JP2004536070A - 抗糖尿病および抗肥満症薬として有用な置換されたアゾール酸誘導体および方法 - Google Patents
抗糖尿病および抗肥満症薬として有用な置換されたアゾール酸誘導体および方法 Download PDFInfo
- Publication number
- JP2004536070A JP2004536070A JP2002592871A JP2002592871A JP2004536070A JP 2004536070 A JP2004536070 A JP 2004536070A JP 2002592871 A JP2002592871 A JP 2002592871A JP 2002592871 A JP2002592871 A JP 2002592871A JP 2004536070 A JP2004536070 A JP 2004536070A
- Authority
- JP
- Japan
- Prior art keywords
- compound
- mmol
- embedded image
- alkyl
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 Cc1c(CCOc2cccc(CC(C(C(O)=O)=[N+])=[N+]Nc3ccc(*)cc3)c2)nc(-c2ccccc2)[o]1 Chemical compound Cc1c(CCOc2cccc(CC(C(C(O)=O)=[N+])=[N+]Nc3ccc(*)cc3)c2)nc(-c2ccccc2)[o]1 0.000 description 9
- NQRYJNQNLNOLGT-UHFFFAOYSA-N C1CCNCC1 Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N C1CCOCC1 Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- VDFVNEFVBPFDSB-UHFFFAOYSA-N C1COCOC1 Chemical compound C1COCOC1 VDFVNEFVBPFDSB-UHFFFAOYSA-N 0.000 description 1
- GLUUGHFHXGJENI-UHFFFAOYSA-N C1NCCNC1 Chemical compound C1NCCNC1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 1
- YNAVUWVOSKDBBP-UHFFFAOYSA-N C1NCCOC1 Chemical compound C1NCCOC1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N C1NCCSC1 Chemical compound C1NCCSC1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- BNBQRQQYDMDJAH-UHFFFAOYSA-N C1Oc2ccccc2OC1 Chemical compound C1Oc2ccccc2OC1 BNBQRQQYDMDJAH-UHFFFAOYSA-N 0.000 description 1
- VELXUKZIJDAYRH-SPYQMNCUSA-N CC1C2(CCOc3ccc(Cc4c[n](C)cc4C(O)=O)cc3)N=C(c3ccccc3)O[C@@H]12 Chemical compound CC1C2(CCOc3ccc(Cc4c[n](C)cc4C(O)=O)cc3)N=C(c3ccccc3)O[C@@H]12 VELXUKZIJDAYRH-SPYQMNCUSA-N 0.000 description 1
- SWHCRARXERKZKL-FLWNBWAVSA-N CCOC(/C(/N)=C(\Cc(cc1)ccc1OCCc1c(C)[o]c(-c2ccccc2)n1)/N(C)Cc1ccccc1)=O Chemical compound CCOC(/C(/N)=C(\Cc(cc1)ccc1OCCc1c(C)[o]c(-c2ccccc2)n1)/N(C)Cc1ccccc1)=O SWHCRARXERKZKL-FLWNBWAVSA-N 0.000 description 1
- YKFKFFKBCJBROH-UHFFFAOYSA-N COc1cccc(C(c2c[n](-c3ccccc3)nc22)OC2=O)c1 Chemical compound COc1cccc(C(c2c[n](-c3ccccc3)nc22)OC2=O)c1 YKFKFFKBCJBROH-UHFFFAOYSA-N 0.000 description 1
- MSSDTZLYNMFTKN-UHFFFAOYSA-N O=CN1CCNCC1 Chemical compound O=CN1CCNCC1 MSSDTZLYNMFTKN-UHFFFAOYSA-N 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Child & Adolescent Psychology (AREA)
- Pulmonology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pain & Pain Management (AREA)
- Dermatology (AREA)
- Reproductive Health (AREA)
- Vascular Medicine (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
構造:
【化1】
[式中、QはCまたはNであり;R2a、R2b、R2c、X1〜X7、R1、R2、R3、R3a、R4、A、Y、m、およびnは本明細書で定義される]を有し、抗糖尿病剤、脂質低下剤、および高肥満症剤として有用な化合物を提供する。本発明はさらに、ヒトを含む哺乳類におけるペルオキシソーム増殖因子活性化受容体−γ(PPARγ)およびペルオキシソーム増殖因子活性化受容体−α(PPARα)の同時阻害による、肥満症および異脂肪血症の治療方法を提供する。
【化1】
[式中、QはCまたはNであり;R2a、R2b、R2c、X1〜X7、R1、R2、R3、R3a、R4、A、Y、m、およびnは本明細書で定義される]を有し、抗糖尿病剤、脂質低下剤、および高肥満症剤として有用な化合物を提供する。本発明はさらに、ヒトを含む哺乳類におけるペルオキシソーム増殖因子活性化受容体−γ(PPARγ)およびペルオキシソーム増殖因子活性化受容体−α(PPARα)の同時阻害による、肥満症および異脂肪血症の治療方法を提供する。
Description
【0001】
本出願は、米国仮出願第60/294,380号(2001年5月30日出願)の優先権を主張するもので、これらは本明細書に引用される。
【0002】
(技術分野)
本発明は、血中ブドウ糖レベル、トリグリセリドレベル、インスリンレベルおよび非エステル型脂肪酸(NEFA)レベルを調節して、糖尿病および肥満症の治療に特に有効となる新規な置換されたアゾール酸誘導体に関するものであり、且つ、かかる置換された酸誘導体単独または他の抗糖尿病剤および/または抗高脂血症剤および/または他の治療剤と組み合わせて用いて、糖尿病(特に2型糖尿病)、並びに高血糖、高インスリン血症、高脂血症、肥満症、アテローム性動脈硬化症および関連する疾患を治療する方法に関するものである。本発明はまた、ヒトを含む哺乳類におけるペルオキシソーム増殖因子活性化受容体−γ(PPARγ)の同時阻害およびペルオキシソーム増殖因子活性化受容体−α(PPARα)の刺激による、肥満症および異脂肪血症の治療方法に関するものである。本発明はさらに、抗肥満症、インスリン感受性および循環器疾患のためになるPPARγアンタゴニスト活性により、遺伝子発現が脂肪組織において変動する標的遺伝子のリストを提供する。
【0003】
(背景技術)
ヒトを含む哺乳類において、脂肪細胞は、栄養過剰の数倍で、トリグリセリドの形状でエネルギーを蓄える(参照:Lowell, Cell, 99: 239-242, 1999)。飢餓の間、蓄えられたトリグリセリドは、栄養およびエネルギーの要求を補充するため、脂肪細胞において脂肪酸に分解される。この条件において、過剰の脂肪組織の蓄積[脂肪細胞(分化)となるように前駆細胞(前脂肪細胞)を補充するか、および/または先に存在する脂肪細胞(肥厚および肥大)の増大によるかのいずれかにより行われる]は、肥満症およびインスリン耐性になる(参照:Lowell, Cell, 99: 239-242, 1999)。その理由は、肥大化した脂肪細胞(これらは比較的低い代謝活性であると考えられる)が過剰量の脂肪酸およびサイトカインを産生し、代わるがわるインスリン情報伝達並びに骨格筋および脂肪細胞(これらは2つの主要なブドウ糖利用組織である)へのブドウ糖取り込みを減少するように作用するためである(参照:Hotamisligil, et al., Science, 259: 87-90, 1993; Lowell, Cell, 99: 239-242, 1999)。肥満の個体は、不適切なエネルギー消費、「骨格筋、肝臓および血漿における高脂質含量」、インスリン耐性、高血圧症、アテローム性動脈硬化症および循環器疾患をしばしば被っている(参照:Rosenbaum et al., New. Eng. J. Med. 337: 396-407, 1997, 参照:Friedman, Nature, 404: 632-634, 2000)。重厚な脂肪貯蔵の枯渇を伴った脂肪異栄養症候群の患者に見られるような症状は、体重の減少、並びに血漿、肝臓および骨格筋における脂質含量の増加をもたらし、代わるがわる該患者にインスリン耐性および2型糖尿病を罹らせることとなる(参照:Arioglu et. al., Annals of Int. Med, 2000, 133: 263-274)。これらの異常の第一の原因は、脂質の安全な貯蔵に有効な比較的少量の脂肪組織のためと思われる。
【0004】
肥満症は、先進国共通の臨床上の問題であり、また急速に先進国における主要な健康の関心事となってきている。体重過剰の個体は、例えば異脂肪血症、インスリン耐性および2型糖尿病のようないくつかの代謝障害をしばしば被っている。これらの個体はまた、高血圧症、アテローム性動脈硬化症および循環器疾患のリスクの増加もしばしば被っている(参照:Friedman, Nature, 404: 632-634, 2000)。
【0005】
ペルオキシソーム増殖因子活性化受容体(PPAR)は、転写因子を調節するリガンドの核ホルモン受容体ファミリーのメンバーである(参照:Willson, et al., J. Med. Chem., 43 : 527-550, 2000, Kersten et al., Nature, 405: 421424, 2000)。3つのPPARイソ型、PPARγ、PPARα、およびPPARδは、ヒトを含む様々な哺乳類から単離されている。これらの受容体は、分類として、その結合相手のRXRαと偏性のヘテロダイマーを形成し、食餌由来の長鎖脂肪酸、脂肪酸代謝物によって、および合成剤によって活性化される(参照:Willson, et al., J. Med. Chem., 43: 527-550, 2000)。ブドウ糖および脂質の代謝経路における遺伝子の調節により、PPARがヒトを含む哺乳類においてブドウ糖および脂質の恒常性を維持する主要な役割をするということが、現在よく報告されている。
【0006】
PPARγは、前脂肪細胞補充並びに成熟した脂肪細胞への分化および成熟した脂肪細胞中の脂質の貯蔵の主要な制御因子である(参照:Tontonoz et al., Current Biology, 571-576, 1995)。PPARγの活性化剤は、前脂肪細胞分化、成熟した脂肪細胞中の脂質貯蔵を促進し、インスリン感受性抗糖尿剤として作用する(参照:Tontonoz et al., Current Biology, 571-576, 1995; Lehmann et al., J. Biol. Chem., 270: 12953-12956, 1995; Nolan et al. New. Eng. J. Med., 331: 1188-1193 ; Inzucchi et al., New Eng. J. Med., 338: 867-872, 1998, Willson, et al., J. Med. Chem.: 43: 527-550, 2000, Kersten et al., Nature, 405: 421424, 2000)。しかしながら、PPARγ誘導型抗糖尿病活性は、しばしば動物モデルおよびヒトにおいていくらかの体重増大を伴う。PPARγ発現は、肥満個体の脂肪組織において十分に高められ(参照:Vidal-Puig et al., J. Clinical Investigation, 99: 2416-2422, 1997)、構造的に活性なPPARγを産生する突然変異は重厚な肥満症につながる(参照:Ristow et al., New England J. Med., 339: 953-959, 1998)。PPARγ発現の部分的な消失は、ヘテロ接合性PPARγノックアウトマウス(参照:Kubota et al. Mol. Cell; 4: 597-609, 1999)における食餌誘導型肥満症への耐性となり、アミノ酸の12位でプロリンのアラニンへの変化を伴ったヒトにおけるより低い肥満度指数となる(参照:Deeb et al Nature Genetics, 20: 284-287, 1998)。ドミナントネガティブ突然変異によるヒトPPARγ活性の比較的より厳しい消失は、受容体に結合するリガンドを破壊し、高脂血症、脂肪肝およびのインスリン耐性となる(参照:Barroso et al. Nature, 402, 860-861, 1999)。該異常の主な原因は、脂質の安全な貯蔵に有用な脂肪組織が比較的少量であるためと思われる。これらのマウスおよびヒトでの知見で、従って、肥満症の該誘導および/または進展におけるPPARγの役割を示し、PPARγの阻害が体脂肪蓄積および肥満症における低下になると示唆される。これらの知見でまた、より高い血漿遊離脂肪酸および高脂血症および脂肪肝の進行およびインスリン耐性にもなるようであると示唆される。
【0007】
PPARαイソ型は、脂肪酸合成、脂肪酸酸化および脂質代謝経路における遺伝子を調節する(参照:Isseman and Green, Nature, 347: 645-649, 1990; Torra et al., Current Opinion in Lipidology, 10 : 151-159, 1999; Kersten et al., Nature, 405: 421424, 2000)。PPARαアゴニスト(例えば、フェノフィブラート、ゲムフィブロジル)の処置は、肝臓および筋肉における脂肪酸酸化を高め、肝臓における脂肪酸およびトリグリセリド合成 を減少させ、血漿トリグリセリドレベルを下げる(参照:Kersten et al., Nature, 405: 421424, 2000)。高トリグリセリドおよび低HDL−コレステロールである患者に、PPARαアゴニストで処置することで、血漿のHDL−コレステロールの増加、血漿のトリグリセリドの減少および第一次および第二次心臓事象(primary and secondary cardiac events)の両方の減少となる(参照:Balfour et al., Drugs. 40: 260-290, 1990; Rubins et al., New Eng. J. Med., 341: 410-418, 1999)。
【0008】
従って、PPARγアンタゴニスト活性およびPPARαアゴニスト活性を、単一の両作用化合物にまたは1つの製剤に組み合わせることで、PPARγを阻害し、高脂血症、脂肪肝およびインスリン耐性を引き起こさずに肥満症を治療することが可能である。本発明は、2つの異なる活性、すなわちPPARγアンタゴニスト活性およびPPARαアゴニスト活性を組み合わせて、高脂血症およびインスリン耐性を引き起こすことなく、体脂肪蓄積および体重を減らす肥満症の新規な治療方法を示す。本発明は、肥満の、高脂血症のおよびインスリン耐性2型糖尿病の患者が、両PPARγアンタゴニスト/PPARαアゴニスト、またはPPARγアンタゴニストおよびPPARαアゴニストを脂質低下剤および抗糖尿病剤と組み合わせて、治療され得ることを提案する。本発明はまた、その発現が抗肥満症、インスリン感受性および循環器疾患のためになるPPARγアンタゴニスト活性により脂肪組織において変動するような、標的遺伝子のリストを提供する。
【0009】
本発明に従い、構造I:
【化1】
[式中、
mは0、1または2であり;n=0、1または2であり;
QはCまたはNであり;
Aは(CH2)x(xは1〜5である)であるか;またはAは(CH2)x 1(x1は2〜5である)で、アルケニル結合またはアルキニル結合が該鎖のいずれかに結合しているか;またはAは−(CH2)x 2−O−(CH2)x 3−(x2は0〜5で、x3は0〜5である、但し少なくともx2およびx3の1つは0でない)であり;
X1はCHまたはNであり;
X2はC、N、OまたはSであり;
X3はC、N、OまたはSであり;
X4はC、N、OまたはSであるが、但しX2、X3およびX4のうち少なくとも1つはNであり;
X5はC、N、OまたはSであり;
X6はCまたはNであり;
X7はC、N、OまたはSであるが、但しX5、X6またはX7のうち少なくとも1つはNであり;
X1〜X7の各々が上記のように定義される場合において、CはCHを含み得;
R1はHまたはアルキルであり;
R2はH、アルキル、アルコキシ、ハロゲン、アミノまたは置換されたアミノであり;
R2a、R2bおよびR2cは同一または異なって、H、アルキル、アルコキシ、ハロゲン、アミノまたは置換されたアミノから選択され;
R3およびR3aは同一または異なって、H、アルキル、アリールアルキル、アリールオキシカルボニル、アルキルオキシカルボニル、アルキニルオキシカルボニル、アルケニルオキシカルボニル、アリールカルボニル、アルキルカルボニル、アリール、ヘテロアリール、シクロヘテロアルキル、ヘテロアリールカルボニル、ヘテロアリール−ヘテロアリールアルキル、アルキルカルボニルアミノ、アリールカルボニルアミノ、ヘテロアリールカルボニルアミノ、アルコキシカルボニルアミノ、アリールオキシカルボニルアミノ、ヘテロアリールオキシカルボニルアミノ、ヘテロアリール−ヘテロアリールカルボニル、アルキルスルホニル、アルケニルスルホニル、ヘテロアリールオキシカルボニル、シクロヘテロアルキルオキシカルボニル、ヘテロアリールアルキル、アミノカルボニル、置換されたアミノカルボニル、アルキルアミノカルボニル、アリールアミノカルボニル、ヘテロアリールアルケニル、シクロヘテロアルキル−ヘテロアリールアルキル、ヒドロキシアルキル、アルコキシ、アルコキシアリールオキシカルボニル、アリールアルキルオキシカルボニル、アルキルアリールオキシカルボニル、アリールヘテロアリールアルキル、アリールアルキルアリールアルキル、アリールオキシアリールアルキル、ハロアルコキシアリールオキシカルボニル、アルコキシカルボニルアリールオキシカルボニル、アリールオキシアリールオキシカルボニル、アリールスルフィニルアリールカルボニル、アリールチオアリールカルボニル、アルコキシカルボニルアリールオキシカルボニル、アリールアルケニルオキシカルボニル、ヘテロアリールオキシアリールアルキル、アリールオキシアリールカルボニル、アリールオキシアリールアルキルオキシカルボニル、アリールアルケニルオキシカルボニル、アリールアルキルカルボニル、アリールオキシアルキルオキシカルボニル、アリールアルキルスルホニル、アリールチオカルボニル、アリールアルケニルスルホニル、ヘテロアリールスルホニル、アリールスルホニル、アルコキシアリールアルキル、ヘテロアリールアルコキシカルボニル、アリールヘテロアリールアルキル、アルコキシアリールカルボニル、アリールオキシヘテロアリールアルキル、ヘテロアリールアルキルオキシアリールアルキル、アリールアリールアルキル、アリールアルケニルアリールアルキル、アリールアルコキシアリールアルキル、アリールカルボニルアリールアルキル、アルキルアリールオキシアリールアルキル、アリールアルコキシカルボニルヘテロアリールアルキル、ヘテロアリールアリールアルキル、アリールカルボニルヘテロアリールアルキル、ヘテロアリールオキシアリールアルキル、アリールアルケニルヘテロアリールアルキル、アリールアミノアリールアルキルまたはアミノカルボニルアリールアリールアルキルから独立して選択され;
YはCO2R4(R4はH若しくはアルキル、またはプロドラッグエステルである)であるか、またはYはC−連結した1−テトラゾール、構造P(O)(OR4a)R5(R4aはHまたはプロドラッグエステルであり、R5はアルキルまたはアリールである)のホスフィン酸または構造P(O)(OR4a)2のホスホン酸であり;
(CH2)x、(CH2)x 1、(CH2)x 2、(CH2)x 3、(CH2)m、および(CH2)nは適宜、1、2または3個の置換基で置換され得る]
を有し、そのすべての立体異性体、そのプロドラッグエステル、およびその医薬的に許容される塩を含む置換された酸誘導体が提供される。
【0010】
好ましくは、構造IA:
【化2】
を有する本発明の式Iの化合物である。
【0011】
より好ましくは、構造IB:
【化3】
を有する本発明の式Iの化合物である。
上記化合物で、最も好ましいのは、
R2a、R2bおよびR2cが各々Hであり;
R1がアルキル(好ましくはCH3)であり;
x2が1〜3で、x3が0であり;
R2がHであり;
mが0であるか、(CH2)mがCH2またはCHOHまたはCH−アルキルであり;
X2、X3、およびX4が、総計1、2または3個の窒素を表し;
(CH3)nが結合またはCH2であり;
R3がアリール、アリールアルキルまたはヘテロアリール(例えば、チオフェンまたはチアゾール、最も好ましくはフェニルまたは以下で置換されたフェニル;該置換基はアルキル、ポリハロアルキル、ハロ、アルコキシ、好ましくはCF3およびCH3である)であり;
R3aが好ましくはHまたはアルキルである。
【0012】
本発明の好ましい化合物には、以下が含まれる。
【化4】
【化5】
【0013】
本発明は、ある単一の分子中に両PPARγアンタゴニスト/PPARαアゴニスト活性を有するという発見について記述する。本発明は、重厚な糖尿病の、高脂血症のおよび肥満のdb/dbマウスへの両PPARγアンタゴニスト/PPARαアゴニストの投与が、ブドウ糖レベルを変化させることなく、血漿のトリグリセリドおよび遊離脂肪酸レベルを減少させるということを示す。本発明は、食餌で誘発された肥満のマウスへの両PPARγアンタゴニスト/PPARαアゴニストの投与が、高脂血症および/またはインスリン耐性を引き起こすことなく、体脂肪含量を減らし、肝臓の脂肪を減らすということを示す。本発明は、抗肥満症、インスリン感受性および循環器疾患のためになるPPARγアンタゴニスト活性により、遺伝子発現が脂肪組織において変動する標的遺伝子のリストを提供する。
【0014】
従って、本発明の1つの目的は、ヒトを含む哺乳類における肥満症の新規な治療方法で、PPARγの阻害およびPPARαの活性化を同時にする単一の化合物または化合物の組み合わせの治療上の有効量を、かかる治療が必要な哺乳類に投与することからなる新規な治療方法を提供する。
【0015】
本発明の別の目的は、ヒトを含む哺乳類での代謝症候群(肥満症、インスリン耐性および異脂肪血症)の治療方法で、以下の化合物、すなわち:PPARγを拮抗し、PPARα活性を活性化する化合物または化合物の組み合わせ;抗糖尿病性化合物(例えば、これらに限らないが、インスリン、メトホルミン、インスリン増感剤、スルホニル尿素、aP2阻害剤、SGLT−2阻害剤);脂質低下剤(例えば、これらに限らないが、スタチン類、フィブラート類、ナイアシンACAT阻害剤、LCAT活性化剤、胆汁酸隔離剤)および体重減少剤(例えば、これらに限らないが、オルリスタット、シブトラミン、aP2阻害剤、アディポネクチン)のうち2個以上からなるいずれかの組み合わせの治療上の有効量を、かかる治療が必要な哺乳類に投与することからなる治療方法を提供する。
【0016】
本発明の別の目的は、標的遺伝子(例えば、HMGic、グリセロール−3−PO4−デヒドロゲナーゼ、Gタンパク質共役受容体26、脂肪酸輸送タンパク質、アジポフィリンおよびケラチノサイト脂肪酸結合タンパク質)のリストを提供することであり、該標的遺伝子の発現は、PPARγアンタゴニストおよび両PPARγアンタゴニスト/PPARαアゴニストの投与により、または他の方法により、抗肥満症の効果を得るよう変えられ得る。
【0017】
本発明の別の目的は、標的遺伝子(例えば、PAI−1、レニン、アンジオテンシノーゲン前駆体)のリストを提供することであり、該標的遺伝子の発現は、PPARγアンタゴニストおよび両PPARγアンタゴニスト/PPARαアゴニストの投与により、または他の方法により、循環器疾患に対する有益な効果を得るよう変えられ得る。
【0018】
本発明の別の目的は、医薬的に許容される担体と、PPARγの阻害およびPPARαの活性化を同時にする化合物または化合物の組み合わせの治療上の有効量とからなる、肥満症を治療する医薬組成物を提供することである。
【0019】
本発明の別の目的は、肥満症、インスリン耐性および/または異脂肪血症の治療用で、医薬的に許容される担体、並びにPPARγの阻害およびPPARαの活性化を同時にする化合物または化合物の組み合わせの治療上の有効量、並びに抗糖尿病性化合物、脂質低下剤および体重減少剤からなる医薬組成物を提供することである。
【0020】
さらに、本発明によって、糖尿病、特に2型糖尿病、および関連する疾患(例えば、インスリン耐性、高血糖、高インスリン血症、脂肪酸またはグリセロールの血中レベルの上昇、高脂血症、肥満症、高トリグリセリド血症、炎症、シンドロームX、糖尿病性合併症、代謝異常症侯群、アテローム性動脈硬化症、および関連する疾患)を治療するための方法で、構造Iの化合物の治療上の有効量を治療が必要な患者に投与する方法を提供する。
【0021】
さらに、本発明によって、初期悪性病変(例えば、乳房のインシトゥーの管の癌および乳房のインシトゥーの小葉癌)、前悪性病変(例えば、乳房の線維腺腫および前立腺上皮内異常増殖(PIN)、脂肪肉腫および様々な他の上皮性腫瘍(乳房、前立腺、大腸、卵巣、胃および肺を含む))、過敏性大腸症候群、クローン病、胃潰瘍、および骨多孔症および増殖性疾患(例えば乾癬)を治療するための方法で、構造Iの化合物の治療上の有効量を治療が必要な患者に投与する方法を提供する。
【0022】
さらに、本発明によって、糖尿病および上記または以下で規定される関連疾患を治療する方法で、構造Iの化合物と別のタイプの抗糖尿病剤および/または抗高脂血症剤、および/または脂質調節剤および/または他のタイプの治療剤との組み合わせの治療上の有効量を治療が必要なヒトの患者に投与する方法を提供する。
【0023】
本発明の上記方法において、構造Iの化合物は、抗糖尿病剤(処置の方法に依存する)に対する重量比が、約0.01:1〜約100:1、好ましくは約0.5:1〜約10:1の範囲内で用いられる。
【0024】
「シンドロームX」または代謝異常症侯群(Johanson, J. Clin. Endocrinol. Metab., 1997, 82, 727-734および他の公知文献に詳述される)に関して集合的に言及される症状、疾病、および疾患には、高血糖および/または前糖尿病性インスリン耐性症候群を含み、高インスリン血症、異脂肪血症、および障害性ブドウ糖耐性(2型糖尿病に進行し得る)を生じる初期のインスリン耐性状態の特徴があり、高血糖(糖尿病性合併症に進行し得る)の特徴がある。
【0025】
用語「糖尿病および関連疾患」とは、2型糖尿病、1型糖尿病、障害性ブドウ糖耐性、肥満症、高血糖、シンドロームX、代謝異常症侯群、糖尿病性合併症および高インスリン血症をいう。
【0026】
「糖尿病性合併症」に関して集合的に言及される症状、疾病、および疾患には、網膜症、神経障害および腎障害、および他の公知の糖尿病の合併症が含まれる。
【0027】
本明細書で用いられる用語「他のタイプの治療剤」とは、1個以上の抗糖尿病剤(式Iの化合物以外)、1個以上の抗肥満症剤、および/または1個以上の脂質低下剤、1個以上の脂質調節剤(抗アテローム性動脈硬化症剤を含む)、および/または1個以上の抗血小板剤、1個以上の高血圧症治療剤、1個以上の抗癌薬、1個以上の関節炎治療剤、1個以上の抗骨多孔症剤、1個以上の抗肥満剤、1個以上の免疫調節性疾患の治療剤、および/または1個以上の拒食症治療剤をいう。
【0028】
本明細書で用いられる用語「脂質調節」剤とは、LDLを低下し、および/またはHDLを上昇させ、および/またはトリグリセリドを低下させ、および/または総コレステロールを低下させる薬剤、および/または脂質障害の治療的な処置の他の公知の機構をいう。
【0029】
(発明の詳細な説明)
PPARγは、前脂肪細胞の補充および成熟した脂肪細胞への分化の主要な調節剤である(参照:Tontonoz et al., Current Biology, 571-576, 1995)。PPARγの活性化剤は、前脂肪細胞の分化、成熟した脂肪細胞中の脂質貯蔵を促進し、インスリンとして作用し、抗糖尿病剤を感作する(参照:Tontonoz et al., Current Biology, 571-576, 1995 ; Lehmann et al., J. Biol. Chem., 270: 12953-12956, 1995; Nolan et al. New. Eng. J. Med., 331: 1188-1193 ; Inzucchi et al., New Eng. J. Med., 338: 867-872, 1998, Willson, et al., J. Med. Chem. : 43: 527-550, 2000, Kersten et al., Nature, 405: 421424, 2000)。しかしながら、PPARγで誘発される抗糖尿病活性は、動物モデルおよびヒトにおいていくらかの体重増大をしばしば伴う。最近の発見で、PPARγの阻害が体脂肪蓄積および肥満症における減少になると示唆されている(参照:Vidal-Puig et al., J. Clinical Investigation, 99: 2416-2422, 1997 ; Deeb et al Nature Genetics, 20: 284-287, 1998 ; Kubota et al. Mol. Cell; 4: 597-609, 1999 ; Barroso et al. Nature; 402, 860-861, 1999)。しかしながら、かかる減少で、より高い血漿遊離脂肪酸および高脂血症および発達脂肪肝およびインスリン耐性になるようである。PPARαのイソ型は、脂肪酸合成、脂肪酸酸化および脂質代謝経路における遺伝子を調節する(参照:Issenman and Green, Nature, 347: 645-649, 1990 ; Torra et al., Current Opinion in Lipidology, 10: 151-159, 1999; Kersten et al., Nature, 405: 421424, 2000)。PPARαアゴニスト(例えば、フェノフィブラート、ゲムフィブロジル)処理は、肝臓および筋肉における脂肪酸酸化を高め、肝臓における脂肪酸およびトリグリセリド合成を減少させ、血漿トリグリセリドを減少させる(参照:Kersten et al., Nature, 405: 421424, 2000)。高トリグリセリドおよび低HDL−コレステロールの患者への、PPARαアゴニストでの処置は、血漿HDL−コレステロールの増加、血漿トリグリセリドの減少および1°および2°両方の心臓病事故の縮小につながる(参照:Balfour et al., Drugs. 40: 260-290, 1990 ; Frick et al., New Eng. J. Med., 317 : 1237-1245 ; Rubins et al., New Eng. J. Med., 341: 410-418, 1999)。従って、PPARγアンタゴニストおよびPPARαアゴニストの単一の両活性化合物または組み合わせにおける、PPARγアンタゴニスト活性およびPPARαアゴニスト活性を合わせることによって、高脂血症、脂肪肝およびインスリン耐性を引き起こすことなく、安全なPPARγ阻害および肥満症の治療が可能である。
【0030】
化合物Yは本明細書の実施例1に概説された反応式で合成された化合物である。図1−A、1−Bに図示されているように、化合物Yは高い親和性(IC50=69 nM)でヒトPPARγリガンド結合ドメインと強く結合した。同様に化合物Yは精製ヒトPPARαリガンド結合ドメインとも強く結合した(IC50=69 nM)。関連するPPARγリガンド結合の研究において、ロシグリタゾン(真正のPPARγアゴニスト)でIC50=250 nM、GW0072(真正のPPARγアンタゴニスト)でIC50=280 nMを得た。PPARαリガンド結合の研究において、GW−2331(PPARα選択的アゴニスト)でIC50=410 nMを得た。このような精製リガンド結合ドメインでのインビトロリガンド結合の研究より、PPARγおよびPPARαの両方に強く結合する化合物Yの能力が示される。しかしながら、強く結合する化合物(すなわちリガンド)が、アゴニスト(活性化リガンド)およびアンタゴニスト(受容体を不活性化するリガンド)として作用し得ることは、転写因子の核ホルモン受容体ファミリーとしてよく知られている(PPARはこのファミリーのメンバーである)。
【0031】
図2に図示するように、化合物Yはマウス前脂肪細胞3T3L−1に加えられる場合、(該細胞からのグリセロール放出によって測定されながら)成熟した脂質を負荷した脂肪細胞への分化を誘発するロシグリタゾン(PPARγアゴニスト)との競合的阻害を示す。マウス3T3−L−前脂肪細胞は、ホルモンシグナル(例えばインスリン、デキサメタゾン )およびPPARγアゴニスト(例えばロシグリタゾン)に応答し、成熟した脂肪細胞へ分化し、および脂質を蓄積することが知られている。PPARγは脂肪細胞の分化過程の主要なトリガーであると考えられている(参照:Tontonoz et al., Current Biology, 571-576, 1995)。化合物YはPPARγの強力なリガンドであるが、ロシグリタゾンで誘発される分化の競合的阻害を示すので、従ってPPARγのアンタゴニストであると示唆される。分化の阻害のED50=9.9μMということは、化合物Yが前脂肪細胞分化の適当な阻害剤であることを示している。対照的に、ED50=0.585μMは、PPARγアンタゴニストであるGW0072として得られた(参照:Oberfield et al, Proc. Nat. Acad. Sci., 96: 6102-6106, 1999)。
【0032】
図3に図示したように、化合物YのPPARγアンタゴニスト活性は、第二の細胞系で確認された。内在性のPPARγの発現を示す確立されたCV−1細胞(霊長類の腎臓起源)は、PPAR応答型内在性アルカリフォスファターゼ(SEAP)レポーター遺伝子と安定にトランスフェクトされた。先の研究でのように、化合物Yはロシグリタゾン(PPARγアゴニスト)依存活性によって、すなわちCV−1細胞におけるSEAPレポーター遺伝子発現の誘導によって競合的に阻害された。SEAP遺伝子のロシグリタゾン誘発性トランス活性化の特異的な阻害であるED50=1.5μMは、化合物YがPPARγのアンタゴニストであることを再び示す。PPARγアンタゴニストであるGW0072の研究(参照:Oberfield et al, Proc. Nat. Acad. Sci., 96: 6102-6106, 1999)において、またCV−1細胞におけるロシグリタゾンで媒介されるSEAP遺伝子の誘発(阻害のED50=0.37μM)を用量依存的に阻害し、データの信頼性を立証した。
【0033】
図4に図示するように、化合物Yはヒト肝臓細胞HepG2中のSEAPレポーター遺伝子のPPARα依存トランス活性化を用量依存的に刺激したことから、それはPPARαのアゴニストであることが示された。内在のPPARα遺伝子を発現するHepG2細胞(ヒト肝臓起源)は、PPAR応答性SEAPレポーター遺伝子で安定にトランスフェクトされた。処置の際の化合物Yは、PPARαトランス活性化のEC50=0.587μMのHepG2細胞において、SEAP遺伝子発現を用量依存的に刺激した。本研究において、BMS−250773(PPARα選択的薬剤)は、EC50=0.063μMでSEAPレポーター遺伝子のPPARα依存性トランス活性化を用量依存的に刺激し、ロシグリタゾン(PPARγアゴニスト)はほとんど活性化を示さなかった。
【0034】
このように、図1、2、3、4に図示するインビトロ PPARγおよびPPARαリガンド結合の研究、並びにPPARγおよびPPARα依存細胞に基づくトランス活性化の研究は、化合物YがPPARγおよびPPARαの両方の強力なリガンドであることを示すが、しかしながら、PPARγへのアンタゴニスト活性およびPPARαへのアゴニスト活性を示すものである。これらの知見は、化合物Yが単一分子中に両方の(両)PPARγアンタゴニスト活性およびPPARαアゴニスト活性を有する分子の新規な分類に属することを示唆する。
【0035】
【表1】
表1に示すように、化合物Yはインビボのいくつかの遺伝子の発現のレベルで、PPARγアンタゴニストおよびPPARαアゴニストの両方の効果を示す。化合物YのインビボのPPARγアンタゴニストおよびPPARαアゴニスト効果を示すために、肥満の糖尿病性db/dbマウスは、化合物Y、ロシグリタゾン(真正のPPARγアゴニスト)およびBMS−化合物Cで処置した。(この化合物はPPARαおよびPPARγの両方へのアゴニスト活性を有する。研究の終結において、白色脂肪組織(WAT)は収集され、全RNAは調製され、標的遺伝子の発現の効果について分析された。これらの分析で、多くの遺伝子の発現が化合物Yの処置によって特異的に変わることが示され、化合物YのインビボのPPARγアンタゴニスト活性が確認された。例えば、(1)脂肪細胞分化を防止するHMGicの発現は化合物Yで誘発されるが、ロシグリタゾンまたはBMS−化合物Cでは誘発されない、(2)脂肪細胞分化を促進するグリセロール3−PO4デヒドロゲナーゼの発現は、化合物Yで阻害されるが、ロシグリタゾンおよびBMS−化合物Cでは阻害されない、(3)脂肪酸の細胞への運搬を促進する脂肪酸輸送タンパク質の発現は、化合物Yでは影響を受けないが、ロシグリタゾンおよびBMS−化合物Cで誘発された、また(4)ボンベシン受容体に関連するオーファンGPCR26の発現は化合物Yで影響を受けないが、ロシグリタゾンおよびBMS−化合物Cで誘発された。これらの分析でまた、多くの他の遺伝子の発現が化合物YおよびBMS−化合物Cによってだけ誘発され、ロシグリタゾンでは誘発されず、インビボの化合物YのPPARαアゴニスト活性が確認された。かかる遺伝子の例には、アジポフィリンおよびケラチノサイト脂肪酸結合タンパク質が含まれ、これらの遺伝子の遺伝子産物は細胞内脂肪酸輸送に含まれる。)
【0036】
このように、遺伝子発現プロフィール研究で、両PPARγアンタゴニスト/PPARαアゴニスト化合物Yの、インビボのPPARγアンタゴニストおよびPPARαアゴニスト活性が確認される。さらに、これらの研究でまた、PPARγアンタゴニストおよび/または両PPARγアンタゴニスト/PPARαアゴニストの投与によって、脂肪の(脂肪)組織中、脂肪細胞分化に影響を与える遺伝子(例えば、HMGic、グリセロール3−PO4デヒドロゲナーゼ、脂肪酸輸送タンパク質および新規なオーファンGタンパク質共役受容体26レベル)を変化させて、肥満症を治療する方法も示される。これらの研究ではまた、PPARαアゴニストおよび/または両PPARγアンタゴニスト/PPARαアゴニストの投与によって、脂肪の(脂肪)組織中、アジポフィリンおよびケラチノサイト脂肪酸結合タンパク質レベルを変化させて、肥満症を治療する方法も示される。
【0037】
【表2】
表2に示すように、両PPARγアンタゴニスト/PPARαアゴニスト化合物Yで処置した肥満の糖尿病性db/dbマウスの白色脂肪組織(WAT)の発現プロフィール分析で、循環器疾患の開発で役割を担うことで知られるいくつかの遺伝子の発現に実質的有益な変化が示される。脂肪の(脂肪)組織は、PAI−1合成の主要な場所、血栓症の危険因子、アンジオテンシノーゲン前駆体、高血圧症およびレニンの危険因子、高血圧症の危険因子である(参照:Ahima and Flier, TEM, 11: 327-332, 2000)。PAI−1およびアンジオテンシノーゲン前駆体遺伝子発現の阻害、およびレニン遺伝子の発現の変化の欠如により、選択的に化合物Yによって、PPARγアンタゴニスト活性が再度確認され、両PPARγアンタゴニスト/PPARαアゴニスト(例えば、化合物Y)で、ヒトを含む肥満の哺乳類を処置する血管性の有益な効果が示される。
【0038】
【表3】
表3に示すように、両PPARγアンタゴニスト/PPARαアゴニスト化合物Yによる肥満の糖尿病性db/dbマウスの処置は、血漿ブドウ糖の大きな変化はなく、血漿トリグリセリドおよび遊離脂肪酸レベルにおいて大きな減少の結果となる。先に述べたように、脂質の変化および糖血症の症状は、PPARγ活性を減らす2つの重要な潜在的な関心事である。本明細書で記載した研究に基づき、肥満の哺乳類が両PPARγアンタゴニスト/PPARαアゴニストで安全に処置され得ると結論される。血漿トリグリセリドおよび遊離脂肪酸の減少は、化合物YのPPARγアゴニスト活性のためであると考えられる。
【0039】
【表4】
表4に示すように、食餌によって誘発した肥満のマウスを1日1回、l0mg/kg/日で3週間、両PPARγアンタゴニスト/PPARαアゴニスト化合物Yで処置して、体脂肪量の十分な15%の減少、および化合物Yの有益な効果に示される除脂肪体重の対応する14%増加の結果となった。両PPARγアンタゴニスト/PPARαアゴニスト化合物Yでの処置による体脂肪量の減少は、脂肪細胞の増大を減少させ、体脂肪量の蓄積を減少させるPPARγ活性の阻害の結果のためであると最も考えられる。体重の大きな減少は本研究では観察されないが、減少した体脂肪量および除脂肪体重の補償する増加は(かかる補償は、脂肪組織蓄積がないヒト脂肪異栄養患者では観察されない)、両PPARγアンタゴニスト/PPARαアゴニスト化合物Yでの重要で有益な治療効果を表している。PPARαアゴニスト活性が除脂肪体重発達の増進に貢献することは可能であり、おそらく脂肪酸代謝経路遺伝子の誘発によってまたは未知のメカニズムで筋肉タンパク質合成の誘発によって起こる。
【0040】
【表5】
表5に示すように、食餌によって誘発した肥満のマウスを両PPARγアンタゴニスト/PPARαアゴニスト化合物Yで処置しても、血漿脂質(遊離脂肪酸、トリグリセリドおよびコレステロール)および糖血症のパラメーター(ブドウ糖およびインスリン)にはほとんど変化が生じなかった。先に述べたように、脂質の変化および糖血症の症状は、PPARγ活性を減らす2つの潜在的な関心事である。本明細書で記載した研究に基づき、肥満の糖尿病哺乳類(ヒトを含む)における体脂肪量の安全な減少は、両PPARγアンタゴニスト/PPARαアゴニストの投与によって可能であると結論される。この特徴は、脂肪異栄養患者およびPPARγ遺伝子における重篤な突然変異を有する患者において観察される高脂血症および高血糖と対照をなす。
【0041】
【表6】
表6に示すように、食餌によって誘発した肥満のマウスを両PPARγアンタゴニスト/PPARαアゴニスト化合物Yで処置すると、肝臓フェノタイプに改善が生じる。肥満のマウスにおいて、肥満のヒトのように、肝脂質レベルが上昇する。しばしば、このことは血漿肝臓酵素ALTレベルの増加を伴い、肝臓の損傷を示唆する。両PPARγアンタゴニスト/PPARαアゴニスト化合物Yによる処置で、肝臓トリグリセリド含量の実質的な減少があり、統計的な有意性には及ばなかったが、血漿肝臓酵素ALTレベルにおける有意な減少を伴った。これらの両変化は、PPARαで媒介される脂肪酸酸化を刺激し、脂質合成を減少させて脂質含量を減らすように刺激するという結果として、肝臓機能における改善を示す(参照:Torra et al., Current Opinion in Lipidology, 10: 151-159, 1999; Kersten et al., Nature, 405: 421424, 2000)。
【0042】
従って、本発明は新規な両活性のPPARγアンタゴニスト/PPARαアゴニスト剤の発見を示すものである。本発明は、両PPARγアンタゴニスト/PPARαアゴニストの投与のより肥満症を治療する原理の医薬的な証明を提供する。本発明によれば、単一分子中のPPARγアンタゴニスト活性およびPPARαアゴニスト活性の組み合わせ、または薬剤中のPPARγアンタゴニスト活性およびPPARαアゴニスト活性の組み合わせは、肥満の個体における脂質および/または血糖のコントロールのさらなる悪化を伴うことなく、肥満症の治療を提供する。
【0043】
本発明は、PPARγアンタゴニスト、または両PPARγアンタゴニスト/PPARαアゴニストまたはPPARαアゴニストによる治療により得られる、抗肥満症を達成するようにその発現が修飾される遺伝子(例えば、HMGic、グリセロール−PO4デヒドロゲナーゼ、脂肪酸輸送タンパク質、Gタンパク質共役受容体26、アジポフィリン、ケラチノサイト脂肪酸結合タンパク質)および心臓血管を達成するようにその発現が修飾される遺伝子(例えば、アンジオテンシノーゲン、PAI−1、レニン)のリストの同定を提供する。
【0044】
本発明はまた、両PPARγアンタゴニスト/PPARαアゴニストまたはPPARαアゴニストの投与により、肝臓障害を治療する方法を提供する。
【0045】
本発明はまた、単一の薬剤を含む薬理的組成物または2つの薬剤の組み合わせの投与により、ヒトを含む哺乳類の肥満症を治療する方法:すなわち同時に、
(1)PPARγタンパク質の活性、または(2)PPARγ遺伝子の発現、(3)活性化補助因子の結合または(4)PPARγ調節される標的遺伝子の発現(または上記のいずれかの組み合わせ)を減少し、並びに
(1)PPARαタンパク質の活性、または(2)PPARα遺伝子の発現、または(3)活性化補助因子の結合または(4)PPARα調節される標的遺伝子の発現(または上記のいずれかの組み合わせ)を増加する方法も提供する。これらの変化の生じた生成物には、これらに限らないが、(1)体重増加の抑制、(2)体重減少、(3)体脂肪量の特異的減少、(4)除脂肪体重の増加、(5)体脂肪量/除脂肪量比の変化、(7)肝臓の脂質の減少および肝臓機能の改善のいずれかの組み合わせを含み得る。
【0046】
本発明はまた、肥満の患者における体重、インスリン耐性、2型糖尿病、高脂血症および循環器疾患をコントロールするために、両PPARγアンタゴニスト/PPARαアゴニストと、以下の薬剤との組み合わせの使用を含む治療方法を提供する:抗糖尿病剤(例えば、これに限らないが、メトホルミン、スルホニル尿素、インスリン、インスリン増感剤、aP2阻害剤、SGLT2阻害剤)、肝臓ブドウ糖排出に影響を与える薬剤、PPARαアゴニスト(例えば、これに限らないが、フェノフィブラートおよびゲムフィブロジル)のような脂質低下剤およびHMG−CoAリダクターゼ阻害剤(例えば、これに限らないが、プラバスタチン、ロバスタチン、シンバスタチンおよびアトルバスタチン)、ナイアシン、ACT阻害剤、LCAT活性化剤、胆汁酸隔離剤および他の抗肥満剤(例えば、これに限らないが、オルリスタット、シブトラミン、P2阻害剤、アディポネクチン)。
【0047】
本発明の式Iの化合物は、以下の一般的合成反応式、並びに当業者に用いられる関連する公開された文献の方法に従って調製し得る。これらの反応の具体的な試薬および方法は、以下の記述および実施例に示されている。以下の反応式にある保護および脱保護は、当該技術分野で一般的に知られている方法で実施し得る(参照:例えば、Greene, T. W. and Wuts, P. G. M., Protecting Groups in Organic Synthesis, 3rd Edition, 1999 [Wiley])。
【0048】
本発明の化合物の合成に要求される鍵中間体の合成は、反応式1に記載されている。アルコール体1(R5(CH2)x 2OH)(最も好ましいものの1つは、2−フェニル−5−メチル−オキサゾール−4−エタノール)は、標準的なミツノブ反応の条件(例えば、Mitsunobu, 0., Synthesis, 1981, 1)下、ヒドロキシアリール−またはヘテロアリール−アルデヒド体2とカップリングして、鍵中間体のアルデヒド3が得られる。あるいは、アルコール体1は標準的な条件下そのメタンスルホン酸エステルに変換され得 ;メシレート体4は次いで、ヒドロキシアリール−またはヘテロアリール−アルデヒド体2をアルキル化して、アルデヒド体3を供給するのに用いられ得る。
【0049】
反応式2に、2−アリール(ヘテロアリール)−4−カルボキシ−トリアゾール化合物Iの一般的合成を記載する。適当に保護されたオキシ安息香酸クロリド体またはオキシフェニル酢酸クロリド体5のメルドラム酸との塩基存在下での処理で、対応するクルードなメルドラム酸付加体6が得られ、アニリンとすぐに反応してβ−ケトアニリド体7が得られる(Synthesis, 1992, 1213-1214)。β−ケトアミド体7は、亜硝酸(塩基/亜硝酸ナトリウムからインシトゥーで産生する)との反応、続く酸処理により、対応するα−オキシム−β−ケトアミド体8を得る(引用文献: Hamanaka, E. S., et al, WO9943663)。β−ケト−アミド体8は次いで、適当に置換されたヒドラジン体9と縮合し、対応するβ−ヒドラゾン−アミド体10を得る。中間体10の酸による処理で、目的の2−置換された−4−カルボキサミド−トリアゾール体11を得る(引用文献: Hamanaka, E. S., et al, WO9943663)。トリアゾール−アニリド体11のフェノール性保護基の脱保護で、対応するフェノール体12を得る。フェノール−トリアゾール体12は次いで、適当なアルコール体1と標準的なミツノブ反応条件下(例えば、Mitsunobu, O., Synthesis, 1981, 1)カップリングして、目的のアルキル化したトリアゾール−アミド体13を得る。あるいは、該フェノール体は塩基性条件下メタンスルホン酸エステル4とカップリングして、アルキル化したトリアゾール−アミド体13を得ることができる(引用文献:Cheng, P. T. W., et. al., WO0121602)。このアニリド体の続く塩基性による脱保護で、本発明の目的の2−置換された4−カルボキシトリアゾール体IIを得る。
【0050】
反応式3には、2−アリール−4−カルボキシトリアゾール体Iの製造のための、反応式2に示すものの補足的なアプローチを示す。適当に保護されたヒドロキシアリールまたはヒドロキシヘテロアリールカルボン酸14は、1)塩基の存在下メシレート体4と、または2)標準的なミツノブ条件下アルコール体1とのいずれかで処理して、カルボン酸への脱保護後、鍵となるアルキル化した酸中間体15を得る。酸化合物15の対応する酸クロリド体16への変換は、シュウ酸クロリドを用いて行う。酸クロリド体16のメルドラム酸との処理で、対応する付加体17を得て、次いですぐにアニリンと反応してβ−ケトアニリド体18を得る。次いでβ−ケトアニリド体18の亜硝酸(塩基/NaNO2からインシトゥーで産生する)による処理で、対応するβ−ケト−α−オキシミノ−アニリド体19を得て、次いで適当に置換されたヒドラジン体9と反応して、中間体β−ヒドラゾン−アミド体20を得る。次いでオキシム−ヒドラゾン体20の酸による環化で、アリールトリアゾールアニリド体21を得る。最終的に、塩基によるアニリド体の加水分解で、本発明の目的の2−置換された−4−カルボキシトリアゾール体IIAを得る。
【0051】
反応式4に、1−置換された4−カルボキシトリアゾール体IIの合成を示す。β−ケトアニリド体18のp−トルエンスルホニルアジド(Padwa, A., et al, J. Org. Chem., 1997, 62, 6842)との処理で、対応するβ−ケト−α−ジアゾ−アニリド体21を得る。β−ケト−α−ジアゾ−アニリド体21の適当に置換されたアミン体22とのルイス酸で介される反応で、対応する1−置換された−4−アミド トリアゾール体23を得る(Ohno, M., et al, Synthesis, 1993, 793)。トリアゾール−アニリド体23のフェノール官能基の脱保護で、フェノール体23を得る。次いでフェノール−トリアゾール体23のアルキル化を、標準的なミツノブ反応条件下(例えば、Mitsunobu, O., Synthesis, 1981, 1)アルコール体1により実施し、対応するアルキル化したトリアゾール−アミド体を得る。あるいはフェノール−トリアゾール体23は、塩基性条件下メタンスルホン酸エステル4とカップリングして、同じアルキル化したトリアゾール−アミド体を得ることができる。その後塩基性によるカルボン酸への脱保護で、本発明の目的の1−置換された−4−カルボキシトリアゾール体IIIを得る。
【0052】
反応式5に、位置異性体の1−置換された−5−カルボキシトリアゾール体IIIおよび1−置換された−4−カルボキシトリアゾール体IVの合成を記載する。アルデヒド体3は、塩基性/アニオン性条件下適当に保護されたプロパルギル酸(propargylic acid)と反応して(J. Org. Chem., 1980, 45, 28)、対応するアセチレンアルコール付加体25が得られる。アセチレンアルコール体25は次いで、標準的な文献の条件下(Czernecki, S., et al, J. Org. Chem., 1989, 54, 610)脱酸素して、アセチレンエステル体26を得る。熱条件下(Can. J. Chem., 1980, 58, 2550)のアセチレンエステル体26の適当に置換されたアリールアジド体27との双極環化付加、その後のカルボン酸官能基への脱保護により、本発明の目的のアリールトリアゾール酸化合物IVおよびVが得られる。
【0053】
反応式6に、トリアゾール酸化合物IVおよびV、並びにヒドロキシトリアゾール酸化合物VIおよびVIIの製造のわずかに修正した工程を示す。アセチレンアルコール付加体25は熱条件下すぐに適当に置換されたアジド体27と双極環化付加反応して、対応する位置異性体のヒドロキシトリアゾールエステル体28および29が得られ、次いで脱保護されて本発明のヒドロキシトリアゾール酸VIおよびVIIがそれぞれ得られる。あるいはヒドロキシトリアゾールエステル体28および29は、脱酸素化および脱保護されて、本発明のトリアゾール酸化合物IVおよびVが得られる。
【0054】
反応式7に、1−置換された−4−カルボキシピラゾール化合物VIIIの合成を記載する。保護されたフェノール−アルコール体30は標準的な文献の方法(Tetrahedron Lett., 1986, 42, 2725)で、対応するクロリド体31に変換される。保護されたシアノ酢酸体32は次いで、塩基の存在下クロリド体31でアルキル化されて、シアノ酢酸体33が得られる。シアノ酢酸体33の脱保護で、シアノ酢酸体34が得られる。亜硝酸(亜硝酸ナトリウムおよび酸からインシトゥーで産生する)の存在下、シアノ酢酸体34の適当に置換されたヒドラジン体9との処理で、対応するシアノ−ヒドラゾン体35が得られる(Skorcz, J. A., et al, J. Med. Chem., 1966, 9, 656)。塩基存在下、シアノヒドラゾン体35の適当に保護したアクリレート体36との反応で、鍵となるアリール−ピラゾールエステル中間体37が得られる(Kim, Y. H., et al, Tetrahedron Lett., 1996, 37, 8771)。1)ピラゾール体37のフェノールの保護基の除去、2)生じたフェノール体の塩基性条件下メシレート体4によるアルキル化、および3)カルボン酸への脱保護を含む3工程で、本発明の1−アリール−3−置換された−4−カルボキシピラゾール体VIIIが得られる。
【0055】
反応式8に、位置異性体の1−置換された−5−置換された−4−カルボキシピラゾール化合物IXの合成を示す。保護されたフェノール−酸クロリド体5は、塩基性条件下メルドラム酸で処理されて、対応する付加体が得られ、適当なアルコール体R3OHと反応して、β−ケトエステル体38が得られる。β−ケト−エステル体38のジメチルホルムアミドジメチルアセタールとの処理で、α−エナミノ−β−ケト−エステル体39が得られる(Almansa, C., et al, J. Med. Chem., 1997, 40, 547)。α−エナミノ−β−ケト−エステル体39の適当に置換されたヒドラジン体9による反応、続く分子内環化反応で、アリール−N−ピラゾールエステル体40が得られる。1)化合物40のフェノールの保護基の除去、2)生じたフェノール体のメシレート体4によるアルキル化、および3)カルボン酸への脱保護を含む3工程で、本発明のN−置換されたピラゾール酸IXが得られる。
【0056】
位置異性体のカルボキシピラゾール化合物Xの合成を、反応式9に示す。アルデヒド体3の(適当に置換されたアルキニル金属試薬41との)処理で、アセチレンアルコール付加体42が得られる。アルコール体42は次いで、熱条件下(Kato, T., et al, Chem. Pharm. Bull., 1975, 20, 2203)ケテン二量体で処理して、アセト酢酸エステル体43が得られる。標準的な条件下(引用文献)のアセト酢酸エステル体43の塩素化で、α−クロロ−β−ケトエステル体44が得られる。熱条件下α−クロロ−β−ケトエステル体44の適当に置換されたジアゾ化合物45による処理で、クロロヒドラゾン体46が得られる(Garantic, L., et al, Synthesis, 1975, 666)。クロロヒドラゾン体46の塩基による熱分子内環化付加反応により、次いでピラゾール−ラクトン体47が得られる(Garantic, L., et al, Synthesis, 1975, 666)。ピラゾール−ラクトン体47の付随する環の開環/脱酸素化は、多くの異なる反応条件下(TMSCl/NaIまたはZn/NH4OH ; Sabitha, G., Synth. Commun., 1998, 28, 3065)実施され、ピラゾール酸化合物48が得られる。1)化合物48のフェノールの保護基の除去、2)生じたフェノール体のメシレート体4によるアルキル化、および3)カルボン酸への脱保護を含む3工程で、本発明のN−置換されたピラゾール酸Xが得られる。
【0057】
N−置換された−ピロール−3−カルボン酸化合物XIへの一般的な工程を、反応式10に示す。アルデヒド体3は塩基性条件下適当に保護されたプロピオール酸エステル化合物49と反応して、アルキン−アルコール体50が得られる(J. Org. Chem., 1980, 45, 28)。標準的な方法(例えば、Et3SiH/酸; Tetrahedron Lett., 1987, 28, 4921)を用いたアルキン体50のアルコール官能基の脱酸素化により、アルキン酸エステル化合物51が得られる。標準的な方法(「Preparation of Alkenes, A Practical Approach」, J. M. J. Williams, Ed., Chapter 6,「Reduction of Alkynes」, J. Howarth. Oxford University Press, 1996)を用いたアルキン酸エステル化合物51の還元により、Z−アルケニルエステル化合物52が得られる。α,β−不飽和エステル化合物52は次いで、標準的な文献の条件で(Van Leusen, A. M., et al, Tetrahedron Lett., 1972, 5337)トシルメチルイソシアネート(TosMIC)と反応し、対応するピロール−エステル化合物53が得られる。標準的な文献の条件で(Lam, P. Y. S., et al, Tetrahedron Lett., 1998, 39, 2941)ピロール−エステル化合物53の適当に置換されたアリールまたはヘテロアリールボロン酸54とのカップリングにより、N−置換されたピロールエステル体55が得られる。次いでN−置換されたピロールエステル体55の脱保護で、本発明のN−置換されたピロール酸XIが得られる。
【0058】
反応式11に、N−置換されたピロール−3−カルボン酸化合物XIIへの合成工程が記載されている。アルデヒド体3は、ホスホラニリデンエステル化合物53とウィッティヒ反応するか(「Preparation of Alkenes, A Practical Approach」, J. M. J. Williams, Ed., Chapter 2,「The Wittig reaction and related methods」, N. J. Lawrence, Oxford University Press, 1996)、またはホスホン酸エステル56とのホーナー・エモンズ反応を行い(J. M. J. Williams, supra and N. J. Lawrence, supra)、有意にE−アルケニルエステル体57が得られる。E−アルケニルエステル体57は次いで、トシルメチルイソシアネート(TosMIC)と反応して、ピロール−エステル化合物58が得られる。ピロール−エステル化合物58は次いで、標準的な文献の条件下(Evansの文献)適当なボロン酸化合物54と反応して、対応するN−置換されたピロール−エステル化合物59が得られる。次いでN−置換されたピロール−エステル化合物59の脱保護により、本発明のN−置換されたピロール酸化合物XIIが得られる。
【0059】
反応式12に、必要な中間体2−アリール(または2−ヘテロアリール)−5−メチル−オキサゾール−4−イルメチルクロリドの製造を示す(Malamas, M. S., et al, J. Med. Chem., 1996, 39, 237-245に記載の一般方法に従う)。置換されたアルデヒド体60は、酸性条件下ブタン−2,3−ジオン モノ−オキシムと縮合して、対応するオキサゾール−N−オキシド体61が得られる。付随する塩素化でオキサゾール−N−オキシド体61が脱酸素化されて、目的のクロロメチルアリール(またはヘテロアリール)−オキサゾール体62が得られる。塩基性条件下のクロロメチルオキサゾール体62の加水分解で、対応するオキサゾール−メタノール体63が得られる。アルコール体63の対応するアルデヒド体への酸化に続いて、対応するジブロモアルケン体64(例えば、Ph3P/CBr4)に転換される。ジブロミド体64は、対応するアルキニル−リチウム体(n−BuLiのような有機リチウム試薬を用いる)に転換され、これは適当な親電子体(例えばホルムアルデヒド)とインシテューで反応して、対応するアセチレンアルコール体が得られる(引用文献:Corey, E. J., et al., Tetrahedron Lett. 1972, 3769, またはGangakhedkar, K. K., Synth. Commun. 1996, 26, 1887-1896)。このアルコール体は次いで、対応するメシレート体65に転換され、適当なフェノール化合物66でアルキル化され、カルボン酸への脱保護後、類似体XIIIが得られる。一般に、フェノール体66は適当な中間体(例えば、11、23および37)のフェノール官能基の脱保護で得られる。本発明のアルキン体XIIIの立体選択的な部分的還元(例えば、H2/リンドラー触媒)で、E−またはZ−アルケニル類似体XIVが得られる。アルケン類似体XIVの完全な還元(水素化)で、本発明のアルキル類似体XVが得られる。あるいは、本発明のアルキン類似体XIIIの完全な還元(例えば、H2/パラジウム炭素触媒)はまた、本発明のアルキル類似体XVも得られる。
【0060】
炭素連結の類似体XVI、XVII、およびXVIIIの合成が、反応式13〜14に示されている。反応工程は反応式2に示されているものに類似する。塩基の存在下適当に保護されたハロ−アリール(またはヘテロアリール)酸クロリド体67をメルドラム酸で処理して、対応するクルードメルドラム酸付加体68が得られ、アニリンと直ちに反応してβ−ケトアニリド体69が得られる。β−ケトアミド体69は亜硝酸(塩基/亜硝酸ナトリウムからインシトゥーで産生する)との反応、続く酸処理により、対応するα−オキシム−β−ケトアミド体70を得る。β−ケト−アミド体70は次いで、適当に置換されたヒドラジン体9と縮合し、対応するβ−ヒドラゾン−アミド体71を得る。中間体71の酸による処理で、目的の2−アリール−4−カルボキサミド−トリアゾール体72を得る。ソノガシラ反応条件下(例えば、「Organocopper Reagents, a Practical Approach」, R. J. K. Taylor, E., Chapter, 10, p 217-236, Campbel, I. B., Oxford University Press, 1994)、アルキン体73のハロ−トリアゾール体72とのカップリングで、対応するアルキニルトリアゾール化合物74が得られる。次いでアニリド体74の加水分解で、本発明のアルキニルトリアゾール酸類似体XVIが得られる。本発明のアルキニルトリアゾール酸化合物XVIの選択的還元で(例えば、H2/リンドラー触媒)、本発明のE−またはZ−アルケニルトリアゾール酸化合物XVIIが得られる。次いで本発明のアルケニルトリアゾール酸化合物XVIIの完全な還元で、本発明の飽和アルキルトリアゾール酸化合物XVIIIが得られる。
【0061】
エーテルを含む類似体XIXおよびXXの合成は、反応式15〜16に示す。
【0062】
反応式15では、適当に保護したハロ−アリールトリアゾール化合物72を金属化試薬(例えば、イソプロピルマグネシウムブロミド,引用文献:P. Knochel et al., Synthesis, 2002, 565-569)で処理して、対応するアリールマグネシウム試薬を得て、次いでホルムアルデヒドで反応して、ベンジルアルコール体75が得られる。塩基存在下アルコール体75をメシレート体VIIIで処理して、対応するエーテルアニリド体を得て、次いで脱保護して、本発明のエーテル酸化合物XIXが得られる。
【0063】
反応式16では、スチルカップリング条件下(引用文献:Farina, V., Krishnamurthy, V., and Scott, W. J., Organic Reactions, 1997, 50, 1)適当に保護されたハロ−アリールトリアゾール化合物72を適当なビニルスズ試薬(例えば、トリブチルビニルスズ)で処理して、対応するビニル中間体を得て、次いでハイドロボレーション(例えば、ボラン−THF)して、アルコール体76が得られる。塩基存在下アルコール体76をメシレート体VIIIで処理して、対応するエーテルアニリド体を得て、次いで脱保護して本発明のエーテル酸化合物XXが得られる。
【0064】
2−置換された−トリアゾール−4−酸化合物XXIの合成を、反応式17に示す。アセチレンエステル化合物26のアジ化ナトリウムとの処理で、双極環化付加反応して、トリアゾール−エステル体77が得られる。標準的な文献の条件(Lam, P. Y. S., et. al., Tetrahedron Lett., 1998, 39, 2941)を用いて、トリアゾール−エステル体77を適当に置換されたアリールまたはヘテロアリールボロン酸化合物54でカップリングして、優先的にN(2)−置換されたトリアゾールエステル化合物78が得られる。次いでトリアゾール−エステル体78脱保護で、本発明のN(2)−置換されたトリアゾール酸XXIが得られる。
【0065】
認められたエーテルを含む類似体XXII〜XXIVの合成は、反応式18〜19に示される。
【0066】
反応式18では、標準的なソノガシラカップリング条件下(例えば、「Organocopper Reagents, a Practical Approach」, R. J. K. Taylor, E., Chapter, 10, p 217-236, Campbell, I. B., Oxford University Press, 1994)、適当に保護したハロ−アリールトリアゾール化合物72を適当に保護したアセチレンアルコール体79(式中、x3=1〜3が好ましい)で処理して、対応するアルキニルトリアゾール化合物80が得られる。化合物80を水素化し、続いてアルコール体へ脱保護して、トリアゾール−アルコール体81が得られる。塩基存在下アルコール化合物81をメシレート体VIIIで処理し、対応するエーテル−アニリド体を得て、次いで脱保護して、本発明のエーテル−酸化合物XXIIが得られる。
【0067】
反応式19では、トリアゾール化合物80を脱保護して、アセチレンアルコール化合物81を得て、塩基存在下メシレート体VIIIと反応して、対応するエーテルアニリド化合物が得られ、次いで脱保護して本発明のエーテル酸化合物XXIIIが得られる。アルキニルトリアゾール酸化合物XXIIIの選択的な還元(例えば、H2/リンドラー触媒)で、本発明のE−またはZ−アルケニルトリアゾール酸XXIVが得られる。
【0068】
反応式20〜21に示したように、トリアゾール−酸類似体の製造のこれらの一般合成スキームは、ピロール酸類似体にも適用される。ピロール酸類似体XXV〜XXIXの製造の合成スキームは、反応式10に記載のアプローチに続く。ハロ−アルデヒド化合物83を塩基性条件下(最も好ましくは18−クラウン−6の存在下フッ化物アニオンを用いる)、トリメチルシリルプロピオール酸エステル化合物84と反応させ、アルキン−アルコール化合物85を得る。標準的な方法を用いて(例えば、Et3SiH/酸; Tetrahedron Lett., 1987, 28, 4921)、アルキン化合物50のアルコール官能基の脱酸素化により、アルキン酸エステル化合物86が得られる。標準的な方法を用いて(「Preparation of Alkenes, A Practical Approach」, J. M. J. Williams, Ed., Chapter 6,「Reduction of Alkynes」, J. Howarth. Oxford University Press, 1996)、アルキン酸エステル化合物86を還元して、Z−アルケニルエステル化合物87が得られる。α,β−不飽和エステル化合物87は次いで、標準的な文献の条件下(Van Leusen, A. M., et al, Tetrahedron Lett., 1972, 5337)トシルメチルイソシアネート(TosMIC)と反応させて、対応するピロール−エステル化合物88が得られる。標準的な文献の条件下(Lam, P. Y. S., et al, Tetrahedron Lett., 1998, 39, 2941)、ピロール−エステル化合物88を適当に置換されたアリールまたはヘテロアリールボロン酸化合物54とカップリングさせて、鍵中間体であるハロ−アリール−N−置換された−ピロールエステル化合物89が得られ、これはハロ−アリールトリアゾール中間体72とのピロール等価体である。ハロアリールピロール化合物89を、トリアゾール化合物72の代わりに反応式15、16、18および19に記載の同一の反応経路に用いることで、反応式21に示されたような本発明のピロール酸化合物XXV〜XXIXが得られる。
【0069】
【化6】
【0070】
【化7】
【0071】
【化8】
【0072】
【化9】
【0073】
【化10】
【0074】
【化11】
【0075】
【化12】
【0076】
【化13】
【0077】
【化14】
【0078】
【化15】
【0079】
【化16】
【0080】
【化17】
【0081】
【化18】
【0082】
【化19】
【0083】
【化20】
【0084】
【化21】
【0085】
【化22】
【0086】
【化23】
【0087】
【化24】
【0088】
【化25】
【0089】
【化26】
【0090】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルキル」、「アルキル」または「アルカ(alk)」には、通常の鎖において1〜20個の炭素原子、好ましくは1〜10個の炭素原子、より好ましくは1〜8個の炭素原子を含む直鎖および分枝鎖の両方の炭化水素が含まれ、通常の鎖において酸素または窒素を適宜含み得、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、t−ブチル、イソブチル、ペンチル、ヘキシル、イソヘキシル、ヘプチル、4,4−ジメチルペンチル、オクチル、2,2,4−トリメチルペンチル、ノニル、デシル、ウンデシル、ドデシル、その様々な分枝鎖の異性体が含まれ、並びに例えば、ハロ(例えば、F、Br、ClまたはIまたはCF3)、アルコキシ、アリール、アリールオキシ、アリール(アリール)またはジアリール、アリールアルキル、アリールアルキルオキシ、アルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルアルキルオキシ、アミノ、ヒドロキシ、ヒドロキシアルキル、アシル、ヘテロアリール、ヘテロアリールオキシ、シクロヘテロアルキル、アリールヘテロアリール、アリールアルコキシカルボニル、ヘテロアリールアルキル、ヘテロアリールアルコキシ、アリールオキシアルキル、アリールオキシアリール、アルキルアミド、アルカノイルアミノ、アリールカルボニルアミノ、ニトロ、シアノ、チオール、ハロアルキル、トリハロアルキルおよび/またはアルキルチオおよび/またはR3基のいずれかの置換基の1〜4個を含むかかる基も同様に含まれる。
【0091】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「シクロアルキル」には、単環式アルキル、二環式アルキルおよび三環式アルキルを含み、環を形成する全部で3〜20個の炭素原子、好ましくは環を形成する3〜10個の炭素原子を含む、1〜3個の環を含む飽和または一部不飽和(1または2個の二重結合を含む)の環状炭化水素基が含まれ、アリールで記載されているような1または2個の芳香環と縮合してもよく、これらにはシクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロデシルおよびシクロドデシル、シクロヘキセニル、
【化27】
、1〜4個の置換基(例えば、ハロゲン、アルキル、アルコキシ、ヒドロキシ、アリール、アリールオキシ、アリールアルキル、シクロアルキル、アルキルアミド、アルカノイルアミノ、オキソ、アシル、アリールカルボニルアミノ、アミノ、ニトロ、シアノ、チオールおよび/またはアルキルチオ)で適宜置換され得る基のいずれかおよび/またはアルキルへの置換基のいずれかが含まれる。
【0092】
本明細書で単独または他の基の一部として用いる用語「シクロアルケニル」とは、3〜12個の炭素原子、好ましくは5〜10個の炭素原子および1または2個の二重結合を含む環状炭化水素をいう。具体的なシクロアルケニル基には、シクロペンテニル、シクロヘキセニル、シクロヘプテニル、シクロオクテニル、シクロヘキサジエニル、およびシクロヘプタジエニルが含まれ、シクロアルキルで定義されているように適宜置換されていてもよい。
【0093】
本明細書で用いる用語「シクロアルキレン」とは、フリーな結合を含み、このように例えば
【化28】
などのような結合基である「シクロアルキル」基をいい、上記「シクロアルキル」で定義されているように適宜置換されていてもよい。
【0094】
本明細書で単独または他の基の一部として用いる用語「アルカノイル」とは、カルボニル基に結合するアルキルをいう。
【0095】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルケニル」または「アルケニル」とは、通常の鎖において1〜6個の二重結合を含む、通常の鎖において2〜20個の炭素原子、好ましくは2〜12個の炭素原子、より好ましくは1〜8個の炭素原子の直鎖および分枝鎖の基をいい、通常の鎖において酸素または窒素を適宜含み得、例えば、ビニル、2−プロペニル、3−ブテニル、2−ブテニル、4−ペンテニル、3−ペンテニル、2−ヘキセニル、3−ヘキセニル、2−ヘプテニル、3−ヘプテニル、4−ヘプテニル、3−オクテニル、3−ノネニル、4−デセニル、3−ウンデセニル、4−ドデセニル、4,8,12−テトラデカトリエニルなどをいい、これらは置換基、すなわち、ハロゲン、ハロアルキル、アルキル、アルコキシ、アルケニル、アルキニル、アリール、アリールアルキル、シクロアルキル、アミノ、ヒドロキシ、ヘテロアリール、シクロヘテロアルキル、アルカノイルアミノ、アルキルアミド、アリールカルボニルアミノ、ニトロ、シアノ、チオール、アルキルチオおよび/または本明細書で定められるアルキルへの置換基のいずれかの置換基の1〜4個で適宜置換されていてもよい。
【0096】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルキニル」または「アルキニル」とは、通常の鎖において1個の三重結合を含む、通常の鎖において2〜20個の炭素原子、好ましくは2〜12個の炭素原子、より好ましくは2〜8個の炭素原子の直鎖および分枝鎖の基をいい、通常の鎖において酸素または窒素を適宜含み得、例えば、2−プロピニル、3−ブチニル、2−ブチニル、4−ペンチニル、3−ペンチニル、2−ヘキシニル、3−ヘキシニル、2−ヘプチニル、3−ヘプチニル、4−ヘプチニル、3−オクチニル、3−ノニニル、4−デシニル、3−ウンデシニル、4−ドデシニルなどをいい、これらは置換基、すなわち、ハロゲン、ハロアルキル、アルキル、アルコキシ、アルケニル、アルキニル、アリール、アリールアルキル、シクロアルキル、アミノ、ヘテロアリール、シクロヘテロアルキル、ヒドロキシ、アルカノイルアミノ、アルキルアミド、アリールカルボニルアミノ、ニトロ、シアノ、チオール、および/またはアルキルチオ、および/または本明細書で定められるアルキルへの置換基のいずれかの置換基の1〜4個で適宜置換されていてもよい。
【0097】
単独または他の基の一部として用いる用語「アリールアルケニル」および「アリールアルキニル」とは、アリール置換基を有する上述のようなアルケニルおよびアルキニル基をいう。
【0098】
上記で定義されるようなアルキル基が2つの異なる炭素原子で他の基をつなぐ単結合を有する場合、それらは「アルキレン」と呼ばれ、「アルキル」として上記で定義されたように適宜置換されていてもよい。
【0099】
上記で定義されるようなアルケニル基および上記で定義されるようなアルキニル基がそれぞれ、2つの異なる炭素原子で結合するための単結合を有する場合、それらは「アルケニレン基」および「アルキニレン基」とそれぞれ呼ばれ、「アルケニル」および「アルキニル」として上記で定義されたように適宜置換されていてもよい。
【0100】
(CH2)x、(CH2)x 1、(CH2)x 2、(CH2)x 3、(CH2)m、または(CH2)nには、本明細書で定義されているように、アルキレン、アレニル、アルケニレンまたはアルキニレン基が含まれ、これらのそれぞれは通常の鎖において酸素または窒素を適宜含み得、アルキル、アルケニル、ハロゲン、シアノ、ヒドロキシ、アルコキシ、アミノ、チオアルキル、ケト、C3〜C6シクロアルキル、アルキルカルボニルアミノまたはアルキルカルボニルオキシの1、2、または3個の置換基が適宜含まれていてもよく;該アルキル置換基は、(CH2)x、(CH2)x 1、(CH2)x 2、(CH2)x 3または(CH2)mまたは(CH2)n基の1または2個の炭素に結合してそれによるシクロアルキル基を形成し得る1〜4個の炭素原子のアルキレン部分でもよい。
【0101】
(CH2)x、(CH2)x 1、(CH2)x 2、(CH2)x 3、(CH2)m、(CH2)n、アルキレン、アルケニレンおよびアルキニレンの例には、
【化29】
が含まれる。
【0102】
本明細書で単独または他の基の一部として用いる用語「ハロゲン」または「ハロ」とは、塩素、臭素、フッ素、およびヨウ素、並びにCF3をいい、,好ましくは塩素またはフッ素である。
【0103】
用語「金属イオン」とは、ナトリウム、カリウムまたはリチウムのようなアルカリ金属イオンおよびマグネシウムおよびカルシウムのようなアルカリ土類金属イオン、並びに亜鉛およびアルミニウムイオンをいう。
【0104】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「アリール」または基:
【化30】
(式中、QはCである)とは、環部分に6〜10個の炭素を含む単環式および二環式芳香基(例えば、フェニル、または1−ナフチルおよび2−ナフチルを含むナフチル)をいい、炭素環またはヘテロ環(例えば、アリール、シクロアルキル、ヘテロアリールまたはシクロヘテロアルキル環)に縮合した1〜3個の付加の環を適宜含んでいてもよく、例えば、
【化31】
が例であり、水素、ハロ、ハロアルキル、アルキル、ハロアルキル、アルコキシ、ハロアルコキシ、アルケニル、トリフルオロメチル、トリフルオロメトキシ、アルキニル、シクロアルキル−アルキル、シクロヘテロアルキル、シクロヘテロアルキルアルキル、アリール、ヘテロアリール、アリールアルキル、アリールオキシ、アリールオキシアルキル、アリールアルコキシ、アルコキシカルボニル、アリールカルボニル、アリールアルケニル、アミノカルボニルアリール、アリールチオ、アリールスルフィニル、アリールアゾ、ヘテロアリールアルキル、ヘテロアリールアルケニル、ヘテロアリールヘテロアリール、ヘテロアリールオキシ、ヒドロキシ、ニトロ、シアノ、アミノ、置換されたアミノ[該アミノには、1または2個の置換基(置換基は、アルキル、アリールまたは定義で記載されている他のアリール化合物のいずれか)が含まれる]、チオール、アルキルチオ、アリールチオ、ヘテロアリールチオ、アリールチオアルキル、アルコキシアリールチオ、アルキルカルボニル、アリールカルボニル、アルキルアミノカルボニル、アリールアミノカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルキルカルボニルアミノ、アリールカルボニルアミノ、アリールスルフィニル、アリールスルフィニルアルキル、アリールスルホニルアミノまたはアリールスルホンアミノカルボニルおよび/または本明細書で定められるアルキルへの置換基のいずれかから選択される1、2、または3個の基を、置換可能な炭素原子に適宜置換してもよい。
【0105】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルコキシ」、「アルコキシ」、「アリールオキシ」、または「アラルコキシ」には、酸素原子結合する上記のいずれかのアルキル、アラルキルまたはアリール基のいずれかを含む。
【0106】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「置換されたアミノ」とは、例えばアルキル、アリール、アリールアルキル、ヘテロアリール、ヘテロアリールアルキル、シクロヘテロアルキル、シクロヘテロアルキルアルキル、シクロアルキル、シクロアルキルアルキル、ハロアルキル、ヒドロキシアルキル、アルコキシアルキルまたはチオアルキルのような1または2個の置換基(同一または異なってもよい)で置換されたアミノをいう。これらの置換基はさらに、カルボン酸および/または上記で定められたようなアルキルへの置換基のいずれかで置換されてもよい。さらに、該アミノ置換基は結合している窒素原子と一緒になって、1−ピロリジニル、1−ピペリジニル、1−アゼピニル、4−モルホリニル、4−チアモルホリニル、1−ピペラジニル、4−アルキル−1−ピペラジニル、4−アリールアルキル−1−ピペラジニル、4−ジアリールアルキル−1−ピペラジニル、1−ピロリジニル、1−ピペリジニル、または1−アゼピニル(適宜アルキル、アルコキシ、アルキルチオ、ハロ、トリフルオロメチルまたはヒドロキシで置換される)を形成していてもよい。
【0107】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルキルチオ」、「アルキルチオ」、「アリールチオ」または「アラルキルチオ」には、硫黄原子に連結する上記のアルキル、アラルキルまたはアリール基のいずれかを含む。
【0108】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルキルアミノ」、「アルキルアミノ」、「アリールアミノ」、または「アリールアルキルアミノ」には、窒素原子に連結する上記のアルキル、アリールまたはアリールアルキル基のいずれかを含む。
【0109】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「アシル」とは、カルボニル:
【化32】
基に連結する有機基をいい:アシル基の例には、カルボニルに結合するR3基のいずれかを含み、例えば、アルカノイル、アルケノイル、アロイル、アラルカノイル、ヘテロアロイル、シクロアルカノイル、シクロヘテロアルカノイルなどがある。
【0110】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「シクロヘテロアルキル」とは、窒素、酸素および/または硫黄のようなヘテロ原子1〜2個を含み、炭素原子またはヘテロ原子を介して連結し、可能なら適宜結合鎖(CH2)p(pは1、2または3)で結合する5−、6−または7員の飽和または一部不飽和の環をいい、例えば、
【化33】
などがある。上記の基には、例えばアルキル、ハロ、オキソおよび/または本明細書で定められるアルキルまたはアリールへの置換基のいずれかのような置換基の1〜4個を含んでいてもよい。さらに、シクロヘテロアルキル環のいずれかは、シクロアルキル、アリール、ヘテロアリールまたはシクロヘテロアルキル環と縮合し得る。
【0111】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「ヘテロアリール」とは、1、2、3または4個のヘテロ原子(例えば、窒素、酸素または硫黄)を含み、
【化34】
(QはNである)を含む5−または6員芳香環をいい、並びにアリール、シクロアルキル、ヘテロアリールまたはシクロヘテロアルキル環(例えば、ベンゾチオフェニル、インドリル)と縮合したかかる環(可能なN−オキシドを含む)をいう。ヘテロアリール基は、上記で定められるアルキルまたはアリールへの置換基のいずれかのような1〜4個の置換基を適宜含んでいてもよい。ヘテロアリール基の例には、以下:
【化35】
などが含まれる。
【0112】
基:
【化36】
の例には、これらに限らないが、
【化37】
が含まれる。
【0113】
基:
【化38】
の例には、これらに限らないが、
【化39】
が含まれる。
【0114】
本明細書で単独または他の基の一部として用いる用語「シクロヘテロアルキルアルキル」とは、C原子またはヘテロ原子を介して(CH2)p鎖に連結する上記で定義されるようなシクロヘテロアルキル基をいう。
【0115】
本明細書で単独または他の基の一部として用いる用語「ヘテロアリールアルキル」または「ヘテロアリールアルケニル」とは、C原子またはヘテロ原子を介して(CH2)p鎖、上記で定義されるようなアルキレンまたはアルケニレンに連結する上記で定義されるようなヘテロアリール基をいう。
【0116】
本明細書で用いる用語「ポリハロアルキル」とは、2〜9個、好ましくは2〜5個のハロ置換基(ハロ置換基は例えばFまたはClであり、好ましくはFである)を含む上記で定義されるような「アルキル」基をいい、例えばCF3CH2、CF3またはCF3CF2CH2がある。
【0117】
本明細書で用いる用語「ポリハロアルキルオキシ」とは、2〜9個、好ましくは2〜5個のハロ置換基(ハロ置換基は例えばFまたはClであり、好ましくはFである)を含む、上記で定義したような「アルコキシ」または「アルキルオキシ」基をいい、例えば、CF3CH2O、CF3OまたはCF3CF2CH2Oがある。
【0118】
本明細書で用いる用語「プロドラッグエステル」には、カルボン酸およびホスホン酸エステル(例えば、メチル、エチル、ベンジルなど)について当該技術分野で公知のプロドラッグエステルが含まれる。R4の他のプロドラッグエステルの例には、以下の基:すなわち
【化40】
[式中、Ra、RbおよびRcは、H、アルキル、アリールまたはアリールアルキルであるが;RaOはHOではあり得ない]のような(1−アルカノイルオキシ)アルキルを含む。かかるプロドラッグエステルR4の例には、
【化41】
が含まれる。適当なプロドラッグエステルR4の他の例には、
【化42】
[式中、Raは、H、アルキル(例えば、メチルまたはt−ブチル)、アリールアルキル(例えば、ベンジル)またはアリール(例えば、フェニル)であり得;Rdは、H、アルキル、ハロゲンまたはアルコキシであり、Reは、アルキル、アリール、アリールアルキルまたはアルコキシルであり、n1は、0、1または2である]が含まれる。
【0119】
構造Iの化合物が酸型である場合、医薬的に許容される塩、例えば、アルカリ金属塩(例えば、リチウム、ナトリウムまたはカリウム)、アルカリ土類金属塩(例えば、カルシウムまたはマグネシウム)、並びに亜鉛またはアルミニウム、および他のカチオン[例えばアンモニウム、コリン、ジエタノールアミン、リジン(DまたはL)、エチレンジアミン、t−ブチルアミン、t−オクチルアミン、トリス(ヒドロキシメチル)アミノメタン(TRIS)、N−メチルグルコサミン(NMG)、トリエタノールアミンおよびデヒドロアビエチルアミン]の塩を形成し得る。
【0120】
本発明の化合物のすべての立体異性体は、混合物または純粋なまたは十分に純粋な形態のいずれかであることと考慮される。本発明の化合物は、それ自身のいずれかまたはR置換基に含まれる炭素原子のいずれかに不斉中心を有し得る。従って、式Iの化合物はエナンチオマー型またはジアステレオマー型またはその混合物で存在し得る。製造の工程には、出発物質としてラセミ体、エナンチオマーまたはジアステレオマーを利用できる。ジアステレオマーまたはエナンチオマー生成物を製造する場合、例えばクロマトグラフィまたは分別結晶のようなありふれた方法で分離できる。
【0121】
所望なら、構造Iの化合物は、1つ以上の抗高脂血症剤または脂質低下剤または脂質調節剤と、および/または抗糖尿病剤、抗肥満剤、降圧剤、血小板凝集阻害剤および/または抗骨多孔症剤を含む治療剤の1以上の他のタイプと組み合わせて用いてもよく、同一の製剤で経口投与されてもよく、別の経口製剤で投与されてもよく、または注射で投与されてもよい。
【0122】
適宜本発明の式Iの化合物と組み合わせて用い得る抗高脂血症剤または脂質低下剤または脂質調節剤は、1、2、3またはそれ以上のMTP阻害剤、HMG CoA還元酵素阻害剤、スクアレン合成酵素阻害剤、フィブリン酸誘導体、ACAT阻害剤、リポキシゲナーゼ阻害剤、コレステロール吸収阻害剤、回腸Na+/胆汁酸共輸送体阻害剤、LDL受容体活性の上方制御剤、胆汁酸金属イオン封鎖剤、および/またはニコチン酸およびその誘導体を含み得る。
【0123】
本明細書に用いられるMTP阻害剤には、米国特許第5,595,872号、米国特許第5,739,135号、米国特許第5,712,279号、米国特許第5,760,246号、米国特許第5,827,875号、米国特許第5,885,983号および米国特許出願第09/175,180号(1998年10月20日出願)、米国特許第5,962,440号に開示されているMTP阻害剤が含まれる。好ましいのは、上記特許および出願のそれぞれに開示されている好ましいMTP阻害剤である。
【0124】
上記の米国特許および出願のすべては、本明細書に引用される。
【0125】
本発明に従って用いられる最も好ましいMTP阻害剤には、米国特許第5,739,135号および第5,712,279号、および米国特許第5,760,246号で述べられるような好ましいMTP阻害剤が含まれる。
【0126】
最も好ましいMTP阻害剤は、9−[4−[4−[[2−(2,2,2−トリフルオロエトキシ)ベンゾイル]アミノ]−1−ピペリジニル]ブチル]−N−(2,2,2−トリフルオロエチル)−9H−フルオレン−9−カルボキサミド:
【化43】
である。
【0127】
抗高脂血症剤は、HMG CoA還元酵素阻害剤であり得、これらに限らないが、米国特許第3,983,140号に開示されるようなメバスタチンおよび関連化合物、米国特許第4,231,938号に開示されるようなロバスタチン(メビノリン)および関連化合物、米国特許第4,346,227号に開示されるようなプラバスタチンおよび関連化合物、米国特許第4,448,784号および第4,450,171号に開示されるようなシンバスタチンおよび関連化合物が含まれる。本明細書で用いられ得る他のHMG CoA還元酵素阻害剤には、これらに限らないが、フルバスタチン(米国特許第5,354,772号に開示)、セリバスタチン(米国特許第5,006,530号および第5,177,080号に開示)、アトルバスタチン(米国特許第4,681,893号、第5,273,995号、第5,385,929号および第5,686,104号に開示)、イタバスタチン(米国特許第5,011,930号に開示されたニッサン/三共のニスバスタチン(NK−104))、塩野義−アストラ/ゼネカのビサスタチン(ZD−4522)(米国特許第5,260,440号に開示)、および関連するスタチン化合物(米国特許第5,753,675号に開示)、メバロノラクトン誘導体のピラゾール類似体(米国特許第4,613,610号に開示)、メバロノラクトン誘導体のインデン類似体(PCT出願WO 86/03488に開示)、6−[2−(置換された−ピロール−1−イル)−アルキル)ピラン−2−オン化合物およびその誘導体(米国特許第4,647,576号に開示)、シアール社のSC−45355(3−置換されたペンタンジオン酸誘導体)ジクロロアセテート、メバロノラクトンのイミダゾール類似体(PCT出願WO 86/07054に開示)、3−カルボキシ−2−ヒドロキシ−プロパンホスホン酸誘導体(フランス特許第2,596,393号に開示)、2,3−二置換されたピロール、フランおよびチオフェン誘導体(欧州特許出願第0221025号に開示)、メバロノラクトンのナフチル類似体(米国特許第4,686,237号に開示)、オクタヒドロナフタレン化合物(米国特許第4,499,289号に開示)、メビノリン(ロバスタチン)のケト類似体(欧州特許出願第0,142,146 A2号に開示)、およびキノリンおよびピリジン誘導体(米国特許第5,506,219号および第5,691,322号に開示)が含まれる。
【0128】
さらに、本明細書で用いるのに適したHMG CoA還元酵素阻害に有用なホスフィン酸化合物は、GB 2205837に開示されている。
【0129】
本明細書で用いるのに適したスクアレン合成酵素阻害剤には、これらに限らないが、イソプレノイド(ホスフィニルメチル)ホスホン酸化合物を含むα−ホスホノスルホネート化合物(米国特許第5,712,396号およびBiller et al, J. Med. Chem., 1988, Vol. 31, No. 10, pp 1869-1871に開示)、並びに他の公知のスクアレン合成酵素阻害剤(例えば、米国特許第4,871,721号および第4,924,024号およびBiller, S. A., Neuenschwander, K., Ponpipom, M. M., and Poulter, C. D., Current Pharmaceutical Design, 2, 1-40 (1996)に開示)が含まれる。
【0130】
さらに、本明細書で用いるのに適した他のスクアレン合成酵素阻害剤には、テルペノイドピロホスフェート化合物(P. Ortiz de Montellano et al, J. Med. Chem., 1977, 20, 243-249に開示)、ファルネシルジホスフェート類似体Aおよびプレスクアレンピロホスフェート(PSQ−PP)類似体(Corey and Volante, J. Am. Chem. Soc., 1976, 98, 1291-1293に開示)、ホスフィニルホスホネート化合物(McClard, R. W. et al, J. A. C. S., 1987, 109, 5544に報告)、およびシクロプロパン化合物(Capson, T. L., PhD dissertation, June, 1987, Dept. Med. Chem. U of Utah, Abstract, Table of Contents, pp 16, 17, 40-43, 48-51, Summaryに報告)が含まれる。
【0131】
本明細書で用いるのに適した他の抗高脂血症剤には、これらに限らないが、フィブリン酸誘導体(例えば、フェノフィブラート、ゲムフィブロジル、クロフィブラート、ベザフィブラート、シプロフィブラート、クリノフィブラートなど)、プロブコールおよび関連化合物(米国特許第3,674,836号に開示)(好ましくはプロブコールおよびゲムフィブロジル)、胆汁酸金属イオン封鎖剤[例えば、コレスチラミン、コレスチポールおよびDEAE−セファデックス(セコレックス(登録商標)、ポリセキシド(登録商標))]およびコレスタゲル(三共/ゲルテックス)、並びに、リポスタビル(ローヌ・プーラン社)、エーザイE−5050(N−置換されたエタノールアミン誘導体)、イマニキシル(HOE−402)、テトラヒドロリプスタチン(THL)、イスチグマスタニルホスホリルコリン(SPC、ロッシュ社)、アミノシクロデキストリン(田辺製薬)、味の素AJ−814 (アズレン誘導体)、メリナミド(住友)、サンド社58−035、アメリカンシアナミド社CL−277,082およびCL−283,546(二置換された尿素誘導体)、ニコチン酸(ナイアシン)、アシピモックス、アシフラン、ネオマイシン、p−アミノサリチル酸、アスピリン、ポリ(ジアリルメチルアミン)誘導体(米国特許第4,759,923号に開示)、四級アミン ポリ(ジアリルジメチルアンモニウムクロリド)およびイオネン(米国特許第4,027,009号に開示)、および他の公知の血清コレステロール低下剤が含まれる。
【0132】
抗高脂血症剤は、以下のように開示されるようなACAT阻害剤であり得る:
Drugs of the Future 24, 9-15 (1999), (Avasimibe);
「The ACAT inhibitor, Cl-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters」, Nicolosi et al, Atherosclerosis (Shannon, Irel). (1998), 137 (1), 77-85;
「The pharmacological profile of FCE 27677: a novel ACAT inhibitor with potent hypolipidemic activity mediated by selective suppression of the hepatic secretion of ApoB100-containing lipoprotein」, Ghiselli, Giancarlo, Cardiovasc. Drug Rev. (1998), 16 (1), 16-30;
「RP 73163: a bioavailable alkylsulfinyl-diphenylimidazole ACAT inhibitor」, Smith, C., et al, Bioorg. Med. Chem. Lett. (1996), 6 (1), 47-50;
「ACAT inhibitors: physiologic mechanisms for hypolipidemic and anti-atherosclerotic activities in experimental animals」, Krause et al, Editor (s): Ruffolo, Robert R., Jr.; Hollinger, Mannfred A., Inflammation : Mediators Pathways (1995), 173-98, Publisher: CRC, Boca Raton, Fla.;
「ACAT inhibitors: potential anti-atherosclerotic agents」, Sliskovic et al, Curr. Med. Chem. (1994), 1 (3), 204-25;
「Inhibitors of acyl-CoA : cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 6. The first water-soluble ACAT inhibitor with lipid-regulating activity. Inhibitors of acyl-CoA: cholesterol acyltransferase (ACAT). 7. Development of a series of substituted N-phenyl-N'-[(1- phenylcyclopentyl) methyl] ureas with enhanced hypocholesterolemic activity」, Stout et al, Chemtracts: Org. Chem. (1995), 8 (6), 359-62, またはTS-962 (大正製薬)。
【0133】
抗高脂血症剤は、MD−700(大正製薬)およびLY295427(イーライリリィ)のような、LD2受容体活性の上方制御剤であり得る。
【0134】
抗高脂血症剤は、コレステロール吸収阻害剤であり得、好ましくはシェーリングプラウのSCH48461、並びにAtherosclerosis 115, 45-63 (1995)およびJ. Med. Chem. 41, 973 (1998)に開示された阻害剤である。
【0135】
抗高脂血症剤は、Drugs of the Future, 24, 425-430 (1999)に開示されているような回腸Na+/胆汁酸共輸送体阻害剤であり得る。
【0136】
脂質調節剤は、ファイザーのCP 529,414(WO/0038722およびEP 818448)およびファルマシアのSC−744およびSC−795のような、コレステリルエステル転送タンパク質(CETP)阻害剤であり得る。
【0137】
本発明の組み合わせて用いられ得るATPクエン酸リアーゼ阻害剤は、例えば米国特許第5,447,954号に開示の阻害剤が含まれ得る。
【0138】
好ましい抗高脂血症剤は、プラバスタチン、ロバスタチン、シンバスタチン、アトルバスタチン、フルバスタチン、セリバスタチン、イタバスタチンおよびビサスタチンおよびZD−4522である。
【0139】
上述の米国特許は本明細書に引用される。用いる量および用量は、フィジシャンズ デスク レファレンス(Physician's Desk Reference)および/または上記で述べた特許に示される。
【0140】
本発明の式Iの化合物は、(存在するなら)抗高脂血症剤に対して重量比が約500:1〜約1:500、好ましくは約100:1〜約1:100の範囲内で用いられる。
【0141】
投与量は年齢、体重、患者の症状、並びに投与経路、投与形態および投与計画および目的の成果に従い、注意深く調整しなければならない。
【0142】
抗高脂血症剤の投与量および処方は、上述の様々な特許および出願に開示されている。
【0143】
適用できるように用いられる他の抗高脂血症剤の投与量および処方は、フィジシャンズ デスク レファレンス(Physician's Desk Reference)の最新版に記載されている。
【0144】
経口投与では、MTP阻害剤を1日1〜4回で、約0.01 mg〜約500 mg、好ましくは約0.1 mg〜約100 mgの範囲内の量で用いると、満足な結果が得られ得る。
【0145】
好ましい経口製剤、例えば錠剤またはカプセル剤には、1日1〜4回で、約1〜約500 mg、好ましくは約2〜約400 mg、より好ましくは約5〜約250 mgの量でMTP阻害剤が含まれる。
【0146】
経口投与では、HMG CoA還元酵素阻害剤、例えばプラバスタチン、ロバスタチン、シンバスタチン、アトルバスタチン、フルバスタチンまたはセリバスタチンを、フィジシャンズ デスク レファレンス(Physician's Desk Reference)で示される投与量、例えば約1〜2000 mg、好ましくは約4〜約200 mgの範囲内の量で用いると、満足な結果が得られ得る。
【0147】
スクアレン合成酵素阻害剤は、約10 mg〜約2000 mg、好ましくは約25 mg〜約200 mgの範囲内の量の投与量で用いられ得る。
【0148】
好ましい経口製剤、例えば錠剤またはカプセル剤には、約0.1〜約100 mg、好ましくは約0.5〜約80 mg、より好ましくは約1〜約40 mgの量でHMG CoA還元酵素阻害剤が含まれる。
【0149】
好ましい経口製剤、例えば錠剤またはカプセル剤には、約10〜約500 mg、好ましくは約25〜約200 mgの量でスクアレン合成酵素阻害剤が含まれる。
【0150】
抗高脂血症剤はまた、リポキシゲナーゼ阻害剤であり得、15−リポキシゲナーゼ(15−LO)阻害剤(例えばベンゾイミダゾール誘導体、WO 97/12615に開示)、15−LO阻害剤(WO 97/12613に開示)、イソチアゾロン化合物(WO 96/38144に開示)、および15−LO阻害剤(Sendobry et al「Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties」, Brit. J. Pharmacology (1997) 120, 1199-1206, およびCornicelli et al,「15-Lipoxygenase and its Inhibition: A Novel Therapeutic Target for Vascular Disease」, Current Pharmaceutical Design, 1999, 5, 11-20に開示)が含まれる。
【0151】
式Iの化合物および抗高脂血症剤は、同一経口製剤で、または別の経口製剤を同時に服用することで、一緒に用い得る。
【0152】
上述の組成物は、上述の製剤形で、1回でまたは1日1〜4回に分割した用量で投与され得る。患者には低用量の組み合わせから開始し、徐々に高用量の組み合わせに上げていくことが望ましい。
【0153】
好ましい抗高脂血症剤は、プラバスタチン、シンバスタチン、ロバスタチン、アトルバスタチン、フルバスタチンまたはセリバスタチン、並びにナイアシンおよび/またはコレスタゲルである。
【0154】
式Iの化合物と組み合わせて適宜用い得る他の抗糖尿病剤は、1、2、3またはそれ以上の抗糖尿病剤、またはインスリン分泌促進物質またはインスリン増感剤を含む抗高血糖症剤、または好ましくは本発明の式Iの化合物と異なるメカニズムを有する他の抗糖尿病剤であり得、それらには、ビグアナイド、スルホニル尿素、グルコシダーゼ阻害剤、PPARγアゴニスト(例えばチアゾリジンジオン化合物)、aP2阻害剤、ジペプチジルペプチダーゼIV(DP4)阻害剤、SGLT2阻害剤、および/またはメグリチナイド化合物、並びにインスリン、および/またはグルカゴン様ペプチド−1(GLP−1)が含まれ得る。
【0155】
他の抗糖尿病剤は、経口抗高血糖症剤、好ましくはビグアナイド類(例えばメトホルミンまたはフェンホルミンまたはその塩、好ましくはメトホルミンHCl)であり得る。
【0156】
抗糖尿病剤がビグアナイドの場合、構造Iの化合物は、ビグアナイドに対する重量比が約0.001:1〜約10:1、好ましくは約0.01:1〜約5:1の範囲内で用いられる。
【0157】
他の抗糖尿病剤はまた好ましくは、スルホニル尿素[例えば、グリブリド(グリベンクラミドとしても知られる)、グリメピリド(米国特許第4,379,785号に開示)、グリピジド、グリクラジドまたはクロロプロパミド]、他の公知のスルホニル尿素またはβ細胞のATP感受性チャンネルに作用する他の抗高血糖症剤であり、グリブリドおよびグリピジドが好ましく、同一または別の経口製剤で投与され得る。
【0158】
構造Iの化合物は、スルホニル尿素に対する重量比が約0.01:1〜約100:1、好ましくは約0.02:1〜約5:1の範囲で用いられる。
【0159】
経口抗糖尿病剤はまた、グルコシダーゼ阻害剤、例えばアカルボース(米国特許第4,904,769号に開示)またはミグリトール(米国特許第4,639,436号に開示)であり得、同一または別の経口製剤で投与され得る。
【0160】
構造Iの化合物は、グルコシダーゼ阻害剤に対する重量比が約0.01:1〜約100:1、好ましくは約0.05:1〜約10:1の範囲内で用いる。
【0161】
式Iの化合物は、チアゾリジンジオン経口抗糖尿病剤または他のインスリン増感剤(NIDDM患者におけるインスリン感受性効果を有する)のようなPPARγアゴニストと組み合わせて用いられ得、例えば、トログリタゾン(ワーナーランバード社のレズリン(登録商標)、米国特許第4,572,912号に開示)、ロシグリタゾン(SKB)、ピオグリタゾン(武田)、三菱のMCC−555(米国特許第5,594,016号に開示)、グラクソウェルカム社のGL−262570、エングリタゾン(CP−68722, ファイザー)またはダルグリタゾン(CP−86325, ファイザー)、イサグリタゾン(MIT/J&J)、JTT−501(JPNT/P&U)、L−895645(メルク)、R−119702(三共/WL)、NN−2344(レディ博士/NN)、またはYM−440(山之内)、好ましくはロシグリタゾンおよびピオグリタゾンがある。
【0162】
構造Iの化合物は、チアゾリジンジオンに対する重量比が約0.01:1〜約100:1、好ましくは約0.05〜約10:1の範囲内の量で用いられる。
【0163】
経口抗糖尿病剤が約150 mg未満の量において、スルホニル尿素およびチアゾリジンジオンは、構造Iの化合物と単一の錠剤に組み込まれ得る。
【0164】
構造Iの化合物はまた、抗高血糖症剤(例えば、インスリン)と、またはグルカゴン様ペプチド−1(GLP−1)[例えば、GLP−1(1−36)アミド、GLP−1(7−36)アミド、GLP−1(7−37)(米国特許第5,614,492号に開示、ハベナーによる、この開示は本明細書に引用する)、並びにAC2993(アミリン社)およびLY−315902(リリィ社)]と組み合わせても用いられ、注射、鼻腔内、吸入を通じて、または経皮またはバッカル装置により投与し得る。
【0165】
存在する場合は、メトホルミン、スルホニル尿素化合物(例えば、グリブリド、グリメピリド、グリピリド、グリピジド、クロロプロパミドおよびグリクラジド)およびグルコシダーゼ阻害剤アカルボースまたはミグリトールまたはインスリン(注射、肺、バッカル、または経口)は、上述のような製剤形で、フィジシャンズ デスク レファレンス(Physician's Desk Reference、PDR)に示されるような量および投薬で用いられ得る。
【0166】
存在する場合は、メトホルミンまたはその塩は、1日当たり約500〜約2000 mgの範囲内の量で用いられ、1回でまたは1日1〜4回に用量を分割して投与され得る。
【0167】
存在する場合は、チアゾリジンジオン 抗糖尿病剤は、約0.01〜約2000 mg/日の範囲内の量で用いられ、1回でまたは1日1〜4回に用量を分割して用いられ得る。
【0168】
存在する場合は、インスリンは、フィジシャンズ デスク レファレンス(Physician's Desk Reference)に示される製剤形、量、投薬で用いられ得る。
【0169】
存在する場合は、GLP−1ペプチドは、経口バッカル製剤で、鼻腔内または非経口(米国特許第5,346,701号(テラテック)、第5,614,492号および第5,631,224号に開示;これらは本明細書に引用される)で投与され得る。
【0170】
他の抗糖尿病剤はまた、PPARα/γ両アゴニスト[例えば、AR−HO39242(アストラ/ゼネカ)、GW−409544(グラクソ−ウェルカム)、KRP297(キョーリン メルク)]、並びにムラカミらの文献[Murakami et al,「A Novel Insulin Sensitizer Acts As a Coligand for Peroxisome Proliferation-Activated Receptor Alpha (PPAR alpha) and PPAR gamma. Effect on PPAR alpha Activation on Abnormal Lipid Metabolism in Liver of Zucker Fatty Rats」, Diabetes 47,1841-1847 (1998)]に開示されるものでもあり得る。
【0171】
抗糖尿病剤は、例えば米国特許出願第09/679,027号(2000年10月4日出願,代理人ファイル番号LA49 NP、ここに述べられた投与量を用いる)に開示されるようなSGLT2阻害剤であり得る。好ましくは上記の出願で好ましいとされる化合物である。
【0172】
抗糖尿病剤は、例えば米国特許出願第09/391,053号(1999年9月7日出願)および米国特許出願第09/519,079号(2000年3月6日出願)(代理人ファイル番号LA27 NP、ここに述べられた投与量を用いる)に開示されるようなaP2阻害剤であり得る。好ましくは上記の出願で好ましいとされる化合物である。
【0173】
抗糖尿病剤は、米国特許出願第09/788,173号(2001年2月16日出願)(代理人ファイル番号LA50)、WO99/38501、WO99/46272、WO99/67279 (プロバイオドラッグ)、WO99/67278 (プロバイオドラッグ)、WO99/61431 (プロバイオドラッグ)、NVP−DPP728A(1−[[[2−[(5−シアノピリジン−2−イル)アミノ]エチル]アミノ]アセチル]−2−シアノ−(S)−ピロリジン)(ノバルティス)(好適化合物)(Hughes et al, Biochemistry, 38 (36), 11597-11603, 1999に開示)、TSL−225(トリプトフィル−1,2,3,4−テトラヒドロイソキノリン−3−カルボン酸(Yamada et al, Bioorg. & Med. Chem. Lett. 8 (1998) 1537-1540に開示)、2−シアノピロリジド化合物および4−シアノピロリジド化合物(Ashworth et al, Bioorg. & Med. Chem. Lett., Vol. 6, No. 22, pp 1163-1166および2745-2748 (1996)に開示)(上記引用文献に述べられた投与量を用いる)に開示されるようなDP4阻害剤であり得る。
【0174】
本発明の式Iの化合物と組み合わせて適宜用いられ得るメグリチナイドは、レパグリニド、ナテグリニド(ノバルティス)またはKAD1229(PF/キッセイ)であり得、レパグリニドが好ましい。
【0175】
式Iの化合物は、メグリチナイド、PPARγアゴニスト、PPARα/γ両アゴニスト、aP2阻害剤、DP4阻害剤またはSGLT2阻害剤との重量比が、約0.01:1〜約100:1、好ましくは約0.05〜約10:1の範囲内で用いられる。
【0176】
式Iの化合物とともに適宜用いられ得る他のタイプの治療剤は、β3アドレナリン作動性アゴニスト、リパーゼ阻害剤、セロトニン(およびドパミン)再取り込み阻害剤、aP2阻害剤、甲状腺受容体アゴニストおよび/または摂食障害剤を含む、1、2、3またはそれ以上の抗肥満剤であり得る。
【0177】
式Iの化合物と組み合わせて適宜用いられ得るβ3アドレナリン作動性アゴニストは、AJ9677(武田/大日本)、L750355(メルク)、またはCP331648(ファイザー)または他の公知のβ3アゴニスト(米国特許第5,541,204号、第5,770,615号、第5,491,134号、第5,776,983号および第5,488,064号に開示)であり得、AJ9677、L750,355およびCP331648が好ましい。
【0178】
式Iの化合物と組み合わせて適宜用いられ得るリパーゼ阻害剤は、オルリスタットまたはATL−962(アリザイム)であり得、オルリスタットが好ましい。
【0179】
式Iの化合物と組み合わせて適宜用いられ得るセロトニン(およびドパミン)再取り込み阻害剤は、シブトラミン、トピラメート(ジョンソンアンドジョンソン)またはアキソキン(リジェネロン)であり得、シブトラミンおよびトピラメートが好ましい。
【0180】
式Iの化合物と組み合わせて適宜用いられ得る甲状腺受容体アゴニストは、W097/21993 (U. Cal SF)、W099/00353(カロバイオ)、GB98/284425(カロバイオ)および米国仮出願第60/183,223号(2000年2月17日出願)に開示される、甲状腺受容体リガンドであり得、カロバイオの出願および上記米国仮出願の化合物が好ましい。
【0181】
式Iの化合物と組み合わせて適宜用いられ得る摂食障害剤は、デキサアンフェタミン、フェンテルミン、フェニルプロパノールアミンまたはマジンドールであり得、デキサアンフェタミンが好ましい。
【0182】
上述される様々な抗肥満剤は、式Iの化合物と同一の投与形態でまたは異なる投与形態で、当該技術分野またはPDRにおいて一般的に公知であるような投与量および投与計画で用いられ得る。
【0183】
本発明の式Iの化合物と組み合わせて用いられ得る降圧剤には、ACE阻害剤、アンジオテンシンII受容体アンタゴニスト、NEP/ACE阻害剤、並びにカルシウムチャンネルブロッカー、β−アドレナリン作動性遮断薬、および利尿剤を含む他のタイプの降圧剤が含まれる。
【0184】
本明細書で用いられ得るアンジオテンシン変換酵素阻害剤には、上述のOndetti et alの米国特許第4,046,889号に開示される誘導体のいずれかのような、置換されたプロリン誘導体のようなメルカプト(−S−)部位を含む誘導体が含まれ、カプトプリル、すなわち、1−[(2S)−3−メルカプト−2−メチルプロピオニル]−L−プロリンが好ましく、ゾフェノプリルで米国特許第4,316,906号に開示の誘導体のいずれかのような置換されたプロリンのメルカプトアシル誘導体が好ましい。
【0185】
本明細書で用いられ得るメルカプトを含むACE阻害剤の他の例には、レンチアプリル(フェンチアプリル、参天)(Clin. Exp. Pharmacol. Physiol. 10: 131 (1983)に開示);並びにピボプリルおよびYS980が含まれる。
【0186】
本明細書で用いられ得るアンジオテンシン変換酵素阻害剤の他の例には、上述の米国特許第4,374,829号に開示されるいずれかが含まれ、N−(1−エトキシカルボニル−3−フェニルプロピル)−L−アラニル−L−プロリン、すなわち、エナラプリルが好ましく、(S)−1−[6−アミノ−2−[[ヒドロキシ−(4−フェニルブチル)ホスフィニル]オキシ]−1−オキソヘキシル]−L−プロリンまたは(セロナプリル)で米国特許第4,452,790号に開示されるホスホン酸で置換されたアミノまたはイミノ酸またはその塩のいずれかが好ましく、フォシノプリルで上述された米国特許第4,168,267号に開示されるホスフィニルアルカノイルプロリン化合物が好ましく、米国特許第4,337,201号に開示されるホスフィニルアルカノイルで置換されたプロリン化合物のいずれか、および上記で述べられている米国特許第4,432,971号に開示されるホスホンアミデート化合物が含まれる。
【0187】
本明細書で用いられ得るACE阻害剤の他の例には、ビーチャムのBRL 36,378(欧州特許出願第80822号および第60668号に開示);中外のMC−838(C. A. 102:72588vおよびJap. J. Pharmacol. 40:373 (1986)に開示);チバ−ガイギィのCGS 14824(3−([1−エトキシカルボニル−3−フェニル−(1S)−プロピル]アミノ)−2,3,4,5−テトラヒドロ−2−オキソ−1−(3S)−ベンゾアゼピン−1−酢酸 HCl)(英国特許第2103614号に開示)およびCGS 16,617(3(S)−[[(1S)−5−アミノ−1−カルボキシペンチル]アミノ]−2,3,4,5−テトラヒドロ−2−オキソ−1H−1−ベンゾアゼピン−1−エタン酸)(米国特許第4,473,575号で開示);セタプリル(アラセプリル、大日本)(Eur. Therap. Res. 39: 671(1986);40: 543 (1986)に開示);ラミプリル(ヘキスト)(欧州特許第79-022号およびCurr. Ther. Res. 40: 74 (1986)に開示);Ru 44570(ヘキスト)(Arzneimittelforschung 34: 1254 (1985)に開示)、シラザプリル(ホフマン−ラロッシュ)(J. Cardiovasc. Pharmacol. 9: 39 (1987)に開示);R 31−2201(ホフマン−ラロッシュ)(FEBS Lett. 165: 201 (1984)に開示);リシノプリル(メルク)、インダラプリル(デラプリル)(米国特許第4,385,051号に開示);インドラプリル(シェーリング)(J. Cardiovasc. Pharmacol. 5: 643, 655 (1983) に開示)、スピラプリル(シェーリング)(Acta. Pharmacol. Toxicol. 59 (Supp. 5): 173 (1986)に開示);ペルインドプリル(セルビエ)(Eur. J. clin. Pharmacol. 31: 519 (1987)に開示);キナプリル(ワーナーランバード)(米国特許第4,344,949号に開示)およびCI925(ワーナーランバード)([3S−[2−[R(*)R(*)]]3R(*)]−2−[2−[[1−(エトキシ−カルボニル)−3−フェニルプロピル]アミノ]−1−オキソプロピル]−1,2,3,4−テトラヒドロ−6,7−ジメトキシ−3−イソキノリンカルボン酸 HCl)(Pharmacologist 26 : 243, 266 (1984)に開示)、WY44221(ワイエス)(J. Med. Chem. 26: 394 (1983)に開示)が含まれる。
【0188】
好ましいACE阻害剤は、カプトプリル、フォシノプリル、エナラプリル、リシノプリル、キナプリル、ベナゼプリル、フェンチアプリル、ラミプリルおよびモエキシプリルである。
【0189】
NEP/ACE阻害剤はまた、それらが中性エンドペプチダーゼ(NEP)阻害剤活性およびアンジオテンシン変換酵素(ACE)阻害剤活性を有するので、本明細書で用いられる。本明細書の使用に適したNEP/ACE阻害剤の例は、米国特許第5,362,727号、第5,366,973号、第5,225,401号、第4,722,810号、第5,223,516号、第4,749,688号、米国特許第5,552,397号、米国特許第5,504,080号、米国特許第5,612,359号、米国特許第5,525,723号、欧州特許出願第0599,444号、第0481,522号、第0599,444号、第0595,610号、欧州特許出願第0534363A2号、第534,396号および第534,492号、および欧州特許出願第0629627A2号に開示されている例が含まれる。
【0190】
好ましくは、上記特許/特許出願で好適として示されるNEP/ACE阻害剤およびその投与量であり(該米国特許は本明細書に引用される);最も好ましくは、オマパトリラット、BMS189,921([S−(R*,R*)]−ヘキサヒドロ−6−[(2−メルカプト−1−オキソ−3−フェニルプロピル)アミノ−2,2−ジメチル−7−オキソ−1H−アゼピン−1−酢酸(ゲモパトリラット))およびCGS30440である。
【0191】
本明細書の使用に適したアンジオテンシンII受容体アンタゴニスト(これらもまたアンジオテンシンIIアンタゴニストまたはAIIアンタゴニストとして本明細書に引用される)には、これらに限らないが、イルベサルタン、ロサルタン、バルサルタン、カンデサルタン、テルミサルタン、タソサルタンまたはエプロサルタンが含まれ、イルベサルタン、ロサルタンまたはバルサルタンが好ましい。
【0192】
錠剤またはカプセル剤のような好ましい経口製剤には、約0.1〜約500 mg、好ましくは約5〜約200 mg、より好ましくは約10〜約150 mgの範囲内の量で、ACE阻害剤またはAIIアンタゴニストが含まれる。
【0193】
非経口投与として、ACE阻害剤、アンジオテンシンIIアンタゴニストまたはNEP/ACE阻害剤は、約0.005 mg/kg〜約10 mg/kg、好ましくは約0.01 mg/kg〜約1 mg/kgの範囲内の量で用いられる。
【0194】
薬剤が静脈内投与される場合、例えば蒸留水、生理食塩水、リンゲル液または他の従前のキャリアのような従前のビークル中製剤化される。
【0195】
ACE阻害剤およびAIIアンタゴニスト、並びに本明細書で開示される他の降圧剤の投与量は、フィジシャンズ デスク レファレンス(Physician's Desk Reference,PDR)の最新版に示されるような量であることが望ましい。
【0196】
本明細書の使用に適した好ましい降圧剤の他の例には、オマパトリラット(バンレブ(Vanlev、登録商標))、アムロジピン ベスィレート(ノルバスク(登録商標))、プラゾシン HCl(ミニプレス(登録商標))、ベラパミル、ニフェジピン、ナドロール、ジルチアゼム、フェロジピン、ニソルジピン、イスラジピン、ニカルジピン、アテノロール、カルベジロール、ソタロール、テラゾシン、ドキサゾシン、プロプラノロール、およびクロニジン HCl(カタプレス(登録商標))が含まれる。
【0197】
式Iの化合物と組み合わせて用いられ得る利尿剤には、ヒドロクロロチアジド、トラセミド、フロセミド、スピロノラクトン、およびインダパミドが含まれる。
【0198】
式Iの化合物と組み合わせて用いられ得る抗血小板剤には、アスピリン、クロピドグレル、チクロピジン、ジピリダモール、アブシキシマブ、チロフィバン、エプチフィバチド、アナグレリド、およびイフェトロバンが含まれ、クロピドグレルおよびアスピリンが好ましい。
【0199】
抗血小板薬は、PDRに示された量を用い得る。イフェトロバンは、米国特許第5,100,889号に示される量で用いられ得る。
【0200】
本発明の式Iの化合物と組み合わせて本明細書での使用に適した抗骨多孔剤には、MK−217(アレンドロネート)(フォサマックス(登録商標))のような副甲状腺ホルモンまたはビスホスホネートが含まれる。用いる投与量は、フィジシャンズ デスク レファレンス(Physician's Desk Reference)に記載されるような量である。
【0201】
本発明の方法を実施する場合、医薬組成物は、構造Iの化合物を含み、他の治療剤を含んだり含まなかったり、医薬のビークルまたは希釈剤と共に用いられる。医薬組成物は、従前の固体または液体のビークルまたは希釈剤、および目的の投与方法に適したタイプの医薬添加剤を用いて製剤化される。本化合物は、ヒト、サル、イヌなどを含む哺乳類に、経口経路(例えば、錠剤、カプセル剤、顆粒剤または散剤)で投与されるか、または注射製剤の形態で非経口経路で投与される。成人への用量は、好ましくは1日50〜2,000 mgの間で、単一用量で、または1日1〜4回の個々の用量の形態で投与され得る。
【0202】
経口投与用の典型的なカプセル剤には、構造Iの化合物(250 mg)、乳糖(75 mg)およびステアリン酸マグネシウム(15 mg)が含まれる。該混合物は60メッシュのふるいを通り、1号ゼラチンカプセルに詰められる。
【0203】
典型的な注射製剤は、バイアルに構造Iの化合物(250 mg)を無菌充填し、無菌凍結乾燥し、封かんして製造される。使用に際しては、バイアルの内容物は生理食塩水(2 mL)と混合し、注射製剤を製造する。
【0204】
以下の実施例は本発明の好ましい態様を示す。
【0205】
以下の略号は実施例に用いられる:
Ph=フェニル
Bn=ベンジル
t−Bu=三級ブチル
Me=メチル
Et=エチル
TMS=トリメチルシリル
TMSN3=トリメチルシリルアジド
TBS=tert−ブチルジメチルシリル
FMOC=フルオレニルメトキシカルボニル
Boc=tert−ブトキシカルボニル
Cbz=カルボベンジルオキシまたはカルボベンゾキシまたはベンジルオキシカルボニル
THF=テトラヒドロフラン
Et2O=ジエチルエーテル
hex=ヘキサン
EtOAc=酢酸エチル
DMF=ジメチルホルムアミド
MeOH=メタノール
EtOH=エタノール
i−PrOH=イソプロパノール
DMSO=ジメチルスルホキシド
DME=1,2−ジメトキシエタン
DCE=1,2 ジクロロエタン
HMPA=ヘキサメチルリン酸トリアミド
HOAcまたはAcOH=酢酸
TFA=トリフルオロ酢酸
TFAA=トリフルオロ無水酢酸
i−Pr2NEt=ジイソプロピルエチルアミン
Et3N=トリエチルアミン
NMM=N−メチルモルホリン
DMAP=4−ジメチルアミノピリジン
NaBH4=水素化ホウ素ナトリウム
NaBH(OAc)3=三アセトキシ水素化ホウ素ナトリウム
DIBALH=水素化ジイソブチルアルミニウム
LiAlH4=水素化アルミニウムリチウム
n−BuLi=n−ブチルリチウム
Pd/C=パラジウム炭素
PtO2=酸化白金
KOH=水酸化カリウム
NaOH=水酸化ナトリウム
LiOH=水酸化リチウム
K2CO3=炭酸カリウム
NaHCO3=炭酸水素ナトリウム
DBU=1,8−ジアザビシクロ[5.4.0]ウンデセ−7−エン
EDC(またはEDC・HCl)またはEDCI(またはEDCI・HCl)またはEDAC=3−エチル−3’−(ジメチルアミノ)プロピル−カルボジイミド塩酸塩(または1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩)
HOBTまたはHOBT・H2O=1−ヒドロキシベンゾトリアゾール水和物
HOAT=1−ヒドロキシ−7−アザベンゾトリアゾール
BOP試薬=ベンゾトリアゾール−1−イルオキシ−トリス(ジメチルアミノ)ホスホニウムヘキサフルオロリン酸塩
NaN(TMS)2=ヘキサメチルジシラジドナトリウムまたはビス(トリメチルシリル)アミドナトリウム
Ph3P=トリフェニルホスフィン
Pd(OAc)2=酢酸パラジウム
(Ph3P)4Pd0=テトラキストリフェニルホスフィンパラジウム
DEAD=ジエチルアゾジカルボキシレート
DIAD=ジイソプロピルアゾジカルボキシレート
Cbz−Cl=ベンジルクロロホルメート
CAN=硝酸セリウムアンモニウム
SAX=強力陰イオン交換体
SCX=強力陽イオン交換体
Ar=アルゴン
N2=窒素
min=分
hまたはhr=時
L=リットル
mL=ミリリットル
μL=マイクロリットル
g=グラム
mg=ミリグラム
mol=モル
mmol=ミリモル
meq=ミリ等量
RT=室温
satまたはsat’d=飽和
aq.=水の
TLC=薄層クロマトグラフィ
HPLC=高速液体クロマトグラフィ
LC/MS=高速液体クロマトグラフィ/マススペクトル
MSまたはMass Spec=マススペクトル
NMR=核磁気共鳴
NMRスペクトルデータ:s=シングレット;d=ダブレット;m=マルチプレット;br=ブロード;t=トリプレット
mp=融点
【0206】
実施例1
【化44】
A.
【化45】
メルドラム酸(9.4 g; 65 mmol)およびピリジン(8.0 g; 100 mmol)の0℃のCH2Cl2溶液に、2時間かけて3−メトキシフェニルアセチルクロリド(10.0 g; 54 mmol)を滴下して加えた。生じた混合物を室温で2時間攪拌し、次いで2N HCl水およびCH2Cl2で分液処理した。有機層を乾燥し(Na2SO4)、減圧濃縮して、油状物としてクルードなパートAの化合物を得た。この物質をさらに精製することなく、次工程に用いた。
【0207】
B.
【化46】
クルードなパートAの化合物およびアニリン(5.0 g; 54 mmol)のトルエン(20 mL)溶液を3時間加熱還流した。反応液を次いで1M HCl水で洗浄し、次いで少量になるまで減圧濃縮し、目的の生成物のパートBの化合物を黄色の固形物として沈殿させた(9.0 g; 59%)。
【0208】
C.
【化47】
0℃のH2SO4水(1.84M 溶液の5 mL)に、パートBの化合物(6.0 g; 14 mmol)、NaNO2(1.38 g; 20 mmol)および1M NaOH水(14 mL)の溶液を、20分かけて滴下して加えた。反応混合物を0℃で30分間攪拌し、生じた沈殿物を濾別し、H2Oで洗浄して、黄色の固形物を得た。この物質をクロマトグラフィで精製して(SiO2;hex:EtOAcを5:1から3:1に段階的に濃度勾配する)、黄色の結晶としてパートCの化合物(3.0 mg; 68%)を得た。
【0209】
D.
【化48】
パートCの化合物(0.100 g; 0.32 mmol)、フェニルヒドラジン(0.060 g; 0.55 mmol)およびMgSO4(200 mg)の溶液を、EtOH(10 mL)中2時間還流し、その時点で出発物質は分析用HPLCで消費されていた。揮発物を減圧留去し、残渣をヘキサン/CH2Cl2(1:1)から再結晶して、黄色の結晶としてパートDの化合物を得た(90 mg; 70%)。
【0210】
E.
【化49】
パートDの化合物(90 mg; 0.22 mmol)、TFAA(1 mL)およびTFA(1 mL)の混合物を、シールしたチューブ中45℃で10時間加熱した。この時点で出発物質は分析用HPLCで消費されていた。揮発物を減圧留去し、該残渣をEtOAcおよびNaHCO3水で分液処理した。有機相を乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2;hex:EtOAc=3:1)、黄色の固形物としてパートEの化合物を得た(30 mg; 35%)。
【0211】
F.
【化50】
パートEの化合物(30 mg; 0.078 mmol)の−70℃のCH2Cl2溶液(2.0 mL)に、BBr3(1M CH2Cl2溶液の1.0 mL)を滴下して加えた。該混合物を0℃まで加温して、0℃で3時間攪拌した。反応液を−20℃に冷却し、NH4Cl水溶液でクエンチした。この混合物を室温まで加温し、30分間攪拌し、次いでEtOAcで抽出した。有機相を1M HCl水および水で連続して洗浄し、次いで乾燥し(Na2SO4)、減圧濃縮して、油状物としてクルードなパートFの化合物(30 mg; 99%)を得て、さらに精製することなく、次工程に用いた。
【0212】
G.
【化51】
パートFの化合物(30 mg; 0.081 mmol)、5−メチル−2−フェニル オキサゾール−4−エタノール メシレート(30 mg; 0.11 mmol;実施例11に記載のように調製)およびK2CO3(500 mg; 3.61 mmol)のDMF混合溶液(3 mL)を、80℃で12時間攪拌した。LC/MSで、出発物質が完全に消費されたのが示唆された。反応混合物を濾過し、濾液を減圧濃縮して、油状物を得て、これをクロマトグラフィで精製して(SiO2;hex:EtOAc=3:1)、淡褐色の固形物としてパートGの化合物を得た(12 mg; 36%)。
【0213】
H.
【化52】
パートGの化合物(38 mg; 0.054 mmol)およびKOH(200 mg; 3.6 mmol)のEtOH(30 mL)溶液を、シールしたチューブ中90℃で24時間加熱した。反応混合物をEtOAcおよび1M HCl水で分液処理した。有機相を水で洗浄し、乾燥し(Na2SO4)、減圧濃縮した。生じた油状物をプレパラティブHPLC(YMC 逆相カラム;B:A=30:70からB=100%に連続的に濃度勾配)で精製して、固形物として標題化合物を得た(8 mg; 31%)。
[M+H]+ = 481.1
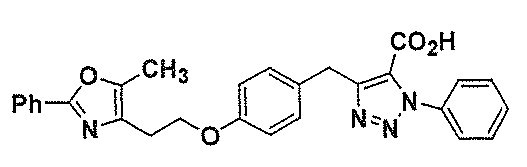
1H NMR (CDCl3 ; 400 MHz)δ : 2.43 (s, 3H), 3.05 (t, 2H; J=Hz), 4.26 (t, 2H; J=Hz), 4.35 (s, 2H), 6.73 (dd, 1H; J=Hz), 6.93 (d, 1H ; J=Hz), 7.14 (dd, 2H; J=Hz), 7.41 (t, 1H ; J=Hz), 7.47-7.54 (m, 5H), 8.05 (dd, 2H; J=Hz), 8.10 (dd, 2H; J=Hz), 11.32 (br s, 1H)
13C NMR (CDCl3 ; 100 MHz)δ: 10.2, 24.5, 31.7, 65.6, 113.6, 114.7, 119.4, 121.6, 124.5, 126.7, 128.4, 129.2, 129.3, 129.5, 130.1, 131.9, 137.5, 139.1, 139.6, 146.8, 151.4, 157.9, 160.2, 163.5
【0214】
実施例1(別の合成方法)
【化53】
A.
【化54】
3−ヒドロキシフェニル酢酸(3.89 g;25 mmol)および濃H2SO4(4滴)のMeOH溶液(30 mL)を、終夜加熱還流し、次いで室温まで冷却し、減圧濃縮した。該残渣をEtOAc(150 mL)および飽和NaHCO3水(20 mL)で分液処理した。有機相を乾燥し(MgSO4)、減圧濃縮して、油状物としてパートAの化合物を得た(3.80 g; 92%)。
【0215】
B.
【化55】
パートAの化合物(5.50 g; 33 mmol)、5−メチル−2−フェニル−オキサゾール−4−エタノール メシレート(5.43 g; 19 mmol;実施例11で記載したように調製)およびK2CO3(5.50 g; 40 mmol)のMeCN(50 mL)混合液を、終夜加熱還流し、次いで室温まで冷却し、濾過した。濾液を減圧濃縮し、次いでEtOAc(150 mL)および1N NaOH水(15 mL)で分液処理した。有機相を1N NaOH水(15 mL)で洗浄し、乾燥し(MgSO4)、減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2;ヘキサンからヘキサン:EtOAc=7:3への10分かけた連続的な濃度勾配;次いで15分間hex:EtOAc=7:3、次いでhex:EtOAc=7:3から2:3への5分間の連続的な濃度勾配、次いで15分間hex:EtOAc=2:3)、粘性の油状物としてパートBの化合物を得た(4.30 g; 64%)。
【0216】
C.
【化56】
パートBの化合物(4.30 g;12 mmol)およびLiOH・H2O(1.02 g;24 mmol)のTHF:H2O=1:1(60 mL)の混合溶液を、室温で終夜攪拌し、その後HCl水(1N 溶液の15 mL)を加えた。有機溶媒を減圧留去し、水相をEtOAc(2×120 mL)で抽出した。有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮した。該残渣をトルエン(50 mL)からストリッピングし、パートCの化合物(4.12 g; 100%)を得て、さらに精製することなく、次工程に用いた。
【0217】
D.
【化57】
パートCの化合物(4.12 g;12 mmol)の無水CH2Cl2溶液に、シュウ酸クロリドのCH2Cl2溶液(2M溶液の15.3 mL; 15 mmol)を滴下して加えた。該混合物を室温で2時間攪拌し、次いで減圧濃縮した。該残渣をトルエン(50 mL)からストリッピングし、黄色の固形物としてパートDの化合物を得て、さらに精製することなく、次工程に用いた。
【0218】
E.
【化58】
メルドラム酸(2.16 g;15 mmol)の0℃の無水CH2Cl2(44 mL)溶液に、15分間かけてピリジン(3.63 mL; 45 mmol)を滴下して加えた。パートDの化合物の無水CH2Cl2(44 mL)溶液を、次いで2時間かけてシリンジポンプで滴下して加えた。該反応液を室温まで加温し、終夜室温で攪拌し、その後EtOAc(300 mL)およびHCl水(1N溶液の30 mL)で分液処理した。有機相を乾燥し(MgSO4)、減圧濃縮して、パートEの化合物を得た。
【0219】
F.
【化59】
クルードなパートEの化合物およびアニリン(1.1 mL;12 mmol)のトルエン(22 mL)溶液を、2時間加熱還流した。該反応液をEtOAc(150 mL)および1M HCl水(20 mL)で分液処理し;有機相を減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2;100%ヘキサンからhex:EtOAc=2:3、hex:EtOAc=2:5に段階的に濃度勾配する)、黄色の固形物として沈殿したパートFの化合物 を得た(4.27 g;3工程の全収率77%)。
【0220】
G.
【化60】
パートFの化合物(4.27 g; 9.40 mmol)、p−トルエンスルホニルアジド(2.50 mg; 12.7 mmol)およびEt3N(1.83 mL; 13.1 mmol)のCH2Cl2(60 mL)溶液を、室温で2.5時間攪拌した。揮発物を減圧留去し、該残渣をクロマトグラフィで精製して(SiO2 ;hex:EtOAc=1:1からEtOAc=100%、EtOAc:MeOH=10:1へと段階的に濃度勾配)、黄色の固形物としてパートGの化合物を得た(3.50 g; 77%)。
【0221】
H.
【化61】
パートGの化合物(3.50 mg; 7.24 mmol)、ベンジルアミン(1.13 mL; 11.1 mmol)およびTiCl4(1M CH2Cl2溶液の7.24 mL;7.24 mmol)のDCE(100 mL)混合溶液を、シールしたチューブ中2時間88℃に加熱した。該反応混合物を室温まで冷却し、EtOAc(200 mL)およびH2O(50 mL)で分液処理した。有機相を乾燥し(MgSO4)、減圧濃縮した。該残渣をクロマトグラフィで精製し(SiO2 ; ヘキサン=100%からhex:EtOAc=1:1に段階的に濃度勾配する)、淡褐色固形泡状物としてパートHの化合物を得た(2.30 g; 55%)。
【0222】
I.
【化62】
パートHの化合物(2.0 g; 3.51 mmol)およびKOH(4.35 g; 77 mmol)の混合物を、EtOH(75 mL)中118℃で3時間加熱した。この時点で、HPLC/MSにより反応終結を確認した。該反応混合物を室温まで冷却し、EtOAc(150 mL)、H2O(20 mL)および過剰の濃HCl(6 mL)で分液処理した。有機相をH2Oで洗浄し、乾燥し(MgSO4)、減圧濃縮して、褐色固形物としてクルードな酸化合物を得た。この物質をHClのMeOH飽和溶液(30 mL)に溶かし、該反応液を室温で4日間攪拌し、次いで減圧濃縮した。該残渣をEtOAc(150 mL)および飽和NaHCO3水(20 mL)で分液処理した。有機相を減圧濃縮し、該残渣をクロマトグラフィで精製して(SiO2;20分かけてヘキサン=100%からhex:EtOAc=1:1に連続的に濃度勾配し、次いで20分間hex:EtOAc=1:1にする)、固形物としてパートIの化合物を得た(1.35 g; 76%)。
【0223】
J.
【化63】
パートIの化合物(1.35 g; 2.65 mmol)および10%パラジウム炭素(1.35 g)のMeOH(60 mL)混合液および飽和HClのMeOH(1 mL)溶液を、常圧下H2中(バルーンで)70時間攪拌した。バルーンを除き、MeOH(60 mL)を加えて、該混合物を加熱還流し、熱濾過した。濾液を減圧濃縮して、白色固形物としてパートJの化合物を得た(1.10 g; 91%)。
【0224】
K.
【化64】
パートJの化合物(25 mg; 0.55 mmol)、フェニルボロン酸(22 mg;1.80 mmol)およびCu(OAc)2(16 mg; 0.88 mmol)の混合物に、ピリジン(50μL)およびEt3N(50μL)を加えた。該混合物を室温で終夜攪拌し、次いでEtOAcおよびH2O(それぞれ10 mL)で分液処理した。有機相を減圧濃縮し、該残渣をクロマトグラフィで精製して(SiO2;ヘキサン:EtOAcを5:1から3:1へ段階的に濃度勾配)、油状物としてパートKの化合物を得た(3 mg;10%)。
【0225】
L.
【化65】
パートKの化合物(3 mg;0.006 mmol)およびLiOH・H2O(2 mg; 0.48 mmol)のTHF:H2O=1:1(0.60 mL)混合溶液を、室温で4時間攪拌し、次いでTHFを減圧留去した。pHが〜3になるまで1N HCl水を加え、該混合物をEtOAc(5 mL)で抽出した。有機相を減圧濃縮し、該残渣をプレパラティブHPLC(YMC逆相ODS 20×100 mmカラム;流速=20 mL/分;B:A=25:75からB=100%の10分間の濃度勾配 + B=100%での5分間のホールド時間,溶媒A;H2O:MeOH:TFA=90:10:0.1および溶媒B;MeOH:H2O:TFA=90:10:0.1)で精製し、無色の油状物として標題化合物を得た(1.2 mg; 41%)。
[M+H]+ = 481
【0226】
実施例2
【化66】
フェニルヒドラジンの代わりに4−メチルフェニルヒドラジンを用いた以外は、実施例1に記載の方法を用いて、標題化合物を製造した。
[M+H]+ = 495.0
【0227】
実施例3
【化67】
A.
【化68】
メルドラム酸(9.4 g; 65 mmol)およびピリジン(8.0 g; 100 mmol)の0℃のCH2Cl2溶液に、2時間かけて4−メトキシフェニルアセチルクロリド(10.0 g; 54 mmol)を滴下して加えた。生じた混合物を室温で2時間攪拌し、次いで2N HCl水およびCH2Cl2で分液処理した。有機層を乾燥し(Na2SO4)、減圧濃縮して、油状物としてクルードなパートAの化合物を得た。この物質をさらに精製することなく、次工程に用いた。
【0228】
B.
【化69】
クルードなパートAの化合物およびアニリン(5.0 g; 54 mmol)のトルエン(20 mL)溶液を、3時間加熱還流した。反応液を次いで1M HCl水で洗浄し、次いで少量になるまで減圧濃縮し、黄色の固形物として沈殿した目的の生成物であるパートBの化合物を得た(7.5 g; 49%)。
【0229】
C.
【化70】
0℃のH2SO4水(1.84M溶液の5 mL)に、20分かけてパートBの化合物(2.0 g; 7.1 mmol)、NaNO2(0.73 g; 10.6 mmol)、1M NaOH水(7.06 mL)およびTHF(50 mL)の溶液を滴下して加えた。該反応混合物を0℃で30分間攪拌し;生じた沈殿物を濾別し、H2Oで洗浄し、黄色の固形物を得た。この物質をクロマトグラフィで精製して(SiO2;hex:EtOAcを5:1から3:1へ段階的に濃度勾配)、黄色の結晶としてパートCの化合物を得た(2.00 g; 91%)。
【0230】
D.
【化71】
パートCの化合物(0.250 g; 0.80 mmol)、フェニルヒドラジン(0.097 g; 0.90 mmol)およびMgSO4(2 g)の溶液を、EtOH(10 mL)中分析用HPLCで出発物質が消費された時点まで2時間還流した。揮発物を減圧留去し、該残渣をクロマトグラフィで精製して(SiO2;hex:EtOAcを3:1から1:1へ段階的に濃度勾配)、黄色の固形物としてパートDの化合物を得た(200 mg; 62%)。
【0231】
E.
【化72】
パートDの化合物(30 mg; 0.075 mmol)、TFAA(1 mL)およびTFA(1 mL)の混合物を、シールしたチューブ中45℃で10時間加熱した。この時点で、分析用HPLCで出発物質の消費を確認した。揮発物を減圧留去し、該残渣をEtOAcおよびNaHCO3水で分液処理した。有機相を乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2;hex:EtOAc=3:1)、黄色の固形物としてパートEの化合物を得た(25 mg; 86%)。
【0232】
F.
【化73】
パートEの化合物(25 mg; 0.065 mmol)の−70℃のCH2Cl2(2.0 mL)溶液に、BBr3(1M CH2Cl2溶液の1.0 mL)を滴下して加えた。該混合物を0℃まで加温し、0℃で3時間攪拌した。反応液を−20℃まで冷却し、NH4Cl水溶液でクエンチした。この混合物を室温まで加温し、30分間攪拌し、次いでEtOAcで抽出した。有機相を1M HCl水および水で連続して洗浄し、次いで乾燥し(Na2SO4)、減圧濃縮して、油状物としてクルードなパートFの化合物を得て(30 mg)、さらに精製することなく、次工程に用いた。
【0233】
G.
【化74】
パートFの化合物(30 mg; 0.081 mmol)、5−メチル−2−フェニルオキサゾール−4−エタノール メシレート(30 mg; 0.11 mmol;実施例11に記載のように調製)およびK2CO3(500 mg; 3.61 mmol)のDMF(3 mL)混合溶液を、80℃で12時間攪拌した。LC/MSで、出発物質が完全に消費されたのが示唆された。反応混合物を濾過し、濾液を減圧濃縮して、油状物を得て、これをクロマトグラフィで精製して(SiO2;hex:EtOAc=3:1)、固形物としてパートGの化合物を得た(13 mg; 2工程で28%)。
【0234】
H.
【化75】
パートGの化合物(0.013 g; 0.023 mmol)およびKOH(200 mg; 3.6 mmol)のEtOH(30 mL)溶液を、シールしたチューブ中90℃で24時間加熱した。該反応混合物をEtOAcおよび1M HCl水で分液処理した。有機相を水で洗浄し、乾燥し(Na2SO4)、減圧濃縮した。生じた油状物をプレパラティブHPLC(BMS−460913の精製で記載したように;以下参照)で精製し、固形物として標題化合物を得た(9 mg; 81%)。
[M+H]+ = 481.1
【0235】
実施例4
【化76】
フェニルヒドラジンの代わりに4−メチルフェニルヒドラジンを用いた以外は、実施例3の方法を用いて、標題化合物を製造した。
[M+H]+ = 495.1
【0236】
実施例5
【化77】
A.
【化78】
4−ヒドロキシフェニル酢酸メチル(2.66 g; 1.6 mmol)、5−フェニル−2−メチルオキサゾール−3−エタノール(3.25 g; 1.6 mmol)およびPh3P(5.0 g; 1.9 mmol)の0℃の無水THF(30 mL)溶液に、DEAD(3.5 g; 2.0 mmol)を滴下して加えた。該反応混合物を0℃で30分間攪拌し、次いで室温まで加温し、室温で終夜攪拌した。揮発物を減圧留去し、該残渣をクロマトグラフィで精製して(SiO2;段階的な濃度勾配;ヘキサン:EtOAc=5:1〜5:2)、白色固形物としてパートAの化合物を得た(3.5 g; 62%)。
【0237】
B.
【化79】
パートAの化合物(2.85 g;0.812 mmol)およびLiOH水(1M溶液の2.0 mL; 2.0 mmol)のTHF(2 mL)溶液を、室温で3時間攪拌した。この時点で、HPLC/MSによりすべての出発物質の消費が示された。揮発物を減圧留去し、反応液を1N HCl水で酸性にした。水相をEtOAc(2×250 mL)で抽出し;有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮して、クルードのフェニル酢酸化合物を得た。該クルードの酸の溶液に、シュウ酸クロリド(2M CH2Cl2溶液の10 mL)を加え、反応混合物を室温で3時間攪拌した。揮発物を減圧留去し、固形物としてパートBの化合物を得て、さらに精製することなく、次反応に用いた。
【0238】
C.
【化80】
メルドラム酸(980 mg; 678 mmol)およびピリジン(1.0 mL; 10 mmol)の0℃の無水CH2Cl2(10 mL)溶液に、パートBの化合物(2.0 g; 5.65 mmol)のCH2Cl2(5 mL)溶液を、2時間かけて滴下して加えた。該反応混合物を室温まで加温し、室温で2時間攪拌した。該混合物を次いで過剰の2N HCl水の添加で酸性にし、CH2Cl2(2×25 mL)で抽出した。有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮して、クルードのメルドラム酸付加体を得た。このクルードの生成物の溶液およびアニリン(600μL)のトルエン(10 mL)溶液を、3時間還流した。該反応液を室温まで冷却し、1N HCl水で洗浄した。 揮発物を減圧留去して、黄色の固形物としてパートCの化合物を得た(2.50 g; 97%)。
【0239】
D.
【化81】
0℃のH2SO4水溶液(1.84M溶液の0.60 mL; 1.10 mmol)に、20分かけてパートCの化合物(300 mg, 0.60 mmol)、NaNO2(64 mg; 1.0 mmol)および1N NaOH水(0.70 mL; 0.70 mmol)のTHF(10 mL)溶液を滴下して加えた。該反応混合物を室温で30分間攪拌し、その後沈殿物を濾別し、H2Oで洗浄して、黄色の固形物を得た。この物質をクロマトグラフィで精製して(SiO2;ヘキサン:EtOAc=5:1〜3:1)、黄色の固形物としてパートDの化合物を得た(250 mg; 84%)。
【0240】
E.
【化82】
ベンジルヒドラジン・2HCl(41 mg; 0.21 mmol)およびナトリウムエトキシド(21%EtOH溶液の200μL; 0.42 mmol)のエタノール(5 mL)溶液を、室温で2時間攪拌した。パートDの化合物(100 mg; 0.21 mmol)および無水MgSO4(200 mg)を次いで加え、該反応混合物を油浴中80℃に16時間加熱した。TLCで、すべての出発物質が消費されたのが示唆された。揮発物を減圧留去し、該残渣(クルードなトリアゾール−アニリド化合物)を2−エトキシエタノール(10 mL)に溶かした。この溶液を、150℃のKOH(1.0 g; 8 mmol)の2−エトキシエタノール(20 mL)溶液に加えた。該反応混合物を150℃で30分間加熱した。HPLC/MSで、アニリド化合物のすべてがこの時点で消費していることが示唆された。該反応混合物を室温まで冷却し、過剰の1N HCl水で酸性にし、EtOAc(3回)で抽出した。有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮した。該残渣をプレパラティブHPLC(YMC 逆相 ODS 30×250 mmカラム; 流速=25 mL/分;B:A=30:70からB=100%の30分間の連続的な濃度勾配 + B=100%での10分間のホールド時間,溶媒A;H2O:MeOH:TFA=90:10:0.1および溶媒B;MeOH:H2O:TFA=90:10:0.1)で精製し、MeOHからストリッピングして、白色固形物として標題化合物を得た(61 mg; 58%)。
[M+H]+ = 495.0
1H NMR (DMSO; 400 MHz) 2.34 (s, 3H), 2.87-2.92 (t, J=6.6 Hz, 2H), 4.14-4.17 (m, 4H), 5.65 (s, 2H), 6.82-6.85 (d, J=8.76 Hz, 2H), 7.09-7.11 (d, J=8.32 Hz, 2H), 7.26-7.40 (m, 5H), 7.42-7.55 (m, 3H), 7.94-7.97 (m, 2H)
【0241】
実施例6
【化83】
出発物質として4−ヒドロキシフェニル酢酸メチルの代わりに3−ヒドロキシフェニル酢酸メチルを用いた以外は、実施例5の方法を用いて、標題化合物を製造した。固形物として標題化合物を得た(6 mg)。
[M+H]+ = 495.2
【0242】
実施例7
【化84】
A.
【化85】
実施例5パートCの化合物(100 mg; 0.22 mmol)、p−トルエンスルホニルアジド(60 mg; 0.3 mmol)およびEt3N(50μL; 0.3 mmol)のCH2Cl2(3 mL)溶液を、室温で3時間攪拌し、この時点でTLCにより反応が完了していた。揮発物を減圧留去し、該残渣をクロマトグラフィで精製して(SiO2;hex:EtOAc=1:1からEtOAcへ、さらにCH2Cl2:MeOH :Et3N=10:1:1の段階的な濃度勾配)、黄色の固形物としてパートAの化合物を得た(100 mg; 95%)。
【0243】
B.
【化86】
パートAの化合物(100 mg; 0.21 mmol)、ベンジルアミン(30μL; 0.30 mmol)およびTiCl4(1M CH2Cl2溶液の300μL; 0.30 mmol)の1,2−ジクロロエタン(5 mL)溶液を、シールしたチューブ中18時間88℃で加熱した。この時点でLC/MSにより、目的のトリアゾール化合物の形成を確認した。該反応混合物を室温まで冷却し、EtOAcおよびH2O(それぞれ100 mL)で分液処理した。有機相を乾燥し(Na2SO4)、減圧濃縮して、油状物としてクルードなトリアゾール−アニリド化合物を得た。このクルードな物質およびKOH(300 mg)の混合物を、EtOH(3 mL)中80℃で3時間加熱した。この時点でHPLC/MSにより、反応が完了していた。該反応混合物を室温まで冷却し、EtOAcおよび過剰の1M HCl水で分液処理した。有機相をH2Oで洗浄し、乾燥し(Na2SO4)、減圧濃縮した。該残渣をプレパラティブHPLC(YMC 逆相 ODS 30×250 mmカラム;流速=25 mL/分;B:A=30:70からB=100%の10分間の連続的な濃度勾配 + B=100%での10分間のホールド時間,溶媒A;H2O:MeOH:TFA=90:10:0.1および溶媒B;MeOH:H2O:TFA=90:10:0.1)で精製して、固形物として標題化合物を得た(48 mg; 46%)。
[M+H]+ = 495.1
【0244】
実施例8
【化87】
標題化合物を製造するのに、実施例7パートAの1,4−置換された中間体の代わりに対応する1,3−置換された中間体 ジアゾ−β−ケトアミド:
【化88】
を用いた以外は、実施例7に記載の合成方法を用いた。この1,3−置換された中間体は、4−ヒドロキシフェニル酢酸メチルの代わりに3−ヒドロキシフェニル酢酸メチルを用いた以外は、実施例5パートCの化合物の合成に記載された方法に従い製造した。
[M+H]+ = 495.2
【0245】
実施例9および10
実施例7および8の調製方法を用いて、以下の類似体を製造した。
【化89】
[M+H]+ = 481.1
【化90】
[M+H]+ = 481.1
【0246】
実施例11
【化91】
A.
【化92】
4−ヒドロキシベンズアルデヒド(1.70 g, 12.3 mmol)、5−フェニル−2−メチル−オキサゾール−4−エタノール(メイブリッジ製; 2.50 g, 14.0 mmol)およびPh3P(4.20 g, 16.0 mmol)の0℃の乾燥THF(30 mL)溶液に、DEAD(3.20 g, 15.0 mmol)を滴下して加えた。該溶液を0℃で0.5時間攪拌し、次いで室温まで加温し、終夜攪拌した。該橙赤色の溶液を減圧濃縮し、該残渣をクロマトグラフィで精製して(hex:EtOAc=5:1から5:2への段階的な濃度勾配)、澄明でわずかに黄色の粘性の油状物としてパートAの化合物を得た(2.47 g, 65%)。
【0247】
パートAのアルデヒドを製造する別法:
(1)
【化93】
5−フェニル−2−メチル−オキサゾール−4−エタノール(20.00 g, 0.098 mol)の−5℃のCH2Cl2(100 mL)溶液に、メタンスルホニルクロリド(12.40 g, 0.108 mol)を1回で加えた(発熱反応)。−5℃に再冷却後、Et3N(11.1 g, 0.110 mol)を30分かけて(内温<3℃)ゆっくり加えた。該反応液を室温まで加温し、1時間(反応を分析用HPLCでモニターして)攪拌し、この時点で出発物質が消費した。反応液をHCl水(2×3N溶液50 mL)で洗浄した。水層を合わせて、CH2Cl2(50 mL)で抽出した。有機抽出物を合わせて、飽和NaHCO3水および食塩水(それぞれ50 mL)で連続して洗浄し、乾燥し(Na2SO4)、〜30 mLの量に濃縮した。メチルtert−ブチルエーテル(120 mL)を加え、該混合物を攪拌し;白色の固形物が形成された。該混合物を完全に結晶化するのに、−20℃に冷却した。生成物を濾過し、真空乾燥して、白色固形物として生成物のメシレートを得た(23.3 g, 85%)。母液を減圧濃縮し、メチルtert−ブチルエーテル/ヘプタンから再結晶して、生成物のメシレートの第二晶を得た(3.3 g, 12% ;全収率=97%)。
【0248】
(2)
【化94】
上記メシレート化合物(13.6 g, 0.048 mol)、4−ヒドロキシベンズアルデヒド(7.09 g, 0.058 mol)およびK2CO3(9.95 g, 0.072 mol)のDMF(110 mL)混合液を、100℃で2時間加熱した(分析用HPLCで反応完了)。該混合物を室温まで冷却し、次いで氷水(400 mL)に注ぎ、30分間攪拌した。固体の生成物を濾過し、冷水(3×25 mL)で洗浄し、50〜60℃で終夜減圧乾燥した。粗生成物をメチルtert−ブチルエーテル/ヘキサンから結晶化して、白色固形物としてパートAの化合物を得た(12.2 g, 82%; 2回)。
【0249】
B.
【化95】
プロピオール酸エチル(256 mg; 2.6 mmol)の−78℃のTHF (12 mL)溶液に、n−ブチルリチウム(2.5Mヘキサン溶液の1.04 mL; 2.6 mmol)を滴下して加えた。該溶液を−78℃で30分間攪拌し;パートAのアルデヒド(800 mg; 2.6 mmol)のTHF(3 mL)溶液を次いで滴下して加えた。該反応液を−70℃で1時間攪拌し、次いで飽和NH4Cl水を滴下して加えてクエンチした。該混合物を室温まで加温し、次いでEtOAcで抽出した。有機相をH2Oで洗浄し、乾燥し(Na2SO4)、減圧濃縮して、油状物としてクルードなパートBの化合物を得て、さらに精製することなく、次工程に用いた。
【0250】
C.
【化96】
上記からのクルードなパートBの化合物の0℃の乾燥MeCN(5 mL)溶液に、Et3SiH(620μL; 3.97 mmol)およびBF3・OEt2(384μL; 3.1 mmol)を連続的に加えた。該反応混合物を室温まで加温し、室温で2時間攪拌し、この時点で分析用HPLCのより、すべての出発物質が消費していた。揮発物を減圧留去し、該残渣をH2OおよびEtOAcで分液処理した。有機相をNaHCO3水で洗浄し、次いで減圧濃縮した。該粗生成物をクロマトグラフィで精製して(SiO2;ヘキサン:EtOAc=4:1)、白色の結晶としてパートCの化合物を得た(514 mg; 2工程で50%)。
【0251】
D.
【化97】
パートCの化合物(233 mg; 0.60 mmol)およびフェニルアジド(2 mL;Organic Syntheses Collective Volume IV,p. 75-77に従い、アニリンから調製)のトルエン(50 mL)混合液を、シールしたチューブ中130℃で18時間加熱した。該混合物を室温まで冷却し、減圧濃縮した。褐色の残渣をクロマトグラフィで精製して(SiO2;ヘキサン:EtOAc=4:1から2:1への段階的な濃度勾配)、固形物としてパートEの化合物の異性体生成物(100 mg; 32%):
【化98】
と同様にパートDの化合物を得た(50 mg; 16%)。
[M+H]+ = 509.0
【0252】
F.
【化99】
パートDの化合物(50 mg; 0.098 mmol)および1M LiOH水(1 mL; 1.0 mmol)のTHF(5 mL)溶液を、室温で終夜攪拌した。該反応液を1M HCl(2 mL; 2.0 mmol)で酸性にし、EtOAcで抽出した(2回)。有機抽出物を合わせて、H2Oで洗浄し、減圧濃縮した。該残渣をプレパラティブHPLCで精製して、白色固形物として標題化合物を得た(38 mg;2工程で13%)。
[M+H]+ = 481.2
【0253】
実施例12
【化100】
実施例11パートEの化合物(50 mg; 0.098 mmol):
【化101】
および1M LiOH水(1 mL; 1.0 mmol)のTHF(5 mL)溶液を室温で終夜攪拌した。反応液を1M HCl(2 mL; 2.0 mmol)で酸性にし、EtOAc(2回)で抽出した。有機抽出物を合わせて、H2Oで洗浄し、減圧濃縮した。該残渣をプレパラティブHPLCで精製し、白色固形物として標題化合物を得た(80 mg;2工程で26%)。
[M+H]+ = 481.1
【0254】
実施例13
【化102】
A.
【化103】
この中間体は、4−ヒドロキシベンズアルデヒドの代わりに出発物質として3−ヒドロキシベンズアルデヒドを用いた以外は、対応する1,4−誘導体の実施例11のパートAの方法を用いて製造した。
【0255】
B.
【化104】
プロピオール酸エチル(256 mg; 2.6 mmol)の−78℃のTHF(12 mL)溶液に、n−ブチルリチウム(2.5Mヘキサン溶液の1.04 mL; 2.6 mmol)を滴下して加えた。該溶液を−78℃で30分間攪拌し;パートAのアルデヒド化合物(800 mg; 2.6 mmol)のTHF溶液(3 mL)を次いで滴下して加えた。反応液を−70℃で1時間攪拌し、次いで飽和NH4Cl水を滴下して加えてクエンチした。該混合液を室温まで加温し、次いでEtOAcで抽出した。有機相をH2Oで洗浄し、乾燥し(Na2SO4)、減圧濃縮して、油状物としてクルードなパートBの化合物を得て、さらに精製することなく、次工程に用いた。
【0256】
C.およびD.
【化105】
パートAの化合物(230 mg; 0.57 mmol)およびフェニルアジド(2 mL; Organic Syntheses Collective Volume IV, p. 75-77の方法に従いアニリンから製造)のトルエン(50 mL)混合液を、シールしたチューブ中130℃で18時間加熱した。該混合物を室温まで冷却し、減圧濃縮した。褐色残渣をクロマトグラフィで精製して(SiO2 ;ヘキサン:EtOAc=4:1から2:1への段階的な濃度勾配)、パートDの化合物の異性体生成物(75 mg; 2工程で25%):
【化106】
と同様にパートCの化合物を得た(70 mg; 23%)。
【0257】
E.
【化107】
パートCの化合物(45 mg; 0.085 mmol)および1M LiOH水(1 mL; 1.0 mmol)のTHF(5 mL)溶液を、室温で24時間攪拌した。反応液を1M HCl(2 mL; 2.0 mmol)で酸性にし、EtOAcで抽出した(2回)。有機抽出物を合わせて、H2Oで洗浄し、減圧濃縮した。該残渣をプレパラティブHPLC(YMC 逆相 ODS 30×250 mmカラム;A:B=70:30からB=100%の30分間の連続的な濃度勾配,溶媒A;H2O:MeOH:TFA=90:10:0.1および溶媒B;MeOH:H2O:TFA=90:10:0.1;流速=25 mL/分)で精製し、白色固形物として標題化合物を得た(34 mg; 80%)。
[M+H]+ = 497.1
【0258】
実施例14
【化108】
実施例13パートDの化合物(45 mg; 0.085 mmol)および1M LiOH水(1 mL; 1.0 mmol)のTHF(5 mL)溶液を、終夜室温で攪拌した。反応液を1M HCl(2 mL; 2.0 mmol)で酸性にし、EtOAc(2回)で抽出した。有機抽出物を合わせて、H2Oで洗浄し、減圧濃縮した。該残渣をプレパラティブHPLC(実施例13の化合物の精製と同様の条件で)で精製し、白色固形物として標題化合物を得た(32 mg; 75%)。
[M+H]+ = 497.1
【0259】
実施例15
【化109】
実施例13パートCの化合物(35 mg; 0.067 mmol)の0℃の乾燥MeCN(2.5 mL)溶液に、Et3SiH(12 mg; 0.10 mmol)およびBF3・OEt2(14 mg; 0.10 mmol)を連続して加えた。該反応混合物を室温まで加温し、室温で2時間攪拌し、この時点で分析用HPLCにより、すべての出発が消費していた。揮発物を減圧留去し、該残渣をH2OおよびEtOAcで分液処理した。有機相をNaHCO3水で洗浄し、次いで減圧濃縮した。粗生成物を実施例13および14の合成で記載されたように1M LiOH水/THFで加水分解し、黄色の固形物として標題化合物を得た(26 mg; 2工程で80%)。
[M+H]+ = 481.1
【0260】
実施例16
【化110】
A.
【化111】
シアノ酢酸メチル(26 g; 256 mmol)およびナトリウムメトキシドのMeOH(0.5M溶液の152 mL; 76 mmol)溶液に、4−メトキシベンジルクロリド(10.0 g; 64 mmol)を室温で1時間かけて加えた。生じた乳濁液を3時間加熱還流し、その後揮発物を減圧留去した。該残渣をH2OおよびEt2Oで分液処理した。有機相をH2Oで洗浄し、乾燥し(Na2SO4)、ある程度減圧濃縮した。白色の固形の沈殿物を濾別し、濾液を減圧濃縮して、油状物を得た。このクルードな物質をクーゲルロール蒸留(沸点=180℃/0.3 mm Hg)で精製し、澄明な油状物としてパートAの化合物を得て(7.6g; 54%)、室温で結晶化して白色固形物として得た。
【0261】
B.
【化112】
パートAの化合物(7.6 g; 35 mmol)およびNaOH(4.4 g; 110 mmol)のH2O(50 mL)溶液を、室温で1時間攪拌した。該反応混合物をEt2O(50 mL)および濃塩酸(12 mL)で分液処理した。有機相を水で洗浄し、減圧濃縮し、乾燥して(Na2SO4)、室温で白色の固体になる残渣としてパートBの化合物を得た(7.1 g; 96%)。
【0262】
C.
【化113】
ジアゾ化アニリンのHCl溶液(Walker, T. K., J. Chem.Soc.,1924,1622-1625の方法に従い製造)を、NaOAc(338 mg; 4.9 mmol;フリーなHClを除去)で処理し、続いてパートBの化合物(1 g; 4.9 mmol)を0℃で加えた(CO2の発生した)。該反応混合物を0℃で24時間攪拌した。黄色シロップを水相から分離し、CH2Cl2に溶かした。該水相をCH2Cl2(2×20 mL)で抽出し;有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮して、油状物としてパートCの化合物を得た(40 mg; 7%)。
【0263】
D.
【化114】
パートCの化合物(20 mg; 0.062 mmol)の−78℃のCH2Cl2(2 mL)溶液に、BBr3(20 mg; 0.079 mmol)を加えた。反応液を−78℃で攪拌し、次いで室温まで加温した。ワークアップ(詳細は欠く)して、クルードな油状物としてパートDの化合物を得て(20 mg)、さらに精製することなく、次反応に用いた。
【0264】
E.
【化115】
パートDの化合物(20 mg; 0.064 mmol)、該メシレート化合物(30 mg; 0.11 mmol):
【化116】
およびK2CO3(100 mg; 0.72 mmol)のMeCN(5 mL)混合液を、80℃に加熱した。ワークアップして、油状物として粗生成物Eを得て(20 mg)、さらに精製することなく、次工程に用いた。
【0265】
F.
【化117】
粗生成物EおよびLiOH水(1M溶液の1 mL)のTHF溶液を、室温で終夜攪拌した。反応液を1M HCl(2 mL)で酸性にし、EtOAcで抽出した(2回)。有機抽出物を合わせて、H2Oで洗浄し、減圧濃縮した。該残渣をプレパラティブHPLC(実施例13の化合物の精製で記載のように)で精製し、白色固形物として標題化合物を得た(7 mg; 22%)。
[M+H]+ =480.2
【0266】
実施例17
以下の化合物は、4−メトキシベンジルクロリドの代わりにパートAの3−メトキシベンジルクロリドを用いた以外は、実施例16の方法を用いて調製した。
【化118】
[M+H]+ = 480.2
【0267】
実施例18
【化119】
A.
【化120】
メルドラム酸(4.33 g; 30 mmol)およびピリジン(7.0 mL; 100 mmol)の0℃のCH2Cl2(100 mL)溶液に、1時間かけて3−メトキシフェニルアセチルクロリド(5.0 g; 27 mmol)を滴下して加えた。生じた混合物を室温で2時間攪拌し、次いで2N HCl水およびCH2Cl2で分液処理した。有機層を乾燥し(Na2SO4)、減圧濃縮して、粗生成物を得た。この残渣をMeOH(20 mL)に溶かし、該溶液を3時間加熱還流した。該反応混合物を室温まで冷却し、揮発物を減圧留去して、澄明な油状物としてパートAの化合物を得た(5.0 g; 83%)。
【0268】
B.
【化121】
パートAの化合物(1.0 g; 4.5 mmol)、ジメチルホルムアミド ジメチルアセタール(600 mg; 5.0 mmol)のCH2Cl2(2.5 mL)溶液を、室温で2時間攪拌した。反応混合物を直接クロマトグラフィで精製し(SiO2 ; ヘキサン:EtOAc=1:1からEtOAcへの段階的な濃度勾配)、油状物としてパートBの化合物を得た(400 mg; 32%)。
【0269】
C.
【化122】
パートBの化合物(100 mg; 0.36 mmol)、フェニルヒドラジン(40 mg,0.38 mmol)および活性化した4Aモレキュラシーブズ(500 mg)の溶液を、100℃で10時間加熱した。この時点で分析用LC−MSにより、反応完了が示唆された。反応液を室温まで冷却し、濾過し、濾液を減圧濃縮した。該残渣をクロマトグラフィで精製し(SiO2 ; ヘキサン:EtOAc=4:1)、澄明な油状物としてパートCの化合物を得た(90 mg; 77%)。
【0270】
D.
【化123】
パートCの化合物(80 mg; 0.25 mmol)の−78℃のCH2Cl2(5 mL)溶液に、BBr3(124 mg; 0.50 mmol)を滴下して加えた。反応混合物を−78℃で30分間攪拌し、次いで室温まで加温し、室温で2時間攪拌した。揮発物を減圧留去し、該残渣をEtOAcおよびH2O(各5 mL)で分液処理した。水相をEtOAcで抽出した(2回)。有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮して、油状物を得た(フェノール酸化合物)。この物質を、室温で2時間MeOH(2 mL)のHClの飽和溶液中攪拌して、再エステル化した。揮発物を減圧留去し、該残渣をクロマトグラフィで精製して(SiO2; ヘキサン:EtOAc=3:1)、澄明な油状物としてパートDの化合物を得た(50 mg; 62%)。
【0271】
E.
【化124】
パートDの化合物(50 mg;0.16 mmol,実施例1パートFの化合物に代わり)、メシレート化合物(68 mg; 0.24 mmol):
【化125】
およびK2CO3(224 mg; 1.6 mmol)を用いてMeCN(5 mL)中、実施例1に記載のような同様のアルキル化を行い、粗生成物として生成物Eを得て(20 mg; 25%)、さらに精製することなく、次工程に用いた。
【0272】
F.
【化126】
クルードな生成物EのTHFおよびLiOH水(1M溶液の2 mL)溶液を、室温で終夜攪拌した。反応液を過剰の1M HCl水でpH〜2に酸性にし;水層をEtOAcで抽出した(3回)。有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮した。該残渣をプレパラティブHPLC(実施例13の化合物の精製で記載のように)で精製して、白色固形物として標題化合物を得た(9 mg; 12%)。
【0273】
実施例19
【化127】
パートAの3−メトキシフェニルアセチルクロリドの代わりに4−メトキシフェニルアセチルクロリドを用いた以外の実施例18の方法を用いて、実施例19の位置異性体を合成した。
[M+H]+ = 480.2
【0274】
実施例20
【化128】
A.
【化129】
0℃のプロパルギルマグネシウムブロミドのTHF溶液(0.5M 溶液の50 mL; 25 mmol)に、N2の常圧下3−アニスアルデヒド(1.36 g; 10 mmol)のTHF(10 mL)溶液を滴下して加えた。反応混合物を0℃で3時間攪拌し、次いで終夜室温まで加温し、その後すべての出発物質が消費した(TLC)。反応混合物を飽和NH4Cl水(30 mL)および氷(30 mL)に注意深く注いでクエンチした。水中の混合物をEtOAcで抽出した(2×150 mL)。有機抽出物を合わせて、H2O(3×150 mL)で洗浄し、乾燥し(Na2SO4)、減圧濃縮して、油状物としてパートAの化合物を得た(1.2 g; 79%)。この物質をさらに精製することなく、次工程に用いた。
【0275】
B.
【化130】
パートAの化合物(500 mg; 3.08 mmol)、Et3N(数滴)およびCH2Cl2(4 mL)の還流した混合液に、ジケテン(ケテン二量体; 336 mg; 4.0 mmol)のCH2Cl2(1 mL)溶液を30分間かけて加えた。添加完了後、還流下の加熱をもう3時間続け、その後反応混合物を室温まで冷却した。揮発物を減圧留去し、粗生成物を減圧蒸留で精製して、無色の油状物としてパートBの化合物を得た(450 mg; 59%)。(沸点=112℃/0.05 mm Hg)
【0276】
C.
【化131】
パートBの化合物(450 mg; 1.83 mmol)の0℃の無水CH2Cl2(3
mL)溶液に、SO2Cl2(161μL ; 2.0 mmol)の無水CH2Cl2(1 mL)溶液を2時間かけて滴下して加えた。この間反応混合物に、連続して窒素を吹き込ん
だ。反応液を室温まで加温し、室温で2時間攪拌した。追加のCH2Cl2(10 mL)
を加えて、反応液を過剰の飽和NaHCO3水を加えてクエンチした。有機相を分液処理
し、H2Oで洗浄し(2回)、乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマト
グラフィで精製して(SiO2; ヘキサン:EtOAc=5:1)、澄明な油状物としてパートCの化合物を得た(380 mg; 68%)。
【0277】
D.
【化132】
パートCの化合物(150 mg; 0.53 mmol)および酢酸ナトリウム(82 mg; 1.0 mmol)の0℃の70% MeOH水溶液(15 mL)に、ベンゼンジアゾニウムクロリド(アニリン50μLおよびNaNO269 mgから生成)の0℃の溶液をゆっくり滴下して加えた。反応液を次いでゆっくり室温まで加温し、室温で終夜攪拌した。反応混合物をEtOAcおよびH2O(各50 mL)で分液処理した。有機相をH2O(2回)で洗浄し、乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2; ヘキサン:EtOAc=3:1)、澄明な油状物としてパートDの化合物を得た(192 mg; 94%)。
【0278】
E.
【化133】
パートDの化合物(192 mg; 0.56 mmol)およびEt3N(1 mmol)の無水トルエン(20 mL)溶液を、すべての出発物質が消費するまで(2時間; TLC)加熱還流した。室温に冷却後、該混合物を1N HCl水(30 mL)およびH2O(3×20 mL)で洗浄し、乾燥し(Na2SO4)、減圧濃縮した。生じた油状物をクロマトグラフィで精製して(SiO2; ヘキサン:EtOAc=3:1)、油状物としてパートEの化合物を得た(120 mg; 69%)。
【0279】
F.
【化134】
TMSCl(25 mg; 0.23 mmol)を、パートEの化合物(20 mg; 0.06 mmol)およびヨウ化ナトリウム(34 mg; 0.23 mmol)の無水アセトニトリル(5 mL)混合液に加えた。該反応混合物をN2雰囲気下2時間加熱還流した。室温に冷却後、水(2 mL)を加え、該混合物を室温で10分間攪拌した。EtOAc(10 mL)を加え、有機相を70% Na2S2O3水(10 mL)および水で洗浄し、乾燥し(Na2SO4)、減圧濃縮した。該残渣をプレパラティブHPLC(実施例13の化合物で記載のように)で精製して、白色固形物としてパートFの化合物を得た(15 mg; 81%)。
【0280】
G.
【化135】
パートFの化合物(15 mg; 0.049 mmol)の−78℃のCH2Cl2(3 mL)溶液に、ニートのBBr3(200μL ; 2.1 mmol)を滴下して加えた。反応混合物をゆっくり室温まで加温し、室温で1時間攪拌した。反応液を次いで−65℃に冷却し、MeOH(0.5 mL)を注意深く加えた。該溶液を室温まで加温し、室温で30分間攪拌した。揮発物を減圧留去し、該残渣をEtOAcおよび水(各10 mL)で分液処理した。有機相を乾燥し(Na2SO4)、減圧濃縮して、油状物としてパートGの化合物を得た(15 mg; 99%)。
【0281】
H.
【化136】
パートGの化合物(15 mg; 0.051 mmol)、K2CO3(28 mg; 0.20 mmol)およびメシレート化合物(34 mg; 0.12 mmol):
【化137】
のMeCN(20 mL)混合液を、100℃で18時間加熱した。この時点でHPLC/MSにより、反応は完了していることが示唆された。反応液を室温まで冷却し、次いでEtOAc(150 mL)およびH2O(100 mL)で分液処理した。有機相をH2O(2×100 mL)で洗浄し、乾燥し(Na2SO4)、減圧濃縮して、粗生成物を得た。この物質をクロマトグラフィ(SiO2;ヘキサン:EtOAc=3:1)で精製して、油状物としてパートHの化合物を得た(20 mg; 59%)。
【0282】
I.
【化138】
パートGの化合物(20 mg; 0.03 mmol)のLiOH水(1.0M溶液の1.0 mL)およびTHF(5 mL)の溶液を、50℃で4時間攪拌した。この時点でHPLC/MSにより、反応完了が示された。反応液をEtOAc(10 mL)およびHCl水(1N溶液の10 mL)で分液処理した。有機相をH2O(3×20 mL)で洗浄し、次いで減圧濃縮した。該残渣をプレパラティブHPLC(BMS−460193の精製で記載のように)で精製して、固形物として標題化合物を得た(12 mg; 83%)。
[M+H]+ = 480.5
【0283】
実施例21
【化139】
A.
【化140】
リンドラー触媒(10% Pd/C)の存在下の実施例11パートCのアセチレンエステル化合物(100 mg; 0.26 mmol):
【化141】
およびキノリン(2μL; 0.014 mmol)のトルエン(5 mL)溶液を、1.5時間H2の常圧下(バルーン)攪拌した。この時点でのHPLC/MSで反応は完了していた。触媒をセライト(登録商標)で濾過して除き、濾液を減圧濃縮して、油状物としてクルードなα,β−不飽和エステル体を得た。この物質をクロマトグラフィで精製して(SiO2 ; hex:EtOAc=3:1)、油状物としてパートAの化合物を得た(50 mg; 49%)。
【0284】
B.
【化142】
パートAの化合物(430 mg; 1.09 mmol)およびトシルメチルイソシアニド(216 mg; 1.09 mmol)のDMSO(3 mL)溶液を、0℃のNaH(60%オイル懸濁の65 mg)のEt2O(2 mL)懸濁液に滴下して加えた。反応液を次いで室温まで加温し、室温で15分間攪拌し、この時点で反応は分析用HPLCで完了していた。反応混合物をEtOAcおよび飽和NH4Cl水で分液処理した。水相をEtOAcで抽出した(2回)。有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮した。粗生成物をクロマトグラフィで精製して(SiO2 ; hex:EtOAc=3:1)、油状物としてパートBの化合物を得た(300 mg; 69%)。
【0285】
C.
【化143】
パートBの化合物(20 mg; 0.047 mmol)、フェニルボロン酸(7 mg; 0.057 mmol)、Cu(OAc)2(5 mg; 0.028 mmol)および4Aモレキュラシーブズ(200 mg)のEt3N:ピリジン:CH2Cl2(1:1:2の混合液の2 mL)混合液を、シールしたチューブ中70℃で3日間加熱した。分析用HPLCにより、反応は60%完了していた。反応液を室温まで冷却し、EtOAcおよび1M HCl水で分液処理した。水相をEtOAcで抽出し(2回);有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮して、油状物としてパートCの化合物を得て、さらに精製することなく、次工程に用いた。
【0286】
D.
【化144】
クルードなパートCの化合物およびLiOH水(1M溶液の2 mL)のTHF:H2Oの溶液を、100℃で24時間攪拌した。反応液を室温まで冷却し、次いで1M HCl水でpH 2まで酸性にした。水相をEtOAcで抽出し(2回);有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮して、粗生成物を得た。この物質をプレパラティブHPLC(実施例13の化合物の精製で記載のように)で精製して、白色固形物として標題化合物を得た(8 mg; 35%)。
[M+H]+ = 479.2
【0287】
実施例22
【化145】
A.
【化146】
3−ヒドロキシフェニルエタノール(500 mg; 3.61 mmol)、メシレート体(990 mg; 3.52 mmol):
【化147】
およびK2CO3(2.0 g; 14 mmol)のMeCN(5 mL)混合液を、90℃で5時間攪拌した。この時点でLC/MSにより、反応が完了していた。反応液を室温まで冷却し、固形物を濾別し、濾液をEtOAc(100 mL)で希釈した。該溶液を連続して1M HCl水(10 mL)、1M NaOH(10 mL)およびH2O(50 mL)で洗浄し、乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2;hex:EtOAc=2:1)、油状物としてパートAの化合物を得た(1.0 g; 87%)。
【0288】
B.
【化148】
パートAの化合物(1.0 g; 3.10 mmol)のCH2Cl2(20 mL)溶液に、デス−マーチン過ヨウ素体(3.0 g; 7.1 mmol)を加え、該混合物を室温で3時間攪拌した。揮発物を減圧留去し、該残渣をEtOAc(25 mL)およびH2O(25 mL)で分液処理した。有機相を乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2; hex:EtOAc=3:1)、油状物としてパートBの化合物を得た(227 mg; 23%)。
【0289】
C.
【化149】
パートBの化合物(86 mg; 0.27 mmol)および(トリフェニルホスホラニリデン)酢酸メチル(110 mg; 0.33 mmol)のトルエン(2 mL)混合液を100℃で2時間加熱した。分析用HPLCにより、反応が完了していた。揮発物を減圧留去し、該残渣をクロマトグラフィで精製して(SiO2; hex:EtOAc=3:1)、油状物としてパートCの化合物を得た(110 mg; 98%)。
【0290】
D.
【化150】
パートCの化合物(101 mg; 0.27 mmol)およびトシルメチルイソシアニド(TosMIC ; 53 mg; 0.27 mmol)のDMSO(1 mL)溶液を、0℃のNaH(60%オイル懸濁の15 mg)のEt2O(1 mL)懸濁液に滴下して加えた。反応液を次いで室温まで加温し、室温で15分間攪拌し、この時点で反応液は分析用HPLCで完了していた。反応混合物をEtOAcおよび飽和NH4Cl水で分液処理した。水相をEtOAcで抽出した(2回)。有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮した。粗生成物をクロマトグラフィで精製して(SiO2 ; hex:EtOAc=3:1)、油状物としてパートDの化合物を得た(20 mg; 18%)。
【0291】
E.
【化151】
パートDの化合物(20 mg; 0.048 mmol)、フェニルボロン酸(7 mg; 0.057 mmol)、Cu(OAc)2(5 mg; 0.028 mmol)および4Aモレキュラシーブズ(200 mg)のEt3N:ピリジン:CH2Cl2(1:1:2の混合液の2 mL)の混合液を、シールしたチューブ中70℃で3日間加熱した。分析用HPLCにより、反応は60%完了していた。反応液を室温まで冷却し、EtOAcおよび1M HCl水で分液処理した。水相をEtOAcで抽出し(2回);有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮して、油状物としてパートEの化合物を得て、さらに精製することなく、次工程に用いた。
【0292】
F.
【化152】
クルードなパートEの化合物およびLiOH水(1M溶液の2 mL)のTHF:H2O溶液を、100℃で24時間攪拌した。反応液を室温まで冷却し、次いで1M HCl水でpH〜2まで酸性にした。水相をEtOAcで抽出し(2回);有機抽出物を合わせて、乾燥し(Na2SO4)、減圧濃縮して、粗生成物を得た。この物質をプレパラティブHPLC(実施例13の化合物の精製で記載したような条件を用いる)で精製し、白色固形物として標題化合物を得た(7 mg; 2工程で30%)。
[M+H]+ = 479.2
【0293】
実施例23〜50
以下のN−アリールピロール酸は、上述の方法の1つを用いて合成した。
【表7】
【表8】
【表9】
【表10】
【0294】
実施例51
【化153】
実施例1に記載の同一の合成工程で(フェニルヒドラジンに代わり3−メチルフェニルヒドラジンを用いたことを除く)、標題化合物を製造した(1.2 mg; 最終3工程の全収率24%)。
[M+H]+ = 495.1
【0295】
実施例52
【化154】
A.
【化155】
実施例3パートEの化合物(50 mg; 0.13 mmol)の−74℃の無水THF(2 mL)溶液に、リチウムジイソプロピルアミド(LDA)(2Mのヘプタン/THF溶液の200μL)を加えた。青色の反応液を−74℃で1時間攪拌し、次いで室温まで加温し、室温で1時間攪拌し、次いで−78℃に冷却した。ヨードメタン(85 mg; 0.6 mmol)のTHF(0.5 mL)溶液を滴下して加え、反応液を−78℃で2時間攪拌し、次いで室温まで加温した。反応液を飽和NH4Cl水(0.5 mL)、H2OおよびEtOAc(各5 mL)で分液処理した。有機相を乾燥し(Na2SO4)、減圧濃縮し;該残渣をクロマトグラフィで精製して(SiO2;hex=100%からhex:EtOAc=3:7の連続的な濃度勾配)、白色の結晶としてパートAの化合物を得た(20 mg; 38%)。
【0296】
B.
【化156】
パートAの化合物(20 mg; 0.05 mmol)の室温のCH2Cl2(2.0 mL)溶液に、BBr3(1MのCH2Cl2溶液の0.2 mL)を滴下して加えた。該混合物を室温で30分間攪拌し、次いで減圧濃縮した。該残渣をMeOH(1 mL)からストリッピングし、クロマトグラフィで精製して(SiO2;hex:EtOAc=3:1)、白色の結晶としてパートBの化合物を得た(13 mg; 68%)。
【0297】
C.
【化157】
パートBの化合物(13 mg; 0.032 mmol)、5−メチル−2−フェニルオキサゾール−4−エタノールメシレート(15 mg; 0.053 mmol;実施例11に記載のように製造する)およびK2CO3(500 mg; 3.6 mmol)のMeCN(2 mL)の混合液を、シールしたチューブ中18時間加熱還流し、次いで室温まで冷却し、濾過した。濾液を減圧濃縮し;該残渣をEtOH(2 mL)に溶解し、KOH(200 mg; 3.6 mmol)を加えた。該混合物をシールしたチューブ中80℃で攪拌し、次いで室温まで冷却し、EtOAc(20 mL)および1N HCl水(5 mL)で分液処理した。有機相をH2O(2×10 mL)で洗浄し、減圧濃縮した。該残渣をプレパラティブHPLC(YMC逆相ODS 20×100 mmカラム;流速=20 mL/分;B:A=25:75からB=100%の10分間の連続した濃度勾配 + B=100%での5分間のホールド時間,溶媒A;H2O:MeOH:TFA=90:10:0.1および溶媒B;MeOH:H2O:TFA=90:10:0.1)で精製して、白色固形物として標題化合物を得た(14.8 mg; 88%)。
[M+H]+ = 495.3
【0298】
実施例53
【化158】
A.
【化159】
実施例52パートAの化合物の合成で記載した方法を用いて(ただし、実施例3パートEの化合物の代わりに実施例1パートEの化合物 [50 mg; 0.13 mmol]を用いた)、油状物としてパートAの化合物を製造した(35 mg; 68%)。
【0299】
B.
【化160】
実施例52の合成で記載した合成工程を用いて(ただし、実施例52パートAの化合物の代わりにパートAの化合物を用いた)、固形物として標題化合物を製造した(24 mg; 3工程の全収率55%)。
[M+H]+ = 495.3
【0300】
実施例54
【化161】
A.
【化162】
実施例22パートBの化合物(150 mg; 0.47 mmol)および(トリフェニルホスホラニリデン)酢酸tert−ブチル(200 mg; 0.53 mmol)のトルエン(10 mL)溶液を、90℃で1時間攪拌した。冷却後、揮発物を減圧留去し、該残渣をクロマトグラフィで精製して(SiO2;hex:EtOAc=3:1)、油状物としてパートAの化合物を得た(200 mg; 99%)。
【0301】
B.
【化163】
パートAの化合物(200 mg; 0.477 mmol)およびトシルメチルイソシアニド(100 mg; 0.512 mmol)のDMSO溶液を、Et2O中のNaH(60%オイル混合物の26 mg;0.65 mmol)のスラリーに、30分間かけて室温で滴下して加えた。反応液を室温で30分間攪拌し、次いでH2OおよびEtOAcで分液処理した。有機相を乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2;hex:EtOAc=3:1)、油状物としてパートBの化合物を得た(60 mg; 27%)。
【0302】
C.
【化164】
パートBの化合物(23 mg; 0.05 mmol)、K2CO3(200 mg; 1.45 mmol)およびヨウ化メチル(10 mg; 0.07 mmol)のDMF(2 mL)混合液を、シールしたチューブ中80℃で2時間攪拌した。反応液を室温まで冷却し、H2OおよびEtOAc(各10 mL)で分液処理した。有機相をH2O(2×10 mL)で洗浄し、乾燥し(Na2SO4)、減圧濃縮した。クルードなN−メチルピロールエステルのTFA/CH2Cl2溶液(1:1溶液の2 mL)を、室温で30分間攪拌し、次いで減圧濃縮した。該残渣をプレパラティブHPLC(実施例52の化合物の条件に従う、ただし,連続的な濃度勾配は、A:B=75:25からB=100%でなく、A:B=70:30からB=100%を用いた)で精製し、白色固形物として標題化合物を得た(7.2 mg; 34%)。
[M+H]+ = 417.2
【0303】
実施例55
【化165】
A.
【化166】
パートAの化合物は、メシレート体:
【化167】
および4−ヒドロキシフェニルエタノール(3−ヒドロキシフェニルエタノールの代わりに用いた)から、実施例22パートBの化合物の合成に記載のように製造した。
【0304】
B.
【化168】
パートAの化合物(150 mg; 0.47 mmol)を用いて、(実施例54パートAの化合物の合成に記載のように)油状物としてパートBの化合物を製造した(200 mg; 99%)。
【0305】
C.
【化169】
パートBの化合物(200 mg; 0.477 mmol)を用いて、(実施例54パートBの化合物の合成に記載のように)油状物としてパートCの化合物を製造した(100 mg; 46%)。
【0306】
D.
【化170】
パートCの化合物(23 mg; 0.05 mmol)を用いて、(実施例54の合成に記載のように)白色固形物として標題化合物を製造した(7.7 mg; 37%)。
[M+H]+ = 417.2
【0307】
実施例56
【化171】
A.
【化172】
実施例54パートBの化合物(20 mg; 0.044 mmol)、2−ブロモチオフェン(8 mg; 0.05 mmol)、CuI(30 mg; 0.157 mmol)、ZnO(10 mg; 0.122 mmol)およびK2CO3(50 mg; 0.36 mmol)の1−メチル−2−ピロリジノン(NMP; 2 mL)混合液を、シールしたチューブ中166℃で18時間加熱した。反応液を室温まで冷却し、EtOAcおよびHCl水(1M溶液の10 mL)で分液処理した。有機相を食塩水(2×10 mL)で洗浄し、乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2;ヘキサン:EtOAc=1:1)、固形物としてパートAの化合物を得た。
【0308】
B.
【化173】
パートAの化合物のTFA/CH2Cl2(1:1溶液の1 mL)溶液を、室温で1時間攪拌し、次いで減圧濃縮した。該残渣をプレパラティブHPLC(実施例54の化合物で記載の条件に従う)で精製して、白色固形物として標題化合物を得た(7 mg ; 2工程で32%)。
[M+H]+ = 485.2
【0309】
実施例57
【化174】
実施例55パートCの化合物(20 mg; 0.044 mmol ;実施例56に記載と同じ合成工程を用いる)を用いて、固形物として標題化合物を合成した(5 mg; 23%)。
[M+H]+ = 485.2
【0310】
実施例58
【化175】
A.
【化176】
実施例54パートBの化合物(20 mg; 0.044 mmol)、2−ブロモチアゾール(10 mg; 0.061 mmol)、CuI(30 mg; 0.157 mmol)、ZnO(10 mg; 0.122 mmol)およびK2CO3(50 mg; 0.36 mmol)の1−メチル−2−ピロリジノン(2 mL)混合液を、シールしたチューブ中166℃で18時間加熱した。反応液を室温まで冷却し、EtOAcおよびHCl水(1M溶液の10 mL)で分液処理した。有機相を食塩水(2×10 mL)で洗浄し、乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2 ; ヘキサン:EtOAc=1:1)、固形物としてパートAの化合物を得た。
【0311】
B.
【化177】
パートAの化合物のTFA/CH2Cl2(1:1溶液の1 mL)溶液を、室温で1時間攪拌し、次いで減圧濃縮した。該残渣をプレパラティブHPLC(実施例54に記載の条件に従う)で精製し、褐色固形物として標題化合物を得た(9 mg; 2工程で42%)。
[M+H]+ = 486.3
【0312】
実施例59
【化178】
実施例55パートCの化合物(20 mg; 0.044 mmol;実施例56に記載と同じ合成工程を用いる)を用いて、褐色固形物として標題化合物を得た(5 mg; 23%)。
[M+H]+ = 486.3
【0313】
実施例60
【化179】
A.
【化180】
実施例11パートCの化合物(176 mg; 0.45 mmol)およびアジ化ナトリウム(32 mg; 0.49 mmol)の無水DMF(1 mL)溶液を、N2の常圧下室温で15分間攪拌し、その後H2O(10 mL)を加えた。固形物を濾取し、減圧乾燥し、次いでクロマトグラフィで精製して(SiO2 ;hex:EtOAc=3:1)、黄色の固形物としてパートAの化合物を得た(138 mg; 71%)。
【0314】
B.
【化181】
パートAの化合物(138 mg; 0.319 mmol)、ベンジルブロミド(118 mg; 0.69 mmol)およびK2CO3(238 mg; 2.05 mmol)のDMF(1 mL)溶液を、室温で18時間攪拌した。反応液をH2OおよびEtOAc(各5 mL)で分液処理し;有機相を乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2 ; hex:EtOAc=3:1)、油状物としてパートBの化合物を得た(25 mg; 15%)。また、他の2つの位置異性体も得た。
パートCの化合物(40 mg; 23%):
【化182】
およびパートDの化合物(12 mg; 7%):
【化183】
【0315】
E.
【化184】
パートBの化合物のTHF(2 mL)およびLiOH水(1M溶液の1 mL)の溶液を、室温で18時間攪拌し、次いでHCl水(1M溶液の2 mL)およびEtOAc(5 mL)で分液処理した。有機相をH2O(2×5 mL)で洗浄し、乾燥し(Na2SO4)、減圧濃縮して、白色固形物として標題化合物を得た(19 mg; 80%)。
[M+H]+ = 495.2
【0316】
実施例61
【化185】
実施例54パートBの化合物を用いて、(実施例54の合成で記載したように、ただしヨウ化メチルの代わりにベンジルブロミドを用いる)、プレパラティブHPLC精製後(実施例54に関する)、黄色の固形物として標題化合物を製造した(7 mg)。
[M+H]+ = 493.1
【0317】
実施例62
【化186】
A.
【化187】
ベンズアルデヒド(23.8 g,234 mmol)のEtOAc(150 mL;あらかじめHClガスを飽和にした)溶液に、2,3−ブタンジオン モノオキシム(25.0 g, 234 mmol)を1回で加え、生じた溶液を室温で12時間攪拌した。分析用HPLCにより、すべての出発物質が消費していることが示唆された。反応混合液を減圧濃縮して、白色固形物としてパートAの化合物を得て、さらに精製することなく、次工程に用いた。
【0318】
B.
【化188】
パートAの化合物のCHCl3(200 mL)溶液に、POCl3(30 mL, 320 mmol)を滴下して加えた。反応液を50℃で12時間攪拌し、次いで減圧濃縮した。該褐色の残渣をEtOAc(300 mL)および1N NaOH水で分液処理した。有機相を食塩水で洗浄し、乾燥し(MgSO4)、減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2 ; Et2O)、淡褐色の固形物としてパートBの化合物を得た(41.5 g ; 86%)(分析用HPLCおよび1H−NMR分析で純度>95%)。
【0319】
C.
【化189】
実施例1パートCの化合物(592 mg; 1.9 mmol)および3−メチルフェニルヒドラジン(330 mg; 2.08 mmol)のEtOH(30 mL)および無水MgSO4(1 g)の混合物を、終夜で加熱還流した。反応液を濾過し、濾液を減圧濃縮した。該残渣をクロマトグラフィで精製して(SiO2;hex=100%からEtOAc=100%への連続的な濃度勾配)、S−シスおよびS−トランスオキシムの混合物としてパートCの化合物を得た(478 mg; 76%)。
【0320】
D.
【化190】
パートCの化合物(103 mg; 0.31 mmol)の室温のトルエン(5 mL)溶液に、PCl5(70 mg; 0.34 mmol)を加え、反応液を室温で2時間攪拌し、次いで減圧濃縮した。該残渣をEtOAcおよびH2Oで分液処理し;有機相を食塩水で洗浄し、乾燥し(Na2SO4)、減圧濃縮して、クルードなパートDの化合物を得て、さらに精製することなく、次工程に用いた。
【0321】
E.
【化191】
クルードなパートDの化合物の室温の無水EtOH(3 mL)溶液に、NaOH水(2M溶液の0.25 mL)を滴下して加えた。該混合物が橙色から暗褐色に変わり、室温で1時間攪拌し、次いで過剰の1N HCl水およびEtOAcで分液処理した。水相をEtOAcで抽出し、有機抽出物を合わせて、食塩水で洗浄し、乾燥し(Na2SO4)、減圧濃縮した。該残渣をプレパラティブHPLC(実施例5で記載のように)で精製して、褐色固形物としてパートEの化合物を得た(10.5 mg;2工程で11%)。
【0322】
F.
【化192】
パートEの化合物(11 mg; 0.033 mmol)の−78℃の溶液に、BBr3(0.02 mL;0.21 mmol)を滴下して加えた。反応液を−78℃で15分間攪拌し、次いで室温まで加温し、室温で5時間攪拌した。0℃まで冷却後、反応液を大過剰の飽和NH4Cl水で注意深くクエンチした。水相をEtOAcで抽出し;有機抽出物を合わせて、食塩水で洗浄し、乾燥し(Na2SO4)、減圧濃縮して、クルードなパートFの化合物を得て、さらに精製することなく、次工程に用いた。
【0323】
G.
【化193】
パートFの化合物(10 mg; 0.033 mmol)、K2CO3(15 mg; 0.11 mmol)およびパートBの化合物(20 mg; 0.096 mmol)のMeCN(2 mL)混合液を、90℃で終夜加熱し、次いで室温まで冷却し、H2OおよびEtOAcで分液処理した。水相をEtOAcで抽出し;有機抽出物を合わせて、食塩水で洗浄し、乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマトグラフィで精製し(SiO2 ;ヘキサン=100%からEtOAc=100%への連続的な濃度勾配)、次いでさらにプレパラティブHPLC(実施例52の精製での条件;ただし、A:B=30:70からB=100%への連続的な濃度勾配を用いることを除く)で精製して、無色の油状物として標題化合物を得た(5.2 mg;2工程で26%)。
[M+H]+ = 481.1
【0324】
上記の実施例および反応式で述べた方法に従い、以下の例示される化合物は製造され得る。
【化194】
【0325】
実施例63
両PPARγアンタゴニスト/PPARαアゴニストのインビトロスクリーニングアッセイ
A.マウス3T3−L1前脂肪細胞におけるPPARγアンタゴニストのスクリーニング
PPARγへの強い結合を示す化合物は、マウス3T3−L1前脂肪細胞の成熟した脂肪細胞への分化を誘発するロシグリタゾン(真正のPPARγアゴニスト)、50nMを阻害する能力でアッセイした。プレートあたり5×105の3T3−L1細胞を、96ウェルプレートに加え、誘発する前に2日間DMEM−高ブドウ糖および10% FBS培地で培養した。細胞は、同一培地中デキサメタゾン(1μm)、インスリン(5μg/ml)、およびイソブチルメチルキサンチン(IBMX)(0.6μm)で48時間誘発した。このとき、連続した希釈の試験化合物を、各ウェルの培地を含んだロシグリタゾン(50 nM)およびDMSO(0.1%)に加えた。培地(インスリン、デキサメタゾンおよびIBMXを含まない)を含む試験化合物、ロシグリタゾン(PPARγアゴニスト)およびDMSOの同一の濃度で、細胞をさらに72時間再フィードした。全部で5日間インキュベートした後、各ウェルから培地(4μl)を集め、96ウェルELISAプレート中のH2O(40μl)に希釈し、トリグリセリドブランク試薬(バイエル診断薬)(300μl)を各ウェルに加えて、室温で5分間インキュベートした。細胞から遊離グリセロールの放出を誘発するロシグリタゾンへの各化合物の阻害%は、スペクトレマックス(Spectremax) 250ELISAリーダーを用いて波長500nMで測定した。データはDMSOだけのコントロールに標準化して、トランス活性化の最大阻害%はロシグリタゾン正コントロール(50nM)と比較して計算した。ED50値は、活性阻害曲線の中点で標準方程式を用いて計算した。
【0326】
B.CV−1霊長類腎臓細胞におけるPPARγアンタゴニストのスクリーニング
PPARγに強い結合を示す化合物は、CV−1細胞のSEAPレポーター遺伝子活性のトランス活性化を誘発するロシグリタゾン(真正のPPARγアゴニスト)(1μM)を阻害する能力としてアッセイした。CV−1細胞(これらの細胞は内在性PPARγ遺伝子を発現する)は、3×PPRE−SEAPレポーター遺伝子DNAコンストラクトでトランスフェクトされ、安定なコロニーが選択され、拡張され、標準的なプロトコールを用いて化合物への応答として試験した。SEAPレポーター遺伝子コンストラクトは、pSEAP2のSV40初期最小促進剤(クロンテック)へ素早く5分で、7ヌクレオチドを含むラットの脂肪酸結合タンパク質PPREの3リピートを挿入して構築した。1.2×106のCV−1/PPRE−SEAP細胞を、化合物を添加する1日前に96ウェルのプレートに培養した。試験化合物の連続した希釈は、DMEM 10% FBS、0.5%(最終的に容積比)のDMSOおよび1μMのロシグリタゾン(PPARγアゴニスト)中調製した。各濃度の150μl分割量を、2つの隣接しないウェルに加えた。各プレートには、0.5% DMSO培地中に1μMのロシグリタゾン(PPARγアゴニスト)の6ウェルが含まれた。培地をフレッシュな96ウェルプレートに集め、続く40時間化合物とともにインキュベートし、SEAP活性としてアッセイした。SEAPは熱に耐性があり、集めた培地の内在性のホスファターゼは、圧力感受性の粘性シールフィルム(コーニング)でプレートをシールし、30分から1時間65℃に加熱して不活性化した。室温(RT)にした後、熱で不活性化した培地の25μl分割量をクリアーな底の96ウェル黒色プレートに加え、蛍光気質のアトフォス試薬(プロメガ)100μlをウェルあたり加えた。プレートを暗所で5分間インキュベートし、次いでサイトフルオラ(CytoFluor)シリーズ4000プレートリーダー(パーセプチブ バイオシステムズ)で蛍光を測定した:励起フィルター,450/50nm;発光フィルター,580/50nm;8周期,1分/周期,3読み取り/ウェル/周期。データはDMSOだけのコントロールに標準化して、トランス活性化の最大阻害%はロシグリタゾン正コントロール(1μM)と比較して計算した。ED50値は、活性阻害曲線の中点で標準方程式を用いて計算した。
【0327】
C.HepG2ヒト肝臓細胞におけるPPARαアゴニストのスクリーニング
PPARαに強い結合を示す化合物は、HepG2、誘導されたヒト肝臓、内在性PPARα遺伝子を発現する細胞、またはGal−4 DNA結合ドメイン−PPARαリガンド結合ドメインキメラ受容体(後述)を安定に発現するHepG2細胞において、レポーター遺伝子活性のPPARα依存の刺激を刺激する能力として試験した。レポーター遺伝子コンストラクトは、素早く5分で7ヌクレオチドを含むラットの脂肪酸結合タンパク質PPREの3リピートを、またはpSEAP2のSV40初期最小促進剤(クロンテック)の上方のGal4応答因子の4リピートを挿入して(それぞれ3×PPRE−SEAPおよびGal4−SEAP)、構築した。キメラ受容体は、哺乳類バイシストロン性発現ベクターpIRESlneo(クロンテック)におけるgal4 DNA結合ドメイン(アミノ酸1−47)、gal4−PPARαへと、フレームおよび3’のヒトPPARαのリガンド結合ドメインをコードするcDNAをクローニングして調製した。安定な細胞系は、リポフェクタミン プラス(ギブコ)を用い、続く製造者の指示で、gal4−SEAPおよびgal4−PPARαの両方で、または3XPPRE−SEAPでトランスフェクトして調製した。細胞は96ウェルプレート上で培養され、終夜付着させた。次の日、0.5%(容積比)DMSOを含む増殖培地(DMEM+10% チャコール/デキストランストリップしたFBS)中の化合物の連続する希釈物を、隣接しないウェルに2倍量加え、24〜40時間37℃で5% CO2中インキュベートした。各プレートには、1μMの標準溶液、正のコントロールとしてのGW−2331(真正のPPARα選択的アゴニスト)、負のコントロールとしてのロシグリタゾン(真正のPPARγアゴニスト)の少なくとも6ウェル、およびコントロールとしてDMSOのみの培地の3ウェルを有した。インキュベートに続いて、培地を除き、内在性ホスファターゼを上記で示唆されたように不活性化し、加工処理した培地の25μl分割量でのSEAP活性を、100μlのアトフォス試薬(プロメガ)を加えて、以下の条件でクリアーな底の黒色96ウェルプレート(ファルコン)中アッセイした:室温暗所で5分間インキュベートし、サイトフルオラ(CytoFluor)シリーズ4000プレートリーダー(パーセプチブ バイオシステムズ,8周期,1分/周期)で蛍光(励起:450nm;発光:580nm)の増加を測定。蛍光発光の相対速度は、DMSOコントロールからの倍増として計算した。内在性の活性は、1μM標準溶液の活性の%としての1μMで試験化合物の活性として定義した。ED50値は、活性曲線の中点で標準方程式を用いて計算した。
【0328】
実施例64:インビボ肥満動物モデル
C57BL/6マウスは、脂肪(40%)およびショ糖(40%)に富んだ食餌(参照:York {肥満の遺伝的モデル} and Sclafani {肥満の食餌的モデル}, both in Obesity, Bjorntorp and Brodoff eds. JB Lippincott Company, 1992; McIntosh and Pederson; McNeill. eds. CRC press LLC, 337-398, 1999; Farrelly et al., Proc. Natl. Acad. Sci. 96: 14511-14516, 1999)が与えられた。これらの食餌の条件下、C57BL/6マウスは、十分に体重が増加し、肥満となる。これらのマウスは、薬理学的に許容されるビークル(例えば、これに限らないが、5% CM−セルロース)中、経口で、静脈内、皮下または門脈内注射で、または食物または水と混ぜて、急性的にまたは時間を延長して、両PPARγアンタゴニスト/PPARαアゴニスト(用量:0.01〜100 mg/kg/日)を投与して処置した。実験過程においては、様々なパラメーター、例えば、水と食物の消費、体重の増加、両放射X線アナライザー(DEXA;この装置は体脂肪量、体除脂肪筋肉量および体骨ミネラル含量を正確に測定する)による身体構成、体温を、標準的な方法で測定した。尾静脈から血液を血液凝固防止のためにヘパリン−EDTAコーティングチューブに集めて、血漿を割けて、COBAS−MIRA器具のロシュ・ダイアグノスティックス社から入手できる試薬キットを用いて、ブドウ糖、遊離脂肪酸、トリグリセリドおよびコレステロールを分析した。インスリンおよびレプチンは、購入可能なELISAキットで測定する。体重を減らすことおよび/またはブドウ糖を減少させることに作用する化合物が選択された。処置の時間の終わりに、動物はCO2に短時間曝して安楽死させ、肝臓および白色脂肪組織のような内部器官を別の分析のために収集した。これらの分析には、これらに限らないが、脂質含量、および様々なPPARγおよびPPARα標的遺伝子の発現の効果の測定が含まれ得る。
【0329】
体脂肪量、除脂肪体重を減少させ、肥満症、インスリン耐性を防止または回復させる試験化合物はまた、抗糖尿病剤(例えば、これらに限らないが、メトホルミンおよびスルホニル尿素)および/またPPARαアゴニストのようなは脂質低下剤(例えば、これらに限らないが、フェノフィブラートおよびゲムフィブロジル)および/またはHMG CoA還元酵素阻害剤(例えば、これらに限らないが、プラバスタチン、ロバスタチン、シンバスタチンおよびアトルバスタチン)と組み合わせて、上述の疾患モデルで試験する。実験過程においては、様々なパラメーター、例えば、水と食物の消費、体重の増加、体温および血漿ブドウ糖、インスリン、遊離脂肪酸、トリグリセリドおよびコレステロールレベルを測定した。体脂肪量および体重を減少(体除脂肪骨格量を増加)させ、および/またはグルコース、および脂質を減少させることに作用する化合物は、さらに特徴付けのために選択された。
【図面の簡単な説明】
【0330】
【図1−A】図1−Aには、標識された真正のPPARγリガンド(BMS−化合物A)のヒトPPARγリガンド結合ドメインへの結合を、競合的に阻害する化合物Yの能力が記載される。
【図1−B】図1−Bには、標識された真正のPPARαリガンド(BMS−化合物B)のヒトPPARα結合ドメインへの結合が記載される。
【図2】図2には、真正のPPARγアゴニスト(例えば、ロシグリタゾン)依存性の、マウス3T3L−1前脂肪細胞(未成熟の脂肪細胞)の成熟した脂肪細胞に負荷した脂質(成熟した脂肪細胞)への分化を、競合的に阻害する化合物Yの能力が記載される。
【図3】図3には、霊長類腎臓細胞CV−1における内在性アルカリフォスファターゼ(SEAP)レポーター遺伝子発現の、真正のPPARγアゴニスト(例えば、ロシグリタゾン)依存性活性を、競合的に阻害する化合物Yの能力が記載される。
【図4】図4には、ヒト肝臓細胞系HepG2(この細胞系は、PPARαの重要な量を示す)中のPPARα依存性SEAPレポーター遺伝子活性を、安定にまとめられたPPARα依存性SEAPレポーターにより、用量依存的に刺激する化合物Yの能力が記載される。
本出願は、米国仮出願第60/294,380号(2001年5月30日出願)の優先権を主張するもので、これらは本明細書に引用される。
【0002】
(技術分野)
本発明は、血中ブドウ糖レベル、トリグリセリドレベル、インスリンレベルおよび非エステル型脂肪酸(NEFA)レベルを調節して、糖尿病および肥満症の治療に特に有効となる新規な置換されたアゾール酸誘導体に関するものであり、且つ、かかる置換された酸誘導体単独または他の抗糖尿病剤および/または抗高脂血症剤および/または他の治療剤と組み合わせて用いて、糖尿病(特に2型糖尿病)、並びに高血糖、高インスリン血症、高脂血症、肥満症、アテローム性動脈硬化症および関連する疾患を治療する方法に関するものである。本発明はまた、ヒトを含む哺乳類におけるペルオキシソーム増殖因子活性化受容体−γ(PPARγ)の同時阻害およびペルオキシソーム増殖因子活性化受容体−α(PPARα)の刺激による、肥満症および異脂肪血症の治療方法に関するものである。本発明はさらに、抗肥満症、インスリン感受性および循環器疾患のためになるPPARγアンタゴニスト活性により、遺伝子発現が脂肪組織において変動する標的遺伝子のリストを提供する。
【0003】
(背景技術)
ヒトを含む哺乳類において、脂肪細胞は、栄養過剰の数倍で、トリグリセリドの形状でエネルギーを蓄える(参照:Lowell, Cell, 99: 239-242, 1999)。飢餓の間、蓄えられたトリグリセリドは、栄養およびエネルギーの要求を補充するため、脂肪細胞において脂肪酸に分解される。この条件において、過剰の脂肪組織の蓄積[脂肪細胞(分化)となるように前駆細胞(前脂肪細胞)を補充するか、および/または先に存在する脂肪細胞(肥厚および肥大)の増大によるかのいずれかにより行われる]は、肥満症およびインスリン耐性になる(参照:Lowell, Cell, 99: 239-242, 1999)。その理由は、肥大化した脂肪細胞(これらは比較的低い代謝活性であると考えられる)が過剰量の脂肪酸およびサイトカインを産生し、代わるがわるインスリン情報伝達並びに骨格筋および脂肪細胞(これらは2つの主要なブドウ糖利用組織である)へのブドウ糖取り込みを減少するように作用するためである(参照:Hotamisligil, et al., Science, 259: 87-90, 1993; Lowell, Cell, 99: 239-242, 1999)。肥満の個体は、不適切なエネルギー消費、「骨格筋、肝臓および血漿における高脂質含量」、インスリン耐性、高血圧症、アテローム性動脈硬化症および循環器疾患をしばしば被っている(参照:Rosenbaum et al., New. Eng. J. Med. 337: 396-407, 1997, 参照:Friedman, Nature, 404: 632-634, 2000)。重厚な脂肪貯蔵の枯渇を伴った脂肪異栄養症候群の患者に見られるような症状は、体重の減少、並びに血漿、肝臓および骨格筋における脂質含量の増加をもたらし、代わるがわる該患者にインスリン耐性および2型糖尿病を罹らせることとなる(参照:Arioglu et. al., Annals of Int. Med, 2000, 133: 263-274)。これらの異常の第一の原因は、脂質の安全な貯蔵に有効な比較的少量の脂肪組織のためと思われる。
【0004】
肥満症は、先進国共通の臨床上の問題であり、また急速に先進国における主要な健康の関心事となってきている。体重過剰の個体は、例えば異脂肪血症、インスリン耐性および2型糖尿病のようないくつかの代謝障害をしばしば被っている。これらの個体はまた、高血圧症、アテローム性動脈硬化症および循環器疾患のリスクの増加もしばしば被っている(参照:Friedman, Nature, 404: 632-634, 2000)。
【0005】
ペルオキシソーム増殖因子活性化受容体(PPAR)は、転写因子を調節するリガンドの核ホルモン受容体ファミリーのメンバーである(参照:Willson, et al., J. Med. Chem., 43 : 527-550, 2000, Kersten et al., Nature, 405: 421424, 2000)。3つのPPARイソ型、PPARγ、PPARα、およびPPARδは、ヒトを含む様々な哺乳類から単離されている。これらの受容体は、分類として、その結合相手のRXRαと偏性のヘテロダイマーを形成し、食餌由来の長鎖脂肪酸、脂肪酸代謝物によって、および合成剤によって活性化される(参照:Willson, et al., J. Med. Chem., 43: 527-550, 2000)。ブドウ糖および脂質の代謝経路における遺伝子の調節により、PPARがヒトを含む哺乳類においてブドウ糖および脂質の恒常性を維持する主要な役割をするということが、現在よく報告されている。
【0006】
PPARγは、前脂肪細胞補充並びに成熟した脂肪細胞への分化および成熟した脂肪細胞中の脂質の貯蔵の主要な制御因子である(参照:Tontonoz et al., Current Biology, 571-576, 1995)。PPARγの活性化剤は、前脂肪細胞分化、成熟した脂肪細胞中の脂質貯蔵を促進し、インスリン感受性抗糖尿剤として作用する(参照:Tontonoz et al., Current Biology, 571-576, 1995; Lehmann et al., J. Biol. Chem., 270: 12953-12956, 1995; Nolan et al. New. Eng. J. Med., 331: 1188-1193 ; Inzucchi et al., New Eng. J. Med., 338: 867-872, 1998, Willson, et al., J. Med. Chem.: 43: 527-550, 2000, Kersten et al., Nature, 405: 421424, 2000)。しかしながら、PPARγ誘導型抗糖尿病活性は、しばしば動物モデルおよびヒトにおいていくらかの体重増大を伴う。PPARγ発現は、肥満個体の脂肪組織において十分に高められ(参照:Vidal-Puig et al., J. Clinical Investigation, 99: 2416-2422, 1997)、構造的に活性なPPARγを産生する突然変異は重厚な肥満症につながる(参照:Ristow et al., New England J. Med., 339: 953-959, 1998)。PPARγ発現の部分的な消失は、ヘテロ接合性PPARγノックアウトマウス(参照:Kubota et al. Mol. Cell; 4: 597-609, 1999)における食餌誘導型肥満症への耐性となり、アミノ酸の12位でプロリンのアラニンへの変化を伴ったヒトにおけるより低い肥満度指数となる(参照:Deeb et al Nature Genetics, 20: 284-287, 1998)。ドミナントネガティブ突然変異によるヒトPPARγ活性の比較的より厳しい消失は、受容体に結合するリガンドを破壊し、高脂血症、脂肪肝およびのインスリン耐性となる(参照:Barroso et al. Nature, 402, 860-861, 1999)。該異常の主な原因は、脂質の安全な貯蔵に有用な脂肪組織が比較的少量であるためと思われる。これらのマウスおよびヒトでの知見で、従って、肥満症の該誘導および/または進展におけるPPARγの役割を示し、PPARγの阻害が体脂肪蓄積および肥満症における低下になると示唆される。これらの知見でまた、より高い血漿遊離脂肪酸および高脂血症および脂肪肝の進行およびインスリン耐性にもなるようであると示唆される。
【0007】
PPARαイソ型は、脂肪酸合成、脂肪酸酸化および脂質代謝経路における遺伝子を調節する(参照:Isseman and Green, Nature, 347: 645-649, 1990; Torra et al., Current Opinion in Lipidology, 10 : 151-159, 1999; Kersten et al., Nature, 405: 421424, 2000)。PPARαアゴニスト(例えば、フェノフィブラート、ゲムフィブロジル)の処置は、肝臓および筋肉における脂肪酸酸化を高め、肝臓における脂肪酸およびトリグリセリド合成 を減少させ、血漿トリグリセリドレベルを下げる(参照:Kersten et al., Nature, 405: 421424, 2000)。高トリグリセリドおよび低HDL−コレステロールである患者に、PPARαアゴニストで処置することで、血漿のHDL−コレステロールの増加、血漿のトリグリセリドの減少および第一次および第二次心臓事象(primary and secondary cardiac events)の両方の減少となる(参照:Balfour et al., Drugs. 40: 260-290, 1990; Rubins et al., New Eng. J. Med., 341: 410-418, 1999)。
【0008】
従って、PPARγアンタゴニスト活性およびPPARαアゴニスト活性を、単一の両作用化合物にまたは1つの製剤に組み合わせることで、PPARγを阻害し、高脂血症、脂肪肝およびインスリン耐性を引き起こさずに肥満症を治療することが可能である。本発明は、2つの異なる活性、すなわちPPARγアンタゴニスト活性およびPPARαアゴニスト活性を組み合わせて、高脂血症およびインスリン耐性を引き起こすことなく、体脂肪蓄積および体重を減らす肥満症の新規な治療方法を示す。本発明は、肥満の、高脂血症のおよびインスリン耐性2型糖尿病の患者が、両PPARγアンタゴニスト/PPARαアゴニスト、またはPPARγアンタゴニストおよびPPARαアゴニストを脂質低下剤および抗糖尿病剤と組み合わせて、治療され得ることを提案する。本発明はまた、その発現が抗肥満症、インスリン感受性および循環器疾患のためになるPPARγアンタゴニスト活性により脂肪組織において変動するような、標的遺伝子のリストを提供する。
【0009】
本発明に従い、構造I:
【化1】
mは0、1または2であり;n=0、1または2であり;
QはCまたはNであり;
Aは(CH2)x(xは1〜5である)であるか;またはAは(CH2)x 1(x1は2〜5である)で、アルケニル結合またはアルキニル結合が該鎖のいずれかに結合しているか;またはAは−(CH2)x 2−O−(CH2)x 3−(x2は0〜5で、x3は0〜5である、但し少なくともx2およびx3の1つは0でない)であり;
X1はCHまたはNであり;
X2はC、N、OまたはSであり;
X3はC、N、OまたはSであり;
X4はC、N、OまたはSであるが、但しX2、X3およびX4のうち少なくとも1つはNであり;
X5はC、N、OまたはSであり;
X6はCまたはNであり;
X7はC、N、OまたはSであるが、但しX5、X6またはX7のうち少なくとも1つはNであり;
X1〜X7の各々が上記のように定義される場合において、CはCHを含み得;
R1はHまたはアルキルであり;
R2はH、アルキル、アルコキシ、ハロゲン、アミノまたは置換されたアミノであり;
R2a、R2bおよびR2cは同一または異なって、H、アルキル、アルコキシ、ハロゲン、アミノまたは置換されたアミノから選択され;
R3およびR3aは同一または異なって、H、アルキル、アリールアルキル、アリールオキシカルボニル、アルキルオキシカルボニル、アルキニルオキシカルボニル、アルケニルオキシカルボニル、アリールカルボニル、アルキルカルボニル、アリール、ヘテロアリール、シクロヘテロアルキル、ヘテロアリールカルボニル、ヘテロアリール−ヘテロアリールアルキル、アルキルカルボニルアミノ、アリールカルボニルアミノ、ヘテロアリールカルボニルアミノ、アルコキシカルボニルアミノ、アリールオキシカルボニルアミノ、ヘテロアリールオキシカルボニルアミノ、ヘテロアリール−ヘテロアリールカルボニル、アルキルスルホニル、アルケニルスルホニル、ヘテロアリールオキシカルボニル、シクロヘテロアルキルオキシカルボニル、ヘテロアリールアルキル、アミノカルボニル、置換されたアミノカルボニル、アルキルアミノカルボニル、アリールアミノカルボニル、ヘテロアリールアルケニル、シクロヘテロアルキル−ヘテロアリールアルキル、ヒドロキシアルキル、アルコキシ、アルコキシアリールオキシカルボニル、アリールアルキルオキシカルボニル、アルキルアリールオキシカルボニル、アリールヘテロアリールアルキル、アリールアルキルアリールアルキル、アリールオキシアリールアルキル、ハロアルコキシアリールオキシカルボニル、アルコキシカルボニルアリールオキシカルボニル、アリールオキシアリールオキシカルボニル、アリールスルフィニルアリールカルボニル、アリールチオアリールカルボニル、アルコキシカルボニルアリールオキシカルボニル、アリールアルケニルオキシカルボニル、ヘテロアリールオキシアリールアルキル、アリールオキシアリールカルボニル、アリールオキシアリールアルキルオキシカルボニル、アリールアルケニルオキシカルボニル、アリールアルキルカルボニル、アリールオキシアルキルオキシカルボニル、アリールアルキルスルホニル、アリールチオカルボニル、アリールアルケニルスルホニル、ヘテロアリールスルホニル、アリールスルホニル、アルコキシアリールアルキル、ヘテロアリールアルコキシカルボニル、アリールヘテロアリールアルキル、アルコキシアリールカルボニル、アリールオキシヘテロアリールアルキル、ヘテロアリールアルキルオキシアリールアルキル、アリールアリールアルキル、アリールアルケニルアリールアルキル、アリールアルコキシアリールアルキル、アリールカルボニルアリールアルキル、アルキルアリールオキシアリールアルキル、アリールアルコキシカルボニルヘテロアリールアルキル、ヘテロアリールアリールアルキル、アリールカルボニルヘテロアリールアルキル、ヘテロアリールオキシアリールアルキル、アリールアルケニルヘテロアリールアルキル、アリールアミノアリールアルキルまたはアミノカルボニルアリールアリールアルキルから独立して選択され;
YはCO2R4(R4はH若しくはアルキル、またはプロドラッグエステルである)であるか、またはYはC−連結した1−テトラゾール、構造P(O)(OR4a)R5(R4aはHまたはプロドラッグエステルであり、R5はアルキルまたはアリールである)のホスフィン酸または構造P(O)(OR4a)2のホスホン酸であり;
(CH2)x、(CH2)x 1、(CH2)x 2、(CH2)x 3、(CH2)m、および(CH2)nは適宜、1、2または3個の置換基で置換され得る]
を有し、そのすべての立体異性体、そのプロドラッグエステル、およびその医薬的に許容される塩を含む置換された酸誘導体が提供される。
【0010】
好ましくは、構造IA:
【化2】
【0011】
より好ましくは、構造IB:
【化3】
上記化合物で、最も好ましいのは、
R2a、R2bおよびR2cが各々Hであり;
R1がアルキル(好ましくはCH3)であり;
x2が1〜3で、x3が0であり;
R2がHであり;
mが0であるか、(CH2)mがCH2またはCHOHまたはCH−アルキルであり;
X2、X3、およびX4が、総計1、2または3個の窒素を表し;
(CH3)nが結合またはCH2であり;
R3がアリール、アリールアルキルまたはヘテロアリール(例えば、チオフェンまたはチアゾール、最も好ましくはフェニルまたは以下で置換されたフェニル;該置換基はアルキル、ポリハロアルキル、ハロ、アルコキシ、好ましくはCF3およびCH3である)であり;
R3aが好ましくはHまたはアルキルである。
【0012】
本発明の好ましい化合物には、以下が含まれる。
【化4】
本発明は、ある単一の分子中に両PPARγアンタゴニスト/PPARαアゴニスト活性を有するという発見について記述する。本発明は、重厚な糖尿病の、高脂血症のおよび肥満のdb/dbマウスへの両PPARγアンタゴニスト/PPARαアゴニストの投与が、ブドウ糖レベルを変化させることなく、血漿のトリグリセリドおよび遊離脂肪酸レベルを減少させるということを示す。本発明は、食餌で誘発された肥満のマウスへの両PPARγアンタゴニスト/PPARαアゴニストの投与が、高脂血症および/またはインスリン耐性を引き起こすことなく、体脂肪含量を減らし、肝臓の脂肪を減らすということを示す。本発明は、抗肥満症、インスリン感受性および循環器疾患のためになるPPARγアンタゴニスト活性により、遺伝子発現が脂肪組織において変動する標的遺伝子のリストを提供する。
【0014】
従って、本発明の1つの目的は、ヒトを含む哺乳類における肥満症の新規な治療方法で、PPARγの阻害およびPPARαの活性化を同時にする単一の化合物または化合物の組み合わせの治療上の有効量を、かかる治療が必要な哺乳類に投与することからなる新規な治療方法を提供する。
【0015】
本発明の別の目的は、ヒトを含む哺乳類での代謝症候群(肥満症、インスリン耐性および異脂肪血症)の治療方法で、以下の化合物、すなわち:PPARγを拮抗し、PPARα活性を活性化する化合物または化合物の組み合わせ;抗糖尿病性化合物(例えば、これらに限らないが、インスリン、メトホルミン、インスリン増感剤、スルホニル尿素、aP2阻害剤、SGLT−2阻害剤);脂質低下剤(例えば、これらに限らないが、スタチン類、フィブラート類、ナイアシンACAT阻害剤、LCAT活性化剤、胆汁酸隔離剤)および体重減少剤(例えば、これらに限らないが、オルリスタット、シブトラミン、aP2阻害剤、アディポネクチン)のうち2個以上からなるいずれかの組み合わせの治療上の有効量を、かかる治療が必要な哺乳類に投与することからなる治療方法を提供する。
【0016】
本発明の別の目的は、標的遺伝子(例えば、HMGic、グリセロール−3−PO4−デヒドロゲナーゼ、Gタンパク質共役受容体26、脂肪酸輸送タンパク質、アジポフィリンおよびケラチノサイト脂肪酸結合タンパク質)のリストを提供することであり、該標的遺伝子の発現は、PPARγアンタゴニストおよび両PPARγアンタゴニスト/PPARαアゴニストの投与により、または他の方法により、抗肥満症の効果を得るよう変えられ得る。
【0017】
本発明の別の目的は、標的遺伝子(例えば、PAI−1、レニン、アンジオテンシノーゲン前駆体)のリストを提供することであり、該標的遺伝子の発現は、PPARγアンタゴニストおよび両PPARγアンタゴニスト/PPARαアゴニストの投与により、または他の方法により、循環器疾患に対する有益な効果を得るよう変えられ得る。
【0018】
本発明の別の目的は、医薬的に許容される担体と、PPARγの阻害およびPPARαの活性化を同時にする化合物または化合物の組み合わせの治療上の有効量とからなる、肥満症を治療する医薬組成物を提供することである。
【0019】
本発明の別の目的は、肥満症、インスリン耐性および/または異脂肪血症の治療用で、医薬的に許容される担体、並びにPPARγの阻害およびPPARαの活性化を同時にする化合物または化合物の組み合わせの治療上の有効量、並びに抗糖尿病性化合物、脂質低下剤および体重減少剤からなる医薬組成物を提供することである。
【0020】
さらに、本発明によって、糖尿病、特に2型糖尿病、および関連する疾患(例えば、インスリン耐性、高血糖、高インスリン血症、脂肪酸またはグリセロールの血中レベルの上昇、高脂血症、肥満症、高トリグリセリド血症、炎症、シンドロームX、糖尿病性合併症、代謝異常症侯群、アテローム性動脈硬化症、および関連する疾患)を治療するための方法で、構造Iの化合物の治療上の有効量を治療が必要な患者に投与する方法を提供する。
【0021】
さらに、本発明によって、初期悪性病変(例えば、乳房のインシトゥーの管の癌および乳房のインシトゥーの小葉癌)、前悪性病変(例えば、乳房の線維腺腫および前立腺上皮内異常増殖(PIN)、脂肪肉腫および様々な他の上皮性腫瘍(乳房、前立腺、大腸、卵巣、胃および肺を含む))、過敏性大腸症候群、クローン病、胃潰瘍、および骨多孔症および増殖性疾患(例えば乾癬)を治療するための方法で、構造Iの化合物の治療上の有効量を治療が必要な患者に投与する方法を提供する。
【0022】
さらに、本発明によって、糖尿病および上記または以下で規定される関連疾患を治療する方法で、構造Iの化合物と別のタイプの抗糖尿病剤および/または抗高脂血症剤、および/または脂質調節剤および/または他のタイプの治療剤との組み合わせの治療上の有効量を治療が必要なヒトの患者に投与する方法を提供する。
【0023】
本発明の上記方法において、構造Iの化合物は、抗糖尿病剤(処置の方法に依存する)に対する重量比が、約0.01:1〜約100:1、好ましくは約0.5:1〜約10:1の範囲内で用いられる。
【0024】
「シンドロームX」または代謝異常症侯群(Johanson, J. Clin. Endocrinol. Metab., 1997, 82, 727-734および他の公知文献に詳述される)に関して集合的に言及される症状、疾病、および疾患には、高血糖および/または前糖尿病性インスリン耐性症候群を含み、高インスリン血症、異脂肪血症、および障害性ブドウ糖耐性(2型糖尿病に進行し得る)を生じる初期のインスリン耐性状態の特徴があり、高血糖(糖尿病性合併症に進行し得る)の特徴がある。
【0025】
用語「糖尿病および関連疾患」とは、2型糖尿病、1型糖尿病、障害性ブドウ糖耐性、肥満症、高血糖、シンドロームX、代謝異常症侯群、糖尿病性合併症および高インスリン血症をいう。
【0026】
「糖尿病性合併症」に関して集合的に言及される症状、疾病、および疾患には、網膜症、神経障害および腎障害、および他の公知の糖尿病の合併症が含まれる。
【0027】
本明細書で用いられる用語「他のタイプの治療剤」とは、1個以上の抗糖尿病剤(式Iの化合物以外)、1個以上の抗肥満症剤、および/または1個以上の脂質低下剤、1個以上の脂質調節剤(抗アテローム性動脈硬化症剤を含む)、および/または1個以上の抗血小板剤、1個以上の高血圧症治療剤、1個以上の抗癌薬、1個以上の関節炎治療剤、1個以上の抗骨多孔症剤、1個以上の抗肥満剤、1個以上の免疫調節性疾患の治療剤、および/または1個以上の拒食症治療剤をいう。
【0028】
本明細書で用いられる用語「脂質調節」剤とは、LDLを低下し、および/またはHDLを上昇させ、および/またはトリグリセリドを低下させ、および/または総コレステロールを低下させる薬剤、および/または脂質障害の治療的な処置の他の公知の機構をいう。
【0029】
(発明の詳細な説明)
PPARγは、前脂肪細胞の補充および成熟した脂肪細胞への分化の主要な調節剤である(参照:Tontonoz et al., Current Biology, 571-576, 1995)。PPARγの活性化剤は、前脂肪細胞の分化、成熟した脂肪細胞中の脂質貯蔵を促進し、インスリンとして作用し、抗糖尿病剤を感作する(参照:Tontonoz et al., Current Biology, 571-576, 1995 ; Lehmann et al., J. Biol. Chem., 270: 12953-12956, 1995; Nolan et al. New. Eng. J. Med., 331: 1188-1193 ; Inzucchi et al., New Eng. J. Med., 338: 867-872, 1998, Willson, et al., J. Med. Chem. : 43: 527-550, 2000, Kersten et al., Nature, 405: 421424, 2000)。しかしながら、PPARγで誘発される抗糖尿病活性は、動物モデルおよびヒトにおいていくらかの体重増大をしばしば伴う。最近の発見で、PPARγの阻害が体脂肪蓄積および肥満症における減少になると示唆されている(参照:Vidal-Puig et al., J. Clinical Investigation, 99: 2416-2422, 1997 ; Deeb et al Nature Genetics, 20: 284-287, 1998 ; Kubota et al. Mol. Cell; 4: 597-609, 1999 ; Barroso et al. Nature; 402, 860-861, 1999)。しかしながら、かかる減少で、より高い血漿遊離脂肪酸および高脂血症および発達脂肪肝およびインスリン耐性になるようである。PPARαのイソ型は、脂肪酸合成、脂肪酸酸化および脂質代謝経路における遺伝子を調節する(参照:Issenman and Green, Nature, 347: 645-649, 1990 ; Torra et al., Current Opinion in Lipidology, 10: 151-159, 1999; Kersten et al., Nature, 405: 421424, 2000)。PPARαアゴニスト(例えば、フェノフィブラート、ゲムフィブロジル)処理は、肝臓および筋肉における脂肪酸酸化を高め、肝臓における脂肪酸およびトリグリセリド合成を減少させ、血漿トリグリセリドを減少させる(参照:Kersten et al., Nature, 405: 421424, 2000)。高トリグリセリドおよび低HDL−コレステロールの患者への、PPARαアゴニストでの処置は、血漿HDL−コレステロールの増加、血漿トリグリセリドの減少および1°および2°両方の心臓病事故の縮小につながる(参照:Balfour et al., Drugs. 40: 260-290, 1990 ; Frick et al., New Eng. J. Med., 317 : 1237-1245 ; Rubins et al., New Eng. J. Med., 341: 410-418, 1999)。従って、PPARγアンタゴニストおよびPPARαアゴニストの単一の両活性化合物または組み合わせにおける、PPARγアンタゴニスト活性およびPPARαアゴニスト活性を合わせることによって、高脂血症、脂肪肝およびインスリン耐性を引き起こすことなく、安全なPPARγ阻害および肥満症の治療が可能である。
【0030】
化合物Yは本明細書の実施例1に概説された反応式で合成された化合物である。図1−A、1−Bに図示されているように、化合物Yは高い親和性(IC50=69 nM)でヒトPPARγリガンド結合ドメインと強く結合した。同様に化合物Yは精製ヒトPPARαリガンド結合ドメインとも強く結合した(IC50=69 nM)。関連するPPARγリガンド結合の研究において、ロシグリタゾン(真正のPPARγアゴニスト)でIC50=250 nM、GW0072(真正のPPARγアンタゴニスト)でIC50=280 nMを得た。PPARαリガンド結合の研究において、GW−2331(PPARα選択的アゴニスト)でIC50=410 nMを得た。このような精製リガンド結合ドメインでのインビトロリガンド結合の研究より、PPARγおよびPPARαの両方に強く結合する化合物Yの能力が示される。しかしながら、強く結合する化合物(すなわちリガンド)が、アゴニスト(活性化リガンド)およびアンタゴニスト(受容体を不活性化するリガンド)として作用し得ることは、転写因子の核ホルモン受容体ファミリーとしてよく知られている(PPARはこのファミリーのメンバーである)。
【0031】
図2に図示するように、化合物Yはマウス前脂肪細胞3T3L−1に加えられる場合、(該細胞からのグリセロール放出によって測定されながら)成熟した脂質を負荷した脂肪細胞への分化を誘発するロシグリタゾン(PPARγアゴニスト)との競合的阻害を示す。マウス3T3−L−前脂肪細胞は、ホルモンシグナル(例えばインスリン、デキサメタゾン )およびPPARγアゴニスト(例えばロシグリタゾン)に応答し、成熟した脂肪細胞へ分化し、および脂質を蓄積することが知られている。PPARγは脂肪細胞の分化過程の主要なトリガーであると考えられている(参照:Tontonoz et al., Current Biology, 571-576, 1995)。化合物YはPPARγの強力なリガンドであるが、ロシグリタゾンで誘発される分化の競合的阻害を示すので、従ってPPARγのアンタゴニストであると示唆される。分化の阻害のED50=9.9μMということは、化合物Yが前脂肪細胞分化の適当な阻害剤であることを示している。対照的に、ED50=0.585μMは、PPARγアンタゴニストであるGW0072として得られた(参照:Oberfield et al, Proc. Nat. Acad. Sci., 96: 6102-6106, 1999)。
【0032】
図3に図示したように、化合物YのPPARγアンタゴニスト活性は、第二の細胞系で確認された。内在性のPPARγの発現を示す確立されたCV−1細胞(霊長類の腎臓起源)は、PPAR応答型内在性アルカリフォスファターゼ(SEAP)レポーター遺伝子と安定にトランスフェクトされた。先の研究でのように、化合物Yはロシグリタゾン(PPARγアゴニスト)依存活性によって、すなわちCV−1細胞におけるSEAPレポーター遺伝子発現の誘導によって競合的に阻害された。SEAP遺伝子のロシグリタゾン誘発性トランス活性化の特異的な阻害であるED50=1.5μMは、化合物YがPPARγのアンタゴニストであることを再び示す。PPARγアンタゴニストであるGW0072の研究(参照:Oberfield et al, Proc. Nat. Acad. Sci., 96: 6102-6106, 1999)において、またCV−1細胞におけるロシグリタゾンで媒介されるSEAP遺伝子の誘発(阻害のED50=0.37μM)を用量依存的に阻害し、データの信頼性を立証した。
【0033】
図4に図示するように、化合物Yはヒト肝臓細胞HepG2中のSEAPレポーター遺伝子のPPARα依存トランス活性化を用量依存的に刺激したことから、それはPPARαのアゴニストであることが示された。内在のPPARα遺伝子を発現するHepG2細胞(ヒト肝臓起源)は、PPAR応答性SEAPレポーター遺伝子で安定にトランスフェクトされた。処置の際の化合物Yは、PPARαトランス活性化のEC50=0.587μMのHepG2細胞において、SEAP遺伝子発現を用量依存的に刺激した。本研究において、BMS−250773(PPARα選択的薬剤)は、EC50=0.063μMでSEAPレポーター遺伝子のPPARα依存性トランス活性化を用量依存的に刺激し、ロシグリタゾン(PPARγアゴニスト)はほとんど活性化を示さなかった。
【0034】
このように、図1、2、3、4に図示するインビトロ PPARγおよびPPARαリガンド結合の研究、並びにPPARγおよびPPARα依存細胞に基づくトランス活性化の研究は、化合物YがPPARγおよびPPARαの両方の強力なリガンドであることを示すが、しかしながら、PPARγへのアンタゴニスト活性およびPPARαへのアゴニスト活性を示すものである。これらの知見は、化合物Yが単一分子中に両方の(両)PPARγアンタゴニスト活性およびPPARαアゴニスト活性を有する分子の新規な分類に属することを示唆する。
【0035】
【表1】
【0036】
このように、遺伝子発現プロフィール研究で、両PPARγアンタゴニスト/PPARαアゴニスト化合物Yの、インビボのPPARγアンタゴニストおよびPPARαアゴニスト活性が確認される。さらに、これらの研究でまた、PPARγアンタゴニストおよび/または両PPARγアンタゴニスト/PPARαアゴニストの投与によって、脂肪の(脂肪)組織中、脂肪細胞分化に影響を与える遺伝子(例えば、HMGic、グリセロール3−PO4デヒドロゲナーゼ、脂肪酸輸送タンパク質および新規なオーファンGタンパク質共役受容体26レベル)を変化させて、肥満症を治療する方法も示される。これらの研究ではまた、PPARαアゴニストおよび/または両PPARγアンタゴニスト/PPARαアゴニストの投与によって、脂肪の(脂肪)組織中、アジポフィリンおよびケラチノサイト脂肪酸結合タンパク質レベルを変化させて、肥満症を治療する方法も示される。
【0037】
【表2】
【0038】
【表3】
【0039】
【表4】
【0040】
【表5】
【0041】
【表6】
【0042】
従って、本発明は新規な両活性のPPARγアンタゴニスト/PPARαアゴニスト剤の発見を示すものである。本発明は、両PPARγアンタゴニスト/PPARαアゴニストの投与のより肥満症を治療する原理の医薬的な証明を提供する。本発明によれば、単一分子中のPPARγアンタゴニスト活性およびPPARαアゴニスト活性の組み合わせ、または薬剤中のPPARγアンタゴニスト活性およびPPARαアゴニスト活性の組み合わせは、肥満の個体における脂質および/または血糖のコントロールのさらなる悪化を伴うことなく、肥満症の治療を提供する。
【0043】
本発明は、PPARγアンタゴニスト、または両PPARγアンタゴニスト/PPARαアゴニストまたはPPARαアゴニストによる治療により得られる、抗肥満症を達成するようにその発現が修飾される遺伝子(例えば、HMGic、グリセロール−PO4デヒドロゲナーゼ、脂肪酸輸送タンパク質、Gタンパク質共役受容体26、アジポフィリン、ケラチノサイト脂肪酸結合タンパク質)および心臓血管を達成するようにその発現が修飾される遺伝子(例えば、アンジオテンシノーゲン、PAI−1、レニン)のリストの同定を提供する。
【0044】
本発明はまた、両PPARγアンタゴニスト/PPARαアゴニストまたはPPARαアゴニストの投与により、肝臓障害を治療する方法を提供する。
【0045】
本発明はまた、単一の薬剤を含む薬理的組成物または2つの薬剤の組み合わせの投与により、ヒトを含む哺乳類の肥満症を治療する方法:すなわち同時に、
(1)PPARγタンパク質の活性、または(2)PPARγ遺伝子の発現、(3)活性化補助因子の結合または(4)PPARγ調節される標的遺伝子の発現(または上記のいずれかの組み合わせ)を減少し、並びに
(1)PPARαタンパク質の活性、または(2)PPARα遺伝子の発現、または(3)活性化補助因子の結合または(4)PPARα調節される標的遺伝子の発現(または上記のいずれかの組み合わせ)を増加する方法も提供する。これらの変化の生じた生成物には、これらに限らないが、(1)体重増加の抑制、(2)体重減少、(3)体脂肪量の特異的減少、(4)除脂肪体重の増加、(5)体脂肪量/除脂肪量比の変化、(7)肝臓の脂質の減少および肝臓機能の改善のいずれかの組み合わせを含み得る。
【0046】
本発明はまた、肥満の患者における体重、インスリン耐性、2型糖尿病、高脂血症および循環器疾患をコントロールするために、両PPARγアンタゴニスト/PPARαアゴニストと、以下の薬剤との組み合わせの使用を含む治療方法を提供する:抗糖尿病剤(例えば、これに限らないが、メトホルミン、スルホニル尿素、インスリン、インスリン増感剤、aP2阻害剤、SGLT2阻害剤)、肝臓ブドウ糖排出に影響を与える薬剤、PPARαアゴニスト(例えば、これに限らないが、フェノフィブラートおよびゲムフィブロジル)のような脂質低下剤およびHMG−CoAリダクターゼ阻害剤(例えば、これに限らないが、プラバスタチン、ロバスタチン、シンバスタチンおよびアトルバスタチン)、ナイアシン、ACT阻害剤、LCAT活性化剤、胆汁酸隔離剤および他の抗肥満剤(例えば、これに限らないが、オルリスタット、シブトラミン、P2阻害剤、アディポネクチン)。
【0047】
本発明の式Iの化合物は、以下の一般的合成反応式、並びに当業者に用いられる関連する公開された文献の方法に従って調製し得る。これらの反応の具体的な試薬および方法は、以下の記述および実施例に示されている。以下の反応式にある保護および脱保護は、当該技術分野で一般的に知られている方法で実施し得る(参照:例えば、Greene, T. W. and Wuts, P. G. M., Protecting Groups in Organic Synthesis, 3rd Edition, 1999 [Wiley])。
【0048】
本発明の化合物の合成に要求される鍵中間体の合成は、反応式1に記載されている。アルコール体1(R5(CH2)x 2OH)(最も好ましいものの1つは、2−フェニル−5−メチル−オキサゾール−4−エタノール)は、標準的なミツノブ反応の条件(例えば、Mitsunobu, 0., Synthesis, 1981, 1)下、ヒドロキシアリール−またはヘテロアリール−アルデヒド体2とカップリングして、鍵中間体のアルデヒド3が得られる。あるいは、アルコール体1は標準的な条件下そのメタンスルホン酸エステルに変換され得 ;メシレート体4は次いで、ヒドロキシアリール−またはヘテロアリール−アルデヒド体2をアルキル化して、アルデヒド体3を供給するのに用いられ得る。
【0049】
反応式2に、2−アリール(ヘテロアリール)−4−カルボキシ−トリアゾール化合物Iの一般的合成を記載する。適当に保護されたオキシ安息香酸クロリド体またはオキシフェニル酢酸クロリド体5のメルドラム酸との塩基存在下での処理で、対応するクルードなメルドラム酸付加体6が得られ、アニリンとすぐに反応してβ−ケトアニリド体7が得られる(Synthesis, 1992, 1213-1214)。β−ケトアミド体7は、亜硝酸(塩基/亜硝酸ナトリウムからインシトゥーで産生する)との反応、続く酸処理により、対応するα−オキシム−β−ケトアミド体8を得る(引用文献: Hamanaka, E. S., et al, WO9943663)。β−ケト−アミド体8は次いで、適当に置換されたヒドラジン体9と縮合し、対応するβ−ヒドラゾン−アミド体10を得る。中間体10の酸による処理で、目的の2−置換された−4−カルボキサミド−トリアゾール体11を得る(引用文献: Hamanaka, E. S., et al, WO9943663)。トリアゾール−アニリド体11のフェノール性保護基の脱保護で、対応するフェノール体12を得る。フェノール−トリアゾール体12は次いで、適当なアルコール体1と標準的なミツノブ反応条件下(例えば、Mitsunobu, O., Synthesis, 1981, 1)カップリングして、目的のアルキル化したトリアゾール−アミド体13を得る。あるいは、該フェノール体は塩基性条件下メタンスルホン酸エステル4とカップリングして、アルキル化したトリアゾール−アミド体13を得ることができる(引用文献:Cheng, P. T. W., et. al., WO0121602)。このアニリド体の続く塩基性による脱保護で、本発明の目的の2−置換された4−カルボキシトリアゾール体IIを得る。
【0050】
反応式3には、2−アリール−4−カルボキシトリアゾール体Iの製造のための、反応式2に示すものの補足的なアプローチを示す。適当に保護されたヒドロキシアリールまたはヒドロキシヘテロアリールカルボン酸14は、1)塩基の存在下メシレート体4と、または2)標準的なミツノブ条件下アルコール体1とのいずれかで処理して、カルボン酸への脱保護後、鍵となるアルキル化した酸中間体15を得る。酸化合物15の対応する酸クロリド体16への変換は、シュウ酸クロリドを用いて行う。酸クロリド体16のメルドラム酸との処理で、対応する付加体17を得て、次いですぐにアニリンと反応してβ−ケトアニリド体18を得る。次いでβ−ケトアニリド体18の亜硝酸(塩基/NaNO2からインシトゥーで産生する)による処理で、対応するβ−ケト−α−オキシミノ−アニリド体19を得て、次いで適当に置換されたヒドラジン体9と反応して、中間体β−ヒドラゾン−アミド体20を得る。次いでオキシム−ヒドラゾン体20の酸による環化で、アリールトリアゾールアニリド体21を得る。最終的に、塩基によるアニリド体の加水分解で、本発明の目的の2−置換された−4−カルボキシトリアゾール体IIAを得る。
【0051】
反応式4に、1−置換された4−カルボキシトリアゾール体IIの合成を示す。β−ケトアニリド体18のp−トルエンスルホニルアジド(Padwa, A., et al, J. Org. Chem., 1997, 62, 6842)との処理で、対応するβ−ケト−α−ジアゾ−アニリド体21を得る。β−ケト−α−ジアゾ−アニリド体21の適当に置換されたアミン体22とのルイス酸で介される反応で、対応する1−置換された−4−アミド トリアゾール体23を得る(Ohno, M., et al, Synthesis, 1993, 793)。トリアゾール−アニリド体23のフェノール官能基の脱保護で、フェノール体23を得る。次いでフェノール−トリアゾール体23のアルキル化を、標準的なミツノブ反応条件下(例えば、Mitsunobu, O., Synthesis, 1981, 1)アルコール体1により実施し、対応するアルキル化したトリアゾール−アミド体を得る。あるいはフェノール−トリアゾール体23は、塩基性条件下メタンスルホン酸エステル4とカップリングして、同じアルキル化したトリアゾール−アミド体を得ることができる。その後塩基性によるカルボン酸への脱保護で、本発明の目的の1−置換された−4−カルボキシトリアゾール体IIIを得る。
【0052】
反応式5に、位置異性体の1−置換された−5−カルボキシトリアゾール体IIIおよび1−置換された−4−カルボキシトリアゾール体IVの合成を記載する。アルデヒド体3は、塩基性/アニオン性条件下適当に保護されたプロパルギル酸(propargylic acid)と反応して(J. Org. Chem., 1980, 45, 28)、対応するアセチレンアルコール付加体25が得られる。アセチレンアルコール体25は次いで、標準的な文献の条件下(Czernecki, S., et al, J. Org. Chem., 1989, 54, 610)脱酸素して、アセチレンエステル体26を得る。熱条件下(Can. J. Chem., 1980, 58, 2550)のアセチレンエステル体26の適当に置換されたアリールアジド体27との双極環化付加、その後のカルボン酸官能基への脱保護により、本発明の目的のアリールトリアゾール酸化合物IVおよびVが得られる。
【0053】
反応式6に、トリアゾール酸化合物IVおよびV、並びにヒドロキシトリアゾール酸化合物VIおよびVIIの製造のわずかに修正した工程を示す。アセチレンアルコール付加体25は熱条件下すぐに適当に置換されたアジド体27と双極環化付加反応して、対応する位置異性体のヒドロキシトリアゾールエステル体28および29が得られ、次いで脱保護されて本発明のヒドロキシトリアゾール酸VIおよびVIIがそれぞれ得られる。あるいはヒドロキシトリアゾールエステル体28および29は、脱酸素化および脱保護されて、本発明のトリアゾール酸化合物IVおよびVが得られる。
【0054】
反応式7に、1−置換された−4−カルボキシピラゾール化合物VIIIの合成を記載する。保護されたフェノール−アルコール体30は標準的な文献の方法(Tetrahedron Lett., 1986, 42, 2725)で、対応するクロリド体31に変換される。保護されたシアノ酢酸体32は次いで、塩基の存在下クロリド体31でアルキル化されて、シアノ酢酸体33が得られる。シアノ酢酸体33の脱保護で、シアノ酢酸体34が得られる。亜硝酸(亜硝酸ナトリウムおよび酸からインシトゥーで産生する)の存在下、シアノ酢酸体34の適当に置換されたヒドラジン体9との処理で、対応するシアノ−ヒドラゾン体35が得られる(Skorcz, J. A., et al, J. Med. Chem., 1966, 9, 656)。塩基存在下、シアノヒドラゾン体35の適当に保護したアクリレート体36との反応で、鍵となるアリール−ピラゾールエステル中間体37が得られる(Kim, Y. H., et al, Tetrahedron Lett., 1996, 37, 8771)。1)ピラゾール体37のフェノールの保護基の除去、2)生じたフェノール体の塩基性条件下メシレート体4によるアルキル化、および3)カルボン酸への脱保護を含む3工程で、本発明の1−アリール−3−置換された−4−カルボキシピラゾール体VIIIが得られる。
【0055】
反応式8に、位置異性体の1−置換された−5−置換された−4−カルボキシピラゾール化合物IXの合成を示す。保護されたフェノール−酸クロリド体5は、塩基性条件下メルドラム酸で処理されて、対応する付加体が得られ、適当なアルコール体R3OHと反応して、β−ケトエステル体38が得られる。β−ケト−エステル体38のジメチルホルムアミドジメチルアセタールとの処理で、α−エナミノ−β−ケト−エステル体39が得られる(Almansa, C., et al, J. Med. Chem., 1997, 40, 547)。α−エナミノ−β−ケト−エステル体39の適当に置換されたヒドラジン体9による反応、続く分子内環化反応で、アリール−N−ピラゾールエステル体40が得られる。1)化合物40のフェノールの保護基の除去、2)生じたフェノール体のメシレート体4によるアルキル化、および3)カルボン酸への脱保護を含む3工程で、本発明のN−置換されたピラゾール酸IXが得られる。
【0056】
位置異性体のカルボキシピラゾール化合物Xの合成を、反応式9に示す。アルデヒド体3の(適当に置換されたアルキニル金属試薬41との)処理で、アセチレンアルコール付加体42が得られる。アルコール体42は次いで、熱条件下(Kato, T., et al, Chem. Pharm. Bull., 1975, 20, 2203)ケテン二量体で処理して、アセト酢酸エステル体43が得られる。標準的な条件下(引用文献)のアセト酢酸エステル体43の塩素化で、α−クロロ−β−ケトエステル体44が得られる。熱条件下α−クロロ−β−ケトエステル体44の適当に置換されたジアゾ化合物45による処理で、クロロヒドラゾン体46が得られる(Garantic, L., et al, Synthesis, 1975, 666)。クロロヒドラゾン体46の塩基による熱分子内環化付加反応により、次いでピラゾール−ラクトン体47が得られる(Garantic, L., et al, Synthesis, 1975, 666)。ピラゾール−ラクトン体47の付随する環の開環/脱酸素化は、多くの異なる反応条件下(TMSCl/NaIまたはZn/NH4OH ; Sabitha, G., Synth. Commun., 1998, 28, 3065)実施され、ピラゾール酸化合物48が得られる。1)化合物48のフェノールの保護基の除去、2)生じたフェノール体のメシレート体4によるアルキル化、および3)カルボン酸への脱保護を含む3工程で、本発明のN−置換されたピラゾール酸Xが得られる。
【0057】
N−置換された−ピロール−3−カルボン酸化合物XIへの一般的な工程を、反応式10に示す。アルデヒド体3は塩基性条件下適当に保護されたプロピオール酸エステル化合物49と反応して、アルキン−アルコール体50が得られる(J. Org. Chem., 1980, 45, 28)。標準的な方法(例えば、Et3SiH/酸; Tetrahedron Lett., 1987, 28, 4921)を用いたアルキン体50のアルコール官能基の脱酸素化により、アルキン酸エステル化合物51が得られる。標準的な方法(「Preparation of Alkenes, A Practical Approach」, J. M. J. Williams, Ed., Chapter 6,「Reduction of Alkynes」, J. Howarth. Oxford University Press, 1996)を用いたアルキン酸エステル化合物51の還元により、Z−アルケニルエステル化合物52が得られる。α,β−不飽和エステル化合物52は次いで、標準的な文献の条件で(Van Leusen, A. M., et al, Tetrahedron Lett., 1972, 5337)トシルメチルイソシアネート(TosMIC)と反応し、対応するピロール−エステル化合物53が得られる。標準的な文献の条件で(Lam, P. Y. S., et al, Tetrahedron Lett., 1998, 39, 2941)ピロール−エステル化合物53の適当に置換されたアリールまたはヘテロアリールボロン酸54とのカップリングにより、N−置換されたピロールエステル体55が得られる。次いでN−置換されたピロールエステル体55の脱保護で、本発明のN−置換されたピロール酸XIが得られる。
【0058】
反応式11に、N−置換されたピロール−3−カルボン酸化合物XIIへの合成工程が記載されている。アルデヒド体3は、ホスホラニリデンエステル化合物53とウィッティヒ反応するか(「Preparation of Alkenes, A Practical Approach」, J. M. J. Williams, Ed., Chapter 2,「The Wittig reaction and related methods」, N. J. Lawrence, Oxford University Press, 1996)、またはホスホン酸エステル56とのホーナー・エモンズ反応を行い(J. M. J. Williams, supra and N. J. Lawrence, supra)、有意にE−アルケニルエステル体57が得られる。E−アルケニルエステル体57は次いで、トシルメチルイソシアネート(TosMIC)と反応して、ピロール−エステル化合物58が得られる。ピロール−エステル化合物58は次いで、標準的な文献の条件下(Evansの文献)適当なボロン酸化合物54と反応して、対応するN−置換されたピロール−エステル化合物59が得られる。次いでN−置換されたピロール−エステル化合物59の脱保護により、本発明のN−置換されたピロール酸化合物XIIが得られる。
【0059】
反応式12に、必要な中間体2−アリール(または2−ヘテロアリール)−5−メチル−オキサゾール−4−イルメチルクロリドの製造を示す(Malamas, M. S., et al, J. Med. Chem., 1996, 39, 237-245に記載の一般方法に従う)。置換されたアルデヒド体60は、酸性条件下ブタン−2,3−ジオン モノ−オキシムと縮合して、対応するオキサゾール−N−オキシド体61が得られる。付随する塩素化でオキサゾール−N−オキシド体61が脱酸素化されて、目的のクロロメチルアリール(またはヘテロアリール)−オキサゾール体62が得られる。塩基性条件下のクロロメチルオキサゾール体62の加水分解で、対応するオキサゾール−メタノール体63が得られる。アルコール体63の対応するアルデヒド体への酸化に続いて、対応するジブロモアルケン体64(例えば、Ph3P/CBr4)に転換される。ジブロミド体64は、対応するアルキニル−リチウム体(n−BuLiのような有機リチウム試薬を用いる)に転換され、これは適当な親電子体(例えばホルムアルデヒド)とインシテューで反応して、対応するアセチレンアルコール体が得られる(引用文献:Corey, E. J., et al., Tetrahedron Lett. 1972, 3769, またはGangakhedkar, K. K., Synth. Commun. 1996, 26, 1887-1896)。このアルコール体は次いで、対応するメシレート体65に転換され、適当なフェノール化合物66でアルキル化され、カルボン酸への脱保護後、類似体XIIIが得られる。一般に、フェノール体66は適当な中間体(例えば、11、23および37)のフェノール官能基の脱保護で得られる。本発明のアルキン体XIIIの立体選択的な部分的還元(例えば、H2/リンドラー触媒)で、E−またはZ−アルケニル類似体XIVが得られる。アルケン類似体XIVの完全な還元(水素化)で、本発明のアルキル類似体XVが得られる。あるいは、本発明のアルキン類似体XIIIの完全な還元(例えば、H2/パラジウム炭素触媒)はまた、本発明のアルキル類似体XVも得られる。
【0060】
炭素連結の類似体XVI、XVII、およびXVIIIの合成が、反応式13〜14に示されている。反応工程は反応式2に示されているものに類似する。塩基の存在下適当に保護されたハロ−アリール(またはヘテロアリール)酸クロリド体67をメルドラム酸で処理して、対応するクルードメルドラム酸付加体68が得られ、アニリンと直ちに反応してβ−ケトアニリド体69が得られる。β−ケトアミド体69は亜硝酸(塩基/亜硝酸ナトリウムからインシトゥーで産生する)との反応、続く酸処理により、対応するα−オキシム−β−ケトアミド体70を得る。β−ケト−アミド体70は次いで、適当に置換されたヒドラジン体9と縮合し、対応するβ−ヒドラゾン−アミド体71を得る。中間体71の酸による処理で、目的の2−アリール−4−カルボキサミド−トリアゾール体72を得る。ソノガシラ反応条件下(例えば、「Organocopper Reagents, a Practical Approach」, R. J. K. Taylor, E., Chapter, 10, p 217-236, Campbel, I. B., Oxford University Press, 1994)、アルキン体73のハロ−トリアゾール体72とのカップリングで、対応するアルキニルトリアゾール化合物74が得られる。次いでアニリド体74の加水分解で、本発明のアルキニルトリアゾール酸類似体XVIが得られる。本発明のアルキニルトリアゾール酸化合物XVIの選択的還元で(例えば、H2/リンドラー触媒)、本発明のE−またはZ−アルケニルトリアゾール酸化合物XVIIが得られる。次いで本発明のアルケニルトリアゾール酸化合物XVIIの完全な還元で、本発明の飽和アルキルトリアゾール酸化合物XVIIIが得られる。
【0061】
エーテルを含む類似体XIXおよびXXの合成は、反応式15〜16に示す。
【0062】
反応式15では、適当に保護したハロ−アリールトリアゾール化合物72を金属化試薬(例えば、イソプロピルマグネシウムブロミド,引用文献:P. Knochel et al., Synthesis, 2002, 565-569)で処理して、対応するアリールマグネシウム試薬を得て、次いでホルムアルデヒドで反応して、ベンジルアルコール体75が得られる。塩基存在下アルコール体75をメシレート体VIIIで処理して、対応するエーテルアニリド体を得て、次いで脱保護して、本発明のエーテル酸化合物XIXが得られる。
【0063】
反応式16では、スチルカップリング条件下(引用文献:Farina, V., Krishnamurthy, V., and Scott, W. J., Organic Reactions, 1997, 50, 1)適当に保護されたハロ−アリールトリアゾール化合物72を適当なビニルスズ試薬(例えば、トリブチルビニルスズ)で処理して、対応するビニル中間体を得て、次いでハイドロボレーション(例えば、ボラン−THF)して、アルコール体76が得られる。塩基存在下アルコール体76をメシレート体VIIIで処理して、対応するエーテルアニリド体を得て、次いで脱保護して本発明のエーテル酸化合物XXが得られる。
【0064】
2−置換された−トリアゾール−4−酸化合物XXIの合成を、反応式17に示す。アセチレンエステル化合物26のアジ化ナトリウムとの処理で、双極環化付加反応して、トリアゾール−エステル体77が得られる。標準的な文献の条件(Lam, P. Y. S., et. al., Tetrahedron Lett., 1998, 39, 2941)を用いて、トリアゾール−エステル体77を適当に置換されたアリールまたはヘテロアリールボロン酸化合物54でカップリングして、優先的にN(2)−置換されたトリアゾールエステル化合物78が得られる。次いでトリアゾール−エステル体78脱保護で、本発明のN(2)−置換されたトリアゾール酸XXIが得られる。
【0065】
認められたエーテルを含む類似体XXII〜XXIVの合成は、反応式18〜19に示される。
【0066】
反応式18では、標準的なソノガシラカップリング条件下(例えば、「Organocopper Reagents, a Practical Approach」, R. J. K. Taylor, E., Chapter, 10, p 217-236, Campbell, I. B., Oxford University Press, 1994)、適当に保護したハロ−アリールトリアゾール化合物72を適当に保護したアセチレンアルコール体79(式中、x3=1〜3が好ましい)で処理して、対応するアルキニルトリアゾール化合物80が得られる。化合物80を水素化し、続いてアルコール体へ脱保護して、トリアゾール−アルコール体81が得られる。塩基存在下アルコール化合物81をメシレート体VIIIで処理し、対応するエーテル−アニリド体を得て、次いで脱保護して、本発明のエーテル−酸化合物XXIIが得られる。
【0067】
反応式19では、トリアゾール化合物80を脱保護して、アセチレンアルコール化合物81を得て、塩基存在下メシレート体VIIIと反応して、対応するエーテルアニリド化合物が得られ、次いで脱保護して本発明のエーテル酸化合物XXIIIが得られる。アルキニルトリアゾール酸化合物XXIIIの選択的な還元(例えば、H2/リンドラー触媒)で、本発明のE−またはZ−アルケニルトリアゾール酸XXIVが得られる。
【0068】
反応式20〜21に示したように、トリアゾール−酸類似体の製造のこれらの一般合成スキームは、ピロール酸類似体にも適用される。ピロール酸類似体XXV〜XXIXの製造の合成スキームは、反応式10に記載のアプローチに続く。ハロ−アルデヒド化合物83を塩基性条件下(最も好ましくは18−クラウン−6の存在下フッ化物アニオンを用いる)、トリメチルシリルプロピオール酸エステル化合物84と反応させ、アルキン−アルコール化合物85を得る。標準的な方法を用いて(例えば、Et3SiH/酸; Tetrahedron Lett., 1987, 28, 4921)、アルキン化合物50のアルコール官能基の脱酸素化により、アルキン酸エステル化合物86が得られる。標準的な方法を用いて(「Preparation of Alkenes, A Practical Approach」, J. M. J. Williams, Ed., Chapter 6,「Reduction of Alkynes」, J. Howarth. Oxford University Press, 1996)、アルキン酸エステル化合物86を還元して、Z−アルケニルエステル化合物87が得られる。α,β−不飽和エステル化合物87は次いで、標準的な文献の条件下(Van Leusen, A. M., et al, Tetrahedron Lett., 1972, 5337)トシルメチルイソシアネート(TosMIC)と反応させて、対応するピロール−エステル化合物88が得られる。標準的な文献の条件下(Lam, P. Y. S., et al, Tetrahedron Lett., 1998, 39, 2941)、ピロール−エステル化合物88を適当に置換されたアリールまたはヘテロアリールボロン酸化合物54とカップリングさせて、鍵中間体であるハロ−アリール−N−置換された−ピロールエステル化合物89が得られ、これはハロ−アリールトリアゾール中間体72とのピロール等価体である。ハロアリールピロール化合物89を、トリアゾール化合物72の代わりに反応式15、16、18および19に記載の同一の反応経路に用いることで、反応式21に示されたような本発明のピロール酸化合物XXV〜XXIXが得られる。
【0069】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【化13】
【化14】
【化15】
【化16】
【化17】
【化18】
【化19】
【化20】
【化21】
【化22】
【化23】
【化24】
【化25】
【化26】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルキル」、「アルキル」または「アルカ(alk)」には、通常の鎖において1〜20個の炭素原子、好ましくは1〜10個の炭素原子、より好ましくは1〜8個の炭素原子を含む直鎖および分枝鎖の両方の炭化水素が含まれ、通常の鎖において酸素または窒素を適宜含み得、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、t−ブチル、イソブチル、ペンチル、ヘキシル、イソヘキシル、ヘプチル、4,4−ジメチルペンチル、オクチル、2,2,4−トリメチルペンチル、ノニル、デシル、ウンデシル、ドデシル、その様々な分枝鎖の異性体が含まれ、並びに例えば、ハロ(例えば、F、Br、ClまたはIまたはCF3)、アルコキシ、アリール、アリールオキシ、アリール(アリール)またはジアリール、アリールアルキル、アリールアルキルオキシ、アルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルアルキルオキシ、アミノ、ヒドロキシ、ヒドロキシアルキル、アシル、ヘテロアリール、ヘテロアリールオキシ、シクロヘテロアルキル、アリールヘテロアリール、アリールアルコキシカルボニル、ヘテロアリールアルキル、ヘテロアリールアルコキシ、アリールオキシアルキル、アリールオキシアリール、アルキルアミド、アルカノイルアミノ、アリールカルボニルアミノ、ニトロ、シアノ、チオール、ハロアルキル、トリハロアルキルおよび/またはアルキルチオおよび/またはR3基のいずれかの置換基の1〜4個を含むかかる基も同様に含まれる。
【0091】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「シクロアルキル」には、単環式アルキル、二環式アルキルおよび三環式アルキルを含み、環を形成する全部で3〜20個の炭素原子、好ましくは環を形成する3〜10個の炭素原子を含む、1〜3個の環を含む飽和または一部不飽和(1または2個の二重結合を含む)の環状炭化水素基が含まれ、アリールで記載されているような1または2個の芳香環と縮合してもよく、これらにはシクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロデシルおよびシクロドデシル、シクロヘキセニル、
【化27】
【0092】
本明細書で単独または他の基の一部として用いる用語「シクロアルケニル」とは、3〜12個の炭素原子、好ましくは5〜10個の炭素原子および1または2個の二重結合を含む環状炭化水素をいう。具体的なシクロアルケニル基には、シクロペンテニル、シクロヘキセニル、シクロヘプテニル、シクロオクテニル、シクロヘキサジエニル、およびシクロヘプタジエニルが含まれ、シクロアルキルで定義されているように適宜置換されていてもよい。
【0093】
本明細書で用いる用語「シクロアルキレン」とは、フリーな結合を含み、このように例えば
【化28】
【0094】
本明細書で単独または他の基の一部として用いる用語「アルカノイル」とは、カルボニル基に結合するアルキルをいう。
【0095】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルケニル」または「アルケニル」とは、通常の鎖において1〜6個の二重結合を含む、通常の鎖において2〜20個の炭素原子、好ましくは2〜12個の炭素原子、より好ましくは1〜8個の炭素原子の直鎖および分枝鎖の基をいい、通常の鎖において酸素または窒素を適宜含み得、例えば、ビニル、2−プロペニル、3−ブテニル、2−ブテニル、4−ペンテニル、3−ペンテニル、2−ヘキセニル、3−ヘキセニル、2−ヘプテニル、3−ヘプテニル、4−ヘプテニル、3−オクテニル、3−ノネニル、4−デセニル、3−ウンデセニル、4−ドデセニル、4,8,12−テトラデカトリエニルなどをいい、これらは置換基、すなわち、ハロゲン、ハロアルキル、アルキル、アルコキシ、アルケニル、アルキニル、アリール、アリールアルキル、シクロアルキル、アミノ、ヒドロキシ、ヘテロアリール、シクロヘテロアルキル、アルカノイルアミノ、アルキルアミド、アリールカルボニルアミノ、ニトロ、シアノ、チオール、アルキルチオおよび/または本明細書で定められるアルキルへの置換基のいずれかの置換基の1〜4個で適宜置換されていてもよい。
【0096】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルキニル」または「アルキニル」とは、通常の鎖において1個の三重結合を含む、通常の鎖において2〜20個の炭素原子、好ましくは2〜12個の炭素原子、より好ましくは2〜8個の炭素原子の直鎖および分枝鎖の基をいい、通常の鎖において酸素または窒素を適宜含み得、例えば、2−プロピニル、3−ブチニル、2−ブチニル、4−ペンチニル、3−ペンチニル、2−ヘキシニル、3−ヘキシニル、2−ヘプチニル、3−ヘプチニル、4−ヘプチニル、3−オクチニル、3−ノニニル、4−デシニル、3−ウンデシニル、4−ドデシニルなどをいい、これらは置換基、すなわち、ハロゲン、ハロアルキル、アルキル、アルコキシ、アルケニル、アルキニル、アリール、アリールアルキル、シクロアルキル、アミノ、ヘテロアリール、シクロヘテロアルキル、ヒドロキシ、アルカノイルアミノ、アルキルアミド、アリールカルボニルアミノ、ニトロ、シアノ、チオール、および/またはアルキルチオ、および/または本明細書で定められるアルキルへの置換基のいずれかの置換基の1〜4個で適宜置換されていてもよい。
【0097】
単独または他の基の一部として用いる用語「アリールアルケニル」および「アリールアルキニル」とは、アリール置換基を有する上述のようなアルケニルおよびアルキニル基をいう。
【0098】
上記で定義されるようなアルキル基が2つの異なる炭素原子で他の基をつなぐ単結合を有する場合、それらは「アルキレン」と呼ばれ、「アルキル」として上記で定義されたように適宜置換されていてもよい。
【0099】
上記で定義されるようなアルケニル基および上記で定義されるようなアルキニル基がそれぞれ、2つの異なる炭素原子で結合するための単結合を有する場合、それらは「アルケニレン基」および「アルキニレン基」とそれぞれ呼ばれ、「アルケニル」および「アルキニル」として上記で定義されたように適宜置換されていてもよい。
【0100】
(CH2)x、(CH2)x 1、(CH2)x 2、(CH2)x 3、(CH2)m、または(CH2)nには、本明細書で定義されているように、アルキレン、アレニル、アルケニレンまたはアルキニレン基が含まれ、これらのそれぞれは通常の鎖において酸素または窒素を適宜含み得、アルキル、アルケニル、ハロゲン、シアノ、ヒドロキシ、アルコキシ、アミノ、チオアルキル、ケト、C3〜C6シクロアルキル、アルキルカルボニルアミノまたはアルキルカルボニルオキシの1、2、または3個の置換基が適宜含まれていてもよく;該アルキル置換基は、(CH2)x、(CH2)x 1、(CH2)x 2、(CH2)x 3または(CH2)mまたは(CH2)n基の1または2個の炭素に結合してそれによるシクロアルキル基を形成し得る1〜4個の炭素原子のアルキレン部分でもよい。
【0101】
(CH2)x、(CH2)x 1、(CH2)x 2、(CH2)x 3、(CH2)m、(CH2)n、アルキレン、アルケニレンおよびアルキニレンの例には、
【化29】
【0102】
本明細書で単独または他の基の一部として用いる用語「ハロゲン」または「ハロ」とは、塩素、臭素、フッ素、およびヨウ素、並びにCF3をいい、,好ましくは塩素またはフッ素である。
【0103】
用語「金属イオン」とは、ナトリウム、カリウムまたはリチウムのようなアルカリ金属イオンおよびマグネシウムおよびカルシウムのようなアルカリ土類金属イオン、並びに亜鉛およびアルミニウムイオンをいう。
【0104】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「アリール」または基:
【化30】
【化31】
【0105】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルコキシ」、「アルコキシ」、「アリールオキシ」、または「アラルコキシ」には、酸素原子結合する上記のいずれかのアルキル、アラルキルまたはアリール基のいずれかを含む。
【0106】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「置換されたアミノ」とは、例えばアルキル、アリール、アリールアルキル、ヘテロアリール、ヘテロアリールアルキル、シクロヘテロアルキル、シクロヘテロアルキルアルキル、シクロアルキル、シクロアルキルアルキル、ハロアルキル、ヒドロキシアルキル、アルコキシアルキルまたはチオアルキルのような1または2個の置換基(同一または異なってもよい)で置換されたアミノをいう。これらの置換基はさらに、カルボン酸および/または上記で定められたようなアルキルへの置換基のいずれかで置換されてもよい。さらに、該アミノ置換基は結合している窒素原子と一緒になって、1−ピロリジニル、1−ピペリジニル、1−アゼピニル、4−モルホリニル、4−チアモルホリニル、1−ピペラジニル、4−アルキル−1−ピペラジニル、4−アリールアルキル−1−ピペラジニル、4−ジアリールアルキル−1−ピペラジニル、1−ピロリジニル、1−ピペリジニル、または1−アゼピニル(適宜アルキル、アルコキシ、アルキルチオ、ハロ、トリフルオロメチルまたはヒドロキシで置換される)を形成していてもよい。
【0107】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルキルチオ」、「アルキルチオ」、「アリールチオ」または「アラルキルチオ」には、硫黄原子に連結する上記のアルキル、アラルキルまたはアリール基のいずれかを含む。
【0108】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「低級アルキルアミノ」、「アルキルアミノ」、「アリールアミノ」、または「アリールアルキルアミノ」には、窒素原子に連結する上記のアルキル、アリールまたはアリールアルキル基のいずれかを含む。
【0109】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「アシル」とは、カルボニル:
【化32】
【0110】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「シクロヘテロアルキル」とは、窒素、酸素および/または硫黄のようなヘテロ原子1〜2個を含み、炭素原子またはヘテロ原子を介して連結し、可能なら適宜結合鎖(CH2)p(pは1、2または3)で結合する5−、6−または7員の飽和または一部不飽和の環をいい、例えば、
【化33】
【0111】
特に断らない限り、本明細書で単独または他の基の一部として用いる用語「ヘテロアリール」とは、1、2、3または4個のヘテロ原子(例えば、窒素、酸素または硫黄)を含み、
【化34】
【化35】
【0112】
基:
【化36】
【化37】
【0113】
基:
【化38】
【化39】
【0114】
本明細書で単独または他の基の一部として用いる用語「シクロヘテロアルキルアルキル」とは、C原子またはヘテロ原子を介して(CH2)p鎖に連結する上記で定義されるようなシクロヘテロアルキル基をいう。
【0115】
本明細書で単独または他の基の一部として用いる用語「ヘテロアリールアルキル」または「ヘテロアリールアルケニル」とは、C原子またはヘテロ原子を介して(CH2)p鎖、上記で定義されるようなアルキレンまたはアルケニレンに連結する上記で定義されるようなヘテロアリール基をいう。
【0116】
本明細書で用いる用語「ポリハロアルキル」とは、2〜9個、好ましくは2〜5個のハロ置換基(ハロ置換基は例えばFまたはClであり、好ましくはFである)を含む上記で定義されるような「アルキル」基をいい、例えばCF3CH2、CF3またはCF3CF2CH2がある。
【0117】
本明細書で用いる用語「ポリハロアルキルオキシ」とは、2〜9個、好ましくは2〜5個のハロ置換基(ハロ置換基は例えばFまたはClであり、好ましくはFである)を含む、上記で定義したような「アルコキシ」または「アルキルオキシ」基をいい、例えば、CF3CH2O、CF3OまたはCF3CF2CH2Oがある。
【0118】
本明細書で用いる用語「プロドラッグエステル」には、カルボン酸およびホスホン酸エステル(例えば、メチル、エチル、ベンジルなど)について当該技術分野で公知のプロドラッグエステルが含まれる。R4の他のプロドラッグエステルの例には、以下の基:すなわち
【化40】
【化41】
【化42】
【0119】
構造Iの化合物が酸型である場合、医薬的に許容される塩、例えば、アルカリ金属塩(例えば、リチウム、ナトリウムまたはカリウム)、アルカリ土類金属塩(例えば、カルシウムまたはマグネシウム)、並びに亜鉛またはアルミニウム、および他のカチオン[例えばアンモニウム、コリン、ジエタノールアミン、リジン(DまたはL)、エチレンジアミン、t−ブチルアミン、t−オクチルアミン、トリス(ヒドロキシメチル)アミノメタン(TRIS)、N−メチルグルコサミン(NMG)、トリエタノールアミンおよびデヒドロアビエチルアミン]の塩を形成し得る。
【0120】
本発明の化合物のすべての立体異性体は、混合物または純粋なまたは十分に純粋な形態のいずれかであることと考慮される。本発明の化合物は、それ自身のいずれかまたはR置換基に含まれる炭素原子のいずれかに不斉中心を有し得る。従って、式Iの化合物はエナンチオマー型またはジアステレオマー型またはその混合物で存在し得る。製造の工程には、出発物質としてラセミ体、エナンチオマーまたはジアステレオマーを利用できる。ジアステレオマーまたはエナンチオマー生成物を製造する場合、例えばクロマトグラフィまたは分別結晶のようなありふれた方法で分離できる。
【0121】
所望なら、構造Iの化合物は、1つ以上の抗高脂血症剤または脂質低下剤または脂質調節剤と、および/または抗糖尿病剤、抗肥満剤、降圧剤、血小板凝集阻害剤および/または抗骨多孔症剤を含む治療剤の1以上の他のタイプと組み合わせて用いてもよく、同一の製剤で経口投与されてもよく、別の経口製剤で投与されてもよく、または注射で投与されてもよい。
【0122】
適宜本発明の式Iの化合物と組み合わせて用い得る抗高脂血症剤または脂質低下剤または脂質調節剤は、1、2、3またはそれ以上のMTP阻害剤、HMG CoA還元酵素阻害剤、スクアレン合成酵素阻害剤、フィブリン酸誘導体、ACAT阻害剤、リポキシゲナーゼ阻害剤、コレステロール吸収阻害剤、回腸Na+/胆汁酸共輸送体阻害剤、LDL受容体活性の上方制御剤、胆汁酸金属イオン封鎖剤、および/またはニコチン酸およびその誘導体を含み得る。
【0123】
本明細書に用いられるMTP阻害剤には、米国特許第5,595,872号、米国特許第5,739,135号、米国特許第5,712,279号、米国特許第5,760,246号、米国特許第5,827,875号、米国特許第5,885,983号および米国特許出願第09/175,180号(1998年10月20日出願)、米国特許第5,962,440号に開示されているMTP阻害剤が含まれる。好ましいのは、上記特許および出願のそれぞれに開示されている好ましいMTP阻害剤である。
【0124】
上記の米国特許および出願のすべては、本明細書に引用される。
【0125】
本発明に従って用いられる最も好ましいMTP阻害剤には、米国特許第5,739,135号および第5,712,279号、および米国特許第5,760,246号で述べられるような好ましいMTP阻害剤が含まれる。
【0126】
最も好ましいMTP阻害剤は、9−[4−[4−[[2−(2,2,2−トリフルオロエトキシ)ベンゾイル]アミノ]−1−ピペリジニル]ブチル]−N−(2,2,2−トリフルオロエチル)−9H−フルオレン−9−カルボキサミド:
【化43】
【0127】
抗高脂血症剤は、HMG CoA還元酵素阻害剤であり得、これらに限らないが、米国特許第3,983,140号に開示されるようなメバスタチンおよび関連化合物、米国特許第4,231,938号に開示されるようなロバスタチン(メビノリン)および関連化合物、米国特許第4,346,227号に開示されるようなプラバスタチンおよび関連化合物、米国特許第4,448,784号および第4,450,171号に開示されるようなシンバスタチンおよび関連化合物が含まれる。本明細書で用いられ得る他のHMG CoA還元酵素阻害剤には、これらに限らないが、フルバスタチン(米国特許第5,354,772号に開示)、セリバスタチン(米国特許第5,006,530号および第5,177,080号に開示)、アトルバスタチン(米国特許第4,681,893号、第5,273,995号、第5,385,929号および第5,686,104号に開示)、イタバスタチン(米国特許第5,011,930号に開示されたニッサン/三共のニスバスタチン(NK−104))、塩野義−アストラ/ゼネカのビサスタチン(ZD−4522)(米国特許第5,260,440号に開示)、および関連するスタチン化合物(米国特許第5,753,675号に開示)、メバロノラクトン誘導体のピラゾール類似体(米国特許第4,613,610号に開示)、メバロノラクトン誘導体のインデン類似体(PCT出願WO 86/03488に開示)、6−[2−(置換された−ピロール−1−イル)−アルキル)ピラン−2−オン化合物およびその誘導体(米国特許第4,647,576号に開示)、シアール社のSC−45355(3−置換されたペンタンジオン酸誘導体)ジクロロアセテート、メバロノラクトンのイミダゾール類似体(PCT出願WO 86/07054に開示)、3−カルボキシ−2−ヒドロキシ−プロパンホスホン酸誘導体(フランス特許第2,596,393号に開示)、2,3−二置換されたピロール、フランおよびチオフェン誘導体(欧州特許出願第0221025号に開示)、メバロノラクトンのナフチル類似体(米国特許第4,686,237号に開示)、オクタヒドロナフタレン化合物(米国特許第4,499,289号に開示)、メビノリン(ロバスタチン)のケト類似体(欧州特許出願第0,142,146 A2号に開示)、およびキノリンおよびピリジン誘導体(米国特許第5,506,219号および第5,691,322号に開示)が含まれる。
【0128】
さらに、本明細書で用いるのに適したHMG CoA還元酵素阻害に有用なホスフィン酸化合物は、GB 2205837に開示されている。
【0129】
本明細書で用いるのに適したスクアレン合成酵素阻害剤には、これらに限らないが、イソプレノイド(ホスフィニルメチル)ホスホン酸化合物を含むα−ホスホノスルホネート化合物(米国特許第5,712,396号およびBiller et al, J. Med. Chem., 1988, Vol. 31, No. 10, pp 1869-1871に開示)、並びに他の公知のスクアレン合成酵素阻害剤(例えば、米国特許第4,871,721号および第4,924,024号およびBiller, S. A., Neuenschwander, K., Ponpipom, M. M., and Poulter, C. D., Current Pharmaceutical Design, 2, 1-40 (1996)に開示)が含まれる。
【0130】
さらに、本明細書で用いるのに適した他のスクアレン合成酵素阻害剤には、テルペノイドピロホスフェート化合物(P. Ortiz de Montellano et al, J. Med. Chem., 1977, 20, 243-249に開示)、ファルネシルジホスフェート類似体Aおよびプレスクアレンピロホスフェート(PSQ−PP)類似体(Corey and Volante, J. Am. Chem. Soc., 1976, 98, 1291-1293に開示)、ホスフィニルホスホネート化合物(McClard, R. W. et al, J. A. C. S., 1987, 109, 5544に報告)、およびシクロプロパン化合物(Capson, T. L., PhD dissertation, June, 1987, Dept. Med. Chem. U of Utah, Abstract, Table of Contents, pp 16, 17, 40-43, 48-51, Summaryに報告)が含まれる。
【0131】
本明細書で用いるのに適した他の抗高脂血症剤には、これらに限らないが、フィブリン酸誘導体(例えば、フェノフィブラート、ゲムフィブロジル、クロフィブラート、ベザフィブラート、シプロフィブラート、クリノフィブラートなど)、プロブコールおよび関連化合物(米国特許第3,674,836号に開示)(好ましくはプロブコールおよびゲムフィブロジル)、胆汁酸金属イオン封鎖剤[例えば、コレスチラミン、コレスチポールおよびDEAE−セファデックス(セコレックス(登録商標)、ポリセキシド(登録商標))]およびコレスタゲル(三共/ゲルテックス)、並びに、リポスタビル(ローヌ・プーラン社)、エーザイE−5050(N−置換されたエタノールアミン誘導体)、イマニキシル(HOE−402)、テトラヒドロリプスタチン(THL)、イスチグマスタニルホスホリルコリン(SPC、ロッシュ社)、アミノシクロデキストリン(田辺製薬)、味の素AJ−814 (アズレン誘導体)、メリナミド(住友)、サンド社58−035、アメリカンシアナミド社CL−277,082およびCL−283,546(二置換された尿素誘導体)、ニコチン酸(ナイアシン)、アシピモックス、アシフラン、ネオマイシン、p−アミノサリチル酸、アスピリン、ポリ(ジアリルメチルアミン)誘導体(米国特許第4,759,923号に開示)、四級アミン ポリ(ジアリルジメチルアンモニウムクロリド)およびイオネン(米国特許第4,027,009号に開示)、および他の公知の血清コレステロール低下剤が含まれる。
【0132】
抗高脂血症剤は、以下のように開示されるようなACAT阻害剤であり得る:
Drugs of the Future 24, 9-15 (1999), (Avasimibe);
「The ACAT inhibitor, Cl-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters」, Nicolosi et al, Atherosclerosis (Shannon, Irel). (1998), 137 (1), 77-85;
「The pharmacological profile of FCE 27677: a novel ACAT inhibitor with potent hypolipidemic activity mediated by selective suppression of the hepatic secretion of ApoB100-containing lipoprotein」, Ghiselli, Giancarlo, Cardiovasc. Drug Rev. (1998), 16 (1), 16-30;
「RP 73163: a bioavailable alkylsulfinyl-diphenylimidazole ACAT inhibitor」, Smith, C., et al, Bioorg. Med. Chem. Lett. (1996), 6 (1), 47-50;
「ACAT inhibitors: physiologic mechanisms for hypolipidemic and anti-atherosclerotic activities in experimental animals」, Krause et al, Editor (s): Ruffolo, Robert R., Jr.; Hollinger, Mannfred A., Inflammation : Mediators Pathways (1995), 173-98, Publisher: CRC, Boca Raton, Fla.;
「ACAT inhibitors: potential anti-atherosclerotic agents」, Sliskovic et al, Curr. Med. Chem. (1994), 1 (3), 204-25;
「Inhibitors of acyl-CoA : cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 6. The first water-soluble ACAT inhibitor with lipid-regulating activity. Inhibitors of acyl-CoA: cholesterol acyltransferase (ACAT). 7. Development of a series of substituted N-phenyl-N'-[(1- phenylcyclopentyl) methyl] ureas with enhanced hypocholesterolemic activity」, Stout et al, Chemtracts: Org. Chem. (1995), 8 (6), 359-62, またはTS-962 (大正製薬)。
【0133】
抗高脂血症剤は、MD−700(大正製薬)およびLY295427(イーライリリィ)のような、LD2受容体活性の上方制御剤であり得る。
【0134】
抗高脂血症剤は、コレステロール吸収阻害剤であり得、好ましくはシェーリングプラウのSCH48461、並びにAtherosclerosis 115, 45-63 (1995)およびJ. Med. Chem. 41, 973 (1998)に開示された阻害剤である。
【0135】
抗高脂血症剤は、Drugs of the Future, 24, 425-430 (1999)に開示されているような回腸Na+/胆汁酸共輸送体阻害剤であり得る。
【0136】
脂質調節剤は、ファイザーのCP 529,414(WO/0038722およびEP 818448)およびファルマシアのSC−744およびSC−795のような、コレステリルエステル転送タンパク質(CETP)阻害剤であり得る。
【0137】
本発明の組み合わせて用いられ得るATPクエン酸リアーゼ阻害剤は、例えば米国特許第5,447,954号に開示の阻害剤が含まれ得る。
【0138】
好ましい抗高脂血症剤は、プラバスタチン、ロバスタチン、シンバスタチン、アトルバスタチン、フルバスタチン、セリバスタチン、イタバスタチンおよびビサスタチンおよびZD−4522である。
【0139】
上述の米国特許は本明細書に引用される。用いる量および用量は、フィジシャンズ デスク レファレンス(Physician's Desk Reference)および/または上記で述べた特許に示される。
【0140】
本発明の式Iの化合物は、(存在するなら)抗高脂血症剤に対して重量比が約500:1〜約1:500、好ましくは約100:1〜約1:100の範囲内で用いられる。
【0141】
投与量は年齢、体重、患者の症状、並びに投与経路、投与形態および投与計画および目的の成果に従い、注意深く調整しなければならない。
【0142】
抗高脂血症剤の投与量および処方は、上述の様々な特許および出願に開示されている。
【0143】
適用できるように用いられる他の抗高脂血症剤の投与量および処方は、フィジシャンズ デスク レファレンス(Physician's Desk Reference)の最新版に記載されている。
【0144】
経口投与では、MTP阻害剤を1日1〜4回で、約0.01 mg〜約500 mg、好ましくは約0.1 mg〜約100 mgの範囲内の量で用いると、満足な結果が得られ得る。
【0145】
好ましい経口製剤、例えば錠剤またはカプセル剤には、1日1〜4回で、約1〜約500 mg、好ましくは約2〜約400 mg、より好ましくは約5〜約250 mgの量でMTP阻害剤が含まれる。
【0146】
経口投与では、HMG CoA還元酵素阻害剤、例えばプラバスタチン、ロバスタチン、シンバスタチン、アトルバスタチン、フルバスタチンまたはセリバスタチンを、フィジシャンズ デスク レファレンス(Physician's Desk Reference)で示される投与量、例えば約1〜2000 mg、好ましくは約4〜約200 mgの範囲内の量で用いると、満足な結果が得られ得る。
【0147】
スクアレン合成酵素阻害剤は、約10 mg〜約2000 mg、好ましくは約25 mg〜約200 mgの範囲内の量の投与量で用いられ得る。
【0148】
好ましい経口製剤、例えば錠剤またはカプセル剤には、約0.1〜約100 mg、好ましくは約0.5〜約80 mg、より好ましくは約1〜約40 mgの量でHMG CoA還元酵素阻害剤が含まれる。
【0149】
好ましい経口製剤、例えば錠剤またはカプセル剤には、約10〜約500 mg、好ましくは約25〜約200 mgの量でスクアレン合成酵素阻害剤が含まれる。
【0150】
抗高脂血症剤はまた、リポキシゲナーゼ阻害剤であり得、15−リポキシゲナーゼ(15−LO)阻害剤(例えばベンゾイミダゾール誘導体、WO 97/12615に開示)、15−LO阻害剤(WO 97/12613に開示)、イソチアゾロン化合物(WO 96/38144に開示)、および15−LO阻害剤(Sendobry et al「Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties」, Brit. J. Pharmacology (1997) 120, 1199-1206, およびCornicelli et al,「15-Lipoxygenase and its Inhibition: A Novel Therapeutic Target for Vascular Disease」, Current Pharmaceutical Design, 1999, 5, 11-20に開示)が含まれる。
【0151】
式Iの化合物および抗高脂血症剤は、同一経口製剤で、または別の経口製剤を同時に服用することで、一緒に用い得る。
【0152】
上述の組成物は、上述の製剤形で、1回でまたは1日1〜4回に分割した用量で投与され得る。患者には低用量の組み合わせから開始し、徐々に高用量の組み合わせに上げていくことが望ましい。
【0153】
好ましい抗高脂血症剤は、プラバスタチン、シンバスタチン、ロバスタチン、アトルバスタチン、フルバスタチンまたはセリバスタチン、並びにナイアシンおよび/またはコレスタゲルである。
【0154】
式Iの化合物と組み合わせて適宜用い得る他の抗糖尿病剤は、1、2、3またはそれ以上の抗糖尿病剤、またはインスリン分泌促進物質またはインスリン増感剤を含む抗高血糖症剤、または好ましくは本発明の式Iの化合物と異なるメカニズムを有する他の抗糖尿病剤であり得、それらには、ビグアナイド、スルホニル尿素、グルコシダーゼ阻害剤、PPARγアゴニスト(例えばチアゾリジンジオン化合物)、aP2阻害剤、ジペプチジルペプチダーゼIV(DP4)阻害剤、SGLT2阻害剤、および/またはメグリチナイド化合物、並びにインスリン、および/またはグルカゴン様ペプチド−1(GLP−1)が含まれ得る。
【0155】
他の抗糖尿病剤は、経口抗高血糖症剤、好ましくはビグアナイド類(例えばメトホルミンまたはフェンホルミンまたはその塩、好ましくはメトホルミンHCl)であり得る。
【0156】
抗糖尿病剤がビグアナイドの場合、構造Iの化合物は、ビグアナイドに対する重量比が約0.001:1〜約10:1、好ましくは約0.01:1〜約5:1の範囲内で用いられる。
【0157】
他の抗糖尿病剤はまた好ましくは、スルホニル尿素[例えば、グリブリド(グリベンクラミドとしても知られる)、グリメピリド(米国特許第4,379,785号に開示)、グリピジド、グリクラジドまたはクロロプロパミド]、他の公知のスルホニル尿素またはβ細胞のATP感受性チャンネルに作用する他の抗高血糖症剤であり、グリブリドおよびグリピジドが好ましく、同一または別の経口製剤で投与され得る。
【0158】
構造Iの化合物は、スルホニル尿素に対する重量比が約0.01:1〜約100:1、好ましくは約0.02:1〜約5:1の範囲で用いられる。
【0159】
経口抗糖尿病剤はまた、グルコシダーゼ阻害剤、例えばアカルボース(米国特許第4,904,769号に開示)またはミグリトール(米国特許第4,639,436号に開示)であり得、同一または別の経口製剤で投与され得る。
【0160】
構造Iの化合物は、グルコシダーゼ阻害剤に対する重量比が約0.01:1〜約100:1、好ましくは約0.05:1〜約10:1の範囲内で用いる。
【0161】
式Iの化合物は、チアゾリジンジオン経口抗糖尿病剤または他のインスリン増感剤(NIDDM患者におけるインスリン感受性効果を有する)のようなPPARγアゴニストと組み合わせて用いられ得、例えば、トログリタゾン(ワーナーランバード社のレズリン(登録商標)、米国特許第4,572,912号に開示)、ロシグリタゾン(SKB)、ピオグリタゾン(武田)、三菱のMCC−555(米国特許第5,594,016号に開示)、グラクソウェルカム社のGL−262570、エングリタゾン(CP−68722, ファイザー)またはダルグリタゾン(CP−86325, ファイザー)、イサグリタゾン(MIT/J&J)、JTT−501(JPNT/P&U)、L−895645(メルク)、R−119702(三共/WL)、NN−2344(レディ博士/NN)、またはYM−440(山之内)、好ましくはロシグリタゾンおよびピオグリタゾンがある。
【0162】
構造Iの化合物は、チアゾリジンジオンに対する重量比が約0.01:1〜約100:1、好ましくは約0.05〜約10:1の範囲内の量で用いられる。
【0163】
経口抗糖尿病剤が約150 mg未満の量において、スルホニル尿素およびチアゾリジンジオンは、構造Iの化合物と単一の錠剤に組み込まれ得る。
【0164】
構造Iの化合物はまた、抗高血糖症剤(例えば、インスリン)と、またはグルカゴン様ペプチド−1(GLP−1)[例えば、GLP−1(1−36)アミド、GLP−1(7−36)アミド、GLP−1(7−37)(米国特許第5,614,492号に開示、ハベナーによる、この開示は本明細書に引用する)、並びにAC2993(アミリン社)およびLY−315902(リリィ社)]と組み合わせても用いられ、注射、鼻腔内、吸入を通じて、または経皮またはバッカル装置により投与し得る。
【0165】
存在する場合は、メトホルミン、スルホニル尿素化合物(例えば、グリブリド、グリメピリド、グリピリド、グリピジド、クロロプロパミドおよびグリクラジド)およびグルコシダーゼ阻害剤アカルボースまたはミグリトールまたはインスリン(注射、肺、バッカル、または経口)は、上述のような製剤形で、フィジシャンズ デスク レファレンス(Physician's Desk Reference、PDR)に示されるような量および投薬で用いられ得る。
【0166】
存在する場合は、メトホルミンまたはその塩は、1日当たり約500〜約2000 mgの範囲内の量で用いられ、1回でまたは1日1〜4回に用量を分割して投与され得る。
【0167】
存在する場合は、チアゾリジンジオン 抗糖尿病剤は、約0.01〜約2000 mg/日の範囲内の量で用いられ、1回でまたは1日1〜4回に用量を分割して用いられ得る。
【0168】
存在する場合は、インスリンは、フィジシャンズ デスク レファレンス(Physician's Desk Reference)に示される製剤形、量、投薬で用いられ得る。
【0169】
存在する場合は、GLP−1ペプチドは、経口バッカル製剤で、鼻腔内または非経口(米国特許第5,346,701号(テラテック)、第5,614,492号および第5,631,224号に開示;これらは本明細書に引用される)で投与され得る。
【0170】
他の抗糖尿病剤はまた、PPARα/γ両アゴニスト[例えば、AR−HO39242(アストラ/ゼネカ)、GW−409544(グラクソ−ウェルカム)、KRP297(キョーリン メルク)]、並びにムラカミらの文献[Murakami et al,「A Novel Insulin Sensitizer Acts As a Coligand for Peroxisome Proliferation-Activated Receptor Alpha (PPAR alpha) and PPAR gamma. Effect on PPAR alpha Activation on Abnormal Lipid Metabolism in Liver of Zucker Fatty Rats」, Diabetes 47,1841-1847 (1998)]に開示されるものでもあり得る。
【0171】
抗糖尿病剤は、例えば米国特許出願第09/679,027号(2000年10月4日出願,代理人ファイル番号LA49 NP、ここに述べられた投与量を用いる)に開示されるようなSGLT2阻害剤であり得る。好ましくは上記の出願で好ましいとされる化合物である。
【0172】
抗糖尿病剤は、例えば米国特許出願第09/391,053号(1999年9月7日出願)および米国特許出願第09/519,079号(2000年3月6日出願)(代理人ファイル番号LA27 NP、ここに述べられた投与量を用いる)に開示されるようなaP2阻害剤であり得る。好ましくは上記の出願で好ましいとされる化合物である。
【0173】
抗糖尿病剤は、米国特許出願第09/788,173号(2001年2月16日出願)(代理人ファイル番号LA50)、WO99/38501、WO99/46272、WO99/67279 (プロバイオドラッグ)、WO99/67278 (プロバイオドラッグ)、WO99/61431 (プロバイオドラッグ)、NVP−DPP728A(1−[[[2−[(5−シアノピリジン−2−イル)アミノ]エチル]アミノ]アセチル]−2−シアノ−(S)−ピロリジン)(ノバルティス)(好適化合物)(Hughes et al, Biochemistry, 38 (36), 11597-11603, 1999に開示)、TSL−225(トリプトフィル−1,2,3,4−テトラヒドロイソキノリン−3−カルボン酸(Yamada et al, Bioorg. & Med. Chem. Lett. 8 (1998) 1537-1540に開示)、2−シアノピロリジド化合物および4−シアノピロリジド化合物(Ashworth et al, Bioorg. & Med. Chem. Lett., Vol. 6, No. 22, pp 1163-1166および2745-2748 (1996)に開示)(上記引用文献に述べられた投与量を用いる)に開示されるようなDP4阻害剤であり得る。
【0174】
本発明の式Iの化合物と組み合わせて適宜用いられ得るメグリチナイドは、レパグリニド、ナテグリニド(ノバルティス)またはKAD1229(PF/キッセイ)であり得、レパグリニドが好ましい。
【0175】
式Iの化合物は、メグリチナイド、PPARγアゴニスト、PPARα/γ両アゴニスト、aP2阻害剤、DP4阻害剤またはSGLT2阻害剤との重量比が、約0.01:1〜約100:1、好ましくは約0.05〜約10:1の範囲内で用いられる。
【0176】
式Iの化合物とともに適宜用いられ得る他のタイプの治療剤は、β3アドレナリン作動性アゴニスト、リパーゼ阻害剤、セロトニン(およびドパミン)再取り込み阻害剤、aP2阻害剤、甲状腺受容体アゴニストおよび/または摂食障害剤を含む、1、2、3またはそれ以上の抗肥満剤であり得る。
【0177】
式Iの化合物と組み合わせて適宜用いられ得るβ3アドレナリン作動性アゴニストは、AJ9677(武田/大日本)、L750355(メルク)、またはCP331648(ファイザー)または他の公知のβ3アゴニスト(米国特許第5,541,204号、第5,770,615号、第5,491,134号、第5,776,983号および第5,488,064号に開示)であり得、AJ9677、L750,355およびCP331648が好ましい。
【0178】
式Iの化合物と組み合わせて適宜用いられ得るリパーゼ阻害剤は、オルリスタットまたはATL−962(アリザイム)であり得、オルリスタットが好ましい。
【0179】
式Iの化合物と組み合わせて適宜用いられ得るセロトニン(およびドパミン)再取り込み阻害剤は、シブトラミン、トピラメート(ジョンソンアンドジョンソン)またはアキソキン(リジェネロン)であり得、シブトラミンおよびトピラメートが好ましい。
【0180】
式Iの化合物と組み合わせて適宜用いられ得る甲状腺受容体アゴニストは、W097/21993 (U. Cal SF)、W099/00353(カロバイオ)、GB98/284425(カロバイオ)および米国仮出願第60/183,223号(2000年2月17日出願)に開示される、甲状腺受容体リガンドであり得、カロバイオの出願および上記米国仮出願の化合物が好ましい。
【0181】
式Iの化合物と組み合わせて適宜用いられ得る摂食障害剤は、デキサアンフェタミン、フェンテルミン、フェニルプロパノールアミンまたはマジンドールであり得、デキサアンフェタミンが好ましい。
【0182】
上述される様々な抗肥満剤は、式Iの化合物と同一の投与形態でまたは異なる投与形態で、当該技術分野またはPDRにおいて一般的に公知であるような投与量および投与計画で用いられ得る。
【0183】
本発明の式Iの化合物と組み合わせて用いられ得る降圧剤には、ACE阻害剤、アンジオテンシンII受容体アンタゴニスト、NEP/ACE阻害剤、並びにカルシウムチャンネルブロッカー、β−アドレナリン作動性遮断薬、および利尿剤を含む他のタイプの降圧剤が含まれる。
【0184】
本明細書で用いられ得るアンジオテンシン変換酵素阻害剤には、上述のOndetti et alの米国特許第4,046,889号に開示される誘導体のいずれかのような、置換されたプロリン誘導体のようなメルカプト(−S−)部位を含む誘導体が含まれ、カプトプリル、すなわち、1−[(2S)−3−メルカプト−2−メチルプロピオニル]−L−プロリンが好ましく、ゾフェノプリルで米国特許第4,316,906号に開示の誘導体のいずれかのような置換されたプロリンのメルカプトアシル誘導体が好ましい。
【0185】
本明細書で用いられ得るメルカプトを含むACE阻害剤の他の例には、レンチアプリル(フェンチアプリル、参天)(Clin. Exp. Pharmacol. Physiol. 10: 131 (1983)に開示);並びにピボプリルおよびYS980が含まれる。
【0186】
本明細書で用いられ得るアンジオテンシン変換酵素阻害剤の他の例には、上述の米国特許第4,374,829号に開示されるいずれかが含まれ、N−(1−エトキシカルボニル−3−フェニルプロピル)−L−アラニル−L−プロリン、すなわち、エナラプリルが好ましく、(S)−1−[6−アミノ−2−[[ヒドロキシ−(4−フェニルブチル)ホスフィニル]オキシ]−1−オキソヘキシル]−L−プロリンまたは(セロナプリル)で米国特許第4,452,790号に開示されるホスホン酸で置換されたアミノまたはイミノ酸またはその塩のいずれかが好ましく、フォシノプリルで上述された米国特許第4,168,267号に開示されるホスフィニルアルカノイルプロリン化合物が好ましく、米国特許第4,337,201号に開示されるホスフィニルアルカノイルで置換されたプロリン化合物のいずれか、および上記で述べられている米国特許第4,432,971号に開示されるホスホンアミデート化合物が含まれる。
【0187】
本明細書で用いられ得るACE阻害剤の他の例には、ビーチャムのBRL 36,378(欧州特許出願第80822号および第60668号に開示);中外のMC−838(C. A. 102:72588vおよびJap. J. Pharmacol. 40:373 (1986)に開示);チバ−ガイギィのCGS 14824(3−([1−エトキシカルボニル−3−フェニル−(1S)−プロピル]アミノ)−2,3,4,5−テトラヒドロ−2−オキソ−1−(3S)−ベンゾアゼピン−1−酢酸 HCl)(英国特許第2103614号に開示)およびCGS 16,617(3(S)−[[(1S)−5−アミノ−1−カルボキシペンチル]アミノ]−2,3,4,5−テトラヒドロ−2−オキソ−1H−1−ベンゾアゼピン−1−エタン酸)(米国特許第4,473,575号で開示);セタプリル(アラセプリル、大日本)(Eur. Therap. Res. 39: 671(1986);40: 543 (1986)に開示);ラミプリル(ヘキスト)(欧州特許第79-022号およびCurr. Ther. Res. 40: 74 (1986)に開示);Ru 44570(ヘキスト)(Arzneimittelforschung 34: 1254 (1985)に開示)、シラザプリル(ホフマン−ラロッシュ)(J. Cardiovasc. Pharmacol. 9: 39 (1987)に開示);R 31−2201(ホフマン−ラロッシュ)(FEBS Lett. 165: 201 (1984)に開示);リシノプリル(メルク)、インダラプリル(デラプリル)(米国特許第4,385,051号に開示);インドラプリル(シェーリング)(J. Cardiovasc. Pharmacol. 5: 643, 655 (1983) に開示)、スピラプリル(シェーリング)(Acta. Pharmacol. Toxicol. 59 (Supp. 5): 173 (1986)に開示);ペルインドプリル(セルビエ)(Eur. J. clin. Pharmacol. 31: 519 (1987)に開示);キナプリル(ワーナーランバード)(米国特許第4,344,949号に開示)およびCI925(ワーナーランバード)([3S−[2−[R(*)R(*)]]3R(*)]−2−[2−[[1−(エトキシ−カルボニル)−3−フェニルプロピル]アミノ]−1−オキソプロピル]−1,2,3,4−テトラヒドロ−6,7−ジメトキシ−3−イソキノリンカルボン酸 HCl)(Pharmacologist 26 : 243, 266 (1984)に開示)、WY44221(ワイエス)(J. Med. Chem. 26: 394 (1983)に開示)が含まれる。
【0188】
好ましいACE阻害剤は、カプトプリル、フォシノプリル、エナラプリル、リシノプリル、キナプリル、ベナゼプリル、フェンチアプリル、ラミプリルおよびモエキシプリルである。
【0189】
NEP/ACE阻害剤はまた、それらが中性エンドペプチダーゼ(NEP)阻害剤活性およびアンジオテンシン変換酵素(ACE)阻害剤活性を有するので、本明細書で用いられる。本明細書の使用に適したNEP/ACE阻害剤の例は、米国特許第5,362,727号、第5,366,973号、第5,225,401号、第4,722,810号、第5,223,516号、第4,749,688号、米国特許第5,552,397号、米国特許第5,504,080号、米国特許第5,612,359号、米国特許第5,525,723号、欧州特許出願第0599,444号、第0481,522号、第0599,444号、第0595,610号、欧州特許出願第0534363A2号、第534,396号および第534,492号、および欧州特許出願第0629627A2号に開示されている例が含まれる。
【0190】
好ましくは、上記特許/特許出願で好適として示されるNEP/ACE阻害剤およびその投与量であり(該米国特許は本明細書に引用される);最も好ましくは、オマパトリラット、BMS189,921([S−(R*,R*)]−ヘキサヒドロ−6−[(2−メルカプト−1−オキソ−3−フェニルプロピル)アミノ−2,2−ジメチル−7−オキソ−1H−アゼピン−1−酢酸(ゲモパトリラット))およびCGS30440である。
【0191】
本明細書の使用に適したアンジオテンシンII受容体アンタゴニスト(これらもまたアンジオテンシンIIアンタゴニストまたはAIIアンタゴニストとして本明細書に引用される)には、これらに限らないが、イルベサルタン、ロサルタン、バルサルタン、カンデサルタン、テルミサルタン、タソサルタンまたはエプロサルタンが含まれ、イルベサルタン、ロサルタンまたはバルサルタンが好ましい。
【0192】
錠剤またはカプセル剤のような好ましい経口製剤には、約0.1〜約500 mg、好ましくは約5〜約200 mg、より好ましくは約10〜約150 mgの範囲内の量で、ACE阻害剤またはAIIアンタゴニストが含まれる。
【0193】
非経口投与として、ACE阻害剤、アンジオテンシンIIアンタゴニストまたはNEP/ACE阻害剤は、約0.005 mg/kg〜約10 mg/kg、好ましくは約0.01 mg/kg〜約1 mg/kgの範囲内の量で用いられる。
【0194】
薬剤が静脈内投与される場合、例えば蒸留水、生理食塩水、リンゲル液または他の従前のキャリアのような従前のビークル中製剤化される。
【0195】
ACE阻害剤およびAIIアンタゴニスト、並びに本明細書で開示される他の降圧剤の投与量は、フィジシャンズ デスク レファレンス(Physician's Desk Reference,PDR)の最新版に示されるような量であることが望ましい。
【0196】
本明細書の使用に適した好ましい降圧剤の他の例には、オマパトリラット(バンレブ(Vanlev、登録商標))、アムロジピン ベスィレート(ノルバスク(登録商標))、プラゾシン HCl(ミニプレス(登録商標))、ベラパミル、ニフェジピン、ナドロール、ジルチアゼム、フェロジピン、ニソルジピン、イスラジピン、ニカルジピン、アテノロール、カルベジロール、ソタロール、テラゾシン、ドキサゾシン、プロプラノロール、およびクロニジン HCl(カタプレス(登録商標))が含まれる。
【0197】
式Iの化合物と組み合わせて用いられ得る利尿剤には、ヒドロクロロチアジド、トラセミド、フロセミド、スピロノラクトン、およびインダパミドが含まれる。
【0198】
式Iの化合物と組み合わせて用いられ得る抗血小板剤には、アスピリン、クロピドグレル、チクロピジン、ジピリダモール、アブシキシマブ、チロフィバン、エプチフィバチド、アナグレリド、およびイフェトロバンが含まれ、クロピドグレルおよびアスピリンが好ましい。
【0199】
抗血小板薬は、PDRに示された量を用い得る。イフェトロバンは、米国特許第5,100,889号に示される量で用いられ得る。
【0200】
本発明の式Iの化合物と組み合わせて本明細書での使用に適した抗骨多孔剤には、MK−217(アレンドロネート)(フォサマックス(登録商標))のような副甲状腺ホルモンまたはビスホスホネートが含まれる。用いる投与量は、フィジシャンズ デスク レファレンス(Physician's Desk Reference)に記載されるような量である。
【0201】
本発明の方法を実施する場合、医薬組成物は、構造Iの化合物を含み、他の治療剤を含んだり含まなかったり、医薬のビークルまたは希釈剤と共に用いられる。医薬組成物は、従前の固体または液体のビークルまたは希釈剤、および目的の投与方法に適したタイプの医薬添加剤を用いて製剤化される。本化合物は、ヒト、サル、イヌなどを含む哺乳類に、経口経路(例えば、錠剤、カプセル剤、顆粒剤または散剤)で投与されるか、または注射製剤の形態で非経口経路で投与される。成人への用量は、好ましくは1日50〜2,000 mgの間で、単一用量で、または1日1〜4回の個々の用量の形態で投与され得る。
【0202】
経口投与用の典型的なカプセル剤には、構造Iの化合物(250 mg)、乳糖(75 mg)およびステアリン酸マグネシウム(15 mg)が含まれる。該混合物は60メッシュのふるいを通り、1号ゼラチンカプセルに詰められる。
【0203】
典型的な注射製剤は、バイアルに構造Iの化合物(250 mg)を無菌充填し、無菌凍結乾燥し、封かんして製造される。使用に際しては、バイアルの内容物は生理食塩水(2 mL)と混合し、注射製剤を製造する。
【0204】
以下の実施例は本発明の好ましい態様を示す。
【0205】
以下の略号は実施例に用いられる:
Ph=フェニル
Bn=ベンジル
t−Bu=三級ブチル
Me=メチル
Et=エチル
TMS=トリメチルシリル
TMSN3=トリメチルシリルアジド
TBS=tert−ブチルジメチルシリル
FMOC=フルオレニルメトキシカルボニル
Boc=tert−ブトキシカルボニル
Cbz=カルボベンジルオキシまたはカルボベンゾキシまたはベンジルオキシカルボニル
THF=テトラヒドロフラン
Et2O=ジエチルエーテル
hex=ヘキサン
EtOAc=酢酸エチル
DMF=ジメチルホルムアミド
MeOH=メタノール
EtOH=エタノール
i−PrOH=イソプロパノール
DMSO=ジメチルスルホキシド
DME=1,2−ジメトキシエタン
DCE=1,2 ジクロロエタン
HMPA=ヘキサメチルリン酸トリアミド
HOAcまたはAcOH=酢酸
TFA=トリフルオロ酢酸
TFAA=トリフルオロ無水酢酸
i−Pr2NEt=ジイソプロピルエチルアミン
Et3N=トリエチルアミン
NMM=N−メチルモルホリン
DMAP=4−ジメチルアミノピリジン
NaBH4=水素化ホウ素ナトリウム
NaBH(OAc)3=三アセトキシ水素化ホウ素ナトリウム
DIBALH=水素化ジイソブチルアルミニウム
LiAlH4=水素化アルミニウムリチウム
n−BuLi=n−ブチルリチウム
Pd/C=パラジウム炭素
PtO2=酸化白金
KOH=水酸化カリウム
NaOH=水酸化ナトリウム
LiOH=水酸化リチウム
K2CO3=炭酸カリウム
NaHCO3=炭酸水素ナトリウム
DBU=1,8−ジアザビシクロ[5.4.0]ウンデセ−7−エン
EDC(またはEDC・HCl)またはEDCI(またはEDCI・HCl)またはEDAC=3−エチル−3’−(ジメチルアミノ)プロピル−カルボジイミド塩酸塩(または1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩)
HOBTまたはHOBT・H2O=1−ヒドロキシベンゾトリアゾール水和物
HOAT=1−ヒドロキシ−7−アザベンゾトリアゾール
BOP試薬=ベンゾトリアゾール−1−イルオキシ−トリス(ジメチルアミノ)ホスホニウムヘキサフルオロリン酸塩
NaN(TMS)2=ヘキサメチルジシラジドナトリウムまたはビス(トリメチルシリル)アミドナトリウム
Ph3P=トリフェニルホスフィン
Pd(OAc)2=酢酸パラジウム
(Ph3P)4Pd0=テトラキストリフェニルホスフィンパラジウム
DEAD=ジエチルアゾジカルボキシレート
DIAD=ジイソプロピルアゾジカルボキシレート
Cbz−Cl=ベンジルクロロホルメート
CAN=硝酸セリウムアンモニウム
SAX=強力陰イオン交換体
SCX=強力陽イオン交換体
Ar=アルゴン
N2=窒素
min=分
hまたはhr=時
L=リットル
mL=ミリリットル
μL=マイクロリットル
g=グラム
mg=ミリグラム
mol=モル
mmol=ミリモル
meq=ミリ等量
RT=室温
satまたはsat’d=飽和
aq.=水の
TLC=薄層クロマトグラフィ
HPLC=高速液体クロマトグラフィ
LC/MS=高速液体クロマトグラフィ/マススペクトル
MSまたはMass Spec=マススペクトル
NMR=核磁気共鳴
NMRスペクトルデータ:s=シングレット;d=ダブレット;m=マルチプレット;br=ブロード;t=トリプレット
mp=融点
【0206】
実施例1
【化44】
【化45】
【0207】
B.
【化46】
【0208】
C.
【化47】
【0209】
D.
【化48】
【0210】
E.
【化49】
【0211】
F.
【化50】
【0212】
G.
【化51】
【0213】
H.
【化52】
[M+H]+ = 481.1
1H NMR (CDCl3 ; 400 MHz)δ : 2.43 (s, 3H), 3.05 (t, 2H; J=Hz), 4.26 (t, 2H; J=Hz), 4.35 (s, 2H), 6.73 (dd, 1H; J=Hz), 6.93 (d, 1H ; J=Hz), 7.14 (dd, 2H; J=Hz), 7.41 (t, 1H ; J=Hz), 7.47-7.54 (m, 5H), 8.05 (dd, 2H; J=Hz), 8.10 (dd, 2H; J=Hz), 11.32 (br s, 1H)
13C NMR (CDCl3 ; 100 MHz)δ: 10.2, 24.5, 31.7, 65.6, 113.6, 114.7, 119.4, 121.6, 124.5, 126.7, 128.4, 129.2, 129.3, 129.5, 130.1, 131.9, 137.5, 139.1, 139.6, 146.8, 151.4, 157.9, 160.2, 163.5
【0214】
実施例1(別の合成方法)
【化53】
【化54】
【0215】
B.
【化55】
【0216】
C.
【化56】
【0217】
D.
【化57】
【0218】
E.
【化58】
【0219】
F.
【化59】
【0220】
G.
【化60】
【0221】
H.
【化61】
【0222】
I.
【化62】
【0223】
J.
【化63】
【0224】
K.
【化64】
【0225】
L.
【化65】
[M+H]+ = 481
【0226】
実施例2
【化66】
[M+H]+ = 495.0
【0227】
実施例3
【化67】
【化68】
【0228】
B.
【化69】
【0229】
C.
【化70】
【0230】
D.
【化71】
【0231】
E.
【化72】
【0232】
F.
【化73】
【0233】
G.
【化74】
【0234】
H.
【化75】
[M+H]+ = 481.1
【0235】
実施例4
【化76】
[M+H]+ = 495.1
【0236】
実施例5
【化77】
【化78】
【0237】
B.
【化79】
【0238】
C.
【化80】
【0239】
D.
【化81】
【0240】
E.
【化82】
[M+H]+ = 495.0
1H NMR (DMSO; 400 MHz) 2.34 (s, 3H), 2.87-2.92 (t, J=6.6 Hz, 2H), 4.14-4.17 (m, 4H), 5.65 (s, 2H), 6.82-6.85 (d, J=8.76 Hz, 2H), 7.09-7.11 (d, J=8.32 Hz, 2H), 7.26-7.40 (m, 5H), 7.42-7.55 (m, 3H), 7.94-7.97 (m, 2H)
【0241】
実施例6
【化83】
[M+H]+ = 495.2
【0242】
実施例7
【化84】
【化85】
【0243】
B.
【化86】
[M+H]+ = 495.1
【0244】
実施例8
【化87】
【化88】
[M+H]+ = 495.2
【0245】
実施例9および10
実施例7および8の調製方法を用いて、以下の類似体を製造した。
【化89】
【化90】
【0246】
実施例11
【化91】
【化92】
【0247】
パートAのアルデヒドを製造する別法:
(1)
【化93】
【0248】
(2)
【化94】
【0249】
B.
【化95】
【0250】
C.
【化96】
【0251】
D.
【化97】
【化98】
[M+H]+ = 509.0
【0252】
F.
【化99】
[M+H]+ = 481.2
【0253】
実施例12
【化100】
【化101】
[M+H]+ = 481.1
【0254】
実施例13
【化102】
【化103】
【0255】
B.
【化104】
【0256】
C.およびD.
【化105】
【化106】
【0257】
E.
【化107】
[M+H]+ = 497.1
【0258】
実施例14
【化108】
[M+H]+ = 497.1
【0259】
実施例15
【化109】
[M+H]+ = 481.1
【0260】
実施例16
【化110】
【化111】
【0261】
B.
【化112】
【0262】
C.
【化113】
【0263】
D.
【化114】
【0264】
E.
【化115】
【化116】
【0265】
F.
【化117】
[M+H]+ =480.2
【0266】
実施例17
以下の化合物は、4−メトキシベンジルクロリドの代わりにパートAの3−メトキシベンジルクロリドを用いた以外は、実施例16の方法を用いて調製した。
【化118】
【0267】
実施例18
【化119】
【化120】
【0268】
B.
【化121】
【0269】
C.
【化122】
【0270】
D.
【化123】
【0271】
E.
【化124】
【化125】
【0272】
F.
【化126】
【0273】
実施例19
【化127】
[M+H]+ = 480.2
【0274】
実施例20
【化128】
【化129】
【0275】
B.
【化130】
【0276】
C.
【化131】
mL)溶液に、SO2Cl2(161μL ; 2.0 mmol)の無水CH2Cl2(1 mL)溶液を2時間かけて滴下して加えた。この間反応混合物に、連続して窒素を吹き込ん
だ。反応液を室温まで加温し、室温で2時間攪拌した。追加のCH2Cl2(10 mL)
を加えて、反応液を過剰の飽和NaHCO3水を加えてクエンチした。有機相を分液処理
し、H2Oで洗浄し(2回)、乾燥し(Na2SO4)、減圧濃縮した。該残渣をクロマト
グラフィで精製して(SiO2; ヘキサン:EtOAc=5:1)、澄明な油状物としてパートCの化合物を得た(380 mg; 68%)。
【0277】
D.
【化132】
【0278】
E.
【化133】
【0279】
F.
【化134】
【0280】
G.
【化135】
【0281】
H.
【化136】
【化137】
【0282】
I.
【化138】
[M+H]+ = 480.5
【0283】
実施例21
【化139】
【化140】
【化141】
【0284】
B.
【化142】
【0285】
C.
【化143】
【0286】
D.
【化144】
[M+H]+ = 479.2
【0287】
実施例22
【化145】
【化146】
【化147】
【0288】
B.
【化148】
【0289】
C.
【化149】
【0290】
D.
【化150】
【0291】
E.
【化151】
【0292】
F.
【化152】
[M+H]+ = 479.2
【0293】
実施例23〜50
以下のN−アリールピロール酸は、上述の方法の1つを用いて合成した。
【表7】
実施例51
【化153】
[M+H]+ = 495.1
【0295】
実施例52
【化154】
【化155】
【0296】
B.
【化156】
【0297】
C.
【化157】
[M+H]+ = 495.3
【0298】
実施例53
【化158】
【化159】
【0299】
B.
【化160】
[M+H]+ = 495.3
【0300】
実施例54
【化161】
【化162】
【0301】
B.
【化163】
【0302】
C.
【化164】
[M+H]+ = 417.2
【0303】
実施例55
【化165】
【化166】
【化167】
【0304】
B.
【化168】
【0305】
C.
【化169】
【0306】
D.
【化170】
[M+H]+ = 417.2
【0307】
実施例56
【化171】
【化172】
【0308】
B.
【化173】
[M+H]+ = 485.2
【0309】
実施例57
【化174】
[M+H]+ = 485.2
【0310】
実施例58
【化175】
【化176】
【0311】
B.
【化177】
[M+H]+ = 486.3
【0312】
実施例59
【化178】
[M+H]+ = 486.3
【0313】
実施例60
【化179】
【化180】
【0314】
B.
【化181】
パートCの化合物(40 mg; 23%):
【化182】
【化183】
E.
【化184】
[M+H]+ = 495.2
【0316】
実施例61
【化185】
[M+H]+ = 493.1
【0317】
実施例62
【化186】
【化187】
【0318】
B.
【化188】
【0319】
C.
【化189】
【0320】
D.
【化190】
【0321】
E.
【化191】
【0322】
F.
【化192】
【0323】
G.
【化193】
[M+H]+ = 481.1
【0324】
上記の実施例および反応式で述べた方法に従い、以下の例示される化合物は製造され得る。
【化194】
実施例63
両PPARγアンタゴニスト/PPARαアゴニストのインビトロスクリーニングアッセイ
A.マウス3T3−L1前脂肪細胞におけるPPARγアンタゴニストのスクリーニング
PPARγへの強い結合を示す化合物は、マウス3T3−L1前脂肪細胞の成熟した脂肪細胞への分化を誘発するロシグリタゾン(真正のPPARγアゴニスト)、50nMを阻害する能力でアッセイした。プレートあたり5×105の3T3−L1細胞を、96ウェルプレートに加え、誘発する前に2日間DMEM−高ブドウ糖および10% FBS培地で培養した。細胞は、同一培地中デキサメタゾン(1μm)、インスリン(5μg/ml)、およびイソブチルメチルキサンチン(IBMX)(0.6μm)で48時間誘発した。このとき、連続した希釈の試験化合物を、各ウェルの培地を含んだロシグリタゾン(50 nM)およびDMSO(0.1%)に加えた。培地(インスリン、デキサメタゾンおよびIBMXを含まない)を含む試験化合物、ロシグリタゾン(PPARγアゴニスト)およびDMSOの同一の濃度で、細胞をさらに72時間再フィードした。全部で5日間インキュベートした後、各ウェルから培地(4μl)を集め、96ウェルELISAプレート中のH2O(40μl)に希釈し、トリグリセリドブランク試薬(バイエル診断薬)(300μl)を各ウェルに加えて、室温で5分間インキュベートした。細胞から遊離グリセロールの放出を誘発するロシグリタゾンへの各化合物の阻害%は、スペクトレマックス(Spectremax) 250ELISAリーダーを用いて波長500nMで測定した。データはDMSOだけのコントロールに標準化して、トランス活性化の最大阻害%はロシグリタゾン正コントロール(50nM)と比較して計算した。ED50値は、活性阻害曲線の中点で標準方程式を用いて計算した。
【0326】
B.CV−1霊長類腎臓細胞におけるPPARγアンタゴニストのスクリーニング
PPARγに強い結合を示す化合物は、CV−1細胞のSEAPレポーター遺伝子活性のトランス活性化を誘発するロシグリタゾン(真正のPPARγアゴニスト)(1μM)を阻害する能力としてアッセイした。CV−1細胞(これらの細胞は内在性PPARγ遺伝子を発現する)は、3×PPRE−SEAPレポーター遺伝子DNAコンストラクトでトランスフェクトされ、安定なコロニーが選択され、拡張され、標準的なプロトコールを用いて化合物への応答として試験した。SEAPレポーター遺伝子コンストラクトは、pSEAP2のSV40初期最小促進剤(クロンテック)へ素早く5分で、7ヌクレオチドを含むラットの脂肪酸結合タンパク質PPREの3リピートを挿入して構築した。1.2×106のCV−1/PPRE−SEAP細胞を、化合物を添加する1日前に96ウェルのプレートに培養した。試験化合物の連続した希釈は、DMEM 10% FBS、0.5%(最終的に容積比)のDMSOおよび1μMのロシグリタゾン(PPARγアゴニスト)中調製した。各濃度の150μl分割量を、2つの隣接しないウェルに加えた。各プレートには、0.5% DMSO培地中に1μMのロシグリタゾン(PPARγアゴニスト)の6ウェルが含まれた。培地をフレッシュな96ウェルプレートに集め、続く40時間化合物とともにインキュベートし、SEAP活性としてアッセイした。SEAPは熱に耐性があり、集めた培地の内在性のホスファターゼは、圧力感受性の粘性シールフィルム(コーニング)でプレートをシールし、30分から1時間65℃に加熱して不活性化した。室温(RT)にした後、熱で不活性化した培地の25μl分割量をクリアーな底の96ウェル黒色プレートに加え、蛍光気質のアトフォス試薬(プロメガ)100μlをウェルあたり加えた。プレートを暗所で5分間インキュベートし、次いでサイトフルオラ(CytoFluor)シリーズ4000プレートリーダー(パーセプチブ バイオシステムズ)で蛍光を測定した:励起フィルター,450/50nm;発光フィルター,580/50nm;8周期,1分/周期,3読み取り/ウェル/周期。データはDMSOだけのコントロールに標準化して、トランス活性化の最大阻害%はロシグリタゾン正コントロール(1μM)と比較して計算した。ED50値は、活性阻害曲線の中点で標準方程式を用いて計算した。
【0327】
C.HepG2ヒト肝臓細胞におけるPPARαアゴニストのスクリーニング
PPARαに強い結合を示す化合物は、HepG2、誘導されたヒト肝臓、内在性PPARα遺伝子を発現する細胞、またはGal−4 DNA結合ドメイン−PPARαリガンド結合ドメインキメラ受容体(後述)を安定に発現するHepG2細胞において、レポーター遺伝子活性のPPARα依存の刺激を刺激する能力として試験した。レポーター遺伝子コンストラクトは、素早く5分で7ヌクレオチドを含むラットの脂肪酸結合タンパク質PPREの3リピートを、またはpSEAP2のSV40初期最小促進剤(クロンテック)の上方のGal4応答因子の4リピートを挿入して(それぞれ3×PPRE−SEAPおよびGal4−SEAP)、構築した。キメラ受容体は、哺乳類バイシストロン性発現ベクターpIRESlneo(クロンテック)におけるgal4 DNA結合ドメイン(アミノ酸1−47)、gal4−PPARαへと、フレームおよび3’のヒトPPARαのリガンド結合ドメインをコードするcDNAをクローニングして調製した。安定な細胞系は、リポフェクタミン プラス(ギブコ)を用い、続く製造者の指示で、gal4−SEAPおよびgal4−PPARαの両方で、または3XPPRE−SEAPでトランスフェクトして調製した。細胞は96ウェルプレート上で培養され、終夜付着させた。次の日、0.5%(容積比)DMSOを含む増殖培地(DMEM+10% チャコール/デキストランストリップしたFBS)中の化合物の連続する希釈物を、隣接しないウェルに2倍量加え、24〜40時間37℃で5% CO2中インキュベートした。各プレートには、1μMの標準溶液、正のコントロールとしてのGW−2331(真正のPPARα選択的アゴニスト)、負のコントロールとしてのロシグリタゾン(真正のPPARγアゴニスト)の少なくとも6ウェル、およびコントロールとしてDMSOのみの培地の3ウェルを有した。インキュベートに続いて、培地を除き、内在性ホスファターゼを上記で示唆されたように不活性化し、加工処理した培地の25μl分割量でのSEAP活性を、100μlのアトフォス試薬(プロメガ)を加えて、以下の条件でクリアーな底の黒色96ウェルプレート(ファルコン)中アッセイした:室温暗所で5分間インキュベートし、サイトフルオラ(CytoFluor)シリーズ4000プレートリーダー(パーセプチブ バイオシステムズ,8周期,1分/周期)で蛍光(励起:450nm;発光:580nm)の増加を測定。蛍光発光の相対速度は、DMSOコントロールからの倍増として計算した。内在性の活性は、1μM標準溶液の活性の%としての1μMで試験化合物の活性として定義した。ED50値は、活性曲線の中点で標準方程式を用いて計算した。
【0328】
実施例64:インビボ肥満動物モデル
C57BL/6マウスは、脂肪(40%)およびショ糖(40%)に富んだ食餌(参照:York {肥満の遺伝的モデル} and Sclafani {肥満の食餌的モデル}, both in Obesity, Bjorntorp and Brodoff eds. JB Lippincott Company, 1992; McIntosh and Pederson; McNeill. eds. CRC press LLC, 337-398, 1999; Farrelly et al., Proc. Natl. Acad. Sci. 96: 14511-14516, 1999)が与えられた。これらの食餌の条件下、C57BL/6マウスは、十分に体重が増加し、肥満となる。これらのマウスは、薬理学的に許容されるビークル(例えば、これに限らないが、5% CM−セルロース)中、経口で、静脈内、皮下または門脈内注射で、または食物または水と混ぜて、急性的にまたは時間を延長して、両PPARγアンタゴニスト/PPARαアゴニスト(用量:0.01〜100 mg/kg/日)を投与して処置した。実験過程においては、様々なパラメーター、例えば、水と食物の消費、体重の増加、両放射X線アナライザー(DEXA;この装置は体脂肪量、体除脂肪筋肉量および体骨ミネラル含量を正確に測定する)による身体構成、体温を、標準的な方法で測定した。尾静脈から血液を血液凝固防止のためにヘパリン−EDTAコーティングチューブに集めて、血漿を割けて、COBAS−MIRA器具のロシュ・ダイアグノスティックス社から入手できる試薬キットを用いて、ブドウ糖、遊離脂肪酸、トリグリセリドおよびコレステロールを分析した。インスリンおよびレプチンは、購入可能なELISAキットで測定する。体重を減らすことおよび/またはブドウ糖を減少させることに作用する化合物が選択された。処置の時間の終わりに、動物はCO2に短時間曝して安楽死させ、肝臓および白色脂肪組織のような内部器官を別の分析のために収集した。これらの分析には、これらに限らないが、脂質含量、および様々なPPARγおよびPPARα標的遺伝子の発現の効果の測定が含まれ得る。
【0329】
体脂肪量、除脂肪体重を減少させ、肥満症、インスリン耐性を防止または回復させる試験化合物はまた、抗糖尿病剤(例えば、これらに限らないが、メトホルミンおよびスルホニル尿素)および/またPPARαアゴニストのようなは脂質低下剤(例えば、これらに限らないが、フェノフィブラートおよびゲムフィブロジル)および/またはHMG CoA還元酵素阻害剤(例えば、これらに限らないが、プラバスタチン、ロバスタチン、シンバスタチンおよびアトルバスタチン)と組み合わせて、上述の疾患モデルで試験する。実験過程においては、様々なパラメーター、例えば、水と食物の消費、体重の増加、体温および血漿ブドウ糖、インスリン、遊離脂肪酸、トリグリセリドおよびコレステロールレベルを測定した。体脂肪量および体重を減少(体除脂肪骨格量を増加)させ、および/またはグルコース、および脂質を減少させることに作用する化合物は、さらに特徴付けのために選択された。
【図面の簡単な説明】
【0330】
【図1−A】図1−Aには、標識された真正のPPARγリガンド(BMS−化合物A)のヒトPPARγリガンド結合ドメインへの結合を、競合的に阻害する化合物Yの能力が記載される。
【図1−B】図1−Bには、標識された真正のPPARαリガンド(BMS−化合物B)のヒトPPARα結合ドメインへの結合が記載される。
【図2】図2には、真正のPPARγアゴニスト(例えば、ロシグリタゾン)依存性の、マウス3T3L−1前脂肪細胞(未成熟の脂肪細胞)の成熟した脂肪細胞に負荷した脂質(成熟した脂肪細胞)への分化を、競合的に阻害する化合物Yの能力が記載される。
【図3】図3には、霊長類腎臓細胞CV−1における内在性アルカリフォスファターゼ(SEAP)レポーター遺伝子発現の、真正のPPARγアゴニスト(例えば、ロシグリタゾン)依存性活性を、競合的に阻害する化合物Yの能力が記載される。
【図4】図4には、ヒト肝臓細胞系HepG2(この細胞系は、PPARαの重要な量を示す)中のPPARα依存性SEAPレポーター遺伝子活性を、安定にまとめられたPPARα依存性SEAPレポーターにより、用量依存的に刺激する化合物Yの能力が記載される。
Claims (20)
- 構造:
mは0、1または2であり;n=0、1または2であり;
QはCまたはNであり;
Aは(CH2)x(xは1〜5である)であるか;またはAは(CH2)x 1(x1は2〜5である)で、アルケニル結合またはアルキニル結合が該鎖に結合しているか;またはAは−(CH2)x 2−O−(CH2)x 3−(x2は0〜5で、x3は0〜5である、但し少なくともx2およびx3の1つは0でない)であり;
X1はCHまたはNであり;
X2はC、N、OまたはSであり;
X3はC、N、OまたはSであり;
X4はC、N、OまたはSであるが、但しX2、X3およびX4のうち少なくとも1つはNであり;
X5はC、N、OまたはSであり;
X6はCまたはNであり;
X7はC、N、OまたはSであるが、但しX5、X6またはX7のうち少なくとも1つはNであり;およびX1〜X7の各々が上記のように定義される場合において、CはCHを含み得;
R1はHまたはアルキルであり;
R2はH、アルキル、アルコキシ、ハロゲン、アミノまたは置換されたアミノであり;
R2a、R2bおよびR2cは同一または異なって、H、アルキル、アルコキシ、ハロゲン、アミノまたは置換されたアミノから選択され;
R3およびR3aは同一または異なって、H、アルキル、アリールアルキル、アリールオキシカルボニル、アルキルオキシカルボニル、アルキニルオキシカルボニル、アルケニルオキシカルボニル、アリールカルボニル、アルキルカルボニル、アリール、ヘテロアリール、アルキル(ハロ)アリールオキシカルボニル、アルキルオキシ(ハロ)アリールオキシカルボニル、シクロアルキルアリールオキシカルボニル、シクロアルキルオキシアリールオキシカルボニル、シクロヘテロアルキル、ヘテロアリールカルボニル、ヘテロアリール−ヘテロアリールアルキル、アルキルカルボニルアミノ、アリールカルボニルアミノ、ヘテロアリールカルボニルアミノ、アルコキシカルボニルアミノ、アリールオキシカルボニルアミノ、ヘテロアリールオキシカルボニルアミノ、ヘテロアリール−ヘテロアリールカルボニル、アルキルスルホニル、アルケニルスルホニル、ヘテロアリールオキシカルボニル、シクロヘテロアルキルオキシカルボニル、ヘテロアリールアルキル、アミノカルボニル、置換されたアミノカルボニル、アルキルアミノカルボニル、アリールアミノカルボニル、ヘテロアリールアルケニル、シクロヘテロアルキルヘテロアリールアルキル、ヒドロキシアルキル、アルコキシ、アルコキシアリールオキシカルボニル、アリールアルキルオキシカルボニル、アルキルアリールオキシカルボニル、アリールヘテロアリールアルキル、アリールアルキルアリールアルキル、アリールオキシアリールアルキル、アルキニルオキシカルボニル、ハロアルコキシアリールオキシカルボニル、アルコキシカルボニルアリールオキシカルボニル、アリールオキシアリールオキシカルボニル、アリールスルフィニルアリールカルボニル、アリールチオアリールカルボニル、アルコキシカルボニルアリールオキシカルボニル、アリールアルケニルオキシカルボニル、ヘテロアリールオキシアリールアルキル、アリールオキシアリールカルボニル、アリールオキシアリールアルキルオキシカルボニル、アリールアルケニルオキシカルボニル、アリールアルキルカルボニル、アリールオキシアルキルオキシカルボニル、アリールアルキルスルホニル、アリールチオカルボニル、アリールアルケニルスルホニル、ヘテロアリールスルホニル、アリールスルホニル、アルコキシアリールアルキル、ヘテロアリールアルコキシカルボニル、アリールヘテロアリールアルキル、アルコキシアリールカルボニル、アリールオキシヘテロアリールアルキル、ヘテロアリールアルキルオキシアリールアルキル、アリールアリールアルキル、アリールアルケニルアリールアルキル、アリールアルコキシアリールアルキル、アリールカルボニルアリールアルキル、アルキルアリールオキシアリールアルキル、アリールアルコキシカルボニルヘテロアリールアルキル、ヘテロアリールアリールアルキル、アリールカルボニルヘテロアリールアルキル、ヘテロアリールオキシアリールアルキル、アリールアルケニルヘテロアリールアルキル、アリールアミノアリールアルキルまたはアミノカルボニルアリールアリールアルキルから独立して選択され;
YはCO2R4(R4はH若しくはアルキル、またはプロドラッグエステルである)であるか、またはYはC−連結した1−テトラゾール、構造P(O)(OR4a)R5(R4aはHまたはプロドラッグエステルであり、R5はアルキルまたはアリールである)のホスフィン酸または構造P(O)(OR4a)2のホスホン酸であり;
(CH2)x、(CH2)x 1、(CH2)x 2、(CH2)x 3、(CH2)m、および(CH2)nは適宜、1、2または3個の置換基で置換され得る]
を有し、そのすべての立体異性体、そのプロドラッグエステル、およびその医薬的に許容される塩を含む化合物。 - 構造:
mは0、1または2であり;n=0、1または2であり;
QはCまたはNであり;
x2は0〜5で、x3は0〜5であるが、但し少なくともx2およびx3の1つは0でなく;
X2はC、N、OまたはSであり;
X3はC、N、OまたはSであり;
X4はC、N、OまたはSであるが、但しX2、X3およびX4のうち少なくとも1つはNであり;およびX2〜X4の各々が上記のように定義される場合において、CはCHを含み得;
R1はHまたはアルキルであり;
R2はH、アルキル、アルコキシ、ハロゲン、アミノまたは置換されたアミノであり;
R2a、R2bおよびR2cは同一または異なって、H、アルキル、アルコキシ、ハロゲン、アミノまたは置換されたアミノから選択され;
R3およびR3aは同一または異なって、H、アルキル、アリールアルキル、アリールオキシカルボニル、アルキルオキシカルボニル、アルキニルオキシカルボニル、アルケニルオキシカルボニル、アリールカルボニル、アルキルカルボニル、アリール、ヘテロアリール、アルキル(ハロ)アリールオキシカルボニル、アルキルオキシ(ハロ)アリールオキシカルボニル、シクロアルキルアリールオキシカルボニル、シクロアルキルオキシアリールオキシカルボニル、シクロヘテロアルキル、ヘテロアリールカルボニル、ヘテロアリール−ヘテロアリールアルキル、アルキルカルボニルアミノ、アリールカルボニルアミノ、ヘテロアリールカルボニルアミノ、アルコキシカルボニルアミノ、アリールオキシカルボニルアミノ、ヘテロアリールオキシカルボニルアミノ、ヘテロアリール−ヘテロアリールカルボニル、アルキルスルホニル、アルケニルスルホニル、ヘテロアリールオキシカルボニル、シクロヘテロアルキルオキシカルボニル、ヘテロアリールアルキル、アミノカルボニル、置換されたアミノカルボニル、アルキルアミノカルボニル、アリールアミノカルボニル、ヘテロアリールアルケニル、シクロヘテロアルキルヘテロアリールアルキル、ヒドロキシアルキル、アルコキシ、アルコキシアリールオキシカルボニル、アリールアルキルオキシカルボニル、アルキルアリールオキシカルボニル、アリールヘテロアリールアルキル、アリールアルキルアリールアルキル、アリールオキシアリールアルキル、アルキニルオキシカルボニル、ハロアルコキシアリールオキシカルボニル、アルコキシカルボニルアリールオキシカルボニル、アリールオキシアリールオキシカルボニル、アリールスルフィニルアリールカルボニル、アリールチオアリールカルボニル、アルコキシカルボニルアリールオキシカルボニル、アリールアルケニルオキシカルボニル、ヘテロアリールオキシアリールアルキル、アリールオキシアリールカルボニル、アリールオキシアリールアルキルオキシカルボニル、アリールアルケニルオキシカルボニル、アリールアルキルカルボニル、アリールオキシアルキルオキシカルボニル、アリールアルキルスルホニル、アリールチオカルボニル、アリールアルケニルスルホニル、ヘテロアリールスルホニル、アリールスルホニル、アルコキシアリールアルキル、ヘテロアリールアルコキシカルボニル、アリールヘテロアリールアルキル、アルコキシアリールカルボニル、アリールオキシヘテロアリールアルキル、ヘテロアリールアルキルオキシアリールアルキル、アリールアリールアルキル、アリールアルケニルアリールアルキル、アリールアルコキシアリールアルキル、アリールカルボニルアリールアルキル、アルキルアリールオキシアリールアルキル、アリールアルコキシカルボニルヘテロアリールアルキル、ヘテロアリールアリールアルキル、アリールカルボニルヘテロアリールアルキル、ヘテロアリールオキシアリールアルキル、アリールアルケニルヘテロアリールアルキル、アリールアミノアリールアルキルまたはアミノカルボニルアリールアリールアルキルから独立して選択され;
(CH2)x 2、(CH2)x 3、(CH2)m、および(CH2)nは適宜、1、2または3個の置換基で置換され得る]
を有し、そのすべての立体異性体、そのプロドラッグエステル、およびその医薬的に許容される塩を含む化合物。 - 構造:
- 構造:
- (CH2)x、(CH2)x 1、(CH2)x 2、(CH2)x 3が、アルキレン、アルケニレン、アレニル、またはアルキニレンである、請求項1の化合物。
- X1がCHである請求項1の化合物。
- XがNである請求項1の化合物。
- 構造:
- R1がCH3、およびR3がフェニル、またはアルキル、ポリハロアルキル、ハロまたはアルコキシで置換されるフェニルである、請求項8の化合物。
- 構造:
- 請求項1の化合物およびそのための医薬的に許容される担体からなる医薬組成物。
- 請求項1の化合物の治療上の有効量を治療が必要な患者に投与することからなる、血中ブドウ糖レベルの低下方法または糖尿病の治療方法。
- 請求項1の化合物の治療上の有効量を治療が必要な患者に投与することからなる、前悪性疾患、初期悪性疾患、悪性疾患、または形成異常疾患の治療方法。
- 請求項1の化合物と、脂質低下剤、脂質調節剤、抗糖尿病剤、抗肥満剤、降圧剤、血小板凝集阻害剤、および/または抗骨多孔剤(該抗糖尿病剤は、1、2、3またはそれ以上のビグアナイド、スルホニル尿素、グルコシダーゼ阻害剤、PPARγアゴニスト、PPARα/γ両アゴニスト、SGLT2阻害剤、DP4阻害剤、aP2阻害剤、インスリン増感剤、グルカゴン様ペプチド−1(GLP−1)、インスリンおよび/またはメグリチナイドであり;該抗肥満剤は、β3アドレナリン作動性アゴニスト、リパーゼ阻害剤、セロトニン(およびドパミン)再取り込み阻害剤、甲状腺受容体アゴニスト、aP2 阻害剤および/または摂食障害剤であり;該脂質低下剤は、MTP阻害剤、HMG CoA還元酵素阻害剤、スクアレン合成酵素阻害剤、フィブリン酸誘導体、LDL受容体活性の上方制御剤、リポキシゲナーゼ阻害剤、またはACAT阻害剤であり;該降圧剤は、ACE阻害剤、アンジオテンシンII受容体アンタゴニスト、NEP/ACE阻害剤、カルシウムチャンネルブロッカーおよび/またはβ−アドレナリン作動性遮断薬である)とからなる組み合わせ医薬。
- 該抗糖尿病剤が、1、2、3またはそれ以上のメトホルミン、グリブリド、グリメピリド、グリピリド、グリピジド、クロロプロパミド、グリクラジド、アカルボース、ミグリトール、ピオグリタゾン、トログリタゾン、ロシグリタゾン、インスリン、Gl−262570、イサグリタゾン、JTT−501、NN−2344、L895645、YM−440、R−119702、AJ9677、レパグリニド、ナテグリニド、KAD1129、AR−HO39242、GW−409544、KRP297、AC2993、LY315902、P32/98および/またはNVP−DPP−728Aであり、
該抗肥満剤が、オルリスタット、ATL−962、AJ9677、L750355、CP331648、シブトラミン、トピラメート、アキソキン、デキサアンフェタミン、フェンテルミン、フェニルプロパノールアミン、および/またはマジンドールであり、
該脂質低下剤が、プラバスタチン、ロバスタチン、シンバスタチン、アトルバスタチン、セリバスタチン、フルバスタチン、イタバスタチン、ビサスタチン、フェノフィブラート、ゲムフィブロジル、クロフィブラート、アバシミベ、TS−962、MD−700、コレスタゲル、ナイアシンおよび/またはLY295427であり、
該降圧剤が、ACE阻害剤(カプトプリル、フォシノプリル、エナラプリル、リシノプリル、キナプリル、ベナゼプリル、フェンチアプリル、ラミプリルまたはモエキシプリル);NEP/ACE阻害剤(オマパトリラット、[S[(R*,R*)]−ヘキサヒドロ−6−[(2−メルカプト−1−オキソ−3−フェニルプロピル)アミノ]−2,2−ジメチル−7−オキソ−1H−アゼピン−1−酢酸(ゲモパトリラット)またはCGS 30440);アンジオテンシンII受容体アンタゴニスト(イルベサルタン、ロサルタンまたはバルサルタン);アムロジピン ベスィレート、プラゾシンHCl、ベラパミル、ニフェジピン、ナドロール、プロプラノロール、カルベジロール、またはクロニジンHClであり、
該血小板凝集阻害剤が、アスピリン、クロピドグレル、チクロピジン、ジピリダモールまたはイフェトロバンである、請求項12の組み合わせ。 - 請求項14の組み合わせ医薬の治療上の有効量を治療が必要な哺乳類に投与することからなる、インスリン耐性、高血糖、高インスリン血症、または遊離脂肪酸またはグリセロールの血中レベルの上昇、高脂血症、肥満症、シンドロームX、代謝異常症侯群、炎症、糖尿病性合併症、障害性ブドウ糖ホメオスタシス、障害性ブドウ糖耐性、高トリグリセリド血症またはアテローム性動脈硬化症の治療方法。
- 該疾患が、脂肪肉腫または上皮性腫瘍である、請求項13の方法。
- 該上皮性腫瘍が、乳房、前立腺、大腸、卵巣、胃または肺の腫瘍である、請求項17の方法。
- 請求項1の化合物の治療上の有効量を治療が必要な哺乳類に投与することからなる、過敏性大腸症候群、クローン病、胃潰瘍または骨粗鬆症、または乾癬の治療方法、または肥満症、インスリン耐性、異脂肪血症、循環器疾患および肝臓異常の治療方法。
- 両PPARγアンタゴニスト/PPARαアゴニストの投与によって、HMGic、グリセロール−PO4デヒドロゲナーゼ、脂肪酸輸送タンパク質、Gタンパク質共役受容体26、アジポフィリン、ケラチノサイト 脂肪酸結合タンパク質、アンジオテンシノーゲン、PAI−1、およびレニンから選択される遺伝子発現の変動に基づく、肥満症および循環器疾患を治療する方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US29438001P | 2001-05-30 | 2001-05-30 | |
| PCT/US2002/016633 WO2002096358A2 (en) | 2001-05-30 | 2002-05-23 | Substituted azole acid derivatives useful as antidiabetic and antiobesity agents and method |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004536070A true JP2004536070A (ja) | 2004-12-02 |
| JP2004536070A5 JP2004536070A5 (ja) | 2005-12-22 |
Family
ID=23133159
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002592871A Pending JP2004536070A (ja) | 2001-05-30 | 2002-05-23 | 抗糖尿病および抗肥満症薬として有用な置換されたアゾール酸誘導体および方法 |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US20030092736A1 (ja) |
| EP (1) | EP1390363A4 (ja) |
| JP (1) | JP2004536070A (ja) |
| AU (1) | AU2002259306B2 (ja) |
| CA (1) | CA2449160A1 (ja) |
| CZ (1) | CZ20033230A3 (ja) |
| DE (1) | DE02729306T1 (ja) |
| ES (1) | ES2214168T1 (ja) |
| HU (1) | HUP0401504A3 (ja) |
| MX (1) | MXPA03010997A (ja) |
| NO (1) | NO327089B1 (ja) |
| PE (1) | PE20030043A1 (ja) |
| PL (1) | PL367066A1 (ja) |
| TR (1) | TR200400650T3 (ja) |
| TW (1) | TWI235061B (ja) |
| UY (1) | UY27316A1 (ja) |
| WO (1) | WO2002096358A2 (ja) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2003256419A1 (en) * | 2002-08-21 | 2004-03-11 | Merck & Co., Inc. | Combination therapy using a dual ppar alpha/gamma agonist and an angiotensin ii type i receptor antagonist |
| EP1424070A1 (en) * | 2002-11-28 | 2004-06-02 | Fournier Laboratories Ireland Limited | Combination of a PPAR alpha agonist and metformin for decreasing the serum triglycerides |
| SE0300988D0 (sv) * | 2003-04-03 | 2003-04-03 | Astrazeneca Ab | New use |
| US7601756B2 (en) * | 2003-06-06 | 2009-10-13 | Snowden Pharmaceuticals, Llc | Method of treatment for irritable bowel syndrome |
| JP2009513523A (ja) | 2003-07-08 | 2009-04-02 | ノバルティス アクチエンゲゼルシャフト | ベンゼンスルホニルアミノ化合物およびそれらを含む医薬組成物 |
| ES2390053T3 (es) * | 2003-09-22 | 2012-11-06 | Ono Pharmaceutical Co., Ltd. | Derivado de ácido fenilacético, procedimiento para producir el mismo y uso |
| EP1734953A4 (en) * | 2004-03-02 | 2008-08-20 | Abeille Pharmaceuticals Inc | CO-PR PARATIONS OF KITS OF BIOACTIVE AGENTS |
| ES2282062T1 (es) | 2004-06-04 | 2007-10-16 | Teva Pharmaceutical Industries Ltd. | Composicion farmaceutica que contiene irbesartan. |
| CN1304393C (zh) * | 2004-07-01 | 2007-03-14 | 中国药科大学 | 取代的吡唑啉酮衍生物及其制备方法与药用组合物 |
| KR100706600B1 (ko) * | 2004-11-25 | 2007-04-12 | 주식회사 엘지생명과학 | PPAR gamma와 PPAR alpha의 활성을항진시키는 신규 화합물, 그것의 제조방법, 및 그것을함유한 약제 조성물 |
| KR20060058632A (ko) * | 2004-11-25 | 2006-05-30 | 주식회사 엘지생명과학 | PPAR gamma와 PPAR alpha의 활성을항진시키는 신규 화합물, 그것의 제조방법, 및 그것을함유한 약제 조성물 |
| WO2007058504A1 (en) * | 2005-11-21 | 2007-05-24 | Lg Life Sciences, Ltd. | Novel compounds as agonist for ppar gamma and ppar alpha, method for preparation of the same, and pharmaceutical composition containing the same |
| US20090048199A1 (en) * | 2007-03-09 | 2009-02-19 | Hiberna Corporation | Hibernation-Related Genes and Proteins, Activators and Inhibitors Thereof and Methods of Use |
| BRPI0822349A2 (pt) * | 2007-11-29 | 2019-09-24 | Neuraltus Pharmaceuticals Inc | composições e métodos para tratar doenças lisossômicas |
| CN102256493A (zh) * | 2008-10-29 | 2011-11-23 | 迪赛孚尔制药有限公司 | 表现出抗癌活性和抗增殖活性的环丙烷酰胺及其类似物 |
| WO2020113094A1 (en) | 2018-11-30 | 2020-06-04 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
| CN109498622A (zh) * | 2018-12-28 | 2019-03-22 | 成都恒瑞制药有限公司 | 一种氯沙坦钾与罗格列酮组合物及其制备方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1003445B (zh) * | 1984-10-03 | 1989-03-01 | 武田药品工业株式会社 | 噻唑烷二酮衍生物,其制备方法和用途 |
| US5591862A (en) * | 1993-06-11 | 1997-01-07 | Takeda Chemical Industries, Ltd. | Tetrazole derivatives, their production and use |
| EP1177176B1 (en) * | 1999-04-28 | 2006-04-19 | Aventis Pharma Deutschland GmbH | Tri-aryl acid derivatives as ppar receptor ligands |
| US6506781B1 (en) * | 1999-09-08 | 2003-01-14 | Smithkline Beecham Corporation | Oxazole PPAR antagonist |
| HK1045991B (en) * | 1999-11-10 | 2004-12-10 | Takeda Pharmaceutical Company Limited | 5-membered n-heterocyclic compounds with hypoglycemic and hypolipidemic activity |
| EP1394154A4 (en) * | 2001-03-23 | 2005-05-18 | Takeda Pharmaceutical | FIVE-GLASS HETEROCYCLIC ALKANIC ACID DERIVATIVE |
-
2002
- 2002-05-22 US US10/153,454 patent/US20030092736A1/en not_active Abandoned
- 2002-05-23 EP EP02729306A patent/EP1390363A4/en not_active Withdrawn
- 2002-05-23 CA CA002449160A patent/CA2449160A1/en not_active Abandoned
- 2002-05-23 MX MXPA03010997A patent/MXPA03010997A/es active IP Right Grant
- 2002-05-23 CZ CZ20033230A patent/CZ20033230A3/cs unknown
- 2002-05-23 TR TR2004/00650T patent/TR200400650T3/xx unknown
- 2002-05-23 ES ES02729306T patent/ES2214168T1/es active Pending
- 2002-05-23 DE DE0001390363T patent/DE02729306T1/de active Pending
- 2002-05-23 JP JP2002592871A patent/JP2004536070A/ja active Pending
- 2002-05-23 HU HU0401504A patent/HUP0401504A3/hu unknown
- 2002-05-23 WO PCT/US2002/016633 patent/WO2002096358A2/en not_active Ceased
- 2002-05-23 PL PL02367066A patent/PL367066A1/xx not_active Application Discontinuation
- 2002-05-23 AU AU2002259306A patent/AU2002259306B2/en not_active Ceased
- 2002-05-24 TW TW091111100A patent/TWI235061B/zh not_active IP Right Cessation
- 2002-05-30 UY UY27316A patent/UY27316A1/es not_active Application Discontinuation
- 2002-05-30 PE PE2002000455A patent/PE20030043A1/es not_active Application Discontinuation
-
2003
- 2003-11-28 NO NO20035312A patent/NO327089B1/no not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| WO2002096358A2 (en) | 2002-12-05 |
| ES2214168T1 (es) | 2004-09-16 |
| PL367066A1 (en) | 2005-02-21 |
| TWI235061B (en) | 2005-07-01 |
| HUP0401504A3 (en) | 2008-05-28 |
| DE02729306T1 (de) | 2004-08-26 |
| PE20030043A1 (es) | 2003-02-05 |
| TR200400650T3 (tr) | 2004-06-21 |
| UY27316A1 (es) | 2002-12-31 |
| EP1390363A4 (en) | 2011-01-05 |
| MXPA03010997A (es) | 2004-02-27 |
| EP1390363A2 (en) | 2004-02-25 |
| NO20035312D0 (no) | 2003-11-28 |
| CA2449160A1 (en) | 2002-12-05 |
| CZ20033230A3 (cs) | 2004-02-18 |
| NO327089B1 (no) | 2009-04-20 |
| HUP0401504A2 (hu) | 2004-11-29 |
| AU2002259306B2 (en) | 2007-02-08 |
| US20030092736A1 (en) | 2003-05-15 |
| WO2002096358A3 (en) | 2003-03-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6673815B2 (en) | Substituted acid derivatives useful as antidiabetic and antiobesity agents and method | |
| EP1401433B1 (en) | Azole acid derivatives, alone or in combination, for treating diabetes and dyslipidemias; and for treating malignant diseases | |
| US7795291B2 (en) | Substituted acid derivatives useful as anti-atherosclerotic, anti-dyslipidemic, anti-diabetic and anti-obesity agents and method | |
| RU2327692C2 (ru) | Окса- и тиазолпроизводные в качестве антидиабетических агентов и агентов против ожирения | |
| US6875782B2 (en) | Substituted heterocyclic derivatives useful as antidiabetic and antiobesity agents and method | |
| US6727271B2 (en) | Substituted acid derivatives useful as antidiabetic and antiobesity agents and method | |
| US20080009534A1 (en) | Substituted acid derivatives useful as antidiabetic and antiobesity agents and method | |
| US6967212B2 (en) | Substituted azole acid derivatives useful as antidiabetic and antiobesity agents and method | |
| AU2002259306B2 (en) | Substituted azole acid derivatives useful as antidiabetic and antiobesity agents and method | |
| AU2002259306A1 (en) | Substituted azole acid derivatives useful as antidiabetic and antiobesity agents and method | |
| US6919323B2 (en) | Pyridazinone inhibitors of fatty acid binding protein and method | |
| AU2002310141B2 (en) | Conformationally constrained analogs useful as antidiabetic and antiobesity agents and method | |
| AU2002310141A1 (en) | Conformationally constrained analogs useful as antidiabetic and antiobesity agents and method |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050510 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050510 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20081125 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20090421 |