ES2872555T3 - Inhibidores de HPK1 y métodos de uso de los mismos - Google Patents
Inhibidores de HPK1 y métodos de uso de los mismos Download PDFInfo
- Publication number
- ES2872555T3 ES2872555T3 ES16813437T ES16813437T ES2872555T3 ES 2872555 T3 ES2872555 T3 ES 2872555T3 ES 16813437 T ES16813437 T ES 16813437T ES 16813437 T ES16813437 T ES 16813437T ES 2872555 T3 ES2872555 T3 ES 2872555T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- heterocyclyl
- membered
- optionally substituted
- cycloalkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000003112 inhibitor Substances 0.000 title claims description 18
- 102100028199 Mitogen-activated protein kinase kinase kinase kinase 1 Human genes 0.000 title description 43
- 238000000034 method Methods 0.000 title description 22
- 101001059991 Homo sapiens Mitogen-activated protein kinase kinase kinase kinase 1 Proteins 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 148
- 150000003839 salts Chemical class 0.000 claims abstract description 31
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 21
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims abstract description 17
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 15
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 15
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 13
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 13
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims abstract description 11
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims abstract description 7
- 125000006714 (C3-C10) heterocyclyl group Chemical group 0.000 claims abstract description 5
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 claims abstract description 4
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims abstract description 4
- 229910003827 NRaRb Inorganic materials 0.000 claims abstract description 3
- 229910052705 radium Inorganic materials 0.000 claims abstract description 3
- 229910052701 rubidium Inorganic materials 0.000 claims abstract description 3
- -1 N-piperazinyl Chemical group 0.000 claims description 72
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 70
- 206010028980 Neoplasm Diseases 0.000 claims description 58
- 201000011510 cancer Diseases 0.000 claims description 43
- 238000011282 treatment Methods 0.000 claims description 17
- 239000002246 antineoplastic agent Substances 0.000 claims description 12
- 239000003814 drug Substances 0.000 claims description 9
- 230000002519 immonomodulatory effect Effects 0.000 claims description 9
- 125000002757 morpholinyl group Chemical group 0.000 claims description 9
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- 239000003795 chemical substances by application Substances 0.000 claims description 7
- 125000002950 monocyclic group Chemical group 0.000 claims description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 6
- 239000003085 diluting agent Substances 0.000 claims description 4
- 208000003837 Second Primary Neoplasms Diseases 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 125000004193 piperazinyl group Chemical group 0.000 claims description 3
- 125000003386 piperidinyl group Chemical group 0.000 claims description 3
- 101001037261 Homo sapiens Indoleamine 2,3-dioxygenase 2 Proteins 0.000 claims description 2
- 101000892398 Homo sapiens Tryptophan 2,3-dioxygenase Proteins 0.000 claims description 2
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 claims description 2
- 102100040061 Indoleamine 2,3-dioxygenase 1 Human genes 0.000 claims description 2
- 102100040062 Indoleamine 2,3-dioxygenase 2 Human genes 0.000 claims description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 claims description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 claims description 2
- 102100040653 Tryptophan 2,3-dioxygenase Human genes 0.000 claims description 2
- 229940127089 cytotoxic agent Drugs 0.000 claims description 2
- 125000001475 halogen functional group Chemical group 0.000 claims description 2
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 claims description 2
- 230000003647 oxidation Effects 0.000 claims description 2
- 238000007254 oxidation reaction Methods 0.000 claims description 2
- 230000005855 radiation Effects 0.000 claims description 2
- 238000001356 surgical procedure Methods 0.000 claims description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 2
- 229940124597 therapeutic agent Drugs 0.000 claims description 2
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims 1
- 125000004043 oxo group Chemical group O=* 0.000 claims 1
- 125000003545 alkoxy group Chemical group 0.000 abstract description 4
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 abstract 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 105
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 80
- 239000000203 mixture Substances 0.000 description 51
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 45
- 108010002838 hematopoietic progenitor kinase 1 Proteins 0.000 description 42
- 238000006243 chemical reaction Methods 0.000 description 41
- 239000000243 solution Substances 0.000 description 40
- 206010012812 Diffuse cutaneous mastocytosis Diseases 0.000 description 39
- 239000007787 solid Substances 0.000 description 38
- 238000005160 1H NMR spectroscopy Methods 0.000 description 34
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 29
- 210000004027 cell Anatomy 0.000 description 24
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 24
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 24
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 23
- 239000011541 reaction mixture Substances 0.000 description 22
- 235000019439 ethyl acetate Nutrition 0.000 description 21
- 230000000694 effects Effects 0.000 description 20
- 241001465754 Metazoa Species 0.000 description 19
- 238000007429 general method Methods 0.000 description 19
- 230000002829 reductive effect Effects 0.000 description 18
- 238000003756 stirring Methods 0.000 description 18
- 230000005764 inhibitory process Effects 0.000 description 17
- 210000001744 T-lymphocyte Anatomy 0.000 description 16
- 239000012043 crude product Substances 0.000 description 16
- 238000003818 flash chromatography Methods 0.000 description 16
- 239000000047 product Substances 0.000 description 15
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 14
- 239000000284 extract Substances 0.000 description 14
- 239000011734 sodium Substances 0.000 description 14
- 239000000725 suspension Substances 0.000 description 14
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 13
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 13
- 239000003921 oil Substances 0.000 description 13
- 125000004195 4-methylpiperazin-1-yl group Chemical group [H]C([H])([H])N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 12
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 12
- 238000000746 purification Methods 0.000 description 12
- 206010006187 Breast cancer Diseases 0.000 description 11
- 208000026310 Breast neoplasm Diseases 0.000 description 11
- 241000699670 Mus sp. Species 0.000 description 11
- 125000003118 aryl group Chemical group 0.000 description 11
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 11
- IUVCFHHAEHNCFT-INIZCTEOSA-N 2-[(1s)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one Chemical compound C1=C(F)C(OC(C)C)=CC=C1C(C1=C(N)N=CN=C11)=NN1[C@@H](C)C1=C(C=2C=C(F)C=CC=2)C(=O)C2=CC(F)=CC=C2O1 IUVCFHHAEHNCFT-INIZCTEOSA-N 0.000 description 10
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 10
- 239000011575 calcium Substances 0.000 description 10
- 201000010099 disease Diseases 0.000 description 10
- 206010009944 Colon cancer Diseases 0.000 description 9
- 239000012270 PD-1 inhibitor Substances 0.000 description 9
- 239000012668 PD-1-inhibitor Substances 0.000 description 9
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 9
- 125000004432 carbon atom Chemical group C* 0.000 description 9
- 229940121655 pd-1 inhibitor Drugs 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 8
- 206010039491 Sarcoma Diseases 0.000 description 8
- 238000002953 preparative HPLC Methods 0.000 description 8
- 238000002560 therapeutic procedure Methods 0.000 description 8
- 239000008346 aqueous phase Substances 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 238000009169 immunotherapy Methods 0.000 description 7
- 230000026731 phosphorylation Effects 0.000 description 7
- 238000006366 phosphorylation reaction Methods 0.000 description 7
- 0 *c1c[s]c(N2)c1C(N*)=C(c1nc(ccc(N(*)*)c3)c3[n]1)C2=O Chemical compound *c1c[s]c(N2)c1C(N*)=C(c1nc(ccc(N(*)*)c3)c3[n]1)C2=O 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 6
- 229940125962 HPK1 kinase inhibitor Drugs 0.000 description 6
- 101001090688 Homo sapiens Lymphocyte cytosolic protein 2 Proteins 0.000 description 6
- 102100034709 Lymphocyte cytosolic protein 2 Human genes 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- 208000032383 Soft tissue cancer Diseases 0.000 description 6
- 125000000217 alkyl group Chemical group 0.000 description 6
- 230000012010 growth Effects 0.000 description 6
- 229960003301 nivolumab Drugs 0.000 description 6
- 229960002621 pembrolizumab Drugs 0.000 description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 6
- 150000003254 radicals Chemical class 0.000 description 6
- 125000006413 ring segment Chemical group 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- 101000932478 Homo sapiens Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 5
- 206010033128 Ovarian cancer Diseases 0.000 description 5
- 206010061535 Ovarian neoplasm Diseases 0.000 description 5
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 5
- 125000003342 alkenyl group Chemical group 0.000 description 5
- 235000019270 ammonium chloride Nutrition 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 229910052799 carbon Inorganic materials 0.000 description 5
- 208000029742 colonic neoplasm Diseases 0.000 description 5
- 238000004440 column chromatography Methods 0.000 description 5
- HYMXUYQKXCHWDC-UHFFFAOYSA-N ethyl 3-ethoxy-3-iminopropanoate;hydrochloride Chemical compound Cl.CCOC(=N)CC(=O)OCC HYMXUYQKXCHWDC-UHFFFAOYSA-N 0.000 description 5
- 239000006260 foam Substances 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 239000012458 free base Substances 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 5
- 208000020816 lung neoplasm Diseases 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 5
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- ZCWXYZBQDNFULS-UHFFFAOYSA-N 5-chloro-2-nitroaniline Chemical compound NC1=CC(Cl)=CC=C1[N+]([O-])=O ZCWXYZBQDNFULS-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- 108010074708 B7-H1 Antigen Proteins 0.000 description 4
- 208000003174 Brain Neoplasms Diseases 0.000 description 4
- 101001047681 Homo sapiens Tyrosine-protein kinase Lck Proteins 0.000 description 4
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- 241000283973 Oryctolagus cuniculus Species 0.000 description 4
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 4
- 206010060862 Prostate cancer Diseases 0.000 description 4
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 102100024036 Tyrosine-protein kinase Lck Human genes 0.000 description 4
- 125000000304 alkynyl group Chemical group 0.000 description 4
- 238000002659 cell therapy Methods 0.000 description 4
- 210000004443 dendritic cell Anatomy 0.000 description 4
- 208000033068 episodic angioedema with eosinophilia Diseases 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 230000028993 immune response Effects 0.000 description 4
- 238000007912 intraperitoneal administration Methods 0.000 description 4
- 201000005202 lung cancer Diseases 0.000 description 4
- 230000003211 malignant effect Effects 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 230000003287 optical effect Effects 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- MPLUORBCRRXCBP-UHFFFAOYSA-N (3,4-diaminophenyl)-(4-methylpiperazin-1-yl)methanone Chemical compound C1CN(C)CCN1C(=O)C1=CC=C(N)C(N)=C1 MPLUORBCRRXCBP-UHFFFAOYSA-N 0.000 description 3
- WENXBQBOYXBODV-UHFFFAOYSA-N (3,4-dinitrophenyl)-(4-methylpiperazin-1-yl)methanone Chemical compound C1CN(C)CCN1C(=O)C1=CC=C([N+]([O-])=O)C([N+]([O-])=O)=C1 WENXBQBOYXBODV-UHFFFAOYSA-N 0.000 description 3
- ZXZLWWJIFPMIJL-UHFFFAOYSA-N (3,4-dinitrophenyl)-morpholin-4-ylmethanone Chemical compound C1=C([N+]([O-])=O)C([N+](=O)[O-])=CC=C1C(=O)N1CCOCC1 ZXZLWWJIFPMIJL-UHFFFAOYSA-N 0.000 description 3
- MOHYOXXOKFQHDC-UHFFFAOYSA-N 1-(chloromethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCl)C=C1 MOHYOXXOKFQHDC-UHFFFAOYSA-N 0.000 description 3
- IHOCPRONZDTJIO-UHFFFAOYSA-N 4-(4-methyl-1,4-diazepan-1-yl)benzene-1,2-diamine Chemical compound C1CN(C)CCCN1C1=CC=C(N)C(N)=C1 IHOCPRONZDTJIO-UHFFFAOYSA-N 0.000 description 3
- HCQIHJAOJIQOLC-UHFFFAOYSA-N 5-(4-methyl-1,4-diazepan-1-yl)-2-nitroaniline Chemical compound C1CN(C)CCCN1C1=CC=C([N+]([O-])=O)C(N)=C1 HCQIHJAOJIQOLC-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- 108091008875 B cell receptors Proteins 0.000 description 3
- 206010053717 Fibrous histiocytoma Diseases 0.000 description 3
- 201000010915 Glioblastoma multiforme Diseases 0.000 description 3
- 101000632319 Homo sapiens Septin-7 Proteins 0.000 description 3
- 102000000588 Interleukin-2 Human genes 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- 102000019149 MAP kinase activity proteins Human genes 0.000 description 3
- 108040008097 MAP kinase activity proteins Proteins 0.000 description 3
- 206010027480 Metastatic malignant melanoma Diseases 0.000 description 3
- 206010029260 Neuroblastoma Diseases 0.000 description 3
- 206010033799 Paralysis Diseases 0.000 description 3
- 239000012980 RPMI-1640 medium Substances 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 102100027981 Septin-7 Human genes 0.000 description 3
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 108091008874 T cell receptors Proteins 0.000 description 3
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 230000003213 activating effect Effects 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 230000006786 activation induced cell death Effects 0.000 description 3
- 150000001412 amines Chemical group 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 210000003719 b-lymphocyte Anatomy 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 230000005754 cellular signaling Effects 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 229950009791 durvalumab Drugs 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 208000005017 glioblastoma Diseases 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 150000003840 hydrochlorides Chemical class 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 239000000543 intermediate Substances 0.000 description 3
- 206010024627 liposarcoma Diseases 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 201000001441 melanoma Diseases 0.000 description 3
- 208000021039 metastatic melanoma Diseases 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 229950010773 pidilizumab Drugs 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 238000007363 ring formation reaction Methods 0.000 description 3
- 239000011593 sulfur Substances 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 3
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 3
- 238000002255 vaccination Methods 0.000 description 3
- 239000011534 wash buffer Substances 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- ZADWXFSZEAPBJS-SNVBAGLBSA-N (2r)-2-amino-3-(1-methylindol-3-yl)propanoic acid Chemical compound C1=CC=C2N(C)C=C(C[C@@H](N)C(O)=O)C2=C1 ZADWXFSZEAPBJS-SNVBAGLBSA-N 0.000 description 2
- MKWJZTFMDWSRIH-UHFFFAOYSA-N (4-fluoro-3-nitrophenyl)methanol Chemical compound OCC1=CC=C(F)C([N+]([O-])=O)=C1 MKWJZTFMDWSRIH-UHFFFAOYSA-N 0.000 description 2
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 description 2
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 2
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 2
- ONTPJHYNVVEOLP-UHFFFAOYSA-N 1-[(4-methoxyphenyl)methyl]thieno[3,2-d][1,3]oxazine-2,4-dione Chemical compound C1=CC(OC)=CC=C1CN1C(=O)OC(=O)C2=C1C=CS2 ONTPJHYNVVEOLP-UHFFFAOYSA-N 0.000 description 2
- FPLKSYDDWQIJCU-UHFFFAOYSA-N 2-amino-4-ethoxythiophene-3-carbonitrile Chemical compound CCOC1=CSC(N)=C1C#N FPLKSYDDWQIJCU-UHFFFAOYSA-N 0.000 description 2
- XVGHZFWFGXDIOU-UHFFFAOYSA-N 2-aminothiophene-3-carbonitrile Chemical compound NC=1SC=CC=1C#N XVGHZFWFGXDIOU-UHFFFAOYSA-N 0.000 description 2
- BQLAISVDXGXTNA-UHFFFAOYSA-N 4-fluoro-5-morpholin-4-yl-2-nitroaniline Chemical compound C1=C([N+]([O-])=O)C(N)=CC(N2CCOCC2)=C1F BQLAISVDXGXTNA-UHFFFAOYSA-N 0.000 description 2
- PFWLVRSDFJSMJU-UHFFFAOYSA-N 4-methyl-5-(4-methylpiperazin-1-yl)-2-nitroaniline Chemical compound C1CN(C)CCN1C1=CC(N)=C([N+]([O-])=O)C=C1C PFWLVRSDFJSMJU-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 108010075944 Erythropoietin Receptors Proteins 0.000 description 2
- 102100036509 Erythropoietin receptor Human genes 0.000 description 2
- 201000008808 Fibrosarcoma Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 description 2
- 208000018142 Leiomyosarcoma Diseases 0.000 description 2
- 206010024612 Lipoma Diseases 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 101150024075 Mapk1 gene Proteins 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 2
- 101100407308 Mus musculus Pdcd1lg2 gene Proteins 0.000 description 2
- CAUBWLYZCDDYEF-UHFFFAOYSA-N N-Nitroso-N-methylurethane Chemical compound CCOC(=O)N(C)N=O CAUBWLYZCDDYEF-UHFFFAOYSA-N 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- 108700030875 Programmed Cell Death 1 Ligand 2 Proteins 0.000 description 2
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 description 2
- 239000007868 Raney catalyst Substances 0.000 description 2
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 2
- 229910000564 Raney nickel Inorganic materials 0.000 description 2
- 206010038389 Renal cancer Diseases 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 230000006044 T cell activation Effects 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 108010018242 Transcription Factor AP-1 Proteins 0.000 description 2
- 102100023132 Transcription factor Jun Human genes 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 210000000577 adipose tissue Anatomy 0.000 description 2
- 229940009456 adriamycin Drugs 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 239000004479 aerosol dispenser Substances 0.000 description 2
- 238000011319 anticancer therapy Methods 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004204 blood vessel Anatomy 0.000 description 2
- 125000002837 carbocyclic group Chemical group 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 230000003828 downregulation Effects 0.000 description 2
- 102000015694 estrogen receptors Human genes 0.000 description 2
- 108010038795 estrogen receptors Proteins 0.000 description 2
- LBAQSKZHMLAFHH-UHFFFAOYSA-N ethoxyethane;hydron;chloride Chemical compound Cl.CCOCC LBAQSKZHMLAFHH-UHFFFAOYSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000012894 fetal calf serum Substances 0.000 description 2
- 206010016629 fibroma Diseases 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 125000004438 haloalkoxy group Chemical group 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 201000010536 head and neck cancer Diseases 0.000 description 2
- 208000014829 head and neck neoplasm Diseases 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 2
- 201000000284 histiocytoma Diseases 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 125000003037 imidazol-2-yl group Chemical group [H]N1C([*])=NC([H])=C1[H] 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 201000010982 kidney cancer Diseases 0.000 description 2
- 238000000021 kinase assay Methods 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 210000001365 lymphatic vessel Anatomy 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- 201000009020 malignant peripheral nerve sheath tumor Diseases 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 208000037819 metastatic cancer Diseases 0.000 description 2
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 2
- 208000007538 neurilemmoma Diseases 0.000 description 2
- 208000029974 neurofibrosarcoma Diseases 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 2
- RFIOZSIHFNEKFF-UHFFFAOYSA-M piperazine-1-carboxylate Chemical compound [O-]C(=O)N1CCNCC1 RFIOZSIHFNEKFF-UHFFFAOYSA-M 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 159000000001 potassium salts Chemical class 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 102000003998 progesterone receptors Human genes 0.000 description 2
- 108090000468 progesterone receptors Proteins 0.000 description 2
- 239000003380 propellant Substances 0.000 description 2
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 2
- 238000010814 radioimmunoprecipitation assay Methods 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 229930195734 saturated hydrocarbon Natural products 0.000 description 2
- 206010039667 schwannoma Diseases 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 235000017550 sodium carbonate Nutrition 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- PUZPDOWCWNUUKD-UHFFFAOYSA-M sodium fluoride Chemical compound [F-].[Na+] PUZPDOWCWNUUKD-UHFFFAOYSA-M 0.000 description 2
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- LESRBHHIRARKMI-UHFFFAOYSA-N tert-butyl 4-(3,4-diaminophenyl)piperazine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCN1C1=CC=C(N)C(N)=C1 LESRBHHIRARKMI-UHFFFAOYSA-N 0.000 description 2
- SQDDRYKOQRWTMK-UHFFFAOYSA-N tert-butyl 4-(3-amino-4-nitrophenyl)piperazine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCN1C1=CC=C([N+]([O-])=O)C(N)=C1 SQDDRYKOQRWTMK-UHFFFAOYSA-N 0.000 description 2
- XVRYCJXJTWMUOK-UHFFFAOYSA-N tert-butyl 4-(5-amino-4-cyanothiophen-3-yl)piperazine-1-carboxylate Chemical compound NC1=C(C(=CS1)N1CCN(CC1)C(=O)OC(C)(C)C)C#N XVRYCJXJTWMUOK-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 125000003831 tetrazolyl group Chemical group 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 2
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- OEKYJLOPJTXIRR-UHFFFAOYSA-N (3,4-diaminophenyl)-morpholin-4-ylmethanone Chemical compound C1=C(N)C(N)=CC=C1C(=O)N1CCOCC1 OEKYJLOPJTXIRR-UHFFFAOYSA-N 0.000 description 1
- VEEGZPWAAPPXRB-BJMVGYQFSA-N (3e)-3-(1h-imidazol-5-ylmethylidene)-1h-indol-2-one Chemical compound O=C1NC2=CC=CC=C2\C1=C/C1=CN=CN1 VEEGZPWAAPPXRB-BJMVGYQFSA-N 0.000 description 1
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 1
- 125000006708 (C5-C14) heteroaryl group Chemical group 0.000 description 1
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 1
- RFWSYCZAGQHTIG-UHFFFAOYSA-N 1-[(4-methoxyphenyl)methyl]-5-methylthieno[2,3-d][1,3]oxazine-2,4-dione Chemical compound COC1=CC=C(CN2C(OC(C3=C2SC=C3C)=O)=O)C=C1 RFWSYCZAGQHTIG-UHFFFAOYSA-N 0.000 description 1
- UTVFVQSOWWTMCW-UHFFFAOYSA-N 1-[(4-methoxyphenyl)methyl]thieno[2,3-d][1,3]oxazine-2,4-dione Chemical compound C1=CC(OC)=CC=C1CN1C(=O)OC(=O)C2=C1SC=C2 UTVFVQSOWWTMCW-UHFFFAOYSA-N 0.000 description 1
- MTJDCKZKPGOCGB-UHFFFAOYSA-N 1-[(4-methoxyphenyl)methyl]thieno[3,4-d][1,3]oxazine-2,4-dione Chemical compound COC1=CC=C(CN2C(OC(C=3C2=CSC=3)=O)=O)C=C1 MTJDCKZKPGOCGB-UHFFFAOYSA-N 0.000 description 1
- FXHRAKUEZPSMLJ-UHFFFAOYSA-N 1-methyl-1,4-diazepane Chemical compound CN1CCCNCC1 FXHRAKUEZPSMLJ-UHFFFAOYSA-N 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- MFJCPDOGFAYSTF-UHFFFAOYSA-N 1H-isochromene Chemical compound C1=CC=C2COC=CC2=C1 MFJCPDOGFAYSTF-UHFFFAOYSA-N 0.000 description 1
- IKHIUUFLWGIGMC-UHFFFAOYSA-N 1h-thieno[3,4-d][1,3]oxazine-2,4-dione Chemical compound O=C1OC(=O)NC2=CSC=C21 IKHIUUFLWGIGMC-UHFFFAOYSA-N 0.000 description 1
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 1
- LWTIGYSPAXKMDG-UHFFFAOYSA-N 2,3-dihydro-1h-imidazole Chemical compound C1NC=CN1 LWTIGYSPAXKMDG-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- KISWVXRQTGLFGD-UHFFFAOYSA-N 2-[[2-[[6-amino-2-[[2-[[2-[[5-amino-2-[[2-[[1-[2-[[6-amino-2-[(2,5-diamino-5-oxopentanoyl)amino]hexanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-(diaminomethylideneamino)p Chemical compound C1CCN(C(=O)C(CCCN=C(N)N)NC(=O)C(CCCCN)NC(=O)C(N)CCC(N)=O)C1C(=O)NC(CO)C(=O)NC(CCC(N)=O)C(=O)NC(CCCN=C(N)N)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=C(O)C=C1 KISWVXRQTGLFGD-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- YPDXBHMAPRUUPZ-AOOOYVTPSA-N 2-nitro-5-[(3S,5R)-3,4,5-trimethylpiperazin-1-yl]aniline Chemical compound [N+](=O)([O-])C1=C(N)C=C(C=C1)N1C[C@H](N([C@H](C1)C)C)C YPDXBHMAPRUUPZ-AOOOYVTPSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- UAIUNKRWKOVEES-UHFFFAOYSA-N 3,3',5,5'-tetramethylbenzidine Chemical compound CC1=C(N)C(C)=CC(C=2C=C(C)C(N)=C(C)C=2)=C1 UAIUNKRWKOVEES-UHFFFAOYSA-N 0.000 description 1
- UZJBHDNDSWAPQK-UHFFFAOYSA-N 3-ethoxy-3-iminopropanoic acid;hydrochloride Chemical compound Cl.CCOC(=N)CC(O)=O UZJBHDNDSWAPQK-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- ROADCYAOHVSOLQ-UHFFFAOYSA-N 3-oxetanone Chemical compound O=C1COC1 ROADCYAOHVSOLQ-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- WQHBFCKTRBCJEV-AOOOYVTPSA-N 4-[(3S,5R)-3,4,5-trimethylpiperazin-1-yl]benzene-1,2-diamine Chemical compound C[C@@H]1CN(C[C@@H](N1C)C)C=1C=C(C(=CC=1)N)N WQHBFCKTRBCJEV-AOOOYVTPSA-N 0.000 description 1
- ZZBIGOMBVGTIJA-UHFFFAOYSA-N 4-amino-5-(6-piperazin-1-yl-1H-benzimidazol-2-yl)-7H-thieno[2,3-b]pyridin-6-one Chemical compound NC=1C2=C(NC(C=1C1=NC3=C(N1)C=C(C=C3)N1CCNCC1)=O)SC=C2 ZZBIGOMBVGTIJA-UHFFFAOYSA-N 0.000 description 1
- HEMRQORZSSSFIX-UHFFFAOYSA-N 4-fluoro-5-morpholin-4-ylbenzene-1,2-diamine Chemical compound C1=C(N)C(N)=CC(F)=C1N1CCOCC1 HEMRQORZSSSFIX-UHFFFAOYSA-N 0.000 description 1
- FIWDORRZJLBDMW-UHFFFAOYSA-N 4-methyl-5-(4-methylpiperazin-1-yl)benzene-1,2-diamine Chemical compound CC1=CC(=C(C=C1N1CCN(CC1)C)N)N FIWDORRZJLBDMW-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical group [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- VRJKEIWZSOHDOH-UHFFFAOYSA-N 5-chloro-4-fluoro-2-nitroaniline Chemical compound NC1=CC(Cl)=C(F)C=C1[N+]([O-])=O VRJKEIWZSOHDOH-UHFFFAOYSA-N 0.000 description 1
- BBWHBSDZHSQECL-UHFFFAOYSA-N 5-chloro-4-methyl-2-nitroaniline Chemical compound CC1=CC([N+]([O-])=O)=C(N)C=C1Cl BBWHBSDZHSQECL-UHFFFAOYSA-N 0.000 description 1
- WBLHZJOTBCBRCB-UHFFFAOYSA-N 5-methyl-1h-thieno[2,3-d][1,3]oxazine-2,4-dione Chemical compound N1C(=O)OC(=O)C2=C1SC=C2C WBLHZJOTBCBRCB-UHFFFAOYSA-N 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- IFBSMSSIEBVQLS-HDICACEKSA-N 7-hydroxy-4-[(4-methoxyphenyl)methyl]-6-[6-[(3R,5S)-3,4,5-trimethylpiperazin-1-yl]-1H-benzimidazol-2-yl]thieno[3,2-b]pyridin-5-one Chemical compound OC=1C2=C(N(C(C=1C1=NC3=C(N1)C=C(C=C3)N1C[C@H](N([C@H](C1)C)C)C)=O)CC1=CC=C(C=C1)OC)C=CS2 IFBSMSSIEBVQLS-HDICACEKSA-N 0.000 description 1
- MQJNETSYXIIIKE-UHFFFAOYSA-N 7h-thieno[2,3-b]pyridin-6-one Chemical compound N1C(=O)C=CC2=C1SC=C2 MQJNETSYXIIIKE-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 208000010507 Adenocarcinoma of Lung Diseases 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 208000037540 Alveolar soft tissue sarcoma Diseases 0.000 description 1
- 201000003076 Angiosarcoma Diseases 0.000 description 1
- 102000004452 Arginase Human genes 0.000 description 1
- 108700024123 Arginases Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 102000019260 B-Cell Antigen Receptors Human genes 0.000 description 1
- 108010012919 B-Cell Antigen Receptors Proteins 0.000 description 1
- 230000003844 B-cell-activation Effects 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 208000035821 Benign schwannoma Diseases 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 101001018362 Bos taurus Myelin basic protein Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- LPHGDWGNISTTDV-UHFFFAOYSA-N CC(OC(C1=C(C)[U]C=C1C)=O)=O Chemical compound CC(OC(C1=C(C)[U]C=C1C)=O)=O LPHGDWGNISTTDV-UHFFFAOYSA-N 0.000 description 1
- UDAVIPFGPZCLEF-UHFFFAOYSA-N CN(CCC1)CCN1c(cc(c([N+]([O-])=O)c1)N)c1F Chemical compound CN(CCC1)CCN1c(cc(c([N+]([O-])=O)c1)N)c1F UDAVIPFGPZCLEF-UHFFFAOYSA-N 0.000 description 1
- 229940045513 CTLA4 antagonist Drugs 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- 206010073140 Clear cell sarcoma of soft tissue Diseases 0.000 description 1
- 235000019750 Crude protein Nutrition 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 1
- 208000008743 Desmoplastic Small Round Cell Tumor Diseases 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 201000005231 Epithelioid sarcoma Diseases 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 201000005618 Glomus Tumor Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 1
- 108010069236 Goserelin Proteins 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 208000002125 Hemangioendothelioma Diseases 0.000 description 1
- 208000006050 Hemangiopericytoma Diseases 0.000 description 1
- 208000001258 Hemangiosarcoma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 1
- 101001037256 Homo sapiens Indoleamine 2,3-dioxygenase 1 Proteins 0.000 description 1
- 101100519206 Homo sapiens PDCD1 gene Proteins 0.000 description 1
- 101000611936 Homo sapiens Programmed cell death protein 1 Proteins 0.000 description 1
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101001052849 Homo sapiens Tyrosine-protein kinase Fer Proteins 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 102000002227 Interferon Type I Human genes 0.000 description 1
- 108010014726 Interferon Type I Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 101150028321 Lck gene Proteins 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 206010025219 Lymphangioma Diseases 0.000 description 1
- 108010002481 Lymphocyte Specific Protein Tyrosine Kinase p56(lck) Proteins 0.000 description 1
- 102000036243 Lymphocyte Specific Protein Tyrosine Kinase p56(lck) Human genes 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 239000007993 MOPS buffer Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 1
- FBKMWOJEPMPVTQ-UHFFFAOYSA-N N'-(3-bromo-4-fluorophenyl)-N-hydroxy-4-[2-(sulfamoylamino)ethylamino]-1,2,5-oxadiazole-3-carboximidamide Chemical compound NS(=O)(=O)NCCNC1=NON=C1C(=NO)NC1=CC=C(F)C(Br)=C1 FBKMWOJEPMPVTQ-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- 241001274216 Naso Species 0.000 description 1
- 201000004404 Neurofibroma Diseases 0.000 description 1
- 208000005890 Neuroma Diseases 0.000 description 1
- DZRUMEJQJCSDEM-UHFFFAOYSA-N OC=1C2=C(N(C(C=1C1=NC3=C(N1)C=C(C=C3)N1CCN(CC1)C)=O)CC1=CC=C(C=C1)OC)SC=C2 Chemical compound OC=1C2=C(N(C(C=1C1=NC3=C(N1)C=C(C=C3)N1CCN(CC1)C)=O)CC1=CC=C(C=C1)OC)SC=C2 DZRUMEJQJCSDEM-UHFFFAOYSA-N 0.000 description 1
- COUBHIVFLSMYQQ-UHFFFAOYSA-N OC=1C2=C(N(C(C=1C1=NC3=C(N1)C=C(C=C3)N1CCN(CC1)C)=O)CC1=CC=C(C=C1)OC)SC=C2C Chemical compound OC=1C2=C(N(C(C=1C1=NC3=C(N1)C=C(C=C3)N1CCN(CC1)C)=O)CC1=CC=C(C=C1)OC)SC=C2C COUBHIVFLSMYQQ-UHFFFAOYSA-N 0.000 description 1
- SBRAGGMAFKQVKR-UHFFFAOYSA-N OC=1C2=C(N(C(C=1C1=NC3=C(N1)C=C(C=C3)N1CCOCC1)=O)CC1=CC=C(C=C1)OC)SC=C2 Chemical compound OC=1C2=C(N(C(C=1C1=NC3=C(N1)C=C(C=C3)N1CCOCC1)=O)CC1=CC=C(C=C1)OC)SC=C2 SBRAGGMAFKQVKR-UHFFFAOYSA-N 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 101150087384 PDCD1 gene Proteins 0.000 description 1
- 239000002033 PVDF binder Substances 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 108010081690 Pertussis Toxin Proteins 0.000 description 1
- 201000010395 Pleomorphic liposarcoma Diseases 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 208000034541 Rare lymphatic malformation Diseases 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- 208000005678 Rhabdomyoma Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- 206010068771 Soft tissue neoplasm Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102100024537 Tyrosine-protein kinase Fer Human genes 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- 208000002495 Uterine Neoplasms Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 150000001243 acetic acids Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 229940124650 anti-cancer therapies Drugs 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 230000000118 anti-neoplastic effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000003101 antineoplastic hormone agonist and antagonist Substances 0.000 description 1
- 230000005975 antitumor immune response Effects 0.000 description 1
- 238000003782 apoptosis assay Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 1
- 125000005160 aryl oxy alkyl group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 208000001119 benign fibrous histiocytoma Diseases 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- OWMVSZAMULFTJU-UHFFFAOYSA-N bis-tris Chemical compound OCCN(CCO)C(CO)(CO)CO OWMVSZAMULFTJU-UHFFFAOYSA-N 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 238000002619 cancer immunotherapy Methods 0.000 description 1
- 238000009566 cancer vaccine Methods 0.000 description 1
- 229940022399 cancer vaccine Drugs 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000004452 carbocyclyl group Chemical group 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000003729 cation exchange resin Substances 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- 238000000633 chiral stationary phase gas chromatography Methods 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- VZWXIQHBIQLMPN-UHFFFAOYSA-N chromane Chemical compound C1=CC=C2CCCOC2=C1 VZWXIQHBIQLMPN-UHFFFAOYSA-N 0.000 description 1
- QZHPTGXQGDFGEN-UHFFFAOYSA-N chromene Chemical compound C1=CC=C2C=C[CH]OC2=C1 QZHPTGXQGDFGEN-UHFFFAOYSA-N 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 201000000292 clear cell sarcoma Diseases 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 235000019784 crude fat Nutrition 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000006317 cyclopropyl amino group Chemical group 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 230000007402 cytotoxic response Effects 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 125000000723 dihydrobenzofuranyl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 1
- 125000004582 dihydrobenzothienyl group Chemical group S1C(CC2=C1C=CC=C2)* 0.000 description 1
- 125000005435 dihydrobenzoxazolyl group Chemical group O1C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000001070 dihydroindolyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004611 dihydroisoindolyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000005045 dihydroisoquinolinyl group Chemical group C1(NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 125000005044 dihydroquinolinyl group Chemical group N1(CC=CC2=CC=CC=C12)* 0.000 description 1
- JMRYOSQOYJBDOI-UHFFFAOYSA-N dilithium;di(propan-2-yl)azanide Chemical compound [Li+].CC(C)[N-]C(C)C.CC(C)N([Li])C(C)C JMRYOSQOYJBDOI-UHFFFAOYSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 1
- 229960002986 dinoprostone Drugs 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 208000029509 elastofibroma dorsi Diseases 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 238000000119 electrospray ionisation mass spectrum Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- YJGVMLPVUAXIQN-UHFFFAOYSA-N epipodophyllotoxin Natural products COC1=C(OC)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YJGVMLPVUAXIQN-UHFFFAOYSA-N 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- SYFFHRPDTQNMQB-UHFFFAOYSA-N ethyl 3-oxopropanoate Chemical compound CCOC(=O)CC=O SYFFHRPDTQNMQB-UHFFFAOYSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 206010049444 fibromatosis Diseases 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- 102000054766 genetic haplotypes Human genes 0.000 description 1
- 201000005626 glomangioma Diseases 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 229960002913 goserelin Drugs 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 210000000777 hematopoietic system Anatomy 0.000 description 1
- 125000005114 heteroarylalkoxy group Chemical group 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 208000027833 hibernoma Diseases 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 125000002962 imidazol-1-yl group Chemical group [*]N1C([H])=NC([H])=C1[H] 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000005746 immune checkpoint blockade Effects 0.000 description 1
- 230000006058 immune tolerance Effects 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 229940127121 immunoconjugate Drugs 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 102000006639 indoleamine 2,3-dioxygenase Human genes 0.000 description 1
- 108020004201 indoleamine 2,3-dioxygenase Proteins 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 229950009034 indoximod Drugs 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000012678 infectious agent Substances 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 108091008042 inhibitory receptors Proteins 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 229960005386 ipilimumab Drugs 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- HEBMCVBCEDMUOF-UHFFFAOYSA-N isochromane Chemical compound C1=CC=C2COCCC2=C1 HEBMCVBCEDMUOF-UHFFFAOYSA-N 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 210000005067 joint tissue Anatomy 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960004891 lapatinib Drugs 0.000 description 1
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 1
- 208000027906 leg weakness Diseases 0.000 description 1
- 201000010260 leiomyoma Diseases 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- 208000010033 lipoblastoma Diseases 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 201000005249 lung adenocarcinoma Diseases 0.000 description 1
- 208000037841 lung tumor Diseases 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- BCVXHSPFUWZLGQ-UHFFFAOYSA-N mecn acetonitrile Chemical compound CC#N.CC#N BCVXHSPFUWZLGQ-UHFFFAOYSA-N 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical class CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- PLLLBSPBFALBFB-UHFFFAOYSA-N methyl 2-amino-4-methylthiophene-3-carboxylate Chemical compound COC(=O)C=1C(C)=CSC=1N PLLLBSPBFALBFB-UHFFFAOYSA-N 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 229940126619 mouse monoclonal antibody Drugs 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- WOOWBQQQJXZGIE-UHFFFAOYSA-N n-ethyl-n-propan-2-ylpropan-2-amine Chemical compound CCN(C(C)C)C(C)C.CCN(C(C)C)C(C)C WOOWBQQQJXZGIE-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 238000003333 near-infrared imaging Methods 0.000 description 1
- 210000000944 nerve tissue Anatomy 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 238000003305 oral gavage Methods 0.000 description 1
- 239000007935 oral tablet Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 229940127084 other anti-cancer agent Drugs 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-M perchlorate Inorganic materials [O-]Cl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-M 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 238000003566 phosphorylation assay Methods 0.000 description 1
- 229960001237 podophyllotoxin Drugs 0.000 description 1
- YJGVMLPVUAXIQN-XVVDYKMHSA-N podophyllotoxin Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H]3[C@@H]2C(OC3)=O)=C1 YJGVMLPVUAXIQN-XVVDYKMHSA-N 0.000 description 1
- YVCVYCSAAZQOJI-UHFFFAOYSA-N podophyllotoxin Natural products COC1=C(O)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YVCVYCSAAZQOJI-UHFFFAOYSA-N 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 210000001948 pro-b lymphocyte Anatomy 0.000 description 1
- 230000005522 programmed cell death Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 description 1
- 239000003531 protein hydrolysate Substances 0.000 description 1
- 229940034080 provenge Drugs 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002206 pyridazin-3-yl group Chemical group [H]C1=C([H])C([H])=C(*)N=N1 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 1
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 210000003289 regulatory T cell Anatomy 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 230000007727 signaling mechanism Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- FQENQNTWSFEDLI-UHFFFAOYSA-J sodium diphosphate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]P([O-])(=O)OP([O-])([O-])=O FQENQNTWSFEDLI-UHFFFAOYSA-J 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 235000013024 sodium fluoride Nutrition 0.000 description 1
- 239000011775 sodium fluoride Substances 0.000 description 1
- 229940048086 sodium pyrophosphate Drugs 0.000 description 1
- 229940054269 sodium pyruvate Drugs 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000012089 stop solution Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000005415 substituted alkoxy group Chemical group 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid group Chemical class S(O)(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 208000016706 superficial Fibromatosis Diseases 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 210000002435 tendon Anatomy 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000005958 tetrahydrothienyl group Chemical group 0.000 description 1
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 1
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 1
- 235000019818 tetrasodium diphosphate Nutrition 0.000 description 1
- 239000001577 tetrasodium phosphonato phosphate Substances 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- IHIXIJGXTJIKRB-UHFFFAOYSA-N trisodium vanadate Chemical compound [Na+].[Na+].[Na+].[O-][V]([O-])([O-])=O IHIXIJGXTJIKRB-UHFFFAOYSA-N 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4355—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4365—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system having sulfur as a ring hetero atom, e.g. ticlopidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5355—Non-condensed oxazines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2818—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD28 or CD152
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Un compuesto representado por la fórmula (I-A): **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en el que: R es H, -F, -CI, -Br, -OH, -alquilo (C1-C4), -haloalquilo (C1-C4), -alcoxilo (C1-C4), -alquilen (C1-C4)-OH o heterociclilo monocíclico de 4-7 miembros opcionalmente sustituido con 1-3 grupos seleccionados de -F, - CI, -Br, -OH, -alquilo (C1-C4), -haloalquilo (C1-C4) o -alcoxilo (C1-C4); R1 es -NRaRb o -ORa1; Ra en cada aparición es independientemente -H, -alquilo (C1-C6), -(CH2)n-cicloalquilo (C3-C7), -(CH2)n- heterociclilo monocíclico de 3-7 miembros, -(CH2)n-cicloalquilo (C6-C12) en puente, -(CH2)n-heteroarilo de 5- 10 miembros opcionalmente sustituido; o -(CH2)n-heterociclilo en puente de 6-12 miembros, en el que el - alquilo (C1-C6), -(CH2)n-cicloalquilo (C3-C7), -(CH2)n-heterociclilo monocíclico de 3-7 miembros, -(CH2)n- cicloalquilo (C6-C12) en puente, -(CH2)n-heteroarilo de 5-10 miembros o -(CH2)n-heterociclilo en puente de 6- 12 miembros está opcionalmente sustituido con 1-3 grupos seleccionados de -F, -CI, -Br, -CN, -NH2, -OH, oxo, -alquilo (C1-C4), -haloalquilo (C1-C4), -alcoxilo (C1-C4), -haloalcoxilo (C1-C4), -alquilen (C1-C4)-OH o - alquilen (C1-C4)-NH2; Rb en cada aparición es independientemente -H o -alquilo (C1-C6); o Ra y Rb, junto con el nitrógeno al que están unidos, forman -heterociclilo (C3-C10); Ra1 en cada aparición es independientemente -H, alquilo (C1-C6), cicloalquilo (C3-C10), heterociclilo de 3-10 miembros, arilo (C6-C10) o heteroarilo de 3-10 miembros; R4 y R5, junto con el nitrógeno al que están unidos, forman heterociclilo monocíclico de 4-7 miembros o heterociclilo en puente de 6-12 miembros, en el que el heterociclilo monocíclico de 4-7 miembros o heterociclilo en puente de 6-12 miembros está opcionalmente sustituido con 1-3 grupos seleccionados de - F, -CI, -Br, -CN, -NH2, -OH, oxo, -alquilo (C1-C4), -haloalquilo (C1-C4), -alcoxilo (C1-C4), -haloalcoxilo (C1-C4), -alquilen (C1-C4)-OH o -alquilen (C1-C4)-NH2; R6 en cada aparición es independientemente -F, -CI, -Br, -CN, -NH2, -OH, -alquilo (C1-C6), - haloalquilo (C1- C6), -alquenilo (C2-C6), -alquinilo (C2-C6), cicloalquilo (C3-C6), -alcoxilo (C1-C6), -haloalcoxilo (C1-C6), - alquilen (C1-C6)-OH o -alquilen (C1-C6)-NH2; m es 0, 1, 2 o 3; y n es 0, 1 o 2.
Description
DESCRIPCIÓN
Inhibidores de HPK1 y métodos de uso de los mismos
Solicitudes relacionadas
Esta solicitud reivindica el beneficio de la solicitud provisional estadounidense n.° 62/184.348, presentada el 25 de junio de 2015.
Antecedentes
La cinasa de progenitores hematopoyéticos 1 (HPK1) es una serina/treonina cinasa Ste20 restringida a células hematopoyéticas. La actividad cinasa de HPK1 puede inducirse mediante la activación de señales generadas por diversos receptores de superficie celular diferentes encontrados en células hematopoyéticas tras la unión al ligando. La unión al ligando o la reticulación mediada por anticuerpo de los receptores de células T (TCR), el receptor del antígeno de células B (BCR) (Liou et al., 2000, Immunity 12:399), el receptor del factor de crecimiento transformante P (TGF-pR) (Wang et al., 1997. J. Biol. Chem. 272:22771; Zhou et al., 1999, J. Biol. Chem. 274:13133), el receptor de eritropoyetina (EPOR) (Nagata et al., 1999, Blood 93:3347) y Fas (Chen et al., 1999, Oncogene 18:7370) puede inducir la actividad cinasa de HPK1. Cada receptor utiliza mecanismos de señalización únicos, pero a veces superpuestos, para activar HPK1. La HPK1 actúa como modulador por disminución de las funciones de células T y B a través de las rutas de AP-1, NFKB, Erk2 y Fos; por ejemplo, HPK1 se ha implicado como regulador negativo de la transducción de señales en células T a través de la fosforilación y la activación de la proteína adaptadora del receptor de células T SLP-76 (Di Bartolo et al., 2007, J. Exp. Med. 204:681), lo que conduce a la posterior regulación por disminución de las rutas de AP-1 y Erk2. En las células B, HPK1 regula por disminución la señalización del receptor de células B (BCR) a través de la fosforilación del parálogo de SLP-76 BLINK (Wang et al., 2012, J. Biol. Chem. 287:11037).
Por tanto, HPK1 se visualiza ahora como posible diana para intervención terapéutica. Por ejemplo, se ha notificado que HPK1 puede ser una diana novedosa para la inmunoterapia contra el cáncer (Sawasdikosol et al., Immunol Res. Diciembre de 2012;54(1-3):262-5). Específicamente, la alteración dirigida de alelos de HPK1 confiere a las células T una elevada producción de citocinas Th1 en respuesta a la unión al TCR. Las células T HPK1 (-/-) proliferan más rápidamente que las homólogas silvestres con el mismo haplotipo y son resistentes a la supresión mediada por la prostaglandina E2 (PGE(2)). Lo más sorprendente es que ratones que recibieron transferencia adoptiva de células T HPK1 (-/-) se volvieron resistentes al crecimiento tumoral del pulmón. Además, la pérdida de HPK1 de las células dendríticas (DC) les otorga capacidad de presentación de antígenos superior, lo que permiten que las DC HPK1 (-/-) provoquen una respuesta inmunitaria antitumoral más potente cuando se usan como vacuna contra el cáncer.
Cuando se evalúa si un inhibidor de molécula pequeña de HPK1 capturaría el fenotipo de ratones con alteración dirigida del gen, es importante considerar los papeles no catalíticos de la proteína. En particular, aunque HPK1 de longitud completa puede fomentar la activación medida por TCR del potenciador de cadena ligera kappa del factor nuclear de la ruta de células B activadas (NF- k B), el producto de escisión catalíticamente inactivo HPK1-C puede suprimir la activación de NF- k B tras la reestimulación del TCR, lo que conduce a la muerte celular inducida por la activación (AICD) (Brenner et al., EMBO J. 2005, 24:4279). Tomando en conjunto los papeles catalíticos y no catalíticos de HPK1, es posible que el bloqueo de la actividad cinasa de HPK1 con un inhibidor de molécula pequeña pueda fomentar la activación de células B y T, lo que conduce a una inmunidad antitumoral superior, aunque también facilita la AICD, ayudando a mantener la tolerancia inmunitaria periférica. Los efectos exactos de un inhibidor de HPK1 se corroborarían mediante pruebas en modelos de ratón de cáncer, tales como xenoinjertos tumorales singénicos. Dado que HPK1 no se expresa en ningún órgano principal, fuera del sistema hematopoyético, es menos probable que un inhibidor de la actividad cinasa de HPK1 provoque algún efecto secundario grave.
En vista de lo anterior, existe la necesidad en la técnica de compuestos novedosos que puedan inhibir HPK1.
Sumario de la invención
El solicitante ha descubierto ahora que determinados compuestos de tienopiridinona son inhibidores de HPK1 (véase el ejemplo B). También tienen actividades inhibidoras contra FLT3 y LCK (véase el ejemplo C). Además, se ha demostrado que determinados compuestos de tienopiridinona, como inhibidores de HPK1 sola, y en combinación con anticuerpos anti-PD-1, son eficaces en modelos preclínicos con determinados tipos de célula cancerosas (véase el ejemplo E). Las terapias de combinación particulares dadas a conocer en el presente documento demuestran una actividad biológica sorprendente con efectos anticancerígenos significativos. Específicamente, con la combinación de inhibidores de HPK1 y anticuerpos anti-PD-1, ahora se han demostrado respuestas significativas tras el bloqueo de PD-1/PD-L1 en carcinoma de colon CT26.WT. Basándose en estos descubrimientos, en el presente documento se dan a conocer compuestos de tienopiridinona, composiciones farmacéuticas de los mismos y métodos de uso de los mismos.
Las realizaciones de la invención son tal como se definen en las reivindicaciones adjuntas. A continuación se describen realizaciones ilustrativas adicionales.
Una realización de la divulgación es un compuesto representado por la fórmula estructural (I):

o una sal farmacéuticamente aceptable del mismo. Los valores para cada una de las variables se proporcionan a continuación.
Otra realización de la divulgación es una composición farmacéutica que comprende un portador o diluyente farmacéuticamente aceptable y un compuesto representado por la fórmula estructural (I) descrita anteriormente o una sal farmacéuticamente aceptable del mismo.
Otra realización de la divulgación es un método de tratamiento de un sujeto con una enfermedad que puede regularse por HPK1, que comprende administrar al sujeto una cantidad eficaz de un compuesto de fórmula estructural (I) o una sal farmacéuticamente aceptable del mismo.
Otra realización es un método de inhibición de la actividad de HPK1 en un sujeto que necesita la inhibición de la actividad de HPK1, que comprende administrar al sujeto una cantidad eficaz de un compuesto representado por la fórmula estructural (I) o una sal farmacéuticamente aceptable del mismo.
Otra realización es un compuesto representado por la fórmula estructural (I) o una sal farmacéuticamente aceptable del mismo para su uso en terapia. En algunas realizaciones, la terapia es para tratar a un sujeto con cáncer. Alternativamente, la terapia es para inhibir la actividad de HPK1 en un sujeto que necesita la inhibición de la actividad de HPK1.
Otra realización es el uso de un compuesto representado por la fórmula estructural (I) o una sal farmacéuticamente aceptable del mismo para la fabricación de un medicamento para tratar a un sujeto con cáncer.
Otra realización es el uso de un compuesto representado por la fórmula estructural (I) o una sal farmacéuticamente aceptable del mismo para la fabricación de un medicamento para inhibir la actividad de HPK1 en un sujeto que necesita la inhibición de la actividad de HPK1.
La presente divulgación también se refiere a un método de tratamiento de un sujeto con cáncer, que comprende administrar al sujeto una cantidad eficaz de un inhibidor de HPK1 (por ejemplo, un compuesto representado por la fórmula estructural (I)), o una sal farmacéuticamente aceptable del mismo, y un segundo tratamiento contra el cáncer eficaz (por ejemplo, un agente quimioterápico, un agente terapéutico dirigido, radiación o cirugía). En un ejemplo, el segundo tratamiento contra el cáncer es un inhibidor de PD-1.
La presente divulgación también se refiere a un método de tratamiento de un sujeto con cáncer, que comprende administrar al sujeto una cantidad eficaz de un inhibidor de HPK1 (por ejemplo, un compuesto representado por la fórmula estructural (I)), o una sal farmacéuticamente aceptable del mismo, y una cantidad eficaz de un agente inmunomodulador, tal como un inhibidor del punto de control (por ejemplo, anticuerpo anti-PD-1, anticuerpo anti-CTLA4 o anticuerpo anti-PD-L1), o un inhibidor de la oxidación del triptófano (por ejemplo, inhibidor de IDO1, IDO2 o TDO2). En un ejemplo, el agente inmunomodulador es anticuerpo anti-PD-1.
En una realización, la presente divulgación proporciona además el uso de un inhibidor de HPK1 (por ejemplo, un compuesto representado por la fórmula estructural (I), o una sal farmacéuticamente aceptable del mismo), para la fabricación de un medicamento para el tratamiento de un sujeto con cáncer, en combinación con un inhibidor de PD-1, tal como nivolumab, pembrolizumab, pidilizumab, BMS 936559, MPDL3280A, MSB0010718C o MEDI4736. Preferiblemente, el inhibidor de PD-1 es nivolumab. Alternativamente, el inhibidor de PD-1 es pembrolizumab. En una realización, el inhibidor de PD-1 es anticuerpo anti-PD1.
En una alternativa, el inhibidor de HPK1 se administra con una cantidad eficaz de una o más de otras terapias contra el cáncer, y preferiblemente en combinación con un inhibidor de PD-1.
Breve descripción de los dibujos
La figura 1 muestra el efecto inhibidor del compuesto ejemplo A30 contra la fosforilación de serina 376 de SLP-76 en células Jurkat E6.1 estimuladas con a -CD3.
La figura 2 es un gráfico que ilustra el porcentaje de inhibición del crecimiento tumoral tras la administración de compuesto A1 solo y en combinación con un anticuerpo anti-PD1.
La figura 3 muestra el efecto del compuesto ejemplo A30 en el modelo de progresión de la enfermedad por EAE. Descripción detallada de la invención
En una primera realización, la divulgación se refiere a un compuesto representado por la fórmula (I):

o a una sal farmacéuticamente aceptable del mismo, en el que:
uno de X1, X2,y X3 es S, los otros dos son cada uno independientemente CR, en el que R es -H, -F, -Cl, -Br, -CN, -NH2, -OH, alquilo (C1-C6) opcionalmente sustituido, alcoxilo (C1-C6) opcionalmente sustituido, -(CH2)n-cicloalquilo (C3-C10) opcionalmente sustituido, -(CH2)n-heterociclilo monocíclico de 3-7 miembros opcionalmente sustituido, -(CH2)n-fenilo opcionalmente sustituido, -(CH2)n-heteroarilo monocíclico de 5-7 miembros opcionalmente sustituido, -(CH2)n-cicloalquilo (C6-C12) en puente opcionalmente sustituido, -(CH2)n-heterociclilo en puente de 6-12 miembros opcionalmente sustituido, -(CH2)n-heteroarilo bicíclico de 7-12 miembros opcionalmente sustituido o -(CH2)nheteroarilo bicíclico de 7-12 miembros opcionalmente sustituido;
Y es un enlace, -CH2-, -C(=O)-;
R1 es -NRaRb o -ORa1;
Ra en cada aparición es independientemente -H, alquilo (Ci-C6) opcionalmente sustituido, -(CH2)n-cicloalquilo (C3-Ci0) opcionalmente sustituido, -(CH2)n-heterociclilo de 3-10 miembros opcionalmente sustituido, -(CH2)n-arilo (C6-C10) opcionalmente sustituido, -(CH2)n-heteroarilo de 5-10 miembros opcionalmente sustituido, -(CH2)ncicloalquilo (C6-C12) en puente opcionalmente sustituido o -(CH2)n-heterociclilo en puente de 6-12 miembros opcionalmente sustituido;
Rb en cada aparición es independientemente -H o -alquilo (C1-C6); o,
Ra y Rb, junto con el nitrógeno al que están unidos, forman -heterociclilo (C3-C10) opcionalmente sustituido;
Ra1 en cada aparición es independientemente -H, alquilo (C1-C6) opcionalmente sustituido, cicloalquilo (C3-C10) opcionalmente sustituido, heterociclilo de 3-10 miembros opcionalmente sustituido, arilo (C6-C10) opcionalmente sustituido o heteroarilo de 3-10 miembros opcionalmente sustituido; o
R2 y R3 son cada uno independientemente -H o -alquilo (C1-C6);
R4 y R5 son cada uno independientemente -H, alquilo (C1-C6) opcionalmente sustituido, cicloalquilo (C3-C10) opcionalmente sustituido, heterociclilo de 3-10 miembros opcionalmente sustituido, arilo (C6-C10) opcionalmente sustituido, heteroarilo de 5-10 miembros opcionalmente sustituido, cicloalquilo (C6-C12) en puente opcionalmente sustituido o heterociclilo en puente de 6-12 miembros opcionalmente sustituido; o
R4 y R5, junto con el nitrógeno al que están unidos, forman heterociclilo de 4-10 miembros opcionalmente sustituido, heteroarilo de 5-10 miembros opcionalmente sustituido o heterociclilo en puente de 6-12 miembros opcionalmente sustituido;
R6 en cada aparición es independientemente -F, -Cl, -Br, -CN, -NH2, -OH, -alquilo (C1-C6), -haloalquilo (C1-C6), -alquenilo (C2-C6), -alquinilo (C2-C6), cicloalquilo (C3-C6), -alcoxilo (C1-C6), -haloalcoxilo (C1-C6), -alquilen (C1-C6)-OH o -alquilen (C1-C6)-NH2;
m es 0, 1, 2 o 3; y
n es 0 , 1 o 2.
En una segunda realización, se da a conocer un compuesto representado por la fórmula estructural (I-A)-(I-C), (II-A)
(II-C) o (III-A)-(MI-O):
Ċ 

o una sal farmacéuticamente aceptable de los mismos. Los valores para las variables en las fórmulas estructurales (I-A)-(I-C), (N-A)-(N-C) y (NI-A)-(NI-C) son tal como se describen para la fórmula estructural (I).
En una tercera realización, se da a conocer un compuesto representado por la fórmula estructural (I), (I-A)-(I-C), (N-A)-(M-C) o (NI-A)-(NI-C), en el que R4 y R5, junto con el nitrógeno al que están unidos, forman heterociclilo monocíclico de 4-7 miembros o heterociclilo en puente de 6-12 miembros, en el que el heterociclilo monocíclico de 4 7 miembros o heterociclilo en puente de 6-12 miembros está opcionalmente sustituido con 1-3 grupos seleccionados de -F, -Cl, -Br, -CN, -NH2, -OH, oxo, -alquilo (C1-C4), -haloalquilo (C1-C4), -alcoxilo (C1-C4), -haloalcoxilo (C1-C4), -alquilen (C1-C4)-OH o -alquilen (C1-C4)-n H2. Los valores para las demás variables son tal como se describen para la fórmula estructural (I).
En una cuarta realización, se da a conocer un compuesto representado por la fórmula estructural (I), (I-A)-(I-C), (N-A)-(M-C) o (IN-A)-(IN-C), en el que Ra en cada aparición es independientemente -H, -alquilo (C1-C6), -(CH2V cicloalquilo (C3-C7), -(CH2)n-heterociclilo monocíclico de 4-7 miembros, -(CH2)n-cicloalquilo (C6-C12) en puente, -(CH2)n-heteroarilo de 5-10 miembros opcionalmente sustituido; o -(CH2)n-heterociclilo en puente de 6-12 miembros, en el que el -alquilo (C1-C6), -(CH2)n-cicloalquilo (C3-C7), -(CH2)n-heterociclilo monocíclico de 4-7 miembros, -(CH2V cicloalquilo (C6-C12) en puente, -(CH2)n-heteroarilo de 5-10 miembros o -(CH2)n-heterociclilo en puente de 6-12 miembros, está opcionalmente sustituido con 1-3 grupos seleccionados de -F, -Cl, -Br, -CN, -NH2, -OH, oxo, -alquilo (C1-C4), -haloalquilo (C1-C4), -alcoxilo (C1-C4), -haloalcoxilo (C1-C4), -alquilen (C1-C4)-OH o -alquilen (C1-C4)-NH2, y los valores para las demás variables son tal como se describieron anteriormente para la fórmula estructural (I), o en la tercera realización.
En una quinta realización, se da a conocer un compuesto representado por la fórmula estructural (I), (I-A)-(I-C), (N-A)-(M-C) o (IN-A)-(m-C), en el que R es H, -F, -Cl, -Br, -OH, -alquilo (C1-C4), -haloalquilo (C1-C4), -alcoxilo (C1-C4), -alquilen (C1-C4)-OH o heterociclilo monocíclico de 4-7 miembros opcionalmente sustituido con 1-3 grupos seleccionados de -F, -Cl, -Br, - OH, -alquilo (C1-C4), -haloalquilo (C1-C4) o -alcoxilo (C1-C4), y los valores para las demás variables son tal como se describieron anteriormente para la fórmula estructural (I), o en la tercera o cuarta realización.
En una sexta realización, se da a conocer un compuesto representado por la fórmula estructural (I), (I-A)-(I-C), (II-A)-(II-C) o (NI-A)-(IN-C), en el que R4 y R5, junto con el nitrógeno al que están unidos, forman -N-alquil-piperazinilo o morfolinilo, en el que el piperazinilo o morfolinilo está opcionalmente sustituido con 1-2 grupos seleccionados de -F, -Cl, -Br, -OH, -alquilo (C1-C4), -haloalquilo (C1-C4) o -alcoxilo (C1-C4), y los valores para las demás variables son tal
como se describieron anteriormente para la fórmula estructural (I), o en la tercera, cuarta o quinta realización.
En una séptima realización, se da a conocer un compuesto representado por la fórmula estructural (I), (I-A)-(I-C), (N-A)-(N-C) o (IM-A)-(IM-C), en el que Ra en cada aparición es independientemente -H, -(CH2)n-cicloalquilo (C3-Ca), -(CH2)n-heterociclilo de 3-6 miembros, en el que el -(CH2)n-cicloalquilo (C3-Ca) o -(CH2)n-heterociclilo de 3-6 miembros está opcionalmente sustituido con 1-3 grupos seleccionados de -F, -Cl, -Br, -CN, -NH2, -OH, -alquilo (C1-C4) o -alcoxilo (C1-C4); y n es 0 o 1, y los valores para las demás variables son tal como se describieron anteriormente para la fórmula estructural (I), o en la tercera, cuarta, quinta o sexta realización.
En una octava realización, se da a conocer un compuesto representado por la fórmula estructural (I), (I-A)-(I-C), (II-A)-(N-C) o (NI-A)-(NI-C), en el que R es H, -alquilo (C1-C4), -alcoxilo (C1-C4), N-piperazinilo opcionalmente sustituido con -CO2-alquilo (C1-C4), y los valores para las demás variables son tal como se describieron anteriormente para la fórmula estructural (I), o en la tercera, cuarta, quinta, sexta o séptima realización. Alternativamente, R es H.
En una novena realización, se da a conocer un compuesto representado por la fórmula estructural (I), (I-A)-(I-C), (II-A)-(N-C) o (IM-A)-(MI-C), en el que R4 y R5, junto con el nitrógeno al que están unidos, forman -N-metil-piperazinilo o morfolinilo, ambos de los cuales están opcionalmente sustituidos con uno o dos metilo, y los valores para las demás variables son tal como se describieron anteriormente para la fórmula estructural (I), o en la tercera, cuarta, quinta, sexta, séptima u octava realización.
En una décima realización, se da a conocer un compuesto representado por la fórmula estructural (I), (I-A)-(I-C), (II-A)-(N-C) o (NI-A)-(NI-C), en el que Ra en cada aparición es independientemente -H; -cicloalquilo (C3-Ca) opcionalmente sustituido con -OH; -(CH2)n-tetrahidro-2H-pirano; morfolinilo; piperidinilo opcionalmente sustituido con -F, -OH o metilo; o tetrahidrofurano; y n es 0 o 1, y los valores para las demás variables son tal como se describieron anteriormente para la fórmula estructural (I), o en la tercera, cuarta, quinta, sexta, séptima, octava o novena realización.
La invención también incluye los compuestos representados por la estructura y/o descritos por su nombre en la ejemplificación. La invención incluye tanto la forma neutra (base libre) de estos compuestos así como sales farmacéuticamente aceptables de los mismos. Los tratamientos con y/o usos de estos compuestos incluyen la forma neutra de estos compuestos así como sales farmacéuticamente aceptables de los mismos.
El término “alquilo”, usado solo o como parte de un resto más grande, tal como “alcoxilo” o “haloalquilo”, y similares, significa radical hidrocarbonado saturado monovalente alifático de cadena lineal o ramificado. A menos que se especifique lo contrario, un grupo alquilo tiene normalmente 1-6 átomos de carbono, es decir, alquilo (C1-Ca). Tal como se usa en el presente documento, un grupo “alquilo (C1-Ca)” significa un radical que tiene desde 1 hasta 6 átomos de carbono en una disposición lineal o ramificada. Los ejemplos incluyen metilo, etilo, n-propilo, /so-propilo, etc.
“Alcoxilo” significa un radical alquilo unido a través de un átomo de unión a oxígeno, representado por-O-alquilo. Por ejemplo, “alcoxilo (C1-C4)” incluye metoxilo, etoxilo, propoxilo y butoxilo.
Los términos “haloalquilo” y “haloalcoxilo” significan alquilo o alcoxilo, según sea el caso, sustituido con uno o más átomos de halógeno. El término “halógeno” significa F, Cl, Br o I. Preferiblemente, el halógeno en un haloalquilo o haloalcoxilo es F.
“Alquenilo” significa radical hidrocarbonado monovalente de cadena lineal o ramificado que contiene al menos un doble enlace. Alquenilo puede ser monoinsaturado o poliinsaturado, y puede existir en la configuración E o Z. A menos que se especifique lo contrario, un grupo alquenilo tiene normalmente 2-6 átomos de carbono, es decir, alquenilo (C2-Ca). Por ejemplo, “alquenilo (C2-Ca)” significa un radical que tiene desde 2 hasta 6 átomos de carbono en una disposición lineal o ramificada.
“Alquinilo” significa radical hidrocarbonado monovalente de cadena lineal o ramificado que contiene al menos un triple enlace. A menos que se especifique lo contrario, un grupo alquinilo tiene normalmente 2-6 átomos de carbono, es decir, alquinilo (C2-Ca). Por ejemplo, “alquinilo (C2-Ca)” significa un radical que tiene desde 2 hasta 6 átomos de carbono en una disposición lineal o ramificada.
“Cicloalquilo” significa un radical hidrocarbonado saturado alifático cíclico, que contiene normalmente desde 3 hasta 8 átomos de carbono de anillo, es decir, cicloalquilo (C3-C8). Cicloalquilo (C3-C8) incluye, pero no se limita a, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo y ciclooctilo.
Tal como se usa en el presente documento, el término “en puente”, usado solo o como parte de un resto más grande como en “cicloalquilo en puente” o “heterociclilo en puente”, se refiere a un sistema de anillos que incluye dos anillos que comparten al menos tres átomos de anillo adyacentes. Cicloalquilo en puente contiene normalmente 6-12 átomos de carbono de anillo. Heterociclilo en puente tiene normalmente 6-12 átomos de anillo seleccionados de carbono y al menos un (normalmente de 1 a 4, más normalmente 1 o 2) heteroátomo (por ejemplo, oxígeno, nitrógeno o azufre).
El término “arilo”, usado solo o como parte de un resto más grande como en “arilalquilo”, “arilalcoxilo” o “ariloxialquilo”, significa un anillo aromático carbocíclico. También incluye un anillo de fenilo condensado con un grupo cicloalquilo. El término “arilo” puede usarse de manera intercambiable con los términos “anillo de arilo” “anillo aromático carbocíclico”, “grupo arilo” y “grupo aromático carbocíclico”. Un grupo arilo tiene normalmente de seis a catorce átomos de anillos. Los ejemplos incluyen fenilo, naftilo, antracenilo, 1,2-dihidronaftilo, 1,2,3,4-tetrahidronaftilo, fluorenilo, indanilo, indenilo y similares. Un “grupo arilo sustituido” está sustituido en uno cualquiera 0 más de un átomo de anillo sustituible, que es un átomo de carbono de anillo unido a un hidrógeno.
El término “heteroarilo”, “heteroaromático”, “anillo de heteroarilo”, “grupo heteroarilo”, “anillo heteroaromático” y “grupo heteroaromático” se usan de manera intercambiable en el presente documento. “Heteroarilo”, cuando se usa solo o como parte de un resto más grande como en “heteroarilalquilo” o “heteroarilalcoxilo”, se refiere a grupos de anillos aromáticos que tienen de cinco a catorce átomos de anillo seleccionados de carbono y al menos un (normalmente de 1 a 4, más normalmente 1 o 2) heteroátomo (por ejemplo, oxígeno, nitrógeno o azufre). “Heteroarilo” incluye anillos monocíclicos y anillos policíclicos en los que un anillo heteroaromático monocíclico se condensa con uno o más de un anillo de arilo, heterociclilo o heteroaromático. Como tal, “heteroarilo de 5-14 miembros” incluye sistemas de anillos monocíclicos, bicíclicos o tricíclicos.
Los ejemplos de grupos heteroarilo de 5-6 miembros monocíclicos incluyen furanilo (por ejemplo, 2-furanilo, 3-furanilo), imidazolilo (por ejemplo, N-imidazolilo, 2-imidazolilo, 4-imidazolilo, 5-imidazolilo), isoxazolilo (por ejemplo, 3- isoxazolilo, 4-isoxazolilo, 5-isoxazolilo), oxadiazolilo (por ejemplo, 2-oxadiazolilo, 5-oxadiazolilo), oxazolilo (por ejemplo, 2-oxazolilo, 4-oxazolilo, 5-oxazolilo), pirazolilo (por ejemplo, 3-pirazolilo, 4-pirazolilo), pirrolilo (por ejemplo, 1 -pirrolilo, 2-pirrolilo, 3-pirrolilo), piridilo (por ejemplo, 2-piridilo, 3-piridilo, 4-piridilo), pirimidinilo (por ejemplo, 2-pirimidinilo, 4-pirimidinilo, 5-pirimidinilo), piridazinilo (por ejemplo, 3-piridazinilo), tiazolilo (por ejemplo, 2-tiazolilo, 4- tiazolilo, 5-tiazolilo), isotiazolilo, triazolilo (por ejemplo, 2-triazolilo, 5-triazolilo), tetrazolilo (por ejemplo, tetrazolilo) y tienilo (por ejemplo, 2-tienilo, 3-tienilo). Los ejemplos de grupos heteroarilo aromáticos policíclicos incluyen carbazolilo, bencimidazolilo, benzotienilo, benzofuranilo, isobenzofuranilo, indolilo, benzotriazolilo, benzotiazolilo, benzoxazolilo, quinolinilo, isoquinolinilo, indazolilo, isoindolilo, acridinilo o bencisoxazolilo. Un “grupo heteroarilo sustituido” está sustituido en uno cualquiera o más de un átomo de anillo sustituible, que es un átomo de carbono de anillo o un átomo de nitrógeno de anillo unido a un hidrógeno.
“Heterociclilo” significa un radical de anillo de 3-12 miembros no aromático saturado o insaturado que contiene opcionalmente uno o más dobles enlaces. Puede ser monocíclico, bicíclico, tricíclico o estar condensado. El heterocicloalquilo contiene de 1 a 4 heteroátomos, que pueden ser iguales o diferentes, seleccionados de N, O o S. El anillo de heterociclilo contiene opcionalmente uno o más dobles enlaces y/o está opcionalmente condensado con uno o más anillos aromáticos (por ejemplo, anillo de fenilo). Se pretende que el término “heterociclilo” incluya todas las formas isoméricas posibles. Los ejemplos de heterocicloalquilo incluyen, pero no se limitan a, azetidinilo, morfolinilo, tiomorfolinilo, pirrolidinonilo, pirrolidinilo, piperidinilo, piperazinilo, hidantoinilo, valerolactamilo, oxiranilo, oxetanilo, dihidroimidazol, dihidrofuranilo, dihidropiranilo, dihidropiridinilo, dihidropirimidinilo, dihidrotienilo, dihidrotiofenilo, dihidrotiopiranilo, tetrahidroimidazol, tetrahidrofuranoílo, tetrahidropiranilo, tetrahidrotienilo, tetrahidropiridinilo, tetrahidropirimidinilo, tetrahidrotiofenilo y tetrahidrotiopiranilo. Los ejemplos de grupos heterocicloalquilo policíclicos incluyen dihidroindolilo, dihidroisoindolilo, dihidrobencimidazolilo, dihidrobenzotienilo, dihidrobenzofuranilo, dihidroisobenzofuranilo, dihidrobenzotriazolilo, dihidrobenzotiazolilo, dihidrobenzoxazolilo, dihidroquinolinilo, tetrahidroquinolinilo, dihidroisoquinolinilo, tetrahidroisoquinolinilo, dihidroindazolilo, dihidroacridinilo, tetrahidroacridinilo, dihidrobencisoxazolilo, cromano, cromeno, isocromano e isocromeno.
Determinados compuestos descritos en el presente documento pueden existir en diversas formas estereoisoméricas o tautoméricas. Los estereoisómeros son compuestos que difieren únicamente en su disposición espacial. Cuando un compuesto dado a conocer se nombra o representa por su estructura sin indicar la estereoquímica, se entiende que el nombre o la estructura abarca todos los posibles estereoisómeros, isómeros geométricos, incluyendo estereoisómeros o isómeros geométricos esencialmente puros, así como una combinación de los mismos.
En determinados casos, existen formas tautoméricas de los compuestos dados a conocer, tal como las estructuras tautoméricas mostradas a continuación:

Debe entenderse que, cuando un compuesto en el presente documento está representado por una fórmula estructural o denominado por un nombre químico en el presente documento, todas las otras formas tautoméricas que pueden existir para el compuesto está abarcadas por la fórmula estructural.
Determinados compuestos dados a conocer pueden existir en diversas formas estereoisoméricas. Los estereoisómeros son compuestos que difieren únicamente en su disposición espacial. Los enantiómeros son pares de estereoisómeros cuyas imágenes especulares no son superponibles, lo más habitualmente debido a que contienen un átomo de carbono asimétricamente sustituido que actúa como centro quiral. “Enantiómero” significa uno de un par de moléculas que son imágenes especulares entre sí y no son superponibles. Los diastereómeros son estereoisómeros que contienen dos o más átomos de carbono asimétricamente sustituidos. Los “isómeros geométricos” son estereoisómeros que difieren en la orientación de los átomos sustituyentes en relación con un doble enlace carbono-carbono, con un anillo de carbociclilo o con un sistema bicíclico en puente.
Cuando un isómero geométrico se representa por su nombre o estructura, debe entenderse que la pureza del isómero geométrico del isómero geométrico nombrado o representado es al menos el 60 %, el 70 %, el 80 %, el 90 %, el 99 % o el 99,9 % en peso puro. La pureza del isómero geométrico se determina dividiendo el peso del isómero geométrico nombrado o representado en la mezcla entre el peso total de todos los isómeros geométricos en la mezcla.
Cuando la estereoquímica de un compuesto dado a conocer se nombra o representa por su estructura, el estereoisómero nombrado o representado es al menos el 60 %, el 70 %, el 80 %, el 90 %, el 99 % o el 99,9 % en peso puro en relación con los demás estereoisómeros. El porcentaje en peso puro en relación con los demás estereoisómeros es la razón del peso de un estereoisómero con respecto al peso de los demás estereoisómeros. Cuando un enantiómero individual se nombra o representa por su estructura, el enantiómero nombrado o representado es al menos el 60 %, el 70 %, el 80 %, el 90 %, el 99 % o el 99,9 % ópticamente en peso puro (también denominado “enantioméricamente puro”). El porcentaje de pureza óptica en peso es la razón del peso del enantiómero con respecto al peso del enantiómero más el peso de su isómero óptico.
Cuando la estereoquímica de un compuesto dado a conocer se nombra o representa por su estructura, y la estructura nombrada o representada abarca más de un estereoisómero (por ejemplo, como en un par diastereomérico), debe entenderse que se incluyen uno de los estereoisómeros abarcados o cualquier mezcla de los estereoisómeros abarcados. También debe entenderse que la pureza estereoisomérica de los estereoisómeros nombrados o representados es al menos el 60 %, el 70 %, el 80 %, el 90 %, el 99 % o el 99,9 % en peso puro en relación con los demás estereoisómeros. La pureza estereoisomérica en este caso se determina dividiendo el peso total en la mezcla de los estereoisómeros abarcados por el nombre o la estructura entre el peso total en la mezcla de todos los estereoisómeros.
Cuando un compuesto dado a conocer se nombra o representa por su estructura sin indicar la estereoquímica y el compuesto tiene un centro quiral, debe entenderse que el nombre o la estructura abarca un enantiómero del compuesto libre del isómero óptico correspondiente, una mezcla racémica del compuesto y mezclas enriquecidas en un enantiómero en relación con su isómero óptico correspondiente.
Cuando un compuesto dado a conocer se nombra o representa por su estructura sin indicar la estereoquímica y, por ejemplo, el compuesto tiene al menos dos centros quirales, debe entenderse que el nombre o la estructura abarca un estereoisómero libre de los demás estereoisómeros, mezclas de estereoisómeros y mezclas de estereoisómeros en las que uno o más estereoisómeros está(n) enriquecido(s) en relación con los demás estereoisómeros. Por ejemplo, el nombre o la estructura puede abarcar un estereoisómero libre de los demás diastereómeros, mezclas de estereoisómeros y mezclas de estereoisómeros en las que uno o más diastereómeros está(n) enriquecido(s) en relación con los demás diastereómeros.
Las mezclas enantioméricas y diastereoméricas pueden resolverse en sus enantiómeros o estereoisómeros componentes mediante métodos bien conocidos, tales como cromatografía de gases de fase quiral, cromatografía de líquidos de alta resolución de fase quiral, cristalización del compuesto como complejo de sal quiral o cristalización del compuesto en un disolvente quiral. Los enantiómeros y diastereómeros también pueden obtenerse a partir de reactivos, productos intermedios y catalizadores diastereomérica o enantioméricamente puros mediante métodos de síntesis asimétrica bien conocidos.
En las presentes enseñanzas se incluyen sales farmacéuticamente aceptables de los compuestos dados a conocer en el presente documento. Los compuestos dados a conocer tienen grupos amina básicos y, por tanto, pueden formar sales farmacéuticamente aceptables con ácidos farmacéuticamente aceptables. Las sales de adición de ácido farmacéuticamente aceptables adecuadas de los compuestos descritos en el presente documento incluyen sales de ácidos inorgánicos (tales como los ácidos clorhídrico, bromhídrico, fosfórico, metafosfórico, nítrico y sulfúrico) y de ácidos orgánicos (tales como los ácidos acético, bencenosulfónico, benzoico, etanosulfónico, metanosulfónico, succínico y trifluoroacético). Los compuestos de las presentes enseñanzas con grupos ácidos, tales como ácidos carboxílicos, pueden formar sales farmacéuticamente aceptables con bases farmacéuticamente aceptables. Las sales básicas farmacéuticamente aceptables adecuadas incluyen sales de amonio, sales de metales alcalinos (tales como sales de sodio y potasio) y sales de metales alcalinotérreos (tales como sales de magnesio y
calcio). Los compuestos con un grupo amonio cuaternario también contienen un contraión, tal como cloruro, bromuro, yoduro, acetato, perclorato y similares. Otros ejemplos de tales sales incluyen clorhidratos, bromhidratos, sulfatos, metanosulfonatos, nitratos, acetatos, succinatos, benzoatos y sales con aminoácidos, tales como ácido glutámico.
Los compuestos descritos en el presente documento puede inhibir HPK1. Por tanto, generalmente, los compuestos descritos en el presente documento son útiles en el tratamiento de enfermedades o estados asociados con tales cinasas.
En una realización, los compuestos descritos en el presente documento son inhibidores de HPK1, y son útiles para el tratamiento de enfermedades, tales como cáncer, asociadas con tal(es) cinasa(s).
Otro aspecto de las presentes enseñanzas se refiere a un método de tratamiento de un sujeto con cáncer, que comprende administrar al sujeto una cantidad eficaz de un compuesto descrito en el presente documento. En una realización, los compuestos descritos en el presente documento inhiben el crecimiento de un tumor.
Los cánceres que pueden tratarse (incluyendo la reducción de la probabilidad de recidiva) mediante los métodos de las presentes enseñanzas incluyen cáncer de mama, cáncer colorrectal, cáncer de pulmón, cáncer de ovario, cáncer de útero, cáncer de próstata, leucemias, linfomas, cáncer de cerebro (incluyendo glioblastoma multiforme y neuroblastoma), cáncer de cabeza y cuello, cáncer de páncreas, melanoma, carcinoma hepatocelular, cáncer de riñón y sarcomas de partes blandas. En una realización, el cáncer es cáncer de mama, cáncer de colon y cáncer de ovario. En una realización, el cáncer se selecciona de leucemia, leucemia mieloide aguda, leucemia mielógena crónica, cáncer de mama, cáncer de cerebro, cáncer de colon, cáncer colorrectal, cáncer de cabeza y cuello, carcinoma hepatocelular, adenocarcinoma de pulmón, melanoma metastásico, cáncer de páncreas, cáncer de próstata, cáncer de ovario y cáncer de riñón. En una realización, el cáncer es cáncer de pulmón, cáncer de colon, cáncer de cerebro, neuroblastoma, cáncer de próstata, melanoma, glioblastoma multiforme o cáncer de ovario. En otra realización, el cáncer es cáncer de pulmón, cáncer de mama, cáncer de colon, cáncer de cerebro, neuroblastoma, cáncer de próstata, melanoma, glioblastoma multiforme o cáncer de ovario. En aún otra realización, el cáncer es cáncer de mama, cáncer de colon y cáncer de pulmón. En otra realización, el cáncer es un cáncer de mama. En aún otra realización, el cáncer es un cáncer de mama de subtipo basal o un cáncer de mama de subtipo luminal B. En aún otra realización, el cáncer es un cáncer de mama de subtipo basal. En aún otra realización, el cáncer de mama de subtipo basal es cáncer de mama negativo para ER (receptor de estrógenos), HER2 y PR (receptor de progesterona). En aún otra realización, el cáncer es un cáncer de partes blandas. Un “cáncer de partes blandas” es un término reconocido en la técnica que abarca tumores derivados de cualquier parte blanda del cuerpo. Tales partes blandas conectan, sostienen o rodean diversas estructuras y órganos del cuerpo, incluyendo, pero sin limitarse a, músculo liso, músculo esquelético, tendones, tejidos fibrosos, tejido graso, vasos sanguíneos y linfáticos, tejido perivascular, nervios, células mesenquimatosas y tejidos sinoviales. Por tanto, los cánceres de partes blandas pueden ser de tejido graso, tejido muscular, tejido nervioso, tejido de las articulaciones, vasos sanguíneos, vasos linfáticos y tejidos fibrosos. Los cánceres de partes blandas pueden ser benignos o malignos. Generalmente, los cánceres de partes blandas malignos de denominan sarcomas, o sarcomas de partes blandas. Existen muchos tipos de tumores de partes blandas, incluyendo lipoma, lipoblastoma, hibernoma, liposarcoma, leiomioma, leiomiosarcoma, rabdomioma, rabdomiosarcoma, neurofibroma, schwannoma (neurilemoma), neuroma, schwannoma maligno, neurofibrosarcoma, sarcoma neurógeno, tenosinovitis nodular, sarcoma sinovial, hemangioma, glomangioma, hemangiopericitoma, hemangioendotelioma, angiosarcoma, sarcoma de Kaposi, linfangioma, fibroma, elastofibroma, fibromatosis superficial, histiocitoma fibroso, fibrosarcoma, fibromatosis, dermatofibrosarcoma protuberante (DFSP), histiocitoma fibroso maligno (HFM), mixoma, tumor de células granulares, mesenquimomas malignos, sarcoma alveolar de partes blandas, sarcoma epitelioide, sarcoma de células claras y tumor desmoplásico de células pequeñas. En una realización particular, el cáncer de partes blandas es un sarcoma seleccionado del grupo que consiste en un fibrosarcoma, un sarcoma gastrointestinal, un leiomiosarcoma, un liposarcoma indiferenciado, un liposarcoma pleomórfico, un histiocitoma fibroso maligno, un sarcoma de células redondas y un sarcoma sinovial.
Las presentes enseñanzas también proporcionan métodos de tratamiento de un sujeto con una enfermedad, que comprenden administrar al sujeto una cantidad eficaz de un compuesto representado por la fórmula estructural (I) en combinación con una terapia inmunomoduladora eficaz (también denominada inmunoterapia). La inmunoterapia es el tratamiento de la enfermedad usando un agente inmunomodulador para reducir, potenciar o suprimir una respuesta inmunitaria. Las inmunoterapias diseñadas para provocar o amplificar una respuesta inmunitaria se clasifican como inmunoterapias de activación, mientras que las inmunoterapias que reducen o suprimen se clasifican como inmunoterapias de supresión. La enfermedad descrita en el presente documento es un cáncer.
Las terapias inmunomoduladoras, usadas solas o en enfoques de combinación, incluyen i) inhibidores del bloqueo del punto de control inmunitario, incluyendo, pero sin limitarse a, anticuerpos anti-CTLA4 (proteína 4 asociadas a linfocitos T citotóxicos) (por ejemplo, ipilimumab), agentes que alteran la interacción de PD-1/PD-L1 y PD-L2, por ejemplo, nivolumab (Opdivo - Bristol Myers Squibb), pembrolizumab (Keytruda, KM-3475, Merck), pidilizumab (Ct -011, Cure Tech), BMS 936559 (BMS) y Mp Dl328OA (Roche); y otros receptores inhibidores de la respuesta inmunitaria, por ejemplo, anticuerpo anti-CD47; ii) terapias basadas en células (incluyendo, pero sin limitarse a, terapia con células dendríticas (por ejemplo, Sipuleucel T (Provenge) y terapias adoptivas con células T, iii)
estrategias de vacunación; iv) terapia adoptiva con células T; v) agentes que impiden la inhibición metabólica de la respuesta inmunitaria, incluyendo inhibidores de indoleamina 2,3-dioxigenasa (por ejemplo, INCB024360 (Incyte), 1-metil-D-triptófano, indoximod (NewLink Genetics)) o arginasa; y vi) terapia basada en citocinas, por ejemplo, interferones (en particular, interferón de tipo I) e interleucinas (por ejemplo, interleucina-2 ).
En una realización, el agente inmunomodulador usado para la terapia inmunomoduladora es un inhibidor de PD-1, por ejemplo, un anticuerpo anti-PD1.
La proteína de muerte celular programada 1, también conocida como PD-1 y CD279 (grupo de diferenciación 279), es una proteína que, en seres humanos, está codificada por el gen PDCD1. PD-1 es un receptor de superficie celular que pertenece a las superfamilia de inmunoglobulinas y se expresa en células T y células pro-B. PD-1 se une a dos ligandos, PD-L1 y PD-L2, ambos de los cuales son miembros de la familia de B7.
PD-1 y sus ligandos desempeñan un papel importante en la regulación por disminución del sistema inmunitario impidiendo la activación de células T, lo que a su vez reduce la autoinmunidad y fomenta la autotolerancia. El efecto inhibidor de PD-1 se logra a través de un mecanismo dual de fomento de la apoptosis (muerte celular programada) en células T específicas de antígeno en ganglios linfáticos, mientras que simultáneamente reduce la apoptosis en células T reguladoras (células T supresoras).
El inhibidor de PD-1 usado en la presente invención incluye, pero no se limita a, nivolumab, pembrolizumab, pidilizumab, BMS 936559, MPDL3280A, MSB0010718C o MEDI4736. Entre ellos, BMS 936559, MPDL3280A, MSB0010718C y MEDI4736 se unen al ligando PD-L1, todos de los cuales son anticuerpos. Tanto nivolumab como pembrolizumab están aprobados por la Administración de Alimentos y Fármacos (f Da ) para el tratamiento de melanoma no resecable o metastásico que ya no responde a otros fármacos.
Las estrategias de vacunación incluyen inmunoterapia antimicrobiana, que incluye vacunación, implica la activación del sistema inmunitario para responder a un agente infeccioso.
La terapia adoptiva con células T usa respuestas citotóxicas basadas en células T para atacar a las células cancerosas. Las células T que tienen una reactividad natural o modificada por ingeniería genética para el cáncer de un paciente se generan in vitro y luego se transfieren de nuevo al paciente con cáncer. Un estudio que usa linfocitos infiltrantes de tumor autólogos fue un tratamiento eficaz para pacientes con melanoma metastásico. Eso puede conseguirse tomando células T que se encuentran con el tumor del paciente, que están entrenadas para atacar a las células cancerosas. A estas células T, que se denominan linfocitos infiltrantes de tumor (TIL), se les estimula luego para multiplicarse in vitro usando altas concentraciones de IL-2, anticuerpo anti-CD3 y células alimentadoras alorreactivas. A continuación, estas células T se transfieren de nuevo al paciente junto con la administración exógena de IL-2 para potenciar adicionalmente su actividad antineoplásica.
Las presentes enseñanzas también proporcionan métodos de tratamiento de un sujeto con un cáncer, que comprenden administrar al sujeto una cantidad eficaz de un compuesto representado por la fórmula estructural (I) en combinación con una terapia contra el cáncer eficaz. En una realización, el cáncer es un cáncer metastásico. Un “cáncer metastásico” es un cáncer que se ha diseminado a partir de su sitio primario a otras partes del cuerpo. La terapia contra el cáncer descrita en el presente documento incluye la administración conjunta de una cantidad eficaz de un segundo agente anticancerígeno junto con un inhibidor de HPK-1 dado a conocer. Un “agente anticancerígeno” es un compuesto que, cuando se administra en una cantidad eficaz a un sujeto con cáncer, puede conseguir, parcial o sustancialmente, uno o más de lo siguiente: detener el crecimiento, reducir el grado de un cáncer (por ejemplo, reducir el tamaño de un tumor), inhibir la velocidad de crecimiento de un cáncer y aliviar o mejorar un síntoma clínico o un indicador asociado con un cáncer (tal como componentes de suero o tejido) o aumentar la longevidad del sujeto.
Los agentes anticancerígenos adecuados para su uso en los métodos descritos en el presente documento incluyen cualquier agente anticancerígeno que haya sido aprobado para el tratamiento de cáncer. En una realización, el agente anticancerígeno incluye, pero no se limita a, un anticuerpo dirigido, un inhibidor de la angiogénesis, un agente alquilante, un antimetabolito, un alcaloide de la vinca, un taxano, una podofilotoxina, un inhibidor de topoisomerasa, un agente anticancerígeno hormonal y otros agentes anticancerígenos. En una realización, el agente anticancerígeno es un inhibidor de PD-1, por ejemplo, un anticuerpo anti-PD1.
En una realización, los agentes anticancerígenos que pueden usarse en los métodos descritos en el presente documento incluyen, pero no se limitan a, paclitaxel, docetaxel, 5-fluorouracilo, trastuzumab, lapatinib, bevacizumab, letrozol, goserelina, tamoxifeno, cetuximab, panitumumab, gemcitabina, capecitabina, irinotecán, oxaliplatino, carboplatino, cisplatino, doxorrubicina, epirrubicina, ciclofosfamida, metotrexato, vinblastina, vincristina, melfalán, citarabina, etopósido, daunorrubicina, bleomicina, mitomicina y adriamicina, y una combinación de los mismos. En una realización, el agente anticancerígeno y el compuesto representado por la fórmula estructural (I) se administran simultáneamente. Cuando se administran simultáneamente, el agente anticancerígeno y el compuesto pueden administrarse en la misma formulación o en formulaciones diferentes. Alternativamente, el compuesto y el agente anticancerígeno adicional se administran por separado en momentos diferentes.
Tal como se usa en el presente documento, “tratar a un sujeto con un cáncer” incluye conseguir, parcial o sustancialmente, uno o más de lo siguiente: detener el crecimiento, reducir el grado del cáncer (por ejemplo, reducir el tamaño de un tumor), inhibir la velocidad de crecimiento del cáncer, aliviar o mejorar un síntoma clínico o un indicador asociado con el cáncer (tal como componentes de suero o tejido) o aumentar la longevidad del sujeto; y reducir la probabilidad de recidiva del cáncer.
El término “cantidad eficaz” significa una cantidad que, cuando se administra al sujeto, da como resultado resultados beneficiosos o deseados, incluyendo resultados clínicos, por ejemplo, inhibe, suprime o reduce el cáncer (por ejemplo, tal como se determina mediante los síntomas clínicos o la cantidad de células cancerosas) en un sujeto en comparación con un control.
Generalmente, una cantidad eficaz de un compuesto enseñado en el presente documento varía dependiendo de diversos factores, tales como el fármaco o compuesto administrado, la formulación farmacéutica, la vía de administración, el tipo de enfermedad o trastorno, la identidad del sujeto o huésped que está tratándose y similares, sin embargo, puede determinarse de manera rutinaria por un experto en la técnica. Una cantidad eficaz de un compuesto de las presentes enseñanzas puede determinarse fácilmente por un experto habitual mediante métodos de rutina conocidos en la técnica.
En una realización, una cantidad eficaz de un compuesto enseñado en el presente documento oscila entre aproximadamente 0,1 y aproximadamente 1000 mg/kg de peso corporal, alternativamente entre aproximadamente 1 y aproximadamente 500 mg/kg de peso corporal. En otra realización, una cantidad eficaz de un compuesto enseñado en el presente documento oscila entre aproximadamente 0,5 y aproximadamente 5000 mg/m2, alternativamente entre aproximadamente 5 y aproximadamente 2500 mg/m2, y en otra alternativa, entre aproximadamente 50 y aproximadamente 1000 mg/m2. El experto en la técnica apreciará que determinados factores pueden influir en la dosificación requerida para tratar eficazmente a un sujeto que padece cáncer o reducir la probabilidad de recidiva de un cáncer. Estos factores incluyen, pero no se limitan a, la gravedad de la enfermedad o del trastorno, tratamientos previos, la salud general y/o la edad del sujeto y otras enfermedades presentes.
Un “sujeto” es un mamífero, preferiblemente un ser humano, pero también puede ser un animal que necesita tratamiento veterinario, por ejemplo, animales de compañía (por ejemplo, perros, gatos y similares), animales de granja (por ejemplo, vacas, ovejas, cerdos, caballos y similares) y animales de experimentación (por ejemplo, ratas, ratones, cobayas y similares).
Los compuestos enseñados en el presente documento pueden administrarse a un paciente en una variedad de formas dependiendo de la vía de administración seleccionada, tal como entenderán los expertos en la técnica. Los compuestos de las presentes enseñanzas pueden administrarse, por ejemplo, mediante administración oral, parenteral, bucal, sublingual, nasal, rectal, en parche, con bomba o transdérmica y las composiciones farmacéuticas formuladas por consiguiente. La administración parenteral incluye los modos de administración intravenoso, intraperitoneal, subcutáneo, intramuscular, transepitelial, nasal, intrapulmonar, intratecal, rectal y tópico. La administración parenteral puede ser mediante infusión continua a lo largo de un periodo de tiempo seleccionado. Los compuestos enseñados en el presente documento pueden formularse de manera adecuada en composiciones farmacéuticas para su administración a un sujeto. Las composiciones farmacéuticas de las presentes enseñanzas incluyen opcionalmente uno o más portadores y/o diluyentes farmacéuticamente aceptables para las mismas, tales como lactosa, almidón, celulosa y dextrosa. También pueden incluirse otros excipientes, tales como agentes saborizantes; edulcorantes; y conservantes, tales como metil, etil, propil y butil parabenos. Un listado más completo de excipientes adecuados puede encontrarse en Handbook of Pharmaceutical Excipients (5a ed., Pharmaceutical Press (2005)). Un experto en la técnica sabrá cómo preparar formulaciones adecuadas para los diversos tipos de vías de administración. Los procedimientos y componentes convencionales para la selección y preparación de formulaciones adecuadas se describen, por ejemplo, en Remington's Pharmaceutical Sciences (2003 - 20a edición) y en la Farmacopea de Estados Unidos: Formulario Nacional (USP 24 NF19) publicado en 1999. Los portadores, diluyentes y/o excipientes son “aceptables” en el sentido de que son compatibles con los demás componentes de la composición farmacéutica y no perjudiciales para el receptor de los mismos.
Normalmente, para la administración terapéutica oral, un compuesto de las presentes enseñanzas puede incorporarse con un excipiente y usarse en forma de comprimidos comestibles, comprimidos orales, trociscos, cápsulas, elixires, suspensiones, jarabes, obleas y similares.
Normalmente, para la administración parenteral, las disoluciones de un compuesto de las presentes enseñanzas pueden prepararse generalmente en agua mezclada de manera adecuada con un tensioactivo, tal como hidroxipropilcelulosa. Las dispersiones también pueden prepararse en glicerol, polietilenglicoles líquidos, DMSO y mezclas de los mismos, con o sin alcohol, y en aceites. En condiciones ordinarias de almacenamiento y uso, estas preparaciones contienen un conservante para impedir el crecimiento de microorganismos.
Normalmente, para el uso inyectable, son apropiados disoluciones acuosas estériles o una dispersión de, y polvos estériles de, un compuesto descrito en el presente documento para la preparación extemporánea de disoluciones o dispersiones inyectables estériles.
Para la administración nasal, los compuestos de las presentes enseñanzas pueden formularse como aerosoles, gotas, geles y polvos. Las formulaciones de aerosol comprenden normalmente una disolución o una suspensión fina del principio activo en un disolvente acuoso o no acuoso fisiológicamente aceptable, y se presentan habitualmente en cantidades monodosis o multidosis en forma estéril en un recipiente sellado, que puede tener la forma de un cartucho o envase reutilizable para su uso con un dispositivo de atomización. Alternativamente, el recipiente sellado puede ser un dispositivo de dispensación unitaria, tal como un inhalador nasal monodosis o un dispensador de aerosol equipado con una válvula dosificadora, que está destinado a desecharse tras su uso. Cuando la forma de dosificación comprende un dispensador de aerosol, contendrá un propelente que puede ser un gas comprimido, tal como aire comprimido o un propelente orgánico, tal como fluoroclorohidrocarburo. Las formas de dosificación en aerosol también pueden tener la forma de un atomizador por bomba.
Para la administración bucal o sublingual, los compuestos de las presentes enseñanzas pueden formularse con un portador, tal como azúcar, goma arábiga, tragacanto, o gelatina y glicerina, como comprimidos, pastillas para chupar o pastillas.
Para la administración rectal, los compuestos descritos en el presente documento pueden formularse en forma de supositorios que contienen una base de supositorio convencional, tal como manteca de cacao.
Los compuestos de invención pueden prepararse mediante métodos conocidos por los expertos en la técnica, tal como se ilustra a continuación mediante los esquemas y procedimientos generales y mediante los siguientes ejemplos de preparación. Todos los materiales de partida o bien están disponibles comercialmente o bien se preparan mediante métodos conocidos por los expertos en la técnica y los procedimientos descritos a continuación. Los enfoques de síntesis generales para los compuestos de las reivindicaciones se proporcionan en la ejemplificación a continuación, tal como se ilustra en los esquemas 1 y 2.
Ejemplificación
Los compuestos que no se encuentran dentro del alcance de las reivindicaciones se consideran como compuestos de referencia únicamente.
Ejemplo A: síntesis
Métodos generales
Los materiales de partida, reactivos y disolventes comercialmente disponibles se usaron tal como se recibieron. En general, se realizaron reacciones anhidras bajo una atmósfera inerte, tal como nitrógeno o argón. PoraPak® Rxn CX se refiere a una resina de intercambio catiónico comercial disponible de Waters.
Se realizaron reacciones de microondas con un reactor de microondas Biotage Initiator. En general, el progreso de la reacción se monitorizó mediante CL-EM (sistema Exquire 4000 de Bruker o Acquity UPLC de Waters). Se realizó la purificación mediante cromatografía ultrarrápida en columna de los productos intermedios o productos finales usando un dispositivo Biotage Isolera con cartuchos de sílice KP-SIL o HP-SIL, o sílice modificada con KP-NH básica y las muestras correspondientes. Se realizó purificación mediante HPLC de fase inversa en un sistema de HPLC PrepStar modelo SD-1 de Varian con una columna de fase inversa Monochrom 10|i C-18 de Varian usando un gradiente del 10 % de MeOH/el 0,05 % de TFA-H2O al 90 % de MeOH/el 0,05 % de TfA en H2O a lo largo de un periodo de 40 min a una velocidad de flujo de 40 ml/min. También se realizó purificación de fase inversa usando un dispositivo Biotage Isolera equipado con una columna KP-C18-H usando entre el 10-95 % de MeOH o CH3CN/el 0,1 % de TFA en H2O. Se registraron las RMN de protón en un espectrómetro de 400 MHz de Bruker y se obtuvieron los espectros de masas usando un espectrómetro Esquire 4000 de Bruker o un sistema Acquity UPLC de Waters. Los nombres de los compuestos se generaron usando el software integrado en ChemBioDraw Ultra versión 12.0 CambridgeSoft-PerkinElmer.
Abreviaturas:
ac. acuoso
anh. anhidro
Ar argón
Boc ferc-butoxicarbonilo
a ancho
calc. calculado
d doblete (sólo cuando se usan en los espectros de 1H-RMN)
DCM diclorometano
e.d. exceso diastereomérico
DIPEA diisopropiletilamina
DMF N,N-dimetilformamida
DMSO dimetilsulfóxido
dppf 1,1 '-bis(difenilfosfino)ferroceno
equiv. equivalente
Flt3 tirosina cinasa 3 relacionada con fms
h hora
HPK1 cinasa de progenitores hematopoyéticos 1
HPLC cromatografía de líquidos de alta resolución
IPA isopropanol
KHMDS hexametildisilazida de potasio
Lck proteína tirosina cinasa específica de linfocitos
CL-EM cromatografía de líquidos acoplada a espectrometría de masas LDA diisopropillamida de litio
LiHMDS hexametildisilazida de litio
min minuto
m multiplete
MeCN acetonitrilo
EM ESI espectro de masas, ionización por electropulverización RMN resonancia magnética nuclear
O/N durante la noche
PMB para-metoxibencilo
prep. preparativa
ta temperatura ambiente
Tr tiempo de retención
RP fase inversa
s singlete
sat. saturado
t triplete
temp. temperatura
TFA ácido trifluoroacético
THF tetrahidrofurano

Método general A1 (ciclización inducida por base usando éster de bencimidazol)
Se trató una disolución de ariloxazin-2,4-diona (1 equiv.), o aminoarilnitrilo y 1H-benzo[d]imidazol-2-il)acetato sustituido (1-1,2 equiv.) en THF con KHMDS, LiHMDS o LDA (3-5 equiv.). Se agitó la reacción a 45 °C durante 4-24 h. A continuación se enfrió la reacción hasta ta y se extinguió con NH4Cl ac. sat. Se extrajo la fase acuosa con EtOAc o DCM y se secaron los extractos orgánicos combinados sobre MgSO4, se filtraron y se concentraron. Se purificó el producto en bruto mediante cromatografía en columna o HPLC prep. para dar el producto deseado.
Método general A2 (ciclización inducida por base en dos etapas usando éster de bencimidazol)
Se trató una disolución de ariloxazin-2,4-diona (1 equiv.), o aminoarilnitrilo y 1H-benzo[d]imidazol-2-il)acetato sustituido (1-1,2 equiv.) en con KHMDS, LiHMDS, KOBu* o lDa (3-5 equiv.) a 45 °C durante 2-4 h. A continuación se enfrió la reacción hasta ta y se extinguió con NH4Cl ac. sat. Se extrajo la fase acuosa con EtOAc o DCM y se secaron los extractos orgánicos combinados sobre MgSO4, se filtraron y se concentraron. Se separó el aducto de adición no ciclado mediante cromatografía en columna, se disolvió en THF y se trató con KHMDS, LiHMDS o LDA (3-5 equiv.). Se agitó la reacción a 45 °C durante 1-4 h. A continuación se enfrió la reacción hasta ta y se extinguió con NH4C ac. sat. Se extrajo la fase acuosa con EtOAc o DCM y se secaron los extractos orgánicos combinados sobre MgSO4, se filtraron y se concentraron. Se purificó el producto en bruto mediante cromatografía en columna o HPLC prep. para dar el producto deseado.
Método general A3 (ciclización inducida por base en dos etapas usando éster de bencimidazol)
Se trató una disolución de aminoarilnitrilo y 1H-benzo[d]imidazol-2-il)acetato sustituido (1 equiv.) en THF con LiHMDS o LDA (5 equiv.) (etapa 1). Se agitó la reacción a 35-40 °C durante 1-1,5 h. A continuación se enfrió la reacción hasta ta y se extinguió con NH4Cl ac. sat. y se concentró. Se purificó el producto en bruto mediante HPLC prep. para dar un producto intermedio no ciclado que se neutralizó, se secó y se sometió a las condiciones descritas en el método general A1 usando LiHMDS (etapa 2).
Método general B (formación de triflato)
Se trató una disolución de derivado de bencimidazol-2-ilarilpiridinona (1 equiv.) y piridina (20 equiv.) en DCM con Tf2O (8 equiv.). Se agitó la reacción a 0 °C durante 2-8 h. A continuación se extinguió la reacción con NaHCO3 ac. sat. Se extrajo la fase acuosa con DCM y se secaron los extractos orgánicos combinados sobre MgSO4, se filtraron y se concentraron. Se usó el producto en bruto en la siguiente etapa sin purificación adicional.
Método general C (sustitución con amina)
Se trató una disolución de derivado de bistriflato de benzoimidazol-2-ilarilpiridinona (1 equiv.) en MeCN, DCM o DMF con amina (1,2-3 equiv.). En el caso en el que la amina sea una sal (por ejemplo, HCl), se disolvió la sal de amina en MeOH o DMF y se hizo pasar a través de una columna de intercambio iónico PoraPak Rxn CX para producir la base libre que se añadió a la mezcla de reacción. Se agitó la mezcla de reacción a ta o hasta 45 °C durante 1-48 h. Se eliminó el disolvente y se purificó el producto en bruto mediante cromatografía en columna o HPLC prep. para dar el producto deseado.
Método general D (desprotección global)
Se calentó una disolución de derivado de benzoimidazol-2-ilarilpiridinona protegido (1 equiv.) en TFA/HCl conc. (7:1 v/v) a 80-100 °C durante 3-24 h. Se eliminó el disolvente y se purificó el producto en bruto mediante cromatografía en columna (base libre) o HPLC prep. (sal de TFA) para dar el producto deseado. Para generar el producto deseado como sal de HCl, se disolvió la base libre en MeOH y se añadió HCl 1 M-Et2O (2-4 equiv.) a ta. Se agitó la disolución durante 5 min y se sometió a destilación azeotrópica dos veces con MeOH.
Método general E (protección con PMB)
Se agitó una disolución de anhídrido tiaisatoico (1 equiv.), 1-(clorometil)-4-metoxibenceno (1-1,2 equiv.), K2CO3 (1-1,2 equiv.) y/o KI (1-1,2 equiv.) en DMF a ta durante 4-24 h. A continuación se añadió lentamente la mezcla de reacción a H2O, se recogió el precipitado mediante filtración a vacío para dar el deseado.
Productos intermedios:
1H-tieno[3,4-d1[1,31oxazin-2,4-diona

A una disolución de ácido 4-fe/c-butoxicarbonilamino-tiofeno-3-carboxílico (2,5 g, 10,2 mmol) en PhMe (25 ml) se le añadió cloruro de oxalilo (1,29 ml, 15,3 mmol) a ta. Se calentó gradualmente la mezcla de reacción hasta 95 °C y se agitó a 95 °C durante 1 h. Después de completarse la reacción, se enfrió la reacción hasta ta y se filtró. Se lavó el sólido con hexanos (2 x 5 ml) y se secó a vacío para proporcionar el compuesto del título como un sólido de color crema (1,61 g, 93 %). 1H-RMN (400 MHz, DMSO-de) 811,57 (s, 1H), 8,64 (d, J=3,2 Hz, 1H), 6,89 (d, J=2,8 Hz, 1H); EM ESI [M H]+ 170,0, calc. para [C6H3NO3S+ H]+ 169,9.
1-(4-metoxibencil)-1H-tieno[3,4-d1[1,31oxazin-2,4-diona

Según el método general E, a una disolución de 1H-tieno[3,4-d][1,3]oxazin-2,4-diona (1,6 g, 9,45 mmol) en DMF anh. (20 ml), se le añadió K2CO3 (1,56 g, 11,3 mmol) seguido por Kl (0,62 g, 3,78 mmol) con agitación a ta. Se añadió PMBCl (1,54 ml, 11,3 mmol) gota a gota a lo largo de 10 min y se agitó la mezcla de reacción durante 2 h adicionales. Después de completarse la reacción, se vertió la mezcla de reacción en H2O (200 ml) para precipitar el producto, que se filtró, se lavó con H2O y se secó para proporcionar el compuesto del título como un sólido blanquecino (2,3 g, 84 %). 1H-RMN (400 MHz, CDC3) 88,35 (d, J=3,2 Hz, 1H), 7,31 (d, J=8,8 Hz, 2H), 6,89 (d, J=8,8 Hz, 2H), 6,62 (d, J=3,2 Hz, 1H), 5,08 (s, 2H), 3,80 (s, 3H); EM ESI [M H]+ 291,2, calc. para [C14H11NO4S+ H]+ 290,0.
7-hidroxi-4-(4-metoxibencil)-6-(6-(4-metilpiperazin-1-il)-1H-benzo[d1imidazol-2-il)tieno[3,2-b1piridin-5(4H)-ona
Según el método general A1, se usaron una disolución de 1-(4-metoxibencil)-1H-tieno[3,2-d][1,3]oxazin-2,4-diona [Tetrahedron (1999) 556167-6174] (2,89 g, 10 mmol), 2-(6-(4-metilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)acetato de etilo [J.Med.Chem. (2009), 52, 278-292] (3,02 g, 10 mmol) y LiHMDS (1 M en THF, 4 ml, 4 mmol) para generar el compuesto del título como un sólido naranja (2,65 g, 51 %). 1H-RMN (400 MHz, CDC3) 813,68 (s a, 1H), 12,57 (s, 1H), 7,55 (dd, J=5,2, 2,0 Hz, 1H), 7,40-7,32 (m, 1H), 7,23 (d, J=8,8 Hz, 2H), 7,04-6,93 (m, 3H), 6,85 (d, J=8,8 Hz, 2H), 5,37 (s, 2H), 3,77 (s, 3H), 3,30-3,19 (m, 4H), 2,69-2,58 (m, 4H), 2,39 (s, 3H); EM ESI [M+H]+ 502,1, calc. para [C27H27N5OsS+H]+ 502,2.
4-hidroxi-7-(4-metoxibencil)-5-(6-(4-metilpiperazin-1-il)-1H-benzo[d1imidazol-2-il)tieno[2,3-b1piridin-6(7H)-ona

Según el método general A2, se usaron una disolución de 1-(4-metoxibencil)-1H-tieno[2,3-d][1,3]oxazin-2,4-diona (0,40 g, 1,4 mmol), 2-(6-(4-metilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)acetato de etilo (0,46 g, 1,5 mmol) y LDA (1 M en THF, 6,2 ml, 4,5 mmol) para generar el compuesto del título como un sólido marrón (0,220 g, 32 %). 1H-RMN (400 MHz, CDCIs) 813,87 (s a, 1H), 12,52 (s, 1H), 7,49 (dd, J = 14,9 Hz, 1H), 7,40-7,24 (m, 3H), 7,03-6,64 (m, 5H), 5,28 (d, J = 13,8 Hz, 2H), 3,76 (s, 3H), 3,21 (d, J = 18,8 Hz, 4H), 2,65 (m, d, J = 19,1 Hz, 4H), 2,41 (s, 3H); EM ESI [M+H]+ 502,3, calc. para [C27H27N5OsS+H]+ 502,2.

3-(4-((4-Metoxibencil)amino)tiofen-3-il)-2-(6-(4-metilpiperazin-1-il)-1H-benzo[d1imidazol-2-il)-3-oxopropanoato de etilo
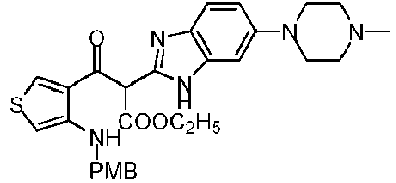
A una disolución de 2-(6-(4-metilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)acetato de etilo (2,58 g, 8,55 mmol) y 1-(4-metoxibencil)-1H-tieno[3,4-d][1,3]oxazin-2,4-diona (2,46 g, 8,55 mmol) en THF anh. (48 ml), se le añadió LDA 1 M (34 ml, 1 M en THF/hexano, 34 mmol) gota a gota a 40 °C bajo Ar. Se agitó la disolución marrón resultante a 40 °C durante 1 h y, a continuación, se extinguió con NH4Cl ac. (50 ml) a ta. Se diluyó la mezcla de reacción con H2O (50 ml) y se extrajo con DCM (2 x 200 ml). Se lavaron las fases orgánicas combinadas una vez con H2O, se secaron sobre Na2SO4 y se concentraron para dar un éster en bruto. Se purificó el producto en bruto mediante cromatografía ultrarrápida (gradiente: el 0-40 % de EtOAc/hex, seguido por el 0-25 % de MeOH/DCM) para dar el compuesto del
título como un sólido de color marrón claro (3,05 g, 65 %). EM ESI [M H]+ 548,2, calc. para [C29H33N5O4S+ H]+ 548,2.
4-hidroxi-1-(4-metoxibencil)-3-(6-(4-metilpiperazin-1-il)-1H-benzo[d1imidazol-2-il)tieno[3,4-b1piridin-2(1H)-ona

Se disolvió 3-(4-((4-metoxibencil)amino)tiofen-3-il)-2-(6-(4-metilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)-3-oxopropanoato de etilo (3,05 g, 5,57 mmol), descrito anteriormente, en THF anh. (30 ml) a ta bajo Ar. Se añadió una disolución de LDA (16,8 ml, 1 M en THF/hexano, 16,71 mmol) gota a gota a 40 °C. Se agitó la disolución marrón resultante a 40 °C durante 1 h y, a continuación, se extinguió con NH4Cl ac. (25 ml) a ta. Se diluyó la mezcla con H2O (25 ml) y se extrajo con dCm (2 x 250 ml). Se lavaron las fases orgánicas combinadas una vez con H2O, se secaron sobre Na2SO4 y se concentraron para dar el producto en bruto. Se purificó el producto en bruto mediante cromatografía ultrarrápida (gradiente: el 0-20 % de MeOH/DCM) para dar el compuesto del título como un sólido de color marrón claro (1,81 g, 65 %). 1H-RMN (400 MHz, DMSO-de) 813,8-13,25 (m, 1H), 8,13 (d, J=3,6 Hz, 1H), 7,58 (d, J=8,8 Hz, 1H), 7,36-7,29 (m, 3H), 7,06-7,02 (m, 1H), 6,97 (d, J=3,6 Hz, 1H), 6,86 (d, J=8,4 Hz, 2H), 5,19 (s, 2H), 3,69 (s, 3H), 3,16 (s a, 4H), 2,60 (s a, 4H), 2,31 (s, 3H); no puede detectarse fácilmente la señal debida al grupo o H. EM ESI 502,1 [M H]+, calc. para [C27H27N5O3S+ H]+ 502,2.
Trifluorometanosulfonato de 7-(4-metoxibencil)-5-(5 y/o 6-(4-metilpiperazin-1-il)-1-((trifluorometil)sulfonil)-1H-benzo[d1imidazol-2-il)-6-oxo-6,7-dihidrotieno[2,3-b1piridin-4-ilo

Sintetizados según el método general B usando 4-hidroxi-7-(4-metoxibencil)-5-(6-(4-metilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)tieno[2,3-b]piridin-6(7H)-ona (0,22 g, 0,44 mmol), Tf2O (0,60 ml, 3,5 mmol) y piridina (0,72 ml, 8,8 mmol). Se usaron los compuestos del título obtenidos como mezcla indeterminada de regioisómeros en la siguiente etapa sin purificación. EM ESI [M+H]+ 766,1, calc. para [C29H25F6N5O7S3+H]+ 766,1.
Trifluorometanosulfonato de1-(4-metoxibencil)-3-(5 y/o 6-(4-metilpiperazin-1-il)-1-((trifluorometil)sulfonil)-1H-benzo[d]-imidazol-2-il)-2-oxo-1,2-dihidrotieno[3,4-b1piridin-4-ilo

Según el método general B, a una disolución de 4-hidroxi-1-(4-metoxibencil)-3-(6-(4-metilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)tieno[3,4-b]piridin-2(1H)-ona (220 mg, 0,43 mmol) y piridina (708 ml, 8,76 mmol) en DCM (12 ml) se le añadió Tf2O (558 ml, 3,50 mmol) a -5 °C. Se agitó la reacción entre -5 y 0 °C durante 1 h. Se extinguió la reacción con NaHCO3 ac. sat. Se extrajo la fase acuosa con DCM y se secaron los extractos orgánicos combinados sobre Na2SO4 y se concentraron a vacío para dar un aceite de color marrón oscuro. Se usó el producto en bruto, obtenido como mezcla indeterminada de regioisómeros, directamente en la siguiente etapa sin purificación adicional. EM ESI [M H]+ 766,0, calc. para [C29H25F6N5O7S3+ H]+ 766,1.
Trifluorometanosulfonato de 7-(4-metoxibencil)-5-(5 y/o 6-morfolino-1-((trifluorometil)sulfonN)-1H-benzo[d]imidazol-2-il)-6-oxo-6,7-dihidrotieno[2,3-b1piridin-4-ilo

Según el método general B, a una disolución de 4-hidroxi-7-(4-metoxibencil)-5-(6-morfolino-1H-benzo[d]imidazol-2-il)-tieno[2,3-b]piridin-6(7H)-ona (200 mg, 0,41 mmol) y piridina (0,66 ml, 8,2 mmol) en DCM (20 ml) se le añadió Tf2O (0,55 ml, 3,28 mmol) a 0 °C. Se agitó la mezcla de reacción at 0 °C durante 2 h y, a continuación, se extinguió con NaHCO3 ac. sat. Se extrajo la fase acuosa con DCM. Se secaron los extractos orgánicos combinados sobre Na2SO4 y se concentraron para dar el compuesto del título en bruto (mezcla de dos regioisómeros) como un aceite marrón que se usó directamente en la etapa posterior sin purificación adicional considerando rendimiento cuantitativo. EM ESI [M H]+ 753,0, calc. para [C28H22F6N4O8S3+ H]+ 752,9.
-Metil-1H-tieno[2,3-d1[1,31oxazin-2,4-diona

A una disolución de KOH (0,49 g, 8,76 mmol) en H2O (20 ml) se le añadió 2-amino-4-metil-3-tiofenocarboxilato de metilo (1,0 g, 5,84 mmol) a ta. Se calentó la reacción resultante hasta 90 °C durante 2 h y, a continuación, se enfrió hasta 0 °C. Se añadió una disolución de trifosgeno (0,866 g, 2,92 mmol) en PhMe (12 ml) gota a gota a lo largo de 10 min. Se calentó gradualmente la disolución resultante hasta ta y se agitó durante 2 h. Se filtró el sólido resultante, se lavó con H2O y se secó para proporcionar el compuesto del título como un sólido de color rosa claro (0,65 g, 61 %). 1H-RMN (400 MHz, CD3 OD) 86,65 (d, J=1,2 Hz,1H), 2,42 (d, J=1,2 Hz, 3H); EM ESI [M H]+ 184,0, calc. para [C7H5NO3S+ H]+ 184,0.
1-(4-metoxibencil)-5-metil-1H-tieno[2,3-d1[1,31oxazin-2,4-diona

A una disolución de 5-metil-1H-tieno[2,3-d][1,3]oxazin-2,4-diona (0,625 g, 3,41 mmol) en DMF anh. (9 ml), se le añadió K2CO3 (0,566 g, 4,09 mmol) seguido por KI (0,142 g, 0,85 mmol) con agitación a ta. Se añadió PMBCl (0,56 ml, 4,06 mmol) gota a gota a la reacción a lo largo de 10 min y se agitó durante 2 h adicionales. Se vertió la mezcla de reacción en H2O (100 ml) para precipitar el producto, que se filtró, se lavó con H2O y se secó para proporcionar el compuesto del título como un sólido de color marrón claro (0,935 g, 91 %). 1H-Rm N (400 MHz, CDC3) 87,38 (d, J=8,8 Hz, 2H), 6,90-6,88 (m, 2H), 6,46 (d, J=1,2 Hz, 1H), 5,05 (s, 2H), 3,80 (s, 3H), 2,42 (d, J=1,2 Hz, 3H); EM ESI [M+H]+ 304,2, calc. para [C15H1sNO4S+H]+ 304,1.
4-hidroxi-7-(4-metoxibencil)-3-metil-5-(6-(4-metilpiperazin-1-il)-1H-benzo[d1imidazol-2-il)tieno[2,3-b1piridin-6(7H)-ona

Se añadió una disolución de LDA (34 ml, 1 M en THF/hexano, 34 mmol) gota a gota a una disolución de 2-(6-(4-metilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)acetato de etilo (922 mg, 3,04 mmol) y 1-(4-metoxibencil)-5-metil-1H-tieno[2,3-d][1,3]oxazin-2,4-diona (925 mg, 3,04 mmol) en Th F anh. (28 ml) a 40 °C bajo Ar. Se agitó la disolución marrón resultante a 40 °C durante 2 h y, a continuación, se extinguió con NH4Cl ac. (25 ml) a ta. Se diluyó la mezcla de reacción con H2O (25 ml) y se extrajo con DCM (2 x 100 ml). Se lavaron las fases orgánicas combinadas una vez con H2O, se secaron sobre Na2SO4 y se concentraron para dar una mezcla del producto y éster no ciclado. Se purificó la masa en bruto mediante cromatografía ultrarrápida (gradiente: el 0-40 % de EtOAc/hex, seguido por el 0 25 % de MeOH/DCM) para dar una mezcla del producto y éster no ciclado (900 mg).
Se disolvió la mezcla del producto y éster no ciclado anterior (900 mg) en THF anh. (9 ml) a ta bajo Ar. Se añadió una disolución de LDA (5 ml, 1 M en THF/hexano) gota a gota a 40 °C. Se agitó la disolución marrón resultante a 40 °C durante 1 h y se sometió a tratamiento final tal como anteriormente para dar un producto en bruto. Se purificó el producto en bruto mediante cromatografía ultrarrápida (gradiente: el 0-20 % de MeOH/DCM) para dar el compuesto del título como un sólido de color crema (325 mg, 21 %). 1H-RMN (400 MHz, CDCI3) 8 12,54 (s, 1H), 7,38 (d, J=8,4 Hz, 1H), 7,33 (d, J=8,4 Hz, 2H), 7,01-6,98 (m, 2H), 6,85 (d, J=8,8 Hz, 2H), 6,40 (s, 1H), 5,26 (s, 2H), 3,80-3,61 (m, 6 H), 3,60-3,51 (m, 4H), 2,89-2,88 (m, 4H), 2,63 (s, 3h); no puede detectarse fácilmente la señal debida al grupo OH. EM ESI [M+H]+ 516,2, calc. para ^s^gNaOsS+Hr 516,2.
Trifluorometanosulfonato de 7-(4-metoxibencil)-3-metil-5-(5 y /o 6-(4-metilpiperazin-1-il)-1-((trifluorometil)sulfonil)-1H-benzo[d1imidazol-2-il)-6-oxo-6,7-dihidrotieno[2,3-b1piridin-4-ilo

Se preparó el compuesto del título según el método general B utilizando 4-hidroxi-7-(4-metoxi-bencil)-3-metil-5-(6-(4-metilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)tieno[2,3-b]piridin-6(7H)-ona (320 mg, 0,62 mmol), piridina (1,0 ml, 12,4 mmol), Tf2O (0,833 ml, 4,96 mmol) en DCM (12 ml) para dar un aceite de color marrón oscuro. Se usó el producto en bruto, obtenido como mezcla indeterminada de 2 regioisómeros, directamente en la siguiente etapa sin purificación adicional. EM ESI [M H]+ 780,0, calc. para [C30H27F6N5O7S3+ H]+ 780,1.
2-(6-((3r,5s)-rel-3,4,5-trimetilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)acetato de etilo
A. 2-nitro-5-((3r,-5s)-rel-3,4,-5-trimetilpiperazin-1-il)anilina

Se irradió una mezcla de 5-cloro-2-nitroanilina (1,73 g, 10mmol), (3r,5s)-rel-1,2,6-trimetilpiperazina (1,41 g, 11 mmol) y K2CO3 (2,72 g, 20 mmol) en microondas a 140 °C durante 4 h. A continuación se añadió H2O (150 ml) con agitación, se filtró por succión, se aclaró con H2O y se secó para dar el compuesto del título como un sólido marrón (2,47 g, 94 %). 1H-RMN (400 MHz, DMSO-de) 87,79 (d, J=10,0 Hz, 1H), 7,23 (s, 2H, NH2), 6,41 (dd, J=9,6, 1,6 Hz, 1H), 6,20 (d, J=2,4 Hz, 1H), 3,77 (d, J=12,4 Hz, 2H), 2,59 (t, J=11,8 Hz, 2H), 2,19-2,11 (m, 2H), 2,16 (s, 3H), 1,05 (d, J=e,0 Hz, 6H); EM ESI [M+H]+ 265,3, calc. para [C13H20N4O2+HI+ 265,2.
B. 4-((3r,5s)-rel-3,4,-5-trimetilpiperazin-1 -il)benceno- 1,2-diamina

A una suspensión de 2-nitro-5-((3r,5s)-rel-3,4,5-trimetilpiperazin-1-il)anilina (2,47 g, 9,4 mmol) en MeOH (30 ml) se le añadió Pd al 10 %/C (247 mg, 10 % en peso). Se hidrogenó la mezcla resultante bajo un globo de H2 O/N. Después se añadió Pd al 10 %/C adicional (124 mg, 5 % en peso), se hidrogenó bajo un globo de H2 O/N, se filtró, se concentró y se secó para dar el compuesto del título como un sólido de color marrón oscuro (2,25 g, cuantitativo).
1H-RMN (400 MHz, CD3OD) 8 6,66 (d, J=8,4 Hz, 1H), 6,47 (d, J=2,4 Hz, 1H), 6,31 (dd, J=8,4, 2,8 Hz, 1H), 3,35-3,25 (m, 2H), 2,47-2,40 (m, 4H), 2,34 (s, 3H), 1,18 (d, J=5,6 Hz, 6H); EM ESI [M+H]+ 235,3, calc. para [C13H22N4+HI+ 235,2.
C. 2-(e-((3r,-5s)-rel-3,4,-5-trimetilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)acetato de etilo

A una disolución de 4-((3r,5s)-rel-3,4,5-trimetilpiperazin-1-il)benceno-1,2-diamina (2,25 g, 9,4 mmol) en EtOH (40 ml) se le añadió clorhidrato de 3-etoxi-3-iminopropionato de etilo (2,93 g, 15 mmol). Se calentó la mezcla resultante a 80 °C durante 2 h. Después de eliminar los disolventes, se diluyó con DCM/MeOH (100 ml/10 ml), se basificó con NaHCO3 ac. sat.(30 ml) y se separó. Se extrajo la fase acuosa con DCM (60 ml x 2) y se concentraron los extractos combinados y se purificaron mediante cromatografía ultrarrápida (gradiente: el 100 % de EtOAc, luego el 0-20 % de MeOH/DCM) para dar el compuesto del título como un sólido de color naranja oscuro (2,32 g, 73 %). 1H-RMN (400 MHz, CDCIs) 810,13 (s a, 1H, NH), 7,53-6,88 (m, 3H), 4,25 (q, J=7,2 Hz, 2H), 4,03 (s, 2H), 3,43 (d, J=11,2 Hz, 2H), 2,61 (t, J=11,2 Hz, 2H), 2,50-2,41 (m, 2H), 2,35 (s, 3H), 1,32 (t, J=7,2 Hz, 3H), 1,19 (d, J=6,0 Hz, 6H); EM ESI [M+H]+ 331,3, calc. para [C18H26N4O2+HI+ 331,2.
7-hidroxi-4-(4-metoxibencil)-6-(6-((3r,5s)-rel-3,4,5-trimetilpiperazin-1-il)-1H-benzo[d1imidazol-2-il)tieno[3,2-b1piridin-5(4H)-ona

A una mezcla de 1-(4-metoxibencil)-1H-tieno[3,2-d][1,3]oxazin-2,4-diona (1,16g, 4 mmol) y 2-(6-((3r,5s)-rel-3,4,5-trimetilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)acetato de etilo (990 mg, 3 mmol) en THF (20 ml) se le añadió LDA (1,0 M en THF/hex, 10 ml, 10 mmol) gota a gota a ta. Después de la adición, se agitó la mezcla resultante a 40 °C durante 1 h, se diluyó con DCM, se extinguió con NH4Cl ac. sat. y se extrajo con DCM. Se concentraron los extractos combinados y se purificaron mediante cromatografía ultrarrápida (gradiente: el 20-100 % de EtOAc/hex, luego el 0-20 % de MeOH (el 0,5 % de NH3)/DCM) para dar una mezcla de producto ciclado y no ciclado como una espuma marrón (1,10 g). Volvió a disolverse la mezcla en THF (15 ml) y se añadió LDA (1,0 M en THF/hex, 6 ml, 6 mmol) gota a gota a ta. El procedimiento y el tratamiento final fueron los mismos que anteriormente. Se obtuvo el compuesto del título como un sólido naranja (630 mg, 40 %). EM ESI [M+H]+ 530,3, calc. para [C29H3iN5O3S+H]+ 530,2.
Trifluorometanosulfonato de 4-(4-metoxibencil)-5-oxo-6-(1-((trifluorometil)sulfonil)-(5 y/o 6-((3r,5s)-rel-3,4,5-trimetil-piperazin-1-il)-1H-benzo[d]imidazol-2-ir)-4,5-dihidrotieno[3,2-b]piridin-7-ilo

Según el método general B, a una disolución de 7-hidroxi-4-(4-metoxibencil)-6-(6-((3r,5s)-rel-3,4,5-trimetil-piperazin-1-il)-1H-benzo[d]imidazol-2-il)tieno[3,2-b]piridin-5(4H)-ona (106 mg, 0,2 mmol) en Dc M (15 ml) a 0 °C se le añadió piridina (0,32 ml, 4 mmol), seguido por Tf2O (0,27 ml, 1,2 mmol). Se agitó la mezcla resultante a 0 °C durante 1 h, se diluyó con DCM (10 ml), se extinguió con NaHCO3 ac. sat. (15 ml), se extrajo con DCM (20 ml x 2) y se concentró para dar el compuesto del título en bruto (una mezcla indeterminada de dos regioisómeros) como un aceite marrón que se usó directamente en las etapas posteriores. EM ESI [M+H]+ 794,1, calc. para [C3iH29F6N5O7S3+H]+ 794,11.
Síntesis de 2-amino-4-etoxitiofeno-3-carbonitrilo

Se agitó la mezcla de MeC(OMe)3 (2,26 ml, 12,3 mmol) y CH2(CN)2 (0,78 ml, 12,3 mmol) a 65 °C durante 3 h antes de enfriarse hasta ta. Se añadió THF (10 ml) y azufre (395 mg) seguido por la adición de Et3N (1,72 ml, 12,3 mmol) gota a gota. Se agitó la mezcla de reacción resultante a 60 °C durante 15 min y se concentró a presión reducida. Se sometió a reparto el residuo entre EtOAc y H2O, se extrajo con EtOAc, se secó sobre Na2SO4, se filtró y se concentró hasta sequedad. Se trituró el residuo con DCM y se filtró para dar el compuesto del título como un sólido marrón (1,23 g, 60 %). 1H-RMN (400 MHz, CD3 OD) 85,27 (s, 1H), 3,99 (q, J=7,0 Hz, 2H), 1,38 (t, J=7,0 Hz, 3H); EM ESI [M H]+ 169,0, calc. para [C7H8N2OS+HI+ 169,0.
4-(5-amino-4-cianotiofen-3-il)piperazin-1-carboxilato de terc-butilo

Se calentó una mezcla de MeC(OMe)3 (1,3 ml, 10 mmol) y CH2(CN)2 (0,66 g, 10 mmol) en un vial cerrado a 80 °C durante 17 h. Se enfrió la reacción hasta ta y se añadió piperazin-1-carboxilato de terc-butilo (2,79 g, 15,0 mmol). Se continuó el calentamiento con agitación a 65 °C durante 5 h. A continuación se concentró la mezcla de reacción a vacío. Se añadieron S8 (0,34 g) y THF anh. (10 ml). Se calentó la suspensión con agitación a 40 °C. Se añadió Et3N (1,3 ml, 9,3 mmol) gota a gota a lo largo de 15 min. Se aumentó la temperatura del baño de aceite hasta 60 °C y se continuó la agitación durante 11 h. A continuación se concentró la reacción a presión reducida y se purificó mediante cromatografía ultrarrápida (SiO2, el 5-50 % de hexanos:EtOAc) para proporcionar 4-(5-amino-4-cianotiofen-3-il)piperazin-1-carboxilato de terc-butilo como un sólido de color naranja claro (0,71 g, 23 %). 1H-RMN (400 MHz, DMSO-de) 87,12 (s, 2H), 5,46 (s, 1H), 3,45-3,37 (m, 4H), 2,90-2,81 (m, 4H), 1,40 (s, 9H). EM ESI [M+H]+ 309,3, calc. para [C14H20N4O2S H]+ 309,1.
4-(4-amino-5-(6-(4-metilpiperazin-1-il)-1H-benzo[d1imidazol-2-il)-6-oxo-6,7-dihidrotieno-[2,3-b1piridin-3-il)piperazin-1-carboxilato de terc-butilo

Se añadió LiHMDS (1,0 M en THF, 2,8 ml, 2,8 mmol) gota a gota a ta a una suspensión con agitación de 2-(6-(4-metilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)acetato de etilo (0,170 g, 0,56 mmol) y 4-(5-amino-4-cianotiofen-3-il)piperazin-1-carboxilato de terc-butilo (0,175 g, 0,56 mmol) en THF anh. (10 ml) bajo Ar. Se agitó la reacción a ta durante 5 min adicionales y, a continuación, se calentó en un baño de aceite a 40 °C durante 1 h. Se enfrió la reacción hasta ta, se extinguió con NH4Cl ac. sat., se concentró a presión reducida y se purificó mediante cromatografía ultrarrápida (el 0-20 % de MeOH/DCM) para dar el compuesto del título como un sólido de color marrón tostado claro (83 mg, 26 %). 1H-RMN (400 MHz, CD3 OD) 87,45 (d, J=8,8 Hz, 1H), 7,10 (d, J=2,3 Hz, 1H), 7,03 (dd, J=8,8, 2,3 Hz, 1H), 6,18 (s, 1H), 3,62-3,50 (m, 4H), 3,24-3,18 (m, 4H), 3,05-2,98 (m, 4H), 2,75-2,67 (m, 4H), 2,41 (s, 3H), 1,49 (s, 9H); EM ESI [M+H]+ 565,3, calc. para [C2sH36NsO3S H]+ 565,4.
2-(6-(4-metilpiperazin-1-carbonil)-1H-benzo[d1imidazol-2-il)acetato de etilo
A. (3,4-dinitrofenil)(4-metilpiperazin-1-il)metanona

A una suspensión de ácido 3,4-dinitrobenzoico (1,23 g, 5,8 mmol) en DCM anh. (20 ml) a ta se le añadió gota a gota cloruro de oxalilo (1,0 ml, 11,7 mmol) seguido por DMF anh. (2 gotas). Se agitó la reacción durante la noche y, a continuación, se concentró a ta. Se disolvió el residuo en THF anh. (40 ml) a 0 °C bajo Ar. Se añadió 1-metilpiperazina (1,3 ml, 11,7 mmol) gota a gota (se agitó la suspensión blanca espesa con agitación intermitente). Después de la adición, se continuó el enfriamiento durante 10 min antes de retirar el baño de enfriamiento. Después de agitar la reacción a ta durante 3 h, se añadió H2O. Se eliminó el THF a presión reducida y se extrajo el residuo acuoso (CH2Cl2; el 2 % de MeOH en CH2CI2; 2x). Se secaron los extractos orgánicos combinados sobre Na2SO4 y se concentraron a presión reducida para proporcionar (3,4-dinitrofenil)(4-metilpiperazin-1-il)metanona como un sólido de color naranja claro (1,77 g, cuant.) . 1H-RMN (400 MHz, DMSO-de) 88,29 (d, J=8,3 Hz, 1H), 8,27 (d, J=1,5 Hz, 1H), 7,97 (dd, J=8,3, 1,8 Hz, 1H), 3,59-3,68 (m, 2H), 3,24-2,53 (m, 2H), 2,42-2,35 (m, 2H), 2,21-2,32 (m, 2H), 2,20 (s, 3H). EM ESI [M+H]+ 295,2, calc. para [C12H14N4O5 + H]+ 295,1.
B. (3,4-diaminofenil)(4-metilpiperazin-1 -il)metanona

Se desgasificó una disolución de (3,4-dinitrofenil)(4-metilpiperazin-1-il)metanona (0,53 g, 1,8 mmol) en THF (25 ml) y EtOH (50 ml) con N2. Se añadió Pd/C (191 mg, 0,18 mmol) y se agitó la reacción bajo H2 (1 atm) durante la noche a ta. A continuación se filtró la mezcla de reacción a través de Celite y se concentró a presión reducida para proporcionar (3,4-diaminofenil)(4-metilpiperazin-1-il)metanona como un sólido morado (0,44 g, cuant.). 1H-Rm N (400 MHz, DMSO-de) 86,61-6,55 (m, 1H), 6,47-6,45 (m, 2H), 4,81 (s a, 2H), 4,58 (s a, 2H), 3,50-3,39 (m, 4H), 2,34 2,22 (m, 4H), 2,18 (s, 3H). EM ESI [M+H]+ 235,1, calc. para [C12H18N4O H]+ 235,1.
C. 2-(e-(4-metilpiperazin-1-carbonil)-1H-benzo[d]imidazol-2-il)acetato de etilo

Se calentaron (3,4-diaminofenil)(4-metilpiperazin-1-il)metanona (0,44 g, 1,8 mmol) y clorhidrato del ácido 3-etoxi-3-iminopropanoico (1,07 g, 5,5 mmol) en EtOH anh. (100 ml) bajo Ar con agitación durante la noche a 65 °C. A continuación se concentró la mezcla de reacción a presión reducida. Se llevó el residuo a H2O (15 ml), se neutralizó con Na2CO3 ac. al 10 %, se extrajo con CH2Cl2 (2x), se lavó (salmuera) y se secó sobre Na2SO4. La purificación mediante cromatografía ultrarrápida (el 0-50 % de MeOH en c H2CI2) proporcionó el compuesto del título como una espuma amarilla (0,31 g, 52 %). 1H-RMN (400 MHz, CD3 OD) 87,71-7,57 (m, 2H), 7,33 (d, J=8,2 Hz, 1H), 4,23 (q, J=7,2 Hz, 2H), 3,91-3,40 (m, 4H), 2,62-2,38 (m, 4H), 2,34 (s, 3H), 1,28 (t, J=7,1 Hz, 3H); las señales debidas al CH2-éster están ausentes en CD3OD. e M ESI [M+h]+ 331,2, calc. para [Ci7H22N4O3+H]+ 331,2.
2-(6-(morfolin-4-carbonil)-1H-benzo[d1imidazol-2-il)acetato de etilo
A. (3,4-dinitrofenil)(morfolino)metanona

A una suspensión de ácido 3,4-dinitrobenzoico (1,30 g, 6,1 mmol) en DCM anh. (50 ml) a ta se le añadió gota a gota (COCl)2 (1,0 ml, 11,7 mmol) seguido por DMF anh. (2 gotas). Se agitó la reacción durante la noche y, a continuación, se concentró a ta. Se disolvió el residuo en THF anh. (24 ml) a 0 °C bajo Ar. Se añadió morfolina (1,0 ml, 11,6 mmol) gota a gota (se agitó la suspensión blanca espesa con agitación intermitente). Después de la adición, se continuó el enfriamiento durante 10 min antes de retirar el baño de enfriamiento. Después de agitar la reacción a ta durante 3 h, se añadió H2O. Se eliminó el THF a presión reducida y se extrajo el residuo acuoso (CH2CI2; 2x). Se secaron los extractos orgánicos combinados (Na2SO4) y se concentraron a presión reducida para proporcionar (3,4-dinitrofenil)(morfolino)metanona como un sólido de color naranja claro (1,8 g, cuant.). 1H-Rm N (400 MHz, DMSO-de) 88,28 - 8,31 (m, 2H), 8,00 (dd, J=8,28, 1,76 Hz, 1H), 3,39 - 3,80 (m, 8H).
B. 2-(6-(morfolin-4-carbonil)-1H-benzo[d]imidazol-2-il)acetato de etilo

Se desgasificó una disolución de (3,4-dinitrofenil)(morfolino)metanona (0,83 g, 2,9 mmol) en THF (30 ml) y EtOH (60 ml) con N2. Se añadió Pd/C (0,31 mg, 0,29 mmol) y se agitó la reacción bajo H2 (1 atm) durante la noche a ta. A continuación se filtró la mezcla de reacción a través de Celite y se concentró a presión reducida para proporcionar (3,4-diaminofenil)(morfolino)metanona como una espuma morada. CL-EM (ESI) m/z calc. para [C11H15N3O2 H]+ 222,1; encontrado 222,2. Se calentaron el material y clorhidrato de 3-etoxi-3-iminopropanoato de etilo (1,2 g, 6,2 mmol) en EtOH anh. (100 ml) bajo Ar con agitación durante la noche a 65 °C. A continuación se concentró la mezcla de reacción a presión reducida. La purificación mediante cromatografía ultrarrápida (el 0-20 % de MeOH en CH2Cl2) proporcionó el compuesto del título como una espuma de color rojo pálido (0,43 g, 47 %). 1H-RMN (400 MHz, CD3OD) 87,52 - 7,76 (m, 2H), 7,33 (dd, J=8,28, 1,51 Hz, 1H), 4,22 (q, J=7,19 Hz, 2H), 4,00 - 4,05 (m, 2h ), 3,70 (s a., 8H), 1,28 (t, J=7,15 Hz, 3h ); las señales debidas al CH2-éster están ausentes en CD3OD; EM ESI [M+H]+ 318,2, calc. para [C17H22N4O3+HI+ 318,1.
2-(5-metil-6-(4-metilpiperazin-1-il)-1H-benzo[d1imidazol-2-il)acetato de etilo
A. 4-metil-5-(4-metilpiperazin-1-il)-2-nitroanilina

Se calentaron 5-cloro-4-metil-2-nitroanilina (0,32 g, 1,7 mmol) y 1-metilpiperazina (1,5 ml, 13,5 mmol) en un tubo sellado a 80 °C durante 30 min seguido por a 105 °C durante 1 d y 120 °C durante 2 d. Posteriormente se enfrió la reacción, se diluyó con H2O y se filtró. Se aclaró el sólido recogido con H2O y se secó a vacío para proporcionar el compuesto del título como un sólido amarillo (0,36 g, 84 %). 1H-RMN (400 MHz, DMSO-de) 87,72 (s, 1H), 7,27 (s, 2H), 6,44 (s, 1H), 2,97-2,86 (m, 4H), 2,49-2,39 (m, 4H), 2,22 (s, 3H), 2,11 (s, 3H). CL-EM (ESI) m/z calc. para [Ci 2Hi 8N4O2 H]+ 251,1; encontrado 235,3.
B. 2-(5-metil-e-(4-metilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)acetato de etilo

Se desgasificaron 4-metil-5-(4-metilpiperazin-1-il)-2-nitroanilina (0,36 g, 1,4 mmol) y Pd/C (10 %, 81 mg, 0,08 mmol) en EtOH (50 ml), THF(25 ml) con N2 y, a continuación, se agitó bajo H2 (1 atm) durante 5 d. Se filtró la mezcla de reacción a través de Celite, se aclaró el lecho con EtOH. Se concentró el filtrado a presión reducida para proporcionar 4-metil-5-(4-metilpiperazin-1-il)benceno-1,2-diamina como un sólido de color marrón tostado amarillento (0,35 g, cuant.). Se calentaron el material (0,35 g) y clorhidrato de 3-etoxi-3-iminopropanoato de etilo (0,81 g, 4,1 mmol) en EtOH anh. (70 ml) bajo Ar con agitación durante la noche a 65 °C. A continuación se concentró la mezcla de reacción a presión reducida, se llevó a H2O (20 ml) y se basificó con Na2CO3 ac. 2 M hasta pH 9. Se extrajo la mezcla con DCM (2x); se secaron los extractos orgánicos (Na2SO4) y se concentraron a presión reducida. La purificación mediante cromatografía ultrarrápida (el 0-30 % de MeOH en Ch2CI2) proporcionó el compuesto del título como una espuma amarilla (0,36 g, 82 %). 1H-RMN (500 MHz, CD3OD) 87,35 (s, 1H), 7,25 (s, 1H), 4,22 (q, J=7,09 Hz, 2H), 2,95 - 3,03 (m, 4H), 2,88-2,58 (m, 4H), 2,43 (s, 3H), 2,41 (s, 3H), 1,28 (t, J=7,09 Hz, 3H); las señales debidas al CH2-éster están ausentes en CD3OD; CL-EM (ESI) m/z calc. para [C17H24N4O2+ H]+ 317,2; encontrado 317,3.
2-(5-fluoro-6-morfolino-1H-benzo[d1imidazol-2-il)acetato de etilo
A. 4-fluoro-5-morfolino-2-nitroanilina

Se calentó una mezcla de 5-cloro-4-fluoro-2-nitroanilina (1,0 g, 5,24 mmol), morfolina (1,37 ml, 15,7 mmol) y DMSO (5 ml) en un baño de aceite a 140 °C durante 3 h. A continuación se añadió H2O (50 ml) con agitación a 80 °C para precipitar el producto y se dejó la suspensión hasta ta, se filtró por succión, se lavó con H2O y se secó para dar el compuesto del título como un sólido amarillo (1,25 g, 94 %). 1H-RMN (400 MHz, CD3 OD) 87,17 (d, J=14,0 Hz, 1H),
6,37 (d, J=8,0 Hz, 1H), 3,83 (t, J=4,4 Hz, 4H), 3,22 (t, J=4,8 Hz, 4H); EM ESI [M+H]+ 242,1, calc. para [Ci0Hi2FNsO3+H]+ 242,1.
B. 4-fluom-5-morfolinobenceno- 1,2-diamina
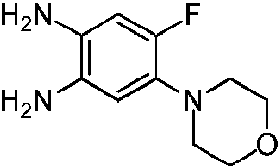
Se cargó un matraz de fondo redondo de 100 ml con 4-fluoro-5-morfolino-2-nitroanilina (1,23 g) y MeOH (37 ml) a ta bajo una manta de Ar. Se añadió níquel Raney (0,123 g) con agitación con precaución a ta. Se calentó lentamente la masa de reacción hasta 60-65° y se añadió hidrazina hidratada (0,86 ml) a la masa de reacción gota a gota en aproximadamente 5 min. Se agitó la reacción a 65-70 °C durante 2 h. Después de completarse la reacción, se enfrió hasta ta y se filtró el catalizador a través de un lecho de Celite bajo Ar y se lavó el lecho de Celite con MeOH (5 ml * 2). Se concentró el filtrado combinado y se purificó mediante cromatografía ultrarrápida (gradiente: el 0-25 % de MeOH/DCM) para dar el compuesto del título como un sólido de color marrón claro (0,615 g, 57 %). 1H-RMN (400 MHz, CDsOD) 86,51- 6,47 (m, 2H), 3,81 (t, J=4,8 Hz, 4H), 2,93 (t, J=4,8 Hz, 4H); EM ESI [M+H]+ 212,0, calc. para [C10H14FN3O+HI+212,1.
C. 2-(5-fíuoro-6-morfolino-1H-benzo[d]imidazol-2-il)acetato de etilo

A una disolución de 4-fluoro-5-morfolinobenceno-1,2-diamina (0,615 g, 2,91 mmol) en EtOH (30 ml) a 65 °C se le añadió clorhidrato de 3-etoxi-3-iminopropionato de etilo (1,14 g, 5,82mmol) en dos lotes iguales en un intervalo de 5 min cada uno. A continuación se agitó la masa de reacción a 65 °C durante 2 h. Después de completarse la reacción, se concentró la masa de reacción a presión reducida para dejar atrás un aceite marrón espeso. Al aceite resultante se le añadió H2O (25 ml) y se ajustó hasta pH ~ 10 usando Na2CO3 ac. 2 M. Se extrajo la mezcla resultante con DCM (30 ml * 2) y se concentraron los extractos combinados y se purificaron mediante cromatografía ultrarrápida (gradiente: el 0-40 % de hex/EtOAc, luego el 0-20 % de MeOH/DCM) para dar el compuesto del título como un sólido marrón (0,786 g, 88 %). 1H-RMN (400 MHz, CD3 OD) 87,26 (d, J=12,4 Hz, 1H), 7,19 (d, J=7,6 Hz, 1H),4,25 - 4,20 (m, 2H), 3,88 (t, J=4,4 Hz, 4H), 3,08 (t, J=4,8 Hz, 4H), 1,28 (t, J=7,2 Hz, 3H); EM ESI [M+H]+ 308,1, calc. para [Ci5Hi8FN3O3+H]+ 308,1.
2-(6-(4-metil-1,4-diazepan-1-il)-1H-benzo[d1imidazol-2-il)acetato de etilo
A. 5-(4-metil-1,4-diazepan-1-il)-2-nitroanilina

Se calentó una mezcla de 5-cloro-2-nitroanilina (8,63 g, 50 mmol), 1-metil-1,4-diazepano (6,85 g, 60 mmol) y K2CO3 (8,28 g, 60 mmol) a 90 °C durante 20 h. Después de diluir con H2O (500 ml), se extrajo con EtOAc (60 ml x 3), se concentró y se secó para dar 5-(4-metil-1,4-diazepan-1-il)-2-nitroanilina en bruto como un aceite de color rojo oscuro (12,50 g). La RMN indicó una mezcla del producto y 5-cloro-2-nitroanilina (2:1). 1H-RMN (400 MHz, CD3 OD) 87,72 (d, J=10,0 Hz, 1H), 6,26 (dd, J=9,8, 2,6 Hz, 1H), 6,02 (d, J=2,4 Hz, 1H), 3,66-3,63 (m, 2H), 3,58 (t, J=6,4 Hz, 2H), 2,77-2,74 (m, 2H), 2,62-2,59 (m, 2H), 2,39 (s, 3H), 2,07-2,00 (m, 2H); EM ESI [M+H]+ 251,3, calc. para [C12H18N4O2+HI+251,15.
B. 4-(4-metil- 1,4-diazepan-1-il)benceno-1,2-diamina

A una mezcla de 5-(4-metil-1,4-diazepan-1-il)-2-nitroanilina en bruto (12,50 g) y níquel Raney (1,25 g) en MeOH (150 ml) a 65 °C se le añadió N2H4-H2O (12,0 ml) a lo largo de 10 min. Después de la adición, se agitó la mezcla resultante a 70 °C durante 30 min. Tras enfriar hasta ta, se filtró a través de Celite y se aclaró con MeOH. Se concentró el filtrado y se secó para dar 4-(4-metil-1,4-diazepan-1-il)benceno-1,2-diamina en bruto como un aceite de color marrón rojizo oscuro (10,57 g). 1H-RMN (400 MHz, CD3 OD) 86,63 (d, J=8,0 Hz, 1H), 6,53 (dd, J=8,4, 2,4 Hz,
1H), 6,26 (d, J=2,4 Hz, 1H), 3,60-3,40 (m, 4H), 2,75-2,71 (m, 2H), 2,62-2,58 (m, 2H), 2,37 (s, 3H), 2,04-1,97 (m, 2H).
C. 2-(6-(4-metil-1,4-diazepan-1-il)-1H-benzo[d]itnidazol-2-il)acetato de etilo

Se calentó una mezcla de 4-(4-metil-1,4-diazepan-1-il)benceno-1,2-diamina en bruto (10,57 g) y clorhidrato de 3-etoxi-3-iminopropionato de etilo (19,50 g, 100 mmol) en EtOH (200 ml) a 90 °C durante 2 h. Después de eliminar los disolventes, se diluyó con H2O (50 ml), se basificó con Na2CO3 ac. 2 M (40 ml) y se extrajo con DCM (60 ml x 3). Se concentraron los extractos combinados y se purificaron mediante cromatografía ultrarrápida (gradiente: el 0-100 % de EtOAc/hexano, luego el 0-25 % de MeOH/DCM) para dar el compuesto del título como un aceite de color marrón oscuro (7,31 g, 46 % a lo largo de 3 etapas). 1H-RMN (400 MHz, CD3OD) 88,38 (d, J=8,8 Hz, 1H), 6,82-6,77 (m, 2H), 4,22 (q, J =6,8 Hz, 2H), 3,66-3,61 (m, 2H), 3,54 (t, J=6,4 Hz, 2H), 2,85-2,80 (m, 2H), 2,68-2,64 (m, 2H), 2,41 (s, 3H), 2,12-2,05 (m, 2H), 1,29 (t, J=7,0 Hz, 3H); EM ESI [M+H]+ 317,3, calc. para [C17H24N4O2+H]+ 317,20.
Ejemplos representativos:
A1: 4-amino-5-(6-(4-metilpiperazin-1-il)-1H-benzo[d1imidazol-2-il)tieno[2,3-b1piridin-6(7H)-ona

A una disolución de 2-(6-(4-metilpiperazin-1-il)-1H-benzo[d]imidazol-2-il)acetato de etilo (2,42 g, 8,05 mmol) y 2-aminotiofeno-3-carbonitrilo (1,0 g, 8,05 mmol) en THF anh. (40 ml) a 40 °C se le añadió LDA (40 ml, 1 M en THF/hexano, 40 mmol) gota a gota a lo largo de 15 min bajo Ar. Se agitó la disolución marrón resultante a 40 °C durante 2 h y, a continuación, se extinguió con NH4Cl ac. (50 ml) a ta. Se diluyó la mezcla con H2O (125 ml) y se extrajo con acetato de etilo (2 x 200 ml). Se lavaron las fases orgánicas combinadas una vez con H2O, se secaron sobre Na2SO4 y se concentraron para dar un producto en bruto. Se trituró el producto en bruto con DCM (20 ml) seguido por MeOH (25 ml) para dar el compuesto del título como un sólido de color marrón claro (1,95 g, 64 %).
Se suspendió la base libre (1,95 g) en MeOH (50 ml) y se añadió HCl 1 M-Et2O (13 ml) a ta. Se agitó la suspensión durante 15 min a ta y se concentró a vacío y se sometió a destilación azeotrópica con MeOH (2 x 25 ml) para dar la sal de HCl como un sólido de color marrón oscuro (2,28 g, 62 %); 1H-RMN (400 MHz, CD3OD) 8 7,69 (d, J=9,2 Hz, 1H), 7,52 (d, J=5,6 Hz, 1H), 7,36 (dd, J=8 ,8 , 2,4 Hz, 1H), 7,30 (d, J=2,4 Hz, 1H), 7,19 (d, J=5,6 Hz, 1H), 3,97-3,93 (m, 2H), 3,70-3,67 (m, 2H), 3,39-3,35 (m, 2H), 3,34-3,18 (m, 2H), 3,01 (s, 3H); EM ESI [M H]+ 381,2, calc. para [C19H20N6OS+HI+ 381,1.








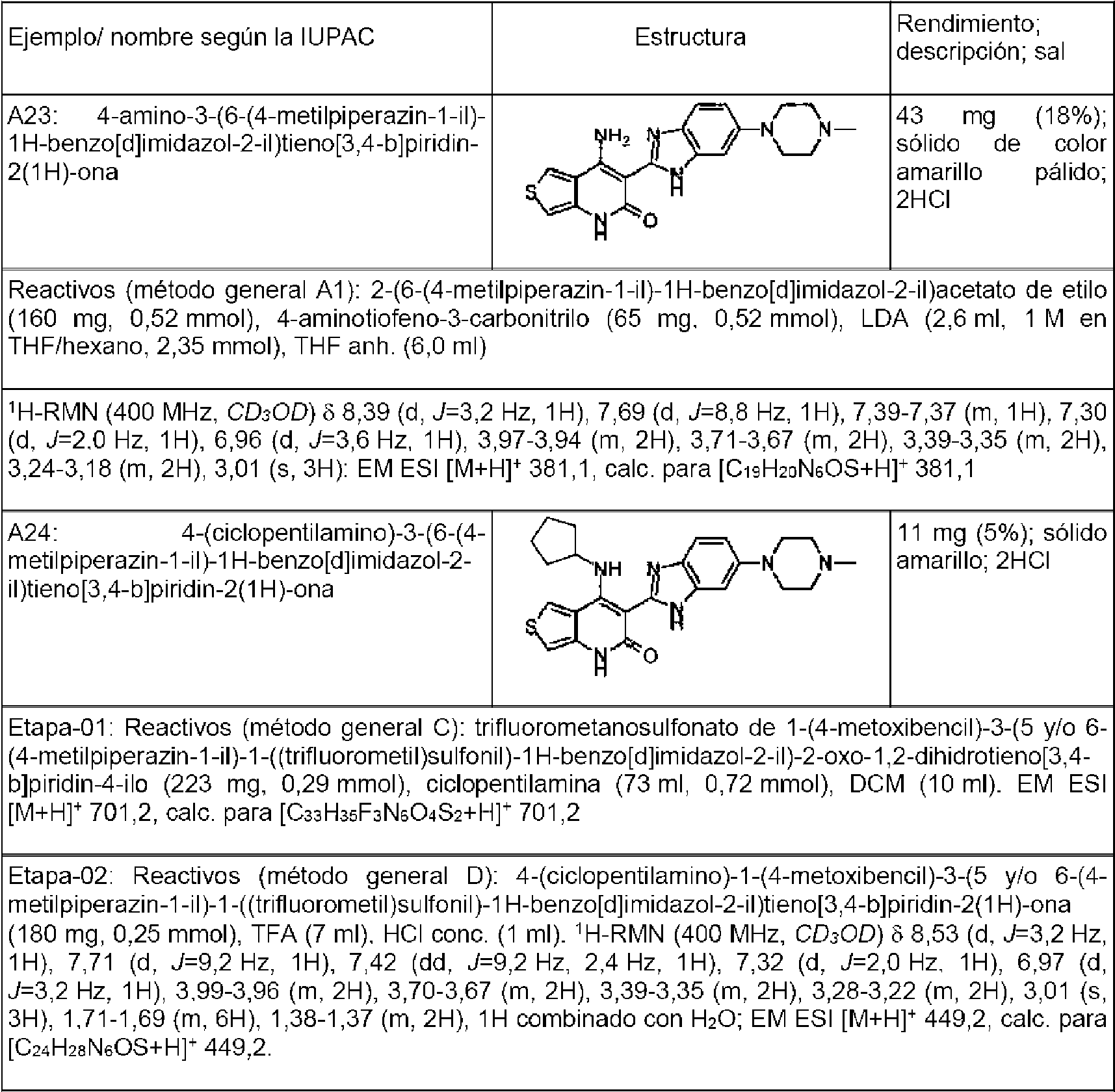

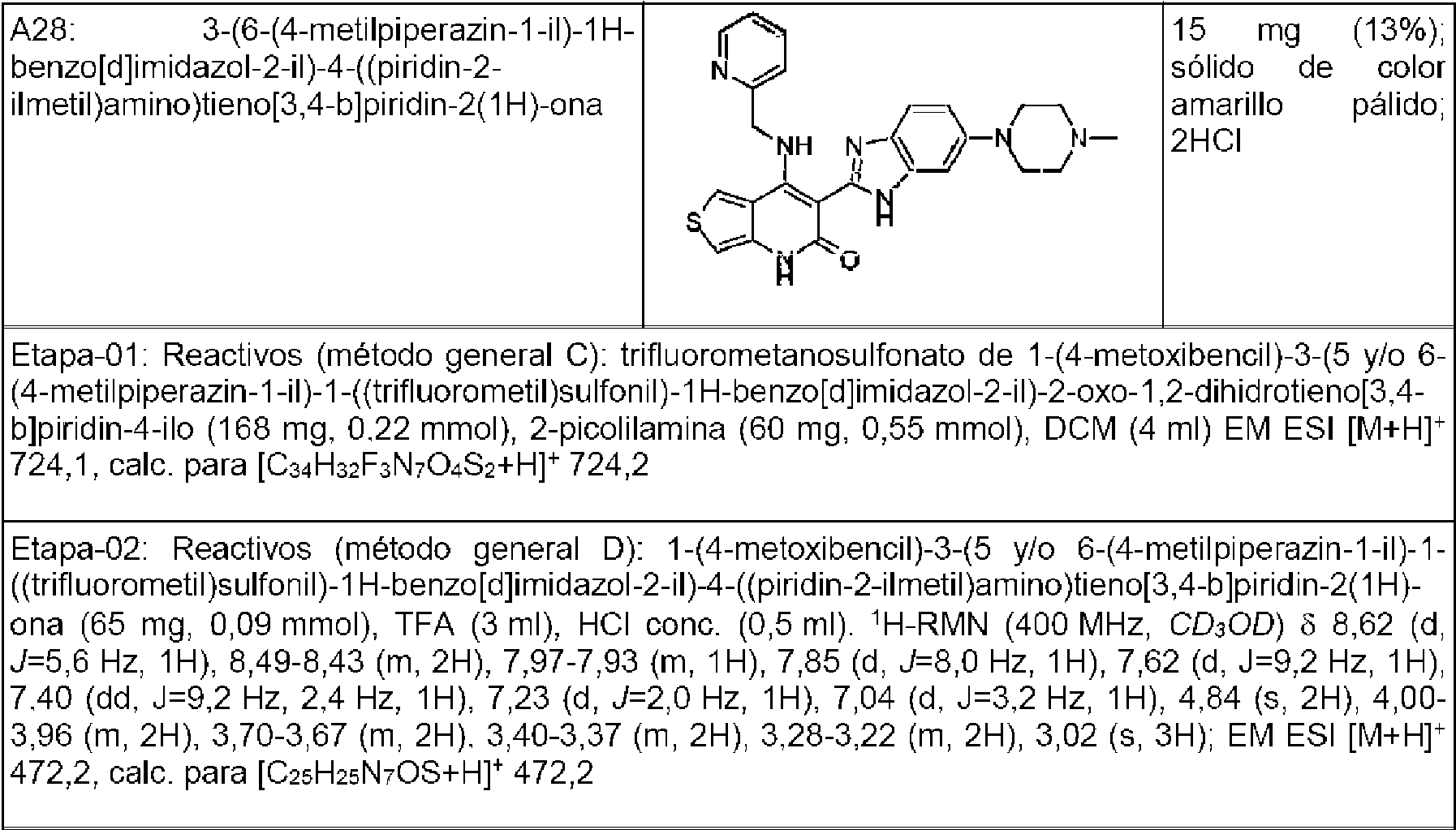
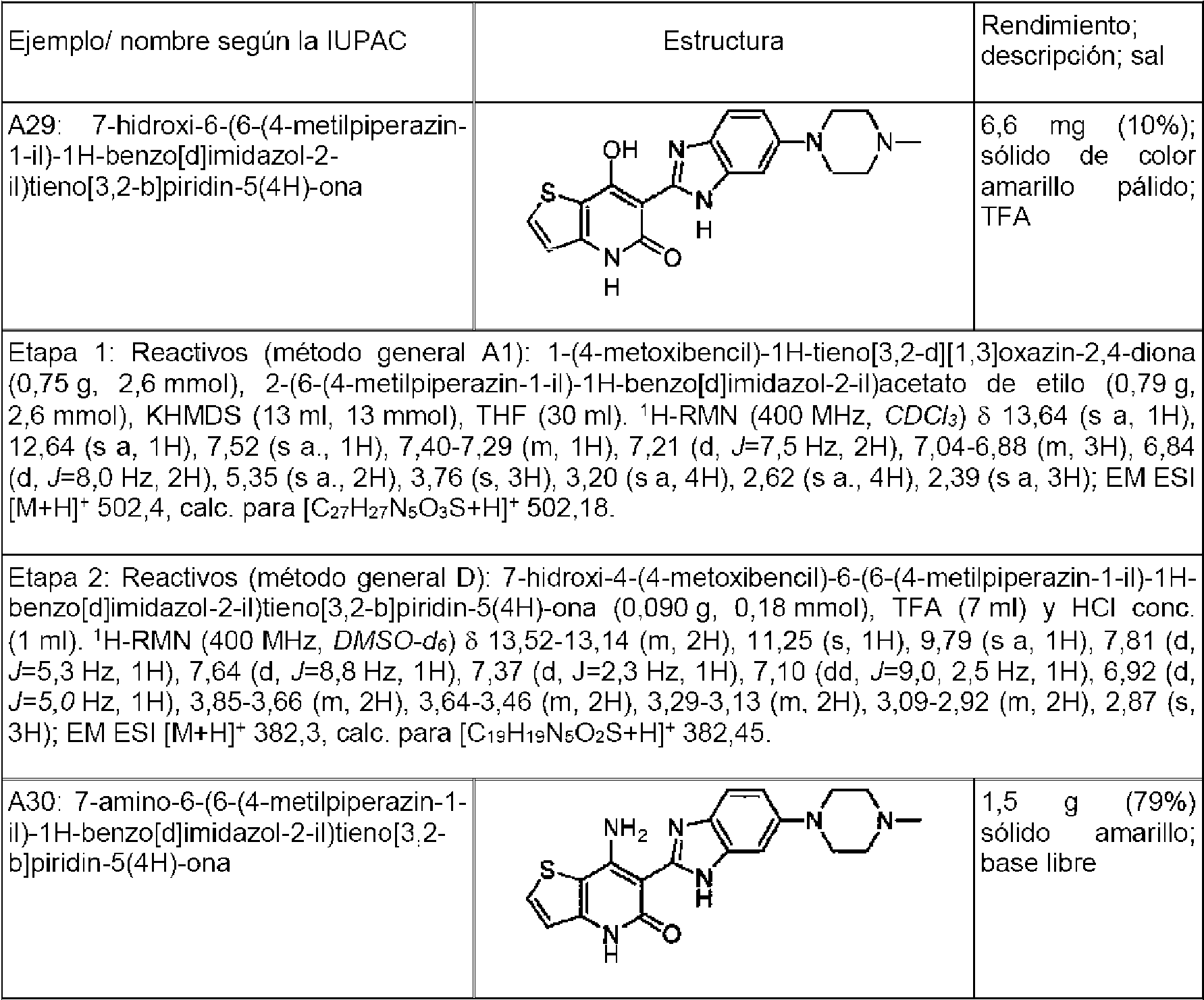











Se prepararon los siguientes compuestos según el método general A3.
A60: 4-amino-5-(5-(4-metilpiperazin-1-carbonin-1H-benzo[d1imidazol-2-il)tieno[2,3-b1-piridin-6(7H)-ona

Se añadió LDA (1,0 M en THF/hexanos, 2,3 ml, 2,3 mmol) gota a gota a lo largo de 15 min a ta a una suspensión con agitación de 2-(6-(4-metilp¡peraz¡n-1-carbon¡l)-1H-benzo[d]¡m¡dazol-2-il)acetato de etilo (0,150 g, 0,45 mmol) y 2-aminotiofeno-3-carbonitrilo (0,056 g, 0,45 mmol) en THF anh. (20 ml) bajo Ar. La adición se realizó inicialmente a ta y después de 5 minutos a 35 °C. Se continuó el calentamiento a 35 °C durante 1 h antes de enfriar la mezcla de reacción hasta ta, se extinguió con NH4Cl ac. y se concentró a presión reducida. La purificación mediante RP-HPLC proporcionó N-(3-cianotiofen-2-il)-2-(5-(4-metilpiperazin-1 -carbonil)-1H-benzo[d]imidazol-2-il)acetamida*TFA como un sólido de color marrón claro (82 mg, 35 %). 1H-RMN (400 MHz, CD3OD) 87,96 (s, 1H), 7,89 (d, J=8,5 Hz, 1H), 7,68 (dd, J=8,4, 1,4 Hz, 1H), 7,09 -7,14 (m, 2H), 3,25 -3,81 (m, 8H), 2,97 (s, 3H).
Etapa 2. Se filtró el producto de la reacción anterior a través de PoraPak (2 g, usando MeOH, luego NH32 M en MeOH) y se secó. Se trató una disolución en THF anh. (12 ml) del material (0,055 g, 0,13 mmol) bajo Ar con LiHMDS (1,0 M en THF, 0,7 ml, 0,7 mmol) a lo largo de 3 min a ta,, se agitó durante 10 min y se calentó a 45 °C durante 95 min. A continuación se enfrió la reacción hasta ta, se extinguió con NH4Cl ac., se concentró a presión reducida y se purificó mediante HPLC prep. La filtración a través de PoraPak (2 g) y la trituración con CH2Cl2 proporcionó el compuesto del título como un sólido de color amarillo claro 3,6 mg (3 %).1H-RMN (400 MHz, CD3OD) 87,58 - 7,77 (m, 2H), 7,51 (d, J=5,80 Hz, 1H), 7,30 (dd, J=8,30, 1,30 Hz, 1H), 7,14 (d, J=5,80 Hz, 1H), 3,53 - 3,92 (m, 4H), 2,48 -2,70 (m, 4H), 2,43 (s, 3H). EM ESI [M H]+ 409,2, calc. para [C20H20N6O2S H]+ 409,2.


A63: 2,2,2-trifluroaceatato de 7-(ciclopropilamino)-6-(6-(4-metilpiperazin-1-il)-1H-benzo[d1imidazol-2-il)tieno[3,2-b1-piridin-5(4H)-ona

A una suspensión de 7-h¡drox¡-6-(6-(4-met¡lp¡peraz¡n-1-il)-1H-benzo[d]¡m¡dazol-2-¡l)t¡eno[3,2-b]p¡r¡d¡n-5(4H)-ona (58 mg, 0,152 mmol) en DCM anhidro (1 ml) se le añadió Tf2O (0,55 ml, 0,916 mmol) gota a gota a ta. Se agitó la mezcla de reacción resultante a ta durante la noche antes de la adición de ciclopropanamina (100 mg, 1,83 mmol) a 0 °C gota a gota. Se agitó la mezcla de reacción resultante a 40 °C durante la noche y se diluyó con DCM seguido por el lavado con NaHCO3 sat. Se secó la fase orgánica sobre Na2SO4, se filtró y se concentró hasta sequedad. Se disolvió el residuo en MeOH y se hizo pasar a través de PoraPak seguido por la eliminación del disolvente a presión
reducida. Se purificó el producto en bruto mediante HPLC prep. para dar el compuesto del título como un sólido amarillo (5 mg, 6 % de rendimiento). 1H-RMN (400 MHz, CD3 OD) 87,98 (d, J=5,5 Hz, 1H), 7,61 (d, J=9,0 Hz, 1H), 7,26 (d, J=2,0 Hz, 1H ), 7,21 (dd, J=8,4, 2,0 Hz, 1H), 7,10 (d, J=5,5 Hz, 1H), 3,92-3,84 (m, 2H), 3,71-3,62 (m, 2H), 3,41-3,36 (m, 2H), 3,20-3,10 (m, 2H), 3,09-3,03 (m, 1H), 3,01 (s, 3H), 1,04-0,96 (m, 2H), 0,93-0,89 (m, 2H); EM ESI [M H]+ 421,2 , calc. para [C22H24N6OS+H]+ 421,2.
A64: 4-amino-5-(6-(piperazin-1-il)-1H-benzo[d1imidazol-2-il)tieno[2,3-b1piridin-6(7H)-ona
4-(3-amino-4-nitrofenil)piperazin-1-carboxilato de terc-butilo

Se agitó una mezcla de 5-cloro-2-nitroanilina (2,5 g, 14,48 mmol), piperazin-1-carboxilato de terc-butilo (3,24 g, 17,38 mmol) y K2CO3 (4,0 g, 28,96 mmol) en DMs O (100 ml) a 100 °C durante 3 días. A continuación se añadió H2O (150 ml) con agitación, se filtró por succión, se aclaró con H2O y se secó para dar el compuesto del título como un sólido marrón (2,6 g, 57 %). 1H-RMN (400 MHz, CDCI3) 8 8,04 (d, J=9,79 Hz, 1H), 6,27 (dd, J=9,66, 2,64 Hz, 1H), 6,21 - 6,11 (m, 2H), 5,95 (d, J=2,51 Hz, 1H), 3,61 - 3,54 (m, 4H), 3,40 - 3,34 (m, 4H), 1,50 (s, 9H); EM ESI [M+H]+ 323,2, calc. para [C15H22N4O4+HI+ 323,2.
4-(3,4-diaminofenil)piperazin-1-carboxilato de terc-butilo

A una suspensión de 4-(3-amino-4-nitrofenil)piperazin-1-carboxilato de terc-butilo (2,6 g, 8,04 mmol) en MeOH (150 ml) se le añadió Pd al 10 %/C (130 mg, 5 % en peso). Se hidrogenó la mezcla resultante bajo un globo de H2 O/N. Se filtró la mezcla de reacción resultante, se concentró y se secó para dar el compuesto del título como un sólido de color marrón oscuro (2,29 g, 97 %). 1H-RMN (400 MHz, CDCI3) 8 6,66 (d, J=8,28 Hz, 1H), 6,39 (d, J=2,51 Hz, 1H), 6,34 (dd, J=8,28, 2,51 Hz, 1H), 3,60 - 3,53 (m, 4H), 3,46 - 3,23 (m, 4H), 3,02- 2,95 (m, 4H), 1,49 (s, 9H); EM ESI [M+H]+ 293,1, calc. para [C15H24N4O2+HI+ 293,2.
4-(2-(2-etoxi-2-oxoetil)-1H-benzo[d]imidazol-6-il)piperazin-1-carboxilato de terc-butilo
A una disolución de 4-(3,4-diaminofenil)piperazin-1-carboxilato de terc-butilo (100 mg, 0,34 mmol) en EtOH (3 ml) se le añadió clorhidrato de 3-etoxi-3-iminopropionato de etilo (190 mg, 0,68 mmol). Se calentó la mezcla resultante a 60 °C durante 3 h. Después de eliminar los disolventes, se diluyó con DCM (10 ml), se ajustó a pH = 8 con NaHCO3 sat. y se separó. Se extrajo la acuosa con DCM (10 ml x 2) y se secaron los extractos combinados sobre NaSO4, luego se concentraron y se purificaron mediante cromatografía ultrarrápida (gradiente: el 100 % de EtOAc, luego el 0-20 % de MeOH/DCM) para dar el compuesto del título como un sólido de color naranja oscuro (116 mg, 87 %). 1H-RMN (400 MHz, CD3 OD) 87,49 - 7,40 (m, 1H), 7,15 - 7,10 (m, 2H), 4,22 (q, J=7,11 Hz, 2H), 3,95 (s, 1H), 3,61 (s a., 4H), 3,11 (s a., 4H), 1,50 (s, 9H), 1,28 (t, J=7,15 Hz, 3H); EM ESI [M+H]+ 389,2, calc. para [C20H28N4O4+HI+ 389,2.
4-(2-(4-amino-6-oxo-6,7-dihidrotieno[2,3-b]piridin-5-il)-1H-benzo[d]imidazol-6-il)piperazin-1-carboxilato de terc-butilo

Según el método general A, se usaron una disolución de 2-amino-4-etoxitiofeno-3-carbonitrilo (64 mg, 0,52 mmol), 4-(2-(2-etoxi-2-oxoetil)-1H-benzo[d]imidazol-6-il)piperazin-1-carboxilato de terc-butilo (200 mg, 0,52 mmol), LiHMDS (1 M en THF, 2,0 ml, 2,06 mmol) para generar el compuesto del título como un sólido de color marrón claro (88 mg, 35 %). 1H-RMN (500 MHz, DMSO-d6) 812,72 - 12,61 (m, 1H), 12,13 - 12,02 (m, 1H), 10,72 - 10,55 (m, 1H), 8,01 -7,93 (m, 1H), 7,57 (d, J=5,62 Hz, 1H), 7,52 - 7,43 (m, 1H), 7,24 - 7,10 (m, 2H), 6,93 - 6,87 (m, 1H), 3,52 - 3,44 (m, 4H), 3,07 - 3,00 (m, 4H), 1,45 - 1,40 (m, 9H); EM ESI [M+H]+ 467,2, calc. para [C23H26^ O 3S+H]+ 467,2.
4-amino-5-(6-(piperazin-1-il)-1H-benzo[d]imidazol-2-il)tieno[2,3-b]piridin-6(7H)-ona
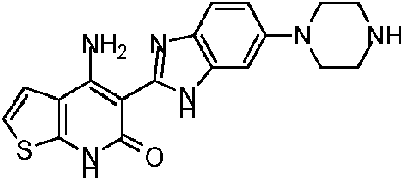
Se agitó una mezcla de 4-(2-(4-amino-6-oxo-6,7-dihidrotieno[2,3-b]piridin-5-il)-1H-benzo[d]imidazol-6-il)piperazin-1-carboxilato de terc-butilo (83 mg, 0,178 mmol) en TFA (1 ml) a ta durante 2 h antes de concentrarse. Se disolvió el
residuo en MeOH (20 ml) y se hizo pasar a través de PoraPak, luego se concentró para dar el compuesto del título como un sólido amarillo (45 mg, 69 %). 1H-RMN (400 MHz, DMSO-^) 812,65 - 12,58 (m, 1H), 10,77 - 10,61 (m, 1H), 8,03 - 7,94 (m, 1H), 7,59 (d, J=5,77 Hz, 1H), 7,54 - 7,42 (m, 1H), 7,19 - 7,10 (m, 2H), 6,92 - 6,86 (m, 1H), 3,09 -3,01 (m, 4H), 2,94 - 2,88 (m, 4H); no puede detectarse fácilmente la señal debida al NH2. e M ESI [M+H]+ 367,2, calc. para [C1sH1sN6OS+H]+ 367,1.
A65: 4-amino-5-(6-(4-(oxetan-3-il)piperazin-1-il)-1H-benzo[d1imidazol-2-il)tieno[2,3-b1piridin-6(7H)-ona

Se agitó una mezcla de 4-amino-5-(6-(piperazin-1-il)-1H-benzo[d]imidazol-2-il)tieno[2,3-b]piridin-6(7H)-ona (45 mg, 0,123 mmol), oxetan-3-ona (8,8 mg, 0,123 mmol) y NaBH(OAc)3 (120 mg, 0,552 mmol) en Dc E (2 ml) a ta durante la noche, luego se filtró. Se concentró el filtrado y se purificó mediante HPLC prep. para dar el compuesto del título como sal de TFA como un sólido amarillo (50 mg, 76 %). 1H-RMN (400 MHz, CD3 OD) 87,66 (d, J=9,03 Hz, 1H), 7,51 (d, J=5,77 Hz, 1H), 7,29 (t, J=8,91 Hz, 2H), 7,18 (d, J=6,02 Hz, 1H), 4,98 - 4,87 (m, 4H), 4,54 - 4,45 (m, 1H), 3,63 -3,40 (m, 8H); EM ESI [M+H]+ 423,2, calc. para [C21H22N6O2S+HI+ 423,2.



dazol--RMN


Ejemplo B: ensayo de inhibición de HPK1
Se adquirió HPK1 activa (MAP4K1) como fusión de GST N-terminal de HPK1 humana (aa 1-346) de Invitrogen (n.° de cat. PV6355). Se midió la actividad de HPK1 usando un sistema de detección indirecto mediante ELISA. Se incubó GST-HPK1 (0,6 nM) en presencia de ATP 12 pM (Sigma n.° de cat. A7699), MOPS 5 mM (pH 7,2), p-glicerolfosfato 2,5 mM, MgCh 5 mM, EDTA 0,4 mM, EgTa 1 mM, DTT 0,05 mM en una placa de microtitulación de 96 pocillos recubierta previamente con 0,5 pg/pocillo de proteína básica de mielina bovina (MBP) (Millipore, n.° de cat.13-110). Se dejó avanzar la reacción durante 30 min, seguido por 5 lavados de la placa con tampón de lavado (solución salina tamponada con fosfato complementada con Tween 20 al 0,2 %) e incubación durante 30 min con una dilución 1:3000 de anticuerpo policlonal de conejo anti-fosfo-treonina (Cell Signaling, n.° de cat. 9381). Se lavó la placa 5 veces con tampón de lavado, se incubó durante 30 min en presencia de conjugado de anticuerpo de cabra anti-peroxidasa del rábano de conejo (BioRad, n.° de cat. 1721019, concentración 1:3000), se lavó 5 veces adicionales con tampón de lavado y se incubó en presencia de sustrato TMB (Sigma, n.° de cat. T0440). Se dejó continuar la reacción colorimétrica durante 5 min, seguido por la adición de disolución de parada (H2SO40,5 N), y se cuantificó mediante detección a 450 nm con un lector de microplacas monocromático (Molecular Devices, M5).
Se determinó la inhibición por parte de los compuestos o bien a una concentración fija (10 pM) o bien a una concentración variable de inhibidor (normalmente de 50 pM a 0,1 pM en una titulación de la respuesta a la dosis de 10 puntos). Se incubaron previamente los compuestos en presencia de enzima durante 15 min antes de la adición de ATP y se cuantificó la actividad restante usando el ensayo de actividad descrito anteriormente. Se determinó el % de inhibición de un compuesto usando la siguiente fórmula; % de inhibición = 100 x (1 - (valor experimental - valor de fondo)/(control de alta actividad - valor de fondo)). Se determinó el valor de CI50 usando un ajuste de la curva logístico de 4 puntos no lineal (XLfit4, IDBS) con la fórmula; (A+(B/(1+((x/C)AD)))), en la que A = valor de fondo, B = rango, C = punto de inflexión, D = parámetro de ajuste de la curva.
Ejemplo C: ensayo de inhibición de FLT3
Se determinaron la inhibición de FLT3 y LCK por parte de los compuestos usando un kit de ensayo de cinasas Z'-LYTE basado en FRET con péptido tirosina 2 como sustrato (Invitrogen, n.° de cat. PV3191). Se realizó el ensayo de cinasa FLT3 según las especificaciones sugeridas por el fabricante con una concentración de ATP de 940 pM y FLT3 1 nM (Invitrogen, n.° de cat. PV3182), y ATP 180 pM y LCK 25 nM (Invitrogen, n.° de cat. P3043) para la reacción de la cinasa LCK. Se determinaron los valores del % de inhibición según las instrucciones del fabricante y se obtuvieron los valores de CI50 usando un ajuste de la curva logístico de 4 puntos no lineal (XLfit4, IDBS).
En la tabla 1 a continuación se proporcionan los intervalos de los valores de CI50 para los compuestos de ejemplo. Los intervalos de CI50 se indican como “A”, “B” y “C” para valores menores de o iguales a 0,05 pM; aquellos mayores de 0,05 pM y menores de o iguales a 0,5 pM; y aquellos mayores de 0,5 pM, respectivamente.
Tabla 1: datos de inhibición de HPK1, Lck y Flt3
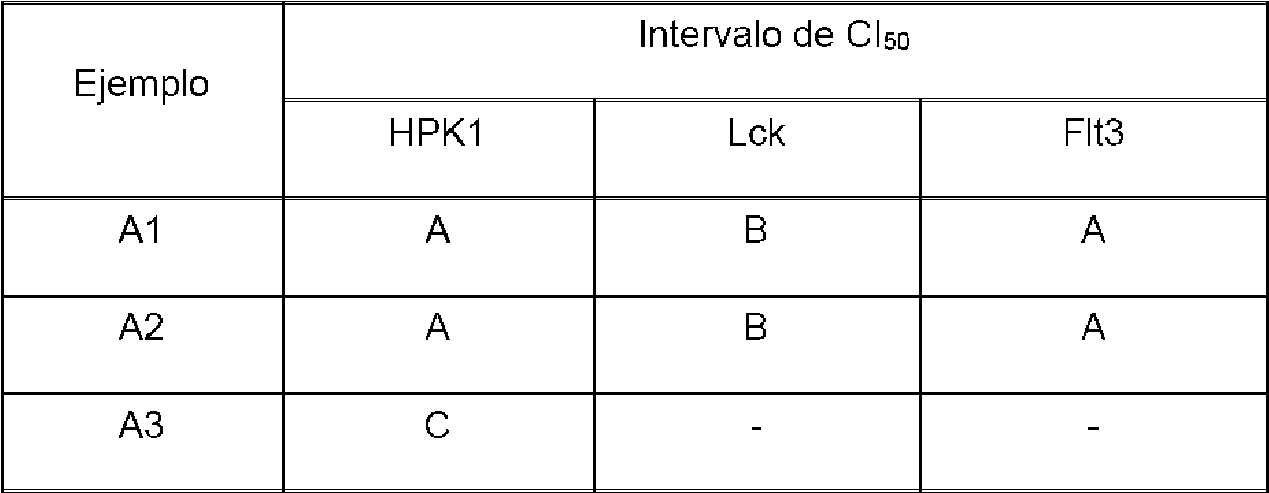



Ejemplo D: ensayos de fosforilación in vitro
Se obtuvieron células Jurkat E6.1 de la Colección Americana de Cultivos Tipo (ATCC, Manassas, VA) y se mantuvieron según las instrucciones del proveedor. Se lavaron las células tres veces y se privaron de nutrientes en medio RPMI 1640 complementado con suero fetal de ternero al 0,5 % durante 18 h a 37 °C. Se trataron previamente las células privadas de suero con la concentración de inhibidor indicada durante 4 horas antes de la estimulación con 10 |i g/ml de anticuerpo anti-a -CD3 (BioLegend, Inc., San Diego, CA) durante 10 min a 37 °C. Se lavaron las células una vez en solución salina tamponada con fosfato (pH 7,4) que contenía pirofosfato de sodio 10 mm, fluoruro de sodio 10 mm, EDTA 10 mm y ortovanadato de sodio 1 mm. Se prepararon lisados de proteínas usando tampón de lisis helado de ensayo de radioinmunoprecipitación (RIPA). Se cargó un total de 100 |i g de lisado celular en geles de Bis-Tris (Life Technologies, Carlsbad, CA) con marcador de peso molecular de intervalo completo como referencia de tamaño y se resolvió mediante electroforesis en SDS-PAGE. Se transfirieron las proteínas a una membrana de PVDF (Millipore, Billerica, MA), se bloquearon y se sondaron con anticuerpos para fosfo-SLP-76 (Ser376) (anticuerpo policlonal de conejo #13177; Cell Signaling Technology Inc., Danvers, MA), SLP-76 (anticuerpo policlonal de conejo #4958; Cell Signaling Technology Inc., Danvers, MA), fosfo-ERK (anticuerpo monoclonal de ratón sc-7383; Santa Cruz Biotechnology Inc., Santa Cruz, CA) y ERK1/2 (anticuerpo policlonal de conejo 06-182; Millipore, Billerica, MA). Se diluyeron 1:15.000 los anticuerpos secundarios y se incubaron durante 1 h a ta. Se visualizaron las bandas de las proteínas y se cuantificaron usando un dispositivo de obtención de imágenes en el infrarrojo cercano Odyssey (LI-COR, Lincoln, NE).
La tabla 2 a continuación enumera los efectos de compuestos representativos de la presente invención contra la fosforilación en serina 376 de SLP-76 y la fosforilación en T202/Y204 de ERK1/2 en células Jurkat E6.1 estimuladas con a -CD3.
Tabla 2. Efectos de inhibidores de HPK1 contra la fosforilación en serina 376 de SLP-76 y la fosforilación en T202/Y204 de ERK1/2 en células Jurkat E6.1 estimuladas con a -CD3.

*>75% de inhibición tal como se estima mediante análisis por inmunotransferencia
Ejemplo E: modelo de xenoinjerto de la línea celular CT26 singénica.
Se obtuvo la línea celular CT26 WT, que es una línea celular de carcinoma de colon indiferenciada, derivada de ratón, inducida por N-nitroso-N-metiluretano (NNMU), de la Colección Americana de Cultivos Tipo (ATCC CRL-2638, Manassas, VA, DC, EE.UU.). Se hicieron crecer las células en medio de Roswell Park Memorial Institute, habitualmente denominado medio RPMI 1640, que contenía 4,5 g/l de glucosa, 0,11 g/l de piruvato de sodio, 1,5 g/l de bicarbonato de sodio, L-glutamina y 2,385 g/l de HEPES más suero fetal bovino al 10 %. Se adquirieron ratones BALB/c hembra de seis a ocho semanas de edad de Jackson Laboratories y se recibieron y aclimataron en el centro de recursos animales MaRS-TMDT durante 1 semana antes del inicio del experimento. Se alimentaron los ratones a voluntad con agua esterilizada en autoclave y dieta de laboratorio para roedores (Harlan Teklad LM-485) que consistía en el 19 % de proteína en bruto, el 5 % de grasa en bruto y el 5 % de fibra en bruto. Se alojaron los ratones en jaulas con microaisladores y se mantuvieron en un entorno con un ciclo de 12 h de luz a 20-22 °C y el 40-60 % de humedad. El día de la implantación, se recogieron células CT26 y se resuspendieron con RPMI1640 libre de suero hasta una concentración de 1x107/ml, y a cada ratón se le inyectó por vía subcutánea un volumen de 0,1 ml que contenía 1x106 células CT26 en el flanco trasero derecho. Después de 6 d, se habían formado tumores palpables con un volumen promedio de ~65 mm3 (calculado usando la fórmula: volumen tumoral = ancho2 x longitud / 2). En este momento, se separaron los animales en cinco grupos de ocho animales por grupo, de tal manera que cada grupo contenía animales que portaban tumores de tamaño promedio similar, y se inició el tratamiento. Para la dosificación, se disolvió el ejemplo A1 en agua hasta una concentración de 7,5 mg/ml o 15 mg/ml para la dosificación de las dosis de 75 mg/kg y 150 mg/kg, respectivamente. Como control positivo y para investigar la actividad combinatoria del ejemplo A1, se usó una rata que se dosificó con anticuerpo IgG2b anti-PD1 (BioXcell (NH, EE.UU.)). Se trataron los cinco grupos con: i) 10ml/kg de agua QD durante 21 d administrados mediante alimentación por sonda oral (v.o.) más 150 pg de anticuerpo de control de isotipo IgG2b de rata dosificado mediante inyección intraperitoneal (i.p.) el día 0, 3, 6 y 10 (el grupo de control); ii) 150 pg de anticuerpo anti-PD-1 dosificado mediante inyección intraperitoneal (i.p.) el día 0, 3, 6 y 10; iii) 75 mg/kg del ejemplo A1 QD durante 21 días administrado por v.o.; iv) 150 mg/kg del ejemplo A1 QD durante 21 días administrado por v.o. v) 150 mg/kg del ejemplo A1 QD durante 21 días administrado por v.o. más 150 pg de anticuerpo anti-PD-1 dosificado mediante inyección intraperitoneal (i.p.) el día 0, 3, 6 y 10. Se evaluó la toxicidad mediante mediciones del peso corporal y observaciones clínicas. Se tomaron mediciones del tumor y los pesos corporales tres veces a la semana. Se calculó el porcentaje de inhibición del crecimiento tumoral (TGI) mediante la fórmula:
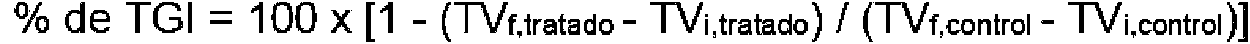
La inhibición del crecimiento tumoral en el día 21 se muestra en la figura 2. Se observó un efecto dependiente de la dosis en respuesta al tratamiento con el ejemplo A1, inhibiendo con 75 mg/kg y 150 mg/kg QD el crecimiento tumoral en un 44 % y 64 %, respectivamente. Aunque el anticuerpo anti-PD-1 solo dio como resultado una TGI promedio del 34 %, cuando se combinó con 150 mg/kg Qd del ejemplo A1, la TGI aumentó hasta el 86 %.
Según los Protocolos de Uso de Animales (AUP) de la University Health Network (UHN), los ratones en experimentos de eficacia deben sacrificarse cuando el tamaño tumoral supera los 1500 mm3 o si el peso corporal del animal disminuye o si los animales muestran signos clínicos que requieren la terminación por motivos compasivos. En este estudio, el compuesto se toleró bien, ganando peso todos los animales a lo largo del transcurso del estudio, y no se sacrificó ningún animal debido a signos clínicos. Se usó un tamaño tumoral de <1500 mm3 en el día 21 como valor de corte para representar la supervivencia. Usando este valor de corte, en el día 21 no sobrevivió ningún animal en el grupo de control, 1 de 8 animales (12,5 %) sobrevivieron en el grupo de anticuerpo anti-PD-1, 2 de 8 animales (25 %) sobrevivieron en el grupo de 75 mg/kg/día del ejemplo A1, 3 de 8 animales (37,5 %) sobrevivieron en el grupo de 150 mg/kg/día del ejemplo A1 y 7 de 8 animales (87,5 %) sobrevivieron en el grupo de 150 mg/kg/día del ejemplo A1 y anticuerpo anti-PD-1. Estos resultados demuestran que los compuestos de la invención, tal como se ejemplifica mediante el compuesto A1, tienen actividad antitumoral in vivo y pueden combinarse eficazmente con otros enfoques inmunomoduladores.
Ejemplo F: modelo de progresión de la enfermedad por EAE
Se obtuvieron ratones C57/BL6 de Jackson Laboratories. El comité institucional de uso y atención de animales de la University Health Network aprobó todos los procedimientos con animales. A los ratones se les inmunizó por vía subcutánea (s.c.) con péptido MOG35-55 emulsionado en adyuvante completo de Freund (CFA) complementado con Mycobacterium tuberculosis. Los días 0 y 2 después de la inmunización, a los ratones se les inyectó por vía intraperitoneal (i.p.) toxina tosferínica. Se monitorizaron los signos clínicos de EAE diariamente, según los siguientes criterios: 0, sin enfermedad; 1, tono de la cola disminuido; 2, debilidad de las patas traseras o parálisis parcial; 3, parálisis completa de las patas traseras; 4, parálisis de las patas delanteras y traseras; 5, muerte, o sacrificio debido a un estado moribundo. Para el tratamiento con el compuesto durante la inducción de EAE, a los ratones se les dosificó por vía oral (v.o.) 50 mg/kg de A30 (n=4) o agua (control de vehículo; n=5) cada día (QD). Los datos son la puntuación media ± EEM. Los resultados de la prueba se muestran en la figura 3.
Claims (14)
- REIVINDICACIONESUn compuesto representado por la fórmula (I-A):
 o una sal farmacéuticamente aceptable del mismo, en el que:R es H, -F, -CI, -Br, -OH, -alquilo (Ci -C4), -haloalquilo (Ci-C4), -alcoxilo (Ci -C4), -alquilen (Ci-C4)-OH o heterociclilo monocíclico de 4-7 miembros opcionalmente sustituido con 1-3 grupos seleccionados de -F, -CI, -Br, -OH, -alquilo (C1-C4), -haloalquilo (C1-C4) o -alcoxilo (C1-C4);R1 es -NRaRb o -ORa1;Ra en cada aparición es independientemente -H, -alquilo (C1-C6), -(CH2)n-cicloalquilo (C3-C7), -(CH2)nheterociclilo monocíclico de 3-7 miembros, -(CH2)n-cicloalquilo (C6-C12) en puente, -(CH2)n-heteroarilo de 5 10 miembros opcionalmente sustituido; o -(CH2)n-heterociclilo en puente de 6-12 miembros, en el que el -alquilo (C1-C6), -(CH2)n-cicloalquilo (C3-C7), -(CH2)n-heterociclilo monocíclico de 3-7 miembros, -(CH2)ncicloalquilo (C6-C12) en puente, -(CH2)n-heteroarilo de 5-10 miembros o -(CH2)n-heterociclilo en puente de 6 12 miembros está opcionalmente sustituido con 1-3 grupos seleccionados de -F, -CI, -Br, -CN, -NH2, -OH, oxo, -alquilo (C1-C4), -haloalquilo (C1-C4), -alcoxilo (C1-C4), -haloalcoxilo (C1-C4), -alquilen (C1-C4)-OH o -alquilen (C1-C4)-NH2;Rb en cada aparición es independientemente -H o -alquilo (C1-C6); oRa y Rb, junto con el nitrógeno al que están unidos, forman -heterociclilo (C3-C10);Ra1 en cada aparición es independientemente -H, alquilo (C1-C6), cicloalquilo (C3-C10), heterociclilo de 3-10 miembros, arilo (C6-C10) o heteroarilo de 3-10 miembros;R4 y R5, junto con el nitrógeno al que están unidos, forman heterociclilo monocíclico de 4-7 miembros o heterociclilo en puente de 6-12 miembros, en el que el heterociclilo monocíclico de 4-7 miembros o heterociclilo en puente de 6-12 miembros está opcionalmente sustituido con 1-3 grupos seleccionados de -F, -CI, -Br, -CN, -NH2, -OH, oxo, -alquilo (C1-C4), -haloalquilo (C1-C4), -alcoxilo (C1-C4), -haloalcoxilo (C1-C4), -alquilen (C1-C4)-OH o-alquilen (C1-C4)-NH2;R6 en cada aparición es independientemente -F, -CI, -Br, -CN, -NH2, -OH, -alquilo (C1-C6), - haloalquilo (C1-C6), -alquenilo (C2-C6), -alquinilo (C2-C6), cicloalquilo (C3-C6), -alcoxilo (C1-C6), -haloalcoxilo (C1-C6), -alquilen (C1-C6)-OH o -alquilen (C1-C6)-NH2;m es 0, 1,
o una sal farmacéuticamente aceptable del mismo, en el que:R es H, -F, -CI, -Br, -OH, -alquilo (Ci -C4), -haloalquilo (Ci-C4), -alcoxilo (Ci -C4), -alquilen (Ci-C4)-OH o heterociclilo monocíclico de 4-7 miembros opcionalmente sustituido con 1-3 grupos seleccionados de -F, -CI, -Br, -OH, -alquilo (C1-C4), -haloalquilo (C1-C4) o -alcoxilo (C1-C4);R1 es -NRaRb o -ORa1;Ra en cada aparición es independientemente -H, -alquilo (C1-C6), -(CH2)n-cicloalquilo (C3-C7), -(CH2)nheterociclilo monocíclico de 3-7 miembros, -(CH2)n-cicloalquilo (C6-C12) en puente, -(CH2)n-heteroarilo de 5 10 miembros opcionalmente sustituido; o -(CH2)n-heterociclilo en puente de 6-12 miembros, en el que el -alquilo (C1-C6), -(CH2)n-cicloalquilo (C3-C7), -(CH2)n-heterociclilo monocíclico de 3-7 miembros, -(CH2)ncicloalquilo (C6-C12) en puente, -(CH2)n-heteroarilo de 5-10 miembros o -(CH2)n-heterociclilo en puente de 6 12 miembros está opcionalmente sustituido con 1-3 grupos seleccionados de -F, -CI, -Br, -CN, -NH2, -OH, oxo, -alquilo (C1-C4), -haloalquilo (C1-C4), -alcoxilo (C1-C4), -haloalcoxilo (C1-C4), -alquilen (C1-C4)-OH o -alquilen (C1-C4)-NH2;Rb en cada aparición es independientemente -H o -alquilo (C1-C6); oRa y Rb, junto con el nitrógeno al que están unidos, forman -heterociclilo (C3-C10);Ra1 en cada aparición es independientemente -H, alquilo (C1-C6), cicloalquilo (C3-C10), heterociclilo de 3-10 miembros, arilo (C6-C10) o heteroarilo de 3-10 miembros;R4 y R5, junto con el nitrógeno al que están unidos, forman heterociclilo monocíclico de 4-7 miembros o heterociclilo en puente de 6-12 miembros, en el que el heterociclilo monocíclico de 4-7 miembros o heterociclilo en puente de 6-12 miembros está opcionalmente sustituido con 1-3 grupos seleccionados de -F, -CI, -Br, -CN, -NH2, -OH, oxo, -alquilo (C1-C4), -haloalquilo (C1-C4), -alcoxilo (C1-C4), -haloalcoxilo (C1-C4), -alquilen (C1-C4)-OH o-alquilen (C1-C4)-NH2;R6 en cada aparición es independientemente -F, -CI, -Br, -CN, -NH2, -OH, -alquilo (C1-C6), - haloalquilo (C1-C6), -alquenilo (C2-C6), -alquinilo (C2-C6), cicloalquilo (C3-C6), -alcoxilo (C1-C6), -haloalcoxilo (C1-C6), -alquilen (C1-C6)-OH o -alquilen (C1-C6)-NH2;m es 0, 1, - 2 o 3; yn es 0 , 1 o 2.El compuesto según la reivindicación 1, en el que el compuesto está representado por la fórmula (II-A) o (III-A):

- 3. El compuesto según la reivindicación 1o 2, en el que R4 y R5, junto con el nitrógeno al que están unidos, forman -N-alquil-piperazinilo o morfolinilo, en el que el piperazinilo o morfolinilo está opcionalmente sustituido con 1-2 grupos seleccionados de -F, -CI, -Br, -OH, -alquilo (Ci-C4), -haloalquilo (Ci-C4) o -alcoxilo (C1-C4).
- 4. El compuesto según una cualquiera de las reivindicaciones 1 a 3, en el que Ra en cada aparición es independientemente -H, -(CH2)n-cicloalquilo (C3-C6), -(CH2)n-heterociclilo monocíclico de 3-6 miembros, en el que el -(CH2)n-cicloalquilo (C3-C6) o -(CH2)n-heterociclilo monocíclico de 3-6 miembros está opcionalmente sustituido con 1-3 grupos seleccionados de -F, -CI, - Br, -CN, -NH2, -OH, -alquilo (C1-C4) o -alcoxilo (C1-C4); y n es 0 o 1.
- 5. El compuesto según una cualquiera de las reivindicaciones 1 a 4, en el que R es H, -alquilo (C1-C4), -alcoxilo (C1-C4), N-piperazinilo.
- 6. El compuesto según una cualquiera de las reivindicaciones 1 a 5, en el que R es H.
- 7. El compuesto según una cualquiera de las reivindicaciones 1 a 6 , en el que R4 y R5, junto con el nitrógeno al que están unidos, forman -N-metil-piperazinilo o morfolinilo, ambos de los cuales están opcionalmente sustituidos con uno o dos metilo.
- 8. El compuesto según una cualquiera de las reivindicaciones 1 a 7, en el que Ra en cada aparición es independientemente -H; -cicloalquilo (C3-C6) opcionalmente sustituido con -OH; -(CH2)n-tetrahidro-2H-pirano; morfolinilo; piperidinilo opcionalmente sustituido con -F, -OH o metilo; o tetrahidrofurano; y n es 0 o 1.
- 9. Un compuesto según la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en el que el compuesto es:

- 10. Una composición farmacéutica, que comprende un compuesto según una cualquiera de las reivindicaciones 1 a 9 y un portador o diluyente farmacéuticamente aceptable.
- 11. Un compuesto según las reivindicaciones 1 a 9, o una sal farmacéuticamente aceptable del mismo, para su uso en medicina.
- 12. Un compuesto según una cualquiera de las reivindicaciones 1 a 9, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer.
- 13. Un compuesto según una cualquiera de las reivindicaciones 1 a 9, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer en combinación con un segundo tratamiento contra el cáncer, tal como un agente quimioterápico, un agente terapéutico dirigido, radiación o cirugía.
- 14. Un compuesto según una cualquiera de las reivindicaciones 1 a 9, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer en combinación con un agente inmunomodulador, tal como un inhibidor del punto de control, tal como anticuerpo anti-PD-1, anticuerpo anti-CTLA4 o anticuerpo anti-PD-L1, o un inhibidor de la oxidación del triptófano, tal como inhibidor de IDO1, IDO2 o TDO2.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562184348P | 2015-06-25 | 2015-06-25 | |
| PCT/CA2016/050734 WO2016205942A1 (en) | 2015-06-25 | 2016-06-23 | Hpk1 inhibitors and methods of using same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2872555T3 true ES2872555T3 (es) | 2021-11-02 |
Family
ID=57584335
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16813437T Active ES2872555T3 (es) | 2015-06-25 | 2016-06-23 | Inhibidores de HPK1 y métodos de uso de los mismos |
Country Status (27)
| Country | Link |
|---|---|
| US (3) | US10501474B2 (es) |
| EP (1) | EP3322711B1 (es) |
| JP (3) | JP6898868B2 (es) |
| KR (2) | KR102614872B1 (es) |
| CN (2) | CN107922431B (es) |
| AU (1) | AU2016282289B2 (es) |
| BR (2) | BR112017027241B1 (es) |
| CA (1) | CA2989684A1 (es) |
| CY (1) | CY1124489T1 (es) |
| DK (1) | DK3322711T3 (es) |
| EA (1) | EA035421B1 (es) |
| ES (1) | ES2872555T3 (es) |
| HK (1) | HK1255833A1 (es) |
| HR (1) | HRP20210895T1 (es) |
| HU (1) | HUE054159T2 (es) |
| IL (2) | IL283353B (es) |
| LT (1) | LT3322711T (es) |
| MA (1) | MA42456B1 (es) |
| MD (1) | MD3322711T2 (es) |
| MX (2) | MX2018000048A (es) |
| PL (1) | PL3322711T3 (es) |
| PT (1) | PT3322711T (es) |
| RS (1) | RS61919B1 (es) |
| SG (1) | SG10202105964RA (es) |
| SI (1) | SI3322711T1 (es) |
| TW (1) | TWI733679B (es) |
| WO (1) | WO2016205942A1 (es) |
Families Citing this family (92)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL283353B (en) * | 2015-06-25 | 2022-09-01 | Univ Health Network | hpk1 inhibitors and methods of using them |
| US20180072718A1 (en) | 2016-09-09 | 2018-03-15 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| AR109595A1 (es) | 2016-09-09 | 2018-12-26 | Incyte Corp | Compuestos de pirazolopirimidina y usos de estos como inhibidores de hpk1 |
| JP7076432B2 (ja) | 2016-09-09 | 2022-05-27 | インサイト・コーポレイション | Hpk1調節薬としてのピラゾロピリジン誘導体及びがんの治療のためのその用法 |
| WO2018049191A1 (en) | 2016-09-09 | 2018-03-15 | Incyte Corporation | Pyrazolopyridone derivatives as hpk1 modulators and uses thereof for the treatment of cancer |
| WO2018152220A1 (en) * | 2017-02-15 | 2018-08-23 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| EP3596075B1 (en) | 2017-03-15 | 2023-10-11 | F. Hoffmann-La Roche AG | Azaindoles as inhibitors of hpk1 |
| CR20190424A (es) * | 2017-03-30 | 2019-11-04 | Hoffmann La Roche | Isoquinolinas como inhibidores de hpk1 |
| AU2018244935A1 (en) * | 2017-03-30 | 2019-08-15 | F. Hoffmann-La Roche Ag | Naphthyridines as inhibitors of HPK1 |
| WO2018228920A1 (en) | 2017-06-13 | 2018-12-20 | Bayer Pharma Aktiengesellschaft | Substituted pyrrolopyridine-derivatives |
| WO2018228923A1 (en) | 2017-06-13 | 2018-12-20 | Bayer Pharma Aktiengesellschaft | Substituted pyrrolopyridine-derivatives as map4k1 modulators for the treatment of cancer diseases |
| US20200246347A1 (en) | 2017-06-13 | 2020-08-06 | Bayer Pharma Aktiengesellschaft | Substituted Pyrrolopyridine-Derivatives |
| US11427578B1 (en) | 2017-07-18 | 2022-08-30 | Bayer Pharma Aktiengesellschaft | Substituted pyrrolopyridine-derivatives |
| WO2019051199A1 (en) * | 2017-09-08 | 2019-03-14 | Incyte Corporation | 6-CYANO-INDAZOLE COMPOUNDS AS HEMATOPOIETIC PROGENITOR KINASE 1 (HPK1) MODULATORS |
| CN109721620B (zh) * | 2017-10-27 | 2022-05-13 | 药捷安康(南京)科技股份有限公司 | Hpk1抑制剂及其用途 |
| JP7167146B2 (ja) * | 2017-11-06 | 2022-11-08 | ブリストル-マイヤーズ スクイブ カンパニー | Hpk1阻害剤として有用なイソフラノン化合物 |
| PE20210397A1 (es) | 2018-02-20 | 2021-03-02 | Incyte Corp | Derivados de n-(fenil)-2-(fenil)pirimidina-4-carboxamida y compuestos relacionados como inhibidores hpki para tratar el cancer |
| US10745388B2 (en) | 2018-02-20 | 2020-08-18 | Incyte Corporation | Indazole compounds and uses thereof |
| WO2019164847A1 (en) | 2018-02-20 | 2019-08-29 | Incyte Corporation | Indazole compounds and uses thereof |
| US11299473B2 (en) | 2018-04-13 | 2022-04-12 | Incyte Corporation | Benzimidazole and indole compounds and uses thereof |
| WO2020023551A1 (en) * | 2018-07-24 | 2020-01-30 | Genentech, Inc. | Naphthyridine compounds and uses thereof |
| TW202019905A (zh) * | 2018-07-24 | 2020-06-01 | 瑞士商赫孚孟拉羅股份公司 | 異喹啉化合物及其用途 |
| US10899755B2 (en) | 2018-08-08 | 2021-01-26 | Incyte Corporation | Benzothiazole compounds and uses thereof |
| ES2973117T3 (es) | 2018-09-25 | 2024-06-18 | Incyte Corp | Compuestos de pirazolo[4,3-d]pirimidina como moduladores de ALK2 y/o FGFR |
| TW202024053A (zh) * | 2018-10-02 | 2020-07-01 | 美商建南德克公司 | 異喹啉化合物及其用途 |
| US11612606B2 (en) | 2018-10-03 | 2023-03-28 | Genentech, Inc. | 8-aminoisoquinoline compounds and uses thereof |
| WO2020072695A1 (en) * | 2018-10-03 | 2020-04-09 | Genentech, Inc. | 8-aminoisoquinoline compounds and uses thereof |
| US11071730B2 (en) | 2018-10-31 | 2021-07-27 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| EP3873903B1 (en) | 2018-10-31 | 2024-01-24 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| WO2020120257A1 (en) | 2018-12-11 | 2020-06-18 | Bayer Aktiengesellschaft | Substituted pyrrolopyridine-derivatives |
| AR117472A1 (es) | 2018-12-21 | 2021-08-11 | Celgene Corp | Inhibidores de tienopiridina de ripk2 |
| EP3902806A4 (en) * | 2018-12-26 | 2022-09-28 | Janssen Pharmaceutica NV | THIENOPYRIDINONE COMPOUNDS |
| EP3947366A1 (en) | 2019-03-26 | 2022-02-09 | Janssen Pharmaceutica NV | Hpk1 inhibitors |
| AU2020249397A1 (en) | 2019-03-26 | 2021-09-30 | Janssen Pharmaceutica Nv | Bicyclic HPK1 inhibitors |
| WO2020235902A1 (ko) * | 2019-05-17 | 2020-11-26 | 주식회사 보로노이 | 헤테로고리 융합 피리미딘 유도체 및 이의 용도 |
| WO2020237025A1 (en) | 2019-05-23 | 2020-11-26 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| CA3142513A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| EP4010338A1 (en) | 2019-08-06 | 2022-06-15 | Incyte Corporation | Solid forms of an hpk1 inhibitor |
| BR112022004451A2 (pt) | 2019-09-13 | 2022-06-21 | Nimbus Saturn Inc | Antagonistas de hpk1 e usos dos mesmos |
| CN112552293A (zh) * | 2019-09-25 | 2021-03-26 | 珠海宇繁生物科技有限责任公司 | 一种protac小分子化合物及其应用 |
| EP4045083B1 (en) | 2019-10-18 | 2024-01-10 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| CN114599392A (zh) | 2019-10-31 | 2022-06-07 | 四十七公司 | 基于抗cd47和抗cd20的血癌治疗 |
| TWI778443B (zh) | 2019-11-12 | 2022-09-21 | 美商基利科學股份有限公司 | Mcl1抑制劑 |
| CN117736207A (zh) | 2019-12-24 | 2024-03-22 | 卡尔那生物科学株式会社 | 二酰基甘油激酶调节化合物 |
| WO2021141106A1 (ja) * | 2020-01-09 | 2021-07-15 | 住友化学株式会社 | 複素環化合物及びそれを含有する有害節足動物防除組成物 |
| CN117964757A (zh) | 2020-02-14 | 2024-05-03 | 吉利德科学公司 | 与ccr8结合的抗体和融合蛋白及其用途 |
| CN115867275A (zh) * | 2020-04-13 | 2023-03-28 | 大学健康网络 | 治疗细胞因子释放综合征的方法 |
| US12110294B2 (en) | 2020-05-01 | 2024-10-08 | Gilead Sciences, Inc. | CD73 compounds |
| AU2021272064A1 (en) * | 2020-05-11 | 2023-01-19 | University Health Network | Salt and crystal forms of 4-amino-5-(6-(4-methylpiperazin-1-yl)-1H-benzo(d)imidazol-2-yl)thieno(2,3-b)pyridin-6(7H) |
| WO2021249913A1 (en) | 2020-06-09 | 2021-12-16 | Bayer Aktiengesellschaft | 2'-(quinolin-3-yl)-5',6'-dihydrospiro[azetidine-3,4'-pyrrolo[1,2-b]pyrazole]-1-carboxylate derivatives and related compounds as map4k1 (hpk1) inhibitors for the treatment of cancer |
| WO2022068848A1 (en) * | 2020-09-30 | 2022-04-07 | Beigene, Ltd. | 3-[(1h-pyrazol-4-yl)oxy]pyrazin-2-amine compounds as hpk1 inhibitor and use thereof |
| EP4240361A4 (en) * | 2020-11-09 | 2024-09-25 | Merck Sharp & Dohme Llc | INHIBITORS, OF THE 7-AZOLE SUBSTITUTED 2-AMINOQUINAZOLINE TYPE, OF HPK1 |
| US20240002394A1 (en) * | 2020-11-24 | 2024-01-04 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | Deuterium-modified thienopyridone compound |
| EP4288437A1 (en) | 2021-02-05 | 2023-12-13 | Bayer Aktiengesellschaft | Map4k1 inhibitors |
| WO2022187856A1 (en) | 2021-03-05 | 2022-09-09 | Nimbus Saturn, Inc. | Hpk1 antagonists and uses thereof |
| TW202246284A (zh) * | 2021-03-10 | 2022-12-01 | 大陸商山東軒竹醫藥科技有限公司 | 三并環類hpk1抑制劑及其用途 |
| WO2022199676A1 (zh) * | 2021-03-26 | 2022-09-29 | 江苏恒瑞医药股份有限公司 | 稠合四环类化合物、其制备方法及其在医药上的应用 |
| JP2024513011A (ja) | 2021-03-29 | 2024-03-21 | ニンバス サターン, インコーポレイテッド | Hpk1アンタゴニスト及びその使用 |
| TW202302145A (zh) | 2021-04-14 | 2023-01-16 | 美商基利科學股份有限公司 | CD47/SIRPα結合及NEDD8活化酶E1調節次單元之共抑制以用於治療癌症 |
| WO2022226667A1 (en) * | 2021-04-30 | 2022-11-03 | Ontario Institute For Cancer Research (Oicr) | Substituted amino pyridine compounds as inhibitors of the haematopoietic progenitor kinase 1 (hpk1) |
| TW202313094A (zh) | 2021-05-18 | 2023-04-01 | 美商基利科學股份有限公司 | 使用FLT3L—Fc融合蛋白之方法 |
| WO2022253252A1 (en) * | 2021-06-03 | 2022-12-08 | Silexon Biotech Co., Ltd. | Heterocyclic compounds useful as hpk1 inhibitors |
| CN115433161A (zh) * | 2021-06-04 | 2022-12-06 | 轶诺(浙江)药业有限公司 | 新型hpk1抑制剂及其制备方法和应用 |
| US11932634B2 (en) | 2021-06-23 | 2024-03-19 | Gilead Sciences, Inc. | Diacylglycerol kinase modulating compounds |
| EP4359415A1 (en) | 2021-06-23 | 2024-05-01 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| CN117396478A (zh) | 2021-06-23 | 2024-01-12 | 吉利德科学公司 | 二酰基甘油激酶调节化合物 |
| EP4359411A1 (en) | 2021-06-23 | 2024-05-01 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2023001794A1 (en) | 2021-07-20 | 2023-01-26 | Astrazeneca Ab | Substituted pyrazine-2-carboxamides as hpk1 inhibitors for the treatment of cancer |
| JP2024528986A (ja) * | 2021-08-03 | 2024-08-01 | ニンバス サターン, インコーポレイテッド | Hpk1アンタゴニスト及びその使用 |
| WO2023023942A1 (en) * | 2021-08-24 | 2023-03-02 | Biofront Ltd (Cayman) | Hpk1 inhibitors, compositions comprising hpk1 inhibitor, and methods of using the same |
| WO2023057882A1 (en) * | 2021-10-05 | 2023-04-13 | Pfizer Inc. | Combinations of azalactam compounds with a pd-1 axis binding antagonist for the treatment of cancer |
| CN118076605A (zh) * | 2021-10-15 | 2024-05-24 | 洛蒙治疗公司 | 经取代的1H-吡唑并[4,3-c]喹啉、制备方法及其用途 |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| MX2024005066A (es) | 2021-10-29 | 2024-05-24 | Gilead Sciences Inc | Compuestos de cd73. |
| US20230220106A1 (en) | 2021-12-08 | 2023-07-13 | Dragonfly Therapeutics, Inc. | Antibodies targeting 5t4 and uses thereof |
| WO2023107956A1 (en) | 2021-12-08 | 2023-06-15 | Dragonfly Therapeutics, Inc. | Proteins binding nkg2d, cd16 and 5t4 |
| WO2023109902A1 (zh) * | 2021-12-17 | 2023-06-22 | 海思科医药集团股份有限公司 | 一种并环杂环衍生物及其在医药上的应用 |
| KR20240136983A (ko) * | 2021-12-22 | 2024-09-19 | 유니버시티 헬스 네트워크 | 급성 골수성 백혈병 또는 림프종의 치료 |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023138612A1 (en) * | 2022-01-19 | 2023-07-27 | Silexon Ai Technology Co., Ltd. | Heterocyclic compounds useful as hpk1 inhibitors |
| WO2023143384A1 (zh) * | 2022-01-27 | 2023-08-03 | 四川海思科制药有限公司 | 一种抑制或降解hpk1激酶的化合物及其在医药中的用途 |
| TW202340168A (zh) | 2022-01-28 | 2023-10-16 | 美商基利科學股份有限公司 | Parp7抑制劑 |
| CN116462685A (zh) * | 2022-02-08 | 2023-07-21 | 和径医药科技(上海)有限公司 | 杂环化合物、包含其的药物组合物及其抗肿瘤应用 |
| IL315083A (en) | 2022-03-17 | 2024-10-01 | Gilead Sciences Inc | The IKAROS family of zinc fingers degrades and uses them |
| US20230355796A1 (en) | 2022-03-24 | 2023-11-09 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| TW202345901A (zh) | 2022-04-05 | 2023-12-01 | 美商基利科學股份有限公司 | 用於治療結腸直腸癌之組合療法 |
| AU2023256670A1 (en) | 2022-04-21 | 2024-10-17 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| US20240116928A1 (en) | 2022-07-01 | 2024-04-11 | Gilead Sciences, Inc. | Cd73 compounds |
| US20240091351A1 (en) | 2022-09-21 | 2024-03-21 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPa DISRUPTION ANTICANCER COMBINATION THERAPY |
| KR20240077873A (ko) | 2022-11-25 | 2024-06-03 | 충남대학교산학협력단 | 이미다조피리딘 유도체, 이를 유효성분으로 포함하는 약제학적 조성물 및 이를 이용하는 암의 치료 방법 |
| US20240254118A1 (en) | 2022-12-22 | 2024-08-01 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU7314200A (en) * | 1999-09-17 | 2001-04-24 | Yamanouchi Pharmaceutical Co., Ltd. | Benzimidazole derivatives |
| ES2235970T3 (es) | 1999-10-19 | 2005-07-16 | MERCK & CO. INC. | Inhibidores de tirosina quinasa. |
| ATE448226T1 (de) | 2000-09-01 | 2009-11-15 | Novartis Vaccines & Diagnostic | Aza heterocyclische derivate und ihre therapeutische verwendung |
| WO2004014375A2 (en) | 2002-08-13 | 2004-02-19 | Warner-Lambert Company Llc | Fused bicyclic metalloproteinase inhibitors |
| US7119205B2 (en) | 2003-05-16 | 2006-10-10 | Abbott Laboratories | Thienopyridones as AMPK activators for the treatment of diabetes and obesity |
| EP2151440A4 (en) | 2007-05-21 | 2010-10-20 | Takeda Pharmaceutical | Heterocyclic compound and its use |
| CA2721025C (en) * | 2008-04-11 | 2016-07-26 | Merck Patent Gmbh | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| KR101648593B1 (ko) | 2008-05-05 | 2016-08-16 | 메르크 파텐트 게엠베하 | Amp-활성화된 단백질 키나아제 (ampk) 활성화제로서의 티에노피리돈 유도체 |
| US8653089B2 (en) | 2011-02-09 | 2014-02-18 | F. Hoffmann-La Roche Ag | Heterocyclic compounds and methods of use |
| GB201114212D0 (en) * | 2011-08-18 | 2011-10-05 | Ucb Pharma Sa | Therapeutic agents |
| IL283353B (en) | 2015-06-25 | 2022-09-01 | Univ Health Network | hpk1 inhibitors and methods of using them |
-
2016
- 2016-06-23 IL IL283353A patent/IL283353B/en unknown
- 2016-06-23 SG SG10202105964RA patent/SG10202105964RA/en unknown
- 2016-06-23 WO PCT/CA2016/050734 patent/WO2016205942A1/en active Application Filing
- 2016-06-23 LT LTEP16813437.7T patent/LT3322711T/lt unknown
- 2016-06-23 BR BR112017027241-5A patent/BR112017027241B1/pt active IP Right Grant
- 2016-06-23 EP EP16813437.7A patent/EP3322711B1/en active Active
- 2016-06-23 CA CA2989684A patent/CA2989684A1/en active Pending
- 2016-06-23 HU HUE16813437A patent/HUE054159T2/hu unknown
- 2016-06-23 BR BR122023006150-0A patent/BR122023006150B1/pt active IP Right Grant
- 2016-06-23 PT PT168134377T patent/PT3322711T/pt unknown
- 2016-06-23 JP JP2017566338A patent/JP6898868B2/ja active Active
- 2016-06-23 PL PL16813437T patent/PL3322711T3/pl unknown
- 2016-06-23 CN CN201680048058.0A patent/CN107922431B/zh active Active
- 2016-06-23 MX MX2018000048A patent/MX2018000048A/es unknown
- 2016-06-23 ES ES16813437T patent/ES2872555T3/es active Active
- 2016-06-23 MA MA42456A patent/MA42456B1/fr unknown
- 2016-06-23 RS RS20210675A patent/RS61919B1/sr unknown
- 2016-06-23 DK DK16813437.7T patent/DK3322711T3/da active
- 2016-06-23 KR KR1020187001327A patent/KR102614872B1/ko active IP Right Grant
- 2016-06-23 EA EA201890059A patent/EA035421B1/ru not_active IP Right Cessation
- 2016-06-23 US US15/738,286 patent/US10501474B2/en active Active
- 2016-06-23 AU AU2016282289A patent/AU2016282289B2/en active Active
- 2016-06-23 MD MDE20180529T patent/MD3322711T2/ro unknown
- 2016-06-23 SI SI201631189T patent/SI3322711T1/sl unknown
- 2016-06-23 KR KR1020237042987A patent/KR20240000617A/ko active IP Right Grant
- 2016-06-23 CN CN202110534553.5A patent/CN113214287B/zh active Active
- 2016-06-24 TW TW105120043A patent/TWI733679B/zh active
-
2017
- 2017-12-11 IL IL256250A patent/IL256250B/en active IP Right Grant
-
2018
- 2018-01-08 MX MX2021011435A patent/MX2021011435A/es unknown
- 2018-11-21 HK HK18114909.1A patent/HK1255833A1/zh unknown
-
2019
- 2019-12-06 US US16/705,458 patent/US11059832B2/en active Active
-
2021
- 2021-06-07 HR HRP20210895TT patent/HRP20210895T1/hr unknown
- 2021-06-08 CY CY20211100498T patent/CY1124489T1/el unknown
- 2021-06-11 JP JP2021097834A patent/JP7241810B2/ja active Active
- 2021-07-12 US US17/372,717 patent/US20220002313A1/en active Pending
-
2023
- 2023-03-07 JP JP2023034474A patent/JP2023071911A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2872555T3 (es) | Inhibidores de HPK1 y métodos de uso de los mismos | |
| TWI827677B (zh) | 作為src同源-2磷酸酶抑制劑之稠合三環衍生物 | |
| RU2743343C2 (ru) | Имидазолонилхинолины и их применение в качестве ингибиторов киназы атм | |
| ES2360933T3 (es) | Derivados de heteroarilo condensados. | |
| ES2676224T3 (es) | Compuestos a base de pirazolo[1,5-a]pirimidina, composiciones que los comprenden y métodos para usarlos | |
| DK2947086T3 (en) | UNKNOWN CONDENSED PYRIMIDINE COMPOUND OR SALT THEREOF | |
| RU2537945C2 (ru) | Триазиновые, пиримидиновые и пиридиновые аналоги и их применение в качестве терапевтических агентов и диагностических проб | |
| BR112020019700A2 (pt) | Compostos heterocíclicos como imunomoduladores | |
| JP5615902B2 (ja) | イミダゾ[2,1−b][1,3,4]チアジアゾール誘導体 | |
| JP2013522286A (ja) | スピロ環化合物ならびに治療薬及び診断プローブとしてのその使用 | |
| JP2019505541A (ja) | Rsk阻害剤として有用なカルボキサミド誘導体 | |
| TW202237568A (zh) | Gcn2及perk激酶抑制劑及其使用方法 | |
| JP2018522867A (ja) | 縮合二環式ピリミジン誘導体およびこれらの使用 | |
| KR20220028075A (ko) | 티로신 키나제 비-수용체 1 (tnk1) 억제제 및 그의 용도 | |
| CN111727044A (zh) | 用于治疗癌症或炎性疾病的杂二环羧酸 | |
| CA3134221A1 (en) | Small-molecule focal adhesion kinase (fak) inhibitors | |
| TW202237101A (zh) | Ctla-4小分子降解劑及其應用 | |
| BR112017020941B1 (pt) | Imidazolonilquinolinas, seus usos, composições farmacêuticas, e kit |