ES2865278T3 - Amantadina para mejorar la marcha en esclerosis múltiple - Google Patents
Amantadina para mejorar la marcha en esclerosis múltiple Download PDFInfo
- Publication number
- ES2865278T3 ES2865278T3 ES15857591T ES15857591T ES2865278T3 ES 2865278 T3 ES2865278 T3 ES 2865278T3 ES 15857591 T ES15857591 T ES 15857591T ES 15857591 T ES15857591 T ES 15857591T ES 2865278 T3 ES2865278 T3 ES 2865278T3
- Authority
- ES
- Spain
- Prior art keywords
- amantadine
- hours
- dose
- composition
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 title claims abstract description 479
- 229960003805 amantadine Drugs 0.000 title claims abstract description 469
- 230000005021 gait Effects 0.000 title claims abstract description 48
- 201000006417 multiple sclerosis Diseases 0.000 title claims abstract description 35
- 150000003839 salts Chemical class 0.000 claims abstract description 139
- 238000000034 method Methods 0.000 claims abstract description 93
- 239000000546 pharmaceutical excipient Substances 0.000 claims abstract description 44
- 239000000203 mixture Substances 0.000 claims description 350
- 239000003814 drug Substances 0.000 claims description 88
- 229940079593 drug Drugs 0.000 claims description 81
- 239000012730 sustained-release form Substances 0.000 claims description 77
- 238000012360 testing method Methods 0.000 claims description 53
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 37
- 238000013265 extended release Methods 0.000 claims description 37
- WOLHOYHSEKDWQH-UHFFFAOYSA-N amantadine hydrochloride Chemical compound [Cl-].C1C(C2)CC3CC2CC1([NH3+])C3 WOLHOYHSEKDWQH-UHFFFAOYSA-N 0.000 claims description 35
- 229960001280 amantadine hydrochloride Drugs 0.000 claims description 35
- NUKYPUAOHBNCPY-UHFFFAOYSA-N 4-aminopyridine Chemical compound NC1=CC=NC=C1 NUKYPUAOHBNCPY-UHFFFAOYSA-N 0.000 claims description 24
- 229960004979 fampridine Drugs 0.000 claims description 24
- 230000009467 reduction Effects 0.000 claims description 15
- -1 tetrabenizine Chemical compound 0.000 claims description 12
- DUGOZIWVEXMGBE-UHFFFAOYSA-N Methylphenidate Chemical compound C=1C=CC=CC=1C(C(=O)OC)C1CCCCN1 DUGOZIWVEXMGBE-UHFFFAOYSA-N 0.000 claims description 8
- 229960001344 methylphenidate Drugs 0.000 claims description 8
- KPYSYYIEGFHWSV-UHFFFAOYSA-N Baclofen Chemical compound OC(=O)CC(CN)C1=CC=C(Cl)C=C1 KPYSYYIEGFHWSV-UHFFFAOYSA-N 0.000 claims description 6
- MKXZASYAUGDDCJ-SZMVWBNQSA-N LSM-2525 Chemical compound C1CCC[C@H]2[C@@]3([H])N(C)CC[C@]21C1=CC(OC)=CC=C1C3 MKXZASYAUGDDCJ-SZMVWBNQSA-N 0.000 claims description 6
- 229960000794 baclofen Drugs 0.000 claims description 6
- 229960001985 dextromethorphan Drugs 0.000 claims description 6
- LDCRTTXIJACKKU-ONEGZZNKSA-N dimethyl fumarate Chemical compound COC(=O)\C=C\C(=O)OC LDCRTTXIJACKKU-ONEGZZNKSA-N 0.000 claims description 6
- 229960004419 dimethyl fumarate Drugs 0.000 claims description 6
- 229960000556 fingolimod Drugs 0.000 claims description 6
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 claims description 6
- 229960000331 teriflunomide Drugs 0.000 claims description 6
- UTNUDOFZCWSZMS-YFHOEESVSA-N teriflunomide Chemical compound C\C(O)=C(/C#N)C(=O)NC1=CC=C(C(F)(F)F)C=C1 UTNUDOFZCWSZMS-YFHOEESVSA-N 0.000 claims description 6
- XFYDIVBRZNQMJC-UHFFFAOYSA-N tizanidine Chemical compound ClC=1C=CC2=NSN=C2C=1NC1=NCCN1 XFYDIVBRZNQMJC-UHFFFAOYSA-N 0.000 claims description 6
- 229960000488 tizanidine Drugs 0.000 claims description 6
- 241000282412 Homo Species 0.000 claims 1
- 239000011248 coating agent Substances 0.000 description 73
- 238000000576 coating method Methods 0.000 description 73
- 230000036470 plasma concentration Effects 0.000 description 71
- 239000002775 capsule Substances 0.000 description 68
- 238000013268 sustained release Methods 0.000 description 63
- 238000009472 formulation Methods 0.000 description 55
- 238000004090 dissolution Methods 0.000 description 51
- 239000002552 dosage form Substances 0.000 description 46
- 238000011282 treatment Methods 0.000 description 39
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 31
- 230000006872 improvement Effects 0.000 description 30
- 201000010099 disease Diseases 0.000 description 28
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 26
- 239000011230 binding agent Substances 0.000 description 25
- 239000004014 plasticizer Substances 0.000 description 24
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 22
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 22
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 22
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 22
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 22
- 238000000338 in vitro Methods 0.000 description 21
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 20
- 241000699670 Mus sp. Species 0.000 description 19
- 239000000243 solution Substances 0.000 description 19
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 18
- 239000003795 chemical substances by application Substances 0.000 description 18
- 229940069328 povidone Drugs 0.000 description 18
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 17
- 206010017577 Gait disturbance Diseases 0.000 description 17
- 239000001856 Ethyl cellulose Substances 0.000 description 16
- 239000012738 dissolution medium Substances 0.000 description 16
- 235000019325 ethyl cellulose Nutrition 0.000 description 16
- 229920001249 ethyl cellulose Polymers 0.000 description 16
- 229940068196 placebo Drugs 0.000 description 16
- 239000000902 placebo Substances 0.000 description 16
- 230000000694 effects Effects 0.000 description 14
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 14
- 241001465754 Metazoa Species 0.000 description 13
- 208000006011 Stroke Diseases 0.000 description 13
- 230000001965 increasing effect Effects 0.000 description 13
- 239000003826 tablet Substances 0.000 description 13
- FLKPEMZONWLCSK-UHFFFAOYSA-N diethyl phthalate Chemical compound CCOC(=O)C1=CC=CC=C1C(=O)OCC FLKPEMZONWLCSK-UHFFFAOYSA-N 0.000 description 12
- 239000008188 pellet Substances 0.000 description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 11
- 201000002491 encephalomyelitis Diseases 0.000 description 11
- 239000010410 layer Substances 0.000 description 11
- 238000004519 manufacturing process Methods 0.000 description 11
- 229920001223 polyethylene glycol Polymers 0.000 description 11
- 208000024891 symptom Diseases 0.000 description 11
- 239000002202 Polyethylene glycol Substances 0.000 description 10
- 238000004458 analytical method Methods 0.000 description 10
- 230000006735 deficit Effects 0.000 description 10
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 10
- 208000019116 sleep disease Diseases 0.000 description 10
- 208000022925 sleep disturbance Diseases 0.000 description 10
- 208000016285 Movement disease Diseases 0.000 description 9
- 238000013270 controlled release Methods 0.000 description 9
- 229920001577 copolymer Polymers 0.000 description 9
- 229920001531 copovidone Polymers 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 8
- 238000000540 analysis of variance Methods 0.000 description 8
- 208000035475 disorder Diseases 0.000 description 8
- 230000003483 hypokinetic effect Effects 0.000 description 8
- 229940016286 microcrystalline cellulose Drugs 0.000 description 8
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 8
- 239000008108 microcrystalline cellulose Substances 0.000 description 8
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 230000008901 benefit Effects 0.000 description 7
- 230000037396 body weight Effects 0.000 description 7
- 239000004359 castor oil Substances 0.000 description 7
- 235000019438 castor oil Nutrition 0.000 description 7
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 7
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 7
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 7
- 229940057917 medium chain triglycerides Drugs 0.000 description 7
- 230000003204 osmotic effect Effects 0.000 description 7
- 239000008194 pharmaceutical composition Substances 0.000 description 7
- 229920000642 polymer Polymers 0.000 description 7
- 235000000346 sugar Nutrition 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- 208000018737 Parkinson disease Diseases 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 230000002411 adverse Effects 0.000 description 6
- 239000001768 carboxy methyl cellulose Substances 0.000 description 6
- 150000001860 citric acid derivatives Chemical class 0.000 description 6
- 238000013461 design Methods 0.000 description 6
- 235000013305 food Nutrition 0.000 description 6
- 125000005456 glyceride group Chemical group 0.000 description 6
- 238000011068 loading method Methods 0.000 description 6
- 239000011148 porous material Substances 0.000 description 6
- 230000001105 regulatory effect Effects 0.000 description 6
- 238000011947 six minute walk test Methods 0.000 description 6
- 238000002560 therapeutic procedure Methods 0.000 description 6
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 5
- 206010008748 Chorea Diseases 0.000 description 5
- 208000023105 Huntington disease Diseases 0.000 description 5
- 206010030312 On and off phenomenon Diseases 0.000 description 5
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 5
- 229960002805 amantadine sulfate Drugs 0.000 description 5
- MYWTWSQFJLXGGQ-UHFFFAOYSA-N amantadine sulfate Chemical compound OS(O)(=O)=O.C1C(C2)CC3CC2CC1(N)C3.C1C(C2)CC3CC2CC1(N)C3 MYWTWSQFJLXGGQ-UHFFFAOYSA-N 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 5
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 5
- 239000011093 chipboard Substances 0.000 description 5
- 150000001875 compounds Chemical class 0.000 description 5
- 230000001186 cumulative effect Effects 0.000 description 5
- 238000002513 implantation Methods 0.000 description 5
- 235000019359 magnesium stearate Nutrition 0.000 description 5
- 238000002483 medication Methods 0.000 description 5
- 229920000058 polyacrylate Polymers 0.000 description 5
- 239000001632 sodium acetate Substances 0.000 description 5
- 235000017281 sodium acetate Nutrition 0.000 description 5
- 238000001356 surgical procedure Methods 0.000 description 5
- 239000000454 talc Substances 0.000 description 5
- 229910052623 talc Inorganic materials 0.000 description 5
- 230000002618 waking effect Effects 0.000 description 5
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 4
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 4
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 4
- 208000000323 Tourette Syndrome Diseases 0.000 description 4
- 208000016620 Tourette disease Diseases 0.000 description 4
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 4
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 4
- 208000029028 brain injury Diseases 0.000 description 4
- 239000011247 coating layer Substances 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 230000003111 delayed effect Effects 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 239000007903 gelatin capsule Substances 0.000 description 4
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 4
- 238000011081 inoculation Methods 0.000 description 4
- 206010022437 insomnia Diseases 0.000 description 4
- 229920000609 methyl cellulose Polymers 0.000 description 4
- 235000010981 methylcellulose Nutrition 0.000 description 4
- 239000001923 methylcellulose Substances 0.000 description 4
- 238000010172 mouse model Methods 0.000 description 4
- 229920003023 plastic Polymers 0.000 description 4
- 239000004033 plastic Substances 0.000 description 4
- 239000000565 sealant Substances 0.000 description 4
- 238000007789 sealing Methods 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 239000004925 Acrylic resin Substances 0.000 description 3
- 229920000178 Acrylic resin Polymers 0.000 description 3
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 3
- 206010003591 Ataxia Diseases 0.000 description 3
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 3
- 229920003157 Eudragit® RL 30 D Polymers 0.000 description 3
- 229920003161 Eudragit® RS 30 D Polymers 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 229930195725 Mannitol Natural products 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- 229920002845 Poly(methacrylic acid) Polymers 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- 206010043118 Tardive Dyskinesia Diseases 0.000 description 3
- 208000030886 Traumatic Brain injury Diseases 0.000 description 3
- 230000005856 abnormality Effects 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 239000011324 bead Substances 0.000 description 3
- 230000036760 body temperature Effects 0.000 description 3
- 239000007963 capsule composition Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 229920001477 hydrophilic polymer Polymers 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 239000007937 lozenge Substances 0.000 description 3
- 238000012423 maintenance Methods 0.000 description 3
- 239000000594 mannitol Substances 0.000 description 3
- 235000010355 mannitol Nutrition 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 239000003703 n methyl dextro aspartic acid receptor blocking agent Substances 0.000 description 3
- 230000004770 neurodegeneration Effects 0.000 description 3
- 208000015122 neurodegenerative disease Diseases 0.000 description 3
- 239000006186 oral dosage form Substances 0.000 description 3
- 239000005022 packaging material Substances 0.000 description 3
- 230000035699 permeability Effects 0.000 description 3
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 230000002035 prolonged effect Effects 0.000 description 3
- 239000008213 purified water Substances 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- CPKVUHPKYQGHMW-UHFFFAOYSA-N 1-ethenylpyrrolidin-2-one;molecular iodine Chemical compound II.C=CN1CCCC1=O CPKVUHPKYQGHMW-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- 229920002126 Acrylic acid copolymer Polymers 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 208000019736 Cranial nerve disease Diseases 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- 229920003152 Eudragit® RS polymer Polymers 0.000 description 2
- 229920003134 Eudragit® polymer Polymers 0.000 description 2
- 201000004311 Gilles de la Tourette syndrome Diseases 0.000 description 2
- 101001068052 Homo sapiens Lysine-specific demethylase hairless Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 2
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 2
- 229940127523 NMDA Receptor Antagonists Drugs 0.000 description 2
- 229940099433 NMDA receptor antagonist Drugs 0.000 description 2
- 208000027089 Parkinsonian disease Diseases 0.000 description 2
- 206010034010 Parkinsonism Diseases 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- 208000001647 Renal Insufficiency Diseases 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- ZFOZVQLOBQUTQQ-UHFFFAOYSA-N Tributyl citrate Chemical compound CCCCOC(=O)CC(O)(C(=O)OCCCC)CC(=O)OCCCC ZFOZVQLOBQUTQQ-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 230000000202 analgesic effect Effects 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 230000001977 ataxic effect Effects 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000002146 bilateral effect Effects 0.000 description 2
- 230000036772 blood pressure Effects 0.000 description 2
- RMRJXGBAOAMLHD-IHFGGWKQSA-N buprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)CN2CC1CC1 RMRJXGBAOAMLHD-IHFGGWKQSA-N 0.000 description 2
- 208000015114 central nervous system disease Diseases 0.000 description 2
- 208000012601 choreatic disease Diseases 0.000 description 2
- 230000001149 cognitive effect Effects 0.000 description 2
- 229960000913 crospovidone Drugs 0.000 description 2
- STORWMDPIHOSMF-UHFFFAOYSA-N decanoic acid;octanoic acid;propane-1,2,3-triol Chemical compound OCC(O)CO.CCCCCCCC(O)=O.CCCCCCCCCC(O)=O STORWMDPIHOSMF-UHFFFAOYSA-N 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 230000001079 digestive effect Effects 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- 208000037765 diseases and disorders Diseases 0.000 description 2
- 238000002565 electrocardiography Methods 0.000 description 2
- 239000002702 enteric coating Substances 0.000 description 2
- 238000009505 enteric coating Methods 0.000 description 2
- 238000001125 extrusion Methods 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 239000007897 gelcap Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 102000053615 human HR Human genes 0.000 description 2
- 229920013821 hydroxy alkyl cellulose Polymers 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 239000002054 inoculum Substances 0.000 description 2
- 239000011229 interlayer Substances 0.000 description 2
- 229960002725 isoflurane Drugs 0.000 description 2
- 201000006370 kidney failure Diseases 0.000 description 2
- 230000003907 kidney function Effects 0.000 description 2
- 238000011545 laboratory measurement Methods 0.000 description 2
- 239000004922 lacquer Substances 0.000 description 2
- 238000004811 liquid chromatography Methods 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 229920003145 methacrylic acid copolymer Polymers 0.000 description 2
- 230000002981 neuropathic effect Effects 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- WEXRUCMBJFQVBZ-UHFFFAOYSA-N pentobarbital Chemical compound CCCC(C)C1(CC)C(=O)NC(=O)NC1=O WEXRUCMBJFQVBZ-UHFFFAOYSA-N 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 229920001282 polysaccharide Polymers 0.000 description 2
- 239000005017 polysaccharide Substances 0.000 description 2
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 2
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 2
- 125000001453 quaternary ammonium group Chemical group 0.000 description 2
- 239000013557 residual solvent Substances 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 230000001953 sensory effect Effects 0.000 description 2
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 2
- 239000001509 sodium citrate Substances 0.000 description 2
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 238000005563 spheronization Methods 0.000 description 2
- 230000002269 spontaneous effect Effects 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 238000007619 statistical method Methods 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 230000009747 swallowing Effects 0.000 description 2
- 238000004885 tandem mass spectrometry Methods 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- DCXXMTOCNZCJGO-UHFFFAOYSA-N tristearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCC DCXXMTOCNZCJGO-UHFFFAOYSA-N 0.000 description 2
- 238000002562 urinalysis Methods 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- MKJIEFSOBYUXJB-HOCLYGCPSA-N (3S,11bS)-9,10-dimethoxy-3-isobutyl-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-one Chemical compound C1CN2C[C@H](CC(C)C)C(=O)C[C@H]2C2=C1C=C(OC)C(OC)=C2 MKJIEFSOBYUXJB-HOCLYGCPSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- VKNASXZDGZNEDA-UHFFFAOYSA-N 2-cyanoethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCC#N VKNASXZDGZNEDA-UHFFFAOYSA-N 0.000 description 1
- SFPNZPQIIAJXGL-UHFFFAOYSA-N 2-ethoxyethyl 2-methylprop-2-enoate Chemical class CCOCCOC(=O)C(C)=C SFPNZPQIIAJXGL-UHFFFAOYSA-N 0.000 description 1
- 229920000856 Amylose Polymers 0.000 description 1
- 206010003497 Asphyxia Diseases 0.000 description 1
- 208000009017 Athetosis Diseases 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- 208000001408 Carbon monoxide poisoning Diseases 0.000 description 1
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 1
- 208000015879 Cerebellar disease Diseases 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- 208000010693 Charcot-Marie-Tooth Disease Diseases 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- PYGXAGIECVVIOZ-UHFFFAOYSA-N Dibutyl decanedioate Chemical compound CCCCOC(=O)CCCCCCCCC(=O)OCCCC PYGXAGIECVVIOZ-UHFFFAOYSA-N 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- 229920003139 Eudragit® L 100 Polymers 0.000 description 1
- 229920003136 Eudragit® L polymer Polymers 0.000 description 1
- 229920003151 Eudragit® RL polymer Polymers 0.000 description 1
- 229920003141 Eudragit® S 100 Polymers 0.000 description 1
- 206010015548 Euthanasia Diseases 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 206010017585 Gait spastic Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 206010018910 Haemolysis Diseases 0.000 description 1
- 208000004547 Hallucinations Diseases 0.000 description 1
- 206010019468 Hemiplegia Diseases 0.000 description 1
- 208000000269 Hyperkinesis Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 241000712431 Influenza A virus Species 0.000 description 1
- 229920003085 Kollidon® CL Polymers 0.000 description 1
- 241000218652 Larix Species 0.000 description 1
- 235000005590 Larix decidua Nutrition 0.000 description 1
- 244000178870 Lavandula angustifolia Species 0.000 description 1
- 235000010663 Lavandula angustifolia Nutrition 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- SPAGIJMPHSUYSE-UHFFFAOYSA-N Magnesium peroxide Chemical compound [Mg+2].[O-][O-] SPAGIJMPHSUYSE-UHFFFAOYSA-N 0.000 description 1
- 229920003091 Methocel™ Polymers 0.000 description 1
- 208000034819 Mobility Limitation Diseases 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- 229920000715 Mucilage Polymers 0.000 description 1
- 208000021642 Muscular disease Diseases 0.000 description 1
- 206010052904 Musculoskeletal stiffness Diseases 0.000 description 1
- 201000009623 Myopathy Diseases 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 208000036110 Neuroinflammatory disease Diseases 0.000 description 1
- 208000011644 Neurologic Gait disease Diseases 0.000 description 1
- 206010029412 Nightmare Diseases 0.000 description 1
- 206010033557 Palpitations Diseases 0.000 description 1
- 206010033799 Paralysis Diseases 0.000 description 1
- 206010056242 Parkinsonian gait Diseases 0.000 description 1
- 108010081690 Pertussis Toxin Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 208000036757 Postencephalitic parkinsonism Diseases 0.000 description 1
- 229920003082 Povidone K 90 Polymers 0.000 description 1
- 229920000153 Povidone-iodine Polymers 0.000 description 1
- 239000004373 Pullulan Substances 0.000 description 1
- 229920001218 Pullulan Polymers 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 208000034189 Sclerosis Diseases 0.000 description 1
- 208000008039 Secondary Parkinson Disease Diseases 0.000 description 1
- 208000027522 Sydenham chorea Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O ammonium group Chemical group [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 210000003484 anatomy Anatomy 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000000420 anogeissus latifolia wall. gum Substances 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 229940035678 anti-parkinson drug Drugs 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 239000000939 antiparkinson agent Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 208000015802 attention deficit-hyperactivity disease Diseases 0.000 description 1
- 230000003190 augmentative effect Effects 0.000 description 1
- DZGUJOWBVDZNNF-UHFFFAOYSA-N azanium;2-methylprop-2-enoate Chemical compound [NH4+].CC(=C)C([O-])=O DZGUJOWBVDZNNF-UHFFFAOYSA-N 0.000 description 1
- 208000018300 basal ganglia disease Diseases 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 235000012216 bentonite Nutrition 0.000 description 1
- 229940064804 betadine Drugs 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 239000000378 calcium silicate Substances 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 239000004203 carnauba wax Substances 0.000 description 1
- 235000013869 carnauba wax Nutrition 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 1
- 229920003086 cellulose ether Polymers 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 210000001638 cerebellum Anatomy 0.000 description 1
- 206010008129 cerebral palsy Diseases 0.000 description 1
- 208000026106 cerebrovascular disease Diseases 0.000 description 1
- 235000011967 chocolate pudding Nutrition 0.000 description 1
- 229920001688 coating polymer Polymers 0.000 description 1
- 230000019771 cognition Effects 0.000 description 1
- 230000003920 cognitive function Effects 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 210000003792 cranial nerve Anatomy 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- FSXVSUSRJXIJHB-UHFFFAOYSA-M ethyl prop-2-enoate;methyl 2-methylprop-2-enoate;trimethyl-[2-(2-methylprop-2-enoyloxy)ethyl]azanium;chloride Chemical compound [Cl-].CCOC(=O)C=C.COC(=O)C(C)=C.CC(=C)C(=O)OCC[N+](C)(C)C FSXVSUSRJXIJHB-UHFFFAOYSA-M 0.000 description 1
- 208000012997 experimental autoimmune encephalomyelitis Diseases 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 239000001087 glyceryl triacetate Substances 0.000 description 1
- 235000013773 glyceryl triacetate Nutrition 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 235000019314 gum ghatti Nutrition 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 230000008588 hemolysis Effects 0.000 description 1
- 239000010903 husk Substances 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 1
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 1
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 description 1
- 230000000642 iatrogenic effect Effects 0.000 description 1
- 208000035231 inattentive type attention deficit hyperactivity disease Diseases 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 150000002484 inorganic compounds Chemical class 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 238000009434 installation Methods 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- 239000008274 jelly Substances 0.000 description 1
- 239000001102 lavandula vera Substances 0.000 description 1
- 235000018219 lavender Nutrition 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 1
- 229910052808 lithium carbonate Inorganic materials 0.000 description 1
- 208000018883 loss of balance Diseases 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- HCWCAKKEBCNQJP-UHFFFAOYSA-N magnesium orthosilicate Chemical compound [Mg+2].[Mg+2].[O-][Si]([O-])([O-])[O-] HCWCAKKEBCNQJP-UHFFFAOYSA-N 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019792 magnesium silicate Nutrition 0.000 description 1
- 229910052919 magnesium silicate Inorganic materials 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 1
- 125000005397 methacrylic acid ester group Chemical group 0.000 description 1
- 125000005395 methacrylic acid group Chemical group 0.000 description 1
- DCUFMVPCXCSVNP-UHFFFAOYSA-N methacrylic anhydride Chemical compound CC(=C)C(=O)OC(=O)C(C)=C DCUFMVPCXCSVNP-UHFFFAOYSA-N 0.000 description 1
- NXMXPVQZFYYPGD-UHFFFAOYSA-N methyl 2-methylprop-2-enoate;methyl prop-2-enoate Chemical compound COC(=O)C=C.COC(=O)C(C)=C NXMXPVQZFYYPGD-UHFFFAOYSA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 210000000274 microglia Anatomy 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 235000013379 molasses Nutrition 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- 230000007659 motor function Effects 0.000 description 1
- 201000006938 muscular dystrophy Diseases 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 230000003959 neuroinflammation Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 208000002040 neurosyphilis Diseases 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- GQPLMRYTRLFLPF-UHFFFAOYSA-N nitrous oxide Inorganic materials [O-][N+]#N GQPLMRYTRLFLPF-UHFFFAOYSA-N 0.000 description 1
- 230000000422 nocturnal effect Effects 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Polymers 0.000 description 1
- 239000003605 opacifier Substances 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 229960001412 pentobarbital Drugs 0.000 description 1
- PNJWIWWMYCMZRO-UHFFFAOYSA-N pent‐4‐en‐2‐one Natural products CC(=O)CC=C PNJWIWWMYCMZRO-UHFFFAOYSA-N 0.000 description 1
- 210000000578 peripheral nerve Anatomy 0.000 description 1
- 208000027232 peripheral nervous system disease Diseases 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 238000009520 phase I clinical trial Methods 0.000 description 1
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920000151 polyglycol Polymers 0.000 description 1
- 239000010695 polyglycol Substances 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 150000004804 polysaccharides Chemical class 0.000 description 1
- 229920000915 polyvinyl chloride Polymers 0.000 description 1
- 239000004800 polyvinyl chloride Substances 0.000 description 1
- 208000000170 postencephalitic Parkinson disease Diseases 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- OTYBMLCTZGSZBG-UHFFFAOYSA-L potassium sulfate Chemical compound [K+].[K+].[O-]S([O-])(=O)=O OTYBMLCTZGSZBG-UHFFFAOYSA-L 0.000 description 1
- 229910052939 potassium sulfate Inorganic materials 0.000 description 1
- 235000011151 potassium sulphates Nutrition 0.000 description 1
- 229960001621 povidone-iodine Drugs 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000019423 pullulan Nutrition 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000028527 righting reflex Effects 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 231100000241 scar Toxicity 0.000 description 1
- 231100000161 signs of toxicity Toxicity 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 239000007974 sodium acetate buffer Substances 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- 235000021058 soft food Nutrition 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 230000001148 spastic effect Effects 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 210000004281 subthalamic nucleus Anatomy 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 208000002025 tabes dorsalis Diseases 0.000 description 1
- 239000008399 tap water Substances 0.000 description 1
- 235000020679 tap water Nutrition 0.000 description 1
- 229960005333 tetrabenazine Drugs 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 229960002622 triacetin Drugs 0.000 description 1
- 201000008827 tuberculosis Diseases 0.000 description 1
- 230000010330 visuo-spatial memory Effects 0.000 description 1
- 230000001755 vocal effect Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5073—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals having two or more different coatings optionally including drug-containing subcoatings
- A61K9/5078—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals having two or more different coatings optionally including drug-containing subcoatings with drug-free core
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5026—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5036—Polysaccharides, e.g. gums, alginate; Cyclodextrin
- A61K9/5042—Cellulose; Cellulose derivatives, e.g. phthalate or acetate succinate esters of hydroxypropyl methylcellulose
- A61K9/5047—Cellulose ethers containing no ester groups, e.g. hydroxypropyl methylcellulose
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Neurology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Una dosis que comprende (i) 220 mg a 600 mg de amantadina o sales farmacéuticamente aceptables de la misma y (ii) al menos un excipiente, para su uso en un método para mejorar la marcha en un sujeto humano con esclerosis múltiple, comprendiendo el método administrar por vía oral la dosis a dicho sujeto una vez al día.
Description
DESCRIPCIÓN
Amantadina para mejorar la marcha en esclerosis múltiple
REFERENCIA CRUZADA
[0001] Esta solicitud reivindica el beneficio de prioridad bajo 35 U.S.C. § 119(e) de la Solicitud Provisional de los Estados Unidos N° 62/075,137, presentada el 4 de noviembre de 2014.
ANTECEDENTES DE LA INVENCIÓN
[0002] La amantadina está indicada para varias condiciones que puede tratarse con antagonistas del receptor NMDA, incluido el tratamiento de la enfermedad de Parkinson idiopática (Parálisis Agitans), el parkinsonismo posencefalítico y el parkinsonismo sintomático que puede seguir a una lesión del sistema nervioso por intoxicación por monóxido de carbono. La amantadina también tiene actividad como inhibidor del canal M2 viral y se usa para la profilaxis y el tratamiento de la infección de enfermedades virales, especialmente el virus de la influenza A.
[0003] Las formas actualmente comercializadas de amantadina son formulaciones de liberación inmediata que se administran típicamente dos o más veces al día. El uso de amantadina está limitado por los efectos secundarios relacionados con la dosis en el SNC que incluyen mareos, confusión, alucinaciones, insomnio y pesadillas. Gracies JM, Olanow CW; Current and Experimental Therapeutics of Parkinson's Disease; Neuropsycho-pharmacology: the Fifth Generation of Progress, págs. 1802; American College of Neuropsycho-Pharmacology 2002), que puede exacerbarse particularmente cuando la amantadina se administra al final del día (Jackson et al., Bull. Pan Am. Health Org., 147, 595 603 (1967); Jackson, JAMA, 235 (25), (1976), pp 2739-2742;. y Hayden, AAC, 23 (3) 1983, pp 458-464
[0004] La amantadina de liberación inmediata ha demostrado una mejora en la velocidad de la marcha en pacientes con Parkinson Parkes et al., Lancet, 1, págs. 259-62 (1970). Los pacientes de Parkinson avanzado con estimulación STN fueron evaluados en un estudio de etiqueta abierta con amantadina de liberación inmediata que mostró mejoras en la marcha. Chan et al., Parkinsonism and Related Disorders, 19, págs. 316-9 (2013). El metilfenidato también mostró una mejora en la marcha en pacientes de Parkinson con trastornos graves de la marcha que estaban recibiendo estimulación STN. Moreau et al., Lancet Neurology 11, págs. 589-96 (2012). Otro estudio de la alteración de la marcha en la enfermedad de Parkinson concluyó que el metilfenidato no era eficaz. Espay et al., Neurology 76 , págs. 1256-1262 (2011). En la esclerosis múltiple, la fampridina (4-aminopiridina; dalfampridina) demostró una mejora en la capacidad para caminar en algunos pacientes. Goodman y col., Lancet, 373, págs. 732-8 (2009). Por el contrario, la fampridina no mostró una mejora en la capacidad de caminar de los pacientes con Parkinson (es decir, la velocidad y la longitud de la zancada) a las cuatro semanas (Luca et al., Neurology 84 (14) Supplement P.6.049 (2015)). Chan y col. describe el uso de amantadina en pacientes con enfermedad de Parkinson avanzada y a quienes se les ha administrado estimulación cerebral profunda del núcleo subtalámico (STN-DBS). El documento US 2011/189273 describe el uso de amantadina en, por ejemplo, la enfermedad de Parkinson y también la esclerosis múltiple.
[0005] Se sabe que la amantadina de liberación inmediata puede actuar como un estimulante, causando insomnio y trastornos del sueño. Por lo tanto, la última dosis de amantadina de liberación inmediata se administra típicamente a más tardar a las 16:00 para minimizar estos efectos secundarios. Tal dosificación de amantadina da como resultado concentraciones plasmáticas máximas de amantadina que se producen por la tarde o por la noche y concentraciones plasmáticas muy bajas por la mañana.
[0006] Formas de liberación prolongada de la amantadina se han descrito en la técnica. La patente de EE. UU. N° 5.358.721 de Guittard et al., La patente de EE. UU. N° 6.217.905 de Edgren et al. y la patente de EE. UU. N° 8.574.626 de Vergez et al., describen cada una una forma de dosificación osmótica oral que comprende un fármaco antivírico o antiparkinsoniano, respectivamente, donde en cada caso la amantadina se enumera como un posible fármaco a utilizar en la forma de dosificación. La patente de EE. UU. N° 6.194.000, de Smith et al., describe composiciones farmacéuticas analgésicas de liberación inmediata y controlada que utilizan antagonistas del receptor de NMDA, tales como amantadina, como agente activo. Cada una de las solicitudes de patente de EE. UU. Los números de publicación US 2006/0252788, US 2006/0189694, US 2006/0142398, US 2008/0227743 y US 2011/0189273 (cada uno de Went et al.) describen la administración de un antagonista del receptor de NMDA, tal como amantadina, opcionalmente en forma de liberación controlada.
[0007] Hay ocho marchas patológicas básicas que se pueden atribuir a las condiciones neurológicas: hemipléjica, espástica dipléjica, neuropática, miopática, parkinsoniana, coreiforme, atáxica (cerebelo) y sensorial. La marcha hemipléjica se observa con mayor frecuencia en los accidentes cerebrovasculares. La marcha dipéjica se observa en lesiones periventriculares bilaterales, como las que se observan en la parálisis cerebral. La marcha neuropática, si es bilateral, se observa en la esclerosis lateral amiotrófica, la enfermedad de Charcot-Marie-Tooth y otras neuropatías periféricas, incluidas las asociadas con la diabetes no controlada. La marcha miopática se observa en pacientes con miopatías, como distrofia muscular. La marcha parkinsoniana se observa en la enfermedad de Parkinson o en cualquier otra afección que cause parkinsonismo, como los efectos secundarios de los medicamentos. La marcha coreiforme se observa con ciertos trastornos de los ganglios basales como la corea de Sydenham, la enfermedad de Huntington y otras
formas de corea, atetosis o distoma. La marcha atáxica se observa con mayor frecuencia en la enfermedad cerebelosa. Los pacientes con esclerosis múltiple con frecuencia expresan síntomas atáxicos, que incluyen caminar descoordinado, control reducido del rango de movimiento, incapacidad para mantener una postura estable, incapacidad para mantener un ritmo constante o pérdida del equilibrio. La marcha sensorial se puede observar en trastornos de las columnas dorsales (p. ej., deficiencia de B 12 o tabes dorsalis) o en enfermedades que afectan a los nervios periféricos, como la diabetes no controlada.
Resumen de la invención
[0008] La presente invención es una dosis que comprende (i) 220 mg a 600 mg de amantadina o sales farmacéuticamente aceptables de la misma y (ii) al menos un excipiente, para uso en un método de mejorar la marcha en un sujeto humano con esclerosis múltiple, comprendiendo el método administrar oralmente la dosis a dicho sujeto una vez al día. Los inventores han descubierto sorprendentemente que la amantadina es útil en el tratamiento de ciertos trastornos hipocinéticos, incluida la alteración de la marcha o la alteración de la marcha en pacientes con esclerosis múltiple o pacientes que han experimentado un accidente cerebrovascular. En algunas formas de realización, las formas de liberación inmediata de amantadina o una sal farmacéuticamente aceptable de la misma o las formas de liberación controlada de amantadina o una sal farmacéuticamente aceptable de la misma son útiles en dichos métodos. En algunas formas de realización, se prefieren las formas de liberación controlada. En una realización, los métodos incluyen la administración de formas de liberación inmediata de amantadina o sales farmacéuticamente aceptables (que incluyen, por ejemplo, clorhidrato de amantadina o sulfato de amantadina) una, dos o tres veces al día. En una realización, los métodos incluyen la administración de composiciones de liberación prolongada de amantadina o sales farmacéuticamente aceptables de la misma una vez al día por la mañana (p. ej., después de que el paciente se despierta y no más tarde de las 24:00) donde la composición de liberación prolongada proporciona una dosis única de Tmax de 5 a 11 horas. En algunas formas de realización, los métodos incluyen la administración de composiciones de liberación prolongada de amantadina o sales farmacéuticamente aceptables de la misma una vez al día por la noche (p. ej., de 0 a 4 horas antes de acostarse) donde la composición de liberación prolongada proporciona una dosis única Tmax de 9 a 19 horas. En algunas formas de realización, se ha encontrado que la invención facilita la administración una vez al día de una dosis relativamente alta de amantadina (o una sal farmacéuticamente aceptable de la misma), mientras que sorprendentemente reduce los efectos secundarios tales como alteraciones del sueño.
[0009] Algunas formas de realización descritas en este documento proporcionan una dosis de la invención para uso en un método de mejorar la marcha (por ejemplo, el aumento de la velocidad de marcha), o el tratamiento de deterioro de la marcha en un paciente (por ejemplo, un paciente con EM o un paciente que ha experimentado un derrame). En algunas formas de realización de la divulgación, el método incluye administrar al paciente de aproximadamente 50 mg a aproximadamente 600 mg por día de un fármaco seleccionado del grupo que consiste en amantadina o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización de la divulgación, el fármaco se administra dos veces al día o tres veces al día. En la invención, el fármaco se administra una vez al día. En algunas formas de realización, el fármaco está en una composición farmacéutica que comprende el fármaco y al menos un excipiente, como un excipiente de liberación sostenida. En algunas formas de realización, el fármaco se administra en una, dos, tres o cuatro formas de dosificación que consisten en el fármaco y al menos un excipiente. En algunas formas de realización, las formas de dosificación una, dos, tres o cuatro son formas de dosificación oral. En algunas formas de realización, las formas de dosificación oral son comprimidos, comprimidos oblongos, cápsulas de gel u otras formas de dosificación unitarias orales adecuadas. En algunas formas de realización, el paciente tiene una función renal normal y el fármaco se administra en una dosis inicial de aproximadamente 100 a aproximadamente 220 mg por día, aproximadamente 120 a aproximadamente 200 mg por día, aproximadamente 140 a aproximadamente 180 mg por día o aproximadamente 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210 o 220 mg por día. En algunas formas de realización, el paciente tiene una función renal normal y el fármaco se administra, después de un período inicial de administración de amantadina, en una dosis de aproximadamente 200 a aproximadamente 440 mg por día, aproximadamente 240 a aproximadamente 400 mg por día, aproximadamente 280 a aproximadamente 360 mg por día, o aproximadamente 160, 170, 180, 200, 220, 240, 260, 280, 300, 320, 340, 360, 380, 400, 420 o 440 mg por día. En algunas formas de realización, el paciente tiene insuficiencia renal y el fármaco se administra en una dosis inicial de aproximadamente 50 a aproximadamente 110 mg por día, aproximadamente 60 a aproximadamente 100 mg por día, aproximadamente 70 a aproximadamente 90 mg por día, o aproximadamente 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105 o 110 mg por día. En algunas de estas realizaciones, el paciente con EM tiene insuficiencia renal y, después de un período inicial de administración de amantadina, la dosis se incrementa a aproximadamente 100 a aproximadamente 220 mg por día, aproximadamente 120 a aproximadamente 200 mg por día, aproximadamente 140 a aproximadamente 180 mg por día, o aproximadamente 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210 o 220 mg por día. En algunas formas de realización, el fármaco se administra en una forma de liberación prolongada que comprende el fármaco y al menos un excipiente de liberación prolongada. En algunas formas de realización, el fármaco se administra por la mañana, por ejemplo, después de despertarse y no más tarde de las 24:00 o entre las 5:00 y las 24:00, 6:00 y 24:00, 7:00 y 24:00, 8:00 y 24:00, y tiene una mediana de Tmax de 5 a 8 horas, 5 a 9 horas, 5 a 10 horas, 5 a 11 horas, 6 a 9 horas, 6 a 10 horas o 6 a 11 horas. En algunas formas de realización, el fármaco se administra antes de acostarse (por ejemplo, 0, 1, 2, 3 o 4 horas antes de acostarse) y tiene un Tmax de 8 a 18 horas, 8 a 20 horas, 10 a 18 horas, 10 a 20 horas, 12 a 18 horas, 12 a 20 horas. Las referencias antes mencionadas a Tmax son valores medianos de Tmax determinados a partir de un estudio farmacocinético humano en ayunas de dosis única. En algunas formas de realización, el fármaco se proporciona en forma de liberación inmediata y se administra dos, tres o cuatro veces al día.
[0010] En algunas formas de realización de la divulgación, se proporciona un método de mejorar la marcha en un sujeto humano, comprendiendo dicho método la administración oral a dicho paciente una vez al día o de noche una dosis de (i) 50 mg a 600 mg de un fármaco seleccionado del grupo que consiste en amantadina y sus sales farmacéuticamente aceptables y (ii) al menos un excipiente, (por ejemplo, en el que la dosis incluye una composición que comprende los componentes (i) y (ii)). En la invención, el sujeto humano es un paciente con EM. En algunas formas de realización, la dosis incluye una composición que comprende (i) 100 mg a 450 mg de amantadina, o una sal farmacéuticamente aceptable de la misma y (ii) al menos un excipiente. En algunas formas de realización, la composición puede comprender aproximadamente 130-210 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En varias realizaciones específicas, una forma de dosificación que contiene la composición comprende (i) 50 a 75 mg, 70 a 95 mg, 90 a 115 mg, 110 a 135 mg, 130 a 155 mg, 150 a 175 mg, 170 a 195 mg, 190 a 215 mg, 210 a 235 mg, 230 a 255 mg, 250 a 275 mg, 270 a 295 mg, 290 a 305 mg, 300 a 315 mg, 310 a 325 mg, 320 a 335 mg, 330 a 345 mg, 340 a 355 mg, a 350 a 365 mg de 360 a 375 mg, 370 a 385 mg 380 a 395 mg, 390 a 405 mg, 400 a 415 mg, 410 a 425 mg, 420 a 435 mg, 430 a 445 mg o 440 a 455 mg de amantadina, o una sal farmacéuticamente aceptable de la misma, y (ii) al menos un excipiente. En algunas formas de realización, la composición comprende aproximadamente 110, 120, 130, 140, 150, 160, 170, 180, 190, 210, 220 o 340 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende 110 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 130 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 170 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 210 mg de hidrocloruro de amantadina. En la invención, el sujeto humano tiene esclerosis múltiple. En algunas formas de realización, la dosis comprende adicionalmente uno o más fármacos seleccionados del grupo que consiste en 4-aminopiridina, baclofeno, dextrometorfano, dimetilfumarato, fingolimod, metilfenidato y teriflunomida, tetrabenizina y tizanidina. En algunas formas de realización, al menos un excipiente es un excipiente que modifica la liberación. En algunas formas de realización, al menos uno de dichos excipientes modifica la liberación de la amantadina o una sal farmacéuticamente aceptable de la misma para proporcionar una forma de liberación prolongada, y en la que la administración de la dosis proporciona a) una Tmax para amantadina de 5 a 18 horas, y/o b) una Cmax de 1,0 a 2,8 ng/ml por mg de amantadina, y/o c) un AUCü.inf de 40 a 75 ng*h/ml por mg de amantadina, según se determina a partir de un estudio farmacocinético humano de dosis única. En algunas formas de realización, al menos uno de dichos excipientes modifica la liberación de la amantadina o una sal farmacéuticamente aceptable de la misma para proporcionar una forma de liberación prolongada, y en la que la administración de la dosis proporciona a) una Tmax para amantadina de 5 a 18 horas, y/o b) una Cmax de 1,0 a 2,8 ng/ml por mg de amantadina, y/o c) un AUCü.inf de 40 a 75 ng*h/ml por mg de amantadina, según se determina a partir de un estudio farmacocinético humano de dosis única. En algunas formas de realización, tal dosificación de amantadina da como resultado concentraciones plasmáticas máximas de amantadina que se producen en la mañana o al principio de la tarde, y concentraciones plasmáticas muy bajas en la noche. En algunas formas de realización, el fármaco se proporciona en forma de liberación prolongada. En realizaciones específicas adicionales, la concentración plasmática máxima de amantadina se logra entre 5, 6 , 7, 8 , 9, 10, 11 o 12 horas hasta aproximadamente 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,22, 23 o 24 horas después de la administración de una dosis única de la composición (por ejemplo, de 8 horas a aproximadamente 18 horas después de la administración). En algunas formas de realización, la composición de amantadina se administra a un paciente de 0 a 4 horas antes de acostarse. En algunas formas de realización, la composición de amantadina se administra a un paciente de 0 a 3, de 0 a 2 o de 0 a 1 horas antes de acostarse. En algunas formas de realización para la administración oral una vez al día de 0 a 4 horas antes de acostarse, la mediana de Tmax para dicha composición es de 9 a 18 horas. En algunas formas de realización para la administración oral una vez al día después de despertar y antes de las 24:00 o entre las 6:00 y las 24:00, la mediana de Tmax para dicha composición es de 5 a 9 horas. En algunas formas de realización, la velocidad o capacidad de caminar se evalúa mediante al menos uno de los siguientes o una combinación de los mismos: prueba de caminata cronometrada de 25 pies (T25FW), prueba de caminata cronometrada (TUG), prueba de caminata de 2 minutos, prueba de caminata cronometrada de seis minutos (6MTW) y/o la Escala para caminar de esclerosis múltiple de doce elementos (MSWS-12) (referencias proporcionadas en el Ejemplo 10). En algunas formas de realización, se mejora significativamente la marcha del sujeto. En algunas formas de realización, se mejoran los síntomas de la EM. En algunas formas de realización, T25FW, TUG, caminata de 2 minutos, 6MTW y/o MSWS-12 se mejoran significativamente (en relación con el placebo). En algunas formas de realización, la administración de la composición a un sujeto con enfermedad de EM da como resultado una reducción significativa en la alteración de la marcha por enfermedad de EM. En una realización específica, la administración de la composición da como resultado aproximadamente un 5%, 10%, 15%, 20%, 25%, 30%, 35% o 40%, 45%, 50%, 55% o 60% de reducción. en la enfermedad de EM, discapacidad para caminar. En una realización específica, la administración de la composición da como resultado al menos 5%, 10%, 15%, 20%, 25%, 30%, 35% o 40%, 45%, 50%, 55% o 60% reducción de la discapacidad para caminar por la enfermedad de EM. En una realización específica, la administración de la composición da como resultado una mejora de aproximadamente el 5%, 10%, 15%, 20%, 25%, 30%, 35% o 40%, 45%, 50%, 55% o 60% en una o más de las puntuaciones de evaluación mencionadas anteriormente. En algunas formas de realización, la reducción de la alteración de la marcha se mide en una escala numérica que es utilizada o aceptada por la FDA u otras agencias reguladoras para evaluar la eficacia y aprobar medicamentos con licencia para el tratamiento de la alteración de la marcha. En algunas formas de realización, la composición de una vez al día o una vez por la noche se administra como una, dos, tres o cuatro formas de dosificación unitarias en unidades divididas de manera desigual o, preferiblemente, igualmente. En algunas formas de realización más específicas, la composición se administra como una, dos o tres formas de dosificación unitarias que comprenden cada una de 75 a 200 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos formas de dosificación unitarias que comprenden
cada una de 100 a 175 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos o tres formas de dosificación unitaria de dosificación desigual, o preferiblemente igual, comprendiendo cada una de 85 a 250 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos formas de dosificación unitarias con dosis iguales (por ejemplo, cada una comprende 130-210 mg de amantadina o una sal farmacéuticamente aceptable de la misma). En algunas formas de realización más específicas, la composición se administra como dos formas de dosificación unitarias, cada una de las cuales comprende de 150 a 180 mg de amantadina, o una sal farmacéuticamente aceptable de la misma.
[0011] Algunas formas de realización de la descripción proporcionan un método para mejorar la marcha en un sujeto humano, comprendiendo dicho método administrar oralmente a dicho paciente una vez al día o una vez cada noche, que comprende una dosis (i) 50 mg a 600 mg de un fármaco seleccionado del grupo que consiste en amantadina y sus sales farmacéuticamente aceptables y (ii) al menos un excipiente (por ejemplo, en el que la dosis incluye una composición que comprende los componentes (i) y (ii)). En la invención, el sujeto humano tiene Em . En algunas formas de realización, la composición puede comprender de 100 mg a 450 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición puede comprender 130-210 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, una forma de dosificación que contiene la composición comprende 50 a 75 mg, 70 a 95 mg, 90 a 115 mg, 110 a 135 mg, 130 a 155 mg, 150 a 175 mg, 170 a 195 mg. mg, 190 a 215 mg, 210 a 235 mg, 230 a 255 mg, 250 a 275 mg, 270 a 295 mg, 290 a 305 mg, 300 a 315 mg, 310 a 325 mg, 320 a 335 mg, 330 a 345 mg, 340 a 355 mg, 350 a 365 mg, 360 a 375 mg, 370 a 385 mg, 380 a 395 mg, 390 a 405 mg, 400 a 415 mg, 410 a 425 mg, 420 a 435 mg, 430 a 445 mg o 440 a 455 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende aproximadamente 110, 120, 130, 140, 150, 160, 170, 180, 190, 210, 220 o 340 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende 110 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 130 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 170 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 210 mg de hidrocloruro de amantadina. En algunas formas de realización, el sujeto humano tiene esclerosis múltiple. En algunas formas de realización, la dosis comprende además uno o más fármacos seleccionados del grupo que consiste en 4-aminopiridina, baclofeno, dextrometorfano, dimetilfumarato, fingolimod, metilfenidato y teriflunomida, tetrabenizina y tizanidina. En algunas formas de realización, el al menos un excipiente es un excipiente que modifica la liberación. En algunas formas de realización, al menos uno de dichos excipientes modifica la liberación de la amantadina o una sal farmacéuticamente aceptable de la misma para proporcionar una forma de liberación prolongada, y en la que la administración de la dosis proporciona a) una Tmax para la amantadina de 5 a 18 horas, b) una Cmax de 1,0 a 2,8 ng/ml por mg de amantadina, y c) un AUCü-inf de 40 a 75 ng*h/ml por mg de amantadina, determinado a partir de un estudio farmacocinético en humanos de dosis única. En algunas formas de realización, tal dosificación de amantadina da como resultado concentraciones plasmáticas máximas de amantadina que se producen por la mañana o por la tarde, y concentraciones plasmáticas muy bajas durante la noche, es decir, mientras el paciente duerme. En algunas formas de realización, el fármaco se proporciona en forma de liberación prolongada. En realizaciones específicas adicionales, la concentración plasmática máxima de amantadina se logra entre 5, 6 , 7, 8 , 9, 10, 11 o 12 horas hasta aproximadamente 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,22, 23 o 24 horas después de la administración de una dosis única de la composición (por ejemplo, de 8 horas a aproximadamente 18 horas después de la administración). En algunas formas de realización descritas en el presente documento, la composición de amantadina se administra a un paciente de 0 a 4 horas antes de acostarse. En algunas formas de realización, la composición de amantadina se administra a un paciente de 0 a 3, de 0 a 2 o de 0 a 1 horas antes de acostarse. En algunas formas de realización de cualquiera de los aspectos anteriores, la administración de la composición a un sujeto con enfermedad de EM da como resultado una reducción significativa en el deterioro de la marcha por enfermedad de EM. En una realización específica, la administración de la composición da como resultado al menos aproximadamente 5%, 10%, 15%, 20%, 25%, 30%, 35% o 40%, 45%, 50%, 55% o 60% de reducción en la alteración de la marcha por enfermedad de EM. En algunas formas de realización, la marcha (o la alteración de la marcha) se mide en una escala numérica que es utilizada o aceptada por la FDA u otras agencias reguladoras para evaluar la eficacia y aprobar los fármacos autorizados para el tratamiento de la marcha. En algunas formas de realización, la administración una vez al día de la composición de amantadina (o combinación que comprende amantadina) proporciona una mejora en un trastorno del movimiento hipocinético que afecta la marcha. La mejora puede determinarse mediante métodos conocidos en la técnica, tales como prueba de caminata cronometrada (TUG), prueba de caminata cronometrada de 25 pies (T25FW) o prueba de caminata cronometrada de seis minutos (6MTW). En algunas formas de realización, la composición de una vez al día o una vez por la noche se administra como una, dos, tres o cuatro formas de dosificación unitarias en unidades divididas de manera desigual o, preferiblemente, igualmente. En algunas formas de realización más específicas, la composición se administra como una forma de dosificación unitaria que comprende de 75 a 350 mg de amantadina o una sal farmacéuticamente aceptable de la misma, dos o tres formas de dosificación unitaria que comprenden cada una de 85 a 175 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos formas de dosificación unitarias que comprenden cada una de 100 a 175 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos o tres formas de dosificación unitaria de dosificación desigual, o preferiblemente igual, comprendiendo cada una de 85 a 250 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización más específicas, la composición se administra como dos formas de dosificación unitarias, cada una de las cuales comprende de 150 a 180 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición
se administra por vía oral, una vez al día.
[0012] La descripción proporciona métodos para mejorar los trastornos del movimiento hipocinéticos, generalmente caracterizados por el movimiento corporal disminuido o rigidez muscular, tal como caminar, y asuntos asociados con esclerosis múltiple y caminar déficits después del accidente cerebrovascular. La divulgación proporciona métodos para mejorar la marcha en sujetos humanos con trastornos del movimiento hipocinético asociados con afecciones tales como apoplejía. Estas condiciones hipocinéticas pueden afectar de manera sustancialmente adversa la deambulación de los sujetos, aumentando la discapacidad de la condición subyacente.
[0013] La descripción proporciona métodos para mejorar los trastornos del movimiento hipercinéticos general se caracterizan por movimientos anormalmente aumentados, a veces incontrolables, tales como la corea de Huntington, discinesia tardía, y síndrome de Tourette.
[0014] Por lo tanto, existe una necesidad en la técnica de métodos mejorados de la terapia de amantadina para el tratamiento de fluctuaciones motores o deficiencias motoras en EM y pacientes con accidente cerebrovascular, que se puede administrar a un paciente poco antes de que desean dormir (por ejemplo, en hora de acostarse) sin causar insomnio o alteraciones del sueño. Además, existe la necesidad de una terapia con amantadina que pueda tomar el paciente antes de irse a dormir y luego proporcione una concentración plasmática adecuada de amantadina cuando se despierte, por ejemplo, por la mañana, después de una noche de sueño completo. Existe una necesidad en la técnica de métodos mejorados de la terapia de amantadina para el tratamiento de fluctuaciones motoras o deficiencias motoras en EM y derrames, que se pueden administrar a un paciente una vez al día, al despertar por la mañana (por ejemplo, antes de mediodía o entre el horas de 6:00 y 24:00) para brindar dicho tratamiento sin causar insomnio o alteración del sueño.
[0015] Cada uno de los trastornos, condiciones y enfermedades anteriormente mencionados resultan en la debilitación del paciente. Por tanto, existe una necesidad en la técnica de métodos y composiciones para el tratamiento de trastornos de la marcha y/o trastornos del movimiento hipocinético y trastornos hipercinéticos.
[0016] En algunos aspectos de la invención, la dosis de la invención es para uso en un método de administración de amantadina a un paciente en necesidad de la misma, comprendiendo dicho método la administración por vía oral de ciertas composiciones de liberación prolongada (LP) que comprenden amantadina, o una sal farmacéuticamente aceptable de la misma, menos de tres horas antes de acostarse (es decir, la hora a la que el sujeto desea irse a dormir por la noche). Este aspecto también incluye el uso de tales composiciones y el uso de amantadina, o una sal farmacéuticamente aceptable de la misma, para la fabricación de un medicamento como se describe a continuación. Alternativamente, la composición se administra menos de aproximadamente 4 horas antes de acostarse. En algunos aspectos, la administración ocurre menos de cuatro, menos de tres y medio, menos de tres, menos de dos y medio, menos de dos, menos de una hora y media, menos de una o menos de media hora antes de acostarse.
[0017] En algunos aspectos de la invención, la dosis de la invención es para uso en un método de administración de amantadina a un paciente en necesidad del mismo, comprendiendo dicho método la administración por vía oral de ciertas composiciones LP que comprenden amantadina,o una sal farmacéuticamente aceptable de la misma, una vez al día por la mañana después de que el paciente se haya despertado, preferiblemente antes del mediodía (por ejemplo, entre las 6:00 y 24:00, 7:00 y 24:00, 8:00 y 24:00). Este aspecto también incluye el uso de tales composiciones y el uso de amantadina, o una sal farmacéuticamente aceptable de la misma, en la fabricación de un medicamento como se describe a continuación.
[0018] En algunos aspectos, la invención proporciona una dosis de la invención para uso en un método para reducir las alteraciones del sueño en un sujeto humano sometido a tratamiento con amantadina, comprendiendo dicho método la administración de una composición de liberación prolongada (LP) que comprende amantadina, o una sal farmacéuticamente aceptable de la misma, menos de aproximadamente tres horas antes de acostarse (es decir, la hora a la que el sujeto desea irse a dormir por la noche). Este aspecto también incluye el uso de tales composiciones y el uso de amantadina para la fabricación de un medicamento como se describe a continuación. Alternativamente, la composición se administra menos de aproximadamente 4 horas antes de acostarse.
[0019] En algunos aspectos, la invención proporciona una dosis de la invención para uso en un método para reducir las alteraciones del sueño en un sujeto humano sometido a tratamiento con amantadina, comprendiendo dicho método la administración de una composición de liberación prolongada (LP) que comprende amantadina, o una sal farmacéuticamente aceptable de la misma, una vez al día por la mañana después de que el sujeto se ha despertado. Este aspecto también incluye el uso de tales composiciones y el uso de amantadina para la fabricación de un medicamento como se describe a continuación.
[0020] La dosis de la invención comprende de 220 mg a 600 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En algunos aspectos de la invención, la amantadina o una sal farmacéuticamente aceptable de la misma (como el hidrocloruro) se administra en una cantidad reducida, es decir, de 85 a 260 mg por día, durante al menos una semana antes de la administración una vez al día de la dosis de mantenimiento. Este período de titulación puede mejorar la tolerabilidad de la dosis de mantenimiento. En un aspecto de la invención, a los pacientes se les administran 85 o 170 mg por día durante al menos una semana antes de aumentar la dosis a 170 o 340 mg por día.
[0021] Además, muchos pacientes en necesidad de la terapia de amantadina tienen dificultad para tragar. Por tanto, existe la necesidad de una terapia con amantadina que suministre una dosis terapéuticamente eficaz del fármaco en una forma de dosificación oral que sea de tamaño pequeño y no aumente indebidamente la carga de píldoras. Preferiblemente, dicha forma de dosificación se administra una vez al día. Preferiblemente, dicha forma de dosificación se puede rociar y consumir con alimentos, por ejemplo, puré de manzana.
[0022] La dosis de la invención es para uso en un método para mejorar la marcha en un sujeto humano con la esclerosis múltiple. En algunos aspectos de la divulgación, se proporciona un método para tratar lesiones cerebrales, traumatismos cerebrales, accidentes cerebrovasculares, enfermedad de Huntington, ELA, enfermedades neurodegenerativas, afecciones cerebrovasculares, trastornos del movimiento, trastornos de los nervios craneales, comprendiendo dicho método la administración de ciertas composiciones de liberación prolongada (LP) que comprende amantadina, o una sal farmacéuticamente aceptable de la misma, menos de aproximadamente tres horas antes de acostarse (es decir, la hora a la que el sujeto desea irse a dormir por la noche). Este aspecto también incluye el uso de tales composiciones y el uso de amantadina para la fabricación de un medicamento como se describe a continuación.
[0023] En algunas formas de realización de cualquiera de los aspectos anteriores, el paciente tiene esclerosis múltiple (por ejemplo, ha sido diagnosticado con esclerosis múltiple). En algunas formas de realización de la divulgación, el paciente ha sufrido un derrame cerebral.
[0024] En algunas formas de realización de cualquiera de los aspectos anteriores, la composición se administra una vez cada noche. En otro aspecto, la dosis diaria supera los 200 mg y, preferiblemente, es de 240 a 420 mg, más preferiblemente de 260 a 340 mg (incluso más preferiblemente, 340 mg). En algunas formas de realización, la dosis diaria de 260 a 340 mg se administra en 1,2 o 3 cápsulas de tamaño 0, 1 o 2, en formatos normal y/o EL.
[0025] En algunas formas de realización de cualquiera de los aspectos anteriores, la administración de la composición a un paciente da como resultado una mejora significativa en la Impresión Clínico Global (CGI) o cualquier otra medición médica de la condición general del paciente. En una realización específica, la administración de la composición da como resultado aproximadamente un 5%, 10%, 15%, 20%, 25%, 30%, 35% o 40% de mejora en la CGI. En realizaciones específicas adicionales, la mejora en CGI se mide en una escala numérica que utiliza la FDA para evaluar la eficacia de los fármacos indicados para tratar trastornos del SNC.
[0026] En algunas formas de realización de cualquiera de los aspectos anteriores, no hay un aumento en la concentración plasmática en estado estacionario de la amantadina para al menos una hora después de la administración de la dosis.
[0027] En algunas formas de realización de cualquiera de los aspectos anteriores, no hay un aumento en la concentración plasmática en estado estacionario de la amantadina durante al menos dos horas después de la administración de la dosis.
[0028] En algunas formas de realización de cualquiera de los aspectos anteriores, la administración de la composición a un sujeto humano a concentraciones plasmáticas de amantadina de estado estacionario aumenta la concentración plasmática de amantadina por menos de 5%, 10%, 15%, 20% o 25% a 1,2, 2,5 o 3 horas después de dicha administración. Por ejemplo, la administración de la composición a un sujeto humano a concentraciones plasmáticas de amantadina en estado estacionario aumenta la concentración plasmática de amantadina en menos del 5% 1, 2, 2,5 o 3 horas después de dicha administración; o en menos del 10% 1,2, 2,5 o 3 horas después de dicha administración; o en menos del 15% a 1, 2, 2,5 o 3 horas después de dicha administración; o en menos del 20% a 1, 2, 2,5 o 3 horas después de dicha administración; o en menos del 25% a 1, 2, 2,5 o 3 horas después de dicha administración.
[0029] En una realización de cualquiera de los aspectos anteriores, la concentración plasmática pico de la amantadina se consigue entre 5 y 16 horas después de la administración de una dosis única de la composición. En una realización más específica, la concentración plasmática máxima de amantadina se alcanza de 8 a 14 horas después de la administración de una dosis única de la composición. En otra realización más específica, la concentración plasmática máxima de amantadina se alcanza de 10 a 12 horas después de la administración de una dosis única de la composición. En realizaciones específicas adicionales, la concentración plasmática máxima de amantadina se logra entre 5, 6 , 7, 8 , 9, 10, 11 o 12 horas hasta aproximadamente 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,22, 23 o 24 horas después de la administración de una dosis única de la composición (por ejemplo, de 8 horas a aproximadamente 18 horas después de la administración).
[0030] En algunas formas de realización de cualquiera de los aspectos anteriores, la amantadina tiene una sola dosis Tmax de 9 a 18 horas. En realizaciones más específicas, la amantadina tiene una Tmax de dosis única de 12 a 18 horas después de la administración.
[0031] En algunas formas de realización de cualquiera de los aspectos anteriores de la amantadina tiene un Tmax de estado estable de 7 a 13 horas. En realizaciones más específicas, la amantadina tiene un Tmax en estado estacionario de 8 a 12 horas después de la administración.
[0032] En algunas formas de realización de cualquiera de los aspectos anteriores, una administración oral de una vez
todas las noches de la composición a un sujeto humano proporciona un perfil de concentración plasmática en estado estacionario que se caracteriza por un aumento de la concentración de la amantadina de menos de 25% en tres horas después de la administración. En realizaciones más específicas, el perfil de concentración plasmática en estado estacionario se caracteriza por un aumento de la concentración de amantadina de menos del 25% cuatro horas después de la administración.
[0033] En algunas formas de realización de cualquiera de los aspectos anteriores, la composición se administra una vez al día y la relación de Cmax a Cmin en estado estacionario es 1,3 a 3,0, 1,3 a 2,6, 1,03 a 2,04, 1,03 a 2,02, 1,4 a 3,0, 1,4 a 2,6, 1,4 a 2,4, 1,4 a 2,2, 1,5 a 3,0, 1,5 a 2,6, 1,5 a 2,4 o 1,5 a 2,2.
[0034] En realizaciones de cualquiera de los aspectos anteriores, el perfil de concentración plasmática en estado estacionario después de múltiples administraciones a un sujeto humano de la composición una vez al día se caracteriza por una concentración media en plasma durante el día ("C-media-día", definida como la concentración plasmática media de amantadina durante el día medida en un estudio de FC humana) que es de 1,4 a 1,7 veces la concentración plasmática media durante la noche ("C-media-noche", definida como la concentración plasmática media de amantadina durante la noche medida en un estudio de PK en humanos). En realizaciones más específicas, el C-media-día es la concentración plasmática promedio de amantadina medida entre las horas de 5:00, 6:00, 7:00, 8:00 o 9:00 hasta las horas 16:00, 17:00, 18:00, 19:00 o 20:00; por ejemplo, entre las 6:00 y las 16:00, entre las 7:00 y las 18:00, o entre las 7:00 y las 17:00. La C-media-noche es la concentración plasmática promedio de amantadina medida entre las 16:00, 17:00, 18:00, 19:00, 20:00, 21:00, 22:00 o 23:00 hasta las 5:00, 6:00, 7:00, 8:00 o 9:00; por ejemplo, entre las 22:00 y las 6:00, entre las 19:00 y las 6 :00, o entre las 20:00 y las 6 :00.
[0035] En algunas formas de realización de cualquiera de los aspectos anteriores, la amantadina se administra como una sal farmacéuticamente aceptable. En una realización más específica, la amantadina se administra como clorhidrato de amantadina o sulfato de amantadina. En algunas formas de realización de cualquiera de los aspectos anteriores, la dosis de amantadina una vez al día o una vez a la noche, o una sal farmacéuticamente aceptable de la misma, puede estar en el intervalo de 260 a 420 mg. En algunas formas de realización de la divulgación, la dosis de amantadina una vez al día o una vez a la noche, o una sal farmacéuticamente aceptable de la misma, puede estar en el intervalo de 50 a 600 mg. En otras realizaciones, la dosis de amantadina una vez al día o una vez a la noche, o una sal farmacéuticamente aceptable de la misma, excede los 300 mg por día, por ejemplo, está entre 320 y 360 mg por día, más específicamente está entre 330 y 350 mg por día. La dosis de la invención comprende de 220 mg a 600 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En varias realizaciones específicas, la dosis diaria de amantadina o una sal farmacéuticamente aceptable de la misma puede ser de 230 a 255 mg, 250 a 275 mg, 270 a 295 mg, 290 a 305, 300 a 315 mg, 310 a 325 mg, 320 a 335 mg., 330 a 345 mg, 340 a 355 mg o 350 a 365 mg. También se describen dosis diarias de 50 a 75 mg, 70 a 95 mg, 90 a 115 mg, 110 a 135 mg, 130 a 155 mg, 150 a 175 mg, 170 a 195 mg, 190 a 215 mg y 210 a 235 mg. En algunas formas de realización particularmente preferidas, la dosis de amantadina una vez al día o una vez a la noche, o una sal farmacéuticamente aceptable de la misma, es de 340 mg. En algunas formas de realización, la dosis de amantadina una vez al día o una vez a la noche, o una sal farmacéuticamente aceptable de la misma, es de 360 a 375 mg, 370 a 385 mg, 380 a 395 mg, 390 a 405 mg, 400 a 415 mg, 410 a 425 mg., 420 a 435 mg, 430 a 445 mg o 440 a 455 mg.
[0036] En algunas formas de realización de cualquiera de los aspectos anteriores, la composición comprende de 50 mg a 600 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición puede comprender de 100 mg a 450 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición puede comprender 130 - 210 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En varias realizaciones específicas, una forma de dosificación que contiene la composición comprende 50 a 75 mg, 70 a 95 mg, 90 a 115 mg, 110 a 135 mg, 130 a 155 mg, 150 a 175 mg, 170 a 195 mg, 190 a 215 mg, 210 a 235 mg, 230 a 255 mg, 250 a 275 mg, 270 a 295 mg, 290 a 305 mg, 300 a 315 mg, 310 a 325 mg, 320 a 335 mg, 330 a 345 mg, 340 a 355 mg, 350 a 365 mg, 360 a 375 mg, 370 a 385 mg, 380 a 395 mg, 390 a 405 mg, 400 a 415 mg, 410 a 425 mg, 420 a 435 mg, 430 a 445 mg o 440 a 455 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende aproximadamente 110, 120, 130, 140, 150, 160, 170, 180, 190, 210 o 220 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende 110 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 130 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 170 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 210 mg de hidrocloruro de amantadina.
[0037] En algunas formas de realización de cualquiera de los aspectos anteriores, una vez al día o una vez cada noche composición se administra como una, dos, tres o cuatro formas de dosificación unitaria en forma desigual o, preferiblemente, unidades igualmente divididas. En algunas formas de realización más específicas, la composición se administra como dos o tres formas de dosificación unitarias que comprenden cada una de 85 a 175 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos formas de dosificación unitarias que comprenden cada una de 100 a 175 mg de amantadina, o una sal farmacéuticamente aceptable de la misma.
[0038] En algunas formas de realización de cualquiera de los aspectos anteriores, la composición se administra como dos
o tres de dosificación unitaria formas de dosificación desigual, o preferiblemente igual, que comprenden cada uno 85 hasta 250 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización más específicas, la composición se administra como dos formas de dosificación unitarias, cada una de las cuales comprende de 150 a 180 mg de amantadina, o una sal farmacéuticamente aceptable de la misma.
[0039] En algunas formas de realización de cualquiera de los aspectos anteriores, la administración oral de una dosis única de la composición a una cohorte de sujetos voluntarios humanos sanos en un estado de ayuno proporciona una concentración plasmática máxima media (Cmax) de 1,1 hasta 2,8 ng/ml por mg de amantadina. En realizaciones más específicas, la administración oral de una dosis única de la composición a una cohorte de sujetos humanos en ayunas proporciona una concentración plasmática máxima promedio (Cmax) de 1,4 a 2,6 ng/ml por mg de amantadina y un AUC0-inf (Área bajo la curva de concentración-curva de t = 0 a t = infinito) de 46 a 60 ng*h/mL por mg de amantadina.
[0040] En algunas formas de realización de cualquiera de los aspectos anteriores, la administración oral diaria de una dosis de la composición a una cohorte de sujetos humanos proporciona un perfil de concentración plasmática en estado estacionario que se caracteriza por al menos uno de: (i) una Cmax media de 2,2 a 2,7 ng/ml por mg de amantadina, (ii) una Cmin media de 1,4 a 1,7 ng/ml por mg de amantadina, y (iii) un AUC0-24 media de 46 a 60 ng*h/ml por mg de amantadina. En ejemplos más específicos, se cumplen los tres criterios de (i), (ii) y (iii).
[0041] En realizaciones más específicas, el perfil de la concentración plasmática en estado estacionario se caracteriza además por: (iv) ningún aumento en la concentración de la amantadina para al menos una hora después de la administración; y (v) relación Cmax/Cmin de 1,3 a 3,0. En realizaciones más específicas, se cumplen ambos criterios de (iv) y (v).
[0042] En algunas formas de realización, el perfil de la concentración plasmática en estado estacionario se caracteriza además por al menos uno de: (iv) ningún aumento en la concentración plasmática de la amantadina durante al menos dos horas después de la administración; y (v) una relación Cmax/Cmin de 1,3 a 3,0. En algunas formas de realización, se cumplen ambos criterios de (iv) y (v).
[0043] En algunas formas de realización, la composición tiene un perfil de disolución in vitro de amantadina que muestra al menos uno de (i) no más de 25% de disolución a las 2 horas, (ii) no más 55-85% de disolución a las 6 horas, y (iii) al menos 80% de disolución a las 12 horas, usando un Aparato II USP (Paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, se cumplen dos de los criterios (i), (ii) y (iii). En algunas formas de realización, se cumplen los tres criterios (i), (ii) y (iii).
[0044] En algunas formas de realización, la composición tiene un perfil de disolución in vitro de amantadina que muestra al menos uno de (i) no más de 25% de disolución a las 2 horas, (ii) no más de 25-55% de disolución en 6 horas y (iii) disolución de al menos 80% a las 12 horas, usando un Aparato II USP (Paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, se cumplen dos de los criterios (i), (ii) y (iii). En algunas formas de realización, se cumplen los tres criterios (i), (ii) y (iii).
[0045] En algunas formas de realización, la composición tiene un perfil de disolución in vitro de amantadina que muestra al menos uno de (i) no más del 20% de disolución a 1 hora, (ii) aproximadamente 25-45% de disolución a las 2 horas, (iii) no más de 50-80% de disolución a las 4 horas, y (iv) al menos 80% de disolución a las 8 horas, usando un Aparato II USP (Paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, se cumplen dos de los criterios (i), (ii), (iii) y (iv). En algunas formas de realización, se cumplen los cuatro criterios (i), (ii), (iii) y (iv).
[0046] En algunas formas de realización, el perfil de disolución in vitro de amantadina se caracteriza además por la liberación de amantadina de: (i) no más de 10% en 1 hora, o (ii) 30-50% a las 4 horas, o (iii) al menos 90% a las 12 horas usando un Aparato II USP (Paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, se cumplen dos de los criterios (i), (ii) y (iii). En algunas formas de realización, se cumplen los tres criterios de (i), (ii) y (iii).
[0047] En algunas formas de realización, la presente invención proporciona una composición farmacéutica que comprende o consiste de un aglomerado en cápsula, en el que un aglomerado comprende un núcleo que comprende una semilla núcleo con una mezcla de amantadina y un aglomerante revestido sobre la semilla núcleo, y un revestimiento de liberación prolongada que rodea el núcleo que comprende etilcelulosa, un agente formador de poros tal como hidroxipropilmetilcelulosa o povidona, y un plastificante.
[0048] En algunas formas de realización, la presente invención proporciona una composición farmacéutica para uso en los métodos de los aspectos descritos anteriormente, en donde dicha composición es para la administración oral y comprende una cápsula para la administración oral de administración, dicha cápsula que comprende una pluralidad de pastillas, comprendiendo cada sedimento: (a) un núcleo de aglomerados que comprende amantadina, o una sal farmacéuticamente aceptable de la misma, y (b) un revestimiento de liberación prolongada que rodea el núcleo de aglomerados.
[0049] En algunas formas de realización, la liberación prolongada de revestimiento comprende acetato de celulosa y al
menos uno de povidona y celulosa de metilo de hidroxipropilo, y un plastificante. En algunas formas de realización, el revestimiento de liberación prolongada comprende etilcelulosa, povidona y un plastificante.
[0050] En algunas formas de realización, el núcleo de aglomerado comprende amantadina y un aglutinante revistieron sobre una semilla núcleo. En algunas formas de realización, la semilla núcleo es una esfera de azúcar (sin igual) o semilla de celulosa microcristalina (por ejemplo, Celphere®). En algunas formas de realización, la semilla del núcleo es un núcleo de celulosa microcristalina. En algunas formas de realización, la semilla del núcleo tiene un diámetro en el intervalo de 100 micrómetros a 1000 micrómetros (por ejemplo, al menos aproximadamente el 95% tiene este diámetro). En algunas formas de realización, la semilla núcleo tiene un diámetro de 100, 200, 300, 400, 500, 600 o 700 micrones. En algunas formas de realización, la semilla núcleo tiene un diámetro de menos de 500 micrómetros (por ejemplo, al menos aproximadamente el 95% tiene este diámetro).
[0051] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, la amantadina, o una sal farmacéuticamente aceptable de la misma, está presente en cantidades de 20 a 80% en peso, con una densidad aparente de 0,3 a 1,2 g/cm3.
[0052] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, la amantadina, o una sal farmacéuticamente aceptable de la misma, está presente en cantidades de 40 a 60% en peso, con una densidad aparente de 0,5 a 1,2 g/cm3.
[0053] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, la amantadina, o una sal farmacéuticamente aceptable de la misma, está presente en cantidades de 60 a 80% en peso, con una densidad aparente de 0,5 a 1,2 g/cm3.
[0054] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, el aglutinante está presente en cantidades de 8 a 25% en peso.
[0055] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, la semilla núcleo está presente en cantidades de 8 a 25% en peso.
[0056] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, la etilcelulosa está presente en cantidades de 10 a 20% en peso.
[0057] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, la povidona está presente en cantidades de 1 a 4% en peso.
[0058] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, y el plastificante está presente en cantidades de 1 a 4% en peso.
[0059] En algunas formas de realización, el aglomerado recubierto tiene un diámetro en el intervalo de 200 micras a 1700 micros. En algunas formas de realización, el aglomerado revestido tiene un diámetro de 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1100, 1200, 1300 o 1500 micrones. En algunas formas de realización, el aglomerado revestido tiene un diámetro de menos de 1000 micrómetros, por ejemplo, de 500 a 1000 micrómetros.
[0060] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, el aglutinante está presente en cantidades de 5 a 25% en peso.
[0061] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, la semilla núcleo está presente en cantidades de 1 a 15% en peso.
[0062] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, la etilcelulosa está presente en cantidades de 5 a 20% en peso.
[0063] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, la povidona está presente en cantidades de 0,25 a 4% en peso.
[0064] En algunas formas de realización, basado en el peso combinado del núcleo de aglomerado y recubrimiento de liberación prolongada, y el plastificante está presente en cantidades de 0,25 a 4% en peso.
[0065] En algunas formas de realización, el sedimento comprende además un sello de revestimiento entre el núcleo del aglomerado y el recubrimiento de liberación extendida. En algunas formas de realización, se puede aplicar un revestimiento inerte al núcleo inerte antes del revestimiento del fármaco o sobre aglomerados revestidos con fármaco o sobre aglomerados revestidos de liberación controlada. En otra realización, se puede aplicar un recubrimiento entérico a los aglomerados recubiertos con fármaco o a los aglomerados de liberación controlada.
[0066] En algunas formas de realización, el núcleo del aglomerado comprende un aglutinante, seleccionado de entre el
grupo constituido por hidroxipropilmetilcelulosa, copovidona, y mezclas de los mismos.
[0067] En algunas formas de realización, la composición anterior se proporciona en un tamaño de 3, tamaño 2, tamaño 1, tamaño 0 o el tamaño 00 de la cápsula, incluyendo formas EL de estos tamaños de cápsulas.
[0068] En algunas formas de realización, la dosis diaria terapéuticamente eficaz de la composición anterior se administra en no más de dos cápsulas. En otra realización, la dosis diaria terapéuticamente eficaz de la composición se administra en no más de tres cápsulas de tamaño 1. En otra realización, la dosis diaria terapéuticamente eficaz de la composición se administra en no más de dos cápsulas de tamaño 0. En una realización aún más preferida, la dosis diaria terapéuticamente eficaz de la composición se administra en no más de dos cápsulas de tamaño 1. En otra realización, la dosis diaria terapéuticamente eficaz de la composición se administra en no más de tres cápsulas de tamaño 2.
[0069] En una realización preferida, la composición anterior proporciona una cantidad de 50 a 110 mg de amantadina o una sal farmacéuticamente aceptable de la misma en una cápsula de tamaño 2 , y en la cantidad de 110 mg a 210 mg de amantadina o una sal farmacéuticamente aceptable de la misma en una cápsula de tamaño 1. En realizaciones adicionales, la composición anterior comprende aglomerados recubiertos de un diámetro de 300 a 1000 micrones (por ejemplo, al menos aproximadamente el 95% tiene este diámetro), con amantadina o una sal farmacéuticamente aceptable de la misma con un contenido de 40-80% en peso y con una densidad aparente de 0,5-1,2 g/cm3. En una realización preferida adicional, la composición anterior tiene un perfil de disolución in vitro de amantadina que muestra al menos uno de (i) no más del 25% de disolución a las 2 horas, (ii) no más del 55-85% de disolución a las 6 horas y (iii) disolución de al menos 80% a las 12 horas, usando un Aparato II USP (Paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, se cumplen dos de los criterios (i), (ii) y (iii). En algunas formas de realización, se cumplen los tres criterios (i), (ii) y (iii).
[0070] En algunas formas de realización, el plastificante se selecciona del grupo que consiste de triglicéridos de cadena media, ftalato de dietilo, ésteres de citrato, polietilenglicol, glicerol, glicéridos acetilados y aceite de ricino. En algunas formas de realización, el plastificante es triglicéridos de cadena media, por ejemplo, Miglyol 812 N.
[0071] En algunas formas de realización, la presente invención proporciona una dosis para uso en un método de administración de amantadina, o una sal farmacéuticamente aceptable de la misma, a un sujeto humano si lo necesita, comprendiendo dicho método la administración oral de una composición de cualquiera de los aspectos anteriores.
[0072] En un aspecto preferido, la presente invención proporciona una dosis para uso en un método de tratamiento de la enfermedad en un ser humano en necesidad de la misma, comprendiendo dicho método administrar oralmente una composición de cualquiera de los aspectos anteriores, una vez al día por la noche, la administración de 1,2 o 3 cápsulas o formas farmacéuticas.
[0073] En un aspecto preferido, la presente invención proporciona una dosis para uso en un método de tratamiento de la enfermedad en un ser humano sujeto en necesidad del mismo, método que comprende administrar oralmente una composición de cualquiera de los aspectos anteriores, una vez dicho día por la mañana, la administración de 1, 2 o 3 cápsulas o formas farmacéuticas.
[0074] Las referencias a la administración de amantadina a un sujeto que lo necesite incluyen el tratamiento de un paciente con una enfermedad o afección, incluida una afección iatrogénica, que puede tratarse, prevenirse o curarse mediante un antagonista de NMDA. Más específicamente, administrar amantadina a un sujeto que lo necesita incluye tratar a un paciente con lesión cerebral, traumatismo cerebral, accidente cerebrovascular (y déficits para caminar asociados con ellos), enfermedad de Huntington (y corea asociada con la misma), ELA, esclerosis múltiple (y problemas para caminar asociados con ellos), enfermedades neurodegenerativas, enfermedades cerebrovasculares, trastornos del movimiento, trastornos de los nervios craneales, síndrome de Tourette, discinesia tardía y otros trastornos del SNC.
[0075] Algunas formas de realización descritas en este documento proporcionan una dosis de la invención para uso en un método de mejora de CGI en un paciente, que comprende administrar a dicho paciente una vez todas las noches, 0 a 4 horas antes de acostarse una composición que comprende 260 a 420 mg de amantadina, o una sal farmacéuticamente aceptable de la misma y al menos un excipiente modificador de la liberación. En algunas de tales realizaciones, la dosis de la invención para uso en un método puede ser para el tratamiento de los trastornos y afecciones descritos en el presente documento. En algunas formas de realización, la composición comprende de 260 a 340 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende 260 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende 340 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, el cambio en CGI se determina en un estudio clínico doble ciego controlado con placebo.
Breve descripción de los dibujos
[0076]
La Figura 1 muestra un gráfico de las concentraciones plasmáticas medias (DE) de amantadina frente al tiempo
programado para la Formulación 1 en un estudio PK en ayunas de dosis única (Ejemplo 4).
La Figura 2 muestra la concentración plasmática media simulada de amantadina frente a las curvas de tiempo después de la administración de dosis múltiples de varias concentraciones de amantadina de LP administradas una vez por la noche. (Ejemplo 5).
La Figura 3 muestra los perfiles de disolución para tres formulaciones de amantadina de LP, A, B y C, a las que se hace referencia en el Ejemplo 7.
La Figura 4 muestra un gráfico de las concentraciones plasmáticas medias (DE) de amantadina frente al tiempo programado para cuatro (4) tratamientos con amantadina.
La Figura 5 muestra una media semilogarítmica (DE) de las concentraciones plasmáticas de amantadina frente al tiempo programado para cuatro (4) tratamientos con amantadina.
La Figura 6 muestra las puntuaciones clínicas de los 4 grupos de prueba utilizados en el ensayo MotoRater. (Ejemplo 9).
La Figura 7 muestra un diagrama de dispersión de las puntuaciones clínicas acumulativas de los 4 grupos de prueba usados en el ensayo MotoRater. (Ejemplo 9).
La Figura 8 muestra un gráfico de dispersión de las puntuaciones clínicas acumuladas de la expresión de Iba-1 por cada uno de los 4 grupos de prueba usados en el ensayo MotoRater. (Ejemplo 9).
La Figura 9 muestra un gráfico de líneas que representa el % de animales en cada grupo de prueba que fueron capaces de completar la prueba de caminata MotoRater durante el estudio de 27 días. (Ejemplo 9).
La Figura 10A proporciona un gráfico de barras que muestra el tiempo necesario para caminar 60 cm el día 14 del estudio. La Figura 10B muestra la velocidad media de cada grupo en el día 14.
Descripción detallada de la invención
[0077] La dosis de la invención es, en una realización, para uso en un método que comprende la administración oral al paciente de una composición de liberación prolongada (LP) de amantadina diseñada para administración nocturna. La composición se toma menos de tres horas antes de acostarse, y preferiblemente menos de dos horas y media, menos de dos, menos de una hora y media o menos de una hora antes de acostarse. Lo más preferiblemente, la composición de amantadina de LP se toma menos de media hora antes de acostarse (es decir, la hora a la que el sujeto desea irse a dormir por la noche). Alternativamente, la composición se administra menos de aproximadamente 4 horas antes de acostarse. La dosis de la invención es, en una realización, para su uso en un método que comprende administrar oralmente al paciente una composición de amantadina de liberación prolongada (LP) diseñada para la administración matutina. La composición se toma después de despertar, preferiblemente antes del mediodía, más preferiblemente entre las 6:00 y 24:00, 7:00 y 24:00, 8:00 y 24:00, 6:00 y 11:00, 7:00 y 11:00.
[0078] La dosis de la invención es, en una realización, para uso en un método que comprende la administración oral al paciente de una composición de liberación inmediata amantadina, una vez por día.
[0079] Como se usa en este documento, se pretende que una referencia a la amantadina abarque las sales farmacéuticamente aceptables de la misma (por ejemplo, clorhidrato de amantadina, sulfato de amantadina, etc.).
[0080] Como se usa en este documento, "liberación prolongada" incluye "liberación controlada", "liberación modificada", "liberación sostenida", "liberación temporizada", "liberación retardada", y también mezclas de liberación retardada, liberación inmediata, recubrimiento entérico, etc., con cada uno de los anteriores.
[0081] El paciente puede ser diagnosticado con cualquier enfermedad o trastorno para el que se prescribe la amantadina. En algunas formas de realización, los presentes métodos son para prevenir las enfermedades y trastornos descritos en el presente documento, por ejemplo, cuando un paciente tiene riesgo de desarrollar enfermedades y trastornos. En algunas formas de realización de la divulgación, el paciente tiene una lesión cerebral, lesión cerebral, trauma cerebral, accidente cerebrovascular, enfermedad de Huntington, ELA, enfermedades neurodegenerativas, afecciones cerebrovasculares, trastornos del movimiento, discinesia tardía, síndrome de Tourette o trastornos de los nervios craneales.
[0082] La invención también proporciona una dosis para su uso en un método para reducir las alteraciones del sueño en un paciente sometido a tratamiento con amantadina. El método comprende administrar amantadina a un paciente que lo necesita, de modo que la amantadina no interfiera con el sueño, pero proporcione el máximo beneficio en las horas de la mañana cuando a menudo se necesitan más por muchos pacientes que toman amantadina y, además, proporciona una cobertura nocturna de los síntomas de la enfermedad. si es necesario. La cobertura nocturna incluye proporcionar beneficios si el paciente se despierta y desea volver a dormir. En algunos de estos métodos, C-media-día es de 1,2 a 1,7 veces C-media-noche; en los métodos preferidos, C-media-día se mide entre las 8:00 y 20:00 y C-media-noche se mide entre las 20:00 y las 8:00. En algunos de estos métodos, la administración de una dosis única de la composición a una cohorte de voluntarios humanos sanos en ayunas proporciona una concentración plasmática máxima promedio (Cmax) de 1,1 a 2,2 ng/ml por mg de amantadina o un AUCü-inf (Área bajo la curva de concentración-curva de t = 0 a t = infinito) de 46 a 56 ng*h/mL por mg de amantadina o ambos. En algunos de estos métodos, la administración oral diaria de una dosis de la composición a una cohorte de sujetos humanos proporciona un perfil de concentración plasmática en estado estacionario caracterizado por al menos uno de: (i) una Cmax media de 2,2 a 3,0 ng/ml por mg de amantadina, (ii) una Cmin media de 1,4 a 1,7 ng/ml por mg de amantadina, y (iii) una AUC media 0-24 de 46 a 56 ng*h/ml por mg de
amantadina; en métodos más específicos, se cumplen los tres criterios de (i), (ii) y (iii).
[0083] Una composición amantadina de LP para su uso en la invención está adaptada para la administración durante la noche, proporcionando un perfil de concentración plasmática que no interfiere con el sueño del sujeto. La composición de la invención, tras la administración a un sujeto humano, dará como resultado un aumento inicial gradual en la concentración plasmática de amantadina de tal manera que, en condiciones de estado estacionario, la administración de una dosis de la composición dará como resultado un aumento en la concentración plasmática de amantadina de menos del 25% a las tres horas después de la administración de la dosis. Por ejemplo, si la concentración plasmática de amantadina en estado estacionario de un sujeto es de 500 ng/ml en el momento en que se administra una dosis de la composición, tres horas después, la concentración plasmática de amantadina del sujeto será inferior a 625 ng/ml. Preferiblemente, el aumento en la concentración plasmática de amantadina tres horas después de la administración es menos del 15% y más preferiblemente menos del 10%. Las composiciones particularmente preferidas tienen un perfil de concentración plasmática caracterizado además por ningún aumento en la concentración plasmática de amantadina, o incluso una disminución (en condiciones de estado estacionario), durante al menos una o, en una realización preferida, dos horas después de la administración. La composición para uso en la invención se adapta además para la administración a la hora de acostarse (es decir, la hora a la que el sujeto desea irse a dormir por la noche) proporcionando una concentración máxima de amantadina (Cmax) en las horas de la mañana. El tiempo para alcanzar Cmax (Tmax), medido después de la administración de una dosis única en ayunas, en algunas formas de realización es de al menos 9 horas y hasta 13, 14, 15, 16, 17 o 18 horas, o al menos 10 horas y hasta 14, 15, 16, 17 o 18 horas, o al menos 12 horas, y hasta 14, 15, 16 o 17 horas. En realizaciones específicas, Tmax es de 9 a 18 horas, lo más preferiblemente de 12 a 18 horas. En algunas formas de realización, Tmax es de 8 a 18 horas. En el estado estacionario, con la administración una vez por la noche de la composición, Tmax puede ser de 7 a 13 horas, lo más preferiblemente de 8 a 12 horas.
[0084] Una composición de amantadina de LP adecuada se puede caracterizar además por tener una relación Cmax/Cmin en estado estacionario de 1,3 a 3,0, y preferiblemente de 1,5 a 2,8, lo que resulta en una composición con un perfil diario que exhibe un pico diurno en las horas diurnas o de vigilia y una caída en las horas de noche o de sueño. Las composiciones de amantadina de LP preferidas proporcionan una mayor variación diurna (es decir, una relación Cmax/Cmin aumentada) mientras proporcionan Cmax en estado estacionario durante las horas del día (por ejemplo, de 9:00 A 18:00, de 9:00 a 17:00, de 9:00 a 16:00, 10:00 a las 18:00, 10:00 a 17:00, 10:00 a 16:00).
[0085] En realizaciones más específicas preferidas, el perfil de concentración de plasma se caracteriza además por tener un perfil de AUC después de la administración de una dosis única de la composición caracterizada por: un AUC fraccional a partir de 0 a 4 horas en que es menor que 5%, y preferiblemente menos del 3% de AUCo-inf; un AUC fraccional de 0 a 8 horas que es de aproximadamente 5 a 15%, y preferiblemente de aproximadamente 8 a 12% de AUCo-inf; un AUC fraccional de 0 a 12 horas que es de aproximadamente 10 a 40%, y preferiblemente de aproximadamente 15 a 30% de AUCo-inf; un AUC fraccional de 0 a 18 horas que es aproximadamente del 25 al 60%, y preferiblemente de aproximadamente el 30 al 50% del AUCü-inf; y un AUC fraccional de 0 a 24 horas que es aproximadamente del 40 al 75%, y preferiblemente de aproximadamente el 50 al 70% del AUCü-inf.
[0086] En una realización preferida adicional, el perfil de concentración plasmática se caracteriza además por tener un perfil AUC después de una vez todas las noches de dosificación de la composición a condiciones de estado estacionario se caracteriza por: AUC fraccional a partir de 0 a 4 horas que es de aproximadamente 2 a 25%, y preferiblemente aproximadamente del 5 al 20% de AUC0-24; un AUC fraccional de 0 a 8 horas que es aproximadamente del 15 al 50%, y preferiblemente de aproximadamente el 20 al 40% del AUC0-24; un AUC fraccional de 0 a 12 horas que es de aproximadamente 30 a 70%, y preferiblemente de aproximadamente 40 a 60% de AUC0-24: y un AUC fraccional de 0 a 18 horas que es aproximadamente 60 a 95%, y preferiblemente aproximadamente 75 al 90% del AUC0-24.
[0087] En algunas formas de realización de cualquiera de los aspectos anteriores, el perfil de concentración plasmática en estado estacionario después de múltiples administraciones a un sujeto humano de la composición a una vez al día se caracteriza por una concentración plasmática promedio durante el día ("C-media-día", definida como la concentración plasmática media de amantadina durante el día medida en un estudio de FC humana) que es de 1,1 a 2,0 veces la concentración plasmática media durante la noche ("C-media-noche", definida como la concentración plasmática media de amantadina durante la noche como medido en un estudio de PK humana). En algunas formas de realización, la relación de C-media-día/C-media-noche en estado estacionario está dentro de uno de los rangos de 1,1 a 1,9, 1,1 a 1,8, 1,1 a 1,7, 1,1 a 1,6, 1,1 a 1,5, 1,1 a 1,4, 1,2 a 1,9, 1,2 a 1,7, 1,2 a 1,6, 1,2 a 1,5, 1,3 a 1,9, 1,3 a 1,8, 1,3 a 1,7, 1,3 a 1,6, 1,4 a 1,9, 1,4 a 1,8, 1,4 a 1,7, 1,5 a 1,9, 1,5 a 1,8, 1,5 a 1,7, 1,6 a 1,8 o 1,6 a 1,9. En algunas formas de realización, la relación de C-media-día/C-media-noche en estado estacionario es 1,1, 1,15, 1,2, 1,25, 1,3, 1,35, 1,4, 1,45, 1,5, 1,55, 1,6, 1,65, 1,7, 1,75, 1,8, 1,85 o 1,9. En algunas formas de realización, C-media-día es la concentración plasmática promedio de amantadina medida entre las horas de 5:00, 6:00, 7:00, 8:00 o 9:00 hasta las 16:00, 17:00, 18:00, 19:00 u 20:00 y C-media-noche es la concentración plasmática promedio de amantadina medida entre las 16:00, 17:00, 18:00, 19:00, 20:00, 21:00, 22:00 o 23:00 hasta las 5:00, 6:00, 7:00, 8:00 o 9:00. En algunas formas de realización, C-media-día es la concentración plasmática promedio de amantadina medida dentro de un período de cuatro a doce horas entre las 5:00 y las 20:00; y C-media-noche es la concentración plasmática promedio de amantadina medida dentro de un período de cuatro a doce horas entre las 20:00 y las 5:00. En algunas formas de realización, el C-media-día es la concentración plasmática media de amantadina medida dentro de cualquier período de cuatro, cinco, seis, siete, ocho, nueve, diez, once o doce horas entre las 5:00 y las 20:00; y C-media-noche es la concentración plasmática promedio de amantadina medida
dentro de cualquier período de cuatro, cinco, seis, siete, ocho, nueve, diez, once o doce horas entre las 20:00 y 5:00.
[0088] En algunas formas de realización descritas aquí una composición de amantadina se administra a un paciente desde 0 a 4 horas antes de la hora de acostarse. En algunas formas de realización, la composición de amantadina se administra a un paciente de 0 a 3, de 0 a 2 o de 0 a 1 horas antes de acostarse. En algunas formas de realización, la composición de amantadina se administra a un paciente de 0 a 240 minutos, de 0 a 180 minutos, por ejemplo, de 0 a 120 minutos, de 0 a 60 minutos, de 0 a 45 minutos, de 0 a 30 minutos, de 0 a 15 minutos o de 0 a 10 minutos antes de acostarse. En algunas formas de realización, la composición de amantadina se administra a un paciente de 60 a 240 minutos, de 60 a 180 minutos, de 60 a 120 minutos o de 60 a 90 minutos antes de acostarse.
[0089] Se ha de entender que la administración a un paciente incluye la administración por un profesional de la salud y la autoadministración por el paciente.
[0090] A menos que se especifique lo contrario en el presente documento, el término "hora de acostarse" tiene el significado normal de un momento cuando una persona se retira para el período primario de sueño durante un período de veinticuatro horas. Mientras que para la población en general, la hora de acostarse ocurre por la noche, hay pacientes, como los que trabajan de noche, para quienes la hora de acostarse ocurre durante el día. Por tanto, en algunas formas de realización, la hora de acostarse puede ser en cualquier momento durante el día o la noche.
[0091] Como se usa en este documento, a menos que se indique otra cosa, la referencia a un perfil de concentración de plasma o una propiedad específica farmacocinética (por ejemplo, Cmax, Cmin, AUC, Tmax, etc.) en un sujeto humano se refiere a un valor medio (o en algunas formas de realización un valor mediano de Tmax) obtenido de adultos sanos determinado en un ensayo clínico de fase I típico diseñado para medir las propiedades farmacocinéticas de un fármaco (ver, por ejemplo, los Ejemplos 2 y 3, a continuación). Las referencias en este documento a Tmax y T1/2 se refieren a valores obtenidos después de la administración de una dosis única en ayunas, a menos que se indique lo contrario.
[0092] En algunas formas de realización de la invención, la dosis de la amantadina administrada de acuerdo con la presente invención está dentro o por encima de los rangos normalmente prescritos para las composiciones de liberación inmediata de la amantadina. Como se describe en el presente documento, las dosis unitarias de amantadina administradas de acuerdo con la presente invención son generalmente más altas que los intervalos normalmente prescritos para las composiciones de amantadina de liberación inmediata. Por ejemplo, la dosis recomendada de amantadina para el tratamiento de la enfermedad de Parkinson es de 100 mg de amantadina de liberación inmediata administrada dos veces al día. En casos limitados en los que el paciente no obtiene un beneficio suficiente con esa dosis y sujeto a que el paciente pueda tolerar dicha dosis más alta, la dosis diaria puede aumentarse a 300 mg o 400 mg, que siempre se administra en dosis divididas. Antes de la presente invención, la dosis más comúnmente prescrita de amantadina es de 200 mg por día, siempre administrada en dosis divididas. Antes de la presente invención, siempre se administraban más de 200 mg (por ejemplo, 300 mg) en dosis divididas. Para la presente invención, se administran dosis de 260 a 420 mg para el tratamiento de pacientes con EM y accidente cerebrovascular, y las dosis, usos y composiciones de la invención pueden comprender la administración una vez por la noche de una dosis definida por cualquiera de estos rangos, particularmente en dosis de 260 mg a 420 mg, y lo más preferiblemente 340 mg, una vez por noche. En algunas de tales realizaciones, la administración de tales dosis más altas es por la noche, es decir, después de las 16:00 y/o dentro de las 4 horas antes de acostarse. En realizaciones adicionales, la administración de tales dosis más altas puede ser en forma de 1, 2 o 3 cápsulas de tamaño 0, 1 o 2 en el formato normal o EL administradas una vez por la noche.
[0093] En algunas formas de realización de cualquiera de los aspectos anteriores, la amantadina se administra como una sal farmacéuticamente aceptable. En una realización más específica, la amantadina se administra como clorhidrato de amantadina o sulfato de amantadina.
[0094] En alguna realización de cualquiera de los aspectos anteriores, una dosis diaria total de 260 mg a 420 mg se administra como una formulación una vez cada noche después de 16:00 y/o dentro de las 4 horas de la hora de acostarse. En algunas formas de realización, la dosis de amantadina o una sal farmacéuticamente aceptable de la misma administrada una vez por la noche supera los 300 mg por día (por ejemplo, de 300 a 420 mg por día). La dosis de la invención comprende de 220 mg a 600 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En diversas realizaciones específicas, la dosis diaria o nocturna de amantadina o una sal farmacéuticamente aceptable de la misma puede ser de 230 a 255 mg, 250 a 275 mg, 260 a 275 mg, 270 a 285 mg, 280 a 295 mg, 290 a 305 mg, 300 a 315 mg, 310 a 325 mg, 320 a 335 mg, 330 a 345 mg, 340 a 355 mg, 350 a 365 mg, 360 a 375 mg, 370 a 385 mg, 380 a 395 mg, 390 a 405 mg, 400 a 415 mg o 410 a 420 mg. También se describen dosis una vez al día o por la noche de 50 a 75 mg, 70 a 95 mg, 90 a 115 mg, 110 a 135 mg, 130 a 155 mg, 150 a 175 mg, 170 a 195 mg, 190 a 215 mg, 210 a 235 mg. En algunas formas de realización preferidas, la dosis diaria o nocturna de amantadina o una sal farmacéuticamente aceptable de la misma es de 260 mg a 360 mg, 300 a 360 mg, 330 a 350 mg o 340 mg, 430 a 445 mg o 440 a 455 mg.
[0095] En algunas formas de realización de cualquiera de los aspectos anteriores, la composición una vez al día o de noche de la amantadina o la sal farmacéuticamente aceptable de la misma comprende de aproximadamente 260 mg, 265 mg, 270 mg, 275 mg, 280 mg, 285 mg, 290 mg, 295 mg, o 300 mg de amantadina, o una sal farmacéuticamente aceptable de la misma hasta aproximadamente 305 mg, 310 mg, 315 mg, 320 mg, 325 mg, 330 mg, 335 mg, 340 mg, 345 mg, 350 mg, 355 mg, 360 mg, 365 mg, 370 mg, 375 mg, 380 mg, 385 mg, 390 mg, 395 mg, 400 mg, 405 mg, 410 mg, 415 mg o
420 mg de amantadina o una sal farmacéuticamente aceptable de la misma.
[0096] En realizaciones específicas descritas en este documento (por ejemplo, cuando la formulación tiene una Tmax media de 8 a 18 horas como se determina a partir de un estudio PK de dosis única en humano en ayunas), toda la dosis diaria de un sujeto de la amantadina se administra una vez, durante un período de menos de aproximadamente cuatro, tres, dos o una horas antes de acostarse (es decir, después de las 16:00 y/o la hora a la que el sujeto desea irse a dormir por la noche).
[0097] En algunas formas de realización descritas en este documento (por ejemplo, cuando la formulación tiene una Tmax media de 5 a 11 horas como se determina a partir de un estudio PK de dosis única en humanos en ayunas), la dosis diaria entera de la amantadina de un sujeto puede ser administrada una vez por la mañana (por ejemplo, después de despertarse y no más tarde de las 24:00).
[0098] En algunas formas de realización de cualquiera de los aspectos anteriores, la administración de la composición a un paciente con EM resulta en una reducción significativa en los síntomas asociados con la enfermedad o condición. En algunas formas de realización específicas, la administración de la composición da como resultado una reducción de aproximadamente el 5%, 10%, 15%, 20%, 25%, 30%, 35% o 40% de los síntomas de la EM o las fluctuaciones motoras. En otras realizaciones específicas, la reducción de los síntomas de la EM o las fluctuaciones motoras se mide en una escala numérica utilizada o aceptada por la FDA u otras agencias reguladoras para evaluar la eficacia y aprobar los medicamentos con licencia para el tratamiento de los síntomas de la EM o las fluctuaciones motoras..
[0099] Algunas formas de realización descritas en este documento proporcionan una dosis para su uso en un método para mejorar la impresión global del clínico sin aumentar los trastornos del sueño en un paciente que comprende administrar a dicho paciente una vez todas las noches, 0 a 4 horas antes de la hora de dormir una composición que comprende 260 a 420 mg de amantadina, o una sal farmacéuticamente aceptable de la misma (por ejemplo, hidrocloruro de amantadina), y al menos un excipiente modificador de la liberación. En algunos de tales métodos, la dosis de amantadina, o una sal farmacéuticamente aceptable de la misma, es de 300 a 360 mg por día, particularmente de 330 a 350 mg por día, y en particular de 340 mg por día.
[0100] Algunas formas de realización descritas en la presente memoria proporcionan un método para mejorar la impresión global del clínico sin aumento de los trastornos del sueño en un paciente que comprende administrar a dicho paciente una vez al día una composición que comprende 260 a 420 mg de amantadina, o una sal farmacéuticamente aceptable de la misma (por ejemplo, clorhidrato de amantadina), y al menos un excipiente modificador de la liberación. En algunos de tales métodos, la dosis de amantadina, o una sal farmacéuticamente aceptable de la misma, es de 300 a 360 mg por día, particularmente de 330 a 350 mg por día, y en particular de 340 mg por día. En algunos de tales métodos, C-media-día es de 1,2 a 1,7 veces la C-media-noche; en los métodos preferidos, C-media-día se mide entre las 9:00 y 17:00 y la C-media-noche se mide entre las 22:00 y 6:00. En algunos de estos métodos, la administración de una dosis única de la composición a una cohorte de voluntarios humanos sanos en ayunas proporciona una concentración plasmática máxima promedio (Cmax) de 1,4 a 2,8 ng/ml por mg de amantadina o un AUCü-inf (Área bajo la curva de concentración-curva de t = 0 a t = infinito) de 46 a 56 ng*h/mL por mg de amantadina o ambos. En algunos de estos métodos, la administración oral diaria de una dosis de la composición a una cohorte de sujetos humanos proporciona un perfil de concentración plasmática en estado estacionario caracterizado por al menos uno de: (i) una Cmax media de 2,2 a 2,9 ng/ml por mg de amantadina, (ii) una Cmin media de 1,4 a 1,7 ng/ml por mg de amantadina, y (iii) una AUC media 0-24 de 46 a 56 ng*h/ml por mg de amantadina; en métodos más específicos, se cumplen los tres criterios de (i), (ii) y (iii).
[0101] En algunas formas de realización de cualquiera de los aspectos anteriores, la administración de la composición a pacientes da como resultado una mejora significativa en la impresión general de los médicos. En algunas formas de realización específicas, la administración de la composición da como resultado una mejora de aproximadamente 0,5, 1,0, 1,5, 2,0, 2,5 o 3,0 puntos en la impresión general de los médicos utilizando una escala de 7 puntos (o cambios proporcionales utilizando una escala diferente). En realizaciones específicas adicionales, la mejora en la impresión general de los médicos se mide en una escala numérica que es utilizada o aceptada por la FDA u otras agencias reguladoras para evaluar la efectividad y aprobar los medicamentos con licencia indicados para pacientes. En otras formas de realización específicas, la escala utilizada para medir la mejora en la impresión general de los médicos podría ser la escala de valoración de la impresión de cambio global de los médicos (CGIC). En otras realizaciones específicas, la mejora en la impresión general de los médicos se mide en relación con el placebo en un ensayo clínico controlado. En otras realizaciones, la mejora en la impresión general de los médicos se mide en relación con la línea de base en un ensayo clínico controlado.
[0102] El objeto de la invención tiene esclerosis múltiple. En una realización de la invención, a un paciente con una anomalía en la marcha se le administra una composición que comprende amantadina y al menos un excipiente. En determinadas realizaciones, se administra una formulación de liberación inmediata de amantadina o una sal farmacéuticamente aceptable de la misma a un paciente con esclerosis múltiple o un paciente después de un accidente cerebrovascular una o dos veces al día para proporcionar una mejora en la marcha. En algunas de estas realizaciones, la dosis diaria de amantadina o una sal farmacéuticamente aceptable de la misma es de 100 mg a 300 mg.
[0103] En ciertas realizaciones, al menos uno de dichos excipientes en la composición para ser administrada es una
versión de la modificación de excipiente.
[0104] En ciertas realizaciones, la composición que va a administrarse comprende una forma de liberación prolongada de la amantadina o una sal farmacéuticamente aceptable de la misma. En algunas de estas realizaciones, la forma de liberación prolongada de amantadina o una sal farmacéuticamente aceptable de la misma se administra por vía oral, una vez al día para proporcionar una mejora en la marcha en un paciente con una anomalía en la marcha. En algunas de estas realizaciones, la anomalía de la marcha está asociada con la esclerosis múltiple en el paciente. En determinadas realizaciones, la dosis diaria de amantadina o una sal farmacéuticamente aceptable de la misma es de 220 mg a 600 mg, 220 mg a 445 mg, 240 mg a 445 mg, 240 mg a 420 mg, 240 mg a 340 mg. En algunas formas de realización, la dosis diaria de amantadina o una sal farmacéuticamente aceptable de la misma es de 260 mg a 420 mg.
[0105] En ciertas realizaciones, también se le administra al paciente por vía oral uno o más segundos agentes seleccionados del grupo que consiste en 4-aminopiridina, baclofeno, dextrometorfano, fumarato de dimetilo, fingolimod, metilfenidato y teriflunomida, tetrabenazina, y tizanidina, incluyendo sales farmacéuticamente aceptable de los mismos.
En determinadas formas de realización, el segundo o los segundos agentes se proporcionan por separado de la composición de amantadina. En determinadas realizaciones, el segundo agente se proporciona en la misma composición que la amantadina o una sal farmacéuticamente aceptable de la misma. En determinadas formas de realización, el segundo agente se proporciona en una forma de liberación controlada. En algunas formas de realización, la dosis diaria total del segundo agente es 25%, 50%, 75%, 100% de la dosis diaria total proporcionada en la etiqueta del producto aprobado para el segundo agente. En algunas formas de realización, el segundo agente se administra por separado, secuencial o simultáneamente con la amantadina o una sal farmacéuticamente aceptable de la misma.
[0106] En ciertas realizaciones, la administración diaria de amantadina a pacientes con déficit de caminar proporciona una mejora (frente a placebo) en al menos una de las siguientes medidas: TUG (prueba de caminata cronometrada), T25FW (prueba de caminata cronometrada de 25 pies), o 6MWT (prueba de caminata de seis minutos), o prueba de caminata de 2 minutos, o MSWS-12 (escala de caminata de esclerosis múltiple de doce ítems).
[0107] En algunas formas de realización de la descripción, se proporciona un método para mejorar la marcha en un sujeto humano, comprendiendo dicho método administrar oralmente a dicho paciente una vez al día por la mañana o por noche, una dosis que comprende (i) 50 mg a 600 mg de un fármaco seleccionado del grupo que consiste en amantadina y sus sales farmacéuticamente aceptables y (ii) al menos un excipiente (por ejemplo, en el que la dosis incluye una composición que comprende los componentes (i) y (ii)). En algunas formas de realización de la invención, la composición puede comprender de 100 mg a 450 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización de la invención, la composición puede comprender 130-210 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, una forma de dosificación que contiene la composición comprende 50 a 75 mg, 70 a 95 mg, 90 a 115 mg, 110 a 135 mg, 130 a 155 mg, 150 a 175 mg, 170 a 195 mg, 190 a 215 mg, 210 a 235 mg, 230 a 255 mg, 250 a 275 mg, 270 a 295 mg, 290 a 305 mg, 300 a 315 mg, mg, 320 a 335 mg, 330 a 345 mg, 340 a 355 mg, 350 a 365 mg, 360 a 375 mg, 370 a 385 mg, 380 a 395 mg, mg, 400 a 415 mg, 410 a 425 mg, 420 a 435 mg, 430 a 445 mg o 440 a 455 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende aproximadamente 110, 120, 130, 140, 150, 160 170, 180, 190, 210, 220 o 340 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende 110 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 130 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 170 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 210 mg de hidrocloruro de amantadina. En la invención, el sujeto humano tiene esclerosis múltiple. En algunas formas de realización, la dosis comprende adicionalmente uno o más fármacos seleccionados del grupo que consiste en 4-aminopiridina, baclofeno, dextrometorfano, dimetilfumarato, fingolimod, metilfenidato y teriflunomida, tetrabenizina y tizanidina. En algunas formas de realización, el al menos un excipiente es un excipiente que modifica la liberación. En algunas formas de realización, al menos uno de dichos excipientes (o el al menos un excipiente) modifica la liberación de la amantadina o una sal farmacéuticamente aceptable de la misma para proporcionar una forma de liberación prolongada, y donde la administración de la dosis proporciona) un Tmax para amantadina de 5 a 18 horas, b) un Cmax de 1,0 a 2,8 ng/ml por mg de amantadina, y c) un A u Cq-m de 40 a 75 ng*h/ml por mg de amantadina medido en un estudio farmacocinético humano de dosis única. En algunas formas de realización, tal dosificación de amantadina da como resultado concentraciones plasmáticas máximas de amantadina que se producen por la mañana o por la tarde, y concentraciones plasmáticas muy bajas por la noche. En algunas formas de realización, el fármaco se proporciona en forma de liberación prolongada. En realizaciones específicas adicionales, la concentración plasmática máxima de amantadina se logra entre 5, 6 , 7, 8 , 9, 10, 11 o 12 horas hasta aproximadamente 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 o 24 horas después de la administración de una dosis única de la composición (por ejemplo, de 8 horas a aproximadamente 18 horas después de la administración). En algunas formas de realización descritas en el presente documento, la composición de amantadina se administra a un paciente de 0 a 4 horas antes de acostarse. En algunas formas de realización, la composición de amantadina se administra a un paciente de 0 a 3, de 0 a 2 o de 0 a 1 horas antes de acostarse. En algunas formas de realización, la composición de amantadina se administra a un paciente de 0 a 240 minutos, de 0 a 180 minutos, por ejemplo, de 0 a 120 minutos, de 0 a 60 minutos, de 0 a 45 minutos, de 0 a 30 minutos, de 0 a 15 minutos o de 0 a 10 minutos antes de acostarse. En algunas formas de realización, la composición de amantadina se administra a un paciente de 60 a 240 minutos, de 60 a 180 minutos, de 60 a 120 minutos o de 60 a 90 minutos antes de acostarse. En algunas formas de realización descritas en el presente
documento, la composición de amantadina se administra a un paciente por la mañana; en realizaciones más específicas, la composición de amantadina se administra a dicho paciente después de que dicho paciente se despierta pero antes de 3, 4, 5 o 6 horas después de despertar. En algunas formas de realización descritas en el presente documento, la composición de amantadina se administra a un paciente después de que el paciente se despierta por la mañana y antes de las 24:00. En algunas formas de realización de cualquiera de los aspectos anteriores, la administración de la composición a sujetos con enfermedad de EM da como resultado una reducción significativa en la alteración de la marcha de la enfermedad de EM. En una realización específica, la administración de la composición da como resultado al menos aproximadamente 5%, 10%, 15%, 20%, 25%, 30%, 35% o 40%, 45%, 50%, 55% o 60% de reducción en la alteración de la marcha por enfermedad de EM. En realizaciones específicas adicionales, la marcha (o alteración de la marcha) se mide en una escala numérica que es utilizada o aceptada por la FDA u otras agencias reguladoras para evaluar la efectividad y aprobar medicamentos con licencia para el tratamiento de alteración de la marcha. En algunas formas de realización, la administración una vez al día de la composición de amantadina (o combinación que comprende amantadina) proporciona una mejora en un trastorno del movimiento hipocinético tal como alteración de la marcha. La mejora puede determinarse mediante métodos conocidos en la técnica, tales como prueba de caminata cronometrada (TUG), prueba de caminata cronometrada de 25 pies (T25FW) o prueba de caminata cronometrada de seis minutos (6MTW). En algunas formas de realización, la composición de una vez al día o una vez por la noche se administra como una, dos, tres o cuatro formas de dosificación unitarias en unidades divididas de manera desigual o, preferiblemente, igualmente. En algunas formas de realización más específicas, la composición se administra como dos o tres formas de dosificación unitarias que comprenden cada una de 85 a 175 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos formas de dosificación unitarias que comprenden cada una de 100 a 175 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos o tres formas de dosificación unitaria de dosificación desigual, o preferiblemente igual, comprendiendo cada una de 85 a 250 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización más específicas, la composición se administra como dos formas de dosificación unitarias, cada una de las cuales comprende de 150 a 180 mg de amantadina, o una sal farmacéuticamente aceptable de la misma.
[0108] En algunas formas de realización de la divulgación, se proporciona un método de mejorar la marcha en un sujeto humano, comprendiendo dicho método la administración oral a dicho paciente una vez al día o de noche una dosis de (i) 50 mg a 600 mg de un fármaco seleccionado del grupo que consiste en amantadina y sus sales farmacéuticamente aceptables y (ii) al menos un excipiente (por ejemplo, en el que la dosis incluye una composición que comprende los componentes (i) y (ii)). En algunas de tales realizaciones, los métodos tratan los trastornos y enfermedades definidos en el presente documento. En algunas formas de realización, la composición puede comprender de 100 mg a 450 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición puede comprender 130-210 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En varias realizaciones específicas, una forma de dosificación que contiene la composición comprende 50 a 75 mg, 70 a 95 mg, 90 a 115 mg, 110 a 135 mg, 130 a 155 mg, 150 a 175 mg, 170 a 195 mg, 190 a 215 mg, 210 a 235 mg, 230 a 255 mg, 250 a 275 mg, 270 a 295 mg, 290 a 305 mg, 300 a 315 mg, 310 a 325 mg, 320 a 335 mg, 330 a 345 mg, 340 a 355 mg, 350 a 365 mg, 360 a 375 mg, 370 a 385 mg, 380 a 395 mg, 390 a 405 mg, 400 a 415 mg, 410 a 425 mg, 420 a 435 mg, 430 a 445 mg o 440 a 455 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende aproximadamente 110, 120, 130, 140, 150, 160, 170, 180, 190, 210, 220 o 340 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende 110 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 130 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 170 mg de hidrocloruro de amantadina. En algunas formas de realización, la composición comprende 210 mg de hidrocloruro de amantadina. En algunas formas de realización, el sujeto humano tiene esclerosis múltiple. En algunas formas de realización, la dosis comprende adicionalmente uno o más fármacos seleccionados del grupo que consiste en 4-aminopiridina, baclofeno, dextrometorfano, dimetilfumarato, fingolimod, metilfenidato y teriflunomida, tetrabenizina y tizanidina. En algunas formas de realización, el al menos un excipiente es un excipiente que modifica la liberación. En algunas formas de realización, al menos uno de dichos excipientes (o al menos un excipiente) modifica la liberación de la amantadina o una sal farmacéuticamente aceptable de la misma para proporcionar una forma de liberación prolongada, y donde la administración de la dosis proporciona a) un Tmax para amantadina de 5 a 18 horas, b) un Cmax de 1,0 a 2,8 ng/ml por mg de amantadina, y c) un AUCü-inf de 40 a 75 ng*h/ml por mg de amantadina, medido en un estudio farmacocinético humano de dosis única. En algunas formas de realización, tal dosificación de amantadina da como resultado concentraciones plasmáticas máximas de amantadina que se producen por la mañana o por la tarde, y concentraciones plasmáticas muy bajas por la noche. En algunas formas de realización, el fármaco se proporciona en forma de liberación prolongada. En realizaciones específicas adicionales, la concentración plasmática máxima de amantadina se logra entre 5, 6, 7, 8, 9, 10, 11 o 12 horas hasta aproximadamente 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 o 24 horas después de la administración de una dosis única de la composición (por ejemplo, de 8 horas a aproximadamente 18 horas después de la administración). En algunas formas de realización, la concentración plasmática máxima de amantadina se alcanza entre 5 y 9 horas, entre 5 y 10 horas, entre 5 y 11 horas, entre 5 y 12 horas, entre 5 y 13 horas, entre 5 y 14 horas, entre 5 y 16 horas. horas, entre 5 y 18 horas, entre 5 y 20 horas, entre 5 y 22 horas, entre 5 y 24 horas, entre 6 y 9 horas, entre 6 y 10 horas, entre 6 y 11 horas, entre 6 y 12 horas, entre 6 y 13 horas, entre 6 y 14 horas, entre 6 y 16 horas, entre 6 y 18 horas, entre 6 y 20 horas, entre 6 y 22 horas, entre 6 y 24 horas, después de una dosis única de la composición, entre 7 y 9 horas, entre 7 y 10 horas, entre 7 y 11 horas, entre 7 y 12 horas, entre 7 y 13 horas, entre 7 y 14 horas, entre 7 y 16 horas, entre 7 y 18 horas, entre 7 y 20 horas, entre 7 y 22 horas, entre 7 y 24 horas, entre 8 y 9 horas, entre 8 y 10 horas, entre 8 y 11 horas, entre 8 y 12 horas, entre n 8 y 13 horas, entre 8 y 14 horas, entre 8 y 16 horas,
entre 8 y 18 horas, entre 8 y 20 horas, entre 8 y 22 horas, entre 8 y 24 horas, entre 9 y 10 horas, entre 9 y 11 horas, entre 9 y 12 horas, entre 9 y 13 horas, entre 9 y 14 horas, entre 9 y 16 horas, entre 9 y 18 horas, entre 9 y 20 horas, entre 9 y 22 horas, entre 9 y 24 horas horas, entre 10 y 11 horas, entre 10 y 12 horas, entre 10 y 13 horas, entre 10 y 14 horas, entre 10 y 16 horas, entre 10 y 18 horas, entre 10 y 20 horas, entre 10 y 22 horas, entre 10 y 24 horas, entre 12 y 13 horas, entre 12 y 14 horas, entre 12 y 16 horas, entre 12 y 18 horas, entre 12 y 20 horas, entre 12 y 22 horas, o entre 12 y 24 horas, después de la administración de una dosis única de la composición. En algunas formas de realización descritas en el presente documento, la composición de amantadina se administra a un paciente de 0 a 4 horas antes de acostarse. En algunas formas de realización, la composición de amantadina se administra a un paciente de 0 a 3, de 0 a 2 o de 0 a 1 horas antes de acostarse. En algunas formas de realización, la composición de amantadina se administra a un paciente de 0 a 240 minutos, de 0 a 180 minutos, por ejemplo, de 0 a 120 minutos, de 0 a 60 minutos, de 0 a 45 minutos, de 0 a 30 minutos, de 0 a 15 minutos o de 0 a 10 minutos antes de acostarse. En algunas formas de realización, la composición de amantadina se administra a un paciente de 60 a 240 minutos, de 60 a 180 minutos, de 60 a 120 minutos o de 60 a 90 minutos antes de acostarse. En algunas formas de realización, la velocidad o la capacidad de caminar se evalúa mediante al menos uno de los siguientes o una combinación de los siguientes: prueba de caminata cronometrada de 25 pies (T25FW), prueba de caminata cronometrada (TUG), prueba de caminata de 2 minutos y/o escala para caminar de esclerosis múltiple de doce ítems (MSWS-12). En algunas formas de realización, se mejora significativamente la marcha del sujeto. En algunas formas de realización, se mejoran los síntomas de la EM. En algunas formas de realización, T25FW, TUG, caminata de 2 minutos y/o MSWS-12 se mejoran significativamente (en relación con el placebo). En algunas formas de realización, la administración de la composición a un sujeto con enfermedad de EM da como resultado una reducción significativa en la alteración de la marcha por enfermedad de EM. En una realización específica, la administración de la composición da como resultado al menos aproximadamente 5%, 10%, 15%, 20%, 25%, 30%, 35% o 40%, 45%, 50%, 55% o 60% de reducción en la discapacidad para caminar por la enfermedad de EM. En una realización específica, la administración de la composición da como resultado aproximadamente un 5%, 10%, 15%, 20%, 25%, 30%, 35% o 40%, 45%, 50%, 55% o 60% de reducción en la dificultad para caminar por la enfermedad de EM. En realizaciones específicas adicionales, la reducción en la discapacidad para caminar se mide en una escala numérica que es utilizada o aceptada por la FDA u otras agencias reguladoras para evaluar la efectividad y aprobar medicamentos con licencia para el tratamiento de la discapacidad para caminar. En algunas formas de realización, la composición de una vez al día o una vez por la noche se administra como formas de dosificación de una, dos, tres o cuatro unidades en unidades divididas de manera desigual o, preferiblemente, igualmente. En algunas formas de realización más específicas, la composición se administra como dos o tres formas de dosificación unitarias que comprenden cada una de 85 a 175 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos formas de dosificación unitarias que comprenden cada una de 100 a 175 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos o tres formas de dosificación unitaria de dosificación desigual, o preferiblemente igual, comprendiendo cada una de 85 a 250 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización más específicas, la composición se administra como dos formas de dosificación unitarias, cada una de las cuales comprende de 150 a 180 mg de amantadina, o una sal farmacéuticamente aceptable de la misma.
Formulaciones de liberación prolongada
[0109] Las composiciones de amantadina de liberación prolongada adecuadas para su uso en la invención se pueden realizar utilizando una variedad de tecnologías de liberación prolongada, tales como las descritas en las publicaciones de patentes referenciadas en la sección de antecedentes arriba. En algunas formas de realización, la invención es un aglomerado en forma de dosificación de cápsulas. En algunas formas de realización, los aglomerados comprenden un núcleo de aglomerados, que está revestido con al menos una capa de fármaco y al menos una capa de revestimiento de liberación prolongada. En algunas formas de realización, los aglomerados se recubren con al menos una capa de fármaco, una capa intermedia tal como un revestimiento de sellado y una capa de revestimiento de liberación prolongada. En algunas formas de realización, el aglomerado, la capa de fármaco o ambos comprenden uno o más aglutinantes.
[0110] En algunas formas de realización, la unidad de dosificación comprende una pluralidad de aglomerados revestidos. En algunas formas de realización, los aglomerados tienen un diámetro de, por ejemplo, 300 a 1700 micrómetros, en algunos casos de 500 a 1200 micrómetros. Los aglomerados comprenderán, por ejemplo, sustratos inertes, tales como esferas de azúcar, esferas de celulosa microcristalina (MCC), aglomerados de almidón. En algunas formas de realización, los aglomerados se pueden preparar mediante otros procesos tales como granulación, extrusión, esferonización, etc., o combinaciones de los mismos. Los aglomerados del núcleo comprenderán hidrocloruro de amantadina y excipientes farmacéuticamente aceptables.
Aglomerados recubiertos
[0111] Los núcleos de aglomerados se recubren con el ingrediente activo, por ejemplo, amantadina o una sal farmacéuticamente aceptable y/o polimorfo de la misma. En algunas formas de realización, además del ingrediente activo, los aglomerados también comprenden uno o más aglutinantes, tales como por ejemplo hidroxipropilmetilcelulosa, copovidona, povidona, hidroxipropilcelulosa, hidroxietilcelulosa, metilcelulosa, carboximetilcelulosa, etc. En algunas formas de realización, los aglomerados también contienen uno o más excipientes adicionales, tales como agentes anti pegajosidad (por ejemplo, talco, estearato de magnesio, etc.).
[0112] En algunas formas de realización, los aglomerados de núcleo se recubren con una capa de fármaco que comprende ingrediente activo, y opcionalmente uno o más aglutinantes, agentes anti-pegajosidad y/o disolventes mediante técnicas de revestimiento convencionales tales como revestimiento en lecho fluidizado, revestimiento en bandeja.
Recubrimiento de capa intermedia
[0113] En algunas formas de realización, los aglomerados están recubiertos con una capa intermedia, tal como un recubrimiento de sellado. En algunas formas de realización, el revestimiento de sellado está adaptado para evitar que los ingredientes del revestimiento de liberación prolongada interactúen con los ingredientes del núcleo del aglomerado, para evitar que la migración de los ingredientes del núcleo del aglomerado se difunda fuera del núcleo del aglomerado hacia la capa de liberación prolongada, etc. Como se describe en este documento, la capa de sellado de la presente invención puede comprender uno o más polímeros formadores de película que incluyen, entre otros, hidroxipropilmetilcelulosa (HPMC), copovidona, povidona, polivinilpirrolidona, hidroxipropilcelulosa, hidroxietilcelulosa, metilcelulosa, carboximetilcelulosa o cualquier combinación de los mismos y similares.
[0114] La capa de sellado puede comprender además otros aditivos como plastificantes, tales como, propilenglicol, triacetina, polietilenglicol, citrato de tributilo y, opcionalmente, agentes antiadherentes, tales como, estearato de magnesio, silicato de calcio, silicato de magnesio y dióxido de silicio coloidal o talco.
[0115] Aparte de plastificantes y agentes antiadherentes como se mencionó anteriormente, la capa de sellado puede contener, opcionalmente, tampones, colorantes, opacificantes, agentes tensioactivos o bases, que son conocidos para los expertos en la técnica.
[0116] El recubrimiento de sellado se puede aplicar al núcleo usando técnicas de recubrimiento convencionales tales como recubrimiento de lecho fluidizado, recubrimiento de bandeja, etc. En algunas formas de realización, los núcleos de aglomerados recubiertos con fármaco se recubren con una capa de recubrimiento de sellado que opcionalmente comprende uno o más aglutinantes, agentes anti-pegajosidad y/o disolventes mediante revestimiento en lecho fluidizado o revestimiento en bandeja.
Aglutinantes
[0117] En algunas formas de realización, los núcleos de aglomerados, la capa de revestimiento intermedia, o ambos pueden comprender uno o más aglutinantes (por ejemplo, polímeros, formadores de película). Los aglutinantes adecuados para su uso en la presente invención incluyen, por ejemplo: ácido algínico y sales del mismo; derivados de celulosa tales como carboximetilcelulosa, metilcelulosa (por ejemplo, Methocel®), hidroxipropilmetilcelulosa, hidroxietilcelulosa, hidroxipropilcelulosa (por ejemplo, Klucel®), etilcelulosa (por ejemplo, Ethocel®) y celulosa microcristalina (por ejemplo, Avicel®); dextrosa microcristalina; amilosa; silicato de magnesio y aluminio; ácidos polisacáridos; bentonitas; gelatina; copolímero de polivinilpirrolidona/acetato de vinilo; crospovidona; povidona; almidón; almidón pregelatinizado; tragacanto, dextrina, un azúcar, como sacarosa (por ejemplo, Dipac®), glucosa, dextrosa, melaza, manitol, sorbitol, xilitol (por ejemplo, Xylitab®) y lactosa; una goma natural o sintética como goma arábiga, tragacanto, goma ghatti, mucílago de cáscaras de isapol, polivinilpirrolidona (p. ej., Polyvidone® CL, Kollidon® CL, Polyplasdone® XL-10), alerce arabogalactano, Veegum®, polietilenglicol, ceras, sodio alginato y similares.
Revestimiento de liberación prolongada
[0118] Los aglomerados se recubren con un revestimiento de liberación prolongada. El recubrimiento de liberación prolongada está adaptado para retrasar la liberación del fármaco de los núcleos de fármaco recubiertos durante un período de tiempo después de la introducción de la forma de dosificación en el entorno de uso. En algunas formas de realización, el recubrimiento de liberación prolongada incluye uno o más excipientes de liberación prolongada dependientes o no dependientes del pH. Ejemplos de polímeros de liberación prolongada que no dependen del pH incluyen etilcelulosa, hidroxipropilmetilcelulosa, hidroxietilcelulosa, hidroxipropilcelulosa, carboximetilcelulosa, copolímero de acrilato de etilo, metacrilato de metilo (por ejemplo, Eudragit RS), etc. Ejemplos de excipientes de liberación prolongada dependientes del pH incluyen copolímeros de ácido metacrílico, hidroxipropilmetilcelulosa acetato succinato, ftalato de hidroxipropilmetilo de celulosa, y ftalato de acetato de celulosa, etc. El recubrimiento de liberación prolongada también puede incluir un formador de poros, tal como povidona, polietilenglicol, hidroxipropilcelulosa, hidroxipropilmetilcelulosa, etc., azúcares tales como sacarosa, manitol, lactosa y sales, como cloruro de sodio, citrato de sodio, etc., un plastificante, como ésteres citratados acetilados, glicéridos acetilados, aceite de ricino, ésteres de citrato, sebacato de dibutilo, monoestearato de glicerilo, ftalato de dietilo, glicerol, triglicéridos de cadena media, propilenglicol, polietilenglicol. El revestimiento de liberación prolongada también puede incluir uno o más excipientes adicionales, tales como lubricantes (por ejemplo, estearato de magnesio, talco, etc.).
[0119] El recubrimiento de liberación prolongada puede aplicarse utilizando técnicas de recubrimiento convencionales tales como recubrimiento en lecho fluidizado, revestimiento en bandeja, etc. Los aglomerados de núcleos recubiertos con medicamento, que comprenden opcionalmente un recubrimiento de sellado, se recubren con el recubrimiento de liberación prolongada por recubrimiento en lecho fluidizado.
Excipientes de liberación prolongada (polímeros de recubrimiento)
[0120] Como se describe aquí, a modo de ejemplo excipientes de liberación prolongada incluyen, pero no se limitan a plásticos insolubles, polímeros hidrofílicos y compuestos grasos. Las matrices de plástico incluyen, pero no se limitan a metacrilato de acrilato-metilo de metilo, cloruro de polivinilo y polietileno. Los polímeros hidrófilos incluyen, entre otros, polímeros celulósicos como metil y etilcelulosa, hidroxialquilcelulosas como hidroxipropilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa de sodio y polímeros de ácido acrílico reticulados como Carbopol® 934, óxidos de polietileno y mezclas de los mismos. Los compuestos grasos incluyen, pero no se limitan a diversas ceras tales como cera de carnauba y triestearato de glicerilo y sustancias de tipo cera que incluyen aceite de ricino hidrogenado o aceite vegetal hidrogenado, o mezclas de los mismos.
[0121] En ciertas realizaciones, el material plástico puede ser un polímero acrílico farmacéuticamente aceptable, incluyendo pero no limitado a ácido acrílico y copolímeros de ácido metacrílico, metacrilato de metilo, copolímeros de metacrilato de metilo, metacrilatos de etoxietilo, metacrilato de cianoetilo, metacrilato de aminoalquilo, copolímero de ácido poli(acrílico), ácido poli(metacrílico), copolímero de alquilamina de ácido metacrílico, poli(metacrilato de metilo), ácido poli(metacrílico) (anhídrido), polimetacrilato, poliacrilamida, poli(anhídrido de ácido metacrílico) y copolímeros de metacrilato de glicidilo.
[0122] En ciertas otras realizaciones, el polímero acrílico está compuesto por uno o más copolímeros de metacrilato de amonio. Los copolímeros de metacrilato de amonio son bien conocidos en la técnica y se describen en NF XVII como copolímeros completamente polimerizados de ésteres de ácido acrílico y metacrílico con un bajo contenido de grupos de amonio cuaternario.
[0123] En todavía otras formas de realización, el polímero acrílico es una laca de resina acrílica como el que es comercialmente disponible en Rohm Pharma con el nombre comercial Eudragit®. En realizaciones adicionales, el polímero acrílico comprende una mezcla de dos lacas de resina acrílica disponibles comercialmente de Rohm Pharma con los nombres comerciales Eudragit® RL30D y Eudragit® RS30D, respectivamente. Eudragit® RL30D y Eudragit® RS30D son copolímeros de ésteres acrílicos y metacrílicos con un bajo contenido de grupos amonio cuaternario, siendo la relación molar de grupos amonio a los ésteres (met)acrílicos neutros restantes 1:20 en Eudragit RL30D y 1:40 en Eudragit® RS30D. El peso molecular medio es de aproximadamente 150.000. Eudragit® S-100 y Eudragit® L-100 también son adecuados para su uso aquí. Las designaciones de código RL (alta permeabilidad) y RS (baja permeabilidad) se refieren a las propiedades de permeabilidad de estos agentes. Las mezclas de Eudragit® Rl/RS son insolubles en agua y en fluidos digestivos. Sin embargo, los sistemas multiparticulados formados para incluir los mismos son hinchables y permeables en soluciones acuosas y fluidos digestivos.
[0124] Los polímeros descritos anteriormente, tales como Eudragit® RL/RS pueden mezclarse juntos en cualquier proporción deseada con el fin de, en última instancia, obtener una formulación de liberación prolongada que tiene un perfil de disolución deseable. Un experto en la técnica reconocerá que también se pueden usar otros polímeros acrílicos, tales como, por ejemplo, Eudragit® L.
Formadores de poros
[0125] En algunas formas de realización, el revestimiento de liberación prolongada incluye un formador de poros. Los formadores de poros adecuados para su uso en el revestimiento de liberación prolongada pueden ser agentes orgánicos o inorgánicos e incluyen materiales que se pueden disolver, extraer o lixiviar del revestimiento en el entorno de uso. Los ejemplos de formadores de poros incluyen, pero no se limitan a compuestos orgánicos tales como mono, oligo y polisacáridos que incluyen sacarosa, glucosa, fructosa, manitol, manosa, galactosa, lactosa, sorbitol, pululano, dextrano; polímeros solubles en el entorno de uso tales como polímeros hidrófilos solubles en agua, tales como povidona, crospovidona, polietilenglicol, hidroxipropilcelulosa, hidroxipropilmetilcelulosa, hidroxialquilcelulosas, carboxialquilcelulosas, éteres de celulosa, resinas acrílicas, polivinilpirrolidona, polivinilpirrolidona reticulada, óxido de polietileno, carbowaxes, Carbopol® y similares, dioles, polioles, alcoholes polihídricos, polialquilenglicoles, polietilenglicoles, polipropilenglicoles o sus polímeros en bloque, poliglicoles, poli(a-Q)alquilendioles; compuestos inorgánicos tales como sales de metales alcalinos, carbonato de litio, cloruro de sodio, bromuro de sodio, cloruro de potasio, sulfato de potasio, fosfato de potasio, acetato de sodio, citrato de sodio, sales de calcio adecuadas y similares. En determinadas formas de realización, también se pueden utilizar plastificantes como formadores de poros.
Cápsulas
[0126] Los aglomerados de liberación prolongada se puede introducir en una cápsula adecuada mediante el uso de un encapsulador equipado con cámara de dosificación de aglomerados. Los tamaños de las cápsulas pueden ser 00, 0, 0EL, 1, 1EL, 2, 2EL, 3, 4 o 5. Una composición particularmente preferida que proporciona propiedades farmacocinéticas ideales y perfiles de concentración plasmática es una composición de aglomerados en cápsulas que comprende una pluralidad de aglomerados, que normalmente tienen un diámetro de aproximadamente 500 pm a 1,2 mm, y preferiblemente de aproximadamente 700 pm a 1000 pm, donde cada aglomerado comprende un núcleo que comprende amantadina y un aglutinante, y un recubrimiento de liberación prolongada que rodea el núcleo que extiende la liberación de la amantadina
para proporcionar las propiedades farmacocinéticas y los perfiles de concentración plasmática de amantadina deseados descritos anteriormente.
[0127] En algunas formas de realización, los aglomerados en el aglomerado en cápsula están en un tamaño de 0 o menor, preferentemente un tamaño de 1 o cápsula más pequeña. Los diámetros medios de los aglomerados en algunas formas de realización pueden estar en un rango de 500 pm a 1200 pm, por ejemplo, de 500 pm a 1100 pm, de 500 pm a 1000 |jm, de 500 pm a 900 pm, de 500 pm a 800 pm, de 500 pm a 700 pm, de 600 pm a 1100 pm, de 600 pm a 1000 pm, de 600 pm a 900 pm, de 600 pm a 800 pm, de 600 pm a 700 pm, de 700 pm a 1100 pm, de 700 pm a 1000 pm, de 700 pm a 900 pm, o de 700 pm a 800 pm. En algunas formas de realización, los diámetros medios de partículas son, ±10%, por ejemplo: 500 pm, 550 pm, 600 pm, 650 pm, 700 pm, 750 pm, 800 pm, 850 pm, 900 pm, 950 pm, 1000 pm, 1050 pm, 1100 pm, 1150 pm o 1200 pm.
[0128] Una composición preferida de la invención es una composición aglomerado en cápsula en la que cada aglomerado comprende un núcleo que comprende una semilla núcleo con una mezcla de amantadina y un aglutinante revestido sobre la semilla núcleo, y un recubrimiento de liberación prolongada que rodea el núcleo que comprende etilcelulosa, un agente formador de poros como hidroxipropilmetilcelulosa o povidona, y un plastificante. En algunas formas de realización, los aglomerados pueden comprender además un revestimiento de sellado entre el núcleo del aglomerado y el revestimiento de liberación prolongada. Los aglomerados se formulan usando métodos conocidos en la técnica, tales como los descritos en el Ejemplo 1 a continuación. En una realización específica, basada en el peso combinado del núcleo del aglomerado y el recubrimiento de liberación prolongada, la amantadina está presente en cantidades de 20-80% en peso, 45-70% en peso, 40-50% en peso, 45-55% en peso, 50-60% en peso, 55-65% en peso, 60-70% en peso, 65-75% en peso, 70-80% en peso, o 40 a 60% en peso, el aglutinante, que es preferiblemente hidroxipropilmetilcelulosa, copovidona o mezclas de los mismos, está presente en cantidades de 1 a 25% en peso, la semilla central, preferiblemente una esfera de azúcar (sin igual) o semilla de celulosa microcristalina (por ejemplo, Celphere®), está presente en cantidades de 8 a 25% en peso, la etilcelulosa está presente en cantidades de 10 a 20% en peso, el agente formador de poros, preferiblemente povidona, está presente en cantidades de 1 a 4% en peso, y el plastificante está presente en cantidades de 1 a 4% en peso. En otra realización específica, basada en el peso combinado del núcleo del aglomerado y el recubrimiento de liberación prolongada, la amantadina está presente en cantidades de 50 a 70% en peso, el aglutinante, que es preferiblemente hidroxipropilmetilcelulosa, copovidona o mezclas de los mismos, presente en cantidades de 1 a 25% en peso, la semilla central, preferiblemente una esfera de azúcar (sin igual) o semilla de celulosa microcristalina (por ejemplo, Celphere®), está presente en cantidades de 5 a 15% en peso, la etilcelulosa está presente en cantidades de 1 a 15% en peso, el agente formador de poros, preferiblemente povidona, está presente en cantidades de 0,25 a 4% en peso, y el plastificante está presente en cantidades de 0,25 a 4% en peso.
[0129] Las realizaciones adicionales de la invención se ilustran en la Tabla 1, a continuación, titulada "Varias formulaciones de amantadina de LP de cápsulas de tamaño 1". Por medio de los métodos y composiciones descritos en el presente documento, se pueden preparar formulaciones que logren las características de disolución deseadas y los perfiles farmacocinéticos diana descritos en el presente documento. Más específicamente, se pueden administrar dosis terapéuticamente eficaces de amantadina una vez por la noche en no más de dos cápsulas de tamaño 1 (o más pequeñas, por ejemplo, tamaño 2 o 3) usando los métodos de fabricación y composiciones que se han descrito en este documento para lograr estos resultados. En particular, se puede lograr una carga de fármaco más alta usando composiciones y métodos de fabricación descritos en este documento. En algunas formas de realización, se puede lograr una carga de fármaco más alta, con el perfil de disolución requerido, usando tamaños de aglomerados de núcleo más pequeños y capas de fármaco aumentadas concomitantemente en núcleos más pequeños, pero sin cambios en la capa de liberación prolongada. En algunas formas de realización, utilizando enfoques de fabricación alternativos descritos en este documento, por ejemplo, extrusión y esferonización, se pueden lograr cargas de fármaco incluso más altas para realizar el perfil de disolución deseado, lo que permite altas cargas de fármaco de amantadina con perfiles farmacocinéticos adecuados, dando como resultado composiciones que son terapéuticamente más eficaces y al menos igualmente bien toleradas, y pueden llenarse en cápsulas de tamaño relativamente pequeño (por ejemplo, tamaño 1, 2 o 3), lo que permite una fácil administración a los pacientes.
Tabla 1: Varias formulaciones de amantadina de LP de cápsulas de tamaño 1

(Continuación)

[0130] En alguna realización, la amantadina, o una sal farmacéuticamente aceptable de la misma, está presente en cantidades de 20 a 80% en peso (basado en el peso combinado del núcleo del aglomerado y el recubrimiento de liberación prolongada), con una densidad aparente de 0,3 a 1,2 g/cm3 En algunas formas de realización, la amantadina o su sal farmacéuticamente aceptable está presente en cantidades del 20 al 77,5% en peso, del 20 al 75% en peso, del 20 al 72,5% en peso, del 20 al 70% en peso, del 20 al 67,5% en peso, del 20 al 65% en peso, del 20 al 62,5% en peso, del 20 al 60% en peso, del 20 al 57,5% en peso, del 20 al 55% en peso, del 20 al 52,5% en peso, del 20 al 50% en peso, del Del 20 al 47,5% en peso, del 20 al 45% en peso, del 20 al 42,5% en peso, del 20 al 40% en peso, del 20 al 37,5% en peso, del 20 al 35% en peso, del 20 al 32,5% en peso, del 20 al 30% en peso, del 30 al 80% en peso, del 30 al 77,5% en peso, del 30 al 75% en peso, del 30 al 72,5% en peso, del 30 al 70% en peso, del 30 al 67,5% en peso, del 30 al 65% en peso, del 30 al 62,5% en peso, del 30 al 60% en peso, del 30 al 57,5% en peso, del 30 al 55% en peso, del 30 al 52,5% en peso, del 30 al 50% en peso, del 30 al 47,5% en peso, del 30 al 45% en peso, del 30 al 42,5% en peso, del 30 al 40% en peso, del 40 al 80% en peso, del 40 al 77,5% en peso, del 40 al 75% en peso, del 40 al 72,5% en peso%, del 40 al 70% en peso, del 40 al 67,5 en peso%, del 40 al 65% en peso, del 40 al 62,5% en peso, del 40 al 60% en peso, del 40 al 57,5% en peso, del 40 al 55% en peso, del 40 al 52,5% en peso, del 40 al 50% en peso, del 40 al 47,5% en peso, del 40 al 45% en peso, del 50 al 80% en peso, del 50 al 77,5% en peso, del 50 al 75% en peso, del 50 al 72,5% en peso, del 50 al 70% en peso, de 50 a 67,5% en peso, de 50 a 65% en peso, de 50 a 62,5% en peso, de 50 a 60% en peso, de 50 a 57,5% en peso, de 50 a 55% en peso, de 60 a 80% en peso, de 60 a 77,5% en peso, 60 a 75% en peso, 60 a 72,5% en peso, 60 a 70% en peso, 60 a 67,5% en peso, 60 a 65% en peso. En algunas formas de realización, la densidad aparente es de 0,3 a 1,2 g/cm3, 0,3 a 1,15 g/cm3, 0,3 a 1,1 g/cm3, 0,3 a 1,05 g/cm3, 0,3 a 1,0 g/cm3, 0,3 a 0,9 g/cm3, 0,3 a 0,8 g/cm3, 0,3 a 0,7 g/cm3, 0,3 a 0,6 g/cm3, 0,3 a 0,5 g/cm3, 0,3 a 0,4 g/cm3, 0,4 a 1,2 g/cm3, 0,4 a 1,15 g/cm3, 0,4 a 1,1 g/cm3, 0,4 a 1,05 g/cm3, 0,4 a 1,0 g/cm3, 0,4 a 0,9 g/cm3, 0,4 a 0,8 g/cm3, 0,4 a 0,7 g/cm3, 0,4 a 0,6 g/cm3, 0,4 a 0,5 g/cm3, 0,5 a 1,2 g/cm3, 0,5 a 1,15 g/cm3, 0,5 a 1,1 g/cm3, 0,5 a 1,05 g/cm3, 0,5 a 1,0 g/cm3, 0,5 a 0,9 g/cm3, 0,5 a 0,8 g/cm3, 0,5 a 0,7 g/cm3, 0,5 a 0,6 g/cm3, 0,6 a 1,2 g/cm3, 0,6 a 1,15 g/cm3, 0,6 a 1,1 g/cm3, 0,6 a 1,05 g/cm3, 0,6 a 1,0 g/cm3, 0,6 a 0,9 g/cm3, 0,6 a 0,8 g/cm3, 0,6 a 0,7 g/cm3, 0,7 a 1,2 g/cm3, 0,7 a 1,15 g/cm3, 0,7 a 1,1 g/cm3, 0,7 a 1,05 g/cm3, 0,7 a 1,0 g/cm3, 0,7 a 0,9 g/cm3, 0,7 a 0,8 g/cm3, 0,5 a 1,2 g/cm3, 0,8 a 1,15 g/cm3, 0,8 a 10,1 g/cm3, 0,8 a 1,05 g/cm3, 0,8 a 1,0 g/cm3, 0,8 a 0,9 g/cm3, 0,9 a 1,2 g/cm3, 0,9 a 1,15 g/cm3, 0,9 a 1,1 g/cm3, 0,9 a 1,05 g/cm3 o 0,9 a 1,0 g/cm3. En algunas formas de realización, la composición está en una unidad de dosificación que comprende un aglomerado en una formulación de cápsula, en donde el tamaño de la cápsula es de tamaño 00, tamaño 0, tamaño 1, tamaño 2 o tamaño 3. En algunas formas de realización preferidas, la unidad de dosificación incluye aglomerados que contienen de 50 a 250 mg de amantadina en un tamaño de 0, 1, 2 o 3 cápsulas. En algunas formas de realización, la unidad de dosificación incluye aglomerados que contienen de 100 a 250 mg, por ejemplo, de 100 a 200 mg de amantadina en una cápsula de tamaño 0, 1, 2 o 3, preferiblemente una cápsula de tamaño 1, 2 o 3. En algunas formas de realización, la unidad de dosificación comprende aproximadamente 110, 120, 130, 140, 150, 160, 170, 180, 190, 210 o 220 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la unidad de dosificación comprende 110 mg de hidrocloruro de amantadina. En algunas formas de realización, la unidad de dosificación comprende 130 mg de hidrocloruro de amantadina. En algunas formas de realización, la unidad de dosificación comprende 170 mg de hidrocloruro de amantadina. En algunas formas de realización, la unidad de dosificación comprende 210 mg de hidrocloruro de amantadina.
[0131] Los plastificantes adecuados incluyen triglicéridos de cadena media, ftalato de dietilo, ésteres de citrato,
polietilenglicol, glicerol, glicéridos acetilados, aceite de ricino y similares. Los aglomerados se envasan en cápsulas para proporcionar la concentración deseada de amantadina. Una ventaja de esta composición es que proporciona las propiedades de liberación deseadas que hacen que la composición sea adecuada para su administración durante dicho período antes de acostarse. Una ventaja adicional es que el recubrimiento de liberación prolongada es lo suficientemente duradero para que la cápsula se pueda abrir y los aglomerados se pueden esparcir sobre los alimentos para su administración a pacientes que tienen dificultad para tragar las píldoras, sin afectar adversamente las propiedades de liberación de la composición. Cuando la composición se administra espolvoreando sobre alimentos, se prefiere utilizar un alimento blando como puré de manzana o pudín de chocolate, que se consume en 30 minutos, y preferiblemente en 15 minutos. Una ventaja adicional más de la composición descrita anteriormente es que tiene muy buena reproducibilidad de lote a lote y estabilidad de vida útil.
[0132] En realizaciones adicionales, 110 mg a 210 mg de amantadina de LP en una cápsula de tamaño 1 de la composición de la invención tiene un perfil de disolución in vitro de la amantadina de no más de 25% a las 2 horas, 55-85% a 6 horas, y al menos el 80% a las 12 horas, medido usando un Aparato II USP (Paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. Más preferiblemente, la disolución in vitro se caracteriza además por una liberación de amantadina de no más del 10% a 1 hora, 30-50% a las 4 horas y al menos 90% a las 12 horas.
[0133] En algunas formas de realización, la composición tiene un perfil de disolución in vitro de amantadina que muestra al menos uno de (i) no más de 25% de disolución a las 2 horas, (ii) no más de 25-55% de disolución en 6 horas y (iii) disolución de al menos 80% a las 12 horas, usando un Aparato II USP (Paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, se cumplen dos de los criterios (i), (ii) y (iii). En algunas formas de realización, se cumplen los tres criterios (i), (ii) y (iii).
[0134] En algunas formas de realización de cualquiera de los aspectos anteriores, la composición tiene un perfil de disolución in vitro de amantadina que muestra al menos uno de (i) no más del 20% de disolución a 1 hora, (ii) aproximadamente 25-45% de disolución a las 2 horas, (iii) no más de 50-80% de disolución a las 4 horas, y (iii) al menos 80% de disolución a las 8 horas, usando un Aparato II USP (Paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, se cumplen dos de los criterios (i), (ii) y (iii). En algunas formas de realización, se cumplen los tres criterios (i), (ii) y (iii).
[0135] En algunas formas de realización de cualquiera de los aspectos anteriores, la composición tiene un perfil de disolución in vitro de amantadina que muestra al menos uno de (i) aproximadamente 35-55% de disolución a las 2 horas, (ii) no más de 60-80% de disolución a las 4 horas, y (iii) al menos 90% de disolución a las 8 horas, usando un Aparato II USP (Paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, se cumplen dos de los criterios (i), (ii) y (iii). En algunas formas de realización, se cumplen los tres criterios (i), (ii) y (iii).
[0136] Una composición de aglomerado en cápsula de la invención se prefiere, además de tener las propiedades anteriores de disolución in vitro y cualquiera de las propiedades farmacocinéticas se ha descrito anteriormente (por ejemplo, perfil de liberación in vivo, Tmax, ratio Cmax/Cmin, etc.) que hacen que la composición sea adecuada para su administración en dicho período antes de acostarse. La composición se caracteriza además por proporcionar un Cmax de 1,6-2,4 ng/ml por mg de amantadina y un AUCü.inf de 40-75 ng*h/ml por mg de amantadina después de la administración oral de una dosis única de la cápsula a un sujeto humano en ayunas. Una composición preferida de aglomerados en cápsulas se caracteriza además por una concentración plasmática en estado estacionario en la que la administración oral una vez por la noche de la cápsula a un sujeto humano proporciona un Cmax de 2,4 a 4,2 ng/ml por mg de amantadina, una Cmin de 1,1 a 2,6 ng/ml por mg de amantadina y un AUC0-24 de 48-73 ng*h/ml por mg de amantadina.
[0137] Las composiciones de aglomerado en cápsula descritas anteriormente pueden proporcionarse en una resistencia adecuada para la terapia de amantadina. Las concentraciones típicas oscilan entre al menos aproximadamente 50 mg y aproximadamente 250 mg. En una realización específica, la concentración de la cápsula es de 70 mg, 80 mg, 85 mg, 90 mg, 110 mg, 120 mg, 125 mg, 130 mg, 140 mg, 150 mg, 160 mg, 160 mg, 170 mg, 180 mg, 190 mg, 210 mg y 220 mg, que proporciona una dosis única de AUCü.inf por mg que equivale a una tableta de 100 mg de una formulación de liberación inmediata de amantadina HC1 (p. ej., Symmetrel® u otra referencia del libro naranja de la FDA incluido fármaco). Se pueden administrar una, dos o tres de tales cápsulas a un sujeto en el período antes de acostarse. En una realización preferida, se administran entre 220 mg y 650 mg de amantadina usando 2 cápsulas de formulaciones LP adecuadas una vez por la noche.
Otras formas de dosificación de liberación prolongada
[0138] La persona experta en la técnica reconocerá que otras formas de realización de composiciones de liberación prolongada pueden estar previstas, además de la formulación de cápsulas descrita anteriormente. Dichas otras realizaciones incluyen formas de dosificación sólidas de liberación prolongada, tales como tabletas, cápsulas, cápsulas de gel, polvos, aglomerados, perlas, etc. Incluidas en tales composiciones de liberación prolongada están aquellas que tienen las características de liberación y el perfil farmacocinético in vivo para ser empleadas en la invención. En algunas formas de realización, la persona experta en la técnica puede emplear, con el ajuste apropiado de las características de diseño para lograr el perfil farmacocinético necesario descrito en este documento, la tecnología de liberación prolongada descrita en la Patente de los EE. UU. N° 5,358,721, de Guittard et al., o la Patente de los EE. UU. N° 6,217,905, de Edgren
et al., o la patente de EE. UU. N° 8,574,626, de Vergez et al., cada una de las cuales describe una forma de dosificación osmótica oral de amantadina. En otras realizaciones, la persona experta en la técnica puede emplear, de nuevo con el ajuste apropiado de las características de diseño, la tecnología descrita en la Patente de EE. UU. N° 6,194,000, de Smith et al., Patente N° 8,741.343 o Solicitud de Patente de EE. UU. Nos de publicación US 2006/0252788, US 2006/0189694, US 2006/0142398, US 2008/0227743 US2011/0189273, US2015/0045446, US2015/0051292 o US2014/0242163, todos a Went et al., cada uno de los cuales describe la administración de un antagonista del receptor de NMDA, tal como amantadina, opcionalmente en forma de liberación controlada.
[0139] Algunas formas de realización proporcionan una dosis de la invención para uso en un método de administración de una vez cada noche de amantadina (o una sal farmacéuticamente aceptable de la misma, tal como hidrocloruro de amantadina) a un sujeto en necesidad del mismo, comprendiendo dicho método administrar oralmente una liberación prolongada (LP) que comprende amantadina, o una sal farmacéuticamente aceptable de la misma, menos de cuatro horas antes de acostarse (y/o después de las 16:00). En algunas formas de realización, la administración se produce menos de cuatro horas antes de acostarse. En algunas formas de realización, el método mejora la impresión global del médico y lo hace sin inducir o aumentar las alteraciones del sueño en el paciente. En algunas formas de realización, la composición se agrega a los alimentos antes de la administración. En algunas formas de realización, no hay aumento en la concentración plasmática de amantadina durante al menos una hora después de la administración. En algunas formas de realización, no hay aumento en la concentración plasmática de amantadina durante al menos dos horas después de la administración. En algunas formas de realización, la amantadina tiene una Tmax de dosis única de 9 a 18 horas y/o una Tmax en estado estacionario de 7 a 13 horas. En algunas formas de realización, la amantadina tiene una Tmax de dosis única de 12 a 18 horas después de la administración, y/o una Tmax en estado estacionario de 8 a 12 horas. En algunas formas de realización, la amantadina tiene una Tmax de dosis única de 12 a 16 horas después de la administración y/o una Tmax en estado estacionario de 9 a 12 horas. En algunas formas de realización, una administración oral una vez por la noche de la composición a un sujeto humano proporciona un perfil de concentración plasmática en estado estacionario caracterizado por un aumento de la concentración de amantadina de menos del 25% a las tres horas de la administración. En algunas formas de realización, la curva PK tiene una relación Cmax/Cmin de 1,3 a 3,0. En algunas formas de realización, la relación C-media-día/C-media-noche en estado estacionario es de 1,4 a 1,7. En algunas formas de realización, la concentración plasmática media de amantadina durante el día (C-media-día) en estado estacionario es de 500-2000 ng/ml. En algunas formas de realización, la amantadina es clorhidrato de amantadina o sulfato de amantadina. En algunas formas de realización, la composición comprende de 260 a 420 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos, tres o cuatro formas de dosificación unitarias que comprenden cada una de 85 a 175 mg de amantadina, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición se administra como dos formas de dosificación unitarias que comprenden cada una de 130 a 210 mg de amantadina de liberación prolongada, o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición está dentro de una cápsula de tamaño de cápsula n° 1. En algunas formas de realización, la composición comprende de 260 mg a 340 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende 340 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición comprende 170 mg de hidrocloruro de amantadina. En algunas formas de realización, la administración oral de una dosis única de la composición a un sujeto humano en ayunas proporciona una concentración plasmática máxima (Cmax) de 1,1 a 1,7 ng/ml por mg de amantadina, y un AUCü-inf de 46 a 56 ng*h/mL por mg de amantadina. En algunas formas de realización, la administración oral una vez por la noche de una dosis de la composición a un sujeto humano proporciona un perfil de concentración plasmática en estado estacionario caracterizado por: (a) un Cmax de 2,0 a 3,1 ng/ml por mg de amantadina; (b) una Cmin de 1,3 a 2,0 ng/ml por mg de amantadina, y (c) un AUC0-24 de 46 a 56 ng*h/ml por mg de amantadina. En algunas formas de realización, el perfil de concentración plasmática en estado estacionario se caracteriza además por: (d) ningún aumento en la concentración plasmática de amantadina durante al menos una hora después de la administración; y (e) una relación Cmax/Cmin de 1,3 a 3,0. En algunas formas de realización, el perfil de concentración plasmática en estado estacionario se caracteriza además por: (f) ningún aumento en la concentración de amantadina durante al menos dos horas después de la administración; y (g) una relación Cmax/Cmin de 1,4 a 2,6.
[0140] En algunas formas de realización, la composición tiene un perfil de disolución in vitro de la amantadina de no más de 25% a las 2 horas, 55-85% a las 6 horas, y al menos 80% a las 12 horas, usando un aparato II USP (Paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, la composición tiene un perfil de disolución in vitro de amantadina de no más del 25% a las 2 horas, 25-55% a las 6 horas y al menos 80% a las 12 horas, usando un Aparato II de la USP (paletas) en 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, la composición tiene un perfil de disolución in vitro de amantadina de no más del 20% a 1 hora, 25-45% a las 2 horas, 50-80% a las 4 horas y al menos 80% a las 8 horas, usando un aparato II USP (paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, el perfil de disolución in vitro de amantadina se caracteriza además por una liberación de amantadina de no más del 10% a 1 hora, 30-50% a las 4 horas y al menos 90% a las 12 horas. En algunas formas de realización, las propiedades de disolución in vitro facilitan las propiedades farmacocinéticas descritas en el presente documento. En algunas formas de realización, la composición tiene un perfil de AUC después de la administración de una dosis única de la composición caracterizado por: un AUC fraccional de 0 a 4 horas que es menos del 5% de AUCo-inf; un AUC fraccional de 0 a 8 horas que es aproximadamente del 5 al 15% del AUCü.inf; un AUC fraccional de 0 a 12 horas que es aproximadamente del 10 al 40% del AUCo-inf; un AUC fraccional de 0 a 18 horas que es aproximadamente del 25 al 60% del AUCo-inf; y un AUC fraccional de 0 a 24 horas
que es aproximadamente del 40 al 75% del AUCo-inf. En algunas formas de realización, la composición tiene un perfil de AUC después de la dosificación una vez por la noche de la composición en condiciones de estado estacionario caracterizado por: un AUC fraccional de 0 a 4 horas que es aproximadamente del 2 al 25% del AUC0-24; un AUC fraccional de 0 a 8 horas que es aproximadamente del 15 al 50% del AUC0-24; un AUC fraccional de 0 a 12 horas que es aproximadamente del 30 al 70% del AUC0-24; y un AUC fraccional de 0 a 18 horas que es aproximadamente del 60 al 95% del AUC0-24.
[0141] Algunas formas de realización proporcionan una composición farmacéutica para cualquiera de los métodos descritos en el presente documento, en donde dicha composición es para la administración oral y comprende al menos una cápsula para administración oral, comprendiendo dicha cápsula una pluralidad de aglomerados, comprendiendo cada aglomerado: (a) un núcleo de aglomerado que comprende amantadina, o una sal farmacéuticamente aceptable del mismo, y (b) un recubrimiento de liberación prolongada que rodea el núcleo del aglomerado. En algunas formas de realización, la composición comprende dos de dichas cápsulas. En algunas formas de realización, el revestimiento de liberación prolongada comprende etilcelulosa, al menos uno de entre povidona e hidroxipropilmetilcelulosa y un plastificante. En algunas formas de realización, el núcleo de la pastilla comprende amantadina, o una sal farmacéuticamente aceptable de la misma, y un aglutinante revestido sobre una semilla de núcleo. En algunas formas de realización, basado en el peso combinado del núcleo del aglomerado y el recubrimiento de liberación prolongada, la amantadina está presente en cantidades del 40 al 60% en peso, el aglutinante está presente en cantidades del 8 al 25% en peso, la semilla del núcleo está presente en cantidades del 1 al 25% en peso, la etilcelulosa está presente en cantidades del 10 al 20% en peso, la povidona está presente en cantidades del 1 al 4% en peso y el plastificante está presente en cantidades del 1 al 4% en peso. En algunas formas de realización, la composición comprende además un revestimiento de sellado entre el núcleo de la pastilla y el revestimiento de liberación prolongada. En algunas formas de realización, el núcleo de aglomerado comprende un aglutinante seleccionado del grupo que consiste en hidroxipropilmetilcelulosa, copovidona y mezclas de los mismos. En algunas formas de realización, el plastificante se selecciona del grupo que consiste en triglicéridos de cadena media, ftalato de dietilo, ésteres de citrato, polietilenglicol, glicerol, glicéridos acetilados y aceite de ricino.
[0142] Algunas formas de realización proporcionan una dosis de la invención para uso en un método de administración de amantadina, o una sal farmacéuticamente aceptable de la misma, a un sujeto humano en necesidad del mismo, comprendiendo dicho método administrar oralmente una composición farmacéutica que comprende amantadina en al menos una cápsula para administración oral (por ejemplo, una cápsula), comprendiendo dicha cápsula una pluralidad de aglomerados, comprendiendo cada aglomerado: (a) un núcleo de aglomerado que comprende amantadina, o una sal farmacéuticamente aceptable de la misma, y (b) un recubrimiento de liberación prolongada que rodea el aglomerado centro. En algunas formas de realización, la composición comprende dos de dichas cápsulas. En algunas formas de realización, el revestimiento de liberación prolongada comprende etilcelulosa, al menos una de entre povidona e hidroxipropilmetilcelulosa y un plastificante. En algunas formas de realización, el núcleo de la pastilla comprende amantadina, o una sal farmacéuticamente aceptable de la misma, y un aglutinante revestido sobre una semilla de núcleo. En algunas formas de realización, basado en el peso combinado del núcleo del aglomerado y el recubrimiento de liberación prolongada, la amantadina está presente en cantidades del 40 al 60% en peso, el aglutinante está presente en cantidades del 8 al 25% en peso, la semilla del núcleo está presente en cantidades del 1 al 25% en peso, la etilcelulosa está presente en cantidades del 10 al 20% en peso, la povidona está presente en cantidades del 1 al 4% en peso y el plastificante está presente en cantidades del 1 al 4% en peso. En algunas formas de realización, la composición comprende además un revestimiento de sellado entre el núcleo de la pastilla y el revestimiento de liberación prolongada. En algunas formas de realización, el núcleo del aglomerado comprende un aglutinante seleccionado del grupo que consiste en hidroxipropilmetilcelulosa, copovidona y mezclas de los mismos. En algunas formas de realización, el plastificante se selecciona del grupo que consiste en triglicéridos de cadena media, ftalato de dietilo, ésteres de citrato, polietilenglicol, glicerol, glicéridos acetilados y aceite de ricino.
[0143] Algunas formas de realización proporcionan una composición farmacéutica adecuada para la administración una vez al día por vía oral a un paciente en necesidad del mismo de dicha composición que comprende una cantidad terapéuticamente eficaz de la amantadina o una sal farmacéuticamente aceptable de la misma en una forma de liberación prolongada que se puede administrar como no más de dos cápsulas de tamaño 0 o más pequeñas en una sola administración diaria. En algunas formas de realización, la composición comprende 110-220 mg de amantadina o una sal farmacéuticamente aceptable de la misma. En algunas formas de realización, la composición tiene un perfil de disolución in vitro de amantadina de no más del 25% a las 2 horas, 40-80% a las 6 horas y al menos 80% a las 12 horas, usando un Aparato II de la USP (paletas) en 50 rpm con 500 ml de agua a 37°C como medio de disolución. En algunas formas de realización, la composición comprende una pluralidad de aglomerados, comprendiendo cada aglomerado: (a) un núcleo de aglomerados que comprende amantadina, o una sal farmacéuticamente aceptable de la misma, y (b) un recubrimiento de liberación prolongada que rodea el núcleo de aglomerados. En algunas formas de realización, el revestimiento de liberación prolongada comprende etilcelulosa, al menos una de entre povidona e hidroxipropilmetilcelulosa y un plastificante. En algunas formas de realización, el núcleo de la pastilla comprende amantadina, o una sal farmacéuticamente aceptable de la misma, y un aglutinante revestido sobre una semilla de núcleo. En algunas formas de realización, la composición comprende amantadina y, basándose en el peso combinado del núcleo de la pastilla y el recubrimiento de liberación prolongada, la amantadina está presente en cantidades de 40 a 70% en peso. En algunas formas de realización, el núcleo de la pastilla comprende una semilla de núcleo que comprende azúcar o celulosa microcristalina que tiene entre 100 y 500 micrómetros de diámetro. En algunas formas de realización, la densidad aparente está comprendida entre 0,5 y 1 g/cm3. En algunas formas de realización, la composición comprende un revestimiento de
sellado entre el núcleo de la pastilla y el revestimiento de liberación prolongada. En algunas formas de realización, el núcleo del aglomerado comprende un aglutinante seleccionado del grupo que consiste en hidroxipropilmetilcelulosa, copovidona y mezclas de los mismos. En algunas formas de realización, el plastificante se selecciona del grupo que consiste en triglicéridos de cadena media, ftalato de dietilo, ésteres de citrato, polietilenglicol, glicerol, glicéridos acetilados y aceite de ricino.
[0144] Algunas formas de realización en el presente documento proporcionan una dosis de la invención para uso en un método para reducir las alteraciones del sueño en un sujeto humano sometido a tratamiento con amantadina, comprendiendo dicho método una vez cada noche la administración de una composición de liberación prolongada (LP) que comprende amantadina, o una sal farmacéuticamente aceptable de la misma, menos de cuatro horas antes de acostarse (y/o después de las 16:00). En algunas formas de realización, la composición se agrega a los alimentos antes de la administración. En algunas formas de realización, no hay aumento en la concentración plasmática de amantadina durante al menos una hora después de la administración. En algunas formas de realización, la composición se agrega a los alimentos antes de la administración. En algunas formas de realización, no hay aumento en la concentración plasmática de amantadina durante al menos una hora después de la administración. En algunas formas de realización, no hay aumento en la concentración plasmática de amantadina durante al menos dos horas después de la administración.
[0145] En algunas formas de realización, una administración diaria de la composición de la amantadina (o combinación que comprende amantadina) proporciona una mejora en un trastorno del movimiento hipocinético, tales como la marcha. La mejora puede determinarse mediante métodos conocidos en la técnica, tales como prueba de caminata cronometrada (TUG), prueba de caminata cronometrada de 25 pies (T25FW) o prueba de caminata cronometrada de seis minutos (6MTW). En pacientes con esclerosis múltiple, la composición o combinación de amantadina que comprende amantadina proporciona una mejora en la fatiga que puede ser determinada por la escala de fatiga cognitiva motora (FSMC). En pacientes con depresión, la composición o combinación de amantadina que comprende amantadina proporciona una mejora en la depresión que puede ser determinada por el Inventario de Depresión de Beck-2 (BDI-2). En pacientes con esclerosis múltiple, la composición o combinación de amantadina que comprende amantadina proporciona una mejora en la marcha que puede determinarse mediante la prueba de marcha cronometrada de 25 pies (T25FW), la prueba de marcha cronometrada (TUG), la prueba de marcha de 2 minutos y/o, escala de caminata de esclerosis múltiple de doce ítems (MSWS-12). En pacientes con esclerosis múltiple, la composición o combinación de amantadina que comprende amantadina proporciona una mejora en la cognición que puede ser determinada por la Prueba de Modalidades de Dígitos de Símbolo (SDMT), Prueba de Aprendizaje Verbal de California segunda edición (CVLT-II) con recuerdo retardado, Prueba Revisada de Memoria Visuoespacial Breve (BVMT-R) con recuperación diferida y/o evaluación cognitiva internacional breve para la esclerosis múltiple (BICAMS). Por ejemplo, en algunas formas de realización la presente invención puede proporcionar una mejora según lo determinado por la evaluación en el ejemplo 10 y/o el ejemplo 11.
[0146] La presente invención puede entenderse mejor por referencia a los siguientes ejemplos.
Ejemplo 1: Formulaciones de aglomerados recubiertos de amantadina de liberación prolongada
[0147] Las composiciones de aglomerados recubiertos de amantadina HCl de liberación prolongada diseñadas para administración nocturna se prepararon usando los componentes y las cantidades relativas mostradas en la Tabla 2, a continuación. Para cada composición, la solución de revestimiento de fármaco se preparó añadiendo HPMC 5 cps y copovidona a alcohol isopropílico con agitación continua. Se añadió agua purificada a esta dispersión y se continuó agitando hasta que se formó una solución transparente. A continuación, se añadió fármaco (Amantadine HCl) a esta solución aglutinante y se continuó agitando hasta que el fármaco se disolvió por completo. Finalmente, se añadió talco y se dispersó uniformemente mediante agitación.
[0148] Se cargaron perlas de Celphere (tamaños de cribado del n° 35 al n° 50, es decir, de 300 a 500 micrómetros) en una unidad de revestimiento Wurster. La dispersión de recubrimiento de fármaco se pulverizó sobre las perlas seguido de un período de secado. Los aglomerados resultantes revestidos con fármaco se tamizaron para retener la fracción entre los tamices n° 18 y n° 24 (aproximadamente 700 pm a 1 mm de diámetro).
[0149] La solución de sellado de recubrimiento se preparó mediante la adición de HPMC 5 cps a alcohol isopropílico con agitación continua. Se añadió agua purificada a esta dispersión y se continuó agitando hasta que se formó una solución transparente. Se añadió talco y se dispersó uniformemente mediante agitación. Los aglomerados recubiertos con fármaco tamizados se cargaron en una unidad de recubrimiento Wurster. La dispersión de revestimiento de sellado se pulverizó sobre los aglomerados revestidos con fármaco seguido de un período de secado para eliminar el disolvente residual y el agua de los aglomerados. Los aglomerados resultantes revestidos con sello se tamizaron para retener la fracción entre los tamices n° 18 y n° 24.
[0150] La solución de LP de recubrimiento se preparó disolviendo etilcelulosa (viscosidad 7 cps) en alcohol isopropílico y agua purificada y agitar hasta que se formó una solución clara. A continuación, se disolvió povidona K-90 en esta solución transparente seguido de la adición de plastificante Miglyol 812N con agitación continua para formar una solución transparente. Los aglomerados revestidos con sello tamizado se cargaron en una unidad de revestimiento Wurster. La solución de revestimiento LP se pulverizó sobre los aglomerados revestidos con sello seguido de un período de secado para afectar al revestimiento LP y eliminar el disolvente residual y el agua en los aglomerados. Después de secar, se
extendió estearato de magnesio sobre el lecho superior de los aglomerados revestidos en la región del anillo seguido de la recirculación de los aglomerados en la unidad Wurster para mezclar el estearato de magnesio con los aglomerados revestidos. Los aglomerados resultantes revestidos con LP se tamizaron para retener la fracción entre los tamices n° 18 y n° 24.
[0151] El peso deseado de los aglomerados revestidos LP que contienen la dosis unitaria se introdujeron en vacío 1 de gelatina dura cubierta de la cápsula (tamaño 1 durante 60 - 140 mg fuerza) utilizando un encapsulador equipado con cámara de dosificación de aglomerados.
Tabla 2: Composición de las cápsulas de amantadina HCI LP

[0152] La disolución in vitro de cápsulas preparada anteriormente se ensayó usando un aparato II USP (paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución. Las cápsulas que cumplen las especificaciones de disolución deseadas liberan no más del 25% del fármaco en 2 horas, 40-80% en 6 horas y al menos 80% a las 12 horas. En un perfil de disolución ejemplar, hubo 0% de liberación de fármaco a 1 hora, 12% de liberación a las 2 horas, 43% de liberación a las 4 horas, 68% de liberación a las 6 horas, 83% de liberación a las 8 horas, 92% de liberación a las 10 horas y 97% de liberación a las 12 horas. Las cápsulas preparadas de acuerdo con el método anterior exhibieron una buena estabilidad en almacenamiento y reproducibilidad de lote a lote al escalar.
Ejemplo 2: La formulación de aglomerados recubiertos de amantadina de liberación prolongada con una mayor carga de fármaco
[0153] Las composiciones de aglomerados recubiertos de amantadina HC1 de liberación prolongada diseñadas para la administración durante la noche se preparan usando los componentes y cantidades relativas que se muestran en la Tabla 3 a continuación y el proceso de fabricación descrito en el Ejemplo 1.
[0154] El diámetro de los núcleos inertes es de 200 a 300 micrones. El diámetro de los aglomerados recubiertos es de 600 a 1200 micrones. La densidad aparente de los aglomerados revestidos es 0,7 -1 ,2 g/cm3.
[0155] El peso deseado de los aglomerados revestidos LP que contienen la dosis unitaria se llenan en una cáscara de cápsula de gelatina dura vacía (tamaño 1 para fuerza de 170 mg) utilizando un encapsulador equipado con cámara de dosificación de aglomerado.
Tabla 3: Composición de las cápsulas de amantadina HCl LP

[0156] La disolución in vitro de las cápsulas preparadas anteriormente se ensayan usando un Aparato II de la USP (paletas) a 50 rpm con 500 ml de agua a 37°C como medio de disolución y liberan no más del 25% del fármaco en 2 horas, 40-80% en 6 horas y al menos 80% a las 12 horas.
Ejemplo 3: Formulaciones de aglomerados recubiertos de amantadina de liberación prolongada
[0157] Las composiciones de aglomerados recubiertas de amantadina HC1 de liberación prolongada se prepararon adecuadas para la administración durante la noche usando los componentes y cantidades relativas que se muestran en la Tabla 4 a continuación y el procedimiento de fabricación descrito en el Ejemplo 1.
Tabla 4: Composición de las cápsulas de amantadina HCI LP

[0158] El peso deseado de los aglomerados revestidos LP que contienen la dosis unitaria se llenó en cubiertas de cápsula de gelatina dura vacía n° 1 (60, fuerzas de 140 mg) utilizando un encapsulador equipado con cámara de dosificación de aglomerados. Estas formas de dosificación se utilizan para proporcionar la amantadina para el estudio descrito en el Ejemplo 4 a continuación de acuerdo a las combinaciones de la Tabla 5, como sigue:
Tabla 5: Cápsulas para dosificación

Ejemplo 4: Medición farmacocinética de una formulación de la amantadina de LP en comparación con la amantadina de LI
[0159] Objetivo: El objetivo primario del estudio es evaluar el perfil farmacocinético, la seguridad y la tolerabilidad de una formulación prototipo de LP de amantadina HCl (Formulación 1), en relación con una tableta de LI de 100 mg de amantadina HCl recubierta con película (SYMMETREL®) administrada como dosis única a sujetos adultos sanos en ayunas.
[0160] Diseño del estudio: Este es un estudio farmacocinético de fase 1, aleatorizado, de dosis única, de etiqueta abierta, de dos períodos, cruzado de dos tratamientos, en ayunas, en donde dosis individuales de 340 mg de la formulación 1 de cápsulas de amantadina de LP se compara a dosis individuales de 100 mg de tabletas LI de amantadina comercializadas (SYMMETREL®).
[0161] Métodos: Los sujetos son admitidos en la unidad para el primer periodo de dosificación dentro de los 21 días de selección para el estudio. Habrá un período de descanso de 7 días entre la dosificación en el período 1 y 2. En cada período de dosificación, los sujetos serán dosificados el día después de registrarse en la unidad y se les dará el alta 72 horas después de la dosis. Se realizará un seguimiento final del estudio dentro de los 14 días posteriores a la dosificación en el segundo período.
[0162] Después de una noche de ayuno, la formulación se administra a los sujetos mientras que en una posición sentada con 240 ml de agua. Se recolectaron muestras de sangre a las 0 (antes de la dosis), 1, 2, 3, 4, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 18, 24, 30, 36, 48, 60, 72 horas después de cada dosis. Las muestras de plasma se analizan para determinar la amantadina mediante un método validado de cromatografía líquida/espectroscopía de masas en tándem (CL/EM/EM). Los parámetros farmacocinéticos se calculan utilizando un análisis no compartimental con el software WinNonlin (versión 5,3 o superior; Pharsight Corporation).
[0163] Un análisis de la varianza (ANOVA) se realiza en los logaritmos naturales de Cmax y AUCü-inf determinados a partir de los datos después de una sola dosis del fármaco del estudio usando el modelo lineal de efectos mixtos. El modelo incluye secuencia, período y régimen como efectos fijos y sujeto con secuencia como efecto aleatorio. Se calcula la relación de LP a LI tanto para el AUC (biodisponibilidad relativa para la formulación de LP) como para la Cmáx. Los eventos adversos se monitorean durante todo el estudio. Los signos vitales (frecuencia del pulso, presión arterial y temperatura corporal), las medidas de laboratorio clínico (bioquímica, hematología y análisis de orina) y los ECG se recogen en varios momentos durante el estudio.
[0164] Resultados esperados: Un total de 20 sujetos que comprenden adultos masculinos y femeninos sanos participan en el estudio.
[0165] Los resultados de PK de este estudio demuestran un Cmax reducido (sobre una base de dosis proporcional) para la amantadina de LP con relación a la forma de LI (alrededor de 1,1 hasta 1,7 ng/ml/mg de amantadina para la forma de LP frente a aproximadamente 2,7 ng/ml/mg de amantadina para la forma de LI). Además, Tmax para la amantadina de LP es de 9 a 18 horas frente a aproximadamente 4 horas para la forma de LI. La exposición total a la amantadina, medida por el AUCü-inf, para la formulación de amantadina de LP es del 80 al 100 por ciento de SYMMETREL® en base a la dosis ajustada). La Figura 1 muestra un gráfico de las concentraciones plasmáticas estimadas de amantadina por mg de amantadina dosificado frente al tiempo programado para la formulación de LP. Las curvas alta y baja delimitan el intervalo de valores medios predichos en varios momentos después de la dosificación. Estos resultados se resumen en la Tabla 6, a continuación.
Tabla 6: Parámetros farmacocinéticos de dosis única de amantadina de LP (formulación 1), en comparación con SYMMETREL® (formulación de LI)

Ejemplo 5: Concentración de amantadina en plasma en estado estacionario (ng/ml) después de la dosificación una vez al día de dosis de 260 mg, 340 mg y 420 mg de amantadina de LP HCl (Formulación 1).
[0166] La concentración de amantadina en plasma en estado estacionario fue predicha por la formulación 1 de amantadina de LP (260 mg, 340 mg y 420 mg) administrada una vez al día sobre la base de un modelo obtenido usando WINNONLIN a partir de los datos observados a partir de un estudio de dosis única anterior (Estudio 5103-C-101). Las predicciones del estado estacionario se realizaron utilizando los principios de superposición utilizando los datos de dosis única observados y se asumió que la cinética lineal generaba los perfiles a varios niveles de dosis (260 mg, 340 mg y 420 mg). La Figura 2 muestra los perfiles para la formulación 1 de amantadina de LP (260 mg, 340 mg y 420 mg) administrada una vez al día.
Ejemplo 6: Composiciones de amantadina de liberación prolongada
[0167] Las composiciones de aglomerados recubiertos de amantadina HCl de liberación prolongada adecuadas para administración nocturna se prepararon a partir de los aglomerados recubiertos de LP preparados como se describe en el Ejemplo 1 y se introducen en cápsulas de gelatina dura vacías como se describe en la Tabla 7, a continuación.
Tabla 7: Cápsulas de LP de amantadina HCl

Ejemplo 7: Formulaciones de aglomerados recubiertos de amantadina de liberación prolongada
[0168] Las composiciones de aglomerados recubiertos de liberación prolongada de amantadina HCl adecuadas para una administración diaria se prepararon usando los componentes y las cantidades relativas mostradas en la Tabla 8, a continuación, y el proceso de fabricación descrito en el Ejemplo 1.
[0169] El peso deseado de los aglomerados revestidos con LP que contienen la dosis unitaria se introdujo en la cubierta de la cápsula de gelatina dura vacía n°1 (concentración de 100 mg) usando un encapsulador equipado con una cámara de dosificación de aglomerados.
Tabla 8: Composición de las cápsulas de LP de amantadina HCl

[0170] La disolución in vitro de las cápsulas anteriormente preparadas se ensayaron usando un aparato II USP (paletas)
a 50 rpm con 500 ml de agua a 37°C como medio de disolución. Los resultados se muestran en la Figura 3.
Ejemplo 8: Medición de farmacocinética de las formulaciones de la amantadina de LP en comparación con la amantadina de LI
[0171] Objetivo: El principal objetivo del estudio era confirmar las propiedades de PK de formulaciones de liberación prolongada en el ejemplo 7, para determinar los perfiles farmacocinéticos, seguridad y tolerabilidad de tres formulaciones prototipo de cápsulas Lp de amantadina HCl descritas con diferentes propiedades de liberación en el Ejemplo 7 en relación con una tableta de LI de 100 mg de amantadina HCl recubierta con película (SYMMETREL®) administrada en dosis únicas a sujetos adultos sanos en ayunas.
[0172] Diseño del estudio: Este fue un estudio farmacocinético de fase 1, aleatorizado, de dosis única, de etiqueta abierta, de cuatro períodos, cruzado, en ayuno, en donde dosis únicas de 100 mg de tres formulaciones de cápsulas de LP de amantadina con diferentes propiedades de liberación se compararon con dosis únicas de 100 mg de tabletas de amantadina de LI comercializadas (SYMMETREL®). Las tres formulaciones de LP diferían en las velocidades de liberación de amantadina in vitro, como se muestra en la Figura 3.
[0173] Métodos: Los sujetos fueron admitidos en la unidad para el primer periodo de dosificación dentro de los 21 días de selección para el estudio. Se administró la dosis a los sujetos el día después de registrarse en la unidad y se les dio de alta a las 24 horas posteriores a la dosis. Se pidió a los sujetos que regresaran después del alta para visitas de seguimiento a las 56 horas y 152 horas después de la dosificación. Cada período de dosificación se separó por al menos 7 días de lavado.
[0174] Después de una noche de ayuno, la formulación se administró a los sujetos después de despertarse por la mañana, mientras que en una posición sentada con 240 ml de agua. Se recolectaron muestras de sangre a las 0 (antes de la dosis), 1,2, 3, 4, 5, 6 , 7, 8 , 9, 10, 11, 12, 13, 14, 15, 16, 18, 24 (alta) y 56 horas después de cada dosis. Las muestras de plasma se analizaron para determinar la amantadina mediante un método validado de cromatografía líquida/espectroscopía de masas en tándem (CL/EM/EM). Los parámetros farmacocinéticos se calcularon mediante un análisis no compartimental con el software WinNonlin (versión 4,1 o superior; Pharsight Corporation).
[0175] Un análisis de la varianza (ANOVA) se realizó en los logaritmos naturales de Cmax y AUCü-~ determinados a partir de los datos después de una sola dosis del fármaco del estudio usando modelo lineal de efectos mixtos. El modelo incluyó efectos por sujeto, secuencia, período y régimen. Los efectos de la secuencia, el período y el régimen fueron fijos, mientras que el efecto del sujeto fue aleatorio. Se calculó la relación de LP a LI tanto para el AUC (biodisponibilidad relativa para las formulaciones de LP) como para Cmax. (Los eventos adversos se monitorizaron durante todo el estudio. Los signos vitales (frecuencia del pulso, presión arterial y temperatura corporal), medidas de laboratorio clínico (bioquímica, hematología y análisis de orina) y ECGs se recogieron en diversos momentos durante el estudio.
[0176] Resultados: Un total de 20 sujetos participaron en el estudio. La edad media fue de 25,5 años (intervalo de 20-38 años). El estudio consistió en 8 hombres (40%) y 12 mujeres (60%) con un índice de masa corporal medio (IMC) de 23,6 kg/m2 ± 2,85. La composición racial era 100% caucásica. Quince sujetos recibieron los 4 tratamientos.
[0177] Los resultados del pK de este estudio mostraron que las tres formulaciones de amantadina de LP redujeron la tasa de absorción, en base a los valores reducidos de Cmax y Tmax aumentado, en comparación con SYMMETREL® (Tabla 9, Figuras 4, 5). La formulación de LI tenía los valores Cmax medios más altos (277 ± 73,9 ng/ml) y valores Tmax medianos más cortos (4 h). las formulaciones A, B y C produjeron valores de Cmax cada vez más bajos y valores de Tmax más largos. Cmax disminuyó de 204 ± 61,4 a 166 ± 34,8 a 149 ± 34,4 ng/ml, y la mediana de Tmax aumentó de 7,0 a 11,0 a 14,0 h para las formulaciones A, B y C, respectivamente. La exposición total a la amantadina, medida por el AUC0-», fue ligeramente más baja en las tres formulaciones de amantadina de LP que en SYMMETREL®, pero las tres formulaciones tenían una biodisponibilidad aceptable (85-95%). La Tabla 10 resume las relaciones Cmax y AUC0-» (LP/LI) de las formulaciones A, B y C de LP en relación con LI.
Tabla 9: Parámetros farmacocinéticos de dosis única de tres formulaciones de amantadina de LP (formulación A, B y C), en comparación con SYMMETREL® (formulación de LI)

Tabla 10: Relación LP/LI para Cmax y AUCq-»

Ejemplo 9: Evaluación de parámetros de la marcha en un modelo de ratón EAE de esclerosis múltiple
[0178] El principal objetivo de este estudio fue determinar la oportunidad y alcance de alteraciones de la marcha en MOG35-55 inducida por la encefalomielitis autoinmune experimental (EAE) en ratones C57BL/6, un modelo animal de esclerosis múltiple y el efecto o efectos de la amantadina sobre dicho modelo. Los criterios de valoración principales fueron la gravedad de la enfermedad y los cambios en los parámetros motores finos medidos por el análisis cinemático de la marcha.
MATERIALES DE ESTUDIO
1. Animales de prueba
[0179] Ratones hembra C57BL/6, de 10-12 semanas de edad y un peso de entre 16-22 gramos de Charles River (Alemania) se utilizaron para este estudio. Los ratones se alojaron 4 por jaula durante al menos 4 días antes de su uso y se mantuvieron con comida estándar para roedores y agua del grifo en la instalación para animales.
2. Artículos de prueba
[0180] La amantadina HCl (AMT) fue fabricada por MOEHS CATALANA, S.L. (Barcelona, España).
[0181] La dalfampridina (4-aminopiridina, DAL) se adquirió mediante CRL de Sigma.
PROCEDIMIENTOS EXPERIMENTALES
1. Grupos de prueba
[0182] Los animales se asignaron a grupos de tratamiento de acuerdo con el diseño del estudio en la Tabla 11. Los animales se asignaron en grupos de tratamiento para asegurar una distribución similar de los pesos corporales en todos los grupos (± 10% diferencia en el peso corporal medio entre los grupos).
Tabla 11: Diseño del estudio

2. Selección de dosis
[0183] La dosis seleccionada de la amantadina enumerada en la Tabla 11 utilizada se basó en un estudio de minibomba osmótica Alzet anterior en ratones, con el fin de alcanzar concentraciones en plasma en estado estacionario de aproximadamente 1200 ng/mL.
[0184] La dosis de dalfampridina enumerada en la Tabla 11 se basó en Gobel et al, para conseguir concentraciones plasmáticas en estado estacionario en ratones de aproximadamente 50 ng/mL.
3. Preparación, administración, análisis y disposición de los artículos de prueba
3.1. Preparación de soluciones de dosificación
[0185] El tampón de acetato de sodio (pH 5,0) se preparó a partir de una solución 25 mM de ácido acético y una solución de 25 mM de acetato de sodio en 0,9% de solución salina estéril mediante la adición de la solución de acetato de sodio en la solución de ácido acético hasta alcanzarse un pH de 5,0. La solución final se filtró sobre un filtro de 0,2 pm antes de su uso.
[0186] La amantadina HCl se pesó en matraces volumétricos estériles y se ajustó a volumen con acetato de sodio 25 mM pH 5,0 en 0,9% de solución salina para producir las concentraciones especificadas en la Tabla 12. El pH se ajustó a pH 5,0 con 4 M de acetato de sodio. La dalfampridina se pesó en matraces volumétricos estériles y se ajustó al volumen con agua estéril para producir las concentraciones especificadas en la Tabla 11. Todas las soluciones se filtraron a través de un filtro de 0,2 pm en condiciones asépticas.
3.2. Recogida de formulación
[0187] Dos alícuotas de 100 pL cada una de cada lote de formulación de compuesto se recogieron después de la preparación. Al final (Día 27), después de retirar la bomba del ratón, se recogió una alícuota de cada bomba. Las muestras de formulación se recogieron en viales de vidrio y se almacenaron a una temperatura de -70 a -80°C hasta el análisis al finalizar el estudio.
3.3. Preparación de la minibomba Alzet
[0188] Se implantó a los animales por vía subcutánea con minibombas osmóticas Alzet n° 2004 (0,25 pL/h, bomba de 28 días) que se habían cebado con amantadina o dalfampridina a las concentraciones especificadas en la Tabla 11. Antes de la cirugía, las minibombas se cebaron mediante el siguiente protocolo. Las minibombas se llenaron con la solución de fármaco adecuada utilizando una aguja y una jeringa estériles. A continuación, se colocaron los tapones osmóticos sobre las minibombas. Las minibombas se limpiaron luego con un pañuelo empapado en isopropanol, se dejaron secar y luego se colocaron en un recipiente de solución salina estéril al 0,9% durante 6-16 horas a 37°C para cebar las bombas. Las bombas del mismo grupo se cebaron juntas en un recipiente.
4. Procedimientos durante la vida
4.1. Cirugía de implantación de minibomba Alzet
[0189] Los animales fueron implantados con bombas osmóticas que contienen AMT o DAL en concentraciones especificadas en la Tabla 11. Los ratones fueron anestesiados con 5% de isoflurano después de lo cual isoflurano contenido en oxígeno/oxígeno nitroso se redujo a 1,5-2% para el mantenimiento durante todo el procedimiento quirúrgico. La temperatura rectal se mantuvo a 36,9 ± 1,0 con una manta de calentamiento homeotérmico y la sonda rectal. Se colocaron ratones anestesiados sobre la mesa quirúrgica y se afeitó la piel de la espalda y se desinfectó con una solución de povidona yodada (Betadine, Leiras, 457028). Antes de la incisión, se administró un analgésico a los ratones (Temgesic, 1 ml/kg, sc, RB Pharmaceuticals Ltd, 2014-07).
[0190] Después de ello, se hizo una pequeña incisión para exponer el espacio subcutáneo para la instalación de minibomba Alzet. La minibomba osmótica cebada se limpió con isopropanol. Después de que se evaporó el alcohol, se insertó la bomba en el bolsillo, con el extremo del portal de entrega primero, minimizando así la interacción entre el compuesto de prueba administrado y la curación de la incisión. La piel se cerró con suturas de monofilamento 7-0 y se permitió que los ratones se recuperaran en jaulas individuales antes de devolverlos a su jaula de origen (3-4 ratones/jaula). Además, los ratones recibieron analgésicos (Temgesic, 1 ml/kg, s.c., RB Pharmaceuticals Ltd, 2014-07) el día 0 (mínimo 6 h después de la cirugía), el día 1 (mañana y tarde) y el día 2 (mañana).
[0191] La cirugía de implantación y la inoculación MOG se realizó simultáneamente en el mismo día (día 0).
4.2. Inducción y puntuación clínica de la EAE
[0192] Los ratones fueron inoculados usando inóculo comercialmente disponible listo para usar (EK-0115, Hooke Laboratories, EE. UU.) que contiene 100 pg de MOG35-55, 200 pg inactivado por calor de tuberculosis de micobacteria en aceite mineral en 100 pL de inóculo. La inoculación se realizó administrando a cada ratón 2 inyecciones de 100 pl por
vía subcutánea en la parte inferior y superior de la espalda. Se administraron inyecciones intraperitoneales de toxina pertussis (4 |jg/ml) de 100 j l cada una en el momento de la inoculación y 24 horas después de la inoculación.
[0193] Los signos clínicos se puntuaron diariamente por investigadores ciegos para los grupos de tratamiento como se describe en la Tabla 12.
Tabla 12: Puntuación clínica de EAE

4.3. Pesos corporales y observaciones clínicas generales
[0194] Los pesos corporales se midieron en el Día 0 (pre y post-implantación de minibomba) y una vez al día a partir de entonces. Se registrarán el tiempo y el peso. Se realizaron y registraron observaciones clínicas generales e inspección del sitio de implantación de los animales una vez al día. Se llevó a cabo diariamente una palpitación suave del sitio de implantación de los animales para evitar que se formara tejido cicatricial alrededor de la bomba. Animales que muestran signos sustanciales de toxicidad (es decir, postrados, fríos a temperatura corporal) y/o en estado moribundo se sacrificaron mediante asfixia con CO2 seguido de decapitación antes del sacrificio programado.
[0195] Las definiciones de criterios de valoración aceptables que requieren eutanasia incluyen: sin movimientos espontáneos e incapacidad para beber o comer en un período de observación de 24 h, sangrado masivo, inflamación espontánea, falta de anatomía, hinchazón o tumores mayores de 20 mm e incapacidad para enderezarse en un período de 30 segundos. Los criterios de valoración específicos del modelo (EAE) que justifican el sacrificio incluyen: puntuación clínica de 4 (debilidad parcial en todas las extremidades o hemiplejía) durante más de 1 día sin mejoría (puntuación inferior a 4), reflejo de enderezamiento >30 segundos y caída del peso corporal superior a 25% de la línea de base.
4.4. Análisis de la marcha cinemática
[0196] En los días 1, 5, 8, 11, 14, 17, 20, 23 y 26 todos los ratones por grupo se pueden ejecutar en el MotoRater (TSE Systems, Hamburgo, Alemania), utilizando el modo de caminar. Los videos capturados de cada ratón se convirtieron primero al software SimiMotion para rastrear los puntos marcados del cuerpo (por ejemplo, patas, caderas, cola, etc.), y el movimiento de estos diferentes puntos del cuerpo en relación con el suelo se registró en coordenadas. Se analizaron diferentes patrones de marcha y movimientos de motricidad fina utilizando un sistema de análisis automatizado hecho a medida. Los criterios de valoración cinemáticos se describen en la Tabla 13.
Tabla 13: Criterios de valoración cinemáticos: modo de marcha
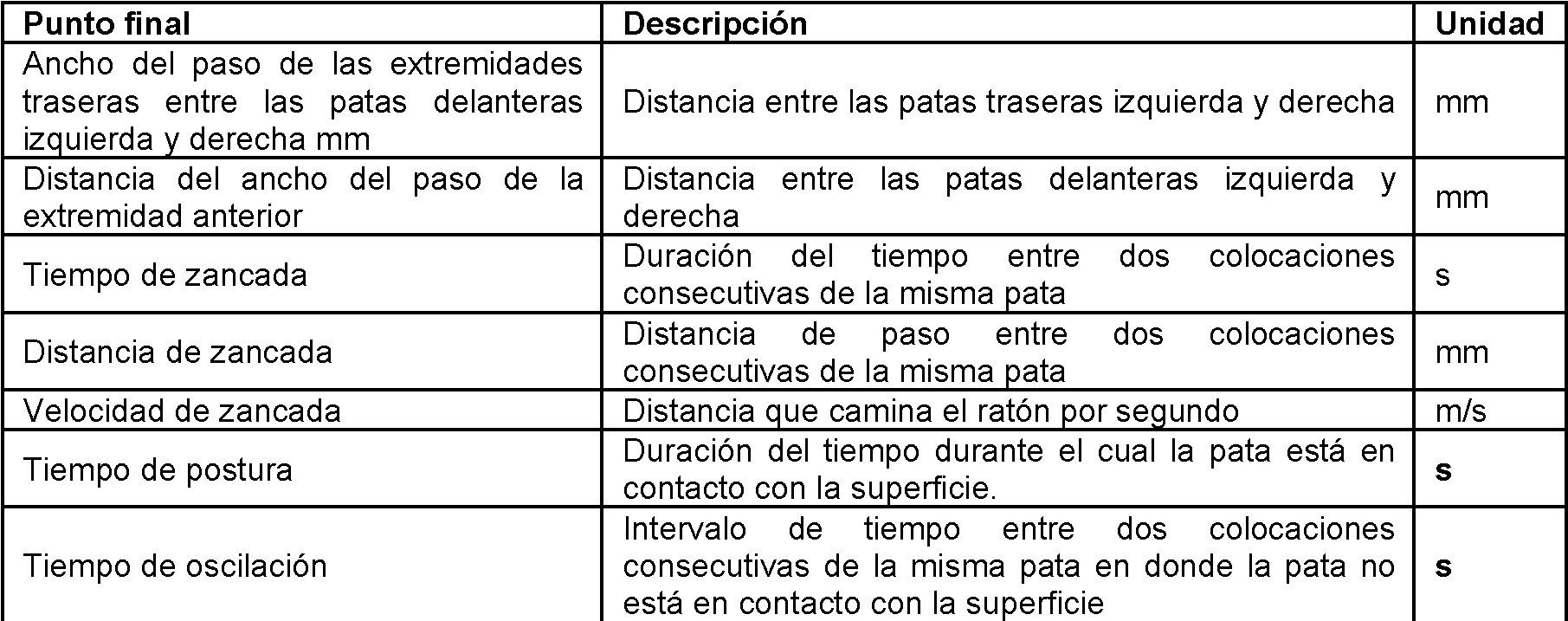
(Continuación)

4.5. Procedimientos terminales y procesamiento de tejido
[0197] En el punto de tiempo de terminación (día 27), todos los ratones fueron profundamente anestesiados con muestras pentobarbitales y de sangre se recogieron mediante punción cardiaca. Se recogieron aproximadamente 0,4-0,5 ml de sangre en tubos anticoagulantes de plástico lavanda K2EDTA de 500 j l y se almacenaron en hielo húmedo hasta la centrifugación. Las muestras de sangre se centrifugaron (dentro de los 15 minutos posteriores a la recolección) a 3000 g durante 10 minutos a 4°C. Se observaron signos de hemólisis (el plasma es rosado o rojo). El plasma se dividió en alícuotas en un solo tubo Eppendorf de 1,5 ml por animal y se colocó en una caja de almacenamiento con separadores y se almacenó entre -70 y -80°C hasta el envío. No se recolectaron tejidos adicionales para este estudio.
7. ANÁLISIS BIOANALÍTICO
[0198] El análisis bioanalítico del plasma y las formulaciones se realizó mediante CL/EM/EM utilizando métodos establecidos para la amantadina y la dalfampridine con el fin de determinar las concentraciones de amantadina y dalfampridina, respectivamente.
8. ANÁLISIS DE DATOS
[0199] La concentración del compuesto en plasma en el punto de tiempo terminal se tabuló para cada animal por grupo, y la concentración media (± DE) se calculó para cada grupo.
[0200] Pesos corporales absolutos individuales y medios (± DE) y cambio de peso corporal a partir del día 1 se tabularon y se representaron gráficamente como una función del tiempo.
[0201] Las puntuaciones clínicas se graficaron como puntuaciones medias (± SEM) como una función del tiempo. La hora de inicio de la enfermedad y las puntuaciones de la enfermedad acumulativas se representaron gráficamente como valores individuales en un gráfico de dispersión y/o como media (± SEM) en un gráfico de barras.
[0202] Los parámetros cinemáticos (Tabla 13) se representaron como valores individuales en un gráfico de dispersión y/o como media (± SEM) en un gráfico de barras.
9. ANÁLISIS ESTADÍSTICO
[0203] Todos los valores se presentan como desviación estándar media ± (DE) o el error estándar de la media (SEM), y las diferencias se consideraron estadísticamente significativas al nivel p <0,05. El análisis estadístico se realizó utilizando el software estadístico StatsDirect. Las diferencias entre las medias se analizan utilizando ANOVA de 1 vía seguido de la prueba de Dunnet (comparación con el grupo de control).
[0204] La comparación dentro del grupo con la línea de base se realiza mediante ANOVA de 2 vías. Los datos no paramétricos se analizan con ANOVA de Kruskal-Wallis o ANOVA de Friedman, respectivamente.
Resultados:
[0205] Un total de 34 ratones fueron utilizados en el estudio, siendo utilizados 10 como controles SHAM-vehículos, y siendo utilizados 10 como controles de vehículos, siendo tratados 10 con amantadina y siendo tratados 10 con dalfampridina.
[0206] Los resultados de este estudio muestran que el tratamiento con amantadina disminuyó la gravedad de los síntomas en un modelo de ratón de EM. La Figura 6 muestra la progresión de la enfermedad en el modelo de ratón EAE para EM, con puntuaciones clínicas que comienzan a aumentar entre los días 12-13, y alcanzan su punto máximo entre los días 18-20 en los ratones EAE. Los ratones tratados con amantadina y dalfampridina muestran puntuaciones clínicas más bajas que los ratones EAE no tratados, lo que indica que los fármacos disminuyeron los síntomas en estos grupos en comparación con el control no tratado. Ninguno de los grupos tratados con el fármaco mostró un retraso en la aparición de la enfermedad en el modelo de ratón EAE para la EM.
[0207] Un gráfico de dispersión de la puntuación acumulativa clínica (Día 18-27) para cada grupo (Figura 7), tanto en el grupo tratado con amantadina, y el grupo tratado con dalfampridina han reducido las puntuaciones clínicas acumulativas
en comparación con el control no tratado.
[0208] La Figura 8 muestra la expresión de la expresión de IBa-1, un marcador de activación de la microglía, en cada uno de los grupos de ensayo. El grupo tratado con amantadina mostró una expresión significativamente menor, lo que indica que la amantadina reduce significativamente la neuroinflamación en un modelo animal para la EM.
[0209] La figura 9 proporciona un gráfico de líneas que muestra el efecto de la progresión de la enfermedad en sujetos animales y el deterioro de su rendimiento en la prueba de marcha. El grupo de control de EAE sin tratar perdió drásticamente la capacidad de completar la prueba de caminata después del día 14, y solo el 40% pudo completar la prueba el día 16. En comparación, el 60% del grupo tratado con amantadina y el 50% del grupo tratado con dalfampridina. grupo, pudieron completar la prueba de la marcha el día 16. La prueba de la marcha del día 14 proporcionó el último momento con los datos de la marcha más completos.
[0210] El análisis adicional de los resultados del estudio de la marcha indicó que la amantadina mejoró la velocidad de marcha en el día 14. Los ratones tratados con amantadina mostraron un tiempo menor en la prueba de tiempo de marcha de 60 cm (Figura 10A), así como una velocidad más rápida media de marcha (Figura 10B) que el control de EAE no tratado así como el grupo tratado con dalfampridina.
Ejemplo 10: Evaluación de amantadina para el tratamiento de trastornos de la marcha en pacientes con esclerosis múltiple
[0211] Se comparan las composiciones de aglomerados recubiertos de liberación prolongada de HCl de amantadina del Ejemplo 2 con placebo en un estudio doble ciego de 2 brazos en sujetos con EM con déficit de la marcha como se define por EDSS (Escala ampliada del estado de discapacidad - Kurtzke 1983). Todos los sujetos reciben un régimen estable de medicamentos para la EM (excluida la 4-aminopiridina) durante al menos 30 días antes de la selección y continúan con las mismas dosis y régimen durante la duración del estudio. Los sujetos incluidos en el estudio se asignan al azar a uno de dos grupos de tratamiento: placebo o 340 mg de amantadina en una formulación de liberación prolongada. Los medicamentos del estudio se administran una vez por la noche antes de acostarse.
[0212] Los sujetos que inicialmente se aleatorizaron para recibir placebo recibieron placebo durante el período de preinclusión inicial de 1 semana y durante todo el período de tratamiento aleatorizado de 4 semanas. Los sujetos que inicialmente se asignaron al azar al tratamiento activo reciben placebo durante el período de preinclusión inicial de 1 semana, seguido de una formulación de liberación prolongada de amantadina de 170 mg durante una semana, seguida de una formulación de liberación prolongada de amantadina de 340 mg durante el período de tratamiento de 3 semanas restante.
[0213] Los eventos adversos se registraron comenzando con la primera dosis del fármaco del estudio y continúan hasta el último estudio de la visita. Los medicamentos concomitantes se registran durante todo el estudio.
[0214] Medidas de resultado:
A. prueba de caminata cronometrada de 25 pies (T25FW - Kiesei er y Pozzili 2012)
B. Prueba de caminata cronometrada (TUG - Learmonth, Paul et al. 2012)
C. Prueba de caminata de seis minutos (6MWT - Goldman, Marrie et al. 2008)
D. Escala de fatiga para funciones motoras y cognitivas (FSMC - Penner Raselli et al., 2009)
E. Inventorio de depresión Beck 2 (BDI-2 - Smarr y Keefer 2011; Watson, Ford et al. 2014)
[0215] El resultado esperado para el brazo de tratamiento activo es una mejora en al menos una de las medidas de resultado mencionadas anteriormente. Se espera que los resultados de T25FW, TUG o 6MWT para el grupo de tratamiento activo sean mejores que los del placebo.
Ejemplo 11: Evaluación de la amantadina para el tratamiento de los déficits a pie en pacientes con esclerosis múltiple
[0216] Las composiciones de aglomerados recubiertas de amantadina HCl de liberación prolongada descritas en el presente documento (por ejemplo, los del Ejemplo 2) se administran a sujetos con EM con déficit para caminar, ya definidas por EDSS (escala de estado de discapacidad expandida - Kurtzke 1983). Todos los sujetos reciben un régimen estable de medicamentos para la EM (excluida la 4-aminopiridina) durante al menos 30 días antes de la selección y continúan con las mismas dosis y régimen durante la duración del estudio. Los sujetos se asignan al azar a uno de dos grupos de tratamiento: placebo o 340 mg de amantadina en una formulación de liberación prolongada. Los medicamentos del estudio se administran una vez por la noche antes de acostarse.
[0217] Los sujetos inicialmente aleatorizados para recibir placebo reciben placebo durante el período de preinclusión inicial de 1 semana y durante todo el período de tratamiento aleatorizado de 4 semanas. Los sujetos inicialmente aleatorizados al tratamiento activo reciben placebo durante el período de preinclusión inicial de 1 semana, seguido de una formulación de liberación prolongada de amantadina de 170 mg durante 1 semana, seguida de una formulación de
liberación prolongada de amantadina de 340 mg durante el período de tratamiento de 3 semanas restante.
[0218] Los eventos adversos se registraron comenzando con la primera dosis del fármaco del estudio y continúan hasta el último estudio de la visita. Los medicamentos concomitantes se registran durante todo el estudio.
[0219] La velocidad o habilidad para caminar se evalúa mediante al menos uno de los siguientes o una combinación de los mismos: prueba de caminata cronometrada de 25 pies (T25FW), prueba de caminata cronometrada (TUG), prueba de caminata de 2 minutos, prueba de caminata cronometrada de seis minutos (6MTW) y/o escala de caminata de esclerosis múltiple de doce ítems (MSWS-12). En algunas formas de realización, se mejora significativamente la marcha del sujeto. El resultado para el grupo de tratamiento activo es una mejora en al menos una de las medidas de resultado mencionadas anteriormente. Los resultados de T25FW, TUG, prueba de caminata de 2 minutos, 6MWT o MSWS-12 para el grupo de tratamiento activo son mejores que los del placebo. Los síntomas de la EM mejoran. T25FW, TUG, 2 minutos de caminata, 6MWT y/o MSWS-12 mejoran significativamente con respecto al placebo.
Claims (14)
1. Una dosis que comprende (i) 220 mg a 600 mg de amantadina o sales farmacéuticamente aceptables de la misma y (ii) al menos un excipiente, para su uso en un método para mejorar la marcha en un sujeto humano con esclerosis múltiple, comprendiendo el método administrar por vía oral la dosis a dicho sujeto una vez al día.
2. La dosis para uso según la reivindicación 1, en donde la dosis comprende adicionalmente uno o más fármacos seleccionados del grupo que consiste en 4-aminopiridina, baclofeno, dextrometorfano, dimetilfumarato, fingolimod, metilfenidato, teriflunomida, tetrabenizina, tizanidina y sales farmacéuticamente aceptable de los mismos.
3. La dosis para su uso de acuerdo con la reivindicación 2, en donde uno o más fármacos se proporcionan en una forma de liberación prolongada.
4. La dosis para su uso de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en donde al menos un excipiente es un excipiente modificador de la liberación.
5. La dosis para su uso de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en donde al menos uno de dichos excipientes modifica la liberación de la amantadina o una sal farmacéuticamente aceptable de la misma para proporcionar una forma de liberación prolongada, y en donde la administración de la dosis proporciona a) a Tmax para amantadina de 5 a 18 horas, b) a Cmax de 1,0 a 2,8 ng/ml por mg de amantadina, y c) un AUCü-inf de 40 a 75 ng*h/ml por mg de amantadina medido en una sola dosis estudio farmacocinético de dosis en humanos.
6. La dosis para su uso de acuerdo con una cualquiera de las reivindicaciones 1 a 5, en donde dicha amantadina o una sal farmacéuticamente aceptable de la misma se proporciona en una forma de liberación prolongada.
7. La dosis para uso de acuerdo con una cualquiera de las reivindicaciones 1 a 6, en donde dicha dosis se administra por la mañana y la composición proporciona una mediana de Tmax de 5 a 9 horas determinada a partir de un estudio farmacocinético humano en ayunas de dosis única.
8. La dosis para su uso de acuerdo con una cualquiera de las reivindicaciones 1 a 6, en donde dicha dosis se administra de 0 a 4 horas antes de acostarse y la composición proporciona una mediana de Tmax de 10 a 18 horas determinada a partir de un estudio farmacocinético en humanos de dosis única en ayunas.
9. La dosis para su uso de acuerdo con una cualquiera de las reivindicaciones 1 a 8, en donde la administración una vez al día de dicha dosis proporciona una relación C-media-día/C-media-noche en estado estacionario de 1,1 a 2,0.
10. La dosis para su uso de acuerdo con una cualquiera de las reivindicaciones 7 y 8, en donde la relación Cmax/Cmin en el estado estacionario es 1,3-3.
11. La dosis para su uso de acuerdo con una cualquiera de las reivindicaciones 1 a 10, en donde el método aumenta la velocidad al caminar.
12. La dosis para su uso de acuerdo con la reivindicación 11, en donde la velocidad o la capacidad de caminar se evalúa mediante al menos uno de los siguientes o una combinación de los mismos: prueba de caminata cronometrada de 25 pies, prueba de caminata cronometrada, prueba de caminata de 2 minutos, prueba de caminata cronometrada de seis minutos y/o escala de caminata de esclerosis múltiple de doce ítems.
13. La dosis para uso de acuerdo con cualquiera de las reivindicaciones 1-12, en donde la administración de la composición da como resultado un 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55% o 60% de reducción en la discapacidad para caminar por la enfermedad de la esclerosis múltiple.
14. La dosis para su uso de acuerdo con una cualquiera de las reivindicaciones 1 a 13, en donde la amantadina se administra como hidrocloruro de amantadina.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462075137P | 2014-11-04 | 2014-11-04 | |
| PCT/US2015/058872 WO2016073510A1 (en) | 2014-11-04 | 2015-11-03 | Methods of administering amantadine compositions |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2865278T3 true ES2865278T3 (es) | 2021-10-15 |
Family
ID=55909712
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15857591T Active ES2865278T3 (es) | 2014-11-04 | 2015-11-03 | Amantadina para mejorar la marcha en esclerosis múltiple |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US20160228388A1 (es) |
| EP (2) | EP3215132B1 (es) |
| JP (1) | JP6735020B2 (es) |
| CN (1) | CN107205950B (es) |
| AU (1) | AU2015343199B2 (es) |
| BR (1) | BR112017009289A2 (es) |
| CA (1) | CA2966195C (es) |
| ES (1) | ES2865278T3 (es) |
| HU (1) | HUE055779T2 (es) |
| MX (1) | MX2017005578A (es) |
| PL (1) | PL3215132T3 (es) |
| PT (1) | PT3215132T (es) |
| SI (1) | SI3215132T1 (es) |
| WO (1) | WO2016073510A1 (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014204933A1 (en) | 2013-06-17 | 2014-12-24 | Adamas Pharmaceuticals, Inc. | Amantadine compositions and methods of use |
| IL272834B1 (en) * | 2017-08-24 | 2024-04-01 | Adamas Pharma Llc | Compositions of amantadine, their preparation, and methods of use |
| TR201720406A2 (tr) | 2017-12-14 | 2019-07-22 | Sanovel Ilac Sanayi Ve Ticaret Anonim Sirketi | Bi̇r i̇skelet kasi gevşeti̇ci̇si̇ ve bi̇r multi̇pl skleroz tedavi̇ edi̇ci̇ ajan i̇çeren kombi̇nasyonlar |
| WO2019245513A2 (en) | 2018-06-21 | 2019-12-26 | Sanovel Ilac Sanayi Ve Ticaret Anonim Sirketi | A combination comprising fingolimod and amantadine |
| TR201818307A2 (tr) * | 2018-11-30 | 2020-06-22 | Sanovel Ilac Sanayi Ve Ticaret Anonim Sirketi | Di̇meti̇l fumarat i̇çeren bi̇r kompozi̇syon |
| TR201818859A2 (tr) | 2018-12-07 | 2020-06-22 | Sanovel Ilac Sanayi Ve Ticaret Anonim Sirketi | Di̇meti̇l fumarat ve en az bi̇r kas gevşeti̇ci̇ ajan i̇çeren bi̇r kombi̇nasyon |
| TR201819202A2 (tr) | 2018-12-12 | 2020-06-22 | Sanovel Ilac Sanayi Ve Ticaret Anonim Sirketi | Bi̇r multi̇pl skleroz ajani ve en az bi̇r kas relaksan ajan i̇çeren bi̇r kombi̇nasyon |
| TR201820976A2 (tr) * | 2018-12-28 | 2020-07-21 | Sanovel Ilac Sanayi Ve Ticaret Anonim Sirketi | Fi̇ngoli̇mod ve bi̇r spazmoli̇ti̇k i̇çeren farmasöti̇k kombi̇nasyonlar |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5221536A (en) | 1990-05-07 | 1993-06-22 | Alza Corporation | Dosage form indicated for the management of abnormal posture, tremor and involuntary movement |
| US5358721A (en) | 1992-12-04 | 1994-10-25 | Alza Corporation | Antiviral therapy |
| AUPN605795A0 (en) | 1995-10-19 | 1995-11-09 | F.H. Faulding & Co. Limited | Analgesic pharmaceutical composition |
| WO2006053067A2 (en) * | 2004-11-09 | 2006-05-18 | Prestwick Pharmaceuticals, Inc. | Combination of amantadine and a tetrabenazine compound for treating hyperkinetic disorders |
| US7619007B2 (en) | 2004-11-23 | 2009-11-17 | Adamas Pharmaceuticals, Inc. | Method and composition for administering an NMDA receptor antagonist to a subject |
| CA2588296A1 (en) | 2004-11-24 | 2006-06-01 | Neuromolecular Pharmaceuticals, Inc. | Composition comprising an nmda receptor antagonist and levodopa and use thereof for treating neurological disease |
| US8574626B2 (en) | 2004-12-03 | 2013-11-05 | Osmotica Kereskedelmi és Szolgáltató KFT | Osmotic device containing amantadine and an osmotic salt |
| WO2006089066A1 (en) * | 2005-02-15 | 2006-08-24 | Neuromolecular Pharmaceuticals, Inc. | Combinations therapy for treatment of demyelinating conditions |
| CA2604052C (en) | 2005-04-06 | 2014-07-08 | Adamas Pharmaceuticals, Inc. | Methods and compositions for the treatment of cns-related conditions |
| US7981930B2 (en) | 2007-03-13 | 2011-07-19 | Adamas Pharmaceuticals, Inc. | Compositions and kits for treating influenza |
| JP5885668B2 (ja) * | 2009-12-02 | 2016-03-15 | アダマス ファーマシューティカルズ, インコーポレイテッド | アマンタジン組成物および使用方法 |
| WO2014093475A1 (en) * | 2012-12-11 | 2014-06-19 | Acorda Therapeutics, Inc. | Methods for treating parkinson's disease using aminopyridines |
| WO2014204933A1 (en) * | 2013-06-17 | 2014-12-24 | Adamas Pharmaceuticals, Inc. | Amantadine compositions and methods of use |
-
2015
- 2015-11-03 EP EP15857591.0A patent/EP3215132B1/en active Active
- 2015-11-03 HU HUE15857591A patent/HUE055779T2/hu unknown
- 2015-11-03 SI SI201531585T patent/SI3215132T1/sl unknown
- 2015-11-03 JP JP2017522637A patent/JP6735020B2/ja active Active
- 2015-11-03 CN CN201580072014.7A patent/CN107205950B/zh active Active
- 2015-11-03 BR BR112017009289A patent/BR112017009289A2/pt not_active Application Discontinuation
- 2015-11-03 MX MX2017005578A patent/MX2017005578A/es active IP Right Grant
- 2015-11-03 AU AU2015343199A patent/AU2015343199B2/en active Active
- 2015-11-03 PT PT158575910T patent/PT3215132T/pt unknown
- 2015-11-03 PL PL15857591T patent/PL3215132T3/pl unknown
- 2015-11-03 CA CA2966195A patent/CA2966195C/en active Active
- 2015-11-03 US US14/931,546 patent/US20160228388A1/en not_active Abandoned
- 2015-11-03 WO PCT/US2015/058872 patent/WO2016073510A1/en active Application Filing
- 2015-11-03 EP EP21153566.1A patent/EP3909569A1/en not_active Withdrawn
- 2015-11-03 ES ES15857591T patent/ES2865278T3/es active Active
Also Published As
| Publication number | Publication date |
|---|---|
| US20160228388A1 (en) | 2016-08-11 |
| HUE055779T2 (hu) | 2021-12-28 |
| AU2015343199A1 (en) | 2017-05-18 |
| EP3215132B1 (en) | 2021-01-27 |
| EP3909569A1 (en) | 2021-11-17 |
| MX2017005578A (es) | 2018-01-11 |
| BR112017009289A2 (pt) | 2017-12-19 |
| CN107205950B (zh) | 2021-06-04 |
| CA2966195A1 (en) | 2016-05-12 |
| EP3215132A4 (en) | 2018-07-11 |
| PT3215132T (pt) | 2021-05-07 |
| SI3215132T1 (sl) | 2021-11-30 |
| CN107205950A (zh) | 2017-09-26 |
| AU2015343199B2 (en) | 2021-04-08 |
| CA2966195C (en) | 2024-04-30 |
| EP3215132A1 (en) | 2017-09-13 |
| JP6735020B2 (ja) | 2020-08-05 |
| PL3215132T3 (pl) | 2021-10-11 |
| WO2016073510A1 (en) | 2016-05-12 |
| JP2017533211A (ja) | 2017-11-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2865278T3 (es) | Amantadina para mejorar la marcha en esclerosis múltiple | |
| ES2525005T3 (es) | Formulación de liberación prolongada que contiene una cera | |
| US11903908B2 (en) | Methods of administering amantadine | |
| US11975103B2 (en) | Compositions of midodrine and methods of using the same | |
| US20060263427A1 (en) | Quinine formulations | |
| JP2008280351A (ja) | テルビナフィンを含有する医薬組成物およびその使用 | |
| KR102381586B1 (ko) | 메살라진의 경구용 약제학적 조성물 | |
| AU2010258345A1 (en) | Novel pharmaceutical compositions containing pregabalin | |
| TW200800155A (en) | Gastric retentive gabapentin dosage forms and methods for using same | |
| BR112020008128A2 (pt) | comprimidos de liberação retardada de deferiprona e métodos para utilização dos mesmos | |
| JP2021152050A (ja) | フマル酸ジメチルを含む医薬ビーズ製剤 | |
| KR20170086053A (ko) | 디메틸 푸마르산염을 포함하는 제약학적 매트릭스 제제 | |
| US20210154161A1 (en) | Modified-release bucillamine compositions, kits, and methods for treating cystinuria, arthritis, gout, and related disorders | |
| Hadi | Development and evaluation of mini-tablets-filed-capsule system for chronotherapeutic delivery of montelukast |