JP2004504347A - ヒストン脱アセチル化酵素阻害剤としてのバルプロ酸およびその誘導体 - Google Patents
ヒストン脱アセチル化酵素阻害剤としてのバルプロ酸およびその誘導体 Download PDFInfo
- Publication number
- JP2004504347A JP2004504347A JP2002513458A JP2002513458A JP2004504347A JP 2004504347 A JP2004504347 A JP 2004504347A JP 2002513458 A JP2002513458 A JP 2002513458A JP 2002513458 A JP2002513458 A JP 2002513458A JP 2004504347 A JP2004504347 A JP 2004504347A
- Authority
- JP
- Japan
- Prior art keywords
- cells
- vpa
- compound
- histone
- histone deacetylase
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images











Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/02—Drugs for disorders of the endocrine system of the hypothalamic hormones, e.g. TRH, GnRH, CRH, GRH, somatostatin
Abstract
Description
(技術分野)
本発明は、ヒストン脱アセチル化活性を有する酵素の阻害剤としての、薬剤バルプロ酸およびその誘導体の使用に関する。本発明はまた、ヒストンの低アセチル化に起因する疾病または、高アセチル化誘導が、例えば形質転換細胞(transformed cell)の分化及び/またはアポトーシスの誘導を引き起こすことによって、有効な効果を奏する疾病の治療剤の製造のための、これら化合物の使用に関する。
【0002】
(背景技術)
局所的なクロマチン再構築は遺伝子の転写活性化において重要な段階である。転写タンパク質が鋳型DNAと接触するには、DNAのヌクレオソームへのパッケージングにおいて大幅な変化が起こらなくてはならない。クロマチン再構築に関与する最も重要な機構のひとつは、アセチル化によるヒストンの翻訳後修飾である。DNAとの間に働く静電気的引力の変化および疎水性アセチル基により引き起こされる立体機能障害により、ヒストンとDNAとの関係が不安定となる。
【0003】
その結果、ヒストンのアセチル化によりヌクレオソームが破壊され、DNAの転写が可能となる。アセチル基の除去によりヒストンがより強固にDNAおよび隣接するヌクレオソームと結合し、これにより転写抑制されたクロマチン構造を保っている。アセチル化はヒストンアセチル化酵素(HAT)活性を有する一連の酵素を介して行われる。逆に、アセチル基は特定のヒストン脱アセチル化酵素(HDAC)によって除去される。これらの機構が破綻すると転写の制御不良が頻発し、白血病性の変異を導く可能性がある。
【0004】
核内ホルモン受容体はリガンド依存性の転写因子であり、遺伝子発現を正負に制御して発生とホメオスタシスを制御する。これら制御過程における異常が多くの疾病の原因の根底にあり、癌の発生に大きく関与している。
【0005】
幾つかの核内受容体のスーパーファミリーが、TFIIBを含む基本的な転写因子と相互に作用することが報告されている。しかしながら、標的遺伝子の活性化と抑制化を調節するには、核内受容体がさらに別な因子と相互に作用しなければならないことが多くの証拠により示唆されている。エストロゲン(ER)、レチノイン酸(RAR)、甲状腺ホルモン(T3R)、レチノイドX(RXR)、および他の核内受容体のリガンド結合領域と結びつく数多くの共同因子が近年確認された。想定されるコアクチベータータンパクにはSRC−1/NCoA−1、GRIP1/TIF2/NCoA−2、p/CIP/ACTR/AIB1、CBPおよび多様な他の因子がある(Xuら、1999年、Current Opinion in Genetics & Development 9, 140−147参照)。興味深い点として、SRCとCBPタンパク質は内因性ヒストンアセチル基転移酵素活性を有し、ヒストンアセチラーゼP/CAFと共に複合体を形成して存在することが明らかにされている。
【0006】
T3R、RARおよびPPARを含む多くの核内受容体は、リガンド非存在下でコリプレッサーN−CoRおよびSMRTと相互作用し、それによって転写を抑制することが出来る。さらに、N−CoRはアンタゴニストと結合したプロゲステロンおよびエストロゲン受容体と相互作用することも報告されている。N−CoRとSMRTは、mSin3タンパク質とヒストン脱アセチル化酵素とともに巨大タンパク質複合体を形成して存在することが明らかにされている。従って、リガンドに誘発されて核内受容体が抑制型から活性型へと転換すると、コリプレッサー複合体が影響を受けてアンタゴニスト酵素活性を有するコアクチベーター複合体に変換される。
【0007】
N−CoRコリプレッサー複合体は、核内受容体による抑制を媒介するだけでなく、Mad−1、BCL−6およびETOを含む付加的な転写因子と相互に関係する。これらタンパク質の多くは、細胞の異常増殖と異常分化に多大に関係している。例えばT3Rは、はじめはその相同性からウィルス腫瘍遺伝子v−erbAと同一であるとされていたが、その野生型受容体と対照的にリガンドと結合せず、構成的転写抑制因子として機能する。さらに、RARの変異が多くのヒト癌、特に急性前骨髄球性白血病(APL)や肝細胞癌と関連があるとされてきた。APL患者において、染色体転位の結果生成されるRAR融合タンパク質は、前骨髄球性白血球タンパク質(PML)または前骨髄ジンクフィンガータンパク質(PLZF)のいずれかを含んでいる。両融合タンパク質はコリプレッサー複合体の構成要素と相互作用することが出来るが、レチノイン酸が加わるとコリプレッサー複合体はPML−RARから離脱し、一方、PLZF−RARとは構成的に相互作用する。
【0008】
これらの知見により、なぜPML−RAR APL患者はレチノイン酸治療後に完全寛解するのに、PLZF−RAR APL患者にはあまり効果がないのかということの説明がつく。コリプレッサーが介在する異常抑制が、偶然APLの病因となるかもしれないという仮説は、ヒストン脱アセチル化活性阻害剤であるトリコスタチンAが、該PLZF−RAR融合タンパク質を含有している細胞内での分化阻害を克服できるという知見により裏付けられる。さらに最近では、レチノイン酸治療後に何度も再発を繰り返すPML−RAR患者に対しては、HDAC阻害フェニル酪酸による治療が行われており、白血病からの完全寛解という成果を挙げている(Warrellら、1998、Journal of the National Cancer Institute、90、1621−1625)。
【0009】
ヒストンのアセチル化が癌に関与していることの更なる証拠として、AML1−ETO腫瘍タンパク質とMLL遺伝子座を含む染色体再配列についての研究がある(Renderら、1999、Blood 94、417−428)
【0010】
公開公報No.99/37150には、レチノイド薬剤およびヒストン脱アセチル化酵素阻害剤を投与することを特徴とする癌の遺伝子転写治療法が開示されている。
【0011】
HDAC阻害剤として複数の化合物が知られている。酪酸または酪酸塩が最初に同定されたHDAC阻害剤であった。ミリモラー濃度では、酪酸塩はHDACに特異的ではなく、核タンパク質のリン酸化およびメチル化、同様にDNAのメチル化を阻害する。それに類似のフェニル酪酸塩も同様に作用する。さらに特異的なのは、トリコスタチンA(TSA)とトラポキシン(TPX)である。TSAとTPXは有効なヒストン脱アセチル化酵素阻害剤として取り上げられてきた。TSAが可逆的に阻害するのに対し、TPXは不可逆的にHDAC酵素に結合して不活化させる。酪酸塩とは異なり、TSAおよびTPXについては他の酵素系を非特異的に阻害することは報告されていない。しかしTSAおよびTPXは相当な毒性を示し、生体ではほとんど利用することが出来ない。したがって、これらの治療上の用途は限られている。
【0012】
(発明の開示)
本発明の1つの目的は、多型の形質転換細胞において分化及び/またはアポトーシスを誘導することができ、それゆえに癌の治療に有用である物質を供給することである。
本発明は、バルプロ酸(VPA;2−n−プロピルペンタン酸)が、ヒストン脱アセチル化酵素(histone deacetylase)を阻害することができるという新規知見に依拠している。
【0013】
バルプロ酸は、様々な分子作用機構に基づく多様な生物活性を有する公知の薬剤である。
VPAは抗てんかん剤である。
VPAは催奇性がある。VPAが妊娠中に抗てんかん剤として使われると、VPAは生まれてきた子供の数%において先天性異常(神経管閉鎖欠損およびその他の奇形)を誘導しうる。マウスでは、VPAが適当に投与されると、マウス胎児の大半で催奇性がみられる。
VPAは、核内ホルモン受容体(PPARδ)を活性化する。また、幾つかの別な転写因子も活性化されるが、顕著に活性化されない因子(グルココルチコイド受容体、PPARδ)もある。
VPAは、コエンザイムAとのエステルが完全に代謝されないことに起因しうる肝細胞毒性を示す。
【0014】
VPA誘導体を使用することにより、異なる活性は異なる分子作用機構により調整されているということを判断することができた。催奇性誘因活性と抗てんかん活性は異なる作用形態に従うが、これは化合物が優先的に催奇性を示すか、又は抗てんかん性を示すのどちらかに分かれうるからである(Nau ら, 1991, Pharmacol. Toxicol. 69, 310−321)。PPARδの活性は催奇性と厳密に関連するということが分かっており(Lampen ら, 1999, Toxicol. Appl. Pharmacol. 160, 238−249)、このことからPPARδの活性と催奇性の両方に同じVPAの分子活性が必要であることが示唆される。また、Lampenらによる示唆(1999)および分化マーカーの分析(Werling ら, 2001, Mol. Pharmacol. 59, 1269−1276)により証明されているように、F9細胞の分化はPPARδの活性および催奇性に厳密に関連する。
【0015】
本明細書には、PPARδの活性化がVPAとその誘導体が有するHDAC阻害活性により引き起こされることが示されている。さらに、従来から用いられているHDAC阻害剤であるTSAが、PPARδを活性化しVPAと同様の形式によりF9細胞の分化を誘導することが示されている。これらの結果より発明者らは、PPARδの活性化のみならずF9細胞の分化誘導、およびVPA又はVPA誘導体による催奇形性も、HDACの阻害により引き起こされる可能性が非常に高いと判断した。
【0016】
本発明は、本明細書に記載されたVPAやその誘導体がヒストン脱アセチル化酵素の阻害剤であるとの知見に基づいている。VPAとVPAから誘導される化合物のこの新規の作用機構、すなわちヒストン脱アセチル化活性を有する酵素の阻害を発見したことにより、発明者らは(1)これらの酵素は全ての細胞中に存在し、(2)酪酸塩やTSAのような本発明で述べられているものとはモデル化合物を用いた予備実験で、HDAC阻害剤が多岐にわたる細胞において分化を誘導することが示された、という2つの理由に基づき、VPAがそのHDAC阻害活性により様々な多型の癌細胞において細胞分化及び/またはアポトーシスを誘導するのに有用であろうと主張するに至った。
【0017】
様々な多型の形質転換細胞において、分化及び/またはアポトーシスを誘導する活性は、単に増殖を抑制するよりもはるかに複雑な生物活性である。単に増殖を抑制するケースでは、なぜ形質転換細胞(癌)の増殖だけが阻害されて健常細胞の増殖が阻害されないのかが明らかではない。アポトーシスや細胞分化、または脱分化した癌細胞における特異的な再分化を誘導するということは、本発明の化合物が細胞分化及び/またはアポトーシスの誘導により多岐にわたる癌において有効な効果を奏するということの理論的根拠となる。この説は、様々な多型の癌細胞において証明された(実施例参照)。
【0018】
抗てんかん性活性と鎮静化活性は、異なる機構の活性の相互作用によるものであり、よって、明らかにHDAC阻害とは異なる本来のVPA活性に起因するものである。
【0019】
肝細胞毒性を示す機構はあまり解明されておらず、VPA−CoAエステルの形成に関係しているかどうかは知られていない。しかしながら、本発明による使用、例えばHDAC阻害では、VPA−CoAエステルを形成する必要はないようである。
【0020】
米国特許No.5,672,746とWO 96/06821では、VPAとその誘導体を神経退化障害や神経増殖障害の治療に用いることが開示されている。
【0021】
(発明を実施するための最良の形態)
本発明の一つの特徴は、VPAおよびその誘導体の、ヒストン脱アセチル化活性を有する酵素の阻害剤としての使用である。VPAの誘導体は、式I;
【化2】

R1およびR2残基が異なっていれば、キラル化合物が生じる。通常、立体異性体の一つは、他方よりも強い催奇性効果を有し(Nauら, 1991, Pharmacol. Toxicol. 69, 310−321)、より催奇性の高い異性体は、より効果的にPPARδを活性する(Lampen ら, 1999)。それゆえこの異性体は、HDACsをより強く阻害することが期待され得る(本発明)。本発明は、それぞれの化合物のラセミ体の混合物、より活性の低い異性体および、特に、より活性の高い異性体を含む。
【0022】
炭化水素鎖R1およびR2は、炭化水素鎖の炭素原子の代わりに、1または数個のヘテロ原子(例えば、O,N,S)を含んでいてもよい。これはヘテロ原子が対応する炭素置換基と同じ型の混成形成型である場合、ヘテロ原子が炭素置換基の構造とよく似た構造をとり得るという事実によるものである。
R1およびR2は、置換されていてもよい。可能な置換基としては、アリール基および複素環基に加え、水酸基、アミノ基、カルボキシル基およびアルコキシ基等が挙げられる。
【0023】
好ましくは、R1およびR2は独立して、2〜10の、より好ましくは3〜10または5〜10の炭素原子を含む。また、R1およびR2は独立して、飽和しているかまたは1個の二重結合または1個の三重結合を含んでいることも好ましい。特に、側鎖(R1)の一つが2位および3位にsp1混成軌道を形成している炭素原子を含むか、または類似の構造をもたらすヘテロ原子を含み得ることが好ましい。この側鎖は3つの炭素もしくはヘテロ原子を含むべきであるが、より長い鎖はまたHDAC−阻害分子を生じさせうる。さらに、HDACタンパク質の触媒部位は様々な結合分子と適合するようなので、HDAC阻害作用を有する化合物の創製においてR2に芳香環またはヘテロ原子を含有させることが検討される。催奇性のVPA誘導体がHDAC阻害剤であるという新規知見によれば、適切な抗てんかん剤としてはこれまで無視されていた化合物もまた、本発明のもとではHDAC阻害剤であると考えられる。特に、R1がプロピニル残基で、R2が7またはそれ以上の炭素を含む残基である化合物が考えられる(Lampen ら、1999)が、これらの化合物に限定されるわけではない。
【0024】
置換基COR3は、カルボキシル基であることが好ましい。また、可能性のあるHDAC阻害活性を有する化合物の創製において、カルボキシル基の誘導体化を検討しなければならない。そのような誘導体は、ハロゲン化物(例えば塩化物)、エステルまたはアミドであってもよい。R3がアルコキシであるときは、該アルコキシ基は1〜25、好ましくは1〜10の炭素原子を含む。R3がモノ−またはジ−アルキルアミノ基であるときは、該アルキル置換基は1〜25、好ましくは1〜10の炭素原子を含む。しかし、置換されていないアミノ基が好ましい。
【0025】
本発明によれば、式Iで示される化合物の医薬上許容され得る塩もまた使用することができる。本発明によれば、人体で式Iで表される化合物に代謝される物質、または例えばエステル加水分解により式Iで表される化合物を放出させる物質も使用することができる。
【0026】
個々の実施態様として、本発明は式Iで表されるα−炭素分岐カルボン酸またはその医薬上許容され得る塩の、ヒストン脱アセチル化活性を有する酵素の阻害剤としての使用に関する。式Iで表されるα−炭素分岐カルボン酸において、R1は直鎖状または分枝状の、飽和または不飽和の炭素数5〜25の脂肪族炭化水素鎖であり、R2は独立して、直鎖状または分枝状の、飽和または不飽和の炭素数2〜25の脂肪族炭化水素鎖であるが、−CH2−CH=CH2、−CH2−C≡CHまたは−CH2−CH2−CH3ではなく、R1およびR2は任意に水酸基、アミノ基、カルボキシル基、アルコキシ基、アリール基及び/または複素環基で置換されていてもよく、R3は水酸基である。
【0027】
さらに他の実施態様として、本発明は式Iで表されるα−炭素分岐カルボン酸またはその医薬上許容される塩の、ヒストン脱アセチル化活性を有する酵素の阻害剤としての使用に関する。式Iで表されるα−炭素分岐カルボン酸において、R1は直鎖状または分枝状の、飽和または不飽和の炭素数3〜25の脂肪族炭化水素鎖であり、R2は独立して、直鎖状または分枝状の、飽和または不飽和の炭素数2〜25の脂肪族炭化水素鎖であって。R1またはR2は、炭化水素鎖において炭素原子の代わりに1または数個のヘテロ原子(例えば、O,N,S)を含んでいてもよく、R1およびR2は任意に水酸基、アミノ基、カルボキシル基、アルコキシ基、アリール基及び/または複素環基で置換されていてもよく、R3は水酸基である。
【0028】
さらに他の実施態様として、R1およびR2はエステル基(−CO−O−)を含まない。R1において、式Iで示されるカルボン酸(誘導体)のα−炭素に隣接する原子および前記α−炭素に共有結合でつながっている原子は、炭素原子であってもよい。R2において、式Iで示されるカルボン酸(誘導体)のα−炭素に隣接する原子および前記α−炭素に共有結合でつながっている原子は、炭素原子であってもよい。R1およびR2は、ヘテロ原子O、NまたはSを含まない炭化水素鎖であってもよい。
【0029】
本発明において、最も好ましく用いられる化合物は、VPA、 S−4−yn VPA、2−EHXA(2−エチルヘキサン酸)である。
【0030】
該化合物は、哺乳類(生体外検定や動物モデル系におけるセルラインの使用で)、および特にヒト(生体内外で)のヒストン脱アセチル化酵素HDAC1〜3(I類)とHDAC4〜8(II類)の阻害に有効である。
【0031】
それらの化合物は未分化癌細胞のような細胞の分化やアポトーシスを誘導するのに用いられ得る。おそらくこれは全般的な機構を示している。なぜなら形態学上の変化や特異的なマーカー遺伝子やタンパク質の発現により評価すると、F9奇形癌細胞、MT450乳癌細胞、HT−29結腸癌細胞およびいくつかの白血病セルラインにおいて分化が誘導され得るからである。更に、例えばMT450細胞では、アポトーシスが誘導され得る(実施例6参照)。
【0032】
本発明はまた、形質転換細胞の分化及び/またはアポトーシス誘導のための式Iの化合物の使用に関する。
【0033】
本発明のもう一つの特徴は、式Iの化合物を、遺伝子特異的なヒストンの低アセチル化に起因する疾病の治療薬の製造に用いることである。局所的な標準レベルよりも低いヒストンのアセチル化レベルと相関する、特定遺伝子の異常抑制に起因する疾患が多数ある。
【0034】
さらに本発明のもう一つの特徴は、式Iの化合物を、ヒストンの高アセチル化が誘導されると結果として患者の癌細胞の分化及び/またはアポトーシスが誘導され、患者の症状の臨床的改善につながるため有効である疾患の治療剤を製造するために用いることである。そのような疾患の例として、皮膚癌、エストロゲン受容体依存型および非依存型乳癌、卵巣癌、前立腺癌、腎臓癌、結腸直腸癌、膵臓癌、頭頸部癌、小細胞肺癌および非小細胞肺癌が挙げられる。また、高アセチル化誘導は、甲状腺抵抗性症候群等のヒストン脱アセチル化酵素活性の異常増に起因する疾患において、不適当な遺伝子発現を正常に戻すため、効果的である。
【0035】
該化合物およびその塩は、少なくともひとつの前述の化合物を単独で、または医薬上許容しうる担体、賦形剤及び/または希釈剤との混合で含むことを特徴とする医薬組成物(例えば粉剤、顆粒剤、錠剤、丸薬、カプセル剤、注射剤、溶液、泡状剤、浣腸剤等)として調整することが出来る。医薬組成物は、公知の方法に従って調整されてよい。患者それぞれに応じた投与量は、投与する化合物の活性、患者の年齢、体重、全体的な健康状態、性別、食事、投与時間、投与経路、排出率、薬の組合せおよび治療をうける特定の病気の重篤さを含む様々な因子によって決定される。有効成分は、例えば1日に経口または静脈注射により、体重1kgあたり約10mg〜100mgの範囲で適切な量で投与されることが好ましい。投与量は、血中濃度が0.05mM〜3mM、好ましくは約0.4mM〜1.2mMに達している限り、特に限定されるものではない。
【0036】
本発明のもう一つの特徴は、ヒストン脱アセチル化酵素阻害活性を有する物質を同定する方法であって、バルプロ酸の誘導体を提供し、そのヒストン脱アセチル化酵素阻害活性を測定し、ヒストン脱アセチル化酵素阻害活性を有する物質を選別する方法である。バルプロ酸は、ヒストン脱アセチル化酵素阻害活性を有する他の化合物を同定するための指示物質として用いることが出来る。それによって、低い投与量及び血中濃度でもHDAC阻害活性が高まり、鎮静効果のような中枢神経系への影響が低い化合物が選別され得る。他の最適なパラメーターは、肝細胞毒性の発現状況をみることである。肝毒性を減少させる化合物が選別され得る。誘導体は追加の及び/または修飾された置換基を有する化合物を合成することにより調整することができる。該HDAC阻害活性は、転写抑制検定法、ヒストンH3及び/またはヒストンH4のアセチル化検出のためのウェスタンブロット法、または酵素検定法等の最新技術により測定される。
【0037】
リプレッサー活性測定のための転写検定には、Gal4依存型リポーター遺伝子の活性化及び抑制解除が活用される。この検定法は、哺乳動物のセルライン(例えばHela、293T、CV−1)を一時的に形質導入すること、または特別に構築された不変セルラインを用いることにより行われうる。甲状腺ホルモン受容体、PPARδ、レチノイン酸受容体、N−CoR及びAML/ETO等の転写因子は、酵母Gal4タンパク質の異種DNA結合領域との融合タンパク質としてUAS要素を有するプロモーターと結合すると、転写を抑制する。Gal4融合タンパク質の非存在下では、リポーター遺伝子は高い基礎転写活性を示すが、これは他の転写因子が結合できる部位がチミジンキナーゼプロモーターに存在するためである。Gal4融合タンパク質は、この活性を140倍抑制する。HDAC阻害剤はこの抑制効果を解除するが、その解除の度合いはリポーター遺伝子活性の増加により検知(例えば、ルシフェラーゼ検定)される。
【0038】
ヒストン脱アセチル化酵素阻害剤は、N末端高アセチル化ヒストンH3及びH4の蓄積を誘導する。これらのアセチル化ヒストンは、全細胞抽出物又はヒストンH3及びH4のアセチル化N末端リジン残基に特異的な抗体を用いて、ヒストン脱アセチル化酵素阻害剤により処理を施した細胞から生産されたヒストン生産物を、ウェスタンブロット分析することにより検出することができる。
【0039】
酵素法によるHDAC活性の測定は、高アセチル化物質からの3H標識酢酸の遊離を測定して行われる。HDAC活性化源は、N−CoR(又はHDACを増加させることが知られている他のリプレッサー)又はヒストン脱アセチル化酵素を有する細胞(例えば、Hela、293T、F9)の粗抽出物に対して直接反応をおこす抗体との共免疫沈殿物であり得る。基質は、ヒストンH3又はH4のN末端に相当する、3Hで化学的に標識されたアセチル化ペプチド又はヒストンタンパク質のどちらかであり、HDAC阻害剤で処理されて代謝的に標識された細胞とは区別される。エチル酢酸で抽出後、遊離した3H標識酢酸が液体シンチレーションカウンターにより検出される。
【0040】
さらに、本発明のもう一つの特徴は、化合物のHDACへの結合及び/またはHDACへの結合の競合を測定することを特徴とする、式Iで表される化合物のHDACイソ酵素特異性を識別する方法である。
【0041】
この方法には次の段階が含まれうる:HDACは、イソ型HDAC特異的な抗体、コリプレッサー複合体に直接反応する抗体あるいは遺伝子組換え細胞において過剰発現した組換えHDACに反応する特異的な抗体のいずれかと反応して免疫沈澱する。さらにこの方法には、ウェスタンブロット分析により、これらの免疫沈澱物中に存在する個々のHDACを同定する方法が含まれていても良い。放射性同位元素で標識されたVPA又は式Iの化合物は、免疫沈澱物中に取り込まれる。取り込まれた化合物の量は、適当な洗浄段階を経た後、化合物中の放射線量を測定することにより求められる。この特徴を変化させたもの、VPA、TSA又はトラポキシン等の標識されたHDAC阻害剤との結合や式Iの化合物による結合の競合を測定して識別する方法がある。別の変化させた方法として、代替の標識方法及び/または検出方法がある。
【0042】
HDACのサブセットのみを特異的に阻害する化合物が選別されることが好ましい。
【0043】
本発明のもう一つの特徴は、ヒトまたはげっ歯類動物の一次細胞、白血性細胞、他の癌細胞や腫瘍細胞のセルラインのような細胞において上記化合物により誘導される遺伝子を特定するために、VPA又はその誘導体を用いることである。VPAにより誘導される遺伝子を特定する方法には、cDNAの長配列、発現配列タグまたはいわゆる単一遺伝子コレクションの公知スクリーニング方法が用いられる。また、サブトラクティブハイブリダイゼーション技術は、VPA又はその誘導体により誘導される遺伝子を特定するのに適している。HDAC阻害作用を利用する医薬を開発する際において、可能性のある目的物を同定するためにこれらの方法を用いること、更に適切な医薬組成物を用いて患者に治療を施すための診断方法を確定するために、これらの方法を用いることは本発明に包含される。他のHDAC阻害剤、特にトリコスタチンAのような他のHDAC阻害剤と比較して、VPAが生体内で一般的に毒性が低いことを考慮すれば、VPAにより選択的に制御を受けるかまた制御を受けない目的遺伝子を特定するためにVPA又はその誘導体を用いることは、本発明に特有の特徴である。
【0044】
また本発明は、式Iで表される化合物による治療に腫瘍が応答するかを試験することを特徴とする、腫瘍同定のための診断方法に関する。この方法には上記したVPAやその誘導体により誘導される遺伝子を特定する方法が包含されていることが好ましい。1つの特別な実施態様として、その診断方法には核酸技術、好ましくはハイブリダイゼーション又はポリメラーゼ連鎖反応の使用が含まれる。しかしながら、その他のタイプの核酸技術も用いることができる。また別の実施態様としては、検出のために異なった制御を受けるタンパク質に対する特異的な抗体を用いてよい。この目的のために、例えば組換え発現システム内で、VPA治療によりその発現が抑制された遺伝子にコードされるタンパク質が発現し、これらのタンパク質に直接反応する抗体が作られる。次いで、治療中及び/または予後判断のために、それらの抗体(又は抗体のパターン)を用いて癌または癌細胞の状態を特徴づけることが出来る。
【0045】
本発明は、多様な癌疾患を治療できるという新規な可能性を提供する。本発明者は、VPAとその誘導体が有効なHDAC阻害剤であることを見出した。これまでに知られているHDAC阻害剤は、酪酸塩のように非特異的なものか、若しくはTSAやTPXのように全ての生体に毒性を示すか、殆ど生体に用いることができない阻害剤であった。VPAは既に認可された薬剤であり、ヒトのてんかん治療のために数十年以上用いられてきたという利点を有する。よって、医薬上の許容性および重篤な副作用がないことに関する莫大な量のデータが入手可能である。ゆえにVPAは、形質転換細胞における分化及び/またはアポトーシスを誘導し、それにより癌を患っている様々な患者に有益な効果を発揮するため、ヒトに対して使用するのに適した医薬である。
【0046】
以下の実施例において、本発明を更に詳しく説明する。
【0047】
<実施例>
実施例1
PPARδ−グルココルチコイド受容体ハイブリッドタンパク質のVPAによる活性化
PPARδリガンド結合領域を活性化するためのリポーター遺伝子セルラインをCHO細胞において構築した。マウス乳癌ウルスのグルココルチコイド受容体依存性LTR−プロモーターの制御下にて、分泌型のヒト胎盤のアルカリ性ホスファターゼを発現する組換えリポーター遺伝子を導入したCHO細胞のサブクローンを使用した(Goettlicher ら、(1992) Proc. Natl. Acad. Sci. USA 89, PP. 4653−4657)。PPARδリガンド結合領域に融合したグルココルチコイド受容体のアミノ末端を有することを特徴とするハイブリッド受容体が、本質的に上述したように対応するPPARαとのハイブリッド受容体の発現に代わって、これらの細胞内において発現した(Goettlicher ら、(1992) ibd)。PPARδリガンド結合領域は、Amriらにより発表された遺伝子配列から推定される138番目のアミノ酸残基からのものが使用された(J. Biol. Chem. 270 (1995) PP. 2367−2371)。これらの細胞内におけるPPARδリガンド結合領域の活性化によって、細胞培養液上清の酵素検定法により検出可能なアルカリ性ホスファターゼリポーターの発現が誘導される。全長グルココルチコイド受容体を発現する同様の細胞が、受容体活性の特定のための対照として用いられた。
【0048】
図1に示す実験のために、PPARδハイブリッド受容体発現細胞を20%コンフルエントで24−ウェル培養皿に接種し、PPARδリガンド炭素環式プロスタグランディンI2(PGI、5μM)、VPA(1または2mM)、またはヒストン脱アセチル化酵素阻害剤である酪酸ナトリウム(0.2−5mM)およびトリコスタチンA(TSA、300nM)で40時間処理した。リポーター遺伝子活性を酵素検定法(アルカリ性ホスファターゼ)により経過測定した。酪酸塩の場合を除いて、数値は2つの独立した実験において3回測定した値をcPGI−誘導活性によって標準化した平均値±標準偏差である(図1)。PPARδリガンドcPGIとともにVPAを用いて処理した場合(P+V)のリポーター遺伝子の高い相乗的活性は、cPGIとともにトリコスタチンAを用いて処理した場合(P+T)の相乗的活性と同程度であり、トリコスタチンとともに用いた場合(T+V)または酪酸塩とともに用いた場合(データ無し)に相乗的活性化が見られないことは、VPAがPPARδへの真のリガンドのように作用しないことを示している。VPAはむしろ、コリプレッサー関連型ヒストン脱アセチル化酵素阻害剤が同様にPPARδの転写活性を誘導するのと同じ系列の事象に存する機構に基づいて、PPARδの活性に影響を及ぼす。
【0049】
実施例2
VPAによる転写抑制因子の活性化
転写因子である甲状腺ホルモン受容体(TR)、ペルオキシソーム増殖因子活性化受容体δ(PPARδ)、レチノイン酸受容体(RAR)、コリプレッサーN−CoRおよびAML/ETO融合タンパク質は、これらが酵母Gal4タンパク質の異種DNA結合領域との融合タンパク質としてUAS部位(Gal4反応要素)を有するプロモーターと結合すると、転写を抑制する。Gal4融合タンパク質非存在下において、ルシフェラーゼリポーター遺伝子は、他の転写因子が結合できる結合部位がチミジンキナーゼ (TK)プロモーター中に存在するため、高い基礎レベルで転写される。UAS TK ルシフェラーゼリポータープラスミド(Heinzel ら、 1997、Nature 387、PP. 43−48)および指標Gal4融合タンパク質発現プラスミドを、リン酸カルシウム沈殿法を用いてHela細胞に導入した。
【0050】
24時間後、培養液を交換し、ヒストン脱アセチル化酵素阻害剤と共にさらに24時間細胞を培養した。Gal4 DNA結合領域のみに関する発現プラスミドを導入した細胞を基準として、転写抑制をルシフェラーゼ活性を測定することにより評価した(図2)。Gal4融合タンパク質はこの基準値の最大140倍の抑制活性を示した。1mM濃度のVPA(治療での使用の間に到達する血清濃度に近い)はこの抑制を解除し、それがリポーター遺伝子活性を指標として示された。抑制の解除は、TRおよびPPARδのそれぞれのリガンドによる部分的活性化後に見られるのと同様に、従来のヒストン脱アセチル化酵素阻害剤(10nMトラポキシン、100nM TSA)による処理後にも見られた。リガンドとHDAC阻害剤(VPAを含む)の組合せにより相乗的効果が得られ、このことは異なる分子機構が関連していることを示唆している。
【0051】
図2は、VPAがいくつかの個別の転写因子および補助因子の活性に影響を及ぼすことを示す。この発見は、VPAが個々の転写因子または受容体に作用する(例えばリガンドとして)よりも、むしろ遺伝子発現の制御において、コリプレッサー関与型ヒストン脱アセチル化酵素のような共通の因子に作用することを示唆している。
【0052】
実施例3
VPA処理細胞中の高アセチル化ヒストンの蓄積
VPAおよび酪酸ナトリウム(NaBu)またはトリコスタチンA(TSA)のような従来のヒストン脱アセチル化酵素阻害剤は、高アセチル化ヒストンH3およびH4の蓄積を誘導する。適当に処理された細胞の抽出物をウェスタンブロット分析することにより、これらのアセチル化ヒストンを検知し得る。図3は代表的な実験からの前述の分析による結果を示す。この実験で、VPAにより誘導された高アセチル化の時間的経過による推移(A)および必要なVPAの濃度(B)を調べた。
【0053】
(A) 時間的経過分析のために、指示された分析時間の30時間前にF9細胞を6−ウェル培養皿に接種した。分析前の指示された時間に、個々の培養細胞に10倍濃度のVPAまたはトリコスタチンA貯蔵溶液を含む培地を添加した。細胞培養液を氷冷したリン酸塩緩衝生理食塩水で2度洗浄をし、変性SDSゲル電気泳動法にかけるために250μl試料緩衝液に溶解して全細胞抽出液を作成した。集めた試料のDNAは音波処理によりせん断され、試料は15%変性ポリアクリルアミドゲル上で分離された。市場で入手可能なアセチル化された型のヒストンに特異的な抗体(Ac−H3、Cat−Nr、: 06−599;Ac−H4、 Cat−Nr、: 06−598)(Upstate Biotechnology社製)を用いて、ウェスタンブロット分析法によりアセチル化されたH3およびH4を検知した。ポリアクリルアミドの一部がクーマシーブルーで染色されることによってレーンの等電点を確認した。
【0054】
(B)VPA必要量を決定するために、設定した濃度のVPA添加前にF9細胞を6−ウェル培養皿で8時間培養した。上述した処理の16時間後に全細胞抽出液を調製した。アセチル化ヒストンH3およびH4の分析を(A)に述べた方法で行った。VPA濃度がVPAをヒトの抗てんかん剤として治療に使用する間に到達する血清濃度の範囲内の時、ヒストンH3およびH4の高アセチル化が誘導される。VPAは、てんかん治療を目的とした血清濃度をほんの僅か上回る血清濃度で、最大の効果を有することが予想される濃度で使用された酪酸ナトリウムまたはトリコスタチンAと同等の効力のヒストン高アセチル化を誘導する。この実験はVPAまたはF9細胞中で産生した代謝産物がヒストン脱アセチル化酵素活性を阻害することを示している。
【0055】
実施例4
VPAおよびその誘導体が、in vitroにおいてヒストン脱アセチル化酵素活性を阻害する。
コリプレッサーN−CoRまたはmSin3に対する抗体を用いて得られた全細胞抽出液からの免疫沈殿物が、ヒストン脱アセチル化酵素活性を抑制する。ヒストンが3H−酢酸塩の存在下で高アセチル化された細胞から得られる放射性アセチル化ヒストン基質とともに、該免疫沈殿物を培養することによって、この酵素活性は測定される。酢酸エチルによる抽出およびそれに次ぐ液体シンチレーションカウンターによって、3H−酢酸塩の遊離を検出して酵素活性の値とした(図4)。in vitroにおいて、ヒストン脱アセチル化酵素阻害剤であるトリコスタチンA(TSA, 10−7M)を反応に加えると、酵素活性が強く阻害された。VPA(左から右へ0.2mM、1mM、5mM)および関連化合物であるヘキサン酸エチル(EHXA、左から右へ0.008mM、0.04mM、0.2mM、1mM、5mM)、R−4−yn VPA(左から右へ0.2mM、1mM、5mM)およびS−4−yn VPA(左から右へ0.2mM、1mM、5mM)のHDAC阻害活性試験を行った。検定は293T細胞のN−CoR免疫沈殿物を用いて、2回ずつ行われた。免疫沈殿物は、基質の添加前にHDAC阻害剤で15分間前処理し、その後37℃で2.5時間培養した(未処理区の酵素活性2,205cpm=100%とする)。前免疫血清の沈殿物を対照とした。EC50値は、VPAに対して0.6mM, EHXAに対して0.2mMおよび S−4−yn VPAに対して0.3mMであるが、S−4−yn VPA立体異性体には活性がなかった。これらのデータは、細胞内で産生された代謝産物というよりはむしろVPAそのものが、ヒストン脱アセチル化酵素活性を阻害することを示す。
【0056】
実施例5
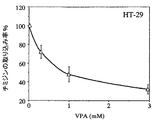
F9奇形癌、HT−29結腸癌およびRenCa腎癌各細胞における細胞分化の誘導
ヒストン脱アセチル化酵素阻害剤および特にVPAが、脱分化した発癌性細胞の分化を誘導する。細胞分化は細胞周期阻害、形態変化および異なった表現型のマーカーの発現状況と関連がある。F9およびHT−29の細胞形態的な変化は顕微鏡観察によって評価した。分化のパラメーターの1つである細胞周期阻害は、F9奇形癌、エストロゲン非依存性MT−450乳癌およびHT−29結腸癌細胞において、3H−チミジンの培養細胞への取り込みの減少によって示された。1mM VPAの存在下または非存在下で、F9およびHT−29細胞を96ウェル培養皿で36時間培養した。37kBqの3H−チミジンを添加し、更に12時間培養した。MT−450を72時間培養し、3H−チミジン標識を1時間行った。自動細胞採取および液体シンチレーションカウンターにより、DNAへの3H−チミジンの取り込みを測定した。VPA前処理によってチミジンの取り込み率が、F9、 MT−450およびHT−29細胞においてそれぞれ48±5%、63±8%、および52±8%低下した。HT−29細胞へのチミジン取り込み低下率の用量−反応関係(図5A)を同様の実験方法により測定した。さらにF9奇形癌細胞において細胞分化マーカーの誘導状況を示した(図5B)。
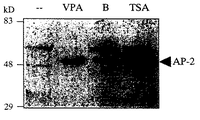
【0057】
F9奇形癌細胞をVPA(1mM)、酪酸ナトリウム(B、1mM)およびトリコスタチンA(TSA、 30nM)で48時間処理した。形態的判定基準、細胞数増加率の減少(例えば細胞周期阻害、データは示さず)、12時間のパルス標識中における3H−チミジンの取り込み率48%低下(上記参照)、およびF9細胞においてヒストン脱アセチル化酵素阻害により誘導される分化の特異的なマーカーである核AP−2タンパク質の出現状況(図5B)によって、分化は追跡調査された。核AP−2タンパク質は、処理または未処理のF9細胞を穏やかな洗剤で溶解し(25mM Tris, pH7.5 ; 1mM EDTA, 0.05%NP40)、遠心分離(3000×g, 5min)で核分画を回収し、変性SDSゲル電気泳動分析に供するためにサンプル緩衝液に溶解することにより作成された核抽出物中で検出された。核抽出物は9%SDSポリアクリルアミドゲル上で分離した。3%脱脂粉乳および0.05%Tween20を含むトリス緩衝生理食塩水で1/1000倍に希釈されたラビットポリクローナル抗体(Santa Cruz社製、Cat. No.: SC−184)を用いたウェスタンブロット分析によってAP−2タンパク質を検出した。VPAおよびトリコスタチンAの両者は核AP−2タンパク質を誘導するが、酪酸塩はここで選ばれた濃度においては活性が弱かった。AP−2の出現がVPA処理の36〜40時間後にのみ検出される遅発効果性であることから、酪酸塩の活性が弱いのは、化合物の効率的な代謝のためである可能性がある。そうとは言え、他のヒストン脱アセチル化酵素阻害剤による分化と区別できない程度に、VPAは上皮F9セルラインの分化を誘導した 。
【0058】
VPAによるRenCa−LacZ細胞の分化誘導を細胞形態学的変化から測定した。1mM VPA存在下または非存在下で、RenCa−LacZ細胞を36時間培養した。位相差顕微鏡により形態的変化を観察し代表的な領域の顕微鏡写真を撮影した(図5C)。
【0059】
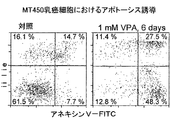
実施例6
MT−450乳癌細胞のアポトーシス誘導
1mM VPAのA存在下または非存在下で、MT−450細胞を72時間培養した。メーカーの取扱説明書に従い、ホスファチジルセリン処理された細胞表面をFITC−接合アネキシンV(Becton Dickinson社製)で染色した後、フローサイトメトリー分析法によりアポトーシス細胞を検出した。プロピジウムイオダイド(propidium iodide)染色により死滅細胞を除いた。アネキシンVに対して陽性およびプロピジウムイオダイドに対して陰性の細胞(図6の右下4区画部分)を判別し、アポトーシスを起こした細胞としてカウントした。
【0060】
実施例7
バルプロ酸処理による生存腫瘍細胞の死滅(MTTテスト)
セルラインおよび細胞培養
ヒトMDA−MB468、MDA−MB453およびSKBR3乳癌細胞、A431扁平上皮細胞癌細胞、およびSKOV3卵巣癌細胞を、加熱により不活化した10%胎児ウシ血清(FBS)、2mM L−グルタミン、100units/mlペニシリン、および100μg/mlストレプトマイシンで補完されたダルベッコ修正イーグル培地(DMEM、 BioWhittaker社、 ベルビエ、 ベルギー)中で継代培養した。上記のように補完したRPMI培地中でヒトMCF乳癌細胞を増殖させた。
【0061】
E.coli由来β−ガラクトシダーゼをコードするプラスミドpZeoSV2/lacZを腎細胞癌細胞(Renca)に安定的に導入した細胞(Renca−lacZ 細胞) (Maurer−Gebhard ら、Cancer Res. 58:2661−2666、1998)を、8%FBS、2mM L−グルタミン、100units/mlペニシリン、100μg/mlストレプトマイシン、0.25mg/mlゼオシンで補完されたRPMI−1640培地中で増殖させた。c−erbB2およびネオマイシン耐性をコードするプラスミドpSV2ErbB2NとpSV2neoとを同時に導入したRenca−lacZ 細胞(Renca−lacZ/ErbB2)(Maurer−Gebhard ら、Cancer Res. 58:2661、1998)、または上皮細胞増殖因子(EGF)受容体、発癌性活性型EGF受容体変異株EGFRvIII、およびネオマイシン耐性をコードするプラスミドPLTR−EGFR又はPLTR−EGFRvIIIとpSV2neo(Schmidt ら、Oncogene 18: 1711−1721、1999)とを同時に導入したRenca−lacZ 細胞(Renca−lacZ/EGFRおよびRenca−lacZ/EGFRvIII)を、さらに0.48mg/ml G418を含む同様の培地にて増殖させた。
【0062】
細胞生存率検定
通常培地に、細胞数が1×104個/ウェルとなるように、腫瘍細胞を96ウェル培養皿に接種した。バルプロ酸を最終濃度1または3mMになるようにサンプル(それぞれの濃度について3サンプル)に添加し、細胞を40時間(Renca−lacZ、Renca−lacZ/ErbB2、 Renca−lacZ/EGFR、Renca−lacZ/EGFRvIII、SKBR3およびSKOV3細胞)または70時間(A431、MCF7、MDA−MB453およびMDA−MB468細胞)培養した。バルプロ酸非存在下で対照細胞を培養した。3−(4,5−ジメチルチアゾール−2−yl)−2,5ジフェニルテトラゾリウムブロマイド(MTT) (Sigma社、 ダイゼンホッフェン、ドイツ国)濃度が10mg/mlであるPBS溶液10μlを各ウェルに添加し、さらに3時間細胞を培養した。90μlの溶解緩衝液(20%SDSを含有する50%ジメチルホルムアミド溶液、pH 4.7)を添加して細胞を溶解した。ホルマザンを溶解後、590nmでの吸光度をマイクロプレートリーダー(Dynatech社、 デンケンドルフ、ドイツ国)で読み取り、バルプロ酸無添加で培養した細胞と比較した生細胞の相対数を算出した。
【0063】
結果
図7に示された結果より、バルプロ酸が、濃度に依存してErbB2またはEGF受容体癌源遺伝子または腫瘍原性活性型EGF受容体変異株EGFRvIIIをハイレベルで発現する乳癌細胞、卵巣癌細胞、扁平上皮癌細胞、腎細胞癌細胞の生存率を低減させることが分かった。これらの結果は、バルプロ酸が、上皮起源の固形癌由来の多種多様な腫瘍細胞の数及び/または生存率を著しく低減することを示している。生存率の低下により細胞分化の誘導及び/または細胞死の誘導に基づく細胞数の減少を示唆できた。細胞の形態的変化が見られたことは、細胞分化が少なくともある程度の影響を与えたことを示唆している。この分化の誘導及び/または細胞死の誘導により、バルプロ酸およびその誘導体がこのような腫瘍の治療に使用が可能であることが示唆された。
【0064】
実施例8
バルプロ酸処理後のヒト癌培養細胞の細胞バイオマスの低減
VPAが一連のヒト癌細胞の分化及び/または細胞死を誘導し、ヒト癌培養細胞の全細胞バイオマスを低減させる。バイオマスの低減により、細胞周期阻害に関連した細胞死及び/または分化による細胞数低減を示唆することが出来た。定量的パラメーター、例えばバイオマスの低減を30のヒト癌セルライン(図8e)について測定し、その結果を12例の用量−反応曲線、例えばBT − 549乳癌細胞(1)、エストロゲン依存ZR − 75乳癌細胞(2)、DSM − 114小細胞肺癌細胞(3)、NCI − H226非小細胞肺癌細胞(4)、SK − MEL − 28皮膚癌細胞(5)、OVCAR − 3卵巣癌細胞(6)、HUP − T3膵臓癌細胞(7)、DU − 145前立腺癌細胞(8)、DETROIT − 562頭頸部癌細胞(9)、LS − 174大腸癌細胞(10)、A − 172脳癌細胞(11)、およびHL − 60白血病細胞(12)(図8a〜d)に示す。すべての細胞について細胞死及び/または細胞分化の形態的兆候を評価した。試験した中で最高のVPA濃度値では、すべての培養細胞で死滅細胞数の増加が見られ、SW−1116大腸癌細胞(図8e)等のいくつかの培養細胞においては、既に1mMの濃度で、試験中にほとんどの細胞が死滅していった。PC − 3(図8e)およびDU − 145(図8c)細胞は、それら本来の丸型形態から長い繊維芽細胞型に変化する。またU87MG(図8e)細胞は長さが伸び、蜘蛛のような糸状の伸びを発達させる。
【0065】
パネル1〜9(図8a〜c)の細胞を、細胞数が1ウェル当り3000〜8000個になるように96ウェル培養皿に接種した。24時間の回復培養後、指示された濃度のVPA存在下又は非存在下にて、48時間細胞を培養した。培養細胞を、各ウェルの培養培地の上に50%冷TCAを50μl添加してTCAで固定し、TCAの最終濃度を10%とした。4℃にて1時間培養後、細胞を水道水で5回水洗して引き続き風乾した。固定細胞を1%酢酸に溶解した0.4%(wt/vol)スルフォホダミン(Sulforhodamine)Bで30分間染色し、次いで1%酢酸で4回洗浄して遊離の染料を除去した。風乾後、旋回シェーカーに5分間かけて、吸着した染料を10mM非緩衝トリス塩基(pH10.5)で溶解した。Titertec Multiskan Plusプレートリーダー上で、550nm単一波長における吸光度を読み取った。細胞当たり、各々の用量−反応関係のために6個のテストウェルを2連で設定し、12個の対照ウェルを同時に設定した。テスト皿に薬剤を添加する直前に、TSAで固定した細胞が入った12個の参照ウェルの経過時間0(T0;薬剤が添加された時間)の細胞群密度を測定した。上記したように固定および染色された5%FBS完全培地のバックグラウンド吸光度も、12個のそれぞれ離れたウェルにて測定した。
【0066】
未処理吸光度データからバックグラウンド吸光度データ(すなわち、完全培地プラス染料の吸光度およびT0における細胞の吸光度)を差し引いて、細胞の全細胞バイオマスの減少値とした。
【0067】
パネル10〜12(図8d)の細胞を、指示された濃度のVPAの非存在下又は存在下で、指示どおり36〜50時間96ウェル皿にて培養した。37kBqの3H−チミジンを添加し、さらに12時間培養した。3H−チミジンのDNAへの組み込みを、自動細胞採取装置および液体シンチレーションカウンターによって判定した。
【0068】
図8a〜dのグラフに、6回の測定値から算出した平均値±標準偏差の値を示す。
【0069】
加えて、さらなる臓器由来の癌細胞を、図8a〜cにて記載した実験と同様の方法でバルプロ酸処理した。図8eに、1mMのVPA処理による様々なヒト癌細胞の全細胞バイオマスの減少についてまとめた。この減少により細胞周期阻害及び/または細胞死の誘導が起こることが示唆される。細胞をVPAで48時間処理した。2連で行った6個の反応テストから阻害の程度を算出し、細胞バイオマスの減少を未処理細胞に対する%として、標準偏差で表した。
【図面の簡単な説明】
【図1】PPARδのヒストン脱アセチル化酵素阻害剤と同様のVPAによる活性を示す(実施例1)。
【図2】VPAがPPARδ以外にもいくつかの転写因子を活性化することを示す(実施例2)。
【図3】VPAにより誘導された高アセチル化ヒストンH3及びH4の蓄積量を示す(実施例3)。
【図4】VPA存在下又は非存在下におけるヒストン脱アセチル化酵素活性の生化学的分析値を示す(実施例4)。
【図5A〜図5C】HT−29結腸癌細胞、F9奇形癌細胞およびRenCa腎癌細胞における、VPAにより誘導された細胞分化の指標を示す。VPAまたはヒストン脱アセチル化酵素トリコスタチンAによって分化したF9奇形癌細胞の表現型には相同性がある(実施例5)。
【図6】MT450乳癌細胞におけるアポトーシス誘導を示す(実施例6)。
【図7】バルプロ酸処理後の生細胞数の減少率を示す。(A)Renca−lacZ、Renca−lacZ/EGFR、Renca−lacZ/EGFRvIII及びRenca−lacZ/ErbB2腎癌細胞または(B)SKOV3卵巣癌細胞、SKBR3、MCF7、MDA−MB453及びMDA−MB468乳癌細胞及びA431扁平上皮細胞癌細胞は、指定濃度のバルプロ酸とともに培養される。生細胞数の相対値は、実施例7に記載された細胞内代謝活性を測定するMTT酵素法により求められる。それぞれの点は、3回試験を繰り返すことにより求められたデータの平均値を表している(実施例7)。
【図8a〜図8e】培養細胞をVPA処理した後の細胞バイオマスの減少を示す。
Claims (23)
- 式Iで表される化合物
- R1およびR2が独立して、1個の二重結合または三重結合を含んでいてもよい直鎖状または分岐状の炭素数2〜10の炭化水素鎖であることを特徴とする請求項1に記載の使用。
- 前記化合物がバルプロ酸(VPA)、S−4−yn VPA、2−EHXA(2−エチルヘキサン酸)およびそれらの医薬上許容される塩から選ばれることを特徴とする請求項1または2に記載の使用。
- ヒストン脱アセチル化活性を有する前記酵素が、哺乳類の、好ましくはヒトのヒストン脱アセチル化酵素であることを特徴とする請求項1〜3のいずれかに記載の使用。
- 前記ヒトのヒストン脱アセチル化酵素が、HDAC1〜HDAC8から選ばれることを特徴とする請求項4に記載の使用。
- 前記化合物が細胞の分化誘導に用いられることを特徴とする請求項1〜5のいずれかに記載の使用。
- 前記化合物が形質転換細胞の分化誘導に用いられることを特徴とする請求項6に記載の使用。
- 前記化合物が形質転換細胞のアポトーシス誘導に用いられることを特徴とする請求項1〜5のいずれかに記載の使用。
- ヒストンの高アセチル化誘導が有効である疾病の治療剤を製造するための請求項1〜3のいずれかに記載の化合物の使用。
- 患者の体内で請求項1〜3のいずれかに記載の化合物へと代謝される物質の、ヒストンの高アセチル化誘導が有効である疾病の治療剤を製造するための使用。
- 前記疾病が皮膚癌、エストロゲン受容体依存型および非依存型乳癌、卵巣癌、前立腺癌、腎臓癌、結腸直腸癌、膵臓癌、頭頸部癌、小細胞肺癌および非小細胞肺癌、および甲状腺抵抗性症候群等のヒストン脱アセチル化酵素の異常増に起因する内分泌性疾患であることを特徴とする請求項9または10に記載の使用。
- 請求項1〜3のいずれかに記載の化合物を提供し、該化合物の誘導体のヒストン脱アセチル化酵素阻害活性を測定し、ヒストン脱アセチル化酵素阻害活性を有する物質を選別することを特徴とするヒストン脱アセチル化酵素阻害活性を有する物質の同定方法。
- 前記方法が、化合物の鎮静効果を評価する段階、およびヒストン脱アセチル化酵素阻害活性およびバルプロ酸より低い鎮静効果を有する化合物を選別する段階を含むことを特徴とする請求項12に記載の方法。
- 前記ヒストン脱アセチル化酵素阻害活性が、転写抑制検定法、ヒストンH3またはH4のアセチル化検出のためのウェスタンブロット法、または脱アセチル化酵素検定法によって測定されることを特徴とする請求項12または13に記載の方法。
- 化合物のHDACへの結合及び/またはHDACへの結合の競合を測定することを特徴とする請求項1〜3のいずれかに記載の化合物のHDACイソ酵素特異性を識別する方法。
- HDACのサブセットのみを特異的に阻害する化合物を選別することを特徴とする請求項15に記載の方法。
- 実質的に同一な2つの細胞群を提供し、該細胞群の1つをVPAまたはその誘導体に接触させ、VPAまたはその誘導体と接触した該細胞群において、VPAまたはその誘導体と接触していない方の細胞群よりも有意に高いレベルで発現する遺伝子または遺伝子産物を検出することを特徴とする、バルプロ酸またはその誘導体に誘導される遺伝子の同定方法。
- サブトラクティブハイブリダイゼーション、またはcDNAサンプルの配列、発現配列タグまたは単一遺伝子コレクションのスクリーニング方法が用いられることを特徴とする請求項17に記載の方法。
- 請求項1〜3に記載の化合物を用いた治療に腫瘍が応答するかを試験する段階を含むことを特徴とする腫瘍同定のための診断方法。
- 請求項17または18に記載の方法を含むことを特徴とする請求項19に記載の診断方法。
- 核酸技術の使用を含むことを特徴とする請求項19または20に記載の診断方法。
- 検出に、ハイブリダイゼーションまたはポリメラーゼ連鎖反応が用いられることを特徴とする請求項21に記載の診断方法。
- 検出に、それぞれ異なった制御を受けるタンパク質に対する特異的抗体が用いられることを特徴とする請求項19または20に記載の診断方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP00114088A EP1170008A1 (en) | 2000-07-07 | 2000-07-07 | Valproic acid and derivatives thereof as histone deacetylase inhibitors |
| PCT/EP2001/007704 WO2002007722A2 (en) | 2000-07-07 | 2001-07-05 | Valproic acid and derivatives thereof as histone deacetylase inhibitors |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004504347A true JP2004504347A (ja) | 2004-02-12 |
| JP2004504347A5 JP2004504347A5 (ja) | 2005-02-03 |
Family
ID=8169134
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002513458A Ceased JP2004504347A (ja) | 2000-07-07 | 2001-07-05 | ヒストン脱アセチル化酵素阻害剤としてのバルプロ酸およびその誘導体 |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US7265154B2 (ja) |
| EP (2) | EP1170008A1 (ja) |
| JP (1) | JP2004504347A (ja) |
| AT (1) | ATE295725T1 (ja) |
| AU (2) | AU2001283925B2 (ja) |
| CA (1) | CA2414967C (ja) |
| DE (1) | DE60110912T2 (ja) |
| ES (1) | ES2239677T3 (ja) |
| PT (1) | PT1301184E (ja) |
| WO (1) | WO2002007722A2 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005506348A (ja) * | 2001-10-16 | 2005-03-03 | スローン − ケタリング・インスティテュート・フォー・キャンサー・リサーチ | 神経変性疾患および脳の癌の処置 |
| JP2007535518A (ja) * | 2004-04-30 | 2007-12-06 | トポターゲット ジャーマニィ アーゲー | ヒストンデアセチラーゼ阻害剤を含んで成る製剤 |
| JP2010511597A (ja) * | 2006-12-06 | 2010-04-15 | 北海道公立大学法人 札幌医科大学 | ヒストン脱アセチル化酵素阻害活性を有する物質を用いた細胞性免疫増強剤 |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1170008A1 (en) | 2000-07-07 | 2002-01-09 | Chemotherapeutisches Forschungsinstitut Georg-Speyer-Haus | Valproic acid and derivatives thereof as histone deacetylase inhibitors |
| GB0024362D0 (en) * | 2000-10-05 | 2000-11-22 | Glaxo Group Ltd | Medicaments |
| WO2003018767A2 (en) * | 2001-08-27 | 2003-03-06 | Advanced Cell Technology, Inc. | Trans-differentiation and re-differentiation of somatic cells and production of cells for cell therapies |
| EP1293205A1 (en) * | 2001-09-18 | 2003-03-19 | G2M Cancer Drugs AG | Valproic acid and derivatives thereof for the combination therapy of human cancers, for the treatment of tumour metastasis and minimal residual disease |
| US6706686B2 (en) | 2001-09-27 | 2004-03-16 | The Regents Of The University Of Colorado | Inhibition of histone deacetylase as a treatment for cardiac hypertrophy |
| US7566535B2 (en) | 2002-03-07 | 2009-07-28 | University Of Delaware | Enhanced oligonucleotide-mediated nucleic acid sequence alteration |
| IL149404A0 (en) * | 2002-04-29 | 2002-11-10 | Yissum Res Dev Co | METHODS AND COMPOSITIONS FOR MODULATING β-CATENIN PHOSPHORYLATION |
| GB0226855D0 (en) | 2002-11-18 | 2002-12-24 | Queen Mary & Westfield College | Histone deacetylase inhibitors |
| JP2006509010A (ja) * | 2002-12-05 | 2006-03-16 | インペリアル・カレッジ・イノベイションズ・リミテッド | アポトーシスの制御 |
| US7244751B2 (en) | 2003-02-14 | 2007-07-17 | Shenzhen Chipscreen Biosciences Ltd. | Histone deacetylase inhibitors of novel benzamide derivatives with potent differentiation and anti-proliferation activity |
| KR20050122210A (ko) * | 2003-03-17 | 2005-12-28 | 다케다 샌디에고, 인코포레이티드 | 히스톤 탈아세틸화 효소 억제제 |
| EP1491188A1 (en) * | 2003-06-25 | 2004-12-29 | G2M Cancer Drugs AG | Topical use of valproic acid for the prevention or treatment of skin disorders |
| WO2005066151A2 (en) * | 2003-12-19 | 2005-07-21 | Takeda San Diego, Inc. | Histone deacetylase inhibitors |
| US20050159470A1 (en) * | 2003-12-19 | 2005-07-21 | Syrrx, Inc. | Histone deacetylase inhibitors |
| KR100659779B1 (ko) | 2004-09-04 | 2006-12-19 | 주식회사 바이넥스 | 히스톤 디아세틸라제 억제제를 함유하는 중간엽줄기세포의 골분화유도용 조성물 및 이를 이용한 골분화증가방법 |
| JP2008524246A (ja) * | 2004-12-16 | 2008-07-10 | タケダ サン ディエゴ インコーポレイテッド | ヒストンデアセチラーゼ阻害剤 |
| JP2008540574A (ja) * | 2005-05-11 | 2008-11-20 | タケダ サン ディエゴ インコーポレイテッド | ヒストンデアセチラーゼ阻害剤 |
| CN101263121A (zh) | 2005-07-14 | 2008-09-10 | 塔克达圣地亚哥公司 | 组蛋白脱乙酰基酶抑制剂 |
| JP2009525955A (ja) * | 2006-01-13 | 2009-07-16 | タケダ サン ディエゴ インコーポレイテッド | ヒストンデアセチラーゼ阻害剤 |
| WO2008011603A2 (en) * | 2006-07-20 | 2008-01-24 | Wisconsin Alumni Research Foundation | Modulating notch1 signaling pathway for treating neuroendocrine tumors |
| WO2008027837A2 (en) * | 2006-08-28 | 2008-03-06 | The Regents Of The University Of California | Small molecule potentiator of hormonal therapy for breast cancer |
| GB0620823D0 (en) | 2006-10-19 | 2006-11-29 | Univ London | Histone deacetylase inhibitors |
| US20090023186A1 (en) * | 2007-07-22 | 2009-01-22 | Excellgene Sa | Use of valproic acid for enhancing production of recombinant proteins in mammalian cells |
| CN101970399A (zh) * | 2007-12-14 | 2011-02-09 | 乔治敦大学 | 组蛋白脱乙酰酶抑制剂 |
| WO2010011700A2 (en) | 2008-07-23 | 2010-01-28 | The Brigham And Women's Hospital, Inc. | Treatment of cancers characterized by chromosomal rearrangement of the nut gene |
| ES2473792T3 (es) | 2009-04-03 | 2014-07-07 | Naturewise Biotech & Medicals Corporation | Compuestos cin�micos y derivados de los mismos para la inhibición de la histona desacetilasa |
| US7994357B2 (en) | 2009-04-03 | 2011-08-09 | Naturewise Biotech & Medicals Corporation | Cinamic compounds and derivatives therefrom for the inhibition of histone deacetylase |
| EP2277387B1 (en) | 2009-07-22 | 2016-10-19 | NatureWise Biotech & Medicals Corporation | New use of histone deacetylase inhibitors in changing mrjp3 protein in royal jelly |
| CN102575236B (zh) * | 2009-09-07 | 2014-10-01 | 贾瓦哈拉尔尼赫鲁高级科学研究中心 | 利用ctk7a抑制组蛋白乙酰转移酶及其方法 |
| US8921533B2 (en) | 2011-07-25 | 2014-12-30 | Chromatin Technologies | Glycosylated valproic acid analogs and uses thereof |
| US10184933B2 (en) | 2013-10-01 | 2019-01-22 | The J. David Gladstone Industries | Compositions, systems and methods for gene expression noise drug screening and uses thereof |
| RU2016146099A (ru) * | 2014-05-02 | 2018-06-05 | Онкоэтикс Гмбх | Способ лечения острого миелоидного лейкоза и/или острого лимфобластного лейкоза с помощью тиенотриазолодиазепиновых соединений |
| CN105622712B (zh) * | 2014-10-27 | 2019-08-27 | 天津金耀集团有限公司 | 一种甾体3-酮-4-烯物合成工艺 |
| TWI794171B (zh) | 2016-05-11 | 2023-03-01 | 美商滬亞生物國際有限公司 | Hdac抑制劑與pd-l1抑制劑之組合治療 |
| US20210386787A1 (en) * | 2018-10-12 | 2021-12-16 | Life Technologies Corporation | Hematopoietic Stem and Progenitor Cell Expansion System |
| CN111358777A (zh) * | 2020-02-24 | 2020-07-03 | 军事科学院军事医学研究院环境医学与作业医学研究所 | 一种提高外周血单个核细胞线粒体功能的方法和应用 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998029114A1 (en) * | 1996-12-30 | 1998-07-09 | Bar-Ilan University | Tricarboxylic acid-containing oxyalkyl esters and uses thereof |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5672746A (en) * | 1994-08-30 | 1997-09-30 | American Biogenetic Sciences, Inc. | Antiproliferative and neurotrophic molecules |
| US6300373B1 (en) * | 1993-09-10 | 2001-10-09 | American Biogenetic Sciences, Inc. | Antiproliferative and neurotrophic molecules |
| RO115498B1 (ro) * | 1993-10-29 | 2000-03-30 | Univ Boston | Metoda de tratament a neoplasmului |
| AU712801B2 (en) * | 1995-09-20 | 1999-11-18 | Merck Sharp & Dohme Corp. | Histone deacetylase as target for antiprotozoal agents |
| US6777217B1 (en) * | 1996-03-26 | 2004-08-17 | President And Fellows Of Harvard College | Histone deacetylases, and uses related thereto |
| WO1997047307A1 (en) * | 1996-06-14 | 1997-12-18 | The Uab Research Foundation | Use of histone deacetylase inhibitors to activate transgene expression |
| US6387673B1 (en) * | 1997-05-01 | 2002-05-14 | The Salk Institute For Biological Studies | Compounds useful for the modulation of processes mediated by nuclear hormone receptors, methods for the identification and use of such compounds |
| AU1395999A (en) * | 1997-11-10 | 1999-05-31 | Salk Institute For Biological Studies, The | Methods for the use of inhibitors of co-repressors for the treatment of neoplastic diseases |
| US6262116B1 (en) * | 1998-01-23 | 2001-07-17 | Sloan-Kettering Institute For Cancer Research | Transcription therapy for cancers |
| EP1170008A1 (en) | 2000-07-07 | 2002-01-09 | Chemotherapeutisches Forschungsinstitut Georg-Speyer-Haus | Valproic acid and derivatives thereof as histone deacetylase inhibitors |
-
2000
- 2000-07-07 EP EP00114088A patent/EP1170008A1/en not_active Withdrawn
-
2001
- 2001-07-05 DE DE60110912T patent/DE60110912T2/de not_active Expired - Lifetime
- 2001-07-05 US US10/332,353 patent/US7265154B2/en not_active Expired - Fee Related
- 2001-07-05 EP EP01962831A patent/EP1301184B1/en not_active Expired - Lifetime
- 2001-07-05 PT PT01962831T patent/PT1301184E/pt unknown
- 2001-07-05 AT AT01962831T patent/ATE295725T1/de active
- 2001-07-05 CA CA002414967A patent/CA2414967C/en not_active Expired - Fee Related
- 2001-07-05 ES ES01962831T patent/ES2239677T3/es not_active Expired - Lifetime
- 2001-07-05 AU AU2001283925A patent/AU2001283925B2/en not_active Ceased
- 2001-07-05 AU AU8392501A patent/AU8392501A/xx active Pending
- 2001-07-05 WO PCT/EP2001/007704 patent/WO2002007722A2/en active IP Right Grant
- 2001-07-05 JP JP2002513458A patent/JP2004504347A/ja not_active Ceased
-
2007
- 2007-01-17 US US11/653,855 patent/US20070232696A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998029114A1 (en) * | 1996-12-30 | 1998-07-09 | Bar-Ilan University | Tricarboxylic acid-containing oxyalkyl esters and uses thereof |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005506348A (ja) * | 2001-10-16 | 2005-03-03 | スローン − ケタリング・インスティテュート・フォー・キャンサー・リサーチ | 神経変性疾患および脳の癌の処置 |
| JP4638148B2 (ja) * | 2001-10-16 | 2011-02-23 | スローン − ケタリング・インスティテュート・フォー・キャンサー・リサーチ | 神経変性疾患および脳の癌の処置 |
| JP2007535518A (ja) * | 2004-04-30 | 2007-12-06 | トポターゲット ジャーマニィ アーゲー | ヒストンデアセチラーゼ阻害剤を含んで成る製剤 |
| JP2010511597A (ja) * | 2006-12-06 | 2010-04-15 | 北海道公立大学法人 札幌医科大学 | ヒストン脱アセチル化酵素阻害活性を有する物質を用いた細胞性免疫増強剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1301184A2 (en) | 2003-04-16 |
| EP1301184B1 (en) | 2005-05-18 |
| US20040087652A1 (en) | 2004-05-06 |
| AU8392501A (en) | 2002-02-05 |
| ATE295725T1 (de) | 2005-06-15 |
| PT1301184E (pt) | 2005-07-29 |
| EP1170008A1 (en) | 2002-01-09 |
| DE60110912D1 (de) | 2005-06-23 |
| AU2001283925B2 (en) | 2005-07-14 |
| CA2414967A1 (en) | 2002-01-31 |
| CA2414967C (en) | 2009-08-25 |
| WO2002007722A2 (en) | 2002-01-31 |
| US20070232696A1 (en) | 2007-10-04 |
| WO2002007722A3 (en) | 2002-07-18 |
| ES2239677T3 (es) | 2005-10-01 |
| DE60110912T2 (de) | 2005-10-20 |
| US7265154B2 (en) | 2007-09-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2004504347A (ja) | ヒストン脱アセチル化酵素阻害剤としてのバルプロ酸およびその誘導体 | |
| AU2001283925A1 (en) | Valproic acid and derivatives thereof as histone deacetylase inhibitors | |
| US7550490B2 (en) | Benzamide derivatives as histone deacetylase inhibitors with potent differentiation and anti-proliferation activity | |
| Clay et al. | Influence of J series prostaglandins on apoptosis and tumorigenesis of breast cancer cells | |
| Nagai et al. | Significance of the transcription factor KLF5 in cardiovascular remodeling | |
| JP2005512961A (ja) | ヒト癌の組合せ療法的処置、及び腫瘍転移並びに微小残存病変処置のためのバルプロ酸及びその誘導体 | |
| Nagpal et al. | Retinoid antagonism of NF-IL6: insight into the mechanism of antiproliferative effects of retinoids in Kaposi’s sarcoma | |
| Matsushima-Nishiwaki et al. | Molecular mechanism for growth suppression of human hepatocellular carcinoma cells by acyclic retinoid | |
| JP4896743B2 (ja) | Prame阻害剤とhdac阻害剤との併用 | |
| Okado et al. | The transcriptional repressor RP58 is crucial for cell-division patterning and neuronal survival in the developing cortex | |
| Bignante et al. | APP/Go protein Gβγ-complex signaling mediates Aβ degeneration and cognitive impairment in Alzheimer's disease models | |
| Lee et al. | Interferon regulatory factor-1 (IRF-1) regulates VEGF-induced angiogenesis in HUVECs | |
| WO2012014936A1 (ja) | 癌幹細胞の分化誘導剤 | |
| Mo et al. | PTPRM Is Critical for Synapse Formation Regulated by Zinc Ion | |
| Tjalkens et al. | The peroxisome proliferator‐activated receptor‐γ agonist 1, 1‐bis (3′‐indolyl)‐1‐(p‐trifluoromethylphenyl) methane suppresses manganese‐induced production of nitric oxide in astrocytes and inhibits apoptosis in cocultured PC12 cells | |
| Tatenhorst et al. | Peroxisome proliferator-activated receptors (PPARs) as potential inducers of antineoplastic effects in CNS tumors | |
| US20070134349A1 (en) | Methods of treating hematological malignancies | |
| US20070104714A1 (en) | Composition and method for the treatment of cancer and other physiologic conditins based on modulation of the ppar-gamma pathway and her-kinase axis | |
| EP4252750A1 (en) | Pharmaceutical preservation of creb activation in the treatment of alzheimer's disease | |
| JP2019089726A (ja) | がん遺伝子産物yap1/taz機能調節剤 | |
| Mkwanazi | Characterising the anticancer effects of a small molecule with potential to inhibit nuclear import via karyopherin beta1 | |
| Coleman | Roles of Peroxisome Proliferator-Activated Receptor-beta/delta and B-cell Lymphoma-6 in Pancreatic Cancer | |
| Capasso et al. | L-NAME prevents EEG and behavioral alterations induced by Morphine and Deltorphin II in the rabbit | |
| US20110190307A1 (en) | Assay | |
| Epping | Functional genetic screens as tools to discover signaling pathways targeted by cancer drugs |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080611 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110809 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20111026 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20111102 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20111130 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20111207 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20111227 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120110 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120208 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120306 |
|
| A045 | Written measure of dismissal of application [lapsed due to lack of payment] |
Free format text: JAPANESE INTERMEDIATE CODE: A045 Effective date: 20120731 |