ES2772689T3 - Derivado de oxa espiro, método de preparación del mismo y aplicaciones del mismo en medicamentos - Google Patents
Derivado de oxa espiro, método de preparación del mismo y aplicaciones del mismo en medicamentos Download PDFInfo
- Publication number
- ES2772689T3 ES2772689T3 ES16854888T ES16854888T ES2772689T3 ES 2772689 T3 ES2772689 T3 ES 2772689T3 ES 16854888 T ES16854888 T ES 16854888T ES 16854888 T ES16854888 T ES 16854888T ES 2772689 T3 ES2772689 T3 ES 2772689T3
- Authority
- ES
- Spain
- Prior art keywords
- cycloalkyl
- heterocyclyl
- alkyl
- group
- heteroaryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 B[C@](*)(CC[C@]1*(CC*2CC3(CCCC3)OCC2)=C)CCc2c1cc*(*)c2 Chemical compound B[C@](*)(CC[C@]1*(CC*2CC3(CCCC3)OCC2)=C)CCc2c1cc*(*)c2 0.000 description 7
- UXYJYULDWRQJHG-UHFFFAOYSA-N CC(C)(C=C1)C=Nc2c1[n](C)nc2 Chemical compound CC(C)(C=C1)C=Nc2c1[n](C)nc2 UXYJYULDWRQJHG-UHFFFAOYSA-N 0.000 description 1
- KSGXWBUPARAZPX-UHFFFAOYSA-N CC1(C)Nc2ccncc2OC1 Chemical compound CC1(C)Nc2ccncc2OC1 KSGXWBUPARAZPX-UHFFFAOYSA-N 0.000 description 1
- VMPITZXILSNTON-UHFFFAOYSA-N COc1ccccc1N Chemical compound COc1ccccc1N VMPITZXILSNTON-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4402—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 2, e.g. pheniramine, bisacodyl
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4433—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4436—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a heterocyclic ring having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Un compuesto de fórmula (I): **(Ver fórmula)** o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, en el que: el anillo A se selecciona del grupo que consiste en cicloalquilo y heterociclilo; R se selecciona del grupo que consiste en arilo y heteroarilo, en el que el arilo y el heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo, heteroarilo, -OR3, - C(O)R3, -C(O)OR3, -S(O)mR3 y -NR4R5; cada R1 es idéntico o diferente y cada uno se selecciona independientemente del grupo que consiste en hidrógeno, alquilo, alcoxi, haloalquilo, halógeno, amino, nitro, hidroxi, ciano, cicloalquilo, heterociclilo, arilo, heteroarilo, -OR3, - C(O)R3, -C(O)OR3, -S(O)mR3 y -NR4R5, en la que el alquilo, haloalquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilo; cada R2 es idéntico o diferente y cada uno se selecciona independientemente del grupo que consiste en hidrógeno, alquilo, alcoxi, haloalquilo, halógeno, amino, nitro, hidroxi, ciano, oxo, alquenilo, cicloalquilo, heterociclilo, arilo, heteroarilo, -OR3, -C(O)R3, -C(O)OR3, -S(O)mR3 y - NR4R5, en la que el alquilo, alcoxi, alquenilo, haloalquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en deuterio, alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio; o dos R2 se toman juntos para formar un cicloalquilo o heterociclilo, en el que el cicloalquilo o heterociclilo está cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio; R3 se selecciona del grupo que consiste en hidrógeno, alquilo, alquilo deuterado, amino, alcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilo, en el que el alquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, halógeno, hidroxi, amino, nitro, ciano, alcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio; R4 y R5 se seleccionan cada uno independientemente del grupo que consiste en que consiste en hidrógeno, alquilo, alcoxi, hidroxialquilo, hidroxi, amino, alcoxicarbonilo, cicloalquilo, heterociclilo, arilo y heteroarilo, en la que el alquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, halógeno, hidroxi, amino, alcoxicarbonilo, nitro, ciano, alcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio; p y q están cada uno independientemente 0, 1, 2, 3 o 4; y m es 0, 1 o 2.
Description
DESCRIPCIÓN
Derivado de oxa espiro, método de preparación del mismo y aplicaciones del mismo en medicamentos
Campo de la invención
La presente invención pertenece al campo de la medicina, y se refiere a un derivado de oxa espiro, un método de preparación del mismo y usos del mismo en medicamentos. En particular, la presente invención se refiere a un derivado de oxa espiro representado por la fórmula (I), un método de preparación del mismo y una composición farmacéutica que comprende el derivado, uso del mismo como agonista del receptor MOR y uso del mismo en la preparación de un medicamento para tratar y/o prevenir el dolor y las enfermedades relacionadas con el dolor.
Antecedentes de la invención
El receptor de opioides es un importante receptor acoplado a proteínas G (GPCR), y es el objetivo de combinar péptidos opioides endógenos y fármacos opioides. Los receptores opioides activados juegan un papel regulador en el sistema inmune del sistema nervioso y endocrino. Los fármacos opioides son los analgésicos centrales más fuertes y de uso común. Los péptidos opioides endógenos son sustancias activas similares a los opioides presentes en los mamíferos. Actualmente, los péptidos opioides endógenos conocidos se pueden dividir aproximadamente en encefalina, endorfina, dinorfina y nociceptina (Pharmacol Rev 2007; 59: 88-123). Hay receptores opioides correspondientes en el sistema nervioso central, esto es, receptores |i (MOR), a (DOR), k (KOR) y similares. Se encuentra que la fuerza del efecto analgésico de los péptidos opioides endógenos depende principalmente del nivel de expresión de los receptores opioides. Los receptores opioides son los objetivos del efecto analgésico de los fármacos opioides y los péptidos opioides endógenos. Zadina et al encontraron que la capacidad de unión del receptor m Or al péptido de morfina 1 es más fuerte (360 pM). Es 4000 veces mayor que el receptor DOR al péptido de morfina 1, 15000 veces mayor que el receptor KOR al péptido de morfina 1. El receptor MOR es el receptor opioide más importante que media el efecto analgésico (Science, 2001, 293: 311-315; Biochem Biophys Res Commun 235:567-570; Life Sci 61:PL409-PL415).
Los estudios actuales sugieren que GPCR media y regula las funciones fisiológicas principalmente a través de dos vías: la ruta de la proteína G y la ruta de la p-arrestina. La ruta de señalización de la proteína G se puede activar mediante la unión del agonista tradicional de GPCR al receptor, e incluye el segundo sistema de mensajería tal como el ion calcio, adenil ciclasa (AC), proteínas quinasas activadas por mitógeno (MAPK) y similares. Mientras que la ruta de p-arrestina se activa principalmente por un ligando sesgado por p-arrestina. La respuesta de GPCR mediada por p-arrestina incluye principalmente tres aspectos: 1) la p-arrestina como un regulador negativo se hace reaccionar con el receptor de la quinasa acoplada a la proteína G (GRK), lo que provoca la desensibilización del receptor en los GPCR y bloquea la transducción de la señalización de la proteína G; 2) la p-arrestina como proteína adaptadora recluta la proteína endocítica e induce la endocitosis de GPCR; 3) p-arrestina como una proteína adaptadora forma un complejo con las moléculas señalizadoras de GPCR aguas abajo, y activa las moléculas de transducción de señales, tales como MAPK, proteína tirosina quinasa Src y Akt, etc. de manera independiente de la proteína G. Las diferencias de la estimulación del ligando en la señalización de la proteína G y/o la señalización de p-arrestina determinan en última instancia los efectos biológicos celulares específicos del ligando de GPCR.
MOR es el objetivo de los fármacos analgésicos opioides tales como la encefalina endógena y la morfina. Los primeros estudios han demostrado que la encefalina endógena y el fármaco opioide etorfina pueden volver agonista la proteína G y causar endocitosis del receptor, pero la morfina no puede causar endocitosis del receptor. Esto se debe a que la capacidad de la morfina para volver agonista la fosforilación de MOR es demasiado débil, y solo se reclutan trazas de p-arrestina en la membrana (Zhang et al., Proc Natl Acad Sci USA, 1998, 95 (12): 7157-7162). Estos ligandos ejercen sus funciones fisiológicas totalmente a través de la ruta de señalización de la proteína G en lugar de la ruta de p-arrestina. El estudio encontró que después de que se inyecta la morfina en ratones deficiencia genética de p-arrestina2, el efecto analgésico mediado por la señalización de la proteína G es más fuerte y la duración es más larga (Bohn et al., Science, 1999). Es previsible que si el sesgo negativo de p-arrestina de tales ligandos es más fuerte, incluso ellos pueden escapar de la desensibilización del receptor mediada por p-arrestina, lo que lleva a una mayor duración de la señalización de la proteína G y efectos analgésicos más potentes.
Las solicitudes de patente que describen agonistas de MOR incluyen las Publicaciones de Solicitud de Patente Internacional Nos. WO2014022733, WO2008009415, WO2009018169, WO2012129495, WO2001049650, WO2002020481, WO2010051476 y WO2013087589 y similares.
El uso a largo plazo de fármacos opioides produce efectos secundarios tales como tolerancia, depresión respiratoria y estreñimiento. Y se ha demostrado que estos efectos secundarios están estrechamente relacionados con la función de p-arrestina. Con el fin de reducir los efectos secundarios de los fármacos opioides, los fármacos se pueden diseñar en base al ligando sesgado por p-arrestina negativo en MOR, reduciendo así los efectos secundarios mediados por p-arrestina y potenciando el efecto terapéutico. En un estudio de los derivados de oxo espiro de la presente invención usados como fármacos MOR selectivos, Trevena Inc. descubrió que la actividad es
menor cuando el sustituyente está en la posición bencílica del arilo (J. Med. Chem. 2013, 56, 8019-8031), pero después de una serie de estudios, el presente inventor ha descubierto que los derivados de oxo espiro tienen una alta actividad después de que se cicla la posición bencílica, Emax también mejoró significativamente, hERG también mejoró significativamente y otros estudios encontraron que el compuesto con una sola configuración tiene una mayor selectividad para el MOR.
Resumen de la invención
La presente invención se dirige a un compuesto de fórmula (I-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos:

o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos,
en el que:
el anillo A se selecciona del grupo que consiste en cicloalquilo y heterociclilo;
R se selecciona del grupo que consiste en arilo y heteroarilo, en el que el arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo, heteroarilo, -OR3, -C(O)R3, -C(O)OR3, -S(O)mR3 y -NR4R5;
cada R1 es idéntico o diferente y cada uno se selecciona independientemente del grupo que consiste en hidrógeno, alquilo, alcoxi, haloalquilo, halógeno, amino, nitro, hidroxi, ciano, cicloalquilo, heterociclilo, arilo, heteroarilo, -OR3, -C(O)R3, -C(O)OR3, -S(O)mR3 y -NR4R5, en el que el alquilo, haloalquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio;
cada R2 es idéntico o diferente y cada uno se selecciona independientemente del grupo que consiste en hidrógeno, alquilo, alcoxi, haloalquilo, halógeno, amino, nitro, hidroxi, ciano, oxo, alquenilo, cicloalquilo, heterociclilo, arilo, heteroarilo, -OR3, -C(O)R3, -C(O)OR3, -S(O)mR3 y -NR4R5, en la que el alquilo, alcoxi, alquenilo, haloalquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en deuterio, alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio;
o dos R2 se toman juntos para formar un cicloalquilo o heterociclilo, en el que el cicloalquilo o heterociclilo está cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio; R3 se selecciona del grupo que consiste en hidrógeno, alquilo, alquilo deuterado, amino, alcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilo, en la que el alquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, halógeno, hidroxi, amino, nitro, ciano, alcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio;
R4 y R5 se seleccionan cada uno independientemente del grupo que consiste en hidrógeno, alquilo, alcoxi, hidroxialquilo, hidroxi, amino, alcoxicarbonilo, cicloalquilo, heterociclilo, arilo y heteroarilo, en el que el alquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, halógeno, hidroxi, amino, alcoxicarbonilo, nitro, ciano, alcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio;
p y q son cada uno independientemente 0, 1, 2, 3 o 4; y
m es 0, 1 o 2.
En una realización preferida de la presente invención, un compuesto de fórmula (I-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, es un compuesto de fórmula (I):

o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos,
en el que:
el anillo A, R, R1, R2, p y q son como se definen en la fórmula (I-A).
En una realización preferida de la presente invención, en un compuesto de fórmula (I) o fórmula (I-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, el anillo A se selecciona del grupo que consiste en heterociclilo de 5 a 6 miembros y cicloalquilo de 5 a 6 miembros.
En una realización preferida de la presente invención, en un compuesto de fórmula (I) o fórmula (I-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, R es piridilo
En una realización preferida de la presente invención, en un compuesto de fórmula (I) o fórmula (I-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, cada R1 es idéntico o diferente y cada uno se selecciona independientemente del grupo que consiste en hidrógeno y halógeno.
En una realización preferida de la presente invención, en un compuesto de fórmula (I) o fórmula (I-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, cada R2 es idéntico o diferente y cada uno se selecciona independientemente del grupo que consiste en hidrógeno, alquilo, oxo, alcoxi, hidroxi, halógeno y -OR3, en el que el alquilo y alcoxi están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en deuterio, alquilo, halógeno, hidroxi, amino, alcoxicarbonilo, nitro, ciano, alcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio; R3 se selecciona del grupo que consiste en hidrógeno, alquilo y cicloalquilo, en la que el alquilo es opcionalmente sustituido con halógeno o cicloalquilo.
En una realización preferida de la presente invención, un compuesto de fórmula (I-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, es un compuesto de fórmula (II-A),

o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos,
en el que:
G se selecciona del grupo que consiste en un enlace, CRaRb, C=O, NR4 y oxígeno;
Ra y Rb se seleccionan cada uno independientemente del grupo que consiste en hidrógeno, alquilo, alcoxi, haloalquilo, halógeno, amino, nitro, hidroxi, ciano, cicloalquilo, heterociclilo, arilo, heteroarilo, -OR3, -C(O)R3, -C(O)OR3, -S(O)mR3 y -NR4R5, en la que el alquilo, haloalquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilo, preferiblemente hidroxi o -OR3;
o Ra y Rb se toman juntos para formar un cicloalquilo o heterociclilo, en el que el cicloalquilo o heterociclilo está cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilo; R1 a R5, p, m y q son como se definen en la fórmula (I-A).
En una realización preferida de la presente invención, un compuesto de fórmula (II-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, es un compuesto de fórmula (II-B):

o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos,
en el que:
G se selecciona del grupo que consiste en un enlace, CRaRb, C = O, NR4 y oxígeno; y
R1, R2, R4, Ra, Rb, p y q son como se definen en la fórmula (II-A).
En una realización preferida de la presente invención, un compuesto de fórmula (II-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, es un compuesto de fórmula (II):

o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos,
en el que:
G se selecciona del grupo que consiste en un enlace, CRaRb, C = O, NR4 y oxígeno; y
Ra, Rb, R1, R2, R4, p y q son como se definen en la fórmula (II-A).
En una realización preferida de la presente invención, un compuesto de fórmula (II-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, es un compuesto de fórmula (IV-A):

o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos,
en la que:
R1, R2 y p son como se definen en la fórmula (II-A).
En una realización preferida de la presente invención, un compuesto de fórmula (II), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, es un compuesto de fórmula (IV):

o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos.
en la que:
R1, R2 y p son como se definen en la fórmula (II).
Los compuestos típicos de fórmula (I-A) incluyen, pero no se limitan a:

continuación

continuación

continuación

continuación

continuación

continuación

o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos.
En otro aspecto, la presente invención también se dirige a un procedimiento de preparación de un compuesto de fórmula (I-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, que comprende una etapa de:

hacer reaccionar un compuesto de fórmula (VA) o clorhidrato del mismo con un compuesto de fórmula (VIA) a través de una aminación reductora para obtener el compuesto de fórmula (I-A);
en el que:
el anillo A, R, R1, R2, p y q son como se definen en la fórmula (I-A).
En otro aspecto, la presente invención también se dirige a un procedimiento de preparación del compuesto de fórmula (I-A), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, que comprende una etapa de:

hacer reaccionar un compuesto de fórmula (VB-A) con un compuesto de fórmula (VIB-A) o clorhidrato del mismo mediante una aminación reductora para obtener el compuesto de fórmula (I-A);
en el que:
el anillo A, R, R1, R2, p y q son como se definen en la fórmula (I-A).
En otro aspecto, la presente invención también se dirige a un procedimiento de preparación del compuesto de fórmula (I), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, que comprende una etapa de:

hacer reaccionar un compuesto de fórmula (VB) con un compuesto de fórmula (VIB) o clorhidrato del mismo mediante una aminación reductora para obtener el compuesto de fórmula (I);
en el que:
el anillo A, R, R1, R2, p y q son como se definen en la fórmula (I).
En otro aspecto, la presente invención también se dirige a una composición farmacéutica que comprende una cantidad terapéuticamente eficaz del compuesto de cada una de las fórmulas mencionadas anteriormente, o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, y uno o más portadores, diluyentes o excipientes farmacéuticamente aceptables. La presente invención también se dirige a un procedimiento de preparación de la composición mencionada anteriormente que comprende una etapa de mezclar un compuesto representado por cada fórmula o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, con uno o más portadores, diluyentes o excipientes farmacéuticamente aceptables.
La presente invención se dirige además a un compuesto de cada fórmula, particularmente la fórmula (I), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, o una composición farmacéutica que comprende lo mismo, para uso del mismo como medicamento para volver agonista o antagonista el receptor MOR.
La presente invención se dirige además a un compuesto de cada fórmula, particularmente la fórmula (I), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, o una composición farmacéutica que comprende lo mismo, para uso del mismo en la prevención y/o tratamiento de una enfermedad mediada y relacionada con un receptor agonista de MOR, en la que la enfermedad se selecciona del grupo que consiste en dolor, disfunción inmune, inflamación, reflujo esofágico, trastornos neurológicos y psiquiátricos, enfermedades urinarias y reproductivas, enfermedades cardiovasculares y enfermedades respiratorias.
La presente invención se dirige además a un compuesto de cada fórmula, particularmente la fórmula (I), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, o una composición farmacéutica que comprende lo mismo, para uso del mismo en la prevención o el tratamiento del dolor y enfermedades relacionadas con el dolor en mamíferos, en el que el dolor puede ser dolor postoperatorio, dolor inducido por cáncer, dolor neuropático, dolor traumático y dolor inflamatorio, etc., en el que el cáncer se puede seleccionar del grupo que consiste en cáncer de mama, cáncer de endometrio, cáncer de cuello uterino, cáncer de piel, cáncer de próstata, cáncer de ovario, tumor de trompa de Falopio, tumor de ovario, hemofilia y leucemia.
La presente invención se dirige a un compuesto de cada fórmula, particularmente la fórmula (I) o un tautómero, mesómero, racemato, enantiómero, diastereómero o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, o la composición farmacéutica que comprende el mismo para uso como medicamento para el tratamiento de la disfunción inmune, inflamación, reflujo esofágico, trastornos neurológicos y psiquiátricos, trastornos urinarios y reproductivos, abuso de drogas y alcohol, gastritis y diarrea, enfermedades cardiovasculares, enfermedades respiratorias y tos.
Las composiciones farmacéuticas que contienen el ingrediente activo pueden estar en una forma apropiada para administración oral, por ejemplo, un comprimido, una pastilla, un comprimido para deshacer en la boca, una suspensión acuosa u oleosa, polvo o gránulo dispersable, emulsión, cápsula dura o blanda, o jarabe o elixir. Las composiciones orales se pueden preparar según cualquier método conocido en la técnica para la preparación de composiciones farmacéuticas. Tales composiciones pueden contener uno o más agentes seleccionados del grupo
que consiste en agentes edulcorantes, agentes aromatizantes, colorantes y conservantes, para proporcionar una formulación farmacéutica agradable y agradable al paladar. El comprimido contiene el ingrediente activo mezclado con excipientes no tóxicos farmacéuticamente aceptables apropiados para la fabricación de un comprimido.
Las formulaciones orales se pueden proporcionar como cápsulas de gelatina dura en las que el ingrediente activo se mezcla con un diluyente sólido inerte tal como carbonato de calcio, fosfato de calcio o caolín, o como cápsulas de gelatina blanda en las que el ingrediente activo se mezcla con un portador soluble en agua tal como el polietilenglicol o un medio oleoso tal como el aceite de maní, parafina líquida o aceite de oliva.
Una suspensión acuosa contiene el ingrediente activo mezclado con excipientes apropiados para la fabricación de una suspensión acuosa.
El ingrediente activo mezclado con los agentes dispersantes o humectantes, el agente de suspensión o uno o más conservantes se puede preparar como un polvo o gránulo dispersable apropiado para la preparación de una suspensión acuosa mediante la adición de agua. Los agentes dispersantes o humectantes apropiados y los agentes de suspensión se ejemplifican por los ya mencionados anteriormente. También se pueden agregar excipientes adicionales, tales como agentes edulcorantes, saborizantes y colorantes. Estas composiciones se pueden conservar agregando un antioxidante tal como ácido ascórbico.
La presente composición farmacéutica también puede estar en forma de una emulsión de aceite en agua.
La composición farmacéutica de la presente invención puede estar en forma de una solución acuosa estéril. Los vehículos o solventes aceptables que se pueden usar son agua, solución de Ringer y solución isotónica de cloruro de sodio.
La composición farmacéutica puede estar en forma de una suspensión acuosa o aceitosa inyectable estéril para administración intramuscular y subcutánea.
Los expertos en el arte saben bien que la dosificación de un fármaco depende de una variedad de factores que incluyen, pero no se limita a, los siguientes factores: actividad de un compuesto específico, edad del paciente, peso del paciente, salud general del paciente, comportamiento del paciente, dieta del paciente, tiempo de administración, ruta de administración, tasa de excreción, combinación de fármacos y similares. Además, el mejor tratamiento, tal como el modo de tratamiento, la dosis diaria del compuesto de fórmula (I) o el tipo de sal farmacéuticamente aceptable del mismo se puede verificar mediante regímenes terapéuticos tradicionales.
Descripción detallada de la invención
A menos que se indique lo contrario, los términos usados en la especificación y las reivindicaciones tienen los significados que se describen a continuación.
"Alquilo" se refiere a un grupo hidrocarburo alifático saturado que incluye grupos de cadena lineal y ramificada de C1 a C20, preferiblemente un alquilo que tiene de 1 a 12 átomos de carbono, y más preferiblemente un alquilo que tiene de 1 a 6 átomos de carbono. Ejemplos no limitantes incluyen metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, tert-butilo, sec-butilo, n-pentilo, 1,1-dimetilpropilo, 1,2-dimetilpropilo, 2,2-dimetilpropilo, 1 -etilpropilo, 2-metilbutilo, 3-metilbutilo, n-hexilo, 1-etil-2-metilpropilo, 1,1,2-trimetilpropilo, 1,1-dimetilbutilo, 1,2-dimetilbutilo, 2,2-dimetilbutilo, 1, 3-dimetilbutilo, 2-etilbutilo, 2-metilpentilo, 3-metilpentilo, 4-metilpentilo, 2,3-dimetilbutilo, n-heptilo, 2-metilhexilo, 3-metilhexilo, 4-metilhexilo, 5-metilhexilo, 2,3-dimetilpentilo, 2,4-dimetilpentilo, 2,2-dimetilpentilo, 3,3-dimetilpentilo, 2-etilpentilo, 3-etilpentilo, n-octilo, 2,3-dimetilhexilo, 2,4-dimetilhexilo, 2,5-dimetilhexilo, 2,2-dimetilhexilo, 3,3-dimetilhexilo, 4,4-dimetilhexilo, 2-etilhexilo, 3-etilhexilo, 4-etilhexilo, 2-metil-2-etilpentilo, 2-metil-3-etilpentilo, n-nonilo, 2-metil-2-etilhexilo, 2-metil-3-etilhexilo, 2,2-dietilpentilo, n-decilo, 3,3-dietilhexilo, 2,2-dietilhexilo, e isómeros ramificados de los mismos. Más preferiblemente, un grupo alquilo es un alquilo inferior que tiene de 1 a 6 átomos de carbono, y ejemplos no limitantes incluyen metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, tert-butilo, sec-butilo, n-pentilo, 1,1-dimetilpropilo, 1,2-dimetilpropilo, 2,2-dimetilpropilo, 1-etilpropilo, 2-metilbutilo, 3-metilbutilo, n-hexilo, 1-etil-2-metilpropilo, 1,1,2-trimetilpropilo, 1,1-dimetilbutilo, 1,2-dimetilbutilo, 2,2-dimetilbutilo, 1,3-dimetilbutilo, 2-etilbutilo, 2-metilpentilo, 3-metilpentilo, 4-metilpentilo, 2,3-dimetilbutilo y similares. El grupo alquilo puede estar sustituido o no sustituido. Cuando está sustituido, el grupo o grupos sustituyentes pueden estar sustituidos en cualquier punto de conexión disponible. El grupo o grupos sustituyentes son preferiblemente uno o más grupos seleccionados independientemente del grupo que consiste en alquilo, alquenilo, alquinilo, alcoxi, alquiltio, alquilamino, halógeno, tiol, hidroxi, nitro, ciano, cicloalquilo, heterociclilo, arilo, heteroarilo, cicloalcoxi, alcoxi heterocíclico, cicloalquiltio, alquiltio heterocíclico, oxo, carboxi y alcoxicarbonilo.
"Alquenilo" se refiere a un alquilo como se definió anteriormente que tiene al menos dos átomos de carbono y al menos un doble enlace carbono-carbono, por ejemplo, etenilo, 1-propenilo, 2-propenilo, 1-, 2- o 3-butenilo y similares. El grupo alquenilo puede estar sustituido o no sustituido. Cuando está sustituido, el grupo o grupos sustituyentes son preferiblemente uno o más grupos seleccionados independientemente del grupo que consiste en
alquilo, alquenilo, alquinilo, alcoxi, alquiltio, alquilamino, halógeno, tiol, hidroxi, nitro, ciano, cicloalquilo, heterocicNlo, arilo, heteroarilo, cicloalcoxi, alcoxi heterocíclico, cicloalquiltio y alquiltio heterocíclico.
"Cicloalquilo" se refiere a un grupo hidrocarburo monocíclico o policíclico saturado o parcialmente insaturado que tiene de 3 a 20 átomos de carbono, preferiblemente de 3 a 12 átomos de carbono, más preferiblemente de 3 a 6 átomos de carbono, y más preferiblemente de 5 a 6 átomos de carbono. Los ejemplos no limitantes de cicloalquilo monocíclico incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclopentenilo, ciclohexilo, ciclohexenilo, ciclohexadienilo, cicloheptilo, cicloheptatrienilo, ciclooctilo y similares. El cicloalquilo policíclico incluye un cicloalquilo que tiene un anillo espiro, anillo condensado o anillo en puente.
"Cicloalquilo de espiro" se refiere a un grupo policíclico de 5 a 20 miembros con anillos conectados a través de un átomo de carbono común (llamado átomo de espiro), en el que uno o más anillos pueden contener uno o más dobles enlaces, pero ninguno de los anillos tiene un sistema de electrones pi completamente conjugado, preferiblemente espiro cicloalquilo de 6 a 14 miembros, y más preferiblemente espiro cicloalquilo de 7 a 10 miembros. Según el número de átomos de espiro compartidos entre los anillos, el espiro cicloalquilo se puede dividir en cicloalquilo monoespiro, cicloalquilo diespiro o cicloalquilo poliespiro, y preferiblemente un cicloalquilo monoespiro o cicloalquilo diespiro, y más preferiblemente cicloalquilo monoespiro de 4 miembros/4 miembros, 4 miembros/5 miembros, 4 miembros/6 miembros, 5 miembros/5 miembros o 5 miembros/6 miembros. Los ejemplos no limitantes de espiro cicloalquilos incluyen:

"Cicloalquilo condensado" se refiere a un grupo policíclico de carbono de 5 a 20 miembros, en el que cada anillo del sistema comparte un par adyacente de átomos de carbono con otro anillo, en el que uno o más anillos pueden contener uno o más dobles enlaces, pero ninguno de los anillos tiene un sistema de electrones pi completamente conjugado, preferiblemente cicloalquilo condensado de 6 a 14 miembros, y más preferiblemente cicloalquilo condensado de 7 a 10 miembros. Según el número de anillos miembros, el cicloalquilo condensado se puede dividir en cicloalquilo condensado bicíclico, tricíclico, tetracíclico o policíclico, preferiblemente cicloalquilo condensado bicíclico o tricíclico, y más preferiblemente cicloalquilo condensado bicíclico de 5 miembros/5 miembros, o 5 miembros/6 miembros. Los ejemplos no limitantes de cicloalquilo condensado incluyen:

"Cicloalquilo en puente" se refiere a un grupo policíclico de carbono de 5 a 20 miembros, en el que cada dos anillos en el sistema comparten dos átomos de carbono desconectados, en el que los anillos pueden tener uno o más dobles enlaces, pero ninguno de los anillos tiene un sistema de electrones pi completamente conjugado, preferiblemente cicloalquilo en puente de 6 a 14 miembros, y más preferiblemente cicloalquilo en puente de 7 a 10 miembros. Según el número de anillos miembros, el cicloalquilo en puente se puede dividir en cicloalquilo en puente bicíclico, tricíclico, tetracíclico o policíclico, preferiblemente cicloalquilo en puente bicíclico, tricíclico o tetracíclico, y más preferiblemente cicloalquilo en puente bicíclico o tricíclico. Los ejemplos no limitantes de cicloalquilos en puente incluyen:

El anillo de cicloalquilo se puede condensar con el anillo de arilo, heteroarilo o heterociclilo, en el que el anillo unido a la estructura original es cicloalquilo. Ejemplos no limitantes incluyen indanilo, tetrahidronaftilo, benzocicloheptilo y similares, preferiblemente benzociclopentilo, tetrahidronaftilo. El cicloalquilo puede estar opcionalmente sustituido o no sustituido. Cuando está sustituido, el grupo o grupos sustituyentes son preferiblemente uno o más grupos seleccionados independientemente del grupo que consiste en alquilo, alquenilo, alquinilo, alcoxi, alquiltio, alquilamino, halógeno, tiol, hidroxi, nitro, ciano, cicloalquilo, heterociclilo, arilo, heteroarilo, cicloalcoxi, alcoxi heterocíclico, cicloalquiltio, alquiltio heterocíclico, oxo, carboxi y alcoxicarbonilo.
"Heterociclilo" se refiere a un grupo hidrocarburo monocíclico o policíclico saturado o parcialmente insaturado de 3 a 20 miembros que tiene uno o más heteroátomos seleccionados del grupo que consiste en N, O, y S(O)m (en el que m es un número entero de 0 a 2) como átomos del anillo, pero excluyendo -O-O-, -O-S- y -S-S- en el anillo, siendo los átomos del anillo restantes átomos de carbono. Preferiblemente, el heterociclilo tiene 3 a 12 átomos en el que 1 a 4 átomos son heteroátomos, más preferiblemente 3 a 8 átomos en el que 1 a 3 átomos son heteroátomos y más preferiblemente 5 a 6 átomos en el que 1 a 2 o 1 a 3 átomos son heteroátomos. Los ejemplos no limitantes de heterociclilo monocíclicos incluyen pirrolidinilo, imidazolidinilo, tetrahidrofuranilo, tetrahidropiranilo, tetrahidrotienilo, dihidroimidazolilo, dihidrofuranilo, dihidropirazolilo, dihidropirrolilo, piperidilo, piperazinilo, morfolinilo, tiomorfolinilo, homopiperazinilo y similares, preferiblemente tetrahidropiranilo, piperidilo o pirrolidinilo. El heterociclilo policíclico incluye un heterociclilo que tiene un anillo espiro, anillo condensado o anillo en puente.
El "espiro heterociclilo" se refiere a un heterociclilo policíclico de 5 a 20 miembros con anillos conectados a través de un átomo común (llamado átomo espiro), en el que los anillos tienen uno o más heteroátomos seleccionados del grupo que consiste en N, O y S(O)m (en el que m es un número entero de 0 a 2) como átomos en el anillo, siendo los átomos del anillo restantes átomos de carbono, en el que uno o más anillos pueden contener uno o más dobles enlaces, pero ninguno de los anillos tiene un sistema de electrones pi completamente conjugado, preferiblemente espiro heterociclilo de 6 a 14 miembros, y más preferiblemente espiro heterociclilo de 7 a 10 miembros. Según el número de átomos de espiro compartidos entre los anillos, el heterociclilo espiro se puede dividir en heterociclilo monoespiro, heterociclilo diespiro o heterociclilo poliespiro, preferiblemente heterociclilo monoespiro o heterociclilo diespiro, y más preferiblemente heterociclilo monoespiro de 4 miembros/4 miembros, 4 miembros/5 miembros, 4 miembros/6 miembros, 5 miembros/5 miembros o 5 miembros/6 miembros. Los ejemplos no limitantes de espiro heterociclilos incluyen:
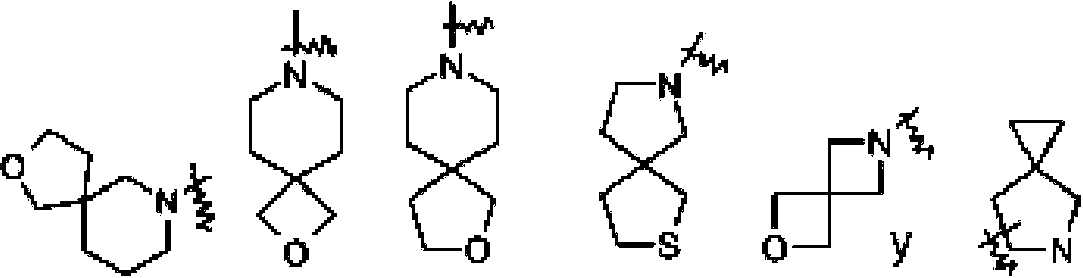
"Heterociclilo condensado" se refiere a un grupo heterociclilo policíclico de 5 a 20 miembros, en el que cada anillo en el sistema comparte un par de átomos adyacentes con otro anillo, en el que uno o más anillos pueden contener uno o más dobles enlaces, pero ninguno los anillos tienen un sistema de electrones pi completamente conjugado, y en el que los anillos tienen uno o más heteroátomos seleccionados del grupo que consiste en N, O y S(O)m (en el que m es un número entero de 0 a 2) como átomos del anillo, siendo los átomos del anillo restantes átomos de carbono; preferiblemente heterociclilo condensado de 6 a 14 miembros, y más preferiblemente heterociclilo condensado de 7 a 10 miembros. Según el número de anillos miembros, el heterociclilo condensado se puede dividir en heterociclilo condensado bicíclico, tricíclico, tetracíclico o policíclico, preferiblemente heterociclilo condensado bicíclico o tricíclico, y más preferiblemente heterociclilo condensado bicíclico de 5 miembros/5 miembros, o 5 miembros/6 miembros. Los ejemplos no limitantes de heterociclilo condensado incluyen:

"Heterociclilo en puente" se refiere a un grupo heterociclilo policíclico de 5 a 14 miembros, en el que cada dos anillos en el sistema comparten dos átomos desconectados, en el que los anillos pueden tener uno o más dobles enlaces, pero ninguno de los anillos tiene un sistema de electrones pi completamente conjugado, y los anillos tienen uno o más heteroátomos seleccionados del grupo que consiste en N, O y S(O)m (en el que m es un número entero de 0 a 2) como átomos del anillo, siendo los átomos del anillo restantes átomos de carbono, preferiblemente heterociclilo en puente de 6 a 14 miembros, y más preferiblemente heterociclilo en puente de 7 a 10 miembros. Según el número de anillos de miembros, el heterociclilo en puente se puede dividir en heterociclilo en puente bicíclico, tricíclico, tetracíclico o policíclico, preferiblemente heterociclilo en puente bicíclico, tricíclico o tetracíclico, y más preferiblemente heterociclilo en puente bicíclico o tricíclico. Los ejemplos no limitantes de heterociclilos en puente incluyen:

El anillo heterociclilo se puede condensar con el anillo de un arilo, heteroarilo o cicloalquilo, en el que el anillo unido a la estructura original es heterociclilo. Los ejemplos no limitantes incluyen:

etc.
El heterociclilo puede estar opcionalmente sustituido o no sustituido. Cuando está sustituido, el grupo o grupos sustituyentes son preferiblemente uno o más grupos seleccionados independientemente del grupo que consiste en alquilo, alquenilo, alquinilo, alcoxi, alquiltio, alquilamino, halógeno, tiol, hidroxi, nitro, ciano, cicloalquilo, heterociclilo, arilo, heteroarilo, cicloalcoxi, alcoxi heterocíclico, cicloalquiltio, alquiltio heterocíclico, oxo, carboxilo y alcoxicarbonilo.
"Arilo" se refiere a un anillo monocíclico de carbono de 6 a 14 miembros o un anillo condensado policíclico (esto es, cada anillo en el sistema comparte un par adyacente de átomos de carbono con otro anillo en el sistema) que tiene un sistema de electrones pi completamente conjugado, preferiblemente arilo de 6 a 10 miembros, y más preferiblemente arilo de 5 a 6 miembros, por ejemplo, fenilo y naftilo. El anillo de arilo se puede condensar con el anillo de heteroarilo, heterociclilo o cicloalquilo, en el que el anillo unido a la estructura original es el anillo de arilo. Los ejemplos no limitantes incluyen:

El arilo puede estar opcionalmente sustituido o no sustituido. Cuando está sustituido, el grupo o grupos sustituyentes son preferiblemente uno o más grupos seleccionados independientemente del grupo que consiste en alquilo, alquenilo, alquinilo, alcoxi, alquiltio, alquilamino, halógeno, tiol, hidroxi, nitro, ciano, cicloalquilo, heterociclilo, arilo, heteroarilo y cicloalcoxi, alcoxi heterocíclico, cicloalquiltio, alquiltio heterocíclico, carboxi, alcoxicarbonilo.
"Heteroarilo" se refiere a un sistema heteroaromático de 5 a 14 miembros que tiene de 1 a 4 heteroátomos seleccionados del grupo que consiste en O, S y N como átomos en el anillo, preferiblemente heteroarilo de 5 a 10 miembros que tiene 1 a 3 heteroátomos, y más preferiblemente heteroarilo de 5 o 6 miembros que tiene 1 a 2 heteroátomos, por ejemplo, imidazolilo, furilo, tienilo, tiazolilo, pirazolilo, oxazolilo, pirrolilo, tetrazolilo, piridilo, pirimidinilo, tiadiazolilo, pirazinilo y similares, preferiblemente imidazolilo, pirazolilo, pirimidinilo o tiazolilo, y más preferiblemente pirazolilo. El anillo heteroarilo se puede condensar con el anillo de un arilo, heterociclilo o cicloalquilo, en el que el anillo unido a la estructura original es el anillo heteroarilo. Los ejemplos no limitantes incluyen:
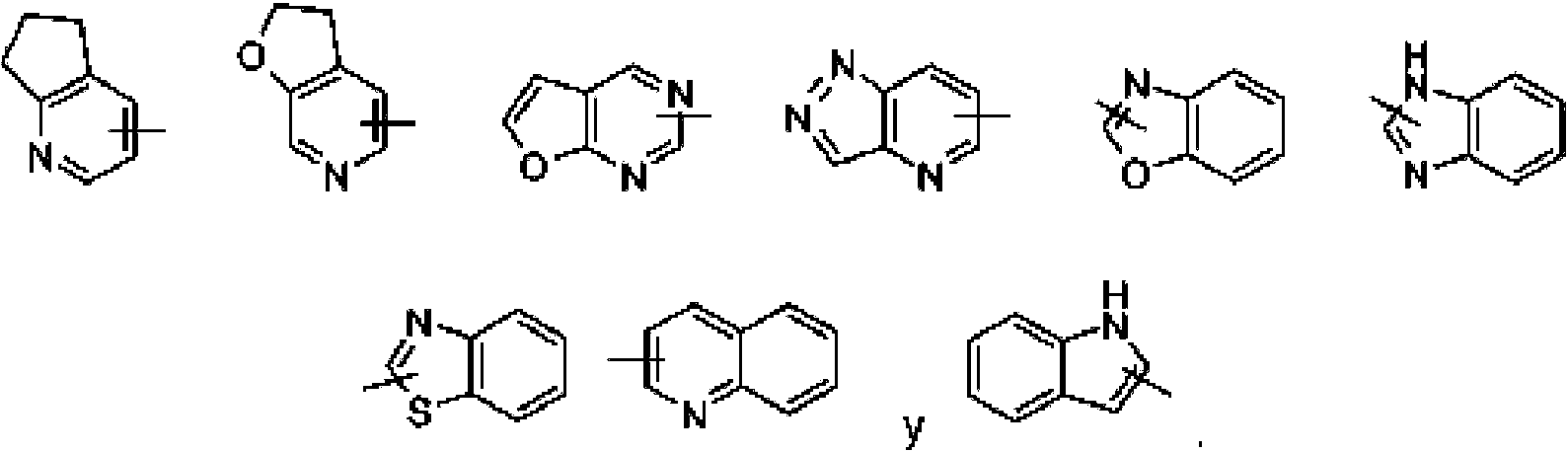
El heteroarilo puede estar opcionalmente sustituido o no sustituido. Cuando está sustituido, el grupo o grupos sustituyentes son preferiblemente uno o más grupos seleccionados independientemente del grupo que consiste en alquilo, alquenilo, alquinilo, alcoxi, alquiltio, alquilamino, halógeno, tiol, hidroxi, nitro, ciano, cicloalquilo, heterociclilo, arilo, heteroarilo, cicloalcoxi, alcoxi heterocíclico, cicloalquiltio, alquiltio heterocíclico, carboxi y alcoxicarbonilo. "Alcoxi" se refiere a un grupo -O-(alquilo) o un grupo -O-(cicloalquilo no sustituido), en el que el alquilo es como se definió anteriormente. Los ejemplos no limitantes incluyen metoxi, etoxi, propoxi, butoxi, ciclopropiloxi, ciclobutiloxi, ciclopentiloxi, ciclohexiloxi y similares. El alcoxi puede estar opcionalmente sustituido o no sustituido. Cuando está sustituido, el sustituyente es preferiblemente uno o más grupos seleccionados independientemente del grupo que consiste en alquilo, alquenilo, alquinilo, alcoxi, alquiltio, alquilamino, halógeno, tiol, hidroxi, nitro, ciano, cicloalquilo, heterociclilo, arilo, heteroarilo, cicloalcoxi, alcoxi heterocíclico, cicloalquiltio, alquiltio heterocíclico, carboxi y alcoxicarbonilo.
"Alquilo deuterado" se refiere a un alquilo sustituido por átomo(s) de deuterio, en el que el alquilo es como se definió anteriormente.
"Hidroxialquilo" se refiere a un alquilo sustituido con hidroxi(s), en el que el alquilo es como se definió anteriormente. "Hidroxi" se refiere a un grupo -OH.
"Halógeno" se refiere a flúor, cloro, bromo o yodo.
"Amino" se refiere a un grupo -NH2.
"Ciano" se refiere a un grupo -CN.
"Nitro" se refiere a un grupo -NO2.
"Carboxi" se refiere a un grupo -C(O)OH.
"Alcoxicarbonilo" se refiere a un grupo -C(O)O(alquilo) o -C(O)O(cicloalquilo), en el que el alquilo y el cicloalquilo son como se definieron anteriormente.
"Haluro de acilo" se refiere a un compuesto que comprende un grupo -C(O)-halógeno.
Todo "X se selecciona del grupo que consiste en A, B o C", "X se selecciona del grupo que consiste en A, B y C", "X es A, B o C", "X es A, B y C" y similares, tienen el mismo significado. Significa que X puede ser uno cualquiera o más de A, B y C. "Opcional" u "opcionalmente" significa que el evento o circunstancia descrita posteriormente puede, pero no necesariamente ocurrir, y esta descripción incluye la situación en la que el evento o las circunstancias ocurren o no. Por ejemplo, "el grupo heterocíclico opcionalmente sustituido por un alquilo" significa que un grupo alquilo puede estar presente, pero no necesariamente, y esta descripción incluye la situación del grupo heterocíclico siendo sustituido por un alquilo y el grupo heterocíclico siendo no sustituido por un alquilo.
"Sustituido" se refiere a uno o más átomos de hidrógeno en un grupo, preferiblemente hasta 5, más preferiblemente de 1 a 3 átomos de hidrógeno, independientemente sustituidos por un número correspondiente de sustituyentes. No hace falta decir que los sustituyentes solo existen en su posible posición química. El experto en el arte puede determinar si la sustitución es posible o imposible mediante experimentos o teoría sin pagar esfuerzos excesivos. Por ejemplo, la combinación de amino o hidroxi que tiene hidrógeno libre y átomos de carbono que tienen enlaces insaturados (tales como olefínicos) puede ser inestable.
Una "composición farmacéutica" se refiere a una mezcla de uno o más de los compuestos según la presente invención o sales o profármacos fisiológicamente/farmacéuticamente aceptables de los mismos y otros componentes químicos, y otros componentes tales como portadores y excipientes fisiológicamente/farmacéuticamente aceptables. El propósito de una composición farmacéutica es facilitar la administración de un compuesto a un organismo, que conduce a la absorción del ingrediente activo, mostrando así la actividad biológica.
Una "sal farmacéuticamente aceptable" se refiere a una sal del compuesto de la presente invención, que es segura y eficaz en mamíferos y tiene la actividad biológica deseada.
Método de síntesis del compuesto de la presente invención
Para alcanzar el objeto de la presente invención, la presente invención aplica las siguientes soluciones técnicas. Un procedimiento de preparación de un compuesto de fórmula (I-A) de la presente invención, o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, comprende la siguiente etapa:

hacer reaccionar un compuesto de fórmula (VA) o clorhidrato del mismo con un compuesto de fórmula (VIA) a través de una aminación reductora para obtener el compuesto de fórmula (I-A);
en el que:
el anillo A, R, R1, R2, p y q son como se definen en la fórmula (I-A).
El compuesto de fórmula (I-A) de la presente invención también se puede preparar de la siguiente manera:

hacer reaccionar un compuesto de fórmula (VB-A) con fórmula (VIB-A) o clorhidrato del mismo mediante una aminación reductora para obtener el compuesto de fórmula (I-A);
en el que:
el anillo A, R, R1, R2, p y q son como se definen en la fórmula (I-A).
En otro aspecto, la presente invención también se dirige a un procedimiento de preparación del compuesto de fórmula (I), o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos, que comprende una etapa de:

hacer reaccionar un compuesto de fórmula (VB) con un compuesto de fórmula (VIB) o clorhidrato del mismo mediante una aminación reductora para obtener el compuesto de fórmula (I);
en el que:
el anillo A, R, R1, R2, p y q son como se definen en la fórmula (I).
Realizaciones preferidas
La presente invención se describirá adicionalmente con referencia a los siguientes ejemplos, pero los ejemplos no deben considerarse como limitativos del alcance de la invención.
Ejemplos
Las estructuras de los compuestos se identifican por resonancia magnética nuclear (RMN) y/o espectrometría de masas (MS). Los desplazamientos químicos de RMN (8) se dan en 10'6 (ppm). La RMN está determinada por una máquina Bruker AVANCE-400. Los disolventes para la determinación son dimetil sulfóxido deuterado (DMSO-d6), cloroformo deuterado (CDCb) y metanol deuterado (CD3OD), y el patrón interno es tetrametilsilano (TMS).
La MS se determina mediante un espectrómetro de masas FINNIGAN LCQAd (ESI) (fabricante: Thermo, tipo: Finnigan LCQ Advantage MAX).
La cromatografía líquida de alta resolución (HPLC) se determina en un espectrómetro de cromatografía líquida de alta presión Agilent 1200DAD (columna cromatográfica Sunfire C18 150X4.6 mm) y un espectrómetro de cromatografía líquida de alta presión Waters 2695-2996 (columna cromatográfica Gimini C18150X4.6 mm).
Las tasas medias de inhibición de la quinasa y los valores de IC50 se determinan mediante un ELISA NovoStar (BMG Co., Alemania).
La placa de gel de sílice Yantai Huanghai HSGF254 o Qingdao GF254 se usa para la cromatografía en gel de sílice (TLC) de capa fina. La dimensión de la placa de gel de sílice usada en TLC es de 0.15 mm a 0.2 mm, y la dimensión de la placa de gel de sílice usada en la purificación del producto es de 0.4 mm a 0.5 mm.
El gel de sílice de malla 200 a 300 de Yantai Huanghai se usa como portador para la cromatografía en columna. Las materias primas conocidas de la presente invención se pueden preparar mediante métodos de síntesis convencionales conocidos en la técnica, o se pueden adquirir de ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc., o Dari chemical Company, etc.
A menos que se indique lo contrario, las reacciones se llevan a cabo bajo atmósfera de nitrógeno o atmósfera de argón.
El término "atmósfera de nitrógeno" o "atmósfera de argón" significa que un matraz de reacción está equipado con un globo de nitrógeno o argón de 1 L.
El término "atmósfera de hidrógeno" significa que un matraz de reacción está equipado con un globo de hidrógeno de 1 L.
Las reacciones de hidrogenación a presión se llevan a cabo con un instrumento de hidrogenación Parr 3916EKX y un generador de hidrógeno QL-500 o un instrumento de hidrogenación HC2-SS.
En las reacciones de hidrogenación, el sistema de reacción generalmente se aspira y se llena con hidrógeno, y la operación anterior se repite tres veces.
El reactor de microondas tipo CEM Discover-S 908860 se usa en la reacción de microondas.
A menos que se indique lo contrario, la solución usada en las reacciones se refiere a una solución acuosa.
A menos que se indique lo contrario, la temperatura de reacción en las reacciones se refiere a la temperatura ambiente, que varía desde 20 °C a 30 °C.
El procedimiento de reacción se controla por cromatografía en capa fina (TLC), y el sistema de disolvente en desarrollo incluye: A: sistema de diclorometano y metanol, B: sistema de n-hexano y acetato de etilo, C: sistema de diclorometano y acetona. La proporción del volumen del disolvente se puede ajustar según la polaridad de los compuestos.
El sistema de elución para la purificación de los compuestos por cromatografía en columna y cromatografía en capa fina incluye: A: sistema de diclorometano y metanol, B: sistema de n-hexano y acetato de etilo, C: sistema de diclorometano y acetona. La proporción del volumen del disolvente se puede ajustar según la polaridad de los compuestos, y algunas veces se puede agregar un poco de reactivo alcalino tal como la trietilamina o reactivo ácido tal como el ácido acético.
Ejemplos 1, 2
(S)-1-etil-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidroquinolin-4-amina 1
(R)-1-etil-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidroquinolin-4-amina 2

(R)-2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 1a (294 mg, 1.135 mmol), preparado mediante un método descrito en la Solicitud de Patente "WO2012129495") y 1-etil-1,2,3,4-tetrahidroquinolin-4-amina 1b (200 mg, 1.135 mmol, preparados mediante un método descrito en la Solicitud de Patente "WO2014078454") se disolvieron en 15 mL de diclorometano y la mezcla se agitó durante 1 hora. Luego, se agregó triacetoxiborohidruro de sodio (1.203 g, 5.675 mmol), y la mezcla resultante se agitó durante 16 horas. Se agregaron 20 mL de agua y la solución de reacción se extrajo con diclorometano (20 mLX3). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro y se filtraron. El filtrado se concentró a presión reducida, y el residuo resultante se purificó por cromatografía líquida de alta resolución para obtener el compuesto base 1-etil-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan- 9-il)etil)-1,2,3,4-tetrahidroquinolin-1 -amina, que luego se separó quiralmente (condiciones de separación: columna preparativa quiral Superchiral S-AD (Chiralway), 2 cm I.D. * 25 cm, 5 |im; fase móvil: CO2: metanol: dietanolamina = 75: 25: 0.05, velocidad de flujo: 50 g/min). Las fracciones correspondientes se recogieron y se concentraron a presión reducida para dar los compuestos base 1 (98 mg, un aceite de color marrón) y 2 (95 mg, un sólido de color amarillo).
Ejemplo 1:
MS m/z (ESI): 420.3 [M+1];
Análisis de HPLC quiral: tiempo de retención 4.028 min, pureza quiral: 99.7% (columna cromatográfica: Superchiral S-AD (Chiralway), 0.46 cm I.D. * 15 cm, 5 |im; fase móvil: CO2: metanol: dietanolamina = 75: 25: 0.05 (v/v/v)) 1H RMN (400 MHz, DMSO-d6) 88.54 (s, 1H), 7.72 (s, 1H), 7.45 (d, 1H), 7.20 (s, 1H), 6.95 (s, 1H), 6.78 (d, 1H), 6.52 (d, 1H), 6.37 (s, 1H), 3.60 (br, 2H), 3.18-3.43 (m, 3H), 2.99 (m, 1H), 2.33-2.45 (m, 3H), 1.77-1.99 (m, 3H), 1.19-1.60 (m, 12H), 1.00-1.06 (m, 4 H), 0.63 (m, 1H).
Ejemplo 2
MS m/z (ESI): 420.3 [M+1];
Análisis de HPLC quiral: tiempo de retención 3.725 minutos, pureza quiral: 99.8% (columna cromatográfica: Superchiral S-AD (Chiralway), 0.46 cm I.D. * 15 cm, 5 |im; fase móvil: CO2: metanol: dietanolamina = 75: 25: 0.05 (v/v/v))
1H RMN (400 MHz, DMSO-d6) 8 8.53 (s, 1H), 7.72 (s, 1H), 7.46 (d, 1H), 7.20 (s, 1H), 6.97 (s, 1H), 6.85 (d, 1H), 6.54 (d, 1H), 6.40 (s, 1H), 3.61 (br, 2H), 3.17-3.25 (m, 3H), 3.00-3.01 (m, 1H), 2.33-2.46 (m, 3H), 1.78-1.97 (m, 3H), 1.24 1.65 (m, 12H), 1.01-1.06 (m, 4 H), 0.61 (m, 1H).
Ejemplo 3
(1R,2R)-1-((2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-2,3-dihidro -1H-inden-2-ol 3

1a (50 mg, 0.193 mmol) y (1R,2R)-1-amino-2,3-dihidro-1H inden-2-ol 3a (31.6 mg, 0.212 mmol, preparado mediante un método descrito en la Solicitud de la Patente "WO2010148191") se disolvieron en 15 mL de diclorometano, se agregó una cantidad apropiada de metanol para mejorar la solubilidad. La mezcla resultante se agitó durante 1 hora a temperatura ambiente, luego se agregó triacetoxiborohidruro de sodio (200 mg, 0.965 mmol). Después de agitar durante 16 horas, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 3 (50 mg, rendimiento 66%) como un sólido de color blanco.
MS m/z (ESI): 393.5 [M+1]
1H RMN (400 MHz, DMSO-d6) 88.51 (d, 1H), 7.73-7.66 (m, 1H), 7.37 (d, 1H), 7.28-7.20 (m, 3H), 7.19-7.12 (m, 2H), 4.75 (d, 1H), 4.61 (d, 1H), 3.82-3.71 (m, 4H), 3.41-3.31 (m, 2H), 2.30-2.89 (m, 2H), 2.41-2.25 (m, 2H), 1.96-1.90 (m, 2H), 1.85-1.61 (m, 4H), 1.61-1.25 (m, 6H).
Ejemplo 4
(1R,2R)-2-metoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-2,3-dihidro-1H-inden-1-amina 4

((1R,2R)-2-metoxi-2,3-dihidro-1H-inden-1-il)carbamato de tert-butilo 4b
((1R,2R)-2-hidroxi-2,3-dihidro-1H-inden-1-il)carbamato de tert-butilo 4a (350 mg, 1.34 mmol, preparado mediante un método bien conocido descrito en "Angewandte Chemie-International Edition, 2012, 51(34), 8495-8499") se disolvió en 15 mL de diclorometano, luego óxido de plata (930 mg, 4.02 mmol), yodometano (0.25 mL, 4.02 mmol) y se agregó una pequeña cantidad de tamices moleculares 4Á. La mezcla resultante se agitó durante 16 horas a temperatura ambiente, se filtró y el filtrado se concentró a presión reducida. El residuo resultante se purificó por cromatografía en capa fina con el sistema de elución B para obtener el compuesto base 4b (200 mg, rendimiento 57%) como un sólido de color blanco.
MS m/z (ESI): 208.2 [M-56+1]
Etapa 2
(1R,2R)-2-metoxi-2,3-dihidro-1H-inden-1-amina 2,2,2-trifluoroacetato 4c
Se disolvieron 4b (60 mg, 0.228 mmol) en 5 mL de diclorometano, luego se agregaron 0.5 mL de ácido trifluoroacético. Después de agitar durante 2 horas, la solución de reacción se concentró a presión reducida para obtener el compuesto base en bruto 4c (66 mg) como un aceite de color amarillo, que se usó directamente en la siguiente etapa sin purificación adicional.
MS m/z (ESI): 164.2 [M+1]
Etapa 3
(1R,2R)-2-metoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-2,3-dihidro-1H-inden-1-amina 4
Se disolvieron 1a (50 mg, 0.193 mmol) y el 4c en bruto (66 mg, 0.228 mmol) en 15 mL de diclorometano. La mezcla resultante se agitó durante 30 minutos a temperatura ambiente, luego se agregó triacetoxiborohidruro de sodio (200 mg, 0.965 mmol). Después de agitar durante 16 horas, la mezcla de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 4 (25 mg, rendimiento 32%) como un aceite de color amarillo claro.
MS m/z (ESI): 407.3 [M+1]
1H RMN (400 MHz, DMSO-d6) 88.55 (d, 1H), 7.71 (d, 1H), 7.58 (d, 1H), 7.40 (d, 1H), 7.28 (d, 1H), 7.25-7.10 (m, 3H), 4.39 (d, 1H), 4.26 (d, 1H), 3.82-3.70 (m, 5H), 3.30 (s, 3H), 2.88-2.30 (m, 2H), 2.40-2.26 (m, 2H), 1.96-1.91 (m, 2H), 1.85-1.62 (m, 4H), 1.61-1.24 (m, 6H).
Ejemplo 5
N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)croman-4-amina

(R) -2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 5a (20 mg, 0.08 mmol, preparado mediante un método descrito en la Solicitud de Patente "WO2012129495") y croman-4-amina 5b (23 mg, 0.15 mmol, preparado mediante un método descrito en " Bioorganic & Medicinal Chemistry Letters, 2011, 21(5), 1338-1341") se disolvieron en 10 mL de diclorometano, y la mezcla se agitó durante 2 horas. Luego, se agregó triacetoxiborohidruro de sodio (65 mg, 0.31 mmol), y la mezcla resultante se agitó durante 12 horas. La solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 5 (6 mg, rendimiento 20%) como un aceite de color amarillo.
MS m/z (ESI): 393.5 [M+1]
1H RMN (400 MHz, Metanol-d4) 88.55 (s, 1H), 7.78 (t, 1H), 7.52 (d, 1H), 7.27 (d, 1H), 7.01-7.12 (m, 2H), 6.66-6.85 (m, 2H), 4.05-4.23 (m, 2H), 3.71-3.86 (m, 2H), 3.59-3.69 (m, 1H), 2.51-2.65 (m, 2H), 2.37-2.47 (m, 1H), 1.98-2.17 (m, 2H), 1.84-1.96 (m, 2H), 1.37-1.83 (m, 9H), 1.24-1.35 (m, 1H), 1.05-1.17 (m, 1H), 0.65-0.71 (m, 1H).
Ejemplo 6
(S) -N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)croman-4-amina

(R)-2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 5a (80 mg, 0.31 mmol) y clorhidrato de (S)-cromano-4-amina 6a (86 mg, 0.46 mmol, preparado mediante un método descrito en " ACS Catalysis, 3(4), 555-559; 2013") se disolvió en 10 mL de una mezcla de diclorometano y metanol (V:V = 5:1), y la mezcla se agitó durante 1 hora. Luego, se agregó triacetoxiborohidruro de sodio (263 mg, 1.24 mmol), y la mezcla resultante se agitó durante 12 horas. La solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 6 (36 mg, rendimiento 32.1%) como un sólido viscoso de color blanco.
MS m/z (ESI): 393.5 [M+1]
1H RMN (400 MHz, Metanol-d4) 88.55 (d, 1H), 7.80-7.76 (m, 1H), 7.53 (d, 1H), 7.26-7.25 (m, 1H), 7.05-7.01 (m, 2H), 6.78-6.70 (m, 2H), 4.17-4.10 (m, 2H), 3.79-3.63 (m, 3H), 2.56-2.42 (m, 3H), 2.19-2.10 (m, 2H), 1.92-1.82 (m, 2H), 1.80-1.44 (m, 12H).
Ejemplo 7
(R)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)croman-4-amina

(R)-2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 5a (80 mg, 0.31 mmol), preparado mediante un método descrito en la Solicitud de Patente "WO2012129495"), clorhidrato de (R)-croman-4-amina 7a (115 mg, 0.62 mmol, preparado mediante un método descrito en "European Journal of Organic Chemistry, 2014(31), 7034-7038, 2014") y triacetoxiborohidruro de sodio (197 mg, 0.93 mmol) se disolvieron en 10 mL de una mezcla de diclorometano y metanol (V:V = 5:1), y la mezcla se agitó durante 12 horas. La solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 7 (30 mg, rendimiento 24.8%) como un aceite de color amarillo claro.
MS m/z (ESI): 393.5 [M+1]
1H RMN (400 MHz, Metanol-d4) 88.63 (d, 1H), 7.93 (t, 1H), 7.64 (d, 1H), 7.39 (t, 1H), 7.29 (t, 1H), 7.19 (d, 1H), 6.81 6.97 (m, 2H), 4.25-4.35 (m, 1H), 4.14-4.24 (m, 1H), 3.79 (d, 2H), 2.47-2.65 (m, 3H), 2.13-2.32 (m, 3H), 1.87-2.03 (m, 2H), 1.72-1.85 (m, 2H), 1.40-1.71 (m, 5H), 1.25-1.35 (m, 2H), 1.06-1.15 (m, 1H), 0.66-0.75 (m, 1H).
Ejemplo 8
6-fluoro-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)croman-4-amina

(R)-2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 5a (30 mg, 0.12 mmol), preparado mediante un método descrito en la Solicitud de Patente "WO2012129495") y 6-fluorocroman-4-amina 8a (39 mg, 0.23 mmol, preparados mediante un método descrito en "Bioorganic & Medicinal Chemistry Letters, 2011, 21(5), 1338-1341") se disolvieron en 20 mL de diclorometano, luego se agregó triacetoxiborohidruro de sodio (74 mg, 0.35 mmol). Después de agitar durante 12 horas, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 8 (10 mg, rendimiento 20.4%) como un sólido de color amarillo claro.
MS m/z (ESI): 411.2 [M+1]
1H RMN (400 MHz, CDCla) 88.56 (d, 1H), 7.67-7.64 (m, 1H), 7.34-7.31 (m, 1H), 7.16-7.14 (m, 1H), 6.84-6.74 (m, 2H), 6.73-6.7 (m, 1H), 4.02-4.08 (m, 2H), 3.78-3.75 (m, 3H), 2.66-2.12 (m, 6H), 2.1-1.59 (m, 9H), 1.35-1.18 (m, 4H). Ejemplo 9
(R)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidronaftalen-1-amina

(R)-2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 5a (35 mg, 0.14 mmol, preparado mediante un método descrito en la Solicitud de Patente "WO2012129495") y (R)-1,2,3,4-tetrahidronaftalen-1-amina 9a (40 mg, 0.27 mmol, preparado mediante un método descrito en "Angewandte Chemie-International Edition, 45(28), 4641-4644, 2006") se disolvieron en 5 mL de diclorometano. La mezcla resultante se agitó durante 1 hora, luego se agregó triacetoxiborohidruro de sodio (144 mg, 0.68 mmol). Después de agitar durante 1 hora, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 9 (15 mg, rendimiento 27.5%) como un sólido de color amarillo.
MS m/z (ESI): 391.2 [M+1]
1H RMN (400 MHz, CDCla) 88.57 (d, 1H), 7.65 (t, 1H), 7.32 (d, 1H), 7.16 (d, 1H), 7.11-7.07 (m, 3H), 7.05 (d, 1H), 3.77 (d, 2H), 3.60-3.57 (br, 1H), 2.73-2.70 (m, 3H), 2.45 (d, 1H), 2.34 (d, 1H), 2.15-2.08 (m, 1H), 2.05-2.02 (m, 1H), 1.91 (d, 1H), 1.75 -1.70 (m, 12H), 1.50-1.44 (m, 3H).
Ejemplo 10
(S)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidronaftalen-1-amina

(R) -2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 5a (20 mg, 0.14 mmol, preparado mediante un método descrito en la Solicitud de Patente "WO2012129495") y (S)-1,2,3,4-tetrahidronaftalen-1-amina 10a (50 mg, 0.272 mmol, preparado mediante un método descrito en "Angewandte Chemie-International Edition, 45(28), 4641-4644, 2006") se disolvieron en 20 mL de diclorometano. La mezcla resultante se agitó durante 1 hora, luego se agregó triacetoxiborohidruro de sodio (144 mg, 0.68 mmol). Después de agitar durante 1 hora, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 10 (15 mg, rendimiento 28.3%) como un sólido de color amarillo.
MS m/z (ESI): 391.2 [M+1]
1H RMN (400 MHz, Metanol-d4) 88.77 (d, 1H), 8.28 (t, 1H), 7.92 (d, 1H), 7.71 (t, 1H), 7.33-7.19 (m, 4H), 4.38 (t, 1H), 3.80-3.74 (m, 2H), 3.23-3.11 (m, 1H), 3.08-2.98 (m, 1H), 2.87-2.82 (m, 2H), 2.56-2.48 (m, 3H), 2.26-2.0 4(m, 5H), 1.85-1.81 (m, 3H), 1.56-1.32 (m, 5H), 1.34-1.31 (m, 1H), 0.82-0.79 (m, 1H).
Ejemplo 11
(S) -4-((2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-3,4-dihidronaftalen-1(2H)-ona

(S)-tert-butil (1,2,3,4-tetrahidronaftalen-1-il)carbamato 11a
(S)-1,2,3,4-tetrahidronaftalen-1-amina 10a (3 g, 20.41 mmol, preparado mediante un método descrito en "Angewandte Chemie-International Edition, 45(28), 4641-4644, 2006") se disolvió en 100 mL de diclorometano, luego se agregaron trietilamina (5.7 mL, 40.82 mmol) y dicarbonato de di-tert-butilo (4.9 g, 22.45 mmol). Después de agitar durante 12 horas, la solución de reacción se lavó con agua (100 mL) y solución saturada de bicarbonato de sodio (100 mL) sucesivamente. La fase orgánica se secó sobre sulfato de sodio anhidro, se filtró y el filtrado se concentró a presión reducida para obtener el compuesto en bruto 11a (5.6 g) como un aceite de color amarillo claro, que se usó directamente en la siguiente etapa sin purificación adicional.
MS m/z (ESI): 248.3 [M+1]
Etapa 2
(S)-tert-butil (4-oxo-1,2,3,4-tetrahidronaftalen-1-il)carbamato 11b
El (S)-tert-butil (1,2,3,4-tetrahidronaftalen-1-il)carbamato en bruto 11a (5.6 g, 20.41 mmol) se disolvió en 90 mL de una mezcla de acetona y agua (V/V = 2: 1), luego se agregó sulfato de magnesio (5.5 g, 45.66 mmol) y se agregó lentamente con agitación permanganato de potasio (7.22 g, 45.66 mmol). El sistema de reacción se agitó durante 12 horas. La solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en columna de gel de sílice con el sistema de elución B para obtener el compuesto base 11b (3.1 g, rendimiento 52%) como un sólido de color blanco crema.
MS m/z (ESI): 262.3 [M+1]
Etapa 3
(S)-4-amino-3,4-dihidronaftalen-1(2H)-ona 11 c
El (S)-tert-butil (4-oxo-1,2,3,4-tetrahidronaftalen-1-il)carbamato 11b (1 g, 3.83 mmol) se disolvió en 20 mL de diclorometano, luego se agregaron 8 mL de cloruro de hidrógeno 4 M en solución de 1,4-dioxano. Después de agitar durante 2 horas, la solución de reacción se concentró a presión reducida, se agregaron 10 mL de etanol al residuo resultante, y se agregó gota a gota amoniaco acuoso al 30% para ajustar el pH a 8. La mezcla se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 11c (400 mg, rendimiento 64.8%) como un material viscoso de color verde.
MS m/z (ESI): 162.3 [M+1]
Etapa 4
(S)-4-((2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-3,4-dihidronaftalen-1(2H)-ona 11
(S)-4-amino-3,4-dihidronaftalen-1(2H)-ona 11c (200 mg, 1.24 mmol) y (R)-2-(9-(piridin-2-il)-6-oxaspiro[ 4.5]decan-9-il)acetaldehído 5a (268 mg, 1.04 mmol, preparado mediante un método descrito en la Solicitud de Patente "WO2012129495") se disolvieron en 20 mL de diclorometano, y la mezcla se agitó durante 1 hora, luego se agregó triacetoxiborohidruro de sodio (1.1 g, 5.18 mmol). Después de agitar durante 2 horas, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 11 (136 mg, rendimiento 32.4%) como un sólido de color blanco.
MS m/z (ESI): 405.6 [M+1]
1H RMN (400 MHz, Metanol-d4) 88.73 (d, 1H), 8.15-8.09 (m, 2H), 7.83 (d, 1H), 7.81-7.69 (m, 3H), 7.47 (d, 1H), 4.45 (t, 1H), 3.77-3.74 (m, 2H), 3.03-2.98 (m, 1H), 2.75-2.68 (m, 3H), 2.51-2.44 (m, 5H), 2.05-2.01 (m, 2H), 1.57-1.48 (m, 7H), 1.20-1.05 (m, 1H), 0.80-0.77 (m, 1H).
Ejemplo 12 y Ejemplo 13
(1S,4S)-4-((2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-1,2,3,4-tetra hidronaftalen-1-ol 12 (1R,4S)-4-((2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-1,2,3,4-tetr ahidronaftalen-1-ol 13

Se disolvió (S)-4-((2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-3,4-dihidronaftalen-1(2H)-ona 11 (50 mg, 0.12 mmol) en 10 mL de diclorometano, se agregaron 0.29 mL de solución de hidróxido de diisobutil aluminio 1 M gota a gota a -78 °C y la mezcla se agitó durante 2 horas a -78 °C. Se agregaron 5 mL de metanol para apagar la reacción. La solución de reacción se calentó a temperatura ambiente y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener los compuestos base 12 (18 mg, rendimiento 35.3%) como un sólido viscoso de color blanco crema y 13 (20 mg, rendimiento 39.2%) como un sólido viscoso de color blanco crema.
12: MS m/z (ESI): 407.6 [M+1],
1H RMN (400 MHz, CDCb) 88.51(d, 1H), 7.50(t, 1H), 7.36(d, 1H), 7.33-7.30(m, 3H), 7.21-7.18 (m, 2H), 4.83(t, 1H), 4.25(t, 1H), 3.81-3.75(m, 2H), 2.85-2.83(m, 1H), 2.36-2.30(m, 5H), 1.98-1.80(m, 2H), 1.78-1.60(m, 9H), 1.48-1.25(m, 5H).
13: MS m/z (ESI): 407.6 [M+1],
1H RMN (400 MHz, CDCla) 88.51(d, 1H), 7.50(t, 1H), 7.36(d, 1H), 7.33-7.30(m, 3H), 7.21-7.18 (m, 2H), 4.83(t, 1H), 4.25(t, 1H), 3.81-3.75(m, 2H), 2.85-2.83(m, 1H), 2.36-2.30(m, 5H), 1.98-1.80(m, 2H), 1.78-1.60(m, 9H), 1.48-1.25(m, 5H).
Ejemplo 14
(1S,4S)-4-metoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidronaftalen-1-amina

Etapa 1
((1S,4S)-4-hidroxi-1,2,3,4-tetrahidronaftalen-1-il)carbamato de tert-butilo 14a
El (S)-tert-butil (4-oxo-1,2,3,4-tetrahidronaftalen-1-il)carbamato 11b (100 mg, 0.883 mmol) se disolvió en 5 mL de tolueno, la reacción se enfrió a 0 °C, se agregó con (R) -2-metil-CBS-oxazaborolidina (0.1 mL, 0.076 mmol), y se agitó durante 5 minutos. Luego, se agregó sulfato de metil borano (0.88 mL, 0.76 mmol) y la reacción se agitó durante 2 horas. La reacción se inactivó mediante la adición de 50 mL de solución saturada de cloruro de sodio y se extrajo con acetato de etilo (30 mL X 3). Las fases orgánicas se combinaron, se lavaron con solución saturada de cloruro de sodio (30 mL X 3), se secaron sobre sulfato de sodio anhidro y se filtraron. El filtrado se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 14a (60 mg, rendimiento 60%) como un sólido de color blanco.
MS m/z (ESI): 208.3 [M-55]
Etapa 2
((1S,4S)-4-metoxi-1,2,3,4-tetrahidronaftalen-1-il)carbamato de tert-butilo 14b
El compuesto en bruto 14a (30 mg, 0.11 mmol) se disolvió en 4 mL de diclorometano, luego se agregaron óxido de plata (76 mg, 0.33 mmol) y yoduro de metilo (62 mg, 0.44 mmol). Después de agitar durante 48 horas, la solución de reacción se filtró. El filtrado se concentró a presión reducida para obtener el compuesto base en bruto 14b (30 mg) como un aceite de color amarillo, que se usó directamente en la siguiente etapa sin purificación adicional.
MS m/z (ESI): 278.4 [M+1].
Etapa 3
Clorhidrato de (1S,4S)-4-metoxi-1,2,3,4-tetrahidronaftalen-1-amina 14c
El compuesto en bruto 14b (30 mg, 0.11 mmol) se disolvió en 0.5 mL de diclorometano, luego se agregó 1 mL de una solución de cloruro de hidrógeno 4 M en 1,4-dioxano. La reacción se agitó durante 2.5 horas. La solución de reacción se concentró a presión reducida para obtener el compuesto base en bruto 14c (24 mg) como un sólido de color blanco, que se usó directamente en la siguiente etapa sin purificación adicional.
MS m/z (ESI): 178.4 [M+1].
Etapa 4
(1S,4S)-4-metoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidronaftalen-1-amina 14 El compuesto 5a (29 mg, 0.11 mmol), el compuesto en bruto 14c (24 mg, 0.11 mmol) y sulfato de sodio se disolvieron en 4 mL de metanol, y la mezcla se agitó durante 12 horas. Luego, se agregó borohidruro de sodio (8 mg, 0.22 mmol), y la mezcla se agitó durante 15 minutos. La solución de reacción se concentró a presión reducida y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 14 (4 mg, rendimiento 8.7%) como un sólido de color blanco.
MS m/z (ESI): 407.6 [M+1]
1H RMN (400 MHz, CDCla) 88.56 (d, 1H), 7.66 (t, 1H), 7.33 (d, 1H), 7.15 (d, 1H), 7.08-7.06 (m, 3H), 7.04 (d, 1H), 3.76 (d, 2H), 3.61-3.58 (br, 1H), 3.41 (s, 3H), 2.74-2.72 (m, 3H), 2.46 (d, 1H), 2.32 (d, 1H), 2.13-2.08 (m, 1H), 2.03 2.00 (m, 1H), 1.90 (d, 1H), 1.75 -1.72 (m, 11H), 1.51-1.46 (m, 3H).
Ejemplo 15
(R)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-2,3-dihidro-1H-inden-1-amina

(R) -2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 5a (20 mg, 0.08 mmol) y (clorhidrato R)-2,3-dihidro- 1H-inden-1-amina 15a (27 mg, 0.16 mmol, preparado mediante un método descrito en "Síntesis, (14), 2283-2287, 2008") se disolvieron en 10 mL de diclorometano, y la mezcla se agitó durante 2 horas, luego se agregó triacetoxiborohidruro de sodio (51 mg, 0.24 mmol). Después de agitar durante 12 horas, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 15 (5 mg, rendimiento 16.7%) como un aceite de color amarillo.
MS m/z (ESI): 377.5 [M+1]
1H RMN (400 MHz, Metanol-d4) 88.62 (d, 1H), 7.91 (t, 1H), 7.60 (d, 1H), 7.37 (s, 4H), 7.35 (d, 1H), 4.64-4.70 (m, 1H), 3.76 (d, 2H), 2.91-3.15 (m, 2H), 2.41-2.60 (m, 4H), 1.85-2.11 (m, 4H), 1.70-1.81 (m, 2H), 1.41-1.69 (m, 5H), 1.31-1.39 (m, 1H), 1.10-1.20 (m, 1H), 0.71-0.80 (m, 1H).
Ejemplo 16
(S) -N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-2,3-dihidro-1H-inden-1 -amina

(R)-2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 5a (20 mg, 0.08 mmol) y clorhidrato de (S)-2,3-dihidro-1H-inden-1-amina 16a (26 mg, 0.15 mmol, preparado mediante un método descrito en "Tetrahedron Asymmetry, 14(22), 3479-3485; 2003") se disolvió en 10 mL de diclorometano, y la mezcla se agitó durante 2 horas, luego se agregó triacetoxiborohidruro de sodio (49 mg, 0.23 mmol). Después de agitar durante 12 horas, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 16 (5 mg, rendimiento 17%) como un aceite de color amarillo MS m/z (ESI): 377.5 [M+1]
1H RMN (400 MHz, Metanol-d4) 88.63 (d, 1H), 7.90 (t, 1H), 7.60 (d, 1H), 7.38 (s, 4H), 7.35 (d, 1H), 4.65-4.70 (m, 1H), 3.76 (d, 2H), 2.90-3.16 (m, 2H), 2.40-2.60 (m, 4H), 1.85-2.10 (m, 4H), 1.70-1.80 (m, 2H), 1.40-1.69 (m, 5H), 1.30-1.39 (m, 1H), 1.10-1.20 (m, 1H), 0.70-0.80 (m, 1H).
Ejemplo 17
(1S,2S)-2-metoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-2,3-dihidro-1H-inden-1-amina

trifluoroacetato de (1S,2S)-2-metoxi- 2,3-dihidro-1H-inden-1-amina 17b
((1S,2S)-2-metoxi-2,3-dihidro-1H-inden-1-il)carbamato de tert-butilo 17a (110 mg, 0.42 mmol, preparado mediante un método descrito en la Solicitud de Patente "WO2008080015") se disolvió en 5 mL de diclorometano, luego se agregó 1 mL de ácido trifluoroacético. Después de agitar durante 2 horas, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en columna de gel de sílice con el sistema de elución A para obtener el compuesto base en bruto 17b (70 mg, rendimiento 60.3%) como un aceite de color amarillo.
MS m/z (ESI): 164.1 [M+1].
Etapa 2
(15.25) -2-metoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-2,3-dihidro-1H-inden-1-amina 17
Se disolvieron (R)-2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 5a (25 mg, 0.96 mmol) y trifluoroacetato de (1S,2S)-2-metoxi- 2,3-dihidro-1H-inden-1-amina 17b (54 mg, 0.19 mmol) en 10 mL de diclorometano, luego se agregó triacetoxiborohidruro de sodio (61 mg, 0.29 mmol). Después de agitar durante 12 horas, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 17 (10 mg, rendimiento 25.5%) como un aceite de color amarillo.
MS m/z (ESI): 407.6 [M+1]
1H RMN (400 MHz, CDCla) 88.58 (d, 1H), 7.71 (t, 1H), 7.57 (d, 1H), 7.40 (d, 1H), 7.29 (d, 1H), 7.16-7.24 (m, 3H), 3.77 (d, 3H), 3.31 (s, 3H), 2.87-3.05 (m, 2H), 2.24-2.50 (m, 4H), 2.14-2.24 (m, 1H), 1.61-1.84 (m, 4H), 1.35-1.51 (m, 5H), 1.24-1.35 (m, 2H), 1.11-1.20 (m, 1H), 0.65-0.75 (m, 1H).
Ejemplo 18
(15.25) -1-((2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-2,3-dihidro-1H -inden-2-ol

Etapa 1
2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 18b
2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetonitrilo 18a (500 mg, 1.95 mmol, preparado mediante un método descrito en la Solicitud de Patente "WO2012129495") se disolvió en 20 mL de tolueno, se agregaron lentamente gota a gota 4.2 mL de solución de hidruro de diisobutilaluminio 1 M a -78 °C, y la reacción se agitó durante 1.5 horas. Luego, se agregaron 18 mL de ácido clorhídrico 2 M y la mezcla se agitó durante 30 minutos. Se agregó solución de hidróxido de sodio 5 M gota a gota hasta que el pH de la reacción de reacción fue de 9 a 10. La mezcla se calentó a temperatura ambiente y se extrajo con acetato de etilo (30 mL X 3). Las fases orgánicas se combinaron, se lavaron con solución saturada de cloruro de sodio (30 mL X 3), se secaron sobre sulfato de sodio anhidro y se filtraron. El filtrado se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en columna de gel de sílice con el sistema de elución A para obtener el compuesto base 18b (270 mg, rendimiento 53.4%) como un aceite de color amarillo.
MS m/z (ESI): 260.5 [M+1].
Etapa 2
(1S,2S)-1-((2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-2,3-dihidro-1H -inden-2-ol 18
Compuesto 18b (20 mg, 0.08 mmol) y (1S,2S)-1-amino-2,3-dihidro-1H-inden-2-ol 18c (23 mg, 0.15 mmol, preparado mediante un método descrito en "Advanced Synthesis & Catalysis, 350 (14 15), 2250-2260; 2008") se disolvieron en 15 mL de una mezcla de diclorometano y metanol (V: V = 5: 1), la mezcla se agitó durante 2 horas, luego se agregó triacetoxiborohidruro de sodio (49 mg, 0.23 mmol). Después de agitar durante 12 horas, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 18 (10 mg, rendimiento 33%) como un aceite de color amarillo.
MS m/z (ESI): 393.5 [M+1]
1H RMN (400 MHz, CDCla) 88.50 (d, 1H), 7.70 (t, 1H), 7.37 (d, 1H), 7.20-7.26 (m, 3H), 7.11-7.19 (m, 2H), 3.76 (d, 3H), 3.36 (d, 1H), 2.88-3.05 (m, 2H), 2.25-2.50 (m, 4H), 2.15-2.24 (m, 1H), 1.60-1.84 (m, 4H), 1.36-1.51 (m, 5H), 1.25-1.35 (m, 2H), 1.10-1.20 (m, 1H), 0.65-0.75 (m, 1H).
Ejemplo 19
(1S,4S)-4-etoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-te trahidronaftalen-1-amina

Etapa 1
((1S,4S)-4-etoxi-1,2,3,4-tetrahidronaftalen-1-il)carbamato de tert-butilo 19a
El compuesto en bruto ((1S)-4-hidroxi-1,2,3,4-tetrahidronaftalen-1-il) carbamato de tert-butilo 14a (850 mg, 3.23 mmol), óxido de plata (76 mg, 0.33 mmol) y yodoetano (1.3 mL, 16.15 mmol) se disolvieron en 30 mL de diclorometano, y la mezcla se agitó durante 48 horas. La solución de reacción se filtró y el filtrado se concentró a presión reducida para obtener el compuesto en bruto 19a (800 mg) como un aceite de color amarillo, que se usó directamente en la siguiente etapa sin purificación adicional.
MS m/z (ESI): 236.1 [M-55].
Etapa 2
(15.45) -4-etoxi-1,2,3,4-tetrahidronaftalen-1-amina 19b
El compuesto en bruto 19a (698 mg, 2.4 mmol) se disolvió en 4 mL de diclorometano, luego se agregaron 8 mL de una solución de cloruro de hidrógeno 4 M en 1,4-dioxano. Después de agitar durante 2 horas, la solución de reacción se concentró a presión reducida, se trituró con acetato de etilo (30 mL) y se filtró. La torta del filtro se disolvió en 20 mL de una mezcla de diclorometano y metanol (V: V = 5: 1). Se agregó solución saturada de bicarbonato de sodio para ajustar el pH de la solución de reacción a 7 a 8. La solución de reacción se concentró a presión reducida, se lavó con una mezcla de diclorometano y metanol (V: V = 5: 1) (30 mL X 2) y filtrado. El filtrado se concentró a presión reducida para obtener el compuesto base en bruto 19b (310 mg) como un líquido de color amarillo, que se usó directamente en la siguiente etapa sin purificación adicional.
MS m/z (ESI): 191.1 [M+1].
Etapa 3
(15.45) -4-etoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-te trahidronaftalen-1-amina 19
Se disolvieron (R)-2-(9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)acetaldehído 5a (500 mg, 1.85 mmol) y el compuesto en bruto 19b (310 mg, 1.85 mmol) en 30 mL de diclorometano, y la mezcla se agitó durante 40 minutos, luego se agregó triacetoxiborohidruro de sodio (980 mg, 4.63 mmol). Después de agitar durante 2 horas, la solución de reacción se lavó sucesivamente con una solución saturada de bicarbonato de sodio (30 mL X 3) y una solución saturada de cloruro de sodio (30 mL X 3). La fase orgánica se secó sobre sulfato de sodio anhidro y se filtró. El filtrado se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 19 (280 mg, rendimiento 35%) como un sólido viscoso de color amarillo.
MS m/z (ESI): 435.3 [M+1]
1H RMN (400 MHz, CDCla) 89.74 (d, 1H), 9.58 (d, 1H), 8.94 (d, 1H), 8.37 (d, 1H), 7.94 (d, 1H), 7.67 (d, 1H), 7.52 (d, 1H), 7.47 (t, 1H), 4.46-4.49 (m, 1H), 4.30-4.33 (m, 1H), 3.84-3.87 (m, 1H), 3.66-3.70 (m, 2H), 3.53-3.56 (m, 2H), 2.82 2.85 (d, 2H), 2.67 (s, 2H), 2.39-2.41 (m, 4H), 2.30-2.33 (m, 4H), 1.85 (s, 2H), 1.48-1.52 (m, 6H), 1.27 (m, 3H).
Ejemplo 20
(1S,4S)-4-(ciclopropilmetoxi)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidronaftalen-1-amina

Etapa 1
((S)-4-oxo-1,2,3,4-tetrahidronaftalen-1-il)(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)carbamato de tert-butilo 20a
El compuesto 11 (220 mg, 0.54 mmol), dicarbonato de di-tert-butilo (173 mg, 0.82 mmol) y trietilamina (0.15 mL, 1.08 mmol) se disolvieron en 20 mL de diclorometano. Después de agitar durante 12 horas, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 20a (100 mg, rendimiento 37%) como un sólido viscoso de color amarillo claro.
MS m/z (ESI): 505.3 [M+1].
Etapa 2
((1S,4S)-4-hidroxi-1,2,3,4-tetrahidronaftalen-1-il)(2-((R)-9-(piridin-2-il)-6-oxaspiro [4.5]decan-9- il)etil) carbamato de tert-butilo 20b
El compuesto 20a (100 mg, 0.2 mmol) y (R)-2-metil-CBS-oxazaborolidina 1M (0.04 mL, 0.4 mmol) se disolvieron en 10 mL de tolueno, la reacción se enfrió a 0 °C, luego se agregó sulfato de metil borano 2 M (0.02 mL, 0.4 mmol). La reacción se calentó a temperatura ambiente y se agitó durante 3 horas. La reacción se inactivó mediante la adición de 10 mL de solución saturada de cloruro de sodio y se extrajo con acetato de etilo (10 mL X 3). Las fases orgánicas se combinaron, se lavaron con solución saturada de cloruro de sodio (10 mL X 3), se secaron sobre sulfato de sodio anhidro y se filtraron. El filtrado se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 20b (10 mg, rendimiento 10%) como un sólido de color blanco.
MS m/z (ESI): 507.3 [M+1].
Etapa 3
((1S,4S)-4-(ciclopropilmetoxi)-1,2,3,4-tetrahidronaftalen-1-il)(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)carbamato de tert-butilo 20c
El compuesto 20b (10 mg, 0.02 mmol) se disolvió en 5 mL de N, N-dimetilformamida, luego se agregó hidruro de sodio (2.2 mg, 0.06 mmol). La mezcla se agitó durante 30 minutos, luego se agregó bromuro de ciclopropilmetilo (6.7 mg, 0.05 mmol). Después de agitar durante 3 horas, la reacción se inactivó mediante la adición de 20 mL de agua y se extrajo con acetato de etilo (10 mL X 3). Las fases orgánicas se combinaron, se lavaron con solución saturada de cloruro de sodio (10 mL X 3), se secaron sobre sulfato de sodio anhidro y se filtraron. El filtrado se concentró a presión reducida para obtener el compuesto base en bruto 20c (5 mg) como un sólido de color blanco, que se usó directamente en la siguiente etapa sin purificación adicional.
MS m/z (ESI): 561.0 [M+1].
Etapa 4
(15.45) -4-(ciclopropilmetoxi)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidronaftalen-1-amina 20d
El compuesto en bruto 20c (5 mg, 0.0089 mmol) se disolvió en 5 mL de diclorometano, luego se agregaron 0.1 mL de una solución de ácido clorhídrico 4 M en 1,4-dioxano. Después de agitar durante 2 horas, la solución de reacción se concentró a presión reducida y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 20d (3 mg, rendimiento 73.2%) como un sólido de color blanco.
MS m/z (ESI): 461.3 [M+1]
1H RMN (400 MHz, CD3OD) 88.59 (d, 1H), 7.84-7.81 (m, 1H), 7.55 (d, 1H), 7.53 (d, 1H), 7.47-7.40 (m, 1H), 7.39 7.29 (m, 2H), 7.25 (d, 1H), 4.48-4.46 (m, 1H), 4.28-4.25 (m, 1H), 3.77-3.75 (m, 2H), 3.45-3.43 (m, 2H), 3.35-3.30 (m, 2H), 2.93-2.92 (m, 1H), 2.53-2.50 (m, 2H), 2.49-2.48 (m, 1H), 2.25-2.13 (m, 2H), 1.95-1.31 (m, 11H), 1.10-1.08 (m, 2H), 0.76-0.73 (m, 1H), 0.55-0.53 (m,2H),0.25-0.23 (m,2H).
Ejemplo 21
(15.45) -4-(2-fluoroetoxi)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidronaftalen-1- amina

Etapa 1
((1S,4S)-4-(2-fluoroetoxi)-1,2,3,4-tetrahidronaftalen-1-il)(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil) carbamato de tert-butilo 21a
Se disolvió 20b (45 mg, 0.088 mmol) en 5 mL de N, N-dimetilformamida, luego se agregó hidruro de sodio (20 mg, 0.44 mmol). La mezcla se agitó durante 20 minutos, luego se agregó 1-bromo-2-fluoroetano (23 mg, 0.176 mmol). Después de agitar durante 16 horas, la reacción se inactivó mediante la adición de 5 mL de agua y se extrajo con acetato de etilo (10 mL X 3). Las fases orgánicas se combinaron, se lavaron con solución saturada de cloruro de sodio (30 mL X 3), se secaron sobre sulfato de sodio anhidro y se filtraron. El filtrado se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 21a (30 mg, rendimiento 61.1%) como un aceite de color amarillo.
MS m/z (ESI): 553.4 [M+1].
Etapa 2
(15.45) -4-(2-fluoroetoxi)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidronaftalen-1- amina 21 El compuesto 21a (30 mg, 0.543 mmol) se disolvió en 10 mL de diclorometano, luego se agregaron 0.3 mL de una solución de ácido clorhídrico 4 M en 1,4-dioxano. La mezcla se agitó durante 1 hora, luego se agregaron 10 mg de carbonato de sodio. La solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 21 (10 mg, rendimiento 40.7%) como un material viscoso de color blanco.
MS m/z (ESI): 453.4 [M+1]
1H RMN (400 MHz, CDCb) 89.76 (d, 1H), 9.61 (d, 1H), 8.89 (d, 1H), 8.34 (d, 1H), 7.94 (d, 1H), 7.69 (d, 1H), 7.58 (d, 1H), 7.44 (t, 1H), 4.43-4.49 (m, 2H), 4.28-4.33 (m, 2H), 3.81-3.87 (m, 1H), 3.61-3.71 (m, 2H), 3.51-3.56 (m, 2H), 2.81 2.89 (d, 2H), 2.67 (s, 2H), 2.39-2.43 (m, 4H), 2.30-2.36 (m, 4H), 1.85 (s, 2H), 1.48-1.61 (m, 6H).
Ejemplos 22, 23
(15.45) -4-(metoximetil)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil) -1,2,3,4-tetrahidronaftalen-1- amina 22 (1S,4R)-4-(metoximetil)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil) -1,2,3,4-tetrahidronaftalen- 1-amina 23

Etapa 1
(4-metileno-1,2,3,4-tetrahidronaftalen-1-il)carbamato de (S)-tert-butilo 22a
Se disolvió bromuro de metiltrifenilfosfonio (2.95 g, 11.5 mmol) en 20 mL de tetrahidrofurano. La reacción se enfrió a 0 °C, se agregó tert-butóxido de potasio (1.29 g, 11.5 mmol) y se agitó durante 30 minutos. Luego, se agregaron 11b (1 g, 7.66 mmol), la reacción se calentó a temperatura ambiente y se agitó durante 12 horas. La solución de reacción se concentró a presión reducida y se disolvió en metanol. El residuo resultante se purificó por cromatografía en capa fina con sistema de elución B para obtener el compuesto base 22a (200 mg, rendimiento 22.2%) como un sólido de color blanco.
MS m/z (ESI): 204.2 [M-55].
Etapa 2
((1S)-4-(hidroximetil)-1,2,3,4-tetrahidronaftalen-1-il)carbamato de tert-butilo 22b
Se disolvieron 22a (780 mg, 3 mmol) en 20 mL de tetrahidrofurano, la reacción se enfrió a 0 °C, luego se agregaron 6 mL de una solución de borano 1 M en tetrahidrofurano. La solución de reacción se agitó durante 5 horas. Se agregaron 12 mL de solución de hidróxido de sodio 3 M, y la mezcla se agitó durante 30 minutos. Después de agregar 12 mL de peróxido de hidrógeno al 30%, la solución de reacción se calentó a temperatura ambiente y se agitó durante 12 horas. La solución de reacción se concentró a presión reducida y se extrajo con diclorometano. Las fases orgánicas se combinaron, se concentraron a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 22b (740 mg, rendimiento 89.2 %) como un sólido de color blanco.
MS m/z (ESI): 222.1 [M-55].
Etapa 3
((1S)-4-(metoximetil)-1,2,3,4-tetrahidronaftalen-1-il)carbamato de tert-butilo 22c
Se disolvieron 22b (200 mg, 0.72 mmol) en 10 mL de tetrahidrofurano, luego se agregó hidruro de sodio (60 mg, 1.4 mmol) y la reacción se agitó durante 1 hora. Luego, se agregó yodometano (123 mg, 0.86 mmol) y la reacción se agitó durante 12 horas. La solución de reacción se concentró a presión reducida y se disolvió en metanol. El residuo
resultante se purificó por cromatografía en capa fina con sistema de elución B para obtener el compuesto base 22c (20 mg, rendimiento 9.5%) como un sólido de color blanco.
MS m/z (ESI): 236.2 [M-55].
Etapa 4
(1S)-4-(metoximetil)-1,2,3,4-tetrahidronaftalen-1-amina 22d
El compuesto en bruto 22c (20 mg, 0.07 mmol) se disolvió en 10 mL de diclorometano, luego se agregaron 10 mL de una solución de ácido clorhídrico 4 M en 1,4-dioxano. Después de agitar durante 2 horas, la solución de reacción se concentró a presión reducida para obtener el compuesto base en bruto 22d (13 mg) como un aceite de color amarillo, que se usó directamente en la siguiente etapa sin purificación adicional.
MS m/z (ESI): 192.2 [M-55].
Etapa 5
(1S,4S)-4-(metoximetil)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil) -1,2,3,4-tetrahidronaftalen-1- amina 22 Se disolvieron 5a (30 mg, 0.116 mmol) y el compuesto en bruto 22d (22 mg, 0.116 mmol) en 20 mL de una mezcla de diclorometano y metanol (V: V = 1: 1), luego se agregó cianoborohidruro de sodio (15 mg, 0.23 mmol). Después de agitar durante 12 horas, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 22 (10 mg, rendimiento 20%) como un sólido de color blanco y el título compuesto 23 (8 mg, rendimiento 16%) como un sólido de color blanco.
MS m/z (ESI): 435.3 [M+1]
Ejemplo 22
1H RMN (400 MHz, CDCla) 88.26-8.25 (d, 1H), 7.76-7.72 (t, 1H), 7.42-7.40 (d, 1H), 7.34-7.29 (m, 3H), 7.29-7.27 (m, 1H), 7.18-7.16 (m, 1H), 4.16 (s, 1H), 3.75-3.70 (m, 2H), 3.47-3.45 (m, 2H), 3.41 (s, 3H), 3.35-3.33 (m, 1H), 3.18-3.17 (m, 1H), 2.80-2.70 (m, 1H), 2.4-2.33 (m, 1H), 2.28-1.95 (m, 7H), 1.81-1.62 (m, 5H), 1.59-1.51 (m, 1H), 1.46-1.20 (m, 4H), 1.22-1.1 (m,1H).
Ejemplo 23
1H RMN (400 MHz, CDCla) 88.47-8.46 (d, 1H), 7.72-7.68 (t, 1H), 7.39-7.37 (d, 1H), 7.37-7.33 (m, 1H), 7.24-7.21 (m, 1H), 7.15-7.05 (m, 2H), 6.93-6.91 (d, 1H),,3.94 (s, 1H), 3.68-3.60 (m, 2H), 3.59-3.57 (m, 2H), 3.22 (s, 3H), 3.22-3.19 (m, 1H), 2.71-2.70 (m, 1H), 2.34-2.30 (m, 5H), 2.28-2.25 (m, 1H), 1.84-1.81(m, 1H), 1.81-1.71 (m, 5H), 1.69-1.51 (m, 2H), 1.45-1.4 (m, 2H), 1.32-1.26 (m,2H), 1.23-1.15 (m, 1H), 1.1-0.95 (m, 1H).
Ejemplo 24
(S)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-3',4'-dihidro-2'H-spiro[ [1,3]ditiolano-2,1'-naftalen]- 4'-amina 24
Se disolvieron 11 (35 mg, 0.0865 mmol), etano-1,2-ditiol 24a (82 mg, 0.865 mmol) y p-toluenosulfonato de piridinio (240 mg, 0.952 mmol) en 15 mL de tolueno, la reacción se calentó a 110 °C y se agitó durante 12 horas. La solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 24 (40 mg, rendimiento 96%) como un sólido de color amarillo claro.
MS m/z (ESI): 481.2 [M+1]
1H RMN (400 MHz, CD3OD) 88.60 (d, 1H), 7.60 (t, 1H), 7.25-7.31 (m, 2H), 7.15-7.20 (m, 4H), 4.26-4.30 (m, 1H), 3.76 (d, 2H), 2.81-3.01 (m, 4H), 2.41-2.60 (m, 2H), 2.21-2.30 (m, 2H), 1.86-2.13 (m, 4H), 1.70-1.81 (m, 2H), 1.41 1.69 (m, 5H), 1.31-1.39 (m, 2H), 1.10-1.20 (m, 2H), 0.71-0.80 (m, 2H).
Ejemplo 25
(1S,4R)-4-etoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-te trahidronaftalen-1-amina 25

Etapa 1
((1S,4R)-4-hidroxi-1,2,3,4-tetrahidronaftalen-1-il)carbamato de tert-butilo 25a
Se disolvió (S)-2-metil-CBS-oxazaborolidina (221.8 mg, 0.8 mmol) en 140 mL de tetrahidrofurano, luego se agregó sulfato de metil borano (2.4 mL, 48 mmol) en una atmósfera de argón. La reacción se calentó a 30 °C, luego se agregaron 80 mL de una solución preparada previamente de 11b (10.5 g, 40 mmol) en tetrahidrofurano gota a gota durante 30 minutos. La mezcla de reacción se agitó durante 1 hora a 30 °C. Se agregaron 100 mL de metanol a 15 °C y se agitó durante 1 hora para inactivar la reacción. La solución de reacción se concentró a presión reducida. Se agregaron 200 mL de acetato de etilo y 5 g de carbón activado. La mezcla se agitó durante 30 minutos bajo microebullición y se filtró. La torta del filtro se lavó con acetato de etilo (100 mL X 3). El filtrado se concentró a presión reducida para obtener el compuesto base en bruto 25a (10.5 g) como un aceite incoloro.
MS m/z (ESI): 264.4 [M+1].
Etapas 2 a 4
(1S,4R)-4-etoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-te trahidronaftalen-1-amina 25
De acuerdo con la ruta de síntesis del ejemplo 19, el material de partida 14a se reemplazó con 25a, de acuerdo con lo anterior, se preparó el compuesto base 25 (7 g) como un aceite de color rojo claro.
MS m/z (ESI): 435.5 [M+1]
1H RMN (400 MHz, DMSO-da) 80.62 (dt, 1H), 0.92 - 1.03 (m, 1H), 1.12 (t, 3H), 1.34 (td, 2H), 1.41-1.69 (m, 9H), 1.79 (d, 1H), 1.82-1.92 (m, 2H), 2.02 (td, 1H), 2.26-2.38 (m, 2H), 2.43 (d, 1H), 3.37-3.48 (m, 2H), 3.52-3.66 (m, 3H), 4.25 (t, 1H), 7.11-7.16 (m, 2H), 7.16-7.20 (m, 1H), 7.21-7.28 (m, 2H), 7.45 (d, 1H), 7.71 (td, 1H), 8.52 (dd, 1H).
Ejemplo 26
(1S,4S)-4-(etoxi-d5)-N-(2-((R)-9-(piridina-2-il)-6-oxaspiro[4.5]dec-9-il)etil) -1,2,3,4-tetrahidronaftalen-1-amina 26

Etapa 1
((1S, 4S) -4-(etoxi-ds) -1,2,3,4-tetrahidronaftalen-1-il) carbamato de tert-butilo 26a
Se disolvió 14a (3.3 g, 12.5 mmol) en 50 mL de N, N-dimetilformamida, luego se agregaron tamices moleculares activados. Después la solución de reacción se enfrío a 0 °C, se agregó hidróxido de sodio (0.75 g, 18.75 mmol) bajo una atmósfera de argón. La reacción se agitó durante 0.5 horas a 0 °C. Luego, se agregó yodoetano-d5 deuterado (0.8 mL, 10 mmol), y la reacción se selló durante 16 horas a 0 °C. Una vez completada la reacción, la solución de reacción se vertió en una mezcla de 50 mL de agua, 50 mL de n-hexano y 5 mL de acetato de etilo, se agitó durante 10 minutos y se filtró. Se eliminaron los insolubles. El filtrado se separó en dos fases, y la fase acuosa se extrajo con una mezcla de n-hexano y acetato de etilo (V: V = 10: 1) (33 mL X 2). Las fases orgánicas se combinaron, se lavaron con solución saturada de cloruro de sodio (30 mL X 2), se secaron sobre sulfato de sodio anhidro y se filtraron. El filtrado se concentró a presión reducida, y el residuo resultante se purificó mediante un instrumento de preparación flash CombiFlash con el sistema de elución B para obtener el compuesto base 26a (1.89 g, rendimiento 64%) como un sólido de color blanco.
MS m/z (ESI): 241.4 [M-56+1].
Etapa 2
(1S,4S)-4-(etoxi-d5)-1,2,3,4-tetrahidronaftalen-1-amina 26b
Se agregaron 8 mL de una solución de cloruro de hidrógeno 4 M en 1,4-dioxano a 26a (1.89 g, 6.38 mmol). La solución de reacción se agitó durante 1 hora y se concentró a presión reducida. Se agregaron 30 mL de acetato de etilo, y la mezcla se concentró a presión reducida. Se agregó 1 mL de solución saturada de carbonato de sodio al residuo resultante, y la mezcla se agitó durante 5 minutos. Se agregaron 30 mL de acetato de etilo, 2 g de carbonato de sodio sólido y 10 g de sulfato de sodio, la solución de reacción se agitó durante 30 minutos hasta que la solución ya no estaba turbia. La mezcla se filtró y el filtrado se concentró a presión reducida para obtener el compuesto base en bruto 26b (1.21 g, un líquido de color marrón claro), que se usó directamente en la siguiente etapa sin purificación adicional.
MS m/z (ESI): 197.4 [M+1].
E ta p a 3
(1 S ,4 S ) -4 -(e to x i-d 5 ) -N -(2 -( (R ) -9 -(p ir id in a -2 - i l) -6 -o x a s p iro [4.5 ]d e c -9 - i l)e t i l) -1 ,2 ,3 ,4 - te t ra h id ro n a fta le n -1 -a m in a 26 Se disolvió 1a (1.37 g, 5.31 mmol) y el compuesto en bruto 26b (1.21 g, 6.16 mmol) en 50 mL de dicloroetano, se agregó una gota de ácido acético y la reacción se agitó durante 1 hora. Se agregó triacetoxiborohidruro de sodio (2.81 g, 13.27 mmol) y la reacción se agitó durante 16 horas. La solución de reacción se agregó con 10 mL de solución saturada de carbonato de sodio y se agitó durante 5 minutos. Se agregaron sucesivamente 10 mL de solución de hidróxido de sodio al 15%, 30 mL de agua, 30 mL de diclorometano y la mezcla se agitó durante 5 minutos. Se separaron dos fases y la fase acuosa se extrajo con diclorometano (50 mL X 2). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro y se filtraron. El filtrado se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en columna de gel de sílice con el sistema de elución A para obtener el compuesto base 26 (1.7 g, rendimiento 73%) como un líquido de color amarillo claro.
MS m/z (ESI): 440.5 [M+1]
1H RMN (400 MHz, CDCla) 80.70 (dt, 1H), 1.09 - 1.16 (m, 1H), 1.45 - 1.55 (m, 4H), 1.62 - 1.84 (m, 6H), 1.86 - 2.04 (m, 4H), 2.23 (td, 1H) 2.34 (dd, 1H), 2.44 (dd, 1H), 2.53 (td, 1H), 3.68 (br. s., 1H), 3.72 - 3.81 (m, 2H), 4.34 (t, 1H), 7.11 (ddd, 1 H), 7.17 (t, 2H), 7.18 - 7.23 (m, 1 H), 7.31 (t, 2 H), 7.62 (td, 1 H), 8.55 (dd, 1H).
Ejemplo 27
(S)-4-etil-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2-dihidronaftalen-1-amina 27

Etapa 1
(S,E)-tert-butil (4-etilideno-1,2,3,4-tetrahidronaftalen-1-il)carbamato 27a
Se disolvió bromuro de etiltrifenilfosfonio (2.1 g, 5.75 mmol) en 20 mL de tetrahidrofurano. Se agregó tert-butóxido de potasio (643 mg, 5.75 mmol) en un baño de agua con hielo, y la reacción se agitó durante 30 minutos en un baño de agua con hielo. Se agregó gota a gota una solución preparada previamente de 11b (1 g, 3.83 mmol) en tetrahidrofurano, y la mezcla se agitó durante 16 horas a 25 °C. La solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución B para obtener el compuesto base 27a (530 mg, rendimiento 51%) como un aceite de color amarillo claro.
Etapa 2
Clorhidrato (S)-4-etil-1,2-dihidronaftalen-1-amina 27b
Se disolvió 27a (273 mg, 1 mmol) en 5 mL de diclorometano, luego se agregaron 2 mL de una solución de cloruro de hidrógeno 4 M en 1,4-dioxano. Después de agitar durante 1 hora, la solución de reacción se concentró a presión reducida para obtener el compuesto base en bruto 27b (173 mg) como un aceite de color marrón, que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa 3
(S)-4-etil-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2-dihidronaftalen-1-amina 27
Se disolvieron 1a (150 mg, 0.58 mmol) y el compuesto en bruto 27b (158 mg, 0.58 mmol) en 30 mL de mezcla de dicloroetano y metanol (V: V = 10: 1), luego se agregó triacetoxiborohidmro de sodio (369 mg, 1.74 mmol). Después de agitar durante 16 horas, la solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con el sistema de elución A para obtener el compuesto base 27 (40 mg, rendimiento 17%) como un sólido de color amarillo claro.
MS m/z (ESI): 417.2 [M+1]
1H RMN (400 MHz, DMSO-d6) 88.58 (d, 1H), 7.83-7.78 (m, 1H), 7.53-7.48 (m, 3H), 7.33-7.29 (m, 2H), 7.21 (d, 1H), 5.84 (t, 1H), 4.25 (t, 1H), 3.73-3.72 (m, 3H), 3.41-3.31 (m, 2H), 2.81-2.80 (m, 2H), 2.41-2.25 (m, 3H), 1.96-1.90 (m, 3H), 1.85-1.61 (m, 8H), 1.25 (t, 3H), 1.23-1.21 (m, 1H), 0.68-0.65 (m, 1H).
Ejemplo 28
(S)-4-metileno-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tet rahidronaftalen-1-amina 28

De acuerdo con la ruta de síntesis del ejemplo 17, el material de partida 17a se reemplazó con 22a, de acuerdo con lo anterior, se preparó el compuesto base 28 (20 mg) como un sólido de color marrón.
MS m/z (ESI): 403.5 [M+1]
1H RMN (400 MHz, DMSO-d6) 88.50 (d, 1H), 7.72 (t, 1H), 7.68-7.21 (m, 6H), 5.95 (d, 1H), 4.09 (d, 1H), 3.71-3.69 (m, 3H), 3.01-2.80 (m, 2H), 2.67-2.63 (m, 2H), 2.11 (d, 1H), 1.74-1.21 (m, 14H), 0.99-0.98 (m, 1H), 0.45-0.42 (m, 1H). Ejemplo 29
2-(((1S,4S)-4-((2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-1,2,3,4-tetrahidronaftalen-1- il)oxi) acetonitrilo 29
E ta p a 1
( (1 S ,4 S ) -4 -(c ia n o m e to x i) -1 ,2 ,3 ,4 - te t ra h id ro n a f ta le n -1 - i l) (2 -( (R ) -9 -(p ir id in -2 - i l) -6 -o x a s p iro [4.5 ]d e c a n -9 - i l) e t i l) c a rb a m a to d e te r t-b u t ilo 29 a
Se disolvió 20b (40 mg, 0.08 mmol) en 10 mL de tetrahidrofurano, luego se agregaron tert-butóxido de potasio (45 mg, 0.4 mmol) y bromoacetonitrilo (20 mg, 0.16 mmol) sucesivamente, y la reacción se agitó durante 16 horas. Se agregaron 20 mL de agua y 20 mL de acetato de etilo y se agitó. La mezcla se dejó en reposo y se separó, y se extrajo con acetato de etilo (30 mL X 2). Las fases orgánicas se combinaron y se concentraron a presión reducida para obtener el compuesto base en bruto 29a (50 mg) como un aceite, que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa 2
2-(((1S,4S)-4-((2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-1,2,3,4-tetrahidronaftalen-1-il)oxi) acetonitrilo 29
El compuesto en bruto 29a (50 mg, 0.1 mmol) se disolvió en 10 mL de diclorometano, luego se agregaron 0.1 mL de una solución de cloruro de hidrógeno 4 M en dioxano. La reacción se agitó durante 0.5 horas. Se agregó amoníaco acuoso hasta que la solución de reacción fue alcalina. La mezcla se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 29 (10 mg, rendimiento 8%) como una cera de color blanco.
MS m/z (ESI): 446.3 [M+1]
1H RMN (400 MHz, DMSO-d6) 88.54 (d, 1H), 7.75-7.72 (m, 1H), 7.43 (d, 1H), 7.37-7.32 (m, 2H), 7.28-7.15 (m, 3H), 4.67 (d, 1H), 4.40 (d, 2H), 4.31 (d, 1H), 3.97 (d, 1H), 3.63-3.51 (m, 2H), 2.41-2.25 (m, 2H), 2.16-2.06 (m, 2H), 2.04 1.87 (m, 2H), 1.86-1.72 (m, 4H), 1.62-1.21 (m, 8H), 1.04-0.94 (m, 1H), 0.68-0.61 (m, 1H).
Ejemplo 30
(1S,4R)-4-metoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidronaftalen-1-amina 30

Etapa 1
((1S,4R)-4-hidroxi-1,2,3,4-tetrahidronaftalen-1-il)(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9- il)etil)carbamato de tert-butilo 30a
El compuesto 13 (46 mg, 0.11 mmol), dicarbonato de di-tert-butilo (27 mg, 0.121 mmol) y trietilamina (23 mg, 0.22 mmol) se disolvieron en 15 mL de diclorometano, y la reacción se agitó durante 16 horas. La solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 30a (46 mg, rendimiento 82%) como un sólido de color blanco.
MS m/z (ESI): 507.3 [M+1].
Etapa 2
((1S,4R)-4-metoxi-1,2,3,4-tetrahidronaftalen-1-il)(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9- il)etil) carbamato de tert-butilo 30b
El compuesto 30a (46 mg, 0.091 mmol) se disolvió en 10 mL de tetrahidrofurano, luego se agregó hidruro de sodio (8 mg, 0.182 mmol). La reacción se agitó durante 30 minutos a temperatura ambiente. Se agregó yodometano (16 mg, 0.11 mmol) y la reacción se agitó durante 16 horas a temperatura ambiente. Se agregaron 50 mL de agua y 50 mL de acetato de etilo, y se separaron dos fases. La fase orgánica se concentró a presión reducida para obtener el compuesto base en bruto 30b (47 mg) como un sólido de color marrón, que se usó directamente en la siguiente etapa sin purificación adicional.
MS m/z (ESI): 521.3 [M+1].
Etapa 4
(1S,4R)-4-metoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-tetrahidronaftalen-1-amina 30 El compuesto en bruto 30b (47 mg, 0.091 mmol) se disolvió en 10 mL de diclorometano, luego se agregaron 0.1 mL de una solución de cloruro de hidrógeno 4 M en 1,4-dioxano, y la reacción se agitó durante 1 hora. La solución de reacción se concentró a presión reducida. Se agregó etanol al residuo, y el pH se ajustó a 8 mediante amoniaco acuoso. La mezcla se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 30 (36 mg, rendimiento 95%) como un material viscoso de color amarillo.
MS m/z (ESI): 421.3 [M+1]
1H RMN (400 MHz, DMSO-d6) 88.55 (d, 1H), 7.75-7.72 (m, 1H), 7.46 (d, 1H), 7.37-7.32 (m, 2H), 7.28-7.15 (m, 3H), 4.67 (d, 1H), 4.30 (d, 1H), 3.97 (d, 1H), 3.64-3.50 (m, 2H), 3.35 (s, 3H), 2.41-2.26 (m, 2H), 2.16-2.06 (m, 2H), 2.04 1.87 (m, 2H), 1.86-1.72 (m, 4H), 1.62-1.21 (m, 8H), 1.04-0.94 (m, 1H), 0.68-0.61 (m, 1H).
Ejemplo 31
2-((S,E)-4-((2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-3,4-dihidronaftalen-1(2H)-ilideno)acetonitrilo 31

Etapa 1
(S,E)-tert-butil (4-(cianometileno)-1,2,3,4-tetrahidronaftalen-1-il)carbamato 31a
Se disolvió cianometilfosfonato de dietilo (200 mg, 0.76 mmol) en 20 mL de tetrahidrofurano. Se agregó hidruro de sodio (61 mg, 1.52 mmol) en un baño de agua con hielo, y la reacción se agitó durante 30 minutos en un baño de
agua con hielo. Se agregó gota a gota una solución preparada previamente de 11b (200 mg, 0.76 mmol) en tetrahidrofurano, y la mezcla se agitó durante 16 horas a 25 °C. La solución de reacción se vertió en agua con hielo y se extrajo con acetato de etilo tres veces. Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro y se filtraron. El filtrado se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución B para obtener el compuesto base 31a (150 mg, rendimiento 69%) como un material viscoso incoloro.
MS m/z (ESI): 285.1 [M+1].
Etapa 2
Clorhidrato de (S,E)-2-(4-amino-3,4-dihidronaftalen-1(2H)-ilideno)acetonitrilo 31b
Se disolvió 31a (150 mg, 0.52 mmol) en 5 mL de diclorometano, luego se agregaron 2 mL de una solución de ácido clorhídrico 1 M en 1,4-dioxano. La reacción se agitó durante 3 horas. La solución de reacción se concentró a presión reducida para obtener el compuesto base en bruto 31b (110 mg) como un sólido de color blanco, que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa 3
2-((S,E)-4-((2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-3,4-dihidronaftalen-1(2H)-ilideno)acetonitrilo 31 Se disolvieron 5a (100 mg, 0.39 mmol) y el compuesto en bruto 31b (85 mg, 0.39 mmol) en 10 mL de una mezcla de dicloroetano y metanol (V: V = 10: 1), luego se agregó triacetoxiborohidruro de sodio (165 mg, 0.78 mmol) y la reacción se agitó durante 16 horas. La solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 31 (30 mg, rendimiento 18%) como un material viscoso de color amarillo claro.
MS m/z (ESI): 428.0 [M+1]
1H RMN (400 MHz, DMSO-d6) 88.57 (d, 1H), 7.86-7.78 (m, 1H), 7.76-7.74 (m, 1H), 7.39-7.22 (m, 3H), 7.26-7.23 (m, 2H), 6.36-6.35 (m,1H), 3.65-3.54 (m, 3H), 2.90-2.60 (m, 2H), 2.42-2.37 (m, 3H), 2.03-1.90 (m, 4H), 1.82-1.78 (m, 2H), 1.51-1.24 (m, 10H).
Ejemplo 32
2-((4S)-4-((2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)amino)-1,2,3,4-tetrahidronaftalen-1-il)acetonitrilo 32

Etapa 1
2-((4S)-4-amino-1,2,3,4-tetrahidronaftalen-1-il)acetonitrilo clorhidrato 32a
Se disolvieron 31b (50 mg, 0.227 mmol) en 5 mL de etanol, luego se agregaron 5 mg de Pd/C, y el sistema de reacción se purgó con hidrógeno tres veces. La reacción se agitó durante 16 horas a temperatura ambiente bajo una atmósfera de hidrógeno. Los insolubles se eliminaron por filtración, y el filtrado se concentró a presión reducida para obtener el compuesto base en bruto 32a (45 mg) como un material viscoso incoloro, que se usó directamente en la siguiente etapa sin purificación adicional.
E ta p a 2
2 -( (4 S ) -4 -( (2 -( (R ) -9 -(p ir id in -2 - i l) -6 -o x a s p iro [4.5 ]d e c a n -9 - i l) e t i l )a m in o ) -1 ,2 ,3 ,4 - te t ra h id ro n a f ta le n -1 - i l) a c e to n it r i lo 32 Se disolvieron 5a (53 mg, 0.2 mmol) y el compuesto en bruto 32b (25 mg, 0.2 mmol) en 10 mL de una mezcla de dicloroetano y metanol (V: V = 10: 1), luego se agrega triacetoxiborohidmro de sodio (80 mg, 0.4 mmol) y la reacción se agitó durante 16 horas. La solución de reacción se concentró a presión reducida, y el residuo resultante se purificó por cromatografía en capa fina con sistema de elución A para obtener el compuesto base 32 (5 mg, rendimiento 5.8%) como un material viscoso de color amarillo claro.
MS m/z (ESI): 430.3 [M+1]
1H RMN (400 MHz, DMSO-d6) 88.55 (d, 1H), 7.85-7.78 (m, 1H), 7.75-7.72 (m, 1H), 7.35-7.20 (m, 3H), 7.25-7.21 (m, 2H), 3.75-3.60 (m, 3H), 2.95-2.80 (m, 2H), 2.70-2.65 (m, 4H), 2.41-2.30 (m, 4H), 1.95-1.89 (m, 4H), 1.85-1.60 (m, 4H), 1.55-1.21 (m, 6H).
Ejemplo 33
(S)-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-3,4-dihidro-2H-spiro[[ 1,3]dioxolano-2,1'-naftalen]-4'- amina 33

De acuerdo con la ruta de síntesis del ejemplo 27, el material de partida 2 se reemplazó con 11, de acuerdo con lo anterior, se preparó el compuesto base 33 (5 mg) como un aceite de color amarillo.
MS m/z (ESI): 449.0[M+1]
1H RMN (400 MHz, DMSO-d6) 88.58 (d, 1H), 7.81 (s, 1H), 7.77-7.70 (m, 1H), 7.51 (d, 1H), 7.27-7.11 (m, 4H), 3.85 (s, 1H), 3.66-3.50 (m, 5H), 3.51-3.42 (m, 1H), 3.42-3.33 (m, 1H), 2.48-2.35 (m, 2H), 2.38-2.32 (m, 1H), 2.20-2.08 (m, 2H), 2.01-1.88 (m, 2H), 1.85-1.75 (m, 3H), 1.71-1.31 (m, 8H), 1.00-0.96 (m, 1H), 0.70-0.62 (m, 1H).
Ejemplo 34
(1S,4S)-4-propoxi-N-(2-((R)-9-(piridin-2-il)-6-oxaspiro[4.5]decan-9-il)etil)-1,2,3,4-t etrahidronaftalen-1-amina 34

De acuerdo con la ruta de síntesis del ejemplo 21, el material de partida 1-bromo-2-fluoroetano se reemplazó con yodopropano, de acuerdo con lo anterior, se preparó el compuesto base 34 (8 mg) como un sólido de color amarillo. MS m/z (ESI): 449.3 [M+1]
1H RMN (400 MHz, DMSO-da) 88.53 (d, 1H), 7.75-7.70 (m, 1H), 7.45 (d, 1H), 7.37-7.33 (m, 2H), 7.28-7.17 (m, 3H), 4.65 (d, 1H), 4.32 (d, 1H), 3.98 (d, 1H), 3.64-3.52 (m, 3H), 3.49-3.40 (m, 1H), 2.62-2.52 (m, 1H), 2.41-2.27 (m, 2H), 2.16-2.06 (m, 1H), 2.04-1.87 (m, 2H), 1.86-1.71 (m, 5H), 1.67 (d, 1H), 1.60-1.20 (m, 8H), 1.13 (t, 3H), 1.03-0.95 (m, 1H), 0.68-0.60 (m, 1H).
Ensayo biológico
La presente invención se describirá adicionalmente con referencia a los siguientes ejemplos de prueba, pero los ejemplos no deben considerarse como limitativos del alcance de la invención.
Ejemplo de prueba 1
1. Objeto experimental
El objeto de este experimento es determinar el efecto agonista de los compuestos de la presente invención sobre MOR, KOR, DOR y evaluar la actividad in vitro de los compuestos según los valores de EC50 y Emax.
2. Prueba de actividad de MOR
2.1 Objeto experimental
Los compuestos de la presente invención pueden activar los receptores opioides p (MOR). El MOR activado puede regular el nivel de AMPc intracelular, y el AMPc ingresa al núcleo y se une a la región CRE de la luciferasa del gen indicador, iniciando así la expresión del gen indicador. La luciferasa puede reaccionar con su sustrato para emitir fluorescencia, y las señales de fluorescencia medidas reflejan la actividad agonista de los compuestos.
2.2 Método experimental
La actividad de los compuestos del ejemplo de prueba sobre volver agonista MOR y afectar el nivel de AMPc aguas abajo se probó mediante el siguiente método.
2.1.1 Materiales experimentales

2.2.2 Procedimiento experimental
1) Obtención de líneas celulares monoclonales HEK293/MOR/CRE
MOR/pcDNA3.1 (+) y CRE/pGL4.29 se transfirieron a células HEK293. Se agregaron G418 e higromicina al medio de cultivo, y las líneas celulares monoclonales HEK293/MOR/CRE se cribaron en una placa de cultivo celular de 96 pocillos.
2) Efecto agonista de los compuestos de ejemplo sobre MOR
Las células monoclonales HEK293/MOR/CRE se cultivaron en un medio de alto contenido en glucosa/DMEM (10% de FBS, 1 mg/mL de G418, 200 |ig/mL de higromicina, mezclado uniformemente) y se pasaron cada 3 días. El día del experimento, se preparó una suspensión celular con un medio celular nuevo, se agregó a una placa de 96 pocillos (BD, # 356692) con 20.000 células/pocillo, y se incubó en CO2 al 5% a 37 °C. El segundo día, el compuesto se disolvió en DMSO puro a una concentración de 20 mM, luego se formuló con DMSO a una primera concentración de 4 mM y se diluyó en un gradiente de concentración de 10 veces a 6 concentraciones. Se agregaron 90 |iL de DMSO a los pocillos en blanco y control; luego 2.5 |iL de las soluciones del compuesto formuladas en DMSO a una concentración de gradiente a 97.5 |iL de un medio de cultivo celular nuevo que contiene foscolina 5 |iM; Se agregaron 10 |iL del compuesto formulado a la placa de cultivo celular para hacer que la concentración final del compuesto sea 10000, 1000, 100, 10, 1, 0.1, 0.01 nM, y la placa se incubó a 37 °C, en CO2 al 5%, durante 5 horas. En una placa de cultivo celular de 96 pocillos, se agregaron 100 |iL de solución de ensayo de luciferasa (Promega, # E6110) a cada pocillo. La placa se colocó en la oscuridad a temperatura ambiente durante 10-15 minutos, se sopló y aspiró 10 veces, y se pipetearon 100 |iL a una placa blanca de 96 pocillos. Los valores de la señal de quimioluminiscencia se leyeron en un lector de microplacas (PE, Victor3), y los datos leídos se procesaron usando un software.
2.3 Resultados de la prueba
La actividad del compuesto de la presente invención sobre volver agonista MOR y afectar el nivel de AMPc aguas abajo se determinó mediante la prueba anterior, y el valor de EC50 se muestra en la tabla 1.1. Emax es el efecto máximo del compuesto de ejemplo sobre la activación de MOR y afecta la ruta de señalización de AMPc (el efecto máximo de TRV-130 es 100%).
3. Prueba de actividad KOR y DOR
3.1 Objeto experimental
El objeto del experimento es determinar la actividad de los compuestos de la presente invención en volver agonista el receptor KOR y el receptor DOR y afectar los niveles de AMPc aguas abajo
3.2 Procedimiento experimental
Se inocularon 90 |iL de células HEK293/KOR/CRE o HEK293/DOR/CRE (ADNc de CRE comprado en Promega, número de producto E8471, ADNc de KOR y ADNc de DOR fueron construidos en la compañía) en una placa de 96 pocillos con una densidad de 1 X 104 células/pocillo, luego las células se incubaron durante la noche a 37 °C, en CO2 al 5%. El fármaco se preparó como una solución madre 20 mM que luego se diluyó con un DMSO al 100% hasta un gradiente de concentración de 200X, y luego se diluyó con un medio DMEM/glucosa alta de 20 veces (SH30243.01B, Hyclone). Se sacó la placa de cultivo celular inoculada el primer día y se agregaron a cada pocillo 10 |iL del fármaco diluido o control (DMSO al 0.5%). La placa se agitó suavemente y se colocó en una incubadora a 37 °C, CO2 al 5% durante 4 horas. Finalmente, se agregaron 100 |iL de un reactivo de detección ONE-Glo (E6120, Promega) a cada pocillo, y la placa se colocó a temperatura ambiente durante 5 minutos. El valor de absorbancia se midió mediante el modelo de luz fría de un lector de microplacas (PE, Victor3). El valor de EC50 del compuesto se calculó mediante el software Graphpad Prism según cada concentración del compuesto y el valor de señal correspondiente. Emax es el efecto máximo del compuesto sobre el cambio de nivel de AMPc.
3.3 Resultados de la prueba
La actividad del compuesto de la presente invención en volver agonista el receptor KOR o el receptor DOR y afectar el nivel de AMPc aguas abajo se determinó mediante la prueba anterior, y el valor de EC50 se muestra en la tabla 1.2. Emax es el efecto máximo del compuesto de ejemplo en afectar el nivel de AMPc (el efecto máximo de la morfina es del 100%).
Tabla 1.1: valores de EC50 y Emax del compuesto de la presente invención para volver agonista el receptor MOR y afectar el nivel de AMPc

Tabla 1.2: valores de EC50 y Emax del compuesto de la presente invención en volver agonista el receptor KOR y el receptor DOR y afectan el nivel de AMPc aguas abajo

Conclusión: la actividad agonista de los compuestos de la presente invención sobre el receptor KOR o el receptor DOR es débil obviamente; y los compuestos de la presente invención tienen alta selectividad para el receptor MOR. Ejemplo de prueba 2
1. Objeto experimental
El objeto del experimento es determinar la actividad del compuesto de la presente invención en la activación de la ruta de señalización p-arrestina del receptor MOR.
2. Método experimental
2.1 Procedimiento del experimento
Se inocularon 90 pL de células CHO-K1 OPRM1 p-Arrestina (93-0213C2, DiscoveRX) en una placa de 96 pocillos con una densidad de 1X104 células/pocillo, luego las células se incubaron durante la noche a 37 °C, en CO2 al 5%. El fármaco se preparó como una solución madre 20 mM que luego se diluyó con un DMSO al 100% hasta un 200 X gradiente de concentración, y luego se diluyó con un medio 20 veces reactivo AssayComplete™ Cell Plating 2 (93-0563R2B, DiscoveRX). Se sacó la placa de cultivo celular inoculada el primer día y se agregaron a cada pocillo 10 |iL del fármaco diluido o control (DMSO al 0.5%). La placa se agitó suavemente y se colocó en una incubadora a 37 °C, CO2 al 5% durante 90 minutos. Finalmente, se agregaron 50 pL de un reactivo de detección (93-0001, DiscoveRX) a cada pocillo, y la placa se colocó a temperatura ambiente durante 60 minutos. El valor de absorbancia se midió mediante el modelo de luz fría de un lector de microplacas (PE, Victor3). El valor de EC50 del compuesto se calculó mediante el software Graphpad Prism según cada concentración del compuesto y el valor de señal correspondiente.
2.2 Resultados de la prueba
La actividad del compuesto de la presente invención sobre la activación de la ruta de señalización de p-arrestina se determinó mediante el ensayo anterior, y el valor de EC50 se muestra en la tabla 2. Emax es el efecto máximo del compuesto que afecta la ruta de señalización de p-arrestina (el efecto máximo de la morfina es del 100%).
Tabla 2: valor EC50 del compuesto de la presente invención en la ruta de señalización de p-arrestina

Conclusión: Los compuestos de la presente invención tienen poco efecto de activación en la ruta de señalización de p-arrestina
Ejemplo de prueba 3
1. Objeto experimental
El efecto de bloqueo del compuesto de la presente invención y el compuesto positivo TRV-130 (Journal of Pharmacology and Experimental Therapeutics, Volume 344, Issue 3, Pages 708-717, 2013) sobre la corriente de potasio hERG se probó en una línea celular estable transfectada con canal de potasio hERG usando un parche automático
2. Método experimental
2.1 Materiales e instrumentos experimentales
2.1.1 Materiales experimentales:

2.1.2 Instrumentos

2.2 Procedimiento experimental de abrazadera de parche automático
Las células estabilizadas con HEK293-hERG se subcultivaron a una densidad de 1: 4 en un medio MEM/EBSS (FBS al 10%, 400 pg/mL de G418, solución de aminoácidos no esenciales de MEM al 1% (100X), solución de piruvato de sodio al 1%) durante 48-72 horas, luego se realizó el experimento de abrazadera de parche automático. Las células se digirieron con tripsina al 0.25% el día del experimento, luego las células se recogieron por centrifugación y se volvieron a suspender con fluido extracelular (NaCl 140 mM, KCl 4 mM, MgCb 1 mM, CaCb 2 mM, monohidrato de D-glucosa 5 mM, Hepes 10 mM, pH 7.4, 298 mOsmol) en una suspensión celular. La suspensión celular se colocó en el banco de células del instrumento Patchliner, y las células se agregaron al chip (NPC-16) por el controlador de presión negativa del instrumento Patchliner, y la presión negativa atrajo células individuales al pequeño orificio del chip. Después de que se forma todo el modelo de células, el instrumento generará la corriente hERG según el programa preestablecido de corriente y voltaje hERG, y luego el instrumento puede perfundir el compuesto de baja concentración a alta concentración automáticamente. HEAK Patchmaster, el amplificador de abrazadera de parche HEAK EPC10 (Nanion), el software Pathliners y un software de análisis de datos proporcionado por el software Pathcontrol HT se usaron para analizar la corriente de los compuestos a diferentes concentraciones y la corriente del control en blanco.
2.3 Resultados de la prueba
El efecto de bloqueo del compuesto de la presente invención sobre la corriente de potasio hERG se determinó mediante la prueba anterior, y los valores de IC50 se muestran en la tabla 3.
Tabla 3: IC50 del compuesto de la presente invención en el bloqueo de la corriente de potasio hERG

Conclusión: Los compuestos de la presente invención tienen un efecto inhibidor más débil sobre hERG que el control positivo, y hay una diferencia significativa.
Claims (14)
1. Un compuesto de fórmula (I):

o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos,
en el que:
el anillo A se selecciona del grupo que consiste en cicloalquilo y heterociclilo;
R se selecciona del grupo que consiste en arilo y heteroarilo, en el que el arilo y el heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo, heteroarilo, -OR23, -C(O)R3, -C(O)OR3, -S(O)mR3 y -NR4R5;
cada R1 es idéntico o diferente y cada uno se selecciona independientemente del grupo que consiste en hidrógeno, alquilo, alcoxi, haloalquilo, halógeno, amino, nitro, hidroxi, ciano, cicloalquilo, heterociclilo, arilo, heteroarilo, -OR3, -C(O)R3, -C(O)OR3, -S(O)mR3 y -NR4R5, en la que el alquilo, haloalquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilo;
cada R2 es idéntico o diferente y cada uno se selecciona independientemente del grupo que consiste en hidrógeno, alquilo, alcoxi, haloalquilo, halógeno, amino, nitro, hidroxi, ciano, oxo, alquenilo, cicloalquilo, heterociclilo, arilo, heteroarilo, -OR3, -C(O)R3, -C(O)OR3, -S(O)mR3 y - NR4R5, en la que el alquilo, alcoxi, alquenilo, haloalquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en deuterio, alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio;
o dos R2 se toman juntos para formar un cicloalquilo o heterociclilo, en el que el cicloalquilo o heterociclilo está cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio; R3 se selecciona del grupo que consiste en hidrógeno, alquilo, alquilo deuterado, amino, alcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilo, en el que el alquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, halógeno, hidroxi, amino, nitro, ciano, alcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio;
R4 y R5 se seleccionan cada uno independientemente del grupo que consiste en que consiste en hidrógeno, alquilo, alcoxi, hidroxialquilo, hidroxi, amino, alcoxicarbonilo, cicloalquilo, heterociclilo, arilo y heteroarilo, en la que el alquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, halógeno, hidroxi, amino, alcoxicarbonilo, nitro, ciano, alcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio;
p y q están cada uno independientemente 0, 1, 2, 3 o 4; y
m es 0, 1 o 2.
2. El compuesto de fórmula (I) según la reivindicación 1, en el que el anillo A se selecciona del grupo que consiste en heterociclilo de 5 a 6 miembros y cicloalquilo de 5 a 6 miembros.
3. El compuesto de fórmula (I) según la reivindicación 1 o 2, en el que R es piridilo.
4. El compuesto de fórmula (I) según la reivindicación 1, que es un compuesto de fórmula (II):

o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos,
en el que:
G se selecciona del grupo que consiste en un enlace, CRaRb, C = O, NR4 y oxígeno;
Ra y Rb se seleccionan cada uno independientemente del grupo que consiste en hidrógeno, alquilo, alcoxi, haloalquilo, halógeno, amino, nitro, hidroxi, ciano, cicloalquilo, heterociclilo, arilo, heteroarilo, -OR3, -C(O)R3, -C(O)OR3, -S(O)mR3 y -NR4R5, en la que el alquilo, haloalquilo, cicloalquilo, heterociclilo, arilo y heteroarilo están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio; o Ra y Rb se toman juntos para formar un cicloalquilo o heterociclilo, en la que el cicloalquilo o heterociclilo está cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en alquilo, haloalquilo, halógeno, amino, nitro, ciano, hidroxi, alcoxi, haloalcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio; y R1 a R5, p, m y q son como se definen en la reivindicación 1.
5. El compuesto de fórmula (I) según las reivindicaciones 1 a 4, en el que R1 es hidrógeno o halógeno.
6. El compuesto de fórmula (I) según las reivindicaciones 1 a 5, en el que R2 se selecciona del grupo que consiste en hidrógeno, alquilo, oxo, alcoxi, hidroxi, halógeno y -OR3, en la que el alquilo y alcoxi están cada uno opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en deuterio, alquilo, halógeno, hidroxi, amino, alcoxicarbonilo, nitro, ciano, alcoxi, hidroxialquilo, cicloalquilo, heterociclilo, arilo y heteroarilio; R3 se selecciona del grupo que consiste en hidrógeno, alquilo y cicloalquilo, en la que el alquilo es opcionalmente sustituido con halógeno o cicloalquilo; preferiblemente, R2 es alquilo, en la que el alquilo es opcionalmente sustituido con alcoxi, cicloalquilo o haloalquilo; preferiblemente, R3 es hidrógeno, alquilo, haloalquilo o cicloalquilo.
7. El compuesto de fórmula (I) según una cualquiera de las reivindicaciones 4, que es un compuesto de fórmula (IV):

o un tautómero, mesómero, racemato, enantiómero, diastereómero del mismo, o una mezcla de los mismos, o una sal farmacéuticamente aceptable de los mismos,
en el que
R1, R2 y p son como se definen en la reivindicación 1.
8. El compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 7, en el que el compuesto se selecciona del grupo que consiste en:

9. Un procedimiento de preparación del compuesto de fórmula (I) según la reivindicación 1, que comprende una etapa de:
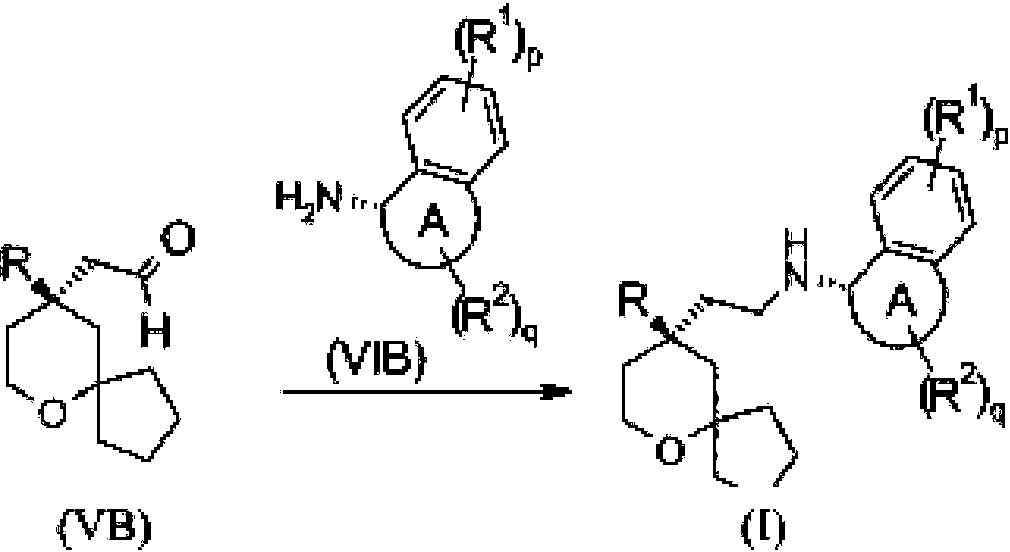
hacer reaccionar un compuesto de fórmula (VB) con un compuesto de fórmula (VIB) o clorhidrato del mismo mediante una aminación reductora para obtener el compuesto de fórmula (I);
en el que:
el anillo A, R, R1, R2, p y q son como se definen en la reivindicación 1.
10. Una composición farmacéutica que comprende una cantidad terapéuticamente eficaz del compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 8, y uno o más portadores, diluyentes o excipientes farmacéuticamente aceptables.
11. Compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 8, o la composición farmacéutica según la reivindicación 10 para uso en la prevención y/o tratamiento de disfunción inmune, inflamación, reflujo esofágico, trastornos neurológicos y psiquiátricos, enfermedades urinarias y reproductivas, enfermedades cardiovasculares y enfermedades respiratorias.
12. Compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 8, o la composición farmacéutica según la reivindicación 10 para uso en la prevención o el tratamiento del dolor y enfermedades relacionadas con el dolor.
13. El compuesto o la composición para uso según la reivindicación 12, en el que el dolor se selecciona del grupo que consiste en dolor postoperatorio, dolor inducido por cáncer, dolor neuropático, dolor traumático y dolor inflamatorio.
14. El compuesto o la composición para uso según la reivindicación 13, en el que el cáncer se selecciona del grupo que consiste en cáncer de mama, cáncer de endometrio, cáncer de cuello uterino, cáncer de piel, cáncer de próstata, cáncer de ovario, tumor de trompa de Falopio, tumor de ovario, hemofilia y leucemia.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201510665328 | 2015-10-15 | ||
| CN201511032876 | 2015-12-31 | ||
| PCT/CN2016/101064 WO2017063509A1 (zh) | 2015-10-15 | 2016-09-30 | 氧杂螺环类衍生物、其制备方法及其在医药上的应用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2772689T3 true ES2772689T3 (es) | 2020-07-08 |
Family
ID=58517778
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16854888T Active ES2772689T3 (es) | 2015-10-15 | 2016-09-30 | Derivado de oxa espiro, método de preparación del mismo y aplicaciones del mismo en medicamentos |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US10442793B2 (es) |
| EP (1) | EP3354649B1 (es) |
| JP (1) | JP6824502B2 (es) |
| KR (1) | KR102703513B1 (es) |
| CN (1) | CN107001347B (es) |
| AU (1) | AU2016339404B2 (es) |
| CA (1) | CA3000761C (es) |
| DK (1) | DK3354649T3 (es) |
| ES (1) | ES2772689T3 (es) |
| HU (1) | HUE048032T2 (es) |
| MX (1) | MX2018004175A (es) |
| PL (1) | PL3354649T3 (es) |
| PT (1) | PT3354649T (es) |
| RU (1) | RU2733373C2 (es) |
| TW (1) | TWI727981B (es) |
| WO (1) | WO2017063509A1 (es) |
Families Citing this family (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2019012251A (es) * | 2017-04-14 | 2019-11-28 | Jiangsu Hengrui Medicine Co | Sal agonista del receptor opioide, su forma de cristal i de sal de fumarato y metodo de preparacion de la misma. |
| CN108727347B (zh) * | 2017-04-14 | 2020-06-16 | 江苏恒瑞医药股份有限公司 | 一种阿片样物质受体(mor)激动剂的晶型及其制备方法 |
| JP2020516591A (ja) | 2017-04-14 | 2020-06-11 | ジエンス ヘンルイ メデイシンカンパニー リミテッドJiangsu Hengrui Medicine Co.,Ltd. | Morアゴニストおよびkorアゴニストを含有する医薬組成物、およびその用途 |
| CN115124520A (zh) * | 2017-07-04 | 2022-09-30 | 四川海思科制药有限公司 | 阿片受体激动剂及其应用 |
| CN109516982B (zh) | 2017-09-18 | 2022-08-23 | 上海华汇拓医药科技有限公司 | μ-阿片受体激动剂及其制备方法和在医药领域的应用 |
| CA3076395A1 (en) | 2017-09-28 | 2019-04-04 | Jiangsu Hengrui Medicine Co., Ltd. | Method for preparing oxaspirocycle derivative, and intermediate thereof |
| AU2018349930A1 (en) | 2017-10-13 | 2020-04-16 | Shandong Luye Pharmaceutical Co., Ltd. | 2,6-dioxaspiro (4,5) decane derivatives and preparation method therefor and pharmaceutical applications thereof |
| CN111065390B (zh) * | 2017-12-06 | 2023-01-24 | 江苏恒瑞医药股份有限公司 | Kor激动剂与mor激动剂联合在制备治疗疼痛的药物中的用途 |
| WO2019205983A1 (zh) * | 2018-04-28 | 2019-10-31 | 四川科伦博泰生物医药股份有限公司 | 氧杂螺环类化合物及其制备方法和用途 |
| CN108424372B (zh) * | 2018-05-11 | 2021-08-10 | 浙江华贝药业有限责任公司 | 2,2,2-三氟-n-[(s)-4-羰基四氢萘-1-基]-乙酰胺的纯化工艺 |
| CN111163778B (zh) * | 2018-05-14 | 2023-01-24 | 江苏恒瑞医药股份有限公司 | 一种mor受体激动剂药物组合物 |
| CN108707086B (zh) * | 2018-05-15 | 2021-08-10 | 浙江华贝药业有限责任公司 | 一种(1s,4s)-n-(4-羟基四氢萘-1-基)叔丁氧基碳酰胺的纯化工艺 |
| WO2020073985A1 (zh) * | 2018-10-12 | 2020-04-16 | 江苏恒瑞医药股份有限公司 | 一种阿片样物质受体(mor)激动剂的结晶形式及制备方法 |
| CN112638906B (zh) * | 2018-10-12 | 2022-05-27 | 江苏恒瑞医药股份有限公司 | 一种阿片样物质受体(mor)激动剂的结晶形式及制备方法 |
| CN112334465B (zh) * | 2019-01-17 | 2024-06-11 | 上海海雁医药科技有限公司 | 三环取代的氧杂螺环衍生物、其制法与医药上的用途 |
| CN111662284B (zh) * | 2019-03-06 | 2021-08-10 | 上海海雁医药科技有限公司 | 双杂环取代的氧杂螺环衍生物、其制法与医药上的用途 |
| WO2021027304A1 (zh) * | 2019-08-14 | 2021-02-18 | 上海海雁医药科技有限公司 | 镇痛化合物、其制法与医药上的用途 |
| CN114269751B (zh) * | 2020-01-17 | 2024-04-16 | 上海海雁医药科技有限公司 | 光学纯的氧杂螺环取代的吡咯并吡唑衍生物、其制法与医药上的用途 |
| JP7442652B2 (ja) * | 2020-01-17 | 2024-03-04 | 上海海雁医薬科技有限公司 | アザビシクロ置換オキサスピロ環誘導体、その調製方法及び医学的使用 |
| CN113214264B (zh) * | 2020-01-21 | 2022-08-09 | 上海海雁医药科技有限公司 | 二氢吡咯并五元杂芳基取代的氧杂螺环衍生物、其制法与医药上的用途 |
| MX2022008474A (es) | 2020-01-22 | 2022-08-02 | Jiangsu Hengrui Medicine Co | Conjugado de anticuerpo anti-trop-2-analogo de exatecan y uso medico del mismo. |
| CN114075141A (zh) * | 2020-08-20 | 2022-02-22 | 上海致根医药科技有限公司 | 阿片受体“偏向性”配体、其制备方法及其在医药上的应用 |
| CN114539257B (zh) * | 2020-11-24 | 2023-08-11 | 江苏恒瑞医药股份有限公司 | 嘧啶二酮类螺环衍生物、其制备方法及其在医药上的应用 |
| WO2022143715A1 (zh) | 2020-12-29 | 2022-07-07 | 上海海雁医药科技有限公司 | 氧杂螺环取代的吡咯并吡唑衍生物及其中间体和制备方法 |
| AU2022312049A1 (en) | 2021-07-13 | 2024-02-22 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Pharmaceutically acceptable salt of mor receptor agonist, and polymorph thereof and use thereof |
| WO2023011422A1 (zh) * | 2021-08-02 | 2023-02-09 | 上海枢境生物科技有限公司 | 氧杂螺环类衍生物、制备方法及其用途 |
| CN117263956A (zh) * | 2022-01-19 | 2023-12-22 | 天地恒一制药股份有限公司 | 阿片受体激动剂及其制备方法和用途 |
| CN116589451B (zh) * | 2022-02-11 | 2024-07-09 | 成都硕德药业有限公司 | 一种6-氧杂螺[4,5]癸烷类化合物的新晶型、用途及其制备方法 |
| WO2024153159A1 (zh) * | 2023-01-18 | 2024-07-25 | 上海森辉医药有限公司 | 一种四氢-1-萘胺及其衍生物的制备方法 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2160534B1 (es) | 1999-12-30 | 2002-04-16 | Vita Invest Sa | Nuevos esteres derivados de (rr,ss)-2-hidroxibenzoato de 3-(2-dimetilaminometil-1-hidroxiciclohexil) fenilo. |
| DE10044649A1 (de) | 2000-09-08 | 2002-07-04 | Gruenenthal Gmbh | Substituierte 4-Phenyl-1-(1-phenyl-cyclohexyl)-1,2,3,6-tetrahydropyridine |
| DE102006033109A1 (de) | 2006-07-18 | 2008-01-31 | Grünenthal GmbH | Substituierte Heteroaryl-Derivate |
| WO2009018169A1 (en) | 2007-07-27 | 2009-02-05 | Auspex Pharmaceuticals, Inc. | Substituted cyclohexanols |
| AU2009308769B2 (en) * | 2008-10-31 | 2015-03-12 | Pain Therapeutics, Inc. | Filamin A-binding anti-inflammatory analgesic |
| WO2010148191A2 (en) | 2009-06-17 | 2010-12-23 | Board Of Regents, The University Of Texas System | Compositions and methods for cyclofructans as separation agents |
| SI2688403T1 (sl) | 2011-03-23 | 2017-08-31 | Trevena, Inc. | Ligandi opioidnih receptorjev in metode uporabe in izdelave istega |
| CN104159902B (zh) | 2011-12-12 | 2016-05-04 | 格吕伦塔尔有限公司 | 制备(1r,4r)-6’-氟-(N,N-二甲基-和N-甲基)-4-苯基-4’,9’-二氢-3’H-螺[环己烷-1,1’-吡喃并[3,4,b]吲哚]-4-胺的方法 |
| HUE042762T2 (hu) | 2012-08-03 | 2019-07-29 | Johnson Matthey Plc | Eljárás oxikodon elõállítására |
| JP6345684B2 (ja) | 2012-11-13 | 2018-06-20 | アレイ バイオファーマ、インコーポレイテッド | 疼痛の治療に有用な二環式尿素、チオ尿素、グアニジン、およびシアノグアニジン化合物 |
-
2016
- 2016-09-30 PT PT168548881T patent/PT3354649T/pt unknown
- 2016-09-30 CA CA3000761A patent/CA3000761C/en active Active
- 2016-09-30 AU AU2016339404A patent/AU2016339404B2/en active Active
- 2016-09-30 RU RU2018115569A patent/RU2733373C2/ru active
- 2016-09-30 MX MX2018004175A patent/MX2018004175A/es unknown
- 2016-09-30 CN CN201680003945.6A patent/CN107001347B/zh active Active
- 2016-09-30 EP EP16854888.1A patent/EP3354649B1/en active Active
- 2016-09-30 PL PL16854888T patent/PL3354649T3/pl unknown
- 2016-09-30 ES ES16854888T patent/ES2772689T3/es active Active
- 2016-09-30 DK DK16854888.1T patent/DK3354649T3/da active
- 2016-09-30 KR KR1020187012600A patent/KR102703513B1/ko active IP Right Grant
- 2016-09-30 US US15/766,985 patent/US10442793B2/en active Active
- 2016-09-30 JP JP2018515975A patent/JP6824502B2/ja active Active
- 2016-09-30 HU HUE16854888A patent/HUE048032T2/hu unknown
- 2016-09-30 WO PCT/CN2016/101064 patent/WO2017063509A1/zh active Application Filing
- 2016-10-14 TW TW105133229A patent/TWI727981B/zh active
Also Published As
| Publication number | Publication date |
|---|---|
| KR102703513B1 (ko) | 2024-09-06 |
| AU2016339404A1 (en) | 2018-05-24 |
| TW201718558A (zh) | 2017-06-01 |
| AU2016339404B2 (en) | 2020-08-27 |
| US10442793B2 (en) | 2019-10-15 |
| JP6824502B2 (ja) | 2021-02-03 |
| EP3354649A1 (en) | 2018-08-01 |
| WO2017063509A1 (zh) | 2017-04-20 |
| PL3354649T3 (pl) | 2020-07-27 |
| RU2018115569A (ru) | 2019-11-18 |
| EP3354649A4 (en) | 2018-08-08 |
| BR112018006448A2 (pt) | 2018-10-16 |
| CA3000761A1 (en) | 2017-04-20 |
| JP2018534257A (ja) | 2018-11-22 |
| KR20180063251A (ko) | 2018-06-11 |
| EP3354649B1 (en) | 2019-12-04 |
| HUE048032T2 (hu) | 2020-05-28 |
| DK3354649T3 (da) | 2020-02-24 |
| CN107001347B (zh) | 2019-09-24 |
| MX2018004175A (es) | 2018-06-06 |
| RU2733373C2 (ru) | 2020-10-01 |
| RU2018115569A3 (es) | 2020-01-20 |
| US20180297988A1 (en) | 2018-10-18 |
| TWI727981B (zh) | 2021-05-21 |
| CA3000761C (en) | 2024-02-13 |
| PT3354649T (pt) | 2020-02-03 |
| CN107001347A (zh) | 2017-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2772689T3 (es) | Derivado de oxa espiro, método de preparación del mismo y aplicaciones del mismo en medicamentos | |
| JP7103952B2 (ja) | Fgfr4阻害剤、その製造方法及び応用 | |
| ES2586213T3 (es) | Compuestos de bencenosulfonamida y su uso como agentes terapéuticos | |
| ES2656496T3 (es) | Triazoles sustituidos útiles como inhibidores de AXL | |
| ES2452039T3 (es) | Sales de derivados azoicos de pirazolona biciclo-sustituidos, procedimiento de preparación y uso de los mismos | |
| BR112021004269A2 (pt) | composto de fórmula (i) ou um sal farmaceuticamente aceitável do mesmo, uso do mesmo, relacionada à proteína crbn e método para tratamento da mesma | |
| TW200808808A (en) | Antagonists of the vanilloid receptor subtype 1 (VR1) and uses thereof | |
| EP3416964A1 (en) | Carboxamide derivatives useful as rsk inhibitors | |
| TW202003495A (zh) | 苯並哌啶或雜芳基並哌啶類衍生物、其製備方法及其在醫藥上的應用 | |
| CN114746424B (zh) | 三环四氢异喹啉类衍生物、其制备方法及其在医药上的应用 | |
| CN113874367B (zh) | 吲唑类衍生物、其制备方法及其在医药上的应用 | |
| CN111499614A (zh) | 四氢异喹啉类衍生物、其制备方法及其在医药上的应用 | |
| BR112021003949A2 (pt) | derivado de anel fundido usado como inibidor de fgfr4 | |
| RU2753036C9 (ru) | Производное триптолида, способ его получения и применение | |
| ES2593057A1 (es) | Moduladores selectivos de la actividad del receptor GPR55: derivados de cromenopirazol | |
| WO2022237825A1 (zh) | 具有egfr抑制活性的氮杂芳基衍生物、其制备方法和应用 | |
| CN112996783B (zh) | 2-氨基嘧啶类衍生物、其制备方法及其在医药上的应用 | |
| BR112018006448B1 (pt) | Derivado de oxaespiro, seu uso e processo de preparação, e composição farmacêutica | |
| CN118056836A (zh) | 取代的氨基嘧啶类大环化合物、其制备方法及其在医药上的应用 | |
| KR20240042486A (ko) | 옥사스피로환계 유도체, 제조 방법 및 이의 용도 | |
| TW202345852A (zh) | 稠雜環類化合物、其製備方法及其在醫藥上的應用 | |
| TW201915000A (zh) | 稠合雜芳基衍生物、其製備方法及其在醫藥上的應用 |