凝縮する。従って、ナノプローブへの1つまたは複数の分子ロッドの添加(例えば、図1Aの分子リンカー20に示されるように)は、重合剤および化学的部分をさらに分離するため、またはフルオロフォアまたはプローブ上の他のタグをさらに分離するために含むことができる。
異なる長さのロッドを持つフルオロフォア間の距離の算定
上記に記載されるように、開示されたナノプローブは、例えば、テザー、例えば、PEGから構成されるテザーを含むロッドなどの分子ロッドを介して化学的部分に結合される重合剤を含むことができる(例えば、図1Aの分子リンカー14、16、18を参照)。しかしながら、それらを分離し続ける力がない、遊離型PEG鎖のそれぞれは、一点周辺の集団を形成するために凝縮する。従って、ナノプローブへの1つまたは複数の分子ロッドの添加(例えば、図1Aの分子リンカー20に示されるように)は、重合剤および化学的部分をさらに分離するため、またはフルオロフォアまたはプローブ上の他のタグをさらに分離するために含むことができる。
本実施例は、FRETシグナルが例えば、dsDNAから構成することができる分子ロッドの長さによりいかに影響されるかを決定するために使用されるコンピュータシュミレーションを記載する。図1Aの分子リンカー20は、シュミレーションの状況を示す。2つのテザー68、70は、ロッド66に結合され、フルオロフォア38と30との距離が測定される。シュミレーションの目的のために、重合剤12、フルオロフォア30、部分28およびフルオロフォア38はすべて点物体であると考えられる。単一テザーのために、テザーは、非常に薄く(それ自体と交差する問題がない)とすれば、テザーの末端の結合点からの距離は三次元において、ランダムウォークである。それぞれの一次元において、これは小さいランダム工程の合計であり、ガウス分布を得る。三次元において、それは、Maxwell球対称ガス分布である(Schneider,J.Theor.Biol,148:83−123,1991)。個別の分子が特定の方向および距離の状態にあるため、所定の分離を有する2つの該分布の共通集合を統合することだけは適切ではない。この理由から、明示シュミレーションを実施した。
該方法を要約するために、それぞれのロッドの長さに対して、2つのテザーを伸長し、チップ間の距離およびその後、FRET効率を算出した。本シュミレーションにおいて、プログラムBite(二重テザー)によりコード化されると、2つのポリマーは、固定ロッドの末端に結合された。それぞれのポリマーは、一連のランダム工程により生成され、ロッド末端から開始する。工程の大きさは、鎖の持続長により与えられる。PEGに対して、持続長は、3.8±0.02Åである。それぞれの工程の方向は、無作為に選ばれた。FRETシグナルは、無作為に延長された鎖のそれぞれの対について算出された。このシグナルは、FRET効率によるテザー鎖末端RおよびFRET半径R0との間の最終距離の関数である。
E=1/((R/R0)6+1)(1)
E=1/((R/R0)6+1)(1)
プログラムにより使用されるパラメータは、持続長、テザーLの長さ、およびテザー点を分離するロッドの長さDを含む。本過程は、図4に示される分布を得るために1,000回繰り返された。
距離がR、フルオロフォア間がR0である場合、転移効率は、50%である。例えば、R0=60Åを有するFRETの対に対して、それは典型的な距離であり、図4A−Fの120Åのテザーの長さは、ロッドの長さの異なる効果を示す。ロッドの長さが0である(図4Eおよび4F)テザーは、共有点周辺に集まり(図4E)、大きいFRETシグナルをもたらす(図4F)。ロッドの長さが60Åに増加する場合(図4C)、FRET分布シグナルは散らばる(図4D)。時々、2つの末端が閉じ、いくつかのFRETを観測される。著しくは、ロッドの長さが120Åである場合(図4Aおよび4B)、FRETシグナルはほぼ完全に排除される(図4B)。テザーの長さがロッドの長さの2倍である場合、これが生じるので、テザーの多くの潜在的な重複があり、テザーが標的に結合することを可能にする。さらに、FRETシグナルは、ほとんど検出できない。これらの結果は、分子リンカーのロッドを含む特定の利点を示す。
実施例9
異なる長さのテザーを持つFRET
本実施例は、FRETシグナル上のテザーの長さを変更する効果を決定するために使用される方法を記載する。
異なる長さのテザーを持つFRET
本実施例は、FRETシグナル上のテザーの長さを変更する効果を決定するために使用される方法を記載する。
Biteプログラムは、種々のテザーの長さで起動された(図5)。左上(図5A)において、非常に短い2Åのテザーは、方程式(1)に与えられる基本FRET曲線をもたらす。テザーの長さが、2から60Å、120Åおよび140Åに増加する場合(図5B、CおよびD)、基本FRET曲線は維持するが、分布は、さらに散らばる。約120Åの分子ロッドの長さおよび120Åのテザーについては、テザーが大いに重複することができたが、FRETはほとんどない。従って、約120Åの分子ロッドおよび120Åのテザーの使用により、2つのテザーが標的分子の結合により1つになるまで、ほとんどFRETがない。
240Åの長さであるテザーでさえ(図5D)、分布の右側は、120Åの長さのテザーを有するものとほぼ同一である(図5C)。つまり、テザーの長さは、FRETの結果にあまり差異をもたらさないが、分子ロッドの長さは、有意な効果を有する。
要約すれば、テザーの長さは、FRETにほとんど効果がないが、分子ロッドの長さは有意に差異を示した。ナノプローブの分子リンカーにおいてロッドを含み、テザーが標的に到達するのに十分過ぎる長さの場合でさえ、FRETは、ほとんど検出できないレベルまで減少する。対照的に、標的の存在下で、FRETシグナルは大きい場合がある。これは、標的の存在または非存在に基づいて出力シグナルに強い分子スイッチを提供する。ナノプローブのための有用なロッド−テザーの結合の実施例は、120Åのロッド、また2つの120Åのテザーを使用する。これらは、都合よくロッドを作製するためのdsDNAの40のヌクレオチド、テザーを作製するためのそれぞれ23Åの5つから6つのPEG18スペーサーで構築される。
分子ナノプローブの作用を理解する上で、2つの時間スケールを考慮することができる。分子振動、ピコ秒の時間スケールに、テザーは、多種多様の可能性を調べ、分子ロッドにより分離される2つのテザーチップの結合部は長時間をかけて出現する。例えば、本過程は、分子振動より長い規模の数桁、例えば、100ミリ秒を取ることができた。100ミリ秒は、分子運動の見解から長い時間ではあるが、それはヒトの時間スケールでは1秒の1/10に過ぎない。従って、分子プローブを使用する検出過程は、極めて迅速であると考えられる。
実施例10
肉眼的なMedusaシーケンサーの使用
本実施例は、本明細書に記載される肉眼的なプローブの使用方法を記載する。例えば、これらの方法は、単一分子検出の報告なしにMedusaプローブの特性を決定するために使用することができる。
肉眼的なMedusaシーケンサーの使用
本実施例は、本明細書に記載される肉眼的なプローブの使用方法を記載する。例えば、これらの方法は、単一分子検出の報告なしにMedusaプローブの特性を決定するために使用することができる。
一時的滞留事象を観測するために、実施例2に記載されるものなどのヘアピンDNAを使用することができる。使用することができるヘアピンオリゴヌクレオチドの概略図を、図7に例示する。図7の上部は、5’突出部(4つの塩基対など)および蛍光供与体標識(円)を有するヘアピンオリゴヌクレオチドを示す。自由拡散dTTPは、FRET受容体(六角形)で標識化される。標識化dTTPが、ポリメラーゼ(楕円)によってヘアピンに保持される場合、下部は、供与体と受容体との間のFRETを示す。ヘアピンは、次に読まれる塩基として「A」を露出する小さい突出部を有するように設計される。オリゴヌクレオチドは、5’末端上に供与体フルオロフォアを有する。一部の例において、この制御DNAは、受容体フルオロフォア標識化されたdTTP(またはdUTP)により埋められる。これは、FRETおよび受容体発光シグナルをもたらす。
次に読まれる塩基として「A」および5’末端上の供与体フルオロフォア(Alexa Fluor488)(SEQ ID NO:31、5’末端上のAlexa Flour488を有する)を露出する小さい5’突出部(4つの塩基)を有する単一ヘアピンDNA分子の測定は、画像相関分光法(ICS)を使用して得られた。Olympus FluoView FV1000共焦点顕微鏡を使用して、このヘアピンオリゴヌクレオチドの1nMの試料を画像化した。標識DNAの低濃度は、共焦点の体積(約1フェムトリットル)あたり1つのみのフルオロフォアであることを確実にした。
次に読まれる塩基として「A」、5’末端上の供与体フルオロフォア(Alexa Fluor488)、および共有結合した3’受容体標識dUTP(Alexa Fluor594−dUTP)(SEQ ID NO:32)を露出する小さい5’突出部(4つの塩基)を有するヘアピンDNA分子の測定が実施された。FRETは、Molecular Devices Spectra Max Gemini EMプレートリーダーを使用して、約200nMのオリゴヌクレオチドの濃度で、Zeiss LSM510Meta NLO共焦点システムを使用してさらに低濃度(10nMオリゴ)で、観測された。従って、この共焦点法を、ヌクレオチド滞留を検出するために使用してもよい。
遊離Alexa Fluor555標識ヌクレオチドおよび次に読まれる塩基として「A」および5’末端上のフルオロフォア(Alexa Fluor555)(SEQ ID NO:33)を露出する小さい突出部(4つの塩基)を有するヘアピンDNA分子の双方の測定が蛍光相関分光法(FCS)を使用して、実施された。標識ヘアピン分子は、ヘアピンオリゴヌクレオチドの質量が高いため、標識ヌクレオチドよりもさらにゆっくりと拡散した。従って、FCS法は、ヌクレオチド滞留を検出するために使用してもよい。
例えば、次に読まれる塩基としてヌクレオチド(「A」または「G」など)および供与体フルオロフォアを露出する小さい突出部(4つの塩基など)を含む3’末端および5’末端上にジデオキシヌクレオチド(ddCなど)を有するヘアピンオリゴヌクレオチドを使用することができる(図7を参照)。このヘアピンオリゴヌクレオチドは、ポリメラーゼにより伸長できず、受容体−標識dNTPは、ヘアピンオリゴヌクレオチドに結合できないため、それは、開示した配列決定のプローブの非加水分解性アーム用のモデルである。そのような伸長できないオリゴヌクレオチドを、ポリメラーゼポケットに滞留するヌクレオチドを観測するために、ポリメラーゼおよび標識ヌクレオチド(標識dNTPなど、例えば、Alexa Fluor647−標識dUTPまたはAlexa Fluor647−標識dCTPなどの受容体−標識dUTP)の存在で画像化することができる。ポケット/鋳型へ結合するヌクレオチドは、溶液中の遊離型ヌクレオチドよりもさらにゆっくりと拡散する。例えば、一致するヌクレオチドと鋳型に相補的でないものとの画像比較を、滞留をモニタリングするために実施することができ、一致したヌクレオチドは、一致しないものよりも長く滞留する。例えば、塩基が特異的に結合するが、非共有結合しないことを示すために、受容体標識dUTPの存在の滞留事象の発生は、dATP、dCTPまたはdGTPによる競争により影響が少ないようであるが、dUTPとの競争により大幅に減少するはずである。
異なる滞留時間は、標識化した非相補的塩基(例えば、同位置に標識を有する塩基)を使用して示すことができる。例えば、Invitrogen/Molecular ProbesからのdCTPおよびdUTP上の蛍光標識はすべて、C5の位置に結合される。
一部の例において、ヘアピンDNAは、離散配置において、個別の分子に滞留するヌクレオチドの測定を許可することにより、表面に結合するためのビオチンを含む。
滞留をモニタリングするために使用することができる別の例示的な構築物を図8に示す。本実施例において、受容体−標識ヌクレオチド三リン酸(dNTPなど)は、カルボジイミド交差−リンカーを介してリンカー上の1級アミンにトリホスフェートの5’γ−ホスフェートを結合することによるなど、リンカーを介してヘアピンオリゴヌクレオチドに結合する。これは、単一リンカーまたはMedusaの「アーム」を生成する。中間設計において、2つまたは3つのテザー化アームを追加することができる。さらに、アームを、ポリメラーゼに結合することができ、ポリメラーゼを、表面に結合することができる。ポリメラーゼは、遊離型ヌクレオチド三リン酸を付加することにより、ヘアピンDNAに沿って進むように誘導されることができる。例えば、付加される第1塩基がdCTPである場合、付加される第2塩基は、dTTPであり、「アーム」は、標識dUTPを含み、dCTPなしにほとんどシグナルがないはずである。しかしながら、dCTPが付加される場合、ポリメラーゼは進み、DNA供与体を伴うFRETにアーム上に標識dUTPを可能にする。複数の「アーム」を有するMedusaシーケンサーを使用して、滞留時間は、テザー化DNAに塩基を連続的に付加することにより測定することができる。それぞれの塩基が溶液に添加される場合、予測した単一分子スペクトルが観測される。本過程を、リアルタイムで観測でき、単一ヌクレオチド工程を示す。例えば、単一分子検出法は、この構築物を利用してヌクレオチド滞留を検出するために使用されることができる。
上記に記載されるヘアピンDNA分子を使用して、FRETは、EcoRIなどの制限酵素(GAATTCは、Fis部位で配列内である)でヘアピンを切断するまたはDNase Iを使用することにより生じることを示すことも可能であり、FRETは減少すべきである。第二オリゴヌクレオチドは、ジデオキシCを伴う3’末端でCを置換するので、次の塩基は、共有結合できない。オリゴヌクレオチドと蛍光dTTPとの混合は、FRETを起こさない。ポリメラーゼの付加は、蛍光dTTPを結合させ、FRETを起こす。dGTP、dATPおよびdCTPは、競合しないが、蛍光dTTPからのFRETは、その後、規定のdTTPとは離れて競合することができる。これらの方法を、dTTPがポリメラーゼにおいて、滞留することを示すのに使用することができる。
進歩的には、より複雑な構築物を検討することができ、完全なMedusaプローブをもたらす。例えば、PEGを、上記に記載されるように、ヘアピンオリゴヌクレオチドの5’末端に付加することができ、フルオロフォアおよびTをもたらす。これは、現在、実験的構築物の一部であるため、遊離型蛍光dTTPを必要としない場合を除いて、本質的には、上記のように使用することができる単一アームのMedusaプローブである。第2アームを、ヘアピンの5’末端から伸長した単一鎖DNAにハイブリダイズされるDNAのストレッチを使用することにより構築物に付加することができる。
完全なMedusaプローブを、フルオロフォアを結合することなく最初のヘアピンを使用して試験することができる。
ヘアピンは、3’末端で付加される塩基が一意的な順において、4つのdNTPから成るように設計される。示される設計において、突出部を埋めるために付加される塩基は、dTTP、dGTP、dCTPおよびdATPである。
ヘアピンは、いかなるdNTPなしに、Medusa分子とともに混合され、蛍光分光計に溶液を置く。付加される次の塩基はTであるため、Medusaプローブは、Tシグナルを生成する。このシグナルは、溶液中で利用可能な追加のヌクレオチドがないため、安定している。すべてのMedusaプローブは、同一のシグナルを与え、結果が肉眼的に読み込まれることを可能にする。正しい操作を実施しないいずれのMedusaプローブは、バックグラウンドシグナルを与えるため、これは個別のMedusaプローブのエラー率を見積もるための手段を提供する。
結果は、鋳型鎖がTを含むことを示す。つまり、Medusaプローブは、配列の一塩基を読む。Tがプローブにより報告される塩基であるため、溶液にdTTPを付加することができ、スペクトルを再度決定することができる。Cは、現在ポケット内にあるため、やがて、スペクトルは、変わり、Gシグナルを与える。従って、Medusaプローブは、配列の第2塩基を報告する。これらの方法を使用して報告された配列は、簡潔である。つまり、突出部が、互いに隣接した同種のいくつかの塩基を含んだ場合、プローブは、単一塩基を報告するのみである。
以下のように方法が進み、初めから再度開始する。
1つの塩基により埋められたヘアピンへのdTTPの付加が、得られる。
この時、Medusaプローブは、Gシグナルを報告する。
2つの塩基により埋められた以下のヘアピンへのdGTPの付加が、得られる。
この時、Medusaプローブは、Cシグナルを報告する。
3つの塩基により埋められたヘアピンへのdCTPの付加は、以下を得る。
最後に、Medusaプローブは、Aシグナルを報告する。
ヘアピンに完全に埋められたものへのdATPの付加は、以下を得る。
すべての4つのヌクレオチドは、この時点で溶液中にある。ヘアピンが完全に埋められたので、Medusaプローブは、シグナルを生成するのを停止する。
従って、これらの方法は、すべて4つのMedusaアームおよび単一分子検出システムを必要とせず、配列を完成する状態もテストすることができる。
実施例11
顕微鏡システム
本実施例は、本明細書に開示されるプローブを使用して標的核酸分子を配列決定するために使用することができる顕微鏡システムを記載する。
顕微鏡システム
本実施例は、本明細書に開示されるプローブを使用して標的核酸分子を配列決定するために使用することができる顕微鏡システムを記載する。
顕微鏡
全反射(TIR)蛍光顕微鏡検査法は、例えば、Pierceら(Nature,388:338,1997;Methods Cell Biol.58:49,1999)、Funatsuら(Nature,374:555,1995)、Weiss(Science,283:1676,1999)およびSchuttら(米国特許第5,017,009号)により記載された方法およびデバイスを使用して、使用することができる。TIRは、屈折率が高い物質を通ってその物質と屈折率がより低い第2物質の接合部分の方へ臨界角未満で光が誘導される場合に生じる光学現象である。この状況において、すべての光は、短距離のみの第2物質に伝播する顕微鏡のエバネセント波を除いて、接合部分から後方へ反射する。特定の例において、TIRおよび単一分子の検出は、通常の蛍光顕微鏡における単一光ファイバーを使用して実施される(例えば、Fang and Tan.Anal.Chem.,71:3101−5,1999、Xu and Yeung.Science,275:1106−9,1997、Ma et al,AnalChem,72:4640−5,2000、およびLevene et al.,Science,299:682−6,2003を参照)。
全反射(TIR)蛍光顕微鏡検査法は、例えば、Pierceら(Nature,388:338,1997;Methods Cell Biol.58:49,1999)、Funatsuら(Nature,374:555,1995)、Weiss(Science,283:1676,1999)およびSchuttら(米国特許第5,017,009号)により記載された方法およびデバイスを使用して、使用することができる。TIRは、屈折率が高い物質を通ってその物質と屈折率がより低い第2物質の接合部分の方へ臨界角未満で光が誘導される場合に生じる光学現象である。この状況において、すべての光は、短距離のみの第2物質に伝播する顕微鏡のエバネセント波を除いて、接合部分から後方へ反射する。特定の例において、TIRおよび単一分子の検出は、通常の蛍光顕微鏡における単一光ファイバーを使用して実施される(例えば、Fang and Tan.Anal.Chem.,71:3101−5,1999、Xu and Yeung.Science,275:1106−9,1997、Ma et al,AnalChem,72:4640−5,2000、およびLevene et al.,Science,299:682−6,2003を参照)。
TIR蛍光顕微鏡検査法において、第1物質は、ガラス基板であり、第2物質は、水またはアッセイ法が実施される別の水媒体である。蛍光標識物質が接合部分に接近する場合、エバネセント波のフィールド内で、蛍光分子は、励起することができ、検出された蛍光は、上部液にその後発散する。TIRは、優勢なシグナル対雑音比を生じ、試料の薄層のみが露出されるため、蛍光分子の光退色を減少する。
光退色を減少する方法は、当技術分野で公知であり、開示された方法は、特定の還元方法に限定されない。一例において、共焦点顕微鏡法を、フルオロフォアの光退色を減少させるために使用することができる。共焦点レーザーの一例は、Leica Confocal Spectrophotometer TCS−SP(Leica、Germany)である。共焦点レーザーは、配列決定用ポリメラーゼに照射し、リザーバの残りの部分を暗いままにしておく。これを達成するために、初めにポリメラーゼのために利用可能なボリューム全体を走査することができ、そのため、機能するポリメラーゼを含むそれらの小さい領域を露出するためのみに顕微鏡のプログラムを作成することができる。共焦点顕微鏡法は、三次元において、反応を配列決定するために使用することができる(米国特許第6,982,146号を参照)。共焦点顕微鏡法は、関心対象ではない平面を含まず、採用される配列の総数を増加することを可能にする。これは、実施され、視野あたりに検出されるより多くの配列決定反応をさらに可能にする。
近接場光学顕微鏡(NSOM)はまた、本明細書に開示された配列決定法に使用することもできる。NSOMのためのいくつかの方法およびデバイスが記載される(米国特許第5,105,305号およびPCT公報国際公開公報第97/30366号)。NSOMにおいて、可視光よりも小さい直径を有する開口は、試料の表面への近接近(1波長未満など)に位置付けられ、表面上を走査される。光は、プローブの末端において、そのような開口により放出されるか、または収集されることができる。機械的手段または圧電手段は、試料に対してプローブを移動するために提供される。試料と相互に作用する光は、例えば、分光光度計、およびCCDカメラなどにより収集され、検出される。検出された光シグナルの強度は、試料に対するプローブ位置に応じて、デジタルデータの形で典型的に格納される。格納データを核酸配列に変換することができる。NSOMは、サブ波長解像による光学的測定を可能にし、FRETを測定でき、溶液中で十分作用する(Ha et al,Proc.Natl.Acad.Sci.USA.93:6264−8,1996)。標準顕微鏡は、Nanonics Ltd.で市販されているデバイス(Malha,Jerusalem,Israel)を使用して近接場光学顕微鏡に変換することができる。
NSOMの一利点は、試料の高解像度を取得できることである。しかしながら、プローブは、基板表面を走査するため、常にモニタリングすることができる配列決定反応数は減少する。この減少の補正に役立てるために、追加のヌクレオチド率を溶液の粘度を増加するか(例えば、PEG、Ficoll、グリセロール、またはその組み合わせを含むことにより、溶液への適切な濃度で)または温度の減少することにより、減少することができる。
Kairos Scientificは、蛍光画像顕微分光光度計(FIMS)を提供する。この顕微鏡は、視野において、すべてのピクセルに対して蛍光発光スペクトルを生成する。従って、一意的な発光スペクトルは、相補的核酸鎖に添加される場合、それぞれのヌクレオチドに対して生成される。
その他の例において、該方法は、例えば、FangおよびTan(Anal.Chem.1999,71:3101−5、参照により本明細書に組み入れられる)により開示されたシステムを使用して、単一分子検出(SMD)を可能にする。簡単に言えば、本システムにおいて、光ファイバーは、溶液に精査するために使用される(例えば、水性の環境または固体表面で)。光ファイバーは、全反射を有し、蛍光分子をエバネント波により励起させるために表面に近づけることを可能にする。フルオロフォアにより生成された蛍光性シグナルは、強化した電荷結合素子(ICCD)ベースの顕微鏡システムにより検出される。光ファイバーを、Newport社から購入することができる(Irvine,CA)。
さらに他の例において、SMDは、Ungerらにより開示される方法を使用して、実施することができる(BioTechniques,1999,27:1008−14、参照により本明細書に組み入れられる)。簡単に言えば、水銀ランプの励起およびCCDカメラを有する標準蛍光顕微鏡を使用して、単一蛍光分子を、分子が希釈により十分に分離される場合、空中および水溶液中で観察することができる。
光退色の減少
光退色を減少させる方法は、当技術分野で公知であり、開示される方法は、特定の還元方法に限定されない。一例において、共焦点顕微鏡法を、フルオロフォアの光退色を減少させるために使用することができる(上記記載)。光退色を減少するために使用することができる別の手段は、例えば、Kitamuraら(Nature,397:129,1999)、OkadaおよびHirokawa(Science,283:1152,1999)、Haradaら(J.Mol.Biol.216:49,1990)に記載されるように、脱酸素剤システムを含む溶液中に試料をインキュベートすることである。溶液の例は、1%のグルコース、0.05mg/mlのグルコースオキシダーゼおよび0.1mg/mlのカタラーゼ、ならびに0.5%の2−メルカプトエタノール、4.5mg/mlのグルコース、216μg/mlのグルコースオキシダーゼ、36μg/mlのカタラーゼ、緩衝液中の2mMのATPを含む。
光退色を減少させる方法は、当技術分野で公知であり、開示される方法は、特定の還元方法に限定されない。一例において、共焦点顕微鏡法を、フルオロフォアの光退色を減少させるために使用することができる(上記記載)。光退色を減少するために使用することができる別の手段は、例えば、Kitamuraら(Nature,397:129,1999)、OkadaおよびHirokawa(Science,283:1152,1999)、Haradaら(J.Mol.Biol.216:49,1990)に記載されるように、脱酸素剤システムを含む溶液中に試料をインキュベートすることである。溶液の例は、1%のグルコース、0.05mg/mlのグルコースオキシダーゼおよび0.1mg/mlのカタラーゼ、ならびに0.5%の2−メルカプトエタノール、4.5mg/mlのグルコース、216μg/mlのグルコースオキシダーゼ、36μg/mlのカタラーゼ、緩衝液中の2mMのATPを含む。
光退色を減少するために使用することができる一方法は、カルシウムリン酸でフルオロフォア(また、分子点として知られる)の表面を覆うことである。例えば、60nmのナノ粒子内に閉じ込められる場合、フルオロフォアは、非常に安定していて、有意には遅延しない。本プローブに関して、表面上に1つのアミノ基を有する約0.5〜2nm(1nm〜2nmなど)の小ナノ粒子(またはその他の一意的な結合点)を使用することができる。例えば、アミノ基を有するテザー化フルオロフォア(ナノプローブ上の所望の位置にフルオロフォアの結合を可能にするため)は、表面を覆われ、ナノプローブに結合することができる。層化は、カルボキシル(−COOH)基とともにフルオロフォアをインキュベートし、その後、カルシウムまたはアルミニウムを付加することにより、達成することができる。ホスフェートH2PO4 −の付加において、別の層が形成される。一部の例において、金は、フルオロフォアの表面を覆うために使用される。得られる粒子は、保護被覆に加えて蛍光を強化すると考えられるプラズモン共鳴を有する(Lakowicz,Anal.Biochem.298:1−24,2001を参照)。
光退色を減少するさらに他の方法は、本明細書に開示されるナノプローブシーケンサーおよび金属島へ隣接した標的核酸分子の設置(Lakowicz,Anal.Biochem.298:1−24,2001)およびTroloxにおけるインキュベーションを含む(Rasnik et al.,Nat.Methods3:891−3,2006)。
電磁放射線源
特定の例では、電磁放射線は、レーザーにより放出される。使用するレーザーの選択は、使用する特定の供与体フルオロフォアによって決まる。供与体フルオロフォアを励起するためにレーザー光線の波長を選択する。例えば、野生型GFPおよびFITCは、488nmでアルゴンレーザーにより励起することができる。H9−40 GFP突然変異体を励起するために、400nmで(Nichia Chemical Industries Ltd.)または404nmで(Power Technology Inc.,Little Rock,AR)放射する青色レーザーダイオードを使用することができる。当業者に公知の他の電磁放射線源、例えば、HeNeレーザーおよび水銀ランプなどを使用することができる。
特定の例では、電磁放射線は、レーザーにより放出される。使用するレーザーの選択は、使用する特定の供与体フルオロフォアによって決まる。供与体フルオロフォアを励起するためにレーザー光線の波長を選択する。例えば、野生型GFPおよびFITCは、488nmでアルゴンレーザーにより励起することができる。H9−40 GFP突然変異体を励起するために、400nmで(Nichia Chemical Industries Ltd.)または404nmで(Power Technology Inc.,Little Rock,AR)放射する青色レーザーダイオードを使用することができる。当業者に公知の他の電磁放射線源、例えば、HeNeレーザーおよび水銀ランプなどを使用することができる。
流体工学
流体走査システムの使用は任意である。簡素化のために、すべての必要な試薬を添加し、次に乾燥を避けるためにガラスのカバースリップまたは1滴の油でチャンバを密閉することを選んでもよい。あるいは、ヌクレオチド含有の溶液の遅いフローは、ヌクレオチドを補給し、生成物(ジホスフェート)を除去するために提供されることができる。そのようなシステムは、ヌクレオチドの使用を増加するが、定常状態条件を維持し、連続する配列の長さを増加することができる。
流体走査システムの使用は任意である。簡素化のために、すべての必要な試薬を添加し、次に乾燥を避けるためにガラスのカバースリップまたは1滴の油でチャンバを密閉することを選んでもよい。あるいは、ヌクレオチド含有の溶液の遅いフローは、ヌクレオチドを補給し、生成物(ジホスフェート)を除去するために提供されることができる。そのようなシステムは、ヌクレオチドの使用を増加するが、定常状態条件を維持し、連続する配列の長さを増加することができる。
蛍光顕微鏡の段階である液体の処理を実施するコンピュータチップを構築することができる。マイクロマシンおよびマイクロ流体デバイスおよびナノリットルの大きさの液体試料の分配の方法は、前述されている(Service,Science282:399−401,1998;Burns et al.Science 282:484−7 1998)。
検出器
検出器は、分光光度計により生成された発光スペクトルの獲得を可能にする。
検出器は、分光光度計により生成された発光スペクトルの獲得を可能にする。
CCDカメラを、画像を獲得するための検出器として使用することができる。分光光度計により生成された発光スペクトルは、CCDカメラにより収集され、この入力を電荷に変換する。電荷は、CCD出力によりシグナルに変換される。得られるシグナルは、それぞれの型のヌクレオチド(A、T/U、CまたはG)に関連する特性シグナルとして、デジタル化され、デジタルデータはコンピュータのハードドライブなどのメモリに獲得される。獲得されたデータの合計は、その後、ヌクレオチド配列に処理される。CCDカメラは、Kodak(Rochester,NY)を含む多くの供給元から市販されている。
1000×1000ピクセルフィールド(例えば、Kodak Professional DCS 520デジタルカメラ)、またはさらに4096×4096ピクセルフィールド(例えば、Kodak 16.8i、KAF16800)以上を含むカラーCCDカメラにより、それは、最大毎秒750塩基の速度で、1000もの数の核酸を同時に配列決定することが可能である。従って、本明細書に開示されるプローブによる分子配列決定は、1日で全体の染色体またはゲノムを配列決定する可能性がある。ポリメラーゼが通常の六角形配列に配置される場合、約17ピクセルがそれぞれのポリメラーゼに使用可能である。
あるいは、フィルターを含む単色CCDまたはスペクトルを含むその他の手段を使用することができる。バックグラウンドノイズを低減するために、いずれのCCDカメラを冷却してもよい。
核酸の配列決定が行われる割合を、多くの因子により制御することができる。温度の上昇により(耐熱性ポリメラーゼおよびロッド用のPNAを使用して)、またはHPLCなどの場合、高圧で反応が進むことにより高速を得ることができる。より粘性のある溶液を生成することにより、反応温度を下げることにより、利用できる反応性ヌクレオチドをほとんど有しないことにより、または利用できる遊離型の非加水分解性、非蛍光dNTPを有することにより、反応速度を落とすことができる。重合率は、CCD統合率およびコンピュータ記録時間を越えないように、このように制御されることがある。従って、重合率は、蛍光性シグナルがCCDによりさらに確実に読み込まれ、コンピュータにより解釈されるように、このように制御される。
一例において、該方法は、直接コンピュータに入力する配列決定シグナルを生成するクローズドチャンバデバイスにおいて実施される。該方法は、核酸分子の大集団を代表する電気泳動ゲル上のパターンなど高分子事象をモニタリングすることにより、核酸を配列決定する代わりに、分子レベルにおいて標的核酸分子のその相補的ヌクレオチドとの化学的部分の対合をモニタリングすることにより、核酸分子を配列決定する。反応が開始すると、さらなる液体の処理は必要ない(が、必要に応じて追加されることができる)。従って、機械は、操作中肉眼で見える可動部がなく、迅速な配列決定を促進することができる。
CCDカメラの代替物として、光電子増倍管または増倍化電荷結合素子(ICCD)を使用することができる。
一例において、超電導物質は、FRETシグナルを検出するために使用される。超電導物質ベースの光検出器は、光子の損失がほとんどない単一分子からの全スペクトルを得る次の高感度検出器である。一部の例において、超電導物質は、その転移温度にもたらされる。これは、超電導物質が超電導を損失する温度である。光子が超電導物質に衝突する場合、それは光子エネルギーに比例して超電導を損失する。従って、それは、個別の光子を検出することができるだけでなく、エネルギーに比例するため、光の頻度を与えることもできる(hvにより)。検出器の個別のピクセルは、デバイスを通して電圧を入れることによる遷移で、維持することができる。デバイスの温度が高すぎる場合、抵抗は増加し、電流は減少し、温度は再度減少する。
シグナルの増加
光退色の減少に加えて、検出したシグナルを増加する方法は、当技術分野で公知である。例えば、フルオロフォア付近の金属島の使用に加えて、高開口数顕微鏡対物レンズを使用することができる(Lakowicz,Anal.Biochem.298:1−24,2001)。金属島は、光退色を減少させ、光子数を約100倍に増加させるライフタイムを減少させる。
光退色の減少に加えて、検出したシグナルを増加する方法は、当技術分野で公知である。例えば、フルオロフォア付近の金属島の使用に加えて、高開口数顕微鏡対物レンズを使用することができる(Lakowicz,Anal.Biochem.298:1−24,2001)。金属島は、光退色を減少させ、光子数を約100倍に増加させるライフタイムを減少させる。
一例において、鏡は、フルオロフォアの上方に配置される。別の例において、試料は、放物面反射鏡により取り囲まれ、それにより焦点でFRETシグナルを増加させる。例えば、Kartalovら(BioTechniques,40:85−90,2006)は、放物線フローチャネルプロファイルを有するマイクロ流体デバイスを提供する。そのようなデバイスは、本明細書に開示される方法において使用することができる。一例において、マイクロ流体デバイスのチャネルは、反応物質を有し、レーザーは、フルオロフォアを励起するためにデバイスの全長に向ける。一例において、全反射(TIR)は、フルオロフォアを励起するために使用される。
実施例12
コンピュータシステム
本明細書に開示される方法は、パソコンで実行するコンピュータプログラムのコンピュータ実行可能命令の一般状況で実施することができる。一般に、プログラムモジュールは、ルーチン、プログラム、コンポーネント、データ構造などを含み、特定のタスクを実施、または特定の抽象データ型を実行する。さらに、該方法が、携帯デバイス、マルチプロセッサシステム、マイクロプロセッサを用いるまたはプログラマブル家庭用電化製品、ミニコン、メインフレームコンピュータなどを含む他のコンピュータシステム構成で実行されることを当業者は、理解するであろう。本明細書に開示される方法はまた、タスクが通信ネットワークを介してリンクされている遠隔処理デバイスにより実施される分散コンピューティング環境において実行することができる。分散コンピューティング環境において、プログラムモジュールは、ローカルおよび遠隔メモリ記憶装置の双方に、位置してもよい。
コンピュータシステム
本明細書に開示される方法は、パソコンで実行するコンピュータプログラムのコンピュータ実行可能命令の一般状況で実施することができる。一般に、プログラムモジュールは、ルーチン、プログラム、コンポーネント、データ構造などを含み、特定のタスクを実施、または特定の抽象データ型を実行する。さらに、該方法が、携帯デバイス、マルチプロセッサシステム、マイクロプロセッサを用いるまたはプログラマブル家庭用電化製品、ミニコン、メインフレームコンピュータなどを含む他のコンピュータシステム構成で実行されることを当業者は、理解するであろう。本明細書に開示される方法はまた、タスクが通信ネットワークを介してリンクされている遠隔処理デバイスにより実施される分散コンピューティング環境において実行することができる。分散コンピューティング環境において、プログラムモジュールは、ローカルおよび遠隔メモリ記憶装置の双方に、位置してもよい。
本明細書に開示される方法の本実装プラットフォームは、ユーザーインターフェースとしてUnixを含む少なくとも1ギガバイトのメインメモリ、1ギガバイトのハードディスクドライブを有するSunコンピュータで実行されるシステムである。アプリケーションソフトは、Pascal、Javaまたは他のコンピュータ言語で書かれている。
特定の例において、Hidden Markov Modeling(McKinney et al.,Biophys.J.91:1941−51,2006)は、獲得したFRETデータを分析し、特性化するために使用される。
特定の例において、線形アンミキシング法は、獲得したFRETデータを分析し、特性化するために使用される。線形アンミキシング法は、そのスペクトルの一因になる個々のフルオロフォア量を決定するために所与のスペクトルから後方へ作用する過程である(例えば、Thaler et al.Biophys.J.89:2736−49,2005を参照)。例えば、99%のA、1%のT、0%のG、0%のCがあることを決定する場合、塩基がAであることを示唆する。
実施例13
プローブを使用する配列決定
本実施例は、本開示のプローブを使用して、種々のソースから核酸を配列決定するための方法を記載する。特定の方法が提供されるが、方法の変化が可能であることを当業者は理解するであろう。
プローブを使用する配列決定
本実施例は、本開示のプローブを使用して、種々のソースから核酸を配列決定するための方法を記載する。特定の方法が提供されるが、方法の変化が可能であることを当業者は理解するであろう。
本実施例において、図1Bに示されるプローブを参照する。しかしながら、本明細書に開示されるいずれかのプローブを、本プローブに置換できることを当業者は理解するであろう。本実施例において、重合剤12は、フルオレセイン−HIV−1−逆転写酵素(従って、タグ30はフルオレセインである)であり、化学的部分22、24、26、28は、非加水分解性dCMPCPP、dGMPCPP、dAMPCPP、およびTMPCPPであり、化学的部分に関連するタグ32、34、36、38は、Cy3、Cy5、テキサスレッド、ローダミンレッドである。
配列決定反応は、5μMのプローブ、5μMのプライマー、1ng−1μgの標的核酸配列(1μgの試料核酸など)、および200μMの各dNTPを含み、その反応は、Ambion HIV RT First Strand Synthesis Buffer(10×):500mM Tris−HCl(pH8.3)、750mM KCl、30mM MgCl2、50mM DTTにおいて実施される。一例において、標的核酸は、対象の生体試料から獲得される。特定の例において、反応は、45℃で進む。
本実施例において、供与体フルオロフォアは、フルオレセインである。従って、配列決定反応は、供与体を励起する光の波長を放射するレーザーなど(アルゴンレーザーの488nmなど)によって、励起することができる。
配列決定反応物は、フルオレセインポリメラーゼ12をプライマー/標的核酸配列に結合することができ、標的核酸鎖上で非加水分解性dCMPCPP、dGMPCPP、dAMPCPP、およびTMPCPP 22、24、26、28を現在露出されたヌクレオチド44と対にすることができる状態下で、インキュベートされる。フルオレセインポリメラーゼがオリゴヌクレオチド42によりプライミングされる標的DNAまたはRNA40に結合する場合、分子リンカー14、16、18、20のそれぞれは、非加水分解性dCMPCPP、dGMPCPP、dAMPCPP、およびTMPCPP 22、24、26、28をフルオレセインポリメラーゼ12の活性部位ポケットに入れるために柔軟に伸長することができる。従って、正確に対にされた非加水分解性dAMPCPP 26は、長い間ポリメラーゼ活性部位ポケットに維持される(非相補的非加水分解性22、24、28と比較して)。対応する受容体フルオロフォア36は、供与体フルオロフォアフルオレセイン30に近いので、2つの間にFRETがあり、それによって、非加水分解性dCMPCPP、dGMPCPP、dAMPCPP、およびTMPCPP 22、24、26、28が、配列決定された標的鎖において、相補的塩基とともに現在対合することを示すスペクトル出力を生成する。
配列決定された標的鎖40において相補的塩基44とともに現在対合する受容体フルオロフォア36からの発光シグナルを検出する。例えば、反応は、蛍光顕微鏡下で、または共焦点顕微鏡により観測することができ、顕微鏡は、使用されるそれぞれの受容体フルオロフォアのスペクトルを獲得することができる。
非加水分解性dCMPCPP、dGMPCPP、dAMPCPP、およびTMPCPP 22、24、26、28(本実施例において26)は、最終的には、加水分解性の非標識ヌクレオチド(この場合、dATP、46)により活性部位において置換され、伸長する相補的鎖に組み込まれる。これは、標的核酸分子40において1塩基先にプローブを進める。これは、標的核酸分子40における次の塩基を露出し、反応は上記に記載されるように反復される。プローブ上の相補的非加水分解性ヌクレオチド類似体が露出した塩基とともに対になる場合、受容体発光シグナルを検出する。従って、プローブは、標的分子のヌクレオチド配列を示すシグナルを変化する時間を生成する。
例えば、得られる発光シグナルは、図6に示されるように、一連のパルスを生成することがある。図は、デジタルエレクトロニクスおよびコンピュータのためのクロックパルスに類似する。時間は、横軸に沿って示し、種々のシグナルの強度は、横線に与えられる。標的配列は、上部の5’ A C G T T C A G T 3’ (SEQ ID NO:39)に沿って与えられる。それぞれの塩基に対する1つの直線がある。4つの蛍光標識非加水分解性ヌクレオチド類似体からの異なったスペクトルシグナルは、標的鎖上の対応する相補的塩基に変換される。ポリメラーゼを閉じる場合に(本明細書に記載されるように、ポリメラーゼは、ポリメラーゼの開口および閉口をモニタリングするタグを含むことができる)、一番下の線CLOSEが示す。標的核酸鎖上のそれぞれの塩基がプローブ上の相補的非加水分解性ヌクレオチド類似体と対になる場合、対応するシグナルが出現する。実際のデータは、示されるものよりも騒々しく、時間の長さは、定期的ではない可能性がある。一対のTが図6に示されるように出くわす場合、それらのシグナルは、ノイズのため、はっきりと識別できないことがある。しかしながら、ポリメラーゼは、第2のTを承認するために開閉しなければならないため、CLOSEシグナルは、2つのTシグナルがある読み込みによりクロッキングを提供する。
一例において、該方法は、対象が疾病に関連する突然変異などの標的核酸配列において突然変異を有するかどうか決定するために使用される。
多くの異なる配列を、同時に決定することができる。開示される方法の1つの応用は、プラスミドの配列決定である。ランダムな切り目をプラスミドに導入後、DNAは、固定したプローブを含む基板に添加される。全プラスミドは、その後、多くの点から配列決定される。コンピュータは、すべての配列のトラックを保持し、完全なプラスミド配列にそれらを自動的に組み立てる。
別の使用法は、無作為に化学的に合成した核酸の領域を配列決定するためのものである。使用されるプライマーは、無作為領域外付近の位置に対して特異的である。無作為化された核酸は、固定されたポリメラーゼの領域に置かれる。本方法は、同時に無作為化実験の全結果を獲得することを可能にし、それにより、時間と費用を節約する。
ニックトランスレーションは、未知の核酸上の配列決定を開始するために使用することができる。
実施例14
一塩基多型の検出
本明細書に開示されるプローブは、未知塩基のちょうど上流の配列を用いるシークエンサーを対象にすることにより、一塩基多型(例えば、Twist et al,Anal Biochem.327:35−44,2004を参照)を決定するために使用することができる。dNTPは、提供されない。Medusaプローブは、プライマーの末端に置き、未知の塩基を報告する。費用削減のために、4つよりも少ないアームを持ち、故に、より少ない塩基を識別する特殊プローブは、多くのアッセイ法が求められる特定の場合に使用することができた。
一塩基多型の検出
本明細書に開示されるプローブは、未知塩基のちょうど上流の配列を用いるシークエンサーを対象にすることにより、一塩基多型(例えば、Twist et al,Anal Biochem.327:35−44,2004を参照)を決定するために使用することができる。dNTPは、提供されない。Medusaプローブは、プライマーの末端に置き、未知の塩基を報告する。費用削減のために、4つよりも少ないアームを持ち、故に、より少ない塩基を識別する特殊プローブは、多くのアッセイ法が求められる特定の場合に使用することができた。
実施例15
臨床的応用
本実施例は、本明細書に開示される方法がいかに病理学的標本の分析のために使用できるかを記載する。対象から得た標本のソースは、末梢血、尿、唾液、組織生検、微細針吸引物、外科標本、羊水穿刺標本、組織切片、頬標本、および剖検材料を含むことがある。
臨床的応用
本実施例は、本明細書に開示される方法がいかに病理学的標本の分析のために使用できるかを記載する。対象から得た標本のソースは、末梢血、尿、唾液、組織生検、微細針吸引物、外科標本、羊水穿刺標本、組織切片、頬標本、および剖検材料を含むことがある。
特定の例において、生体試料は、試料中の存在する核酸分子を保存する状態で、スライドガラスなどの基板に取り付けられている。あるいは、核酸は、試料から分離し、その後、実施例13に記載される方法に従うことができる。例えば、細菌染色体および突然変異体を含むヒト遺伝子を配列決定する本方法を使用することができる。本明細書に記載される技術を使用して、ウイルス性の病原菌および/または細菌性の病原菌の存在は、ウイルス性の核酸配列および/または細菌性の核酸配列の存在により検出することができる。さらに、本明細書に開示される方法は、プライマー、プローブ、および4つの非標識加水分解性ヌクレオチドを薄い組織切片に付加することにより元の場所で核酸配列を可能にする。
一例において、該方法は、正常、前癌状態、および1つまたは複数の癌に対する癌細胞の生成した遺伝的分析結果に使用される。例えば、これらのそれぞれの細胞型からのcDNAライブラリは、癌細胞の遺伝的分析結果を決定するために配列決定される。開示された方法は、クローニングのない、従来の配列、またはマイクロアレイに直接遺伝子発現データを獲得するために使用することができる。これは、時間と費用を削減することができ、従って決定される組織の型を追加することができる。
一例において、生体試料は、例えば、多数の標本からの多数の標本を含む組織マイクロアレイなどの組織を含む。そのようなアレイは、多数の対象から多数の標本、または同一の対象から多数の標本のスクリーニングを可能にする。
本開示の原則を適用できる多数の可能な実施形態を考慮して、例示した実施例が特定の実施例のみであることを認識し、開示の範囲の制限としてとらえるべきではない。むしろ、開示の範囲は、添付の特許請求の範囲により定義される。従って、これらの特許請求の範囲およびその精神に付随する本発明としてすべて特許請求する。
Claims (68)
- 核酸分子を配列決定するためのプローブであって、
標的核酸分子に結合すること、および相補的ヌクレオチドが相補的核酸分子に組み込まれるように、伸長する前記相補的核酸分子の合成を促進することができる、活性部位を有する重合剤と、
前記重合剤上で離間している1つまたは複数の分子リンカーであって、前記リンカーのうちの1つ以上は、前記標的核酸分子内の相補的ヌクレオチドに特異的に結合することによって、前記リンカーから分離することなく、前記標的核酸分子に可逆的に結合することができる化学的部分を有しており、前記標的核酸分子内の相補的ヌクレオチドと前記リンカー上の前記化学的部分との特異的結合は、相補的ヌクレオチドと前記リンカー上の前記化学的部分との対合を示す特性シグナルの放出によって示される、1つまたは複数の分子リンカー
とを含む、プローブ。 - 前記化学的部分がヌクレオチド類似体を含む、請求項1記載のプローブ。
- 前記ヌクレオチド類似体が非加水分解性ヌクレオチド類似体を含む、請求項2記載のプローブ。
- 前記非加水分解性ヌクレオチド類似体が、非加水分解性ヌクレオチド三リン酸類似体を含む、請求項2記載のプローブ。
- 前記非加水分解性ヌクレオチド三リン酸類似体が、非加水分解性であるα−β結合を有する非加水分解性ヌクレオチド三リン酸類似体を含む、請求項4記載のプローブ。
- 前記化学的部分がモノヌクレオチドである、請求項1記載のプローブ。
- 前記モノヌクレオチドが、塩基、3’リボース炭素、2’リボース炭素、またはαリン酸上の前記リンカーに結合される、請求項6記載のプローブ。
- 前記1つまたは複数の分子リンカーが、少なくとも4つの独立したリンカーを含み、それぞれが、前記標的核酸分子内の異なるヌクレオチドに特異的に結合することができる、異なるヌクレオチド類似体を有する、請求項2記載のプローブ。
- 前記1つまたは複数の分子リンカーが分岐構造を形成し、それぞれの分岐が、前記標的核酸分子内の異なるヌクレオチドに特異的に結合することができる異なるヌクレオチド類似体を有する、請求項2記載のプローブ。
- 前記分岐構造が少なくとも4つの分岐を含み、それぞれの分岐が、前記標的核酸分子内の異なるヌクレオチドに特異的に結合することができる異なるヌクレオチド類似体を有する、請求項9記載のプローブ。
- 前記重合剤がタグに関連し、前記ヌクレオチド類似体のそれぞれが、前記リンカーが有する特定のヌクレオチド類似体を識別するタグに関連し、前記重合剤に関連する前記タグと、前記ヌクレオチド類似体に関連する前記タグとの相互作用が、前記標的核酸分子内の相補的ヌクレオチドとの前記リンカー上の前記ヌクレオチド類似体の対合を示す前記特性シグナルの放出を誘発する、請求項2記載のプローブ。
- 前記重合剤に関連する前記タグが、それぞれのヌクレオチド類似体に関連する前記タグにより供与体−受容体の対を形成し、それによって、前記供与体−受容体の対の相互作用が、前記特性シグナルの放出を刺激する、請求項11記載のプローブ。
- 前記供与体−受容体の対が、前記受容体が反応して前記特性シグナルを放出する刺激を放出する外部刺激を与えることによって刺激される供与体を含む、請求項12記載のプローブ。
- それぞれのタグがフルオロフォアを含む、請求項11記載のプローブ。
- 前記リンカーが有する特定のヌクレオチド類似体を識別する前記タグのそれぞれが、一意的な放出シグナルを放出する1つまたは複数のフルオロフォアを含む、請求項11記載のプローブ。
- 前記1つまたは複数の分子リンカーが4つの分子リンカーを含み、それぞれが、前記リンカーから分離することなく前記標的核酸分子の鋳型鎖に可逆的に結合することができる異なる化学的部分と、それぞれの化学的部分に関連する受容体タグとを有し、ここで前記受容体タグが、前記リンカーが有する前記特定の化学的部分を識別し、前記重合剤が供与体タグに関連し、前記標的核酸分子への前記化学的部分の可逆的結合が、前記リンカーが有する前記化学的部分の識別性を示す前記受容体タグの特性シグナルの放出を誘発するように、前記供与体および受容体タグを十分に接近させる、請求項1記載のプローブ。
- 前記1つまたは複数の分子リンカーが、前記分子リンカーの絡み合いを抑制するために十分な距離をおいて前記重合剤の周囲に離間し、前記分子リンカーが、前記重合剤の前記活性部位に到達するほど十分に長い、請求項1記載のプローブ。
- 前記分子リンカーが線状ポリマーを含む、請求項17記載のプローブ。
- 前記ポリマーが核酸を含む、請求項18記載のプローブ。
- 前記1つまたは複数の分子リンカーが、前記標的核酸分子の非存在下で、前記重合剤および前記化学的部分との実質上の絡み合いを回避するように、前記重合剤および前記化学的部分を互いに十分な距離をおいて維持する、請求項1記載のプローブ。
- 前記分子リンカーの少なくとも一部は、前記標的核酸分子の非存在下で、前記重合剤と前記化学的部分との相互作用を減少させるように十分な剛性を有する、請求項1記載のプローブ。
- 前記標的核酸分子の非存在下で、前記重合剤と前記化学的部分との相互作用を減少させるように十分な剛性を有する前記分子リンカーの少なくとも一部が、分子ロッドを含む、請求項21記載のプローブ。
- 前記分子リンカーが、テザー(tether)、分子ロッド、またはそれらの組み合わせを含む、請求項1記載のプローブ。
- 前記分子ロッドが、少なくとも10ヌクレオチドの二本鎖DNA(dsDNA)を含む、請求項23記載のプローブ。
- 十分な剛性を有する前記分子リンカーが、分子ロッドによって連結される少なくとも2つのテザーを含む、請求項24記載のプローブ。
- 前記テザーが、ポリエチレングリコール(PEG)を含む、請求項23記載のプローブ。
- 前記テザーがEGから成る、請求項26記載のプローブ。
- 前記テザーが長さが187Å未満である、請求項26記載のプローブ。
- 前記分子ロッドが40ヌクレオチドのdsDNA分子を含む、請求項24記載のプローブ。
- 前記dsDNA分子が10〜150のヌクレオチドを含む、請求項24記載のプローブ。
- 前記重合剤が、DNAポリメラーゼ、RNAポリメラーゼ、リボソーム、または逆転写酵素を含む、請求項1記載のプローブ。
- 前記重合剤が、供与体フルオロフォアに関連する逆転写酵素を含む、請求項31記載のプローブ。
- 前記標的核酸分子がDNAであり、かつ前記重合剤がDNAまたはRNAポリメラーゼである、請求項31記載のプローブ。
- 前記標的核酸分子がRNAであり、かつ前記重合剤が逆転写酵素である、請求項31記載のプローブ。
- 前記重合剤がDNAポリメラーゼIのクレノウフラグメントである、請求項31記載のプローブ。
- 標的核酸分子に結合すること、および相補的ヌクレオチドが相補的核酸分子に組み込まれるように、伸長する前記相補的核酸分子の合成を促進することができる活性部位と、
絡み合いを抑制するために前記重合剤上で離間している1つまたは複数の分子リンカーであって、それぞれのリンカーが、前記標的核酸分子内の相補的ヌクレオチドに特異的に結合することによって、前記リンカーから分離することなく核酸分子の前記鋳型鎖に可逆的に結合することができる異なる非加水分解性ヌクレオチド類似体を有している、1つまたは複数の分子リンカーと、
核酸分子の前記鋳型鎖に可逆的に結合することができる前記リンカーが有する前記非加水分解性ヌクレオチド類似体を識別するそれぞれの非加水分解性ヌクレオチド類似体に関連するタグと、
前記リンカーが有する前記非加水分解性ヌクレオチド類似体を識別する特性シグナルを放出するために、前記非加水分解性ヌクレオチド類似体に関連する前記タグと相互に作用する前記ポリメラーゼに関連するタグ
とを含む、重合剤。 - 標的核酸分子の核酸配列を決定する方法であって、
オリゴヌクレオチドプライマーと、前記標的核酸分子内の相補的ヌクレオチドとハイブリダイズし、前記鋳型核酸分子に可逆的に結合する前記化学的部分を置換することによって、伸長している核酸分子に組み込むことができる加水分解性ヌクレオチドの混合物との存在下で、前記標的核酸分子を請求項1記載のプローブに曝露する工程と、
相補的ヌクレオチドと前記分子リンカー上の前記化学的部分との対合を示す複数の前記特性シグナルの放出を含む、一連のシグナルの放出を検出する工程
とを含む、方法。 - 前記重合剤がタグに関連し、前記化学的部分のそれぞれもまた、前記リンカーが有する特定の化学的部分を識別するタグに関連し、前記重合剤に関連する前記タグと、前記化学的部分に関連する前記タグとの相互作用が、相補的ヌクレオチドと前記リンカー上の前記化学的部分との対合を示す前記特性シグナルの放出を誘発する、請求項37記載の方法。
- 前記重合剤に関連する前記タグが、供与体フルオロフォアを含み、特定の化学的部分を識別する前記タグが、1つまたは複数の受容体フルオロフォアを含み、前記重合剤と、前記標的核酸分子内の前記相補的ヌクレオチドに特異的に結合する前記化学的部分との相互作用が、供与体フルオロフォアによって前記受容体フルオロフォアの励起が可能となるように、前記受容体フルオロフォアを前記供与体フルオロフォアに接近させる、請求項38記載の方法。
- 前記シグナルを検出する工程が、前記受容体フルオロフォアから放出される蛍光性シグナルを検出する工程を含むか、または前記供与体フルオロフォアから放出される蛍光性シグナル内の減少を検出する工程を含む、請求項39記載の方法。
- 一連のシグナルの前記放出が核酸配列に変換される、請求項37記載の方法。
- 一連のシグナルの前記放出が発光共鳴エネルギー移動(LRET)または蛍光共鳴エネルギー移動(FRET)によって生成される、請求項37記載の方法。
- 相補的ヌクレオチドと前記リンカー上の前記化学的部分との対合を示す前記特性シグナルを放出するために、前記供与体フルオロフォアを励起して前記1つまたは複数の受容体フルオロフォアを励起するシグナルを放出する工程をさらに含む、請求項39記載の方法。
- 前記供与体フルオロフォアが緑色蛍光タンパク質(GFP)である、請求項43記載の方法。
- 前記受容体フルオロフォアが、BODIPY、フルオレセイン、ローダミングリーン、オレゴングリーン、またはそれらの誘導体である、請求項43記載の方法。
- 前記供与体フルオロフォアが発光分子によって励起される、請求項39記載の方法。
- 前記供与体フルオロフォアがGFPであり、かつ前記発光分子が化学発光エクオリンである、請求項46記載の方法。
- 前記供与体フルオロフォアが発光分子である、請求項39記載の方法。
- 前記発光分子がエクオリンである、請求項48記載の方法。
- 前記重合剤がGFP−ポリメラーゼである、請求項39記載の方法。
- 前記プローブが基板に固定されている、請求項37記載の方法。
- 前記重合剤がリンカーによって前記基板に固定されている、請求項51記載の方法。
- 前記リンカーが、ストレプトアビジン−ビオチン、ヒスチジン−Ni、S−タグ−S−タンパク質、またはグルタチオン−グルタチオン−S−トランスフェラーゼ(GST)である、請求項52記載の方法。
- 前記標的核酸分子または前記オリゴヌクレオチドプライマーが基板に固定されている、請求項37記載の方法。
- 実質上同時に複数の配列決定反応を行う工程と、前記複数の配列決定反応から一連のシグナルを検出する工程とを含む、請求項37記載の方法。
- 複数の重合剤、標的核酸分子、またはオリゴヌクレオチドプライマーが、所定のパターンで前記基板に直接的または間接的に固定され、一連のシグナルを検出する工程が、そのようなパターン内の所定の位置に対応する核酸分子と前記シグナルとを相関させる工程をさらに含む、請求項55記載の方法。
- 前記重合剤、標的核酸分子、またはオリゴヌクレオチドプライマーが、規則的なアレイにエッチングされたチャネル内で、前記所定のパターンで前記基板に固定される、請求項56記載の方法。
- 前記重合剤、標的核酸分子、またはオリゴヌクレオチドプライマーが、基板上に液滴をマイクロピペットで滴下することによって、前記所定のパターンで前記基板に固定される、請求項56記載の方法。
- 基板上に液滴をマイクロピペットで滴下する前記工程が、手動または自動アレイヤーを使用して行われる、請求項56記載の方法。
- 一連のシグナルが電荷結合素子(CCD)カメラによって検出され、前記核酸配列に変換される、請求項37記載の方法。
- 一連のシグナルがコンピュータ可読媒体に保存される、請求項37記載の方法。
- 前記標的核酸分子が、対象から取得された生体試料に存在している、請求項37記載の方法。
- 前記標的核酸分子が細胞内に存在し、前記鋳型核酸分子をオリゴヌクレオチドプライマーおよび前記プローブに曝露する工程が、前記オリゴヌクレオチドプライマーおよび前記プローブの前記細胞への導入を含む、請求項37記載の方法。
- 前記細胞が対象内に存在し、前記オリゴヌクレオチドプライマーおよび前記プローブの前記細胞への導入が、前記オリゴヌクレオチドプライマーおよび前記プローブの対象への投与を含む、請求項63記載の方法。
- 前記標的核酸鎖が、疾病に関連する1つまたは複数の突然変異を含む、請求項37記載の方法。
- 高ストリンジェンシー条件下で前記標的核酸配列に特異的にハイブリダイズするプライマーをさらに含むプローブであって、前記プライマーが、分子リンカーを介して前記重合剤に結合される、請求項1記載のプローブ。
- 前記重合剤が重合剤−Tus融合タンパク質を含み、前記分子リンカーがter配列を含む、請求項1記載のプローブ。
- 前記ヌクレオチド類似体に関連する前記タグが、エラーの検出および修正を可能にする、コード化したフルオロフォアの集まりである、請求項11記載のプローブ。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US74985805P | 2005-12-12 | 2005-12-12 | |
| US74972905P | 2005-12-12 | 2005-12-12 | |
| PCT/US2006/047534 WO2007070572A2 (en) | 2005-12-12 | 2006-12-12 | Probe for nucleic acid sequencing and methods of use |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009519041A true JP2009519041A (ja) | 2009-05-14 |
| JP2009519041A5 JP2009519041A5 (ja) | 2010-01-28 |
Family
ID=38051529
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008545768A Pending JP2009519041A (ja) | 2005-12-12 | 2006-12-12 | 核酸配列決定のためのプローブおよび使用方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (3) | US8344121B2 (ja) |
| EP (2) | EP2311982A1 (ja) |
| JP (1) | JP2009519041A (ja) |
| AT (1) | ATE481505T1 (ja) |
| DE (1) | DE602006016984D1 (ja) |
| WO (2) | WO2007070542A2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019518466A (ja) * | 2016-06-24 | 2019-07-04 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 繋ぎ止められたヌクレオシド三リン酸を用いた核酸合成および配列決定 |
Families Citing this family (104)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10337054B2 (en) | 2004-02-02 | 2019-07-02 | Quantum-Si Incorporated | Enrichment of nucleic acid targets |
| US7968287B2 (en) | 2004-10-08 | 2011-06-28 | Medical Research Council Harvard University | In vitro evolution in microfluidic systems |
| US7998717B2 (en) | 2005-12-02 | 2011-08-16 | Pacific Biosciences Of California, Inc. | Mitigation of photodamage in analytical reactions |
| DE602006016984D1 (de) * | 2005-12-12 | 2010-10-28 | Us Gov Health & Human Serv | Sonde zum sequenzieren von nukleinsäure und verwendungsverfahren |
| EP1984738A2 (en) | 2006-01-11 | 2008-10-29 | Raindance Technologies, Inc. | Microfluidic devices and methods of use in the formation and control of nanoreactors |
| US9562837B2 (en) | 2006-05-11 | 2017-02-07 | Raindance Technologies, Inc. | Systems for handling microfludic droplets |
| JP2010506136A (ja) | 2006-05-11 | 2010-02-25 | レインダンス テクノロジーズ, インコーポレイテッド | 微小流体デバイス |
| US9074242B2 (en) | 2010-02-12 | 2015-07-07 | Raindance Technologies, Inc. | Digital analyte analysis |
| WO2007146158A1 (en) * | 2006-06-07 | 2007-12-21 | The Trustees Of Columbia University In The City Of New York | Dna sequencing by nanopore using modified nucleotides |
| US8772046B2 (en) | 2007-02-06 | 2014-07-08 | Brandeis University | Manipulation of fluids and reactions in microfluidic systems |
| WO2008130623A1 (en) | 2007-04-19 | 2008-10-30 | Brandeis University | Manipulation of fluids, fluid components and reactions in microfluidic systems |
| EP3543357A1 (en) | 2007-05-08 | 2019-09-25 | Trustees of Boston University | Chemical functionalization of solid-state nanopores and nanopore arrays and applications thereof |
| ES2717354T3 (es) | 2007-09-14 | 2019-06-20 | Biomadison Inc | Ensayo de transferencia de energía por resonancia con secuencia de escisión y espaciador |
| EP2215115A1 (en) * | 2007-11-08 | 2010-08-11 | The University of Chicago | Molecular affinity clamp technology and uses thereof |
| US8652781B2 (en) | 2008-02-12 | 2014-02-18 | Pacific Biosciences Of California, Inc. | Cognate sampling kinetics |
| EP2255014A4 (en) | 2008-02-12 | 2011-05-18 | Pacific Biosciences California | COMPOSITIONS AND METHODS FOR USE IN ANALYTICAL REACTIONS |
| US8236499B2 (en) | 2008-03-28 | 2012-08-07 | Pacific Biosciences Of California, Inc. | Methods and compositions for nucleic acid sample preparation |
| US8628940B2 (en) | 2008-09-24 | 2014-01-14 | Pacific Biosciences Of California, Inc. | Intermittent detection during analytical reactions |
| US8143030B2 (en) | 2008-09-24 | 2012-03-27 | Pacific Biosciences Of California, Inc. | Intermittent detection during analytical reactions |
| US8153375B2 (en) | 2008-03-28 | 2012-04-10 | Pacific Biosciences Of California, Inc. | Compositions and methods for nucleic acid sequencing |
| ES2614078T3 (es) * | 2008-03-31 | 2017-05-29 | Pacific Biosciences Of California, Inc. | Generación de polimerasas modificadas para precisión mejorada en secuenciación de una única molécula |
| US8420366B2 (en) * | 2008-03-31 | 2013-04-16 | Pacific Biosciences Of California, Inc. | Generation of modified polymerases for improved accuracy in single molecule sequencing |
| EP2274446B1 (en) * | 2008-03-31 | 2015-09-09 | Pacific Biosciences of California, Inc. | Two slow-step polymerase enzyme systems and methods |
| US8999676B2 (en) | 2008-03-31 | 2015-04-07 | Pacific Biosciences Of California, Inc. | Recombinant polymerases for improved single molecule sequencing |
| EP4047367A1 (en) | 2008-07-18 | 2022-08-24 | Bio-Rad Laboratories, Inc. | Method for detecting target analytes with droplet libraries |
| WO2010027484A2 (en) * | 2008-09-05 | 2010-03-11 | Pacific Biosciences Of California, Inc. | Engineering polymerases and reaction conditions for modified incorporation properties |
| US8383369B2 (en) | 2008-09-24 | 2013-02-26 | Pacific Biosciences Of California, Inc. | Intermittent detection during analytical reactions |
| WO2010117420A2 (en) | 2009-03-30 | 2010-10-14 | Pacific Biosciences Of California, Inc. | Fret-labeled compounds and uses therefor |
| US8674080B2 (en) | 2009-04-09 | 2014-03-18 | Roche Molecular Systems, Inc. | Dye composition for liquid transfer control |
| US8501405B2 (en) * | 2009-04-27 | 2013-08-06 | Pacific Biosciences Of California, Inc. | Real-time sequencing methods and systems |
| US8409802B2 (en) | 2009-08-14 | 2013-04-02 | Roche Molecular Systems, Inc. | Format of probes to detect nucleic acid differences |
| US9566335B1 (en) | 2009-09-25 | 2017-02-14 | The Governing Council Of The University Of Toronto | Protein sequencing method and reagents |
| AU2010301128B2 (en) | 2009-09-30 | 2014-09-18 | Quantapore, Inc. | Ultrafast sequencing of biological polymers using a labeled nanopore |
| US8324914B2 (en) | 2010-02-08 | 2012-12-04 | Genia Technologies, Inc. | Systems and methods for characterizing a molecule |
| US9678055B2 (en) | 2010-02-08 | 2017-06-13 | Genia Technologies, Inc. | Methods for forming a nanopore in a lipid bilayer |
| US9605307B2 (en) | 2010-02-08 | 2017-03-28 | Genia Technologies, Inc. | Systems and methods for forming a nanopore in a lipid bilayer |
| US9366632B2 (en) | 2010-02-12 | 2016-06-14 | Raindance Technologies, Inc. | Digital analyte analysis |
| US9399797B2 (en) | 2010-02-12 | 2016-07-26 | Raindance Technologies, Inc. | Digital analyte analysis |
| US10351905B2 (en) | 2010-02-12 | 2019-07-16 | Bio-Rad Laboratories, Inc. | Digital analyte analysis |
| WO2012009206A2 (en) | 2010-07-12 | 2012-01-19 | Pacific Biosciences Of California, Inc. | Sequencing reactions with alkali metal cations for pulse width control |
| US8834847B2 (en) | 2010-08-12 | 2014-09-16 | Pacific Biosciences Of California, Inc. | Photodamage mitigation compounds and systems |
| WO2012045012A2 (en) | 2010-09-30 | 2012-04-05 | Raindance Technologies, Inc. | Sandwich assays in droplets |
| WO2012088341A2 (en) | 2010-12-22 | 2012-06-28 | Genia Technologies, Inc. | Nanopore-based single dna molecule characterization, identification and isolation using speed bumps |
| US9581563B2 (en) | 2011-01-24 | 2017-02-28 | Genia Technologies, Inc. | System for communicating information from an array of sensors |
| US9110478B2 (en) | 2011-01-27 | 2015-08-18 | Genia Technologies, Inc. | Temperature regulation of measurement arrays |
| US9364803B2 (en) | 2011-02-11 | 2016-06-14 | Raindance Technologies, Inc. | Methods for forming mixed droplets |
| EP3736281A1 (en) | 2011-02-18 | 2020-11-11 | Bio-Rad Laboratories, Inc. | Compositions and methods for molecular labeling |
| EP3709018A1 (en) | 2011-06-02 | 2020-09-16 | Bio-Rad Laboratories, Inc. | Microfluidic apparatus for identifying components of a chemical reaction |
| US8658430B2 (en) | 2011-07-20 | 2014-02-25 | Raindance Technologies, Inc. | Manipulating droplet size |
| EP2745360A4 (en) | 2011-08-01 | 2015-07-08 | Univ Columbia | CONJUGATES OF NANODIAMANT AND MAGNETIC OR METALLIC PARTICLES |
| WO2013040446A1 (en) | 2011-09-16 | 2013-03-21 | The Trustees Of Columbia University In The City Of New York | High-precision ghz clock generation using spin states in diamond |
| US9632045B2 (en) | 2011-10-19 | 2017-04-25 | The Trustees Of Columbia University In The City Of New York | Systems and methods for deterministic emitter switch microscopy |
| EP2769202A4 (en) * | 2011-10-20 | 2015-11-18 | Exxonmobil Upstream Res Co | NANOPARTICLE PROBES, METHODS AND SYSTEMS FOR USE THEREOF |
| EP2594942A1 (en) * | 2011-11-16 | 2013-05-22 | Koninklijke Philips Electronics N.V. | Long rigid spacers to enhance binding kinetics in immunoassays |
| US8986629B2 (en) | 2012-02-27 | 2015-03-24 | Genia Technologies, Inc. | Sensor circuit for controlling, detecting, and measuring a molecular complex |
| GB2502306A (en) * | 2012-05-22 | 2013-11-27 | Univ Singapore | Microparticle sensor |
| US9817002B2 (en) * | 2012-05-23 | 2017-11-14 | University Of Calcutta | Molecular discriminators using carbon nanotubes |
| GB2517875A (en) | 2012-06-08 | 2015-03-04 | Pacific Biosciences California | Modified base detection with nanopore sequencing |
| JP2015525077A (ja) | 2012-06-15 | 2015-09-03 | ジェニア・テクノロジーズ・インコーポレイテッド | チップの構成および高精度な核酸配列決定 |
| US9399766B2 (en) | 2012-10-01 | 2016-07-26 | Pacific Biosciences Of California, Inc. | Recombinant polymerases for incorporation of protein shield nucleotide analogs |
| US9651539B2 (en) | 2012-10-28 | 2017-05-16 | Quantapore, Inc. | Reducing background fluorescence in MEMS materials by low energy ion beam treatment |
| US9605309B2 (en) | 2012-11-09 | 2017-03-28 | Genia Technologies, Inc. | Nucleic acid sequencing using tags |
| US9759711B2 (en) | 2013-02-05 | 2017-09-12 | Genia Technologies, Inc. | Nanopore arrays |
| EP2971071B1 (en) * | 2013-03-15 | 2018-02-28 | Illumina, Inc. | Enzyme-linked nucleotides |
| EP2971051A4 (en) | 2013-03-15 | 2017-03-01 | The Trustees of Columbia University in the City of New York | Method for detecting multiple predetermined compounds in a sample |
| EP3004385B1 (en) | 2013-05-24 | 2018-11-28 | Quantapore Inc. | Nanopore-based nucleic acid analysis with mixed fret detection |
| US11901041B2 (en) | 2013-10-04 | 2024-02-13 | Bio-Rad Laboratories, Inc. | Digital analysis of nucleic acid modification |
| US9551697B2 (en) | 2013-10-17 | 2017-01-24 | Genia Technologies, Inc. | Non-faradaic, capacitively coupled measurement in a nanopore cell array |
| US9322062B2 (en) | 2013-10-23 | 2016-04-26 | Genia Technologies, Inc. | Process for biosensor well formation |
| CN105723222B (zh) | 2013-10-23 | 2019-01-22 | 吉尼亚科技公司 | 使用纳米孔的高速分子感测 |
| AU2014348306B2 (en) | 2013-11-17 | 2020-11-12 | Quantum-Si Incorporated | Active-source-pixel, integrated device for rapid analysis of biological and chemical specimens |
| US9944977B2 (en) | 2013-12-12 | 2018-04-17 | Raindance Technologies, Inc. | Distinguishing rare variations in a nucleic acid sequence from a sample |
| CA2957543A1 (en) | 2014-08-08 | 2016-02-11 | Quantum-Si Incorporated | Optical system and assay chip for probing, detecting and analyzing molecules |
| EP3471402B1 (en) | 2014-08-08 | 2023-05-31 | Quantum-Si Incorporated | Integrated device for temporal binning of received photons |
| MX2017001808A (es) | 2014-08-08 | 2018-02-08 | Quantum Si Inc | Dispositivo integrado con fuente de luz externa para el sondeo, deteccion y analisis de moleculas. |
| WO2016057829A1 (en) | 2014-10-10 | 2016-04-14 | Quantapore, Inc. | Nanopore-based polymer analysis with mutually-quenching fluorescent labels |
| EP3209800B1 (en) | 2014-10-24 | 2022-02-16 | Quantapore, Inc. | Efficient optical analysis of polymers using arrays of nanostructures |
| EP3508853A1 (en) | 2014-11-12 | 2019-07-10 | Technische Universiteit Eindhoven | Dynamic switching biosensor |
| EP3265812A2 (en) * | 2015-03-04 | 2018-01-10 | University College Dublin National University Of Ireland, Dublin | Molecular sensors |
| DK3294885T3 (da) | 2015-05-08 | 2020-08-10 | Curevac Real Estate Gmbh | Fremgangsmåde til at fremstille rna |
| US10174363B2 (en) | 2015-05-20 | 2019-01-08 | Quantum-Si Incorporated | Methods for nucleic acid sequencing |
| US11130986B2 (en) | 2015-05-20 | 2021-09-28 | Quantum-Si Incorporated | Method for isolating target nucleic acid using heteroduplex binding proteins |
| EP3744843A1 (en) | 2015-05-29 | 2020-12-02 | CureVac Real Estate GmbH | A method for producing and purifying rna, comprising at least one step of tangential flow filtration |
| US10647981B1 (en) | 2015-09-08 | 2020-05-12 | Bio-Rad Laboratories, Inc. | Nucleic acid library generation methods and compositions |
| AU2017219894B2 (en) | 2016-02-17 | 2021-12-09 | Tesseract Health, Inc. | Sensor and device for lifetime imaging and detection applications |
| CN109477813A (zh) | 2016-07-05 | 2019-03-15 | 昆塔波尔公司 | 基于光学的纳米孔测序 |
| US20190276881A1 (en) * | 2016-11-08 | 2019-09-12 | President And Fellows Of Harvard College | Multiplexed imaging using merfish, expansion microscopy, and related technologies |
| KR20230052995A (ko) | 2016-12-19 | 2023-04-20 | 퀀텀-에스아이 인코포레이티드 | 시퀀싱 반응을 위한 중합 효소 |
| CN110168732B (zh) | 2016-12-22 | 2024-03-08 | 宽腾矽公司 | 具有直接合并像素的整合式光电侦测器 |
| CA3059211A1 (en) | 2017-05-05 | 2018-11-08 | Quantum-Si Incorporated | Substrates having modified surface reactivity and antifouling properties in biological reactions |
| WO2018218150A1 (en) | 2017-05-26 | 2018-11-29 | President And Fellows Of Harvard College | Systems and methods for high-throughput image-based screening |
| EP3658682A4 (en) | 2017-07-24 | 2021-06-02 | Quantum-si Incorporated | HIGH INTENSITY LABEL REAGENT COMPOSITIONS AND SEQUENCING METHODS |
| GB201715684D0 (en) | 2017-09-28 | 2017-11-15 | Univ Gent | Means and methods for single molecule peptide sequencing |
| WO2019165017A1 (en) | 2018-02-23 | 2019-08-29 | The University Of Chicago | Methods and composition involving thermophilic fibronectin type iii (fn3) monobodies |
| CN112534803A (zh) | 2018-06-22 | 2021-03-19 | 宽腾矽公司 | 具有不同检测时间的电荷存储仓的集成光电检测器 |
| WO2020018739A1 (en) * | 2018-07-18 | 2020-01-23 | University Of Florida Research Foundation | Rna silencing nanozymes |
| US10704094B1 (en) | 2018-11-14 | 2020-07-07 | Element Biosciences, Inc. | Multipart reagents having increased avidity for polymerase binding |
| US10876148B2 (en) | 2018-11-14 | 2020-12-29 | Element Biosciences, Inc. | De novo surface preparation and uses thereof |
| EP3914603A1 (en) | 2019-01-23 | 2021-12-01 | Quantum-Si Incorporated | High intensity labeled reactant compositions and methods for sequencing |
| WO2020242901A1 (en) * | 2019-05-24 | 2020-12-03 | Element Biosciences, Inc. | Polymerase-nucleotide conjugates for sequencing by trapping |
| US11287422B2 (en) | 2019-09-23 | 2022-03-29 | Element Biosciences, Inc. | Multivalent binding composition for nucleic acid analysis |
| CN114829669A (zh) | 2019-10-11 | 2022-07-29 | 宽腾矽公司 | 气相表面改性 |
| JP2023512634A (ja) | 2020-01-21 | 2023-03-28 | クアンタム-エスアイ インコーポレイテッド | C末端の選択的標識のための化合物および方法 |
| US11198121B1 (en) | 2020-06-10 | 2021-12-14 | Element Biosciences, Inc. | Flow cell systems and devices |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004074503A2 (en) * | 2003-02-21 | 2004-09-02 | Hoser Mark J | Nucleic acid sequencing methods, kits and reagents |
Family Cites Families (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4711955A (en) * | 1981-04-17 | 1987-12-08 | Yale University | Modified nucleotides and methods of preparing and using same |
| US5821058A (en) * | 1984-01-16 | 1998-10-13 | California Institute Of Technology | Automated DNA sequencing technique |
| US5360523A (en) | 1984-03-29 | 1994-11-01 | Li-Cor, Inc. | DNA sequencing |
| US5064754A (en) * | 1984-12-14 | 1991-11-12 | Mills Randell L | Genomic sequencing method |
| US5221518A (en) * | 1984-12-14 | 1993-06-22 | Mills Randell L | DNA sequencing apparatus |
| US5017009A (en) | 1986-06-26 | 1991-05-21 | Ortho Diagnostic Systems, Inc. | Scattered total internal reflectance immunoassay system |
| US5242796A (en) | 1986-07-02 | 1993-09-07 | E. I. Du Pont De Nemours And Company | Method, system and reagents for DNA sequencing |
| CA1340806C (en) | 1986-07-02 | 1999-11-02 | James Merrill Prober | Method, system and reagents for dna sequencing |
| US5047519A (en) | 1986-07-02 | 1991-09-10 | E. I. Du Pont De Nemours And Company | Alkynylamino-nucleotides |
| US5151507A (en) | 1986-07-02 | 1992-09-29 | E. I. Du Pont De Nemours And Company | Alkynylamino-nucleotides |
| US5654176A (en) * | 1987-05-28 | 1997-08-05 | Amrad Corporation Limited | Fusion proteins containing glutathione-s-transferase |
| US4962037A (en) * | 1987-10-07 | 1990-10-09 | United States Of America | Method for rapid base sequencing in DNA and RNA |
| US4793705A (en) * | 1987-10-07 | 1988-12-27 | The United States Of America As Represented By The United States Department Of Energy | Single molecule tracking |
| US4917462A (en) * | 1988-06-15 | 1990-04-17 | Cornell Research Foundation, Inc. | Near field scanning optical microscopy |
| DE3841565C2 (de) | 1988-12-09 | 1998-07-09 | Europ Lab Molekularbiolog | Verfahren zur Sequenzierung von Nukleinsäuren |
| US5302509A (en) | 1989-08-14 | 1994-04-12 | Beckman Instruments, Inc. | Method for sequencing polynucleotides |
| US5105305A (en) | 1991-01-10 | 1992-04-14 | At&T Bell Laboratories | Near-field scanning optical microscope using a fluorescent probe |
| US5817462A (en) | 1995-02-21 | 1998-10-06 | Applied Spectral Imaging | Method for simultaneous detection of multiple fluorophores for in situ hybridization and multicolor chromosome painting and banding |
| US5405747A (en) * | 1991-09-25 | 1995-04-11 | The Regents Of The University Of California Office Of Technology Transfer | Method for rapid base sequencing in DNA and RNA with two base labeling |
| CH683667A5 (fr) | 1992-02-26 | 1994-04-29 | Nestle Sa | Matière première aromatique. |
| CA2153387A1 (en) * | 1993-01-07 | 1994-07-21 | Hubert Koester | Dna sequencing by mass spectrometry |
| CA2155186A1 (en) * | 1993-02-01 | 1994-08-18 | Kevin M. Ulmer | Methods and apparatus for dna sequencing |
| US5354985A (en) * | 1993-06-03 | 1994-10-11 | Stanford University | Near field scanning optical and force microscope including cantilever and optical waveguide |
| US5389779A (en) * | 1993-07-29 | 1995-02-14 | At&T Corp. | Method and apparatus for near-field, scanning, optical microscopy by reflective, optical feedback |
| US5470710A (en) * | 1993-10-22 | 1995-11-28 | University Of Utah | Automated hybridization/imaging device for fluorescent multiplex DNA sequencing |
| US5886177A (en) * | 1994-01-11 | 1999-03-23 | Isis Pharmaceuticals, Inc. | Phosphate linked oligomers |
| US5654419A (en) | 1994-02-01 | 1997-08-05 | The Regents Of The University Of California | Fluorescent labels and their use in separations |
| US6682886B1 (en) * | 1994-04-28 | 2004-01-27 | Gilead Sciences, Inc. | Bivalent binding molecules of 7 transmembrane G protein-coupled receptors |
| US5763594A (en) * | 1994-09-02 | 1998-06-09 | Andrew C. Hiatt | 3' protected nucleotides for enzyme catalyzed template-independent creation of phosphodiester bonds |
| US5625048A (en) | 1994-11-10 | 1997-04-29 | The Regents Of The University Of California | Modified green fluorescent proteins |
| US5777079A (en) * | 1994-11-10 | 1998-07-07 | The Regents Of The University Of California | Modified green fluorescent proteins |
| US5556790A (en) * | 1994-12-05 | 1996-09-17 | Pettit; John W. | Method for Automated DNA sequencing |
| US5614386A (en) * | 1995-06-23 | 1997-03-25 | Baylor College Of Medicine | Alternative dye-labeled primers for automated DNA sequencing |
| US5661028A (en) * | 1995-09-29 | 1997-08-26 | Lockheed Martin Energy Systems, Inc. | Large scale DNA microsequencing device |
| US6766183B2 (en) * | 1995-11-22 | 2004-07-20 | Medtronic Minimed, Inc. | Long wave fluorophore sensor compounds and other fluorescent sensor compounds in polymers |
| US5796909A (en) | 1996-02-14 | 1998-08-18 | Islam; Mohammed N. | All-fiber, high-sensitivity, near-field optical microscopy instrument employing guided wave light collector and specimen support |
| US5800996A (en) | 1996-05-03 | 1998-09-01 | The Perkin Elmer Corporation | Energy transfer dyes with enchanced fluorescence |
| US5780232A (en) * | 1996-05-28 | 1998-07-14 | Atom Sciences, Inc. | DNA sequencing, mapping, and diagnostic processes using hybridization and stable isotope labels of DNA |
| US5866336A (en) * | 1996-07-16 | 1999-02-02 | Oncor, Inc. | Nucleic acid amplification oligonucleotides with molecular energy transfer labels and methods based thereon |
| US5804386A (en) * | 1997-01-15 | 1998-09-08 | Incyte Pharmaceuticals, Inc. | Sets of labeled energy transfer fluorescent primers and their use in multi component analysis |
| WO1998033939A1 (fr) | 1997-01-31 | 1998-08-06 | Hitachi, Ltd. | Procede pour determiner une sequence de base d'acide nucleique et appareil correspondant |
| US6197928B1 (en) * | 1997-03-14 | 2001-03-06 | The Regents Of The University Of California | Fluorescent protein sensors for detection of analytes |
| US5998204A (en) * | 1997-03-14 | 1999-12-07 | The Regents Of The University Of California | Fluorescent protein sensors for detection of analytes |
| WO1998040477A1 (en) | 1997-03-14 | 1998-09-17 | The Regents Of The University Of California | Fluorescent protein sensors for detection of analytes |
| US5866366A (en) | 1997-07-01 | 1999-02-02 | Smithkline Beecham Corporation | gidB |
| DE69808661T3 (de) | 1997-07-28 | 2012-05-03 | Gen-Probe Inc. | Sequenzanalyse von nukleinsäuren |
| AU2460399A (en) * | 1998-01-20 | 1999-08-02 | Packard Bioscience Company | Gel pad arrays and methods and systems for making them |
| US20010008766A1 (en) * | 1998-03-17 | 2001-07-19 | Sylvia Daunert | Quantitative binding assays using green fluorescent protein as a label |
| US6780591B2 (en) * | 1998-05-01 | 2004-08-24 | Arizona Board Of Regents | Method of determining the nucleotide sequence of oligonucleotides and DNA molecules |
| US6287765B1 (en) * | 1998-05-20 | 2001-09-11 | Molecular Machines, Inc. | Methods for detecting and identifying single molecules |
| US6140132A (en) * | 1998-06-09 | 2000-10-31 | The Regents Of The University Of California | Fluorescent protein sensors for measuring the pH of a biological sample |
| US6150176A (en) * | 1998-06-09 | 2000-11-21 | The Regents Of The University Of California | Fluorescent protein sensors for measuring the pH of a biological sample |
| US6495664B1 (en) * | 1998-07-24 | 2002-12-17 | Aurora Biosciences Corporation | Fluorescent protein sensors of post-translational modifications |
| US6210896B1 (en) * | 1998-08-13 | 2001-04-03 | Us Genomics | Molecular motors |
| DK1159453T3 (da) | 1999-03-10 | 2008-10-06 | Asm Scient Inc | Fremgangsmåde til direkte nukleinsyresekventering |
| US7056661B2 (en) * | 1999-05-19 | 2006-06-06 | Cornell Research Foundation, Inc. | Method for sequencing nucleic acid molecules |
| US6982146B1 (en) * | 1999-08-30 | 2006-01-03 | The United States Of America As Represented By The Department Of Health And Human Services | High speed parallel molecular nucleic acid sequencing |
| AU7086800A (en) | 1999-08-30 | 2001-03-26 | Government Of The United States Of America, As Represented By The Secretary Of The Department Of Health And Human Services, The | High speed parallel molecular nucleic acid sequencing |
| US6908736B1 (en) * | 1999-10-06 | 2005-06-21 | Medical Biosystems, Ltd. | DNA sequencing method |
| US6627396B1 (en) * | 1999-10-28 | 2003-09-30 | The Regents Of The University Of California | Influenza sensor |
| GB9926336D0 (en) | 1999-11-05 | 2000-01-12 | Isis Innovation | Universal flourescent sensors |
| US6936702B2 (en) | 2000-06-07 | 2005-08-30 | Li-Cor, Inc. | Charge-switch nucleotides |
| AU2001282881B2 (en) | 2000-07-07 | 2007-06-14 | Visigen Biotechnologies, Inc. | Real-time sequence determination |
| US7211414B2 (en) * | 2000-12-01 | 2007-05-01 | Visigen Biotechnologies, Inc. | Enzymatic nucleic acid synthesis: compositions and methods for altering monomer incorporation fidelity |
| US6890556B1 (en) * | 2001-02-14 | 2005-05-10 | Northwestern University | Controlled surface-associated delivery of genes and oligonucleotides |
| JP2004523243A (ja) * | 2001-03-12 | 2004-08-05 | カリフォルニア インスティチュート オブ テクノロジー | 非同期性塩基伸長によってポリヌクレオチド配列を分析するための方法および装置 |
| GB0111459D0 (en) | 2001-05-10 | 2001-07-04 | Isis Innovation | Universal fluorescent sensors |
| US6544746B2 (en) * | 2001-08-13 | 2003-04-08 | St. Louis University | Rapid and sensitive proximity-based assay for the detection and quantification of DNA binding proteins |
| US7172865B2 (en) * | 2001-08-13 | 2007-02-06 | Saint Louis University | Rapid and sensitive assay for the detection and quantification of coregulators of nucleic acid binding factors |
| US20050267326A1 (en) * | 2001-10-02 | 2005-12-01 | Alfred E. Mann Institute For Biomedical Eng. At The University Of Southern California | Percutaneous chemical sensor based on fluorescence resonant energy transfer (FRET) |
| US6790632B2 (en) * | 2002-06-17 | 2004-09-14 | Stephen Eliot Zweig | Membrane receptor reagent and assay |
| WO2005077065A2 (en) | 2004-02-09 | 2005-08-25 | The Regents Of The University Of California | Selective high-affinity polydentate ligands and methods of making such |
| EP1869206A4 (en) * | 2005-03-02 | 2011-08-10 | Einstein Coll Med | ENZYME SENSORS WITH ENVIRONMENTAL SENSITIVE OR FLUORESCENT LABELS AND APPLICATIONS THEREOF |
| DE602006016984D1 (de) * | 2005-12-12 | 2010-10-28 | Us Gov Health & Human Serv | Sonde zum sequenzieren von nukleinsäure und verwendungsverfahren |
-
2006
- 2006-12-12 DE DE602006016984T patent/DE602006016984D1/de active Active
- 2006-12-12 WO PCT/US2006/047456 patent/WO2007070542A2/en active Application Filing
- 2006-12-12 JP JP2008545768A patent/JP2009519041A/ja active Pending
- 2006-12-12 AT AT06847604T patent/ATE481505T1/de not_active IP Right Cessation
- 2006-12-12 WO PCT/US2006/047534 patent/WO2007070572A2/en active Application Filing
- 2006-12-12 EP EP10176710A patent/EP2311982A1/en not_active Withdrawn
- 2006-12-12 US US11/638,160 patent/US8344121B2/en not_active Expired - Fee Related
- 2006-12-12 EP EP06847604A patent/EP1960550B1/en not_active Not-in-force
- 2006-12-12 US US12/095,973 patent/US7871777B2/en not_active Expired - Fee Related
-
2010
- 2010-12-29 US US12/980,802 patent/US20110111975A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004074503A2 (en) * | 2003-02-21 | 2004-09-02 | Hoser Mark J | Nucleic acid sequencing methods, kits and reagents |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019518466A (ja) * | 2016-06-24 | 2019-07-04 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 繋ぎ止められたヌクレオシド三リン酸を用いた核酸合成および配列決定 |
| JP7058228B2 (ja) | 2016-06-24 | 2022-04-21 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 繋ぎ止められたヌクレオシド三リン酸を用いた核酸合成および配列決定 |
| CN114874337A (zh) * | 2016-06-24 | 2022-08-09 | 加利福尼亚大学董事会 | 使用栓系的核苷三磷酸的核酸合成和测序 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1960550B1 (en) | 2010-09-15 |
| WO2007070572A3 (en) | 2008-04-17 |
| US20100227913A1 (en) | 2010-09-09 |
| WO2007070572A2 (en) | 2007-06-21 |
| WO2007070542A3 (en) | 2008-04-03 |
| US7871777B2 (en) | 2011-01-18 |
| US20110111975A1 (en) | 2011-05-12 |
| US8344121B2 (en) | 2013-01-01 |
| US20080299565A1 (en) | 2008-12-04 |
| EP2311982A1 (en) | 2011-04-20 |
| ATE481505T1 (de) | 2010-10-15 |
| DE602006016984D1 (de) | 2010-10-28 |
| WO2007070542A2 (en) | 2007-06-21 |
| EP1960550A2 (en) | 2008-08-27 |
| WO2007070572A8 (en) | 2007-09-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7871777B2 (en) | Probe for nucleic acid sequencing and methods of use | |
| US11807904B2 (en) | FRET-labeled compounds and uses therefor | |
| US6982146B1 (en) | High speed parallel molecular nucleic acid sequencing | |
| EP1226271B1 (en) | Dna sequencing method | |
| US6908736B1 (en) | DNA sequencing method | |
| WO2001016375A2 (en) | High speed parallel molecular nucleic acid sequencing | |
| JP2004513619A (ja) | リアルタイム配列決定 | |
| JP2022513574A (ja) | ポリヌクレオチドを配列決定する方法 | |
| Rubens et al. | Schneider et al. | |
| WO2011108344A1 (ja) | 基板上に固定化された複数の核酸検体の識別方法及び装置 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20091202 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20091202 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120820 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20130130 |