ES2945905T3 - Compuestos y composiciones terapéuticos - Google Patents
Compuestos y composiciones terapéuticos Download PDFInfo
- Publication number
- ES2945905T3 ES2945905T3 ES15746142T ES15746142T ES2945905T3 ES 2945905 T3 ES2945905 T3 ES 2945905T3 ES 15746142 T ES15746142 T ES 15746142T ES 15746142 T ES15746142 T ES 15746142T ES 2945905 T3 ES2945905 T3 ES 2945905T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- compound
- aryl
- heteroaryl
- cycloalkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 384
- 239000000203 mixture Substances 0.000 title claims abstract description 149
- 230000001225 therapeutic effect Effects 0.000 title description 12
- 125000000217 alkyl group Chemical group 0.000 claims description 256
- -1 C 1-10 perhaloalkyl Chemical group 0.000 claims description 157
- 125000003118 aryl group Chemical group 0.000 claims description 79
- 125000001072 heteroaryl group Chemical group 0.000 claims description 74
- 125000000623 heterocyclic group Chemical group 0.000 claims description 73
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 65
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 46
- 150000003839 salts Chemical class 0.000 claims description 45
- 125000000304 alkynyl group Chemical group 0.000 claims description 40
- 125000001424 substituent group Chemical group 0.000 claims description 39
- 230000000302 ischemic effect Effects 0.000 claims description 29
- 125000003342 alkenyl group Chemical group 0.000 claims description 28
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 claims description 24
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 19
- 206010047249 Venous thrombosis Diseases 0.000 claims description 18
- 208000006011 Stroke Diseases 0.000 claims description 17
- 229910052739 hydrogen Inorganic materials 0.000 claims description 17
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 16
- 239000001257 hydrogen Substances 0.000 claims description 16
- 208000010378 Pulmonary Embolism Diseases 0.000 claims description 15
- 206010051055 Deep vein thrombosis Diseases 0.000 claims description 14
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 13
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 claims description 13
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 12
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 12
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 claims description 11
- 229910052736 halogen Inorganic materials 0.000 claims description 11
- 125000006714 (C3-C10) heterocyclyl group Chemical group 0.000 claims description 10
- 125000004429 atom Chemical group 0.000 claims description 10
- 150000002367 halogens Chemical class 0.000 claims description 10
- 239000008194 pharmaceutical composition Substances 0.000 claims description 10
- 125000003545 alkoxy group Chemical group 0.000 claims description 9
- 125000006708 (C5-C14) heteroaryl group Chemical group 0.000 claims description 8
- 125000004475 heteroaralkyl group Chemical group 0.000 claims description 8
- 125000004415 heterocyclylalkyl group Chemical group 0.000 claims description 8
- 210000003169 central nervous system Anatomy 0.000 claims description 7
- 125000004076 pyridyl group Chemical group 0.000 claims description 7
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 claims description 6
- 208000005189 Embolism Diseases 0.000 claims description 6
- 150000002431 hydrogen Chemical class 0.000 claims description 6
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 claims description 5
- 206010030113 Oedema Diseases 0.000 claims description 5
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 5
- 230000009885 systemic effect Effects 0.000 claims description 5
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 4
- 125000006590 (C2-C6) alkenylene group Chemical group 0.000 claims description 3
- 125000006591 (C2-C6) alkynylene group Chemical group 0.000 claims description 3
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 3
- 238000011321 prophylaxis Methods 0.000 claims description 3
- 125000001475 halogen functional group Chemical group 0.000 claims 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 2
- 125000005418 aryl aryl group Chemical group 0.000 claims 1
- 238000000034 method Methods 0.000 abstract description 145
- 108010080805 Factor XIa Proteins 0.000 abstract description 52
- 108060005987 Kallikrein Proteins 0.000 abstract description 25
- 102000001399 Kallikrein Human genes 0.000 abstract description 25
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 187
- 238000004128 high performance liquid chromatography Methods 0.000 description 124
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 113
- 239000000243 solution Substances 0.000 description 111
- 238000002360 preparation method Methods 0.000 description 109
- 238000001819 mass spectrum Methods 0.000 description 106
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 102
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 97
- 239000011541 reaction mixture Substances 0.000 description 88
- 235000019439 ethyl acetate Nutrition 0.000 description 78
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 71
- 238000006243 chemical reaction Methods 0.000 description 70
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 70
- 230000014759 maintenance of location Effects 0.000 description 64
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 59
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 59
- 239000007787 solid Substances 0.000 description 54
- 239000000460 chlorine Substances 0.000 description 52
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 50
- 229920006395 saturated elastomer Polymers 0.000 description 48
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 46
- 235000002639 sodium chloride Nutrition 0.000 description 42
- 239000003921 oil Substances 0.000 description 41
- 235000019198 oils Nutrition 0.000 description 41
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 39
- 239000000543 intermediate Substances 0.000 description 36
- 238000005481 NMR spectroscopy Methods 0.000 description 35
- 239000012267 brine Substances 0.000 description 35
- 239000012074 organic phase Substances 0.000 description 35
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 35
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 34
- 239000000741 silica gel Substances 0.000 description 34
- 229910002027 silica gel Inorganic materials 0.000 description 34
- 239000011734 sodium Substances 0.000 description 34
- 230000015572 biosynthetic process Effects 0.000 description 33
- 239000003814 drug Substances 0.000 description 33
- 125000004404 heteroalkyl group Chemical group 0.000 description 32
- 239000002253 acid Substances 0.000 description 31
- 238000003786 synthesis reaction Methods 0.000 description 31
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 30
- 238000003818 flash chromatography Methods 0.000 description 28
- 230000002829 reductive effect Effects 0.000 description 28
- 238000011282 treatment Methods 0.000 description 28
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 27
- 239000007858 starting material Substances 0.000 description 27
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 26
- 229910052757 nitrogen Inorganic materials 0.000 description 26
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 25
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 23
- 239000003112 inhibitor Substances 0.000 description 23
- 238000001990 intravenous administration Methods 0.000 description 23
- 229940124597 therapeutic agent Drugs 0.000 description 23
- 239000013078 crystal Substances 0.000 description 22
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 21
- 238000004587 chromatography analysis Methods 0.000 description 21
- 238000000746 purification Methods 0.000 description 21
- 239000002904 solvent Substances 0.000 description 21
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 20
- 239000013058 crude material Substances 0.000 description 20
- 125000006239 protecting group Chemical group 0.000 description 20
- 125000005842 heteroatom Chemical group 0.000 description 19
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 19
- 125000004432 carbon atom Chemical group C* 0.000 description 18
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N DMSO Substances CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 17
- 208000007536 Thrombosis Diseases 0.000 description 17
- 229910052805 deuterium Inorganic materials 0.000 description 17
- 206010019860 Hereditary angioedema Diseases 0.000 description 16
- 239000000047 product Substances 0.000 description 16
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 15
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 15
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 15
- 210000004369 blood Anatomy 0.000 description 15
- 239000008280 blood Substances 0.000 description 15
- 208000035475 disorder Diseases 0.000 description 15
- 230000000694 effects Effects 0.000 description 15
- 238000001802 infusion Methods 0.000 description 15
- 208000028185 Angioedema Diseases 0.000 description 14
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 14
- 239000003480 eluent Substances 0.000 description 14
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 14
- 230000001052 transient effect Effects 0.000 description 14
- 229940122036 Factor XIa inhibitor Drugs 0.000 description 13
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 13
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 13
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 13
- 208000028867 ischemia Diseases 0.000 description 13
- 101000975003 Homo sapiens Kallistatin Proteins 0.000 description 12
- 101001077723 Homo sapiens Serine protease inhibitor Kazal-type 6 Proteins 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- 229940122920 Kallikrein inhibitor Drugs 0.000 description 12
- 102100023012 Kallistatin Human genes 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 12
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 12
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 12
- 238000012360 testing method Methods 0.000 description 12
- 238000004809 thin layer chromatography Methods 0.000 description 12
- 239000003795 chemical substances by application Substances 0.000 description 11
- 239000012043 crude product Substances 0.000 description 11
- 201000010099 disease Diseases 0.000 description 11
- 235000019441 ethanol Nutrition 0.000 description 11
- 239000006260 foam Substances 0.000 description 11
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 10
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 10
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- 238000003556 assay Methods 0.000 description 10
- 230000015271 coagulation Effects 0.000 description 10
- 238000005345 coagulation Methods 0.000 description 10
- 229940079593 drug Drugs 0.000 description 10
- 238000010348 incorporation Methods 0.000 description 10
- 239000012948 isocyanate Substances 0.000 description 10
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 9
- 208000032843 Hemorrhage Diseases 0.000 description 9
- 108090000190 Thrombin Proteins 0.000 description 9
- 208000034158 bleeding Diseases 0.000 description 9
- 230000000740 bleeding effect Effects 0.000 description 9
- 229910052799 carbon Inorganic materials 0.000 description 9
- 239000012230 colorless oil Substances 0.000 description 9
- 239000002552 dosage form Substances 0.000 description 9
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 9
- 229910052737 gold Inorganic materials 0.000 description 9
- 239000010931 gold Substances 0.000 description 9
- 125000005843 halogen group Chemical group 0.000 description 9
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 9
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 9
- 230000002401 inhibitory effect Effects 0.000 description 9
- 208000014674 injury Diseases 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 9
- 230000009424 thromboembolic effect Effects 0.000 description 9
- JJSCUXAFAJEQGB-MRVPVSSYSA-N [(1r)-1-isocyanatoethyl]benzene Chemical compound O=C=N[C@H](C)C1=CC=CC=C1 JJSCUXAFAJEQGB-MRVPVSSYSA-N 0.000 description 8
- 239000003146 anticoagulant agent Substances 0.000 description 8
- 239000012298 atmosphere Substances 0.000 description 8
- 125000001188 haloalkyl group Chemical group 0.000 description 8
- 230000005764 inhibitory process Effects 0.000 description 8
- 238000002347 injection Methods 0.000 description 8
- 239000007924 injection Substances 0.000 description 8
- 150000002513 isocyanates Chemical class 0.000 description 8
- 239000010410 layer Substances 0.000 description 8
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 8
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 8
- 208000010125 myocardial infarction Diseases 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 239000002953 phosphate buffered saline Substances 0.000 description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 8
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 8
- 239000000377 silicon dioxide Substances 0.000 description 8
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 8
- 229960004072 thrombin Drugs 0.000 description 8
- 239000003039 volatile agent Substances 0.000 description 8
- 102000004190 Enzymes Human genes 0.000 description 7
- 108090000790 Enzymes Proteins 0.000 description 7
- 206010028980 Neoplasm Diseases 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N acetone Substances CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- 229940088598 enzyme Drugs 0.000 description 7
- 239000000706 filtrate Substances 0.000 description 7
- 238000002513 implantation Methods 0.000 description 7
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- 150000003254 radicals Chemical class 0.000 description 7
- 230000035484 reaction time Effects 0.000 description 7
- 150000003384 small molecules Chemical class 0.000 description 7
- 239000000758 substrate Substances 0.000 description 7
- 208000024891 symptom Diseases 0.000 description 7
- 210000001519 tissue Anatomy 0.000 description 7
- 238000005160 1H NMR spectroscopy Methods 0.000 description 6
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 6
- 102000004506 Blood Proteins Human genes 0.000 description 6
- 108010017384 Blood Proteins Proteins 0.000 description 6
- 101800004538 Bradykinin Proteins 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 6
- QXZGBUJJYSLZLT-UHFFFAOYSA-N H-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH Natural products NC(N)=NCCCC(N)C(=O)N1CCCC1C(=O)N1C(C(=O)NCC(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CO)C(=O)N2C(CCC2)C(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CCCN=C(N)N)C(O)=O)CCC1 QXZGBUJJYSLZLT-UHFFFAOYSA-N 0.000 description 6
- 102100035792 Kininogen-1 Human genes 0.000 description 6
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 6
- 208000027418 Wounds and injury Diseases 0.000 description 6
- 210000004204 blood vessel Anatomy 0.000 description 6
- QXZGBUJJYSLZLT-FDISYFBBSA-N bradykinin Chemical compound NC(=N)NCCC[C@H](N)C(=O)N1CCC[C@H]1C(=O)N1[C@H](C(=O)NCC(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CO)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)CCC1 QXZGBUJJYSLZLT-FDISYFBBSA-N 0.000 description 6
- 201000011510 cancer Diseases 0.000 description 6
- 238000009833 condensation Methods 0.000 description 6
- 230000005494 condensation Effects 0.000 description 6
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical compound [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 6
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 5
- 206010003658 Atrial Fibrillation Diseases 0.000 description 5
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 5
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 5
- 239000008346 aqueous phase Substances 0.000 description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 5
- PVXOUGHWLCBJOW-UHFFFAOYSA-N azetidine-1-carboxamide Chemical compound NC(=O)N1CCC1 PVXOUGHWLCBJOW-UHFFFAOYSA-N 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 230000023555 blood coagulation Effects 0.000 description 5
- 150000001721 carbon Chemical group 0.000 description 5
- 230000001684 chronic effect Effects 0.000 description 5
- 238000004440 column chromatography Methods 0.000 description 5
- 230000006378 damage Effects 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000005457 ice water Substances 0.000 description 5
- 238000007918 intramuscular administration Methods 0.000 description 5
- 230000000155 isotopic effect Effects 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 230000002503 metabolic effect Effects 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 5
- 239000001301 oxygen Substances 0.000 description 5
- 230000036961 partial effect Effects 0.000 description 5
- 230000037361 pathway Effects 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- PJVWKTKQMONHTI-UHFFFAOYSA-N warfarin Chemical compound OC=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 PJVWKTKQMONHTI-UHFFFAOYSA-N 0.000 description 5
- 150000003952 β-lactams Chemical class 0.000 description 5
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 4
- DDFHBQSCUXNBSA-UHFFFAOYSA-N 5-(5-carboxythiophen-2-yl)thiophene-2-carboxylic acid Chemical compound S1C(C(=O)O)=CC=C1C1=CC=C(C(O)=O)S1 DDFHBQSCUXNBSA-UHFFFAOYSA-N 0.000 description 4
- 206010002388 Angina unstable Diseases 0.000 description 4
- 201000001320 Atherosclerosis Diseases 0.000 description 4
- 229920000858 Cyclodextrin Polymers 0.000 description 4
- 206010020751 Hypersensitivity Diseases 0.000 description 4
- 229940127379 Kallikrein Inhibitors Drugs 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- 235000019502 Orange oil Nutrition 0.000 description 4
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 4
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 4
- 208000032109 Transient ischaemic attack Diseases 0.000 description 4
- 208000007814 Unstable Angina Diseases 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 230000001154 acute effect Effects 0.000 description 4
- 125000002947 alkylene group Chemical group 0.000 description 4
- CWNKMHIETKEBCA-UHFFFAOYSA-N alpha-Ethylaminohexanophenone Chemical compound CCCCC(NCC)C(=O)C1=CC=CC=C1 CWNKMHIETKEBCA-UHFFFAOYSA-N 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- 229910052796 boron Inorganic materials 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 239000006227 byproduct Substances 0.000 description 4
- 208000026106 cerebrovascular disease Diseases 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 4
- 238000010828 elution Methods 0.000 description 4
- 210000003414 extremity Anatomy 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 238000004108 freeze drying Methods 0.000 description 4
- 230000023597 hemostasis Effects 0.000 description 4
- 125000001183 hydrocarbyl group Chemical group 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 201000004332 intermediate coronary syndrome Diseases 0.000 description 4
- 238000001361 intraarterial administration Methods 0.000 description 4
- 150000002500 ions Chemical class 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 230000003287 optical effect Effects 0.000 description 4
- 239000010502 orange oil Substances 0.000 description 4
- 210000000056 organ Anatomy 0.000 description 4
- 230000002265 prevention Effects 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 4
- 238000007920 subcutaneous administration Methods 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 238000001356 surgical procedure Methods 0.000 description 4
- WUEIYIPUVOLPDP-UHFFFAOYSA-N tert-butyl N-[4-(bromomethyl)pyridin-2-yl]-N-[(4-methoxyphenyl)methyl]carbamate Chemical compound C(C)(C)(C)OC(N(CC1=CC=C(C=C1)OC)C1=NC=CC(=C1)CBr)=O WUEIYIPUVOLPDP-UHFFFAOYSA-N 0.000 description 4
- SWKKMNWZFJJIGZ-UHFFFAOYSA-N tert-butyl n-[(4-methoxyphenyl)methyl]-n-(4-methylpyridin-2-yl)carbamate Chemical compound C1=CC(OC)=CC=C1CN(C(=O)OC(C)(C)C)C1=CC(C)=CC=N1 SWKKMNWZFJJIGZ-UHFFFAOYSA-N 0.000 description 4
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 4
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 4
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 4
- 201000010875 transient cerebral ischemia Diseases 0.000 description 4
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- 229960005080 warfarin Drugs 0.000 description 4
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical compound NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 description 3
- MOHYOXXOKFQHDC-UHFFFAOYSA-N 1-(chloromethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCl)C=C1 MOHYOXXOKFQHDC-UHFFFAOYSA-N 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- 208000004998 Abdominal Pain Diseases 0.000 description 3
- 208000004476 Acute Coronary Syndrome Diseases 0.000 description 3
- 206010002383 Angina Pectoris Diseases 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- 206010008088 Cerebral artery embolism Diseases 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- 102000010911 Enzyme Precursors Human genes 0.000 description 3
- 108010062466 Enzyme Precursors Proteins 0.000 description 3
- 108010074860 Factor Xa Proteins 0.000 description 3
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 208000002193 Pain Diseases 0.000 description 3
- 208000003251 Pruritus Diseases 0.000 description 3
- 206010040047 Sepsis Diseases 0.000 description 3
- 102000012479 Serine Proteases Human genes 0.000 description 3
- 108010022999 Serine Proteases Proteins 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 230000029936 alkylation Effects 0.000 description 3
- 238000005804 alkylation reaction Methods 0.000 description 3
- 208000026935 allergic disease Diseases 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 229950003476 aminothiazole Drugs 0.000 description 3
- 208000003455 anaphylaxis Diseases 0.000 description 3
- 150000001450 anions Chemical class 0.000 description 3
- 239000003416 antiarrhythmic agent Substances 0.000 description 3
- 229940127219 anticoagulant drug Drugs 0.000 description 3
- 229940030600 antihypertensive agent Drugs 0.000 description 3
- 239000002220 antihypertensive agent Substances 0.000 description 3
- 125000004567 azetidin-3-yl group Chemical group N1CC(C1)* 0.000 description 3
- MRIWDWQIZATGCC-WIYYLYMNSA-N benzyl (2S,3R)-3-[2-(4-methoxyphenyl)ethyl]-3-methyl-4-oxoazetidine-2-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)[C@H]1NC([C@]1(C)CCC1=CC=C(C=C1)OC)=O MRIWDWQIZATGCC-WIYYLYMNSA-N 0.000 description 3
- FPHKPVOSWOTUGR-RSXGOPAZSA-N benzyl (2S,3R)-3-[[2-[(4-methoxyphenyl)methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]pyridin-4-yl]methyl]-4-oxoazetidine-2-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)[C@H]1NC([C@@H]1CC1=CC(=NC=C1)N(CC1=CC=C(C=C1)OC)C(=O)OC(C)(C)C)=O FPHKPVOSWOTUGR-RSXGOPAZSA-N 0.000 description 3
- 235000019445 benzyl alcohol Nutrition 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 208000029078 coronary artery disease Diseases 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000002270 dispersing agent Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- AEZAOIZKMOJRLY-UHFFFAOYSA-N ethyl 2-[2-[(4-methoxyphenyl)methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]pyridin-4-yl]acetate Chemical compound C(C)(C)(C)OC(=O)N(C1=NC=CC(=C1)CC(=O)OCC)CC1=CC=C(C=C1)OC AEZAOIZKMOJRLY-UHFFFAOYSA-N 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 230000020764 fibrinolysis Effects 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 3
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 3
- 238000011540 hip replacement Methods 0.000 description 3
- 239000008240 homogeneous mixture Substances 0.000 description 3
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 3
- 230000004054 inflammatory process Effects 0.000 description 3
- 238000007917 intracranial administration Methods 0.000 description 3
- 201000010849 intracranial embolism Diseases 0.000 description 3
- 238000007919 intrasynovial administration Methods 0.000 description 3
- 238000007913 intrathecal administration Methods 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 210000001853 liver microsome Anatomy 0.000 description 3
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 3
- TZCLGBJFBGZAQO-UHFFFAOYSA-N methyl 2-[(2-methylpropan-2-yl)oxycarbonylamino]pyridine-4-carboxylate Chemical compound COC(=O)C1=CC=NC(NC(=O)OC(C)(C)C)=C1 TZCLGBJFBGZAQO-UHFFFAOYSA-N 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 230000036407 pain Effects 0.000 description 3
- 206010033675 panniculitis Diseases 0.000 description 3
- 235000021317 phosphate Nutrition 0.000 description 3
- 238000012746 preparative thin layer chromatography Methods 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 230000002685 pulmonary effect Effects 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 210000003491 skin Anatomy 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 239000012258 stirred mixture Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000011550 stock solution Substances 0.000 description 3
- 210000004304 subcutaneous tissue Anatomy 0.000 description 3
- 125000000547 substituted alkyl group Chemical group 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 230000008961 swelling Effects 0.000 description 3
- NAXQLPZECGNMSJ-UHFFFAOYSA-N tert-butyl 2-[(4-methoxyphenyl)methylamino]-4-methylpyridine-3-carboxylate Chemical compound COC1=CC=C(CNC2=C(C(=O)OC(C)(C)C)C(=CC=N2)C)C=C1 NAXQLPZECGNMSJ-UHFFFAOYSA-N 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 229960000187 tissue plasminogen activator Drugs 0.000 description 3
- 230000008733 trauma Effects 0.000 description 3
- 238000000825 ultraviolet detection Methods 0.000 description 3
- 230000002792 vascular Effects 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- CGCLCGPCTZEREL-NUBCRITNSA-N (1R)-1-(2,2-difluoro-1,3-benzodioxol-5-yl)ethanamine hydrochloride Chemical compound Cl.FC1(OC2=C(O1)C=CC(=C2)[C@@H](C)N)F CGCLCGPCTZEREL-NUBCRITNSA-N 0.000 description 2
- CSFOVXWGDRENON-INIZCTEOSA-N (2S)-1-[tert-butyl(diphenyl)silyl]-4-oxoazetidine-2-carbaldehyde Chemical compound C(C)(C)(C)[Si](N1[C@@H](CC1=O)C=O)(C1=CC=CC=C1)C1=CC=CC=C1 CSFOVXWGDRENON-INIZCTEOSA-N 0.000 description 2
- OKDMJXVHXRPNNZ-UHFFFAOYSA-N (4-nitrophenoxy)carbonyloxymethyl 2-methylpropanoate Chemical compound CC(C)C(=O)OCOC(=O)OC1=CC=C([N+]([O-])=O)C=C1 OKDMJXVHXRPNNZ-UHFFFAOYSA-N 0.000 description 2
- YFSSTNJIVXGSMO-INIZCTEOSA-N (4S)-1-[tert-butyl(diphenyl)silyl]-4-(hydroxymethyl)azetidin-2-one Chemical compound C(C)(C)(C)[Si](N1C(C[C@H]1CO)=O)(C1=CC=CC=C1)C1=CC=CC=C1 YFSSTNJIVXGSMO-INIZCTEOSA-N 0.000 description 2
- TVPUYRAEEYNDMC-KRWDZBQOSA-N (4S)-1-[tert-butyl(diphenyl)silyl]-4-(methoxyiminomethyl)azetidin-2-one Chemical compound CON=C[C@H]1N(C(C1)=O)[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C TVPUYRAEEYNDMC-KRWDZBQOSA-N 0.000 description 2
- XEDRFRAQMLFZEB-IBGZPJMESA-N (NE,S)-N-[(2,2-difluoro-1,3-benzodioxol-5-yl)methylidene]-2-methylpropane-2-sulfinamide Chemical compound FC1(OC2=C(O1)C=CC(=C2)C=N[S@@](=O)C(C)(C)C)F XEDRFRAQMLFZEB-IBGZPJMESA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- YSPMLLKKKHCTBN-UHFFFAOYSA-N 4-oxoazetidine-2-carboxylic acid Chemical class OC(=O)C1CC(=O)N1 YSPMLLKKKHCTBN-UHFFFAOYSA-N 0.000 description 2
- 239000005541 ACE inhibitor Substances 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 2
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 2
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 description 2
- 102000015081 Blood Coagulation Factors Human genes 0.000 description 2
- 108010039209 Blood Coagulation Factors Proteins 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- 206010008132 Cerebral thrombosis Diseases 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 206010053567 Coagulopathies Diseases 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- 206010015769 Extradural haematoma Diseases 0.000 description 2
- 108010074864 Factor XI Proteins 0.000 description 2
- 108010049003 Fibrinogen Proteins 0.000 description 2
- 102000008946 Fibrinogen Human genes 0.000 description 2
- 208000018522 Gastrointestinal disease Diseases 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 229910004373 HOAc Inorganic materials 0.000 description 2
- 206010019280 Heart failures Diseases 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 201000001429 Intracranial Thrombosis Diseases 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 208000029027 Musculoskeletal and connective tissue disease Diseases 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- SOSGOLAMRAQPIW-PMLQENADSA-N N-[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-yl)ethyl]-2-methylpropane-2-sulfinamide Chemical compound FC1(OC2=C(O1)C=CC(=C2)[C@@H](C)NS(=O)C(C)(C)C)F SOSGOLAMRAQPIW-PMLQENADSA-N 0.000 description 2
- AHVYPIQETPWLSZ-UHFFFAOYSA-N N-methyl-pyrrolidine Natural products CN1CC=CC1 AHVYPIQETPWLSZ-UHFFFAOYSA-N 0.000 description 2
- 229910017855 NH 4 F Inorganic materials 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 2
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 108010022233 Plasminogen Activator Inhibitor 1 Proteins 0.000 description 2
- 102100039418 Plasminogen activator inhibitor 1 Human genes 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 2
- 108010056373 SK potentiator Proteins 0.000 description 2
- 108010023197 Streptokinase Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 2
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 2
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 2
- 208000024780 Urticaria Diseases 0.000 description 2
- 206010053648 Vascular occlusion Diseases 0.000 description 2
- YWSNPTBCJATDQX-GHTZIAJQSA-N [(2R,3R)-3-[[2-[bis[(2-methylpropan-2-yl)oxycarbonyl]amino]pyridin-4-yl]methyl]-4-oxoazetidin-2-yl] 3-chlorobenzoate Chemical compound ClC=1C=C(C(=O)O[C@H]2NC([C@@H]2CC2=CC(=NC=C2)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C)=O)C=CC1 YWSNPTBCJATDQX-GHTZIAJQSA-N 0.000 description 2
- 230000003187 abdominal effect Effects 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 229960001138 acetylsalicylic acid Drugs 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 206010000891 acute myocardial infarction Diseases 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 150000001350 alkyl halides Chemical class 0.000 description 2
- 230000007815 allergy Effects 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 150000003927 aminopyridines Chemical class 0.000 description 2
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 2
- 239000001099 ammonium carbonate Substances 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 2
- 230000002253 anti-ischaemic effect Effects 0.000 description 2
- 230000002785 anti-thrombosis Effects 0.000 description 2
- 229960004676 antithrombotic agent Drugs 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- JGUNLPMKKUKVDJ-MSOLQXFVSA-N benzyl (2S,3R)-1-[tert-butyl(dimethyl)silyl]-4-oxo-3-(4-oxobutyl)azetidine-2-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)[C@H]1N(C([C@@H]1CCCC=O)=O)[Si](C)(C)C(C)(C)C JGUNLPMKKUKVDJ-MSOLQXFVSA-N 0.000 description 2
- VSUZULBODGJMAV-OLZOCXBDSA-N benzyl (2S,3R)-3-[2-(2-amino-1,3-thiazol-5-yl)ethyl]-4-oxoazetidine-2-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)[C@H]1NC([C@@H]1CCC1=CN=C(S1)N)=O VSUZULBODGJMAV-OLZOCXBDSA-N 0.000 description 2
- 208000015294 blood coagulation disease Diseases 0.000 description 2
- 239000003114 blood coagulation factor Substances 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- 229940045348 brown mixture Drugs 0.000 description 2
- 230000005587 bubbling Effects 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 235000013877 carbamide Nutrition 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 125000006244 carboxylic acid protecting group Chemical group 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 206010008118 cerebral infarction Diseases 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- PDTWCUYBIVJSTL-UHFFFAOYSA-N chloromethyl (4-nitrophenyl) carbonate Chemical compound [O-][N+](=O)C1=CC=C(OC(=O)OCCl)C=C1 PDTWCUYBIVJSTL-UHFFFAOYSA-N 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 229960003009 clopidogrel Drugs 0.000 description 2
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 description 2
- 230000035602 clotting Effects 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 210000004351 coronary vessel Anatomy 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 150000001975 deuterium Chemical group 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 208000010643 digestive system disease Diseases 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 230000002526 effect on cardiovascular system Effects 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 238000013171 endarterectomy Methods 0.000 description 2
- 201000007219 factor XI deficiency Diseases 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 229940012952 fibrinogen Drugs 0.000 description 2
- 230000003480 fibrinolytic effect Effects 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 238000005194 fractionation Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 125000005456 glyceride group Chemical group 0.000 description 2
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 2
- 210000002216 heart Anatomy 0.000 description 2
- 210000003709 heart valve Anatomy 0.000 description 2
- 229920000669 heparin Polymers 0.000 description 2
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 2
- KIWBRXCOTCXSSZ-UHFFFAOYSA-N hexyl carbonochloridate Chemical compound CCCCCCOC(Cl)=O KIWBRXCOTCXSSZ-UHFFFAOYSA-N 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- NZKQUJFOPJXEFF-UHFFFAOYSA-N iodomethyl (4-nitrophenyl) carbonate Chemical compound [O-][N+](=O)C1=CC=C(OC(=O)OCI)C=C1 NZKQUJFOPJXEFF-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- 230000007803 itching Effects 0.000 description 2
- 238000013150 knee replacement Methods 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 210000000867 larynx Anatomy 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 210000004324 lymphatic system Anatomy 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000002483 medication Methods 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- HTMFEPXINZZLPZ-UHFFFAOYSA-N methyl 2-[methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]pyridine-4-carboxylate Chemical compound C(C)(C)(C)OC(=O)N(C=1C=C(C(=O)OC)C=CN1)C HTMFEPXINZZLPZ-UHFFFAOYSA-N 0.000 description 2
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 2
- 230000003228 microsomal effect Effects 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 210000000214 mouth Anatomy 0.000 description 2
- 210000004877 mucosa Anatomy 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 150000002825 nitriles Chemical class 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 150000002923 oximes Chemical class 0.000 description 2
- 125000000636 p-nitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)[N+]([O-])=O 0.000 description 2
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- 229920000728 polyester Polymers 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 229960003712 propranolol Drugs 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- 238000005956 quaternization reaction Methods 0.000 description 2
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 239000002464 receptor antagonist Substances 0.000 description 2
- 229940044551 receptor antagonist Drugs 0.000 description 2
- 208000037803 restenosis Diseases 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- GYEMIEGAEOIJQR-UHFFFAOYSA-M silver;2-methylpropanoate Chemical compound [Ag+].CC(C)C([O-])=O GYEMIEGAEOIJQR-UHFFFAOYSA-M 0.000 description 2
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 2
- 229960002855 simvastatin Drugs 0.000 description 2
- 238000002603 single-photon emission computed tomography Methods 0.000 description 2
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 2
- 235000019345 sodium thiosulphate Nutrition 0.000 description 2
- LYPGDCWPTHTUDO-UHFFFAOYSA-M sodium;methanesulfinate Chemical compound [Na+].CS([O-])=O LYPGDCWPTHTUDO-UHFFFAOYSA-M 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 229960005202 streptokinase Drugs 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- SYPWAYLRMLSQAY-GHTZIAJQSA-N tert-butyl N-[(4-methoxyphenyl)methyl]-N-[4-[[(2R,3S)-2-methylsulfonyl-4-oxoazetidin-3-yl]methyl]pyridin-2-yl]carbamate Chemical compound C(C)(C)(C)OC(N(C1=NC=CC(=C1)C[C@@H]1[C@H](NC1=O)S(=O)(=O)C)CC1=CC=C(C=C1)OC)=O SYPWAYLRMLSQAY-GHTZIAJQSA-N 0.000 description 2
- UAUPKDHYFYBJOK-UHFFFAOYSA-N tert-butyl N-[4-(2-bromoethyl)pyridin-2-yl]-N-[(4-methoxyphenyl)methyl]carbamate Chemical compound C(C)(C)(C)OC(N(CC1=CC=C(C=C1)OC)C1=NC=CC(=C1)CCBr)=O UAUPKDHYFYBJOK-UHFFFAOYSA-N 0.000 description 2
- OBJUZMSJCALFLI-UHFFFAOYSA-N tert-butyl N-[4-(2-hydroxyethyl)pyridin-2-yl]-N-[(4-methoxyphenyl)methyl]carbamate Chemical compound C(C)(C)(C)OC(N(CC1=CC=C(C=C1)OC)C1=NC=CC(=C1)CCO)=O OBJUZMSJCALFLI-UHFFFAOYSA-N 0.000 description 2
- NXHZKRQCFVCPST-MJGOQNOKSA-N tert-butyl N-[4-[[(2S,3R)-2-carbamoyl-4-oxoazetidin-3-yl]methyl]pyridin-2-yl]-N-[(4-methoxyphenyl)methyl]carbamate Chemical compound C(C)(C)(C)OC(N(CC1=CC=C(C=C1)OC)C1=NC=CC(=C1)C[C@@H]1[C@H](NC1=O)C(N)=O)=O NXHZKRQCFVCPST-MJGOQNOKSA-N 0.000 description 2
- DRZJRXUZGPFSIP-UHFFFAOYSA-N tert-butyl N-[[4-(bromomethyl)pyridin-2-yl]methyl]carbamate Chemical compound C(C)(C)(C)OC(NCC1=NC=CC(=C1)CBr)=O DRZJRXUZGPFSIP-UHFFFAOYSA-N 0.000 description 2
- DAASPRONXUABDC-UHFFFAOYSA-N tert-butyl N-[[4-(hydroxymethyl)pyridin-2-yl]methyl]carbamate Chemical compound C(C)(C)(C)OC(NCC1=NC=CC(=C1)CO)=O DAASPRONXUABDC-UHFFFAOYSA-N 0.000 description 2
- SABMAMDNSRZJBK-UHFFFAOYSA-N tert-butyl n-(4-methylpyridin-2-yl)carbamate Chemical compound CC1=CC=NC(NC(=O)OC(C)(C)C)=C1 SABMAMDNSRZJBK-UHFFFAOYSA-N 0.000 description 2
- NYAFXJAKULRGTA-UHFFFAOYSA-N tert-butyl n-[4-(hydroxymethyl)pyridin-2-yl]-n-[(4-methoxyphenyl)methyl]carbamate Chemical compound C1=CC(OC)=CC=C1CN(C(=O)OC(C)(C)C)C1=CC(CO)=CC=N1 NYAFXJAKULRGTA-UHFFFAOYSA-N 0.000 description 2
- 229960000278 theophylline Drugs 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 2
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 2
- 150000003672 ureas Chemical class 0.000 description 2
- 208000021331 vascular occlusion disease Diseases 0.000 description 2
- 230000002861 ventricular Effects 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- XWTYSIMOBUGWOL-UHFFFAOYSA-N (+-)-Terbutaline Chemical compound CC(C)(C)NCC(O)C1=CC(O)=CC(O)=C1 XWTYSIMOBUGWOL-UHFFFAOYSA-N 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- GILTYZBJXXHWGV-FYZOBXCZSA-N (1R)-1-[3-(trifluoromethoxy)phenyl]ethanamine hydrochloride Chemical compound Cl.FC(OC=1C=C(C=CC1)[C@@H](C)N)(F)F GILTYZBJXXHWGV-FYZOBXCZSA-N 0.000 description 1
- DZCAUMADOBDJJH-ZETCQYMHSA-N (1s)-2,2,2-trifluoro-1-phenylethanamine Chemical compound FC(F)(F)[C@@H](N)C1=CC=CC=C1 DZCAUMADOBDJJH-ZETCQYMHSA-N 0.000 description 1
- BLFRMOOGAICNSZ-UHFFFAOYSA-N (2,4,6-trimethoxyphenyl)methanamine;hydrochloride Chemical compound Cl.COC1=CC(OC)=C(CN)C(OC)=C1 BLFRMOOGAICNSZ-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 1
- CLPHYCGWFVWVQT-AUJRIZCKSA-N (2S,3R)-1-[[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-yl)ethyl]carbamoyl]-3-[[2-(2-methylpropanoyloxymethoxycarbonylamino)pyridin-4-yl]methyl]-4-oxoazetidine-2-carboxylic acid Chemical compound FC1(OC2=C(O1)C=CC(=C2)[C@@H](C)NC(=O)N2[C@@H]([C@H](C2=O)CC2=CC(=NC=C2)NC(=O)OCOC(C(C)C)=O)C(=O)O)F CLPHYCGWFVWVQT-AUJRIZCKSA-N 0.000 description 1
- PYXWAVPVSFZNJY-WMBPSLEXSA-N (2S,3R)-1-[[(1R)-1-cyclohexylethyl]carbamoyl]-3-[[2-[(4-methoxyphenyl)methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]pyridin-4-yl]methyl]-4-oxoazetidine-2-carboxylic acid Chemical compound C(C)(C)(C)OC(=O)N(C1=NC=CC(=C1)C[C@@H]1[C@H](N(C1=O)C(N[C@H](C)C1CCCCC1)=O)C(=O)O)CC1=CC=C(C=C1)OC PYXWAVPVSFZNJY-WMBPSLEXSA-N 0.000 description 1
- CFCGNTDHZXOPHT-GZJHNZOKSA-N (2S,3R)-3-[(2-aminopyridin-4-yl)methyl]-N-benzhydryl-2-cyano-4-oxoazetidine-1-carboxamide 2,2,2-trifluoroacetic acid Chemical compound FC(C(=O)O)(F)F.NC1=NC=CC(=C1)C[C@@H]1[C@H](N(C1=O)C(=O)NC(C1=CC=CC=C1)C1=CC=CC=C1)C#N CFCGNTDHZXOPHT-GZJHNZOKSA-N 0.000 description 1
- KPLNXUBQWUYJDZ-NFHUAXAMSA-N (2S,3R)-3-[2-(4-methoxyphenyl)ethyl]-3-methyl-4-oxo-1-[[(1R)-1-phenylethyl]carbamoyl]azetidine-2-carboxylic acid Chemical compound COC1=CC=C(C=C1)CC[C@@]1([C@H](N(C1=O)C(N[C@H](C)C1=CC=CC=C1)=O)C(=O)O)C KPLNXUBQWUYJDZ-NFHUAXAMSA-N 0.000 description 1
- DNISEZBAYYIQFB-PHDIDXHHSA-N (2r,3r)-2,3-diacetyloxybutanedioic acid Chemical compound CC(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(C)=O DNISEZBAYYIQFB-PHDIDXHHSA-N 0.000 description 1
- LIEWITJXZYCDLE-ZETCQYMHSA-N (2s)-1-[tert-butyl(dimethyl)silyl]-4-oxoazetidine-2-carboxylic acid Chemical compound CC(C)(C)[Si](C)(C)N1[C@H](C(O)=O)CC1=O LIEWITJXZYCDLE-ZETCQYMHSA-N 0.000 description 1
- QVHJQCGUWFKTSE-YFKPBYRVSA-N (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound OC(=O)[C@H](C)NC(=O)OC(C)(C)C QVHJQCGUWFKTSE-YFKPBYRVSA-N 0.000 description 1
- JOUXPQKPKJSQDD-SFYZADRCSA-N (2s,3r)-1-[tert-butyl(dimethyl)silyl]-3-methyl-4-oxoazetidine-2-carboxylic acid Chemical compound C[C@@H]1[C@@H](C(O)=O)N([Si](C)(C)C(C)(C)C)C1=O JOUXPQKPKJSQDD-SFYZADRCSA-N 0.000 description 1
- NGFPJGCOTJXEKD-MJGOQNOKSA-N (2s,3r)-3-[[2-[bis[(2-methylpropan-2-yl)oxycarbonyl]amino]pyridin-4-yl]methyl]-1-[tert-butyl(dimethyl)silyl]-4-oxoazetidine-2-carboxylic acid Chemical compound C1=NC(N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C)=CC(C[C@H]2C(N([C@@H]2C(O)=O)[Si](C)(C)C(C)(C)C)=O)=C1 NGFPJGCOTJXEKD-MJGOQNOKSA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 1
- QHFKWIKCUHNXAU-UHFFFAOYSA-N (4-nitrophenyl) carbamate Chemical class NC(=O)OC1=CC=C([N+]([O-])=O)C=C1 QHFKWIKCUHNXAU-UHFFFAOYSA-N 0.000 description 1
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 1
- 125000006716 (C1-C6) heteroalkyl group Chemical group 0.000 description 1
- UOORRWUZONOOLO-OWOJBTEDSA-N (E)-1,3-dichloropropene Chemical compound ClC\C=C\Cl UOORRWUZONOOLO-OWOJBTEDSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- IWXIVQYETPJJTB-KBPBESRZSA-N 1,3-bis[(1S)-2,2,2-trifluoro-1-phenylethyl]urea Chemical compound FC([C@H](C1=CC=CC=C1)NC(=O)N[C@H](C(F)(F)F)C1=CC=CC=C1)(F)F IWXIVQYETPJJTB-KBPBESRZSA-N 0.000 description 1
- OXHPTABOQVHKLN-UHFFFAOYSA-N 1-(2-bromoethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCBr)C=C1 OXHPTABOQVHKLN-UHFFFAOYSA-N 0.000 description 1
- SRQMOFTXBPTVER-SSDOTTSWSA-N 1-[(1R)-1-isocyanatoethyl]-3-(trifluoromethoxy)benzene Chemical compound N(=C=O)[C@H](C)C1=CC(=CC=C1)OC(F)(F)F SRQMOFTXBPTVER-SSDOTTSWSA-N 0.000 description 1
- YVUDEEVLSJCCFC-BFHBGLAWSA-N 1-[[(1R)-1-phenylethyl]carbamoyl]azetidine-2-carboxylic acid Chemical compound C1(=CC=CC=C1)[C@@H](C)NC(=O)N1C(CC1)C(=O)O YVUDEEVLSJCCFC-BFHBGLAWSA-N 0.000 description 1
- AVFZOVWCLRSYKC-UHFFFAOYSA-N 1-methylpyrrolidine Chemical compound CN1CCCC1 AVFZOVWCLRSYKC-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- GGERGLKEDUUSAP-UHFFFAOYSA-N 2,2-difluoro-1,3-benzodioxole-5-carbaldehyde Chemical compound C1=C(C=O)C=C2OC(F)(F)OC2=C1 GGERGLKEDUUSAP-UHFFFAOYSA-N 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- QKYFPVCTQRWPDV-UHFFFAOYSA-N 2-[(4-methoxyphenyl)methylamino]-4-methylpyridine-3-carboxylic acid Chemical compound C1=CC(OC)=CC=C1CNC1=NC=CC(C)=C1C(O)=O QKYFPVCTQRWPDV-UHFFFAOYSA-N 0.000 description 1
- 125000004174 2-benzimidazolyl group Chemical group [H]N1C(*)=NC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- KMGUEILFFWDGFV-UHFFFAOYSA-N 2-benzoyl-2-benzoyloxy-3-hydroxybutanedioic acid Chemical compound C=1C=CC=CC=1C(=O)C(C(C(O)=O)O)(C(O)=O)OC(=O)C1=CC=CC=C1 KMGUEILFFWDGFV-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- CESUXLKAADQNTB-ZETCQYMHSA-N 2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](N)=O CESUXLKAADQNTB-ZETCQYMHSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 1
- DDYUBCCTNHWSQM-UHFFFAOYSA-N 3-(3-cyclopentyloxy-4-methoxyphenyl)-3-(1,3-dioxoisoindol-2-yl)propanamide Chemical compound COC1=CC=C(C(CC(N)=O)N2C(C3=CC=CC=C3C2=O)=O)C=C1OC1CCCC1 DDYUBCCTNHWSQM-UHFFFAOYSA-N 0.000 description 1
- FQEVHRCPXFKJHF-UHFFFAOYSA-N 3-(trifluoromethoxy)benzaldehyde Chemical compound FC(F)(F)OC1=CC=CC(C=O)=C1 FQEVHRCPXFKJHF-UHFFFAOYSA-N 0.000 description 1
- AVGJKVAGEJJZJT-UHFFFAOYSA-N 3-[(2-amino-1,3-thiazol-5-yl)methyl]azetidin-2-one Chemical class NC=1SC(=CN=1)CC1C(NC1)=O AVGJKVAGEJJZJT-UHFFFAOYSA-N 0.000 description 1
- SCEQLUUSIXAYGZ-UHFFFAOYSA-N 3-[2-(2-amino-1,3-thiazol-5-yl)ethyl]azetidin-2-one Chemical class NC=1SC(=CN=1)CCC1C(NC1)=O SCEQLUUSIXAYGZ-UHFFFAOYSA-N 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000006041 3-hexenyl group Chemical group 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-M 3-phenylpropionate Chemical compound [O-]C(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-M 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- MNHXYNNKDDXKNP-UHFFFAOYSA-N 4-(3-chlorophenyl)-1,7-diethyl-2-pyrido[2,3-d]pyrimidinone Chemical compound N=1C(=O)N(CC)C2=NC(CC)=CC=C2C=1C1=CC=CC(Cl)=C1 MNHXYNNKDDXKNP-UHFFFAOYSA-N 0.000 description 1
- WMRACESMZQRVQS-BHAWFMJASA-N 4-[(2R,3R)-3-[[2-[bis[(2-methylpropan-2-yl)oxycarbonyl]amino]pyridin-4-yl]methyl]-4-oxo-1-[[(1R)-1-phenylethyl]carbamoyl]azetidin-2-yl]oxybenzoic acid Chemical compound C(C)(C)(C)OC(=O)N(C1=NC=CC(=C1)C[C@@H]1[C@H](N(C1=O)C(N[C@H](C)C1=CC=CC=C1)=O)OC1=CC=C(C(=O)O)C=C1)C(=O)OC(C)(C)C WMRACESMZQRVQS-BHAWFMJASA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- KYWCWBXGRWWINE-UHFFFAOYSA-N 4-methoxy-N1,N3-bis(3-pyridinylmethyl)benzene-1,3-dicarboxamide Chemical compound COC1=CC=C(C(=O)NCC=2C=NC=CC=2)C=C1C(=O)NCC1=CC=CN=C1 KYWCWBXGRWWINE-UHFFFAOYSA-N 0.000 description 1
- ORLGLBZRQYOWNA-UHFFFAOYSA-N 4-methylpyridin-2-amine Chemical compound CC1=CC=NC(N)=C1 ORLGLBZRQYOWNA-UHFFFAOYSA-N 0.000 description 1
- BTJIUGUIPKRLHP-UHFFFAOYSA-N 4-nitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1 BTJIUGUIPKRLHP-UHFFFAOYSA-N 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical group [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- 102000056834 5-HT2 Serotonin Receptors Human genes 0.000 description 1
- 108091005479 5-HT2 receptors Proteins 0.000 description 1
- LSLYOANBFKQKPT-DIFFPNOSSA-N 5-[(1r)-1-hydroxy-2-[[(2r)-1-(4-hydroxyphenyl)propan-2-yl]amino]ethyl]benzene-1,3-diol Chemical compound C([C@@H](C)NC[C@H](O)C=1C=C(O)C=C(O)C=1)C1=CC=C(O)C=C1 LSLYOANBFKQKPT-DIFFPNOSSA-N 0.000 description 1
- LPNANKDXVBMDKE-UHFFFAOYSA-N 5-bromopent-1-ene Chemical compound BrCCCC=C LPNANKDXVBMDKE-UHFFFAOYSA-N 0.000 description 1
- 125000006163 5-membered heteroaryl group Chemical group 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- BZCQGAVADFKHAF-UHFFFAOYSA-N 7-(methylamino)chromen-2-one Chemical compound C1=CC(=O)OC2=CC(NC)=CC=C21 BZCQGAVADFKHAF-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 206010000087 Abdominal pain upper Diseases 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- 201000010000 Agranulocytosis Diseases 0.000 description 1
- 102100022712 Alpha-1-antitrypsin Human genes 0.000 description 1
- 102100035991 Alpha-2-antiplasmin Human genes 0.000 description 1
- 229920001450 Alpha-Cyclodextrin Polymers 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- ITPDYQOUSLNIHG-UHFFFAOYSA-N Amiodarone hydrochloride Chemical compound [Cl-].CCCCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(I)=C(OCC[NH+](CC)CC)C(I)=C1 ITPDYQOUSLNIHG-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 206010002198 Anaphylactic reaction Diseases 0.000 description 1
- 206010002199 Anaphylactic shock Diseases 0.000 description 1
- 206010002329 Aneurysm Diseases 0.000 description 1
- 102100030988 Angiotensin-converting enzyme Human genes 0.000 description 1
- 208000031104 Arterial Occlusive disease Diseases 0.000 description 1
- 206010003162 Arterial injury Diseases 0.000 description 1
- 206010003178 Arterial thrombosis Diseases 0.000 description 1
- 208000022211 Arteriovenous Malformations Diseases 0.000 description 1
- 206010003497 Asphyxia Diseases 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 239000005528 B01AC05 - Ticlopidine Substances 0.000 description 1
- 208000008035 Back Pain Diseases 0.000 description 1
- SESFRYSPDFLNCH-UHFFFAOYSA-N Benzyl benzoate Natural products C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 1
- MOZDKDIOPSPTBH-UHFFFAOYSA-N Benzyl parahydroxybenzoate Chemical compound C1=CC(O)=CC=C1C(=O)OCC1=CC=CC=C1 MOZDKDIOPSPTBH-UHFFFAOYSA-N 0.000 description 1
- 206010005949 Bone cancer Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 201000006474 Brain Ischemia Diseases 0.000 description 1
- 206010048962 Brain oedema Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 1
- PCBZRNYXXCIELG-WYFCWLEVSA-N COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 Chemical compound COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 PCBZRNYXXCIELG-WYFCWLEVSA-N 0.000 description 1
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 1
- CKDWPUIZGOQOOM-UHFFFAOYSA-N Carbamyl chloride Chemical compound NC(Cl)=O CKDWPUIZGOQOOM-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 108090000201 Carboxypeptidase B2 Proteins 0.000 description 1
- 102100035023 Carboxypeptidase B2 Human genes 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 206010008092 Cerebral artery thrombosis Diseases 0.000 description 1
- 206010008111 Cerebral haemorrhage Diseases 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- 206010008635 Cholestasis Diseases 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 1
- 244000223760 Cinnamomum zeylanicum Species 0.000 description 1
- 102100026735 Coagulation factor VIII Human genes 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 206010011091 Coronary artery thrombosis Diseases 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 238000006646 Dess-Martin oxidation reaction Methods 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- OIFBSDVPJOWBCH-UHFFFAOYSA-N Diethyl carbonate Chemical compound CCOC(=O)OCC OIFBSDVPJOWBCH-UHFFFAOYSA-N 0.000 description 1
- 206010014513 Embolism arterial Diseases 0.000 description 1
- 206010014522 Embolism venous Diseases 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- 108010080865 Factor XII Proteins 0.000 description 1
- 102000000429 Factor XII Human genes 0.000 description 1
- 229940123583 Factor Xa inhibitor Drugs 0.000 description 1
- 102000009123 Fibrin Human genes 0.000 description 1
- 108010073385 Fibrin Proteins 0.000 description 1
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 208000035451 General disorders and administration site conditions Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 102000018899 Glutamate Receptors Human genes 0.000 description 1
- 108010027915 Glutamate Receptors Proteins 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- 206010058423 Haemangioma-thrombocytopenia syndrome Diseases 0.000 description 1
- 206010062506 Heparin-induced thrombocytopenia Diseases 0.000 description 1
- 101000911390 Homo sapiens Coagulation factor VIII Proteins 0.000 description 1
- 101001091365 Homo sapiens Plasma kallikrein Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 208000013016 Hypoglycemia Diseases 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000026350 Inborn Genetic disease Diseases 0.000 description 1
- 206010061216 Infarction Diseases 0.000 description 1
- 102100025306 Integrin alpha-IIb Human genes 0.000 description 1
- 101710149643 Integrin alpha-IIb Proteins 0.000 description 1
- 102100032999 Integrin beta-3 Human genes 0.000 description 1
- 108010020950 Integrin beta3 Proteins 0.000 description 1
- 206010022562 Intermittent claudication Diseases 0.000 description 1
- 206010048858 Ischaemic cardiomyopathy Diseases 0.000 description 1
- 208000032382 Ischaemic stroke Diseases 0.000 description 1
- 206010023126 Jaundice Diseases 0.000 description 1
- 208000010299 Kasabach-Merritt syndrome Diseases 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- 238000005684 Liebig rearrangement reaction Methods 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 208000001344 Macular Edema Diseases 0.000 description 1
- 206010025415 Macular oedema Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000029001 Mediastinal disease Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- GMPKIPWJBDOURN-UHFFFAOYSA-N Methoxyamine Chemical compound CON GMPKIPWJBDOURN-UHFFFAOYSA-N 0.000 description 1
- 208000011682 Mitral valve disease Diseases 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 208000007101 Muscle Cramp Diseases 0.000 description 1
- 208000021642 Muscular disease Diseases 0.000 description 1
- 201000009623 Myopathy Diseases 0.000 description 1
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000008457 Neurologic Manifestations Diseases 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 206010068319 Oropharyngeal pain Diseases 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 102100037600 P2Y purinoceptor 1 Human genes 0.000 description 1
- 108050008996 P2Y purinoceptor 1 Proteins 0.000 description 1
- 229940127424 P2Y12 Receptor Antagonists Drugs 0.000 description 1
- 206010033425 Pain in extremity Diseases 0.000 description 1
- 208000007542 Paresis Diseases 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 208000005764 Peripheral Arterial Disease Diseases 0.000 description 1
- CXOFVDLJLONNDW-UHFFFAOYSA-N Phenytoin Chemical compound N1C(=O)NC(=O)C1(C=1C=CC=CC=1)C1=CC=CC=C1 CXOFVDLJLONNDW-UHFFFAOYSA-N 0.000 description 1
- 229940121836 Phosphodiesterase 1 inhibitor Drugs 0.000 description 1
- 229940121828 Phosphodiesterase 2 inhibitor Drugs 0.000 description 1
- 229940123263 Phosphodiesterase 3 inhibitor Drugs 0.000 description 1
- 229940123932 Phosphodiesterase 4 inhibitor Drugs 0.000 description 1
- 229940123304 Phosphodiesterase 7 inhibitor Drugs 0.000 description 1
- 102000001938 Plasminogen Activators Human genes 0.000 description 1
- 108010001014 Plasminogen Activators Proteins 0.000 description 1
- 208000005374 Poisoning Diseases 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 206010057765 Procedural complication Diseases 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108010094028 Prothrombin Proteins 0.000 description 1
- 102100027378 Prothrombin Human genes 0.000 description 1
- 206010037437 Pulmonary thrombosis Diseases 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 206010063544 Renal embolism Diseases 0.000 description 1
- 208000037656 Respiratory Sounds Diseases 0.000 description 1
- 206010038980 Retroperitoneal haemorrhage Diseases 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- RUOGJYKOQBFJIG-UHFFFAOYSA-N SCH-351591 Chemical compound C12=CC=C(C(F)(F)F)N=C2C(OC)=CC=C1C(=O)NC1=C(Cl)C=[N+]([O-])C=C1Cl RUOGJYKOQBFJIG-UHFFFAOYSA-N 0.000 description 1
- GIIZNNXWQWCKIB-UHFFFAOYSA-N Serevent Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1 GIIZNNXWQWCKIB-UHFFFAOYSA-N 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 208000005392 Spasm Diseases 0.000 description 1
- 208000032562 Spinal haematoma Diseases 0.000 description 1
- 206010042033 Stevens-Johnson syndrome Diseases 0.000 description 1
- 231100000168 Stevens-Johnson syndrome Toxicity 0.000 description 1
- 206010042241 Stridor Diseases 0.000 description 1
- 208000002667 Subdural Hematoma Diseases 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 208000008253 Systolic Heart Failure Diseases 0.000 description 1
- 208000001871 Tachycardia Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- GUGOEEXESWIERI-UHFFFAOYSA-N Terfenadine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GUGOEEXESWIERI-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- 229940122388 Thrombin inhibitor Drugs 0.000 description 1
- 102000003790 Thrombin receptors Human genes 0.000 description 1
- 108090000166 Thrombin receptors Proteins 0.000 description 1
- 102000003938 Thromboxane Receptors Human genes 0.000 description 1
- 108090000300 Thromboxane Receptors Proteins 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108060005989 Tryptase Proteins 0.000 description 1
- 102000001400 Tryptase Human genes 0.000 description 1
- 201000004810 Vascular dementia Diseases 0.000 description 1
- 208000030451 Vascular dementia disease Diseases 0.000 description 1
- 206010047139 Vasoconstriction Diseases 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- 206010052428 Wound Diseases 0.000 description 1
- YEEZWCHGZNKEEK-UHFFFAOYSA-N Zafirlukast Chemical compound COC1=CC(C(=O)NS(=O)(=O)C=2C(=CC=CC=2)C)=CC=C1CC(C1=C2)=CN(C)C1=CC=C2NC(=O)OC1CCCC1 YEEZWCHGZNKEEK-UHFFFAOYSA-N 0.000 description 1
- HGJXLMLUXYBSKY-JYFHCDHNSA-N [(2R,3R)-3-[[2-[(4-methoxyphenyl)methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]pyridin-4-yl]methyl]-4-oxoazetidin-2-yl] 3-chlorobenzoate Chemical compound ClC=1C=C(C(=O)O[C@H]2NC([C@@H]2CC2=CC(=NC=C2)N(CC2=CC=C(C=C2)OC)C(=O)OC(C)(C)C)=O)C=CC1 HGJXLMLUXYBSKY-JYFHCDHNSA-N 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- WKWLUUTWWLTZPZ-UHFFFAOYSA-N [(dimethoxyamino)-(triazolo[4,5-b]pyridin-3-yloxy)methylidene]-dimethoxyazanium Chemical compound CO[N+](=C(ON1N=NC2=C1N=CC=C2)N(OC)OC)OC WKWLUUTWWLTZPZ-UHFFFAOYSA-N 0.000 description 1
- CPLTWWPCZUZQEZ-UHFFFAOYSA-N [2-[(4-methoxyphenyl)methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]pyridin-4-yl] acetate Chemical compound C(C)(=O)OC1=CC(=NC=C1)N(CC1=CC=C(C=C1)OC)C(=O)OC(C)(C)C CPLTWWPCZUZQEZ-UHFFFAOYSA-N 0.000 description 1
- YPFLFUJKZDAXRA-UHFFFAOYSA-N [3-(carbamoylamino)-2-(2,4-dichlorobenzoyl)-1-benzofuran-6-yl] methanesulfonate Chemical compound O1C2=CC(OS(=O)(=O)C)=CC=C2C(NC(N)=O)=C1C(=O)C1=CC=C(Cl)C=C1Cl YPFLFUJKZDAXRA-UHFFFAOYSA-N 0.000 description 1
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 1
- REKQCRSUOJRCDV-UHFFFAOYSA-N [isocyano(phenyl)methyl]benzene Chemical compound C=1C=CC=CC=1C([N+]#[C-])C1=CC=CC=C1 REKQCRSUOJRCDV-UHFFFAOYSA-N 0.000 description 1
- GOEMGAFJFRBGGG-UHFFFAOYSA-N acebutolol Chemical compound CCCC(=O)NC1=CC=C(OCC(O)CNC(C)C)C(C(C)=O)=C1 GOEMGAFJFRBGGG-UHFFFAOYSA-N 0.000 description 1
- 229960002122 acebutolol Drugs 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 208000005707 acquired angioedema Diseases 0.000 description 1
- 229940099983 activase Drugs 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000009056 active transport Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 239000000808 adrenergic beta-agonist Substances 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 150000008431 aliphatic amides Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 125000004419 alkynylene group Chemical group 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- 108010050122 alpha 1-Antitrypsin Proteins 0.000 description 1
- 229940024142 alpha 1-antitrypsin Drugs 0.000 description 1
- 108090000183 alpha-2-Antiplasmin Proteins 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- HFHDHCJBZVLPGP-RWMJIURBSA-N alpha-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO HFHDHCJBZVLPGP-RWMJIURBSA-N 0.000 description 1
- 229940043377 alpha-cyclodextrin Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 229960005260 amiodarone Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 238000002399 angioplasty Methods 0.000 description 1
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 229940065524 anticholinergics inhalants for obstructive airway diseases Drugs 0.000 description 1
- 229940127090 anticoagulant agent Drugs 0.000 description 1
- 239000003524 antilipemic agent Substances 0.000 description 1
- 206010002906 aortic stenosis Diseases 0.000 description 1
- 208000021328 arterial occlusion Diseases 0.000 description 1
- 230000005744 arteriovenous malformation Effects 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 125000005165 aryl thioxy group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 230000003143 atherosclerotic effect Effects 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- MNFORVFSTILPAW-UHFFFAOYSA-N azetidin-2-one Chemical class O=C1CCN1 MNFORVFSTILPAW-UHFFFAOYSA-N 0.000 description 1
- PPVWKLLLMBEYBH-UHFFFAOYSA-N azetidine-1-carboxamide 2,2,2-trifluoroacetic acid Chemical compound FC(C(=O)O)(F)F.N1(CCC1)C(=O)N PPVWKLLLMBEYBH-UHFFFAOYSA-N 0.000 description 1
- IADUEWIQBXOCDZ-UHFFFAOYSA-N azetidine-2-carboxylic acid Chemical compound OC(=O)C1CCN1 IADUEWIQBXOCDZ-UHFFFAOYSA-N 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 229940092705 beclomethasone Drugs 0.000 description 1
- NBMKJKDGKREAPL-DVTGEIKXSA-N beclomethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O NBMKJKDGKREAPL-DVTGEIKXSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- JMZBBLOIKVEPNC-DEOSSOPVSA-N benzyl (2S)-1-[tert-butyl(diphenyl)silyl]-4-oxoazetidine-2-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)[C@H]1N(C(C1)=O)[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C JMZBBLOIKVEPNC-DEOSSOPVSA-N 0.000 description 1
- MAGAKJWBEKOCEB-MOPGFXCFSA-N benzyl (2S,3R)-1-[tert-butyl(dimethyl)silyl]-4-oxo-3-pent-4-enylazetidine-2-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)[C@H]1N(C([C@@H]1CCCC=C)=O)[Si](C)(C)C(C)(C)C MAGAKJWBEKOCEB-MOPGFXCFSA-N 0.000 description 1
- WGLLBHSIXLWVFU-UHFFFAOYSA-N benzyl 4-oxoazetidine-2-carboxylate Chemical compound C1C(=O)NC1C(=O)OCC1=CC=CC=C1 WGLLBHSIXLWVFU-UHFFFAOYSA-N 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-N beta-phenylpropanoic acid Natural products OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 1
- 229960004853 betadex Drugs 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 125000000319 biphenyl-4-yl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 229960004620 bitolterol Drugs 0.000 description 1
- FZGVEKPRDOIXJY-UHFFFAOYSA-N bitolterol Chemical compound C1=CC(C)=CC=C1C(=O)OC1=CC=C(C(O)CNC(C)(C)C)C=C1OC(=O)C1=CC=C(C)C=C1 FZGVEKPRDOIXJY-UHFFFAOYSA-N 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 230000036770 blood supply Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 208000006752 brain edema Diseases 0.000 description 1
- 210000000133 brain stem Anatomy 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 150000001649 bromium compounds Chemical class 0.000 description 1
- 229960004436 budesonide Drugs 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 206010061592 cardiac fibrillation Diseases 0.000 description 1
- 229940082638 cardiac stimulant phosphodiesterase inhibitors Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 1
- 229960004195 carvedilol Drugs 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 230000019522 cellular metabolic process Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229960005110 cerivastatin Drugs 0.000 description 1
- SEERZIQQUAZTOL-ANMDKAQQSA-N cerivastatin Chemical compound COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 SEERZIQQUAZTOL-ANMDKAQQSA-N 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- JQXXHWHPUNPDRT-YOPQJBRCSA-N chembl1332716 Chemical compound O([C@](C1=O)(C)O\C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)/C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CCN(C)CC1 JQXXHWHPUNPDRT-YOPQJBRCSA-N 0.000 description 1
- CFBUZOUXXHZCFB-OYOVHJISSA-N chembl511115 Chemical compound COC1=CC=C([C@@]2(CC[C@H](CC2)C(O)=O)C#N)C=C1OC1CCCC1 CFBUZOUXXHZCFB-OYOVHJISSA-N 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 230000035606 childbirth Effects 0.000 description 1
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- JYWJULGYGOLCGW-UHFFFAOYSA-N chloromethyl chloroformate Chemical compound ClCOC(Cl)=O JYWJULGYGOLCGW-UHFFFAOYSA-N 0.000 description 1
- 231100000359 cholestasis Toxicity 0.000 description 1
- 230000007870 cholestasis Effects 0.000 description 1
- 239000000812 cholinergic antagonist Substances 0.000 description 1
- 208000023819 chronic asthma Diseases 0.000 description 1
- 229950001653 cilomilast Drugs 0.000 description 1
- RRGUKTPIGVIEKM-UHFFFAOYSA-N cilostazol Chemical compound C=1C=C2NC(=O)CCC2=CC=1OCCCCC1=NN=NN1C1CCCCC1 RRGUKTPIGVIEKM-UHFFFAOYSA-N 0.000 description 1
- 229960004588 cilostazol Drugs 0.000 description 1
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 1
- 235000017803 cinnamon Nutrition 0.000 description 1
- KSPYMJJKQMWWNB-UHFFFAOYSA-N cipamfylline Chemical compound O=C1N(CC2CC2)C(=O)C=2NC(N)=NC=2N1CC1CC1 KSPYMJJKQMWWNB-UHFFFAOYSA-N 0.000 description 1
- 229950002405 cipamfylline Drugs 0.000 description 1
- 230000004087 circulation Effects 0.000 description 1
- 230000001427 coherent effect Effects 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 1
- 238000007887 coronary angioplasty Methods 0.000 description 1
- 208000002528 coronary thrombosis Diseases 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 229940072645 coumadin Drugs 0.000 description 1
- 229960000265 cromoglicic acid Drugs 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 238000011461 current therapy Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 210000004207 dermis Anatomy 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000011903 deuterated solvents Substances 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 1
- 229960000452 diethylstilbestrol Drugs 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- GXGAKHNRMVGRPK-UHFFFAOYSA-N dimagnesium;dioxido-bis[[oxido(oxo)silyl]oxy]silane Chemical compound [Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O GXGAKHNRMVGRPK-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 229960002768 dipyridamole Drugs 0.000 description 1
- IZEKFCXSFNUWAM-UHFFFAOYSA-N dipyridamole Chemical compound C=12N=C(N(CCO)CCO)N=C(N3CCCCC3)C2=NC(N(CCO)CCO)=NC=1N1CCCCC1 IZEKFCXSFNUWAM-UHFFFAOYSA-N 0.000 description 1
- 230000009266 disease activity Effects 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- VLARUOGDXDTHEH-UHFFFAOYSA-L disodium cromoglycate Chemical compound [Na+].[Na+].O1C(C([O-])=O)=CC(=O)C2=C1C=CC=C2OCC(O)COC1=CC=CC2=C1C(=O)C=C(C([O-])=O)O2 VLARUOGDXDTHEH-UHFFFAOYSA-L 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 208000009190 disseminated intravascular coagulation Diseases 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- IXTMWRCNAAVVAI-UHFFFAOYSA-N dofetilide Chemical compound C=1C=C(NS(C)(=O)=O)C=CC=1CCN(C)CCOC1=CC=C(NS(C)(=O)=O)C=C1 IXTMWRCNAAVVAI-UHFFFAOYSA-N 0.000 description 1
- 229960002994 dofetilide Drugs 0.000 description 1
- 238000003182 dose-response assay Methods 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- XGXOSJSGDNPEEF-NRFANRHFSA-N dsstox_cid_27291 Chemical compound N([C@@H]1N=C(C=2C=3N(C1=O)CCC=3C=C(C=2)N)C=1C=CC=CC=1)C(=O)C1=CC=CN=C1 XGXOSJSGDNPEEF-NRFANRHFSA-N 0.000 description 1
- 201000006549 dyspepsia Diseases 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 229960000610 enoxaparin Drugs 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- KAQKFAOMNZTLHT-VVUHWYTRSA-N epoprostenol Chemical compound O1C(=CCCCC(O)=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)CCCCC)[C@H](O)C[C@@H]21 KAQKFAOMNZTLHT-VVUHWYTRSA-N 0.000 description 1
- 229960001123 epoprostenol Drugs 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- HDMPLRFXVMUUFN-QBIMZIAESA-N ethyl (2s,3r)-3-[(2-aminopyridin-4-yl)methyl]-4-oxo-1-[[(1r)-1-phenylethyl]carbamoyl]azetidine-2-carboxylate Chemical compound C([C@@H]1[C@H](N(C1=O)C(=O)N[C@H](C)C=1C=CC=CC=1)C(=O)OCC)C1=CC=NC(N)=C1 HDMPLRFXVMUUFN-QBIMZIAESA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 229960001022 fenoterol Drugs 0.000 description 1
- 230000002600 fibrillogenic effect Effects 0.000 description 1
- 229950003499 fibrin Drugs 0.000 description 1
- 239000002319 fibrinogen receptor antagonist Substances 0.000 description 1
- 239000003527 fibrinolytic agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229960000676 flunisolide Drugs 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 229960002714 fluticasone Drugs 0.000 description 1
- MGNNYOODZCAHBA-GQKYHHCASA-N fluticasone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(O)[C@@]2(C)C[C@@H]1O MGNNYOODZCAHBA-GQKYHHCASA-N 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 229960002848 formoterol Drugs 0.000 description 1
- BPZSYCZIITTYBL-UHFFFAOYSA-N formoterol Chemical compound C1=CC(OC)=CC=C1CC(C)NCC(O)C1=CC=C(O)C(NC=O)=C1 BPZSYCZIITTYBL-UHFFFAOYSA-N 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- GDSRMADSINPKSL-HSEONFRVSA-N gamma-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO GDSRMADSINPKSL-HSEONFRVSA-N 0.000 description 1
- 229940080345 gamma-cyclodextrin Drugs 0.000 description 1
- 208000018685 gastrointestinal system disease Diseases 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- YRSVDSQRGBYVIY-GJZGRUSLSA-N gemopatrilat Chemical compound O=C1N(CC(O)=O)C(C)(C)CCC[C@@H]1NC(=O)[C@@H](S)CC1=CC=CC=C1 YRSVDSQRGBYVIY-GJZGRUSLSA-N 0.000 description 1
- 229950006480 gemopatrilat Drugs 0.000 description 1
- 208000016361 genetic disease Diseases 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 210000004392 genitalia Anatomy 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 238000001631 haemodialysis Methods 0.000 description 1
- 229960003878 haloperidol Drugs 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 206010019465 hemiparesis Diseases 0.000 description 1
- 230000000322 hemodialysis Effects 0.000 description 1
- 208000031169 hemorrhagic disease Diseases 0.000 description 1
- 230000002008 hemorrhagic effect Effects 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 208000024557 hepatobiliary disease Diseases 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 125000004474 heteroalkylene group Chemical group 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003707 hexyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- XNXVOSBNFZWHBV-UHFFFAOYSA-N hydron;o-methylhydroxylamine;chloride Chemical compound Cl.CON XNXVOSBNFZWHBV-UHFFFAOYSA-N 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- BBPRUNPUJIUXSE-DXKRWKNPSA-N ifetroban Chemical compound CCCCCNC(=O)C1=COC([C@H]2[C@H]([C@@H]3CC[C@H]2O3)CC=2C(=CC=CC=2)CCC(O)=O)=N1 BBPRUNPUJIUXSE-DXKRWKNPSA-N 0.000 description 1
- 229950004274 ifetroban Drugs 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- BCGWQEUPMDMJNV-UHFFFAOYSA-N imipramine Chemical compound C1CC2=CC=CC=C2N(CCCN(C)C)C2=CC=CC=C21 BCGWQEUPMDMJNV-UHFFFAOYSA-N 0.000 description 1
- 229960004801 imipramine Drugs 0.000 description 1
- 230000003100 immobilizing effect Effects 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000007574 infarction Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 208000021156 intermittent vascular claudication Diseases 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 208000003243 intestinal obstruction Diseases 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 229960001361 ipratropium bromide Drugs 0.000 description 1
- KEWHKYJURDBRMN-ZEODDXGYSA-M ipratropium bromide hydrate Chemical compound O.[Br-].O([C@H]1C[C@H]2CC[C@@H](C1)[N@@+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 KEWHKYJURDBRMN-ZEODDXGYSA-M 0.000 description 1
- 229960002198 irbesartan Drugs 0.000 description 1
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- FPCCSQOGAWCVBH-UHFFFAOYSA-N ketanserin Chemical compound C1=CC(F)=CC=C1C(=O)C1CCN(CCN2C(C3=CC=CC=C3NC2=O)=O)CC1 FPCCSQOGAWCVBH-UHFFFAOYSA-N 0.000 description 1
- 229960005417 ketanserin Drugs 0.000 description 1
- 229960004125 ketoconazole Drugs 0.000 description 1
- 210000003127 knee Anatomy 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 108010051044 lanoteplase Proteins 0.000 description 1
- 229950010645 lanoteplase Drugs 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- RLAWWYSOJDYHDC-BZSNNMDCSA-N lisinopril Chemical compound C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 RLAWWYSOJDYHDC-BZSNNMDCSA-N 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 229940118179 lovenox Drugs 0.000 description 1
- 229940127215 low-molecular weight heparin Drugs 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 201000010230 macular retinal edema Diseases 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 229910001425 magnesium ion Inorganic materials 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 description 1
- XQFBZUMJYZUKKG-UHFFFAOYSA-N methyl 2-[(4-methoxyphenyl)methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]pyridine-4-carboxylate Chemical compound C(C)(C)(C)OC(=O)N(C=1C=C(C(=O)OC)C=CN1)CC1=CC=C(C=C1)OC XQFBZUMJYZUKKG-UHFFFAOYSA-N 0.000 description 1
- SVWWNEYBEFASMP-UHFFFAOYSA-N methyl 2-aminopyridine-4-carboxylate Chemical compound COC(=O)C1=CC=NC(N)=C1 SVWWNEYBEFASMP-UHFFFAOYSA-N 0.000 description 1
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 1
- 229960002237 metoprolol Drugs 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000002297 mitogenic effect Effects 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 125000006682 monohaloalkyl group Chemical group 0.000 description 1
- 125000004572 morpholin-3-yl group Chemical group N1C(COCC1)* 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 210000004165 myocardium Anatomy 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- VHDUUXNHZLBGHQ-XLVZBRSZSA-N n-cyclohexyl-n-methyl-2-[(e)-[(2-oxo-5,10-dihydro-3h-imidazo[2,1-b]quinazolin-7-yl)-phenylmethylidene]amino]oxyacetamide Chemical compound C=1C=CC=CC=1\C(C=1C=C2CN3CC(=O)N=C3NC2=CC=1)=N/OCC(=O)N(C)C1CCCCC1 VHDUUXNHZLBGHQ-XLVZBRSZSA-N 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- VWPOSFSPZNDTMJ-UCWKZMIHSA-N nadolol Chemical compound C1[C@@H](O)[C@@H](O)CC2=C1C=CC=C2OCC(O)CNC(C)(C)C VWPOSFSPZNDTMJ-UCWKZMIHSA-N 0.000 description 1
- 229960004255 nadolol Drugs 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 1
- 229960004398 nedocromil Drugs 0.000 description 1
- RQTOOFIXOKYGAN-UHFFFAOYSA-N nedocromil Chemical compound CCN1C(C(O)=O)=CC(=O)C2=C1C(CCC)=C1OC(C(O)=O)=CC(=O)C1=C2 RQTOOFIXOKYGAN-UHFFFAOYSA-N 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 230000009251 neurologic dysfunction Effects 0.000 description 1
- 230000007996 neuronal plasticity Effects 0.000 description 1
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- OKXGHXHZNCJMSV-UHFFFAOYSA-N nitro phenyl carbonate Chemical compound [O-][N+](=O)OC(=O)OC1=CC=CC=C1 OKXGHXHZNCJMSV-UHFFFAOYSA-N 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 210000001331 nose Anatomy 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- JPAWFIIYTJQOKW-UHFFFAOYSA-N olprinone Chemical compound N1C(=O)C(C#N)=CC(C2=CN3C=CN=C3C=C2)=C1C JPAWFIIYTJQOKW-UHFFFAOYSA-N 0.000 description 1
- 229950005421 olprinone Drugs 0.000 description 1
- LVRLSYPNFFBYCZ-VGWMRTNUSA-N omapatrilat Chemical compound C([C@H](S)C(=O)N[C@H]1CCS[C@H]2CCC[C@H](N2C1=O)C(=O)O)C1=CC=CC=C1 LVRLSYPNFFBYCZ-VGWMRTNUSA-N 0.000 description 1
- 229950000973 omapatrilat Drugs 0.000 description 1
- 229940127234 oral contraceptive Drugs 0.000 description 1
- 239000003539 oral contraceptive agent Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 239000012285 osmium tetroxide Substances 0.000 description 1
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 1
- 201000008482 osteoarthritis Diseases 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 238000005895 oxidative decarboxylation reaction Methods 0.000 description 1
- 210000002741 palatine tonsil Anatomy 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- VQJKHLNJLBDYFV-SNVBAGLBSA-N phenyl N-[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-yl)ethyl]carbamate Chemical compound C1(=CC=CC=C1)OC(N[C@H](C)C1=CC2=C(OC(O2)(F)F)C=C1)=O VQJKHLNJLBDYFV-SNVBAGLBSA-N 0.000 description 1
- BSCCSDNZEIHXOK-UHFFFAOYSA-N phenyl carbamate Chemical class NC(=O)OC1=CC=CC=C1 BSCCSDNZEIHXOK-UHFFFAOYSA-N 0.000 description 1
- AHWALFGBDFAJAI-UHFFFAOYSA-N phenyl carbonochloridate Chemical compound ClC(=O)OC1=CC=CC=C1 AHWALFGBDFAJAI-UHFFFAOYSA-N 0.000 description 1
- 229960002036 phenytoin Drugs 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000002570 phosphodiesterase III inhibitor Substances 0.000 description 1
- 239000002587 phosphodiesterase IV inhibitor Substances 0.000 description 1
- 239000002606 phosphodiesterase VII inhibitor Substances 0.000 description 1
- 239000002571 phosphodiesterase inhibitor Substances 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- RRRUXBQSQLKHEL-UHFFFAOYSA-N piclamilast Chemical compound COC1=CC=C(C(=O)NC=2C(=CN=CC=2Cl)Cl)C=C1OC1CCCC1 RRRUXBQSQLKHEL-UHFFFAOYSA-N 0.000 description 1
- 229950005184 piclamilast Drugs 0.000 description 1
- 229960001006 picotamide Drugs 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 229960002164 pimobendan Drugs 0.000 description 1
- GLBJJMFZWDBELO-UHFFFAOYSA-N pimobendane Chemical compound C1=CC(OC)=CC=C1C1=NC2=CC=C(C=3C(CC(=O)NN=3)C)C=C2N1 GLBJJMFZWDBELO-UHFFFAOYSA-N 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-M pivalate Chemical compound CC(C)(C)C([O-])=O IUGYQRQAERSCNH-UHFFFAOYSA-M 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 1
- 230000007505 plaque formation Effects 0.000 description 1
- 229940012957 plasmin Drugs 0.000 description 1
- 229940127126 plasminogen activator Drugs 0.000 description 1
- 231100000572 poisoning Toxicity 0.000 description 1
- 230000000607 poisoning effect Effects 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229940113116 polyethylene glycol 1000 Drugs 0.000 description 1
- 125000006684 polyhaloalkyl group Polymers 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 229910001414 potassium ion Inorganic materials 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- DTGLZDAWLRGWQN-UHFFFAOYSA-N prasugrel Chemical compound C1CC=2SC(OC(=O)C)=CC=2CN1C(C=1C(=CC=CC=1)F)C(=O)C1CC1 DTGLZDAWLRGWQN-UHFFFAOYSA-N 0.000 description 1
- 229960004197 prasugrel Drugs 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000035935 pregnancy Effects 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 229950008679 protamine sulfate Drugs 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 229940121649 protein inhibitor Drugs 0.000 description 1
- 239000012268 protein inhibitor Substances 0.000 description 1
- 229940039716 prothrombin Drugs 0.000 description 1
- 230000003331 prothrombotic effect Effects 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000005344 pyridylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- MIXMJCQRHVAJIO-TZHJZOAOSA-N qk4dys664x Chemical compound O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O MIXMJCQRHVAJIO-TZHJZOAOSA-N 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 229960001404 quinidine Drugs 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 108010051412 reteplase Proteins 0.000 description 1
- 229960002917 reteplase Drugs 0.000 description 1
- 210000001525 retina Anatomy 0.000 description 1
- 229950004118 revizinone Drugs 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- MNDBXUUTURYVHR-UHFFFAOYSA-N roflumilast Chemical compound FC(F)OC1=CC=C(C(=O)NC=2C(=CN=CC=2Cl)Cl)C=C1OCC1CC1 MNDBXUUTURYVHR-UHFFFAOYSA-N 0.000 description 1
- 229960002586 roflumilast Drugs 0.000 description 1
- HJORMJIFDVBMOB-UHFFFAOYSA-N rolipram Chemical compound COC1=CC=C(C2CC(=O)NC2)C=C1OC1CCCC1 HJORMJIFDVBMOB-UHFFFAOYSA-N 0.000 description 1
- 229950005741 rolipram Drugs 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- 229960004017 salmeterol Drugs 0.000 description 1
- 108010073863 saruplase Proteins 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000005659 seminal clot liquefaction Effects 0.000 description 1
- 230000035807 sensation Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 208000007056 sickle cell anemia Diseases 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- VFWRGKJLLYDFBY-UHFFFAOYSA-N silver;hydrate Chemical compound O.[Ag].[Ag] VFWRGKJLLYDFBY-UHFFFAOYSA-N 0.000 description 1
- 238000006884 silylation reaction Methods 0.000 description 1
- 201000009890 sinusitis Diseases 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 206010042772 syncope Diseases 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 230000006794 tachycardia Effects 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 239000006068 taste-masking agent Substances 0.000 description 1
- 229960000195 terbutaline Drugs 0.000 description 1
- 229960000351 terfenadine Drugs 0.000 description 1
- ZWOZOZQQLLDRPH-RTBURBONSA-N tert-butyl N-[4-[[(2S,3R)-2-cyano-4-oxoazetidin-3-yl]methyl]pyridin-2-yl]-N-[(4-methoxyphenyl)methyl]carbamate Chemical compound C(C)(C)(C)OC(N(CC1=CC=C(C=C1)OC)C1=NC=CC(=C1)C[C@@H]1[C@H](NC1=O)C#N)=O ZWOZOZQQLLDRPH-RTBURBONSA-N 0.000 description 1
- XCAQIUOFDMREBA-UHFFFAOYSA-N tert-butyl n-[(2-methylpropan-2-yl)oxycarbonyl]carbamate Chemical compound CC(C)(C)OC(=O)NC(=O)OC(C)(C)C XCAQIUOFDMREBA-UHFFFAOYSA-N 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 1
- SFLXUZPXEWWQNH-UHFFFAOYSA-K tetrabutylazanium;tribromide Chemical compound [Br-].[Br-].[Br-].CCCC[N+](CCCC)(CCCC)CCCC.CCCC[N+](CCCC)(CCCC)CCCC.CCCC[N+](CCCC)(CCCC)CCCC SFLXUZPXEWWQNH-UHFFFAOYSA-K 0.000 description 1
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical compound C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 125000005309 thioalkoxy group Chemical group 0.000 description 1
- 210000000115 thoracic cavity Anatomy 0.000 description 1
- 206010048627 thoracic outlet syndrome Diseases 0.000 description 1
- 229960000103 thrombolytic agent Drugs 0.000 description 1
- 230000003558 thrombophilic effect Effects 0.000 description 1
- 201000005060 thrombophlebitis Diseases 0.000 description 1
- 230000001732 thrombotic effect Effects 0.000 description 1
- RZWIIPASKMUIAC-VQTJNVASSA-N thromboxane Chemical compound CCCCCCCC[C@H]1OCCC[C@@H]1CCCCCCC RZWIIPASKMUIAC-VQTJNVASSA-N 0.000 description 1
- 229960005001 ticlopidine Drugs 0.000 description 1
- PHWBOXQYWZNQIN-UHFFFAOYSA-N ticlopidine Chemical compound ClC1=CC=CC=C1CN1CC(C=CS2)=C2CC1 PHWBOXQYWZNQIN-UHFFFAOYSA-N 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- 230000019432 tissue death Effects 0.000 description 1
- 230000009092 tissue dysfunction Effects 0.000 description 1
- AOBORMOPSGHCAX-DGHZZKTQSA-N tocofersolan Chemical compound OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-DGHZZKTQSA-N 0.000 description 1
- 229960000984 tocofersolan Drugs 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 208000004371 toothache Diseases 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000008736 traumatic injury Effects 0.000 description 1
- 229960005294 triamcinolone Drugs 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 102000033691 triglyceride binding proteins Human genes 0.000 description 1
- 108091009665 triglyceride binding proteins Proteins 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 210000001635 urinary tract Anatomy 0.000 description 1
- 208000019206 urinary tract infection Diseases 0.000 description 1
- 229960005356 urokinase Drugs 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- 210000005167 vascular cell Anatomy 0.000 description 1
- 208000019553 vascular disease Diseases 0.000 description 1
- 238000007631 vascular surgery Methods 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 230000025033 vasoconstriction Effects 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 208000004043 venous thromboembolism Diseases 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 229960004764 zafirlukast Drugs 0.000 description 1
- MWLSOWXNZPKENC-SSDOTTSWSA-N zileuton Chemical compound C1=CC=C2SC([C@H](N(O)C(N)=O)C)=CC2=C1 MWLSOWXNZPKENC-SSDOTTSWSA-N 0.000 description 1
- 229960005332 zileuton Drugs 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
- 239000002076 α-tocopherol Substances 0.000 description 1
- 235000004835 α-tocopherol Nutrition 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/397—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having four-membered rings, e.g. azetidine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/06—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D205/08—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with one oxygen atom directly attached in position 2, e.g. beta-lactams
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/443—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4436—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a heterocyclic ring having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
- A61K31/4725—Non-condensed isoquinolines, e.g. papaverine containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Pulmonology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pyridine Compounds (AREA)
- Medicinal Preparation (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
La presente invención proporciona compuestos y composiciones que inhiben el Factor XIa o la calicreína y métodos para usar estos compuestos y composición. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Compuestos y composiciones terapéuticos
Antecedentes de la invención
La coagulación sanguínea es la primera línea de defensa contra la pérdida de sangre tras una lesión. En la "cascada" de la coagulación sanguínea intervienen una serie de zimógenos de serina proteasa circulantes, cofactores reguladores e inhibidores. Cada enzima, una vez generada a partir de su zimógeno, escinde específicamente el siguiente zimógeno de la cascada para producir una proteasa activa. Este proceso se repite hasta que, por último, la trombina escinde los fibrinopéptidos del fibrinógeno para producir fibrina que se polimeriza para formar un coágulo sanguíneo. Aunque una coagulación eficaz limita la pérdida de sangre en el lugar del traumatismo, también plantea el riesgo de una coagulación sistémica que provoque una trombosis masiva. En circunstancias normales, la hemostasia mantiene un equilibrio entre la formación de coágulos (coagulación) y su disolución (fibrinólisis). Sin embargo, en determinados estados patológicos, tales como el infarto agudo de miocardio y la angina inestable, la rotura de una placa aterosclerótica establecida da lugar a la formación de trombos anómalos en la vasculatura arterial coronaria. Las enfermedades que tienen su origen en la coagulación sanguínea, tales como el infarto de miocardio, la angina inestable, la fibrilación auricular, el ictus, la embolia pulmonar y la trombosis venosa profunda, se encuentran entre las principales causas de muerte en los países desarrollados. Los tratamientos anticoagulantes actuales, tales como la heparina inyectable no fraccionada y de bajo peso molecular (HBPM) y la warfarina administrada por vía oral (cumadina), conllevan el riesgo de episodios hemorrágicos y presentan una variabilidad de un paciente a otro que hace necesario un estrecho seguimiento y ajuste de las dosis terapéuticas. En consecuencia, existe una gran necesidad médica de nuevos fármacos anticoagulantes que carezcan de algunos o todos los efectos secundarios de los fármacos actualmente disponibles.
El factor XIa es una diana terapéutica atractiva implicada en la vía asociada a estas enfermedades. Se ha observado un aumento de los niveles del factor XIa o de su actividad en varios trastornos tromboembólicos, tales como la trombosis venosa (Meijers et al., N. Engl. J. Med. 342:696, 2000), el infarto agudo de miocardio (Minnema et al., Arterioscler. Thromb. Vase. Biol., 20:2489, 2000), el síndrome coronario agudo (Butenas et al., Thromb. Haemost., 99:142, 2008), la enfermedad coronaria (Butenas et al., Thromb. Haemost., 99:142, 2008), la enfermedad pulmonar obstructiva crónica (Jankowski et al., Thromb. Res., 127:242, 2011), la estenosis aórtica (Blood Coagul., Fibrinolysis, 22:473, 2011), la isquemia cerebrovascular aguda (Undas et al., Eur. J. Clin. Invest., 42:123, 2012) y la insuficiencia cardiaca sistólica debida a miocardiopatía isquémica (Zabcyk et al., Pol. Arch. Med. Wewn., 120:334, 2010). Los pacientes que carecen de factor XI debido a una deficiencia genética del mismo presentan pocos o ningún ictus isquémico (Salomon et al., Blood, 111:4113, 2008). Al mismo tiempo, la pérdida de actividad del factor XIa, que deja intacta una de las vías que inician la coagulación, no altera la hemostasia. En seres humanos, la deficiencia del factor XI puede provocar un trastorno hemorrágico de leve a moderado, especialmente en tejidos con altos niveles de actividad fibrinolítica local, tales como las vías urinarias, la nariz, la cavidad oral y las amígdalas. Además, la hemostasia es casi normal en ratones deficientes en factor XI (Gailani, Blood Coagul. Fibrinolysis, 8:134, 1997). Por consiguiente, los compuestos que inhiben el factor XIa tienen el potencial de prevenir o tratar una amplia gama de trastornos tromboembólicos, evitando al mismo tiempo los efectos secundarios y las dificultades terapéuticas que afectan a los fármacos que inhiben otros componentes de la vía de coagulación. Además, debido a la eficacia limitada y a los efectos secundarios adversos de algunas terapias actuales para la inhibición de trombosis indeseables (por ejemplo, trombosis venosa profunda e ictus), se necesitan compuestos y procedimientos mejorados (por ejemplo, los asociados al factor XIa) para prevenir o tratar trombosis indeseables. El documento WO2006/108039 describe azetidinonas sustituidas útiles como inhibidores de triptasa, trombina, tripsina, factor Xa, factor VIla, factor XIa y activador del plasminógeno de tipo urocinasa que pueden emplearse en la prevención y/o el tratamiento del asma, asma crónica, rinitis alérgica y trastornos trombóticos.
Otra diana terapéutica es la enzima calicreína. La calicreína plasmática humana es una serina proteasa que puede ser responsable de la activación de varios factores derivados (por ejemplo, la bradicinina y la plasmina) que son fundamentales para la coagulación y el control, por ejemplo, de la presión arterial, la inflamación y el dolor. Las calicreínas se expresan, por ejemplo, en la próstata, la epidermis y el sistema nervioso central (SNC) y pueden participar, por ejemplo, en la regulación de la licuefacción del semen, la escisión de proteínas de adhesión celular y la plasticidad neuronal en el SNC. Además, las calicreínas pueden estar implicadas en la tumorigénesis y el desarrollo de cáncer y angioedema, por ejemplo, el angioedema hereditario. La sobreactivación de la vía de calicreína-quinina puede dar lugar a una serie de trastornos, incluido el angioedema, por ejemplo, el angioedema hereditario (Schneider et al., J. Allergy Clin. Immunol., 120:2, 416, 2007). Hasta la fecha, las opciones de tratamiento del AEH son limitadas (por ejemplo, el documento WO2003/076458). Por ello, se necesitan terapias para prevenir o tratar estas enfermedades.
Sumario de la invención
La presente invención presenta compuestos que inhiben el factor XIa o la calicreína, que pueden utilizarse para prevenir o tratar la trombosis no deseada o el angiodema (por ejemplo, angiodema hereditario) administrando uno o más de estos compuestos solos o junto con otras moléculas a un mamífero. Se proporcionan procedimientos para diseñar o seleccionar inhibidores adicionales del factor XIa o de la calicreína utilizando estas estructuras. De modo deseable, estos compuestos tienen determinadas características estructurales, físicas y espaciales que permiten a los compuestos interactuar con residuos específicos del sitio activo del factor XIa o calicreína.
En un aspecto, la invención reivindicada en el presente documento se dirige a un compuesto de fórmula (II):

o una sal farmacéuticamente aceptable del mismo, en la que,
R1 es H o -alquilo C1-6;
R2 es -CO2R5, H, -alquilo C1-6, -C(O)NR9R10, -CN,-CHN(OR5) o un heteroarilo;
R3 es H o -alquilo C1-6;
A es alquileno C1-6, un enlace, alquenileno C2-6 o alquinileno C2-6;
R4 es cicloalquilo, arilo, heteroarilo o heterociclilo, cada uno de los cuales está sustituido con 0 a 3 apariciones de -NH2 o R6;
cada R5 es independientemente H, -alquilo C1-6, aralquilo o arilo sustituido con 0 a 3 apariciones de -NH2 o R6; cada R6 es independientemente halo, hidroxi, ciano, nitro, alquilo C1-6, alcoxi C1-6, -NR9R10, -NHR10, -C(O)R11, -C(O)OR11, -C(O)NR9R10, -C(NR8)(N(R8)2), -SOqR11, -SO2NR9R10, -NHC(O)OR11, -NHC(O)R11, arilo, heteroarilo, aralquilo, cicloalquilo, heteroaralquilo, heterociclilo o heterociclilalquilo; o
dos grupos R6 tomados conjuntamente con los átomos a los que están unidos forman un anillo de 5 a 7 miembros;
X es -C(O)N(R5)-, C(O)O-, -OC(O)-, -C(O)S(O)2-, -S(O)2C(O)- o -N(R5)C(O)-;
Y es cicloalquilo;
R7 es -alquilo C1-6, H, cicloalquilo, arilo, heteroarilo o heterociclilo, cada uno de los cuales está sustituido con 0 a 3 apariciones de -NH2 o R6;
cada R8 es independientemente H, -alquilo C1-6, -C(O)R5, -C(O)OR5, arilo, heteroarilo, aralquilo, heteroaralquilo, heterociclilo o heterociclilalquilo;
cada uno de R9 y R10 es independientemente -alquilo C1-6, cicloalquilo, heterociclilo, arilo o heteroarilo, o R9 y R10 juntos forman un anillo de 5 a 7 miembros opcionalmente sustituido.
cada R11 es independientemente H, -alquilo C1-10, aralquilo o arilo;
q es un número entero de 0 a 2; y n es un número entero de 0 a 2;
en el que cualquier grupo alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo o heteroarilo, salvo que se defina de otro modo, está opcionalmente sustituido con uno o más sustituyentes seleccionados independientemente entre halógeno, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -ORaa, -ON(Rbb)2, -N(Rbb)2, -N(Rbb)3 +X-, -N(ORcc)Rbb, -SH, -SRaa, -SSRcc, -C(=O)Raa, -CO2H, -CHO, -C(ORcc)2, -CO2Raa, -OC(=O)Raa, -OCO2Raa, -C(=O)N(Rbb)2, -OC(=O)N(Rbb)2, -NRbbC(=O)Raa, -NRbbCO2Raa, -NRbbC(=O)N(Rbb)2, -C(=NRbb)Raa, -C(=NRbb)ORaa, -OC(=NRbb)Raa, -OC(=NRbb)ORaa, -C(=NRbb)N(Rbb)2, -OC(=NRbb)N(Rbb)2, -NRbbC(=NRbb)N(Rbb)2, -C(=O)NRbbSO2Raa, -NRbbSO2Raa, -SO2N(Rbb)2, -SO2Raa, -SO2OR33, -OSO2Raa, -S(O)Raa, -S(=O)Raa, -OS(=O)Raa, -Si(Raa)3, -OSi(Raa)3, -C(=S)N(Rbb)2, -C(=O)SRaa, -C(=S)SRaa, -SC(=S)SRaa, -SC(=O)SRaa, -OC(=O)SRaa, -SC(=O)ORaa, -SC(=O)Raa, -P(=O)2Raa, -OP(=O)2Raa, -P(=O)(Raa)2, -OP(=O)(Raa)2, -OP(=O)(ORcc)2, -P(=O)2N(Rbb)2, -OP(=O)2N(Rbb)2, -P(=O)(NRbb)2, -OP(=O)(NRbb)2, -NRbbP(=O)(ORcc)2, -NRbbP(=O)(NRbb)2, -P(Rcc)2, -P(Rcc)3, -OP(Rcc)2, -OP(Rcc)3, -B(Raa)2, -B(ORcc)2, -BRaa(ORcc), alquilo C1-10 , perhaloalquilo C1-10 , alquenilo C2-10 , alquinilo C2-10 , cicloalquilo C3-10 , heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, en los que cada alquilo,
alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está sustituido independientemente con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Raa se selecciona independientemente entre alquilo C1-10, perhaloalquilo C1-10, alquenilo C2-10, alquinilo C2-10, cicloalquilo C3-10, heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, o dos grupos Raa se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Rbb se selecciona, independientemente, entre hidrógeno, -OH, -ORaa, -N(Rcc)2, -CN, -C(=O)Raa, -C(=O)N(Rcc)2, -CO2Raa, -SO2R33, -C(=NRcc)ORaa, -C(=NRcc)N(Rcc)2, -SO2N(Rcc)2, -SO2Rcc, -SO2ORcc, -SORaa, -C(=S)N(Rcc)2, -C(=O)SRcc, -C(=S)SRcc, -P(=O)2Raa, -P(=O)(Raa)2, -P(=O)2N(Rcc)2, -P(=O)(NRcc)2, alquilo C1-10, perhaloalquilo C1-10, alquenilo C2-10, alquinilo C2-10, cicloalquilo C3-10, heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, o dos grupos Rbb se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está sustituido independientemente con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Rcc se selecciona, independientemente, entre hidrógeno, alquilo C1-10, perhaloalquilo C1-10, alquenilo C2-10, alquinilo C2-10, cicloalquilo C3-10, heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, o dos grupos Rcc se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Rdd se selecciona, independientemente, entre halógeno, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -ORee, -ON(Rff)2, -N(Rff)2, -N(Rff)3 +X-, -N(ORee)Rff, -SH, -SRee, -SSRee, -C(=O)Ree, -CO2H, -CO2Ree, -OC(=O)Ree, -OCO2Ree, -C(=O)N(Rff)2, -OC(=O)N(Rff)2, -NRffC(=O)Ree, -NRffCO2Ree, -NRffC(=O)N(Rff)2, -C(=NRff)ORee, -OC(=NRff)Ree, -OC(=NRff)ORee, -C(=NRff)N(Rff)2, -OC(=NRff)N(Rff)2, -NRffC(=NRff)N(Rff)2,-NRffSO2Ree, -SO2N(Rff)2, -SO2Ree, -SO2ORee, -OSO2Ree, -S(=O)Ree, -Si(Ree)3, -OSi(Ree)3, -C(=S)N(Rff)2, -C(=O)SRee, -C(=S)SRee, -SC(=S)SRee, -P(=O)2Ree, -P(=O)(Ree)2, -OP(=O)(Ree)2, -OP(=O)(ORee)2, alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, arilo C6-10, heteroarilo de 5 a 10 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rgg, o dos sustituyentes geminales Rdd pueden unirse para formar =O o =S;
cada aparición de Ree se selecciona, independientemente, entre alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, arilo C6-10, heteroarilo de 3 a 10 miembros y heterociclilo de 3 a 10 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rgg;
cada aparición de Rff se selecciona, independientemente, entre hidrógeno, alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, arilo C6-10 y heteroarilo de 5 a 10 miembros, o dos grupos Rff se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rgg; y
cada aparición de Rgg es, independientemente, halógeno, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -O-alquilo C1-6, -ON(alquilo C1-6)2, -N(alquilo C1-6)2, -N(alquilo C1-6)3 +X-, -NH(alquilo C1-6)2 +X-, -NH2(alquilo C1-6)+X-, -NH3 +X-, -N(O-alquil C1-6)(alquilo C1-6), -N(OH)(alquilo C1-6), -NH(OH), -SH, -S-alquilo C1-6, -SS(alquilo C1-6), -C(=O)(alquilo C1-6), -CO2H, -CO2(alquilo C1-6), -OC(=O)(alquilo C1-6), -OCO2(alquilo C1-6), -C(=O)NH2, -C(=O)N(alquilo C1-6)2, -OC(=O)NH(alquilo C1-6), -NHC(=O)(alquilo C1-6), -N(alquilo C1-6)C(=O)(alquilo C1-6), -NHCO2(alquilo C1-6), -NHC(=O)N(alquilo C1-6)2, -NHC(=O)NH(alquilo C1-6), -NHC(=O)NH2, -C(=NH)O(alquilo C1-6), -OC(=NH)(alquilo C1-6), -OC(=NH)O-alquilo C1-6, -C(=NH)N(alquilo C1-6)2, -C(=NH)NH(alquilo C1-6), -C(=NH)NH2, -OC(=NH)N(alquilo C1-6)2, -OC(NH)NH(alquilo C1-6), -OC(NH)NH2, -NHC(NH)N(alquilo C1-6)2, -NHC(=NH)NH2, -NHSO2(alquilo C1-6), -SO2N(alquilo C1-6)2, -SO2NH(alquilo C1-6), -SO2NH2,-SO2-alquilo C1-6, -SO2O-alquilo C1-6 -OSO2-alquilo C1-6, -SO-alquilo C1-6, -Si(alquilo C1-6)3, -OSi(alquilo C1-6)3 -C(=S)N(alquilo C1-6)2, C(=S)NH(alquilo C1-6), C(=S)NH2, -C(=O)S(alquilo C1-6), -C(=S)S-alquilo C1-6, -SC(=S)S-alquilo C1-6, -P(=O)2(alquilo C1-6), -P(=O)(alquilo C1-6)2, -OP(=O)(alquilo C1-6)2, -OP(=O)(O-alquilo C1-6)2, alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, arilo C6-10, heterociclilo de 3 a 10 miembros, heteroarilo de 5 a 10 miembros; o dos sustituyentes geminales Rgg pueden unirse para formar =O o =S; en los que X- es un contraión.
En algunas realizaciones, el compuesto es un compuesto de fórmula (II):

o una sal farmacéuticamente aceptable del mismo, en el que R1 es H o -alquilo C1-6; R2 es H, -alquilo C1-6, -CO2R5, -C(O)NR9R10, -CN,-CHN(OR5) o un heteroarilo; R3 es H o -alquilo C1-6; A es un enlace, alquileno C1-6, alquenileno C2-6 o alquinileno C2-6; R4 es cicloalquilo, arilo, heteroarilo o heterociclilo, cada uno de los cuales está sustituido con 0 a 3 apariciones de -NH2 o R6; cada R5 es independientemente H, -alquilo C1-6, aralquilo o arilo sustituido con 0 a 3 apariciones de -NH2 o R6; cada R6 es independientemente halo, hidroxi, ciano, nitro, alquilo C1-6 (por ejemplo, metilo, etilo, haloalquilo (por ejemplo, -CF3)),alcoxi C1-6 (por ejemplo, haloalcoxi (por ejemplo, -OCF3)), -NR9R10, -NHR10, -C(O)R11, -C(O)OR11, -C(O)NR9R10, -C(NR8)(N(R8)2), -SOqR11, -SO2NR9R10, -NHC(O)OR11, -NHC(O)R11, arilo, heteroarilo, aralquilo, cicloalquilo, heteroaralquilo, heterociclilo o heterociclilalquilo, o dos grupos R6 junto con los átomos a los que están unidos forman un anillo de 5 a 7 miembros (por ejemplo,

); X es -C(O)O-, -OC(O)-, -C(O)S(O)2-, -S(O)2C(O)-, -C(O)N(R5)- o -N(R5)C(O) -; Y es cicloalquilo (por ejemplo, cicloalquilo de 3 a 8 miembros, por ejemplo, cicloalquilo de 5 a 7 miembros); R7 es H, -alquilo C1-6 ( por ejemplo, metilo, haloalquilo (por ejemplo, -CF3)), cicloalquilo, arilo, heteroarilo o heterociclilo, cada uno de los cuales está sustituido con 0 a 3 apariciones de -NH2 o R6; cada R8 es independientemente H, -alquilo C1-6 (por ejemplo, haloalquilo( por ejemplo, -CF3)), -C(O)R5, -C(O)OR5, arilo, heteroarilo, aralquilo, heteroaralquilo, heterociclilo o heterociclilalquilo; cada uno de R9 y R10 es independientemente -alquilo C1-6 , cicloalquilo, heterociclilo, arilo o heteroarilo, o R9 y R10 juntos forman un anillo de 5 a 7 miembros opcionalmente sustituido; cada R11 es independientemente H, -alquilo C1-6 (por ejemplo, alquilo sustituido (por ejemplo,

)), aralquilo o arilo; q es un número entero de 0 a 2; y n es un número entero de 0 a 2.
En algunas realizaciones, R1 es H.
En algunas realizaciones, R2 es -CO2R5 y R5 es H o -alquilo C1-6. En algunas realizaciones, R5 es H, metilo, etilo o isopropilo. En algunas realizaciones, R5 es H. En algunas realizaciones, R5 es etilo. En algunas realizaciones, A es alquileno C1-6 (por ejemplo, etileno o propileno).
En algunas realizaciones, R4 es arilo o heteroarilo. En algunas realizaciones, R4 es fenilo sustituido con 1 aparición de R6. En algunas realizaciones, R6 es halo, alcoxi C1-6 o -C(NR8)(N(R8)2). En algunas realizaciones, R6 es -C(NR8)(N(R8)2) y cada R8 es H. En algunas realizaciones, R8 es independientemente H o -CO2R5. En algunas realizaciones, R5 es -alquilo C1-6 (por ejemplo, hexilo). En algunas realizaciones, R4 es un heteroarilo (por ejemplo, un heteroarilo de 6 miembros o un heteroarilo de 5 miembros) sustituido con 0 a 3 apariciones de -NH2 o R6. En algunas realizaciones, R4 es un heteroarilo de 6 miembros (por ejemplo, piridilo) sustituido con 0 a 3 apariciones de -NH2 o R6. En algunas realizaciones, R4 es un heteroarilo que contiene nitrógeno (por ejemplo, piridilo) sustituido con 0 a 3
apariciones de -NH2 o R6. En algunas realizaciones, R4 es piridilo sustituido con 1 aparición de R6. En algunas realizaciones, R6 es halo (por ejemplo, cloro, bromo, fluoro). En algunas realizaciones, R4 es piridilo sustituido con 1 aparición de -NH2. En algunas realizaciones, R6 es -NHR10 y R10 es -alquilo C1-6. En algunas realizaciones, R6 es -NHC(O)OR11 y R11 es -alquilo C1-6 (por ejemplo, alquilo sustituido (por ejemplo,

En algunas realizaciones, X es -C(O)N(R5)- o -N(R5)C(O)-. En algunas realizaciones, X es -C(O)N(R5)- y R5 es H. En algunas realizaciones, n es 0.
En algunas realizaciones, R7 es -alquilo C1-6 (por ejemplo, metilo, etilo, propilo, -CF3). En algunas realizaciones, R7 es metilo. En algunas realizaciones, R7 es -CF3.
En algunas realizaciones, Y es cicloalquilo no sustituido. En algunas realizaciones, Y es ciclohexilo.
En algunas realizaciones, R6 es haloalcoxi (por ejemplo, -OCF3). En algunas realizaciones, dos grupos R6 tomados junto con los átomos a los que están unidos forman un anillo de 5 a 7 miembros. En algunas realizaciones, dos grupos R6 tomados junto con los átomos a los que están unidos forman un anillo de 5 a 7 miembros y el anillo se selecciona entre:

En algunas realizaciones, el compuesto de la fórmula (II) se selecciona de un compuesto de fórmula (IIa):

en la que R1, R2, R3, R4, R7 e Y son como se describió para la fórmula (II), y m es un número entero de 1 a 6.
En algunas realizaciones, el compuesto de la fórmula (IIa) se selecciona de un compuesto de fórmula (IIb):

en la que R1, R2, R3, R4, R7, Y y m son como se describió para la fórmula (IIa).
En algunas realizaciones, el compuesto de fórmula (IIb) es:



En algunas realizaciones, el compuesto de fórmula (IIb) se selecciona de un compuesto de fórmula (IIc):

En un aspecto, la invención reivindicada en el presente documento se dirige a una composición farmacéutica que comprende un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo y uno o más excipientes farmacéuticamente aceptables.
En algunas realizaciones, la composición se proporciona como una solución.
En un aspecto, la invención reivindicada en el presente documento se dirige a un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o a una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)) para su uso en un procedimiento de reducción del riesgo de ictus (por ejemplo, isquemia, por ejemplo, un acontecimiento isquémico transitorio) en un sujeto que ha sufrido un acontecimiento isquémico (por ejemplo, un acontecimiento isquémico transitorio), que comprende administrar al sujeto una cantidad eficaz de un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o de una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)).
En algunas realizaciones, la administración reduce el riesgo de ictus en un sujeto en comparación con un sujeto al que no se le administra el compuesto.
En un aspecto, la invención reivindicada en el presente documento se dirige a un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o a una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)) para su uso en un procedimiento de reducción de la embolia sistémica del sistema nervioso no central (por ejemplo, isquemia, por ejemplo, un acontecimiento isquémico transitorio) en un sujeto que ha sufrido un acontecimiento isquémico (por ejemplo, un acontecimiento isquémico transitorio), que comprende administrar al sujeto una cantidad eficaz de un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o de una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)).
En algunas realizaciones, la administración reduce la embolia sistémica del sistema nervioso no central en un sujeto en comparación con un sujeto al que no se le administra el compuesto.
En un aspecto, la invención reivindicada en el presente documento se dirige a un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o a una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)) para su uso en un procedimiento de tratamiento de la trombosis venosa profunda que comprende administrar al sujeto que ha sufrido un acontecimiento isquémico (por ejemplo, un acontecimiento isquémico transitorio) una cantidad eficaz de un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o de una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)).
En un aspecto, la invención reivindicada en el presente documento se dirige a un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o a una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)) para su uso en un procedimiento de reducción del riesgo de recurrencia de la trombosis venosa profunda que comprende administrar al sujeto que ha sufrido trombosis venosa profunda una cantidad eficaz de un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o de una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)). En algunas realizaciones, la administración reduce el riesgo de recurrencia de la trombosis venosa profunda en un sujeto en comparación con un sujeto al que no se le administra el compuesto. En un aspecto, la invención reivindicada en el presente documento se dirige a un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o a una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)) para su uso en un procedimiento de reducción del riesgo de recurrencia de la embolia pulmonar (por ejemplo, embolia pulmonar sintomática) que comprende administrar al sujeto que ha sufrido una embolia pulmonar una cantidad eficaz de un compuesto de la fórmula (II) o de una sal farmacéuticamente aceptable del mismo, o de una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)).
En algunas realizaciones, la administración reduce el riesgo de recurrencia de la embolia pulmonar en un sujeto en comparación con un sujeto al que no se le administra el compuesto.
En un aspecto, la invención reivindicada en el presente documento se dirige a un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o a una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)) para su uso en un procedimiento de profilaxis de la embolia pulmonar en un sujeto que ha sufrido una embolia pulmonar, que comprende administrar al sujeto una cantidad eficaz de un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o de una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)). Un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)) puede ser útil en un procedimiento de tratamiento de un sujeto que ha sufrido un acontecimiento isquémico (por ejemplo, isquemia transitoria), que comprende: administrar un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)) al sujeto. En algunas realizaciones, el compuesto se administra al sujeto dentro de las 24 horas o menos, por ejemplo, 12, 10, 9, 8, 7, 6 horas o menos, después del inicio del acontecimiento isquémico en el sujeto. En algunas realizaciones, el compuesto se administra al sujeto en un plazo de más de 2 horas a 12 horas, por ejemplo, de más de 2 horas a 10 horas o menos, de más de 2 horas a 8 horas o menos, después del inicio del acontecimiento isquémico en el sujeto.
Un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)) puede ser útil en un procedimiento de inhibición del factor XIa en un sujeto, que comprende administrar al sujeto que ha sufrido isquemia una cantidad eficaz de un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o de una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)).
En determinadas realizaciones, el sujeto es un mamífero (por ejemplo, un ser humano). En algunas realizaciones, el sujeto se somete a una intervención quirúrgica (por ejemplo, la implantación de una prótesis de rodilla o de cadera). En algunas realizaciones, el sujeto es un sujeto con fibrilación auricular no valvular. En algunas realizaciones, el sujeto tiene uno o más de los siguientes factores de riesgo de ictus: un ictus previo (por ejemplo, isquémico, desconocido, hemorrágico), un accidente isquémico transitorio o una embolia sistémica no relacionada con el SNC. En algunas realizaciones, el sujeto tiene uno o más de los siguientes factores de riesgo de ictus: 75 años o más de edad, hipertensión, insuficiencia cardiaca o fracción de eyección del ventrículo izquierdo (por ejemplo, menor o igual al 35%), o diabetes mellitus.
En algunas realizaciones, el compuesto se administra por vía oral o parenteral (por ejemplo, intravenosa).
En algunas realizaciones, el compuesto se administra antes de un acontecimiento isquémico (por ejemplo, a un sujeto en riesgo de un acontecimiento isquémico).
En algunas realizaciones, el compuesto se administra después de un acontecimiento isquémico (por ejemplo, un acontecimiento isquémico transitorio).
En algunas realizaciones, el compuesto se administra aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 o 14 días o más, después de un acontecimiento isquémico (por ejemplo, un acontecimiento isquémico transitorio).
En algunas realizaciones, el compuesto se administra aproximadamente 1, 2, 3, 4, 5, 6, 7 u 8 semanas o más, después de un acontecimiento isquémico (por ejemplo, un acontecimiento isquémico transitorio).
En determinadas realizaciones, el compuesto se administra junto con un agente terapéutico adicional. En algunas realizaciones, el agente terapéutico adicional se administra después de la administración del compuesto. En algunas realizaciones, el agente terapéutico adicional se administra por vía oral. En algunas realizaciones, el agente terapéutico adicional se administra al menos 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 14, 16, 18, 20 o 24 horas o más, después de la administración del compuesto. En algunas realizaciones, el agente terapéutico adicional se administra al menos 1, 2, 3, 4, 5, 6, 7, 14, 21 o 28 días o más, después de la administración del compuesto. En algunas realizaciones, el agente terapéutico adicional se administra aproximadamente 1 día, aproximadamente 2 días, aproximadamente 3 días, aproximadamente 4 días, aproximadamente 5 días, aproximadamente 6 días, aproximadamente 7 días o más, después de la administración del compuesto.
En algunas realizaciones, el agente terapéutico adicional se administra crónicamente (por ejemplo, durante aproximadamente 1 día, aproximadamente 2 días, aproximadamente 3 días, aproximadamente 4 días, aproximadamente 5 días, aproximadamente 6 días, aproximadamente 7 días, aproximadamente 8 días, aproximadamente 9 días, aproximadamente 10 días, aproximadamente 11 días, aproximadamente 12 días, aproximadamente 13 días o aproximadamente 14 días o más) después de la administración del compuesto.
En algunas realizaciones, el agente terapéutico adicional trata un efecto secundario (por ejemplo, hemorragia patológica activa o reacciones de hipersensibilidad graves (por ejemplo, reacciones anafilácticas), hematoma espinal y/o epidural, trastorno gastrointestinal (por ejemplo, dolor del abdomen superior, dispepsia, dolor de muelas), trastornos generales y afecciones del lugar de administración (por ejemplo, fatiga), infecciones e infestaciones (por ejemplo, sinusitis, infección urinaria), trastornos musculoesqueléticos y del tejido conjuntivo (por ejemplo, dolor de espalda, artrosis), trastornos respiratorios, torácicos y mediastínicos (por ejemplo, dolor orofaríngeo), lesiones, intoxicaciones y complicaciones de procedimientos (por ejemplo, secreción de heridas), trastornos musculoesqueléticos y del tejido conjuntivo (por ejemplo, dolor en extremidades, espasmo muscular), trastornos del sistema nervioso (por ejemplo, síncope), trastornos de la piel y del tejido subcutáneo (por ejemplo, prurito, ampollas), trastornos de la sangre y del sistema linfático (por ejemplo, agranulocitosis), trastornos gastrointestinales (por ejemplo, hemorragia retroperitoneal), trastornos hepatobiliares (por ejemplo, ictericia, colestasis, hepatitis citolítica), trastornos del sistema inmunitario (por ejemplo, hipersensibilidad, reacción anafiláctica, shock anafiláctico, angioedema), trastornos del sistema nervioso (por ejemplo, hemorragia cerebral, hematoma subdural, hematoma epidural, hemiparesia), trastornos de la piel y del tejido subcutáneo (por ejemplo, síndrome de Stevens-Johnson).
En algunas realizaciones, el agente terapéutico adicional es un AINE (por ejemplo, aspirina, naproxeno), un inhibidor de la agregación plaquetaria (por ejemplo, clopidogrel) o un anticoagulante (por ejemplo, warfarina, enoxaparina). En algunas realizaciones, el agente terapéutico adicional produce un efecto terapéutico aditivo.
En algunas realizaciones, el agente terapéutico adicional produce un efecto terapéutico sinérgico.
En otro aspecto, la invención reivindicada en el presente documento presenta una composición farmacéutica que comprende un compuesto descrito en el presente documento (por ejemplo, un compuesto de fórmula (II)) y un excipiente farmacéuticamente aceptable.
Un compuesto descrito en el presente documento (por ejemplo, un compuesto de fórmula (II)) puede ser útil en un procedimiento de modulación (por ejemplo, inhibición) del factor XIa en un paciente. El procedimiento comprende la
etapa de administrar una cantidad eficaz de un compuesto descrito en el presente documento (por ejemplo, un compuesto de fórmula (II)) a un paciente que lo necesite, modulando así (por ejemplo, inhibiendo) el factor XIa. Un compuesto descrito en el presente documento (por ejemplo, un compuesto de fórmula (II)) puede ser útil en un procedimiento de tratamiento de un sujeto que lo necesite para un trastorno tromboembólico. El procedimiento comprende administrar al sujeto una cantidad terapéuticamente eficaz de un compuesto descrito en el presente documento (por ejemplo, un compuesto de fórmula (II)).
El trastorno tromboembólico puede consistir en trastornos tromboembólicos cardiovasculares arteriales, trastornos tromboembólicos cardiovasculares venosos y trastornos tromboembólicos en las cavidades del corazón; incluye angina inestable, síndrome coronario agudo, primer infarto de miocardio, infarto de miocardio recurrente, isquemia (por ejemplo, muerte súbita isquémica o accidente isquémico transitorio), ictus, aterosclerosis, enfermedad arterial oclusiva periférica, trombosis venosa, trombosis venosa profunda, tromboflebitis, embolia arterial, trombosis arterial coronaria, trombosis arterial cerebral, embolia cerebral, embolia renal embolia pulmonar y trombosis resultante de (a) válvulas protésicas u otros implantes, (b) catéteres permanentes, (c) endoprótesis vasculares, (d) circulación extracorporal, (e) hemodiálisis, u (f) otros procedimientos en los que la sangre se expone a una superficie artificial que favorece la trombosis.
Un compuesto descrito en el presente documento (por ejemplo, un compuesto de fórmula (II)) puede ser útil en un procedimiento de tratamiento de un sujeto que se ha identificado que está en riesgo de ictus o trombosis, reduciendo así la probabilidad de ictus o trombosis en el sujeto. En algunas realizaciones, además se ha identificado que el sujeto está en riesgo de hemorragia (por ejemplo, hemorragia excesiva) o sepsis. En algunas realizaciones, el tratamiento es eficaz sin riesgo de sangrado. En algunas realizaciones, el tratamiento es eficaz para mantener la comunicación de los puertos y líneas de infusión. Además, los compuestos descritos en el presente documento (por ejemplo, compuestos de fórmula (II)) son útiles en el tratamiento y prevención de otras enfermedades en las que se ha determinado que la generación de trombina desempeña un papel fisiológico. Por ejemplo, se ha implicado a la trombina en la contribución a la morbilidad y la mortalidad de enfermedades crónicas y degenerativas, tales como el cáncer, la artritis, la aterosclerosis, la demencia vascular y la enfermedad de Alzheimer, por su capacidad para regular muchos tipos diferentes de células mediante la escisión específica y la activación de un receptor de trombina de la superficie celular, efectos mitogénicos, diversas funciones celulares, tales como la proliferación celular, por ejemplo, la proliferación anómala de células vasculares que da lugar a reestenosis o angiogénesis, la liberación de PDGF y la síntesis de ADN. La inhibición del factor XIa bloquea eficazmente la generación de trombina y, por tanto, neutraliza cualquier efecto fisiológico de la trombina sobre diversos tipos de células. Las indicaciones representativas analizadas anteriormente incluyen algunas, pero no todas, las posibles situaciones clínicas susceptibles de tratamiento con un inhibidor del factor XIa.
En otro aspecto, la invención reivindicada en el presente documento presenta un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)) para su uso en un procedimiento de tratamiento de un sujeto que tiene edema (por ejemplo, angioedema, por ejemplo, angioedema hereditario), que comprende administrar al sujeto un compuesto de la fórmula (II) o una sal farmacéuticamente aceptable del mismo, o una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de la fórmula (II)).
Un compuesto descrito en el presente documento (por ejemplo, un compuesto de fórmula (II)) puede ser útil en un procedimiento de inhibición de la calicreína en un sujeto, que comprende administrar al sujeto con edema (por ejemplo, angioedema, por ejemplo, angioedema hereditario), una cantidad eficaz de un compuesto de fórmula (II) o una sal farmacéuticamente aceptable del mismo, o de una composición descrita en el presente documento (por ejemplo, una composición que comprende un compuesto de fórmula (II)) al sujeto.
Breve descripción de las figuras
Las figuras 1A-1M representan la tabla A y la síntesis de las estructuras A-8 y A-9.
La figura 2 representa la tabla B y la síntesis de la estructura B-5.
La figura 3 representa las tablas C y D y la síntesis de la estructura D-8.
La figura 4 representa las tablas E y la síntesis de la estructura E-6.
La figura 5 representa la tabla F y la síntesis de la estructura F-8.
La figura 6 representa la tabla G y la síntesis de la estructura G-4.
La figura 7 representa la tabla H y la síntesis de la estructura H-9.
La figura 8 representa la tabla I y la síntesis de las estructuras I-4 a I-7.
Las figuras 9A-9B representan la tabla J y la síntesis de las estructuras J-3 y J-4.
Las figuras 10A-10B representan la tabla K y la síntesis de la estructura K-9.
Descripción detallada
Definiciones
El término "alquilo", por sí mismo o como parte de otro sustituyente, significa, a menos que se indique lo contrario, un radical hidrocarburo de cadena lineal o ramificada, o cíclico, o una combinación de los mismos, el cual puede estar totalmente saturado, mono- o poliinsaturado y puede incluir radicales di- y multivalentes, que tiene el número de átomos de carbono indicado (por ejemplo, C1-10 significa de uno a diez carbonos). Los ejemplos de radicales hidrocarburos saturados incluyen grupos, tales como metilo, etilo, n-propilo, isopropilo, n-butilo, t-butilo, isobutilo, secbutilo, ciclohexilo, (ciclohexilo)metilo, ciclopropilmetilo, los homólogos e isómeros, por ejemplo, de n-pentilo, n-hexilo, n-heptilo, n-octilo y similares. Un grupo alquilo insaturado es aquel que tiene uno o más dobles o triples enlaces. Los ejemplos de grupos alquilos insaturados incluyen vinilo, 2-propenilo, crotilo, 2-isopentenilo, 2-(butadienilo), 2,4-pentadienilo, 3-(1,4-pentadienilo), etilo, 1- y 3-propinilo, 3-butinilo y los homólogos e isómeros superiores. El término "alquilo", a menos que se indique lo contrario, también incluye los derivados de alquilo definidos con más detalle a continuación, tales como "heteroalquilo" Los grupos alquilo que se limitan a grupos hidrocarburo se denominan "homoalquilo". A menos que se especifique lo contrario, cada aparición de un grupo alquilo está independientemente sustituido opcionalmente, es decir, no está sustituido (un "alquilo no sustituido") o está sustituido (un "alquilo sustituido") con uno o más sustituyentes, por ejemplo, de 1 a 5 sustituyentes, de 1 a 3 sustituyentes, o 1 sustituyente. En determinadas realizaciones, el grupo alquilo es alquilo C1-10 no sustituido (por ejemplo, -CH3). En determinadas realizaciones, el grupo alquilo es alquilo C1-10 sustituido. Las abreviaturas comunes de alquilo incluyen Me (-CH3), Et (-CH2CH3), iPr (-CH(CH3)2), nPr (-CH2CH2CH3), n-Bu (-CH2CH2CH2CH3) o i-Bu (-CH2CH(CH3)2).
El término "alquileno", por sí mismo o como parte de otro sustituyente, significa un radical divalente derivado de un alcano, como se ilustra con --CH2CH2CH2CH2--, e incluye además aquellos grupos descritos a continuación como "heteroalquileno". Generalmente, un grupo alquilo (o alquileno) tendrá de 1 a 24 átomos de carbono, prefiriéndose en la presente invención aquellos grupos que tengan 10 o menos átomos de carbono. Un "alquilo inferior" o "alquileno inferior" es un grupo alquilo o alquileno de cadena más corta, que generalmente tiene ocho átomos de carbono o menos.
El término "alquenilo" se refiere a una cadena hidrocarbonada lineal o ramificada que contiene de 2 a 12 átomos de carbono (a menos que se indique lo contrario) y que tiene uno o más dobles enlaces. Algunos ejemplos de grupos alquenilo son los grupos alilo, propenilo, 2-butenilo, 3-hexenilo y 3-octenilo. Uno de los carbonos de doble enlace puede ser opcionalmente el punto de unión del sustituyente alquenilo.
El término "alquenileno" se refiere a un alquenilo divalente, por ejemplo, -CH=CH-,-CH2-CH=CH- y -CH=CH-CH2-. El término "alquinilo" se refiere a una cadena hidrocarbonada lineal o ramificada que contiene de 2 a 12 átomos de carbono (a menos que se indique lo contrario) y se caracteriza por tener uno o más triples enlaces. Algunos ejemplos de grupos alquinilo son etinilo, propargilo y 3-hexinilo. Uno de los carbonos del triple enlace puede ser opcionalmente el punto de unión del sustituyente alquinilo.
El término "alquinileno" se refiere a un alquinilo divalente, por ejemplo, -CH≡CH-, -CH2-CH≡CH- y -CH≡CH-CH2-. Los términos "alcoxi", "alquilamino" y "alquiltio" (o tioalcoxi) se utilizan en su sentido convencional, y se refieren a aquellos grupos alquilo unidos al resto de la molécula a través de un átomo de oxígeno, un grupo amino o un átomo de azufre, respectivamente.
Los términos "ciano" y "nitrilo" se refieren al radical -CN.
Los términos "cicloalquilo", "heterocicloalquilo" o "heterociclilo", por sí mismos o en combinación con otros términos, representan, a menos que se indique lo contrario, versiones cíclicas de "alquilo" y "heteroalquilo", respectivamente. Además, para el heterocicloalquilo o heterociclilo, un heteroátomo puede ocupar la posición en la que el heterociclo se une al resto de la molécula. Los ejemplos de cicloalquilo incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, 1-ciclohexenilo, 3-ciclohexenilo, cicloheptilo, ciclooctanilo y similares. Los ejemplos de heterocicloalquilo y heterociclilo incluyen 1-(1,2,5,6-tetrahidropiridilo), 1-piperidinilo, 2-piperidinilo, 3-piperidinilo, 4-morfolinilo, 3-morfolinilo, tetrahidrofuran-2-ilo, tetrahidrofuran-3-ilo, tetrahidrotien-2-ilo, tetrahidrotien-3-ilo, 1-piperazinilo, 2-piperazinilo y similares.
El término "heteroalquilo", tal como se utiliza en el presente documento, se refiere a un grupo alquilo, tal como se define en el presente documento, que comprende además 1 o más (por ejemplo, 1, 2, 3 o 4) heteroátomos (por ejemplo, oxígeno, azufre, nitrógeno, boro, silicio, fósforo) dentro de la cadena parental, en el que dichos uno o más heteroátomos se insertan entre átomos de carbono adyacentes dentro de la cadena de carbono parental y/o uno o más heteroátomos se insertan entre un átomo de carbono y la molécula parental, es decir, entre el punto de unión. En determinadas realizaciones, un grupo heteroalquilo se refiere a un grupo saturado que tiene de 1 a 10 átomos de carbono y 1, 2, 3 o 4 heteroátomos ("heteroalquilo C1-10"). En algunas realizaciones, un grupo heteroalquilo es un grupo saturado que tiene de 1 a 9 átomos de carbono y 1, 2, 3 o 4 heteroátomos ("heteroalquilo C1-9"). En algunas
realizaciones, un grupo heteroalquilo es un grupo saturado que tiene de 1 a 8 átomos de carbono y 1, 2, 3 o 4 heteroátomos ("heteroalquilo C1-8"). En algunas realizaciones, un grupo heteroalquilo es un grupo saturado que tiene de 1 a 7 átomos de carbono y 1, 2, 3 o 4 heteroátomos ("heteroalquilo C1-7"). En algunas realizaciones, un grupo heteroalquilo es un grupo que tiene de 1 a 6 átomos de carbono y 1, 2 o 3 heteroátomos ("heteroalquilo C1-6"). En algunas realizaciones, un grupo heteroalquilo es un grupo saturado que tiene de 1 a 5 átomos de carbono y 1 o 2 heteroátomos ("heteroalquilo C1-5"). En algunas realizaciones, un grupo heteroalquilo es un grupo saturado que tiene de 1 a 4 átomos de carbono y 1 o 2 heteroátomos ("heteroalquilo C1-4"). En algunas realizaciones, un grupo heteroalquilo es un grupo saturado que tiene de 1 a 3 átomos de carbono y 1 heteroátomo ("heteroalquilo C1-3"). En algunas realizaciones, un grupo heteroalquilo es un grupo saturado que tiene de 1 a 2 átomos de carbono y 1 heteroátomo ("heteroalquilo C1-2"). En algunas realizaciones, un grupo heteroalquilo es un grupo saturado que tiene 1 átomo de carbono y 1 heteroátomo ("heteroalquilo C1"). En algunas realizaciones, un grupo heteroalquilo es un grupo saturado que tiene de 2 a 6 átomos de carbono y 1 o 2 heteroátomos ("heteroalquilo C2-6"). A menos que se especifique lo contrario, cada aparición de un grupo heteroalquilo es independientemente un grupo heteroalquilo no sustituido (un "heteroalquilo no sustituido") o sustituido (un "heteroalquilo sustituido") con uno o más sustituyentes. En determinadas realizaciones, el grupo heteroalquilo es un heteroalquilo C1-10 no sustituido. En determinadas realizaciones, el grupo heteroalquilo es un heteroalquilo C1-10 sustituido.
Los términos "heterociclilo", cuando se utilizan en combinación con otros términos (por ejemplo, heterociclilalquilo), incluyen anillos heterociclilo como los definidos anteriormente. Por lo tanto, el término "heterociclilalquilo" incluye aquellos radicales en los que un grupo heterociclilo está unido a un grupo alquilo, incluidos aquellos grupos alquilo en los que un átomo de carbono (por ejemplo, un grupo metileno) ha sido sustituido, por ejemplo, por un átomo de oxígeno.
Los términos "halo" o "halógeno", por sí mismos o como parte de otro sustituyente, significan, a menos que se indique lo contrario, un átomo de flúor, cloro, bromo o yodo.
El término "haloalquilo", tal como se utiliza en el presente documento, se refiere a un grupo alquilo, tal como se define en el presente documento, que comprende además 1 o más (por ejemplo, 1, 2, 3 o 4) átomos de halógeno (por ejemplo, flúor, cloro, bromo o yodo), en el que el grupo alquilo está sustituido con uno o más átomos de halógeno. En determinadas realizaciones, un grupo haloalquilo se refiere a un grupo saturado que tiene de 1 a 10 átomos de carbono y 1, 2, 3 o 4 átomos de halógeno ("haloalquilo C1-10"). Además, el término "haloalquilo" incluye monohaloalquilo y polihaloalquilo. Por ejemplo, el término "haloalquilo" incluye trifluorometilo, 2,2,2-trifluoroetilo, 4-clorobutilo, 3-bromopropilo y similares.
Los términos "haloalcoxi" o "haloalcoxilo", tal como se utilizan en el presente documento, se refieren a un grupo alcoxi, tal como se define en el presente documento, que además comprende 1 o más (por ejemplo, 1, 2, 3 o 4) átomos de halógeno (por ejemplo, flúor, cloro, bromo o yodo), en el que el grupo alcoxi está sustituido con uno o más átomos de halógeno.
El término "hidroxi" se refiere al radical -OH.
El término "arilo" significa, a menos que se indique lo contrario, un sustituyente hidrocarbonado poliinsaturado, aromático, que puede ser un anillo único o múltiples anillos (preferiblemente de 1 a 3 anillos), que están condensados o unidos covalentemente. El término "heteroarilo" se refiere a grupos (o anillos) arilo que contienen de uno a cuatro heteroátomos seleccionados entre N, O y S, en los que los átomos de nitrógeno y azufre están opcionalmente oxidados, y el átomo o átomos de nitrógeno están opcionalmente cuaternizados. Un grupo heteroarilo puede unirse al resto de la molécula a través de un heteroátomo. Algunos ejemplos de grupos arilo y heteroarilo son fenilo, 1-naftilo, 2-naftilo, 4-bifenilo, 1-pirrolilo, 2-pirrolilo, 3-pirrolilo, 3-pirazolilo, 2-imidazolilo, 4-imidazolilo, pirazinilo, 2-oxazolilo, 4-oxazolilo, 2-fenil-4-oxazolilo, 5-oxazolilo, 3-isoxazolilo, 4-isoxazolilo, 5-isoxazolilo, 2-tiazolilo, 4-tiazolilo, 5-tiazolilo, 2-furilo, 3-furilo, 2-tienilo, 3-tienilo, 2-piridilo, 3-piridilo, 4-piridilo, 2-pirimidilo, 4-pirimidilo, 5-benzotiazolilo, purinilo, 2-bencimidazolilo, 5-indolilo, 1-isoquinolilo, 5-isoquinolilo, 2-quinoxalinilo, 5-quinoxalinilo, 3-quinolilo y 6-quinolilo. Los sustituyentes de cada uno de los sistemas de anillos arilo y heteroarilo mencionados anteriormente se seleccionan del grupo de sustituyentes aceptables descritos a continuación.
Por brevedad, el término "arilo", cuando se utiliza en combinación con otros términos (por ejemplo, ariloxi, ariltioxi, arilalquilo, aralquilo, heteroaralquilo) incluye tanto anillos arilo como heteroarilo, tal como se han definido anteriormente. Por lo tanto, los términos "arilalquilo", "aralquilo" y "heteroaralquilo" incluyen aquellos radicales en los que un grupo arilo o heteroarilo está unido a un grupo alquilo (por ejemplo, bencilo, fenetilo, piridilmetilo y similares), incluidos los grupos alquilo en los que un átomo de carbono (por ejemplo, un grupo metileno) se ha sustituido, por ejemplo, por un átomo de oxígeno (por ejemplo, fenoximetilo, 2-piridiloximetilo, 3-(1-naftiloxi)propilo y similares). El término "nitro" se refiere al radical -NO2.
Un "grupo protector", tal como se utiliza en el presente documento, se refiere a una porción de un sustrato que es sustancialmente estable en una condición de reacción concreta, pero que se escinde del sustrato en una condición de reacción diferente. También puede seleccionarse un grupo protector tal que participe en la oxidación directa del
componente de anillo aromático de los compuestos de la invención. Para ejemplos de grupos protectores útiles, véase, por ejemplo, Greene et al., Protective Groups in Organic Synthesis, John Wiley & Sons, Nueva York, 1991.
Los grupos alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo, tal como se definen en el presente documento, están opcionalmente sustituidos (por ejemplo, un grupo alquilo "sustituido" o "no sustituido", alquenilo "sustituido" o "no sustituido", alquinilo "sustituido" o "no sustituido", cicloalquilo "sustituido" o "no sustituido", heterociclilo "sustituido" o "no sustituido", arilo "sustituido" o "no sustituido" o heteroarilo "sustituido" o "no sustituido"). En general, el término "sustituido", precedido o no del término "opcionalmente", significa que al menos un hidrógeno presente en un grupo (por ejemplo, un átomo de carbono o de nitrógeno) se sustituye por un sustituyente permitido, por ejemplo, un sustituyente que, al actuar como sustituyente, da lugar a un compuesto estable, por ejemplo, un compuesto que no sufre espontáneamente una transformación, tal como por reordenación, ciclación, eliminación u otra reacción. A menos que se indique lo contrario, un grupo "sustituido" tiene un sustituyente en una o más posiciones sustituibles del grupo, y cuando se sustituye más de una posición en cualquier estructura concreta, el sustituyente es el mismo o diferente en cada posición. El término "sustituido" incluye la sustitución con todos los sustituyentes permitidos de los compuestos orgánicos descritos en el presente documento que den lugar a la formación de un compuesto estable. Para los fines de esta invención, los heteroátomos, tales como el nitrógeno, pueden tener sustituyentes de hidrógeno y/o cualquier sustituyente adecuado como se describe en el presente documento que satisfaga las valencias de los heteroátomos y de como resultado la formación de un resto estable.
Los ejemplos de sustituyentes de átomo de carbono incluyen halógeno, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -ORaa, -ON(Rbb)2, -N(Rbb)2, -N(Rbb)3 +X-, -N(ORcc)Rbb, -SH, -SRaa, -SSRcc, -C(=O)Raa, -CO2H, -CHO, -C(ORcc)2, -CO2Raa, -OC(=O)Raa, -OCO2Raa, -C(=O)N(Rbb)2, -OC(=O)N(Rbb)2, -NRbbC(=O)Raa, -NRbbCO2Raa, -NRbbC(=O)N(Rbb)2, -C(=NRbb)Raa, -C(=NRbb)ORaa, -OC(=NRbb)Raa, -OC(=NRbb)ORaa, -C(=NRbb)N(Rbb)2, -OC(=NRbb)N(Rbb)2, -NRbbC(=NRbb)N(Rbb)2, -C(=O)NRbbSO2Raa, -NRbbSO2Raa, -SO2N(Rbb)2, -SO2Raa, -SO2ORaa, -OSO2Raa, -S(O)Raa -S(=O)Raa, -OS(=O)Raa, -Si(Raa)3, -OSi(Raa)3, -C(=S)N(Rbb)2, -C(=O)SRaa, -C(=S)SRaa, -SC(=S)SRaa, -SC(=O)SRaa, -OC(=O)SRaa, -SC(=O)ORaa, -SC(=O)Raa, -P(=O)2Raa, -OP(=O)2Raa, -P(=O)(Raa)2, -OP(=O)(Raa)2,-OP(=O)(ORcc)2, -P(=O)2N(Rbb)2, -OP(=O)2N(Rbb)2, -P(=O)(NRbb)2, -OP(=O)(NRbb)2, -NRbbP(=O)(ORcc)2, -NRbbP(=O)(NRbb)2, -P(Rcc)2, -P(Rcc)3, -OP(Rcc)2, -OP(Rcc)3, -B(Raa)2, -B(ORcc)2, -BRaa(ORcc), alquilo C1-10, perhaloalquilo C1-10, alquenilo C2-10, alquinilo C2-10,cicloalquilo C3-10, heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Raa se selecciona, independientemente, entre alquilo C1-10, perhaloalquilo C1-10, alquenilo C2-10, alquinilo C2-10, cicloalquilo C3-10, heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, o dos grupos Raa se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Rbb se selecciona, independientemente, entre hidrógeno, -OH, -ORaa, -N(Rcc)2, -CN, -C(=O)Raa, -C(=O)N(Rcc)2, -CO2Raa, -SO2Raa, -C(=NRcc)ORaa, -C(=NRcc)N(Rcc)2, -SO2N(Rcc)2, -SO2Rcc, -SO2ORcc, -SORaa, -C(=S)N(Rcc)2, -C(=O)SRcc, -C(=S)SRcc, -P(=O)2Raa, -P(=O)(Raa)2, -P(=O)2N(Rcc)2, -P(=O)(NRcc)2, alquilo C1-10, perhaloalquilo C1-10, alquenilo C2-10, alquinilo C2-10, cicloalquilo C3-10, heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, o dos grupos Rbb se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está sustituido independientemente con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Rcc se selecciona, independientemente, entre hidrógeno, alquilo C1-10, perhaloalquilo C1-10, alquenilo C2-10, alquinilo C2-10, cicloalquilo C3-10, heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, o dos grupos Rcc se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Rdd se selecciona, independientemente, entre halógeno, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -ORee, -ON(Rff)2, -N(Rff)2, -N(Rff)3 +X-, -N(ORee)Rff, -SH, -SRee, -SSRee, -C(=O)Ree, -CO2H, -CO2Ree, -OC(=O)Ree, -OCO2Ree, -C(=O)N(Rff)2, -OC(=O)N(Rff)2, -NRffC(=O)Ree, -NRffCO2Ree, -NRffC(=O)N(Rff)2, -C(=NRff)ORee, -OC(=NRff)Ree, -OC(=NRff)ORee, -C(=NRff)N(Rff)2, -OC(=NRff)N(Rff)2, -NRffC(=NRff)N(Rff)2,-NRffSO2Ree, -SO2N(Rff)2, -SO2Ree, -SO2ORee, -OSO2Ree, -S(=O)Ree, -Si(Ree)3, -OSi(Ree)3, -C(=S)N(Rff)2, -C(=O)SRee, -C(=S)SRee, -SC(=S)SRee, -P(=O)2Ree, -P(=O)(Ree)2, -OP(=O)(Ree)2, -OP(=O)(ORee)2, alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, arilo C6-10, heteroarilo de 5 a 10 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rgg, o dos sustituyentes geminales Rdd pueden unirse para formar =O o =S;
cada aparición de Ree se selecciona, independientemente, entre alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, arilo C6-10, heteroarilo de 3 a 10 miembros y heterociclilo de 3 a 10 miembros,
en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rgg;
cada aparición de Rff se selecciona, independientemente, entre hidrógeno, alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, arilo C6-10 y heteroarilo de 5 a 10 miembros, o dos grupos Rff se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rgg; y
cada aparición de Rgg es, independientemente, halógeno, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -O-alquilo C1-6, -ON(alquilo C1-6)2, -N(alquilo C1-6)2, -N(alquilo C1-6)3 +X-, -NH(alquilo C1-6)2 +X-, -NH2(alquilo C1-6) +X-, -NH3 +X-, -N(O-alquil C1-6)(alquilo C1-6), -N(OH)(alquilo C1-6), -NH(OH), -SH, -S-alquilo C1-6, -SS(alquilo C1-6), -C(=O)(alquilo C1-6), -CO2H, -CO2(alquilo C1-6), -OC(=O)(alquilo C1-6), -OCO2(alquilo C1-6), -C(=O)NH2, -C(=O)N(alquilo C1-6)2, -OC(=O)NH(alquilo C1-6), -NHC(=O)(alquilo C1-6), -N(alquilo C1-6)C(=O)(alquilo C1-6), -NHCO2(alquilo C1-6), -NHC(=O)N(alquilo C1-6)2, -NHC(=O)NH(alquilo C1-6), -NHC(=O)NH2, -C(=NH)O(alquilo C1-6), -OC(=NH)(alquilo C1-6), -OC(=NH)O-alquilo C1-6, -C(=NH)N(alquilo C1-6)2, -C(=NH)NH(alquilo C1-6), -C(=NH)NH2, -OC(=NH)N(alquilo C1-6)2, -OC(NH)NH(alquilo C1-6), -OC(NH)NH2, -NHC(NH)N(alquilo C1-6)2, -NHC(=NH)NH2, -NHSO2(alquilo C1-6), -SO2N(alquilo C1-6)2, -SO2NH(alquilo C1-6), -SO2NH2, -SO2-alquilo C1-6, -SO2O-alquilo C1-6, -OSO2-alquilo C1-6, -SO-alquilo C1-6, -Si(alquilo C1-6)3, -OSi(alquilo C1-6)3 -C(=S)N(alquilo C1-6)2, C(=S)NH(alquilo C1-6), C(=S)NH2, -C(=O)S(alquilo C1-6), -C(=S)S-alquilo C1-6, -SC(=S)S-alquilo C1-6, -P(=O)2(alquilo C1-6), -P(=O)(alquilo C1-6)2, -OP(=O)(alquilo C1-6)2, -OP(=O)(O-alquilo C1-6)2, alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, arilo C6-10, heterociclilo de 3 a 10 miembros, heteroarilo de 5 a 10 miembros; o dos sustituyentes geminales Rgg pueden unirse para formar =O o =S; en los que X- es un contraión.
Estos y otros ejemplos de sustituyentes se describen con más detalle en la descripción detallada, los ejemplos y las reivindicaciones. La invención no pretende estar limitada en modo alguno por la enumeración de ejemplos de sustituyentes anterior.
Compuestos
Se describen en el presente documento compuestos que inhiben el factor XIa o la calicreína, por ejemplo, los compuestos descritos en el presente documento (por ejemplo, un compuesto de fórmula (II)).
Los ejemplos de compuestos incluyen, entre otros, los compuestos descritos en la siguiente tabla 1 a continuación: Tabla 1: Ejemplos de compuestos de la invención reivindicada en el presente documento. Los compuestos marcados con un asterisco (∗) se proporcionan como referencia.




















En algunas realizaciones, un compuesto descrito en el presente documento se forme como una sal. Un compuesto descrito en el presente documento puede administrarse como un ácido libre, un ion bipolar o como una sal. También puede formarse una sal entre un catión y un sustituyente cargado negativamente en un compuesto descrito en el presente documento. Los contraiones catiónicos adecuados incluyen iones sodio, iones potasio, iones magnesio, iones calcio e iones amonio (por ejemplo, un catión tetraalquilamonio, tal como el ion tetrametilamonio). En compuestos que incluyen un sustituyente cargado positivamente o un sustituyente básico, puede formarse una sal entre un anión y un sustituyente cargado positivamente (por ejemplo, un grupo amino) o un sustituyente básico (por ejemplo, piridilo) en un compuesto descrito en el presente documento. Los aniones adecuados incluyen cloruro, bromuro, yoduro, sulfato, nitrato, fosfato, citrato, metanosulfonato, trifluoroacetato y acetato.
Las sales farmacéuticamente aceptables de los compuestos descritos en el presente documento (por ejemplo, un compuesto de fórmula (II)) también incluyen las derivadas de ácidos y bases inorgánicos y orgánicos farmacéuticamente aceptables. Los ejemplos de sales ácidas adecuadas son acetato, adipato, alginato, aspartato, benzoato, bencenosulfonato, bisulfato, butirato, citrato, canforato, canforsulfonato, digluconato, dodecilsulfato, etanosulfonato, formiato, fumarato, glucoheptanoato, glicolato, hemisulfato, heptanoato, hexanoato, clorhidrato, bromhidrato, yodhidrato, 2-hidroxietanosulfonato, lactato, maleato, malonato, metanosulfonato, 2-naftalenosulfonato, nicotinato, nitrato, palmoato, pectinato, persulfato, 3-fenilpropionato, fosfato, picrato, pivalato, propionato, salicilato, succinato, sulfato, tartrato, tiocianato, tosilato, trifluoroacetato y undecanoato.
Las sales derivadas de bases apropiadas incluyen sales de metales alcalinos (por ejemplo, sodio), metales alcalinotérreos (por ejemplo, magnesio), amonio y N-(alquilo)4 +. Esta invención también considera la cuaternización de cualquier grupo que contenga nitrógeno básico de los compuestos que descritos en el presente documento. Se pueden obtener productos solubles o dispersables en agua o en aceite mediante esta cuaternización.
Tal como se utilizan en el presente documento, los compuestos de esta invención, incluidos los compuestos de fórmula (II), se definen para incluir derivados o profármacos farmacéuticamente aceptables de los mismos. La expresión "derivado o profármaco farmacéuticamente aceptable" significa cualquier sal, éster, sal de un éster u otro derivado farmacéuticamente aceptable de un compuesto de la presente invención que, tras su administración a un receptor, es capaz de proporcionar (directa o indirectamente) un compuesto de la presente invención. Los derivados y profármacos especialmente preferidos son aquellos que aumentan la biodisponibilidad de los compuestos de esta invención cuando dichos compuestos se administran a un mamífero (por ejemplo, permitiendo que un compuesto administrado por vía oral se absorba más fácilmente hacia la sangre), o que mejoran el transporte del compuesto original a un compartimento biológico (por ejemplo, el cerebro o el sistema linfático) en relación con la especie original. Los profármacos preferidos incluyen derivados en los que un grupo que mejora la solubilidad acuosa o el transporte activo a través de la membrana intestinal se adjunta a la estructura de las fórmulas descritas en el presente documento.
Cualquier fórmula o un compuesto descrito en el presente documento también pretende representar formas no marcadas, así como formas isotópicamente marcadas de los compuestos, y los compuestos isotópicamente marcados tienen estructuras representadas por las fórmulas ofrecidas en el presente documento, excepto que uno o más átomos se sustituyen por un átomo que tiene una masa atómica o número de masa seleccionados. Los ejemplos de isótopos que pueden incorporarse en los compuestos de la invención incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro, tales como 2H, 3H, 11C, 13C, 14C, 15N, 18F 51P, 32P, 35S, 36Cl y 125I, respectivamente. La invención incluye diversos compuestos marcados isotópicamente, tal como se define en el presente documento, por ejemplo, aquellos en los que están presentes isótopos radiactivos, tales como 3H, 13C y 14C. Dichos compuestos marcados isotópicamente son útiles en estudios metabólicos (con 14C), estudios cinéticos de reacción (por ejemplo, con 'H o 3H), técnicas de detección o formación de imágenes, tales como la tomografía por emisión de positrones
("positron emission tomography", PET) o la tomografía computarizada por emisión monofotónica ("·single-photon emission computed tomography", SPECT), incluidos los ensayos de distribución tisular de fármacos o sustratos, o en el tratamiento radiactivo de pacientes. En concreto, un compuesto con 18F o marcado puede ser especialmente deseable para estudios PET o SPECT, y los compuestos isotópicamente marcados de esta invención y los profármacos de los mismos pueden prepararse generalmente llevando a cabo los procedimientos divulgados en los esquemas o en los ejemplos y preparaciones descritos a continuación mediante la sustitución de un reactivo isotópicamente marcado fácilmente disponible por un reactivo no isotópicamente marcado.
Además, la sustitución por isótopos más pesados, en especial, el deuterio (es decir 2 H o D) puede proporcionar determinadas ventajas terapéuticas que surgen de la mayor estabilidad metabólica (por ejemplo, una semivida in vivo incrementada o necesidades de dosis menores) o una mejora del índice terapéutico. Se entiende que, en este contexto, el deuterio se considera un sustituyente de un compuesto de una fórmula descrita en el presente documento. La concentración de dicho isótopo más pesado, concretamente de deuterio, puede definirse mediante el factor de enriquecimiento isotópico. La expresión "factor de enriquecimiento isotópico", tal como se utiliza en el presente documento, significa la proporción entre la abundancia isotópica y la abundancia natural de un isótopo específico. Si un sustituyente en un compuesto de esta invención se designa como deuterio, dicho compuesto tiene un factor de enriquecimiento isotópico para cada átomo de deuterio designado de al menos 3500 (un 52,5 % de incorporación de deuterio en cada átomo de deuterio designado), al menos 4000 (un 60 % de incorporación de deuterio), al menos 4500 (un 67,5 % de incorporación de deuterio), al menos 5000 (un 75 % de incorporación de deuterio), al menos 5500 (un 82,5 % de incorporación de deuterio), al menos 6000 (un 90 % de incorporación de deuterio), al menos 6333,3 (un 95 % de incorporación de deuterio), al menos 6466,7 (un 97 % de incorporación de deuterio), al menos 6600 (un 99 % de incorporación de deuterio) o al menos 8633,3 (un 99,5 % de incorporación de deuterio).
Los compuestos marcados isotópicamente descritos en el presente documento pueden prepararse generalmente mediante técnicas convencionales conocidas por los expertos en la materia o mediante procesos análogos a los descritos en los ejemplos y preparaciones adjuntos, utilizando un reactivo marcado isotópicamente apropiado en lugar del reactivo no marcado empleado previamente. Los solvatos farmacéuticamente aceptables según la invención incluyen aquellos en los que el disolvente de cristalización puede estar isotópicamente sustituido, por ejemplo, D2O, D2-acetona o D2-DMSO.
Cualquier átomo asimétrico (por ejemplo, carbono o similar) del compuesto o compuestos de la presente invención puede estar presente en una configuración racémica o enriquecida enantioméricamente, por ejemplo, la configuración (R), (S) o (RS), y, en determinadas realizaciones, cada átomo asimétrico tiene al menos un 50 % de exceso enantiomérico, al menos un 60 % de exceso enantiomérico, al menos un 70 % de exceso enantiomérico, al menos un 80 % de exceso enantiomérico, al menos un 90 % de exceso enantiomérico, al menos un 95 % de exceso enantiomérico, o al menos un 99 % de exceso enantiomérico en la configuración (R) o (S). Los sustituyentes en átomos con enlaces insaturados pueden, si es posible, estar presentes en forma cis (Z) o trans (E). Por consiguiente, tal como se utiliza en el presente documento, un compuesto de la presente invención puede estar en forma de uno de los posibles isómeros, rotámeros, atropisómeros, tautómeros o mezclas de los mismos, por ejemplo, como isómeros geométricos sustancialmente puros (cis o trans), diastereómeros, isómeros ópticos (antípodas), racematos o mezclas de los mismos. Las mezclas de isómeros resultantes pueden separarse, basándose en las diferencias fisicoquímicas de los constituyentes, en isómeros geométricos u ópticos puros o sustancialmente puros, diastereómeros, racematos, por ejemplo, mediante cromatografía o cristalización fraccionada.
Cualquier racemato resultante de los productos finales o intermedios puede resolverse en las antípodas ópticas por procedimientos conocidos, por ejemplo, por separación de las sales diastereoméricas de los mismos, obtenidas con un ácido o base ópticamente activos, y liberando el compuesto ácido o básico ópticamente activo. En concreto, puede emplearse una fracción básica para resolver los compuestos de la presente invención en sus antípodas ópticas, por ejemplo, mediante cristalización fraccionada de una sal formada con un ácido ópticamente activo, por ejemplo, ácido tartárico, ácido dibenzoiltartárico, ácido diacetiltartárico, ácido (+)-O,O'-di-p-toluoil-D-tartárico, ácido mandélico, ácido málico o ácido canfor-10-sulfónico. Los productos racémicos también pueden resolverse mediante cromatografía quiral, por ejemplo, cromatografía líquida de alta presión ("high pressure liquid chromatography", HPLC) utilizando un adsorbente quiral.
Los compuestos descritos en el presente documento (por ejemplo, un compuesto de fórmula (II)) también pueden representarse en múltiples formas tautoméricas, por ejemplo, un compuesto de fórmula (II). En tales casos, la invención incluye expresamente todas las formas tautoméricas de los compuestos descritos en el presente documento. Todas las formas cristalinas de los compuestos descritos en el presente documento se incluyen de forma expresa en la presente invención.
Se puede evaluar un compuesto descrito en el presente documento (por ejemplo, un compuesto de fórmula (II)) para determinar su capacidad para modular (por ejemplo, inhibir) el factor XIa o la calicreína.
Procedimientos de síntesis de compuestos
Los compuestos descritos en el presente documento pueden sintetizarse por procedimientos convencionales utilizando materiales de partida y reactivos disponibles en el mercado. Por ejemplo, los compuestos pueden
sintetizarse utilizando los procedimientos expuestos en la patente de EE. UU. n.º 7501404 o como se describe en los procedimientos descritos en el presente documento.
Procedimientos de tratamiento
Los compuestos descritos en el presente documento (por ejemplo, los compuestos de fórmula (I)-(VII)) pueden inhibir el factor XIa o la calicreína. En algunas realizaciones, un compuesto descrito en el presente documento puede inhibir tanto el factor XIa como la calicreína. Como resultado, estos compuestos pueden ser útiles en el tratamiento o la prevención de un trastorno descrito en el presente documento. Los ejemplos de trastornos incluyen los acontecimientos trombóticos asociados a enfermedades coronarias y cerebrovasculares, trombosis venosa o arterial, síndromes de coagulación, isquemia y angina de pecho (estable e inestable), trombosis venosa profunda (TVP), coagulopatía intravascular diseminada, síndrome de Kasabach-Merritt, embolia pulmonar, infarto de miocardio, infarto cerebral, trombosis cerebral, accidentes isquémicos transitorios, fibrilación auricular, embolia cerebral, las complicaciones tromboembólicas de la cirugía (tales como la implantación de una prótesis de cadera o rodilla, la introducción de válvulas cardíacas artificiales y la endarterectomía) y la oclusión arterial periférica, y también pueden ser útiles para tratar o prevenir el infarto de miocardio, el ictus, la angina de pecho y otras consecuencias de la rotura de placas ateroscleróticas. Los compuestos de la invención que poseen actividad inhibidora del factor XIa o de la calicreína también pueden ser útiles para prevenir la trombosis en pacientes con cáncer y para prevenir acontecimientos tromboembólicos durante o después de la restauración mecánica o basada en el activador tisular del plasminógeno de la permeabilidad de los vasos sanguíneos. Los compuestos de la invención que poseen actividad inhibidora del factor XIa o de la calicreína también pueden ser útiles como inhibidores de la coagulación sanguínea, por ejemplo, durante la preparación, la conservación y el fraccionamiento de la sangre total.
La inhibición del factor XIa, según la presente invención, puede ser un procedimiento más eficaz y seguro de inhibir la trombosis en comparación con la inhibición de otras serina proteasas de la coagulación, tales como la trombina o el factor Xa. La administración de un inhibidor de molécula pequeña del factor XIa debería tener el efecto de inhibir la generación de trombina y la formación de coágulos, sin efecto alguno o prácticamente sin efecto alguno sobre los tiempos de hemorragia y con escasa o nula alteración de la hemostasia. Estos resultados difieren sustancialmente de los de otros inhibidores de la proteasa de la coagulación de "acción directa" (por ejemplo, inhibidores del sitio activo de la trombina y del factor Xa), que demuestran una prolongación del tiempo de hemorragia y una menor separación entre la eficacia antitrombótica y la prolongación del tiempo de hemorragia. Un tratamiento preferido comprende administrar a un mamífero una composición farmacéutica que contenga al menos un compuesto de la invención. Los compuestos descritos en el presente documento pueden inhibir la calicreína, por ejemplo, un compuesto de fórmula (II). Como resultado, estos compuestos pueden ser útiles en el tratamiento o la prevención de enfermedades implicadas en la inflamación, tales como el edema (por ejemplo, edema cerebral, edema macular y angioedema (por ejemplo, angioedema hereditario)). En algunas realizaciones, los compuestos de la invención pueden ser útiles en el tratamiento o la prevención del angioedema hereditario. Los compuestos descritos en el presente documento también pueden ser útiles en el tratamiento, por ejemplo, del ictus, la isquemia y la pérdida de sangre perioperatoria, por ejemplo, un compuesto de fórmula (II).
Los compuestos de la presente invención son útiles para tratar o prevenir aquellas afecciones que implican la acción del factor XIa o la calicreína. En consecuencia, los compuestos de la presente invención son útiles en el tratamiento de las consecuencias de la rotura de la placa aterosclerótica, incluidas las enfermedades cardiovasculares asociadas con la activación de la cascada de la coagulación en estados trombóticos o trombofílicos. Tal como se utilizan en el presente documento, los términos "tratar" o "tratamiento" abarcan medidas de respuesta o profilaxis, por ejemplo, medidas diseñadas para inhibir o retrasar la aparición de la enfermedad, lograr una reducción total o parcial de los síntomas o del grado de actividad de la enfermedad, o aliviar, disminuir o curar la enfermedad o el trastorno o sus síntomas.
Más concretamente, los compuestos de la presente invención pueden utilizarse para tratar síndromes coronarios agudos, tales como la enfermedad arterial coronaria, el infarto de miocardio, la angina inestable (incluida la angina progresiva), la isquemia (por ejemplo, la isquemia resultante de la oclusión vascular) y el infarto cerebral. Los compuestos de la presente invención pueden ser útiles además en el tratamiento del ictus y de las enfermedades vasculares cerebrales relacionadas (incluido el accidente cerebrovascular, la demencia vascular y el accidente isquémico transitorio); la trombosis venosa y la tromboembolia, tal como la trombosis venosa profunda (TVP) y la embolia pulmonar; la trombosis asociada a la fibrilación auricular, el agrandamiento ventricular, la miopatía cardiaca dilatada o la insuficiencia cardiaca; la enfermedad arterial periférica y la claudicación intermitente; la formación de placas ateroscleróticas y la aterosclerosis por trasplante; la reestenosis tras una lesión arterial inducida de forma endógena (por rotura de una placa aterosclerótica) o exógena (por procedimientos cardiológicos invasivos, tales como una lesión de la pared vascular resultante de una angioplastia); la coagulopatía intravascular diseminada, el síndrome de Kasabach-Merritt, la trombosis cerebral y la embolia cerebral.
Además, los compuestos de la presente invención pueden ser útiles en el tratamiento de las consecuencias o las complicaciones tromboembólicas asociadas con el cáncer; cirugía (tal como implantación de una prótesis de cadera, endarterectomía, introducción de válvulas cardíacas artificiales, injertos vasculares, órganos mecánicos e implantación o trasplante de órgano, tejido o células); medicamentos (tales como el activador tisular del plasminógeno o agentes
similares y la repermeabilización quirúrgica de los vasos sanguíneos) en pacientes que sufren infarto de miocardio, ictus, embolia pulmonar y afecciones similares; medicamentos (tales como anticonceptivos orales, reposición hormonal y heparina, por ejemplo, para tratar la trombocitopenia inducida por heparina); sepsis (tal como la sepsis relacionada con la coagulación intravascular diseminada); y embarazo o parto. Los compuestos de la presente invención pueden utilizarse para tratar la trombosis debida al confinamiento (es decir, inmovilización, hospitalización, reposo en cama, inmovilización de extremidades, por ejemplo, con yesos inmovilizadores, etc.).
Los compuestos de la presente invención también pueden utilizarse para mantener la permeabilidad de los vasos sanguíneos, por ejemplo, en pacientes sometidos a angioplastia coronaria transluminal, o en relación con la cirugía vascular, tal como injertos de revascularización, reconstrucción arterial, aterectomía, injertos vasculares, permeabilidad de endoprótesis vasculares, e implantación y trasplante de órganos, tejidos o células. Los compuestos de la invención pueden utilizarse para inhibir la coagulación sanguínea en relación con la preparación, la conservación, el fraccionamiento o el uso de sangre total. Por ejemplo, los compuestos de la invención pueden utilizarse para mantener la sangre total y fraccionada en la fase fluida, tal como se requiere para pruebas analíticas y biológicas, por ejemplo, para estudios ex vivo de plaquetas y otras funciones celulares, procedimientos bioanalíticos y cuantificación de componentes que contienen sangre, o para mantener circuitos sanguíneos extracorpóreos, tales como en diálisis o cirugía (por ejemplo, cirugía de revascularización arterial coronaria).
Además, los compuestos de la presente invención pueden ser útiles para tratar y prevenir las complicaciones protrombóticas del cáncer. Los compuestos pueden ser útiles en el tratamiento del crecimiento tumoral, como tratamiento complementario a la quimioterapia, para prevenir la angiogénesis y para tratar el cáncer, más concretamente, el cáncer de pulmón, próstata, colon, mama, ovarios y hueso.
Isquemia
La "isquemia" o un "acontecimiento isquémico" es una enfermedad vascular que generalmente implica la oclusión vascular o una restricción en el suministro de sangre a los tejidos. La isquemia puede provocar una escasez de oxígeno y glucosa necesarios para el metabolismo celular. La isquemia suele estar causada por vasos sanguíneos problemáticos que provocan daños o disfunciones en los tejidos. La isquemia también puede referirse a una pérdida local de sangre u oxígeno en una parte determinada del cuerpo como consecuencia de una congestión (por ejemplo, vasoconstricción, trombosis o embolia). Las causas incluyen embolia, trombosis de una arteria aterosclerótica, traumatismo, problemas venosos, aneurisma, afecciones cardiacas (por ejemplo, infarto de miocardio, valvulopatía mitral, fibrilación arterial crónica, miocardiopatías y prótesis), traumatismo o lesión traumática (por ejemplo, en una extremidad que produzca la oclusión parcial o total de un vaso), síndrome de salida torácica, aterosclerosis, hipoglucemia, taquicardia, hipotensión, compresión externa de un vaso sanguíneo (por ejemplo, por un tumor), anemia falciforme, frío extremo localizado (por ejemplo , por congelación), aplicación de torniquetes, estimulación de receptores de glutamato, malformaciones arteriovenosas, rotura de vasos sanguíneos importantes que irrigan un tejido u órgano, y anemia.
Un acontecimiento isquémico transitorio generalmente se refiere a un episodio transitorio (por ejemplo, de corta duración) de disfunción neurológica causada por la pérdida de flujo sanguíneo (por ejemplo, en el cerebro focal, la médula espinal o la retina) sin infarto agudo (por ejemplo, muerte tisular). En algunas realizaciones, el acontecimiento isquémico transitorio dura menos de 72 horas, 48 horas, 24 horas, 12 horas, 10 horas, 8 horas, 4 horas, 2 horas, 1 hora, 45 minutos, 30 minutos, 20 minutos, 15 minutos, 10 minutos, 5 minutos, 4 minutos, 3 minutos, 2 minutos o 1 minuto.
Angioedema
El angioedema es la hinchazón rápida de la dermis, el tejido subcutáneo, la mucosa y los tejidos submucosos. El angioedema suele clasificarse como hereditario o adquirido.
El "angioedema adquirido" puede ser inmunológico, no inmunológico o idiopático; causado, por ejemplo, por alergia, como efecto secundario de medicamentos, por ejemplo, medicamentos inhibidores de la ECA.
El "angioedema hereditario" o "AEH" hace referencia a un trastorno genético que provoca periodos agudos de edema (por ejemplo, hinchazón) que pueden producirse en casi todas las partes del cuerpo, incluida la cara, las extremidades, el cuello, la garganta, la laringe, las extremidades, el tracto gastrointestinal y los genitales. Los ataques de AEH a menudo pueden ser mortales, y su gravedad depende de la zona afectada; por ejemplo, los ataques abdominales pueden provocar una obstrucción intestinal, mientras que la inflamación de la laringe y de las vías respiratorias superiores puede conducir a la asfixia. La patogénesis del angioedema hereditario puede estar relacionada con la activación sin oposición de la vía de contacto por la generación inicial de calicreína o factores de coagulación (por ejemplo, el factor XII).
Los signos y síntomas incluyen hinchazón, por ejemplo, de la habilidad de la cara, mucosa de la boca o garganta y lengua. El picor, el dolor, la disminución de la sensibilidad en las zonas afectadas, la urticaria o el estridor de las vías respiratorias también pueden ser signos de angioedema. Sin embargo, puede no haber picor asociado ni urticaria, por ejemplo, en el angioedema hereditario. Los sujetos con AEH pueden sufrir dolor abdominal (por ejemplo, dolor
abdominal que dura de uno a cinco días, ataques abdominales que aumentan el recuento de glóbulos blancos del sujeto), vómitos, debilidad, diarrea acuosa o erupción cutánea.
La bradicinina desempeña un papel importante en el angioedema, en especial en el angioedema hereditario. La bradicinina es liberada por diversos arios tipos de células en respuesta a numerosos estímulos diferentes y es un mediador del dolor. Interferir en la producción o la degradación de la bradicinina puede provocar angioedema. En el angioedema hereditario, la producción continua de la enzima calicreína puede facilitar la formación de bradicinina. La inhibición de la calicreína puede interferir con la producción de bradicinina y tratar o prevenir el angioedema.
Composiciones farmacéuticas
Las composiciones descritas en el presente documento incluyen el compuesto descrito en el presente documento (por ejemplo, un compuesto de fórmula (II), así como agentes terapéuticos adicionales, si están presentes, en cantidades eficaces para lograr el tratamiento de una enfermedad o síntomas de enfermedad (por ejemplo, una enfermedad asociada con el factor XIa o la calicreína).
Los vehículos, adyuvantes y soportes farmacéuticamente aceptables que pueden usarse en las composiciones farmacéuticas proporcionadas junto la presente invención incluyen, entre otros, intercambiadores iónicos, alúmina, estearato de aluminio, lecitina, sistemas transporte de fármacos autoemulgentes ("self-emulsifying drug delivery systems", SEDDS), tales como succinato de d-α-tocoferol polietilenglicol 1000, tensioactivos usados en formas farmacéuticas, tales como Tweens u otras matrices poliméricas de transporte similares, proteínas de suero, tales como albúmina de suero humana, tampones, tales como fosfatos, glicina, ácido sórbico, sorbato de potasio, mezclas de glicéridos parciales de ácidos grasos vegetales saturados, agua, sales o electrolitos, tales como sulfato de protamina, hidrógeno fosfato de disodio, hidrógeno fosfato de potasio, cloruro de sodio, sales de zinc, sílice coloidal, trisilicato de magnesio, polivinilpirrolidona, sustancias a base de celulosa, polietilenglicol, carboximetilcelulosa de sodio, poliacrilatos, ceras, polímeros en bloque de polietileno-polioxipropileno, polietilenglicol y lanolina. Las ciclodextrinas, tales como α-, β-, y γ-ciclodextrina, o los derivados químicamente modificados, tales como hidroxialquilciclodextrinas, incluidas 2- y 3-hidroxipropil-β-ciclodextrinas, u otros derivados solubilizados también pueden utilizarse ventajosamente para mejorar la administración de compuestos de las fórmulas descritas en el presente documento. Vías de administración
Las composiciones farmacéuticas proporcionadas en el presente documento pueden administrarse por vía oral, rectal o parenteral (por ejemplo, infusión intravenosa, inyección intravenosa en embolada, inhalación, implantación). El término parenteral, tal como se utiliza en el presente documento, incluye la inyección subcutánea, intracutánea, intravenosa (por ejemplo, infusión intravenosa, inyección intravenosa en embolada), intranasal, por inhalación, pulmonar, transdérmica, intramuscular, intraarticular, intraarterial, intrasinovial, intraesternal, intratecal, intralesional e intracraneal u otras técnicas de infusión. Las composiciones farmacéuticas proporcionadas en el presente documento pueden contener cualquier portador, adyuvante o vehículo convencional no tóxico farmacéuticamente aceptable. En algunos casos, el pH de la formulación puede ajustarse con ácidos, bases o tampones farmacéuticamente aceptables para mejorar la estabilidad del compuesto formulado o su forma de administración.
Las composiciones farmacéuticas pueden presentarse en forma de preparación inyectable estéril, por ejemplo, como solución o suspensión acuosa u oleaginosa inyectable estéril. Estas suspensiones se pueden formular según las técnicas conocidas en la técnica utilizando agentes de dispersión o humectantes adecuados (tal como, por ejemplo, Tween 80) y agentes de suspensión. El preparado inyectable estéril también puede ser una solución o suspensión inyectable estéril en un diluyente o disolvente parenteralmente aceptable no tóxico, por ejemplo, tal como una solución en 1,3-butanodiol. Entre los vehículos y disolventes aceptables que se pueden emplear se incluyen el manitol, agua, solución de Ringer y solución isotónica de cloruro de sodio. Además, los aceites fijos y estériles se utilizan convencionalmente como disolvente o medio de suspensión. Para este fin, se puede emplear cualquier aceite fijo suave, incluidos mono- o diglicéridos sintéticos. Los ácidos grasos, tales como ácido oleico y sus derivados de glicérido, son útiles en la preparación de sustancias inyectables, así como los aceites naturales farmacéuticamente aceptables, tales como aceite de olivo o aceite de ricino, especialmente en sus versiones polioxietiladas. Estas soluciones o suspensiones de aceite también pueden contener un diluyente o dispersante de alcohol de cadena larga o carboximetilcelulosa o agentes dispersantes similares que se utilizan habitualmente en la formulación de formas farmacéuticas farmacéuticamente aceptables, tales como emulsiones y suspensiones. Otros tensioactivos que se emplean habitualmente, tales como Tweens o Spans u otros agentes de emulsión similares o mejoradores de la biodisponibilidad, los cuales se usan habitualmente en la fabricación de formas farmacéuticas líquidas o sólidas farmacéuticamente aceptables u otras formas farmacéuticas también se pueden usar para la formulación.
Las composiciones farmacéuticamente aceptables proporcionadas en el presente documento se pueden administrar por vía oral en cualquier forma farmacéutica oralmente aceptable incluidas, entre otras, cápsulas, comprimidos, emulsiones y suspensiones, dispersiones o soluciones acuosas. En el caso de los comprimidos para uso oral, los vehículos que habitualmente se usan incluyen lactosa y almidón de maíz. También se agregan generalmente agentes lubricantes, tales como el estearato de magnesio. Para la administración oral en una forma de cápsula, los diluyentes útiles incluyen lactosa y almidón de maíz secado. Cuando se administran suspensiones acuosas o emulsiones por vía oral, el principio activo se puede suspender o disolver en una fase aceitosa combinada con agentes de emulsión o
suspensión. Si se desea, se pueden agregar determinados agentes edulcorantes, aromatizantes o colorantes o de enmascaramiento del sabor.
Los compuestos descritos en el presente documento pueden administrarse, por ejemplo, mediante inyección, por vía intravenosa (por ejemplo, infusión intravenosa, inyección intravenoso en embolada), por vía intraarterial, subdérmica, intraperitoneal, intramuscular o subcutánea; o por vía oral, bucal, nasal, transmucosa, tópica, en una dosis que oscila entre aproximadamente 0,5 y aproximadamente 100 mg/kg de peso corporal, como alternativa, en dosis de entre 1 mg y 1000 mg/dosis, cada 4 a 120 horas, o según los requisitos del fármaco en particular. Los procedimientos del presente documento contemplan la administración de una cantidad eficaz de compuesto o la composición del compuesto para lograr el efecto deseado o expuesto. Generalmente, las composiciones farmacéuticas proporcionadas en la presente invención se administrarán de aproximadamente 1 a aproximadamente 6 veces al día (por ejemplo, mediante inyección intravenosa en embolada) o, como alternativa, como una infusión continua. Tal administración se puede usar como un tratamiento crónico o agudo. La cantidad de principio activo que se puede combinar con los materiales de vehículo para producir una forma farmacéutica individual variará dependiendo del hospedante tratado y el modo concreto de administración. Un preparado típico contendrá desde aproximadamente un 5 % hasta aproximadamente un 95 % del compuesto activo (p/p). Como alternativa, tales preparados contienen desde aproximadamente un 20 % hasta aproximadamente un 80 % del compuesto activo.
Combinaciones
Cuando se ponga en práctica la presente invención, puede desearse administrar los compuestos de la presente invención (inhibidores del factor XIa o de la calicreína) en combinación entre sí y con uno o más agentes para lograr un beneficio terapéutico, tales como agentes antitrombóticos o anticoagulantes, agentes antihipertensivos, agentes antiisquémicos, agentes antiarrítmicos, inhibidores de la función plaquetaria, etc. Por ejemplo, la presente invención puede llevarse a cabo administrando los inhibidores de molécula pequeña del factor XIa o de la calicreína junto con un inhibidor de molécula pequeña del factor XIa o de la calicreína. Más concretamente, la invención puede llevarse a cabo administrando los inhibidores de molécula pequeña del factor XIa o de la calicreína junto con aspirina, clopidogrel, ticlopidina o CS-747, warfarina, heparinas de bajo peso molecular (tal como LOVENOX), bloqueantes de GPIIb/GPIIIa, inhibidores de PAI-1, tales como XR-330 y T-686, antagonistas de los receptores P2Y1 y P2Y12; antagonistas de los receptores de tromboxano (tal como ifetrobán), miméticos de la prostaciclina, inhibidores de la tromboxano A sintetasa (tal como picotamida), antagonistas de los receptores de serotonina-2 (tal como ketanserina); compuestos que inhiben otros factores de coagulación, tales como FVII, FVIII, FIX, FX, protrombina, TAFI y fibrinógeno, u otros compuestos que inhiben el FXI o la calicreína; fibrinolíticos, tales como TPA, estreptoquinasa, inhibidores de PAI-1 e inhibidores de α-2-antiplasmina, tales como antagonistas de los receptores de fibrinógeno de anticuerpos anti-α-2-antiplasmina, inhibidores de α-1-antitripsina, agentes hipolipidémicos, tales como inhibidores de HMG-CoA reductasa (por ejemplo, pravastatina, simvastatina, atorvastatina, fluvastatina, cerivastatina, AZ4522, e itavastatina) e inhibidores de la proteína microsómica transportadora de triglicéridos (tales como los descritos en las patentes de EE. UU. n.os 5 739135, 5712279 y 5760246); agentes antihipertensivos, tales como inhibidores de la enzima convertidora de angiotensina (por ejemplo, captopril, lisinopril o fosinopril); antagonistas de los receptores de angiotensina-II (por ejemplo, irbesartán, losartán o valsartán); inhibidores de la ECA/NEP (por ejemplo, omapatrilat y gemopatrilat); o βbloqueantes (tales como propranolol, nadolol y carvedilol). La invención puede llevarse a cabo administrando los inhibidores de molécula pequeña del factor XIa o de la calicreína junto con agentes antiarrítmicos, tal como para la fibrilación auricular, por ejemplo, amiodarona o dofetilida.
Al llevar a cabo la presente invención, puede desearse administrar los compuestos de la invención (inhibidores del factor XIa o de la calicreína) junto con agentes que aumentan los niveles de AMPc o GMPc en las células para obtener un beneficio terapéutico. Por ejemplo, los compuestos de la invención pueden tener efectos ventajosos cuando se utilizan junto con inhibidores de la fosfodiesterasa, incluidos los inhibidores de la PDE1 (tales como los descritos en Journal of Medicinal Chemistry, vol.40, págs.2196-2210 [1997]), inhibidores de la PDE2, inhibidores de la PDE3 (tales como revizinona, pimobendán u olprinona), inhibidores de la PDE4 (como rolipram, cilomilast o piclamilast), inhibidores de la PDE7 u otros inhibidores de la PDE, tales como dipiridamol, cilostazol, sildenafilo, denbutilina, teofilina (1,2-dimetilxantina), ARIFLOT.TM. (es decir, ácido cis-4-ciano-4-[3-(ciclopentiloxi)-4-metoxifenil]ciclohexan-1-carboxílico), arofilina, roflumilast, C-11294A, CDC-801, BAY-19-8004, cipamfilina, SCH351591, YM-976, PD-189659, mesiopram, pumafentrina, CDC-998, IC-485 y KW-4490.
La invención puede llevarse a cabo administrando los compuestos de la invención junto con agentes protrombolíticos, tales como el activador del plasminógeno tisular (natural o recombinante), estreptoquinasa, reteplasa, activasa, lanoteplasa, urocinasa, prourocinasa, complejo activador del plasminógeno de estreptoquinasa anisolado ("anisolated streptokinase plasminogen activator complex", ASPAC), activadores del plasminógeno de glándulas salivales animales y similares.
La invención puede llevarse a cabo administrando los compuestos de la invención junto con agonistas β-adrenérgicos, tales como albuterol, terbutalina, formoterol, salmeterol, bitolterol, pilbuterol o fenoterol; anticolinérgicos, tales como bromuro de ipratropio; corticoesteroides antiinflamatorios, tales como beclometasona, triamcinolona, budesonida, fluticasona, flunisolida o dexametasona; y agentes antiinflamatorios, tales como cromolina, nedocromil, teofilina, zileutón, zafirlukast, monteleukast y pranleukast.
Los inhibidores de molécula pequeña del factor XIa o de la calicreína pueden actuar sinérgicamente con uno o más de los agentes anteriores. De este modo, pueden utilizarse dosis reducidas del agente o agentes trombolíticos, obteniendo así los beneficios de la administración de estos compuestos, al tiempo que se minimizan los posibles efectos hemorrágicos y otros efectos secundarios.
Desarrollo del tratamiento
Las composiciones descritas en el presente documento incluyen una cantidad terapéuticamente eficaz de un compuesto de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína) junto con uno o más agentes (por ejemplo, un agente terapéutico adicional), tales como agentes antitrombóticos o anticoagulantes, agentes antihipertensivos, agentes antiisquémicos, agentes antiarrítmicos, inhibidores de la función plaquetaria, etc., para lograr un beneficio terapéutico.
En algunas realizaciones, el agente terapéutico adicional se administra tras la administración del compuesto de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína). En algunas realizaciones, el agente terapéutico adicional se administra 15 minutos, 30 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 8 horas, 10 horas, 12 horas, 14 horas, 18 horas, 24 horas, 48 horas, 72 horas o más después de la administración del compuesto de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína). En algunas realizaciones, el agente terapéutico adicional se administra (por ejemplo, por vía oral) tras el alta de un centro médico (por ejemplo, un hospital).
En algunas realizaciones, el compuesto de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína) y el agente terapéutico adicional se coformulan en una única composición o dosis. En algunas realizaciones, el compuesto de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína) y el agente terapéutico adicional se administran por separado. En algunas realizaciones, el compuesto de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína) y el agente terapéutico adicional se administran secuencialmente. En algunas realizaciones, el compuesto de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína) y el agente terapéutico adicional se administran por separado y secuencialmente. En general, al menos uno de los compuestos de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína) y el agente terapéutico adicional se administra por vía parenteral (por ejemplo, intranasal, intramuscular bucal, inhalación, implantación, transdérmica, intravenosa (por ejemplo, infusión intravenosa, inyección intravenosa en embolada), subcutánea, intracutánea, intranasal, pulmonar, transdérmica, intraarticular, intraarterial, intrasinovial, intraesternal, intratecal, intralesional e intracraneal u otras técnicas de infusión); por vía oral; o por vía rectal, por ejemplo, inyección intramuscular o intravenosa (por ejemplo, infusión intravenosa, inyección intravenosa en embolada)). En algunas realizaciones, el compuesto de la invención se administra por vía parenteral (por ejemplo, intranasal, bucal, intravenosa (por ejemplo, infusión intravenosa, inyección intravenosa en embolada) o intramuscular). En algunas realizaciones, el agente terapéutico adicional se administra por vía oral. En algunas realizaciones, el compuesto de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína) se administra por vía parenteral (por ejemplo, intranasal, bucal, intravenosa (por ejemplo, infusión intravenosa, inyección intravenosa en embolada) o intramuscular) y el agente terapéutico adicional se administra por vía oral.
En algunas realizaciones, el compuesto de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína) puede administrarse una o varias veces al día. La duración del tratamiento puede ser, por ejemplo, de una vez al día durante un período de aproximadamente 1, 2, 3, 4, 5, 6, 7 días o más. En algunas realizaciones, el tratamiento es crónico (por ejemplo, de por vida). En algunas realizaciones, el tratamiento se administra por vía oral. En algunas realizaciones, se administra una dosis única en forma de una forma farmacéutica unitaria individual o varias formas farmacéuticas unitarias más pequeñas o mediante administraciones múltiples de dosis subdivididas en determinados intervalos. Por ejemplo, una forma farmacéutica unitaria puede administrarse de aproximadamente 0 horas a aproximadamente 1 hora, de aproximadamente 1 hora a aproximadamente 24 horas, de aproximadamente 1 hora a aproximadamente 72 horas, de aproximadamente 1 hora a aproximadamente 120 horas, o de aproximadamente 24 horas al menos a aproximadamente 120 horas después de la lesión. Como alternativa, la unidad de dosificación puede administrarse a partir de aproximadamente 0,5, 1, 1,5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 30, 40, 48, 72, 96, 120 horas o más después de la lesión. Las formas farmacéuticas unitarias posteriores pueden administrarse en cualquier momento tras la administración inicial, de forma que se consiga un efecto terapéutico. En algunas realizaciones, la dosis inicial se administra por vía oral. En algunas realizaciones, las dosis posteriores a la dosis inicial se administran por vía parenteral (por ejemplo, intranasal, intramuscular bucal, inhalación, implantación, transdérmica, intravenosa (por ejemplo, infusión intravenosa, inyección intravenosa en embolada), subcutánea, intracutánea, intranasal, pulmonar, transdérmica, intraarticular, intraarterial, intrasinovial, intraesternal, intratecal, intralesional e intracraneal u otras técnicas de infusión); por vía oral; o por vía rectal.
Cuando un sujeto sometido a terapia presenta una respuesta parcial o una recaída después de completar el primer ciclo del tratamiento, pueden necesitarse ciclos posteriores de tratamiento para lograr una respuesta terapéutica parcial o completa (por ejemplo, tratamiento crónico, por ejemplo, de por vida).
En algunas realizaciones, el compuesto de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína) se administra por vía intravenosa, por ejemplo, como una infusión intravenosa o inyección intravenosa en embolada, durante aproximadamente 5 minutos a aproximadamente 1 semana; durante aproximadamente 30 minutos a aproximadamente 24 horas, durante aproximadamente 1 hora a aproximadamente 12 horas, durante
aproximadamente 2 horas a aproximadamente 12 horas, durante aproximadamente 4 horas a aproximadamente 12 horas, durante aproximadamente 6 horas a aproximadamente 12 horas, durante aproximadamente 6 horas a aproximadamente 10 horas; durante aproximadamente 5 minutos a aproximadamente 1 hora, durante aproximadamente 5 minutos a aproximadamente 30 minutos; durante aproximadamente 12 horas a aproximadamente 1 semana, durante aproximadamente 24 horas a aproximadamente 1 semana, durante aproximadamente 2 días a aproximadamente 5 días, o durante aproximadamente 3 días a aproximadamente 5 días. En una realización, el compuesto de la invención (por ejemplo, un inhibidor del factor XIa o de la calicreína) se administra como una infusión intravenosa durante aproximadamente 5, 10, 15, 30, 45 o 60 minutos o más; durante aproximadamente 1, 2, 4, 6, 8, 10, 12, 16 o 24 horas o más; durante aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9 o 10 días o más.
Dosis y pauta posológica
La cantidad eficaz del inhibidor de molécula pequeña del factor XIa o de la calicreína administrada según la presente invención puede ser determinada por un experto en la materia. El nivel de dosis específico y la frecuencia de dosificación para cualquier sujeto concreto se pueden variar y dependerán de una diversidad de factores que incluyen la actividad del compuesto específico empleado, la estabilidad metabólica y la duración de la acción de ese compuesto, la especie, la edad, el peso corporal, la salud general, el sexo y la dieta del sujeto, el modo y el momento de la administración, la tasa de excreción, la combinación de fármacos y la gravedad de la afección concreta.
Con la mejora de la condición del paciente, puede administrarse, en caso necesario, una dosis de mantenimiento del compuesto, la composición o la combinación proporcionados en el presente documento. Posteriormente, la dosis o la frecuencia de administración, o ambas, pueden reducirse, en función de los síntomas, hasta un nivel en el que se conserve la condición mejorada una vez se han aliviado los síntomas hasta el nivel deseado. Sin embargo, los pacientes pueden necesitar un tratamiento intermitente a largo plazo si aparece recurrencia de los síntomas de la enfermedad.
Ejemplos
Los compuestos de la presente invención se preparan como se describe en las tablas, los esquemas, la preparación de intermedios y los ejemplos a continuación, o se preparan por procedimientos análogos a los mismos, que son pueden conocerse con facilidad y están disponibles para los expertos en la técnica de la síntesis orgánica. Los compuestos que no se incluyen en el alcance de las reivindicaciones adjuntas se proporcionan a modo de referencia y no forman parte de la invención reivindicada.
Síntesis de compuestos:
Las temperaturas se expresan en grados Celsius (°C). Salvo indicación contraria, los procedimientos se efectúan a temperatura ambiente, es decir, a una temperatura comprendida entre 18 y 25 °C, en atmósfera inerte y sin humedad. La cromatografía se realiza mediante cromatografía en columna rápida sobre gel de sílice como se describe en Still, W.C, Kahn, M., Mitra, A., J. Org. Chem.1978, 43, 2923.; la cromatografía en capa fina (TLC) se realiza en placas de gel de sílice. Los datos de RMN se ofrecen en partes por millón (ppm) en relación con la señal de bloqueo de deuterio del disolvente deuterado utilizado. Se utilizan las abreviaturas convencionales para la forma de la señal. En los espectros de masas (MS), se indica el ion principal de menor masa para las moléculas en las que la división isotópica da lugar a múltiples picos espectrales de masas. Las composiciones de las mezclas de disolventes se indican como porcentajes o proporciones de volumen. En los casos en que los espectros de RMN son complejos, solo se notifican las señales de diagnóstico.
HPLC analítica
Procedimiento A: HPLC Agilent 1100, columna Zorbax Eclipse XDB-C1850 × 4,6 mm, 1,5 ml/min, disolvente A: Agua (TFA al 0,1 %), disolvente B: Acetonitrilo (TFA al 0,07 %), gradiente: 5 min de A al 95 %A a B al 90 %; 1 min de mantenimiento; luego reciclado (hasta A al 95 % durante 1 min), detección UV a 214 y 254 nm.
Procedimiento B: HPLC Agilent 1100, columna Zorbax Eclipse XDB-C1850 × 4,6 mm, 1,5 ml/min, disolvente A: Agua (TFA al 0,1 %), disolvente B: Acetonitrilo (TFA al 0,07 %), gradiente: 5 min de A al 95 %A a B al 90 %; 1 min de mantenimiento; luego reciclado (hasta A al 95 % durante 1 min), detección UV a 210 y 254 nm.
Procedimiento C: HPLC Agilent 1100, columna Zorbax Eclipse XDB-C1850 × 4,6 mm, 1,5 ml/min, disolvente A: Agua (TFA al 0,1 %), disolvente B: Acetonitrilo (TFA al 0,07 %), gradiente: 6 min de A al 95 % a B al 95 %; 1 min de mantenimiento; luego reciclado (hasta A al 95 % durante 1 min), detección UV a 210 y 254 nm.
Términos y abreviaturas:
ACN = acetonitrilo,
BOC = Boc = terc-butoxicarbonilo,
a = ancho,
t-BuOH = alcohol terc-butílico,
Cat. = catalítico,
Conc. = concentrado,
d = doblete,
dd = doblete de dobletes,
ddd = doblete de dobletes de dobletes,
dt = doblete de tripletes,
DCM = diclorometano
Peryodinano de Dess-Martin = 1,1,1-tris(acetiloxi)-1,1-dihidro-1,2-benziodoxol-3(1H)-ona, DIAD = azodicarboxilato de diisopropilo,
DMF = N,N-dimetilformamida,
DMSO = dimetilsulfóxido,
Et2O = éter etílico,
Et3N = trietilamina,
EtOAc = acetato de etilo,
EtOH = alcohol etílico,
equiv. = equivalentes,
h = horas,
H2O = agua,
HCl = ácido clorhídrico;
HPLC = cromatografía líquida de alta presión,
HOAc = ácido acético,
IPA = alcohol isopropílico,
ISCO = cartuchos de gel de sílice de fase normal suministrados por Teledyne ISCO, K2CO3 = carbonato de potasio,
LiBH4 = tetrahidroborato de litio,
LiBr = bromuro de litio,
LiCl = cloruro de litio,
LAH = tetrahidroaluminato de litio,
m = multiplete,
min = minutos,
MgCl2 = cloruro de magnesio,
MeOH = metanol,
MsCl = cloruro de metanosulfonilo
MTBE = metil terc-butil éter,
NaHCO3 = bicarbonato de sodio,
Na2SO4 = sulfato de sodio,
NH4OH = hidróxido de amonio,
NH4OAc = acetato de amonio,
NH4Cl = cloruro de amonio,
RMN = resonancia magnética nuclear,
NMP = N-metilpirrolidina
Pd-C = paladio sobre carbono activado
p = pentete,
PMB = p-metoxibencilo,
PMBC1 = cloruro de p-metoxibencilo,
ret. = retención,
ta = temperatura ambiente
s = singulete,
sat. = saturado,
t = triplete,
TFA = ácido trifluoroacético,
TBDPS = t-butildifenilsililo,
TBS = t-butildimetilsililo,
THF = tetrahidrofurano
THP = tetrahidropirano,
TLC = cromatografía en capa fina
Ejemplo 1. Esquemas y procedimientos sintéticos generales
En general, en los casos en que el compuesto contiene un sustituyente básico (por ejemplo, un grupo amino, piridilo), un sustituyente ácido (por ejemplo, un ácido carboxílico), o es bipolar (es decir, que contiene tanto un sustituyente básico como un sustituyente ácido), para facilitar el aislamiento y la manipulación, se prefiere aislar el compuesto como una sal. Esta forma de sal facilita la caracterización y se utiliza directamente en ensayos biológicos.
Esquema 1. Síntesis de compuestos con las estructuras A-8 y A-9 como se muestra en la tabla A.

El esquema 1 describe un procedimiento general para la preparación de 4-oxoazetidin-2-carboxilatos (2S,3R)-trans-disustituidos de estructura general A-8 y A-9. La alquilación del dianión de la azetidinona N-protegida A-1 (por ejemplo, R1 = TBS) con un halogenuro de alquilo de estructura general A-2 disponible en el mercado o preparado con facilidad proporciona el intermedio deseado como un producto predominantemente trans y con retención de la estereoquímica en la posición 2- (Baldwin, J.E., et al., Tetrahedron, 1990, 46, 4733). Para los compuestos que contienen aminopiridina, se prefiere el bromuro descrito en la preparación del intermedio 1 como agente alquilante. Además, la secuencia sintética descrita para la preparación del intermedio 1 es de utilidad general para la preparación de una serie de bromuros necesarios de estructura general A-2 a partir de ésteres y alcoholes disponibles en el mercado. La esterificación del ácido carboxílico en condiciones convencionales de condensación produce el intermedio A-4 , que se purifica con facilidad por cromatografía en fase normal. Para la protección temporal del ácido carboxílico, son especialmente útiles los ésteres bencílicos y 4-metoxibencílicos. La retirada del grupo N-protector de la beta-lactama produce el intermedio A-5 que se acila con facilidad con un isocianato A-6 disponible en el mercado o preparado con facilidad (por ejemplo, Tsai, M.H., Takaoka, L.R., Powell, N.A., Nowick, J.S. J. Org. Syn., 2002, 78, 220) para obtener la beta-lactama intermedia A-8. El intermedio A-5 también puede convertirse en A-8 mediante su reacción con otros reactivos conocidos que reaccionan con aminas para producir ureas, tales como carbamatos de fenilo (A-7, en el que Ar = fenilo, por ejemplo: Thavonekham, B., Síntesis, 1997, 1189) y carbamatos de 4-nitrofenilo (A-7, en el que Ar es 4-nitrofenilo, por ejemplo: Hutchins, S.M., Chapman, K.T., Tet. Lett., 1995, 38, 2449). Además, la reacción de A-5 con un cloruro de carbamoílo (A-10, en el que R5 = Me, por ejemplo: Holmes, D.L., Smith, E. M., Nowick, J. S., J. Am. Chem. Soc., 1997, 119(33), 7665) produce las ureas tetrasustituidas A-8 (R5 = Me). Las aminas disponibles en el mercado o preparadas con facilidad R4-NH2 (para un análisis reciente sobre la síntesis asimétrica de aminas de especial interés, véase Robak, M.T., Herbage, M.A., Ellman, J.A., Chem. Rev., 2010, 110, 3600) actúan como materiales de partida adecuados para la preparación de los isocianatos A-6 y los carbamatos A-7. La retirada del grupo protector temporal del carboxilato (ya sea por hidrogenación en los casos en que R3 es bencilo o mediante acidólisis con TFA cuando R3 es 4-metoxibencilo) y la retirada de cualquier otro grupo protector presente en la molécula produce los ejemplos de estructura general A-9. En los ejemplos en los que se conserva el éster CO2R3, la retirada de cualquier otro grupo protector presente en las moléculas proporciona ejemplos de la estructura general A-8. Para algunas estructuras A-9, el ácido carboxílico también puede esterificarse utilizando procedimientos conocidos por un experto en la materia para obtener otros ejemplos de la estructura general A-8. Los ejemplos de compuestos sintetizados según la ruta descrita en el esquema 1 se resumen en la tabla A (figuras 1A-1M).
Esquema 2. Síntesis de compuestos con la estructura B-5 como se muestra en la tabla B.

El esquema 2 describe un procedimiento general para la preparación de 4-oxoazetidinas 2,3-disustituidas que contienen un grupo 2-ciano. La conversión del ácido carboxílico del intermedio B-1 (preparado como se describe para el intermedio A-3 en el esquema 1) en la carboxamida (por ejemplo, por la acción de di-terc-butildicarbonato y bicarbonato de amonio en presencia de piridina, como se describe en Pozdnev, V., Tet. Lett., 1995, 36, 7115) produce el intermedio B-2 con la retirada concomitante del grupo protector de sililo en N-1. La deshidratación de la amida alifática proporciona el intermediario de nitrilo B-3 (para un análisis general, véase Mowry, D., Chem. Rev., 1948, 42, 189). La reacción con un isocianato B-4 y, si es necesario, la retirada de los grupos protectores restantes proporciona ejemplos de la fórmula general B-5. Los ejemplos de compuestos sintetizados según la ruta descrita en el esquema 2 se resumen en la tabla B (figura 2).
Esquema 3. Síntesis de compuestos con la estructura C-8 como se muestra en la tabla C y D.

El esquema 3 describe la síntesis de 3-[2-(2-amino-1,3-tiazol-5-il)etil]azetidin-2-onas de estructura general C-8. La oxidación del alqueno C-2 (preparado con facilidad mediante los procedimientos descritos en el esquema 1) produce
al aldehído C-3 que se trata sucesivamente con bromuro de tetrabutilamonio, seguido de tiourea, para obtener el intermedio de aminotiazol C-4 con la retirada concomitante del grupo protector en N-1. La protección del aminotiazol con un grupo protector adecuado, tal como BOC, produce al intermedio C-5. La reacción con un isocianato C-6, seguida de la retirada de cualquier grupo protector, produce ejemplos de la fórmula general C-8. Los ejemplos de compuestos sintetizados según la ruta descrita en el esquema 3 se resumen en las tablas C y D (figura 3).
Esquema 4. Síntesis de compuestos con la estructura D-8 como se muestra en la tabla C y D.

El esquema 4 describe la síntesis de 3-(2-aminotiazol-5-ilmetil)azetidin-2-onas de estructura general D-8. El tratamiento del haluro de vinilo D-2 (preparado con facilidad mediante los procedimientos descritos en el esquema 1) con MCPBA, seguido de la condensación del cloro epóxido intermedio con tiourea, produce el intermedio de aminotiazol D-3. La protección del aminotiazol con un grupo adecuado, tal como BOC, produce el intermedio D-4. La desprotección del nitrógeno beta-lactámico y la reacción con un isocianato D-6 produce el intermedio D-7. La retirada de los grupos protectores restantes proporciona ejemplos de la fórmula general D-8. Los ejemplos de compuestos sintetizados según la ruta descrita en el esquema 4 se resumen en las tablas C y D (figura 3).
Esquema 5. Síntesis de compuestos con la estructura E-8 como se muestra en la tabla E.

El Esquema 5 describe un procedimiento general para la preparación de 2-carboxamidas de estructura general E-6. La condensación del ácido E-1 (preparado como se ha descrito para A-3 del esquema 1) produce la amida E-2. La retirada del grupo protector N-1, seguida de la condensación del isocianato E-4 produce el intermedio E-5. La retirada del grupo protector amida y de cualquier otro grupo protector presente en la molécula produce ejemplos de la fórmula general E-6. Los ejemplos de compuestos sintetizados según la ruta descrita en el esquema 5 se resumen en la tabla E (figura 3).
Esquema 6. Síntesis de compuestos con la estructura F-8 como se muestra en la tabla F.

El esquema 6 describe la síntesis de éteres (2R,3R)-trans-disustituidos de fórmula general F-8. La descarboxilación oxidativa de F-1 (preparado como se describe para A-3 del esquema 1) produce el benzoato F-2 (por ejemplo, véase Shiozaki, M., Synthesis, 1990, 691). La retirada del grupo protector N-1, seguida del desplazamiento del benzoato con un nucleófilo de oxígeno, F-5, produce el éter F-6. La reacción con un isocianato F-7 y la retirada de cualquier grupo protector presente en la molécula produce ejemplos de la fórmula general F-8. Los ejemplos de compuestos sintetizados según la ruta descrita en el esquema 6 se resumen en la tabla F (figura 5).
Esquema 7. Síntesis de compuestos con la estructura G-4 como se muestra en la tabla G.

.
El esquema 7 describe la síntesis de sulfonas de fórmula general G-4. La reacción del benzoato G-1 (preparado como se describe para F-3, esquema 6) con metanosulfinato de sodio produce la sulfona G-2 (Clauss, K., et al., Liebigs Ann. Chem., 1974, 539). La reacción con un isocianato G-3 y la posterior retirada de cualquier grupo protector presente en la molécula produce ejemplos de la fórmula general G-4. Los ejemplos de compuestos sintetizados según la ruta descrita en el esquema 7 se resumen en la tabla G (figura 3).
Esquema 8. Síntesis de compuestos con la estructura H-9 como se muestra en la tabla H.

El esquema 8 describe un procedimiento general para la preparación de oximas de estructura general H-9. La reducción de la azetidinona N-protegida H-1 (por ejemplo, R1 = TBDPS) produce el intermedio de hidroximetilo H-2 que se oxida con facilidad al aldehído H-3. La condensación con metoxiamina produce la oxima H-4 como una mezcla de isómeros geométricos. La alquilación del anión de la oxima H-4 con un haluro de alquilo de estructura general H-5 disponible en el mercado o preparado con facilidad produce el producto 2,3-trans H-6. La retirada del grupo protector de N de la beta-lactama produce el intermedio H-7. La reacción con un isocianato H-8 y la retirada de cualquier grupo protector presente en la molécula produce ejemplos de la fórmula general H-9. Los ejemplos de compuestos sintetizados según la ruta descrita en el esquema 8 se resumen en la tabla H (figura 3).
Esquema 9. Síntesis de compuestos con las estructuras I-4 a I-7 como se muestra en la tabla I.

El esquema 9 describe un procedimiento general para la preparación de ejemplos de la estructura general I-4 a I-7. La retirada de los grupos protectores Boc y PMB de I-1 (preparado como se describe para A-8, esquema 1) produce la aminopiridina I-2. La reacción con un cloroformiato u otro derivado adecuado del ácido carbónico produce el carbamato de estructura general I-4. La eliminación del grupo ácido carboxílico, si se desea, produce compuestos de estructura general I-5. Además, la reacción de I-2 con un ácido carboxílico R7CO2H en presencia de un agente de condensación adecuado proporciona compuestos de estructura general I-6. La retirada del grupo protector de ácido carboxílico y la condensación con un alcohol diferente R3OH en presencia de un agente condensador adecuado permite la interconversión del grupo R3 de I-6. En aquellos casos en los que se desee el ácido carboxílico libre, la retirada del grupo protector del ácido carboxílico y de cualquier otro grupo protector presente en la molécula produce compuestos de estructura general I-7. Los ejemplos de compuestos sintetizados según la ruta descrita en el esquema 9 se resumen en la tabla I (figura 3).
Esquema 10. Síntesis de compuestos con las estructuras J-3 y J-4 como se muestra en la tabla J.

El esquema 10 describe un procedimiento general para la preparación de ejemplos de estructura general J-3 y J-4. La sililación transitoria de los compuestos de estructura general J-1 (preparados como se describe en el esquema 1), seguida de la reacción con el carbonato de nitrofenilo J-2 (R5 = H o Me), produce el compuesto de estructura general J-3. La reacción de J-3 con un alcohol (R3-OH) en presencia de un agente condensador adecuado o un bromuro (R3-Br)
en presencia de una base adecuada proporciona ésteres de estructura general J-4. Los ejemplos de compuestos sintetizados según la ruta descrita en el esquema 10 se resumen en la tabla J (figuras 9A-9B).
Esquema 11. Síntesis de compuestos con la estructura K-9 como se muestra en la tabla K.

El esquema 11 describe un procedimiento general para la preparación de 4-oxoazetidin-2-carboxilatos (2S, 3R)-trisustituidos de estructura general K-9. La química es completamente análoga al procedimiento descrito en el esquema 1, en el que el material de partida utilizado es la conocida lactama K-1 (véase Finke, P. E., et al., J. Med. Chem.1995, 38, 2449). Los ejemplos de compuestos sintetizados según la ruta descrita en el esquema 11 se resumen en la tabla K (figuras 10A-10B).
Ejemplo 2. Preparación de intermedios.
Intermedio 1: [4-(Bromometil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo

Etapa 1. Preparación de 2-[(terc-butoxicarbonil)amino]isonicotinato de metilo

Una solución de di-terc-butildicarbonato (47,8 g, 219 mmol) en t-BuOH (650 ml, 6,80 mol) se calentó a 33 °C y se trató con 2-aminoisonicotinato de metilo (30,4 g, 200 mmol) en porciones durante 1 h [se añadieron aproximadamente 5 g cada 10 min]. La mezcla de reacción se agitó a 33 °C durante la noche (18 h); la HPLC/LC MS indicó una conversión casi completa. Los sólidos se recogieron por filtración al vacío [embudo de vidrio sinterizado con frita gruesa] y se lavaron con Et2O (aproximadamente 200 ml). El sólido de color canela claro se secó a un vacío elevado durante 3,5 h hasta conseguir un peso constante (39,65 g, 79 %): MS (ESI+) para C12H16N2O4 m/z 253,2 [M+H]+, 275,2 [M+Na]+; tiempo de retención en HPLC: 3,25 min (procedimiento B).
Etapa 2. Preparación de [4-(hidroximetil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo

Una suspensión de 2-[(terc-butoxicarbonil)amino]isonicotinato de metilo (41,5 g, 164 mmol) en DMF (610 ml, 7,90 mol) se enfrió hasta aproximadamente 0 °C (hielo/NaCl) y se trató con una solución 1,0 M de NaHMDS en THF (197 ml, 197 mmol) gota a gota durante 2 h para obtener una solución marrón clara. La mezcla de reacción se agitó durante 30 minutos a 0 °C y se trató con PMBCl (24,5 ml, 181 mmol) gota a gota durante 11 minutos. La mezcla de reacción se agitó durante 10 min para obtener una mezcla amarilla/marrón, se retiró el baño frío y la mezcla de reacción se agitó a temperatura ambiente durante 1 h y 40 min; la HPLC indicó una conversión casi completa. La mezcla de reacción se enfrió hasta aproximadamente 0 °C (hielo/NaCl) y la reacción se inactivó añadiendo NH4Cl acuoso saturado (100 ml). La mezcla se dejó calentar hasta la temperatura ambiente y se extrajo con Et2O (1 × 200 ml). La capa acuosa separada se diluyó con agua (100 ml) y se extrajo con Et2O (2 × 100 ml). Las fases orgánicas reunidas
se lavaron con NaHCO3 acuoso saturado (1 × 400 ml) y la capa acuosa separada se filtró [vidrio sinterizado de grano grueso] y se extrajo con Et2O (2 × 100 ml). Las fases orgánicas reunidas se lavaron con LiCl acuoso al 10 % (2 × 600 ml), se secaron (Na2SO4) y se concentraron al vacío para producir un aceite naranja. El material bruto se volvió a secar a un vacío elevado durante la noche para obtener un sólido de color claro/aceite naranja (80,86 g), que se utilizó sin manipulación posterior: MS (ESI+) para C20H24N2O5 m/z 373,2 [M+H]+; tiempo de retención en HPLC: 4,53 min (procedimiento B); una RMN de 1H indicó que el material estaba contaminado con hexametildisilazano.
El 2-[(terc-butoxicarbonil)(4-metoxibencil)amino]isonicotinato de metilo bruto anterior (80,86 g) se dividió en dos partes iguales y cada una se trató como sigue: Una solución del material bruto en THF (400 ml) se enfrió hasta aproximadamente 0 °C (hielo/NaCl) y se trató con LiBH4 (3,94 g, 181 mmol) en una porción. La mezcla de reacción se agitó durante 5 min, se retiró el baño frío y se dejó que la mezcla de reacción se calentara hasta la temperatura ambiente con agitación durante 30 min. El matraz se transfirió a un baño de aceite precalentado a 50 °C y la mezcla de reacción se agitó a 40 °C durante 2,5 h; la HPLC indicó una conversión completa. La mezcla de reacción se enfrió hasta aproximadamente 0 °C y la reacción se inactivó cuidadosamente mediante la adición lenta y gota a gota de NaHCO3 acuoso saturado (100 ml). La mezcla se diluyó con agua (150 ml) y se extrajo con EtOAc (1 × 200 ml, 2 × 100 ml). Las fases orgánicas reunidas se lavaron con NH4Cl acuoso saturado (1 × 200 ml), agua (1 × 200 ml) y salmuera (1 × 200 ml), se secaron (Na2SO4) y se concentraron al vacío para obtener el compuesto del título como un aceite amarillo viscoso (55,6 g, 89 %), que se utilizó sin manipulación posterior: MS (ESI+) para C19H24N2O4 m/z 345,3 [M+H]+; tiempo de retención en HPLC: 3,19 min (procedimiento B).
Etapa 3. Preparación de [4-(bromometil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo

A una solución agitada de [4-(hidroximetil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo (23,14 g, 67,19 mmol) en THF (300 ml) enfriada en un baño de hielo/agua se le añadió Et3N (13,6 ml, 97,8 mmol). Se añadió lentamente MsCl (7,20 ml, 93,1 mmol) durante 15 min y la mezcla de reacción se agitó durante 1 h a 0 °C; la HPLC indicó una conversión casi completa. Se añadió LiBr (8,54 g, 98,3 mmol) en una porción y se retiró el baño frío. La mezcla de reacción se agitó durante la noche a temperatura ambiente; la HPLC indicó una conversión completa al producto deseado. La mezcla de reacción se repartió entre EtOAc (250 ml) y agua (250 ml). La fase acuosa separada se extrajo con EtOAc (200 ml) y las fases orgánicas reunidas se lavaron con agua (300 ml), se secaron (MgSO4) y se evaporaron al vacío producir un aceite naranja viscoso (12,05 g) La mezcla bruta se adsorbió sobre sílice y se purificó mediante cromatografía en columna rápida (500 g de sílice, cargada con hexanos, eluida con EtOAc al 15 %/hexanos) para obtener el compuesto del título como un aceite amarillo pálido (11,59 g, 42 %): MS (ESI+) para C19H23BrN2O3 m/z 407,1, 409,1 [M+H]+; tiempo de retención en HPLC: 4,48 min (procedimiento B); RMN de 1H (CDCl3, 300 MHz) δ 8,42-8,30 (m, 1H), 7,73 (s a, 1H), 7,26-7,19 (m, 2H), 7,07-6,99 (m, 1H), 6,85-6,77 (m, 2H), 5,14 (s, 2H), 4,38 (s, 2H), 3,78 (s, 3H), 1,44 (s, 9H).
Intermedio 2: 1-[(1R)-1-Isocianatoetil]-3-(trifluorometoxi)benceno

A una solución enfriada (0-5 °C) y agitada vigorosamente de clorhidrato de (1R)-1-[3-(trifluorometoxi)fenil]etanamina (preparado como se describe en las etapas 1 a 3 del ejemplo 4 a partir de 3-trifluorometoxibenzaldehído) (0,197 g de 0,815 mmol) en cloruro de metileno (4 ml) y NaHCO3 sat. (4 ml) se le añadió trifosgeno (0,080 g, 0,269 mmol) en una porción. Tras 30 min, la mezcla se transfirió a un embudo de decantación y se recogió la capa orgánica. La capa acuosa se lavó con DCM y las capas orgánicas reunidas se secaron con sulfato de sodio anhidro, se filtraron y se concentraron para obtener el compuesto del título (155 mg, 82 %) como un aceite viscoso que se utilizó inmediatamente sin purificación posterior. RMN de 1H (300 MHz, CDCl3) δ 1,63 (d, J = 7 Hz, 3H), 4,82 (q, J = 7 Hz, 1H), 7,16-7,29 (m, 3H), 7,41 (t, J = 8 Hz, 1H).
Intermedio 3: (2S,3R)-3-({2-[(Terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-4-oxo-1-{[(1S)-2,2,2-trifluoro-1-feniletil]carbamoil}azetidin-2-carboxilato de 4-metoxibencilo

A una disolución de 4-nitrofenilcloroformato (0,378 g, 2,22 mmol) en tetrahidrofurano (20 ml) a temperatura ambiente se le añadió una disolución de (S)-2,2,2-trifluoro-1-feniletilamina (0,390 g, 2,22 mmol) y piridina (0,180 ml, 2,22 mmol) en THF (20 ml) gota a gota durante 45 minutos. Después de 15 min, la mezcla se diluyó con acetato de etilo y se lavó con HCl acuoso 0,1 N , salmuera, se secó con sulfato de sodio anhidro, se filtró y se concentró para obtener ((1S)-2,2,2-trifluoro-1-feniletil]carbamato de 4-nitrofenilo que se utilizó sin purificación posterior. MS (ESI+) para C15H11F3N2O4 m/z 341,3 (M+H)+; tiempo de retención en HPLC: 4,4 min (procedimiento C). A una solución del carbamato descrito anteriormente en THF (5 ml) se le añadió (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-4-oxoazetidin-2-carboxilato de 4-metoxibencilo (185 mg, 0,329 mmol), trietilamina (0,080 ml, 0,57 mmol) y 4-dimetilaminopiridina (0,002 g, 0,016 mmol) a temperatura ambiente. Después de tres días, los volátiles se eliminaron a presión reducida y el residuo se purificó por cromatografía en columna rápida usando hexanos y acetato de etilo (al 15-30 %) como eluyente para obtener el compuesto del título (220 mg, 74 %) como un sólido blanco que contenía aproximadamente un 15 % de (S,S)-1,3-bis-(2,2,2-trifluoro-1-feniletil)urea que se eliminó fácilmente por cromatografía después de retirar los grupos protectores. RMN de 1H (300 MHz, CDCl3) δ 1,43 (s, 9H), 3,02-3,24 (m, 2H), 3,50-3,56 (m, 1H), 3,78 (s, 3H), 3,80 (s, 3H), 4,29 (d, J = 3 Hz, 1H), 5,50-5,62 (m, 1H), 6,79-6,85 (m solapado, 5H), 7,10 (d, J = 9 Hz, 2H), 7,21-7,23 (m solapado, 3H), 7,39-7,45 (m solapado, 6H), 7,64 (s, 1H), 8,27 (d, J = 5 Hz, 1H); MS (ESI+) para C40H41F3N4O8 m/z 763,0 (M+H)+; tiempo de retención en HPLC: 5,54 min (procedimiento C).
Intermedio 4. [4-(2-Bromoetil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo

Etapa 1. Preparación de (4-metilpiridin-2-il)carbamato de terc-butilo

Una solución de 4-metil-2-piridinamina (0,60 g, 5,5 mmol) en t-BuOH (35 ml) se trató con di-terc-butildicarbonato (1,3 g, 6,1 mmol). La mezcla de reacción se agitó a 30 °C durante 2 d y 17 h; la HPLC/TLC indicó la conversión al producto. El disolvente se eliminó al vacío para obtener un sólido cristalino de color canela, que se recristalizó en isopropanol caliente para proporcionar el compuesto del título como un sólido cristalino blanco (0,87 g, 75 %): (RMN de 1H coherente, no muestra ninguna contaminación, lo que concuerda con la LC/MS, aunque la HPLC muestra dos picos). MS (ESI+) para C11H16N2O2 m/z 153,2 [M-tBu+H]+; tiempo de retención en HPLC: 2,24 min (segundo pico a 2,11 min) (procedimiento B).
Etapa 2. Preparación de (4-metoxibencil)(4-metilpiridin-2-il)carbamato de terc-butilo

Una solución de (4-metilpiridin-2-il)carbamato de terc-butilo (81 mg, 0,39 mmol) en DMF (1,2 ml) se enfrió hasta -10 °C (hielo/salmuera) y se trató con hidruro de sodio (al 60 % en aceite mineral, 22 mg, 0,55 mmol). La mezcla de reacción se agitó enérgicamente durante 20 min a -10 °C para obtener una solución casi homogénea, que se trató con PMBCl (0,07 ml, 0,5 mmol). La mezcla de reacción se agitó durante 40 min para obtener una mezcla rosa, se retiró el baño frío y la mezcla se agitó a temperatura ambiente durante 17 h para obtener una mezcla naranja; la HPLC/LC MS indicó una conversión completa al producto. La reacción se inactivó añadiendo agua (1 ml) y se diluyó con agua (5 ml) y Et2O (40 ml). La capa orgánica separada se lavó con agua (10 ml), HCl acuoso 0,1 N (10 ml), NaHCO3 acuoso saturado (10 ml) y salmuera (10 ml), se secó (Na2SO4) y se concentró al vacío para obtener un aceite naranja claro. La purificación mediante cromatografía en columna (2 × 8 cm sílice; hex., EtOAc al 5 %, al 10 %, al 20 %/hex.)
proporcionó el compuesto del título como un aceite transparente e incoloro (91 mg, 71 %); MS (ESI+) para C19H24N2O3 m/z 329,2 [M+H]+; tiempo de retención en HPLC: 3,66 min (procedimiento B).
Etapa 3. Preparación de {2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}acetato de etilo y 2-[(4-metoxibencil)amino]-4-metilnicotinato de terc-butilo

Una disolución de (4-metoxibencil)(4-metilpiridin-2-il)carbamato de terc-butilo (3,3 g, 10,0 mmol) y carbonato de dietilo (6,1 ml, 50,0 mmol) en THF (82 ml) se enfrió hasta -78 °C (acetona/CO2) y se trató gota a gota con una disolución 1,33 M de LDA en hexanos/THF/etilbenceno (9,1 ml, 12 mmol) durante 15 min. La mezcla de reacción de color amarillo anaranjado se agitó durante 30 min a -78 °C; la HPLC indicó una mezcla 1,1:1:0,5 de (4-metoxibencil)(4-metilpiridin-2-il)carbamato de terc-butilo, el producto deseado {2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}acetato de etilo y el producto secundario 2-[(4-metoxibencil)amino]-4-metilnicotinato de terc-butilo. A los 45 min se inactivó la reacción añadiendo ácido acético (0,68 ml, 12 mmol) y la mezcla se diluyó con agua (75 ml) y EtOAc (36 ml). Se dejó que la mezcla se calentase hasta la temperatura ambiente y la capa acuosa separada EtOAc (2 x 36 ml). Las fases orgánicas reunidas se secaron (Na2SO4) y se concentraron al vacío para obtener un aceite amarillo. La purificación mediante cromatografía en columna (sílice de 5 × 18 cm; hex., EtOAc al 5 %, al 10 %, al 15 %, al 20 % EtOAc/hex.) proporcionó una mezcla 70:30 de {2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}acetato de etilo y 2-[(4-metoxibencil)amino]-4-metilnicotinato de terc-butilo como un aceite transparente e incoloro (1,77 g, 33 %) y se recuperó el (4-metoxibencil)(4-metilpiridin-2-il)carbamato de terc-butilo como un aceite transparente e incoloro (1,78 g, 54 %): la RMN1Hindicó una mezcla 2,9:1 de acetato de etilo {2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}acetato de etilo y 2-[(4-metoxibencil)amino]-4-metilnicotinato de terc-butilo; MS (ESI+) para C22H28N2O5 m/z 401,3 [M+H]+; MS (ESI+) para C19H24N2O3 m/z 329,3 [M+H]+; tiempo de retención en HPLC para {2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}acetato de etilo: 4,22 min (procedimiento B), tiempo de retención en HPLC para 2-[(4-metoxibencil)amino]-4-metilnicotinato de terc-butilo: 3,24 min (procedimiento B).
Etapa 4. Preparación de [4-(2-hidroxietil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo

Una mezcla aproximadamente 2:1 de {2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}acetato de etilo (1,77 g, 3,27 mmol) y 2-[(4-metoxibencil)amino]-4-metilnicotinato de terc-butilo en THF (16 ml) se enfrió hasta 0 °C (hielo/salmuera) y se trató con LiBH4 (0,21 g, 9,6 mmol). Se retiró el baño frío y se dejó que la mezcla de reacción se calentase hasta la temperatura ambiente durante 20 min, seguido de calentamiento a 50 °C durante 3 h; la HPLC/LC MS indicó el consumo completo del acetato de {2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-ilo}. La mezcla de reacción se dejó enfriar hasta la temperatura ambiente y la reacción se inactivó cuidadosamente añadiendo NaHCO3 acuoso saturado (30 ml). La mezcla se diluyó con agua (60 ml) y se extrajo con EtOAc (3 x 60 ml). Los extractos orgánicos reunidos se secaron (Na2SO4) y se concentraron al vacío para obtener un aceite amarillo brillante. La purificación mediante cromatografía en columna (4 × 14 cm sílice; hex. EtOAc al 10 %, al 30 %, al 50 %/hex.) proporcionó el compuesto del título como un aceite transparente e incoloro (0,91 g, 78 %): MS (ESI+) para C20H26N2O4 m/z 359,3 [M+H]+; tiempo de retención en HPLC: 3,17 min (procedimiento B).
Etapa 5. Preparación de [4-(2-bromoetil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo

Una mezcla de [4-(2-hidroxietil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo (0,91 g, 2,5 mmol) y tetrabromuro de carbono (0,93 g, 2,8 mmol) en CH2Cl2 (15 ml) se enfrió hasta 0 °C (hielo/agua). Se añadió trifenilfosfina (0,73 g, 2,8 mmol) en una porción y la mezcla de reacción de color amarillo brillante se agitó durante 30 min a 0 °C. Se retiró el baño frío y la mezcla de reacción se agitó a temperatura ambiente durante 1 h; la HPLC indicó una conversión casi completa al producto. A las 2 h, la HPLC indicó que no había cambios. La mezcla de reacción se diluyó con CH2Cl2 (120 ml) y se lavó con NaHCO3 acuoso saturado (70 ml). La capa acuosa separada se extractó con CH2Cl2 (2 × 70 ml) y las fases orgánicas reunidas se secaron (Na2SO4) y se concentraron al vacío para obtener un aceite transparente e incoloro. La purificación mediante cromatografía en columna (4 × 16 cm sílice; hex. EtOAc al 10 %, al 20 %, al 30 %/hex.) proporcionó el compuesto del título como un aceite transparente e incoloro (0,69 g, 64 %): MS (ESI+) para
C20H25BrN2O3 m/z 421,2 [M+H]+; tiempo de retención en HPLC: 4,29 min (procedimiento B); RMN de 1H (CDCl3, 300 MHz) δ 8,37-8,28 (m, 1H), 7,54 (s a, 1H), 7,26-7,16 (m, 2H), 6,90-6,84 (m, 1H), 6,84-6,74 (m, 2H), 5,38-4,88 (m, 2H), 3,77 (s, 3H), 3,62-3,51 (m, 2H), 3,19-3,09 (m, 2H), 1,44 (s, 9H).
Intermedio 5: [4-(bromometil)piridin-2-il]metilcarbamato de terc-butilo
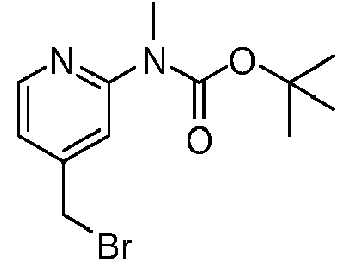
Etapa 1. Preparación de 2-[(terc-butoxicarbonil)(metil)amino]isonicotinato de metilo
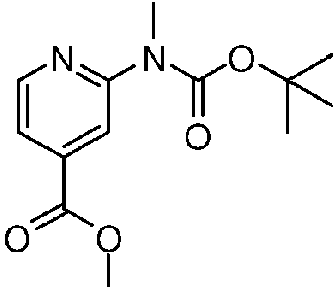
Una suspensión de 2-[(terc-butoxicarbonil)amino]isonicotinato de metilo (5,0 g, 2,0E1 mmol) en DMF (75 ml) se enfrió hasta aproximadamente 0 °C (hielo/NaCl) y se trató con una solución 1,0 M de hexametildisilazano de sodio en THF (24 ml, 24 mmol) gota a gota durante 25 min para obtener una solución transparente de color amarillo/marrón. La mezcla de reacción se agitó durante 30 min a 0 °C y se trató con yoduro de metilo (1,4 ml, 22 mmol). La mezcla de reacción se agitó durante 20 min, se retiró el baño frío y la mezcla de reacción se agitó a temperatura ambiente durante 2 h; la HPLC/LC MS indicó una conversión completa al producto. La mezcla de reacción se enfrió hasta aproximadamente ~0 °C y la reacción se inactivó añadiendoNH4Cl acuoso saturado (25 ml). Se dejó que la mezcla se calentase hasta la temperatura ambiente y se extrajo con Et2O (3 × 50 ml). Las fases orgánicas reunidas se lavaron con NaHCO3 acuoso saturado (1 × 50 ml) y LiCl acuoso al 10 % (2 × 50 ml), se secaron (Na2SO4) y se concentraron al vacío para obtener un aceite amarillo brillante con precipitado. El material bruto se secó en un vacío elevado durante la noche para obtener el compuesto del título bruto como un sólido de color claro/aceite amarillo (5,29 g). MS (ESI+) para C13H18N2O4LC m/z 267,1 (M+H)+; tiempo de retención en HPLC: 3,78 min (procedimiento B).
Etapa 2. Preparación de [4-(hidroximetil)piridin-2-il]metilcarbamato de terc-butilo

Una solución de 2-[(terc-butoxicarbonil)(metil)amino]isonicotinato de metilo bruto (5,29 g) en THF (50 ml) se enfrió hasta aproximadamente 0 °C (hielo/salmuera) y se trató con tetrahidroborato de litio (0,47 g, 22 mmol) en una porción. La mezcla de reacción se agitó durante 10 min, se retiró el baño frío y se dejó que la mezcla de reacción se calentara hasta la temperatura ambiente durante 30 min. El matraz se transfirió a un baño de aceite precalentado a 40 °C y la mezcla de reacción se agitó a 40 °C durante 3,5 h; la HPLC indicó una mezcla de producto y material de partida. La temperatura del baño de aceite aumentó hasta 50 °C durante 50 min; la HPLC indicó una conversión casi completa al producto. La mezcla de reacción se dejó enfriar hasta la temperatura ambiente y se conservó a 0-5 °C durante la noche. La mezcla de reacción se enfrió hasta aproximadamente 0 °C y la reacción se inactivó cuidadosamente mediante la adición lenta y gota a gota de NaHCO3 acuoso saturado (15 ml). La mezcla se diluyó con agua (20 ml) y se extrajo con EtOAc (3 x 20 ml). Los extractos orgánicos reunidos se lavaron con NH4Cl acuoso saturado (1 × 20 ml), agua (1 × 20 ml) y salmuera (1 × 30 ml), se secaron (Na2SO4) y se concentraron al vacío para obtener el compuesto del título bruto como un aceite de color canela (4,49 g). MS (ESI+) para C12H18N2O3LC m/z 239,2 (M+H)+; tiempo de retención en HPLC: 2,10 min (procedimiento B).
Etapa 3. Preparación de [4-(bromometil)piridin-2-il]metilcarbamato de terc-butilo

Se suspendió una mezcla de [4-(hidroximetil)piridin-2-il]metilcarbamato de terc-butilo bruto (4,49 g) y tetrabromuro de carbono (7,2 g, 22 mmol) en CH2Cl2 (110 ml) y se enfrió hasta aproximadamente 0 °C (agua helada). Se añadió trifenilfosfina (5,7 g, 22 mmol) en una porción y la mezcla de reacción de color amarillo brillante se agitó durante 20 min, se retiró el baño frío y la mezcla de color rojo anaranjado se agitó a temperatura ambiente durante 1 h; la HPLC/LC MS indicó una conversión completa al producto. La mezcla de reacción se diluyó con CH2Cl2 (50 ml) y se lavó con NaHCO3 acuoso saturado (1 × 50 ml). La capa acuosa separada se extrajo con CH2Cl2 (2 × 20 ml) y las fases orgánicas reunidas se secaron (Na2SO4) y se concentraron al vacío para obtener un aceite rojo anaranjado, que se conservó a 0-5 °C durante la noche. La purificación mediante cromatografía en columna (5 × 14 cm sílice; hex. EtOAc al 10 %, al 20 %, al 30 %/hex.) proporcionó el compuesto del título como un aceite transparente, casi incoloro (4,74 g, 80 %). MS (ESI+) para C12H17BrN2O2 m/z 301,1, 303,1 (M+H)+; tiempo de retención en HPLC: 3,52 min (procedimiento B); RMN de 1H (300 MHz, CDCl3) δ 8,43-8,27 (m, 1H), 7,84-7,66 (m, 1H), 7,12-6,92 (m, 1H), 4,39 (s a, 2H), 3,41 (s, 3H), 1,54 (s, 9H).
Ejemplo 3, esquema 1: Trifluoroacetato del ácido (2S,3R)-3-[(2-aminopiridin-4-il)metil]-1-{[(1R)-1-ciclohexiletil]carbamoil}-4-oxoazetidin-2-carboxílico

Etapa 1. Preparación del ácido (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxílico

Se enfrió en una atmosfera de nitrógeno un matraz de 3 bocas secado en horno, se cargó con ácido (2S)-1-[tercbutil(dimetil)silil]-4-oxoazetidin-2-carboxílico (3,00 g, 13,1 mmol) y THF seco (110 ml) y se enfrió la mezcla hasta -70 °C. Se añadió diisopropilamida de litio en THF [1,25 M (valorado), 21,4 ml, 26,8 mmol] gota a gota durante 8 min, manteniendo la temperatura interna por debajo de -55 °C, y la mezcla de color canela resultante se agitó a -78 °C durante 35 min y después se situó en un baño de hielo-salmuera-MeOH y se agitó por debajo de -15 °C durante 30 min más. A continuación, se añadió una solución de [4-(bromometil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo (5,86 g, 14,4 mmol) en THF seco (21 ml) que se había enfriado hasta 0 °C durante 5 min., manteniendo la temperatura interna por debajo de -5 °C, y la mezcla oscura resultante se agitó a esta temperatura durante 1 h, se inactivó con ácido cítrico acuoso (0,5 M, 120 ml), se diluyó con agua (120 ml) y se extrajo con EtOAc (3 × 100 ml). La fase orgánica reunida se lavó con salmuera (60 ml), se secó sobre sulfato de magnesio anhidro, se filtró y se concentró a presión reducida para proporcionar el producto bruto como un residuo transparente y pegajoso que se utilizó tal cual en la etapa siguiente. MS (ESI+) para C29H41N3O6Si m/z 556,5 (M+H)+.
Etapa 2. Preparación de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxilato de bencilo

Una disolución agitada de ácido (2S,3R)-3-({2-[terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il }metil)-1-[terc-butil(dimetil) silil]-4-oxoazetidin-2-carboxílico bruto en DCM (60 ml) en una atmósfera de nitrógeno se trató con clorhidrato de N-(3-dimetilaminopropil)-N'-etilcarbodiimida (3,26 g, 17,0 mmol), seguido de alcohol bencílico (1,62 ml, 15,7 mmol) y 4-dimetilaminopiridina (160 mg, 1,31 mmol), y la mezcla marrón resultante se agitó a temperatura ambiente durante 2 h, momento en el que la HPLC indicó que la reacción se había completado. La mezcla se diluyó con agua (60 ml) y se extrajo con DCM (2 × 60 ml), y la fase orgánica reunida se lavó con agua (60 ml) y salmuera (50 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. La purificación mediante cromatografía en gel de sílice [cartucho de 120 g; elución con un gradiente de EtOAc del 10 % al 20 %/hexanos] proporcionó 4,16 g del compuesto del título (contaminado con aproximadamente un 15 % en peso de alcohol bencílico; rendimiento del 42 %
en las dos primeras etapas) como un aceite viscoso de color canela que se utilizó sin purificación posterior. MS (ESI+) para C36H47N3O6Si m/z 646,7 (M+H)+.
Etapa 3. Preparación de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-4-oxoazetidin-2-carboxilato de bencilo

Una disolución agitada de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-[tercbutil(dimetil)silil]-4-oxoazetidin-2-carboxilato de bencilo (2,98 g, 3,92 mmol) en MeOH (39 ml) en una atmósfera de nitrógeno se trató con ácido acético (0,780 ml, 13,7 mmol), seguido de una disolución de fluoruro de amonio en MeOH (0,5 M, 10,2 ml, 5,10 mmol), y la mezcla homogénea resultante se agitó a temperatura ambiente durante 1 h, momento en el que la HPLC indicó que la reacción se había completado. La mezcla se concentró a presión reducida y el residuo se suspendió en DCM (120 ml), se lavó con NaHCO3 acuoso saturado (80 ml), agua (60 ml) y salmuera (40 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. La purificación mediante cromatografía en gel de sílice [cartucho de 40 g; elución con EtOAc del 20 % al 50 %/hexanos] proporcionó 1,95 g (94 %) del compuesto del título como un sólido blanco. MS (ESI+) para C30H33N3O6 m/z 532,4 (M+H)+.
Etapa 4. Preparación de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-{[(1R)-1-ciclohexiletil]carbamoil}-4-oxoazetidin-2-carboxilato de bencilo

Una disolución agitada de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-4-oxoazetidin-2-carboxilato de bencilo (2,72 g, 5,12 mmol) en DCM (51 ml) en una atmósfera de nitrógeno se trató con Et3N (2,50 ml, 17,9 mmol), seguido de una disolución de [(1R)-1-isocianatoetil]ciclohexano del mercado (1,02 g, 6,65 mmol) en DCM (5 ml) y la mezcla se agitó a temperatura ambiente durante la noche, momento en el que la HPLC indicó que el material de partida se había consumido casi por completo. A las 17 h, la mezcla se concentró a presión reducida y el residuo se purificó por cromatografía en gel de sílice [120 g; elución con EtOAc del 15 % al 25 %/hexanos] para obtener 2,31 g (66 %) del compuesto del título como una espuma blanca. MS (ESI+) para C39H48N4O7 m/z 685,7 (M+H)+.
Etapa 5. Preparación del ácido (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il]metil)-1-{[(1R)-1-ciclohexiletil]carbamoil}-4-oxoazetidin-2-carboxílico

Se trató una mezcla de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-{[(1R)-1-ciclohexiletil]carbamoil}-4-oxoazetidin-2-carboxilato de bencilo (1,94 g, 2,83 mmol) en MeOH/EtOAc 1:1 (56 ml) en una atmósfera de nitrógeno se trató con paladio al 10 % sobre carbono (301 mg, 0,283 mmol de Pd), y la mezcla se sometió a vacío y se llenó de nitrógeno dos veces y se agitó en una atmósfera de hidrógeno (globo de doble capa) durante 70 min, momento en el que la HPLC indicó que la reacción se había completado. El catalizador se retiró por filtración a través de Solka-Floc, enjuagando con MeOH/EtOAc 1:1, y el filtrado se concentró a presión reducida para proporcionar 1,67 g (99 %) del compuesto del título como un sólido blanco y pegajoso. MS (ESI+) para C32H42N4O7 m/z 595,5 (M+H)+.
Etapa 6. Preparación del trifluoroacetato del ácido (2S,3R)-3-[(2-aminopiridin-4-il)metil]-1-{[(1R)-1-ciclohexiletilcarbamoil-4-oxoazetidin-2-carboxílico

Una disolución agitada de ácido (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-{[(1R)-1-ciclohexiletil]carbamoil}-4-oxoazetidin-2-carboxílico (78,0 mg, 0,131 mmol) en DCM (2,1 ml) en una atmósfera de nitrógeno se enfrió en un baño de agua helada y se trató gota a gota con TFA (0,7 ml, 9 mmol). La mezcla resultante se agitó a 0-5 °C durante 30 min y después se calentó hasta la temperatura ambiente y se agitó durante la noche, momento en el que la HPLC indicó que la reacción se había completado. La mezcla se concentró a presión reducida y el residuo se purificó por HPLC preparativa en un sistema CombiFlash Rf [columna C18 Gold de 30 g; elución con un gradiente de acetonitrilo al 10 % (TFA al 0,07 %)/agua (TFA al 0,1 %) a acetonitrilo al 100 % (TFA al 0,07 %)]. La liofilización de las fracciones del producto proporcionó 45 mg (70 %) del compuesto del título como sólido amorfo blanco. RMN de 1H(400 MHz, MeOD) δ 7,79 (d, J = 6,4 Hz, 1H), 6,98 (s, 1H), 6,91 (d, J = 6,8 Hz, 1H), 6,64 (bd, J = 9,2 Hz, 1H), 4,27 (d, J = 2,8 Hz, 1H), 3,70 (m, 2H), 3,24 (m, 2H), 1,85-1,65 (m, 5H), 1,42 (m, 1H), 1,36-1,15 (m, 3H), 1,17 (d, J = 6,8 Hz, 3H), 1,15-0,95 (m, 2H); MS (ESI+) para C19H26N4O4 (parental) m/z 375,3 (M+H)+; tiempo de retención en HPLC: 3,21 min (procedimiento A).
Ejemplo 4, esquema 1: Trifluoroacetato del ácido (2S,3R)-3-[(2-aminopiridin-4-il)metil]-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-4-oxoazetidin-2-carboxílico (proporcionado como referencia)

Etapa 1. Preparación de (S)-N-[(1E)-(2,2-difluoro-1,3-benzodioxol-5-il)metilen]-2-metilpropano-2-sulfinamida

A una solución de la amida del ácido (S)-2-metil-propano-2-sulfínico (0,207 g, 1,71 mmol) en DCM (5 ml) se le añadió sulfato de cobre(II) (0,8186 g, 5,129 mmol), seguido de una solución de 2,2-difluoro-1,3-benzodioxol-5-carbaldehído (0,350 g, 1,88 mmol) en DCM (5 ml). La mezcla se agitó a temperatura ambiente durante 16 h, se filtró a través de Celite y se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna rápida (hexano con acetato de etilo (al 2-8 %) como eluyente) para obtener el compuesto del título (0,468 g, 95 %) en forma de aceite. MS (ESI+) para C13H17F2NO3S m/z 290,1 (M+H)+; tiempo de retención en HPLC: 4,73 min (procedimiento C).
Etapa 2. Preparación de N-[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]-2-metilpropano-2-sulfinamida

A una solución enfriada (-20 °C) de (S)-N-[(1E)-(2,2-difluoro-1,3-benzodioxol-5-il)metilen]-2-metilpropano-2-sulfinamida (0,274 g, 0,947 mmol) en THF (9 ml) se le añadió bromuro de metilmagnesio 3 M (3 M en éter etílico, 3,157 ml, 9,471 mmol) durante 15 min a una velocidad lo suficientemente lenta como para mantener la temperatura de reacción por debajo de -15 °C. Después de 60 min a -20 °C, la reacción se inactivó añadiendo NH4Cl acuoso saturado. La mezcla se diluyó con acetato de etilo y se lavó con NaHCO3 saturado, salmuera, se secó con sulfato de sodio anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía en columna rápida (hexanos con acetato de etilo al 20-30 % como eluyente) para obtener el producto del título (el diastereómero mayoritario y de elución más lenta, 0,234 g, 81 %) en forma de aceite: MS (ESI+) para C13H17F2NO3S m/z 306,2 (M+H)+; tiempo de retención en HPLC: 4,20 min (procedimiento C).
Etapa 3. Preparación de clorhidrato de (1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etanamina

A una solución enfriada (0-5 °C) de N-[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]-2-metilpropano-2-sulfinamida (0,234 g, 0,766 mmol) en metanol (4,6 ml) se le añadió cloruro de hidrógeno en 1,4-dioxano (4 M, 0,958 ml, 3,83 mmol). Después de 5 min, se retiró el baño de hielo y la mezcla de reacción se agitó a temperatura ambiente durante 1 h. Los volátiles se eliminaron al vacío y el residuo se concentró dos veces en metanol, dos veces en éter etílico y se secó al vacío para obtener el compuesto del título (0,175 g, 96 %) como un sólido blanco: RMN de 1H (300 MHz, CD3OD) δ 1,67 (t, J = 7 Hz, 3H), 4,55 (q, J = 7 Hz, 2H), 7,29-7,36 (m, 2H), 7,43-7,44 (m, 1H); tiempo de retención en HPLC: 2,69 min (procedimiento C).
Etapa 4. Preparación de [(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamato de fenilo

A una solución enfriada (0-5 °C) de clorhidrato de (1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etanamina (100 mg, 0,421 mmol) en cloruro de metileno (2 ml) se le añadió trietilamina (0,132 ml, 0,947 mmol), seguida de cloroformiato de fenilo (0,054 ml, 0,433 mmol) gota a gota. Después de 15 min se retiró el baño de hielo y la mezcla de reacción se agitó a temperatura ambiente durante 3 horas. La mezcla se diluyó con acetato de etilo y se lavó con HCl 0,1 N, NaHCO3 saturado, salmuera, se secó con sulfato de sodio anhidro, se filtró y se concentró. El residuo se purificó por cromatografía en columna rápida (hexanos con acetato de etilo al 5 % como eluyente) para obtener el compuesto del título (140 mg, 93 %) como un sólido blanco: MS (ESI+) para C16H13F2NO4 m/z 322,2 (M+H)+; tiempo de retención en HPLC: 4,75 min (procedimiento C).
Etapa 5. Preparación de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-4-oxoazetidin-2-carboxilato de 4-metoxibencilo

A una solución de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-4-oxoazetidin-2-carboxilato de 4-metoxibencilo (preparada como se describe en el ejemplo 3 sustituyendo el alcohol bencílico por alcohol 4-metoxibencílico en la etapa 2, 0,161 g de 0,287 mmol) en dimetilsulfóxido (2 ml) se le añadió [(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamato de fenilo (0,120 g, 0,374 mmol). Tras 16 h, la mezcla se diluyó con acetato de etilo y se lavó con HCl 0,1 N, NaHCO3 saturado, salmuera, se secó con sulfato de sodio anhidro, se filtró y se concentró. El residuo se purificó por cromatografía en columna rápida (hexanos con acetato de etilo al 5-20 % como eluyente) para obtener el compuesto del título (156 mg, 69 %) como un sólido blanco: MS (ESI+) para C41H42F2N4O10 m/z 789,5 (M+H)+; tiempo de retención en HPLC: 5,71 min (procedimiento C).
Etapa 6. Preparación de trifluoroacetato del ácido (2S,3R)-3-[(2-aminopiridin-4-il)metil]-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-4-oxoazetidin-2-carboxílico

A una solución enfriada (0-5 °C) de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}-metil)-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-4-oxoazetidin-2-carboxilato de 4-metoxibencilo (0,160 g, 0,203 mmol) en cloruro de metileno (6 ml) se le añadió trietilsilano (1 ml), seguido de ácido trifluoroacético (3 ml). Tras 2 h, se retiró el baño de hielo y la mezcla de reacción se agitó a temperatura ambiente durante 16 h. Se eliminaron los volátiles y el residuo se purificó mediante cromatografía CombiFlash [cartucho de gel de sílice RediSep C-18 Gold de 30 g, gradiente de disolvente: de acetonitrilo al 10 % (TFA al 0,07 %)/agua (TFA al 0,1 %) a acetonitrilo al 100 % (TFA
al 0,07 %)] y se liofilizó para obtener el compuesto del título (80 mg, 70 %) como un sólido blanco: RMN de 1H(300 MHz, CD3OD) δ 1,54 (d, J = 7 Hz, 3H), 3,20-3,29 (m, 2H), 3,69-3,75 (m, 1H), 4,30 (d, J = 3 Hz, 1H), 4,94-5,00 (m, 1H), 6,90-6,98 (m, 2H), 7,13-7,26 (m, 3H); MS (ESI+) para C20H18F2N4O6 m/z 449,2 (M+H)+; tiempo de retención en HPLC: 3,39 min (procedimiento C).
Ejemplo 5, esquema 1: Trifluoroacetato del ácido (2S,3R)-3-[(2-aminopiridin-4-il)metil]-1-{[(1S)-1-ciclohexil-2,2,2-trifluoroetil]carbamoil}-4-oxoazetidin-2-carboxílico

Etapa 1. Preparación de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-{[(1S)-1-ciclohexil-2,2,2-trifluoroetil]carbamoil}-4-oxoazetidin-2-carboxilato de bencilo

A una disolución de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-4-oxoazetidin-2-carboxilato de bencilo (100 mg, 0,19 mmol, preparado mediante los procedimientos generales descritos en el esquema 1) en dimetilsulfóxido (0,58 ml) se le añadió [(1S)-1-ciclohexil-2,2,2-trifluoroetil]carbamato de fenilo (74 mg, 0,24 mmol), seguido de trietilamina (28 µl, 0,21 mol). La mezcla se agitó a temperatura ambiente durante la noche, se diluyó con acetato de etilo, se lavó con agua, se secó con sulfato de sodio anhidro, se filtró y se concentró. El residuo se purificó por cromatografía en columna rápida utilizando hexanos/acetato de etilo (al 5-15 %) como eluyente para obtener el compuesto del título (70 mg, 50 %) como un sólido de color canela: RMN de 1H (CDCl3) δ 0,97-1,33 (m, 5H), 1,42 (s, 9H), 1,59-1,86 (m, 6H), 3,05-3,25 (m, 2H), 3,53-3,59 (m, 1H), 3,78 (s, 3H), 4,31-4,39 (m solapado, 2H), 5,09-5,25 (m solapado, 4H), 6,64 (d, J = 10 Hz, 1H), 6,78-6,86 (m solapado, 3H), 7,19-7,23 (m, 4H), 7,32-7,36 (m, 3H), 7,65 (s, 1H), 8,27 (d, J = 5 Hz, 1H); MS (ESI+) para C39H45F3N4O7 m/z 739,3 (M+H)+. Tiempo de retención en HPLC: 5,99 min (procedimiento C).
Etapa 2. Preparación de trifluoroacetato del ácido (2S,3R)-3-[(2-aminopiridin-4-il)metil]-1-{[(1S)-1-ciclohexil-2,2,2-trifluoroetil]carbamoil}-4-oxoazetidin-2-carboxílico

A un matraz que contenía Pd/C (al 10 %, 10 mg) se le añadió una solución de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-{[(1S)-1-ciclohexil-2,2,2-trifluoroetil]carbamoil}-4-oxoazetidin-2-carboxilato de bencilo (70 mg, 0,095 mmol) en etanol (40 ml). La mezcla se agitó en 1 atmósfera de H2 durante 3 h, se filtró a través de un lecho corto de Solka-Floc y se concentró a presión reducida. A una solución enfriada (0-5 °C) del residuo obtenido anteriormente en cloruro de metileno (3 ml) se le añadió trietilsilano (0,5 ml), seguido de ácido trifluoroacético (1,5 ml). Tras 2 h, se retiró el baño de hielo y la mezcla de reacción se agitó a temperatura ambiente durante 16 h. Los volátiles se eliminaron a presión reducida y el residuo se purificó mediante cromatografía CombiFlash [cartucho de gel de sílice RediSep C-18 Gold de 30 g, gradiente de disolvente: acetonitrilo al 10 % (TFA al 0,07 %)/agua (TFA al 0,1 %) a acetonitrilo al 100 % (TFA al 0,07 %)] y se liofilizó para obtener el compuesto del título (40 mg, 78 %) como un sólido blanco: RMN de 1H (CDCl3) δ 1,08-1,39 (m, 5H), 1,68-1,92 (m, 6H), 3,23-3,29 (m, 2H), 3,76-3,82 (m, 1H), 4,34-4,39 (m solapado, 2H), 6,90-6,93 (m, 1H), 6,98 (s, 1H), 7,78-7,80 (m, 1H); MS (ESI+) para C19H23F3N4O4 m/z 429,1 (M+H)+; tiempo de retención en HPLC: 3,89 min (procedimiento C).
Ejemplo 6, esquema 1: Trifluoroacetato del ácido (2S,3R)-1-{[(1S)-1-ciclohexil-2,2,2-trifluoroetil]carbamoil}-4-oxo-3-(piridin-4-ilmetil)azetidin-2-carboxílico

A una solución enfriada (0-5 °C) de (2S,3R)-3-[(2-cloropiridin-4-il)metil]-1-{[(1S)-1-ciclohexil-2,2,2-trifluoroetil]carbamoil}-4-oxoazetidin-2-carboxilato de 4-metoxibencilo (0,190 g de 0,33 mmol, preparada por los procedimientos generales descritos en el esquema 1) en CH2Cl2 (6 ml) se añadió ácido trifluoroacético (3 ml). Tras 5 min, se retiró el baño de hielo y la reacción se agitó a temperatura ambiente durante 30 min más. Los volátiles se eliminaron a presión reducida y el residuo se concentró dos veces en éter. El residuo se disolvió en etanol (20 ml) y se añadió a un matraz que contenía Pd/C (al 10 %, 20 mg). Se añadió trietilamina (0,102 ml, 0,74 mmol) y la mezcla se agitó en 1 atmósfera de H2 durante 30 min. La mezcla se filtró a través de Celite y se concentró a presión reducida. El residuo se purificó mediante cromatografía CombiFlash [cartucho de gel de sílice RediSep C-18 Gold de 30 g, gradiente de disolvente: acetonitrilo al 10 % (TFA al 0,07 %)/agua (TFA al 0,1 %) a acetonitrilo al 100 % (TFA al 0,07 %)] y se liofilizó para obtener el compuesto del título (39 mg, 22 %) como un sólido blanco. RMN de 1H (CD3OD) δ 1,07-1,39 (m, 5H), 1,68-1,93 (m, 6H), 3,46-3,61 (m, 2H), 3,89-3,95 (m, 1H), 4,36-4,44 (m solapado, 2H), 8,02 (d, J = 7 Hz, 2H), 8,76 (d, J = 7 Hz, 2H); MS (ESI+) para C19H22F3N3O4 m/z 414,0 (M+H)+; tiempo de retención en HPLC: 3,45 min (procedimiento C).
Ejemplo 7, esquema 2: Trifluoroacetato de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-2-ciano-4-oxo-N-[(1R)-1-feniletil]azetidin-1-carboxamida (proporcionado como referencia)

Etapa 1. Preparación del ácido (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxílico

El compuesto del título se preparó a partir de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxilato de bencilo según el procedimiento general descrito en la etapa 5 del ejemplo 3 con un 98 % de rendimiento como un sólido blanco pegajoso. MS (ESI+) para C29H41N3O6Si m/z 556,5 (M+H)+.
Etapa 2. Preparación de (4-{[(2S,3R)-2-carbamoil-4-oxoazetidin-3-il]metil}piridin-2-il)(4-metoxibencil)carbamato de terc-butilo

Una disolución agitada de ácido (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxílico (etapa 1, 417 mg, 0,750 mmol) en DMF seco (3,5 ml) en una atmósfera de nitrógeno se trató secuencialmente con piridina (38 µl, 0,47 mmol) y di-terc-butildicarbonato (205 mg, 0,938 mmol) y bicarbonato de amonio (71,2 mg, 0,900 mmol), y la mezcla tan resultante se agitó a temperatura ambiente durante la noche. La HPLC a las 24 h indicó que la reacción se había completa, de modo que la mezcla se diluyó con agua (30 ml) y se extrajo con DCM (3 × 30 ml). La fase orgánica reunida se lavó con salmuera (5 ml), se secó sobre Na2SO4 y se concentró a presión reducida. La purificación por cromatografía radial [rotor de gel de sílice de 2000 micrómetros; eluyente de MeOH al 5-10 %/DCM] produjo 225 mg (68 %) del compuesto del título como un sólido vítreo. MS (ESI+) para C23H28N4O5 m/z 441,4 (M+H)+.
Etapa 3. Preparación de (4-{[(2S,3R)-2-ciano-4-oxoazetidin-3-il]metil}piridin-2-il)(4-metoxibencil)carbamato de tercbutilo

Una solución agitada de (4-{[(2S,3R)-2-carbamoil-4-oxoazetidin-3-il]metil}piridin-2-il)(4-metoxibencil)carbamato de terc-butilo (etapa 2, 75,0 mg, 0,170 mmol) y piridina (33,0 µl, 0,409 mmol) en THF seco (2 ml) en una atmósfera de nitrógeno se enfrió en un baño de hielo-salmuera-MeOH a -10 °C y se trató con anhídrido trifluoroacético (28,8 µl, 0,204 mmol) lentamente gota a gota durante aproximadamente 3 min. La mezcla homogénea resultante se dejó que se calentase lentamente hasta 0 °C durante 25 min, momento en el que la HPLC indicó que la reacción estaba se había completado fundamentalmente. La mezcla se diluyó con agua (15 ml) y se extrajo con EtOAc (3 × 15 ml), y la fase orgánica reunida se lavó con salmuera (10 ml), se secó sobre MgSO4 y se concentró a presión reducida para proporcionar 75 mg de una película transparente. Este producto bruto se combinó con otros 72 mg de producto bruto de otra reacción y se purificó por cromatografía radial [rotor de gel de sílice de 2000 micrómetros; eluyente de MeOH al 5%/DCM] para proporcionar 119 mg (un 86 % para las reacciones reunidas) del compuesto del título como un sólido blanco. MS (ESI+) para C23H26N4O4 m/z 423,4 (M+H)+.
Etapa 4. Preparación de (4-{[(2S,3R)-2-ciano-4-oxo-1-{[(1R)-1-feniletil]-carbamoil}azetidin-3-il]metil}piridin-2-il)(4-metoxibencil)carbamato de terc-butilo

El compuesto del título se preparó a partir de (4-{[(2S,3R)-2-ciano-4-oxoazetidin-3-il]metil}piridin-2-il)(4-metoxibencil)carbamato de terc-butilo y [(1R)-1-isocianatoetil]benceno según el procedimiento general de la etapa 4 del ejemplo 3 en un 52 % como un sólido vítreo. MS (ESI+) para C32H35N5O5 m/z 570,5 (M+H)+.
Etapa 5. Preparación de trifluoroacetato de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-2-ciano-4-oxo-N-[(1R)-1-feniletil]azetidin-1-carboxamida

El compuesto del título se preparó a partir de (4-{[(2S,3R)-2-ciano-4-oxo-1-{ [(1R)-1-feniletil]carbamoil}azetidin-3-il]metil}piridin-2-il)(4-metoxibencil)carbamato de terc-butilo según el procedimiento general descrito en la etapa 6 del ejemplo 3 en un 51 % como un sólido blanco. RMN de 1H (400 MHz, MeOD) δ 7,82 (d, J = 6,8 Hz, 1H), 7,38 (m, 4H), 7,29 (m, 1H), 7,09 (bd, J = 7,6 Hz, 1H), 6,97 (s, 1H), 6,91 (dd, J = 6,8, 1,6 Hz, 1H), 5,00 (m, 1H), 4,70 (d, J = 3,2 Hz, 1H), 4,17 (td, J = 8,0, 3,2 Hz, 1H), 3,27 (d, J = 8,0 Hz, 2H), 1,55 (d, J = 6,8 Hz, 3H); MS (ESI+) para C19H19N5O2 (parental) m/z 350,3 (M+H)+; tiempo de retención en HPLC: 2,85 min (procedimiento A).
Ejemplo 8, esquema 2: Trifluoroacetato de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-2-ciano-N-(difenilmetil)-4-oxoazetidin-1-carboxamida (proporcionado como referencia)

Etapa 1. Preparación de [4-({(2S,3R)-2-ciano-1-[(difenilmetil)carbamoil]-4-oxoazetidin-3-il}metil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo

El compuesto del título se preparó a partir de (4-{[(2S,3R)-2-ciano-4-oxoazetidin-3-il]metil}piridin-2-il)(4-metoxibencil)carbamato de terc-butilo y 1,1'-(isocianatometileno)dibenceno según el procedimiento general de la etapa 4 del ejemplo 3 usando un tiempo de reacción de 1,5 h en un 84 % como un sólido vítreo. MS (ESI+) para C37H37N5O5 m/z 632,7 (M+H)+.
Etapa 2. Preparación de trifluoroacetato de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-2-ciano-N-(difenilmetil)-4-oxoazetidin-1-carboxamida

[0218] El compuesto del título se preparó a partir de [4-({(2S,3R)-2-ciano-1-[(difenilmetil)carbamoil]-4-oxoazetidin-3-il}metil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo según el procedimiento general de la etapa 6 del ejemplo 3 en un 54 % como un sólido blanco. RMN de 1H (400 MHz, MeOD) δ 7,81 (d, J = 6,8 Hz, 1H), 7,44 (bd, J = 7,6 Hz, 1H), 7,28-7,40 (m, 10H), 6,96 (m, 1H), 6,91 (dd, J = 6,8, 1,6 Hz, 1H), 6,15 (d, J = 8,0 Hz, 1H), 4,73 (d, J = 3,2 Hz, 1H), 4,22 (td, J = 8,0, 3,2 Hz, 1H), 3,27 (d, J = 8,0 Hz, 2H); MS (ESI+) para C24H21N5O2 (parental) m/z 412,4 (M+H)+; tiempo de retención en HPLC: 3,36 min (procedimiento A).
Ejemplo 9, esquema 3: Trifluoroacetato de ácido (2S,3R)-3-[2-(2-amino-1,3-tiazol-5-il)etil]-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxílico (proporcionado como referencia)

Etapa 1. Preparación de (2S,3R)-1-[terc-butil(dimetil)silil]-4-oxo-3-pent-4-en-1-ilazetidin-2-carboxilato de bencilo

El compuesto del título se preparó siguiendo el procedimiento general de las etapas 1 y 2 del ejemplo 3, pero usando 5-bromopent-1-eno en la etapa 1 y permitiendo que la alquilación se calentara hasta 0 °C. El compuesto del título se obtuvo en un 29 % como un aceite viscoso. MS (ESI+) para C22H33NO3Si m/z 388,3 (M+H)+.
Etapa 2. Preparación de (2S,3R)-1-[terc-butil(dimetil)silil]-4-oxo-3-(4-oxobutil)azetidin-2-carboxilato de bencilo

Una mezcla agitada de (2S,3R)-1-[terc-butil(dimetil)silil]-4-oxo-3-pent-4-en-1-ylazetidin-2-carboxilato de bencilo (412 mg, 1,06 mmol) en 1,4-dioxano (12 ml)/agua (3 ml) se trató con N-óxido de N-metilmorfolina (156 mg, 1,33 mmol), seguido de tetróxido de osmio (al 2,5 % en peso en 2-metil-2-propanol, 670 µl, 0,053 mmol) y la mezcla resultante se agitó a temperatura ambiente en una atmósfera de nitrógeno durante 2,5 h, momento en el que se completó la conversión al intermedio de (2S,3R)-1-[terc-butil(dimetil)silil]-3-(4,5-dihidroxipentil)-4-oxoazetidin-2-carboxilato de bencilo [MS (ESI+) para C22H35NO5Si m/z 422,2 (M+H)+] como indicó la HPLC. Se añadió metaperyodato de sodio (284 mg, 1,33 mmol) y la mezcla blanca heterogénea resultante se agitó a temperatura ambiente durante 2,5 h, momento en el que la HPLC indicó que el intermedio se había consumido. La mezcla se inactivó con tiosulfato de sodio acuoso semisaturado (15 ml), se diluyó con agua (15 ml) y se extrajo con EtOAc (2 × 40 ml). La fase orgánica reunida se lavó con salmuera (15 ml), se secó sobre MgSO4, se filtró, se concentró y se secó a presión reducida para proporcionar 418 mg (100 %) del compuesto del título como un aceite viscoso que se utilizó sin purificación posterior. MS (ESI+) para C21H31NO4Si m/z 390,3 (M+H)+.
Etapa 3. Preparación de (2S,3R)-3-[2-(2-amino-1,3-tiazol-5-il)etil]4-oxoazetidin-2-carboxilato de bencilo

Una solución agitada de (2S,3R)-1-[terc-butil(dimetil)silil]-4-oxo-3-(4-oxobutil)azetidin-2-carboxilato de bencilo (414 mg, 1,06 mmol) en acetonitrilo (11 ml) en una atmósfera de nitrógeno se trató con tribromuro de tetrabutilamonio (512 mg, 1,06 mmol) en una porción y la mezcla homogénea de color amarillo intenso resultante se agitó a temperatura ambiente durante 75 min, momento en el que la HPLC indicó que se había consumido el material de partida. La mezcla se diluyó con agua (40 ml) y se extrajo con EtOAc (2 × 40 ml), y la fase orgánica reunida se lavó con agua (2 × 40 ml) y salmuera (20 ml), se secó sobre MgSO4, se filtró y se concentró a presión reducida para proporcionar 302 mg del intermedio bruto de (2S,3R)-3-(3-bromo-4-oxobutil)-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxilato de bencilo [MS (ESI+) para C21H30BrNO4Si m/z 468 (M+H)+] como un aceite viscoso de color canela. Una solución de este intermedio en EtOH (11 ml) se trató con tiourea (89,0 mg, 1,17 mmol) y la mezcla se calentó a reflujo suave durante 2 h, se enfrió hasta la temperatura ambiente, se diluyó con DCM (45 ml), se lavó con NaHCO3 acuoso sat (30 ml), agua (30 ml) y salmuera (20 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. La purificación mediante cromatografía en gel de sílice [40 g, eluyente de MeOH al 2,5-7,5 %/DCM] proporcionó 60 mg (17 %) del compuesto del título como un sólido de color canela. MS (ESI+) para C16H17N3O3S m/z 332,2 (M+H)+. También se aisló el subproducto protegido con TBS, (2S,3R)-3-[2-(2-amino-1,3-tiazol-5-il)etil]-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxilato de bencilo (145 mg, 24 %, un 80 % de pureza). MS (ESI+) para C22H31N3O3SSi m/z 446,2 (M+H)+.
Etapa 4. Preparación de (2S,3R)-3-(2-{2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}-etil)-4-oxoazetidin-2-carboxilato de bencilo

Una solución agitada de (2S,3R)-3-[2-(2-amino-1,3-tiazol-5-il)etil]-4-oxoazetidin-2-carboxilato de bencilo (57,0 mg, 0,172 mmol) en acetonitrilo (5 ml) en una atmósfera de nitrógeno se trató con di-terc-butildicarbonato (48,8 mg, 0,224 mmol) y se agitó a temperatura ambiente durante aproximadamente 2 días, durante los cuales se añadió en una porción adicional de di-terc-butildicarbonato (13 mg, 0,060 mmol). La mezcla se concentró a presión reducida y se purificó mediante cromatografía radial [rotor de gel de sílice de 2000 micras, eluyente de MeOH al 2,5-10 %/DCM] para proporcionar 74 mg (81 %) del compuesto del título como una película vítrea (contaminada con el subproducto de bis-BOC (2S,3R)-3-(2-{2-[bis(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}etil)-4-oxoazetidin-2-carboxilato de bencilo). MS (ESI+) para C21H25N3O5S m/z 432,2 (M+H)+.
Etapa 5. Preparación de (2S,3R)-3-(2-{2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}etil)-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxilato de bencilo

El compuesto del título se preparó a partir de (2S,3R)-3-(2-{2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}etil)-4-oxoazetidin-2-carboxilato y 1,1'-(isocianatometileno)dibenceno siguiendo el procedimiento general de la etapa 4 del ejemplo 3 y utilizando un tiempo de reacción de 5 h en un 82 % como un sólido vítreo. MS (ESI+) para C35H36N4O6S m/z 641,3 (M+H)+.
Etapa 6. Preparación del ácido (2S,3R)-3-(2-{2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il]etil)-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxílico

Una mezcla de (2S,3R)-3-(2-{2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}etil)-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxilato de bencilo (85,0 mg, 0,133 mmol) en MeOH/EtOAc 1:1 (6 ml) en una atmósfera de nitrógeno se trató con paladio al 10 % sobre carbono (35 mg, 0,013 mmol de Pd) y la mezcla se sometió a vacío y se llenó de nitrógeno dos veces y después se agitó bajo un atmósfera de hidrógeno (globo de doble capa) durante aproximadamente 6 h, durante las cuales se añadió más paladio al 10 % sobre carbono (35 mg, 0,013 mmol de Pd) en una porción. El catalizador se retiró por filtración a través de Solka-Floc, aclarando con 1:1 MeOH/EtOAc, y el filtrado se concentró a presión reducida para proporcionar 71 mg (97 %) del compuesto del título como una espuma blanquecina que se utilizó sin purificación posterior. MS (ESI+) para C28H30N4O6S m/z 551,2 (M+H)+.
Etapa 7. Preparación del trifluoroacetato del ácido (2S,3R)-3-[2-(2-amino-1,3-tiazol-5-il)etil]-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxílico

El compuesto del título se preparó a partir del ácido (2S,3R)-3-(2-{2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}etil)-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxílico siguiendo el procedimiento general de la etapa 6 del ejemplo 3 y usando un tiempo de reacción de 5 h en un 47 % como un sólido blanco. RMN de 1H (400 MHz, MeOD) δ 7,26-7,39 (m, 10H), 6,96 (s a., 1H), 6,11 (s a, 1H), 4,31 (s a, 1H), 3,40 (m, 1H), 2,88 (m, 2H), 2,16 (m, 2H); MS (ESI+) para C23H22N4O4S (parental) m/z 451,0 (M+H)+; tiempo de retención en HPLC: 3,25 min (procedimiento A).
Ejemplo 10, esquema 3: Trifluoroacetato del ácido (2S,3R)-3-[2-(2-amino-1,3-tiazol-5-il)etil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico (proporcionado como referencia)

Etapa 1. Preparación de (2S,3R)-3-(2-{2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}etil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de bencilo

El compuesto del título se preparó a partir de (2S,3R)-3-(2-{2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}etil)-4-oxoazetidin-2-carboxilato de bencilo y [(1R)-1-isocianatoetil]benceno siguiendo el procedimiento general de la etapa 4 del ejemplo 3 en un 66 % como un sólido vítreo. MS (ESI+) para C30H34N4O6S m/z 579,3 (M+H)+.
Etapa 2. Preparación del ácido (2S,3R)-3-(2-{2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}etil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico

El compuesto del título se preparó a partir de (2S,3R)-3-(2-{2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}etil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de bencilo siguiendo el procedimiento general de la etapa 6 del ejemplo 9 en un 82 % como un sólido blanco. MS (ESI+) para C23H28N4O6S m/z 489,1 (M+H)+.
Etapa 3. Preparación de trifluoroacetato del ácido (2S,3R)-3-[2-(2-amino-1,3-tiazol-5-il)etil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico

El compuesto del título se preparó a partir del ácido (2S,3R)-3-(2-{2-[(terc-butoxicarbonil)amino] -1,3-tiazol-5-il }etil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico siguiendo el procedimiento general de la etapa 6 del ejemplo 3 y usando un tiempo de reacción de 5 h en un 66 % como un sólido blanco. RMN de 1H (400 MHz, MeOD) δ 7,36 (m, 4H), 7,27 (m, 1H), 6,96 (s a, 1H), 4,98 (q, J = 6,8 Hz, 1H), 4,26 (s a, 1H), 3,35 (m, 1H), 2,89 (t, J = 7,2 Hz, 2H), 2,16 (m, 2H), 1,53 (d, J = 6,8 Hz, 3H); MS (ESI+) para C18H20N4O4S (parental) m/z 389,1 (M+H)+; tiempo de retención en HPLC: 2,71 min (procedimiento A).
Ejemplo 11, esquema 4: Trifluoroacetato del ácido (2S,3R)-3-[(2-amino-1,3-tiazol-5-il)metil]-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxílico (proporcionado como referencia)

Etapa 1. Preparación de (2S,3R)-1-[terc-butil(dimetil)silil]-3-[(2E)-3-cloroprop-2-en-1-il]-4-oxoazetidin-2-carboxilato de bencilo

El compuesto del título se preparó siguiendo los procedimientos generales de las etapas 1-2 del ejemplo 3, pero usando (1E)-1,3-dicloroprop-1-eno en lugar de [4-(bromometil)piridin-2-il](4-metoxibencil)carbamato de terc-butilo en la etapa 1 y permitiendo que la temperatura de reacción se calentara hasta 0 °C. El compuesto del título se obtuvo en un 39 % (mezcla de isómeros E/Z ) como un aceite viscoso. MS (ESI+) para C20H28ClNO3Si m/z 394,2 (M+H)+.
Etapa 2. Preparación de (2S,3R)-3-[(2-amino-1,3-tiazol-5-il)metil]-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxilato de bencilo

Una solución agitada de (2S,3R)-1-[terc-butil(dimetil)silil]-3-[(2E)-3-cloroprop-2-en-1-il]-4-oxoazetidin-2-carboxilato de bencilo (220 mg, 0,558 mmol) en 1,2-dicloroetano seco (2,8 ml) en un vial con tapón de rosca se trató con ácido mcloroperbenzoico (289 mg, 1,68 mmol), se lavó y se secó (pureza >90 %) y 2,6-di-terc-butil-4-metilfenol (7,0 mg, 0,032 mmol), se calentó rápidamente hasta 60°C y se agitó a esta temperatura durante 18 h. La mezcla se enfrió hasta la temperatura ambiente, se diluyó con EtOAc (20 ml), se lavó con NaHCO3 acuoso saturado (2 × 20 ml), tiosulfato de sodio acuoso semisaturado (2 × 15 ml), agua (20 ml) y salmuera (10 ml), se secó sobre MgSO4 y se concentró a presión reducida para proporcionar el intermedio bruto (2S,3R)-1-[terc-butil(dimetil)silil]-3-[(3-cloroxiran-2-il)metil]-4-oxoazetidin-2-carboxilato de bencilo [MS (ESI+) paraC20H28ClNO4Si m/z 410,2 (M+H)+] como un aceite viscoso que se utilizó sin purificación posterior. Una mezcla agitada de este intermedio en 1,2-dimetoxietano seco (5,5 ml) en una atmósfera de nitrógeno se trató con tiourea (53,1 mg, 0,698 mmol), se calentó hasta 60 °C y se agitó a esta temperatura durante 2,5 h, momento en el que la HPLC y la LC-MS indicaron que el intermedio se había consumido. La mezcla se enfrió hasta la temperatura ambiente, se diluyó con EtOAc (30 ml), agua (8 ml) y NaHCO3 acuoso saturado (8 ml) y se separaron las capas. La fase orgánica se lavó con NaHCO3 acuoso saturado (15 ml), agua (15 ml) y salmuera (10 ml), se secó sobre MgSO4 y se concentró a presión reducida para proporcionar 240 mg de producto bruto, que se reunió con 100 mg de producto bruto de una reacción anterior. La purificación por cromatografía radial [rotor de gel de sílice de 2000 micrómetros; eluyente de MeOH al 5-10 %/DCM] proporcionó 172 mg (un 49 % para las reacciones reunidas) del compuesto del título como un aceite viscoso. MS (ESI+) para C21H29N3O3SSi m/z 432,3 (M+H)+.
Etapa 3. Preparación de (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxilato de bencilo

Una solución agitada de (2S,3R)-3-[(2-amino-1,3-tiazol-5-il)metil]-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxilato de bencilo (168 mg, 0,389 mmol) en acetonitrilo (6 ml) en una atmósfera de nitrógeno se trató con di-terc-butildicarbonato(170 mg, 0,778 mmol), y la mezcla resultante se agitó a temperatura ambiente durante aproximadamente 24 h, momento en el que la HPLC indicó que la reacción se había completado. La mezcla se concentró a presión reducida y se purificó por cromatografía radial [rotor de gel de sílice de 2000 micrómetros; eluyente EtOAc al 30-50 %/hexanos] para proporcionar 242 mg (100 %) del compuesto del título como una película aceitosa
(contaminada con el subproducto de bis-BOC (2S,3R)-3-({2-[bis(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxilato de bencilo). MS (ESI+) para C26H37N3O5SSim/z 532,3 (M+H)+. Etapa 4. Preparación de (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}-metil)-4-oxoazetidin-2-carboxilato de bencilo

El compuesto del título se preparó a partir de (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxilato de bencilo siguiendo el procedimiento general de la etapa 3 del ejemplo 3 en 99 % (contaminado con el subproducto de bis-BOC (2S,3R)-3-({2-[bis(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-4-oxoazetidin-2-carboxilato de bencilo). MS (ESI+) para C20H23N3O5S m/z 418,2 (M+H)+.
Etapa 5. Preparación de (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxilato de bencilo

El compuesto del título se preparó a partir de (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-4-oxoazetidin-2-carboxilato de bencilo y 1,1'-(isocianometileno)dibenceno siguiendo el procedimiento general de la etapa 4 del ejemplo 3 usando un tiempo de reacción de 3 h en un 88% como un sólido vítreo. MS (ESI+) para C34H34N4O6S m/z 627,4 (M+H)+.
Etapa 6. Preparación del ácido (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxílico

El compuesto del título se preparó a partir de (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxilato de bencilo siguiendo el procedimiento general de la etapa 6 del ejemplo 9 en rendimiento cuantitativo como un sólido blanco. MS (ESI+) para C27H28N4O6S m/z 537,3 (M+H)+.
Etapa 7. Preparación de trifluoroacetato del ácido (2S, R)-3-[(2-amino-1,3-tiazol-5-il)metil]-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxílico

El compuesto del título se preparó a partir del ácido (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-1-[(difenilmetil)carbamoil]-4-oxoazetidin-2-carboxílico siguiendo el procedimiento general de la etapa 6 del ejemplo 3 y
usando un tiempo de reacción de 3 h en un 53 % como un sólido blanco. RMN de 1H (400 MHz, MeOD) δ 7,49 (d, J = 8,4 Hz, 1H), 7,40-7,27 (m, 10H), 7,14 (s, 1H), 6,12 (m, 1H), 4,34 (d, J = 2,8 Hz, 1H), 3,69 (td, J = 7,2, 2,8 Hz, 1H), 3,25 (m, 2H); MS (ESI+) para C22H20N4O4S (parental) m/z 437,1 (M+H)+; tiempo de retención en HPLC: 3,11 min (procedimiento A).
Ejemplo 12, esquema 4: Trifluoroacetato del ácido (2S,3R)-3-[(2-amino-1,3-tiazol-5-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico (proporcionado como referencia)

Etapa 1. Preparación de (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de bencilo

El compuesto del título se preparó a partir de (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il }-metil)-4-oxoazetidin-2-carboxilato de bencilo y [(1R)-1-isocianatoetil]benceno siguiendo el procedimiento general de la etapa 4 del ejemplo 3, en aproximadamente un 95 % como un sólido vítreo. MS (ESI+) para C29H32N4O6S m/z 565,2 (M+H)+. Etapa 2. Preparación del ácido (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico

El compuesto del título se preparó a partir de (2S,3R)-3-({2-[(terc-butoxicarbonil)amino] -1,3-tiazol-5-il }metil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de bencilo siguiendo el procedimiento general de la etapa 6 del ejemplo 9 en un 75 % como un sólido blanco. MS (ESI+) para C22H26N4O6S m/z 475,2 (M+H)+.
Etapa 3. Preparación de trifluoroacetato del ácido (2S,3R)-3-[(2-amino-1,3-tiazol-5-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico

El compuesto del título se preparó a partir del ácido (2S,3R)-3-({2-[(terc-butoxicarbonil)amino]-1,3-tiazol-5-il}metil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico siguiendo el procedimiento general de la etapa 6 del ejemplo 3 usando un tiempo de reacción de 5 h en un 54 % como un sólido blanco. RMN de 1H (400 MHz, MeOD) δ 7,37 (m, 4H), 7,28 (m, 1H), 7,16 (s, 1H), 4,98 (q, J = 6,8 Hz, 1H), 4,32 (d, J = 2,8 Hz, 1H) 3,65 (td, J = 7,2, 2,8 Hz, 1H), 3,25 (m, 2H), 1,54 (d, J = 7,2 H, 3H); MS (ESI+) para C17H18N4O4S (parental) m/z 375,3 (M+H)+; tiempo de retención en HPLC: 2,58 min (procedimiento A).
Ejemplo 13, esquema 5: Trifluoroacetato de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-4-oxo-N-1-[(1R)-1-feniletillazetidin-1,2-dicarboxamida (proporcionado como referencia)

Etapa 1. Preparación de [4-({(3R,4S)-1-[terc-butil(dimetil)silil]-2-oxo-4-[(2,4,6-trimetoxibencil)carbamoil]azetidin-3-il}metil]piridin-2-il]imidodicarbonato de di-terc-butilo

Una solución de ácido (2S,3R)-3-({2-[bis(tert-butoxicarbonil)amino]piridin-4-il}metil)-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxílico (170 mg, 0,32 mmol) en N,N-dimetilformamida (2,0 ml) se trató con clorhidrato de (2,4,6-trimetoxifenil)metanamina (81,6 mg, 0,349 mmol) y hexafluorofosfato de N,N,N',N'-tetrametil-O-(7-azabenzotriazol-1-il)uronio (145 mg, 0,381 mmol), seguido de la adición gota a gota de N,N-diisopropiletilamina (182 µl, 1,05 mmol) y la mezcla de reacción se agitó a 0 °C. El análisis por HPLC después de 15 min indicó que se había consumido el material de partida. La mezcla de reacción se diluyó con 25 ml de H2O, se extrajo con dos porciones de 25 ml de CH2Cl2 y la fase orgánica reunida se lavó con porciones de 20 ml de H2O y salmuera. La fase orgánica se secó sobre Na2SO4, se filtró y se concentró para obtener un líquido de color ligeramente tostado que se situó a un vacío elevado para eliminar el DMF. El material bruto se purificó mediante cromatografía en columna rápida (60 g de gel de sílice, EtOAc al 15-50 %/CH2Cl2) para obtener el compuesto del título (130 mg, 60 %) como un cristal de color ligeramente amarillo: Tiempo de retención en HPLC: 5,05 min (procedimiento A); MS (ESI+) para C36H54N4O9Si m/z 715,5 (M+H)+.
Etapa 2. Preparación de [4-(1(3R,4S)-2-oxo-4-[(2,4,6-trimetoxibencil)carbamoil]azetidin-3-il}metil]piridin-2-il]imidodicarbonato de di-terc-butilo

Una solución de [4-({(3R,4S)-1-[terc-butil(dimetil)silil]-2-oxo-4-[(2,4,6-trimetoxibencil)carbamoil]azetidin-3-il}metil)piridin-2-il]imidodicarbonato de di-terc-butilo (137 mg, 0,192 mmol) en metanol (3,3 ml) se trató gota a gota con ácido acético (38 µl, 0,67 mmol), seguido de fluoruro de amonio 0,5 M en metanol (0,46 ml, 0,23 mmol) y la mezcla de reacción se agitó a temperatura ambiente. La HPLC después de 1,5 h indicó que la reacción se había completado. La mezcla de reacción se concentró, el residuo se diluyó con 7 ml de tolueno y se concentró. El residuo se recogió en 20 ml de CH2Cl2 y se lavó con porciones de 10 ml de H2O y NaHCO3 saturado. La fase orgánica se secó sobre Na2SO4, se filtró y se concentró para producir el compuesto del título (70 mg, 95 %) como una espuma rígida de color amarillo claro: Tiempo de retención en HPLC, 3,84 min (procedimiento A); MS (ESI+) para C30H40N4O9 m/z 601,3 (M+H)+. Etapa 3. Preparación de [4-({(3R,4S)-2-oxo-1-{[(1R)-1-feniletil]carbamoil}-4-[(2,4,6-trimetoxibencil)carbamoil]azetidin-3-il}metil]piridin-2-il]imidodicarbonato de di-terc-butilo.

Una solución de [4-({(3R,4S)-2-oxo-4-[(2,4,6-trimetoxibencil)carbamoil]azetidin-3-il}-metil)piridin-2-il]imidodicarbonato de di-terc-butilo (94 mg, 0,16 mmol) en cloruro de metileno (2,5 ml) se trató gota a gota con trietilamina (87 µl), seguida de [(1R)-1-isocianatoetil]benceno (29 µl, 0,20 mmol) y la mezcla de reacción se agitó a temperatura ambiente. Tras 4 h, la HPLC indicó que la reacción se había completado. La mezcla de reacción se concentró y el residuo se suspendió
en 5 ml de CH2Cl2 y se concentró hasta obtener un cristal de color ligeramente amarillo. El material bruto se purificó mediante cromatografía en columna rápida (30 g de gel de sílice, EtOAc al 40-70 %/hex.) para obtener el compuesto del título (94 mg, 80 %) como un cristal incoloro: Tiempo de retención en HPLC, 4,88 min (procedimiento A); MS (ESI+) para C39H49N5O10 m/z 748,9 (M+H)+; MS (ESI-) para C39H49N5O10 m/z 746,5 (M-H)-.
Etapa 4. Preparación de trifluoroacetato de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-4-oxo-N-1-[(1R)-1-feniletil]azetidin-1,2-dicarboxamida

Una solución de [4-({(3R,4S)-2-oxo-1-{[(1R)-1-feniletil]carbamoil}-4-[(2,4,6-trimetoxibencil)carbamoil]azetidin-3-il}-metil)piridin-2-il]imidodicarbonato de di-terc-butilo (94 mg, 0,12 mmol) en cloruro de metileno (4,0 ml) se enfrió a 0 °C, se trató gota a gota con ácido trifluoroacético (1,0 ml, 13 mmol) y la mezcla se agitó a 0 °C durante 60 minutos. Tras 24 h de agitación, se descubrió que la reacción se había completado por HPLC. La mezcla de reacción se concentró y el material bruto se purificó mediante cromatografía CombiFlash [cartucho de gel de sílice RediSep C-18 Gold de 30 g, gradiente de disolvente: acetonitrilo al 10 % (TFA al 0,07 %)/agua (TFA al 0,1 %) a acetonitrilo al 100 % (TFA al 0,07 %)] para producir el compuesto del título (31 mg, 51 %) como un sólido blanco tras la liofilización: Tiempo de retención en HPLC: 2,58 min (procedimiento A); MS (ESI+) para C19H21N5O3 m/z 368,1 (M+H)+; RMN de 1H (400 MHz, MeOD) δ 7,96 (1H, s a), 7,77 (1H, d, J = 6,6 Hz), 7,40 (1H, s a), 7,35 (4H, m), 7,26 (1H, m), 7,09 (1H, d, J = 7,8 Hz), 6,96 (1H, s), 6,89 (1H, dd, J = 6,7, 1,6 Hz), 4,96 (1H, m), 4,28 (1H, d, J = 2,8 Hz), 3,67 (1H, dt, J = 8,0, 2,8 Hz), 3,23 (2H, m), 1,52 (3H, d, J = 7,1 Hz).
Ejemplo 14, esquema 6: Trifluoroacetato del ácido 4-{[(2R,3R)-3-[(2-aminopiridin-4-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-il]oxi}benzoico (proporcionado como referencia)

Etapa 1. Preparación de 3-clorobenzoato de (2R,3R)-3-({2-[bis(terc-butoxicarbonil)amino]piridin-4-il}metil)-1-[tercbutil(dimetil)silil]-4-oxoazetidin-2-ilo

Una solución de ácido (2S,3R)-3-({2-[bis(terc-butoxicarbonil)amino]piridin-4-il}metil)-1-[terc-butil(dimetil)silil]-4-oxoazetidin-2-carboxílico (201 mg, 0,375 mmol) en cloruro de metileno (7,0 ml) se enfrió a 0 °C con un baño de hielo y se trató con ácido m-cloroperbenzoico (79 mg, 0,41 mmol), seguido de N,N'-diclohexilcarbodiimida (85 mg, 0,41 mmol) y la mezcla de reacción se agitó a 0 °C durante 25 min, momento en el que la HPLC indicó que se había consumido el material de partida. La mezcla de reacción se filtró, el sólido se lavó con 20 ml de Et2O/CH2Cl21/1 y el filtrado se lavó con 15 ml de H2O y salmuera. La fase orgánica se secó sobre Na2SO4, se filtró y se concentró para producir un vidrio de color amarillo claro. El material bruto se purificó mediante cromatografía en columna rápida (45 g de gel de sílice, EtOAc al 10-30 %/hex.) para obtener el compuesto del título (131 mg, 54 %) como un cristal incoloro: Tiempo de retención en HPLC, 5,95 min (procedimiento A); MS (ESI+) para C32H44ClN3O7Si m/z 646,3/648,3 (M+H)+. Etapa 2. Preparación de 3-clorobenzoato de (2R,3R)-3-({2-[bis(terc-butoxicarbonil)amino]piridin-4-il}metil)-4-oxoazetidin-2-ilo

Una solución de 3-clorobenzoato de (2R,3R)-3-({2-[bis(terc-butoxicarbonil)amino]piridin-4-il}metil)-1-[tercbutil(dimetil)silil]-4-oxoazetidin-2-ilo (130,0 mg, 0,201 mmol) en metanol (3,0 ml, 74 mmol) se trató con ácido acético (40 µl, 0,70 mmol), seguido de fluoruro de amonio 0,5 M en metanol (0,48 ml, 0,24 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 1 h, momento en el que la HPLC indicó que se había consumido el material de partida. La mezcla de reacción se concentró al vacío, el residuo se suspendió en 5 ml de tolueno y la solución se concentró. Este proceso se repitió una vez, el residuo se suspendió en 25 ml de CH2Cl2 y la solución se lavó con 15 ml de H2O y una solución saturada de NaHCO3. La fase orgánica se secó sobre Na2SO4, se filtró y se concentró para obtener el compuesto del título (106 mg, 96 %) como un cristal de color ligeramente amarillo: Tiempo de retención en HPLC: 4,43 min (procedimiento A); MS (ESI+) para C26H30ClN3O7 m/z 532,1/534,2 (M+H)+.
Etapa 3. Preparación de 4-{[(2R,3R)-3-({2-[bis(terc-butoxicarbonil)amino1piridin-4-il}-metil)-4-oxoazetidin-2-il]oxi}benzoato de bencilo

Una disolución de 3-clorobenzoato de (2R,3R)-3-({2-[bis(terc-butoxicarbonil)amino]piridin-4-il}-metil)-4-oxoazetidin-2-ilo (105 mg, 0,197 mmol) en acetonitrilo (13 ml) y agua (1 ml) se trató con p-hidroxibenzoato de bencilo (45 mg, 0,20 mmol), seguido de carbonato de cesio (96 mg, 0,30 mmol) y la mezcla se agitó a temperatura ambiente durante 70 min, momento en el que la HPLC indicó que se había consumido el material de partida. La mezcla de reacción se diluyó con 25 ml de H2O y se extrajo con tres porciones de 25 ml de EtOAc. Se lavó la fase orgánica con 25 ml de salmuera y se secó sobre Na2SO4. La solución se filtró y se concentró para producir un cristal incoloro que se purificó por cromatografía en columna rápida (30 g de gel de sílice, EtOAc al 50-60 %/hex.) para producir el compuesto del título (85 mg, 71 %) como cristal incoloro: Tiempo de retención en HPLC: 4,71 min (procedimiento A); MS (ESI+) para C33H37N3O8 m/z 604,3 (M+H)+; MS (ESI-) para C33H37N3O8 m/z 604,6 (M-H)-.
Etapa 4. Preparación de 4-{[(2R,3R)-3-({2-[bis(terc-butoxicarbonil)amino]piridin-4-il}-metil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-il]oxi}benzoato de bencilo

Una solución de 4-{[(2R,3R)-3-({2-[bis(tert-butoxicarbonil)amino]piridin-4-il}metil)-4-oxoazetidin-2-il]oxi}benzoato de bencilo (82 mg, 0,14 mmol) en cloruro de metileno (2,2 ml) se trató gota a gota con trietilamina (76 µl, 0,54 mmol), seguida de [(1R)-1-isocianatoetil]benceno (25 µl, 0,18 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 4 h, momento en el que la HPLC indicó que se había consumido casi todo el material de partida. Se añadieron otros 10 µl de isocianato y se continuó la reacción durante otra hora. La mezcla de reacción se concentró, el residuo se suspendió en 5 ml de CH2Cl2 y la solución se concentró para proporcionar una espuma rígida blanca. El material bruto se purificó mediante cromatografía en columna rápida (25 g de gel de sílice, EtOAc al 25-45 %/hex.) para obtener el compuesto del título (87 mg, 85 %) como un cristal incoloro: Tiempo de retención en HPLC: 5,54 min (procedimiento A); MS (ESI+) para C42H46N4O m/z 751,4 (M+H)+.
Etapa 5. Preparación del ácido 4-{[(2R,3R)-3-({2-[bis(terc-butoxicarbonil)amino]piridin-4-il}metil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-il]oxi}benzoico

Una solución de 4-{[(2R,3R)-3-({2-[bis(terc-butoxicarbonil)amino]piridin-4-il}metil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-il]oxi}benzoato de bencilo (87 mg, 0,12 mmol) en metanol (3,0 ml) y acetato de etilo (3,0 ml) se trató cuidadosamente con paladio al 10 % sobre carbono (12 mg). El matraz de reacción se sometió al vacío y se rellenó con gas hidrógeno tres veces y la mezcla se agitó bajo una atmósfera de hidrógeno durante 4,5 h, momento en el que la TLC indicó que se había consumido el material de partida. La mezcla de reacción se filtró a través de un lecho corto Solka-Floc y el lecho corto se lavó con 40 ml de EtOAc/MeOH 1/1. El filtrado se concentró para producir el compuesto del título (77 mg, 100 %) como un cristal incoloro: Tiempo de retención en HPLC, 4,56 min (procedimiento A); MS (ESI+) para C35H40N4O9 m/z 661,3 (M+H)+; MS (ESI-) para C35H40N4O9 m/z 559,8 (M-H)-.
Etapa 6. Preparación del trifluoroacetato del ácido 4-{[(2R,3R)-3-[(2-aminopiridin-4-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-il]oxi}benzoico

Una solución de ácido 4-{[(2R,3R)-3-({2-[bis(terc-butoxicarbonil)amino]piridin-4-il}metil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-il]oxi}benzoico (76 mg, 0,12 mmol) en cloruro de metileno (3,3 ml) se enfrió a 0 °C y se trató gota a gota con ácido trifluoroacético (0,66 ml, 8,6 mmol). La mezcla de reacción se agitó a 0 °C durante 1 h, tras lo cual se dejó que el baño de hielo se calentase lentamente. Tras 7 h, la HPLC indicó que se había consumido el material de partida. La mezcla de reacción se concentró al vacío. El residuo se suspendió en 5 ml de CH2Cl2 y se concentró para obtener el producto bruto como un cristal rosa claro. El material se purificó mediante cromatografía CombiFlash [cartucho de gel de sílice RediSep C-18 Gold de 30 g, gradiente de disolvente: acetonitrilo al 10 % (TFA al 0,07 %)/agua (TFA al 0,1 %) a acetonitrilo al 100 % (TFA al 0,07 %)] para producir el compuesto del título (32 mg, 48 %) como un polvo blanco: Tiempo de retención en HPLC: 3,13 min (procedimiento A); MS (ESI+) para C25H24N4O5 m/z 461,1 (M+H)+; RMN de 1H (400 MHz, MeOD) δ 7,94 (2H, m), 7,72 (1H, d, J = 6,6 Hz), 7,34 (4H, m), 7,27 (2H, m), 7,13 (2H, m), 6,91 (1H, s), 6,86 (1H, dd, J = 6,8, 1,5 Hz), 6,08 (1H, d, J = 1,5 Hz), 4,96 (1H, m), 3,79 (1H, dt, J = 8,2, 1,5 Hz), 3,24 (2H, d, J = 8,1 Hz), 1,53 (3H, d, J = 7,1 Hz).
Ejemplo 15, esquema 7: Trifluoroacetato de (2R,3S)-3-[(2-aminopiridin-4-il)metil]-2-(metilsulfonil)-4-oxo-N-[(1R)-1-feniletil]azetidin-1-carboxamida (proporcionado como referencia)

Etapa 1. Preparación de (4-metoxibencil)(4-{[(2R,3S)-2-(metilsulfonil)-4-oxoazetidin-3-il]metil}piridin-2-il)carbamato de terc-butilo

Una solución de 3-clorobenzoato de (2R,3R)-3-({2-[(tert-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-4-oxoazetidin-2-ilo (100 mg, 0,181 mmol) en acetonitrilo (4,0 ml) y agua (1,1 ml) se trató con metanosulfinato de sodio (87 mg, 0,72 mmol). La mezcla se agitó a temperatura ambiente durante 4 h, momento en el que la HPLC indicó que se había consumido el material de partida. La mezcla de reacción se concentró para eliminar la mayor parte del CH3CN y se diluyó con 10 ml de H2O. La fase acuosa se extrajo con tres porciones de 10 ml de EtOAc. La fase orgánica
reunida se secó sobre MgSO4, se filtró y se concentró para obtener un cristal incoloro. El material bruto se purificó mediante TLC preparativa (placa de TLC preparativa de 20 cm x 10 cm x 1,0 mm, EtOAc al 65 %/hex.) para obtener el compuesto del título (52 mg, 60 %) como un cristal incoloro: Tiempo de retención en HPLC, 3,50 min (procedimiento A); MS (ESI+) para C23H29N3O6S m/z 476,4 (M+H)+; MS (ESI-) para C23H29N3O6S m/z 474,3 (M-H)-.
Etapa 2. Preparación de (4-metoxibencil)(4-{[(2R,3S)-2-(metilsulfonil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-3-il]metil}piridin-2-il)carbamato de terc-butilo

Una solución de (4-metoxibencil)(4-{[(2R,3S)-2-(metilsulfonil)-4-oxoazetidin-3-il]metil}piridin-2-il)carbamato de tercbutilo (52,0 mg, 0,109 mmol) en cloruro de metileno (2,4 ml) se trató gota a gota con trietilamina (61 µl, 0,44 mmol), seguida de [(1R)-1-isocianatoetil]benceno (20 µl, 0,14 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 2 h, momento en el que la HPLC indicó que se había consumido el material de partida. La mezcla de reacción se concentró para producir un aceite incoloro. El material se suspendió en 5 ml de CH2Cl2 y se concentró y el producto bruto resultante se purificó mediante TLC preparativa (placa de TLC preparativa de 20 cm x 20 cm x 1,0 mm, EtOAc al 50 %/hex.) para obtener el compuesto del título (63 mg, 91 %) como una espuma rígida incolora: Tiempo de retención en HPLC: 5,02 min (procedimiento A); MS (ESI+) para C32H38N4O7S m/z 623,5 (M+H)+; MS (ESI+) para C32H38N4O7S m/z 621,5 (M+H)+.
Etapa 3. Preparación de trifluoroacetato de (2R,3S)-3-[(2-aminopiridin-4-il)metil]-2-(metilsulfonil)-4-oxo-N-[(1R)-1-feniletil]azetidin-1-carboxamida

Una solución de (4-metoxibencil)(4-{[(2R,3S)-2-(metilsulfonil)-4-oxo-1-{ [(1R)-1-feniletil]carbamoil}azetidin-3-il]metil}piridin-2-il)carbamato de terc-butilo (62 mg, 0,10 mmol) en cloruro de metileno (3 ml) se enfrió a 0 °C y se trató con ácido trifluoroacético (1,0 ml, 13 mmol). La solución se agitó a 0°C durante 30 minutos y luego se dejó calentar hasta temperatura ambiente. La HPLC de la mezcla de reacción después de 23 h indicó que el material de partida se había consumido. La mezcla de reacción bruta se concentró. El residuo se suspendió en 5 ml de CH2Cl2 y se concentró para obtener el producto bruto como un cristal rosa. El material bruto se purificó mediante cromatografía CombiFlash [cartucho de gel de sílice RediSep C-18 Gold de 30 g, gradiente de disolvente: acetonitrilo al 10 % (TFA al 0,07 %)/agua (TFA al 0,1 %) a acetonitrilo al 100 % (TFA al 0,07 %)] para producir el compuesto del título (31 mg, 60 %) como un sólido blanco tras liofilización: Tiempo de retención en HPLC: 2,99 min (procedimiento A); MS (ESI+) para C19H22N4O4S m/z 403,2 (M+H)+; RMN de 1H (400 MHz, MeOD) δ 7,77 (1H, d, J = 6,8 Hz), 7,35 (4H, d, J = 4,3 Hz), 7,27 (1H, m), 7,19 (1H, d, J = 7,6 Hz), 6,98 (1H, s), 6,91 (1H, dd, J = 6,8, 1,5 Hz), 5,30 (1H, d, J = 2,8 Hz), 4,97 (1H, m), 4,02 (1H, m), 3,34 (1H, m), 3,21 (1H, m), 3,17 (3H, s), 1,54 (3H, d, J = 7,1 Hz).
Ejemplo 16, esquema 8: Trifluoroacetato de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-2-[(metoxiimino)metil]-4-oxo-N-[(1R)-1-feniletil]azetidin-1-carboxamida (proporcionado como referencia)

Etapa 1. Preparación de (4S)-1-[terc-butil(difenil)silil]-4-(hidroximetil)azetidin-2-ona

Una solución de (2S)-1-[terc-butil(difenil)silil]-4-oxoazetidin-2-carboxilato de bencilo (2,20 g, 4,96 mmol), en metanol (20 ml) se enfrió hasta 0 °C (agua helada) y se trató con tetrahidroborato de sodio (0,55 g, 15 mmol) en una porción. La mezcla de reacción se agitó y dejó enfriar lentamente hasta la temperatura ambiente. Tras 18 h, la TLC indicó que se había consumido el material de partida. La mezcla de reacción se inactivó con 10 ml de solución saturada de NaHCO3 y la mezcla se concentró para eliminar el disolvente. La suspensión lechosa resultante se diluyó con 20 ml de H2O y se extrajo con tres porciones de 25 ml de MTBE. Se lavó la fase orgánica con 20 ml de salmuera y se secó sobre MgSO4. La fase orgánica se filtró y se concentró para obtener un aceite viscoso incoloro. El aceite amarillo se purificó por cromatografía en columna rápida (EtOAc al 15-50 % en hexanos) para obtener S1 (0,82 g, 48 %) como un sólido blanco. Tiempo de retención en HPLC: 4,47 min (procedimiento A).
Etapa 2. Preparación de (2S)-1-[terc-butil(difenil)silil]-4-oxoazetidin-2-carbaldehído

Una solución de (4S)-1-[terc-butil(difenil)silil]-4-(hidroximetil)azetidin-2-ona (530 mg, 1,56 mmol) en cloruro de metileno (39 ml) se trató con peryodinano de Dess-Martin (1,32 g, 3,12 mmol) y la mezcla de reacción se agitó a temperatura ambiente produciendo una solución transparente. Después de 1,5 h a temperatura ambiente, la mezcla de reacción se diluyó con 40 ml de Et2O y se concentró al vacío para obtener un aceite transparente, que se suspendió en 50 ml de Et2O y se añadió de una mezcla 1/1 de 40 ml de Na2S2O3 acuoso al 10 % y 40 ml de NaHCO3 saturado. La mezcla se agitó enérgicamente hasta que se disolvieron todos los sólidos. La capa acuosa separada se extrajo con dos porciones de 20 ml de Et2O y la fase orgánica reunida se secó sobre Na2SO4, se filtró y se concentró para obtener el compuesto del título (520 mg, 99 %) como un aceite viscoso incoloro que se utilizó tal cual en la etapa siguiente: RMN de 1H (300 MHz, CDCl 3 ) δ 9,12 (1H, d, J = 4,1 Hz), 7,68 (2H, m), 7,59 (2H, m), 7,45 (6 H, m), 3,76 (1H, m), 3,39 (1H, dd, J = 15,8, 6,3 Hz), 3,02 (1H, dd, J = 15,8, 3,1 Hz), 1,25 (9 H, s).
Etapa 3. Preparación de la O-metiloxima del (2S)-1-[terc-butil(difenil)silil]-4-oxoazetidin-2-carbaldehído

Se suspendió (2S)-1-[terc-butil(difenil)silil]-4-oxoazetidin-2-carbaldehído (520 mg, 1,54 mmol) en etanol (14 ml) y se trató con piridina (177 µl, 2,19 mmol), seguida de clorhidrato de metoxiamina (154 mg, 1,87 mmol) en una porción. La solución se agitó a temperatura ambiente durante 19 h, momento en el que la TLC (AE al 30 %/hex.) indicó que la reacción se había completado. La mezcla de reacción se concentró para eliminar la mayor parte del disolvente y el líquido incoloro resultante se diluyó con 20 ml de H2O y se extrajo con tres porciones de 20 ml de MTBE. La capa orgánica se lavó con 20 ml de H2O, se secó sobre Na2SO4, se filtró y se concentró para producir un aceite viscoso incoloro. El material bruto se purificó mediante cromatografía en columna rápida (50 g, EtOAc al 10-20 %/hex.) para producir el compuesto del título (469 mg, 82 %) como un aceite viscoso incoloro que era una mezcla de isómeros geométricos E/Z: Tiempo de retención en HPLC: 4,93 min (procedimiento A); MS (ESI+) para C21H26N2O2Si m/z 367,4 (M+H)+, m/z 389,3 (M+Na)+.
Etapa 4. Preparación de [4-({(2S,3R)-1-[terc-butil(difenil)silil]-2-[(metoxiimino)metil]-4-oxoazetidin-3-il]metil]piridin-2-il](4-metoxibencil)carbamato de terc-butilo

Una disolución de la O-metiloxima del (2S)-1-[terc-butil(difenil)silil]-4-oxoazetidin-2-carbaldehído (469 mg, 1,28 mmol) en tetrahidrofurano (9,6 ml) se enfrió hasta -78 °C y se trató gota a gota con una disolución 1,2 M de diisopropilamida de litio en hexanos/THF/etilbenceno (1,2 ml, 1,4 mmol). La mezcla de reacción de color amarillo pálido se agitó durante 15 min y se transfirió por cánula a una solución preenfriada (-78 °C) de [4-(bromometil)piridin-2-il](4
metoxibencil)carbamato de terc-butilo (570 mg, 1,4 mmol) en tetrahidrofurano (9,6 ml) gota a gota durante 15 min para obtener una solución de color marrón amarillento. La mezcla de reacción se agitó durante 90 min a -78 °C, momento en el que la HPLC indicó que se había consumido el material de partida. La reacción se apagó añadiendo 10 ml de NH4Cl acuoso saturado. Se retiró el baño de enfriamiento y la mezcla se agitó durante 5 min. La mezcla de reacción se diluyó con 20 ml de H2O y se extrajo con dos porciones de 40 ml de EtOAc. La fase orgánica reunida se lavó con porciones de 20 ml de H2O y salmuera y se secó sobre MgSO4. La solución se filtró y se concentró para obtener un aceite viscoso de color granate. El material bruto se purificó mediante cromatografía en columna rápida (80 g de gel de sílice, EtOAc al 10-30 %/hex.) para producir el compuesto del título (553 mg, 62 %) como una espuma rígida de color amarillo claro que era una mezcla de isómeros geométricos E/Z: Tiempo de retención en HPLC: 5,77/5,82 min (procedimiento A); MS (ESI+) para C40H48N4O5 m/z 693,6 (M+H)+.
Etapa 5. Preparación de (4-metoxibencil)[4-({(2S,3R)-2-[(metoxiimino)metil]-4-oxoazetidin-3-il}metil)piridin-2-il]carbamato de terc-butilo

Una solución de [4-({(2S,3R)-1-[terc-butil(difenil)silil]-2-[(metoxiimino)metil]-4-oxoazetidin-3-il}metil)piridin-2-il] (4-metoxibencil)carbamato de terc-butilo (553 mg, 0,678 mmol) en metanol (10 ml) se trató gota a gota con ácido acético (130 µl, 2,4 mmol), seguido de fluoruro de amonio 0,5 M en metanol (1,6 ml, 0,81 mmol). La solución se agitó a temperatura ambiente durante 1 h, momento en el que la HPLC indicó que se había consumido el material de partida. La mezcla de reacción se concentró para eliminar el MeOH y el aceite resultante se recogió en 50 ml de CH2Cl2. La fase orgánica se lavó con porciones de 25 ml de NaHCO3 saturado y H2O, se secó sobre Na2SO4, se filtró y se concentró hasta obtener un aceite amarillo claro. El material bruto se purificó mediante cromatografía en columna rápida (60 g de gel de sílice, EtOAc al 40-80 %/hex.) para obtener el compuesto del título (250 mg, 81 %) como un cristal ligeramente amarillo que era una mezcla de isómeros E/Z: Tiempo de retención en HPLC: 3,59/3,66 min (procedimiento A); MS (ESI+) para C24H30N4O5 m/z 455,3 (M+H)+; MS (ESI-) para C24H30N4O5 m/z 453,3 (M-H)-.
Etapa 6. Preparación de (4-metoxibencil)(4-{[(2S,3R)-2-[(metoxiimino)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-3-il]metil}piridin-2-il)carbamato de terc-butilo

Una solución de (4-metoxibencil)[4-({(2S,3R)-2-[(metoxiimino)metil]-4-oxoazetidin-3-il}metil)piridin-2-il]carbamato de terc-butilo (131 mg, 0,288 mmol) en cloruro de metileno (4,6 ml) se trató gota a gota con trietilamina (160 µl, 1,2 mmol), seguida de [(1R)-1-isocianatoetil]benceno (53 µl, 0,37 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 21 h, momento en el que la HPLC indicó que se había consumido el material de partida. La reacción se concentró, el residuo se suspendió en 5 ml de CH2Cl2 y se concentró hasta un residuo de color canela. El material se suspendió en Et2O/hex.1/1 y la suspensión se filtró a través de una frita fina. Los sólidos se lavaron con más Et2O/hex.
1/1 y el filtrado se concentró hasta obtener una espuma rígida de color canela. El material bruto se purificó mediante cromatografía en columna rápida (25 g de gel de sílice, EtOAc al 25-40 %/hex.) para obtener el compuesto del título (145 mg, 83 %) como un cristal incoloro que era una mezcla de isómeros E/Z: Tiempo de retención en HPLC: 4,84 min (procedimiento A); MS (ESI+) para C33H39N5O6 m/z 602,5 (M+H)+; MS (ESI-) para C33H39N5O6 m/z 600,4 (M-H)-.
Etapa 7. Preparación de trifluoroacetato de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-2-[(metoxiimino)metil]-4-oxo-N-[(1R)-1-feniletil]azetidin-1-carboxamida

Una disolución de (4-metoxibencil)(4-{[(2S,3R)-2-[(metoxiimino)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-3-il]metil}piridin-2-il)carbamato de terc-butilo (145 mg, 0241 mmol) en cloruro de metileno (4,5 ml) se enfrió a 0 °C y se trató con ácido trifluoroacético (1,5 ml, 20,0 mmol). La solución se agitó a 0°C durante 30 minutos y luego se dejó calentar hasta temperatura ambiente. La mezcla de reacción se agitó durante 24 h, momento en el que la HPLC indicó que se había consumido el material de partida. La mezcla de reacción se concentró. El residuo se recogió en 10 ml de CH2Cl2 y se concentró para proporcionar un cristal de color canela. El material bruto se purificó mediante cromatografía CombiFlash [cartucho de gel de sílice RediSep C-18 Gold de 30 g, gradiente de disolvente: acetonitrilo al 10 % (TFA al 0,07 %)/agua (TFA al 0,1 %) a acetonitrilo al 100 % (TFA al 0,07 %)] para producir el compuesto del título (71 mg, 59 %) como un sólido blanco tras liofilización: (los datos corresponden a la mezcla E/Z). Tiempo de retención HPLC: 2,88/2,93 min (procedimiento A); MS (ESI+) para C20H23N5O3 m/z 382,3 (M+H)+; RMN de 1H (400 MHz, MeOD) δ 7,78 (1H, m), 7,48 (0,6H, d, J = 6,6 Hz), 7,34 (4H, m), 7,26 (1H, m), 6,96 (0,4H, d, J = 4,8 Hz), 6,94 (1H, s), 6,88 (1H, m), 4,94 (1H, m), 4,73 (0,4H, dd, J = 4,9, 3,2 Hz), 4,43 (0,6H, dd, J = 6,4, 2,9 Hz), 3,79 (1,2 H, s), 3,78 (1,8 H, s), 3,73 (0,6H, dt, J = 8,0, 3,0 Hz), 3,58 (0,4H, dt, J = 7,6, 3,2 Hz), 3,21 (2H, m), 1,51 (3H, d, J = 7,1 Hz)
Ejemplo 17, esquema 9: (2S,3R)-3-[(2-{[(hexiloxi)carbonil]amino}piridin-4-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de etilo (proporcionado como referencia)

Etapa 1. Preparación de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-4-oxo-1-{[(1R)-1-feniletil]-2-carboxilato de etilo

Una solución de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]-piridin-4-il}metil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de etilo (474 mg, 0,715 mmol) en cloruro de metileno (9,0 ml) se enfrió a 0 °C y se trató gota a gota con ácido trifluoroacético (3,0 ml). La mezcla de reacción agitó a 0 °C durante 60 min y después se calentó hasta la temperatura ambiente. Tras 24 h, la HPLC indicó que se había consumido el material de partida. El aceite púrpura se suspendió en 15 ml de CH2Cl2 y se concentró. El residuo se disolvió en 11 ml de CH2Cl2, se trató con 10 ml de NaHCO3 saturado y se agitó a temperatura ambiente hasta que cesó el burbujeo. La mezcla se vertió en un embudo de decantación y se separaron las fases. La fase acuosa se lavó con 20 ml de CH2Cl2 y las fases orgánicas reunidas se secaron sobre Na2SO4. La solución se filtró y se concentró para obtener una espuma rígida. El material bruto se purificó mediante cromatografía en columna rápida (35 g de gel de sílice, MeOH al 6-8 %/CH2Cl2) para producir el compuesto del título (254 mg, 89 %) como una espuma rígida incolora: Tiempo de retención en HPLC: 3,01 min (procedimiento A); MS (ESI+) para C21H24N4O4 m/z 397,3 (M+H)+; MS (ESI-) para C21H24N4O4 m/z 395,3 (M-H)-.
Etapa 2. Preparación de (2S,3R)-3-[(2-{(hexiloxi)carbonil)amino}piridin-4-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de etilo

Una solución agitada de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de etilo (50,0 mg, 0,126 mmol) en cloruro de metileno seco (0,50 ml) en una atmósfera de nitrógeno se enfrió en un baño de agua helada y se trató con piridina (22 µl, 0,28 mmol), seguida de cloroformiato de hexilo (23 µl, 0,14 mmol) gota a gota. La mezcla de reacción se agitó a 0-5 °C durante 1 h y, a continuación, se calentó lentamente (durante 2,5 h) hasta alcanzar la temperatura ambiente. La mezcla de reacción se diluyó con 10 ml de H2O y se extrajo con dos porciones de 15 ml de CH2Cl2. La fase orgánica se lavó con porciones de 15 ml de H2O y salmuera y se secó sobre Na2SO4. La solución se concentró hasta obtener un aceite viscoso casi incoloro. El material bruto se purificó mediante cromatografía en columna rápida (25 g de gel de sílice, EtOAc al 30-50 %/hex.) para obtener el compuesto del título (43 mg, 66 %) como un cristal incoloro: Tiempo de retención en HPLC: 4,27 min (procedimiento A); MS (ESI+)
para C28H36N4O6 m/z 525,3 (M+H)+; MS (ESI-) para C28H36N4O6 m/z 523,4 (M-H)-; RMN de 1H (400 MHz, CDCl3) δ 8,21 (1H, d, J = 5,1 Hz), 7,93 (1H, s), 7,73 (1H, s a), 7,35 (4H, m), 7,28 (1H, m), 6,89 (1H, dd, J = 5,1, 1,5 Hz), 6,67 (1H, d, J = 8,1 Hz), 5,03 (1H, m), 4,16 (5 H, m), 3,54 (1H, ddd, J = 8,3, 6,8, 2,7 Hz), 3,20 (1H, m), 3,09 (1H, m), 1,70 (2H, m), 1,55 (3H, d, J = 7,1 Hz), 1,37 (6 H, m), 1,16 (3H, t, J = 7,2 Hz), 0,91 (3H, m).
Ejemplo 18, esquema 9: Ácido (2S,3R)-3-[(2-{[(hexiloxi)carbonil]amino}piridin-4-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico (proporcionado como referencia)

Etapa 1. Preparación de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato

Una solución de (2S,3R)-3-({2-[(terc-butoxicarbonil)(4-metoxibencil)amino]piridin-4-il}metil)-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de bencilo (344 mg, 0,507 mmol) en cloruro de metileno (6,4 ml) se enfrió a 0 °C y se trató gota a gota con ácido trifluoroacético (2,1 ml, 28 mmol). La mezcla de reacción agitó a 0 °C durante 60 min y después se calentó hasta la temperatura ambiente. Tras 24 h, la HPLC indicó que la reacción se había completado. El aceite púrpura se suspendió en 15 ml de CH2Cl2 y se concentró. El residuo se disolvió en 20 ml de CH2Cl2, se trató con 10 ml de NaHCO3 saturado y se agitó a temperatura ambiente hasta que cesó el burbujeo. La mezcla se vertió en un embudo de decantación y se separaron las fases. La fase acuosa se lavó con 10 ml de CH2Cl2 y las fases orgánicas reunidas se secaron sobre Na2SO4. La solución se filtró y se concentró para producir una espuma rígida de color amarillo claro. El material bruto se purificó mediante cromatografía en columna rápida (30 g de gel de sílice (MeOH al 4-6 %/CH2Cl2) para producir el compuesto del título (206 mg, 89 %) como una espuma rígida incolora: Tiempo de retención en HPLC: 3,48 min (procedimiento A); MS (ESI+) para C26H26N4O4 m/z 459,3 (M+H)+; MS (ESI-) para C26H26N4O4 m/z 457,2 (M-H)-.
Etapa 2. Preparación de (2S,3R)-3-[(2-{[(hexiloxi)carbonil]amino}piridin-4-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de bencilo

Una solución agitada de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de bencilo (206 mg, 0,449 mmol) en cloruro de metileno seco (3,0 ml) en una atmósfera de nitrógeno se enfrió en un baño de agua helada y se trató con piridina (95 µl, 1,2 mmol), seguida de cloroformiato de hexilo (110 µl, 0,67 mmol) gota a gota. La mezcla de reacción se agitó a 0-5 °C durante 1 h, momento en el que se dejó que el baño de enfriamiento se calentara lentamente hasta la temperatura ambiente durante 2,5 h. En ese momento, la HPLC indicó que se había consumido el material de partida. La mezcla de reacción se diluyó con 20 ml de H2O y se extrajo con dos porciones de 20 ml de CH2Cl2. Se lavó la fase orgánica con 15 ml de H2O y se secó sobre Na2SO4. La solución se concentró hasta obtener un cristal de color amarillo claro. El material bruto se purificó mediante cromatografía en columna rápida (30 g de gel de sílice, EtOAc al 30-50 %/hex.) para obtener el compuesto del título (242 mg, 92 %) como un cristal incoloro: Tiempo de retención en HPLC: 4,51 min (procedimiento A); MS (ESI+) para C33H38N4O6 m/z 587,4 (M+H)+.
Etapa 3. Preparación del ácido (2S,3R)-3-{(2-{[(hexiloxi)carbonil]amino}piridin-4-il)metil]-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico

Una solución de (2S,3R)-3-[(2-{[(hexiloxi)carbonil]amino}piridin-4-il)metil]-4-oxo-1-{(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de bencilo (242 mg, 0,412 mmol) en metanol (4,0 ml) y acetato de etilo (4,0 ml) se trató cuidadosamente con catalizador Pd al 10 %-C (44 mg). El matraz de reacción se sometió al vacío y se rellenó con gas hidrógeno tres veces y la reacción se agitó bajo una atmósfera de hidrógeno durante 1,5 h, momento en el que la HPLC indicó que se había consumido el material de partida. La mezcla de reacción se filtró a través de un lecho corto Solka-Floc y el lecho corto se lavó con 40 ml de EtOAc/MeOH 1/1. El filtrado se concentró para obtener el compuesto del título (197 mg, 96 %) como sólido incoloro: Tiempo de retención en HPLC: 3,77 min (procedimiento A); MS (ESI+) para C26H32N4O6 m/z 497,3 (M+H)+; MS (ESI-) para C26H32N4O6 m/z 495,2 (M-H)-; RMN DE 1H (400 MHz, CDCl3) δ 9,34 (1H, s a), 8,04 (2H, s), 7,33 (4H, s), 7,28 (1H, m), 6,95 (1H, d, J = 4,5 Hz), 6,78 (1H, d, J = 7,8 Hz), 5,05 (1H, m), 4,31 (1H, s a), 4,15 (2H, t, J = 6,7 Hz), 3,63 (1H, ddd, J = 8,8, 6,1, 2,8 Hz), 3,22 (1H, m), 3,09 (1H, m), 1,67 (2H, m), 1,56 (3H, d, J = 6,8 Hz), 1,33 (6 H, m), 0,88 (3H, m).
Ejemplo 19, esquema 9: Trifluoroacetato de (2S,3R)-3-{[2-(L-alanilamino)piridin-4-il]metil}-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-4-oxoazetidin-2-carboxilato de etilo

A una solución de (2S,3R)-3-[(2-aminopiridin-4-il)metil]-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-4-oxoazetidin-2-carboxilato de bencilo (150 mg, 0,28 mmol) y N-(terc-butoxicarbonil)-L-alanina (79 mg, 0,42 mmol) en N,N-dimetilformamida (1,7 ml) se le añadió N,N-diisopropiletilamina (0,194 ml, 1,11 mmol), seguida de hexafluorofosfato de N,N,N',N'-tetrametil-O-(7-azabenzotriazol-1-il)uronio (159 mg, 0,42 mmol). Tras 72h, la reacción se diluyó con acetato de etilo y se lavó con NaHCO3 saturado, salmuera, se secó con sulfato de sodio anhidro, se filtró y se concentró. El residuo se purificó parcialmente mediante cromatografía en columna rápida utilizando hexanos/acetato de etilo (al 30-40 %) como eluyente para obtener (2S,3R)-3-[(2-{[N-(tert-butoxicarbonil)-L-alanil]amino}piridin-4-il)metil]-1-{[(1R)-1-(2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-4-oxoazetidin-2-carboxilato de bencilo (80 mg, aproximadamente un 80 % de pureza, 40 %) como sólido de color canela que se utilizó sin purificación posterior. MS (ESI+) para C35H37F2N5O9 m/z 710,2 (M+H)+.
A un matraz que contenía Pd/C (al 10 %, 13 mg) se le añadió una solución del intermedio anterior (130 mg, una combinación de dos lotes de pureza similar) en etanol (5 ml). La mezcla se agitó en 1 atmósfera de H2 durante 5 h. Se añadió más Pd/C adicional (al 10 %, 5 mg) en una atmósfera de nitrógeno y la mezcla se agitó 3 h más en 1 atmósfera de H2. La mezcla se filtró a través de Celite y se concentró a presión reducida para obtener ácido (2S,3R)-3-[(2-{[N-(tert-butoxicarbonil)-L-alanil]amino}piridin-4-il)metil]-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-4-oxoazetidin-2-carboxílico (100 mg) como un sólido blanquecino que se utilizó sin purificación posterior. MS (ESI+) para C28H31F2N5O9 m/z 620,2 (M+H)+.
A una solución de ácido (2S,3R)-3-[(2-{[[N-(tert-butoxicarbonil)-L-alanil]amino}piridin-4-il)metil]-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-4-oxoazetidin-2-carboxílico (100 mg, 0,16 mmol) en CH2Cl2 (1 ml) se le añadió etanol (0,28 ml, 4,84 mmol), seguido de clorhidrato de N-(3-dimetilaminopropil)-N'-etilcarbodiimida (0,120 g, 0,62 mmol) y DMAP (1 mg). Tras 16 h, la mezcla se diluyó con acetato de etilo y se lavó con HCl acuoso 0,1, NaHCO3 saturado, salmuera, se secó con sulfato de sodio anhidro, se filtró y se concentró. El residuo se disolvió en CH2Cl2 (4 ml) y se enfrió hasta 0-5 °C. A la solución enfriada se le añadió ácido trifluoroacético (2 ml). La reacción se agitó a 0-5 °C durante 30 min y después a temperatura ambiente durante 1 h. Los volátiles se eliminaron a presión reducida y el residuo se purificó mediante cromatografía CombiFlash [cartucho de gel de sílice RediSep C-18 Gold de 30 g, gradiente de disolvente: acetonitrilo al 10 % (TFA al 0,07 %)/agua (TFA al 0,1 %) a acetonitrilo al 100 % (TFA al 0,07 %)] y se liofilizó para obtener el compuesto del título (24 mg, 22 %) como un sólido blanco. RMN de 1H (CD3OD) δ 1,09 (t, J = 7 Hz, 3H), 1,54 (d, J = 7 Hz, 3H), 1,62 (d, J = 7 Hz, 3H), 3,15-3,29 (m, 2H), 3,69-3,74 (m, 1H), 4,05-4,18 (m, 3H), 4,28 (d, J = 3 Hz, 1H), 4,95-4,98 (m, 1H), 7,14-7,24 (m solapado, 4H), 8,07 (s a, 1H), 8,28 (d, J = 5 Hz, 1H); MS (ESI+) para C25H27F2N5O7 m/z 548,1 (M+H)+; tiempo de retención en HPLC: 3,58 min (procedimiento C).
Ejemplo 20, esquema 10: (2S,3R)-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-3-{[2-({[(isobutiriloxi)metoxi]carbonil}amino)piridin-4-il]metil}-4-oxoazetidin-2-carboxilato de etilo (proporcionado como referencia)

Etapa 1. Preparación del éster 4-nitrofenílico éster clorometílico del ácido carbónico

A una solución de 4-nitrofenol (3,81 g, 0,027 mol) en THF (50 ml) se LE añadió cloroformiato de clorometilo (4,00 g, 0,030 mol), seguido de N,N-diisopropiletilamina (5,29 ml, 0,030 mol). La mezcla se agitó durante 2 h, se diluyó con acetato de etilo y se lavó con NaHCO3 saturado, salmuera, se secó con MgSO4 anhidro, se filtró y se concentró para obtener el compuesto del título (6,10 g, 96 %) como un sólido amarillo que se utilizó sin purificación posterior. RMN de 1H (CDCl3) δ 5,88 (s, 2H), 7,44 (d, J = 9 Hz, 2H), 8,32 (d, J = 9 Hz, 2H).
Etapa 2. Preparación del éster 4-nitrofenílico éster yodometílico del ácido carbónico

A una solución del éster 4-nitrofenílico éster clorometílico del ácido carbónico (3,00 g, 0,013 mol) en acetona (60 ml) se le añadió yoduro de sodio (5,82 g, 0,039 mol) y tamices moleculares de 4 Å (3,00 g). La mezcla se calentó a 40 °C hasta que se consideró completa mediante el examen de una parte alícuota de la mezcla de reacción por RMN de 1H (aproximadamente 6 h). La mezcla se enfrió hasta la temperatura ambiente y se filtró a través de un lecho corto de Celite. Los volátiles se eliminaron a presión reducida y el residuo se disolvió en CH2Cl2, se lavó con NaHCO3 saturado, agua, salmuera, se secó con sulfato de sodio anhidro, se filtró y se concentró para obtener el compuesto del título (3,82 g, 91 %) como un sólido que se utilizó sin purificación adicional. RMN de 1H (CDCl3) δ 6,08 (s, 2 H), 7,44 (d, J = 9 Hz, 2 H), 8,32 (d, J = 9 Hz, 2 H).
Etapa 3. Preparación de 2-metilpropionato de plata
A una solución de ácido 2-metilpropiónico (2,65 g, 0,030 mol) en acetonitrilo (100 ml) se le añadió óxido de plata(I) (4,12 g, 0,018 mol). El matraz se protegió de la luz y se calentó a 70 °C durante 90 min. La mezcla se enfrió a temperatura ambiente y se filtró a través de un lecho corto de Celite. Los volátiles se eliminaron al vacío para obtener el compuesto del título (5,65 g, 96 %) como un sólido de color canela que se utilizó sin purificación ni caracterización posteriores.
Etapa 4. Preparación del éster 4-nitrofenoxicarboniloximetílico del ácido 2-metilpropiónico
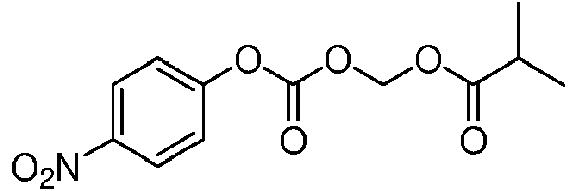
A una solución del éster 4-nitrofenílico éster yodometílico del ácido carbónico (2,10 g, 6,50 mmol) en tolueno (30 ml) se le añadió 2-metilpropanoato de plata (2,53 g, 13,0 mmol). La mezcla se calentó a 55 °C hasta que se consideró completa mediante el examen de una parte alícuota de la mezcla de reacción por RMN de 1H (aproximadamente 5 h). La mezcla se enfrió hasta la temperatura ambiente, se filtró a través de un lecho corto de Celite y se lavó con K2CO3 acuoso al 10 %, agua, salmuera, se secó con sulfato de sodio anhidro, se filtró y se concentró. El residuo se purificó por cromatografía en columna rápida utilizando hexanos y acetato de etilo (al 5 %) como eluyente para obtener el compuesto del título (1,51 g, 82 %) en forma de aceite: RMN de 1H (CDCl3) δ 1,25 (d, J = 7 Hz, 6H), 2,68 (heptete, J = 7 Hz, 1H), 5,91 (s, 2H), 7,42 (d, J = 9 Hz, 2H), 8,31 (d, J = 9 Hz, 2H).
Etapa 5. Preparación del ácido (2S,3R)-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-3-{[2-({[isobutiriloxi)metoxi]carbonil}amino)piridin-4-ilmetil-4-oxoazetidin-2-carboxílico

A una mezcla agitada de trifluoroacetato del ácido (2S,3R)-3-[(2-aminopiridin-4-il)metil]-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-4-oxoazetidin-2-carboxílico (0,200 g, 0,36 mmol) en CH2Cl2 (0,5 ml) se le añadió clorotrimetilsilano (0,180 ml, 1,42 mmol) y N,N-diisopropiletilamina (0,217 ml, 1,24 mmol). La mezcla se calentó a 40 °C durante 1 hora y después se enfrió hasta la temperatura ambiente. Se añadió una solución del éster 4-nitrofenoxicarboniloximetílico del ácido 2-metilpropiónico (0,201 g, 0,72 mmol) en CH2Cl2 (0,36 ml), seguida de N,N-diisopropiletilamina (0,124 ml, 0,72 mmol). La mezcla se calentó a 40 °C durante 2 horas y después se enfrió hasta la temperatura ambiente. La reacción se inactivó añadiendo HCl acuoso 0,1 (pH aproximadamente 3), se agitó durante 15 minutos y se extrajo con acetato de etilo. Las capas orgánicas reunidas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó por cromatografía en columna rápida utilizando hexanos y acetato de etilo (al 20 %), seguido de CH2Cl2/metanol (al 1-2 %) como eluyente para obtener el compuesto del título (0,080 g, 38 %) como un sólido blanco vidrioso: RMN de 1H (CDCl3) δ 1,19 (d, J = 6 Hz, 6H), 1,56 (d, J = 8 Hz, 3H), 2,61 (heptete, J = 7 Hz, 1H), 3,11-3,31 (m, 2H), 3,68-3,74 (m, 1H), 4,32 (d, J = 2 Hz, 1H), 5,01 (pentete, J = 7 Hz, 1H), 5,83-5,88 (m, 2H), 6,75 (d, J = 8 Hz, 1H), 7,04-7,08 (m, 4H), 8,06-8,12 (m solapado, 2H), 9,79 (s muy a, 1H); MS (ESI+) para C26H26F2N4O10 m/z 593,2 (M+H)+; tiempo de retención en HPLC: 3,98 min (procedimiento C).
Etapa 6. Preparación de (2S,3R)-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-3{[2-({(isobutiriloxi)metoxi]carbonil}amino)piridin-4-il]metil}-4-oxoazetidin-2-carboxilato de etilo

A una solución de ácido (2S,3R)-1-{[(1R)-1-(2,2-difluoro-1,3-benzodioxol-5-il)etil]carbamoil}-3-{[2-({[(isobutiriloxi)metoxi]carbonil}amino)piridin-4-il]metil}-4-oxoazetidin-2-carboxílico (60 mg, 0,10 mmol) en CH2Cl2 (1 ml) se le añadió etanol (0,118 ml, 2,02 mmol), seguido de clorhidrato de N-(3-dimetilaminopropil)-N'-etilcarbodiimida (23 mg, 0,12 mmol) y 4-dimetilaminopiridina (0,6 mg, 0,006 mmol). La mezcla se agitó a temperatura ambiente durante la noche, se diluyó con acetato de etilo y se lavó con HCl acuoso 0,25 N, agua, salmuera, se secó con sulfato de sodio anhidro, se filtró y se concentró. El residuo se purificó por cromatografía en columna rápida utilizando hexanos/acetato de etilo (al 20-40 %) como eluyente para obtener el compuesto del título (35 mg, 60 %) como un sólido blanco: RMN de 1H (CDCl3) δ 1,14-1,22 (tripletes solapados, 9H), 1,54 (d, J = 7 Hz, 3H), 2,63 (heptete, J = 7 Hz, 1H), 3,10-3,27 (m, 2H), 3,54-3,60 (m, 1H), 4,13-4,24 (m solapado, 3H), 4,99 (pentete, J = 6 Hz, 1H), 5, (s, 2H), 6,65 (d, J = 7 Hz, 1H), 6,98-7,07 (m solapado, 4H), 8,02 (s, 1H), 8,02 (s, 1H), 5,00 (d, J = 7 Hz, 1H).89 (s, 2H), 6,65 (d, J = 7 Hz, 1H), 6,98-7,07 (m solapado, 4H), 8,02 (s, 1H), 8,31 (d, J = 5 Hz, 1H), 9,66 (s, 1H); MS (ESI+) para C28H30F2N4O10 m/z 621,1 (M+H)+; tiempo de retención en HPLC: 5,01 min (procedimiento C).
Ejemplo 21, esquema 11: Ácido (2S,3R)-3-[2-(4-metoxifenil)etil]-3-metil-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico (proporcionado como referencia)

Etapa 1. Preparación de bencil-(2S,3R)-1-[terc-butil(dimetil)silil]-3-[2-(4-metoxifenil)etil]-3-metil-4-oxoazetidin-2-carboxilato de bencilo

Se cargó un matraz de fondo redondo de 2 bocas de 50 ml con ácido (2S,3R)-1-[terc-butil(dimetil)silil]-3-metil-4-oxoazetidin-2-carboxílico (450,0 mg, 1,849 mmol) preparado por el procedimiento de Finke, P. E., et al., J. Med. Chem., 1995, 38, 2449. El matraz se sometió al vacío y se rellenó de nitrógeno tres veces. Se añadió tetrahidrofurano (4,2 ml) y la solución se enfrió a 0 °C en un baño de hielo. Se añadió gota a gota una disolución 1,45 M de LDA en heptano/THF/etilbenceno (2,8 ml, 1,76 mmol) y la disolución se agitó durante 55 min a 0 °C. Se añadió gota a gota una disolución de 1-bromo-2-(4-metoxifenil)etano (0,54 ml, 3,5 mmol) en tetrahidrofurano (2 ml) y se agitó a 0°C durante 2 h, calentando después la mezcla de reacción hasta la temperatura ambiente. Después de 6 h a temperatura ambiente, la mezcla de reacción se diluyó con 25 ml de acetato de etilo y se vertió en 14 ml de disolución helada de KHSO40,5 M que estaba en un embudo de decantación. Se extrajo la mezcla y se separaron las fases. La fase acuosa se lavó con dos porciones de 20 ml de acetato de etilo y la fase orgánica reunida se lavó con 20 ml de salmuera. La fase orgánica se secó sobre Na2SO4, se filtró y se concentró para producir un aceite de color canela. El producto bruto se suspendió en cloruro de metileno (11 ml) y se trató con clorhidrato de N-(3-dimetilaminopropil)-N'-etilcarbodiimida (390 mg, 2,03 mmol), seguido de alcohol bencílico (210 µl, 2,03 mmol) y 4-dimetilaminopiridina (11 mg, 0,094 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 7 h. La mezcla de reacción se diluyó con 35 ml de CH2Cl2 y se lavó con dos porciones de 25 ml de H2O y 20 ml de salmuera. La fase orgánica se secó sobre Na2SO4, se filtró y se concentró para producir un aceite de color canela. El material bruto se purificó mediante cromatografía en columna rápida (75 g de gel de sílice; acetato de etilo al 5-20 %/hex.) para obtener 168 mg del compuesto del título contaminado con una impureza no identificada en forma de un aceite amarillo. La pureza fue del 57-62 % por HPLC; tiempo de retención HPLC: 5,77 min (procedimiento A).
Etapa 2. Preparación de (2S,3R)-3-[2-(4-metoxifenil)etil]-3-metil-4-oxoazetidin-2-carboxilato de bencilo

Una solución de (2S,3R)-3-[2-(4-metoxifenil)etil]-3-metil-4-oxoazetidin-2-carboxilato de bencilo (260 mg, 0,35 mmol) en metanol (5,3 ml) se trató con ácido acético (72 µl, 1,3 mmol), seguido de NH4F 0,5 M en metanol (1,0 ml, 0,52 mmol) gota a gota y la mezcla se agitó a temperatura ambiente durante 20 h, momento en el que la HPLC indicó que el material de partida seguía presente. La mezcla de reacción se trató con 16 µl de HOAc y 190 µl de una solución de NH4F 0,5 M y se dejó en agitación a temperatura ambiente durante 20 h más, momento en el que la HPLC indicó que la reacción se había completado. La mezcla de reacción se concentró y el residuo se suspendió en 10 ml de tolueno y se concentró. El proceso se repitió una vez y el residuo se suspendió en 25 ml de CH2Cl2 y se lavó con porciones de 20 ml de H2O y una solución deNaHCO3. La fase orgánica se secó sobre Na2SO4, se filtró y se concentró para producir un aceite viscoso de color amarillo claro. El producto bruto se purificó mediante cromatografía en columna rápida (25 g de gel de sílice; acetato de etilo al 20-40 %/hex.) para obtener el compuesto del título (80 mg) como un cristal incoloro: Tiempo de retención en HPLC: 3,81 min (procedimiento A); RMN de 1H (400 MHz, CDCl3) δ 7,39 (m, 5H), 7,11 (d, J = 8,59 Hz, 2H), 6,83 (m, 2H), 6,22 (s a, 1H), 5,23 (m, 2H), 4,08 (s, 1H), 3,79 (s, 3H), 2,68 (m, 2H), 2,00 (m, 2H), 1,19 (s, 3H).
Etapa 3. Preparación de (2S,3R)-3-[2-(4-metoxifenil)etil]-3-metil-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de bencilo

Una solución de (2S,3R)-3-[2-(4-metoxifenil)etil]-3-metil-4-oxoazetidin-2-carboxilato de bencilo (60,0 mg, 0,170 mmol) en cloruro de metileno (1,8 ml) se trató con trietilamina (95 µl, 0,679 mmol), seguida de [(1R)-1-isocianatoetil]benceno (31 µl, 0,221 mmol) gota a gota y la mezcla se agitó a temperatura ambiente durante 18 h, momento en el que la HPLC indicó que se había consumido el material de partida. La mezcla de reacción se concentró, el residuo se suspendió en 10 ml de CH2Cl2 y se concentró hasta obtener un aceite/sólido viscoso incoloro. El producto bruto se purificó mediante TLC preparativa (placa de TLC preparativa de 20 cm x 20 cm x 1,0 mm; acetato de etilo al 30 %/hex.) para obtener el compuesto del título (72 mg) como un cristal incoloro: Tiempo de retención en HPLC: 4,84 min (procedimiento A); RMN de 1H (400 MHz, CDCl3) δ 7,35 (m, 9H), 7,29 (m, 1H), 7,10 (d, J = 8,59 Hz, 2H), 6,84 (m, 2H), 6,71 (d, J = 7,83 Hz, 1H), 5,27 (m, 1H), 5,18 (m, 1H), 5,09 (m, 1H), 4,43 (s, 1H), 3,80 (s, 3H), 2,72 (m, 1H), 2,62 (m, 1H), 2,04 (m, 2H), 1,57 (d, J = 6,82 Hz, 3H), 1,17 (s, 3H).
Etapa 4. Preparación del ácido (2S,3R)-3-[2-(4-metoxifenil)etil]-3-metil-4-oxo-1-{[(1R)-1-feniletil]carbamoil}azetidin-2-carboxílico

Una solución de (2S,3R)-3-[2-(4-metoxifenil)etil]-3-metil-4-oxo-1-{ [(1R)-1-feniletil]carbamoil}azetidin-2-carboxilato de bencilo (72 mg, 0,14 mmol) en metanol (2,3 ml) y acetato de etilo (2,3 ml) se trató cuidadosamente con paladio al 10 % sobre carbono (13 mg). El matraz de reacción se sometió al vacío y se llenó con gas hidrógeno tres veces y la mezcla de reacción se agitó a temperatura ambiente bajo una atmósfera de hidrógeno durante 3,5 h, momento en el que la TLC (acetato de etilo al 25 %/hex.) indicó que el SM se había consumido. La mezcla de reacción se filtró a través de un lecho corto de Solka-Floc y el lecho corto se lavó con 30 ml de acetato de etilo/MeOH 1/1. El filtrado se concentró hasta un cristal incoloro que se suspendió en metanol acuoso y se liofilizó para obtener el compuesto del título (56 mg) como un sólido blanco: Tiempo de retención en HPLC 4,19 min (procedimiento A); MS (ESI+) para C23H26N2O5 m/z 411,2 (M+H)+; MS (ESI-) para C23H26N2O5 m/z 409,2 (M-H)-; RMN de 1H (400 MHz, MeOD) δ 7,35 (m, 4H), 7,26 (m, 1H), 7,15 (m, 2H), 7,11 (d, J = 7,83 Hz, 1H), 6,84 (m, 2H), 4,97 (m, 1H), 4,43 (s, 1H), 3,76 (s, 3H), 2,77 (m, 1H), 2,65 (m, 1H), 2,05 (m, 2H), 1,52 (d, J = 7,07 Hz, 3H), 1,31 (s, 3H).
Ejemplo 22. Ensayo de respuesta a la dosis para inhibidores de la proteasa
Materiales:

Procedimientos:
1. Diluir el sustrato hasta 100 µM en tampón de ensayo (30 µl/6 ml). La enzima se diluye hasta 0,5 nM justo antes de su uso (12 µl/6 ml para el factor XIa; 15 µl/6 ml para el resto).
2. Pipetear 50 µl de sustrato en cada pocillo de la placa de microtitulación de 96 pocillos (12 x 8) (la columna 1 se utiliza como control de actividad al 100 % y no recibe ningún compuesto, y la columna 12 es el blanco y no recibe ninguna enzima). Añadir 46 µl más a la columna 2.
3. Pipetear 4 µl de cada compuesto en el pocillo apropiado de la columna 2 de la placa (las incógnitas se ensayan por triplicado, el patrón se ensaya por duplicado). La concentración final del compuesto será 1/50 de la disolución madre.
4. Diluir el compuesto dos veces en serie mezclando la muestra en la columna 2, retirar 50 µl al pocillo siguiente (columna 3), mezclar y extraer a la columna 4, etc., hasta la columna 11. Después de mezclar la columna 11, extraer 50 µl y desechar.
5. Pipetear 50 µl de tampón en la columna 12. Iniciar la reacción añadiendo 50 µl de solución enzimática a cada pocillo de las columnas 1 a 11 lo más rápidamente posible.
6. Leer la placa en un espectrofotómetro (SpectraMax) a 30 °C, midiendo cada pocillo cada 60 s durante 30 min. Para los ensayos del factor Xa, cada pocillo se mide cada 1 min durante un total de 60 min.
7. Para los ensayos del factor XIa, los compuestos se ensayan, por duplicado, a 3 concentraciones iniciales diferentes, 20, 2 y 0,2 µM; dilución de 1:10, 1:100 y 1:1000 de una disolución madre 10 mM. Todos los conjuntos de datos se reúnen para su representación gráfica y ajuste.
8. Los datos pueden utilizarse tanto para el cálculo de la CI50 como para el cálculo de la Kon.
Ejemplo 23. Estabilidad de β-lactamas en plasma de rata
Reactivos madre:
Plasma normal de rata, conservado a -80 °C
Protocolo:
1. Colocar 6 µl de cada compuesto en tubos de microcentrífuga de 0,5 ml o en placas de microtitulación de polipropileno de 96 pocillos con fondo en U.
2. Introducir 40 µl de acetonitrilo (AcN) en tubos de microcentrífuga de 0,5 ml etiquetados con los números 1 a 9.
3. Añadir 114 µl de plasma a cada compuesto.
4. Tomar muestras de 10 µl de compuesto/plasma en AcN a 2, 10, 20, 30, 60, 90, 120, 240 y 480 min de incubación a temperatura ambiente.
5. Mezclar cada punto temporal después de añadir la muestra y situar en hielo.
6. Centrifugar los puntos temporales (12000 rpm, 3 min), extraer 15 µl del sobrenadante y mezclar con 15 µl de TFA al 0,1% en una placa de microtitulación de fondo en V.
7. Situar la placa de microtitulación en el automuestreador de HPLC y analizar en la columna Restek Pinnacle C18 (2,1 x 100 mm).
8. Generar el cromatograma de iones extraídos para el compuesto original y el compuesto original 18 (H2O). Integrar los cromatogramas de iones extraídos ("extracted ion chromatograms", EIC) y representar gráficamente el porcentaje de área de pico total para el compuesto original y el aducto en función del tiempo. Ajustar a un modelo de desintegración exponencial monofásica [Y=A0∗exp(k*x) C]. T½ es igual a ln(2)/k. Ejemplo 24. Ensayos de inhibición enzimática del factor XIa
La capacidad de los compuestos de la presente invención para inhibir el factor XIa se evaluó determinando la concentración de inhibidor que dio lugar a una reducción del 50 % en la actividad enzimática (CI50) utilizando enzima purificada. Los inhibidores potenciales del factor XIa se evaluaron mediante el siguiente ensayo.
S-2366, piroGlu-Pro-Arg-7-metilaminocumarina (AMC), disponible en CPC Scientific, Inc., se basa en el sustrato piro-Glu-Pro-Arg-pAN, disponible en Diapharma Group, Inc. (Columbus, Ohio), donde el grupo p-nitroanalina se sustituye por 7-metilaminocumarina (AMC).
La concentración final del sustrato en el ensayo fue de 50 µM y la concentración final de la enzima fue de 0,25 nM. Los inhibidores se ensayaron mediante diluciones seriadas en un intervalo adecuado para obtener una curva de dosisrespuesta que permitiera determinar el valor de CI50 de los inhibidores. La mezcla de ensayo se leyó cada minuto durante 30 minutos para generar curvas de progreso. Las placas se leyeron en un lector de placas multimodo Spectramax m5 (Molecular Devices LLC, Sunnyvale, CA). Las curvas de dosis-respuesta se ajustaron a la siguiente ecuación 1, en la que A es la inhibición máxima, B es la inhibición mínima, C es la CI50 y D es el coeficiente de Hill.
[(A-B)/(1 (X/C)D)] B (Ecuación 1)
Los compuestos deseables tienen un valor de CI50 para inhibir el factor XIa de menos de 1 micromolar, 100 nanomolar, 10 nanomolar o 1 nanomolar (en orden de preferencia creciente).
Tabla 2. Potencia, selectividad y estabilidad de ejemplos de compuestos





Para la tabla 2: FXIa y hFXIa se refieren al factor XIa y al factor XIa humano, respectivamente. Potencia: "A" indica <10 nM, "B" indica 10-100 nM, "C" indica 100-1000 nM, "D" indica >1000 nM, y "E" indica que los datos no están disponibles o no se han determinado. Selectividad: "F" indica <1, "G" indica 1-500, "H" indica 500-1000; "I" >1000, y "J" indica que los datos no están disponibles o no se han determinado. Estabilidad en plasma de rata: "K" indica 0-100 min; "L" indica >100 min; "M" indica que los datos no están disponibles o no se han determinado.
Ejemplo 25. Ensayos de solubilidad
Se utilizó el siguiente procedimiento para determinar la solubilidad acuosa de un compuesto de ensayo en solución salina tamponada con fosfato (PBS - NaCl 137 mM, KCl 2,7 mM, Na2HPO48,1 mM, KH2PO41,5 mM, pH 7,4) en formato de placa de 96 pocillos mediante análisis por HPLC-UV/VIS. El compuesto de ensayo se preparó a 200 µM en PBS a partir de una solución madre de DMSO 10 mM. La concentración final de DMSO fue del 2 %. Las muestras de tampón PBS se mezclaron a fondo y se incubaron a temperatura ambiente durante 24 h. Al final de la incubación, las muestras de tampón PBS se centrifugaron y los sobrenadantes se analizaron por HPLC. La solubilidad acuosa (µM) del compuesto de ensayo en PBS se determinó comparando el área del pico principal en el patrón de calibración (200 µM) con el área del pico correspondiente en cada una de las muestras de PBS. El intervalo del ensayo fue aproximadamente de 0,5 µM a 200 µM. Los compuestos de referencia utilizados en cada ensayo fueron metoprolol, rifampicina, ketoconazol, fenitoína, haloperidol, simvastatina, dietilestilbestrol y tamoxifeno, clasificados de totalmente solubles (200 µM) a poco solubles (<1 µM).
Ejemplo 26. Ensayos de estabilidad metabólica
Se utilizó el siguiente procedimiento para determinar la estabilidad de un compuesto de ensayo en microsomas hepáticos agrupados de humanos (género mixto) en formato de placa de 96 pocillos. El compuesto de ensayo se cuantificó en cinco puntos temporales mediante análisis por HPLC-MS/MS. La concentración final de proteína microsomal en el ensayo fue de 0,1 mg/ml. Cada compuesto se ensayó a 0,1 µM con DMSO al 0,01%, acetonitrilo al 0,25% y metanol al 0,25%. El compuesto de ensayo se preincubó con microsomas de hígado humano en tampón fosfato (pH 7,4) durante 5 min en un baño de agua con agitación a 37 °C. La reacción se inició añadiendo el sistema generador de NADPH y se incubó durante 0, 15, 30, 45 y 60 minutos. La reacción se detuvo transfiriendo la mezcla de incubación a acetonitrilo/metanol. A continuación, las muestras se mezclaron y centrifugaron, y los sobrenadantes
se utilizaron para el análisis por HPLC-MS/MS. Se registraron las áreas de los picos correspondientes al compuesto de ensayo. El compuesto restante se calculó comparando el área del pico en cada punto temporal con el tiempo cero. Se ensayaron cuatro compuestos de referencia en cada ensayo; el propranolol y la imipramina son relativamente estables, mientras que el verapamilo y la terfenadina se metabolizan fácilmente en los microsomas hepáticos humanos.
Ejemplo 27. Ensayos de unión a proteínas plasmáticas
Se utilizó el siguiente procedimiento para determinar la unión a proteínas plasmáticas de un compuesto de ensayo en plasma agrupado de humanos (género mixto) mediante diálisis de equilibrio en un formato de placa de 96 pocillos. El compartimento del dializado se carga con solución salina tamponada con fosfato (pH 7,4) y el lado de la muestra se carga con plasma enriquecido con el compuesto de ensayo a una concentración de 10 µM. Tras la carga, se cubren las muestras y se incuban durante 4 horas a 37 °C. Tras la incubación, se toman muestras de cada compartimento, se diluyen con acetonitrilo/tampón y se centrifugan. Los sobrenadantes se analizan por HPLC-MS/MS. La cantidad medida en el compartimento plasmático incluye tanto el fármaco libre como el unido, mientras que la del lado del tampón representa solo el fármaco libre; las diferencias se utilizan para calcular el porcentaje de proteína plasmática unida. En cada ensayo se ensayaron tres compuestos de referencia: acebutolol, quinidina y warfarina. Estos compuestos producen unos valores de unión a proteínas que representan una unión baja, media y alta a proteínas plasmáticas humanas, respectivamente.
Tabla 3. Unión a proteínas plasmáticas, solubilidad y estabilidad metabólica de ejemplos de compuestos

Claims (20)
1. Un compuesto de fórmula (II):

o una sal farmacéuticamente aceptable del mismo, en la que,
R1 es H o -alquilo C1-6;
R2 es -CO2R5, H, -alquilo C1-6, -C(O)NR9R10, -CN,-CHN(OR5) o un heteroarilo;
R3 es H o -alquilo C1-6;
A es alquileno C1-6, un enlace, alquenileno C2-6 o alquinileno C2-6;
R4 es cicloalquilo, arilo, heteroarilo o heterociclilo, cada uno de los cuales está sustituido con 0 a 3 apariciones de -NH2 o R6;
cada R5 es independientemente H, -alquilo C1-6alquilo, aralquilo o arilo sustituido con 0-3 apariciones de -NH2 o R6;
cada R6 es independientemente halo, hidroxi, ciano, nitro, alquilo C1-6, alcoxi C1-6, -NR9R10, -NHR10, -C(O)R11, -C(O)OR11, -C(O)NR9R10, -C(NR8)(N(R8)2), -SOqRn, -SO2NR9R10, -NHC(O)OR11, -NHC(O)R11, arilo, heteroarilo, aralquilo, cicloalquilo, heteroaralquilo, heterociclilo o heterociclilalquilo; o
dos grupos R6 tomados conjuntamente con los átomos a los que están unidos forman un anillo de 5 a 7 miembros;
X es -C(O)N(R5)-, C(O)O-, -OC(O)-, -C(O)S(O)2-, -S(O)2C(O)- o -N(R5)C(O)-;
Y es cicloalquilo;
R7 es -alquilo C1-6, H, cicloalquilo, arilo, heteroarilo o heterociclilo, cada uno de los cuales está sustituido con 0-3 apariciones de -NH2 o R6;
cada R8 es independientemente H, -alquilo C1-6, -C(O)R5, -C(O)OR5, arilo, heteroarilo, aralquilo, heteroaralquilo, heterociclilo o heterociclilalquilo;
cada uno de R9 y R10 es independientemente -alquilo C1-6, cicloalquilo, heterociclilo, arilo o heteroarilo, o R9 y R10 juntos forman un anillo de 5 a 7 miembros opcionalmente sustituido.
cada R11 es independientemente H, -alquilo C1-10, aralquilo o arilo;
q es un número entero de 0 a 2; y
n es un número entero de 0 a 2;
en el que cualquier grupo alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo o heteroarilo, a menos que se defina de otro modo, está opcionalmente sustituido con uno o más sustituyentes seleccionados independientemente entre halógeno,-CN, -NO2, -N3, -SO2H, -SO3H, -OH, -ORaa, -ON(Rbb)2, -N(Rbb)2, -N(Rbb)3 +X-, -N(ORcc)Rbb, -SH, -SRaa, -SSRcc, -C(=O)Raa, -CO2H, -CHO, -C(ORcc)2, -CO2Raa, -OC(=O)Raa, -OCO2Raa, -C(=O)N(Rbb)2, -OC(=O)N(Rbb)2, -NRbbC(=O)Raa, -NRbbCO2Raa, -NRbbC(=O)N(Rbb)2, -C(=NRbb)Raa, -C(=NRbb)ORaa, -OC(=NRbb)Raa, -OC(=NRbb)ORaa, -C(=NRbb)N(Rbb)2, -OC(=NRbb)N(Rbb)2, -NRbbC(=NRbb)N(Rbb)2, -C(=O)NRbbSO2Raa, -NRbbSO2Raa, -SO2N(Rbb)2, -SO2Raa, -SO2ORaa, -OSO2Raa, -S(O)Raa-S(=O)Raa, -OS(=O)Raa, -Si(Raa)3, -OSi(Raa)3 -C(=S)N(Rbb)2, -C(=O)SRaa, -C(=S)SRaa, -SC(=S)SRaa, -SC(=O)SRaa, -OC(=O)SRaa, -SC(=O)ORaa, -SC(=O)Raa, -P(=O)2Raa, -OP(=O)2Raa, -P(=O)(Raa)2, -OP(=O)(Raa)2, -OP(=O)(ORcc)2, -P(=O)2N(Rbb)2, -OP(=O)2N(Rbb)2, -P(=O)(NRbb)2, -OP(=O)(NRbb)2, -NRbbP(=O)(ORcc)2, -NRbbP(=O)(Nbb)2, -P(Rcc)2, -P(Rcc)3, -OP(Rcc)2, -OP(Rcc)3, -B(Raa)2, -B(ORcc)2, -BRaa(ORcc), alquilo C1-10, perhaloalquilo C1-10, alquenilo C2-10, alquinilo C2-10, cicloalquilo C3-10, heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está sustituido independientemente con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Raa se selecciona, independientemente, entre alquilo C1-10, perhaloalquilo C1-10, alquenilo C2-10, alquinilo C2-10, cicloalquilo C3-10, heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, o dos grupos Raa se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Rbb se selecciona, independientemente, entre hidrógeno, -OH, -ORaa, -N(Rcc)2, -CN, -C(=O)Raa, -C(=O)N(Rcc)2, -CO2Raa, -SO2Raa, -C(=NRcc)ORaa, -C(=NRcc)N(Rcc)2, -SO2N(Rcc)2, -SO2Rcc, -SO2ORcc, -SORaa, -C(=S)N(Rcc)2, -C(=O)SRcc, -C(=S)SRcc, -P(=O)2Raa, -P(=O)(Raa)2, -P(=O)2N(Rcc)2, -P(=O)(NRcc)2, alquilo C1-10, perhaloalquilo C1-10, alquenilo C2-10, alquinilo C2-10, cicloalquilo C3-10, heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, o dos grupos Rbb se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está sustituido independientemente con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Rcc se selecciona, independientemente, entre hidrógeno, alquilo C1-10, perhaloalquilo C1-10, alquenilo C2-10, alquinilo C2-10, cicloalquilo C3-10, heterociclilo de 3 a 14 miembros, arilo C6-14 y heteroarilo de 5 a 14 miembros, o dos grupos Rcc se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rdd;
cada aparición de Rdd se selecciona, independientemente, entre halógeno, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -ORee, -ON(Rff)2, -N(Rff)2, -N(Rff)3 +X-, -N(ORee)Rff, -SH, -SRee, -SSRee, -C(=O)Ree, -CO2H, -CO2Ree, -OC(=O)Ree, -OCO2Ree, -C(=O)N(Rff)2, -OC(=O)N(Rff)2, -NRffC(=O)Ree, -NRffCO2Ree, -NRffC(=O)N(Rff)2, -C(=NRff)ORee, -OC(=NRff)Ree, -OC(=NRff)ORee, -C(=NRff)N(Rff)2, -OC(=NRff)N(Rff)2, -NRffC(=NRff)N(Rff)2,-NRffSO2Ree, -SO2N(Rff)2, -SO2Ree, -SO2ORee, -OSO2Ree, -S(=O)Ree, -Si(Ree)3, -OSi(Ree)3, -C(=S)N(Rff)2, -C(=O)SRee, -C(=S)SRee, -SC(=S)SRee, -P(=O)2Ree, -P(=O)(Ree)2, -OP(=O)(Ree)2, -OP(=O)(ORee)2, alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, arilo C6-10, heteroarilo de 5 a 10 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rgg, o dos sustituyentes germinales Rdd pueden unirse para formar =O o =S;
cada aparición de Ree se selecciona, independientemente, entre alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, arilo C6-10, heteroarilo de 3 a 10 miembros y heterociclilo de 3 a 10 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rgg;
cada aparición de Rff se selecciona, independientemente, entre hidrógeno, alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, arilo C6-10 y heteroarilo de 5 a 10 miembros, o dos grupos Rff se unen para formar un anillo heterociclilo de 3 a 14 miembros o heteroarilo de 5 a 14 miembros, en los que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo y heteroarilo está independientemente sustituido con 0, 1, 2, 3, 4 o 5 grupos Rgg; y
cada aparición de Rgg es, independientemente, halógeno, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -O-alquilo C1-6, -ON(alquilo C1-6)2, -N(alquilo C1-6)2, -N(alquilo C1-6)3 +X-, -NH(alquilo C1-6)2 +X-, -NH2(alquilo C1-6)+X-, -NH3 +X-, -N(O-alquil C1-6)(alquilo C1-6), -N(OH)(alquilo C1-6), -NH(OH), -SH, -S-alquilo C1-6, -SS(alquilo C1-6), -C(=O)(alquilo C1-6), -CO2H, -CO2(alquilo C1-6), -OC(=O)(alquilo C1-6), -OCO2(alquilo C1-6), -C(=O)NH2, -C(=O)N(alquilo C1-6)2, -OC(=O)NH(alquilo C1-6), -NHC(=O)(alquilo C1-6), -N(alquilo C1-6)C(=O)(alquilo C1-6), -NHCO2(alquilo C1-6), -NHC(=O)N(alquilo C1-6)2, -NHC(=O)NH(alquilo C1-6), -NHC(=O)NH2, -C(=NH)O(alquilo C1-6), -OC(=NH)(alquilo C1-6), -OC(=NH)O-alquilo C1-6, -C(=NH)N(alquilo C1-6)2, -C(=NH)NH(alquilo C1-6), -C(=NH)NH2, -OC(=NH)N(alquilo C1-6)2, -OC(NH)NH(alquilo C1-6), -OC(NH)NH2, -NHC(NH)N(alquilo C1-6)2, -NHC(=NH)NH2, -NHSO2(alquilo C1-6), -SO2N(alquilo C1-6)2, -SO2NH(alquilo C1-6), -SO2NH2,-SO2-alquilo C1-6, -SO2O-alquilo C1-6, -OSO2-alquilo C1-6, -SO-alquilo C1-6, -Si(alquilo C1-6)3, -OSi(alquilo C1-6)3, -C(=S)N(alquilo C1-6)2, C(=S)NH(alquilo C1-6), C(=S)NH2, -C(=O)S(alquilo C1-6), -C(=S)S-alquilo C1-6, -SC(=S)S-alquilo C1-6, -P(=O)2(alquilo C1-6), -P(=O)(alquilo C1-6)2, -OP(=O)(alquilo C1-6)2, -OP(=O)(O-alquilo C1-6)2,alquilo C1-6, perhaloalquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-10, arilo C6-10 arilo, heterociclilo de 3 a 10 miembros, heteroarilo de 5 a 10 miembros; o dos sustituyentes geminales Rgg pueden unirse para formar =O o =S; en los que X- es un contraión.
2. El compuesto de la reivindicación 1, en el que:
a) R1 es H; y/o
b) R2 es -CO2R5, en el que R5 es H o -alquilo C1-6.
3. El compuesto de la reivindicación 1, en el que Y es cicloalquilo no sustituido.
4. El compuesto de la reivindicación 1 en el que A es alquileno C1-6
5. El compuesto de la reivindicación 1, en el que R4 es arilo o heteroarilo, opcionalmente en el que R4 es fenilo sustituido con 1 aparición de R6, opcionalmente en el que R6 es halo, alcoxi C1-6 o -C(NR8)(N(R8)2), opcionalmente en el que:
a) R6 es -C(NR8)(N(R8)2) y cada R8 es H; o
b) R8 es independientemente H o -C(O)OR5 , opcionalmente en el que R5 es -alquilo C1-6.
6. El compuesto de la reivindicación 1, en el que R4 es heteroarilo sustituido con 0 a 3 apariciones de -NH2 o R6, opcionalmente en el que R4 es un heteroarilo que contiene nitrógeno, opcionalmente en el que R4 es piridilo sustituido con 1 aparición de R6.
7. El compuesto de la reivindicación 1, en el que R4 es un heteroarilo de 6 miembros sustituido con 0 a 3 apariciones de -NH2 o R6, opcionalmente en el que R6 es halo o hidroxi.
8. El compuesto de la reivindicación 1, en el que:
a) X es -C(O)N(R5)- o -N(R5)C(O)-, opcionalmente en el que X es -C(O)N(R5)- y R5 es H; y/o
b) n es 0.
9. El compuesto de la reivindicación 1, en el que R7 es -alquilo C1-6 sustituido con 0 a 3 apariciones de NH2 o R6, opcionalmente en el que R7 es metilo o -CF3.
10. El compuesto de la reivindicación 1, en el que:
a) R6 e alcoxi o haloalcoxi; o
b) R6 es -NHC(O)OR11, y R11 es

11. El compuesto de la reivindicación 1, en el que el compuesto de fórmula (II) se selecciona de un compuesto de fórmula (IIa):

en la que
R1, R2, R3, R4, R7 e Y son como se describió para la fórmula (II), y m es un número entero de 1 a 6, opcionalmente, en el que el compuesto de fórmula (IIa) se selecciona entre un compuesto de fórmula (IIb):

en la que R1, R2, R3, R4, R7, Y y m son como se describió para la fórmula (IIa).
12. El compuesto de la reivindicación 11, en el que el compuesto de fórmula (IIb) es:



13. El compuesto de la reivindicación 1, en el que el compuesto de fórmula (II) es:

o una sal farmacéuticamente aceptable del mismo.
14. El compuesto de la reivindicación 11, en el que el compuesto de fórmula (IIb) se selecciona de un compuesto de fórmula (IIc):

15. Un compuesto de una cualquiera de las reivindicaciones 1 a 14, en el que el compuesto es una sal farmacéuticamente aceptable.
16. Una composición farmacéutica que comprende un compuesto de una cualquiera de las reivindicaciones 1 a 5, o una sal farmacéuticamente aceptable del mismo, y uno o más excipientes farmacéuticamente aceptables.
17. Un compuesto de una cualquiera de las reivindicaciones 1-15 o una sal farmacéuticamente aceptable del mismo, o la composición de la reivindicación 16 para su uso en:
a) reducir el riesgo de ictus en un sujeto que ha sufrido un acontecimiento isquémico;
b) reducir la embolia sistémica del sistema nervioso no central en un sujeto que ha sufrido un acontecimiento isquémico;
c) tratar la trombosis venosa profunda en un sujeto que ha sufrido un acontecimiento isquémico;
d) reducir el riesgo de recurrencia de la trombosis venosa profunda en un sujeto que haya sufrido una trombosis venosa profunda;
e) reducir el riesgo de recurrencia de la embolia pulmonar en un sujeto que ha sufrido una embolia pulmonar; f) la profilaxis de la embolia pulmonar en un sujeto que ha sufrido una embolia pulmonar; o
g) tratar a un sujeto que presenta edema.
18. El compuesto de la reivindicación 1, en el que el compuesto de fórmula (II) es:

o una sal farmacéuticamente aceptable del mismo.
19. El compuesto de la reivindicación 1, en el que el compuesto de fórmula (II) es:

o una sal farmacéuticamente aceptable del mismo.
20. El compuesto de la reivindicación 1, en el que el compuesto de fórmula (II) es:

o una sal farmacéuticamente aceptable del mismo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461937031P | 2014-02-07 | 2014-02-07 | |
| PCT/US2015/014478 WO2015120062A2 (en) | 2014-02-07 | 2015-02-04 | Therapeutic compounds and compositions |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2945905T3 true ES2945905T3 (es) | 2023-07-10 |
Family
ID=53774361
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15746142T Active ES2945905T3 (es) | 2014-02-07 | 2015-02-04 | Compuestos y composiciones terapéuticos |
Country Status (15)
| Country | Link |
|---|---|
| US (5) | US9499532B2 (es) |
| EP (2) | EP4309653A1 (es) |
| JP (4) | JP6382997B2 (es) |
| KR (2) | KR102256242B1 (es) |
| CN (3) | CN106029064B (es) |
| AU (3) | AU2015214251B2 (es) |
| BR (1) | BR112016018062B1 (es) |
| CA (1) | CA2938884C (es) |
| DK (1) | DK3102200T3 (es) |
| ES (1) | ES2945905T3 (es) |
| FI (1) | FI3102200T3 (es) |
| HK (1) | HK1232137A1 (es) |
| IL (2) | IL247128B (es) |
| RU (2) | RU2020131276A (es) |
| WO (1) | WO2015120062A2 (es) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014160668A1 (en) | 2013-03-25 | 2014-10-02 | Bristol-Myers Squibb Company | Tetrahydroisoquinolines containing substituted azoles as factor xia inhibitors |
| NO2760821T3 (es) | 2014-01-31 | 2018-03-10 | ||
| RS57659B1 (sr) | 2014-01-31 | 2018-11-30 | Bristol Myers Squibb Co | Makrociklusi sa heterocikličnim p2' grupama kao inhibitori faktora xia |
| ES2945905T3 (es) * | 2014-02-07 | 2023-07-10 | Exithera Pharmaceuticals Inc | Compuestos y composiciones terapéuticos |
| US10081623B2 (en) | 2014-09-04 | 2018-09-25 | Bristol-Myers Squibb Company | Diamide macrocycles that are FXIa inhibitors |
| US9453018B2 (en) | 2014-10-01 | 2016-09-27 | Bristol-Myers Squibb Company | Pyrimidinones as factor XIa inhibitors |
| EP3371162B1 (en) | 2015-10-29 | 2022-01-26 | Merck Sharp & Dohme Corp. | Macrocyclic spirocarbamate derivatives as factor xia inhibitors, pharmaceutically acceptable compositions and their use |
| US10143681B2 (en) | 2016-08-22 | 2018-12-04 | Merck Sharp & Dohme Corp. | Factor XIa inhibitors |
| DK3532057T3 (da) | 2016-10-31 | 2023-02-20 | Biocryst Pharm Inc | Prodrugs af kallikreininhibitorer |
| WO2018118705A1 (en) * | 2016-12-23 | 2018-06-28 | Exithera Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| EP3749662A4 (en) * | 2018-02-07 | 2021-07-14 | Exithera Pharmaceuticals Inc. | COMPOUNDS AND THERAPEUTIC COMPOSITIONS |
| WO2019185046A1 (zh) * | 2018-03-30 | 2019-10-03 | 上海美悦生物科技发展有限公司 | 四元内酰胺类化合物及其药学用途 |
| AU2019373237A1 (en) * | 2018-10-30 | 2021-04-22 | Exithera Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| BR112021008462A2 (pt) * | 2018-10-30 | 2021-08-03 | Exithera Pharmaceuticals, Inc. | compostos e composições terapêuticas |
| CN113784951A (zh) * | 2019-01-29 | 2021-12-10 | 艾克赛特赫拉制药有限责任公司 | 治疗化合物和组合物 |
| EP4213857A1 (en) * | 2020-09-17 | 2023-07-26 | Exithera Pharmaceuticals Inc. | Therapeutic compounds, compositions, and methods of use thereof |
Family Cites Families (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4595532A (en) * | 1983-02-02 | 1986-06-17 | University Of Notre Dame Du Lac | N-(substituted-methyl)-azetidin-2-ones |
| US5739135A (en) | 1993-09-03 | 1998-04-14 | Bristol-Myers Squibb Company | Inhibitors of microsomal triglyceride transfer protein and method |
| SE9602263D0 (sv) * | 1996-06-07 | 1996-06-07 | Astra Ab | New amino acid derivatives |
| US5760246A (en) | 1996-12-17 | 1998-06-02 | Biller; Scott A. | Conformationally restricted aromatic inhibitors of microsomal triglyceride transfer protein and method |
| CA2317761A1 (en) | 1998-01-26 | 1999-07-29 | Basf Aktiengesellschaft | Thrombin inhibitors |
| SE9802206D0 (sv) | 1998-06-22 | 1998-06-22 | Astra Pharma Inc | Novel compounds |
| US6335324B1 (en) * | 1998-06-25 | 2002-01-01 | Bristol-Myers Squibb Co. | Beta lactam compounds and their use as inhibitors of tryptase |
| PL345067A1 (en) * | 1998-06-25 | 2001-12-03 | Bristol Myers Squibb Co | Amidino and guanidino azetidinone tryptase inhibitors |
| WO2000005204A1 (fr) * | 1998-07-23 | 2000-02-03 | Shionogi & Co., Ltd. | COMPOSES DE β-LACTAME MONOCYCLIQUES ET INHIBITEURS DE LA CHYMASE LES RENFERMANT |
| GB0205527D0 (en) | 2002-03-08 | 2002-04-24 | Ferring Bv | Inhibitors |
| ATE381947T1 (de) | 2003-04-14 | 2008-01-15 | Wyeth Corp | Zusammensetzungen enthaltend piperacillin und tazobactam zur injektion |
| PA8603801A1 (es) * | 2003-05-27 | 2004-12-16 | Janssen Pharmaceutica Nv | Derivados de la quinazolina |
| WO2005018672A1 (ja) * | 2003-08-22 | 2005-03-03 | Teijin Pharma Limited | キマーゼ阻害剤を有効成分として含有する薬剤 |
| CA2549869C (en) * | 2003-12-18 | 2015-05-05 | Janssen Pharmaceutica N.V. | Pyrido- and pyrimidopyrimidine derivatives as anti- proliferative agents |
| JO3088B1 (ar) * | 2004-12-08 | 2017-03-15 | Janssen Pharmaceutica Nv | مشتقات كوينازولين كبيرة الحلقات و استعمالها بصفتها موانع كينيز متعددة الاهداف |
| US7501404B2 (en) | 2005-04-04 | 2009-03-10 | Daimed | Substituted azetidinones |
| PE20070171A1 (es) * | 2005-06-30 | 2007-03-08 | Boehringer Ingelheim Int | GLICINAMIDAS SUSTITUIDAS CON EFECTO ANTITROMBOTICO E INHIBIDOR DEL FACTOR Xa |
| TW200745084A (en) | 2006-03-08 | 2007-12-16 | Astrazeneca Ab | Novel compounds |
| EP1873157A1 (en) * | 2006-06-21 | 2008-01-02 | Bayer Schering Pharma Aktiengesellschaft | Pyrazolopyrimidines and salts thereof, pharmaceutical compositions comprising same, methods of preparing same and uses of same |
| WO2008124757A1 (en) | 2007-04-10 | 2008-10-16 | Bristol-Myers Squibb Company | Thiazolyl compounds useful as kinase inhibitors |
| DK2170827T3 (da) * | 2007-06-21 | 2013-11-18 | Janssen Pharmaceutica Nv | Indolin-2-oner og aza-indolin-2-oner |
| WO2009114677A1 (en) * | 2008-03-13 | 2009-09-17 | Bristol-Myers Squibb Company | Pyridazine derivatives as factor xia inhibitors |
| BRPI0919873B8 (pt) * | 2008-10-22 | 2021-05-25 | Array Biopharma Inc | compostos de pirazol[1,5-a]pirimidina substituídos como inibidores da trk quinase, seus processos de preparação e composições farmacêuticas |
| EP3786165A1 (en) | 2010-02-11 | 2021-03-03 | Bristol-Myers Squibb Company | Synthetic intermediates for producing macrocycles as factor xia inhibitors |
| WO2012120128A1 (en) | 2011-03-09 | 2012-09-13 | Csl Behring Gmbh | Factor xii inhibitors for the administration with medical procedures comprising contact with artificial surfaces |
| CA2851810C (en) | 2011-10-14 | 2020-01-07 | Bristol-Myers Squibb Company | Substituted tetrahydroisoquinoline compounds as factor xia inhibitors |
| SG11201406142XA (en) * | 2012-03-27 | 2014-10-30 | Univ Duke | Compositions and methods for the prevention and treatment of mast cell-induced vascular leakage |
| CA2923844C (en) | 2013-09-11 | 2022-07-26 | Arsia Therapeutics, Inc. | Liquid protein formulations containing organophosphates |
| ES2945905T3 (es) * | 2014-02-07 | 2023-07-10 | Exithera Pharmaceuticals Inc | Compuestos y composiciones terapéuticos |
| DK2926805T3 (en) | 2014-03-31 | 2016-07-25 | Vasopharm Gmbh | Solid pharmaceutical compositions comprising biopterine derivatives and uses of such compositions |
| WO2018118705A1 (en) | 2016-12-23 | 2018-06-28 | Exithera Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| EP3749662A4 (en) | 2018-02-07 | 2021-07-14 | Exithera Pharmaceuticals Inc. | COMPOUNDS AND THERAPEUTIC COMPOSITIONS |
| AU2019373237A1 (en) | 2018-10-30 | 2021-04-22 | Exithera Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| BR112021008462A2 (pt) | 2018-10-30 | 2021-08-03 | Exithera Pharmaceuticals, Inc. | compostos e composições terapêuticas |
| CN113784951A (zh) | 2019-01-29 | 2021-12-10 | 艾克赛特赫拉制药有限责任公司 | 治疗化合物和组合物 |
-
2015
- 2015-02-04 ES ES15746142T patent/ES2945905T3/es active Active
- 2015-02-04 FI FIEP15746142.7T patent/FI3102200T3/fi active
- 2015-02-04 DK DK15746142.7T patent/DK3102200T3/da active
- 2015-02-04 CN CN201580007721.8A patent/CN106029064B/zh active Active
- 2015-02-04 JP JP2016550776A patent/JP6382997B2/ja active Active
- 2015-02-04 CN CN201810794986.2A patent/CN108892661B/zh active Active
- 2015-02-04 CN CN202310159569.1A patent/CN116444506A/zh active Pending
- 2015-02-04 CA CA2938884A patent/CA2938884C/en active Active
- 2015-02-04 US US14/614,169 patent/US9499532B2/en active Active
- 2015-02-04 RU RU2020131276A patent/RU2020131276A/ru unknown
- 2015-02-04 WO PCT/US2015/014478 patent/WO2015120062A2/en active Application Filing
- 2015-02-04 KR KR1020187034865A patent/KR102256242B1/ko active IP Right Grant
- 2015-02-04 KR KR1020167021085A patent/KR101927114B1/ko active IP Right Grant
- 2015-02-04 AU AU2015214251A patent/AU2015214251B2/en active Active
- 2015-02-04 EP EP23166345.1A patent/EP4309653A1/en active Pending
- 2015-02-04 RU RU2016135922A patent/RU2733405C2/ru active
- 2015-02-04 EP EP15746142.7A patent/EP3102200B1/en active Active
- 2015-02-04 BR BR112016018062-3A patent/BR112016018062B1/pt active IP Right Grant
-
2016
- 2016-08-04 IL IL247128A patent/IL247128B/en active IP Right Grant
- 2016-10-11 US US15/290,565 patent/US9994521B2/en active Active
-
2017
- 2017-06-12 HK HK17105777.9A patent/HK1232137A1/zh unknown
- 2017-06-30 JP JP2017128851A patent/JP2017165782A/ja not_active Withdrawn
- 2017-10-20 AU AU2017248572A patent/AU2017248572B2/en active Active
-
2018
- 2018-04-11 US US15/950,545 patent/US10259785B2/en active Active
- 2018-07-06 JP JP2018129299A patent/JP6785824B2/ja active Active
-
2019
- 2019-02-27 US US16/287,222 patent/US11198673B2/en active Active
- 2019-12-18 AU AU2019283876A patent/AU2019283876B2/en active Active
-
2020
- 2020-04-16 IL IL274006A patent/IL274006A/en unknown
- 2020-09-16 JP JP2020155300A patent/JP2020203939A/ja not_active Withdrawn
-
2021
- 2021-10-20 US US17/506,276 patent/US12084414B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2945905T3 (es) | Compuestos y composiciones terapéuticos | |
| KR102090780B1 (ko) | 혈전색전성 질환의 치료를 위한 인자 xia 억제제로서 치환된 피롤리딘 | |
| BR112017016193B1 (pt) | Derivados de 4h-pirrolo[3,2-c]piridin-4-ona | |
| WO2018118705A1 (en) | Therapeutic compounds and compositions | |
| KR20160064100A (ko) | 치환된 페닐알라닌 유도체 | |
| EP2934536B1 (en) | FACTOR IXa INHIBITORS | |
| AU2019217366B2 (en) | Therapeutic compounds and compositions | |
| WO2017006295A1 (en) | Hydroxy formamide derivatives and their use | |
| US20180362485A1 (en) | N-HYDROXYFORMAMIDE COMPOUNDS AND COMPOSITIONS COMPRISING THEM FOR USE AS BMP l, TLL1 AND/OR TLL2 INHIBITORS |