EP1027331B1 - Verfahren zur herstellung von arylmercaptanen durch hydrierung von diaryldisulfiden - Google Patents
Verfahren zur herstellung von arylmercaptanen durch hydrierung von diaryldisulfiden Download PDFInfo
- Publication number
- EP1027331B1 EP1027331B1 EP98955433A EP98955433A EP1027331B1 EP 1027331 B1 EP1027331 B1 EP 1027331B1 EP 98955433 A EP98955433 A EP 98955433A EP 98955433 A EP98955433 A EP 98955433A EP 1027331 B1 EP1027331 B1 EP 1027331B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- process according
- carried out
- catalytic hydrogenation
- alcoholic medium
- hydrogenation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 8
- 150000001504 aryl thiols Chemical class 0.000 title abstract 2
- 238000005984 hydrogenation reaction Methods 0.000 claims abstract description 13
- 230000001476 alcoholic effect Effects 0.000 claims abstract description 12
- 238000009903 catalytic hydrogenation reaction Methods 0.000 claims abstract description 6
- -1 aryl mercaptans Chemical class 0.000 claims description 22
- 238000000034 method Methods 0.000 claims description 20
- 239000003054 catalyst Substances 0.000 claims description 11
- 229910052739 hydrogen Inorganic materials 0.000 claims description 10
- 239000001257 hydrogen Substances 0.000 claims description 10
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 10
- 239000011541 reaction mixture Substances 0.000 claims description 6
- 239000002904 solvent Substances 0.000 claims description 6
- 229910052751 metal Inorganic materials 0.000 claims description 5
- 239000002184 metal Substances 0.000 claims description 5
- 150000002739 metals Chemical class 0.000 claims description 5
- 239000002253 acid Substances 0.000 claims description 4
- 150000008044 alkali metal hydroxides Chemical class 0.000 claims description 3
- 229910052736 halogen Inorganic materials 0.000 claims description 3
- 150000002367 halogens Chemical class 0.000 claims description 3
- 150000002431 hydrogen Chemical class 0.000 claims description 3
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 2
- 125000001931 aliphatic group Chemical group 0.000 claims description 2
- 150000001341 alkaline earth metal compounds Chemical class 0.000 claims description 2
- 239000002585 base Substances 0.000 claims description 2
- 239000003795 chemical substances by application Substances 0.000 claims description 2
- YCWSUKQGVSGXJO-NTUHNPAUSA-N nifuroxazide Chemical group C1=CC(O)=CC=C1C(=O)N\N=C\C1=CC=C([N+]([O-])=O)O1 YCWSUKQGVSGXJO-NTUHNPAUSA-N 0.000 claims 3
- 229910052783 alkali metal Inorganic materials 0.000 claims 1
- 229910000288 alkali metal carbonate Inorganic materials 0.000 claims 1
- 150000008041 alkali metal carbonates Chemical class 0.000 claims 1
- 230000007704 transition Effects 0.000 claims 1
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 23
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 9
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 7
- 239000007868 Raney catalyst Substances 0.000 description 7
- 229910000564 Raney nickel Inorganic materials 0.000 description 7
- 239000004305 biphenyl Substances 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 239000012071 phase Substances 0.000 description 5
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 239000003513 alkali Substances 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 239000012065 filter cake Substances 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 150000002736 metal compounds Chemical class 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 239000012279 sodium borohydride Substances 0.000 description 3
- 229910000033 sodium borohydride Inorganic materials 0.000 description 3
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- RAHZWNYVWXNFOC-UHFFFAOYSA-N Sulphur dioxide Chemical compound O=S=O RAHZWNYVWXNFOC-UHFFFAOYSA-N 0.000 description 2
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 229910017052 cobalt Inorganic materials 0.000 description 2
- 239000010941 cobalt Substances 0.000 description 2
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- OJWYYSVOSNWCCE-UHFFFAOYSA-N 2-methoxyethyl hypofluorite Chemical compound COCCOF OJWYYSVOSNWCCE-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000012868 active agrochemical ingredient Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- HTZCNXWZYVXIMZ-UHFFFAOYSA-M benzyl(triethyl)azanium;chloride Chemical compound [Cl-].CC[N+](CC)(CC)CC1=CC=CC=C1 HTZCNXWZYVXIMZ-UHFFFAOYSA-M 0.000 description 1
- 125000000319 biphenyl-4-yl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 210000003298 dental enamel Anatomy 0.000 description 1
- 238000007323 disproportionation reaction Methods 0.000 description 1
- 150000002019 disulfides Chemical class 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- 150000003333 secondary alcohols Chemical class 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 150000003509 tertiary alcohols Chemical class 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/02—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of thiols
- C07C319/06—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of thiols from sulfides, hydropolysulfides or polysulfides
Definitions
- the present invention relates to a particularly advantageous method of manufacture of aryl mercaptans by hydrogenation of diaryl disulfides.
- Aryl mercaptans are important intermediates for the manufacture of pharmaceuticals and agrochemical active ingredients (see e.g. EP-A 100 172, BE-A 887 423, US-A 3 912 757 and WO 96/25936).
- a useful method for making aryl mercaptans is in the reaction sequence the reduction of arylsulfochlorides with sodium sulfite to arylsulfinic acid salts, further reduction with sulfurous acid to the diaryl disulfide and finally cleavage of the disulfide with sodium borohydride (DE-A 44 20 777).
- Unsatisfactory here is the cleavage of the disulfide with sodium borohydride, although in good Yield proceeds, but with the sodium borohydride it is a complex process and therefore requires expensive reagent.
- R and R ' are preferably independently of one another for hydrogen, methyl, ethyl, methoxy, ethoxy, fluorine or chlorine. Especially R and R 'are preferably hydrogen.
- the diaryl disulfides required as the starting product are accessible in various ways, e.g. according to DE-A 44 20 777.
- a process of interest in which S- (4-biphenyl) sulfinic acid or their salts are reacted with an aqueous bisulfite solution at pH values from 2 to 7, so receives the corresponding S- (4-biphenyl) thiosulfuric acids or their salts and heated in the presence of a strong aqueous acid.
- Suitable solvents for forming the alcoholic medium required according to the invention are, for example, aliphatic C 1 -C 5 alcohols, which can be straight-chain or branched. It can be primary, secondary or tertiary alcohols. Methanol, ethanol and isopropanol are preferred.
- the alcoholic medium can optionally also contain water, for example up to 20% by weight, preferably up to 10% by weight. For example, it is not necessary to use the diaryl disulfide in dried form. It can also be used in the form of the water-moist product that is produced during its manufacture. Mixtures of different alcohols can also be used as the alcoholic medium.
- diaryl disulfide Based on 1 mole of diaryl disulfide, e.g. Use 200 to 10,000 ml of alcohol. This amount is preferably 500 to 5,000 ml.
- Alkali hydroxides alkali carbonates or alkali alcoholates, in particular alkali C 1 -C 4 alkyl alcoholates or corresponding alkaline earth metal compounds, for example, can be used as agents for generating a basic character in the alcoholic medium.
- Alkali hydroxides in particular sodium and potassium hydroxide, are preferred.
- diaryl disulfide Based on 1 mol of diaryl disulfide, for example 0.5 to 5 equivalents of base can be used deploy. This amount is preferably 1.5 to 2.5 equivalents.
- catalysts for the hydrogenation according to the invention are Metals of 8. Subgroup of the PSE in question, especially nickel and palladium. such Metals can be used as such or as metal compounds. metals or metal compounds can, if necessary, on carrier materials such as activated carbon, Alumina, alkaline earth carbonate or alkaline earth sulfate can be arranged. The Catalysts can be doped with other metals or metal compounds, for example those from subgroup 4 or 8 of the PSE. You can also in Form of skeletal catalysts, e.g. in Raney form. Preferred catalysts are palladium on carbon, palladium black, Raney nickel and with cobalt and / or iron-doped Raney nickel. Raney nickel is particularly preferred.
- the catalytic hydrogenation according to the invention can e.g. at temperatures from 20 to 200 ° C and pressures up to 50 bar. Temperatures are preferred from 40 to 150 ° C and pressures up to 25 bar, especially temperatures from 60 to 120 ° C and pressures up to 15 bar.
- Working up the reaction mixture present after the hydrogenation according to the invention can e.g. so that it is cooled, for example to 20 to 65 ° C, the catalyst separates, e.g. by filtration, which is then present alcoholic solution of the aryl mercaptan produced with acid and optionally Water is added and the product which then precipitates is filtered off and dried.
- aryl mercaptans in particular Diaryl mercaptans of the formula (I) obtained in good yields and purities.
- the method according to the invention has surprising advantages. So it doesn't need any reagents that are difficult to manufacture. It can be used at lower temperatures and what can be done especially at lower pressures than other processes means less equipment and handling. After all, it avoids a cumbersome way of working with two liquid that are next to each other Phases.
- Houben-Weyl, Methods of Organic Chemistry, Volume IX, p. 77 (1955) in basic medium Disproportionation of disulfides to sulfinates and thiols is known. This unwanted So far, the reaction has been through the use of water-immiscible Tries to suppress solvents. Surprisingly, has shown that this is not necessary.
- the reaction mixture also contained unreacted bis (4-diphenyl) disulfide as a solid.
- the water phase was with 37% by weight aqueous hydrochloric acid acidified, the precipitated product is suctioned off and washed with 100 ml of water. After drying, 21.4 g of 4-mercaptodiphenyl obtained with a content of 93.6 wt .-%. That corresponds to a yield of 53.8% of theory
- reaction mixture was then placed in a 3 liter 80 ml enamel autoclave Sulfur dioxide added and heated to 130 ° C for 3 hours. The was 6 hours Approach stirred at 130 ° C and a pressure of 4.4 to 6.9 bar. Then cooled one to room temperature and relaxed. The suspension obtained was suction filtered. The filter cake was dried. 237.4 g of solid with a content was obtained of bis (4-diphenyl) disulfide at 78.3%. This corresponds to a yield of 91.2% theory
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
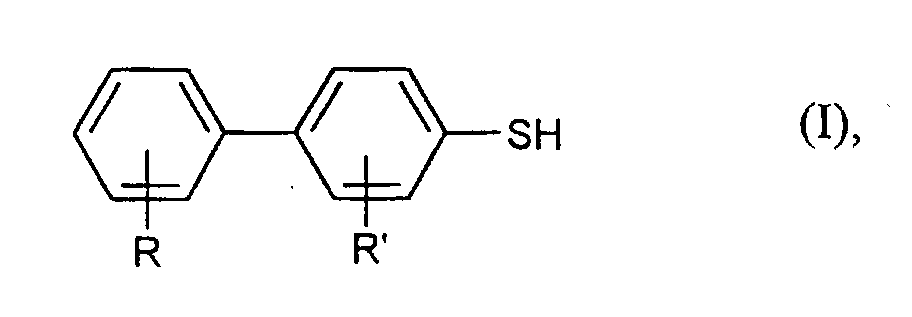
- R und R'
- unabhängig voneinander jeweils für Wasserstoff, C1-C6-Alkyl, C1-C6-Alkoxy oder Halogen stehen,
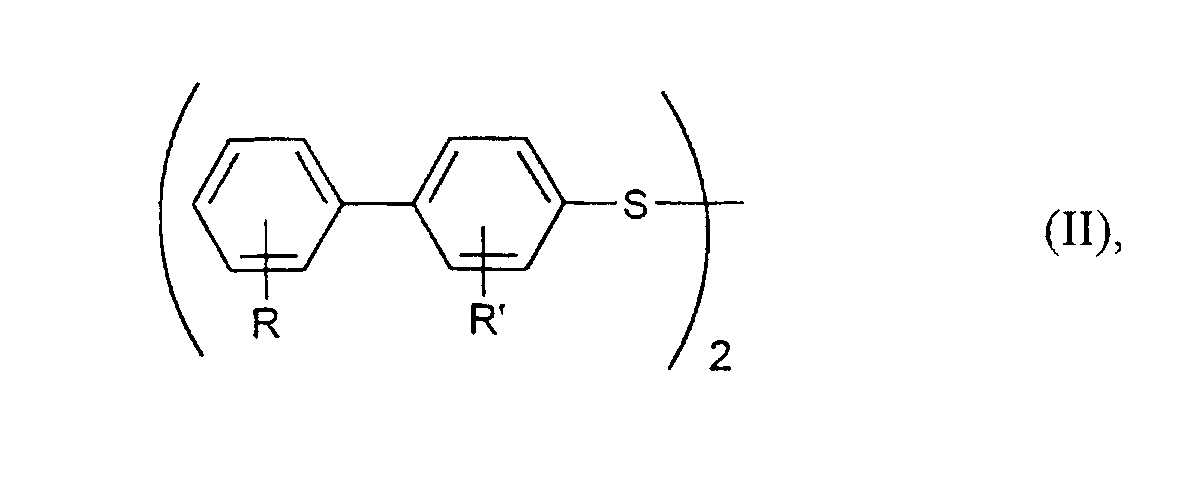
- R und R'
- die bei Formel (I) angegebene Bedeutung haben.
Claims (10)
- Verfahren zur Herstellung von Arylmercaptanen durch katalytische Hydrierung von Diaryldisulfiden, dadurch gekennzeichnet, daß man die Hydrierung in einem basischen alkoholischen Medium durchführt.
- Verfahren nach Anspruch 1, dadurch gekennzeichnet, daß man Arylmercaptane der Formel (I)in der
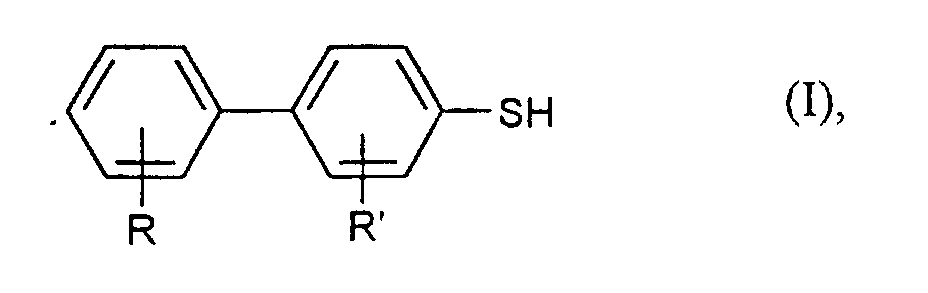
- R und R'
- unabhängig voneinander jeweils für Wasserstoff, C1-C6-Alkyl, C1-C6-Alkoxy oder Halogen stehen,
in der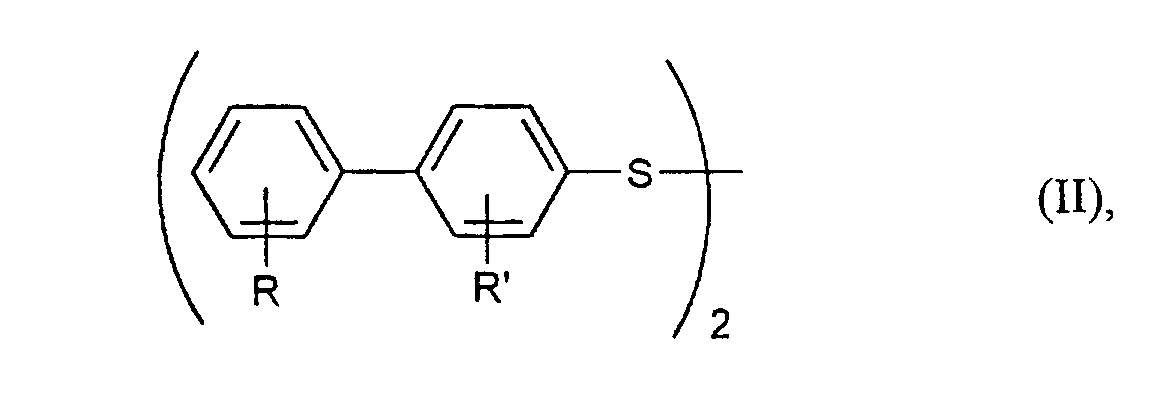
- R und R'
- die bei Formel (I) angegebene Bedeutung haben.
- Verfahren nach Ansprüchen 1 und 2, dadurch gekennzeichnet, daß man als Lösungsmittel zur Bildung des alkoholischen Mediums aliphatische C1-C5-Alkohole einsetzt.
- Verfahren nach Ansprüchen 1 bis 3, dadurch gekennzeichnet, daß das alkoholische Medium bis zu 20 Gew.-% Wasser enthält.
- Verfahren nach Ansprüchen 1 bis 4, dadurch gekennzeichnet, daß man als Mittel zur Erzeugung eines basischen Charakters im alkoholischen Medium Alkalihydroxide, Alkalicarbonate oder Alkalialkoholate oder entsprechende Erdalkaliverbindungen einsetzt.
- Verfahren nach Ansprüchen 1 bis 5, dadurch gekennzeichnet, daß man auf I Mol Diaryldisulfid 0,5 bis 5 Äquivalente Base einsetzt.
- Verfahren nach Ansprüchen 1 bis 6, dadurch gekennzeichnet, daß man als Katalysatoren Metalle der 8. Nebengruppe des PSE einsetzt.
- Verfahren nach Ansprüchen 1 bis 7, dadurch gekennzeichnet, daß man die katalytische Hydrierung bei Temperaturen von 20 bis 200°C und Drucken bis zu 50 bar durchführt.
- Verfahren nach Ansprüchen 1 bis 8, dadurch gekennzeichnet, daß man die katalytische Hydrierung bei 40 bis 150°C und Drucken bis zu 25 bar durchführt.
- Verfahren nach Ansprüchen 1 bis 9, dadurch gekennzeichnet, daß man das nach Durchführung der katalytischen Hydrierung vorliegende Reaktionsgemisch aufarbeitet, indem man es abkühlt, den Katalysator abtrennt, die dann vorliegende alkoholische Lösung des hergestellten Arylmercaptans mit Säure versetzt und das dann ausfallende Produkt abfiltriert und trocknet.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19746512A DE19746512A1 (de) | 1997-10-22 | 1997-10-22 | Verfahren zur Herstellung von Arylmercaptanen durch Hydrierung von Diaryldisulfiden |
| DE19746512 | 1997-10-22 | ||
| PCT/EP1998/006450 WO1999020602A1 (de) | 1997-10-22 | 1998-10-12 | Verfahren zur herstellung von arylmercaptanen durch hydrierung von diaryldisulfiden |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP1027331A1 EP1027331A1 (de) | 2000-08-16 |
| EP1027331B1 true EP1027331B1 (de) | 2002-10-09 |
Family
ID=7846194
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP98955433A Expired - Lifetime EP1027331B1 (de) | 1997-10-22 | 1998-10-12 | Verfahren zur herstellung von arylmercaptanen durch hydrierung von diaryldisulfiden |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US6500988B1 (de) |
| EP (1) | EP1027331B1 (de) |
| JP (1) | JP2001520218A (de) |
| CN (1) | CN1277602A (de) |
| AT (1) | ATE225769T1 (de) |
| AU (1) | AU1227799A (de) |
| DE (2) | DE19746512A1 (de) |
| IL (1) | IL135114A0 (de) |
| WO (1) | WO1999020602A1 (de) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2651882B1 (de) * | 2010-12-16 | 2018-11-14 | F.Hoffmann-La Roche Ag | Verfahren zur herstellung von aromatischen thiolderivaten durch hydrogenierung von disulfiden |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2402641A (en) * | 1940-10-31 | 1946-06-25 | Du Pont | Production of aryl thiols |
| US3326981A (en) * | 1964-02-04 | 1967-06-20 | Universal Oil Prod Co | Process for the preparation of aromatic thiols |
| US3331205A (en) * | 1964-06-22 | 1967-07-18 | Consolidation Coal Co | Preparation of chlorothiophenols |
| DE1768421A1 (de) * | 1968-05-11 | 1971-10-07 | Bayer Ag | Verfahren zur Herstellung von halogensubstituierten aromatischen Mercaptoverbindungen |
| US4006186A (en) * | 1971-04-07 | 1977-02-01 | Bayer Aktiengesellschaft | Process for the preparation of thiophenols |
| US3912757A (en) | 1972-08-28 | 1975-10-14 | Sterling Drug Inc | 1,2,3,4-Tetrahydrodibenzothiophene-1-carboxylates |
| JPS5681541A (en) * | 1979-12-06 | 1981-07-03 | Mitsui Petrochem Ind Ltd | Reduction process |
| AU542420B2 (en) | 1980-02-07 | 1985-02-21 | Richardson-Merrell Inc. | Anti-rhinovirus agents |
| EP0100172B1 (de) | 1982-07-23 | 1987-08-12 | Imperial Chemical Industries Plc | Amide-Derivate |
| JPS60199871A (ja) * | 1984-03-26 | 1985-10-09 | Nippon Kayaku Co Ltd | チオフエノ−ル類の製造法 |
| JP3332561B2 (ja) * | 1994-03-17 | 2002-10-07 | イハラケミカル工業株式会社 | チオアリール化合物の製造方法 |
| DE4420777A1 (de) | 1994-06-15 | 1995-12-21 | Bayer Ag | Verfahren zur Herstellung von 4-Fluorthiophenol |
| US5670504A (en) | 1995-02-23 | 1997-09-23 | Merck & Co. Inc. | 2,6-diaryl pyridazinones with immunosuppressant activity |
| US5728887A (en) * | 1996-04-10 | 1998-03-17 | E. I. Du Pont De Nemours And Company | Catalytic hydrogenolysis of organic thiocyanates and disulfides to thiols |
-
1997
- 1997-10-22 DE DE19746512A patent/DE19746512A1/de not_active Withdrawn
-
1998
- 1998-10-12 WO PCT/EP1998/006450 patent/WO1999020602A1/de not_active Ceased
- 1998-10-12 CN CN98810528A patent/CN1277602A/zh active Pending
- 1998-10-12 DE DE59805923T patent/DE59805923D1/de not_active Expired - Fee Related
- 1998-10-12 IL IL13511498A patent/IL135114A0/xx unknown
- 1998-10-12 US US09/529,875 patent/US6500988B1/en not_active Expired - Fee Related
- 1998-10-12 JP JP2000516944A patent/JP2001520218A/ja active Pending
- 1998-10-12 AT AT98955433T patent/ATE225769T1/de not_active IP Right Cessation
- 1998-10-12 AU AU12277/99A patent/AU1227799A/en not_active Abandoned
- 1998-10-12 EP EP98955433A patent/EP1027331B1/de not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| CN1277602A (zh) | 2000-12-20 |
| US6500988B1 (en) | 2002-12-31 |
| WO1999020602A1 (de) | 1999-04-29 |
| JP2001520218A (ja) | 2001-10-30 |
| EP1027331A1 (de) | 2000-08-16 |
| AU1227799A (en) | 1999-05-10 |
| DE19746512A1 (de) | 1999-04-29 |
| ATE225769T1 (de) | 2002-10-15 |
| DE59805923D1 (de) | 2002-11-14 |
| IL135114A0 (en) | 2001-05-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69508298T2 (de) | Modifizierte Ionaustauscherharze und ihre Verwendung | |
| CH626866A5 (en) | Process for preparing aromatic or heteroaromatic ethers | |
| DE2800324C2 (de) | ||
| DE60108668T2 (de) | Verfahren zur herstellung von phenethylaminderivaten | |
| DE3639752A1 (de) | Verfahren zur herstellung von amphoteren oberflaechenaktiven agentien | |
| EP0671382B1 (de) | Fluorhaltige Carboxylbetaine und Alkylsulfobetaine sowie deren Mischungen mit gesättigten Fluoralkyaminen | |
| EP1027331B1 (de) | Verfahren zur herstellung von arylmercaptanen durch hydrierung von diaryldisulfiden | |
| EP0134200B1 (de) | Verfahren zur Herstellung von Merkaptanen | |
| EP0557835A2 (de) | Verfahren zur Herstellung von Betainen | |
| CH677663A5 (de) | ||
| EP0687671B1 (de) | Verfahren zur Herstellung von 4-Fluorthiophenol | |
| DE3856098T2 (de) | Verfahren zur selektiven Reduktion von 2-Chinoxalinol-4-Oxiden zu 2-Chinoxalinolen | |
| EP0212301B1 (de) | Verfahren zur Herstellung von 2-Aminophenyl-thioethern | |
| EP0051782B1 (de) | Verfahren zur Herstellung von m-halogensubstituierten Anilinen | |
| EP1025081B1 (de) | S-(4-biphenyl)-thioschwefelsäuren und ihre salze, verfahren zu deren herstellung und die herstellung von 4-mercaptobiphenylen | |
| EP0146889A1 (de) | Verfahren zur Herstellung von aromatischen Aminosulfonsäuren | |
| EP0497210B1 (de) | Verfahren zur Herstellung von Dialkylaminopropandiol | |
| EP0290886B1 (de) | Verfahren zur Herstellung von 2,5-Dichlorphenylthioglykolsäure | |
| EP0010244A1 (de) | Verfahren zur Herstellung von 4-Nitro-4'-amino-diphenylamin-2-sulfonsäure und 2-Nitro-4'-amino-diphenylamin-4-sulfonsäure | |
| EP0002691A1 (de) | Verfahren zur Herstellung von 1-Aminonaphthalin-3,6,8-trisulfonsäure | |
| EP0519246B1 (de) | Verfahren zur Herstellung von N,N'-bis-[7-(4-hydroxy-2-sulfo)naphthyl]-harnstoff | |
| DD255941A1 (de) | Verfahren zur herstellung von 5-aminosalicylsaeure | |
| DE3025654A1 (de) | Verfahren zur herstellung von mercaptanderivaten der kohlensaeure | |
| DE2424372B2 (de) | Verfahren zur Herstellung von Isonitrosoacetaniliden | |
| DE3225605A1 (de) | Verfahren zur herstellung von cyclopropanderivaten |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 20000522 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE CH DE ES FI FR GB IE IT LI NL PT SE |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| 17Q | First examination report despatched |
Effective date: 20020306 |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20020913 Year of fee payment: 5 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH DE ES FI FR GB IE IT LI NL PT SE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20021009 Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20021009 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20021009 Year of fee payment: 5 |
|
| REF | Corresponds to: |
Ref document number: 225769 Country of ref document: AT Date of ref document: 20021015 Kind code of ref document: T |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: AT Payment date: 20021010 Year of fee payment: 5 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: NV Representative=s name: E. BLUM & CO. PATENTANWAELTE Ref country code: CH Ref legal event code: EP |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 20021021 Year of fee payment: 5 |
|
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) |
Effective date: 20021009 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: IE Payment date: 20021030 Year of fee payment: 5 Ref country code: BE Payment date: 20021030 Year of fee payment: 5 |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4C Free format text: NOTIFICATION RECEIVED FROM THE EUROPEAN PATENT OFFICE THAT THE PUBLICATION LANGUAGE IS ACTUALLY GERMAN. |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 20021031 Year of fee payment: 5 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D Free format text: GERMAN |
|
| REF | Corresponds to: |
Ref document number: 59805923 Country of ref document: DE Date of ref document: 20021114 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20030109 Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20030109 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20030429 |
|
| EN | Fr: translation not filed | ||
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed |
Effective date: 20030710 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20031012 Ref country code: AT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20031012 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20031013 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20031031 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20031031 Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20031031 |
|
| BERE | Be: lapsed |
Owner name: *BAYER A.G. Effective date: 20031031 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20040501 Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20040501 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20031012 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee |
Effective date: 20040501 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: MM4A |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20051012 |