ES2942642T3 - Composiciones para suministro yeyunoileal de fármacos - Google Patents
Composiciones para suministro yeyunoileal de fármacos Download PDFInfo
- Publication number
- ES2942642T3 ES2942642T3 ES15834774T ES15834774T ES2942642T3 ES 2942642 T3 ES2942642 T3 ES 2942642T3 ES 15834774 T ES15834774 T ES 15834774T ES 15834774 T ES15834774 T ES 15834774T ES 2942642 T3 ES2942642 T3 ES 2942642T3
- Authority
- ES
- Spain
- Prior art keywords
- dosage form
- oral dosage
- solid oral
- pharmaceutically acceptable
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0002—Galenical forms characterised by the drug release technique; Application systems commanded by energy
- A61K9/0004—Osmotic delivery systems; Sustained release driven by osmosis, thermal energy or gas
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2886—Dragees; Coated pills or tablets, e.g. with film or compression coating having two or more different drug-free coatings; Tablets of the type inert core-drug layer-inactive layer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2813—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/282—Organic compounds, e.g. fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
- A61K9/2846—Poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/2853—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, polyethylene oxide, poloxamers, poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
- A61K9/2866—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5026—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5073—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals having two or more different coatings optionally including drug-containing subcoatings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Inorganic Chemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Pyridine Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
La invención actual proporciona nuevas formulaciones que administran inhibidores de quinasa covalentes reversibles e irreversibles, en particular inhibidores de BTK, en el intestino delgado y específicamente en el íleon y el yeyuno del intestino delgado. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Composiciones para suministro yeyunoileal de fármacos
Referencia cruzada
Esta solicitud reivindica el beneficio de prioridad de la solicitud de patente provisional de Estados Unidos número de serie 62/096.812, presentada el 24 de diciembre de 2014.
Campo
La presente divulgación proporciona formas farmacéuticas de dosificación oral que comprenden moléculas de fármaco covalentes reversibles o irreversibles que no liberan el ingrediente activo en el estómago y liberan sustancialmente la molécula de fármaco en el íleon y yeyuno del intestino delgado y el uso de estas formulaciones para el tratamiento de enfermedades tratables mediante dichos compuestos, tales como cáncer y enfermedades autoinmunitarias.
Antecedentes
El tratamiento dirigido ha recibido mayor atención particularmente en el área de oncología, debido al éxito clínico de los inhibidores de cinasa como agentes antineoplásicos. Las dificultades continuadas al desarrollo de tratamientos dirigidos incluyen conseguir alta selectividad por la diana principal e inhibición prolongada para maximizar su eficacia terapéutica. Los fármacos covalentes se han convertido en una estrategia muy atractiva para diseñar tratamientos dirigidos de última generación, debido a su capacidad potenciada de conseguir alta selectividad, así como inhibición prolongada incluso con exposición sistémica significativamente reducida de los fármacos. Los fármacos covalentes consiguen su alta selectividad y potencia excepcional debido a la interacción covalente con un residuo de cisteína específico en el sitio activo de proteínas a las que se une la molécula de fármaco. Esta unión covalente proporciona adicionalmente eficacia prolongada con duración aumentada de acción que dura más que la exposición sistémica del fármaco. Los fármacos que contienen un resto de acrilamida como aceptadores de Michael en general reaccionan de forma irreversible con tioles como glutatión y también pueden reaccionar de forma irreversible con proteínas distintas de la diana deseada, especialmente proteínas con cisteínas hiperreactivas.
Las moléculas de fármaco covalentes reversibles, es decir, los fármacos que contienen un aceptados de Michael con un segundo grupo que extrae electrones, pueden presentar poca biodisponibilidad o retardo en absorción sistémica cuando el fármaco se administra por vía oral, lo que puede manifestarse por unos bajos valores de ABC y/o Cmáx en plasma, provocando eficacia subóptima in vivo. La poca biodisponibilidad de esta nueva clase de fármacos puede atribuirse, en parte, a la reactividad de restos aceptadores de Michael covalentes reversibles en estos fármacos. Por consiguiente, limitando la exposición de los fármacos covalentes reversibles al estómago, donde se produce la combinación de bajo pH y enzimas digestivas o metabólicas y otras fuentes de tioles, puede obtenerse un aumento significativo en la exposición sistémica del fármaco.
Además, limitar la exposición de moléculas de fármaco covalentes irreversibles al estómago, también puede dar lugar a un aumento significativo en la exposición sistémica del fármaco y una reducción en los posibles efectos secundarios adversos, tales como diarrea, náuseas o vómitos, y mareos. Por ejemplo, cuando se administra ibrutinib, una molécula de fármaco unida de forma covalente irreversible, por vía intraduodenal, la biodisponibilidad aumentó inesperadamente de un 21 % a un 100 % en comparación con la administración oral directa, que se determina por ABC. (D. M. Goldstein, Formulations Comprising Ibrutinib, documento WO 2014/004707 publicado el 3 de enero de 2014) La derivación gástrica de ibrutinib debe aumentar la biodisponibilidad y/o reducir o eliminar totalmente los posibles efectos secundarios adversos de este fármaco, tales como diarrea, náuseas o vómitos, y mareos.
Además, la expresión de enzimas metabolizantes, tales como cisteína proteasas, mucinas, transportadores y moléculas que contienen tiol reactivo en el estómago, tales como glutatión, también pueden contribuir a la baja biodisponibilidad oral de fármacos que contienen aceptador de Michael covalentes reversibles e irreversibles (véase, por ejemplo, Johnson D. S., et al., Future Med Chem. 1 de junio de 2010; 2(6):949-964 y Potashman M. H. et al. J. Med. Chem., Vol 52, N.° 5. Pág. 1231-1246). Por ejemplo, la combinación de enzimas digestivas, tales como la cisteína proteasa, pepsina, transportadores y enzimas metabolizantes tales como enzimas CYP en la mucosa gástrica, puede provocar un bajo pH en la elevada transformación química y/o metabólica de los aceptadores de Michael covalentes reversibles e irreversibles. Por consiguiente, evitando la exposición de los fármacos covalentes reversibles e irreversibles al estómago, donde se produce la combinación de bajo pH y enzimas digestivas o metabólicas y otras fuentes de tioles, puede obtenerse un aumento significativo en la exposición sistémica de estos fármacos. Además, evitar la exposición al estómago puede reducir o eliminar completamente los posibles efectos secundarios adversos de estos fármacos, tales como diarrea y vómitos, habitualmente denominados vómitos.
El documento WO 2013/191965 describe formulaciones farmacéuticas orales que comprenden compuestos covalentes reversibles que tienen un resto aceptador de Michael. El documento WO 2014/039899 describe inhibidores covalentes reversibles de tirosina cinasas, en particular BTK.
Sumario
Ahora se ha demostrado que hay un aumento inesperado en la absorción de moléculas de fármaco divulgadas en este documento en el yeyuno y el íleon en comparación con el duodeno y la biodisponibilidad puede mejorarse más con formas farmacéuticas que liberen la molécula de fármaco principalmente distal al duodeno y óptimamente en el íleon y el yeyuno. Este resultado es sorprendente ya que la liberación del fármaco más adelante en el tubo gastrointestinal reduce la longitud de trayecto para la absorción del fármaco y se esperaría que redujera la biodisponibilidad.
La presente invención proporciona una forma farmacéutica oral sólida, que es un comprimido o cápsula, que comprende:
1. (A) un núcleo que comprende un compuesto elegido de:


y/o una sal farmacéuticamente aceptable del mismo;
2. (B) un recubrimiento entérico;
3. (C) un subrecubrimiento entre el recubrimiento entérico y el núcleo, en donde el subrecubrimiento es:
(1) un polímero erosionable soluble en agua o hidrófilo, siendo dicho polímero un polímero de bajo peso molecular seleccionado de hidroximetilcelulosa (HPMC), hidroxietilcelulosa, hidroximetilcelulosa, hidroxipropilcelulosa, celulosa microcristalina, polivinilpirrolidionas, polisacáridos (o un derivado de polisacárido), poli(alcoholes vinílicos), polietilenglicol (PEG), polipropilenglicol (PPG) y copolímeros de bloque de PEG-PPG; o
(2) una composición insoluble en agua, que comprende: (i) partículas de compuesto soluble en agua que pueden formar canales en la composición insoluble en agua; o (ii) partículas hidrófilas insolubles en agua que provocan el hinchamiento del subrecubrimiento cuando está en contacto con un medio acuoso; y
4. (D) un excipiente farmacéuticamente aceptable,
en donde la forma farmacéutica oral sólida tiene un inicio de liberación del compuesto y/o dicha sal farmacéuticamente aceptable del mismo del núcleo (A) en la parte yeyunoileal del intestino delgado de un mamífero después de ingesta oral de la forma farmacéutica por parte del mamífero; y
en donde la forma farmacéutica oral sólida puede liberar al menos un 80 % en peso de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo en 120 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4.
La forma farmacéutica oral sólida puede liberar una molécula de fármaco (es decir, el compuesto y/o sal farmacéuticamente aceptable del mismo del núcleo (A)) en una ubicación predeterminada en el intestino después de la ingesta oral de la forma farmacéutica por parte de un mamífero.
La forma farmacéutica oral sólida suministra una molécula de fármaco a la parte yeyunoileal del intestino delgado de un mamífero.
En un aspecto, la forma farmacéutica oral sólida es una forma farmacéutica oral sólida de liberación retardada que libera una molécula de fármaco cuando la forma farmacéutica encuentra un pH específico en el tubo gastrointestinal de un mamífero.
La forma farmacéutica oral sólida comprende al menos un recubrimiento entérico que cubre el núcleo, cuyo recubrimiento entérico permanece intacto en el estómago de un mamífero.
La forma farmacéutica oral sólida puede liberar la molécula de fármaco en el íleon y/o el yeyuno de un mamífero, provocando una biodisponibilidad potenciada de la molécula de fármaco en comparación con la liberación inmediata de la molécula de fármaco después de la ingesta.
En otro aspecto, la forma farmacéutica oral sólida libera la molécula de fármaco como un bolo en el íleon y/o el yeyuno del intestino delgado de un mamífero, provocando una biodisponibilidad potenciada de la molécula de fármaco en comparación con la liberación inmediata de la molécula de fármaco después de la ingesta.
La forma farmacéutica oral sólida comprende un núcleo que contiene una molécula de fármaco en combinación con uno o más subrecubrimientos adicionales, diluyentes o excipientes diseñados para liberar sustancialmente la molécula de fármaco en el íleon y/o yeyuno del intestino delgado de un mamífero.
En otro aspecto, la forma farmacéutica oral sólida comprende un núcleo cubierto por un subrecubrimiento insoluble en agua que puede hincharse o formar canales que permiten en flujo entrante de agua al núcleo, lo que provoca el hinchamiento del núcleo, la rotura del subrecubrimiento y la liberación de los contenidos del núcleo como un bolo en el intestino.
En otro aspecto, la forma farmacéutica oral sólida contiene un núcleo cubierto por un subrecubrimiento que puede formar canales que provocan el flujo entrante de agua al núcleo y el posterior flujo saliente de la molécula de fármaco a través de los canales según pasa el núcleo a través del intestino.
En otro aspecto, la forma farmacéutica oral sólida contiene un núcleo cubierto por un polímero soluble en agua que se disuelve a un pH predeterminado en el intestino.
La invención proporciona además una forma farmacéutica oral sólida como se define en este documento para su uso en un método de tratamiento de una enfermedad tratable mediante inhibición de BTK, en un paciente que lo necesita reconocidamente, en donde dicho método comprende administrar al paciente una cantidad terapéuticamente eficaz del compuesto y/o la sal farmacéuticamente aceptable del mismo del núcleo (A) en una sola o múltiples dosis. Dicha forma farmacéutica oral sólida típicamente tiene biodisponibilidad mejorada en comparación con formulaciones que provocan la liberación inmediata de la molécula de fármaco después de la ingesta.
Breve descripción de las figuras
La figura 1 representa los resultados de un experimento farmacocinético que compara la dosificación por sonda oral e intraduodenal de (R)-2-(3-(4-amino-3-(2-fluoro-4-fenoxifenil)-1H-pirazolo[3,4-d]pirimidin-1-il)piperidina-1-carbonil)-4-metil-4-(4-(oxetan-3-il)piperazin-1-il)pent-2-enonitrilo que tiene una relación de E/Z de aproximadamente 9:1. Están dibujadas el área bajo la curva (ABC) y la Cmáx.
La figura 2 representa los resultados de un estudio de la permeabilidad de compuestos farmacológicamente activos que son aceptadores de la reacción de Michael en diversas regiones del tubo GI, incluyendo el estómago, el duodeno, el íleon, el yeyuno y el colon. Los fármacos estudiados incluyen (R)-2-(3-(4-amino-3-(2-fluoro-4-fenoxifenil)-1 H-pirazolo[3,4-d]pirimidin-1-il)piperidina-1-carbonil)-4-metil-4-(4-(oxetan-3-il)piperazin-1-il)pent-2-enonitrilo que tiene una relación de E/Z de aproximadamente 9:1 (P10), (R,E)-2-(3-(4-amino-3-(2-fluoro-4-fenoxifenil)-1 H-pirazolo[3,4-d]pirimidin-1-il)piperidina-1-carbonil)-4,4-dimetilpent-2-enonitrilo(P47), (R,E)-2-(3-(4-amino-3-(2-fluoro-4-fenoxifenil)-1 H-pirazolo[3,4-d]pirimidin-1-il)piperidina-1-carbonil)-4-metil-4-(4-metilpiperazin-1 -il)pent-2-enonitrilo (P61), 8-(3-(4-acriloilpiperazin-1 -il)propil)-6-(2,6-dicloro-3,5-dimetoxifenil)-2-(dimetilamino)pirido[2,3-d]pirimidin-7(8H)-ona (P13), 1 -[(3R)-3-[4-amino-3-(4-fenoxifenil)-1 H-pirazolo[3,4-d]pirimidin-1 -il]piperidin-1 -il]prop-2-en-1 -ona (ibrutinib), N-(3-((5-fluoro-2-((4-(2-metoxietoxi)fenil)amino)pirimidin-4-il)amino)fenil)acrilamida (CC-292), (2E)-2-ciano-3-(3,4-dihidroxi-5-nitrofenil)-N,N-dietilprop-2-enamida (entacapona) y 2-ciano-3,12-dioxooleana-1,9(11)dien-28-oato de metilo (bardoxolona metilo).
Descripción detallada
El artículo indefinido "un/o" o "una" entidad, como se usa en este documento, se refiere a uno o más de esa entidad; por ejemplo, un compuesto se refiere a uno o más compuestos o al menos un compuesto salvo que se indique de otro modo. Por tanto, las expresiones "un/o" (o "una"), "uno o más" y "al menos uno" pueden usarse indistintamente en esta documento.
El término "independientemente" se usa en este documento para indicar que una variable se aplica en un caso cualquiera independientemente de la presencia o ausencia de una variable que tiene la misma definición o una diferente dentro del mismo compuesto. Por tanto, en un compuesto en que R'' aparece dos veces y se define como "independientemente carbono o nitrógeno", ambos R'' pueden ser carbono, ambos R'' pueden ser nitrógeno, o un R'' puede ser carbono y el otro nitrógeno.
Cuando cualquier variable (por ejemplo, R1, R4a, Ar, X1 o Het) existe más de una vez en cualquier resto o fórmula que represente y que describa compuestos empleados o reivindicados en la presente invención, su definición en cada aparición es independiente de su definición en cada otra aparición. Además, son permisibles combinaciones de sustituyentes y/o variables solamente si dichos compuestos dan como resultado compuestos estables.
Una línea dibujada a través de un enlace, "
", se refiere al punto de fijación de un grupo funcional u otro resto químico al resto de la molécula de la que forma parte. Por tanto, por ejemplo:

Un enlace dibujado en sistema de anillo (en oposición a conectado en un vértice distinto) indica que el enlace puede fijarse a cualquiera de los átomos adecuados en el anillo.
El término "opcional" u "opcionalmente", como se usa en este documento, significa que un acontecimiento o circunstancia descrita posteriormente puede producirse, pero no necesariamente, y que la descripción incluye casos donde el acontecimiento o circunstancia se produce y casos en que no. Por ejemplo, "opcionalmente sustituido" significa que el resto opcionalmente sustituido puede incorporar un hidrógeno o un sustituyente.
El término "aproximadamente" se usa en este documento para indicar aproximado, en la región de, más o menos, o alrededor de. Cuando el término "aproximadamente" se usa junto con un intervalo numérico, modifica ese intervalo ampliando los límites por encima y por debajo de los valores numéricos expuestos. En general, el término "aproximadamente" se usa en este documento para modificar un valor numérico por encima y por debajo del valor indicado en una variación de un 5 %.
Como se usa en este documento, la enumeración de un intervalo numérico para una variable pretende transmitir que la invención puede ponerse en práctica con la variable igual a cualquiera de los valores dentro de ese intervalo. Por tanto, para una variable que es inherentemente discontinua, la variable puede ser igual a cualquier valor entero del intervalo numérico, incluyendo los valores extremos del intervalo. Asimismo, para una variable que es inherentemente continua, la variable puede ser igual a cualquier valor real del intervalo numérico, incluyendo los valores extremos del intervalo. Como un ejemplo, una variable que se describe por tener valores entre 0 y 2, puede ser 0, 1 o 2 para variables que son inherentemente discontinuas, y puede ser 0,0, 0,1, 0,01, 0,001, o cualquier otro valor real para variables que son inherentemente continuas.
Un aspecto de esta divulgación se refiere a una forma farmacéutica oral sólida para su uso en un método de tratamiento de una enfermedad tratable mediante inhibición de una tirosina cinasa tal como BLK, BMX, EGFR, HER2, HER4, ITK, TEC, BTK y TXK en un paciente, comprendiendo dicho método administrar al paciente que lo necesita reconocidamente, una formulación farmacéutica oral sólida divulgada en este documento. En una realización, la tirosina cinasa es BTK.
BTK (número de acceso UniProt Q06187) es un miembro de la familia Tec de tirosina cinasas, y ha demostrado ser un regulador crucial del desarrollo temprano de linfocitos B y la activación y supervivencia de linfocitos B maduros (Khan et al. Immunity 19953:283; Ellmeier et al. J. Exp. Med. 2000 192:1611).
Para evitar dudas, la expresión "molécula de fármaco" o "inhibidor de BTK", como se usa en este documento, se refiere a la base libre de un compuesto presente en el núcleo (A) y/o a una sal farmacéuticamente aceptable de un compuesto presente en el núcleo (A) salvo que se limite explícitamente a la base libre o una sal de la misma.
En una realización, la molécula de fármaco es un inhibidor de cinasa covalente reversible o irreversible. Por ejemplo, en una realización no limitante, un inhibidor de cinasa covalente reversible o irreversible es un inhibidor de BTK. En una realización, la molécula de fármaco es un inhibidor de BTK covalente reversible. Se ha revisado el uso de modificación covalente para modular dianas farmacológicas. (I. M. Serfimova etal., Nature Chem. Biol. 20128:471; R. Mah et al. Bioorg. Med. Chem. Lett. 201424:33-39; J. Sing, et al., Nat. Rev. Drug Discov. 2011 10:307).
En otra realización más, la molécula de fármaco es un inhibidor de BTK covalente irreversible.
En otra realización, la molécula de fármaco es un inhibidor de cinasa covalente reversible o irreversible elegido de ibrutinib, ACP196, acalabrutinib, BGB3111, ONO-4059, CC-292, X-022, afatinib, mereletinib, osimertinib, rociletinib, neratinib, dacomitinib, poziotinib, spebrutinib, tarloxotinib, selinexor, verdinexor, PF-06747775, BLU-554, NSC-687852, VLX-1570, NT-113, BLU-9931, KPT-350 y AZ-13767370.
En otra realización, la molécula de fármaco es un inhibidor de cinasa covalente reversible o irreversible elegido de:
(R)-1 -(3-(4-amino-3-(4-fenoxifenil)-1 H-pirazolo[3,4-d]pirimidin-1 -il)piperidin-1 -il)prop-2-en-1 -ona,
(R) -4-(8-amino-3-(1-(but-2-inoil)pirrolidin-2-il)imidazo[1,5-a]pirazin-1-il)-N-(piridin-2-il)benzamida, (S) -7-(1-acriloilpiperidin-4-il)-2-(4-fenoxifenil)-4,5,6,7-tetrahidropirazolo[1,5-a]pirimidina-3-carboxamida, (S)-7-(1-(but-2-inoil)piperidin-4-il)-2-(4-fenoxifenil)-4,5,6,7-tetrahidropirazolo[1,5-a]pirimidina-3-carboxamida, N-(3-((2-((2-metoxi-4-(4-metilpiperazin-1-il)fenil)amino)tieno[3,2-d]pirimidin-4-il)oxi)fenil)acrilamida, 1 -(3-((2-((4-(4-metilpiperazin-1 -il)fenil)amino)-[1,2,4]triazolo[1,5-a]piridin-8-il)oxi)fenil)prop-2-en-1 -ona, (R)-6-amino-9-(1-(but-2-inoil)pirrolidin-3-il)-7-(4-fenoxifenil)-7H-purin-8(9H)-ona,
N-(2-((6-((5-(5-fluoro-2-(hidroximetil)-3-(4-oxo-6,7,8,9-tetrahidrobenzo[4,5]tieno[2,3-d]piridazin-3(4H)-il)fenil)-1-metil-2-oxo-1,2-dihidropiridin-3-il)amino)piridin-2-il)amino)etil)acrilamida,
N-(3-((5-fluoro-2-((4-(2-metoxietoxi)fenil)amino)pirimidin-4-il)amino)fenil)acrilamida,
6-(1 -acriloilpiperidin-4-il)-2-(4-fenoxifenil)nicotinam ida,
(R)-1-(1-acriloilpiperidin-3-il)-4-amino-N-(5-clorobenzo[d]oxazol-2-il)-1 H-pirazolo[3,4-d]pirimidina-3-carboxamida,
(S,E)-N-(4-((3-cloro-4-fluorofenil)amino)-7-((tetrahidrofurano-3-il)oxi)quinazolin-6-il)-4-(dimetilamino)but-2-enamida,
N-(2-((2-(dimetilamino)etil)(metil)amino)-4-metoxi-5-((4-(1-metil-1 H-indol-3-il)pirimidin-2-il)amino)fenil)acrilamida,
N-(3-((2-((4-(4-acetilpiperazin-1-il)-2-metoxifenil)amino)-5-(trifluorometil)pirimidin-4-il)amino)fenil)acrilamida, (E)-N-(4-((3-cloro-4-(piridin-2-ilmetoxi)fenil)amino)-3-ciano-7-etoxiquinolin-6-il)-4-(dimetilamino)but-2-enamida,
(E)-N-(4-((3-cloro-4-fluorofenil)amino)-7-metoxiquinazolin-6-il)-4-(piperidin-1-il)but-2-enamida,
1-(4-((4-((3,4-dicloro-2-fluorofenil)amino)-7-metoxiquinazolin-6-il)oxi)piperidin-1-il)prop-2-en-1-ona, N-(3-((5-fluoro-2-((4-(2-metoxietoxi)fenil)amino)pirimidin-4-il)amino)fenil)acrilamida,
bromuro de (E)-4-((4-((3-bromo-4-clorofenil)amino)pirido[3,4-d]pirimidin-6-il)amino)-N,N-dimetil-N-((1-metil-4-nitro-1 H-imidazol-5-il)metil)-4-oxobut-2-en-1 -aminio,
(Z)-3-(3-(3,5-bis(trifluorometil)fenil)-1 H-1,2,4-triazol-1-il)-N'-(pirazin-2-il)acrilohidrazida,
(Z)-3-(3-(3,5-bis(trifluorometil)fenil)-1 H-1,2,4-triazol-1-il)-N'-(piridin-2-il)acrilohidrazida,
N-((3R,4R)-4-fluoro-1-(6-((3-metoxi-1-metil-1 H-pirazol-4-il)amino)-9-metil-9H-purin-2-il)pirrolidin-3-il)acrilamida,
N-((3S,4S)-3-((6-(2,6-dicloro-3,5-dimetoxifenil)quinazolin-2-il)amino)tetrahidro-2H-piran-4-il)acrilamida, (3E,5E)-1-acriloil-3,5-bis(4-nitrobencilideno)piperidin-4-ona,
(E)-N-(7-((1 R,5S,6s)-3-oxabiciclo[3.1.0]hexan-6-iletinil)-4-((3-cloro-4-fluorofenil)amino)quinazolin-6-il)-4-(dimetilamino)but-2-enamida,
(3E,5E)-1-acriloil-3,5-bis(4-fluoro-3-nitrobencilideno)azepan-4-ona,
N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)quinazolin-2-il)amino)-3-metilfenil)acrilamida,
(Z)-3-(3-(3,5-bis(trifluorometil)fenil)-1 H-1,2,4-triazol-1 -il)-N'-pivaloilacrilohidrazida y
N-(2-((2-((tetrahidro-2H-piran-4-il)amino)-5-(trifluorometil)pirimidin-4-il)amino)fenil)acrilamida.
En otra realización, la molécula de fármaco es un inhibidor de cinasa covalente reversible o irreversible elegido de:


Salvo que se indique de otro modo, los siguientes términos usados en la memoria descriptiva y reivindicaciones se definen para los propósitos de esta solicitud y tienen el significado:
"Alquilo", como se usa en este documento, significa un radical hidrocarbonado monovalente saturado lineal de uno a seis átomos de carbono o un radical hidrocarbonado monovalente saturado ramificado de tres a seis átomos de carbono, por ejemplo, metilo, etilo, propilo, 2-propilo, butilo (incluyendo todas las formas isoméricas), pentilo (incluyendo todas las formas isoméricas) y similares.
"Alquilo sustituido" significa grupo alquilo como se define en este documento, que está sustituido con uno, dos o tres sustituyentes seleccionados independientemente de hidroxilo, alcoxi, carboxi, ciano, carboxi, alcoxicarbonilo, alquiltio, alquilsulfonilo, halo, -CONRR' o -NRR' (donde cada R es hidrógeno, alquilo, cicloalquilo, hidroxialquilo o alcoxialquilo, y cada R' es hidrógeno, alquilo o cicloalquilo) o heterociclilo (por ejemplo, heterocicloamino) que está opcionalmente sustituido con uno o dos grupos seleccionados independientemente de alquilo, hidroxilo, alcoxi, alquiltio, alquilsulfonilo, halo o -CONRR' donde R y R' son como se definen anteriormente.
"Alquinilo", como se usa en este documento, significa un radical hidrocarbonado monovalente saturado lineal de dos a seis átomos de carbono o un radical hidrocarbonado monovalente saturado ramificado de tres a seis átomos de carbono que contiene un triple enlace, por ejemplo, etinilo, propinilo, 2-propinilo, butinilo (incluyendo todas las formas isoméricas), pentinilo (incluyendo todas las formas isoméricas) y similares.
"Alquileno", como se usa en este documento, significa un radical hidrocarbonado divalente saturado lineal de uno a seis átomos de carbono o un radical hidrocarbonado divalente saturado ramificado de tres a seis átomos de carbono salvo que se indique de otro modo, por ejemplo, metileno, etileno, propileno, 1-metilpropileno, 2-metilpropileno, butileno, pentileno y similares.
"Alquiltio", como se usa en este documento, significa un radical -SR donde R es alquilo como se define anteriormente, por ejemplo, metiltio, etiltio y similares.
"Alquilsulfonilo", como se usa en este documento, significa un radical -SO2 R donde R es alquilo como se define anteriormente, por ejemplo, metilsulfonilo, etilsulfonilo y similares.
"Amino" significa un -NH2.
"Alquilamino", como se usa en este documento, significa un radical -NHR donde R es alquilo como se define anteriormente, por ejemplo, metilamino, etilamino, propilamino o 2-propilamino y similares.
"Alcoxi", como se usa en este documento, significa un radical -OR donde R es alquilo como se define anteriormente, por ejemplo, metoxi, etoxi, propoxi o 2-propoxi, n-, ¡so - o ferc-butoxi y similares.
"Alcoxialquilo", como se usa en este documento, significa un radical hidrocarbonado monovalente lineal de uno a seis átomos de carbono o un a radical hidrocarbonado monovalente ramificado de tres a seis carbonos sustituidos con al menos un grupo alcoxi, por ejemplo, uno o dos grupos alcoxi, como se definen anteriormente, por ejemplo, 2-metoxietilo, 1-, 2- o 3-metoxipropilo, 2-etoxietilo y similares.
"Alcoxicarbonilo", como se usa en este documento, significa un radical -C(O)OR donde R es alquilo como se define anteriormente, por ejemplo, metoxicarbonilo, etoxicarbonilo y similares.
"Aminocarbonilo", como se usa en este documento, significa un radical -CONRR' donde R es independientemente hidrógeno, alquilo o alquilo sustituido, cada uno como se define en este documento y R' es hidrógeno, alquilo, cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heteroarilo, heteroaralquilo, heterociclilo, heterociclilalquilo o alquilo sustituido, cada uno como se define en este documento y donde el arilo, heteroarilo o anillo heterociclilo en solitario o como parte de otro grupo, por ejemplo, aralquilo, está opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente de alquilo, hidroxi, alcoxi, halo, haloalquilo, haloalcoxi, carboxi, alcoxicarbonilo, alquilcarbonilo, ciano, -CONH2 , alquilaminocarbonilo, dialquilaminocarbonilo o alquilaminocarbonilo sustituido, por ejemplo, -CONH2 , metilaminocarbonilo, 2-dimetilaminocarbonilo y similares. Cuando R es hidrógeno y R' es alquilo en -CONRR', el grupo también se denomina en este documento alquilaminocarbonilo y cuando R y R' son ambos alquilo en -CONRR', el grupo también se denomina en este documento dialquilaminocarbonilo. Cuando R es hidrógeno y R' es alquilo sustituido en -CONRR', el grupo también se denomina en este documento alquilaminocarbonilo sustituido.
"Aminosulfonilo", como se usa en este documento, significa un radical -SO2NRR' donde R es independientemente hidrógeno, alquilo o alquilo sustituido, cada uno como se define en este documento y R' es hidrógeno, alquilo, cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heteroarilo, heteroaralquilo, heterociclilo, heterociclilalquilo o alquilo sustituido, cada uno como se define en este documento y en donde el arilo, heteroarilo o anillo heterociclilo en solitario o como parte de otro grupo, por ejemplo, aralquilo, está opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente de alquilo, hidroxi, alcoxi, halo, haloalquilo, haloalcoxi, carboxi, alcoxicarbonilo, alquilcarbonilo, ciano, -CONH2 , alquilaminocarbonilo, dialquilaminocarbonilo, alquilaminocarbonilo sustituido, por ejemplo, -SO2NH2 , metilaminosulfonilo, dimetilaminosulfonilo y similares. Cuando R es hidrógeno y R' es alquilo en -SO2NRR', el grupo también se denomina en este documento alquilaminosulfonilo y cuando R y R' son ambos alquilo en -SO2NRR', el grupo también se denomina en este documento dialquilaminosulfonilo.
"Acilo", como se usa en este documento, significa un radical -COR donde R es alquilo, haloalquilo, cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heteroarilo, heteroaralquilo, heterociclilo o heterociclilalquilo, cada uno como se define en este documento, y en donde el arilo, heteroarilo o anillo heterociclilo en solitario o como parte de otro grupo, por ejemplo, aralquilo, está opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente de alquilo, hidroxi, alcoxi, halo, haloalquilo, haloalcoxi, carboxi, alcoxicarbonilo, alquilcarbonilo, ciano, -CONH2 , alquilaminocarbonilo, dialquilaminocarbonilo o alquilaminocarbonilo sustituido, por ejemplo, acetilo, propionilo, benzoílo, piridinilcarbonilo y similares. Cuando R es alquilo, el radical también se denomina en este documento alquilcarbonilo.
"Arilo", como se usa en este documento, significa un radical hidrocarbonado aromático monocíclico o bicíclico monovalente de 6 a 10 átomos en el anillo, por ejemplo, fenilo o naftilo.
"Aralquilo", como se usa en este documento, significa un radical -(alquileno)-R donde R es arilo como se define anteriormente.
"Cicloalquilo", como se usa en este documento, significa un radical hidrocarbonado monovalente saturado cíclico de tres a diez átomos de carbono, en donde uno o dos átomos de carbono pueden remplazarse por un grupo oxo, por ejemplo, ciclopropilo, ciclobutilo, ciclopentilo o ciclohexilo y similares.
"Cicloalquilalquilo", como se usa en este documento, significa un radical -(alquileno)-R donde R es cicloalquilo, como se define anteriormente; por ejemplo, ciclopropilmetilo, ciclobutilmetilo, ciclopentiletilo o ciclohexilmetilo y similares.
"Cicloalquileno", como se usa en este documento, significa un radical hidrocarbonado divalente saturado cíclico de tres a diez átomos de carbono, en donde uno o dos átomos de carbono pueden remplazarse por un grupo oxo, por ejemplo, ciclopropileno, ciclobutileno, ciclopentileno o ciclohexileno y similares.
Cicloalquileno(alquileno), como se usa en este documento, significa un radical -(alquileno)-R donde R es cicloalquileno como se define en este documento y un punto de fijación del radical cicloalquilo divalente es al resto alquileno.
"Carboxi" significa -COOH.
"Amino disustituido" significa un radical -NRR' donde R y R' son independientemente alquilo, cicloalquilo, cicloalquilalquilo, acilo, arilo, aralquilo, heteroarilo, heteroaralquilo, heterociclilo, heterociclilalquilo o alquilo sustituido, cada uno como se define en este documento, y en donde el arilo, heteroarilo o anillo heterociclilo en solitario o como parte de otro grupo, por ejemplo, aralquilo, está opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente de alquilo, hidroxi, alcoxi, halo, haloalquilo, haloalcoxi, carboxi, alcoxicarbonilo, alquilcarbonilo, ciano, -CONH2 , alquilaminocarbonilo, dialquilaminocarbonilo o alquilaminocarbonilo sustituido, por ejemplo, dimetilamino, fenilmetilamino y similares. Cuando los grupos R y R' son alquilo, el grupo amino disustituido puede denominarse en este documento dialquilamino.
"Halo", como se usa en este documento, significa fluoro, cloro, bromo o yodo, por ejemplo, fluoro o cloro.
"Haloalquilo", como se usa en este documento, significa radical alquilo como se define en este documento, que está sustituido con uno o más átomos de halógeno, por ejemplo, de uno a cinco átomos de halógeno, por ejemplo, flúor o cloro, incluyendo los sustituidos con diferentes halógenos, por ejemplo, -CH2Cl, -CF3 , -CHF2 , -CH2CF3 , -CF2CF3 , -CF(CH3)2 y similares. Cuando el alquilo está sustituido con solamente fluoro, se denomina en esta solicitud fluoroalquilo.
"Haloalcoxi", como se usa en este documento, significa un radical -OR donde R es haloalquilo como se define anteriormente, por ejemplo, -OCF3 , -OCHF2 y similares. Cuando R es haloalquilo donde el alquilo está sustituido con solamente fluoro, se denomina en esta solicitud fluoroalcoxi.
"Hidroxialquilo", como se usa en este documento, significa un radical hidrocarbonado monovalente lineal de uno a seis átomos de carbono o un radical hidrocarbonado monovalente ramificado de tres a seis carbonos sustituido con uno o dos grupos hidroxi, con la condición de que si dos grupos hidroxi están presentes, no estén ambos en el mismo átomo de carbono. Ejemplos representativos incluyen, aunque sin limitación, hidroximetilo, 2-hidroxietilo, 2-hidroxipropilo, 3-hidroxipropilo, 1-(hidroximetil)-2-metilpropilo, 2-hidroxibutilo, 3-hidroxibutilo, 4-hidroxibutilo, 2,3-dihidroxipropilo, 1-(hidroximetil)-2-hidroxietilo, 2,3-dihidroxibutilo, 3,4-dihidroxibutilo y 2-(hidroximetil)-3-hidroxipropilo, por ejemplo, 2-hidroxietilo, 2,3-dihidroxipropilo y 1-(hidroximetil)-2-hidroxietilo.
"Heterociclilo", como se usa en este documento, significa un grupo monocíclico monovalente saturado o insaturado de 4 a 8 átomos en el anillo en que uno o dos átomos en el anillo son heteroátomos seleccionados de N, O o S(O)n, donde n es un número entero de 0 a 2, siendo los átomos restantes en el anillo C. El anillo heterociclilo está opcionalmente condensado a un (uno) arilo o anillo heteroarilo como se define en este documento, con la condición de que los anillos arilo y heteroarilo sean monocíclicos. El anillo heterociclilo condensado a anillos arilo o heteroarilo monocíclico también se denomina en esta solicitud anillo "heterociclilo bicíclico". Además, uno o dos átomos de carbono del anillo en el anillo heterociclilo pueden remplazarse opcionalmente por un grupo -CO-. Más específicamente, el término heterociclilo incluye, aunque sin limitación, pirrolidino, piperidino, homopiperidino, 2-oxopirrolidinilo, 2-oxopiperidinilo, morfolino, piperazino, tetrahidropiranilo, tiomorfolino y similares. Cuando el anillo heterociclilo está insaturado, puede contener uno o dos dobles enlaces en el anillo, con la condición de que el anillo no sea aromático. Cuando el grupo heterociclilo contiene al menos un átomo de nitrógeno, también se denomina en este documento heterocicloamino y es un subconjunto del grupo heterociclilo. Cuando el grupo heterociclilo es un anillo saturado y no está condensado a anillo arilo o heteroarilo como se indica anteriormente, también se denomina en este documento heterociclilo monocíclico saturado.
"Heterociclilalquilo", como se usa en este documento, significa un radical -(alquileno)-R donde R es anillo heterociclilo como se define anteriormente, por ejemplo, tetrahidrofuranilmetilo, piperazinilmetilo, morfoliniletilo y similares.
"Heterocicloamino", como se usa en este documento, significa un grupo monocíclico monovalente saturado o insaturado de 4 a 8 átomos en el anillo en que uno o dos átomos en el anillo son heteroátomos seleccionados de N, O o S(O)n, donde n es un número entero de 0 a 2, siendo los átomos restantes en el anillo C, con la condición de que al menos uno de los átomos en el anillo sea N. Además, uno o dos átomos de carbono del anillo en el anillo heterocicloamino pueden remplazarse opcionalmente por un grupo -CO-. Cuando el anillo heterocicloamino está insaturado, puede contener uno o dos dobles enlaces en el anillo, con la condición de que el anillo no sea aromático.
"Heteroarilo", como se usa en este documento, significa un radical aromático monocíclico o bicíclico monovalente de 5 a 10 átomos en el anillo donde uno o más, por ejemplo, uno, dos o tres átomos en el anillo son heteroátomos seleccionados de N, O o S, siendo los átomos restantes en el anillo carbono. Ejemplos representativos incluyen,
aunque sin limitación, pirrolilo, tienilo, tiazolilo, imidazolilo, furanilo, indolilo, isoindolilo, oxazolilo, isoxazolilo, benzotiazolilo, benzoxazolilo, quinolinilo, isoquinolinilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo, triazolilo, tetrazolilo y similares.
"Heteroaralquilo", como se usa en este documento, significa un radical -(alquileno)-R donde R es heteroarilo como se define en este documento."Arilo sustituido o heteroarilo sustituido" significa arilo o heteroarilo como se definen anteriormente, que está sustituido con uno, dos o tres sustituyentes seleccionados independientemente de hidrógeno, alquilo, alquilo sustituido, cicloalquilo, cicloalquilalquilo, haloalquilo, haloalcoxi, halógeno, amino, amino monosustituido, amino disustituido, acilo, aminocarbonilo, aminosulfonilo, -OR', -SR', -OC(O)R', -CO2 R', -NR''C(O)R', -NR''C(O)NR'R", -NR''C(O)2 R', -SO2 R', -NR''SO2 R', -CN, -NO2 , arilo, aralquilo, heteroarilo, heteroaralquilo, heterociclilo y heterociclilalquilo donde R' es hidrógeno, alquilo, haloalquilo, alquilo sustituido, cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heteroarilo, heteroaralquilo, heterociclilo o heterociclilalquilo y R'' es hidrógeno, alquilo o alquilo sustituido; o R' y R'' junto con el átomo de nitrógeno al que están unidos forman heterocicloamino; con la condición de que al menos uno de los tres sustituyentes no sea hidrógeno y, además, en donde cada uno del arilo (excepto en "arilo sustituido"), heteroarilo (excepto en "arilo sustituido"), cicloalquilo, heterocicloamino y anillo heterociclilo en cualquiera de los grupos anteriores, está opcionalmente sustituido con:
(i) uno, dos o tres sustituyentes seleccionados independientemente de hidrógeno, alquilo, alquilo sustituido, cicloalquilo, cicloalquilalquilo, haloalquilo, haloalcoxi, halógeno, amino, amino monosustituido, amino disustituido, acilo, aminocarbonilo, aminosulfonilo, -OR', -SR', -OC(O)R', -CO2 R', -NR''C(O)R', -NR''C(O)NR'R'', -NR''C(O)2 R', -SO2 R', -NR''SO2 R', -CN, -NO2 , arilo, aralquilo, heteroarilo, heteroaralquilo, heterociclilo o heterociclilalquilo donde R' es hidrógeno, alquilo, haloalquilo, alquilo sustituido, cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heteroarilo, heteroaralquilo, heterociclilo o heterociclilalquilo y R'' es hidrógeno, alquilo o alquilo sustituido; o R' y R'' junto con el átomo de nitrógeno al que están unidos forman heterocicloamino; y, además, en donde el arilo, heteroarilo, cicloalquilo, heterocicloamino o anillo heterociclilo en cualquiera de los grupos anteriores en (i) está sustituido con uno, dos o tres sustituyentes seleccionados independientemente de hidrógeno, alquilo, hidroxi, alcoxi, halo, haloalquilo, haloalcoxi, carboxi, alcoxicarbonilo, alquilcarbonilo, ciano, -CONH2 , alquilaminocarbonilo, dialquilaminocarbonilo, alquilaminocarbonilo sustituido, amino o amino monosustituido o disustituido.
"Heteroalquileno", como se usa en este documento, significa un radical -(alquileno)- donde uno, dos o tres carbonos en la cadena alquileno están remplazados por -O-, N(H, alquilo o alquilo sustituido), S, SO, SO2 o CO.
"Amino monosustituido", como se usa en este documento, significa un radical -NHR donde R es alquilo, cicloalquilo, cicloalquilalquilo, acilo, arilo, aralquilo, heteroarilo, heteroaralquilo, heterociclilo, heterociclilalquilo o alquilo sustituido, cada uno como se define en este documento, y en donde el arilo, heteroarilo o anillo heterociclilo en solitario o como parte de otro grupo, por ejemplo, aralquilo, está opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente de alquilo, hidroxi, alcoxi, halo, haloalquilo, haloalcoxi, carboxi, alcoxicarbonilo, alquilcarbonilo, ciano, -CONH2 , alquilaminocarbonilo, dialquilaminocarbonilo o alquilaminocarbonilo sustituido, por ejemplo, metilamino, fenilamino, hidroxietilamino y similares. Cuando R es alquilo, el grupo amino monosustituido puede denominarse en este documento alquilamino.
La expresión "forma farmacéutica oral sólida" se refiere a la forma de farmacéutica oral administrada a un paciente, que comprende un recubrimiento entérico que rodea un núcleo que contiene la molécula de fármaco, cuyo núcleo opcionalmente comprende excipientes, diluyentes, vehículos y recubrimientos.
La expresión "liberación retardada", como se usa en este documento, significa la liberación de la molécula de fármaco desde la forma farmacéutica hasta que se alcanza un tiempo o sitio predeterminado en el tubo GI por la forma farmacéutica.
La expresión "derivado de celulosa" o "derivado de polisacárido" se refiere a un polímero de celulosa o polisacárido, en donde al menos una parte de los hidroxilos en las unidades de repetición de sacárido se han reaccionado para formar un enlace éter o éster. Ejemplos incluyen y no se limitan a hidroxialquilcelulosas, hidroxialquilalquilcelulosas, y ésteres de carboxialquilcelulosa, tales como hidroxipropilmetilcelulosas (hipromelosas o HPMC), hidroxipropilcelulosas (HPC) y similares.
El término "hidrófilo", para los propósitos de la presente divulgación, se refiere a materiales que tienen afinidad hacia agua.
La expresión "soluble en agua", para los propósitos de la presente divulgación, se refiere a materiales que se disuelven al grado requerido, en un medio acuoso a un pH de aproximadamente 1 a aproximadamente 8, y no está particularmente limitado.
La expresión "hinchable en agua", para los propósitos de la presente divulgación, se refiere a materiales que son relativamente insolubles en agua, pero que pueden absorber agua.
Materiales hidrófilos adecuados comprenden materiales solubles en agua o hinchables en agua. Ejemplos de dichos materiales incluyen sales, glúcidos y polímeros tales como hidroxialquilcelulosas, hidroxialquilalquilcelulosas y ésteres de carboxialquilcelulosa, por ejemplo, hidroxipropilmetilcelulosas (hipromelosas o HPMC), hidroxipropilcelulosas (HPC) y combinaciones que comprenden uno o más de los materiales anteriores. Las hidroxipropilmetilcelulosas que son de naturaleza hidrófila y pueden usarse en la presente divulgación se venden en diferentes grados de viscosidad, tales como las vendidas con la marca comercial Methocel™ disponible en Dow Chemical Co. Ejemplos de hidroxipropilmetilcelulosas de un bajo grado de viscosidad incluyen las disponibles con las marcas comerciales Methocel E5, Methocel E-15 LV, Methocel E50 LV, Methocel K100 LV y Methocel F50 LV, cuyas soluciones acuosas al 2 % en peso tienen viscosidades de 5 cP, 15 cP, 50 cP, 100 cP y 50 cP, respectivamente. Ejemplos de hidroxipropilmetilcelulosas que tienen una viscosidad media incluyen las disponibles con las marcas comerciales Methocel E4M y Methocel K4M, cuyas soluciones acuosas al 2 % en peso de ambas tienen una viscosidad de 4000 cP. Ejemplos de polímeros de hidroxipropilmetilcelulosa que tienen alta viscosidad incluyen los disponibles con las marcas comerciales Methocel K15M y Methocel K100M, cuyas soluciones acuosas al 2 % en peso tienen viscosidades de 15000 cP y 100000 cP, respectivamente. Los polímeros de hidroxipropilmetilcelulosa pueden estar presentes en las composiciones farmacéuticas de la presente divulgación en cantidades de aproximadamente un 0,1 % a un 50 % en peso.
Los polímeros de hidroxipropilcelulosa que pueden usarse en la presente divulgación también incluyen, por ejemplo, polímeros disponibles con la marca comercial Klucel™, disponibles en Nippon Soda Co. Los polímeros de hidroxipropilcelulosa disponibles con las marcas comerciales Klucel EF, Klucel LF, Klucel JF y Klucel GF, cuyas soluciones acuosas al 2 % en peso tienen viscosidades menores de 1000 cP, son ejemplos de polímeros hidrófilos de baja viscosidad. Un polímero de hidroxipropilcelulosa disponible con la marca comercial Klucel ME, cuya solución acuosa al 2 % en peso tiene una viscosidad en el intervalo de 4000-6500 cP es un polímero hidrófilo de viscosidad media. Los polímeros de hidroxipropilcelulosa disponibles vendidos como HPC-SL, HPC-L, y HPC-M, cuyas soluciones acuosas al 2 % en peso tienen viscosidades de 3-6 cP, 6-10 cP y 150-400 cP, respectivamente, son ejemplos de polímeros hidrófilos de baja viscosidad, mientras que HPC-H tiene una viscosidad de 1000-4000 cP y es un ejemplo de un polímero hidrófilo de viscosidad media. Los polímeros de hidroxipropilcelulosa pueden estar presentes en una cantidad de aproximadamente un 0,1 % a un 50 % en peso.
Los materiales hinchables en agua adecuados para preparar formas de dosificación de liberación retardada son compuestos que pueden expandirse cuando se exponen a líquidos acuosos, tales como líquidos gastrointestinales. Uno o más compuestos hinchables en agua pueden estar presentes en un recubrimiento y opcionalmente uno o más excipientes farmacéuticamente aceptables.
Compuestos adecuados que pueden usarse como sustancias hinchables en agua incluyen, por ejemplo, hidroxipropilcelulosas con baja sustitución, por ejemplo, L-HPC, polivinilpirrolidonas reticuladas, por ejemplo, PVP-XL, Kollidone™ CL y Polyplasdone™ XL, carboximetilcelulosa de sodio, carboximetilcelulosa de sodio reticulada, por ejemplo, Ac-di-sol™ y Primellose™, almidón glicolato de sodio, por ejemplo, Primojel™, carboximetilcelulosas de sodio, por ejemplo, Nymcel™ ZSB10, carboximetil almidones de sodio, por ejemplo, Explotab™, resinas de intercambio iónico, por ejemplo, productos Dowex™ o Amberlite™, celulosa microcristalina, por ejemplo, productos Avicel™, almidones y almidones pregelatinizados, por ejemplo, Starch 1500™ y Sepistab ST200™, formalina-caseína, por ejemplo, Plas-Vita™, y combinaciones que comprenden una o más de las sustancias hinchables en agua anteriores.
En algunas realizaciones, los materiales hidrófilos incluyen óxidos de polialquileno, gomas de polisacárido y ácidos poliacrílicos reticulados. Óxidos de polialquileno adecuados, tales como polímeros lineales de óxido de etileno sin sustituir, incluyen productos Polyox™ de The Dow Chemical Company, EE. UU., que tienen pesos moleculares de aproximadamente 100000-7000000. Otros polímeros de óxido de polialquileno útiles se preparan a partir de óxido de propileno, o mezclas de óxido de etileno y óxido de propileno.
Pueden usarse gomas de polisacárido, tanto naturales como modificadas (semisintéticas). Ejemplos son dextrano, goma xantana, goma gellan, goma welan y goma rhamsan.
Ácidos poliacrílicos reticulados que pueden usarse incluyen los que tienen propiedades similares a las descritas anteriormente para celulosa sustituida con alquilo y polímeros de óxido de polialquileno. Ácidos poliacrílicos reticulados útiles incluyen aquellos con viscosidades de aproximadamente 4000 a aproximadamente 40000 cP (para una solución acuosa al 1 % a 25 °C). Tres ejemplos específicos son CARBOPOL™ grados 971 P, 974P y 934P (vendidos por The Lubrizol Corporation, Cleveland, Ohio, EE. UU.). Ejemplos adicionales son polímeros conocidos como WATER LOCK™, que son copolímeros de almidón/acrilato/acrilamida disponibles en Grain Processing Corporation, Muscatine, Iowa, EE. UU.
La hidrofilia y la capacidad de hincharse de agua de estos polímeros provocan que el subrecubrimiento se hinche de tamaño después de administración oral, debido a la entrada de agua. La tasa de liberación del agente activo desde el subrecubrimiento depende principalmente de la tasa de inhibición de agua y la tasa a la que el agente activo se disuelve y difunde desde el polímero hinchado, que a su vez está relacionada con la solubilidad y tasa de disolución del agente activo, el tamaño de partícula del agente activo y/o la concentración de agente activo en la forma farmacéutica.
Materiales "hidrófobos" adecuados son ceras neutras o sintéticas insolubles en agua, alcoholes grasos tales como alcohol laurílico, miristílico, estearílico, cetílico o cetoestearílico, ácidos grasos y derivados de los mismos, incluyendo ésteres de ácido graso tales como monoestearato de glicerilo, monooleato de glicerol, monoglicéridos acetilados, estearina, palmitina, laurina, miristina, cera de ésteres cetílicos, palmitoestearato de glicerilo, behenato de glicerilo, aceites de ricino hidrogenados, aceites de semilla de algodón, glicéridos (mono-, di- y triglicéridos) de ácido graso, grasas hidrogenadas, hidrocarburos, ceras normales, ácido esteárico, alcohol estearílico, materiales que tienen cadenas principales hidrocarbonadas y combinaciones que comprenden uno o más de los materiales anteriores. Ceras adecuadas incluyen, aunque sin limitación, cera de abejas, Glycowax® (una N,N'-diestearoiletielendiamina, de Lonza), cera de ricino, cera de carnauba y sustancias similares a cera.
La expresión "forma farmacéutica de liberación inmediata", como se usa en este documento, se refiere a una formulación de dosificación, en forma líquida o sólida, que libera fármaco en el estómago y no tiene un recubrimiento protector para retardar el contacto del fármaco con la mucosa intestinal. Un recubrimiento entérico o enmascarador del sabor puede incluirse en la "forma farmacéutica de liberación inmediata".
El término "núcleo", como se usa en este documento, se refiere a todos los componentes de la forma farmacéutica oral sólida que están rodeados por el subrecubrimiento (C) y el recubrimiento entérico exterior (B) que es estable en pH ácido en el estómago y sirve para minimizar la exposición del núcleo que contiene la molécula de fármaco al estómago. El núcleo comprende la molécula de fármaco y opcionalmente otros excipientes, diluyentes y vehículos. El núcleo también puede comprender opcionalmente uno o más recubrimientos adicionales distintos del subrecubrimiento (C) y el recubrimiento entérico exterior (B).
La expresión "recubrimiento entérico", como se usa en este documento, se refiere a un recubrimiento polimérico sensible al pH que evita o minimiza la liberación o disolución de la molécula de fármaco en el estómago, pero permite la liberación en el intestino delgado. Los recubrimientos entéricos se usan para proteger la molécula de fármaco de la degradación completa o parcial en el entorno ácido del estómago.
La expresión "líquido intestinal simulado", como se usa en este documento, se refiere al medio de disolución descrito en la Farmacopea Estadounidense 33-28NF (2010) y la Farmacopea Europea 7.0 (2010).
Los términos "recubrimiento", "subrecubrimiento", "revestimiento", "película", "capa", "cobertura", "membrana" y similares son intercambiable.
El término "canal", como se usa en este documento, se refiere a una trayectoria en un recubrimiento o subrecubrimiento que permite la captación de agua en el núcleo y/o el flujo saliente de la molécula de fármaco a través del recubrimiento o subrecubrimiento. En algunas realizaciones, la captación en agua puede provocar el hinchamiento del núcleo, que permite el flujo entrante de agua o el flujo saliente de la molécula de fármaco. En realizaciones en que el subrecubrimiento es un polímero insoluble en agua que permite el flujo entrante de agua que provoca suficiente hinchamiento del núcleo para romper el subrecubrimiento que cubre el núcleo, liberando de ese modo la molécula de fármaco inhibidora como un bolo.
Cmáx y ABC son parámetros usados para evaluar la biodisponibilidad de una molécula de fármaco. El término Cmáx, como se usa en este documento, significa le concentración máxima en plasma de la molécula de fármaco conseguida después de la administración de una sola dosis de la forma farmacéutica. La expresión "área bajo la curva" (ABC), como se usa en este documento, se refiere al área bajo una curva (es decir, la integral) de un diagrama de concentración de fármaco en plasma sanguíneo frente al tiempo. La ABC (desde cero al infinito) representa la exposición de fármaco total a lo largo del tiempo y es proporcional a la cantidad total de fármaco absorbido por el organismo (es decir, la cantidad total de fármaco que alcanza la circulación sanguínea) suponiendo una farmacocinética lineal.
La expresión "farmacéuticamente aceptable" indica que la sustancia o composición es compatible química y/o toxicológicamente, con los otros ingredientes que comprende una formulación, y/o el mamífero que se está tratando con la misma.
Mamífero, como se usa en este documento, significa animales domesticados tales como perros, gatos, caballos y seres humanos. Preferiblemente un paciente humano.
La expresión "sal farmacéuticamente aceptable" de un compuesto significa una sal que es farmacéuticamente aceptable y que posee la actividad farmacológica deseada del compuesto precursor. Dichas sales incluyen: (1) sales de adición de ácido, formadas con ácidos inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico y similares; o formadas con ácidos orgánicos tales como ácido acético, ácido propiónico, ácido hexanoico, ácido ciclopentanopropiónico, ácido glicólico, ácido pirúvico, ácido láctico, ácido malónico, ácido succínico, ácido málico, ácido maleico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido 3-(4-hidroxibenzoil)benzoico, ácido cinámico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido 1,2-etano-disulfónico, ácido 2-hidroxietanosulfónico, ácido bencenosulfónico, ácido 4-clorobencenosulfónico, ácido 2
naftalenosulfónico, ácido 4-toluenosulfónico, ácido alcanforsulfónico, ácido 4-metilbiciclo[2.2.2]-oct-2-eno-1-carboxílico, ácido glucoheptónico, ácido 3-fenilpropiónico, ácido trimetilacético, ácido ferc-butilacético, ácido laurilsulfúrico, ácido glucónico, ácido glutámico, ácido hidroxinaftoico, ácido salicílico, ácido esteárico, ácido mucónico y similares; o (2) sales formadas cuando un protón ácido presente en el compuesto precursor se remplaza por un ion metálico, por ejemplo, un ion de metal alcalino, un ion alcalinotérreo o un ion de aluminio; o se coordina con una base orgánica tal como etanolamina, dietanolamina, trietanolamina, trometamina, N-metilglucamina y similares.
La expresión "recubrimiento entérico", como se usa en este documento, se refiere un recubrimiento polimérico sensible al pH que evita o minimiza la liberación o disolución de la molécula de fármaco en el estómago, pero permite la liberación en el intestino delgado e incluye, aunque sin limitación, polímeros y copolímeros de acrilato, metacrilato y etacrilato, derivados de celulosa y acetatos de polivinilo. Las propiedades de los polímeros de acrilato, metacrilato y etacrilato (principalmente su solubilidad en líquidos biológicos) pueden variar en función del grado y tipo de sustitución. Ejemplos de polímeros acrílicos adecuados incluyen copolímeros de ácido metacrílico y copolímeros de metacrilato de amonio. Las series Eudragit L, S y RS (fabricadas por Rohm Pharma y conocidas como Evonik®) están disponibles solubilizadas en un disolvente orgánico, dispersión acuosa o polvos secos. Las series Eudragit RL, NE y RS son insoluble en el tubo gastrointestinal, pero son permeable y se usan principalmente para dirección al colon. Las series Eudragit L, L-30D y S son insolubles en el estómago y se disuelven en el intestino. El término poli(met)acrilato significa polímeros de poliacrilato, polímeros o copolímeros de polimetacrilato que contienen tanto ácido acrílico como metacrílico.
Ejemplos de derivados de celulosa adecuados incluyen etilcelulosa y mezclas de reacción de ésteres parciales de acetato de celulosa con anhídrido ftálico. El rendimiento puede variar en función del grado y tipo de sustitución. El acetato ftalato de celulosa (CAP) se disuelve en pH >6. Aquateric (FMC) es un sistema de base acuosa y es un seudolátex CAP secado por pulverización con partículas <1 ^m. Otros componentes de Aquateric pueden incluir Pluronics®, Tweens® y monoglicéridos acetilados. Otros derivados de celulosa adecuados incluyen; acetato trimelitato de celulosa (CAT, Eastman); acetato succinato de celulosa (CAS), metilcelulosa (Pharmacoat, Methocel™); ftalato de hidroxipropilmetilcelulosa (HPMCP); succinato de hidroxipropilmetilcelulosa (HPMCS); y acetato succinato de hidroxipropilmetilcelulosa (HPMCAS, por ejemplo, AQOAT (Shin Etsu)). El rendimiento puede variar en función del grado y tipo de sustitución. Por ejemplo, son adecuados HPMCP tales como los grados HP-50, HP-55, HP-55S, HP-55F. Los grados adecuados de acetato succinato de hidroxipropilmetilcelulosa incluyen, aunque sin limitación, AS-LG (LF), que se disuelve a pH 5, AS-MG (MF), que se disuelve a pH 5,5 y AS-HG (HF), que se disuelve a pH mayor. Estos polímeros se ofrecen como gránulos, o como polvos finos para dispersiones acuosas.
El acetato ftalato de polivinilo (PVAP) se disuelve en pH mayor de 5, y es mucho menos permeable al vapor de agua y los líquidos gástricos. Puede encontrarse descripción detallada de los polímeros anteriores y su solubilidad dependiente del pH en el artículos titulado "Enteric coated hard gelatin capsules" del Profesor Karl Thoma y Karoline Bechtold en http://pop.www.capsugel.com/media/library/enteric-coated-hard-gelatin-capsules.pdf.
La goma-laca, también denominada laca purificada, es un producto refinado obtenido de la secreción resinosa de un insecto. Este recubrimiento se disuelve en medios de pH >7. La zeína es una clase de proteína prolamina encontrada en maíz. La zeína es transparente, inodora, insípida, dura, insoluble en agua y comestible, y se usa como recubrimiento en formulaciones farmacéuticas.
En una realización, el recubrimiento entérico se prepara a partir de polímeros o copolímeros de ácido acrílico, ácido metacrílico o ácido etacrílico, acetato de celulosa (y sus derivados de succinato y ftalato), ftalato de hidroxipropilmetilcelulosa, acetato ftalato de polivinilo, ftalato de hidroxietiletilcelulosa, acetato tetrahidroftalato de celulosa, resina acrílica o goma-laca. En otra realización, el polímero se elige de acetato ftalato de celulosa (CAP; se disuelve por encima de pH 6), acetato ftalato de polivinilo (PVAP, se disgrega a pH 5), ftalato de hidroxipropilmetilcelulosa (HPMCP, el grado HP50 se disgrega a pH 5 y HP50 se disgrega a 5,5), copolímeros de ácido metilacrílico (Eudragit L 100 y L12.5 se disgregan entre aproximadamente 6 y aproximadamente 7, Eudragit L-30 y L100-55 se disgregan a pH mayor de 5,5 y Eudragit S100, S12.5 y FS 30D se disgregan a pH mayor de 7).
El recubrimiento entérico puede contener un plastificante y, habitualmente, lo contiene. Plastificantes adecuados incluyen citrato de trietilo (Citroflex 2), triacetina (triacetato de glicerilo), citrato de acetiltrietilo (Citroflec A2), Carbowax 400 (polietilenglicol 400), ftalato de dietilo, citrato de tributilo, monoglicéridos acetilados, glicerol, ésteres de ácido graso, propilenglicol y ftalato de dibutilo. En particular, los polímeros acrílicos carboxílicos aniónicos habitualmente contendrán un 10-25 % en peso de un plastificante, especialmente ftalato de dibutilo, polietilenglicol (PEG), citrato de trietilo y triacetina. La cantidad de plastificante se optimiza para cada fórmula de capa de recubrimiento entérico, en relación con el polímero o polímeros de capa de recubrimiento entérico seleccionados, el plastificante o plastificantes seleccionados y la cantidad aplicada de dicho polímero o polímeros, de tal manera que las propiedades mecánicas, es decir, la flexibilidad y dureza de la capa o capas de recubrimiento entérico, por ejemplo, ejemplificadas como dureza Vickers, se ajusten de modo que si se desea un comprimido, la resistencia a ácido de los gránulos cubiertos con una o más capas de recubrimiento entérico no disminuya significativamente durante la compresión de los gránulos en comprimidos. La cantidad de plastificante habitualmente es por encima de un 5 % en peso del polímero o polímeros de capa de recubrimiento entérico (en una realización la cantidad de plastificante es de un 15-50%. En otra realización
la cantidad de plastificante es de un 20-50 %). El grosor máximo del recubrimiento entérico aplicado normalmente está limitado únicamente por las condiciones de procesamiento y el perfil de disolución deseado.
Las formulaciones divulgadas en este documento contienen, salvo que se indique de otro modo, uno o más excipientes farmacéuticamente aceptables, tales como aglutinantes, tensioactivos, diluyentes, agentes tamponantes, antiadherentes, emolientes, polímeros hidrófilos o hidrófobos, retardantes, agentes etabilizantes o estabilizadores, disgregantes o superdisgregantes, dispersantes, antioxidantes, agentes antiespumantes, rellenos, aromas, colorantes, lubricantes, absorbentes, conservantes, plastificantes o edulcorantes, o mezclas de los mismos, que facilitan el procesamiento de la molécula de fármaco (o realizaciones del mismo divulgadas en este documento) o una sal farmacéuticamente aceptable del mismo en preparaciones que pueden usarse farmacéuticamente. Los excipientes farmacéuticamente puede estar en el recubrimiento y/o el núcleo. Cualquiera de las técnicas bien conocidas y excipientes pueden usarse como adecuados y como entendidos en la técnica, véase, por ejemplo, Remington: The Science and Practice of Pharmacy, vigesimoprimera Ed., (Pharmaceutical Press, 2005); Liberman, H. A., Lachman, L., y Schwartz, J.B. Eds., Pharmaceutical Dosage Forms, Vol. 1-2 Taylor y Francis 1990; y R.I. Mahato, Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems, segunda Ed. (Taylor y Francis, 2012).
También pueden incluirse aditivos tales como dispersantes, colorantes, polímeros de pigmentos (por ejemplo, poli(acrilato de etilo, metacrilato de metilo), agentes antiadherentes y antiespumantes en la capa o capas de recubrimiento entérico. Pueden añadirse otros compuestos para aumentar el grosor de la película y para disminuir la difusión de los jugos gástricos ácidos en el material susceptible a ácido.
En algunas realizaciones, las formulaciones pueden incluir uno o más agentes de ajuste del pH o agentes tamponantes, por ejemplo, ácidos tales como ácido acético, bórico, cítrico, láctico, fosfórico y clorhídrico; bases tales como hidróxido de sodio, fosfato de sodio, borato de sodio, citrato de sodio, acetato de sodio, lactato de sodio y tris-hidroximetilaminometano; y tampones tales como citrato/dextrosa, bicarbonato de sodio, cloruro de amonio y similares. Dichos ácidos, bases y tampones se incluyen en una cantidad requerida para mantener el pH de la composición en un intervalo aceptable.
En algunas realizaciones, las formulaciones también pueden incluir una o más sales en una cantidad requerida para llevar la osmolalidad de la composición a un intervalo aceptable. Dichas sales incluyen las que tienen cationes de sodio, potasio o amonio y aniones de cloruro, citrato, ascorbato, borato, fosfato, bicarbonato, sulfato, tiosulfato o bisulfito; sales adecuadas incluyen cloruro de sodio, cloruro de potasio, tiosulfato de sodio, bisulfito de sodio y sulfato de amonio.
En algunas realizaciones, las formulaciones también pueden incluir uno o más agentes antiespumantes para reducir la formación de espuma durante el procesamiento, que puede provocar la coagulación de dispersiones acuosas, burbujas en la película acabada o en general alterar el procesamiento. Agentes antiespumantes ejemplares incluyen emulsiones de silicio o sesquioleato de sorbitán.
En algunas realizaciones, las formulaciones también pueden incluir uno o más antioxidantes, tales como antioxidantes sin tiol, por ejemplo, hidroxitolueno butilado (BHT), ascorbato de sodio, ácido ascórbico y tocoferol. En determinadas realizaciones, los antioxidantes potencian la estabilidad química cuando sea necesario.
En algunas realizaciones, las formulaciones también pueden incluir uno o más conservantes para inhibir la actividad microbiana. Conservantes adecuados incluyen sustancias que contienen mercurio tales como merfen y tiomersal; dióxido de cloro estabilizado; y compuestos de amonio cuaternario tales como cloruro de benzalconio, bromuro de cetiltrimetilamonio y cloruro de cetilpiridinio.
En algunas realizaciones, las formulaciones también pueden incluir uno o más aglutinantes. Los aglutinantes confieren cualidades cohesivas e incluyen, por ejemplo, ácido algínico y sales del mismo; derivados de celulosa tales como carboximetilcelulosa, metilcelulosa (por ejemplo, Methocel®), hidroxipropilmetilcelulosa, hidroxietilcelulosa, hidroxipropilcelulosa (por ejemplo, Klucel®), etilcelulosa (por ejemplo, Ethocel®) y celulosa microcristalina (por ejemplo, Avicel®); dextrosa microcristalina; amilosa; silicato de magnesio y aluminio; ácidos de polisacáridos; bentonitas; gelatina; copolímero de polivinilpirrolidona/acetato de vinilo; crospovidona; povidona; almidón; almidón pregelatinizado; tragacanto, dextrina, un glúcido, tal como sacarosa (por ejemplo, Dipac®), glucosa, dextrosa, melazas, manitol, sorbitol, xilitol (por ejemplo, Xylitab®) y lactosa; una goma natural o sintética tal como goma arábiga, tragacanto, goma ghatti, mucílago de cáscaras de isapol, polivinilpirrolidona (por ejemplo, Polyvidone® CL, Kollidon® CL, Polyplasdone® XL-10), arabogalactano de alerce, Veegum®, polietilenglicol, óxido de polietileno, ceras, alginato de sodio y similares.
En general, se usan niveles de aglutinante de aproximadamente un 10 a aproximadamente un 70 % en formulaciones de cápsula de gelatina rellena de polvo. El nivel de uso de aglutinante en formulaciones de comprimido varía según sea por compresión directa, granulación en húmedo, compactación con rodillo o según el uso de otros excipientes tales como rellenos que, por sí mismo, pueden actuar como aglutinante moderado. Los formuladores expertos en la materia pueden determinar el nivel de aglutinante para las formulaciones, pero el nivel de uso de aglutinante de hasta un 70 % en formulaciones de comprimido es habitual.
En algunas realizaciones, las formulaciones también pueden incluir agentes dispersantes y/o agentes moduladores de la viscosidad. Los agentes dispersantes y/o agentes moduladores de la viscosidad incluyen materiales que controlan la difusión y homogeneidad de un fármaco a través de medios líquidos o un método de granulación o método de mezcla. En algunas realizaciones, estos agentes también facilitan la eficacia de un recubrimiento o matriz erosiva. Facilitadores de la dispersión/agentes dispersantes ejemplares incluyen, por ejemplo, polímeros hidrófilos, electrólitos, Tween® 20, 60 o 80, PEG, polivinilpirrolidona (PVP; disponible en el mercado como Plasdone®) y los agentes de dispersión basados en carbohidrato tales como, por ejemplo, hidroxipropilcelulosas (por ejemplo, HPC, HPC-SL y HPC-L), hidroxipropilmetilcelulosas (por ejemplo, HPMC K100, r Pm C K4M, Hp Mc K15M y HPMC K100M), carboximetilcelulosa de sodio, metilcelulosa, trietilcelulosa, hidroxietilcelulosa, hidroxipropilcelulosa, ftalato de hidroxipropilmetilcelulosa, acetato estearato de hidroxipropilmetilcelulosa (HPMCAS), celulosa no cristalina, silicato de magnesio y aluminio, trietanolamina, poli(alcohol vinílico) (PVA), copolímero de vinilpirrolidona/acetato de vinilo (S630), polímero de 4-(1,1,3,3-tetrametilbutil)-fenol con óxido de etileno y formaldehído (también conocido como tiloxapol), poloxámeros (por ejemplo, Pluronics® F68, F88 y F108, que son copolímeros de bloque de óxido de etileno y óxido de propileno); y poloxaminas (por ejemplo, Tetronic® 908, también conocida como Poloxamine® 908, que es un copolímero de bloque tetrafuncional derivado de la adición secuencial de óxido de propileno y óxido de etileno a etilendiamina (BASF Corporation, Parsippany, N.J.)), polivinilpirrolidona K12, polivinilpirrolidona K17, polivinilpirrolidona K25 o polivinilpirrolidona K30, copolímero de polivinilpirrolidona/acetato de vinilo (S-630), polietilenglicol, por ejemplo, el polietilenglicol puede tener un peso molecular de aproximadamente 300 a aproximadamente 6000, o de aproximadamente 3350 a aproximadamente 4000, o de aproximadamente 7000 a 5400, carboximetilcelulosa de sodio, metilcelulosa, polisorbato-80, alginato de sodio, gomas, tales como, por ejemplo, goma de tragacanto y goma arábiga, goma guar, xantanos, incluyendo goma xantana, glúcidos, productos celulósicos, tales como, por ejemplo, carboximetilcelulosa de sodio, metilcelulosa, carboximetilcelulosa de sodio, polisorbato-80, alginato de sodio, monolaurato de sorbitán polietoxilado, monolaurato de sorbitán polietoxilado, povidona, carbómeros, poli(alcohol vinílico) (PVA), alginatos, quitosanos y combinaciones de los mismos.
En algunas realizaciones, las formulaciones también pueden incluir uno o más "diluyentes", que se refiere a compuestos químicos que se usan para diluir el compuesto de interés antes de su suministro. Los diluyentes también pueden usarse para estabilizar compuestos, porque pueden proporcionar un entorno más estable. Se utilizan sales disueltas en soluciones tamponadas (que también pueden proporcionar control o mantenimiento del pH) como diluyentes en la técnica, incluyendo, aunque sin limitación, una solución salina tamponada con fosfato. En determinadas realizaciones, los diluyentes aumentan el volumen de la composición para facilitar la compresión o crear suficiente volumen para una mezcla homogénea para el llenado de cápsulas. Dichos compuestos incluyen, por ejemplo, lactosa, almidón, manitol, sorbitol, dextrosa, celulosa microcristalina tal como Avicel®; fosfato de calcio dibásico, fosfato de dicalcio dihidrato; fosfato de tricalcio, fosfato de calcio; lactosa anhidra, lactosa secada por pulverización; almidón pregelatinizado, azúcar comprimible, tal como Di-Pac® (Amstar); hidroxipropilmetilcelulosa, acetato estearato de hidroxipropilmetilcelulosa, diluyentes basados en sacarosa, azúcar de pastelería; sulfato de calcio monobásico monohidrato, sulfato de calcio dihidrato; lactato de calcio trihidrato, dextratos; sólidos de cereal hidrolizados, amilosa; celulosa en polvo, carbonato de calcio; glicina, caolín; manitol, cloruro de sodio; inositol, bentonita y similares.
En algunas realizaciones, la formulación puede contener agentes tensioactivos o tensioactivos que son moléculas de cadena larga que pueden acumularse en superficies de contacto hidrófilas/hidrófobas (agua/aceite) y reducir la tensión superficial en la superficie de contacto. Como resultado, pueden estabilizar una emulsión. En algunas realizaciones, el tensioactivo puede comprender: La familia Tween® (sorbato de polioxietileno) de tensioactivos, la familia Span® (ésteres de ácido carboxílico de cadena larga de sorbitán) de tensioactivos, la familia Pluronic® (copolímeros de bloque de óxido de etileno o propileno) de tensioactivos, las familias Labrasol®, Labrafil® y Labrafac® (cada una glicéridos poliglicolizados) de tensioactivos, ésteres de sorbitán de oleato, estearato, laurato u otros ácidos carboxílicos de cadena larga, poloxámeros (copolímeros de bloqueo de polietileno-polipropilenglicol o Pluronic®), otros ésteres de ácido carboxílico de cadena larga de sorbitán o sacarosa, mono- y diglicéridos, derivados de PEG de triglicéridos caprílicos/cápricos y mezclas de los mismos o mezcla de dos o más de los anteriores. En algunas realizaciones, la fase de tensioactivo puede comprender una mezcla de monooleato de polioxietilén (20) sorbitán (Tween 80®) y monooleato de sorbitán (Span 80®).
En algunas realizaciones, las formulaciones también pueden incluir uno o más "disgregantes", que incluyen tanto la disolución como la dispersión de la forma farmacéutica cuando se ponen en contacto con líquido gastrointestinal. Los "agentes de disgregación o disgregantes" facilitan la descomposición o disgregación de una sustancia. Ejemplos de agentes de disgregación incluyen un almidón, por ejemplo, un almidón natural tal como almidón de maíz o almidón de patata, un almidón pregelatinizado tal como National 1551 o almidón glicolato de sodio tal como Promogel® o Explotab®, una celulosa tal como un producto de madera, metilcelulosa cristalina, por ejemplo, Avicel®, Avicel® PH101, Avicel® PH102, Avicel® PH105, Elceme® P100, Emcocel®, Vivacel® y Solka-Floc®, metilcelulosa, croscarmelosa o una celulosa reticulada, tal como carboximetilcelulosa de sodio reticulada (Ac-Di-Sol®), carboximetilcelulosa reticulada o croscarmelosa reticulada, un almidón reticulado tal como almidón glicolato de sodio, un polímero reticulado tal como crospovidona, una polivinilpirrolidona reticulada, alginato tal como ácido algínico o una sal de ácido algínico, tal como alginato de sodio, una arcilla tal como Veegum® HV (silicato de magnesio y aluminio), una goma tal como agar, guar, garrofín, Karaya, pectina o tragacanto, almidón glicolato de sodio, bentonita, una esponja natural, un tensioactivo, una resina tal como una resina de intercambio catiónico, pulpa de cítrico, lauril sulfato de sodio, lauril sulfato de sodio en combinación con almidón y similares.
En algunas realizaciones, las formulaciones también pueden incluir facilitadores de la erosión. Los "facilitadores de la erosión" incluyen materiales que controlan la erosión de un material particular en líquido gastrointestinal. Los facilitadores de erosión en general son conocidos por los expertos en la materia. Facilitadores de la erosión ejemplares incluyen, por ejemplo, polímeros hidrófilos, electrólitos, proteínas, péptidos y aminoácidos.
En algunas realizaciones, las formulaciones también pueden incluir uno o más agentes de relleno que incluyen compuestos tales como lactosa, carbonato de calcio, fosfato de calcio, fosfato de calcio dibásico, sulfato de calcio, celulosa microcristalina, polvo de celulosa, dextrosa, dextratos, dextrano, almidones, almidón pregelatinizado, sacarosa, xilitol, lactitol, manitol, sorbitol, cloruro de sodio, polietilenglicol y similares.
En algunas realizaciones, las formulaciones también pueden incluir uno o más agentes aromatizantes y/o "edulcorantes", por ejemplo, jarabe de gorma arábiga, acesulfamo K, alitamo, anís, manzana, aspartamo, plátano, crema bávara con frambuesas, grosella negra, caramelo duro, citrato de calcio, alcanfor, caramelo, cereza, crema de chocolate con cerezas, canela, chicle, cítrico, ponche de cítricos, crema de cítricos, algodón de azúcar, cacao, refresco de cola, cereza fría, cítrico frío, ciclamato, cilamato, dextrosa, eucalipto, eugenol, fructosa, ponche de frutas, jengibre, glicirretinato, jarabe de glicirriza (regaliz), uva, pomelo, miel, isomaltitol, limón, lima, crema de limón, glirrizinato de monoamonio, maltol, manitol, arce, malvavisco, mentol, crema de menta, bayas mixtas, neohesperidina DC, neotame, naranja, pera, melocotón, hierbabuena, crema de hierbabuena, polvo, frambuesa, cerveza de raíz, ron, sacarina, safrol, sorbitol, menta verde, crema de menta verde, fresa, crema de fresa, stevia, sucralosa, sacarosa, sacarina sódica, sacarina, aspartamo, acesulfamo de potasio, manitol, talina, silitol, sucralosa, sorbitol, crema suiza, tagatosa, mandarina, taumatina, macedonia, vainilla, nuez, sandía, cereza silvestre, gaulteria, xilitol, o cualquier combinación de estos ingredientes aromatizantes, por ejemplo, anís-mentol, cereza-anís, canela-naranja, cereza-canela, chocolatementa, miel-limón, limón-lima, limón-menta, mentol-eucalipto, naranja-crema, vainilla-menta, y mezclas de los mismos.
En algunas realizaciones, las formulaciones también pueden incluir uno o más lubricantes y emolientes que son compuestos que evitan, reducen o inhiben la adhesión o fricción de los materiales. Lubricantes ejemplares incluyen, por ejemplo, ácido esteárico, hidróxido de calcio, talco, estearilfumarato de sodio, un hidrocarburo tal como aceite de vaselina, o aceite vegetal hidrogenado tal como aceite de soja hidrogenado, ácidos grasos superiores y sus sales de metal alcalino y metal alcalinotérreo, tales como aluminio, calcio, magnesio, cinc, ácido esteárico, estearatos de sodio, glicerol, talco, ceras, ácido bórico, benzoato de sodio, acetato de sodio, cloruro de sodio, leucina, un polietilenglicol (por ejemplo, PEG4000) o un metoxipolietilenglicol tal como Carbowax®, oleato de sodio, benzoato de sodio, behenato de glicerilo, polietilenglicol, lauril sulfato de magnesio o sodio, sílice coloidal tal como Syloid®, Cab-O-Sil®, un almidón tal como almidón de maíz, aceite siliconado, un tensioactivo y similares.
En algunas realizaciones, las formulaciones también pueden incluir uno o más solubilizantes que incluyen compuestos tales como triacetina, citrato de trietilo, oleato de etilo, caprilato de etilo, lauril sulfato de sodio, docusato de sodio, vitamina E TPGS, dimetilacetamida, N-metilpirrolidona, N-hidroxietilpirrolidona, polivinilpirrolidona, hidroxipropilmetilcelulosa, hidroxipropilciclodextrinas, por ejemplo, Captisol®, etanol, n-butanol, alcohol isopropílico, colesterol, sales biliares, polietilenglicol 200-600, glicofurol, transcutol, propilenglicol y dimetilisosorbida y similares. En una realización, el solubilizante es vitamina E TPGS y/o Captisol®.
En algunas realizaciones, las formulaciones también pueden incluir uno o más agentes de suspensión que incluyen compuestos tales como polivinilpirrolidona, por ejemplo, polivinilpirrolidona K112, polivinilpirrolidona K17, polivinilpirrolidona K25 o polivinilpirrolidona K30, copolímero de vinilpirrolidona/acetato de vinilo (S630), polietilenglicol, por ejemplo, el polietilenglicol puede tener un peso molecular de aproximadamente 300 a aproximadamente 6000, o de aproximadamente 3350 a aproximadamente 4000, o de aproximadamente 7000 a aproximadamente 5400, carboximetilcelulosa de sodio, metilcelulosa, hidroxipropilmetilcelulosa, acetato estearato de hidroximetilcelulosa, polisorbato-80, hidroxietilcelulosa, alginato de sodio, gomas, tales como, por ejemplo, goma de tragacanto y goma arábiga, goma guar, xantanos, incluyendo goma xantana, glúcidos, productos celulósicos, tales como, por ejemplo, carboximetilcelulosa de sodio, metilcelulosa, carboximetilcelulosa de sodio, hidroxipropilmetilcelulosa, hidroxietilcelulosa, polisorbato-80, alginato de sodio, monolaurato de sorbitán polietoxilado, monolaurato de sorbitán polietoxilado, povidona y similares.
En algunas realizaciones, las formulaciones también pueden incluir uno o más agentes humectantes que incluyen compuestos tales como ácido oleico, monoestearato de glicerilo, monooleato de sorbitán, monolaurato de sorbitán, oleato de trietanolamina, monooleato de polioxietilensorbitán, monolaurato de polioxietilensorbitán, docusato de sodio, oleato de sodio, lauril sulfato de sodio, docusato de sodio, triacetina, Tween 80, vitamina E TPGS, sales de amonio y similares.
En una realización, la forma farmacéutica oral sólida puede liberar menos de un 10 % en peso del compuesto y/o dicha sal farmacéuticamente aceptable del mismo del núcleo (A) en 1,5 horas en un recipiente de disolución que comprende una solución acuosa a un pH de menos de 3 o un pH de 4,5 a 5,5, y la forma farmacéutica oral sólida puede liberar no menos de un 80 % en peso de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo del núcleo (A), de veinte minutos a dos horas en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4.
En un aspecto de esta realización, el núcleo (A) puede liberar no menos de un 80 % en peso del compuesto y/o la sal farmacéuticamente aceptable del mismo del núcleo (A) de veinte minutos a dos horas en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7.
En otro aspecto de esta realización, la forma farmacéutica oral sólida puede liberar menos de un 10 % en peso del compuesto y/o dicha sal farmacéuticamente aceptable del mismo del núcleo (A) en 1,5 horas en un recipiente de disolución que comprende una solución acuosa a un pH de 5,1 a 5,5. La forma farmacéutica oral sólida puede liberar menos de un 20 % en 1,5 horas en un recipiente de disolución que comprende una solución acuosa a un pH de 5,1 a 5,5. La forma farmacéutica oral sólida puede liberar menos de un 25 % en 1,5 horas en un recipiente de disolución que comprende una solución acuosa a un pH de 5,1 a 5,5. La forma farmacéutica oral sólida puede liberar menos de un 10 % en 1,5 horas en un recipiente de disolución que comprende una solución acuosa a un pH por debajo de 5,1. La forma farmacéutica oral sólida puede liberar menos de un 25 % en peso de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo en 15 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4. La forma farmacéutica oral sólida puede liberar menos de un 25 % de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo en 15 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4. La forma farmacéutica oral sólida puede liberar menos de un 35 % de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo en 15 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4.
En otro aspecto de esta realización, la forma farmacéutica oral sólida puede liberar menos de un 80 % en peso del compuesto y/o dicha sal farmacéuticamente aceptable del mismo del núcleo (A) en 30 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4. La forma farmacéutica oral sólida puede liberar menos de un 80 % de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo en 30 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4. La forma farmacéutica oral sólida puede liberar menos de un 85 % de dicha molécula de fármaco en 30 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4.
En otro aspecto de esta realización, la forma farmacéutica oral sólida puede liberar menos de un 80 % en peso del compuesto y/o dicha sal farmacéuticamente aceptable del mismo del núcleo (A) en 45 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4. La forma farmacéutica oral sólida puede liberar menos de un 80 % de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo en 45 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4.
En otro aspecto de esta realización, la forma farmacéutica oral sólida puede liberar menos de un 80 % del compuesto y/o dicha sal farmacéuticamente aceptable del mismo del núcleo (A) en 60 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4. La forma farmacéutica oral sólida puede liberar menos de un 80 % de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo en 60 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4.
En otro aspecto de esta realización, la forma farmacéutica oral sólida puede liberar al menos un 80 % del compuesto y/o dicha sal farmacéuticamente aceptable del mismo del núcleo (A) en 120 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4. La forma farmacéutica oral sólida puede liberar al menos un 80 % de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo en 60 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4.
En otro aspecto de esta realización, en cada uno de los ensayos descritos en los aspectos anteriores, la solución acuosa es un líquido intestinal simulado a un pH de 6,4 a 7,4.
En otro aspecto de esta realización, la forma farmacéutica oral sólida comprende una sal de ácido farmacéuticamente aceptable del compuesto del núcleo (A). Como alternativa, la forma farmacéutica oral sólida puede comprender la base libre del compuesto del núcleo (A).
En otro aspecto de esta realización, el núcleo (A) de la forma farmacéutica oral sólida comprende una sal farmacéuticamente aceptable del compuesto y suficiente ácido farmacéuticamente aceptable adicional para potenciar la disolución de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo.
En otro aspecto de esta realización, el núcleo (A) de la forma farmacéutica oral sólida comprende una sal farmacéuticamente aceptable del compuesto y un ácido farmacéuticamente aceptable en cantidad suficiente para producir una solución acuosa ácida dentro de la forma farmacéutica oral sólida antes de la liberación del compuesto y/o la sal farmacéuticamente aceptable del mismo.
En otro aspecto de esta realización, el núcleo (A) de la forma farmacéutica oral sólida comprende una sal farmacéuticamente aceptable del compuesto y un tensioactivo presente en una concentración por encima de su concentración micelar crítica tras la disgregación en 50 ml de medio acuoso. El tensioactivo puede estar presente en una concentración por encima de su concentración micelar crítica tras la disgregación en 20 ml de medio acuoso.
En otro aspecto de esta realización, el compuesto y/o la sal farmacéuticamente aceptable del mismo del núcleo (A) es un sólido en donde el tamaño medio de partícula es de 0,3 micrómetros a 100 micrómetros.
En otro aspecto de esta realización, el compuesto y/o la sal farmacéuticamente aceptable del mismo del núcleo (A) es un sólido en donde el tamaño medio de partícula es de 1 micrómetros a 50 micrómetros.
En otro aspecto de esta realización, el compuesto y/o la sal farmacéuticamente aceptable del mismo del núcleo (A) es un sólido en donde el tamaño medio de partícula es menor de o igual a 15 micrómetros.
En otro aspecto de esta realización, el recubrimiento entérico (B) es de un 10 % a un 150 % del peso del núcleo.
En otro aspecto de esta realización, el recubrimiento entérico (B) es de un 20 % a un 100 % del peso del núcleo.
En otro aspecto de esta realización, el recubrimiento entérico (B) es de un 30 % a un 60 % del peso del núcleo.
En otro aspecto de esta realización, el recubrimiento entérico (B) es de 5 a 500 micrómetros de grosor.
En otro aspecto de esta realización, el recubrimiento entérico (B) es de 8 a 150 micrómetros de grosor.
En otro aspecto de esta realización, el recubrimiento entérico (B) es de 50 a 100 micrómetros de grosor. En otro aspecto de esta realización, el recubrimiento entérico (B) se selecciona de gelatina polimerizada, goma-laca, copolímero de ácido metacrílico de tipo CNF, butirato ftalato de celulosa, hidrogenoftalato de celulosa, propionato ftalato de celulosa, acetato ftalato de polivinilo (PVAP), acetato ftalato de celulosa (CAP), acetato trimelitato de celulosa (CAT), ftalato de hidroxipropilmetilcelulosa, acetato de hidroxipropilmetilcelulosa, succinato de dioxipropilmetilcelulosa, carboximetiletilcelulosa (CMEC), acetato succinato de hidroxipropilmetilcelulosa (HPMCAS) y polímeros y copolímeros de ácido (met)acrílico, en donde dichos polímeros se preparan a partir de un monómero y dichos copolímeros se preparan a partir de dos o más monómeros, en donde dichos monómeros se seleccionan de cualquiera de acrilato de metilo, acrilato de etilo, metacrilato de metilo y metacrilato de etilo.
100 micrómetros de grosor. En otro aspecto de esta realización, el recubrimiento entérico (B) se selecciona de gelatina polimerizada, goma-laca, copolímero de ácido metacrílico de tipo CNF, butirato ftalato de celulosa, hidrogenoftalato de celulosa, propionato ftalato de celulosa, acetato ftalato de polivinilo (PVAP), acetato ftalato de celulosa (CAP), acetato trimelitato de celulosa (CAT), ftalato de hidroxipropilmetilcelulosa, acetato de hidroxipropilmetilcelulosa, succinato de dioxipropilmetilcelulosa, carboximetiletilcelulosa (CMEC), acetato succinato de hidroxipropilmetilcelulosa (HPMCAS) y polímeros y copolímeros de ácido (met)acrílico, en donde dichos polímeros se preparan a partir de un monómero y dichos copolímeros se preparan a partir de dos o más monómeros, en donde dichos monómeros se seleccionan de cualquiera de acrilato de metilo, acrilato de etilo, metacrilato de metilo y metacrilato de etilo.
En otro aspecto de esta realización, el recubrimiento entérico comprende un polímero de poli(met)acrilato. En otro aspecto de esta realización, el recubrimiento entérico comprende un polímero de la serie Eudragit® L o S. En otro aspecto de esta realización, el recubrimiento entérico comprende un Eudragit® L100, L12.5, S100, S12.5 o FS 30D.
En otro aspecto de esta realización, el recubrimiento entérico comprende un derivado de celulosa.
En otro aspecto de esta realización, el derivado de celulosa se selecciona de metilcelulosa, acetato ftalato de celulosa, ftalato de hidroximetilcelulosa (HPMCP), succinato de hidroxipropilmetilcelulosa (HPMCS) y acetato succinato de hidroximetilcelulosa (HPMCAS).
En otro aspecto de esta realización, el recubrimiento entérico comprende un polímero de acetato ftalato de polivinilo (PVAP).
En otro aspecto de esta realización, el subrecubrimiento (C) es un polímero erosionable soluble en agua o hidrófilo, siendo dicho polímero un polímero de bajo peso molecular seleccionado de hidroximetilcelulosa (HPMC), hidroxietilcelulosa, hidroximetilcelulosa, hidroxipropilcelulosa, celulosa microcristalina, polivinilpirrolidionas, polisacáridos (o un derivado de polisacárido), poli(alcoholes vinílicos), polietilenglicol (PEG), un polipropilenglicol (PPG) y un copolímero de bloque de PEG-PPG.
En otro aspecto de esta realización, el subrecubrimiento (C) es una composición insoluble en agua que comprende (i) partículas de compuesto soluble en agua que pueden formar canales en la composición insoluble en agua, o (ii) partículas hidrófilas insolubles en agua que provocan el hinchamiento del subrecubrimiento cuando está en contacto con un medio acuoso.
En otro aspecto de esta realización, el subrecubrimiento (C) comprende partículas de un compuesto soluble en agua que puede formar canales.
La expresión "partículas hidrófilas insolubles en agua", como se usa en este documento, incluye, aunque sin limitación, polisacáridos que incluyen partículas de pectinato de calcio, alginato de calcio, xantato de calcio, cualquier sal metálica de un polisacárido que contiene un grupo ácido donde la sal hace que el polisacárido sea insoluble en agua, almidón microcristalino, almidón insoluble, cualquier polisacárido insoluble en agua (por ejemplo, celulosa o celulosa microcristalina), cualquier polisacárido reticulado covalentemente donde dicha reticulación hace que el polisacárido
sea insoluble en agua. Dichos agentes reticulantes incluyen, aunque sin limitación, glutaraldehído, formaldehído, epiclorhidrina, cloruros de diácido, diisociananatos, anhídridos de diácido y diaminas.
En otro aspecto de esta realización, el subrecubrimiento (C) comprende partículas hidrófilas insolubles en agua que provocan el hinchamiento del subrecubrimiento cuando está en contacto con un medio acuoso.
En otro aspecto de esta realización, el subrecubrimiento (C) es una composición insoluble en agua y comprende partículas de un compuesto soluble en agua que forma canales que permiten el flujo entrante de agua en la forma farmacéutica oral sólida y la difusión del compuesto y/o dicha sal farmacéuticamente aceptable del mismo en el intestino.
En otro aspecto de esta realización, el subrecubrimiento (C) es una composición insoluble en agua tal como un polímero que comprende partículas de un compuesto soluble en agua que puede formar canales que son impermeables a dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo, pero permite la entrada de agua y el hinchamiento y rotura del subrecubrimiento y que provoca la liberación del compuesto y/o la sal farmacéuticamente aceptable del mismo.
La forma farmacéutica oral sólida es un comprimido o es una cápsula.
La forma farmacéutica oral sólida puede comprender un excipiente farmacéuticamente aceptable seleccionado de aglutinantes, tensioactivos, diluyentes, tampones, antiadherentes, emolientes, disgregantes, antioxidantes, agentes antiespumantes, rellenos, aromas, colores, lubricantes, absorbentes, conservantes, plastificantes y edulcorantes. En una realización, la ABC resultante de la administración de la dosificación oral sólida es al menos aproximadamente un 500 % mayor que el ABC resultante de la administración de una forma farmacéutica de liberación inmediata que tiene una cantidad equivalente del compuesto y/o dicha sal farmacéuticamente aceptable del mismo.
En una realización, la ABC resultante de la administración de la dosificación oral sólida es al menos aproximadamente un 100 % mayor que el ABC resultante de la administración de una forma farmacéutica de liberación inmediata que tiene una cantidad equivalente del compuesto y/o dicha sal farmacéuticamente aceptable del mismo.
En una realización, la ABC resultante de la administración de la dosificación oral sólida es al menos aproximadamente un 50 % mayor que el ABC resultante de la administración de una forma farmacéutica de liberación inmediata que tiene una cantidad equivalente del compuesto y/o dicha sal farmacéuticamente aceptable del mismo.
En una realización, la forma farmacéutica oral sólida de la invención es para su uso en un método de tratamiento de una enfermedad tratable mediante inhibición de BTK, en un paciente que lo necesita reconocidamente, en donde dicho método comprende administrar a dicho paciente en una sola o múltiples dosis, una cantidad terapéuticamente eficaz del compuesto y/o la sal farmacéuticamente aceptable del mismo del núcleo (A).
En un aspecto de esta realización, la enfermedad se selecciona de una enfermedad autoinmunitaria, cáncer y una enfermedad inflamatoria.
En otro aspecto de esta realización, la enfermedad es una leucemia o linfoma.
En otro aspecto de esta realización, la enfermedad se selecciona de leucemia linfocítica crónica (CLL), leucemia linfocítica pequeña (SLL), mieloma múltiple, linfoma de células del manto y linfoma no hodgkiniano de linfocitos B. En una realización, la forma farmacéutica oral sólida de la invención es para su uso en un método de tratamiento de una enfermedad tratable mediante inhibición de BTK, en un paciente que lo necesita reconocidamente, en donde dicho método comprende administrar a dicho paciente una molécula de fármaco del núcleo (A) y que tiene un inicio de liberación de dicha BTK en el yeyuno o el íleon del paciente, en donde la biodisponibilidad sistémica promedio de la molécula de fármaco mediante por la ABC en plasma resultante de la administración de dicha forma farmacéutica de dosificación es de aproximadamente un 10 %, 20 %, 30 %, 40 %, 50 %, 60 % o 70 % más que la biodisponibilidad sistémica promedio de una formulación de liberación inmediata que tiene una cantidad equivalente de dicha molécula de fármaco o una sal farmacéutica del mismo.
La expresión "formulación de liberación inmediata", como se usa en este documento, se refiere a una formulación que está biodisponible tan pronto como se administra y no tiene recubrimientos protectores para retardar el contacto con la mucosa intestinal.
En una realización, la forma farmacéutica oral sólida de la invención es para su uso en un método de tratamiento de una enfermedad tratable mediante inhibición de BTK, en un paciente que lo necesita reconocidamente, en donde dicho método comprende administrar a dicho paciente una molécula de fármaco del núcleo (A) y que tiene un inicio de liberación de dicha BTK en el yeyuno y/o el íleon del paciente, en donde la biodisponibilidad sistémica promedio de la molécula de fármaco mediante por la ABC en plasma resultante de la administración de dicha forma farmacéutica de
dosificación es de aproximadamente un 10 %, 20 %, 30 %, 40 %, 50 %, 60 % o 70 % más que la biodisponibilidad sistémica promedio de una formulación de liberación inmediata que tiene una cantidad equivalente de dicha molécula de fármaco o una sal farmacéutica del mismo administrada al paciente después de un ayuno de 4 horas.
En una realización, pueden usarse semillas o microesferas (por ejemplo, SODAS®) estratificadas con el agente activo, opcionalmente mezclado con sustancias alcalinas o tampón, como el material de núcleo para procesamiento adicional. Las semillas que se tienen que estratificar con el agente activo pueden ser semillas insolubles en agua que comprenden diferentes óxidos, celulosas, polímeros orgánicos y otros materiales, en solitario o en mezclas o semillas solubles en agua que comprende diferentes sales inorgánicas, glúcidos, no semejantes y otros materiales, en solitario o en mezclas. Además, las semillas pueden comprender el agente activo en forma de cristales, aglomerados, compactos etc. El tamaño de las semillas no es esencial para la presente divulgación, pero puede variar entre aproximadamente 0,1 y 4 mm, tal como menos de 2 mm. Las semillas estratificadas con el agente activo se producen por estratificación en polvo o solución/suspensión usando, por ejemplo, granulación o equipo de estratificación de recubrimiento por pulverización. Las semillas pueden cubrirse por un recubrimiento entérico u otro subrecubrimiento.
Antes de estratificar las semillas, el ingrediente activo puede mezclarse con componentes adicionales. Dichos componentes pueden ser aglutinantes, tensioactivos, rellenos, agentes disgregantes, aditivos alcalinos u otros y/o ingredientes farmacéuticamente aceptables en solitario o en mezclas.
En otra realización, la molécula de fármaco puede mezclarse opcionalmente con diluyentes, vehículos y excipientes adecuados para obtener propiedades de manipulación y procesamiento preferidas y una concentración adecuada del agente activo y el núcleo entonces se produce por extrusión/esferización, formación de bolas o compresión utilizando un equipo de proceso convencional. El tamaño del material de núcleo formulado es aproximadamente entre 0,1 y 4 mm, tal como entre 0,1 y 2 mm. El material de núcleo fabricado puede estratificarse además con ingredientes adicionales que comprenden el agente activo y/o usarse para procesamiento adicional. Como alternativa, el material de núcleo mencionado anteriormente puede prepararse usando secado por pulverización o técnica de coagulación por pulverización.
El núcleo opcionalmente puede cubrirse con una o más capas de separación que comprenden excipientes farmacéuticos antes de aplicar la capa o capas de recubrimiento entérico sobre el material de núcleo en forma de gránulos individuales. Esta o estas capas de separación se formulan para producir propiedades deseadas específicas a la formulación sólida y contienen, por ejemplo, azúcar, PEG, polivinilpirrolidona, poli(alcohol vinílico), acetato de polivinilo, hidroxipropilcelulosa, metilcelulosa, etilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa de sodio, sales solubles en agua de polímeros de recubrimiento entérico y otros, usados en solitario o en mezclas. También pueden incluirse aditivos tales como plastificantes, colorantes, pigmentos, rellenos, agentes antiadherentes y antiestáticos, tales como, por ejemplo, estearato de magnesio, dióxido de titanio, talco y otros aditivos en la capa o capas de separación.
Cuando la capa de separación opcional se aplica al material de núcleo, puede constituir un grosor variable. El grosor máximo de la capa o capas de separación normalmente se limita únicamente por las condiciones de procesamiento. La capa de separación puede servir como barrera de difusión y puede actuar como una zona tamponante del pH. La capa o capas de separación aplicadas opcionalmente no son esenciales. Sin embargo, la capa o capas de separación pueden mejorar la estabilidad química de la sustancia activa y/o las propiedades físicas de la forma de dosificación en comprimido de múltiples unidades novedosa.
Como alternativa, la capa de separación puede formarse in situ mediante una reacción entre una capa polimérica de recubrimiento entérico aplicada sobre el material de núcleo y un compuesto de reacción alcalina en el material de núcleo. Por tanto, la capa de separación formada comprende una sal soluble en agua formada entre el polímero o polímeros de la capa de recubrimiento entérico y un compuesto de reacción alcalina que está en la posición para formar una sal.
El recubrimiento y/o las capas de separación se aplican sobre el material de núcleo o sobre el material de núcleo cubierto con la capa o capas de separación usando cualquier técnica de recubrimiento adecuada. La capa o capas de recubrimiento opcionalmente pueden cubrirse además con una o más capas de sobrerrecubrimiento. La capa o capas de sobrerrecubrimiento pueden aplicarse a los gránulos estratificados de recubrimiento entérico por procedimientos de recubriendo o estratificación en un equipo adecuado tal como bandeja de recubrimiento, granulador de recubrimiento o en un aparato de lecho fluido usando agua y/o disolventes orgánicos para el proceso de recubrimiento o estratificación. Como alternativa la capa o capas de separación pueden aplicarse al material de núcleo usando técnica de recubrimiento en polvo. La capa de sobrerrecubrimiento puede evitar adicionalmente la posible aglomeración de gránulos estratificados de recubrimiento entérico, además puede proteger la capa de recubrimiento entérico del agrietamiento durante el proceso de compactación y potenciar el proceso de formación de comprimidos. El grosor máximo de la capa o capas de sobrerrecubrimiento aplicadas normalmente se limita por las condiciones de procesamiento y el perfil de disolución deseado. La capa de sobrerrecubrimiento también puede usarse como capa de recubrimiento de película para comprimidos.
Las preparación farmacéuticas divulgadas en este documento también incluyen cápsulas hechas de gelatina, así como cápsulas selladas blandas hechas de gelatina y un plastificante, tal como glicerol o sorbitol. Las cápsulas también pueden hacerse de polímeros tales como hipromelosa. Las cápsulas pueden contener los ingredientes activos en el núcleo recubierto como se describe anteriormente. La cápsula puede contener adicionalmente lactosa, aglutinantes tales como almidones y/o lubricantes tales como talco o estearato de magnesio y, opcionalmente, estabilizantes. En cápsulas blandas, los compuestos activos pueden disolverse o suspenderse en líquidos adecuados, tales como aceites grasos, parafina líquida, lípidos, solubilizantes o polietilenglicoles líquidos. Además, pueden añadirse estabilizantes. Todas las formulaciones para administración oral deben estar en dosificaciones adecuadas para dicha administración.
Puede ser deseable incorporar el fármaco en la fase acuosa de una emulsión. Dicha emulsión de "agua en aceite" proporciona un entorno biofísico adecuado para el fármaco y puede proporcionar un superficie de contacto de aceiteagua que puede proteger el fármaco de los efectos adversos del pH o las enzimas que pueden degradar el fármaco. Además, dichas formulaciones de agua en aceite pueden proporcionar una capa lipídica, que puede interactuar favorablemente con lípidos de las células del organismo, y puede aumentar la separación de la formulación en las membranas de las células. Dicha separación puede aumentar la absorción de fármacos en dichas formulaciones en la circulación y, por lo tanto, puede aumentar la biodisponibilidad del fármaco. La fase acuosa puede comprender opcionalmente el agente activo suspendido en agua y un tampón.
En algunas realizaciones, la emulsión de agua en aceite contiene una fase oleosa compuesta de ácidos carboxílicos saturados C8-22 o ácido carboxílicos insaturados o ésteres con hasta tres enlaces insaturados (también ramificaciones) o ésteres o alcoholes de los mismos, un tensioactivo o un agente tensioactivo, y una fase acuosa que contiene principalmente agua y el agente activo.
En algunas realizaciones, las formas farmacéuticas sólidas descritas en este documento son formas farmacéuticas de liberación retardada en el tiempo no entérica. La expresión "liberación retardada en el tiempo no entérica", como se usa en este documento, se refiere al suministro de modo que la liberación del fármaco puede realizarse en alguna ubicación en general predecible en el tubo intestinal más distal a la que se conseguiría si no hubiera alteraciones de liberación retardada. En algunas realizaciones, el método para retardar la liberación es un recubrimiento que se vuelve permeable, se disuelve, se rompe y/o ya no está intacto después de una duración indicada.
El recubrimiento en las formas farmacéuticas de liberación retardada en el tiempo puede tener un tiempo fijo para erosionarse, tras lo que se libera el fármaco (recubrimientos adecuados incluyen recubrimiento polimérico tal como HPMC y similares) o tiene un núcleo compuesto de uno o más disgregantes o uno o más agentes osmóticos tales como una sal, polímero hidrófilo, típicamente óxido de polietileno o una alquilcelulosa, glúcido o similares, que extraen agua a través de una membrana o un agente que genera gas tal como ácido cítrico y bicarbonato de sodio. La membrana puede romperse después de que la presión de hinchamiento exceda un determinado umbral sobre un tiempo de retardo deseado. Como alternativa, una membrana podría llegar a ser porosa por lixiviación de un extraíble acuoso sobre un tiempo de retardo deseado. Las formas farmacéuticas retardadas en el tiempo a veces se administran en un estado en ayunas para evitar la variabilidad en el vaciado gástrico en el estado alimentado.
En un aspecto de la invención, la forma farmacéutica oral sólida contiene un núcleo con un recubrimiento rígido insoluble en agua o relativamente insoluble en agua alrededor de un núcleo hinchable que contiene fármaco. El recubrimiento consiste en un polímero hidrófobo que resiste la entrada de agua en el comprimido. El recubrimiento se integra con partículas hidrófilas solubles en agua o insolubles en agua que pueden hincharse o formar canales a través de lo que entra solución acuosa al comprimido. El diseño es tal que el recubrimiento determina la tasa de captación de agua mientras se hincha el núcleo, lo que depende de la tasa de captación de agua y de las propiedades de hinchamiento del propio núcleo, determina el tiempo de filtración del recubrimiento.
Las propiedades del núcleo le dan además la característica de que se disgrega después de la filtración del recubrimiento, aportando un estallido de liberación de fármaco en un sitio predeterminado en un tubo gastrointestinal. El fármaco puede incrustarse en el material de núcleo o asociarse de otro modo con el material de núcleo, por ejemplo, por mezcla en seco o granulación en húmedo. El núcleo con recubrimiento entérico puede estar en forma de un comprimido de matriz o una cápsula que contiene el fármaco. El núcleo puede estar en forma de gránulos del fármaco puro. Como alternativa, el núcleo puede contener gránulos del fármaco estratificados en un material de núcleo separado. Como alternativa, el núcleo puede contener microcápsulas que contienen el material de fármaco. Más de una de estas formas puede estar presente y puede suministrarse más de un fármaco en el mismo sistema de suministro. En todas estas formas, la liberación de fármaco desde el núcleo es eficaz. El núcleo tiene las características esenciales de poder absorber suficiente líquido de modo que se hinche considerablemente, y se disgregue rápidamente después de que el recubrimiento se filtre. Por "hincharse considerablemente" se entiende que se produce suficiente hinchamiento para conseguir y producir una presión que inicie y/o facilite de otro modo la disgregación. Por "disgregarse rápidamente" se entiende que la disgregación se produce esencialmente en un estallido, siendo el estallido suficiente para liberar cantidades eficaces del fármaco desde la forma farmacéutica oral sólida.
En diversas realizaciones, los métodos y composiciones referidas al suministro yeyunoileal de moléculas de fármaco covalentes reversibles se proporcionan en forma de formulaciones de liberación mantenida y pueden acoplarse con
un componente de liberación inmediata en una forma farmacéutica unitaria. El componente de liberación inmediata puede formularse por cualquier método conocido tal como una capa que envuelve el componente de liberación mantenida o similares.
En algunas realizaciones, las composiciones descritas en este documento mejoran la tolerabilidad de los inhibidores de cinasa covalentes reversibles o irreversibles. Por ejemplo, en una realización no limitante, las composiciones descritas en este documento reducen o eliminan los posibles efectos secundarios adversos de los inhibidores de cinasa covalentes reversibles o irreversibles tales como diarrea y vómitos, habitualmente denominados vómitos. Los siguientes ejemplos ilustran la preparación de composiciones dentro del alcance de la invención. Estas composiciones se proporcionan para posibilitar que los expertos en la materia entiendan más claramente y pongan en práctica la presente invención. No deben considerarse limitantes del alcance de la invención, sino simplemente ilustrativos y representativos de la misma.
Ejemplo 1:
Forma farmacéutica activada osmóticamente para suministro yeyunoileal
Los siguientes ingredientes se usan para preparar una forma farmacéutica yeyunoileal para el inhibidor de BTK, (R)-2-(3-(4-amino-3-(2-fluoro-4-fenoxifenil)-1 H-pirazolo[3,4-d]pirimidin-1-il)piperidina-1-carbonil)-4-metil-4-(4-(oxetan-3-il)piperazin-1 -il)pent-2-enonitrilo:
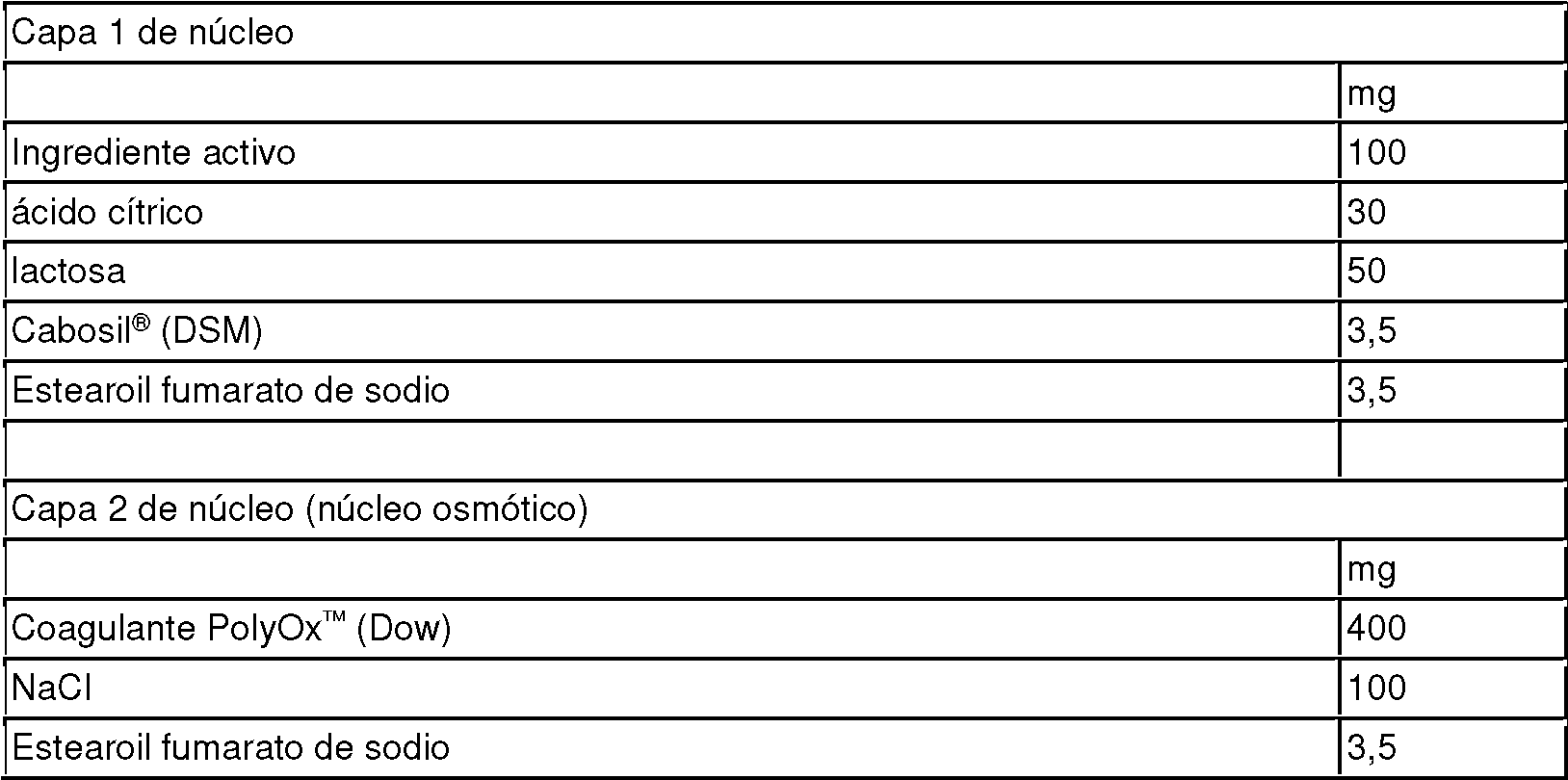

La mezcla para cada capa de núcleo se dimensiona y mezcla en seco con el lubricante añadido el último con mezcla adicional. Las 2 mezclas se forman en comprimidos mediante técnicas de formación de comprimidos convencional en una prensa de comprimidos de bicapa giratoria. Opcionalmente también puede aplicarse un prerrecubrimiento de solución de hipromelosa a los núcleos sin recubrir. Las mezclas para los comprimidos se estratifican en la prensa de comprimidos y se comprimen en un comprimido con una sola compresión.
Los ingredientes para la membrana semipermeable se añade a una solución de cloruro de metileno:metanol (4:1 p/p) usando una mezcladora de propulsor. Los comprimidos no recubiertos se recubren por pulverización y se secan.
A estos comprimidos recubiertos entonces se les aplica recubrimiento entérico con la solución acuosa para diferentes intervalos hasta que los comprimidos resisten la liberación en medio ácido durante 2 horas, típicamente con aumentos de peso de un 6 a un 15 % y variando el grosor de recubrimiento de aproximadamente 50 a aproximadamente 150 micrómetros.
Ejemplo 2:
Comprimidos con recubrimiento entérico para suministro yeyunoileal
En primer lugar se dimensionan 200 mg de ingrediente activo para cada comprimido recubierto y después se mezclan en seco con 7 mg de croscarmelosa de sodio (Ac-Disol®) y 143 mg de celulosa microcristalina (Avicel® PH101) en una mezcladora en V durante 20 a 30 minutos. Después de esto, se añaden 5 mg de estearil fumarato de sodio como lubricante y se mezclan durante 4 a 5 minutos. La mezcla es la que se forma en comprimidos en una prensa de comprimidos giratoria usando perforaciones cóncavas convencionales de 12,7/40,64 cm (5/16 pulgadas). Se prepara una mezcla de recubrimiento a partir de 250 g de Eudragit® L-30 D-55, 7,5 g de citrato de trietilo, 37,5 g de talco y 205 g de agua desionizada. Los núcleos de comprimido se colocan en un recubridor de bandeja perforada que gira a 15 rpm a 40 °C. Esta mezcla se pulveriza con una temperatura de aire de entrada de 44 a 48 °C, una temperatura de aire de salida de 29 a 32 °C, una temperatura de producto de 26 °C, girando a 30 hasta 32 rpm, presión de pulverización de 137,89 kPa (20 psi), y un flujo de aire de 30-32 CFM. Después de curar durante 30 minutos con una temperatura de entrada de aire de 60 °C y rotación a 15 rpm, el calor se desactiva y los comprimidos se enfrían hasta temperatura ambiente mientras se gira. La cantidad de aumento de peso después del recubrimiento es de aproximadamente un 5 a aproximadamente un 15 %, y típicamente de aproximadamente un 10 %. Los tiempos de disolución para el recubrimiento entérico a pH 6,8 estaban dirigidos a más de un 80 % en 1 hora.
Ejemplo 3:
Gránulos con recubrimiento entérico
Se incorporan gránulos con recubrimiento entérico de un intervalo de tamaño de 300 a 500 micrómetros para su inclusión en cápsulas, en cápsulas de gelatina o hipromelosa, en almidones o en cartuchos, o en una suspensión oral. El ingrediente activo y la hipromelosa de baja viscosidad (aproximadamente un 2 %) se dimensionan y mezclan en una mezcladora en V, y después se añaden a un granulador de lecho fluido. Se forman gránulos pulverizando solución de polivinilpirrolidona acuosa diluida sobre el polvo, y después secando los gránulos en el lecho fluido a 45 °C. En el lecho fluido, estos gránulos secados se recubren entonces con una solución transparente de Opadry® en agua para proporcionar un recubrimiento de sellado y se secan. Se combina Eudragit® L-30 D-55 como una dispersión acuosa de un 30 % de polímero, un 0,7 % de lauril sulfato de sodio y un 2,3 % de Tween® 20 con los plastificantes, citrato de trietilo y monoestearato de glicerilo, y se recubren sobre el polvo en el lecho fluido. Después de secar, la composición final de los gránulos con recubrimiento entérico es de aproximadamente un 81,8 % de ingrediente activo, aproximadamente un 1,5 % de hipromelosa, aproximadamente un 0,5 % de Opadry Clear, aproximadamente un 14,5 % de copolímero de ácido metacrílico, un 1,45 % de citrato de trietilo y un 0,25 % de monoestearato de glicerilo. Los gránulo secados se llenan en cápsulas de hipromelosa de tamaño 0. La disolución de las cápsulas a pH 2 muestra menos de un 2 % de liberación a las 2 horas y la cápsula libera más de un 80 % de la carga teórica de fármaco en aproximadamente 45 minutos.
Ejemplo 4:
Dosificación yeyunoileal de (R)-2-(3-(4-amino-3-(2-fluoro-4-fenoxifenil)-1H-pirazolo[3,4-d]pirimidin-1-il)piperidina-1-carbonil)-4-metil-4-(4-(oxetan-3-il)piperazin-1-il)pent-2-enonitrilo en ratas
Se dosificó (R)-2-(3-(4-amino-3-(2-fluoro-4-fenoxifenil)-1 H-pirazolo[3,4-d]pirimidin-1 -il)piperidina-1 -carbonil)-4-metil-4-(4-(oxetan-3-il)piperazin-1-il)pent-2-enonitrilo en 10 mg/ml de solución de citrato acuosa a 20 mg/kg a ratas Wister-Hans. Se añadió ácido cítrico anhidro (0,5 equivalentes) a la base libre de (R)-2-(3-(4-amino-3-(2-fluoro-4-fenoxifenil)-1 H-pirazolo[3,4-d]pirimidin-1 -il)piperidina-1 -carbonil)-4-metil-4-(4-(oxetan-3-il)piperazin-1 -il)pent-2-enonitrilo. El experimento se repitió 3 veces cada uno con 2 grupos separados, y se incluyó un grupo de sonda oral en cada estudio. La dosificación intrayeyunal (IJ) e intraduodenal se hizo en ratas sondadas. En el primer estudio, hubo un aumento de 40 veces en la ABC para el grupo de dosificación U en comparación con el grupo PO, y el metabolito presente en el grupo U se redujo 10 veces. Los 3 estudios se resumen en la figura 1. Se muestra que la dosificación ID aumentaba la ABC y Cmáx de (R)-2-(3-(4-amino-3-(2-fluoro-4-fenoxifenil)-1H-pirazolo[3,4-d]pirimidin-1-il)piperidina-1-carbonil)-4-metil-4-(4-(oxetan-3-il)piperazin-1-il)pent-2-enonitrilo en comparación con la dosificación PO, y que la dosificación IJ aumentaba sustancialmente la ABC y Cmáx en comparación con la dosificación PO en ambos estudios.
Ejemplo 5:
Filtración in vitro en cámaras Ussing a través de diferentes regiones intestinales de rata
Reactivos: Tampón de Krebs-Ringer (KRB) 10X compuesto de NaCI 1,26 M (Promega, Madison, WI), KCl 25 mM, NaHCÜ3250 mM, NaH2PÜ4 12 mM, MgCl2 12 mM, CaCl225 mM y 18 g/l de D-glucosa (todos de Sigma Aldrich, St. Louis, MO) preparado y almacenado individualmente para evitar la cristalización. Se diluye hasta 1X de concentración de trabajo antes de su uso y se ajusta el pH con perfusión de carbógeno durante 20 min a pH 7,4 (KRB oxigenado) o HCl 1 M (Alfar Aesar, Ward Hill, MA) hasta pH 3,5, 6,5 o 7,4. Se adquirieron rojo fenol al 0,1 %, antipirina y atenolol de Sigma Aldrich, St. Louis, MO.
Recolección y preparación de tejido intestinal: Se aclimataron ratas Sprague-Drawley hembra vírgenes adultas de 4 a 9 meses de edad (Harlan Laboratories, Livermore,CA) en el propio laboratorio durante un mínimo de 3 días antes de la experimentación. Deben evitarse animales adolescentes y geriátricos para evitar las diferencias relacionadas con la edad en la morfología y función gastrointestinal. Los animales se alojaron según protocolos de cría aprobados por IACUC, que incluyen ciclos de 12 horas de luz y 12 horas de oscuridad, y se dejó pienso convencional y agua ad libitum. La recolección del tejido debe realizarse un animal cada vez para mantener la viabilidad del tejido. Una cada vez, las ratas se anestesiaron en una cámara de gas sellada perfundida lentamente con un 10 % (v/v)/min de dióxido de carbono para minimizar el malestar en la mucosa nasal durante aproximadamente 3-5 minutos o hasta la sedación completa evidenciada por la ausencia de respuesta a pellizco en el dedo y respiración superficial. Las ratas entonces se sacrificaron rápidamente por dislocación cervical.
Se utilizó un protocolo modificado de la bibliografía (D. I. Kosik-Bogacka, et al., (2011) "The effect of L-ascorbic acid and/or tocopherol supplementation on electrophysiological parameters of the colon of rats chronically exposed to lead" Med. Sci. Monit. 2011 17(1):BR16-26). Se realizó una incisión en la línea media para exponer la cavidad abdominal. La longitud del tejido gastrointestinal desde el estómago a través del colon ascendente se diseccionó en una pieza y se colocó en una bandeja de disección preenfriada llena de KRB oxigenado enfriado en hielo a pH 7,4. Los tejidos se movieron rápidamente a una bandeja de disección nueva llena de KRB enfriado en hielo a pH 7,4 y se burbujeó continuamente con gas carbógeno (95 % de O2 , 5 % de CO2) (CryoSpec, South San Francisco, CA). Los diferentes segmentos del intestino entonces se separaron con un bisturí de acuerdo con los esquemas. El estómago comprendía aprox. 3,81 cm (1,5 pulgadas), el duodeno aprox. 5,08 cm (2 pulgadas), el yeyuno aprox. 20,32-25,4 cm (8-10 pulgadas), el íleon aprox. 2,54 cm (1 pulgada) y el colon ascendente aprox. 2,54 cm (1 pulgada).
Cada segmento tubular entonces se cortó longitudinalmente a lo largo del límite mesentérico para exponer la superficie luminal. Los contenidos intestinales se enjuagaron suavemente con cuidado para no alterar el epitelio luminal. Cada segmento se cortó adicionalmente en piezas más pequeñas que medían aproximadamente 8 mm x 10 mm, se estiraron suavemente y se montaron en los pernos de una mitad de una cámara Ussing de difusión vertical (Navicyte®, Harvard Apparatus Inc., Holliston, MA) con un área superficial eficaz de 0,49 cm2. Los segmentos montados se inspeccionaron visualmente para desgarros antes de fusionarla con la segunda mitad de la cámara. Las cámaras se llenaron inmediatamente con 5 ml de KRB con pH fisiológico coincidente (cámaras mucosas: estómago pH 3,5, duodeno pH 6,5, yeyuno pH 6,5, íleon pH 7,4, colon pH 6,5; cámaras serosas: todas las secciones a pH 7,4). Cada ensamblaje de cámara Ussing completado se montó en serie en el bloque térmico termocirculado mantenido a 37 °C y fijado a un colector de gas rociado continuamente con gas carbógeno a una tasa de 3-5 burbujas/segundo. Se añadió rojo fenol (10 ul al 0,1 %) a cada cámara mucosa para comprobar el equilibrado del pH y las evidencias de desgarros microscópicos en el tejido.
Protocolo experimental: Todos los compuestos (muestras experimentales y muestras de control) se prepararon inicialmente como una solución madre 10 mM en DMSO al 100 %. El esquema experimental se realizó de acuerdo con (S. Haslam, et al., "Intestinal Ciprofloxacin Efflux: The Role of Breast Cancer Resistance Protein (ABCG2)" Drug Met. Disp. 2011 39(12):2321-28). En resumen, entonces se retiró un volumen idéntico de KRB y se remplazó por el volumen de cada compuesto para obtener una concentración de partida de 100 uM. Se incluyeron antipirina y atenolol en cada experimento como controles de referencia internos con un máximo de 5 compuestos en total (incluyendo controles de referencia) en cada experimento. Las muestras (100 ul) se retiraron de la cámara mucosa en t = 0 y t = 150 min, mientras que se retiraron muestras de 100 ul de la cámara serosa en t = 30, 60, 90 y 150 min. Las muestras se colocaron en una placa de 96 pocillos, se congelaron a -80 °C para análisis por RapidFire-LC-MS/MS. La permeabilidad aparente, Papp, se expresó como Papp= (dQ/dt).(1/(A.Cü)) donde dQ/dt es la tasa de transporte desde la cámara mucosa a la serosa, A es el área superficial eficaz de tejido y C0 es la concentración mucosa inicial (A. Sjoberg, et al., "Comprehensive study on regional human intestinal permeability and prediction of fraction absorbed of drugs using the Ussing chamber technique" Eur. J Pharm. Sci. 201348: 166-180).
Los resultados para una serie de inhibidores covalentes reversibles se muestran en la figura 2. Hay una permeabilidad en general aumentada en las regiones distales del tubo GI en comparación con el estómago o el duodeno y, en particular, las permeabilidades estadísticamente aumentadas para estos inhibidores de BTK y algunos análogos en las regiones yeyunal, ileal y colónica.
Ejemplo 6:
Ensayo de disolución
El ensayo de disolución se realiza de acuerdo con la sección 711 de la Farmacopea Estadounidense (http://www.pharmacopeia.cn/v29240/usp29nf24s0_c711 h.html) usando aparato 2 (palas a 75 rpm) con pesa de plomo alternativa 2A. Baños que contiene 900 ml tanto para ensayos en ácido (pH 2 o 3) como a pH 6,8 a temperatura 37 °C (±0,5 °C).
Los rasgos característicos divulgados en la descripción anterior, o las siguientes reivindicaciones, expresados en sus formas específicas o en términos de un medio para realizar la función reivindicada, o un método o proceso para obtener el resultado divulgado, según lo apropiado, pueden utilizarse por separado, o en cualquier combinación de dichos rasgos característicos, para lograr la invención en diversas formas de la misma.
La invención anterior se ha descrito en cierto detalle a modo de ilustración y ejemplo, por motivos de claridad y comprensión. Será obvio para un experto en la materia que pueden ponerse en práctica cambios y modificaciones dentro del alcance de las reivindicaciones adjuntas. Por lo tanto, debe entenderse que la descripción anterior pretende ser ilustrativa y no restrictiva.
Cualquier conflicto entre cualquier referencia citada en este documento y los contenidos específicos de esta memoria descriptiva se resolverá en favor de la última. Asimismo, cualquier conflicto entre una definición entendida en la técnica de una palabra o frase y una definición de la palabra o frase mostrada específicamente en esta memoria descriptiva se resolverá en favor de la última.
Claims (15)
1. Una forma farmacéutica oral sólida, que es un comprimido o cápsula, que comprende:
A un núcleo que comprende un compuesto ele ido de:


y/o una sal farmacéuticamente aceptable del mismo;
(B) un recubrimiento entérico;
(C) un subrecubrimiento entre el recubrimiento entérico y el núcleo, en donde el subrecubrimiento es:
(1) un polímero erosionable soluble en agua o hidrófilo, siendo dicho polímero un polímero de bajo peso molecular seleccionado de hidroximetilcelulosa (HPMC), hidroxietilcelulosa, hidroximetilcelulosa, hidroxipropilcelulosa, celulosa microcristalina, polivinilpirrolidionas, polisacáridos (o un derivado de polisacárido), poli(alcoholes vinílicos), polietilenglicol (PEG), polipropilenglicol (PPG) y copolímeros de bloque de PEG-PPG; o
(2) una composición insoluble en agua, que comprende: (i) partículas de compuesto soluble en agua que pueden formar canales en la composición insoluble en agua; o (ii) partículas hidrófilas insolubles en agua que provocan el hinchamiento del subrecubrimiento cuando está en contacto con un medio acuoso; y
(D) un excipiente farmacéuticamente aceptable,
en donde la forma farmacéutica oral sólida tiene un inicio de liberación del compuesto y/o dicha sal farmacéuticamente aceptable del mismo del núcleo (A) en la parte yeyunoileal del intestino delgado de un mamífero después de ingesta oral de la forma farmacéutica por parte del mamífero; y
en donde la forma farmacéutica oral sólida puede liberar al menos un 80 % en peso de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo en 120 minutos en un recipiente de disolución que comprende una solución acuosa a un pH de 6,4 a 7,4.
2. La forma farmacéutica oral sólida de acuerdo con la reivindicación 1, en donde la forma farmacéutica oral sólida puede liberar menos de un 10 % en peso de dicho compuesto y/o dicha sal farmacéuticamente aceptable del mismo del núcleo (A) en 1,5 horas en un recipiente de disolución que comprende una solución acuosa a un pH de menos de 3 o un pH de 4,5 a 5,5.
3. La forma farmacéutica oral sólida de acuerdo con la reivindicación 1 o reivindicación 2, en donde la forma farmacéutica oral sólida libera el compuesto y/o dicha sal farmacéuticamente aceptable del mismo del núcleo (A) como un bolo en el íleon y/o el yeyuno del intestino delgado del mamífero.
4. La forma farmacéutica oral sólida de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en donde el compuesto del núcleo (A) se elige de:



y/o una sal farmacéuticamente aceptable del mismo.
5. La forma farmacéutica oral sólida de acuerdo con una cualquiera de las reivindicaciones 1 a 4, en donde el subrecubrimiento (C) comprende: (i) partículas de un compuesto soluble en agua; o (ii) partículas hidrófilas insolubles en agua que provocan el hinchamiento del subrecubrimiento cuando está en contacto con un medio acuoso.
6. La forma farmacéutica oral sólida de acuerdo con una cualquiera de las reivindicaciones 1 a 4, en donde el subrecubrimiento (C) es una composición insoluble en agua que comprende partículas de un compuesto soluble en agua que forma canales que permiten el flujo entrante de agua en la forma farmacéutica oral sólida y la difusión del compuesto y/o la sal farmacéuticamente aceptable del mismo en el intestino.
7. La forma farmacéutica oral sólida de acuerdo con una cualquiera de las reivindicaciones 1 a 4, en donde el subrecubrimiento (C) es una composición insoluble en agua que comprende partículas de un compuesto soluble en agua que puede formar canales que son impermeables al compuesto y/o la sal farmacéuticamente aceptable del mismo, pero permiten la entrada de agua y el hinchamiento y rotura del subrecubrimiento, provocando la liberación del compuesto y/o la sal farmacéuticamente aceptable del mismo.
8. La forma farmacéutica oral sólida de acuerdo con una cualquiera de las reivindicaciones 1 a 7, en donde la solución acuosa es un líquido intestinal simulado a un pH de 6,4 a 7,4.
9. La forma farmacéutica oral sólida de acuerdo con una cualquiera de las reivindicaciones 1 a 8, en donde la forma farmacéutica oral sólida comprende una sal de ácido farmacéuticamente aceptable del compuesto.
10. La forma farmacéutica oral sólida de acuerdo con una cualquiera de las reivindicaciones 1 a 9, en donde el núcleo (A) comprende además:
(a) un ácido farmacéuticamente aceptable en una cantidad suficiente para producir una solución acuosa ácida dentro de la forma farmacéutica oral sólida antes de la liberación del compuesto y/o la sal farmacéuticamente aceptable del mismo del núcleo desde la forma farmacéutica oral sólida; y/o
(b) un tensioactivo que está presente a una concentración por encima de su concentración micelar crítica tras la disgregación en 50 ml de medio acuoso.
11. La forma farmacéutica oral sólida de acuerdo con una cualquiera de las reivindicaciones 1 a 10, en donde el tamaño medio de partícula del compuesto y/o la sal farmacéuticamente aceptable del mismo es de 0,3 micrómetros a 100 micrómetros.
12. La forma farmacéutica oral sólida de acuerdo con una cualquiera de las reivindicaciones 1 a 11, en donde el recubrimiento entérico (B):
(a) es de un 10 % a un 150 % del peso del núcleo;
(b) es de 5 a 500 micrómetros de grosor;
(c) se selecciona de gelatina polimerizada, goma-laca, copolímero de ácido metacrílico de tipo CNF, butirato ftalato de celulosa, hidrogenoftalato de celulosa, propionato ftalato de celulosa, acetato ftalato de polivinilo (PVAP), acetato ftalato de celulosa (CAP), acetato trimelitato de celulosa (CAT), ftalato de hidroxipropilmetilcelulosa, acetato de hidroxipropilmetilcelulosa, succinato de dioxipropilmetilcelulosa, carboximetiletilcelulosa (CMEC), acetato succinato de hidroxipropilmetilcelulosa (HPMCAS) y polímeros y copolímeros de ácido (met)acrílico, en donde dichos polímeros se preparan a partir de un monómero y dichos copolímeros se preparan a partir de dos o más monómeros, en donde
dichos monómeros se seleccionan de acrilato de metilo, acrilato de etilo, metacrilato de metilo y metacrilato de etilo, preferiblemente en donde el recubrimiento entérico comprende un polímero de poli(met)acrilato, opcionalmente en donde el recubrimiento entérico es una serie Eudragit® L o S, y opcionalmente además en donde el recubrimiento entérico es un Eudragit® L100, L12,5, S100, S12.5 o FS 30D;
(d) comprende un derivado de celulosa, preferiblemente en donde el derivado de celulosa se selecciona de metilcelulosa, acetato ftalato de celulosa, ftalato de hidroximetilcelulosa (HPMCP), succinato de hidroxipropilmetilcelulosa (HPMCS) y acetato succinato de hidroximetilcelulosa (HPMCAS); y/o
(e) comprende un polímero de acetato ftalato de polivinilo (PVAP).
13. La forma farmacéutica oral sólida de acuerdo con una cualquiera de las reivindicaciones 1 a 12, en donde el excipiente farmacéuticamente aceptable (D) se selecciona independientemente de aglutinantes, tensioactivos, diluyentes, tampones, antiadherentes, emolientes, disgregantes, antioxidantes, agentes antiespumantes, rellenos, aromas, colores, lubricantes, absorbentes, conservantes, plastificantes y edulcorantes.
14. Una forma farmacéutica oral sólida de acuerdo con una cualquiera de las reivindicaciones 1 a 13 para su uso en un método de tratamiento de una enfermedad tratable mediante inhibición de BTK, en un paciente que lo necesita reconocidamente, en donde dicho método comprende administrar al paciente una cantidad terapéuticamente eficaz del compuesto y/o la sal farmacéuticamente aceptable del mismo del núcleo (A) en una sola o múltiples dosis.
15. La forma farmacéutica oral sólida para su uso de acuerdo con la reivindicación 14, en donde la enfermedad se selecciona de una enfermedad autoinmunitaria, cáncer y una enfermedad inflamatoria, preferiblemente una leucemia o linfoma, y opcionalmente además en donde la leucemia se selecciona de leucemia linfocítica crónica (CLL), leucemia linfocítica pequeña (SLL), mieloma múltiple, linfoma de células del manto y linfoma no hodgkiniano de linfocitos B.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462096812P | 2014-12-24 | 2014-12-24 | |
| PCT/US2015/000515 WO2016105582A1 (en) | 2014-12-24 | 2015-12-23 | Compositions for ileo-jejunal drug delivery |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2942642T3 true ES2942642T3 (es) | 2023-06-05 |
Family
ID=55361929
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15834774T Active ES2942642T3 (es) | 2014-12-24 | 2015-12-23 | Composiciones para suministro yeyunoileal de fármacos |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US20180193274A1 (es) |
| EP (1) | EP3236943B1 (es) |
| JP (1) | JP6687248B2 (es) |
| DK (1) | DK3236943T3 (es) |
| ES (1) | ES2942642T3 (es) |
| FI (1) | FI3236943T3 (es) |
| HK (1) | HK1245071A1 (es) |
| HR (1) | HRP20230305T1 (es) |
| HU (1) | HUE061809T2 (es) |
| LT (1) | LT3236943T (es) |
| MA (1) | MA41265B1 (es) |
| PL (1) | PL3236943T3 (es) |
| PT (1) | PT3236943T (es) |
| RS (1) | RS64139B1 (es) |
| SI (1) | SI3236943T1 (es) |
| WO (1) | WO2016105582A1 (es) |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX361815B (es) | 2012-09-10 | 2018-12-17 | Principia Biopharma Inc | Compuestos pirazolopirimidinicos como inhibidores de cinasas. |
| MX367918B (es) | 2013-04-25 | 2019-09-11 | Beigene Ltd | Compuestos heterociclicos fusionados como inhibidores de proteina quinasa. |
| SI3702373T1 (sl) | 2013-09-13 | 2022-11-30 | Beigene Switzerland Gmbh | Protitelesa proti PD-1 in njihova uporaba kot terapevtiki in diagnostiki |
| KR20220027271A (ko) | 2014-02-21 | 2022-03-07 | 프린시피아 바이오파마, 인코퍼레이티드 | Btk 억제제의 염 및 고체 형태 |
| KR102003754B1 (ko) | 2014-07-03 | 2019-07-25 | 베이진 엘티디 | Pd-l1 항체와 이를 이용한 치료 및 진단 |
| WO2016100914A1 (en) | 2014-12-18 | 2016-06-23 | Gourlay Steven | Treatment of pemphigus |
| TW201718572A (zh) | 2015-06-24 | 2017-06-01 | 普林斯匹亞生物製藥公司 | 酪胺酸激酶抑制劑 |
| AR110038A1 (es) | 2016-05-26 | 2019-02-20 | Kalyra Pharmaceuticals Inc | Compuestos inhibidores de egfr; composición farmacéutica que lo comprende; métodos para mejorar o tratar un cáncer; método para inhibir la replicación de un crecimiento maligno o un tumor; métodos para inhibir la actividad del egfr; y usos de los compuestos |
| IL293621B2 (en) | 2016-06-29 | 2023-09-01 | Principia Biopharma Inc | Modified release formulations of 2-[3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[4,3-d]pyrimidin-1-yl]piperidine-1-carbonyl]-4 -Methyl-4-[4-(oxane-3-yl)piperazine-1-yl)penta-2-ananitrile |
| NZ749997A (en) | 2016-07-05 | 2022-11-25 | Beigene Ltd | Combination of a pd-l antagonist and a raf inhibitor for treating cancer |
| TW202233628A (zh) * | 2016-08-16 | 2022-09-01 | 英屬開曼群島商百濟神州有限公司 | (s)-7-(1-丙烯醯基六氫吡啶-4-基)-2-(4-苯氧基苯基)-4,5,6,7-四氫吡唑並[1,5-a]嘧啶-3-甲醯胺的晶型、其製備和用途 |
| ES2971881T3 (es) | 2016-08-19 | 2024-06-10 | Beigene Switzerland Gmbh | Combinación de zanubrutinib con un anticuerpo anti-cd20 o anti-pd-1 para su uso en el tratamiento del cáncer |
| CN106588937B (zh) * | 2017-01-16 | 2018-09-21 | 东莞市真兴贝特医药技术有限公司 | 咪唑并吡嗪类化合物及其制备方法和应用 |
| EP3573989A4 (en) | 2017-01-25 | 2020-11-18 | Beigene, Ltd. | CRYSTALLINE FORMS OF (S) -7- (1- (BUT-2-YNOYL) -PIPERIDINE-4-YL) -2- (4-PHENOXYPHENYL) -4,5,6,7-TETRAHYDROPYRAZOLO [1,5-A ] PYRIMIDINE-3-CARBOXAMIDE, MANUFACTURING AND USES THEREOF |
| TW201906866A (zh) | 2017-06-26 | 2019-02-16 | 英屬開曼群島商百濟神州有限公司 | 酸性鞘磷脂酶缺乏症患者中異常骨狀況的治療 |
| WO2019034009A1 (en) | 2017-08-12 | 2019-02-21 | Beigene, Ltd. | BTK INHIBITOR WITH ENHANCED DOUBLE SELECTIVITY |
| JP6922085B2 (ja) * | 2017-09-28 | 2021-08-18 | シャンハイ ハイヤン ファーマシューティカル テクノロジー カンパニー リミテッドShanghai Haiyan Pharmaceutical Technology Co., Ltd. | 4,6,7−三置換 1,2−ジヒドロピロロ[3,4−c]ピリジン/ピリミジン−3−オン誘導体及びその使用 |
| CN111801334B (zh) | 2017-11-29 | 2023-06-09 | 百济神州瑞士有限责任公司 | 使用包含btk抑制剂的组合治疗惰性或侵袭性b-细胞淋巴瘤 |
| CA3089537A1 (en) | 2018-01-20 | 2019-07-25 | Natco Pharma Limited | Pharmaceutical compositions comprising ibrutinib |
| SG11202102116XA (en) * | 2018-09-18 | 2021-04-29 | Beijing Innocare Pharma Tech Co Ltd | Crystalline forms of 6- (1-acryloylpiperidin-4-yl) -2- (4-phenoxyphenyl) nicotinamide |
| WO2020093896A1 (en) * | 2018-11-06 | 2020-05-14 | Beijing Innocare Pharma Tech Co., Ltd. | Amorphous solid dispersion comprising 6- (1-acryloylpiperidin-4-yl) -2- (4-phenoxyphenyl) nicotinamide |
| WO2020102304A1 (en) * | 2018-11-13 | 2020-05-22 | Rain Therapeutics Inc. | Combination of a kinase inhibitor and an electrolyte, compositions and methods comprising the same |
| CN109851725A (zh) * | 2018-12-29 | 2019-06-07 | 华南理工大学 | 纳米纤维素-聚丙烯酰胺-明胶复合水凝胶、其制备方法及应用 |
| TW202235083A (zh) * | 2020-11-02 | 2022-09-16 | 美商黑鑽醫療公司 | 使用經炔取代之喹唑啉衍生物類以治療癌症之方法 |
| CN113855695B (zh) * | 2021-11-01 | 2023-06-06 | 上海理工大学 | 一种结肠释药口服复方药物组合物及其制备方法 |
| CN114181159B (zh) * | 2021-12-28 | 2024-02-20 | 南通大学 | 含2,4,5-三取代嘧啶酰肼衍生物及其制备方法与应用 |
| US11786531B1 (en) | 2022-06-08 | 2023-10-17 | Beigene Switzerland Gmbh | Methods of treating B-cell proliferative disorder |
| CN116473923B (zh) * | 2023-05-29 | 2024-05-14 | 杭州剂泰医药科技有限责任公司 | 一种奈拉替尼固体分散体组合物及其制备方法和应用 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140303191A1 (en) * | 2011-10-19 | 2014-10-09 | Pharmacyclics, Inc. | Use of inhibitors of bruton's tyrosine kinase (btk) |
| CA2874461C (en) * | 2012-06-18 | 2021-10-12 | Principia Biopharma Inc. | Formulations containing reversible covalent compounds |
| WO2014004707A1 (en) | 2012-06-29 | 2014-01-03 | Principia Biopharma Inc. | Formulations comprising ibrutinib |
| MX361815B (es) * | 2012-09-10 | 2018-12-17 | Principia Biopharma Inc | Compuestos pirazolopirimidinicos como inhibidores de cinasas. |
| EP2941245B1 (en) * | 2013-01-05 | 2024-06-26 | Anji Pharmaceuticals Inc. | Delayed-release composition comprising biguanide |
-
2015
- 2015-12-23 PL PL15834774.0T patent/PL3236943T3/pl unknown
- 2015-12-23 ES ES15834774T patent/ES2942642T3/es active Active
- 2015-12-23 MA MA41265A patent/MA41265B1/fr unknown
- 2015-12-23 US US15/539,100 patent/US20180193274A1/en not_active Abandoned
- 2015-12-23 HR HRP20230305TT patent/HRP20230305T1/hr unknown
- 2015-12-23 EP EP15834774.0A patent/EP3236943B1/en active Active
- 2015-12-23 HU HUE15834774A patent/HUE061809T2/hu unknown
- 2015-12-23 PT PT158347740T patent/PT3236943T/pt unknown
- 2015-12-23 WO PCT/US2015/000515 patent/WO2016105582A1/en active Application Filing
- 2015-12-23 FI FIEP15834774.0T patent/FI3236943T3/fi active
- 2015-12-23 LT LTEPPCT/US2015/000515T patent/LT3236943T/lt unknown
- 2015-12-23 JP JP2017533932A patent/JP6687248B2/ja active Active
- 2015-12-23 RS RS20230281A patent/RS64139B1/sr unknown
- 2015-12-23 SI SI201531939T patent/SI3236943T1/sl unknown
- 2015-12-23 DK DK15834774.0T patent/DK3236943T3/da active
-
2018
- 2018-03-26 HK HK18104152.6A patent/HK1245071A1/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| FI3236943T3 (fi) | 2023-04-27 |
| HRP20230305T1 (hr) | 2023-05-26 |
| JP2018500353A (ja) | 2018-01-11 |
| LT3236943T (lt) | 2023-04-11 |
| WO2016105582A1 (en) | 2016-06-30 |
| MA41265B1 (fr) | 2023-05-31 |
| US20180193274A1 (en) | 2018-07-12 |
| SI3236943T1 (sl) | 2023-06-30 |
| EP3236943B1 (en) | 2023-01-25 |
| DK3236943T3 (da) | 2023-04-11 |
| PL3236943T3 (pl) | 2023-05-29 |
| EP3236943A1 (en) | 2017-11-01 |
| RS64139B1 (sr) | 2023-05-31 |
| JP6687248B2 (ja) | 2020-04-22 |
| MA41265A (fr) | 2017-10-31 |
| PT3236943T (pt) | 2023-04-20 |
| HUE061809T2 (hu) | 2023-08-28 |
| HK1245071A1 (zh) | 2018-08-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2942642T3 (es) | Composiciones para suministro yeyunoileal de fármacos | |
| ES2873389T3 (es) | Composición farmacéutica de olaparib oral de liberación prolongada y controlada y usos de la misma | |
| ES2754573T3 (es) | Formulación de comprimido recubierto y método | |
| ES2525005T3 (es) | Formulación de liberación prolongada que contiene una cera | |
| JP2019123738A (ja) | ジアルキルフマレート含有薬剤調合物 | |
| ES2547226T3 (es) | Formulaciones de dosificación oral de liberación controlada que comprenden un núcleo y una o más capas de barrera | |
| ES2958616T3 (es) | Formulaciones farmacéuticas de floroglucinol y trimetilfloroglucinol | |
| ES2885437T3 (es) | Formulación de combinación de tesofensina y metoprolol | |
| US20170231927A1 (en) | Pharmaceutical compositions of memantine | |
| BR112012024019B1 (pt) | Forma de dosagem de liberação controlada para administração oral | |
| WO2003039519A2 (es) | Forma de dosificación de liberación controlada dual | |
| KR20070043806A (ko) | pH에 매우 의존적인 용해도를 가지는 활성 성분의 제어방출을 위한 약제학적 다층 정제 | |
| ES2865278T3 (es) | Amantadina para mejorar la marcha en esclerosis múltiple | |
| WO2007106960A1 (en) | Controlled-release floating dosage forms | |
| US20110287096A1 (en) | Modified gastroretentive drug delivery system for amine drugs | |
| RU2010109408A (ru) | Композиции с пролонгированным высвобождением, включающие микофенолят натрия, и их действия | |
| JP2018538289A (ja) | アプレミラスト徐放性製剤 | |
| BRPI0715959A2 (pt) | composiÇço farmacÊutica de liberaÇço modificada de cloridrato de bupropion | |
| KR101925590B1 (ko) | 개선된 생체이용률을 갖는 페노피브릭산 제제 | |
| JP2023509754A (ja) | リオチロニンを含む持続放出組成物 | |
| JP2023181218A (ja) | 持続放出ミドドリン塩酸塩組成物及び使用方法 | |
| KR102248114B1 (ko) | 메트포르민 함유 중공형 위체류 제제 | |
| KR20170086053A (ko) | 디메틸 푸마르산염을 포함하는 제약학적 매트릭스 제제 | |
| ES2973959T3 (es) | Composición de liberación sostenida que comprende oxalato de tapentadol y método de preparación de la misma | |
| ES2896150T3 (es) | Composiciones líquidas de liberación prolongada de guaifenesina |