ES2806460T3 - Proceso para la preparación de derivados de 4-aminoindano y amidas de aminoindano relacionadas - Google Patents
Proceso para la preparación de derivados de 4-aminoindano y amidas de aminoindano relacionadas Download PDFInfo
- Publication number
- ES2806460T3 ES2806460T3 ES16717473T ES16717473T ES2806460T3 ES 2806460 T3 ES2806460 T3 ES 2806460T3 ES 16717473 T ES16717473 T ES 16717473T ES 16717473 T ES16717473 T ES 16717473T ES 2806460 T3 ES2806460 T3 ES 2806460T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- group
- groups
- acyl
- aminoindan
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 aminoindan amides Chemical class 0.000 title claims abstract description 55
- 238000000034 method Methods 0.000 title claims abstract description 43
- 238000002360 preparation method Methods 0.000 title claims abstract description 30
- RXTJLDXSGNEJIT-UHFFFAOYSA-N 2,3-dihydro-1h-inden-4-amine Chemical class NC1=CC=CC2=C1CCC2 RXTJLDXSGNEJIT-UHFFFAOYSA-N 0.000 title claims abstract description 29
- LBUJPTNKIBCYBY-UHFFFAOYSA-N 1,2,3,4-tetrahydroquinoline Chemical compound C1=CC=C2CCCNC2=C1 LBUJPTNKIBCYBY-UHFFFAOYSA-N 0.000 claims abstract description 45
- 125000005843 halogen group Chemical group 0.000 claims abstract description 22
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 17
- IRFSXVIRXMYULF-UHFFFAOYSA-N 1,2-dihydroquinoline Chemical compound C1=CC=C2C=CCNC2=C1 IRFSXVIRXMYULF-UHFFFAOYSA-N 0.000 claims abstract description 15
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N benzocyclopentane Natural products C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 claims abstract description 14
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims abstract description 13
- 150000003839 salts Chemical class 0.000 claims abstract description 12
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims abstract description 8
- 125000005947 C1-C6 alkylsulfonyloxy group Chemical group 0.000 claims abstract description 6
- 230000002378 acidificating effect Effects 0.000 claims abstract description 6
- 125000005279 aryl sulfonyloxy group Chemical group 0.000 claims abstract description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 6
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 claims abstract description 3
- 125000002252 acyl group Chemical group 0.000 claims abstract description 3
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical class CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 77
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 30
- 239000011541 reaction mixture Substances 0.000 claims description 27
- 239000002904 solvent Substances 0.000 claims description 20
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 20
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 18
- 239000000725 suspension Substances 0.000 claims description 17
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 16
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 15
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 12
- 239000003960 organic solvent Substances 0.000 claims description 12
- 239000003054 catalyst Substances 0.000 claims description 10
- 150000001875 compounds Chemical class 0.000 claims description 10
- 238000004821 distillation Methods 0.000 claims description 10
- 239000000203 mixture Substances 0.000 claims description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 9
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 9
- 238000005984 hydrogenation reaction Methods 0.000 claims description 9
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 7
- 125000000217 alkyl group Chemical group 0.000 claims description 7
- 125000000623 heterocyclic group Chemical group 0.000 claims description 7
- 150000007522 mineralic acids Chemical class 0.000 claims description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 6
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 claims description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 5
- 239000007864 aqueous solution Substances 0.000 claims description 5
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 5
- 125000004429 atom Chemical group 0.000 claims description 5
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 5
- 125000005842 heteroatom Chemical group 0.000 claims description 5
- 229910052760 oxygen Inorganic materials 0.000 claims description 5
- 229910052717 sulfur Inorganic materials 0.000 claims description 5
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 claims description 4
- 150000007524 organic acids Chemical class 0.000 claims description 4
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 3
- 150000001298 alcohols Chemical class 0.000 claims description 3
- 150000008280 chlorinated hydrocarbons Chemical class 0.000 claims description 3
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 3
- 229910052736 halogen Inorganic materials 0.000 claims description 3
- 150000002367 halogens Chemical class 0.000 claims description 3
- 229930195733 hydrocarbon Natural products 0.000 claims description 3
- 150000002430 hydrocarbons Chemical class 0.000 claims description 3
- 230000007062 hydrolysis Effects 0.000 claims description 3
- 238000006460 hydrolysis reaction Methods 0.000 claims description 3
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 2
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 claims description 2
- 239000012346 acetyl chloride Substances 0.000 claims description 2
- 125000001931 aliphatic group Chemical group 0.000 claims description 2
- 125000003342 alkenyl group Chemical group 0.000 claims description 2
- 125000003545 alkoxy group Chemical group 0.000 claims description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 claims description 2
- 150000002148 esters Chemical class 0.000 claims description 2
- 150000002334 glycols Chemical class 0.000 claims description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 2
- 230000003301 hydrolyzing effect Effects 0.000 claims description 2
- 235000011007 phosphoric acid Nutrition 0.000 claims description 2
- 239000002798 polar solvent Substances 0.000 claims 1
- ODIGIKRIUKFKHP-UHFFFAOYSA-N (n-propan-2-yloxycarbonylanilino) acetate Chemical compound CC(C)OC(=O)N(OC(C)=O)C1=CC=CC=C1 ODIGIKRIUKFKHP-UHFFFAOYSA-N 0.000 abstract description 2
- 238000006243 chemical reaction Methods 0.000 description 34
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 30
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 20
- 239000000243 solution Substances 0.000 description 14
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- BHJHVXBWRQTCRB-UHFFFAOYSA-N 1-(6-fluoro-2,2,4-trimethyl-3,4-dihydroquinolin-1-yl)ethanone Chemical compound CC1CC(C)(C)N(C(C)=O)c2ccc(F)cc12 BHJHVXBWRQTCRB-UHFFFAOYSA-N 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- 239000002253 acid Substances 0.000 description 9
- WOWUXBKVJPNKRG-UHFFFAOYSA-N 6-fluoro-2,2,4-trimethyl-1h-quinoline Chemical compound C1=C(F)C=C2C(C)=CC(C)(C)NC2=C1 WOWUXBKVJPNKRG-UHFFFAOYSA-N 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 239000000047 product Substances 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- XJEVHMGJSYVQBQ-UHFFFAOYSA-N 2,3-dihydro-1h-inden-1-amine Chemical class C1=CC=C2C(N)CCC2=C1 XJEVHMGJSYVQBQ-UHFFFAOYSA-N 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- KRZCOLNOCZKSDF-UHFFFAOYSA-N 4-fluoroaniline Chemical compound NC1=CC=C(F)C=C1 KRZCOLNOCZKSDF-UHFFFAOYSA-N 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- 229910052799 carbon Inorganic materials 0.000 description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical class CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 5
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 5
- LQRAKGPRTLXFBL-UHFFFAOYSA-N S(=O)(=O)(O)O.FC=1C=CC(=C2C(CC(C12)(C)C)C)N Chemical compound S(=O)(=O)(O)O.FC=1C=CC(=C2C(CC(C12)(C)C)C)N LQRAKGPRTLXFBL-UHFFFAOYSA-N 0.000 description 5
- 239000003377 acid catalyst Substances 0.000 description 5
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 5
- 150000001448 anilines Chemical class 0.000 description 5
- 238000009833 condensation Methods 0.000 description 5
- 230000005494 condensation Effects 0.000 description 5
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 238000010907 mechanical stirring Methods 0.000 description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 5
- 238000005917 acylation reaction Methods 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 238000002425 crystallisation Methods 0.000 description 4
- 230000008025 crystallization Effects 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 238000001556 precipitation Methods 0.000 description 4
- 238000005292 vacuum distillation Methods 0.000 description 4
- OTAFYGOGVVSKOO-UHFFFAOYSA-N 2,3-dihydro-1h-inden-1-amine;sulfuric acid Chemical compound OS(O)(=O)=O.C1=CC=C2C(N)CCC2=C1 OTAFYGOGVVSKOO-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 0 CCC1(C)N(C(*)=O)C2=CCCC=C2C(C)C1 Chemical compound CCC1(C)N(C(*)=O)C2=CCCC=C2C(C)C1 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 230000010933 acylation Effects 0.000 description 3
- 239000003637 basic solution Substances 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 238000011068 loading method Methods 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- 125000004767 (C1-C4) haloalkoxy group Chemical group 0.000 description 2
- XCGBHLLWJZOLEM-UHFFFAOYSA-N 3-(difluoromethyl)-N-(7-fluoro-1,1,3-trimethyl-2,3-dihydro-1H-inden-4-yl)-1-methyl-1H-pyrazole-4-carboxamide Chemical compound CC1CC(C)(C)C(C(=CC=2)F)=C1C=2NC(=O)C1=CN(C)N=C1C(F)F XCGBHLLWJZOLEM-UHFFFAOYSA-N 0.000 description 2
- USXBDIZEJAVICB-UHFFFAOYSA-N 7-fluoro-1,1,3-trimethyl-2,3-dihydroinden-4-amine Chemical compound NC1=CC=C(F)C2=C1C(C)CC2(C)C USXBDIZEJAVICB-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 2
- 230000002051 biphasic effect Effects 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- 238000004508 fractional distillation Methods 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- MGNPLIACIXIYJE-UHFFFAOYSA-N n-fluoroaniline Chemical compound FNC1=CC=CC=C1 MGNPLIACIXIYJE-UHFFFAOYSA-N 0.000 description 2
- 230000008707 rearrangement Effects 0.000 description 2
- 238000004064 recycling Methods 0.000 description 2
- 239000012265 solid product Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- LEMQFBIYMVUIIG-UHFFFAOYSA-N trifluoroborane;hydrofluoride Chemical compound F.FB(F)F LEMQFBIYMVUIIG-UHFFFAOYSA-N 0.000 description 2
- KPZGRMZPZLOPBS-UHFFFAOYSA-N 1,3-dichloro-2,2-bis(chloromethyl)propane Chemical compound ClCC(CCl)(CCl)CCl KPZGRMZPZLOPBS-UHFFFAOYSA-N 0.000 description 1
- RRWLNRQGJSQRAF-UHFFFAOYSA-N 1-(3,4-dihydro-2h-quinolin-1-yl)ethanone Chemical compound C1=CC=C2N(C(=O)C)CCCC2=C1 RRWLNRQGJSQRAF-UHFFFAOYSA-N 0.000 description 1
- SSHJBHQGONOUQG-UHFFFAOYSA-N 1-(6-fluoro-2,2,4-trimethylquinolin-1-yl)ethanone Chemical compound CC(=O)N1c2ccc(F)cc2C(C)=CC1(C)C SSHJBHQGONOUQG-UHFFFAOYSA-N 0.000 description 1
- RLOHOBNEYHBZID-UHFFFAOYSA-N 3-(difluoromethyl)-1-methylpyrazole-4-carboxylic acid Chemical compound CN1C=C(C(O)=O)C(C(F)F)=N1 RLOHOBNEYHBZID-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- KYJMCSGXJLHJBD-UHFFFAOYSA-N 6-fluoro-2,2,4-trimethyl-3,4-dihydro-1h-quinoline Chemical compound C1=C(F)C=C2C(C)CC(C)(C)NC2=C1 KYJMCSGXJLHJBD-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- KYWYAWFSWFSPAY-UHFFFAOYSA-N CC(CC(C)(C)c1ccc2)c1c2N Chemical compound CC(CC(C)(C)c1ccc2)c1c2N KYWYAWFSWFSPAY-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- 239000003929 acidic solution Substances 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 1
- 150000008041 alkali metal carbonates Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 125000004775 chlorodifluoromethyl group Chemical group FC(F)(Cl)* 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000004850 cyclobutylmethyl group Chemical group C1(CCC1)C* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000004851 cyclopentylmethyl group Chemical group C1(CCCC1)C* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 125000006003 dichloroethyl group Chemical group 0.000 description 1
- 125000004772 dichloromethyl group Chemical group [H]C(Cl)(Cl)* 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 230000000855 fungicidal effect Effects 0.000 description 1
- 239000000417 fungicide Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000002638 heterogeneous catalyst Substances 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 150000002468 indanes Chemical class 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 238000003760 magnetic stirring Methods 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004998 naphthylethyl group Chemical group C1(=CC=CC2=CC=CC=C12)CC* 0.000 description 1
- 125000004923 naphthylmethyl group Chemical group C1(=CC=CC2=CC=CC=C12)C* 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- FWFGVMYFCODZRD-UHFFFAOYSA-N oxidanium;hydrogen sulfate Chemical compound O.OS(O)(=O)=O FWFGVMYFCODZRD-UHFFFAOYSA-N 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004346 phenylpentyl group Chemical group C1(=CC=CC=C1)CCCCC* 0.000 description 1
- 125000004344 phenylpropyl group Chemical group 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 239000003495 polar organic solvent Substances 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- KOPFEFZSAMLEHK-UHFFFAOYSA-N pyrazolecarboxylic acid Natural products OC(=O)C=1C=CNN=1 KOPFEFZSAMLEHK-UHFFFAOYSA-N 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000006462 rearrangement reaction Methods 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000011269 tar Substances 0.000 description 1
- 125000006337 tetrafluoro ethyl group Chemical group 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 125000004205 trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/62—Preparation of compounds containing amino groups bound to a carbon skeleton by cleaving carbon-to-nitrogen, sulfur-to-nitrogen, or phosphorus-to-nitrogen bonds, e.g. hydrolysis of amides, N-dealkylation of amines or quaternary ammonium compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/68—Preparation of compounds containing amino groups bound to a carbon skeleton from amines, by reactions not involving amino groups, e.g. reduction of unsaturated amines, aromatisation, or substitution of the carbon skeleton
- C07C209/70—Preparation of compounds containing amino groups bound to a carbon skeleton from amines, by reactions not involving amino groups, e.g. reduction of unsaturated amines, aromatisation, or substitution of the carbon skeleton by reduction of unsaturated amines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/02—Preparation of carboxylic acid amides from carboxylic acids or from esters, anhydrides, or halides thereof by reaction with ammonia or amines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/08—Preparation of carboxylic acid amides from amides by reaction at nitrogen atoms of carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/14—Preparation of carboxylic acid amides by formation of carboxamide groups together with reactions not involving the carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/18—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/04—One of the condensed rings being a six-membered aromatic ring
- C07C2602/08—One of the condensed rings being a six-membered aromatic ring the other ring being five-membered, e.g. indane
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
Abstract
Un proceso para la preparación de derivados de 4-aminoindano de Fórmula (I), sales y enantiómeros de los mismos **(Ver fórmula)** que comprende las siguientes etapas: a) hidrogenar una 1,2-dihidroquinolina de Fórmula (IV) **(Ver fórmula)** para dar una tetrahidroquinolina correspondiente de Fórmula (V) **(Ver fórmula)** b) acilar la tetrahidroquinolina de Fórmula (V) con un derivado de ácido carboxílico de Fórmula RC(O)LG para obtener un compuesto de derivado de acilo correspondiente de Fórmula (VI) **(Ver fórmula)** c) reordenar el compuesto de derivado de acilo de Fórmula (VI) en condiciones ácidas para dar un compuesto de indano de acilo de Fórmula (VII) **(Ver fórmula)** d) hidrolizar el grupo acilo del compuesto de indano de acilo de Fórmula (VII) para obtener el compuesto de 4- aminoindano de Fórmula (I), en donde, en dichas Fórmulas: - n es un número entero seleccionado dentro del intervalo de 0 a 3; - R se selecciona de un grupo alquilo C1-C6 o un grupo arilo C6-C10, estando estos grupos opcionalmente sustituidos con uno o más de: grupos alquilo C1-C6, átomos de halógeno; - LG es un grupo saliente seleccionado de: (i) un grupo hidroxi; (ii) un átomo de halógeno; (iii) un grupo alquilsulfoniloxi C1-C6; (iv) un grupo arilsulfoniloxi C6-C10, (v) un grupo RaCOO, en donde Ra es un grupo alquilo C1-C6, estando los grupos (iii) - (v) opcionalmente sustituidos con uno o más átomos de halógeno.
Description
DESCRIPCIÓN
Proceso para la preparación de derivados de 4-aminoindano y amidas de aminoindano relacionadas
La presente invención se refiere a un proceso para la preparación de derivados de 4-aminoindano de Fórmula (I)

La presente invención también se refiere a un proceso para la preparación de amidas de aminoindano de Fórmula (II), que tienen una actividad fungicida, a partir de un producto intermedio de derivado de 4-aminoindano de Fórmula (I) obtenido a través del proceso mencionado anteriormente.

Antecedentes de la invención
Las amidas de aminoindano, así como los procesos para la preparación de las mismas, se han indicado ampliamente en la técnica anterior, tal como en los documentos JP1070479, JP1117864, JP1313402, JP2157266, JP2249966, JP3077381, JP62096471, EP199822, EP256503, EP276177, EP280275, EP569912, US5093347, WO01/53259, WO2004/018438, WO2004/039789, WO2004/072023, WO2004/103975, WO2005/075452, WO2012/084812 y WO2013/186325.
En particular, el documento WO2013/186325 desvela que el compuesto 3-difluorometil-N-(7-fluoro-1,1,3-trimetil-4-indanil)-1-metil-4-pirazolcarboxamida se puede preparar en cuatro etapas a partir de 4-fluoroanilina y acetona. Estos dos compuestos se condensan en primer lugar en conjunto para formar una dihidroquinolina sustituida, que se hidrogena, a continuación, para proporcionar la tetrahidroquinolina correspondiente. La tetrahidroquinolina se hace reaccionar, a continuación, con un derivado de ácido carboxílico de pirazol y el compuesto resultante se somete a un reordenamiento de ácido para proporcionar el derivado de amida de aminoindano correspondiente. La preparación de 4 etapas se indica, a continuación, en el Esquema 1.

Sin embargo, dicho proceso no es satisfactorio, dado que, desde un punto de vista químico, la amina secundaria en el anillo de tetrahidroquinolina es bastante difícil de acilar; por lo tanto, se necesitan condiciones de reacción forzadas, tales como la adición de un exceso de una base y el uso de un disolvente orgánico clorado, para obtener la tetrahidroquinolina de acilo correspondiente. Además, los rendimientos globales de este proceso son relativamente bajos y conducen a una pérdida significativa del derivado de cloruro de ácido de pirazol, que es un material costoso. También se sabe que los derivados de 4-aminoindano se pueden usar como productos intermedios clave para la sintetización del derivado de amida de aminoindano. Un ejemplo de tal síntesis se puede hallar en el documento EP199822, que desvela que los derivados de amida de aminoindano se pueden obtener a través de una reacción de condensación entre un haluro de ácido carboxílico de pirazol y un derivado de 4-aminoindano, tal como se indica, a continuación, en el Esquema 2.

Sin embargo, en el documento EP199822 no hay ninguna indicación sobre la vía sintética para la preparación del derivado de 4-aminoindano.
Pocos documentos de la técnica anterior describen realmente un proceso para la preparación de tales derivados de 4-aminoindano.
Por ejemplo, el documento EP654464 desvela que los derivados de 4-aminoindano en forma enriquecida diastereoisoméricamente se pueden obtener en cuatro etapas, tal como se indica en el Esquema 3: i) condensación entre una dihidroquinolina y un derivado de ácido carboxílico que tiene tanto un centro quiral (indicado con * en el Esquema, a continuación) como un grupo saliente terminal LG; ii) hidrogenación catalítica para proporcionar la tetrahidroquinolina correspondiente; iii) adición de un ácido fuerte para obtener el derivado de 4-aminoindano correspondiente; y iv) hidrólisis del enlace de amida.

Sin embargo, el proceso mencionado anteriormente para la preparación de los derivados de 4-aminoindano no es satisfactorio desde un punto de vista industrial, dado que este requiere usar un disolvente diferente para cada etapa (es decir, tetrahidrofurano, metanol y ácido sulfúrico agua ácido acético en el Ejemplo 1, Vía N. 1, página 9). Por lo tanto, se han de realizar operaciones adicionales al final de cualquier etapa de reacción individual con el fin de evitar, por ejemplo, la contaminación de la mezcla de reacción posterior mediante un producto químico o disolvente restante.
Además, la dihidroquinolina de acilo y la tetrahidroquinolina correspondiente son escasamente solubles en disolventes apolares, tales como hidrocarburos alifáticos. Por consiguiente, con el fin de convertir completamente la dihidroquinolina de acilo en la tetrahidroquinolina correspondiente, se requieren temperaturas de reacción más altas o la dilución de la mezcla de reacción.
Por lo tanto, resulta deseable proporcionar un proceso más fácil para la preparación de derivados de aminoindano, en particular, amidas de aminoindano a gran escala, lo que reduciría los costes y tiempos de producción.
Descripción de la invención
Actualmente, se ha hallado, sorprendentemente, que, mediante la inversión de las etapas de hidrogenación y condensación del proceso desvelado en el documento EP654464, resulta posible preparar derivados de 4-aminoindanos y las amidas de aminoindano correspondientes de una manera más sencilla y rentable.
Por lo tanto, un primer objeto de la presente invención es un proceso para la preparación de derivados de 4-aminoindano de Fórmula (I), sales y enantiómeros de los mismos

que comprende las siguientes etapas:
a) hidrogenar una 1,2-dihidroquinolina de Fórmula (IV)

para dar una tetrahidroquinolina correspondiente de Fórmula (V)

b) acilar la tetrahidroquinolina de Fórmula (V) con un derivado de ácido carboxílico de Fórmula RC(O)LG para proporcionar un compuesto de derivado de acilo de Fórmula (VI)

c) reordenar el compuesto de derivado de acilo de Fórmula (VI) en condiciones ácidas para dar un compuesto de indano de acilo de Fórmula (VII)

d) hidrolizar el grupo acilo del compuesto de indano de acilo de Fórmula (VII) para proporcionar el 4-aminoindano deseado de Fórmula (I),
en donde, en dichas Fórmulas:
- n es un número entero seleccionado dentro del intervalo de 0 a 3;
- R es un grupo alquilo C1-C6, un grupo arilo C6-C10, estando estos grupos opcionalmente sustituidos con uno o más de: grupo alquilo C1-C6 , átomo de halógeno;
- LG es un grupo saliente seleccionado de: (i) un grupo hidroxi; (ii) un átomo de halógeno; (iii) un grupo alquilsulfoniloxi C1-C6 ; (iv) un grupo arilsulfoniloxi C6-C10; (v) un grupo RaCOO, en donde Ra es un grupo alquilo C1-C6 , estando los grupos (iii)-(v) opcionalmente sustituidos con uno o más átomos de halógeno. El proceso objeto de la presente invención comprende al menos las cuatro etapas indicadas anteriormente, que se llevan a cabo en el orden indicado.
Tal como lo demuestran los datos experimentales incluidos en la presente descripción, el solicitante ha hallado, sorprendentemente, que mediante la inversión de las etapas de hidrogenación y condensación (acilación) del proceso desvelado en el documento EP654464, resulta posible usar solo un tipo de disolvente orgánico (por ejemplo, un hidrocarburo alifático, tal como heptano) en todo el proceso de producción de los derivados de 4-amnoindano de Fórmula (I), simplificando, por tanto, el proceso y reduciendo sus costes y tiempos de producción. Además, el proceso de preparación de acuerdo con la presente invención se puede llevar a cabo, ventajosamente, mediante la realización de las Etapas (a) a (c) sin ser necesario aislar y/o purificar los productos intermedios de las Fórmulas (VI) y (VII) al final de sus respectivas etapas de formación.
La presente invención proporciona, por tanto, una ruta más rentable para la preparación de derivados de 4-aminoindano de Fórmula (I), así como de otros compuestos de interés industrial y comercial, que se preparan típicamente a partir de estos derivados de 4-aminoindano, tales como las amidas de aminoindano de Fórmula (II), que se pueden usar como fungicidas. De acuerdo con el proceso actualmente reivindicado, en la Etapa (a) una 1,2-dihidroquinolina de Fórmula (IV) se somete en primer lugar a hidrogenación catalítica, tal como se indica, a continuación, en el Esquema 4.

El compuesto de Fórmula (IV) está disponible en el mercado o se puede preparar, por ejemplo, tal como se describe en Organic Synthesis, vol. III, página 329. De acuerdo con un aspecto preferido de la presente invención, un derivado de anilina de Fórmula (III) se condensa con acetona en presencia de un catalizador ácido para proporcionar la dihidroquinolina correspondiente de Fórmula (IV), tal como se indica, a continuación, en el Esquema 5.

El derivado de anilina de Fórmula (III) se hace reaccionar con acetona, en una cantidad comprendida entre 1 y 15 equivalentes molares, preferentemente entre 3 y 10 equivalentes molares, más preferentemente entre 4 y 7 equivalentes molares, con respecto a la anilina de partida de Fórmula (III).
Preferentemente, la acetona se añade a dicho derivado de anilina de Fórmula (III) durante un tiempo comprendido entre 1 y 15 horas, más preferentemente entre 2 y 12 horas, incluso más preferentemente entre 5 y 10 horas. A la mezcla así formada, se añade un catalizador ácido, preferentemente un catalizador ácido seleccionado de un ácido orgánico, un ácido inorgánico o una mezcla de los mismos. Los ejemplos no limitantes de catalizadores ácidos adecuados de acuerdo con la presente invención son: ácido metanosulfónico, ácido paratoluenosulfónico, ácido acético, ácido tetrafluobórico, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico o mezclas de los mismos.
Los catalizadores ácidos más preferidos se seleccionan de ácido tetrafluobórico y ácido paratoluenosulfónico.
Durante la adición de la acetona al derivado de anilina de Fórmula (III), la mezcla de reacción se mantiene a una temperatura comprendida entre 80 °C y 200 °C, preferentemente entre 100 °C y 180 °C, más preferentemente entre 125 °C y 145 °C.
Cuando se completa la reacción, el derivado de dihidroquinolina de Fórmula (IV) así obtenido se puede aislar y
purificar de acuerdo con métodos bien conocidos por aquellos expertos en la materia. Por ejemplo, la mezcla de reacción se puede someter a tratamiento con una base, tal como una base inorgánica, para retirar la acidez libre y se puede extraer mediante su mezclado con un disolvente orgánico ligeramente miscible o inmiscible con agua. El producto de 1,2-dihidroquinolina deseado se puede recuperar, por ejemplo, mediante destilación fraccionada.
De acuerdo con un aspecto preferido de la presente invención, el derivado de anilina residual de Fórmula (III) también se puede recuperar mediante destilación fraccionada y se puede usar de nuevo, ventajosamente, en el lote de producción posterior.
De acuerdo con la presente invención, en la Etapa (a), el compuesto de Fórmula (IV) se disuelve en un disolvente orgánico, preferentemente se añade un disolvente orgánico polar y un catalizador de metal a la mezcla de reacción. Dicho catalizador de metal es, preferentemente, un catalizador heterogéneo, más preferentemente, seleccionado de paladio sobre carbón vegetal, hidróxido de paladio sobre carbón vegetal, níquel Raney y óxido de platino; incluso más preferentemente, el catalizador de metal es paladio sobre carbón vegetal.
De acuerdo con la presente invención, la carga de catalizador está comprendida entre el 0,05 y el 0,7 %, preferentemente comprendida entre el 0,1 y el 0,6 %, más preferentemente la carga es de aproximadamente el 0,5 %, con respecto a la cantidad molar de dihidroquinolina de Fórmula (IV).
Los ejemplos no limitantes de disolventes que se pueden usar en la reacción de hidrogenación son: hidrocarburos alifáticos o cicloalifáticos (por ejemplo, éter de petróleo, hexano, ciclohexano, heptano), hidrocarburos clorados (por ejemplo, cloruro de metileno, cloroformo, tetracloruro de carbono, dicloroetano), hidrocarburos aromáticos (por ejemplo, benceno, tolueno, xileno, clorobenceno), alcoholes y glicoles (por ejemplo, metanol, etanol, isopropanol, etilen glicol), ésteres (por ejemplo, acetato de etilo, acetato de butilo) o mezclas de los mismos.
Entre estos, los disolventes preferidos son: hidrocarburos alifáticos, tales como hexano y heptanos; hidrocarburos clorados, tales como cloruro de metileno y dicloroetano; alcoholes, tales como metanol, etanol e isopropanol; tolueno; acetato de etilo.
El heptano, dicloroetano, metanol y tolueno son particularmente preferidos.
Dentro del significado de la presente invención, el término "heptano" se refiere a n-heptano o a una mezcla de isómeros.
Tal como conoce bien la persona experta, la reacción de hidrogenación de la Etapa a) se puede llevar a cabo a una presión mayor de 100 kPa (1 bar) o a presión atmosférica.
Preferentemente, la Etapa a) de la presente invención se lleva a cabo a presión atmosférica.
Cuando la Etapa a) se lleva a cabo a presión atmosférica, la carga de catalizador es preferentemente de aproximadamente el 0,5 % y la mezcla de reacción se deja reaccionar durante un tiempo comprendido preferentemente entre 1 y 5 horas a temperatura ambiente.
Cuando la Etapa a) se lleva a cabo a una sobrepresión de hidrógeno, dicha sobrepresión preferentemente está comprendida entre 500 y 900 kPa (5 y 9 bares).
Cuando la Etapa a) se lleva a cabo a una sobrepresión de hidrógeno, la carga de catalizador está comprendida preferentemente entre el 0,1 y el 0,6 % y la mezcla de reacción se deja reaccionar durante un tiempo comprendido preferentemente entre 10 y 18 horas a una temperatura comprendida preferentemente entre 35 y 50 °C.
Al final de la Etapa (a), el catalizador se recupera y se usa de nuevo preferentemente en lotes de producción posteriores.
La tetrahidroquinolina de Fórmula (V) obtenida en la Etapa a) de hidrogenación se aísla de la mezcla de reacción mediante métodos bien conocidos por aquellos expertos en la materia, por ejemplo, mediante concentración de disolvente.
Preferentemente, el compuesto aislado de Fórmula (V) no se purifica y se usa como tal en la etapa posterior del proceso.
La Etapa b) de acilación se realiza mediante la adición de un derivado de ácido carboxílico de Fórmula RC(O)LG a la tetrahidroquinolina de Fórmula (V), tal como se indica, a continuación, en el Esquema 6.
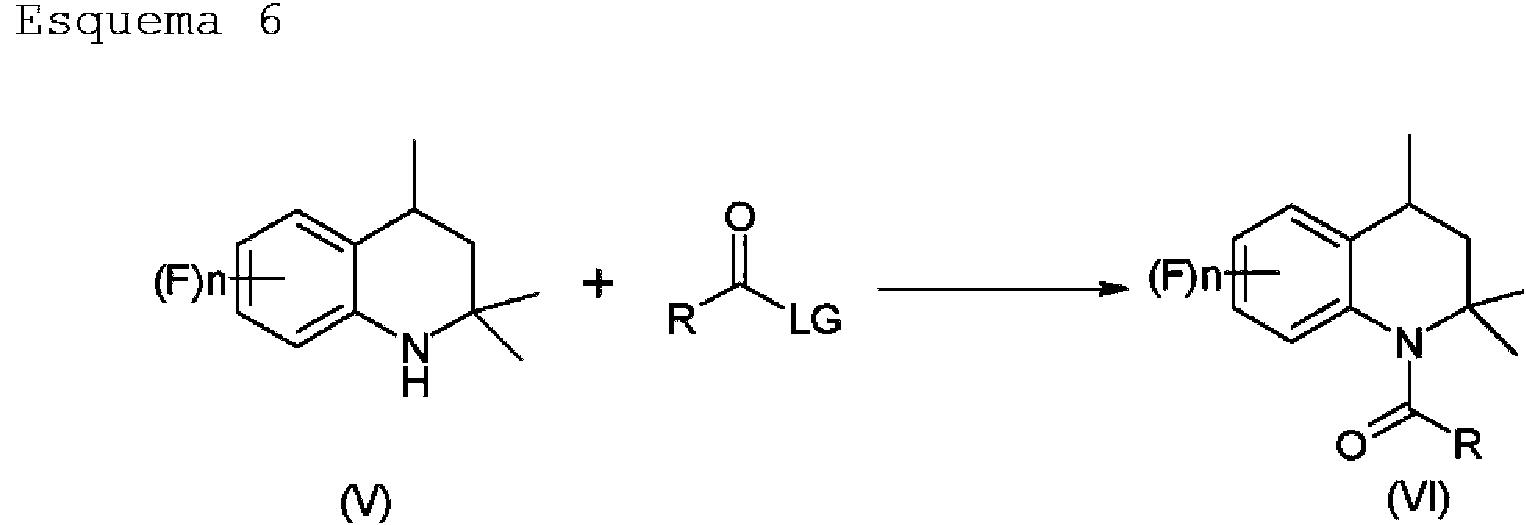
en el que:
- R es un grupo alquilo C1-C6, un grupo arilo Ca-C-io, estando estos grupos opcionalmente sustituidos por uno o más de: grupos alquilo C1-C6 , átomos de halógeno;
- LG es un grupo saliente seleccionado de: (i) un grupo hidroxilo; (ii) un átomo de halógeno; (iii) un grupo alquilsulfoniloxi C1-C6; (iv) un grupo arilsulfoniloxi C6-C10; (v) un grupo RaCOO, en donde Ra es grupo alquilo C1-Ce, estando los grupos (iii)-(v) opcionalmente sustituidos con uno o más átomos de halógeno.
Preferentemente, el derivado de ácido carboxílico se añade en una cantidad comprendida entre el 5 y el 30 %, más preferentemente entre el 10 y el 25 %, con respecto a la cantidad molar de la tetrahidroquinolina de partida de Fórmula (V). De acuerdo con un aspecto más preferido, el derivado de ácido carboxílico se selecciona de cloruro de acetilo y anhídrido acético, incluso más preferentemente el derivado de ácido carboxílico es anhídrido acético.
La reacción de acilación se puede llevar a cabo en un disolvente orgánico o en ausencia de un disolvente. De acuerdo con la presente invención, dicha reacción se lleva a cabo preferentemente en ausencia de cualquier disolvente añadido.
En la Etapa c), la mezcla de reacción se mantiene a una temperatura comprendida entre 80 °C y 200 °C, preferentemente comprendida entre 100 °C y 150 °C, más preferentemente de aproximadamente 130 °C.
Si se usa el anhídrido acético, una vez que se completa la conversión, preferentemente se añade algo de agua para causar la descomposición del anhídrido acético residual en ácido acético.
Con el fin de retirar completamente el ácido acético residual, se puede añadir un disolvente inmiscible con agua a la mezcla de reacción, preferentemente un hidrocarburo alifático, tal como hexano o heptano.
De acuerdo con un aspecto preferido de la presente invención, el ácido acético se retira de la mezcla de reacción mediante destilación al vacío/destilación azeotrópica con heptano.
La tetrahidroquinolina de acilo de Fórmula (VI) obtenida al final de la etapa de acilación se puede purificar de acuerdo con métodos bien conocidos por la persona experta, por ejemplo, mediante precipitación, cristalización y similares.
Preferentemente, dicha tetrahidroquinolina de acilo de Fórmula (VI) se somete a cristalización con un disolvente orgánico formando, por tanto, una suspensión con ese disolvente.
Ventajosamente, de acuerdo con la presente invención, el disolvente usado para la cristalización es del mismo tipo que el disolvente usado para la retirada del ácido acético residual.
La tetrahidroquinolina de acilo de Fórmula (VI) contenida en dicha suspensión preferentemente no está aislada. La suspensión se puede usar como tal en la siguiente etapa.
Posteriormente, la tetrahidroquinolina de acilo de Fórmula (VI) se somete a reordenamiento en un entorno ácido, tal como se indica, a continuación, en el Esquema 7.

La condición de pH ácido que permite el reordenamiento de la tetrahidroquinolina de Fórmula (VI) para dar el derivado de indano correspondiente de Fórmula (VII) se puede obtener mediante la adición de un ácido orgánico o inorgánico al compuesto de Fórmula (VI).
Preferentemente, se añade un ácido inorgánico, más preferentemente, un ácido inorgánico seleccionado de ácido ortofosfórico y ácido sulfúrico, incluso más preferentemente, el ácido inorgánico es ácido sulfúrico.
Dicho ácido inorgánico u orgánico se añade en una cantidad comprendida entre 3 y 10 equivalentes molares, preferentemente entre 4 y 9 equivalentes molares, más preferentemente entre 6 y 7 equivalentes molares, con respecto a la tetrahidroquinolina de Fórmula (VI). De acuerdo con un aspecto más preferido de la presente invención, la concentración del ácido está comprendida entre el 80 % y el 98 %, incluso más preferentemente entre el 90 % y el 97 %.
Dado que la disolución ácida es exotérmica, la temperatura de la mezcla de reacción se ha de controlar.
Por lo tanto, de acuerdo con el proceso actualmente reivindicado, la mezcla de reacción se mantiene preferentemente a una temperatura comprendida entre 10 °C y 60 °C, más preferentemente entre temperatura ambiente (25 °C) y 40 °C.
Preferentemente, la mezcla de reacción se deja reaccionar durante un tiempo comprendido entre 10 y 30 horas, más preferentemente entre 15 y 25 horas, incluso más preferentemente de aproximadamente 20 horas, con el fin de obtener una conversión sustancialmente completa de la tetrahidroquinolina en el derivado de indano correspondiente de Fórmula (VII).
Ventajosamente, una vez que se completa la reacción de reordenamiento de la Etapa c), el indano de acilo de Fórmula (VII) no se aísla y la mezcla de reacción se usa como tal en la siguiente Etapa d) de hidrólisis, tal como se indica, a continuación, en el Esquema 8.

En la Etapa d), la mezcla de reacción de la Etapa c) que contiene el indano de acilo de Fórmula (VII) se diluye con agua para obtener preferentemente una concentración de ácido en la mezcla de reacción comprendida entre el 30 % y el 70 % en peso, más preferentemente entre el 40 % y el 60 %, incluso más preferentemente de aproximadamente el 50 %.
La mezcla de reacción se lleva, a continuación, hasta una temperatura comprendida preferentemente entre 60 °C y
la temperatura de reflujo, más preferentemente entre 95 °C y 110 °C. Después de unas horas a la temperatura seleccionada, se forma el 4-aminoindano correspondiente, como sal de adición de ácido de Fórmula (VIII).
Dicha sal de Fórmula (VIII) se puede aislar y purificar de acuerdo con métodos bien conocidos por aquellos expertos en la materia, tales como precipitación o cristalización.
De acuerdo con un aspecto preferido de la presente invención, la sal se puede precipitar mediante la adición de agua y las impurezas orgánicas se pueden retirar mediante la adición de un disolvente orgánico, preferentemente un disolvente inmiscible con agua, tal como un hidrocarburo alifático, más preferentemente heptano.
Posteriormente, la sal de adición de ácido de 4-aminoindano (VIII) se suspende en agua y se añade una solución básica para obtener el 4-aminoindano deseado de Fórmula (I) en forma libre. La solución básica se añade preferentemente en una cantidad comprendida entre el 10 % y el 80 % de exceso molar, más preferentemente comprendida entre el 40 % y el 60 % de exceso molar, con respecto a la cantidad de la sal de Fórmula (VIII).
Los ejemplos no limitantes de bases adecuadas para la presente invención son los hidróxidos de metales alcalinos, tales como hidróxido de sodio e hidróxido de potasio, o los carbonatos de metales alcalinos.
Las bases particularmente preferidas de acuerdo con la presente invención son los hidróxidos de metales alcalinos. Después de la adición de la solución básica, la mezcla de reacción se deja reaccionar a una temperatura comprendida entre 35 °C y 70 °C, preferentemente entre 40 °C y 60 °C, más preferentemente de aproximadamente 55 °C.
Tal como conoce bien la persona experta, se pueden usar diferentes técnicas con el fin de aislar el producto deseado de Fórmula (I); por ejemplo, la mezcla de reacción se puede extraer mediante su mezclado con un disolvente orgánico ligeramente miscible o inmiscible con agua, preferentemente heptano, y la capa orgánica se puede filtrar para retirar los residuos sólidos.
De acuerdo con la presente invención, la solución orgánica que contiene el 4-aminoindano de Fórmula (I) obtenida al final de la Etapa (d) preferentemente no está concentrada y, ventajosamente, se puede usar como tal para la preparación de las amidas de aminoindano de Fórmula (II).
Un objeto adicional de la presente invención es un proceso para la preparación de amidas de aminoindano de Fórmula (II),
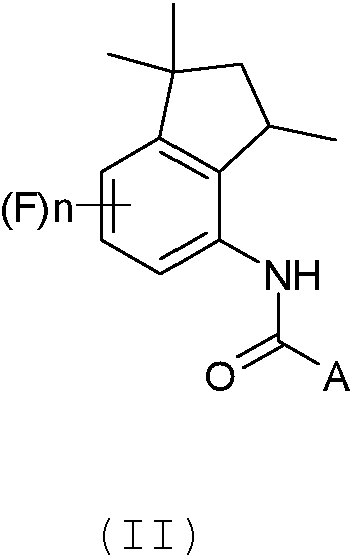
que comprende las etapas de:
- preparar al menos un derivado de 4-aminoindano de Fórmula (I) llevando a cabo las Etapas (a)-(d) del proceso descrito anteriormente;
- condensar dicho derivado de 4-aminoindano de Fórmula (I) con al menos un compuesto de Fórmula AC(O)X en donde, en dichas Fórmulas:
- A representa un grupo arilo C6-C10 o un anillo heterocíclico con 5 o 6 átomos que contienen de 1 a 3 heteroátomos seleccionados de N, O, S, estando estos grupos opcionalmente sustituidos por uno o más grupos R1 y R2;
- R1 representa un grupo alquilo C1-C6 o un grupo haloalquilo C1-C6 , estando dichos grupos opcionalmente sustituidos con uno o más grupos seleccionados de R', OR', S(O)mR'; o R1 representa un grupo cicloalquilo C3-Ca, un grupo cicloalquilalquilo C4-C9 , un grupo alquenilo C2-C6 , un grupo alquinilo C2-C6 , un grupo arilo C6-C10, un grupo arilalquilo C7-C12, un anillo heterocíclico con 5 o 6 átomos que contienen de 1 a 3 heteroátomos seleccionados de N, O, S, estando estos grupos opcionalmente sustituidos por uno o más grupos seleccionados
de átomos de halógeno, R', OR', NR'R", S(O)mR', CONR'R", COR', CO2R', CN, NO2;
- R2 representa un grupo alquilo C1-C6 o un grupo haloalquilo C1-C6 , estando dichos grupos opcionalmente sustituidos con uno o más grupos seleccionados de R', OR', S(O)mR'; o R2 representa un grupo cicloalquilo C3-Ca, un grupo cicloalquilalquilo C4-C9, un grupo arilo C6-C10, un grupo arilalquilo C7-C12, estando estos grupos opcionalmente sustituidos por uno o más grupos seleccionados de átomos de halógeno, R', OR', S(O)mR', NR'R", CONR'R", COR', CO2R', NO2, CN;
- R' y R'' representan un átomo de hidrógeno, un grupo alquilo C1-C4 , un grupo haloalquilo C1-C4;
- X representa un grupo hidroxilo, halógeno, un grupo alcoxi C1-C6 , un grupo alquilsulfoniloxi C1-C6, un grupo arilsulfoniloxi C6-C10, estando estos grupos opcionalmente sustituidos por uno o más átomos de halógeno;
- n es un número entero seleccionado dentro del intervalo de 0 a 3;
- m es un número entero seleccionado dentro del intervalo de 0 a 2.
Los ejemplos de un grupo alquilo C1-C6 son metilo, etilo, propilo, butilo, pentilo, hexilo.
Los ejemplos de un grupo haloalquilo C1-C6 son diclorometilo, difluorometilo, triclorometilo, trifluorometilo, clorodifluorometilo, dicloroetilo, trifluoroetilo, tetra-fluoroetilo, pentafluoroetilo, tetrafluoropropilo, pentafluoropropilo, diclorobutilo, difluorobutilo, dicloropentilo, difluoropentilo, diclorohexilo, difluorohexilo.
Los ejemplos de un grupo cicloalquilo C3-C6 son ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo.
Los ejemplos de un grupo cicloalquilalquilo C4-C9 son ciclopropilmetilo, ciclobutilmetilo, ciclopentilmetilo, ciclohexilmetilo, ciclohexiletilo, ciclohexilpropilo. Los ejemplos de un grupo alquenilo C2-C6 son etenilo, propenilo, butenilo, pentenilo, hexenilo.
Los ejemplos de un grupo alquinilo C2-C6 son etinilo, propinilo, butinilo, pentinilo, hexinilo.
Los ejemplos de un grupo arilo C6-C10 son fenilo, naftilo. Los ejemplos de un grupo arilalquilo C7-C12 son bencilo, feniletilo, fenilpropilo, fenilbutilo, fenilpentilo, fenilhexilo, naftilmetilo, naftiletilo.
Los ejemplos de un anillo heterocíclico con 5 o 6 átomos que contienen de 1 a 3 heteroátomos seleccionados de N, O, S son pirrolilo, pirazolilo, imidazolilo, triazolilo, tiadiazolilo, oxadiazolilo, furanilo, tiofenilo, piridilo, pirimidilo, triazinilo.
Los ejemplos de un anillo nitrogenado heterocíclico con 5 o 6 átomos son pirrolidilo, piperidilo, morfolilo.
Los ejemplos de átomos de halógeno son flúor, cloro, bromo, yodo.
Entre las amidas de aminoindano que tienen la Fórmula (II) general que se pueden preparar con el proceso de la presente invención, se prefieren aquellas en donde:
- A representa uno de los siguientes heterociclos A1-A5 :


- Ri representa un grupo alquilo C1-C6 , un grupo haloalquilo C1-C6 o un grupo fenilo opcionalmente sustituido con átomos de halógeno, grupos alquilo C1-C4, grupos haloalquilo C1-C4, grupos alcoxilo C1-C4 , grupos haloalcoxilo C1-C4 ,
- R2 representa un grupo alquilo C1-C6 , un grupo haloalquilo C1-C6 o un grupo fenilo opcionalmente sustituido con átomos de halógeno, grupos alquilo C1-C4, grupos haloalquilo C1-C4, grupos alcoxilo C1-C4 , grupos haloalcoxilo C1-C4 ;
particularmente preferidos son los productos que tienen la Fórmula (II), en donde:
- R1 representa un grupo alquilo C1-C6 o un fenilo opcionalmente sustituido con átomos de halógeno;
- R2 representa un metilo, un difluorometilo, un trifluorometilo o un fenilo opcionalmente sustituido con átomos de halógeno;
incluso más preferidos son los productos que tienen la Fórmula (II) general, en donde:
- A representa A1;
- R1 representa un metilo;
- R2 representa un metilo, un difluorometilo, un trifluorometilo.
De acuerdo con un aspecto preferido de la presente invención, se añade un derivado de ácido carboxílico de Fórmula AC(O)X a la solución de 4-aminoindano de Fórmula (I) obtenida en la Etapa d), preferentemente en una relación molar comprendida entre 0,9 y 1,1, más preferentemente entre 0,95 y 1,05, incluso más preferentemente en cantidad equimolar, con respecto a la cantidad de 4-aminoindano de Fórmula (I).
Preferentemente, dicho derivado de ácido carboxílico de Fórmula AC(O)X es un cloruro de ácido, es decir, X representa un átomo de cloro.
Después de la adición del derivado de ácido, la mezcla de reacción se lleva hasta una temperatura comprendida entre 60 °C y la temperatura de reflujo del disolvente de hidrocarburo, preferentemente entre 95 °C y 100 °C. Al final de la etapa de condensación, la mezcla de reacción se puede enfriar y se puede añadir una solución acuosa alcalina con el fin de neutralizar la acidez residual.
La amida de aminoindano de Fórmula (II) formada al final de la etapa de condensación se puede aislar y, posiblemente, purificar posteriormente de acuerdo con técnicas bien conocidas, por ejemplo, mediante precipitación y posterior filtración y lavado del producto sólido. El hecho de que el producto final de amida de aminoindano se pueda aislar mediante filtración a partir de la masa de reacción representa una ventaja adicional de la presente invención con respecto al proceso de preparación de la técnica anterior.
Los siguientes Ejemplos se proporcionan con fines ilustrativos de la presente invención.
PARTE EXPERIMENTAL
Ejemplo 1
a) Preparación de 6-fluoro-2,2,4-trimetil-1,2-dihidroquinolina (IV)
Reacción
En un matraz de fondo redondo de un litro con recubriente de nitrógeno se carga 4-fluoroanilina (III) (445 gramos, 4,0 mol), junto con HBF4 (solución acuosa al 48 %, 56 gramos, 0,32 mol) y 50 ml de acetona.
El matraz está equipado con un termómetro y un sistema de destilación de vidrio, que comprende una columna vertical llena de anillos de vidrio grueso y un condensador. Se emplea una agitación magnética eficaz.
El matraz se calienta en un baño externo, ajustado a la temperatura de 150 a 155 °C.
Cuando la temperatura interna alcanza los 120 °C y el condensado comienza a recogerse en el extremo de destilación, se inicia la adición de acetona. La acetona se alimenta a través de tubos de PTFE, terminando cerca de la parte inferior de la masa de reacción, a la velocidad constante de aproximadamente 150-175 ml/hora, para mantener una velocidad de destilación muy lenta. La temperatura interna de la reacción se mantiene en el intervalo de 132 a 140 °C. Se emplea un tiempo global de 8-10 horas para el suministro de acetona (1.500 ml), tras lo que la reacción se agita durante 20 min adicionales a 140 °C; a continuación, la masa de reacción se enfría por debajo de 40 °C.
Destilación
La destilación se realiza en el mismo recipiente de reacción, manteniendo la misma columna de fraccionamiento. La masa de reacción se somete a evaporación preliminar a 50 °C a 2-4 kPa (20-40 mbar) para completar la retirada de acetona y agua.
El vacío se aplica en el intervalo 0,2 a 0,3 kPa (2,0 a 3,0 mbar). El matraz se calienta, con la temperatura del baño externo aumentada progresivamente y alcanzando el máximo final de 170 °C, en el que se completa la recogida del producto, con el vacío aumentado hasta 0,1 kPa (1,0 mbar).
Se separan cuatro fracciones principales.
1. Destilado a 49-52 °C (temperatura de cabeza), constituido principalmente por fluoroanilina (aproximadamente el 98 %).
2. Destilado a 52-90 °C, pequeña cantidad de fracciones mixtas (4 gramos).
3. Destilado a 92-102 °C, que constituye el producto dihidroquinolina.
4. Residuo en el matraz: material denso oscuro (alquitranes), que se solidifica al enfriarse.
Los resultados del experimento representativo, incluyendo la recuperación de fluoroanilina, en términos de peso y rendimientos molares fueron:
- dihidroquinolina (como material destilado con aproximadamente el 95 % de pureza de GCA): 340 gramos, aproximadamente el 42 %
- rendimiento de dihidroquinolina (disponible en general): aproximadamente el 43 %
- rendimiento de 4-fluoroanilina recuperada (disponible en general): aproximadamente el 42 %
- rendimiento de dihidroquinolina, calculado sobre la 4-fluoroanilina consumida: aproximadamente el 72 %
La 4-fluoroanilina destilada y cualquier fracción mixta se envían a reciclar en un lote sucesivo.
b) Preparación de 6-fluoro-2,2,4-trimetil-1,2,3,4-tetrahidroquinolina (V)
La dihidroquinolina (IV) de líquido en bruto (96 % de pureza, 106 gramos, 0,530 mol) se disuelve en 400 ml de heptano y se extrae con 200 ml de ácido clorhídrico acuoso al 1 %. La capa acuosa se desecha y la solución orgánica se transfiere a un autoclave de hidrogenación de 1 litro. Se carga paladio catalítico sobre carbono (10 %, 2,5 gramos), se introduce gas de hidrógeno a una presión de 300 kPa (3 bares) y la reacción se realiza a 30 °C durante 2 horas.
Después de la filtración del catalizador, el disolvente se retira completamente mediante destilación, obteniendo así 100,0 gramos de tetrahidroquinolina (V) en bruto, que tiene el 98,5 % de pureza.
c) Preparación de 1-acetil-6-fluoro-2,2,4-trimetil-1,2,3,4-tetrahidroquinolina (VI)
En un matraz de fondo redondo de un litro con agitación mecánica con recubriente de nitrógeno se mezcla la tetrahidroquinolina (V) (98 % de pureza, 100,0 gramos, 0,506 mol) con anhídrido acético (60,0 gramos, 0,58 mol). La mezcla de reacción se calienta en un baño externo, ajustado a la temperatura de 150 °C, alcanzando la temperatura interna de 134 a 138 °C, a la que se mantiene durante 5 horas. Se puede añadir anhídrido acético en exceso adicional (1,0 g) para lograr la conversión completa.
La mezcla de reacción se refrigera hasta aproximadamente 40 °C y se añade agua (2,0 ml) para descomponer el exceso de anhídrido acético.
El líquido (ácido acético) se destila del recipiente de reacción a presión reducida (6 kPa (60 mbar) y, a continuación, 2 kPa (20 mbar); con la temperatura en el intervalo de 70-90 °C).
Por consiguiente, el residuo semisólido se recoge con heptano (300 ml) y se somete, de nuevo, a destilación completa a presión atmosférica, con la temperatura interna de 98-124 °C, con el fin de retirar completamente el ácido acético.
Se añade más heptano (150 ml), para obtener una solución completa a 95 °C. A continuación, la masa se deja enfriar lentamente hasta 20 °C con agitación, con el fin de causar la precipitación de un producto sólido finamente dividido.
d) Preparación de sulfato de 7-fluoro-1,1,3-trimetilindan-4-ilamina (VIII)
En un matraz de fondo redondo de un litro con agitación mecánica se carga inicialmente ácido sulfúrico (concentración del 93 %, 375 gramos, 3,50 mol).
La suspensión de heptano de acetil-tetrahidroquinolina (VI) (119 gramos, 0,50 mol) de la Etapa c se añade lentamente a la capa de ácido sulfúrico con agitación eficaz, al tiempo que la temperatura de masa se controla entre 15 y 20 °C. A continuación, la suspensión bifásica resultante se mantiene con agitación a 34-36 °C durante 20 horas. Se permite una hora adicional con la temperatura aumentada hasta 48-50 °C para completar la conversión.
A la masa de reacción se añade lentamente agua (320 ml) con agitación con fuerte exotermia (con el fin de obtener aproximadamente el 50 % de concentración de H2SO4). La masa de reacción se calienta progresivamente y el heptano se retira por destilación, con la recogida de la capa orgánica (170 ml) y un poco de agua.
A continuación, la solución se calienta hasta reflujo (110-111 °C, temperatura interna) y se mantiene 5 horas.
La masa de reacción se refrigera hasta 40 °C y se vierte lentamente en agua enfriada con hielo (1.000 gramos) con exotermia evidente (con el fin de obtener aproximadamente el 20 % de concentración de H2SO4) en un recipiente de 2 litros con agitación mecánica. La temperatura final se ajusta a aproximadamente 20 °C y la suspensión resultante de sal de sulfato de indanamina (VIII) se filtra. El sólido se lava en el filtro con 150 ml de agua, seguido de heptano (250 ml).
La torta de filtración se aspira con el filtro durante un tiempo suficiente, a continuación, el sólido húmedo (aproximadamente 180 gramos) se envía al desbloqueo de sal.
e) Preparación de 7-fluoro-1,1,3-trimetilindan-4-ilamina (I)
La sal sólida de sulfato de indanamina (cantidad total, 0,450 mol) se añade a 400 gramos de solución acuosa que contiene NaOH (28 gramos, 0,68 mol) en un matraz de 2 litros. A la suspensión alcalina se añade heptano (400 ml) y el conjunto se agita con calentamiento hasta 55 °C. Después de la disolución completa de sólidos, las fases se separan. La capa acuosa se extrae de nuevo con heptano (300 ml) a 55 °C. La solución de heptano caliente combinada (540 gramos) se filtra en una capa de Celite para retirar algo de material no disuelto.
Parte del heptano se retira por destilación a presión reducida (27,5 kPa (275 mbar), 57 °C), con el fin de retirar azeotrópicamente algunas trazas de agua y alcanzar el volumen adecuado para su uso en la siguiente Etapa. El peso final de la solución es de aproximadamente 460 gramos, que contiene aproximadamente 89,0 gramos de indanamina (I).
Ejemplo 2
a) Preparación de cloruro de 1-metil-3-(difluorometil-1H-pirazol-4-carbonilo (A = A1. en el que R1 = metilo, R2 =
difluorometilo)
El cloruro de ácido de pirazol se prepara a partir de ácido 3-difluorometil-1-metil-1H-pirazol-4-carboxílico y cloruro de tionilo en heptano poco antes de su uso.
En un matraz de fondo redondo de 500 ml provisto de un depurador alcalino se suspende ácido de pirazol (74,0 gramos, 0,42 mol) en heptano (170 ml). Se añaden dimetilformamida (0,70 gramos, 0,009 mol) y cloruro de tionilo (55,0 gramos, 0,462 mol) y la mezcla bifásica se agita y calienta a 42-45 °C.
Después de la conversión completa del ácido de pirazol (2,5 horas), el disolvente y el exceso de cloruro de tionilo se retiran por completo mediante destilación al vacío.
Se obtiene cloruro de ácido de pirazol líquido en forma de residuo (aproximadamente 81,0 gramos).
b) Preparación de 3-difluorometil-N-(7-fluoro-1,1,3-trimetil-4-indanil)-1-metil-4-pirazolcarboxamida (II), A = A1, en el que R1 = metilo, R2 = difluorometilo
Se emplea un reactor de vidrio de 2 litros con agitador mecánico, condensador de reflujo y depurador alcalino eficaces.
A la solución de heptano de indanamina (I) (cantidad total, 460 gramos, 0,46 mol de indanamina) a 50 °C se añade el cloruro de ácido de pirazol líquido obtenido en la Etapa a) durante aproximadamente 10 min a 50-70 °C con reacción exotérmica y formación de un precipitado. Se usa más heptano (20 ml) para el enjuague.
La reacción se agita a reflujo (temperatura interna de 95-97 °C) durante 4 horas, tras lo que se completa la reacción. El desprendimiento de HCl cesa en 3 horas.
La suspensión de reacción resultante se refrigera por debajo de 30 °C, a continuación, se añade solución acuosa de NaOH (215 ml, concentración del 2,5 %) y la mezcla se agita al menos durante 30 min, al tiempo que la temperatura se ajusta hasta 22 °C.
La suspensión se filtra en un filtro de vidrio sinterizado plano y el sólido se lava a 45 °C con agua (250 ml) después de volver a suspender y agitar.
El sólido resultante se filtra de nuevo y se lava en el filtro con agua (250 ml, o hasta que el pH resultante sea neutro) y sucesivamente con heptano (250 ml).
Después de la aspiración en el filtro durante 1 hora, el sólido húmedo (165 gramos) se seca en un horno de vacío a 55 °C.
Se obtiene el producto deseado (aproximadamente 140 gramos, con análisis del 98 %).
Los licores madre de heptano contienen exceso de indanamina, que se envía a reciclar.
Ejemplo 3 (comparativo)
a) Preparación de 6-fluoro-2,2,4-trimetil-1,2-dihidroquinolina

La Etapa 1 se realiza de acuerdo con el Ejemplo 1, Etapa a). Además, la dihidroquinolina destilada resultante se disuelve en un disolvente adecuado y se agita con una solución acuosa de ácido clorhídrico al 1 %, con el fin de retirar cualquier 4-fluoroanilina residual y alcanzar aproximadamente el 98 % de pureza. Dicho disolvente se retira mediante destilación al vacío antes de realizar la siguiente etapa.
b) Preparación de 1-acetil-6-fluoro-2,2,4-trimetil-1,2-dihidroquinolina
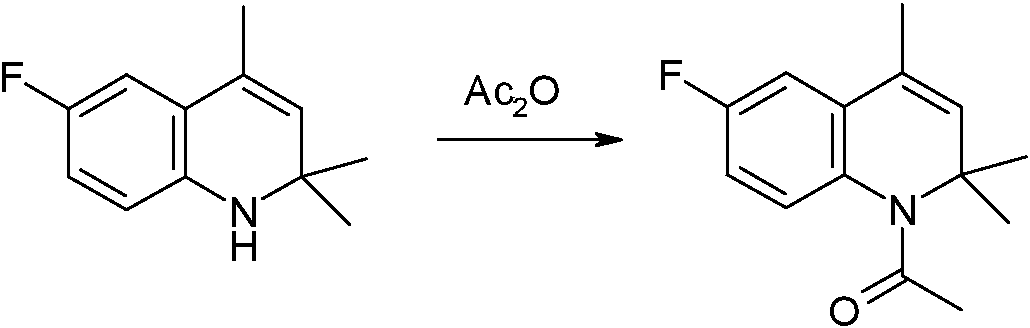
En un matraz de fondo redondo de un litro con agitación mecánica con recubriente de nitrógeno se mezcla dihidroquinolina (98 % de pureza, 104,0 gramos, 0,533 mol) con anhídrido acético (65,0 gramos, 0,635 mol).
La mezcla de reacción se calienta en un baño externo, ajustado a la temperatura de 150 °C, alcanzando la temperatura interna aproximadamente 138 °C, a la que se mantiene durante 5 horas para lograr la conversión completa.
La mezcla de reacción se refrigera hasta aproximadamente 40 °C y se añade agua (3,0 ml) para descomponer el exceso de anhídrido acético.
El líquido (ácido acético) se destila del recipiente de reacción a presión reducida (6 kPa (60 mbar) y, a continuación, 2 kPa (20 mbar); con la temperatura en el intervalo de 70-90 °C).
Por consiguiente, el residuo oleoso se recoge con heptano (300 ml) y se somete, de nuevo, a destilación completa a presión atmosférica, con la temperatura interna de 96-125 °C, con el fin de retirar completamente el ácido acético. Se añade más heptano (500 ml), para obtener una solución completa a 65 °C. La masa se mantiene a la temperatura de aproximadamente 50 °C y se transfiere a la siguiente fase.
c) Preparación de 1-acetil-6-fluoro-2,2,4-trimetil-1,2,3,4-tetrahidroquinolina

La solución orgánica se transfiere a un autoclave de hidrogenación de 1 litro con nitrógeno. Se carga paladio catalítico sobre carbono (10 %, 2,5 gramos), se introduce gas de hidrógeno a una presión de 300 kPa (3 bares) y la reacción se realiza a 50 °C durante 2 horas.
Después de la filtración del catalizador, el disolvente se retira parcialmente mediante destilación al vacío y, finalmente, se refrigera hasta 20 °C, obteniendo así 119 gramos de acetil-THQ en bruto, que tiene el 98 % de pureza, en forma de la suspensión de aproximadamente 240 gramos de heptano.
d) Preparación de 7-fluoro-1,1,3-trimetilindan-4-ilamina

En un matraz de fondo redondo de un litro con agitación mecánica se carga inicialmente ácido sulfúrico (concentración del 93 %, 375 gramos, 3,50 mol).
La suspensión de heptano de acetil-THQ (119 gramos, 0,50 mol) de la Etapa 3 se añade lentamente a la capa de ácido sulfúrico con agitación eficaz, al tiempo que la temperatura de masa se controla entre 15 y 20 °C. A continuación, la suspensión bifásica resultante se mantiene con agitación a 34-36 °C durante 20 horas.
Se permite una hora adicional con la temperatura aumentada hasta 48-50 °C para completar la conversión.
A la masa de reacción se añade lentamente agua (320 ml) con agitación con fuerte exotermia (con el fin de obtener aproximadamente el 50 % de concentración de H2SO4). La masa de reacción se calienta progresivamente y el heptano se retira por destilación, con la recogida de la capa orgánica (170 ml) y un poco de agua.
A continuación, la solución se calienta hasta reflujo (110-111 °C, temperatura interna) y se mantiene 5 horas.
La masa de reacción se refrigera hasta 40 °C y se vierte lentamente en agua enfriada con hielo (1.000 gramos) con exotermia evidente en un recipiente de 2 litros con agitación mecánica. La temperatura final se ajusta a aproximadamente 20 °C y la suspensión resultante de sal de sulfato de indanamina se filtra. El sólido se lava en el filtro con 150 ml de agua, seguido de heptano (250 ml).
A continuación, la torta de filtración en húmedo se añade a 400 gramos de solución acuosa que contiene NaOH (28 gramos, 0,68 mol) en un matraz de 2 litros. A la suspensión alcalina se añade heptano (400 ml) y el conjunto se agita con calentamiento hasta 55 °C. Después de la disolución completa de sólidos, las fases se separan. La capa acuosa se extrae de nuevo con heptano (300 ml) a 55 °C. La solución de heptano caliente combinada (540 gramos) se filtra en una capa de Celite para retirar algo de material no disuelto.
Cuando el heptano se retira por destilación completamente a presión reducida, se obtiene un residuo sólido oscuro, constituido por aproximadamente 89,0 gramos de base libre de indanamina, que tiene una pureza de por encima del 99 %.
Claims (13)
1. Un proceso para la preparación de derivados de 4-aminoindano de Fórmula (I), sales y enantiómeros de los mismos

que comprende las siguientes etapas:
a) hidrogenar una 1,2-dihidroquinolina de Fórmula (IV)

para dar una tetrahidroquinolina correspondiente de Fórmula (V)

b) acilar la tetrahidroquinolina de Fórmula (V) con un derivado de ácido carboxílico de Fórmula RC(O)LG para obtener un compuesto de derivado de acilo correspondiente de Fórmula (VI)

c) reordenar el compuesto de derivado de acilo de Fórmula (VI) en condiciones ácidas para dar un compuesto de indano de acilo de Fórmula (VII)
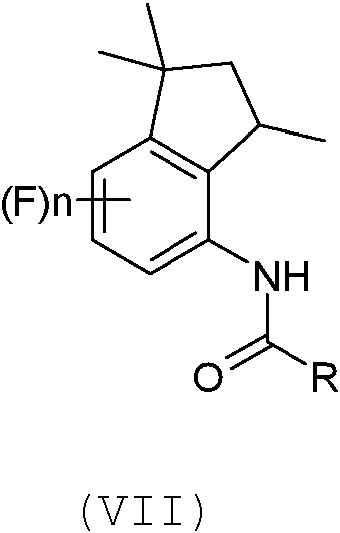
d) hidrolizar el grupo acilo del compuesto de indano de acilo de Fórmula (VII) para obtener el compuesto de 4-aminoindano de Fórmula (I),
en donde, en dichas Fórmulas:
- n es un número entero seleccionado dentro del intervalo de 0 a 3;
- R se selecciona de un grupo alquilo C1-C6 o un grupo arilo C6-C10, estando estos grupos opcionalmente sustituidos con uno o más de: grupos alquilo C1-C6, átomos de halógeno;
- LG es un grupo saliente seleccionado de: (i) un grupo hidroxi; (ii) un átomo de halógeno; (iii) un grupo alquilsulfoniloxi C1-C6 ; (iv) un grupo arilsulfoniloxi C6-C10, (v) un grupo RaCOO, en donde Ra es un grupo alquilo C1-C6 , estando los grupos (iii) -(v) opcionalmente sustituidos con uno o más átomos de halógeno.
2. El proceso de acuerdo con la reivindicación anterior, en donde dicha Etapa (a) de hidrogenación comprende poner en contacto dicha 1,2-dihidroquinolina de Fórmula (IV) disuelta en un disolvente orgánico, preferentemente un disolvente polar, con hidrógeno gaseoso en presencia de un catalizador de hidrogenación para obtener dicha tetrahidroquinolina de Fórmula (V).
3. El proceso de acuerdo con cualquiera de las reivindicaciones anteriores, en donde dicho derivado de ácido carboxílico de Fórmula RC(O)LG se selecciona preferentemente de cloruro de acetilo, anhídrido acético o una mezcla de los mismos.
4. El proceso de acuerdo con cualquiera de las reivindicaciones anteriores, en donde dicha Etapa (b) se lleva a cabo en ausencia de un disolvente añadido.
5. El proceso de acuerdo con cualquiera de las reivindicaciones anteriores, en donde la mezcla de reacción obtenida al final de dicha Etapa (b) se añade con un disolvente orgánico y, a continuación, se somete a destilación para retirar el exceso de dicho derivado de ácido carboxílico y para formar una suspensión que contiene el compuesto de derivado de acilo de Fórmula (VI).
6. El proceso de acuerdo con las reivindicaciones anteriores, en donde dicha suspensión que contiene dicho compuesto de derivado de acilo de Fórmula (VI) se alimenta a dicha Etapa (c).
7. El proceso de acuerdo con cualquiera de las reivindicaciones anteriores, en donde dicha Etapa (c) comprende suspender dicho compuesto de derivado de acilo de Fórmula (VI) en un disolvente orgánico y poner en contacto la suspensión así obtenida con un ácido orgánico o un ácido inorgánico, preferentemente seleccionado de ácido sulfúrico o ácido ortofosfórico, para obtener dicho compuesto de indano de acilo de Fórmula (VII) en forma de una sal de adición.
8. El proceso de acuerdo con cualquiera de las reivindicaciones anteriores, en donde dicha Etapa (d) de hidrólisis comprende poner en contacto dicho compuesto de indano de acilo de Fórmula (VII) con agua.
9. El proceso de acuerdo con la reivindicación 7, en donde dicha suspensión que contiene dicho compuesto de indano de acilo de Fórmula (VII) se alimenta a dicha Etapa (d).
10. El proceso de acuerdo con la reivindicación 7, en donde dicho compuesto de indano de acilo de Fórmula (VII) en forma de dicha sal de adición se pone en contacto con una solución acuosa alcalina para obtener dicho derivado de 4-aminoindano de Fórmula (I).
11. El proceso de acuerdo con cualquiera de las reivindicaciones 2, 5 y 7, en donde dicho disolvente orgánico se selecciona de: hidrocarburos alifáticos o cicloalifáticos; hidrocarburos clorados; hidrocarburos aromáticos; alcoholes; glicoles; ésteres o mezclas de los mismos.
12. El proceso de acuerdo con la reivindicación anterior, en donde dicho disolvente orgánico se selecciona de: hexano, heptanos, cloruro de metileno, dicloroetano, metanol, etanol, isopropanol, tolueno, acetato de etilo y mezclas de los mismos, preferentemente de heptano, dicloroetano, metanol, tolueno y mezclas de los mismos.
13. Un proceso para la preparación de amidas de aminoindano de Fórmula (II),

que comprende las etapas de:
- preparar al menos un derivado de 4-aminoindano de Fórmula (I) llevando a cabo las Etapas (a)-(d) del proceso de acuerdo con las reivindicaciones anteriores
- condensar dicho derivado de 4-aminoindano de Fórmula (I) con al menos un compuesto de Fórmula AC(O)X
en donde, en dichas Fórmulas:
- A representa un grupo arilo C6-C10 o un anillo heterocíclico con 5 o 6 átomos que contienen de 1 a 3 heteroátomos seleccionados de N, O, S, estando estos grupos opcionalmente sustituidos por uno o más grupos R1 y R2;
- R1 representa un grupo alquilo C1-C6 o un grupo haloalquilo C1-C6, estando dichos grupos opcionalmente sustituidos con uno o más grupos seleccionados de R', OR', S(O)mR'; o R1 representa un grupo cicloalquilo C3-C6, un grupo cicloalquilalquilo C4-C9 , un grupo alquenilo C2-C6 , un grupo alquinilo C2-C6 , un grupo arilo C6-C10, un grupo arilalquilo C7-C12, un anillo heterocíclico con 5 o 6 átomos que contienen de 1 a 3 heteroátomos seleccionados de N, O, S, estando estos grupos opcionalmente sustituidos por uno o más grupos seleccionados de átomos de halógeno, R', OR', NR'R", S(O)mR', CONR'R", COR', CO2R', CN, NO2;
- R2 representa un grupo alquilo C1-C6 o un grupo haloalquilo C1-C6, estando dichos grupos opcionalmente sustituidos con uno o más grupos seleccionados de R', OR', S(O)mR'; o R2 representa un grupo cicloalquilo C3-C6, un grupo cicloalquilalquilo C4-C9, un grupo arilo C6-C10, un grupo arilalquilo C7-C12, estando estos grupos opcionalmente sustituidos por uno o más grupos seleccionados de átomos de halógeno, R', OR', S(O)mR', NR'R", CONR'R", COR', CO2R', NO2, CN;
- R' y R'' representan un átomo de hidrógeno, un grupo alquilo C1-C4, un grupo haloalquilo C1-C4 ;
- X representa un grupo hidroxilo, halógeno, un grupo alcoxi C1-C6 , un grupo alquilsulfoniloxi C1-C6, un grupo arilsulfoniloxi C6-C10, estando estos grupos opcionalmente sustituidos por uno o más átomos de halógeno;
- n es un número entero seleccionado dentro del intervalo de 0 a 3;
- m es un número entero seleccionado dentro del intervalo de 0 a 2.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IB2016/052169 WO2017178868A1 (en) | 2016-04-15 | 2016-04-15 | Process for the preparation of 4-aminoindane derivatives and related aminoindane amides |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2806460T3 true ES2806460T3 (es) | 2021-02-17 |
Family
ID=55795012
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16717473T Active ES2806460T3 (es) | 2016-04-15 | 2016-04-15 | Proceso para la preparación de derivados de 4-aminoindano y amidas de aminoindano relacionadas |
Country Status (10)
| Country | Link |
|---|---|
| US (3) | US10640454B2 (es) |
| EP (1) | EP3442944B1 (es) |
| JP (1) | JP6780011B2 (es) |
| CN (1) | CN109071439B (es) |
| BR (1) | BR112018070345B1 (es) |
| DK (1) | DK3442944T3 (es) |
| ES (1) | ES2806460T3 (es) |
| HU (1) | HUE051536T2 (es) |
| PL (1) | PL3442944T3 (es) |
| WO (1) | WO2017178868A1 (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL3442944T3 (pl) * | 2016-04-15 | 2020-09-21 | Stichting I-F Product Collaboration | Sposób wytwarzania pochodnych 4-aminoindanu i pokrewnych aminoindanoamidów |
| CN108276329B (zh) * | 2018-04-02 | 2020-05-08 | 金凯(辽宁)化工有限公司 | 一种6-氟-2,2,4-三甲基-1,2-二氢喹啉的制备方法 |
| CN112119061A (zh) | 2018-05-23 | 2020-12-22 | 拜耳公司 | 制备取代的4-氨基茚满的方法 |
| IT201900017330A1 (it) | 2019-09-26 | 2021-03-26 | Isagro Spa | Processo per la preparazione di (r)-4-amminoindani e corrispondenti ammidi. |
| WO2022014414A1 (ja) * | 2020-07-17 | 2022-01-20 | 住友化学株式会社 | 光学活性な化合物の製造方法 |
| WO2024224297A1 (ko) * | 2023-04-25 | 2024-10-31 | 제이더블유중외제약 주식회사 | 4-아미노인단의 제조 방법 및 디하이드로인데닐-피라졸로[3,4-b]피리딘 아민 화합물의 제조 방법 |
Family Cites Families (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR920003893B1 (ko) * | 1984-10-29 | 1992-05-18 | 스미또모가가꾸고오교 가부시끼가이샤 | 피라졸 카르복스아미드 유도체의 제조방법 |
| DK383086A (da) | 1985-08-21 | 1987-02-22 | Rohm & Haas | (2-cyano-2-arylethyl)pyridiner, deres fremstilling og deres anvendelse som fungicider |
| ATE82966T1 (de) | 1986-08-12 | 1992-12-15 | Mitsubishi Chem Ind | Pyridincarboxamid-derivate und ihre verwendung als fungizides mittel. |
| US4837242A (en) | 1987-01-20 | 1989-06-06 | Sumitomo Chemical Company, Limited | Thiazoles and pyrazoles as fungicides |
| US4914097A (en) | 1987-02-25 | 1990-04-03 | Mitsubishi Kasei Corporation | N-indanyl carboxamide derivative and agricultural/horticultural fungicide containing the derivative as active ingredient |
| JP2582863B2 (ja) | 1988-06-13 | 1997-02-19 | 三菱化学株式会社 | N−インダニルカルボン酸アミド誘導体を有効成分とする灰色かび病防除剤 |
| JP2717852B2 (ja) | 1988-12-09 | 1998-02-25 | 株式会社紀文食品 | 冷凍すり身の鮮度測定法 |
| JPH02157266A (ja) | 1988-12-12 | 1990-06-18 | Mitsubishi Kasei Corp | N−インダニルカルボン酸アミド誘導体およびこれを有効成分とする農園芸用殺菌剤 |
| US5093347A (en) | 1991-01-28 | 1992-03-03 | Monsanto Company | 3-difluoromethylpyrazolecarboxamide fungicides, compositions and use |
| JP3077381B2 (ja) | 1992-05-14 | 2000-08-14 | 住友化学工業株式会社 | 木材防腐剤 |
| HUT63941A (en) | 1992-05-15 | 1993-11-29 | Hoechst Ag | Process for producing 4-alkyl-substituted pyrimidine-5-carboxanilide derivatives, and fungicidal compositions comprising same |
| EP0654464A1 (en) | 1993-10-22 | 1995-05-24 | Shell Internationale Researchmaatschappij B.V. | Processes for the preparation of pesticides and intermediates |
| US5521317A (en) | 1993-10-22 | 1996-05-28 | American Cyanamid Co. | Processes for the preparation of pesticides and intermediates |
| JPH1070479A (ja) | 1996-08-29 | 1998-03-10 | Nippon Seiki Co Ltd | Fm多重受信機 |
| JP3675112B2 (ja) | 1997-06-23 | 2005-07-27 | 富士写真フイルム株式会社 | 画像読取装置及び情報処理装置 |
| GB0001447D0 (en) | 2000-01-21 | 2000-03-08 | Novartis Ag | Organic compounds |
| KR20050037584A (ko) | 2002-08-22 | 2005-04-22 | 신젠타 파티서페이션즈 아게 | 살미생물성(예를 들어, 살진균성)1,2,3-트리아졸 유도체 |
| DE10250110A1 (de) | 2002-10-28 | 2004-05-13 | Bayer Cropscience Ag | Thiazol-(bi)cycloalkyl-carboxanilide |
| EP1599460B1 (de) | 2003-02-14 | 2007-11-07 | Bayer CropScience Aktiengesellschaft | Oxathiincarboxamide |
| KR20060009917A (ko) | 2003-05-21 | 2006-02-01 | 바이엘 크롭사이언스 아게 | 요오도피라졸릴 카복스아닐리드 |
| DE102004005785A1 (de) | 2004-02-06 | 2005-08-25 | Bayer Cropscience Ag | 2-Halogenfuryl/thienyl-3-carboxamide |
| ITMI20090488A1 (it) | 2009-03-27 | 2010-09-28 | Isagro Ricerca Srl | Composti benzammidici ad elevata attivita' fungicida e relativo uso |
| IT1403275B1 (it) | 2010-12-20 | 2013-10-17 | Isagro Ricerca Srl | Indanilanilidi ad elevata attività fungicida e loro composizioni fitosanitarie |
| ITMI20121045A1 (it) | 2012-06-15 | 2013-12-16 | Isagro Ricerca Srl | Composizioni sinergiche per la protezione di colture agrarie e relativo uso |
| EP2940001B1 (en) * | 2012-12-27 | 2017-05-03 | Sumitomo Chemical Company, Limited | Method for producing purified form of amine compound |
| PL3442944T3 (pl) * | 2016-04-15 | 2020-09-21 | Stichting I-F Product Collaboration | Sposób wytwarzania pochodnych 4-aminoindanu i pokrewnych aminoindanoamidów |
-
2016
- 2016-04-15 PL PL16717473T patent/PL3442944T3/pl unknown
- 2016-04-15 HU HUE16717473A patent/HUE051536T2/hu unknown
- 2016-04-15 EP EP16717473.9A patent/EP3442944B1/en active Active
- 2016-04-15 JP JP2018553223A patent/JP6780011B2/ja active Active
- 2016-04-15 WO PCT/IB2016/052169 patent/WO2017178868A1/en not_active Ceased
- 2016-04-15 ES ES16717473T patent/ES2806460T3/es active Active
- 2016-04-15 BR BR112018070345-1A patent/BR112018070345B1/pt active IP Right Grant
- 2016-04-15 DK DK16717473.9T patent/DK3442944T3/da active
- 2016-04-15 US US16/091,764 patent/US10640454B2/en active Active
- 2016-04-15 CN CN201680084469.5A patent/CN109071439B/zh active Active
-
2020
- 2020-03-31 US US16/836,038 patent/US11148995B2/en active Active
-
2021
- 2021-09-15 US US17/476,101 patent/US11851388B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| US20190119195A1 (en) | 2019-04-25 |
| DK3442944T3 (da) | 2020-07-13 |
| WO2017178868A1 (en) | 2017-10-19 |
| CN109071439A (zh) | 2018-12-21 |
| BR112018070345A2 (pt) | 2019-01-29 |
| EP3442944A1 (en) | 2019-02-20 |
| BR112018070345B1 (pt) | 2022-01-18 |
| US11851388B2 (en) | 2023-12-26 |
| US10640454B2 (en) | 2020-05-05 |
| CN109071439B (zh) | 2022-07-15 |
| US11148995B2 (en) | 2021-10-19 |
| EP3442944B1 (en) | 2020-04-15 |
| US20220002225A1 (en) | 2022-01-06 |
| JP2019511524A (ja) | 2019-04-25 |
| US20200231533A1 (en) | 2020-07-23 |
| HUE051536T2 (hu) | 2021-03-01 |
| JP6780011B2 (ja) | 2020-11-04 |
| PL3442944T3 (pl) | 2020-09-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2806460T3 (es) | Proceso para la preparación de derivados de 4-aminoindano y amidas de aminoindano relacionadas | |
| JP5817079B2 (ja) | 6−(7−((1−アミノシクロプロピル)メトキシ)−6−メトキシキノリン−4−イルオキシ)−n−メチル−1−ナフトアミド、またはそれの薬学的に許容される塩を調製するための方法 | |
| AU2015341788B2 (en) | Synthesis of copanlisib and its dihydrochloride salt | |
| KR20170039121A (ko) | 3-(3-클로로-1h-피라졸-1-일)피리딘의 제조 방법 | |
| TW201609694A (zh) | 用於製備3-(3-氯-1h-吡唑-1-基)吡啶的方法(一) | |
| JP2021504418A (ja) | 2−(5−メトキシイソクロマン−1−イル)−4,5−ジヒドロ−1h−イミダゾールおよびその硫酸水素塩の製造方法 | |
| EP4073056B1 (en) | Process for the preparation of lasmiditan and of a synthesis intermediate | |
| EP3351532A1 (en) | Method for preparing pimavanserin | |
| KR100574350B1 (ko) | 2-아미노피리딘 유도체의 제조방법 | |
| ES2227807T3 (es) | Proceso para la fabricacion de cloruro de arilsulfonilo. | |
| US8183385B2 (en) | SNAR process for preparing benzimidazole compounds | |
| CN109776498B (zh) | 一种西洛他唑的制备方法 | |
| US6426417B1 (en) | Processes and intermediates useful to make antifolates | |
| KR20120113263A (ko) | 인다졸-3-카르복실산 및 n-(s)-1-아자바이사이클로[2.2.2]옥트-3-일-1h-인다졸-3-카르복사미드 하이드로클로라이드 염의 제조 방법 | |
| JPH11269148A (ja) | 3,4−ジヒドロカルボスチリル類の製造方法 | |
| Ukrainets et al. | 4-Hydroxyquinol-2-ones. 86. Synthesis of Methyl (Ethyl) Esters of 1-Substituted 4-Amino-2-oxoquinoline-3-carboxylic Acids. | |
| CA3214107A1 (en) | New process for the synthesis of 5-{5-chloro-2-[(3s)-3- [(morpholin-4-yl)methyl]-3,4-dihydroisoquinoline-2(1h)- carbonyl]phenyl}-1,2-dimethyl-1h-pyrrole-3-carboxylic acid derivatives and its application for the production of pharmaceutical compounds | |
| CN105111209A (zh) | 一种氮杂吲哚啉化合物及其制备方法 | |
| EP0641782A1 (en) | Process for producing quinolonecarboxylic acid derivative | |
| HK1231871A1 (en) | A process for the preparation of compound and synthetic intermediates thereof | |
| DE1179943A (de) | Verfahren zur Herstellung von 5,6-Dihydro-6-oxo-llHpyrido [2,3-b] [1,4] - benzodiazepinen | |
| DE1179943B (de) | Verfahren zur Herstellung von 5,6-Dihydro-6-oxo-HH-pyrido-[2,3-b] [1,4] benzodiazepines | |
| PL85526B1 (es) | ||
| JP2004277353A (ja) | ピリジルピペリドン化合物の製造法 |