EP3303522B1 - Procede de conversion de charges comprenant une etape d'hydrocraquage, une etape de precipitation et une etape de separation des sediments pour la production de fiouls - Google Patents
Procede de conversion de charges comprenant une etape d'hydrocraquage, une etape de precipitation et une etape de separation des sediments pour la production de fiouls Download PDFInfo
- Publication number
- EP3303522B1 EP3303522B1 EP16719813.4A EP16719813A EP3303522B1 EP 3303522 B1 EP3303522 B1 EP 3303522B1 EP 16719813 A EP16719813 A EP 16719813A EP 3303522 B1 EP3303522 B1 EP 3303522B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- fraction
- hydrocracking
- process according
- weight
- separation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G67/00—Treatment of hydrocarbon oils by at least one hydrotreatment process and at least one process for refining in the absence of hydrogen only
- C10G67/02—Treatment of hydrocarbon oils by at least one hydrotreatment process and at least one process for refining in the absence of hydrogen only plural serial stages only
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G31/00—Refining of hydrocarbon oils, in the absence of hydrogen, by methods not otherwise provided for
- C10G31/06—Refining of hydrocarbon oils, in the absence of hydrogen, by methods not otherwise provided for by heating, cooling, or pressure treatment
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G31/00—Refining of hydrocarbon oils, in the absence of hydrogen, by methods not otherwise provided for
- C10G31/09—Refining of hydrocarbon oils, in the absence of hydrogen, by methods not otherwise provided for by filtration
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G47/00—Cracking of hydrocarbon oils, in the presence of hydrogen or hydrogen- generating compounds, to obtain lower boiling fractions
- C10G47/24—Cracking of hydrocarbon oils, in the presence of hydrogen or hydrogen- generating compounds, to obtain lower boiling fractions with moving solid particles
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G67/00—Treatment of hydrocarbon oils by at least one hydrotreatment process and at least one process for refining in the absence of hydrogen only
- C10G67/02—Treatment of hydrocarbon oils by at least one hydrotreatment process and at least one process for refining in the absence of hydrogen only plural serial stages only
- C10G67/12—Treatment of hydrocarbon oils by at least one hydrotreatment process and at least one process for refining in the absence of hydrogen only plural serial stages only including oxidation as the refining step in the absence of hydrogen
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/10—Feedstock materials
- C10G2300/1037—Hydrocarbon fractions
- C10G2300/1048—Middle distillates
- C10G2300/1059—Gasoil having a boiling range of about 330 - 427 °C
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/10—Feedstock materials
- C10G2300/1077—Vacuum residues
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/20—Characteristics of the feedstock or the products
- C10G2300/201—Impurities
- C10G2300/202—Heteroatoms content, i.e. S, N, O, P
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/20—Characteristics of the feedstock or the products
- C10G2300/201—Impurities
- C10G2300/205—Metal content
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/20—Characteristics of the feedstock or the products
- C10G2300/201—Impurities
- C10G2300/205—Metal content
- C10G2300/206—Asphaltenes
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/20—Characteristics of the feedstock or the products
- C10G2300/30—Physical properties of feedstocks or products
- C10G2300/301—Boiling range
Definitions
- the present invention relates to the refining and the conversion of heavy hydrocarbon fractions containing, inter alia, sulfur-containing impurities. It relates more particularly to a process for converting heavy petroleum feeds of the atmospheric residue type and / or vacuum residue for the production of heavy fractions that can be used as fuel bases, in particular bunker oil bases, with a low sediment content.
- the process according to the invention also makes it possible to produce atmospheric distillates (naphtha, kerosene and diesel), vacuum distillates and light gases (C1 to C4).
- sediment content after aging is a measurement carried out according to the method described in the ISO 10307-2 standard (also known to those skilled in the art under the name of IP390). In the rest of the text will therefore read "sediment content after aging", the sediment content measured according to the ISO 10307-2 method.
- the reference to IP390 will also indicate that the measurement of the sediment content after aging is performed according to the ISO 10307-2 method.
- the sediment content according to ISO 10307-1 (also known as IP375) is different from the sediment content after aging according to ISO 10307-2 (also known as IP390).
- the sediment content after aging according to ISO 10307-2 is a much more stringent specification and corresponds to the specification for bunker fuels.
- terrestrial fuel oils in particular fuel oils that can be used for the production of heat and / or electricity, may also be subject to stability specifications, in particular maximum sediment contents, the thresholds of which vary according to the places of production. Because there is no international harmonization as in the case of maritime transport. There is, however, an interest in reducing the sediment content of terrestrial fuel oils.
- Residue hydrocracking processes convert low value residues to higher value added distillates.
- the resulting heavy fraction corresponding to the unconverted residual cut is generally unstable. It contains sediments that are mainly precipitated asphaltenes. This unstable residual cut can not therefore be efficiently be efficiently converted as fuel oil, especially as bunker oil without a specific treatment since the hydrocracking is operated under severe conditions leading to a high conversion rate.
- the patent US6447671 discloses a process for converting heavy petroleum fractions comprising a first bubbling bed hydrocracking step, a step of removing the catalyst particles contained in the hydrocracking effluent, and then a fixed bed hydrotreating step.
- the demand US2014 / 0034549 discloses a residue conversion process using a bubbling bed hydrocracking step and a step with an upflow reactor associated with a so-called "stripper" reactor.
- the sediment content of the final effluent is reduced relative to the effluent of the boiling bed stage.
- the sediment content after aging is not less than 0.1% by weight, as required for marketing as a residual type marine fuel.
- the patent FR2981659 discloses a petroleum heavy fraction conversion process comprising a first bubbling bed hydrocracking step and a fixed bed hydrotreating step comprising permutable reactors.
- the hydrocracking process partially converts heavy feeds to produce atmospheric distillates and / or vacuum distillates.
- ebullated bed technology is known to be suitable for heavy loads loaded with impurities, the bubbling bed inherently produces catalyst fines and sediments that must be removed to meet product quality such as heating oil. hold. The fines come mainly from the attrition of the catalyst in the bubbling bed.
- the sediments may be precipitated asphaltenes.
- the hydrocracking conditions and in particular the temperature cause them to undergo reactions (dealkylation, polycondensation, etc.) leading to their precipitation.
- reactions dealkylation, polycondensation, etc.
- conversion rates for compounds boiling above 540 ° C: 540 + ° C, for example greater than 30, 40 or 50% depending of the nature of the charge.
- An advantage of the method according to the invention is to avoid in particular the risk of clogging of boat engines.
- Another advantage of the process of the invention is to avoid the risk of fouling, in the case of any processing steps carried out downstream of the hydrocracking step of avoiding clogging of the bed (s) ( s) catalytic (s) implemented.
- the heavy fractions obtained by the present process can be mixed with fluxing bases so as to achieve the target viscosity of the desired fuel grade as well as the specification in sediment content after aging.
- Another point of interest of the process is the partial conversion of the feedstock making it possible to produce, particularly by hydrocracking, atmospheric distillates or vacuum distillates (naphtha, kerosene, diesel, vacuum distillate), which can be used as bases in plants.
- fuel pools directly or after passing through another refining process such as hydrotreating, reforming, isomerization, hydrocracking or catalytic cracking.
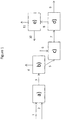
- the figure 1 illustrates a schematic view of the process according to the invention showing a hydrocracking zone, a separation zone, a precipitation zone, a physical separation zone of the sediments and a recovery zone of the fraction of interest.
- the feedstocks treated in the process according to the invention are advantageously chosen from atmospheric residues, vacuum residues from direct distillation, crude oils, crude head oils, deasphalted oils, deasphalting resins, asphalts or pitches. deasphalting, residues resulting from conversion processes, aromatic extracts from lubricant base production lines, oil sands or their derivatives, oil shales or their derivatives, whether alone or as a mixture.
- fillers can advantageously be used as they are or else diluted by a hydrocarbon fraction or a mixture of hydrocarbon fractions which may be chosen from products resulting from a fluid catalytic cracking process (FCC according to the initials of the English name of "Fluid Catalytic Cracking"), a light cutting oil (LCO), a heavy cutting oil (HCO), a decanted oil (OD according to the initials of the English name “Decanted Oil”), a residue of FCC , or which may come from the distillation, gas oil fractions including those obtained by atmospheric or vacuum distillation, such as vacuum gas oil.
- the heavy charges can also advantageously comprise cuts resulting from the process of liquefying coal or biomass, aromatic extracts, or any other hydrocarbon cuts or non-petroleum fillers such as pyrolysis oil from lignocellulosic biomasses.
- the fillers according to the invention generally have a sulfur content of at least 0.1% by weight, an initial boiling point of at least 340 ° C. and a final boiling point of at least 440 ° C. preferably a final boiling temperature of at least 540 ° C.
- the feedstock may contain at least 1% C7 asphaltenes and at least 5 ppm metals, preferably at least 2% C7 asphaltenes and at least 25 ppm metals.
- the fillers according to the invention are preferably atmospheric residues or residues under vacuum, or mixtures of these residues.
- Step a) Hydrocracking in a bubbling bed
- the filler according to the invention is subjected to a hydrocracking step which is carried out in at least one reactor containing a catalyst supported in a bubbling bed and preferably operating with an upward flow of liquid and gas.
- the objective of the hydrocracking step is to convert the heavy fraction into lighter cuts while partially refining the charge.
- Bubbling bed technologies use extruded bed catalysts supported in the form of extrudates, the diameter of which is generally of the order of 1 mm or less than 1 mm.
- the catalysts remain inside the reactors and are not evacuated with the products.
- the temperature levels are high in order to obtain high conversions while minimizing the amounts of catalysts used.
- the catalytic activity can be kept constant by replacing the catalyst in line. It is therefore not necessary to stop the unit to change the spent catalyst, nor to increase the reaction temperatures along the cycle to compensate for the deactivation.
- operating at constant operating conditions provides consistent yields and product qualities along the cycle. Also, because the catalyst is kept agitated by a large recycling of liquid, the pressure drop on the reactor remains low and constant.
- the conditions of hydrocracking step a) in the presence of hydrogen are usually conventional bubbling bed hydrocracking conditions of a liquid hydrocarbon fraction. It is advantageously carried out under a hydrogen partial pressure of 5 to 35 MPa, often 8 to 25 MPa and usually 12 to 20 MPa at a temperature of 330 to 500 ° C and often 350 to 450 ° C.
- the hourly space velocity (VVH) and the hydrogen partial pressure are important factors that are chosen according to the characteristics of the product to be treated and the desired conversion.
- the VVH defined as the volumetric flow rate of the feed divided by the total volume of the reactor, is generally in a range from 0.05 h -1 to 5 h -1 , preferably from 0.1 h -1 to 2 h -1 and more preferably from 0.2 h -1 to 1 h -1 .
- the amount of hydrogen mixed with the feedstock is usually 50 to 5000 Nm 3 / m 3 (normal cubic meters (Nm 3 ) per cubic meter (m 3 ) of liquid charge) and most often from 100 to 1000 Nm 3 / m 3 and preferably from 200 to 500 Nm 3 / m 3 .
- a conventional granular hydrocracking catalyst comprising, on an amorphous support, at least one metal or metal compound having a hydrodehydrogenating function.
- This catalyst may be a catalyst comprising Group VIII metals, for example nickel and / or cobalt, most often in combination with at least one Group VIB metal, for example molybdenum and / or tungsten.
- a catalyst comprising from 0.5 to 10% by weight of nickel and preferably from 1 to 5% by weight of nickel (expressed as nickel oxide NiO) and from 1 to 30% by weight of molybdenum of preferably from 5 to 20% by weight of molybdenum (expressed as MoO 3 molybdenum oxide) on an amorphous mineral support.
- This support will for example be chosen from the group formed by alumina, silica, silica-aluminas, magnesia, clays and mixtures of at least two of these minerals.
- This support may also contain other compounds and for example oxides chosen from the group formed by boron oxide, zirconia, titanium oxide and phosphoric anhydride. Most often an alumina support is used and very often a support of alumina doped with phosphorus and possibly boron.
- phosphorus pentoxide P 2 O 5 When phosphorus pentoxide P 2 O 5 is present, its concentration is usually less than 20% by weight and most often less than 10% by weight.
- the concentration of boron trioxide B 2 O 3 is usually from 0 to 10% by weight.
- the alumina used is usually a gamma or eta alumina. This catalyst is most often in the form of extrudates.
- the total content of metal oxides of groups VI and VIII is often from 5 to 40% by weight and in general from 7 to 30% by weight and the weight ratio expressed as metal oxide between metal (or metals) of Group VI on metal (or metals) of group VIII is usually 20 to 1 and most often 10 to 2.
- the spent catalyst is partly replaced by fresh catalyst, generally by withdrawal at the bottom of the reactor and introduction to the top of the fresh or new catalyst reactor at a regular time interval, that is to say for example by puff or almost keep on going.
- the catalyst can also be introduced from below and withdrawn from the top of the reactor. For example, fresh catalyst can be introduced every day.
- the replacement rate of spent catalyst with fresh catalyst may be, for example, from about 0.05 kilograms to about 10 kilograms per cubic meter of charge. This withdrawal and this replacement are performed using devices allowing the continuous operation of this hydrocracking step.
- the unit usually includes a recirculation pump allowing the maintaining the catalyst in a bubbling bed by continuously recycling at least a portion of the liquid withdrawn at the top of the reactor and reinjected at the bottom of the reactor. It is also possible to send the spent catalyst withdrawn from the reactor into a regeneration zone in which the carbon and the sulfur contained therein are eliminated before it is reinjected in the hydrocracking step a).
- hydrocracking step a) is carried out under the conditions of the H-OIL® process as described, for example, in US6270654 .
- the hydrocracking can be carried out in a single reactor or in several (generally two) reactors arranged in series.
- the use of at least two ebullated bed reactors in series makes it possible to obtain products of better quality and with a better yield, thus limiting the energy and hydrogen needs in possible post-treatments.
- the hydrocracking into two reactors makes it possible to have improved operability in terms of the flexibility of the operating conditions and of the catalytic system.
- the temperature of the second reactor is preferably at least 5 ° C higher than that of the first bubbling bed reactor.
- the pressure of the second reactor is 0.1 to 1 MPa lower than for the first reactor to allow the flow of at least a portion of the effluent from the first step without pumping is necessary.
- the different operating conditions in terms of temperature in the two hydrocracking reactors are selected to be able to control the hydrogenation and the conversion of the feedstock into the desired products in each reactor.
- the effluent obtained at the end of the first hydrocracking reactor is subjected to a separation of the light fraction and at least a portion, preferably all, of the residual effluent is treated in the second hydrocracking reactor .
- This separation can be performed in an inter-floor separator as described in the patent US 6270654 and in particular makes it possible to avoid excessive hydrocracking of the light fraction in the second hydrocracking reactor.
- the hydrocracking step may also be carried out in at least one reactor operating in a hybrid bed mode, that is to say operating in a bubbling bed with a supported catalyst associated with a dispersed catalyst consisting of very fine catalyst particles. forming a suspension with the charge to be treated.
- a hybrid bed has two populations of catalyst, a population of bubbling bed catalyst to which is added a population of "dispersed” type catalyst.
- the term “dispersed” refers to an implementation of the reactor in which the catalyst is in the form of very fine particles, that is to say generally a size of between 1 nanometer (ie 10 -9 m) and 150 microns, preferably between 0.1 and 100 microns, and even more preferably between 10 and 80 microns.
- the hydrocracking stage may comprise a first bubbling bed reactor followed by a second hybrid bed type reactor (that is to say bubbling bed type with "dispersed" type catalyst injection).
- the hydrocracking step may comprise a first hybrid bed type reactor followed by a second hybrid type reactor.
- the hydrocracking step may comprise a single hybrid bed type reactor.
- the "disperse" catalyst used in the hybrid bed reactor may be a sulfide catalyst preferably containing at least one member selected from the group consisting of Mo, Fe, Ni, W, Co, V, Ru. These catalysts are generally monometallic or bimetallic (by combining, for example, a non-noble group VIIIB element (Co, Ni, Fe) and a group VIB element (Mo, W) .
- the catalysts used may be heterogeneous solid powders (such as natural ores, iron sulphate, etc.), dispersed catalysts derived from water-soluble precursors such as phosphomolybdic acid, ammonium molybdate, or a mixture of Mo or Ni oxide. With aqueous ammonia, the catalysts used are preferably derived from soluble precursors in an organic phase (oil-soluble catalysts).
- the precursors are generally organometallic compounds such as the naphthenates of Mo, Co, Fe, or Ni, or the Mo octoates, or the multi-carbonyl compounds of these metals, for example 2-ethyl hexanoates of Mo or Ni , Mo or Ni acetylacetonates, C7-C12 fatty acid salts of Mo or W, etc. They can be used in the presence a surfactant to improve the dispersion of the metals, when the catalyst is bimetallic.
- the catalysts are in the form of dispersed particles, colloidal or otherwise depending on the nature of the catalyst. Such precursors and catalysts that can be used in the process according to the invention are widely described in the literature.
- the catalysts are prepared before being injected into the feed.
- the preparation process is adapted according to the state in which the precursor is and of its nature. In all cases, the precursor is sulfided ( ex-situ or in-situ ) to form the catalyst dispersed in the feedstock.
- the precursor is advantageously mixed with a carbonaceous feedstock (which may be a part of the feedstock to be treated, an external feedstock, a recycled fraction, etc.), the mixture is then sulphurized. by addition of a sulfur compound (preferred hydrogen sulphide or optionally an organic sulphide such as DMDS in the presence of hydrogen) and heated.
- a sulfur compound preferred hydrogen sulphide or optionally an organic sulphide such as DMDS in the presence of hydrogen
- the preparations of these catalysts are described in the literature.
- the "dispersed" catalyst particles as defined above generally have a size of between 1 nanometer and 150 microns, preferably between 0.1 and 100 microns, and even more preferably between 10 and 80 microns.
- the content of catalytic compounds (expressed as weight percentage of metal elements of group VIII and / or of group VIB) is between 0 and 10% by weight, preferably between 0 and 1% by weight.
- Additives may be added during the preparation of the catalyst or to the "dispersed" catalyst before it is injected into the reactor. These additives are described in the literature.
- the preferred solid additives are inorganic oxides such as alumina, silica, mixed Al / Si oxides, supported spent catalysts (for example, on alumina and / or silica) containing at least one group VIII element (such as Ni, Co) and / or at least one group VIB element (such as Mo, W).
- group VIII element such as Ni, Co
- group VIB element such as Mo, W
- the catalysts described in the application US2008 / 177124 Carbonaceous solids with a low hydrogen content (for example 4% hydrogen), such as coke or ground activated carbon, optionally pretreated, can also be used. Mixtures of such additives can also be used.
- the particle size of the additive is generally between 10 and 750 microns, preferably between 100 and 600 microns.
- the content of any solid additive present at the inlet of the reaction zone of the "dispersed" hydrocracking process is between 0 and 10% by weight, preferably between 1 and 3% by weight, and the content of catalytic compounds (expressed as weight percentage of Group VIII and / or Group VIB metal elements) is between 0 and 10% by weight, preferably between 0 and 1% by weight.
- the hybrid bed reactor (s) used in the hydrocracking zone therefore consist of two populations of catalysts, a first population using supported catalysts in the form of extrudates whose diameter is advantageously between 0.8 and 1.2 mm. , generally equal to 0.9 mm or 1.1 mm and a second population of "dispersed" type catalyst discussed above.
- the fluidization of the catalyst particles in the bubbling bed is enabled by the use of a boiling pump which allows a recycle of liquid, generally inside the reactor.
- the flow rate of liquid recycled by the boiling pump is adjusted so that the supported catalyst particles are fluidized but not transported, so that these particles remain in the bubbling bed reactor (with the exception of catalyst fines that can be formed by attrition and entrained with the liquid since these fines are small).
- the "dispersed" type catalyst is also entrained with the liquid since the "dispersed” type catalyst consists of particles of very small size.
- Step b) Separation of the hydrocracking effluent
- the effluent obtained at the end of the hydrocracking step a) undergoes at least one separation step, optionally supplemented by further additional separation steps, making it possible to separate at least one light hydrocarbon fraction containing bases. fuels and a heavy fraction containing boiling compounds at least 350 ° C.
- the separation step may advantageously be carried out by any method known to those skilled in the art such as, for example, the combination of one or more high and / or low pressure separators, and / or distillation stages and / or or high and / or low pressure stripping.
- the separation step b) makes it possible to obtain a gaseous phase, at least a light fraction of hydrocarbons of the naphtha, kerosene and / or diesel type, a vacuum distillate fraction and a vacuum residue fraction and / or a fraction of atmospheric residue.
- the heavy fraction sent in the precipitation step c) corresponds at least in part to an atmospheric residue fraction.
- the separation may be carried out in a fractionation section which may first comprise a high temperature high pressure separator (HPHT), and optionally a low temperature high pressure separator (HPBT), and / or atmospheric distillation and / or distillation under empty.
- HPHT high temperature high pressure separator
- HPBT low temperature high pressure separator
- the effluent obtained at the end of step a) is separated (generally in an HPHT separator) into a light fraction and a heavy fraction containing predominantly boiling compounds at least 350 ° C.
- the cutting point of the separation is advantageously between 200 and 400 ° C.
- the effluent from the hydrocracking may also undergo a succession of flashes comprising at least one high temperature high pressure balloon (HPHT) and a low pressure balloon high temperature (BPHT) for separating a heavy fraction which is sent in a steam stripping step for removing from said heavy fraction at least a light fraction rich in hydrogen sulfide.
- HPHT high temperature high pressure balloon
- BPHT low pressure balloon high temperature
- the heavy fraction recovered at the bottom of the stripping column contains compounds boiling at least 350 ° C. but also atmospheric distillates.
- said heavy fraction separated from the light fraction rich in hydrogen sulphide is then sent to the precipitation step c) and then to the sediment separation step d).
- At least a portion of the so-called heavy fraction from step b) is fractionated by atmospheric distillation into at least one atmospheric distillate fraction containing at least one light fraction of naphtha, kerosene and / or diesel type hydrocarbons. and an atmospheric residue fraction.
- At least a part of the fraction atmospheric residue, corresponding at least in part to the heavy fraction resulting from step b), can be sent in the precipitation step c) and then in step d) of physical separation of the sediments .
- the atmospheric residue may also be at least partially fractionated by vacuum distillation into a vacuum distillate fraction containing vacuum gas oil and a vacuum residue fraction.
- Said fraction vacuum residue corresponding at least in part to the heavy fraction from step b), is advantageously sent at least partly in the precipitation step c) and then in step d) of physical separation of the sediments d).
- At least a portion of the vacuum distillate and / or the vacuum residue may also be recycled to the hydrocracking step a).
- the light fraction (s) obtained may (may) undergo further separation steps, possibly in the presence of the light fraction obtained from the internal separator. stage between the two hydrocracking reactors.
- it (s) is (are) subject (s) to atmospheric distillation to obtain a gaseous fraction, at least a light fraction of naphtha, kerosene and / or diesel type hydrocarbons and a vacuum distillate fraction.
- Part of the atmospheric distillate and / or the vacuum distillate from the separation step b) may constitute a part of a fuel oil as a fluxing agent. These cuts can also be marine fuels with low viscosity (MGO or MGO, Marine Diesel Oil or Marine Gas Oil according to English terminology). Another part of the vacuum distillate can still be upgraded by hydrocracking and / or catalytic cracking in a fluidized bed.
- the gaseous fractions resulting from the separation step preferably undergo a purification treatment to recover the hydrogen and recycle it to the hydrocracking reactors (step a)). Part of the purified hydrogen can be used during the precipitation step.
- the recovery of different fuel base cuts (LPG, naphtha, kerosene, diesel and / or vacuum gas oil) obtained from the present invention is well known to those skilled in the art.
- the products obtained can be integrated in fuel tanks (also called “pools" fuels according to the English terminology) or undergo additional refining steps.
- the fraction (s) naphtha, kerosene, gas oil and vacuum gas oil may be subjected to one or more treatments (hydrotreatment, hydrocracking, alkylation, isomerization, catalytic reforming, catalytic cracking or thermal or other) to bring them to the specifications. required (sulfur content, smoke point, octane, cetane, etc ...) separately or in mixture.
- the vacuum distillate leaving the bubbling bed after separation can be hydrotreated.
- This hydrotreated vacuum distillate may be used as a fluxing agent for the fuel oil pool having a sulfur content of less than or equal to 0.5% by weight or may be used directly as oil with a sulfur content of less than or equal to 0.1% by weight.
- Part of the atmospheric residue, vacuum distillate and / or vacuum residue may undergo further refining steps, such as hydrotreatment, hydrocracking, or fluidized catalytic cracking.
- the heavy fraction obtained at the end of the separation step b) contains organic sediments which result from the hydrocracking conditions and the catalyst residues.
- Part of the sediments consist of asphaltenes precipitated under hydrocracking conditions and are analyzed as existing sediments (IP375).
- the process according to the invention comprises a precipitation step making it possible to improve the sediment separation efficiency and thus to obtain stable fuel oils or bases, that is to say a sediment content after aging less than or equal to 0.1% by weight.
- the precipitation step according to the invention makes it possible to form all the existing and potential sediments (by converting the potential sediments into existing sediments) so as to separate them more effectively and thus respect the sediment content after aging (measured according to the ISO 10307-2 method) of 0.1% maximum weight.
- the precipitation step according to the invention comprises bringing the heavy fraction resulting from the separation step b) into contact with a distillate cut of which at least 20% by weight has a boiling temperature greater than or equal to 100 ° C. C, preferably greater than or equal to 120 ° C, more preferably greater than or equal to 150 ° C.
- the distillate cut is characterized in that it comprises at least 25% by weight having a boiling point greater than or equal to 100 ° C. preferably greater than or equal to 120 ° C, more preferably greater than or equal to 150 ° C.
- At least 5% by weight or even 10% by weight of the distillate cut according to the invention has a boiling point of at least 252 ° C.
- At least 5 wt.% Or even 10 wt.% Of the distillate cut according to the invention has a boiling point of at least 255 ° C.
- the precipitation step c) according to the invention is advantageously carried out for a residence time of less than 500 minutes, preferably less than 300 minutes, more preferably less than 60 minutes, at a temperature between 25 and 350 °. C, preferably between 50 and 350 ° C, preferably between 65 and 300 ° C and more preferably between 80 and 250 ° C.
- the pressure of the precipitation step is advantageously less than 20 MPa, preferably less than 10 MPa, more preferably less than 3 MPa and even more preferably less than 1.5 MPa.
- the distillate cut according to the invention advantageously comprises hydrocarbons having more than 12 carbon atoms, preferably hydrocarbons having more than 13 carbon atoms, more preferably hydrocarbons having between 13 and 40 carbon atoms.
- Said distillate fraction can partly or entirely from the separation step b) of the invention or another refining process or another chemical process.
- Said distillate cut may be used in a mixture with a naphtha-type cut and / or a vacuum-type gas oil cut and / or vacuum residue.
- Said distillate cut may be used in a mixture with the light fraction obtained after step b), the atmospheric distillate fraction resulting from step b) and / or the vacuum distillate fraction from step b ) of seperation.
- the distillate cut according to the invention is mixed with another cut, a light fraction and / or a heavy fraction as indicated above, the proportions are chosen so that the resulting mixture respects the characteristics of the the distillate cut according to the invention.
- distillate cut according to the invention has the advantage of avoiding the majority use of high value added cuts such as petrochemical cuts, naphtha ...
- the mass ratio between the distillate cut according to the invention and the heavy fraction obtained at the end of the separation step b) is between 0.01 and 100, preferably between 0.05 and 10, more preferably between 0, 1 and 5, and even more preferably between 0.1 and 2.
- the distillate cut according to the invention is at least drawn from the process, it is possible to accumulate this cut during a start-up period so as to reach the desired ratio.
- the precipitation step can be carried out using an exchanger or a heating furnace followed by one or more capacity (s) in series or in parallel such (s) as a horizontal or vertical balloon, optionally with a settling function to remove some of the heavier solids, and / or a piston reactor.
- a stirred and heated tank may also be used, and may be provided with a bottom draw to remove some of the heavier solids.
- the precipitation step can be carried out online, without buffer capacity, possibly using a static mixer.
- step c) of precipitation of the heavy fraction resulting from step b) is carried out in the presence of an inert gas and / or an oxidizing gas and / or an oxidizing liquid and / or hydrogen, preferably from the separation steps of the process of the invention, in particular from the separation step b).
- the precipitation step c) can be carried out in the presence of an inert gas such as dinitrogen, or in the presence of an oxidizing gas such as dioxygen, ozone or nitrogen oxides, or in the presence of a mixture containing an inert gas and an oxidizing gas such as air or air depleted by nitrogen, or in the presence of an oxidizing liquid to accelerate the precipitation process.
- oxidizing liquid means an oxygenated compound, for example a peroxide such as hydrogen peroxide, or an inorganic oxidizing solution such as a solution of potassium permanganate or a mineral acid such as sulfuric acid. According to this variant, the oxidizing liquid is then mixed with the heavy fraction from the separation step b) and the distillate cut according to the invention during the implementation of step c).
- Step d) Separation of sediments
- the method according to the invention further comprises a step d) of physical separation of sediments and catalyst residues.
- the heavy fraction obtained at the end of the precipitation step c) contains precipitated asphaltene-type organic sediments which result from hydrocracking and precipitation conditions. This heavy fraction may also contain catalyst fines resulting from the attrition of extruded type catalysts in the implementation of hydrocracking reactor. This heavy fraction may optionally contain "dispersed" catalyst residues in the case of the implementation of a hybrid reactor.
- At least a portion of the heavy fraction from step c) of precipitation is subjected to a physical separation of sediments and catalyst residues, by means of at least one physical separation means selected from a filter, a separation membrane, a bed of organic or inorganic type filtering solids, electrostatic precipitation, a centrifugation system, decantation, auger withdrawal.
- a combination, in series and / or in parallel, of several separation means of the same type or different type can be used during this step d) separation of sediments and catalyst residues.
- One of these solid-liquid separation techniques may require the periodic use of a light rinsing fraction, resulting from the process or not, allowing for example the cleaning of a filter and the evacuation of sediments.
- the heavy fraction (with a sediment content after aging of less than or equal to 0.1% by weight) is obtained, comprising a portion of the distillate cut according to US Pat. invention introduced in step c).
- Step e) Recovery of the heavy fraction at the end of step d) of separation
- the mixture resulting from stage d) is advantageously introduced into a stage e) of recovery of the heavy fraction having a sediment content after aging less than or equal to 0.1% by weight, said step consisting in separating the heavy fraction from step d) of the distillate cut introduced in step c).
- Step e) is a separation step similar to separation step b).
- Step e) can be implemented by means of separator balloon and / or distillation distillation equipment so as to separate on the one hand at least part of the distillate cup introduced during step c) of precipitation and on the other hand the heavy fraction having a sediment content after aging less than or equal to 0.1% by weight.
- a part of the distillate cut separated from step e) is recycled in step c) of precipitation.
- Said recovered heavy fraction may advantageously be used as a base of fuel oil or as fuel oil, especially as a base of bunker oil or as bunker oil, having a sediment content after aging less than 0.1% by weight.
- said heavy fraction is mixed with one or more fluxing bases selected from the group consisting of catalytic cracking light cutting oils, catalytic cracking heavy cutting oils, catalytic cracking residue, kerosene, a gas oil, a vacuum distillate and / or a decanted oil.
- one or more fluxing bases selected from the group consisting of catalytic cracking light cutting oils, catalytic cracking heavy cutting oils, catalytic cracking residue, kerosene, a gas oil, a vacuum distillate and / or a decanted oil.
- part of the distillate cut according to the invention can be left in the sediment-reduced heavy fraction so that the viscosity of the mixture is directly that of a desired fuel grade, for example 180 or 380 cSt at 50 ° C.
- the sulfur content of the heavy fraction resulting from stage d) or e), and containing predominantly compounds boiling at least 350 ° C., is a function of the operating conditions of the hydrocracking stage but also of the content of the hydrocracking stage. sulfur of the original charge.
- a step f) of hydrotreatment in a fixed bed is made necessary in the case where the refiner wishes to reduce the sulfur content, in particular for a bunker oil base or a bunker oil intended to be burned on a ship without smoke treatment.
- Stage f) of hydrotreatment in a fixed bed is carried out on at least a part of the heavy fraction resulting from stage d) or e).
- the heavy fraction from step f) can advantageously be used as a base of fuel oil or as fuel oil, especially as a base of bunker oil or as bunker oil, having a sediment content after aging less than 0.1% by weight.
- said heavy fraction is mixed with one or more fluxing bases selected from the group consisting of catalytic cracking light cutting oils, catalytic cracking heavy cutting oils, catalytic cracking residue, kerosene, a gas oil, a vacuum distillate and / or a decanted oil.
- the heavy fraction resulting from step d) or e) is sent to the hydrotreatment step f) comprising one or more hydrotreatment zones in fixed beds.
- the sending in a fixed bed of a heavy fraction devoid of sediment is an advantage of the present invention since the fixed bed will be less subject to clogging and increased pressure drop.
- Hydroprocessing is understood to mean, in particular, hydrodesulphurization (HDS) reactions, hydrodenitrogenation (HDN) reactions and hydrodemetallation (HDM) reactions, but also hydrogenation, hydrodeoxygenation, hydrodearomatization, hydrodenetration, hydroisomerization, hydrodealkylation, hydrocracking, hydro-deasphalting and Conradson carbon reduction.
- HDS hydrodesulphurization
- HDN hydrodenitrogenation
- HDM hydrodemetallation
- Such a method of hydrotreating heavy cuts is widely known and can be related to the process known as HYVAHL-F TM described in US Pat. US5417846 .
- hydrodemetallation reactions are mainly carried out but also part of the hydrodesulfurization reactions.
- hydrodesulphurization reactions are mainly carried out but also part of the hydrodemetallation reactions.
- a co-charge may be introduced with the heavy fraction in the hydrotreatment step f).
- This co-charge can be chosen from atmospheric residues, vacuum residues from direct distillation, deasphalted oils, aromatic extracts from lubricant base production lines, hydrocarbon fractions or a mixture of hydrocarbon fractions that can be chosen. from products resulting from a fluid-bed catalytic cracking process: a light cutting oil (LCO), a heavy cutting oil (HCO), a decanted oil, or possibly derived from distillation, the gas oil fractions, in particular those obtained by distillation atmospheric or vacuum, such as vacuum gas oil.
- LCO light cutting oil
- HCO heavy cutting oil
- decanted oil or possibly derived from distillation
- the gas oil fractions in particular those obtained by distillation atmospheric or vacuum, such as vacuum gas oil.
- the hydrotreatment step can advantageously be carried out at a temperature of between 300 and 500 ° C., preferably 350 ° C. to 420 ° C. and under a hydrogen partial pressure advantageously between 2 MPa and 25 MPa, preferably between 10 and 20 MPa, an overall hourly space velocity (VVH) which is defined as the volumetric flow rate of the feed divided by the total volume of the catalyst, being in a range from 0.1 hr-1 to 5 hr -1 and preferably from 0.1 hr -1 to 2 hr -1, a quantity of hydrogen mixed with the load usually of 100 to 5000 Nm3 / m3 (normal cubic meters (Nm3) per cubic meter (m3) of filler liquid), most often from 200 to 2000 Nm3 / m3 and preferably from 300 to 1500 Nm3 / m3.
- VVH hourly space velocity
- the hydrotreating step is carried out industrially in one or more liquid downflow reactors.
- the hydrotreatment temperature is generally adjusted according to the desired level of hydrotreatment.
- the hydrotreatment catalysts used are preferably known catalysts and are generally granular catalysts comprising, on a support, at least one metal or metal compound having a hydrodehydrogenating function. These catalysts are advantageously catalysts comprising at least one Group VIII metal, generally selected from the group consisting of nickel and / or cobalt, and / or at least one Group VIB metal, preferably molybdenum and / or tungsten. .
- a catalyst comprising from 0.5 to 10% by weight of nickel and preferably from 1 to 5% by weight of nickel (expressed as nickel oxide NiO) and from 1 to 30% by weight of molybdenum, preferably from 5 to 20% by weight of molybdenum (expressed as molybdenum oxide MoO 3 ) on a mineral support.
- This support will, for example, be selected from the group formed by alumina, silica, silica-aluminas, magnesia, clays and mixtures of at least two of these minerals.
- this support contains other doping compounds, in particular oxides chosen from the group formed by boron oxide, zirconia, ceria, titanium oxide, phosphoric anhydride and a mixture of these oxides.
- an alumina support is used and very often a support of alumina doped with phosphorus and possibly boron.
- concentration of phosphorus pentoxide P 2 O 5 is usually between 0 or 0.1% and 10% by weight.
- concentration of trioxide boron B 2 O 5 is usually from 0 or 0.1% to 10% by weight.
- the alumina used is usually a ⁇ or ⁇ alumina. This catalyst is most often in the form of extrudates.
- the total content of metal oxides of groups VIB and VIII is often from 5 to 40% by weight and in general from 7 to 30% by weight and the weight ratio expressed as metal oxide between metal (or metals) of Group VIB on metal (or metals) of group VIII is usually 20 to 1 and most often 10 to 2.
- hydrotreatment step including a hydrodemetallation step (HDM), then a hydrodesulfurization step (HDS), it is most often used specific catalysts adapted to each step.
- HDM hydrodemetallation step
- HDS hydrodesulfurization step
- Catalysts that can be used in the hydrodemetallation (HDM) stage are for example indicated in the patents EP113297 , EP113284 , US5221656 , US5827421 , US7119045 , US5622616 and US5089463 .
- Hydrodemetallation (HDM) catalysts are preferably used in the reactive reactors.
- Catalysts that can be used in the hydrodesulfurization (HDS) step are, for example, indicated in the patents EP113297 , EP113284 , US6589908 , US4818743 or US6332976 . It is also possible to use a mixed catalyst that is active in hydrodemetallization and in hydrodesulfurization for both the hydrodemetallation (HDM) section and the hydrodesulfurization (HDS) section as described in the patent. FR2940143 .
- the catalysts used in the process according to the present invention are preferably subjected to an in-situ or ex-situ sulphurization treatment .
- Step g) Optional step of separation of the hydrotreatment effluent
- the process according to the invention may comprise a step g) of separating the effluents from the hydrotreating step f).
- the optional separation step g) may advantageously be carried out by any method known to those skilled in the art such as, for example, the combination of one or more high and / or low pressure separators, and / or distillation and / or high and / or low pressure stripping.
- This optional separation step g) is similar to the separation step b) and will not be further described.
- the effluent obtained in step f) may be at least partly, and often entirely, sent to a separation step g), comprising atmospheric distillation and / or vacuum distillation.
- the effluent of the hydrotreatment step is fractionated by atmospheric distillation into a gaseous fraction, at least one atmospheric distillate fraction containing the fuels bases (naphtha, kerosene and / or diesel) and an atmospheric residue fraction. At least a portion of the atmospheric residue can then be fractionated by vacuum distillation into a vacuum distillate fraction containing vacuum gas oil and a vacuum residue fraction.
- the vacuum residue fraction and / or the vacuum distillate fraction and / or the atmospheric residue fraction may be at least partly the bases of low-sulfur fuel oils having a sulfur content of less than or equal to 0.5% by weight and a sediment content after aging less than or equal to 0.1%.
- the vacuum distillate fraction can constitute a fuel oil base having a sulfur content of less than or equal to 0.1% by weight.
- Part of the vacuum residue and / or the atmospheric residue can also be recycled to the hydrocracking step a).
- the heavy fractions resulting from steps d) and / or e) and / or f) and / or g) can be mixed with one or more fluxing bases chosen from the group consisting of light cutting oils.
- kerosene, gas oil and / or vacuum distillate produced in the process of the invention will be used.
- use will be kerosene, gas oil and / or vacuum distillate obtained (s) in the separation steps b) or g) of the process.
- the figure 1 schematically describes an example of implementation of the invention without limiting the scope.
- the hydrocarbon feedstock (1) and hydrogen (2) are contacted in a bubbling bed hydrocracking zone a).
- the effluent (3) from the hydrocracking zone a) is sent to a separation zone b) to obtain at least a light hydrocarbon fraction (4) and a heavy liquid fraction (5) containing compounds boiling at at least 350 ° C.
- This heavy fraction (5) is brought into contact with a distillate cut (6) during a precipitation step in zone c).
- the effluent (7) consists of a heavy fraction and sediment is treated in a physical separation step in zone d) to remove a sediment-containing fraction (9) and recover a sediment-reduced liquid hydrocarbon fraction (8).
- the liquid hydrocarbon fraction (8) is then treated in a recovery step in zone e) firstly of the liquid hydrocarbon fraction (11) having a sediment content after aging less than or equal to 0.1% by weight, and on the other hand a fraction (10) containing at least a portion of the distillate cut introduced during step c).
- a vacuum residue feedstock (RSV Ural) containing 84% by weight of compounds boiling at a temperature above 520 ° C, having a density of 9.5 ° API and a sulfur content of 2.6% by weight is treated.
- the feedstock was subjected to a hydrocracking step comprising two successive bubbling bed reactors.
- the operating conditions of the hydrocracking step are given in Table 1.
- Table 1 Operating conditions of the hydrocracking section ⁇ / u> 2 bubbling beds catalysts NiMo on alumina Temperature R1 (° C) 430 Temperature R2 (° C) 430 H2 partial pressure (MPa) 15 VVH "reactors" (h-1, Sm3 / h fresh load / m3 of reactors) 0.35 VVH "bubbling bed catalysts" (h-1, Sm3 / h fresh load / m3 bubbling bed catalysts) 0.65 H2 / HC entry hydrocracking section excluding H2 consumption (Nm3 / m3 fresh load) 600
- NiMo catalyst on Alumina used is sold by the company Axens under the reference HOC-458.
- the effluent of the hydrocracking step is then subjected to a separation step for separating a gaseous fraction and a heavy liquid fraction by means of separators.
- the heavy liquid fraction is then distilled in an atmospheric distillation column so as to recover distillates and an atmospheric residue.
- the atmospheric residue corresponding to the 350 ° C + fraction of the effluent in the proportion 44% by weight of DSV and 56% by weight of RSV of the hydrocracking step of the invention is characterized by a sediment content ( IP375) of 0.4% w / w and sediment content after aging (IP390) of 0.9% w / w.
- the mixture is then subjected to a step of separating the sediments and catalyst residues by means of a Pall® brand porous metal filter.
- This step of physical separation of the sediments is followed by a distillation step of the mixture making it possible to recover, on the one hand, the atmospheric residue with a reduced sediment content, and on the other hand the distillate cut.
- the operating conditions of the hydrocracking step coupled with the different treatment variants (separation of sediments with precipitation step (B) according to the invention or without precipitation step (A)) of the atmospheric residue (RA) have an impact on the stability of the effluents obtained. This is illustrated by the post-aging sediment concentrations measured in RA atmospheric residues (350 ° C + cut) before and after the sediment precipitation and separation step.
- the atmospheric residue obtained according to the invention is an excellent fuel oil base, especially a bunker oil base having a sediment content after aging (IP390) less than 0.1% by weight.
- the RA atmospheric residue treated according to the mixture of Table 4 has a sediment content after aging of less than 0.1%, a sulfur content of 0.93% w / w and a viscosity of 380 cSt at 50 ° C.
- This atmospheric residue thus constitutes a quality bunker oil, which can be sold according to the RMG or IFO 380 grade, with low sediment content.
- it may be burned in the ECA zone or outside the ECA zones by 2020-25 provided that the vessel is equipped with flue-gas scrubbers to cut down the sulfur oxides.
Description
- La présente invention concerne le raffinage et la conversion des fractions lourdes d'hydrocarbures contenant entre autre des impuretés soufrées. Elle concerne plus particulièrement un procédé de conversion de charges lourdes pétrolières de type résidu atmosphérique et/ou résidu sous vide pour la production de fractions lourdes utilisables comme bases de fiouls, notamment de bases de fiouls de soute, à basse teneur en sédiments. Le procédé selon l'invention permet également de produire des distillats atmosphériques (naphta, kérosène et diesel), des distillats sous vide et des gaz légers (C1 à C4).
- Les exigences de qualité des combustibles marins sont décrites dans la norme ISO 8217. La spécification concernant le soufre s'attache désormais aux émissions de SOx (Annexe VI de la convention MARPOL de l'Organisation Maritime Internationale) et se traduit par une recommandation en teneur en soufre inférieure ou égale à 0,5% poids en dehors des Zones de Contrôle des Emissions de Soufre (ZCES ou Emissions Control Areas / ECA en anglais) à l'horizon 2020-2025, et inférieure ou égale à 0,1% poids dans les ZCES. Selon l'Annexe VI de la convention MARPOL, les teneurs en soufre mentionnées précédemment sont des teneurs équivalentes conduisant à des émissions de SOx. Un navire pourra donc utiliser un fioul soufré dès lors que le navire est équipé d'un système de traitement des fumées permettant de réduire des émissions d'oxydes de soufre.
- Une autre recommandation très contraignante est la teneur en sédiments après vieillissement selon ISO 10307-2 (également connue sous le nom d'IP390) qui doit être inférieure ou égale à 0,1%. La teneur en sédiments après vieillissement est une mesure réalisée selon la méthode décrite dans la norme ISO 10307-2 (également connu de l'homme du métier sous le nom de IP390). Dans la suite du texte en entendra donc par « teneur en sédiments après vieillissement», la teneur en sédiment mesurée selon la méthode ISO 10307-2. La référence à IP390 indiquera également que la mesure de la teneur en sédiments après vieillissement est réalisée selon la méthode ISO 10307-2.
- La teneur en sédiments selon ISO 10307-1 (également connue sous le nom d'IP375) est différente de la teneur en sédiments après vieillissement selon ISO 10307-2 (également connue sous le nom d'IP390). La teneur en sédiments après vieillissement selon ISO 10307-2 est une spécification beaucoup plus contraignante et correspond à la spécification s'appliquant aux fiouls de soute.
- D'autre part, les fiouls terrestres, notamment des fiouls utilisables pour la production de chaleur et/ou d'électricité peuvent également être soumis à des spécifications de stabilité, notamment des teneurs maximales en sédiments dont les seuils varient en fonction des lieux de production car il n'y a pas d'harmonisation internationale comme dans le cas du transport maritime. Il y a toutefois un intérêt à réduire la teneur en sédiments des fiouls terrestres.
- Les procédés d'hydrocraquage de résidus permettent de convertir des résidus à faible valeur en des distillats à plus forte valeur ajoutée. La fraction lourde qui en résulte correspondant à la coupe résiduelle non convertie est généralement instable. Elle contient des sédiments qui sont principalement des asphaltènes précipités. Cette coupe résiduelle instable ne peut donc pas être valorisée comme fioul, notamment comme fioul de soute sans un traitement spécifique dès lors que l'hydrocraquage est opéré dans des conditions sévères conduisant à un taux de conversion élevée.
- Le brevet
US6447671 décrit un procédé de conversion de fractions lourdes pétrolières comprenant une première étape d'hydrocraquage en lit bouillonnant, une étape d'élimination des particules de catalyseur contenues dans l'effluent de l'hydrocraquage, puis une étape d'hydrotraitement en lit fixe. - La demande
US2014/0034549 décrit un procédé de conversion de résidus mettant en oeuvre une étape d'hydrocraquage en lit bouillonnant et une étape avec un réacteur dit « upflow » associé à un réacteur dit « stripper ». La teneur en sédiments de l'effluent final est réduite par rapport à l'effluent de l'étape en lit bouillonnant. Toutefois, la teneur en sédiment après vieillissement n'est pas inférieure à 0,1% poids, telle qu'exigée pour la commercialisation comme combustible marin de type résiduel. - Le brevet
FR2981659 - Le procédé d'hydrocraquage permet de convertir partiellement les charges lourdes afin de produire des distillats atmosphériques et/ou de distillats sous vide. Bien que la technologie en lit bouillonnant soit connue pour être adaptée à des charges lourdes chargées en impuretés, le lit bouillonnant produit de par sa nature des fines de catalyseurs et des sédiments qui doivent être enlevés pour satisfaire une qualité de produit tel que le fioul de soute. Les fines proviennent principalement de l'attrition du catalyseur dans le lit bouillonnant.
- Les sédiments peuvent être des asphaltènes précipités. Initialement dans la charge, les conditions d'hydrocraquage et notamment la température font qu'ils subissent des réactions (déalkylation, polycondensation...) conduisant à leur précipitation. Ces phénomènes interviennent généralement lors de mise en oeuvre de conditions sévères donnant lieu à des taux de conversion (pour les composés bouillant à plus de 540°C : 540+°C) élevés, par exemple supérieurs à 30, 40 ou 50% en fonction de la nature de la charge.
- La demanderesse dans ses recherches a mis au point un nouveau procédé intégrant une étape de précipitation et de séparation physique des sédiments en aval d'une étape d'hydrocraquage. De manière surprenante, il a été trouvé qu'un tel procédé permettait d'obtenir des fractions lourdes présentant une basse teneur en sédiments après vieillissement, lesdites fractions lourdes pouvant avantageusement être utilisées totalement ou en partie comme fioul ou comme base de fioul, notamment comme fioul de soute ou base de fioul de soute répondant aux spécifications, à savoir et une teneur en sédiments après vieillissement (mesurée selon la méthode ISO 10307-2) inférieure ou égale à 0,1% en poids.
- Un avantage du procédé selon l'invention est d'éviter notamment les risques d'encrassement des moteurs de bateaux. Un autre avantage du procédé de l'invention est d'éviter les risques d'encrassement, dans le cas d'éventuelles étapes de traitement mises en oeuvre en aval de l'étape d'hydrocraquage d'éviter un bouchage du ou des lit(s) catalytique(s) mis en oeuvre.
- Plus particulièrement, l'invention concerne un procédé de conversion d'une charge hydrocarbonée contenant au moins une fraction d'hydrocarbures ayant une teneur en soufre d'au moins 0,1 % poids, une température initiale d'ébullition d'au moins 340°C et une température finale d'ébullition d'au moins 440°C, ledit procédé comprenant les étapes suivantes :
- a) une étape d'hydrocraquage de la charge en présence d'hydrogène dans au moins un réacteur contenant un catalyseur supporté en lit bouillonnant,
- b) une étape de séparation de l'effluent obtenu à l'issue de l'étape a) en au moins une fraction légère d'hydrocarbures contenant des bases carburants et une fraction lourde contenant des composés bouillant à au moins 350°C,
- c) une étape de précipitation des sédiments dans laquelle la fraction lourde issue de l'étape de séparation b) est mise en contact avec une coupe de distillat dont au moins 20% poids présente une température d'ébullition supérieure ou égale à 100°C, pendant une durée inférieure à 500 minutes, à une température comprise entre 25 et 350°C, et une pression inférieure à 20 MPa,
- d) une étape de séparation physique des sédiments de la fraction lourde issue de l'étape c) de précipitation pour obtenir une fraction lourde séparée des sédiments,
- e) une étape de récupération d'une fraction lourde ayant une teneur en sédiments, mesurée selon la méthode ISO 10307-2, inférieure ou égale à 0,1% en poids consistant à séparer la fraction lourde issue de l'étape d) de la coupe de distillat introduite lors de l'étape c).
- Afin de constituer le fioul répondant aux recommandations de la viscosité et de la teneur en sédiments après vieillissement, les fractions lourdes obtenues par le présent procédé peuvent être mélangées avec des bases fluxantes de manière à atteindre la viscosité cible du grade de fioul désiré ainsi que la spécification en teneur en sédiments après vieillissement.
- Un autre point d'intérêt du procédé est la conversion partielle de la charge permettant de produire, notamment par l'hydrocraquage, des distillats atmosphériques ou des distillats sous vide (naphta, kérosène, diesel, distillat sous vide), valorisables comme bases dans les pools carburants directement ou après passage dans un autre procédé de raffinage tel que l'hydrotraitement, le reformage, l'isomérisation, l'hydrocraquage ou le craquage catalytique.
- La
figure 1 illustre une vue schématique du procédé selon l'invention faisant apparaitre une zone d'hydrocraquage, une zone de séparation, une zone de précipitation, une zone de séparation physique des sédiments et une zone de récupération de la fraction d'intérêt. - Les charges traitées dans le procédé selon l'invention sont avantageusement choisies parmi les résidus atmosphériques, les résidus sous vide issus de distillation directe, des pétroles bruts, des pétroles bruts étêtés, les huiles désasphaltées, des résines de désasphaltage, les asphaltes ou brais de désasphaltage, les résidus issus des procédés de conversion, des extraits aromatiques issus des chaînes de production de bases pour lubrifiants, des sables bitumineux ou leurs dérivés, des schistes bitumineux ou leurs dérivés, pris seuls ou en mélange.
- Ces charges peuvent avantageusement être utilisées telles quelles ou encore diluées par une fraction hydrocarbonée ou un mélange de fractions hydrocarbonées pouvant être choisies parmi les produits issus d'un procédé de craquage catalytique en lit fluide (FCC selon les initiales de la dénomination anglo-saxonne de « Fluid Catalytic Cracking »), une huile de coupe légère (LCO), une huile de coupe lourde (HCO), une huile décantée (DO selon les initiales de la dénomination anglo-saxonne de « Decanted Oil »), un résidu de FCC, ou pouvant venir de la distillation, les fractions gazoles notamment celles obtenues par distillation atmosphérique ou sous vide, comme par exemple le gazole sous vide. Les charges lourdes peuvent aussi avantageusement comprendre des coupes issues du procédé de liquéfaction du charbon ou de la biomasse, des extraits aromatiques, ou toutes autres coupes hydrocarbonées ou encore des charges non pétrolières comme de l'huile de pyrolyse de biomasses lignocellulosiques.
- Les charges selon l'invention ont généralement une teneur en soufre d'au moins 0,1 % poids, une température initiale d'ébullition d'au moins 340°C et une température finale d'ébullition d'au moins 440°C, de manière préférée une température finale d'ébullition d'au moins 540°C. Avantageusment, la charge peut contenir au moins 1% d'asphaltènes C7 et au moins 5 ppm de métaux, de préférence au moins 2% d'asphaltènes C7 et au moins 25 ppm de métaux.
- Les charges selon l'invention sont de préférence des résidus atmosphériques ou des résidus sous vide, ou des mélanges de ces résidus.
- La charge selon l'invention est soumise à une étape d'hydrocraquage qui est réalisée dans au moins un réacteur contenant un catalyseur supporté en lit bouillonnant et de préférence fonctionnant à courant ascendant de liquide et de gaz. L'objectif de l'étape d'hydrocraquage est de convertir la fraction lourde en coupes plus légères tout en raffinant partiellement la charge.
- La technologie en lit bouillonnant étant largement connue, on ne reprendra ici que les principales conditions opératoires.
- Les technologies à lits bouillonnants utilisent des catalyseurs à lits bouillonnants supportés sous forme d'extrudés dont le diamètre est généralement de l'ordre de 1mm ou inférieur à 1mm. Les catalyseurs restent à l'intérieur des réacteurs et ne sont pas évacués avec les produits. Les niveaux de température sont élevés afin d'obtenir des conversions élevées tout en minimisant les quantités de catalyseurs mises en oeuvre. L'activité catalytique peut être maintenue constante grâce au remplacement en ligne du catalyseur. Il n'est donc pas nécessaire d'arrêter l'unité pour changer le catalyseur usagé, ni d'augmenter les températures de réaction le long du cycle pour compenser la désactivation. De plus, le fait de travailler à des conditions opératoires constantes permet d'obtenir des rendements et des qualités de produits constants le long du cycle. Aussi, du fait que le catalyseur est maintenu en agitation par un recyclage important de liquide, la perte de charge sur le réacteur reste faible et constante.
- Les conditions de l'étape a) d'hydrocraquage de la charge en présence d'hydrogène sont habituellement des conditions classiques d'hydrocraquage en lit bouillonnant d'une fraction hydrocarbonée liquide. On opère avantageusment sous une pression partielle d'hydrogène de 5 à 35 MPa, souvent de 8 à 25 MPa et le plus souvent de 12 à 20 MPa à une température de 330 à 500 °C et souvent de 350 à 450 °C. La vite sse spatiale horaire (VVH) et la pression partielle d'hydrogène sont des facteurs importants que l'on choisit en fonction des caractéristiques du produit à traiter et de la conversion souhaitée. La VVH, défini comme étant le débit volumétrique de la charge divisée par le volume total du réacteur, se situe généralement dans une gamme allant de 0,05 h-1 à 5 h-1, de préférence de 0,1 h-1 à 2 h-1 et de manière plus préférée de 0,2 h-1 à 1 h-1. La quantité d'hydrogène mélangé à la charge est habituellement de 50 à 5000 Nm3/m3 (normaux mètres cube (Nm3) par mètre cube (m3) de charge liquide) et le plus souvent de 100 à 1000 Nm3/m3 et de préférence de 200 à 500 Nm3/m3.
- On peut utiliser un catalyseur granulaire classique d'hydrocraquage comprenant, sur un support amorphe, au moins un métal ou composé de métal ayant une fonction hydrodéshydrogénante. Ce catalyseur peut être un catalyseur comprenant des métaux du groupe VIII par exemple du nickel et/ou du cobalt le plus souvent en association avec au moins un métal du groupe VIB par exemple du molybdène et/ou du tungstène. On peut par exemple employer un catalyseur comprenant de 0,5 à 10 % en poids de nickel et de préférence de 1 à 5 % en poids de nickel (exprimé en oxyde de nickel NiO) et de 1 à 30 % en poids de molybdène de préférence de 5 à 20 % en poids de molybdène (exprimé en oxyde de molybdène MoO3) sur un support minéral amorphe. Ce support sera par exemple choisi dans le groupe formé par l'alumine, la silice, les silices-alumines, la magnésie, les argiles et les mélanges d'au moins deux de ces minéraux. Ce support peut également renfermer d'autres composés et par exemple des oxydes choisis dans le groupe formé par l'oxyde de bore, la zircone, l'oxyde de titane, l'anhydride phosphorique. On utilise le plus souvent un support d'alumine et très souvent un support d'alumine dopée avec du phosphore et éventuellement du bore. Lorsque l'anhydride phosphorique P2O5 est présent, sa concentration est habituellement inférieure à 20 % en poids et le plus souvent inférieure à 10 % en poids. La concentration du trioxyde de bore B2O3 est habituellement de 0 à 10 % en poids. L'alumine utilisée est habituellement une alumine gamma ou êta. Ce catalyseur est le plus souvent sous forme d'extrudés. La teneur totale en oxydes de métaux des groupes VI et VIII est souvent de 5 à 40 % en poids et en général de 7 à 30 % en poids et le rapport pondéral exprimé en oxyde métallique entre métal (ou métaux) du groupe VI sur métal (ou métaux) du groupe VIII est en général de 20 à 1 et le plus souvent de 10 à 2.
- Le catalyseur usagé est en partie remplacé par du catalyseur frais, généralement par soutirage en bas du réacteur et introduction en haut du réacteur de catalyseur frais ou neuf à intervalle de temps régulier c'est-à-dire par exemple par bouffée ou de façon quasi continue. On peut également introduire le catalyseur par le bas et le soutiré par le haut du réacteur. On peut par exemple introduire du catalyseur frais tous les jours. Le taux de remplacement du catalyseur usé par du catalyseur frais peut être par exemple d'environ 0,05 kilogramme à environ 10 kilogrammes par mètre cube de charge. Ce soutirage et ce remplacement sont effectués à l'aide de dispositifs permettant le fonctionnement continu de cette étape d'hydrocraquage. L'unité comporte habituellement une pompe de recirculation permettant le maintien du catalyseur en lit bouillonnant par recyclage continu d'au moins une partie du liquide soutiré en tête du réacteur et réinjecté en bas du réacteur. Il est également possible d'envoyer le catalyseur usé soutiré du réacteur dans une zone de régénération dans laquelle on élimine le carbone et le soufre qu'il renferme avant son réinjection dans l'étape a) d'hydrocraquage.
- Le plus souvent l'étape a) d'hydrocraquage est mise en oeuvre dans les conditions du procédé H-OIL® tel que décrit par exemple dans
US6270654 . - L'hydrocraquage peut se faire dans un seul réacteur ou dans plusieurs réacteurs (généralement deux) disposées en série. L'utilisation d'au moins deux réacteurs en lit bouillonnant en série permet d'obtenir des produits de meilleure qualité et avec un meilleur rendement, limitant ainsi les besoins d'énergie et d'hydrogène dans des post-traitements éventuels. En plus, l'hydrocraquage en deux réacteurs permet d'avoir une opérabilité améliorée au niveau de la flexibilité des conditions opératoires et du système catalytique. Généralement, la température du deuxième réacteur est de préférence au moins 5°C plus élevée que celle du premier réacteur en lit bouillonnant. La pression du deuxième réacteur est de 0,1 à 1 MPa plus faible que pour le premier réacteur pour permettre l'écoulement d'au moins une partie de l'effluent issue de la première étape sans qu'un pompage soit nécessaire. Les différentes conditions opératoires en termes de température dans les deux réacteurs d'hydrocraquage sont sélectionnées pour pouvoir contrôler l'hydrogénation et la conversion de la charge en produits souhaités dans chaque réacteur. Éventuellement, l'effluent obtenu à l'issue du premier réacteur d'hydrocraquage est soumis à une séparation de la fraction légère et au moins une partie, de préférence la totalité, de l'effluent résiduel est traitée dans le deuxième réacteur d'hydrocraquage.
- Cette séparation peut être réalisée dans un séparateur inter-étage tel que décrit dans le brevet
US 6270654 et permet notamment d'éviter un hydrocraquage trop poussé de la fraction légère dans le deuxième réacteur d'hydrocraquage. - Il est également possible de transférer en totalité ou en partie le catalyseur usagé soutiré du premier réacteur d'hydrocraquage, opérant à plus basse température, directement dans le deuxième réacteur d'hydrocraquage, opérant à température plus élevée ou de transférer en totalité ou en partie le catalyseur usagé soutiré du deuxième réacteur d'hydrocraquage directement au premier réacteur d'hydrocraquage. Ce système de cascade est décrit dans le brevet
US4816841 . - L'étape d'hydrocraquage peut aussi se faire dans au moins un réacteur fonctionnant en mode lit hybride, c'est-à-dire fonctionnant en lit bouillonnant avec un catalyseur supporté associé à un catalyseur dispersé constitué de particules de catalyseur très fines le tout formant une suspension avec la charge à traiter.
- Un lit hybride comporte deux populations de catalyseur, une population de catalyseur de type lit bouillonnant à laquelle s'ajoute une population de catalyseur de type "dispersé ". Le terme "dispersé " désigne une mise en oeuvre du réacteur dans laquelle le catalyseur est sous forme de particules très fines, c'est à dire généralement une taille comprise entre 1 nanomètre (soit 10-9 m) et 150 micromètres, de manière préférée entre 0,1 et 100 micromètres, et de manière encore plus préférée entre 10 et 80 microns.
- Dans une première variante, l'étape d'hydrocraquage peut comporter un premier réacteur de type lit bouillonnant suivi d'un second réacteur de type lit hybride (c'est à dire de type lit bouillonnant avec injection de catalyseur de type "dispersé").
- Dans une seconde variante, l'étape d'hydrocraquage peut comporter un premier réacteur de type lit hybride suivi d'un second réacteur de type hybride.
- Dans une troisième variante, l'étape d'hydrocraquage peut comporter un seul réacteur de type lit hybride.
- Le catalyseur "dispersé" utilisé dans le réacteur en lit hybride peut être un catalyseur sulfure contenant de préférence au moins un élément choisi dans le groupe forme par Mo, Fe, Ni, W, Co, V, Ru. Ces catalyseurs sont généralement monométalliques ou bimétalliques (en combinant par exemple un élément du groupe VIIIB non-noble (Co, Ni, Fe) et un élément du groupe VIB (Mo, W). Les catalyseurs utilisés peuvent être des poudres de solides hétérogènes (tels que des minerais naturels, du sulfate de fer, etc...), des catalyseurs dispersés issus de précurseurs solubles dans l'eau tels que l'acide phosphomolybdique, le molybdate d'ammonium, ou un mélange d'oxyde Mo ou Ni avec de l'ammoniaque aqueux. De préférence, les catalyseurs utilisés sont issus de précurseurs solubles dans une phase organique (catalyseurs solubles dans l'huile).
- Les précurseurs sont généralement des composés organométalliques tels que les naphténates de Mo, de Co, de Fe, ou de Ni, ou les octoates de Mo, ou des composés multi-carbonyl de ces métaux, par exemple 2-ethyl hexanoates de Mo ou Ni, acétylacétonates de Mo ou Ni, sels d'acides gras C7-C12 de Mo ou W, etc. Ils peuvent être utilisés en présence d'un agent tensio-actif pour améliorer la dispersion des métaux, lorsque le catalyseur est bimétallique. Les catalyseurs se trouvent sous forme de particules dispersées, colloïdales ou non selon la nature du catalyseur. De tels précurseurs et catalyseurs utilisables dans le procédé selon l'invention sont largement décrits dans la littérature.
- En général, les catalyseurs sont préparés avant d'être injectes dans la charge. Le procédé de préparation est adapté en fonction de l'état dans lequel se trouve le précurseur et de sa nature. Dans tous les cas, le précurseur est sulfuré (ex-situ ou in-situ) pour former le catalyseur dispersé dans la charge.
- Pour le cas des catalyseurs dits solubles dans l'huile, le précurseur est avantageusement mélangé à une charge carbonée (qui peut être une partie de la charge à traiter, une charge externe, une fraction recyclée...), le mélange est ensuite sulfuré par addition d'un compose soufré (hydrogène sulfuré préféré ou éventuellement un sulfure organique tel du DMDS en présence d'hydrogène) et chauffé. Les préparations de ces catalyseurs sont décrites dans la littérature. Les particules de catalyseurs "dispersées" telles que définies ci-dessus (poudres de composés minéraux métalliques ou issus de précurseurs solubles dans l'eau ou dans l'huile) ont généralement une taille comprise entre 1 nanomètre et 150 micromètres, de manière préférée entre 0,1 et 100 micromètres, et de manière encore plus préférée entre 10 et 80 microns. La teneur en composés catalytiques (exprimée en pourcentage poids d'éléments métalliques du groupe VIII et/ou du groupe VIB) est comprise entre 0 et 10% pds, de préférence entre 0 et 1% poids.
- Des additifs peuvent être ajoutés lors de la préparation du catalyseur ou au catalyseur en "dispersé" avant qu'il soit injecté dans le réacteur. Ces additifs sont décrits dans la littérature.
- Les additifs solides préférés sont des oxydes minéraux tels que l'alumine, la silice, des oxydes mixtes Al/Si, des catalyseurs usagés supportés (par exemple, sur alumine et/ou silice) contenant au moins un élément du groupe VIII (tel que Ni, Co) et/ou au moins un élément du groupe VIB (tel que Mo, W). On citera par exemple les catalyseurs décrits dans la demande
US2008/177124 . Des solides carbonés à faible teneur d'hydrogène (par exemple 4% d'hydrogène) tels que du coke ou du charbon actif broyé, éventuellement prétraités, peuvent être également utilisés. On peut également utiliser des mélanges de tels additifs. La taille de particules de l'additif est généralement comprise entre 10 et 750 microns, de manière préférée entre 100 et 600 microns. La teneur en éventuel additif solide présent à l'entrée de la zone réactionnelle du procédé d'hydrocraquage en "dispersé" est comprise entre 0 et 10% pds, préférentiellement entre 1 et 3% pds, et la teneur en composés catalytiques (exprimée en pourcentage poids d'éléments métalliques du groupe VIII et/ou du groupe VIB) est comprise entre 0 et 10% poids, de préférence entre 0 et 1% poids. - Le ou les réacteurs à lit hybride utilisés dans la zone d'hydrocraquage sont donc constitués par deux populations de catalyseurs, une première population utilisant des catalyseurs supportés sous forme d'extrudés dont le diamètre est avantageusement compris entre 0,8 et 1,2 mm, généralement égal à 0,9 mm ou 1,1 mm et une seconde population de catalyseur de type « dispersé » dont il a été question plus haut.
- La fluidisation des particules de catalyseurs dans le lit bouillonnant est permise par l'utilisation d'une pompe d'ébullition qui permet un recyclage de liquide, généralement à l'intérieur du réacteur. Le débit de liquide recyclé par la pompe d'ébullition est ajusté de telle sorte à ce que les particules de catalyseurs supportés soient fluidisées mais pas transportées, de manière donc à ce que ces particules restent dans le réacteur en lit bouillonnant (à l'exception des fines de catalyseurs qui peuvent être formées par attrition et entrainées avec le liquide puisque ces fines sont de petite taille). Dans le cas d'un lit hybride, le catalyseur de type « dispersé » est également entrainé avec le liquide puisque le catalyseur de type « dispersé » est constitué de particules de très petite taille.
- L'effluent obtenu à l'issue de l'étape a) d'hydrocraquage subit au moins une étape de séparation, éventuellement complétée par d'autres étapes de séparation supplémentaires, permettant de séparer au moins une fraction légère d'hydrocarbures contenant des bases carburants et une fraction lourde contenant des composés bouillants à au moins 350°C.
- L'étape de séparation peut avantageusement être mise en oeuvre par toute méthode connue de l'homme du métier telle que par exemple la combinaison d'un ou plusieurs séparateurs haute et/ou basse pression, et/ou d'étapes de distillation et/ou de strippage haute et/ou basse pression. De préférence, l'étape de séparation b) permet d'obtenir une phase gazeuse, au moins une fraction légère d'hydrocarbures de type naphta, kérosène et/ou diesel, une fraction distillat sous vide et une fraction résidu sous vide et/ou une fraction résidu atmosphérique. Dans un tel cas, la fraction lourde envoyée dans l'étape c) de précipitation correspond au moins en partie à une fraction résidu atmosphérique.
- La séparation peut être effectuée dans une section de fractionnement qui peut d'abord comprendre un séparateur haute pression haute température (HPHT), et éventuellement un séparateur haute pression basse température (HPBT), et/ou une distillation atmosphérique et/ou une distillation sous vide. L'effluent obtenu à l'issue de l'étape a) est séparé (généralement dans un séparateur HPHT) en une fraction légère et une fraction lourde contenant majoritairement des composés bouillants à au moins 350°C. Le point de coupe de la séparation se situe avantageusement entre 200 et 400°C.
- Dans une variante du procédé de l'invention, lors de l'étape b), l'effluent issu de l'hydrocraquage peut également subir une succession de flash comprenant au moins un ballon haute pression haute température (HPHT) et un ballon basse pression haute température (BPHT) pour séparer une fraction lourde qui est envoyée dans une étape de stripage à la vapeur permettant d'éliminer de ladite fraction lourde au moins une fraction légère riche en hydrogène sulfuré. La fraction lourde récupérée en fond de colonne de stripage contient des composés bouillants à au moins 350°C mais aussi des distillats atmosphériques. Selon le procédé de l'invention, ladite fraction lourde séparée de la fraction légère riche en hydrogène sulfuré est ensuite envoyée dans l'étape de précipitation c) puis dans l'étape de séparation de sédiments d).
- Dans une variante, au moins une partie de la fraction dite lourde issue de l'étape b) est fractionnée par distillation atmosphérique en au moins une fraction distillat atmosphérique contenant au moins une fraction légère d'hydrocarbures de type naphta, kérosène et/ou diesel et une fraction résidu atmosphérique. Au moins une partie de la fraction résidu atmosphérique, correspondant au moins en partie à la fraction lourde issue de l'étape b), peut être envoyée dans l'étape de précipitation c) puis dans l'étape d) de séparation physique des sédiments.
- Le résidu atmosphérique peut également au moins en partie être fractionné par distillation sous vide en une fraction distillat sous vide contenant du gazole sous vide et une fraction résidu sous vide. Ladite fraction résidu sous vide, correspondant au moins en partie à la fraction lourde issue de l'étape b), est avantageusement envoyée au moins en partie dans l'étape de précipitation c) puis dans l'étape d) de séparation physique des sédiments d).
- Au moins une partie du distillat sous vide et/ou du résidu sous vide peut également être recyclée dans l'étape d'hydrocraquage a).
- Quelle que soit la méthode de séparation mise en oeuvre, la ou les fraction(s) légère(s) obtenue(s) peut(peuvent) subir d'autres étapes de séparation, éventuellement en présence de la fraction légère issue du séparateur inter-étage entre les deux réacteurs d'hydrocraquage. Avantageusement, elle(s) est(sont) soumise(s) à une distillation atmosphérique permettant d'obtenir une fraction gazeuse, au moins une fraction légère d'hydrocarbures de type naphta, kérosène et/ou diesel et une fraction distillat sous vide.
- Une partie du distillat atmosphérique et/ou du distillat sous vide issue de l'étape de séparation b) peut constituer une partie d'un fioul comme fluxant. Ces coupes peuvent également constituer des combustibles marins à faible viscosité (MGO ou MGO, Marine Diesel Oil ou Marine Gas Oil selon les terminologies anglo-saxonnes). Une autre partie du distillat sous vide peut encore être valorisée par hydrocraquage et/ou par craquage catalytique en lit fluidisé.
- Les fractions gazeuses issues de l'étape de séparation subissent de préférence un traitement de purification pour récupérer l'hydrogène et le recycler vers les réacteurs d'hydrocraquage (étape a)). Une partie de l'hydrogène purifié peut être utilisée lors de l'étape de précipitation.
- La valorisation des différentes coupes de bases carburants (GPL, naphta, kérosène, diesel et/ou gazole sous vide) obtenues de la présente invention est bien connue de l'Homme du métier. Les produits obtenus peuvent être intégrés à des réservoirs carburants (aussi appelé "pools" carburants selon la terminologie anglo-saxonne) ou subir des étapes de raffinage supplémentaires. Le(s) fraction(s) naphta, kérosène, gazole et le gazole sous vide peuvent être soumises à un ou plusieurs traitements (hydrotraitement, hydrocraquage, alkylation, isomérisation, reformage catalytique, craquage catalytique ou thermique ou autres) pour les amener aux spécifications requises (teneur en soufre, point de fumée, octane, cétane, etc...) de façon séparée ou en mélange.
- Avantageusement, le distillat sous vide sortant du lit bouillonnant après séparation peut subir un hydrotraitement. Ce distillat sous vide hydrotraité peut être utilisé comme fluxant au pool fioul ayant une teneur en soufre inférieure ou égale à 0,5 % poids ou être valorisé directement comme fioul ayant une teneur en soufre inférieure ou égale à 0,1 % pds.
- Une partie du résidu atmosphérique, du distillat sous vide et/ou du résidu sous vide peut subir d'autres étapes de raffinage supplémentaire, telles qu'un hydrotraitement, un hydrocraquage, ou un craquage catalytique en lit fluidisé.
- La fraction lourde obtenue à l'issue de l'étape b) de séparation contient des sédiments organiques qui résultent des conditions d'hydrocraquage et des résidus de catalyseurs. Une partie des sédiments est constituée d'asphaltènes précipités dans les conditions d'hydrocraquage et sont analysés comme des sédiments existants (IP375).
- En fonction des conditions d'hydrocraquage, la teneur en sédiments dans la fraction lourde varie. D'un point de vue analytique, on distingue les sédiments existants (IP375) et les sédiments après vieillissement (mesurée selon la méthode ISO 10307-2) qui incluent les sédiments potentiels. Or, des conditions d'hydrocraquage poussées, c'est-à-dire lorsque le taux de conversion est par exemple supérieur à 30, 40 ou 50% en fonction de la charge, provoquent la formation de sédiments existants et de sédiments potentiels.
- Afin d'obtenir un fioul ou une base de fioul à teneur réduite en sédiments, notamment un fioul de soute ou une base de fioul de soute répondant aux recommandations d'une teneur en sédiments après vieillissement (mesurée selon la méthode ISO 10307-2) inférieure ou égale à 0,1%, le procédé selon l'invention comprend une étape de précipitation permettant d'améliorer l'efficacité de séparation des sédiments et ainsi d'obtenir des fiouls ou des bases de fiouls stables, c'est à dire une teneur en sédiments après vieillissement inférieure ou égale à 0,1% en poids.
- L'étape de précipitation selon l'invention permet de former l'ensemble des sédiments existants et potentiels (en convertissant les sédiments potentiels en sédiments existants) de manière à les séparer plus efficacement et ainsi respecter la teneur en sédiments après vieillissement (mesurée selon la méthode ISO 10307-2) de 0,1% poids maximum.
- L'étape de précipitation selon l'invention comprend la mise en contact de la fraction lourde issue de l'étape de séparation b) avec une coupe de distillat dont au moins 20% poids présente une température d'ébullition supérieure ou égale à 100°C, de préférence supérieure ou égale à 120°C, de manière plus préférée supérieure ou égale à 150°C. Dans une variante selon l'invention, la coupe de distillat se caractérise en ce qu'elle comprend au moins 25% poids ayant une température d'ébullition supérieure ou égale à 100°C, de préférence supérieure ou égale à 120°C, de manière plus préférée supérieure ou égale à 150°C.
- De manière avantageuse, au moins 5% poids, voire 10% poids de la coupe de distillat selon l'invention présente une température d'ébullition d'au moins 252°C.
- De manière plus avantageuse, au moins 5% poids, voire 10% poids de la coupe de distillat selon l'invention présente une température d'ébullition d'au moins 255°C.
- L'étape c) de précipitation selon l'invention est avantageusement mise en oeuvre pendant un temps de séjour inférieur à 500 minutes, de préférence inférieur à 300 minutes, de manière plus préférée inférieur à 60 minutes, à une température entre 25 et 350°C, de préférence entre 50 et 350°C, de préférence entre 65 et 300°C et de manière plus préférée entre 80 et 250°C. La pression de l'étape de précipitation est avantageusement inférieure à 20 MPa, de préférence inférieure à 10 MPa, plus préférentiellement inférieure à 3 MPa et encore plus préférentiellement inférieure à 1,5 MPa.
- La coupe de distillat selon l'invention comprend avantageusement des hydrocarbures ayant plus de 12 atomes de carbones, de préférence des hydrocarbures ayant plus de 13 atomes de carbones, de manière plus préférée des hydrocarbures ayant entre 13 et 40 atomes de carbones.
- Ladite coupe de distillat peut en partie, voire en totalité, provenir de l'étape b) de séparation de l'invention ou d'un autre procédé de raffinage ou encore d'un autre procédé chimique.
- Ladite coupe de distillat peut être utilisée en mélange avec une coupe de type naphta et/ou une coupe de type gazole sous vide et/ou résidu sous vide. Ladite coupe de distillat peut être utilisée en mélange avec la fraction légère obtenue à l'issue de l'étape b), la fraction distillat atmosphérique issue de l'étape b) et/ou la fraction distillat sous vide provenant de l'étape b) de séparation. Dans le cas où la coupe de distillat selon l'invention est mélangée avec une autre coupe, une fraction légère et/ou une fraction lourde telle que indiquée ci-dessus, les proportions sont choisies de telle sorte que le mélange résultant respecte les caractéristiques de la coupe de distillat selon l'invention.
- L'utilisation de la coupe de distillat selon l'invention présente l'avantage de s'affranchir de l'utilisation majoritaire de coupes à fortes valeurs ajoutées telles que les coupes pétrochimiques, naphta...
- Le ratio massique entre la coupe de distillat selon l'invention et la fraction lourde obtenue à l'issue de l'étape b) de séparation est compris entre 0.01 et 100, de préférence entre 0.05 et 10, de manière plus préférée entre 0,1 et 5, et de manière encore plus préférée entre 0,1 et 2. Lorsque la coupe de distillat selon l'invention est au moins tirée du procédé, il est possible d'accumuler cette coupe pendant une période de démarrage de manière à atteindre le ratio désiré.
- L'étape de précipitation peut être réalisée à l'aide d'un échangeur ou d'un four de chauffe suivi d'une ou plusieurs capacité(s) en série ou en parallèle telle(s) qu'un ballon horizontal ou vertical, éventuellement avec une fonction de décantation pour éliminer une partie des solides les plus lourds, et/ou un réacteur piston. Une cuve agitée et chauffée peut également être utilisée, et peut être munie d'un soutirage en fond pour éliminer une partie des solides les plus lourds. Avantageusement, l'étape de précipitation peut être réalisée en ligne, sans capacité tampon, éventuellement à l'aide d'un mélangeur statique.
- Selon une variante, l'étape c) de précipitation de la fraction lourde issue de l'étape b) est réalisée en présence d'un gaz inerte et/ou d'un gaz oxydant et/ou d'un liquide oxydant et/ou de l'hydrogène, de préférence issu des étapes de séparation du procédé de l'invention, notamment de l'étape de séparation b).
- L'étape c) de précipitation peut être réalisée en présence d'un gaz inerte tel que le diazote, ou en présence d'un gaz oxydant tel que le dioxygène, l'ozone ou les oxydes d'azotes, ou en présence d'un mélange contenant un gaz inerte et un gaz oxydant tel que l'air ou l'air appauvri par de l'azote, ou en présence d'un liquide oxydant permettant d'accélérer le processus de précipitation. On entend par « liquide oxydant » un composé oxygéné, par exemple un peroxyde tel que l'eau oxygénée, ou encore une solution oxydante minérale telle qu'une solution de permanganate de potassium ou un acide minéral tel que l'acide sulfurique. Selon cette variante, le liquide oxydant est alors mélangé avec la fraction lourde issue de l'étape b) de séparation et la coupe de distillat selon l'invention lors de la mise en oeuvre de l'étape c).
- A l'issue de l'étape c) de précipitation, on obtient au moins une fraction hydrocarbonée à teneur enrichie en sédiments existants qui est envoyée dans l'étape d) de séparation des sédiments.
- Le procédé selon l'invention comprend en outre une étape d) de séparation physique des sédiments et des résidus de catalyseurs.
- La fraction lourde obtenue à l'issue de l'étape c) de précipitation contient des sédiments organiques de type asphaltènes précipités qui résultent des conditions d'hydrocraquage et de précipitation. Cette fraction lourde peut aussi contenir des fines de catalyseurs issues de l'attrition de catalyseurs de type extrudés dans la mise en oeuvre de réacteur d'hydrocraquage. Cette fraction lourde peut éventuellement contenir des résidus de catalyseur « dispersés » dans le cas de la mise en oeuvre d'un réacteur hybride.
- Ainsi, au moins une partie de la fraction lourde issue de l'étape c) de précipitation est soumise à une séparation physique des sédiments et des résidus de catalyseurs, au moyen d'au moins un moyen de séparation physique choisi parmi un filtre, une membrane de séparation, un lit de solides filtrant de type organique ou inorganique, une précipitation électrostatique, un système de centrifugation, une décantation, un soutirage par vis sans fin. Une combinaison, en série et/ou en parallèle, de plusieurs moyens de séparation du même type ou de type différent peut être utilisée lors de cette étape d) de séparation des sédiments et résidus de catalyseurs. Une de ces techniques de séparation solide-liquide peut nécessiter l'utilisation périodique d'une fraction légère de rinçage, issue du procédé ou non, permettant par exemple le nettoyage d'un filtre et l'évacuation des sédiments.
- A l'issue de l'étape d) de séparation physique des sédiments, on obtient la fraction lourde (à teneur en sédiments après vieillissement inférieure ou égale à 0,1% en poids) comprenant une partie de la coupe de distillat selon l'invention introduite lors de l'étape c).
- Selon l'invention, le mélange issu de l'étape d) est avantageusement introduit dans une étape e) de récupération de la fraction lourde ayant une teneur en sédiments après vieillissement inférieure ou égale à 0,1% en poids, ladite étape consistant à séparer la fraction lourde issue de l'étape d) de la coupe de distillat introduite lors de l'étape c).
- L'étape e) est une étape de séparation similaire à l'étape de séparation b). L'étape e) peut être mise oeuvre au moyen d'équipements de type ballons séparateurs et/ou colonnes de distillations de manière à séparer d'une part au moins une partie de la coupe de distillat introduite lors de l'étape c) de précipitation et d'autre part la fraction lourde ayant une teneur en sédiments après vieillissement inférieure ou égale à 0,1% en poids.
- Avantageusement, une partie de la coupe de distillat séparée de l'étape e) est recyclée dans l'étape c) de précipitation.
- Ladite fraction lourde récupérée peut avantageusement servir comme base de fioul ou comme fioul, notamment comme base de fioul de soute ou comme fioul de soute, ayant une teneur en sédiments après vieillissement inférieure à 0,1% poids.
- Avantageusement, ladite fraction lourde est mélangée avec une ou plusieurs bases fluxantes choisies dans le groupe constitué par les huiles de coupe légère d'un craquage catalytique, les huiles de coupe lourde d'un craquage catalytique, le résidu d'un craquage catalytique, un kérosène, un gazole, un distillat sous vide et/ou une huile décantée.
- Selon un mode de réalisation particulier, une partie de la coupe de distillat selon l'invention peut être laissée dans la fraction lourde à teneur réduite en sédiments de manière à ce que la viscosité du mélange soit directement celle d'un grade de fioul souhaité, par exemple 180 ou 380 cSt à 50°C.
- La teneur en soufre de la fraction lourde issue de l'étape d) ou e), et contenant majoritairement des composés bouillant à au moins 350°C, est fonction des conditions opératoires de l'étape d'hydrocraquage mais aussi de la teneur en soufre de la charge d'origine.
- Ainsi, pour les charges à faible teneur en soufre, généralement inférieure à 1,5% poids, il est possible d'obtenir directement une fraction lourde avec moins de 0,5% poids en soufre telle qu'exigée pour les navires dépourvus de traitement des fumées et opérant en dehors des ZCES à l'horizon 2020-2025.
- Pour les charges plus soufrées, dont la teneur en soufre est généralement supérieure à 1,5% poids, la teneur en soufre de la fraction lourde peut excéder 0,5% poids. Dans un tel cas, une étape f) d'hydrotraitement en lit fixe est rendue nécessaire dans le cas où le raffineur souhaite diminuer la teneur en soufre, notamment pour une base de fioul de soute ou un fioul de soute destiné à être brulé sur un navire dépourvu de traitement de fumées. L'étape f) d'hydrotraitement en lit fixe est mise en oeuvre sur une partie au moins de la fraction lourde issue de l'étape d) ou e).
- La fraction lourde issue de l'étape f) peut avantageusement servir comme base de fioul ou comme fioul, notamment comme base de fioul de soute ou comme fioul de soute, ayant une teneur en sédiments après vieillissement inférieure à 0,1% poids. Avantageusement, ladite fraction lourde est mélangée avec une ou plusieurs bases fluxantes choisies dans le groupe constitué par les huiles de coupe légère d'un craquage catalytique, les huiles de coupe lourde d'un craquage catalytique, le résidu d'un craquage catalytique, un kérosène, un gazole, un distillat sous vide et/ou une huile décantée.
- La fraction lourde issue de l'étape d) ou e) est envoyée dans l'étape f) d'hydrotraitement comprenant une ou plusieurs zones d'hydrotraitement en lits fixes. L'envoi dans un lit fixe d'une fraction lourde dépourvue de sédiments constitue un avantage de la présente invention puisque le lit fixe sera moins sujet au bouchage et à l'augmentation de la perte de charge.
- On entend par hydrotraitement (HDT) notamment des réactions d'hydrodésulfuration (HDS), des réactions d'hydrodésazotation (HDN) et des réactions d'hydrodémétallation (HDM), mais aussi l'hydrogénation, l'hydrodéoxygénation, l'hydrodéaromatisation, l'hydroisomérisation, l'hydrodéalkylation, hydrocraquage, l'hydrodéasphaltage la réduction du carbone Conradson.
- Un tel procédé d'hydrotraitement de coupes lourdes est largement connu et peut s'apparenter au procédé connu sous le nom de HYVAHL-F™ décrit dans le brevet
US5417846 . - L'homme du métier comprend aisément que dans l'étape d'hydrodémétallation, on effectue majoritairement des réactions d'hydrodémétallation mais parallèlement aussi une partie des réactions d'hydrodésulfuration. De même, dans l'étape d'hydrodésulfuration, on effectue majoritairement des réactions d'hydrodésulfuration mais parallèlement aussi une partie des réactions d'hydrodémétallation.
- Selon une variante, une co-charge peut être introduite avec la fraction lourde dans l'étape d'hydrotraitement f). Cette co-charge peut être choisie parmi les résidus atmosphériques, les résidus sous vide issus de distillation directe, les huiles désasphaltées, des extraits aromatiques issus des chaînes de production de bases pour lubrifiants, des fractions hydrocarbonées ou un mélange de fractions hydrocarbonées pouvant être choisies parmi les produits issus d'un procédé de craquage catalytique en lit fluide : une huile de coupe légère (LCO), une huile de coupe lourde (HCO), une huile décantée, ou pouvant venir de la distillation, les fractions gazoles notamment celles obtenues par distillation atmosphérique ou sous vide, comme par exemple le gazole sous vide.
- L'étape d'hydrotraitement peut avantageusement être mise en oeuvre à une température comprise entre 300 et 500°C, de préférence 350°C à 420°C et sous une pression partielle d'hydrogène avantageusement comprise entre 2 MPa et 25 MPa, de préférence entre 10 et 20 MPa, une vitesse spatiale horaire globale (VVH) qui se définit comme étant le débit volumétrique de la charge divisé par le volume total du catalyseur, se situant dans une gamme allant de 0,1 h-1 à 5 h-1 et de préférence de 0,1 h-1 à 2 h-1, une quantité d'hydrogène mélangée à la charge habituellement de 100 à 5000 Nm3/m3 (normaux mètres cube (Nm3) par mètre cube (m3) de charge liquide), le plus souvent de 200 à 2000 Nm3/m3 et de préférence de 300 à 1500 Nm3/m3.
- Habituellement, l'étape d'hydrotraitement est effectuée industriellement dans un ou plusieurs réacteurs à courant descendant de liquide. La température d'hydrotraitement est généralement ajustée en fonction du niveau souhaité d'hydrotraitement.
- Les catalyseurs d'hydrotraitement utilisés sont de préférence des catalyseurs connus et sont généralement des catalyseurs granulaires comprenant, sur un support, au moins un métal ou composé de métal ayant une fonction hydrodéshydrogénante. Ces catalyseurs sont avantageusement des catalyseurs comprenant au moins un métal du groupe VIII, choisi généralement dans le groupe formé par le nickel et/ou le cobalt, et/ou au moins un métal du groupe VIB, de préférence du molybdène et/ou du tungstène. On emploiera par exemple un catalyseur comprenant de 0,5 à 10 % en poids de nickel et de préférence de 1 à 5 % en poids de nickel (exprimé en oxyde de nickel NiO) et de 1 à 30 % en poids de molybdène, de préférence de 5 à 20 % en poids de molybdène (exprimé en oxyde de molybdène MoO3) sur un support minéral. Ce support sera, par exemple, choisi dans le groupe formé par l'alumine, la silice, les silices-alumines, la magnésie, les argiles et les mélanges d'au moins deux de ces minéraux. Avantageusement, ce support renferme d'autres composés dopants, notamment des oxydes choisis dans le groupe formé par l'oxyde de bore, la zircone, la cérine, l'oxyde de titane, l'anhydride phosphorique et un mélange de ces oxydes. On utilise le plus souvent un support d'alumine et très souvent un support d'alumine dopée avec du phosphore et éventuellement du bore. La concentration en anhydride phosphorique P2O5 est habituellement comprise entre 0 ou 0,1 % et 10% poids. La concentration en trioxyde de bore B2O5 est habituellement comprise entre 0 ou 0,1 % et 10 % en poids. L'alumine utilisée est habituellement une alumine γ ou η. Ce catalyseur est le plus souvent sous forme d'extrudés. La teneur totale en oxydes de métaux des groupes VIB et VIII est souvent de 5 à 40 % en poids et en général de 7 à 30 % en poids et le rapport pondéral exprimé en oxyde métallique entre métal (ou métaux) du groupe VIB sur métal (ou métaux) du groupe VIII est en général de 20 à 1 et le plus souvent de 10 à 2.
- Dans le cas d'une étape d'hydrotraitement incluant une étape d'hydrodémétallation (HDM), puis une étape d'hydrodésulfuration (HDS), on utilise le plus souvent des catalyseurs spécifiques adaptés à chaque étape.
- Des catalyseurs utilisables dans l'étape d'hydrodémétallation (HDM) sont par exemple indiqués dans les brevets
EP113297 EP113284 US5221656 ,US5827421 ,US7119045 ,US5622616 etUS5089463 . On utilise de préférence des catalyseurs d'hydrodémétallation (HDM) dans les réacteurs permutables. Des catalyseurs utilisables dans l'étape d'hydrodésulfuration (HDS) sont par exemple indiqués dans les brevetsEP113297 EP113284 US6589908 ,US4818743 ouUS6332976 . On peut également utiliser un catalyseur mixte étant actifs en hydrodémétallation et en hydrodésulfuration à la fois pour la section hydrodémétallation (HDM) et pour la section hydrodésulfuration (HDS) tel que décrit dans le brevetFR2940143 - Préalablement à l'injection de la charge, les catalyseurs utilisés dans le procédé selon la présente invention sont de préférence soumis à un traitement de sulfuration in-situ ou ex-situ.
- Le procédé selon l'invention peut comprendre une étape g) de séparation des effluents de l'étape f) d'hydrotraitement. L'étape g) optionnelle de séparation peut avantageusement être mise en oeuvre par toute méthode connue de l'homme du métier telle que par exemple la combinaison d'un ou plusieurs séparateurs haute et/ou basse pression, et/ou d'étapes de distillation et/ou de strippage haute et/ou basse pression. Cette étape optionnelle g) de séparation est similaire à l'étape b) de séparation et ne sera pas décrite davantage.
- Dans une variante de mise en oeuvre de l'invention l'effluent obtenu à l'étape f) peut au moins en partie, et souvent en totalité, être envoyé dans une étape de séparation g), comprenant une distillation atmosphérique et/ou une distillation sous vide. L'effluent de l'étape d'hydrotraitement est fractionné par distillation atmosphérique en une fraction gazeuse, au moins une fraction distillat atmosphérique contenant les bases carburants (naphta, kérosène et/ou diesel) et une fraction résidu atmosphérique. Au moins une partie du résidu atmosphérique peut ensuite être fractionnée par distillation sous vide en une fraction distillat sous vide contenant du gazole sous vide et une fraction résidu sous vide.
- La fraction résidu sous vide et/ou la fraction distillat sous vide et/ou la fraction résidu atmosphérique peuvent constituer en partie au moins les bases de fiouls à basse teneur en soufre ayant une teneur en soufre inférieure ou égale à 0,5 % poids et une teneur en sédiments après vieillissement inférieure ou égale à 0,1%. La fraction distillat sous vide peut constituer une base de fioul ayant une teneur en soufre inférieure ou égale à 0,1 % poids.
- Une partie du résidu sous vide et/ou du résidu atmosphérique peut également être recyclée dans l'étape d'hydrocraquage a).
- Pour obtenir un fioul, les fractions lourdes issues des étapes d) et/ou e) et/ou f) et/ou g) peuvent être mélangées avec une ou plusieurs bases fluxantes choisies dans le groupe constitué par les huiles de coupe légère d'un craquage catalytique, les huiles de coupe lourde d'un craquage catalytique, le résidu d'un craquage catalytique, un kérosène, un gazole, un distillat sous vide et/ou une huile décantée, la coupe de distillat selon l'invention. De préférence, on utilisera du kérosène, du gazole et/ou du distillat sous vide produit dans le procédé de l'invention. Avantageusement, on utilisera du kérosène, du gazole et/ou du distillat sous vide obtenu(s) dans les étapes de séparation b) ou g) du procédé.
- La
figure 1 décrit schématiquement un exemple de mise en oeuvre de l'invention sans en limiter la portée. - La charge hydrocarbonée (1) et de l'hydrogène (2) sont mis en contact dans une zone d'hydrocraquage en lit bouillonnant a). L'effluent (3) issu de la zone a) d'hydrocraquage est envoyée dans une zone de séparation b) pour obtenir au moins une fraction légère d'hydrocarbures (4) et une fraction liquide lourde (5) contenant des composés bouillant à au moins 350°C. Cette fraction lourde (5) est mise en contact avec une coupe de distillat (6) lors d'une étape de précipitation dans la zone c). L'effluent (7) constitué d'une fraction lourde et de sédiments est traité dans une étape de séparation physique dans la zone d) permettant d'éliminer une fraction comprenant des sédiments (9) et de récupérer une fraction hydrocarbonée liquide (8) à teneur réduite en sédiments. La fraction hydrocarbonée liquide (8) est ensuite traitée dans une étape de récupération dans la zone e) d'une part de la fraction hydrocarbonée liquide (11) ayant une teneur en sédiments après vieillissement inférieure ou égale à 0,1% en poids, et d'autre part d'une fraction (10) contenant au moins une partie de la coupe de distillat introduite lors de l'étape c).
- L'exemple suivant illustre l'invention sans toutefois en limiter la portée. On traite une charge résidu sous vide (RSV Oural) contenant 84% en poids de composés bouillant à une température supérieure à 520°C, ayant une densité de 9,5°API et une teneur en soufre de 2,6% en poids.
- La charge a été soumise à une étape d'hydrocraquage comprenant deux réacteurs successifs en lit bouillonnant. Les conditions opératoires de l'étape d'hydrocraquage sont données dans le tableau 1.
Tableau 1 : Conditions opératoires de la section d'hydrocraquage 2 lits bouillonnants Catalyseurs NiMo sur alumine Température R1 (°C) 430 Température R2 (°C) 430 Pression partielle H2 (MPa) 15 VVH "réacteurs" (h-1, Sm3/h charge fraîche /m3 de réacteurs) 0,35 VVH "catalyseurs lit bouillonnant" (h-1, Sm3/h charge fraîche /m3 de catalyseurs lit bouillonnant) 0,65 H2 / HC entrée section d'hydrocraquage hors consommation H2 (Nm3 / m3 de charge fraîche) 600 - Le catalyseur NiMo sur Alumine utilisé est vendu par la société Axens sous la référence HOC-458.
- L'effluent de l'étape d'hydrocraquage est ensuite soumis à une étape de séparation permettant de séparer une fraction gazeuse et une fraction liquide lourde au moyen de séparateurs. La fraction liquide lourde est ensuite distillée dans une colonne de distillation atmosphérique de manière à récupérer des distillats et un résidu atmosphérique.
- Des prélèvements, pesés et analysés permettent d'établir un bilan matière global de l'hydrocraquage en lit bouillonnant. Les rendements et les teneurs en soufre de chaque fraction obtenue dans l'effluent en sortie de l'hydrocraquage en lit bouillonnant sont donnés dans le tableau 2 ci-dessous :
Tableau 2 : Rendement (Rdt) et teneur en soufre (S) de l'effluent en sortie d'hydrocraquage (% pds / charge) Hydrocraquage 2 lits bouillonnants (430/430°C) Produits Rdt (%pds) S (%pds) NH3 0,2 H2S 2,3 C1-C4 (gaz) 4,7 Naphta léger (PI - 100°C) 3,2 0,06 Naphta lourd (100-150°C) 7,8 0,05 Kérosène (150°C-225°C) 9,8 0,08 Diesel (225°C - 350°C) 18,1 0,13 Distillat sous vide (350°C - 520°C) 36,5 0,51 Résidu sous vide (520°C+) 19,2 1,27 - Le résidu atmosphérique RA est une coupe 350°C+ com posée d'une partie du distillat sous vide (DSV) de l'effluent et de la totalité du résidu sous vide (RSV) de l'effluent dans la proportion 44% poids de DSV et 56% en poids de RSV. Ce résidu atmosphérique présente une viscosité de 38 cSt à 100°C. Ce résidu atmosphérique RA est soumis à un traitement selon plusieurs variantes :
- A) une variante A (non-conforme à l'invention) dans laquelle on filtre le résidu atmosphérique RA au moyen d'un filtre poreux métallique de marque Pall®. La teneur en sédiments après vieillissement est mesurée sur le résidu atmosphérique récupéré après séparation des sédiments ;
- B) une variante B dans laquelle une étape de précipitation (conforme à l'invention) est réalisée en mélangeant sous agitation à 80°C pendan t 1 minute le résidu atmosphérique RA et une coupe de distillat selon l'invention selon un ratio décrits au tableau 3 :
On mélange 50% poids de résidu atmosphérique (RA) et 50% poids de la coupe de distillat selon l'invention. - Le résidu atmosphérique qui correspond à la coupe 350°C+ de l'effluent dans la proportion 44% en poids de DSV et 56% en poids de RSV de l'étape d'hydrocraquage de l'invention est caractérisé par une teneur en sédiments (IP375) de 0,4% m/m et une teneur en sédiments après vieillissement (IP390) de 0,9% m/m.
- La coupe de distillat caractérisée par la distillation simulée qui traduit le pourcentage distillé en fonction de la température, contient plus de 5% poids des composés qui bouillent à plus de 255°C (tableau 3).
Tableau 3. Courbes de distillation simulée de la coupe de distillat % en poids distillé Température d'ébullition (°C) 5% 191 10% 202 20% 222 30% 240 40% 258 50% 276 60% 293 70% 309 80% 325 90% 340 95% 347 - Le mélange est ensuite soumis à une étape de séparation des sédiments et résidus de catalyseurs au moyen d'un filtre poreux métallique de marque Pall®. Cette étape de séparation physique des sédiments est suivie d'une étape de distillation du mélange permettant de récupérer d'une part le résidu atmosphérique à teneur réduite en sédiments, et d'autre part la coupe de distillat.
- La teneur en sédiments après vieillissement est mesurée sur le résidu atmosphérique récupéré après l'étape de distillation. L'ensemble des données de précipitation et de séparation des sédiments est résumé dans le tableau 4.
Tableau 4 : Résumé des performances avec ou sans précipitation, séparation des sédiments et récupération de la fraction lourde Pas de précipitation (non-conforme) Mélange, précipitation et séparation des sédiments (invention) Proportions du mélange (% m/m) Proportions du mélange (% m/m) Proportion du Résidu atmosphérique (RA) dans le mélange (% m/m) 100 50 Proportion de la coupe de distillat dans le mélange (% m/m) - 50 Teneur en sédiments du mélange (mesure selon IP375a % m/m) - 0,79a Teneur en sédiments du résidu atmosphérique RA récupéré après séparation physique des sédiments (mesure selon IP390 b %m/m) 0,5b < 0,1b - Les conditions opératoires de l'étape d'hydrocraquage couplées aux différentes variantes de traitement (séparation des sédiments avec étape de précipitation (B) selon l'invention ou sans étape de précipitation(A)) du résidu atmosphérique (RA) ont un impact sur la stabilité des effluents obtenus. Ceci est illustré par les teneurs en sédiments après vieillissement mesurées dans les résidus atmosphériques RA (coupe 350°C+) avant et après l'étape de précipitation et de séparation des sédiments.
- Ainsi, le résidu atmosphérique obtenu selon l'invention, constitue une excellente base de fioul, notamment une base de fioul de soute ayant une teneur en sédiments après vieillissement (IP390) inférieure à 0,1% poids.
- Le résidu atmosphérique RA traité selon le mélange du tableau 4 présente une teneur en sédiments après vieillissement inférieure à 0,1%, une teneur en soufre de 0,93% m/m et une viscosité de 380 cSt à 50°C. Ce résidu atmosphérique constitue ainsi un fioul de soute de qualité, pouvant être vendu selon le grade RMG ou IFO 380, à basse teneur en sédiments. Il pourra par exemple être brulé en zone ECA ou en dehors des zones ECA à l'horizon 2020-25 sous réserve que le navire soit équipé de lavage de fumées pour abattre les oxydes de soufre.
Claims (15)
- Procédé de conversion d'une charge hydrocarbonée contenant au moins une fraction d'hydrocarbures ayant une teneur en soufre d'au moins 0,1 % poids, une température initiale d'ébullition d'au moins 340°C et une température finale d'ébullition d'au moins 440°C, ledit procédé comprenant les étapes suivante s :a) une étape d'hydrocraquage de la charge en présence d'hydrogène dans au moins un réacteur contenant un catalyseur supporté en lit bouillonnant,b) une étape de séparation de l'effluent obtenu à l'issue de l'étape a) en au moins une fraction légère d'hydrocarbures contenant des bases carburants et une fraction lourde contenant des composés bouillant à au moins 350°C,c) une étape de précipitation des sédiments dans laquelle la fraction lourde issue de l'étape de séparation b) est mise en contact avec une coupe de distillat dont au moins 20% poids présente une température d'ébullition supérieure ou égale à 100°C, pendant une durée inférieure à 500 minutes, à une température comprise entre 25 et 350°C, et une pression inférieure à 20 MPa,d) une étape de séparation physique des sédiments de la fraction lourde issue de l'étape c) de précipitation pour obtenir une fraction lourde séparée des sédiments,e) une étape de récupération d'une fraction lourde ayant une teneur en sédiments, mesurée selon la méthode ISO 10307-2, inférieure ou égale à 0,1% en poids consistant à séparer la fraction lourde issue de l'étape d) de la coupe de distillat introduite lors de l'étape c).
- Procédé selon la revendication 1 comprenant une étape f) d'hydrotraitement en lit fixe mise en oeuvre sur une partie au moins de la fraction lourde issue de l'étape d) ou e).
- Procédé selon la revendication 1 ou 2 dans lequel la coupe de distillat comprend au moins 25% poids ayant une température d'ébullition supérieure ou égale à 100°C.
- Procédé selon l'une des revendications précédentes dans lequel au moins 5% poids de la coupe de distillat présente une température d'ébullition d'au moins 252°C.
- Procédé selon l'une des revendications précédentes dans lequel la coupe de distillat comprend des hydrocarbures ayant plus de 12 atomes de carbones.
- Procédé selon l'une des revendications précédentes dans lequel la coupe de distillat provient en partie, voire en totalité, de l'étape b) de séparation ou d'un autre procédé de raffinage ou encore d'un autre procédé chimique.
- Procédé selon l'une des revendications précédentes dans lequel une partie de la coupe de distillat séparée de l'étape e) est recyclée dans l'étape c) de précipitation.
- Procédé selon l'une des revendications précédentes dans lequel l'étape a) d'hydrocraquage est opérée sous une pression partielle d'hydrogène de 5 à 35 MPa, à une température de 330 à 500 °C, une vitesse spatia le allant de 0,05 h-1 à 5 h-1 et la quantité d'hydrogène mélangé à la charge est de 50 à 5000 Nm3/m3.
- Procédé selon l'une des revendications précédentes dans lequel l'étape d'hydrocraquage est réalisée dans au moins un réacteur fonctionnant en mode lit hybride.
- Procédé selon l'une des revendications précédentes dans lequel l'étape c) de précipitation de la fraction lourde issue de l'étape b) est réalisée en présence d'un gaz inerte et/ou d'un gaz oxydant et/ou d'un liquide oxydant et/ou de l'hydrogène, de préférence issu de l'étape de séparation b).
- Procédé selon l'une des revendications précédentes dans lequel l'étape d) de séparation physique est réalisée au moyen d'au moins un moyen de séparation choisi parmi un filtre, une membrane de séparation, un lit de solides filtrant de type organique ou inorganique, une précipitation électrostatique, un système de centrifugation, une décantation, un soutirage par vis sans fin.
- Procédé selon l'une des revendications précédentes dans lequel au moins une partie de la fraction dite lourde issue de l'étape b) est fractionnée par distillation atmosphérique en au moins une fraction distillat atmosphérique contenant au moins une fraction légère d'hydrocarbures de type naphta, kérosène et/ou diesel et une fraction résidu atmosphérique.
- Procédé selon l'une des revendications précédentes dans lequel la charge traitée est choisie parmi les résidus atmosphériques, les résidus sous vide issus de distillation directe, des pétroles bruts, des pétroles bruts étêtés, les huiles désasphaltées, des résines de désasphaltage, les asphaltes ou brais de désasphaltage, les résidus issus des procédés de conversion, des extraits aromatiques issus des chaînes de production de bases pour lubrifiants, des sables bitumineux ou leurs dérivés, des schistes bitumineux ou leurs dérivés, pris seuls ou en mélange.
- Procédé selon la revendication 13 dans lequel la charge contient au moins 1% d'asphaltènes C7 et au moins 5 ppm de métaux.
- Procédé selon l'une des revendications précédentes dans lequel les fractions lourdes issues des étapes d) et/ou e) et/ou f) sont mélangées avec une ou plusieurs bases fluxantes choisies dans le groupe constitué par les huiles de coupe légère d'un craquage catalytique, les huiles de coupe lourde d'un craquage catalytique, le résidu d'un craquage catalytique, un kérosène, un gazole, un distillat sous vide et/ou une huile décantée la coupe de distillat selon les revendications 1 à 5 pour obtenir un fioul.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR1554962A FR3036703B1 (fr) | 2015-06-01 | 2015-06-01 | Procede de conversion de charges comprenant une etape d'hydrocraquage, une etape de precipitation et une etape de separation des sediments pour la production de fiouls |
| PCT/EP2016/058747 WO2016192892A1 (fr) | 2015-06-01 | 2016-04-20 | Procede de conversion de charges comprenant une etape d'hydrocraquage, une etape de precipitation et une etape de separation des sediments pour la production de fiouls |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP3303522A1 EP3303522A1 (fr) | 2018-04-11 |
| EP3303522B1 true EP3303522B1 (fr) | 2019-03-06 |
Family
ID=54199789
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP16719813.4A Active EP3303522B1 (fr) | 2015-06-01 | 2016-04-20 | Procede de conversion de charges comprenant une etape d'hydrocraquage, une etape de precipitation et une etape de separation des sediments pour la production de fiouls |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US11702603B2 (fr) |
| EP (1) | EP3303522B1 (fr) |
| JP (1) | JP6670855B2 (fr) |
| KR (1) | KR102529350B1 (fr) |
| CN (1) | CN107849466A (fr) |
| ES (1) | ES2728566T3 (fr) |
| FR (1) | FR3036703B1 (fr) |
| PT (1) | PT3303522T (fr) |
| SA (1) | SA517390453B1 (fr) |
| TW (1) | TWI700361B (fr) |
| WO (1) | WO2016192892A1 (fr) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE202016007978U1 (de) | 2016-12-27 | 2018-03-28 | Rudolf King | GPS-basierte Methode zur effektiven, mobilen Nachbarschaftshilfe im Notfall mittels statischen und/oder dynamischen und zeitlich unbegrenzten oder definierten sozialen Netzwerken (SENI) und mobilen Endgeräten |
| US10604709B2 (en) | 2017-02-12 | 2020-03-31 | Magēmā Technology LLC | Multi-stage device and process for production of a low sulfur heavy marine fuel oil from distressed heavy fuel oil materials |
| US20190233741A1 (en) | 2017-02-12 | 2019-08-01 | Magēmā Technology, LLC | Multi-Stage Process and Device for Reducing Environmental Contaminates in Heavy Marine Fuel Oil |
| US11788017B2 (en) | 2017-02-12 | 2023-10-17 | Magëmã Technology LLC | Multi-stage process and device for reducing environmental contaminants in heavy marine fuel oil |
| US10696906B2 (en) | 2017-09-29 | 2020-06-30 | Marathon Petroleum Company Lp | Tower bottoms coke catching device |
| TWI756504B (zh) * | 2017-12-29 | 2022-03-01 | 大陸商中國石油化工科技開發有限公司 | 一種蠟油加氫裂化方法和系統 |
| FR3084372B1 (fr) * | 2018-07-24 | 2020-08-07 | Ifp Energies Now | Procede de traitement d'une charge hydrocarbonee lourde comprenant un hydrotraitement en lit fixe, deux desasphaltages et un hydrocraquage en lit bouillonnant de l'asphalte |
| FR3084371B1 (fr) * | 2018-07-24 | 2020-08-07 | Ifp Energies Now | Procede de traitement d'une charge hydrocarbonee lourde comprenant un hydrotraitement en lit fixe, un desasphaltage et un hydrocraquage en lit bouillonnant de l'asphalte |
| KR20210072217A (ko) * | 2019-12-06 | 2021-06-17 | 현대오일뱅크 주식회사 | 안정화된 연료유의 제조방법 및 그로부터 얻는 안정화된 연료유 |
| US11352578B2 (en) | 2020-02-19 | 2022-06-07 | Marathon Petroleum Company Lp | Low sulfur fuel oil blends for stabtility enhancement and associated methods |
| US11898109B2 (en) | 2021-02-25 | 2024-02-13 | Marathon Petroleum Company Lp | Assemblies and methods for enhancing control of hydrotreating and fluid catalytic cracking (FCC) processes using spectroscopic analyzers |
| US20220268694A1 (en) | 2021-02-25 | 2022-08-25 | Marathon Petroleum Company Lp | Methods and assemblies for determining and using standardized spectral responses for calibration of spectroscopic analyzers |
| US11905468B2 (en) | 2021-02-25 | 2024-02-20 | Marathon Petroleum Company Lp | Assemblies and methods for enhancing control of fluid catalytic cracking (FCC) processes using spectroscopic analyzers |
| CA3188122A1 (fr) | 2022-01-31 | 2023-07-31 | Marathon Petroleum Company Lp | Systemes et methodes de reduction des points d'ecoulement de gras fondus |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2988501A (en) * | 1958-08-18 | 1961-06-13 | Union Oil Co | Hydrorefining of crude oils |
| US4883581A (en) * | 1986-10-03 | 1989-11-28 | Exxon Chemical Patents Inc. | Pretreatment for reducing oxidative reactivity of baseoils |
| US5108580A (en) * | 1989-03-08 | 1992-04-28 | Texaco Inc. | Two catalyst stage hydrocarbon cracking process |
| FR2753984B1 (fr) * | 1996-10-02 | 1999-05-28 | Inst Francais Du Petrole | Procede de conversion d'une fraction lourde d'hydrocarbures impliquant une hydrodemetallisation en lit bouillonnant de catalyseur |
| FR2866897B1 (fr) * | 2004-03-01 | 2007-08-31 | Inst Francais Du Petrole | Utilisation de gaz pour le preraffinage de petrole conventionnel et optionnellement sequestration de co2 |
| US7811444B2 (en) * | 2006-06-08 | 2010-10-12 | Marathon Oil Canada Corporation | Oxidation of asphaltenes |
| FR2933709B1 (fr) * | 2008-07-10 | 2011-07-22 | Inst Francais Du Petrole | Procede de conversion comprenant une hydroconversion d'une charge, un fractionnement, puis un desasphatage de la fraction residu sous vide |
| EA025489B1 (ru) * | 2009-11-17 | 2016-12-30 | Эйч А Ди Корпорейшн | Способ удаления асфальтенов из тяжелой нефти путем воздействия с высокой скоростью сдвига |
| PL2348091T3 (pl) * | 2010-01-12 | 2013-04-30 | Ifp Energies Now | Sposób bezpośredniego hydroupłynniania biomasy obejmujący dwa etapy hydrokonwersji na złożu wrzącym |
| US8790508B2 (en) | 2010-09-29 | 2014-07-29 | Saudi Arabian Oil Company | Integrated deasphalting and oxidative removal of heteroatom hydrocarbon compounds from liquid hydrocarbon feedstocks |
| US9493710B2 (en) * | 2011-07-29 | 2016-11-15 | Saudi Arabian Oil Company | Process for stabilization of heavy hydrocarbons |
| US10400184B2 (en) * | 2011-08-31 | 2019-09-03 | Exxonmobil Research And Engineering Company | Hydroprocessing of heavy hydrocarbon feeds using small pore catalysts |
| FR2981659B1 (fr) * | 2011-10-20 | 2013-11-01 | Ifp Energies Now | Procede de conversion de charges petrolieres comprenant une etape d'hydroconversion en lit bouillonnant et une etape d'hydrotraitement en lit fixe pour la production de fiouls a basse teneur en soufre |
| FR2983866B1 (fr) * | 2011-12-07 | 2015-01-16 | Ifp Energies Now | Procede d'hydroconversion de charges petrolieres en lits fixes pour la production de fiouls a basse teneur en soufre |
| FR3000097B1 (fr) * | 2012-12-20 | 2014-12-26 | Ifp Energies Now | Procede integre de traitement de charges petrolieres pour la production de fiouls a basse teneur en soufre |
| FR3000098B1 (fr) * | 2012-12-20 | 2014-12-26 | IFP Energies Nouvelles | Procede avec separation de traitement de charges petrolieres pour la production de fiouls a basse teneur en soufre |
| KR20170034908A (ko) * | 2014-07-25 | 2017-03-29 | 사우디 아라비안 오일 컴퍼니 | 아스팔트, 석유 그린 코크스, 및 액체 및 기체 코킹 유닛 생성물을 제조하기 위한 통합된 공정 |
-
2015
- 2015-06-01 FR FR1554962A patent/FR3036703B1/fr not_active Expired - Fee Related
-
2016
- 2016-04-20 JP JP2017562068A patent/JP6670855B2/ja active Active
- 2016-04-20 EP EP16719813.4A patent/EP3303522B1/fr active Active
- 2016-04-20 WO PCT/EP2016/058747 patent/WO2016192892A1/fr active Application Filing
- 2016-04-20 KR KR1020177037761A patent/KR102529350B1/ko active IP Right Grant
- 2016-04-20 CN CN201680032073.6A patent/CN107849466A/zh active Pending
- 2016-04-20 PT PT16719813T patent/PT3303522T/pt unknown
- 2016-04-20 US US15/578,467 patent/US11702603B2/en active Active
- 2016-04-20 ES ES16719813T patent/ES2728566T3/es active Active
- 2016-06-01 TW TW105117261A patent/TWI700361B/zh active
-
2017
- 2017-11-30 SA SA517390453A patent/SA517390453B1/ar unknown
Non-Patent Citations (1)
| Title |
|---|
| None * |
Also Published As
| Publication number | Publication date |
|---|---|
| FR3036703A1 (fr) | 2016-12-02 |
| US11702603B2 (en) | 2023-07-18 |
| JP2018520228A (ja) | 2018-07-26 |
| CN107849466A (zh) | 2018-03-27 |
| ES2728566T3 (es) | 2019-10-25 |
| EP3303522A1 (fr) | 2018-04-11 |
| WO2016192892A1 (fr) | 2016-12-08 |
| FR3036703B1 (fr) | 2017-05-26 |
| US20180134974A1 (en) | 2018-05-17 |
| KR102529350B1 (ko) | 2023-05-04 |
| JP6670855B2 (ja) | 2020-03-25 |
| SA517390453B1 (ar) | 2021-06-28 |
| TW201715033A (zh) | 2017-05-01 |
| KR20180014776A (ko) | 2018-02-09 |
| TWI700361B (zh) | 2020-08-01 |
| PT3303522T (pt) | 2019-06-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3303522B1 (fr) | Procede de conversion de charges comprenant une etape d'hydrocraquage, une etape de precipitation et une etape de separation des sediments pour la production de fiouls | |
| EP3018187B1 (fr) | Procede de conversion de charges petrolieres comprenant une etape d'hydrocraquage en lit bouillonnant, une etape de maturation et une etape de separation des sediments pour la production de fiouls a basse teneur en sediments | |
| EP3026097B1 (fr) | Procede de production de combustibles de type fuel lourd a partir d'une charge hydrocarbonee lourde utilisant une separation entre l'etape d'hydrotraitement et l'etape d'hydrocraquage | |
| EP3303523B1 (fr) | Procede de conversion de charges comprenant une etape d'hydrotraitement, une etape d'hydrocraquage, une etape de precipitation et une etape de separation des sediments pour la production de fiouls | |
| EP2788458B1 (fr) | Procede d'hydroconversion de charges petrolieres en lits fixes pour la production de fiouls a basse teneur en soufre | |
| EP3018188B1 (fr) | Procede de conversion de charges petrolieres comprenant une etape d'hydrotraitement en lit fixe, une etape d'hydrocraquage en lit bouillonnant, une etape de maturation et une etape de separation des sediments pour la production de fiouls a basse teneur en sediments | |
| CA3021600A1 (fr) | Procede de conversion comprenant des lits de garde permutables d'hydrodemetallation, une etape d'hydrotraitement en lit fixe et une etape d'hydrocraquage en reacteurs permutables | |
| WO2015091033A1 (fr) | Nouveau procede integre de traitement de charges petrolieres pour la production de fiouls a basse teneur en soufre et en sediments | |
| WO2014096704A1 (fr) | Procédé avec separation de traitement de charges petrolieres pour la production de fiouls a basse teneur en soufre | |
| WO2013057389A1 (fr) | Procédé de conversion de charges petrolieres comprenant une etape d'hydroconversion en lit bouillonnant et une etape d'hydrotraitement en lit fixe pour la production de fiouls a basse teneur en soufre | |
| EP3018189B1 (fr) | Procede de conversion de charges petrolieres comprenant une etape de viscoreduction, une etape de maturation et une etape de separation des sediments pour la production de fiouls a basse teneur en sediments | |
| FR3027909A1 (fr) | Procede integre de production de combustibles de type fuel lourd a partir d'une charge hydrocarbonee lourde sans separation intermediaire entre l'etape d'hydrotraitement et l'etape d'hydrocraquage | |
| WO2012085407A1 (fr) | Procède de conversion de charge hydrocarbonate comprenant une huile de schiste par hydre conversion en lit bouillonnant, fractionnement par distillation atmosphérique, et hydrocraquage | |
| WO2012085406A1 (fr) | Procede de conversion de charge hydrocarbonee comprenant une huile de schiste par hydroconversion en lit bouillonnant, fractionnement par distillation atmospherique et extraction liquide/liquide de la fraction lourde. | |
| WO2012085408A1 (fr) | Procede de conversion de charge hydrocarbonee comprenant une huile de schiste par decontamination, hydroconversion en lit bouillonnant, et fractionnement par distillation atmospherique | |
| FR3084371A1 (fr) | Procede de traitement d'une charge hydrocarbonee lourde comprenant un hydrotraitement en lit fixe, un desasphaltage et un hydrocraquage en lit bouillonnant de l'asphalte | |
| FR3084372A1 (fr) | Procede de traitement d'une charge hydrocarbonee lourde comprenant un hydrotraitement en lit fixe, deux desasphaltages et un hydrocraquage en lit bouillonnant de l'asphalte | |
| WO2016192893A1 (fr) | Procédé de conversion de charges comprenant une étape de viscoréduction, une étape de précipitation et une étape de séparation des sédiments pour la production de fiouls |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE INTERNATIONAL PUBLICATION HAS BEEN MADE |
|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: REQUEST FOR EXAMINATION WAS MADE |
|
| 17P | Request for examination filed |
Effective date: 20180102 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| AX | Request for extension of the european patent |
Extension state: BA ME |
|
| DAV | Request for validation of the european patent (deleted) | ||
| DAX | Request for extension of the european patent (deleted) | ||
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: GRANT OF PATENT IS INTENDED |
|
| RIC1 | Information provided on ipc code assigned before grant |
Ipc: C10G 31/06 20060101ALI20180913BHEP Ipc: C10G 67/12 20060101ALI20180913BHEP Ipc: C10G 47/24 20060101AFI20180913BHEP Ipc: C10G 31/09 20060101ALI20180913BHEP Ipc: C10G 67/02 20060101ALI20180913BHEP |
|
| INTG | Intention to grant announced |
Effective date: 20180928 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE PATENT HAS BEEN GRANTED |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D Free format text: NOT ENGLISH |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP Ref country code: AT Ref legal event code: REF Ref document number: 1104530 Country of ref document: AT Kind code of ref document: T Effective date: 20190315 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602016010745 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D Free format text: LANGUAGE OF EP DOCUMENT: FRENCH |
|
| REG | Reference to a national code |
Ref country code: PT Ref legal event code: SC4A Ref document number: 3303522 Country of ref document: PT Date of ref document: 20190612 Kind code of ref document: T Free format text: AVAILABILITY OF NATIONAL TRANSLATION Effective date: 20190603 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: FP |
|
| REG | Reference to a national code |
Ref country code: LT Ref legal event code: MG4D |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190606 Ref country code: LT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: SE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: HR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: RS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: LV Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190606 Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190607 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MK05 Ref document number: 1104530 Country of ref document: AT Kind code of ref document: T Effective date: 20190306 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2728566 Country of ref document: ES Kind code of ref document: T3 Effective date: 20191025 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: AL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: IT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: RO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: CZ Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SM Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: PL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 602016010745 Country of ref document: DE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190420 Ref country code: IS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190706 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190430 Ref country code: DK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190430 Ref country code: MC Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 |
|
| 26N | No opposition filed |
Effective date: 20191209 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: TR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190420 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20200420 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20200420 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CY Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 Ref country code: HU Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT; INVALID AB INITIO Effective date: 20160420 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190306 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 20230424 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: PT Payment date: 20230406 Year of fee payment: 8 Ref country code: FR Payment date: 20230421 Year of fee payment: 8 Ref country code: ES Payment date: 20230515 Year of fee payment: 8 Ref country code: DE Payment date: 20230427 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20230424 Year of fee payment: 8 |