ES2909797T3 - Composición farmacéutica de derivado de aminodihidrotiazina condensado - Google Patents
Composición farmacéutica de derivado de aminodihidrotiazina condensado Download PDFInfo
- Publication number
- ES2909797T3 ES2909797T3 ES15848771T ES15848771T ES2909797T3 ES 2909797 T3 ES2909797 T3 ES 2909797T3 ES 15848771 T ES15848771 T ES 15848771T ES 15848771 T ES15848771 T ES 15848771T ES 2909797 T3 ES2909797 T3 ES 2909797T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- salt
- dissolution
- saturated aliphatic
- pharmaceutical composition
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/542—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/20—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing sulfur, e.g. dimethyl sulfoxide [DMSO], docusate, sodium lauryl sulfate or aminosulfonic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
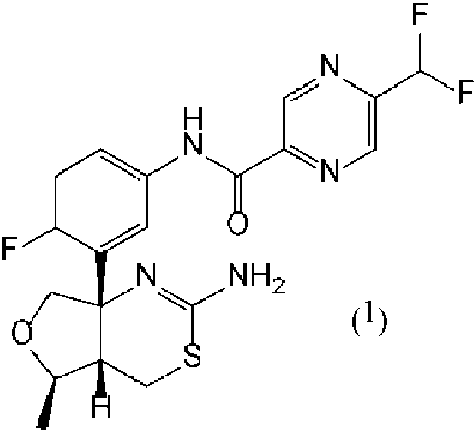
Composición farmacéutica que comprende un compuesto representado por la fórmula (1): **(Ver fórmula)** o N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazin-7a-il)-4-fluorofenil]-5- difluorometilpirazin-2-carboxamida, o una sal farmacéuticamente aceptable de lo anterior, y una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22.
Description
DESCRIPCIÓN
Composición farmacéutica de derivado de aminodihidrotiazina condensado
Campo técnico
La presente invención se refiere a una composición farmacéutica de un derivado de aminodihidrotiazina condensado. Más particularmente, la presente invención se refiere a una composición farmacéutica que comprende un derivado de aminodihidrotiazina condensado en forma de un compuesto representado por la fórmula (1):

o N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazin-7a-il)-4-fluorofenil]-5-difluorometilpirazin-2-carboxamida, o una sal farmacéuticamente aceptable de lo anterior.
Antecedentes de la técnica
El inhibidor de BACE1 (Enzima 1 que escinde la Proteína Precursora de Amiloide del Sitio Beta), N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazin-7a-il)-4-fluorofenil]-5-difluorometilpirazin-2-carboxamida, es un compuesto que tiene la estructura representada por la fórmula (1) (que se denominará simplemente como “Compuesto Farmacológico 1”).

Se espera que el Compuesto Farmacológico 1 o una sal farmacéuticamente aceptable del mismo se use como agente terapéutico para la demencia y MCI (deterioro cognitivo leve) (Documento 1 de Patente).
Lista de citas
Documento de patente
Documento 1 de Patente: Patente US No. 8158620 (Memoria descriptiva)
Las publicaciones Study Record NCT 01716897, ClinicalTrials.gov, 20 de marzo de 2012, study record NCT 02222324, ClinicalTrials.gov, 19 de agosto de 2014, US 2014/271911 y EP 2233 474 describen formas de dosificación sólidas de un compuesto de fórmula (I), pero ninguno de los documentos anteriores describe una formulación farmacéutica que comprende un compuesto de fórmula (I) y una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22.
Sumario
Problema técnico
El problema a resolver por la presente invención es proporcionar una composición farmacéutica que comprende un compuesto representado por la fórmula (1), N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazin-7a-il)-4-fluorofenil]-5-difluorometilpirazin-2-carboxamida, o una sal farmacéuticamente aceptable de la
anterior, que no provoca un cambio sustancial en el perfil de disolución incluso después del almacenamiento durante un cierto período de tiempo. Solución al problema
Como resultado de la realización de extensos estudios para resolver el problema mencionado anteriormente, los inventores de la presente invención encontraron que el problema mencionado anteriormente se resuelve mediante una composición farmacéutica que comprende un compuesto representado por la fórmula (1), N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazin-7a-il)-4-fluorofenil]-5-difluorometilpirazin-2-carboxamida, o una sal farmacéuticamente aceptable de la anterior, y un aditivo que no provoca un cambio sustancial en el perfil de disolución incluso después del almacenamiento durante un cierto período de tiempo, lo que conduce a la finalización de la presente invención.
La presente invención es como se indica a continuación.
1. [1] Una composición farmacéutica que comprende un compuesto representado por la fórmula (1):

o N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazin-7a-il)-4-fluorofenil]-5-difluorometilpirazin-2-carboxamida, o una sal farmacéuticamente aceptable de lo anterior, y
una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22.
2. [2] La composición farmacéutica descrita en [1], en la que la sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 es al menos una seleccionada del grupo que consiste en una sal metálica de un ácido orgánico alifático saturado de C12-22, una sal de monometal alcalino de un fumarato de monoalquilo de C12-22, y una sal de monometal alcalino-térreo de un fumarato de monoalquilo de C12-22.
3. [3] La composición farmacéutica descrita en [1], en la que la sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 es al menos una seleccionada del grupo que consiste en estearilfumarato de sodio, laurilsulfato de sodio, estearato de calcio, y estearato de magnesio.
Efectos ventajosos de la invención
Según la presente invención, se puede proporcionar una composición farmacéutica que contiene un compuesto representado por la fórmula (1), N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazin-7ail)-4-fluorofenil]-5-difluorometilpirazin-2-carboxamida, o una sal farmacéuticamente aceptable de la anterior, que no provoca un cambio sustancial en el perfil de disolución incluso después del almacenamiento durante un cierto período de tiempo.
Breve descripción de los dibujos
La Fig. 1 muestra un perfil de disolución inicial y perfiles de disolución después del almacenamiento durante 2 semanas y 1 mes a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz en una disolución acuosa de ácido clorhídrico 0,1 mol/l del ejemplo 1.
La Fig. 2 muestra un perfil de disolución inicial y perfiles de disolución después del almacenamiento durante 2 semanas y 1 mes a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz en una disolución acuosa de ácido clorhídrico 0,1 mol/l del ejemplo 4.
La Fig. 3 muestra un perfil de disolución inicial y perfiles de disolución después del almacenamiento durante 2 semanas y 1 mes a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz en una disolución acuosa de ácido clorhídrico 0,1 mol/l del ejemplo comparativo 1. La Fig. 4 muestra un perfil de disolución inicial y perfiles de disolución después del almacenamiento durante 2 semanas y 1 mes a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz en una disolución acuosa de ácido clorhídrico 0,1 mol/l del ejemplo comparativo 2.
La Fig. 5 muestra cambios en las tasas de disolución en 15 minutos atribuibles al almacenamiento a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz entre sales de compuestos que tienen un grupo hidrocarbonado alifático saturado de C12-22. Cada muestra contiene además una hidroxipropilcelulosa poco sustituida.
La Fig. 6 muestra cambios en las tasas de disolución en 15 minutos atribuibles al almacenamiento a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz entre sales de compuestos que tienen un grupo hidrocarbonado alifático saturado de C12-22 de la misma manera que la Fig. 5. Cada muestra contiene además una hidroxipropilcelulosa poco sustituida.
La Fig. 7 muestra cambios en las tasas de disolución en 15 minutos atribuibles al almacenamiento a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz entre sales de compuestos que tienen un grupo hidrocarbonado alifático saturado de C12-22. Cada muestra contiene además carmelosa cálcica.
La Fig. 8 muestra cambios en las tasas de disolución en 15 minutos atribuibles al almacenamiento a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz entre sales de compuestos que tienen un grupo hidrocarbonado alifático saturado de C12-22. Cada muestra contiene además carboximetilalmidón sódico.
La Fig. 9 muestra cambios en las tasas de disolución en 15 minutos atribuibles al almacenamiento a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz entre sales de compuestos que tienen un grupo hidrocarbonado alifático saturado de C12-22. Cada muestra contiene además carmelosa.
La Fig. 10 muestra cambios en las tasas de disolución en 15 minutos atribuibles al almacenamiento a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz entre muestras que tienen diferentes contenidos de Compuesto Farmacológico 1.
La Fig. 11 muestra cambios en las tasas de disolución en 15 minutos atribuibles al almacenamiento a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz entre muestras en las que otros aditivos difieren cuando la sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 es estearilfumarato de sodio.
La Fig. 12 muestra cambios en las tasas de disolución en 15 minutos atribuibles al almacenamiento a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz entre muestras en las que otros aditivos difieren cuando la sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 es estearato de magnesio.
Descripción de las realizaciones
A continuación, la presente invención se describirá con más detalle.
La presente invención se refiere a una composición farmacéutica que comprende un compuesto representado por la fórmula (1)

o N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazin-7a-il)-4-fluorofenil]-5-difluorometilpirazin-2-carboxamida, o sal farmacéuticamente aceptable de lo anterior, y
una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22.
Un compuesto representado por la fórmula (1) o N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazina-7a-il)-4-fluorofenil]-5-difluorometilpirazina-2-carboxamida utilizada en la presente invención se describe como Compuesto 43 del documento WO 2012/100179.
El compuesto representado por la fórmula (1) o N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]t¡az¡na-7a-¡l)-4-fluorofen¡l]-5-d¡fluoromet¡lp¡razina-2-carboxam¡da puede estar en forma libre o en forma de una sal farmacéuticamente aceptable. Está preferiblemente en forma libre.
Ejemplos de sales farmacéuticamente aceptables incluyen sales de ácidos inorgánicos tales como sulfatos, nitratos, percloratos, fosfatos, carbonatos, bicarbonatos, hidrofluoruros, hidrocloruros, hidrobromuros o hidroyoduros, sales de ácidos orgánicos tales como acetatos, oxalatos, maleatos, tartratos, fumaratos, citratos, metanosulfonatos, trifluorometanosulfonatos, etanosulfonatos, bencenosulfonatos, toluenosulfonatos, o canfosulfonatos, sales de aminoácidos tales como aspartatos o glutamatos, sales de aminas cuaternarias, y sales metálicas tales como sales de sodio, sales de potasio, sales de magnesio, o sales de calcio.
El Compuesto Farmacológico 1 o una sal farmacéuticamente aceptable del mismo puede obtenerse mediante el método descrito en el Documento 1 de Patente, una modificación del mismo, o un método que es obvio para los expertos normales en la técnica.
Una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 contenido en la composición farmacéutica según la presente invención se refiere a una sal compuesta por un compuesto que tiene un grupo hidrocarbonado alifático saturado que tiene 12 a 22 átomos de carbono en una molécula del mismo, y un grupo funcional que tiene una carga positiva o carga negativa en una parte del mismo distinta del grupo hidrocarbonado alifático saturado de C12-22, y un contraión enlazado iónicamente al mismo.
Se puede seleccionar un compuesto preferible para la sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 desde el punto de vista de tener una estabilidad que no provoque un cambio sustancial en el perfil de disolución incluso después del almacenamiento durante un cierto período de tiempo. Además, también se puede seleccionar un compuesto preferible desde los puntos de vista de mejorar la fluidez del polvo y de evitar la adhesión del polvo con respecto a la moldeabilidad en una preparación sólida.
Los ejemplos de una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 incluyen sales de amonio cuaternario que tienen un grupo alquilo de C12-22 tal como cloruro de cetilpiridinio, sales metálicas de un ácido orgánico alifático saturado de C12-22 tales como laurilsulfato de sodio o estearato de magnesio, sales de mono-alquilo de C12-22 y monometal alcalino de ácido fumárico tal como estearilfumarato de sodio, y sales de monoalquilo de C12-22 y monometal alcalino-térreo de ácido fumárico tal como estearilfumarato de magnesio.
Dicha sal se puede usar sola, o se pueden usar dos o más tipos de sales.
Un ácido orgánico alifático saturado de C12-22 se refiere a un ácido orgánico que tiene una cadena alifática que tiene 12 a 22 átomos de carbono, pero que no tiene un enlace insaturado.
La cadena alifática puede ser lineal o ramificada. Los ejemplos de ácidos orgánicos incluyen ácido carboxílico y ácido sulfúrico.
Una sal metálica de un ácido orgánico alifático saturado de C12-22 se refiere a una sal en la que un metal está enlazado a un ácido orgánico alifático saturado de C12-22, y sus ejemplos incluyen sales de metales alcalinos tales como sales de sodio o sales de potasio, sales de metales alcalino-térreos tales como sales de magnesio o sales de calcio, sales de zinc, y sales de aluminio.
Los ejemplos de sales de metales alcalinos de ácidos orgánicos alifáticos saturados de C12-22 incluyen laurilsulfato de potasio, laurilsulfato de sodio, miristato de sodio, palmitato de potasio, estearato de potasio, y estearato de sodio.
Los ejemplos de sales de metales alcalino-térreos de ácidos orgánicos alifáticos saturados de C12-22 incluyen laurilsulfato de magnesio, miristato de magnesio, palmitato de calcio, estearato de magnesio, y estearato de calcio.
Ejemplos de sales de zinc y sales de aluminio de ácidos orgánicos alifáticos saturados de C12-22 incluyen estearato de zinc y estearato de aluminio.
Una sal de monometal alcalino de un fumarato de monoalquilo de C12-22 se refiere a una sal que tiene una estructura de éster monoalquílico de ácido fumárico en la que uno de los dos ácidos carboxílicos del ácido fumárico está enlazado mediante éster a un alcohol alquílico que tiene 12 a 22 átomos de carbono, mientras que el otro ácido carboxílico está enlazado a un metal alcalino.
El alcohol alquílico que tiene 12 a 22 átomos de carbono puede ser un alcohol alquílico lineal o un alcohol alquílico ramificado, y sus ejemplos incluyen alcohol laurílico, alcohol miristílico, alcohol palmitílico, y alcohol estearílico.
Los ejemplos de una sal de monometal alcalino de un fumarato de monoalquilo de C12-22 incluyen estearilfumarato de sodio, laurilfumarato de potasio, y miristilfumarato de sodio.
Una sal de monometal alcalino-térreo de un fumarato de monoalquilo de C12-22 se refiere a una sal que tiene una estructura de éster monoalquílico de ácido fumárico en la que uno de los dos ácidos carboxílicos del ácido fumárico está enlazado mediante éster a un alcohol alquílico que tiene 12 a 22 átomos de carbono, mientras que el otro ácido carboxílico está enlazado a un metal alcalino-térreo.
Los ejemplos de una sal de monometal alcalino-térreo de un fumarato de monoalquilo de C12-22 incluyen estearilfumarato de magensio, laurilfumarato de calcio, y palmitilfumarato de magnesio.
La sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 contenida en la composición farmacéutica según la presente invención es preferiblemente estearilfumarato de sodio, laurilsulfato de sodio, estearato de calcio, o estearato de magnesio, y más preferiblemente estearilfumarato de sodio.
La relación de mezclamiento de la sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 a la masa total de la composición farmacéutica según la presente invención es normalmente 0,1% en masa a 10% en masa, preferiblemente 0,2% en masa a 5% en masa, y más preferiblemente 0,3% en masa a 3% en masa.
En la presente invención, un “metal alcalino-térreo” incluye berilio, magnesio, calcio, estroncio, y bario, y es preferiblemente magnesio o calcio.
La composición farmacéutica según la presente invención puede contener preferiblemente uno o más compuestos seleccionados del grupo de compuestos indicados a continuación desde el punto de vista de la disolución rápida del Compuesto Farmacológico 1 o una sal farmacéuticamente aceptable del mismo.
Los ejemplos de estos compuestos incluyen hidroxipropilcelulosa poco sustituida, almidón parcialmente gelatinizado, almidón (tal como almidón de arroz, almidón de trigo, almidón de patata, o almidón de maíz), crospovidona, carmelosa, carmelosa cálcica, croscarmelosa sódica, y carboximetilalmidón sódico. Los ejemplos preferibles de estos compuestos incluyen hidroxipropilcelulosa poco sustituida, almidón, crospovidona, carmelosa cálcica, carmelosa, y carboximetilalmidón sódico.
La composición farmacéutica según la presente invención también puede contener celulosa cristalina, agar, gelatina, carbonato cálcico, bicarbonato sódico, citrato cálcico, dextrina, o pectina, además de los compuestos ejemplificados anteriormente.
La composición farmacéutica según la presente invención tiene una estabilidad que no provoca un cambio sustancial en el perfil de disolución del Compuesto Farmacológico 1 o una sal farmacéuticamente aceptable del mismo, incluso después del almacenamiento durante un cierto período de tiempo.
En la presente invención, “no provocar un cambio sustancial en el perfil de disolución incluso después del almacenamiento durante un cierto período de tiempo” significa que el cambio en las tasas de disolución en 15 minutos (o 30 minutos) después del almacenamiento durante un cierto período de tiempo con respecto a las tasas de disolución inicial en 15 minutos (o 30 minutos) antes del almacenamiento pueden caer dentro de ±20%.
Aunque no existen limitaciones particulares en las condiciones de disolución siempre que permitan medir la disolución del Compuesto Farmacológico 1, como se describirá posteriormente en los ejemplos, preferiblemente se lleva a cabo un ensayo de disolución según el método de ensayo de disolución descrito en la 16a edición de la Farmacopea Japonesa utilizando 900 ml de una disolución acuosa de ácido clorhídrico 0,1 mol/l ajustada a una temperatura de 372C±0,52C y a una velocidad de rotación de las paletas de 50 rpm.
En la presente invención, es preferible una composición farmacéutica en la que el Compuesto Farmacológico 1 o una sal farmacéuticamente aceptable del mismo se disuelve rápidamente incluso después del almacenamiento a 60°C y 75% de humedad relativa normalmente durante 2 semanas, y preferiblemente 1 mes, en condiciones de estar abierto a la atmósfera y protegido de la luz de la misma manera que antes del almacenamiento.
En la presente invención, “se disuelve rápidamente” significa que un promedio de la tasa de disolución es normalmente 75% o más, preferiblemente 80%, o más y más preferiblemente 85% o más.
La tasa de disolución promedio a los 15 minutos después del almacenamiento durante 2 semanas a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz es normalmente 75% o más, preferiblemente 80% o más, y más preferiblemente 85% o más. La tasa de disolución promedio a los 15 minutos después del almacenamiento durante 1 mes a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz es normalmente 75% o más, preferiblemente 80% o más, y más preferiblemente 85% o más. La tasa de disolución promedio a los 15 minutos después del almacenamiento durante 3 meses a 40°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz es normalmente 75% o más, preferiblemente 80% o más, y más preferiblemente 85% o más. La tasa de disolución promedio a los 15 minutos después del almacenamiento durante 6 meses a 40°C y 75% de humedad relativa en condiciones de estar abierto a
la atmósfera y protegido de la luz es normalmente 75% o más, preferiblemente 80% o más, y más preferiblemente 85% o más.
En la presente invención, la dosificación diaria del Compuesto Farmacológico 1 o una sal farmacéuticamente aceptable del mismo es normalmente 0,1 mg a 1000 mg, preferiblemente 1 mg a 250 mg, y más preferiblemente 2 mg a 150 mg. La relación de mezclamiento del Compuesto Farmacológico 1 o una sal farmacéuticamente aceptable del mismo a la masa total de la composición farmacéutica según la presente invención es normalmente 0,1% en masa a 80% en masa, preferiblemente 0,5% en masa a 60% en masa, y más preferiblemente 0,9% en masa a 50% en masa.
La composición farmacéutica según la presente invención también puede contener un excipiente, aglutinante, agente disgregante, lubricante, colorante, corrector, auxiliar de la disolución, antioxidante, emulsionante, promotor de absorción, tensioactivo, o agente de recubrimiento, además de la sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22.
Los ejemplos de excipientes incluyen lactosa, sacarosa, glucosa, manitol, sorbitol, dextrina, celulosa cristalina, anhídrido silícico ligero, silicato de aluminio, silicato de calcio, metasilicato de aluminato magnesio, e hidrogenofosfato de calcio.
Los ejemplos de aglutinantes incluyen polivinilpirrolidona, etilcelulosa, metilcelulosa, goma arábiga, hidroxipropilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa sódica, y alcohol polivinílico.
Los ejemplos de colorantes incluyen sesquióxido de hierro, sesquióxido de hierro amarillo, carmín, caramelo, pcaroteno, óxido de titanio, talco, fosfato de riboflavina de sodio, y laca de aluminio amarilla.
Los ejemplos de correctores incluyen cacao en polvo, aceite de menta piperita, y canela en polvo.
Los ejemplos de auxiliares de la disolución incluyen polietilenglicol, propilenglicol, benzoato de bencilo, etanol, colesterol, trietanolamina, carbonato de sodio, citrato de sodio, polisorbato 80, y amida de ácido nicotínico.
Los ejemplos de antioxidantes incluyen ácido ascórbico, a-tocoferol, etoxiquina, dibutilhidroxitolueno, y butilhidroxianisol.
Los ejemplos de emulsionantes, promotores de absorción, y tensioactivos incluyen esteariltrietanolamina, ácido laurilaminopropiónico, lecitina, monoestearato de glicerina, ésteres de ácidos grasos de sacarosa, y ésteres de ácidos grasos de glicerina.
Los ejemplos de agentes de recubrimiento incluyen hipromelosa, hidroxipropilcelulosa, metilcelulosa, copolímero E de metacrilato de aminoalquilo, y alcohol polivinílico.
La composición farmacéutica según la presente invención puede estar en forma de comprimido sin recubrir, comprimido recubierto, comprimido de disolución oral, comprimido masticable, cápsula dura, cápsula blanda, gránulos, granos, o polvo. Cada una de estas formas de preparación de la composición farmacéutica se puede producir mediante un método conocido en la técnica anterior.
En la presente invención, los gránulos que contienen el Compuesto Farmacológico 1 o una sal farmacéuticamente aceptable del mismo se preparan preferiblemente y después se formulan desde el punto de vista de la moldeabilidad. No existen limitaciones particulares sobre una composición farmacéutica en forma de gránulos que contienen el Compuesto Farmacológico 1 o una sal farmacéuticamente aceptable del mismo, y puede producirse mediante un método conocido.
Los gránulos también pueden contener un excipiente, aglutinante, agente disgregante, lubricante, colorante, corrector, auxiliar de la disolución, antioxidante, emulsionante, promotor de absorción, tensioactivo, o agente de recubrimiento según sea necesario, además del Compuesto farmacológico 1 o una sal farmacéuticamente aceptable del mismo. En el caso de contener gránulos, la composición farmacéutica de la presente invención es una composición farmacéutica que contiene gránulos que contienen un compuesto representado por la fórmula (1):
o N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazin-7a-il)-4-fluorofenil]-5-difluorometilpirazin-2-carboxamida, o una sal farmacéuticamente aceptable de lo anterior, y
una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22.
Además, debido a su moldeabilidad superior, la composición farmacéutica de la presente invención es una preparación sólida, y más específicamente, un comprimido, tal como un comprimido sin recubrir, un comprimido recubierto, un comprimido de disolución oral, o un comprimido masticable.
EJEMPLOS
[Ejemplo 1]
2500 mg del Compuesto Farmacológico 1,2285 mg de lactosa (DFE Pharma), 330 mg de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.), y 165 mg de hidroxipropilcelulosa (SL, Nippon Soda Co., Ltd.) se mezclaron en un mortero. Se añadió una cantidad adecuada de disolución acuosa de etanol (30% p/p) a la mezcla resultante, y después se sometió a granulación húmeda en el mortero. Después de secar los gránulos resultantes usando un baño a temperatura constante, los gránulos se clasificaron por tamaño usando un tamiz que tenía aberturas de 1 mm.
Se añadieron 33 mg de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) y 11 mg de estearilfumarato de sodio (JRS Pharma) por 1056 mg de gránulos clasificados, y se mezclaron en un vial. La mezcla resultante se comprimió a 9 kN utilizando una máquina de formación de comprimidos de un solo punzón para obtener comprimidos que tenían un diámetro de 6,5 mm y una masa de 110 mg.
[Ejemplos 2 a 18 y Ejemplos Comparativos 1 a 5]
Se obtuvieron comprimidos con un diámetro de 6,5 mm y una masa de 110 mg usando el mismo método que en el Ejemplo 1 para tener las formulaciones indicadas en las Tablas 1 a 4.
Las Tablas 1 a 4 indican las cantidades constituyentes de cada componente por comprimido. Además, cada uno de los componentes utilizados son los que se indican a continuación.
Laurilsulfato de sodio (Nikko Chemicals Co., Ltd.), estearato de calcio (Merck KGaA), estearato de magnesio (Mallinckrodt plc), ácido esteárico (Mallinckrodt plc), éster de ácido graso de sacarosa (J-1811F, Mitsubishi-Kagaku Foods Corporation), almidón parcialmente gelatinizado (Asahi Kasei Chemicals Corp.), almidón de maíz (Nihon Shokuhin Kako Co., Ltd.), crospovidona (XL-10, ISP), carmelosa cálcica (Gotoku Chemical Company Ltd.), croscarmelosa sódica (FMC), carboximetilalmidón sódico (JRS Pharma), carmelosa (Gotoku Chemical Company Ltd.).
[Tabla 1]


[Tabla 4]

[Método de ensayo de disolución]
Se llevó a cabo un ensayo de disolución según el método de ensayo de disolución descrito en la 16a edición de la Farmacopea Japonesa utilizando 900 ml de una disolución acuosa de ácido clorhídrico 0,1 mol/l ajustada a una temperatura de 372C±0,52C a una velocidad de rotación de las paletas de 50 rpm. Para la disolución patrón usada para calcular la tasa de disolución, se usó una disolución obtenida disolviendo el Compuesto Farmacológico 1 en una disolución acuosa de ácido clorhídrico 0,1 mol/l. La disolución patrón se preparó de manera que la concentración del Compuesto Farmacológico 1 estuviera cerca de una concentración equivalente a una tasa de disolución del 100%. Las muestras de medida se recogieron del recipiente del analizador de la disolución en el momento especificado después del inicio del ensayo, y después se filtraron usando un filtro que tenía un diámetro de poro de 0,45 gm. La tasa de disolución se evaluó utilizando el método de medida de la absorbancia o el método de cromatografía de líquidos de altas prestaciones que se indica a continuación.
(Método de medida de la absorbancia)
La absorbancia se midió usando una celda que tenía una longitud de recogido óptico de 10 mm en condiciones de una longitud de onda de medida de 300 nm y una longitud de onda de referencia de 650 nm. La tasa de disolución se calculó en base a los resultados de la medida de la absorbancia y la concentración de la disolución patrón preparada.
(Método de cromatografía de líquidos de altas prestaciones)
La medida por cromatografía de líquidos de altas prestaciones se llevó a cabo en las condiciones de ensayo que se indican a continuación. La tasa de disolución se calculó en base al área del pico correspondiente al Compuesto Farmacológico 1 y la concentración de la disolución patrón preparada.
Columna analítica: Columna con un diámetro interior de 4,6 mm y una longitud de 150 mm rellena con gel de sílice octadecilsililado de 5 gm (Mightysil RP18-GP, Kanto Chemical Co., Inc.)
Temperatura de columna: 40°C
Temperatura de la muestra: 25°C
Volumen de inyección: 50 gl
Caudal: aprox. 1 ml/min
Fase móvil: Mezcla de agua, metanol, ácido perclórico al 70%, y monohidrato de perclorato de sodio en una relación de 600/400/1/7 (v/v/v/p).
Longitud de onda de detección: 270 nm
[Resultado 1 de ensayo de disolución]
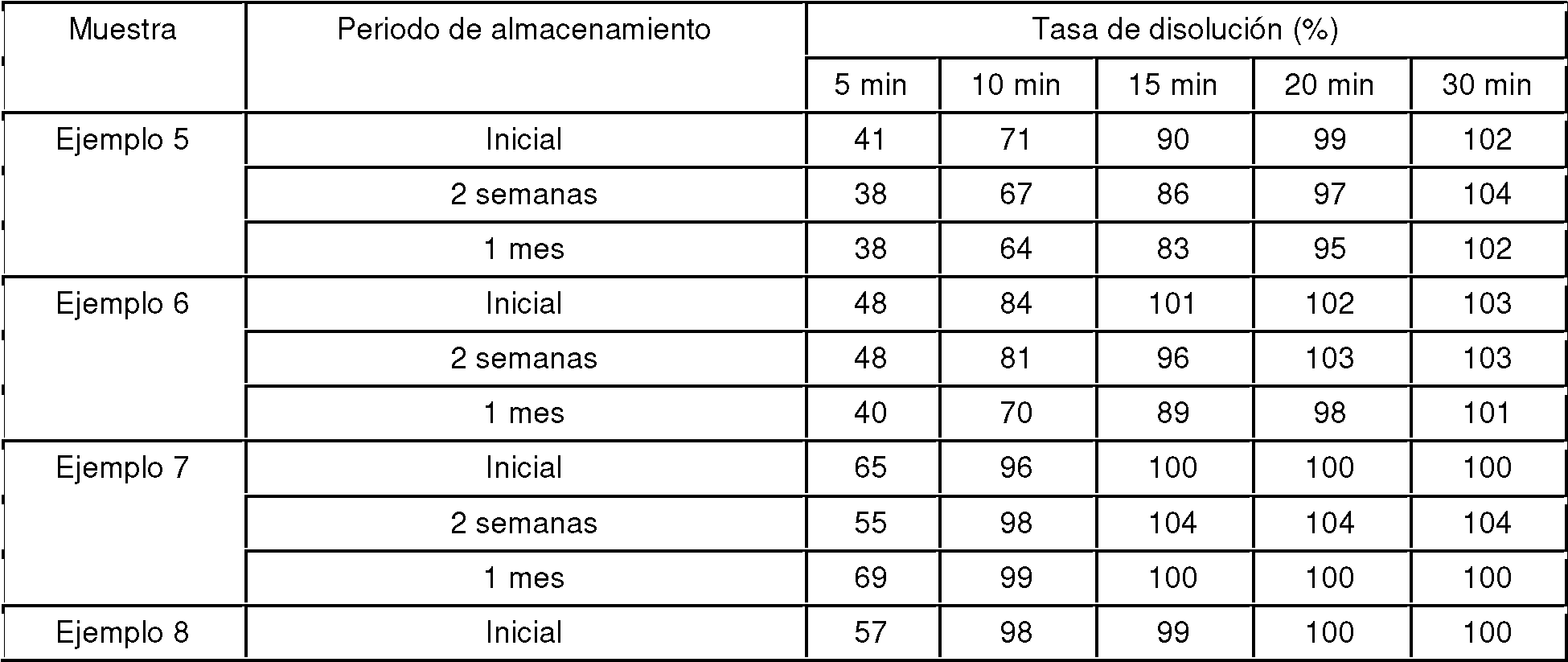
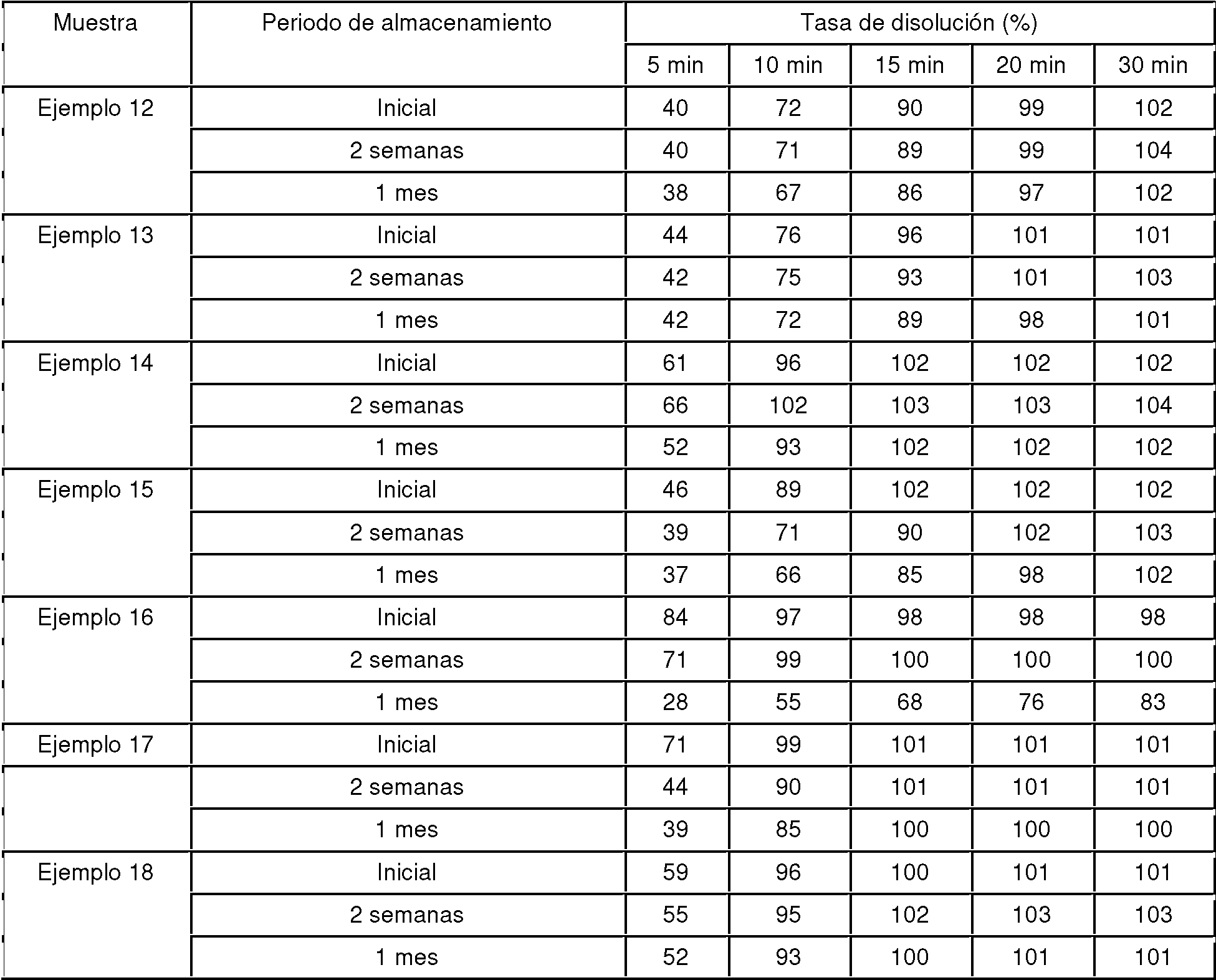
Las muestras preparadas en los Ejemplos 1 a 18 y los Ejemplos Comparativos 1 a 5 se colocaron en viales y se almacenaron durante 2 semanas y 1 mes a 60°C y 75% de humedad relativa en condiciones de estar abiertas a la atmósfera y protegidas de la luz. Los perfiles de disolución de las muestras antes y después del almacenamiento se midieron dos veces en una disolución acuosa de ácido clorhídrico 0,1 mol/l. La tasa de disolución se evaluó usando el método de medida de la absorbancia descrito en la sección Método de Ensayo de la Disolución. Los resultados se muestran en las Tablas 5 a 8 como valores promedio de las tasas de disolución.
[Tabla 5]

[Tabla 6]

[Tabla 7]
[Tabla 8]

[Ejemplo 19]
4,5 g del Compuesto Farmacológico 1, 394,0 g de lactosa (DFE Pharma), y 49,5 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) se mezclaron con un granulador agitador. Se añadieron a la mezcla resultante 14,9 g de hidroxipropilcelulosa (SL, Nippon Soda Co., Ltd.) disuelta en una cantidad adecuada de disolución acuosa de etanol (35% p/p), seguido de la granulación con un granulador agitador. Después de secar los gránulos resultantes usando un lecho fluidizado, los gránulos se clasificaron por tamaño usando un tamiz que tenía un diámetro de abertura de 1 mm.
Se añadieron 5,0 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) y 1,5 g de estearilfumarato de sodio (JRS Pharma), y se mezclaron por 93,5 g de gránulos clasificados. La mezcla resultante se comprimió usando una máquina giratoria de formación de comprimidos para obtener comprimidos que tenían un diámetro de 6,5 mm y una masa de 110 mg.
Los comprimidos resultantes se recubrieron con una película soluble en agua utilizando Opadry Pink a base de hipromelosa (que contiene propilenglicol, Colorcon Japan LLC) para obtener comprimidos recubiertos con película (cantidad de recubrimiento: 7 mg/comprimido).
[Ejemplo 20]
120.0 g del Compuesto Farmacológico 1,2005,2 g de lactosa (DFE Pharma), y 264,0 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) se mezclaron con un granulador agitador. Se añadieron a la mezcla resultante 79,2 g de hidroxipropilcelulosa (SL, Nippon Soda Co., Ltd.) disuelta en una cantidad adecuada de disolución acuosa de etanol (35% p/p), seguido de la granulación con un granulador agitador. Después de secar los gránulos resultantes usando un lecho fluidizado, los gránulos se clasificaron por tamaño usando un tamiz que tenía un diámetro de abertura de 1 mm.
Se añadieron 5,0 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) y 1,5 g de estearilfumarato de sodio (JRS Pharma), y se mezclaron por 93,5 g de gránulos clasificados. La mezcla resultante se comprimió usando una máquina giratoria de formación de comprimidos para obtener comprimidos que tenían un diámetro de 6,5 mm y una masa de 110 mg.
Los comprimidos resultantes se recubrieron con una película soluble en agua utilizando Opadry Red a base de hipromelosa (que no contiene propilenglicol, Colorcon Japan LLC) propilenglicol (Adeka Corporation) para obtener comprimidos recubiertos con película (cantidad de recubrimiento: 7 mg/comprimido, relación másica de Opadry Red a propilenglicol = 9:1).
[Ejemplo 21]
240.0 g del Compuesto Farmacológico 1, 1885,2 g de lactosa (DFE Pharma), y 264,0 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) se mezclaron con un granulador agitador. Se añadieron a la mezcla resultante 79,2 g de hidroxipropilcelulosa (SL, Nippon Soda Co., Ltd.) disuelta en una cantidad adecuada de disolución acuosa de etanol (35% p/p), seguido de la granulación con un granulador agitador. Después de secar los gránulos
resultantes usando un lecho fluidizado, los gránulos se clasificaron por tamaño usando un tamiz que tenía un diámetro de abertura de 1 mm.
Se añadieron 5,0 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) y 1,5 g de estearilfumarato de sodio (JRS Pharma), y se mezclaron por 93,5 g de gránulos clasificados. La mezcla resultante se comprimió usando una máquina giratoria de formación de comprimidos para obtener comprimidos que tenían un diámetro de 6,5 mm y una masa de 110 mg.
Los comprimidos resultantes se recubrieron con una película soluble en agua utilizando Opadry Red a base de hipromelosa (que no contiene propilenglicol Colorcon Japan LLC), y propilenglicol para obtener comprimidos recubiertos con película (cantidad de recubrimiento: 7 mg/comprimido, relación másica de Opadry Red a propilenglicol = 9:1).
[Ejemplo 22]
67,5 g del Compuesto Farmacológico 1, 331,0 g de lactosa (DFE Pharma), y 49,5 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) se mezclaron con un granulador agitador. Se añadieron a la mezcla resultante 14,9 g de hidroxipropilcelulosa (SL, Nippon Soda Co., Ltd.) disuelta en una cantidad adecuada de disolución acuosa de etanol (35% p/p), seguido de la granulación con un granulador agitador. Después de secar los gránulos resultantes usando un lecho fluidizado, los gránulos se clasificaron por tamaño usando un tamiz que tenía un diámetro de abertura de 1 mm.
Se añadieron 5,0 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) y 1,5 g de estearilfumarato de sodio (JRS Pharma), y se mezclaron por 93,5 g de gránulos clasificados. La mezcla resultante se comprimió usando una máquina giratoria de formación de comprimidos para obtener comprimidos que tenían un diámetro de 6,5 mm y una masa de 110 mg.
Los comprimidos resultantes se recubrieron con una película soluble en agua utilizando Opadry Pink a base de hipromelosa (que contiene propilenglicol, Colorcon Japan LLC) para obtener comprimidos recubiertos con película (cantidad de recubrimiento: 7 mg/comprimido).
[Ejemplo 23]
600.0 g del Compuesto Farmacológico 1, 1525,2 g de lactosa (DFE Pharma), y 264,0 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) se mezclaron con un granulador agitador. Se añadieron a la mezcla resultante 79,2 g de hidroxipropilcelulosa (SL, Nippon Soda Co., Ltd.) disuelta en una cantidad adecuada de disolución acuosa de etanol (35% p/p), seguido de la granulación con un granulador agitador. Después de secar los gránulos resultantes usando un lecho fluidizado, los gránulos se clasificaron por tamaño usando un tamiz que tenía un diámetro de abertura de 1 mm.
Se añadieron 5,0 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) y 1,5 g de estearilfumarato de sodio (JRS Pharma), y se mezclaron por 93,5 g de gránulos clasificados. La mezcla resultante se comprimió usando una máquina giratoria de formación de comprimidos para obtener comprimidos que tenían un diámetro de 6,5 mm y una masa de 110 mg.
Los comprimidos resultantes se recubrieron con una película soluble en agua utilizando Opadry Red a base de hipromelosa (que no contiene propilenglicol, Colorcon Japan LLC) propilenglicol (Adeka Corporation) para obtener comprimidos recubiertos con película (cantidad de recubrimiento: 7 mg/comprimido, relación másica de Opadry Red a propilenglicol = 9:1).
[Ejemplo 24]
225.0 g del Compuesto Farmacológico 1, 173,5 g de lactosa (DFE Pharma), y 49,5 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) se mezclaron con un granulador agitador. Se añadieron a la mezcla resultante 14,9 g de hidroxipropilcelulosa (SL, Nippon Soda Co., Ltd.) disuelta en una cantidad adecuada de disolución acuosa de etanol (35% p/p), seguido de la granulación con un granulador agitador. Después de secar los gránulos resultantes usando un lecho fluidizado, los gránulos se clasificaron por tamaño usando un tamiz que tenía un diámetro de abertura de 1 mm.
Se añadieron 5,0 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) y 1,5 g de estearilfumarato de sodio (JRS Pharma), y se mezclaron por 93,5 g de gránulos clasificados. La mezcla resultante se comprimió usando una máquina giratoria de formación de comprimidos para obtener comprimidos que tenían un diámetro de 6,5 mm y una masa de 110 mg.
Los comprimidos resultantes se recubrieron con una película soluble en agua utilizando Opadry Pink a base de hipromelosa (que contiene propilenglicol, Colorcon Japan LLC) para obtener comprimidos recubiertos con película (cantidad de recubrimiento: 7 mg/comprimido).
[Ejemplo 25]
225,0 g del Compuesto Farmacológico 1, 173,5 g de lactosa (DFE Pharma), y 49,5 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) se mezclaron con un granulador agitador. Se añadieron a la mezcla resultante 14,9 g de hidroxipropilcelulosa (SL, Nippon Soda Co., Ltd.) disuelta en una cantidad adecuada de disolución acuosa de etanol (35% p/p), seguido de la granulación con un granulador agitador. Después de secar los gránulos resultantes usando un lecho fluidizado, los gránulos se clasificaron por tamaño usando un tamiz que tenía un diámetro de abertura de 1 mm.
Se añadieron 5,0 g de hidroxipropilcelulosa poco sustituida (LH21, Shin-Etsu Chemical Co., Ltd.) y 1,5 g de estearilfumarato de sodio (JRS Pharma), y se mezclaron por 93,5 g de gránulos clasificados. La mezcla resultante se comprimió usando una máquina giratoria de formación de comprimidos para obtener comprimidos que tenían un diámetro de 8,0 mm y una masa de 220 mg.
Los comprimidos resultantes se recubrieron con una película soluble en agua utilizando Opadry Pink a base de hipromelosa (que contiene propilenglicol, Colorcon Japan LLC) para obtener comprimidos recubiertos con película (cantidad de recubrimiento: 11 mg/comprimido).

[Resultado 2 de ensayo de disolución]
Las muestras preparadas en los Ejemplos 19 a 25 se colocaron en viales y se almacenaron durante 1 mes a 602C y 75% de humedad relativa en condiciones de estar abiertas a la atmósfera y protegidas de la luz. Los perfiles de disolución de las muestras antes y después del almacenamiento se midieron en una disolución acuosa de ácido clorhídrico 0,1 mol/l. La tasa de disolución en el Ejemplo 19 se evaluó utilizando el método de cromatografía de líquidos de altas prestaciones descrito en la sección Método de Ensayo de disolución. Las tasas de disolución en los Ejemplos 20 a 25 se evaluaron usando el método de medida de la absorbancia descrito en la sección Método de Ensayo de Disolución. Los perfiles de disolución en los Ejemplos 19, 24 y 25 se midieron 3 veces, mientras que los perfiles de disolución en los Ejemplos 20 a 23 se midieron 6 veces. Los resultados se muestran en la Tabla 10 como valores promedio de las tasas de disolución.
[Tabla 10]

[Ejemplo 26 y Ejemplos Comparativos 6 a 9]
Se obtuvieron comprimidos con un diámetro de 6,5 mm y una masa de 110 mg usando el mismo método que en el Ejemplo 1 para tener las formulaciones indicadas en la Tabla 11.
La Tabla 11 indica las cantidades constituyentes de cada componente por comprimido. A continuación se indican aquellos materiales de partida utilizados que no se describieron en el Ejemplo 1.
Cloruro de cetilpiridinio (Wako Pure Chemical Industries, Ltd.), alcohol estearílico (Kolliwax SA, BASF SE), lecitina de soja refinada (Basis LP-2013, The Nisshin OilliO Group, Ltd.), éster de ácido graso de glicerina (P-100, Riken Vitamin Co., Ltd.), éster de ácido graso de sorbitán (Nikkol SS-10MV, Nikko Chemicals Co., Ltd.).
[Tabla 11]

[Resultado 3 de ensayo de disolución]
Las muestras preparadas en el Ejemplo 26 y los Ejemplos Comparativos 6 a 9 se colocaron en viales y se almacenaron durante 2 semanas y 1 mes a 60°C y 75% de humedad relativa en condiciones de estar abiertas a la atmósfera y protegidas de la luz. Los perfiles de disolución de las muestras antes y después del almacenamiento se midieron dos veces en una disolución acuosa de ácido clorhídrico 0,1 mol/l. Las tasas de disolución se evaluaron usando el método de medida de la absorbancia descrito en la sección Método de Ensayo de la Disolución. Los resultados se muestran en la Tabla 12 como valores promedio de las tasas de disolución.
[Tabla 12]

No se observaron cambios en los perfiles de disolución atribuibles al almacenamiento en los Ejemplos 1 y 4 que incorporaron una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 en forma de
estearilfumarato de sodio o estearato de magnesio (Fig. 1 y Fig. 2). Por otro lado, se observaron retrasos considerables en los perfiles de disolución atribuibles al almacenamiento en los ejemplos comparativos 1 y 2 que incorporaron ácido esteárico o éster de ácido graso de sacarosa, que no es una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 (Fig. 3 y Fig. 4).
Las tasas de disolución a los 15 minuto en los Ejemplos 1 a 4 y el Ejemplo 26, que incorporaron una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22, fueron 75% o más durante los periodos de almacenamiento de 2 semanas y 1 mes. Por otro lado, las tasas de disolución a los 15 minutos disminuyeron por debajo de 75% después de 2 semanas de almacenamiento en el caso de los Ejemplos Comparativos 1 y 2 y los Ejemplos Comparativos 6 a 9, que incorporaron compuestos en forma de ésteres de ácido esteárico o ácido graso que no son sales de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22.
Se muestra que la incorporación de una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 puede inhibir los retrasos en la disolución atribuibles al almacenamiento de una preparación que contiene el Compuesto Farmacológico 1 (Fig. 5 y Fig. 6).
Las tasas de disolución a los 15 minuto en los Ejemplos 8, 15, 10, 17, 11 y 18, que incorporaron una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22, fueron 75% o más durante los periodos de almacenamiento de 2 semanas y 1 mes. Por otro lado, las tasas de disolución a los 15 minutos disminuyeron hasta alrededor de 70% después de 1 mes de almacenamiento en el caso de los Ejemplos Comparativos 3 a 5, que incorporaron compuestos que no son una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22.
Se muestra que la incorporación de una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 puede inhibir los retrasos en la disolución atribuibles al almacenamiento de una preparación que contiene el Compuesto Farmacológico 1 (Fig. 7 a Fig. 9).
[Efecto del contenido del Compuesto Farmacológico 1]
Los ejemplos 19 a 25 son muestras que tienen diferentes contenidos de Compuesto Farmacológico 1, y cada uno de estos ejemplos contiene estearilfumarato de sodio. Lo siguiente describe el efecto del contenido del Compuesto Farmacológico 1 sobre el perfil de disolución basado en el Resultado 2 de Ensayo de disolución.
En la Fig. 10 se muestra el efecto del almacenamiento a 60°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz sobre la tasa de disolución 15 minutos después del inicio del ensayo de disolución. En el caso de usar estearilfumarato de sodio, las tasas de disolución después del almacenamiento durante 1 mes fueron 85% o más, incluso si el contenido del Compuesto Farmacológico 1 en un solo comprimido varió dentro de un intervalo de 1 mg a 100 mg (Fig. 10).
Los resultados de este estudio indican que la utilidad de una sal de metal alcalino de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 en forma de estearilfumarato de sodio no depende del contenido del Compuesto Farmacológico 1.
[Ejemplos Comparativos 10 a 19]
Se obtuvieron comprimidos que tenían un diámetro de 6,5 mm y las masas que se muestran en las Tablas 13 y 14 usando el mismo método como el Ejemplo 1 para tener las formulaciones indicadas en las Tablas 13 y 14.
Las Tablas 13 y 14 indican las cantidades constituyentes de cada componente por comprimido. A continuación se indican aquellos materiales de partida utilizados que no se describieron en el Ejemplo 1 o en el Ejemplo 26.
Aceite hidrogenado (Lubriwax 103, Freund Corporation), Macrogol (Macrogol 6000, Sanyo Chemical Industries, Ltd.).

[Resultados de la observación de la superficie del comprimido]
Los resultados de la observación de la superficie del comprimido, cuando se prepararon tres de cada uno de los comprimidos de los Ejemplos 1 a 4, Ejemplo 26 y Ejemplos Comparativos 10 a 19, se muestran en las Tablas 13 y 14. Como resultado, las superficies de los comprimidos de los Ejemplos Comparativos 10 a 19, que no contienen una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 en la formulación de los mismos, demostraron una rugosidad superficial provocada por la pegajosidad que era inaceptable en términos de aspecto. Por otro lado, se obtuvieron comprimidos con una superficie lisa en los Ejemplos 1 a 4 y el Ejemplo 26, que contienen una sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 en la formulación de los mismos, y no se observó que tuvieran ningún problema en términos de aspecto.
[Resultado 4 de ensayo de disolución]
Las muestras preparadas en los Ejemplos 20 a 23 se colocaron en viales y se almacenaron durante 6 mes a 40°C y 75% de humedad relativa en condiciones de estar abiertas a la atmósfera y protegidas de la luz. Los perfiles de disolución de las muestras antes y después del almacenamiento se midieron en una disolución acuosa de ácido clorhídrico 0,1 mol/l. Las tasas de disolución en los Ejemplos 20 y 23 se evaluaron 6 veces usando el método de medida de la absorbancia descrito en la sección Método de Ensayo de Disolución. Los resultados se muestran en la Tabla 15 como valores promedio de las tasas de disolución.
[Tabla 15]

[Efecto del almacenamiento a 40°C sobre el comportamiento de disolución]
Los Ejemplos 20 y 23 son muestras que tienen diferentes contenidos del Compuesto Farmacológico 1, y cada uno de estos ejemplos contiene estearilfumarato de sodio. En el caso de usar estearilfumarato de sodio, las tasas de disolución promedio a los 15 minutos después del almacenamiento durante 6 meses a 40°C y 75% de humedad relativa en condiciones de estar abierto a la atmósfera y protegido de la luz fueron 85% o más (Tabla 15).
Los resultados de este estudio indican que una sal de metal alcalino de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 en forma de estearilfumarato de sodio puede inhibir los retrasos en la disolución de una preparación que contiene el Compuesto Farmacológico 1, incluso si se almacena a 40°C.
Aplicabilidad Industrial
La presente invención demuestra aplicabilidad industrial en el campo de los productos farmacéuticos como resultado de poder proporcionar una composición farmacéutica estable que no provoca un cambio sustancial en el perfil de disolución incluso después del almacenamiento durante un cierto período de tiempo.
Claims (3)
- REIVINDICACIONES
 o N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazin-7a-il)-4-fluorofenil]-5-difluorometilpirazin-2-carboxamida, o una sal farmacéuticamente aceptable de lo anterior, yuna sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22.
o N-[3-((4aS,5R,7aS)-2-amino-5-metil-4a,5,7,7a-tetrahidro-4H-furo[3,4-d][1,3]tiazin-7a-il)-4-fluorofenil]-5-difluorometilpirazin-2-carboxamida, o una sal farmacéuticamente aceptable de lo anterior, yuna sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22. - 2. La composición farmacéutica según la reivindicación 1, en la que la sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 es al menos una seleccionada del grupo que consiste en una sal metálica de un ácido orgánico alifático saturado de C12-22, una sal de monometal alcalino de un fumarato de monoalquilo de C12-22, y una sal de monometal alcalino-térreo de un fumarato de monoalquilo de C12-22.
- 3. La composición farmacéutica según la reivindicación 1, en la que la sal de un compuesto que tiene un grupo hidrocarbonado alifático saturado de C12-22 es al menos una seleccionada del grupo que consiste en estearilfumarato de sodio, laurilsulfato de sodio, estearato de calcio, y estearato de magnesio.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014209289 | 2014-10-10 | ||
| PCT/JP2015/078688 WO2016056638A1 (ja) | 2014-10-10 | 2015-10-08 | 縮合アミノジヒドロチアジン誘導体の医薬組成物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2909797T3 true ES2909797T3 (es) | 2022-05-10 |
Family
ID=55653242
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15848771T Active ES2909797T3 (es) | 2014-10-10 | 2015-10-08 | Composición farmacéutica de derivado de aminodihidrotiazina condensado |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US10117876B2 (es) |
| EP (1) | EP3205343B1 (es) |
| JP (3) | JP6590822B2 (es) |
| KR (1) | KR102524217B1 (es) |
| CN (1) | CN107106568B (es) |
| AU (1) | AU2015329027B2 (es) |
| BR (1) | BR112017007293B1 (es) |
| CA (1) | CA2963761C (es) |
| ES (1) | ES2909797T3 (es) |
| IL (1) | IL251559B (es) |
| MX (1) | MX370338B (es) |
| RU (1) | RU2710227C2 (es) |
| SG (1) | SG11201702632QA (es) |
| WO (1) | WO2016056638A1 (es) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102524217B1 (ko) * | 2014-10-10 | 2023-04-21 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 축합 아미노디하이드로티아진 유도체의 의약 조성물 |
| CA3042020A1 (en) | 2016-10-27 | 2018-05-03 | Eisai R&D Management Co., Ltd. | Composition comprising an anti-abeta protofibril antibody and a beta-secretase bace1 inhibitor for the treatment of alzheimer's disease |
| JP6326114B2 (ja) * | 2016-11-01 | 2018-05-16 | エルメッド エーザイ株式会社 | レベチラセタム含有医薬組成物及びその製造方法、並びにレベチラセタム含有医薬組成物の崩壊及び溶出の少なくともいずれかの遅延防止方法、及びレベチラセタム含有医薬組成物の崩壊及び溶出の少なくともいずれかの遅延防止剤 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4568426B2 (ja) * | 1998-04-08 | 2010-10-27 | 協和発酵キリン株式会社 | 錠剤の製造方法及び錠剤 |
| CN101068546A (zh) * | 2004-10-06 | 2007-11-07 | 卫材R&D管理有限公司 | 医药组合物及其制造方法以及医药组合物中的二氢吡啶类化合物的稳定化方法 |
| JP5158922B2 (ja) * | 2006-02-24 | 2013-03-06 | 塩野義製薬株式会社 | ビタミンb12類含有組成物 |
| UA101352C2 (uk) * | 2008-01-18 | 2013-03-25 | Ейсей Р Енд Д Менеджмент Ко., Лтд. | Конденсоване похідне амінодигідротіазину |
| EP2251012A4 (en) * | 2008-02-11 | 2013-03-06 | Dainippon Sumitomo Pharma Co | TABLET HAVING ENHANCED ELUTING PROPERTIES |
| JP5993875B2 (ja) * | 2011-01-21 | 2016-09-14 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 縮合アミノジヒドロチアジン誘導体の合成に有用な方法および化合物 |
| JP2014037356A (ja) * | 2012-08-13 | 2014-02-27 | Takada Seiyaku Kk | カンデサルタンシレキセチル経口製剤 |
| US20140271911A1 (en) * | 2014-03-26 | 2014-09-18 | James Wallace | Lithium and a beta-secretase inhibitor for the treatment of alzheimer's disease |
| KR102524217B1 (ko) * | 2014-10-10 | 2023-04-21 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 축합 아미노디하이드로티아진 유도체의 의약 조성물 |
-
2015
- 2015-10-08 KR KR1020177009221A patent/KR102524217B1/ko active IP Right Grant
- 2015-10-08 JP JP2016553160A patent/JP6590822B2/ja active Active
- 2015-10-08 MX MX2017004418A patent/MX370338B/es active IP Right Grant
- 2015-10-08 BR BR112017007293-9A patent/BR112017007293B1/pt active IP Right Grant
- 2015-10-08 WO PCT/JP2015/078688 patent/WO2016056638A1/ja active Application Filing
- 2015-10-08 CN CN201580055101.1A patent/CN107106568B/zh active Active
- 2015-10-08 ES ES15848771T patent/ES2909797T3/es active Active
- 2015-10-08 SG SG11201702632QA patent/SG11201702632QA/en unknown
- 2015-10-08 EP EP15848771.0A patent/EP3205343B1/en active Active
- 2015-10-08 RU RU2017111801A patent/RU2710227C2/ru active
- 2015-10-08 US US15/516,567 patent/US10117876B2/en active Active
- 2015-10-08 AU AU2015329027A patent/AU2015329027B2/en active Active
- 2015-10-08 CA CA2963761A patent/CA2963761C/en active Active
-
2017
- 2017-04-04 IL IL251559A patent/IL251559B/en active IP Right Grant
-
2019
- 2019-07-26 JP JP2019137458A patent/JP2019196395A/ja active Pending
-
2020
- 2020-04-21 JP JP2020075659A patent/JP2020125330A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| KR102524217B1 (ko) | 2023-04-21 |
| CA2963761A1 (en) | 2016-04-14 |
| SG11201702632QA (en) | 2017-05-30 |
| WO2016056638A1 (ja) | 2016-04-14 |
| BR112017007293B1 (pt) | 2022-12-13 |
| IL251559A0 (en) | 2017-05-29 |
| JP6590822B2 (ja) | 2019-10-16 |
| JPWO2016056638A1 (ja) | 2017-07-20 |
| EP3205343A1 (en) | 2017-08-16 |
| CN107106568A (zh) | 2017-08-29 |
| RU2017111801A (ru) | 2018-11-13 |
| US10117876B2 (en) | 2018-11-06 |
| RU2710227C2 (ru) | 2019-12-25 |
| JP2020125330A (ja) | 2020-08-20 |
| CN107106568B (zh) | 2023-09-26 |
| AU2015329027A1 (en) | 2017-04-27 |
| RU2017111801A3 (es) | 2019-02-12 |
| IL251559B (en) | 2021-02-28 |
| US20180263998A1 (en) | 2018-09-20 |
| BR112017007293A2 (pt) | 2017-12-26 |
| CA2963761C (en) | 2023-10-17 |
| KR20170066397A (ko) | 2017-06-14 |
| AU2015329027B2 (en) | 2020-04-30 |
| MX370338B (es) | 2019-12-10 |
| EP3205343A4 (en) | 2018-06-06 |
| JP2019196395A (ja) | 2019-11-14 |
| MX2017004418A (es) | 2017-06-26 |
| EP3205343B1 (en) | 2022-01-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2954514T3 (es) | Compuestos terapéuticos útiles para el tratamiento profiláctico o terapéutico de una infección por el virus del VIH | |
| ES2947819T3 (es) | Método para el tratamiento del cáncer con N-((4,6-dimetil-2-oxo-1,2-dihidropiridin-3-il)metil)-5-(etil(tetrahidro-2H-piran-4-il)amino)-4-metil-4'-(morfolinometil)-[1,1'-bifenil]-3-carboxamida | |
| ES2429160T3 (es) | (R)-N-metilnaltrexona, procesos para su síntesis y su uso farmacéutico | |
| ES2397746T3 (es) | Métodos de administración de tetrahidrobiopterina, composiciones asociadas y métodos de medición | |
| ES2820536T3 (es) | Composición farmacéutica novedosa | |
| ES2909797T3 (es) | Composición farmacéutica de derivado de aminodihidrotiazina condensado | |
| EP2524689B1 (en) | Method for producing donepezil tablets | |
| WO2014027334A2 (es) | Composicion farmacéutica oral en forma de microesferas y proceso de elaboración | |
| ES2881317T3 (es) | Composición farmacéutica que comprende sacubitrilo y valsartán | |
| ES2535106T3 (es) | Composiciones farmacéuticas de rosuvastatina cálcica | |
| ES2359647T3 (es) | Formulación oral de olanzapina anhídra forma i. | |
| ES2959243T3 (es) | Nuevo tratamiento de enfermedades mitocondriales | |
| ES2908924T3 (es) | Composiciones orales sólidas de liberación lenta | |
| EP2568957A1 (en) | Pharmaceutical composition comprising cefixime and clavulanic acid derivative compound | |
| JPWO2013172297A1 (ja) | 6,7−不飽和−7−カルバモイルモルヒナン誘導体含有製剤 | |
| JP4808246B2 (ja) | 医薬組成物 | |
| ES2953126T3 (es) | Un método para fabricar una composición farmacéutica que comprende Nefopam y Acetaminofén, y la composición farmacéutica obtenida de esta manera | |
| JP2020117475A (ja) | ラメルテオン含有フィルムコーティング錠剤 | |
| JP7210794B2 (ja) | リナグリプチン含有製剤及びリナグリプチン含有口腔内崩壊性錠剤 | |
| ES2313578T3 (es) | Formulacion estable que comprende una combinacion de un farmaco sensible a la humedad y un segundo farmaco y procedimiento de preparacion de la misma. | |
| JPWO2004078173A1 (ja) | 溶出性の改善された錠剤 | |
| ES2533613T3 (es) | Método de producción de comprimidos farmacéuticos | |
| US20190070167A1 (en) | Pitavastatin containing preparation and method for producing same | |
| JP2013216644A (ja) | イマチニブメシル酸塩経口投与製剤 |