JP2008120695A - ピレン−1,6−ジカルボン酸の製造方法 - Google Patents
ピレン−1,6−ジカルボン酸の製造方法 Download PDFInfo
- Publication number
- JP2008120695A JP2008120695A JP2006303030A JP2006303030A JP2008120695A JP 2008120695 A JP2008120695 A JP 2008120695A JP 2006303030 A JP2006303030 A JP 2006303030A JP 2006303030 A JP2006303030 A JP 2006303030A JP 2008120695 A JP2008120695 A JP 2008120695A
- Authority
- JP
- Japan
- Prior art keywords
- pyrene
- dicarboxylic acid
- isomer mixture
- dialkyl
- dibromopyrene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 *[N+](c1c(ccc(c2c3cc4)ccc3[N+](*)[O-])c2c4cc1)[O-] Chemical compound *[N+](c1c(ccc(c2c3cc4)ccc3[N+](*)[O-])c2c4cc1)[O-] 0.000 description 1
- SCWCBWXJSFHLEL-UHFFFAOYSA-N COC(c(cc1)c(cc2)c3c1ccc1c3c2ccc1C(OC)=O)=O Chemical compound COC(c(cc1)c(cc2)c3c1ccc1c3c2ccc1C(OC)=O)=O SCWCBWXJSFHLEL-UHFFFAOYSA-N 0.000 description 1
- IJDIPICFINEEDN-UHFFFAOYSA-N OC(c(cc1)c(cc2)c3c1ccc1c3c2ccc1C(O)=O)=O Chemical compound OC(c(cc1)c(cc2)c3c1ccc1c3c2ccc1C(O)=O)=O IJDIPICFINEEDN-UHFFFAOYSA-N 0.000 description 1
- RQJSAJJBBSJUFF-UHFFFAOYSA-N Oc1ccc(cc2)c3c1ccc(cc1)c3c2c1O Chemical compound Oc1ccc(cc2)c3c1ccc(cc1)c3c2c1O RQJSAJJBBSJUFF-UHFFFAOYSA-N 0.000 description 1
Images



Landscapes
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
【課題】ピレン誘導体であるピレンジカルボン酸のうち、ピレン−1,6−ジカルボン酸を容易にかつ確実に製造する方法を提供する。
【解決手段】再結晶により、ピレン−1,6−ジカルボン酸エステルと、ピレン−1,3−ジカルボン酸エステル又はピレン−1,8−ジカルボン酸エステルの一方又は双方と、を含有するピレンジカルボン酸エステル異性体混合物から前記ピレン−1,6−ジカルボン酸エステルを分離する分離工程と、ピレン−1,6−ジカルボン酸エステルを加水分解してピレン−1,6−ジカルボン酸を得る反応工程とを備えるピレン−1,6−ジカルボン酸の製造方法である。
【選択図】なし
【解決手段】再結晶により、ピレン−1,6−ジカルボン酸エステルと、ピレン−1,3−ジカルボン酸エステル又はピレン−1,8−ジカルボン酸エステルの一方又は双方と、を含有するピレンジカルボン酸エステル異性体混合物から前記ピレン−1,6−ジカルボン酸エステルを分離する分離工程と、ピレン−1,6−ジカルボン酸エステルを加水分解してピレン−1,6−ジカルボン酸を得る反応工程とを備えるピレン−1,6−ジカルボン酸の製造方法である。
【選択図】なし
Description
本発明は、ピレン−1,6−ジカルボン酸の製造方法に関する。
有機合成において、得られた反応混合物が構造異性体や光学異性体の混合物である場合、各成分の分子量が同一で化学的性質が近似していることから各成分を分離するのは一般的に困難である。
ピレン誘導体もその構造異性体の混合物から目的とする異性体を分離することが困難な化合物の一つである。例えば、下記非特許文献1には、ピレンをN−ブロモスクシンイミド2モル当量と反応させると、ピレン誘導体として1,3−ジブロモピレン、1,6−ジブロモピレン、および1,8−ジブロモピレンの混合物が得られることが記載されているが、かかる異性体混合物から特定の異性体を分離する手法は未だ確立されていない。
なお、非特許文献2には、トルエンを溶媒とした再結晶により、ジブロモピレン異性体混合物から1,6−ジブロモピレンを分離できることが記載されているが、1,6−ジブロモピレンを高収率又は高純度で得るためには再結晶操作を何度も繰り返す必要があり、工業化に適しているとは言い難い。
M.Minabe,S.Takashige,Y.Soeda,T.Kimura,and M.Tsubota,Bull.Chem.Soc.Jpn.,67,172(1994). J.Grimshaw and J.Trocha−Grismshaw,J.Chem.Soc.,Perkin Trans.1,1972,1622.
M.Minabe,S.Takashige,Y.Soeda,T.Kimura,and M.Tsubota,Bull.Chem.Soc.Jpn.,67,172(1994). J.Grimshaw and J.Trocha−Grismshaw,J.Chem.Soc.,Perkin Trans.1,1972,1622.
上記のような事情の下、本発明は、ピレン誘導体であるピレンジカルボン酸のうち、ピレン−1,6−ジカルボン酸を容易にかつ確実に製造する方法を提供することを目的とする。
本発明者は、上記目的を達成するために、先ず、ピレンジカルボン酸の異性体混合物からピレン−1,6−ジカルボン酸を直接分離することを試みた。しかし、ピレンジカルボン酸の異性体混合物は有機溶媒に対してほとんど溶解性を示さず、シリカゲルカラムやリサイクル分取HPLCなどの一般的な分離方法が適用できないことが判明した。
そこで、本発明者らは、上記知見に基づいてさらに検討を重ねた結果、ピレンジカルボン酸のカルボキシル基をエステル化又はアルキル化した異性体混合物に再結晶を施せば、エステル化物又はアルキル化物としての1,6−置換体を十分に分離可能であることを見出した。そして、このようにして分離された1,6−置換体のエステル化物又はアルキル化物を加水分解又は酸化させることによって、目的物であるピレン−1,6−ジカルボン酸が容易にかつ確実に得られることを見出し、本発明を完成させるに至った。
すなわち、本発明は、再結晶により、下記一般式(1)で表されるピレン−1,6−ジカルボン酸エステルと、下記一般式(2)で表されるピレン−1,3−ジカルボン酸エステル又は下記一般式(3)で表されるピレン−1,8−ジカルボン酸エステルの一方又は双方と、を含有するピレンジカルボン酸エステル異性体混合物からピレン−1,6−ジカルボン酸エステルを分離する分離工程と、上記分離工程で得られたピレン−1,6−ジカルボン酸エステルを加水分解してピレン−1,6−ジカルボン酸を得る反応工程とを備える、ピレン−1,6−ジカルボン酸の製造方法(以下、便宜的に「第1の製造方法」という。)を提供する。
[式中、R1及びR2は各々独立に炭素数1〜4のアルキル基を示す。]
[式中、R3及びR4は各々独立に炭素数1〜4のアルキル基を示す。]
[式中、R5及びR6は各々独立に炭素数1〜4のアルキル基を示す。]
上記第1の製造方法において、ピレンジカルボン酸エステル異性体混合物は、ピレンジカルボン酸の異性体混合物に比べて、有機溶媒に対する溶解性が十分に改善されたものである。そして、かかるピレンジカルボン酸エステル異性体混合物に再結晶を施すことによってピレン−1,6−ジカルボン酸エステルを容易にかつ確実に分離することができる。したがって、このようにして得られたピレン−1,6−ジカルボン酸エステルを加水分解することによって、目的物であるピレン−1,6−ジカルボン酸を有効に得ることができる。
なお、本発明者らの検討によれば、有機溶媒に対する溶解性が改善されたピレンジカルボン酸エステル異性体混合物であっても、これにシリカゲルカラムやリサイクル分取HPLCなどの一般的な異性体分離方法を適用して1,6−置換体を分離することは困難であり、本発明による上述の効果はピレンジカルボン酸エステル異性体混合物からのピレン−1,6−ジカルボン酸エステルの分離手段として再結晶が適しているという本発明者らの独自の知見に基づくものである。
また、再結晶は、通常、目的化合物を主成分として含有する反応混合物から少量の副生成物を除去するために用いられる分離方法であり、その原理から化合物相互間の極性や溶解性に差がある混合物からの分離に適していると考えられている。このため目的化合物以外の成分が多く、かつ極性や溶解性に差が殆ど観られないことの多い異性体の混合物から再結晶によって特定の異性体を分離できることは稀である。本発明においてピレンジカルボン酸エステル異性体混合物からピレン−1,6−ジカルボン酸エステルを再結晶によって分離できる理由は必ずしも明確ではないが、本発明による上述の効果はこのような技術常識を鑑みれば極めて予想外の効果といえる。
上記第1の製造方法におけるピレンジカルボン酸エステル異性体混合物は、ピレンの臭素化反応により1,6−ジブロモピレンと1,3−ジブロモピレンと1,8−ジブロモピレンとを含有するジブロモピレン異性体混合物を得る工程と、ジブロモピレン異性体混合物のカルボキシル化反応によりピレン−1,6−ジカルボン酸とピレン−1,3−ジカルボン酸とピレン−1,8−ジカルボン酸とを含有するピレンジカルボン酸異性体混合物を得る工程と、ピレンジカルボン酸異性体混合物のエステル化反応により上記一般式(1)で表されるピレン−1,6−ジカルボン酸エステルと、上記一般式(2)で表されるピレン−1,3−ジカルボン酸エステルと、上記一般式(3)で表されるピレン−1,8−ジカルボン酸エステルと、を含有するピレンジカルボン酸エステル異性体混合物を得る工程と、を経ることによって、好適に得ることができる。
また、上記第1の製造方法における分離工程は、ピレンジカルボン酸エステル異性体混合物をジオキサンに溶解させた後、再結晶により前記ピレン−1,6−ジカルボン酸エステルを析出させて分離する工程であることが好ましい。
ジオキサンを用いることによって、ピレンジカルボン酸エステル異性体混合物を容易に溶解し、再結晶によってピレン−1,6−ジカルボン酸エステルを容易に分離することができる。
また、本発明は、再結晶により、下記一般式(4)で表されるピレン−1,6−ジアルキルと、下記一般式(5)で表されるピレン−1,3−ジアルキル又は下記一般式(6)で表されるピレン−1,8−ジアルキルの一方又は双方と、を含有するピレンジアルキル異性体混合物からピレン−1,6−ジアルキルを分離する分離工程と、上記分離工程で得られたピレン−1,6−ジアルキルの酸化反応によりピレン−1,6−ジカルボン酸を得る酸化工程と、を備える、ピレン−1,6−ジカルボン酸の製造方法(以下、便宜的に「第2の製造方法」という。)を提供する。
[式中、R7及びR8は各々独立に炭素数3又は4のアルキル基を示す。]
[式中、R9及びR10は各々独立に炭素数3又は4のアルキル基を示す。]
[式中、R11及びR12は各々独立に炭素数3又は4のアルキル基を示す。]
上記第2の製造方法において、ピレンジアルキル異性体混合物は、ピレンジカルボン酸の異性体混合物に比べて、有機溶媒に対する溶解性が十分に改善されたものである。そして、かかるピレンジアルキル異性体混合物に再結晶を施すことによってピレン−1,6−ジアルキルを容易にかつ確実に分離することができる。したがって、このようにして得られたピレン−1,6−ピレンジアルキルを酸化することによって、目的物であるピレン−1,6−ジカルボン酸を有効に得ることができる。
なお、本発明者らの検討によれば、有機溶媒に対する溶解性が改善されたピレンジアルキル異性体混合物であっても、これにシリカゲルカラムやリサイクル分取HPLCなどの一般的な異性体分離方法を適用して1,6−置換体を分離することは困難であり、本発明による上述の効果はピレンジアルキル異性体混合物からのピレン−1,6−ジアルキルの分離手段として再結晶が適しているという本発明者らの独自の知見に基づくものである。
また、上述の通り、再結晶によって、目的化合物以外の成分が多くかつ極性や溶解性に差が殆ど観られないことの多い異性体の混合物から特定の異性体を分離できることは稀である。本発明においてピレンジアルキル異性体混合物からピレン−1,6−ジアルキルを再結晶によって分離できる理由は必ずしも明確ではないが、本発明による上述の効果はこのような技術常識を鑑みれば極めて予想外の効果といえる。
上記第2の製造方法におけるピレンジアルキル異性体混合物は、ピレンの臭素化反応により1,6−ジブロモピレンと1,3−ジブロモピレンと1,8−ジブロモピレンとを含有するジブロモピレン異性体混合物を得る工程と、ジブロモピレン異性体混合物のアルキル化反応により上記一般式(4)で表されるピレン−1,6−ジアルキルと上記一般式(5)で表されるピレン−1,3−ジアルキルと上記一般式(6)で表される1,8−ピレンジアルキルとを含有するピレンジアルキル異性体混合物を得る工程と、を経ることによって好適に得ることができる。
また、上記第2の製造方法における分離工程は、ピレンジアルキル異性体混合物をジオキサンに溶解させた後、再結晶により前記ピレン−1,6−ジアルキルを析出させて分離する工程であることが好ましい。
ジオキサンを用いることによって、ピレンジアルキル異性体混合物を容易に溶解し、再結晶によってピレン−1,6−ジアルキルを容易に分離することができる。
本発明によれば、ピレン誘導体であるピレンジカルボン酸のうち、ピレン−1,6−ジカルボン酸を容易にかつ確実に製造する方法を提供することができる。
以下、本発明の好適な実施形態について詳細に説明する。
本発明の第1の製造方法は、再結晶によりピレンジカルボン酸エステル異性体混合物からピレン−1,6−ジカルボン酸エステルを得る分離工程と、加水分解によりピレン−1,6−ジカルボン酸エステルからピレン−1,6−ジカルボン酸を得る反応工程とを備えるピレン−1,6−ジカルボン酸の製造方法である。
第1の製造方法におけるピレンジカルボン酸エステル異性体混合物は、臭素化反応によりピレンからジブロモピレン異性体混合物を得る工程と、カルボキシル化反応によりジブロモピレン異性体混合物からピレンジカルボン酸異性体混合物を得る工程と、エステル化反応によりピレンジカルボン酸異性体混合物からピレンジカルボン酸エステル異性体混合物を得る工程とによって得られる。
ピレンからピレンジカルボン酸エステル異性体混合物を得る工程について、以下に説明する。
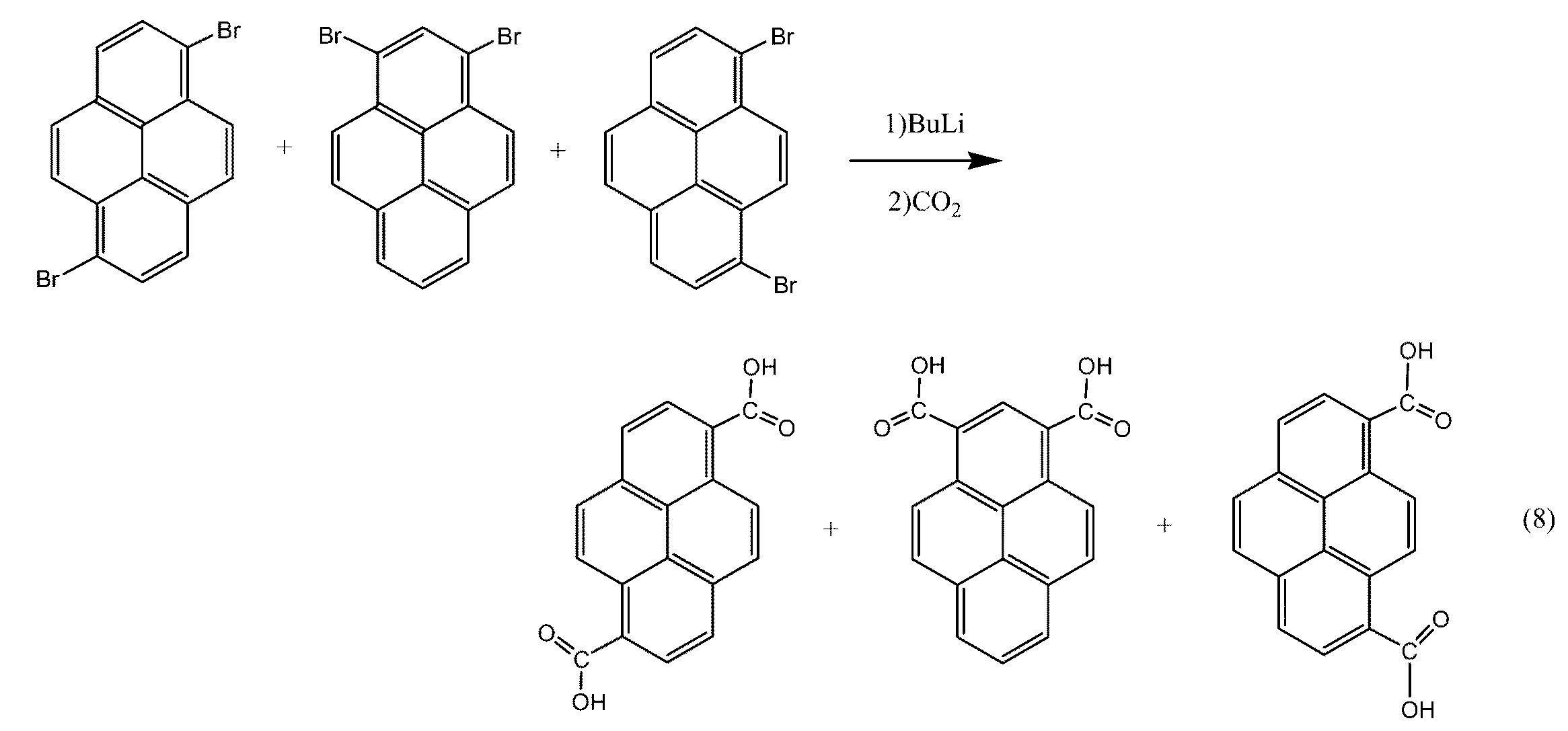
まず、下記の反応式(7)によって表される臭素化反応によりピレンからジブロモピレン異性体混合物を得ることができる。
ピレンの臭素化は、有機溶媒に溶解したピレンの溶液にN−ブロモスクシンイミドを添加することによって行なうことができる。有機溶媒としてはクロロホルム、四塩化炭素などを使用することができる。このうち、製造コストの観点からクロロホルムが好ましい。ピレン1質量部に対して有機溶媒は10〜500質量部とすることができる。
ピレンとN−ブロモスクシンイミドとは、上記の反応式(7)の反応により1,6−ジブロモピレンと1,3−ジブロモピレンと1,8−ジブロモピレンとを含有するジブロモピレン異性体混合物を反応液中に生成する。N−ブロモスクシンイミド添加時の温度は0〜40℃とすることができる。N−ブロモスクシンイミドの添加量は、ピレン1molに対して1.9〜2.1molであることが好ましく、1.95〜2.05molであることがより好ましい。N−ブロモスクシンイミドの添加量が、ピレン1molに対して1.9mol未満又は2.1molを越える場合には、副生成物が生成しやすくなる傾向がある。
該反応液から有機溶媒を留去すると、固形分としてジブロモピレン異性体混合物とスクシンイミドの混合物を得ることができる。この混合物を塩酸、次いでエタノールで洗浄することにより、混合物からスクシンイミドを除去することができる。有機溶媒の留去は0〜60℃で大気圧以下で行うことができる。
洗浄に用いる無機酸として例えば塩酸を、有機溶媒として例えばエタノールを用いることができる。
該ジブロモピレン異性体混合物は、通常、乾燥質量基準で40〜50質量%の1,6−ジブロモピレンと、5〜15質量%の1,3−ジブロモピレンと、40〜50質量%の1,8−ジブロモピレンとを含有する。
次に、下記の反応式(8)によって表されるカルボキシル化反応によってジブロモピレン異性体混合物からピレンジカルボン酸異性体混合物を得ることができる。
まず、ジブロモピレン異性体混合物を有機溶媒に懸濁して懸濁液を作製することができる。有機溶媒としてはジエチルエーテル、テトラヒドロフランなどを使用できる。このうち、ろ過のしやすさの観点からジエチルエーテルが好ましい。有機溶媒の量は、ジブロモピレン異性体混合物1質量部に対して例えば10〜500質量部とすることができる。
次に、作製した懸濁液にアルキルリチウム含有溶液を徐々に滴下し、ジブロモピレンとアルキルリチウムとを反応させて、反応溶液を得ることができる。反応の際、反応溶液の温度は0〜40℃とすることが好ましく、0〜20℃とすることがより好ましい。反応溶液の温度が0℃未満又は40℃を超える場合は副反応が進行しやすい傾向がある。
アルキルリチウムとしては、n−ブチルリチウム、メチルリチウム、sec−ブチルリチウム、tert−ブチルリチウムなどを用いることができる。このうち、製造コストの観点からn−ブチルリチウムが好ましい。アルキルリチウムの添加量は、ジブロモピレン1.0molに対して2.0〜2.5molとすることが好ましく、2.0〜2.3molとすることがより好ましく、2.0〜2.1molとすることがさらに好ましい。アルキルリチウムの添加量がピレン1.0molに対して2.0mol未満又は2.5molを越える場合には、未反応の原料が増加する傾向がある。
該反応溶液にCO2を加え、さらに無機酸を加えると、黄色のピレンジカルボン酸異性体混合物が沈殿した分散液が得られる。CO2としては、CO2ガスまたはドライアイスを使用できる。このうち取り扱い性の観点からドライアイスが好ましい。無機酸としては、塩酸、硝酸などを使用できる。このうち取り扱い性の観点から塩酸が好ましい。
該分散液から沈殿物を分離することによって、ピレンジカルボン酸異性体混合物が得られる。分離方法としては、ろ過等の通常の分離手段を用いることができる。
得られたピレンジカルボン酸異性体混合物は、不純物を除去するために無機酸や有機溶剤等で洗浄することが好ましい。
該ピレンジカルボン酸異性体混合物は、通常、乾燥質量基準で40〜50質量%のピレン−1,6−ジカルボン酸と、5〜15質量%のピレン−1,3−ジカルボン酸と、40〜50質量%のピレン−1,8−ジカルボン酸とを含有する。
なお、ピレンジカルボン酸異性体混合物は有機溶媒に殆ど溶解しない。このため、ピレンジカルボン酸異性体混合物から直接ピレン−1,6−ジカルボン酸を分離することは困難である。
次に、下記の反応式(9)によって表されるエステル化反応によって、ピレンジカルボン酸異性体混合物からピレンジカルボン酸ジメチルエステル異性体混合物を得ることができる。
ピレンジカルボン酸異性体混合物に塩化チオニルを加えると、反応して黄色のピレン酸塩化物異性体混合物が懸濁した懸濁液が得られる。反応時の温度は20〜60℃とすることができる。また、塩化チオニルの量は、ピレンジカルボン酸異性体混合物1質量部に対して5〜50質量部とすることができる。該懸濁液をメチルアルコールとトリエチルアミンとの混合溶液に加えることによって、反応液を得ることができる。メチルアルコールの使用量は、ピレンジカルボン酸異性体混合物1質量部に対して2〜100質量部、トリエチルアミンの使用量は、ピレンジカルボン酸異性体混合物1質量部に対して2〜100質量部とすることができる。該反応液を濃縮して溶媒を留去することによってピレンジカルボン酸ジメチルエステル異性体混合物が得られる。濃縮は0〜70℃の温度で大気圧以下で行うことができる。
なお、本実施形態ではメチルアルコールを用いたが、メチルアルコールの代わりにエチルアルコール、イソプロピルアルコール、n−ブタノールなどの炭素数2〜4の直鎖または分岐アルキルアルコールを使用することができる。この場合、使用するアルコールに応じて得られるピレンジカルボン酸エステルが変化する。例えば、エチルアルコールを使用した場合に得られるピレンジカルボン酸エステルは、ピレンジカルボン酸ジエチルエステルである。
ピレンジカルボン酸エステルの結晶性の観点から、エステル化に使用する上述のアルコールは炭素数が少ない方が好ましく、メチルアルコールが最も好ましい。エステル構造の炭素鎖が長くなると、ピレンジカルボン酸エステルの結晶性が悪くなる傾向がある。
得られたピレンジカルボン酸ジメチルエステル異性体混合物は、不純物を除去するために無機酸や有機溶媒等で洗浄することが好ましい。
エステル化反応により、ピレン1質量部に対して、例えば1.0〜1.5質量部のピレンジカルボン酸ジメチルエステル異性体混合物を得ることができる。得られたピレンジカルボン酸ジメチルエステル異性体混合物は、通常、乾燥質量基準で40〜50質量%のピレン−1,6−ジカルボン酸ジメチルエステルと、5〜15質量%のピレン−1,3−ジカルボン酸ジメチルエステルと、40〜50質量%のピレン−1,8−ジカルボン酸ジメチルエステルとを含有する。
<分離工程>
再結晶によってピレンジカルボン酸ジメチルエステル異性体混合物からピレン−1,6−ジカルボン酸ジメチルエステルを分離する分離工程について、以下に説明する。
再結晶によってピレンジカルボン酸ジメチルエステル異性体混合物からピレン−1,6−ジカルボン酸ジメチルエステルを分離する分離工程について、以下に説明する。
分離工程では、まず有機溶媒にピレンジカルボン酸ジメチルエステル異性体混合物を溶解する。ピレンジカルボン酸ジメチルエステル異性体を完全に溶解した後、得られた溶液を冷却するとピレン−1,6−ジカルボン酸ジメチルエステルが析出する。この析出物を冷却した液から分離することによって、ピレン−1,6−ジカルボン酸ジメチルエステルが得られる。分離には、ろ過などの通常の分離方法を用いることができる。
再結晶に用いる有機溶媒のうち特に好ましいものの一つとして、ジオキサンが挙げられる。この理由は、ピレンジカルボン酸エステル異性体混合物の溶解性が良く、経験的にピレンジカルボン酸エステル異性体混合物の再結晶溶媒として相性が良いためと推察する。
ピレンジカルボン酸ジメチルエステル異性体混合物1質量部に対して再結晶に用いられる有機溶媒の量は、使用する溶媒の種類にもよるが、ジオキサンを使用する場合は、ピレンジカルボン酸ジメチルエステル異性体混合物1質量部に対して3〜10質量部とすることが好ましい。ピレンジカルボン酸ジメチルエステル異性体混合物1質量部に対してジオキサンが3質量部未満であると、ピレンジカルボン酸ジメチルエステル異性体混合物が溶解しにくい傾向がある。ピレンジカルボン酸ジメチルエステル異性体混合物1質量部に対してジオキサンが10質量部を超えると、析出するピレン−1,6−ジカルボン酸ジメチルエステルの量が少なくなり歩留まりが低下する傾向がある。
ピレンジカルボン酸ジメチルエステル異性体混合物を溶解する際の温度は、再結晶に用いる有機溶媒の種類に応じて適宜選択されるが、例えばジオキサンを用いる場合、40〜100℃が好ましい。40℃未満であると、ピレンジカルボン酸ジメチルエステル異性体混合物を十分に溶解できない傾向がある。また、100℃は、ほぼジオキサンの沸点に相当するため、これを超える温度に上げるのは実用上好ましくない。
ピレン−1,6−ジカルボン酸ジメチルエステルを析出させるための冷却温度は、再結晶に用いる有機溶媒の種類に応じて適宜選択されるが、例えばジオキサンを用いる場合、10〜30℃が好ましい。10℃未満であるとジオキサンが凝固する傾向があり、30℃を超えるとピレン−1,6−ジカルボン酸ジメチルエステルの析出量が低下する傾向にある。
通常、分離工程における再結晶の回数は1回とすることができる。なお、用途に応じて再結晶を2回以上繰り返してピレン−1,6−ジカルボン酸エステルの純度を上げることができる。
<反応工程>
下記の反応式(10)で表される加水分解によって、ピレン−1,6−ジカルボン酸ジメチルエステルからピレン−1,6−ジカルボン酸を得る反応工程について以下に説明する。
下記の反応式(10)で表される加水分解によって、ピレン−1,6−ジカルボン酸ジメチルエステルからピレン−1,6−ジカルボン酸を得る反応工程について以下に説明する。
反応工程では、分離工程で得られるピレン−1,6−ジカルボン酸ジメチルエステルをアルカリ含有溶液に添加して、ピレン−1,6−ジカルボン酸ジメチルエステルを加水分解する。アルカリ含有溶液用のアルカリとしては、水酸化ナトリウム、水酸化カリウムなどが使用できる。このうち、製造コストの観点から水酸化カリウムが好ましい。アルカリ含有溶液の溶媒としてはアルコールと水との混合溶媒を使用できる。
ピレン−1,6−ジカルボン酸ジメチルエステルを加水分解する際の温度は、円滑に加水分解を進行させる観点から、80〜100℃とすることが好ましい。
ピレン−1,6−ジカルボン酸ジメチルエステルを加水分解した後、無機酸を加えると黄色い沈殿物(ピレン−1,6−ジカルボン酸)が析出する。無機酸としては、濃塩酸、硝酸などが使用できる。このうち、取り扱い性の観点から濃塩酸が好ましい。無機酸は、沈殿物が析出した分散液のpHが1以下になるまで加えることが好ましい。
沈殿物(ピレン−1,6−ジカルボン酸)が析出した分散液から沈殿物である固形分を分離することによって、ピレン−1,6−ジカルボン酸が得られる。分離方法は、ろ過などの通常の分離方法を用いることができる。ピレン1質量部に対して得られるピレン−1,6−ジカルボン酸の量は、例えば0.10〜0.30質量部である。
次に、第2の製造方法について説明する。
本発明の第2の製造方法は、再結晶によりピレンジアルキル異性体混合物からピレン−1,6−ジアルキルを得る分離工程と、酸化反応によりピレン−1,6−ジアルキルからピレン−1,6−ジカルボン酸を得る酸化工程とを備えるピレン−1,6−ジカルボン酸の製造方法である。
第2の製造方法において、ピレンジアルキル異性体混合物は、臭素化反応によりピレンからジブロモピレン異性体混合物を得る工程と、アルキル化反応によりジブロモピレン異性体混合物からピレンジアルキル異性体混合物を得る工程とによって得られる。
臭素化反応によりピレンからジブロモピレン異性体混合物を得る工程は、上述の第1の製造方法と同一であるので、アルキル化反応によりジブロモピレン異性体混合物からピレンジアルキル異性体混合物を得る工程について以下に説明する。
下記の反応式(11)によって表されるアルキル化反応によって、ジブロモピレン異性体混合物からピレンジプロピル異性体混合物を得ることができる。
まず、ジブロモピレン異性体混合物を有機溶媒に懸濁して懸濁液を作製することができる。有機溶媒としては、ジエチルエーテル、テトラヒドロフランなどを使用できる。このうち、ろ過のしやすさの観点から、ジエチルエーテルが好ましい。有機溶媒の量は、ジブロモピレン異性体混合物1質量部に対して10〜500質量部とすることができる。
次に、作製した懸濁液にアルキルリチウム含有溶液を徐々に滴下し、ジブロモピレンとアルキルリチウムとを反応させると、反応溶液を得ることができる。反応の際、反応溶液の温度は0〜40℃とすることが好ましく、0〜20℃とすることがより好ましい。反応温度が0℃未満又は40℃を超えると、副反応が増加する傾向がある。
アルキルリチウムとしては、n−ブチルリチウム、メチルリチウム、sec−ブチルリチウム、tert−ブチルリチウムなどを用いることができる。このうち、製造コストの観点からn−ブチルリチウムが好ましい。アルキルリチウムの添加量は、未反応の原料を少なくするという観点から、ジブロモピレン1.0molに対して2.0〜2.5molとすることが好ましく、2.0〜2.3molとすることがより好ましく、2.0〜2.1molとすることがさらに好ましい。アルキルリチウムの添加量が、ピレン1.0molに対して2.0mol未満又は2.5molを超える場合には、未反応の原料が増加する傾向がある。
該反応溶液にハロゲン化アルキルを加えて溶液を濃縮すると、ピレンジアルキル異性体混合物を得ることができる。ハロゲン化アルキルとしては、炭素数3〜4の塩化アルキル、臭化アルキルなどを用いることができる。
得られたピレンジアルキル異性体混合物は、不純物を除去するために有機溶剤等で洗浄することが好ましい。
アルキル化反応により、ピレン1質量部に対して、例えば1.0〜1.5質量部のピレンジアルキル異性体混合物を得ることができる。得られたピレンジアルキル異性体混合物は、通常、乾燥質量基準で40〜50質量%のピレン−1,6−ジアルキルと、5〜15質量%のピレン−1,3−ジアルキルと、40〜50質量%のピレン−1,8−ジアルキルとを含有する。
<分離工程>
再結晶によって、ピレンジアルキル異性体混合物からピレン−1,6−ジアルキルを分離する分離工程について以下に説明する。
再結晶によって、ピレンジアルキル異性体混合物からピレン−1,6−ジアルキルを分離する分離工程について以下に説明する。
分離工程では、まず有機溶媒にピレンジアルキル異性体混合物を溶解する。ピレンジアルキル異性体を完全に溶解した後、得られた溶液を冷却するとピレン−1,6−ジアルキルが析出する。この析出物を冷却した液から分離することによって、ピレン−1,6−ジアルキルが得られる。分離には、ろ過などの通常の分離方法を用いることができる。
再結晶に用いる有機溶媒のうち特に好ましいものの一つとして、ジオキサンが挙げられる。この理由は、ピレンジアルキル異性体混合物の溶解性が良く、経験的にピレンジアルキル異性体混合物の再結晶溶媒として相性が良いためと推察する。
ピレンジアルキル異性体混合物1質量部に対して再結晶に用いられる有機溶媒の量は、使用する溶媒の種類にもよるが、ジオキサンを使用する場合は3〜10質量部とすることが好ましい。ピレンジアルキル異性体混合物1質量部に対して3質量部未満であると、ピレンジアルキル異性体混合物が溶解しにくい傾向がある。ピレンジアルキル異性体混合物1質量部に対してジオキサンが10質量部を超えると、析出するピレン−1,6−ジアルキルの量が少なくなり、歩留まりが低下する傾向がある。
ピレンジアルキル異性体混合物を溶解する際の温度は、再結晶に用いる有機溶媒の種類に応じて適宜選択されるが、例えばジオキサンを用いる場合、40〜100℃が好ましい。40℃未満であると、ピレンジアルキル異性体混合物を十分に溶解できない傾向がある。また、100℃は、ほぼジオキサンの沸点に相当するため、これを超える温度に上げるのは実用上好ましくない。
ピレン−1,6−ジアルキルを析出させるための冷却温度は、再結晶に用いる有機溶媒の種類に応じて適宜選択されるが、例えばジオキサンを用いる場合、10〜30℃が好ましい。10℃未満であるとジオキサンが凝固する傾向があり、30℃を超えるとピレン−1,6−ジアルキルが十分に析出しない傾向がある。
通常、分離工程における再結晶の回数は1回とすることができる。なお、用途に応じて再結晶を2回以上繰り返してピレン−1,6−ジアルキルの純度を上げることができる。
<酸化工程>
下記の反応式(12)によって表される酸化反応によって、ピレン−1,6−ジアルキルからピレン−1,6−ジカルボン酸を得る酸化工程について以下に説明する。
下記の反応式(12)によって表される酸化反応によって、ピレン−1,6−ジアルキルからピレン−1,6−ジカルボン酸を得る酸化工程について以下に説明する。
酸化工程では、分離工程で得られるピレン−1,6−ジアルキルを酸化することによって、ピレン−1,6−ジカルボン酸を得ることができる。酸化剤、有機溶媒、塩化トリカプリルメチルアンモニウムを水に混合して溶液を調製し、その溶液にピレン−1,6−ジアルキルを加えると、懸濁液を得ることができる。懸濁液から、例えばろ過によって不純物を除去し、得られた溶液に亜硫酸ナトリウム及び無機酸を加えるとピレン−1,6−ジカルボン酸の沈殿が得られる。
酸化剤、有機溶媒、塩化トリカプリルメチルアンモニウムを混合して調製した溶液に、ピレン−1,6−ジアルキルを加えて懸濁液を生成するための温度は30〜50℃とすることができる。
酸化剤としては、製造コスト及び反応性の観点から、過マンガン酸カリウムを好適に用いることができる。有機溶媒としては、例えばベンゼン、ジクロロメタンなどを用いることができる。
本実施形態では塩化トリカプリルメチルアンモニウムを用いたが、代わりに臭化テトラブチルアンモニウムなどを用いることができる。また、本実施形態では亜硫酸ナトリウムを用いたが、代わりにチオ硫酸ナトリウムなどを用いることができる。無機酸としては、塩酸、硝酸などを用いることができる。
得られた沈殿物(ピレン−1,6−ジカルボン酸)を分離することによって、ピレン−1,6−ジカルボン酸が得られる。分離方法は、ろ過などの通常の分離方法を用いることができる。ピレン1質量部に対して得られるピレン−1,6−ジカルボン酸の量は、例えば0.01〜0.30質量部である。
以上、本発明の好適な実施形態について説明したが、本発明は上記実施形態に何ら限定されるものではない。
以下、実施例及び比較例に基づき本発明をさらに具体的に説明するが、本発明は以下の実施例に何ら限定されるものではない。
(実施例1)
ピレン(東京化成製)6.0gを200mlのクロロホルムに溶かし、溶液を調製した。この溶液にN−ブロモスクシンイミド(和光純薬工業製)10.7gを30分かけて加え、室温で30分攪拌した。溶媒を減圧留去して、得られた反応混合物を1Nの塩酸200mlで2回、次いでエタノール200mlで2回洗浄し、ジブロモピレン異性体混合物10.0gを得た。
ピレン(東京化成製)6.0gを200mlのクロロホルムに溶かし、溶液を調製した。この溶液にN−ブロモスクシンイミド(和光純薬工業製)10.7gを30分かけて加え、室温で30分攪拌した。溶媒を減圧留去して、得られた反応混合物を1Nの塩酸200mlで2回、次いでエタノール200mlで2回洗浄し、ジブロモピレン異性体混合物10.0gを得た。
臭素化反応で得られたジブロモピレン異性体混合物10.0gをジエチルエーテル400mlに懸濁し、その結果得られた懸濁液を氷浴で0℃に冷却した。冷却した懸濁液に、1.6Mのn−ブチルリチウムヘキサン溶液(和光純薬工業製)35mlを30分かけて滴下し、室温で1時間攪拌した。この懸濁液に細かく砕いたドライアイス(10g)を徐々に加えて室温で30分攪拌した。攪拌後、1N塩酸200mlを加えると黄色い沈殿物が析出した。この沈殿物をろ過して採取し、200mlのエタノール、次いで200mlクロロホルムで洗浄し、ピレンジカルボン酸異性体混合物7.1gを得た。
カルボキシル化反応で得られたピレンジカルボン酸異性体混合物7.0gに塩化チオニル(和光純薬工業製)100mlを加えて2時間加熱還流し、溶液を得た。得られた溶液から塩化チオニルを減圧留去してクロロホルム100mlを加え、黄色懸濁液を得た。メタノール10gとトリエチルアミン(和光純薬工業製)10gとを混合した溶液に、上記の黄色懸濁液を10分間かけて滴下し、反応液を得た。この反応液を減圧留去して濃縮し、それによって得られた固形分を1N塩酸200ml、次いでエタノール200mlで洗浄し、ピレンジカルボン酸ジメチルエステル異性体混合物6.0gを得た。このエステル化反応を複数回実施した。
<分離工程>
エステル化反応で得られたピレンジカルボン酸ジメチルエステル異性体混合物8.0gを30mlのジオキサン(和光純薬工業製)に加えて加熱還流させながら1時間混合し、ピレンジカルボン酸ジメチルエステル異性体混合物が完全に溶解した溶液を得た。この溶液を室温で一晩放置して固形分を得た。この固形分をろ過して分離し、2.1gの固形分を得た。
エステル化反応で得られたピレンジカルボン酸ジメチルエステル異性体混合物8.0gを30mlのジオキサン(和光純薬工業製)に加えて加熱還流させながら1時間混合し、ピレンジカルボン酸ジメチルエステル異性体混合物が完全に溶解した溶液を得た。この溶液を室温で一晩放置して固形分を得た。この固形分をろ過して分離し、2.1gの固形分を得た。
得られた固形分の構造を確認するため、該固形分を重水素クロロホルム(和光純薬工業製)に溶解し、600MHzの1H−NMR(VARIAN社製、商品名:INOVA600)を用いて分析した。その結果、得られた固形分の主成分はピレン−1,6−ジカルボン酸ジメチルエステルであることが確認できた。
図1は、実施例1で得られたピレン−1,6−ジカルボン酸ジメチルエステルの1H−NMRチャート図である。なお、横軸はテトラメチルシラン基準の化学シフト(ppm)である。
<反応工程>
水酸化ナトリウム50gを溶解した水50mlとメタノール50mlとの混合溶液に、分離工程で得られたピレン−1,6−ジカルボン酸ジメチルエステルを2.1g加え、10時間加熱還流することによって溶液を得た。得られた溶液にpHが1以下になるまで濃塩酸を加え、黄色の沈殿(固形物)を得た。この固形物を溶液から分離して、水100ml、エタノール100mlおよびクロロホルム100mlで順番に洗浄した。固形物の質量は1.5gであった。
水酸化ナトリウム50gを溶解した水50mlとメタノール50mlとの混合溶液に、分離工程で得られたピレン−1,6−ジカルボン酸ジメチルエステルを2.1g加え、10時間加熱還流することによって溶液を得た。得られた溶液にpHが1以下になるまで濃塩酸を加え、黄色の沈殿(固形物)を得た。この固形物を溶液から分離して、水100ml、エタノール100mlおよびクロロホルム100mlで順番に洗浄した。固形物の質量は1.5gであった。
得られた固形物を重ジメチルスルホキサイド(和光純薬工業製)に溶解して、600MHzの1H−NMR(VARIAN社製、商品名:INOVA600)を用いて分析したところ、固形物はピレン−1,6−ジカルボン酸であることが確認された。
図2は、実施例1で得られたピレン−1,6−ジカルボン酸の1H−NMRチャート図である。なお、横軸はテトラメチルシラン基準の化学シフト(ppm)である。なお、図2のチャート中、1.0ppm及び3.4ppm付近のシグナルは、ピレン−1,6−ジカルボン酸中に微量に残存していたエタノールに、2.5ppm及び3.3ppm付近のシグナルは測定溶媒に含まれるジメチルスルホキサイドにそれぞれ帰属される。図3は、図2のピレン−1,6−ジカルボン酸の1H−NMRチャート図の8.3〜9.5ppmの範囲を拡大した図である。
(比較例1)
エステル化反応まで実施例1と同様に行って、ピレンジカルボン酸ジメチルエステル異性体混合物を得た。ピレンジカルボン酸ジメチルエステル異性体混合物8.0gをジオキサン30mlに溶解して溶液を調整した。
エステル化反応まで実施例1と同様に行って、ピレンジカルボン酸ジメチルエステル異性体混合物を得た。ピレンジカルボン酸ジメチルエステル異性体混合物8.0gをジオキサン30mlに溶解して溶液を調整した。
シリカゲルカラム(富士シリアル化学(株)製、商品名:BW−820MH)を用いて該溶液からピレン−1,6−ジカルボン酸ジメチルエステルの分離を試みた。展開溶媒はクロロホルムを用いた。しかし、ピレン−1,6−ジカルボン酸ジメチルエステルは分離できなかった。
(比較例2)
エステル化反応まで実施例1と同様に行って、ピレンジカルボン酸ジメチルエステル異性体混合物を得た。ピレンジカルボン酸ジメチルエステル異性体混合物8.0gをジオキサン30mlに溶解して溶液を調整した。
エステル化反応まで実施例1と同様に行って、ピレンジカルボン酸ジメチルエステル異性体混合物を得た。ピレンジカルボン酸ジメチルエステル異性体混合物8.0gをジオキサン30mlに溶解して溶液を調整した。
リサイクル分取HPLC用カラム(日本分析工業(株)製、商品名:JAIGEL−SIL−SH)を用いて、該溶液からピレン−1,6−ジカルボン酸ジメチルエステルの分離を試みた。展開溶媒はクロロホルムを用いた。しかし、ピレン−1,6−ジカルボン酸ジメチルエステルは分離できなかった。
(比較例3)
エステル化反応まで実施例1と同様に行って、ピレンジカルボン酸ジメチルエステル異性体混合物を得た。ピレンジカルボン酸ジメチルエステル異性体混合物80mgをエタノール3mlに混合した。混合液を加熱還流下で1時間混合し、溶解性を評価した。また、ピレンジカルボン酸ジメチルエステル異性体混合物を溶解後、室温で一晩放置して結晶析出性を評価した。溶解性及び結晶析出性は表1に示すとおりであった。
エステル化反応まで実施例1と同様に行って、ピレンジカルボン酸ジメチルエステル異性体混合物を得た。ピレンジカルボン酸ジメチルエステル異性体混合物80mgをエタノール3mlに混合した。混合液を加熱還流下で1時間混合し、溶解性を評価した。また、ピレンジカルボン酸ジメチルエステル異性体混合物を溶解後、室温で一晩放置して結晶析出性を評価した。溶解性及び結晶析出性は表1に示すとおりであった。
(比較例4、5)
有機溶媒の種類を表1に示す通りに変更したこと以外は、比較例3と同様にして溶解性を評価した。溶解性及び結晶析出性は表1に示すとおりであった。
有機溶媒の種類を表1に示す通りに変更したこと以外は、比較例3と同様にして溶解性を評価した。溶解性及び結晶析出性は表1に示すとおりであった。
ピレンジカルボン酸ジメチルエステル異性体混合物はジオキサンを用いた場合に完全に溶解し、アセトニトリルを用いた場合に半分程度溶解した。そのうち、室温で一晩放置後に結晶(ピレン−1,6−ジカルボン酸ジメチルエステル)が析出したのは、ジオキサンを用いた場合だけであった。
(比較例6)
カルボキシル化反応まで実施例1と同様に行って、ピレンジカルボン酸異性体混合物を得た。ピレンジカルボン酸異性体混合物1.0gをジオキサン30mlに混合した。混合液を加熱還流下で1時間混合して溶解性を評価した。溶解性は表2に示すとおりであった。
カルボキシル化反応まで実施例1と同様に行って、ピレンジカルボン酸異性体混合物を得た。ピレンジカルボン酸異性体混合物1.0gをジオキサン30mlに混合した。混合液を加熱還流下で1時間混合して溶解性を評価した。溶解性は表2に示すとおりであった。
(比較例7〜9)
有機溶媒の種類を表2に示す通りに変更したこと以外は、比較例6と同様にして溶解性を評価した。溶解性は表2に示すとおりであった。
有機溶媒の種類を表2に示す通りに変更したこと以外は、比較例6と同様にして溶解性を評価した。溶解性は表2に示すとおりであった。
ピレンジカルボン酸異性体混合物は、いずれの有機溶媒にも殆ど溶解しなかった。
(実施例2)
実施例1と同様にしてジブロモピレン異性体混合物10.0gを得た。臭素化反応で得られたジブロモピレン異性体混合物10.0gをジエチルエーテル400mlに懸濁し、その結果得られた懸濁液を氷浴で0℃に冷却した。冷却した懸濁液に、1.6Mのn−ブチルリチウムヘキサン溶液(和光純薬工業製)35mlを30分かけて滴下して室温で1時間攪拌し、さらに2−ブロモプロパン(東京化成製)7.0gを10分間かけて滴下して室温で3時間攪拌し、溶液を得た。該溶液を1N塩酸(和光純薬工業製)200ml、飽和食塩水200mlで順に洗浄した。洗浄済みの溶液を濃縮して得られた固形物をヘキサン(和光純薬製)で洗浄し、褐色固体のピレンジイソプロピル異性体混合物8.0gを得た。
実施例1と同様にしてジブロモピレン異性体混合物10.0gを得た。臭素化反応で得られたジブロモピレン異性体混合物10.0gをジエチルエーテル400mlに懸濁し、その結果得られた懸濁液を氷浴で0℃に冷却した。冷却した懸濁液に、1.6Mのn−ブチルリチウムヘキサン溶液(和光純薬工業製)35mlを30分かけて滴下して室温で1時間攪拌し、さらに2−ブロモプロパン(東京化成製)7.0gを10分間かけて滴下して室温で3時間攪拌し、溶液を得た。該溶液を1N塩酸(和光純薬工業製)200ml、飽和食塩水200mlで順に洗浄した。洗浄済みの溶液を濃縮して得られた固形物をヘキサン(和光純薬製)で洗浄し、褐色固体のピレンジイソプロピル異性体混合物8.0gを得た。
<分離工程>
アルキル化反応で得られたピレンジイソプロピル異性体混合物8.0gを30mlのジオキサン(和光純薬工業製)に加えて加熱還流させながら1時間混合し、ピレンジイソプロピル異性体混合物が完全に溶解した溶液を得た。この溶液を室温で一晩放置して固形分を得た。この固形分をろ過して分離し、2.2gのピレン−1,6−ジイソプロピルを得た。
アルキル化反応で得られたピレンジイソプロピル異性体混合物8.0gを30mlのジオキサン(和光純薬工業製)に加えて加熱還流させながら1時間混合し、ピレンジイソプロピル異性体混合物が完全に溶解した溶液を得た。この溶液を室温で一晩放置して固形分を得た。この固形分をろ過して分離し、2.2gのピレン−1,6−ジイソプロピルを得た。
<酸化工程>
ベンゼン(和光純薬製)50mlと塩化トリカプリルメチルアンモニウム(東京化成製)0.10gと過マンガン酸カリウム(和光純薬製)6.0gとを100mlの水に加えて40℃で1時間攪拌し、溶液を得た。この溶液に、分離工程で得られたピレン−1,6−ジイソプロピル2.2gを10分間かけて加えて40℃で5時間攪拌し、それによって得られた懸濁液をろ過してろ液を得た。得られたろ液に亜硫酸ナトリウム(和光純薬製)10gおよび1N−塩酸100mlを加えて沈殿(固形物)を得た。得られた固形物をろ過して分離し、0.20gの固形物を得た。
ベンゼン(和光純薬製)50mlと塩化トリカプリルメチルアンモニウム(東京化成製)0.10gと過マンガン酸カリウム(和光純薬製)6.0gとを100mlの水に加えて40℃で1時間攪拌し、溶液を得た。この溶液に、分離工程で得られたピレン−1,6−ジイソプロピル2.2gを10分間かけて加えて40℃で5時間攪拌し、それによって得られた懸濁液をろ過してろ液を得た。得られたろ液に亜硫酸ナトリウム(和光純薬製)10gおよび1N−塩酸100mlを加えて沈殿(固形物)を得た。得られた固形物をろ過して分離し、0.20gの固形物を得た。
得られた固形物を重ジメチルスルホキサイド(和光純薬工業製)に溶解し、600MHzの1H−NMR(VARIAN社製、商品名:INOVA600)を用いて分析したところ、固形物はピレン−1,6−ジカルボン酸であることが確認された。
(比較例10)
アルキル化反応まで実施例2と同様に行って、ピレンジイソプロピル異性体混合物を得た。得られたピレンジイソプロピル異性体混合物1.0gを、エタノール30mlに混合した。混合液を、加熱還流下で1時間混合して溶解性を評価した。また、ピレンジイソプロピル異性体混合物を溶解後、室温で一晩放置して結晶析出性を評価した。溶解性及び結晶析出性は表3に示すとおりであった。
アルキル化反応まで実施例2と同様に行って、ピレンジイソプロピル異性体混合物を得た。得られたピレンジイソプロピル異性体混合物1.0gを、エタノール30mlに混合した。混合液を、加熱還流下で1時間混合して溶解性を評価した。また、ピレンジイソプロピル異性体混合物を溶解後、室温で一晩放置して結晶析出性を評価した。溶解性及び結晶析出性は表3に示すとおりであった。
(比較例11〜13)
有機溶媒の種類を表3に示す通りに変更したこと以外は、比較例10と同様にして溶解性及び結晶析出性を評価した。溶解性及び結晶析出性は、表3に示すとおりであった。
有機溶媒の種類を表3に示す通りに変更したこと以外は、比較例10と同様にして溶解性及び結晶析出性を評価した。溶解性及び結晶析出性は、表3に示すとおりであった。
ピレンジイソプロピル異性体混合物は、ジオキサンを用いた場合及びクロロホルムを用いた場合に完全に溶解し、アセトニトリルを用いた場合に半分程度溶解した。そのうち、室温で一晩放置後に結晶(ピレン−1,6−ジイソプロピル)が析出したのは、ジオキサンを用いた場合だけであった。
Claims (6)
- 再結晶により、下記一般式(1)で表されるピレン−1,6−ジカルボン酸エステルと、下記一般式(2)で表されるピレン−1,3−ジカルボン酸エステル又は下記一般式(3)で表されるピレン−1,8−ジカルボン酸エステルの一方又は双方と、を含有するピレンジカルボン酸エステル異性体混合物から前記ピレン−1,6−ジカルボン酸エステルを分離する分離工程と、
前記ピレン−1,6−ジカルボン酸エステルを加水分解してピレン−1,6−ジカルボン酸を得る反応工程と
を備える、ピレン−1,6−ジカルボン酸の製造方法。
[式中、R1及びR2は各々独立に炭素数1〜4のアルキル基を示す。]
[式中、R3及びR4は各々独立に炭素数1〜4のアルキル基を示す。]
[式中、R5及びR6は各々独立に炭素数1〜4のアルキル基を示す。] - 前記ピレンジカルボン酸エステル異性体混合物は、
ピレンの臭素化反応により1,6−ジブロモピレンと1,3−ジブロモピレンと1,8−ジブロモピレンとを含有するジブロモピレン異性体混合物を得る工程と、
前記ジブロモピレン異性体混合物のカルボキシル化反応によりピレン−1,6−ジカルボン酸とピレン−1,3−ジカルボン酸とピレン−1,8−ジカルボン酸とを含有するピレンジカルボン酸異性体混合物を得る工程と、
前記ピレンジカルボン酸異性体混合物のエステル化反応により前記一般式(1)で表されるピレン−1,6−ジカルボン酸エステルと、前記一般式(2)で表されるピレン−1,3−ジカルボン酸エステルと、前記一般式(3)で表されるピレン−1,8−ジカルボン酸エステルと、を含有するピレンジカルボン酸エステル異性体混合物を得る工程と、
を経て得られたものである、請求項1に記載のピレン−1,6−ジカルボン酸の製造方法。 - 前記分離工程は、前記ピレンジカルボン酸エステル異性体混合物をジオキサンに溶解させた後、再結晶により前記ピレン−1,6−ジカルボン酸エステルを析出させて分離する工程である、請求項1又は2に記載のピレン−1,6−ジカルボン酸の製造方法。
- 再結晶により、下記一般式(4)で表されるピレン−1,6−ジアルキルと、下記一般式(5)で表されるピレン−1,3−ジアルキル又は下記一般式(6)で表されるピレン−1,8−ジアルキルの一方又は双方と、を含有するピレンジアルキル異性体混合物から前記ピレン−1,6−ジアルキルを分離する分離工程と、
前記ピレン−1,6−ジアルキルの酸化反応によりピレン−1,6−ジカルボン酸を得る酸化工程と、を備えるピレン−1,6−ジカルボン酸の製造方法。
[式中、R7及びR8は各々独立に炭素数3又は4のアルキル基を示す。]
[式中、R9及びR10は各々独立に炭素数3又は4のアルキル基を示す。]
[式中、R11及びR12は各々独立に炭素数3又は4のアルキル基を示す。] - 前記ピレンジアルキル異性体混合物は、
ピレンの臭素化反応により1,6−ジブロモピレンと1,3−ジブロモピレンと1,8−ジブロモピレンとを含有するジブロモピレン異性体混合物を得る工程と、
前記ジブロモピレン異性体混合物のアルキル化反応により前記一般式(4)で表されるピレン−1,6−ジアルキルと前記一般式(5)で表されるピレン−1,3−ジアルキルと前記一般式(6)で表されるピレン−1,8−ジアルキルとを含有するピレンジアルキル異性体混合物を得る工程と、
を経て得られたものである、請求項4に記載のピレン−1,6−ジカルボン酸の製造方法。 - 前記分離工程は、前記ピレンジアルキル異性体混合物をジオキサンに溶解させた後、再結晶により前記ピレン−1,6−ジアルキルを析出させて分離する工程である、請求項4又は5に記載のピレン−1,6−ジカルボン酸の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006303030A JP2008120695A (ja) | 2006-11-08 | 2006-11-08 | ピレン−1,6−ジカルボン酸の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006303030A JP2008120695A (ja) | 2006-11-08 | 2006-11-08 | ピレン−1,6−ジカルボン酸の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2008120695A true JP2008120695A (ja) | 2008-05-29 |
Family
ID=39505840
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006303030A Pending JP2008120695A (ja) | 2006-11-08 | 2006-11-08 | ピレン−1,6−ジカルボン酸の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2008120695A (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102295523A (zh) * | 2011-07-25 | 2011-12-28 | 天津市科莱博瑞化工有限公司 | 1,6-二溴芘的制备方法 |
| CN105523881A (zh) * | 2016-01-15 | 2016-04-27 | 中节能万润股份有限公司 | 一种1,6-二烷基芘的制备方法 |
| CN108164391A (zh) * | 2018-01-19 | 2018-06-15 | 李现伟 | 一种联苯类化合物及其在制备1,3-二溴芘中的应用 |
-
2006
- 2006-11-08 JP JP2006303030A patent/JP2008120695A/ja active Pending
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102295523A (zh) * | 2011-07-25 | 2011-12-28 | 天津市科莱博瑞化工有限公司 | 1,6-二溴芘的制备方法 |
| CN105523881A (zh) * | 2016-01-15 | 2016-04-27 | 中节能万润股份有限公司 | 一种1,6-二烷基芘的制备方法 |
| CN108164391A (zh) * | 2018-01-19 | 2018-06-15 | 李现伟 | 一种联苯类化合物及其在制备1,3-二溴芘中的应用 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2011042690A (ja) | ナプロキセンのニトロキシアルキルエステル | |
| EP2462098B1 (en) | Process for the preparation of derivatives of 1-(2-halobiphenyl-4-yl)-cyclopropanecarboxylic acid | |
| RU2711358C1 (ru) | Простой способ получения авибактама | |
| JP4852206B2 (ja) | シクロブタンテトラカルボン酸二無水物化合物の製造法 | |
| JP2008120695A (ja) | ピレン−1,6−ジカルボン酸の製造方法 | |
| JP2005104964A (ja) | アダマンチルエステル類の精製方法 | |
| JP2011042647A (ja) | 光学活性ニペコタミドの製造方法 | |
| CA2903708A1 (en) | A process for the preparation of 2-amino-1,3-propane diol compounds and salts thereof | |
| JP2001233854A (ja) | N−ヒドロキシ環状イミドの製造方法 | |
| JP2009073739A (ja) | 医薬品中間体として許容しうる高純度な光学活性1−アリール−1,3−プロパンジオールの製造方法 | |
| EP1280775A1 (en) | TOSYLATE SALTS OF 4-(p-FLUOROPHENYL)-PIPERIDINE-3-CARBINOLS | |
| JP2013119518A (ja) | (s)−2−ベンジル―3−(シス−ヘキサヒドロ−2−イソインドリニルカルボニル)プロピオン酸ベンジルの製造方法 | |
| JPS59186942A (ja) | 粗製1,4−ジヒドロキシ−2−ナフトエ酸又はその塩の精製法 | |
| JP2000344696A (ja) | アダマンタンジオール類の製造方法 | |
| JP2007254293A (ja) | α−メチレン−β−アルキル−γ−ブチロラクトンの製造法 | |
| JP2000198779A (ja) | 3―アルキルフラバノノ―ル誘導体の精製法 | |
| EP4067331A1 (en) | Preparation method for 2,2'-bis(carboxymethoxy)-1,1'-binaphthyl | |
| JP2000212136A (ja) | 1,3―ビス(アミノフェノキシベンゼン)の再結晶法 | |
| JP5395989B2 (ja) | 六員環ラクトン(メタ)アクリル酸エステルの精製方法 | |
| JP2019214542A (ja) | β−ヒドロキシラクトン(メタ)アクリル酸エステルの製造方法 | |
| JPH093059A (ja) | α−メチル−2−チオフェン酢酸誘導体の製法 | |
| JP2001122823A (ja) | 5−ブロモ−イソフタル酸ジアルキル化合物の製造方法 | |
| JPS61103853A (ja) | 2―フェニルプロピオン酸エステル類の光学分割法 | |
| JP2006273731A (ja) | 2−(10、11−ジヒドロ−10−オキシジベンゾ〔b、f〕チエピン−2−イル)プロピオン酸の製造方法 | |
| JPH0267237A (ja) | 光学活性1,1,1−トリフルオロ−2−アルカノールの製造法 |