ES2950453T3 - Compuestos y composiciones farmacéuticas para su uso en el tratamiento de enfermedades asociadas a la retina utilizando inhibidores de la CCR3 - Google Patents
Compuestos y composiciones farmacéuticas para su uso en el tratamiento de enfermedades asociadas a la retina utilizando inhibidores de la CCR3 Download PDFInfo
- Publication number
- ES2950453T3 ES2950453T3 ES18720836T ES18720836T ES2950453T3 ES 2950453 T3 ES2950453 T3 ES 2950453T3 ES 18720836 T ES18720836 T ES 18720836T ES 18720836 T ES18720836 T ES 18720836T ES 2950453 T3 ES2950453 T3 ES 2950453T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- compound
- acid
- retina
- compounds
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/143—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/145—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2059—Starch, including chemically or physically modified derivatives; Amylose; Amylopectin; Dextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2813—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/282—Organic compounds, e.g. fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/2853—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, polyethylene oxide, poloxamers, poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
- A61K9/2866—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Inorganic Chemistry (AREA)
- Ophthalmology & Optometry (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines Containing Plant Substances (AREA)
- Quinoline Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pyridine Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Se proporcionan métodos para mejorar los criterios de valoración visuales relacionados con la enfermedad asociada a la retina con agentes moduladores de CCR3. Un ejemplo de tal criterio de valoración es la agudeza visual. Las enfermedades asociadas a la retina en las que se puede mejorar la agudeza visual y otros criterios de valoración visuales incluyen la retinopatía del prematuro, la degeneración macular relacionada con la edad, la oclusión de la vena central de la retina y la retinopatía diabética. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Compuestos y composiciones farmacéuticas para su uso en el tratamiento de enfermedades asociadas a la retina utilizando inhibidores de la CCR3
REFERENCIA CRUZADA A SOLICITUDES RELACIONADAS
La presente solicitud reivindica prioridad con respecto a la Solicitud de patente estadounidense n.° 62/482,134, presentada el 5 de abril de 2017.
CAMPO TÉCNICO DE LA INVENCIÓN
La invención se refiere a materiales para mejorar la agudeza visual en un sujeto que los necesite.
ANTECEDENTES DE LA INVENCIÓN
Entre la variedad de enfermedades asociadas a la retina, las hay que se manifiestan al principio de la vida, así como las que se manifiestan en relación con el envejecimiento. Un ejemplo del primer tipo de enfermedad es la retinopatía del prematuro (ROP). Algunos ejemplos de enfermedades de la retina asociadas a la edad son: la degeneración macular asociada a la edad (DMAe ), que es la enfermedad degenerativa más común de la mácula; la oclusión de la vena central de la retina (OVCR) y la retinopatía diabética. Si no se trata, la enfermedad asociada a la retina puede provocar ceguera legal.
La DMAE es la principal causa de ceguera irreversible en personas de 50 años o más en el mundo desarrollado. (Jager, R. et al., The New England Journal of Medicine, 358(2606-17), 2008). DMAE es un término que se utiliza para describir una familia de enfermedades que se caracterizan todas ellas por una pérdida progresiva de la visión central asociada a anomalías de la membrana de Bruch, la coroides, la retina neural y/o el epitelio pigmentario de la retina. En las primeras fases de la DMAE, que suele denominarse maculopatía asociada a la edad (ARM), suele observarse acumulación de drusas (subproductos bioquímicos de las células fotorreceptoras que se acumulan en la membrana de Bruch y que se clasifican por su aspecto) y alteraciones del epitelio pigmentario de la retina (EPR).
La DMAE que se vuelve clínicamente avanzada se clasifica en dos formas: DMAE "seca", no exudativa o atrófica y DMAE exudativa "húmeda" o neovascular. La DMAE seca se da en aproximadamente el 15 % de los pacientes con DMAE y la DMAE húmeda en aproximadamente el 10 %. La DMAE húmeda se considera la forma más debilitante de DMAE y se cree que está causada por el crecimiento de membranas neovasculares coroideas (MNVC) anormales. Estos nuevos vasos sanguíneos crecen a partir de la coriocapilar, creciendo bajo el EPR o la retina, y dejan escapar suero y sangre. Este fluido se acumula en los espacios sub-REP y subretiniano junto con la retina neurosensorial, y a su vez provoca un engrosamiento medible de la mácula. Si la fóvea se ve afectada por la MNVC, el edema y la hemorragia resultantes pueden afectar significativamente a la agudeza visual (AV) y provocar una pérdida drástica de visión.
Se calcula que aproximadamente el 10 % de las personas de 65 a 74 años y el 30 % de las de 75 a 85 años presentan signos de Dm a E. El tratamiento estándar actual para la DMAE húmeda son las terapias antiangiogénicas como ranibizumab (Lucentis®) y aflibercept (Eylea®) mediante administración intravítrea (IVT) (es decir, inyección directa en el ojo). Estas terapias se dirigen a los factores de crecimiento del endotelio vascular (VEGF, VEGF-A) y a sus propiedades promotoras de la angiogénesis. Sin embargo, las inyecciones mensuales ITV se han asociado a los efectos adversos de la atrofia geográfica. (Desai, SJ, et al., Curr Opthalmol. Rep. (01 de febrero de 2017)). En la actualidad, no existen terapias eficaces y menos invasivas, lo que subraya la necesidad insatisfecha de una terapia no basada en anti-VEGF de administración oral para el tratamiento de la DMAE. Además de aliviar e invertir los síntomas y los dramáticos efectos perjudiciales sobre la visión de los pacientes, una terapia de este tipo tendría la ventaja añadida de aumentar el cumplimiento terapéutico. Las inyecciones IVT conllevan un mayor riesgo para los pacientes y son una carga tanto para ellos como para los cuidadores.
La base mecanicista de las terapias anti-VEGF también presenta riesgos. El VEGF, en particular el VEGF-A, tiene una función fisiológica citoprotectora en la retina. La modulación de la expresión y la actividad del VEGF puede ser tóxica para múltiples tipos de células. (Ambati, J., et al., Neuron 75(1):26-39, julio de 2012). Las pruebas demuestran que el tratamiento con anti-VEGF-A también puede contribuir a alteraciones fisiológicas en la vasculatura retiniana a corto plazo, así como a la toxicidad del EPR a largo plazo. (Papadopoulou DN, et al., Oftalmología 116(9): 1755-61 (2009); Sacu S, et al., Invest. Ophthalmol. Vis. Sci. 52(6):3046-50 (2011); y Rofagha S, et al. Am. J. Ophthalmol. 159(5):915-24 (2015)).
Por el contrario, el tratamiento con antagonistas del receptor 3 de quimiocina con motivo C-C (CCR3) se produce sin alterar los niveles de VEGF-A ni afectar ampliamente al sistema inmunitario. Además, los antagonistas de CCR3 pueden presentarse en forma de moléculas pequeñas y orgánicas, y pueden prepararse en formulaciones orales. Los compuestos, co-cristales, sales y formulaciones de la invención proporcionan moduladores de molécula pequeña altamente específicos y potentes del receptor humano de quimiocina C-C tipo 3, que es el receptor principal de la eotaxina-1. El eje CCR3/eotaxina es un factor quimiotáctico clave para los eosinófilos, los mastocitos y (en el contexto
retiniano) las células endoteliales de la vasculatura retiniana, y los estudios realizados en roedores han resultado prometedores para aliviar la neovascularización asociada a la enfermedad retiniana.
A pesar de esta promesa, ha habido un fracaso en la industria para desarrollar un antagonista de CCR3 para el tratamiento humano de la enfermedad asociada a la retina. Sin embargo, los compuestos, cocristales, sales y formulaciones aquí divulgados, que modulan/antagonizan específicamente el CCR3, son eficaces para mejorar la agudeza visual de un número significativo de sujetos, incluso cuando los efectos sobre la neovascularización son insignificantes.
El documento WO 2013/149986A1 se refiere a compuestos para su uso en el tratamiento de enfermedades, por ejemplo degeneración macular seca asociada a la edad (dAMD), poliposis nasal, gastroenteritis eosinofílica y síndrome de Chrug Strauss.
SUMARIO DE LA INVENCIÓN
La invención se define mediante las reivindicaciones adjuntas.
Se proporcionan compuestos para su uso en la mejora de la agudeza visual en un sujeto diagnosticado con una enfermedad asociada a la retina. El uso puede incluir la modulación de CCR3, el receptor principal de CCL11/eotaxina-1 mediante la administración de una cantidad eficaz de antagonistas de CCR3 de la invención. El uso puede incluir la administración de dosis terapéuticas eficaces de antagonistas de CCR3 (por ejemplo, un compuesto de Fórmula 1 descrito en el presente documento) a sujetos o pacientes, así como la monitorización de criterios de valoración clínicos específicos.
La presente invención proporciona un compuesto que es un cocristal de fórmula 2, como se define en la reivindicación 1, para su uso en la mejora de la agudeza visual en un sujeto diagnosticado con una enfermedad asociada a la retina. En una realización, la enfermedad asociada a la retina se selecciona del grupo que consiste en degeneración macular seca asociada a la edad, degeneración macular húmeda asociada a la edad, oclusión de la vena central de la retina, retinopatía del prematuro y retinopatía diabética. En una realización, el compuesto es como se define en la reivindicación 3. En otra realización, el compuesto es como se define en la reivindicación 4. En otra realización, el compuesto es como se define en la reivindicación 5. En otra realización, el compuesto es como se define en la reivindicación 6. En otra realización, el compuesto es como se define en la reivindicación 7. En una realización, el compuesto es como se define en la reivindicación 8. En una realización, el compuesto de fórmula 2 es:

En una realización, el compuesto es una sal cristalina de la fórmula:

En otra realización, el compuesto es una sal cristalina de la fórmula:

En una realización, la sal cristalina se caracteriza porque los cuatro picos más altos de difracción de rayos X en polvo se producen a 3,72, 13,60, 16,89 y 19,34 grados 20 (±0,05 grados 20) cuando se miden utilizando radiación CuKa. En otra realización, la sal cristalina se caracteriza porque los cuatro picos más altos de difracción de rayos X en polvo se producen a 16,02, 16,86, 19,45 y 19,71 grados 20 (±0,05 grados 20) cuando se miden utilizando radiación CuKa. En una realización, el compuesto de fórmula 2 está en forma de los isómeros ópticos individuales, una mezcla de los enantiómeros individuales, un racemato o en forma de los compuestos enantioméricamente puros. En una realización, se administrará además una cantidad terapéuticamente eficaz de una terapia anti-VEGF.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
La figura 1 es una tabla de potencia y selectividad de especies para un producto en investigación de la invención.
La figura 2 muestra el diseño y el plan general del ensayo clínico con el producto en investigación de la invención, incluidos los periodos de cribado, tratamiento y seguimiento.
La figura 3 muestra el grosor medio de la retina central de 1 mm en los pacientes a lo largo del ensayo clínico descrito en la figura 2.
La figura 4 es una tabla de estadísticas descriptivas con respecto a las fugas neovasculares a lo largo del calendario del ensayo clínico descrito en la figura 2.
La figura 5 muestra la agudeza visual mejor corregida (AVMC) según el número de letras leídas por los pacientes durante el tiempo del ensayo descrito en la figura 2. La BCVA se comprobó utilizando la tabla ETDRS para la agudeza visual.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
La invención se define mediante las reivindicaciones adjuntas.
Una realización de la invención comprende compuestos para su uso en un procedimiento de mejora de la agudeza visual en sujetos con enfermedad asociada a la retina, comprendiendo el procedimiento la administración de una cantidad terapéuticamente eficaz de un compuesto. El compuesto se presenta en forma de co-cristales de fórmula 2. El compuesto puede presentarse en forma de isómeros ópticos individuales, una mezcla de los enantiómeros individuales, un racemato o compuestos enantioméricamente puros. El compuesto puede presentarse en forma de las composiciones y formulaciones farmacéuticas que se exponen más adelante.
Una cantidad terapéuticamente eficaz de una combinación de un compuesto de fórmula química 2 puede administrarse junto con el tratamiento estándar actual en los Estados Unidos para la enfermedad asociada a la retina. Una cantidad terapéuticamente eficaz de una combinación de un compuesto de fórmula química 2 puede administrarse junto con una terapia anti-VEGF-A, como anticuerpos contra el VEGF-A (p. ej. ranibizumab (Lucentis®), bevacizumab (Avastin®), proteínas de fusión recombinantes que se unen a uno o más tipos de receptores de VEGF (p. ej., aflibercept,Eylea®), o pequeñas moléculas orgánicas que se unen a VEGF-A o a uno o más de sus tipos de receptores (p. ej., receptor 1 o 2 de VEGF).
Por "tratamiento" se entiende al menos una mejoría de uno o más síntomas asociados con la enfermedad relacionada con la retina que aflige al paciente, donde mejoría se utiliza en un sentido amplio para referirse al menos a una reducción en la magnitud de un parámetro, por ejemplo, un síntoma asociado con la enfermedad que se está tratando. Como tal, el tratamiento también incluye situaciones en las que una condición patológica, o al menos los síntomas asociados a ella, se inhiben por completo, por ejemplo, se evita que ocurra, o se detiene, por ejemplo, se termina, de tal manera que el paciente ya no sufre de la deficiencia, o al menos los síntomas que caracterizan a la deficiencia. En algunos casos, "tratamiento", "tratar" y similares se refieren a la obtención de un efecto farmacológico y/o fisiológico deseado. El efecto puede ser profiláctico en términos de prevención total o parcial de una enfermedad o síntoma de la misma y/o puede ser terapéutico en términos de curación parcial o completa de una enfermedad y/o efecto adverso
atribuible a la enfermedad. "Tratamiento" puede ser cualquier tratamiento de una enfermedad en un sujeto, e incluye: (a) prevenir la aparición de la enfermedad en un sujeto que puede estar predispuesto a padecerla pero al que aún no se le ha diagnosticado; (b) inhibir la enfermedad, es decir, detener su desarrollo; o (c) aliviar la enfermedad, es decir, provocar su regresión. El tratamiento puede dar lugar a diversas manifestaciones físicas, como la modulación de la expresión génica, el aumento de la neurogénesis, el rejuvenecimiento de tejidos u órganos, etc. El tratamiento de la enfermedad en curso, en el que el tratamiento estabiliza o reduce los síntomas clínicos indeseables del paciente, se produce en algunas realizaciones. Dicho tratamiento puede realizarse antes de la pérdida completa de función en los tejidos afectados. La terapia del sujeto puede administrarse durante la fase sintomática de la enfermedad y, en algunos casos, después de la fase sintomática de la enfermedad.
En algunos casos, el sujeto es un mamífero. Las especies de mamíferos que pueden tratarse con los presentes procedimientos incluyen caninos y felinos; equinos; bovinos; ovinos; etc., y primates, incluidos los humanos. Los procedimientos, composiciones y reactivos en cuestión también pueden aplicarse a modelos animales, incluidos pequeños mamíferos, por ejemplo, murinos, lagomorfos, etc., por ejemplo, en investigaciones experimentales.
a. Compuestos
En los grupos, radicales o fracciones definidas en esta sección de "Compuestos", el número de átomos de carbono se especifica a menudo sucede al grupo, por ejemplo, alquilo C1-6 significa un grupo o radical alquilo que tiene de 1 a 6 átomos de carbono. En general, para los grupos que comprenden dos o más subgrupos que se divulgan en esta sección "Compuestos", el último grupo nombrado es el punto de unión radical, por ejemplo, "tioalquilo" significa un radical monovalente de la fórmula HS-Alk-. A menos que se especifique lo contrario a continuación, las definiciones convencionales de los términos de control y las valencias convencionales de los átomos estables se presumen y se alcanzan en todas las fórmulas y grupos.
b. Co-cristales y sales
Las realizaciones de la presente invención comprenden los cocristales de los compuestos de fórmula 2 (a continuación) para su uso en la mejora de la agudeza visual en un sujeto diagnosticado con una enfermedad asociada a la retina. En general, para los grupos que comprenden dos o más subgrupos en esta sección "C0-Cristales y Sales", el primer subgrupo nombrado es el punto de unión radical, por ejemplo, el sustituyente "alquilo C1 -3-arilo" significa un grupo arilo que está unido a un grupo alquilo C1 -3 , este último unido al núcleo o al grupo al que está unido el sustituyente.

donde
R1 se selecciona entre alquilo C1-6 , haloalquilo C1 -6 , O-haloalquilo C1 -6 , halógeno;
m es 1, 2 o 3, en algunos casos 1 o 2;
R2a y R2b se seleccionan cada uno independientemente entre H, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, COO-alquilo C1-6 , O-alquilo C1 -6 , CONR2 b 1 R2 b 2 , halógeno;
R2b-1 se selecciona entre H, alquilo C1-6 , alquilo Cü.4-cicloalquilo C3-6, haloalquilo C1-6 ;
R2 b 2 se selecciona entre H, alquilo C1 -6 ;
o R2 b 1 y R2 b 2 son conjuntamente un grupo alquileno C3-6 que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno;
R3 se selecciona entre H, alquilo C1 -6 ;
X es un anión seleccionado del grupo que consiste en cloruro, bromuro, yoduro, sulfato, fosfato, metanosulfonato, nitrato, maleato, acetato, benzoato, citrato, salicilato, fumarato, tartrato, dibenzoiltartrato, oxalato, succinato, benzoato y p-toluenosulfonato; en algunos casos cloruro o dibenzoiltartrato;
J es 0, 0,5, 1, 1,5 o 2; en algunos casos 1 o 2;
con un formador de co-cristales seleccionado del grupo que consiste en ácido orótico, ácido hipúrico, ácido L-piroglutámico, ácido D-piroglutámico, ácido nicotínico, ácido L-(+)-ascórbico, sacarina, piperazina, ácido 3-hidroxi-2-naftoico, ácido múcico (galactárico), ácido pamoico (embónico), ácido esteárico, ácido cólico, ácido desoxicólico, nicotinamida, isonicotinamida, succinamida, uracilo, L-lisina, L-prolina, D-valina, L-arginina, glicina, en algunos casos ácido ascórbico, ácido múcico, ácido pamóico, succinamida, ácido nicotínico, nicotinamida, isonicotinamida, l-lisina o 1 -prolina.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2
R2a se selecciona entre H, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, O-alquilo C1.6 , CONR2 a 1 R2 a 2 ;
R2a1 se selecciona entre H, alquilo C1 -6 , haloalquilo C1.6 ;
R2 a 2 se selecciona entre H, alquilo C1 -6 ;
R2b se selecciona entre H, alquilo C1.6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, COO-alquilo C1.6 , O alquilo C1.6 , CONR2 b 1 R2 b 2 , halógeno;
R2b-1 se selecciona entre H, alquilo C1-6 , alquilo Cü.4-cicloalquilo C3-6, haloalquilo C1.6 ;
R2 b 2 se selecciona entre H, alquilo C1 -6 ;
o R2b1 y R2b 2 son conjuntamente un grupo alquileno C3-6que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno y los residuos restantes se definen como anteriormente.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2
R2a se selecciona entre H, alquilo C1 -6 , alquinilo C2-6, cicloalquilo C3-6, O-alquilo C1 -6 , CONR2 a 1R2 a 2 ;
R2a1 es alquilo C1 -6 ;
R2 a 2 es H;
R2b se selecciona entre H, alquilo C1 -6 , O-alquilo C1.6 , CONR2 b 1R2 b 2 ;
R2b-1 se selecciona entre alquilo C1 -6 , alquilo Cü.4-cicloalquilo C3-6, haloalquilo C1.6 ;
R2 b 2 se selecciona entre H, alquilo C1 -6 ;
o R2b1 y R2b 2 son conjuntamente un grupo alquileno C3-6que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno y los residuos restantes se definen como anteriormente.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2
R2a se selecciona entre H, alquilo C1 -4 , alquinilo C2-4, cicloalquilo C3-6, O-alquilo C1 -4 , CONR2 a 1R2 a 2 ;
R2a1 es alquilo C1 -4 ;
R2 a 2 es H;
R2b se selecciona entre H, alquilo C1 -4 , O-alquilo C1.4 , CONR2 b 1R2 b 2 ;
R2b-1 se selecciona entre alquilo C1 -4 , alquilo Cü-4-cicloalquilo C3-6, haloalquilo C1.4 ;
R2 b 2 se selecciona entre H, alquilo C1 -4 ;
o R2b1 y R2b 2 son conjuntamente un grupo alquileno C3-6que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno y los residuos restantes se definen como anteriormente.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2
R2a se selecciona entre H, alquilo C1 -4 ,
R2b se selecciona entre H, CONR2 b 1 R2 b 2 ;
R2b1 se selecciona entre alquilo C1 -4 , alquilo Cü-4-cicloalquilo C3-6, haloalquilo C1 -4 ;
R2 b 2 se selecciona entre H, alquilo C1 -4 ;
o R2 b 1 y R2 b 2 son conjuntamente un grupo alquileno Ca i que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno y los residuos restantes se definen como anteriormente.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2
R1 se selecciona entre alquilo C1-6 , haloalquilo C1 -6 , O-haloalquilo C1 -6 , halógeno;
m es 1 o 2;
R2a se selecciona entre H, alquilo C1 -4 ;
R2b se selecciona entre H, CONR2 b 1 R2 b 2 ;
R2 b 1 se selecciona entre alquilo C1 -4 , alquilo Co-4-cicloalquilo C3-6, haloalquilo C1 -4 ;
R2 b 2 se selecciona entre H, alquilo C1 -4 ;
o R2 b 1 y R2 b 2 son conjuntamente un grupo alquileno C3-6que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno R3 se selecciona entre H, alquilo C1 -6 ;
X es un anión seleccionado del grupo que consiste en cloruro o dibenzoiltartrato
J es 1 o 2.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2
R2a se selecciona entre H, alquilo C1 -4 ; en algunos casos metilo, etilo, propilo;
R2b se selecciona entre H, CONR2 b 3 R2 b 2 ;
R2 b 1 es alquilo C1 -4 ; en algunos casos metilo, etilo, propilo;
R2 b 2 es alquilo C1 -4 ; en algunos casos metilo, etilo, propilo;
y el resto de residuos se definen como arriba.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2
R2a se selecciona entre H, alquilo C1 -4 ; en algunos casos metilo, etilo, propilo;
R2b se selecciona entre H, CONR2 b 1 R2 b 2 ;
R2b-1 es alquilo Co-4-cicloalquilo C3-6;
R2 b 2 es H, alquilo C1 -4 ; en algunos casos H, metilo, etilo, propilo;
y el resto de residuos se definen como arriba.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2
R2a se selecciona entre H, alquilo C1 -4 ; en algunos casos metilo, etilo, propilo;
R2b se selecciona entre H, CONR2 b 1 R2 b 2 ;
R2 b 1 es haloalquilo C1 -4 ;
R2 b 2 se selecciona entre H, alquilo C1 -4 ; en algunos casos H, metilo, etilo, propilo;
y el resto de residuos se definen como arriba.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2
R2 b 1 y R2b 2 son conjuntamente un grupo alquileno C3-6que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno y los residuos restantes se definen como arriba.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2 R1, m, R2a, R2b, R3, X y j se definen como arriba y el formador del cocristal se selecciona del grupo que consiste en ácido ascórbico, ácido múcico, ácido pamóico, succinamida, ácido nicotínico, nicotinamida, isonicotinamida, I-lisina, 1-prolina, o hidratos o hidrocloruros de los mismos.
En otro aspecto de la presente invención, los co-cristales de los compuestos son de fórmula 2a, en la que R2a, R2b, R3, X y j se definen como arriba

En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2a R2a se selecciona entre H, alquilo C1-4; en algunos casos metilo, etilo, propilo;
R2b se selecciona entre H, CONR2b1R2b2;
R2b1 es alquilo C1-4; en algunos casos metilo, etilo, propilo;
R2b2 es alquilo C1-4; en algunos casos metilo, etilo, propilo;
y el resto de residuos se definen como arriba.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2a
R2a se selecciona entre H, alquilo C1-4; en algunos casos metilo, etilo, propilo;
R2b se selecciona entre H, CONR2b1R2b2;
R2b-1 es alquilo C0-4-cicloalquilo C3-6;
R2b2 es H, alquilo C1-4; en algunos casos H, metilo, etilo, propilo;
y el resto de residuos se definen como arriba.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2a
R2a se selecciona entre H, alquilo C1-4; en algunos casos metilo, etilo, propilo;
R2b se selecciona entre H, CONR2b1R2b2;
R2b1 es haloalquilo C1-4;
R2b2 se selecciona entre H, alquilo C1-4; en algunos casos H, metilo, etilo, propilo;
y el resto de residuos se definen como arriba.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2a
R2b-1 y R2b2 se encuentran juntos un grupo alquileno C3-6formando con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno, y los residuos restantes se definen como arriba.
Las bases libres de los compuestos de fórmula 2 (j = 0) son a menudo amorfas y se utilizan para un proceso de fabricación de co-cristal, y las sales de los compuestos de fórmula 2 pueden emplearse como se desee para un proceso de fabricación de co-cristal. Así, otro aspecto de la invención son sales de compuestos de fórmula 2 en los que R1, m, R2a, R2b, R3 se definen como para los cocristales anteriores y
X es un anión seleccionado del grupo que consiste en cloruro, bromuro, yoduro, sulfato, fosfato, metanosulfonato, nitrato, maleato, acetato, benzoato, citrato, salicilato, fumarato, tartrato, dibenzoiltartrato, oxalato, succinato, benzoato y p-toluenosulfonato; tal como cloruro, o dibenzoiltartrato; y
j es 0, 0,5, 1, 1,5 o 2; como 1 o 2.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2 R1, m, R2a, R2b, R3 se definen como para los cocristales anteriores y
X es un anión seleccionado del grupo que consiste en cloruro o dibenzoiltartrato;
j es 1 o 2.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2 R1, m, R2a, R2b, R3 se definen como para las sales anteriores y X es cloruro y j es 2.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2 R1, m, R2a, R2b, R3 se definen como para las sales anteriores y X es dibenzoiltartrato y j es 1.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2a R2a, R2b, R3, X y j se definen como se ha indicado anteriormente

En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2a R2a se selecciona entre H, alquilo C1-4; en algunos casos metilo, etilo, propilo;
R2b se selecciona entre H, CONR2b3R2b2;
R2b1 es alquilo C1-4; en algunos casos metilo, etilo, propilo;
R2b2 es alquilo C1-4; en algunos casos metilo, etilo, propilo;
y el resto de residuos se definen como arriba.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2a
R2a se selecciona entre H, alquilo C1-4; en algunos casos metilo, etilo, propilo;
R2b se selecciona entre H, CONR2b3R2b2;
R2b1 es alquilo C0-4-cicloalquilo C3-6;
R2b2 es H, alquilo C1-4; en algunos casos H, metilo, etilo, propilo;
y el resto de residuos se definen como arriba.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2a
R2a se selecciona entre H, alquilo C1-4; en algunos casos metilo, etilo, propilo;
R2b se selecciona entre H, CONR2b1R2b2;
R2b1 es haloalquilo C1-4;
R2b2 es H, alquilo C1-4; en algunos casos H, metilo, etilo, propilo;
y el resto de residuos se definen como arriba.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2a
R2b-1 y R2b2 son conjuntamente un grupo alquileno C3-6 que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno y los residuos restantes se definen como arriba.
En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2a R1, m, R2a, R2b, R3 se definen como para las sales anteriores y X es cloruro y j es 2.
En otro aspecto de la presente invención, en los co-cristales de los compuestos de fórmula 2a R1, m, R2a, R2b, R3 se definen como para las sales anteriores y X es dibenzoiltartrato y j es 1. En otro aspecto de la presente invención, en los cocristales de los compuestos de fórmula 2a R1, m, R2a, R2b, R3 se definen como para las sales anteriores y X es (S)-(S)-(+)-2,3-dibenzoil-tartrato y j es 1.
c. Formulaciones
Otras realizaciones de la presente invención comprenden además una composición farmacéutica para su uso en la mejora de la agudeza visual en un sujeto diagnosticado de una enfermedad asociada a la retina, conteniendo la composición farmacéutica compuestos de fórmula 3

en los que
R1 se selecciona entre H, alquilo C1-6 , alquilo Cü-4-cicloalquilo C3-6, haloalquilo C 1-6 ;
R2 se selecciona entre H, alquilo C1-6 ;
X es un anión seleccionado del grupo que consiste en cloruro o ^ dibenzoiltartrato
j es 1 o 2.
En una realización de la presente invención, en la composición farmacéutica que contiene compuestos de fórmula 3 R1 se selecciona entre H, alquilo C1-6 ;
R2 se selecciona entre H, alquilo C1-6 ;
X es un anión seleccionado del grupo que consiste en cloruro o ^ dibenzoiltartrato
j es 1 o 2.
En una realización de la presente invención, en la composición farmacéutica que contiene compuestos de fórmula 3 R1 se selecciona entre H, metilo, etilo, propilo, butilo;
R2 se selecciona entre H, Metilo, Etilo, Propilo, Butilo;
X es un anión seleccionado del grupo que consiste en cloruro o ^ dibenzoiltartrato, como el cloruro;
j es 1 o 2, en algunos casos 2.
En una realización de la presente invención, en la composición farmacéutica que contiene compuestos de fórmula 3 R1 se selecciona entre H, metilo, etilo, propilo, butilo;
R2 se selecciona entre H, Metilo;
X es un anión seleccionado del grupo que consiste en cloruro o ^ dibenzoiltartrato, como el cloruro;
j es 1 o 2, en algunos casos 2.
En una realización de la presente invención, en la composición farmacéutica que contiene compuestos de fórmula 3 R1 se selecciona entre H, Metilo;
R2 se selecciona entre H, Metilo;
X es un anión seleccionado del grupo que consiste en cloruro o ^ dibenzoiltartrato, como el cloruro;
j es 1 o 2, en algunos casos 2.
Una realización de la presente invención comprende además una composición farmacéutica que contiene compuestos descritos en la Tabla 2 como clorhidrato para su uso en la mejora de la agudeza visual en un sujeto diagnosticado con una enfermedad asociada a la retina. Una realización de la presente invención comprende además una composición farmacéutica que contiene los compuestos descritos en la Tabla 2 como dihidrocloruro para su uso en la mejora de la agudeza visual en un sujeto diagnosticado con una enfermedad asociada a la retina.
Tabla 2


Otro aspecto de la presente invención es una forma de dosificación farmacéutica de los compuestos descritos anteriormente para su uso en la mejora de la agudeza visual en un sujeto diagnosticado con una enfermedad asociada a la retina, en el que la dosificación es una forma de dosificación de administración oral.
Otro aspecto de la presente invención es una forma de dosificación farmacéutica de los compuestos descritos anteriormente para su uso en la mejora de la agudeza visual en un sujeto diagnosticado con una enfermedad asociada a la retina, que se presenta en forma de comprimido, cápsula, gránulos, polvo o gránulos.
d. Formas de dosificación/Inaredientes
Las composiciones farmacéuticas sólidas listas para su uso/ingestión elaboradas a partir de un compuesto de fórmula 3 comprenden, por ejemplo, polvos, gránulos, pellas, comprimidos, cápsulas, comprimidos masticables, tablas dispersables, troches y pastillas para chupar.
Las formulaciones en cápsula según la invención comprenden el producto intermedio en polvo de un compuesto de fórmula 3, una mezcla intermedia que comprende el producto intermedio en polvo, gránulos o pellas obtenidos por granulación convencional en húmedo, en seco o por fusión en caliente o extrusión en caliente o secado por pulverización de una mezcla intermedia adecuada, rellenados en cápsulas convencionales, por ejemplo, cápsulas de gelatina dura o HPMC.
Las formulaciones en cápsula anteriores también pueden comprender el intermedio en polvo de un compuesto de fórmula 3 en forma compactada.
Las formulaciones en cápsula según la invención también pueden comprender el compuesto de fórmula 3 suspendido o diluido en un líquido o mezcla de líquidos.
Las formulaciones en comprimidos según la invención comprenden dichos comprimidos obtenidos, por ejemplo, por compresión directa de una mezcla final adecuada o por tableteado de pellas o gránulos obtenidos por granulación convencional húmeda, seca o por fusión en caliente o extrusión por fusión en caliente o secado por pulverización de una mezcla intermedia adecuada.
Otro aspecto de la presente invención es una forma de dosificación en la que se añade un agente de ajuste del pH o tampón para mejorar la estabilidad del principio activo. El agente regulador/tampón del pH puede ser un aminoácido básico, que tiene un grupo amino y características alcalinas (punto isoeléctrico, pI: 7,59-10,76), como, por ejemplo, L-arginina, L-lisina o L-histidina. Un agente tampón en el sentido de la presente invención es la L-arginina. La L-arginina tiene un efecto estabilizador particularmente adecuado en las composiciones de esta invención, por ejemplo, suprimiendo la degradación química de los compuestos de fórmula 3.
Así, en una realización, la presente invención se dirige a una composición farmacéutica (por ejemplo, una forma de dosificación sólida oral, particularmente un comprimido) que comprende un compuesto de fórmula 3 y L-arginina para estabilizar la composición, particularmente contra la degradación química; así como uno o más excipientes farmacéuticos.
De manera adecuada, los excipientes farmacéuticos utilizados en esta invención son materiales convencionales como celulosa y sus derivados, D-manitol, almidón de maíz, almidón pregelatinizado como relleno, copovidona como aglutinante, crospovidona como desintegrante, estearato de magnesio como lubricante, sílice coloidal anhidra como deslizante, hipromelosa como agente de recubrimiento, crospovidona como desintegrante, estearato de magnesio como lubricante, sílice coloidal anhidra como deslizante, hipromelosa como agente de recubrimiento, polietilenglicol como plastificante, dióxido de titanio, óxido de hierro rojo/amarillo como pigmento, talco, etc.
Los excipientes farmacéuticos pueden ser un primer y segundo diluyente, un aglutinante, un desintegrante y un lubricante; un desintegrante adicional y un deslizante adicional son una opción adicional.
Los diluyentes adecuados para una composición farmacéutica según la invención son, por ejemplo, celulosa en polvo, celulosa microcristalina, lactosa en diversas modificaciones cristalinas, calcifosfato dibásico anhidro, calcifosfato dibásico dihidrato, eritritol, hidroxipropilcelulosa poco sustituida, manitol, almidón o almidón modificado (por ejemplo, pregelatinizado o parcialmente hidrolizado) o xilitol. Entre esos diluyentes se emplean en algunos casos el manitol y la celulosa microcristalina.
Los diluyentes que pueden emplearse en algunos casos como segundo diluyente son los diluyentes mencionados anteriormente, manitol y celulosa microcristalina.
Los lubricantes adecuados para una composición farmacéutica según la invención son, por ejemplo, talco, polietilenglicol, behenato de calcio, estearato de calcio, estearilfumarato de sodio, aceite de ricino hidrogenado o estearato de magnesio, donde en algunos casos se emplea estearato de magnesio.
Los aglutinantes adecuados para una composición farmacéutica según la invención incluyen, entre otros, copovidona (copolimerizados de vinilpirrolidón con otros vinilderivados), hidroxipropilmetilcelulosa (HPMC) hidroxipropilcelulosa (HPC), polivinilpirrolidón (povidona), almidón pregelatinizado, ácido esteárico-palmítico, hidroxipropilcelulosa de baja sustitución (L-HPC), copovidona y almidón pregelatinizado. Los aglutinantes antes mencionados, almidón pregelatinizado y L-HPC, presentan propiedades diluyentes y desintegrantes adicionales y también pueden utilizarse como segundo diluyente o desintegrante.
Los desintegrantes adecuados para una composición farmacéutica según la presente invención son, por ejemplo, almidón de maíz, crospovidona, polacrilina potásica, croscarmelosa sódica, hidroxipropilcelulosa de baja sustitución (L-HPC) o almidón pregelatinizado, como la croscarmelosa sódica.
Como deslizante opcional puede utilizarse dióxido de silicio coloidal.
Una composición ejemplar según la presente invención comprende el diluyente manitol, celulosa microcristalina como diluyente con propiedades desintegradoras adicionales, el aglutinante copovidona, el desintegrante croscarmelosa sódica y estearato de magnesio como lubricante.
Las composiciones farmacéuticas típicas comprenden ( % en peso):
10-50 % principio activo
20-88 % diluyente 1,
5-50 % diluyente 2,
1-5 % aglutinante,
1-15 % desintegrante, y
0,1-5 % lubricante.
Las composiciones farmacéuticas según algunas realizaciones comprenden ( % en peso):
10-50 % principio activo
20-75 % diluyente 1,
5-30 % diluyente 2,
2-30 % aglutinante,
1-12 % desintegrante, y
0,1-3 % lubricante
Las composiciones farmacéuticas según algunas realizaciones comprenden ( % en peso)
10-90 % principio activo
5-70 % diluyente 1,
5-30 % diluyente 2,
0-30 % aglutinante,
1-12 % desintegrante, y
0,1-3 % lubricante.
Las composiciones farmacéuticas según algunas realizaciones comprenden ( % en peso)
10-50 % principio activo
20-75 % diluyente 1,
5-30 % diluyente 2,
2-30 % aglutinante,
0,5-20 % agente tampón,
1-12 % desintegrante, y
0,1-3 % lubricante
Las composiciones farmacéuticas según algunas realizaciones comprenden ( % en peso)
30-70 % principio activo
20-75 % diluyente 1,
5-30 % diluyente 2,
2-30 % aglutinante,
0,5-20 % agente tampón,
1-12 % desintegrante, y
0,1-3 % lubricante.
En algunos casos se emplean composiciones farmacéuticas que contienen 10-90 % de principio activo, preferentemente 30-70 % de principio activo ( % en peso).
Una formulación de comprimidos según la invención puede estar sin recubrir o recubierta, por ejemplo, recubierta con película, utilizando recubrimientos adecuados que se sabe que no afectan negativamente a las propiedades de disolución de la formulación final. Por ejemplo, los comprimidos pueden recubrirse con una capa de sellado para proteger el entorno de los pacientes y al personal clínico, así como para protegerlos de la humedad, disolviendo un polímero de alto peso molecular, como polivinilpirrolidona o hidroxipropilmetilcelulosa, junto con plastificantes, lubricantes y, opcionalmente, pigmentos y tensioactivos en agua o disolvente orgánico, como acetona, y pulverizando esta mezcla sobre los núcleos de los comprimidos dentro de un equipo de recubrimiento, como un recubridor de bandeja o un recubridor de lecho fluidizado con inserto Wurster.
Además, pueden aplicarse a los comprimidos agentes como cera de abejas, goma laca, ftalato de acetato de celulosa, ftalato de acetato de polivinilo, zeína, polímeros formadores de película como hidroxipropilcelulosa, etilcelulosa y metacrilatos poliméricos, siempre que el recubrimiento no tenga un efecto sustancial sobre la desintegración/disolución de la forma farmacéutica y que la forma farmacéutica recubierta no vea afectada su estabilidad.
Después de recubrir la forma farmacéutica con película, puede aplicarse un recubrimiento de azúcar sobre la forma farmacéutica sellada. El recubrimiento de azúcar puede comprender sacarosa, dextrosa, sorbitol y similares o mezclas de los mismos. Si se desea, pueden añadirse colorantes u opacificantes a la solución azucarada.
Las formulaciones sólidas de la presente invención tienden a ser higroscópicas. Pueden envasarse utilizando blísteres de PVC, blísteres de PVDC o un material de envasado a prueba de humedad como blísteres de papel de aluminio, blísteres de aluminio/alu, blísteres de polímero transparente u opaco con bolsa, tubos de polipropileno, botellas de vidrio y botellas de polietileno de alta densidad que contengan opcionalmente un dispositivo de seguridad para niños o puedan ser a prueba de manipulaciones. El material de envasado primario puede incluir un desecante, como tamiz molecular o gel de sílice, para mejorar la estabilidad química del API. Se pueden utilizar envases opacos, como blísteres de colores, tubos, botellas de vidrio marrón o similares, para prolongar la vida útil del API mediante la reducción de la fotodegradación.
e. Dosificaciones
Un intervalo de dosificación del compuesto de fórmula 3 suele estar comprendido entre 100 y 1000 mg, en particular entre 200 y 900 mg, 300 y 900 mg, o 350 y 850 mg, o 390 y 810 mg. Es posible administrar uno o dos comprimidos, y en algunos casos se emplean dos comprimidos para una dosis oral diaria de 100, 200, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850 o 900 mg, como 350, 400, 450, 750, 800 u 850 mg.
El intervalo de dosificación puede lograrse con un comprimido o con dos comprimidos; por ejemplo, cuando se administran dos comprimidos, cada uno de los cuales contiene la mitad de la dosificación.
La aplicación del principio activo puede realizarse hasta tres veces al día, por ejemplo una o dos veces al día. Las concentraciones de dosificaciones especiales son 400 mg u 800 mg.
f. Términos utilizados y definiciones
A los términos no definidos específicamente en el presente documento se les debe dar el significado que les daría un experto en la materia a la luz de la divulgación y el contexto. Sin embargo, tal y como se utilizan en la especificación, a menos que se especifique lo contrario, los siguientes términos tienen el significado que se indica y se respetan las siguientes convenciones.
El término "aproximadamente" significa un 5 % más o menos del valor especificado. Así, aproximadamente 100 minutos también podrían leerse como de 95 a 105 minutos.
Cuando un compuesto de la presente invención se represente en forma de nombre químico y como fórmula, en caso de discrepancia prevalecerá la fórmula. Se puede utilizar un asterisco en las subfórmulas para indicar el enlace que está conectado a la molécula central tal y como se ha definido.
A menos que se indique específicamente, a lo largo de la especificación y las reivindicaciones anexas, una fórmula o nombre químico dado abarcará los tautómeros y todos los isómeros estéreo, ópticos y geométricos (por ejemplo, enantiómeros, diastereómeros, isómeros E/Z, etc.) y racematos de los mismos, así como mezclas en diferentes proporciones de los enantiómeros separados, mezclas de diastereómeros, o mezclas de cualquiera de las formas anteriores cuando existan dichos isómeros y enantiómeros, así como sales, incluidas sales farmacéuticamente aceptables de los mismos y solvatos de los mismos, como por ejemplo hidratos, incluidos solvatos de los compuestos libres o solvatos de una sal del compuesto.
El término "sustituido", tal como se utiliza en el presente documento, significa que uno o más hidrógenos del átomo designado se sustituyen por una selección del grupo indicado, siempre que no se supere la valencia normal del átomo designado y que la sustitución dé lugar a un compuesto estable.
Por el término "opcionalmente sustituido" se entiende dentro del alcance de la invención el grupo mencionado anteriormente, opcionalmente sustituido por un grupo molecular inferior. Ejemplos de grupos moleculares inferiores considerados químicamente significativos son los grupos formados por 1-200 átomos. Son interesantes los grupos que no tienen ningún efecto negativo sobre la eficacia farmacológica de los compuestos. Por ejemplo, los grupos pueden incluir:
cadenas de carbono rectas o ramificadas, opcionalmente interrumpidas por heteroátomos, opcionalmente sustituidas por anillos, heteroátomos u otros grupos funcionales comunes;
sistemas de anillos aromáticos o no aromáticos formados por átomos de carbono y, opcionalmente, heteroátomos, que a su vez pueden estar sustituidos por grupos funcionales; o
una serie de sistemas de anillos aromáticos o no aromáticos formados por átomos de carbono y opcionalmente heteroátomos que pueden estar unidos por una o más cadenas de carbono, opcionalmente interrumpidas por heteroátomos, opcionalmente sustituidas por heteroátomos u otros grupos funcionales comunes.
Los compuestos aquí divulgados pueden existir como sales terapéuticamente aceptables. La presente invención incluye los compuestos enumerados anteriormente en forma de sales, incluidas las sales de adición ácida. Las sales adecuadas incluyen las que se forman con ácidos orgánicos e inorgánicos. Dichas sales de adición ácida serán normalmente farmacéuticamente aceptables. Sin embargo, las sales no farmacéuticamente aceptables pueden ser de utilidad en la preparación y purificación del compuesto en cuestión. También pueden formarse sales de adición básicas y ser farmacéuticamente aceptables. Para un análisis más completo de la preparación y selección de sales, consulte Sales farmacéuticas: Propiedades, selección y uso (Stahl, P Heinrich. Wiley- VCHA, Zúrich, Suiza, 2002).
El término "sal terapéuticamente aceptable", tal como se utiliza en el presente documento, representa sales o formas zwitteriónicas de los compuestos divulgados en el presente documento que son solubles en agua o aceite o dispersables y terapéuticamente aceptables tal como se define en el presente documento. Las sales pueden prepararse durante el aislamiento final y la purificación de los compuestos o por separado haciendo reaccionar el compuesto apropiado en forma de base libre con un ácido adecuado. Las sales de adición de ácido representativas incluyen acetato, adipato, alginato, L-ascorbato, aspartato, benzoato, bencenosulfonato (besilato), bisulfato, butirato, canforato, canforsulfonato, citrato, digluconato, formiato, fumarato, gentisato, glutarato, glicerofosfato, glicolato, hemisulfato, heptanoato, hexanoato, hipurato, hidrocloruro, hidrobromuro, hidroyoduro, 2-hidroxietansulfonato (isetionato), lactato, maleato, malonato, DL-mandelato, mesitilenosulfonato, metanosulfonato, naftilenosulfonato, nicotinato, 2-naftalenosulfonato, oxalato, pamoato, pectinato, persulfato, 3-fenilproprionato, fosfonato, picrato, pivalato, propionato, piroglutamato, succinato, sulfonato, tartrato, L-tartrato, tricloroacetato, trifluoroacetato, fosfato, glutamato, bicarbonato, para- toluenosulfonato (p-tosilato) y undecanoato. Además, los grupos básicos de los compuestos aquí descritos pueden cuaternizarse con cloruros, bromuros y yoduros de metilo, etilo, propilo y butilo; sulfatos de dimetilo, dietilo, dibutilo y diamilo; cloruros, bromuros y yoduros de decilo, laurilo, miristilo y esterilo; y bromuros de bencilo y fenetilo. Los ejemplos de ácidos que pueden emplearse para formar sales de adición terapéuticamente aceptables incluyen ácidos inorgánicos como el clorhídrico, el bromhídrico, el sulfúrico y el fosfórico, y ácidos orgánicos como el oxálico, el maleico, el succínico y el cítrico. Las sales también pueden formarse por coordinación de los compuestos con un metal alcalino o un ion alcalinotérreo. Por lo tanto, la presente invención contempla sales de sodio, potasio, magnesio y calcio de los compuestos aquí divulgados, y similares.
Las sales básicas de adición pueden prepararse durante el aislamiento final y la purificación de los compuestos haciendo reaccionar un grupo carboxi con una base adecuada como el hidróxido, carbonato o bicarbonato de un catión metálico o con amoníaco o una amina orgánica primaria, secundaria o terciaria. Los cationes de las sales terapéuticamente aceptables incluyen litio, sodio, potasio, calcio, magnesio y aluminio, así como cationes de amina cuaternaria no tóxicos como amonio, tetrametilamonio, tetraetilamonio, metilamina, dimetilamina, trimetilamina, trietilamina, dietilamina, etilamina, tributilamina, piridina, N,N-dimetilanilina, N-metilpiperidina, N-metilmorfolina, diciclohexilamina, procaína, dibencilamina, N, N-dibencilfenetilamina, 1-efenamina y N,P-dibenciltilendiamina. Otras aminas orgánicas representativas útiles para la formación de sales de adición de bases son la etilendiamina, la etanolamina, la dietanolamina, la piperidina y la piperazina.
Si bien es posible que los compuestos de la invención en cuestión se administren como producto químico en bruto, también es posible presentarlos como formulación farmacéutica. En consecuencia, se proporcionan en el presente documento formulaciones farmacéuticas que comprenden uno o más de ciertos compuestos divulgados en el presente documento, o una o más sales, ésteres, profármacos, amidas o solvatos farmacéuticamente aceptables de los mismos, junto con uno o más portadores farmacéuticamente aceptables de los mismos y, opcionalmente, uno o más ingredientes terapéuticos. El portador o portadores deben ser "aceptables" en el sentido de ser compatibles con los demás ingredientes de la formulación y no perjudiciales para el receptor de la misma. La formulación adecuada depende de la vía de administración elegida. Se puede utilizar cualquiera de las técnicas, soportes y excipientes conocidos según convenga y se entienda en la técnica; por ejemplo, en Remington's Pharmaceutical Sciences. Las composiciones farmacéuticas aquí divulgadas pueden fabricarse de cualquier manera conocida en la técnica, por ejemplo, mediante procesos convencionales de mezcla, disolución, granulación, drageificación, levigación, emulsión, encapsulación, atrapamiento o compresión.
Por anillos heterocíclicos ("het") se entienden anillos heterocíclicos saturados o insaturados de cinco, seis o siete miembros o heteroanillos bicíclicos de 5-10 miembros que pueden contener uno, dos o tres heteroátomos, seleccionados entre oxígeno, azufre y nitrógeno; el anillo puede estar unido a la molécula por un átomo de carbono o, si está presente, por un átomo de nitrógeno. Los siguientes son ejemplos de anillos heterocíclicos de cinco, seis o siete miembros, saturados o insaturados:

A menos que se indique lo contrario, un anillo heterocíclico puede estar provisto de un grupo ceto. Algunos ejemplos son:

Ejemplos de heteroanillos bicíclicos de 5-10 miembros son pirrolizina, indol, indolizina, isoindol, indazol, purina, quinoleína, isoquinolina, bencimidazol, benzofurano, benzopirano, benzotiazol, benzoisotiazol, piridopirimidina, pteridina, pirimidopirimidina,

Aunque el término anillos heterocíclicos incluye grupos aromáticos heterocíclicos, el término grupos aromáticos heterocíclicos ("hetaril") denota grupos aromáticos heterocíclicos de cinco o seis miembros o anillos heterocíclicos bicíclicos de 5-10 miembros que pueden contener uno, dos o tres heteroátomos, seleccionados entre oxígeno, azufre y nitrógeno, que contienen suficientes dobles enlaces conjugados como para que se forme un sistema aromático. El anillo puede estar unido a la molécula a través de un átomo de carbono o, si está presente, a través de un átomo de nitrógeno. Los siguientes son ejemplos de grupos aromáticos heterocíclicos de cinco o seis miembros:

Ejemplos de anillos heterómeros bicíclicos de 5-10 miembros incluyen pirrolizina, indol, indolizina, isoindol, indazol, purina, quinoleína, isoquinolina, bencimidazol, benzofurano, benzopirano, benzotiazol, benzoisotiazol, piridopirimidina, pteridina, pirimidopirimidina.
El término "halógeno", tal como se utiliza aquí, significa un sustituyente halógeno seleccionado entre fluoro, cloro, bromo o yodo.
Por el término "alquilo C W (incluidos los que forman parte de otros grupos) se entienden los grupos alquilo ramificados y no ramificados con 1 a 6 átomos de carbono, y por el término "alquilo C i-4 " se entienden los grupos alquilo ramificados y no ramificados con 1 a 4 átomos de carbono. En algunos casos están presentes grupos alquilo con 1 a 4 átomos de carbono. Algunos ejemplos son: metilo, etilo, n-pmpilo, iso-propilo, n-butilo, iso-butilo, sec-butilo, tercbutilo, n-pentilo, iso-pentilo, neo-pentiloo hexilo. Las abreviaturas Me, Et, n-Pr, i-Pr, n-Bu, i-Bu, t-Bu, etc. también pueden utilizarse opcionalmente para los grupos mencionados. Salvo que se indique lo contrario, las definiciones propilo, butilo, pentilo y hexilo incluyen todas las formas isoméricas posibles de los grupos en cuestión. Así, por ejemplo, el propilo incluye el n-propiloy el iso-propilo, el butilo incluye el iso-butilo, el sec-butilo y el terc-butilo, etc.
Por el término "alquileno C1-6" (incluidos los que forman parte de otros grupos) se entienden los grupos alquileno ramificados y no ramificados con 1 a 6 átomos de carbono y por el término "alquileno C1-4" se entienden los grupos alquileno ramificados y no ramificados con 1 a 4 átomos de carbono. En algunos casos están presentes grupos alquileno con 1 a 4 átomos de carbono. Ejemplos: metileno, etileno, propileno, 1-metiletileno, butileno, 1-metilpropileno,
1.1- dimetiletileno, 1,2-dimetiletileno, pentileno, 1,1-dimetilpropileno, 2,2-dimetilpropileno, 1,2-dimetilpropileno, 1,3-dimetilpropileno o hexileno. Salvo que se indique lo contrario, las definiciones propileno, butileno, pentileno y hexileno incluyen también todas las formas isoméricas posibles de los grupos correspondientes con el mismo número de carbonos. Así, por ejemplo, el propileno también incluye el 1-metiletileno y el butileno incluye el 1-metilpropileno, el 1.1- dimetiletileno, el 1,2-dimetiletileno.
El término "alquenilo C2-6" (incluidos los que forman parte de otros grupos) denota grupos alquenilo ramificados y no ramificados con 2 a 6 átomos de carbono y el término "alquenilo C2-4" denota grupos alquenilo ramificados y no ramificados con 2 a 4 átomos de carbono, siempre que tengan al menos un doble enlace. En algunas realizaciones son interesantes los grupos alquenilo con 2 a 4 átomos de carbono. Algunos ejemplos son: etenilo o vinilo, propenilo, butenilo, pentenilo o hexenilo. Salvo que se indique lo contrario, las definiciones propenilo, butenilo, pentenilo y hexenilo incluyen todas las formas isoméricas posibles de los grupos en cuestión. Así, por ejemplo, el propenilo incluye el 1-propenilo y el 2-propenilo, el butenilo incluye el 1-, 2- y 3-butenilo, el 1-metil-1-propenilo, el 1-metil-2-propenilo, etc.
Por el término "alquenileno C2-6" (incluidos los que forman parte de otros grupos) se entienden los grupos alquenileno ramificados y no ramificados con 2 a 6 átomos de carbono y por el término "alquenileno C2-4" se entienden los grupos alquenileno ramificados y no ramificados con 2 a 4 átomos de carbono. En algunos casos están presentes grupos alquenileno con 2 a 4 átomos de carbono. Ejemplos incluyen: etenileno, propenileno, 1-metiletenileno, butenileno, 1-metilpropenileno, 1,1- dimetiletenileno, 1,2-dimetiletenileno, pentenileno, 1,1-metilpropenileno, 2,2-metilpropenileno, 1.2- metilpropenileno, 1,3-metilpropenileno o hexenileno. Salvo que se indique lo contrario, las definiciones propenileno, butenileno, pentenileno y hexenileno incluyen todas las formas isoméricas posibles de los grupos respectivos con el mismo número de carbonos. Así, por ejemplo, el propenilo también incluye el 1-metiletenileno y el butenileno incluye el 1-metilpropenileno, el 1,1- dimetiletenileno, el 1,2- dimetiletenileno.
Por el término "alquinilo C2-6" (incluidos los que forman parte de otros grupos) se entienden los grupos alquinilo ramificados y no ramificados con 2 a 6 átomos de carbono y por el término "alquinilo C2-4" se entienden los grupos alquinilo ramificados y no ramificados con 2 a 4 átomos de carbono, siempre que tengan al menos un triple enlace. En algunos casos están presentes grupos alquinilo con 2 a 4 átomos de carbono. Algunos ejemplos son: etinilo, propinilo, butinilo, pentinilo, o hexinilo. Salvo que se indique lo contrario, las definiciones propinilo, butinilo, pentinilo y hexinilo incluyen todas las formas isoméricas posibles de los grupos respectivos. Así, por ejemplo, el propinilo incluye el 1-propinilo y el 2-propinilo, el butinilo incluye el 1-, 2- y 3-butinilo, el 1 -metil-1-propinilo, el 1 -metil-2-propinilo, etc.
Por el término "alquinileno C2-6" (incluidos los que forman parte de otros grupos) se entienden los grupos alquinileno ramificados y no ramificados con 2 a 6 átomos de carbono y por el término "alquinileno C2-4" se entienden los grupos alquileno ramificados y no ramificados con 2 a 4 átomos de carbono. En algunos casos están presentes grupos alquinileno con 2 a 4 átomos de carbono. Los ejemplos incluyen: etinileno, propinileno, 1 -metiletinileno, butinileno, 1-metilpropinileno, 1,1-dimetiletinileno, 1,2-dimetiletinileno, pentinileno, 1,1-dimetilpropinileno, 2,2-dimetilpropinileno, 1.2- dimetilpropinileno, 1,3-dimetilpropinileno o hexinileno. Salvo que se indique lo contrario, las definiciones propinileno, butinileno, pentinileno y hexinileno incluyen todas las formas isoméricas posibles de los grupos respectivos con el mismo número de carbonos. Así, por ejemplo, el propinileno incluye también el 1 -metiletinileno y butinileno incluye 1-metilpropinileno, 1,1-dimetiletinileno, 1,2-dimetiletinileno
El término "cicloalquilo C3-6" (incluidos los que forman parte de otros grupos) tal como se utiliza en el presente documento significa grupos alquilo cíclicos con 3 a 8 átomos de carbono, de interés en ciertas realizaciones son los grupos alquilo cíclicos con 5 a 6 átomos de carbono. Ejemplos: ciclopropilo, ciclobutilo, ciclopentilo o ciclohexilo.
Por el término "haloalquilo C1-6" (incluidos los que forman parte de otros grupos) se entienden grupos alquilo ramificados y no ramificados con 1 a 6 átomos de carbono en los que uno o más átomos de hidrógeno se sustituyen por un átomo de halógeno seleccionado entre flúor, cloro o bromo, como flúor y cloro, donde en algunos casos el átomo de halógeno es flúor. Por el término "haloalquilo C1-4" se entienden grupos alquilo ramificados y no ramificados con 1 a 4 átomos de carbono, en los que uno o más átomos de hidrógeno se sustituyen de forma análoga a lo indicado anteriormente. El haloalquilo C1-4 está presente en algunos casos. Ejemplos incluyen: CH2 F, CHF2 , CF3.
El término "alquilo C-i-n", donde n es un número entero de 2 a n, solo o en combinación con otro radical denota un radical hidrocarburo acíclico, saturado, ramificado o lineal con 1 a n átomos de C. Por ejemplo, el término alquilo C1-5 abarca los radicales H3C-, H3C-CH2-, H3C-CH2-CH2-, H3C-CH(CH3 )-, H3C-CH2-CH2-CH2-, H3C-CH2-CH(CH3 )-, H3C-CH(CH3)-CH2-, H3C-C(CH3)2-, H3C-CH2-CH2-CH2-CH2-, H3C-CH2-CH2-CH(CH3)-, H3C-CH2-CH(CH3)-CH2-, H3C-CH(CH3 )-CH2-CH2-, H3C-CH2-C(CH3 )2-, H3C-C(CH3 )2-CH2-, H3C-CH(CH3 )-CH(CH3)- y H3C-CH2-CH(CH2CH3 )-.
El término "haloalquilo C-un", donde n es un número entero de 2 a n, solo o en combinación con otro radical denota un radical hidrocarburo acíclico, saturado, ramificado o lineal con 1 a n átomos de C en el que uno o más átomos de hidrógeno se sustituyen por un átomo de halógeno seleccionado entre flúor, cloro o bromo, como flúor y cloro, donde en algunos casos el halógeno es flúor. Algunos ejemplos son: CH2 F, CHF2 , CF3
El término "alquileno C-un" donde n es un número entero de 2 a n, solo o en combinación con otro radical, denota un radical alquilo divalente acíclico, de cadena recta o ramificada que contiene de 1 a n átomos de carbono. --CH2-, -CH2CH2-, -CH(CHa)-, -CH2-CH2-CH2-, -C(CHa)2-, -CH(CH2CHs )-, -CH( CH3 K H 2-, -CH2-CH(CHs )-, -CH2-CH2-CH2-CH2-, -CH2-CH2-CH(CHa)-, -CH(CHs )-CH2-C H2-, -CH2-CH(CHa)-CH2-, -CH2-C(CHs )2-, -C(CHs )2-CH2-, -CH(CHa )-CH(CHa)-, -CH2-CH( CH2CH3)-, -CH(CH2CH3)-CH2-, -CH(CH2CH2CH3)-, -CH(CH(CH3))2- y -C(CH3)(CH2CH 3)- .
El término "alquenilo C2-n", se utiliza para un grupo como el definido en la definición de "alquilo Ci -n " con al menos dos átomos de carbono, si al menos dos de esos átomos de carbono de dicho grupo están unidos entre sí por un doble enlace.
El término "alquinilo C2-n", se utiliza para un grupo como el definido en la definición de "alquilo C1-n" con al menos dos átomos de carbono, si al menos dos de esos átomos de carbono de dicho grupo están unidos entre sí por un triple enlace.
El término "cicloalquilo C3-n", en el que n es un número entero de 4 a n, solo o en combinación con otro radical denota un radical hidrocarburo cíclico, saturado, no ramificado con 3 a n átomos de C. Por ejemplo, el término cicloalquilo C3-7 incluye ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo y cicloheptilo.
g. Combinaciones
Los compuestos de fórmula general 2 pueden utilizarse solos o combinados con otras sustancias activas de fórmula 2 según la invención. Los compuestos de fórmula general 2 también pueden combinarse opcionalmente con otras sustancias farmacológicamente activas. Entre ellos, agonistas de los receptores adrenérgicos p2 (de acción corta y prolongada), anticolinérgicos (de acción corta y prolongada), esteroides antiinflamatorios (corticosteroides orales y tópicos), cromoglicato, metilxantina, miméticos de glucocorticoides disociados, inhibidores de la PDE3, inhibidores de la PDE4, inhibidores de la PDE7, antagonistas de la LTD4, inhibidores del EGFR, Agonistas de la dopamina, antagonistas del PAF, derivados de la lipoxina A4, moduladores del FPRL1, antagonistas del receptor LTB4 (BLT1, BLT2), antagonistas del receptor de la histamina H1, antagonistas del receptor de la histamina H4, antagonistas duales del receptor de la histamina H1/H3, inhibidores de la PI3-quinasa, inhibidores de tirosina quinasas no receptoras como, por ejemplo, LYN, LCK, SYK, ZAP-70, FYN, BTK o ITK, inhibidores de MAP quinasas como por ejemplo p38, ERK1, ERK2, JNK1, JNK2, JNK3 o SAP, inhibidores de la vía de señalización NF-kB como por ejemplo inhibidores de la quinasa IKK2, inhibidores de iNOS, inhibidores de MRP4, inhibidores de la biosíntesis de leucotrienos como por ejemplo inhibidores de 5-Lipoxigenasa (5-LO), inhibidores de cPLA2, Inhibidores de la leucotrieno A4 hidrolasa o inhibidores de FLAP, agentes antiinflamatorios no esteroideos (AINE), antagonistas de CRTH2, moduladores del receptor DP1, antagonistas del receptor de tromboxano, antagonistas de CCR3, antagonistas de CCR4 , antagonistas de CCR1, antagonistas de CCR5, antagonistas de CCR6, antagonistas de CCR7, antagonistas de CCR8, antagonistas de CCR9, Antagonistas de CCR30,, antagonistas de CXCR3 , antagonistas de CXCR4 , antagonistas de CXCR2, antagonistas de CXCR1, antagonistas de CXCR5, antagonistas de CXCR6, antagonistas de CX3CR3 , antagonistas de neurocinina (NK1, NK2), moduladores de los receptores de esfingosina 1-fosfato, inhibidores de la esfingosina 1-fosfato liasa, moduladores de los receptores de adenosina como por ejemplo los agonistas A2a, moduladores de los receptores purinérgicos como por ejemplo los inhibidores de P2X7, activadores de la histona desacetilasa (HDAC), antagonistas de la bradicinina (BK1, BK2), inhibidores de la TACE, moduladores de PPAR gamma, inhibidores de la Rho-quinasa, inhibidores de la enzima convertidora de la interleucina 1-beta (ICE), moduladores de los receptores Toll-Like (TLR), inhibidores de la HMG-CoA reductasa, Antagonistas de VLA-4, inhibidores de ICAM-1, agonistas de SHIP, antagonistas de receptores GABAa, inhibidores de ENaC, moduladores de receptores de melanocortina (MC1R, MC2R, MC3R, MC4R, MC5R), antagonistas de CGRP, antagonistas de endotelina, antagonistas de TNFa, anticuerpos anti-TNF, anticuerpos anti-GM-CSF, anticuerpos anti-CD46, anticuerpos anti-IL-1, anticuerpos anti-IL-2, anticuerpos anti-II,-4, anticuerpos anti-II,-5, anticuerpos anti-II,-13, anticuerpos anti-IL-4/IL-13, anticuerpos anti-TSLP, anticuerpos anti-OX40, mucorreguladores, agentes inmunoterapéuticos, compuestos contra la inflamación de las vías respiratorias, compuestos contra la tos, inhibidores del VEGF, pero también combinaciones de dos o tres sustancias activas.
También se contemplan los betamiméticos, los anticolinérgicos, los corticosteroides, los inhibidores de la PDE4, los antagonistas de la LTD4, los inhibidores del EGFR, los inhibidores de la CRTH2, los inhibidores de la 5-LO, los antagonistas de los receptores de la histamina y los inhibidores de la SYK, pero también las combinaciones de dos o tres sustancias activas, por ejemplo betamiméticos con corticosteroides, inhibidores de la PDE4, inhibidores de la CRTH2 o antagonistas de la LTD4; anticolinérgicos con betamiméticos, corticosteroides, inhibidores de la PDE4, inhibidores de la CRTH2 o antagonistas de la LTD4, corticosteroides con inhibidores de la PDE4, inhibidores de la CRTH2 o antagonistas de la LTD4; Inhibidores de la PDE4 con inhibidores de la CRTH2 o antagonistas de la LTD4; e inhibidores de la CRTH2 con antagonistas de la LTD4.
También se contemplan en la invención los compuestos de fórmula general 2 más una terapia anti-VEGF (por ejemplo, uno o más moduladores e inhibidores de VEGF) para su uso en un tratamiento combinado de las indicaciones aquí divulgadas. En varios aspectos, el tratamiento comprende mejorar la agudeza visual administrando un compuesto de fórmula 1 y administrando además una terapia anti-VEGF que reduce el efecto biológico del VEGF en el sitio diana. Los moduladores/inhibidores de VEGF incluyen, entre otros, anticuerpos e inhibidores de VEGF-A de molécula pequeña, como ranibizumab, bevacizumab y aflibercept, que se enumeran a modo de ejemplo y no de limitación. También se contemplan constructos similares a anticuerpos anti-VEGF, como el fragmento de anticuerpo de cadena única Inhibidor del VEGF brolucizumab (también conocido como RTH258). Además, los compuestos de fórmula
general 2 pueden utilizarse en combinación con moduladores o inhibidores del receptor de VEGF, como anticuerpos e inhibidores de moléculas pequeñas del receptor de VEGF o de mecanismos de señalización descendentes. Los ejemplos, a título enunciativo y no limitativo, incluyen sunitnib, sorafenib, cabozantinib, ponantinib y axitinib. La divulgación contempla un régimen terapéutico que comprende la administración del compuesto de fórmula 2 y la administración de uno o más de los siguientes: un aptámero que inhibe la expresión o función de VEGF, como pegaptanib sódico (Macugen, Eyetech Pharmaceuticals, Cedar Knolls, NJ; un aptámero que se une selectivamente a la isoforma VEGFA 165); un inhibidor de tirosina quinasa (por ejemplo, PAN-90806, un colirio tópico anti-VEGF, PanOptica, Bernardsville, NJ); ARNip (por ejemplo, Bevasiranib); VEGf soluble o un vector de terapia génica que codifica un VEGF soluble, como RGX-314 (Regenxbio Inc); o una proteína receptora de VEGF soluble, como conbercept (una fusión del dominio 2 de VEGFR-1, los dominios 3 y 4 de VEGFR-2, y la región Fc de IgG1).
En estos tratamientos, los compuestos que forman la combinación se coadministran a un sujeto. Los términos "coadministración" y "en combinación con" incluyen la administración de dos o más agentes terapéuticos de forma simultánea, concurrente o secuencial sin límites de tiempo específicos. Los agentes pueden estar presentes en la célula o en el cuerpo del sujeto al mismo tiempo o ejercer su efecto biológico o terapéutico al mismo tiempo. En una realización, los agentes terapéuticos se encuentran en la misma composición o forma farmacéutica unitaria. En otras realizaciones, los agentes terapéuticos se encuentran en composiciones separadas o formas de dosificación unitarias. Un primer agente puede administrarse antes de (por ejemplo, minutos, 15 minutos, 30 minutos, 45 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 12 horas, 24 horas, 48 horas, 72 horas, 96 horas, 1 semana, 2 semanas, 3 semanas, 4 semanas, 5 semanas, 6 semanas, 8 semanas o 12 semanas antes), concomitantemente con, o posteriormente a (por ejemplo, 5 minutos, 15 minutos, 30 minutos, 45 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 12 horas, 24 horas, 48 horas, 72 horas, 96 horas, 1 semana, 2 semanas, 3 semanas, 4 semanas, 5 semanas, 6 semanas, 8 semanas o 12 semanas después) de la administración de un segundo agente terapéutico. "Administración concomitante" de un fármaco terapéutico conocido con una composición farmacéutica de la presente divulgación significa la administración del compuesto y del segundo agente en el momento en que tanto el fármaco conocido como la composición de la presente invención tendrán un efecto terapéutico. Dicha administración concomitante puede implicar la administración concurrente (es decir, al mismo tiempo), previa o posterior del fármaco con respecto a la administración de un compuesto sujeto. Las vías de administración de los dos agentes pueden variar, y las vías de administración representativas se describen con más detalle a continuación. Una persona experta en la materia no tendría dificultad en determinar el momento, la secuencia y las dosis de administración adecuados para determinados fármacos y compuestos de la presente divulgación. Los compuestos (por ejemplo, un compuesto del sujeto y al menos un compuesto adicional) pueden administrarse al sujeto con un intervalo de veinticuatro horas, por ejemplo, con un intervalo de 12 horas, 6 horas, 3 horas o 1 hora. Los compuestos pueden administrarse con 1 hora de diferencia. Los compuestos pueden administrarse sustancialmente de forma simultánea. Por administrados sustancialmente de forma simultánea se entiende que los compuestos se administran al sujeto con una diferencia de aproximadamente 10 minutos o menos entre sí, como 5 minutos o menos, o 1 minuto o menos entre sí.
h. Formas farmacéuticas
Los preparados adecuados para administrar las formas cocristalinas de las fórmulas 2 y 2a incluyen, por ejemplo, comprimidos, cápsulas, supositorios, soluciones y polvos, etc. El contenido de compuesto(s) farmacéuticamente activo(s) debe estar en el intervalo de 0,05 a 90 % en peso, como de 0,1 a 50 % en peso de la composición en su conjunto. Pueden obtenerse comprimidos adecuados, por ejemplo, mezclando la(s) sustancia(s) activa(s) con excipientes conocidos, por ejemplo diluyentes inertes como carbonato cálcico, fosfato cálcico o lactosa, desintegrantes como almidón de maíz o ácido algínico, aglutinantes como almidón o gelatina, lubricantes como estearato de magnesio o talco y/o agentes para retardar la liberación, como carboximetilcelulosa, ftalato de acetato de celulosa o acetato de polivinilo. Los comprimidos también pueden constar de varias capas.
Los comprimidos recubiertos pueden prepararse en consecuencia recubriendo los núcleos producidos de forma análoga a los comprimidos con sustancias utilizadas normalmente para recubrir comprimidos, por ejemplo, colidona o goma laca, goma arábiga, talco, dióxido de titanio o azúcar. Para conseguir una liberación retardada o evitar incompatibilidades, el núcleo también puede constar de varias capas. Del mismo modo, el recubrimiento del comprimido puede consistir en una serie o capas para lograr la liberación retardada, posiblemente utilizando los excipientes mencionados anteriormente para los comprimidos.
Los jarabes o elixires que contienen las sustancias activas o combinaciones de las mismas según la invención pueden contener además un edulcorante como sacarina, ciclamato, glicerol o azúcar y un potenciador del sabor, por ejemplo, un aromatizante como vainillina o extracto de naranja. También pueden contener adyuvantes de suspensión o espesantes como la carboximetilcelulosa sódica, agentes humectantes como, por ejemplo, productos de condensación de alcoholes grasos con óxido de etileno, o conservantes como los p-hidroxibenzoatos.
Las soluciones se preparan de la manera habitual, por ejemplo, con la adición de agentes isotónicos, conservantes como p-hidroxibenzoatos o estabilizantes como sales de metales alcalinos del ácido etilendiaminotetraacético, utilizando opcionalmente emulsionantes y/o dispersantes, mientras que si se utiliza agua como diluyente, por ejemplo, pueden utilizarse opcionalmente disolventes orgánicos como solubilizantes o auxiliares de disolución, y las soluciones pueden transferirse a viales o ampollas de inyección o frascos de infusión.
Las cápsulas que contienen una o más sustancias activas o combinaciones de sustancias activas pueden prepararse, por ejemplo, mezclando las sustancias activas con portadores inertes como la lactosa o el sorbitol y envasándolas en cápsulas de gelatina.
Los supositorios adecuados pueden elaborarse, por ejemplo, mezclándolos con portadores previstos para este fin, como grasas neutras o polietilenglicol o sus derivados.
Los excipientes que pueden utilizarse incluyen, por ejemplo, agua, disolventes orgánicos farmacéuticamente aceptables como parafinas (por ejemplo, fracciones de petróleo), aceites vegetales (por ejemplo, aceite de cacahuete o de sésamo), alcoholes mono o polifuncionales (por ejemplo, etanol o glicerol), portadores como, por ejemplo, polvos minerales naturales (por ejemplo, caolines, arcillas, talco, creta), polvos minerales sintéticos (por ejemplo, ácido silícico y silicatos muy dispersos), azúcares (por ejemplo, azúcar de caña, lactosa y glucosa), emulgentes (por ejemplo, lignina, licores de sulfito usados, metilcelulosa, almidón y polivinilpirrolidona) y lubricantes (por ejemplo, estearato de magnesio, talco, ácido esteárico y laurilsulfato de sodio).
Para uso oral, los comprimidos pueden contener obviamente, además de los portadores especificados, aditivos como citrato de sodio, carbonato de calcio y fosfato dicálcico, junto con diversas sustancias adicionales como almidón, por ejemplo almidón de patata, gelatina y similares. También pueden utilizarse lubricantes como estearato de magnesio, laurilsulfato de sodio y talco para producir los comprimidos. En el caso de las suspensiones acuosas, las sustancias activas pueden combinarse con diversos potenciadores del sabor o colorantes, además de los excipientes mencionados.
Para administrar las formas cocristalinas de las fórmulas 2 y 2a en algunos casos se emplean preparaciones o formulaciones farmacéuticas que son adecuadas para la inhalación. Los preparados inhalables incluyen polvos inhalables, aerosoles de dosis medida con propelente o soluciones inhalables sin propelente. Dentro del ámbito de la presente invención, el término soluciones inhalables sin propelente también incluye concentrados o soluciones inhalables estériles listas para su uso. Las formulaciones que pueden utilizarse dentro del ámbito de la presente invención se describen con más detalle en la siguiente parte de la especificación.
Los polvos inhalables que pueden utilizarse de acuerdo con la invención pueden contener una forma cocristalina de las fórmulas 2 y 2a, ya sea sola o mezclada con excipientes adecuados fisiológicamente aceptables.
Si las sustancias activas de las formas cocristalinas de las fórmulas 2 y 2a están presentes en mezcla con excipientes fisiológicamente aceptables, pueden usarse los siguientes excipientes fisiológicamente aceptables para preparar estos polvos inhalables según la invención: monosacáridos (p. ej. glucosa o arabinosa), disacáridos (p. ej. lactosa, sacarosa, maltosa), oligo- y polisacáridos (p. ej., dextranos), polialcoholes (por ejemplo, sorbitol, manitol, xilitol), sales (por ejemplo, cloruro de sodio, carbonato de calcio) o mezclas de estos excipientes. En algunos casos, se utilizan mono o disacáridos, como la lactosa o la glucosa, por ejemplo, en forma de sus hidratos. En algunos casos, la lactosa, por ejemplo en forma de lactosa monohidrato, se emplea como excipiente.
En el ámbito de los polvos inhalables según la invención, los excipientes tienen un tamaño medio de partícula máximo de hasta 250 μm, tal como entre 10 y 150 μm, e incluyendo entre 15 y 80 μm. Aveces puede parecer apropiado añadir fracciones de excipiente más finas con un tamaño medio de partícula de 1 a 9 μm al excipiente mencionado anteriormente. Estos excipientes más finos también se seleccionan del grupo de posibles excipientes enumerados anteriormente. Por último, para preparar los polvos inhalables según la invención, se añade a la mezcla de excipientes la sustancia activa micronizada de las formas cocristalinas de las fórmulas 2 y 2a, por ejemplo con un tamaño medio de partícula de 0,5 a 10 μm, incluyendo de 1 a 5 μm. Los procesos para producir los polvos inhalables según la invención moliendo y micronizando y finalmente mezclando los ingredientes son conocidos en la técnica anterior.
Los polvos inhalables según la invención pueden administrarse utilizando inhaladores conocidos en la técnica anterior.
Los aerosoles de inhalación que contienen gas propulsor según la invención pueden contener una forma cocristalina de las fórmulas 2 y 2a disuelta en el gas propulsor o en forma dispersa. Las formas cocristalinas de las fórmulas 2 y 2a pueden estar contenidas en formulaciones separadas o en una formulación común, en la que ambas están disueltas, ambas dispersas o, en cada caso, sólo un componente está disuelto y el otro disperso. Los gases propulsores que pueden utilizarse para preparar los aerosoles de inhalación son conocidos en la técnica anterior. Los gases propulsores adecuados se seleccionan entre hidrocarburos como el n-propanol, el n-butano o el isobutano y los halohidrocarburos como los derivados fluorados del metano, etano, propano, butano, ciclopropano o ciclobutano. Los gases propulsores mencionados pueden utilizarse solos o mezclados entre sí. Los gases propulsores específicos que pueden emplearse son derivados de alcanos halogenados seleccionados de TG134a y TG227 y sus mezclas.
Los aerosoles de inhalación accionados por propelente también pueden contener otros ingredientes como co solventes, estabilizantes, tensioactivos, antioxidantes, lubricantes y ajustadores del pH. Todos estos ingredientes son conocidos en la técnica.
Los aerosoles de inhalación accionados por propelente según la invención mencionada anteriormente pueden administrarse utilizando inhaladores conocidos en la técnica (MDI = inhaladores dosificadores).
Además, las sustancias activas de las formas cocristalinas de las fórmulas 2 y 2a según la invención pueden administrarse en forma de soluciones y suspensiones inhalables sin propelente. El disolvente utilizado puede ser acuoso o alcohólico, como una solución etanólica. El disolvente puede ser agua sola o una mezcla de agua y etanol. La proporción relativa de etanol en comparación con el agua no está limitada, pero el máximo puede ser, en algunos casos, de hasta el 70 por ciento en volumen, como hasta el 60 por ciento en volumen e incluso hasta el 30 por ciento en volumen. El resto del volumen puede estar constituido por agua. Las soluciones o suspensiones que contienen una forma cocristalina de las fórmulas 2 y 2a se ajustan a un pH de 2 a 7, por ejemplo de 2 a 5, utilizando ácidos adecuados. El pH puede ajustarse utilizando ácidos seleccionados entre ácidos inorgánicos u orgánicos. Algunos ejemplos de ácidos inorgánicos especialmente adecuados son el ácido clorhídrico, el ácido bromhídrico, el ácido nítrico, el ácido sulfúrico y/o el ácido fosfórico. Algunos ejemplos de ácidos orgánicos especialmente adecuados son el ácido ascórbico, el ácido cítrico, el ácido málico, el ácido tartárico, el ácido maleico, el ácido succínico, el ácido fumárico, el ácido acético, el ácido fórmico y/o el ácido propiónico, etc. Los ácidos inorgánicos que se emplean en algunos casos son los ácidos clorhídrico y sulfúrico. También es posible utilizar los ácidos que ya han formado una sal de adición ácida con una de las sustancias activas. De los ácidos orgánicos, el ácido ascórbico, el ácido fumárico y el ácido cítrico se emplean en algunos casos. Si se desea, pueden utilizarse mezclas de los ácidos anteriores, especialmente en el caso de ácidos que tengan otras propiedades además de sus cualidades acidificantes, por ejemplo, como aromatizantes, antioxidantes o agentes complejantes, como el ácido cítrico o el ácido ascórbico, por ejemplo. Según la invención, en algunos casos ácido clorhídrico para ajustar el pH.
Si se desea, puede omitirse en estas formulaciones la adición de ácido edítico (EDTA) o una de sus sales conocidas, edetato sódico, como estabilizador o agente complejante. Otras realizaciones pueden contener este compuesto o estos compuestos. En una realización, el contenido basado en edetato sódico es inferior a 100 mg/l00ml, como por ejemplo inferior a 50mg/l00ml, incluyendo menos de 20mg/l00ml. En algunos casos se emplean soluciones inhalables con un contenido de edetato sódico de 0 a 10 mg/l00 ml. A las soluciones inhalables sin propelente pueden añadirse co-solventes y/u otros excipientes. Los co-disolventes que pueden emplearse son los que contienen grupos hidroxilo u otros grupos polares, por ejemplo, alcoholes -en particular alcohol isopropílico-, glicoles -en particular propilenglicol, polietilenglicol, polipropilenglicol-, glicoléter, glicerol, alcoholes polioxietilénicos y ésteres polioxietilénicos de ácidos grasos. Los términos excipientes y aditivos en este contexto denotan cualquier sustancia farmacológicamente aceptable que no es una sustancia activa pero que puede formularse con la sustancia o sustancias activas en el disolvente fisiológicamente adecuado para mejorar las propiedades cualitativas de la formulación de la sustancia activa. En algunos casos, estas sustancias no tienen ningún efecto farmacológico o, en relación con la terapia deseada, ningún efecto farmacológico apreciable o al menos indeseable. Los excipientes y aditivos incluyen, por ejemplo, tensioactivos como lecitina de soja, ácido oleico, ésteres de sorbitán, como polisorbatos, polivinilpirrolidona, otros estabilizantes, agentes complejantes, antioxidantes y/o conservantes que garanticen o prolonguen la vida útil de la formulación farmacéutica acabada, aromatizantes, vitaminas y/u otros aditivos conocidos en la técnica. Los aditivos también incluyen sales farmacológicamente aceptables, como el cloruro sódico, como agentes isotónicos.
Los excipientes empleados en algunas realizaciones incluyen antioxidantes como el ácido ascórbico, por ejemplo, siempre que no se haya utilizado ya para ajustar el pH, vitamina A, vitamina E, tocoferoles y vitaminas y provitaminas similares presentes en el cuerpo humano.
Pueden utilizarse conservantes para proteger la formulación de la contaminación con agentes patógenos. Los conservantes adecuados son los conocidos en la técnica, en particular el cloruro de cetilpiridinio, el cloruro de benzalconio o el ácido benzoico o benzoatos como el benzoato sódico en la concentración conocida en la técnica anterior. Los conservantes mencionados anteriormente están presentes, en algunos casos, en concentraciones de hasta 50 mg/l00 ml, más preferentemente entre 5 y 20 mg/l00 ml. Las formulaciones de algunas realizaciones contienen, además del agua disolvente y las formas cocristalinas de las fórmulas 2 y 2a, sólo cloruro de benzalconio y edetato sódico. En una realización, no hay edetato de sodio presente.
Naturalmente, la dosificación de los compuestos según la invención depende en gran medida del procedimiento de administración y de la dolencia que se esté tratando. Cuando se administran por inhalación, las formas cocristalinas de las fórmulas 2 y 2a se caracterizan por una elevada potencia incluso a dosis en el intervalo de los |jg. Las formas cocristalinas de las fórmulas 2 y 2a también pueden utilizarse eficazmente por encima del intervalo de jg . En ese caso, la dosis puede ser de un gramo, por ejemplo.
En otro aspecto, la presente invención se refiere a las formulaciones farmacéuticas mencionadas anteriormente que se caracterizan porque contienen una forma cocristalina de las fórmulas 2 y 2a, en particular las formulaciones farmacéuticas mencionadas anteriormente que pueden administrarse por inhalación.
Los siguientes ejemplos de formulaciones ilustran la presente invención sin restringir su alcance.
Ejemplos de formulaciones farmacéuticas
A)

g
Se mezclan la sustancia activa finamente molida, la lactosa y parte del almidón de maíz. La mezcla se tamiza, se humedece con una solución de polivinilpirrolidona en agua, se amasa, se granula en húmedo y se seca. Los gránulos, el almidón de maíz restante y el estearato de magnesio se tamizan y se mezclan. La mezcla se prensa en comprimidos de forma y tamaño adecuados.
B)

La sustancia activa finamente molida, parte del almidón de maíz, la lactosa, la celulosa microcristalina y la polivinilpirrolidona se mezclan, la mezcla se tamiza y se trabaja con el resto del almidón de maíz y el agua para formar un granulado que se seca y se tamiza. Se añaden y mezclan el carboximetilalmidón sódico y el estearato de magnesio, y la mezcla se comprime para formar comprimidos de un tamaño adecuado.
C)

La sustancia activa se disuelve en agua a su propio pH u opcionalmente a un pH de 5,5 a 6,5 y se añade cloruro sódico para que la solución sea isotónica. La solución resultante se filtra para eliminar los pirógenos y el filtrado se transfiere en condiciones asépticas a ampollas que se esterilizan y termosellan. Las ampollas contienen 5 mg, 25 mg y 50 mg de principio activo.
D)

La suspensión se transfiere a un recipiente de aerosol convencional con válvula dosificadora. Preferentemente se liberan 50 μl de suspensión en cada accionamiento. Si se desea, el principio activo también puede liberarse en dosis más elevadas (por ejemplo, 0,02 % en peso).
E)

Esta solución puede prepararse de la forma habitual.
F)

El polvo inhalable se prepara de la forma habitual mezclando los distintos ingredientes.
i. Indicaciones
Se proporcionan procedimientos, que no forman parte de la invención, para mejorar la agudeza visual, incluyendo procedimientos para mejorar la agudeza visual mediante el tratamiento de un sujeto/paciente con enfermedades asociadas a la retina. En algunos aspectos, el sujeto padece (o se le ha diagnosticado que padece) una enfermedad asociada a la retina que provoca una pérdida de agudeza visual. La enfermedad asociada a la retina puede estar asociada a la neovascularización, o la enfermedad asociada a la retina puede no incluir un componente vascular (es decir, puede no derivarse de la neovascularización ni estar asociada a ella). Por ejemplo, el paciente puede, en varias realizaciones, sufrir una pérdida de agudeza visual que no esté causada por la neovascularización ocular. Aspectos de los procedimientos incluyen modular CCR3, por ejemplo, con un agente modulador de CCR3 de manera suficiente para mejorar la agudeza visual y, en varios aspectos, tratar al paciente de la enfermedad asociada a la retina. Los procedimientos incluyen la mejora de la agudeza visual (por ejemplo, mediante el tratamiento de la enfermedad asociada a la retina) con una composición biodisponible y administrable por vía oral, incluyendo una composición de un cocristal de las fórmulas 2 o 2a, o una formulación de la fórmula 3, descrita anteriormente. La composición, que modula CCR3, puede administrarse a un paciente/sujeto que necesite mejorar su agudeza visual, incluidos los sujetos diagnosticados con la enfermedad asociada a la retina, como la degeneración macular asociada a la edad (formas seca o húmeda), la oclusión de la vena central de la retina, la oclusión de la arteria central de la retina, el edema macular (como el edema macular diabético), el glaucoma, la enfermedad de Stargardt, la retinopatía del prematuro o la retinopatía diabética que se describen más adelante. Los procedimientos pueden comprender además el seguimiento de la mejora en la progresión de la enfermedad asociada a la retina mediante la agudeza visual u otras pruebas. A continuación se describen con más detalle varias indicaciones ejemplares de enfermedades asociadas a la retina.
j. Enfermedades asociadas a la retina Indicaciones
i. Degeneración macular
Degeneración macular. La degeneración macular es un término clínico que se utiliza para describir una familia de enfermedades que se caracterizan por una pérdida progresiva de la visión central asociada a anomalías de la membrana de Bruch, la coroides, la retina neural y/o el epitelio pigmentario de la retina. Estos trastornos incluyen dolencias muy comunes que afectan a sujetos de edad avanzada -como la degeneración macular asociada a la edad (DMAE)-, así como distrofias más raras y de aparición más temprana que, en algunos casos, pueden detectarse en la primera década de vida. Otras maculopatías son la distrofia macular de Carolina del Norte, la enfermedad de Best y la Malattia leventinese.
La DMAE es la principal causa de pérdida permanente de visión en personas mayores de 65 años y afecta actualmente a aproximadamente 15 millones de estadounidenses. La DMAE afecta a las células fotorreceptoras sensibles a la luz y a las células epiteliales pigmentadas de la mácula, el centro de la retina del ojo. Aunque puede no causar ceguera total, la enfermedad destruye la visión central, imposibilitando la lectura, la observación de pantallas de monitores electrónicos y la conducción. No tiene cura documentada, nunca ha demostrado remisión espontánea y los tratamientos eficaces son limitados, con una carga sustancial para el paciente y el cuidador, así como efectos secundarios.
La retina, o porción neural del ojo, es una complicada red de células nerviosas que transforma la luz en impulsos nerviosos que viajan hasta el cerebro, donde se interpretan como imágenes visuales. Existen cinco tipos de neuronas en la retina. Entre ellas se encuentran los fotorreceptores, las células bipolares, las células ganglionares, las células horizontales y las células amacrinas. La parte central de la retina, denominada mácula, es responsable de la visión
necesaria para leer y realizar otros trabajos detallados. Los daños en la mácula provocan una visión deficiente. El proceso patológico más común que afecta a la mácula es la DMAE. En los pacientes con DMAE, las células fotorreceptoras de la retina y las células epiteliales pigmentarias de la mácula mueren a lo largo de varios años. La muerte celular y la pérdida gradual de visión no suelen comenzar hasta los 60 años o más, de ahí el nombre de degeneración macular asociada a la edad.
Existen dos tipos de DMAE: degeneración macular seca y degeneración macular húmeda. La degeneración macular seca, aunque es más frecuente, suele provocar una pérdida de visión menos grave y más gradual. Los pacientes afectados por DMAE seca sufren una pérdida gradual de la visión central debido a la muerte de las células fotorreceptoras y de sus estrechas colaboradoras, las células epiteliales pigmentadas de la retina (EPR), con la deposición de una compleja mezcla amiloide cerosa, denominada "drusas". Los fotorreceptores, las células de la retina que realmente "ven" la luz, son esenciales para la visión. Las células macrófagas del EPR son necesarias para la supervivencia, la función y la renovación de los fotorreceptores. Los pacientes con degeneración macular húmeda desarrollan nuevos vasos sanguíneos bajo la retina. A medida que las células fotorreceptoras y del EPR degeneran lentamente, existe una tendencia a que los vasos sanguíneos crezcan desde su ubicación normal en la coroides hacia una ubicación anormal bajo la retina. Este crecimiento anormal de nuevos vasos sanguíneos se denomina neovascularización coroidea (NVC). Los vasos sanguíneos anormales tienen fugas y sangran, provocando hemorragias, hinchazón, tejido cicatricial y pérdida grave de la visión central. Sólo el 10 % de los pacientes con DMAE tienen el tipo húmedo, pero es responsable del 90 % de todas las cegueras derivadas de la DMAE.
ii. Oclusión de la vena central de la retina (OVCR)
La oclusión de la vena central de la retina, también conocida como OVCR, se produce cuando la oclusión venosa impide que la sangre desoxigenada fluya fuera de la vasculatura del ojo. Como consecuencia de la reducción del flujo de sangre desoxigenada en el ojo, se impide que la sangre rica en oxígeno llegue a las capas superficiales de la retina, lo que provoca un estado de hipoxia. En respuesta, las capas superficiales de la retina producen factores proangiogénicos que contribuyen al desarrollo de edema macular anormal y neovascularización. Una de las utilidades de los compuestos de fórmula 1 es el tratamiento del edema macular y la neovascularización resultantes de la OVCR.
iii. Retinopatía del prematuro
Afectando a bebés prematuros, la Retinopatía del Prematuro (ROP) es una enfermedad ocular asociada tanto a la toxicidad del oxígeno como a la hipoxia local. Se cree que estas condiciones contribuyen al desarrollo de la retinopatía del prematuro. La fisiopatología subyacente de la enfermedad es que las condiciones de hipoxia conducen a la estimulación de factores proangiogénicos que causan un crecimiento desorganizado de los vasos sanguíneos con el resultado de cicatrización y desprendimiento de retina. Algunos pacientes con retinopatía del prematuro pueden padecerla de forma leve y recuperarse totalmente sin intervención terapéutica, pero en otros puede conducir a la ceguera permanente. Se desconoce la causa exacta de la enfermedad, pero las principales hipótesis son que el oxígeno suplementario provoca una hipoxia local de la retina a través de la vasoconstricción que desencadena la neovascularización, o que los procesos vasculares normales se ven atenuados por el oxígeno suplementario, pero cuando se retira repentinamente se produce una rápida proliferación de la enfermedad vascular y fibrovascular. La cirugía y la intervención terapéutica son las terapias actuales para tratar la enfermedad en su forma grave. La terapia quirúrgica puede incluir la abrochadura de la esclerótica y/o la vitrectomía para el desprendimiento de retina. Sin embargo, la fotocoagulación inducida por láser es el pilar del tratamiento actual de la retinopatía del prematuro. Los compuestos de fórmula 1 tienen utilidad en la prevención de la neovascularización asociada a la retinopatía del prematuro. Los compuestos aquí descritos son útiles para mejorar la agudeza visual asociada a la retinopatía del prematuro.
iv. Retinopatía diabética
La retinopatía diabética es una complicación de la diabetes que puede causar ceguera. El mecanismo por el que se produce es el daño a los vasos sanguíneos del tejido retiniano. La retinopatía diabética puede desarrollarse en sujetos con diabetes de tipo 1 o de tipo 2, asociada a la pérdida de control del contenido de azúcar en sangre. Los síntomas incluyen manchas o moscas volantes en la visión del sujeto, visión borrosa, visión fluctuante, alteración de la visión de los colores, áreas oscuras/vacías de la visión y pérdida de visión, que suele afectar a ambos ojos. La causa de esta enfermedad es un exceso de azúcar en la sangre del sujeto, lo que conduce a la obstrucción de los vasos sanguíneos que nutren la retina y provoca el bloqueo del riego sanguíneo retiniano. En respuesta, la retina intenta hacer crecer nuevos vasos sanguíneos, produciendo factores proangiogénicos. Se produce una regulación inadecuada del crecimiento de estos vasos sanguíneos, lo que da lugar a vasos que gotean con facilidad.
Existen dos tipos de Retinopatía Diabética: Retinopatía diabética precoz o neuropatía diabética no proliferativa (NPDR) y retinopatía diabética avanzada. La NPDR se produce cuando no crecen nuevos vasos sanguíneos, lo que provoca el debilitamiento de las paredes de los vasos sanguíneos de la retina y la aparición de microaneurismas. Estos microaneurismas pueden sobresalir y filtrar líquido y sangre a la retina. A medida que se obstruyen más vasos sanguíneos, la NPDR se agrava. Las fibras nerviosas de la retina y la mácula (parte central de la retina) pueden inflamarse, lo que se conoce como edema macular. En la retinopatía diabética avanzada (o retinopatía diabética proliferativa), los vasos sanguíneos dañados se cierran, lo que provoca el crecimiento de vasos sanguíneos nuevos y
anormales, con la consiguiente filtración al vitreo del ojo. El tejido cicatricial resultante del crecimiento de nuevos vasos sanguíneos puede causar desprendimiento de retina, así como aumento de la presión ocular, lo que a la larga provoca daños en el nervio óptico y glaucoma.
k. Procedimientos de diagnóstico y seguimiento para mejorar las enfermedades asociadas a la retina y la agudeza visual (no forma parte de la invención)
i. Introducción
Los procedimientos comprenden además procedimientos de diagnóstico de enfermedades asociadas a la retina. Dichos procedimientos pueden incluir, a título de ejemplo y no limitativo, pruebas de agudeza visual (AV), degeneración macular o rejillas de Amsler, examen de la retina con pupilas dilatadas, fotografía del fondo de ojo, angiografía fluoresceínica o tomografía de coherencia óptica (OCT), que puede determinar puntos finales como el grosor de la retina central (CRT).
ii. Agudeza visual (AV)
Un procedimiento que puede diagnosticar o determinar la progresión/mejora de la enfermedad es la prueba de agudeza visual. Los procedimientos de comprobación de la agudeza visual son bien conocidos por los expertos en la materia. La agudeza visual evalúa la agudeza de la visión del sujeto, a menudo utilizando una "tabla optométrica", la más común de las cuales es la tabla optométrica de Snellen. Otros procedimientos de comprobación de la agudeza visual incluyen el uso de la tabla del Estudio de Retinopatía Diabética de Tratamiento Precoz (ETDRS), que, al igual que otras pruebas de AV, puede utilizarse para diagnosticar y medir la progresión/mejora de la agudeza visual de sujetos con enfermedades asociadas a la retina como, a modo de ejemplo y no limitativo, la degeneración macular, la oclusión de la vena central de la retina, la retinopatía del prematuro y la retinopatía diabética. (Véase Bokinni, Y, et al., Ojo 29: 1085-91 (2015)).
Un procedimiento para determinar la mejora de la agudeza visual en un paciente consiste en determinar si el sujeto, después del tratamiento, es capaz de identificar más letras en las tablas de Snellen, ETDRS u otras similares de lo que era capaz antes del tratamiento. Dado que estas pruebas de agudeza visual requieren la comunicación entre el sujeto y el profesional médico (por ejemplo, la lectura de letras en voz alta), es difícil obtener lecturas análogas de la agudeza visual cuando se realizan pruebas en modelos animales en estudios preclínicos.
La agudeza visual tiene la ventaja de ser un criterio de valoración clínico que puede ser independiente de otras pruebas visuales que dependen de la observación de la vascularización o neovascularización retiniana, como la fotografía/observación del fondo de ojo, la angiografía con fluoresceína o incluso la tomografía de coherencia óptica. Es decir, si una mejora de la agudeza visual no se debe a un mecanismo que afecte a la vasculatura retiniana, esta prueba puede revelar la eficacia de un tratamiento. Aunque la magnitud de la mejora puede variar para un paciente determinado, en algunos casos la magnitud de la mejora determinada mediante una prueba de agudeza visual, por ejemplo, como se ha descrito anteriormente, es del 5 % o más, como el 10 % o más, incluido el 20 % o más.
iii. Degeneración macular/rejillas de Amsler
Un procedimiento que se utiliza comúnmente para diagnosticar la degeneración macular y para determinar la progresión de la enfermedad es el uso de rejillas de Amsler (degeneración macular), cuyos procedimientos son bien conocidos por aquellos que tienen habilidad normal en el arte. La cuadrícula consta de un cuadrado de aspecto similar al papel milimetrado, con líneas oscuras que forman una cuadrícula cuadrada y un punto oscuro en el centro del cuadrado. Cubriendo cada ojo sucesivamente, el sujeto enfoca cada ojo individualmente en el punto oscuro, y toma nota de si alguna de las líneas de la cuadrícula está rota, distorsionada, ondulada o borrosa.
iv. Examen completo de retina con dilatación pupilar
El examen exhaustivo de la retina con dilatación de la pupila es un procedimiento mediante el cual la retina puede ser observada directamente por un profesional, como un optometrista o un oftalmólogo, y es bien conocido por quienes tienen conocimientos ordinarios en la materia. El profesional administra al sujeto gotas dilatadoras para los ojos. Las gotas pueden ser de dos tipos de medicamentos midriáticos, administrados juntos o por separado. Uno estimula la contracción de los músculos que dilatan la pupila (por ejemplo, la fenilefrina) y el otro relaja los músculos que hacen que la pupila se contraiga (por ejemplo, el ciclopentolato). La dilatación de la pupila permite al profesional observar mejor un campo mayor de la retina durante el examen ocular.
El examen exhaustivo de la retina con dilatación de la pupila permite a los oftalmólogos diagnosticar y determinar la progresión de diversas enfermedades oculares y asociadas a la retina como, a título de ejemplo y no limitativo, el glaucoma, la retinopatía diabética, la retinopatía del prematuro, la oclusión de la vena central de la retina y la degeneración macular asociada a la edad. Entre los signos reveladores de estas enfermedades que pueden determinarse mediante la fotografía del fondo de ojo figuran la inflamación o fuga de los vasos sanguíneos de la retina, el crecimiento anormal de los vasos sanguíneos dentro o debajo de la retina y el deterioro de la mácula de la retina.
v. Fotografía del fondo del ojo
Similar al examen de la retina con dilatación de la pupila, la fotografía del fondo de ojo es un procedimiento a través del cual la retina puede ser fotografiada directamente y es bien conocido por aquellos que tienen habilidad normal en el arte. (Saine, PJ, et al., Fundus Photography Overview, OPHTHALMIC PHOTOGRAPHY: RETINAL PHOTOGRAPHY, ANGIOGRAPHY, AND ELECTRONIC IMAGING, Butterworth-Heinemann Medical (2a ed.).)). El procedimiento incluye la dilatación de la pupila, con el paciente sentado ante la cámara del fondo del ojo. Un flash envía luz al ojo del paciente, creando una fotografía del fondo de ojo o imagen de la retina. La fotografía puede realizarse con diversos filtros de color, o bien pueden administrarse al paciente colorantes como la fluoresceína para facilitar la obtención de imágenes.
Una cámara de fondo de ojo es un microscopio especializado de baja potencia acoplado a una cámara. El ángulo de aceptación de la lente puede crear diferentes resultados. Los expertos en la materia consideran que un ángulo de 30 grados es la visión normal de la retina. Las cámaras de fondo de ojo gran angular son capaces de captar imágenes de entre 45 y 140 grados, y las cámaras de fondo de ojo angular tienen ángulos de visión de 20 grados o menos.
Al igual que el examen completo de la retina con dilatación pupilar, la fotografía del fondo de ojo permite a los oftalmólogos diagnosticar y determinar la progresión de diversas enfermedades oculares y asociadas a la retina como, a título de ejemplo y no limitativo, el glaucoma, la retinopatía del prematuro, la retinopatía diabética, la oclusión de la vena central de la retina y la degeneración macular asociada a la edad. Entre los signos reveladores de estas enfermedades que pueden determinarse mediante la fotografía del fondo de ojo figuran la inflamación o fuga de los vasos sanguíneos de la retina, el crecimiento anormal de los vasos sanguíneos dentro o debajo de la retina y el deterioro de la mácula de la retina.
vi. Angiografía con fluoresceína
La angiografía con fluoresceína es un procedimiento mediante el cual se pueden evaluar los vasos sanguíneos de la retina y es bien conocido por los expertos en la materia. Se utiliza sobre todo para diagnosticar o medir la progresión de la degeneración macular húmeda/neovascularización coroidea.
Se inyecta colorante de fluoresceína en la vena de un sujeto (cuyos ojos han sido dilatados previamente) para que el colorante viaje hasta el ojo y la vasculatura de la retina. Antes de inyectar el tinte, se toman fotografías de referencia de la retina. Cuando se determina que el colorante ha penetrado en la vasculatura retiniana, se toman fotografías adicionales de la retina durante uno o varios minutos. Al ver las fotografías, el oftalmólogo puede determinar si se ha producido alguna fuga de colorante de los vasos, lo que le ayuda a comprender dónde se han desarrollado vasos sanguíneos nuevos y frágiles.
vii. Tomografía de coherencia óptica (OCT)
La OCT es una prueba no invasiva que proporciona imágenes transversales de alta resolución de la retina y emplea ondas de luz para producir las imágenes. (Fujimoto, JG, et al., Neoplasia, 2(1-2):9-25 (enero de 2000)). La OCT permite obtener imágenes de cada una de las capas distintivas de la retina. En consecuencia, un oftalmólogo dispone de medios que le permiten cartografiar la retina y determinar su grosor. A título de ejemplo, y no limitativo, puede medirse con precisión el espesor retiniano central (CRT) de la retina del sujeto. Los procedimientos para realizar una prueba OCT y determinar la CRT son bien conocidos por los expertos en la materia.
La OCT puede realizarse utilizando gotas para los ojos que dilatan las pupilas y permiten un mejor examen de las retinas del sujeto. Una vez que las pupilas están completamente dilatadas, el escáner OCT puede explorar los ojos del sujeto de forma no invasiva. La o Ct puede ayudar a diagnosticar muchas enfermedades asociadas a la retina, como el edema macular, la degeneración macular asociada a la edad, el glaucoma, la retinopatía diabética y la retinopatía del prematuro.
l. Reactivos, dispositivos y kits (no forman parte de la invención)
También se proporcionan reactivos, dispositivos y kits de los mismos para practicar uno o más de los procedimientos descritos anteriormente. Los reactivos, dispositivos y kits correspondientes pueden variar enormemente. Los reactivos y dispositivos de interés incluyen los mencionados anteriormente con respecto a los procedimientos de administración de los compuestos descritos en el presente documento (por ejemplo, los compuestos de fórmula 2) al sujeto.
Además de los componentes anteriores, los kits del sujeto incluirán instrucciones para practicar los procedimientos del sujeto. Estas instrucciones pueden estar presentes en los kits en cuestión en una variedad de formas, una o más de las cuales pueden estar presentes en el kit. Una forma en la que pueden estar presentes estas instrucciones es como información impresa en un medio o sustrato adecuado, por ejemplo, una o varias hojas de papel en las que esté impresa la información, en el embalaje del kit, en un prospecto, etc. Otro medio sería un soporte legible por ordenador, por ejemplo, un disquete, un CD, una unidad flash portátil, etc., en el que se haya grabado la información. Otro medio que puede estar presente es una dirección de sitio web que puede utilizarse a través de Internet para acceder a la información en un sitio remoto. En los kits puede haber cualquier medio conveniente.
Los siguientes ejemplos se proporcionan a título ilustrativo y no limitativo.
EJEMPLOS
a. Preparación farmacéutica
Las composiciones farmacéuticas que se administran a sujetos con enfermedad asociada a la retina que comprenden los compuestos, cocristales y sales descritos anteriormente pueden sintetizarse, hacerse y formularse usando los ejemplos divulgados en Publicación de solicitud de patente de EE. UU. n.° 2013/0266646, 2016/0081998, N.° de patente de EE.UU. 8.278.302, 8.653.075, RE 45323, 8.742.115, 9.233.950 y 8.680.280. Además, las composiciones farmacéuticas pueden prepararse como se describe en los ejemplos siguientes:
1. Formulación de comprimidos - granulación húmeda
La copovidona se disuelve en etanol a temperatura ambiente para producir un líquido de granulación. Un ingrediente activo antagonista de CCR3, lactosa y parte de la crospovidona se mezclan en un mezclador adecuado, para producir una premezcla. La premezcla se humedece con el líquido de granulación y posteriormente se granula. El granulado húmedo se tamiza opcionalmente a través de un tamiz con un tamaño de malla de 1,6-3,0 mm. El granulado se seca a 45 °C en un secador adecuado hasta alcanzar un contenido de humedad residual correspondiente a una pérdida por desecación del 1-3 %. El granulado seco se tamiza a través de un tamiz con un tamaño de malla de 1,0 mm. El granulado se mezcla con parte de la crospovidona y la celulosa microcristalina en un mezclador adecuado. El estearato de magnesio se añade a esta mezcla después de pasarla por un tamiz de 1,0 mm para eliminar los grumos. Posteriormente, la mezcla final se produce mediante una mezcla final en un mezclador adecuado y se comprime en comprimidos. Se puede obtener la siguiente composición de comprimidos:
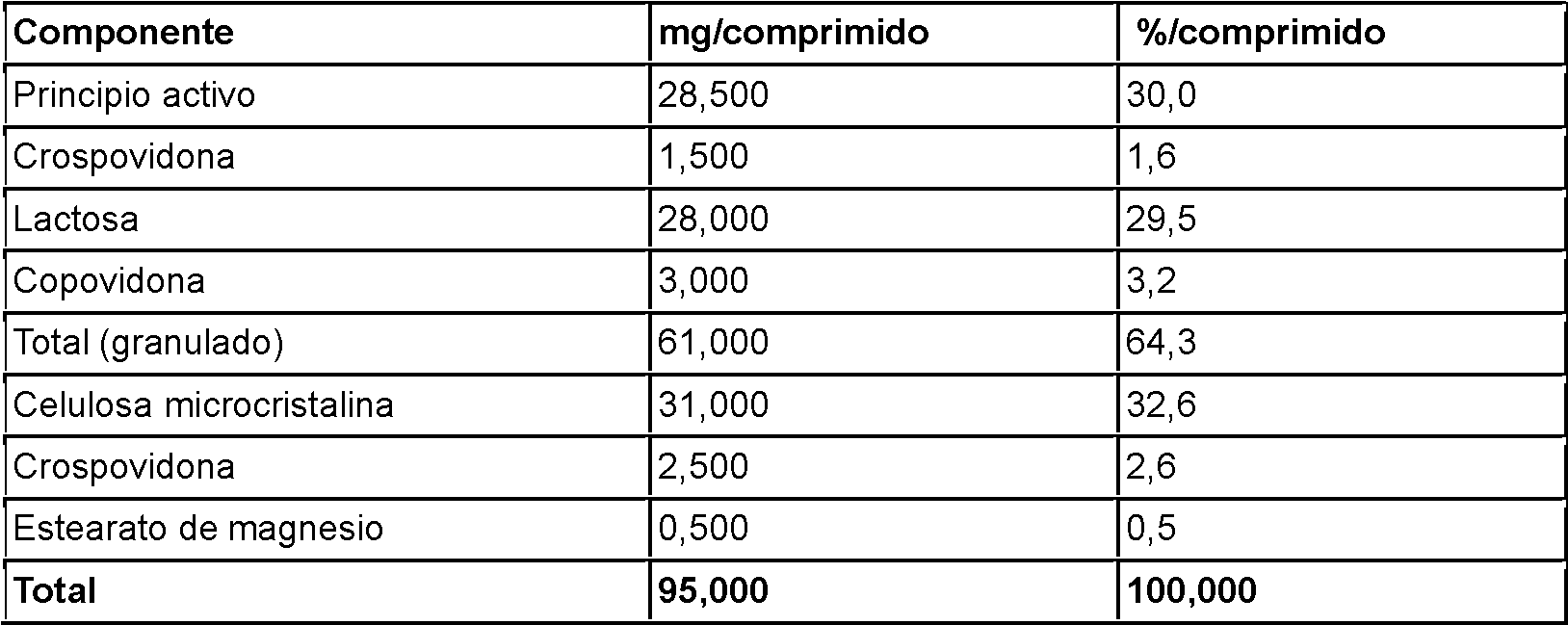
2. Formulación de comprimidos - granulación fundida
Un ingrediente activo antagonista de CCR3, lactosa, parte del mcc, polietilenglicol, lactosa y parte de la crospovidona se mezclan en un mezclador adecuado, para producir una premezcla. La premezcla se calienta en una mezcladora de alto cizallamiento y posteriormente se granula. El granulado caliente se enfría a temperatura ambiente y se tamiza a través de un tamiz con un tamaño de malla de 1,0 mm. El granulado se mezcla con parte de la crospovidona y la celulosa microcristalina en un mezclador adecuado. El estearato de magnesio se añade a esta mezcla después de pasarla por un tamiz de 1,0 mm para eliminar los grumos. Posteriormente, la mezcla final se produce mediante una mezcla final en un mezclador adecuado y se comprime en comprimidos. Se puede obtener la siguiente composición de comprimidos:

3. Formulación de comprimidos - granulación por fusión en caliente
Un ingrediente activo antagonista de CCR3, manit, polietilenglicol y parte de la crospovidona se mezclan en un mezclador adecuado, para producir una premezcla. La premezcla se calienta en una mezcladora de alto cizallamiento y posteriormente se granula. El granulado caliente se enfría a temperatura ambiente y se tamiza a través de un tamiz con un tamaño de malla de 1,0 mm. El granulado se mezcla con parte de la crospovidona y el manit en un mezclador adecuado. El estearato de magnesio se añade a esta mezcla después de pasarla por un tamiz de 1,0 mm para eliminar los grumos. Posteriormente, la mezcla final se produce mediante una mezcla final en un mezclador adecuado y se comprime en comprimidos. Se puede obtener la siguiente composición de comprimidos:

4. Formulación de comprimidos - extrusión en caliente
Un ingrediente activo antagonista de CCR3 y ácido esteárico-palmítico se mezclan en un mezclador adecuado, para producir una premezcla. La premezcla se extruye en una extrusora de doble husillo y posteriormente se granula. El granulado se tamiza a través de un tamiz con un tamaño de malla de 1,0 mm. El granulado se mezcla con manit y crospovidona en un mezclador adecuado. El estearato de magnesio se añade a esta mezcla después de pasarla por un tamiz de 1,0 mm para eliminar los grumos. Posteriormente, la mezcla final se produce mediante una mezcla final en un mezclador adecuado y se comprime en comprimidos. Se puede obtener la siguiente composición de comprimidos:

5. Formulación de comprimidos - extrusión en caliente
Un ingrediente activo antagonista de CCR3 y ácido esteárico-palmítico se mezclan en un mezclador adecuado, para producir una premezcla. La premezcla se extruye en una extrusora de doble husillo y posteriormente se granula. El granulado se tamiza a través de un tamiz con un tamaño de malla de 1,0 mm. El granulado se introduce directamente en cápsulas duras. Se puede obtener la siguiente composición de cápsulas:

6. Formulación de comprimidos - compactación con rodillo
Un ingrediente activo antagonista de CCR3, parte de manit y crospovidona y estearato de magnesio se mezclan en un mezclador adecuado, para producir una premezcla. La premezcla se compacta con un rodillo compactador y posteriormente se granula. Opcionalmente, el granulado se tamiza a través de un tamiz con un tamaño de malla de 0,8 mm. El granulado se mezcla con una parte de manit y crospovidona en un mezclador adecuado. El estearato de magnesio se añade a esta mezcla después de pasarla por un tamiz de 1,0 mm para eliminar los grumos. Posteriormente, la mezcla final se produce mediante una mezcla final en un mezclador adecuado y se comprime en comprimidos. Se puede obtener la siguiente composición de comprimidos:

7. Formulación de comprimidos - compactación con rodillo
Un ingrediente activo antagonista de CCR3 y estearato de magnesio se mezclan en un mezclador adecuado, para producir una premezcla. La premezcla se compacta con un rodillo compactador y posteriormente se granula. Opcionalmente, el granulado se tamiza a través de un tamiz con un tamaño de malla de 0,8 mm. El granulado se mezcla con manit y croscarmelosa sódica en un mezclador adecuado. El estearato de magnesio se añade a esta mezcla después de pasarla por un tamiz de 1,0 mm para eliminar los grumos. Posteriormente, la mezcla final se produce mediante una mezcla final en un mezclador adecuado y se comprime en comprimidos. Se puede obtener la siguiente composición de comprimidos:

8. Formulación de comprimidos - compactación con rodillo
Un ingrediente activo antagonista de CCR3 y estearato de magnesio se mezclan en un mezclador adecuado, para producir una premezcla. La premezcla se compacta con un rodillo compactador y posteriormente se granula. Opcionalmente, el granulado se tamiza a través de un tamiz con un tamaño de malla de 0,8 mm. El granulado se mezcla con celulosa microcristalina y crospovidona en un mezclador adecuado. El estearato de magnesio se añade a esta mezcla después de pasarla por un tamiz de 1,0 mm para eliminar los grumos. Posteriormente, la mezcla final se produce mediante una mezcla final en un mezclador adecuado y se comprime en comprimidos. Se puede obtener la siguiente composición de comprimidos:

continuación

9. Formulación de comprimidos recubiertos
Los núcleos de comprimidos según las formulaciones mencionadas pueden utilizarse para producir comprimidos recubiertos con película. La hidroxipropilmetilcelulosa, el polietilenglicol, el talco, el dióxido de titanio y el óxido de hierro se suspenden en agua purificada en un mezclador adecuado a temperatura ambiente para producir una suspensión de recubrimiento. Los núcleos de los comprimidos se recubren con la suspensión de recubrimiento hasta un aumento de peso de aproximadamente el 3 % para producir comprimidos recubiertos con película. Se puede obtener la siguiente composición de recubrimiento pelicular:

b. Formulación y administración de fármacos
El producto en investigación de la invención se ajustaba a la siguiente estructura química:

Aquellos que tengan habilidad normal en el arte relevante reconocerían que los compuestos, co-cristales, sales, y formulaciones descritas previamente en las secciones anteriores también podrían usarse en estos ejemplos. El producto en investigación de la invención estuvo disponible en forma de comprimidos recubiertos con película de 100 mg, 200 mg y 400 mg con forma biconvexa, redonda u ovalada y color rojo apagado. Los comprimidos se fabricaron mediante un proceso de granulación en seco y contenían celulosa microcristalina, fosfato de hidrógeno, croscarmelosa sódica, estearato de magnesio, alcohol polivinílico, dióxido de titanio, polietilenglicol, talco, óxido de hierro rojo y óxido de hierro amarillo como ingredientes inactivos. Los comprimidos de placebo iguales al producto en investigación se fabricaron mediante un proceso de compresión directa y contenían los mismos ingredientes inactivos. c. Ejemplos preclínicos
La potencia antagonista del producto en investigación de la invención se determinó en varios ensayos humanos dependientes de CCR3 (Figura 1). La potencia del producto en investigación de la invención se determinó mediante un ensayo de unión a receptores, midiéndose el IC50 en 4,0 ± 1,8 nM y el Ki en 3,2 ± 0,6 nM. Se determinó que el IC50 para un ensayo de afluencia de calcio utilizando células humanas CHEM1-Ga15 transfectadas con CCR3 era de 0,9
± 0,2 nM. El antagonismo por el producto en investigación de la invención del cambio de forma de los eosinófilos inducido por la eotaxina-1 humana en sangre total humana se consiguió con una IC50 de 42,5 ± 43,5 nM.
También se determinaron las potencias para otras especies de mamíferos en diferentes ensayos. Las especies incluían monos cynomolgus (macacos), ratones, ratas y caninos. Con respecto a los ensayos de unión a receptores, el Ki para el fármaco en investigación de la invención en CCR3 de ratón fue de 124,3 ± 0,9 nM, y la IC50 de 87,3 ± 5,6 nM. Para CCR3 de rata, la Ki para el fármaco en investigación de la invención fue de 1488,6 ± 127,6 nM y la IC50 de 1719,0 ± 129,9 nM.
d. Diseño del ensayo clínico (sin tratamiento, régimen de 4 semanas)
Los pacientes con NVC subfoveal recién diagnosticada (es decir, sin tratamiento) secundaria a la DMMAo fueron incluidos en un estudio abierto de un solo brazo. Los pacientes tomaron por vía oral 400 mg del medicamento en investigación de la invención b.o.d. (dos veces al día) durante 4 semanas y se programaron visitas semanales con el médico. El régimen consistía en dos comprimidos de 200 mg por la mañana y dos comprimidos de 200 mg por la noche. Tras el final del tratamiento (EoT) o la retirada de la medicación del estudio, los pacientes elegibles podrían recibir el tratamiento estándar (terapia anti-VEGF). Los pacientes fueron sometidos a dos seguimientos en las 4 semanas siguientes a la EoT La figura 2 muestra el diseño del ensayo clínico. Las casillas V1, V2, V3, V4 y V5 indican las visitas de los pacientes 1 a 5, respectivamente. Los espacios entre las casillas indican el tiempo en días entre las visitas. La casilla EoT indica la visita 6, es decir, el final del tratamiento. Las visitas 7 y 8 tuvieron lugar durante las visitas de seguimiento (FU).
1. Criterio de valoración primario - Grosor central de la retina (GCR)
El criterio de valoración primario fue el cambio con respecto al valor basal en el grosor central de la retina (GCR) determinado mediante tomografía de coherencia óptica de dominio espectral (SD-OCT) el día 29 (visita n° 6). Para todos los criterios de valoración primarios y secundarios, el valor de la última evaluación antes de la primera toma de la medicación del estudio se utilizó como valor basal el día 1 (visita n° 2).
La figura 3 muestra la CRT a lo largo del tiempo. La CRT se presentó como media (SEM) en unidades de μm y el tiempo como número de visita. Las capas y el grosor de la retina se visualizaron y midieron mediante SD-OCT, y para aquellos con conocimientos ordinarios en la materia y entrenados en el uso de equipos de OCT, la visualización y medición de las capas de la retina implican técnicas bien conocidas. (Véase, por ejemploFujimoto, JG, et al., Neoplasia 2(1-2)9-25 (2000); y Keane, PA, et al., Investigative Ophthalmology & Visual Science, 50(7):3378-85 (2009)). En general, no hubo cambios significativos desde el inicio en la mediana o la media de CRT a lo largo de la fase de tratamiento.
2. Criterios de valoración secundarios
(a) Fugas neovasculares evaluadas con fluoresceína
Angiografía y fotografía del fondo de ojo
La vasculatura retiniana del ojo de estudio se evaluó mediante angiografía fluoresceínica (AF). El cambio en la fuga neovascular por AF se determinó el día 29 (visita 6) en comparación con el valor basal. Enla figura 4 se presentan los valores absolutos y la variación de los valores de referencia a lo largo del tiempo como valores medios y medianos. N es el número de pacientes evaluados. En general, no se produjeron cambios significativos respecto a los valores iniciales en la mediana o la media a lo largo de la fase de tratamiento.
(b) Agudeza visual
La agudeza visual se determinó utilizando tablas ETDRS a lo largo del tratamiento. Durante las visitas se evaluó el cambio absoluto en el número de letras leídas por los pacientes con respecto a sus niveles iniciales. La figura 5 representa este cambio (eje x) a lo largo del tiempo (eje y) durante el tratamiento con el medicamento en investigación de la invención (Comp. Inv.). De un total de 14 pacientes, un subgrupo de 8 mostró una mejora significativa de la agudeza visual mejor corregida (AVMC) en comparación con el control simulado y con el grupo de pacientes más amplio en general. La mejora de la agudeza visual fue sorprendente, al menos en parte, dado que la terapia no afectó significativamente al grosor central de la retina. En el subgrupo de 8 pacientes, la agudeza visual mejor corregida se comparó favorablemente con el tratamiento anti-VEGF (inyección intravítrea de ranibizumab), tal y como se informó previamente en los estudios MARINAy VIEW (Rosenfeld Pj , et al., N. Engl. J. Med. 355(14):1419-31 (2006); y Heier JS, et al., Ophthalmology 119(12):2537-48 (2012)). Así, de forma notable, el procedimiento aquí descrito consiguió una mejora de la agudeza visual a niveles observados en relación con la terapia anti-VEGF, pero empleando un mecanismo de acción que difiere del estándar actual de tratamiento.
A la luz de la observación de que el compuesto en investigación de la invención no logró alterar significativamente el grosor central de la retina o la fuga neovascular, esta mejora significativa de la agudeza visual en más de la mitad de los pacientes fue sorprendente e inesperada. Sugiere que el compuesto en investigación tiene un efecto neuroprotector sobre las neuronas relacionadas con la retina y las enfermedades retinianas.
d. Diseño del ensayo clínico (sin tratamiento, régimen de 6 semanas)
Los pacientes con NVC subfoveal recién diagnosticada (es decir, sin tratamiento) secundaria a la DMMAo se incorporan a un estudio abierto de un solo brazo. Durante cada visita, se evalúa la seguridad y tolerabilidad de los pacientes. En visitas específicas, se determina la BCVA mediante ETDRS y se realizan evaluaciones morfológicas utilizando SD-OCT y fotografía de fondo de ojo/FA. Los pacientes se autoadministran 800 mg al día p.o. (400 mg b.i.d.) del fármaco en investigación de la invención. El ensayo dura 10 semanas, de las cuales 6 de tratamiento y 4 de seguimiento.
1. Criterio de valoración primario (BCVA)
El criterio de valoración primario es el cambio medio en la puntuación de la letra BCVA determinada por el procedimiento de prueba ETDRS (Early Treatment Diabetic Retinopathy Study). La BCVA se mide durante las visitas 1 (visita de cribado), 2-7 (visitas de tratamiento), 9-10 (visitas de seguimiento).
2. Criterios de valoración exploratorios (cambios morfológicos)
Los criterios de valoración exploratorios investigan los efectos morfológicos oculares relacionados con la administración b.i.d. del fármaco en investigación de la invención. Las mediciones del grosor central de la retina (CRT), el líquido intrarretiniano (IRF), el líquido subretiniano (SRF) y el desprendimiento del epitelio pigmentario (PED) se realizan mediante SD-OCT y fotografía del fondo de ojo/FA (angiografía fluoresceínica).
e. Diseño del ensayo clínico (DMMAo refractaria, régimen de 6 semanas)
Los pacientes con NVC refractaria secundaria a DMMAo tras un tratamiento mensual (durante al menos 3 meses) con terapias intravítreas (IVT) contra el factor de crecimiento endotelial vascular (anti-VEGR) participan en un estudio abierto de un solo brazo. Durante cada visita, se evalúa la seguridad y tolerabilidad de los pacientes. En visitas específicas, se determina la BCVA mediante ETDRS y se realizan evaluaciones morfológicas utilizando SD-OCT y fotografía de fondo de ojo/FA. Los pacientes se autoadministran 800 mg al día p.o. (400 mg b.i.d.) del fármaco en investigación de la invención. El ensayo dura 10 semanas, de las cuales 6 de tratamiento y 4 de seguimiento.
1. Criterio de valoración primario (BCVA)
El criterio de valoración primario es el cambio medio en la puntuación de la letra BCVA determinada por el procedimiento de prueba ETDRS (Early Treatment Diabetic Retinopathy Study). La BCVA se mide durante las visitas 1 (visita de cribado), 2-7 (visitas de tratamiento), 9-10 (visitas de seguimiento).
2. Criterios de valoración exploratorios (cambios morfológicos)
Los criterios de valoración exploratorios investigan los efectos morfológicos oculares relacionados con la administración b.i.d. del fármaco en investigación de la invención. Las mediciones del grosor central de la retina (CRT), el líquido intrarretiniano (IRF), el líquido subretiniano (SRF) y el desprendimiento del epitelio pigmentario (PED) se realizan mediante SD-OCT y fotografía del fondo de ojo/FA (angiografía fluoresceínica).
f. Ensayo de terapia combinada (DMMAo refractaria, régimen de 1,5 años)
Los pacientes con NVC subfoveal recién diagnosticada (es decir, sin tratamiento) secundaria a la DMMAo se incorporan a un estudio aleatorizado de doble brazo. El primer brazo se compone de la inyección de un agente anti-VEGF (por ejemplo, un tratamiento estándar actual, como ranibizumab) administrado como inyección intravítrea más el fármaco en investigación de la invención administrado por vía oral. El segundo brazo se compone de la inyección del agente anti-VEGF más placebo oral (es decir, un tratamiento estándar activo).
Durante cada visita, se evalúa la seguridad y tolerabilidad de los pacientes. En visitas específicas, se determina la BCVA mediante ETDRS y se realizan evaluaciones morfológicas utilizando SD-OCT y fotografía de fondo de ojo/FA. Los pacientes del primer brazo se autoadministran 800 mg al día p.o. (400 mg b.i.d.) del fármaco en investigación de la invención y recibir un agente anti-VEGF como ranibizumab por inyecciones de visita a una dosis de 10 mg/ml como 0. 5 mg en una jeringa precargada. Los pacientes del segundo brazo se autoadministran una cápsula de placebo dos veces al día y reciben un agente anti-VEGF como ranibizumab en forma de inyecciones mensuales a una dosis de 10 mg/ml como 0,5 mg en una jeringa precargada.
1. Criterio de valoración primario (BCVA)
El criterio de valoración primario es el cambio medio en la puntuación de la letra BCVA determinada por el procedimiento de prueba ETDRS (Early Treatment Diabetic Retinopathy Study). La BCVA se mide durante las visitas de cribado, las visitas de tratamiento con el agente anti-VEGF y durante las visitas de seguimiento. El cambio medio en la BCVA entre los dos grupos de tratamiento se determina a los 18 y 24 meses.
2. Criterios de valoración secundarios (número de inyecciones)
Se determina el número de inyecciones intravítreas por año administradas en los pacientes del primer brazo de tratamiento. Los dos brazos de tratamiento se comparan con esos resultados para determinar cuántas inyecciones al año producen resultados equivalentes de BCVA.
Los criterios de valoración adicionales investigan los efectos morfológicos oculares en cada brazo de tratamiento. Las mediciones del grosor central de la retina (CRT), el líquido intrarretiniano (IRF), el líquido subretiniano (SRF) y el desprendimiento del epitelio pigmentario (PED) se realizan mediante SD-OCT y fotografía del fondo de ojo/FA (angiografía fluoresceínica). Estos resultados ayudan a determinar el grado de sinergia entre el tratamiento estándar anti-VEGF y la administración del fármaco en investigación de la invención, elucidando aún más las comparaciones en el mecanismo de acción entre las dos terapias.
Claims (15)
1. Un compuesto que es un cocristal de fórmula 2,

en el que
R1 es alquilo C1 -6 , haloalquilo C1 -6 , O-haloalquilo C1 -6 , halógeno;
m e s 1, 2 o 3 ;
R2a y R2b se seleccionan cada uno independientemente entre H, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, COO-alquilo C1-6 , O-alquilo C1 -6 , CONR2 b 1 R2b2 o halógeno;
R2b1 es H, alquilo C1 -6 , alquilo Cü.4-cicloalquilo C3-6, o haloalquilo C1.6 ;
R2 b 2 es H, o alquilo C1 -6 ;
o R2b1 y R2 b 2 son conjuntamente un grupo alquileno C3-6 que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno; R23 es H, o alquilo C1 -6 ;
X es un anión seleccionado del grupo que consiste en cloruro, bromuro, yoduro, sulfato, fosfato, metanosulfonato, nitrato, maleato, acetato, benzoato, citrato, salicilato, fumarato, tartrato, dibenzoiltartrato, oxalato, succinato, benzoato y p-toluenosulfonato;
j es 0, 0,5,1, 1,5 o 2;
con un formador de co-cristales seleccionado del grupo que consiste en ácido orótico, ácido hipúrico, ácido L-piroglutámico, ácido D-piroglutámico, ácido nicotínico, ácido L-(+)-ascórbico, sacarina, piperazina, ácido 3-hidroxi-2-naftoico, ácido múcico (galactárico), ácido pamoico (embónico), ácido esteárico, ácido cólico, ácido desoxicólico, nicotinamida, isonicotinamida, succinamida, uracilo, L-lisina, L-prolina, D-valina, L-arginina, glicina; para su uso en la mejora de la agudeza visual en un sujeto diagnosticado de una enfermedad asociada a la retina.
2. El compuesto para el uso de la reivindicación 1, en el que la enfermedad asociada a la retina se selecciona del grupo que consiste en degeneración macular seca asociada a la edad, degeneración macular húmeda asociada a la edad, oclusión de la vena central de la retina, retinopatía del prematuro y retinopatía diabética.
3. El compuesto para el uso de la reivindicación 1 o 2, en el que
R2a es H, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, O-alquilo C1-6 , o
CONR2 a 1 R2 a 2 ;
R2a1 es H, alquilo C1-6 o haloalquilo C1 -6 ;
R2 a 2 es H, o alquilo C1 -6 ;
R2b es H, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, COO-alquilo C1 -6 , O-alquilo C1 -6 , CONR2 b 1 R2 b 2 o halógeno;
R2b1 es H, alquilo C1 -6 , alquilo Cü-4-cicloalquilo C3-6, o haloalquilo C1 -6 ;
R2 b 2 es H, o alquilo C1 -6 ;
o R2b1 y R2b 2 son conjuntamente un grupo alquileno C3-6que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno.
4. El compuesto para el uso de la reivindicación 1 o 2, en el que
R1 es alquilo C i -6 , haloalquilo C i -6, O-haloalquilo C i -6 o halógeno;
m es 1 o 2;
R2a es H, o alquilo C1-4;
R2b es H, o CONR2b1R2b2;
R2b1 es alquilo C1-4, alquilo C0-4-cicloalquilo C3-6, o haloalquilo C1-4;
R2b2 es H, o alquilo C1-4;
o R2b1 y R2b2 son conjuntamente un grupo alquileno C3-6 que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno; R3 es H, o alquilo C1 -6 ;
X es un anión seleccionado del grupo que consiste en cloruro o dibenzoiltartrato; y
j es 1 o 2.
5. El compuesto para el uso de la reivindicación 1 o 2, en el que
R2a es H, o alquilo C1 -4 ;
R2b es H, o CONR2b1R2b2;
R2b1 es alquilo C1 -4 ;
R2b2 es alquilo C1 -4.
6. El compuesto para el uso de la reivindicación 1 o 2, en el que
R2a es H, o alquilo C1-4;
R2b es H, o CONR2b1R2b2;
R2b1 es alquilo C0-4-cicloalquilo C3-6;
R2b2 es H, o alquilo C1-4.
7. El compuesto para el uso de la reivindicación 1 o 2, en el que
R2a es H, o alquilo C1 -4 ;
R2b es H, o CONR2b1R2b2;
R2b1 es haloalquilo C1-4;
R2b2 es H, alquilo C1-4.
8. El compuesto para el uso de la reivindicación 1 o 2, en el que el compuesto es un cocristal de la fórmula según la reivindicación 1, en el que R2b1 y R2b2 son conjuntamente un grupo alquileno C3-6que forma con el átomo de nitrógeno un anillo heterocíclico, en el que opcionalmente un átomo de carbono del anillo se sustituye por un átomo de oxígeno.
9. El compuesto para el uso de la reivindicación 1 o 2, en el que el compuesto de fórmula 2 es

10. El compuesto para el uso de la reivindicación 1 o 2, en el que el compuesto es una sal cristalina de la fórmula siguiente

11. El compuesto para el uso de la reivindicación 1 o 2, en el que el compuesto es una sal cristalina de la fórmula siguiente

12. El compuesto para el uso de la reivindicación 10, en el que la sal cristalina se caracteriza por que los cuatro picos más altos de difracción de rayos X en polvo se producen a 3,72, 13,60, 16,89 y 19,34 grados 20 (±0,05 grados 20) cuando se miden utilizando radiación CuKa.
13. El compuesto para el uso de la reivindicación 11, en el que la sal cristalina se caracteriza por que los cuatro picos más altos de difracción de rayos X en polvo se producen a 16,02, 16,86, 19,45 y 19,71 grados 20 (±0,05 grados 20) cuando se miden utilizando radiación CuKa.
14. El compuesto para el uso de la reivindicación 1 o 2, en el que el compuesto de fórmula 2 está en forma de los isómeros ópticos individuales, una mezcla de los enantiómeros individuales, un racemato o en forma de los compuestos enantioméricamente puros.
15. El compuesto para el uso de cualquiera de las reivindicaciones precedentes, en el que debe administrarse además una cantidad terapéuticamente eficaz de un tratamiento anti-VEGF.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762482134P | 2017-04-05 | 2017-04-05 | |
| PCT/US2018/026091 WO2018187473A1 (en) | 2017-04-05 | 2018-04-04 | Methods and compositions for treating retina-associated disease using ccr3-inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2950453T3 true ES2950453T3 (es) | 2023-10-10 |
Family
ID=62067800
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18720836T Active ES2950453T3 (es) | 2017-04-05 | 2018-04-04 | Compuestos y composiciones farmacéuticas para su uso en el tratamiento de enfermedades asociadas a la retina utilizando inhibidores de la CCR3 |
Country Status (18)
| Country | Link |
|---|---|
| US (2) | US11590118B2 (es) |
| EP (2) | EP4245295A3 (es) |
| JP (1) | JP7179755B2 (es) |
| KR (1) | KR102658724B1 (es) |
| CN (1) | CN110650740A (es) |
| AU (2) | AU2018248425B2 (es) |
| BR (1) | BR112019020430A2 (es) |
| CA (1) | CA3058654A1 (es) |
| CL (1) | CL2019002839A1 (es) |
| CO (1) | CO2019012336A2 (es) |
| EA (1) | EA201992356A1 (es) |
| ES (1) | ES2950453T3 (es) |
| IL (1) | IL269743B (es) |
| MX (1) | MX2019012012A (es) |
| SG (1) | SG11201909207VA (es) |
| TW (1) | TW201841636A (es) |
| UA (1) | UA125658C2 (es) |
| WO (1) | WO2018187473A1 (es) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20190131078A (ko) * | 2017-04-05 | 2019-11-25 | 알카헤스트 인코포레이티드 | Ccr3-억제제를 사용한 노화 관련 장애 치료를 위한 방법 및 조성물 |
| BR112021004938A2 (pt) * | 2018-09-26 | 2021-06-01 | Alkahest, Inc. | métodos e composições para tratamento de danos associados com o envelhecimento usando inibidores de ccr3 |
| AU2021287906A1 (en) * | 2020-06-11 | 2022-12-15 | Alkahest Inc. | Methods of improving retina-associated disease outcome using CCR3-inhibitors |
| CN112322719A (zh) * | 2020-11-06 | 2021-02-05 | 温州医科大学 | 循环血外泌体miRNA作为视网膜静脉阻塞疾病诊断标志物中的应用 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2002112800A (ru) | 2002-05-16 | 2004-03-10 | Евгений Ильич Зарайский (RU) | Способ лечения диабетической ретинопатии |
| US8008092B2 (en) | 2007-10-09 | 2011-08-30 | University Of Kentucky Research Foundation | CCR3 inhibition for ocular angiogenesis and macular degeneration |
| US8278302B2 (en) | 2009-04-08 | 2012-10-02 | Boehringer Ingelheim International Gmbh | Substituted piperidines as CCR3 antagonists |
| UA109290C2 (uk) | 2010-10-07 | 2015-08-10 | Спільні кристали і солі інгібіторів ccr3 | |
| JP2015500221A (ja) | 2011-12-01 | 2015-01-05 | グラクソ グループ リミテッドGlaxo Group Limited | 眼疾患の治療および予防方法 |
| AR090566A1 (es) | 2012-04-02 | 2014-11-19 | Boehringer Ingelheim Int | Proceso para la produccion de inhibidores de crr |
| US20130261153A1 (en) | 2012-04-03 | 2013-10-03 | Boehringer Ingelheim International Gmbh | Use of ccr3-inhibitors |
| US10213421B2 (en) | 2012-04-04 | 2019-02-26 | Alkahest, Inc. | Pharmaceutical formulations comprising CCR3 antagonists |
| KR20190131078A (ko) | 2017-04-05 | 2019-11-25 | 알카헤스트 인코포레이티드 | Ccr3-억제제를 사용한 노화 관련 장애 치료를 위한 방법 및 조성물 |
-
2018
- 2018-04-04 AU AU2018248425A patent/AU2018248425B2/en active Active
- 2018-04-04 SG SG11201909207V patent/SG11201909207VA/en unknown
- 2018-04-04 WO PCT/US2018/026091 patent/WO2018187473A1/en unknown
- 2018-04-04 US US16/603,084 patent/US11590118B2/en active Active
- 2018-04-04 JP JP2019554738A patent/JP7179755B2/ja active Active
- 2018-04-04 EA EA201992356A patent/EA201992356A1/ru unknown
- 2018-04-04 CN CN201880023759.8A patent/CN110650740A/zh active Pending
- 2018-04-04 UA UAA201910857A patent/UA125658C2/uk unknown
- 2018-04-04 IL IL269743A patent/IL269743B/en unknown
- 2018-04-04 EP EP23168381.4A patent/EP4245295A3/en active Pending
- 2018-04-04 BR BR112019020430-0A patent/BR112019020430A2/pt not_active IP Right Cessation
- 2018-04-04 ES ES18720836T patent/ES2950453T3/es active Active
- 2018-04-04 CA CA3058654A patent/CA3058654A1/en active Pending
- 2018-04-04 EP EP18720836.8A patent/EP3606521B1/en active Active
- 2018-04-04 KR KR1020197032819A patent/KR102658724B1/ko active IP Right Grant
- 2018-04-04 MX MX2019012012A patent/MX2019012012A/es unknown
- 2018-04-09 TW TW107112125A patent/TW201841636A/zh unknown
-
2019
- 2019-10-04 CL CL2019002839A patent/CL2019002839A1/es unknown
- 2019-11-01 CO CONC2019/0012336A patent/CO2019012336A2/es unknown
-
2023
- 2023-02-16 US US18/170,384 patent/US11951102B2/en active Active
-
2024
- 2024-06-18 AU AU2024204133A patent/AU2024204133A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| JP2020513004A (ja) | 2020-04-30 |
| US20240024305A1 (en) | 2024-01-25 |
| EA201992356A1 (ru) | 2020-03-23 |
| EP4245295A3 (en) | 2023-11-01 |
| CN110650740A (zh) | 2020-01-03 |
| US11590118B2 (en) | 2023-02-28 |
| JP7179755B2 (ja) | 2022-11-29 |
| SG11201909207VA (en) | 2019-11-28 |
| CL2019002839A1 (es) | 2020-06-05 |
| EP4245295A2 (en) | 2023-09-20 |
| MX2019012012A (es) | 2019-12-11 |
| EP3606521B1 (en) | 2023-04-19 |
| US20210106577A1 (en) | 2021-04-15 |
| BR112019020430A2 (pt) | 2021-03-30 |
| TW201841636A (zh) | 2018-12-01 |
| AU2018248425B2 (en) | 2024-03-28 |
| IL269743A (en) | 2019-11-28 |
| KR20190131121A (ko) | 2019-11-25 |
| KR102658724B1 (ko) | 2024-04-18 |
| AU2024204133A1 (en) | 2024-07-04 |
| UA125658C2 (uk) | 2022-05-11 |
| US11951102B2 (en) | 2024-04-09 |
| AU2018248425A1 (en) | 2019-10-31 |
| IL269743B (en) | 2022-09-01 |
| WO2018187473A1 (en) | 2018-10-11 |
| EP3606521A1 (en) | 2020-02-12 |
| CA3058654A1 (en) | 2018-10-11 |
| CO2019012336A2 (es) | 2020-04-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2950453T3 (es) | Compuestos y composiciones farmacéuticas para su uso en el tratamiento de enfermedades asociadas a la retina utilizando inhibidores de la CCR3 | |
| AU2018250214B2 (en) | Methods and compositions for treating aging-associated impairments using CCR3-inhibitors | |
| TW200911265A (en) | Pharmaceutical compositions and methods of treating dry eye disorders | |
| US20200054622A1 (en) | Methods and Compositions for Treating Aging-Associated Impairments Using CCR3-Inhibitors | |
| JP2012006918A (ja) | イソキノリンスルホニル誘導体を有効成分として含有する網脈絡膜変性疾患の予防または治療剤 | |
| JP2020536929A (ja) | Ccr3阻害剤を用いた、そう痒症、乾皮症、及び関連疾患を治療するための方法及び組成物 | |
| WO2016006621A1 (ja) | Pgd2拮抗剤を含有するアレルギー性疾患に伴う症状の治療用医薬 | |
| WO2014133072A1 (ja) | テトラヒドロピラニルアミノシクロペンチルカルボニルテトラヒドロピリドピリジン誘導体を有効成分として含有する後眼部疾患の予防または治療剤 | |
| TW201919597A (zh) | 利用新斯的明(neostigmine)及nk-1拮抗劑治療重症肌無力之醫藥組合物及方法 | |
| EA041892B1 (ru) | Способы и композиции для лечения ассоциированных со старением нарушений с применением ccr3-ингибиторов | |
| CA3111433A1 (en) | Methods and compositions for treating aging-associated impairments using ccr3-inhibitors |